
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 07/37/47. The contractual start date was in April 2010. The draft report began editorial review in March 2015 and was accepted for publication in July 2015. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Dr Tiffany M Osborn declares grants from ImaCor Inc. and Cheetah Medical during the trial.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2015. This work was produced by Mouncey et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Background and rationale
The incidence of infections severe enough to cause systemic sepsis and septic shock in adults is estimated to range from 56 to 91 per 100,000 population per year. 1 Affected patients have high mortality, morbidity and resource utilisation. 2–5 Efforts to improve care for these patients have been hampered by multiple factors including limited evidence regarding the timing and delivery of therapies. It has been suggested that there are ‘golden hours’ in the initial management of emerging septic shock during which prompt, rigorous, protocolised care may reduce unwanted consequences and improve clinical outcomes.
In 2001, Rivers et al. 6 reported the results of a single-centre, randomised controlled trial, which took place in the USA. This trial investigated the delivery of 6 hours of early goal-directed therapy (EGDT), with pre-determined haemodynamic goals, to patients presenting at an emergency department (ED) with emerging septic shock. EGDT, compared with usual resuscitation, significantly reduced hospital mortality (from 46.5% to 30.5%) and shortened hospital length of stay for survivors. The rationale for EGDT is that many patients with emerging septic shock have global tissue hypoxia that is not adequately identified using traditional resuscitation end points (such as blood pressure) and that rapid correction of occult tissue hypoxia leads to improved survival. Accordingly, resuscitation incorporating EGDT incorporates the invasive measurement of central venous oxygen saturation (ScvO2) to detect occult global tissue hypoxia. EGDT aims to optimise tissue oxygen transport by continuous monitoring of pre-specified physiological targets – central venous pressure, mean arterial pressure and ScvO2 – to guide delivery of intravenous fluids, vasoactive drugs and packed red blood cell transfusions.
The plausible biological rationale for EGDT, combined with the results of the Rivers et al. 6 trial and some observational studies,7–12 led to its recommendation for the initial management of patients with septic shock by the Surviving Sepsis Campaign guidelines for the resuscitation and management of severe sepsis13–15 and incorporation into the associated ‘bundles’ of care. 16 However, adoption of, and compliance with, these resuscitation and management bundles has been limited. 2,17
The lack of adoption of EGDT has primarily been due to concerns about the external validity of results from a single-centre trial, generalisability into other health-care settings, the complexity of delivery of EGDT, potential risks of the components of EGDT and the resources required for implementation. 18,19
Reports of successful implementation of EGDT have identified important enablers, including leadership (local champion); communication, education and training; buy-in to the protocol; provision for protocol transition from ED to the intensive care unit (ICU); and locally determined delivery. 20,21
Resuscitation practice in the UK, though not standardised across hospitals, usually involves intravenous fluid and vasoactive drug administration, with the intensity of resuscitation typically being determined by clinical assessment. Therapeutic strategies to improve ScvO2 are not routinely employed during resuscitation in UK hospitals.
Despite its promising results, the Rivers et al. 6 trial can be considered only as ‘proof of concept’, and it is necessary to establish whether or not these results are generalisable to the UK NHS. The sample size was small (n = 263 patients) and single-centre studies often reflect local, and sometimes unique, processes of care. It may not be possible to replicate the results of single-centre studies in larger, multicentre studies, and important examples of this have recently been reported in the critical care literature. 22
To address these concerns, three research teams collaborated to conduct multicentre trials of EGDT in the USA (Protocolized Care for Early Septic Shock: ProCESS),23 Australasia (Australasian Resuscitation In Sepsis Evaluation: ARISE)24 and England (Protocolised Management In Sepsis: ProMISe). The three trials employed harmonised methods25 and, following full reporting, data will be pooled into one individual patient data meta-analysis. 26 Both ProCESS23 and ARISE24 have published their results (in March 2014 and October 2014, respectively) and reported no benefit of EGDT. However, both trials reported mortality in the usual-resuscitation group that was lower than anticipated (ProCESS, 60-day in-hospital mortality, 18.9% observed, 30–46% anticipated; ARISE, 90-day mortality, 18.8% observed, 38% anticipated). Consequently, neither trial could exclude, with 95% confidence, the potential for a 20% relative reduction in 90-day mortality for EGDT compared with usual resuscitation [ProCESS, relative risk 0.94, 95% confidence interval (CI) 0.77 to 1.15; ARISE, relative risk 0.98, 95% CI 0.80 to 1.21].
Aim
The overall aim of the ProMISe trial was to test the hypothesis that EGDT is superior, in terms of both its clinical effectiveness and its cost-effectiveness, to usual resuscitation in patients presenting with early septic shock to NHS EDs in England.
Objectives
Primary
The primary objectives of the ProMISe trial were:
-
to estimate the effect of EGDT compared with usual resuscitation on all-cause mortality at 90 days
-
to compare incremental cost-effectiveness at 1 year of EGDT with usual resuscitation.
Secondary
The secondary objectives of the ProMISe trial were to compare EGDT with usual resuscitation for:
-
requirement for, and duration of, critical care unit organ support
-
length of stay in the ED, critical care unit and acute hospital
-
health-related quality of life at 90 days and at 1 year
-
resource use and costs at 90 days and at 1 year
-
all-cause mortality at 28 days, at acute hospital discharge and at 1 year
-
estimated lifetime incremental cost-effectiveness.
Chapter 2 Methods
Trial design
ProMISe was a pragmatic, open, multicentre, parallel-group randomised controlled trial with an integrated economic evaluation.
Research governance
ProMISe was sponsored by the Intensive Care National Audit & Research Centre (ICNARC) and co-ordinated by the ICNARC Clinical Trials Unit (CTU). An ethics application was made to the North West London Research Ethics Committee 1 on 4 May 2010 and a favourable opinion was received on 2 August 2010 (reference number 10/H0722/42).
Global NHS permissions were obtained from London North West Comprehensive Local Research Network (CLRN) on 8 September 2010 and local NHS permissions were obtained from each participating NHS hospital trust. A clinical trial site agreement, based on the model agreement for non-commercial research in the NHS, was signed by each participating NHS hospital trust and the sponsor (ICNARC).
The National Institute for Health Research (NIHR) Clinical Research Network (CRN) Portfolio details high-quality clinical research studies that are eligible for support from the NIHR CRN in England. The trial was adopted onto the NIHR CRN Portfolio on 11 July 2011.
To ensure transparency, the trial was registered for an International Standard Randomised Controlled Trial Number (ISRCTN). Registration was confirmed on 19 November 2009 (ISRCTN36307479).
Following guidelines from the NIHR, a Trial Steering Committee (TSC), with a majority of independent members, was convened to oversee the trial on behalf of the funder (NIHR) and the sponsor (ICNARC). The TSC met at least annually during the trial and comprised an independent chair (an experienced triallist); independent lay members (representing patient perspectives); independent clinicians (specialising in critical care medicine and emergency care medicine); the chief investigator (KR); and a co-investigator (JB) representing the Trial Management Group (TMG).
Additionally, an independent Data Monitoring and Ethics Committee (DMEC) was convened to monitor trial data and ensure the safety of trial participants. The DMEC met at least annually during the trial and comprised two expert clinicians specialising in critical care medicine and emergency care medicine, and was chaired by an experienced statistician.
Management of the trial
The trial manager (PM) was responsible for the day-to-day management of the trial with support from the research assistant (RJ), data manager (JT) and trial statistician (SP). The TMG was responsible for overseeing the day-to-day management of the trial and comprised the chief investigator (KR), SH, TO and the co-investigators (DB, JB, TC, DH, MS and DY). The TMG met regularly throughout the trial to ensure adherence to the trial protocol and to monitor the conduct and progress of the trial.
Network support
To maintain the profile of the trial, regular updates on trial progress were provided at quarterly meetings of the NIHR CRN Critical Care Specialty Group and at local CLRN meetings. In addition, updates were provided at national meetings, such as the Annual Meeting of the Case Mix Programme and the UK Critical Care Research Forum.
Design and development of the protocol
As part of the international collaboration to evaluate the effectiveness of EGDT for managing patients with septic shock, the ProMISe TMG worked closely with the ProCESS and ARISE TMGs in developing the trial protocol to ensure common standards, design elements and the data variables collected across the three trials. This will enable a prospective individual patient data meta-analysis to be conducted on completion and publication of all three trials. 25
Individuals representing emergency medicine, acute medicine and critical care medicine from NHS hospitals across the UK were invited to attend a meeting to discuss the trial protocol and the proposed intervention, EGDT. The meeting took place on 16 March 2010 and was attended by 91 clinicians from 54 NHS hospitals. The chairperson of the ARISE TMG also attended the meeting to share experiences in the set-up and ongoing delivery of the ARISE trial in Australasia.
Following the meeting, minor changes were made to Rivers’ EGDT protocol6 as follows:
-
arterial catheter – insertion of an arterial catheter was changed from being mandated to recommended
-
physiological goals – rather than a range for physiological goals, clinicians agreed that they preferred a minimum physiological goal, with no upper limit, for both central venous pressure and blood pressure
-
blood pressure – a minimum physiological goal was agreed for systolic blood pressure as well as for mean arterial pressure to allow for variation in practice across NHS hospital trusts.
The trial protocol was approved by the TSC and DMEC.
Amendments to the trial protocol
Following receipt of a favourable opinion of the trial protocol from the research ethics committee on 2 August 2010, five substantial amendments were submitted and received favourable opinion. In summary, these were as follows.
Amendment 1 (March 2011): the consultee consent form was amended to the consultee agreement form to clarify that, in accordance with the Mental Capacity Act 2005,27 personal/professional consultees were being asked for their agreement, rather than their consent, for the patient to participate in the trial. A telephone agreement form was added to document cases where personal/professional consultee agreement was obtained via telephone and an emergency consent form was added to document cases where emergency consent was obtained from an independent clinician.
Amendment 2 (September 2011): in consultation with the research ethics committee, guidance was added for situations where a patient did not regain the mental capacity to provide informed consent (retrospectively) to continue participating in the trial; where possible, agreement was to be sought from a personal consultee. The exclusion criterion – immunosuppressive agents for uncured cancer or immunosuppression for organ transplantation or from systemic disease – was removed following review by the trial clinicians, who felt that this was an important group of patients who potentially might benefit from an intervention for septic shock. In addition, minor semantic changes were made to the trial protocol and the patient follow-up letter.
Amendment 3 (January 2012): the letter to the patient’s general practitioner informing them of the patient’s participation in the trial was amended for use in cases where the patient was known to have died. The patient follow-up letters were amended to be specific to the follow-up time point, namely 90 days and 1 year post randomisation. Following feedback from patients, relatives and clinicians, a short version of the patient information sheet was produced which provided salient information about the trial.
Amendment 4 (November 2012): the exclusion criterion ‘known to be participating in an interventional study’ was removed following review by the TMG; it was agreed that patients could be co-enrolled into two interventional studies if, after careful consideration, there were no concerns about patient safety, risk of biological interaction or the scientific integrity of the trial. Local principal investigators (PIs) were advised to contact the trial on a case-by-case basis to discuss the co-enrolment of patients. In addition, minor semantic changes were made to the trial protocol and the consent/consultee agreement forms.
Amendment 5 (November 2013): a newsletter for patients participating in the trial was produced and sent with the follow-up questionnaires at 90 days and at 1 year post randomisation. Permission was also sought from the research ethics committee to e-mail follow-up questionnaires to patients, if requested.
NHS support costs
Trials in emergency and critical care are challenging and expensive to conduct. Unlike in other areas of health care, such as oncology, recruitment cannot take place solely within usual office hours. Resources are needed to enable screening and recruitment 24 hours per day, 7 days per week. Patients with severe sepsis and emerging septic shock are more likely to present at the ED in the afternoon through to late at night. Another challenge of emergency and critical care research is the informed consent process, which often has to be completed within a very short time frame, as treatments are often time limited. For ProMISe, consent and randomisation occurred within 2 hours of the patient meeting eligibility. Critically ill patients usually lack the mental capacity to be able to provide informed consent prior to randomisation, in which case it is necessary to involve a personal or professional consultee in accordance with the Mental Capacity Act 2005. 27 Senior, experienced staff are needed to be able to assess the patient’s mental capacity and to be able to effectively communicate information about the trial to the patient and/or their relatives in a stressful situation.
To this end, resources equivalent to 0.9 whole-time equivalent band 8 research nurse NHS support costs were successfully agreed with the London North West CLRN on 3 December 2010. Resources were based on an estimated 22 eligible admissions per site per year, of whom 14 would be recruited and 7 would be randomised to receive EGDT. Using these recommendations, participating sites, assisted by the TMG, negotiated resources required locally for the trial with their respective research and development departments and CLRNs.
Trial equipment
The central venous catheters with ScvO2 monitoring capability (PreSepTM central venous oximetry catheter), for use in patients allocated to the EGDT (intervention) group, were purchased from Edwards Lifesciences Ltd (Newbury, Berkshire) and distributed to participating sites by the ICNARC CTU. Edwards Lifesciences loaned the VigileoTM monitor required for continuous monitoring of ScvO2 to each participating site for the duration of the trial. Both the PreSepTM central venous oximetry catheter and the VigileoTM monitor are manufactured by Edwards Lifesciences and are commercially available and licensed for use in the UK. Each participating site received training in the use of the PreSepTM central venous oximetry catheter and the VigileoTM monitor, provided free of charge by Edwards Lifesciences. In addition, Edwards Lifesciences provided 24-hour, 7-days-per-week telephone support for any technical queries. Edwards Lifesciences had no further role in the trial.
Patient and public involvement
Engagement with patients was vital to the successful conduct of the trial. Two former critical care patients were independent members of the TSC and provided input into the conduct of the trial, including reviewing literature to be given to patients and their families (e.g. patient information sheets and patient newsletters).
Participants: sites
The trial aimed to recruit a representative sample of 48 NHS hospitals in the UK. The criteria for inclusion were:
-
EGDT, including continuous monitoring of ScvO2, was not already part of usual resuscitation for patients presenting with severe sepsis/septic shock
-
agreement from senior clinical staff in emergency care, acute care and critical care to recruit eligible patients and to adhere to the trial protocol – sites were asked to identify a ‘champion’ from each specialty to promote the trial locally
-
identification of a local PI and a dedicated research nurse to take responsibility for the local conduct of the trial
-
provision of timely data on recruited patients entered onto a secure, dedicated, electronic case report form.
Invitations for expressions of interest were sent to lead clinicians in acute medicine, emergency medicine and critical care medicine at NHS hospitals throughout the UK. Invitations were also circulated via the College of Emergency Medicine, the Society of Acute Medicine and the Intensive Care Society. The trial was promoted through presentations at national meetings of all three organisations.
Site initiation
Prior to opening sites to recruitment, regional site initiation meetings were held across England. The purpose of these meetings was to present the background and rationale for the ProMISe trial and to discuss delivery of the protocol, including screening and recruiting patients; delivery of the intervention, EGDT; data collection and validation; and safety monitoring. The operational challenges of conducting the trial at sites were discussed in detail, including strategies for ensuring effective communication between the ED, the acute care units/ward and the critical care unit. The PI from each participating site was required to attend the meeting. A representative from Edwards Lifesciences also attended the meeting to provide training in the use of the PreSepTM central venous oximetry catheter and the VigileoTM monitor to be used as part of delivery of the intervention, EGDT.
Investigator site file
An investigator site file was provided to all participating sites. This contained all essential documents for the conduct of the trial and included the approved trial protocol; all relevant approvals (e.g. local NHS permissions); a signed copy of the clinical trial site agreement; the delegation of trial duties log; copies of the approved patient information sheets, patient consent form and personal/professional consultee agreement forms; and all standard operating procedures, for example for screening participants, for obtaining informed consent or consultee agreement, for randomising patients, for delivery of the intervention and for collecting and entering data onto the secure, dedicated, electronic case report form. The site PI was responsible for maintaining the investigator site file.
Site management
Communication
The trial manager (PM), with support from the data manager (JT) and research assistant (RJ), maintained close contact with the PI and trial team at participating sites by e-mail and telephone throughout the trial.
Teleconferences were held, initially every month and then every 2 months, with trial teams at participating sites. The purpose of these was to provide updates on trial progress and to provide a forum for site teams to ask questions, discuss local barriers and challenges to the conduct of the trial and to share successes and best practice. Notes, including ‘hints and tips’, from the teleconferences were distributed to all participating sites. The ICNARC CTU team facilitated communication between sites via an e-mail forum for research nurses.
Teleconferences were also held with individual site teams, as required, to address site-specific issues in the conduct of the trial and/or to support training new staff.
Site monitoring visits
At least one routine monitoring visit was conducted at all participating sites during the trial. During the site visit, the investigator site file was checked for completeness, that is that all essential documents were present; the patient consent forms, personal/professional consultee agreement forms and emergency consent forms were checked to ensure that the relevant completed form was present for every patient recruited into the trial; and a random sample of patient case report forms were checked against the source data for accuracy and completeness. After the visit, the PI and the site team were provided with a report summarising the trial documents that had been reviewed and actions required by the site team. The site PI was responsible for addressing the actions and reporting back to the ICNARC CTU.
Maintenance and motivation
During the trial, an e-mail was sent each week to site teams with an update on patient recruitment and a newsletter was sent every quarter. These provided an opportunity to clarify any issues related to the conduct of the trial and to share ideas for maximising recruitment, as well as maintaining motivation and involvement through regular updates on progress.
To maintain the profile of the trial at participating sites, posters were displayed in staff areas and at relevant locations within the ED, for example beside the blood gas machine; pocket cards summarising the eligibility criteria were distributed; and certificates were given to clinical staff in recognition of their contribution to the trial. Other promotional materials distributed to staff included pens and lanyards.
Support
A 24-hour, 7-days-per-week telephone support service was available to site teams for advice on screening and recruitment of patients and on delivery of the intervention. In addition, Edwards Lifesciences provided a 24-hour, 7-days-per-week telephone support service for queries relating to the ScvO2 monitoring equipment.
Collaborators’ meeting
A collaborators’ meeting was held on 30 May 2013 to provide an update on trial progress and to provide a forum for site teams and investigators to discuss operational challenges to the trial and identify possible solutions, and to share successes and best practice.
Participants: patients
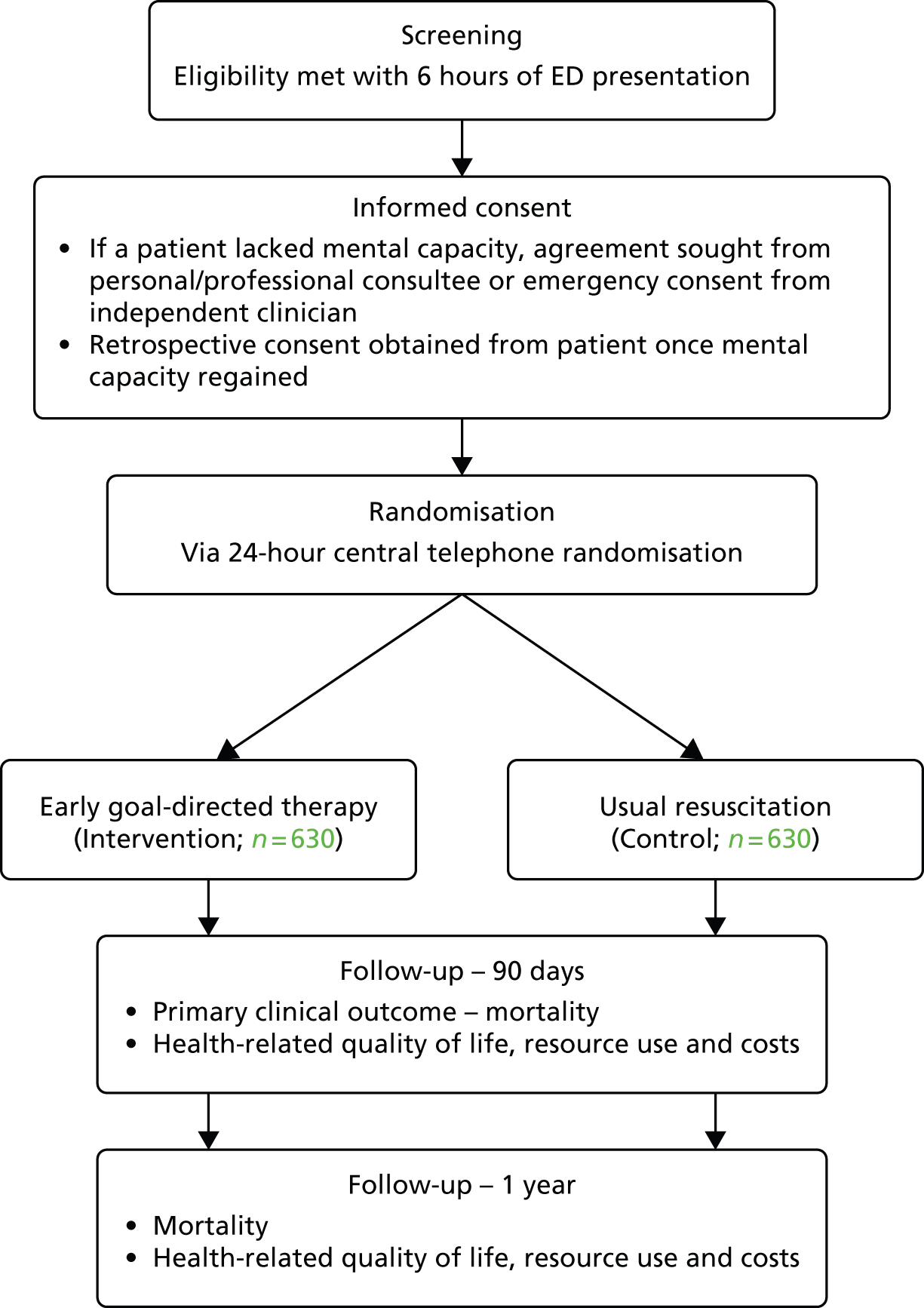
The trial procedures for recruitment and follow-up of patients are summarised in Figure 1.
FIGURE 1.
Summary of trial procedures for the recruitment and follow-up of patients. HSCIC, Health and Social Care Information Centre.
Eligibility
Eligibility was confirmed within 6 hours of the patient presenting at the ED. Patients were eligible for inclusion in the trial if they met all of the following criteria:
-
known or presumed infection
-
refractory hypotension – defined as a systolic blood pressure of < 90 mmHg or a mean arterial pressure of < 65 mmHg, despite an intravenous fluid challenge of a minimum of 1 l (fixed bolus) within 60 minutes (including intravenous fluids administered pre hospital), or hyperlactataemia – defined as a venous or arterial blood lactate concentration of ≥ 4 mmol/l
-
two or more of the following systemic inflammatory response syndrome (SIRS) criteria:28
-
core temperature of ≤ 36 °C or of ≥ 38 °C
-
heart rate of ≥ 90 beats/minute
-
respiratory rate of ≥ 20 breaths/minute [or hyperventilation indicated by either a partial pressure of carbon dioxide (PaCO2) of < 4.3 kPa or mechanical ventilation for an acute process]
-
white blood cell count of ≤ 4 × 109/l or of ≥ 12 × 109/l [or the presence of > 10% immature neutrophils (bands)].
-
Patients were excluded from the trial if they met any of the following criteria:
-
were aged < 18 years
-
had a known pregnancy
-
had a primary diagnosis of:
-
acute cerebral vascular event
-
acute coronary syndrome
-
acute pulmonary oedema
-
status asthmaticus
-
major cardiac arrhythmia (as part of primary diagnosis)
-
seizure
-
drug overdose
-
injury from burns or trauma
-
-
had haemodynamic instability due to active gastrointestinal haemorrhage
-
had a requirement for immediate surgery
-
had a known history of acquired immunodeficiency syndrome
-
had a do-not-attempt-resuscitation order
-
had advanced directives restricting implementation of the EGDT resuscitation protocol
-
had a contraindication to central venous catheterisation
-
had a contraindication to blood transfusion
-
the attending clinician deemed aggressive resuscitation unsuitable
-
had been transferred from another in-hospital setting
-
were not able to commence the EGDT resuscitation protocol within 1 hour of randomisation or complete 6 hours of EGDT from commencement.
The first dose of intravenous antimicrobial therapy had to be initiated prior to the patient being randomised.
During the trial, on the advice of the research ethics committee, patients who were known to have a pre-existing condition, such as dementia, which would have precluded them from providing informed consent at any point during the trial were also excluded.
Screening and recruitment
Following attendance at a site initiation meeting, screening and recruitment was commenced at participating sites once the clinical trial site agreement had been signed and all necessary approvals were in place.
To promote awareness of the trial and facilitate recruitment, posters providing information about ProMISe were displayed in the ED and in family/visitor waiting rooms.
Potentially eligible patients were identified and approached by authorised members of staff about taking part in the trial. Information about the trial was provided to the patient; this included the purpose of the trial, the consequences of taking part or not, data security and funding of the trial. This information was also provided in a patient information sheet (see Appendix 1), along with the name and contact details of the local PI, which was given to the patient to read before making the decision whether or not to take part in the trial. A short version of the patient information sheet, summarising the salient information about the trial, was also provided (see Appendix 2).
If the patient lacked mental capacity (because of their acute illness) to understand the information about the trial, then, in accordance with the UK Mental Capacity Act 2005,27 a personal consultee, who could be a relative or close friend, was identified with whom the patient’s participation in the trial could be discussed. If there was no personal consultee available, the patient was provided with a professional consultee – an independent mental capacity advocate appointed by the NHS hospital trust – with whom the patient’s participation in the trial could be discussed. If there was neither a personal nor a professional consultee immediately available in person or via the telephone, an independent clinician (senior doctor or nurse) was consulted in person or via telephone for emergency consent. The personal/professional consultee or independent clinician was provided with the same information as patients (see Appendix 1) along with an explanation that they were being asked for their agreement for the patient taking part in the trial. Patients, personal/professional consultees and independent clinicians were provided with an opportunity to ask questions before being invited to sign the consent form, personal/professional consultee agreement form or emergency consent form, as appropriate.
Informed consent
Staff members who had received training on the background, rationale and purpose of ProMISe and on the principles of the International Conference on Harmonisation Good Clinical Practice guidelines were authorised to take informed consent from patients, informed agreement from a personal or professional consultee or emergency consent from an independent clinician.
Once the staff member taking informed consent, consultee agreement or emergency consent was satisfied that the patient, personal/professional consultee or independent clinician had read and understood the patient information sheet and all their questions about the trial had been answered, the patient, personal/professional consultee or independent clinician was invited to sign the consent form, personal/professional consultee agreement form or emergency consent form, as appropriate.
For patients who had lacked mental capacity prior to randomisation, informed consent to continue participating in the trial was sought as soon as possible after the patient had regained mental capacity. If a patient did not regain mental capacity, then, if possible, agreement from a personal consultee was obtained for the patient to continue participating in the trial.
Randomisation and allocation procedure
Following informed consent from the patient, agreement from a personal/professional consultee or emergency consent from an independent clinician, eligible patients were randomised within 2 hours of meeting eligibility via a central 24-hour, 7-days-per-week, telephone randomisation service hosted by Sealed Envelope Ltd. Patients were randomly allocated 1 : 1 to either the EGDT group or the usual-resuscitation group, by computer-generated randomised permuted blocks (with variable block lengths of 4, 6 and 8) stratified by recruiting site. A manual randomisation list was prepared a priori by the trial statistician in case the central telephone randomisation service was not available for any reason. Staff at participating sites were advised to call the 24-hours-per-day, 7-days-per-week telephone support service if they experienced any problems with the central telephone randomisation service. Manual randomisation was carried out, as required, by the on-call member of the TMG.
Screening log
To enable full and transparent reporting for the trial, brief details of all patients who met eligibility criteria or who met all inclusion criteria plus one or more of the exclusion criteria were recorded in the screening log. The reasons for eligible patients not being recruited were recorded, which included the patient declining the invitation to take part, the patient being excluded by the treating clinician, logistical reasons, etc. No patient identifiers were recorded in the screening log.
Treatment groups
Early, goal-directed therapy (intervention)
For patients randomised to the EGDT group, during the first hour (defined as the next whole hour, e.g. if randomised at 09.24, then by 11.00), a PreSepTM central venous oximetry catheter was inserted into either a subclavian or an internal jugular vein using standard techniques for central venous access and calibrated against a sample aspirated from the catheter and analysed by co-oximetry. Central venous catheters were managed according to the guidelines of the Central Venous Catheter Care Bundle. 29 If not already initiated, supplemental oxygen was administered, with intubation and mechanical ventilation as needed, to maintain an arterial oxygen saturation (SpO2) of ≥ 93%. An arterial catheter was recommended, but not mandated.
The EGDT resuscitation protocol (Figure 2) was followed for 6 hours (intervention period) with personnel involved and treatment location decided by each site. At least one trained member of staff was available throughout the 6-hour intervention period. All other treatment, during the intervention period and after, was at the discretion of the treating clinician(s).
FIGURE 2.
Early goal-directed therapy resuscitation protocol. CVC, central venous catheter; CVP, central venous pressure; Hb, haemoglobin; i.v., intravenous; MAP, mean arterial pressure; PRBC, packed red blood cells; SBP, systolic blood pressure.
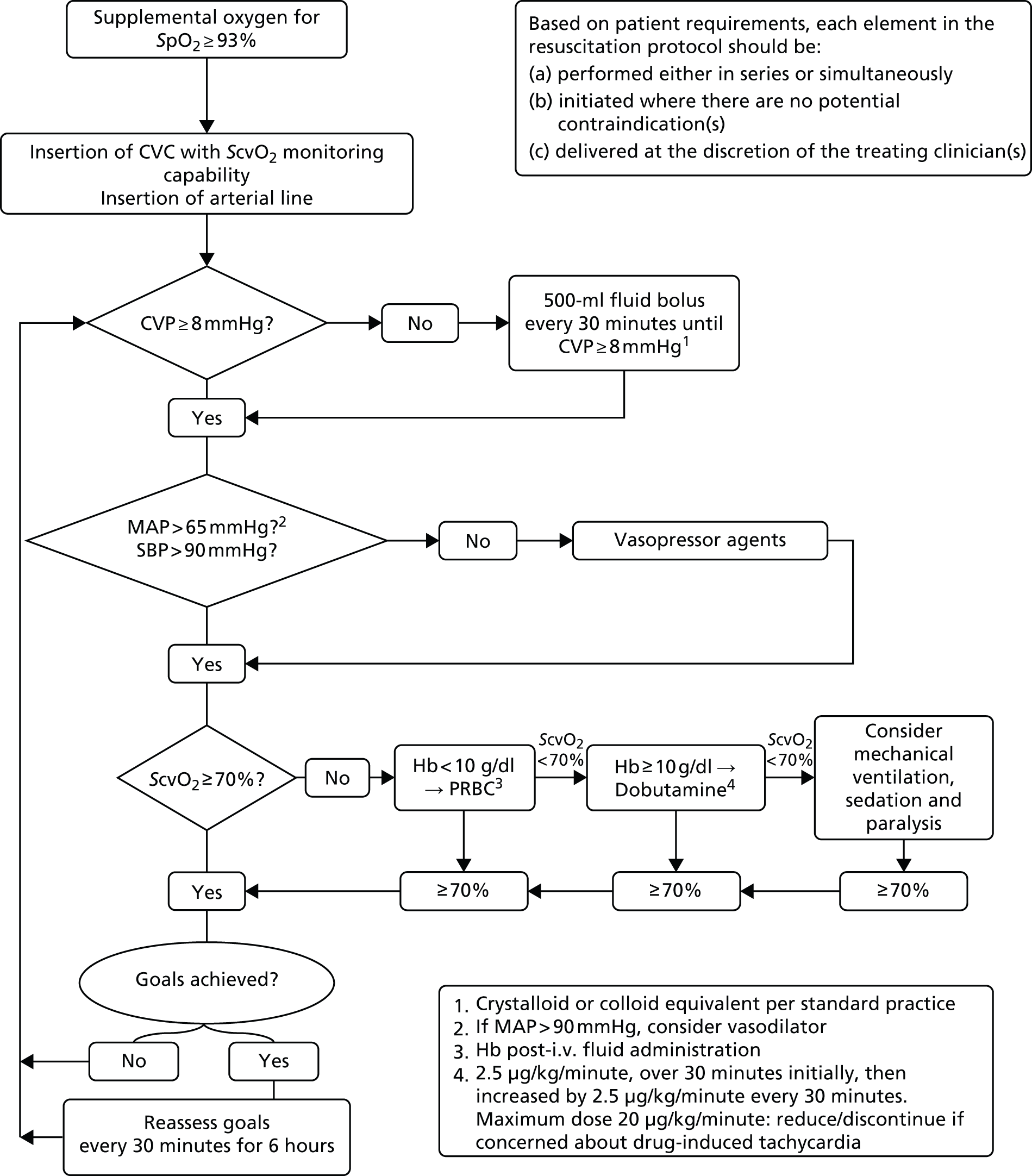
Each element of the resuscitation protocol was administered in series or simultaneously, depending on the clinical assessment of the patient’s requirements. For example, the clinical team could choose to administer intravenous fluids in conjunction with vasopressors if a patient was in extremis.
Central venous pressure
Intravenous fluid boluses in half-litre or equivalent increments were given every 30 minutes until a minimum central venous pressure of 8 mmHg was achieved, unless the treating clinician discerned a risk to patient safety. The type of intravenous fluid and the rate of administration were at the discretion of the treating clinician(s).
Blood pressure
If the mean arterial pressure was < 65 mmHg or the systolic blood pressure was < 90 mmHg and the central venous pressure was at least 8 mmHg, vasopressors were administered and titrated to a achieve a minimum mean arterial pressure of 65 mmHg or a systolic blood pressure of 90 mmHg. The choice of vasopressor was at the discretion of the treating clinician(s) based on best evidence, the patient’s clinical needs and local policy. If the mean arterial pressure was > 90 mmHg, clinicians could consider administering a vasodilator agent to reduce afterload, if clinically indicated.
Central venous oxygen saturation
Once the central venous pressure was at least 8 mmHg and the mean arterial pressure was at least 65 mmHg or the systolic blood pressure at least 90 mmHg, treatment was initiated, if necessary, to achieve a minimum ScvO2 of 70%. If the ScvO2 was < 70% and the post-fluid resuscitation haemoglobin was < 10 g/dl, packed red blood cells were transfused. If the ScvO2 was < 70% and the haemoglobin was at least 10 g/dl, an infusion of dobutamine was commenced, at an initial rate of 2.5 µg/kg/minute for 30 minutes, and then increased by 2.5 µg/kg/minute every 30 minutes, to a maximum dose of 20 µg/kg/minute, until a ScvO2 of ≥ 70% was achieved. The dose of dobutamine was reduced or the infusion discontinued if there was concern about drug-induced tachycardia or arrhythmia. If the ScvO2 remained < 70%, the clinician could consider mechanical ventilation (with sedation and paralysis) to decrease oxygen consumption.
Monitoring
Once all physiological goals for central venous pressure, blood pressure and ScvO2 were met, the patient was monitored continuously for the remainder of the intervention period (a total of 6 hours). If the central venous pressure, blood pressure or ScvO2 fell below its physiological goal during the 6-hour intervention period, the EGDT resuscitation protocol recommenced. At the end of 6 hours, continuous ScvO2 monitoring was no longer mandated and the patient returned to standard care.
Usual resuscitation (control)
For patients randomised to usual resuscitation, all investigations, monitoring and treatment were determined by the treating clinician(s). Although ScvO2 could be measured intermittently, continuous monitoring of ScvO2 was not permitted in control group patients.
Outcome measures
The primary clinical effectiveness outcome was all-cause mortality at 90 days following randomisation and the primary cost-effectiveness outcome was incremental net monetary benefit (INB) gained at 1 year, at a willingness to pay of £20,000 per quality-adjusted life-year (QALY). Secondary outcomes were as follows:
-
Sequential Organ Failure Assessment (SOFA) score30 at 6 and 72 hours
-
receipt of and days alive and free (up to 28 days) from advanced cardiovascular, advanced respiratory or renal support31
-
ED, critical care and acute hospital length of stay
-
duration of survival
-
all-cause mortality at 28 days, at acute hospital discharge and at 1 year
-
health-related quality of life, resource use and costs at 90 days and at 1 year
-
lifetime incremental cost-effectiveness.
Safety monitoring
Patients were monitored for adverse events that occurred between randomisation and 30 days following randomisation. Specified adverse events were defined as follows:
-
pneumothorax – defined as any new pneumothorax requiring insertion of a chest drain (intercostal catheter)
-
haemo-pneumothorax – defined as any new haemo-pneumothorax requiring insertion of a chest drain
-
bleeding – defined as any new, overt blood loss requiring transfusion of one or more units of blood
-
thrombosis – defined as any new clinical and radiographic evidence of a deep-vein thrombus
-
pulmonary emboli – defined as any new evidence from computed tomography pulmonary angiogram with appropriate clinical history
-
vascular catheter infection – defined as any new vascular catheter-related infection in which a vascular catheter, such as a central venous catheter, was identified as the primary source of infection and associated with signs and symptoms of infection requiring antimicrobials
-
pulmonary oedema – defined as any new radiographic evidence consistent with pulmonary oedema
-
blood transfusion reaction – defined as any allergic reaction to blood transfusion, haemolysis related to incompatible blood type or alteration of the immune system related to blood transfusion
-
myocardial ischaemia – defined as any new acute electrocardiogram changes with appropriate clinical findings and changes in cardiac troponins or non-ST segment elevation myocardial infarction with appropriate increases in cardiac troponins but without electrocardiogram changes
-
peripheral ischaemia – defined as any new sustained depression or loss of arterial pulse (as determined by palpation or Doppler ultrasonography) resulting in symptoms consistent with ischaemia or obvious gangrene.
Unspecified adverse events were defined as an unfavourable symptom or disease temporally associated with the use of the trial treatment, whether or not it was related to the trial treatment, that was not deemed to be a direct result of the patient’s medical condition and/or standard critical care treatment.
All adverse events were recorded in the electronic case report form and reported, as part of routine reporting throughout the trial, to the DMEC and the research ethics committee. Adverse events that were assessed to be serious (i.e. prolonging hospitalisation or resulting in persistent or significant disability/incapacity), life-threatening or fatal – collectively termed serious adverse events – were reported to the ICNARC CTU and reviewed by a clinical member of the TMG. Serious adverse events that were unspecified and considered to be possibly, probably or definitely related to the trial treatment were reported to the research ethics committee within 15 calendar days of the event being reported.
Data collection
A secure, dedicated electronic case report form, hosted by ICNARC, was set up to enable trial data to be entered by staff at participating sites. The electronic case report form was accessible only to authorised users, and access was approved centrally by the trial manager or the data manager (after cross-checking the site delegation of trial duties log). Each individual was provided with a unique username and password, and had access to data only for the patients recruited at their site.
The data set for ProMISe included the minimum data required to confirm patient eligibility, to describe the patient population, to monitor and describe delivery of the intervention, to assess primary and secondary outcomes and to enable linkage to the ICNARC Case Mix Programme, the national clinical audit of adult critical care32 (see Appendix 3).
Randomisation
Data were collected to enable the patient to be randomised, and included confirmation that the patient met all of the inclusion criteria and none of the exclusion criteria and that the first dose of intravenous antimicrobial(s) had been initiated (see Appendix 3).
Baseline
The following data were collected at baseline to enable follow-up and to describe the patient population:
-
full name and address of the patient and their general practitioner
-
date of birth
-
sex
-
raw physiology data to enable calculation of the of the following severity of illness scores:
-
SOFA score30 (see Appendix 4)
-
Acute Physiology And Chronic Health Evaluation version II (APACHE II) score and predicted risk of hospital death33 (see Appendix 4)
-
Mortality in Emergency Department Sepsis (MEDS) score34 (see Appendix 4)
-
-
severe comorbidities defined according to APACHE II33 which were present and documented in the past medical history within the 6 months prior to presentation at the ED (see Appendix 4).
Intervention period
Data were collected hourly throughout the 6-hour intervention period to monitor adherence to the treatment allocation (EGDT resuscitation or usual resuscitation) and to describe and cost delivery of the EGDT resuscitation protocol compared with usual resuscitation. During the 6-hour intervention period, data were collected prospectively for the EGDT group and retrospectively for the usual-resuscitation group in order to avoid data collection influencing treatment delivery. The data collected comprised:
-
interventions delivered during the previous hour, for example supplemental oxygen, mechanical ventilation, intravenous fluids, blood products and vasoactive drugs
-
physiology, for example central venous pressure, blood pressure, ScvO2 and haemoglobin.
At 6 hours
At 6 hours post randomisation, the following data were collected:
-
interventions delivered during the previous hour, for example supplemental oxygen, mechanical ventilation, intravenous fluids, blood products and vasoactive drugs
-
physiology, for example central venous pressure, blood pressure, ScvO2 and haemoglobin
-
raw physiology data to enable calculation of the SOFA score30 (see Appendix 4).
Ancillary care
Data were collected to describe and cost interventions delivered after the end of the 6-hour intervention period up to discharge from the acute hospital.
At 24 hours
At 24 hours post randomisation, the following data were collected:
-
interventions delivered between 6 and 24 hours, for example supplemental oxygen, mechanical ventilation, intravenous fluids, blood products and vasoactive drugs
-
raw physiology data to enable calculation of the SOFA score30 (see Appendix 4).
At 72 hours
At 72 hours post randomisation, the following data were collected:
-
interventions delivered between 24 and 72 hours, for example supplemental oxygen, mechanical ventilation, intravenous fluids, blood products and vasoactive drugs
-
raw physiology data (48–72 hours) to enable calculation of the SOFA score30 (see Appendix 4)
-
site of infection and causative organism.
At acute hospital discharge
At the time of discharge from the acute hospital, the following data were collected:
-
the locations of care during the patient’s stay in the acute hospital, for example ED, critical care unit or ward
-
date of discharge from, or death in, the acute hospital
-
discharge location, for example home, nursing home or other hospital
-
organ support, as defined by the UK Department of Health Critical Care Minimum Data Set31 (see Appendix 5) during the critical care unit stay, if applicable
-
co-interventions for the source of sepsis, for example surgery, steroids or activated protein C.
Longer-term follow-up
Following randomisation, a letter was sent to the patient’s general practitioner informing them of the patient’s participation in the trial and issuing a request for assistance with follow-up, if required. All patients who survived to leave hospital were followed up at 90 days for the primary clinical effectiveness outcome (all-cause mortality) and secondary outcomes (health-related quality of life and resource use), and at 1 year for secondary outcomes (all-cause mortality, duration of survival, health-related quality of life and resource use) and to calculate the primary cost-effectiveness outcome (INB).
Data linkage with death registration
Follow-up of patients was carefully monitored to prevent any potential distress to those who care for the patient receiving a letter addressed to a deceased relative, partner or friend. The follow-up process started at 75 days for the 90-day follow-up and at 350 days for the 1-year follow-up to allow for the administrative processes. Each week a list of all patients who had been discharged alive from hospital and who were either 75 days or 350 days post randomisation was sent to the Health and Social Care Information Centre Data Linkage and Extract Service to confirm their mortality status. Patients indicated as having died were logged and the follow-up process ended.
Follow-up procedure
Patients identified by the Health and Social Care Information Centre Data Linkage and Extract Service as not having died started the follow-up process, as summarised in Figure 3. A questionnaire pack was sent from the ICNARC CTU, by post, to the patient. Following evidence-based practice for maximising responses to postal surveys,35 the questionnaire pack included a cover letter (see Appendix 6); the patient information sheet (see Appendix 1) or patient newsletter (which replaced the patient information sheet in November 2013); two questionnaires – the Health Questionnaire (see Appendix 7) and the Health Services Questionnaire (see Appendix 7); a stamped, addressed return envelope; and a pen. The Health Questionnaire (see Appendix 7) included the required questions from the European Quality of Life-5 Dimensions-5-Level (EQ-5D-5L) questionnaire to evaluate health-related quality of life and the Health Services Questionnaire (see Appendix 7) included questions about the patient’s use of health services following discharge from the acute hospital and was used to cost subsequent use of health services. The cover of the questionnaires included a ‘do not wish to participate’ tick box.
FIGURE 3.
Patient follow-up process at 90 days and at 1 year. HSCIC, Health and Social Care Information Centre.
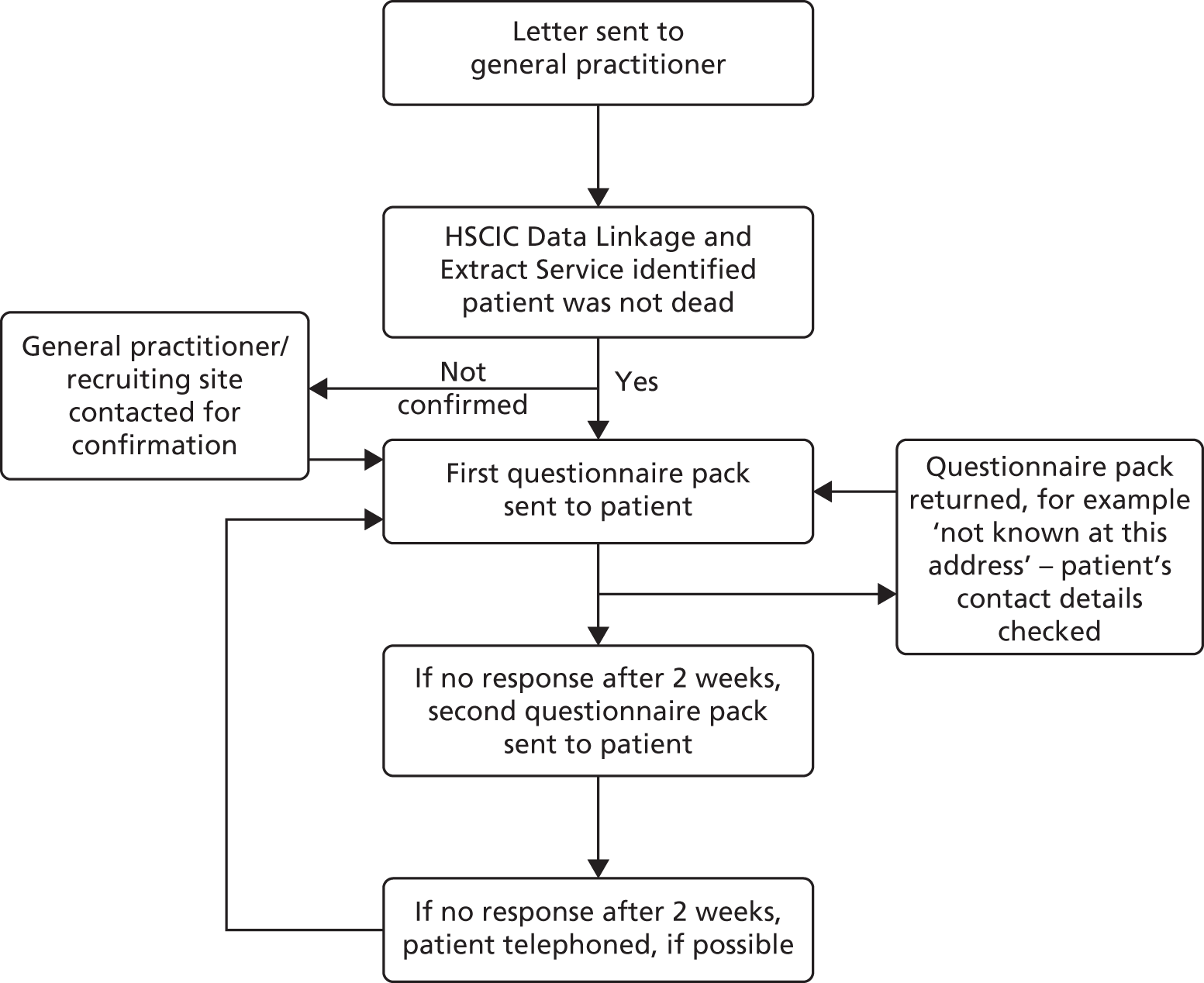
If no response was received after 2 weeks, a reminder letter was sent with another questionnaire pack. If no response was received after a further 2 weeks, the patient was telephoned, if his or her contact details were available. Telephone calls were made at various times from Monday to Friday between 08.30 and 20.30 to maximise the chances of contacting the patient. Patients who were successfully contacted by telephone were asked if they had received the questionnaire pack and were invited to complete the questionnaires over the telephone, if this was convenient. In addition, patients were reminded about completing the questionnaire when they attended hospital follow-up appointments.
Follow-up ended on receipt of a completed (or blank) questionnaire; on receipt of a questionnaire with a ticked ‘do not wish to participate’ box; on notification to the ICNARC CTU by telephone or e-mail that the patient wished to withdraw from the trial; or if there was no response to the telephone follow-up. For questionnaire packs returned indicating that the recipient was not known at the address, the contact details for the patient were checked with the recruiting hospital and/or general practitioner.
For patients who were identified as being either a hospital inpatient or resident in a care home or rehabilitation centre, the relevant institution was contacted to establish the status of the patient and the most appropriate way to proceed with follow-up. If the patient had the mental capacity to consent but required assistance in reading and/or completing the questionnaire, health-care professionals usually assisted the patient. For patients who lacked the mental capacity to consent, institutions advised on the most appropriate person to contact to complete the questionnaires.
If patients were identified as having no fixed abode but were registered with a general practitioner or had regular contact with a homeless shelter, the questionnaire pack was sent to be given (when appropriate) to them at their next appointment or visit.
Data linkage with the Case Mix Programme
The linkage of patient identifiable trial data to the ICNARC Case Mix Programme database provided information on subsequent admission to adult, general, critical care following discharge from the acute hospital. 32
Data for the CMP are collected by trained data collectors to precise rules and definitions. The data then undergo extensive local and central validation for completeness, illogicalities and inconsistencies prior to pooling.
Data management
Data management was an ongoing process. Data entered by sites onto the electronic case report form were monitored and checked throughout the recruitment period to ensure that they were as complete and accurate as possible.
Two levels of data validation were incorporated into the electronic case report form. The first was to prevent obviously erroneous data from being entered, for example entering a date of birth that occurred after the date of randomisation. The second level involved checks for data completeness and any unusual data entered, for example a physiological variable, such as blood pressure, that was outside the pre-defined range. Site staff could generate data validation reports, listing all outstanding data queries, at any time via the electronic case report form. The site PI was responsible for ensuring that all data queries were resolved. Ongoing data entry and validation at sites were closely monitored by the data manager (JT) and any concerns were raised with the site PI.
The contact details for patients and their general practitioners (name and postal address) were checked weekly for completeness to avoid unnecessary delays in sending out questionnaire packs at 90 days and at 1 year.
Adherence to the trial protocol was closely monitored, including adherence to all elements of the EGDT resuscitation protocol. Any queries relating to adherence were generated in a separate report which was sent to the site PI.
Data received from completed European Quality of Life-5 Dimensions (EQ-5D) and Health Services Questionnaires were entered centrally into a secure database at the ICNARC CTU following a standard operating procedure. All identifiable information, such as names (e.g. of patients, family members or hospital staff members), was removed. All queries relating to data entry were reviewed by two members of the TMG (SH/PM) and any disagreement was reviewed and discussed with a third member (KR).
To ensure that data were entered accurately, all questionnaire data entered into the database were cross-checked by a second member of the CTU team. Any errors found were logged and corrected on the database.
Sample size
Estimates for baseline mortality in the usual-resuscitation group were based on the ICNARC Case Mix Programme database. 32 Between 1 January 2005 and 31 December 2006, there were 24,155 patients admitted to 156 participating adult general ICUs direct from the ED. Of these, 6671 (28%) met at least two SIRS criteria during the first 24 hours following ICU admission and had evidence of infection. Acute hospital mortality for these patients was 35%. To allow for additional deaths after discharge from hospital and before 90 days, sample size calculations were based on an anticipated mortality at 90 days of 40% in the usual-resuscitation group. To achieve 80% power to detect a 20% relative reduction in mortality at 90 days (corresponding to an 8% absolute reduction) from 40% to 32% associated with EGDT compared with usual resuscitation (p < 0.05, two-sided) required a sample size of 589 patients per treatment group (Stata/SE version 10.1, StataCorp LP, College Station, TX, USA). Allowing for 6% of patients refusing consent to follow-up (in the PAC-Man trial, 2% of patients refused consent after randomisation36) or being lost to follow-up before 90 days, our aim was to recruit 630 patients per group (1260 patients in total). This sample size provided > 99% power to detect an absolute risk reduction of the magnitude observed in the Rivers et al. trial (i.e. 16%). 6
Interim analysis
Unblinded comparative data on recruitment, withdrawal, adherence with the trial protocol and serious adverse events were regularly reviewed by the DMEC. Without specific analysis of the primary outcome, the DMEC reviewed data from the first 50 trial participants and continued to review data at least 6-monthly to assess potential safety issues and to review adherence with the trial protocol. A single planned formal interim analysis was performed once 90-day outcome data from the first 500 patients enrolled were available. A Haybittle–Peto stopping rule (p < 0.001) was used to guide recommendations for early termination owing to harm.
Analysis principles
All analyses were based on the intention-to-treat principle. Patients were analysed according to the treatment group they were randomised to, irrespective of whether or not the allocated treatment was received (i.e. regardless of whether or not they adhered to the EGDT algorithm). All tests were two-sided with significance levels set at p < 0.05 and with no adjustment for multiplicity. All a priori subgroup analyses were carried out irrespective of whether or not there was strong evidence of a treatment effect associated with the primary outcome. As missing data for the clinical effectiveness primary outcome were anticipated to be minimal, a sensitivity approach was taken when the primary outcome was missing (see Secondary analyses of the primary outcome). Missing data for the cost-effectiveness analysis, as well as missing baseline data for adjusted analysis of clinical outcomes, were handled by multiple imputation.
Multiple imputation
Missing data in baseline covariates, resource use and health-related quality of life variables at 90 days and 1 year were handled with multivariate imputation by chained equations. 37 Under this approach each variable was imputed conditional on fully observed baseline variables such as age, sex, past medical history, site of sepsis, SOFA score, MEDS score, admitted from nursing home, length of stay in critical care and general medical wards up to 90 days and 1 year, and all other imputed variables. Patients who were eligible for 90-day follow-up (i.e. alive at 90 days) but did not return or fully complete the EQ-5D questionnaire administered at 90 days, had their EQ-5D utility scores imputed from those survivors who did fully complete the questionnaire. Similarly, for those eligible patients who did not return the Health Services Questionnaire, information on the use of outpatient services up to 90 days following randomisation, was imputed from those patients who did complete this questionnaire. In the same way, patients who were eligible for 1-year follow-up but did not return or fully complete the EQ-5D questionnaire or the Health Services Questionnaire administered at 1 year also had their information imputed from those survivors who did fully complete the questionnaire. When addressing the missing data, multiple imputation assumes that the data are missing at random conditional on the observed data.
The same multiple imputation approach was used to address the administrative censoring, which applied to the total costs, vital status and quality of life at 1 year for patients randomised after 12 November 2013. In this case it was assumed that the data were censored completely at random, which was plausible as the censoring was administrative, that is it is unlikely that there would be systematic differences between those whose end points (cost, vital status and quality of life) were observed and those who were censored. One-year cost and quality-of-life end points were conditional on survival status; as such, the imputation was conducted in 2 stages. In the first stage, imputation models were specified for mortality at 1 year according to baseline covariates and auxiliary variables, including duration of the initial inpatient stay, and costs at 90 days. In the second stage, for each of the imputed data sets from stage 1, imputation models were specified for costs and quality of life at 1 year for those patients who were missing these but were known to be alive at 1 year, or were predicted to be alive by the first-stage imputation model. These imputation models included those variables in the first-stage imputation model but also information on costs and quality of life at 1 year for those individuals for whom this end point was observed. Each of the resultant estimates was combined with Rubin’s rules, which recognise uncertainty both within and between imputations. All multiple imputation models were implemented in the statistical package R (The R Foundation for Statistical Computing, Vienna, Austria).
Statistical analysis: clinical effectiveness
Statistical analyses were conducted according to a pre-specified, published statistical analysis plan. 38 The final analyses were conducted using Stata/SE version 13.0.
Baseline characteristics
Baseline demographic and clinical data were summarised by treatment group but not subjected to statistical testing. Discrete variables were summarised as numbers and percentages, which were calculated according to the number of patients for whom data were available; where values were missing, the denominator was reported. Continuous variables were summarised by standard measures of central tendency and dispersion: mean and standard deviation (SD) and/or median and interquartile range (IQR), as specified below.
-
Inclusion criteria
-
refractory hypotension, n (%)
-
systolic blood pressure or mean arterial pressure value at which criterion for refractory hypotension was met, mean (SD)
-
-
hyperlactataemia, n (%)
-
blood lactate value at which criterion for hyperlactataemia was met, mean (SD).
-
-
-
Age, mean (SD) and median (IQR).
-
Sex, n (%).
-
Severe comorbidities (as defined by APACHE II33), n (%).
-
severe liver disease
-
severe renal disease
-
severe respiratory disease
-
severe cardiovascular disease
-
immunocompromised.
-
-
Pre-randomisation treatment, n (%) received and median (IQR) volume of
-
intravenous fluids (total before admission to hospital and total from ED presentation to randomisation)
-
blood products (total from ED presentation to randomisation).
-
-
Acute severity of illness.
-
Time from ED presentation to inclusion criteria met, mean (SD) and median (IQR).
-
Time from ED presentation to randomisation, mean (SD) and median (IQR).
-
Patient likely to be admitted directly to ICU from ED if not enrolled in ProMISe, n (%).
-
Infection, n (%).
-
site
-
organism
-
antimicrobial change between ED presentation and 72 hours.
-
Adherence
Non-adherence with the allocated treatment was reported as:
-
insertion of a central venous catheter with ScvO2 monitoring capability to a patient allocated to usual resuscitation
-
failure to insert a central venous catheter with ScvO2 monitoring capability to a patient allocated to EGDT
-
failure to act on a goal in the EGDT algorithm for a patient allocated to EGDT, defined as
-
no fluid resuscitation when central venous pressure < 8 mmHg
-
no administration of vasopressors when mean arterial pressure < 65 mmHg or systolic blood pressure < 90 mmHg and the central venous pressure goal was met
-
no administration of packed red blood cells when ScvO2 < 70% and haemoglobin < 10 g/dl and the central venous pressure and blood pressure goals were met
-
no administration of dobutamine when ScvO2 < 70% and haemoglobin ≥ 10 g/dl and the central venous pressure and blood pressure goals were met
-
-
early (< 6 hours) termination of EGDT in a patient allocated to EGDT (other than due to death or discharge from hospital).
For comparison, adherence in ProMISe was also assessed according to the criteria used in the published reports of ProCESS23 and ARISE. 24
Delivery of care
Delivery of care was summarised by treatment group but not subjected to statistical testing. As with baseline characteristics, discrete variables were summarised as numbers and percentages. Percentages were calculated according to the number of patients for whom data were available; where values were missing, the denominator was reported. Continuous variables were summarised by mean (SD) and/or median (IQR).
Intervention data were summarised as the total over the 6-hour intervention period (hour 0 to hour 6); the total from the end of the 6-hour intervention period to the end of the first 72 hours (hour 6 to hour 72); and from randomisation to the end of the first 72 hours (hour 0 to hour 72). Where measurements were recorded, baseline values were also reported. Catheter insertion and location of care details were included in the hour 0 to hour 6 table. The following were reported:
-
catheter insertion, n (%), and time from randomisation to insertion, mean (SD) and median (IQR)
-
central venous catheter with ScvO2 monitoring capability
-
any central venous catheter
-
arterial catheter
-
-
interventions, n (%) received
-
supplemental oxygen
-
mechanical ventilation
-
-
fluids, n (%) received and mean (SD) and median (IQR) volume of
-
any intravenous fluid
-
intravenous colloid
-
intravenous crystalloid
-
packed red blood cell transfusion
-
platelets
-
fresh-frozen plasma
-
-
drugs, n (%) received
-
vasopressors
-
dobutamine
-
sedatives
-
neuromuscular blocking agent
-
-
co-interventions for the source of sepsis, n (%) received
-
surgery
-
activated protein C
-
steroids
-
-
location of care
-
critical care admission, n (%), and mean (SD) and median (IQR) time from randomisation to admission
-
location of protocol delivery, n (%)
-
review by consultant, n (%)
-
specialty of most senior doctor to review the patient, n (%).
-
The mean volume of intravenous fluids and the number and percentage receiving vasopressors, packed red blood cell transfusions, dobutamine, sedatives, mechanical ventilation and neuromuscular blocking agents were additionally reported hourly for the duration of the 6-hour intervention period.
Physiology data were summarised as the total over the 6-hour intervention period (hour 0 to hour 6); the total from the end of the 6-hour intervention period to the end of the first 24 hours (hour 6 to hour 24); and from the end of the first 48 hours to the end of the first 72 hours (hour 48 to hour 72). Where measurements were recorded, baseline values were also reported. The following values were reported:
-
lowest mean arterial pressure, mean (SD)
-
lowest systolic blood pressure, mean (SD)
-
haemoglobin value at the end of the time period, mean (SD)
-
blood lactate value at the end of the time period, mean (SD)
-
lowest partial pressure of oxygen (PaO2)/fraction of inspired oxygen (FiO2), mean (SD)
-
highest creatinine, mean (SD)
-
highest bilirubin, mean (SD)
-
lowest platelets, mean (SD)
-
lowest Glasgow Coma Scale (GCS) score, mean (SD)
-
individual SOFA score components, n (%).
Mean (SD) of central venous pressure, mean arterial pressure, systolic blood pressure and ScvO2 were additionally reported hourly for the duration of the 6-hour intervention period.
Primary outcome: clinical effectiveness
The number and percentage of deaths at 90 days following randomisation due to any cause were reported for each treatment group. The primary effect estimate was the relative risk of all-cause mortality at 90 days, reported with a 95% CI. The absolute risk reduction and 95% CI were also reported. Deaths at 90 days after randomisation were compared between the treatment groups, unadjusted, using Fisher’s exact test. A secondary analysis of the primary outcome, adjusted for baseline variables, was conducted using multilevel logistic regression. Baseline variables adjusted for in the multilevel logistic regression model were the components of the MEDS score (age, metastatic cancer, nursing home residence, altered mental status, septic shock, respiratory difficulty, low platelet count, high bandforms and low neutrophil count) and a site-level random effect. Baseline variables were selected for inclusion in the adjusted analysis according to anticipated relationship with outcome. The results of the multilevel logistic regression model were reported as an adjusted odds ratio with 95% CI. The unadjusted odds ratio was presented for comparison.
Secondary outcomes: clinical effectiveness
The mean SOFA score at 6 hours and 72 hours after randomisation was reported for each treatment group. Differences in the mean SOFA score at 6 hours and 72 hours after randomisation were compared, adjusted for baseline SOFA score, using analysis of covariance.
The number and percentage of patients receiving advanced cardiovascular, advanced respiratory and renal support were reported for each treatment group. Differences in receipt of advanced cardiovascular, advanced respiratory and renal support were compared, unadjusted, using Fisher’s exact test. The mean (SD) of the number of days alive and free from advanced cardiovascular, advanced respiratory and renal support, up to 28 days, in each treatment group were reported. Patients who died within the first 28 days were assigned 0 days alive and free of each organ support. Differences between the treatment groups were tested using the t-test, using the non-parametric bootstrap to account for anticipated non-normality in the distributions. 39 A total of 1000 bootstrap replications were taken, stratified by treatment group, with bias-corrected and accelerated CIs reported.
The median (IQR) of the length of stay in the ED, in critical care and in acute hospital was reported for each treatment group. Differences in length of stay between the treatment groups were tested using the Wilcoxon rank-sum test, stratified by survival at end of ED stay, critical care discharge and acute hospital discharge, respectively.
Kaplan–Meier curves by treatment group were plotted up to 90 days and 1 year after randomisation and compared using the log-rank test. An adjusted comparison was performed using a Cox proportional hazards model adjusted for the same baseline variables as the primary outcome, including shared frailty within sites (gamma-distributed latent random effects). The appropriateness of the proportional hazards assumption was assessed graphically by plotting –log[−log(survival)] against log(time) within treatment groups. The number and percentage of deaths at acute hospital discharge and by 28 days, 90 days and 1 year after randomisation were reported for the treatment groups. Differences in all-cause mortality at each time point were compared, unadjusted, using Fisher’s exact test and adjusted using multilevel logistic regression, adjusted for the same baseline variables as the primary outcome.
Safety monitoring
The number and percentage of patients experiencing each serious adverse event (occurring between randomisation and 30 days) were reported for each treatment group. The total number of patients experiencing one or more serious adverse events was compared between treatment groups using Fisher’s exact test and summarised as a relative risk with 95% CI.
Subgroup analyses of the primary outcome
Subgroup analyses were conducted using the likelihood ratio test to assess interactions between treatment group and pre-specified subgroups in multilevel logistic regression models for all-cause mortality at 90 days, adjusted for the same baseline variables as the analysis of the primary outcome. The subgroups compared were degree of protocolised care for the usual-resuscitation group; age; MEDS score; SOFA score; and time from ED presentation to randomisation. Degree of protocolised care for the usual-resuscitation group was assessed based on established guidelines14,16,40 as the proportion of patients allocated to the usual-resuscitation group that had lactate measured at baseline and, if ≥ 4 mmol/l at baseline, remeasured within 6 hours. Sites were categorised as having a higher degree of protocolised care if the proportion of patients in the usual-resuscitation group who met this condition was > 50%. Sites with fewer than three patients allocated to the usual-resuscitation group were excluded from this subgroup analysis. The remaining subgroups were analysed in quartiles.
Secondary analyses of the primary outcome
Sensitivity analyses for missing data in the primary outcome
The primary analysis was repeated once, assuming that all patients allocated to EGDT with missing data in the primary outcome survived and all patients allocated to usual resuscitation with missing data in the primary outcome did not survive. The analysis was then repeated again with the opposite assumptions. This gave the absolute range of how much the results could change if the primary outcome was complete.
Learning curve analysis
The delivery of a complex intervention may improve with time as those delivering the intervention gain experience and familiarity. Typically, such improvements will be more rapid at first and then tail off over time to reach a steady state; this relationship is termed a ‘learning curve’. Modelling the learning curve enables estimation of the treatment effect for an experienced team (the asymptotic value to which the curve trends over time). A site-level learning curve for patients allocated to EGDT was modelled by repeating the multilevel logistic regression on the primary outcome and including a power curve (aX−b) for the sequential observation number (X) for each EGDT patient within each site. 41 The power curve model was estimated by direct maximisation of the log-likelihood function using a modified Newton–Raphson algorithm. 42 A single estimate for each of the parameters a and b was fitted across all sites.
Adherence-adjusted analysis
While the intention-to-treat analysis gives the best estimate of the clinical effectiveness of EGDT as delivered, it is also of interest to estimate what the efficacy of this intervention may be if all elements of the protocol were delivered as intended. In a randomised controlled trial, the allocated treatment can be used as an ‘instrumental variable’, that is, a variable associated with receipt of the intervention and only associated with the outcome through its association with the intervention. 43 This relationship enables us to estimate what the treatment effect would be for patients who were fully adherent to the protocol. The primary analysis was repeated, adjusting for adherence using a structural mean model with an instrumental variable of allocated treatment, assuming a linear relationship between the degree of adherence (proportion of the 6 hours that the patient was adherent to the EGDT protocol) and treatment effect. 37,44
Cost-effectiveness analysis
A full cost-effectiveness analysis was undertaken to assess which treatment strategy, EGDT or usual resuscitation, was more cost-effective. This analysis assessed whether or not any intervention costs associated with EGDT were offset by any subsequent reduction in morbidity costs, for example from reduced use of critical care, and whether there were improvements in either mortality or health-related quality of life. The cost-effectiveness analysis was reported for three time periods: randomisation to 90 days, randomisation to 1 year and lifetime. For each time period the cost-effectiveness analysis took a health and personal health services perspective,45 using information on health-related quality of life collected at 90-day and 1-year follow-up, combined with information on vital status, to report QALYs. Each QALY was valued using the National Institute for Health and Care Excellence (NICE)-recommended threshold of willingness to pay for a QALY gain (£20,000), in conjunction with the costs of each treatment strategy to report the INBs of EGDT versus usual resuscitation.
The primary objective of the cost-effectiveness analysis was to compare incremental cost-effectiveness at 1 year between the treatment groups. There were also a number of secondary objectives:
-
to compare health-related quality of life at 90 days and 1 year between the treatment groups
-
to compare resource use and costs at 90 days and 1 year between the treatment groups
-
to estimate the lifetime incremental cost-effectiveness between the treatment groups.
The main assumptions of the cost-effectiveness analysis were subjected to extensive sensitivity analyses.
Resource use
The resource use categories considered were chosen a priori, where differences between the treatment groups were judged as being possible and likely to drive incremental costs, and were reported for each treatment group. Data for interventions, staff time and acute hospital stay for the index hospital admission were collected as part of the ProMISe data set. Readmissions to acute hospital including a critical care stay were identified from the Case Mix Programme database. 32 Readmission to acute hospital not involving critical care as well as hospital outpatient and community services use were collected as part of the Health Services Questionnaires completed at 90 days and 1 year.
Interventions
The type of catheter inserted (central venous catheter capable of ScvO2 monitoring, standard central venous catheter and/or arterial catheter) as well as the use of other catheter insertion-related consumables including pressure transducers to measure intravascular pressures, and the consumables (saline infusion, cleaning packs, sterile gloves) associated with each type of catheter insertion were considered (Table 1). The use of packed red blood cells, platelets, fresh-frozen plasma and dobutamine was also considered. The costs associated with other clinical interventions such as intravenous crystalloid, intravenous colloid, albumin, other blood products and other vasoactive drugs were not anticipated to differ across treatment groups. As such, these were not considered as separate items; however, their costs were included within the unit cost per critical care bed-day according to the Healthcare Resource Group definition. The duration for which EGDT was delivered (up to 6 hours) in the ED and in total was reported.
| Catheter | Equipment | Doctor time (catheter insertion) | Nurse time (monitor set-up) | Consumables |
|---|---|---|---|---|
| PreSepTM central venous oximetry catheter | Monitor | 30 minutes | 20 minutesa + 30 minutesb | Transducer,a saline, consumables pack for insertion |
| Standard CVC | – | 30 minutes | 20 minutesa | Transducer,a saline, consumables pack for insertion |
| Arterial catheter | – | 20 minutes | 20 minutesa | Transducer,a saline, skin cleaning device and dressing |
Staff time
The EGDT protocol required additional staff time for central venous catheter insertion (doctors’ time); monitor set-up (nurses’ time); monitoring patients in ED (nurses’ time); and staff training (nurses’ and doctors’ time). The level of additional staff time for EGDT was estimated according to expert opinion (see Table 1), with alternative levels considered in the sensitivity analyses. It was assumed that in the ED at least one trained nurse was available for the duration of delivery of EGDT. The base-case analysis assumed that, when delivered in the ED, each patient in the EGDT group required an additional 10 minutes of nurses’ monitoring time per hour of EGDT. To provide EGDT in the ED as part of routine practice required additional formal or informal training beyond the existing hospital education program. It was assumed that at each site each clinical member of ED staff required 20 minutes’ additional training to deliver EGDT. The total training time for introducing the EGDT protocol into the ED was then calculated for each site in the trial. The average mix of ED staff was assumed to be seven (attending) consultants, 23 junior doctors and 75 nurses46 over the life cycle of EGDT, which was assumed to be 5 years. Where EGDT was delivered in ICU, it was assumed that no additional staff training time or monitoring time for patients was required.
Acute hospital length of stay
Length of stay in ED, critical care and general medical wards within the index acute hospital admission (i.e. the hospital in which a patient was randomised to the trial) were reported. For critical care stays, Healthcare Resource Groups were assigned according to the maximum number of organs supported during the stay. 31 An acute hospital readmission was defined as a further hospital admission, for any reason, following discharge from the index admission. Length of stay in critical care and general medical wards within acute hospital readmissions were taken from the Case Mix Programme database and the Health Services Questionnaires.
Hospital outpatient visits and community service use
The number of hospital outpatient visits and community service use for any reason were reported. Items of community service use included visits to the general practitioner (family doctor), nurse, health visitor, occupational therapist, physiotherapist and psychologist. The levels of resource use were taken from responses to the Health Services Questionnaire.
Unit costs
The unit costs required for valuing the resource use data listed in Table 2 were taken from three sources: manufacturers’ list and procurement prices, national unit cost databases and published sources. The unit costs of the additional monitor and central venous catheter required for delivering EGDT were obtained from the manufacturer and the procurement department of a participating hospital. The fixed unit costs of the monitor were assigned to an individual patient, according to the assumed 5-year life cycle of the monitor, and assuming that the volume of eligible patients was the annual average recorded in the trial screening logs. In calculating the unit cost per patient, it was also assumed that to provide EGDT in routine practice each site would require two monitors, which would have an average lifespan of 5 years. The monitor costs per patient were calculated by dividing the total costs of the monitors by the expected number of eligible patients over 5 years. Unit costs for blood products and other drugs were taken from NHS Blood and Transplant47 and the British National Formulary. 48
| Items | Unit costs (£) | Source |
|---|---|---|
| Equipment and consumables | ||
| Monitora | 70 | Manufacturer’s price |
| PreSepTM central venous oximetry catheter | 130 | Manufacturer’s price |
| Standard CVC | 24 | Local NHS finance department |
| Arterial catheter | 13 | Local NHS finance department |
| Other equipment/consumables | ||
| Transducer | 13 | NHS supply chain |
| Insertion pack for CVCb | 22 | Local NHS finance department |
| Cleaning device for arterial catheterb | 5 | Local NHS finance department |
| Blood products | ||
| PRBC (280 ml) | 122 | NHSBT47 |
| Platelets (200 ml) | 208 | NHSBT47 |
| Frozen fresh plasma (250 ml) | 28 | NHSBT47 |
| Drugs | ||
| Dobutamine (250 mg)c | 9 | BNF48 |
| Staff time | ||
| Doctor: consultant (per hour) | 139 | Curtis49 |
| Doctor: registrar level (per hour) | 59 | Curtis49 |
| Nurse: grade 6 (per hour) | 49 | Curtis49 |
| Staff training costs (per patient)d | 11 | ProMISe data and assumption |
| Hospital costs (bed-day) | ||
| ED (per hour) | 27 | Dixon et al. 200950 |
| Critical care bed-day: 0 organs supported | 619 | Department of Health51 |
| Critical care bed-day: 1 organ supported | 852 | Department of Health51 |
| Critical care bed-day: 2 organs supported | 1236 | Department of Health51 |
| Critical care bed-day: 3 organs supported | 1422 | Department of Health51 |
| Critical care bed-day: 4 organs supported | 1573 | Department of Health51 |
| Critical care bed-day: 5 organs supported | 1697 | Department of Health51 |
| Critical care bed-day: 6+ organs supported | 1867 | Department of Health51 |
| General ward bed-day | 265 | Department of Health51 |
| Outpatient and community health services | ||
| Hospital outpatient (per visit) | 135 | Curtis49 |
| GP practice visit (per visit) | 45 | Curtis49 |
| GP home visit (per visit) | 114 | Curtis49 |
| GP practice nursee | 10 | Curtis49 |
| Hospital staff nursee | 12 | Curtis49 |
| Health visitore | 13 | Curtis49 |
| Occupational therapiste | 9 | Curtis49 |
| Psychologiste | 15 | Curtis49 |
| Speech and language therapiste | 9 | Curtis49 |
| Physiotherapiste | 9 | Curtis49 |
| Dietitiane | 9 | Curtis49 |
The unit costs associated with the additional staff training required to deliver EGDT were taken from national sources. The total additional training cost per site was calculated by valuing the time of the average mix of ED staff who required training to deliver EGDT. The average additional staff training cost per patient was calculated by dividing the total training costs per site, by the volume of eligible patients per site over 5 years, the assumed life cycle of EGDT. The costs per critical care bed-day, by Healthcare Resource Group, and per general medical bed-day were taken from the ‘Payment by Results’ database. 51 Unit costs for hospital outpatient visits and community service use were obtained from a recommended published source for Health and Social Care costs. 49 All unit costs were reported in 2012–13 prices.
Health-related quality of life
The responses to the EQ-5D questionnaire were used to report each patient’s described health, which was then valued according to health state preferences from the general population to calculate EQ-5D utility scores, anchored on a scale from 0 (death) to 1 (perfect health). 52 The number and percentage of patients in each level of each dimension were reported by treatment group.
Sensitivity analysis
The main assumptions of the cost-effectiveness analysis were subjected to extensive sensitivity analyses. The main assumptions made in the base-case scenario, and how each was relaxed in sensitivity analyses, are detailed below and summarised in Table 3.
| Assumption | Base case | Sensitivity analysis |
|---|---|---|
| Equipment costs for the intervention | Unit costs as per business deal option | Manufacturer’s list price |
| Staff monitoring time | 10 minutes per hour of protocol | 5–15 minutes per hour of protocol |
| Staff training time | 20 minutes’ training time for all ED staff | 15–30 minutes’ training time for all ED staff |
| Location of protocol delivery | Protocol delivered in both ED and ICU | Protocol delivered exclusively either in ED or in ICU |
| Readmissions from Health Services Questionnaires | Included in the analysis | Excluded from the analysis |
| Baseline covariates | Unadjusted analysis | Adjusted for components of MEDS score |
| Distributional assumptions | Costs and QALYs normally distributed | Costs and QALYs gamma distributed |
Equipment costs for the intervention
In the base case, unit costs for the monitor and central venous catheter for ScvO2 monitoring were taken from the manufacturer’s discounted costs, which were judged to be those which would be paid by NHS providers if EGDT was introduced into routine clinical practice. These unit costs imply discounts of over 50% from list prices. In the sensitivity analysis, full list prices were applied for the requisite monitor and catheters.
Staff monitoring time during delivery of the early goal-directed therapy resuscitation protocol
The intervention requires intensive monitoring of patients for the duration of EGDT (up to 6 hours). In the base case, it was assumed that this monitoring would require an additional 10 minutes of nurses’ time per hour of the resuscitation protocol. In the sensitivity analysis, the additional nurses’ time was varied from 5 to 15 minutes per hour over the duration of EGDT.
Staff training time for delivery of the early goal-directed therapy resuscitation protocol
The base-case analysis assumed that when EGDT was provided in the ED, each member of staff would require 20 minutes of training. In the sensitivity analysis, training time was varied between 15 and 30 minutes.
Location of delivery of the early goal-directed therapy resuscitation protocol
The base-case analysis incorporated the relative time that each patient in the EGDT group received the protocol in the ED versus an ICU. In practice, EGDT may be exclusively delivered in either setting. The sensitivity analysis allowed the costs of monitoring and training to reflect either extreme, namely EGDT delivered entirely in the ED or EGDT delivered entirely in ICU. All other aspects of staff time required to deliver the EGDT protocol were assumed to be the same across location (ED or ICU). As with the preceding scenarios, only the costs were allowed to change in the sensitivity analysis; it was assumed that the relative effectiveness of EGDT versus usual care was the same as in the original base-case analysis.
Readmissions from Health Services Questionnaire
The base-case analysis included hospital readmissions including a critical care stay recorded in the Case Mix Programme database but also hospital readmissions recorded from responses to the Health Services Questionnaire. To consider the possible impact of double-counting the same readmissions across both sources, in the sensitivity analysis only the readmissions from the Case Mix Programme database were included.
Baseline covariates
The base-case analysis reported incremental costs and QALYs without any covariate adjustment, assuming randomisation had ensured no imbalances in key prognostic factors such as components of the MEDS score. 34 In the sensitivity analysis, any chance imbalances in components of the MEDS score were adjusted for using seemingly unrelated regression.
Distributional assumptions for costs and quality-adjusted life-years
The base-case analysis assumed that costs and QALYs were normally distributed when reporting the 95% CIs around incremental costs and QALYs. In sensitivity analyses the robustness of the cost-effectiveness results to alternative distributional assumptions about both outcomes were assessed. Following methodological guidance, the sensitivity analysis considered a gamma distribution for costs as they had a right-skewed distribution. For QALYs, the sensitivity analysis also considered a gamma distribution because a large proportion of decedents had zero QALYs, and the remainder of the distribution was again right-skewed. In this sensitivity analysis, costs and QALYs were modelled as univariate regression models assuming a gamma distribution for each end point (i.e. ignoring possible correlation between the end points).
Cost-effectiveness at 90 days following randomisation
Mean EQ-5D utility scores, QALYs, total costs and INBs up to 90 days were reported for each treatment group. Unadjusted mean differences between the treatment groups in quality of life, QALYs, incremental costs and INBs at 90 days were reported with 95% CIs. These were reported both overall and by each of the pre-specified subgroups, and tested using the t-test.
For survivors at 90 days, QALYs were calculated by valuing each patient’s survival time by their health-related quality of life according to the ‘area under the curve’ approach,53 assuming an EQ-5D utility score of zero at randomisation, and a linear interpolation between randomisation and 90 days. Zero QALYs were assumed for decedents between randomisation and 90 days. Total costs up to 90 days were calculated by combining the resource use with unit costs. The differences in average costs and QALYs between the treatment groups were used to calculate the INBs of EGDT versus usual resuscitation. The incremental QALY was valued according to the NICE recommended threshold of willingness to pay for a QALY gain (£20,000); the incremental cost was then subtracted from this.
The uncertainty around the differences in average costs and QALYs between the treatment groups was illustrated on the cost-effectiveness plane. The incremental costs and QALYs were estimated with a seemingly unrelated regression model. To express the uncertainty in the estimation of the incremental costs and QALYs, the estimates of the means, variances and the covariance from the regression model were used to generate 500 estimates of incremental costs and QALYs from the joint distribution of these end points, assuming asymptotic normality. These incremental costs and QALYs were then plotted on the cost-effectiveness plane. A cost-effectiveness acceptability curve was also plotted by calculating the probability that, compared with usual resuscitation, EGDT is cost-effective given the data, at alternative levels of willingness to pay for a QALY gain.
As sensitivity analyses, the mean INB at 90 days with corresponding 95% CIs was also reported for each of the alternative assumptions (see Table 3).
Cost-effectiveness at 1 year following randomisation (primary outcome)
Mean EQ-5D utility scores, QALYs, total costs and INBs up to 1 year were reported for each treatment group. Unadjusted mean differences between the treatment groups in quality of life, QALYs, incremental costs and INBs at 1 year were reported with 95% CIs. These were reported both overall and by each of the pre-specified subgroups, and tested using the t-test. The incremental costs and QALYs at 1 year were plotted on the cost-effectiveness plane and the cost-effectiveness acceptability curve was plotted. As sensitivity analyses, the mean INB at 1 year with corresponding 95% CI was also reported for each of the alternative assumptions (see Table 3).
All analyses followed the same approach as that at 90 days, although for survivors at 1 year QALYs were calculated assuming an EQ-5D utility score of zero at randomisation, and using the quality-of-life scores at 90 days and 1 year, applying linear interpolation between each pair of time points. Quality-of-life scores at 90 days were applied for decedents between 90 days and 1 year.
Lifetime incremental cost-effectiveness
The cost and outcome data reported at 1 year were used to estimate the effect of EGDT versus usual resuscitation on longer-term costs and outcomes. The chosen time horizon of 20 years was judged a reasonable time frame over which to fully assess the relative impact of EGDT versus usual resuscitation, and exceeded those taken in previous studies. 54 The maximum available survival data from the trial was used to plot Kaplan–Meier survival curves out to the date of censoring (12 November 2014). Alternative parametric functions were considered for extrapolating mortality by fitting commonly recommended alternatives to the survival data, excluding that for the first 30 days, as the event rate during this early period was atypical and did not provide an appropriate basis for subsequent extrapolation. Although the relative fit of the alternative curves to the observed data was reported, the one applied gave the most plausible extrapolation according to the previous literature. 55,56 After 15 years following randomisation, it was assumed that all-cause death rates were those of the age-/sex-matched general population. The parametric extrapolation for years 2–15 was combined with applying all-cause death rates for years 16–20 to report life expectancy for each patient observed to survive at 1 year.
The lifetime analysis allowed for the mean differences in estimated survival at 1 year, but these differences were judged small and unlikely to be maintained, and were not statistically significant, and therefore, after 1 year, the same mortality rates were applied to both treatment groups. For calculating lifetime QALYs, it was judged plausible to assume that the mean differences in quality of life reported at 1 year, although not statistically significant, were maintained. For each treatment group, the level of the quality-of-life decrement observed at 1 year versus the age-/sex-matched general population57 was maintained for years 2–15, which was the same duration as the period of assumed excess mortality, after which quality-of-life values for the age-/sex-matched general population were applied. To project lifetime costs attributable to the initial episode of severe sepsis, it was assumed that the average inpatient (general medical not critical care), outpatient and community service costs reported up to 1 year following randomisation applied annually for years 2–15 (period of excess mortality). For years 16–20, it was assumed that there were no further costs attributable to the initial episode. Long-term INB over 20 years was calculated by valuing each QALY at £20,000 per QALY. All future costs and life-years were discounted at the recommended rate of 3.5%. 45
Mean lifetime QALYs, total costs and INBs were reported for each treatment group. Unadjusted mean differences between the treatment groups in lifetime QALYs, incremental costs and INBs were reported with 95% CIs. These were reported both overall and by each of the pre-specified subgroups, and tested using the t-test. The lifetime cost-effectiveness acceptability curves were also plotted.
The sensitivity analyses for lifetime INB considered the scenario from Table 3 that was judged most relevant, that of providing EGDT exclusively in the ED versus in ICU. The following additional scenarios pertinent to the lifetime analysis were also reported:
-
allowing for excess mortality versus the general population to be maintained for a shorter (10 years) and a longer (20 years) period of time than the base case (15 years)
-
allowing for a larger (30%) and smaller (10%) decrement in quality of life over years 2–15 versus the general population than the base case (20%)
-
allowing for the excess costs attributable to the initial episode to be maintained for a shorter (10 years) and a longer (20 years) period of time than the base case (15 years).
Chapter 3 Results: sites and patients
Participants: sites
Expressions of interests were received from 83 NHS hospitals in the UK. A total of 57 hospitals in England obtained local NHS permissions and opened to recruitment between 15 February 2011 and 25 March 2013. Forty-four sites were opened within the first 9 months of the trial’s opening on 15 February 2011 (Figure 4).
FIGURE 4.
Sites open to recruitment during the trial recruitment period.
The rate at which sites were opened for the ProMISe trial was higher than for ProCESS and ARISE. Within 12 months of the trial opening, 47 sites had been opened for ProMISe, compared with 20 each for ProCESS and ARISE (Figure 5).
FIGURE 5.
Recruitment of sites for ProMISe compared with ProCESS and ARISE.

The median time from local NHS permission to the trial opening at sites (i.e. start of screening) was 83 (IQR 51–151) days (Figure 6). Reasons for delays in opening were issues related to the confirmation of NHS support costs from the CLRN and delays in the local set-up of the trial, for example training staff.
FIGURE 6.
Time (in days) from local NHS permission to start of screening.
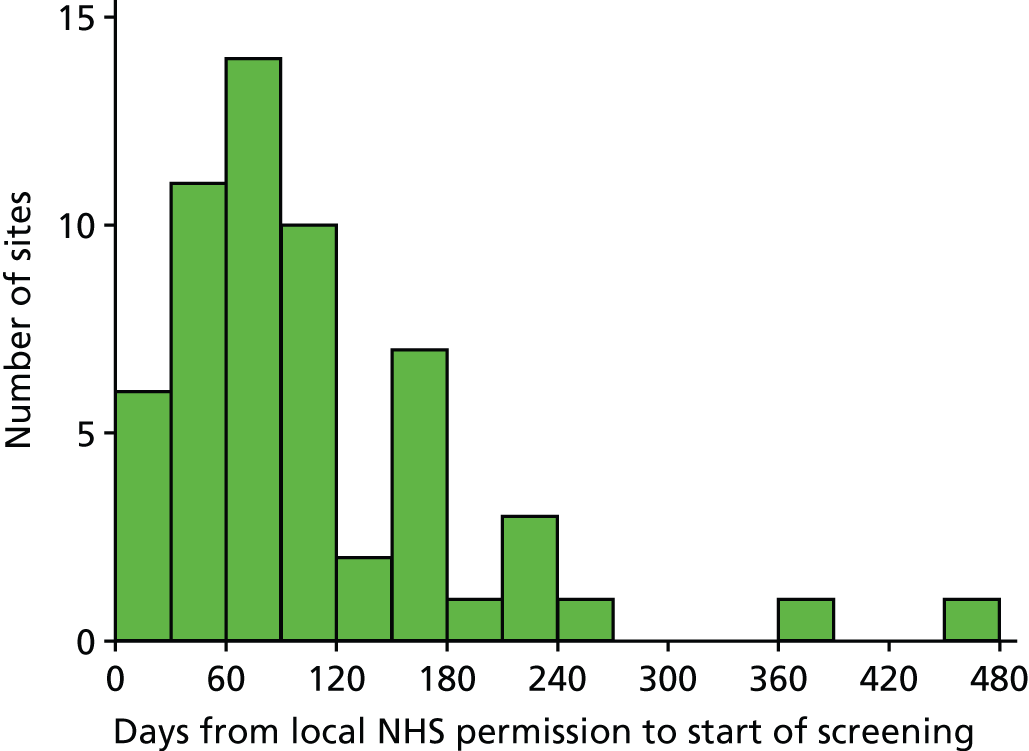
Overall, sites participated in the ProMISe trial for a median of 30 (IQR 19–35) months. Of the 57 sites that opened, seven were closed early because of poor recruitment (one site recruited no patients), two were closed because of insufficient resources locally for screening and recruitment and two were closed for other local logistical reasons. As part of the staggered close-down of the trial, nine sites were closed in October 2013 and a further eight were closed in April 2014, with 29 sites remaining open until the end of recruitment in July 2014 (Figure 7).
FIGURE 7.
Duration of participation of sites.
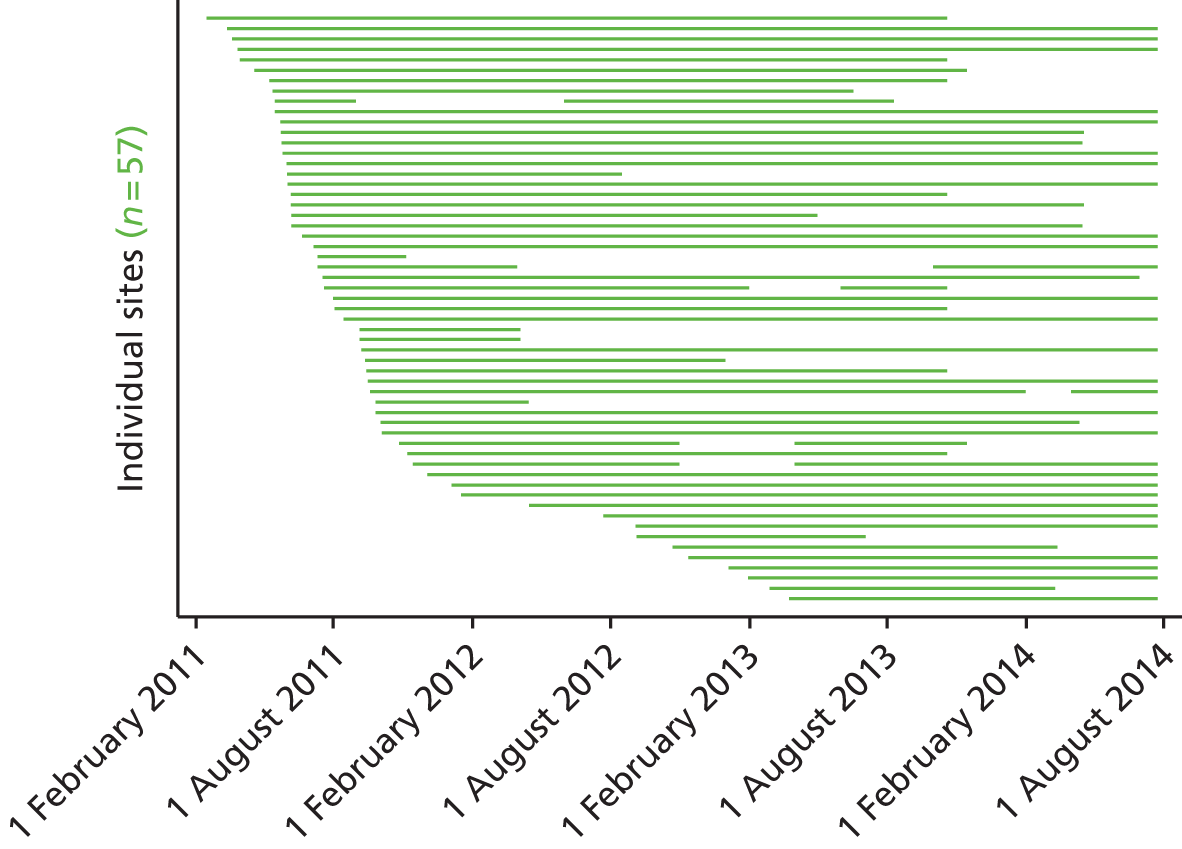
There were eight sites that had at least one period when screening and recruitment was suspended either because of insufficient resources (n = 6) or to enable new staff to be trained in delivery of the trial protocol (n = 2) (see Figure 7).
Characteristics of participating sites
A slightly higher proportion of the hospitals that participated in ProMISe were university teaching hospitals [defined as the main hospital(s) linked with each medical school] than all acute hospitals in England with an ED (Table 4).
| Type of hospital | Hospitals in ProMISe, n (%) | All hospitals in England with an ED, n (%) |
|---|---|---|
| Teaching | 16 (28.1) | 36 (19.9) |
| Non-teaching | 41 (71.9) | 145 (80.1) |
The characteristics of the 57 participating sites are presented in Table 5. There was considerable variation in the number of hospital beds, ranging from 234 to 1313, and in the annual number of ED presentations, ranging from 40,000 to 185,000. The number of patients recruited ranged from 1 to 83 patients per site in the 56 sites that recruited one or more patients.
| Site | Type of hospital | Recruitment period | Hospital beds | Annual ED presentations | Total patients randomised |
|---|---|---|---|---|---|
| Addenbrooke’s Hospital | Teaching | November 2011–October 2012 and April 2013–July 2014 | 950 | 100,000 | 30 |
| Arrowe Park Hospital | Non-teaching | June 2011–July 2014 | 750 | 93,000 | 31 |
| Barnsley Hospital | Non-teaching | September 2011–January 2014 and April 2014–July 2014 | 450 | 80,000 | 28 |
| Bedford Hospital | Non-teaching | November 2011–October 2013 | 380 | 70,000 | 11 |
| Birmingham Heartlands Hospital | Non-teaching | May 2011–October 2013 | 730 | 112,171 | 14 |
| Blackpool Victoria Hospital | Non-teaching | November 2012–July 2014 | 769 | 92,000 | 9 |
| Bristol Royal Infirmary | Teaching | September 2011–December 2012 | 450 | 65,000 | 6 |
| Broomfield Hospital | Non-teaching | May 2011–April 2014 | 521 | 81,513 | 15 |
| Chelsea and Westminster Hospital | Non-teaching | May 2011–July 2014 | 430 | 114,695 | 16 |
| Derriford Hospital | Teaching | April 2011–November 2013 | 900–1000 | 87,000 | 12 |
| Dorset County Hospital | Non-teaching | October 2011–April 2014 | 292 | 40,000 | 17 |
| Frenchay Hospital | Non-teaching | April 2012–July 2014 | 526 | 88,000 | 25 |
| Good Hope Hospital | Non-teaching | September 2011–April 2012 | 480 | 78,713 | 0 |
| Hinchingbrooke Hospital | Non-teaching | May 2011–April 2014 | 247 | 44,962 | 19 |
| Hull Royal Infirmary | Teaching | May 2011–July 2014 | 709 | 122,000 | 30 |
| John Radcliffe Hospital | Teaching | July 2011–January 2013 and June 2013–October 2013 | 832 | 137,766 | 8 |
| Kettering General Hospital | Non-teaching | February 2011–October 2013 | 580 | 88,000 | 15 |
| King’s College Hospital | Teaching | July 2012–July 2014 | 1000 | 140,000 | 33 |
| Leicester Royal Infirmary | Teaching | December 2011–July 2014 | 1000 | 150,000 | 41 |
| Leighton Hospital | Non-teaching | June 2011–October 2013 | 460 | 82,000 | 12 |
| Manchester Royal Infirmary | Teaching | July 2011–July 2014 | 650 | 100,000 | 41 |
| Medway Maritime Hospital | Non-teaching | June 2011–April 2014 | 550 | 90,000 | 27 |
| Musgrove Park Hospital | Non-teaching | August 2011–July 2014 | 700 | 56,000 | 29 |
| New Cross Hospital | Non-teaching | September 2011–October 2013 | 700 | 111,000 | 8 |
| Newham University Hospital | Non-teaching | September 2012–July 2013 | 234 | 125,000 | 10 |
| North Devon District Hospital | Non-teaching | September 2012–July 2014 | 281 | 40,000 | 20 |
| North Tyneside General Hospital | Non-teaching | September 2011–April 2012 | 450 | 60,000 | 1 |
| Peterborough City Hospital | Non-teaching | March 2013–July 2014 | 611 | 90,475 | 24 |
| Poole Hospital | Non-teaching | May 2011–June 2013 | 623 | 67,000 | 42 |
| Queen Elizabeth Hospital, Birmingham | Teaching | July 2011–March 2012 and October 2013–July 2014 | 1313 | 102,000 | 21 |
| Queen Elizabeth Hospital, Gateshead | Non-teaching | September 2011–July 2014 | 600 | 87,000 | 30 |
| Queen’s Medical Centre | Teaching | January 2013–July 2014 | 1300 | 185,000 | 25 |
| Royal Berkshire Hospital | Non-teaching | August 2011–July 2014 | 660 | 100,000 | 55 |
| Royal Bournemouth Hospital | Non-teaching | July 2011–June 2014 | 607 | 71,316 | 23 |
| Royal Lancaster Infirmary | Non-teaching | May 2011–July 2014 | 428 | 56,000 | 21 |
| Royal Preston Hospital | Non-teaching | June 2011–July 2014 | 708 | 74,852 | 22 |
| Royal Surrey County Hospital | Non-teaching | March 2011–October 2013 | 550 | 71,175 | 15 |
| Royal Sussex County Hospital | Teaching | September 2011–July 2014 | 850 | 110,000 | 29 |
| Royal Victoria Infirmary | Teaching | May 2011–August 2011 and June 2012–August 2013 | 1000 | 130,756 | 2 |
| Salford Royal Hospital | Non-teaching | January 2012–July 2014 | 661 | 88,000 | 53 |
| South Tyneside District Hospital | Non-teaching | June 2011–August 2012 | 400 | 74,000 | 4 |
| Southend University Hospital | Non-teaching | July 2011–November 2011 | 700 | 89,965 | 1 |
| Stafford Hospital | Non-teaching | June 2011–May 2013 | 299 | 46,761 | 15 |
| The Great Western Hospital | Non-teaching | October 2011–October 2012 and April 2013–November 2013 | 400 | 70,000 | 15 |
| The Ipswich Hospital | Non-teaching | June 2011–April 2014 | 500 | 80,000 | 18 |
| The James Cook University Hospital | Non-teaching | January 2012–July 2014 | 1000 | 104,000 | 28 |
| The Queen Elizabeth Hospital, King’s Lynn | Non-teaching | May 2011–July 2014 | 489 | 55,000 | 71 |
| The Royal Blackburn Hospital | Non-teaching | October 2012–March 2014 | 693 | 177,901 | 8 |
| The Royal London Hospital | Teaching | September 2011–July 2014 | 680 | 150,000 | 49 |
| Torbay Hospital | Non-teaching | February 2013–March 2014 | 400 | 117,896 | 3 |
| University College Hospital | Teaching | March 2011–July 2014 | 665 | 129,000 | 33 |
| University Hospital of North Staffordshire | Teaching | March 2011–July 2014 | 1180 | 128,000 | 21 |
| Wansbeck General Hospital | Non-teaching | September 2011–April 2012 | 350 | 60,000 | 1 |
| Whipps Cross University Hospital | Non-teaching | January 2013–July 2014 | 450 | 110,000 | 8 |
| Whiston Hospital | Non-teaching | March 2011–July 2014 | 646 | 100,895 | 83 |
| Worthing Hospital | Non-teaching | August 2011–October 2013 | 500 | 58,000 | 17 |
| York Hospital | Teaching | October 2011–July 2014 | 700 | 85,000 | 15 |
Participants: patients
In total, 6192 patients were screened between 15 February 2011 and 24 July 2014. Of these, 2415 (39.0%) met one or more exclusion criteria. There were 2517 (40.6%) patients who, although eligible for inclusion in the trial, were not recruited. The most frequently reported reason for not recruiting eligible patients was logistical issues, mainly no research staff being available, for example if the patient presented at the ED outside usual office hours. Other reported reasons included refusal by the treating clinician to recruit the patient; the patient declined to take part; or the patient was identified as eligible for the trial outside the 2-hour window for obtaining consent and randomising (Figure 8).
FIGURE 8.
Screening, randomisation and follow-up. AIDS, acquired immune deficiency syndrome.
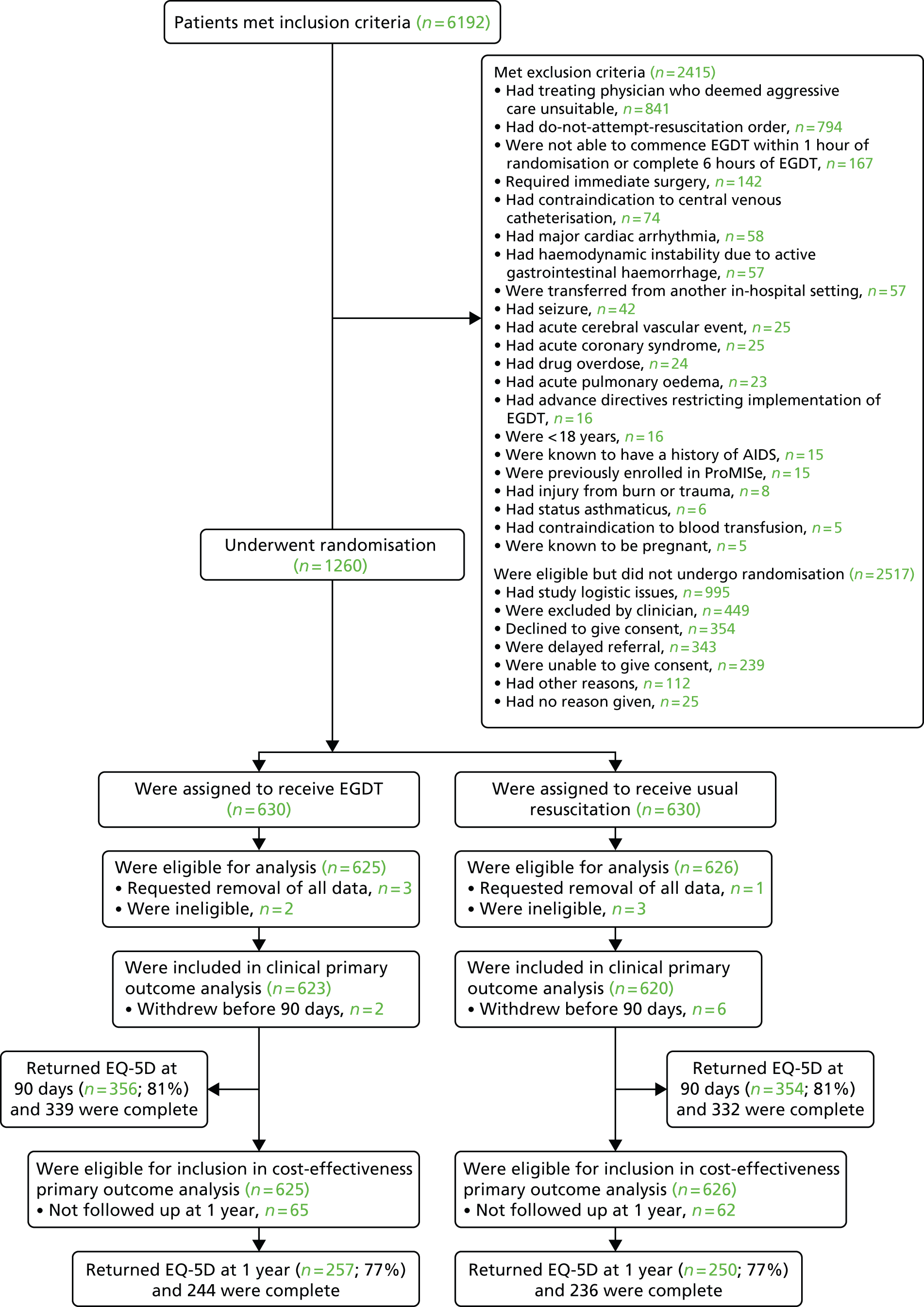
The 1260 (20.3%) patients were recruited between 16 February 2011 and 24 July 2014, with 630 randomised to the EGDT group and 630 randomised to the usual-resuscitation group (Figure 9). There was variation across the 56 sites in the rate of recruitment (Figure 10), the overall median recruitment rate being 0.15 (IQR 0.10–0.22) patients per site per week, with a highest recruitment rate of 0.48 patients per week. Patients were recruited over a relatively shorter time period than in ProCESS and ARISE (Figure 11). Manual randomisation was required for three patients.
FIGURE 9.
Patient recruitment.

FIGURE 10.
Patient recruitment rate.

FIGURE 11.
Recruitment of patients for ProMISe compared with ProCESS and ARISE.
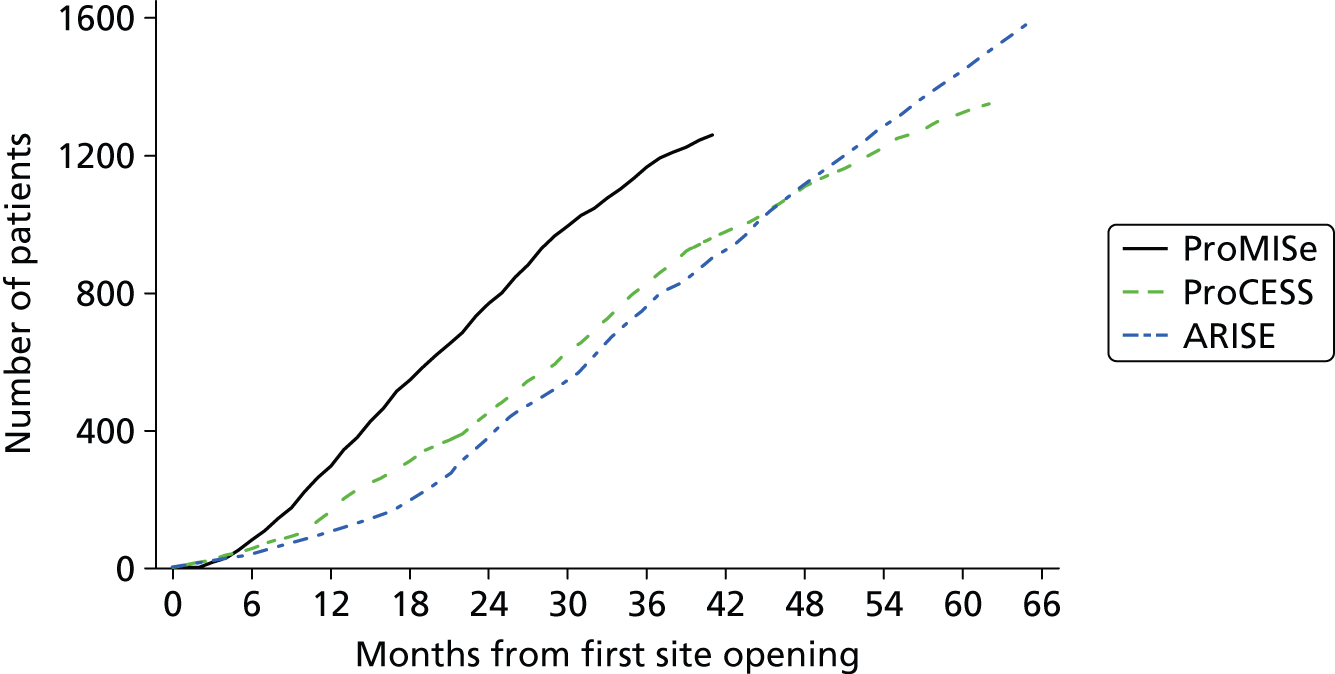
Patients were generally recruited into ProMISe during weekdays (Monday to Friday) and during usual office hours (Figure 12); most of the recruiting sites reported having insufficient resources to enable screening and recruitment at weekends and outside usual office hours.
FIGURE 12.
Randomisation by (a) day of the week and (b) time of day.
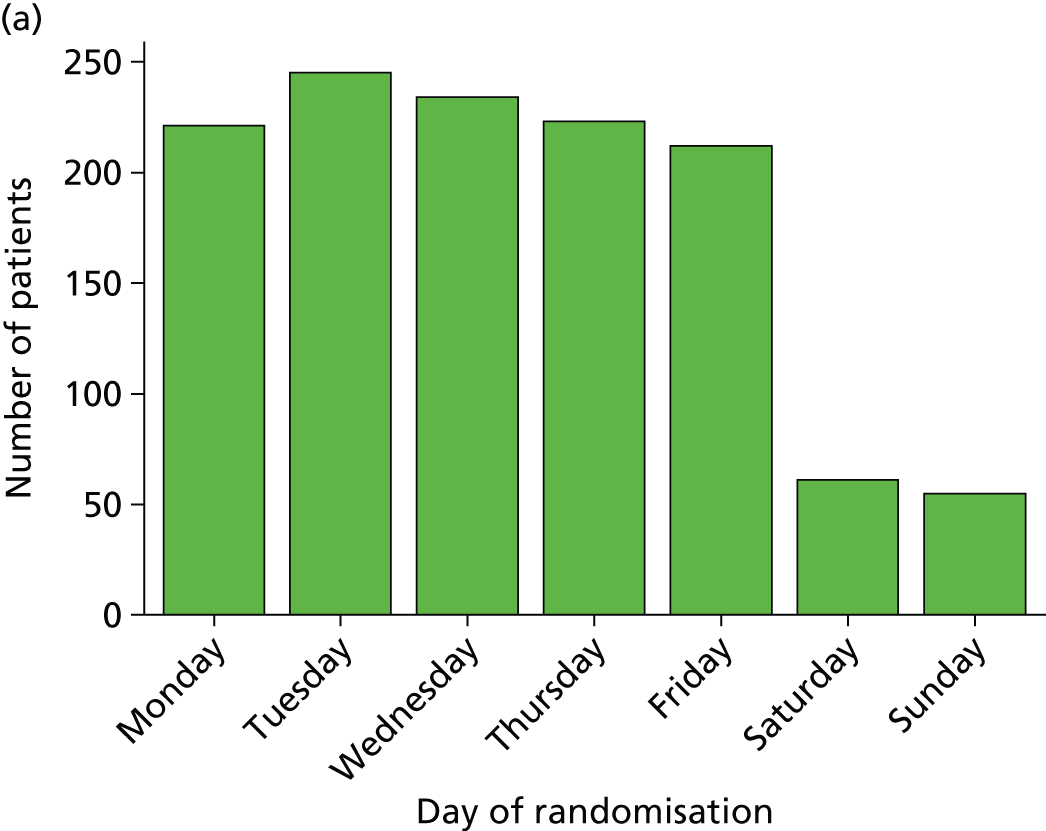
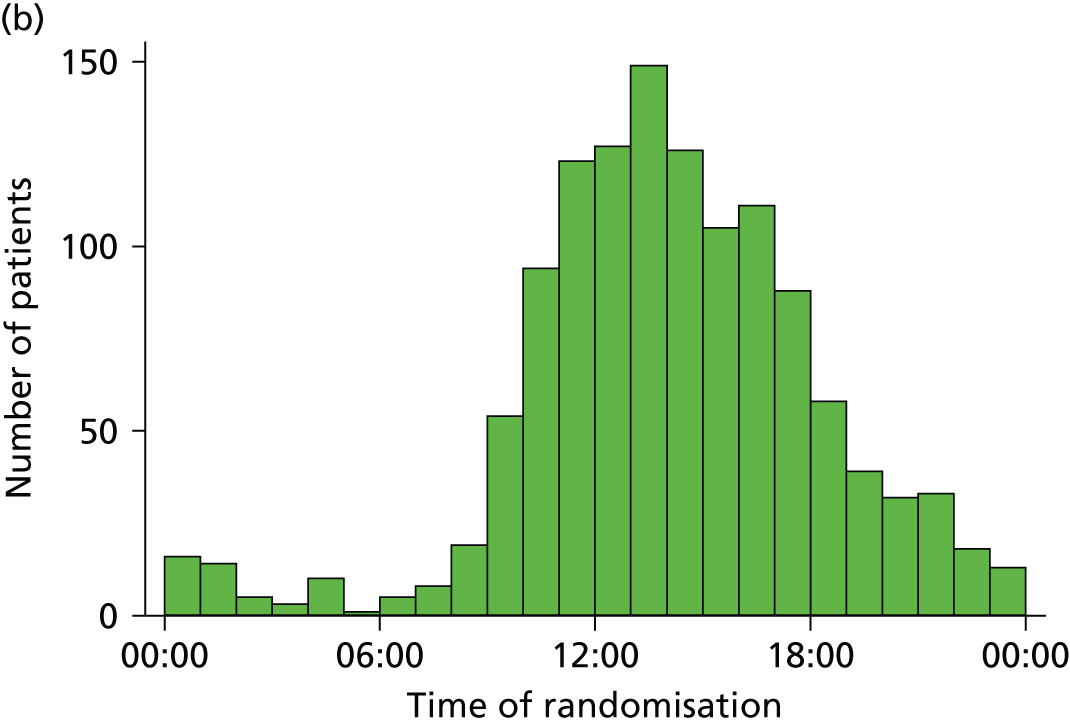
Almost half of patients provided informed consent prior to randomisation (n = 624, 49.5%). For the remaining patients, agreement was obtained from a personal (34.8%) or professional (2.9%) consultee or from an independent clinician using emergency consent (12.8%) (Table 6). Four patients withdrew from the trial, requesting the removal of all of their data from the analysis, and five patients were ineligible and recruited in error, resulting in data on 1251 for initial analysis (n = 625 EGDT; n = 626 usual resuscitation). Eight patients subsequently withdrew before 90 days, resulting in 1243 patients for analysis of outcomes (n = 623, 99.7% EGDT; n = 620, 99.0% usual resuscitation). Owing to the truncation of follow-up, 127 patients were not followed up at 1 year (n = 65 EGDT; n = 62 usual resuscitation) (see Figure 8 and Table 6). Follow-up was completed on 30 October 2014.
| Type of consent/agreement | Patients, n (%) | Requested removal of all data, n | Ineligible: excluded from analysis, n | Withdrew before 90 days, n |
|---|---|---|---|---|
| Informed consent from patient prior to randomisation | 624 (49.5) | 0 | 5 | 1 |
| Agreement from a personal consultee | 439 (34.8) | 1 | 0 | 4 |
| Agreement from a professional consultee | 36 (2.9) | 1 | 0 | 0 |
| Agreement via emergency consent | 161 (12.8) | 2 | 0 | 3 |
| Total | 1260 (100) | 4 | 5 | 8 |
Characteristics of patients at baseline
The groups were well matched at baseline (Table 7). The criterion for refractory hypotension was met in 338 (54.1%) EGDT and 348 (55.6%) usual-resuscitation patients and for hyperlactataemia in 409 (65.4%) EGDT and 399 (63.7%) usual-resuscitation patients. Intravenous fluid volume prior to randomisation was similar [median 1950 ml (IQR 1000–2500 ml) EGDT, 2000 ml (IQR 1000–2500 ml) usual resuscitation]. The median time from ED presentation to meeting inclusion criteria [1.3 (IQR 0.5–2.3) hours for EGDT and 1.3 (IQR 0.6–2.4) hours for usual resuscitation] and from ED presentation to randomisation [2.5 (IQR 1.8–3.5) hours EGDT and usual resuscitation] was the same in both groups. Only two-thirds of patients in either group were deemed as likely to be admitted to an ICU from ED (if not enrolled in the trial); those deemed unlikely to be admitted were less severely ill.
| Characteristics | EGDT (N = 625) | Usual resuscitation (N = 626) |
|---|---|---|
| Refractory hypotension, n (%) | 338 (54.1) | 348 (55.6) |
| SBP (mmHg), mean (SD) | 77.7 (11.0) | 78.4 (10.2) |
| MAP (mmHg), mean (SD) | 58.8 (15.8) | 59.0 (10.7) |
| Hyperlactataemia, n (%) | 409 (65.4) | 399 (63.7) |
| Blood lactate concentration (mmol/l), mean (SD) | 7.0 (3.5) | 6.8 (3.2) |
| Intravenous fluids pre hospital to randomisation,a n/N (%) | 612/625 (97.9) | 606/625 (97.0) |
| Intravenous fluids pre hospital to randomisation (ml), median (IQR) | 1950 (1000–2500) | 2000 (1000–2500) |
| Intravenous fluids pre hospital,b n/N (%) | 119/616 (19.3) | 128/617 (20.7) |
| Intravenous fluids pre hospital (ml), median (IQR) | 500 (250, 500) | 500 (255, 500) |
| Intravenous fluids ED presentation to randomisation,b n/N (%) | 607/625 (97.1) | 599/625 (95.8) |
| Intravenous fluids ED presentation to randomisation (ml), median (IQR) | 1600 (1000–2500) | 1790 (1000–2500) |
| Blood products ED presentation to randomisation, n/N (%) | 4/614 (0.7) | 10/616 (1.6) |
| Blood products ED presentation to randomisation (ml), median (IQR) | 922 (559–1000) | 919 (500–1000) |
| Supplemental O2,c n/N (%) | 397/539 (73.7) | 407/542 (75.1) |
| Time from ED presentation to inclusion criteria met (hours), mean (SD) | 1.6 (1.3) | 1.7 (1.4) |
| Time from ED presentation to inclusion criteria met (hours), median (IQR) | 1.3 (0.5–2.3) | 1.3 (0.6–2.4) |
| Time from ED presentation to randomisation (hours), mean (SD) | 2.7 (1.3) | 2.8 (1.4) |
| Time from ED presentation to randomisation (hours), median (IQR) | 2.5 (1.8–3.5) | 2.5 (1.8–3.5) |
| Would have been admitted direct to ICU from ED if not enrolled into ProMISe, n (%) | 419 (67.0) | 427 (68.2) |
| Yes (APACHE II scored), mean (SD) | 20.5 (6.9) | 19.0 (7.1) |
| No (APACHE II scored), mean (SD) | 15.0 (6.1) | 15.8 (6.5) |
| Age (years), mean (SD) | 66.4 (14.6) | 64.3 (15.5) |
| Age (years), median (IQR) | 68 (58–78) | 67 (54–76) |
| Male sex, n (%) | 356 (57.0) | 367 (58.6) |
| APACHE II score,d mean (SD) | 18.7 (7.1) | 18.0 (7.1) |
| APACHE II score,d median (IQR) | 18 (13–23) | 17 (13–22) |
| MEDS score,e mean (SD) | 8.0 (3.4) | 7.9 (3.3) |
| MEDS score,e median (IQR) | 8 (6–10) | 8 (6–10) |
| MEDS terminal illness, n (%) | 11/622 (1.8) | 14/626 (2.2) |
| MEDS respiratory difficulties, n (%) | 510/620 (82.3) | 499/618 (80.7) |
| MEDS septic shock, n (%) | 277/624 (44.4) | 305/622 (49.0) |
| MEDS platelets < 150 × 109/l, n (%) | 144/585 (24.6) | 144/585 (24.6) |
| MEDS bandforms > 5%, n (%) | 52/54 (96.3) | 64/70 (91.4) |
| MEDS lower respiratory infection, n (%) | 220 (35.2) | 196 (31.3) |
| MEDS nursing home resident, n (%) | 18/622 (2.9) | 14/626 (2.2) |
| MEDS altered mental status, n (%) | 206/608 (33.9) | 208/602 (34.6) |
| MEDS age > 65 years, n (%) | 363 (58.1) | 329 (52.6) |
| SOFA score,f mean (SD) | 4.2 (2.4) | 4.3 (2.4) |
| SOFA score,f median (IQR) | 4 (2–5) | 4 (3–6) |
| SOFA respiratory dysfunction, n (%) | 323 (51.7) | 357 (57.0) |
| SOFA neurological dysfunction, n (%) | 196 (31.4) | 200 (31.9) |
| SOFA cardiovascular dysfunction, n (%) | 410 (65.6) | 433 (69.2) |
| SOFA coagulation dysfunction, n (%) | 144 (23.0) | 144 (23.0) |
| SOFA hepatic dysfunction, n (%) | 211 (33.8) | 199 (31.8) |
| SOFA renal dysfunction,f n (%) | 426 (68.2) | 406 (64.9) |
| Any severe condition in the past medical history,g n/N (%) | 181/622 (29.1) | 161/626 (25.7) |
| Severe liver disease | 11/622 (1.8) | 11/626 (1.8) |
| Severe renal disease | 4/622 (0.6) | 3/626 (0.5) |
| Severe respiratory disease | 93/622 (15.0) | 81/626 (12.9) |
| Severe cardiovascular disease | 22/622 (3.5) | 17/626 (2.7) |
| Immunocompromised | 84/622 (13.5) | 70/626 (11.2) |
| Site of infection, n (%) | ||
| Lungs | 228 (36.5) | 207 (33.1) |
| Abdomen | 40 (6.4) | 51 (8.1) |
| Blood | 97 (15.5) | 86 (13.7) |
| Central nervous system | 12 (1.9) | 9 (1.4) |
| Soft tissue | 39 (6.2) | 39 (6.2) |
| Urinary tract | 108 (17.3) | 117 (18.7) |
| Other | 21 (3.4) | 37 (5.9) |
| Not sepsish | 4 (0.6) | 3 (0.5) |
| Unknown | 76 (12.2) | 77 (12.3) |
| Organism causing infection, n (%) | ||
| Gram positive | 138 (22.1) | 141 (22.5) |
| Gram negative | 175 (28.0) | 171 (27.3) |
| Fungus/yeast | 14 (2.2) | 19 (3.0) |
| Parasite | 0 (0.0) | 2 (0.3) |
| Virus | 12 (1.9) | 9 (1.4) |
| Mixed growth | 7 (1.1) | 12 (1.9) |
| Not sepsish | 4 (0.6) | 3 (0.5) |
| Unknown (not reported or no growth) | 275 (44.0) | 269 (43.0) |
| Antimicrobial change between ED admission and 72 hours, n/N (%) | 359/615 (58.4) | 342/617 (55.4) |
The mean age of patients was similar in both groups (EGDT, 66.4 years; usual resuscitation, 64.3 years) and more than half were male (57.0% EGDT, 58.6% usual resuscitation). The site of infection (most commonly lungs) was well balanced. All patients received antimicrobials prior to randomisation.
Multiple imputation
Table 8 reports all the variables considered for multiple imputation and, for each variable, the number of missing values and the imputation model chosen.
| Variable | Missing values,a n (%) | Imputation model |
|---|---|---|
| Baseline variables | ||
| Treatment group | 0 (0) | None required |
| Age | 0 (0) | None required |
| Sex | 0 (0) | None required |
| Past medical history | 3 (0.2) | None requiredb |
| Site of infection | 0 (0) | None required |
| SOFA score | 0 (0) | None required |
| MEDS score | 0 (0) | None required |
| Admitted from nursing home | 3 (0.2) | None requiredb |
| Shortness of breath with light activity | 3 (0.2) | None requiredb |
| Altered mental status | 41(3.3) | Logistic regression |
| Septic shock | 5 (0.4) | Logistic regression |
| Respiratory difficulty | 13 (1.0) | Logistic regression |
| Low platelet count | 81 (6.5) | Logistic regression |
| Volume of intravenous fluid ED presentation to randomisation | 3 (0.2) | Predictive mean matching |
| Baseline blood lactate concentration | 32 (2.6) | Predictive mean matching |
| Baseline respiratory rate | 5 (0.4) | Predictive mean matching |
| Baseline heart rate | 1 (0.1) | Predictive mean matching |
| Baseline haemoglobin | 31 (2.5) | Predictive mean matching |
| Baseline white blood cell count | 49 (3.9) | Predictive mean matching |
| Resource use variables | ||
| Length of stay in critical care | 0 (0) | None required |
| Length of stay on general medical ward | 0 (0) | None required |
| Outpatient visits at 90 days | 242 (27.3) | Predictive mean matching |
| Acute hospital readmissions, 90 days to 1 year | ||
| Length of stay in critical care | 135 (10.8) | Predictive mean matching |
| Length of stay on general medical ward | 135 (10.8) | Predictive mean matching |
| Outpatient visits at 1 year | 306 (38.5) | Predictive mean matching |
| Mortality and quality-of-life variables | ||
| EQ-5D at 90 days | 215 (24.3) | Predictive mean matching |
| Mortality at 1 year | 135 (10.8) | Logistic regression |
| EQ-5D at 1 year | 314 (39.5) | Predictive mean matching |
Chapter 4 Results: clinical effectiveness
Adherence to the protocol
Most patients randomised to EGDT (n = 545, 87.3%) had timely insertion of a PreSepTM central venous oximetry catheter (Table 9). Two (0.3%) patients in the usual-resuscitation group had one inserted in error but these were not used for monitoring ScvO2. The reasons for failure of insertion in the EGDT group were that patients were determined either to no longer meet inclusion criteria or to now meet exclusion criteria (n = 22); process of care (lack of equipment, staff, beds, communication, error; n = 20); technical or patient difficulties (n = 18); clinician decision (n = 9); refusal by the patient (without withdrawal from the trial; n = 5); and death before insertion (n = 2). No reason was provided for four patients. The mean first ScvO2 value recorded after catheterisation (at hour 1) was 70% (SD 12%). Standard central venous catheters (not mandated) were inserted in 318 (50.9%) of the usual-resuscitation group and measurement of ScvO2 from aspirated blood samples occurred in six patients. Arterial catheters (not mandated) were inserted in the majority of patients (n = 462, 74.2% EGDT; n = 389, 62.2% usual resuscitation). EGDT was stopped prematurely in 21 patients (median time to cessation, 3 hours) owing to withdrawal of active treatment (n = 9); patient no longer considered to be septic (n = 5); error (n = 3); transfer to operating theatre (n = 1); and refusal by the patient (n = 1). No reason was provided for two patients. Of the patients who died within 6 hours (n = 17 EGDT; n = 18 usual resuscitation), five in the EGDT group and six in the usual-resuscitation group had active treatment withdrawn prior to death.
| Interventions | EGDT (N = 625) | Usual resuscitation (N = 626) |
|---|---|---|
| Supplemental O2, n/N (%) | 558/623 (89.6) | 557/625 (89.1) |
| PreSep™ central venous oximetry catheter insertion, n/N (%) | 545/624 (87.3) | 2/625 (0.3) |
| Timing of insertion, n (%) | ||
| Before hour 1 | 459 (84.5) | – |
| Hour 1 to hour 2 | 67 (12.3) | – |
| Hour 2 to hour 3 | 15 (2.8) | – |
| Hour 3 to hour 4 | 2 (0.4) | – |
| Hour 4 to hour 5 | 0 (0.0) | – |
| Hour 5 to hour 6 | 0 (0.0) | – |
| Any CVC insertion, n/N (%) | 575/624 (92.1) | 318/625 (50.9) |
| Time from randomisation to insertion (hours), mean (SD) | 1.2 (0.9) | 1.8 (1.7) |
| Time from randomisation to insertion (hours), median (IQR) | 1.1 (0.8–1.5) | 1.4 (0.6–2.9) |
| Arterial catheter insertion, n/N (%) | 462/623 (74.2) | 389/625 (62.2) |
| Time from randomisation to insertion (hours), mean (SD) | 1.3 (1.6) | 1.2 (1.7) |
| Time from randomisation to insertion (hours), median (IQR) | 1.1 (0.4–1.9) | 1.0 (0.2–1.9) |
| Any intravenous fluid,a n/N (%) | 609/623 (97.8) | 604/625 (96.6) |
| Volume (ml), mean (SD) | 2226.0 (1443.3) | 2022.3 (1271.4) |
| Volume (ml), median (IQR) | 2000 (1150–3000) | 1784 (1075–2775) |
| Intravenous colloid,a n/N (%) | 197/623 (31.6) | 180/625 (28.8) |
| Volume (ml), mean (SD) | 1061.5 (800.5) | 913.4 (626.8) |
| Volume (ml), median (IQR) | 1000 (500–1500) | 750 (500–1000) |
| Intravenous crystalloid,a n/N (%) | 584/623 (93.7) | 597/625 (95.5) |
| Volume (ml), mean (SD) | 1963.2 (1356.9) | 1766.7 (1178.4) |
| Volume (ml), median (IQR) | 1750 (999–2750) | 1500 (900–2380) |
| Vasopressors, n/N (%) | 332/623 (53.3) | 291/625 (46.6) |
| Packed red blood cell transfusion, n/N (%) | 55/623 (8.8) | 24/625 (3.8) |
| Volume (ml), mean (SD) | 426.3 (209.4) | 539.5 (294.2) |
| Volume (ml), median (IQR) | 309 (285–577) | 535 (305–607) |
| Dobutamine, n/N (%) | 113/623 (18.1) | 24/625 (3.8) |
| Mechanical ventilation, n/N (%) | 126/623 (20.2) | 119/625 (19.0) |
| Sedatives, n/N (%) | 138/623 (22.2) | 130/625 (20.8) |
| Neuromuscular blocking agent, n/N (%) | 53/623 (8.5) | 40/625 (6.4) |
| Critical care admission, n/N (%) | 551/625 (88.2) | 467/626 (74.6) |
| Time from randomisation to admission (hours), mean (SD) | 2.0 (2.3) | 2.5 (5.7) |
| Time from randomisation to admission (hours), median (IQR) | 1.2 (0.4–2.8) | 1.2 (0.3–2.8) |
| Location of protocol delivery, n (%) | ||
| ED | 64 (10.2) | – |
| ICU | 275 (44.0) | – |
| Ward | 10 (1.6) | – |
| ED and ICU | 235 (37.6) | – |
| ED and ward | 37 (5.9) | – |
| ICU and ward | 2 (0.3) | – |
| ED, ICU and ward | 1 (0.2) | – |
| Review by consultant, n/N (%) | 520/624 (83.3) | 494/625 (79.0) |
| Specialty of most senior doctor to review the patient, n (%) | ||
| Emergency medicine | 181 (29.0) | 211 (33.8) |
| Critical care medicine | 388 (62.2) | 304 (48.6) |
| Acute medicine | 39 (6.3) | 92 (14.7) |
| Other | 16 (2.6) | 18 (2.9) |
Figure 13 shows the adherence with each element of the EGDT protocol across the 6-hour intervention period. The bars plotted on each hour report the percentage of patients (of those meeting the previous targets) who did not meet or met each physiological target at that hour, or for whom the relevant physiological value was not recorded (within 15 minutes either side of the hour). The bars plotted between each hour report the percentage of patients (of those who did not meet the physiological target at the previous hour) that received the associated action either during the intervention period or after the intervention period, or who no longer required the action as the target was subsequently met. Adherence with the protocol was generally good, particularly for delivery of fluid and vasopressors, but there were delays in obtaining packed red blood cells and dobutamine, such that the ScvO2 value had often resolved spontaneously before these were delivered.
FIGURE 13.
Adherence to the EGDT protocol. (a) Target: CVP; action: fluids; (b) target: MAP/SBP; action: vasopressors; and (c) target: ScVO2; action: PRBC transfusion (L) or dobutamine (R). Numbers at the foot of each bar represent the denominator for the percentages in that bar. Each target is considered sequentially (i.e. those meeting the target for CVP become the denominator for MAP/SBP and those meeting the target for MAP/SBP become the denominator for ScvO2). Of the two smaller bars, (L) denotes the left-hand bar and (R) denotes the right-hand bar. Thirty-two patients who no longer met eligibility criteria or declined the intervention were excluded from the evaluation of adherence. CVP, central venous pressure; MAP, mean arterial pressure; PRBC, packed red blood cells; SBP, systolic blood pressure.


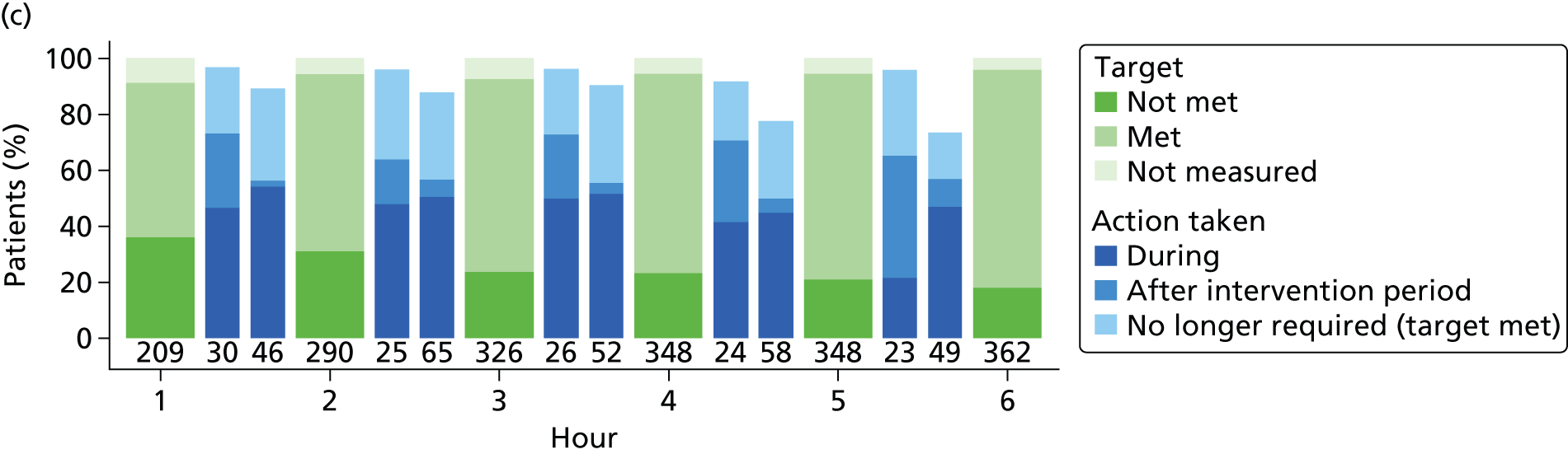
Figure 14 reports the protocol adherence compared with ProCESS and ARISE, according to the adherence algorithms reported in their respective publications. 23,24 For the comparison with ProCESS, adherence was assessed at the end of the 6-hour intervention period only. Patients who died, were discharged or were withdrawn from the intervention prior to 6 hours were excluded (47/625, 7.5%, for ProMISe; 35/439, 8.0%, for ProCESS). Overall adherence among the evaluable patients was similar for the two trials (85.6% for ProMISe vs. 88.1% for ProCESS). The greatest difference was on failure to insert a central venous catheter with ScvO2 monitoring capability; on all other measures, non-adherence for ProMISe was lower than for ProCESS. For the comparison with ARISE, adherence was assessed hourly as the percentage of patients meeting each physiological target (of those for whom the relevant physiological value was recorded) and, for those with physiological values recorded at 2 consecutive hours, the percentage who either met the target at the start or the end of the hour or received the associated action during the hour. The percentage of patients meeting the physiological targets at each hour, for ProMISe compared with ARISE, was similar for central venous pressure, higher for mean arterial pressure/systolic blood pressure and lower for ScvO2. When reported as the percentage either meeting the target or receiving the associated action, adherence was extremely high (and similar to ARISE) for both receipt of intravenous fluids and receipt of vasopressors, but somewhat lower for receipt of packed red blood cells or dobutamine (although this did reach a level of 89% by the final hour of the intervention period).
FIGURE 14.
Protocol adherence compared with ProCESS (a) and ARISE [(b), CVP; (c) i.v. fluids; (d) MAP or SBP; (e) vasopressors; (f) ScvO2; and (g) PRBC and/or dobutamine]. For the comparison with ProCESS, the numbers of evaluable patients were 578 (ProMISe) and 404 (ProCESS). For the comparison with ARISE, numbers at the foot of each bar represent the denominator for the percentages in that bar. CVP, central venous pressure; i.v., intravenous; MAP, mean arterial pressure; PRBC, packed red blood cells; SBP, systolic blood pressure.



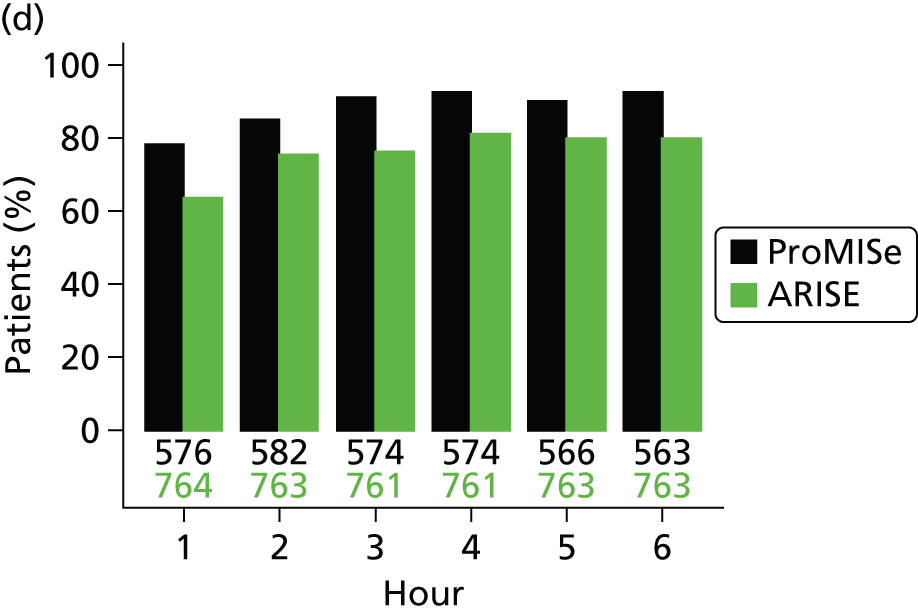
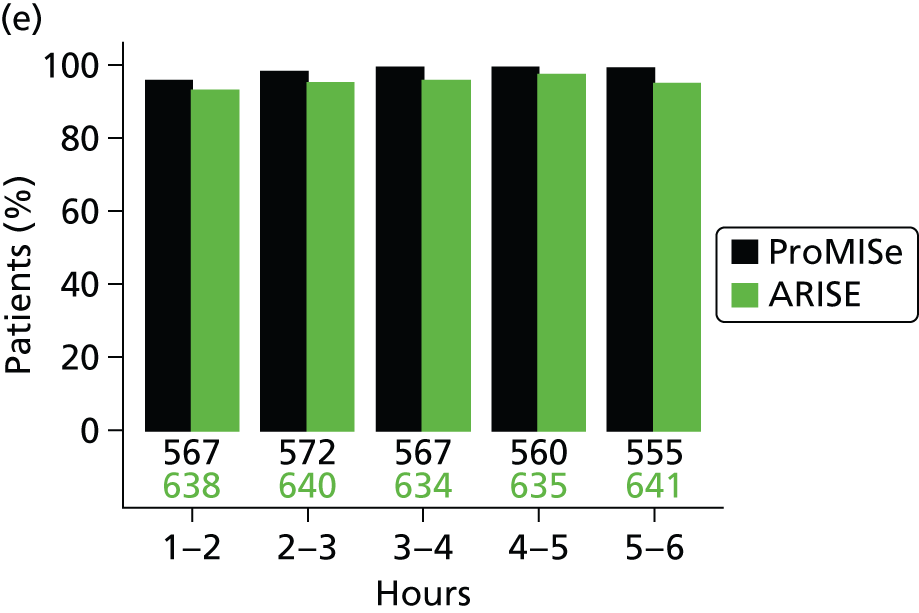

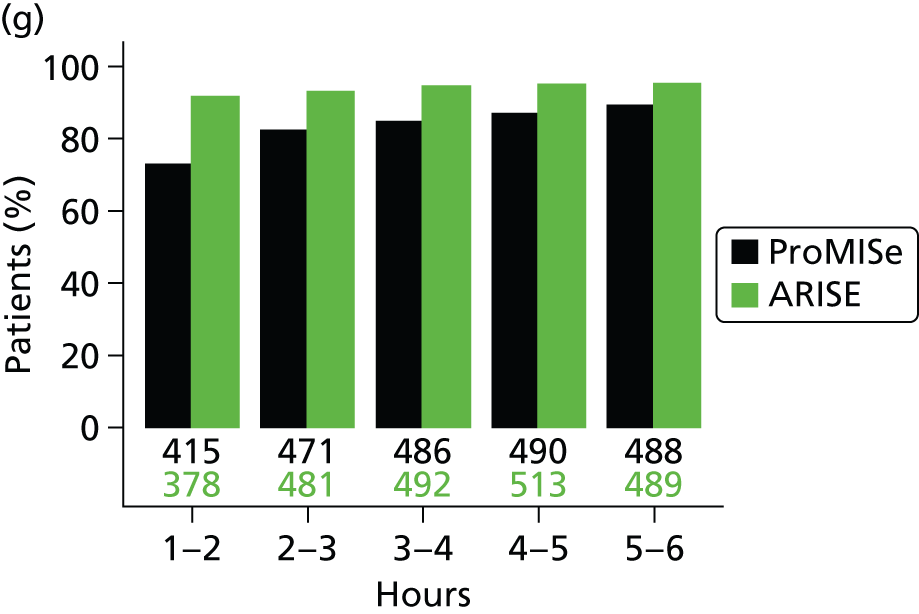
Delivery of care by treatment group
During the 6-hour intervention period, patients randomised to EGDT received more intravenous fluid than those randomised to usual resuscitation (see Table 9). Hourly fluid volume decreased over the 6 hours but patients in the usual-resuscitation group received a greater volume initially (Figure 15). In both groups, crystalloid was used more frequently than colloid. More patients in the EGDT group received vasopressors and dobutamine. Although more patients in the EGDT group received packed red blood cell transfusions, larger volumes were transfused in the usual-resuscitation group. Administration of platelets and fresh-frozen plasma was similar, although volumes of both were higher in the EGDT group. Physiological values normalised slightly over the 6-hour intervention period in both groups (Figure 16). After 6 hours, central venous pressure, mean arterial pressure, systolic blood pressure and haemoglobin, where measured (with greater frequency in the EGDT group), were similar (Table 10).
FIGURE 15.
Interventions delivered during the intervention period. The values reported are the mean and 95% CI (volume of i.v. fluid) or percentage of patients receiving the intervention (all other panels) during each hour of the intervention period. The numbers at the foot of each bar represent the denominators for the means/percentages in that bar. (a) Volume of i.v. fluid; (b) vasopressors; (c) PRBC transfusion; (d) dobutamine; (e) sedatives; (f) neuromuscular blocking agents; and (g) mechanical ventilation. i.v., intravenous; PRBC, packed red blood cells.
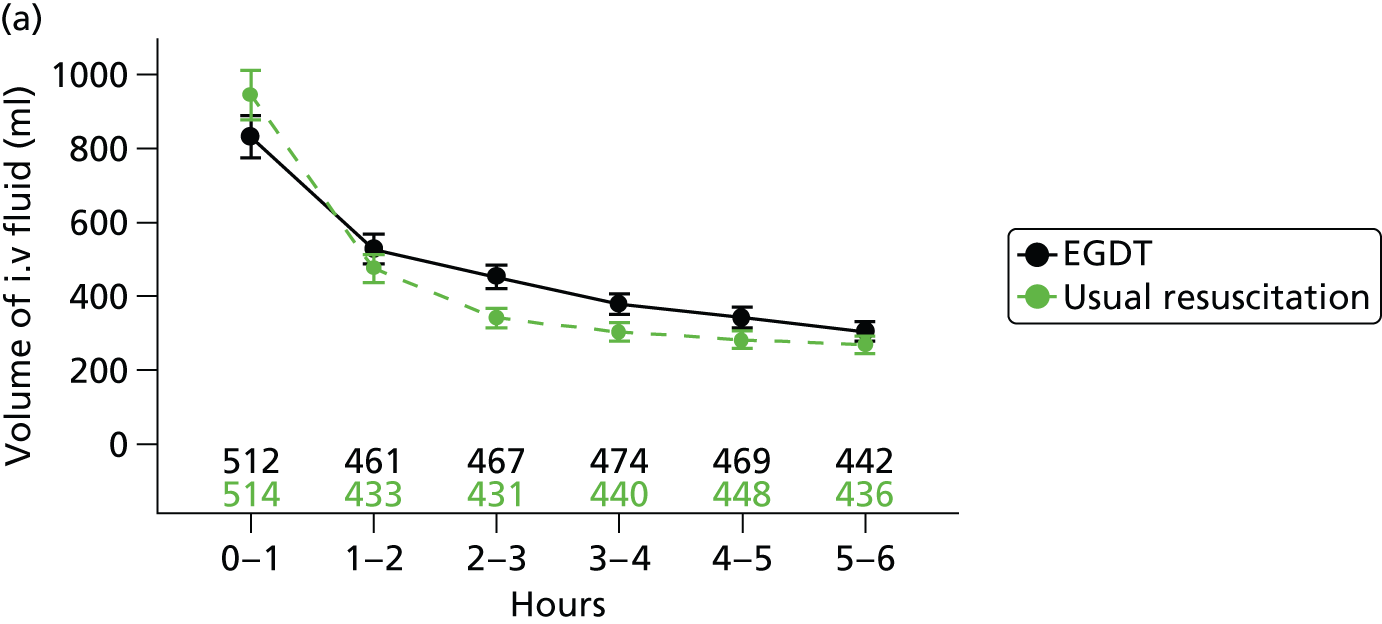
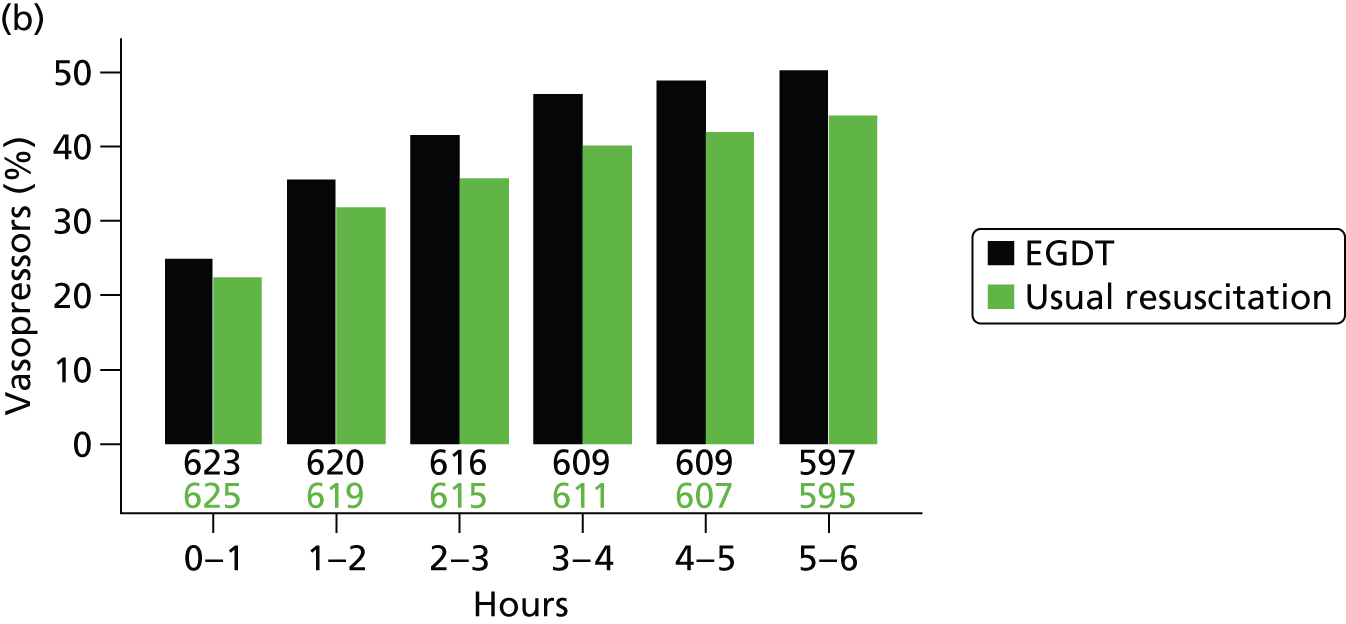
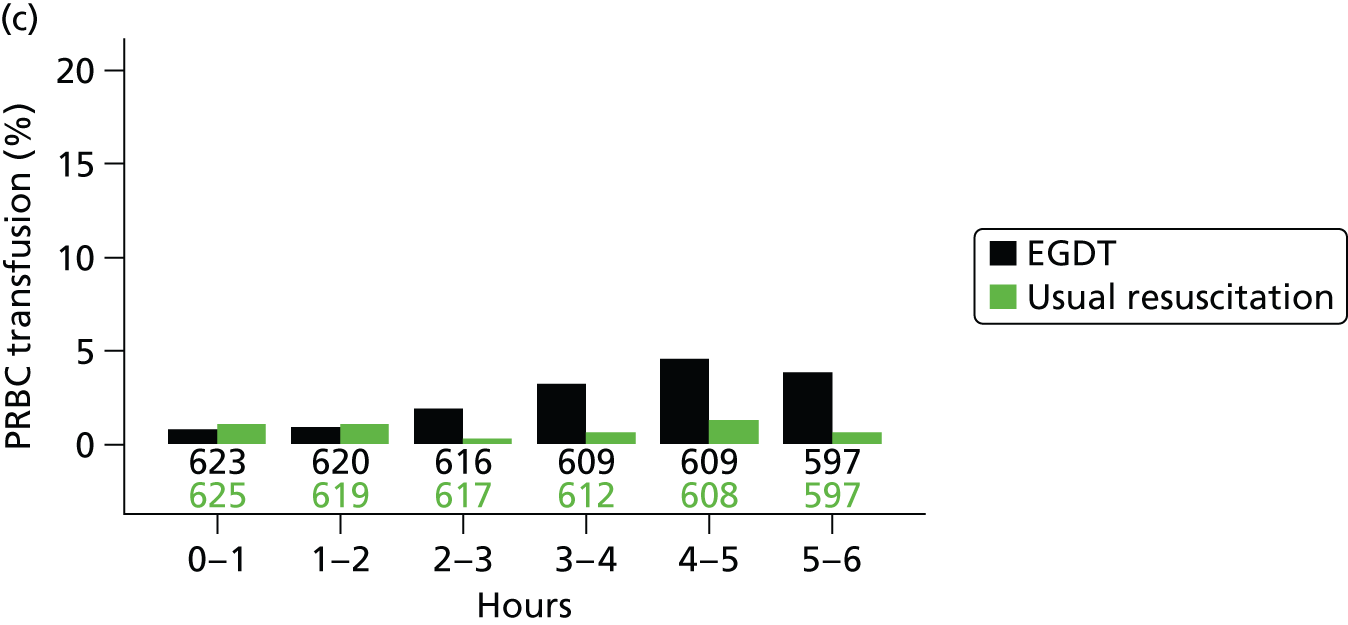
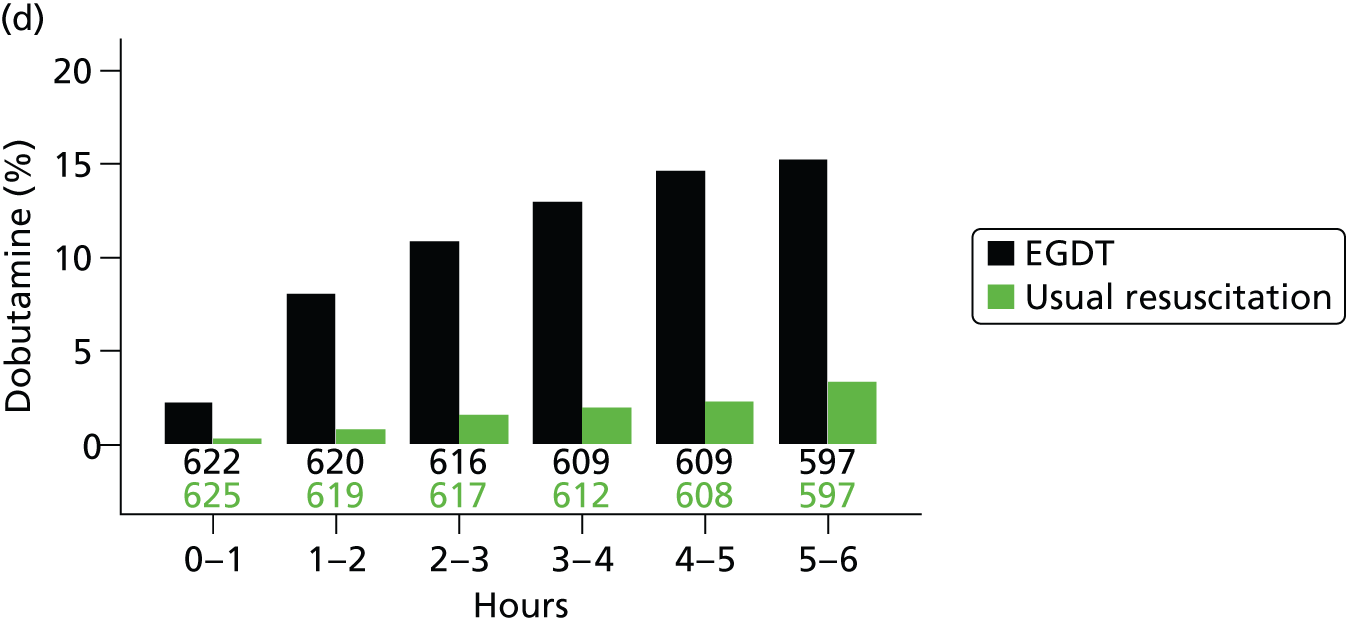
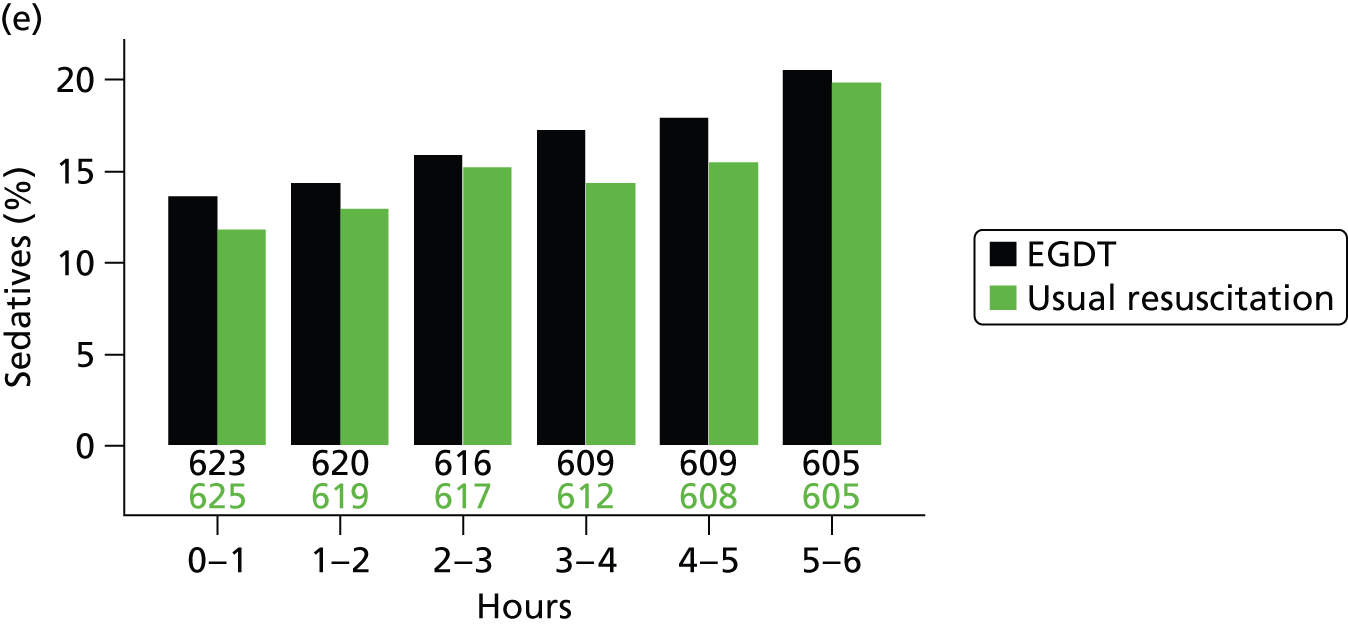

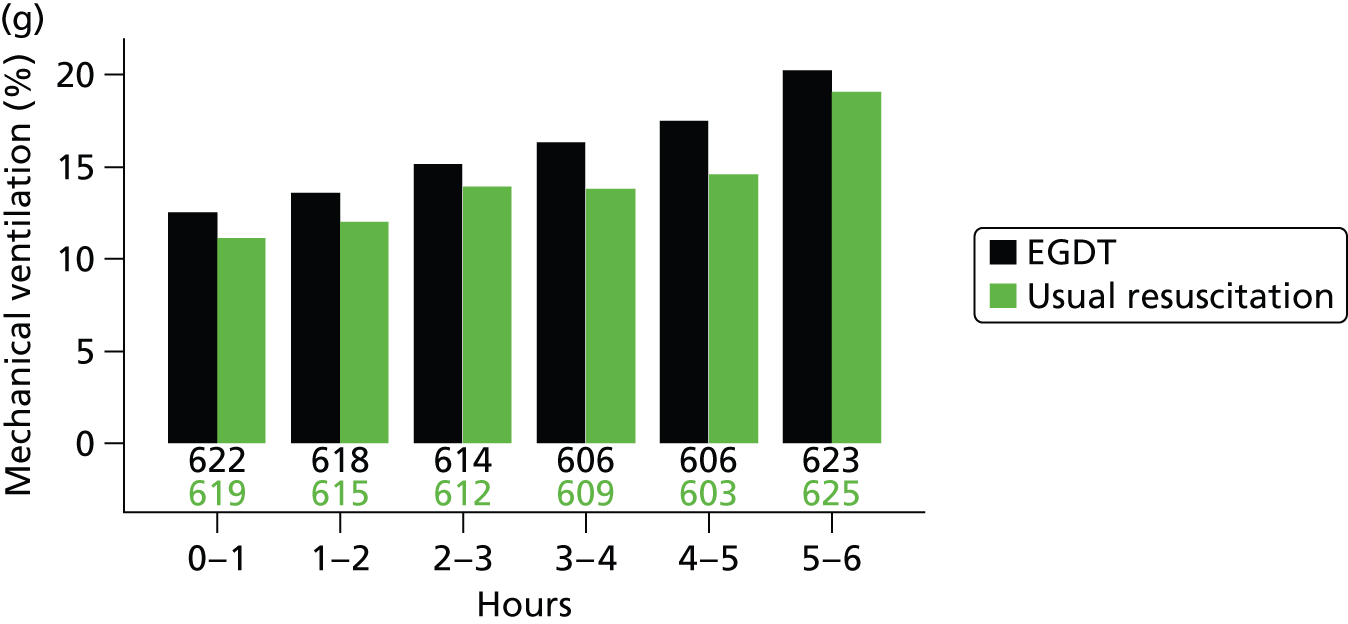
FIGURE 16.
Physiology during the intervention period. (a) CVP; (b) MAP; (c) SBP; and (d) ScvO2. The values reported are the mean plus/minus SD for values recorded within 15 minutes either side of the specified time point. For the EGDT group, first recorded values after insertion of the PreSepTM central venous oximetry catheter are at hour 1. ScvO2 measurements are not reported for the usual-resuscitation group owing to very small numbers of patients for whom these were recorded. CVP, central venous pressure; MAP, mean arterial pressure; SBP, systolic blood pressure.
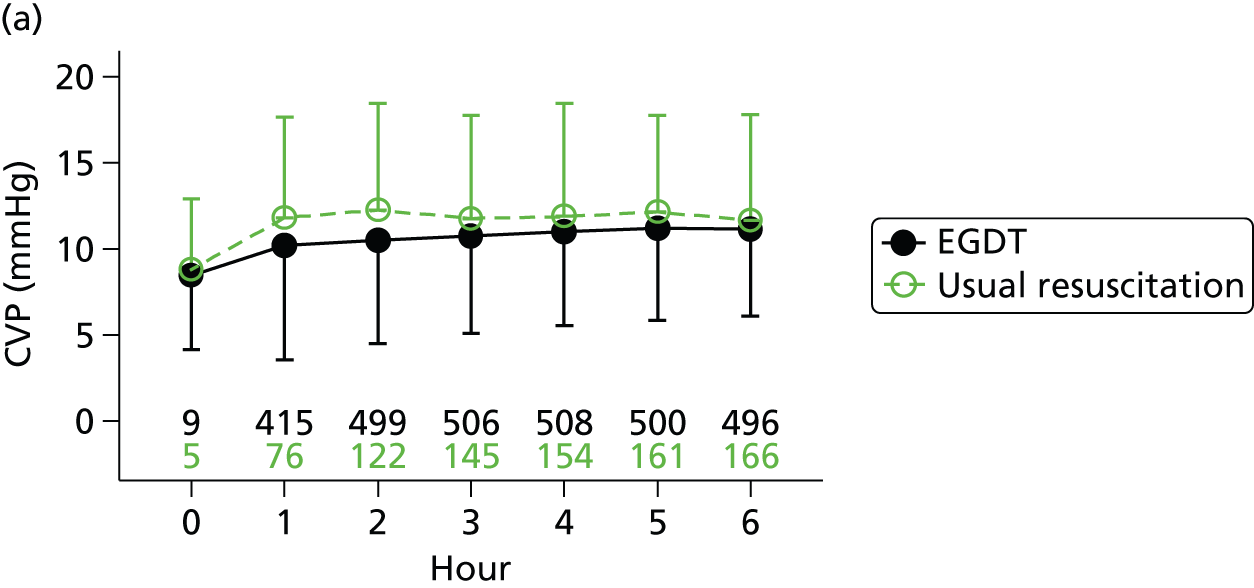


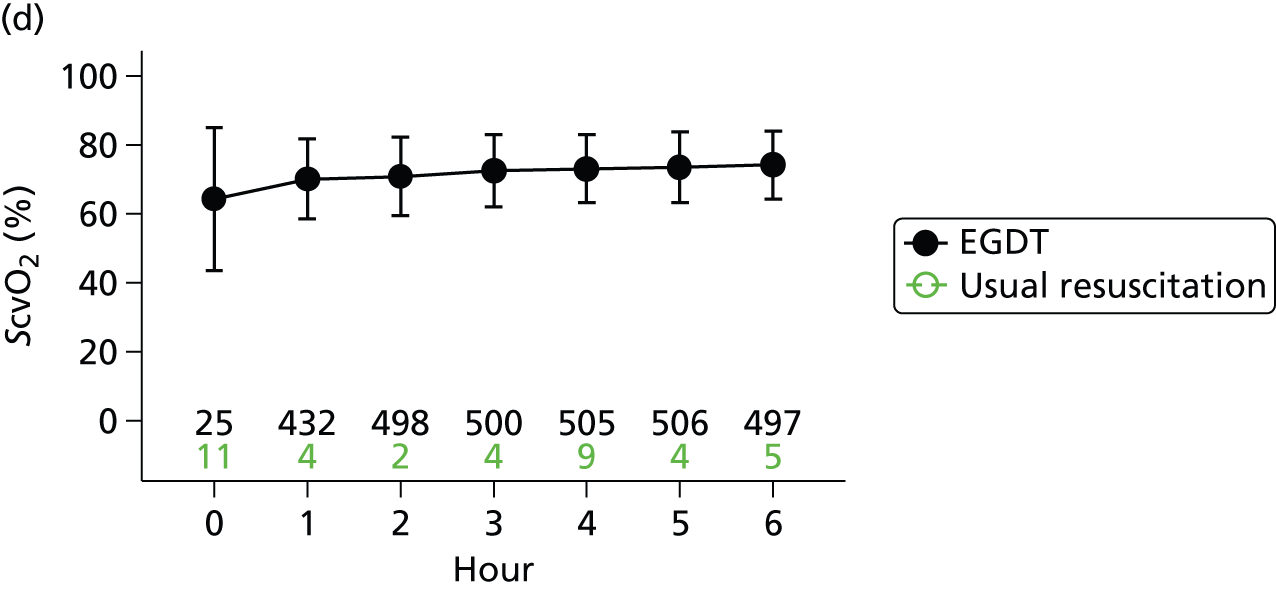
| Physiology | EGDT (n = 625) | Usual resuscitation (n = 626) |
|---|---|---|
| CVP (mmHg), mean (SD) | 11.2 (5.1) [496] | 11.7 (6.1) [166] |
| MAP (mmHg), mean (SD) | 76.5 (13.9) [518] | 76.5 (14.3) [394] |
| SBP (mmHg), mean (SD) | 113.1 (21.0) [573] | 110.7 (22.4) [508] |
| ScvO2 (%), mean (SD) | 74.2 (9.8) [497] | – |
| Haemoglobin (g/dl), mean (SD) | 11.0 (2.0) [384] | 11.3 (2.3) [163] |
Between 6 and 72 hours, use of intravenous fluids was similar but usual-resuscitation patients received higher volumes (Table 11). Intravenous colloid use was higher in the EGDT group but volumes were similar in the two groups, intravenous crystalloid use was similar but volumes were higher in the usual-resuscitation group and use of packed red blood cell transfusions was higher in the EGDT group but the volumes delivered were higher in the usual-resuscitation group. Although the use of platelets and fresh-frozen plasma was similar, the volume of platelets transfused was higher in the EGDT group and the volume of fresh-frozen plasma was higher in the usual-resuscitation group. Vasopressor and dobutamine use remained higher in the EGDT group. At 72 hours, physiological, biochemistry and SOFA values were similar (Table 12).
| Interventions | Baseline | Hour 0 to hour 6 | Hour 6 to hour 72 | Hour 0 to hour 72 | ||||
|---|---|---|---|---|---|---|---|---|
| EGDT (N = 625) | Usual resuscitation (N = 626) | EGDT (N = 625) | Usual resuscitation (N = 626) | EGDT (N = 608) | Usual resuscitation (N = 607) | EGDT (N = 625) | Usual resuscitation (N = 626) | |
| Supplemental O2,a n/N (%) | 397/539 (73.7) | 407/542 (75.1) | 558/623 (89.6) | 557/625 (89.1) | 520/603 (86.2) | 515/603 (85.4) | 577/623 (92.6) | 581/625 (93.0) |
| Any i.v. fluid,b n/N (%) | 612/625 (97.9) | 606/625 (97.0) | 609/623 (97.8) | 604/625 (96.6) | 546/603 (90.5) | 548/603 (90.9) | 615/623 (98.7) | 618/625 (98.9) |
| Volume (ml), mean (SD) | 1890 (1105) | 1965 (1149) | 2226 (1443) | 2022 (1271) | 4215 (3068) | 4366 (3114) | 5946 (3740) | 5844 (3651) |
| Volume (ml), median (IQR) | 1950 (1000–2500) | 2000 (1000–2500) | 2000 (1150–3000) | 1784 (1075–2775) | 3623 (1800–6060) | 3981 (1895–6291) | 5587 (2915–8150) | 5410 (3000–7970) |
| i.v. colloid,b n/N (%) | – | – | 197/623 (31.6) | 180/625 (28.8) | 171/603 (28.4) | 150/603 (24.9) | 260/623 (41.7) | 240/625 (38.4) |
| Volume (ml), mean (SD) | – | – | 1062 (801) | 913 (627) | 1207 (1042) | 1093 (1012) | 1598 (1391) | 1369 (1150) |
| Volume (ml), median (IQR) | – | – | 1000 (500–1500) | 750 (500–1000) | 750 (500–1750) | 750 (500–1500) | 1000 (575–2000) | 1000 (500–1750) |
| i.v. crystalloid,b n/N (%) | – | – | 584/623 (93.7) | 597/625 (95.5) | 537/603 (89.1) | 543/603 (90.0) | 609/623 (97.8) | 617/625 (98.7) |
| Volume (ml), mean (SD) | – | – | 1963 (1357) | 1767 (1178) | 3909 (2869) | 4136 (2914) | 5323 (3518) | 5317 (3435) |
| Volume (ml), median (IQR) | – | – | 1750 (999–2750) | 1500 (900–2380) | 3403 (1576–5647) | 3694 (1832–5911) | 4864 (2520–7241) | 4900 (2700–7408) |
| Vasopressors, n/N (%) | 15/625 (2.4) | 21/626 (3.4) | 332/623 (53.3) | 291/625 (46.6) | 349/603 (57.9) | 317/603 (52.6) | 377/623 (60.5) | 344/625 (55.0) |
| Dobutamine, n/N (%) | 2/625 (0.3) | 0/626 (0.0) | 113/623 (18.1) | 24/625 (3.8) | 107/603 (17.7) | 39/603 (6.5) | 139/623 (22.3) | 44/625 (7.0) |
| PRBC transfusion, n/N (%) | – | – | 55/623 (8.8) | 24/625 (3.8) | 76/603 (12.6) | 51/603 (8.5) | 107/623 (17.2) | 65/625 (10.4) |
| Volume (ml), mean (SD) | – | – | 426 (209) | 540 (294) | 487 (335) | 606 (403) | 565 (393) | 674 (506) |
| Volume (ml), median (IQR) | – | – | 309 (285–577) | 535 (305–607) | 351 (291–579) | 552 (317–620) | 529 (298–602) | 562 (317–660) |
| Platelets, n/N (%) | – | – | 11/623 (1.8) | 10/625 (1.6) | 23/603 (3.8) | 25/603 (4.1) | 31/623 (5.0) | 30/625 (4.8) |
| Volume (ml), mean (SD) | – | – | 286 (72) | 242 (131) | 314 (167) | 278 (162) | 325 (194) | 315 (207) |
| Volume (ml), median (IQR) | – | – | 315 (200–340) | 180 (163–342) | 274 (182–366) | 187 (172–357) | 290 (191–366) | 250 (173–418) |
| Fresh-frozen plasma, n/N (%) | – | – | 15/623 (2.4) | 14/625 (2.2) | 28/603 (4.6) | 30/603 (5.0) | 41/623 (6.6) | 39/625 (6.2) |
| Volume (ml), mean (SD) | – | – | 847 (383) | 769 (285) | 836 (721) | 869 (507) | 881 (658) | 945 (533) |
| Volume (ml), median (IQR) | – | – | 1007 (539–1095) | 793 (526–1085) | 587 (483–1000) | 846 (528–1057) | 791 (516–1095) | 1025 (528–1140) |
| Mechanical ventilation, n/N (%) | 40/625 (6.4) | 28/626 (4.5) | 126/623 (20.2) | 119/625 (19.0) | 147/603 (24.4) | 153/603 (25.4) | 171/623 (27.4) | 178/625 (28.5) |
| Sedatives, n/N (%) | – | – | 138/623 (22.2) | 130/625 (20.8) | 161/603 (26.7) | 172/603 (28.5) | 191/623 (30.7) | 200/625 (32.0) |
| Neuromuscular blocking agent, n/N (%) | – | – | 53/623 (8.5) | 40/625 (6.4) | 39/603 (6.5) | 34/603 (5.6) | 74/623 (11.9) | 60/625 (9.6) |
| Co-interventions for the source of sepsis, n/N (%) | ||||||||
| Surgery | – | – | 9/625 (1.4) | 12/626 (1.9) | 32/608 (5.3) | 36/607 (5.9) | 41/625 (6.6) | 48/626 (7.7) |
| Activated Protein C | – | – | 0/625 (0.0) | 1/626 (0.2) | 2/608 (0.3) | 4/607 (0.7) | 2/625 (0.3) | 4/626 (0.6) |
| Steroids | 31/625 (5.0) | 25/626 (4.0) | 73/625 (11.7) | 72/626 (11.5) | 133/608 (21.9) | 128/607 (21.1) | 142/625 (22.7) | 136/626 (21.7) |
| Physiology | Baselinea | Hour 0 to hour 6 | Hour 6 to hour 24 | Hour 48 to hour 72 | ||||
|---|---|---|---|---|---|---|---|---|
| EGDT (N = 625) | Usual resuscitation (N = 626) | EGDT (N = 625) | Usual resuscitation (N = 626) | EGDT (N = 608) | Usual resuscitation (N = 607) | EGDT (N = 541) | Usual resuscitation (N = 529) | |
| Lowest MAP (mmHg), mean (SD) | 69.0 (20.3) [145] | 64.7 (17.2) [164] | 64.7 (11.5) [566] | 65.0 (14.3) [475] | 64.0 (11.1) [439] | 64.3 (11.9) [369] | 68.9 (11.6) [282] | 68.5 (13.7) [260] |
| Lowest SBP (mmHg), mean (SD) | 99.6 (26.0) [609] | 97.0 (25.5) [602] | 92.2 (19.3) [619] | 91.4 (19.9) [616] | 97.1 (19.1) [300] | 97.9 (20.3) [344] | 107.3 (19.5) [312] | 107.9 (18.3) [308] |
| Haemoglobinb (g/dl), mean (SD) | 12.5 (2.5) [607] | 12.7 (2.5) [613] | 11.0 (2.0) [384] | 11.3 (2.3) [163] | 11.0 (1.8) [422] | 10.9 (1.9) [374] | 10.7 (1.7) [346] | 10.7 (1.8) [331] |
| Blood lactate concentrationb (mmol/l), mean (SD) | 5.2 (3.5) [608] | 5.1 (3.5) [611] | 3.3 (3.0) [392] | 3.8 (3.2) [187] | 2.7 (2.7) [382] | 2.7 (2.6) [316] | 2.0 (2.6) [229] | 1.8 (1.7) [217] |
| Lowest PaO2/FiO2 (kPa), mean (SD) | 32.6 (26.1) [383] | 33.9 (29.1) [416] | 30.3 (25.2) [464] | 30.9 (20.6) [432] | 30.7 (20.3) [436] | 30.4 (16.7) [393] | 30.9 (21.7) [255] | 28.4 (14.5) [225] |
| Highest creatinine (µmol/l), mean (SD) | 183.8 (141.5) [591] | 192.7 (192.0) [586] | 176.3 (135.0) [462] | 196.3 (190.8) [422] | 149.5 (102.1) [511] | 174.4 (145.5) [476] | 129.1 (98.9) [425] | 140.8 (120.4) [413] |
| Highest bilirubin (µmol/l), mean (SD) | 24.7 (25.1) [491] | 28.0 (37.6) [492] | 24.2 (25.6) [408] | 26.0 (37.5) [389] | 26.6 (36.4) [436] | 26.2 (39.6) [385] | 24.8 (42.0) [324] | 23.0 (37.2) [314] |
| Lowest platelet count (× 109/l), mean (SD) | 239.0 (131.5) [585] | 236.1 (123.0) [585] | 203.3 (113.5) [455] | 208.7 (119.6) [415] | 182.3 (100.2) [503] | 181.7 (108.7) [469] | 163.9 (98.1) [421] | 164.9 (102.1) [411] |
| Lowest GCS score, mean (SD) | 13.8 (2.8) [593] | 14.0 (2.2) [588] | 13.3 (3.5) [578] | 13.4 (3.4) [533] | 13.2 (3.8) [492] | 13.3 (3.7) [462] | 13.4 (3.5) [422] | 13.8 (3.2) [393] |
| SOFA respiratory dysfunction, n (%) | 323 (51.7) | 357 (57.0) | 416 (66.6) | 375 (59.9) | 403 (66.3) | 359 (59.1) | 241 (44.5) | 214 (40.5) |
| SOFA neurological dysfunction, n (%) | 196 (31.4) | 200 (31.9) | 226 (36.2) | 186 (29.7) | 169 (27.8) | 144 (23.7) | 126 (23.3) | 93 (17.6) |
| SOFA cardiovascular dysfunction, n (%) | 410 (65.6) | 433 (69.2) | 477 (76.3) | 384 (61.3) | 434 (71.4) | 358 (59.0) | 342 (63.2) | 317 (59.9) |
| SOFA coagulation dysfunction, n (%) | 144 (23.0) | 144 (23.0) | 152 (24.3) | 134 (21.4) | 209 (34.4) | 201 (33.1) | 197 (36.4) | 203 (38.4) |
| SOFA hepatic dysfunction, n (%) | 211 (33.8) | 199 (31.8) | 223 (35.7) | 218 (34.8) | 218 (35.9) | 184 (30.3) | 98 (18.1) | 106 (20.0) |
| SOFA renal dysfunction,c n (%) | 426 (68.2) | 406 (64.9) | 430 (68.8) | 415 (66.3) | 347 (57.1) | 332 (54.7) | 199 (36.8) | 196 (37.1) |
| SOFA score,c median (IQR) | 4 (2–5) | 4 (3–6) | 6 (3–9) | 5 (3–8) | 6 (3–9) | 5 (2–9) | 3 (1–6) | 3 (1–6) |
Primary outcome: clinical effectiveness
At 90 days following randomisation, 184 (29.5%) patients randomised to EGDT had died, compared with 181 (29.2%) patients randomised to usual resuscitation, corresponding to an absolute risk reduction of −0.3 (95% CI −5.4 to 4.7, p = 0.90) and a relative risk of 1.01 (95% CI 0.85 to 1.20; Table 13). This difference remained non-significant after adjustment for baseline characteristics (adjusted odds ratio 0.95, 95% CI 0.74 to 1.24; p = 0.73; unadjusted odds ratio 1.02, 95% CI 0.80 to 1.30).
| Outcome | EGDT (N = 625) | Usual resuscitation (N = 626) | Effect estimate (95% CI) | p-value |
|---|---|---|---|---|
| Primary outcome | ||||
| All-cause mortality at 90 days, n/N (%) | 184/623 (29.5) | 181/620 (29.2) | 1.01 (0.85 to 1.20)a | 0.90 |
| −0.3 (−5.4 to 4.7)b | ||||
| 1.02 (0.80 to 1.30)c | ||||
| 0.95 (0.74 to 1.24)d | 0.73 | |||
| Secondary outcomes | ||||
| SOFA score at 6 hours,e mean (SD) | 6.4 (3.8) | 5.6 (3.8) | 0.8 (0.5 to 1.1)f,g | < 0.001 |
| SOFA score at 72 hours,e mean (SD) | 4.0 (3.8) | 3.7 (3.6) | 0.4 (−0.0 to 0.8)f,g | 0.056 |
| Receipt of advanced cardiovascular support, n/N (%) | 230/622 (37.0) | 190/614 (30.9) | 1.19 (1.02 to 1.40)a | 0.026 |
| Receipt of advanced respiratory support, n/N (%) | 179/620 (28.9) | 175/615 (28.5) | 1.01 (0.85 to 1.21)a | 0.90 |
| Receipt of renal support, n/N (%) | 88/620 (14.2) | 81/614 (13.2) | 1.08 (0.81 to 1.42)a | 0.62 |
| Days alive and free from advanced cardiovascular support up to 28 days, mean (SD) | 20.3 (11.9) | 20.6 (11.8) | −0.3 (−1.6 to 1.0)f | 0.65 |
| Days alive and free from advanced respiratory support up to 28 days, mean (SD) | 19.6 (12.1) | 19.8 (12.0) | −0.2 (−1.5 to 1.1)f | 0.75 |
| Days alive and free from renal support up to 28 days, mean (SD) | 20.6 (12.1) | 20.6 (11.9) | 0.0 (−1.3 to 1.4)f | 0.96 |
| ED length of stay (hours), median (IQR) | 1.5 (0.4–3.1) | 1.3 (0.4–2.9) | – | 0.34 |
| Survivors | 1.4 (0.4–3.1) | 1.3 (0.3–2.9) | – | 0.38 |
| Non-survivors | 3.7 (2.4–4.1) | 2.4 (1.2–5.9) | – | 0.25 |
| Critical care length of stay (days), median (IQR) | 2.6 (1.0–5.8) | 2.2 (0.0–5.3) | – | 0.006 |
| Survivors | 2.9 (1.0–6.1) | 2.8 (0.0–5.9) | – | 0.008 |
| Non-survivors | 1.6 (0.6–3.1) | 1.2 (0.5–4.3) | – | 0.85 |
| Acute hospital length of stay (days), median (IQR) | 9 (4–21) | 9 (4–18) | – | 0.46 |
| Survivors | 11 (7–25) | 11 (7–22) | – | 0.42 |
| Non-survivors | 2 (1–8) | 2 (1–7) | – | 0.44 |
| All-cause mortality at 28 days, n/N (%) | 155/625 (24.8) | 152/621 (24.5) | 1.01 (0.83 to 1.23)a | 0.90 |
| 0.95 (0.73 to 1.25)d | 0.73 | |||
| All-cause mortality at acute hospital discharge, n/N (%) | 160/625 (25.6) | 154/625 (24.6) | 1.04 (0.86 to 1.26)a | 0.74 |
| 0.98 (0.75 to 1.29)d | 0.90 | |||
| All-cause mortality at 1 year, n/N (%) | 224/558 (40.1) | 233/558 (41.8) | 0.96 (0.83 to 1.11)a | 0.63 |
| 0.85 (0.66 to 1.09)d | 0.20 | |||
Secondary outcomes: clinical effectiveness
For patients in the EGDT group, both the mean SOFA score at 6 hours (p < 0.001) and the proportion receiving advanced cardiovascular support (p = 0.026) were significantly higher than for patients in the usual-resuscitation group. The median total length of stay in critical care was significantly longer for patients in the EGDT group than the usual-resuscitation group (p = 0.006). There were no significant differences between the groups in any of the other secondary outcomes, including duration of survival (log-rank test, p = 0.63; Cox proportional hazards model, adjusted hazard ratio 0.94, 95% CI 0.79 to 1.11; p = 0.46) (Figure 17 and see Table 13).
FIGURE 17.
Kaplan–Meier curves for survival to (a) 90 days and (b) 1 year following randomisation.


Safety monitoring
Thirty (4.8%) patients in the EGDT group and 26 (4.2%) patients in the usual-resuscitation group experienced one or more serious adverse events within 30 days following randomisation (relative risk 1.16, 95% CI 0.69 to 1.93; p = 0.58; Table 14). The most commonly reported serious adverse events were pulmonary oedema (four patients in the EGDT group and seven patients in the usual-resuscitation group) and myocardial ischaemia (seven patients in the EGDT group and four patients in the usual-resuscitation group). Three serious adverse events associated with EGDT (two pulmonary oedema and one arrhythmia) were reported as probably related to the intervention, compared with four (in three patients) serious adverse events (two pneumothoraces and one pulmonary oedema probably related, and one ventricular fibrillation definitely related) reported as associated with usual resuscitation.
| Serious adverse events | EGDT (N = 625) | Usual resuscitation (N = 626) |
|---|---|---|
| Any serious adverse event, n (%) | 30 (4.8) | 26 (4.2) |
| Specified serious adverse events, n (%) | ||
| Pneumothorax | 0 (0.0) | 4 (0.6) |
| Haemo-pneumothorax | 0 (0.0) | 0 (0.0) |
| Bleeding | 2 (0.3) | 2 (0.3) |
| Thrombosis | 2 (0.3) | 0 (0.0) |
| Pulmonary embolus | 4 (0.6) | 2 (0.3) |
| Vascular catheter infection | 0 (0.0) | 0 (0.0) |
| Pulmonary oedema | 4 (0.6) | 7 (1.1) |
| Blood transfusion reaction | 0 (0.0) | 1 (0.2) |
| Myocardial ischaemia | 7 (1.1) | 4 (0.6) |
| Peripheral ischaemia | 0 (0.0) | 1 (0.2) |
| Unspecified serious adverse events, n (%) | ||
| Cardiac arrest | 5 (0.8) | 4 (0.6) |
| Cerebrovascular event | 4 (0.6) | 1 (0.2) |
| Arrhythmia | 1 (0.2) | 2 (0.3) |
| Othera | 5 (0.8) | 5 (0.8) |
Subgroup analyses of the primary outcome
There was no difference in the effect of EGDT on the primary outcome of all-cause mortality at 90 days following randomisation according to the pre-specified subgroups defined by the degree of protocolised care/usual resuscitation, age, MEDS score, SOFA score and time from ED presentation to randomisation (p-values for test of interaction 0.39 to 0.72; Figure 18).
FIGURE 18.
Subgroup analyses of the primary outcome. p-values are for tests of interaction. The x-axis is presented on a log scale. The solid line represents no difference between the groups and the dashed line represents the overall effect estimate (adjusted odds ratio).

Secondary analyses of the primary outcome
Eight patients were missing the primary outcome of all-cause mortality at 90 days following randomisation (two in the EGDT group and six in the usual-resuscitation group). The effect of missing data on the results was minimal. Sensitivity analyses making alternative extreme assumptions for the missing outcomes reported relative risks of 0.99 and 1.03 (Table 15). Although the learning curve analysis suggested increased odds of mortality for the first patient randomised to EGDT in each site, this effect was not significant (p = 0.56; Figure 19 and see Table 15). Adjusting for non-adherence to the EGDT protocol resulted in minimal change to the relative risk (see Table 15).
| Analysis | All-cause mortality at 90 days, n/N (%) | Incremental effect (95% CI) | p-value | |
|---|---|---|---|---|
| EGDT (n = 625) | Usual resuscitation (n = 626) | |||
| Sensitivity analyses for missing data in the primary outcome | ||||
| EGDT survive, usual resuscitation die | 184/625 (29.4) | 187/626 (29.9) | 0.99 (0.83 to 1.17)a | 0.90 |
| EGDT die, usual resuscitation survive | 186/625 (29.8) | 181/626 (28.9) | 1.03 (0.87 to 1.22)a | 0.76 |
| Learning curve analysis | 0.56b | |||
| Asymptotic adjusted odds ratio | – | – | 0.89 (0.69 to 1.15)c | 0.34 |
| Adherence-adjusted analysis | – | – | 1.02 (0.78 to 1.32)a | 0.90 |
FIGURE 19.
Learning curve for delivery of EGDT. The y-axis is presented on a log scale.

Comparison with other early goal-directed therapy studies
Comparing the usual-resuscitation group for ProMISe with that from the original randomised controlled trial of EGDT by Rivers et al. ,6 patients in ProMISe were randomised, on average, slightly later than those in the Rivers et al. trial (Table 16). Demographics were similar. Patients in ProMISe had slightly lower blood pressure, blood lactate measurements and APACHE II scores. Patients in the usual-resuscitation group in the Rivers et al. 6 trial received considerably more fluid and packed red blood cell transfusions, and were more likely to be mechanically ventilated; however, those in ProMISe received more vasopressors and dobutamine. Hospital mortality was substantially higher in the Rivers et al. 6 trial than in ProMISe.
| Characteristics | Rivers et al.6 | ProMISe |
|---|---|---|
| Timing | ||
| ED presentation to randomisation (hours), mean (SD) | 1.5 (1.7) | 2.8 (1.4) |
| Baseline characteristics | ||
| Age (years), mean (SD) | 64.4 (17.1) | 64.3 (15.5) |
| Male (%) | 50.4 | 58.6 |
| SBP (mmHg), mean (SD) | 109 (34) | 97.0 (25.5) |
| MAP (mmHg), mean (SD) | 76 (24) | 64.7 (17.2) |
| Blood lactate concentration (mmol/l), mean (SD) | 6.9 (4.5) | 5.1 (3.5) |
| APACHE II score, mean (SD) | 20.4 (7.4) | 18.0 (7.1) |
| Interventions hour 0 to hour 6 | ||
| Total intravenous fluids (ml), mean (SD) | 3499 (2438) | 2022 (1271) |
| Vasopressors (%) | 30.3 | 46.6 |
| Packed red blood cell transfusion (%) | 18.5 | 3.8 |
| Dobutamine (%) | 0.8 | 3.8 |
| Mechanical ventilation (%) | 53.8 | 19.0 |
| Outcomes | ||
| Hospital mortality (%) | 46.5 | 24.6 |
Comparing the usual-resuscitation group for ProMISe with those from ProCESS23 and ARISE,24 time from ED presentation to randomisation was similar but patients in ProMISe had a shorter length of stay in the ED than those in ARISE (Table 17). Demographics were similar. The mean volume of fluid received prior to randomisation was higher for ARISE than for ProCESS or ProMISe. Blood pressure and blood lactate measurements were similar for ProMISe and ProCESS, but mean arterial pressure was slightly higher and blood lactate was slightly lower for ARISE. This lower severity of illness for patients in ARISE was also reflected in APACHE II scores, which were lowest for ARISE and highest for ProCESS. A greater proportion of patients in ProMISe met both the refractory hypotension and the hyperlactataemia inclusion criteria. To explore the hypothesis that the combination of both refractory hypotension and hyperlactataemia was associated with higher mortality than either alone, we examined data from the Case Mix Programme, the national clinical audit for adult critical care. Among 12,004 patients admitted to 183 adult general ICUs in England between February 2011 and June 2014 (the recruitment period of ProMISe) direct from the ED with infection, meeting two or more SIRS criteria during the first 24 hours following admission, and with hypotension (lowest systolic blood pressure < 90 mmHg) and/or hyperlactataemia (highest blood lactate ≥ 4 mmol/l), acute hospital mortality was around 30% for patients meeting a single criterion but almost double for patients meeting both the refractory hypotension and the hyperlactataemia criteria (Table 18).
| Characteristics | ProCESS23 | ARISE24 | ProMISe |
|---|---|---|---|
| Timing | |||
| ED presentation to randomisation (hours), mean (SD) | 3.0 (1.6) | – | 2.8 (1.4) |
| ED presentation to randomisation (hours), median (IQR) | – | 2.7 (2.0–3.9) | 2.5 (1.8–3.5) |
| ED length of stay (hours), median (IQR) | – | 2.0 (1.0–3.8) | 1.3 (0.4–2.9) |
| Baseline characteristics | |||
| Age (years), mean (SD) | 62.0 (16.0) | 63.1 (16.5) | 64.3 (15.5) |
| Male (%) | 57.9 | 59.3 | 58.6 |
| Pre-randomisation fluidsa (l), mean (SD) | 2.1 (1.4) | 2.6 (1.3) | 2.0 (1.1) |
| SBP (mmHg), mean (SD) | 99.9 (29.5) | – | 97.0 (25.5) |
| MAP (mmHg), mean (SD) | 64.7 (15.6) | 70.5 (16.0) | 64.7 (17.2) |
| Blood lactate concentration (mmol/l), mean (SD) | 4.9 (3.1) | 4.2 (2.8) | 5.1 (3.5) |
| Refractory hypotension only (%) | 39.3 | 53.5 | 36.3 |
| Hyperlactataemia only (%) | 46.7 | 30.2 | 44.4 |
| Both refractory hypotension and hyperlactataemia (%) | 14.0 | 16.3 | 19.3 |
| APACHE II score, mean (SD) | 20.7 (7.5) | 15.8 (6.5) | 18.0 (7.1) |
| Interventions hour 0 to hour 6 | |||
| Intravenous fluidsb (ml), mean (SD) | 2279 (1881) | 1713 (1401) | 2022 (1271) |
| Vasopressorsc (%) | 44.1 | 57.8 | 46.6 |
| Dobutamine (%) | 0.9 | 2.6 | 3.8 |
| Packed red blood cell transfusion (%) | 7.5 | 7.0 | 3.8 |
| Mechanical ventilationd (%) | 21.7 | 22.4 | 19.0 |
| CVC insertione (%) | 57.9 | 61.9 | 50.9 |
| Outcomes | |||
| Hospital mortality (%) | – | 15.7 | 24.6 |
| Discharge homef (%) | 51.5 | 79.6 | 82.2 |
| 28-day mortality (%) | – | 15.9 | 24.5 |
| 90-day mortality (%) | 33.7 | 18.8 | 29.2 |
| Criteria met | Admissions, n (%) | Acute hospital mortality, n (%) |
|---|---|---|
| Refractory hypotension only | 2186 (18.2) | 687 (31.4) |
| Hyperlactataemia only | 5339 (44.5) | 1397 (26.2) |
| Both refractory hypotension and hyperlactataemia | 4479 (37.3) | 2485 (55.5) |
During the intervention period, the mean volume of fluid received by patients in the usual-resuscitation group was highest for ProCESS and lowest for ARISE (see Table 17); however, a greater proportion of patients in ARISE received vasopressors. Dobutamine use was highest in ProMISe and lowest in ProCESS and the proportions of usual-resuscitation group patients receiving packed red blood cell transfusions in ProCESS and ARISE were approximately double that in ProMISe. In all three trials, around 20% of patients received mechanical ventilation, and the central venous catheter insertion rates varied from 51% in ProMISe to 62% in ARISE. Reflecting the pattern seen in severity of illness scores, 90-day mortality for usual-resuscitation group patients in ProCESS was slightly higher than in ProMISe (34% vs. 29%), whereas for those in ARISE it was substantially lower (19%). Mortality in ARISE was similarly lower at other comparable time points. Of patients discharged alive from hospital in both ProMISe and ARISE, approximately 80% were discharged home, compared with only just over 50% of similar patients in ProCESS.
Chapter 5 Results: cost-effectiveness
Cost-effectiveness at 90 days following randomisation
Resource use up to 90 days
The average duration for the delivery of the EGDT protocol for the EGDT group was 5.8 hours in total, of which 2.0 were in the ED. The delivery of the EGDT protocol used resources specific to the intervention with respect to catheter insertion (PreSepTM central venous oximetry catheter and arterial catheter), packed red blood cell transfusion, infusion of dobutamine and additional staff time required to implement the protocol in the ED (Table 19). For the index hospital episode, the mean length of stay in critical care and on general medical wards was higher in the EGDT group than the usual-resuscitation group. The proportion of patients who were readmitted and the mean length of stay following readmission were similar between the treatment groups (see Table 19). The mean total length of stay in acute hospital up to 90 days following randomisation was 16.7 days in the EGDT group versus 15.5 days in the usual-resuscitation group.
| Resource use up to 90 days | EGDT (n = 625) | Usual resuscitation (n = 626) |
|---|---|---|
| Interventionsa | ||
| PreSep™ central venous oximetry catheter, n (%) | 545 (87) | 2 (0) |
| Standard CVC, n (%) | 48 (8) | 316 (50) |
| Arterial catheter, n (%) | 462 (74) | 389 (62) |
| Blood products | ||
| Packed red blood cells (ml) | 97 (267) | 70 (262) |
| Platelets (ml) | 16 (82) | 15 (79) |
| Fresh-frozen plasma (ml) | 58 (275) | 59 (264) |
| Dobutamine total dose (mg) | 183 (592) | 88 (489) |
| Duration of protocol delivered in ED (hours) | 2.0 (1.9) | – |
| Duration of protocol delivered in total (hours) | 5.8 (0.8) | – |
| Additional staff time | ||
| Catheter insertion and monitor set-up (hours) | 1.2 (0.3) | 0.5 (0.4) |
| Monitoring (hours) | 0.3 (0.3) | – |
| Training (hours) | 0.3 (0) | – |
| Acute hospital length of stay | ||
| Index hospital admissiona | ||
| Length of stay in ED (hours) | 2.3 (3.2) | 1.9 (2.1) |
| Length of stay in critical care (days) | 4.9 (7.8) | 4.7 (8.9) |
| Length of stay on general medical ward (days) | 10.5 (15.0) | 9.6 (13.5) |
| Acute hospital readmissions,b n (%) | 28 (4) | 30 (5) |
| Length of stay in critical care (days) | 0.3 (2.5) | 0.4 (3.2) |
| Length of stay on general medical ward (days) | 0.7 (4.2) | 0.7 (4.5) |
| Total acute hospital length of stay up to 90 days | 16.7 (19.2) | 15.5 (17.8) |
Table 20 summarises the resource use reported from responses to the Health Services Questionnaire administered at 90 days following randomisation for all patients randomised to each treatment group. The mean number of inpatient days reported from admissions other than those involving critical care was 4.6 days for the EGDT group and 3.8 days for the usual-resuscitation group. The mean numbers of outpatient visits and community care contacts up to 90 days were similar between the groups. Patients in both groups reported low use of community health services over the 90 days following randomisation.
| Resource use | EGDT (N = 625), mean (SD) | Usual resuscitation (N = 626), mean (SD) |
|---|---|---|
| Inpatient days (general medical) | 4.6 (8.5) | 3.8 (7.0) |
| Outpatient visits | 0.9 (2.6) | 1.1 (2.4) |
| GP contacts | 1.2 (2.4) | 1.1 (2.3) |
| Nurse contacts | 0.7 (2.0) | 0.7 (2.0) |
| Occupational therapist contacts | 0.2 (1.4) | 0.3 (2.2) |
| Health visitor contacts | 0.2 (1.6) | 0.4 (2.4) |
| Clinical psychologist contacts | 0.02 (0.2) | 0.01 (0.3) |
| Speech therapist contacts | 0.01 (0.3) | 0.04 (2.7) |
| Physiotherapist contacts | 0.4 (2.3) | 0.4 (2.7) |
| Dietitian contacts | 0.1 (0.9) | 0.1 (0.5) |
Total costs up to 90 days
The net effect of the higher average length of stay in critical care and on general medical wards was that the EGDT group had higher mean total costs per patient than the usual-resuscitation group (Table 21). At 90 days, the mean total costs per patient were £12,414 for the EGDT group and £11,424 for the usual-resuscitation group.
| Resource use categories | EGDT (N = 625), mean (SD) | Usual resuscitation (N = 626), mean (SD) |
|---|---|---|
| Interventiona | ||
| Monitor and consumables | 206 (70) | 33 (26) |
| Blood products | 83 (208) | 66 (207) |
| Drugs (dobutamine) | 8 (24) | 4 (19) |
| Additional staff time costs | ||
| Catheter insertion and monitor set-up | 64 (18) | 29 (21) |
| Monitoring | 16 (16) | – |
| Training | 17 (0) | – |
| Hospital costs | ||
| Index hospital admissiona | ||
| ED | 62 (85) | 53 (56) |
| Critical care | 7255 (12,045) | 6852 (13,529) |
| General medical ward | 2788 (3983) | 2532 (3586) |
| Readmission costs | ||
| Critical careb | 467 (3577) | 626 (4500) |
| General medicalb,c | 196 (1132) | 178 (1187) |
| Outpatient and community costsc,d | 1252 (2848) | 1051 (2660) |
| Total costs up to 90 daysd | 12,414 (14,970) | 11,424 (15,727) |
Health-related quality of life at 90 days
The distribution of responses to each dimension of the EQ-5D questionnaires, administered at 90 days following randomisation, is reported by treatment group in Table 22. The distribution of responses was similar between the groups. The resultant mean EQ-5D utility scores and QALYs were also similar between the treatment groups (Table 23).
| EQ-5D-5L dimension | EGDT (N = 339a), n (%) | Usual resuscitation (N = 332a), n (%) |
|---|---|---|
| Mobility | ||
| No problems | 101 (30) | 102 (31) |
| Slight problems | 44 (13) | 51 (15) |
| Moderate problems | 86 (25) | 71 (21) |
| Severe problems | 75 (22) | 74 (22) |
| Extreme problems | 33 (10) | 34 (10) |
| Self-care | ||
| No problems | 173 (51) | 171 (52) |
| Slight problems | 44 (13) | 40 (12) |
| Moderate problems | 68 (20) | 71 (21) |
| Severe problems | 30 (9) | 25 (8) |
| Extreme problems | 24 (7) | 25 (8) |
| Usual activities | ||
| No problems | 81 (24) | 87 (26) |
| Slight problems | 61 (18) | 62 (19) |
| Moderate problems | 83 (24) | 82 (25) |
| Severe problems | 62 (18) | 51 (15) |
| Extreme problems | 52 (15) | 50 (15) |
| Pain/discomfort | ||
| No problems | 93 (27) | 95 (29) |
| Slight problems | 91 (27) | 81 (24) |
| Moderate problems | 81 (24) | 89 (27) |
| Severe problems | 50 (15) | 53 (16) |
| Extreme problems | 24 (7) | 14 (4) |
| Anxiety/depression | ||
| No problems | 152 (45) | 146 (44) |
| Slight problems | 74 (22) | 79 (24) |
| Moderate problems | 72 (21) | 70 (21) |
| Severe problems | 23 (7) | 22 (7) |
| Extreme problems | 18 (5) | 15 (5) |
| End point | EGDT (n = 625), mean (SD) | Usual resuscitation (n = 626), mean (SD) | Incremental effect (unadjusted), mean (95% CI) | p-value |
|---|---|---|---|---|
| EQ-5D-5L utility score (survivors) | 0.609 (0.319) | 0.613 (0.312) | −0.004 (−0.051 to 0.044) | 0.88 |
| QALYs | 0.054 (0.048) | 0.054 (0.048) | −0.001 (−0.006 to 0.005) | 0.85 |
| Costs (£) | 12,414 (14,970) | 11,424 (15,727) | 989 (−726 to 2705) | 0.26 |
| Incremental net benefit (£)a | −1000 (−2720 to 720) | 0.25 |
Cost-effectiveness at 90 days
The incremental QALY gain for EGDT versus usual resuscitation was negative, but with 95% CIs that included zero (see Table 23). The average costs were higher for the EGDT group, but this difference was not statistically significant. The INB for EGDT versus usual resuscitation was negative at −£1000 (95% CI −£2720 to £720; see Table 23).
When the uncertainty in the incremental costs and QALYs is represented on the cost-effectiveness plane, the majority of the points are in those quadrants that show EGDT has, on average, higher costs (Figure 20). The probability that EGDT is more cost-effective than usual resuscitation, given the data, is never greater than 20%, irrespective of how much society is willing to pay for a QALY gain (Figure 21).
FIGURE 20.
Uncertainty in the mean costs (£) and QALY differences at 90 days and their distribution for EGDT vs. usual resuscitation.
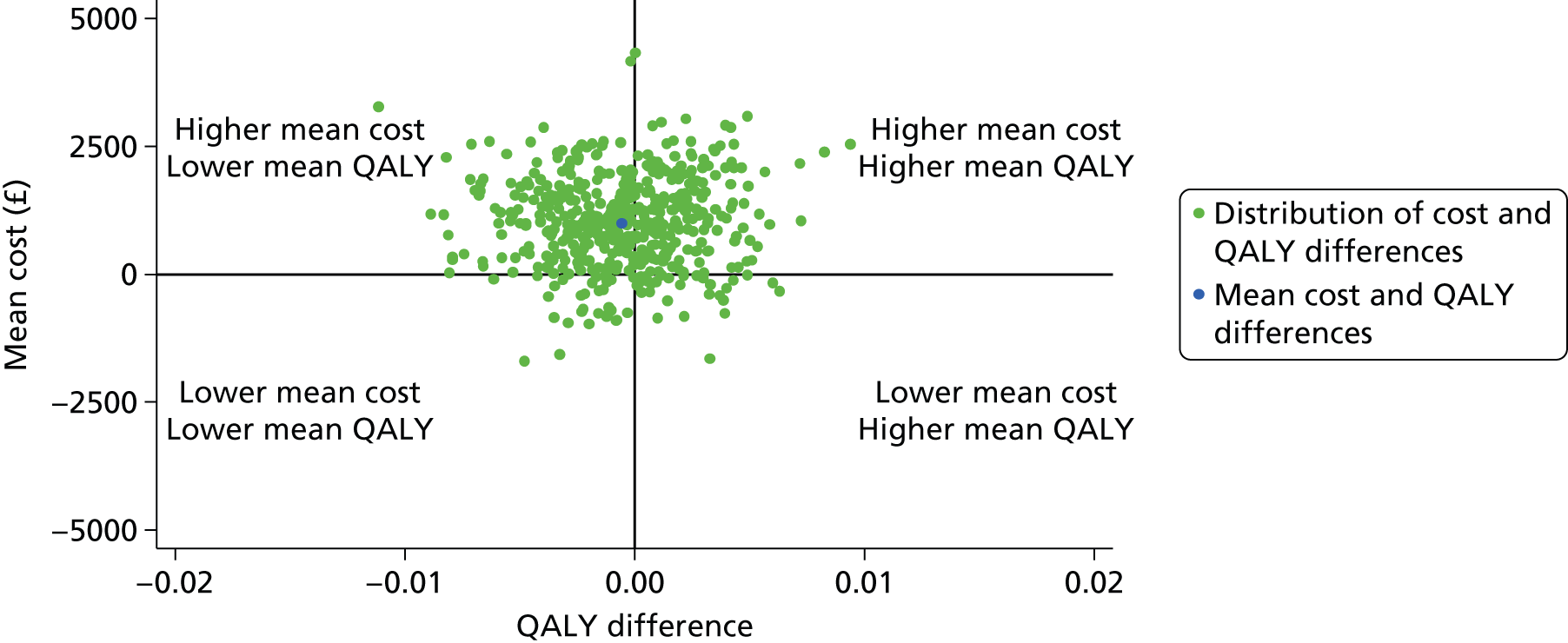
FIGURE 21.
Cost-effectiveness acceptability curve reporting the probability that EGDT is cost-effective at 90 days at alternative willingness to pay.

The estimated INB were similar for the scenarios considered in the sensitivity analyses (Figure 22). For example, the INB remains around −£1000 whether EGDT is provided in the ED or in critical care. Similarly, excluding readmissions that were reported from responses to the Health Services Questionnaire, to avoid any risk of double counting, had only a small impact on the mean INB (−£800 vs. −£1000).
FIGURE 22.
Sensitivity analyses for the incremental net benefit at 90 days following randomisation according to alternative assumptions, compared with the base case. The vertical dashed line indicates incremental net benefit in the base-case analysis. The solid vertical line indicates no difference in net monetary benefits between the treatment groups.
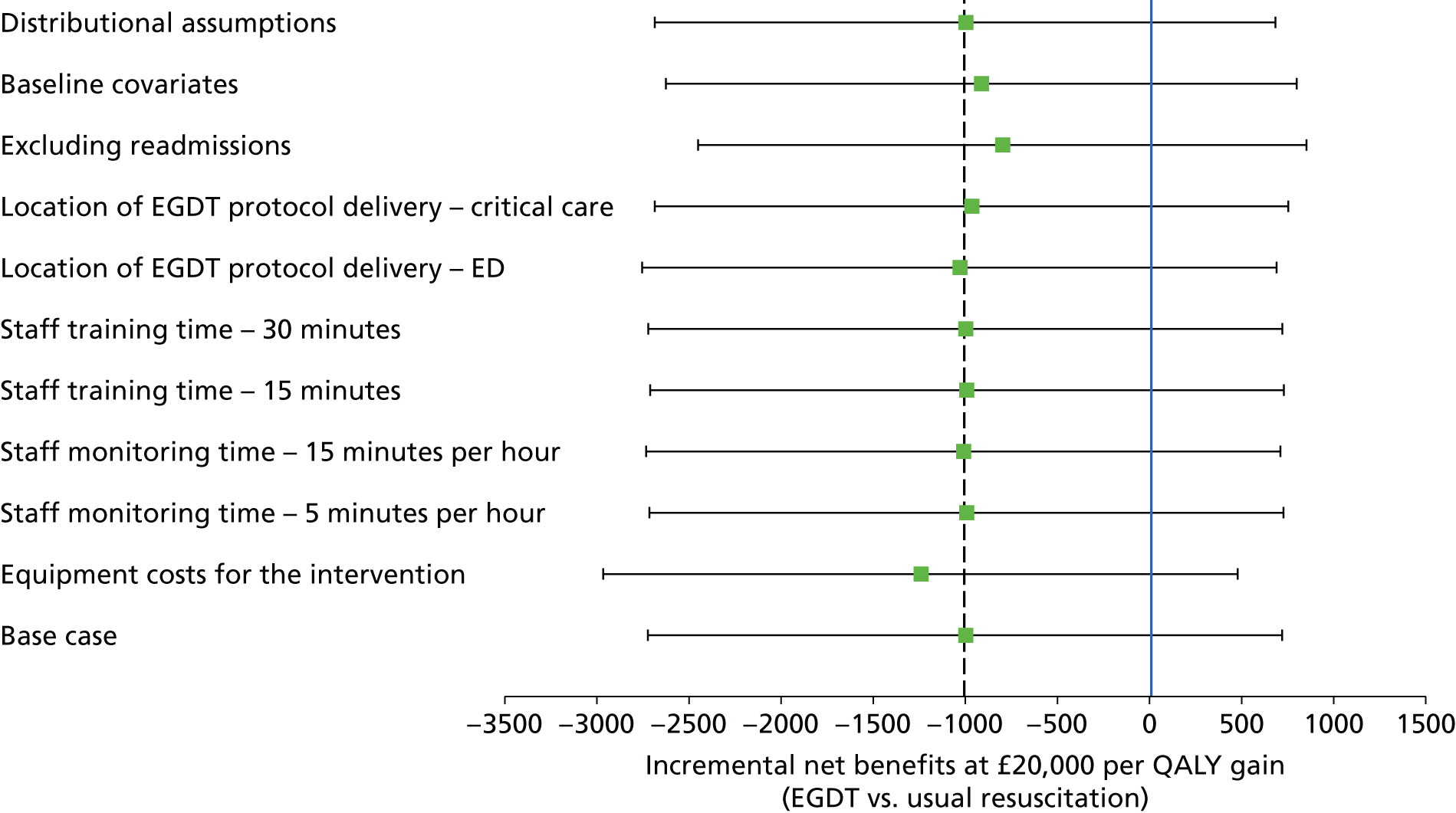
The results of the subgroup analyses are presented in Table 24, and show that the incremental QALYs were similar across all subgroups. Although there were some subgroups for which the incremental costs of EGDT were negative and hence the INBs were positive, the statistical uncertainty surrounding these findings was high. Hence for each subgroup, as for the overall results, the 95% CIs around the INB included zero. Adjusting for adherence to the EGDT protocol decreased the INB to −£1438, but the 95% CI still included zero (see Table 24).
| Subgroup | Incremental cost (£) (95% CI) | Incremental QALYs (95% CI) | Incremental net benefit (£) (95% CI) |
|---|---|---|---|
| Degree of protocolised resuscitation in usual-resuscitation group | |||
| Low | 806 (−1213 to 2825) | 0.002 (−0.004 to 0.009) | −765 (−2789 to 1259) |
| High | 1655 (−1822 to 5131) | −0.005 (−0.017 to 0.006) | −1764 (−5247 to 1719) |
| Age (years) | |||
| 18–56 | 3253 (−155 to 6662) | −0.001 (−0.012 to 0.011) | −3265 (−6684 to 154) |
| 57–67 | 398 (−3021 to 3818) | 0.003 (−0.008 to 0.014) | −329 (−3758 to 3100) |
| 68–77 | −1511 (−4884 to 1862) | −0.003 (−0.015 to 0.008) | 1444 (−1943 to 4831) |
| 78–95 | 2359 (−1112 to 5830) | 0.003 (−0.008 to 0.015) | −2296 (−5777 to 1185) |
| MEDS score | |||
| 0–4 | 2129 (−2644 to 6902) | 0.002 (−0.014 to 0.018) | −2089 (−6864 to 2686) |
| 5–6 | 2700 (−815 to 6215) | 0.002 (−0.009 to 0.014) | −2652 (−6173 to 869) |
| 7–9 | −250 (−3308 to 2807) | 0.005 (−0.005 to 0.015) | 351 (−2715 to 3417) |
| 10–20 | 196 (−2934 to 3326) | −0.009 (−0.019 to 0.001) | −377 (−3514 to 2760) |
| SOFA score | |||
| 0–2 | 1947 (−1482 to 5375) | 0.002 (−0.009 to 0.014) | −1898 (−5327 to 1531) |
| 3–4 | 623 (−2351 to 3598) | 0.001 (−0.009 to 0.011) | −603 (−3580 to 2374) |
| 5 | −1506 (−5705 to 2692) | −0.007 (−0.021 to 0.006) | 1359 (−2848 to 5566) |
| 6–14 | 2658 (−701 to 6016) | −0.004 (−0.014 to 0.007) | −2736 (−6099 to 627) |
| Time from ED presentation to randomisation (hours) | |||
| 0.2–1.8 | 1291 (−2114 to 4697) | −0.002 (−0.013 to 0.009) | −1322 (−4734 to 2090) |
| 1.8–2.5 | 2849 (−515 to 6214) | 0.004 (−0.007 to 0.015) | −2776 (−6147 to 595) |
| 2.5–3.5 | 1123 (−2344 to 4590) | −0.003 (−0.014 to 0.009) | −1179 (−4655 to 2297) |
| 3.5 + | −1453 (−4882 to 1976) | −0.001 (−0.012 to 0.01) | 1426 (−2008 to 4860) |
| Adherence adjusted analysis | 1423 (−1042 to 3888) | −0.001 (−0.009 to 0.007) | −1438 (−3909 to 1033) |
Cost-effectiveness at 1 year following randomisation (primary outcome)
Resource use up to 1 year
Acute hospital length of stay up to 1 year following randomisation is presented in Table 25. A higher proportion of patients in the EGDT group had an index hospital admission or readmission that continued beyond day 90. Between 90 days and 1 year following randomisation, the mean number of days in critical care, on general medical wards and in total was lower for the EGDT group than for the usual-resuscitation group. The mean total acute hospital length of stay up to 1 year following randomisation was 18.7 days in the EGDT group, compared with 18.2 days in the usual-resuscitation group.
| Acute hospital length of stay up to 1 year | EGDT (N = 625) | Usual resuscitation (N = 626) |
|---|---|---|
| Total acute hospital length of stay up to 90 daysa,b | 16.7 (19.2) | 15.5 (17.8) |
| Acute hospital length of stay 90 days to 1 year | ||
| Continuing index hospital admission,a n (%) | 13 (2.1) | 9 (1.4) |
| Length of stay in critical care (days) | 0 (0) | 0.01 (0.1) |
| Length of stay on general medical ward (days) | 0.5 (5.6) | 0.6 (10.0) |
| Acute hospital readmissions,b n (%) | 40 (6.4) | 46 (7.3) |
| Length of stay in critical carec (days) | 0.2 (1.5) | 0.3 (2.0) |
| Length of stay on general medical wardc (days) | 1.3 (6.6) | 1.8 (10.3) |
| Total acute hospital length of stay up to 1 year | 18.7 (24.5) | 18.2 (26.8) |
Table 26 reports results from responses to the Health Services Questionnaire administered at 1 year following randomisation, concerning resource use between 90 days and 1 year. The mean number of inpatient days reported from admissions other than those involving critical care was 6.3 days for the EGDT group and 6.6 days for the usual-resuscitation group. The mean numbers of outpatient visits and community care contacts between 90 days and 1 year were similar between the groups. Overall, both groups reported low use of community health services over 1 year following randomisation.
| Resource use | EGDT (n = 625), mean (SD) | Usual resuscitation (n = 626), mean (SD) |
|---|---|---|
| Inpatient days (general medical) | 6.3 (9.0) | 6.6 (11.9) |
| Outpatient visits | 1.6 (3.7) | 1.8 (4.5) |
| GP contacts | 1.7 (3.3) | 1.6 (3.4) |
| Nurse contacts | 1.5 (4.1) | 1.7 (4.8) |
| Occupational therapist contacts | 0.2 (1.0) | 0.3 (1.2) |
| Health visitor contacts | 0.3 (4.1) | 0.2 (1.6) |
| Clinical psychologist contacts | 0.03 (0.3) | 0.04 (0.4) |
| Speech therapist contacts | 0.05 (0.6) | 0.05 (0.5) |
| Physiotherapist contacts | 0.4 (2.0) | 0.6 (3.2) |
| Dietitian contacts | 0.1 (0.7) | 0.2 (1.1) |
Total costs up to 1 year
Table 27 reports the total costs up to 1 year following randomisation, across all of the resource use items recorded. At 1 year, the mean total costs per patient were £15,139 for the EGDT group and £14,375 for the usual-resuscitation group.
| Resource use categories | EGDT (n = 625), mean (SD) | Usual resuscitation (n = 626), mean (SD) |
|---|---|---|
| Total costs up to 90 daysa,b,c,d | 12,414 (14,970) | 11,424 (15,727) |
| Hospital costs 90 days to 1 year | ||
| Continuing index hospital admissiona | ||
| Critical care | 0 (0) | 15 (281) |
| General medical ward | 144 (1471) | 148 (2666) |
| Acute hospital readmissions | ||
| Critical careb | 607 (2233) | 575 (2678) |
| General medical wardb,c | 340 (1740) | 490 (2727) |
| Outpatient and community costsc,d | 1634 (3546) | 1722 (4406) |
| Total costs up to 1 yeard | 15,139 (18,345) | 14,375 (19,179) |
Health-related quality of life at 1 year
The distribution of responses to each dimension of the EQ-5D questionnaires, administered at 1 year following randomisation, is reported by treatment group in Table 28. At 1 year, a lower proportion of patients in the EGDT group than in the usual-resuscitation group reported ‘no problems’ for each dimension of the EQ-5D, with a higher proportion of patients in the EGDT group reporting ‘severe problems’ or ‘extreme problems’ for each dimension of health. The mean EQ-5D utility score of those patients who were alive at 1 year post randomisation was higher in the usual-resuscitation group (0.653) than in the EGDT group (0.620; Table 29).
| EQ-5D-5L dimension | EGDT (N = 244a), n (%) | Usual resuscitation (N = 236a), n (%) |
|---|---|---|
| Mobility | ||
| No problems | 65 (26.6) | 81 (34.3) |
| Slight problems | 40 (16.4) | 38 (16.1) |
| Moderate problems | 69 (28.3) | 47 (19.9) |
| Severe problems | 49 (20.1) | 55 (23.3) |
| Extreme problems | 21 (8.6) | 15 (6.4) |
| Self-care | ||
| No problems | 124 (50.8) | 141 (59.8) |
| Slight problems | 33 (13.5) | 27 (11.4) |
| Moderate problems | 49 (20.1) | 38 (16.1) |
| Severe problems | 21 (8.6) | 22 (9.3) |
| Extreme problems | 17 (7.0) | 8 (3.4) |
| Usual activities | ||
| No problems | 69 (28.3) | 88 (37.3) |
| Slight problems | 49 (20.1) | 42 (17.8) |
| Moderate problems | 55 (22.5) | 50 (21.2) |
| Severe problems | 46 (18.9) | 39 (16.5) |
| Extreme problems | 25 (10.3) | 17 (7.2) |
| Pain/discomfort | ||
| No problems | 71 (29.1) | 81 (34.3) |
| Slight problems | 50 (20.5) | 45 (19.1) |
| Moderate problems | 71 (29.1) | 57 (24.2) |
| Severe problems | 41 (16.8) | 43 (18.2) |
| Extreme problems | 11 (4.5) | 10 (4.2) |
| Anxiety/depression | ||
| No problems | 104 (42.6) | 120 (50.9) |
| Slight problems | 57 (23.4) | 50 (21.2) |
| Moderate problems | 55 (22.5) | 39 (16.5) |
| Severe problems | 19 (7.8) | 22 (9.3) |
| Extreme problems | 9 (3.7) | 5 (2.1) |
| End point | EGDT (n = 625) | Usual resuscitation (n = 626) | Incremental effect (unadjusted), mean (95% CI) | p-value |
|---|---|---|---|---|
| EQ-5D-5L utility score (survivors) | 0.620 (0.307) | 0.653 (0.323) | −0.032 (−0.085 to 0.020) | 0.23 |
| QALYs | 0.352 (0.323) | 0.351 (0.329) | 0.002 (−0.036 to 0.040) | 0.92 |
| Costs (£) | 15,139 (18,345) | 14,375 (19,179) | 764 (−1402 to 2930) | 0.49 |
| Incremental net benefit (£)a | −725 (−3000 to 1550) | 0.53 |
Cost-effectiveness at 1 year
At 1 year following randomisation, a slightly higher proportion of patients in the EGDT group were alive than in the usual-resuscitation group (see Secondary outcomes: clinical effectiveness). The net effect of the EGDT group having higher survival but a lower average EQ-5D utility score resulted in similar 1-year QALYs between the treatment groups (see Table 29). The mean total cost was higher in the EGDT group, with an incremental cost of £764 (95% CI −£1402 to £2930). Hence the INB for EGDT versus usual resuscitation was negative at −£725 (95% CI −£3000 to £1550). The distribution of incremental costs and QALYs in the cost-effectiveness plane is shown in Figure 23.
FIGURE 23.
Uncertainty in the mean costs (£) and QALY differences at 1 year and their distribution for EGDT vs. usual resuscitation.
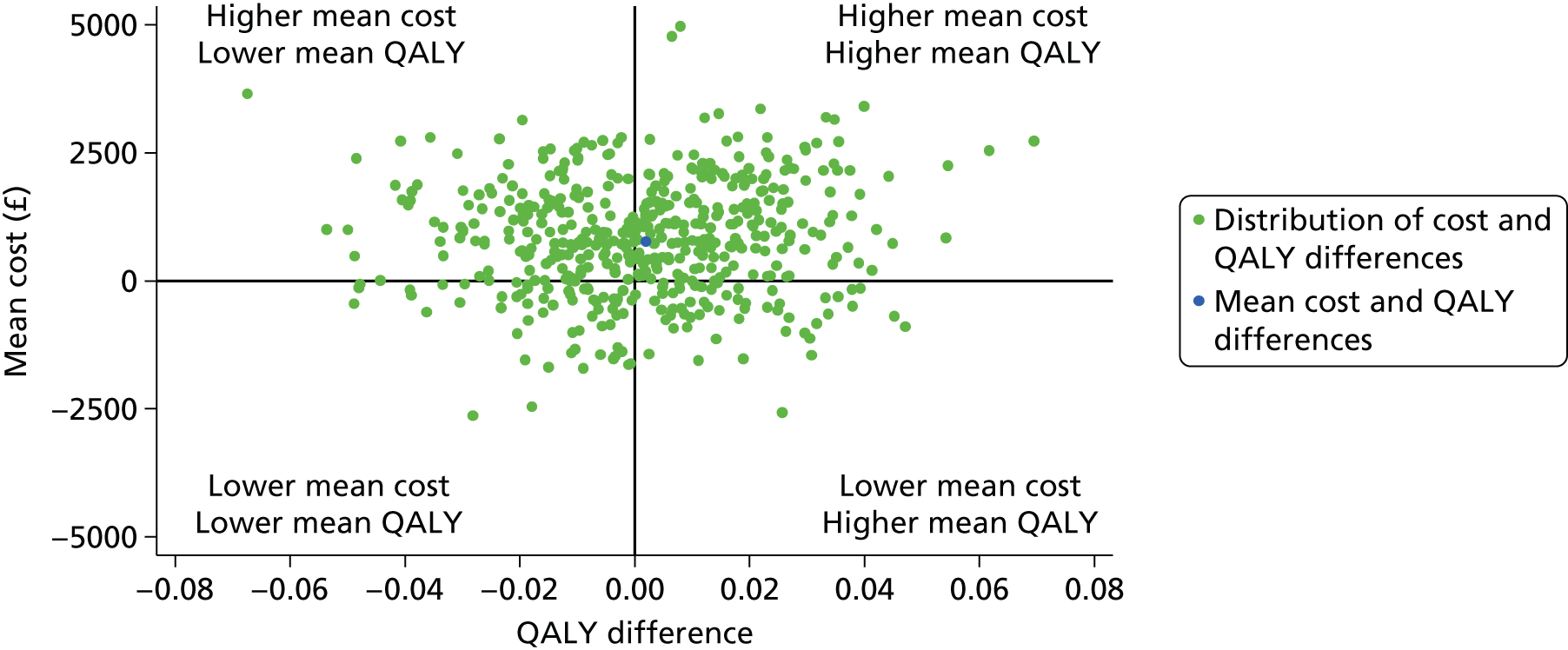
The cost-effectiveness acceptability curve (Figure 24) shows that at 1 year the probability that EGDT is more cost-effective than usual resuscitation, given the data, is below 30% at the £20,000 willingness-to-pay threshold stipulated by NICE.
FIGURE 24.
Cost-effectiveness acceptability curve reporting the probability that EGDT is cost-effective at 1 year at alternative levels of willingness to pay.

The estimated INBs were similar for the scenarios considered in the sensitivity analyses (Figure 25). This shows that the base-case results are robust to alternative assumptions.
FIGURE 25.
Sensitivity analyses for the incremental net benefit at 1 year following randomisation according to alternative assumptions, compared with the base case. The vertical dashed line indicates incremental net benefit in the base-case analysis. The solid vertical line indicates no difference in net monetary benefits between the treatment groups.
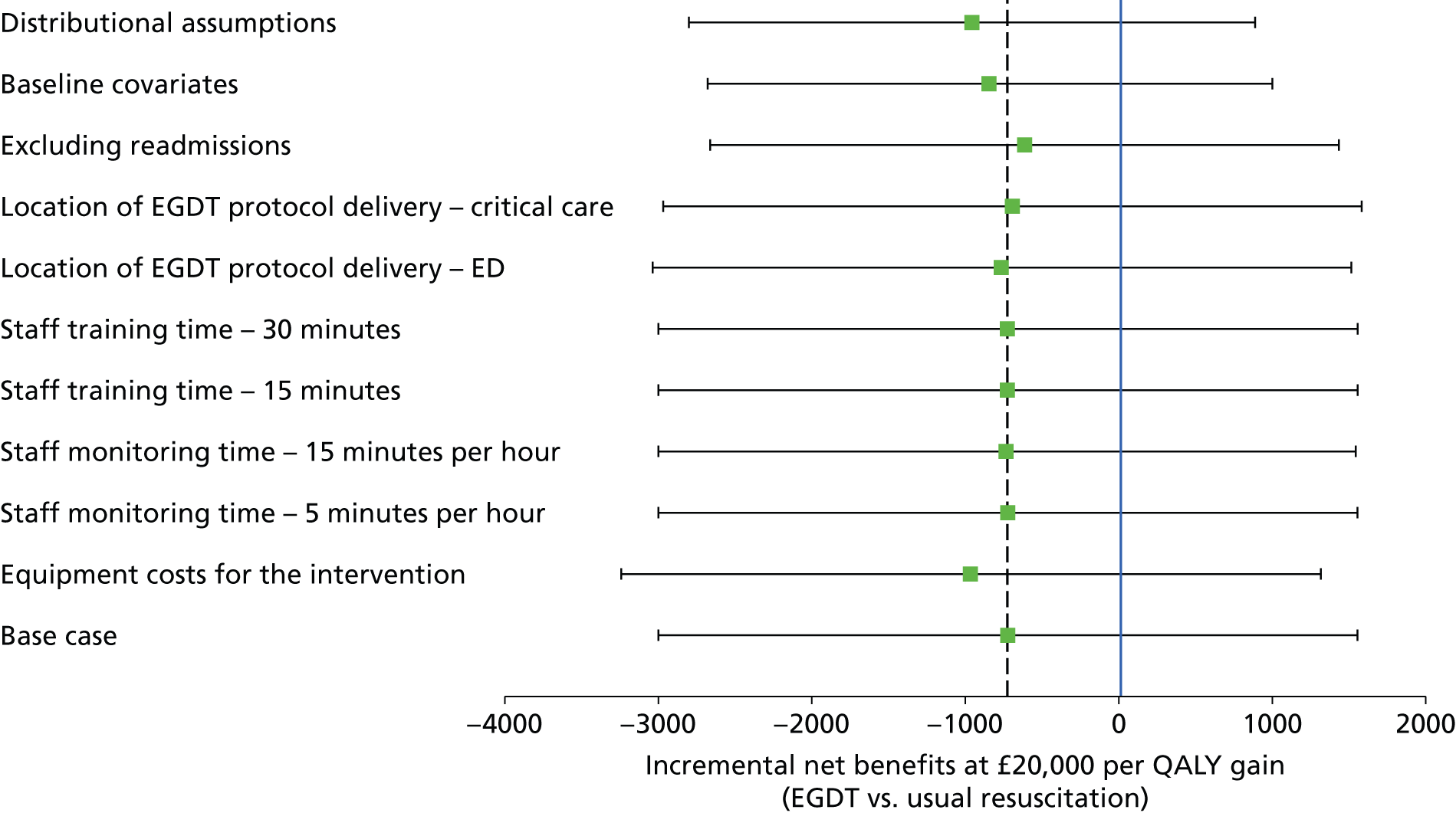
The estimated INBs were similar across all pre-specified subgroups (Table 30). Although there were some subgroups for whom EGDT was cost-saving and hence their INBs were positive, there was high statistical uncertainty around surrounding these findings. Hence for each subgroup, as for the overall results, there is high statistical uncertainty surrounding INBs.
| Subgroup | Incremental cost (£) (95% CI) | Incremental QALYs (95% CI) | Incremental net benefit (£) (95% CI) |
|---|---|---|---|
| Degree of protocolised resuscitation in usual-resuscitation group | |||
| Low | 801 (−1718 to 3319) | 0.014 (−0.03 to 0.058) | −525 (−3172 to 2122) |
| High | 739 (−3562 to 5040) | −0.017 (−0.093 to 0.059) | −1084 (−5578 to 3410) |
| Age (years) | |||
| 18–56 | 3643 (−626 to 7913) | 0.011 (−0.064 to 0.086) | −3422 (−7932 to 1088) |
| 57–67 | 257 (−3977 to 4490) | 0.025 (−0.049 to 0.098) | 238 (−4234 to 4710) |
| 68–77 | −1924 (−6101 to 2252) | −0.028 (−0.104 to 0.047) | 1357 (−3042 to 5756) |
| 78–95 | 2038 (−2227 to 6302) | 0.041 (−0.035 to 0.116) | −1226 (−5720 to 3268) |
| MEDS score | |||
| 0–4 | 1972 (−3966 to 7909) | 0.015 (−0.088 to 0.118) | −1670 (−7866 to 4526) |
| 5–6 | 2401 (−1931 to 6733) | 0.008 (−0.068 to 0.084) | −2241 (−6789 to 2307) |
| 7–9 | −909 (−4684 to 2866) | 0.045 (−0.022 to 0.112) | 1806 (−2160 to 5772) |
| 10–20 | 615 (−3305 to 4534) | −0.047 (−0.114 to 0.021) | −1551 (−5636 to 2534) |
| SOFA score | |||
| 0–2 | 1579 (−2755 to 5913) | 0.020 (−0.055 to 0.095) | −1183 (−5694 to 3328) |
| 3–4 | 696 (−3033 to 4425) | 0.007 (−0.059 to 0.072) | −566 (−4450 to 3318) |
| 5 | −2836 (−8057 to 2384) | −0.036 (−0.127 to 0.056) | 2118 (−3332 to 7568) |
| 6–14 | 2769 (−1401 to 6939) | −0.016 (−0.087 to 0.055) | −3097 (−7446 to 1252) |
| Time from ED presentation to randomisation (hours) | |||
| 0.2–1.8 | 1739 (−2507 to 5985) | −0.010 (−0.085 to 0.065) | −1940 (−6387 to 2507) |
| 1.8–2.5 | 3576 (−633 to 7786) | 0.032 (−0.043 to 0.107) | −2934 (−7350 to 1482) |
| 2.5–3.5 | 1111 (−3225 to 5446) | −0.005 (−0.082 to 0.071) | −1215 (−5785 to 3355) |
| 3.5 + | −3535 (−7762 to 692) | −0.007 (−0.083 to 0.068) | 3388 (−1037 to 7813) |
| Adherence adjusted analysis | 1099 (−2013 to 4211) | 0.003 (−0.051 to 0.057) | −1042 (−4312 to 2228) |
Lifetime incremental cost-effectiveness
Long-term survival
The Kaplan–Meier survival curves show that when the time horizon was extended beyond 1 year, for those with survival data available, the probability of survival remained similar between treatment groups (Figure 26).
FIGURE 26.
Kaplan–Meier survival curves for long-term survival.

To calculate QALYs over 20 years, the long-term survival for each patient was estimated by combining the observed survival for each patient up to 1 year with their predicted survival from 1 year to 20 years. We compared alternative parametric extrapolation approaches to predict longer-term survival of patients recruited to ProMISe. Figure 27 considers alternative parametric extrapolations for survival, using the observed survival data after day 30. The survival data were pooled across the treatment groups, given that there was no evidence of an effect of treatment group on survival. Of the alternative survival functions, log-normal appeared to fit the observed data best in that it reported the lowest Akaike information criteria and Bayesian information criteria (Table 31). However, the Gompertz function offered the most plausible projections of future survival (see Figure 27 and Table 31), in that the levels of survival remained constant over time from 5 years following randomisation onwards. The parametric models predicted excess mortality in patients recruited to ProMISe compared with the age-/sex-matched general population. In the base case, we applied death rates according to the most plausible parametric model (i.e. Gompertz) between year 2 and year 15. At year 16, predicted survival overlaps with age-/sex-matched survival and, therefore, from year 16 onwards we applied age-/sex-matched general population death rates.
FIGURE 27.
Comparison of alternative parametric extrapolations of survival.

| Distribution | AIC | BIC |
|---|---|---|
| Gompertz | 1658.0 | 1687.1 |
| Log-normal | 1642.4 | 1671.5 |
| Logistic | 1644.5 | 1673.5 |
| Weibull | 1645.8 | 1674.8 |
| Exponential | 1691.0 | 1715.2 |
Long-term health-related quality of life
The lifetime cost-effectiveness analysis required health-related quality of life to be estimated over time. We used health-related quality of life from ProMISe and also from the age-/sex-matched general population to predict the long-term quality of life of patients recruited to ProMISe. There was a difference in the mean quality of life between the treatment groups at 1 year (0.62 for EGDT and 0.65 for usual resuscitation), which was maintained for the period over which the excess rate of mortality was applied (years 2–15). In the base case, we applied age-/sex-matched general population quality of life for years 2–15, but applying decrements of 21% (EGDT) and 17% (usual resuscitation) to allow for the relative differences in quality of life observed between the treatment groups at 1 year to be maintained.
Long-term costs
To project lifetime costs attributable to the initial episode of severe sepsis, we considered mean inpatient, outpatient and community costs up to 1 year estimated from the Health Services Questionnaires.
The mean cost for each intervention group was calculated for those patients who survived at least up to 1 year. These mean costs were used to impute mean costs between year 1 and year 15. For each group, these mean costs were similar (£4221 for EGDT and £4216 for usual resuscitation). After year 15 it was assumed that there were no further readmission costs that were attributable to the original episode of severe sepsis.
Lifetime incremental cost-effectiveness
Table 32 presents the resultant lifetime QALYs, lifetime costs and INB according to the base-case assumptions. Overall, at the NICE-stipulated threshold of £20,000 per QALY the INB was negative, but with a wide 95% CI that included zero. The distribution of incremental costs and QALYs in the cost-effectiveness plane is shown in Figure 28.
| End point | EGDT (n = 625) | Usual resuscitation (n = 626) | Incremental effect (unadjusted), mean (95% CI) | p-value |
|---|---|---|---|---|
| QALYs | 4.584 (3.546) | 4.582 (3.720) | 0.002 (−0.411 to 0.414) | 0.99 |
| Costs (£) | 33,620 (25,012) | 32,142 (25,798) | 1478 (−1434 to 4390) | 0.32 |
| Incremental net benefit (£)a | −1446 (−8102 to 5210) | 0.67 |
FIGURE 28.
Uncertainty in the mean costs (£) and QALY differences at 1 year and their distribution for EGDT vs. usual resuscitation.

The cost-effectiveness acceptability curve shows that that the probability of EGDT of being cost-effective compared with usual resuscitation is never > 50% irrespective of how much society is willing to pay for a QALY gain (Figure 29).
FIGURE 29.
Cost-effectiveness acceptability curve reporting the probability that the EGDT is cost-effective (at lifetime) at alternative levels of willingness to pay.

The sensitivity analyses on the long-term results suggest that these findings are robust to alternative assumptions including those applied to extrapolation of long-term survival, quality of life for survivors and costs (Figure 30). For example, a large versus smaller decrement in quality of life over 15 years had only marginal impact on the mean INB.
FIGURE 30.
Sensitivity analyses for lifetime incremental net benefit according to alternative assumptions, compared with the base case. The vertical dashed line indicates incremental net benefit in the base-case analysis. The solid vertical line indicates no difference in net monetary benefits between the treatment groups. QOL, quality of life.

The results of the subgroup analysis presented in Table 33 show that there were some differences in the direction of mean incremental effects but high statistical uncertainty surrounds these findings. Across subgroups, incremental QALYs were small. Although there were some subgroups of patients for whom the incremental costs of EGDT were negative, and hence the INBs were positive, 95% CIs around these INBs included zero. Hence for each subgroup, as for the overall result, cost-effectiveness estimates were surrounded by high statistical uncertainty.
| Subgroup | Incremental cost (£) (95% CI) | Incremental QALYs (95% CI) | Incremental net benefit (£) (95% CI) |
|---|---|---|---|
| Degree of protocolised resuscitation in usual-resuscitation group | |||
| Low | 1905 (−1495 to 5305) | 0.104 (−0.38 to 0.587) | 168 (−7630 to 7966) |
| High | 965 (−4874 to 6805) | −0.131 (−0.968 to 0.706) | −3589 (−17,056 to 9878) |
| Age (years) | |||
| 18–56 | 4881 (−736 to 10,498) | 0.096 (−0.715 to 0.907) | −2957 (−16,236 to 10,322) |
| 57–67 | 1357 (−4289 to 7003) | 0.114 (−0.692 to 0.919) | 915 (−12,166 to 13,996) |
| 68–77 | −1945 (−7570 to 3681) | −0.201 (−1.007 to 0.606) | −2069 (−15,097 to 10,959) |
| 78–95 | 4635 (−1113 to 10,383) | 0.521 (−0.309 to 1.351) | 5787 (−7640 to 19,214) |
| MEDS score | |||
| 0–4 | 3344 (−4654 to 11,343) | 0.139 (−1.006 to 1.284) | −560 (−19,050 to 17,930) |
| 5–6 | 3759 (−2120 to 9638) | 0.139 (−0.700 to 0.977) | −985 (−14,470 to 12,500) |
| 7–9 | 819 (−4262 to 5900) | 0.280 (−0.441 to 1.001) | 4788 (−6840 to 16,416) |
| 10–20 | −140 (−5456 to 5176) | −0.361 (−1.104 to 0.383) | −7077 (−18,979 to 4825) |
| SOFA score | |||
| 0–2 | 3008 (−2798 to 8814) | 0.151 (−0.661 to 0.963) | 4 (−13,077 to 13,085) |
| 3–4 | 1886 (−3143 to 6915) | 0.114 (−0.591 to 0.819) | 386 (−10,938 to 11,710) |
| 5 | −3279 (−10,351 to 3793) | −0.312 (−1.300 to 0.677) | −2952 (−18,774 to 12,870) |
| 6–14 | 1792 (−3854 to 7439) | −0.369 (−1.163 to 0.426) | −9164 (−21,917 to 3589) |
| Time from ED presentation to randomisation (hours) | |||
| 0.2–1.8 | 2471 (−3280 to 8223) | 0.005 (−0.819 to 0.829) | −2371 (−15,643 to 10,901) |
| 1.8–2.5 | 6390 (707 to 12,073) | 0.523 (−0.276 to 1.323) | 4074 (−8777 to 16,925) |
| 2.5–3.5 | 693 (−5140 to 6525) | −0.279 (−1.130 to 0.571) | −6275 (−20,079 to 7529) |
| 3.5 + | –3759 (–9472 to 1953) | –0.230 (–1.052 to 0.591) | –847 (–14,099 to 12,405) |
| Adherence adjusted analysis | 2126 (–2056 to 6307) | 0.002 (–0.591 to 0.595) | –2079 (–11,654 to 7496) |
Chapter 6 Discussion and conclusions
Principal findings
Among adults identified with early signs of septic shock presenting to the ED of one of 56 NHS hospitals in England and receiving 6 hours of protocolised resuscitation, there was no significant difference in mortality at 90 days when compared with usual resuscitation (relative risk 1.01, 95% CI 0.85 to 1.20). Although mortality was lower than anticipated, our results rule out, with 95% confidence, a relative risk reduction with EGDT of > 15%. On average, EGDT increased costs and, given similar QALYs across groups, INB at 1 year was negative (−£725, 95% CI −£3000 to £1550). The probability that EGDT is cost-effective (at a willingness to pay of £20,000 per QALY) is below 30%. Sensitivity analyses found that this conclusion is robust to alternative assumptions to those made in the base-case analysis.
There was no significant interaction between treatment effect and mortality at 90 days across pre-specified subgroups on the basis of degree of protocolisation of usual resuscitation, age, MEDS score, SOFA score or time to randomisation. More patients receiving EGDT were admitted to ICU, resulting in significantly more days spent in critical care in this group. Treatment intensity was greater for the EGDT group, driven by adherence to the protocol, and indicated by the increased use of central venous catheters, intravenous fluids, vasoactive drugs and packed red blood cell transfusions. Increased treatment intensity was reflected by significantly higher SOFA scores and more advanced cardiovascular support days in critical care for the EGDT group. There were no significant differences in any other secondary outcomes including health-related quality of life, measured in health-state utility values, which was substantially poorer in this severely ill patient group at both 90 days (0.61) and 1 year (0.62–0.65) than for the age-/sex-matched general population (0.80). 58 At 12 months post randomisation, approximately 30% of responders reported ‘severe’ or ‘extreme’ problems with mobility or undertaking usual activities, indicating substantial ongoing morbidity for this patient group.
Strengths
ProMISe was set in a real-world context, in a large, representative, mixed sample of approximately one-quarter of NHS hospitals in England, and was pragmatic, with staff and locations for delivery of the protocol determined locally, as would be the case if the intervention were to be adopted into routine NHS care. Site set-up was rapid, resulting in our trial recruiting its full sample of 1260 patients over a relatively shorter time period than the two similar studies already reported from the USA and Australasia; minimising the potential for other changes in treatments to impact on the trial.
Loss to follow-up was minimal and all analyses were conducted according to a pre-specified, published statistical analysis plan including, given the complex intervention, adjusted analyses to address the degree of adherence to EGDT and the possibility of the existence of a learning curve for its delivery.
Unlike the previous studies, our trial reported on quality of life at 90 days and 1 year post randomisation and our results include an integrated analysis of the cost-effectiveness of EGDT. This prospectively designed economic evaluation ensured that resource use data were collected on both primary admissions and readmissions for each patient randomised. The resource use measurement harnessed information from three sources: the case report forms, linked data from the Case Mix Programme database and responses to follow-up Health Service Questionnaires. This approach enabled detailed resource use measurement for those events that were anticipated to be the key drivers of the incremental costs of EGDT versus usual resuscitation. The cost-effectiveness analysis also measured quality of life with the EQ-5D-5L;59 this version of the EQ-5D instrument was anticipated to be sensitive to differences in health status between the treatment groups. To address missing data, we undertook the recommended approach of multiple imputation and imputed missing values, conditional on all the information observed.
Limitations
As with all studies enrolling patients presenting as emergencies, recruitment was more challenging at weekends and out-of-office hours. As a result of this, together with other logistical issues, only around one-third of eligible patients were recruited. However, there were no important differences in baseline characteristics between patients recruited during usual working hours or at weekends and out-of-office hours. In addition, exclusion by a clinician was comparatively rare.
With recruitment rates much lower – and eligible patients missed – at nights and at weekends, alongside a number of sites undergoing a period of suspension or closing early to recruitment owing to lack of available resources, the trial recruited behind the planned schedule. Owing to the short time-windows to recruit and commence treatment in randomised controlled trials in emergency and critical care settings, it is vital that research infrastructure within the NHS is delivered 24 hours per day, 7 days per week.
The intervention could not be blinded to those caring for patients but the risk of bias was minimised through central randomisation to ensure allocation concealment and the use of a primary outcome not subject to observer bias. The mortality observed in the usual-resuscitation group was lower than anticipated as the basis for the sample size calculation (29.2% vs. 40%). This was also true for both ProCESS (60-day in-hospital mortality, 18.9% observed, 30–46% anticipated23) and ARISE (90-day mortality, 18.8% observed, 38% anticipated24). However, unlike both ProCESS and ARISE, based on the 95% CI, our results were able to rule out with 95% confidence, in our setting of NHS EDs, the relative risk reduction in 90-day mortality of 20% that was the basis for our sample size calculation. Although able to provide, relatively, the most precise overall estimate of effect, we have limited power to address many important subgroups for either the clinical effectiveness or the cost-effectiveness.
The long-term cost-effectiveness analysis was inevitably required to make assumptions, in particular about the mortality, quality of life and cost in the time period beyond the observed data. The study made maximum use of the available trial data to inform these assumptions. For example, the analysis of the mortality data found that mortality was similar between the treatment groups at each time point, and that there was excess mortality versus the age-/sex-matched general population for up to 15 years post randomisation. These findings informed the assumptions made in the base-case analysis concerning the long-term survival extrapolation. The study made these and other requisite structural assumptions transparent and subjected them to extensive sensitivity analyses.
Our findings in context
Rivers et al.6
Unlike the original Rivers et al. trial,6 we did not observe a significant reduction in mortality at hospital discharge. There are many possible reasons for this. First, there may be bias in a small, single-centre trial, leading to an inflated effect. Second, in the intervening years, both presenting patients and usual resuscitation has changed; all patients in our trial received antibiotics prior to randomisation and, comparing the usual-resuscitation groups, patients in our trial appeared less sick (with lower baseline serum lactate, lower APACHE II scores and a lower rate of initiation of mechanical ventilation in the first 6 hours), received much lower volumes of fluid, more vasoactive drugs and experienced lower hospital mortality.
ProCESS and ARISE
Our results, both for adherence and outcomes, are comparable with those from the ProCESS23 and ARISE24 studies from the USA and Australasia, respectively. Of note is that a greater proportion of patients in our trial met the inclusion criteria for both refractory hypotension and hyperlactataemia, associated with higher mortality, than met either criterion alone. The rate of death at 90 days in our trial was slightly lower than ProCESS but higher than ARISE.
Economic evaluation
Previous cost-effectiveness analyses have reported that EGDT is cost-effective relative to usual resuscitation. 9,11,54,60,61 However, almost all these studies relied solely on observational data,9,11,60,61 and so the finding that EGDT was associated with improved survival, and higher QALYs, could reflect confounding by indication. Neither ProCESS nor ARISE undertook a fully integrated cost-effectiveness analysis. A key advantage is that individual-level data on quality of life and resource use were collected prospectively. The quality-of-life data were collected at 90 days and 1 year following randomisation with EQ-5D-5L59 and hence the cost-effectiveness analysis was able to incorporate any differences in quality of life between the treatment groups into the final measures of cost-effectiveness.
A previous cost-effectiveness analysis, based on the mortality reduction reported in the Rivers’ trial,6 projected that EGDT would lead to a gain in QALYs and a reduction in hospital costs of 22%. 54 In contrast, our analysis of the individual patient resource use data from the trial found that EGDT led to a small average increase in both intervention costs and the use of critical care and general medical ward resources. Our cost analysis also allowed for the additional training and monitoring costs of providing EGDT in the NHS – relatively minor costs. When combined with no difference in patient outcomes this led to a very low probability of EGDT being cost-effective. The quality-of-life estimates were similar to previous estimates for patients surviving sepsis and sepsis shock,62,63 and to a previous study of general ICU survivors. 64
Trends in outcomes
Mortality for patients with severe sepsis has been reported to be decreasing in a number of settings. 3,65–67 Although this may, in part, be a dilution effect due to increased recognition and changes in clinical coding,68,69 it is also likely that increased global awareness and a focus on early identification and treatment, for example, early administration of antibiotics, have contributed to improved outcomes. It is of note, however, that the reported downwards trend in mortality has been ongoing since before the Rivers et al. 6 trial and Surviving Sepsis Campaign,70 and that similar trends have been reported among critically ill patients without sepsis. 3
Implications for health care
Our results suggest that usual resuscitation has evolved over the fifteen years since the Rivers et al. 6 trial; NHS hospitals now achieve similar levels of in-hospital survival to those achieved with EGDT in the Rivers et al. 6 trial for patients with septic shock identified early and receiving intravenous antibiotics and adequate fluid resuscitation. The addition of continuous ScvO2 monitoring and strict protocolisation of care was, on average, more costly and did not improve outcomes.
Although adherence to the EGDT protocol in ProMISe was similar in most respects with both ProCESS and ARISE, it is of note that the adherence with administration of packed red blood cells was generally low and occurred considerably slower than for other interventions. The lower adherence may reflect concerns over the relatively high haemoglobin threshold for blood transfusion in the EGDT protocol, as more recent research evidence suggests that lower transfusion thresholds may be preferable,71 but the long time lag from reaching the transfusion threshold to administration of blood may warrant local investigation to ensure adequate processes are in place for rapid provision of blood products when required.
Recommendations for research
Recommendation 1: an individual patient data meta-analysis of the three completed trials should be conducted
Our results complete the planned trio of evaluations of EGDT across the USA, Australasia and England. These three large studies, each with their own strengths and weaknesses, have indicated that EGDT is not superior to usual resuscitation. Recognising that each of the three individual, large trials has limited power for evaluating potentially important subgroups, the harmonised approach adopted provides the opportunity to conduct an individual patient data meta-analysis, enhancing both knowledge and generalisability.
Recommendation 2: further research to consider alternative definitions of adherence to the resuscitation protocol should be conducted
Both the clinical effectiveness and cost-effectiveness analyses reported estimates that were adherence-adjusted as part of pre-specified secondary analyses. However, further research to consider alternative definitions of adherence to the EGDT resuscitation protocol are warranted. In particular, future research could apply differential weights for adherence to the different elements of the EGDT resuscitation protocol, or to particular time points within the 6-hour intervention period. Hence subsequent research could report whether EGDT was clinically effective or cost-effective when these alternative definitions of adherence were met.
Acknowledgements
We are grateful to the NIHR Health Technology Assessment programme for funding this project. We wish to thank all the patients and staff from all the sites that participated in the trial.
We wish to thank Edwards Lifesciences for providing trial equipment and technical support, and general practitioners and health-care professionals for their assistance in patient follow-up. A thank you also to all the staff at ICNARC, with special thanks to Catrina Adams; Kimberley Anderson; Ruth Canter; Blair McLennan; Alvin Richards-Belle; Steven Saunders; and Emma Walmsley for their assistance with patient follow-up.
Research staff at participating sites
We acknowledged that there have been many other individuals who made a contribution within the participating units. It is impossible to thank everyone personally; however, we would like to thank the following research staff:
Addenbrooke’s Hospital (Vazeer Ahmed, Adrian Boyle and Andy Scott-Donkin); Arrowe Park Hospital (Heather Black, Christopher Smalley, Reni Jacob and Andrea Wooten); Barnsley Hospital (Julian Humphrey, Sally Anne Pearson and James Griffiths); Bedford Hospital (Devasena Subramanyam, David Niblett and Sunil Krishnanankutty); Birmingham Heartlands Hospital (Fang Gao-Smith, Teresa Melody and Keith Couper); Blackpool Victoria Hospital (Raj Nichani, Emma Brennan and Simon Tucker); Bristol Royal Infirmary (Jonathan Benger, Judith Edwards and Kathryn Pollock); Broomfield Hospital (Dilshan Arawwawala, Alex Hieatt and Fiona McNeela); Chelsea and Westminster Hospital (Derek Bell, Theresa Weldring and Jaime Carungcong); Derriford Hospital (Peter MacNaughton, Helen McMillan and Kate Tantam); Dorset County Hospital (Tony Doyle, Sarah Moreton and Stephanie Jones); Frenchay Hospital (Jason Kendall, Ruth Worner and Anna Gilbertson); Hinchingbrooke Hospital (Colin Borland, Suzanne Boys and Shashank Ranjan); Hull Royal Infirmary (Ian Smith, Neil Smith, Victoria Mendham and Paul Smith); John Radcliffe Hospital (Duncan Young, Roser Farras-Araya and David Vallance); Kettering General Hospital (Phil Watt, Parizade Raymode and Laszlo Hollos); King’s College Hospital (Phil Hopkins, Paul Riozzi, Harriet Couper and Sinead Helyar); Leicester Royal Infirmary (Jonathan Thompson, Dawn Hales, Zubeir Essat and Prem Andreou); Leighton Hospital (Susan Gilby, Phil Chilton and Richard Miller); Manchester Royal Infirmary (John Butler, Alison Jefferies and Richard Clark); Medway Maritime Hospital (Graeme Sanders, Nuno Pinto and Catherine Plowright); Musgrove Park Hospital (Richard Innes, Dawn Bayford and Pippa Richards); New Cross Hospital (Shameer Gopal, Jagtar Singh Pooni and Hazel Spencer); Newham University Hospital (James Napier and Ena Warrington); North Devon District Hospital (Liam Kevern, Jane Hunt and Colin Barrett); North Tyneside General Hospital (Eliot Sykes, Karen Connelly and Bryan Yates); Peterborough City Hospital (Coralie Carle, Critical Care Outreach Team and Theresa Croft); Poole Hospital (Nick Jenkins, Henrik Reschreiter, Julie Camsooksai and Helena Barcraft-Barnes); Queen Elizabeth Hospital Birmingham (Catherine Snelson, Colin Bergin and Frank Keats); Queen Elizabeth Hospital, Gateshead (Vanessa Linnett, Jenny Ritzema and Steve Christian); Queen’s Medical Centre (Daniel Harvey, Philip Miller, Claudia Woodford and Anna Bolland); Royal Berkshire Hospital (Liza Keating, David Mossop and Carys Jones); Royal Bournemouth Hospital (David Martin, Emma Willett and Peter Swallow); Royal Lancaster Infirmary (Sam McBride, Asim Ijaz, Jay Datta and Jayne Craig); Royal Preston Hospital (Thomas Owen, Alex Williams, Sean McMullan and Jackie Baldwin); Royal Surrey County Hospital (Mehrun Zuleika and Peter Carvalho); Royal Sussex County Hospital (Dan Agranoff, Fiona Ingoldby, Laura Ortiz-Ruiz De Gordoa and Carrie Ridley); Royal Victoria Infirmary (Ian Clement and Charley Higham); Salford Royal Hospital (Bruce Martin, Kate Clayton and Julie Chadwick); South Tyneside District Hospital (Christian Frey, Diane Miller and Philippa Laverick); Southend University Hospital (Khurram Iftikhar, David Higgins and Victoria Katsande); Stafford Hospital (Moses Chikungwa and Clare Jackson); The Great Western Hospital (Malcolm Watters, Paul Liddiard and Kate Gannon); The Ipswich Hospital (Richard Howard-Griffin, Stephanie Bell and Heather Blaylock); The James Cook University Hospital (Isabel Gonzalez, Emmanuel Cirstea and Stephen Bonner); The Queen Elizabeth Hospital, King’s Lynn (Parvez Moondi, Kate Wong and Joseph Carter); The Royal Blackburn Hospital (Stephen Hartley, Iain Crossingham, Joanne Hinchcliffe and Leanne Phoenix); The Royal London Hospital (Tim Harris, Jason Pott and Geoffrey Bellhouse); Torbay Hospital (Michael Mercer, Pauline Mercer and Hazel Robinson); University College Hospital (David Brealey, Jung Ryu, Georgia Becardes and the Critical Care Trials Team); University Hospital of North Staffordshire (Anne-Marie Morris, Mark Poulson, Loretta Barnett and Ian Massey); Wansbeck General Hospital (Eliot Sykes, Karen Connelly and Bryan Yates); Whipps Cross University Hospital (Tim Harris and Imogen Skene); Whiston Hospital (Patrick Nee, Susan Dowling and Amanda McCairn); Worthing Hospital (Roger Duckitt, Richard Venn and Jordi Margalef); and York Hospital (Jonathan Redman, Helen Milner and Sara Ma).
Contributions of authors
Paul R Mouncey (senior trial manager) managed the trial, contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Dr Tiffany M Osborn (associate professor) contributed to the design of the trial, the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
G Sarah Power (statistician) contributed to the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Dr David A Harrison (senior statistician and honorary senior lecturer in medical statistics) contributed to the design of the trial, the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Dr M Zia Sadique (lecturer in health economics) contributed to the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor Richard D Grieve (professor of health economics methodology) contributed to the design of the trial, the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Rahi Jahan (research assistant) contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Jermaine CK Tan (trials data manager) contributed to the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Dr Sheila E Harvey (CTU manager and senior research fellow) contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor Derek Bell (professor of acute medicine) contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor Julian F Bion (professor of intensive care medicine) contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor Timothy J Coats (professor of emergency medicine) contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor Mervyn Singer (professor of intensive care medicine) contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor J Duncan Young (professor of intensive care medicine) contributed to the design of the trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Professor Kathryn M Rowan (Director of Scientific & Strategic Development at ICNARC and honorary professor) conceived and designed the trial, contributed to acquisition, analysis and interpretation of the data, and drafted and critically revised the manuscript.
Trial Management Team
Paul Mouncey (senior trial manager); Sarah Corlett (previous trial administrator); Dr David Harrison (senior statistician); Dr Sheila Harvey (CTU manager/senior research fellow); Rahi Jahan (research assistant); Hannah Muskett (previous assistant trials manager); Sarah Power (trial statistician); Professor Kathryn Rowan (chief investigator); Dr Rachael Scott (previous senior trial manager); and Jermaine Tan (trials data manager).
Trial Management Group
Professor Derek Bell (co-investigator); Professor Julian Bion (co-investigator); Professor Timothy Coats (co-investigator); Dr David Harrison (senior statistician); Dr Sheila Harvey (CTU manager/senior research fellow); Dr Tiffany Osborn (co-investigator); Professor Kathryn Rowan (chief investigator); Professor Mervyn Singer (co-investigator); and Professor Duncan Young (co-investigator).
Trial Steering Committee
Professor Steve Goodacre (independent chairperson); Professor David Bennett (previous independent); Professor Julian Bion; Caroline Bowers (independent); Phillip Crow (independent); Professor Rupert Pearse (independent); Professor Kathryn Rowan; and Professor David Yates (independent).
Independent Data Monitoring and Ethics Committee
Professor Jon Nicholl (chairperson); Professor Alasdair Gray; and Professor Timothy Walsh.
Publications
Power GS, Harrison DA, Mouncey PR, Osborn TM, Harvey SE, Rowan KM. The Protocolised Management in Sepsis (ProMISe) trial statistical analysis plan. Crit Care Resusc 2013;15:311–17.
Huang DT, Angus DC, Barnato A, Gunn SR, Kellum JA, Stapleton DK, et al. Harmonizing international trials of early goal directed resuscitation for severe sepsis and septic shock: methodology of ProCESS, ARISE, and ProMISe. Intensive Care Med 2013;39:1760–75.
Mouncey PR, Osborn RM, Power GS, Harrison DA, Sadique MZ, Grieve RD, et al. Trial of early, goal-directed resuscitation for septic shock. N Engl J Med 2015;372:1301–11.
Angus DC, Barnato AE, Bell D, Bellomo R, Chong CR, Coats TJ, et al. A systematic review and meta-analysis of early goal-directed therapy for septic shock: the ARISE, ProCESS and ProMISe Investigators. Intensive Care Med 2015;41:1549–60.
Data sharing statement
Data can be obtained from the corresponding author.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Jawad I, Luksic I, Rafnsson SB. Assessing available information on the burden of sepsis: global estimates of incidence, prevalence and mortality. J Glob Health 2012;2. http://dx.doi.org/10.7189/jogh.01.010404.
- Levy MM, Rhodes A, Phillips GS, Townsend SR, Schorr CA, Beale R, et al. Surviving sepsis campaign: association between performance metrics and outcomes in a 7.5-year study. Crit Care Med 2015;43:3-12. http://dx.doi.org/10.1097/CCM.0000000000000723.
- Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA 2014;311:1308-16. http://dx.doi.org/10.1001/jama.2014.2637.
- Cuthbertson BH, Elders A, Hall S, Taylor J, MacLennan G, Mackirdy F, et al. Mortality and quality of life in the five years after severe sepsis. Crit Care 2013;17. http://dx.doi.org/10.1186/cc12616.
- Torio CM, Andrews RM. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer, 2011: Statistical Brief #160. Rockville, MD: Agency for Health Care Policy and Research (US); 2013.
- Rivers E, Nguyen B, Havstad S, Ressler J, Muzzin A, Knoblich B, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001;345:1368-77. http://dx.doi.org/10.1056/NEJMoa010307.
- Kortgen A, Niederprum P, Bauer M. Implementation of an evidence-based ‘standard operating procedure’ and outcome in septic shock. Crit Care Med 2006;34:943-9. http://dx.doi.org/10.1097/01.CCM.0000206112.32673.D4.
- Sebat F, Johnson D, Musthafa AA, Watnik M, Moore S, Henry K, et al. A multidisciplinary community hospital program for early and rapid resuscitation of shock in nontrauma patients. Chest 2005;127:1729-43. http://dx.doi.org/10.1378/chest.127.5.1729.
- Shapiro NI, Howell MD, Talmor D, Lahey D, Ngo L, Buras J, et al. Implementation and outcomes of the Multiple Urgent Sepsis Therapies (MUST) protocol. Crit Care Med 2006;34:1025-32. http://dx.doi.org/10.1097/01.CCM.0000206104.18647.A8.
- Micek ST, Roubinian N, Heuring T, Bode M, Williams J, Harrison C, et al. Before–after study of a standardized hospital order set for the management of septic shock. Crit Care Med 2006;34:2707-13. http://dx.doi.org/10.1097/01.CCM.0000241151.25426.D7.
- Shorr AF, Micek ST, Jackson WL, Kollef MH. Economic implications of an evidence-based sepsis protocol: can we improve outcomes and lower costs?. Crit Care Med 2007;35:1257-62. http://dx.doi.org/10.1097/01.CCM.0000261886.65063.CC.
- Nguyen HB, Corbett SW, Steele R, Banta J, Clark RT, Hayes SR, et al. Implementation of a bundle of quality indicators for the early management of severe sepsis and septic shock is associated with decreased mortality. Crit Care Med 2007;35:1105-12. http://dx.doi.org/10.1097/01.CCM.0000259463.33848.3D.
- Dellinger RP, Carlet JM, Masur H, Gerlach H, Calandra T, Cohen J, et al. Surviving Sepsis Campaign guidelines for management of severe sepsis and septic shock. Crit Care Med 2004;32:858-73. http://dx.doi.org/10.1097/01.CCM.0000117317.18092.E4.
- Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 2008;36:296-327. http://dx.doi.org/10.1097/01.CCM.0000298158.12101.41.
- Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med 2013;41:580-637. http://dx.doi.org/10.1097/CCM.0b013e31827e83af.
- Levy MM, Pronovost PJ, Dellinger RP, Townsend S, Resar RK, Clemmer TP, et al. Sepsis change bundles: converting guidelines into meaningful change in behavior and clinical outcome. Crit Care Med 2004;32:S595-7. http://dx.doi.org/10.1097/01.CCM.0000147016.53607.C4.
- Levy MM, Artigas A, Phillips GS, Rhodes A, Beale R, Osborn T, et al. Outcomes of the Surviving Sepsis Campaign in intensive care units in the USA and Europe: a prospective cohort study. Lancet Infect Dis 2012;12:919-24. http://dx.doi.org/10.1016/S1473-3099(12)70239-6.
- Ho BC, Bellomo R, McGain F, Jones D, Naka T, Wan L, et al. The incidence and outcome of septic shock patients in the absence of early-goal directed therapy. Crit Care 2006;10. http://dx.doi.org/10.1186/cc4918.
- Chapman M, Gattas D, Suntharalingam G. Why is early goal-directed therapy successful – is it the technology?. Crit Care 2005;9:307-8. http://dx.doi.org/10.1186/cc3726.
- Jones AE, Shapiro NI, Roshon M. Implementing early goal-directed therapy in the emergency setting: the challenges and experiences of translating research innovations into clinical reality in academic and community settings. Acad Emerg Med 2007;14:1072-8. http://dx.doi.org/10.1111/j.1553-2712.2007.tb02391.x.
- Turi SK, Von Ah D. Implementation of early goal-directed therapy for septic patients in the emergency department: a review of the literature. J Emerg Nurs 2013;39:13-9. http://dx.doi.org/10.1016/j.jen.2011.06.006.
- Ioannidis JP. Contradicted and initially stronger effects in highly cited clinical research. JAMA 2005;294:218-28. http://dx.doi.org/10.1001/jama.294.2.218.
- Yealy DM, Kellum JA, Huang DT, Barnato AE, Weissfeld LA, Pike F, et al. A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014;370:1683-93. http://dx.doi.org/10.1056/NEJMoa1401602.
- Peake SL, Delaney A, Bailey M, Bellomo R, Cameron PA, Cooper DJ, et al. Goal-directed resuscitation for patients with early septic shock. N Engl J Med 2014;371:1496-506. http://dx.doi.org/10.1056/NEJMoa1404380.
- Huang DT, Angus DC, Barnato A, Gunn SR, Kellum JA, Stapleton DK, et al. Harmonizing international trials of early goal-directed resuscitation for severe sepsis and septic shock: methodology of ProCESS, ARISE, and ProMISe. Intensive Care Med 2013;39:1760-75. http://dx.doi.org/10.1007/s00134-013-3024-7.
- Reade MC, Delaney A, Bailey MJ, Harrison DA, Yealy DM, Jones PG, et al. Prospective meta-analysis using individual patient data in intensive care medicine. Intensive Care Med 2010;36:11-2. http://dx.doi.org/10.1007/s00134-009-1650-x.
- Mental Capacity Act 2005. London: The Stationery Office; 2005.
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992;101:1644-55. http://dx.doi.org/10.1378/chest.101.6.1644.
- NHS and Department of Health . Saving Lives: Reducing Infection, Delivering Clean and Safe Care. High Impact Intervention No 1. Central Venous Catheter Care Bundle n.d. http://webarchive.nationalarchives.gov.uk/+/www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_078134 (accessed 6 August 2014).
- Vincent JL, Moreno R, Takala J, Willatts S, De Mendonca A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996;22:707-10. http://dx.doi.org/10.1007/BF01709751.
- Critical Care Minimum Data Set Full Specification. London: Information Standards Board for Health and Social Care; 2010.
- Harrison DA, Brady AR, Rowan K. Case mix, outcome and length of stay for admissions to adult, general critical care units in England, Wales and Northern Ireland: the Intensive Care National Audit & Research Centre Case Mix Programme Database. Crit Care 2004;8:R99-111. http://dx.doi.org/10.1186/cc2834.
- Knaus WA, Draper EA, Wagner DP, Zimmerman JE. APACHE II: a severity of disease classification system. Crit Care Med 1985;13:818-29. http://dx.doi.org/10.1097/00003246-198510000-00009.
- Shapiro NI, Wolfe RE, Moore RB, Smith E, Burdick E, Bates DW. Mortality in Emergency Department Sepsis (MEDS) score: a prospectively derived and validated clinical prediction rule. Crit Care Med 2003;31:670-5. http://dx.doi.org/10.1097/01.CCM.0000054867.01688.D1.
- Edwards PJ, Roberts I, Clarke MJ, Diguiseppi C, Wentz R, Kwan I, et al. Methods to increase response to postal and electronic questionnaires. Cochrane Database Syst Rev 2009;3. http://dx.doi.org/10.1002/14651858.mr000008.pub4.
- Harvey SE, Elbourne D, Ashcroft J, Jones CM, Rowan K. Informed consent in clinical trials in critical care: experience from the PAC-Man Study. Intensive Care Med 2006;32:2020-5. http://dx.doi.org/10.1007/s00134-006-0358-4.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. http://dx.doi.org/10.1002/sim.4067.
- Power GS, Harrison DA, Mouncey PR, Osborn TM, Harvey SE, Rowan KM. The Protocolised Management in Sepsis (ProMISe) trial statistical analysis plan. Crit Care Resusc 2013;15:311-17.
- Thompson SG, Barber JA. How should cost data in pragmatic randomised trials be analysed?. BMJ 2000;320:1197-200. http://dx.doi.org/10.1136/bmj.320.7243.1197.
- Survive Sepsis . The Sepsis Six n.d. https://survivesepsis.org/the-sepsis-six/ (accessed 24 December 2014).
- Ramsay CR, Grant AM, Wallace SA, Garthwaite PH, Monk AF, Russell IT. Statistical assessment of the learning curves of health technologies. Health Technol Assess 2001;5. http://dx.doi.org/10.3310/hta5120.
- Gould WW, Pitblado J, Poi BP. Maximum Likelihood Estimation with Stata. College Station, TX: Stata Press; 2010.
- Greenland S. An introduction to instrumental variables for epidemiologists. Int J Epidemiol 2000;29:722-9. http://dx.doi.org/10.1093/ije/29.4.722.
- Goetghebeur E, Lapp K. The effect of treatment compliance in a placebo-controlled trial: regression with unpaired data. J R Stat Soc Ser C Appl Stat 1997;46:351-64. http://dx.doi.org/10.1111/1467-9876.00074.
- Guide to the Methods of Technology Appraisal. London: NICE; 2013.
- Hassan T, McMillan P, Walsh C, Higginson I. The Drive for Quality: How to Achieve Safe, Sustainable Care in our Emergency Departments?. London: College of Emergency Medicine; 2013.
- National Blood and Blood Products Price List 2011–2012. London: NHS Blood and Transplant; 2012.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2012.
- Curtis L. Unit Costs of Health and Social Care 2012–13. Canterbury: Personal Social Services Research Unit, University of Kent; 2012.
- Dixon S, Mason S, Knowles E, Colwell B, Wardrope J, Snooks H, et al. Is it cost effective to introduce paramedic practitioners for older people to the ambulance service? Results of a cluster randomised controlled trial. Emerg Med J 2009;26:446-51. http://dx.doi.org/10.1136/emj.2008.061424.
- National Schedule of Reference Costs, 2012–2013. London: Department of Health; 2013.
- van Hout B, Devlin N. An EQ-5D-5L Value Set for England: Final Model Results. London: Office of Health Economics; 2014.
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96. http://dx.doi.org/10.1002/hec.944.
- Huang DT, Clermont G, Dremsizov TT, Angus DC. Implementation of early goal-directed therapy for severe sepsis and septic shock: a decision analysis. Crit Care Med 2007;35:2090-100. http://dx.doi.org/10.1097/01.CCM.0000281636.82971.92.
- Latimer NR. Survival analysis for economic evaluations alongside clinical trials – extrapolation with patient-level data: inconsistencies, limitations, and a practical guide. Med Decis Making 2013;33:743-54. http://dx.doi.org/10.1177/0272989X12472398.
- Soares MO, Welton NJ, Harrison DA, Peura P, Shankar-Hari M, Harvey SE, et al. An evaluation of the feasibility, cost and value of information of a multicentre randomised controlled trial of intravenous immunoglobulin for sepsis (severe sepsis and septic shock): incorporating a systematic review, meta-analysis and value of information analysis. Health Technol Assess 2012;16. http://dx.doi.org/10.3310/hta16070.
- Cuthbertson BH, Roughton S, Jenkinson D, Maclennan G, Vale L. Quality of life in the five years after intensive care: a cohort study. Crit Care 2010;14. http://dx.doi.org/10.1186/cc8848.
- Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health 2010;13:509-18. http://dx.doi.org/10.1111/j.1524-4733.2010.00700.x.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. http://dx.doi.org/10.1007/s11136-011-9903-x.
- Talmor D, Greenberg D, Howell MD, Lisbon A, Novack V, Shapiro N. The costs and cost-effectiveness of an integrated sepsis treatment protocol. Crit Care Med 2008;36:1168-74. http://dx.doi.org/10.1097/CCM.0b013e318168f649.
- Trzeciak S, Dellinger RP, Abate NL, Cowan RM, Stauss M, Kilgannon JH, et al. Translating research to clinical practice: a 1-year experience with implementing early goal-directed therapy for septic shock in the emergency department. Chest 2006;129:225-32. http://dx.doi.org/10.1378/chest.129.2.225.
- Manns BJ, Lee H, Doig CJ, Johnson D, Donaldson C. An economic evaluation of activated protein C treatment for severe sepsis. N Engl J Med 2002;347:993-1000. http://dx.doi.org/10.1056/NEJMsa020969.
- Fowler RA, Hill-Popper M, Stasinos J, Petrou C, Sanders GD, Garber AM. Cost-effectiveness of recombinant human activated protein C and the influence of severity of illness in the treatment of patients with severe sepsis. J Crit Care 2003;18:181-91. http://dx.doi.org/10.1016/j.jcrc.2003.08.009.
- Cuthbertson BH, Scott J, Strachan M, Kilonzo M, Vale L. Quality of life before and after intensive care. Anaesthesia 2005;60:332-9. http://dx.doi.org/10.1111/j.1365-2044.2004.04109.x.
- Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Crit Care Med 2013;41:1167-74. http://dx.doi.org/10.1097/CCM.0b013e31827c09f8.
- Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Intensive Care Med 2010;36:222-31. http://dx.doi.org/10.1007/s00134-009-1738-3.
- Stevenson EK, Rubenstein AR, Radin GT, Wiener RS, Walkey AJ. Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis. Crit Care Med 2014;42:625-31. http://dx.doi.org/10.1097/CCM.0000000000000026.
- Durston W. Does adoption of a regional sepsis protocol reduce mortality?. Am J Emerg Med 2014;32:280-1. http://dx.doi.org/10.1016/j.ajem.2013.11.035.
- Rhee C, Gohil S, Klompas M. Regulatory mandates for sepsis care – reasons for caution. N Engl J Med 2014;370:1673-6. http://dx.doi.org/10.1056/NEJMp1400276.
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med 2003;348:1546-54. http://dx.doi.org/10.1056/NEJMoa022139.
- Hebert PC, Wells G, Blajchman MA, Marshall J, Martin C, Pagliarello G, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med 1999;340:409-17. http://dx.doi.org/10.1056/NEJM199902113400601.
Appendix 1 Patient information sheet
Appendix 2 Short patient information sheet
Appendix 3 Case report form
Appendix 4 Severity of illness scores
Acute Physiology And Chronic Health Evaluation version II
The APACHE II Acute Physiology Score consists of weightings for 12 physiological parameters to give a total score ranging from 0 to 60, with higher scores indicating greater severity of illness. 33 The 12 physiological parameters are as follows:
-
temperature
-
mean arterial pressure
-
heart rate
-
respiratory rate
-
alveolar–arterial gradient (if FiO2 ≥ 0.5) or PaO2 (if FiO2 < 0.5)
-
arterial pH (or serum bicarbonate if no arterial blood gas recorded)
-
serum sodium
-
serum potassium
-
serum creatinine (with double weighting for acute renal failure)
-
haematocrit (estimated from haemoglobin)
-
white blood cell count
-
GCS score (assumed to be normal for patients sedated or paralysed).
The APACHE II score comprises the Acute Physiology score plus additional weightings for age and severe comorbidities in the past medical history to give a total score ranging from 0 to 71. Severe comorbidities must have been present and documented in the past medical history within the 6 months prior to presentation at hospital and are defined as follows:
-
severe liver condition – presence of portal hypertension, biopsy proven cirrhosis or hepatic encephalopathy
-
severe cardiovascular condition – presence of fatigue, claudication, dyspnoea or angina at rest (New York Heart Association Functional Class IV)
-
severe respiratory condition – presence of permanent shortness of breath with light activity due to pulmonary disease, or on home ventilation
-
renal condition – receiving chronic renal replacement therapy (haemodialysis, haemofiltration and peritoneal dialysis)
-
immunological condition – receiving chemotherapy, radiotherapy or daily high-dose steroid treatment (≥ 0.3 mg/kg, prednisolone or equivalent) for 6 months, or diagnosis of human immunodeficiency virus (HIV)/acquired immunodefiency syndrome (AIDS), lymphoma, acute or chronic myelogenous/lymphocytic leukaemia, multiple myeloma and active metastatic disease.
Sequential Organ Failure Assessment
The SOFA score consists of weightings for six organ systems to give a total score ranging from 0 to 24, with higher scores indicating a greater degree of organ failure. 30 Organ dysfunction is defined as follows:
-
respiratory dysfunction, defined as PaO2/FiO2 < 400 mmHg
-
cardiovascular dysfunction, defined as mean arterial pressure < 70 mmHg (irrespective of vasopressor use)
-
renal dysfunction, defined as creatinine of ≥ 1.2 mg/dl (110 µmol/l)
-
neurological dysfunction, defined as GCS score of ≤ 14
-
hepatic dysfunction, defined as bilirubin of ≥ 1.2 mg/dl (20 µmol/l)
-
coagulation dysfunction, defined as platelets < 150 × 109/l.
Mortality in Emergency Department Sepsis
The MEDS score is derived from nine variables to give a total score ranging from 0 to 33, with higher scores indicating a greater risk of death. 34 The nine variables are as follows:
-
terminal illness, defined as presence of metastatic disease [distant (not regional lymph node) metastases documented by surgery, imaging or biopsy]
-
respiratory difficulties, defined as tachypnea (respiratory rate of > 20 breaths per minute) or hypoxia (SpO2 < 90% or FiO2 of ≥ 0.4)
-
septic shock, defined as severe sepsis plus hypotension (systolic blood pressure < 90 mmHg) that persists after initial fluid challenge of 20–30 ml/kg body weight of intravenous crystalloid
-
low platelet count, defined as < 150 × 109/l
-
bandemia, defined as baseline immature neutrophils (band forms) < 5%
-
age > 65 years
-
suspected lower respiratory tract infection
-
nursing home residence
-
altered mental status, defined as a recent change in sensorium (confusion, disorientation, drowsiness, obtundation, stupor or coma) by history or physical examination or GCS score of ≤ 14.
Appendix 5 Critical Care Minimum Data Set criteria
Definitions
Duration of organ support in the critical care unit was defined as the number of days alive and free from support of each of the following organ systems, as defined by the UK Department of Health Critical Care Minimum Data Set,31 during the first 28 days following randomisation. Patients that died within the first 28 days were assigned 0 days alive and free from organ support. Organ support definitions were as follows:
-
advanced respiratory – indicated by one or more of invasive mechanical ventilatory support through a translaryngeal tube or tracheostomy; bilevel positive airway pressure through a trans-laryngeal tube or tracheostomy; continuous positive airway pressure through a trans-laryngeal tube; or extracorporeal respiratory support
-
advanced cardiovascular – indicated by one or more of receipt of multiple intravenous and/or rhythm controlling drugs (of which at least one must be vasoactive) when used simultaneously to support or control arterial pressure, cardiac output or organ/tissue perfusion; continuous observation of cardiac output and derived indices; an intra-aortic balloon pump or other assist device; or a temporary cardiac pacemaker
-
renal – indicated by receipt of acute renal replacement therapy (e.g. haemodialysis, haemofiltration, etc.) or receipt of renal replacement therapy for chronic renal failure where other acute organ support is received.
Appendix 6 Patient follow-up cover letter
Appendix 7 Patient follow-up questionnaire
List of abbreviations
- APACHE II
- Acute Physiology and Chronic Health Evaluation version II
- ARISE
- Australasian Resuscitation In Sepsis Evaluation
- CI
- confidence interval
- CLRN
- Comprehensive Local Research Network
- CRN
- Clinical Research Network
- CTU
- Clinical Trials Unit
- DMEC
- Data Monitoring and Ethics Committee
- ED
- emergency department
- EGDT
- early goal-directed therapy
- EQ-5D
- European Quality of Life-5 Dimensions
- EQ-5D-5L
- European Quality of Life-5 Dimensions-5 Levels
- FiO2
- fraction of inspired oxygen
- GCS
- Glasgow Coma Scale
- ICNARC
- Intensive Care National Audit & Research Centre
- ICU
- intensive care unit
- INB
- incremental net monetary benefit
- IQR
- interquartile range
- MEDS
- Mortality in Emergency Department Sepsis
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- PaO2
- partial pressure of oxygen
- PI
- principal investigator
- ProCESS
- Protocolized Care for Early Septic Shock
- ProMISe
- Protocolised Management in Sepsis
- QALY
- quality-adjusted life-year
- ScvO2
- central venous oxygen saturation
- SD
- standard deviation
- SIRS
- systemic inflammatory response syndrome
- SOFA
- Sequential Organ Failure Assessment
- SpO2
- arterial oxygen saturation
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee