
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 07/36/01. The contractual start date was in July 2007. The draft report began editorial review in January 2014 and was accepted for publication in November 2014. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Usha Chakravarthy, Simon P Harding and Andrew J Lotery are principal investigators of trials sponsored by Novartis, the manufacturers of ranibizumab. Usha Chakravarthy has attended and been remunerated for attendance at advisory boards for Novartis, Bayer, Neovista, Oraya, Allergan, and Bausch and Lomb, and her employing institution has received payments from Novartis, Bayer, Neovista, Oraya, Alcon and Pfizer. Chris A Rogers has received an honorarium from Novartis for a lecture. The employing institutions of Susan Downes and Andrew J Lotery have received payments from Novartis. Susan Downes and Andrew J Lotery have received honoraria from Novartis for lectures. Andrew J Lotery has attended and been remunerated for attendance at advisory boards for Novartis and Bayer. Barnaby C Reeves has received a fee for teaching from Janssen-Cilag and is a member of the National Institute of Health Research (NIHR) Health Technology Assessment commissioning board and the NIHR Systematic Reviews Programme Advisory Group. James Raftery is a member of the NIHR Editorial Board and the NIHR Journals Library Editorial Group. He was previously Director of the Wessex Institute and Head of the NIHR Evaluation, Trials and Studies Coordinating Centre.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2015. This work was produced by Chakravarthy et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Background and rationale
Introduction
The onset of neovascularisation in age-related macular degeneration (AMD) is accompanied by central distortion and blurring which, when left untreated, intensifies into a dense central scotoma. 1 These visually disabling effects of neovascular age-related macular degeneration (nAMD) are monitored by measuring distance visual acuity (VA), which is a surrogate for central visual function; a drop of ≥ 15 letters in the number of letters read on a logMAR [log(minimum angle of resolution)] letter chart (equivalent to the loss of three lines of letters) is considered to be a visually significant event. 2 Until 2005, all treatments that were considered beneficial in the management of nAMD merely limited VA loss relative to untreated control subjects or natural history. 3,4 Thus, at that time successful treatment was defined in terms of slowing down the rate of VA loss. The most effective treatment for nAMD was verteporfin (Visudyne, Novartis) photodynamic therapy (PDT) which resulted in a reduction of the proportion of patients who suffered a 15-letter loss [three lines on the Early Treatment of Diabetic Retinopathy Study (ETDRS) VA chart] from 69% to 54%. 3,5 The most impressive change that occurred around this time was the introduction of ranibizumab (Lucentis®, Novartis). Ranibizumab is a monoclonal antibody to vascular endothelial growth factor (VEGF), which is a potent mitogen and inducer of permeability in blood vessels. 6 Two pivotal ANCHOR (ANti-vascular endothelial growth factor antibody for the treatment of predominantly Classic CHORoidal neovascularization in age-related macular degeneration) and MARINA (Minimally classic/occult trial of the Anti-VEGF antibody Ranibizumab In the treatment of Neovascular Age-related macular degeneration) clinical trials that established the superiority of ranibizumab as a treatment for nAMD reported their findings. 7,8 At 12 and 24 months, > 90% of eyes treated with ranibizumab (0.5 mg) avoided the loss of three lines of VA compared with < 64% of eyes treated with PDT (the comparator in the ANCHOR trial) or 62% of eyes treated with sham injections (the comparator in the MARINA trial). Even more importantly, eyes treated with ranibizumab showed on average an increase in acuity of between 5 and 10 letters read on an ETDRS vision chart with some 30% achieving 70 letters (Snellen equivalent of 6/12), a level of vision which is compatible with visually demanding tasks such as fluent reading and driving. These results exceeded all expectations, as trials of other therapeutic agents, including the VEGF inhibitor pegaptanib (Macugen, Pfizer),3,4 had shown on average a reduction in acuity in treated eyes of between two and three lines over 24 months.
Although the outcome after ranibizumab therapy was clearly impressive, this was achieved using an intensive dosing schedule of monthly injections of the drug into the vitreous cavity of the eye. The method of administration is an invasive procedure with the attendant risks of infection and iatrogenic eye trauma. In addition, the requirement for monthly attendance over many years poses serious challenges for elderly patients. Furthermore, such intensive treatment regimens also create difficulties in terms of resource implications for health service providers. A small study [PIER: a study of rhuFAB V2 (ranibizumab) in subjects with subfoveal choroidal neovascularization secondary to age-related macular degeneration], which used a less intensive dosing schedule of three 4-weekly injections of ranibizumab 0.5 mg followed by retreatment at fixed 3-monthly intervals, did not yield equivalent VA results as those observed in the MARINA and ANCHOR trials. 9 Although mean VA improved in the PIER study in the first 3 months in a manner similar to that seen in ANCHOR and MARINA, it gradually decreased thereafter, dropping back by 12 months to the mean observed at baseline. By contrast, PrONTO [Prospective Optical Coherence Tomography (OCT) Imaging of Patients with Neovascular Age-Related Macular Degeneration Treated with intraOcular Ranibizumab], another small clinical trial of some 40 patients, suggested that a reduction in treatment frequency could be achieved through rigorous tailoring of treatment to morphological parameters without compromising VA outcomes. 9,10 Taken together, these findings implied that there was a variable need for retreatment among patients, and that a reduction in treatment frequency or alteration of dosing interval would require continuous monitoring and tailoring of therapy. The study design of the ANCHOR and MARINA and other smaller randomised controlled trials (RCTs) that investigated effectiveness of ranibizumab also did not permit conclusions to be drawn about the total duration of treatment required.
Although the role of ranibizumab in the management of nAMD was being investigated, another drug [bevacizumab (Avastin®, Roche)] was identified by a number of small uncontrolled case series as having equivalent visual benefits. 11 Remarkable improvements in acuity and morphological findings following intravitreal injection of bevacizumab were reported by investigators from countries across the world,12,13 primarily because it was available and the cost was more affordable. Bevacizumab (a pan-VEGF monoclonal antibody, as is pegaptanib) is the parent molecule from which ranibizumab was derived and the same manufacturer holds the patents and licences for both drugs. Bevacizumab was licensed for use in colorectal cancer, and the therapeutic dose for systemic administration for this condition is approximately 1000 times greater than that required for intraocular use. Thus clinicians were able to offer a cheap alternative to ranibizumab through unlicensed use of bevacizumab. The IVAN (Inhibit VEGF in Age-related choroidal Neovascularisation) trial was developed to test whether or not visual results obtained with bevacizumab were as good as those obtained with ranibizumab.
By virtue of their ability to inhibit all classes of VEGF, both ranibizumab and bevacizumab have the potential to induce serious ocular and systemic side effects. VEGF is known to have an important growth-promoting role in the retina14 and is also thought to maintain the fenestrated phenotype of the choroidal vasculature. 15 Therefore, there is concern that pan inhibition of VEGF over long periods of time can cause atrophic changes in neural, retinal pigment epithelial (RPE) and vascular cells and tissues, with serious consequences for visual function.
Although no such adverse effects have been detected in clinical trials using VEGF inhibition strategies,4,7–10,16 the data available from trials relate to relatively short periods of follow-up, with few patients having been followed beyond 2 years. Repeated intraocular penetration for drug delivery carries a risk of endophthalmitis, traumatic cataract or retinal detachment. However, as shown by the VISION (pegaptanib for neovascular age-related macular degeneration) clinical trials, adherence to protocols that emphasise sterility and administration of the drug by experienced personnel reduces these risks to acceptable levels. 4
Systemic side effects remain a concern as the pooled findings from ANCHOR and MARINA trials revealed a slight excess of thromboembolic events in the highest dose of ranibizumab groups and a small increase in non-ocular haemorrhages in the treatment groups. 7–9 In addition, circulating antibodies to ranibizumab were discernible in serum samples in a significant proportion of patients who received ocular administration of the drug. 7,8 As bevacizumab had not been tested in a controlled trial environment, there was no orderly collection of information on its potential to cause systemic drug toxicity. Therefore, the IVAN trial also proposed the collection of serum samples from participants immediately prior to the Trial, and at the first post-injection visit, for the assay of VEGF, the levels of VEGF inhibitors themselves and to detect circulating antibodies to the inhibitors.
The IVAN trial also offered the opportunity for the creation of an accompanying biobank of serum and deoxyribonucleic acid (DNA). The recent advances in pharmacogenomics have revealed that genetic variation modifies the therapeutic response to drugs in a number of disease conditions. 17 Thus, there is increasing enthusiasm for linking DNA biobanks to RCTs in which participants are extremely well phenotyped. Trials using VEGF inhibition strategies in cancer have shown that both survival and toxicity are influenced by genetic variation. 18 It is plausible that similar mechanisms may influence visual outcomes following therapeutic VEGF inhibition in choroidal neovascularisation (CNV). The IVAN trial therefore proposed the construction of a DNA biobank to test pharmacogenetic associations between treatment responsiveness and key genetic polymorphisms.
Summary of existing evidence
There was no systematic review of VEGF inhibitors in the treatment of nAMD due to AMD in 2007 when the IVAN trial was conceived. Neither was there a head-to-head comparison of the two main inhibitors of VEGF that were available at that time. There were no data on the minimum treatment frequency/duration that is required to maintain the maximal visual benefit achieved with either of the drugs studied in IVAN, and no trial had compared continuing monthly treatment with early cessation of VEGF inhibition with treatment being restarted if signs of lesion reactivation were detected. Therefore, the research questions that the IVAN study set out to address had not been investigated directly previously.
Importance of the health problem to the NHS
Epidemiological studies have shown that there are some 25,000 incident cases of nAMD each year in the UK. 1 RCTs had demonstrated the substantial benefit of ranibizumab for nAMD. Bevacizumab is considerably cheaper than ranibizumab. The drug costs alone for monthly administration of ranibizumab were estimated to be about £11,000 per patient per year and the cost of assessments and treatment delivery about £2500 per year, with a potential annual cost to the UK NHS of up to £300M per year. The cost-effectiveness of VEGF inhibitor treatment is influenced greatly by the difference in drug cost, with ranibizumab being 20 times the price of bevacizumab. The costs of administering treatment are also high and there were no recommendations about the likely duration of treatment required.
The absence of robust information about the safety of bevacizumab, and uncertainty about treatment frequency for either drug, formed the basis for the alternative treatments in the IVAN trial. 4 Accumulating evidence that susceptibility to nAMD is influenced by the carriage of specific polymorphisms in a number of genes which encode proteins involved in immune mediation and regulation,19–21 and the fact that antibodies to the VEGF inhibitors had been found to develop over time, provided a strong rationale for the establishment of a DNA and serum biobanks in the IVAN trial. While the IVAN trial was being designed, The Comparison of Age-related macular degeneration Treatments Trials (CATT)22,23 was developed in parallel in the USA. With the acquiescence of the funding organisations of the two trials, agreement was reached to share information about the design and conduct of the trials, such as protocols and methods for the collection of adverse events (AEs). This exchange of information allowed CATT22 and IVAN investigators to design the trials to facilitate future meta-analyses of the outcomes of the two trials.
Aims and objectives
The aim of the IVAN trial was to investigate alternative VEGF inhibition treatment regimens for the treatment of nAMD. We hypothesised that:
-
Bevacizumab is not inferior to ranibizumab with respect to the benefits of VEGF inhibition in maintaining/improving VA in eyes with nAMD.
-
Treatment with VEGF inhibition can be ‘safely’ withdrawn at 3 months with monthly review to detect reactivation, that is, criteria for restarting treatment can be prespecified to prevent any difference in average VA compared with continuing monthly treatment.
The IVAN trial was designed as a non-inferiority trial because it was intended to show that bevacizumab was not worse than the active control ranibizumab, as existing historical data suggested that both drugs resulted in similar functional outcomes. We did not hypothesise that bevacizumab would be more effective than ranibizumab with respect to VA. It was clearly not practicable to design an equivalence trial. We chose the non-inferiority margin based on evidence of the effect of the active control (namely ranibizumab), as well as existing data from another study. The drug effects are known to wear off over a 4-week period24 and our study design therefore mandated monthly visits for assessment and retreatment decision-making. Evidence from the pivotal trials and subsequent studies had demonstrated that average VA improved steadily from treatment inception over a period of 3 months. To maximise this benefit, we chose to have a loading phase of 3 months, after which we introduced the opportunity to discontinue treatment if the macula was judged to be fluid free.
The trial had three specific inter-related objectives:
-
To estimate the:
-
relative effectiveness of two VEGF inhibitors, namely ranibizumab and bevacizumab, on visual outcome in patients with nAMD; existing evidence of the benefit of VEGF inhibitors compared with sham treatment precludes inclusion of a sham VEGF inhibition arm
-
effectiveness of more frequent compared with less frequent VEGF inhibition in improving or maintaining visual function, with stringent criteria for restarting treatment to prevent VA loss in patients receiving less frequent treatment
-
cost-effectiveness of the alternative treatment strategies outlined above.
-
In addition to estimating the effectiveness of the treatments on visual outcome, the trial was designed to estimate differences in lesion morphology, quality of life and safety as secondary outcomes.
Chapter 2 Methods
Study design
The IVAN study was a multicentre, randomised controlled factorial trial. The trial is registered as ISRCTN92166560. The research objectives were addressed by randomising participants to one of four combinations of two treatment factors (Table 1). Participants, clinicians and trial personnel were masked to the VEGF inhibitor to which a participant was assigned. Pharmacies dispensed the appropriate drug to the ophthalmic clinic, as a pre-filled syringe for bevacizumab or in the commercially available vial for ranibizumab. We aimed to achieve masking of the drug by using unmasked study teams (ophthalmologists and nurses) who had no role in the outcome assessments in the trial. This method for masking had been used extensively in trials that involve invasive methods of treatment delivery, as it is ethically unjustified to inject a placebo into the eye because of the potential for serious adverse events (SAEs) related to the procedure itself. All assessments and treatment management decisions were made by other trial personnel who were masked to allocation to drug throughout the trial, and to allocation to treatment frequency until after the third treatment. We chose not to mask participants, clinicians and trial personnel to whether or not patients were allocated to continuous (i.e. monthly treatment) or discontinuous treatment (stop at 3 months with reinitiation of therapy if disease reactivation occurred).
| Treatment regimen | Ranibizumab | Bevacizumab |
|---|---|---|
| Continue treatment @ 3 months | A | B |
| Stop treatment @ 3 months | C | D |
Changes to study design after commencement of the study
The only change to the study design was a change to the method of masking the study drug, made in October 2007, before recruitment to the trial began. Bevacizumab was supplied in a pre-filled syringe and ranibizumab was supplied in a commercially manufactured vial. We originally intended that unmasked injectors, with no other role in the trial, would ensure masking. However, nine of the smaller sites did not have enough ophthalmologists to be able to guarantee the availability of an unmasked injector at every visit. For these sites, an unmasked ophthalmic nurse was allowed to draw up ranibizumab into a syringe identical to the pre-filled syringe of bevacizumab. Thus the injecting ophthalmologist was kept masked to the identity of the drug and could therefore be allowed to perform other study procedures. All other trial personnel involved in treatment decisions and outcome assessments remained masked. In December 2007, we further clarified that the injector must be an ophthalmologist; it was always the case that the injections should be performed by an ophthalmologist but we were asked to make this explicit in the protocol by the IVAN Data Monitoring and Safety Committee (DMSC).
Participants
Eligibility criteria
Inclusion criteria:
-
adults of either sex, aged ≥ 50 years
-
newly referred for the treatment of nAMD in the first or second eye
-
best corrected distance visual acuity (BCVA) of ≥ 25 letters measured on a standard ETDRS chart
-
any component of the neovascular lesion (CNV, blood, serous pigment epithelial detachment, elevated blocked fluorescence) involving the centre of the fovea.
Exclusion criteria:
-
previous treatment with PDT or a VEGF inhibitor in the eye being considered for inclusion
-
argon laser treatment to the proposed study eye within the last 6 months
-
long-standing CNV evidenced by the presence of fibrosis in excess of 50% of the total lesion
-
greatest linear diameter of > 6000 μm (equivalent to about 12 disc diameters)
-
presence of thick blood involving the centre of the fovea
-
presence of other active ocular disease causing concurrent vision loss, for example diabetic retinopathy
-
patients with eight or more dioptres of myopia
-
pregnant and/or lactating women
-
women with childbearing potential (i.e. not sterilised or not post-menopausal) who are unwilling to use contraception
-
men with a spouse or partner with childbearing potential unless the participant has agreed to use condoms.
A past medical history of cardiovascular disease (CVD) or cardiovascular comorbidity, for example previous myocardial infarction (MI), stroke or current angina, was not an exclusion criterion. However, such conditions were documented carefully at the time of recruitment, and the potential benefits and harms of treatment were discussed carefully with potential participants.
The eligibility criteria included some items that related to the proposed study eye and some items that related to the person being screened for eligibility. The trial studied only one eye of each participant (eligible ‘study eye’ in an eligible person). If a fellow eye developed nAMD during the trial, it was treated with the optimum locally available treatment.
Settings
Patients were recruited to the IVAN trial from 23 ophthalmic units in NHS hospital trusts.
Interventions
After obtaining written informed consent, participants were allocated to one of four combinations of the two treatment factors, intravitreal injections with ranibizumab or bevacizumab, on either ‘continuous’ or ‘discontinuous’ treatment regimens.
Drug doses were ranibizumab 0.5 mg or bevacizumab 1.25 mg. 8,11,25 Ranibizumab and bevacizumab were procured commercially. Bevacizumab was repackaged in pre-filled syringes in an aseptic manufacturing facility in the Liverpool University Hospitals pharmacy. The potency, sterility and stability of the repackaged bevacizumab aliquots were established prior to the start of the study.
The protocol required all participants to attend monthly (28- to 35-day interval) for clinical examination, OCT and fundus photography. All participants were treated at visits 0, 1 and 2. Participants randomised to the continuous regimen were treated monthly thereafter. Participants randomised to the discontinuous regimen were not retreated after visit 2 unless prespecified clinical and OCT criteria for active disease were met. 26 If retreatment was needed, a further cycle of three doses delivered monthly was required.
The criteria for retreatment were divided into three levels arranged hierarchically (Figure 1).
FIGURE 1.
Criteria for treatment failure/restarting treatment.

If any of these criteria were met, participants in the discontinuous treatment arm were considered to have failed stability criteria and treatment initiated. If treatment was reinitiated a further block of three treatments, given at monthly intervals, was mandated (Figure 2).
FIGURE 2.
Treatment over time by treatment regimen. In continuous groups, VEGF inhibitor is administered at each of 24 visits (from baseline, month 0, to month 23). In discontinuous groups, VEGF inhibitor is administered at each of three visits (from baseline, month 0, to month 2). The patient is then reviewed monthly, receiving a further cycle of three treatments (hatching indicates initiation of a further cycle of treatment) if the patient satisfies any of the criteria for ‘treatment failure’ (see Figure 1). Three different possible treatment courses over time are shown for patients in discontinuous groups. Asterisks (month 24) indicate stopping of treatment in the trial; at this time, the need for treatment will be assessed against guidance in the forthcoming National Institute for Health and Care Excellence (NICE) technology appraisal and evidence from the trial. This figure shows only three potential treatment regimens in the discontinuous treatment arm of the study, any combination from ‘no treatment after the initial 3-month cycle’ to ‘continuous treatment from 24 months’ is possible in this arm of the trial.

Outcomes
Primary outcome
The primary outcome was BCVA, measured as the number of letters read on a standard ETDRS chart. 27 A full assessment of BCVA was carried out at 3-, 6-, 12-, 18- and 24-month visits. BCVA was also measured at every visit but a refraction – to check that the optical correction was carried out only at intermediate visits if the VA had dropped by ≥ 15 letters or a refractive change – was suspected. The primary end point was BCVA after 2 years of follow-up.
Secondary outcomes
-
Clinical measures of vision, namely:
-
Contrast sensitivity Contrast sensitivity is a global measure of macular function. It has been suggested that it represents a better surrogate marker for visual function than BCVA by virtue of the fact that some studies have observed better correlation with patient-reported outcomes. 28
-
Near visual acuity (NVA) NVA is measured in logMAR units using charts that utilise words of specific character sizes and lengths. Therefore, unlike BCVA, which measures VA only at the point of fixation, NVA is also thought to be a better metric of overall macular function. As BCVA and NVA are not perfectly correlated, more information than just acuity is obtained by measuring both. 29
-
Reading index This metric is a derivative of reading speed. Reading speed is a psychophysical test, which measures ability to read a string of words without reference to context. It tests the ability of the eye to scan along a line of words, and this function is impaired if visual deficits are present in the parafoveal retina. Reading speed is measured using a print size subtending a visual angle that is 0.1 logMAR larger than the threshold NVA and expressed in units of words read per minute. The reading index is the reading speed divided by the size of print read and thus makes allowance for the visual angle. 30,31
-
-
Lesion morphology and metrics from angiograms and OCTs: a series of morphological outcomes were generated from independent grading of colour fundus, fluorescein angiographic and tomographic images. The outcomes were either binary (presence or absence) or continuous measures (area or height). The features and measurements recorded are listed by imaging modality, and the accompanying table (Table 2) provides definitions.
-
Colour:
-
presence of haemorrhage
-
presence of fibrosis
-
presence of geographic atrophy (GA).
-
-
Fundus fluorescein angiography (FFA):
-
presence of dye leakage
-
area of active neovascularisation
-
total lesion area
-
area of fibrosis
-
area of atrophy.
-
-
OCT:
-
presence of fluid on OCT
-
foveal:
-
neuroretinal thickness
-
height of subretinal fluid (SRF)
-
height of pigment epithelial detachment (PED).
-
-
At site of maximum retinal pathology:
-
neuroretinal thickness
-
height of SRF
-
height of PED.
-
-
Colour, FFA and OCT:
-
presence of a RPE tear.
-
-
-
Generic and vision-specific health-related quality of life (HRQoL), namely:
-
European Quality of Life-5 dimensions (EQ-5D), three-level version32
-
Health Utilities Index, version 3 (HUI3)33
-
a measure of the impact of macular degeneration on quality of life [Macular disease Dependent Quality of Life (MacDQoL)]34
-
a measure of treatment satisfaction in patients with macular degeneration [Macular disease Treatment Satisfaction Questionnaire (MacTSQ)]. 35
-
-
Survival free from treatment failure (i.e. satisfying one or more of the criteria for retreatment).
-
Resource use.
Best corrected visual acuity was recorded at every visit; other clinical measures of vision were measured at baseline and visits 3, 6, 12, 18 and 24 only. Research OCT and colour images were done every 3 months and FFA was done at baseline, visits 12 and 24. EQ-5D was measured at baseline, and visits 3, 12 and 24, and MacDQoL and MacTSQ at visits 3, 12 and 24.
Adverse events
Adverse events, both serious and non-serious, were recorded at each visit and coded using Medical Dictionary for Regulatory Activities (MedDRA, McLean, VA, USA) version 14.1. All SAEs were reviewed by senior clinicians (UC, SPH, SD and AJL), masked to treatment allocation. The primary safety outcome was the occurrence of an arteriothromboembolic event [arterial thrombotic event (ATE), as defined by the Antiplatelet Trialists’ Collaboration36] or hospital admission for heart failure (HF).
Changes to study outcomes after commencement of the study
Heart failure was added as a SAE at the request of the DMSC in February 2008, and RPE tear, which is part of the natural history of AMD and had been inadvertently missed, was added as an expected ocular adverse event (AE) in June 2008.
The lesion morphology variables and metrics from angiograms and OCTs were not specified explicitly in the study protocol, but the measures to be compared, their definition and derivation were defined prospectively, before any analyses of these measures were undertaken.
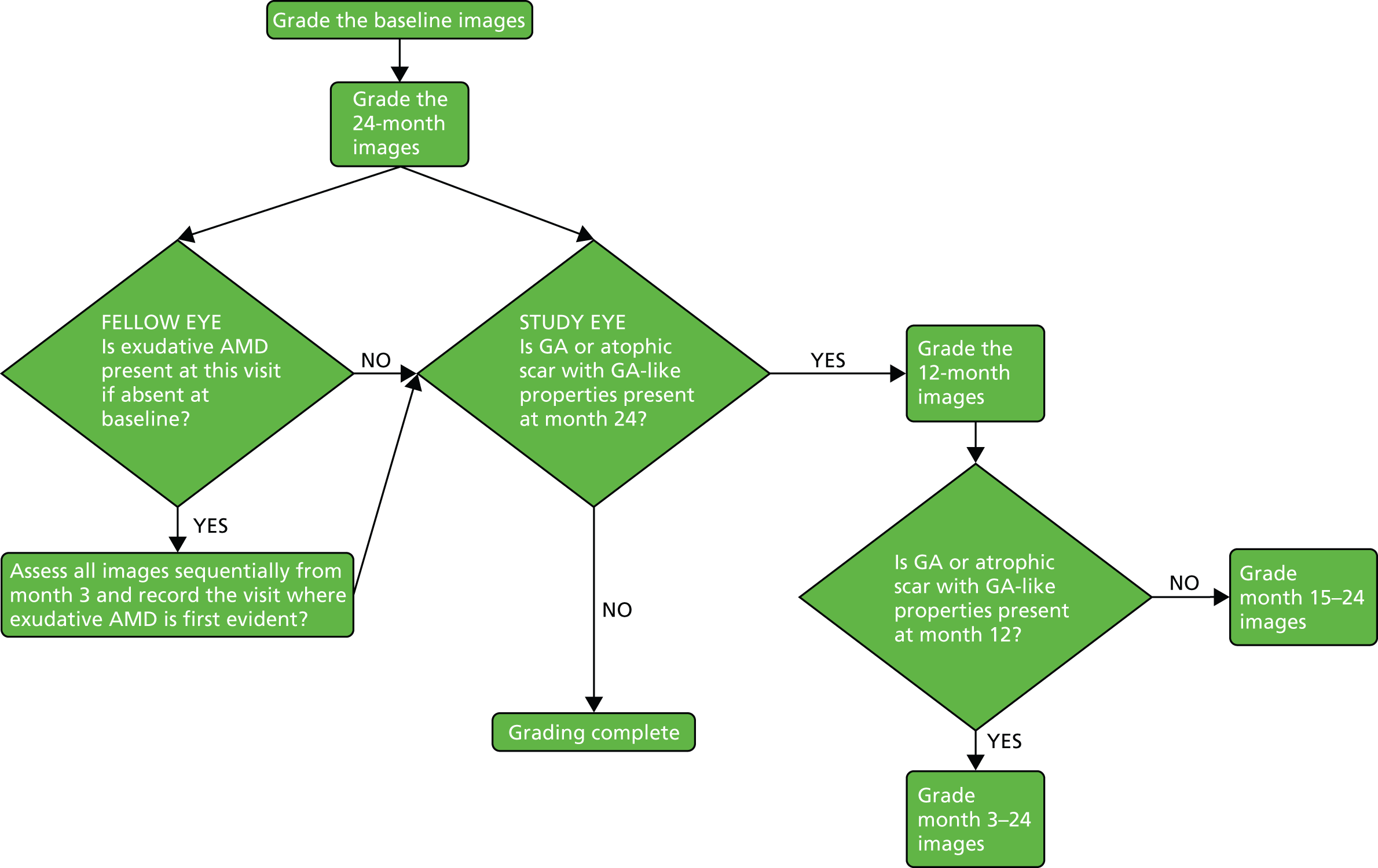
Prompted by the findings from the CATT at 2 years,22 colour and OCT images at baseline and most recent available follow-up were regraded specifically to identify any new GA lesions – in either the study or fellow eye – that had developed during the trial (Figure 3). This outcome was not specified in the study protocol.
FIGURE 3.
New GA assessment diagram.
Sample size
With respect to the primary outcome (BCVA), the trial is designed to answer non-inferiority questions.
Objective I: to compare the clinical effectiveness of the drugs
A small target difference was required to determine whether or not bevacizumab is inferior to ranibizumab. We considered non-inferiority margins of 3 or 4 letters [0.20 to 0.30 standard deviations (SDs), considered ‘small’37]. To help contextualise these differences, the 95% limits of agreement for test–retest of VA among people with impaired but stable vision is 10 letters. 38 We also considered analyses using one or two VA measures (i.e. two post-randomisation repeated measures), adjusted for a baseline measure of VA in both cases. We made the following additional assumptions:
-
The drug comparison (ranibizumab vs. bevacizumab) would combine data from continuous and discontinuous groups.
-
SD for BCVA = 14 letters [based on baseline visual acuities for treated eyes in the Verteporfin PhotoDynamic Therapy cohort study (VPDT), restricting to acuities ranging from 85 (6/6) to 25 (6/60) letters].
-
90% power, 2.5% significance (appropriate when testing two dimensions of the factorial design), one-sided test (appropriate for a non-inferiority research question).
-
Correlations between baseline (pre-) and follow-up (post) acuities = 0.5, and between-follow-up acuities (post 1 and post 2) = 0.8; both of these correlations were calculated from longitudinal VA data collected at 0, 6 and 12 months for patients treated with PDT at baseline in the VPDT.
-
Table 3 gives the sample size for each of the ranibizumab and bevacizumab groups being compared [i.e. for cells (A + C) and (B + D) in Table 1]. On the basis of these calculations, we planned to recruit 150 participants to each of the four cells described in Table 1, providing a sample size of 300 vs. 300 (fewer participants lost to follow-up) for the comparison between drugs. No adjustment was made for dropout, estimated to be < 10% per year. 7,8 Mixed models for analysis of repeated measures can include any patient with at least one ‘post’ outcome measure, although missing data increase the standard errors (SEs) for parameters estimated by the models.
| Equivalence margin | One pre measure, one post measure | One pre measure, two post measures |
|---|---|---|
| < 3 letters | 688 | 596 |
| < 4 letters | 388 | 168 |
The same sample size calculations and assumptions applied to other clinical visual function measures, for which non-inferiority was hypothesised. The calculations and assumptions also applied to HRQoL, treatment satisfaction, and resource use/cost (see Objective III: to estimate the cost-effectiveness of the treatment strategies) except that the comparisons were two-sided, that is, the study was able to detect a difference of 0.20–0.30 SDs with 90% power and 5% significance, as we hypothesised superiority of bevacizumab for these outcomes.
Objective II: to compare the clinical effectiveness of the treatment regimens
Like objective I, this objective was also one of non-inferiority, as the main concern was that people allocated to discontinuous treatment at 3 months do not suffer a visual function disadvantage. The comparison of discontinuous vs. continuous VEGF inhibition (after the first 3 months) combined data for groups allocated to ranibizumab and bevacizumab [i.e. cells (A + B) vs. (C + D) in Table 1]. Other assumptions set out above for objective I also applied to this objective. Therefore, these comparisons had the same sample size as for objective I and hence the same power.
Objective III: to estimate the cost-effectiveness of the treatment strategies
This objective had the same sample size as for objective I and hence the same power but at a significance level of 5% (two sided; see above).
Review of the sample size
In October 2009, when recruitment to IVAN was significantly slower than anticipated and 198 of the planned 600 participants had been recruited, a review of the assumptions underpinning the sample size calculation was undertaken (without reference to the group allocation). The results of the review (which suggested that, for the continuously scaled outcomes a total sample size of 400 could be sufficient) were presented to the Trial Steering Committee (TSC) and DMSC. After much discussion, both committees recommended that the trial should continue to the planned recruitment of 600 patients, and accepted the implications of extended time and costs. A request for a costed extension was subsequently submitted and approved by the National Institute for Health Research (NIHR).
Interim analyses
A formal prespecified interim analysis was undertaken when all participants had been followed up to 1 year. 26 No adjustment to the sample size and significance levels were made for this interim analysis.
Randomisation
Randomisation to one of the four treatment arms was stratified by centre, and blocked to ensure approximately equal numbers of participants per group within a centre. Allocations were generated by computer, in advance of starting the trial, by the coordinating centre, and concealed using an internet-based system provided by Sealed Envelope Ltd (London, UK). Staff in participating centres gained limited access to the system using a password. Information to identify a participant uniquely and to confirm eligibility had to be entered before the system assigned a study number and hence the randomised treatment allocation was determined. This study number was then written onto all patient case report forms (CRFs) and also on the prescription for study drug issued to the local pharmacy. The prescription for each participant, together with a printout from the randomisation system, was then sent to the local pharmacy. The local pharmacy staff took the prescription and looked up the prescription log sheet for that participant according to study number. The prescription log, which linked study number to drug allocation, was prepared by Bristol Clinical Trials and Evaluation Unit (CTEU) and sent to the local pharmacy as part of site initiation, before recruitment at the site started.
Investigators and patients were masked to treatment regimen (continuous/discontinuous) until all data for visit 2 had been submitted to Bristol CTEU. CRFs for visit 3 onwards were customised for a participant, depending on the allocated treatment regimen. Participant-specific CRFs folders were made up by the Bristol CTEU and sent to the centre at which a participant was being treated. Stickers were also added to the spine of the CRF folder, identifying the treatment regimen to which a patient had been allocated (green for continuous treatment, red for discontinuous regimen).
Masking
Participants, clinicians and trial personnel were masked to the VEGF inhibitor to which a participant was assigned throughout the trial, and to the allocation of treatment frequency until after the third treatment. We chose not to mask participants, clinicians and trial personnel to the continuous or discontinuous treatment regimen after the first 3 months, on grounds of pragmatism and practicability. We were interested in the effects of discontinuous treatment on treatment satisfaction. Masking treatment regimens would have required sham injections to have been ‘given’ to patients allocated to the discontinuous regimen on visits when treatment was not required.
Local pharmacies dispensed the appropriate drug to the ophthalmic clinic, as a pre-filled syringe for bevacizumab or in the commercially available vial for ranibizumab. We intended that drug allocation should be concealed by having separate masked assessment and unmasked treating teams. The study drug was transported to clinic in a concealed container, which was then opened and the drug prepared by the injecting ophthalmologist, out of sight of other study staff and participants. The injecting ophthalmologist was not involved in any aspect of the study, and the drug allocation was not revealed to the participant or to any other members of the study team. This system was achieved by 14 sites.
At the other nine sites, staffing levels could not support this system and an unmasked staff member (usually the pharmacist or a research nurse) prepared the injection. Ranibizumab was prepared in a syringe identical to those containing bevacizumab, and any drug labels were replaced with a label containing the participant’s IVAN study number. The unmasked staff member presented this to the masked injector, while ensuring that the original packaging and labels remained out of sight. The syringes were supplied by the IVAN trial, obtained from the same source supplying syringes to the manufacturing pharmacy preparing pre-filled syringes of bevacizumab. Those responsible for preparing the injections did not perform assessments.
To assess the adequacy of masking, ophthalmologists and participants stated at visits 3, 12 and 24 (and at exit visits if participants withdrew early), whether or not they knew the allocated drug (don’t know/Lucentis/Avastin).
Lesion morphology was assessed by independent graders, masked to drug and treatment regimen, in the UK Network of Ophthalmic Reading Centres. Because independent assessment of lesions could not be done immediately, some randomised participants were subsequently found to be ineligible.
Data collection
Data collection in the IVAN study was performed over a 2-year period, during which time patients attended 25 monthly visits. Most data were collected from patients using purpose-designed CRFs at these visits (Table 4 shows the schedule of data collection).
| Treatment/assessment | Follow-up month/visit number | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | Exit | |
| Demography | ✓ | |||||||||||||||||||||||||
| Height | ✓ | |||||||||||||||||||||||||
| Weight | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ||||||||||||||||
| Past medical history | ✓ | |||||||||||||||||||||||||
| Cardiovascular symptoms (CCS, NYHA, ankle swelling/HF) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a |
| Blood pressure | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ||||||||||||||||
| Binocular acuity | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ||||||||||||||||||
| Refraction (sphere, cylinder, axis) | ✓ | ✓b | ✓b | ✓ | ✓b | ✓b | ✓ | ✓b | ✓b | ✓b | ✓b | ✓b | ✓ | ✓b | ✓b | ✓b | ✓b | ✓b | ✓ | ✓b | ✓b | ✓b | ✓b | ✓b | ✓ | ✓a |
| Distance BCVA (letters read)c | ✓ B | ✓ S | ✓ S | ✓ B | ✓ S | ✓ S | ✓ B | ✓ S | ✓ S | ✓ S | ✓ S | ✓ S | ✓ B | ✓ S | ✓ S | ✓ S | ✓ S | ✓ S | ✓ B | ✓ S | ✓ S | ✓ S | ✓ S | ✓ S | ✓ B | ✓a B |
| NVA (logMAR)c | ✓ B | ✓ B | ✓ B | ✓ B | ✓ B | ✓ B | ✓a B | |||||||||||||||||||
| Reading index (words/minute/print size)c | ✓ B | ✓ B | ✓ B | ✓ B | ✓ B | ✓ B | ✓a B | |||||||||||||||||||
| Contrast sensitivity (letters)c | ✓ B | ✓ B | ✓ B | ✓ B | ✓ B | ✓ B | ✓a B | |||||||||||||||||||
| Imaging (OCT, photos) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a |
| Imaging (FFA) | ✓ | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓ | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓d | ✓ | ✓a | ||
| Medications | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a |
| Ocular history | ✓ | |||||||||||||||||||||||||
| Ocular examination (IOP, slit lamp, cataract grading, posterior segment abnormalities) | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a |
| Ocular examination (anterior segment, lens) | ✓ | |||||||||||||||||||||||||
| Eligibility for IVAN | ✓ | |||||||||||||||||||||||||
| Serum sample | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||||||
| Blood sample (genetics) | ✓ | |||||||||||||||||||||||||
| Injection (date, see CF, IOP, antibiotics) | ✓ | ✓ | ✓ | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ✓e | ||
| Adverse reaction | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| AEs | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓e |
| Stopping rules/retreatment criteria | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓e | |||
| EQ-5Df | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||||||||
| HUI3f | ✓ | ✓ | ✓ | ✓ | ||||||||||||||||||||||
| MacDQoL | ✓ | ✓ | ✓ | |||||||||||||||||||||||
| MacTSQ | ✓ | ✓ | ✓ | |||||||||||||||||||||||
| Ocular symptoms – use of medical services | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | |
| Non-ocular symptoms – use of medical services | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | |
| Travel arrangements | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | |
| Masking | ✓ | ✓ | ✓ | ✓a | ||||||||||||||||||||||
| Reasons for withdrawal | ✓ | ✓ | ✓a | |||||||||||||||||||||||
Blood samples (1 × 10-ml ethylenediaminetetraacetic acid tube) for genetic analysis were collected at visit 0 (baseline) and sent to the Gift of Sight Research Centre in Southampton for storage and subsequent analysis. Serum samples (2 × 6-ml serum separator tubes) were collected at visits 0, 1, 6, 11, 12, 18, 23 and 24; the samples at visits 6 and 18 were instituted only for participants recruited later in the trial after observing marked changes in mean VEGF levels between visits 1 and 11 in participants recruited earlier in the trial. 39 If the participant did not attend one of these visits, samples collected at the following visit were sent instead. These samples were sent to the Centre for Vision Science at Queen’s University Belfast for serological analysis. All samples were sent using the Royal Mail Safebox system (guaranteed next-day delivery).
Colour photographs and OCTs were captured every 3 months, and FFA was captured at baseline, 12 and 24 months. Images were submitted to the Central Angiographic Resource Facility (CARF) for analysis. If the criteria for treatment failure were met and a decision to restart treatment was made (for participants allocated to the discontinuous treatment regimen) at an intervening visit between the 3-monthly visits, the colour photographs and OCT images captured at the visit were submitted to CARF. The criteria for treatment failure were also applied to participants who were allocated to the continuous treatment regimen, even though the resulting data did not influence the decision to treat (as these participants were treated at every visit); the protocol required the colour photographs and OCT images captured for these participants at these visits also to be submitted to CARF. If a FFA was required to complete the assessment of criteria for retreatment, the FFA was also submitted to CARF. If a patient withdrew from the study during a study visit, an exit visit was undertaken. As part of this exit visit, OCT and FFA were taken and submitted to CARF.
Participants were asked to complete four HRQoL questionnaires and one questionnaire about symptoms since the last visit (to ascertain adverse effects and resource use relating to the symptoms). The EQ-5D, HUI3 and symptoms questionnaire were completed during the study visit. The EQ-5D and HUI3 were administered at visits 0, 3, 12 and 24, and when a SAE had occurred since the previous visit. Collection of participant responses to MacDQoL and MacTSQ questionnaires was coordinated centrally. A member of the coordinating centre study team contacted participants by telephone approximately 2 weeks after visits 3, 12 and 24.
A bespoke trial database was designed using Microsoft SQL Server 2008 (Microsoft Corporation, Redmond, WA, USA). The database was intended to act as both a data storage facility and a trial management resource. For example, the sites scheduled appointments using the database and the database issued reminders when the telephone questionnaires were due. Activity reports were generated based on data entered into the database, enabling the management of payments to sites (see Contractual and financial arrangements). The database also provided a facility for tracking the progress of SAE reporting.
Owing to the large sample size and number of scheduled visits, a considerable amount of data validation was applied to the database. The validation rules were determined as a result of detailed discussions between clinical trial coordinators, research nurses, statisticians and database developers working on the study, and were refined following any feedback from sites. Validation broadly included rules such as ensuring that:
-
the VA was recorded at all required time points and entered correctly; for example, the average value of VA was calculated at each time point and any value entered that deviated significantly from the average was queried to confirm it was correct
-
injections were given in accordance to the trial protocol; for example, a query would be raised if a patient on discontinuous treatment entered a 3-month treatment cycle and then failed to complete it
-
AEs and SAEs were raised when required and that the event was followed through to completion.
Automated links between the IVAN database and the CARF system were set up so that queries could be raised for any missing images and to check that images received at CARF were matched with the correct event.
Statistical methods
Analyses of the primary and secondary efficacy outcomes were carried out on the basis of treatment allocation, which was consistent with the analysis of CATT. 22,23 The analysis population (and the safety population) consisted of all randomised patients who received at least one injection. Patients who were not treated or withdrew and were unwilling for data already collected to be used were excluded. Reporting guidelines recommend that non-inferiority hypotheses and safety data are analysed by the treatment received. 40,41 As IVAN is a masked trial with respect to drug allocation, the drug treatment received should equal the treatment allocated. If the drug received differed by visit (i.e. the wrong treatment was given on one or more occasions), the patient was grouped according to the drug received with greatest frequency. Grouping patients according to the amount of treatment received was not straightforward, so for consistency of reporting, and on the recommendation of the IVAN DMSC, patients were grouped according to the allocated treatment frequency.
Most outcomes were collected for the study eye (e.g. BCVA, other measures of visual function, lesion morphology) but other outcomes were collected for the participant, notably data concerning systemic AEs/SAEs and HRQoL. The unit of analysis for the former was, therefore, study eye, and for the latter it was participant.
Continuous variables were summarised using the mean and SD [or median and interquartile range (IQR) if the distribution is skewed], and categorical data were summarised as a number and percentage. The primary outcome, BCVA, and other continuously scaled outcomes measured at multiple time points, were analysed using linear mixed-effects methods, incorporating parameter estimates for the mean baseline response where measured and for each treatment at each follow-up time (i.e. saturated model). Baseline and subsequent values were modelled jointly to avoid the necessity to exclude or impute values for cases with missing pre-treatment measures. Binary outcomes were compared using logistic regression, adjusted for baseline values where measured, with treatment estimates presented as odds ratios (ORs). Formal statistical comparisons were performed only if at least 10 participants in total experienced the outcome. Time from completion of the loading dose of three treatments to the first treatment failure was analysed using Cox proportional hazards regression, with treatment comparisons presented as hazard ratios (HRs) and described graphically using Kaplan–Meier plots. The Cox model was also adjusted for whether or not the patient had completed the full cycle in the loading phase (i.e. received three injections at visits 0, 1 and 2). Assumptions underpinning the statistical models were checked using standard methods (e.g. residual plots, tests for normality or for proportional hazards). If the assumptions were not satisfied then transformations were explored. Outcomes analysed on a logarithmic scale were transformed back to the original scale after analysis, and results presented as geometric mean ratios (GMRs). Outlying observations that meant models did not fit the data adequately were excluded from analyses.
For EQ-5D, lesion area and MacTSQ at 2 years, no suitable transformation could be found and so data were dichotomized (EQ-5D score, 1 vs. < 1; lesion present vs. absent; MacTSQ < median TSQ score over all time points vs. ≥ median TSQ score over all time points). For MacDQoL, the outcome was transformed from original scale of –9 to +3 to a scale of –3 to +9), and analysed using a log transformation. The GMR is, therefore, interpreted as the GMR of the MacDQoL score, and not of the MacDQoL score directly.
The interaction of VEGF inhibitor and treatment frequency was tested, and differences between ranibizumab and bevacizumab were to be reported separately for the continuous and discontinuous treatment arms only if the interaction term reached statistical significance (two sided) at the 5% level for outcomes for which the results from the CATT22 suggested possible interaction (i.e. OCT measures of total retinal thickness at the fovea and fluid, and presence of fluid on OCT) or at the 1% level for other outcomes (chosen to reduce the type I error rate); however, this level of significance was not reached in any of the models. As the interaction was not statistically significant, the main effects of ranibizumab vs. bevacizumab and of continuous vs. discontinuous treatment after 2 years were reported. Likelihood ratio tests were used in preference to Wald tests for hypothesis testing.
All treatment comparisons at 2 years are presented as effect sizes with 95% CIs. For tests of superiority, two-sided p-values of < 0.05 are considered to be statistically significant. For tests of non-inferiority, bevacizumab was considered inferior to ranibizumab, and discontinuous treatment inferior to continuous treatment if the lower limit of the 95% confidence interval (CI) for the difference between groups was < –3.5 letters (inferiority margin set at 3–4 letters). No formal adjustment was made for multiple testing. When interpreting the results, consideration has been given to the number of statistical tests performed.
The intention was to adjust all models for study centre. However, for some low-frequency outcomes (e.g. safety) it was anticipated that this would not be feasible, so, for consistency, analyses were adjusted for centre size, fitted as a fixed effect (see Appendix 5).
Best corrected distance visual acuity in the study eye, and several of the secondary outcomes, were measured at all visits (see Table 4). However, the analysis of BCVA included only the ‘main’ study visits, namely visits 0, 3, 6, 9, 12, 15, 18, 21 and 24. If the outcome was missing for one of these visits, but was recorded at the next scheduled visit (i.e. 1 month later), this measurement was used in place of the missing value. When this occurred, an indicator was included in the model (and retained, if statistically significant at the 5% level) to denote that the measurement was taken 1 month later than intended.
For BCVA, if a participant could not read any letters from the chart, the following scores were assigned;
-
‘counting fingers’ 0 (which equates to no letters read at 1 m)
-
‘hand movements’ –15
-
‘perception of light’ –30.
The scores of –15 and –30, which equate to a doubling of the visual angle, were chosen to allow the assignment of arbitrary points in deteriorating visual function to these categories.
The first step in assessing reading speed is to choose a Belfast chart that is appropriate for the patient’s near vision. On occasions the incorrect chart was used. When a chart one size larger or smaller than the correct size (based on the recorded NVA) was used, the data were used to calculate the reading index and were included in the analysis. If the chart was more than one size larger or smaller than the correct size, the data were treated as missing and the reading index was not calculated. Participants with a NVA logMAR of 1.6 have vision, which is too poor for their reading ability to be measured, and, therefore, in the majority of cases reading ability was not assessed. The reading index for these participants was imputed between 0.4 and 2.5 on the lognormal scale. This imputation method was chosen based on the distribution of values for participants with a NVA logMAR of 1.6, who did have their reading ability assessed.
Several morphological outcomes were derived from colour, FFA and/or OCT measurements. Details of how these measures were derived are described in Appendix 5. For all patient-reported outcome data, standard rules have been used to derive outcome measures (see Appendix 5). EQ-5D scores are reported in two sections: the first is the EQ-5D utility index, derived from responses to the five ordinal questions, and the second is the visual analogue (‘thermometer’) scale. Using the MacDQoL questionnaire, an average weighted impact score is derived from 22 out of the 23 questions (the question regarding work was excluded, as it was irrelevant to the majority of participants). The MacTSQ questionnaire was used to derive a single treatment satisfaction score comprising 12 questions.
Pre-planned subgroup analyses were defined as follows:
-
Baseline VA in study eye (< 55 vs. ≥ 55 letters read).
-
Baseline CNV size (< 6 vs. ≥ 6 disc areas).
-
Proportion of classic CNV (< 50% vs. ≥ 50%).
-
Presence of retinal angiomatous proliferation (RAP).
-
Fellow eye status (< 75 vs. ≥ 75 letters read). Differences between subgroups were tested by adding subgroup by treatment interaction terms to the model.
Two pre-planned sensitivity analyses of the primary outcome were carried out. First, we excluded measurements taken 1 month later, when the main study visit was missed. Second, we included data only for the study visits at which all functional outcomes were assessed (visits 0, 3, 6, 12, 18 and 24).
In all tables missing data are indicated by footnotes. Imputation for missing data, except as described above, was not used, as few data were < 20% as prespecified in the statistical analysis plan (SAP).
Fixed-effects meta-analyses were performed to combine the results from the IVAN trial with other head-to-head trials comparing bevacizumab and ranibizumab. There was no intention to perform a systematic review. These meta-analyses were carried out to place the IVAN trial findings in the context of the wider evidence base. Other head-to-head trials – namely the CATT,22 GEFAL (French Evaluation Group Avastin versus Lucenti),42 MANTA (Multicenter ANti-VEGF Trial in Austria),43 LUCAS (LUcentis Compared to Avastin Study), Subramanian et al. ,44 and BRAMD [comparison of bevacizumab (Avastin) and ranibizumab (Lucentis) in exudative age-related macular degeneration; unpublished data, presented at the 2013 meeting of European Society of Retina Specialists] trials – were identified from non-systematic searches during the course of the IVAN trial, through contacts with chief investigators of these trials and expert knowledge of the ophthalmologists in the research team.
Fixed-effects models were used because we judged that the populations (people/eyes with nAMD), the settings (specialist ophthalmology outpatient clinics) and the interventions (ranibizumab and bevacizumab) being studied across trials were relatively homogeneous. This was not the case for comparisons of treatment regimen, but, as only the IVAN trial and CATT22 contributed to these analyses, the fixed-/random-effects distinction is irrelevant. No sensitivity or subgroup analyses were planned within the meta-analyses.
The availability of outcome data differed across the trials. Where available, 2-year data have been used, otherwise data up to the 1-year point have been included. The following meta-analyses were performed: change in BCVA from baseline, safety outcomes (mortality, ATEs, one or more SAE and gastrointestinal disorders), change in total retinal thickness at the fovea from baseline, and new GA. Safety outcomes by treatment regimen were available only for the CATT22 up to 1 year (after this time point, participants allocated to monthly treatment in year 1 were rerandomised to monthly or pro re naba (prn) treatment in year 2).
All statistical models were fitted in SAS version 9.3 (SAS Institute Inc., Cary, NC, USA). All other analyses and data management were performed in Stata version 12.0 (StataCorp LP, College Station, TX, USA).
Health economics
Aims and research questions
The economic evaluation aimed to estimate the incremental cost and incremental cost-effectiveness of continuous and discontinuous regimens of bevacizumab and ranibizumab. Cost-minimisation analysis (CMA) was used to compare bevacizumab and ranibizumab in the absence of a clinically meaningful difference in quality-adjusted life-years (QALYs), while cost–utility analysis (CUA) was used to compare continuous and discontinuous treatment.
Analysis perspective
The economic evaluation took a NHS cost perspective, in accordance with recommendations by the National Institute for Health and Care Excellence (NICE). 45 The perspective for outcomes comprised the patients undergoing treatment. Costs incurred by patients, their families or employers, and QALYs accrued by carers or families, were, therefore, excluded from the analysis.
Factorial design
As the IVAN trial is factorial (see Table 1), it is important to consider the likelihood of interactions between anti-VEGF drug and treatment regimen, that is to evaluate whether or not the difference between the two treatment regimens is likely to differ between bevacizumab and ranibizumab. A priori, there is no reason to expect interactions for VA, particularly given the non-inferiority design and previous research suggesting that interactions are generally unlikely unless both factors influence outcomes. 46,47 Nonetheless, interactions for AEs, HRQoL, survival or costs could occur if the number of injections required in the discontinuous groups differs between drugs (e.g. due to differences in pharmacokinetic properties48–50).
However, the effect of treatment regimen will differ between drugs for anti-VEGF cost, as total drug cost equals cost per dose multiplied by number of doses. When drug costs are analysed on a natural scale, this produces a very large interaction for total costs and cost-effectiveness. For example, if bevacizumab costs £49 per dose and ranibizumab costs £742.17 per dose,51 and, on average, patients receiving discontinuous treatment received 10 doses over the 2-year trial period vs. 24 for continuous treatment, the interaction would equal £9704 (£17,812 – £7422 – £1176 + £490), indicating that the cost of continuous ranibizumab is £9704 higher than what we would expect if the effect of drug and treatment regimen were additive (i.e. had no interaction).
In contrast with the clinical analysis, mean costs and QALYs for each cell in the factorial design are interpreted on an ‘inside the table’ basis,52 considering the four cells of the factorial design (see Table 1) as mutually exclusive alternatives. However, when estimating costs and QALYs, we included only interaction terms that were either statistically significant (p < 0.05) or larger than the main effect of drug or treatment regimen. The estimated costs and QALYs for each cell are used to draw conclusions about relative costs and cost-effectiveness for the following four pairwise comparisons:
-
continuous ranibizumab compared with discontinuous ranibizumab
-
continuous bevacizumab compared with discontinuous bevacizumab
-
continuous ranibizumab compared with continuous bevacizumab
-
discontinuous ranibizumab compared with discontinuous bevacizumab.
Form of analysis and primary outcome measure for economic analyses
Cost–utility analysis and CMA are commonly used frameworks for economic evaluation. 53,54 In CUA, health outcomes are measured in QALYs and the difference in cost between two treatments is divided by the difference in the number of QALYs accrued, to calculate the cost per QALY gained. In CMA, it is implicitly assumed that the treatments have identical health outcomes; treatments are, therefore, compared based on cost alone, and the cheapest strategy is considered best value for money.
The IVAN trial was designed as a non-inferiority study and no differences in BCVA between drugs or treatment regimens were expected. However, it was anticipated that differences in side effects or patient experience between different injection frequencies could translate into QALY differences between drugs or between treatment regimens, even in the absence of differences in VA. Furthermore, even where non-inferiority has been demonstrated, conducting CMA (and therefore assuming that the difference in QALYs is exactly zero) can bias the conclusions and estimates of uncertainty. 54 However, such bias is unlikely if the difference in cost is so large that no plausible difference in efficacy could cause the more costly treatment to be cost-effective.
In the IVAN trial, the large difference in drug costs is likely to drive conclusions about the incremental cost-effectiveness of ranibizumab compared with bevacizumab. Using CMA to compare ranibizumab and bevacizumab is, therefore, highly unlikely to bias conclusions or uncertainty estimates if bevacizumab were non-inferior to ranibizumab for QALYs. We therefore prespecified a non-inferiority margin for the comparison between ranibizumab and bevacizumab that determined whether or not conclusions would be based on a comparison of costs (CMA) or a full economic evaluation (CUA). The non-inferiority margin comprised 0.025 EQ-5D QALYs per patient-year, which is the smallest difference in health-state valuations that could be measured in the study estimating the tariff for the EQ-5D utility measure. 55,56 Conclusions about whether or not ranibizumab is cost-effective compared with bevacizumab were, therefore, based on CMA, unless the mean QALY difference between continuous (or discontinuous) ranibizumab and continuous (or discontinuous) bevacizumab was ≥ 0.05 QALYs over the 2-year trial period.
However, the cost-effectiveness of continuous vs. discontinuous therapy was evaluated for each drug using CUA regardless of the magnitude or direction of the QALY difference. This was because the magnitude of cost differences for this comparison were not known a priori and could have been sufficiently small that differences in health outcomes could bias estimates of uncertainty and/or give misleading conclusions. Therefore, the primary outcome measure for economic analyses comparing treatment regimens comprised the cost per QALY gained.
Conclusions about whether or not continuous treatment is cost-effective compared with discontinuous treatment and about which treatment regimen maximises net benefits were based on a £20,000 per QALY ‘ceiling ratio’. In other words, we assumed that the maximum the NHS is willing to pay to gain one QALY was £20,000 and that the NHS is also willing to accept the loss of one QALY to achieve savings of £20,000. 57 Net benefits were also calculated as QALYs multiplied by ceiling ratio minus cost.
The primary research questions and objectives of the IVAN economic evaluation were:
-
To estimate the incremental cost-effectiveness of continuous ranibizumab compared with discontinuous ranibizumab, and of continuous bevacizumab compared with discontinuous bevacizumab. For each drug, continuous therapy would be considered cost-effective relative to discontinuous therapy if it cost < £20,000 per QALY gained, or saved > £20,000 per QALY lost. 57
-
And:
-
either – if the incremental QALY gain for ranibizumab vs. bevacizumab is ≤ 0.05 QALYs – to estimate the incremental cost of continuous ranibizumab compared with continuous bevacizumab, and of discontinuous ranibizumab compared with discontinuous bevacizumab. For each dosing regimen, ranibizumab would be considered good value for money if the mean total cost was lower than for bevacizumab
-
or – if the incremental QALY gain for ranibizumab vs. bevacizumab is > 0.05 QALYs – to assess the incremental cost-effectiveness of continuous ranibizumab compared with continuous bevacizumab, and of discontinuous ranibizumab compared with discontinuous bevacizumab. Ranibizumab would be considered good value for money if it cost < £20,000 per QALY gained compared with bevacizumab.
-
A secondary presentation of results used the same estimates of costs and QALYs in each arm, but presented the four cells (see Table 1) as mutually exclusive strategies. This analysis followed the established decision rules and methods for presenting uncertainty, including calculating incremental cost-effectiveness ratios (ICERs) for each strategy relative to the next most effective non-dominated alternative58 and presenting cost-effectiveness acceptability curves showing the probability that each treatment maximises net benefits. 59 This approach gives identical conclusions to the primary study question but provides a global representation of uncertainty.
Economic evaluation overview
Data on HRQoL (measured using EQ-5D and HUI3) and health service resource use were collected prospectively in the trial. Table 5 summarises the methods used in the economic evaluation, which are discussed in more detail in subsequent sections.
| Aspect of methodology | Strategy used in base-case analysis | Alternative strategies used in sensitivity analysis |
|---|---|---|
| Data set |
|
|
| Time horizon |
|
|
| Form of economic evaluation |
|
|
| Approach to factorial design |
|
|
| Utility measurement |
|
|
| QALY calculations |
|
|
| Costs included in analysis |
|
|
| Missing data |
|
|
| QALYs lost due to deaths unrelated to study medication |
|
|
| Adjustment for baseline utility |
|
|
Measurement of patient-reported health status and quality adjusted life-years
Two multiattribute utility measures were used in the IVAN trial: the three-level EQ-5D56,60,61 and HUI3. 33,62,63 EQ-5D was the main utility measure for the economic evaluation, as it has several advantages over HUI3. First, the EQ-5D tariff is based on ‘time trade-off’ valuations by around 3000 members of the UK general population. 60,64 By contrast, the HUI3 value set is based on a mixture of visual analogue and ‘standard gamble’ valuations by 256 members of the Canadian general population. 33,63 The HUI3 tariff is, therefore, less precise and less relevant to a UK setting than EQ-5D. Second, EQ-5D is recommended by NICE45 and is used more widely than HUI3. 65 ICERs calculated using the EQ-5D can, therefore, be directly compared with ICERs calculated in a large number of other UK economic evaluations to help decision-makers ensure that the most cost-effective treatments are provided. However, HUI3 was used in sensitivity analyses, as it includes questions specifically relating to vision and may be more sensitive or responsive to changes in eye disease than EQ-5D. 66,67
The EQ-5D and HUI3 were both completed at 0, 3, 12 and 24 months. In addition, to assess the impact of SAEs on utility, both instruments were administered at the next assessment attendance after any SAE or a reduction in BCVA ≥ 15 letters. EQ-5D and HUI3 were also completed at/after withdrawal if the patient opted to attend a full exit assessment. For EQ-5D, the published UK time trade-off valuation tariff was used to value each health state64 and calculate ‘utilities’. The Canadian valuation tariff set33,68,69 was used for HUI3. Missing utility data were imputed by using multiple imputation (see below).
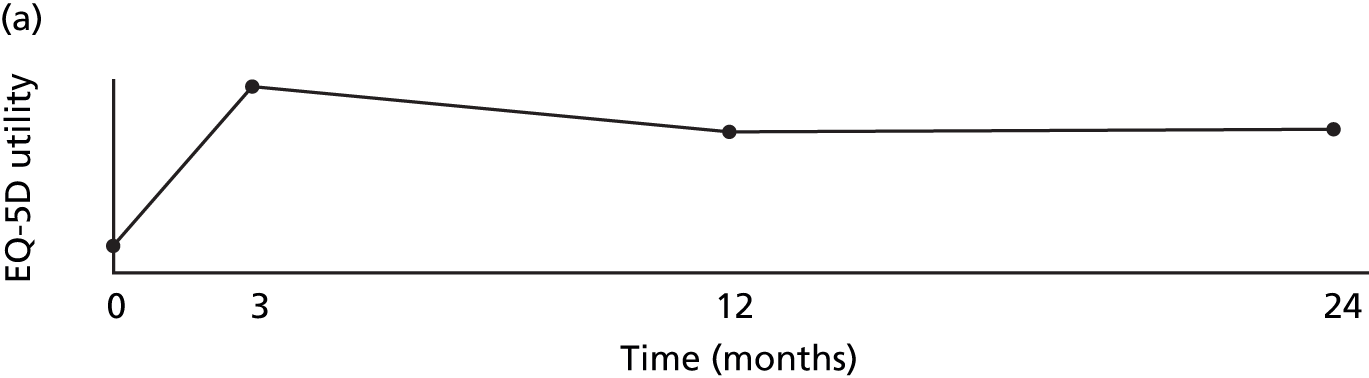
Quality adjusted life-years were calculated as the area under the utility curve, i.e. EQ-5D utility multiplied by length of time spent at that utility. In the absence of SAEs, utility was assumed to change linearly between baseline and 3 months, between 3 and 12 months, and between 12 and 24 months (Figure 4), which is supported by the trends for BCVA (see Figure 15).
FIGURE 4.
How QALYs were calculated. (a) In the absence of SAEs, EQ-5D utility was assumed to change linearly between EQ-5D measurements. (b) As the EQ-5D measurement after this patient’s first set of SAEs is higher than the line joining the baseline and 3-month measurements, we assumed that utility rose linearly between baseline and the post-SAE measurement and between this measurement and 3 months. As utility is lower after SAE 2, we draw a line through the post-SAE 2 measurement with a gradient equal to the recovery rate estimated in the mixed model. This line was used to estimate the utility on the day SAE 2 starts, and the time and the utility at which the patient is expected to have recovered from SAE 2 and returned to the utility trend observed between visits three and 12. The patient died 5 days after SAE 3; the utility was, therefore, assumed to follow the linear trend observed between visit 12 and the value imputed at visit 24 up until the day before SAE 3. Utility was assumed then to fall linearly to 0 over the last 5 days of life.
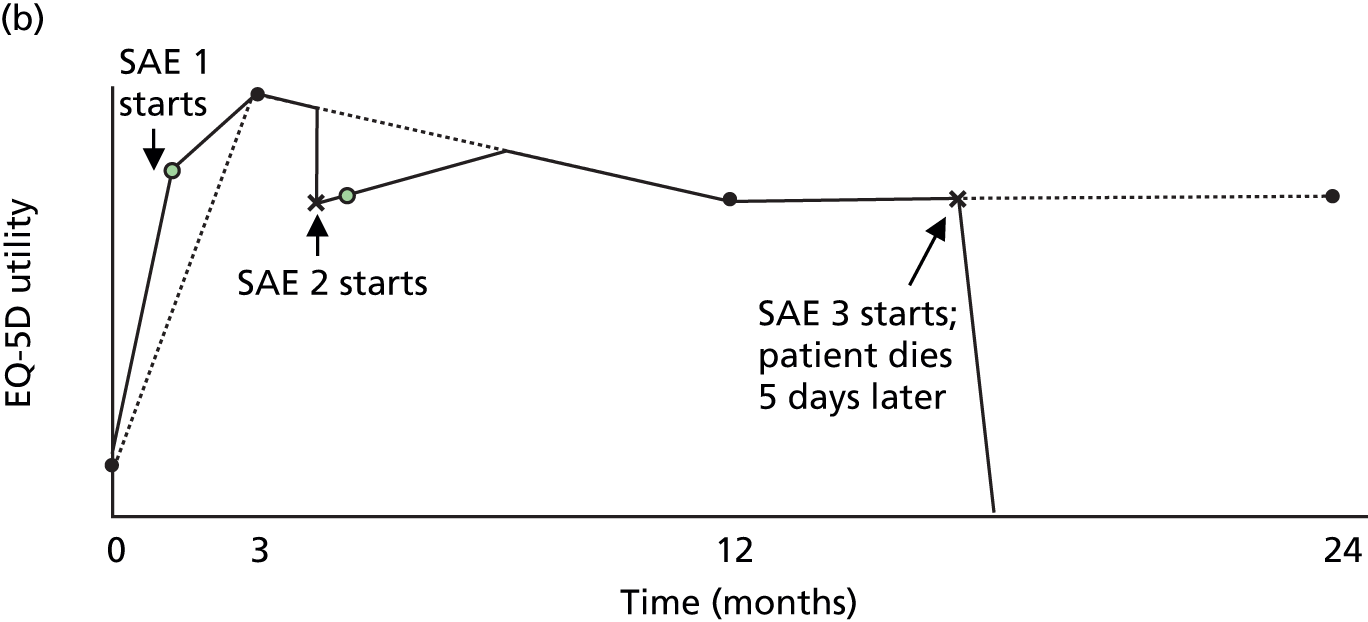
As many patients had several different SAEs starting in a short space of time (e.g. angina, MI and bypass graft in the same admission), SAEs occurring less than 1 week apart were grouped into a set associated with a single post-SAE measurement and with a single onset date equal to the onset date of the first SAE in that set (see Appendix 4). Of the 183 trial participants with SAEs starting before they left the trial or attended visit 24, 129 had one set of SAEs, 37 had two, 15 had three and 2 had four.
For patients who experienced a SAE that reduced utility, the base-case analysis assumed that utility fell on the day of the SAE and rose linearly afterwards. Similar profiles have been used previously to model recovery from acute hepatitis70 and chronic obstructive pulmonary disease exacerbations. 71 We used linear profiles to simplify subsequent calculations and because models with quadratic terms did not fit as well. As post-SAE utility measurements were taken, on average, 56 (range 0–182) days after a SAE, we used mixed models to estimate the rate at which utility rose after each type of SAE (see Appendix 4). Coefficients estimated in the mixed models were used to estimate utility on the day of the SAE and identify the point at which utility returned to the level that would be expected from the utility measurements that were not taken after SAEs (see Figure 4). However, some post-SAE measurements were higher than would have been expected from other measurements for the patient (e.g. see Figure 4, SAE 1); in these cases, we assumed that utility changed linearly between the routine measurements (see Figure 4). For patients dying 1–7 days after the latest SAE, utility was assumed to fall linearly to 0 between the date of the SAE and the date of death. Full details of the methods are described in Appendix 4.
Collection of resource use and cost data
Data were collected on all major NHS resource uses for each patient using the trial CRFs. We included resources associated with only the study eye or expected AEs or SAEs to avoid catastrophic episodes involving high health-care costs unrelated to treatment (e.g. renal failure, cancer or extended psychiatric admissions) swamping the main effect of treatment on costs. 72 Expected SAEs and AEs comprise those previously linked to bevacizumab or ranibizumab treatment, which are listed in Appendix 4. For the purposes of the costing, death was not considered to be an expected SAE unless the primary cause of death comprised one of the expected SAEs or AEs; this was done to ensure that costs unrelated to anti-VEGF treatment (e.g. the cost of treating cancer) are excluded from the analysis, regardless of whether or not the patient died during the study period.
Unit costs (see Appendix 4) were combined with resource volumes to obtain the total cost per patient per 3-month period or quarter. Three categories of cost were considered: anti-VEGF study medication; drug administration and monthly monitoring visits; and NHS costs associated with expected AEs and expected SAEs. An index year of 2011 was used for costs. All costing analyses followed guidelines for economic evaluation. 53,73 Following NICE guidance,45 value added tax (VAT) was excluded from the base-case economic evaluation but was included in budget impact estimates. The boundaries between quarters were based on study visits rather than calendar months to minimise random variations in costs between quarters: for example, quarter 1 included the first three injections and all resource use accrued before visit 3 regardless of whether or not this visit was conducted early or late.
Following NICE guidance,45 the base-case analysis used the list price for ranibizumab (£742.17 per dose51). Although the Department of Health and Novartis have agreed a reduced price that the NHS pays for ranibizumab,74 this discount is commercially sensitive and confidential, and may vary between hospitals. The cost of bevacizumab syringes for intravitreal injection (£49 per dose) was based on the price typically charged in other trials by the not-for-profit NHS compounding pharmacy where bevacizumab was repackaged. This cost includes transport and delivery and was based on micro-costing work conducted by this pharmacy for a subsequent trial. The cost of both drugs was varied in the sensitivity analysis. In the IVAN trial, some anti-VEGF was wasted as a result of it exceeding the shelf life or being booked out of pharmacy and not used. However, most of this in-trial wastage was due to the trial protocol, and our pilot resource use questionnaires demonstrated that the amount of wastage in routine clinical practice is likely to be minimal. Our base-case analysis therefore assumed that no bevacizumab or ranibizumab would be wasted, although a sensitivity analysis allowed for the same amount of wastage observed in the trial.
Accurate estimation of the cost of administering anti-VEGF inhibitors and monitoring outcomes of treatment required micro-costing work for several reasons. First, the only gross costs available at present include all ophthalmology outpatient consultations,75 not just those for nAMD, whereas anti-VEGF injections are covered by local tariffs rather than the Healthcare Resource Group (HRG)76 Payment by Results scheme. Second, no available gross cost or HRG tariff distinguishes between consultations in which an anti-VEGF drug is administered and consultations to monitor outcomes without treatment. The difference between such consultations is likely to be one of the main drivers of the cost-effectiveness of continuous treatment compared with discontinuous treatment. As a result, the base-case analysis used micro-costing to estimate the cost of consultations to administer bevacizumab/ranibizumab and to monitor outcomes. The impact of using reference cost and HRG estimates was evaluated in sensitivity analyses.
Data on the cost associated with drug administration and monitoring were collected using two questionnaires (see Appendix 4). These were completed by the staff at IVAN centres, who were responsible for running clinics in which bevacizumab and/or ranibizumab are administered in routine clinical practice, with additional data being provided by clinicians and other staff at each centre. To exclude protocol-driven resource use, centres were asked about the resources used for routine NHS consultations, rather than IVAN trial visits. The questionnaires were developed following discussions with a consultant at one IVAN centre, observational work at another centre and a pilot of draft questionnaires at three centres (including the two mentioned previously). The set-up cost questionnaire (see Appendix 4) collected data on the resource use and costs associated with purchasing equipment and setting up a clinic to monitor nAMD patients and administer intravitreal injections. The operating cost questionnaire (see Appendix 4) collected data on: the number of staff and rooms needed to run each clinic session (separating out those staff and rooms involved in intravitreal injections and those involved in FFA); the cost of any pre-prepared injection packs used; overheads; and the tests and investigations routinely conducted to monitor the outcomes of anti-VEGF therapy. As set-up costs were unlikely to be major cost drivers, the set-up cost questionnaire was completed by six IVAN centres. Other IVAN centres provided basic details on the number of rooms of each type that were set up or modified in order to provide anti-VEGF therapy, which were valued using the cost per room estimates obtained from the six centres completing the set-up cost questionnaire. Total set-up costs were annuitised over the lifetime of the facilities and divided by the total number of patients likely to use such facilities. The operating cost questionnaire was sent to all 19 centres recruiting ≥ 10 patients and was completed by 12 centres. Estimates of the number of staff hours and costs were returned to each centre to check their accuracy.
Hourly costs were estimated for each grade and type of staff by replicating the costing calculations used by the Personal Social Services Research Unit77 using Agenda for Change salaries78 (see Appendix 4). Total staff costs per clinic were divided by the average number of patients typically attending each clinic to give the average staff cost per patient visit. The costs of reagents used to administer intravitreal injections or conduct FFA (£1.29 per intravitreal injection and £1.91 per FFA; see Appendix 4) were estimated from resource-use estimates provided by two centres and were assumed to be the same for all centres. Total consultation costs per intravitreal injection, per monitoring consultation and per FFA were calculated for each clinic as the sum of staff costs; set-up costs; the cost of pre-prepared injection packs; and the mean cost of reagents. The percentage of overheads applicable to each centre was then added to the total cost. Costs were then averaged over all centres to obtain five unit costs (Table 6).
| Type of cost estimate | Description |
|---|---|
| F: cost per FFA | Included only reagents, consumables, staff and facilities directly involved in FFA |
| M: cost per monitoring consultation excluding FFA and any resources associated with the intravitreal injection | Although seven centres ran separate injection and monitoring clinics as well as, or instead of, a one-stop service by which patients attend clinic for monitoring and receive injections that day if they need them, the cost of monitoring (excluding resources attributable to FFA and injections) was similar for one-stop and monitoring-only clinics |
| MF: cost per monitoring consultation including a fraction of patients receiving FFA | Estimated by multiplying the cost of FFA at each centre by the proportion of patients who typically have FFA done in any given monitoring consultation, and adding it to the monitoring consultation cost excluding FFA |
| IM: cost per intravitreal injection given alongside monitoring | Calculated as the incremental cost of giving an intravitreal injection in a consultation in which monitoring (e.g. OCT) was also provided, which was based on the average across one-stop clinics |
| IO: cost of intravitreal injection not alongside monitoring consultation | The cost of separate injection consultations at two-stop services was typically slightly higher than the incremental cost of giving an injection at a one-stop clinic. We therefore estimated the average cost of injection consultations across all injection-only clinics to reflect the cost of a consultation in which VA is assessed before the injection, but OCT is not conducted |
In the trial, all participants attended monthly clinic consultations including study questionnaires (when scheduled), VA assessment, OCT and (if necessary) FFA and intravitreal injections. However, costing analyses excluded protocol-driven resource use by assuming that patients would not require OCT or FFA unless this would affect treatment decisions. As such, patients on discontinuous treatment were assumed not to require OCT at the second or third visit in a course of three injections (Table 7). Similarly, patients on continuous treatment were assumed to require monitoring consultations (with OCT and/or FFA) only once every 3 months. Although IVAN participants had FFA at visit 12, this is no longer done in routine clinical practice. We therefore excluded any FFAs undergone at visit 12, unless FFA was needed to evaluate the treatment failure criteria for patients in the discontinuous group. In the base-case analysis, missed visits were assumed to accrue no NHS cost, as many clinics overbook patients; however, this assumption was varied in sensitivity analysis.
| Patient | Visit | 0a | 1a | 2a | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient A: continuous treatmentb | Injection | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ |
| IM | IO | IO | IM | IO | IO | IO | IO | IM | IO | IO | |||
| Monitoring consult | ✓ | ✓ | ✗ | ✓ | |||||||||
| MF | MF | MF | |||||||||||
| FFA | ✓ | ✗ | |||||||||||
| F | |||||||||||||
| Patient B: discontinuous treatmentc | Injection | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||||
| IM | IO | IO | IM | IO | IO | IM | |||||||
| Monitoring consult | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | |||||
| MF | M | M | M | M | M | M | |||||||
| FFA | ✓ | F | ✗ | ||||||||||
| F | |||||||||||||
As the number of patients recruited to IVAN does not reflect the number of patients that each centre treats in routine clinical practice, average costs were applied to all patients regardless of which centre they attended. However, to allow for the substantial variability in unit costs between centres, we drew repeated values from the distribution of clinic costs for each patient in each multiple imputation (see Appendix 4).
At each monthly visit, all IVAN participants were asked to give details of all hospital stays, contacts with community health-care providers and hospital outpatient attendances and all changes in their medications. The base-case analysis included only those resources directly attributable to AEs or SAEs that have previously been linked with bevacizumab or ranibizumab and were classed as ‘expected’ in the trial protocol (see Appendix 4). The dates of any hospital admissions were reviewed to identify which SAE they related to; AE dates were reviewed if the admission dates did not match any SAE. Free text from the SAE form was also reviewed to identify hospitalisations and admission/discharge dates associated with SAEs that were not reported at monthly visits (e.g. those occurring between a patient’s last visit and their death). Hospitalisations linked to expected SAEs (or expected AEs) were valued using Department of Health reference costs75 (see Appendix 4), whereas those linked to other events were excluded from the base-case analysis but included in sensitivity analyses.
Consultations with general practitioners (GPs), GP nurses, district nurses, hospital doctors or nurses and other health-care professionals were valued using published cost data,76,77 (see Appendix 4) and were included in the analysis if they were either marked as being related to ocular conditions or if they occurred < 30 days after the onset of an expected AE or an expected SAE. Consultations with community optometrists or dentists were excluded, as study medication is not expected to change the frequency and not all such consultations will be funded by the NHS. The 30-day cut-off was determined through discussions with clinicians.
The cost of concomitant medications applied to the study eye and those that are licensed for the treatment of expected AEs or expected SAEs were included in the analysis and costed using list prices. 51 All drugs for prevention or treatment of CVD, any antibiotic, any influenza medication (other than vaccination) and all medications specifically licensed for an expected AE or expected SAE were included in the analysis. Drugs for unrelated conditions (e.g. cancer or arthritis) were excluded from the analyses. The total cost of medication changes was calculated relative to baseline.
Costs associated with reduced vision were excluded from the analysis. The cost of NHS-funded transport to clinic for treatment/monitoring or for management of AEs was excluded from the analysis, as there was no statistically significant difference between drugs or treatment regimens in the number of NHS-funded journeys accrued during the study period (p ≥ 0.169). In particular, both continuous and discontinuous treatment require monthly consultations for monitoring and/or intravitreal injection, and so reducing injection frequency would not per se be expected to reduce the number of patient journeys required.
Handling missing values
Multiple imputation using a series of chained regression equations was used to impute missing data on utilities. 79–84 Following recent guidelines,81 multiple imputation was conducted using the ice command in Stata (version 1.9.4). 85
Multiple imputation was not required for resource use, as such data were missing for only 2% (330 out of 13,398) of attended visits; methods for imputing such data are described in Appendix 4.
The EQ-5D and HUI3 utilities were imputed at scheduled time points (0, 3, 12 and 24 months), at study exit and after SAEs (see Appendix 4). Although only 3.6% (82 out of 2305) of EQ-5D measurements at scheduled visits were missing, EQ-5D utilities were missing for 54% (32 out of 59) of exit visits and 41% (89 out of 231) of SAEs. Overall, 152 patients (25%) had missing EQ-5D data at ≥ 1 time points. White et al. 81 recommend that the number of imputed data sets is at least equal to the percentage of patients with missing data; however, we generated and analysed 100 imputed data sets to maximise robustness. See Appendix 4 for further details.
Handling censoring and mortality
Given the elderly trial population, it was anticipated that some patients would die during the study, either from events previously linked to study medication (e.g. MI or stroke) or from other causes (e.g. cancer). Although deaths unrelated to study medication were expected to be randomly distributed across study groups, it was anticipated that one or more study group could, by chance, have substantially more deaths unrelated to treatment than the other groups. Although differences are unlikely to be statistically significant, chance differences in unrelated deaths could have substantially affected incremental QALY estimates and reduced statistical power as the patients dying early in the trial would have been assigned zero utility for the remainder of the trial period. The base-case economic evaluation therefore excluded any between-group differences in deaths unrelated to study medication using an adaptation of Kaplan–Meier sample averaging (KMSA). Deaths were categorised as either drug related (i.e. classified by a study clinician as definitely/probably/possibly related to study medication) or not (i.e. classified by a study clinician as unlikely to be/not related to study medication).
Using KMSA, the mean costs or QALYs accrued by the total sample in any given time interval are calculated by multiplying the mean costs for those patients who are alive and non-censored in that period by the Kaplan–Meier estimate of the proportion of patients alive at the start of that time period. 58,86,87 The Kaplan–Meier survival functions used in KMSA are normally based on the number of deaths occurring in each study group. Instead, we used a novel adaptation of this technique, whereby the probability of being alive at time ‘t’ was based on (1) the number of potentially drug-related deaths observed in the study group in question and (2) the number of unrelated deaths that would be expected based on the average risk of unrelated deaths across all four study groups. For each of the four treatment groups, the costs and QALYs accrued in each quarter were averaged across all patients who were alive at the start of that quarter and who did not withdraw from the trial by the end of that quarter (see Appendix 4). Quarterly outcomes were then multiplied by the probability of being alive at the start of the quarter. This method provides an unbiased estimate of total costs and QALYs providing that treatment allocation has no impact on deaths considered unlikely to be/not related to study medication.
Statistical methods and analysis for the within-trial economic evaluation
Patient-level data on costs and QALYs were analysed in Stata version 12. Non-parametric bootstrapping was used to quantify the degree of uncertainty around costs, QALYs and ICERs. The analysis also propagated uncertainty around imputed values, excluded chance differences in deaths unrelated to study medication and adjusted for any imbalance in baseline utility and any interactions for costs or QALYs that are statistically significant or could change the conclusions of the analysis.
The final estimates of costs and QALYs for each of the four groups are interpreted ‘inside the table’52 as mutually exclusive alternatives. However, estimation of mean costs and QALYs for each study group allowed for important interactions to avoid bias46,88 but ignored negligible interactions to maximise statistical power. 46,52 Costs were divided into drug costs, administration/monitoring costs and medication/medical service costs to enable different assumptions about interactions to be used for different types of cost.
Interactions were included for those components of costs or QALYs where interactions were either statistically significant or had an absolute magnitude larger than either the main effect for treatment regimen or the main effect for drug (see Appendix 4). However, interactions that were both non-significant and smaller than both main effects were excluded from the analysis to avoid loss of statistical power. Analyses of the costs and QALYs accrued in the first quarter assumed zero interaction and zero difference between continuous and discontinuous therapy, as all patients received monthly injections in that quarter.
Baseline utility was included as a covariate in all analyses of QALYs to adjust for any imbalance in baseline utility. 89 This was important, as imbalances in baseline utility introduce substantial bias into economic evaluations as baseline utility is directly included in QALY calculations and normally strongly predicts on-treatment utility. 89 Such bias could be particularly influential for IVAN as the differences in post-treatment utility are small and even very small imbalances in baseline utility could change the conclusions. Regression adjustment also allows for regression to the mean and increases precision.
The costs and QALYs accrued in each group of the trial were estimated using regression analyses conducted on data for each quarter, which were repeated for 130 bootstrap replicates on each of the 100 imputed data sets (see Appendix 4). Regression predictions from each bootstrap replicate on each data set were multiplied by the Kaplan–Meier survival estimates and summed over all quarters to give total costs and QALYs in each group. Costs and QALYs accrued in year 2 were discounted at 3.5% per year, with a sensitivity analysis at 1.5%. 45,90 The bootstrapping process outlined in Appendix 4 was also repeated on the resource use quantities shown later in Table 28; interaction terms were included in analyses of those resource use items that fell into cost categories for which interactions were large or statistically significant. Bootstrap analyses were also replicated for the sensitivity analyses shown later in Table 31.
The impact of the five prespecified clinical subgrouping variables (see Statistical methods) and an additional variable indicating whether or not VA in the study eye was > 5 letters better than the fellow eye at baseline was also evaluated post hoc using the methods described in Appendix 4.
Unless otherwise specified, all means reported in Chapter 8 represent the predictions from the regression analyses outlined in Appendix 4, adjusted for mortality and discounting, and averaged across the 100 imputed data sets. Similarly, all SEs reported in Chapter 8 were calculated from the same regression analyses on 130 bootstrap replicates on each of 100 imputed data sets, combined using Rubin’s rule. The p-values and 95% CIs were calculated from the SEs.
Patient and public involvement
The IVAN trial continued a long-standing association with the Macular Society (previously the Macular Disease Society). The Macular Society is ‘the national UK charity for anyone affected by central vision loss’ (www.macularsociety.org/How-we-help/About-us). Although the charity raises funding from corporate sponsors, it receives no government funding or donations from pharmaceutical companies. Professor Chakravarthy and other members of the research team approached the Macular Society in 2003 in relation to the VPDT for the UK. As the IVAN trial grew out of the VPDT, it was natural to continue the collaboration when we were seeking funding for the IVAN trial. The collaboration continued once funding was awarded, when the trial was being set up and throughout its conduct through the Society’s membership of the TSC. Mr Bremridge, the Society’s chief executive at the time, was a member of the TSC at inception. Following his retirement, Helen Jackman (the current chief executive) or Cathy Yelf, the Society’s head of communications and external relations, have represented the Society on the TSC.
We consulted with the Macular Society, directly with Tom Bremridge, Helen Jackman and Cathy Yelf, but also (through the Society) with some of their members, on several aspects of the trial:
-
the need for such a trial
-
the design of the trial
-
information given to patients at the outset who were being approached about the trial (construction of the patient information leaflet that was approved by a NHS research ethics committee) and subsequent revisions to this information
-
information about the first-year findings of the IVAN trial and those of the CATT35
-
information about possible risks that emerged during the trial and which were communicated to participants who had not completed the study
-
on study completion, letters to participants thanking them for their involvement and unmasking them to the drug assignment.
The advice of the Macular Society was most important with respect to (a), (b) and (d). The decision to use a discontinuous treatment regimen requiring cycles of three injections given monthly was substantially based on discussions with Mr Bremridge about the need to minimise the risk of participating in the trial. Mr Bremridge also advised in detail about how to describe the possible risks and benefits of treatment with each drug.
It was vital to have views about how to communicate the trial objectives fairly and to describe what the trial involved in the patient information leaflet. In this context, it should be remembered that the first few months of recruitment to the trial straddled NICE’s publication of its final appraisal determination for ranibizumab; ranibizumab was not widely available at the time that the trial started to recruit and, once ranibizumab was recommended by NICE, the possibility of being treated with bevacizumab was extremely controversial.
In the spring of 2012, the chairperson of the DMSC asked the trial statistician to combine the most up-to-date information from the IVAN trial with information available from the CATT. 35 This synthesis found a statistically significant increased risk of having a systemic SAE with bevacizumab compared with ranibizumab. At the next meeting, the committee reviewed the information and recommended that the trial should continue but the information should be disseminated to the minority of patients who were still continuing in the trial. This recommendation was discussed at length, during the following TSC, with Cathy Yelf, who concurred. The research team drafted a letter to patients (describing the information, the recommendation and giving the opportunity to withdraw) and sought edits and comments on the content from the Macular Society before finalising it. Given the sensitive nature of the information, it is testament to the quality of the patient and public involvement in the IVAN trial that none of the participants remaining in the trial asked to withdraw.
Management of investigational medicinal product
All drugs were purchased centrally by the Liverpool pharmacy, that is standard commercially available ranibizumab 0.23-ml vials and bevacizumab 4-ml vials. Ranibizumab vials as commercially supplied were labelled for use as investigational medicinal product (IMP) in the trial. Pre-filled bevacizumab syringes were manufactured in the aseptic manufacturing facility, packaged in a box and also labelled for use as IMP in the trial. Both drugs were distributed by courier to each centre. Drugs were dispatched bearing study labels approved by the Medicines and Healthcare products Regulatory Agency (MHRA). Each ranibizumab vial ordered and each bevacizumab syringe manufactured by the Liverpool pharmacy was logged on the appropriate product log sheet. The fate of each dose was recorded against each batch (e.g. which hospital they were dispatched to, used for quality control, etc.).
The central coordinating centre managed drug stock at each site. When drug supplies were required by a site, the coordinating centre communicated to the Liverpool pharmacy the drug type, quantity and destination to which they should be dispatched. The product log was completed, stating to which hospital the drug was to be dispatched and the date of dispatch. Drugs were packed for dispatch with cool packs to maintain a temperature of 2 °C to 8 °C. Packs included a temperature monitor to allow temperature breaches during transit to be identified. The Liverpool pharmacy confirmed that drugs had been dispatched via the trial database.
Each batch of study drug received at a site was logged in the IVAN study drug receipt log. On arrival the package was checked for temperature breaches and/or damage to study drugs; if either had occurred, the coordinating centre was contacted to arrange redelivery. All shipments were transferred immediately to a fridge (2 °C to 8 °C) for storage.
Any study medication that exceeded its expiry date before being used was logged and kept until it was checked by IVAN monitors. Expired drugs were recorded on the drug receipt log by pharmacies at local sites; if drugs expired prior to dispatch to site, this was recorded on the appropriate product log sheet by the Liverpool pharmacy.
On receipt of a study prescription, the correct drug was taken from a fridge and the expiry date checked to ensure that the drug was within shelf life. The IVAN study number and visit number were completed on the study label on the outside packaging of the study drug. The prescription log was completed, detailing the expiry date and batch number for each patient and visit number. The drug was concealed in an unmarked bag ready for collection by clinic staff/delivery to clinic.
Used vials and syringes were returned from clinic to pharmacy at the end of each clinic and checked and stored until after monitoring had taken place and accountability completed, when instructions to destroy them would be issued by the trial monitoring team. The trial monitoring team inspected returned packaging to check that the correct drug had been dispensed and that the drug had been given (i.e. that the vial/syringe was empty).
Emergency unmasking
Emergency unmasking was provided by the coordinating centre during office hours (Monday to Friday, 9 a.m. to 5 p.m.). Outside office hours, the pharmacy prescribing the study drug provided unmasking. An unmasking pro forma was provided for the recording of the event.
Study drug recall
If a batch of bevacizumab needed to be recalled, the Liverpool pharmacy notified the co-ordinating centre, which then notified the pharmacy contact at each participating site. The pharmacy was asked to check its stock for any syringes in the recalled batch, notify the coordinating centre and label each box as recalled. The coordinating centre arranged collection of the recalled batch from sites, which were returned to the central pharmacy. Recall of a batch of ranibizumab could come from the Liverpool pharmacy or through local recall procedures. If this happened, they were asked to notify the central coordinating centre. The procedure followed following notification was the same for both drugs.
Contractual and financial arrangements
At the time funding was being sought and the trial was being set up, the costs of the trial had to be attributed as research, service support and treatment (including excess treatment) costs (www.gov.uk/government/news/attributing-the-costs-of-health-social-care-research-development-acord). The task was complex for the IVAN trial because trial set-up and initial recruitment spanned a period of time when (1) ranibizumab had not been recommended for use in the NHS91 and (2) the dispersion of service support costs to participating hospitals through the Comprehensive Research Network had not been completely implemented. Consequently, the attribution of the costs of the trial changed dramatically before and after NICE issued guidance.
Before NICE issued guidance, treatment with ranibizumab represented an estimated excess treatment cost of £15,000–30,000 per patient over the 2-year duration of follow-up (depending on whether a patient was allocated to continuous or discontinuous treatment); the corresponding range for bevacizumab was estimated to be £6000–7000 per patient. After NICE issued guidance, discontinuous ranibizumab approximately represented ‘usual care’, continuous ranibizumab represented an excess treatment cost of £15,000 per patient and bevacizumab a saving in treatment cost of about £8000–9000.
Service support costs (additional tests and other NHS resources used in the course of the trial that were related to the patient’s care but which were not required purely for the research) were estimated to represent a further £2830 per patient over 2 years. These costs were unaffected by the NICE guidance but still had to be reimbursed to hospitals to cover the full cost of the hospitals taking part.
The previous experience of several members of the research team including the chief investigator in evaluating an earlier technology for nAMD (VPDT)92 meant that, in the IVAN trial, the research team was determined to ensure that all costs for the trial were truly in place, namely research, service support and treatment costs, before starting to recruit. The team was also committed to ensuring that reimbursement to hospitals for trial activity related to reviewing and treating trial participants was conditional on carrying out the trial according to the protocol and providing valid data; these two conditions were written into contracts between the sponsor and participating hospitals.
Several unusual features of the trial followed from these decisions:
-
There was a need to put in place service level agreements with commissioning organisations (63 primary care trusts in England and four health boards in Northern Ireland; some agreements were negotiated with consortia of commissioning organisations) to provide the treatment costs (as, at the time, these were excess treatment costs). These agreements required ‘up front’ payment (at the start of the financial year) to enable trial activity to happen.
-
A mechanism had to be established for treatment costs to be paid to hospitals separately from the usual contracts with commissioners, to ensure that the funding for treatment in trial ‘followed the patient’, that is it was made available to the hospital ophthalmology department delivering the trial.
-
A mechanism had to be established to ensure that hospitals did not ‘double count’ activity relating to trial participants, that is, charge their treatment to the trial and also to commissioners under the usual contract for nAMD services. This mechanism ensured that ‘savings’ from using bevacizumab were truly reflected in the funding commissioners paid for treatment of trial participants after ranibizumab was recommended by NICE.
-
There was a need to establish a ‘bank’ to manage service level agreements with primary care trusts, hold their payments in advance on credit against future trial-related activity, and manage reimbursement for trial activity by participating trusts.
These features and requirements were further complicated by:
-
The requirement for the IMP to be provided from a single, central, manufacturing pharmacy, which necessitated splitting the clinic/hospital costs from the drug costs. Contracts with participating hospitals to reimburse for trial activity were restricted to the clinic/hospital costs, with the IMP being provided to hospitals ‘free’ for use in the trial. The bank separately contracted with the central manufacturing pharmacy to supply the IMP to participating trusts. The central, manufacturing pharmacy was unwilling to bear the uncertainty of the costs of cold-chain transport (which were expensive, with cost per dose dependent on the number of doses being transported) in this contract, so the bank also had to pay separately for the transport costs.
-
The fact that an element of the clinic/hospital costs represented research costs, which required the grant holder [Queen’s University Belfast (QUB)] to pay this sum to the bank, to be passed on to participating hospitals pro rata to trial activity.
-
A research partnership consisting of four universities and two NHS trusts who were collaborating to deliver the research. The grant holder (QUB) was responsible for establishing non-commercial contracts with each partner, defining the role and responsibilities of each partner and the proportion of the research costs the partner would receive in return.
The NHS sponsor for the trial (Belfast Health and Social Care Trust) agreed to take on the risks and responsibilities (to administer income from primary care trusts and expenditure in relation to trial activity) associated with being the bank, in addition to its responsibilities for research governance, trial oversight and contracts with participating hospitals. The trial benefited from advance, in-depth discussions with the National Specialised Commissioning Group about the likelihood of NICE recommending ranibizumab and the future potential benefit to commissioners of having evidence about the cost-effectiveness of bevacizumab. These discussions were instrumental in persuading commissioning organisations to sign the service-level agreements required. The trial also benefited from the services of some very committed individuals involved in commissioning when drafting the service-level agreement and in negotiating these agreements on behalf of the trial with several primary care trusts. The contracting and monitoring arrangements, and the flows of finances and data in the trial, are shown in Figures 5–7.
The importance of this infrastructure to the ultimate success of the trial cannot be overemphasised. The trial team extends its gratitude in equal measure to the individuals and organisations that made the trial possible.
FIGURE 5.
Contracts and monitoring. TCC, Trial Coordinating Centre.

FIGURE 6.
Finance flow chart. HTA, Health Technology Assessment; ICH-GCP, International Conference on Harmonisation Good Clinical Practice guidelines.


Chapter 3 Trial cohort
Screened patients
Patients were prescreened to exclude those with no interest in the trial; because of the different ways in which centres did this, data describing all patients approached were not available. Screening data were provided for 693 patients. Of these, 65 patients were excluded; 28 were found to be ineligible (reasons include BCVA of < 25 letters and presence of other ocular disease causing concurrent vision loss) (Figure 8) and the other 37 were excluded for unknown reasons. The remaining 628 patients were randomised into the trial.
FIGURE 8.
Recruitment rate. Recruitment is cumulative. Only participants included in the IVAN cohort are represented on this graph.

Recruitment
Patients were recruited to the IVAN trial between 27 March 2008 and 15 October 2010, and the last patient follow-up visit was on 7 November 2012. The 628 randomised participants were recruited from 23 centres (see Appendix 1 for details). Five of these 628 participants were randomised in error and a further 13 were not treated, leaving 610 cases who received at least one injection. These 610 participants make up the IVAN study cohort. Recruitment was steady throughout the course of the trial (Figure 9). The number of participants in the IVAN cohort recruited at each of the centres is shown in Figure 10.
FIGURE 9.
Number of participants recruited by centre. Only participants included in the IVAN cohort are represented on this graph.
FIGURE 10.
Flow of participants. a, More patients are likely to have been considered for the trial, and the exclusions section is therefore likely to be incomplete, because some sites did not enter full screening data. b, Some patients may be ineligible for more than one reason. c, Details of other withdrawals (i.e. patients that withdrew but data collection will still continue) will be given as footnotes. d, Of the patients who did not drop out, not all of them completed all three treatments. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
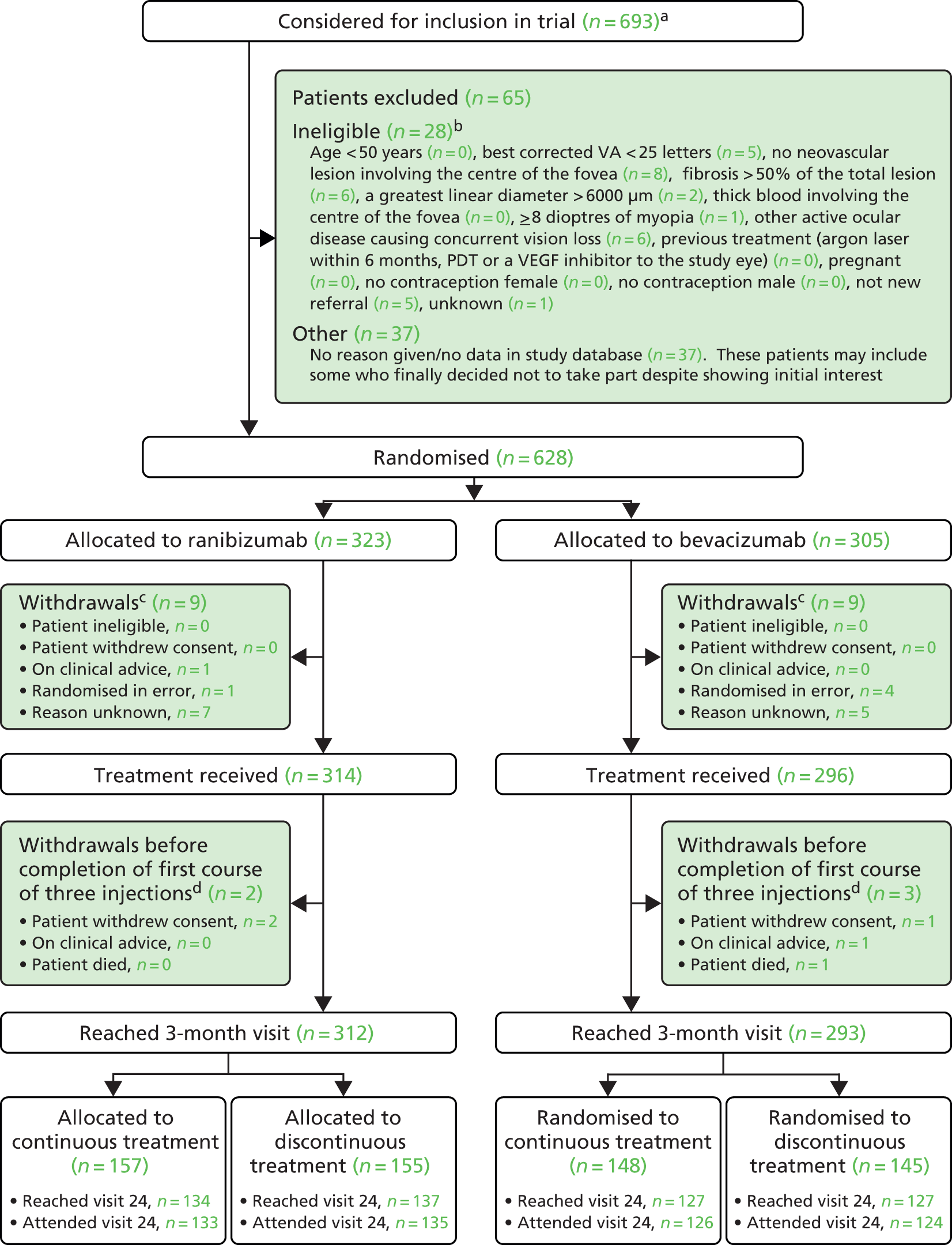
Patient withdrawals
There were 60 participants who withdrew from follow-up after their first injection and before visit 24, that is the scheduled end of the trial. The rate of withdrawal was constant over the first 18 months of the follow-up period (Figure 11). The most common reason for withdrawal was that the patient was too ill to attend (participant-reported reason) or had experienced a SAE (clinician-reported reason) (Table 8).
FIGURE 11.
Time from randomisation to withdrawal (excluding deaths).

| Reason for withdrawal | Randomised to | Overall (n = 60) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 30) | Bevacizumab (n = 30) | Continuous (n = 38) | Discontinuous (n = 22) | |||||||
| n | % | n | % | n | % | n | % | n | % | |
| Any withdrawala | 30 | 30 | 38 | 22 | 60 | |||||
| Clinician’s advice | 9 | 9 | 10 | 8 | 18 | |||||
| Very poor VA | 0/9 | 0 | 2/9 | 22 | 1/10 | 10 | 1/8 | 13 | 2/18 | 11 |
| SAE | 6/9 | 67 | 4/9 | 44 | 6/10 | 60 | 4/8 | 50 | 10/18 | 56 |
| Other reason | 3/9 | 33 | 4/9 | 44 | 3/10 | 30 | 4/8 | 50 | 7/18 | 39 |
| Patient’s choice | 24 | 22 | 31 | 15 | 46 | |||||
| Too ill to attend | 10/24 | 42 | 8/22 | 36 | 9/31 | 29 | 9/15 | 60 | 18/46 | 39 |
| Unhappy with trial procedures | 4/24 | 17 | 3/22 | 14 | 7/31 | 23 | 0/15 | 0 | 7/46 | 15 |
| Difficulties with transport or support from friends/relatives | 0/24 | 0 | 2/22 | 9 | 1/31 | 3 | 1/15 | 7 | 2/46 | 4 |
| Wanted treatment outside trial | 1/24 | 4 | 1/22 | 5 | 1/31 | 3 | 1/15 | 7 | 2/46 | 4 |
| Reason not given | 1/24 | 4 | 3/22 | 14 | 2/31 | 6 | 2/15 | 13 | 4/46 | 9 |
| Other | 11/24 | 46 | 5/22 | 23 | 14/31 | 45 | 2/15 | 13 | 16/46 | 35 |
| NHS/private treatment to continue? | ||||||||||
| No | 11/30 | 37 | 5/30 | 17 | 9/38 | 24 | 7/22 | 32 | 16/60 | 27 |
| Yes | 18/30 | 60 | 22/30 | 73 | 26/38 | 68 | 14/22 | 64 | 40/60 | 67 |
| Missing | 1/30 | 3 | 3/30 | 10 | 3/38 | 8 | 1/22 | 5 | 4/60 | 7 |
| Willing to attend study follow-up at 3, 12 and/or 24 months | ||||||||||
| No | 19/30 | 63 | 16/30 | 53 | 21/38 | 55 | 14/22 | 64 | 35/60 | 58 |
| Yes | 7/30 | 23 | 8/30 | 27 | 11/38 | 29 | 4/22 | 18 | 15/60 | 25 |
| Missing | 4/30 | 13 | 6/30 | 20 | 6/38 | 16 | 4/22 | 18 | 10/60 | 17 |
| Months to last attended visit (median, IQR) | 9.7 | (5.7–13.6) | 11.3 | (6.0–15.6) | 8.7 | (4.9–13.6) | 12.3 | (6.9–16.3) | 10.5 | (5.8–14.5) |
Protocol deviations
Protocol deviations in the IVAN trial included visits on which a participant did not receive the allocated drug, non-adherence to the treatment regimen, missing visits and visits outside the 28- to 35-day window. Table 9 shows the number of times each deviation occurred, and in how many participants. The wrong study drug was administered on 2 out of 12,761 follow-up visits. The allocated treatment regimen was not adhered to on 133 out of 12,761 visits (1.0%) (see Table 9). Further details on protocol deviations are given in Appendix 3. Overall, 57% of participants missed at least one visit, excluding visits missed because a participant died or withdrew early.
| Type of deviation | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| Any protocol deviation (patients) | 211 | 67 | 224 | 76 | 209 | 68 | 226 | 75 | 435 | 71 |
| Did not receive allocated drug | 1/1 | 0 | 1/1 | 0 | 2/2 | 1 | 0/0 | 0 | 2/2 | 0 |
| Patient did not receive allocated regimen | 15/1 | 0 | 0/0 | 0 | 0/0 | 0 | 15/1 | 0 | 15/1 | 0 |
| Did not meet eligibility criteria but was treated | 4/4 | 1 | 5/5 | 2 | 2/2 | 1 | 7/7 | 2 | 9/9 | 1 |
| Patient attended the clinic but treatment was not givena | 50/38 | 12 | 40/31 | 10 | 44/35 | 11 | 46/34 | 11 | 90/69 | 11 |
| Treatment was restarted but the criteria for retreatment were not met | 7/7 | 2 | 6/6 | 2 | 0/0 | 0 | 13/13 | 4 | 13/13 | 2 |
| Treatment was not restarted but the criteria for retreatment were met | 2/2 | 1 | 1/1 | 0 | 0/0 | 0 | 3/3 | 1 | 3/3 | 0 |
| Treatment was extended beyond 3 months but the criteria for retreatment were not assessed | 5/5 | 2 | 7/6 | 2 | 0/0 | 0 | 12/11 | 4 | 12/11 | 2 |
| Time between two consecutive visits was < 28 days or > 35 days: | 123/75 | 24 | 119/86 | 29 | 76/59 | 19 | 166/102 | 34 | 242/161 | 26 |
| < 28 daysb | 59/42 | 13 | 54/45 | 15 | 25/22 | 7 | 88/65 | 22 | 113/87 | 14 |
| > 35 daysc | 64/52 | 17 | 65/56 | 19 | 51/45 | 15 | 78/63 | 21 | 129/108 | 18 |
| Missed visit | 342/166 | 53 | 412/184 | 62 | 372/179 | 58 | 382/171 | 57 | 754/350 | 57 |
Patient follow-up
Of the 610 study participants, 525 completed the trial. Overall, 87% of all scheduled visits were attended (12,761 out of 14,640). Over 60% of patients attended 23 or more visits and 37% attended all 25 visits. Of the 525 participants who completed the trial, 518 attended the 24-month final visit; the other seven did not attend the 24-month visit, but did not withdraw, and survived beyond the scheduled 24-month visit date. The pattern of missed visits was similar for the two drugs and two treatment regimens (Figure 12). The distribution of number of visits attended, by drug and treatment frequency, is shown in Figure 13. Appendix 3 shows the number of participants at each centre, along with the breakdown of the number of visits attended, the number of injections administered and the number of missed visits at each centre.
FIGURE 12.
Distribution of the number of missed visits per visit (a) by drug and (b) by treatment regimen. Note that all patients attended visit 0, as this was a requirement for inclusion in the IVAN cohort.
FIGURE 13.
Distribution of the number of visits attended per participant (a) by drug and (b) by treatment regimen. The minimum number of visits attended was 1 and the maximum was 25. This does not include exit visits.
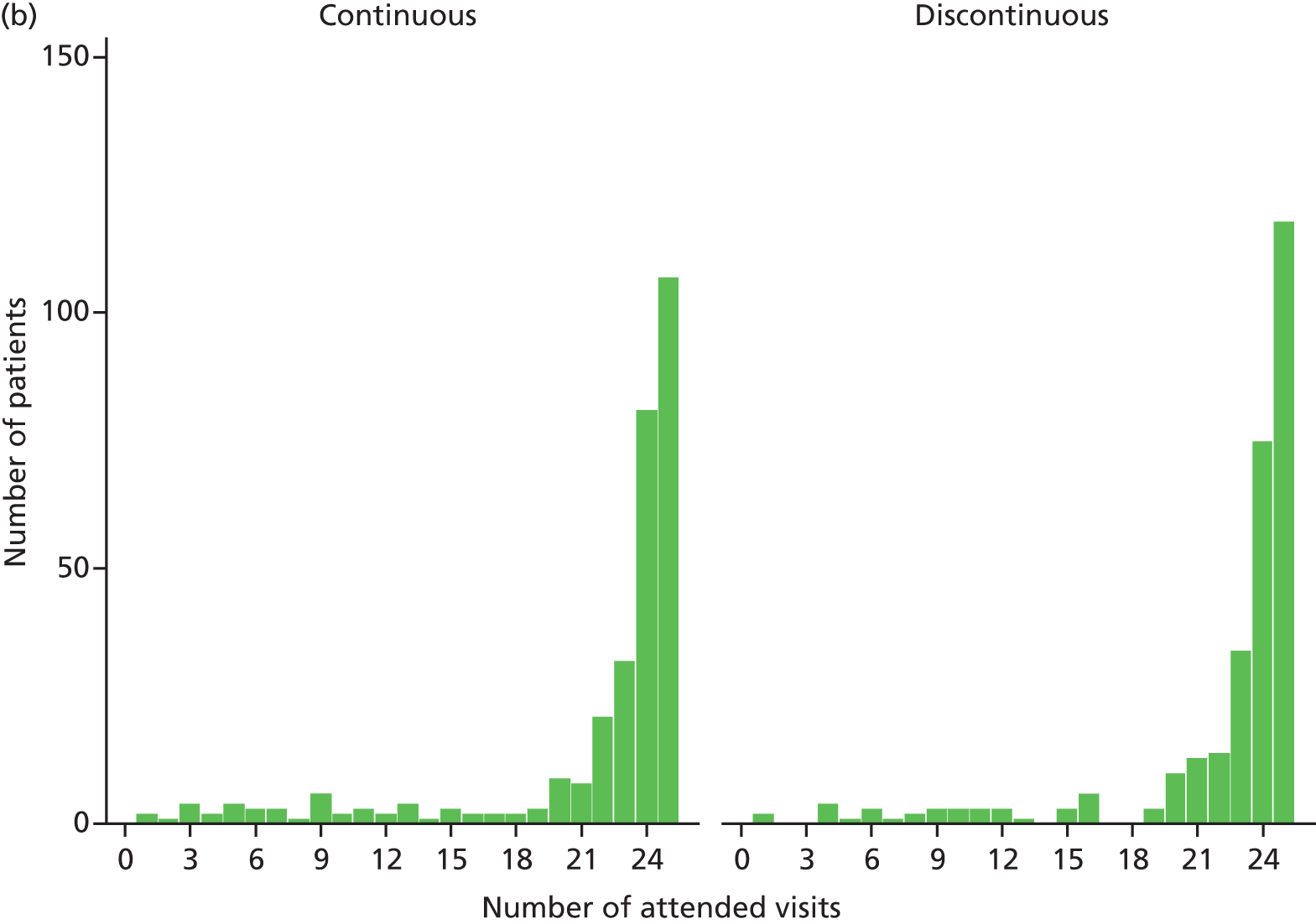
Numbers analysed
A total of 610 participants in the analysis population were included in tables of demographic characteristics, analyses of the primary outcome and secondary outcomes measured at multiple time points, time to treatment failure and of safety outcomes. The 525 participants who were classified as completing the trial (with BCVA at 2 years) are included in the descriptive outcome tables. Of the 525, 518 attended the month 24 exit visit; the remaining seven missed the month 24 exit visit (see Patient follow-up, above).
The 90 participants who withdrew (60 withdrawals) or who died (30 deaths) are reconciled with the 525 with data at 2 years as follows:
-
One participant was recorded as having withdrawn, who then died within 105 days; this participant was classified both as a withdrawal and a death.
-
Two participants completed visit 24 and died within 105 days; these participants are included in the 2-year cohort in the summary outcome tables and are also classified as deaths.
-
Two participants missed visit 24 (but did not withdraw: see above) and died within 105 days of what would have been their visit-24 date; these participants are also included in the 2-year cohort and are classified as deaths.
Baseline data
Baseline characteristics of the trial cohort by drug and treatment regimen are shown in Table 10. The mean age was 77.7 years (SD 7.4 years) and 244 (40%) patients were male. Many (64%) were current or past smokers and almost one-fifth had a history of dyspnoea (19%). Characteristics were similar between the two groups, although slightly more participants in the bevacizumab group than the ranibizumab group had a history of angina (17% vs. 11%). Baseline visual function is described in Chapter 4. Baseline characteristics of nAMD lesions are described in Chapter 5.
| Patient characteristic | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Demography | ||||||||||
| Age, years | 77.8 | 7.6 | 77.7 | 7.3 | 77.8 | 8.0 | 77.6 | 6.8 | 77.7 | 7.4 |
| Male gender (n, %) | 129/314 | 41 | 115/296 | 39 | 126/308 | 41 | 118/302 | 39 | 244/610 | 40 |
| Blood pressure, mmHg | ||||||||||
| Systolic | 141.9 | 19.5 | 143.0 | 19.5 | 143.2 | 19.8 | 141.7 | 19.1 | 142.5 | 19.5 |
| Diastolic | 76.4 | 10.2 | 77.1 | 9.9 | 77.4 | 10.1 | 76.2 | 10.0 | 76.8 | 10.1 |
| Non-ocular past history (n, %) | ||||||||||
| Angina | 35/314 | 11 | 51/296 | 17 | 45/308 | 15 | 41/302 | 14 | 86/610 | 14 |
| Dyspnoea | 57/313 | 18 | 60/295 | 20 | 57/306 | 19 | 60/302 | 20 | 117/608 | 19 |
| MI | 24/314 | 8 | 22/296 | 7 | 26/308 | 8 | 20/302 | 7 | 46/610 | 8 |
| Transient ischaemic attack | 20/294 | 7 | 9/282 | 3 | 15/291 | 5 | 14/285 | 5 | 29/576 | 5 |
| Stroke | 7/314 | 2 | 7/295 | 2 | 4/308 | 1 | 10/301 | 3 | 14/609 | 2 |
| DVT/PE | 16/313 | 5 | 18/295 | 6 | 16/308 | 5 | 18/300 | 6 | 34/608 | 6 |
| Current or past smoker | 200/309 | 65 | 185/295 | 63 | 194/305 | 64 | 191/299 | 64 | 385/604 | 64 |
Treatment received
Over 10,000 injections were given and, overall, participants received a median of 18 injections during the trial (IQR 12–23 injections). The number of injections received was similar by drug, the ranibizumab group receiving a median of 18 injections (IQR 11–23 injections) and the bevacizumab group receiving a median of 19 injections (IQR 12–23 injections). As anticipated, participants randomised to continuous treatment received a higher number of injections [median 23 injections (IQR 21–24 injections)] than those randomised to the discontinuous regimen [median 13 injections (IQR 8–17 injections); Figure 14].
FIGURE 14.
Distribution of the number of injections received per participant (a) by drug and (b) by treatment regimen. The minimum number of injections given was 1 and the maximum was 24 (patients were not given an injection at the 24-month visit). This does not include exit visits.
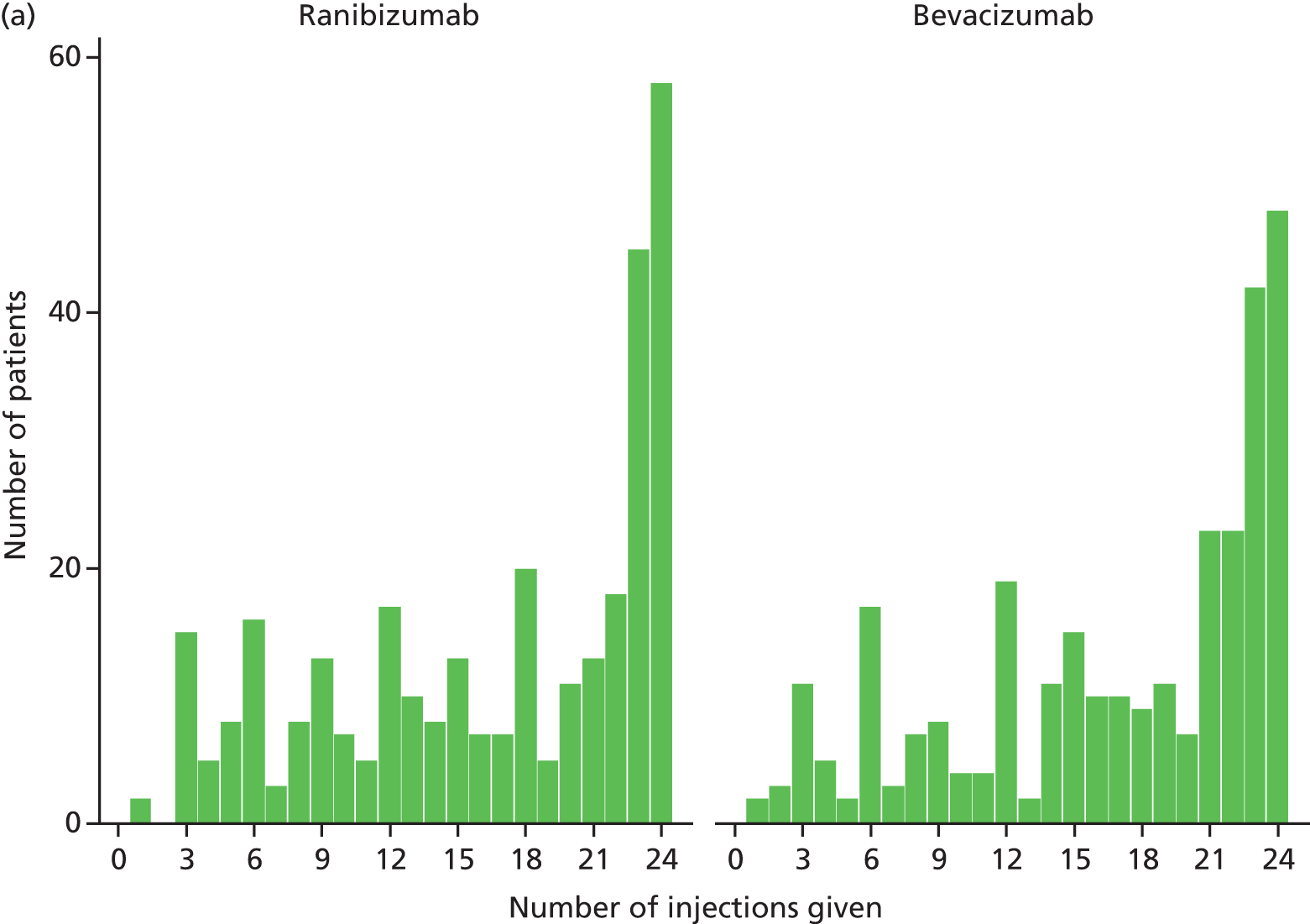

Success of masking
Regarding adequacy of masking, ophthalmologists reported not knowing which drug participants were receiving on 97.9% (555 out of 567) of visits 3, 98.7% (514 out of 521) of visits 12, 98.8% (506 out of 512) of visits 24, and 100% (22 out of 22) of exit visits. At visit 3, 4 out of 12 ophthalmologists who reported the treatment that they thought the patient received were incorrect, with 2 out of 7 incorrect and 2 out of 5 incorrect at visits 12 and 24, respectively.
Participants reported not knowing which drug they were receiving on 99.3% (560 out of 564) of visits 3, 98.7% (509 out of 516) of visits 12, 97.8% (499 out of 510) of visits 24, and 100% (21 out of 21) of exit visits. At visit 3, 2 out of 4 participants who reported the treatment that they thought they were receiving were incorrect, with 4 out of 7 incorrect and 5 out of 11 incorrect at visits 12 and 24, respectively.
Drug accountability and unmasking
Drug accountability
Drug stocks and usage was checked during monitoring visits by the central coordinating centre and from stock returned at the end of the trial. In total, 288 vials of ranibizumab were wasted over the total duration of the trial. There were two main reasons. A pharmacy refrigerator broke down at one site (so all drugs were lost because the required temperature was not maintained). The pharmacy at another site refused to reissue vials that had been dispensed but not used for a previous participant (e.g. participant in the discontinuous group who did not require treatment to be restarted); unfortunately, the pharmacy’s refusal to reissue the drug was not communicated to the coordinating centre until a large number of ranibizumab vials had been wasted.
Unmasking
There were occasions when unmasking was considered but, after discussion with staff at the centre, on each occasion it was agreed that unmasking was not required, usually because it was accepted that knowing the drug allocation would not influence the management of the participant.
Chapter 4 Visual function
Best corrected distance visual acuity
The mean baseline BCVA in the study eye was 61.4 letters (SD 15.3 letters). The BCVA was similar for ranibizumab and bevacizumab groups (mean 61.1 vs. 61.8 letters), but the average BCVA was almost 3 letters higher in the discontinuous group than in the continuous group (62.9 vs. 60.1 letters) (Table 11). For the majority of participants the study eye had poorer vision than the fellow eye at recruitment (see Table 11).
| BCVA (letters) | Randomised to | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Study eye | ||||||||||
| Baseline | 61.8 | 15.0 | 61.1 | 15.5 | 60.1 | 15.5 | 62.9 | 15.0 | 61.4 | 15.3 |
| 1 year | 69.1 | 15.7 | 66.2 | 17.1 | 66.9 | 17.0 | 68.5 | 15.9 | 67.7 | 16.5 |
| 2 years | 67.8 | 17.0 | 66.1 | 18.4 | 66.6 | 17.9 | 67.3 | 17.5 | 67.0 | 17.7 |
| Change from baseline at 1 year | 6.4 | 12.5 | 4.7 | 12.3 | 6.1 | 13.8 | 5.1 | 10.9 | 5.6 | 12.4 |
| Change from baseline at 2 years | 4.9 | 15.0 | 4.1 | 13.5 | 5.5 | 15.3 | 3.5 | 13.1 | 4.5 | 14.3 |
| Fellow eye | ||||||||||
| Baseline | 65.9 | 25.1 | 65.1 | 24.8 | 67.3 | 23.9 | 63.7 | 26.3 | 65.5 | 25.1 |
| > 5 letters worse than study eye, n (%) | 81/306 | 26 | 78/287 | 27 | 69/301 | 23 | 90/292 | 61 | 159/593 | 27 |
| ± 5 letters difference, n (%) | 50/306 | 16 | 39/287 | 14 | 42/301 | 14 | 47/292 | 16 | 89/593 | 15 |
| > 5 letters better than study eye, n (%) | 175/306 | 57 | 170/287 | 59 | 190/301 | 63 | 155/292 | 53 | 345/593 | 58 |
The BCVA in the study eye by treatment group and visit is shown in Figure 15. The mean BCVA at 1 year was 69.1 and 66.2 letters in ranibizumab and bevacizumab groups, respectively, and 66.9 and 68.5 in continuous and discontinuous groups. On average, there was little change in BCVA from 1 to 2 years; the mean BCVA at 2 years was 67.8 and 66.1 letters in ranibizumab and bevacizumab groups, respectively, and 66.6 and 67.3 letters in continuous and discontinuous groups. Change in BCVA in the study eye from baseline by treatment group and visit is shown in Figure 16. Bands of VA at baseline and 2 years by treatment group are shown in Appendix 3.
FIGURE 15.
Best corrected distance visual acuity in the study eye by visit (a) by drug and (b) by treatment regimen. The circles indicate the mean and the bars indicate one SD either side of the mean. The numbers in parentheses are the number of observations. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.


FIGURE 16.
Change from baseline in BCVA in the study eye by visit (a) by drug and (b) by treatment regimen.
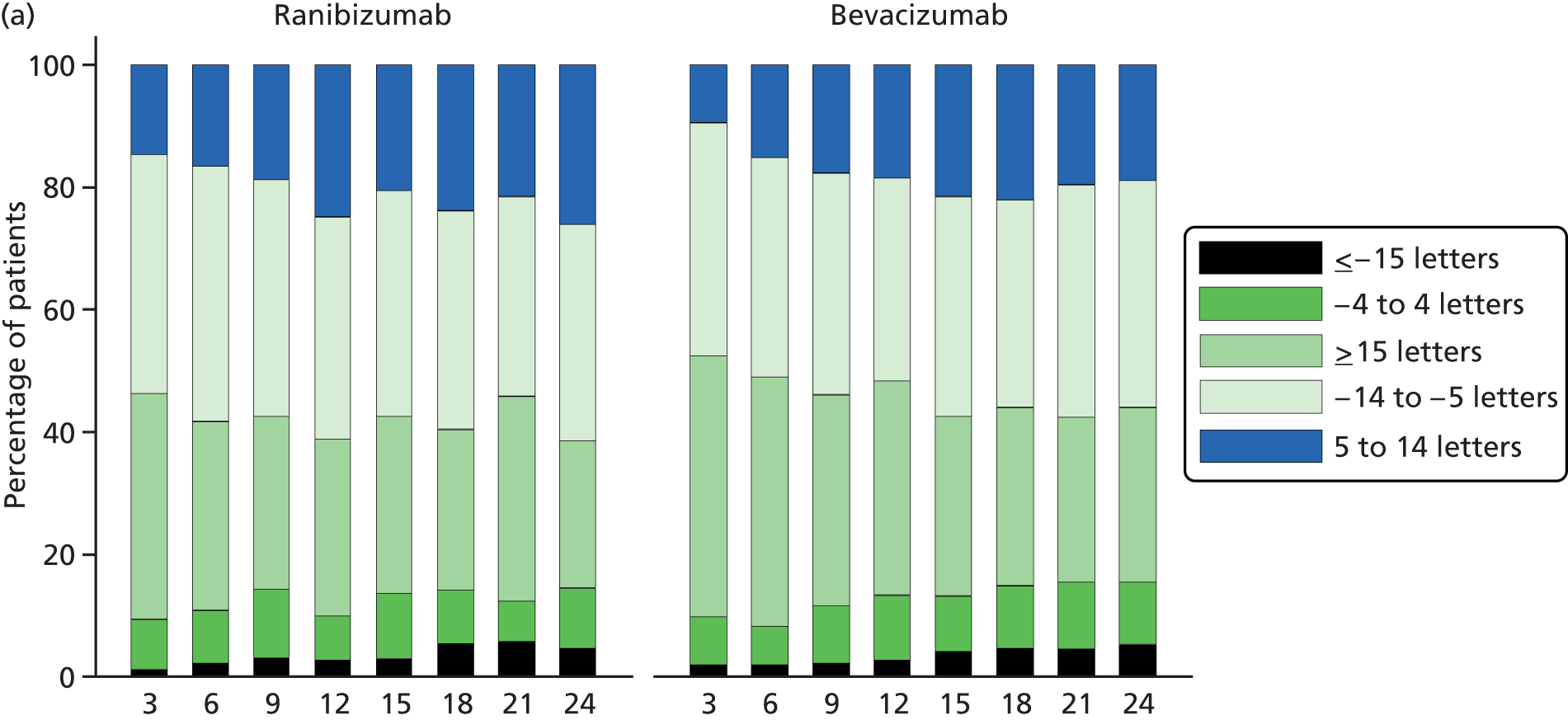
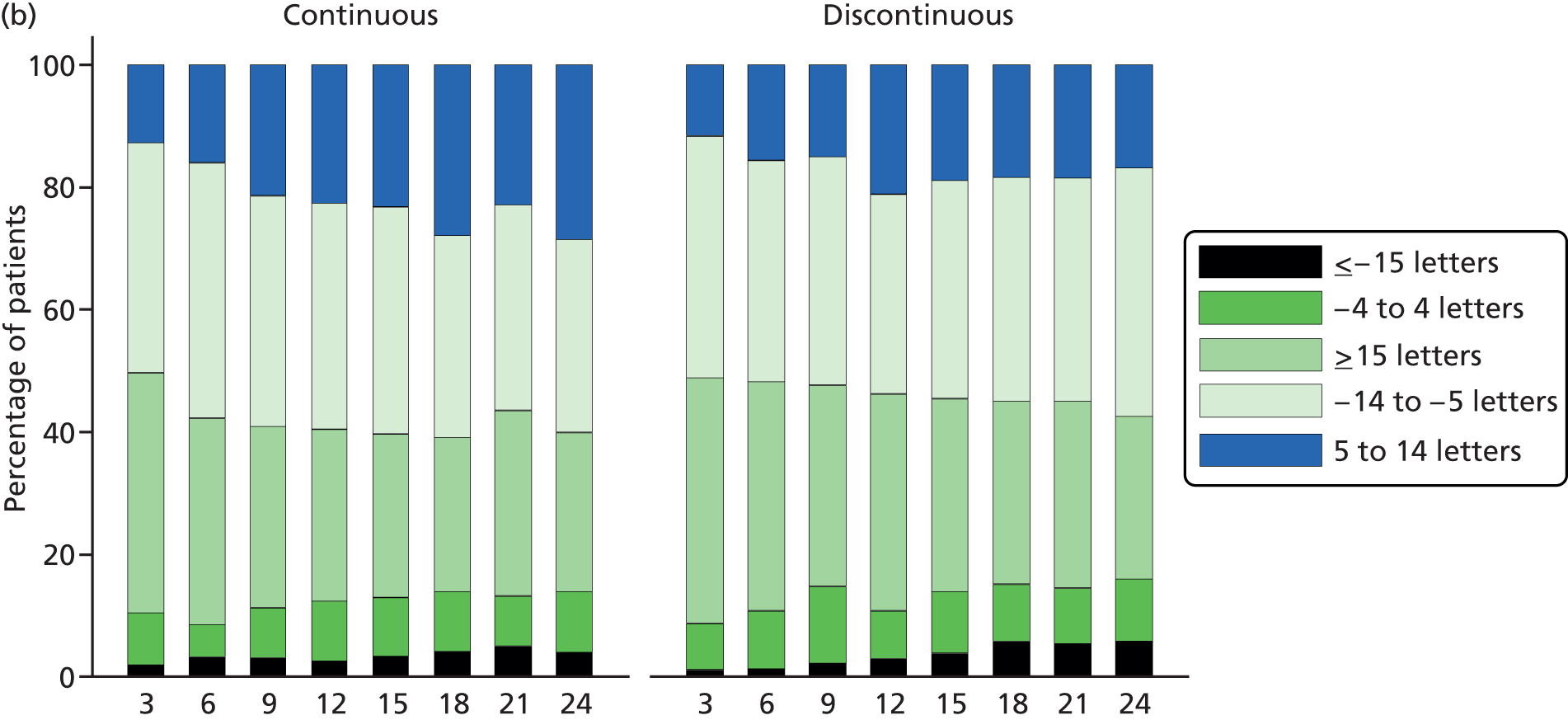
At 1 year, the estimated difference between drugs (bevacizumab minus ranibizumab, estimated from the mixed regression model) was –1.99 letters (95% CI –4.04 to +0.06 letters; p = 0.056) and between treatment regimens (discontinuous minus continuous) –0.35 letters (95% CI –2.40 to +1.70 letters; p = 0.74). 26 The comparison by drug was inconclusive; bevacizumab was neither inferior (because the CI includes zero) nor non-inferior (because the CI includes the non-inferiority margin) to ranibizumab using the 3.5-letter limit, but discontinuous treatment was equivalent to continuous treatment. At the primary outcome point of 2 years, mean BCVA did not differ significantly either by drug or by regimen. The difference between drugs (bevacizumab minus ranibizumab) was –1.37 letters (95% CI –3.75 to +1.01 letters; p = 0.26) and between treatment regimens (discontinuous minus continuous) –1.63 letters (95% CI – 4.01 to +0.75 letters: p = 0.18). Bevacizumab was neither inferior nor non-inferior to ranibizumab, and discontinuous was neither inferior nor non-inferior to continuous treatment, using the 3.5-letter non-inferiority margin (Figure 17).
FIGURE 17.
Difference in BCVA at 2 years. Negative values reflect a better mean VA in the ranibizumab or continuous groups. CIs extending beyond the non-inferiority limit of –3.5 letters indicate that the comparison of the two groups is inconclusive (ranibizumab vs. bevacizumab and continuous vs. discontinuous). Difference is estimated using data from visits 0, 3, 6, 9, 12, 15, 18, 21 and 24 adjusted for centre size. Circles = MD. 95% CIs are illustrated by horizontal bars. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.

The sensitivity analyses outlined in Chapter 2 (see Statistical methods) gave results consistent with the main analysis.
In the analysis restricting visits to those during which other functional outcomes were assessed, the mean difference (MD) for the drug effect was estimated at –1.70 letters (95% CI –4.08 to +0.68 letters; p = 0.16). The analysis excluding deferred visits gave an estimate of –1.35 letters (95% CI –3.73 to +1.03 letters; p = 0.26). The corresponding estimated effects for the treatment regimen comparison are –1.59 letters (95% CI –3.97 to +0.79 letters; p = 0.19) for the sensitivity analysis restricting the visits included, and –1.60 letters (95% CI –3.98 to +0.78 letters; p = 0.19) for the analysis excluding deferred visits.
Secondary measures of visual function
Visual function at baseline was similar for the two drug groups, but, as was observed for BCVA, the discontinuous group had slightly better reading ability compared with the continuous group (40.0 vs. 33.1; Table 12).
| Visual outcome | Randomised to | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| NVA, logMAR (median, IQR) | ||||||||||
| Baseline | 0.6 | 0.4 to 0.8 | 0.6 | 0.4 to 0.9 | 0.6 | 0.4 to 0.9 | 0.6 | 0.4 to 0.8 | 0.6 | 0.4 to 0.9 |
| 1 year | 0.4 | 0.3 to 0.7 | 0.5 | 0.3 to 0.8 | 0.5 | 0.3 to 0.8 | 0.4 | 0.3 to 0.7 | 0.4 | 0.3 to 0.8 |
| 2 years | 0.4 | 0.3 to 0.7 | 0.5 | 0.3 to 0.9 | 0.4 | 0.3 to 0.8 | 0.4 | 0.3 to 0.8 | 0.4 | 0.3 to 0.8 |
| Change from baseline at 1 year | –0.1 | –0.2 to 0.1 | –0.1 | –0.2 to 0.1 | –0.1 | –0.3 to 0.1 | –0.1 | –0.2 to 0.1 | –0.1 | –0.2 to 0.1 |
| Change from baseline at 2 years | –0.1 | –0.3 to 0.1 | –0.1 | –0.3 to 0.1 | –0.1 | –0.3 to 0.1 | 0.0 | –0.2 to 0.1 | –0.1 | –0.3 to 0.1 |
| Belfast reading index (median, IQR) | ||||||||||
| Baseline | 36.9 | 15.6 to 65.3 | 35.0 | 14.0 to 69.6 | 33.1 | 13.6 to 67.7 | 40.0 | 16.3 to 69.6 | 36.5 | 14.6 to 68.4 |
| 1 year | 57.5 | 23.4 to 94.4 | 51.8 | 11.5 to 94.6 | 52.4 | 12.6 to 90.7 | 56.1 | 24.2 to 99.2 | 54.9 | 16.8 to 94.6 |
| 2 years | 50.9 | 22.8 to 93.7 | 52.5 | 9.7 to 90.6 | 46.3 | 11.4 to 84.0 | 55.4 | 19.0 to 97.6 | 52.00 | 15.6 to 93.6 |
| Test not performed due to poor vision (n, %) | ||||||||||
| Baseline | 5/298 | 2 | 0/283 | 0 | 3/290 | 1 | 2/291 | 1 | 5/581 | 1 |
| 1 year | 5/267 | 2 | 5/250 | 2 | 4/259 | 2 | 6/258 | 2 | 10/517 | 2 |
| 2 years | 8/256 | 3 | 2/241 | 1 | 3/250 | 1 | 7/247 | 3 | 10/497 | 2 |
| Change from baseline at 1 year | 8.8 | –7.1 to 33.0 | 5.3 | –10.1 to 39.5 | 7.8 | –11.4 to 36.8 | 7.9 | –6.5 to 33.9 | 7.8 | –8.4 to 35.5 |
| Change from baseline at 2 years | 5.6 | –16.1 to 37.8 | 1.3 | –11.8 to 32.2 | 1.3 | –12.3 to 32.0 | 4.6 | –14.5 to 38.7 | 3.3 | –12.6 to 35.2 |
| Contrast sensitivity, letters (mean, SD) | ||||||||||
| Baseline | 26.2 | 6.2 | 26.3 | 5.7 | 26.1 | 6.0 | 26.4 | 5.9 | 26.2 | 6.0 |
| 1 year | 28.4 | 4.9 | 28.7 | 5.4 | 28.7 | 5.1 | 28.4 | 5.1 | 28.6 | 5.1 |
| 2 years | 28.1 | 6.0 | 28.3 | 5.8 | 28.7 | 5.4 | 27.7 | 6.3 | 28.2 | 5.9 |
| Change from baseline at 1 year | 2.1 | 4.9 | 2.1 | 5.0 | 2.4 | 5.1 | 1.8 | 4.8 | 2.1 | 5.0 |
| Change from baseline at 2 years | 1.5 | 5.9 | 1.7 | 5.1 | 2.2 | 5.2 | 1.0 | 5.8 | 1.6 | 5.5 |
Near visual acuity in the study eye by treatment group and visit is shown in Figure 18. Corresponding data for reading index and contrast sensitivity are shown in Figures 19 and 20. Binocular vision is shown in Appendix 3. At 1 year, NVA was 8% worse in the bevacizumab group (GMR 0.92, 95% CI 0.84 to 1.00; p = 0.058) but did not differ by treatment regimen. 26 By the end of year 2, the average difference by drug was no longer significant (GMR 0.94, 95% CI 0.85 to 1.04; p = 0.23). However, NVA was better when the treatment had been administered monthly for 2 years (GMR 0.90, 95% CI 0.82 to 0.99; p = 0.04). The reading index which takes into account the reading speed and the NVA threshold (by adjusting for size of the print being read) was, however, similar between both drug and regimen groups (see Table 12). Contrast sensitivity did not differ significantly by either drug or regimen groups at 1 year. However, although the drugs were similar, continuous treatment resulted in better outcomes at 2 years (MD –1.07, 95% CI –1.90 to –0.25; p = 0.011; Figure 21).
FIGURE 18.
Near visual acuity in the study eye by visit (a) by drug and (b) by treatment regimen. The direction of the logMAR scale for NVA is opposite to that for BCVA (letters), i.e. small logMAR values represent better vision. The circles indicate the median and the bars indicate the IQR. The numbers in parentheses are the number of observations.

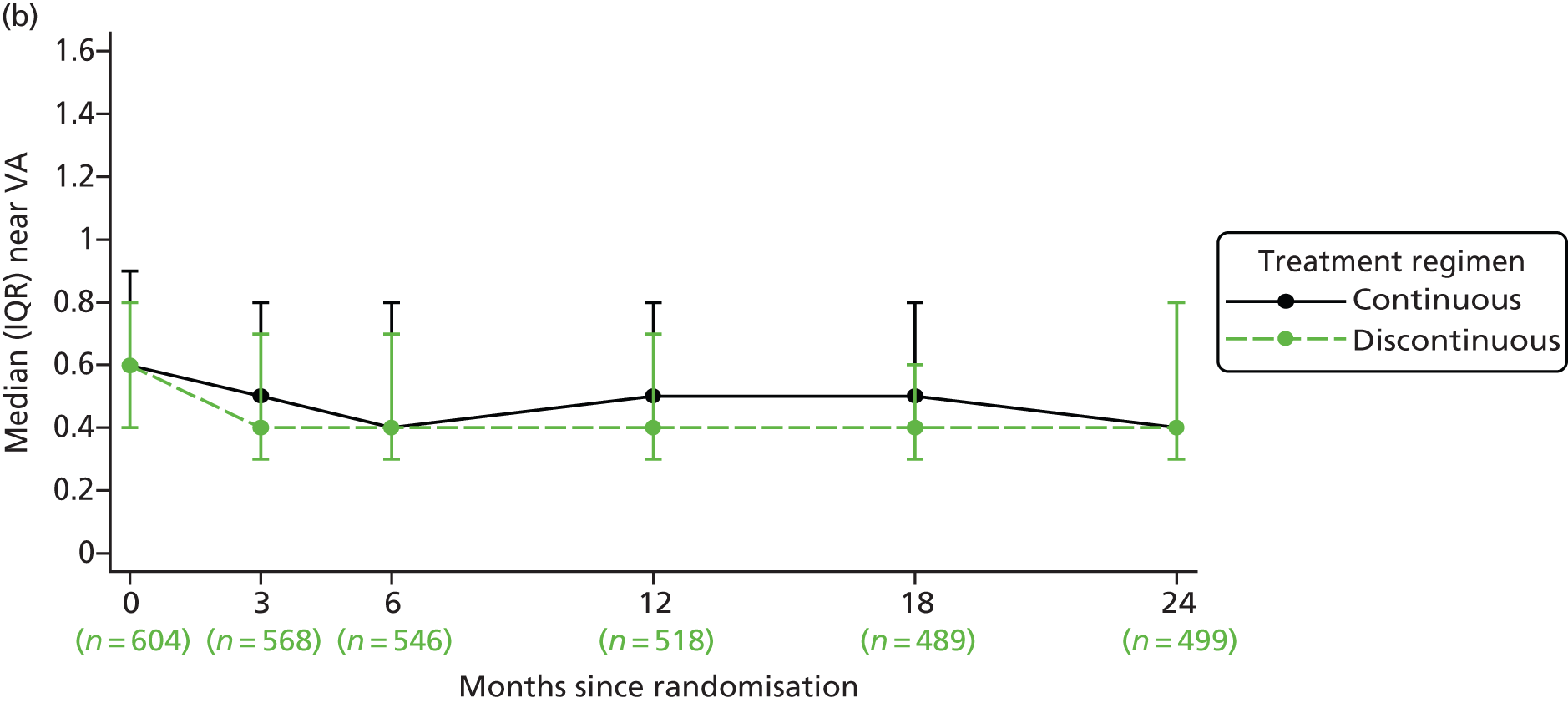
FIGURE 19.
Reading index in the study eye by visit (a) by drug and (b) by treatment regimen. The circles indicate the median and the bars indicate the IQR. The numbers in parentheses are the number of observations.
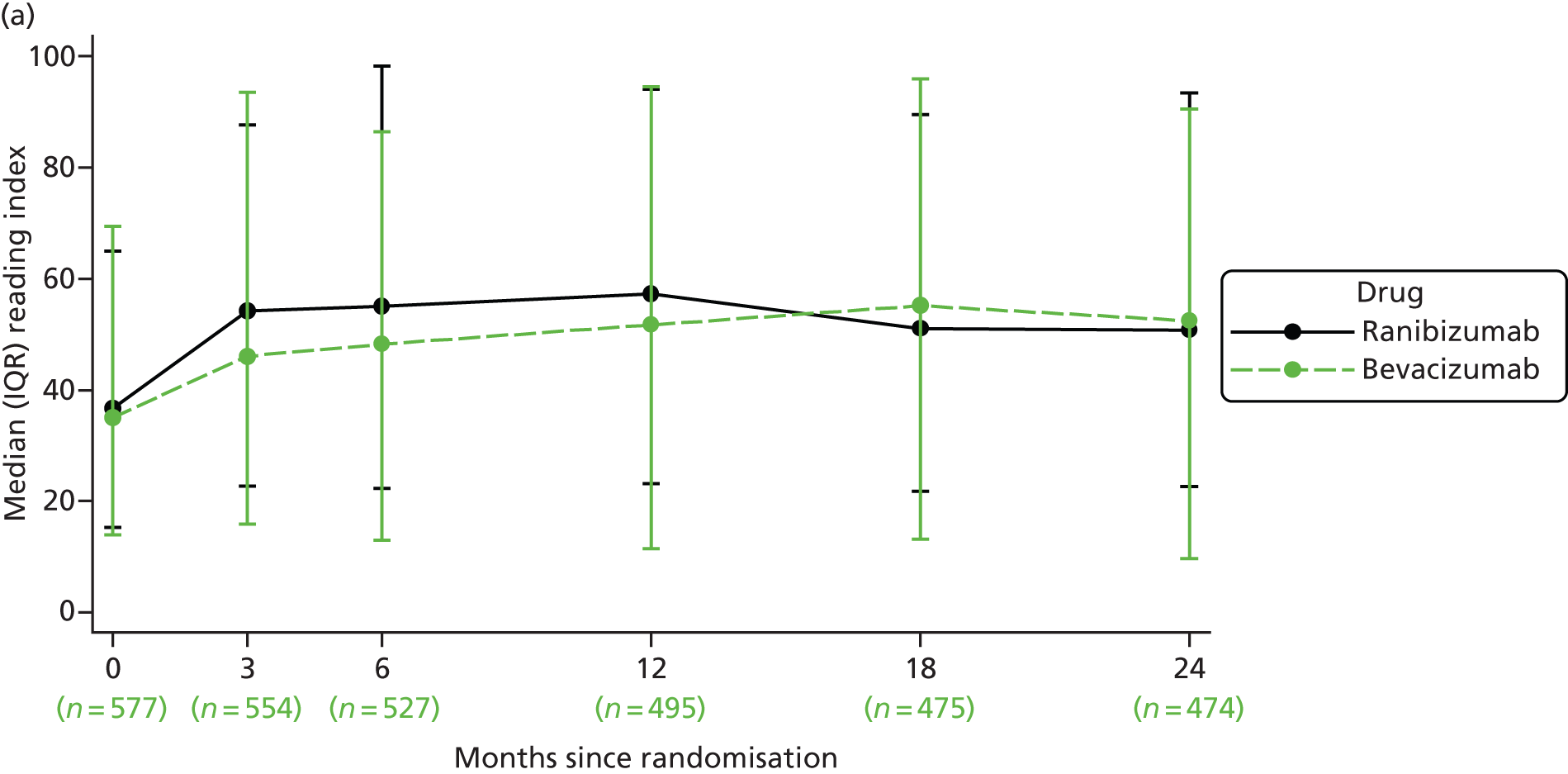

FIGURE 20.
Contrast sensitivity in the study eye by visit (a) by drug and (b) by treatment regimen. The circles indicate the mean and the bars indicate one SD either side of the mean. The numbers in parentheses are the number of observations.
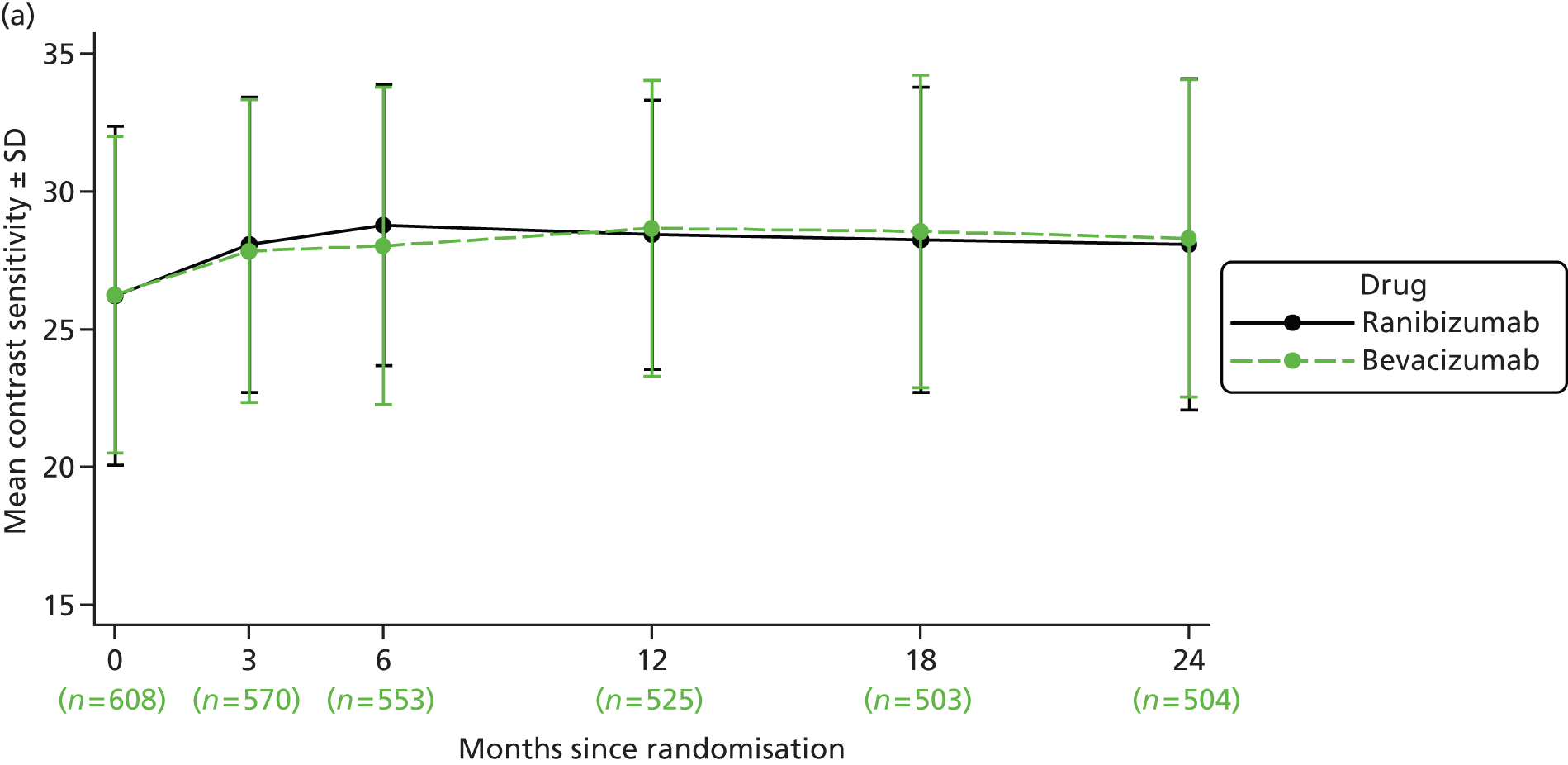
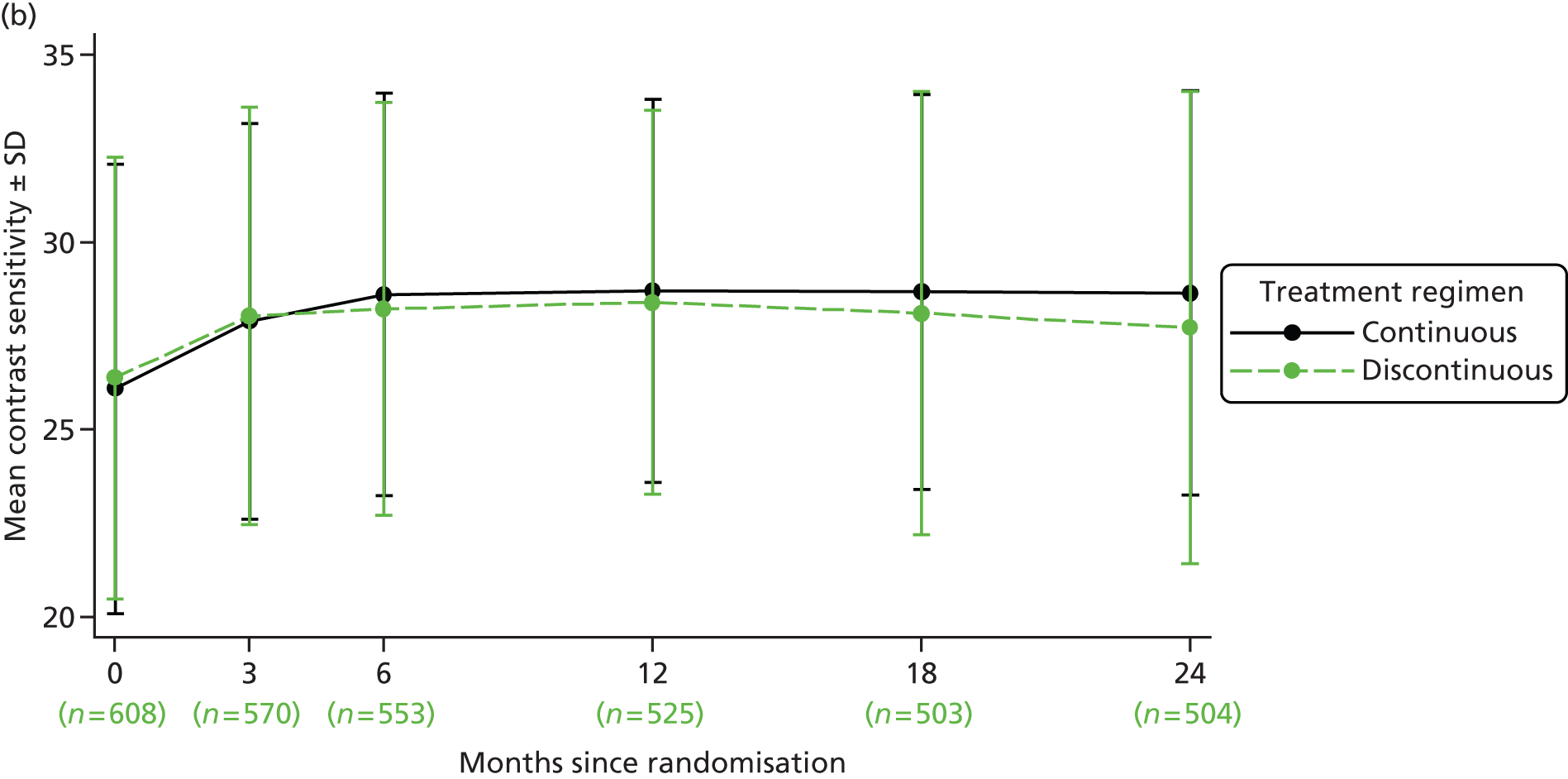
FIGURE 21.
Difference in secondary measures of visual function at 2 years (a) by drug and (b) by treatment regimen. Negative values of MD and GMR values of < 1 reflect a better outcome in the ranibizumab or continuous groups. Difference is estimated using data from visits 0, 3, 6, 12, 18 and 24 adjusted for centre size. Circles = MD or GMR. 95% CIs are illustrated by horizontal bars. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
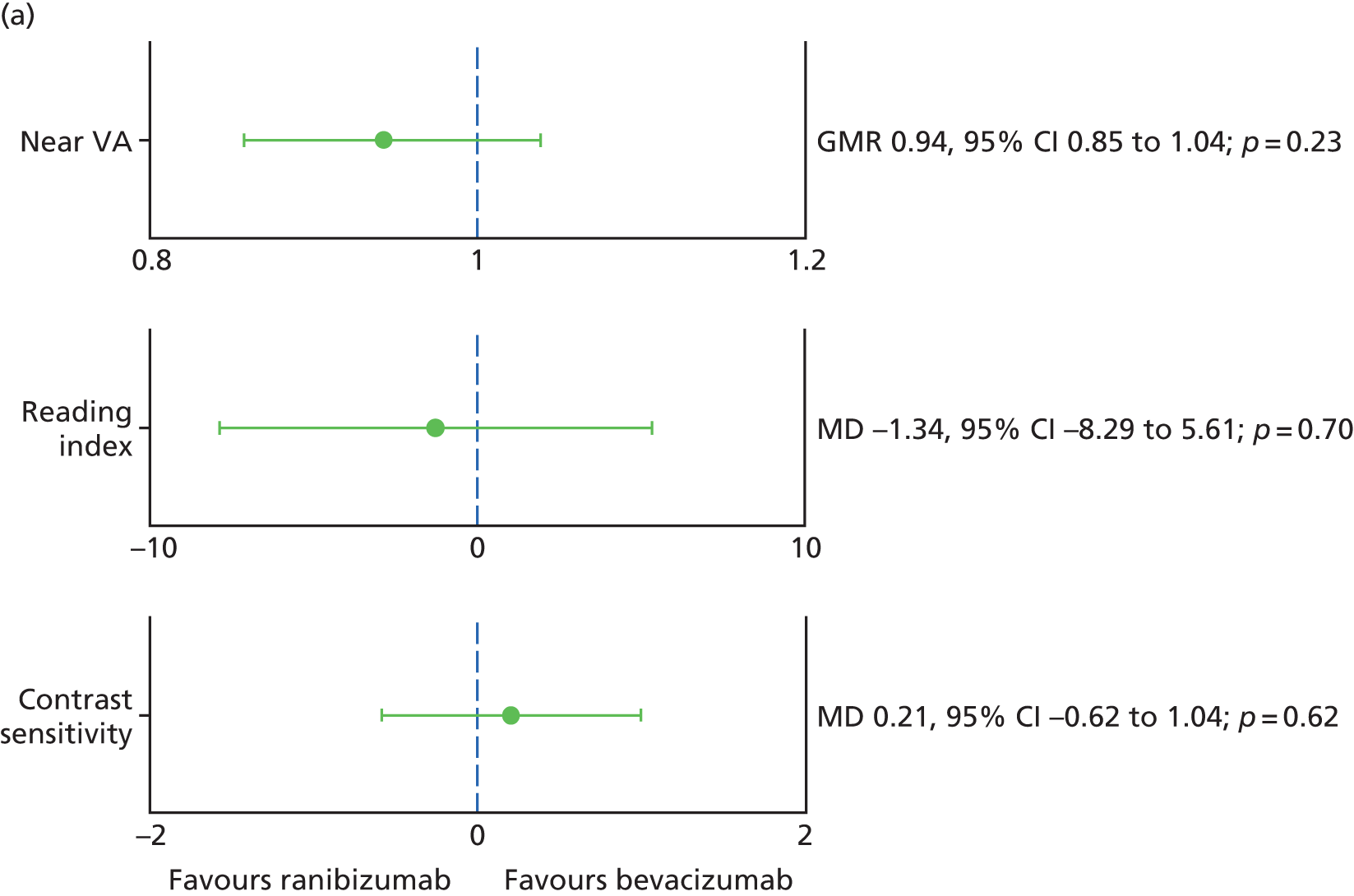

Treatment failure
Overall, 475 (78%) participants met the criteria for retreatment (treatment failure) during their participation in the trial. The median time from randomisation to first treatment failure was 5.1 months (IQR 3.7–16.8 months) in the ranibizumab group and 4.9 months (3.2 to 14.0 months) in the bevacizumab group. For those randomised to continuous treatment the median time to first treatment failure was 7.6 months (lower quartile 3.2 months) compared with 4.4 months in the discontinuous group (IQR 3.2–6.9 months; Figure 22).
FIGURE 22.
Time to first treatment failure (a) by drug and (b) by treatment regimen.

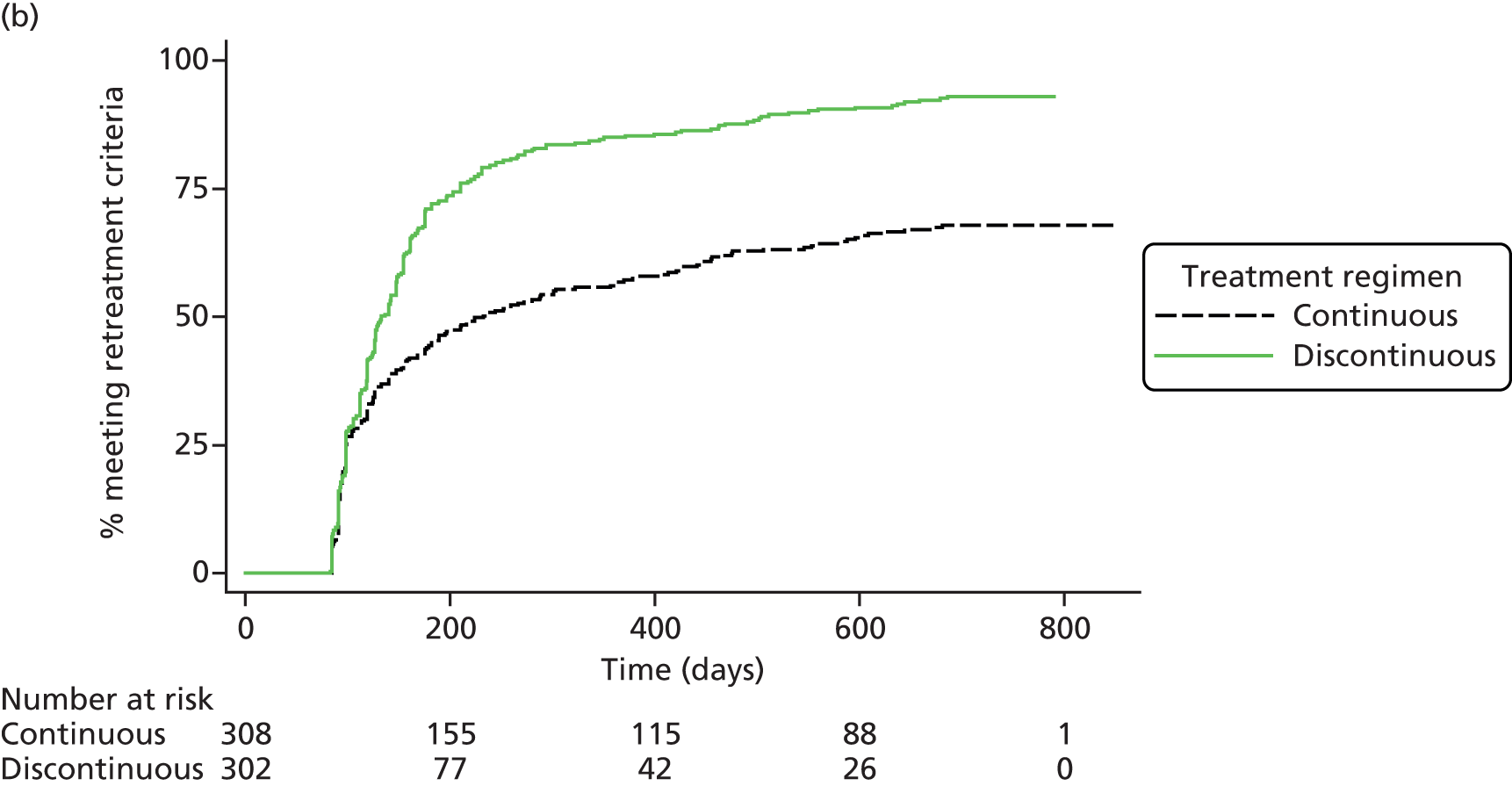
Comparing the time to first treatment failure from visit 2 (end of loading dose phase of the trial), no difference in the risk of treatment failure was found between the drugs [HR (bevacizumab–ranibizumab) 1.13, 95% CI 0.94 to 1.36; p = 0.18). In contrast, the risk of treatment failure was significantly higher for participants randomised to discontinuous treatment rather than continuous treatment [HR (discontinuous–continuous) 1.95, 95% CI 1.61 to 2.35; p < 0.001].
Subgroup analyses
Differences in BCVA by drug and treatment regimen in the five pre-planned subgroups are shown in Figure 23. No statistically significant differences by subgroup were found for the drug or treatment regimen comparisons, that is all tests for interaction were not significant (p ≥ 0.26). Two additional subgroups, which were not specified in the protocol, were also considered: (1) whether or not the vision in study eye was at least 5 letters better than in the fellow eye at baseline and (2) whether or not a haemorrhage was present at baseline. No statistically significant differences between these subgroups were found for the drug or treatment regimen comparisons (see Appendix 3).
FIGURE 23.
Best corrected distance visual acuity in the study eye at 2 years by subgroup (a) by drug and (b) by treatment regimen. Negative values of MD reflect a better mean VA in the ranibizumab or continuous groups. Difference is estimated using data from visits 0, 3, 6, 9, 12, 15, 18, 21 and 24, adjusted for centre size. Circles = MD. 95% CIs are illustrated by horizontal bars. Circle size is relative to the size of the subgroup.
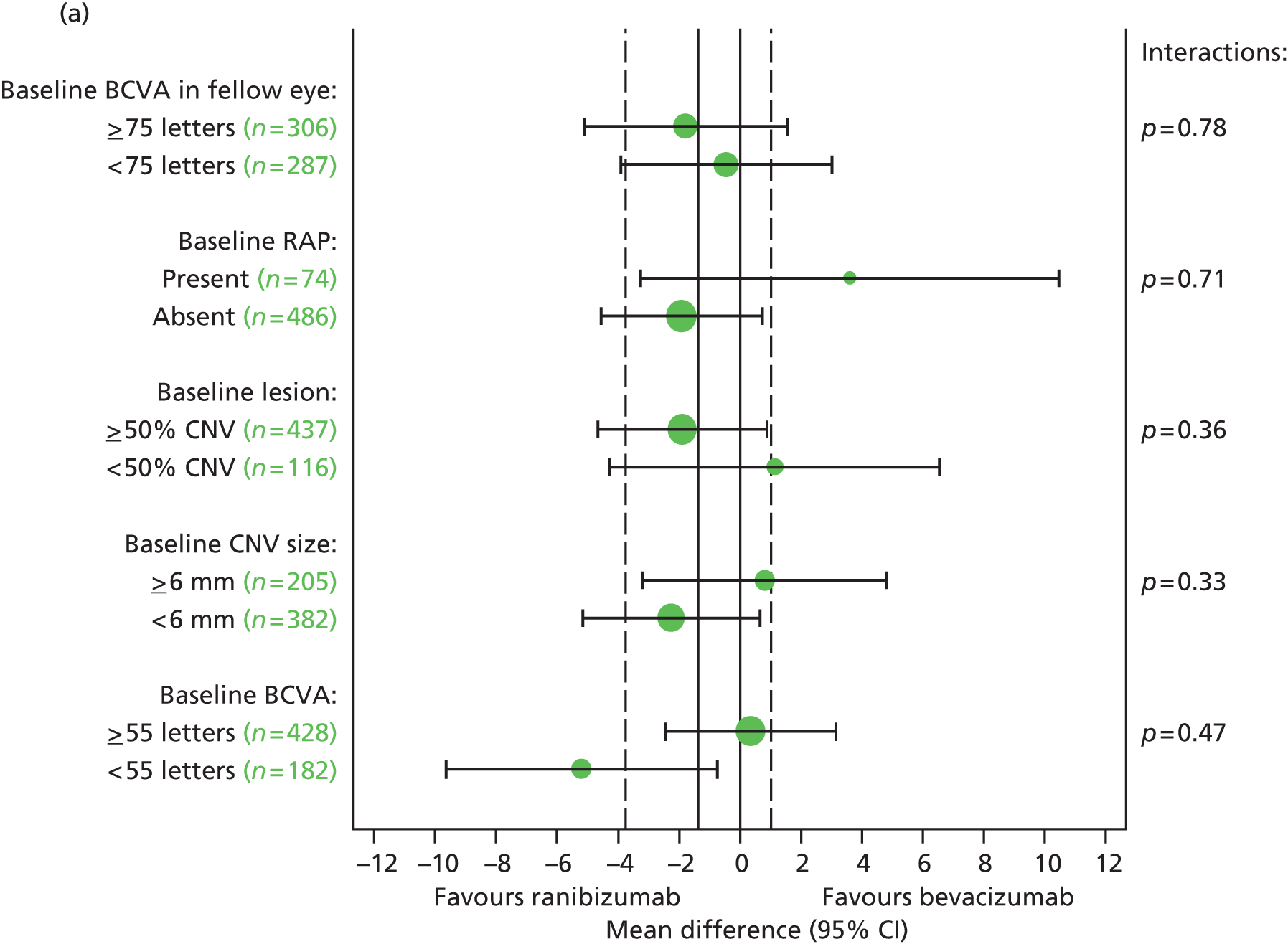

Meta-analysis
As described in Chapter 2 (see Statistical methods), meta-analyses were carried out to place the IVAN trial findings in the context of the wider evidence base. There was no intention to carry out a formal systematic review and findings from these analyses are, therefore, not reported in line with PRISMA guidelines.
The meta-analysis of BCVA was complicated by the fact that triallists have used different ways to analyse BCVA data and to report their findings. With the aim of being as inclusive as possible, we have performed a single meta-analysis including trials that had 1- or 2-year follow-up, using the findings for the longest duration (as BCVA changes very little between 1 and 2 years). We used results from presentations at international conferences when final journal publications were not available.
Figure 24 shows meta-analyses of the trial findings for seven trials [including IVAN, CATT, BRAMD (presented at the 2013 meeting of EURETINA) GEFAL, LUCAS (presented at the 2013 meeting of the American Association for Ophthalmology), MANTA and Subramanian et al. ]22,42–44 for changes in distance BCVA by drug and of the CATT22 and IVAN trial findings for changes in distance BCVA by treatment regimen. The analyses use the 2-year findings for the CATT22 and IVAN, and 1-year findings for all other studies. The pooled BCVA point estimate shows that bevacizumab was statistically non-inferior to ranibizumab judged by the stricter IVAN margin [–0.38 letters; 95% CI –1.47 to +0.70 letters; p = 0.49; heterogeneity chi-squared (χ2) = 4.08; degrees of freedom (df) = 6; p = 0.666; I2 = 0.0%]. Acknowledging the slight difference between the CATT22 and IVAN trial in their ‘as needed’ treatment regimens, discontinuous treatment was significantly inferior to continuous (–2.23 letters; 95% CI –3.93 to –0.53 letters; p = 0.010).
FIGURE 24.
Meta-analysis of change in BCVA from baseline (a) by drug and (b) by treatment regimen. Change in BCVA = 2 years minus baseline or 1 year minus baseline, as indicated in the figures. Negative values for differences reflect better outcomes in the ranibizumab or continuous groups. WMD, weighted mean difference. 95% CIs are given in parentheses and illustrated by horizontal bars. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
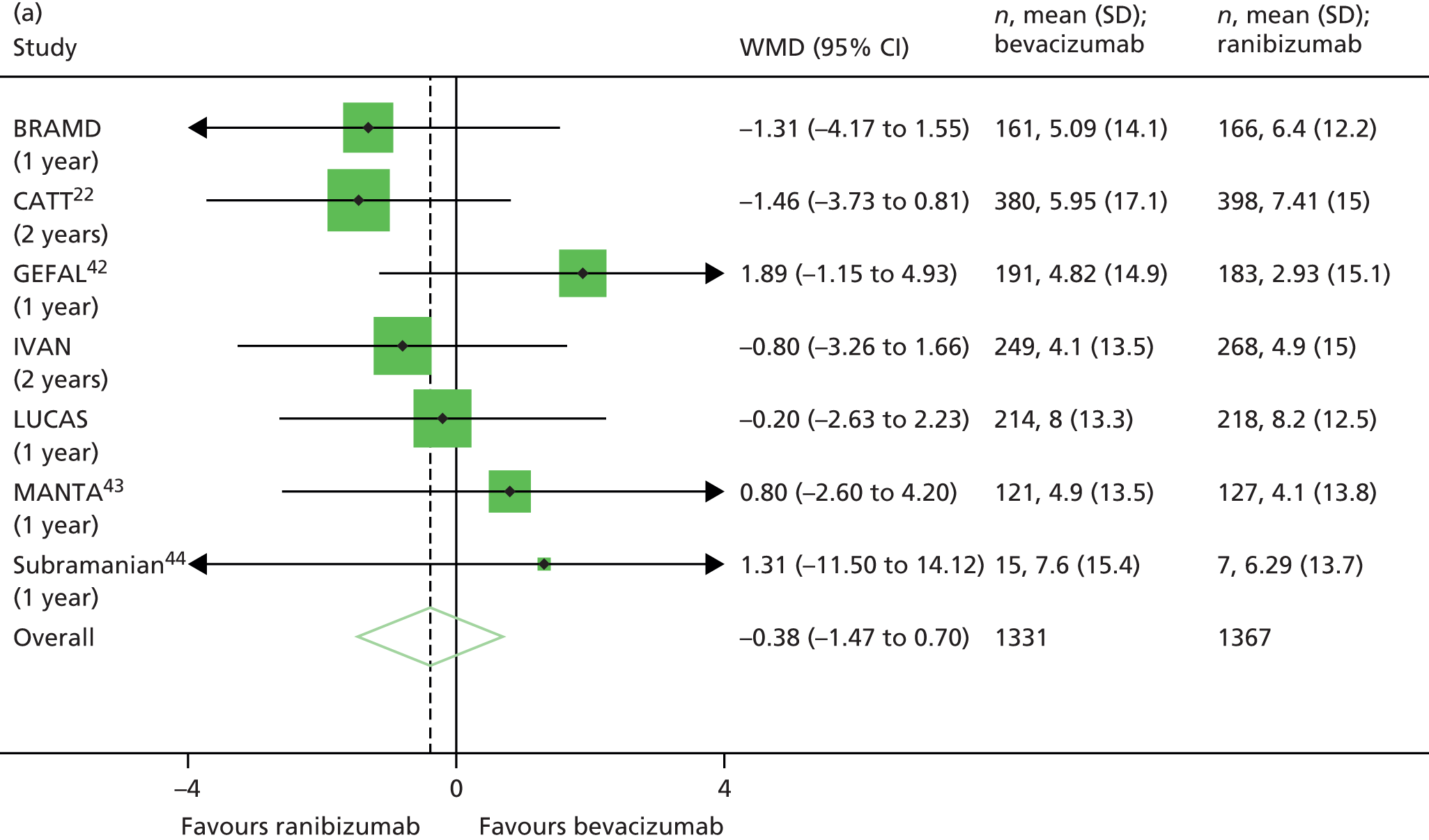

Summary
The visual function at baseline was similar by drug, but the discontinuous group had slightly better BCVA and reading ability than the continuous group. For the majority of participants their vision was poorer in their study eye than their fellow eye. At the primary outcome point of 2 years, the BCVA was, on average, 1.37 letters worse with bevacizumab than ranibizumab, and 1.63 letters worse with discontinuous than continuous treatment. However, bevacizumab was neither inferior nor non-inferior to ranibizumab, and discontinuous was neither inferior nor non-inferior to continuous treatment, using the 3.5-letter non-inferiority margin. At 2 years, NVA and contrast sensitivity were similar by drug, but better in the group receiving continuous treatment. Reading index was similar by drug and treatment regimen at 2 years. Time to first treatment failure was similar for the two drugs, but the risk of failure was significantly higher with discontinuous treatment than with continuous treatment. No differences by subgroup were found. Meta-analyses of the IVAN data with other available trials comparing bevacizumab with ranibizumab show that acuity outcomes are equivalent for the two drugs, and that the confidence limits and point estimates of the observed difference lies well within the IVAN trial prespecified non-inferiority margin of 3.5 letters. Only the CATT22 and IVAN trials compared continuous and discontinuous treatment regimens; meta-analyses of the findings of these trials showed that discontinuous treatment is significantly inferior to continuous treatment.
Chapter 5 Lesion morphology
Morphological characteristics of lesions
At baseline, 76% of participants had a lesion with foveal centre involvement and 7% had GA. The discontinuous group, as well as having better BCVA and reading ability than the continuous group (see Chapter 4) also had fewer participants with a lesion with foveal centre involvement (73% vs. 78%; Table 13). Other morphological characteristics were balanced across the groups at baseline, with the exception of fibrosis, which was more common in the ranibizumab group than in the bevacizumab group (29% vs. 24%; see Table 13).
| Morphological characteristic | Randomised to | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Randomised to bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Randomised to continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Randomised to discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| n | % | n | % | n | % | n | % | n | % | |
| Foveal centre involvement at baseline | 230/309 | 74 | 223/289 | 77 | 235/301 | 78 | 218/297 | 73 | 453/598 | 76 |
| CNV | 156/305 | 51 | 162/287 | 56 | 170/299 | 57 | 148/293 | 51 | 318/592 | 54 |
| Haemorrhage | 86/308 | 28 | 88/292 | 30 | 89/303 | 29 | 85/297 | 29 | 174/600 | 29 |
| Other | 47/306 | 15 | 30/292 | 10 | 39/301 | 13 | 38/297 | 13 | 77/598 | 13 |
| No CNV or unable to grade | 7/309 | 2 | 8/294 | 3 | 4/304 | 1 | 11/299 | 4 | 15/603 | 2 |
| Blood present | ||||||||||
| Baseline | 187/308 | 61 | 180/293 | 61 | 189/303 | 62 | 178/298 | 60 | 367/601 | 61 |
| 1 year | 23/273 | 8 | 28/253 | 11 | 25/268 | 9 | 26/258 | 10 | 51/526 | 10 |
| 2 years | 20/266 | 8 | 17/242 | 7 | 13/255 | 5 | 24/253 | 9 | 37/508 | 7 |
| RPE tear present | ||||||||||
| Baseline | 3/282 | 1 | 2/263 | 1 | 2/275 | 1 | 3/270 | 1 | 5/545 | 1 |
| 1 year | 6/231 | 3 | 5/214 | 2 | 5/227 | 2 | 6/218 | 3 | 11/445 | 2 |
| 2 years | 4/236 | 2 | 4/226 | 2 | 4/234 | 2 | 4/228 | 2 | 8/462 | 2 |
| Fibrosis presenta | ||||||||||
| Baseline | 88/305 | 29 | 69/291 | 24 | 78/299 | 26 | 79/297 | 27 | 157/596 | 26 |
| 1 year | 199/260 | 77 | 197/246 | 80 | 210/256 | 82 | 186/250 | 74 | 396/506 | 78 |
| 2 years | 208/263 | 79 | 191/239 | 80 | 203/253 | 80 | 196/249 | 79 | 399/502 | 79 |
| Dye leakage present on FFA | ||||||||||
| Baseline | 275/307 | 90 | 265/289 | 92 | 272/300 | 91 | 268/296 | 91 | 540/596 | 91 |
| 1 year | 90/241 | 37 | 95/229 | 41 | 75/233 | 32 | 110/237 | 46 | 185/470 | 39 |
| 2 years | 101/247 | 41 | 89/234 | 38 | 86/241 | 36 | 104/240 | 43 | 190/481 | 40 |
| Fluid present on OCT | ||||||||||
| Baseline | 271/286 | 95 | 252/264 | 95 | 265/280 | 95 | 258/270 | 96 | 523/550 | 95 |
| 1 year | 139/268 | 52 | 142/248 | 57 | 119/256 | 46 | 162/260 | 62 | 281/516 | 54 |
| 2 years | 127/254 | 50 | 141/241 | 59 | 112/247 | 45 | 156/248 | 63 | 268/495 | 54 |
| GA present at baseline | 25/309 | 8 | 18/295 | 6 | 24/304 | 8 | 19/300 | 6 | 43/604 | 7 |
| Within lesion | 15/25 | 60 | 7/18 | 39 | 13/24 | 54 | 9/19 | 47 | 22/43 | 51 |
| Contiguous to lesion | 10/25 | 40 | 9/18 | 50 | 9/24 | 38 | 10/19 | 53 | 19/43 | 44 |
| Remote from lesion | 2/25 | 8 | 4/18 | 22 | 4/24 | 17 | 2/19 | 11 | 6/43 | 14 |
| New GA developed during trialb | 86/305 | 28 | 91/291 | 31 | 101/301 | 34 | 76/295 | 26 | 177/596 | 30 |
There were no significant differences between drugs for dye leakage on FFA, a measure of lesion activity, at both years 1 and 2. However, dye leakage on FFA was observed less frequently in the continuous treatment group than in the discontinuous treatment group at 1 year (OR 0.51, 95% CI 0.33 to 0.78; p = 0.0017). 26 This difference was not maintained into year 2, when the OR increased to 0.74 (95% CI 0.51 to 1.07; p = 0.11; Figure 25). At 1 year, fewer participants had fluid on OCT in the ranibizumab group (52% vs. 57%), and at 2 years a similar pattern was observed (50% vs. 58%; p = 0.065). When compared by treatment regimen, significantly fewer participants had fluid on OCT in the continuous group at both 1 and 2 years (45% vs. 62%; OR 0.47, 95% CI 0.33 to 0.67; p < 0.001 at 2 years; see Figure 25).
FIGURE 25.
Secondary morphological outcomes at 2 years (a) by drug and (b) by treatment regimen. Ratios of < 1 reflect better outcomes in the ranibizumab or continuous groups. Circles = GMR or OR. 95% CIs are given in parentheses and illustrated by horizontal bars.

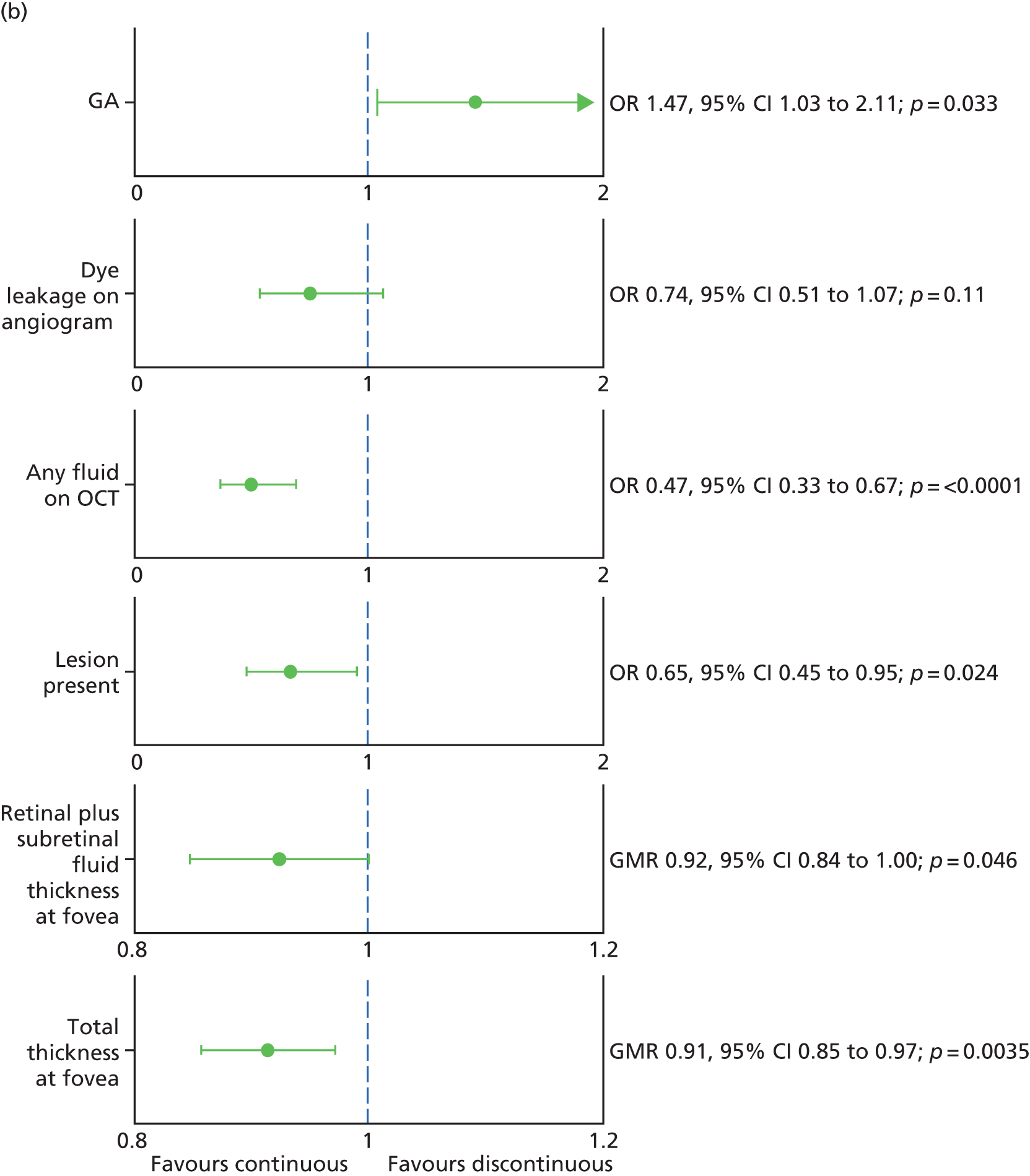
The choice of drug had no effect on the odds of an active neovascular lesion being present in year 1 or year 2. However, compared with discontinuous treatment, continuous treatment protected against an active lesion being present at both times (52% vs. 65% at 1 year, 51% vs. 61% at 2 years; OR 0.65, 95% CI 0.45 to 0.95; p = 0.024 at 2 years; see Figure 25). Despite this finding, the median neovascular lesion area – when an active lesion was judged to be present – was similar in the two groups (Table 14).
| Morphological characteristics | Randomised to | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| n/median | %/IQR | n/median | %/IQR | n/median | %/IQR | n/median | %/IQR | n/median | %/IQR | |
| Active neovascularisation | ||||||||||
| Present at baseline | 270/303 | 89 | 259/284 | 91 | 269/298 | 90 | 260/289 | 90 | 529/587 | 90 |
| Area at baseline | 2.55 | 0.86–6.20 | 2.82 | 1.21–6.71 | 2.76 | 1.14–6.44 | 2.69 | 0.92–6.49 | 2.75 | 0.98–6.44 |
| Present at 1 year | 86/241 | 36 | 93/229 | 41 | 71/233 | 30 | 108/237 | 46 | 179/470 | 38 |
| Area at 1 year | 3.11 | 0.92–7.34 | 3.58 | 1.14–7.27 | 5.21 | 1.14–7.81 | 2.44 | 0.92–7.23 | 3.44 | 1.03–7.34 |
| Present at 2 years | 100/246 | 41 | 85/235 | 36 | 82/241 | 34 | 103/240 | 43 | 185/481 | 38 |
| Area at 2 years | 4.22 | 1.58–9.28 | 4.37 | 1.87–8.34 | 4.22 | 1.83–9.10 | 4.50 | 1.52–9.18 | 4.30 | 1.67–9.14 |
| Total lesion area | ||||||||||
| Present at baseline | 286/303 | 94 | 267/284 | 94 | 285/298 | 96 | 268/289 | 93 | 553/587 | 94 |
| Area at baseline | 3.67 | 1.38–7.94 | 4.46 | 1.67–8.47 | 3.87 | 1.45–7.93 | 4.10 | 1.60–8.75 | 3.97 | 1.50–8.27 |
| Present at 1 year | 136/241 | 56 | 141/229 | 62 | 122/233 | 52 | 155/237 | 65 | 277/470 | 59 |
| Area at 1 year | 1.59 | 0.61–6.20 | 1.88 | 0.55–5.54 | 1.78 | 0.54–6.39 | 1.73 | 0.62–5.84 | 1.75 | 0.60–6.11 |
| Present at 2 years | 142/246 | 58 | 127/234 | 54 | 123/241 | 51 | 146/239 | 61 | 269/480 | 56 |
| Area at 2 years | 2.73 | 0.75–8.54 | 2.89 | 0.85–7.10 | 2.56 | 0.82–7.60 | 2.87 | 0.82–7.93 | 2.79 | 0.82–7.78 |
| Area of SRF | ||||||||||
| Present at baseline | 248/304 | 82 | 239/286 | 84 | 244/298 | 82 | 243/292 | 83 | 487/590 | 83 |
| Area at baseline | 6.74 | 3.41–11.42 | 7.13 | 3.81–12.81 | 6.81 | 3.59–11.93 | 6.99 | 3.58–12.04 | 6.95 | 3.58–11.99 |
| Present at 1 year | 60/239 | 25 | 73/227 | 32 | 48/230 | 21 | 85/236 | 36 | 133/466 | 29 |
| Area at 1 year | 7.20 | 3.82–13.43 | 7.34 | 3.87–12.04 | 6.91 | 4.51–12.32 | 7.34 | 3.31–13.54 | 7.25 | 3.87–13.39 |
| Present at 2 years | 62/245 | 25 | 59/233 | 25 | 57/241 | 24 | 64/237 | 27 | 121/478 | 25 |
| Area at 2 years | 7.90 | 4.44–13.36 | 7.96 | 4.15–13.78 | 7.96 | 4.98–14.03 | 7.85 | 4.01–13.28 | 7.96 | 4.38–13.38 |
| Area of fibrosisa | ||||||||||
| Present at baseline | 44/306 | 14 | 36/292 | 12 | 45/301 | 15 | 35/297 | 12 | 80/598 | 13 |
| Area at baseline | 3.59 | 1.24–5.88 | 2.63 | 1.82–5.55 | 2.49 | 1.14–5.80 | 3.48 | 2.24–5.79 | 3.10 | 1.54–5.80 |
| Present at 1 year | 129/243 | 53 | 119/230 | 52 | 138/236 | 58 | 110/237 | 46 | 248/473 | 52 |
| Area at 1 year | 3.52 | 1.25–6.93 | 4.02 | 1.38–7.43 | 3.27 | 1.24–6.82 | 4.15 | 1.40–7.50 | 3.72 | 1.33–7.18 |
| Present at 2 years | 121/249 | 49 | 124/235 | 53 | 128/243 | 53 | 117/241 | 49 | 245/484 | 51 |
| Area at 2 years | 3.81 | 1.63–8.77 | 4.04 | 1.65–6.78 | 4.02 | 1.44–7.18 | 4.02 | 2.09–7.66 | 4.02 | 1.65–7.28 |
| Present at baseline | 16/307 | 5 | 13/292 | 4 | 15/302 | 5 | 14/297 | 5 | 29/599 | 5 |
| Area at baseline | 3.86 | 1.81–8.96 | 4.29 | 2.02–9.06 | 4.25 | 2.00–8.99 | 3.95 | 1.78–9.06 | 3.95 | 2.00–8.99 |
| Present at 1 year | 52/243 | 21 | 41/230 | 18 | 40/236 | 17 | 53/237 | 22 | 93/473 | 20 |
| Area at 1 year | 3.93 | 1.81–9.66 | 4.03 | 1.08–6.25 | 3.93 | 1.27–6.69 | 4.27 | 1.44–7.96 | 3.94 | 1.38–7.89 |
| Present at 2 years | 59/249 | 24 | 63/235 | 27 | 63/244 | 26 | 59/240 | 25 | 122/484 | 25 |
| Area at 2 years | 5.13 | 2.34–9.35 | 4.60 | 1.80–7.62 | 5.63 | 1.73–10.06 | 4.60 | 1.94–7.29 | 4.72 | 1.93–8.17 |
Several OCT metrics were measured and they are summarised in Table 15. Treatment effects were only estimated for total thickness at the fovea and retinal plus subfoveal fluid thickness at fovea (see Figure 25). Neither of these metrics differed by drug but both were statistically significantly better (smaller mean thickness) in the continuous group compared with discontinuous group at 2 years; they were, on average, 9% and 8% less thick for participants receiving continuous treatment (GMR = 0.91, 95% CI 0.85 to 0.97; p = 0.004; and GMR = 0.92, 95% CI 0.84 to 1.00; p = 0.046; see Figure 25).
| OCT metrics | Randomised to: | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Neuroretinal foveal thickness (median, IQR) | ||||||||||
| Baseline | 0.20 | (0.15 to 0.28) | 0.19 | (0.15 to 0.29) | 0.20 | (0.15 to 0.28) | 0.20 | (0.16 to 0.28) | 0.20 | (0.15 to 0.28) |
| 1 year | 0.15 | (0.12 to 0.19) | 0.16 | (0.13 to 0.20) | 0.15 | (0.12 to 0.19) | 0.16 | (0.13 to 0.19) | 0.15 | (0.12 to 0.19) |
| 2 years | 0.15 | (0.11 to 0.18) | 0.14 | (0.11 to 0.18) | 0.14 | (0.11 to 0.18) | 0.15 | (0.12 to 0.19) | 0.14 | (0.11 to 0.18) |
| Change from baseline at 1 year | –0.04 | (–0.12 to 0.00) | –0.04 | (–0.12 to 0.02) | –0.04 | (–0.12 to 0.01) | –0.04 | (–0.12 to 0.00) | –0.04 | (–0.12 to 0.01) |
| Change from baseline at 2 years | –0.06 | (–0.13 to 0.00) | –0.05 | (–0.14 to 0.01) | –0.05 | (–0.15 to 0.00) | –0.06 | (–0.12 to 0.00) | –0.05 | (–0.13 to 0.00) |
| Retinal plus subfoveal fluid thickness | ||||||||||
| Baseline | 271.9 | 128.6 | 264.0 | 131.6 | 263.9 | 126.9 | 272.4 | 133.2 | 268.1 | 130.0 |
| 1 year | 173.9 | 80.6 | 178.7 | 90.7 | 170.3 | 80.0 | 182.1 | 90.4 | 176.2 | 85.6 |
| 2 years | 163.5 | 77.7 | 172.7 | 95.7 | 161.7 | 84.2 | 174.4 | 89.4 | 168.0 | 87.0 |
| Change from baseline at 1 year | –101.7 | 141.0 | –81.6 | 144.2 | –93.5 | 138.0 | –90.7 | 147.6 | –92.1 | 142.8 |
| Change from baseline at 2 years | –108.1 | 129.3 | –94.9 | 162.7 | –105.6 | 153.7 | –97.9 | 138.7 | –101.7 | 146.3 |
| Total thickness at the fovea | ||||||||||
| Baseline | 471.6 | 192.5 | 465.6 | 183.1 | 473.2 | 187.9 | 464.1 | 188.2 | 468.7 | 187.9 |
| 1 year | 324.1 | 142.9 | 325.5 | 135.5 | 310.8 | 130.9 | 338.5 | 145.9 | 324.8 | 139.2 |
| 2 years | 322.4 | 137.3 | 331.0 | 144.2 | 314.7 | 137.1 | 338.5 | 143.3 | 326.6 | 140.6 |
| Change from baseline at 1 year | –150.3 | 179.0 | –136.2 | 183.6 | –167.5 | 186.1 | –120.0 | 173.4 | –143.6 | 181.2 |
| Change from baseline at 2 years | –146.9 | 177.4 | –133.8 | 205.0 | –167.3 | 211.9 | –113.9 | 163.8 | –140.6 | 191.1 |
| Maximal retinal thickness | ||||||||||
| Baseline | 0.40 | 0.11 | 0.40 | 0.10 | 0.40 | 0.09 | 0.40 | 0.11 | 0.40 | 0.10 |
| 1 year | 0.31 | 0.06 | 0.31 | 0.07 | 0.30 | 0.06 | 0.32 | 0.07 | 0.31 | 0.06 |
| 2 years | 0.31 | 0.06 | 0.31 | 0.07 | 0.30 | 0.06 | 0.32 | 0.06 | 0.31 | 0.06 |
| Change from baseline at 1 year | –0.09 | 0.10 | –0.08 | 0.11 | –0.10 | 0.10 | –0.07 | 0.11 | –0.09 | 0.11 |
| Change from baseline at 2 years | –0.09 | 0.10 | –0.09 | 0.10 | –0.10 | 0.10 | –0.07 | 0.10 | –0.09 | 0.10 |
| Height of PED (median, IQR) | ||||||||||
| Baseline | 0.14 | (0.05 to 0.25) | 0.15 | (0.07 to 0.29) | 0.15 | (0.06 to 0.27) | 0.14 | (0.06 to 0.29) | 0.14 | (0.06 to 0.28) |
| 1 year | 0.11 | (0.06 to 0.21) | 0.12 | (0.06 to 0.20) | 0.11 | (0.06 to 0.19) | 0.12 | (0.07 to 0.21) | 0.12 | (0.06 to 0.21) |
| 2 years | 0.13 | (0.06 to 0.20) | 0.13 | (0.08 to 0.20) | 0.12 | (0.07 to 0.19) | 0.13 | (0.07 to 0.22) | 0.13 | (0.07 to 0.20) |
| Change from baseline at 1 year | –0.01 | (–0.10 to 0.05) | –0.03 | (–0.12 to 0.03) | –0.03 | (–0.13 to 0.04) | –0.01 | (–0.10 to 0.04) | –0.02 | (–0.12 to 0.04) |
| Change from baseline at 2 years | 0.00 | (–0.11 to 0.07) | 0.00 | (–0.12 to 0.05) | –0.01 | (–0.12 to 0.06) | 0.00 | (–0.10 to 0.06) | 0.00 | (–0.11 to 0.06) |
At baseline, GA was present in 7% of participants and in 51% of cases it was within the lesion. All colour and FFA images were regraded (again, masked to treatment allocation) after the end of the study in order to identify whether or not new GA had developed during the course of the study. It was found that 30% of participants had developed new GA during the trial. There was no difference in the frequency of new GA between the drug groups (28% for ranibizumab vs. 31% for bevacizumab; OR 0.87, 95% CI 0.61 to 1.25; p = 0.46), but significantly more participants receiving continuous treatment developed new GA (34% vs. 26%; OR 1.47, 95% CI 1.03 to 2.11; p = 0.03).
Meta-analyses of lesion morphology characteristics
Figure 26 shows meta-analyses of the BRAMD, CATT,22 GEFAL42 and IVAN trial findings for changes in total retinal thickness at the fovea. The analyses use the 2-year findings for the CATT22 and IVAN trials and 1-year findings for the BRAMD (presented at the 2013 meeting of EURETINA) and GEFAL42 trials. All trials contributed to the analysis by drug; the pooled point estimate suggests a non-statistically significant difference in favour of ranibizumab (p = 0.12; heterogeneity χ2 = 0.12, df = 3; p = 0.99; I2 = 0.0%). Only the CATT22 and IVAN trials contributed to the analysis by treatment regimen, for which the pooled point estimate showed a statistically significant difference in favour of continuous treatment (p = 0.001; see Figure 26).
FIGURE 26.
Meta-analysis of change in total retinal thickness at the fovea from baseline (a) by drug and (b) by treatment regimen. Change in total retinal thickness at the fovea = 2 years minus baseline or 1 year minus baseline, as indicated in the figures. Negative values for differences reflect better outcomes in the ranibizumab or continuous groups. 95% CIs are given in parentheses and illustrated by horizontal bars. WMD, weighted mean difference. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.

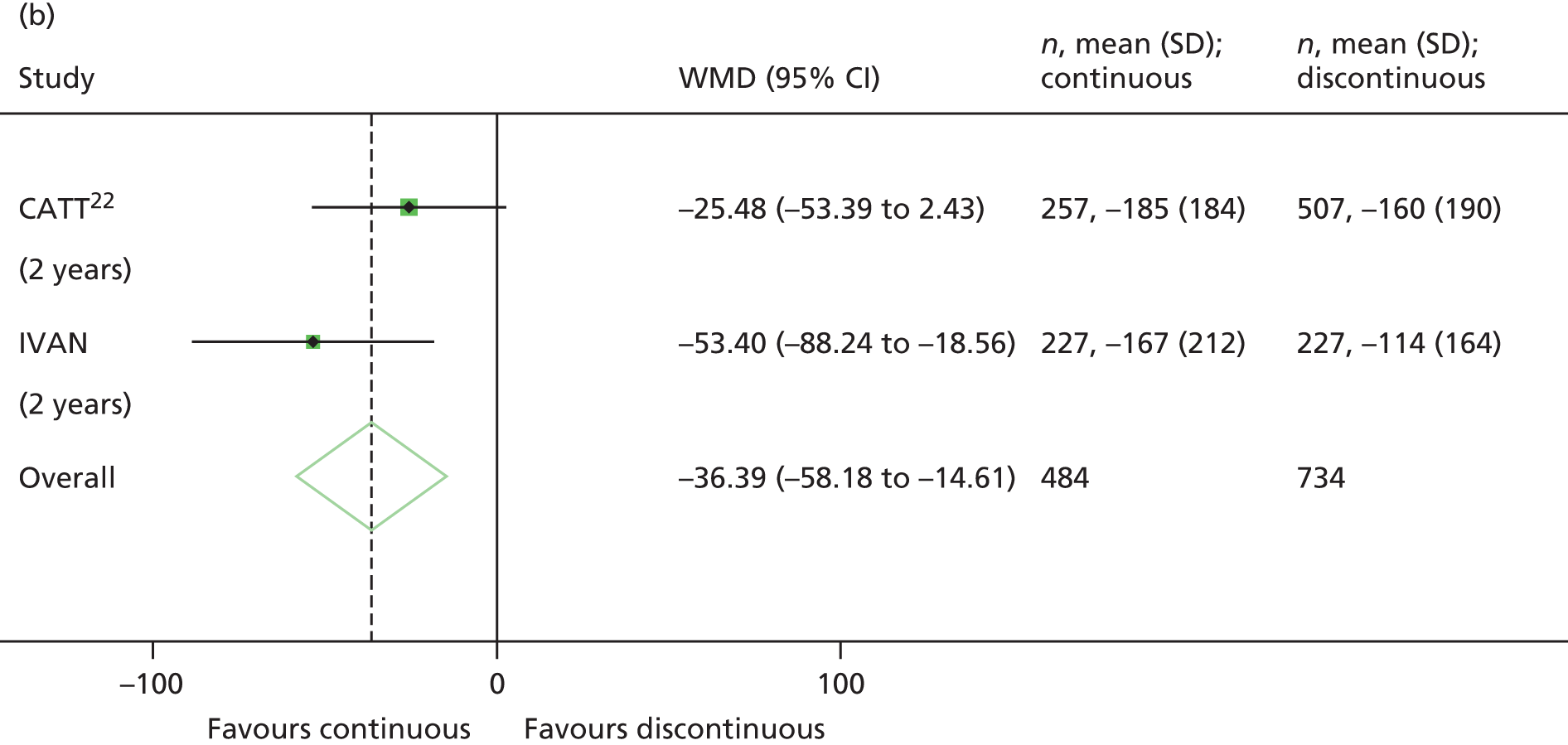
Only the CATT22 and IVAN trials contributed to the meta-analysis of development of new GA over 2 years. The point estimates for the two trials were substantially different for the drug comparison (pooled OR 1.05, 95% CI 0.81 to 1.37; p = 0.70) but similar for the comparison by treatment regimen, showing that new GA developed significantly more often during follow-up among participants treated continuously (OR 1.56, 95% CI 1.20 to 2.03; p = 0.001; Figure 27).
FIGURE 27.
Meta-analysis of GA (a) by drug and (b) by treatment regimen. Ratios of ≤ 1 reflect better outcomes in the ranibizumab or continuous groups. 95% CIs are given in parentheses and illustrated by horizontal bars. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.


Summary
Drug comparisons showed that both ranibizumab and bevacizumab were similar with respect to morphological outcomes. Regimen comparisons found that OCT thickness measurements were lower and an active neovascular lesion was less likely to be present in the continuous group than in the discontinuous group at 2 years. These findings were unchanged in the meta-analysis of the CATT22 and IVAN results. In total, 30% of participants developed new GA during the trial, and this occurred more frequently when receiving continuous treatment, a finding that was also confirmed in the meta-analysis of the CATT22 and IVAN results.
Chapter 6 Safety
Serious adverse events
Over the course of the trial, at least one SAE – defined as an event that is life-threatening, causes hospitalisation (initial or prolonged), disability or permanent damage, a congenital anomaly or birth defect, or death – was reported for 171 participants and, of these, 30 participants died. The frequency of the primary safety end point, an ATE or HF or death from a vascular cause, did not differ significantly between the drugs (OR 1.69, 95% CI 0.80 to 3.57; p = 0.16) and between the treatment regimens drugs (OR 0.56; 95% CI 0.27 to 1.19; p = 0.13) (Table 16 and Figure 28). Of the 30 deaths, 15 participants were the ranibizumab group (4.8%) and 15 in the bevacizumab group (5.1%). By treatment regimen, there were more deaths in the discontinuous treatment group [20 (6.6%) vs. 10 (3.2%), OR 0.47, 95% CI 0.22 to 1.03; p = 0.05] (see Figure 28). Causes of death, determined from the participant death certificates are summarised in Table 17.
| Serious systemic event | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| Death by any cause | 15/15 | 5 | 15/15 | 5 | 10/10 | 3 | 20/20 | 7 | 30/30 | 5 |
| Arteriothrombotic event | 13/13 | 4 | 12/10 | 3 | 7/7 | 2 | 18/16 | 5 | 25/23 | 4 |
| Non-fatal MI | 4/4 | 1 | 5/4 | 1 | 2/2 | 1 | 7/6 | 2 | 9/8 | 1 |
| Non-fatal stroke | 6/6 | 2 | 3/3 | 1 | 4/4 | 1 | 5/5 | 2 | 9/9 | 1 |
| Death from vascular causes | 3/3 | 1 | 4/4 | 1 | 1/1 | 0 | 6/6 | 2 | 7/7 | 1 |
| Arteriothrombotic event or HF | 20/20 | 6 | 14/12 | 4 | 12/12 | 4 | 22/20 | 7 | 34/32 | 5 |
| HF | 7/7 | 2 | 2/2 | 1 | 5/5 | 2 | 4/4 | 1 | 9/9 | 1 |
| Venous thrombotic event | 3/3 | 1 | 4/4 | 1 | 3/3 | 1 | 4/4 | 1 | 7/7 | 1 |
| DVT | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| PE | 3/3 | 1 | 3/3 | 1 | 3/3 | 1 | 3/3 | 1 | 6/6 | 1 |
| Hospitalised for angina | 7/7 | 2 | 3/3 | 1 | 6/6 | 2 | 4/4 | 1 | 10/10 | 2 |
| Hospitalised for non-ocular haemorrhage | 3/3 | 1 | 1/1 | 0 | 2/2 | 1 | 2/2 | 1 | 4/4 | 1 |
| Transient ischaemic attack | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Any of the above excluding non-vascular deaths | 31/31 | 10 | 21/19 | 6 | 21/21 | 7 | 31/29 | 10 | 52/50 | 8 |
| Any of the above including non-vascular deaths | 40/38 | 12 | 31/28 | 9 | 28/27 | 9 | 43/39 | 13 | 71/66 | 11 |
| Ranibizumab monthly | Ranibizumab as needed | Bevacizumab monthly | Bevacizumab as needed |
|---|---|---|---|
| Left ventricular failure Pulmonary oedema Metastatic neoplasm Bladder transitional cell carcinoma Ovarian cancer Metastatic neoplasm |
Subdural haematoma Cerebrovascular accident Haemorrhage Cardiac failure Lower respiratory tract infection Pneumonia Lung neoplasm malignant Prostate cancer metastatic Colon cancer |
PE Pneumonia Multiorgan failure Metastatic neoplasm |
Pericardial haemorrhage Cardiac arrest Cardiac arrest Acute pulmonary oedema Pulmonary oedema Cor pulmonale Lung neoplasm malignant Mantle cell lymphoma Hepatic neoplasm malignant Colon cancer Ovarian cancer |
FIGURE 28.
Safety outcomes at 2 years (a) by drug and (b) by treatment regimen. ORs of < 1 reflect fewer SAEs during the 2 years of follow-up in the ranibizumab or continuous groups. Circles indicate ORs and bars indicate 95% CIs. Vascular events include arteriothrombotic events, HF, deep-vein thrombosis, pulmonary embolism, transient ischaemic attack, hospitalisation for angina, and non-ocular haemorrhage. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
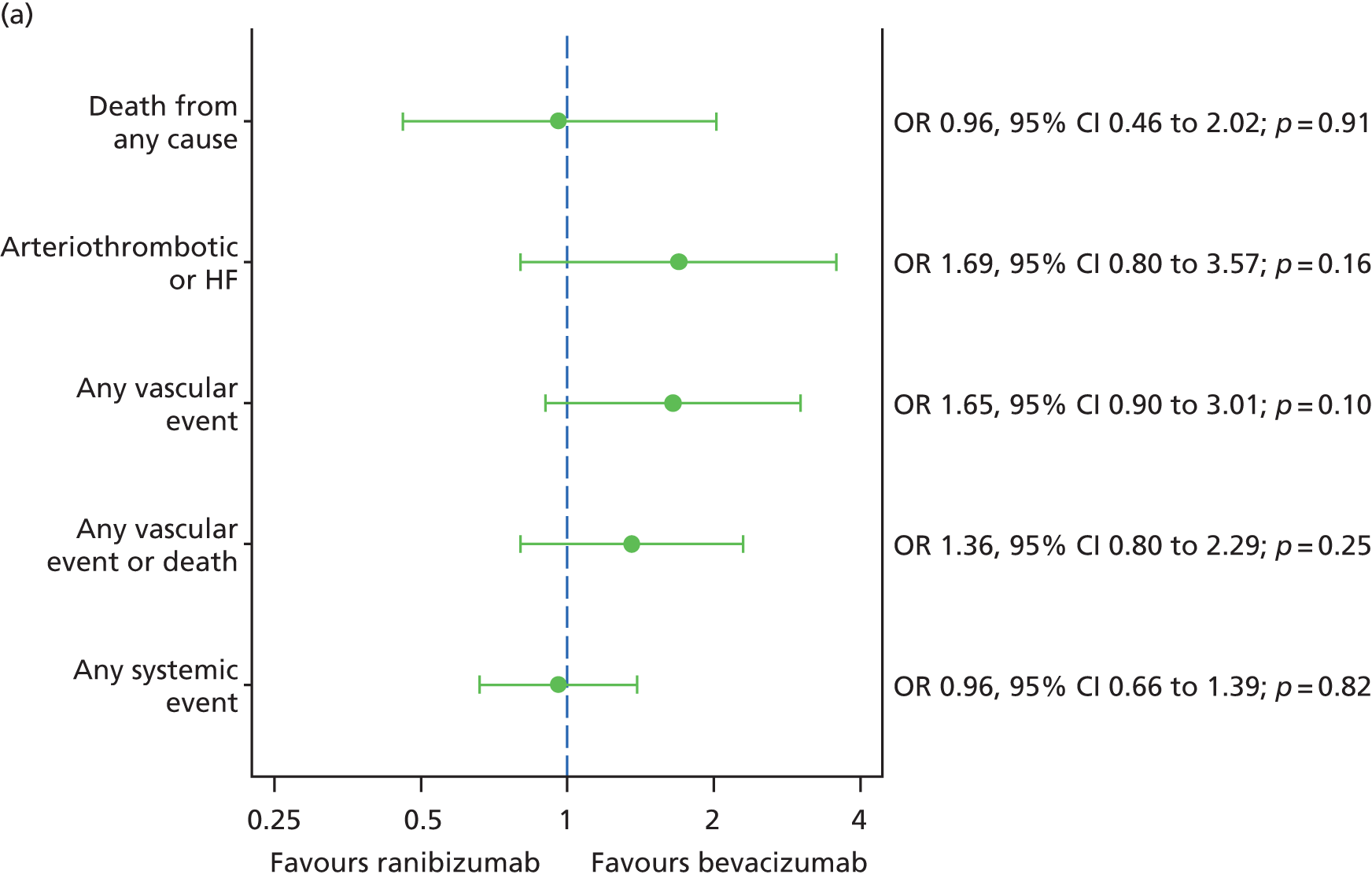
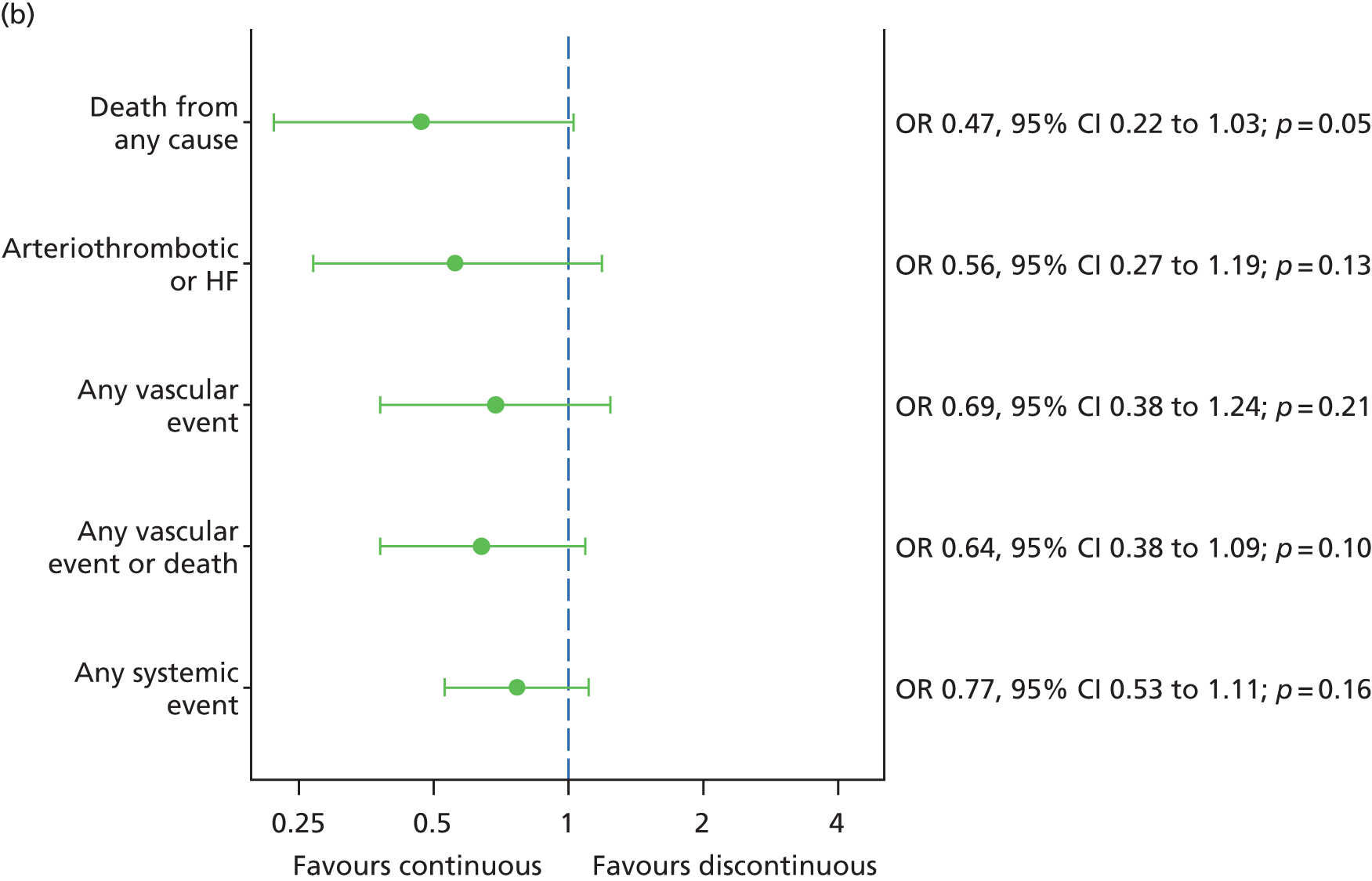
Serious adverse events grouped by MedDRA system organ class are shown in Table 18. The most commonly reported type of SAE was cardiac disorders. Tests of the frequency of SAEs for different organ systems (when there were more than 10 participant-specific events) showed a statistically significant difference in SAEs coded as general disorders and administration site conditions (which includes all deaths) by treatment regimen (continuous 3.2%; discontinuous 7.0%; p = 0.03) but not for any other organ system by drug or treatment regimen (Figure 29). SAEs coded as gastrointestinal disorders were more frequent with bevacizumab but the difference was less marked than at 1 year. 26 Serious ocular AEs in the study eye were extremely rare (14 participants; see Table 18), as were events in the non-study eye (five participants; Table 19).
| MedDRA system organ class | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| Blood and lymphatic system disorders | 0/0 | 0 | 2/2 | 1 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Microcytic anaemia | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Thrombocytopenia | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cardiac disorders | 20/20 | 6 | 20/19 | 6 | 16/16 | 5 | 24/23 | 8 | 40/39 | 6 |
| Angina pectoris | 7/7 | 2 | 3/3 | 1 | 6/6 | 2 | 4/4 | 1 | 10/10 | 2 |
| Arrhythmia | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Atrial fibrillation | 1/1 | 0 | 2/2 | 1 | 1/1 | 0 | 2/2 | 1 | 3/3 | 0 |
| Cardiac arrest | 0/0 | 0 | 3/3 | 1 | 0/0 | 0 | 3/3 | 1 | 3/3 | 0 |
| Cardiac failure | 7/7 | 2 | 2/2 | 1 | 5/5 | 2 | 4/4 | 1 | 9/9 | 1 |
| Cor pulmonale | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Left ventricular failure | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Mitral valve incompetence | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| MI | 4/4 | 1 | 5/4 | 1 | 2/2 | 1 | 7/6 | 2 | 9/8 | 1 |
| Palpitations | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Pericardial haemorrhage | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Supraventricular tachycardia | 0/0 | 0 | 2/2 | 1 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Ear and labyrinth disorders | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Ménière’s disease | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Sudden hearing loss | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Gastrointestinal disorders | 3/3 | 1 | 10/9 | 3 | 7/6 | 2 | 6/6 | 2 | 13/12 | 2 |
| Abdominal pain | 2/2 | 1 | 1/1 | 0 | 2/2 | 1 | 1/1 | 0 | 3/3 | 0 |
| Abdominal pain upper | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Colitis | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Crohn’s disease | 0/0 | 0 | 2/1 | 0 | 2/1 | 0 | 0/0 | 0 | 2/1 | 0 |
| Duodenal ulcer | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Dyspepsia | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Faecaloma | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Intestinal obstruction | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Intestinal perforation | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Pancreatitis | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Pancreatitis chronic | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Vomiting | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| General disorders and administration site conditions | 15/15 | 5 | 16/16 | 5 | 10/10 | 3 | 21/21 | 7 | 31/31 | 5 |
| Death | 15/15 | 5 | 15/15 | 5 | 10/10 | 3 | 20/20 | 7 | 30/30 | 5 |
| Multi-organ failure | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Swelling | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Hepatobiliary disorders | 1/1 | 0 | 3/3 | 1 | 0/0 | 0 | 4/4 | 1 | 4/4 | 1 |
| Cholecystitis | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cholelithiasis | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Jaundice | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Immune system disorders | 2/2 | 1 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Drug hypersensitivity | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Hypersensitivity | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Infections and infestations | 10/9 | 3 | 16/12 | 4 | 13/10 | 3 | 13/11 | 4 | 26/21 | 3 |
| Abdominal infection | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Cellulitis | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cholecystitis infective | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Gastroenteritis | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Localised infection | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Lower respiratory tract infection | 5/4 | 1 | 5/5 | 2 | 5/5 | 2 | 5/4 | 1 | 10/9 | 1 |
| Pneumonia | 3/3 | 1 | 5/5 | 2 | 3/3 | 1 | 5/5 | 2 | 8/8 | 1 |
| Upper respiratory tract infection | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Urinary tract infection | 1/1 | 0 | 2/2 | 1 | 3/3 | 1 | 0/0 | 0 | 3/3 | 0 |
| Injury, poisoning and procedural complications | 12/12 | 4 | 10/10 | 3 | 10/10 | 3 | 12/12 | 4 | 22/22 | 4 |
| Ankle fracture | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Clavicle fracture | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Fall | 7/7 | 2 | 2/2 | 1 | 3/3 | 1 | 6/6 | 2 | 9/9 | 1 |
| Femur fracture | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Foot fracture | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Gastrointestinal stoma complication | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Hip fracture | 1/1 | 0 | 3/3 | 1 | 2/2 | 1 | 2/2 | 1 | 4/4 | 1 |
| Joint dislocation | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Multiple fractures | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Pelvic fracture | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Road traffic accident | 2/2 | 1 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Subdural haematoma | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Upper limb fracture | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Wrist fracture | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Investigations | 2/2 | 1 | 2/2 | 1 | 3/3 | 1 | 1/1 | 0 | 4/4 | 1 |
| Biopsy | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Biopsy vulva | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Endoscopy upper gastrointestinal tract | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| IOP increased | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Metabolism and nutrition disorders | 2/2 | 1 | 0/0 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Hypoglycaemia | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Hyponatraemia | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Musculoskeletal and connective tissue disorders | 3/3 | 1 | 2/2 | 1 | 1/1 | 0 | 4/4 | 1 | 5/5 | 1 |
| Arthralgia | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Back pain | 2/2 | 1 | 1/1 | 0 | 0/0 | 0 | 3/3 | 1 | 3/3 | 0 |
| Musculoskeletal pain | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 11/11 | 4 | 16/14 | 5 | 11/11 | 4 | 16/14 | 5 | 27/25 | 4 |
| Bladder neoplasm | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Bladder transitional cell carcinoma | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Breast cancer | 0/0 | 0 | 2/1 | 0 | 0/0 | 0 | 2/1 | 0 | 2/1 | 0 |
| Colon cancer | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Gallbladder cancer | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Gastrointestinal carcinoma | 0/0 | 0 | 2/2 | 1 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Hepatic neoplasm malignant | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Lung neoplasm malignant | 5/5 | 2 | 2/2 | 1 | 3/3 | 1 | 4/4 | 1 | 7/7 | 1 |
| Mantle cell lymphoma | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Mesothelioma | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Metastatic neoplasm | 2/2 | 1 | 1/1 | 0 | 3/3 | 1 | 0/0 | 0 | 3/3 | 0 |
| Oesophageal carcinoma | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Ovarian cancer | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Pancreatic carcinoma | 0/0 | 0 | 2/2 | 1 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Prostate cancer | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Prostate cancer metastatic | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Nervous system disorders | 9/9 | 3 | 8/8 | 3 | 5/5 | 2 | 12/12 | 4 | 17/17 | 3 |
| Cerebrovascular accident | 7/7 | 2 | 3/3 | 1 | 4/4 | 1 | 6/6 | 2 | 10/10 | 2 |
| Facial paresis | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Frontotemporal dementia | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Presyncope | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Syncope | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Transient ischaemic attack | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Unresponsive to stimuli | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Renal and urinary disorders | 1/1 | 0 | 4/4 | 1 | 3/3 | 1 | 2/2 | 1 | 5/5 | 1 |
| Haematuria | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Nephrolithiasis | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Renal failure | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Urinary bladder polyp | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Reproductive system and breast disorders | 1/1 | 0 | 1/1 | 0 | 2/2 | 1 | 0/0 | 0 | 2/2 | 0 |
| Ovarian cyst | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Postmenopausal haemorrhage | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Respiratory, thoracic and mediastinal disorders | 9/8 | 3 | 7/7 | 2 | 9/8 | 3 | 7/7 | 2 | 16/15 | 2 |
| Acute pulmonary oedema | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Chronic obstructive pulmonary disease | 3/2 | 1 | 0/0 | 0 | 3/2 | 1 | 0/0 | 0 | 3/2 | 0 |
| Dyspnoea | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Emphysema | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Nasal polyps | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| PE | 3/3 | 1 | 3/3 | 1 | 3/3 | 1 | 3/3 | 1 | 6/6 | 1 |
| Pulmonary oedema | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Social circumstances | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Convalescent | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Surgical and medical procedures | 16/16 | 5 | 14/14 | 5 | 14/14 | 5 | 16/16 | 5 | 30/30 | 5 |
| Analgesic therapy | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Aortic aneurysm repair | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Cardiac pacemaker insertion | 2/2 | 1 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Cataract operation | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Chemotherapy | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cholecystectomy | 0/0 | 0 | 2/2 | 1 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Cholelithotomy | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cochlea implant | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Coronary artery bypass | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Enterostomy | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Gastrostomy tube insertion | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Hip arthroplasty | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Hospitalisation | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Hysterectomy | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Knee arthroplasty | 1/1 | 0 | 2/2 | 1 | 1/1 | 0 | 2/2 | 1 | 3/3 | 0 |
| Knee operation | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Lung lobectomy | 2/2 | 1 | 0/0 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| Mastectomy | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Peripheral nerve operation | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Sebaceous cyst excision | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Skin neoplasm excision | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Surgery | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Transurethral bladder resection | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Transurethral prostatectomy | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Vulval operation | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Vascular disorders | 6/5 | 2 | 6/6 | 2 | 4/4 | 1 | 8/7 | 2 | 12/11 | 2 |
| Circulatory collapse | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 2/2 | 1 | 2/2 | 0 |
| DVT | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Haematoma | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Haemorrhage | 3/3 | 1 | 1/1 | 0 | 2/2 | 1 | 2/2 | 1 | 4/4 | 1 |
| Orthostatic hypotension | 2/1 | 0 | 0/0 | 0 | 0/0 | 0 | 2/1 | 0 | 2/1 | 0 |
| Poor peripheral circulation | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Vasculitis | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| 1+ serious systemic event a | 81 | 26 | 80 | 27 | 74 | 24 | 87 | 29 | 161 | 26 |
| Ocular event in the study eye | ||||||||||
| Amaurosis fugax | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Blindness | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Cataract traumatic | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Diplopia | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Keratitis | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Retinal detachment | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Retinal haemorrhage | 2/1 | 0 | 0/0 | 0 | 0/0 | 0 | 2/1 | 0 | 2/1 | 0 |
| RPE tear | 3/3 | 1 | 1/1 | 0 | 2/2 | 1 | 2/2 | 1 | 4/4 | 1 |
| Retinal vein occlusion | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Uveitis | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Vitreous haemorrhage | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| 1+ ocular event | 8 | 3 | 6 | 2 | 6 | 2 | 8 | 3 | 14 | 2 |
| Any unexpected SAE (defined as any event that is not included in Table 16) | 67/51 | 16 | 89/66 | 22 | 68/54 | 18 | 88/63 | 21 | 156/117 | 19 |
| Any SAE | 87 | 28 | 84 | 28 | 79 | 26 | 92 | 30 | 171 | 28 |
FIGURE 29.
Safety outcomes classified by MedDRA system organ class at 2 years (a) by drug and (b) by treatment regimen. ORs of < 1 reflect fewer SAEs during the 2 years of follow-up in the ranibizumab or continuous groups. Circles indicate ORs and bars indicate 95% CIs. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
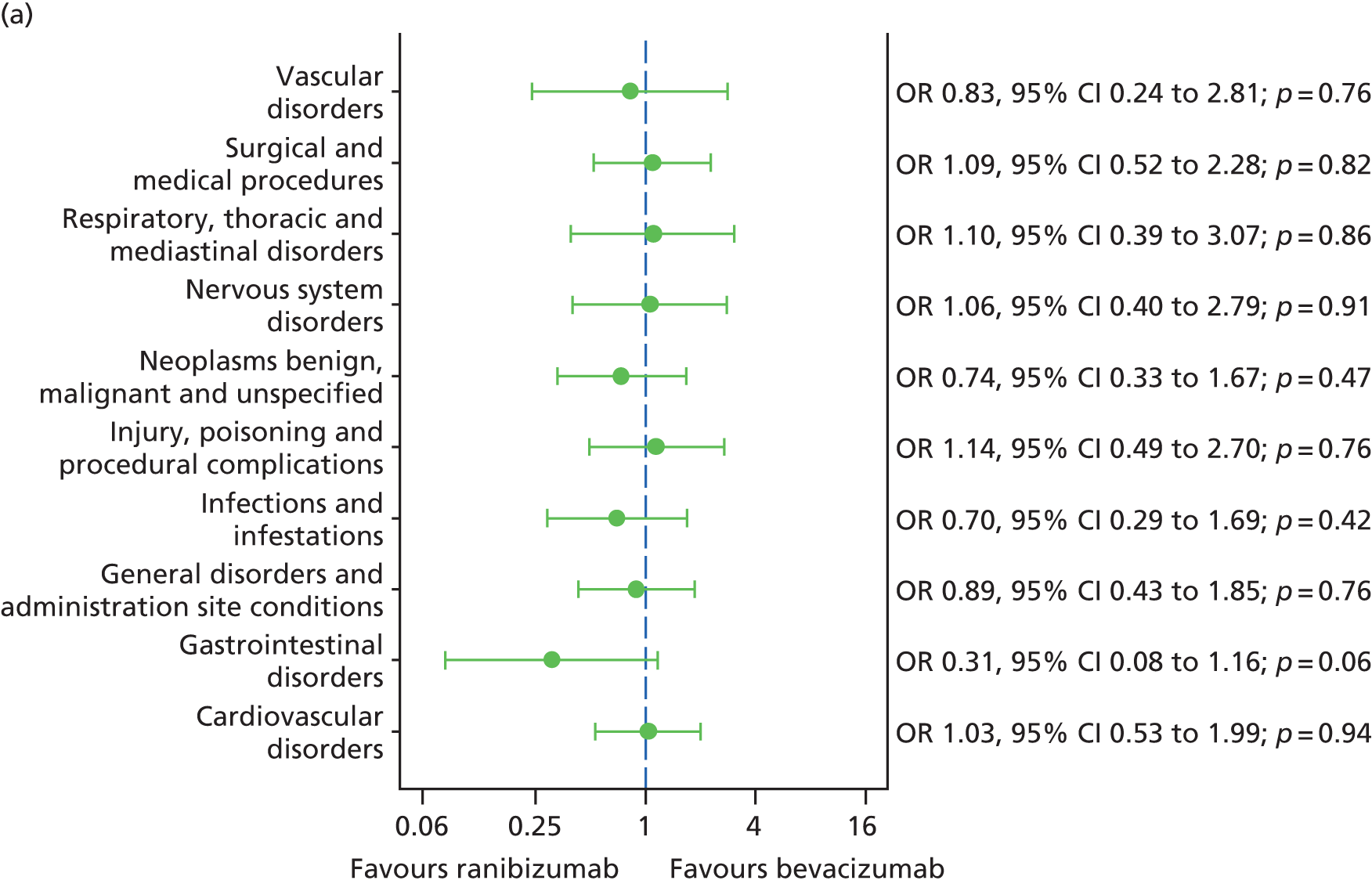
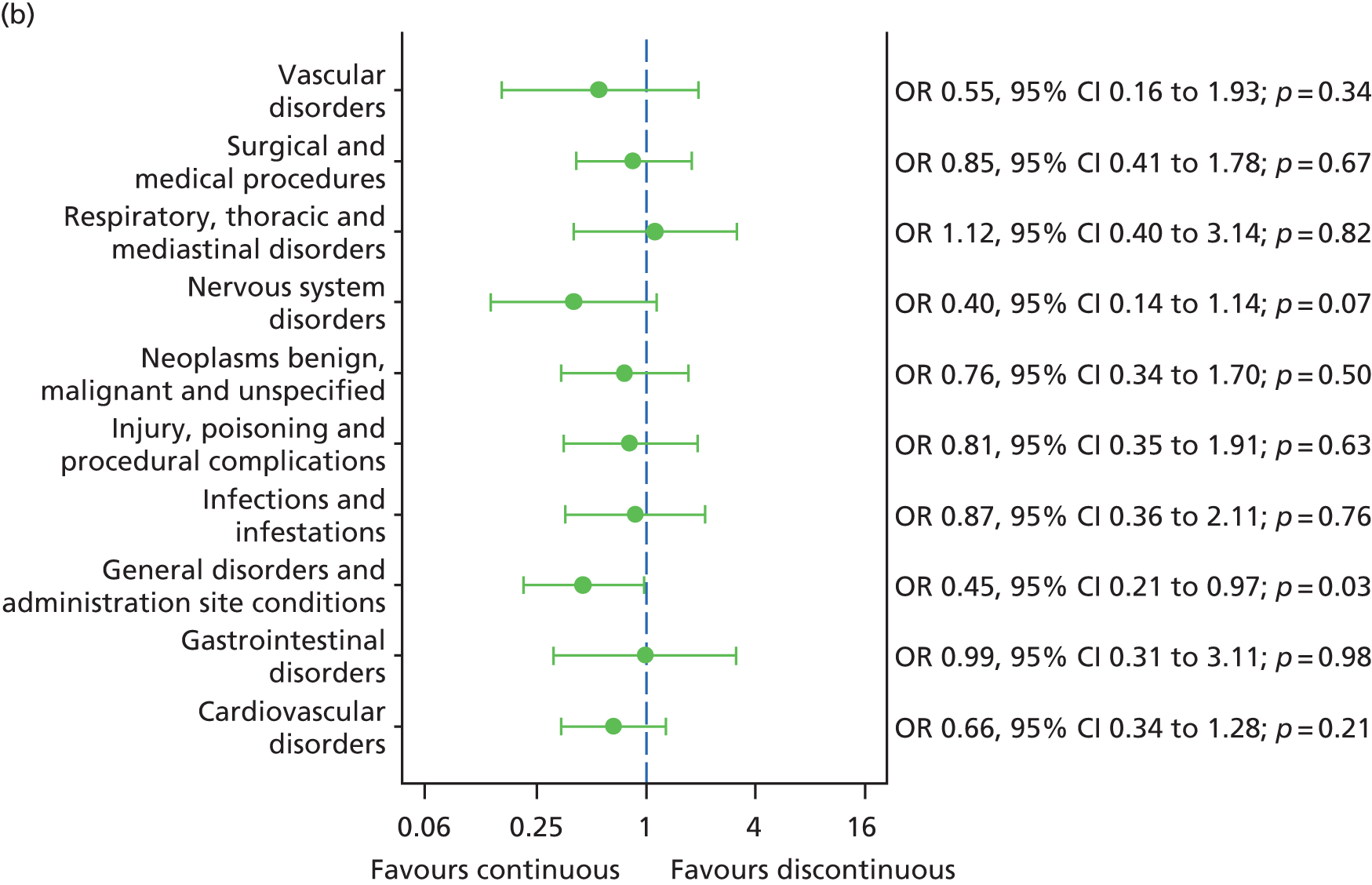
| Event | Randomised to: | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| AMD | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Cataract | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Endophthalmitis | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Herpes zoster | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Wound evisceration | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Any ocular in non-study eye | 2 | 1 | 3 | 1 | 3 | 1 | 2 | 1 | 5 | 1 |
The percentages of patients having any systemic SAE were very similar by drug (ranibizumab 26%; bevacizumab 27%; OR 0.96; 95% CI 0.66 to 1.39; p = 0.82) and by treatment regimen (continuous 24%; discontinuous 29%; OR 0.77; 95% CI 0.53 to 1.11; p = 0.16). In total, 39 non-ocular SAEs were classified as possibly, probably or definitely related to treatment (Table 20).
| Event | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Randomised to ranibizumab (n = 314) | Randomised to bevacizumab (n = 296) | Randomised to continuous (n = 308) | Randomised to discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| Non-ocular | ||||||||||
| Angina pectoris | 4/4 | 1 | 2/2 | 1 | 3/3 | 1 | 3/3 | 1 | 6/6 | 1 |
| Atrial fibrillation | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cardiac arrest | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Cerebrovascular accident | 6/6 | 2 | 2/2 | 1 | 3/3 | 1 | 5/5 | 2 | 8/8 | 1 |
| Death | 4/4 | 1 | 3/3 | 1 | 2/2 | 1 | 5/5 | 2 | 7/7 | 1 |
| Haemorrhage | 2/2 | 1 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Left ventricular failure | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| MI | 4/4 | 1 | 3/2 | 1 | 2/2 | 1 | 5/4 | 1 | 7/6 | 1 |
| Pericardial haemorrhage | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Pneumonia | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| PE | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 1/1 | 0 | 2/2 | 0 |
| Subdural haematoma | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Transient ischaemic attack | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Ocular event in the study eye | ||||||||||
| RPE tear | 1/1 | 0 | 0/0 | 0 | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 |
| Retinal vein occlusion | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
| Uveitis | 0/0 | 0 | 1/1 | 0 | 1/1 | 0 | 0/0 | 0 | 1/1 | 0 |
Non-serious adverse events
Over the course of the trial, non-serious AEs were reported for 570 (93.4%) of the 610 participants. Almost 83% of participants had one or more non-ocular event and 69% had at least one ocular AE. Frequencies were similar by drug and treatment regimen (Table 21). Details of the events, by MedDRA system organ class, are given in Table 21. Ocular AEs in the non-study eye are given in Table 22.
| Adverse events | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| Non-ocular | ||||||||||
| Blood and lymphatic system disorders | 5/5 | 1.6 | 11/10 | 3.4 | 13/12 | 3.9 | 3/3 | 1.0 | 16/15 | 2.5 |
| Cardiac disorders | 16/13 | 4.1 | 15/14 | 4.7 | 17/14 | 4.5 | 14/13 | 4.3 | 31/27 | 4.4 |
| Angina pectoris | 5/4 | 1.3 | 4/4 | 1.4 | 4/4 | 1.3 | 5/4 | 1.3 | 9/8 | 1.3 |
| Arrhythmia | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Atrial fibrillation | 4/4 | 1.3 | 4/4 | 1.4 | 4/4 | 1.3 | 4/4 | 1.3 | 8/8 | 1.3 |
| Bradycardia | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Cardiac disorder | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Cardiac flutter | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.6 | 0/0 | 0.0 | 2/2 | 0.3 |
| Cardiac valve disease | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Mitral valve incompetence | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Palpitations | 4/2 | 0.6 | 0/0 | 0.0 | 4/2 | 0.6 | 0/0 | 0.0 | 4/2 | 0.3 |
| Supraventricular tachycardia | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Tachycardia | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Congenital, familial and genetic disorders | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Ear and labyrinth disorders | 23/18 | 5.7 | 23/20 | 6.8 | 26/21 | 6.8 | 20/17 | 5.6 | 46/38 | 6.2 |
| Endocrine disorders | 1/1 | 0.3 | 7/6 | 2.0 | 4/3 | 1.0 | 4/4 | 1.3 | 8/7 | 1.1 |
| Gastrointestinal disorders | 130/84 | 26.8 | 99/74 | 25.0 | 121/76 | 24.7 | 108/82 | 27.2 | 229/158 | 25.9 |
| Abdominal discomfort | 2/2 | 0.6 | 4/4 | 1.4 | 2/2 | 0.6 | 4/4 | 1.3 | 6/6 | 1.0 |
| Abdominal distension | 2/2 | 0.6 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Abdominal hernia | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Abdominal pain | 2/2 | 0.6 | 5/5 | 1.7 | 5/5 | 1.6 | 2/2 | 0.7 | 7/7 | 1.1 |
| Abdominal pain upper | 7/5 | 1.6 | 3/3 | 1.0 | 8/6 | 1.9 | 2/2 | 0.7 | 10/8 | 1.3 |
| Abdominal symptom | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Anal fissure | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Anorectal discomfort | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Change of bowel habit | 2/2 | 0.6 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Colitis | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Colitis ulcerative | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Constipation | 4/4 | 1.3 | 7/7 | 2.4 | 6/6 | 1.9 | 5/5 | 1.7 | 11/11 | 1.8 |
| Crohn’s disease | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Diarrhoea | 28/23 | 7.3 | 13/13 | 4.4 | 17/15 | 4.9 | 24/21 | 7.0 | 41/36 | 5.9 |
| Dry mouth | 1/1 | 0.3 | 2/2 | 0.7 | 1/1 | 0.3 | 2/2 | 0.7 | 3/3 | 0.5 |
| Dyspepsia | 3/3 | 1.0 | 5/5 | 1.7 | 4/4 | 1.3 | 4/4 | 1.3 | 8/8 | 1.3 |
| Dysphagia | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Faecal incontinence | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Faecal vomiting | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Food poisoning | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Gastric disorder | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Gastritis | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Gastro-oesophageal reflux disease | 0/0 | 0.0 | 3/3 | 1.0 | 2/2 | 0.6 | 1/1 | 0.3 | 3/3 | 0.5 |
| Gingival disorder | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Gingival pain | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Gingivitis | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Glossitis | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Haemorrhoids | 2/2 | 0.6 | 1/1 | 0.3 | 0/0 | 0.0 | 3/3 | 1.0 | 3/3 | 0.5 |
| Hiatus hernia | 2/2 | 0.6 | 3/3 | 1.0 | 4/4 | 1.3 | 1/1 | 0.3 | 5/5 | 0.8 |
| Intestinal obstruction | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Irritable bowel syndrome | 3/1 | 0.3 | 0/0 | 0.0 | 3/1 | 0.3 | 0/0 | 0.0 | 3/1 | 0.2 |
| Lip swelling | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Mouth ulceration | 4/4 | 1.3 | 2/2 | 0.7 | 4/4 | 1.3 | 2/2 | 0.7 | 6/6 | 1.0 |
| Nausea | 51/38 | 12.1 | 36/27 | 9.1 | 46/31 | 10.1 | 41/34 | 11.3 | 87/65 | 10.7 |
| Oedema mouth | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Oesophagitis | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Pancreatic cyst | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Pancreatitis acute | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Rectal haemorrhage | 1/1 | 0.3 | 2/2 | 0.7 | 1/1 | 0.3 | 2/2 | 0.7 | 3/3 | 0.5 |
| Rectal lesion | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Rectal prolapse | 2/2 | 0.6 | 0/0 | 0.0 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Regurgitation | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Sensitivity of teeth | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Toothache | 3/3 | 1.0 | 1/1 | 0.3 | 2/2 | 0.6 | 2/2 | 0.7 | 4/4 | 0.7 |
| Vomiting | 11/8 | 2.5 | 5/5 | 1.7 | 12/9 | 2.9 | 4/4 | 1.3 | 16/13 | 2.1 |
| General disorders and administration site conditions | 52/40 | 12.7 | 61/49 | 16.6 | 56/46 | 14.9 | 57/43 | 14.2 | 113/89 | 14.6 |
| Hepatobiliary disorders | 3/2 | 0.6 | 1/1 | 0.3 | 1/1 | 0.3 | 3/2 | 0.7 | 4/3 | 0.5 |
| Immune system disorders | 10/10 | 3.2 | 8/7 | 2.4 | 10/9 | 2.9 | 8/8 | 2.6 | 18/17 | 2.8 |
| Infections and infestations | 448/194 | 61.8 | 389/165 | 55.7 | 401/167 | 54.2 | 436/192 | 63.6 | 837/359 | 58.9 |
| Injury, poisoning and procedural complications | 88/64 | 20.4 | 72/51 | 17.2 | 86/61 | 19.8 | 74/54 | 17.9 | 160/115 | 18.9 |
| Accident | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Animal bite | 2/2 | 0.6 | 1/1 | 0.3 | 2/2 | 0.6 | 1/1 | 0.3 | 3/3 | 0.5 |
| Ankle fracture | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Arthropod bite | 1/1 | 0.3 | 2/1 | 0.3 | 3/2 | 0.6 | 0/0 | 0.0 | 3/2 | 0.3 |
| Cartilage injury | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Contusion | 13/11 | 3.5 | 3/3 | 1.0 | 12/10 | 3.2 | 4/4 | 1.3 | 16/14 | 2.3 |
| Epicondylitis | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Excoriation | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Eye injury | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Face injury | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Fall | 40/31 | 9.9 | 37/27 | 9.1 | 40/31 | 10.1 | 37/27 | 8.9 | 77/58 | 9.5 |
| Foot fracture | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Foreign body | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Fracture | 3/2 | 0.6 | 6/6 | 2.0 | 2/2 | 0.6 | 7/6 | 2.0 | 9/8 | 1.3 |
| Hand fracture | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Injury | 2/2 | 0.6 | 3/3 | 1.0 | 2/2 | 0.6 | 3/3 | 1.0 | 5/5 | 0.8 |
| Joint dislocation | 2/2 | 0.6 | 2/2 | 0.7 | 2/2 | 0.6 | 2/2 | 0.7 | 4/4 | 0.7 |
| Joint injury | 2/2 | 0.6 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Joint sprain | 4/4 | 1.3 | 3/3 | 1.0 | 2/2 | 0.6 | 5/5 | 1.7 | 7/7 | 1.1 |
| Laceration | 6/5 | 1.6 | 3/3 | 1.0 | 5/4 | 1.3 | 4/4 | 1.3 | 9/8 | 1.3 |
| Ligament sprain | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Limb injury | 7/7 | 2.2 | 5/5 | 1.7 | 9/9 | 2.9 | 3/3 | 1.0 | 12/12 | 2.0 |
| Mouth injury | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Muscle strain | 4/4 | 1.3 | 3/3 | 1.0 | 4/4 | 1.3 | 3/3 | 1.0 | 7/7 | 1.1 |
| Rib fracture | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Road traffic accident | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Thermal burn | 3/2 | 0.6 | 0/0 | 0.0 | 0/0 | 0.0 | 3/2 | 0.7 | 3/2 | 0.3 |
| Vascular injury | 2/2 | 0.6 | 0/0 | 0.0 | 2/2 | 0.6 | 0/0 | 0.0 | 2/2 | 0.3 |
| Investigations | 21/18 | 5.7 | 28/24 | 8.1 | 22/19 | 6.2 | 27/23 | 7.6 | 49/42 | 6.9 |
| Metabolism and nutrition disorders | 20/14 | 4.5 | 8/8 | 2.7 | 15/12 | 3.9 | 13/10 | 3.3 | 28/22 | 3.6 |
| Musculoskeletal and connective tissue disorders | 124/91 | 29.0 | 144/99 | 33.4 | 118/88 | 28.6 | 150/102 | 33.8 | 268/190 | 31.1 |
| Neoplasms benign, malignant and unspecified (including cysts and polyps) | 15/15 | 4.8 | 10/9 | 3.0 | 12/11 | 3.6 | 13/13 | 4.3 | 25/24 | 3.9 |
| Nervous system disorders | 157/90 | 28.7 | 108/77 | 26.0 | 121/76 | 24.7 | 144/91 | 30.1 | 265/167 | 27.4 |
| Psychiatric disorders | 14/10 | 3.2 | 11/10 | 3.4 | 17/13 | 4.2 | 8/7 | 2.3 | 25/20 | 3.3 |
| Renal and urinary disorders | 10/9 | 2.9 | 15/12 | 4.1 | 11/8 | 2.6 | 14/13 | 4.3 | 25/21 | 3.4 |
| Reproductive system and breast disorders | 7/6 | 1.9 | 7/7 | 2.4 | 3/3 | 1.0 | 11/10 | 3.3 | 14/13 | 2.1 |
| Respiratory, thoracic and mediastinal disorders | 218/125 | 39.8 | 199/122 | 41.2 | 199/119 | 38.6 | 218/128 | 42.4 | 417/247 | 40.5 |
| Skin and subcutaneous tissue disorders | 46/38 | 12.1 | 47/41 | 13.9 | 43/38 | 12.3 | 50/41 | 13.6 | 93/79 | 13.0 |
| Social circumstances | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Surgical and medical procedures | 23/19 | 6.1 | 16/15 | 5.1 | 14/13 | 4.2 | 25/21 | 7.0 | 39/34 | 5.6 |
| Vascular disorders | 72/55 | 17.5 | 66/51 | 17.2 | 79/58 | 18.8 | 59/48 | 15.9 | 138/106 | 17.4 |
| Ocular | ||||||||||
| Abnormal sensation in eye | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Blepharitis | 9/9 | 2.9 | 15/10 | 3.4 | 12/10 | 3.2 | 12/9 | 3.0 | 24/19 | 3.1 |
| Blepharoplasty | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Blepharospasm | 2/2 | 0.6 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.7 | 3/3 | 0.5 |
| Cataract | 7/7 | 2.2 | 7/7 | 2.4 | 8/8 | 2.6 | 6/6 | 2.0 | 14/14 | 2.3 |
| Cataract cortical | 3/3 | 1.0 | 1/1 | 0.3 | 3/3 | 1.0 | 1/1 | 0.3 | 4/4 | 0.7 |
| Cataract nuclear | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Cataract operation | 3/3 | 1.0 | 6/5 | 1.7 | 8/7 | 2.3 | 1/1 | 0.3 | 9/8 | 1.3 |
| Cataract traumatic | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Chalazion | 2/2 | 0.6 | 5/4 | 1.4 | 3/3 | 1.0 | 4/3 | 1.0 | 7/6 | 1.0 |
| Colour blindness acquired | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Conjunctival cyst | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Conjunctival haemorrhage | 77/59 | 18.8 | 72/56 | 18.9 | 86/67 | 21.8 | 63/48 | 15.9 | 149/115 | 18.9 |
| Conjunctival hyperaemia | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Conjunctivitis | 9/8 | 2.5 | 9/8 | 2.7 | 11/9 | 2.9 | 7/7 | 2.3 | 18/16 | 2.6 |
| Conjunctivitis allergic | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Corneal abrasion | 13/8 | 2.5 | 11/10 | 3.4 | 16/12 | 3.9 | 8/6 | 2.0 | 24/18 | 3.0 |
| Corneal deposits | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Corneal disorder | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Corneal dystrophy | 2/2 | 0.6 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Corneal erosion | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Corneal perforation | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Dacryocystitis | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Diplopia | 4/4 | 1.3 | 2/2 | 0.7 | 1/1 | 0.3 | 5/5 | 1.7 | 6/6 | 1.0 |
| Dry eye | 13/10 | 3.2 | 5/4 | 1.4 | 9/8 | 2.6 | 9/6 | 2.0 | 18/14 | 2.3 |
| Episcleritis | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Exophthalmos | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Eye discharge | 9/8 | 2.5 | 4/2 | 0.7 | 7/5 | 1.6 | 6/5 | 1.7 | 13/10 | 1.6 |
| Eye haemorrhage | 0/0 | 0.0 | 2/2 | 0.7 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Eye infection | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Eye inflammation | 3/3 | 1.0 | 9/6 | 2.0 | 3/3 | 1.0 | 9/6 | 2.0 | 12/9 | 1.5 |
| Eye irritation | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Eye pain | 146/91 | 29.0 | 150/91 | 30.7 | 166/97 | 31.5 | 130/85 | 28.1 | 296/182 | 29.8 |
| Eye pruritus | 2/2 | 0.6 | 3/2 | 0.7 | 2/2 | 0.6 | 3/2 | 0.7 | 5/4 | 0.7 |
| Eye swelling | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Eyelid cyst | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.6 | 0/0 | 0.0 | 2/2 | 0.3 |
| Eyelid irritation | 0/0 | 0.0 | 2/2 | 0.7 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Eyelid oedema | 0/0 | 0.0 | 4/4 | 1.4 | 1/1 | 0.3 | 3/3 | 1.0 | 4/4 | 0.7 |
| Eyelid pain | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Eyelid ptosis | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Fibrosis | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Foreign body sensation in eyes | 3/3 | 1.0 | 0/0 | 0.0 | 1/1 | 0.3 | 2/2 | 0.7 | 3/3 | 0.5 |
| Glaucoma | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Glaucomatous optic disc atrophy | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Hallucination, visual | 8/8 | 2.5 | 2/2 | 0.7 | 5/5 | 1.6 | 5/5 | 1.7 | 10/10 | 1.6 |
| Hordeolum | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Intraocular lens implant | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| IOP increased | 154/79 | 25.2 | 130/66 | 22.3 | 188/87 | 28.2 | 96/58 | 19.2 | 284/145 | 23.8 |
| Iridocyclitis | 0/0 | 0.0 | 2/2 | 0.7 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Iridotomy | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Keratitis | 2/1 | 0.3 | 1/1 | 0.3 | 2/1 | 0.3 | 1/1 | 0.3 | 3/2 | 0.3 |
| Lacrimal mucocoele | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Lacrimation increased | 37/30 | 9.6 | 53/41 | 13.9 | 43/35 | 11.4 | 47/36 | 11.9 | 90/71 | 11.6 |
| Laser therapy | 2/2 | 0.6 | 2/2 | 0.7 | 2/2 | 0.6 | 2/2 | 0.7 | 4/4 | 0.7 |
| Lenticular opacities | 2/2 | 0.6 | 1/1 | 0.3 | 2/2 | 0.6 | 1/1 | 0.3 | 3/3 | 0.5 |
| Macular oedema | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Maculopathy | 0/0 | 0.0 | 2/2 | 0.7 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Metamorphopsia | 3/2 | 0.6 | 3/3 | 1.0 | 2/2 | 0.6 | 4/3 | 1.0 | 6/5 | 0.8 |
| Mydriasis | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Optic disc haemorrhage | 3/3 | 1.0 | 1/1 | 0.3 | 3/3 | 1.0 | 1/1 | 0.3 | 4/4 | 0.7 |
| Paracentesis eye | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Periorbital haematoma | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Photophobia | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.6 | 0/0 | 0.0 | 2/2 | 0.3 |
| Photopsia | 2/2 | 0.6 | 3/3 | 1.0 | 5/5 | 1.6 | 0/0 | 0.0 | 5/5 | 0.8 |
| Pigment dispersion syndrome | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Posterior capsule opacification | 1/1 | 0.3 | 2/2 | 0.7 | 1/1 | 0.3 | 2/2 | 0.7 | 3/3 | 0.5 |
| Posterior capsulotomy | 2/2 | 0.6 | 1/1 | 0.3 | 0/0 | 0.0 | 3/3 | 1.0 | 3/3 | 0.5 |
| Punctate keratitis | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Pupillary reflex impaired | 2/1 | 0.3 | 0/0 | 0.0 | 2/1 | 0.3 | 0/0 | 0.0 | 2/1 | 0.2 |
| Retinal artery embolism | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Retinal artery occlusion | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Retinal artery spasm | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Retinal detachment | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Retinal haemorrhage | 10/8 | 2.5 | 16/15 | 5.1 | 12/10 | 3.2 | 14/13 | 4.3 | 26/23 | 3.8 |
| Retinal oedema | 2/2 | 0.6 | 3/3 | 1.0 | 0/0 | 0.0 | 5/5 | 1.7 | 5/5 | 0.8 |
| RPE tear | 5/5 | 1.6 | 2/2 | 0.7 | 3/3 | 1.0 | 4/4 | 1.3 | 7/7 | 1.1 |
| Retinal tear | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Retinal vein occlusion | 3/3 | 1.0 | 3/3 | 1.0 | 3/3 | 1.0 | 3/3 | 1.0 | 6/6 | 1.0 |
| Skin lesion excision | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Superficial injury of eye | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Uveitis | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Vascular occlusion | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Vision blurred | 5/5 | 1.6 | 6/6 | 2.0 | 6/6 | 1.9 | 5/5 | 1.7 | 11/11 | 1.8 |
| VA reduced | 5/4 | 1.3 | 3/3 | 1.0 | 1/1 | 0.3 | 7/6 | 2.0 | 8/7 | 1.1 |
| Visual field defect | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Visual impairment | 17/14 | 4.5 | 17/17 | 5.7 | 14/14 | 4.5 | 20/17 | 5.6 | 34/31 | 5.1 |
| Vitreal cells | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Vitreous adhesions | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Vitreous detachment | 19/19 | 6.1 | 15/14 | 4.7 | 17/16 | 5.2 | 17/17 | 5.6 | 34/33 | 5.4 |
| Vitreous disorder | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Vitreous floaters | 81/65 | 20.7 | 74/60 | 20.3 | 88/71 | 23.1 | 67/54 | 17.9 | 155/125 | 20.5 |
| Vitreous haemorrhage | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Any non-ocular event | 254 | 80.9 | 251 | 84.8 | 247 | 80.2 | 258 | 85.4 | 505 | 82.8 |
| Any ocular event | 222 | 70.7 | 199 | 67.2 | 218 | 70.8 | 203 | 67.2 | 421 | 69.0 |
| Any event | 294 | 93.6 | 276 | 93.2 | 289 | 93.8 | 281 | 93.0 | 570 | 93.4 |
| Ocular adverse events | Randomised to | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | |||||||
| Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | Events/patients | % | |
| Cataract | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Cataract operation | 0/0 | 0.0 | 4/3 | 1.0 | 1/0 | 0.0 | 3/3 | 1.0 | 4/3 | 0.5 |
| CNV | 1/1 | 0.3 | 2/2 | 0.7 | 1/1 | 0.3 | 2/2 | 0.7 | 3/3 | 0.5 |
| Conjunctivitis | 2/2 | 0.6 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 2/2 | 0.3 |
| Corneal abrasion | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Eye inflammation | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Eye irritation | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Macular degeneration | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Metamorphopsia | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.2 |
| Ocular hyperaemia | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Retinal artery embolism | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Retinal haemorrhage | 2/2 | 0.6 | 1/1 | 0.3 | 2/2 | 0.6 | 1/1 | 0.3 | 3/3 | 0.5 |
| RPE tear | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Venous stasis retinopathy | 0/0 | 0.0 | 1/1 | 0.3 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| Vision blurred | 1/1 | 0.3 | 0/0 | 0.0 | 0/0 | 0.0 | 1/1 | 0.3 | 1/1 | 0.2 |
| VA reduced | 1/1 | 0.3 | 1/1 | 0.3 | 0/0 | 0.0 | 2/2 | 0.7 | 2/2 | 0.3 |
| Any ocular event | 12 | 3.8 | 11 | 3.7 | 8 | 2.6 | 15 | 5.0 | 23 | 3.8 |
Meta-analyses
Figure 30 shows meta-analyses of safety outcomes from the CATT, GEFAL, IVAN, LUCAS (presented at the 2013 meeting of the American Association for Ophthalmology), MANTA and Subramanian et al. trials comparing ranibizumab and bevacizumab22,42–44 (available information for the BRAMD trial reported only by MedDRA System Organ Class and did not distinguish deaths from other SAEs). The CATT22 and IVAN trials followed participants for 2 years, and the other four trials reported data to 1 year after recruitment. Pooled estimates of safety outcomes showed no differences by drug for deaths (heterogeneity χ2 = 0.76; df = 5; p = 0.98; I2 = 0%) or ATEs (heterogeneity χ2 = 3.21; df = 3; p = 0.36; I2 = 7%) but a significantly increased risk of any systemic SAE for bevacizumab (OR 0.77, 95% CI 0.64 to 0.92; p = 0.004; heterogeneity χ2 = 1.85; df = 3; p = 0.61; I2 = 0%) (see Figure 30). The comparison by treatment regimen (IVAN 2-year data and CATT 1-year data only22) showed consistent increases in mortality (OR 0.49, 95% CI 0.27 to 0.86; p = 0.014) and the risk of any systemic SAE (OR 0.81, 95% CI 0.65 to 1.01; p = 0.063) with discontinuous treatment (see Figure 30).
FIGURE 30.
Meta-analysis of safety outcomes (a) by drug and (b) by treatment regimen. Ratios of < 1 reflect better outcomes in the ranibizumab or continuous groups. 95% CIs are given in parentheses and illustrated by horizontal bars. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
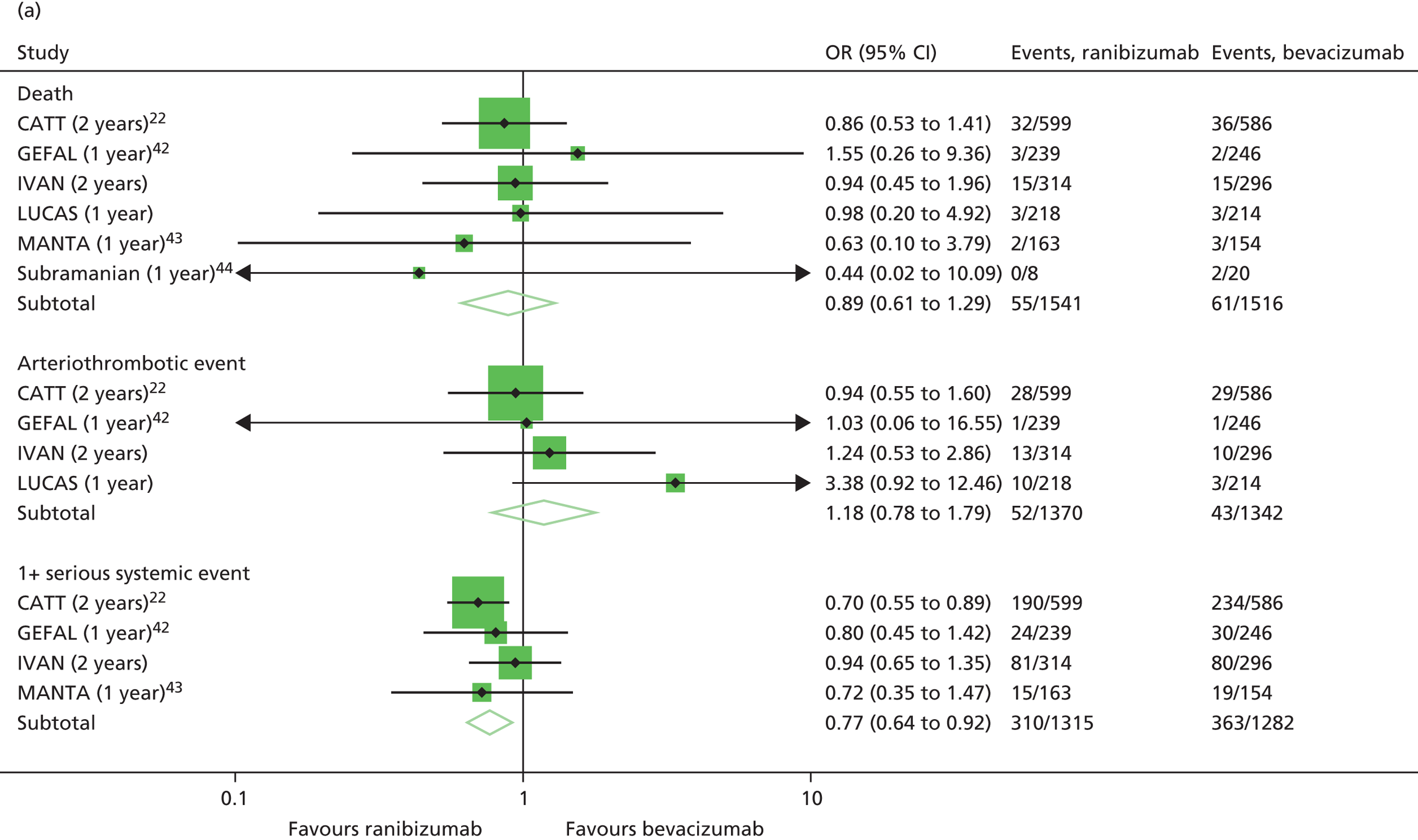

A post hoc meta-analysis of gastrointestinal SAEs, including the studies listed above, with the addition of BRAMD for the drug comparison, was performed. This meta-analysis was prompted by a report from the CATT22 that such events occurred more often when participants were treated with bevacizumab than when treated with ranibizumab; the IVAN interim analysis was also consistent with this observation. 26 The pooled estimate for the drug comparison showed a significantly increased risk of gastrointestinal SAEs in the bevacizumab group (OR 0.53, 95% CI 0.33 to 0.85; p = 0.009; heterogeneity χ2 = 5.13, df = 5; p = 0.40; I2 = 3%) but no difference by treatment regimen (OR 0.88, 95% CI 0.44 to 1.78; p = 0.73) (Figure 31).
FIGURE 31.
Meta-analysis of gastrointestinal SAEs (a) by drug and (b) by treatment regimen. Ratios of < 1 reflect better outcomes in the ranibizumab or continuous groups. 95% CIs are given in parentheses and illustrated by horizontal bars.
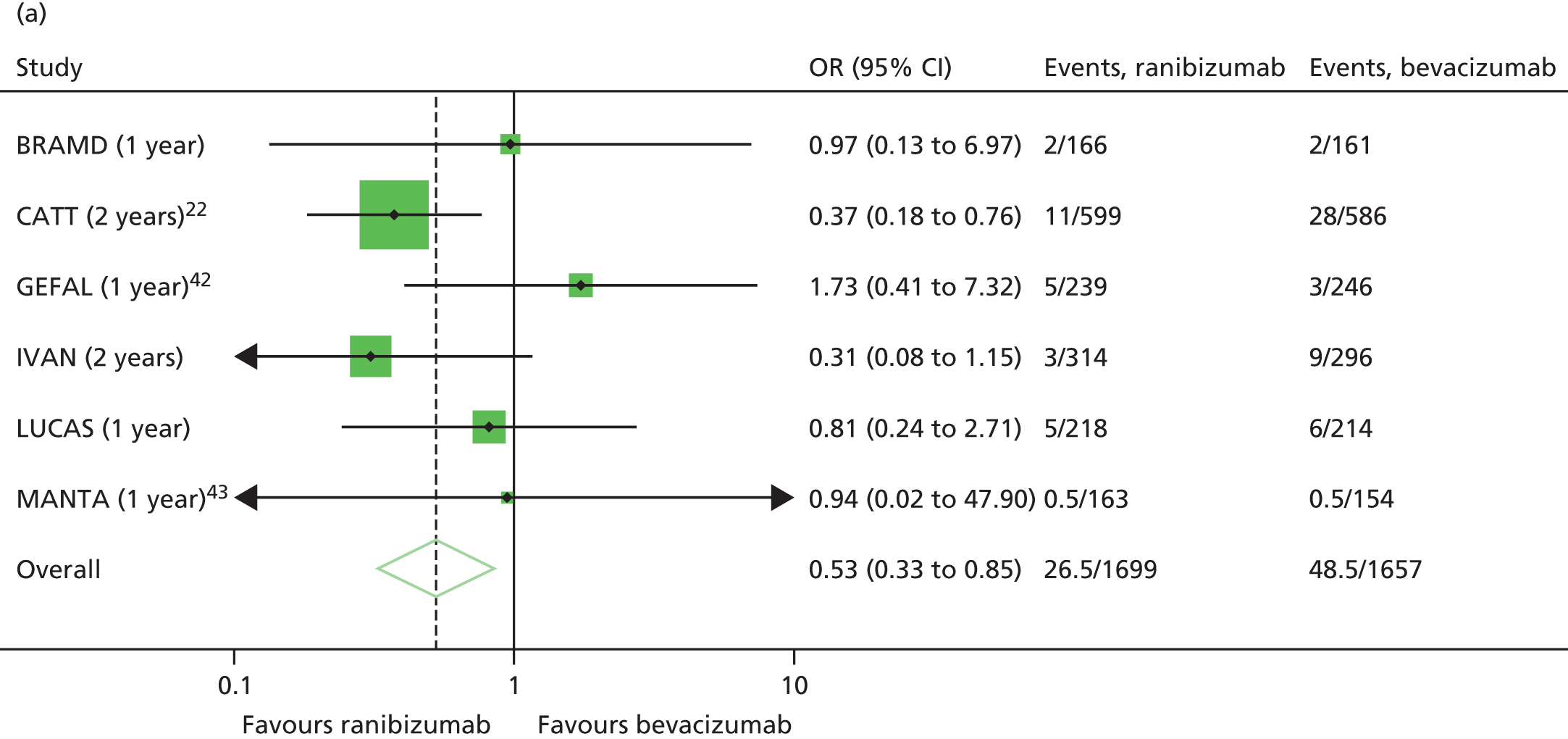

Summary
At least one SAE was reported for 171 participants, and 30 participants died. The frequency of the primary safety end point did not differ by drug or treatment regimen. There were 15 deaths in each of the ranibizumab and bevacizumab groups, but twice as many deaths with discontinuous as continuous treatment regimens (20 deaths compared with 10). We examined the reasons for death by both drug and regimen and we did not observe any clustering of cause for mortality by drug or by regimen. There was also a statistically significant difference in the frequency of SAEs coded as general disorders and administration site conditions (which includes all deaths), with about twice as many of these SAEs observed to occur in the discontinuous treatment group. SAEs coded as ‘gastrointestinal’ were more frequent with bevacizumab in the IVAN trial. Serious ocular AEs were extremely rare. The number of participants having any systemic SAE was similar across the groups. Over 90% of participants reported at least one non-SAE.
Meta-analyses of safety outcomes showed a significantly increased risk of any systemic SAEs and gastrointestinal events for bevacizumab, and consistent increases in mortality and the risk of any systemic SAE with discontinuous treatment across trials.
Chapter 7 Patient-reported outcomes and other information
Patient-reported outcomes are secondary outcomes. Other information, not specified as secondary outcomes, was collected and monitored by the DMSC. This information is reported here.
Patient-reported outcomes
For the EQ-5D utility index and the MacTSQ, an instrument designed to assess patients’ satisfaction with treatment for nAMD, higher summary scores represent better utility and a higher treatment satisfaction, respectively. For the MacDQol, an instrument designed to assess macular disease-specific quality of life, lower scores represent less impact of nAMD on quality of life. HUI3 data were also collected, but have been used only for the health economics analysis. Baseline median EQ-5D utilities were similar by drug and treatment frequency (Table 23). Median MacDQoL and MacTSQ scores and EQ-5D utilities were also very similar at 1 and 2 years (see Table 23). The distribution of responses to individual EQ-5D questions at baseline and 2 years by drug and treatment regimen is given in Appendix 3. EQ-5D was dichotomised as ‘perfect health’ (EQ-5D score of 1) compared with less than perfect health, and MacTSQ was dichotomised at the median (i.e. < median vs. ≥ median). There were no significant differences in the odds of a patient having perfect health by either drug or treatment regimen (p = 0.51 and 0.64, respectively), or of a patient having ‘good’ treatment satisfaction compared with ‘low’ treatment satisfaction (p = 0.23 and p = 0.47) (Figure 32). When analysing the MacDQoL score, this outcome was analysed on a reverse scale as outlined in Chapter 2 (see Statistical methods). As with EQ-5D score, there were no significant differences between groups (p = 0.74 for the drug comparison and p = 0.73 for the treatment regimen comparison) (see Figure 32).
| Quality of life score | Randomised to: | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| Median | IQR | Median | IQR | Median | IQR | Median | IQR | Median | IQR | |
| EQ-5D utility | ||||||||||
| Baseline | 0.81 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 |
| 1 year | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 |
| 2 years | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 | 0.85 | 0.73 to 1.00 |
| Change from baseline at 1 year | –0.12 | –0.24 to 0.00 | –0.13 | –0.26 to 0.00 | –0.12 | –0.24 to 0.00 | –0.12 | –0.27 to 0.00 | –0.12 | –0.26 to 0.00 |
| Change from baseline at 2 years | –0.15 | –0.27 to 0.00 | –0.15 | –0.27 to 0.00 | –0.15 | –0.24 to 0.00 | –0.15 | –0.27 to 0.00 | –0.15 | –0.27 to 0.00 |
| MacDQoL | ||||||||||
| 1 year | –1.27 | –2.76 to –0.36 | –1.18 | –3.14 to –0.39 | –1.27 | –2.91 to –0.36 | –1.23 | –3.00 to –0.36 | –1.23 | –2.91 to –0.36 |
| 2 years | –1.45 | –2.77 to –0.27 | –1.39 | –2.73 to –0.41 | –1.27 | –2.70 to –0.32 | –1.59 | –3.00 to –0.41 | –1.41 | –2.73 to –0.32 |
| MacTSQ | ||||||||||
| 1 year | 66.00 | 61.00 to 69.00 | 66.00 | 59.50 to 69.00 | 65.00 | 59.00 to 69.00 | 67.00 | 61.00 to 70.00 | 66.00 | 61.00 to 69.00 |
| 2 years | 66.00 | 61.50 to 70.00 | 65.00 | 60.00 to 69.00 | 65.50 | 61.00 to 69.00 | 66.00 | 60.00 to 69.00 | 66.00 | 61.00 to 69.00 |
FIGURE 32.
The EQ-5D utility, macular disease-specific quality of life and treatment satisfaction scores (a) by drug and (b) by treatment regimen. Ratios of < 1 reflect better score in the ranibizumab or continuous groups. Circles = GMR or OR. Point estimates and 95% Cls are quantified on the right-hand side of the figure and 95% CIs are illustrated by horizontal bars. Source: © 2013 Chakravarthy et al. 93 Open Access article distributed under the terms of CC-BY-NC-SA (http://creativecommons.org/licenses/by-nc-sa/3.0/). Published by Elsevier Ltd.
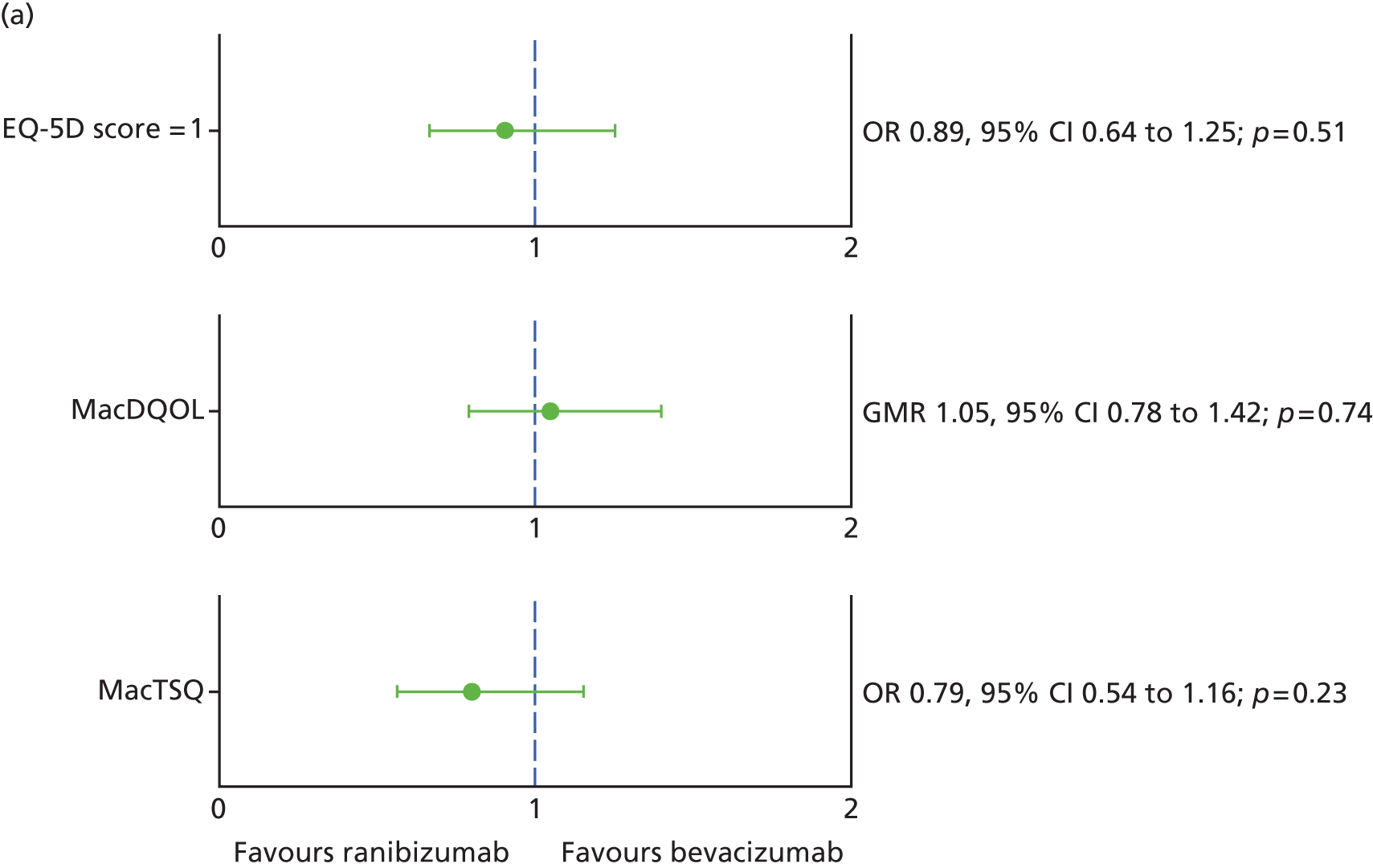
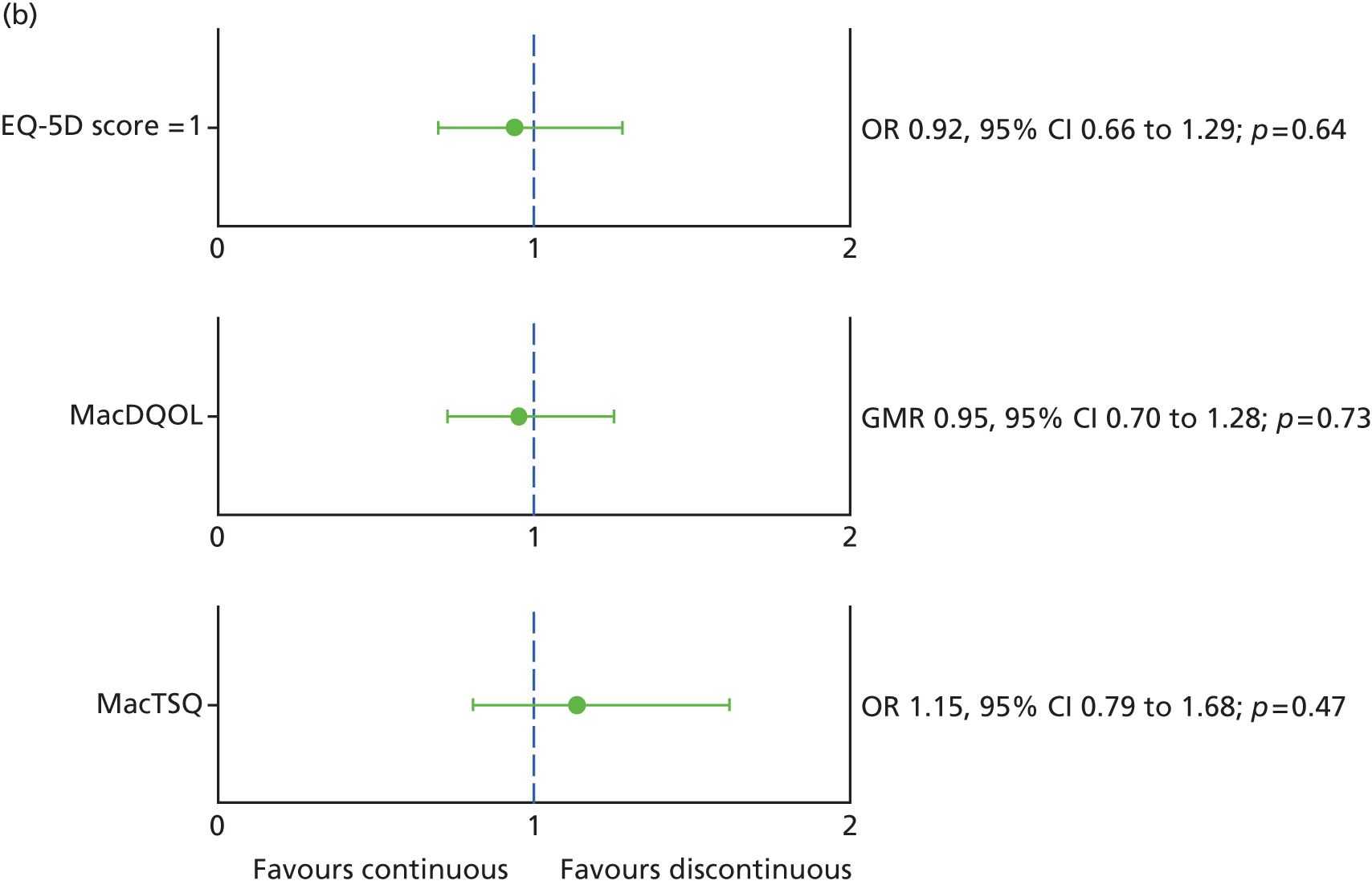
In addition to comparing scores across the cohort as a whole, we also investigated whether or not utility, macular disease-specific quality of life and treatment satisfaction differed between the subgroups of participants when vision in the study eye was > 5 letters better than in the non-study eye (i.e. vision in the study eye better than the fellow eye vs. the same or worse). The results of this post hoc subgroup analysis are shown in Figure 33. For all three patient-reported outcomes, no statistically significant difference between the subgroups was found.
FIGURE 33.
The EQ-5D utility, macular disease-specific quality of life and treatment satisfaction scores for subgroups of participants with vision in the study eye > 5 letters better and ≤ 5 letters better (a) by drug and (b) by treatment frequency. Ratios of < 1 reflect better scores in the ranibizumab or continuous groups. Circles = GMR or OR. 95% CIs are illustrated by horizontal bars. The circle size reflects the number of patients in the subgroup.
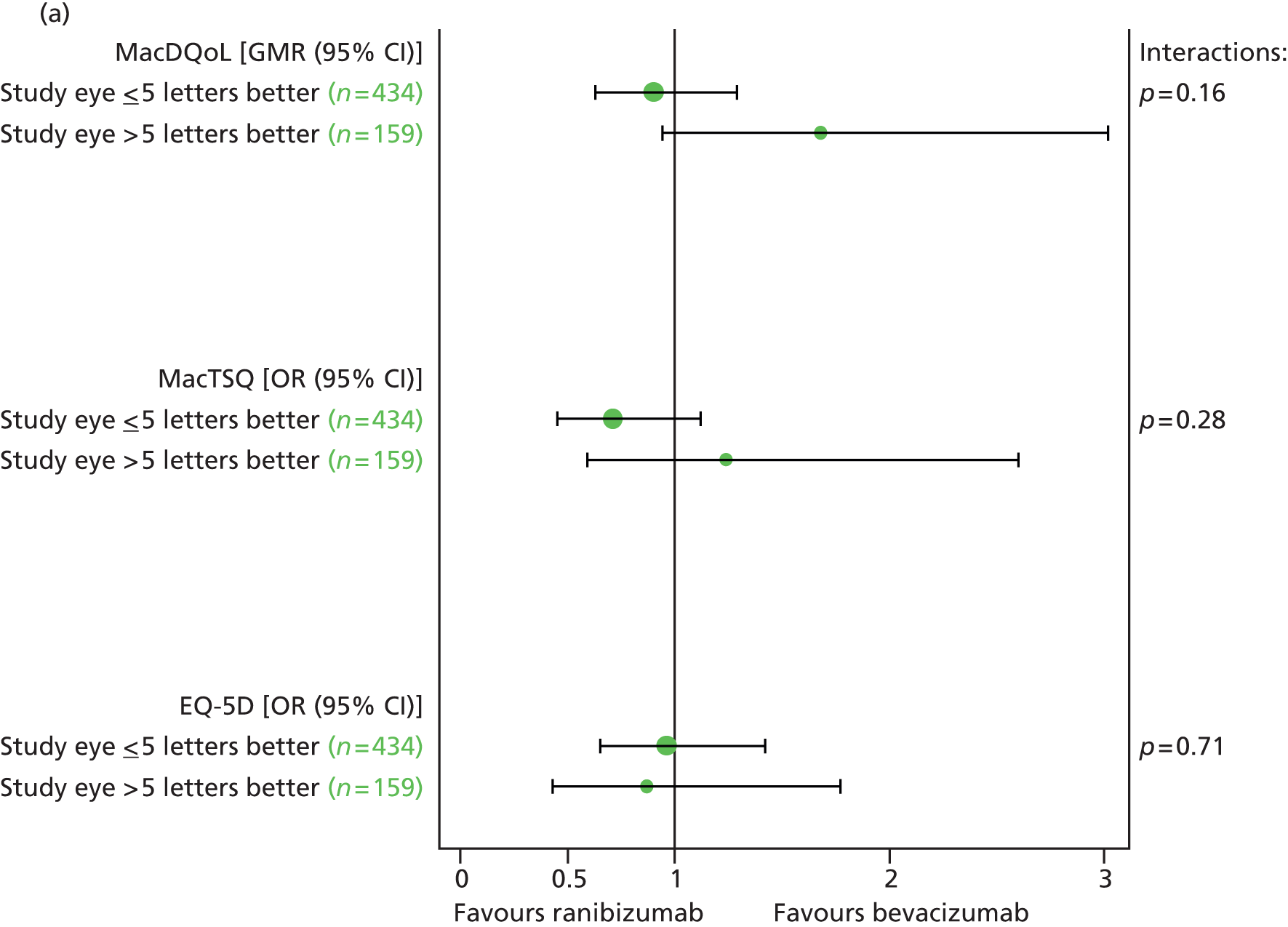

Blood pressure
Descriptive summaries of blood pressure measurements were regularly reviewed by the DMSC throughout the trial. Average diastolic and systolic blood pressure at baseline, 1 year and 2 years are given in Table 24. Serial measurements by group are shown in Figures 34 and 35; there did not appear to be any differences in blood pressure over time by drug or by treatment regimen. These data were not formally compared at 2 years.
| Blood pressure | Randomised to: | Overall (n = 610 at baseline, n = 563 at 1 year, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 288 at 1 year, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 275 at 1 year, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 276 at 1 year, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 287 at 1 year, n = 264 at 2 years) | |||||||
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Systolic blood pressure (mmHg) | ||||||||||
| Baseline | 141.9 | 19.5 | 143.0 | 19.5 | 143.2 | 19.8 | 141.7 | 19.1 | 142.5 | 19.5 |
| 1 year | 138.1 | 17.3 | 138.8 | 18.0 | 138.3 | 18.2 | 138.5 | 17.1 | 138.4 | 17.6 |
| 2 years | 137.5 | 18.3 | 139.9 | 18.3 | 139.4 | 18.8 | 137.9 | 17.9 | 138.6 | 18.4 |
| Change from baseline at 1 year | –3.8 | 17.1 | –4.8 | 18.1 | –5.6 | 16.1 | –3.0 | 18.9 | –4.3 | 17.6 |
| Change from baseline at 2 years | –3.8 | 20.1 | –3.1 | 19.7 | –4.1 | 20.3 | –2.8 | 19.6 | –3.4 | 19.9 |
| Diastolic blood pressure (mmHg) | ||||||||||
| Baseline | 76.4 | 10.2 | 77.1 | 9.9 | 77.4 | 10.1 | 76.2 | 10.0 | 76.8 | 10.1 |
| 1 year | 74.4 | 9.7 | 75.0 | 9.6 | 74.9 | 9.2 | 74.5 | 10.1 | 74.7 | 9.6 |
| 2 years | 74.0 | 9.9 | 74.8 | 10.3 | 75.3 | 9.8 | 73.5 | 10.4 | 74.4 | 10.1 |
| Change from baseline at 1 year | –1.9 | 10.0 | –2.3 | 10.7 | –2.8 | 10.0 | –1.4 | 10.6 | –2.1 | 10.3 |
| Change from baseline at 2 years | –2.6 | 11.1 | –2.5 | 10.4 | –2.5 | 10.9 | –2.6 | 10.7 | –2.5 | 10.8 |
FIGURE 34.
Systolic blood pressure by visit (a) by drug and (b) by treatment regimen.
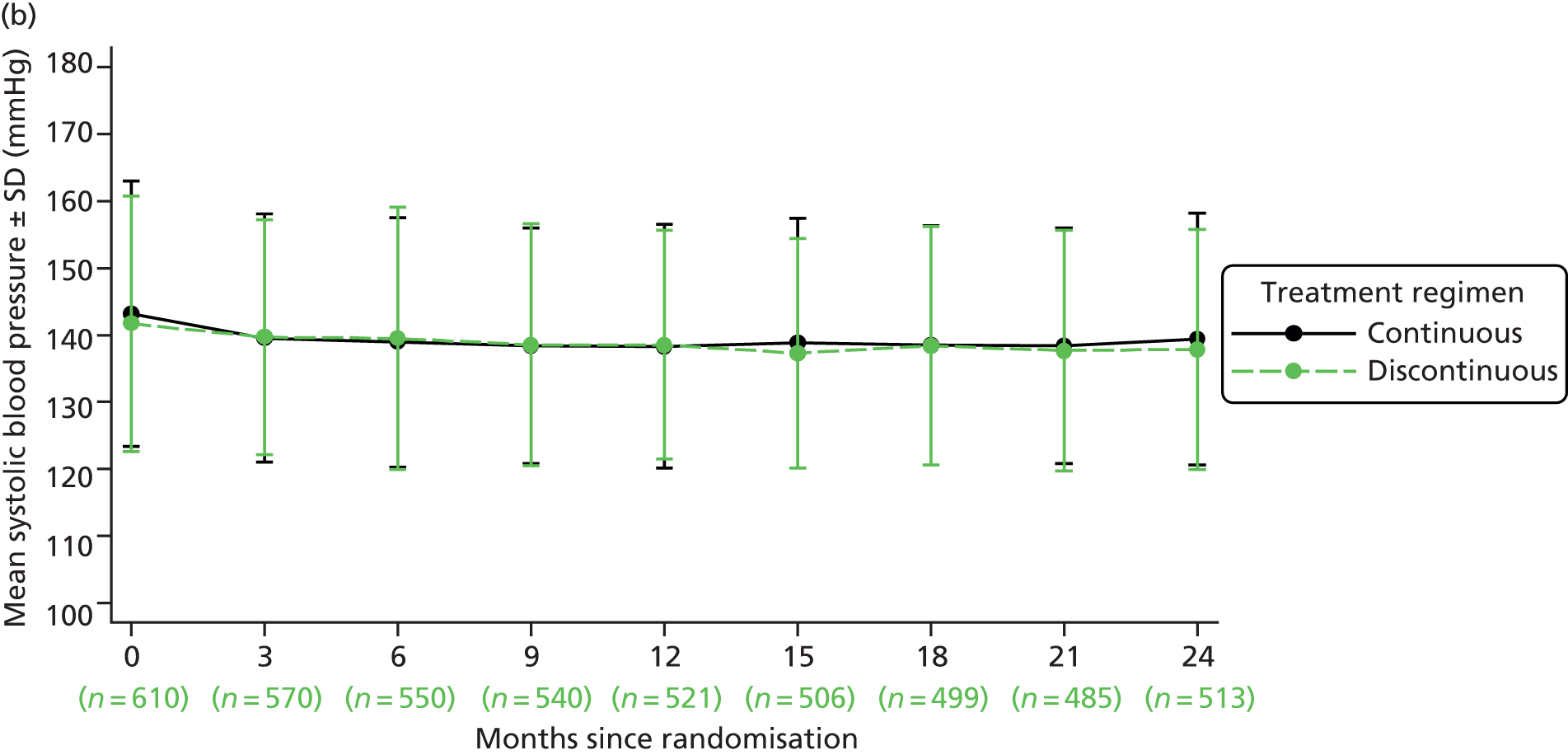
FIGURE 35.
Diastolic blood pressure by visit (a) by drug and (b) by treatment regimen.
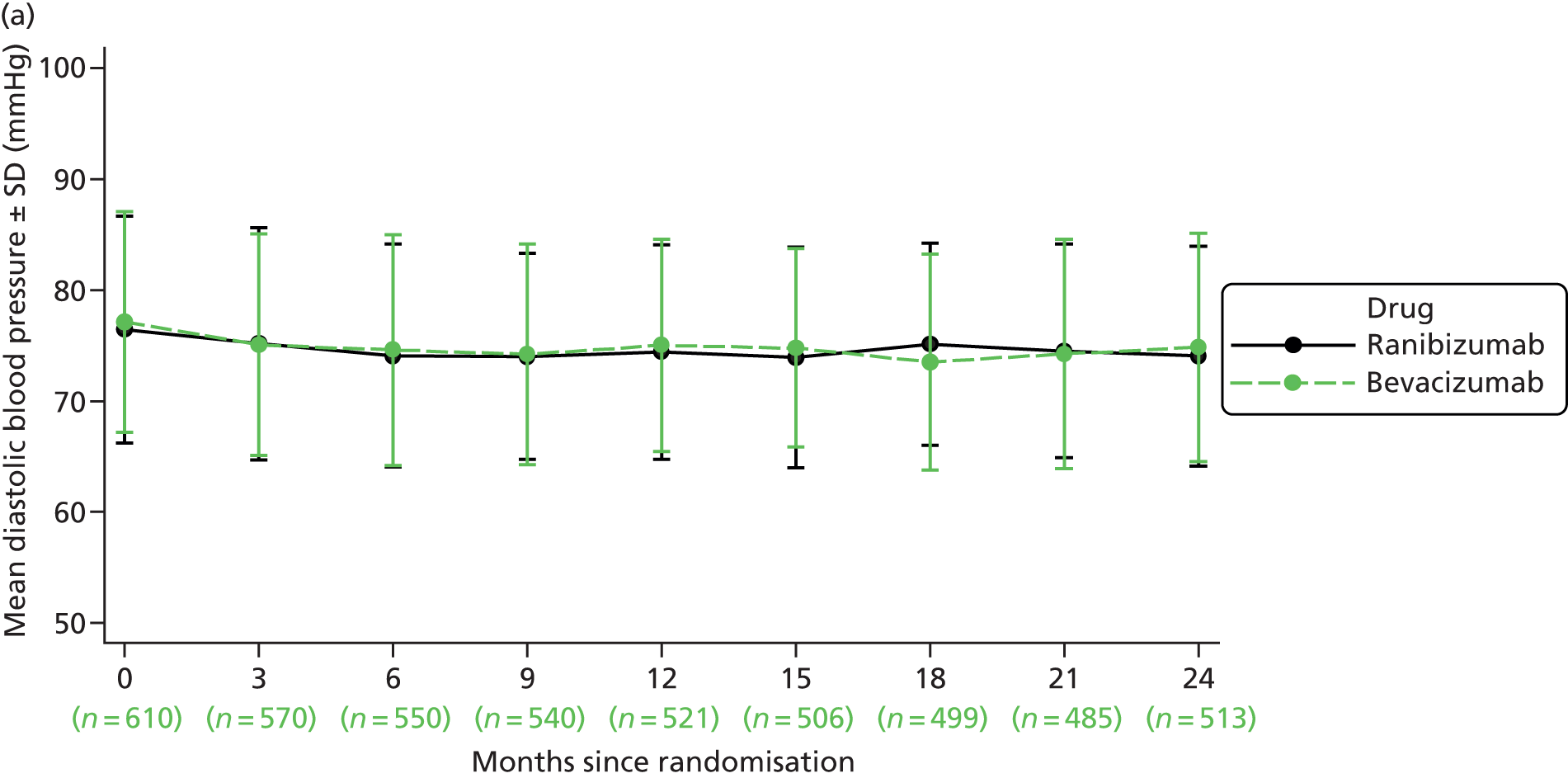
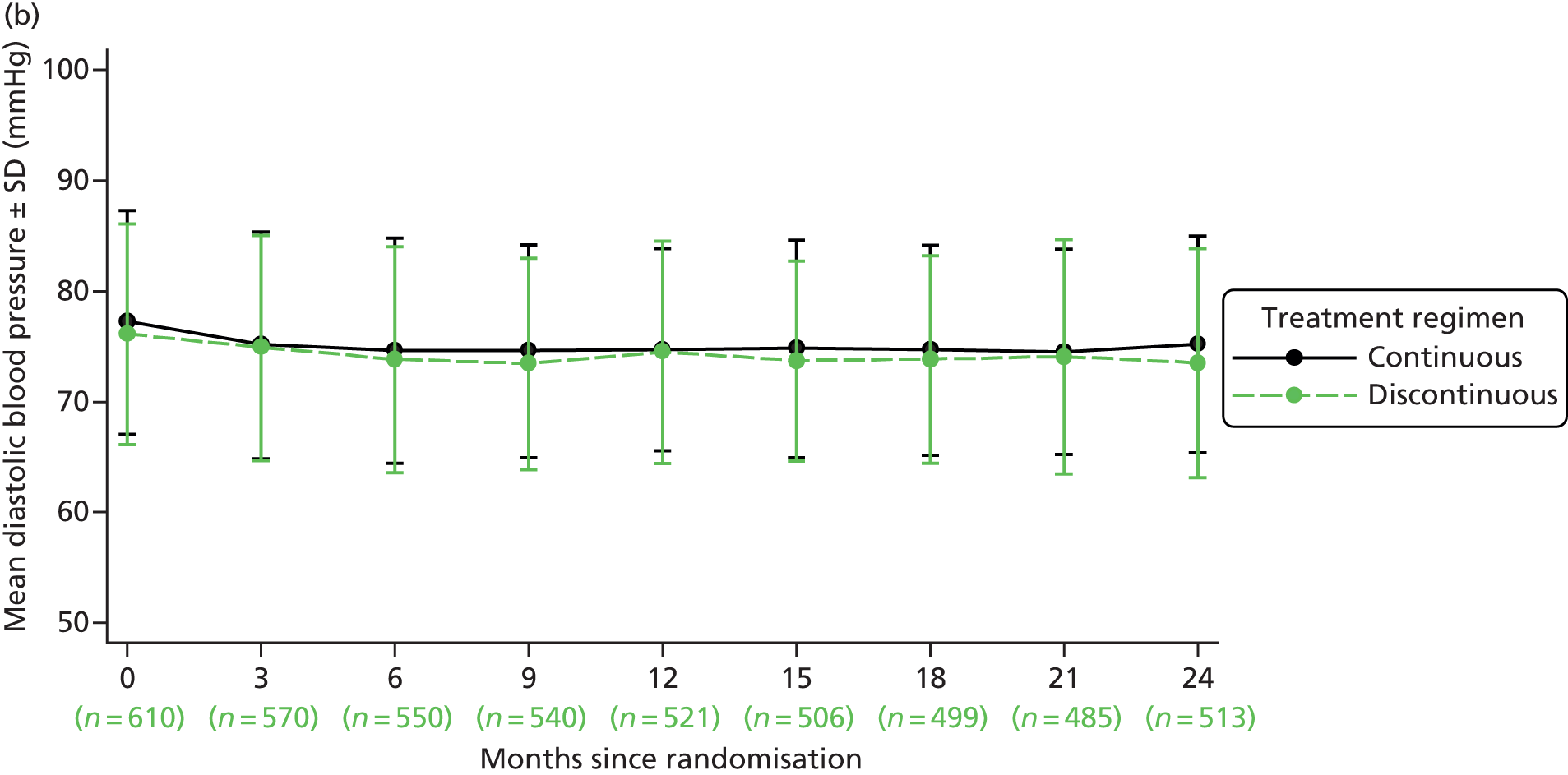
Changes in best corrected distance visual acuity between consecutive visits
Changes in BCVA between consecutive visits were monitored throughout the trial. In particular, a decrease of ≥ 15 letters in the number of letters read on a ETDRS chart (equivalent to the loss of three lines of letters) was considered to be a visually significant event. BCVA was measured at every visit but a refraction to check the optical correction was carried out at intermediate visits only if the VA dropped by ≥ 15 letters or a refractive change was suspected. Overall, 127 participants (21%) experienced a drop of ≥ 15 letters between visits on at least one occasion. A breakdown by group is shown in Table 25.
| Randomised to: | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | ||||||
| n | % | n | % | n | % | n | % | n | % |
| 54/314 | 17 | 73/296 | 25 | 55/308 | 18 | 72/302 | 24 | 127/610 | 21 |
Worsening angina
Incident or worsening angina, defined as a change of two or more classes, or from class 3 to hospital admission for angina, using the Canadian Cardiovascular Society (CCS) classification, was also monitored. However, incident or worsening of angina was not defined as a SAE. (Hospital admission for angina was, by definition, considered to be a SAE.) Worsening angina was reported for just eight participants (Table 26).
| Randomised to: | Overall (n = 610) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314) | Bevacizumab (n = 296) | Continuous (n = 308) | Discontinuous (n = 302) | ||||||
| n | % | n | % | n | % | n | % | n | % |
| 4/314 | 1 | 4/296 | 1 | 5/308 | 2 | 3/302 | 1 | 8/610 | 1 |
Summary
Scores for generic and disease-/treatment-specific HRQoL were similar by drug and treatment regimen at both 1 and 2 years. No differences for subgroups of participants with better or worse vision in the study eye were found for any of the three HRQoL measures. No differences over time or between groups were observed for blood pressure. Overall, 127 participants (21%) experienced a drop of ≥ 15 letters between consecutive visits at some point during follow-up.
Chapter 8 Results of the economic evaluation
Microcosting of consultation costs
Twelve of the 23 IVAN trial centres completed operating cost questionnaires providing data on the resource use and costs associated with managing patients receiving intravitreal anti-VEGF injections for nAMD. Five centres ran a ‘one-stop’ service in which patients attended clinic to have monitoring and (if required) intravitreal injection in the same visit, three centres ran ‘two-stop’ services with separate monitoring and injection clinics, and four centres used a mixture of two-stop and one-stop clinics. The 12 centres ran a total of 40 different types of clinic that differed in staffing levels.
Monitoring consultations without FFA were found to cost an average of £72 per attendance, and each intravitreal injection cost on average £61 (Table 27 and Figure 36). Consultations (such as visit 0 in IVAN) involving monitoring, intravitreal injection and FFA therefore cost £171.92 (£71.83 + £60.65 + £39.44) in total.
| Cost per evaluation or injection | Number of clinicsa | Weighted average (£) | SD (£) | Range (£) |
|---|---|---|---|---|
| Monitoring consultation excluding FFA | 33 | 71.83 | 40.84 | 12.94–284.01 |
| Monitoring consultation including the cost of FFA for the 0–30% of patients who receive FFA | 33 | 75.95 | 40.90 | 13.39–288.62 |
| Intravitreal injection given alongside monitoring, in one-stop clinics | 16 | 60.65 | 17.57 | 34.30–145.25 |
| Intravitreal injection in a separate injection-only consultation without OCT | 7 | 60.93 | 11.28 | 43.56–83.93 |
| FFA: includes reagents, consumables, staff and facilities directly involved in FFA | 33 | 39.44 | 15.61 | 20.46–102.92 |
FIGURE 36.
Distribution of consultation costs across IVAN centres. (a) Monitoring consultations excluding FFA; (b) injection consultations alongside monitoring; (c) injection consultations without monitoring; and (d) FFA.

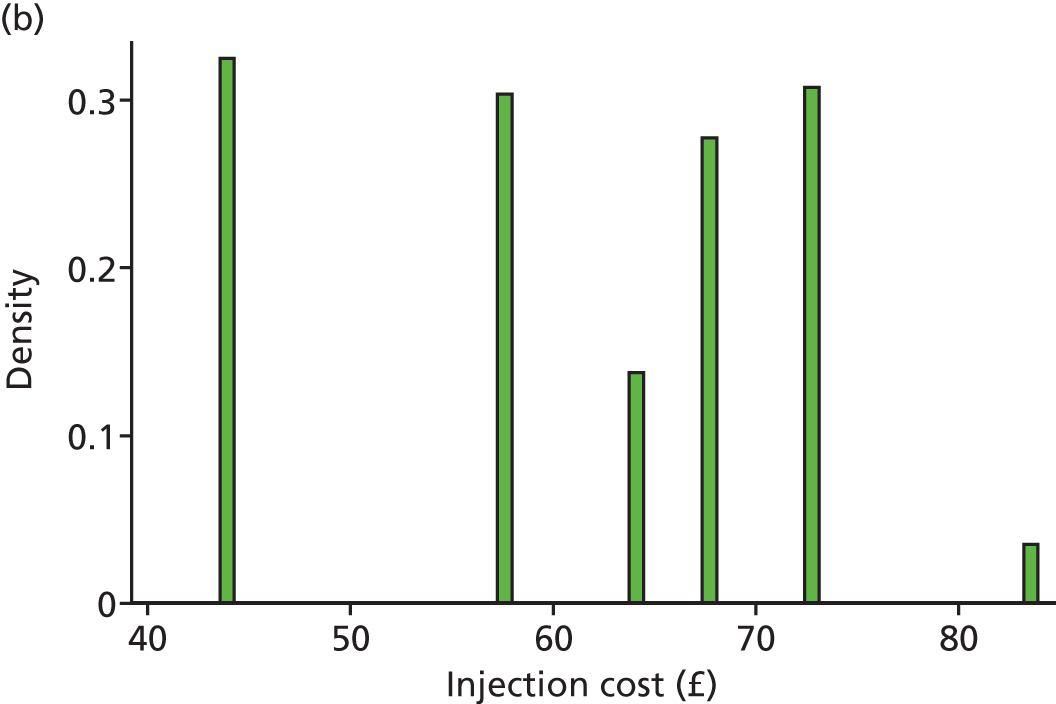
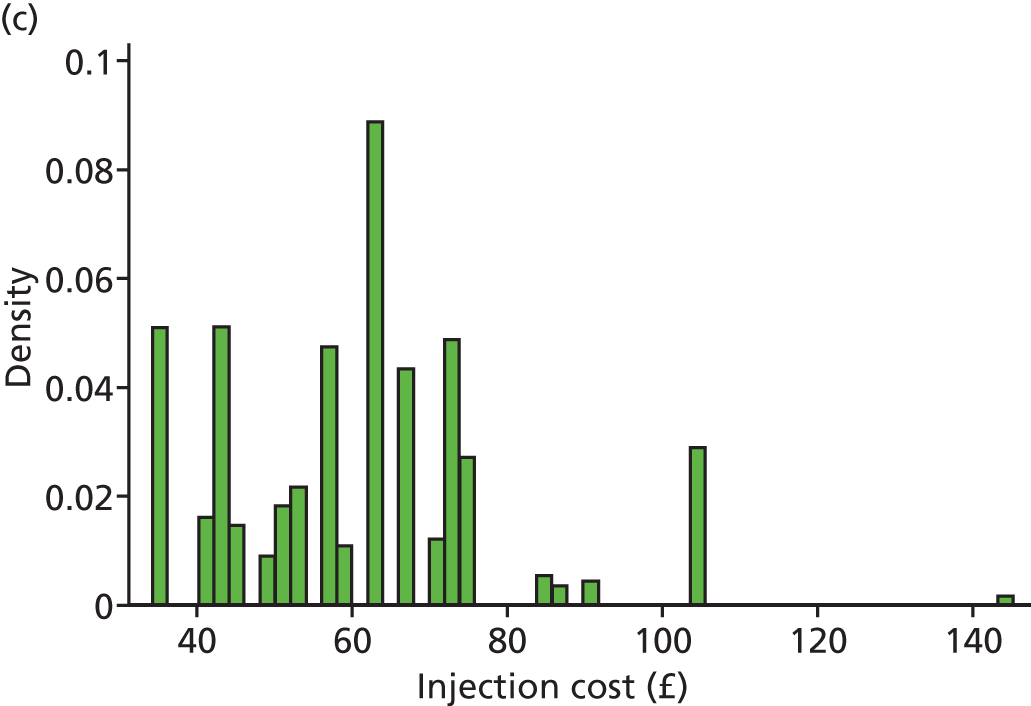

Analysis of resource use and costs
Resource use (Table 28) and costs (Tables 29 and 30) were compared between randomised groups. Unless otherwise stated, all figures represent the mean cost or resource use per patient over the entire 2-year trial and include discounting to adjust for time preference.
| NHS resource | Mean (95% CI) | Difference (95% CI) between | Interaction (95% CI) | ||||
|---|---|---|---|---|---|---|---|
| Continuous ranibizumab | Discontinuous ranibizumab | Continuous bevacizumab | Discontinuous bevacizumab | Ranibizumab vs. bevacizumaba | Continuous vs. discontinuous treatmenta | ||
| Injection consultations | 21.7 (21.2 to 22.1) | 12.7 (12.0 to 13.4) | 22.0 (21.6 to 22.4) | 13.0 (12.3 to 13.8) | –0.3 (–1.0 to 0.4) | 9.0 (8.3 to 9.7)b | 0.0 (–0.3 to 0.3) |
| Monitoring consultations | 7.6 (7.4 to 7.9) | 13.7 (13.1 to 14.2) | 7.1 (6.9 to 7.4) | 13.2 (12.7 to 13.8) | 0.5 (0.0 to 0.9) | –6.1 (–6.5 to -5.6)b | 0.1 (–0.2 to 0.3) |
| Bed-days linked to expected SAEsc | 0.3 (0.0 to 0.6) | 0.7 (0.2 to 1.2) | 0.8 (0.1 to 1.4) | 1.0 (–0.2 to 2.1) | Continuous: –0.4 (–1.2 to 0.3); discontinuous: –0.3 (–1.5 to 1.0) | Ranibizumab: –0.4 (–1.0 to 0.3); bevacizumab: –0.2 (–1.5 to 1.1) | –0.1 (–1.6 to 1.3) |
| Ambulatory consultations within 30 days of expected (S)AEsc | 3.1 (2.4 to 3.9) | 2.8 (2.3 to 3.3) | 3.1 (2.4 to 3.7) | 2.8 (2.2 to 3.5) | Continuous: 0.1 (–0.9 to 1.1); discontinuous: 0. (–0.9 to 0.8) | Ranibizumab: 0.3 (–0.5 to 1.2); bevacizumab: 0.2 (–0.7 to 1.1) | 0.1 (–1.1 to 1.4) |
| Changes in medications associated with expected (S)AEsc | 3.0 (2.4 to 3.6) | 2.8 (2.2 to 3.4) | 2.6 (2.2 to 3.1) | 3. (2.5 to 3.6) | Continuous: 0.4 (–0.4 to 1.1); discontinuous: –0.2 (–1.0 to 0.6) | Ranibizumab: 0.2 (–0.6 to 1.0); bevacizumab: –0.4 (–1.1 to 0.3) | 0.6 (–0.4, 1.6) |
| Outcome | Quarter | Mean (SE), £ | Interaction (95% CI), £ | |||
|---|---|---|---|---|---|---|
| Continuous ranibizumab | Discontinuous ranibizumab | Continuous bevacizumab | Discontinuous bevacizumab | |||
| Drug cost | 1 | 2189 (15) | 2207 (9) | 144 (1) | 145 (1) | –18 (–52 to 17) |
| 2 | 2113 (23) | 953 (69) | 137 (2) | 78 (5) | 1101 (957 to 1245)a | |
| 3 | 2097 (27) | 1164 (66) | 137 (2) | 80 (5) | 877 (736 to 1017)a | |
| 4 | 2110 (26) | 890 (69) | 136 (2) | 77 (5) | 1160 (1016 to 1304)a | |
| 5b | 1994 (31) | 1084 (68) | 128 (2) | 75 (5) | 857 (710 to 1003)a | |
| 6b | 1927 (38) | 997 (65) | 128 (2) | 64 (5) | 866 (721 to 1011)a | |
| 7b | 1936 (34) | 1018 (66) | 126 (2) | 68 (5) | 859 (716 to 1002)a | |
| 8b | 1912 (41) | 926 (69) | 128 (2) | 65 (5) | 924 (768 to 1081)a | |
| Administration and monitoring | 1 | 296 (6) | 298 (6) | 295 (7) | 296 (7) | 0 (–25 to 24) |
| 2 | 251 (6) | 246 (12) | 246 (7) | 239 (12) | –3 (–40 to 35) | |
| 3 | 250 (6) | 224 (10) | 246 (7) | 227 (11) | 6 (–29 to 41) | |
| 4 | 251 (6) | 220 (11) | 245 (7) | 221 (11) | 7 (–29 to 44) | |
| 5 | 238 (7) | 221 (11) | 230 (7) | 215 (11) | 2 (–33 to 37) | |
| 6 | 229 (7) | 212 (10) | 230 (7) | 209 (12) | –4 (–40 to 32) | |
| 7 | 232 (7) | 208 (11) | 228 (7) | 210 (11) | 5 (–30 to 40) | |
| 8 | 226 (7) | 209 (11) | 230 (7) | 209 (12) | –4 (–41 to 32) | |
| Medications and medical service use | 1 | 43 (13) | 27 (16) | 62 (26) | 19 (5) | –26 (–91 to 39) |
| 2 | 31 (9) | 72 (23) | 106 (84) | 36 (9) | –111 (–283 to 61) | |
| 3 | 18 (4) | 102 (71) | 21 (4) | 42 (18) | –63 (–208 to 81) | |
| 4 | 34 (8) | 26 (6) | 52 (32) | 49 (24) | 4 (–76 to 84) | |
| 5b | 75 (34) | 23 (4) | 45 (21) | 22 (5) | 29 (–51 to 109) | |
| 6b | 58 (25) | 81 (45) | 32 (8) | 230 (186) | 175 (–204 to 554) | |
| 7b | 47 (12) | 55 (18) | 223 (133) | 74 (49) | –157 (–441 to 127) | |
| 8b | 36 (8) | 38 (8) | 65 (29) | 32 (13) | –34 (–100 to 32) | |
| QALYs | 1 | 0.206 (0.002) | 0.207 (0.002) | 0.206 (0.002) | 0.204 (0.003) | –0.003 (–0.010 to 0.004) |
| 2 | 0.207 (0.003) | 0.209 (0.004) | 0.209 (0.003) | 0.205 (0.004) | –0.006 (–0.019 to 0.006) | |
| 3 | 0.205 (0.004) | 0.206 (0.004) | 0.208 (0.003) | 0.204 (0.004) | –0.005 (–0.018 to 0.007) | |
| 4 | 0.205 (0.004) | 0.204 (0.004) | 0.205 (0.003) | 0.203 (0.004) | –0.001 (–0.015 to 0.013) | |
| 5b | 0.198 (0.004) | 0.194 (0.004) | 0.199 (0.003) | 0.194 (0.004) | –0.001 (–0.015 to 0.012) | |
| 6b | 0.197 (0.003) | 0.190 (0.004) | 0.197 (0.003) | 0.194 (0.003) | 0.005 (–0.008 to 0.017) | |
| 7b | 0.195 (0.003) | 0.187 (0.004) | 0.192 (0.004) | 0.192 (0.004) | 0.008 (–0.006 to 0.021) | |
| 8b | 0.195 (0.004) | 0.185 (0.005) | 0.189 (0.004) | 0.188 (0.004) | 0.009 (–0.007 to 0.024) | |
| Treatment/difference | Mean (95% CI) QALYsa | Mean (95% CI) drug cost,a £ | Mean (95% CI) administration and monitoring cost, £ | Mean (95% CI) medications/medical service cost,a £ | Mean (95% CI) total cost,a £ | Mean (95% CI) net benefits,a,b £ |
|---|---|---|---|---|---|---|
| Discontinuous bevacizumab | 1.584 (1.538 to 1.630) | 651 (605 to 698) | 1825 (1708 to 1941) | 526 (144 to 908) | 3002 (2601 to 3403) | 28,683 (27,707 to 29,658) |
| Continuous bevacizumab | 1.604 (1.563 to 1.645) | 1065 (1048 to 1081) | 1952 (1860 to 2043) | 585 (250 to 919) | 3601 (3259 to 3943) | 28,480 (27,548 to 29,412) |
| Discontinuous ranibizumab | 1.582 (1.530 to 1.634) | 9229 (8584 to 9875) | 1838 (1724 to 1952) | 432 (253 to 611) | 11,500 (10,798 to 12,202) | 20,142 (18,963 to 21,321) |
| Continuous ranibizumab | 1.608 (1.565 to 1.651) | 16,286 (16,011 to 16,562) | 1970 (1883 to 2057) | 334 (215 to 452) | 18,590 (18,258 to 18,922) | 13,576 (12,769 to 14,383) |
| Difference: ranibizumab vs. bevacizumab | Continuous: 0.004 (–0.046 to 0.054) | Continuous: 15,222 (14,948 to 15,495)c | 16 (–109 to 141) | Continuous: –251 (–604 to 102) | Continuous: 14,989 (14,522 to 15,456)c | Continuous: –14,904 (–15,995 to –13,813)c |
| Discontinuous: –0.002 (–0.064 to 0.060) | Discontinuous: 8578 (7932 to 9225)c | Discontinuous: –94 (–514 to 326) | Discontinuous: 8498 (7700 to 9295)c | Discontinuous: –8541 (–9939 to –7144)c | ||
| Difference: continuous vs. discontinuous | Ranibizumab: 0.026 (–0.032 to 0.085) | Ranibizumab: 7057 (6364 to 7750)c | 130 (20 to 239)c | Ranibizumab: –98 (–310 to 113) | Ranibizumab: 7090 (6337 to 7844)c | Ranibizumab: –6566 (–7861 to –5271)c |
| Bevacizumab: 0.020 (–0.032 to 0.071) | Bevacizumab: 413 (365 to 462)c | Bevacizumab: 59 (–438 to 556) | Bevacizumab: 599 (91 to 1107)c | Bevacizumab: –203 (–1372 to 967) | ||
| Interaction | 0.006 (–0.071 to 0.084) | 6643 (5949 to 7338)c | 5 (–31 to 42) | –157 (–696 to 381) | 6491 (5604 to 7379)c | –6363 (–8088 to –4638)c |
After allowing for censoring and discounting, discontinuous treatment involved nine fewer injections over the 2-year trial period than continuous treatment (p < 0.001; Table 28). However, the discontinuous regimen evaluated in IVAN also required six additional monitoring consultations (p < 0.001) assuming that discontinuous patients receive monitoring consultations on all visits when they did not meet treatment failure criteria the previous month or the month before, while continuous patients receive monitoring every 3 months regardless of outcomes. Bevacizumab-treated patients received 0.3 more injections (p = 0.354) and 0.5 fewer monitoring consultations (p = 0.055) than those randomised to ranibizumab; this trend may be due to chance, particularly as it is seen in the groups randomised to continuous treatment as well as those on discontinuous treatment. There were no significant differences between groups for inpatient stays (p ≥ 0.338), ambulatory consultations with GPs, nurses or hospital clinicians (p ≥ 0.359), or changes in medication (p ≥ 0.718).
Quarterly costs and QALYs were examined to evaluate the statistical significance of interactions between drug and treatment regimen and to explore how costs and QALYs varied over time (see Table 29). Figures were divided into quarters (rather than years or months) to match the KMSA analyses used to adjust for censoring. Within this table and Table 30, ‘drug costs’ comprise the cost of bevacizumab or ranibizumab; ‘administration/monitoring costs’ comprise the cost of injection consultations, monitoring consultations and FFA; and ‘medication/medical service costs’ comprise the cost of hospitalisations for expected SAEs, changes in medications licensed for expected AEs or expected SAEs, and outpatient/GP visits that were for ocular conditions or occurred within 30 days of the date of onset of an expected AE or expected SAE.
In the continuous treatment groups, costs and QALYs were similar across all eight quarters (see Table 29); the small decrease in costs and QALYs over time is due to mortality and discounting of values in year 2 to allow for time preference. By contrast, in the groups randomised to discontinuous treatment, the cost of anti-VEGF drugs was around twice as high in quarter 1 (when patients received monthly injections) and fluctuated over time, depending on the number of patients meeting treatment failure criteria. The cost of administering intravitreal injections and monitoring outcomes to evaluate treatment failure criteria also fell slightly after quarter 1. Conversely, SEs around drug costs and drug administration/monitoring were smallest in quarter 1 (when almost all patients receive three injections) and were smaller for continuous and discontinuous treatment. The combined cost of medication changes, ambulatory consultations and hospitalisations linked to expected AEs or SAEs (referred to hereafter as medication/medical service costs) fluctuated substantially in all groups, being greatly affected by costly hospitalisations in a minority of patients.
Statistically significant interactions between drug and treatment regimen were observed for drug costs in quarters 2–8 (p < 0.001; see Table 29 and Figure 37); as expected, these interactions were larger than the difference between continuous and discontinuous treatment and were super-additive in that the difference between continuous ranibizumab and discontinuous bevacizumab is larger than the sum of the individual treatment effects. Analyses of drug cost, therefore, allowed for interactions in quarters 2–8 (see Chapter 2). Allowing for interactions, drug costs differed significantly between all four groups (p < 0.001). The continuous ranibizumab group accrued the highest drug cost (£16,286, 95% CI £16,011 to £16,562), which was £15,222 (95% CI £14,948 to £15,495; p < 0.001) higher than the cost in the continuous bevacizumab group and £7057 (95% CI £6364 to £7750; p < 0.001) higher than the cost for the discontinuous ranibizumab group (see Table 29 and Figure 37). Although the difference was substantially smaller, the continuous bevacizumab group also accrued significantly higher costs than the discontinuous bevacizumab group (difference £413, 95% CI £365 to £462; p < 0.001).
FIGURE 37.
Breakdown of costs for each treatment. Error bars show SEs around total cost.
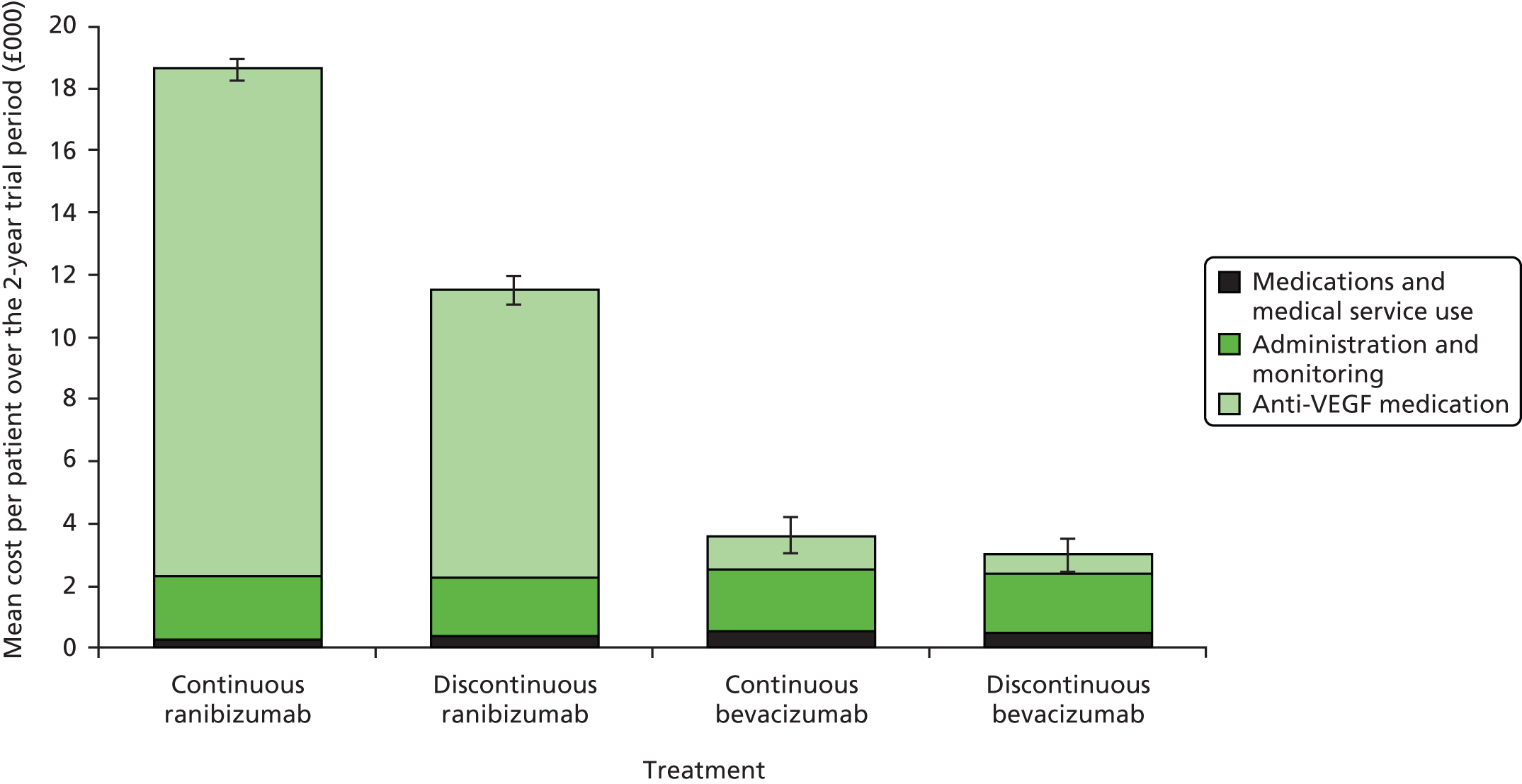
For administration and monitoring costs, interactions were non-significant (p ≥ 0.70) and substantially smaller than the main effects of drug or treatment regimen; drug and treatment regimen were therefore assumed to have additive effects on administration and monitoring costs and differences between drugs are therefore averaged across treatment regimens (see Chapter 2). Administration and monitoring costs were £130 (95% CI £20 to £239; p = 0.021) higher with continuous treatment than with discontinuous treatment, as continuous treatment increased the number of injection consultations more than it decreased the number of monitoring consultations (see Table 28). There was no statistically significant difference in administration and monitoring costs between ranibizumab and bevacizumab (MD £16, 95% CI –£109 to £141; p = 0.80).
Interactions for the medication/medical service costs associated with expected AEs or expected SAEs were non-significant (p ≥ 0.206), although the overall interaction across all eight quarters was larger than the difference between drugs (see Table 30). These interactions were qualitative in that the sign (not just the magnitude) of the effect of treatment regimen changes depending on which drug was used: for example, the continuous ranibizumab group accrued substantially lower medication/medical service costs than the discontinuous ranibizumab group, whereas the continuous bevacizumab group accrued higher medication/medical service costs than the discontinuous ranibizumab group. Interactions were, therefore, included in the base-case analysis of medication/medical service costs in quarters 2–8, as they could change conclusions about which treatment is most cost-effective (see Chapter 2). Medication/medical service costs were highest for the continuous bevacizumab group £585 (95% CI £250 to £919) and lowest for the continuous ranibizumab group £334 (95% CI £215 to £452). The variability within each group was large, mainly due to the high cost of hospitalisations in a handful of patients. As a result, neither drug (p ≥ 0.163) nor treatment regimen (p ≥ 0.363) had a statistically significant effect on medication/medical service costs.
Anti-VEGF drug costs accounted for 80–88% of costs among patients randomised to ranibizumab, compared with 22–30% for the bevacizumab groups (Table 31 and see Figure 37). Among patients randomised to bevacizumab, however, administration and monitoring costs were the greatest cost driver, accounting for 54% of costs with continuous therapy and 61% of costs with discontinuous (vs. 11–16% for the ranibizumab groups).
| Analysis | Expected NB at a 20,000 per QALY ceiling ratio (SE) | Incremental cost Cont ran vs. Cont bev (SE, probability cost-saving, %) | Cost per QALY Cont bev vs. Disc bev (probability cost-effective, %) | Strategy with highest NB (probability maximises NB, %) | |||
|---|---|---|---|---|---|---|---|
| Continuous ranibizumab (Cont ran), £ | Discontinuous ranibizumab (Disc ran), £ | Continuous bevacizumab (Cont bev), £ | Discontinuous bevacizumab (Disc bev), £ | ||||
| Base-case analysis | 13,576 (412) | 20,142 (602) | 28,480 (475) | 28,683 (498) | 14,989 (238, 0) | 30,220 (37) | Disc bev (63) |
| 1-year time horizon | 6780 (218) | 10,075 (298) | 14,741 (237) | 14,814 (242) | 7868 (122, 0) | 26,846 (39) | Disc bev (61) |
| Complete case analysis: excluding all patients with any missing data | 14,340 (424) | 21,196 (684) | 29,310 (475) | 29,828 (495) | 15,343 (197, 0) | 98,691 (18) | Disc bev (82) |
| Including differences in deaths unrelated to study medication | 13,580 (421) | 20,183 (612) | 28,591 (486) | 28,503 (554) | 14,979 (265, 0) | 17,577 (55) | Cont bev (55) |
| Allowing for interactions in all cost and QALY calculations | 13,575 (412) | 20,143 (604) | 28,481 (475) | 28,682 (499) | 14,991 (239, 0) | 30,116 (37) | Disc bev (63) |
| Including only statistically significant interactions | 13,530 (386) | 20,186 (555) | 28,529 (439) | 28,634 (452) | 15,069 (214, 0) | 25,110 (41) | Disc bev (59) |
| Bevacizumab costs £100/dose | 13,576 (412) | 20,142 (602) | 27,372 (473) | 28,005 (494) | 13,881 (237, 0) | 51,922 (14) | Disc bev (86) |
| Bevacizumab costs £30/dose | 13,576 (412) | 20,142 (602) | 28,893 (476) | 28,935 (499) | 15,402 (238, 0) | 22,136 (47) | Disc bev (53) |
| Use one 4-ml vial of bevacizumab per patient (£242.6651), with the remainder thrown away | 13,576 (412) | 20,142 (602) | 24,273 (468) | 26,109 (492) | 10,782 (237, 0) | 112,625 (0) | Disc bev (100) |
| Reducing the price of ranibizumab by 25% | 17,648 (415) | 22,449 (571) | 28,480 (475) | 28,683 (498) | 10,918 (219, 0) | 30,220 (37) | Disc bev (63) |
| Reducing the price of ranibizumab by 50% | 21,719 (422) | 24,756 (550) | 28,480 (475) | 28,683 (498) | 6846 (203, 0) | 30,220 (37) | Disc bev (63) |
| Increasing the price of ranibizumab by 25% | 9505 (411) | 17,834 (642) | 28,480 (475) | 28,683 (498) | 19,061 (261, 0) | 30,220 (37) | Disc bev (63) |
| Excluding the cost of ranibizumab beyond the 14th injection to each eye. This analysis reflects the original 2008 NICE patient access scheme in which the manufacturer paid for all injections beyond the 14th dose91 | 19,649 (425) | 21,368 (562) | 28,480 (475) | 28,683 (498) | 8916 (197, 0) | 30,220 (37) | Disc bev (63) |
| Assuming that 5.3% of ranibizumab vials and 15.9% of bevacizumab syringes are wasted (based on the proportions observed in IVAN) | 12,669 (411) | 19,628 (610) | 28,278 (475) | 28,559 (497) | 15,694 (243, 0) | 34,176 (32) | Disc bev (68) |
| Applying HRG for ophthalmology consultation (£6576) to all visits instead of using microcosting estimates | 14,084 (410) | 20,543 (597) | 28,989 (475) | 29,085 (495) | 14,989 (229, 0) | 24,866 (44) | Disc bev (56) |
| HRG for minor vitreous retinal procedure for injection and monitoring (£14576) | 12,285 (409) | 18,775 (593) | 27,213 (472) | 27,335 (488) | 15,013 (234, 0) | 26,159 (42) | Disc bev (58) |
| Applying reference cost price for ophthalmology consultation (£117.4375) to all visits instead of using microcosting estimates | 12,905 (410) | 19,384 (594) | 27,825 (473) | 27,938 (490) | 15,005 (232, 0) | 25,714 (43) | Disc bev (57) |
| Doubling administration/monitoring costs | 11,606 (417) | 18,304 (608) | 26,529 (477) | 26,858 (501) | 15,008 (267, 0) | 36,625 (30) | Disc bev (70) |
| Halving administration/monitoring costs | 14,561 (411) | 21,061 (600) | 29,456 (476) | 29,595 (499) | 14,980 (229, 0) | 27,018 (41) | Disc bev (59) |
| Including the cost of protocol-driven resource use (monthly OCT and visit 12 FFA in all patients) | 12,505 (418) | 19,511 (612) | 27,399 (480) | 28,041 (500) | 14,978 (258, 0) | 52,389 (14) | Disc bev (86) |
| Assuming no FFA done beyond visit 0 | 14,999 (413) | 20,995 (593) | 29,921 (479) | 29,556 (501) | 15,008 (233, 0) | 1586 (72) | Cont bev (72) |
| Assuming that missed visits accrue the same administration/monitoring cost as attended visits (but zero drug cost) | 13,487 (411) | 20,065 (603) | 28,369 (476) | 28,584 (500) | 14,967 (237, 0) | 30,870 (36) | Disc bev (64) |
| Including ambulatory consultation costs within 30 days of any AE or SAE and including hospitalisations for any SAE (expected or otherwise) | 13,074 (456) | 19,130 (685) | 27,574 (546) | 27,824 (552) | 14,585 (346, 0) | 32,602 (36) | Disc bev (64) |
| Including all hospitalisation and ambulatory consultation costs regardless of their proximity to any (S)AEs | 12,921 (460) | 18,959 (687) | 27,390 (552) | 27,669 (553) | 14,554 (355, 0) | 34,073 (35) | Disc bev (65) |
| Including 20% VAT on all costs | 9858 (415) | 17,842 (639) | 27,760 (493) | 28,082 (513) | 17,987 (286, 0) | 36,265 (31) | Disc bev (69) |
| Discounting costs and QALYs in year 2 at 1.5% (not 3.5%) | 13,710 (416) | 20,340 (608) | 28,751 (481) | 28,956 (504) | 15,129 (241, 0) | 30,256 (37) | Disc bev (63) |
| Making no adjustment for baseline utility | 13,736 (488) | 19,951 (667) | 28,713 (544) | 29,063 (582) | 14,989 (238, 0) | 48,114 (32) | Disc bev (68) |
| Using HUI3 utilities in place of EQ-5D | 9291 (644) | 14,495 (783) | 22,524 (754) | 22,346 (750) | 14,989 (238, 0) | 15,423 (58) | Cont bev (58) |
| Simply interpolating between scheduled EQ-5D measurements (see Appendix 4) | 13,534 (424) | 20,216 (604) | 28,451 (487) | 28,689 (510) | 14,989 (238, 0) | 33,199 (35) | Disc bev (65) |
| Excluding the post-SAE EQ-5D measurements and interpolating between the non-SAE measurements (see Appendix 4) | 13,543 (422) | 20,195 (604) | 28,472 (486) | 28,679 (509) | 14,989 (238, 0) | 30,610 (37) | Disc bev (63) |
| Linear interpolation between all EQ-5D measurements (see Appendix 4) | 13,618 (408) | 20,190 (599) | 28,420 (478) | 28,637 (501) | 14,989 (238, 0) | 31,400 (37) | Disc bev (64) |
| Subtracting QALY loss from expected SAEs but not unexpected SAEs (see Appendix 4) | 13,563 (420) | 20,178 (604) | 28,507 (478) | 28,670 (501) | 14,989 (238, 0) | 27,506 (39) | Disc bev (61) |
| Doubling the QALY impact of SAEs for which there is no EQ-5D measurement within 2 months of the SAE (see Appendix 4) | 13,547 (426) | 20,064 (613) | 28,460 (480) | 28,680 (501) | 14,989 (238, 0) | 31,599 (36) | Disc bev (64) |
| Doubling the QALY impact of all SAEs and doubling the total medication/medical service costs (see Appendix 4) | 13,276 (435) | 19,656 (642) | 27,904 (580) | 28,160 (597) | 14,738 (390, 0) | 32,770 (37) | Disc bev (63) |
Best case analysis for ranibizumab:
|
21,217 (468) | 23,745 (634) | 26,054 (542) | 26,894 (549) | 4922 (323, 0) | 62,375 (11) | Disc bev (89) |
Analysis of health outcomes
Mean EQ-5D baseline utility was 0.82 (SD 0.21) across all groups: slightly higher than the national average utility for 65- to 74-year-olds (0.78; SD 0.2694) or people aged ≥ 75 years (0.73; SD 0.2794), which may be because patients with serious/life-threatening conditions and those who could not be expected to attend monthly consultations were excluded from the trial. Ordinary least squares (OLS) regression, using the mim command to combine results over the 100 imputed data sets demonstrated that mean EQ-5D utility rose by around 0.018 (SE 0.008; p = 0.02) between visits 0 and 3, and fell by 0.025 (SE 0.009; p = 0.006) between visits 3 and 24. There was marked, but non-significant (p ≤ 0.429), imbalance in EQ-5D utility at baseline, with lower utility in the group randomised to discontinuous ranibizumab and higher utility in the group randomised to discontinuous bevacizumab. The base-case analysis therefore used regression analyses to adjust for this imbalance, which is likely to have arisen by chance but could (in the absence of appropriate adjustment) have introduced bias into QALY calculations.
Mixed models demonstrated that EQ-5D utility was significantly lower after cardiovascular (p = 0.002) and cancer (p < 0.001) SAEs, although the effect of ocular (p = 0.491) or other (p = 0.248) SAEs was non-significant (see Appendix 4). However, averaged across all study arms, taking account of post-SAE EQ-5D measurements and the profile of EQ-5D utility changes around SAEs reduced the mean QALYs accrued over the 2-year trial period by just 0.0001.
Interactions between drug and treatment regimen were non-significant for QALYs (p ≥ 0.274), although the overall interaction across all eight quarters was larger than the difference between drugs (see Table 29). As for medication/medical service costs, the interactions for QALYs was qualitative, with the continuous ranibizumab group accruing more QALYs across all eight quarters than the continuous bevacizumab group, whereas the discontinuous ranibizumab group accrued marginally fewer QALYs than discontinuous bevacizumab. The base-case analysis therefore allowed for interactions on QALYs for quarters 2–8 (see Chapter 2).
All four treatment groups accrued around 1.6 QALYs over the 2-year trial period (see Table 30), reflecting the mean utility of 0.8. All treatment groups accrued fewer QALYs during year 2 as a result of discounting and mortality (see Table 29). There were no statistically significant differences in QALYs between drugs (p ≥ 0.87) or treatment regimens (p ≥ 0.361). However, there was a trend towards patients randomised to continuous therapy accruing around 0.02 additional QALYs than those randomised to discontinuous therapy over the 2-year study period. This trend was consistent across all quarters other than quarter 1, in which all patients receive monthly injections (see Table 29). However, this difference was very small compared with the SEs around QALY differences and could have arisen by chance.
Results of the base-case economic evaluation
Base-case comparison between ranibizumab and bevacizumab
The SAP prespecified that CMA would be used to compare ranibizumab and bevacizumab unless the absolute difference in EQ-5D QALYs was > 0.05 (see Chapter 2). The observed MD was 0.004 for continuous therapy and 0.002 for discontinuous therapy (see Table 30), which is substantially smaller than this non-inferiority margin and is so small in relation to cost differences that assuming equivalent efficacy is highly unlikely to bias the conclusions. 54 Conclusions about the cost-effectiveness of ranibizumab and bevacizumab were, therefore, primarily based on differences in cost, assuming that there was no difference in health outcomes.
The total cost accrued by patients randomised to continuous ranibizumab was £18,590 (95% CI £18,258 to £18,922), £14,989 (95% CI £14,522 to £15,456; p < 0.001) more than continuous bevacizumab, which equates to a fivefold difference in cost. The average cost per patient in the group randomised to discontinuous ranibizumab was £11,500 (95% CI £10,798 to £12,202), nearly four times higher than the cost in the discontinuous bevacizumab group (£3002, 95% CI £2601 to £3403), giving an absolute difference of £8498 (95% CI £7700 to £9295; p < 0.001). None of the 13,000 bootstrap replicates found total costs to be lower among patients randomised to ranibizumab than among those randomised to bevacizumab, indicating that we can be more than 99.99% confident that substituting bevacizumab for ranibizumab would save the NHS money and (based on CMA) would be cost-effective.
Broadening the analysis to consider both costs and QALYs demonstrates that continuous ranibizumab costs £3.5M per QALY gained compared with continuous bevacizumab. This ICER is 175 times higher than the £20,000 per QALY ceiling ratio below which treatments are generally considered to be cost-effective,57 indicating that continuous ranibizumab represents very poor value for money.
This conclusion is confirmed by the scatter graph plotting bootstrap estimates of the joint distribution of incremental costs and QALYs (Figure 38). This indicates that, for both continuous and discontinuous treatment, the entire distribution of costs lies well above the x-axis (indicating significantly higher costs for ranibizumab), whereas the incremental QALYs are evenly distributed either side of zero.
FIGURE 38.
Differences in costs for ranibizumab against the difference in QALYs for (a) continuous treatment and (b) discontinuous treatment. The black square represents the MD in cost and QALYs and the green dots represent individual bootstrap replicates.


Bootstrapping results were also used to estimate cost-effectiveness acceptability curves plotting the probability that ranibizumab is cost-effective compared with bevacizumab at different ceiling ratios representing the maximum that the NHS is willing or able to pay to gain one QALY (Figure 39). The probability of ranibizumab being cost-effective can be read off the curve at any ceiling ratio of interest. For example, if the NHS were willing to pay £200,000 per QALY gained (10 times higher than NICE’s stated threshold57), the probability of ranibizumab being cost-effective is 8% for discontinuous treatment and 0.3% for continuous treatment. The probability of ranibizumab being cost-effective compared with bevacizumab remained < 0.01% at all ceiling ratios of < £80,000 per QALY gained: four times higher than the £20,000 per QALY ceiling ratio typically used in NHS decision-making. 57 This analysis demonstrates that ignoring QALY differences has no impact on the conclusions, as the probability that ranibizumab is cost-effective is < 0.01% at a £20,000 per QALY ceiling ratio as well as in a CMA that assumes no QALY gains.
FIGURE 39.
Pairwise cost-effectiveness acceptability curves for ranibizumab vs. bevacizumab. The vertical dotted line represents a ceiling ratio of £20,000 per QALY gained.

Base-case comparison between continuous and discontinuous treatment
As prespecified in the SAP, continuous and discontinuous treatment were compared using CUA. It is important to take account of non-significant differences in QALYs and evaluate the joint distribution of costs and QALYs, as assuming no difference in health outcomes has been shown to give misleading conclusions unless the difference in cost is so large that no plausible difference in health effects could change the conclusions. 53,54,95 For both ranibizumab and bevacizumab, the point estimate (the black squares in Figure 40) lay in the north-east quadrant of the cost-effectiveness plane, as continuous therapy was significantly more costly (p ≥ 0.021) and non-significantly more effective (p ≥ 0.381) than discontinuous therapy.
FIGURE 40.
Incremental costs and outcomes for continuous vs. discontinuous treatment for (a) ranibizumab and (b) bevacizumab. The black squares represent the MD in costs and QALYs and the green dots represent individual bootstrap replicates.


Dividing the incremental cost for continuous vs. discontinuous bevacizumab (£599) by the incremental QALY gain (0.02) gave an ICER of £30,220 per QALY gained. This figure is above the £20,000 per QALY ceiling ratio below which treatments are generally considered to be cost-effective,57 suggesting that continuous bevacizumab is not cost-effective, although it is only just above the £20,000–30,000 per QALY range above which additional considerations are required to justify adoption. 57 To facilitate statistical analysis, costs and QALYs were also combined to estimate net benefits, which equal QALYs multiplied by the ceiling ratio minus costs. At a £20,000 per QALY ceiling ratio, discontinuous bevacizumab had £203 (95% CI –£967 to £1372; p = 0.734; see Table 30) higher net benefits than continuous bevacizumab, indicating that continuous bevacizumab is not cost-effective. However, this difference was not significant and there was substantial uncertainty around both incremental costs and QALYs (see Figure 40).
However, as the higher ranibizumab drug costs substantially increased the difference in cost between continuous and discontinuous therapy, continuous ranibizumab cost £270,217 per QALY gained compared with discontinuous ranibizumab (£7090 ÷ 0.026). This is 13 times higher than the amount the NHS would normally consider cost-effective, indicating that continuous ranibizumab is not cost-effective. Discontinuous ranibizumab was associated with significantly higher net benefits than continuous ranibizumab (p < 0.001).
Bootstrapping results were also used to estimate cost-effectiveness acceptability curves, which demonstrated that there is a 37% chance that continuous bevacizumab is cost-effective compared with discontinuous bevacizumab at a £20,000 per QALY ceiling ratio (Figure 41). This figure rises to 50% at a £30,000 per QALY ceiling ratio and asymptotes towards 77% (the probability that continuous bevacizumab generates more QALYs than discontinuous bevacizumab). By contrast, the probability of continuous ranibizumab being cost-effective compared with discontinuous ranibizumab remained at < 0.001% unless the NHS were willing to pay at least £60,000 per QALY gained.
FIGURE 41.
Pairwise cost-effectiveness acceptability curves for comparison between continuous and discontinuous treatment. The vertical dashed line represents a ceiling ratio of £20,000 per QALY gained.
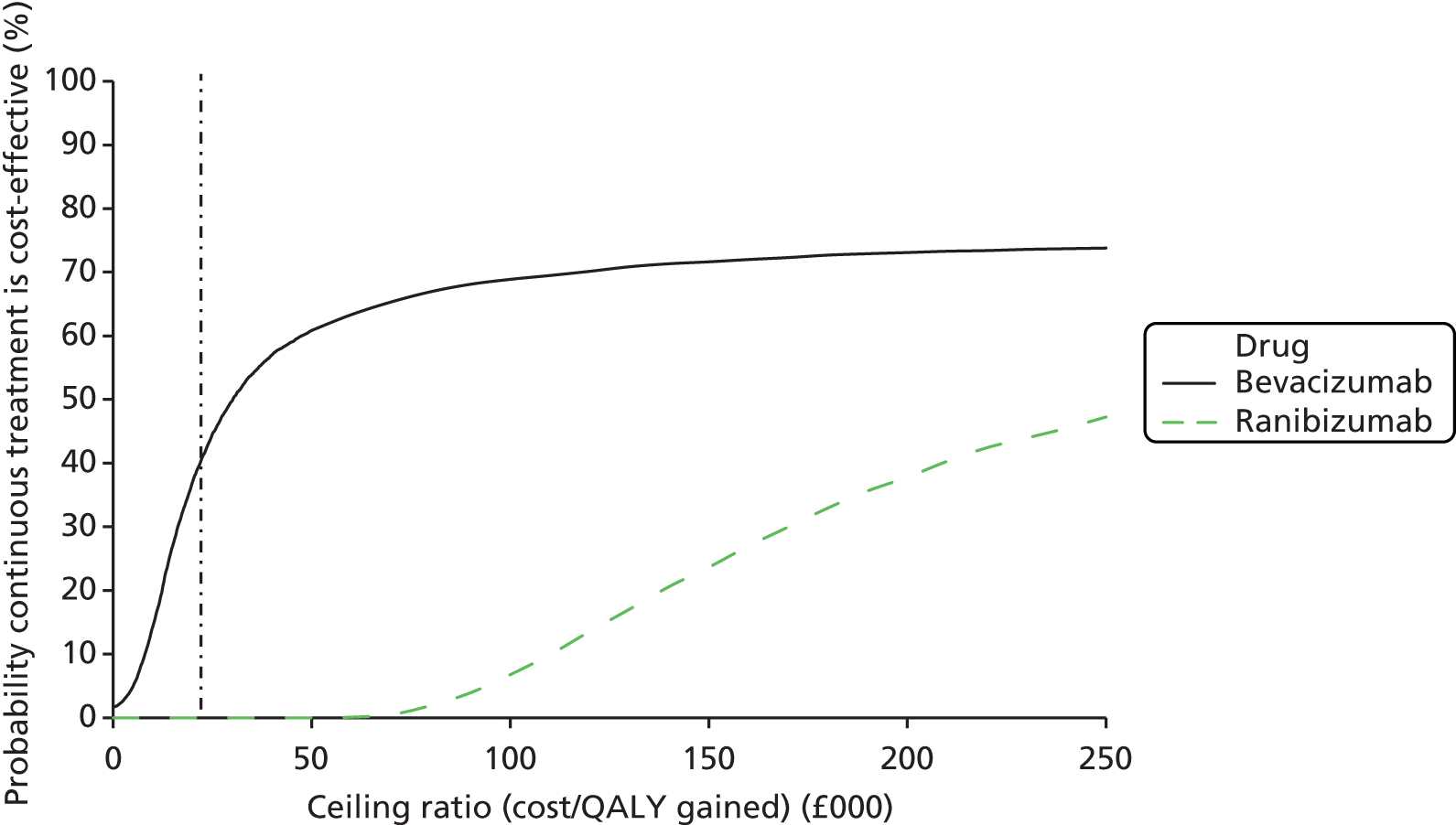
Base-case four-way comparison
Considering the four cells of the factorial design as mutually exclusive alternative strategies for managing nAMD gives the same conclusions as the base-case analysis, but enables us to consider the joint distribution around the entire decision, considering both drug and treatment regimen simultaneously.
Considering the four treatments simultaneously demonstrates that discontinuous ranibizumab is ‘strongly dominated’ by both continuous and discontinuous bevacizumab, that is patients in this group accrued (slightly) fewer QALYs and (substantially) higher costs (Figure 42). Of the three non-dominated treatments that lie on the cost-effectiveness frontier (the black line in Figure 42), continuous bevacizumab costs £30,220 per QALY gained compared with discontinuous bevacizumab, whereas continuous ranibizumab costs £3.5M per QALY gained compared with continuous bevacizumab. Discontinuous bevacizumab is, therefore, the most cost-effective of the four treatments evaluated in IVAN if the NHS is willing to pay no more than £20,000–30,000 per QALY gained, although continuous bevacizumab would be considered the most cost-effective strategy at ceiling ratios between £30,220 and £3.5M per QALY gained. However, there remains substantial uncertainty around differences in QALYs between the four treatment strategies, as shown by the substantial overlap in the distribution of QALYs in Figure 42; by contrast, the differences in costs are substantial and statistically significant.
FIGURE 42.
Total costs against total QALYs for each of the four treatments. The pale crosses represent the mean costs and mean QALYs for each of the four treatment strategies, while the smaller dots represent individual bootstrap replicates. Bootstrap results for discontinuous bevacizumab are shown with blue dots to distinguish them from the overlapping cluster representing continuous bevacizumab. The black line represents the cost-effectiveness frontier.
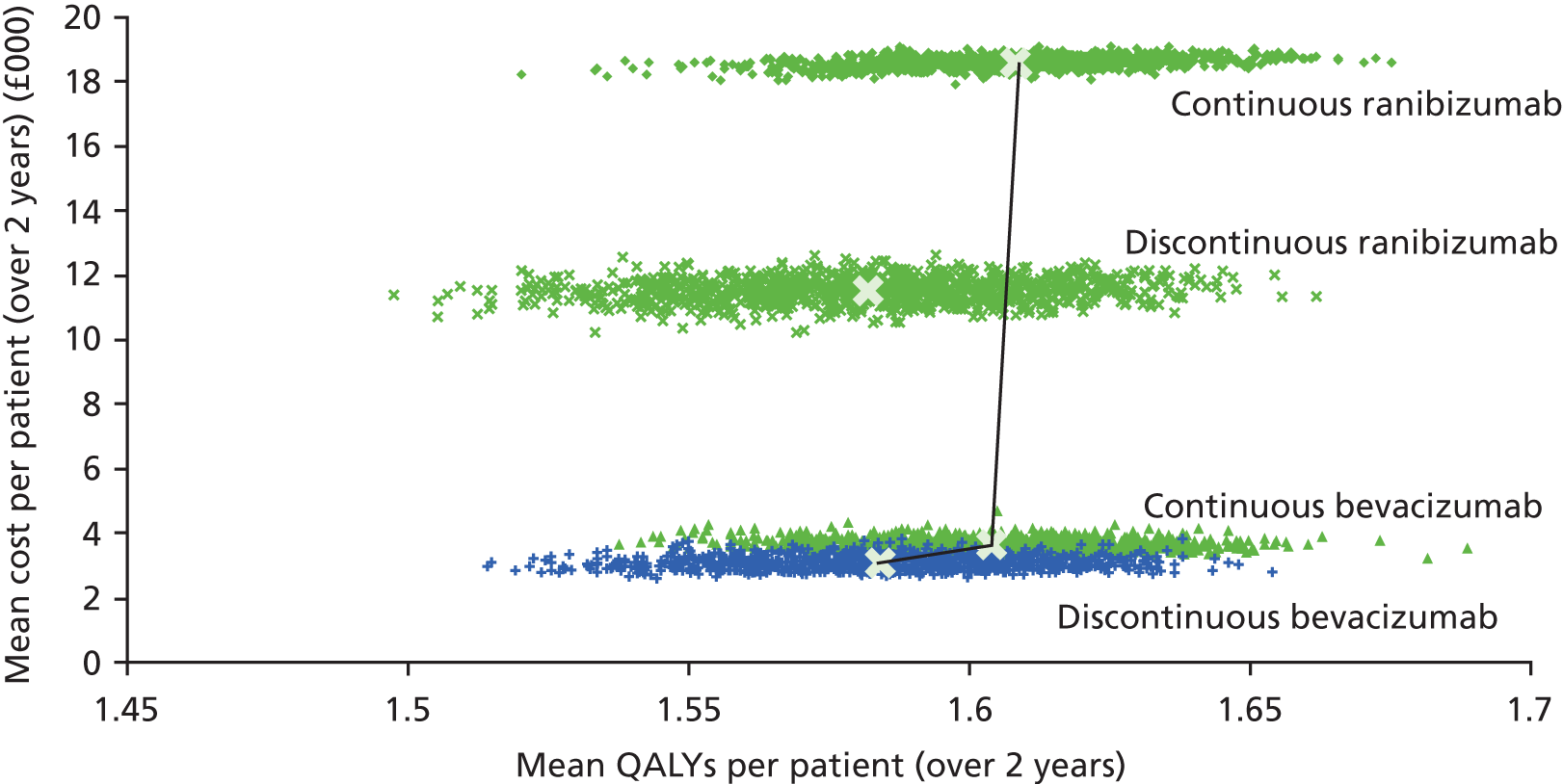
Figure 43 plots the probability that each treatment has highest net benefits across all four treatment strategies against the ceiling ratio. The probability of each treatment having highest net benefits out of the four strategies evaluated in IVAN can be read off the graph at any ceiling ratio. For example, at a £20,000 per QALY ceiling ratio, there is a 63% chance that continuous bevacizumab has highest net benefits and a 37% chance that discontinuous bevacizumab is best, whereas the probability of either ranibizumab strategy having the highest net benefits is < 0.01%. Indeed, this analysis demonstrates that we can be > 99.99% confident that neither continuous nor discontinuous ranibizumab is good value for money unless the NHS are willing to pay > £60,000 per QALY gained.
FIGURE 43.
Cost-effectiveness acceptability curves for multiple comparisons based on comparisons between all four treatment strategies. The thin dashed blue line that follows the curve for discontinuous bevacizumab at ceiling ratios below £30,220 per QALY and then follows the curve for continuous bevacizumab (and is shifted slightly upwards in the figure for clarity) represents the cost-effectiveness frontier, i.e. the probability that the treatment maximising expected net benefits is best value for money. The vertical dashed line represents a ceiling ratio of £20,000 per QALY gained.
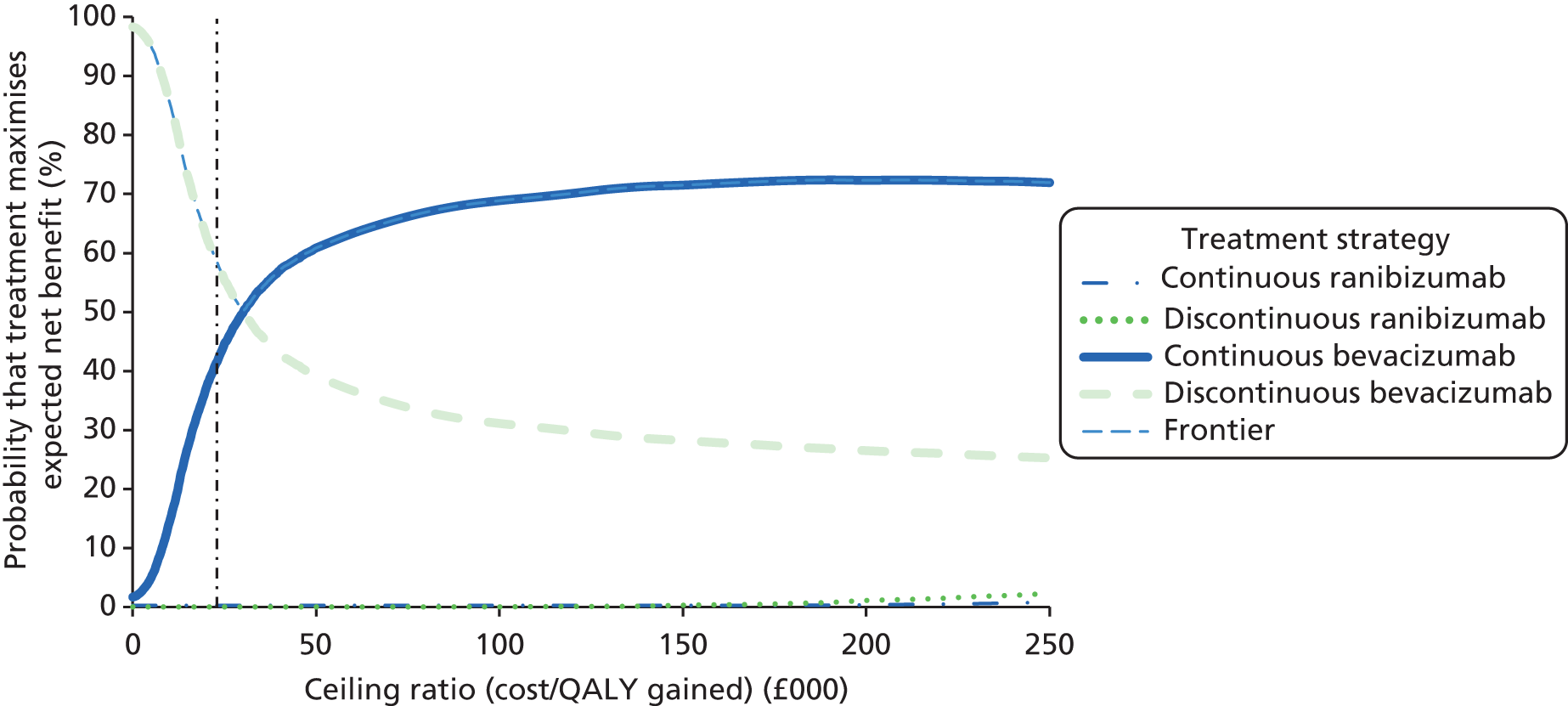
Sensitivity analyses
Thirty-four different sensitivity analyses were conducted to evaluate the impact of changing the assumptions or methods used in the base-case analysis (see Table 31). The rationale and results of each sensitivity analysis are discussed in turn.
The impact of a shorter time horizon was evaluated to assess whether or not costs and/or benefits changed over time. Taking a 1-year time horizon and excluding costs and benefits accrued in year 2 reduced net benefits (as only 1 year of QALYs is included) but had minimal effect on the conclusions. A complete case analysis excluding the 59 patients withdrawing from the study early and the 114 other patients with missing data on any EQ-5D utilities (except after death) confirmed the base-case conclusion that discontinuous bevacizumab is the most cost-effective strategy evaluated in IVAN. This analysis suggests that the conclusions are not sensitive to the methods used to impute missing data, although this analysis should be interpreted with caution, as it excludes 54% of patients with SAEs and over-represents patients who died early in the study.
The base-case analysis used a variant on KMSA to exclude differences in deaths that investigators considered not or unlikely to be related to study medication. If the Kaplan–Meier survival estimates were based solely on the number of deaths observed in each treatment arm in each quarter (rather than being based on the number of potentially drug-related deaths and a common rate of unrelated deaths), the ICER for continuous bevacizumab versus discontinuous bevacizumab fell to £17,577 per QALY, making continuous bevacizumab cost-effective.
Allowing for interactions in administration and monitoring costs as well as drug costs, medication/medical service costs and QALYs had minimal effect, although including only statistically significant interactions (namely those for drug cost but not those for QALYs or medication/medical service costs) decreased the ICER for continuous bevacizumab compared with discontinuous bevacizumab to £25,110 per QALY gained.
The price of bevacizumab (£49 per dose in the base case) was varied over the range of prices charged by different suppliers. Varying the cost over this range did not change the conclusions, although the ICER for continuous bevacizumab compared with discontinuous bevacizumab was found to be £22,136 per QALY if bevacizumab cost £30 per dose and £112,625 per QALY if bevacizumab cost £243 per dose (the list price for a 4-ml vial used to treat colorectal cancer). Similarly, the price of ranibizumab was varied by ±25% and reduced by 50% to evaluate the impact of different discounts that hospitals may receive off the list price. Reducing the price of ranibizumab increased net benefits for ranibizumab-treated groups but did not change the conclusion that we can be more than 99.99% confident that ranibizumab is poor value for money compared with bevacizumab at a £20,000 per QALY ceiling ratio. Although there is a nationally agreed discount price for ranibizumab, this is commercially sensitive, so could not be evaluated as a sensitivity analysis; however, it is unlikely that the discount is > 50%. However, we were able to evaluate the patient access scheme previously negotiated by NICE in the original 2008 NICE appraisal,91 in which the manufacturer supplied ranibizumab free of charge after patients’ 14th injection; this increased net benefits for continuous ranibizumab, but had less effect on discontinuous ranibizumab, as fewer patients on discontinuous therapy had more than 14 injections and did not change the conclusions.
One-way sensitivity analysis was conducted to evaluate the impact of varying the difference in cost between the two drugs on the ICER for continuous ranibizumab versus continuous bevacizumab (Figure 44). This suggested that although reducing the price difference between the two drugs could reduce the ICER from the base-case estimate of £3.5M per QALY gained, the price of ranibizumab would need to cost just £63.46 per dose (£14.46 more than bevacizumab) to be cost-effective at a £20,000 per QALY ceiling ratio.
FIGURE 44.
Two-way sensitivity analysis showing the impact of drug cost on the incremental cost-effectiveness of continuous ranibizumab vs. continuous bevacizumab.
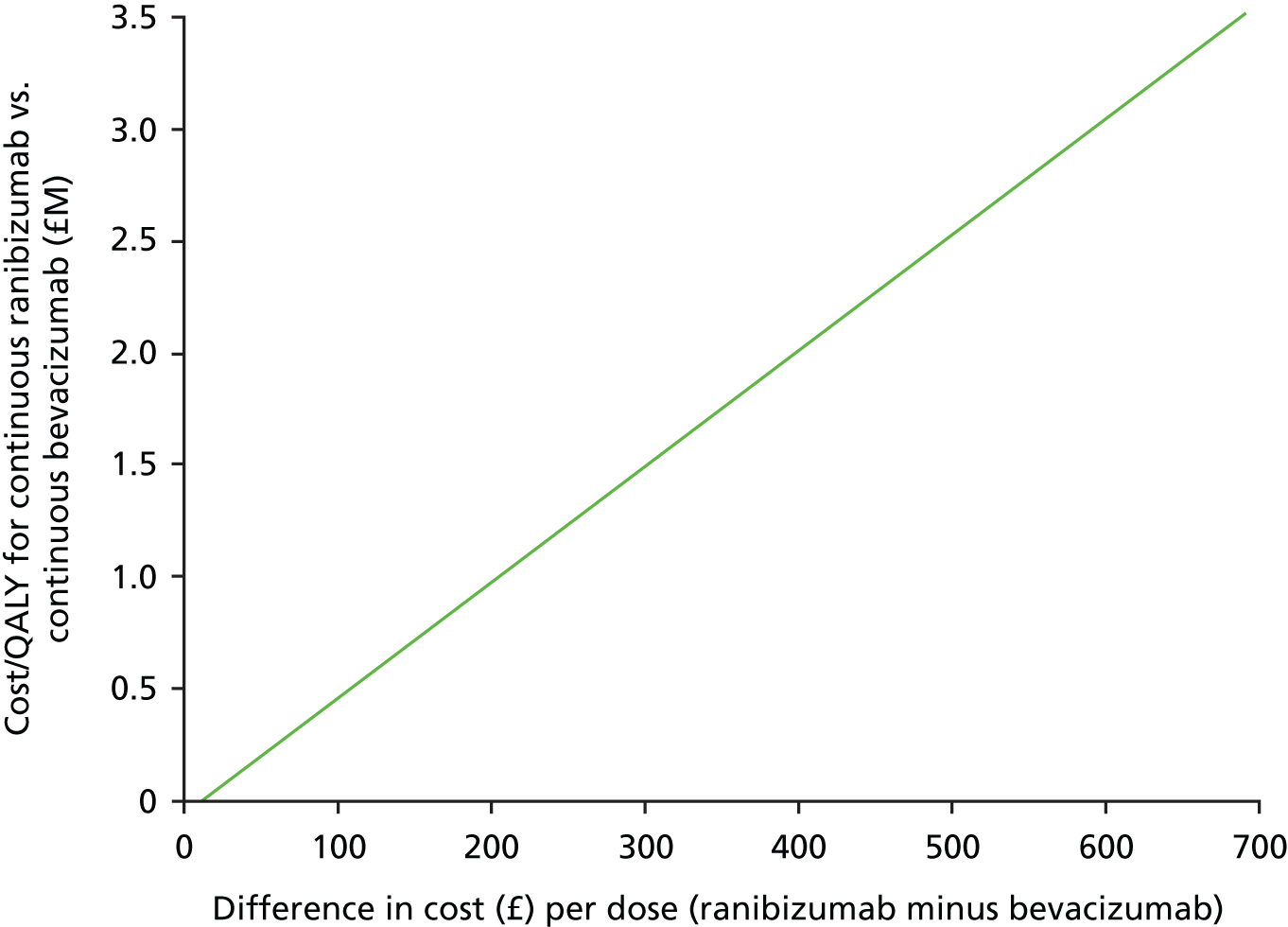
The base-case analysis assumed, based on discussions with ophthalmologists, that no anti-VEGF medication was wasted. However, within the trial 5.3% of ranibizumab doses and 15.9% of bevacizumab doses dispensed were not administered to patients. The main reason for such wastage was that the medication was dispensed by the pharmacy for patients who did not attend or did not need treatment, and could not be given to other patients: either because of pharmacy regulations or preparation of the medication for masked administration. Wastage also resulted from the careful monitoring of temperature and storage conditions during transportation. The shelf life of the bevacizumab syringes after repackaging by the compounding pharmacy at Liverpool also caused wastage early in the trial: initially the product had an expiry time from manufacture of 14 days. After 15 months, this expiry time was increased to 90 days. A sensitivity analysis using the wastage rates observed within the trial increased costs and reduced net benefits, but did not change the conclusions. Furthermore, drug wastage in routine clinical practice is likely to be minimal compared with that observed in the trial, as bevacizumab now has a longer shelf life and drugs dispensed in routine clinics can more easily be administered to other patients if the intended patient does not attend or does not need an injection.
We also conducted a sensitivity analysis using the HRG and reference cost estimates of the cost of ophthalmology consultations or the HRG for minor vitreous procedures in place of the micro-costing estimates used in the base-case analysis; this changed the costs for continuous treatment more than discontinuous treatment, although discontinuous bevacizumab remained the most cost-effective treatment. Similarly, neither halving nor doubling administration/monitoring costs changed the conclusions.
We also varied the assumptions used to identify which resources incurred in follow-up visits were driven by the protocol rather than reflecting routine clinical practice. Including the cost of conducting OCT monthly, and assuming that all patients would have FFA at visit 12, increased costs and reduced net benefits in all treatment arms, but had a disproportionately greater effect in the discontinuous arm, increasing the ICER for continuous bevacizumab compared with discontinuous bevacizumab to £52,000 per QALY. The base-case analysis included the cost of FFAs conducted in the discontinuous arm to evaluate treatment failure criteria, in addition to including the cost of FFA for a proportion of patients in the continuous arm at each visit. Excluding the cost of all post-baseline FFAs increased the net benefits accrued in the continuous groups more than the discontinuous groups, which meant that continuous bevacizumab (not discontinuous) became the most cost-effective treatment, costing £1600 per QALY compared with discontinuous bevacizumab. Including the cost of consultations (but not anti-VEGF medication) for visits that patients missed increased costs and decreased net benefits, but did not change the conclusions.
The base-case analysis focused on hospitalisations and ambulatory consultations within 30 days of expected AEs or SAEs; sensitivity analyses including a wider range of medical service costs reduced net benefits slightly, but did not change the conclusions. Similarly, including VAT or changing the rate at which costs and QALYs accrued in year 2 were discounted to adjust for time preference had no effect on the conclusions.
However, measuring utilities using HUI3 rather than EQ-5D substantially changed the conclusions and reduced all net benefits, as HUI3 utilities were around 16% lower than EQ-5D utilities, but followed a very similar profile. When HUI3 utilities were used in place of EQ-5D, the difference in QALYs between continuous and discontinuous bevacizumab was larger, and the ICER was decreased to £15,423 per QALY, suggesting that continuous bevacizumab is the most cost-effective treatment considered in the analysis. The difference in HUI3 QALYs between continuous ranibizumab and bevacizumab (0.088) was larger than the non-inferiority margin (0.05) determining whether or not the comparison between drugs was based on CMA or CUA; however, continuous ranibizumab, nonetheless, cost £170,742 per QALY gained compared with continuous bevacizumab. Six other sensitivity analyses varying the methods used to estimate QALYs were also conducted (see Appendix 4), although these had negligible effects.
The incidence of SAEs within IVAN was substantially lower than that observed in CATT. 22 We therefore evaluated the impact of doubling the impact of SAEs on QALYs and (simultaneously) doubling the total cost of medication changes and medical service use costs associated with expected AEs and expected SAEs. However, this had negligible impact on the results.
A final analysis using the cost assumptions most favourable to ranibizumab in combination confirmed the base-case analysis finding that ranibizumab is highly unlikely to be good value for money.
Overall, none of the 34 sensitivity analyses changed the conclusion that we can be more than 99% confident that ranibizumab is not cost-effective compared with bevacizumab, although three analyses (using HUI3 utilities, including differences in deaths unrelated to treatment and assuming that FFA would not be done after baseline) found continuous bevacizumab to be cost-effective compared with discontinuous bevacizumab.
Subgroup analyses
The impact of six subgrouping variables was evaluated: baseline VA in the study eye ≥ 55 letters read; baseline CNV size of six or more disc areas; ≥ 50% classic CNV; RAP present; ≥ 75 letters read by the fellow eye at baseline; and VA in the study eye of > 5 letters better than the fellow eye at baseline. None of these subgrouping variables had a statistically significant effect on treatment effects for drug costs, administration/monitoring costs, medication/medical service costs or QALYs accrued in any quarter at the 0.0001 level that was identified using a Bonferroni correction. Incremental costs and QALYs for each subgroup are therefore not shown.
However, some statistically significant effects were seen at the 0.01 level. In particular, in two 3-month periods, the difference in drug cost between continuous and discontinuous treatment was greater in patients with ≥ 55 letters VA in the study eye (p ≤ 0.008), whereas in three quarters the difference in drug cost between continuous and discontinuous treatment was smaller for patients with ≥ 50% classic CNV (p ≤ 0.01). However, these findings should be interpreted with caution as they could have arisen as a result of chance.
Chapter 9 Discussion
Main findings, including meta-analyses
Visual function
After 2 years in the IVAN trial, the prespecified primary time point, the comparison of bevacizumab and ranibizumab for BCVA, was inconclusive when judged against the prespecified non-inferiority margin of 3.5 letters. The comparison of continuous and discontinuous treatment regimens for BCVA was also inconclusive. However, the MDs between groups, tending to favour ranibizumab and continuous treatment, were small (< 2 letters) and estimated to within 2.4 letters.
Non-inferiority for both comparisons would have been established had we used the CATT22 non-inferiority margin of 5 letters. Moreover, the treatment effects for BCVA for the drug comparison were homogeneous across several head-to-head trials. The increased precision gained when all of the comparative effectiveness trials that have been reported were included in a meta-analysis showed clearly that bevacizumab is non-inferior to ranibizumab. However, only IVAN and CATT22 had follow-up to 2 years and are also the only trials to have included arms for drug and treatment regimen comparisons. The treatment effects for BCVA were consistent even when data from only IVAN and CATT22 were included for the drug comparisons. However, the meta-analysis found that discontinuous treatment was inferior to continuous treatment.
The secondary visual function outcomes did not differ by drug. However, the differences in NVA and contrast sensitivity by treatment regimen indicated that discontinuous treatment was inferior to continuous treatment, consistent with the meta-analysis finding for BCVA. Taken together, these findings show that continuous treatment every month gives slightly better visual function than discontinuous treatment. The importance of these findings and their relevance to the patient is unclear, as the differences in secondary visual function outcomes were not reflected in self-reported HRQoL and the difference in BCVA was apparent only in the meta-analysis of IVAN and CATT22 data. Our view is that, although discontinuous treatment is inferior to continuous treatment from a statistical perspective, the magnitude of the observed difference is not clinically important or may not be perceived by patients.
Morphology
With respect to lesion morphology, there were no significant differences between drugs at 2 years, but consistent differences for several morphology indices (lesion present; total lesion thickness at the fovea; retinal plus SRF thickness at the fovea; any fluid on OCT; dye leakage on angiogram) favouring continuous treatment. The meta-analysis of total lesion thickness at the fovea estimated this parameter more precisely.
The CATT 2-year report22 suggested differences between drugs and treatment regimens in the development of new GA in the study eye during follow-up and we regraded colour and FA images in an attempt to replicate this finding. We found no difference in the odds of developing new GA in the study eye between drugs in the IVAN trial alone or when CATT22 and IVAN data were combined. However, our analysis showed a consistent and substantial increase in the risk of developing new GA with continuous compared with discontinuous treatment.
Patient-reported outcomes
There was no evidence at all of any clinically important differences in EQ-5D utility, macular disease-specific quality of life or treatment satisfaction by drug or treatment regimen. The former two findings are consistent with our view that the differences in BCVA and secondary visual function found in the comparisons between treatment regimens may not translate into perceptible differences. The similarity of treatment satisfaction with continuous and discontinuous treatment regimens implies that the differing frequency of injection did not markedly influence treatment satisfaction.
Safety
In the IVAN trial, there were no differences in the frequencies of deaths or SAEs between drugs. However, we observed that mortality was higher at 2 years with discontinuous treatment than continuous treatment.
In meta-analyses of available safety data, there was a statistically significant difference in the risk of any systemic SAE by drug, with bevacizumab tending to confer a higher risk. Although the pooled estimate is strongly weighted by the CATT data (as in a previous meta-analysis of CATT and IVAN data93), the GEFAL42 and MANTA43 trials reported non-significant tendencies consistent with the CATT. 22,96 The finding that SAEs classified as gastrointestinal (a subset of any systemic SAE) occurred more often with bevacizumab than ranibizumab, observed in IVAN and CATT,22,93 was not seen in four other trials that also compared the two drugs42,43 (LUCAS, presented at the 2013 meeting of the American Association for Ophthalmology; BRAMD, presented at the 2013 meeting of EURETINA). Nevertheless, in a post hoc meta-analysis including all of the trials, the finding persisted because the CATT22 and IVAN trials contributed most of the information to the analysis.
The rerandomisation that was undertaken in the CATT22 at the end of year 1 introduced an additional layer of complexity in the study, which resulted in 2-year CATT22 data on SAE and deaths not being reported by treatment regimen. Therefore, the meta-analysis of different treatment regimens that we performed included only the 2-year IVAN data and 1-year data from CATT. 22 This meta-analysis confirmed the finding that the odds of mortality were approximately half for continuous compared with discontinuous treatment, with the OR for the CATT 1-year data22 being very similar to the OR for the IVAN 2-year data. Interestingly, the meta-analysis of the odds of any systemic SAE by treatment regimen also tended to favour continuous treatment, again with the estimates from the two trials being similar. The comparisons of treatment regimens were not masked in either trial, but it seems implausible that bias should lead to an increased frequency of SAEs with discontinuous treatment. Given the almost universal implementation of some form of discontinuous treatment, these findings are worrying and appear counterintuitive when viewed in a conventional dose–response framework. However, in the context of biological therapies, the possibility of immunological sensitisation with intermittent dosing needs to be considered. 97 In addition, it is important to remember that one cannot easily relate the relative risk between treatment regimens to a ‘control’ risk of mortality for the trial populations without any anti-VEGF treatment; the estimated OR could imply a doubling in baseline risk with discontinuous treatment, or a halving in baseline risk with continuous treatment.
Economic evaluation
The main finding from the economic evaluation is that ranibizumab is not cost-effective compared with bevacizumab if the NHS is willing to pay up to £20,000–30,000 per QALY gained. Continuous ranibizumab cost £3.5M per QALY gained compared with continuous bevacizumab. Discontinuous ranibizumab was dominated by bevacizumab, being both more costly and accruing fewer QALYs. The conclusion that ranibizumab is not cost-effective was robust across all sensitivity analyses, including an analysis halving the ranibizumab price, doubling the bevacizumab price and allowing for the cost of managing unexpected AEs and SAEs. The price of ranibizumab would need to be well under £70 per dose in order for it to be cost-effective compared with bevacizumab. Our findings confirm previous threshold analyses by Raftery et al. ,98,99 which showed that the price of ranibizumab would have to be drastically reduced for it to be cost-effective.
Assuming about 17,295 eyes are likely to require treatment for nAMD each year,100 and extrapolating from the difference in cost between discontinuous ranibizumab and discontinuous bevacizumab at 1 year, we estimate that switching all patients from ranibizumab to bevacizumab would save about £102M per year (including 20% VAT).
The economic evaluation suggested that discontinuous bevacizumab is better value for money than continuous bevacizumab unless the NHS is willing to pay ≥ £30,200 to gain a QALY. However, there was substantial uncertainty around this estimate and we can at present be only 63% confident that discontinuous bevacizumab represents best value for money at a £20,000 per QALY ceiling ratio. In particular, this analysis was sensitive to the assumptions used in the analysis. The cost-effectiveness of continuous bevacizumab compared with discontinuous bevacizumab is likely to be particularly sensitive to the way that clinics monitor patients and assess treatment failure, and to the relative cost of consultations for monitoring disease progression and those for administering injections, which vary substantially between clinics.
Strengths and limitations
Applicability and validity of the IVAN trial findings
There are important strengths of the IVAN trial. It was pragmatic, having been carried out in the usual care setting in many hospitals in the NHS, and so directly informs the use of anti-VEGF drugs in similar settings. The factorial design was efficient and provided high statistical power for the primary outcome, despite the fact that the IVAN trial had only half as many participants as CATT. 22 We studied a range of secondary functional outcomes that support the VA findings. Participants, professionals and research staff remained masked to the drug being used with very few exceptions. Adherence to the randomised allocation of drug and treatment regimen was excellent. Retention overall was good given the elderly population, with only 10% withdrawing over the course of the 2 years. Although over half of the participants missed one or more visits, missed visits did not differ by group and most scheduled visits were attended. All participants who were randomised and who had at least one injection in the trial were included in the analysis of the primary outcome. We assessed the resources used to administer treatment and carried out a full health-economic evaluation based on prospectively collected data on costs and QALYs.
We procured bevacizumab for the trial from a compounding pharmacy that manufactured single dose, pre-filled syringes from aliquoting the drug from large-volume commercially available vials. Manufacturing adhered to protocols for tests of potency and sterility approved by the MHRA. As many hospitals that treat patients with bevacizumab do so by dispensing bevacizumab locally for immediate use (as opposed to procuring a ‘manufactured’ product), our findings should be generalised only to bevacizumab sourced from a manufacturing pharmacy. The manufacturing pharmacy should have quality control processes in place to validate stability, potency and sterility that have been approved by a drug regulatory agency.
The interpretation of the meta-analyses is limited by the appropriateness of pooling of available trials. 101 The IVAN and CATT22 were planned to be similar in design, and we described our intention from the outset to pool the findings from the two trials. One-year BCVA and safety results for other trials have been published42–44 or presented at major international meetings [LUCAS, presented at the 2013 meeting of the American Association for Ophthalmology; BRAMD, presented at the 2013 meeting of EURETINA] in recent months. We reasoned that the methods of data collection for these outcomes should be comparable across trials (BCVA is a standard clinical assessment and recording of SAEs is subject to standard regulatory conditions across jurisdictions) so have included data from these trials to ensure that our analyses are as relevant as possible. We have included findings for the longest duration of follow-up available (1 or 2 years) because all trial results show that VA remains effectively stable over this period. We would like to acknowledge that the meta-analyses of safety were prompted by the IVAN and CATT22 data monitoring committees while the trial was still in progress.
Unique design features of the IVAN trial: the ‘IVAN treatment regimen’
The IVAN trial included two design features that were unusual at the time and differed from the CATT22 (which was designed in parallel).
-
All patients were treated (monthly) for the first 3 months.
-
After the first 3 months, participants allocated to discontinuous treatment restarted treatment if the lesion in the study eye met one or more of the retreatment criteria. The decision to restart treatment initiated a further cycle of three injections over 3 months.
We made the first decision because the ANCHOR and MARINA trials had shown clearly that most of the benefit from ranibizumab was achieved in the first 3 months of treatment. 7,8 In the CATT,22 all participants had treatment on their first visit (when an active lesion had to be present in order for a participant to be eligible) but, subsequently, a decision about the need for retreatment was made at each visit for participants allocated to the prn treatment group.
We made the second decision because we wanted to ensure that participants had the least possible risk of disadvantage from discontinuous treatment. We felt that this would remove some of the uncertainty about treatment (for clinicians and patients) and reduce the need for diagnostic investigations compared with higher drug cost/over treatment. Interestingly, it had the unforeseen benefit of avoiding the need to make a decision about retreatment on the subsequent two monthly visits when treatment was mandated as part of the 3-month cycle. At the outset, we were concerned that the IVAN discontinuous treatment regimen might result in more treatment than strictly necessary, i.e. compared with the CATT22 prn treatment regimen.
In the event, the total number of injections required were very similar in the IVAN discontinuous (median = 13 injections) and CATT22 prn (mean 13.3 injections) treatment regimens, suggesting that it takes 3 months of treatment to make a reactivated lesion quiescent or that the cycle of three mandatory injections over 3 months in some way defers lesion reactivation that would have occurred if only a single treatment had been given. It is also plausible that clinicians withhold treatment when very subtle signs of lesion activity are present. An alternative explanation may be that the tomographic outputs do not reveal very low levels of lesion activity.
Our findings suggest that longer periods of lesion inactivity may arise as a consequence of using the modified prn approach in IVAN, where retreatment decision making was restricted to a limited number of visits. We had hypothesised that clinicians might not recognise subtle signs of lesion activity when present and that this could induce variability in decision-making. Thus the IVAN design, which mandated treatment in cycles of three on detecting signs of lesion activation, supports our aforementioned hypothesis. However, it is important to point out that this discussion relates to an indirect (non-randomised) comparison and the trial-specific differences between treatment regimens are within the precision of the estimated differences in numbers of treatments. Anecdotally, the benefit of avoiding the need to review patients for two visits appears to be attractive to ophthalmologists, as several have adopted 3-month cycles of treatment for reactivated lesions, calling this the ‘IVAN regimen’.
Avoiding the need to review an eye for 2 months is one minor way to relieve some of the workload of reviewing and treating patients with nAMD in busy clinics. The design of the CATT22 tested a different aspect of the cost of treatment by rerandomising participants allocated to monthly treatment during year 1 to prn treatment during year 2. This intervention largely failed, in that the VA of participants treated monthly in year 1 and prn in year 2 converged during year 2 to the VA of participants treated prn in both years. A continuing focus on workload and cost is vital in this area, as the total number of patients with nAMD being treated or monitored has not yet reached a steady state.
Other benefits of the IVAN trial
The IVAN trial has many unique and important features. The IVAN trial is among the first trials of an interventional medicinal product in the field of retina with a non-commercial sponsor, for which the entire budget was obtained from NHS sources. It compared two biological agents made by the same manufacturer yet with widely differing costs. It has had a plethora of spin-off benefits. It facilitated the establishment of a network of investigators and clinical sites, who had previously never engaged in research of this nature, increasing research expertise in several ophthalmology departments that had not previously taken part in a RCT. The strength of the network is evidenced by excellent attendance of IVAN principal investigators at a special session of the last investigators’ meeting to identify and prioritise new trials that this network could undertake. Through their involvement in the construction of the study protocol, development of the algorithms for retreatment and participation in the conduct of the trial, many of the IVAN investigators have enhanced both their research skills and their understanding of the disease itself. This process continues as IVAN investigators contribute to the scientific outputs arising from this study.
The IVAN trial also helped to increase clinical expertise directly. As in the case of the VPDT cohort study,92 the IVAN trial required investment in training of investigators and their teams. Although intravitreal injection of a drug is a standard clinical technique, expertise in the detailed assessment of retinal images in order to make decisions about the need for retreatment was not. We also believe that the attention to detail required when doing research and providing training for non-medical staff, for example in photography and visual function assessment, enhanced general professional skills. Training was undertaken through initial meetings when developing the protocol, visits to hospitals when initiating and monitoring and a programme of regular investigators’ meetings. In these meetings, comparing clinical and reading centre image assessments became a standard tool to improve consensus about retreatment decisions.
The pragmatism of the IVAN trial drew on the unique nature of the NHS. Some participating sites were teaching hospitals and others were not. About half of the local principal investigators had experience of participating in commercial trials but very few had participated in a non-commercial trial. About half of the local research teams were assembled de novo to carry out the IVAN trial. Not surprisingly, therefore, participants were managed in diverse ways at participating sites, for example being reviewed and treated in usual care nAMD/retina clinics or in dedicated research clinics.
The status of the IVAN trial as a NIHR-funded study was also hugely beneficial, as the concurrent development of the Ophthalmology specialty group of the UK Clinical Research Network was instrumental in ensuring appropriate resourcing and targeting of support to research-naive and less experienced clinical sites. The arrangements and structure of IVAN led to enhanced collaborative working across the devolved nations and was instrumental in expanding the network activities.
The complex contracting and financing arrangements were developed both as a consequence of our previous experience in the VPDT cohort study92 and as a matter of necessity for research governance. Although they may appear complicated, in general the arrangements worked despite the need for good communication across organisations. During the course of the trial, because some sites were able to recruit and manage more participants than others, service level agreements with health board/primary care trusts often needed renegotiation with respect to the number of patients for which the provider organisations were willing to pay. This typically involved the CTEU requesting approval to invoice for a higher number than originally contracted, and involved the contract team in Belfast issuing a revised agreement and receiving the signed copy back. At times, this caused delays. If we were to use a similar system again, we would want to find a way of managing contracts that was less cumbersome. Nevertheless, the overall success of the arrangement is demonstrated by the fact that the bank obtained all of the treatment and service support costs to reimburse participating trusts, purchase the drugs for the trial, and pay for other costs, such as drug transport through the service-level agreements.
Making all reimbursement for treatment in the trial (research, service support and treatment costs) conditional on supplying the data and complying with the protocol was also a success, probably because the treatment costs represented a large amount of money that was due to the participating trusts. Through the trial database, the CTEU was able to produce regular reports of activity by participating trust; these reports were shared both with the participating trusts and the bank, and provided the basis for invoices to the bank. The criteria for classifying activity as complete (and hence payment due) were not very sophisticated, and activity was sometimes reimbursed before the data for a visit was complete. For example, imaging for a particular visit may have been deferred because of an equipment failure but payment for the visit was authorised. The investment in information and technology made by the CTEU during the course of the trial has allowed the CTEU to improve the sophistication of the checks before authorising payment in a more recent trial (requiring that all data validation queries for a particular trial activity must be resolved).
Research in context
Comparisons of outcomes by drug
Overall, the findings for the IVAN trial and CATT,22 and other recent trials, consistently show that bevacizumab has very similar efficacy to ranibizumab. With seven trials of the head-to-head comparison between ranibizumab and bevacizumab having now reported, the lower confidence limit of the pooled estimate for the difference is < 2 letters.
With respect to safety, even with data from most of the seven head-to-head trials, it is still not possible to rule out clinically important differences. What is clear is that ocular SAEs are extremely rare and do not differ by drug. There is also no differential risk of an ATE by drug, although there is still uncertainty about whether or not regular intraocular injections of an anti-VEGF drug increase this risk compared with not having injections. In the first year of the IVAN trial, such events appeared more frequent in patients treated with ranibizumab, but this finding disappeared over the course of year 2; the GEFAL42 and LUCAS (presented at 2013 meeting of the American Association for Ophthalmology) trials contributed relatively little information, so the pooled estimate is mainly influenced by the CATT and IVAN data.
However, there is uncertainty about SAEs that were not expected at the outset and which were not generally considered to be related to the drugs by IVAN investigators at the time they arose. Two persistent safety signals emerge from the meta-analyses, namely an increased risk of (1) any systemic SAE and (2) a gastrointestinal SAE (i.e. a SAE classified as MedDRA gastrointestinal system organ class). These two findings are complicated by the fact that gastrointestinal SAEs are a subset of all systemic SAEs and, hence, we discuss the gastrointestinal SAEs first.
The findings for gastrointestinal SAEs appear to differ in CATT and IVAN compared with the more recent trials (see Figure 31), with a clear safety signal from the former but not the latter. The combined size of the CATT and IVAN trials means that the fixed effects combined estimate is statistically significant. Although the data for CATT and IVAN are for 2 years’ duration of follow-up, not 1 year, this does not explain the difference because the increased risk of these events was apparent at 1 year in both IVAN and CATT. However, the events are collectively quite rare (1–2% per year, less frequent than events in some other system organ classes; gastrointestinal SAEs have incidence of about 2%, with incidences of other system organ classes ranging from 0% to 5%),23,26 and do not appear to increase the risk of death (as neither CATT or IVAN found any suggestion that bevacizumab increased mortality) and are extremely diverse (see Table 18). 23 To the extent that an underlying mechanism for these events has not been advanced, it is difficult to interpret their clinical importance or speculate about the possible underlying mechanisms.
The finding that the risk of any SAE is increased by bevacizumab is similarly complicated to understand. In IVAN, gastrointestinal events were the main driver of the difference (see Figure 29). This does not appear to have been so clearly the case in the CATT, where the incidences of SAEs classed as ‘infections and infestations’ and ‘surgical and medical procedures’ were also higher in participants treated with bevacizumab. 23 Incidences by system organ class in the GEFAL42 and MANTA43 trials were less clearly reported but do not appear to show any pattern that fits with the CATT data.
Two earlier systematic reviews investigating the safety of anti-VEGF drugs do not include most of the current evidence from head-to-head trials. 102,103 Other findings about the relative safety of ranibizumab and bevacizumab have been reported, based on large analyses of routinely obtained data. 104–106 However, these reports focused on cardiovascular arterial thrombotic events. Their findings vary and are at risk of confounding. 101
Comparisons of outcomes by treatment regimens
Only the CATT and IVAN trials compared continuous/monthly treatment with discontinuous/prn treatment regimens. Nevertheless, the estimates of efficacy were remarkably consistent for both visual function and lesion morphology outcomes, showing that discontinuous treatment is slightly, but statistically significantly, inferior to continuous treatment. The only exception to this pattern of findings was for the lesion morphology outcome of new GA, the risk of which is consistently higher with continuous treatment. As described above, with respect to safety, continuous treatment appears to protect against all-cause mortality; this finding is counterintuitive but was observed, also consistently, in both the IVAN trial and CATT.
We have already reported our opinion that the slightly better visual function with continuous compared with discontinuous treatment is unlikely to be of importance to patients. The finding that continuous treatment carries a 50% increase in the risk of developing new GA reinforces this view; as GA is itself potentially sight threatening, the visual benefit from continuous treatment may not anyway be maintained in the long term.
The finding that mortality was higher at 2 years with discontinuous treatment than continuous treatment is worrying. The uncertainty of this finding is about the same as for the possible association between bevacizumab and SAEs classified as gastrointestinal22,26 (see Figures 28 and 29). [The fact that there was an increase in the risk of any systemic SAE (0.81; p = 0·063) with discontinuous treatment is partly explained by the fact that deaths were included as systemic SAEs.] The comparisons of discontinuous and continuous regimens were not masked in either trial, but it seems implausible that bias should lead to an increased frequency of SAEs with discontinuous treatment. However, as previously stated, the possibility of intermittent dosing with a biological therapy causing immunological sensitisation has been suggested.
One might argue – given that most of the data described in this report are in the public domain and have been widely debated by ophthalmologists – that adoption into clinical practice is a pragmatic ‘test’ of safety. Use of bevacizumab to treat nAMD clearly passes this test, as there is no apparent reluctance to use the drug in many countries in the world. In the USA, bevacizumab was first used to treat nAMD before ranibizumab was licensed, and its use continued at about the same level as ranibizumab up to the end of 2006. 104 If anything, use of bevacizumab has increased recently following changes in the arrangements for reimbursing ophthalmologists under Medicare.
Limitations of findings about the safety of drugs and treatment regimens
When reviewing the safety data, it is important to bear in mind that none of the trials had satisfactory power to detect differences in safety outcomes. Trials, even those carried out to support marketing authorisations, are usually unable to rule out the possibility of a clinically important difference in a safety outcome. Even with data from multiple trials, the total sample size is still relatively small in relation to detecting a clinically important difference in a rare SAE. Nevertheless, there are now many more data for trial participants with nAMD randomised to the head-to-head comparison between ranibizumab and bevacizumab (n > 3300) than for the comparisons between ranibizumab and sham treatment or VPDT (ANCHOR + MARINA + FOCUS* + PIER = 1485) or between aflibercept (Eylea, Bayer) and ranibizumab [VIEW** 1 + VIEW 2 (n = 1240) = 2457]. 7,8,107–109 Interpretation of safety data may have been clouded by the emergence of possible signals not consistent with expectation, which has prompted many statistical comparisons. It is difficult to assign an appropriate level of significance to these statistical tests and the possible safety signals that have arisen need to be interpreted in the knowledge that a large number of comparisons have been carried out which were not prespecified.
*Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration; **VEGF Trap-Eye: Investigation of Efficacy and Safety in Wet AMD.
Lessons learned and implications for practice
The IVAN trial aimed to be pragmatic and achieved this at least with respect to the setting of usual NHS care and the inclusiveness of the population. However, in another respect it was probably too closely modelled on commercially sponsored Phase III trials carried out to obtain evidence to support applications for marketing authorisation. This model was largely adopted because it was that which several IVAN investigators and their local teams were familiar and because the trial was considered at the outset to be ‘high risk’, requiring extreme diligence and care from a research governance perspective. We were also aware that, when the trial was being set up, a campaign of disinformation was mounted with a view to discrediting the trial among potential investigators (www.ivan-trial.co.uk). Future retina trials seeking to answer important uncertainties about clinical practice face difficult choices. Decisions will inevitably be influenced by the precise research question and funder [e.g. NIHR Efficacy and Mechanism Evaluation (EME) vs. Health Technology Assessment (HTA) programmes] but the ophthalmological community has not shifted much towards the pragmatic end of the spectrum by taking part in IVAN. The familiarity of commercially sponsored Phase III trials is likely to continue to influence future Chief Investigators to adopt this template (regular monitoring, intensive scrutiny of multiple outcomes), which may not always be appropriate.
A crucial design decision that ophthalmology investigators need to make concerns the choice between recruiting only one or both eyes. Most commercially sponsored trials recruit only a single study eye. This decision can be justified on two main grounds: first, there is always a risk to the treated eye when using an intervention about which relatively little is known and it is sensible to limit this risk exposure to only one eye; second, in the context of an unlicensed medicinal product, the intervention is very unlikely to be available to treat the fellow eye outside the trial (such treatment of the fellow eye is one risk of studying a single eye).
These grounds are less applicable to non-commercial trials that are usually studying an intervention about which quite a lot is known, which may be already being used in clinical practice or at least be available if requested by a patient and in comparison with an active comparator. Neither option is straightforward. If studying only one eye, decisions need to be made in advance about the number of data to be collected for the fellow eye (e.g. depending on whether an intervention is topical or systemic), how to manage incident disease in fellow eye, etc. A disadvantage of studying one eye is that it can lead to undue emphasis on the effect of treatment on the eye, rather than the patient. If studying both eyes, decisions need to be made in advance about whether or not patients with uniocular disease are eligible, whether patient or eye should be randomised, whether or not there should be special considerations about safety when treating both eyes (e.g. again depending on whether an intervention is topical or systemic). This last point is extremely pertinent to the evaluation of anti-VEGF drugs. The commercial trials carried out for licensing purposes treated only one eye and were careful not to inject ranibizumab more than once every 28 days. By contrast, in usual care, ophthalmologists were quick to adopt the practice of treating both eyes, when indicated, at the same visit without having carefully collected data about safety on which to base this strategy. It seems likely that this decision was driven by convenience to patients and health services, as the disadvantages of the alternative, namely alternate injection of each eye at 2-weekly intervals, are clear. Our experience suggests that, if patients are appropriately counselled, they would wish to receive the same treatment to both eyes if the second eye were to develop neovascular AMD during time on study; in terms of monitoring patient outcomes, it would also make it far simpler if both eyes were randomised to the same treatment regimen. In the IVAN trial, patients who developed nAMD in the fellow eye were treated with the standard of care which was usually ranibizumab. The use of different drugs in the two eyes of some patients made it difficult to interpret systemic AEs and data on quality of life.
Two major NIHR projects (this trial and the VPDT cohort study92) have now reported data to inform the association between VA and HRQoL/utility in this population. Both have highlighted the extent to which economic models used by NICE in technology appraisals of VPDT and ranibizumab1,91 have underestimated the cost per QALY of the respective technologies by relying on a small US study about the disutility of sight loss. 110 Brown et al. 110,111 elicited preferences directly from patients with AMD. Preferences were markedly skewed in contrast to scores on preference-based utility measures derived from societal valuations, an observation that is consistent with some patients refusing to trade years of life for improved vision, and raises concern about the validity of the method. When making comparisons of HRQoL across interventions in different disease areas, it is more appropriate to value health states with preference weights from the general population rather than specific groups. 45,112
Using methods of determining utility recommended by NICE itself, it is now clear that the gradient of this association is much shallower than previously reported. We strongly recommend that more robust recently reported associations estimated from large data sets are used in future appraisals of interventions for nAMD and, possibly, for other eye conditions. The finding reported here about the risk of developing new GA, and evidence of deteriorating vision emerging from other long-term follow-up studies in trial participants’ treatment with anti-VEGF drugs,113,114 also suggest that longer-term follow-up of IVAN participants may be warranted, and future model-based economic evaluations assessing the cost-effectiveness of interventions for chronic eye conditions should use a long time horizon and avoid making unrealistic assumptions about the durability of treatment response.
Future research
Research to improve the clinical effectiveness and cost-effectiveness of treatment for neovascular age-related macular degeneration
It seems unlikely that an inexpensive treatment (other than unlicensed use of bevacizumab) for nAMD will emerge in the next 10 years. The most recent anti-VEGF option, aflibercept,108 has been priced at a very similar level to ranibizumab, taking into account a halving of the dosing interval. Combinations of anti-VEGF injections and intraocular brachytherapy (requiring vitrectomy) have been evaluated with disappointing results115 (http://public.ukcrn.org.uk/search/StudyDetail.aspx?StudyID = 7680). An innovative, stereotactic, low-voltage X-ray system is currently of interest. 116 It is likely to require a hub-and-spoke model of delivery (newly diagnosed patients attending a first visit at a hub centre and returning to a local spoke centre for subsequent review and further anti-VEGF if required) because of the high capital cost and maintenance of the device. If this method of delivering radiotherapy were to reduce markedly the need for further anti-VEGF injections it could be a cost-effective way of reducing both drug costs and the workload of managing patients with nAMD. However, the device has not yet been trialled and the manufacturer is likely to price the device to undercut anti-VEGF drugs only marginally. Other interventions that have shown promise include an inhibitor of platelet derived growth factor used in combination with ranibizumab, or angiostatin (to induce vessel regression) used in combination with ranibizumab.
Not surprisingly, commercial interest in new ways to treat nAMD is focused on new products, as this is how manufacturers generate profits. However, it is clear that there are major opportunities to achieve savings for health services, and improved convenience and possibly HRQoL for patients, through research on the delivery of nAMD care. Key research needs to be carried out to investigate different models of service provision with a view to reducing workload to the Hospital Eye Service and inconvenience to patients, and improving cost-effectiveness.
The current licence for ranibizumab describes a prn treatment regimen that was not evaluated in the two trials carried out to support the application for marketing authorisation (both evaluated continuous monthly injections over 2 years). 7,8 Moreover, although the manufacturer of ranibizumab evaluated an alternative regimen (three doses given over consecutive months at the start of treatment followed by treatment once every 3 months), showing that this regimen had poorer efficacy,117 other dosing frequencies have not been investigated in RCTs, only in single group studies. 118 Manufacturers have a vested interest in treatment regimens that maximise the use of treatment for tolerable safety, rather than regimens that minimise treatment for non-inferior effectiveness. 92 The opportunities are clear; simply extending the treatment/review interval to 6 weeks rather than 4 weeks would reduce the number of outpatient appointments by one-third. As many NHS hospitals have struggled to provide monthly appointments since the introduction of anti-VEGF therapy, nAMD care in the NHS over the last 5 years represents a natural experiment on extending the treatment interval (by necessity). Therefore, there may be an opportunity to investigate the effect of extending the treatment interval on BCVA observationally using routinely collected data. Data on time-to-treatment failure (time to lesion reactivation), and BCVA at this time, for IVAN participants allocated to the discontinuous treatment regimen (and for CATT participants allocated to the prn treatment) are also likely to be useful to inform this question.
Ophthalmology departments in some NHS hospitals have experimented with ‘treat-and-extend’ regimens to manage their workloads. This type of regimen is based on the assumption that the probability of lesion reactivation reduces as the time since the lesion became quiescent increases, extending the time interval to the next review appointment if the nAMD lesion remains inactive. The principle behind this strategy has not been researched. Again data on time-to-treatment failure for IVAN and CATT participants allocated to the discontinuous/prn treatment regimens are likely to be able to provide a test of this principle.
Continuous treatment regimens for nAMD are unlikely to be cost-effective in the UK as long as treatment using ranibizumab or aflibercept remains mandated. However, a continuous regimen using bevacizumab could be more cost-effective than discontinuous treatment, depending on the treatment interval (see above) and the frequency of review visits, for example if review using retinal imaging technologies were required only annually; if treatment decision-making were not required, there may be additional opportunities to streamline the provision of treatment only, perhaps offering such clinics in locations more convenient for patients.
A final important area of uncertainty concerns when to give up treating nAMD, which has not been researched. This decision could be based on lesion morphology or visual function. There will always be some patients who continue to lose vision despite treatment (estimated to be 5–10%7,8). The question is: Under what circumstances is it no longer ‘worth’ (both in terms of inconvenience to the patient and cost to health services) continuing to treat an eye that will not deteriorate much further? It may be that health services should switch their effort to preserving vision in the fellow eye. Clearly, this issue needs to be investigated with great sensitivity, focusing on the views of patients.
Research on the long-term consequences of antivascular endothelial growth factor treatment for neovascular age-related macular degeneration
There is a dearth of information about the long-term consequences of anti-VEGF treatment for nAMD. The importance of such information is highlighted by our findings, and those of the CATT, about the development of new GA; although this adverse effect did not appear to impact on BCVA during 2 years after starting treatment, it would be expected to reduce BCVA eventually.
Two open-label industry-sponsored studies have followed up participants involved in the clinical trials that were undertaken to support marketing authorisations for ranibizumab, namely HORIZON114 (following up participants in the ANCHOR, MARINA and FOCUS trials)7,8,107 and SECURE113 (following up participants in the SUSTAIN and EXCITE trials118,119). These extension studies observed a decrease in the VA benefit of ranibizumab in the long term. Given the findings about the development of new GA in the course of the CATT and IVAN trial, we hypothesise that this gradual fall-off in the benefit can be attributed to atrophy of the macular tissue, which is a component of the disease process itself but could also be accentuated and accelerated by anti-VEGF treatment. Unfortunately, both HORIZON and SECURE suffer from a number of limitations: (1) criteria for treatment during follow-up were inconsistently applied; (2) treatment regimens used in these initial trials were ‘continuous’ (monthly) and therefore there is no opportunity to examine for possible preconditioning by discontinuously administered treatment at disease inception; (3) there was low recruitment to the follow-up studies (< 40% in SECURE) and or high attrition during follow-up (> 30% among recruits to the follow-up studies in HORIZON); and (4) the primary objective of these follow-up studies was to document long-term safety.
Research to validate standard measures of utility in people with vision loss
The lack of impact of effective treatment on utility (measured using methods recommended by NICE) is a key lesson learned in the trial. It remains unclear whether or not this finding reflects inadequacies in the method, for example lack of sensitivity of the EQ-5D to loss of visual function, or truly represents a relatively minor impact on utility compared with treatments for other conditions commonly associated with pain or loss of mobility, for example coronary artery bypass grafting120 or total knee or hip replacement. 121 Given the importance of valid measures of utility in economic models to estimate the cost-effectiveness of treatments for nAMD, and the likelihood of new technologies emerging in the near future, we consider that investigating which of these explanations is correct should be a research priority.
Further health-economic research
There are three areas of further health-economic research that would be worthwhile.
First, the consultation costs micro-costed within the trial varied between centres and differed from readily available costs, such as reference costs or HRGs. Although sensitivity analyses showed that the conclusions were unaffected by the source of consultation costs, further research to identify the reasons for these differences would be informative.
Second, the analysis in this report focused on costs incurred by the NHS. In reality, treatment for nAMD is also likely to improve working patients’ productivity, reduce their need for personal and social care, and impose costs on patients and their families when attending treatment, especially in centres running a ‘two-stop’ service with separate injection and monitoring clinics. Therefore, it would be useful for future studies to explicitly examine the non-NHS costs associated with different models of clinic organisation.
Third, we measured quality of life using the three-level EQ-5D questionnaire because the five-level EQ-5D was not available when IVAN participants were recruited. One of the reasons for developing the five-level version was because of the concern that the three-level version may be too insensitive for some conditions, such as eye disease. A vision-specific bolt-on to the three-level EQ-5D has also been developed. 122 It would, therefore, be interesting to investigate the five-level questionnaire and vision-specific bolt-on in current eye trials to see whether or not these discriminate changes in HRQoL better than the three-level questionnaire.
Chapter 10 Conclusion
Anti-VEGF drugs look set to remain the mainstay treatment for nAMD for the foreseeable future, despite the rapid increase in potential new treatments. Concerns have been raised about the safety of aflibercept,123 despite evidence of efficacy,108 and the results of brachytherapy in combination with anti-VEGF drugs have so far been disappointing. 115 Photodynamic therapy or radiotherapy options used in combination might yet allow reductions in treatment frequency, but it will be important to study these treatment strategies in comparison with monthly treatment, in view of our finding of possible risks of discontinuous treatment.
It is instructive to reflect on the choice of drug and treatment regimen if there were no resource constraints. With respect to drug, given the possibility of poorer safety of bevacizumab, ranibizumab would probably be preferred. With respect to treatment regimen, the choice is not so obvious, because of the possibility of increased mortality with discontinuous treatment and the increased risk of developing new GA with continuous treatment, as well as the risks of many more intraocular injections. However, within a health-care system that has a fixed budget, such as the NHS, the resources required to provide treatment have to be considered in the wider context of competing demands on resources, both to treat other eye diseases and non-eye diseases, such as cancer and CVD. It is therefore essential that treatment decisions take account of cost and cost-effectiveness, as well as clinical efficacy and safety. Our economic evaluation demonstrates that ranibizumab represents very poor value for money, costing £3.5M per QALY gained compared with bevacizumab. The analysis also suggested that discontinuous bevacizumab is likely to be better value for money than continuous bevacizumab.
In conclusion, the IVAN trial and meta-analyses of data from other trials show that the choice of anti-VEGF treatment strategy is less straightforward than previously thought. Bevacizumab and ranibizumab have similar efficacy and can be considered equivalent in this respect in the treatment of nAMD, although bevacizumab provides much better value for money. The possibility of an increased risk of death with discontinuous treatment should probably outweigh the increased risk of new GA that was observed with continuous treatment. The slightly better functional outcomes with continuous treatment are a bonus. Continuous treatment also avoids the need to monitor disease activity on every visit. An important consideration when choosing to give treatment continuously is the high cost of ranibizumab which, at a cost of £270,000 per QALY gained compared with discontinuous ranibizumab, may be unaffordable for publicly funded health systems.
It is disappointing that the findings of the IVAN trial have not changed practice in the UK, where it has been argued that legislation prohibits the use of an unlicensed treatment to treat a condition for which another product has a licensed indication. Despite the investment in the IVAN trial, the UK has one of the lowest uses of bevacizumab for ophthalmic conditions among comparable developed countries. Nevertheless, the findings of the IVAN trial may have indirectly contributed to benefits to the NHS, for example through patient access schemes that are being offered to the NHS.
Acknowledgements
Contributions of authors
Usha Chakravarthy, Simon P Harding, Chris A Rogers, Susan Downes, Andrew J Lotery, Sarah Wordsworth, James Raftery and Barnaby C Reeves obtained funding for the trial and were core members of the Trial Management Group overseeing trial conduct.
Usha Chakravarthy was the chief investigator, with overall responsibility for the trial.
Usha Chakravarthy, Simon P Harding and Tunde Peto established the UK Network of Ophthalmic Reading Centres, which was managed by Alyson Muldrew and Jayashree Sahni.
Usha Chakravarthy, Simon P Harding and Barnaby C Reeves designed the trial.
Barnaby C Reeves and Chris A Rogers led the Trial Management Centre.
Chris A Rogers specified the statistical analyses, and supervised Rachel L Nash and Lauren J Scott in executing them.
Usha Chakravarthy, Simon P Harding, Susan Downes and Andrew J Lotery led recruitment, site selection, addressed clinical queries and reviewed AEs.
Helen A Dakin and Sarah Wordsworth designed and carried out the economic evaluation, with advice from James Raftery.
Lucy Culliford and Jodi Taylor were the trial managers and implemented the trial protocol, developing and refining trial procedures to optimise its conduct.
Chris A Rogers, Usha Chakravarthy, Helen Dakin, Sarah Wordsworth and Barnaby C Reeves wrote the first draft of this report. All authors contributed to the final draft and approved its submission for publication.
Data sharing statement
Anonymised individual patient data will be made available for secondary research, conditional on assurance from the secondary researcher that the proposed use of the data is compliant with the Medical Research Council Policy on Data Sharing regarding scientific quality, ethical requirements and value for money. A minimum requirement with respect to scientific quality will be a publicly available pre-specified protocol describing the purpose, methods and analysis of the secondary research, for example a protocol for a Cochrane systematic review.
However, potential users of the IVAN data set should note that the raw data are extremely complicated, spanning a large number of visits per patient. For example, the database functionality did not support a decision made during the conduct of the trial to allow deferral of collection of some data to a later visit if visit was missed. Code to manage these issues is, typically, contained within existing analysis programmes. Hence there is no single ‘flat’ file (or multiple flat files) with a defined data dictionary; expert knowledge of the data structure is required to interrogate the database. It would be possible to create a set of definitive files for export to a third party, if funding were available (this would be required as the data do not exist in such a format). The Clinical Trials and Evaluation Unit would prefer to enter into a formal collaboration with a third party wanting to investigate a new research question using the IVAN data.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Photodynamic Therapy for Age-related Macular Degeneration. London: NICE; 2003.
- Dong LM, Marsh MJ, Hawkins BS. Measurement and analysis of visual acuity in multicenter randomized clinical trials in the United States: findings from a survey. Ophthalmic Epidemiol 2003;10:149-56. http://dx.doi.org/10.1076/opep.10.3.149.15080.
- Bressler NM. Treatment of Age-Related Macular Degeneration with Photodynamic Therapy Study G. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: two-year results of 2 randomized clinical trials-tap report 2. Arch Ophthalmol 2001;119:198-207.
- Gragoudas ES, Adamis AP, Cunningham ET, Feinsod M, Guyer DR. Group VISiONCT . Pegaptanib for neovascular age-related macular degeneration. N Engl J Med 2004;351:2805-16. http://dx.doi.org/10.1056/NEJMoa042760.
- Treatment of age-related macular degeneration with photodynamic therapy (TAP) Study Group . Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials – TAP report. Arch Ophthalmol 1999;117:1329-45. http://dx.doi.org/10.1001/archopht.117.10.1329.
- Ferrara N, Damico L, Shams N, Lowman H, Kim R. Development of ranibizumab, an anti-vascular endothelial growth factor antigen binding fragment, as therapy for neovascular age-related macular degeneration. Retina 2006;26:859-70. http://dx.doi.org/10.1097/01.iae.0000242842.14624.e7.
- Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med 2006;355:1432-44. http://dx.doi.org/10.1056/NEJMoa062655.
- Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med 2006;355:1419-31. http://dx.doi.org/10.1056/NEJMoa054481.
- Rosenfeld PJ, Rich RM, Lalwani GA. Ranibizumab: Phase III clinical trial results. Ophthalmol Clin North Am 2006;19:361-72.
- Anon . Variable Dosing of Ranibizumab Investigated for Treatment of Neovascular AMD Ophthalmology Times n.d. http://ophthalmologytimes.modernmedicine.com/ophthalmologytimes/news/clinical/ophthalmology/variable-dosing-ranibizumab-investigated-treatment-ne (accessed 18 June 2015).
- Spaide RF, Laud K, Fine HF, Klancnik JM, Meyerle CB, Yannuzzi LA, et al. Intravitreal bevacizumab treatment of choroidal neovascularization secondary to age-related macular degeneration. Retina 2006;26:383-90. http://dx.doi.org/10.1097/00006982-200604000-00001.
- Moshfeghi AA, Rosenfeld PJ, Puliafito CA, Michels S, Marcus EN, Lenchus JD, et al. Systemic bevacizumab (Avastin) therapy for neovascular age-related macular degeneration: twenty-four-week results of an uncontrolled open-label clinical study. Ophthalmology 2006;113:e1-12.
- Avery RL, Pieramici DJ, Rabena MD, Castellarin AA, Nasir MA, Giust MJ. Intravitreal bevacizumab (Avastin) for neovascular age-related macular degeneration. Ophthalmology 2006;113:363-72. http://dx.doi.org/10.1016/j.ophtha.2005.11.019.
- Kilic U, Kilic E, Jarve A, Guo Z, Spudich A, Bieber K, et al. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating ERK-1/2 and Akt pathways. J Neurosci 2006;26:12439-46. http://dx.doi.org/10.1523/JNEUROSCI.0434-06.2006.
- Saint-Geniez M, Maldonado AE, D’Amore PA. VEGF expression and receptor activation in the choroid during development and in the adult. Invest Ophthalmol Vis Sci 2006;47:3135-42. http://dx.doi.org/10.1167/iovs.05-1229.
- Chakravarthy U, Adamis AP, Cunningham ET, Goldbaum M, Guyer DR, Katz B, et al. Year 2 efficacy results of 2 randomized controlled clinical trials of pegaptanib for neovascular age-related macular degeneration. Ophthalmology 2006;113.
- Shah RR. Pharmacogenetics in drug regulation: promise, potential and pitfalls. Philos Trans R Soc Lond B Biol Sci 2005;360:1617-38. http://dx.doi.org/10.1098/rstb.2005.1693.
- Pasqualetti G, Danesi R, Del Tacca M, Bocci G. Vascular endothelial growth factor pharmacogenetics: a new perspective for anti-angiogenic therapy. Pharmacogenomics 2007;8:49-66. http://dx.doi.org/10.2217/14622416.8.1.49.
- Maller J, George S, Purcell S, . Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet 2006;38:1055-9. http://dx.doi.org/10.1038/ng1873.
- Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet 2006;38:1173-7. http://dx.doi.org/10.1038/ng1890.
- Dewan A, Liu M, Hartman S, Zhang SS, Liu DT, Zhao C, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 2006;314:989-92. http://dx.doi.org/10.1126/science.1133807.
- Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology 2012;119:1388-98. http://dx.doi.org/10.1016/j.ophtha.2012.03.053.
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, . CATT Research Group . Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011;364:1897-908. http://dx.doi.org/10.1056/NEJMoa1102673.
- Gaudreault J, Fei D, Rusit J, Suboc P, Shiu V. Preclinical pharmacokinetics of ranibizumab (rhuFabV2) after a single intravitreal administration. Invest Ophthalmol Vis Sci 2005;46:726-33. http://dx.doi.org/10.1167/iovs.04-0601.
- Lynch SS, Cheng CM. Bevacizumab for neovascular ocular diseases. Ann Pharmacother 2007;41:614-25. http://dx.doi.org/10.1345/aph.1H316.
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, . The IVAN Study Investigators . Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology 2012;119:1399-411. http://dx.doi.org/10.1016/j.ophtha.2012.04.015.
- Ferris FL, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol 1982;94:91-6. http://dx.doi.org/10.1016/0002-9394(82)90197-0.
- Pelli DG, Robson JG, Wilkins AJ. The design of a new letter chart for measuring contrast sensitivity. Clin Vis Sci 1988;2:187-99.
- Bailey IL, Lovie JE. The design and use of a new near-vision chart. Am J Optom Physiol Opt 1980;57:378-87. http://dx.doi.org/10.1097/00006324-198006000-00011.
- Hart PM, Chakravarthy U, Mackenzie G, Chisholm IH, Bird AC, Stevenson MR, et al. Visual outcomes in the subfoveal radiotherapy study: a randomized controlled trial of teletherapy for age-related macular degeneration. Arch Ophthalmol 2002;120:1029-38. http://dx.doi.org/10.1001/archopht.120.8.1029.
- McClure ME, Hart PM, Jackson AJ, Stevenson MR, Chakravarthy U. Macular degeneration: do conventional measurements of impaired visual function equate with visual disability?. Br J Ophthalmol 2000;84:244-50. http://dx.doi.org/10.1136/bjo.84.3.244.
- Williams AE. A new facility for the measurement of health-related quality-of-life. Health Policy 1990;16:199-208. http://dx.doi.org/10.1016/0168-8510(90)90421-9.
- Feeny D, Furlong W, Torrance GW, Goldsmith CH, Zhu Z, De Pauw S, et al. Multiattribute and single-attribute utility functions for the health utilities index mark 3 system. Med Care 2002;40:113-28. http://dx.doi.org/10.1097/00005650-200202000-00006.
- Mitchell J, Bradley C. Design of an individualised measure of the impact of macular disease on quality of life (the MacDQoL). Qual Life Res 2004;13:1163-75. http://dx.doi.org/10.1023/B:QURE.0000031348.51292.4a.
- Mitchell J, Brose L, Bradley C. Design of a measure of satisfaction with treatment for macular degeneration (MacTSQ). Qual Life Res 2007;A120.
- Antiplatelet Trialists’ Collaboration . Collaborative overview of randomised trials of antiplatelet therapy – I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ 1994;308:81-106. http://dx.doi.org/10.1136/bmj.308.6921.81.
- Cohen J. Statistical Power Analysis for the Behavioral Sciences. Hillsdale, NJ: Lawrence Erlbaum; 1988.
- Reeves BC, Wood JM, Hill AR. Reliability of high- and low-contrast letter charts. Ophthalmic Physiol Opt 1993;13:17-26. http://dx.doi.org/10.1111/j.1475-1313.1993.tb00421.x.
- The Association for Research in Vision and Ophthalmology (ARVO) Annual Conference 2011 . Fluctuating Levels of Circulating VEGF in a Subset of Patients As Part of the Multicentre IVAN Study 2011. www.abstractsonline.com/plan/ViewAbstract.aspx?mID=2684&sKey=9009b5e8-692c-4edb-9d05-f2eeb2af05eb&cKey=bd842624-68d2-4b65-b3ba-5a13c840fc5d&mKey=6f224a2d-af6a-4533-8bbb-6a8d7b26edb3 (accessed 3 June 2015).
- Piaggio G, Elbourne DR, Pocock SJ, Evans SJ, Altman DG, Group C. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA 2012;308:2594-604. http://dx.doi.org/10.1001/jama.2012.87802.
- International Conference on Harmonisation (ICH) . ICH Harmonised Tripartite Guideline Topic E9: Statistical Principles for Clinical Trials n.d. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf (accessed 16 January 2014).
- Kodjikian L, Souied EH, Mimoun G, Mauget-Faÿsse M, Behar-Cohen F, Decullier E, et al. Ranibizumab versus bevacizumab for neovascular age-related macular degeneration: results from the GEFAL noninferiority randomized trial. Ophthalmology 2013;120:2300-9. http://dx.doi.org/10.1016/j.ophtha.2013.06.020.
- Krebs I, Schmetterer L, Boltz A, Told R, Vécsei-Marlovits V, Egger S, et al. A randomised double-masked trial comparing the visual outcome after treatment with ranibizumab or bevacizumab in patients with neovascular age-related macular degeneration. Br J Ophthalmol 2013;97:266-71. http://dx.doi.org/10.1136/bjophthalmol-2012-302391.
- Subramanian ML, Abedi G, Ness S, Ahmed E, Fenberg M, Daly MK, et al. Bevacizumab vs. ranibizumab for age-related macular degeneration: 1-year outcomes of a prospective, double-masked randomised clinical trial. Eye 2010;24:1708-15. http://dx.doi.org/10.1038/eye.2010.147.
- Guide to the Methods of Technology Appraisal 2013. London: NICE; 2013.
- Brittain E, Wittes J. Factorial designs in clinical trials: the effects of non-compliance and subadditivity. Stat Med 1989;8:161-71. http://dx.doi.org/10.1002/sim.4780080204.
- Byar DP, Piantadosi S. Factorial designs for randomized clinical trials. Cancer Treat Res 1985;69:1055-63. http://dx.doi.org/10.1016/0197-2456(85)90010-8.
- Van Wijngaarden P, Qureshi SH. Inhibitors of vascular endothelial growth factor (VEGF) in the management of neovascular age-related macular degeneration: a review of current practice. Clin Exp Optom 2008;91:427-37. http://dx.doi.org/10.1111/j.1444-0938.2008.00305.x.
- La Cour M. Intravitreal VEGF-inhibitors: is Avastin a generic substitute for Lucentis?. Acta Ophthalmol Scand 2007;85:2-4. http://dx.doi.org/10.1111/j.1600-0420.2006.00869.x.
- Penha FM, Rodrigues EB, Maia M, Furlani BA, Regatieri C, Melo GB, et al. Retinal and ocular toxicity in ocular application of drugs and chemicals – part II: retinal toxicity of current and new drugs. Ophthalmic Res 2010;44:205-24. http://dx.doi.org/10.1159/000316695.
- British National Formulary. London: BMA and RPS; 2011.
- McAlister FA, Straus SE, Sackett DL, Altman DG. Analysis and reporting of factorial trials: a systematic review. JAMA 2003;289:2545-53. http://dx.doi.org/10.1001/jama.289.19.2545.
- Drummond MF, Sculpher MJ, Torrance GW, O’Brien B, Stoddart GL. Methods for the Economic Evaluation of Health Care Programmes. New York, NY: Oxford University Press; 2005.
- Dakin H, Wordsworth S. Cost-minimisation analysis versus cost-effectiveness analysis, revisited. Health Econ 2013;22:22-34. http://dx.doi.org/10.1002/hec.1812.
- Gudex C. Time Trade-off User Manual: Props and Self-completion Methods. York: University of York, Centre for Health Economics; 1994.
- Williams A. The Measurement and Valuation of Health: A Chronicle. York: University of York; 1995.
- Social Value Judgements: Principles for the Development of NICE Guidance. London: NICE; 2008.
- Gray A, Clarke P, Wolstenholme J, Wordsworth S. Applied Methods of Cost-Effectiveness Analysis in Health Care. Oxford: Oxford University Press; 2011.
- Fenwick E, Claxton K, Sculpher M. Representing uncertainty: the role of cost-effectiveness acceptability curves. Health Econ 2001;10:779-87. http://dx.doi.org/10.1002/hec.635.
- Dolan P. Modeling valuations for EuroQol health states. Med Care 1997;35:1095-108. http://dx.doi.org/10.1097/00005650-199711000-00002.
- Szende A, Williams A. Measuring Self-Reported Population Health: An International Perspective Based on EQ-5D. Rotterdam: EuroQol Group; 2004.
- Feeny D, Furlong W, Boyle M, Torrance GW. Multi-attribute health status classification systems. Health Utilities Index. PharmacoEconomics 1995;7:490-502. http://dx.doi.org/10.2165/00019053-199507060-00004.
- Torrance GW, Furlong W, Feeny D, Boyle M. Multi-attribute preference functions. Health Utilities Index. PharmacoEconomics 1995;7:503-20. http://dx.doi.org/10.2165/00019053-199507060-00005.
- Dolan P, Gudex C, Kind P, Williams A. A Social Tariff for Euroqol: Results from a UK General Population Survey. York: Centre for Health Economics, University of York; 1995.
- Rasanen P, Roine E, Sintonen H, Semberg-Konttinen V, Ryynanen OP, Roine R. Use of quality-adjusted life years for the estimation of effectiveness of health care: a systematic literature review. Int J Technol Assess Health Care 2006;22:235-41. http://dx.doi.org/10.1017/S0266462306051051.
- Espallargues M, Czoski-Murray CJ, Bansback NJ, . The impact of age-related macular degeneration on health status utility values. Invest Ophthalmol Vis Sci 2005;46:4016-23. http://dx.doi.org/10.1167/iovs.05-0072.
- Tosh J, Brazier J, Evans P, Longworth L. A Review of Generic Preference-based Measures of Health-related Quality of Life in Visual Disorders. Sheffield: Health Economics and Decision Science, University of Sheffield; 2011.
- Feeny DH, Torrance GW, Furlong WJ, Spilker B. Quality of Life and Pharmacoeconomics in Clinical Trials. Philadelphia, PA: Lippincott-Raven Press; 1996.
- Furlong W, Feeny D, Torrance GW, Goldsmith CH, DePauw S, Zhu Z, et al. Multiplicative Multi-Attribute Utility Function for the Health Utilities Index Mark 3 (HUI3) System: A Technical Report. Hamilton, ON: McMaster University Centre for Health Economics and Policy Analysis; 1998.
- Luyten J, Marais C, Hens N, De Schrijver K, Beutels P. Imputing QALYs from single time point health state descriptions on the EQ-5D and the SF-6D: a comparison of methods for hepatitis a patients. Value Health 2011;14:282-90. http://dx.doi.org/10.1016/j.jval.2010.10.004.
- Spencer M, Briggs AH, Grossman RF, Rance L. Development of an economic model to assess the cost effectiveness of treatment interventions for chronic obstructive pulmonary disease. PharmacoEconomics 2005;23:619-37. http://dx.doi.org/10.2165/00019053-200523060-00008.
- Jonsson B, Weinstein MC. Economic evaluation alongside multinational clinical trials. Study considerations for GUSTO IIb. Int J Technol Assess Health Care 1997;13:49-58. http://dx.doi.org/10.1017/S0266462300010229.
- Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. The BMJ Economic Evaluation Working Party. BMJ 1996;313:275-83. http://dx.doi.org/10.1136/bmj.313.7052.275.
- Ranibizumab and Pegaptanib for the Treatment of Age-Related Macular Degeneration TA155. London: NICE; 2012.
- National Schedule of Reference Costs 2010–11 for NHS Trusts and PCTs Combined. London: DH; 2011.
- PbR Operational Guidance and Tariffs: 2011–12 Tariff Information Spreadsheet. London: DH; 2011.
- Curtis L. Unit Costs of Health and Social Care 2011. Canterbury: PSSRU, University of Kent; 2011.
- Health and Social Care Information Centre (HSCIC) . NHS Staff Earnings January – March 2011 2011. www.hscic.gov.uk/catalogue/PUB00825/nhs-staf-earn-jan-mar-2011-tab.xls (accessed 16 January 2011).
- Yu LM, Burton A, Rivero-Arias O. Evaluation of software for multiple imputation of semi-continuous data. Stat Methods Med Res 2007;16:243-58. http://dx.doi.org/10.1177/0962280206074464.
- Briggs A, Clark T, Wolstenholme J, Clarke P. Missing . . . presumed at random: cost-analysis of incomplete data. Health Econ 2003;12:377-92. http://dx.doi.org/10.1002/hec.766.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. http://dx.doi.org/10.1002/sim.4067.
- Royston P. Multiple imputation of missing values. STATA J 2004;4:227-41.
- Royston P. Multiple imputation of missing values: update of ice. STATA J 2005;5:527-36.
- Royston P. Multiple imputation of missing values: further update of ice, with an emphasis on interval censoring. STATA J 2007;7:445-64.
- De Clercq C, Snyers B, Guagnini AP, Kozyreff A. Treatment of age-related macular degeneration with intravitreous injections of bevacizumab (Avastin): short-term results. Bull Soc Belge Ophthalmol n.d.:15-21.
- Hallstrom AP, Sullivan SD. On estimating costs for economic evaluation in failure time studies. Med Care 1998;36:433-6. http://dx.doi.org/10.1097/00005650-199803000-00019.
- Lin DY, Feuer EJ, Etzioni R, Wax Y. Estimating medical costs from incomplete follow-up data. Biometrics 1997;53:419-34. http://dx.doi.org/10.2307/2533947.
- Montgomery AA, Peters TJ, Little P. Design, analysis and presentation of factorial randomised controlled trials. BMC Med Res Methodol 2003;3. http://dx.doi.org/10.1186/1471-2288-3-26.
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96. http://dx.doi.org/10.1002/hec.944.
- The Green Book: Appraisal and Evaluation in Central Government. London: TSO; 2003.
- Ranibizumab and Pegaptanib for the Treatment of Age-related Macular Degeneration. London: NICE; 2008.
- Reeves BC, Harding SP, Langham J, Grieve R, Tomlin K, Walker J, et al. Verteporfin photodynamic therapy for neovascular age-related macular degeneration: cohort study for the UK. Health Technol Assess 2012;16. http://dx.doi.org/10.3310/hta16060.
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet 2013;382:1258-67. http://dx.doi.org/10.1016/S0140-6736(13)61501-9.
- Kind P, Hardman G, Macran S. UK Population Norms for EQ-5D. York: Centre for Health Economics; 1999.
- Briggs AH, O’Brien BJ. The death of cost-minimization analysis?. Health Econ 2001;10:179-84. http://dx.doi.org/10.1002/hec.584.
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011;364:1897-908. http://dx.doi.org/10.1056/NEJMoa1102673.
- Alsheikh-Ali AA, Karas RH. The safety of niacin in the US Food and Drug Administration adverse event reporting database. Am J Cardiol 2008;101:9-13B. http://dx.doi.org/10.1016/j.amjcard.2008.02.027.
- Raftery J, Clegg A, Jones J, Tan SC, Lotery A. Ranibizumab (Lucentis) versus bevacizumab (Avastin): modelling cost effectiveness. Br J Ophthalmol 2007;91:1244-6. http://dx.doi.org/10.1136/bjo.2007.116616.
- Raftery J, Dent L. Should avastin be used to treat age-related macular degeneration in the NHS? – Yes. Eye 2009;23:1247-9. http://dx.doi.org/10.1038/eye.2009.85.
- Ranibizumab and Pegaptanib for the Treatment of Age-related Macular Degeneration TA155: Costing Template and Report. London: NICE; 2008.
- Williams T, Reeves BC, Foss AJ, Fell G. Risks of adverse events with therapies for age-related macular degeneration: a response. Arch Ophthalmol 2012;130:124-5. http://dx.doi.org/10.1001/archopht.130.1.124.
- Schmucker C, Ehlken C, Agostini HT, Antes G, Ruecker G, Lelgemann M, et al. A safety review and meta-analyses of bevacizumab and ranibizumab: off-label versus goldstandard. PLOS ONE 2012;7. http://dx.doi.org/10.1371/journal.pone.0042701.
- Schmucker C, Ehlken C, Hansen LL, Antes G, Agostini HT, Lelgemann M. Intravitreal bevacizumab (Avastin) vs. ranibizumab (Lucentis) for the treatment of age-related macular degeneration: a systematic review. Curr Opin Ophthalmol 2010;21:218-26. http://dx.doi.org/10.1097/ICU.0b013e3283386783.
- Curtis LH, Hammill BG, Schulman KA, Cousins SW. Risks of mortality, myocardial infarction, bleeding, and stroke associated with therapies for age-related macular degeneration. Arch Ophthalmol 2010;128:1273-9. http://dx.doi.org/10.1001/archophthalmol.2010.223.
- Campbell RJ, Gill SS, Bronskill SE, Paterson JM, Whitehead M, Bell CM. Adverse events with intravitreal injection of vascular endothelial growth factor inhibitors: nested case-control study. BMJ 2012;345. http://dx.doi.org/10.1136/bmj.e4203.
- Kemp A, Preen DB, Morlet N, Clark A, McAllister IL, Briffa T, et al. Myocardial infarction after intravitreal vascular endothelial growth factor inhibitors: a whole population study. Retina 2013;33:920-7. http://dx.doi.org/10.1097/IAE.0b013e318276e07b.
- Heier JS, Boyer DS, Ciulla TA, Ferrone PJ, Jumper JM, Gentile RC, et al. Ranibizumab combined with verteporfin photodynamic therapy in neovascular age-related macular degeneration: year 1 results of the FOCUS Study. Arch Ophthalmol 2006;124:1532-42. http://dx.doi.org/10.1001/archopht.124.11.1532.
- Heier JS, Brown DM, Chong V, Korobelnik JF, Kaiser PK, Nguyen QD, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012;119:2537-48. http://dx.doi.org/10.1016/j.ophtha.2012.09.006.
- Abraham P, Yue H, Wilson L. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER study year 2. Am J Ophthalmol 2010;150:315-24.e1. http://dx.doi.org/10.1016/j.ajo.2010.04.011.
- Brown GC, Sharma S, Brown MM, Kistler J. Utility values and age-related macular degeneration. Arch Ophthalmol 2000;118:47-51. http://dx.doi.org/10.1001/archopht.118.1.47.
- Brown GC. Vision and quality-of-life. Trans Am Ophthalmol Soc 1999;97:473-511.
- Brazier J, Roberts J, Deverill M. The estimation of a preference-based measure of health from the SF-36. J Health Econ 2002;21:271-92. http://dx.doi.org/10.1016/S0167-6296(01)00130-8.
- Silva R, Axer-Siegel R, Eldem B, Kirchhof B, Papp A, Seres A, et al. The SECURE study: long-term safety of ranibizumab 0.5 mg in neovascular age-related macular degeneration. Ophthalmology 2013;120:130-9. http://dx.doi.org/10.1016/j.ophtha.2012.07.026.
- Singer MA, Awh CC, Sadda S, Freeman WR, Antoszyk AN, Wong P, et al. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology 2012;119:1175-83. http://dx.doi.org/10.1016/j.ophtha.2011.12.016.
- Dugel PU, Bebchuk JD, Nau J, Smith KR, Petrarca R, Slakter JS, et al. Epimacular brachytherapy for neovascular age-related macular degeneration: a randomized, controlled trial (CABERNET). Ophthalmology 2013;120:317-27. http://dx.doi.org/10.1016/j.ophtha.2012.07.068.
- Petrarca R, Jackson TL. Radiation therapy for neovascular age-related macular degeneration. Clin Ophthalmol 2011;5:57-63. http://dx.doi.org/10.2147/OPTH.S16444.
- Regillo CD, Brown DM, Abraham P, Yue H, Ianchulev T, Schneider S, et al. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol 2008;145:239-48. http://dx.doi.org/10.1016/j.ajo.2007.10.004.
- Holz FG, Amoaku W, Donate J, Guymer RH, Kellner U, Schlingemann RO, et al. Safety and efficacy of a flexible dosing regimen of ranibizumab in neovascular age-related macular degeneration: the SUSTAIN study. Ophthalmology 2011;118:663-71. http://dx.doi.org/10.1016/j.ophtha.2010.12.019.
- Schmidt-Erfurth U, Eldem B, Guymer R, Korobelnik JF, Schlingemann RO, Axer-Siegel R, et al. Efficacy and safety of monthly versus quarterly ranibizumab treatment in neovascular age-related macular degeneration: the EXCITE study. Ophthalmology 2011;118:831-9. http://dx.doi.org/10.1016/j.ophtha.2010.09.004.
- Griffin SC, Barber JA, Manca A, Sculpher MJ, Thompson SG, Buxton MJ, et al. Cost effectiveness of clinically appropriate decisions on alternative treatments for angina pectoris: prospective observational study. BMJ 2007;334. http://dx.doi.org/10.1136/bmj.39129.442164.55.
- Browne J, Jamieson L, Lewsey J, van der Meulen J, Black N, Cairns J, et al. Patient Reported Outcome Measures (PROMs) in Elective Surgery: Report to the Department of Health. London: London School of Hygiene and Tropical Medicine; 2007.
- Longworth L, Yang YL, Young T, Mulhern B, Hernández Alava M, Mukuria C, et al. Use of generic and condition-specific measures of health-related quality of life in NICE decision-making: a systematic review, statistical modelling and survey. Health Technol Assess 2014;18. http://dx.doi.org/10.3310/hta18090.
- CHMP Assessment Report: Eylea aflibercept. London: European Medicines Agency; 2012.
- Ganiats TG, Browner DK, Kaplan RM. Comparison of two methods of calculating quality-adjusted life years. Qual Life Res 1996;5:162-4. http://dx.doi.org/10.1007/BF00435981.
- Billingham LJ, Abrams KR, Jones DR. Methods for the analysis of quality-of-life and survival data in health technology assessment. Health Technol Assess 1999;3.
- Tosh J, Brazier J, Evans P, Longworth L. A review of generic preference-based measures of health-related quality of life in visual disorders. Value Health 2012;15:118-27. http://dx.doi.org/10.1016/j.jval.2011.08.002.
- Curtis L. Unit Costs of Health and Social Care 2010. Canterbury: PSSRU, University of Kent; 2010.
- Smith D. Community pharmacists and health promotion: a study of consultations between pharmacists and clients. Health Promot Int 1992;7:249-55. http://dx.doi.org/10.1093/heapro/7.4.249.
- O’Hagan A, Stevens JW. On estimators of medical costs with censored data. J Health Econ 2004;23:615-25. http://dx.doi.org/10.1016/j.jhealeco.2003.06.006.
- Lubsen J, Pocock SJ. Factorial trials in cardiology: pros and cons. Eur Heart J 1994;15:585-8.
- Claxton K. The irrelevance of inference: a decision-making approach to the stochastic evaluation of health care technologies. J Health Econ 1999;18:341-64. http://dx.doi.org/10.1016/S0167-6296(98)00039-3.
- Briggs AH, Wonderling DE, Mooney CZ. Pulling cost-effectiveness analysis up by its bootstraps: a non-parametric approach to confidence interval estimation. Health Econ 1997;6:327-40. http://dx.doi.org/10.1002/(SICI)1099-1050(199707)6:4<327::AID-HEC282>3.0.CO;2-W.
Appendix 1 IVAN study investigators
Trial sites
Aintree University Hospital
Investigators: Mr Ahmed Kamal, Thomas Papathomas and Ahmed Khalil.
Research team: Pauline Guinness, Richard Hancock, Maria Dangler-Harles, Jane Blocksage and Samantha Lorenson.
Aston University, Birmingham;a Optegra Birmingham Eye Hospitalb
Investigators: Mr JM Gibson,a Katie Pedwella and Jane Pitt. b
Research team: Ajith Kumar, S Al-Husainy, S Sreekantam, M Hanratty and Tara Clark.
Queen’s University Belfast;a Belfast Health and Social Care Trustb
Investigators: Professor Usha Chakravarthy,a Dr Lisa Kellyb and Dr Karen Gilvray. b
Research team: Ms Pamela Jamison, Mrs Georgina Sterret, Mr Vittorio Silvestri, Dr Deirdre Burns, Ms Rebecca Denham, Mrs Joanne Grattan, Mrs Teresa Rice and Mrs Lenore Ponisi.
Aston University, Birmingham;a Birmingham and Midland Eye Centreb
Investigators: Mr JM Gibson,a Katherine Brownb and Bethan Swain. b
Research team: A Kumar, G Bliss, P Cikatricis and K Damer.
Royal Blackburn Hospital
Investigators: Mrs Salwa Abugreen, Mohamed Alarbie and Debra Myerscough.
Research team: H Patel, N Nixon, M Anderson, T Thompson and P Richardson.
Bradford Teaching Hospitals NHS Foundation Trust
Investigators: Mr Faruque Ghanchi, Mrs Helen Devonport and Miss Nicci Atkinson.
Research team: Julie Dixon, Tony Dook, Cara Phillips, Tomas Cudrnak, Charlotte Hazel and Mary Elliott.
Sussex Eye Hospital
Investigators: Mr Anthony Casswell, Dr Gek Ong and Mr Edward Hughes.
Research team: Mrs Susan Bennett, Mr Nick White, Mrs Catriona Gardiner, Mr Stephen Turner and Mrs Tenesa Sargent.
Bristol Eye Hospital
Investigators: Miss Claire Bailey.
Cambridge University Hospitals NHS Foundation Trust
Investigators: Mr Douglas Newman and Mr Kevin McNally.
Research team: Haris Papanikolaou, Dawn Russell-Hermanns, Katherine Martin and Jo Fielden.
Frimley Park Hospital NHS Foundation Trust
Investigators: Mrs Geeta Menon, Mrs Manju Chandran and Mr Gulrez Ansari.
Research team: Mrs Bhavani Mathapati, Mr Nitin Jain, Mrs Lorraine North, Mrs Jincy Jose and Nadeem Rob.
The Queen Elizabeth Hospital, King’s Lynn NHS Foundation Trust
Investigators: Mr R J Pushapanathan and Mr I Ali.
Department of Eye and Vision Science, Institute of Ageing and Chronic Disease, University of Liverpool;a St Paul’s Eye Unit, Royal Liverpool and Broadgreen University Hospitals NHS Trustb
Investigators: Professor Simon P Harding,a Sandra Taylorb and Valerie Tompkin. b
Research team: Karen Hawkins, Jerry Sharp, Stephen Pearson, Martin Hodgson, William Hooley and Gillian Lewis.
Maidstone Hospital
Investigators: Mr Frank Ahfat and Mr Luke Membrey.
Research team: Mrs Margaret Gurney, Mr Clive Wood, Dr Shabeeba Hannan, Mr Syed Idris Haider and Mr Paul Adley.
Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Sciences Centre & School of Biomedicine, University of Manchester
Investigators: Mr Paul N Bishop and Tariq M Aslam.
Research team: Ekaterina Varimezova-Georgieva.
Newcastle upon Tyne Hospital Foundation Trust
Investigators: Mr S James Talks and Rajeev Kak.
Research team: Alan Branon, Kapka Nenova and Kevin Gales.
Nottingham University Hospitals NHS Trust
Investigator: Mr Alexander Foss.
Research team: Karen Armstrong-Owen.
Oxford Eye Hospital, Oxford University Hospitals
Investigators: Miss Susan Downes, Miss Shahrnaz Izadi and Mr Robert Purbrick.
Research team: Mrs Alexina Fantato, Mrs Ivy Samuel, Miss Vicky Hart, Mrs Anna Rudenko, Mr Lewis Smith, Mr Charles Cottriall and Miss Paula Hedges.
Sheffield Teaching Hospitals NHS Foundation Trust;a South Warwickshire NHS Foundation Trustb
Investigators: Mr Christopher Brand,a Dr Hibba Abdulkarim,a Mrs Uma Thakur. b
Research team: Mrs Helen Pokora, Mr Andy Jubb, Mrs Katy Kelly, Mr Fahd Quhill, Mrs Mary Freeman.
Southend University Hospital NHS Foundation Trust
Investigators: Mr Niral Karia and Mr A Krishnan.
Research team: Ms Maria Shipman, Mr John Williams and Mr Chris Johnson.
Faculty of Medicine, University of Southampton;a Wellcome Trust Clinical Research Facility, University Hospital Southampton NHS Foundation Trust;b Southampton Eye Unit, University Hospital Southampton NHS Foundation Trustc
Investigators: Professor Andrew John Lotery,a Marie Anne Nelsonb and Suresh Thulasidharan. c
Research team: Claire O’Brien, Kevin Oxlade, Caitrin Watkins, Maria Gemenetzi and Gabriella De Salvo.
Institute of Genetic Medicine, Newcastle University;a Sunderland Eye Infirmary, Sunderlandb
Investigators: Mr David Steel,a,b Eoghan Millarb and Vinna Manjunath. b
Research team: Steve Dodds, Shelagh Thomson, Martyn Hallowell, Hugh Harris and Paula Foley.
Torbay Hospital
Investigators: Mr Mick Cole, Yinka Osoba and Sanjay Dhir.
Research team: Annette Field, Sharon Criddle, Karin Tilley, Eddy Doyle and Debbie Knowles.
Royal Wolverhampton NHS Trust
Investigators: Mr Yit C Yang, Nirodhini Narendran and Swathi Paneerselvam.
Research team: Jas Purewal, Mary Stott, Bhogal Bhogal, Sharon Hughes and Gurminder Sahota, Jenny Nosek.
Resource centres
Trial Management Centre, Clinical Trials and Evaluation Unit, University of Bristol: Professor Barnaby C Reeves, Dr Lucy A Culliford, Dr Chris A Rogers, Dr Jodi Taylor, David Hutton, Rachel Nash, Lauren Scott, Dr Alessandro Cardinali, Dr Michael Arnott, Ms Wendy Underwood and Jon Williams.
Health Economics Research Centre, University of Oxford: Dr Sarah Wordsworth, Ms Helen Dakin and Ms Giselle Abangma.
Health Economist consultant to the trial: Professor James Raftery.
Macular disease-specific Patient-reported Outcome consultants to the trial: Professor Clare Bradley and Dr Jan Mitchell.
Centre for Experimental Medicine, Queen’s University Belfast: Josephine Glenn and Philip Earle.
Reading Centre, Belfast; Queen’s University Belfast: Professor Usha Chakravarthy, Dr Alyson Muldrew, Dr Liam Patton, Mrs Barbra Hamill, Mr Frank Picton and Mr Graham Young.
Reading Centre; Department of Eye and Vision Science, University of Liverpool; and St Paul’s Eye Unit, Royal Liverpool University Hospitals Trust: Professor Simon Harding, Jayashree N Sahni, Pauline M Lenfestey, David Parry and John Deane.
Reading Centre, London; NIHR Biomedical Research Centre for Ophthalmology, at Moorfields Eye Hospital NHS Foundation Trust; and UCL Institute of Ophthalmology: Dr Tunde Peto, Irene Leung and Peter Blows.
Manufacturing Pharmacy, Royal Liverpool and Broadgreen University Hospitals Trust: Shakeel Herwitker.
National Health Service (NHS) Public Health Consultant, negotiating contracts for treatment costs with Primary Care NHS Trusts: Dr Daphne Austin.
Comprehensive Clinical Research Network and Health and Social Care Research and Development Northern Ireland: provision of research support costs.
Participating primary care NHS trusts
Ashton Leigh & Wigan.
Bath & North Somerset.
Birmingham East & North.
Blackburn with Darwen.
Bradford & Airedale.
Brighton & Hove.
Bristol.
Bury.
Cambridge.
Central & Eastern Cheshire.
Central Lancashire.
Cornwall & Isles of Scilly.
County Durham.
Devon.
Dudley.
East & North Hertfordshire.
East Berkshire.
East Coast & Kent.
East Health & Social Services Board, Northern Ireland (HSSB, NI).
East Lancashire.
East Sussex Downs & Weald.
Halton & St Helens.
Hampshire.
Hastings & Rother.
Heart of Birmingham.
Knowsley.
Liverpool.
Manchester (East Lancashire).
Medway.
Newcastle.
Norfolk.
North Cumbria.
North HSSB, NI.
North Somerset.
North Tyneside.
North Yorkshire & Yorkshire.
Northumberland.
Nottingham.
Oxford.
Sandwell.
Sefton.
Sheffield (Barnsley).
Shropshire.
Solihull.
South Birmingham.
South East Essex.
South Gloucestershire.
South HSSB, NI.
South Staffordshire.
Southampton City.
Sunderland.
Surrey.
Torbay Care Trust.
Walsall.
Warrington.
Warwickshire.
West Berkshire.
West Cheshire.
West HSSB, NI.
West Kent.
West Sussex.
Wirral.
Wolverhampton City.
Appendix 2 IVAN committees
IVAN Data Monitoring and Safety Committee
Professor Sheila Bird (chairperson), Dr Hugh McIntyre, Mr Richard Wormald and Mr Roger Gray.
IVAN Trial Steering Committee
Mr John Sparrow (chairperson), Ms Helen Jackman, Mr Robert Coates, Professor Andrew Dick, Dr Sue Richards, Professor Usha Chakravarthy, Professor Simon Harding, Dr Barnaby Reeves, Dr Lucy Culliford, Dr Peter Davidson and Professor Ian Young.
Appendix 3 Additional data tables
| Type of breach | Allocated treatment group | Further details |
|---|---|---|
| Did not receive allocated drug | Bevacizumab continuous | Patient given ranibizumab instead of bevacizumab at visit 11 |
| Ranibizumab continuous | Patient given bevacizumab instead of ranibizumab at visit 9 | |
| Patient did not receive allocated regimen | Ranibizumab discontinuous | Patient treated as continuous throughout the study |
| Patient ineligible but treated | Ranibizumab continuous | Exudative AMD not present at visit 0 |
| Ranibizumab discontinuous | Exudative AMD not present at visit 0 | |
| Ranibizumab discontinuous | Exudative AMD not present at visit 0 | |
| Ranibizumab discontinuous | Exudative AMD not present at visit 0 | |
| Bevacizumab continuous | Exudative AMD not present at visit 0 | |
| Bevacizumab discontinuous | Exudative AMD not present at visit 0 | |
| Bevacizumab discontinuous | Exudative AMD not present at visit 0 | |
| Bevacizumab discontinuous | Exudative AMD not present at visit 0 | |
| Bevacizumab discontinuous | Patient had BCVA of < 25 letters at visit 0 |
Eligibility criteria were not met in nine patients. One patient read < 25 letters and eight patients did not meet the angiographic eligibility criteria, although two of these had fluid on OCT, indicative of an active lesion. Of the remaining six patients, two had early drusen – but did not have exudative lesions – and four were misdiagnosed.
| Centre name | Number of patients | Number of visits if there were no deaths/exits | Number of visits attended | Number of injections given | Number of missed visits: events/patients | Number of missed visits due to deaths/exits: events/patients |
|---|---|---|---|---|---|---|
| Aintree University Hospitals NHS Foundation Trust | 25 | 600 | 516 | 387 | 34/16 | 50/5 |
| Optegra Birmingham Eye Hospital, Aston | 2 | 48 | 45 | 16 | 3/2 | 0/0 |
| Royal Victoria Hospital, Belfast | 42 | 1008 | 890 | 698 | 55/23 | 63/4 |
| The Birmingham and Midland Eye Centre | 21 | 504 | 472 | 344 | 32/15 | 0/2 |
| Blackburn Royal Infirmary | 26 | 624 | 529 | 365 | 30/13 | 65/3 |
| Bradford Royal Infirmary | 35 | 840 | 709 | 548 | 71/24 | 60/6 |
| Brighton and Sussex University Hospital | 35 | 840 | 763 | 577 | 27/16 | 50/4 |
| Bristol Eye Hospital | 29 | 696 | 602 | 446 | 40/18 | 54/4 |
| Cambridge University Hospitals NHS Foundation Trust | 4 | 96 | 92 | 78 | 4/3 | 0/0 |
| Frimley Park Hospital, Surrey | 43 | 1032 | 920 | 688 | 43/25 | 69/4 |
| Queen Elizabeth Hospital, King’s Lynn | 8 | 192 | 154 | 98 | 10/5 | 28/2 |
| St Paul’s Eye Unit, Liverpool | 70 | 1680 | 1314 | 1023 | 83/35 | 283/21 |
| Maidstone Hospital | 30 | 720 | 656 | 518 | 24/14 | 40/4 |
| Manchester Royal Eye Hospital | 1 | 24 | 23 | 5 | 1/1 | 0/0 |
| Royal Victoria Infirmary, Newcastle | 10 | 240 | 229 | 201 | 11/6 | 0/0 |
| Queen’s Medical Centre, Nottingham | 29 | 696 | 605 | 388 | 18/10 | 73/6 |
| Oxford Eye Hospital | 24 | 576 | 492 | 376 | 24/14 | 60/4 |
| The Royal Hallamshire Hospital, Sheffield | 25 | 600 | 535 | 400 | 41/18 | 24/3 |
| Southampton University Hospital Trust | 47 | 1128 | 993 | 710 | 47/23 | 88/7 |
| Southend Hospital | 6 | 144 | 123 | 67 | 2/1 | 19/1 |
| Sunderland Eye Hospital | 10 | 240 | 219 | 188 | 19/7 | 2/1 |
| Torbay Hospital | 33 | 792 | 701 | 554 | 37/21 | 54/4 |
| New Cross Hospital, Wolverhampton | 55 | 1320 | 1179 | 875 | 98/40 | 43/4 |
| BCVA | Randomised to: | Overall (n = 610 at baseline, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 264 at 2 years) | |||||||
| n | % | n | % | n | % | n | % | n | % | |
| BCVA at baseline, lettersa | ||||||||||
| 83–99 | 28 | 9 | 34 | 11 | 36 | 12 | 26 | 9 | 62 | 10 |
| 68–82 | 50 | 16 | 49 | 17 | 56 | 18 | 43 | 14 | 99 | 16 |
| 53–67 | 103 | 33 | 97 | 33 | 103 | 33 | 97 | 32 | 200 | 33 |
| 38–52 | 122 | 39 | 99 | 33 | 101 | 33 | 120 | 40 | 221 | 36 |
| ≤ 37 | 11 | 4 | 17 | 6 | 12 | 4 | 16 | 5 | 28 | 5 |
| BCVA at 2 years, lettersb | ||||||||||
| 83–99 | 26 | 10 | 30 | 12 | 27 | 10 | 29 | 11 | 56 | 11 |
| 68–82 | 21 | 8 | 26 | 10 | 26 | 10 | 21 | 8 | 47 | 9 |
| 53–67 | 50 | 19 | 42 | 17 | 46 | 18 | 46 | 18 | 92 | 18 |
| 38–52 | 121 | 45 | 113 | 45 | 117 | 45 | 117 | 45 | 234 | 45 |
| ≤ 37 | 50 | 19 | 38 | 15 | 43 | 17 | 45 | 17 | 88 | 17 |
FIGURE 45.
Binocular vision by visit (a) by drug and (b) by treatment regimen. The circles indicate the median and the bars indicate the IQR. The numbers in parentheses are the number of observations.

FIGURE 46.
Best corrected distance visual acuity in the study eye at 2 years by subgroup, with baseline BCVA in study eye better than fellow eye vs. the same or worse (a) by drug and (b) by treatment regimen. Negative values of MD reflect a better mean VA in the ranibizumab or continuous groups. Difference is estimated using data from visits 0, 3, 6, 9, 12, 15, 18, 21 and 24, adjusted for centre size. Circles = MD. 95% CIs are illustrated by horizontal bars. The circle size is relative to the size of the subgroup.

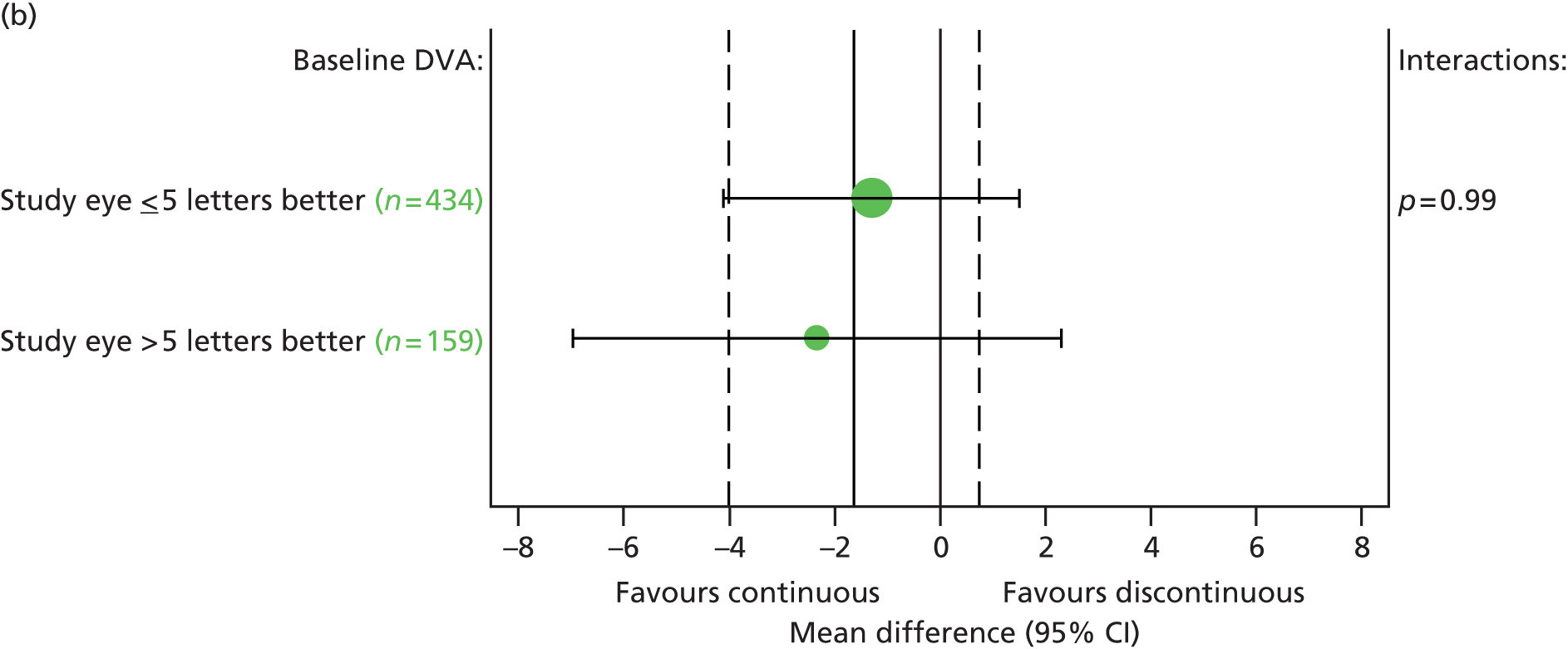
FIGURE 47.
Best corrected distance visual acuity in the study eye at 2 years by subgroup with haemorrhage in the study eye at baseline present vs. absent (a) by drug and (b) by treatment regimen. Negative values of MD reflect a better mean VA in the ranibizumab or continuous groups. Difference is estimated using data from visits 0, 3, 6, 9, 12, 15, 18, 21 and 24, adjusted for centre size. Circles = MD. 95% CIs are illustrated by horizontal bars. The circle size is relative to the size of the subgroup.
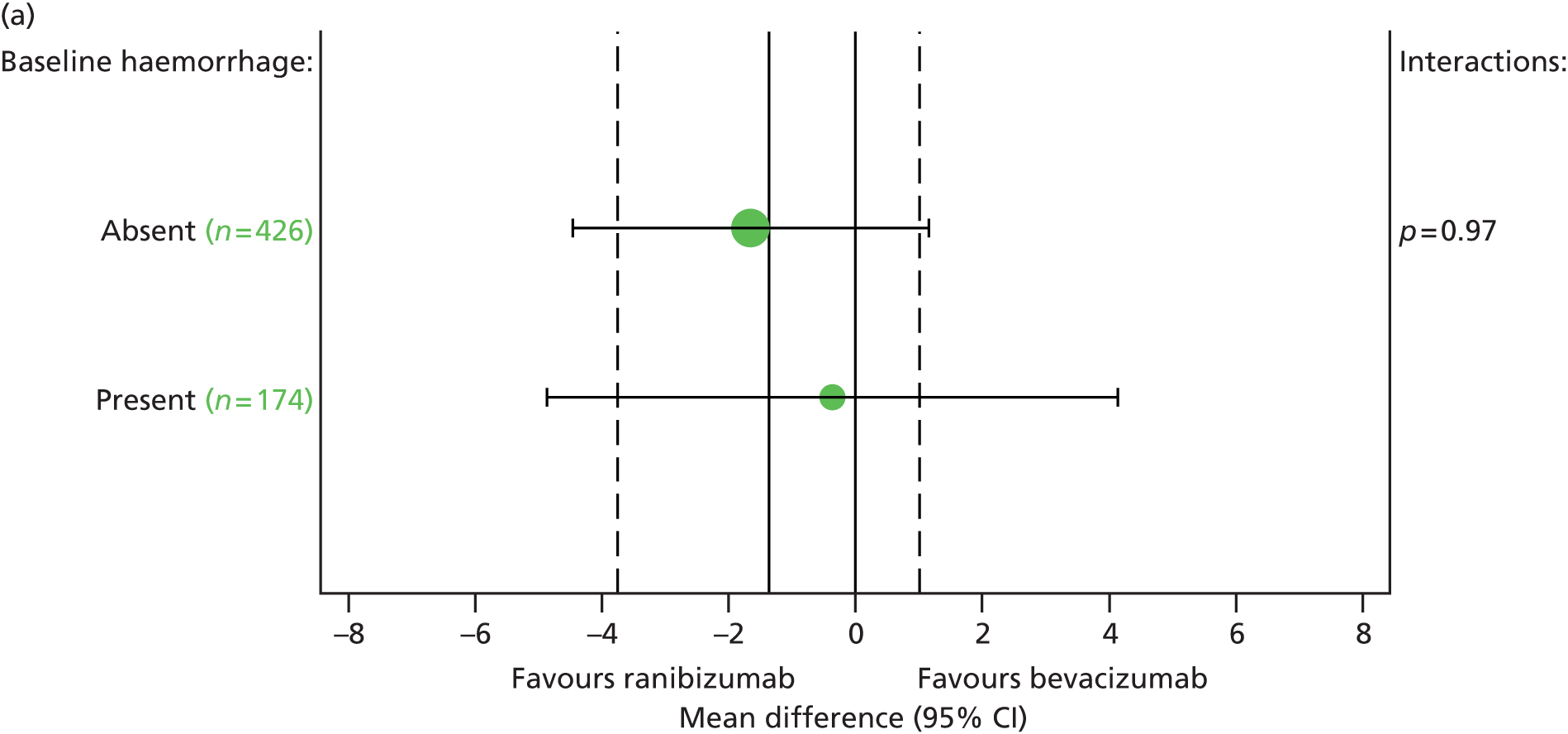

| EQ-5D questions | Randomised to: | Overall (n = 610 at baseline, n = 525 at 2 years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ranibizumab (n = 314 at baseline, n = 271 at 2 years) | Bevacizumab (n = 296 at baseline, n = 254 at 2 years) | Continuous (n = 308 at baseline, n = 261 at 2 years) | Discontinuous (n = 302 at baseline, n = 264 at 2 years) | |||||||
| n | % | n | % | n | % | n | % | n | % | |
| Mobility at baseline | ||||||||||
| No problems | 196 | 62 | 187 | 63 | 191 | 62 | 192 | 64 | 383 | 63 |
| Some problems | 118 | 38 | 108 | 37 | 117 | 38 | 109 | 36 | 226 | 37 |
| Confined to bed | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Mobility at 2 years | ||||||||||
| No problems | 176 | 66 | 171 | 69 | 174 | 68 | 173 | 67 | 347 | 68 |
| Some problems | 91 | 34 | 76 | 31 | 82 | 32 | 85 | 33 | 167 | 32 |
| Confined to bed | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Self-care at baseline | ||||||||||
| No problems | 292 | 93 | 267 | 90 | 286 | 93 | 273 | 90 | 559 | 92 |
| Some problems | 21 | 7 | 29 | 10 | 21 | 7 | 29 | 10 | 50 | 8 |
| Unable to wash/dress | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 |
| Self-care at 2 years | ||||||||||
| No problems | 247 | 93 | 218 | 88 | 238 | 93 | 227 | 88 | 465 | 90 |
| Some problems | 19 | 7 | 28 | 11 | 17 | 7 | 30 | 12 | 47 | 9 |
| Unable to wash/dress | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 2 | 0 |
| Usual activities at baseline | ||||||||||
| No problems | 214 | 68 | 218 | 74 | 227 | 74 | 205 | 68 | 432 | 71 |
| Some problems | 96 | 31 | 75 | 25 | 76 | 25 | 95 | 31 | 171 | 28 |
| Unable to perform usual activities | 4 | 1 | 3 | 1 | 5 | 2 | 2 | 1 | 7 | 1 |
| Usual activities at 2 years | ||||||||||
| No problems | 197 | 74 | 179 | 72 | 187 | 73 | 189 | 73 | 376 | 73 |
| Some problems | 66 | 25 | 67 | 27 | 68 | 27 | 65 | 25 | 133 | 26 |
| Unable to perform usual activities | 4 | 1 | 1 | 0 | 1 | 0 | 4 | 2 | 5 | 1 |
| Pain/discomfort at baseline | ||||||||||
| None | 193 | 62 | 179 | 60 | 188 | 61 | 184 | 61 | 372 | 61 |
| Moderate | 105 | 34 | 109 | 37 | 109 | 35 | 105 | 35 | 214 | 35 |
| Extreme | 15 | 5 | 8 | 3 | 11 | 4 | 12 | 4 | 23 | 4 |
| Pain/discomfort at 2 years | ||||||||||
| None | 154 | 58 | 145 | 59 | 151 | 59 | 148 | 57 | 299 | 58 |
| Moderate | 104 | 39 | 93 | 38 | 95 | 37 | 102 | 40 | 197 | 38 |
| Extreme | 9 | 3 | 9 | 4 | 10 | 4 | 8 | 3 | 18 | 4 |
| Anxiety/depression at baseline | ||||||||||
| None | 244 | 78 | 233 | 79 | 245 | 80 | 232 | 77 | 477 | 78 |
| Moderate | 66 | 21 | 62 | 21 | 60 | 19 | 68 | 23 | 128 | 21 |
| Extreme | 4 | 1 | 1 | 0 | 3 | 1 | 2 | 1 | 5 | 1 |
| Anxiety/depression at 2 years | ||||||||||
| None | 220 | 82 | 203 | 82 | 219 | 86 | 204 | 79 | 423 | 82 |
| Moderate | 44 | 16 | 43 | 17 | 35 | 14 | 52 | 20 | 87 | 17 |
| Extreme | 3 | 1 | 1 | 0 | 2 | 1 | 2 | 1 | 4 | 1 |
Appendix 4 Additional information about the health-economic evaluation
MedDRA codes for the adverse events and serious adverse events classed as expected in costing analyses
MedDRA codes counted as expected serious adverse events within the economic evaluation
-
Angina pectoris.
-
Arthralgia.
-
Cardiac arrest.
-
Cardiac failure.
-
Cardiovascular disorder.
-
Cataract traumatic.
-
Cerebrovascular accident.
-
Coronary artery bypass.
-
Deep-vein thrombosis (DVT).
-
Endophthalmitis.
-
Haemorrhage.
-
Intraocular pressure (IOP) increased.
-
Left ventricular failure.
-
MI.
-
Nausea.
-
Pulmonary embolism.
-
Retinal detachment.
-
Retinal pigment epithelial tear.
-
Retinal vein occlusion.
-
Transient ischaemic attack.
-
Upper respiratory tract infection.
-
Urinary tract infection.
-
Uveitis.
For the purposes of the costing analysis, death was not considered to be an expected SAE unless the primary cause of death comprised one of the expected SAEs or AEs listed, to ensure that costs unrelated to treatment (e.g. the cost of treating cancer) are excluded from the analysis regardless of whether or not the patient died during the study period. Events with a MedDRA code of ‘Visual acuity reduced’ were not counted as SAEs in any costing analysis.
MedDRA codes counted as expected adverse events within the economic evaluation
-
Angina pectoris.
-
Arthralgia.
-
Bronchitis.
-
Cardiac disorder.
-
Cataract.
-
Cataract cortical.
-
Cataract nuclear.
-
Cataract operation.
-
Cataract traumatic.
-
Conjunctival haemorrhage.
-
Cough.
-
Eye inflammation.
-
Eye irritation.
-
Eye pain.
-
Haemorrhage.
-
Hallucination, visual.
-
Headache.
-
Hypertension.
-
Influenza.
-
IOP increased.
-
Lacrimation increased.
-
Nasopharyngitis.
-
Nausea.
-
Pulmonary embolism.
-
Retinal detachment.
-
Retinal pigment epithelial tear.
-
Retinal vein occlusion.
-
Sinusitis.
-
Transient ischaemic attack.
-
Upper respiratory tract infection.
-
Urinary tract infection.
-
Uveitis.
-
Visual impairment.
-
Vitreous detachment.
-
Vitreous floaters.
Events with a MedDRA code of ‘Visual acuity reduced’ were not counted as AEs in any costing analysis.
Methods used to estimate the utility profile around serious adverse events and estimate quality-adjusted life-years for each patient
Within IVAN, EQ-5D and HUI3 utilities were measured at:
-
0, 3, 12 and 24 months (referred to hereafter as ‘scheduled’ time points)
-
at study exit (if the patient chose to attend a full exit visit)
-
at the next visit after SAEs or 15-letter drop in VA (referred to hereafter as ‘post-SAE measurements’)
-
around 24 patients also completed EQ-5D at other times not planned in the trial protocol (referred to hereafter as ’unscheduled visits’).
As the base-case analysis calculates QALYs using EQ-5D utilities, this appendix refers to EQ-5D measurements and utilities throughout. However, analyses were replicated using HUI3 utilities in a sensitivity analysis utilising identical methodology.
Most trial-based economic evaluations measure utility at fixed time points and either assume that utility changes linearly between assessments, or assume that utility remains constant at the level last observed until the next measurement. 124,125 A third assumption (assuming that utility changes midway between EQ-5D measurements) is also cited in the literature,124,125 but is equivalent to assuming linear changes in utility between measurements.
In the absence of SAEs, we assumed that EQ-5D utility changed linearly between the baseline and 3 months, between 3 and 12 months and between 12 and 24 months (Figure 48a), which is supported by the trends for VA (see Figure 15). However, within IVAN, patients also completed EQ-5D and HUI3 after SAEs or ≥ 15-letter reductions in VA in order to more accurately estimate the impact of these events on QALYs. We are not aware of any previous studies making such additional measurements, which introduce unique challenges into the estimation of QALYs, although some papers have explicitly modelled the profile of changes after specific events (e.g. hepatitis70 or chronic obstructive pulmonary disease exacerbations71).
FIGURE 48.
Possible assumptions for estimating the EQ-5D profile around SAEs. (a) No SAE: linear interpolation between routine HRQoL measurements – used in base-case and sensitivity analyses for patient with no SAEs; (b) SAE: assume linear interpolation including point where SAE occurred (the ‘average’ assumption outlined by Ganiats et al. 124 used in sensitivity analyses; and (c) SAE: assume sudden drop in quality of life on the day when SAE occurred, followed by a linear increase in quality of life between SAE and the time when the SAE effect ends – base-case assumption.
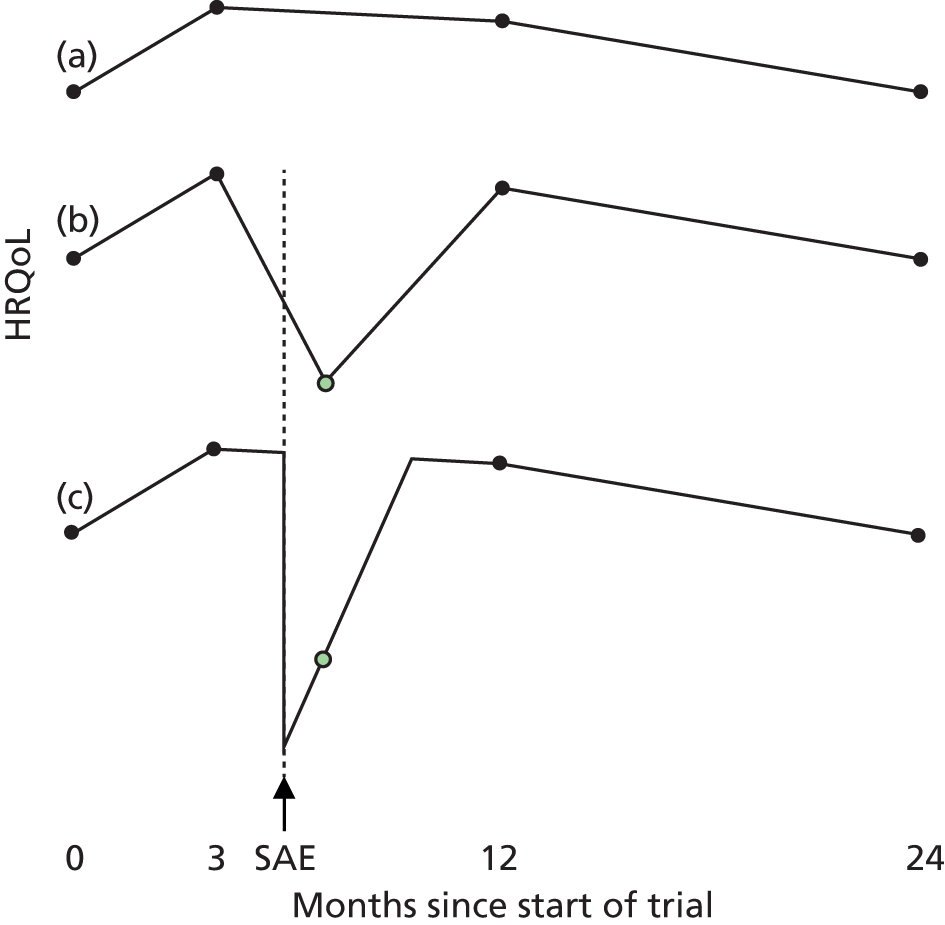
Assuming that EQ-5D utility changed linearly between all observed utility measurements (see Figure 48b), including those after SAEs, would not be clinically realistic and would overestimate the impact of SAEs by assuming that utility began to fall immediately after the measurement preceding the SAE. Similarly, applying a constant utility between the onset and resolution dates would be unrealistic and is likely to underestimate the impact of SAEs. Patients’ estimates of the date their SAE resolved may also not necessarily be reliable: particularly for intermittent symptoms, permanent conditions and cases in which patients continue to have worse health than before the SAE until long after the main symptoms resolve.
For many SAEs, the EQ-5D profile is instead likely to be more similar to Figure 48c, with a sudden fall in utility at the start of the event, followed by gradual recovery. EQ-5D utility is, therefore, at its worst point on the date of onset. This profile is similar to that proposed by Luyten et al. 70 and Spencer et al. 71 after acute hepatitis or chronic obstructive pulmonary disease exacerbations, although both studies also consider logarithmic recovery curves that were not explored in the current study owing to their complexity.
As EQ-5D is rarely completed on the day of onset, as patients are generally too sick to attend the IVAN clinic and the true resolution date is generally unknown, we used mixed models to estimate the rate of recovery after different types of SAE. These recovery rates were then used to back-calculate the EQ-5D utility on the day of SAE onset, and to identify the time and utility at which the line showing recovery from the SAE crosses the line joining two other EQ-5D measurements. The base-case analysis allowed for the reductions in quality of life associated with all SAEs, whether or not expected or unexpected, in order to use all available EQ-5D data.
This appendix first outlines the methods used to estimate mixed models, followed by the methods used to group SAEs into sets, then the methods used to impute missing data, and, finally, the methods and assumptions used to calculate EQ-5D profiles and QALYs.
Mixed models of quality of life after serious adverse events
We defined a basic model of how SAEs affect EQ-5D utility and identified 19 prespecified variations on this model, which were evaluated to assess whether or not they improved model fit [i.e. reduced Akaike information criterion (AIC)]. The basic model assumed that each type of SAE that patients had experienced reduced EQ-5D utility, but that utility rose by a fixed amount with each day that passed after each type of SAE to allow for patients’ recovery. EQ-5D utility was also assumed to be a function of time, as randomisation, treatment and baseline EQ-5D utility (centred by subtracting the mean baseline utility across all patients):
where terms are defined in Appendix 4, Table 38.
| Variable | Definition | Rationale |
|---|---|---|
| EQ-5D | EQ-5D utility | Dependent variable |
| Time | Date EQ-5D measured minus randomisation date | Allows for any systematic rise or fall in utility over time. For simplicity, the basic mixed model assumed that utility rose or fell at a constant rate for all patients. The effect of time since randomisation was assumed to be linear to simplify QALY calculations |
| BaselineEQ5DCentred | EQ-5D at baseline minus the mean EQ-5D at baseline across the trial population | Strong predictor of on-treatment utility. Centred to improve convergence and make predictions easier |
| Ever Had Type X SAE | 1 = Patient has experienced a SAE of type X in the trial to date 0 = Patient has not experienced a SAE of this type In the basic model, three types of SAE were considered: ocular, cardiovascular and other, although these categories were broken down further in additional analyses |
Coefficient reflects the drop in utility on the day of onset of this SAE, but is not used in QALY calculations |
| Time Since Type X SAE | Days since date of onset of the most recent SAE of type X. In the basic model, three types of SAE were considered: ocular, cardiovascular and other, although these categories were broken down further in additional analyses | Coefficient gives the rate of recovery. Used to calculate utility on day of SAE onset and day of recovery in QALY calculations |
| Bevacizumab | 1 = randomised to bevacizumab; 0 = ranibizumab | Treatment indicators allow for the fact that patients’ quality of life may be higher with some treatment regimens than others, and ensure that imputed utilities are not biased by omission of treatment indicators included in subsequent analyses.81 For consistency with the main analysis, all patients were treated as having continuous therapy up to visit 3 |
| Discontinuous | 0 = randomised to continuous treatment, or this measurement is before visit 3, when all patients have continuous therapy 1 = randomised to discontinuous arm, if visit 3 or beyond |
|
| Interaction | = bevacizumab × discontinuous |
In the basic model, random parameters were applied to the constant term and the falls in utility associated with each type of SAE to allow for variations between patients. However, random slopes were not estimated, as it would not be possible to estimate both random slopes and random intercepts for patients with only one post-SAE measurement. The model assumed that different classes of SAE have additive effects on EQ-5D utility and on recovery rates: for example, if CVD events reduce utility by 0.2, with a recovery rate of 0.01/day, while ocular events reduce utility by 0.1 with a recovery rate of 0.005/day, having had angina and endophthalmitis 10 days ago will reduce utility by 0.24 (0.1 + 0.2–0.01 × 10–0.005 × 10). Which event is most recent is assumed to have no effect on utility, except for the number of days of recovery time that are applied. However, for simplicity having two events in the same class on the same date is assumed to have the same impact on utility as having just one event; nonetheless, having a second event of the same type some time after the first will reduce utility to the same level observed immediately after the first event. The model assumed that utility improved linearly to simplify subsequent calculations, although we did evaluate the impact of non-linear functions in sensitivity analysis.
Serious adverse events were initially divided into three main categories:
-
‘Cardiovascular events’, which included all events in the MedDRA category of ‘Cardiac disorders’, plus the systemic expected SAEs listed in the previous section [cerebrovascular accident, coronary artery bypass (which was accompanied by MI), DVT, haemorrhage, pulmonary embolism and transient ischaemic attack]. There were 68 events in this category, after which there were 78 EQ-5D measurements in 47 patients.
-
‘Ocular events’, which included all events in the MedDRA category of ‘Eye disorders’, plus IOP increased. This category included 48 events, after which there were 72 EQ-5D measurements in 36 patients. Subsequent analyses explored the impact of considering VA reduced as a separate type of event, as this was not classed as a SAE (27 events, with 42 EQ-5D measurements).
-
‘Other’ included all SAEs not included in either of the above categories, giving 168 events with 170 EQ-5D measurements in 93 patients afterwards. In the basic model, this category was considered to be one homogeneous group, although subsequent analyses explored the impact of dividing it into smaller MedDRA categories:
-
– ‘Neoplasms benign, malignant and unspecified (including cysts and polyps)’: 24 events, with 14 EQ-5D measurements in 10 patients afterwards.
-
– ‘Infections and infestations’: 25 events, with 29 EQ-5D measurements in 13 patients.
-
– ‘Gastrointestinal or hepatobiliary disorders’: 19 events, with 29 EQ-5D measurements in 14 patients.
-
– ‘Surgical and medical procedures’ and ‘Investigations’ (excluding evaluation of IOP, which was classed as ocular, or coronary artery bypass): 32 events, with 51 EQ-5D measurements in 29 patients.
-
– ‘Injury, poisoning and procedural complications’ or ‘Musculoskeletal and connective tissue disorders’: 28 events, with 31 EQ-5D measurements in 22 patients.
-
– Other unexpected events (those not falling into any of the above categories): 40 events, with 49 EQ-5D measurements in 27 patients.
-
Mixed models were fitted in Stata version 12 using xtmixed on all post-baseline utility measurements in the 2-year data set. Models were run initially on EQ-5D utilities (including 1732 EQ-5D measurements on 601 patients) to inform base-case analyses. The final model specification was rerun using HUI3 utilities (including 1306 HUI3 measurements on 556 patients) to inform sensitivity analyses. Mixed models were fitted only on observed (not imputed) utility measurements to simplify the analysis and ensure that mixed-model estimates were not affected by the assumptions made in the imputation model. However, to avoid omitting observations that had post-baseline EQ-5D but were missing baseline EQ-5D, we assumed that the centred baseline EQ-5D utility for that patient was equal to the difference between their 3-month EQ-5D utility and the mean 3-month EQ-5D utility if this patient had no SAEs before an observed 3-month EQ-5D measurement. This was applied to three patients missing baseline EQ-5D and 146 patients missing baseline HUI3.
The basic model used autocorrelated residuals to allow for the fact that utility measurements for the same patient are correlated and that these correlations are likely to reduce over time. Visit number was used as a measure of the time of EQ-5D measurement, with exit visit utilities being assigned the visit number closest to the date of exit. The basic model also assumed that the random parameters are independent (zero covariance): i.e. that the amount that EQ-5D utility falls after a certain type of SAE is not correlated with the constant term for this patient before the event, or with the utility reduction that the same patient had before a different event. This assumption was necessary as only 19 patients had two different types of SAE during the trial and only one had all three types; as a result, a model with unstructured covariance and three random SAE parameters did not converge.
We evaluated 19 variations on the basic model in four stages to assess whether or not any improved model fit (i.e. reduced AIC by any amount); details are available on request. At each stage, we adopted any variations that reduced model fit and conducted the next step using the best-fitting model from the previous step. We also conducted sensitivity analyses to evaluate the impact of two variations, which were not considered candidates for the final model.
The model with the lowest AIC divided SAEs into ‘ocular disease’, ‘CVD’, ‘cancer’ and ‘other’, and included random effects parameters for the time since randomisation, time since ocular events and the time since cancer or other SAEs. CVD and cancer significantly decreased EQ-5D utility, although the impact of ocular or other SAEs was non-significant (Table 37). The non-significant coefficient for ocular disease may reflect previous evidence suggesting a weak correlation between EQ-5D and VA,126 and the tendency towards EQ-5D correlating best with patients’ better-seeing eye,91 which may not necessarily be the one affected by an ocular SAE or the study eye that shows a ≥ 15-letter drop in VA. The rate of recovery after each type of SAE was positive (indicating that EQ-5D utility is worst immediately after the event and improves over time), but was not statistically significant for any class of SAE other than cancer. Model predictions suggested that EQ-5D utility declined with each class of SAEs and increased gradually over time, with different classes of SAEs having an additive effect on EQ-5D utility (Figure 49).
| Variable | Mean (SE) coefficient | Interpretation | |
|---|---|---|---|
| EQ-5D | HUI3 | ||
| Fixed coefficients | |||
| Time since randomisation | –0.00003 (0.00001) | –0.00007 (0.00002)a | Change in utility per day |
| Baseline utility (centred) | 0.56 (0.025)a | 0.636 (0.043)a | Impact of increasing baseline utility from 0 to 1 |
| Ever had ocular SAE or VA drop | –0.019 (0.028) | –0.066 (0.044) | Decrease in utility on the day this event begins |
| Ever had CVD SAE | –0.084 (0.027)a | –0.263 (0.042)a | |
| Ever had cancer | –0.311 (0.07)a | –0.245 (0.116)a | |
| Ever had other SAE | –0.027 (0.024) | –0.148 (0.038)a | |
| Time since most recent ocular event | 0.00003 (0.00008) | 0.00009 (0.00013) | Increase in utility with every day that passes after an event of this type |
| Time since most recent CVD event | 0.0002 (0.0001) | 0.00062 (0.00016)a | |
| Time since most recent cancer SAE | 0.0011 (0.00034)a | 0.00084 (0.00043) | |
| Time since most recent other event | 0.00003 (0.00007) | 0.00028 (0.0001)a | |
| Bevacizumab | –0.002 (0.014) | –0.065 (0.024)a | Effect of treatment |
| Discontinuous | –0.002 (0.014) | –0.047 (0.024) | |
| Interaction | –0.00036 (0.02045) | 0.05737 (0.03427) | |
| Constant | 0.842 (0.011)a | 0.771 (0.018)a | Constant: predicted utility on day 1 without SAEs for patient with the mean utility |
| Random-effects parameters | |||
| SD: Time since randomisation | 0.0001 (0.00002) | 0.00012 (0.00004) | Between-patient variability in temporal trend |
| SD: Ever had ocular SAE or VA drop | 0.027 (0.08) | 0.07 (0.078) | Between-patient variability in fall in utility associated with each of the main types of SAE |
| SD: Ever had cancer OR another SAE | 0.116 (0.022) | 0.168 (0.034) | |
| SD: Constant | 0.088 (0.006) | 0.161 (0.009) | Between-patient variability in constant |
| Residual: AR(1) | |||
| rho | 0.027 (0.129) | 0.467 (0.128) | Correlation between errors for consecutive visits |
| sd(e) | 0.132 (0.003) | 0.161 (0.005) | Residual |
FIGURE 49.
The effect of model coefficients on EQ-5D utility for a hypothetical patient who experienced a cardiovascular SAE on day 90, an ocular SAE on day 150, an unexpected SAE on day 205, a second cardiovascular SAE on day 330 and a cancer SAE on day 629.

Grouping serious adverse events into sets
The analysis of QALYs excluded six SAEs that started after the patient withdrew from the study, and 14 SAEs that occurred after the patient attended visit 24. These SAEs were excluded because they cannot influence the EQ-5D measurements observed within the 24-month trial period.
A minority of patients within IVAN had up to seven SAEs within the trial, although many of these events began at the same time or in close succession. For example, a patient may have broken their wrist and clavicle in the same fall during year 1, and the following year have experienced a worsening of angina followed by a MI 3 days after angina symptoms worsened, which resulted in death 6 days later. No patients completed EQ-5D or HUI3 between SAEs that occurred < 7 days apart. We therefore grouped SAEs with onset dates < 7 days after the last SAE into the same ‘set’. In the aforementioned example, the broken wrist and clavicle would be classed as this patient’s first set of SAEs, whereas angina, MI and death would be classed as their second set of SAEs (with an onset date equal to the date the angina symptoms worsened). After grouping SAEs in this way (and after excluding SAEs occurring after withdrawal or visit 24), 129 trial participants were considered to have one set of SAEs, 37 had two sets, 15 had three sets and two had four sets. A set of SAEs was considered to be ‘fatal’ if death was one of the SAEs included within it and was considered ‘non-fatal’ if the patient survived for > 7 days after the last SAE in that set. For the purposes of QALY calculations and costing analyses, the onset date for death was assumed to be equal to the date of death, rather than the date of onset for the cause of death. The next EQ-5D (or HUI3) measurement taken after each set of SAEs was considered to be the post-SAE measurement for that SAE set, providing that the next measurement was within 6 months of the SAE onset and before the next set of SAEs began.
Outline of approach to missing data
Missing data are frequently more common for economic evaluation than for clinical end points16,23 and cost-effectiveness is measured using a composite of numerous types of resource use and multiple utility measurements. As a result, the approach used in the primary clinical analysis (mixed models of data on all patients with at least one post-baseline outcome measure) could not be used for the economic evaluation. Although only 3.6% (82/2305) of EQ-5D measurements at scheduled visits, 54% (32/59) of exit visit EQ-5D measurements and 41% (88/216) of EQ-5D measurements after patients’ first or second set of SAEs were missing. Missing utility data were imputed using multiple imputation using a series of chained regression equations (see Chapter 2). This standard, validated approach imputes missing data based on a wide range of covariates and allows for the uncertainty around imputed values by generating multiple imputations sampled from the posterior distribution of predicted values for each patient.
As discussed in Chapter 2, utilities at 0, 3, 12 and 24 months were imputed for all patients, whereas exit visit utilities were imputed for all patients who withdrew from the study within 2 years of randomisation. As some patients who missed visit 3 or 12 completed EQ-5D and/or HUI3 at the following visit, five measurements taken at visit 4 and one measurement taken at visit 5 were moved to visit 3 before multiple imputation was conducted, and seven measurements taken at visit 13 and three measurements from visit 14 were moved to visit 12; measurements were moved only if the patient had not experienced any SAEs in the last 30 days.
Post-SAE utilities were imputed for patients who had no EQ-5D measurement that was within 6 months of the onset of each SAE set, and which occurred before their next set of SAEs began. Each missing post-SAE utility was assigned a time interval between the SAE and the post-SAE measurement. This interval was equal to the date when the next scheduled measurement or exit visit was due minus the SAE onset date, if this was within 6 months and before the patient’s next SAE. When there was no scheduled visit within this interval, the post-SAE measurement was assumed to take place X days after the SAE, where X is whichever is smallest out of the following: the day before the next set of SAEs; and the average interval between SAE onset and the next EQ-5D measurement across patients who experienced that type of SAE.
The EQ-5D and HUI3 utilities after patients’ first set of SAEs were imputed using multiple imputation for all patients who had at least one non-fatal set of SAEs, as were EQ-5D utilities after patients’ second set of SAEs for those patients with at least two non-fatal sets of SAEs. Multiple imputation models included variables indicating the types of SAEs and the time since each type of SAEs to mirror the final mixed model. However, only one patient was missing EQ-5D after a third set of SAEs. As only 15 patients had three or more sets of non-fatal SAEs, EQ-5D utility after this patient’s third set of SAEs (CVD) was imputed by adding to the estimated EQ-5D utility the day before the SAE, the mixed-model estimate of the change in EQ-5D on the day of a cardiovascular SAE plus the product of the time interval between the SAE and the post-SAE measurement, multiplied by the rate of recovery associated with the SAEs that this patient had experienced. As there were insufficient HUI3 measurements to use multiple imputation to impute HUI3 utilities after patients’ second or third sets of SAEs, the utility imputed for the next scheduled/exit visit was used as a measure of post-SAE utility in 11 of these 33 measurements with scheduled/exit visit measurements due within 6 months of the SAE. For the remaining 22 missing measurements, HUI utility was imputed from EQ-5D values using a simple OLS mapping model that estimated HUI3 utility as a function of EQ-5D utility and a constant term, which was estimated on data from all time points for which both HUI3 and EQ-5D utilities were available. This model suggested that HUI3 utility = –0.006 + 0.844 × EQ-5D utility.
In the 15 cases in which the post-SAE measurement was missing but was assumed to have occurred on the same date as the next scheduled/exit measurement, utility at the scheduled/exit visit and the post-SAE measurement were imputed independently in multiple imputation. To ensure that each patient had only one utility value on each day, utility at the scheduled measurement was changed to equal the utility imputed for the post-SAE measurement. For example, if a patient had SAE 1 on day 60 and was missing – the visit 3 utility assigned as their post-SAE measurement – utility at visit 3 and post-SAE 1 utilities were imputed independently, and this patient’s visit 3 utility was changed to equal the post-SAE utility imputed for the same date. Post-SAE utilities were used in preference to the scheduled measurements because they are imputed specifically for the subset of patients who had SAEs and are therefore likely to better represent post-SAE utility.
In addition to EQ-5D and HUI3 utilities, the imputation model included demographic characteristics, VA, costs, indicators of random assignment to bevacizumab, discontinuous therapy and an interaction term as additional covariates to avoid bias and improve the accuracy of imputations. For consistency and to best reflect the available data, the imputation model allowed for the effect of SAEs using the same functional form that was used in the mixed model. The model therefore also included variables capturing the time from randomisation to death, time from randomisation to withdrawal, dummies for which SAE types had occurred before each measurement and variables capturing the time since each type of SAE. For simplicity and as indicators of utility on the day before death, utilities at visits 0, 3, 12 and 24 were imputed for all patients, regardless of whether or not they had died or withdrawn before that time point. Ten imputation cycles were used and predictive mean matching was used to allow for the non-normal distribution of utilities and ensure that plausible utility values (between –0.594 and 1 for EQ-5D) were imputed.
Calculating quality-adjusted life-years
The number of QALYs accrued by each patient in each quarter was approximated by estimating EQ-5D utilities at different time points, joining them with straight lines and calculating the area under the curve. All EQ-5D measurements taken within IVAN and those imputed at visits 0, 3, 12 or 24, at study exit or after SAEs were used in QALY calculations, including those not scheduled in the trial protocol.
Quality-adjusted life-year calculations assumed that EQ-5D measurements taken at visit ‘v’ were measured exactly v × (365/12) days after randomisation, regardless of the exact date of utility measurement: for example, baseline utilities were assumed to have been taken on the day of randomisation, whereas visit 3 utilities were assumed to have been measured 91.25 days later. Exit visit utilities were assumed to have been measured exactly on the date of exit. These assumptions were made to simplify calculations and ensure that utility measurements covered the entire period from randomisation to exit or 24 months. They also avoid the variability and bias that could result from using the exact timing of utility measurements and (as a result) assuming that patients who happen to have had visit 3 utility measured on day 105 have a more gradual increase in utility (and therefore accrue fewer QALYs) than those who happen to have the visit on day 84. [The trial protocol permitted intervals of 28–35 days between study visits; as a result, patients following the protocol could have visit 3 as late as 105 days or as early as 84 days after randomisation.] To ensure that SAEs were correctly ordered with respect to the utility measurement either side of them and that the time interval between the SAE and the subsequent EQ-5D measurement was maintained, QALY calculations used adjusted measures of the time to each SAE, whereby non-fatal SAEs with observed (not imputed) post-SAE EQ-5D measurements were assumed to start on day v × (365/12) – X, where ‘v’ indicates the visit number for the post-SAE measurement and ‘X’ indicates the interval between SAE onset and the post-SAE measurement. The exact interval between randomisation and SAE onset was used for fatal SAEs and those with imputed EQ-5D measurements.
After adjusting the visit number for 15 patients completing visit 3 or visit 12 EQ-5D questionnaires a month or two late and imputing utilities at scheduled time points, study exit and after SAEs, QALY calculations used a complete set of:
-
2305 EQ-5D measurements taken at scheduled time points (of which 2223 were observed)
-
59 at study exit (of which 27 were observed and five coincided with scheduled visits), 231 measurements after non-fatal SAEs (of which 142 were observed and 28 coincided with scheduled time points or exit visits)
-
37 measurements at time points not scheduled in the trial protocol; of these, 16 were from one small IVAN centre, which initially conducted monthly EQ-5D and HUI measurements; the remaining 21 unscheduled measurements could have been made in error or measured by staff who were unsure whether or not a patient had experienced a SAE. However, all measurements (scheduled or otherwise) were included in base-case QALY calculations to make use of available data.
The base-case analysis assumed that utility changed linearly between all EQ-5D measurements that were not the next measurement after a SAE. However, the analysis divided SAE sets into four types determined by the effect of the SAEs on EQ-5D utility:
-
SAEs for which the post-SAE EQ-5D measurement is lower than the profile of the other measurements (e.g. SAEs 1–3 in Figure 50): utility was assumed to drop suddenly on the day of the SAE, before recovering at the rate estimated in the mixed model. A total of 136 SAEs fell into this category.
-
SAEs for which the post-SAE EQ-5D measurement is above the profile of other measurements: for example, SAEs 4–5 in Figure 50: utility was assumed to change linearly from the measurement before the SAE up to the post-SAE measurement and then from the post-SAE measurement to the next measurement. This category included 56 sets of SAEs for which the post-SAE measurement was above the straight line joining patients’ previous measurement and the next measurement that was not post-SAE (e.g. SAE 5 in Figure 50), in addition to 39 sets of SAEs for which the post-SAE measurement was no higher than the trajectory of other measurements, but where extrapolating back to the day of the SAE using the gradient from the mixed model would suggest that the utility the day of the SAE was higher than that the day before (e.g. SAE 4 in Figure 50).
-
SAE sets in which the patient died on the day of onset. In these cases, patients’ utility was assumed to drop immediately to zero as soon as the SAE starts and remain at zero for the rest of the trial. There were 19 SAEs within 2 years of randomisation in which the patient died immediately.
-
SAEs sets in which the patient died 1–7 days after the most recent SAE in that set began (e.g. SAE 6 in Figure 50): utility was assumed to fall linearly from the value estimated the day before the SAE to 0 between the date of onset and the date of death. This simplifying assumption avoided the need to impute utilities in patients’ last week of life and allowed for the poor quality of life experienced by patients who are dying. This simplification is unlikely to have any significant effect on the results, as it affected only EQ-5D estimates for a few days in six patients. Four deaths occurring > 2 years after randomisation were excluded from the QALY calculations, as they were outside the time horizon.
FIGURE 50.
Illustration of the methods used to infer the EQ-5D profile and calculate QALYs for a hypothetical patient with six SAEs and three unscheduled measurements.

The coefficients for the interaction between time and SAE type that were estimated in the mixed model were used as a measure of the rate at which utility recovered after each type of SAE. To allow for uncertainty around the mixed model coefficients, 100 sets of correlated model coefficients were drawn from the distribution estimated within the model by using the Cholesky decomposition of the variance–covariance matrix and assuming that coefficients were normally distributed. Each of the 100 sets of correlated coefficients was used for one of the 100 multiply imputed data sets, such that, for example, imputation 1 was assigned the first set of coefficients and the rate of recovery after ocular SAEs was the same for all patients at any given imputation, but differed between imputations. This approach preserved the correlations between coefficients and propagated the uncertainty within the mixed-model results. As the recovery rates for ocular, cardiovascular and other SAEs were not statistically significantly different from zero, > 2.5% of imputed data sets have negative slopes, suggesting that EQ-5D utility decreases over time. Following the additive assumption made within the mixed model, the recovery rate after each set of SAEs was estimated for each patient in each imputation by adding up the recovery rates for all types of SAEs that each patient had had up until the end of the current set of SAEs. For example, a patient who had ocular and cardiovascular events within SAE set 1 and experienced a cardiovascular event and cancer within SAE set 2 would have a gradient after SAE set 2 equal to the slope for ocular events, plus the slope for cardiovascular events, plus the slope for cancer. The coefficients estimating the fall in utility after each type of SAE were also sampled from the variance–covariance matrix, but were not used in the analysis except to impute EQ-5D utility after one patient’s third set of SAEs.
To estimate utility profiles around SAEs, three additional EQ-5D utilities were estimated for each set of SAEs:
-
EQ-5D utility on the day before the SAE (points a1–6 on Figure 50):
-
For fatal SAEs and SAEs reducing utility, this was assumed to lie on a straight line joining the EQ-5D measurement before the SAE with the next measurement after the SAE that was at scheduled time point, at study exit or at an unscheduled time point that was not the next measurement after a SAE. Imputed values were used for the measurement before the SAE or after the SAE when either of these measurements was missing. The next EQ-5D measurement was also imputed after fatal SAEs; for example, Figure 50, SAE 6, the utility the day before death is assumed to lie on the line between the observed visit 12 utility and the imputed utility at visit 24. In cases where the measurement before SAE set s was the one taken after SAE Set s-1 (e.g. for SAE 3 in Figure 50), the utility the day before SAE s (e.g. point a3) was estimated by extrapolating from the post-SAE s-1 measurement using the recovery rate estimated in the mixed model.
-
For SAEs where the post-SAE EQ-5D measurement was higher than would be expected from the profile of other measurements (e.g. points a4 or a5 in Figure 50), utility the day before the SAE was assumed to lie on a straight line joining the most recent measurement before the SAE with the post-SAE measurement.
-
-
EQ-5D utility on the day of the SAE (points b1–6 on Figure 50):
-
For SAEs reducing utility, utility on the day of the SAE was estimated from the first post-SAE measurement by extrapolating backwards using the recovery rate associated with that set of SAEs (e.g. points b1–3). Values above 1 (which arose in cases for which the post-SAE utility was close to 1 and the recovery rate was negative) were set to 1.
-
For SAEs increasing utility, utility on the day of the SAE was assumed to lie on the straight line joining the most recent measurement before the SAE with the post-SAE measurement (e.g. points b4 and b5).
-
For SAEs when the patient died immediately, EQ-5D utility on the day of the SAE was zero. For SAEs where the patient died 1–7 days after the start of the latest SAE, EQ-5D utility on the first day was assumed to lie on the line between the utility the day before the SAE set began and zero utility on the day of death (e.g. point b6).
-
-
The time when the patient recovered from the SAE and the EQ-5D utility on that day (points c1–6 on Figure 50):
-
For SAEs increasing utility, the effect of that SAE on utility was assumed to end at the next measurement after the post-SAE measurement (e.g. points c4 and c5).
-
The effect of fatal SAEs was assumed to end on the day of death with a utility of zero (e.g. point c6).
-
For non-fatal SAEs decreasing utility, the effect of the SAE was assumed to end at whichever was soonest out of the following:
-
–The day before the next set of SAEs started (e.g. point c2). In cases for which the effect of one SAE set ended the day before the next SAE set, the utility on the day of recovery was estimated from the post-SAE measurement using the recovery rate estimated in mixed model.
-
–At the time of the next EQ-5D measurement that was not post SAE, whether that was a routine time point, study exit or an unscheduled visit (e.g. point c1).
-
–At the point where a straight line running through the post-SAE measurement with a gradient equal to the recovery rate from the mixed model crossed the straight line joining the patient’s previous non-post-SAE measurement with their next non-post-SAE measurement. The time at which the two lines crossed was estimated by solving the simultaneous equations for the two lines (e.g. point c3).
-
-
After estimating points a–c for each SAE, utility at any point in time could be estimated by joining points a–c and all imputed or observed EQ-5D measurements with straight lines. We then estimated utility at the interval for each quarter by interpolation, and calculated the number of QALYs accrued between each pair of points by taking the average between the EQ-5D measurements at the start and end of each period and multiplying that by the duration of each period in years.
In addition to the base-case analysis, seven other estimates of the number of QALYs accrued by each patient were generated for sensitivity analysis, of which three make different assumptions about the EQ-5D profile (Figure 51). These analyses differ in the types of EQ-5D measurements included in QALY calculations and the way in which utility was assumed to change between measurements. In the absence of SAEs, utility was assumed to change linearly between measurements. All sensitivity analysis used the same run of multiple imputation. The EQ-5D profile and the number of QALYs accrued for the 392 patients who did not exit the study early/had no SAEs/had no unscheduled EQ-5D measurements was identical in all analyses except (1), although the different analysis gave different profiles and different QALY estimates for the other 218 patients.
FIGURE 51.
Assumptions made in the base-case analysis and the three main sensitivity analyses about the EQ-5D profile.

-
HUI3 Replicating all analyses in the same way as the base case, but using HUI3 utilities in place of EQ-5D.
-
Simply interpolating between scheduled EQ-5D measurements (see Figure 51, row 2) This analysis included only EQ-5D measurements at 0, 3, 12 and 24 months, and assumed that EQ-5D utility changed linearly between these measurements. Utility the day before death or study exit was assumed to lie on the straight line between the patient’s last measurement and the next scheduled measurement that was imputed after the patient had died/withdrawn. EQ-5D utility was assumed to be zero after death and QALYs that ended after the patient had withdrawn from the study were excluded from the analysis. This analysis indicates the QALYs that would have been accrued within the study if data were collected only at scheduled time points.
-
Excluding the post-SAE measurements and interpolating between the non-SAE measurements (including exit visits and unscheduled measurements (see Figure 51, row 3) Measurements identified as the first one after a SAE were excluded from this analysis (unless they coincided with a scheduled visit or an exit visit), although EQ-5D utilities measured at unscheduled time points or measured/imputed at exit visits were included. Utility was assumed to change linearly between all measurements. Exit visit utilities were assumed to indicate utility on the day of study withdrawal, although utility the day before death was assumed to lie on the straight line between the patient’s last measurement and the next scheduled measurement that was imputed after the patient had died/withdrawn. This analysis indicates the QALYs that would have been accrued if post-SAE measurements had not been taken.
-
Linear interpolation between all EQ-5D measurements (see Figure 51, row 4) This analysis included all observed EQ-5D measurements and those imputed at 0, 3, 12 and 24 months, study exit and after SAEs. EQ-5D utility was assumed to change linearly between all measurements. However, this analysis included only those post-SAE measurements actually observed after SAEs and excluded post-SAE measurements that were imputed at time points other than scheduled/exit visits. This was done to evaluate the impact of imputing post-SAE utilities and modelling changes in EQ-5D utility. This analysis is therefore likely to overestimate the impact of SAEs by assuming that EQ-5D utility starts to decrease long before the SAE starts.
-
Subtracting the QALY loss from expected SAEs but not unexpected SAEs (compare with the base-case analysis, which included the impact of all SAEs, whether or not expected or unexpected) Within this analysis, the number of QALYs accrued in any given quarter was assumed to be equal to the estimate from the base-case analysis if there were either no SAEs that started, ended or spanned that quarter or if there were any expected SAEs, and equalled the number of QALYs accrued in analysis (3) if the only SAEs starting, ending or spanning that quarter were unexpected. This analysis aims to mirror the costing analysis, which included only the costs of expected SAEs, not unexpected events.
-
Doubling the QALY impact of SAEs for which there is no EQ-5D measurement within 2 months of the SA Within this analysis, the QALY impact of SAEs within each quarter was doubled and added on to the QALYs accrued in analysis (3) for any quarters in which there were any SAEs lacking observed post-SAE EQ-5D measurements, or any SAEs with a delay of > 2 months before the next observed EQ-5D measurement. This analysis aims to evaluate the impact of missing not at random data on post-SAE utilities and the impact of underestimating recovery rates for severe SAEs.
-
Doubling the QALY impact of all SAEs The impact of SAEs on the number of QALYs accrued in each quarter was calculated by subtracting the number of QALYs accrued in each quarter in analysis (3) from the number accrued in the base-case analysis. Within this analysis, the QALY impact of SAEs within each quarter was doubled and added on to the QALYs accrued in analysis (3) for all patients. This analysis aims to evaluate what the cost-effectiveness results might be if the analysis underestimated the number or QALY impact of SAEs.
Strengths and limitations of the base-case approach
The base-case analysis takes account of all observed EQ-5D measurements, allows for uncertainty around imputed values and attempts to make clinically realistic assumptions about changes in utility before and after SAEs that account for the frequently unobserved poor utility on the day of the SAE without overestimating the period for which the SAE affects utility.
However, this analysis assumes that the interval between the SAE and utility measurements varies at random and that there was no correlation between this interval and the severity of the SAE. In practice, this assumption may not be the case as patients with SAEs that have the greatest impact on utility may miss more IVAN clinic visits than those with mild SAEs and extremely minor SAEs (e.g. reductions in VA) may not be noticed until the patient attends the clinic for a VA check and completes EQ-5D on the same day. A positive relationship between the interval between the event and measurement would cause the mixed models to underestimate recovery rates, which would mean that QALY calculations overestimate EQ-5D utility on the day the SAE starts and (in many cases) overestimate the time until the patient recovers from the SAE. In many cases, these effects would cancel out, although for very severe SAEs, it may have the effect of underestimating the impact of SAEs on QALYs.
Furthermore, the analysis uses only the available data to impute missing post-SAE utilities and assumes that data are missing at random after controlling for the type and timing of SAEs and other variables. In practice, post-SAE utilities may be missing not at random, as, patients with more severe SAEs within any given category may be less likely to return to the IVAN clinic to complete EQ-5D. For example, within the CVD category, only seven of the 10 patients who had permanent stroke had any EQ-5D measurements after the stroke.
The analysis is also limited by missing data on post-SAE utility. Out of 230 non-fatal SAEs, there were only 62 EQ-5D measurements that occurred within 30 days and before the patient’s next set of SAEs.
Unit costs
| Resource (units) | Unit cost (£) | Reference |
|---|---|---|
| Ranibizumab (per 0.5-mg dose) | 742.17 | List price, British National Formulary51 |
| Bevacizumab (per 1.25-mg dose) | 49.00 | Price typically charged by the not-for-profit NHS manufacturer for bevacizumab in other trials, which was based on micro-costing work conducted by the manufacturer to estimate actual costs, including transport and delivery |
| Antibiotic drops (per injection in which antibiotic drops were given) | 1.75 | One 10-ml bottle of chloramphenicol 0.5%.51 Given the 28-day shelf life, a new bottle will be needed for each injection |
| Reagents and consumables used for FFA (per FFA) | 1.91 | The types and quantities of reagents and consumables used were obtained from two IVAN centres. Based on their responses, it was assumed that all patients required: 1 minim of 1–2% fluorescein (£0.3851); a 2-ml ampoule of sterile water (£0.1851); 1% tropicamide dilating drops (£0.47/injection51); and a Venflon (£0.63 based on price from manufacturer). In addition, 52.5% of patients (all those at one centre and 5% at the other) also receive a minimum of 2.5% phenylepinephrine (£0.4951) |
| Reagents used for injection (per injection) | 1.29 | The types and quantities of reagents and consumables used were obtained from two IVAN centres. Based on their responses, it was assumed that all patients receive a minim of 1% tropicamide dilating drops (£0.47/injection51); 52.5% of patients (all those that one centre and 5% at the other) also receive a minim of 2.5% phenylepinephrine (£0.4951); all require an average of 13 (range 0.8–25) ml povidone–iodine antiseptic solution (£0.0951), diluted in a 25-ml sachet of 0.9% normasol (£0.2551) in 50% of cases (all those at one centre); one minim of 0.4% benzoate (£0.4651) used in 50% of cases (all those at one centre); one minim of 0.5% proxymetacaine (£0.4951) used in 25% of cases (50% at one centre) |
| Staff involved in setting up and running clinics to administer and monitor VEGF injections | ||
| Administrator grade 3 (per hour) | 13.55 | Gross annual salary78 December 2010: £17,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577), but with no staff overheads or qualifications |
| Administrator grade 5 (per hour) | 18.94 | Gross annual salary78 December 2010: £24,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577), but with no staff overheads or qualifications |
| Assistant practitioner/unqualified nurse/HCA grade 4 (per hour) | 15.93 | Gross annual salary78 December 2010: £20,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on assistant therapy worker band 3 (hospital) (p. 18777) |
| Associate specialist grade (per hour) | 87.67 | PSSRU, 2011,77 p. 202 |
| Clerical officer grade 2 (per hour) | 12.39 | Gross annual salary78 December 2010: £16,100; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577), but with no staff overheads or qualifications |
| Clerk grade 2 (per hour) | 12.39 | Gross annual salary78 December 2010: £16,100; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577), but with no staff overheads or qualifications |
| Clerk grade 3 (per hour) | 13.55 | Gross annual salary78 December 2010: £17,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577), but with no staff overheads or qualifications |
| Consultant (medical) (per hour) | 109.40 | PSSRU 2011,77 p. 203 |
| Consultant microbiologist grade 8a–c (per hour) | 51.37 | Gross annual salary78 December 2010: £55,033; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on hospital pharmacist band 6,77 p. 186 |
| Eye clinic liaison officer grade 4 (per hour) | 15.93 | Gross annual salary78 December 2010: £20,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on assistant therapy worker band 3 (hospital) (p. 18777) |
| Eye clinic liaison officer grade 6 (per hour) | 22.81 | Gross annual salary78 December 2010: £29,500; ratio of salary to on-costs, qualifications, overhead and capital overheads based on assistant therapy worker band 3 (hospital) (p. 18777) |
| Eye clinic liaison officer grade 3 (per hour) | 13.61 | Gross annual salary78 December 2010: £17,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on assistant therapy worker band 3 (hospital) (p. 18777) |
| Foundation house officer 1 grade (per hour) | 28.86 | PSSRU, 2011,77 p. 199 |
| Foundation house officer 2 grade (per hour) | 35.46 | PSSRU, 2011,77 p. 200 |
| HCA/clinical support worker grade 2 (per hour) | 11.90 | PSSRU, 2011,77 p. 195 |
| HCA/clinical support worker grade 3 (per hour) | 13.76 | PSSRU, 2011,77 p. 187 |
| Hospital pharmacist grade 6 (per hour) | 29.88 | PSSRU, 2011,77 p. 186 |
| Macular coordinator grade 4 (per hour) | 16.32 | Gross annual salary78 December 2010: £21,200; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Maintenance and works employee grade 1 (per hour) | 11.09 | Gross annual salary78 December 2010: £14,400; ratio of salary to on-costs, qualifications, overhead and capital overheads based on maintenance: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Maintenance and works employee grade 2 (per hour) | 12.01 | Gross annual salary78 December 2010: £15,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on maintenance: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Maintenance and works employee grade 3 (per hour) | 14.01 | Gross annual salary78 December 2010: £18,200; ratio of salary to on-costs, qualifications, overhead and capital overheads based on maintenance: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Maintenance and works employee grade 4 (per hour) | 16.78 | Gross annual salary78 December 2010: £21,800; ratio of salary to on-costs, qualifications, overhead and capital overheads based on maintenance: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Microbiologist grade 6 (per hour) | 29.88 | Assumed to be equivalent to Hospital pharmacist |
| Microbiologist/pharmacist grade 5 (per hour) | 22.96 | Gross annual salary78 December 2010: £24,600; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on hospital pharmacist band 677 (p. 186) |
| Microbiologist/pharmacist grade 7 (per hour) | 33.88 | Gross annual salary78 December 2010: £36,300; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on hospital pharmacist band 677 (p. 186) |
| Nurse advanced grade 8a (per hour) | 42.92 | Gross annual salary78 December 2010: £44,600; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on nurse team manager band 777 (p. 191) |
| Nurse advanced grade 8b (per hour) | 49.75 | Gross annual salary78 December 2010: £51,700; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on nurse team manager band 777 (p. 191) |
| Nurse advanced grade 8c (per hour) | 58.89 | Gross annual salary78 December 2010: £61,200; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on nurse team manager band 777 (p. 191) |
| Nurse advanced grade 8d (per hour) | 70.63 | Gross annual salary78 December 2010: £73,400; ratio of salary to on-costs, qualifications, overhead and capital overheads and direct–indirect time based on nurse team manager band 777 (p. 191) |
| Ophthalmic Imaging Specialists grade 5 (per hour) | 21.52 | Gross annual salary78 December 2010: £22,700; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer grade 8a (per hour) | 42.95 | Gross annual salary78 December 2010: £45,300; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer grade 8b (per hour) | 51.67 | Gross annual salary78 December 2010: £54,500; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer grade 8c (per hour) | 61.91 | Gross annual salary78 December 2010: £65,300; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer/technician grade 4 (per hour) | 19.72 | Gross annual salary78 December 2010: £20,800; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer/technician grade 5 (per hour) | 21.52 | Gross annual salary78 December 2010: £22,700; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer/technician grade 6 (per hour) | 28.92 | Gross annual salary78 December 2010: £30,500; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Ophthalmic photographer/technician grade 7 (per hour) | 38.11 | Gross annual salary78 December 2010: £40,200; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Optician grade 7 (per hour) | 34.23 | Gross annual salary78 December 2010: £36,300; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Optometrist grade 6 (per hour) | 28.76 | Gross annual salary78 December 2010: £30,500; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Optometrist grade 7 (per hour) | 34.23 | Gross annual salary78 December 2010: £36,300; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Optometrist grade 8a (per hour) | 42.71 | Gross annual salary78 December 2010: £45,300; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Optometrist grade 8b (per hour) | 51.38 | Gross annual salary78 December 2010: £54,500; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Optometrist grade 8c (per hour) | 61.57 | Gross annual salary78 December 2010: £65,300; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Optometrist grade 8d (per hour) | 76.18 | Gross annual salary78 December 2010: £80,800; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Orthoptist grade 5 (per hour) | 23.19 | Gross annual salary78 December 2010: £24,600; ratio of salary to on-costs, qualifications, overhead and capital overheads based on dietitian band 577 (p. 184) |
| Orthoptist or optometry assistant grade 3 (per hour) | 14.04 | Calculated from salary – see AFC sheet |
| Photographer grade 5 (per hour) | 21.52 | Gross annual salary78 December 2010: £22,700; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Photographer grade 7 (per hour) | 38.11 | Gross annual salary78 December 2010: £40,200; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Receptionist grade 2 (per hour) | 12.39 | Gross annual salary78 December 2010: £16,100; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Registrar (per hour) | 47.70 | PSSRU 2011,77 p. 201 |
| Secretary/administrative coordinator grade 4 (per hour) | 16.32 | Gross annual salary78 December 2010: £21,200; ratio of salary to on-costs, qualifications, overhead and capital overheads based on administrator: clinical support worker band 2 (hospital) (p. 19577) band 2, but with no staff overheads or qualifications |
| Senior ophthalmic science practitioner grade 6 (per hour) | 28.92 | Gross annual salary78 December 2010: £30,500; ratio of salary to on-costs, qualifications, overhead and capital overheads based on radiographer band 577 (p. 185) |
| Sister grade 7 (per hour) | 37.45 | PSSRU 2011,77 p. 191 |
| Staff nurse grade 5 (per hour) | 26.62 | PSSRU 2011,77 p. 193 |
| Staff nurse grade 6 (per hour) | 32.48 | PSSRU 2011,77 p. 192 |
| Surgeon (per hour) | 108.72 | PSSRU 2011,77 p. 204 |
| Resources from the medical services form | ||
| GP (per surgery consultation lasting 11.17 minutes) | 31.00 | PSSRU 2011,77 pp. 148–9. Includes qualifications, but excluded direct staff costs (as GP nurses are costed separately) |
| General practice nurse (per consultation) | 13.18 | PSSRU 2011,77 p. 146. Corrected, based on revised PSSRU downloaded 1 February 2012 |
| District nurse (per consultation) | 20.00 | PSSRU 2011,77 p. 141. PSSRU 2011 does not give length of consultation, but PSSRU 2010 states that this is 20 minutes per home visit,127 so this figure is used to calculate the cost per district nurse consultation |
| GP home visit (per home visit lasting 23.4 minutes, including travel time) | 104.00 | PSSRU 2011,77 pp. 148–9. Includes qualifications, but excluded direct staff costs (as GP nurses are costed separately) |
| GP telephone consultation (per phone call) | 19.00 | PSSRU 2011,77 pp. 148–9. Includes qualifications, but excluded direct staff costs (as GP nurses are costed separately) |
| Nurse practitioner (per surgery consultation) | 25.00 | p. 147:77 cost per surgery consultation for nurse advanced. Text in table demonstrates that this includes nurse practitioners |
| General practice nurse telephone consultation (per phone call) | 6.04 | PSSRU 2011,77 p. 146. Calculated based on a 7.1-minute telephone call (the same as GPs average, based on p. 149 of PSSRU) |
| Average non-consultant outpatient consultation: mean cost across all face-to-face outpatient attendances recorded as non-consultant led (first and follow-up) | 59.36 | DH reference costs 2010–1175 sheet total – OPATT |
| Non-ophthalmology outpatient consultation: consultant (averaged across consultant led first or follow-up if non-admitted face to face) | 116.84 | DH reference costs 2010–1175 sheet total – OPATT |
| Non-ophthalmology outpatient consultation: consultant plus multiprofessional (consultant led: first or follow-up attendance multiprofessional non-admitted) | 147.89 | DH reference costs 2010–1175 sheet total – OPATT |
| Non-ophthalmology outpatient consultation: non-consultant (non-consultant led first or follow-up attendance non-admitted face to face) | 58.82 | DH reference costs 2010–1175 sheet total – OPATT |
| Non-ophthalmology outpatient consultation: non-consultant plus multiprofessional (non-consultant led: first or follow-up attendance multiprofessional non-admitted) | 81.87 | DH reference costs 2010–1175 sheet total – OPATT |
| Ophthalmology appointment (mean cost across all non-consultant-led ophthalmology outpatient attendances) | 62.55 | DH reference costs 2010–1175 sheet total – OPATT |
| Ophthalmology outpatient consultation: consultant (consultant-led follow-up attendance non-admitted face to face) | 77.21 | DH reference costs 2010–1175 sheet total – OPATT |
| Ophthalmology outpatient consultation: consultant plus multiprofessional (consultant led: follow-up attendance multiprofessional non-admitted) | 117.43 | DH reference costs 2010–1175 sheet total – OPATT |
| Ophthalmology outpatient consultation: non-consultant (non-consultant led follow-up attendance non-admitted face to face) | 56.54 | DH reference costs 2010–1175 sheet total – OPATT |
| Ophthalmology outpatient consultation: non-consultant plus multiprofessional (non-consultant led: follow-up attendance multiprofessional non-admitted) | 79.75 | DH reference costs 2010–1175 sheet total – OPATT |
| Ophthalmology outpatient consultation: average (total–weighted average across all face-to-face follow-up attendances) | 75.30 | DH reference costs 2010–1175 sheet total – OPATT |
| Accident and emergency attendance | 106.00 | PSSRU 2011,77 p. 91: accident and emergency treatments (not admitted) |
| Counsellor (per consultation) | 60.00 | PSSRU 2011,77 p. 41: cost per counselling session |
| CT scan (per attendance) | 110.12 | Weighted average cost across all non-inpatient CT scans. DH reference costs 2010–1175 sheets TPCTDIAGIM_OP, TPCTDIAGIM_Oth and TPCTDIAGIM_DA |
| DXA scan (per scan) | 71.76 | Mean cost of non-inpatient DXA scan in reference costs. DH reference costs 2010–1175 sheet TPCTDIAGIM_OP, TPCTDIAGIM_Oth and TPCTDIAGIM_DA |
| Echocardiography (per attendance) | 48.68 | Weighted average cost across all echocardiogram scans in reference costs. DH reference costs 2010–11,75 average across sheets |
| Hospital physiotherapy (per consultation) | 36.00 | PSSRU77 p. 181: using data from the NHS reference costs |
| MRI scan (per scan) | 173.22 | Weighted average cost across all outpatient MRI scans in reference costs. DH reference costs 2010–1175 sheets TPCTDIAGIM_OP, TPCTDIAGIM_Oth and TPCTDIAGIM_DA |
| Occupational therapist | 60.00 | p. 182:77 using data from the NHS reference costs, the mean average cost for a non-consultant led (non-admitted) follow-up occupational therapy attendance in 2010 was £60, with the minimum for 25% of services being £38 and the maximum being £65. Costs have been uprated using the HCHS Pay and Prices Inflator |
| PET scan (per attendance) | 232.15 | Weighted average across all nuclear medicine scans in reference costs, across direct access, outpatient and other. DH reference costs 2010–1175 sheets TPCTDIAGIM_OP, TPCTDIAGIM_DA and TPCTDIAGIM_Oth |
| Pharmacist (per consultation) | 4.69 | Cost was based on the crude average cost per hour of direct patient contact between community and hospital pharmacist. Assumed consultation lasts for 2 minutes 30 seconds, based on a survey in 1992128 |
| Physiotherapist (per consultation): community or hospital | 41.50 | Mean price across hospital and community physiotherapy consultations: PSSRU77 pp. 133 and 181 |
| Podiatrist or chiropodist (per consultation) | 39.00 | PSSRU 2011,77 p. 136: 9.4 community chiropodist/podiatrist using data from the NHS reference costs,75 the mean average cost for a contact in chiropody/podiatry services for 2010–11 was £39, with the minimum range for 25% of services being £31 and the maximum £45. Costs have been inflated using the HCHS Pay and Prices Inflator |
| Practice nurse home visit | 19.89 | Assumed to last 11.4 minutes, plus 12 minutes’ travel time, like GP visit, but costed based on practice nurse time |
| Scan, unspecified (per attendance) | 96.51 | Weighted average cost across all scans in DH reference costs 2010–1175 sheets TPCTDIAGIM_OP, TPCTDIAGIM_Oth and TPCTDIAGIM_DA |
| Ultrasound (per attendance) | 54.73 | Weighted average cost across all outpatient ultrasound scans in DH reference costs 2010–1175 sheets TPCTDIAGIM_OP, TPCTDIAGIM_Oth and TPCTDIAGIM_DA |
| Walk-in clinic (per attendance) | 41.00 | PSSRU 2011,77 p. 91: walk-in services (not admitted) |
| X-ray (per consultation) | 34.48 | X-rays have been removed from reference costs since 2007–8. Cost was based on the weighted average of codes RA28Z, RA29Z, RA30Z and RA31Z across direct access (TPCTRADGYDA), outpatient (TPCTRADGYOP) and other (TPCTRADGYOth) from 2006 to 2007, NHS and trusts combined (www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/documents/digitalasset/dh_118336.xls), inflated to 2010–11 values using HCHS77 |
The total cost of medication changes was calculated relative to baseline: patient’s daily medication cost on day 0 was set to zero and any changes to medication that occurred during the trial increased or decreased the daily medication cost. Regardless of the timing of visit 24, medication costs were calculated over a 730-day period and quarterly costs of medication changes were calculated based on exact dates of changes. The cost of short courses of drugs licensed for expected AEs/SAEs (e.g. antibiotics) that started and ended in the same month was included in the analysis, regardless of date, with any short courses prescribed after day 730 being included in quarter 8.
| (Serious) AE name | Cost/bed-day,a £ | Cost/admission,b £ | Reference |
|---|---|---|---|
| Endophthalmitis | 413.27 | 1040.80 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for non-surgical ophthalmology, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Uveitis | 413.27 | 1040.80 | |
| Retinal detachment | 413.27 | 1040.80 | |
| RPE tear | 413.27 | 1040.80 | |
| Retinal vein occlusion | 413.27 | 1040.80 | |
| Other ocular admission | 413.27 | 1040.80 | |
| Cataract traumatic | 631.79 | 1239.82 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for cataract procedures, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| IOP increased | 767.28 | 1475.20 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for glaucoma, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Angina pectoris | 996.68 | 3582.99 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for percutaneous coronary interventions or CABG, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Coronary artery bypass | 996.68 | 3582.99 | |
| Cerebrovascular accident | 316.04 | 2267.47 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for stroke-related HRGs, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Cardiac arrest | 336.41 | 1007.69 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for cardiac arrest, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Cardiac failure | 301.13 | 1641.66 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for HF, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Left-ventricular failure | 301.13 | 1641.66 | |
| Cardiovascular disorder | 301.13 | 1641.66 | |
| DVT | 314.22 | 1170.18 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for pulmonary embolus or DVT, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Pulmonary embolism | 314.22 | 1170.18 | |
| Transient ischaemic attack | 347.76 | 780.17 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for TIA, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| MI | 331.88 | 1236.05 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for confirmed or suspected acute MI, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Haemorrhage | 35.69 | 968.81 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for gastrointestinal bleeds, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Urinary tract infection | 415.81 | N/A | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for kidney or urinary tract infections, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Accident and Emergency admission < 1 day | 140.95 | N/A | DH reference costs 2010–11,75 Index sheet. Unit cost for Accident and Emergency Services: leading to Admitted |
| Upper respiratory tract infection | 470.55 | 535.03 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of related HRGs for non-elective inpatient stays for acute upper respiratory tract infection and common cold, across sheets TPCTNEI_L, TPCTNEI_L_XS and TPCTNEI_S |
| Unexpected SAE | 506.49 | 1667.78 | DH reference costs 2010–11.75 Mean cost/bed-day and mean cost per hospital admission of all elective and non-elective admissions: TPCTEI, TPCTEIXS, TPCTNEI_L, TPCTNEI_L_XS, TPCTNEI_S |
| Arthralgia | 506.49 | 1667.78 | |
| Nausea | 506.49 | 1667.78 |
Economic analysis: additional detail on the methods for analysing costs and quality-adjusted life-years
Methods for assigning consultation costs
Microcosting work gave estimates of the cost of different types of consultation for administering/monitoring anti-VEGF therapy for a sample of clinics of differing sizes (see Chapter 2). As the number of patients recruited to IVAN does not reflect the number of patients that each centre treats in routine clinical practice, average costs were applied to all patients regardless of which centre they attended. However, to allow for the substantial variability in unit costs between centres, we drew repeated values from the distribution of clinic costs for each patient in each of the 100 imputed data sets. This was conducted by assigning a weight to each clinic equal to the number of patients attending that clinic in the average week, divided by the total number of patients attending all clinics in the average week. We then generated random numbers from a uniform distribution for each patient in each imputed data set for each type of clinic. Random numbers were compared against the weights to determine which clinic cost each patient was assigned in that imputed data set. As patients generally attend the same clinic throughout their course of treatment, unit costs were assumed to be constant over the 24 visits within each patient for any given imputed data set. For example, if the weight assigned to Liverpool’s one-stop clinic was 5% and the weight assigned to Bristol’s monitoring-only clinic was 2%, the patient would be assigned the cost from Liverpool in imputation 1 if their random number was < 0.05, and assigned to the cost from Bristol in imputation 2 if their random number was between 0.07 and 0.05.
Methods for dealing with missing resource-use data
The amount of missing data on resource use was minimised by asking patients to provide details of all medication changes and ambulatory consultations since their last visit at every IVAN consultation and by collecting additional information from SAE forms and by telephone. However, these measures also meant that it was generally not possible to identify which patients had accrued NHS costs but not reported them at clinic visits. In general it was therefore assumed that the ‘Use of Medical Services’ form captured all ambulatory consultations occurring during the trial period regardless of missed visits or the timing of visit 24. However, if the patient answered ‘yes’ to the question ‘Has the patient made any visits to any hospital, accident and emergency department, GP or NHS walk-in centre regarding (non-)ocular symptoms since their last visit?’ but did not complete the ‘Use of Medical Services’ form at that visit, one episode of missing resource-use data was imputed by randomly drawing costs from another medical services form for that patient (or across all patients if this patient had no other medical services use), with independent draws made for each patient in each of the 100 imputed data sets. Multiple imputation was not used for missing resource-use data, as resource-use data were missing for only 2% (330/13,398) of attended visits.
Handling censoring using Kaplan–Meier sample averaging
Kaplan–Meier sample averaging assumes that censoring occurs at discrete time points on the boundaries of each time interval. As IVAN participants could withdraw from the trial at any time (including part of the way through a quarter), we excluded patients withdrawing during quarter ‘q’ from the mean costs for this quarter, in order to avoid the bias that can arise from including partially observed quarters. 84 This recognised84 variant of KMSA captures one of the main advantages of another censoring analysis method [inverse probability weighting (IPW129)], while excluding only 45 partially observed patient quarters (0.9%).
For QALYs and medication/medical service costs, quarters were excluded from the analysis if the patient withdrew before the end of the quarter; for example, if a patient withdrew 0.45 years after randomisation then the costs and medication/medical service costs they accrued during quarter 1 were included in the analysis, but those accrued in quarter 2 were excluded from averages, as they were incomplete. Quarterly drug costs and administration/monitoring costs cover the three visits included in that quarter; for example, quarter 11 includes resource use from visits 0, 1 and 2, whereas quarter 2 includes resource use from visits 3–5. Patients withdrawing after the third visit of any given quarter was due but before the end of that quarter (e.g. those withdrawing between visits 5 and 6), were considered to have drug and administration/monitoring costs fully observed for that quarter in order to maximise the amount of data used in the analysis. For example, the patient withdrawing 0.45 years after randomisation would be included in the analysis of drug and administration/monitoring costs for quarters 1 and 2, although only their data for quarter 1 were included in the analysis of QALYs and medication/medical service costs.
Methods for estimating mean costs and quality-adjusted life-years
The analysis outlined below was initially conducted with interactions for all four components of net benefit: drug cost; administration/monitoring costs; medication and medical service use costs; and QALYs. The analysis including interactions was used to assess the magnitude and statistical significance of interactions, and identify for which of the four components of net benefit it could be assumed that drug and treatment regimen have additive effects. This approach avoids reducing statistical power by allowing for interactions unnecessarily,43,49 while ensuring unbiased43,85 estimation of costs and QALYs for each treatment regimen.
Within the base-case analysis, interactions were included for those components of costs or QALYs where interactions were either statistically significant or had an absolute magnitude larger than either the main effect for treatment regimen or the main effect for drug. However, interactions that were both non-significant and smaller than both main effects were excluded from the analysis to avoid loss of statistical power. Lack of statistical significance was not considered sufficient grounds to ignore interactions, as ignoring genuine interactions introduces bias43,85 and can give misleading conclusions. 130 We evaluated different methods for identifying important interactions on simulated trial data, which demonstrated that including interactions that are larger than main effects outperformed using statistical significance or Bayesian/Akaike information criteria for identifying influential interactions. The approach used had a lower probability and a lower opportunity cost from failing to adopt the treatment that maximises true net benefits.
As the economic evaluation focuses on estimation rather than hypothesis testing and inference has been argued to be irrelevant to decision-making based on the results of economic evaluation,131 no adjustment was made for multiple comparisons.
The base-case economic evaluation and/or sensitivity analyses were analysed using the algorithm shown below:
-
Multiple imputation was used to impute missing utility data (see Chapter 2), generating 100 imputed data sets. The 100 data sets also had different randomly sampled values for missing resource use, mixed-model recovery rate coefficients, and costs for injection and monitoring consultations.
-
Bootstrapping132 was used to allow for uncertainty and correlations between quarterly costs and QALYs and to avoid parametric assumptions. The following steps were initially conducted for the original trial sample in each of the 100 imputed data sets, as estimates for the original sample represent the best estimate of likely outcomes. 132 One hundred and thirty bootstrap samples were then drawn with replacement from each of the 100 data sets (giving a total of 13,000 bootstraps) and the following steps were repeated on each bootstrap of each imputed data set to provide an empirical estimate of the distribution of costs and QALYs for each study arm.
-
Quarterly QALYs were estimated using OLS regression across all patients alive at the start of that quarter, controlling for baseline utility and treatment allocation. Analysis of QALYs in quarter 1 included a dummy for drug but not treatment regimen or an interaction term. QALYs in subsequent quarters included both treatment dummies, plus an interaction term if interactions for QALYs were statistically significant or were larger than one or more main effect. Baseline utility was included in all analyses of QALYs to adjust for any imbalance in baseline utility, thereby avoiding bias, allowing for regression to the mean and increasing precision. 86
-
Mean quarterly drug costs, administration/monitoring costs and medication/medical service costs were analysed using OLS regression for each of the four arms, across all patients alive at the start of that quarter. Interaction terms were included for those cost components with significant interactions for at least one quarter and those where interactions change the conclusions, although the interaction term and the term for treatment regimen were omitted for quarter 1.
-
A Kaplan–Meier survival curve for all-cause mortality in each group was calculated as described in Chapter 2 (assuming a common rate of deaths unrelated to study medication across all treatment arms). The resulting proportions of patients alive at the start of each quarter was multiplied by the quarterly costs and QALYs for each arm in each quarter (KMSA) to give overall mean quarterly costs and QALYs in each arm.
-
Costs and QALYs accrued in the second year of the trial were discounted at 3.5% per annum to allow for time preference, in line with current recommendations. 42,87
-
Steps a–d were repeated for each imputed data set to provide point estimates. An empirical estimate of the distribution of costs and effects for each arm were obtained by repeating steps a–d for the 130 bootstrap replicates from each of the 130 data sets.
-
-
The 100 imputed data sets were combined using Rubin’s rule77 to estimate mean costs and QALYs for each of the four study arms and SEs around each mean. The empirical distribution across the 13,000 replicates was used to generate cost-effectiveness acceptability curves and scattergraphs on the cost-effectiveness plane and estimate the value of collecting additional information. Cost-effectiveness acceptability curves are presented for the four pairwise comparisons defined in Chapter 2 by plotting the proportion of the 13,000 replicates that find each treatment to have greater net benefits than its comparator against ceiling ratio. Cost-effectiveness acceptability curves were also produced using a net benefit framework to present the likelihood that each intervention produces highest net benefits across all four arms at different ceiling ratios. In the construction of cost-effectiveness acceptability curves, it was assumed that the NHS has symmetrical preferences for gains and losses – i.e. that the minimum savings that they would require to accept the loss of one QALY are equal to the maximum cost they would be willing to pay to gain one QALY.
-
Conclusions were drawn using the framework described in Chapter 2.
-
The impact of heterogeneity was evaluated by exploring the impact of the five prespecified clinical subgrouping variables (see Chapter 2) and an additional variable indicating whether or not VA in the study eye was > 5 letters better than the fellow eye at baseline. The impact of subgroups was evaluated using OLS regression predicting drug costs, administration/monitoring costs, medication/medical service costs and QALYs accrued in each quarter, conditional on drug, treatment regimen, subgroup, an interaction between drug and subgroup, an interaction between treatment regimen and subgroup and (for variables in which interactions were included in the base case) the interaction between drug and treatment regimen and an interaction between drug, treatment regimen and subgroup; the impact of treatment regimen and interactions were omitted for quarter 1 and imputed data sets were combined using the min command. A Bonferroni correction was made to allow for the fact that multiple p-values were reviewed across the six subgroups, four variables and eight quarters, which meant that statistical significance was evaluated at the 0.0001 level. All subgroup analyses should be considered hypothesis generating rather than confirmatory in nature.
Appendix 5 Statistical analysis plan: analysis of 2-year results
List of abbreviations
- AE
- adverse event
- AIC
- Akaike information criterion
- AMD
- age-related macular degeneration
- ANCHOR
- ANti-vascular endothelial growth factor antibody for the treatment of predominantly Classic CHORoidal neovascularization in age-related macular degeneration
- ATE
- arterial thrombotic event
- BCVA
- best corrected distance visual acuity
- BRAMD
- comparison of bevacizumab (Avastin) and ranibizumab (Lucentis) in exudative age-related macular degeneration
- CARF
- Central Angiographic Resource Facility
- CATT
- Comparison of Age-related macular degeneration Treatment Trials
- CCS
- Canadian Cardiovascular Society
- CI
- confidence interval
- CMA
- cost-minimisation analysis
- CNV
- choroidal neovascularisation
- CRF
- case report form
- CTEU
- Clinical Trials and Evaluation Unit
- CUA
- cost–utility analysis
- CVD
- cardiovascular disease
- df
- degree of freedom
- DMSC
- Data Monitoring and Safety Committee
- DNA
- deoxyribonucleic acid
- DVT
- deep-vein thrombosis
- EQ-5D
- European Quality of Life-5 Dimensions
- ETDRS
- Early Treatment of Diabetic Retinopathy Study
- FFA
- fundus fluorescein angiography
- GA
- geographic atrophy
- GEFAL
- French Evaluation Group Avastin versus Lucentis
- GMR
- geometric mean ratio
- GP
- general practitioner
- HF
- heart failure
- HR
- hazard ratio
- HRG
- Healthcare Resource Group
- HRQoL
- health-related quality of life
- HUI3
- Health Utilities Index, version 3
- ICER
- incremental cost-effectiveness ratio
- IMP
- investigational medicinal product
- IOP
- intraocular pressure
- IQR
- interquartile range
- IVAN
- Inhibit VEGF in Age-related choroidal Neovascularisation
- KMSA
- Kaplan–Meier sample averaging
- logMAR
- log(minimum angle of resolution)
- LUCAS
- LUcentis Compared to Avastin Study
- M
- cost per monitoring consultation excluding FFA and any resources associated with the intravitreal injection
- MacDQoL
- Macular disease Dependent Quality of Life
- MacTSQ
- Macular disease Treatment Satisfaction Questionnaire
- MANTA
- Multicenter ANti-VEGF Trial in Austria
- MARINA
- Minimally classic/occult trial of the Anti-VEGF antibody Ranibizumab In the treatment of Neovascular Age-related macular degeneration
- MD
- mean difference
- MedDRA
- Medical Dictionary for Regulatory Activities
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MI
- myocardial infarction
- nAMD
- neovascular age-related macular degeneration
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NVA
- near visual acuity
- NYHA
- New York Heart Association
- OCT
- optical coherence tomography
- OLS
- ordinary least squares
- OR
- odds ratio
- PDT
- photodynamic therapy
- PED
- pigment epithelial detachment
- PIER
- a study of rhuFAB V2 [ranibizumab] in subjects with subfoveal choroidal neovascularization secondary to age-related macular degeneration
- prn
- pro re nata (as needed)
- QALY
- quality-adjusted life-year
- QUB
- Queen’s University Belfast
- RAP
- retinal angiomatous proliferation
- RCT
- randomised controlled trial
- RPE
- retinal pigment epithelium
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SE
- standard error
- SRF
- subretinal fluid
- TCC
- Trial Coordinating Centre
- TSC
- Trial Steering Committee
- VA
- visual acuity
- VAT
- value added tax
- VEGF
- vascular endothelial growth factor
- VIEW
- VEGF Trap-Eye: Investigation of
- (1 and 2)
- Efficacy and Safety in Wet AMD
- VISION
- pegaptanib for neovascular age-related macular degeneration
- VPDT
- Verteporfin PhotoDynamic Therapy cohort study