
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 08/38/01. The contractual start date was in October 2009. The draft report began editorial review in January 2015 and was accepted for publication in August 2015. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Annemieke Hoek declares research awards from Merck Sharp & Dohme, Ferring B.V. and the Netherlands Organisation for Health Research and Development (ZoNMW), and personal fees from Merck Sharp & Dohme, all unrelated to the PROMISE trial.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2016. This work was produced by Coomarasamy et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
This chapter outlines the physiological importance of progesterone in pregnancy, the individual and societal burdens of pregnancy loss, and the rationale to infer a role for progesterone in reducing the risk of miscarriage.
Existing knowledge
Progesterone in pregnancy
Progesterone is essential to achieve and maintain a healthy pregnancy. It is an endogenous hormone, secreted naturally by the corpus luteum (the remnants of the ovarian follicle that enclosed a developing ovum) during the second half of the menstrual cycle, and by the corpus luteum and placenta during early pregnancy. Progesterone prepares the tissue lining of the womb (endometrium) to allow implantation, and stimulates glands in the endometrium to secrete nutrients for the early embryo. During the first 8 weeks of pregnancy, progesterone is produced by the corpus luteum, but between 8 and 12 weeks the placenta takes over this role and maintains the pregnancy thereafter.
The importance of progesterone in pregnancy has prompted many clinicians to infer that progesterone deficiency may be aetiologically linked to recurrent miscarriage (RM), and that progesterone therapy in the first trimester of pregnancy may reduce the risk of miscarriage. In 2003 a Royal College of Obstetricians and Gynaecologists (RCOG) guideline1 and a Cochrane review2 called for a definitive trial to test whether or not progesterone therapy in the first trimester could reduce the risk of miscarriage in women with a history of unexplained RM.
Burden of disease
Miscarriage is the commonest complication of pregnancy: one in six clinically recognised pregnancies ends in a miscarriage. 3 RM, the loss of three or more pregnancies, is a distinct clinical entity. The prevalence of RM (1%) is significantly higher than that expected by chance alone (0.4%). Even after comprehensive investigations, a cause for RM is identified in fewer than 50% of cases. 3 The majority of couples are, therefore, labelled as having unexplained RM.
Recurrent miscarriages affect over 6000 couples in the UK every year, and frequently result in substantial adverse physical and psychological consequences for women as well as impacting their families. For example, miscarriage has the potential to cause physical harm, including severe haemorrhage, infection, perforation of the womb during surgery for miscarriage and, rarely, maternal death. The eighth triennial Confidential Enquiry into Maternal Deaths identified several women who had died from complications related to miscarriage. 4 Moreover, qualitative studies have shown the level of distress and the bereavement reaction associated with miscarriage to be equivalent to the impact of the stillbirth of a term baby. 3
Costs to the NHS
It is estimated that RM costs the NHS £28M per year. This value includes the costs of diagnosis (blood tests and ultrasonography), management of miscarriages (expectant, medical or surgical), investigations for causes of miscarriages (e.g. antiphospholipid syndrome, parental karyotype and uterine cavity tests) and hospital inpatient costs. However, it does not include the management of the complications following treatment of miscarriages (such as uterine perforation, infection, bleeding or visceral damage) or any long-term health consequences of miscarriages or miscarriage management (including complications of intrauterine infections and adhesions). Thus, the true NHS perspective costs are likely to be higher than the estimated £28M per year. The societal costs, including days lost from work and out-of-pocket expenses for patients and partners, can be expected to be far greater.
Progesterone in clinical use for recurrent miscarriages
The PROMISE study was conceived to address the possibility that progesterone therapy in the first trimester of pregnancy may reduce the risk of RM. In 2007 we conducted a clinician survey in the UK (n = 114; response rate 102/114; 89.5%), and found that 2% (2/102) of clinicians use progesterone routinely and 3% (3/102) use it selectively in pregnant women with a history of RM. Over 95% (97/102) reported that they do not use progesterone for this indication and the vast majority of these (92/102; 90.2%) were willing to recruit to a trial evaluating the role of progesterone treatment for the prevention of RM.
We also carried out separate systematic reviews to examine (a) the effectiveness of progesterone in RM5 and (b) the safety of progesterone in pregnancy.
Effectiveness of progesterone in recurrent miscarriages
Two previously conducted systematic reviews2,6 had examined the role of progesterone therapy in RM, but, since these reviews were published and at the time of designing the PROMISE trial, new evidence7 had emerged. Therefore, we conducted a fresh systematic review with a meta-analysis.
We searched the Cochrane Controlled Trials Register, Cochrane Database of Systematic Reviews, Database of Abstracts of Reviews of Effects, ISI Web of Science Proceedings, International Standard Randomised Controlled Trial Number Register, metaRegister of Controlled Trials database, MEDLINE and EMBASE resources, from database inception to January 2008, for the following search terms: (‘progesterone’ OR ‘progestagen’ OR ‘progestogen’ OR ‘progestin’ OR ‘progestational [hormone or agent] ‘ OR ‘progest$’). We considered outcomes of miscarriage (as defined by the primary authors), live birth, gestation at delivery, pregnancy and neonatal outcomes.
Four randomised trials7–10 were identified. There were 14 trials assessing the effects of progesterone in miscarriages including spontaneous (one-time) events2 but these trials should not be confused with trials assessing the effects of progesterone in RM. The quality of the four trials was poor (modified Jadad quality scores between 0/5 and 2/5; Table 1), and participant numbers were small even when the trials were combined in meta-analysis, with only 132 women actively treated with progestogens.
| Features | El-Zibdeh 20057 (n = 130) | Goldzieher 19648 (n = 18) | Levine 19649 (n = 30) | Swyer and Daley 195310 (n = 47) |
|---|---|---|---|---|
| Population | Unexplained RM (three consecutive miscarriages, and conditions such as antiphospholipid syndrome excluded) | Analysis restricted to those with a history of three or more miscarriages | History of three consecutive miscarriages | Analysis restricted to those with a history of three or more miscarriages |
| Intervention | Dydrogesterone 10 mg twice daily (oral) | Medroxyprogesterone 10 mg daily (oral) | Hydroxyprogesterone caproate 500 mg per week (intramuscular) | Progesterone pellets 6 × 25 mg inserted into gluteal muscle |
| Comparison | No treatment | Placebo | Placebo | No treatment |
| Duration of treatment | From diagnosis of pregnancy to 12 weeks | Unclear | Until miscarriage or 36 weeks | Unclear |
| Randomisation method | ‘Randomised’; method not given | ‘Sequentially numbered bottles’ | Alternation | Alternation |
| Allocation concealment | Unclear | Unclear | Unclear | Inadequate |
| Blinding | No | Double | Double | No |
| ITT analysis | Yes | Unreported | No | Unreported |
| Follow-up rates | 100% | 100% | 54% (26/56 excluded) | 100% |
| Jadad score | 0/5 | 2/5 | 0/5 | 1/5 |
Our review and a subsequent review conducted for Cochrane in 201311 found that all four trials showed a trend towards benefit of progesterone, but confidence intervals (CIs) were wide and differences were not statistically significant for all but one of the four trials. A meta-analysis showed a statistically significant reduction in miscarriages (odds ratio 0.39, 95% CI 0.21 to 0.72; Figure 1). There was no evidence of statistical heterogeneity in the results (heterogeneity p-value 0.98).
FIGURE 1.
Meta-analysis of the four pre-existing trials of progestogen use in RM (outcome: miscarriage). df, degrees of freedom.

Although this evidence would be graded level 1a in the evidence hierarchy (because it is a systematic review of randomised trials), our survey of clinicians showed that it did not result in the use of progesterone for RM by clinical practitioners, owing to the weak methods and small sample sizes employed in the four published trials. One example of weak methodology was in lack of concealment, which has been shown to exaggerate effect sizes by up to 41%,12 although there is some evidence that this exaggeration may not be a concern when the outcome is objective. 13 Small sample sizes increase the likelihood of random error (generating the wide CIs in Figure 1). Nonetheless, the existing evidence presented a powerful reason to proceed with a trial of progesterone in RM, especially in consideration of the size of the effect observed and the low cost, widespread availability and convenience of the intervention, in addition to its safety profile.
Safety of progesterone supplementation in pregnancy
At the time of designing this study there was substantial evidence from in vitro fertilisation (IVF) practice that progestogen supplementation is safe to the mother and the fetus (at the proposed dose for the trial of 400 mg twice daily). 14–16
To further explore the question of safety, we conducted a review using the following search terms in MEDLINE (1966–2007) and EMBASE (1988–2007): (‘progesterone’ OR ‘progestational agents’ OR ‘progest$’) AND (‘adverse effects’ OR ‘complications’ OR ‘side effects’ OR ‘harm’) AND ‘pregnancy’. A systematic review of observational studies (both cohort and case–control studies) of first-trimester sex hormone exposure identified 14 studies, comprising 65,567 women. 17 The sex hormone in several of these studies was progestogens alone or with other steroids.
Most of the evidence in our review did not show harm, particularly any external genital malformation in the offspring, but one case–control study suggested an association between hypospadias and progestogen use. 18 Although findings from a case–control study represented weaker evidence than the better-quality evidence from larger cohort studies which did not substantiate this association, we decided to document all of the effects of progesterone in the PROMISE trial. More specifically, we decided to collect information about any neonatal genital abnormalities.
Meta-analyses of progesterone use in RM, in miscarriage2 and in the prevention of preterm birth19 did not identify any evidence of short-term safety concerns in women. However, it was not clear if these trials sought to document maternal side effects prospectively. In one study, intramuscular 17-OHP (hydroxyprogesterone) caused maternal adverse events (AEs) in 50% of women, largely due to injection site reactions. 20 This concern did not apply to the PROMISE trial, in which the route of administration was vaginal. Side effects were not reported in studies of vaginal progesterone in the context of prevention of preterm births. 21,22
Rationale
A trial of progesterone therapy in the treatment of unexplained RM was required for the following reasons:
-
The existing trials, although small and of poor quality, suggested a large benefit in a condition with substantial morbidity and costs.
-
A guideline by the RCOG and a Cochrane review called for a definitive trial to evaluate this research question. 12
-
Participants in two unpublished surveys [one of women with RM (n = 88) and the other of gynaecologists (n = 102) treating women with RM] demonstrated an interest in progesterone therapy and a willingness to participate in a potential trial (Arri Coomarasamy, University of Birmingham, 2008, unpublished data).
-
If proven to be effective, the intervention would represent a low-cost, safe and easily deliverable therapy.
Specific objectives
Primary objective
-
To test the hypothesis that, in women with unexplained RM, progesterone (400-mg vaginal capsules, twice daily), started as soon as possible after a positive urinary pregnancy test (and no later than 6 weeks of gestation) and continued to 12 weeks of gestation, compared with placebo, would increase live births beyond 24 completed weeks of pregnancy by at least 10%.
Secondary objectives
-
To test the hypothesis that progesterone would improve various pregnancy and neonatal outcomes (such as reduced miscarriage rates and improvements in survival at 28 days of neonatal life).
-
To test the hypothesis that progesterone, compared with placebo, would not incur serious adverse events (SAEs) in either the mother or the neonate (such as genital abnormalities in the neonate).
-
To explore differential or subgroup effects of progesterone in various prognostic subgroups, including subgroups of:
-
maternal age (≤ 35 years or > 35 years)
-
number of previous miscarriages (3 or ≥ 4)
-
presence or absence of polycystic ovaries.
-
-
To perform an economic evaluation for cost-effectiveness.
Chapter 2 Methods
This chapter reports the methods used to conduct the PROMISE trial. It describes the study design and protocol to progress potential participants from enrolment to completion of treatment, data analysis plans, quality assurance and governance.
Design
The PROMISE trial was conducted as a randomised, double-blind, placebo-controlled, international multicentre study, with health economic evaluation. Participants were randomised to receive progesterone or placebo in a 1 : 1 ratio.
Participants
The participants in the PROMISE trial were recruited in hospital settings located across the UK (36 sites) and in the Netherlands (nine sites). These sites (Figure 2) included RM clinics and early pregnancy units in secondary or tertiary care hospitals.
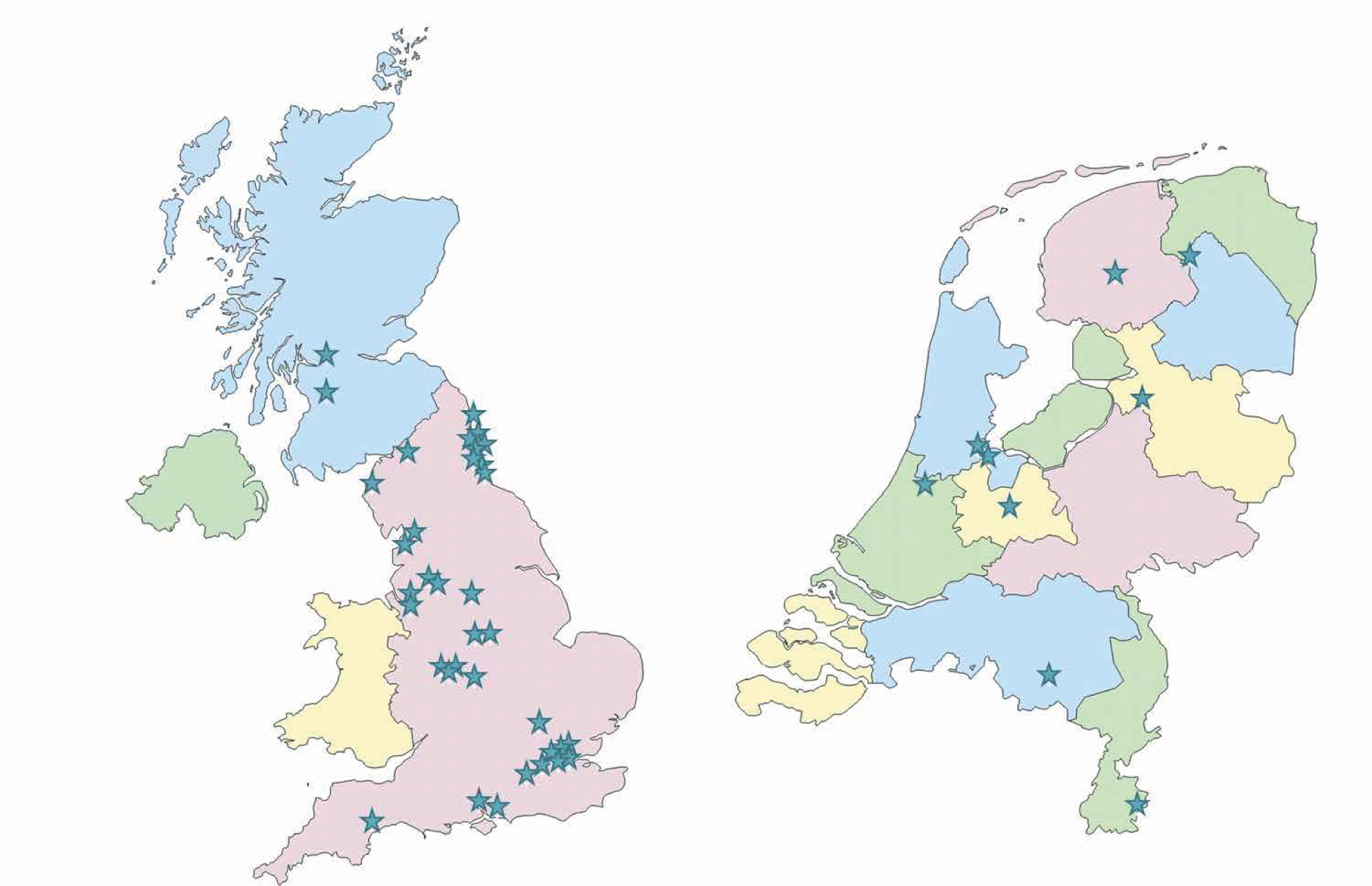
All sites in the UK and the Netherlands with local investigators of appropriate capability and experience in conducting clinical trials were eligible to take part. Primary care settings were not utilised as either research sites or patient identification centres.
Participant eligibility for the trial was assessed according to the criteria listed below.
Inclusion criteria
In order to be eligible for the study, it was necessary for participants to meet all of the following criteria:
-
having a diagnosis of unexplained RM (three or more consecutive or non-consecutive first-trimester losses)
-
being aged 18–39 years at randomisation (the likelihood of miscarriages due to random chromosomal aberrations is higher in older women3,25 and such miscarriages are unlikely to be prevented by progesterone therapy)
-
trying to conceive naturally
-
willing and able to give informed consent.
Exclusion criteria
Participants could not be included in the study if any of the following criteria were applicable:
-
they were unable to conceive naturally (as confirmed by urinary pregnancy tests) within 1 year of recruitment or before the end of the randomisation period in the trial, whichever came earlier
-
they had antiphospholipid syndrome [lupus anticoagulant and/or anticardiolipin antibodies (immunoglobulin G or immunoglobulin M)]; other recognised thrombophilic conditions (testing according to usual clinic practice)
-
they had uterine cavity abnormalities (as assessed by ultrasound, hysterosonography, hysterosalpingogram or hysteroscopy)
-
they had abnormal parental karyotype
-
they had other identifiable causes of RM (tests initiated only if clinically indicated) such as diabetes, thyroid disease or systemic lupus erythematosus
-
they were on current heparin therapy
-
they had any contraindications to progesterone use (see the following section).
Contraindications to progesterone use
Women to whom any of the following applied were not eligible to take part in the trial:
-
they had a history of liver tumours
-
they had severe liver impairment
-
they had genital tract or breast cancer
-
they had severe arterial disease
-
they had undiagnosed vaginal bleeding
-
they had acute porphyria
-
they had a history during pregnancy of:
-
idiopathic jaundice
-
severe pruritus
-
pemphigoid gestationis
-
-
they were taking any of the following drugs, because these products interact with progesterone:
-
bromocriptine
-
cyclosporine
-
rifamycin
-
ketoconazole.
-
Recruitment
Potential participants were identified from dedicated RM clinics, or other hospital clinics in which the caseload included a substantial number of women with RM. Potential participants were identified and approached by clinic doctors, research nurses and midwives, after having received appropriate training relating to the trial. This training included the development of sensitivity in answering questions about the risks of pregnancy loss, and the importance of attention to early signs of possible miscarriage such as spotting or discharge.
The participant eligibility pathway to recruitment and randomisation is illustrated by Figure 3. Eligible women were given verbal and written explanations about the trial. They were informed clearly that participation in the trial was entirely voluntary, with the option of withdrawing at any stage, and that participation or non-participation would not affect their usual care. They were provided with a participant information sheet (PIS) (see Appendix 1). Eligible women were then given the opportunity to decide if they wished to participate, or if they needed more time to consider their decision, or if they did not wish to participate. In all three scenarios, the decision of the woman was respected. If a woman needed more time to consider her potential involvement, she was asked to call the research nurse or midwife when she had decided. If an undecided woman had not called within 14 days, the research nurse or midwife contacted her. If an initially undecided woman decided to participate later, the research nurse or midwife arranged a mutually convenient opportunity for the woman to be consented.
FIGURE 3.
Eligibility pathway to recruitment and randomisation. APS, antiphospholipid syndrome; CRF, case report form; LMWH, low-molecular-weight heparin; M, miscarriage.

A written consent form (see Appendix 2) was provided to each woman who agreed to participate in the trial. The investigator and the participant both signed the consent form. The original copy was kept in the investigator site file, one copy was given to the participant and one copy was retained in the woman’s hospital records. Baseline demographic and medical data were collected, anonymised and stored in an electronic integrated trial management system (ITMS). Any identifying information was collected and stored in a password-protected local database on a secure computer with restricted access.
The first PROMISE participant was enrolled in June 2010 and randomised in October 2010. The last PROMISE participant was recruited and randomised in October 2013 (see Chapter 3, Recruitment).
Non-English speakers
We made provision for translation, if necessary, to communicate with non-English speakers and accommodate any special communications requirements of potential study participants. The PISs and consent forms (see Appendices 1 and 2) were translated from English into Dutch for use in the Netherlands.
Randomisation
Each woman consenting in advance of pregnancy was given instructions to notify the local research nurse or midwife by telephone as soon as she experienced a positive urinary pregnancy test. We expected most pre-consenting participants to notify us early in pregnancy, at approximately 4 weeks of gestation (4 weeks from their last menstrual period).
On receiving notification of pregnancy and confirming the woman’s willingness to participate in the trial (in either order of occurrence), the local research nurse or midwife reverified other aspects of eligibility according to inclusion and exclusion criteria, and obtained details of gestational age. Participants were randomised online to receive the trial intervention (either progesterone or placebo), via a purpose-designed ITMS. Each authorised member of the research team was provided with a unique username and password to the ITMS for this purpose. Online randomisation was available 24 hours per day, 7 days per week, apart from during short periods of scheduled maintenance.
Sequence generation
Computer-generated random numbers were used, and participants were randomised online via a secure internet facility. This third-party independent ITMS was designed, developed and delivered by MedSciNet® (Stockholm, Sweden) according to standards of the International Organisation for Standardisation 9001:2000 and the requirements of the US Food and Drug Administration CFR21:11. 26
Participants were randomised to receive progesterone or placebo in a 1 : 1 ratio. A ‘minimisation’ procedure using a computer-based algorithm was used to avoid chance imbalances in important stratification variables. The stratification variables used for minimisation were as follows:
-
number of previous miscarriages (3 or > 3)
-
maternal age (≤ 35 or > 35 years)
-
polycystic ovaries or not
-
body mass index (BMI) (≤ 30.0 or > 30.0 kg/m2).
Allocation and minimisation
After all of the eligibility criteria and baseline data items were entered online, the ITMS generated a code number which took into account the minimisation variables recorded for the individual, and which was linked to a specific trial intervention pack. The code number was advised via e-mail to the local principal investigator (PI), the relevant trial pharmacist (see Distribution and Investigational medicinal product supply, Storage, dispensing and return) and the research nurse or midwife performing the randomisation.
Blinding
Participants, investigators, research nurses, midwives and other attending clinicians remained unaware of the trial drug allocation throughout the duration of the trial.
In the case of any SAE, the general recommendation was to initiate management and care of the participant as though the woman was taking progesterone. If the drug allocation was specifically requested to assist the medical management of a participant, clinicians could contact the trial manager or the trial co-ordinator for this purpose, 24 hours per day, 7 days per week (see Safety monitoring, Adverse events). Cases that were considered serious, unexpected and possibly, probably or definitely related to the trial intervention (see Appendix 3) were unblinded as appropriate. In any other circumstances, investigators and research nurses and midwives remained blind to drug allocation while the participant remained in the trial.
Distribution
On randomisation, the research nurse or midwife arranged the dispatch of a trial intervention pack to the local participating centre or the home address of the participant within 72 hours of receiving the telephone call informing of pregnancy. Nurses in the UK contacted the trial pharmacy at St Mary’s Hospital in London and nurses in the Netherlands contacted the trial pharmacy in Utrecht (see Investigational medicinal product supply, Storage, dispensing and return). Each trial intervention pack contained either progesterone or placebo.
Instructions to participants
The research nurse or midwife also provided the participant with instructions. In cases of delivery directly to the home address, the research nurse or midwife contacted the participant to ensure receipt and understanding of how to use the supplied capsules.
Each participant was expected to commence the trial intervention on the day it was received, and continue until it was finished, at around 12 completed weeks of gestation, unless the pregnancy ended before this time. The research nurse or midwife also telephoned each woman in the days immediately after the trial medicine was supplied, to ensure that the participant had started taking the medicine. In the event of the capsules being mislaid, the participant was instructed to telephone the research nurse or midwife, who would liaise with trial manager or the trial co-ordinator to arrange a further supply of the same type of intervention.
Each participant was asked for consent to notify the primary care provider (in the UK, the general practitioner) by letter that she was participating in the trial (see Appendix 4). Moreover, each participant was given a business card and a fridge magnet with contact details of local PROMISE investigators and the central Trial Co-ordinating Centre (TCC), to inform any directing clinicians, in case of potential drug interactions.
Interventions
Each participant in the PROMISE trial received either micronised progesterone or placebo capsules, to be administered vaginally. Both products were supplied by Besins Healthcare (Mountrouge, France), a global pharmaceutical company with a manufacturer’s licence for tablets and capsules, in compliance with good manufacturing practice standards27 and good clinical practice (GCP) requirements. 28,29
Progesterone capsules
The investigational medicinal product (IMP) was micronised progesterone at a dose of 400 mg (that is, two capsules of Utrogestan® 200 mg) taken vaginally twice daily (every morning and every evening) for the duration of treatment.
The anatomical therapeutic chemical classification code for the pharmacotherapeutic group of the IMP was G03D and the chemical abstract service number was 57-83-0. The product had all the properties of endogenous progesterone, with induction of a full secretory endometrium and in particular gestagenic, antiestrogenic, slightly antiandrogenic and antialdosterone effects.
Besins Healthcare held the manufacturing authorisation (France) for Utrogestan®, including for the indication of threatened miscarriage or prevention of habitual miscarriage due to luteal phase deficiency up until the 12th week of pregnancy.
Placebo capsules
Placebo capsules were vaginal capsules, composed of sunflower oil, soybean lecithin, gelatin, glycerol, titanium dioxide and purified water, encapsulated in the same form as the IMP, and identical in colour, shape and weight, for use in the control arm of the PROMISE trial. The dose, route and timing of administration were also identical to those in the active progesterone arm of the study.
Dose
The ideal dose of progesterone for the potential prevention of RM was unknown. The biologically effective dosage of micronised progesterone capsules ranged from 200 mg once daily to 400 mg twice daily according to the summary of product characteristics (SmPC)30 and the British National Formulary (BNF). 31 Our choice of 400 mg twice daily was made after a careful review of the existing literature and an extensive survey of clinicians in the UK (see Chapter 1, Existing knowledge, Progesterone in clinical use for recurrent miscarriages). We also reviewed other related evidence. For example, progesterone vaginal capsules are commonly used for luteal support in assisted conception at a treatment dose of 400 mg twice daily, with no specific safety concerns raised on this dose. 16,32
After evaluating the evidence, we considered the dosage of 400-mg vaginal progesterone twice daily to be an acceptable regimen to ensure a clinically effective dose, and to minimise the risk of a negative trial result from therapy with a suboptimal dose.
Route
An immunomodulatory effect of progesterone at the trophoblastic–decidual interface is the key presumed mechanism for preventing RM. 3,33–35 Our choice to use the vaginal route of administration was, therefore, rational to deliver a greater proportion of drug to the relevant site (the uterus) using the ‘first uterine pass’ effect. 36,37 Furthermore, studies that have used vaginal progesterone in the prevention of preterm birth have reported its effectiveness when given via this route. 19,21,22 For example, 14 out of 36 studies of second- and/or third-trimester progesterone to prevent preterm birth (identified by a recent systematic review) used vaginal progesterone, with significant improvements being observed for various clinical outcomes, confirming the biological effects of vaginal progesterone. 38
The acceptability and availability of interventional drugs were also important considerations supporting the vaginal route of drug delivery. Our discussions with consumer representatives confirmed that a vaginal formulation would be more acceptable to women than an intramuscular preparation. These findings were further supported by a study in which 12% of participants were unable to tolerate the intramuscular progesterone preparation and declined participation or withdrew from that trial. 20 Of those who did continue, 34% complained of localised soreness around the injection site. Finally, in our survey of women with RM conducted at St Mary’s Hospital and Guy’s and St Thomas’ Hospitals in London, a very high acceptability of the vaginal route (81/88; 92%) was identified (Arri Coomarasamy, University of Birmingham, 2008, unpublished data).
The capsule formulation of the PROMISE trial is widely available in the UK and worldwide.
Timing
Treatment commenced as soon as possible after a positive pregnancy test and no later than 6 weeks of pregnancy, and continued until the gestational age of 12 weeks. Our rationale to discontinue the treatment at 12 weeks was that production of progesterone by the corpus luteum becomes less important than the placental production of progesterone after 12 weeks of gestation. Furthermore, in the only clinical trial of progesterone treatment for RM to have shown a statistically significant reduction in miscarriage rates, progesterone was given until 12 weeks of gestation (odds ratio 0.37, 95% CI 0.15 to 0.90). 7
Compliance assessment
Our previous experience of research and clinical care for women with RM demonstrated that they would be highly motivated and compliant with therapy advice. However, compliance in the PROMISE trial was evaluated by ‘pill-counting’.
Participants were asked to return completed, partially used and unused treatment packs to the trial centres. The research nurses and midwives at each study centre documented the capsules returned by each participant, while the trial pharmacists kept their own accountability logs.
In an effort to improve compliance, women who failed to return the blister packs from the previous 4 weeks, whether or not these were empty, using the envelope provided were contacted by the local research nurse or midwife by telephone or e-mail to be offered advice and support.
Non-compliance was defined as missing more than 20% of trial medicines for the gestational age at randomisation. Non-compliant participants were interviewed (face to face or via telephone) in an attempt to establish the reason(s) for their non-compliance.
Investigational medicinal product supply
Manufacture, packaging and labelling
All arrangements for trial drug supply, labelling, storage and preparation were undertaken as per the requirements of the Medicines for Human Use (Clinical Trials) Regulations 2004. 39,40
Active (Utrogestan® vaginal 200 mg) and placebo capsules were manufactured and packaged (assembled) by Besins Healthcare, in compliance with good manufacturing practice (EU Directive 2003/94/EC)27 and GCP (Clinical Trials Directive 2001/20/EC)28 requirements. Besins Healthcare also provided qualified person release of the trial drug under the requirements of the Medicines for Human Use (Clinical Trials) Regulations 2004. 39,40
Each trial intervention pack contained the entire supply required for the treatment period of up to 8 weeks. As the treatment regimen was two Utrogestan® 200 mg or placebo capsules twice daily, the drug package for each participant contained 224 capsules: 2 (capsules) × 2 (twice daily) × 7 (7 days per week) × 8 (8 weeks) = 224.
The drug packages were labelled in compliance with the UK Medicines for Human Use (Clinical Trials) Regulations 2004,39,40 ensuring the protection of each participant, traceability and proper identification of the IMP and trial.
Storage, dispensing and return
At study initiation, supplies of progesterone and placebo capsules were delivered by Besins Healthcare to two study pharmacies, where the products were stored and whence they were dispensed to all participants.
The pharmacies were located at St Mary’s Hospital in London and the University Medical Centre of Utrecht. Both sites complied with the relevant guidelines and regulations, including (in the UK) the Duthie report41 and the Royal Pharmaceutical Society of Great Britiain’s practice guidance on pharmacy services for clinical trials42 as well as the appropriate standard operating procedures of Imperial College London. 43
At the pharmacies, the PROMISE trial medications were stored separately from other stock, in areas with restricted access. Drugs expired or returned by participants were stored separately from unallocated trial medicines. Dispensing was undertaken against prescription forms, each entitled ‘The PROMISE trial, EudraCT Number 2009-011208-42’ and labelled with the name, date of birth and study identification number of the relevant participant and a unique code number provided by the ITMS for randomisation (see Randomisation, Allocation and minimisation). Each trial pharmacy kept detailed dispensing records including participant study numbers and names, code numbers, batch numbers, expiry dates, doses and dates of dispensing.
The trial co-ordinator monitored the quantity of supplies held by each dispensing pharmacy in comparison with the number and rate of randomisations undertaken, and liaised with Besins Healthcare to ensure adequate supplies of the IMP in the UK and the Netherlands.
Each participant in the PROMISE trial was provided with freepost envelopes to return the unused or used packets to the local study centre. All unused study drugs, including undispensed supplies and supplies returned by participants, were returned to the trial pharmacies for accountability and destruction. Both dispensing pharmacies were involved in the reconciliation of medicines returned by trial participants and the disposal of unused medication, in compliance with appropriate regulatory guidance.
Outcomes
Primary outcome
Live birth beyond 24 completed weeks of gestation.
Secondary outcomes
-
Clinical pregnancy at 6–8 weeks (defined as the presence of a gestational sac, with or without a yolk sac or fetal pole).
-
Ongoing pregnancy at 12 weeks (range 11–13 weeks) (defined as the presence of a fetal heartbeat).
-
Miscarriage (defined as loss of pregnancy before 24 weeks of gestation).
-
Gestation at delivery.
-
Survival at 28 days of neonatal life.
-
Congenital anomalies, and specifically genital abnormalities.
Exploratory outcomes
-
Antenatal complications such as pre-eclampsia, small for gestational age (< 10th birthweight centile), preterm prelabour rupture of membranes and antepartum haemorrhage.
-
For live births at beyond 24 completed weeks of gestation: mode of delivery, birthweight, arterial and venous cord pH, APGAR (appearance, pulse, grimace, activity, respiration) score and resuscitation.
-
For neonates: surfactant use, ventilation support (days on intermittent positive pressure ventilation, continuous positive airway pressure and oxygen, and discharge on oxygen) and neonatal complications (such as infection, respiratory distress syndrome, necrotising enterocolitis, intraventricular haemorrhages and pneumothorax).
Resource use outcomes
The resource use data listed below were collected to estimate the costs associated with the provision of progesterone for RM:
-
antenatal, outpatient or emergency visits
-
inpatient admissions (nights in hospital)
-
maternal admissions to high-dependency units (HDUs) or intensive care units (ICUs) (nights)
-
neonatal admissions to special care baby units (SCBUs) or neonatal units (NNUs) (nights).
Future outcomes
Women were asked to consent for future evaluation of themselves and their child and the health records of both. Although long-term follow-up was outside the scope of the PROMISE trial, we recognised the value of data collected in this study to inform further studies on outcomes such as the composite end point of death or neurodevelopmental impairment at 2 years of age, cognitive scale scores at 2 years of chronological age, and disability classified into domains according to professional consensus. The hospital number and (in the UK) NHS number of each baby within the PROMISE trial were recorded to facilitate future follow-up studies.
Outcome assessment
The ITMS was utilised to capture baseline and outcome data, for contemporaneous data cleaning, to produce reports for the independent Data Monitoring Committee (DMC) and to maintain an audit trail. Relevant trial data were transcribed directly into the ITMS. Source data comprised the research clinic notes, hospital notes, hand-held pregnancy notes, laboratory results and self-reports.
First outcome assessment (6–8 weeks of pregnancy)
The research nurse or midwife at each study site telephoned every participant at between 6 and 7 weeks of gestation, to ensure there were arrangements for an ultrasound appointment with her usual carers, before 8 weeks of gestation (Figure 4). If an appointment had not been booked, the research nurse or midwife assisted with booking. The research nurse or midwife telephoned each participant again between 3 and 5 days after the scheduled date of the ultrasound appointment, to obtain details of the observations of a gestational sac.
FIGURE 4.
Participant care pathway and outcome assessment.
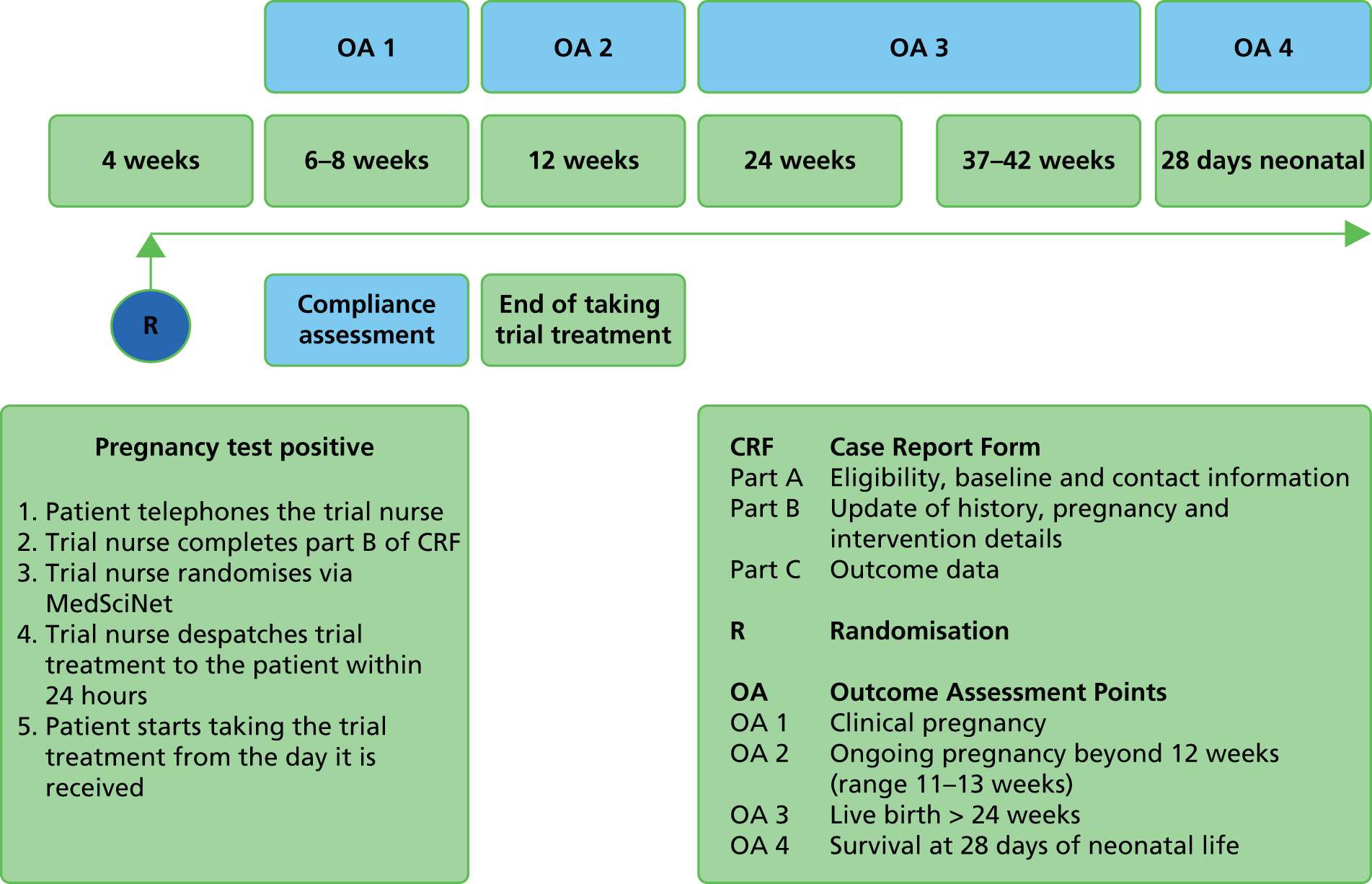
Second outcome assessment (12 weeks of pregnancy)
The research nurse or midwife at each study site telephoned each participant between 10 and 12 weeks of gestation, to ensure there were arrangements for an ultrasound appointment with her usual carers, at between 12 and 14 weeks of gestation (see Figure 4). As previously, the research nurse or midwife assisted with booking an appointment if necessary, and telephoned the participant afterwards to obtain details of variables such as fetal heartbeat. The research nurse or midwife also recorded the expected date of delivery at this stage.
Third outcome assessment
The third outcome assessment was conducted at or after birth (see Figure 4). The research nurse or midwife at each study site telephoned every participant 2 weeks after the expected date of delivery to obtain pregnancy outcome data such as the mode of delivery, gestation, weight and APGAR score at birth. The ITMS generated automated prompts to alert the research nurse or midwife at the time of expected date of delivery. The research nurse or midwife also checked birth registers and inpatient records to track hospital admissions and pregnancy outcomes.
Fourth outcome assessment
The fourth and final outcome assessment was conducted to gather neonatal outcomes at 28 days after birth (see Figure 4). The research nurse or midwife at each study site telephoned every participant to obtain postnatal and neonatal outcome data including any nights of hospital admission or requirements for ventilation support, and complications such as early infection. The ITMS generated automated prompts to alert the research nurse or midwife at the appropriate time. Using the full repertoire of evidence-based methods to maximise data collection,44 the research nurse or midwife also checked birth registers and inpatient records to track hospital admissions and pregnancy outcomes.
Continuity
From previous experience of research and clinical care for women with RM, we expected high rates of compliance with therapy advice. Moreover, the time interval between randomisation and final outcome assessment in the PROMISE trial was short (e.g. if delivery occurred at 40 weeks of gestation, the interval was 40 weeks), so we expected loss to follow-up to be minimal. Participants in the study continued to be managed by their clinical teams throughout their pregnancies, according to local protocols.
Withdrawal
Following discussion with the Trial Management Group (TMG), participants in the PROMISE trial could be withdrawn from trial treatment if it became medically necessary in the opinion of the investigator(s) or clinician(s) providing patient care. In the event of such premature treatment cessation, study nurses and midwives made every effort to obtain and record information about the reasons for discontinuation, and to follow up all safety and efficacy outcomes as appropriate.
Participants in the PROMISE trial could voluntarily decide to cease taking the study treatment at any time. If a participant did not return for a scheduled visit, attempts were made to contact her and (where possible) to review compliance and AEs. We documented the reason(s) for self-withdrawal where possible. Each woman remained able to change her mind about withdrawal, and reconsent to participate in the trial, at any time. Clear distinction was made between withdrawals from trial treatments while allowing further follow-up, and any participants who refused any follow-up. If a woman withdrew from taking the trial treatment but permitted further data collection, she was followed up and outcome assessments were undertaken for the remainder of the study.
If a participant explicitly withdrew consent to any further data recording, this decision was respected and recorded via the ITMS. All communications surrounding the withdrawal were noted in the study records and no further data were collected for such participants.
Concomitant non-trial treatments
Concomitant therapy was provided at the discretion of the care-providing clinicians, and all concomitant treatment and medications were documented via the ITMS. Post-randomisation use of heparin was discouraged unless there was a clear and recognised indication for it (heparin therapy at the time of randomisation made a woman ineligible to participate in the trial). Other than identified contraindicated drugs (see Participants, Exclusion criteria) and other progestogen preparations, the initiation of treatment for another indication did not necessitate withdrawal from the PROMISE study.
Safety monitoring
A review conducted in 2007, before the PROMISE trial commenced, showed no clear or consistent evidence of SAEs on the mother or the baby as a result of progesterone treatment during pregnancy (see Chapter 1, Existing knowledge, Progesterone in clinical use for recurrent miscarriages). Moreover, there is substantial evidence from other studies to indicate that progestogen supplementation at the dose administered in the PROMISE trial is safe to the mother and the fetus. 14–16
The SmPC for progesterone (vaginal capsules)30 states that ‘preclinical data revealed no special hazard for humans based on conventional studies of safety pharmacology and toxicity’ (© Datapharm).
Known side effects
The SmPC for progesterone (vaginal capsules)30 also states that ‘local intolerance (burning, pruritus or fatty discharge) has been observed during the different clinical trials and reported in the literature but incidences were extremely low’ (© Datapharm).
Overdose
The symptoms of progesterone overdose may include somnolence, dizziness and euphoria. In case of overdose, the care-providing clinicians of the PROMISE trial were prepared to undertake observation and reporting, and provide symptomatic and supportive measures, as required.
Dose modification for toxicity
The PROMISE trial included provision that, for participants experiencing non-serious side effects, the dosage could be reduced to 200 mg twice daily (one capsule in the morning and one at bedtime) at the discretion of the care-providing clinician, without unblinding treatment allocation (see the following section). The dose modification was noted on the case report form, along with the gestational age and the date on which such change was implemented.
Adverse events
The pharmacovigilance procedures of the PROMISE trial, including documentation, validation, evaluation and reporting, and responsibilities for the performance of these requirements, were based on the contemporaneously available literature to guide good practice. 28,45–47
Assessment
All of the trial participants were asked to report any hospitalisations, consultations with other medical practitioners, disability, incapacity or any other AEs to their local research team; if the local study nurse or midwife was unavailable for any reason, they were able to report the events to the trial manager or trial co-ordinator via telephone at any time. Moreover, at the time of each outcome assessment, investigators, research nurses and midwives at each study centre proactively asked each participant about any AEs in the preceding weeks. AEs were assessed by clinical investigators and further reported as appropriate, and in any case recorded in the ITMS for scheduled interim analyses to standard formats47 by the independent DMC. If a local clinical investigator was unavailable, initial AE reports without causality and expectedness assessment were submitted to the TCC by a health-care professional within 24 hours, and followed up by medical assessment as soon as possible thereafter, ideally within the following 24 hours.
Regardless of treatment allocation, the expectedness, seriousness and causality of AEs were assessed according to standardised definitions (see Appendix 3) as though the participant had received the active drug (progesterone).
Reporting
Adverse events were reported by local clinical investigators to the TCC, and thence to the sponsor (Imperial College London/Imperial College Healthcare NHS Trust Joint Research Compliance Office). The sponsor (or the chief investigator on behalf of the sponsor) reported to the Medicines and Healthcare products Regulatory Agency (MHRA). The sponsor (or the trial co-ordinator on behalf of the sponsor) also reported to the Research Ethics Committee (REC), and equivalent bodies in the Netherlands, as appropriate. If information was incomplete at the time of initial reporting, or if the event was ongoing, local investigators forwarded follow-up information as soon as possible. If there was a difference between the expectedness, seriousness and causality assessments of the local investigator, the TCC and the sponsor, then the worst-case assessment was used for reporting.
Serious adverse events and serious adverse reactions were recorded on a purpose-designed SAE form and notified by local investigators to the TCC within 24 hours of the local investigators becoming aware of these events. Local investigators were responsible for additionally reporting SAEs to their host institutions, according to local regulations, and instituting supplementary investigations as appropriate based on clinical judgement of the causative factors. Any SAE or serious adverse reaction that was outstanding at the end of the trial treatment period was followed up at least until the final outcome was determined, even if this provision necessitated follow-up beyond 28 days postpartum. The TCC reported all SAEs to the independent DMC approximately 6-monthly. The DMC viewed data blinded to treatment, but was able to review unblinded data if necessary.
Suspected unexpected serious adverse reactions (SUSARs) were unblinded, as appropriate, reviewed by the trial manager within 24 hours of reporting, and further reported to the MHRA and the REC, or the equivalent bodies in the Netherlands, by the TCC as soon as possible, and in any event within 15 days (or 7 days in the case of fatal or life-threatening SUSARs).
Unblinding
Unblinding was undertaken only in the event of a medical emergency requiring knowledge of the drug received. In the event that an investigator or the care-providing clinician required disclosure of the treatment allocation, the ITMS allowed the trial manager or the trial pharmacy to break the randomisation code. For this purpose, the TCC could be contacted between 09.00 and 16.00 every weekday; otherwise, the trial manager or designee could be contacted directly via a 24-hour trial mobile phone. The investigator or care-providing clinician communicating the alert was asked to provide the date of the requirement, the name of person requesting unblinding, the reason for unblinding and any other relevant information.
Sample size
The PROMISE trial investigators believed that it was important to ensure that the study was large enough to detect reliably moderate but clinically important treatment effects. Our calculations indicated that, to detect a minimally important difference (MID) of 10% in rates of live birth after at least 24 weeks (from 60% to 70%, odds ratio 1.56), for an alpha error rate of 5% and beta error rate of 20% (i.e. 80% power), it would be necessary to randomise 376 participants to the intervention arm and 376 participants to the control arm (752 participants in total). However, assuming and adjusting for a worst-case scenario of a loss to follow-up rate of 5%, the total number of participants required would be 790 (395 each in the progesterone and placebo arms). The sample size of the study was planned accordingly.
The MID of 10% was defined following consultations among health-care practitioners, patients and representatives of patient bodies. However, we noted that this difference was much smaller than that expected from the contemporaneously existing literature (see Chapter 1, Existing knowledge, Progesterone in clinical use for recurrent miscarriages), which showed that the odds of miscarriage could be more than halved with progesterone therapy (odds ratio 0.39, 95% CI 0.21 to 0.72). Hence, assuming a conservative actual absolute difference of 15% in rates of live birth beyond 24 weeks, 790 participants (after accounting for 5% attrition) provided a power of 99%.
The 60% baseline (control) event rate was derived from a comprehensive audit carried out at the largest RM unit in the UK (St Mary’s Hospital in London) covering the period between 1998 and 2005, that showed the chances of live birth to be 61.8% (698/1129) after three miscarriages, 60.3% (350/580) after four miscarriages, 47.6% (109/229) after five miscarriages and 42.3% (82/194) after at least six miscarriages. However, because we had identified previously published evidence48 to suggest a higher control event rate, we performed a sensitivity analysis on the power calculations in which we assumed a higher control event rate of 70%. For a 10% absolute difference in rates of live birth after at least 24 weeks, and for an alpha error rate of 5%, 790 participants (after accounting for 5% attrition) gave a power of 89% (higher than 80% power when the control event rate was estimated to be 60%). We prudently adopted a lower control event rate for power calculation, to make provision for a worst-case scenario. All the power calculations noted above used two-sided binominal testing.
Statistical methods
Our data analysis plan was drawn up by the trial statistician and the study team, and approved by the Trial Steering Committee (TSC) and independent DMC prior to any analysis. The analysis was undertaken, using Stata® software, version 12 or later (StataCorp LP, College Station, TX, USA), based on treatment code and following the intention-to-treat principle. Only after the analysis was completed were the actual treatment arms corresponding to the treatment codes revealed. The components of analysis comprised (a) summarising baseline data, (b) intergroup comparisons, (c) subgroup analysis, and (d) adjustments and sensitivity analyses, such as to recognise the implications of missing data. The authors of this report had full access to all data collected in the study.
Summarising trial data
We planned to summarise the recruitment numbers, those lost to follow-up, protocol violations and other relevant data, using a Consolidated Standards Of Reporting Trials (CONSORT) diagram. Baseline data and outcome data were separately summarised. For categorical data, we planned to provide proportions (or percentages). For continuous variables, we planned to examine the distribution of the observations and, if normally distributed, we planned to summarise them as means with standard deviations (SDs). If they were not normally distributed, we planned to report medians and interquartile ranges (IQRs); additionally, we planned to use geometric means and SDs for data where distributions appeared to be log-normal.
We planned to use diagnostic plots to assess the severity of deviations from normality, using log-transformations where necessary, assessing results as estimates with 95% CIs, and using bootstrapping with bias correction and acceleration where non-parametric methods were indicated.
Following previously published CONSORT recommendations,49–51 significance tests were not given between randomised treatment arms.
Intergroup comparisons
The primary analysis was undertaken on the basis of intention to treat. This approach was intended to avoid any potentially misleading artefacts of the study (such as non-random attrition). Every attempt was made to collect a complete data set about each pregnancy and women were encouraged to allow continued data collection even if withdrawing from trial treatment, because the exclusion of withdrawn participants from data analysis could bias results and reduce the power of the study to detect important differences. In particular, participants were followed up even after any protocol treatment deviation or violation. However, if a participant explicitly withdrew consent to any further data recording then all communications surrounding the withdrawal were noted and no further data were collected for such participants (see Outcomes, Withdrawal).
Participants were analysed according to the original randomised allocation, irrespective of compliance and crossovers. Binary regression with a log-link was used to assess relative risks (RRs) for the primary outcomes and for other binary outcomes such as clinical pregnancy at 6–8 weeks; ongoing pregnancy at 12 weeks (range 11–13 weeks), miscarriage rate and survival at 28 days of neonatal life, adjusting for minimisation variables.
Significance tests were, in general, carried out only for estimates of treatment effects, as separate tests for changes over time in the two groups might have resulted in entirely false and misleading conclusions about the differences between the groups when comparing p-values.
Notwithstanding our best efforts to collect a complete data set about each pregnancy, in a small number of cases it was not possible to determine the primary outcome of the study (see Chapter 3, Numbers analysed). We conducted an analysis whereby participants with missing primary outcome data were not included in the primary analysis. This presented a risk of bias, and secondary sensitivity analyses were undertaken to assess the possible impact of the risk, considering alternative assumptions both in favour and against the effect of the intervention. Other sensitivity analyses involved simulating missing responses using multiple imputation, using those baseline variables that are significantly related to the outcome as predictors.
Subgroup analysis
Three subgroup analyses were planned:
-
number of previous miscarriages (3 or ≥ 4)
-
maternal age (≤ 35 or > 35 years)
-
polycystic ovaries or not.
Subgroup analyses were conducted only for the primary end point, and multivariate logistic regression was used. In each case, a test for interaction was first used to determine whether or not treatment was particularly effective in individual subgroups; our performance of subgroup analyses was dependent on sufficient data. Because of the well-known risk of false positives, both main effects and tests for interaction were performed and assessed before we considered results for subgroups. In addition, post-hoc subgroup analysis was performed only for the purpose of hypothesis generation.
Adjustments and sensitivity analyses
The process of randomisation with minimisation is designed to result in comparison groups that are highly similar at baseline, even more so than would be expected by chance. However, if randomisation failed to achieve balanced groups, we planned linear or logistic regression to adjust for the imbalance. We planned to adjust for missing data using multiple imputation. In cases of difference, we planned to give greater weighting to the primary analysis of intergroup comparisons than to sensitivity analyses.
Interim analyses
Interim analyses of principal safety and effectiveness end points were conducted on behalf of the independent DMC. These were considered together with scheduled reports of SAEs. The trial statistician was unblinded to the level of groups ‘A’ and ‘B’. The meaning of ‘A’ and ‘B’ was made known to the DMC separately. The first interim analysis was undertaken after the primary outcome data became available for the first 100 participants, and thereafter at annual intervals.
We were prepared to consider early termination of the PROMISE trial in case of interim analyses showing overwhelming evidence of effectiveness or significant harm (see Termination). Effectiveness and futility criteria were defined by the DMC (see Governance, Trial oversight bodies) with regard to the Peto principle that a trial should be stopped only when there is overwhelming evidence against one treatment or another52,53 with a nominal interim alpha set at 0.001 using O’Brien and Fleming alpha spending rules. 54 Under these conditions, no adjustment to the overall level of significance was needed. 55,56
Long-term analyses
Although the development of the babies born to participants in the PROMISE trial was of interest to the investigators, this was outside the scope and time frame of the study. Nevertheless, we recognised the value of data collected in this study to inform further studies on outcomes such as the composite end point of death or neurodevelopmental impairment at 2 years of age, cognitive scale scores at 2 years of chronological age, and disability classified into domains according to professional consensus. Women were asked to consent to future evaluation of themselves and their child and the health records of both, and could be traced through NHS Strategic Tracing Services. The hospital number and NHS number of each baby in the PROMISE trial were also recorded to facilitate future follow-up studies.
Health economic methods (see also Chapter 4)
An economic evaluation was conducted alongside the PROMISE trial, pragmatically comparing the costs and consequences of treatment using progesterone versus those of usual care. To examine the effect of treatment of each woman (and baby) on health resource use, data were collected from enrolment until the trial end point (hospital discharge). The analysis considered the number of days that treatment was received and three categories of heath service resources [antenatal contacts, how the pregnancy ended (preterm pregnancy loss management or mode of delivery) and postnatal admissions]. This perspective assumed the most salient costs to be those accrued by perinatal services; the assumption was tested by model-based extrapolation and a sensitivity analysis.
The primary outcome of our economic analysis was incremental cost per additional live birth after at least 24 weeks of gestation, with data collected up to 28 days of neonatal life. In order to provide additional information about the wider resource implications to the NHS and longer-term implications to health status, a systematic search identified models employed previously in similar trial-based economic evaluations to extend the time horizon of the cost and generic health gains of the two arms of the PROMISE trial end point. Furthermore, strategies were identified to attribute variation relating to the surrogate outcome of intervention (gestational age) to estimate health-care costs and associated generic health gains in the longer term.
Data access and quality assurance
Data management
The trial was designed and conducted to meet the requirements of:
Information about participants in the PROMISE trial was collected directly from trial participants and hospital notes, and was considered confidential. The trial manager was responsible for overseeing data custody, but all of the staff involved in the study (clinical, academic and support personnel) shared the same duty of care to prevent any unauthorised disclosure of personal information. For this purpose, each trial participant was allocated a unique study number at recruitment, and all study documents used this reference as the identifier. Personal data and contact details were held in separate NHS or university password-protected databases at local sites (in compliance with local and national confidentiality and data protection standards), which were linked to the secure ITMS via the unique study number. All data were analysed and reported in summary format (individuals remaining unidentifiable).
Data were collected and stored on secure NHS or university computers. Access to data was restricted by usernames and passwords at two levels (to gain access to NHS and university computers and then to access the ITMS). Only when strictly necessary and after anonymisation were trial data encrypted and transmitted outside the NHS or university settings (e.g. to the DMC). No study data were retained in handheld media, laptops, personal computers or other similar media.
The online ITMS was maintained according to the security policies of the Women’s Health Unit of King’s College London. These policies included provision for password assignment, encryption, immediate back-up, off-site back-up and disaster recovery processes. Electronic data were backed up to both local and remote media in encrypted format every 24 hours. Paper-based data (such as signed consent forms) were kept in locked filing cabinets at each site.
The data generated during the trial were available for inspection on request by the participating physicians, representatives of the sponsor, the REC, host institutions and the regulatory authorities. These data-handling arrangements were clearly conveyed to participants in the PIS (see Appendix 1), and permission was obtained in the consent form (see Appendix 2).
On completion of data collection, the site files from each study centre were to be securely archived at the sites. Electronic study data remained securely stored within the ITMS. The trial master file was to be securely stored by the sponsor when all study activities were completed. In accordance with the Medicines for Human Use (Clinical Trials) Amendment Regulations 2006 (sections 18 and 28),40 all the study data will remain securely stored for 25 years, to enable review, reappraisal and resolution of any queries or concerns and to facilitate further follow-up research. After this time the data will be securely destroyed.
Data quality assurance
The PROMISE trial co-ordinator performed hospital site visits as part of trial monitoring activities (see Governance, Trial monitoring). This quality assurance activity occasionally involved source data verification. The research and development (R&D) departments of participating study centres also performed routine monitoring audits at least annually. The PROMISE trial was additionally selected for MHRA inspection at two locations (Liverpool Women’s Hospital and Luton and Dunstable Hospital).
The trial also adopted a centralised approach to monitoring data quality and compliance, using the ITMS.
Governance
At all times during the study, the PROMISE trial was conducted strictly in accordance with the most recent version of the authorised PROMISE trial protocol. The PROMISE trial protocol was developed in accordance with the ethical principles originating in the Declaration of Helsinki,60 the principles of GCP, the Medicines for Human Use Regulations 2004 and its subsequent amendments39,40 and the Department of Health’s 2005 Research Governance Framework for Health and Social Care. 61
Ethical implications
In designing the PROMISE trial and considering the ethical implications of the study, the issues indicated below were considered and resolved or safeguarded.
Administration of a drug without full knowledge of its effects (although the existing evidence suggested a large potential benefit)
Existing evidence suggested a potential benefit in reducing the risk of miscarriage but the clinical community was in equipoise (see Chapter 1, Existing knowledge, Progesterone for clinical use for recurrent miscarriages). There was overwhelming support for the study from clinicians, patients and representatives of patient bodies (see Chapter 1, Rationale).
Furthermore, we identified substantial evidence of the safety of progesterone in pregnancy (see Chapter 1, Existing knowledge, Progesterone for clinical use for recurrent miscarriages), from widespread use in pregnancy for other indications such as IVF practice16 and prevention of preterm birth. 19
Moreover, we put in place robust mechanisms to address potential AEs (see Safety monitoring, Adverse events), in an effort to minimise harm. Overall, the balance of potential benefit versus harm was felt to be ethically acceptable to proceed with the trial.
Potential for distress, discomfort and inconvenience to trial participants
During our interviews and consultations with patients and representatives of patient societies at the time of designing the trial, it emerged that any distress or discomfort to the participants would be limited, and of an acceptable level to most women. We also designed the study to reduce any potential inconvenience to the participants, and put in place accommodating provisions wherever possible.
Face-to-face interviews at the time of recruitment could prolong a hospital visit by approximately 30 minutes, but patients and patient society representatives felt that this delay was well within an acceptable time frame. Furthermore, although five or more telephone interviews could be viewed as intrusive, precautions were incorporated into the study protocol to minimise inconvenience to participants.
These precautions included:
-
enquiring at the beginning of the telephone call if it was a convenient time to conduct the interview and, if not, arranging to call at an alternative time
-
specifying the purpose and the expected duration of the call
-
not leaving any messages if the telephone was answered by an automated machine or anyone other than the index patient
-
not telephoning a participant at her place of work if this could be avoided.
Interestingly, most patients and patient society representatives welcomed the telephone calls because the research nurse or midwife was likely to be able to assist standard care (e.g. by helping to arrange ultrasound scans).
Ethical governance
Following a favourable opinion of the National Research Ethics Service via the West Midlands REC and before recruitment commenced at each participating centre, the TCC obtained favourable site-specific assessments and R&D approvals as required.
The PI of each participating study site was responsible for liaison with administrative and managerial representatives of the local institution, and obtaining any necessary permissions from trust authorities. On behalf of the local institution, the PI was also required to sign an Investigator’s Agreement in respect of accrual, compliance, GCP, confidentiality and publication. Deviations from the Investigator’s Agreement were monitored and remedial action taken as appropriate by the TMG.
In addition, and in compliance with the International Conference on the Harmonisation of Good Clinical Practice, all institutions participating in the trial completed a delegation log and supplied this document to the TCC. The delegation log listed the responsibilities of each member of the local study team, and an up-to-date copy was stored in the site file at the institution and also at the TCC. A curriculum vitae of the PI, and confirmation of GCP training and honorary or substantive employment with the participating trust, was also verified by the trial manager and retained.
Clinical trial authorisation
Clinical trial authorisation for the PROMISE trial was obtained from the MHRA before recruitment started.
Changes to the protocol
It was agreed that if any amendments to the study protocol required regulatory approval (from the MHRA, REC or local R&D offices), these changes would not be instituted until the amendment had been reviewed and received favourable opinion from the relevant bodies. However, a protocol amendment intended to eliminate an apparent immediate hazard to participants would be implemented immediately, with notification and request for approval to the MHRA, REC and R&D offices as soon as possible.
There were no significant changes to the methods after trial commencement. All amendments to the study protocol were in the domain of clarifications to wording and intentions (see Appendix 5, Table 16).
Trial monitoring
The PROMISE trial was monitored according to the standard operating procedures of Imperial College London/Imperial College Healthcare NHS Trust Joint Research Compliance Office. 43
The purpose of monitoring was to:
-
protect the rights and well-being of trial participants
-
ensure that the reported trial data were accurate, complete and verifiable from source documents
-
ensure that the trial remained compliant with GCP and other regulatory and good practice guidance.
Participating centres were monitored by the TCC by checking incoming electronic forms for compliance with the protocol, consistency of data and missing data. The trial co-ordinator and trial manager remained in regular telephone or e-mail contact with centre personnel to check on progress and resolve any queries. In addition, periodic site monitoring was undertaken as required by the TCC or the sponsor to:
-
review understanding of the protocol and trial procedures by the trial staff
-
verify that the trial staff had access to the necessary documents
-
verify the existence of participants against clinic records and other sources
-
verify selected data items and SAEs recorded, compared with data in clinical records, to identify errors of omission as well as inaccuracies.
Monitoring visits were followed by a monitoring report, summarising the findings of the visit and recommending remedial actions as necessary. Investigator meetings were held at least annually for the purpose of learning, updating and sharing.
Trial oversight bodies
The PROMISE trial was overseen by the TMG, the DMC and the TSC (Figure 5).
FIGURE 5.
Reporting relationships of trial oversight bodies. HTA, Health Technology Assessment; NIHR, National Institute for Health Research.

Trial Management Group
The TMG directed the management of the trial from the TCC, which was located in a secure office in the University of Birmingham. The TCC comprised the trial manager, trial co-ordinator and a research nurse, with support from the trial statistician, data manager, health economist and trial advisors. The day-to-day co-ordination of the trial was the responsibility of the trial manager (Professor Arri Coomarasamy) and the trial co-ordinator (Dr Ewa Truchanowicz), who maintained regular contact with local collaborators, research nurses and midwives at participating study sites. The trial manager and the trial co-ordinator reported to the TMG, and the TMG reported to the TSC (or directly to the DMC if necessary) any issues relating to the monitoring and auditing of the research (see Figure 5).
The TMG conducted meetings face to face or via teleconference, and action points were implemented via the TCC. The meetings were held on a weekly basis during the early stages of the trial, and regularly as required thereafter, but at least monthly. The TMG disseminated any relevant feedback to investigators and other stakeholders through various approaches, including investigator meetings.
Trial Steering Committee
The TSC provided overall supervision of the trial, affording protection for participants by ensuring that it was conducted in accordance with International Conference on the Harmonisation of Good Clinical Practice guidelines and other relevant regulations. The TSC agreed and authorised the trial protocol and subsequent (minor) amendments (see Appendix 5, Table 16), and provided advice to investigators on all aspects of the trial. The role of the TSC also included reviewing recruitment, protocol deviations and recommendations from the DMC.
The TSC was chaired by an independent representative (Professor Siladitya Bhattacharya) and conducted meetings face to face or via teleconference on a 6-monthly basis, or more often if required.
Independent Data Monitoring Committee (see also Safety monitoring, Adverse events, and Statistical methods, Interim analyses)
The primary role of the PROMISE DMC consisted of periodic reviews of accruing data and assessments of safety, to make recommendations to the TSC about whether the trial should continue, or be modified or terminated. The DMC also reviewed interim analyses of major end points in addition to emerging data from other trials using progesterone in RM patients, to ensure that the continuation of the trial remained ethical.
The DMC additionally examined rates of recruitment, loss to follow-up, compliance and protocol violation data to ensure that the continuation of the trial was not futile.
The membership of the PROMISE DMC (chaired by Professor Jennifer Kurinczuk, and including Professor Javier Zamora and Professor Nick Raine-Fenning) was independent (none of the members had any financial or intellectual conflict of interest). The initiation meeting of the DMC, to review the study protocol and operating procedures, was conducted face to face and subsequent meetings were held either face to face or via teleconference.
Each DMC meeting comprised four consecutive components:
-
For DMC members only, to review rates of recruitment, baseline characteristics, effectiveness, safety, missing data and protocol violations; these data were prepared by the trial statistician, blinded to treatment allocation (identified only as ‘A’ and ‘B’).
-
For DMC members and the chairperson of the TSC, chief investigator, trial manager, sponsor or funder, as appropriate, to access relevant information.
-
For DMC members only, to review the issues arising from the open component above.
-
Discussions of the DMC with the chairperson of the TSC, the chief investigator and/or the trial manager, to convey the results and recommendations of the meeting.
The minutes from each open meeting were made available to all investigators and relevant stakeholders. The minutes from each closed meeting were archived by the DMC chairperson and the trial statistician.
Site responsibilities
To ensure the smooth running of the trial and to minimise the overall procedural workload, each participating centre designated appropriately trained and qualified local individuals to be responsible for the institutional co-ordination of clinical and administrative arrangements.
Local principal investigators
Each participating study centre nominated a local PI to oversee the conduct of PROMISE research at the particular institution. Each PI signed an Investigator’s Agreement to acknowledge these responsibilities, including:
-
adherence to the protocol of the trial
-
helping local colleagues to ensure that study participants received appropriate care throughout the period of research enrolment
-
protecting the integrity and confidentiality of clinical and other records generated by the research
-
reporting any failures in these respects, adverse drug reactions and other events or suspected misconduct through the appropriate systems.
Local nursing co-ordinators
Each participating centre also designated a local nurse or midwife to ensure that all eligible patients were considered for the study, provided with a PIS and offered an opportunity to discuss the study if required. This person also took responsibility for the collection of baseline and outcome data and the co-ordination of follow-up evaluations.
Patient and public involvement
Patient and public involvement (PPI) in research is not only a moral imperative (in consideration of the rights of service users and taxpayers to be involved in activities commissioned for them by government) but also a practical opportunity to ensure relevance, feasibility and accuracy. 62,63 Recognition of the importance of PPI at all stages of the research process and across the clinical spectrum is increasing64 but the emotive implications of pregnancy and miscarriage particularly heighten the importance of PPI in ensuring a sensitive study design. Therefore, the PROMISE trial team sought and drew extensively on the contributions of lay stakeholders to conceive and develop the project.
The nature of PPI may be described as consultation, collaboration or user-controlled research,65 and the participants may be existing or potential patients or recipients of health services, informal or unpaid carers, family members, disabled people, members of the public, groups asking for research because they believe they have been exposed to potential harm or denied potential benefit from health services, and/or organisations to represent any of these people. 65 The PROMISE trial team undertook PPI primarily as consultation, with RM service users and organisations representing a wider public.
Information
From the RM clinics at St Mary’s Hospital and Guy’s and St Thomas’ Hospitals in London, 88 women with a history of RM were surveyed with a set of structured and semi-structured questions to identify their opinions regarding the rationale for the trial and its ethical implications (see Governance, Ethical implications), the route of progesterone administration (vaginal, rectal or intramuscular), the duration of therapy, the suitability of courier transport to deliver study medications directly to study participants, and the relative importance of different outcomes assessed in the PROMISE trial. The interviews were conducted on a one-to-one basis by physicians with experience in early pregnancy care and training in listening to patient concerns. The information collected enabled the PROMISE trial team to understand that self-administration of vaginal progesterone twice daily would be acceptable to most potential participants in the study, and to select meaningful and important outcomes for data collection.
Communication
Five champions from among the participants in our survey volunteered to help the central organisers of the study to develop clear and concise literature for participants (see Appendices 1 and 2). During the development, the provisional documents were reviewed by other patients with a history of RM, as well as the independent service user representatives listed below:
-
Ms Ruth Bender Atik (National Director of the Miscarriage Association)
-
Ms Liz Campbell (Director of the Wellbeing of Women research charity)
-
Officers of the RCOG consumers’ forum.
These members of the group were selected to represent professional and charitable communities with an interest in pregnancy and miscarriage. They brought experience of working with not only women who had previously suffered RM, but also stakeholders such as women trying to conceive, women in pregnancy and women who had previously suffered miscarriage, and their family members, from across the UK.
All of the lay stakeholders expressed appreciation for their involvement in PROMISE activities and enthusiasm for ongoing involvement in the PROMISE trial, whether as a result of inherent personal interest in the topic under investigation or the value they placed on empowerment to influence service improvements. They expressed opinions about their previous experiences of clinical consultations and identified strategies to improve existing services. Their views were valued by researchers and directly informed the development of literature for participants (see Appendices 1 and 2).
Oversight
Ms Ruth Bender Atik, the National Director of the Miscarriage Association, was also involved in overseeing the PROMISE trial as a member of the TSC throughout the duration of the study (see Governance, Trial oversight bodies). Although Ms Bender Atik was enthusiastic and dedicated, it is important to note that her voluntary contribution to the PROMISE trial took her time and attention away from competing commitments; in retrospect, it would have been helpful to ensure that such a time requirement was recognised financially.
Dissemination
During the years of recruitment and follow-up, various research team members facilitated peer group sessions and seminars, and delivered presentations about study activities and developmental concepts of the intervention. The composition of these groups varied, for example from closed meetings of senior clinical practitioners at each of the participating study centres to a large public audience for an inaugural lecture that was also shown online. The contents of the presentations were tailored accordingly.
Monthly newsletters were circulated to boost the morale of the trial staff at study sites, highlight important tasks and encourage recruitment. When recruitment was complete, the newsletters were no longer disseminated, but targeted, site-specific communications were used instead.
Timelines and targets
The duration of the PROMISE trial was anticipated as 3 years, commencing in February 2010. Audits of RM clinics at the seven study sites originally anticipated to participate in the PROMISE trial showed that over 2200 new RM patients per year were seen in these centres, and that over half of these women would be found to be eligible for the trial (i.e. they had a diagnosis of unexplained RM). Based on our previous experience, we expected up to 75% of eligible women to agree to participate, but we adopted a conservative target recruitment rate of only 50% of eligible patients. Recruitment targets were carefully scrutinised by local PIs and their teams to ensure the anticipated numbers were feasible within the anticipated timeline of the trial. However, owing to a delay in procuring placebo capsules, the trial commenced later than expected, incurring a ‘no-cost’ time-only extension by approximately 1 year.
Termination
The interventional phase of the trial ended when the last recruited participant had taken her last dose of the trial intervention. The observational phase of the trial ceased when the 28-day follow-up was completed for the baby of the last participant recruited.
However, we were prepared to consider early termination of the PROMISE trial in case of:
-
interim analysis (see Statistical methods, Interim analyses) showing overwhelming evidence of effectiveness or significant harm with a nominal interim alpha likely to be set at 0.001 using O’Brien and Fleming alpha spending rules54 and with regard to the Peto principle that a trial should be stopped only when there is overwhelming evidence against one treatment or another52,53
-
major safety concerns
-
insurmountable issues with IMP supply
-
a change in the opinion of the REC
-
a regulatory decision or sponsor withdrawal.
We were prepared to terminate recruitment at a particular study site for reasons of low recruitment or compliance issues. The sponsor also reserved the right, only after taking advice from the TSC and the independent DMC, to discontinue the PROMISE trial at any time for safety, overwhelming evidence of effectiveness or significant harm or any other reasonable reasons.
Chapter 3 Results
This chapter commences with a CONSORT diagram that describes the flow of participants through each stage of the trial (see Figure 6). This is followed by demographic information, primary and secondary outcomes, ancillary analyses and AEs.
Participant flow
Participant flow is illustrated by Figure 6. A total of 1568 participants were screened for eligibility and consented to take part in the PROMISE trial. Of these, 732 participants were excluded from randomisation, the most common reasons being that they did not conceive naturally within 1 year (515 women) or that they subsequently decided to withdraw from the study before conceiving a pregnancy (202 women). Of those participants who conceived naturally within 1 year and remained willing to participate in the trial, the gestational age of 10 pregnancies could not be confirmed to ensure eligibility, and a further four women were not eligible for other reasons when reassessed. These women were excluded. The remaining participants were randomised to receive either progesterone (404 women) or placebo (432 women).
FIGURE 6.
Flow of participants through the PROMISE trial. GA, gestational age.
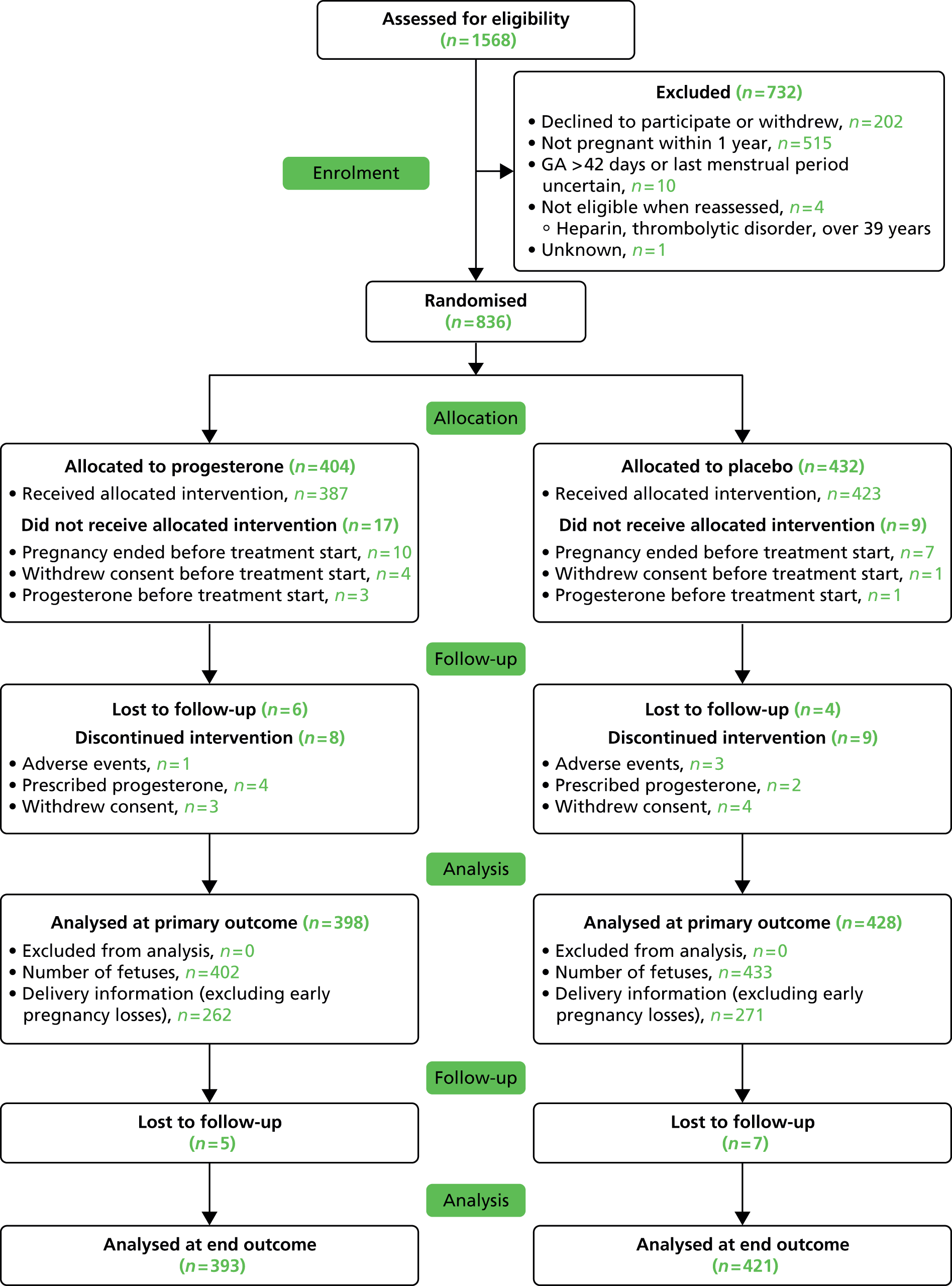
Recruitment
Recruitment and randomisation took place over 41 months (Figure 7). A total of 836 participants were randomised, exceeding the planned target of 790 participants, from 45 active centres (36 in the UK and nine in the Netherlands). Four centres contributed more than 100 enrolled participants each (Table 2). Rates of conversion from enrolment to randomisation varied between 0% and 100%.
| Institution | Site | PI | Recruited (N = 1568), n (%) | Randomised (N = 836), n (%) |
|---|---|---|---|---|
| UK | ||||
| Imperial College Healthcare NHS Trust | St Mary’s Hospital | Dr Rajendra Rai | 232 (14.8) | 137 (16.4) |
| Heart of England NHS Foundation Trust | Birmingham Heartlands Hospital | Professor Siobhan Quenby and Dr Pratima Gupta | 148 (9.4) | 64 (7.7) |
| University Hospital Southampton NHS Foundation Trust | Princess Anne Hospital | Dr Ying Cheong | 87 (5.5) | 61 (7.3) |
| Liverpool Women’s NHS Foundation Trust | Liverpool Women’s Hospital | Dr Feroza Dawood | 115 (7.3) | 53 (6.3) |
| Birmingham Women’s NHS Foundation Trust | Birmingham Women’s Hospital | Professor Mark Kilby | 105 (6.7) | 48 (5.7) |
| University Hospitals Coventry and Warwickshire NHS Trust | University Hospital (Coventry) | Professor Siobhan Quenby | 71 (4.5) | 42 (5.0) |
| Sheffield Teaching Hospitals NHS Foundation Trust | Sheffield Royal Hallamshire Hospital | Professor Tin-Chiu Li and Dr Shehnaz Jivraj | 75 (4.8) | 37 (4.4) |
| Guy’s and St Thomas’ NHS Foundation Trust | St Thomas’ Hospital | Mr Yacoub Khalaf | 64 (4.1) | 23 (2.8) |
| Portsmouth Hospitals NHS Trust | Queen Alexandra Hospital | Dr Nirmala Vaithilingam | 39 (2.5) | 23 (2.8) |
| Sandwell and West Birmingham Hospitals NHS Teaching Trust | Birmingham City Hospital | Mr Ayman Ewies | 38 (2.4) | 21 (2.5) |
| Newcastle upon Tyne Hospitals NHS Foundation Trust | Royal Victoria Infirmary | Dr Jane Stewart | 26 (1.7) | 20 (2.4) |
| NHS Ayrshire and Arran | Ayrshire Maternity Unit | Dr Marjory MacLean | 30 (1.9) | 19 (2.3) |
| Nottingham University Hospitals NHS Trust | Queen’s Medical Centre | Dr Judith Moore | 42 (2.7) | 18 (2.2) |
| King’s College Hospital NHS Foundation Trust | King’s College Hospital | Dr Jackie Ross | 30 (1.9) | 16 (1.9) |
| South Tees Hospitals NHS Foundation Trust | James Cook University Hospital | Dr Gamal Sayed and Dr Padma Manda | 15 (1.0) | 10 (1.2) |
| Central Manchester University Hospitals NHS Foundation Trust | St Mary’s Hospital Manchester | Dr Edmond Edi-Osagie | 17 (1.1) | 9 (1.1) |
| City Hospitals Sunderland NHS Foundation Trust | Sunderland Royal Hospital | Dr Amna Ahmed | 10 (0.6) | 8 (1.0) |
| Northumbria Healthcare NHS Foundation Trust | Wansbeck Hospital | Dr Shonag Mackenzie | 13 (0.8) | 7 (0.8) |
| Luton and Dunstable NHS Foundation Trust | Luton and Dunstable University Hospital | Dr Neela Mukhopadhaya | 12 (0.8) | 7 (0.8) |
| South Tyneside NHS Foundation Trust | South Tyneside District Hospital | Dr Shamma Al-Inizi | 11 (0.7) | 6 (0.7) |
| Frimley Park Hospital NHS Foundation Trust | Frimley Park Hospital | Dr Manisha Chandra | 9 (0.6) | 6 (0.7) |
| Royal Devon and Exeter Hospitals NHS Trust | Royal Devon and Exeter Hospital | Dr Lisa Joels | 9 (0.6) | 5 (0.6) |
| Blackpool Teaching Hospitals NHS Foundation Trust | Blackpool Victoria Hospital | Dr Elizabeth Haslett | 5 (0.3) | 4 (0.5) |
| Derby Hospitals NHS Foundation Trust | Royal Derby Hospital | Dr Gillian Scothern and Mr Jayaprakasan Kanna | 5 (0.3) | 4 (0.5) |
| Chelsea and Westminster Hospital NHS Foundation Trust | Chelsea and Westminster Hospital | Mr Guy Thorpe-Beeston | 12 (0.8) | 3 (0.4) |
| Bolton NHS Foundation Trust | Royal Bolton Hospital | Dr Sangeeta Das | 3 (0.2) | 3 (0.4) |
| Homerton University Hospital NHS Foundation Trust | Homerton University Hospital | Dr Sandra Watson | 7 (0.4) | 2 (0.2) |
| West Middlesex University Hospital NHS Trust | West Middlesex University Hospital | Dr Maysoon Backos | 9 (0.6) | 2 (0.2) |
| Countess of Chester Hospital NHS Foundation Trust | Countess of Chester Hospital | Mr Simon Wood | 8 (0.5) | 2 (0.2) |
| North Tees and Hartlepool NHS Foundation Trust | University Hospital of North Tees | Dr Iona MacLeod | 5 (0.3) | 2 (0.2) |
| NHS Greater Glasgow and Clyde | Southern General Hospital | Dr Judith Roberts | 7 (0.4) | 1 (0.1) |
| North Cumbria University Hospitals NHS Trust | Cumberland Infirmary | Professor Nalini Munjuluri | 6 (0.4) | 1 (0.1) |
| University Hospitals of Morecambe Bay NHS Foundation Trust | Royal Lancaster Infirmary | Mr Faisal Basama | 4 (0.3) | 1 (0.1) |
| North Cumbria University Hospitals NHS Trust | West Cumberland Hospital, Whitehaven | Mr Steve Bober | 2 (0.1) | 1 (0.1) |
| County Durham and Darlington Foundation Trust | University Hospital of North Durham | Dr Seema Sen | 4 (0.3) | 0 (0.0) |
| Gateshead Health NHS Foundation Trust | Queen Elizabeth Hospital | Dr Isaac Evbuomwan | 2 (0.1) | 0 (0.0) |
| Netherlands | ||||
| University Medical Centre Utrecht | Dr Yvonne Koot | 60 (3.8) | 43 (5.1) | |
| Leiden University Medical Centre | Dr Kitty Bloemenkamp | 85 (5.4) | 41 (4.9) | |
| Academic Medical Centre (Amsterdam) | Dr Mariette Goddijn | 58 (3.7) | 24 (2.9) | |
| Maxima Medical Centre (Veldhoven) | Dr Carolien Koks | 26 (1.7) | 20 (2.4) | |
| Onze Lieve Vrouwe Gasthuis (Amsterdam) | Dr Eugenie Kaaijk | 23 (1.5) | 13 (1.6) | |
| University Medical Centre Groningen | Dr Annemieke Hoek | 19 (1.2) | 13 (1.6) | |
| Medical Centre Leeuwarden | Dr Denise Perquin | 9 (0.6) | 8 (1.0) | |
| Isala Klinieken Zwolle | Dr Walter Kuchenbecker | 8 (0.5) | 8 (1.0) | |
| Atrium Medical Centre (Heerlen) | Dr Patricia Mercelina | 3 (0.2) | 0 (0.0) | |
FIGURE 7.
Rates of recruitment to the PROMISE trial.

Baseline data
The randomised participants in the PROMISE trial were aged between 18 and 39 years at the time of randomisation, with a median age of 32.7 years (IQR 29.1–36.1 years). The mean BMI at the time of randomisation was 25.4 kg/m2 (SD 5.1 kg/m2). Of the 823 (98.4%) randomised participants who provided ethnic group data, 682 (82.9%) were white, 35 (4.3%) were black, 68 (8.3%) were Asian and 38 (4.6%) were from other ethnic groups. Most of the participants were non-smokers (702/836; 84.0%).
Of the 836 randomised participants, 375 (44.9%) had experienced more than three previous miscarriages and 55 (6.6%) had previously experienced ectopic pregnancy. The median number of preceding miscarriages was 3.0. Women with previous live births constituted 346 (41.4%) of the 836 participants randomised. Cases of comorbidities included 58 (6.9%) participants with polycystic ovarian syndrome, 29 (3.5%) with a fibroid uterus, 19 (2.3%) with endometriosis and 49 (5.9%) with an arcuate uterus. Furthermore, 29 (3.5%) participants had previously undergone large loop excision of the cervical transformation zone, five (0.6%) had previously undergone myomectomy, seven (0.8%) had previously undergone endometriosis surgery, 32 (3.8%) had previously undergone tubal surgery and 14 (1.7%) had previously undergone ovarian cystectomy. Study records of concurrent medications showed that six (0.7%) of the randomised participants were taking metformin at the time of participation, and 75 (9.0%) were taking low-dose aspirin.
The baseline demographic characteristics of participants in the progesterone and placebo groups were comparable (Table 3).
| Descriptive characteristic | Progesterone | Placebo | All |
|---|---|---|---|
| Maternal agea 18 to 35 years,b n/N (%) | 261/404 (64.6) | 294/432 (68.1) | 555/836 (66.4) |
| Maternal agea > 35 years,b n/N (%) | 143/404 (35.4) | 138/432 (31.9) | 281/836 (33.6) |
| Median age (years) (IQR)b | 32.9 (29.3–36.3) | 32.5 (28.9–35.9) | 32.7 (29.1–36.1) |
| Mean maternal height (m) (SD) | 164.4 (7.4) | 165.3 (7.1) | 164.9 (7.2) |
| Mean maternal weight (kg) (SD) | 68.9 (14.2) | 69.2 (14.4) | 69.0 (14.3) |
| Mean maternal BMI (kg/m2) (SD)b | 25.5 (5.1) | 25.3 (5.1) | 25.4 (5.1) |
| Maternal BMI > 30.0 kg/m2b | 63 (15.6) | 65 (15.0) | 128 (15.3) |
| Maternal ethnicity, n/N (%) | |||
| White | 316/399 (79.2) | 366/424 (86.3) | 682/823 (82.9) |
| Black | 16/399 (4.0) | 19/424 (4.5) | 35/823 (4.3) |
| Asian | 39/399 (9.8) | 29/424 (6.8) | 68/823 (8.3) |
| Other, including mixed | 28/399 (7.0) | 10/424 (2.4) | 38/823 (4.6) |
| Partner’s ethnicity, n/N (%) | |||
| White | 293/371 (79.0) | 327/395 (82.8) | 620/766 (80.9) |
| Black | 24/371 (6.5) | 18/395 (4.6) | 42/766 (5.5) |
| Asian | 39/371 (10.5) | 33/395 (8.4) | 72/766 (9.4) |
| Other, including mixed | 15/371 (4.0) | 17/395 (4.3) | 32/766 (4.2) |
| Maternal smoking (cigarettes per day), n/N (%) | |||
| Non-smoker | 339/404 (83.9) | 363/432 (84.0) | 702/836 (84.0) |
| < 10 | 28/404 (6.9) | 34/432 (7.9) | 62/836 (7.4) |
| 10–19 | 31/404 (7.7) | 27/432 (6.3) | 58/836 (6.9) |
| ≥ 20 | 6/404 (1.5) | 8/432 (1.9) | 14/836 (1.7) |
| Partner smoking (cigarettes per day), n/N (%) | |||
| Non-smoker | 318/404 (78.7) | 348/432 (80.6) | 666/836 (79.7) |
| < 10 | 30/404 (7.4) | 32/432 (7.4) | 62/836 (7.4) |
| 10–19 | 41/404 (10.1) | 33/432 (7.6) | 74/836 (8.9) |
| ≥ 20 | 15/404 (3.7) | 19/432 (4.4) | 34/836 (4.1) |
| Alcohol (units per day), n/N (%) | |||
| None | 229/404 (56.7) | 260/432 (60.2) | 489/836 (58.5) |
| ≤ 3 | 92/404 (22.8) | 89/432 (20.6) | 181/836 (21.7) |
| > 3 and ≤ 20 | 82/404 (20.3) | 83/432 (19.2) | 165/836 (19.7) |
| > 20 | 1/404 (0.2) | 0/432 (0.0) | 1/836 (0.1) |
| Parity, n/N (%) | |||
| Previous live birth | 167/404 (41.3) | 179/432 (41.4) | 346/836 (41.4) |
| ≥ 4 previous miscarriagesb | 183/404 (45.3) | 192/432 (44.4) | 375/836 (44.9) |
| Previous ectopic pregnancy | 26/402 (6.5) | 29/431 (6.7) | 55/833 (6.6) |
| Median number of preceding losses (IQR) | 3.0 (3.0–5.0) | 3.0 (3.0–4.0) | 3.0 (3.0–4.0) |
| Clinical risk factors, n/N (%) | |||
| Polycystic ovariesb | 30/404 (7.4) | 28/432 (6.5) | 58/836 (6.9) |
| Fibroids | 15/404 (3.7) | 14/432 (3.2) | 29/836 (3.5) |
| Endometriosis | 8/404 (2.0) | 11/432 (2.5) | 19/836 (2.3) |
| Arcuate uterus | 24/404 (5.9) | 25/432 (5.8) | 49/836 (5.9) |
| Gynaecological surgeries, n/N (%) | |||
| LLETZ | 10/404 (2.5) | 19/432 (4.4) | 29/836 (3.5) |
| Myomectomy | 1/404 (0.2) | 4/432 (0.9) | 5/836 (0.6) |
| Endometriosis surgery | 4/404 (1.0) | 3/432 (0.7) | 7/836 (0.8) |
| Tubal surgery | 17/404 (4.2) | 15/432 (3.5) | 32/836 (3.8) |
| Ovarian cystectomy | 6/404 (1.5) | 8/432 (1.9) | 14/836 (1.7) |
| Family history of RM | 55/368 (14.9) | 63/391 (16.1) | 118/759 (15.5) |
| Concurrent medications, n/N (%) | |||
| Metformin | 4/404 (1.0) | 2/432 (0.5) | 6/836 (0.7) |
| Aspirin | 38/404 (9.4) | 37/432 (8.6) | 75/836 (9.0) |
Numbers analysed
A total of 836 participants were randomised, of whom 404 were randomised to progesterone and 432 were randomised to placebo. However, some of these participants (17 women receiving progesterone and nine women receiving placebo) did not receive the allocated intervention (most often as a result of pregnancy loss before treatment could commence), and 10 participants (six women receiving progesterone and four women receiving placebo) were lost to follow-up. Primary outcome data were available for 826 out of 836 (98.8%) participants [398 out of 404 (98.5%) randomised to progesterone, and 428 out of 432 (99.1%) randomised to placebo].
Outcomes and estimation
The PROMISE trial found no evidence of significant differences in the rates of primary or secondary outcomes between the group randomised to receive progesterone and the group randomised to placebo during the study.
Primary outcome
Overall, 533 out of 826 women (64.5%) experienced a live birth after at least 24 weeks of gestation. Table 4 shows that the live birth rate in the progesterone group was 65.8% (262/398), and in the placebo group it was 63.3% (271/428), giving a RR of 1.04 (95% CI 0.94 to 1.15; p = 0.45).
| Outcome | Progesterone, n/N (%) | Placebo, n/N (%) | Comparisons, RR (95% CI) | Significance |
|---|---|---|---|---|
| Clinical pregnancy 6–8 weeks (presence of gestational sac)a | 326/398 (81.9) | 334/428 (78.0) | 1.05 (0.98 to 1.12) | p = 0.16 |
| Ongoing pregnancy 12 weeks (presence of fetal heartbeat)a | 267/398 (67.1) | 277/428 (64.7) | 1.04 (0.94 to 1.14) | p = 0.47 |
| Ectopic pregnancya | 6/398 (1.5) | 7/428 (1.6) | 0.92 (0.31 to 2.72) | p = 0.88 |
| Miscarriagea | 128/398 (32.2) | 143/428 (33.4) | 0.96 (0.79 to 1.17) | p = 0.70 |
| Stillbirtha | 1/398 (0.3) | 2/428 (0.5) | 0.54 (0.05 to 5.92) | p = 0.61 |
| Median gestational age at miscarriage (IQR)a | 7.3 (6.0 to 8.7) | 7.1 (6.0 to 8.5) | 0.0 (–0.6 to 0.4) | p = 0.87 |
| Live births (≥ 24 + 0 weeks)a | 262/398 (65.8) | 271/428 (63.3) | 1.04 (0.94 to 1.15) | p = 0.45 |
| Gestation at delivery of live birtha | ||||
| < 28 + 0 weeks | 1/262 (0.4) | 1/271 (0.4) | 1.03 (0.06 to 16.49) | p = 0.98 |
| < 30 + 0 weeks | 1/262 (0.4) | 2/271 (0.7) | 0.52 (0.05 to 5.68) | p = 0.59 |
| < 34 + 0 weeks | 10/262 (3.8) | 10/271 (3.7) | 1.03 (0.44 to 2.45) | p = 0.94 |
| < 37 + 0 weeks | 27/262 (10.3) | 25/271 (9.2) | 1.12 (0.67 to 1.87) | p = 0.68 |
| Twin live births (≥ 24 + 0 weeks)a | 4/398 (1.0) | 5/428 (1.2) | 0.86 (0.23 to 3.18) | p = 0.82 |
| Survival to 28 daysb | 260/261 (99.6) | 269/269 (100.0) | 1.00 (0.99 to 1.00) | p = 0.32 |
Of the 533 pregnancies that proceeded to live birth beyond 24 weeks, 524 (98.3%) were singleton pregnancies and nine (1.7%) were twin pregnancies. None of the women participating in the PROMISE trial delivered more than two babies, and, overall, 1.1% of the trial participants [1.0% (4/398) in the progesterone group and 1.2% (5/428) in the placebo group] experienced a twin birth.
Secondary outcomes
Clinical pregnancy at 6–8 weeks
During the study, we were able to confirm that 660 (79.9%) out of all the randomised participants for whom primary outcome data were available experienced a clinical pregnancy (defined as the presence of a gestational sac) at 6–8 weeks. The clinical pregnancy rates observed were not significantly different between the two arms of the trial; we recorded 326 out of 398 (81.9%) clinical pregnancies in the progesterone group, versus 334 out of 428 (78.0%) clinical pregnancies in the placebo group. We observed a RR of 1.05 (95% CI 0.98 to 1.12) between the progesterone and placebo groups, with a p-value of 0.16 (see Table 4).
Ongoing pregnancy at 12 weeks
We were also able to confirm the ongoing pregnancy (defined as the presence of a fetal heartbeat at 12 weeks) of 544 (65.9%) out of all the randomised participants for whom primary outcome data were available. The ongoing pregnancy rates observed were not significantly different between the two groups; we were able to confirm the ongoing pregnancy of 267 out of 398 (67.1%) women in the progesterone group, versus 277 out of 428 (64.7%) women in the placebo group. We observed a RR of 1.04 (95% CI 0.94 to 1.14) between the progesterone and placebo groups, with a p-value of 0.47 (see Table 4).
Miscarriage
The rates of miscarriage in each study group (progesterone 128/398, 32.2%, vs. placebo 143/428, 33.4%) are given in Table 4. Rates of miscarriage were not significantly different between the groups randomised to receive progesterone or placebo. We observed a RR of 0.96 (95% CI 0.79 to 1.17) between the progesterone and placebo groups, with a p-value of 0.70.
Of the 128 pregnancies that were lost among participants receiving progesterone, the median age of gestation at miscarriage diagnosis was 7.3 weeks (IQR 6.0–8.7 weeks). Of the 143 pregnancies that were lost among participants receiving placebo, the median gestational age was 7.1 weeks (IQR 6.0–8.5 weeks) (see Table 4). We observed a RR of 0.00 (95% CI –0.6 to 0.4) between the progesterone and placebo groups, with a p-value of 0.87.
Ectopic pregnancy
The rates of ectopic pregnancy in each study group (progesterone 6/398, 1.5%, vs. placebo 7/428, 1.6%) are given in Table 4. We observed a RR of 0.92 (95% CI 0.31 to 2.72) between the progesterone and placebo groups, with a p-value of 0.88.
Stillbirth
The rates of stillbirth in each study group (progesterone 1/398, 0.3% vs. placebo 2/428, 0.5%) are given in Table 4. We observed a RR of 0.54 (95% CI 0.05 to 5.92) between the progesterone and placebo groups, with a p-value of 0.61.
Gestational age at live birth
The distributions of gestational age at delivery for the progesterone and placebo groups are illustrated by Figure 8. They show a hazard ratio of 1.04 (95% CI 0.91 to 1.19; p = 0.59).
FIGURE 8.
Distribution of gestational age by randomised treatment: pregnancies continuing beyond 24 weeks only. Hazard ratio 1.04 (95% CI 0.91 to 1.19; p = 0.59).

Preterm birth: < 28 weeks
Of the 533 pregnancies continuing to live birth after at least 24 weeks, two participants (0.4%, one woman in each arm of the trial) were delivered at < 28 weeks. We observed a RR of 1.03 (95% CI 0.06 to 16.49) between the progesterone and placebo groups, with a p-value of 0.98 (see Table 4).
Preterm birth: < 30 weeks
Of the 533 pregnancies continuing to live birth after at least 24 weeks, three participants (0.6%) were delivered at < 30 weeks. The rates of delivery at < 30 weeks were not significantly different between the groups randomised to receive progesterone (1/262, 0.4%) or placebo (2/271, 0.7%). We observed a RR of 0.52 (95% CI 0.05 to 5.68) between the progesterone and placebo groups, with a p-value of 0.59 (see Table 4).
Preterm birth: < 34 weeks
Of the 533 pregnancies continuing to live birth after at least 24 weeks, 20 participants (3.8%, 10 women in each arm of the trial) were delivered at < 34 weeks. Between the progesterone and placebo groups, we observed a RR of 1.03 (95% CI 0.44 to 2.45), with a p-value of 0.94 (see Table 4).
Preterm birth: < 37 weeks
Of the 533 pregnancies continuing to live birth after at least 24 weeks, 52 participants (9.8%) were delivered at < 37 weeks. Rates of delivery at < 37 weeks were not significantly different between the groups randomised to receive progesterone (27/262, 10.3%) or placebo (25/271, 9.2%). We observed a RR of 1.12 (95% CI 0.67 to 1.87) between the progesterone and placebo groups, with a p-value of 0.68 (see Table 4).
Neonatal survival
We were able to collect neonatal survival data from 530 of the 542 babies born alive to 533 women after at least 24 weeks of gestation. The rates of neonatal survival in each study group (progesterone 260/261, 99.6%, vs. placebo 269/269, 100.0%) are given in Table 4.
Congenital anomalies
Congenital anomalies observed in babies born to PROMISE participants are listed in Table 5. These outcomes were rare, both overall and in each arm of the trial, so our power to identify truly significant rates of difference between the groups is low. Among the pregnancies randomised to receive active treatment, we observed two cases of talipes, two heart anomalies (one ventricular septal defect and one transposition of great arteries), one case of tongue tie, one case of hernia, two chromosomal anomalies (one case of Pierre Robin syndrome and one case of Down syndrome duodenal atresia) and one genital abnormality (hypospadias). Among those randomised to placebo, we observed one case of talipes, three heart anomalies (all ventricular septal defects), two cases of ventriculomegaly, two cases of tongue tie, one case of hernia, one chromosomal anomaly (Turner syndrome) and one genital abnormality (urachal cyst). Overall, 8 out of 266 (3.0%) babies in the progesterone group and 11 out of 276 (4.0%) babies in the placebo group were affected by congenital anomalies, giving a RR of 0.75 (95% CI 0.31 to 1.85), with a p-value of 0.54.
| Type of abnormality | Progesterone (N = 266), n (%) | Placebo (N = 276), n (%) | All (N = 542), n (%) |
|---|---|---|---|
| No congenital abnormalities | 258 (97.0) | 265 (96.0) | 523 (96.5) |
| Talipes | 2 (0.8) | 1 (0.4) | 3 (0.6) |
| Heart anomaly | 2 (0.8) | 3 (1.1) | 5 (0.9) |
| Ventriculomegaly | 0 (0.0) | 2 (0.7) | 2 (0.4) |
| Tongue tie | 1 (0.4) | 2 (0.7) | 3 (0.6) |
| Hernia | 1 (0.4) | 1 (0.4) | 2 (0.4) |
| Chromosomal anomaly | 2 (0.8) | 1 (0.4) | 3 (0.6) |
| Genital abnormality | 1 (0.4) | 1 (0.4) | 2 (0.4) |
Ancillary analyses
Subgroup analyses
We planned a priori to conduct subgroup analyses by maternal age (≤ 35 or > 35 years at recruitment), number of previous miscarriages (3 or > 3) and the presence (or not) of polycystic ovaries (Table 6). At the end of the trial, we performed additional post-hoc comparisons between the outcomes of women commencing treatment at earlier and later stages of the first trimester, and between women in the UK and in the Netherlands.
| Subgroup | Progesterone, n/N (%) | Placebo, n/N (%) | Comparisons, RR (95% CI) | Significance |
|---|---|---|---|---|
| Age | ||||
| ≤ 35 yearsa | 171/258 (66.3) | 191/300 (63.7) | 1.04 (0.92 to 1.18) | p = 0.52 |
| > 35 yearsa | 91/140 (65.0) | 80/128 (62.5) | 1.04 (0.87 to 1.25) | p = 0.67 |
| Chi-squared test for interaction | 0.00 | p = 0.98b | ||
| Previous miscarriages | ||||
| 3 | 148/218 (67.9) | 159/236 (67.4) | 1.01 (0.89 to 1.14) | p = 0.91 |
| ≥ 4 | 114/180 (63.3) | 112/192 (58.3) | 1.09 (0.92 to 1.28) | p = 0.32 |
| Chi-squared test for interaction | 0.41 | p = 0.52b | ||
| Polycystic ovaries | ||||
| Absent | 245/369 (66.4) | 252/401 (62.8) | 1.06 (0.95 to 1.17) | p = 0.30 |
| Present | 17/29 (58.6) | 19/27 (70.4) | 0.83 (0.56 to 1.23) | p = 0.36 |
| Chi-squared test for interaction | 1.34 | p = 0.25b | ||
| Gestation at treatment startc,d | ||||
| < 5 + 0 weeks | 120/182 (65.9) | 144/212 (67.9) | 0.97 (0.84 to 1.12) | p = 0.68 |
| ≥ 5 + 0 weeks | 122/176 (69.3) | 113/184 (61.4) | 1.13 (0.97 to 1.31) | p = 0.12 |
| Chi-squared test for interaction | 2.03 | p = 0.15b | ||
| Geographical locationd | ||||
| UK | 212/312 (67.9) | 207/326 (63.5) | 1.07 (0.96 to 1.20) | p = 0.24 |
| Netherlands | 50/86 (58.1) | 64/102 (62.7) | 0.93 (0.73 to 1.17) | p = 0.52 |
| Chi-squared test for interaction | 1.20 | p = 0.27b | ||
Maternal age
For the purposes of a subgroup analysis, we evaluated the effects of progesterone in women aged ≤ 35 years at the time of recruitment, compared with women who were > 35 years of age (see Table 6). We observed that women aged ≤ 35 years experienced a rate of live birth after at least 24 weeks (362/558, 64.9%) similar to older women (171/268, 63.8%). This trend was common to both the progesterone and placebo groups. In the subgroup of women aged ≤ 35 years, the RR of live birth after at least 24 weeks was 1.04 (95% CI 0.92 to 1.18) when progesterone was compared with placebo. In the subgroup of women who were > 35 years of age, the RR of live birth after at least 24 weeks was 1.04 (95% CI 0.87 to 1.25). A chi-squared test for interaction did not find a significant subgroup effect (χ2 = 0.00; p = 0.98).
Previous miscarriages
Our study population was divided into groups of women with a history of three miscarriages and women with a history of more than three miscarriages (see Table 6). We observed that women who had experienced three previous miscarriages experienced a higher rate of live birth after at least 24 weeks (307/454, 67.6%) than women who had experienced more than three previous miscarriages (226/372, 60.8%). This trend was observed among women randomised to receive progesterone and women randomised to receive placebo. In the subgroup of women with three previous miscarriages, the RR of live birth after at least 24 weeks was 1.01 (95% CI 0.89 to 1.14) when progesterone was compared with placebo. In the subgroup of women with more than three previous miscarriages, the RR of live birth after at least 24 weeks was 1.09 (95% CI 0.92 to 1.28) when progesterone was compared with placebo. A chi-squared test for interaction was not significant (χ2 = 0.41; p = 0.52).
Polycystic ovaries
Another subgroup analysis was performed by dividing our population into those women with polycystic ovaries and those without (see Table 6). We observed that women without polycystic ovaries who received progesterone experienced a higher rate of live birth after at least 24 weeks (245/369, 66.4%) than those with polycystic ovaries who received progesterone (17/29, 58.6%). However, we observed that women without polycystic ovaries who received placebo experienced a lower rate of live birth after at least 24 weeks (252/401, 62.8%) than those with polycystic ovaries who received placebo (19/27, 70.4%). In the subgroup of women without polycystic ovaries, the RR of live birth after at least 24 weeks was 1.06 (95% CI 0.95 to 1.17) when progesterone was compared with placebo. In the subgroup of women with polycystic ovaries, the RR of live birth after at least 24 weeks was 0.83 (95% CI 0.56 to 1.23) when progesterone was compared with placebo. A chi-squared interaction test for interaction was not significant (χ2 = 1.34; p = 0.25).
Gestation at treatment start
In order to explore any possible effect of gestational age at the time of commencing treatment, we conducted a post-hoc analysis comparing the outcomes of participants randomised at < 5 weeks with those of participants randomised at ≥ 5 weeks (see Table 6). We found no significant indication that women commencing treatment in the earlier stages of the first trimester experienced higher or lower rates of live birth after at least 24 weeks than those commencing treatment in the later stages. In the subgroup of women randomised at < 5 weeks, the RR of live birth after at least 24 weeks was 0.97 (95% CI 0.84 to 1.12) when progesterone was compared with placebo. In the subgroup of women randomised at ≥ 5 weeks, the RR of live birth after at least 24 weeks was 1.13 (95% CI 0.97 to 1.31) when progesterone was compared with placebo. A chi-squared test for interaction was not significant (χ2 = 2.03; p = 0.15).
Geographical location
We observed (see Table 6) that women in the UK experienced a slightly higher rate of live birth after at least 24 weeks (419/638, 65.7%) than those in the Netherlands (114/188, 60.6%). This trend was particularly observed in women randomised to receive progesterone [212/312 (67.9%) live births after at least 24 weeks vs. 50/86 (58.1%) live births after at least 24 weeks]. However, no significant subgroup effects were found (chi-squared test for interaction 1.20; p = 0.27).
Exploratory analyses
Maternal complications
Most of the PROMISE trial participants did not experience any antenatal or obstetric complications (Table 7). Overall, 17 women (2.1%) were diagnosed with pre-eclampsia. The rate observed in the progesterone group was 1.8% (7/398) and in the placebo group was 2.3% (10/428), giving a RR of 0.75 (95% CI 0.29 to 1.96), with a p-value of 0.56.
| Indicator | Progesterone | Placebo | Comparisons | Significance |
|---|---|---|---|---|
| Maternal complicationsa | ||||
| Pre-eclampsia | 7/398 (1.8) | 10/428 (2.3) | 0.75 (0.29 to 1.96) | p = 0.56 |
| Antepartum haemorrhage | 9/398 (2.3) | 14/428 (3.3) | 0.69 (0.30 to 1.58) | p = 0.38 |
| PPROM | 11/398 (2.8) | 9/428 (2.1) | 1.31 (0.55 to 3.14) | p = 0.54 |
| Mode of birthb | ||||
| Unassisted vaginal | 126/262 (48.1) | 158/274 (57.7) | 0.83 (0.71 to 0.98) | p = 0.03 |
| Instrumental vaginal | 44/262 (16.8) | 32/274 (11.7) | 1.43 (0.94 to 2.19) | p = 0.09 |
| Elective caesarean | 41/262 (15.6) | 36/274 (13.1) | 1.19 (0.79 to 1.80) | p = 0.41 |
| Emergency caesarean | 51/262 (19.5) | 48/274 (17.5) | 1.11 (0.78 to 1.59) | p = 0.56 |
| Birthweightb | ||||
| Live birthweight (g), mean (SD)c | (n = 260) 3213.65 (707.11) |
(n = 274) 3328.87 (635.40) |
Mean difference –115.23 (–229.71 to –0.74) | p = 0.05 |
| Adjusted live birthweight centile, mean (SD)c | (n = 260) 44.08 (30.58) |
(n = 274) 46.58 (28.91) |
Mean difference –2.50 (–7.57 to 2.56) | p = 0.33 |
| Small for gestational ageb | ||||
| Small for gestational aged | 45/260 (17.3) | 35/274 (12.8) | 1.35 (0.90 to 2.04) | p = 0.14 |
| Very small for gestational agee | 24/260 (9.2) | 18/274 (6.6) | 1.41 (0.78 to 2.53) | p = 0.26 |
| Other neonatal outcomesb | ||||
| Arterial cord pH of < 7.00 | 2/58 (3.4) | 1/54 (1.9) | 1.86 (0.17 to 20.0) | p = 0.61 |
| Venous card pH of < 7.00 | 1/55 (1.8) | 0/51 (0.0) | – | – |
| APGAR score at 1 minute < 7 | 22/257 (8.6) | 15/270 (5.6) | 1.54 (0.82 to 2.90) | p = 0.18 |
| APGAR score at 5 minutes < 7 | 3/257 (1.2) | 4/271 (1.5) | 0.79 (0.18 to 3.50) | p = 0.76 |
| Early infection | 9/260 (3.5) | 8/269 (3.0) | 1.16 (0.46 to 2.97) | p = 0.75 |
| Necrotising enterocolitis | 0/261 (0.0) | 0/270 (0.0) | – | – |
| Intraventraventricular haemorrhage (level 2) | 0/261 (0.0) | 1/270 (0.4) | – | – |
| Pneumothorax | 0/261 (0.0) | 3/270 (1.1) | – | – |
| Additional neonatal support requiredb | ||||
| Surfactant | 2/260 (0.8) | 3/269 (1.1) | 0.69 (0.12 to 4.09) | p = 0.68 |
| Ventilator support | 8/260 (3.1) | 8/269 (3.0) | 1.03 (0.39 to 2.72) | p = 0.94 |
| Discharge on oxygen | 0/260 (0.0) | 1/269 (0.4) | – | – |
| Median days of additional neonatal support required (if any)b | ||||
| IPPV | (n = 3) 2.0 (IQR 1.0–3.0) |
(n = 3) 3.0 (IQR 1.0–30.0) |
– | – |
| CPAP | (n = 5) 2.0 (IQR 2.0–3.0) |
(n = 8) 2.5 (IQR 1.0–4.0) |
– | – |
| Oxygen | (n = 6) 1.0 (IQR 1.0–3.0) |
(n = 7) 30.0 (IQR 1.0–80.0) |
– | – |
Antepartum haemorrhage was similarly observed; the rate ocurring in the progesterone group was 2.3% (9/398) and in the placebo group was 3.3% (14/428), giving a RR of 0.69 (95% CI 0.30 to 1.58), with a p-value of 0.38 (see Table 7).
Overall, 20 (2.4%) cases were diagnosed with preterm prelabour rupture of membrane. The rate observed in the progesterone group was 2.8% (11/398) and in the placebo group was 2.1% (9/428), giving a RR of 1.31 (95% CI 0.55 to 3.14), with a p-value of 0.54 (see Table 7).
Mode of delivery
Table 7 shows that, overall, more than half of the babies born alive after at least 24 weeks were delivered vaginally without instrumental assistance (284/536, 53.0%). In the group randomised to receive progesterone, 126 out of 262 (48.1%) were delivered in this way. The corresponding proportion in the placebo group was 57.7% (158/274). We observed a RR of 0.83 (95% CI 0.71 to 0.98) between the progesterone and placebo groups (p = 0.03).
A higher proportion (44/262, 16.8%) of the babies born to women receiving progesterone than to those receiving placebo (32/274, 11.7%) underwent an instrumental vaginal delivery. We observed a RR of 1.43 (95% CI 0.94 to 2.19) between the progesterone and placebo groups (p = 0.09).
Among 536 babies born alive after at least 24 weeks and for whom appropriate delivery data were collected, 77 (14.4%) were delivered by elective caesarean section. In the progesterone group, 41 out of 262 (15.6%) were delivered by elective caesarean section. In the placebo group, 36 out of 274 (13.1%) were delivered by elective caesarean section. We observed a RR of 1.19 (95% CI 0.79 to 1.80) between the progesterone and placebo groups (p = 0.41).
In the progesterone group, 51 out of 262 (19.5%) babies were born by emergency caesarean section, while in the placebo group 48 out of 274 (17.5%) babies were delivered in this way. The RR between the progesterone and placebo groups experiencing emergency caesarean section was 1.11 (95% CI 0.78 to 1.59; p = 0.56).
Birthweight and small for gestational age
Table 7 shows that among 260 babies born alive after at least 24 weeks of gestation to participants who were randomised to receive progesterone in the PROMISE trial, the mean birthweight was 3214 g (SD 707 g). Among 274 babies born to participants who were randomised to receive placebo, we observed a mean birthweight of 3329 g (SD 635 g). After adjustment for maternal height, weight (within the healthy range of BMI 18.5–30.0 kg/m2), ethnicity, parity and neonatal gender and gestational age at delivery,66 there were no significant differences between the mean birthweights of babies born in the different arms of the study.
Among the 534 neonates from whom appropriate data were collected, 80 (15.0%) were small for gestational age (see Table 7). The rate observed in the progesterone group was 17.3% (45/260) and in the placebo group was 12.8% (35/274), giving a RR of 1.35 (95% CI 0.90 to 2.04), with a p-value of 0.14. Across the same cohort, 42 (7.9%) were very small for gestational age. The proportion observed in the progesterone group was 9.2% (24/260) and in the placebo group was 6.6% (18/274), giving a RR of 1.41 (95% CI 0.78 to 2.53), with a p-value of 0.26.
Neonatal outcomes
Other neonatal outcomes of babies born during the PROMISE trial, including arterial and venous cord pH measurements and APGAR scores, are listed in Table 7. Outcomes of clinical concern were rare, both overall and within each arm of the trial.
Among the babies from whom neonatal data were immediately collected, the rate of arterial cord pH of < 7 was 3.4% (2/58) in the progesterone group and 1.9% (1/54) in the placebo group, giving a RR of 1.86 (95% CI 0.17 to 20.0), with a p-value of 0.61.
Among the same cohort, the proportion of babies with venous cord pH of < 7 was 1.8% (1/55) in the progesterone group and 0.0% (0/51) in the placebo group.
Among the live babies born to PROMISE participants receiving progesterone and from whom APGAR scores were collected, the rate of APGAR score < 7 at 1 minute was 8.6% (22/257). The rate of APGAR score < 7 at 1 minute observed in the placebo group was 5.6% (15/270), giving a RR of 1.54 (95% CI 0.82 to 2.90), with a p-value of 0.18.
Among the live babies born to PROMISE participants receiving progesterone and from whom APGAR scores were collected, the rate of APGAR score < 7 at 5 minutes was 1.2% (3/257). The rate observed in the placebo group was 1.5% (4/271), giving a RR of 0.79 (95% CI 0.18 to 3.50), with a p-value of 0.76.
Neonatal complications
We did not observe any significant differences in the rates of early infection or other complications experienced by neonates born to PROMISE participants who received progesterone compared with neonates born to those who received placebo (see Table 7). In the former group, 9 out of 260 (3.5%) babies from whom data were available acquired early infection, and no babies were diagnosed with necrotising enterocolitis, intraventraventricular haemorrhage (level 2) or pneumothorax. In the latter group, 8 out of 269 (3.0%) babies from whom data were available acquired early infection, no babies (out of 270) were diagnosed with necrotising enterocolitis, 1 baby out of 270 (0.4%) was diagnosed with intraventraventricular haemorrhage (level 2) and 3 out of 270 babies (1.1%) suffered pneumothorax. When considering early infection, we observed a RR between the progesterone and placebo groups of 1.16 (95% CI 0.46 to 2.97), with a p-value of 0.75.
There were few requirements for neonatal support among our study population (see Table 7). From the trial arm allocated to receive progesterone, 2 out of 260 (0.8%) babies required surfactant. From the trial arm allocated to receive placebo, 1.1% (3/269) babies required this support. Thus, we calculated a RR of 0.69 (95% CI 0.12 to 4.09), with a p-value of 0.68.
Among the same cohort of neonates, 16 (3.0%, eight babies in each group) required ventilator support. We calculated a RR of 1.03 (95% CI 0.39 to 2.72; p = 0.94).
Altogether, six babies born to participants in the PROMISE trial required intermittent positive pressure ventilation support. Among the three infants in the progesterone group, the median duration of the requirement was 2 days (IQR 1.0–3.0 days). Among the three infants in the placebo group, the median duration of the requirement was 3 days (IQR 1.0–30.0 days). Altogether, 13 babies born to participants in the PROMISE trial required continuous positive airway pressure support: among the five babies born to women allocated to receive progesterone, the median duration of the requirement was 2 days (IQR 2.0–3.0 days); among the eight babies born to women allocated to receive placebo, the median duration of the requirement was 2.5 days (IQR 1.0–4.0 days). We also observed that 13 babies born to participants in the PROMISE trial required oxygen support: among the six infants in the progesterone group, the median duration of the requirement was a single day (IQR 1.0–3.0 days); among the seven infants in the placebo group, the median duration of the requirement was 30 days (IQR 1.0–80.0 days). One baby in the placebo group was also discharged on oxygen.
Harms
The PROMISE trial incurred only one SUSAR, namely development of a rash by a single participant for whom study medication was discontinued and antihistamines were prescribed. The event was reported to regulatory authorities as appropriate and the rash subsided within 48 hours. Otherwise, AEs were few in both arms of the trial and not in excess of expected complications of pregnancy among women with a history of RM. Based on these records (Table 8), it would appear that progesterone in the form of vaginal capsules and at the dose level of 400 mg twice daily is a safe drug to use in early pregnancy.
| Category | Progesterone (N =404), n (%) | Placebo (N = 432), n (%) |
|---|---|---|
| Allergy | 2 (0.5) | 0 (0.0) |
| Dermatological | 1 (0.2) | 3 (0.7) |
| Gastrointestinal | 20 (5.0) | 14 (3.2) |
| Haematological | 0 (0.0) | 1 (0.2) |
| Neurological | 7 (1.7) | 4 (0.9) |
| Urological | 3 (0.7) | 4 (0.9) |
| Miscellaneous | 8 (2.0) | 4 (0.9) |
Chapter 4 Health economics
This chapter reports the health economic evaluation that was conducted alongside the PROMISE trial.
To ascertain the total financial cost associated with treatment, an economic analysis was conducted alongside the PROMISE trial, with data to show the number of days that treatment (progesterone) was received, and three categories of heath service resource use captured within the trial (antenatal contacts, pregnancy loss management for pregnancies ending before 24 weeks of gestation and mode of delivery for pregnancies proceeding beyond 24 weeks).
Following randomisation, women in the first trimester (and with a history of unexplained RM) were allocated either progesterone therapy (400-mg capsules twice daily) or placebo. The initiation of treatment was intended to occur following a positive pregnancy test (and no later than 6 weeks of gestation), to be continued until 12 weeks of gestation. The number of days of receiving capsules (progesterone or placebo) was calculated as the difference between the trial intervention start date and the trial intervention end date per participant.
Resource usage by treatment group was recorded during the gestation period, at labour and for postnatal admissions in the trial records. At gestational age 6–8 weeks, a standard ultrasound scan to confirm clinical pregnancy was undertaken. At circa 12 weeks, a specialised ‘booking’ ultrasound scan recorded viability and other variables. Other specific types of service contact were captured during the gestation period, including antenatal visits, day assessments and emergency visits (additional ultrasound scans, routine observation and diagnostic procedures, respectively). Participants’ total numbers of antenatal inpatient admissions and total antenatal lengths of stay were also recorded.
Before 24 weeks of gestation, the management of pregnancy loss was categorised as spontaneous resolution, expectant management, medical management or surgical management. After 24 weeks of gestation, the end of each pregnancy was recorded with a mode of delivery as either unassisted vaginal, instrumental vaginal, elective caesarean or emergency caesarean. The onset of delivery was recorded as spontaneous, induced, augmented or pre-labour caesarean. Other variables indicated whether or not there were any intrapartum complications, which for the purpose of this analysis were treated as a binary outcome to indicate whether or not there were any complications. To reflect modes of birth reported in the NHS Reference Costs,67 mode of delivery, onset of delivery and intrapartum complications were utilised to form 12 categories of Healthcare Resource Groups (HRGs) of modes of birth (see Valuation of resource use).
Our analysis of HRGs related to mode of delivery included costs of general postnatal admissions. Therefore, more specific information about general postnatal admissions was not considered necessary. However, above general admissions, we considered maternal admissions into critical care, recorded as admissions to HDUs or ICUs. Neonatal admissions were calculated as nights recorded in SCBUs or NNUs by each pregnancy.
The average resource requirements across the items considered for the economic analysis (such as days receiving treatment, antenatal contacts, pregnancy loss management and mode of delivery) are reported in Table 9. Outcome collection was expected at 6–8 weeks of gestation and at 12 weeks of gestation but, for various reasons (such as variation in date of enrolment and miscarriage before these contacts), the table shows the proportions reported as receiving these contacts.
| Resource items | Progesterone | Placebo | ||
|---|---|---|---|---|
| Mean (SD) | n | Mean (SD) | n | |
| Days receiving capsules (progesterone or placebo) | 39.2 (20.0) | 397 | 39.3 (20.9) | 423 |
| Outcome point 1 (6–8 weeks) | 0.8 (0.4) | 397 | 0.8 (0.4) | 423 |
| Outcome point 2 (12 weeks) | 0.7 (0.5) | 397 | 0.7 (0.5) | 423 |
| Antenatal visits | 6.9 (6.7) | 397 | 6.5 (6.4) | 423 |
| Day assessments | 0.8 (2.0) | 397 | 0.9 (2.6) | 423 |
| Emergency visits | 0.4 (0.9) | 397 | 0.4 (1.1) | 423 |
| Number of antenatal admissions | 0.3 (0.6) | 397 | 0.2 (0.7) | 423 |
| Antenatal length of stay (nights) | 0.8 (2.8) | 397 | 0.7 (2.6) | 423 |
| Miscarriage management | ||||
| Spontaneous resolution | 0.6 (0.5) | 134 | 0.7 (0.5) | 152 |
| Expectant management | 0.0 (0.1) | 134 | 0.0 (0.1) | 152 |
| Medical management | 0.1 (0.3) | 134 | 0.1 (0.3) | 152 |
| Surgical management | 0.2 (0.4) | 134 | 0.2 (0.4) | 152 |
| Mode of delivery | ||||
| Normal delivery with CC | 0.0 (0.2) | 259 | 0.1 (0.3) | 271 |
| Normal delivery without CC | 0.3 (0.5) | 259 | 0.3 (0.5) | 271 |
| Normal delivery with induction, with CC | 0.0 (0.2) | 259 | 0.0 (0.2) | 271 |
| Normal delivery with induction, without CC | 0.1 (0.3) | 259 | 0.1 (0.4) | 271 |
| Assisted delivery with CC | 0.0 (0.2) | 259 | 0.0 (0.2) | 271 |
| Assisted delivery without CC | 0.0 (0.2) | 259 | 0.0 (0.2) | 271 |
| Assisted delivery with induction, with CC | 0.0 (0.2) | 259 | 0.0 (0.2) | 271 |
| Assisted delivery with induction, without CC | 0.0 (0.2) | 259 | 0.0 (0.2) | 271 |
| Planned lower-uterine caesarean with CC | 0.2 (0.4) | 259 | 0.1 (0.3) | 271 |
| Planned lower-uterine caesarean without CC | 0.0 (0.2) | 259 | 0.0 (0.2) | 271 |
| Emergency or upper-uterine caesarean with CC | 0.0 (0.1) | 259 | 0.0 (0.1) | 271 |
| Emergency or upper-uterine caesarean without CC | 0.1 (0.3) | 259 | 0.1 (0.3) | 271 |
| Postnatal emergency admissions | ||||
| Maternal admission to HDU (nights) | 0.1 (1.0) | 393 | 0.1 (0.4) | 420 |
| Maternal admission to ICU (nights) | 0.0 (0.0) | 393 | 0.0 (0.6) | 420 |
| NNU or SCBU admission (nights) | 0.8 (4.4) | 394 | 0.8 (4.8) | 421 |
Valuation of resource use
Table 10 provides the unit costs identified to attribute values to items of resource use as observed within the PROMISE trial.
| Resource items | Unit cost (£) | Reference |
|---|---|---|
| Progesterone (Utrogestan®) 200 mg, 21 vaginal capsules | 21.00 | BNF 201431 |
| Antenatal contacts | ||
| Standard ultrasound scan (6–8 weeks) | 109.00 | NHS reference costs 2011–12 (NZ21Z)67 |
| Specialised ultrasound scan (12 weeks) | 121.00 | NHS reference costs 2011–12 (NZ22Z)67 |
| Antenatal visits (standard ultrasound scan) | 109.00 | NHS reference costs 2011–12 (NZ21Z)67 |
| Day assessments (routine observation) | 571.00 | NHS reference costs 2011–12 (NZ16Z)67 |
| Emergency visits (antenatal diagnostic procedures) | 133.00 | NHS reference costs 2011–12 (NZ23Z)67 |
| Antenatal inpatient admissions (per night) | 264.00 | NHS reference costs 2011–12 (extra bed-day)67 |
| Miscarriage management | ||
| Spontaneous resolution | 554.00 | NHS reference costs 2011–12 (MB08Z)67 |
| Expectant management | 1033.97 | aNICE guidelines 201268 |
| Medical management | 1421.88 | aNICE guidelines 201268 |
| Surgical management | 1706.82 | aNICE guidelines 201268 |
| No details available | 554.00 | Assumed spontaneous resolution |
| HRG mode of birth | ||
| Normal delivery with CC | 1603.03 | NHS reference costs 2011–12 (NZ11A)67 |
| Normal delivery without CC | 1292.04 | NHS reference costs 2011–12 (NZ11B)67 |
| Normal delivery with induction, with CC | 2315.42 | NHS reference costs 2011–12 (NZ11E)67 |
| Normal delivery with induction, without CC | 1750.61 | NHS reference costs 2011–12 (NZ11F)67 |
| Assisted delivery with CC | 2233.05 | NHS reference costs 2011–12 (NZ12A)67 |
| Assisted delivery without CC | 1762.08 | NHS reference costs 2011–12 (NZ12B)67 |
| Assisted delivery with induction, with CC | 2924.16 | NHS reference costs 2011–12 (NZ12E)67 |
| Assisted delivery with induction, without CC | 2293.84 | NHS reference costs 2011–12 (NZ12F)67 |
| Planned lower-uterine caesarean with CC | 3041.26 | NHS reference costs 2011–12 (NZ13A)67 |
| Planned lower-uterine caesarean without CC | 2596.22 | NHS reference costs 2011–12 (NZ13B)67 |
| Emergency or upper-uterine caesarean with CC | 3789.24 | NHS reference costs 2011–12 (NZ14A)67 |
| Emergency or upper-uterine caesarean without CC | 3288.24 | NHS reference costs 2011–12 (NZ14B)67 |
| Postnatal emergency admissions | ||
| Maternal admission to HDU | 629.91 | NHS reference costs 2011–12 (XC07Z)67 |
| Maternal admission to ICU | 1209.28 | NHS reference costs 2011–12 (XC06Z to XXC01Zb)67 |
| Neonatal admission (NNU/SCBU) | 440.00 | NHS reference costs 2011–12 (XA05Z)67 |
The costs of treatment using progesterone (Utrogestan®) capsules were identified in the BNF31 as £21.00 for progesterone 200 mg, 21 vaginal capsules (or £1.00 per 200 mg capsule). The PROMISE treatment protocol stated that two capsules of 200 mg (400 mg) were to be taken twice daily. The cost per participant per day was, therefore, £4.00.
The costs of antenatal contacts were obtained from the NHS Reference Costs 2011–12, and for the cost of each antenatal admission night, the average cost of an excess bed-day was provided in a summary report by the NHS Reference Costs 2011–12. 67
Costs of pregnancy-loss management were not provided in the NHS Reference Costs 2011–12,67 with the exception of spontaneous resolution. Therefore, the other costs of pregnancy-loss management were identified from other sources. 68 Specifically, costs for expectant management, medical management and surgical management in the UK for the price year 2001–2 were estimated by another study in 2006, and a 3.5% time discount rate was adopted to estimate values for the 2011–12 price year. 69
Modes of delivery in the NHS Reference Costs 2011–12 were covered by 12 HRGs, broadly categorising each birth as a normal vaginal delivery, an assisted vaginal delivery or a caesarean section, with each category further divided by whether or not there were complications. Normal and assisted vaginal deliveries were further divided by whether or not pregnancy was induced.
As postnatal maternal admissions were recorded as either to HDU or to ICU, together amalgamated into the single entity of ‘critical care’,71 assumptions were required to associate correct types of service use with these new HRGs. HDU was proxied by the HRG ‘adult critical care, zero organs supported’ and for ICU a weighted average was taken across the six higher-intensity ‘adult critical care’ HRGs, where one or more organs were supported.
Table 11 provides (for each treatment group) mean costs of all items of resource use, with the subtotal costs for each of five categories of resources (treatment, antenatal contacts, miscarriage management, mode of delivery and postnatal emergency admissions) and the average total cost per group.
| Resource items | Progesterone | Placebo | ||
|---|---|---|---|---|
| Mean (£) | n | Mean (£) | n | |
| Treatment using vaginal capsules | 156.88 | 397 | 0.00 | 423 |
| Antenatal contacts | ||||
| Outcome point 1 (6–8 weeks) | 87.86 | 397 | 84.00 | 423 |
| Outcome point 2 (12 weeks) | 84.12 | 397 | 82.10 | 423 |
| Antenatal visits | 752.02 | 397 | 703.48 | 423 |
| Day assessments | 474.63 | 397 | 517.00 | 423 |
| Emergency visits | 52.93 | 397 | 54.39 | 423 |
| Antenatal admissions | 208.14 | 397 | 181.62 | 423 |
| Subtotal for all antenatal contacts | 1659.71 | 397 | 1622.59 | 423 |
| Miscarriage management | ||||
| Spontaneous resolution | 359.69 | 134 | 379.05 | 152 |
| Expectant management | 15.43 | 134 | 20.41 | 152 |
| Medical management | 159.17 | 134 | 177.74 | 152 |
| Surgical management | 382.13 | 134 | 291.96 | 152 |
| Subtotal for all miscarriage management | 916.41 | 134 | 869.15 | 152 |
| HRG mode of birth | ||||
| Normal delivery with CC | 61.89 | 259 | 130.14 | 271 |
| Normal delivery without CC | 389.11 | 259 | 414.79 | 271 |
| Normal delivery with induction, with CC | 53.64 | 259 | 93.98 | 271 |
| Normal delivery with induction, without CC | 223.05 | 259 | 245.47 | 271 |
| Assisted delivery with CC | 103.46 | 259 | 90.64 | 271 |
| Assisted delivery without CC | 68.03 | 259 | 39.01 | 271 |
| Assisted delivery with induction, with CC | 124.19 | 259 | 75.53 | 271 |
| Assisted delivery with induction, without CC | 88.57 | 259 | 59.25 | 271 |
| Planned lower-uterine caesarean with CC | 504.92 | 259 | 404.01 | 271 |
| Planned lower-uterine caesarean without CC | 70.17 | 259 | 95.80 | 271 |
| Emergency or upper-uterine caesarean with CC | 73.15 | 259 | 55.93 | 271 |
| Emergency or upper-uterine caesarean without CC | 431.66 | 259 | 388.28 | 271 |
| Subtotal for all HRG modes of delivery | 2191.84 | 259 | 2092.83 | 271 |
| Postnatal emergency admissions | ||||
| Cost of maternity HDU | 120.00 | 393 | 63.34 | 420 |
| Cost of maternity ICU | 0.00 | 393 | 28.50 | 420 |
| Cost of neonatal (NNU or SCBU) admissions | 357.36 | 394 | 345.94 | 421 |
| Total cost | 4062.26 | 393 | 3730.10 | 420 |
Based on the average number of days for which participants received treatment with progesterone capsules, the average cost of care was calculated to be £156.82 per pregnancy. Treatment according to the study protocol commenced at no later than 6 weeks’ gestation and continued until 12 weeks of gestation unless miscarriage occurred before this time, which would equate to 42 days and an expected cost of £168.00 per continuing pregnancy. The mean cost indicated by our trial data is lower because it is a calculation of the cost up to either 12 weeks or the time of miscarriage. Although this provides an accurate indication of the consumption cost of treatment, in a real-world setting capsules could be allocated for the expected treatment period (from 4–6 weeks until 12 weeks) and therefore prescription costs could be considered fixed; such scenarios are explored later in this chapter using a sensitivity analysis.
Costs associated with antenatal contacts appeared marginally higher overall among women receiving progesterone than among women receiving placebo. For all items (except day assessments and emergency visits) mean costs were higher in the progesterone group than in the placebo group.
Miscarriage management was, except in the case of surgical management, less expensive in the progesterone group. Surgical miscarriage of management showed a significantly higher average cost in the progesterone group. Costs associated with HRG modes of delivery were, overall, comparable, but with some notable variations by individual HRGs.
Few emergency admissions for mothers were observed, but each of these cases did incur a high tariff. There were no significant differences between treatment groups in the rates of neonatal admissions (progesterone 9.74% vs. placebo 9.73%), although newborns in the progesterone group experienced slightly higher lengths of stay in hospital than those in the placebo group.
Overall, mean total costs were higher in the progesterone group than in the placebo group (progesterone £4062.26 vs. placebo £3730.10). The incremental costs of treatment using progesterone were calculated to be £332.17, which represents 8.9% higher costs than usual care.
Health benefits: measure of effectiveness
The primary health outcome adopted by the economic analysis of the PROMISE trial was live birth after at least 24 weeks of gestation. The incremental effect was, therefore, the incremental probability of an additional live birth after at least 24 weeks of gestation. Table 12 summarises the expected number of live births beyond 24 weeks by treatment group. To estimate cost-effectiveness from the joint distributions of individuals’ total costs and outcomes, the health benefits were estimated in individuals where total cost was not missing.
| Treatment group | Mean | Median | SD | Minimum | Maximum | n |
|---|---|---|---|---|---|---|
| Unadjusted primary outcome | ||||||
| Placebo | 0.633 | 1 | 0.483 | 0 | 1 | 428 |
| Progesterone | 0.658 | 1 | 0.475 | 0 | 1 | 398 |
| Adjusted primary outcome for complete-case analysisa | ||||||
| Placebo (EC) | 0.638 | 1 | 0.481 | 0 | 1 | 420 |
| Progesterone (EI) | 0.657 | 1 | 0.476 | 0 | 1 | 393 |
where Δ represents change, E represents the effects, and subscripts I and C refer to the intervention and control arms, respectively.
The adjusted incremental difference in the mean probability of a live birth beyond 24 weeks of gestation was 0.0184.
Logistic regression indicated that the adjusted ΔE had a p-value of 0.583 (illustrating substantial uncertainty in the expected incremental benefit); this highlights the importance of referring to the analysis of uncertainty [such as with a cost-effectiveness acceptability curve (CEAC)] while interpreting estimates of cost-effectiveness (see the following section).
Cost-effectiveness and uncertainty
The primary cost-effectiveness outcome of the PROMISE study was cost per additional live birth after at least 24 weeks of gestation. To inform the decision-maker, the incremental cost-effectiveness ratio (ICER) is the ratio of the difference in cost (C) to the differences in difference in effect (E), denoted by the formula:
where Δ represents change, C represents the costs, E represents the effects, and subscripts I and C refer to the intervention and control arms, respectively.
The cost to achieve an additional live birth after at least 24 weeks was calculated to be £18,053. In the UK, a value of £20,000–30,000 per quality-adjusted life-year (QALY) is considered to be an acceptable value for adopting a health-care technology. 70 However, there remains unsettled debate about the valuation of prenatal life in economic evaluations. 72 The generic health gains in terms of QALYs are reported in Sensitivity analyses, Estimation of quality-adjusted life-years and costs beyond the trial end point.
Uncertainty around the mean statistic for both costs and outcomes is a commonplace feature of economic evaluations. A key component of any evaluation is to demonstrate this uncertainty and the robustness of the cost-effectiveness ratio calculated from the initial main trial analysis. A non-parametric bootstrap (with 50,000 replications) was performed to demonstrate uncertainty in the joint distribution of cost and effects in the PROMISE trial. Figure 9 provides a visual representation of the degree of uncertainty in the ICER on a cost-effectiveness plane.
FIGURE 9.
Cost-effectiveness plane (50,000 bootstrap replications). GBP, Great British pound.
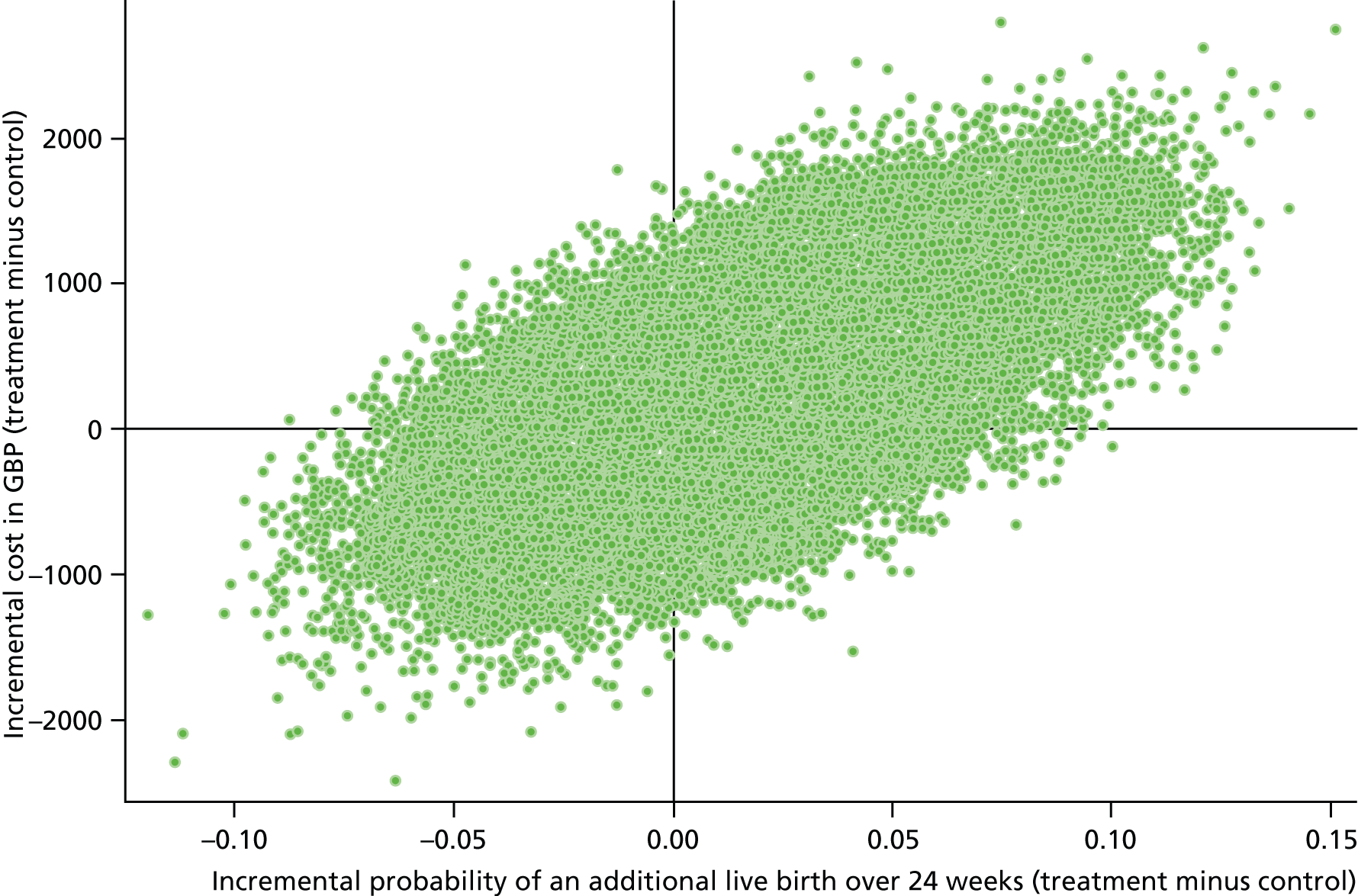
The bootstrap estimates that the 95% CI of the incremental probability of an additional live birth beyond 24 weeks of gestation ranges from –0.0477 to 0.0845. Based on the current level of uncertainty, it might also be concluded that there is a 29.30% chance that progesterone may result in a decrease in the probability of a live birth beyond 24 weeks of gestation, compared with usual care. Nevertheless, 17.16% of the bootstrap replications fell within the bottom-left quadrant, where less effect also resulted in lower cost.
The top-right quadrant of the cost-effectiveness plane contained 59.31% of all replications. Although overall there is substantial uncertainty, this may suggest that treatment was most likely to cost more and have some additional benefit.
Given the uncertainty in the current estimates of the cost per additional live birth beyond 24 weeks of gestation, a decision-maker may wish to examine how the probability that progesterone would be the most cost-effective option will vary with an increasing willingness to pay.
Figure 10 presents the CEAC to illustrate how the probability of treatment being cost-effective changes as the willingness to pay increases.
FIGURE 10.
Cost-effectiveness acceptability curve.
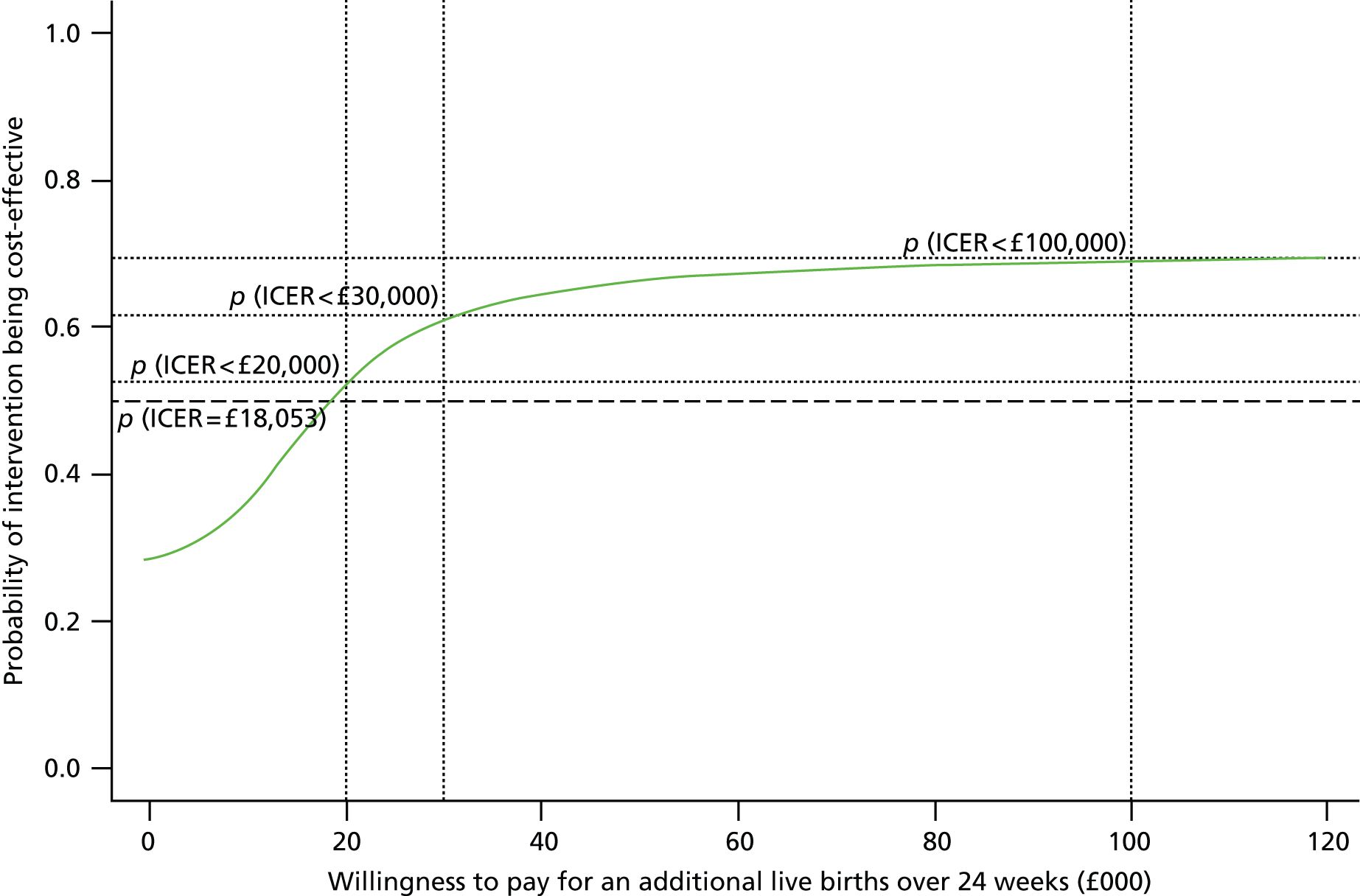
At a willingness to pay of £100,000 per additional live birth beyond 24 weeks of gestation, the probability that treatment would be cost-effective asymptotes [p(ICER < 100,000) = 0.7203]. This raises the question as to what society would be willing to pay to increase the likelihood of an additional live birth beyond 24 weeks of gestation.
Sensitivity analyses
Fixed treatment cost until 12 weeks
In the base-case cost-effectiveness analysis, the direct cost of treatment with progesterone accurately accounted for the total number of days of treatment received by individuals (assuming, as was the case during the trial, that surplus capsules would be returned either at 12 weeks or in the event of miscarriage). Figure 11 illustrates trends in the variation of stages of pregnancy that treatment started and stopped. The variable trialstop illustrates a bimodal distribution relating to whether pregnancy proceeded beyond 12 weeks or treatment was withdrawn as a result of miscarriage.
FIGURE 11.
Distribution in points of gestation where treatment (a) started; and (b) stopped.

If this trial was to be replicated in a real-world setting, it is unlikely that surplus medication prescribed up to an anticipated treatment end point (12 weeks) would be returned and reused. Our sensitivity analysis illustrates the implications that treatment would be prescribed up to a fixed end point (12 weeks of gestation) while still allowing the observed variation in treatment initiation (trialstart).
Repeating the non-parametric bootstrap procedure to re-estimate the ICER and CEAC with respect to this change in the expected cost of treatment generated a predictable increase in the ICER to £22,256 per additional live birth beyond 24 weeks of gestation. Figure 12 illustrates a marginal decrease in the likelihood that treatment would be cost-effective as willingness to pay increases.
FIGURE 12.
Cost-effectiveness acceptability curve: treatment costs fixed until 12 weeks.

Estimation of quality-adjusted life-years and costs beyond the trial end point
The base-case analysis adopted a condition-specific end point to inform clinical choices of alternative strategies intended to increase the likelihood of a viable birth (live birth beyond 24 weeks of gestation). However, there are limitations of condition-specific outcomes to inform health-care policy. Furthermore, it could be argued that the binary outcome of live birth (or not) beyond 24 weeks of gestation, although providing some indication of life-years gained, does not provide any information on the quality of life achieved from treatment.
Expressing the health benefits in terms of QALYs provides more information to guide the allocation of a limited pool of health-care resources based on explicit monetary values per QALY. To explore methods by which the clinical outcomes might be translated into generic health benefits (such as QALYs), a systematic search was undertaken to identify and project the longer-term costs and consequences, and several potential modelling strategies were explored.
A Finnish study provided the most suitable basis for a strategy to express gains in generic health terms (such as QALYs);73 to our knowledge this is the only study to date to evaluate the effects of gestational age on QALYs. It estimated QALYs up to 4 years of age using the 17-dimension parental questionnaire, reported with expected variations in the 4-year QALY by gestational age at birth. 74
By utilising gestational age at delivery within the PROMISE trial to serve as a surrogate outcome for QALYs, QALYs per participant were deduced. However, the estimates of QALYs reported by the Finnish study truncated QALYs at 32 weeks of gestation. Consultation with clinical content experts suggested that generic health gains (QALYs) could be most reasonably assumed to linearly increase up to a gestational age of 34 weeks at birth and, based on this assumption, linear predictors of the expected QALYs for weeks 24–34 were estimated using an ordinary least squares regression. QALYs after 34 weeks showed no additional increase. Table 13 summarises the data on expected QALYs that were used to inform projected health gains up to 4 years of age by gestational age at birth.
| Gestational age at birth (weeks) | Linear predictor of QALYs up to 4 years of age |
|---|---|
| 22 | 3.6058 |
| 23 | 3.6250 |
| 24 | 3.6441 |
| 25 | 3.6633 |
| 26 | 3.6824 |
| 27 | 3.7016 |
| 28 | 3.7207 |
| 29 | 3.7399 |
| 30 | 3.7590 |
| 31 | 3.7782 |
| 32 | 3.7973 |
| 33 | 3.8165 |
| 34 | 3.8356 |
| 35 | 3.8356 |
| 36 | 3.8356 |
| 37 | 3.8356 |
| 38 | 3.8356 |
| 39 | 3.8356 |
| 40 | 3.8356 |
The expected QALYs were applied to PROMISE records of gestational age at birth. In a situation of postnatal infant mortality (up to 28 days), the trial captured mortality date and adjusted individual QALYs to the date of mortality.
Costs beyond initial hospitalisation (at birth) were projected by utilising a data set of individual records supplied from the Oxford Records Linkage Study by Dr Stavros Petrou. 75 This data set provided a sample of 95,635 births in the UK, and their associated costs for the first year after initial hospitalisation (excluding costs associated with the initial admission related to pregnancy). Price years recorded in the data set ranged between 1979 and 1988; a time discount rate of 3.5% was applied to obtain 2011 values. Dummy variables representing weeks of gestation at birth from 23 to 44 weeks were generated. The cost for the first year after initial hospitalisation was regressed against these variables to estimate the magnitude of first year costs by gestational age at birth.
Table 14 provides regression outputs in which coefficients of each gestational age at birth show the expected additional costs following initial hospitalisation.
| Gestation at birth (weeks) | Coefficient | 95% CI | Standard error | p-value |
|---|---|---|---|---|
| 23 | 121,636.10 | 18,538.74 to 798,077.00 | 116,746.20 | < 0.001 |
| 24 | 39,776.66 | 21,247.04 to 74,466.03 | 12,725.98 | < 0.001 |
| 25 | 57,774.52 | 36,609.02 to 91,176.86 | 13,449.13 | < 0.001 |
| 26 | 55,784.84 | 39,094.91 to 79,599.82 | 10,118.58 | < 0.001 |
| 27 | 61,814.74 | 47,116.21 to 81,098.68 | 8563.54 | < 0.001 |
| 28 | 46,018.71 | 36,168.45 to 58,551.65 | 5655.27 | < 0.001 |
| 29 | 44,602.67 | 36,281.46 to 54,832.37 | 4699.00 | < 0.001 |
| 30 | 34,573.18 | 28,697.57 to 41,651.77 | 3285.67 | < 0.001 |
| 31 | 29,243.02 | 24,680.64 to 34,648.79 | 2530.79 | < 0.001 |
| 32 | 25,121.59 | 22,007.13 to 28,676.80 | 1696.52 | < 0.001 |
| 33 | 21,830.22 | 19,561.84 to 24,361.64 | 1222.01 | < 0.001 |
| 34 | 15,688.96 | 14,354.70 to 17,147.23 | 711.46 | < 0.001 |
| 35 | 12,204.82 | 11,321.47 to 13,157.09 | 467.84 | < 0.001 |
| 36 | 10,072.11 | 9397.77 to 10,794.83 | 356.11 | < 0.001 |
| 37 | 7851.10 | 7419.87 to 8307.40 | 226.29 | < 0.001 |
| 38 | 7303.05 | 6994.23 to 7625.50 | 160.99 | < 0.001 |
| 39 | 5793.92 | 5585.30 to 6010.33 | 108.41 | < 0.001 |
| 40 | 5766.30 | 5575.62 to 5963.5 | 98.93 | < 0.001 |
| 41 | 5456.47 | 5246.34 to 5675.02 | 109.33 | < 0.001 |
| 42 | 6446.60 | 6046.69 to 6872.95 | 210.64 | < 0.001 |
| 43 | 6927.92 | 6199.75 to 7741.61 | 392.53 | < 0.001 |
| 44 | 6077.39 | 5260.80 to 7020.73 | 447.42 | < 0.001 |
| n = 95,635 | R2 = 0.2130 | |||
Gestational age at delivery served as a surrogate outcome to estimate QALYs (over 4 years) and additional health-care costs (over the first year), allowing the treatment effect to be estimated using regression analysis. Cost and QALY distributions were jointly estimated using Zellner’s seemingly unrelated regression,76 thereby allowing analysis to also account for correlation in the residuals. Table 15 presents the output of the regression.
| Variables | Coefficient | 95% CI | Standard error | p-value |
|---|---|---|---|---|
| Total cost | ||||
| Progesterone | 366.532 | –960.663 to 1693.728 | 677.153 | 0.588 |
| Constant | 8185.186 | 7257.909 to 9112.462 | 473.109 | < 0.001 |
| QALY | ||||
| Progesterone | 0.099 | –0.176 to 0.374 | 0.140 | 0.482 |
| Constant | 2.246 | 2.053 to 2.438 | 0.098 | < 0.001 |
| Correlation matrix of residuals | Total cost | QALY | ||
| Total cost | 1.000 | |||
| QALY | 0.649 | 1.000 | ||
First, examining the regression coefficients for progesterone indicated the incremental cost to be £366.53 and incremental QALY to be 0.0987, generating an ICER of £3712 per QALY. Subject to certain assumptions required to estimate the utility function (discussed below), this ICER could be considered to be highly cost-effective with respect to explicit decision rules for the reimbursement of health technologies in the UK; it falls well below the benchmark of between £20,000 and £30,000 that is suggested as an appropriate threshold by the National Institute for Health and Care Excellence (NICE). 70
Furthermore, significant correlation was observed between cost and QALY distribution within the estimated utility function (0.6487; p < 0.0001). To an extent, this correlation could be attributed to the common surrogate outcome of gestational age at delivery; lower gestational age would decrease neonatal health status and that would be correlated to higher health-care costs.
Figure 13 illustrates changes in probability that treatment may be cost-effective as the willingness to pay increases. The CEAC indicates a probability of between 0.7145 and 0.7341 to express the likelihood that prescription progesterone therapy in women with a history of unexplained RM falls within the NICE threshold.
FIGURE 13.
Cost-effectiveness acceptability curve: QALYs and cost projected beyond the trial end point.

Although the estimation of QALYs and first-year cost consequences to the NHS may be useful to policy-makers and decision-makers allocating global health-care budgets,77 it should be noted that there are several unavoidable limitations to consider when interpreting these results. First, the extrapolation of QALYs utilised data from Finland without accounting for any variation in the preferences for health state that may exist between countries. Second, incorporation of QALYs into the utility function essentially places values on prenatal life (zero QALYs for miscarriage) that may raise moral and ethical debates. Finally, no reliable method was identified to model the implications of miscarriage on individual health, or indeed health-care, utilisation.
Summary
-
The average total cost in the progesterone group was higher than in the placebo group (progesterone £4062.26 vs. placebo £3730.10), representing an incremental cost of £332.17 (8.9% higher costs than usual care).
-
The incremental difference in the mean probability of a live birth beyond 24 weeks of gestation (adjusted for complete case cost-effectiveness analysis) was 0.0184 (p = 0.583).
-
The ICER for the base case was £18,053 per additional live birth beyond 24 weeks of gestation.
-
Estimates of cost-effectiveness are highly uncertain (the probability of treatment being cost-effective should the decision-maker be willing to pay £100,000 per additional live birth beyond 24 weeks of gestation is 0.7203).
-
Using external data sources, the treatment ICER was estimated in terms of QALYs to be £3679 per QALY. This analysis suggests the probability that progesterone would fall within the NICE threshold (£20,000–30,000 per QALY) as between 0.7145 and 0.7341.
Chapter 5 Discussion
In the following discussion, we focus on the findings of the trial, highlighting the strengths and weaknesses of the study and generalisability of the findings in the context of available evidence. We conclude with our recommendations for clinical practice and further research.
Study strengths
This study is the largest-ever randomised placebo-controlled clinical trial to report on the treatment effects of first-trimester progesterone therapy for pregnant women with a history of unexplained RM. It is, in fact, the largest randomised clinical trial ever conducted on the subject of recurrent pregnancy loss. It has demonstrated that a large, high-quality, randomised trial of IMP can be successfully conducted in the challenging and emotive context of RM, with the help of hospital teams and patients from maternity services reflecting a wide range of clinical practice and research experience across the UK and in the Netherlands.
Internal validity
Randomisation and minimisation were effective in achieving balanced treatment allocations at baseline. Thus, the PROMISE trial optimised statistical power and eliminated selection and allocation bias. 78 A computer-generated allocation sequence, allocation concealment and blinding prevented investigators from knowing the assignment of the next participant based on prior treatment assignments. Possible confounding factors such as the number of previous miscarriages, maternal age and polycystic ovaries were similarly distributed between treatment groups. As a study with allocations blinded to participants and care providers, the PROMISE trial also avoided performance bias.
The research team believed that it was important to ensure that the study was large enough to detect clinically important treatment effects, and, therefore, the target sample size of the study was calculated to enable detection of a MID of 10% in rates of live birth after at least 24 weeks of gestation. We planned to randomise 790 women in total (395 participants in each of the progesterone and placebo arms) and we actually exceeded this number. Consequently, we consider the findings of this trial to be methodologically and statistically robust.
Our trial design offered a number of other strengths with respect to data collection and analysis. The treatment of participants by a large number of study centres and practitioners allowed intervention impact to be evaluated without confounding by individual variance in clinical practice and local technology. The outcome indicators that we measured were variables of routine familiarity in pregnancy care, so they were well understood and easily recorded by clinical researchers. Almost all of the outcome data recorded during the PROMISE study were objective outcomes (rather than subjective descriptions), and the study was blinded, so there was no risk of incurring assessor bias.
Contextual suitability
The PROMISE trial was built on a plausible biological basis and encouraging preliminary trial data, and directly answered calls from national bodies to address the possibility that progesterone therapy in early pregnancy could be beneficial to women with a history of unexplained RM. Although clinical equipoise existed, the lack of definitive knowledge or policy guidance for health services practitioners allowed considerable variations in care. The PROMISE trial answered an important question.
Other strengths
There is increasing recognition of the need for clinical research to embrace the views of both lay and professional stakeholders. 64 In the PROMISE trial, PPI occurred at stages of design, development and monitoring. For example, patients were surveyed to assess the acceptability of the intervention, and engaged in the development of literature for participants, and the TSC included representatives of patient advocacy organisations. We believe these roles were important to ensure appropriate communication with study participants and project oversight throughout the duration of the research.
The trial intervention was deliverable within the context of customary care without major impacts on health service structure. The mode of administration of IMP was designed to reflect the preferences expressed by patients, and most of our data collection could be performed during routine antenatal and postnatal appointments of the study participants.
Limitations and critique
This study is the largest randomised, placebo-controlled trial of progesterone ever conducted in RM, and we consider the trial to be methodologically robust. Nevertheless, it remains possible to observe some limitations.
The potential criticisms of our trial include (1) non-measurement of serum progesterone levels at the time of randomisation, (2) possibly suboptimal dose, (3) possible mistiming of treatment, (4) possibly insufficient duration of treatment, (5) possibly inappropriate pharmacological preparation or route of administration, (6) dilution of treatment effect by factors such as breaches of protocol and loss to follow-up, and (7) narrow and uncertain economic analyses. We examine these issues individually below.
-
Our collection of baseline data for the study population did not include serum progesterone levels at the time of randomisation. None of the existing four trials of first-trimester progesterone therapy for pregnant women with a history of unexplained RM had checked progesterone levels before treatment. Furthermore, serum or salivary progesterone measurements may not accurately reflect progesterone levels and effects at the feto–maternal interface. Finally, PROMISE investigators believed that it would be important to know whether or not progesterone therapy could assist women with RM regardless of their progesterone levels before commencing therapy.
-
The dosage of vaginal progesterone that was adopted by the PROMISE trial (400 mg twice daily) sits at the higher end of the spectrum of biological effect according to the SmPC and the BNF. Our choice was made after a careful review of the existing literature, an extensive survey of clinicians in the UK (see Chapter 1, Existing knowledge, Progesterone in clinical use for recurrent miscarriages) and other related evidence such as recommended dosages for luteal support in assisted conception. 16 Therefore, we believe that the outcome of the study is unlikely to be affected by the possibility of suboptimal dosage.
-
Some have suggested that progesterone supplementation could be more effective in reducing the risk of miscarriage if it is administered during the luteal phase of reproduction, prior to confirmation of pregnancy. 6,79–81 Although plausible, this was not the research question that the PROMISE trial set out to address. When the research question for the PROMISE trial was being discussed and debated, the evidence overwhelmingly suggested that the most useful question to answer would be on the effects of progesterone in the first trimester (not during the luteal phase). The evidence was summarised by a review from Cochrane82 and our update of this review,5 which identified four trials of progesterone supplementation during the first trimester, but none during the luteal phase. All four first-trimester trials suggested promising beneficial effects on miscarriage reduction (see Chapter 1, Existing knowledge, Progesterone in clinical use for recurrent miscarriages). Thus, the pursuit of this question was the most rational option, endorsed by a wide body of clinicians, patients and other stakeholders.
-
As many as 80% of miscarriages occur before 12 weeks,83 and pregnancies that continue beyond 12 weeks rarely miscarry, as observed in the PROMISE trial itself (see Chapter 3, Outcomes and estimation, Secondary outcomes). Corpus luteal function is replaced by placental production of progesterone before the end of the first trimester. Therefore, we consider that progesterone supplementation beyond the first trimester of pregnancy is unlikely to be associated with any further clinical benefit than may be postulated during this early stage. Moreover, the median age of gestation among PROMISE participants who experienced miscarriage was < 8 weeks (7.3 weeks in the progesterone group and 7.1 weeks in the placebo group).
-
An immunomodulatory effect of progesterone at the trophoblastic–decidual interface is the key presumed mechanism for preventing RM. 3,33–35 Our choice of administration via the vaginal route was, therefore, rational to deliver a greater proportion of drug to the relevant site (the uterus) using the ‘first uterine pass’ effect. 36,37 Furthermore, studies of vaginal progesterone in the prevention of preterm birth have shown its effectiveness when given via this route. 19,21,22 Therefore, we believe that any alternative preparation or route of administration is unlikely to result in a greater effect.
-
We recruited and followed up to completion more PROMISE participants than were calculated as necessary to detect a MID between the study treatment groups. Moreover, although we found pill counting to be a poor tool for compliance assessment, deviations were insufficient to compromise the validity of the study findings. The pragmatic nature of the trial intervention was also important to test its effect in everyday clinical practice.
-
The economic analysis has two main limitations. First, it could be suggested that the perspective taken on costs was narrow by solely focusing on perinatal resource use. Second, the economic modelling utilised to estimate wider costs and QALYs related to treatment rested on underlying assumptions, so the results should be interpreted with caution.
Interpretation
Our findings show that women with a history of unexplained RM do not benefit from first-trimester progesterone therapy for any of the key clinical outcomes that we observed. Importantly, our findings are not consistent with the findings of several smaller and poor-quality controlled studies that reported benefit from progesterone supplementation during the first trimester of pregnancy.
An updated review11 from Cochrane recognised the weaknesses of the trials that pre-dated the PROMISE trial. The review reported that the four trials in which participants had a previous history of three or more miscarriages (the same as participants in the PROMISE trial) ‘were of poorer methodological quality’, and called for further research.
The PROMISE trial has added to the available safety data regarding the use of progesterone during early pregnancy. The low rate of AEs observed among mothers and neonates of the PROMISE trial, consistent with the background rate, suggests that progesterone therapy in early pregnancy is safe. This is an important evidence finding because progesterone therapy is commonly used as part of assisted conception treatment.
Generalisability
Centres participating in the study were geographically spread across the UK and in the Netherlands, improving the generalisability of the results for women suffering RM. In PROMISE, exclusion criteria were kept to a minimum and the heterogeneity of the RM population was well reflected by trial participants. The study is, therefore, better applied to assess effectiveness than efficacy.
Chapter 6 Conclusions
In this chapter, we address considerations for the delivery of clinical services and recommendations for future research.
Implications for health care
The key findings of the PROMISE trial are clear and sufficiently generalisable to inform clinical practice. On the basis of the results of this study, first-trimester progesterone therapy does not appear to have clinically significant benefits in pregnant women with a history of unexplained RM. However, it is evident that progesterone at a dose of 400 mg twice daily appears safe to the mother and the fetus. This reassuring information is useful to women who may be taking progesterone for other reasons, for example as part of assisted conception treatment. Indeed, our finding that progesterone does not reduce the risk of miscarriage in pregnant women with a history of RM should not be construed as evidence of the performance of progesterone for other indications such as IVF treatment.
Our economic analysis indicated that prescribing progesterone increases mean total costs by £327.64. This may suggest that the marginal benefit observed justifies the marginal cost. However, the uncertainties must be considered. For example, should a decision-maker be willing to pay £100,000 to increase the chances of an additional live birth after at least 24 weeks of gestation, the probability that this strategy would be cost-effective is < 70%. Alternatively, extrapolating health gains in terms of QALYs suggests the probability that progesterone would fall within the NICE threshold of £20,000–£30,000 per QALY as between 0.7145 and 0.7341. As the magnitude of health gain seems much lower than previously anticipated and remains uncertain, further research may be advised.
Recommendations for research
In our opinion, no further research is necessary to evaluate the role of first-trimester progesterone in the prevention of miscarriage for women with a history of unexplained RM. The PROMISE trial has not addressed questions such as the effectiveness of progesterone therapy during the luteal phase of the menstrual cycle, or whether or not progesterone therapy for patients who have threatened miscarriage could be beneficial. 68 Future large randomised controlled trials, specifically designed to answer these questions, are required.
Our economic analysis demonstrated uncertainty in the expected gains of treatment, given the available sample. However, the magnitude of the estimated health benefits, although non-significant and lower than previously anticipated, may raise an empirical question of the value that the NHS or society might associate with decreasing the level of statistical uncertainty surrounding the current point estimate. Future research might also consider estimating the value of further research using value of information analysis. 84,85
Acknowledgements
Contributions of authors
All of the following named authors contributed substantially to the development of the research question and study design, implementation, analysis and/or interpretation of data and submission of the final report.
Particular contributions are denoted below:
Professor Arri Coomarasamy (Professor of Gynaecology and PROMISE Trial Manager) designed the study, directed the conduct of the study, chaired the TMG and contributed to the TSC and other meetings, and produced the final draft of the report.
Miss Helen Williams (Research Associate, Women’s Health) was responsible for completion of data gathering, providing data quality assurance, co-ordination of the analysis and the writing groups, and preparation of the initial draft of the final report.
Dr Ewa Truchanowicz (Clinical Trial Co-ordinator) co-ordinated the practical conduct of the study including the management of follow-up, attended meetings of the TSC and contributed to the TMG and the final report.
Mr Paul T Seed (Senior Lecturer, Medical Statistics) designed and conducted the statistical analysis of primary and secondary trial outcomes, produced reports for the DMC and contributed to the TMG.
Ms Rachel Small (Miscarriage Midwife Specialist) conducted PROMISE recruitment and data collection at Birmingham Heartlands Hospital, UK.
Professor Siobhan Quenby (Professor of Obstetrics) was responsible for leading PROMISE recruitment and data collection at Birmingham Heartlands Hospital and Coventry University Hospital, UK.
Dr Pratima Gupta (Consultant in Obstetrics and Gynaecology) was responsible for co-ordinating PROMISE recruitment and data collection at Birmingham Heartlands Hospital, UK.
Dr Feroza Dawood (Consultant Obstetrician and Gynaecologist) was responsible for leading PROMISE recruitment and data collection at Liverpool Women’s Hospital, UK.
Dr Yvonne E Koot (Obstetrics and Gynaecology) was responsible for leading PROMISE recruitment and data collection at University Medical Centre Utrecht, Utrecht, the Netherlands, and assisting in the co-ordination of investigators from the Netherlands.
Ms Ruth Bender Atik (National Director of the Miscarriage Association) provided advice and oversight to conduct PPI activities and contributed to the TSC.
Dr Kitty WM Bloemenkamp (Obstetrics and Gynaecology) was responsible for leading PROMISE recruitment and data collection at Leiden University Medical Centre in Leiden, the Netherlands.
Ms Rebecca Brady (Research Midwife) conducted PROMISE recruitment and data collection at St Mary’s Hospital in London, UK, and participated in the TMG.
Dr Annette Briley (Consultant Midwife and Clinical Trials Manager) provided advice and assistance on the design of the study and commented on the final report.
Ms Rebecca Cavallaro (Research Midwife) conducted PROMISE recruitment and data collection at St Mary’s Hospital in London, UK, and participated in the TMG.
Dr Ying C Cheong (Consultant in Obstetrics and Gynaecology) was responsible for leading PROMISE recruitment and data collection at Princess Anne Hospital in Southampton, UK.
Dr Justin Chu (Research Fellow, Obstetrics and Gynaecology) assisted in the preparation of the final report.
Dr Abey Eapen (Research Fellow, Obstetrics and Gynaecology) assisted in the preparation of the final report.
Dr Holly Essex (Research Fellow, Health Sciences) provided advice and assistance to conduct the study, with a focus on health economic elements.
Mr Ayman Ewies (Consultant Gynaecological Surgeon) was responsible for leading PROMISE recruitment and data collection at Birmingham City Hospital, UK.
Dr Annemieke Hoek (Reproductive Medicine and Gynaecology) was responsible for leading PROMISE recruitment and data collection at University Medical Centre Groningen, the Netherlands.
Dr Eugenie M Kaaijk (Obstetrics and Gynaecology) was responsible for leading PROMISE recruitment and data collection at Onze Lieve Vrouwe Gasthuis, Amsterdam, the Netherlands.
Dr Carolien A Koks (Obstetrics and Gynaecology) was responsible for leading PROMISE recruitment and data collection at Maxima Medical Centre, Veldhoven, the Netherlands.
Professor Tin-Chiu Li (Consultant Obstetrician and Gynaecologist) provided advice and assistance to design the study, and took responsibility for leading PROMISE recruitment and data collection at Sheffield Royal Hallamshire Hospital, UK.
Dr Marjory MacLean (Consultant Obstetrician) was responsible for leading PROMISE recruitment and data collection at Ayrshire Maternity Unit, UK.
Dr Ben W Mol (Professor of Obstetrics and Gynaecology) provided advice and assistance on the design of the study.
Dr Judith Moore (Consultant Obstetrician and Gynaecologist) was responsible for leading PROMISE recruitment and data collection at Queen’s Medical Centre in Nottingham, UK.
Dr Steve Parrott (Senior Research Fellow, Health Sciences) provided advice and assistance on the conduct of the study, and commented on the final report, with a focus on health economic elements.
Dr Jackie A Ross (Consultant Gynaecologist) was responsible for leading PROMISE recruitment and data collection at King’s College Hospital in London, UK.
Ms Lisa Sharpe (Research Midwife and Deputy Manager of the Women’s Health Research Centre at Imperial College London) conducted PROMISE recruitment and data collection at St Mary’s Hospital in London, UK, and participated in the TMG.
Dr Jane Stewart (Consultant in Reproductive Medicine and Gynaecology) was responsible for leading PROMISE recruitment and data collection at the Royal Victoria Infirmary in Newcastle upon Tyne, UK.
Dr Dominic Trépel (Research Fellow, Health Economics) designed and conducted the health economic analysis and contributed to the TMG.
Dr Nirmala Vaithilingam (Consultant in Obstetrics and Gynaecology) was responsible for leading PROMISE recruitment and data collection at the Queen Alexandra Hospital in Portsmouth, UK.
Dr Roy G Farquharson (Consultant Gynaecologist) provided methodological support, strategic advice and oversight to conceive, design and deliver the study.
Professor Mark David Kilby (Professor of Fetal Medicine) was responsible for leading PROMISE recruitment and data collection at Birmingham Women’s Hospital, UK, and contributed to the final report.
Mr Yacoub Khalaf (Consultant in Gynaecology and Reproductive Medicine) provided strategic advice on the design and delivery of the study, and was responsible for leading PROMISE recruitment and data collection at St Thomas’ Hospital in London, UK.
Dr Mariëtte Goddijn (Consultant Gynaecologist) was responsible for leading PROMISE recruitment and data collection at the Academic Medical Centre in Amsterdam, administering regulatory activities in the Netherlands and co-ordinating investigators from the Netherlands.
Professor Lesley Regan (Head of Obstetrics and Gynaecology at St Mary’s Hospital Campus) provided methodological support, strategic advice and oversight to conceive, design and deliver the study.
As the lead applicant, Dr Rajendra Rai (Consultant Obstetrician and Gynaecologist and PROMISE Chief Investigator) contributed to the design of the study, TSC and TMG, and took overall responsibility for the project, in addition to leading PROMISE recruitment and data collection at St Mary’s Hospital in London, UK.
The report authors gratefully acknowledge the substantial contributions of the following collaborators in the conduct and delivery of the PROMISE study.
Principal investigators
Dr Amna Ahmed (Consultant Obstetrician and Gynaecologist), Dr Shamma Al-Inizi (Consultant Gynaecologist and Obstetrician), Dr Maysoon Backos (Consultant Gynaecologist and Obstetrician), Mr Faisal Basama (Consultant Obstetrician and Gynaecologist), Dr Kitty Bloemenkamp (Obstetrics and Gynaecology), Mr Steve Bober (Consultant Obstetrician and Gynaecologist), Dr Manisha Chandra (Consultant Obstetrician and Gynaecologist), Dr Ying Cheong (Consultant in Obstetrics and Gynaecology), Dr Sangeeta Das (Consultant Gynaecologist), Dr Feroza Dawood (Consultant Obstetrician and Gynaecologist), Dr Edmond Edi-Osagie (Consultant Gynaecologist), Dr Isaac Evbuomwan (Consultant Gynaecologist and Reproductive Medicine and Surgery Specialist), Mr Ayman Ewies (Consultant Gynaecological Surgeon), Dr Mariette Goddijn (Consultant Gynaecologist), Dr Elizabeth Haslett (Consultant Obstetrician and Gynaecologist), Dr Pratima Gupta (Consultant in Obstetrics and Gynaecology), Dr Annemieke Hoek (Reproductive Medicine and Gynaecology), Dr Shehnaz Jivraj (Consultant Obstetrician and Gynaecologist), Dr Lisa Joels (Consultant in Obstetrics and Gynaecology), Dr Eugenie Kaaijk (Obstetrics and Gynaecology), Mr Jayaprakasan Kanna (Consultant Gynaecologist), Mr Yacoub Khalaf (Consultant in Gynaecology and Reproductive Medicine), Professor Mark Kilby (Professor of Fetal Medicine), Dr Carolien Koks (Obstetrics and Gynaecology), Dr Yvonne Koot (Obstetrics and Gynaecology), Dr Walter Kuchenbecker (Obstetrics and Gynaecology), Professor Tin-Chiu Li (Consultant Obstetrician and Gynaecologist), Dr Shonag Mackenzie (Consultant Obstetrician and Gynaecologist), Dr Marjory MacLean (Consultant Obstetrician), Dr Iona MacLeod (Consultant Obstetrician and Gynaecologist), Dr Padma Manda (Consultant Gynaecologist), Dr Patricia Mercelina (Obstetrics and Gynaecology), Dr Judith Moore (Consultant Obstetrician and Gynaecologist), Dr Neela Mukhopadhaya (Consultant Gynaecologist), Professor Nalini Munjuluri (Professor of Obstetrics and Gynaecology), Dr Denise Perquin (Obstetrics and Gynaecology), Professor Siobhan Quenby (Professor of Obstetrics), Dr Raj Rai (Consultant Obstetrician and Gynaecologist), Dr Judith Roberts (Consultant Obstetrician and Gynaecologist), Dr Jackie Ross (Consultant Gynaecologist), Mr Gamal Sayed (Consultant in Obstetrics and Gynaecology), Miss Gillian Scothern (Consultant Gynaecologist), Dr Seema Sen (Consultant Obstetrician and Gynaecologist), Dr Jane Stewart (Consultant in Reproductive Medicine and Gynaecology), Mr Guy Thorpe-Beeston (Consultant Obstetrician and Gynaecologist), Dr Nirmala Vaithilingam (Consultant in Obstetrics and Gynaecology), Dr Sandra Watson (Consultant Obstetrician and Gynaecologist) and Mr Simon Wood (Consultant Gynaecologist) were each responsible for leading PROMISE recruitment and data collection at their local institutions.
Clinicians, nurses and fellows
Dr Adjoa Appiah (King’s College Hospital, London), Miss Mohammad Aziz (St Mary’s Hospital, London), Ms Sarah Bailey (Princess Anne Hospital, Southampton), Ms Tessa de Vries (Academic Medical Centre, Amsterdam), Ms Jane Forbes (Princess Anne Hospital, Southampton), Dr Joanna Fuller (King’s College Hospital, London), Ms Rosemary Gebhardt (St Mary’s Hospital, London), Ms Ineke Hamming (University Medical Centre Groningen), Mr Etienne Horner (St Mary’s Hospital, London), Dr Anjali Kalaskar (King’s College Hospital, London), Miss Polytimi Katsafourou (St Mary’s Hospital, London), Jose Keurentjes (University Medical Centre Groningen), Mrs Fiona Kinney (Birmingham City Hospital, Birmingham), Ms Clara Kolster-Bijdevaate (Leiden University Medical Centre), Ms Winnie Lo (St Mary’s Hospital, London), Mrs Kuldip Manak (Birmingham City Hospital, Birmingham), Ms Victoria Murtha (Royal Victoria Infirmary, Newcastle upon Tyne), Ms Lida Ulkeman (University Medical Centre Groningen and Medical Centre Leeuwarden), Ms Ingrid van Hooff (Maxima Medical Centre, Veldhoven), Ms Nitolanda van Rijn (Isala Klinieken Zwolle) and Ms Fiona Yelnoorkar (Royal Victoria Infirmary, Newcastle upon Tyne).
Methodological support
Emeritus Professor Christine Godfrey (Health Economics, University of York) assisted the design of the health economic evaluation. Emeritus Professor Peter Braude (Obstetrics and Gynaecology, King’s College London) provided advice and assistance to design the study.
Administrative and other support
M Kruyt (Trial Officer) provided additional support at the Academic Medical Centre in Amsterdam. Ms Marieke van Erven provided additional support at the Maxima Medical Centre in Veldhoven.
Members of the Trial Steering Committee
Professor Siladitya Bhattacharya (Chair in Obstetrics and Gynaecology, University of Aberdeen) chaired the committee. Other members were Ms Ruth Bender Atik (National Director of the Miscarriage Association), Professor Arri Coomarasamy (Professor of Gynaecology and PROMISE Trial Manager, University of Birmingham) and Dr Rajendra Rai (Consultant Obstetrician and Gynaecologist and PROMISE Chief Investigator, Imperial College London).
Members of the Data Monitoring Committee
Professor Jennifer Kurinczuk (Perinatal Epidemiology, University of Oxford) chaired the committee. Other members were Professor Javier Zamora (Clinical Biostatistician, Hospital Ramón y Cajal, Madrid, Spain) and Professor Nick Raine-Fenning (Consultant Gynaecologist, Queen’s Medical Centre, University of Nottingham).
Members of the Trial Management Group
Professor Arri Coomarasamy (Professor of Gynaecology and PROMISE Trial Manager, University of Birmingham) chaired the group. Other members were Ms Rebecca Brady (Research Midwife, Imperial College London), Ms Rebecca Cavallaro (Research Midwife, Imperial College London), Dr Rajendra Rai (Consultant Obstetrician and Gynaecologist and PROMISE Chief Investigator, Imperial College London), Mr Paul Seed (Senior Lecturer, Medical Statistics, King’s Health Partners), Ms Lisa Sharpe (Research Midwife and Deputy Manager of the Women’s Health Research Centre at Imperial College London), Dr Dominic Trépel (Research Fellow, Health Economics, University of York) and Dr Ewa Truchanowicz (Clinical Trial Co-ordinator, University of Birmingham).
Pharmacists
Ms Victoria Latham (Clinical Trials Pharmacist, St Mary’s Hospital, London), Miss Severine Rey (Clinical Trial Pharmacy Assistant, St Mary’s Hospital, London) and Ms Sonjya El Yandouzi (Clinical Trials Pharmacist, University Medical Centre Utrecht).
Support groups
We thank the representatives of the local clinical research networks participating in the trial, the West Midlands Reproductive and Childbirth Research Network (REACH), the European Society for Human Reproduction and Embryology, and the RCOG Early Pregnancy Clinical Study Group for assistance in promoting awareness of the study among clinical practitioners.
Service user representatives
Ms Ruth Bender Atik (National Director of the Miscarriage Association), Ms Liz Campbell (Director of Wellbeing Research) and officers of the RCOG consumers’ forum.
Other contributors to programme development and implementation
Students and staff at the University of Birmingham.
Investigational medicinal product production
We are grateful to Besins Healthcare for producing and delivering IMP supplies safely.
Institutional support
We acknowledge with thanks the trial funders, the National Institute for Health Research Health Technology Assessment programme in the UK.
We thank all those not otherwise mentioned above who have contributed to the PROMISE study.
Publications
Coomarasamy A, Truchanowicz EG, Rai R. Does first trimester progesterone prophylaxis increase the live birth rate in women with unexplained recurrent miscarriages? BMJ 2011;342:d1914. http://dx.doi.org/10.1136/bmj.d1914
Coomarasamy A, Williams H, Truchanowicz E, Seed P, Small R, Quenby S, et al. A randomized trial of progesterone in women with recurrent miscarriages. N Eng J Med 2015;373:2141–8. http://dx.doi.org/10.1056/NEJMoa1504927
Data sharing statement
Non-identifiable data will be made available as appropriate for research purposes following an application to the corresponding author.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Regan L, Backos MJ, Rai R. The Investigation and Treatment of Couples with Recurrent Miscarriage. London: Royal College of Obstetricians and Gynaecologists; 2003.
- Oates-Whitehead RM, Haas DM, Carrier JA. Progestogen for preventing miscarriage. Cochrane Database Syst Rev 2003;4. http://dx.doi.org/10.1002/14651858.cd003511.
- Rai R, Regan L. Recurrent miscarriage. Lancet 2006;368:601-11. http://dx.doi.org/10.1016/S0140-6736(06)69204-0.
- Cantwell R, Clutton-Brock T, Cooper G, Dawson A, Drife J, Garrod D, et al. Saving Mothers’ Lives: reviewing maternal deaths to make motherhood safer: 2006–2008. The Eighth Report of the Confidential Enquiries into Maternal Deaths in the United Kingdom. BJOG An Int J Obstet Gynaecol 2011;118. http://dx.doi.org/10.1111/j.1471-0528.2010.02847.x.
- Coomarasamy A, Truchanowicz EG, Rai R. Does first trimester progesterone prophylaxis increase the live birth rate in women with unexplained recurrent miscarriages?. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.d1914.
- Daya S. Efficacy of progesterone support for pregnancy in women with recurrent miscarriage. A meta-analysis of controlled trials. Br J Obs Gynaecol 1989;96:275-80. http://dx.doi.org/10.1111/j.1471-0528.1989.tb02386.x.
- El-Zibdeh MY. Dydrogesterone in the reduction of recurrent spontaneous abortion. J Steroid Biochem Mol Biol 2005;97:431-4. http://dx.doi.org/10.1016/j.jsbmb.2005.08.007.
- Goldzieher JW. Double-blind trial of a progestin in habitual abortion. JAMA 1964;188:651-4. http://dx.doi.org/10.1001/jama.1964.03060330031008.
- Levine L. Habitual abortion. A controlled study of progestational therapy. West J Surg Obs Gynecol 1964;72:30-6.
- Swyer GI, Daley D. Progesterone implantation in habitual abortion. Br Med J 1953;1:1073-77. http://dx.doi.org/10.1136/bmj.1.4819.1073.
- Haas DM, Ramsey PS. Progestogen for preventing miscarriage. Cochrane Database Syst Rev 2013;10. http://dx.doi.org/10.1002/14651858.cd003511.pub3.
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408-12. http://dx.doi.org/10.1001/jama.1995.03520290060030.
- Wood L, Egger M, Gluud LL, Jüni P, Altman D, Gluud C, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008;336:601-5. http://dx.doi.org/10.1136/bmj.39465.451748.AD.
- Soliman S, Daya S, Collins J, Hughes EG. The role of luteal phase support in infertility treatment: a meta-analysis of randomized trials. Fertil Steril 1994;61:1068-76.
- Pritts EA, Atwood AK. Luteal phase support in infertility treatment: a meta-analysis of the randomized trials. Hum Reprod 2002;17:2287-99. http://dx.doi.org/10.1093/humrep/17.9.2287.
- Nosarka S, Kruger T, Siebert I, Grove D. Luteal phase support in in vitro fertilization: meta-analysis of randomized trials. Gynecol Obs Invest 2005;60:67-74. http://dx.doi.org/10.1159/000084546.
- Raman-Wilms L, Tseng AL, Wighardt S, Einarson TR, Koren G. Fetal genital effects of first-trimester sex hormone exposure: a meta-analysis. Obs Gynecol 1995;85:141-9. http://dx.doi.org/10.1016/0029-7844(94)00341-A.
- Carmichael SL, Shaw GM, Laurent C, Croughan MS, Olney RS, Lammer EJ. Maternal progestin intake and risk of hypospadias. Arch Pediatr Adolesc Med 2005;159:957-62. http://dx.doi.org/10.1001/archpedi.159.10.957.
- Coomarasamy A, Thangaratinam S, Gee H, Khan KS. Progesterone for the prevention of preterm birth: a critical evaluation of evidence. Eur J Obs Gynecol Reprod Biol 2006;129:111-18. http://dx.doi.org/10.1016/j.ejogrb.2006.05.013.
- Meis PJ, Klenabnoff M, Thom E, Dombrowski MP, Sibai B, Moawad AH, et al. Prevention of recurrent preterm delivery by 17 alpha-hydroxyprogesterone caproate. N Engl J Med 2003;348:2379-85. http://dx.doi.org/10.1056/NEJMoa035140.
- Da Fonseca EB, Bittar RE, Carvalho MH, Zugaib M. Prophylactic administration of progesterone by vaginal suppository to reduce the incidence of spontaneous preterm birth in women at increased risk: a randomized placebo-controlled double-blind study. Am J Obs Gynecol 2003;188:419-24. http://dx.doi.org/10.1067/mob.2003.41.
- Fonseca EB, Celik E, Parra M, Singh M, Nicolaides KH. Fetal Medicine Foundation Second Trimester Screening Group . Progesterone and the risk of preterm birth among women with a short cervix. N Engl J Med 2007;357:462-9. http://dx.doi.org/10.1056/NEJMoa067815.
- d-maps.com . United Kingdom United Kingdom of Great Britain and Northern Ireland: Outlines, Nations, Color (White) n.d. http://d-maps.com/m/europa/uk/royaumeuni/royaumeuni80.gif (accessed 9 May 2015).
- d-maps.com . Netherlands/Koninkrijk/Der/Nederlanden:/Outline,/Provinces,/Color/(White) n.d. http://d-maps.com/m/europa/netherlands/paysbas/paysbas50.gif (accessed 9 May 2015).
- Stephenson MD, Awartani KA, Robinson WP. Cytogenetic analysis of miscarriages from couples with recurrent miscarriage: a case–control study. Hum Reprod 2002;17:446-51. http://dx.doi.org/10.1093/humrep/17.2.446.
- Briefing Information for the September 29, 2003 Meeting of the Advisory Committee for Reproductive Health Drugs. Silver Spring, MD: US Food and Drug Administration Advisory Committee for Reproductive Health Drugs; 2003.
- Commission Directive 2003/94/EC of 8 October 2003. Brussels: European Commission; 2003.
- Commission Directive 2001/20/EC of 4 April 2001. Brussels: European Commission; 2001.
- The Rules Governing Medicinal Products in the European Union, Volume 4: Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use – Annex 13, Manufacture of Investigational Medicinal Products. Brussels: European Commission; 2010.
- Utrogestan Vaginal 200 mg Capsules – Summary of Product Characteristics. Datapharm and Besins Healthcare; n.d.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2014.
- Van der Linden M, Buckingham K, Farquhar C, Kremer JA, Metwally M. Luteal phase support for assisted reproduction cycles. Cochrane Database Syst Rev 2011;10. http://dx.doi.org/10.1002/14651858.cd009154.
- Li TC, Spuijbroek MD, Tuckerman E, Anstie B, Loxley M, Laird S. Endocrinological and endometrial factors in recurrent miscarriage. BJOG 2000;107:1471-9. http://dx.doi.org/10.1111/j.1471-0528.2000.tb11670.x.
- Li TC, Tuckerman EM, Laird SM. Endometrial factors in recurrent miscarriage. Hum Reprod Updat 2002;8:43-52. http://dx.doi.org/10.1093/humupd/8.1.43.
- Szekeres-Bartho J, Balasch J. Progestagen therapy for recurrent miscarriage. Hum Reprod Updat 2008;14:27-35. http://dx.doi.org/10.1093/humupd/dmm035.
- Bulletti C, de Ziegler D, Flamigni C, Giacomucci E, Polli V, Bolelli G, et al. Targeted drug delivery in gynaecology: the first uterine pass effect. Hum Reprod 1997;12:1073-9. http://dx.doi.org/10.1093/humrep/12.5.1073.
- Cicinelli E, Cignarelli M, Sabatelli S, Romano F, Schonauer LM, Padovao R, et al. Plasma concentrations of progesterone are higher in the uterine artery than in the radial artery after vaginal administration of micronized progesterone in an oil-based solution to postmenopausal women. Fertil Steril 1998;69:471-3. http://dx.doi.org/10.1016/S0015-0282(97)00545-1.
- Dodd JM, Jones L, Flenady VJ, Cincotta R, Crowther CA. Prenatal administration of progesterone for preventing preterm birth in women considered to be at risk of preterm birth. Cochrane Database Syst Rev 2013;7. http://dx.doi.org/10.1002/14651858.cd004947.pub3.
- The Medicines for Human Use (Clinical Trials) Regulations 2004. London: The Stationery Office; 2004.
- The Medicines for Human Use (Clinical Trials) Amendment Regulations 2006. London: The Stationery Office; 2006.
- The Safe and Secure Handling of Medicines: A Team Approach. London: Royal Pharmaceutical Society of Great Britain; 2005.
- Professional Guidance on Pharmacy Services for Clinical Trials, Version 1. London: Royal Pharmaceutical Society of Great Britain; 2013.
- Standard Operating Procedures. London: Imperial College of Science Technology and Medicine; 2014.
- Campbell M, Snowdon C, Francis D, Elbourne D, McDonald AM, Knight R, et al. Recruitment to randomised trials: strategies for trial enrollment and participation study: the STEPS study. Health Technol Assess 2007;11. http://dx.doi.org/10.3310/hta11480.
- Imperial College London/Imperial College Healthcare NHS Trust Joint Research Compliance Office . Recording Managing and Reporting Adverse Events in the UK Imperial College of Science Technology and Medicine, Version 7.0 2015. https://workspace.imperial.ac.uk/clinicalresearchgovernanceoffice/Public/JRCO_SOP_001_Safety_Reporting%20Final%202015.pdf (accessed 14 October 2015).
- Medical Research Council/Department of Health . Clinical Trials Toolkit 2007. Work Stream 6: Pharmacovigilance (Updated 2012) n.d. www.ct-toolkit.ac.uk/_data/assets/pdf_file/0010/35956/joint-project-pharmacovigilance.pdf.
- Detailed Guidance on the Collection, Verification and Presentation of Adverse Reaction Reports arising from Clinical Trials on Medicinal Products for Human Use: Revision 2. Brussels: European Commission; 2006.
- Brigham SA, Conlon C, Farquharson RG. A longitudinal study of pregnancy outcome following idiopathic recurrent miscarriage. Hum Reprod 1999;14:2868-71. http://dx.doi.org/10.1093/humrep/14.11.2868.
- Altman DG, Dore CJ. Baseline comparisons in clinical trials. Lancet 1990;335. http://dx.doi.org/10.1016/0140-6736(90)91515-C.
- Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c332.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c869.
- Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. Br J Cancer 1976;34:585-612. http://dx.doi.org/10.1038/bjc.1976.220.
- Freedman B. Equipoise and the ethics of clinical research. N Engl J Med 1987;317:141-5. http://dx.doi.org/10.1056/NEJM198707163170304.
- O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics 1979;35:549-56. http://dx.doi.org/10.2307/2530245.
- Haybittle JL. Repeated assessment of results in clinical trials of cancer treatment. Br J Radiol 1971;44:793-7. http://dx.doi.org/10.1259/0007-1285-44-526-793.
- Geller NL, Pocock SJ. Interim analyses in randomized clinical trials: ramifications and guidelines for practitioners. Biometrics 1987;43:213-23. http://dx.doi.org/10.2307/2531962.
- Data Protection Act 1998. London: The Stationery Office; 1998.
- Confidentiality: NHS Code of Practice. London: Department of Health; 2003.
- Report on the Review of Patient-Identifiable Information. London: Department of Health; 1997.
- Declaration of Helsinki: Ethical Principles for Medical Research Involving Human Subjects. Ferney-Voltaire: World Medical Association; 1964.
- Research Governance Framework for Health and Social Care, Second Edition. London: The Stationery Office; 2005.
- Entwistle VA, Renfrew MJ, Yearley S, Forrester J, Lamont T. Lay perspectives: advantages for health research. BMJ 1998;316:463-6. http://dx.doi.org/10.1136/bmj.316.7129.463.
- Hanley B, Truesdale A, King A, Elbourne D, Chalmers I. Involving consumers in designing conducting and interpreting randomised controlled trials: questionnaire survey. BMJ 2001;322:519-23. http://dx.doi.org/10.1136/bmj.322.7285.519.
- National Institute for Health Research . Public and Patient Involvement n.d. www.nets.nihr.ac.uk/ppi (accessed 15 September 2015).
- Involve . Public Involvement in the Management for the Research Design Service 2009.
- Gardosi J, Francis A. Customised Weight Centile Calculator 2013. www.gestation.net/grow_documentation.pdf (accessed 14 October 2015).
- Department of Health . Reference Costs 2011–12 2012. www.gov.uk/government/publications/nhs-reference-costs-financial-year-2011-to-2012 (accessed 15 September 2015).
- Ectopic Pregnancy and Miscarriage: Diagnosis and Initial Management in Early Pregnancy of Ectopic Pregnancy and Miscarriage. London: NICE; 2012.
- Petrou S, Trinder J, Brocklehurst P, Smith L. Economic evaluation of alternative management methods of first-trimester miscarriage based on results from the MIST trial. BJOG 2006;113:879-89. http://dx.doi.org/10.1111/j.1471-0528.2006.00998.x.
- Providing Equity of Critical and Maternity Care for the Critically Ill Pregnant or Recently Pregnant Woman. London: Royal College of Anaesthetists; 2011.
- Guide to the Methods of Technology Appraisal 2013. London: NICE; 2013.
- Simon J, Petrou S, Gray A. The valuation of prenatal life in economic evaluations of perinatal interventions. Health Econ 2009;18:487-94. http://dx.doi.org/10.1002/hec.1375.
- Korvenranta E, Linna M, Rautava L, Andersson S, Gissler M, Hallman M, et al. Hospital costs and quality of life during 4 years after very preterm birth. Arch Pediatr Adolesc Med 2010;164:657-63. http://dx.doi.org/10.1001/archpediatrics.2010.99.
- Apajasalo M, Rautonen J, Holmberg C, Sinkkonen J, Aalberg V, Pinko H, et al. Quality of life in pre-adolescence: a 17-dimensional health-related measure (17D). Qual Life Res 1996;5:532-8. http://dx.doi.org/10.1007/BF00439227.
- Petrou S. The economic consequences of preterm birth during the first 10 years of life. BJOG 2005;112. http://dx.doi.org/10.1111/j.1471-0528.2005.00577.x.
- Zellner A. An efficient method of estimating seemingly unrelated regressions and tests for aggregation bias. J Am Stat Assoc 2012;57:348-68. http://dx.doi.org/10.1080/01621459.1962.10480664.
- Loomes G, McKenzie L. The use of QALYs in health care decision making. Soc Sci Med 1989;28:299-308. http://dx.doi.org/10.1016/0277-9536(89)90030-0.
- Lachin JM. Statistical properties of randomization in clinical trials. Control Clin Trials 1988;9:289-311. http://dx.doi.org/10.1016/0197-2456(88)90045-1.
- Ozlü T, Güngör AC, Dönmez ME, Duran B. Use of progestogens in pregnant and infertile patients. Arch Gynecol Obstet 2012;286:495-503. http://dx.doi.org/10.1007/s00404-012-2340-4.
- Sonntag B, Ludwig M. An integrated view on the luteal phase: diagnosis and treatment in subfertility. Clin Endocrinol (Oxf) 2012;77:500-7. http://dx.doi.org/10.1111/j.1365-2265.2012.04464.x.
- Shah D, Nagarajan N. Luteal insufficiency in first trimester. Ind J Endocrinol Metab 2013;17:44-9. http://dx.doi.org/10.4103/2230-8210.107834.
- Haas DM, Ramsey PS. Progestogen for preventing miscarriage. Cochrane Database Syst Rev 2008;2. http://dx.doi.org/10.1002/14651858.cd003511.pub2.
- Dante G, Vaccaro V, Facchinetti F. Use of progestagens during early pregnancy. Facts Views Vis ObGyn 2013;5:66-71.
- McKenna C, Claxton KP. Addressing adoption and research design decisions simultaneously: the role of value of sample information analysis. Med Decis Making 2011;6:853-65. http://dx.doi.org/10.1177/0272989X11399921.
- Griffin S, Welton NJ, Claxton K. Exploring the research decision space: the expected value of information for sequential research designs. Med Decis Making 2010;30:155-62. http://dx.doi.org/10.1177/0272989X09344746.
{kind=link}
{kind=link}
Appendix 1 Sample participant information sheet
Appendix 2 Sample consent form
Appendix 3 Definitions of adverse events, seriousness and causality
Adverse event
Any untoward medical occurrence in a participant, which does not necessarily have a causal relationship with the trial intervention.
In the context of the PROMISE trial, an AE was considered to be:
-
any unintentional, unfavourable clinical sign or symptom, including complications of miscarriage (but not miscarriage itself)
-
any new illness or disease or the deterioration of existing disease or illness
-
any clinically significant deterioration in any laboratory assessments or clinical tests.
In the context of the PROMISE trial, the circumstances listed below were NOT considered to be AEs:
-
miscarriage or intrauterine death
-
a pre-existing condition (unless it worsened significantly during treatment)
-
routine diagnostic and therapeutic procedures likely in a normal pregnancy.
Adverse reaction
Any untoward and unintended responses to the trial intervention, at any dose administered, including all AEs judged by either the reporting investigator or the sponsor as having a reasonable causal relationship to the trial intervention.
Unexpected adverse reaction
An adverse reaction, the nature or severity of which is not consistent with the applicable product information.
Seriousness
Any AE, adverse reaction or unexpected adverse reaction that at any dose:
-
results in death or immediately threatens life (NOT an event which hypothetically might have caused death if it were more severe)
-
results in hospitalisation or longer than anticipated stay in hospital
-
results in persistent or significant disability or incapacity
-
results in a congenital anomaly or birth defect.
Medical judgement may be exercised in deciding seriousness in other situations. Important AEs or reactions that are not immediately life-threatening or do not result in death or hospitalisation may be considered serious if they jeopardise the subject or require intervention to prevent one of the other outcomes listed in the definition above.
In the context of the PROMISE trial, events NOT considered to be SAEs were hospitalisations for events that were expected, such as:
-
routine treatment or monitoring of miscarriage or threatened preterm birth, not associated with any deterioration in condition
-
elective or pre-planned treatment for a pre-existing condition that was unrelated to the indication under study, and did not worsen, including elective caesarean section
-
admission to a hospital or other institution for general care, not associated with any deterioration in condition, including:
-
hospitalisation for rest
-
hospitalisation for observation or monitoring of pregnancy
-
hospitalisation for maternal discomfort
-
-
hyperemesis which is quickly resolved
-
outpatient treatment for an event not fulfilling any of the definitions of serious given above and not resulting in hospital admission
-
hospital admission for complications of pregnancy unlikely to be related to progesterone use (such as pre-eclampsia, urinary tract infection, pyelonephritis).
Unrelated causality
No evidence of any causal relationship with the trial intervention.
Unlikely causal relationship
Little evidence to suggest a causal relationship (e.g. the event did not occur within a reasonable time after administration of the trial medication) or another reasonable explanation for the event (e.g. another clinical condition or other concomitant treatment).
Possible causal relationship
Some evidence to suggest a causal relationship (e.g. occurrence within a reasonable time after administration of the trial medication), but other factors may have contributed to the event (e.g. another clinical condition or other concomitant treatment).
Probable causal relationship
Evidence to suggest a causal relationship; the influence of other factors is unlikely.
Definite causal relationship
Some evidence to suggest a causal relationship (e.g. occurrence within a reasonable time after administration of the trial medication), but other factors may have contributed to the event (e.g. another clinical condition or other concomitant treatment).
Causal relationship not assessable
Insufficient or incomplete evidence to make a clinical judgement of the causal relationship.
Appendix 4 Sample general practitioner letter
Appendix 5 Other information
This appendix provides miscellaneous details of study registration and development, to ensure transparent identification and adherence of the PROMISE trial to appropriate governance.
Registration
Current Controlled Trials ISRCTN92644181; EudraCT number: 2009-011208-42; REC 09/H1208/44.
Funding
This study was funded by the National Institute for Health Research Health Technology Assessment programme, in a research award to Imperial College London/Imperial College Healthcare NHS Trust. No commercial funding was sought for this trial. Participants were not paid to take part in the trial, nor did they receive any incentives or other benefits. The funders of the study had no role in the study design, data collection, data analysis, data interpretation or writing of the report.
Insurance and indemnity
Imperial College London provided negligent and non-negligent harm insurance for the trial, and arranged insurance and/or indemnity to meet the potential legal liability of the sponsor for harm to participants arising from the design, conduct and management of the research. Imperial College London held a policy with Zurich Municipal in case of harm with no legal liability.
Protocol versions
Table 16 demonstrates the development of the PROMISE trial protocol.
| Version | Description |
|---|---|
| 1.0 | Circulation for internal and external peer-review before submission to the Medical Research Council |
| 2.0 | Outline submission to Medical Research Council (which was shortlisted by Medical Research Council and handed over to the NIHR HTA programme for processing) |
| 3.0 | Submission to the NIHR HTA (incorporating recommendations of Medical Research Council and NIHR HTA reviewers) |
| 4.0 | Submission to the REC (dated 15 July 2009) |
| 5.0 | Acceptance by the REC (dated 28 September 2009) |
| 6.0 | Addition and amendment of research sites (dated 26 November 2010) |
| 6.1 | Clarification that ‘delivery to participant’ included ‘delivery to participants’ home addresses’ added (dated 15 April 2011) |
| 6.2 | Amendment of the date to end recruitment to 31 July 2013 (dated 1 November 2012; URL: www.nets.nihr.ac.uk/__data/assets/pdf_file/0018/52911/PRO-08–38–01.pdf) |
| 6.3 | Clarification that recruited participants will not be eligible for randomisation if they are unable to conceive within a year of recruitment or before the end of randomisation period, whichever comes earlier (dated 30 May 2013) |
List of abbreviations
- AE
- adverse event
- APGAR
- appearance, pulse, grimace, activity, respiration
- BMI
- body mass index
- BNF
- British National Formulary
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- DMC
- Data Monitoring Committee
- GCP
- good clinical practice
- HDU
- high-dependency unit
- HRG
- Healthcare Resource Group
- ICER
- incremental cost-effectiveness ratio
- ICU
- intensive care unit
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ITMS
- integrated trial management system
- IVF
- in vitro fertilisation
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MID
- minimally important difference
- NICE
- National Institute for Health and Care Excellence
- NNU
- neonatal unit
- PI
- principal investigator
- PIS
- participant information sheet
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RCOG
- Royal College of Obstetricians and Gynaecologists
- REC
- Research Ethics Committee
- RM
- recurrent miscarriage
- RR
- relative risk
- SAE
- serious adverse event
- SCBU
- special care baby unit
- SD
- standard deviation
- SmPC
- summary of product characteristics
- SUSAR
- suspected unexpected serious adverse reaction
- TCC
- Trial Co-ordinating Centre
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee