
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 09/55/33. The contractual start date was in July 2011. The draft report began editorial review in July 2015 and was accepted for publication in January 2016. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Andrew Bentley reports honoraria and travel subsistence from BioMarin to attend clinical advisory group in Berlin (April 2015) on respiratory management of mucoploysaccharoidoses. Anita K Simonds reports being on the steering committee of trial of adaptive servo ventilation in heart failure patients with predominant obstructive sleep apnoea (SERVE-HF). Chin Maguire reports grants from Motor Neuron Disease Association and non-financial support from Synapse Biomedical Inc. during the conduct of the study. Christopher J McDermott reports grants from the National Institute for Health Research Health Technology Assessment programme, grants from the Motor Neurone Disease Association and non-financial support from Synapse Biomedical Inc. during the conduct of the study and separate to the Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis (DiPALS) trial. Lyn Taylor reports personal fees from PAREXEL International Corporation not associated with the DiPALS trial. Roger Leek reports membership of the Motor Neurone Disease Association. John Stradling reports personal fees from ResMed UK not associated with the DiPALS trial. Martin Turner reports grants from Medical Research Council, personal fees from Biogen Idec, personal fees from GlaxoSmithKline, personal fees from Neuraltus Pharmaceuticals, personal fees from Ontario Brain Institute, personal fees from GLG Consulting and personal fees from Vertex Pharmaceuticals not associated with the DiPALS trial. Richard W Orrell reports grants from the Motor Neurone Disease Association of England, Wales and Northern Ireland during the conduct of the study. Wisia Wedzicha reports grants from Johnson and Johnson, grants from Vifor Pharma, grants from Takeda, grants from GlaxoSmithKline, personal fees from Novartis, personal fees from GlaxoSmithKline, personal fees from Astra Zeneca, personal fees from Boehringer, personal fees from Takeda and personal fees from Vifor Pharma outside the submitted work.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2016. This work was produced by McDermott et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease with an annual incidence of 2–3 in 100,000 and a prevalence of 5–8 per 100,000. 1–3 Affected individuals experience increasing weakness affecting the limbs, speech and swallowing, and breathing. There is no cure for ALS and patients usually succumb to the illness within 2–3 years.
Management of amyotrophic lateral sclerosis
Management is largely aimed at easing symptoms and supporting patients to maximise their function through a multidisciplinary approach. 4 One treatment, riluzole, can marginally slow down disease progression, prolonging survival, usually by around 3 months. 5,6 The exact mechanism by which riluzole affects the disease course is unclear, although it is known to modulate glutamate release, among other effects. However, the greatest impact on the disease course comes from the use of non-invasive ventilation (NIV). A randomised controlled trial (RCT) demonstrated an improvement in quality of life (QoL) and a median survival benefit of approximately 7 months (p = 0.006), in ALS patients using NIV who had good bulbar function. 7 NIV, however, is not without its problems, as some individuals are unable to tolerate it because of problems with claustrophobia and mask interface issues. Furthermore, there comes a point in the course of the disease when NIV is no longer effective.
Phrenic nerve stimulation, in which the diaphragm is stimulated into contracting, is a potential alternative or complementary method of providing respiratory support. The approach originated in patients with spinal cord injury, and historically it required direct phrenic nerve stimulation. The challenges with this approach have been the significant risk of iatrogenic phrenic nerve injury and, until recently, the need to undertake a thoracotomy. 8 More recently, phrenic nerve stimulation has been achieved using less invasive techniques. The NeuRX® RA/4 diaphragm pacing system (DPS)™ (Synapse Biomedical, Oberlin, OH, USA) is a four-channel percutaneous neuromuscular stimulation system. DPS has an advantage over the earlier direct approach, in that the stimulating electrodes can be inserted into the under-surface of the diaphragm using a minimally invasive laparoscopic technique. 9 The leads are then tunnelled to an exit site on the abdomen and an external stimulator delivers the stimulus pulses and provides respiratory timing. Initial experience with the NeuRX RA/4 DPS in the spinal cord injury population10 suggested diaphragm pacing (DP) could reduce time spent on mechanical ventilation. 11 The NeuRX RA/4 DPS is now licensed for use in spinal cord injury in many countries, including the USA, and within the European Union.
Evidence for diaphragm pacing in amyotrophic lateral sclerosis
To date, the evidence for DPS in the ALS population is limited to case series and one uncontrolled multicentre cohort study. Their findings are consistent with those from the spinal cord patient population, and highlighted the apparent simplicity and operative safety of the NeuRX RA/4 DPS. 7,10 The US Food and Drug Administration (FDA) approved the NeuRX RA/4 DPS as a humanitarian-use device (HUD) in ALS following the submission of a humanitarian device exemption (HDE) application. The FDA summary of safety and probable benefit document (SSPB) summarises the evidence on which the HDE approval is based, which is largely on data from the aforementioned cohort study. 11 The main inclusion criteria for the study were patients with ALS with evidence of residual bilateral phrenic nerve function and a forced vital capacity (FVC) of less than 85% at screening and above 45% at DPS implantation. The full data from this study, including baseline characteristics, Consolidated Standards of Reporting Trials (CONSORT) diagram, survival, etc., on the 144 patients enrolled have not yet been published. Of the 144, 106 were implanted with the NeuRX RA/4 DPS and survival data on 84 are presented in the SSPB. 11
Median survival data are reported as 39 months from ALS diagnosis and 19 months post implantation in the SSPB on 84 implanted patients. As the data are from a cohort study there is no randomised control population to compare survival with. Instead, a subgroup of the HUD patients (n = 43) was compared with a historical control group of NIV users. The DP HUD group patients demonstrated survival of 37.5 months from diagnosis, compared with 21.4 months from diagnosis for the historical NIV control group (p < 0.001).
Safety data have been published in the SSPB for 86 implanted patients from the cohort study. In these patients, there were no reported deaths and none who needed a tracheostomy with permanent ventilation within the 30-day postoperative period. Four serious adverse events (SAEs) were reported relating to implantation [capnothorax (n = 2), respiratory failure due to complications of surgery (n = 1) and serious anaesthetic reaction (n = 1)]. Therefore, the implantation procedure itself appeared to be relatively safe. Important adverse events (AEs) over the 12-month protocol reported were capnothoraces in 16 patients (19%), percutaneous site infection in eight patients (9%) and pacing-related discomfort, which was described as mild in 20 patients (23%) and moderate in two patients (2%). In the 86 patients implanted, there were no SAEs involving malfunctioning device components. There were 18 reports of anode malfunction in 18 patients (21%) and 44 reports of electrode malfunction in 28 patients (33%). Although there was relatively frequent electrode breakage, there was no need to reimplant any electrodes, as all occurred external to the body at the connector holder.
Following the HUD approval of the NeuRX RA/4 DPS for ALS, there has been increasing use of this therapeutic option worldwide. 12 The promising survival data, lack of apparent harm and absence of alternatives have made this an appealing option, especially among patients who are unable to tolerate NIV, who may account for up to 50% of patients with ALS. 2,3,13,14 The evidence base for the NeuRX RA/4 DPS for ALS is limited. Moreover, pacing is expensive and it is not known whether or not DPS would meet the National Institute for Health and Care Excellence’s threshold for cost-effective interventions for end-of-life care. At the time of conducting the Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis (DiPALS) trial, the cost of DPS and implantation procedures (the excess treatment costs) was approximately £16,500 per participant. Therefore, although the preliminary data are promising, DPS is unlikely to be widely introduced without robust, randomised evidence together with a formal analysis of cost-effectiveness. This was our motivation for undertaking the DiPALS trial.
Research objectives
The aim of this study was to perform a definitive RCT on the efficacy and safety of the NeuRX RA/4 DPS when used in addition to NIV, compared with standard care of NIV alone, in patients with ALS. Specifically, we wished to assess whether or not the use of DP in addition to NIV prolongs survival, and also to quantify its impact on:
-
QoL of the participant
-
QoL of the main carer
-
safety (AEs) and tolerability (withdrawal from treatment)
-
quality-adjusted life-years
-
views and perceptions of patients and family carers regarding acceptability and impact on life.
Chapter 2 Methods
The original protocol for DiPALS was submitted to BMC Neurology15 and was published 8 months into the recruitment phase of the trial, prior to any analyses being performed. The methods described in this chapter are consistent with those as published and with the conduct of the trial in practice, but changes were made to the protocol over the course of the project. These are detailed at the end of this chapter, and tabulated in Appendix 1. None of the changes affected the conduct of the trial with respect to the intervention, the primary outcome measures or their statistical analyses.
This report has been prepared in accordance with the CONSORT statement (2010)16 and the Template for Intervention Description and Replication checklist and guide. 17
Trial design
The DiPALS trial was a multicentre, parallel-group, open-label RCT incorporating health economic analyses and a qualitative longitudinal substudy. The primary objective was to assess survival over the study duration of patients with ALS with respiratory failure, allocated to either standard care (NIV alone) or standard care (NIV) plus DPS using the NeuRX/4 DPS. Participants from seven UK hospitals were allocated to treatment using minimisation methods (see Appendices 2 and 3).
The trial was non-commercial, with input from Synapse Biomedical for quality assurance purposes, specifically for the training of surgeons and trial staff on implantation and technical aspects of the NeuRX/4 DPS.
Recruitment took place over 24 months (from December 2011 to December 2013).
Participants and eligibility criteria
Patients aged ≥ 18 years with a confirmed diagnosis of ALS were identified and screened at participating sites or patient referral sites against the following eligibility criteria.
Inclusion criteria
-
Aged ≥ 18 years.
-
Familial or sporadic ALS diagnosed as laboratory-supported probable, probable, or definite according to the World Federation of Neurology El Escorial criteria. 18
-
Stabilised on riluzole therapy for at least 30 days.
-
Respiratory insufficiency as determined by one or more of:
-
FVC < 75% predicted
-
supine vital capacity < 75% of sitting or standing vital capacity
-
sniff nasal inspiratory pressure (SNIP) < 65 cmH2O for men or 55 cmH2O for women in the presence of symptoms
-
SNIP < 40 cmH2O (see exclusion criterion 9).
-
partial pressure of carbon dioxide (PaCO2) > 6 kPa (daytime) or PaCO2 > 6.5 kPa (overnight)
-
significant overnight O2 desaturation (> 5% of night with SpO2 < 90% during overnight oximetry).
-
-
Bilateral phrenic nerve function clinically acceptable as defined by either:
-
absence of paradoxical abdominal wall movement during a sniff manoeuvre (sharp inhalation through the nose) and recording < 10% decline of FVC when moving from sitting to supine position, or
-
on ultrasound: evidence of at least 1 cm of downward diaphragm movement independent of thoracic or abdominal wall movement during the patient performing a sniff manoeuvre.
-
Exclusion criteria
-
Prior NIV prescription.
-
Pre-existing implanted electrical device such as pacemaker or cardiac defibrillator.
-
Underlying cardiac, pulmonary diseases or other disorders that would affect pulmonary tests independently of motor neurone disease/ALS (increased risk of general anaesthesia or adverse effect on survival over the course of the study).
-
Current pregnancy or breastfeeding.
-
Significant decision-making incapacity preventing informed consent by the subject because of a major mental disorder such as major depression, schizophrenia or dementia.
-
Marked obesity affecting surgical access to diaphragm or significant scoliosis/chest wall deformity.
-
The involvement in any respiratory trial that can influence the safety or outcome measures of this study within 3 months of the planned implantation of the device or during the year of follow-up.
-
Pre-existing diaphragm abnormality such as a hiatus hernia or paraoesophageal hernia of abdominal contents ascending into the thoracic cavity.
-
FVC < 50% predicted or SNIP < 30 cmH2O in patients unable to perform FVC (bulbar patients) (because of potential anaesthetic risk).
Participant identification
Potentially eligible patients were identified from NHS hospital clinics at lead research sites or participant identification centres by neurology or respiratory clinicians or a study research nurse (see Appendix 4).
All patients aged ≥ 18 years with a confirmed diagnosis of ALS underwent a ‘prescreen’ whereby their last routine respiratory measure was checked to determine whether or not:
-
patients were ineligible because of poor respiratory criteria or other reason
-
patients were potentially eligible
-
patients may be eligible in the future as their respiratory measure was currently too good.
All those potentially eligible were approached at clinic to discuss the trial in detail. If they were identified from the clinic list prior to attendance the study information was sent to them by post.
Recruitment
Patients were approached by a member of the research team at sites when they attended routine clinic appointments. The study patient and carer information sheets formed a basis for a discussion with potential participants in clinics. Patients were given as long as they required to consider taking part in the trial, with further discussion by telephone or at another clinic offered. Informed consent was taken only if the researcher was satisfied that the patient fully understood the study procedures, was willing to undergo screening and participate in the trial, and was capable of giving full informed consent. Consent was obtained using participant and carer consent forms approved by the Research Ethics Committee (REC), as either full written consent, verbal consent given or consent given via the use of a communication aid (for REC-approved patient information sheets see Appendices 5–8 and for REC-approved consent forms see Appendices 9–12). When non-written consent was obtained an independent witness was asked to sign the consent form to verify that consent was taken.
If participants were willing to provide reasons for declining participation in the trial, these were entered onto the screening log. Anonymised basic details (age, sex and reason for exclusion) were collected on all eligible patients to allow completion of the CONSORT flow chart.
Participants were free to withdraw from the trial at any point without giving a reason, but data collected up to the point of study withdrawal were maintained and used in study analyses. Patients who asked to withdraw from treatment were encouraged to complete follow-up for QoL and safety to reduce attrition bias. All participants were followed up for survival until 12 months after the last participant was recruited, unless they specifically requested otherwise (Figure 1).
FIGURE 1.
Participant flow diagram. a, At 9 months, only ED-5D-3L, DPS/NIV use, AEs and resource use collected. CBI, Caregiver Burden Inventory; EQ-5D-3L, European Quality of Life-5 Dimensions, three levels; SAQLI, Sleep Apnoea Quality of Life Index questionnaire; SF-36, Short Form questionnaire-36 items.

Screening
Once patient consent had been confirmed, a member of the site study team initiated the screening process. Patient eligibility was assessed against both non-clinical and clinical criteria, and baseline assessments. Eligibility for the study was based on all the inclusion and none of the exclusion criteria being met. To confirm eligibility, a combination of techniques was used. Researchers checked medical notes/data to assess past medical history, current prescriptions and any other known information relating to eligibility criteria. At the screening visit, participants were asked to perform a respiratory test to determine their FVC or other measure. An assessment was made of the participant’s phrenic nerve function – a measure intended to ensure that, if the patient were randomised to DPS, it was likely that it would be possible to sufficiently stimulate the participant’s diaphragm once the system was fitted. All criteria were checked and signed off by the recruiting clinician. Once eligibility was established, participants were asked to complete baseline assessments and could proceed to randomisation and study procedures (see Figure 1).
Randomisation and blinding
Once eligibility had been confirmed and consent acquired, the participants were randomly allocated to a treatment group. The recruiting clinician accessed a centralised web-based randomisation system provided by the Sheffield Clinical Trials Research Unit (CTRU) in partnership with a University of Sheffield wholly owned subsidiary software development company, epiGenesys. Sites were able to log on to the system using a site-specific username and password. Researchers were prompted to enter patient details (identification number, date of birth and the minimisation factors) and to confirm that consent and eligibility was complete. Following this, the randomisation system notified the user and the study manager of the treatment allocation. Patients were informed of their treatment allocation within 1 week of randomisation by telephone or letter.
The study was open label; it was not considered feasible to blind participants, carers or site staff as the study intervention involves a surgical procedure and ongoing use of an implanted medical device. A sham surgical procedure was considered but discounted as being an unnecessary burden to participants, given the objectivity of the primary outcome measure.
Patients were allocated their treatment (NIV alone or NIV plus DPS) by method of minimisation, using baseline bulbar function, baseline FVC, age and sex as the minimisation factors. Factors were categorised as follows: bulbar function (mild, moderate or severe); FVC (50–59%, 60–69% or ≥ 70%); age (≤ 39 years, 40–79 years or ≥ 80 years); and sex (male or female). The allocation incorporated a burn-in period of 10 participants (i.e. the first 10 participants were allocated using simple randomisation). Thereafter, allocation was performed using minimisation incorporating a random probability element of 80% into the allocation algorithm. That is each participant was allocated to the arm that reduced treatment imbalance with 80% probability and to the opposite arm with 20% probability.
The minimisation system was prepared by the study statistician; the study team did not have access to the allocation list in order to maintain allocation concealment. The study team and governance committees were blinded during the course of the study with the following exceptions: (1) the trial statistician and data management team had full access to unblinded outcome data; (2) the Data Monitoring and Ethics Committee (DMEC) had access to unblinded summaries of outcome data; (3) site staff were aware of the treatment allocation of their own participants; and (4) the chief investigator, study manager and site monitor were aware of individual participants’ allocations and AEs but did not see unblinded comparative summaries. The trial unblinded statistician was responsible for providing the DMEC with the accumulating unblinded data summaries, but did not attend the closed meetings during which the DMEC reached its decisions.
The original analysis plan was signed off after recruitment start, by which point one participant had died. The updated analysis plan was written by the unblinded trial statistician and endorsed with minimal suggestions by the unblinded DMEC; all other reviewers and approvers did so blinded to outcome data.
Interventions
Participants were randomly allocated to receive either standard care (NIV) or standard care in addition to DPS using the NeuRX/4 DPS.
Non-invasive ventilation
Non-invasive ventilation was initiated as per usual clinical practice at the study site after randomisation.
Diaphragm pacing system
The NeuRX DPS is licensed for use in patients with ALS (Conformité Européenne certificate number 518356). The FDA approved the device under HDE for patients with ALS who had stimulatable diaphragms and were experiencing chronic hypoventilation but who had not progressed to a FVC of < 45% of their predicted capacity.
The system is a four-channel intramuscular, motor point stimulation system. Intramuscular diaphragm electrodes are implanted laparoscopically into the diaphragm, with leads tunnelled to an exit site on the abdomen. The external part of the electrodes are connected to an external pulse generator (EPG), which is a small, white, portable box. A clinical station is used to programme each of the parameters (pulse frequency, pulse duration, inspiration time, pulse amplitude, pulse ramp and respiratory rate) on the EPG, which controls the stimulation delivered to the diaphragm via the implanted wires electrodes. The EPG provides repetitive electrical stimulation to the implanted electrodes allowing the diaphragm to contract in a manner that mimics breathing.
The Motor Neurone Disease Association provided funding for the purchasing of the NeuRX DPSs from the manufacturers of the device, Synapse Biomedical, which supplied them at reduced cost. The DPS system consists of components to both allow implantation of the electrodes into the diaphragm and its subsequent stimulation (Table 1).
| Item | Description |
|---|---|
| Clinical station | Clinical station used to map the diaphragm during surgery (by surgeon) to find the point of maximal contraction. Enables programming of the EPG unit to adjust wire parameters at subsequent visits (by PI/research nurse) |
| Electrode delivery instrument | To enable surgical implantation of the device. Used during the laparoscopic procedure by surgeon |
| Clinical implantation kit | Electrodes and instruments required for implantation. Used in theatre by the surgeon |
| Patient kit | Includes EPG control unit and cables required for each patient. Used by PI/research nurse at sites |
Members of the surgical team were responsible for clinical implantation of the system (see Surgical implantation). Theatre staff processed the electrode delivery instrument to allow its use in theatre including sterilisation and maintenance. Local procedures at each hospital for implanting the device were followed in order to complete documentation to allow sites to receive the system. Hospital medical equipment departments checked equipment prior to use following local hospital procedures.
Surgical implantation
On randomisation, the research nurse made arrangements for the participant to have DPS fitted. The time elapsed between randomisation and surgery varied according to availability, but the protocol encouraged investigators to fit the DPS within 8 weeks of randomisation. Participants were booked for a preoperative assessment up to 1 week prior to the operation to ensure their respiratory criteria remained within the safe range (FVC ≥ 45% and SNIP ≥ 30 cmH2O) and to check they were otherwise able to undergo the procedure safely. The research nurse liaised with the trial manager, surgeons and anaesthetists and members of Synapse Biomedical to organise a theatre slot and medical equipment for the procedure. A member of Synapse Biomedical was present at each operation to provide verbal assistance to the surgeon including mapping the diaphragm and implantation of electrodes at the point of maximal stimulation, as per current standard practice. Synapse Biomedical technical staff received honorary contracts to attend each surgical procedure. Guidance was also provided by Synapse Biomedical in setting the DPS parameters postoperatively.
The clinicians performing the procedure in the treatment arm were experienced surgeons who received training in the DPS implantation technique and were competent to perform the procedure. The training process was straightforward as the technique is a modification of a standard abdominal laparoscopic procedure, and mirrored that employed in the previous DP cohort study. 11 Training was provided by a surgeon from Synapse Biomedical. Given the simplicity of the implantation of electrodes for an experienced laparoscopic surgeon, all surgeons were judged competent by Synapse Biomedical after observation of one implantation procedure. Again this reflects the training of surgeons in the previous multicentre cohort study. A single surgeon undertook procedures in six of the centres; the seventh (London) referred participants in the intervention arm to undergo surgery at the Oxford site. At all surgeries, Synapse Biomedical technical staff were present to advise on implantation.
During the implantation procedure, incisions of 0.5–1 inch long were made in the abdomen. More than one incision was made to enable instruments to be passed through the abdominal wall as per standard laparoscopic procedure.
The surgeon identified the best location to place the electrodes within the diaphragm. A probe was used to temporarily place an electrode on the surface of the diaphragm and to stimulate the diaphragm muscle at several locations. Once the best locations were identified, the probe was removed and two electrodes were placed in each side of the diaphragm muscle. The lead wires from these electrodes travelled under the skin to the abdominal wall. The wires were trimmed so that the ends sticking out of the skin were only 2–6 inches in length. A radiograph was taken following the surgery to check the position of the wires and to make sure that no air had travelled above the diaphragm and into the chest. At the end of surgery the clinical station read out was printed for surgical quality control, which displayed the functioning stimulus connection for each electrode wire.
An assessment of phrenic nerve function formed part of the screening procedures, as described in this section. The purpose of this was to assess whether or not there was enough residual phrenic nerve function to enable meaningful stimulation of the diaphragm with the NeuRX DPS. However, it is only at the time of the operation when it is possible to know for certain if the diaphragm is stimulatable. If during the implantation procedure the diaphragm was not stimulatable (no contraction observed during mapping), then the operation was stopped and the device was not inserted.
Evaluation of electrodes and diaphragm pacing system training
Evaluation of the electrodes and DPS was performed prior to discharge from hospital. A system check of the wires was completed. Using the clinical station electrode evaluation was performed, by adjusting individual stimulus parameters (pulse amplitude, width and frequency) to achieve a comfortable level of stimulation for the diaphragm conditioning sessions. During the initial stimulation period, the participant’s vital signs were monitored for any abnormalities. The patient was given a daily target for the number and length of DP sessions, which was recorded by the study team member in the patient diary for the patient to refer to at home.
Training of the participant and their caregiver took place prior to discharge. This included instruction in the care and use of the stimulator and data collection in the patient diary. Verbal and written instruction was provided in a patient/caregiver instruction manual.
Prior to discharge, the participant/or carer was required to show proficiency to the treating clinician in the following:
-
cleaning and care of skin, wires and exit site
-
care and use of the stimulator
-
attachment and detachment of all components
-
completion of patient diary.
It was accepted that initiation of pacing could be deferred until the 1-week postoperative appointment to allow patients to adjust to having the device fitted in the immediate postoperative period. This was to match practice at all sites, as it was recognised that this enables the patient to recover after their operation. Patients were able to turn the EPG control unit ‘on’ or ‘off’ after programming to provide an adequate stimulus. The initial target for pacing sessions for ALS patients was five times per day, with each session lasting at least 30 minutes. Patients were asked to build up to this target over the first month. In the second month, patients were required to gradually lengthen the training sessions. When pacing was being performed for 6–7 hours a day, patients were instructed to switch from pacing during the day to using the pacing device overnight while asleep. At this stage patients could additionally use the pacing device during the day if they felt it benefited them, but this was their decision. Patients were asked to continue to use their NIV as advised by their study doctor.
A patient diary was given to the participant (on NIV initiation) to take home to record the amount of time spent on DPS and/or NIV.
Outcomes
Primary outcome
The primary outcome was overall survival.
Secondary outcomes
-
Quality-adjusted life-years, calculated from the European Quality of Life-5 Dimensions, three levels (EQ-5D-3L) health utility questionnaire.
-
QoL, measured by the EQ-5D-3L, Short Form questionnaire-36 items (SF-36) and Sleep Apnoea Quality of Life Index questionnaire (SAQLI).
-
QoL of the main carer, measured by the EQ-5D-3L and Caregiver Burden Inventory (CBI).
-
Safety and tolerability of the device.
-
Health economic objectives and resource use.
-
Perceptions of patients and carers regarding acceptability and impact of the device.
Initially, all of these outcome measures were assessed as per the original protocol; however, as a consequence of the trial’s early termination (see Chapter 3, Recruitment suspension), the above was amended. First, both the funder and sponsor agreed that given the substantial cost of the device and the apparent reduction in life-years in the pacing arm, the planned health economic analyses were now unnecessary. Second, the Trial Steering Committee (TSC) requested widening the scope of the statistical analyses to address usage of both NIV and DPS in relation to survival.
We also reported tracheostomy-free survival (TFS), defined as the time from randomisation to either death or the placement of tracheostomy, whichever occurred first. Although not preplanned, this outcome was added to aid comparability with other studies that have reported TFS.
Follow-up visits
Follow-up visits were conducted at clinic (at 1 week and at 2, 3, 6, 9 and 12 months). Some questionnaires were completed via post/e-mail when participants were unable to come into clinic.
Data collection and management
Data collection forms
Data for all participants were captured on the following key sources:
-
screening log – prescreening details containing reasons for ineligibility or non-participation
-
participant and carer consent forms – informed consent
-
case report form – all other study forms including eligibility, baseline assessments, randomisation, surgery, follow-ups (at 1 week and at 2, 3, 6, 9 and 12 months) study completion (withdrawal), AEs/SAEs, concomitant medications and, unscheduled DPS visits
-
patient diary – DPS and/or NIV usage.
Database
Trial data were entered into a validated bespoke web-based database system (Prospect) managed by the Sheffield CTRU in partnership with a University of Sheffield wholly owned subsidiary software development company, epiGenesys. Prospect stores all data in a PostgreSQL 9.1 (open-source software from The PostgreSQL Global Development Group) database on virtual servers hosted by Corporate Information and Computing Services at the University of Sheffield. Prospect uses industry standard techniques to provide security, including password authentication and encryption using Secure Sockets Layer/Transport Layer Security. Access to Prospect was controlled by usernames and encrypted passwords, and a comprehensive privilege management feature was used to ensure that users had access to only the minimum number of data required to complete their tasks. An automated audit trail recorded when (and by which user) records were created, updated or deleted. Prospect provides validation and verification features which were used to monitor study data quality, in line with CTRU’s standard operating procedures (SOPs).
Methods used for treatment allocation, sequence concealment and blinding
Patients were allocated their treatment (NIV alone or NIV plus DPS) by a method of minimisation, using baseline bulbar function, baseline FVC, age and sex as the minimisation factors. Factors were categorised as follows: bulbar function (mild, moderate or severe); FVC (50–59%, 60–69% or ≥ 70%); age (≤ 39 years, 40–79 years or ≥ 80 years); and sex (male or female). The minimisation was non-deterministic and incorporated a burn-in period of 10 participants and a random probability element of 80% into the allocation algorithm. In other words, the first 10 participants were allocated using simple randomisation; thereafter, each participant was allocated to the arm that reduced treatment imbalance with 80% probability and to the opposite with 20% probability. A centralised, web-based randomisation system hosted by the CTRU was used to allocate treatment allocations. Sites were able to log on to the system using a site-specific username and password. Researchers were prompted to enter patient details (identification number, date of birth and the minimisation factors) and to confirm consent and eligibility were complete. Following this, the randomisation system notified the user and the study manager of the treatment allocation.
The study was open label: it was not considered feasible to blind participants, carers or site staff.
Sample size
The sample size calculation was based on log-rank test, using Simpson’s rule19 as implemented in Stata version 11.1 (StataCorp LP, College Station, TX, USA) to allow for the unequal length of follow-up. The study duration comprised an 18-month recruitment period and a 12-month follow-up period, giving a maximum follow-up of 30 months and a minimum of 12 months. Assuming control group survival proportions of 45%, 20% and 10% at the minimum, average and maximum follow-up times, respectively, a hazard ratio (HR) of 0.45 and an additional 10% loss to follow-up, a total of 108 patients (54 per group) were needed to ensure a power of 85% using a two-sided type I error of 5%. The control group figures were conservative estimates based on the sole RCT of NIV,1 which is now considered standard care in the UK. We estimated the sample size on a conservative (but clinically important) 1-year difference in survival of 45% versus 70%, which produced the estimated HR of 0.45. It was expected that complete survival data would be available on all participants recruited, based on previous experience in ALS trials. We did, however, allow for a 10% loss to follow-up in the sample size calculation.
With regard to QoL data, we expected a low level of missing data due to loss to follow-up. We reviewed the patients who were initiated on NIV between July 2008 and June 2009 and had maintained contact with 100% of those patients surviving at 12 months. The appointment of a research nurse at each study site enabled home visits to collect the QoL data when necessary.
Early stopping
No interim analyses or early stopping was foreseen. However, in December 2013 the DMEC recommended that recruitment to DiPALS should cease on safety grounds (discrepancy in survival between the two treatment arms), and a final decision to stop the trial was made in June 2014. The recommendations and actions taken are reported fully in results (see Chapter 3).
Statistical methods
Survival
The primary end point was overall survival, defined as the time from randomisation to death of any cause. Participants were followed up after the last participant’s last visit to determine their final status, and participants who remained alive were censored on the date last known to be alive. TFS was defined as the time from randomisation to either death or the placement of tracheostomy, whichever occurred first.
The Kaplan–Meier method was used to visualise survival data and to derive summary statistics of median survival, interquartile range (IQR) and 95% confidence interval (CI). The median survival was defined as the point the Kaplan–Meier curve first reached 0.5; if the survival curve did not drop this far, then the median survival is stated as ‘not reached’. The IQR and the CI for the median are defined analogously. Overall survival and TFS were compared between the groups using the log-rank test and modelled using Cox proportional hazards regression using the Efron adjustment for tied survival times and with the minimisation factors as covariates. The primary analysis was the Cox regression (i.e. adjusted) analysis. Pretrial modelling found Cox proportional hazards to be the best fit to previous data, but the Cox proportional hazards assumption was checked by adding time-dependent covariates and graphing scaled Schoenfeld residuals against time. 20 If Cox proportional hazards were found not to fit the data adequately, an accelerated failure time alternative was fitted and the adequacy of its fit assessed using Q–Q plots. 21 Finally, if this too did not fit, the non-parametric restricted mean survival analysis,22 derived from the area under the Kaplan–Meier survival curves, was used as the basis for summarising the treatment effect.
The overall survival was also reported as of the point at which the DMEC made the decision to (1) suspend the trial; and (2) terminate the trial with advice to stop pacing. In both of these additional analyses, participants who were randomised to pacing but did not receive it as a result of the DMEC decision (two patients) were excluded.
Quality of life
Quality of life was analysed both longitudinally (i.e. over the duration of the trial rather than at individual time points) and at the end of the study follow-up (12 months). Longitudinal analyses were performed using generalised least squares regression, with the baseline value and minimisation factors as covariates and the patient as a random intercept. End-of-study values were analysed using ordinary least squares regression, with the baseline value and minimisation factors as covariates. For each participant measure, a complete case analysis was followed by an analysis based on imputed data. First, in the case of participants for whom data for any visit were missing but data on either side were available (e.g. no month 2 data but baseline and month 3 data available), the value was imputed using the trapezoid rule. When absence of data persisted (e.g. because the participant withdrew from the study), the missing data were imputed using multiple imputation with chained estimation23 using Rubin’s rules and 50 imputations; the imputation model used the participant’s age, sex, rate of prerandomisation Amyotrophic Lateral Sclerosis Functional Rating Scale – Revised (ALSFRS-R) score decline, FVC, treatment group and any data at other time points for the instrument. Missing data arising because of participant death were not imputed other than for EQ-5D-3L, as the primary objective was to assess QoL among participants while they remained alive. No attempt was made to impute missing data for the carer QoL.
The following QoL instruments and measures were collected for participants:
-
European Quality of Life-5 Dimensions, three levels: health utility/tariff score and health status (‘thermometer scale’): the EQ-5D-3L questionnaire comprises six questions. Questions 1–5 are 3-point scales covering mobility, self-care, usual activities, pain/discomfort and anxiety/depression. The responses to these questions map onto a health state in which 1 corresponds to perfect health and 0 to death. Negative values are possible; these are interpreted as a state worse than death. Question 6 is a standalone question that asks the participant to rate their overall state of health today on a continuous scale between 0 and 10.
-
Short Form questionnaire-36 items (version 1): aggregate physical health and aggregate mental health: the SF-36 was used to derive overall physical and mental health among the participants. Both scores are scaled such that the general population has a mean score of 50 and a standard deviation of 10, with higher scores indicating a better QoL.
-
Sleep Apnoea Quality of Life Index questionnaire: the SAQLI is a single-domain questionnaire comprising 14 questions, each scored from 1 to 7: the SAQLI score is the average. When the questionnaire is incomplete (i.e. < 14 questions are answered), the overall score was defined as the average, provided at least half of the questions (7 of the 14) had been answered. Higher scores indicate a better QoL.
The following QoL instruments and measures were collected for carers:
-
European Quality of Life-5 Dimensions, three levels: health utility/tariff score and health status (‘thermometer scale’): the questionnaire was identical to that provided to participants.
-
Caregiver Burden Inventory: the CBI is a one-domain questionnaire comprising 24 questions, each of which is scored from 0 to 4; the overall score is the total of these. Incomplete questionnaires are scaled up proportionate to the level of missing data unless 12 or fewer questions had been answered. Higher values indicate better QoL.
All questionnaires were completed at the screening visit and the at subsequent visits (2, 3, 6 and 12 months), the EQ-5D-3L (for participants and carer) was also completed at 9 months.
The analysis of EQ-5D-3L was conducted in two ways. First, an analysis of health status was conducted using data (possibly imputed) over the duration for which the participant was still alive. Second, the analysis was performed for all time points but with a score of 0 (which corresponding to a state of death) used following participant death. The two analyses answer different questions: the first is the health among survivors; and the second is the health of the population as a whole. The SF-36, SAQLI and CBI measures were analysed only for the duration in which the participant remained alive.
The EQ-5D-3L was further reported by subgroups (NIV tolerance and bulbar function). Testing for differential treatment effect between subgroups would necessitate a three-way interaction (treatment group × subgroup × time), which given the sample size would produce potentially unstable coefficients. Therefore, the focus here was on within-group summary statistics and graphical displays, separately by treatment group.
Non-invasive ventilation and diaphragm pacing usage
The original analyses of DPS and NIV usage were based primarily on diary data, augmented by participant recall at each visit when diary data were incomplete. Later on in the trial we were able to collect NIV usage data directly from the NIV machine itself, and this was the preferred source of average usage when available. Average NIV usage was defined as the average number of hours used from the date of NIV initiation onwards, and average DPS usage was defined as the average daily use from the date of procedure onwards.
The relationship between NIV and DPS usage by time point was also assessed. As the relationship between typical adherence (in hours) and survival was not expected to be linear, fractional polynomials were used to assess the fits of quadratic and other non-linear relationships. 24 Finally, usage was defined in categories. For NIV we followed the approach of Kleopa et al.,13 who characterised participants as non-adherent (typical usage < 1 hour per day), low-adherent (typical usage 1 to < 4 hours per day) and good adherence (typically ≥ 4 hours per day). Adherence to pacing was not categorised as (unlike NIV) normative data on optimal usage are not available, and also because of the small numbers within each category. Participants whose NIV or DPS adherence could not be determined based on the available data were excluded from these exploratory analyses.
Health resource use
Health resource use was summarised as the use of each of the following:
-
health service use – hospital admission, emergency department attendance, minor injury clinic or walk-in centre or general practitioner (GP)
-
respiratory device – cough assist machine, breath stacking, suction
-
health and social care – physiotherapist, occupational therapist, other
-
additional care/support – formal (e.g. home help) and informal (e.g. family/friends).
Additional outcomes: Amyotrophic Lateral Sclerosis Functional Rating Scale and respiratory function
In the light of the early stopping, the TSC requested additional respiratory function data be collected to augment that which was already collected at baseline (and for DPS, immediately pre surgery). Specifically, we wished to assess (1) whether or not the groups were comparable at baseline; (2) whether or not decline among the NIV plus DPS group appeared more pronounced in the postsurgery period than in the NIV group; and (3) the trajectory across time in general to see if it offered any other clues with which to explain the findings. We were able to obtain data at some, but not all, sites for FVC, arterial carbon dioxide and ALSFRS-R.
Adverse events
Adverse events were coded by the chief investigator blind to the participant’s treatment group. AEs were summarised for each AE category and, overall, as the number and percentage of patients affected and as the number of events in total (as a patient may have more than one occurrence of the same AE). The summary was repeated for SAEs. All AEs that were adjudged related to pacing (either probably or definitely) are listed as recorded. Summaries are presented based on the randomised group (i.e. intention to treat), but any NIV plus DPS group AEs reported by non-implanted participants were highlighted.
General analysis considerations
All treatment comparisons use the NIV-only group as the reference (comparator); all statistical exploratory tests of main effects were two-tailed with α = 0.05; and all CIs were two-sided, with 95% intervals. A permutation test was used to confirm the p-value from the primary end point. 25 As interaction tests have low statistical power, consideration was given to p-values below 0.1 when testing interactions (treatment × centre and treatment × subgroups).
Analyses were by intention to treat, with preplanned secondary analyses of overall survival based on protocol adherence and NIV usage. Analyses were undertaken using Stata version 12.1 and SAS 9.4 (SAS Institute Inc., Cary, NC, USA).
Economic evaluation
Prior to the trial commencing, a modelling exercise was undertaken to assess the feasibility that pacing could be cost-effective at standard willingness-to-pay thresholds for end-of-life care. This modelling confirmed that pacing would be cost-effective if the trial were to demonstrate a treatment effect of similar magnitude as demonstrated by the SSPB cohort in comparison with historical data. A full cost–utility analysis was planned for this trial, but following the early termination of the trial the chief investigator and funder agreed that this was no longer necessary.
Research governance
Sponsorship
Sheffield Teaching Hospitals NHS Foundation Trust sponsored the trial (reference STH15625).
Oversight committees
Oversight committees were established to govern study conduct: Trial Management Group (TMG), TSC and DMEC. The trial was conducted in accordance with CTRU’s SOPs, with committees convening at appropriate intervals as dictated by both study requirements and SOPs (see Acknowledgements).
The TMG consisted of the chief (chairperson) and principal investigators (PIs) and key staff within the CTRU, and this committee met monthly via teleconference during trial recruitment and follow-up. An independent consultant neurologist chaired the TSC, other external members comprising a respiratory clinical expert, an independent statistician and two lay representative/patient and public involvement (PPI) members. All TSC members were appointed by the Health Technology Assessment programme. The DMEC consisted of an independent statistician, clinical neurologist (chairperson) and consultant neurologist. The TSC received formal recommendations from the DMEC.
Research Ethics Committee
Cambridge Central National Research Ethics Service committee (reference 11/EE/0026) approved the trial and all subsequent amendments.
Medicines and Healthcare products Regulatory Agency
The intervention was used within its licence for intended use with appropriate Conformité Européenne mark documentation. Medicines and Healthcare products Regulatory Agency approval was not applicable.
Serious adverse events
Adverse events were reported in accordance with the CTRU’s AEs and SAEs SOP (PM004) and supplementary study-specific guidance approved by the sponsor.
Expected disease progression was specified in the protocol as an expected AE that did not need to be reported. Other expected AEs were listed with the requirement to report: chest infection requiring the use of antibiotics, an infection at the site where DPS was fitted and revision of the DPS.
Protocol non-compliances
Protocol non-compliances were reported in accordance with CTRU’s non-compliance SOP (PM011), but also additional study-specific guidance approved by the sponsor. Non-compliance categories were prespecified with the trial sponsor and are listed below. Major non-compliances were reported to the sponsor and immediate action taken; minor non-compliances were recorded by the CTRU and reported periodically to trial oversight committees and the sponsor.
Prespecified major non-compliances:
-
Participants found to be ineligible following randomisation.
-
Action: participant withdrawn.
-
-
Consent procedure or good clinical practice not followed correctly (e.g. patient not consented).
-
Action: participant reconsented.
-
Prespecified minor non-compliances:
-
completion of any baseline data post randomisation
-
minor errors on the consent form
-
patient did not receive allocated treatment
-
time-specific windows stated in the protocol missed (e.g. participant not randomised or informed of the arm allocated to within 7 days of screening).
Monitoring and reporting
The level and type of monitoring was informed by a risk assessment conducted during the set-up period of the study. A Data Monitoring and Management Plan and a Monitoring Plan were written and agreed with the sponsor prior to the start of trial recruitment. On-site and central data monitoring activities were completed to ensure participant safety, protocol compliance and data integrity.
Central monitoring
Data were centrally monitored based on parameters specified in the Data Monitoring and Management Plan. Checks included point of entry and post-entry validation checks and verification of data entry.
Site monitoring
The trial study manager completed a site initiation and training visit with research staff at sites prior to participant recruitment. Subsequent visits were conducted after sites had started recruitment to check the ongoing suitability of the site and to perform source data verification. Monitors checked data recorded on the case report form against medical records, discussed recruitment and issues with intervention delivery, SAEs/AEs, resolution of data queries and maintenance of the site file. A final closeout visit was performed at each site at the end of participant recruitment, scheduled once data collection was complete.
Monitoring issues were initially highlighted and discussed with sites, and remedial actions sent to the research nurse and PI. Any problem themes identified and specific issues with sites were discussed with the chief investigator and when required escalated to the sponsor, TSC and DMEC.
Reporting
The trial team were required to submit annual reports on trial progress, data completion rates, and safety and protocol compliance to the REC; and 6-monthly reports to the funding body.
Important changes to methods after trial commencement
Details of substantial amendments submitted to the REC, which were important changes to trial methodology, are listed below.
November 2012: amendment 5 (minor)
Protocol version 3 specified that participant be initiated onto NIV (in both arms) within 1 to 2 weeks of randomisation. The protocol was amended to allow sites to initiate NIV as per usual clinical practice after the participants enrol onto the trial and not necessarily before DPS implantation. This distinction was necessary as, although participants were experiencing respiratory insufficiency based on their clinical assessments, clinicians wished to initiate NIV when participants were more symptomatic in line with their standard practice. The trial TSC were in agreement with this rationale. The protocol was also amended to allow DPS implantation to ideally occur within 8 weeks of randomisation based on the practical feasibility of getting participants into theatre.
June 2013: amendment 6 (substantial)
The protocol was clarified to state that blood gases were required only to assess PaCO2 levels (inclusion criteria 4e); if alternative respiratory measures were used for eligibility assessment, blood gases were not required. A member of Synapse Biomedical (manufacturers of the device) was present at each operation as stated in the protocol; however, as sites (surgeons, research nurses and clinicians) gained experience and became competent in the use of the device, it was felt appropriate that the protocol be amended. This change was also reflective of standard practice for having DPS fitted worldwide. The wording in the protocol was amended to:
A member of Synapse will attend each procedure until sites become competent with use of the device to manage patients independently. The local site PI will be responsible, after liaising with local site staff, deciding when site staff are competent in performing the intervention without any input from Synapse. The Surgeon at the site will self-certify their competency to perform the operation independently at this stage.
Although this change was approved by the REC, none of the surgeries was performed independently and a member of Synapse Biomedical was present at each operation to provide verbal assistance during the procedure. The protocol was amended to allow patients not to start pacing until the 1-week postoperative appointment to allow patients to adjust to having the device fitted in the immediate postoperative period and to ensure standardisation of the process across sites. This amendment also added both postal or e-mail options to optimise data collection when it was difficult for patients/carers to attend in person.
October 2013: amendment 7 (substantial)
The protocol was amended to allow respiratory tests up to 2 weeks pre consent to be used for eligibility assessment. This was felt an appropriate cut-off point to not overburden participants with repeat tests while still ensuring that respiratory function would not have significantly changed. As part of this amendment, the instructions on the participant diaries were changed and a newsletter template was approved to both provide more clarity on how to collect the data but also to emphasise the importance of doing so.
November 2013: amendment 8 (minor)
The previous protocol allowed self-reported NIV data to be collected from participants. As the study progressed, it was apparent that more detail was required to build a picture a more accurate picture of NIV use including NIV data collected directly from the NIV machines. The protocol was amended to allow NIV usage data from the machines to be collected.
January 2014: amendment 9 (substantial)
Following the DMEC’s recommendation to the TSC, recruitment into the trial was halted and participants randomised to DPS but who had not undergone surgical implantation were not to do so. Otherwise, participant follow-up was to continue as planned for those already in the trial.
June 2014: amendment 10 (substantial)
Researchers clarified the protocol to allow sites to initiate NIV post consent rather than post randomisation. As the screening process could take a few weeks, it was accepted that NIV could be initiated based on clinical need for participants in this period between consent and randomisation. The collection of routine respiratory and ALSFRS-R data was added to the protocol during the 12 months of participants’ involvement in the trial. This was to aid the analysis about the rate of participant deterioration over the course of the trial.
June 2014: amendment 11 (substantial)
Following unblinded data review on 23 June 2014, the DMEC recommended that participants in the pacing arm should be advised to discontinue using the DPS unless they specifically requested otherwise. All participants should remain under follow-up as scheduled. A specific SOP detailing discontinuation of DP at sites was written and submitted as part of the amendment, together with a GP letter and letters for participants still in the trial (one for each arm) to inform them of this decision and what to do next. Provision was made such that any participant wishing to continue using the DPS would be allowed to do so.
September 2014: amendment 12 (substantial)
A substantial amendment was submitted to allow central University of Sheffield researchers to interview site staff in relation to running the DiPALS trial locally. The interviews explored the experiences of the research staff recruiting participants, particularly with regard to the population (ALS participants at a later stage of disease); intervention (surgical intervention involving general anaesthesia, compliance with the intervention and standard care); and other barriers and facilitators to recruitment.
Patient and public involvement
Patient and public involvement was sought throughout the trial. The Sheffield Motor Neurone Disease Research Advisory Group (SMNDRAG), an independent research advisory group, was approached and agreed to be involved at an early stage. The SMNDRAG was established in 2008 following the principles of INVOLVE. 26 The group comprises members of the public, including several carers of ALS patients, patients and other lay individuals who responded to the call for members. Research training is provided to all members of the public that volunteer for the SMNDRAG as part of their induction and ongoing support. The SMNDRAG has been a valuable part of the DiPALS trial, collaborating as part of the research team at all stages of the research process.
Proposal development
The SMNDRAG was consulted about the concept, research question and design at the proposal application stage. SMNDRAG members collaborated in writing the proposal, with particular input to the lay summary. They also suggested modifications to the protocol; an example of this was the addition of methods to capture carer experience, using both CBI and qualitative interviews.
During the trial
Following the funding decision, the SMNDRAG was involved during the development of the trial protocol and associated essential documents. Members were asked periodically to comment on key changes to participant materials when required.
Study oversight
A PPI member from the SMNDRAG was invited to attend the monthly TMGs for day-to-day running of the trial. The trial TSC had two independent members (PPI and lay person) to assist in governing trial conduct and provide advice from the patient/lay perspective.
Study dissemination
At the end of the study, the SMNDRAG members assisted in dissemination of the trial results through networks that have been established regionally and nationally, and in reviewing the plain English summary.
Chapter 3 Early stopping
Trial recruitment was stopped earlier than planned on safety grounds. The recommendations from the DMEC were accepted in full, and the subsequent changes to trial conduct and recruitment are detailed in this chapter.
Recruitment suspension
The fourth DMEC meeting was held on 16 December 2013. The ‘open’ part of the meeting discussed trial progress with both internal and external DMEC members. This was followed by a ‘closed’ meeting when the external, independent members reviewed unblinded safety data.
Following the closed meeting, the chairperson of the DMEC contacted the chief investigator on 18 December to recommend that the trial suspend recruitment temporarily based on safety grounds, citing a discrepancy in survival between the two arms. In doing so, the DMEC acknowledged that the sample size was relatively small (74 randomised, 24 deaths at the point of this recommendation), and that their decision would be reviewed as additional data became available.
The DMEC provided the following response:
The DMEC has reviewed the unblinded data on survival in the DiPALS study and the members of the Committee are unanimous in recommending that recruitment should cease as soon as practicable for reasons of safety, that monitoring of safety and survival should continue, and that the DMEC should review the updated survival data at the end of February 2014, and periodically thereafter.
Suspension of recruitment did not constitute an ‘urgent safety measure’.
In summary, the DMEC recommended that:
-
recruitment be suspended with immediate effect
-
implantation of new pacing devices be suspended
-
other aspects of the trial remain unaltered; in particular, patients in the pacing arm should be encouraged to continue using their device.
The CTRU suspended the online randomisation system on the same day to ensure that no further patients were recruited to the trial. The TMG (PIs, co-investigators, research nurses and relevant members of the CTRU) were all notified of this decision immediately (18 December 2013). All planned surgeries for the DPS were cancelled. The PIs and chief investigator discussed the key message to give to any concerned trial participants while still remaining blinded to the data.
The TSC convened on 20 December 2013 to discuss the implications of this decision and their concerns that the data had not been thoroughly scrutinised. These included the consideration of centre effects; the effect of patient withdrawal and non-implantation with the device; chance; and non-compliance with either NIV or DPS. A second unblinded report was produced and circulated to only the external, independent DMEC members that incorporated analyses to ensure all these issues were considered.
A joint TSC and DMEC meeting was held on 13 January 2014 to allow all members to present any remaining concerns before responding to the DMEC. The TSC and study team remained blinded to trial results. The DMEC upheld its initial decision by providing the following response to the additional data analyses:
The DiPALS DMEC members, having reviewed the updated unblinded report and having considered the points raised during discussion with the TSC, remain unanimous in their advice that recruitment into the DiPALS trial should cease, but that follow-up should continue. This advice is based on analysis of the primary end point (patient survival during the course of the trial). The DMEC considers that secondary analyses are unlikely to alter the safety concerns raised by the primary intention-to-treat analysis. The DMEC will be happy to consider further reports and to provide advice as required.
The TSC jointly agreed and communicated its agreement with the DMEC to suspend recruitment to the trial team late on 15 January 2014. The TSC requested that the DMEC continue to review unblinded data every 3 months to capture any further changes that would warrant discontinuation of the use of the DPS for participants in follow-up.
Informing the Research Ethics Committee
The acting trial manager informed the REC on 6 January 2014 of the initial decision to suspend recruitment, with a further notification to the REC on 15 January 2015 after the final TSC decision. A formal substantial amendment was submitted to the REC on 5 February 2014. A full account (see Recruitment suspension) was provided to the REC as part of the amendment. The sponsors of the trial (Sheffield Teaching Hospitals) approved the amendment, and sought direct assurance from the chairperson of the DMEC that participants already in the trial would continue to be followed up as per protocol, that is would continue to receive the intervention as prescribed. The CTRU informed the REC that as the central study team (chief investigator, and study and data managers) and all site study staff (PIs and research nurses) remain blinded to the data disclosed in the closed part of DMEC they would be unable to provide any further detail than that provided. The REC was asked to contact the chairperson of the DMEC for any further assurance regarding the decisions made.
As trial participants remained in follow-up and continued to receive the DPS intervention, they were not informed of the decision to halt recruitment of new participants into the trial.
Withdrawal of participants from diaphragm pacing system
The DMEC continued to review unblinded data and the next meeting was convened on 24 March 2014. The DMEC reviewed further deaths in each group and other safety and tolerability data. The DMEC requested that the chief investigator consider a pathway for formal participant withdrawal from the intervention, which would need to be ethically approved should this be required. Following this meeting there was no change in the study status.
The subsequent DMEC meeting was held on the 23 June 2014. The following recommendations were made by the DMEC after the closed session:
-
As the survival data suggest that DP poses an ongoing safety risk compared with standard care, in the interest of safety, DPS should be stopped in all participants using the approved process.
-
Participants, however, subject to consent, should continue to be followed up and data acquisition should continue until the planned end of the trial (December 2014).
The DMEC also wished to review the statistical analysis plan (see Appendix 13) prior to database lock and advised that the trial team (chief investigator, PIs and other central study staff) remain blinded to the data prior to final database lock and analysis. The DMEC advised that the study group inform the REC and discuss the withdrawal procedure with PIs and then withdraw patients as soon as practicably possible.
Formal stopping of recruitment
The recommendation to formally stop recruitment was made by the DMEC in June 2014. This time, its recommendations went further:
-
Participants in the pacing arm were to be informed of the concern and advised to cease use of their device forthwith (unless the patient and their clinician believed there were just grounds to do otherwise).
-
Trial follow-up was to continue until all participants had either died or completed the 12-month follow-up.
-
The TSC and trial team were to remain blind to the outcome data until this time.
Informing Research Ethics Committee, participants and investigators
The chief investigator and study manager drafted the following documentation to inform trial participants still in follow-up within the trial about the decision to stop pacing (see Appendices 14–17):
-
stop pacing participant letter – DPS arm
-
stop pacing participant letter – NIV arm
-
stop pacing GP letter – DPS arm
-
pacing discontinuation SOP.
A TMG meeting was held with study PIs the following day (24 June 2014) to review all drafted documents and agree the process. It is important to note that all the study team remained blinded to the data at this stage. Any modifications suggested to the process or documents were agreed at the meeting. Amended documents were submitted to the REC as part of a substantial amendment on 27 June 2014. Prior to this, the study manager sought confirmation about the nature of risk posed by the intervention; if there was an immediate risk, then urgent safety measures would be required. The DMEC confirmed that there was no evidence to suggest any immediate risk to participants in the DPS arm and, therefore, a more gradual approach was appropriate.
Research Ethics Committee approval was received on 4 July 2014. Individual research and development departments are required to approve a substantial amendment or raise no objections within 35 days of its valid receipt. Researchers did not wish to wait in order to implement this amendment and, therefore, the trial manager e-mailed, telephoned and spoke with each research and development department to approve the amendment to avert any delays in its implementation. The trial manager informed each PI and research nurse as soon as local approval was received. The chief investigator asked site PIs to use the approved documentation and process to directly inform participants of the study status and answer any questions that arose. The study manager asked to be notified of the status of each participant in the DPS arm after the conversation with the participant via the return of the participant discontinuation checklist.
The Motor Neurone Disease Association was informed of the advice to stop pacing. This was communicated to motor neurone disease patients via their website. The DiPALS trial website was updated to inform potential participants that the study was no longer in recruitment.
Chapter 4 Trial results
Recruitment and participant flow
In total, 74 participants (37 per arm) were randomised between 5 December 2011 and 18 December 2013, when the DMEC recommended that recruitment cease. Study follow-up concluded in December 2014, by which time 47 patients had died; one patient was last followed up in August 2014, with the remaining 26 known to be alive in December 2014 (Figure 2).
FIGURE 2.
Trial profile (CONSORT diagram). MND, motor neurone disease; NK, not known.

Baseline data
A total of 74 participants were allocated (37 to each arm). The characteristics of the participants are presented in Table 2. Participants were predominantly male with sporadic (usually limb) onset and with mild bulbar impairment (74%). Despite age being included as a minimisation factor, the NIV plus DPS arm was slightly older (average 60 vs. 54 years), a consequence of most participants falling into the middle category of age 40–79 years; this imbalance was addressed in all comparisons, however, as all prespecified analyses included age as a covariate (continuous, rather than the categorisation used for the purpose of minimisation).
| Variable | Trial arm | |
|---|---|---|
| NIV plus DPS (N = 37) | NIV (N = 37) | |
| Centre, n (%) | ||
| Leeds | 2 (5) | 5 (14) |
| London | 1 (3) | 1 (3) |
| Manchester | 6 (16) | 4 (11) |
| Newcastle | 6 (16) | 2 (5) |
| Oxford | 11 (30) | 13 (35) |
| Plymouth | 3 (8) | 3 (8) |
| Sheffield | 8 (22) | 9 (24) |
| Age (years)a | ||
| Mean (SD) | 60 (9.7) | 54 (12.0) |
| Median (range) | 61 (34–83) | 53 (23–76) |
| Trial subgroup, n (%) | ||
| < 40 years | 1 (3) | 3 (8) |
| 40–79 years | 35 (95) | 34 (92) |
| ≥ 80 years | 1 (3) | 0 |
| Sex, n (%)a | ||
| Male | 29 (78) | 29 (78) |
| Female | 8 (22) | 8 (22) |
| Bulbar score (%)a | ||
| Mild (9–12) | 26 (70) | 29 (78) |
| Moderate (5–8) | 8 (22) | 6 (16) |
| Severe (0–4) | 3 (8) | 2 (6) |
| FVC (%)a | ||
| Mean (SD) | 66.1 (12.3) | 64.6 (12.1) |
| Median (range) | 62.5 (51–105) | 62.5 (42–97) |
| Trial subgroup, n (%) | ||
| 50–59% | 13 (35) | 16 (43) |
| 60–69% | 12 (32) | 10 (27) |
| ≥ 70% | 11 (30) | 10 (27) |
| Missingb | 1 (3) | 1 (3) |
| ALS onset type, n (%) | ||
| Sporadic | 34 (92) | 35 (95) |
| Familial | 3 (8) | 2 (5) |
| ALS onset site, n (%) | ||
| Limb | 26 (70) | 28 (76) |
| Bulbar | 10 (27) | 6 (16) |
| Respiratory | 1 (3) | 1 (3) |
| Mixed | 0 | 2 (5) |
| ALS diagnosis, n (%) | ||
| Clinically definite | 26 (70) | 22 (59) |
| Clinically probable | 7 (19) | 9 (24) |
| Clinically probable, laboratory supported | 4 (11) | 6 (16) |
| Time from symptom onset to randomisation (months) | ||
| Mean (SD) | 22 (18) | 22 (15) |
| Median (range) | 17 (4–89) | 18 (3–66) |
| Trial subgroup, n (%) | ||
| < 12 months | 12 (32) | 14 (38) |
| 12–24 months | 14 (38) | 12 (32) |
| ≥ 24 months | 11 (30) | 11 (30) |
| ALSFRS-R score | ||
| Mean (SD) | 32.9 (7.4) | 33.7 (6.5) |
| Median (range) | 33 (21–46) | 35 (21–46) |
| Rate of ALSFRS-R score decline/monthc | ||
| Mean (SD) | 0.99 (0.68) | 0.94 (0.71) |
| Median (range) | 0.80 (0.02–2.92) | 0.92 (0.20–3.72) |
Primary outcome (overall survival)
Primary overall survival analyses
The Kaplan–Meier curve for overall survival is presented in Figure 3. The median survival was 11.0 months (95% CI 8.3 to 13.6 months) in the pacing arm and 22.5 months in the control arm. As the upper bound of the Kaplan–Meier survival curve never reached 50%, the upper limit of the 95% CI is unknown: the lower bound is 13.6 months. The HR (adjusting for minimisation covariates) was 2.27 (95% CI 1.22 to 4.25; p = 0.01), indicating the risk of death at any point in time was higher in the NIV plus pacing arm than in the control arm. A permutation test approach, as recommended by Scott et al. ,25 produced a p-value of 0.013 confirming the statistical significance of this difference.
FIGURE 3.
Overall survival.
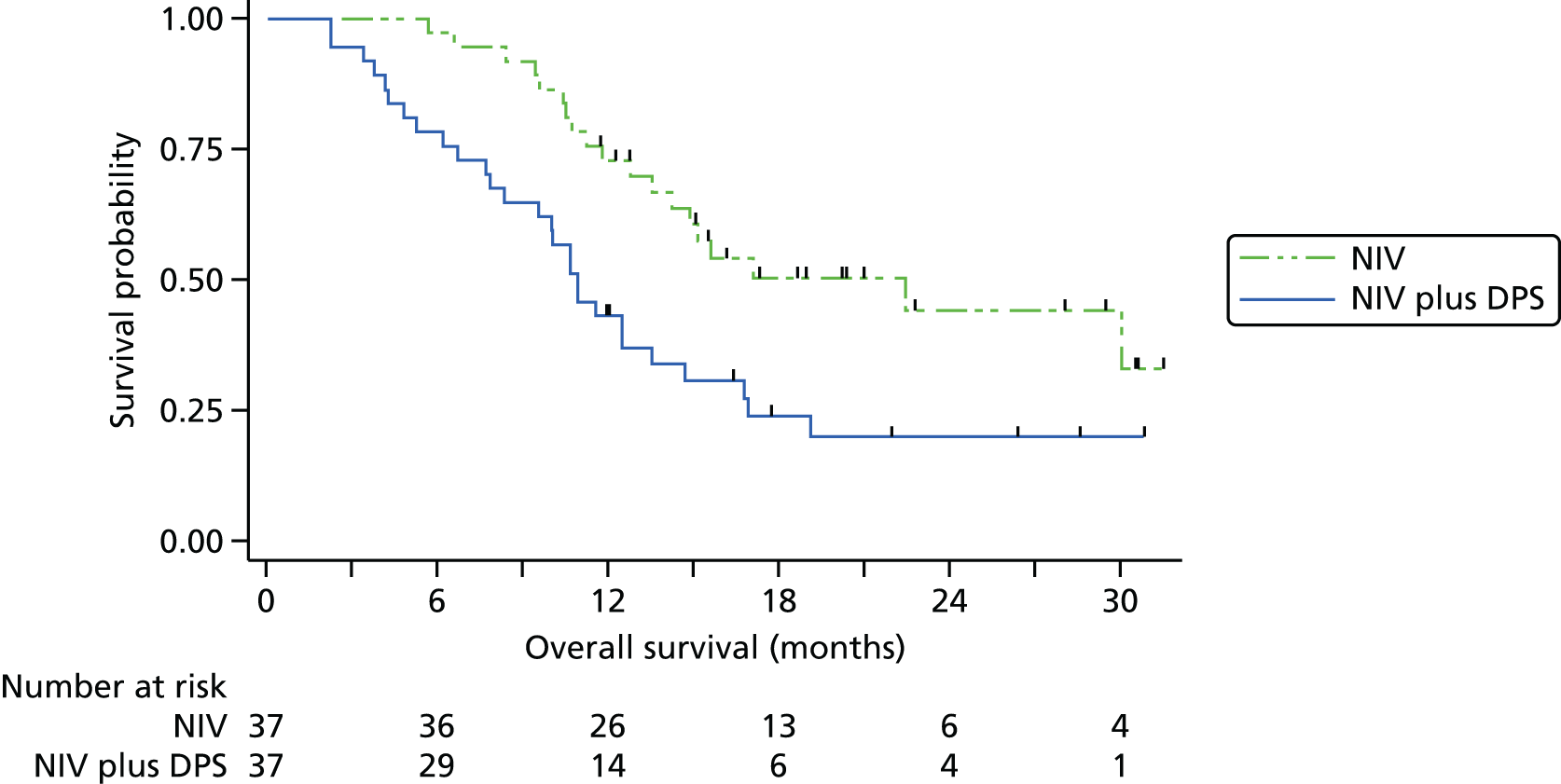
Sensitivity analyses were undertaken to assess the consistency of findings to different covariates and to different models. First, we allowed for between-site differences via a stratified log-rank test in which the strata comprised the seven hospitals. The findings from doing this were not materially changed. There was, however, evidence of non-proportionality of the hazards of the two groups, as detected by the Grambsch–Therneau test20 for the correlation between residuals and time (ρ = –0.31; p = 0.03). In other words, although overall NIV plus DPS was associated with a twofold increase in hazard, the impact was greater in early months and smaller among longer-term survivors. Two alternative modelling approaches were fitted: first, an accelerated failure time model (in which the time, rather than hazard, is modelled); and, second, a non-parametric restricted mean survival time, in which the difference in mean survival is estimated based on the area between the Kaplan–Meier curves. 22 The findings from the sensitivity analyses analysis are presented in Table 3. Overall, the magnitude of the survival deficit in the NIV plus DPS group was remarkably consistent, with survivorship always significantly lower in the NIV plus DPS group.
| Method of analysis | Trial arm | Comparison | ||
|---|---|---|---|---|
| NIV plus DPS | NIV | |||
| Cox proportional hazards regression | Deaths per person-year follow-up | HR (95% CI) | p-value | |
| Log-rank (univariate) | 0.74 | 0.37 | 2.24 (1.30 to 4.19) | 0.005 |
| Log-rank (stratified by centre) | – | – | 2.02 (1.21 to 3.84) | 0.012 |
| Cox regression (univariate) | – | – | 2.28 (1.27 to 4.10) | 0.006 |
| Cox regression (including minimisation factors as covariates) | – | – | 2.27 (1.22 to 4.25) | 0.012 |
| Accelerated failure time regression | Median survival time (months) | Time ratio (95% CI) | p-value | |
| Gehan–Wilcoxon test (univariate) | 11.0 | 22.8 | – | 0.002 |
| Gehan–Wilcoxon test (stratified by centre) | – | – | – | 0.029 |
| Log-normal regression (univariate) | – | – | 0.52 (0.36 to 0.77) | 0.001 |
| Log-normal regression (including minimisation factors as covariates) | – | – | 0.52 (0.35 to 0.77) | 0.001 |
| Restricted mean failure time | Mean survival time over first 30 months | Difference (95% CI) | p-value | |
| Univariate | 13.7 | 20.6 | –6.9 (–11.4 to –2.4) | 0.003 |
| Including minimisation factors as covariates | – | – | –6.8 (–11.5 to –2.2) | 0.005 |
Overall survival at Data Monitoring and Ethics Committee intervention
The DMEC did not prespecify rules for stopping because of safety, preferring to use clinical judgement about participant outcomes as a whole. Nevertheless, its recommendations were based primarily on overall survival, and for reference we present overall survival as of the following dates:
-
10 December 2013 (the date that data were seen by the DMEC, when it recommended suspension of recruitment and implantation)
-
10 June 2014 (the date that data were seen by the DMEC, when it recommended participants in the NIV plus DPS arm should cease pacing).
The statistical significance of the overall survival difference varied markedly during the data collection phase, both across time and also according to which of the aforementioned models were used to derive it. Nevertheless, as shown in Table 4 and Figures 4 and 5, overall survival was significantly worse in the NIV plus DPS group at both time points.
| Data collection time point | Trial arm, number (%) of deaths | Intention-to-treat comparison (univariate) | |||
|---|---|---|---|---|---|
| NIV plus DPS | NIV | ||||
| All patients (n = 37) | Excluding non-implanted (n = 32) | All patients (n = 37) | HR (95% CI)a | p-value | |
| As of 10 December 2013 | 16 (44b) | 14 (44) | 8 (22b) | 2.45 (1.05 to 5.74) | 0.039 |
| As of 10 June 2014 | 23 (62) | 21 (66) | 11 (30) | 2.73 (1.33 to 5.60) | 0.006 |
| At end of study | 28 (76) | 26 (81) | 19 (49) | 2.28 (1.27 to 4.10) | 0.006 |
FIGURE 4.
Survival at 10 December 2013.
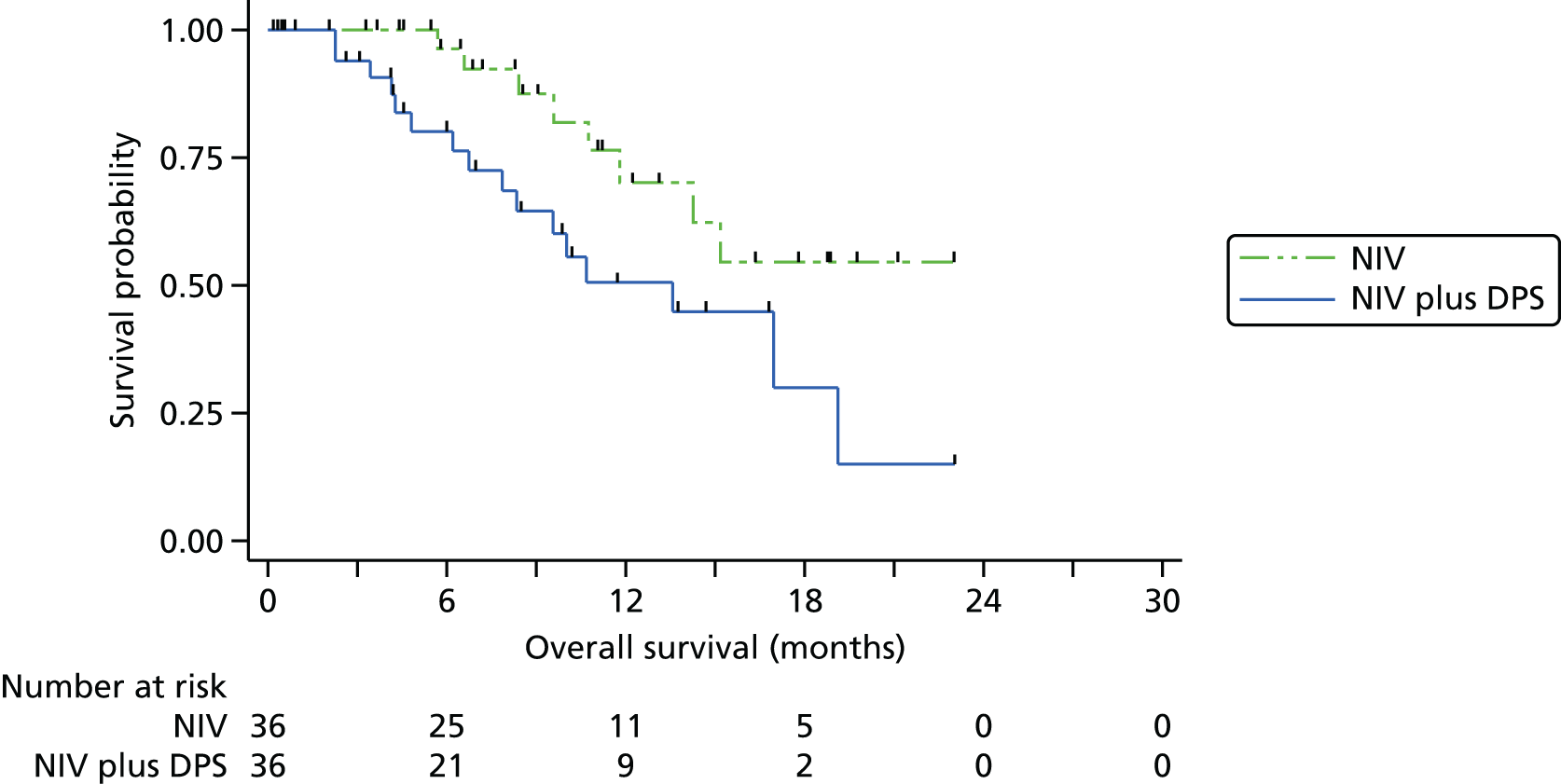
FIGURE 5.
Survival at 10 June 2014.
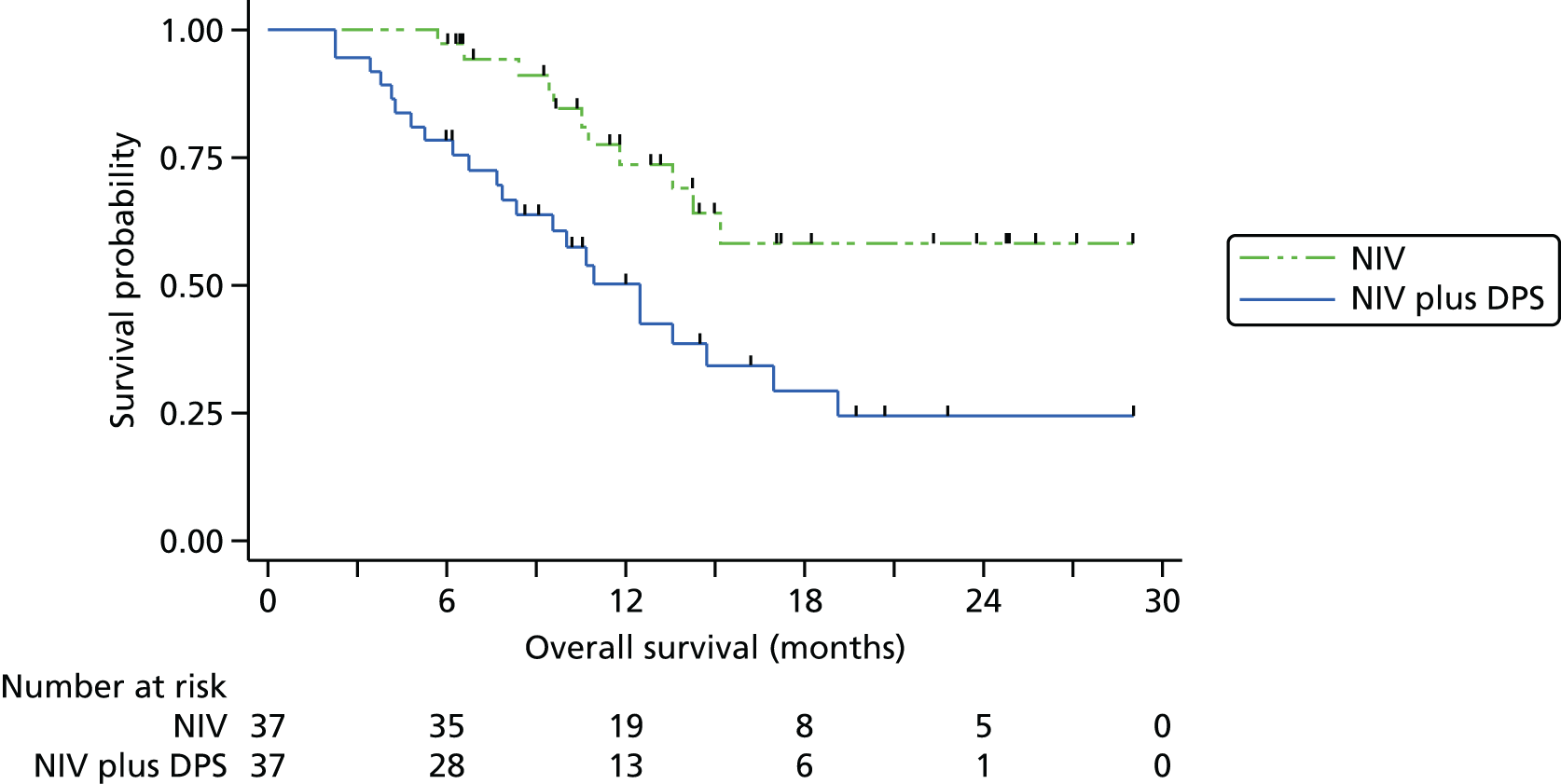
Subgroup analyses of overall survival (preplanned)
We prespecified three subgroup analyses: (1) by NIV tolerance, based on estimated average NIV usage; (2) by bulbar function, as derived from the ALSFRS-R questionnaire; and (3) a per-protocol analysis excluding participants who were inappropriately randomised or who did not adhere to NIV and/or pacing (if that was their randomised allocation).
The results of these are given in this section. In all cases the results were not adjusted for other covariates, as the small number of participants in each subgroup means that more complex models are likely to be unstable. Subgroup analyses were by univariate Cox regression, and tests of interactions were derived by adding treatment, subgroup and their interaction to the model. It should also be noted that small subgroup sizes results in wide CIs and low statistical power both for the main effects and interactions.
Overall survival by baseline bulbar function
The subgroup analysis by bulbar function was originally defined to look at survival in severe (functional score of 0–4) and mild/moderate (functional score of 5–12) separately. As all bar five participants were in the latter group, we have chosen to split the mild and moderate subgroups out, but retain severe bulbar function as its own (small) category.
Overall survival was inferior in the NIV plus DPS arm in both mild and moderate subgroups, although the difference was statistically significant only in the former. There were too few participants in the severe subgroup to allow a meaningful interpretation to be made. Overall, there was little evidence of a differential effect of DP according to bulbar function with a non-significant p-value for the interaction between bulbar function and group (likelihood ratio test χ2(2) = 3.44; p = 0.18) (Figure 6).
FIGURE 6.
Overall survival by bulbar function. (a) Mild bulbar impairment; (b) moderate bulbar impairment; and (c) severe mild bulbar impairment.



Overall survival by non-invasive ventilation tolerance
The subgroup analysis by NIV use was again originally defined to look at survival in two subgroups: NIV tolerant (≥ 4 hours typical daily use) and NIV intolerant (< 4 hours). Post hoc, we have split the latter category further into low NIV use (1–3.9 hours daily, which might confer some benefit) and non-NIV use (< 1 hour daily, which confers virtually no benefit). The rationale is to allow better assessment of the relationship between pacing and outcome. Pacing has been suggested as possibly having particular benefit among patients who do not tolerate NIV, and this analysis allows us to put that hypothesis to the test. Moreover, the difference in survival may (theoretically) be attributed to participants in the NIV plus DPS arm using pacing in place of NIV; this allows us to compare users within each NIV use subgroup.
We categorised average daily NIV use into < 1 hour, 1–3.9 hours or ≥ 4 hours in order to assess whether or not the difference between groups in overall survival was similar. The Kaplan–Meier graphs are shown in Figure 7. In six participants there were inadequate data from which to assess NIV usage, and small numbers hamper interpretation, but survival in the NIV plus DPS was not superior to NIV alone in any subgroup, with the largest difference (in favour of NIV) among the subgroup of participants who did not use NIV. As with bulbar function there was no evidence of a differential effect according to NIV use at conventional statistical levels (likelihood ratio test χ2(2) = 4.14; p = 0.13).
FIGURE 7.
Overall survival by NIV use. (a) Non-use (< 1 hour); (b) low use (1–3 hours); and (c) NIV tolerant (≥ 4 hours).



Per-protocol analysis of overall survival
Protocol compliance was based on excluding participants who:
-
were randomised in error (i.e. in breach of inclusion/exclusion criteria)
-
did not tolerate NIV (< 4 hours’ average daily use)
-
did not undergo successful DP implantation
-
were non-users of DP.
This subgroup was very similar to that shown in the ‘tolerant’ (≥ 4 hours) subset in subgroup analysis. No participants were erroneously randomised and only one non-user of the pacing device was NIV tolerant, meaning that the total number included reduced from 34 to 33. The HR for NIV plus DPS compared with NIV in this subgroup was 1.92 (95% CI 0.81 to 4.56; p = 0.141).
Non-invasive ventilation and pacing usage
Non-invasive ventilation use
Non-invasive ventilation was initiated in 70 out of 74 patients. In total, 57 patients were initiated within 2 weeks of randomisation, a further six within 1 month and the remaining 7 between 3 and 11 months. NIV usage was similar between the two groups (Table 5 and Figure 8).
| Variable | Trial arm | |
|---|---|---|
| NIV plus pacing (n = 37) | NIV (n = 37) | |
| Number of patients with data | 34 | 34 |
| Average daily usage (hours) | ||
| Mean | 5.2 | 4.8 |
| Median (IQR) | 3.2 (0.5–8.2) | 4.6 (0.0–7.8) |
| Number (%) by subgroup | ||
| < 1 hour | 9 (26%) | 11 (32%) |
| 1–3.9 hours | 9 (26%) | 5 (15%) |
| ≥ 4 hours | 16 (47%) | 18 (53%) |
FIGURE 8.
Average daily NIV usage after initiation.

Pacing use
Five pacing group participants did not undergo surgery because of a rapid decline in respiratory function below the safety threshold for surgery (n = 1), patient choice (n = 2) and the DMEC intervention (n = 2); and a sixth patient chose to stop pacing within 1 month of implant following technical problems with the device. All participants who went to surgery had a successful implantation, with a stimulatable diaphragm. When used, the median daily usage was 4.6 hours (IQR 3.0–8.4 hours). Participants pacing were largely able to achieve the target pacing settings within 15 days of surgery and continued to successfully titrate over the course of the study as per the study protocol. Pacing was well tolerated, with only two patients choosing to formally discontinue pacing after implantation, one at 6 months and one at 12 months. The DP settings and usage are described in Table 6–8.
| Variable | First recorded setting | Last recorded setting | ||||||
|---|---|---|---|---|---|---|---|---|
| NIV plus pacing | NIV | NIV plus pacing | NIV | |||||
| Median (IQR) | Minimum–maximum | Median (IQR) | Minimum–maximum | Median (IQR) | Minimum–maximum | Median (IQR) | Minimum–maximum | |
| Target daily usage (hours) | 5 (4–8) | 0–8 | 4 (4–5) | 1–8 | 8 (5–8) | 0–24 | 5 (4–8) | 1–248 |
| Minimum volume assured pressure support (cmH2O) | 5 (5–5) | 5–12 | 5 (5–5) | 5–7 | 5 (5–5) | 5–12 | 5 (5–5) | 5–9 |
| Maximum volume assured pressure support (cmH2O) | 20 (20–25) | 16–25 | 25 (25–25) | 20–25 | 20 (18–25) | 16–30 | 25 (20–25) | 20–25 |
| Inspiratory positive airways pressure (cmH2O) | 12 (10–14) | 5–16 | 12 (10–12) | 5–14 | 12 (12–16) | 5–18 | 12 (10–12) | 5–30 |
| Expiratory positive airways pressure (cmH2O) | 4 (4–4) | 2–8 | 4 (4–4) | 2–6 | 4 (4–4) | 2–6 | 4 (4–4) | 2–8 |
| Target tidal volume (ml) | 500 (500–550) | 400–550 | 500 (470–500) | 450–530 | 550 (500–570) | 500–670 | 500 (450–500) | 420–600 |
| Respiratory rate (b.p.m.) | 12 (10–12) | 6–16 | 12 (10–12) | 8–15 | 12 (10–12) | 8–16 | 12 (10–12) | 8–15 |
| Variable | Initial recommended setting | First recorded setting | Last recorded setting | ||||
|---|---|---|---|---|---|---|---|
| Median (IQR) | Minimum–maximum | Number (%) with settings ≥ initial recommended setting | Median (IQR) | Minimum–maximum | Number (%) with settings ≥ initial recommended setting | ||
| Amperes (mA)a | 10 | 10.8 (10–13) | 6–19 | 26 (81%) | 12.4 (10–16) | 6–23.3 | 27 (84%) |
| Pulse (microseconds)a | 100 | 100 (98–110) | 83–150 | 23 (72%) | 119 (100–145) | 83–160 | 25 (78%) |
| Respiratory rate (b.p.m.) | 12 | 12 (12–12) | 10–14 | 29 (91%) | 12 (12–12) | 10–16 | 30 (94%) |
| Inspiratory interval (seconds) | 1.1 | 1.1 (1.1–1.1) | 1.1–1.2 | 32 (100%) | 1.1 (1.1–1.1) | 1.1–1.2 | 32 (100%) |
| Pulse frequency (Hz) | 12 | 15 (12–15) | 12–20 | 32 (100%) | 15 (12–15) | 10–20 | 31 (97%) |
| Pulse ramp (seconds) | 10 | 10 (0–10) | 0–10 | 27 (84%) | 10 (10–10) | 10–10 | 32 (100%) |
| Variable | NIV plus DPS (n = 37) |
|---|---|
| Number of patients using pacing | 31 |
| Non-users | 6 |
| Did not undergo surgery | 5 |
| Withdrew with minimal usage following technical problems | 1 |
| Time to surgery (n = 32 implanted), n (%) | |
| Within 14 days | 5 (16%) |
| 15–28 days | 12 (38%) |
| 29–56 days | 10 (31%) |
| > 56 days | 5 (16%) |
| Average daily usage when used (hours) | |
| Mean | 6.2 |
| Median (IQR) | 4.6 (3.0 to 8.4) |
Overall survival by non-invasive ventilation and pacing
The relationship between overall survival and NIV use (categorised as non-use, low use and tolerant over the study period) is covered further in this section. Furthermore, the use of overall survival and NIV use in relation to pacing is investigated in more detail. The role of post hoc and exploratory analyses is to attempt to understand the unexpectedly poor survival experienced by participants in the NIV plus DPS group.
Non-invasive ventilation use and overall survival
The problems in evaluating the association between therapeutic regimens and patient outcomes in an uncontrolled, observational setting are well documented. Using NIV is generally accepted as beneficial,27 but the extent to which a participant uses it depends, to a degree, on prognosis. A patient who uses NIV at low levels may do so because their respiratory physician considers them well enough, or they consider themselves well enough, to have no need for high levels of use. Conversely, ALS patients often use NIV at high levels (≥ 12 hours per day) towards the end of their life. There are other factors besides this which impact on NIV use, but the aforementioned factors illustrate how the true impact of NIV is confounded by prognosis and can thereby be underestimated. With this in mind, the overall lack of association between NIV use and survival in the remainder of this section should not be taken as evidence against NIV being beneficial. Rather, this section attempts to assess whether or not NIV use gave rise to differential treatment effect and, crucially, whether or not some participants in the NIV plus DPS group used pacing in place of NIV.
We also note the difficulties in quantifying NIV use. In some cases there was a lag between NIV initiation and regular use, and in a handful of cases several months passed between initiation and regular use. It could be argued that the analysis should take NIV use starting from the date of first regular use, but the limitation of doing so is that ‘regular’ use is not unequivocally defined. Some patients started NIV at low levels (1–2 hours per day), whereas others used NIV on trial basis, only to stop (sometimes for several months) before recommencing. Furthermore, the participant diary data were in many cases not recorded in sufficient detail (completeness) to allow a date of ‘regular’ use to be determined.
Non-invasive ventilation use in the 12-month follow-up period
We first looked at NIV from the point of initiation to the end of the 12 months’ follow-up or death, whichever occurred first. Data were available for 68 of the 74 participants over the study duration.
The primary motivation, however, is not to quantify the association between NIV and survival per se, but to investigate whether or not the difference between the randomised NIV plus DPS and NIV arms may be affected by NIV rather than pacing alone.
The problem in visualising survival data is that the survival times are censored for a proportion of the participants. The relationship between NIV use and survival can be displayed graphically only if some assumptions are made as to what the censored times would have been, had they been observed. There is necessarily considerable uncertainty in doing so. For the purpose of displaying the data in graphical form we followed the approach proposed by Royston and Parmar22 to estimate the underlying (but unobserved) survival times based on each individual’s censored survival time, treatment group and prognostic covariates. A Q–Q plot of the data showed the log-normal survival distribution to give a reasonable fit to the data; therefore, a truncated log-normal regression model was fitted. From this, the overall survival for each individual was being estimated based on their predicted mean survival, their censored survival time, and the root-mean-square error of the model. We fitted two models with minimisation covariates also included in both: first, for all patients and then second, within each group separately. The estimated survival was taken as the average of these two predicted times.
It is important to note that we estimate the survival times only for the purpose of visualising the relationship between NIV, and not for the analysis itself. In all cases, the graphical display distinguishes estimated survival times from observed survival times.
Overall, no relationship was evident between typical NIV use and survival, using either the level of NIV or its categorised version (< 1 hour, 1–3.9 hours or 4 hours). These are demonstrated graphically for the NIV use measured in hours (Figure 9), using the estimated/extrapolated survival for censored survival and in a more conventional Kaplan–Meier graph for the categorisation of NIV use (Figure 10).
FIGURE 9.
Survival by NIV use: NIV use in hours. Censored survival times are estimated based on a censored log-normal distribution using treatment and minimisation covariates. The line is a locally weighted scatterplot smoother (lowess) with a bandwidth of 0.6. Circles denote actual survival times; stars are estimated times.
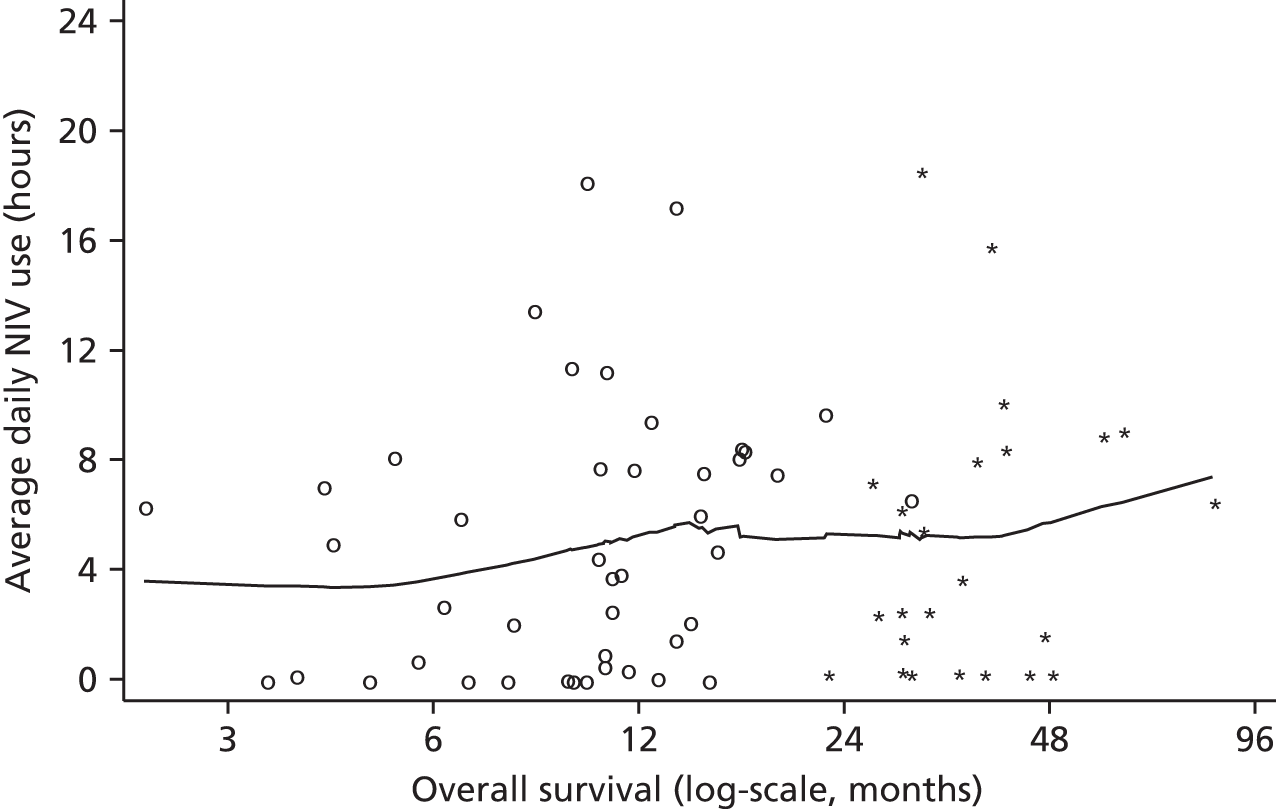
FIGURE 10.
Survival by NIV use: NIV use category. Censored survival times are estimated based on a censored log-normal distribution using treatment and minimisation covariates. The line is a locally weighted scatterplot smoother (lowess) with a bandwidth of 0.6.

In the categorised version of NIV there was no association between average daily usage and overall survival, using either the standard log-rank test (χ2(2) = 0.64; p = 0.72) or the log-rank test for trend across the categories (χ2(1) = 0.64; p = 0.46). As shown in Table 9, the largest difference between NIV plus DPS and NIV alone was observed in the low-use subgroup (n = 20 participants), but the interaction term between treatment group and NIV category was not statistically significant at conventional levels in a Cox regression model (likelihood ratio test χ2(2) = 4.14; p = 0.13).
| Variable | Trial arm, number of participants (deaths) | Comparison | ||
|---|---|---|---|---|
| HR (95% CI) | p-value | |||
| NIV plus DPS | NIV | |||
| Overall survival by bulbar function | ||||
| Overall test of interaction between treatment and bulbar function | 0.179 | |||
| Mild (9–12) | 26 (21) | 29 (14) | 2.99 (1.49 to 6.01) | 0.002 |
| Moderate (5–8) | 8 (6) | 6 (3) | 1.95 (0.48 to 7.92) | 0.349 |
| Severe (0–4) | 3 (1) | 2 (2) | 0.62 (0.05 to 7.00) | 0.697 |
| Overall survival by NIV use | ||||
| Overall test of interaction between treatment and NIV use | 0.126 | |||
| Non-use (< 1 hour daily) | 9 (8) | 11 (5) | 3.90 (1.23 to 12.4) | 0.021 |
| Low use (1–3.9 hours daily) | 9 (5) | 5 (3) | 1.50 (0.35 to 6.38) | 0.584 |
| Tolerant (≥ 4 hours daily) | 16 (12) | 18 (11) | 1.63 (0.70 to 3.78) | 0.253 |
No relationship was found when looking at the average number of hours (i.e. as a continuous measure) and overall survival. The linear association as derived from a univariate Cox regression model showed a small and non-statistically significant increase in survival with increasing hourly use of NIV (HR 0.98, 95% CI 0.92 to 1.04; p = 0.52). The possibility of a non-linear relationship was assessed using fractional polynomials regression, but the best non-linear model still failed to find any association (likelihood ratio test χ2(2) = 1.26; p = 0.53) or of any interaction between treatment and NIV use (likelihood ratio test χ2(1) = 0.68; p = 0.41).
Non-invasive ventilation use in the first 3 and 6 months post randomisation
We also looked at whether overall survival is associated with NIV use in (1) the first 3 months post randomisation or (2) the first 6 months post randomisation. Although this does not use the more complete NIV data over the 12 months’ follow-up, focusing on short-/medium-term use might be seen as a surrogate for intention-to-use NIV, as opposed to the level of use necessitated by disease progression. Participants for whom NIV initiation was delayed are included; a participant who was initiated at 4 months would be defined as having zero use within the 0–3 months period.
However, the findings were similar to those reported for 12-month NIV use above. Overall survival was weakly associated with NIV use, but was not statistically significant: the HR for 3-month use was 0.96 (95% CI 0.87 to 1.05; p = 0.33) and for 6-month use was 0.98 (95% CI 0.91 to 1.07; p = 0.68). Non-linear associations derived from fractional polynomial regression also failed to reach statistical significance (likelihood ratio test for 3-month follow-up: χ2(2) = 2.95; p = 0.23; 6-month follow-up likelihood ratio test χ2(1) = 0.89; p = 0.64). Again, no interaction was found between treatment group and NIV use over either period.
Pacing use and overall survival
We assessed the relationship between pacing use and overall survival. Unlike NIV, no normative data exist for pacing use; consequently, we did not categorise pacing usage into groups. The caveats that apply when assessing the relationship between NIV and survival also apply here.
Figure 11 shows average pacing use against overall survival, again estimating survival times as described in Non-invasive pacing and overall survival. As with NIV, no discernible relationship was evident. The HR for each additional hour of use was 1.01 (95% CI 0.94 to 1.09; p = 0.707); if non-implanted patients were excluded, the HR was 1.00 (95% CI 0.92 to 1.09; p = 0.931). Non-linear models, again derived using fractional polynomials, did not improve the fit of the model.
FIGURE 11.
Survival by pacing use. Censored survival times are estimated based on a censored log-normal distribution using treatment and minimisation covariates. The line is a locally weighted scatterplot smoother (lowess) with a bandwidth of 0.6. Circles denote actual survival times; stars are estimated times. N, not implanted.

Non-invasive ventilation, pacing and overall survival
The final analyses looked at the association between NIV and pacing use within the 37 participants randomised to receive the intervention. A potential theory was that participants in the NIV plus pacing arm may have used pacing in place of NIV. If so, this could partly explain the between-group differences, as NIV use is known to prolong survival.
Nevertheless, this theory was not borne out by the data. The average use of pacing and NIV is shown in Figure 12; the relationship between the two measures was positive rather than negative, suggesting participants who use NIV more also tended to use pacing more. We also split the NIV plus DPS arm into NIV-tolerant users (≥ 4 hours per day on average) and NIV-non-tolerant users (< 4 hours per day on average), with the Kaplan–Meier curves as shown in Figure 13. Participants in the NIV plus DPS arm who used NIV for an average ≥ 4 hours a day had a better survival than those who did not, but their survival remained lower than the NIV alone group. Neither comparison was statistically significant at the 5% significance level; however, the small numbers in each subgroup mean that the statistical power is low for this comparison.
FIGURE 12.
Relationship between NIV and pacing use. N, not implanted.
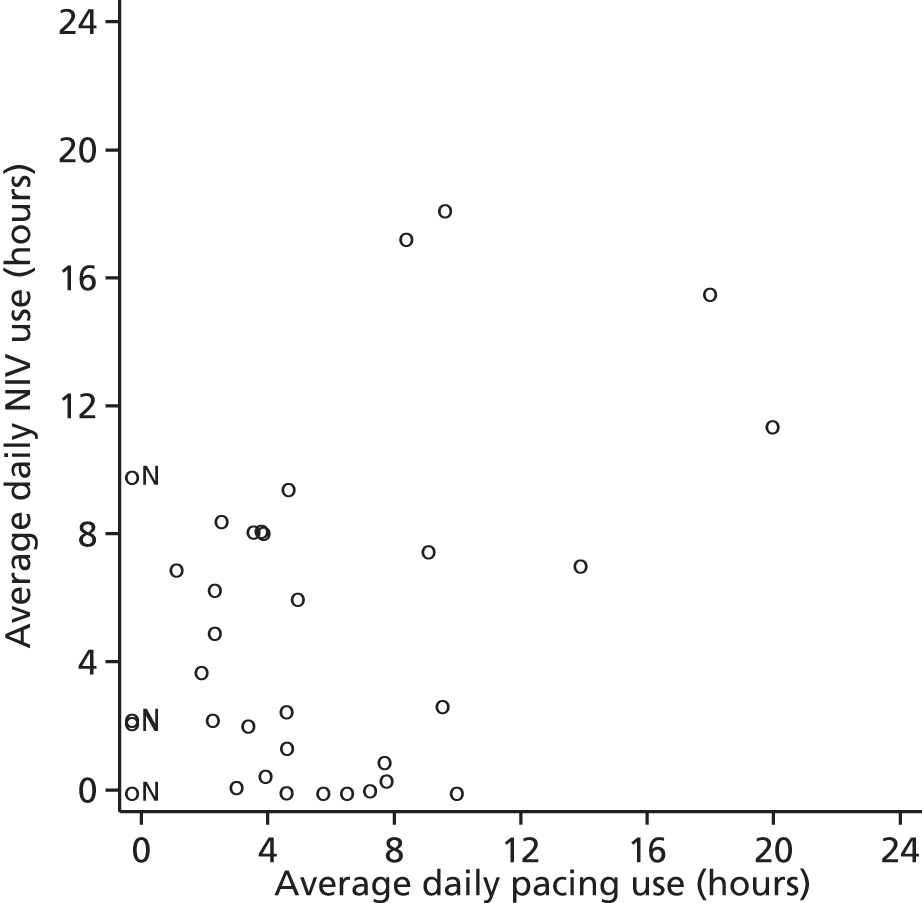
FIGURE 13.
Overall survival by pacing and NIV use.

Secondary outcomes
Tracheostomy and tracheostomy-free survival
Tracheostomy-free survival is defined as the time to death or tracheostomy, whichever comes first. One tracheostomy was reported: one participant in the NIV plus DPS arm underwent a tracheostomy 31 months after randomisation. The TFS is, therefore, very similar to overall survival, with a HR adjusted for minimisation covariates being 2.42 (95% CI 1.28 to 4.59; p = 0.007).
Participant quality of life
European Quality of Life-5 Dimensions, three levels
The participant health utility as measured by EQ-5D-3L is shown in Figures 14 and 15. Among those surviving to 12 months and with complete data, the difference in EQ-5D-3L scores was –0.18 (95% CI –0.44 to 0.08; p = 0.164). However, over the follow-up period as a whole, the EQ-5D-3L score declined quicker in the NIV plus DPS arm than in the NIV alone arm, with the longitudinal analysis providing an average difference across time of –0.13 (95% CI –0.25 to –0.00; p = 0.042). Imputing missing data made no material difference to the conclusions. The preplanned analysis of EQ-5D-3L scores according to NIV tolerance and bulbar function was not undertaken because of small numbers (especially in the NIV plus DPS arm) and the apparent deficit in overall survival.
FIGURE 14.
The EQ-5D-3L health utility among surviving participants over time: EQ-5D-3L health tariff.
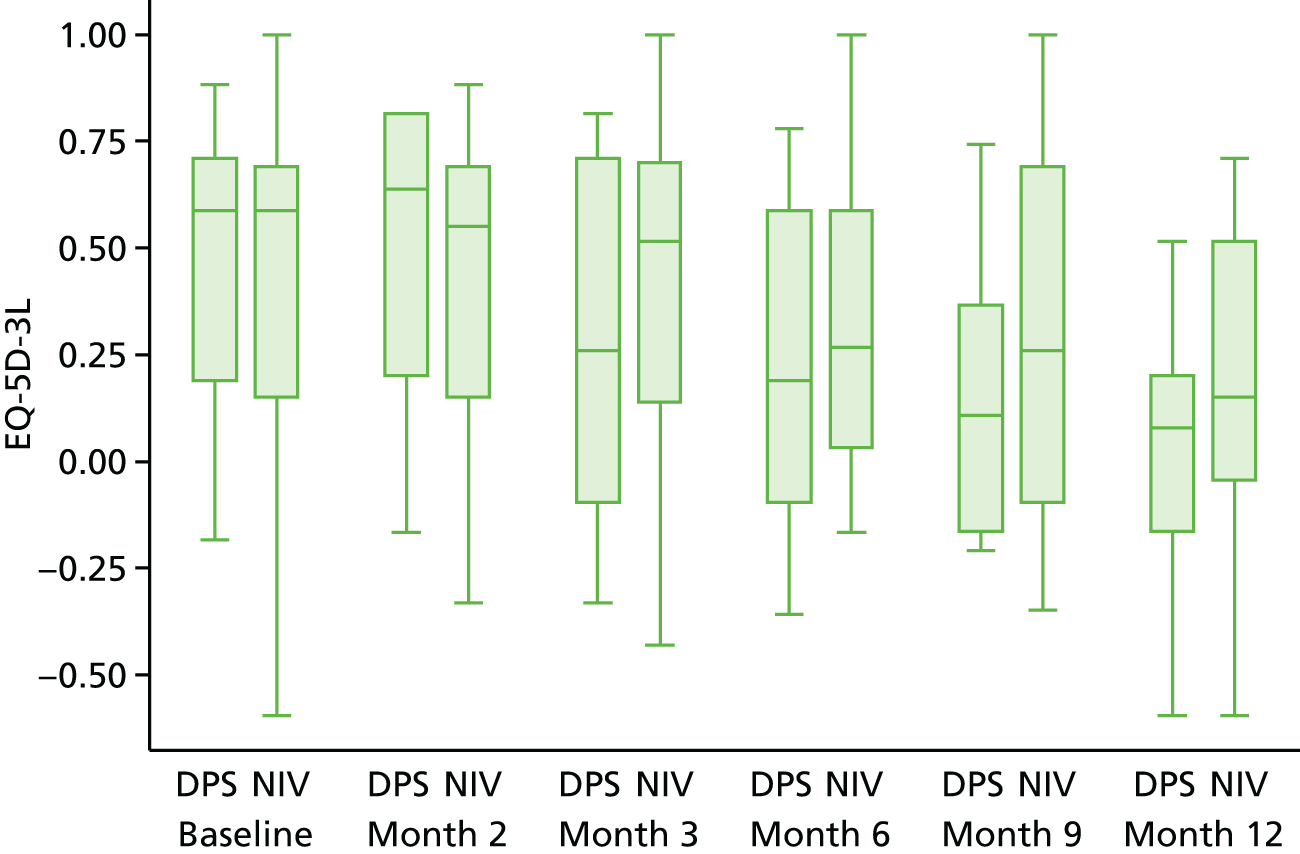
FIGURE 15.
The EQ-5D-3L health utility among surviving participants over time: patient EQ-5D-3L health tariff, deaths imputed as zero.

A further analysis of EQ-5D-3L scores included both surviving and non-surviving participants, with a score of 0 assigned to the latter from the point of death onwards. Doing so demonstrated a lower QoL both at 12 months (mean difference –0.12, 95% CI –0.24 to –0.01; p = 0.040) and also longitudinally (mean difference –0.14, 95% CI –0.24 to –0.04; p = 0.001).
The findings of the EQ-5D-3L thermometer scale were consistent with those for the main EQ-5D-3L questionnaire (Table 10). All longitudinal analyses were repeated using an unstructured correlation matrix, as the random intercept model makes the assumption of a common correlation between repeated measures, irrespective of their proximity in time. Although correlation decreased with time, the estimated difference between intervention and control was virtually unaltered.
| Instrument | End of follow-up analysis | Longitudinal analysis | ||||
|---|---|---|---|---|---|---|
| Trial arm | Comparison | Comparison | ||||
| NIV plus DPS (n = 37) | NIV (n = 37) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| Number (%) of participants alive at 12 months | 16 (43) | 27 (73) | – | – | – | – |
| Patient QoL | ||||||
| EQ-5D-3L health state (% completea) | 131/178 (74) | 161/209 (77) | – | – | – | – |
| Complete case, mean (standard error) | 0.02 (0.09) | 0.19 (0.08) | –0.18 (–0.44 to 0.08) | 0.164 | –0.13 (–0.25 to 0.00) | 0.042 |
| With imputation for surviving participants, mean (standard error) | 0.02 (0.09) | 0.13 (0.08) | –0.11 (–0.39 to 0.16) | 0.410 | –0.12 (–0.24 to 0.00) | 0.056 |
| With imputations for all participants,b mean (standard error) | 0.01 (0.03) | 0.11 (0.05) | –0.12 (–0.24 to 0.01) | 0.040 | –0.14 (–0.24 to 0.04) | 0.001 |
| EQ-5D-3L thermometer scale (% completea) | 132/178 (74) | 160/209 (77) | – | – | – | – |
| Complete case, mean (standard error) | 34.4 (6.3) | 42.3 (5.1) | 3.4 (–14.5 to 21.2) | 0.699 | –8.3 (–17.4 to 0.8) | 0.074 |
| With imputation for surviving participants, mean (standard error) | 36.0 (6.6) | 40.0 (5.2) | 1.3 (–17.6 to 20.1) | 0.893 | –5.6 (–14.5 to 3.2) | 0.212 |
| With imputation for all participants,b mean (standard error) | 14.8 (3.9) | 27.4 (4.8) | –13.4 (–25.9 to 0.9) | 0.036 | –12.0 (–20.8 to 3.1) | 0.008 |
| SF-36 aggregate physical health score (% completea) | 110/154 (72) | 133/174 (76) | – | – | – | – |
| Complete case, mean (standard error) | 25.4 (4.2) | 21.8 (1.8) | 7.4 (–1.9 to 16.8) | 0.110 | 0.2 (–2.2 to 2.5) | 0.900 |
| With imputation, mean (standard error) | 23.8 (3.2) | 20.6 (2.4) | 8.7 (–1.3 to 18.7) | 0.089 | 0.3 (–2.0 to 2.7) | 0.780 |
| SF-36 aggregate mental health score (% completea) | 110/154 (72) | 133/174 (76) | – | – | – | – |
| Complete case, mean (standard error) | 44.1 (3.7) | 47.0 (4.2) | –7.8 (–20.5 to 4.9) | 0.210 | –4.1 (–8.6 to 0.3) | 0.066 |
| With imputation, mean (standard error) | 42.7 (4.2) | 47.4 (3.5) | –8.6 (–22.2 to 5.1) | 0.218 | –3.5 (–7.9 to 0.8) | 0.112 |
| SAQLI (% completea) | 110/154 (72) | 132/174 (76) | – | – | – | – |
| Complete case, mean (standard error) | 4.1 (0.5) | 4.2 (0.3) | –0.2 (–1.3 to 1.0) | 0.751 | –0.3 (–0.7 to 0.1) | 0.163 |
| With imputation, mean (standard error) | 3.9 (0.4) | 4.5 (0.3) | –0.6 (–1.7 to 0.6) | 0.329 | –0.3 (–0.7 to 0.1) | 0.117 |
| Carer QoL | ||||||
| EQ-5D-3L health state (% completea) | 109/178 (61) | 148/209 (71) | – | – | – | – |
| 0.78 (0.11) | 0.82 (0.06) | –0.08 (–0.38 to 0.23) | 0.600 | –0.08 (–0.17 to 0.01) | 0.077 | |
| EQ-5D-3L thermometer scale (% completea) | 110/178 (62) | 149/209 (71) | – | – | – | – |
| 81.3 (7.1) | 71.0 (6.4) | 12.1 (–13.1 to 37.2) | 0.329 | –0.2 (–7.4 to 7.1) | 0.966 | |
| CBI (% completea) | 93/154 (60) | 121/174 (70) | – | – | – | – |
| 28.0 (3.2) | 29.6 (3.2) | 3.1 (–7.5 to 13.8) | 0.536 | 1.2 (–2.7 to 5.0) | 0.558 | |
Short Form questionnaire-36 items
As expected, overall physical health (as derived from SF-36) is considerably lower than population norms in both groups, although mental health was comparable. No differences were evident between the two groups over the duration of the study. As with EQ-5D-3L, the difference in aggregate mental health scores between the groups was not statistically significant among surviving participants at 12 months (mean difference –7.8, 95% CI –20.5 to 4.9; p = 0.210), but was of borderline significance over the duration of the study (mean difference –4.1, 95% CI –8.6 to 0.3; p = 0.066). Physical health appeared similar between groups on both scales (Figures 16 and 17). Relaxing the assumption of a common correlation in the longitudinal analyses made virtually no difference to the findings, with the correlation being relatively constant with time.
FIGURE 16.
The SF-36 aggregate physical health component.
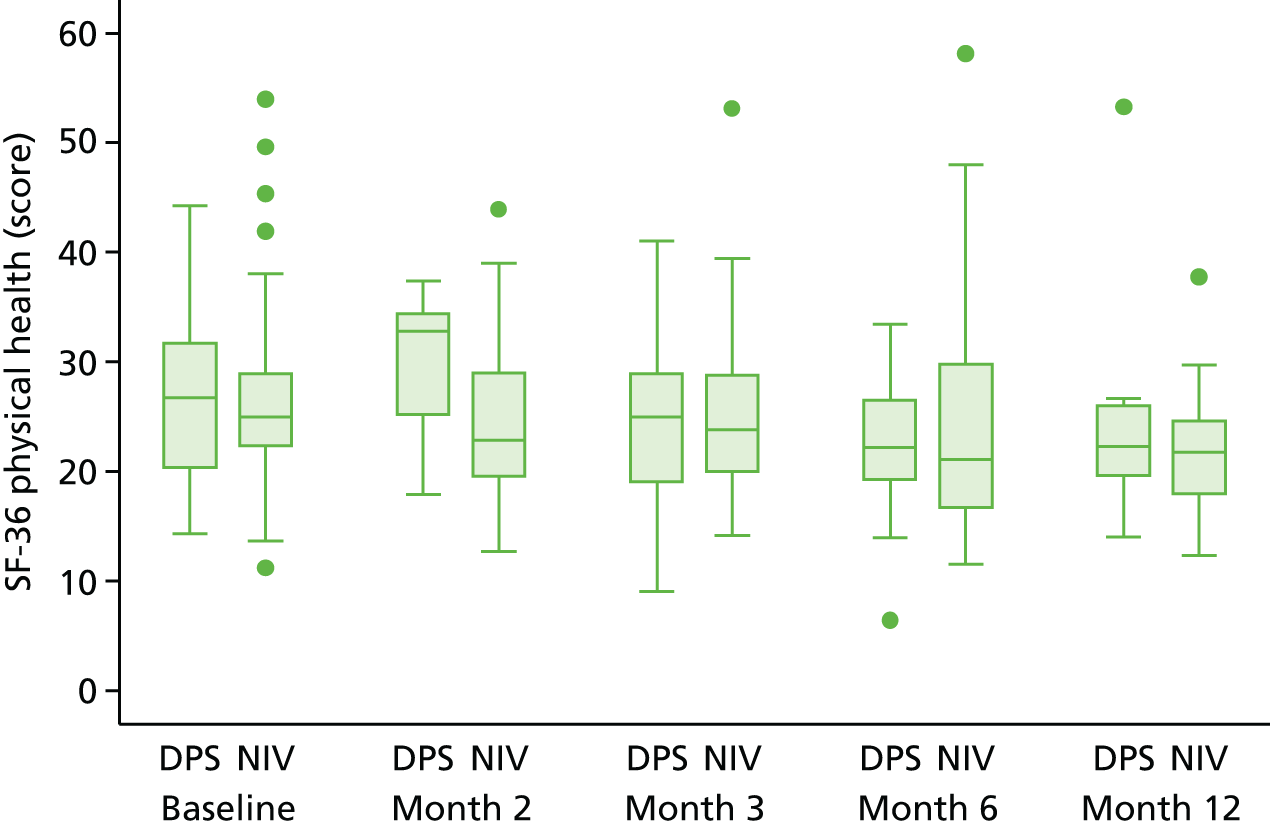
FIGURE 17.
The SF-36 aggregate mental health component.
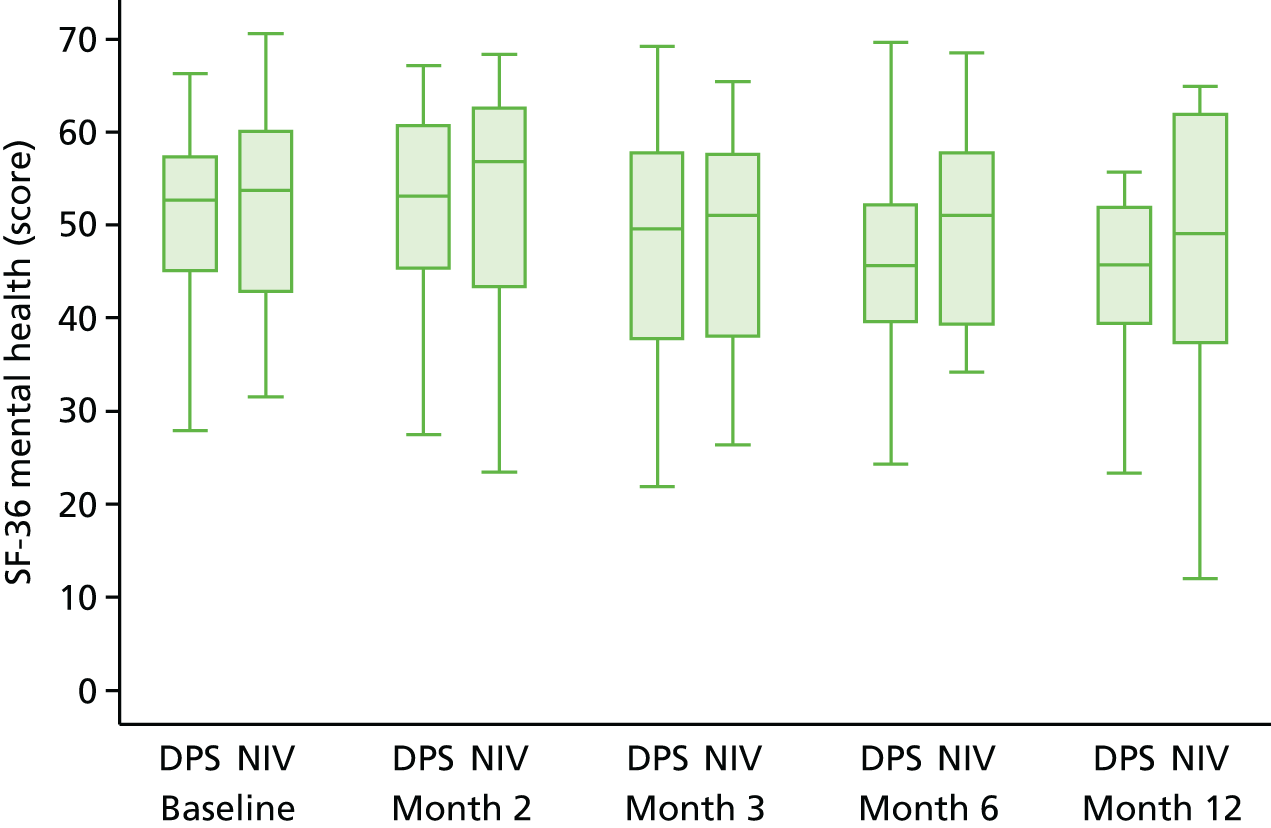
Sleep Apnoea Quality of life Index
Sleep apnoea appeared similar at all time points between the two groups (Figure 18).
FIGURE 18.
Sleep Apnoea Quality of Life Index.

Carer quality of life
The carer QoL is presented in Table 10 and, graphically, in Figures 19 and 20. There was little in the way of difference between the groups.
FIGURE 19.
Carer EQ-5D-3L health tariff.

FIGURE 20.
Caregiver Burden Inventory.
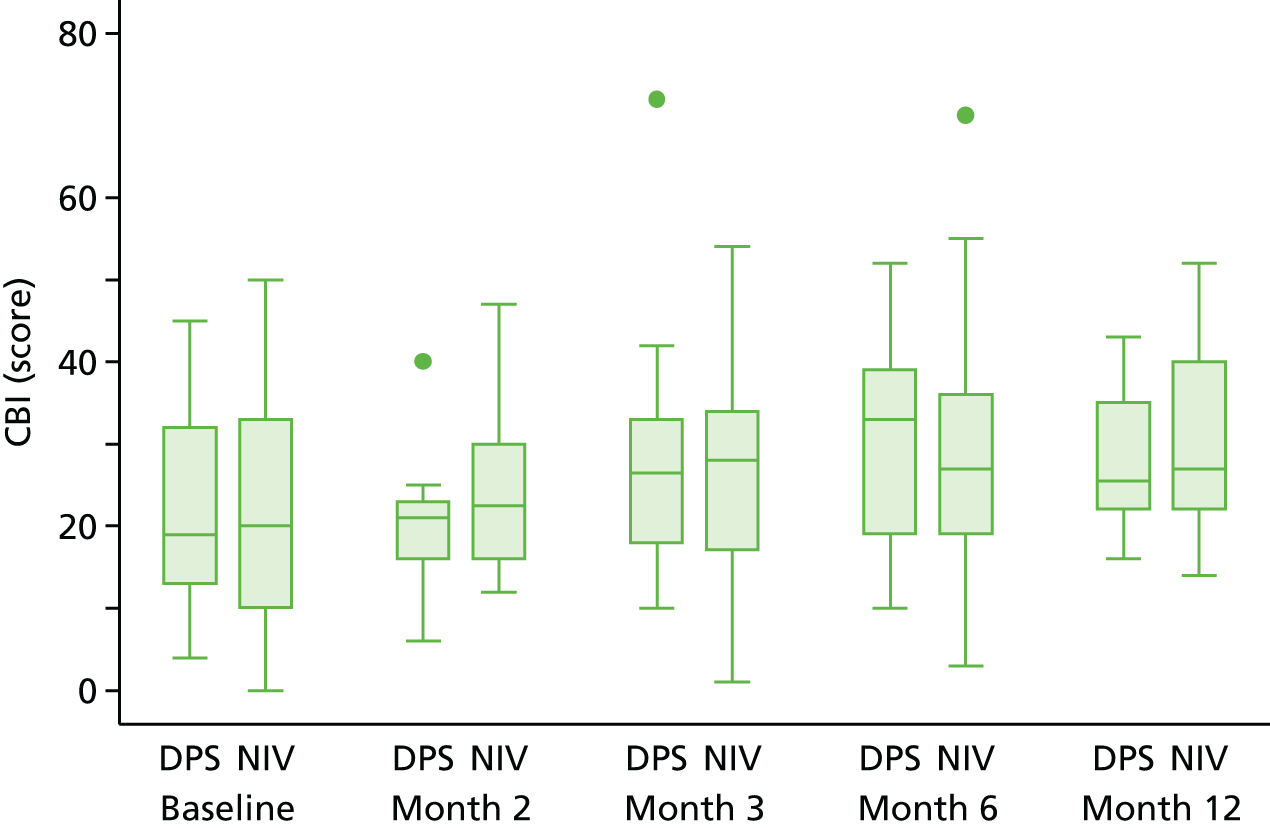
Relationship between quality of life, pacing and baseline symptoms
We have compared baseline scores with 12-month averages in EQ-5D-3L, SF-36 physical component summary score, SF-36 mental component summary score and total SAQLI and see no relationship between symptoms present and QoL scores. Figures 21 and 22 show these for SAQLI; similar patterns were observed on other measures.
FIGURE 21.
Change (12 months – baseline) in SAQLI score in relation to baseline SAQLI score (Q12–Q14). (a) NIV plus DPS; and (b) NIV.

FIGURE 22.
Change (12 months – baseline) in SAQLI score in relation to baseline ALSRFS-R sum (Q11–Q12). (a) NIV plus DPS; and (b) NIV.

Resource use
Resource use is shown in Table 11. As can be seen, the groups generally used similar levels of resources. The exception was respiratory devices, specifically the cough assist device and suction. The number of users was similar in both groups but the level at which they were used was somewhat greater in the NIV alone group.
| Variables | Trial arm | |||
|---|---|---|---|---|
| NIV plus pacing (n = 37) | NIV (n = 37) | |||
| Number (%) of patients | Total days | Number (%) of patients | Total days | |
| Admissions | ||||
| Admissions following surgerya | ||||
| ITU | 12 (32%) | 13 | – | – |
| Non-ITU | 32 (86%) | 86 | – | – |
| Admissions within 1 week of surgery | 2 (5%) | 12 | – | – |
| All other admissions | 9 (24%) | 67 | 13 (35%) | 109 |
| GP/emergency use | ||||
| Number (%) of patients | Total number | Number (%) of patients | Total number | |
| GP appointments | 29 (78%) | 165 | 30 (81%) | 152 |
| A&E attendances | 17 (46%) | 23 | 11 (30%) | 14 |
| Minor injuries unit | 6 (16%) | 9 | 3 (8%) | 11 |
| Respiratory device | ||||
| Number (%) of patients | Monthly use (when usedb) | Number (%) of patients | Monthly use (when usedb) | |
| Cough assist use | 12 (32%) | 4.2 | 10 (27%) | 10.0 |
| Breath stacking | 5 (14%) | 8.6 | 6 (16%) | 6.7 |
| Suction | 7 (19%) | 2.4 | 5 (14%) | 9.5 |
| Other resource use | ||||
| Number (%) of patients | Total number | Number (%) of patients | Total number | |
| Physiotherapy | 22 (59%) | 144 | 24 (65%) | 165 |
| Occupational therapy | 22 (59%) | 127 | 24 (65%) | 99 |
| Other | 13 (35%) | 57 | 16 (43%) | 86 |
Safety
Adverse events
A total of 243 AEs were observed, 162 of which were in the NIV plus DPS arm. Taking into account the differential follow-up owing to survival differences, this equates to 5.9 events per person-year in the NIV plus pacing group, compared with 2.5 events per person-year in the control group (Table 12). Eight of the events occurred in patients who had not been implanted.
| Category | Trial arm | |||
|---|---|---|---|---|
| NIV plus pacing | NIV | |||
| Number of AEsa | Number (%) of patients with an event | Number of AEs | Number (%) of patients with an event | |
| Any AE | 162 (8) | 29 (78%) | 81 | 23 (62%) |
| Respiratory | 45 (5) | 25 (68%) | 19 | 14 (38%) |
| Type of respiratory event | ||||
| Chest infection | 20 | 12 (32%) | 11 | 7 (19%) |
| Respiratory failure | 10 (2) | 10 (27%) | 5 | 5 (14%) |
| Breathless – unclassified | 5 | 4 (11%) | 3 | 2 (5%) |
| Pneumothorax/capnothorax | 5 | 5 (14%) | 0 | – |
| Blocked airway | 3 (3) | 1 (3%) | 0 | – |
| Cough | 1 | 1 (3%) | 0 | – |
| Pulmonary embolism | 1 | 1 (3%) | 0 | – |
| Pain | 23 | 10 (27%) | 10 | 6 (16%) |
| Gastrointestinal | 17 | 10 (27%) | 12 | 9 (24%) |
| ALS symptoms | 18 (3) | 8 (22%) | 7 | 3 (8%) |
| Insertion/removal of PEG/PIG | 9 | 5 (14%) | 10 | 9 (24%) |
| Genitourinary | 7 | 3 (8%) | 8 | 3 (8%) |
| Infection of PEG/PIG | 10 | 3 (8%) | 2 | 1 (3%) |
| Dermatological | 6 | 3 (8%) | 4 | 4 (11%) |
| Wire problems | 8 | 5 (14%) | 0 | n/a |
| Cardiovascular system | 4 | 4 (11%) | 2 | 2 (5%) |
| Psychiatric | 5 | 4 (11%) | 0 | – |
| NIV specific | 3 | 3 (8%) | 2 | 2 (5%) |
| Wire infection | 4 | 3 (8%) | 0 | n/a |
| Central nervous system | 1 | 1 (3%) | 1 | 1 (3%) |
| Other | 2 | 2 (5%) | 4 | 3 (8%) |
Serious adverse events
Serious adverse events are presented in Table 13. Again, more SAEs were observed in the NIV plus pacing arm.
| Category | Trial arm | |||
|---|---|---|---|---|
| NIV plus pacing | NIV | |||
| Number of SAEsa | Number (%) of patients with an event | Number of SAEs | Number (%) of patients with an event | |
| Any SAE | 46 (5) | 27 (73) | 31 | 19 (51) |
| Respiratory | 29 (5) | 21 (57) | 13 | 11 (30) |
| Type of respiratory event | ||||
| Chest infection | 10 | 9 (24) | 6 | 5 (14) |
| Respiratory failure | 10 (2) | 10 (27) | 5 | 5 (14) |
| Breathless – unclassified | 2 | 2 (5) | 2 | 1 (3) |
| Pneumothorax/capnothorax | 3 | 3 (8) | 0 | – |
| Blocked airway | 3 (3) | 1 (3) | 0 | – |
| Pulmonary embolism | 1 | 1 (3) | 0 | – |
| Insertion/removal of PEG/PIG | 6 | 4 (11) | 9 | 8 (22) |
| Gastrointestinal | 3 | 3 (8) | 2 | 2 (5) |
| Cardiovascular system | 3 | 3 (8) | 1 | 1 (3) |
| Pain | 1 | 1 (3) | 3 | 2 (5) |
| Wire problems | 2 | 2 (5) | 0 | n/a |
| ALS symptoms | 1 | 1 (3) | 0 | – |
| Dermatological | 0 | – | 1 | 1 (3) |
| Genitourinary | 0 | – | 1 | 1 (3) |
| Other | 1 | 1 (3) | 1 | 1 (3) |
Adverse events related to surgery or pacing
In total, 31 events (in 15 different participants) were adjudged to have had an AE with either a ‘definite’ or a ‘probable’ relationship to pacing. These AEs are listed in Box 1 and Table 14. Seven surgical complications or device-related adverse events were recorded on the date of procedure.
Bilateral pneumothorax.
Capno/pneumothorax.
Tension pneumothorax.
Right-sided pneumothorax requiring chest drain.
Lines 1- + 2-captured electrocardiogram trace. Maximum settings established.
Small nick to spleen which caused bleeding. Pressure was applied and bleeding stopped after 5 minutes.
Abdominal pain.
| Event | Frequency (if > 1) |
|---|---|
| Respiratory events related to pacing (n = 5) | |
| Bilateral pneumothoraxa (D) | |
| Tension pneumothoraxa (D) | |
| Right-sided pneumothorax requiring chest draina (D) | |
| Capno/pneumothorax (D) | |
| Difficulty with breathing (P) | |
| Wire infections (n = 4) | |
| Infection to electrode site (D) | 4 |
| Wire problems (n = 4) | |
| Anode wire pulled/fell outa (D) | 4 (2 SAE) |
| Anode replaced as the anode was causing the abdominal pain (D) | |
| Diaphragmatic pacing wire number 1 showing X on pacer. This will need replacing (D) | |
| Patient feels his skin is being burnt by anode bypass (P) | |
| Sore pacing wire insertion site (D) | |
| Pain (n = 12) | |
| Anode replaced as the anode was causing the abdomen pain (D) | |
| Pain in back and shoulders (P) | |
| Pain in ribs (P) | |
| Shoulder pain (on contraction) (P) | |
| Shoulder pain on pacing (D) | |
| Pain to left side of chest on pacing (D) | |
| Discomfort during pacing (D) | |
| Back and shoulder pain post operation (D) | |
| Pain – abdominal (D) | |
| Pain in left shoulder and stomach when pacer switched on (D) | |
| Intermittent shoulder pain on pacing (D) | |
| Right and left shoulder pain on pacing (D) | |
| Dermatological (n = 1) | |
| Bypass pad causing contact dermatitis (D) | |
| Gastrointestinal (n = 1) | |
| Acute gastric dilatation responded well to decompression with nasogastric tube. Well described following upper gastrointestinal surgery although rare – was slightly higher risk because of bleeding from spleen during surgerya (P) | |
Cause of death
The causes of death are presented in Table 15. Although participants in the NIV plus pacing arm had shortened overall survival, mortality was not obviously attributable to the procedure or to the pacing. One participant in the NIV plus DPS arm died 45 days after surgery; none of the remaining participants died within 3 months of the procedure.
| Cause of death | Trial arm | |
|---|---|---|
| NIV plus DPS (n = 37) | NIV (n = 37) | |
| Total deaths | 28 | 19 |
| Causes of death | ||
| Respiratory failure | 16 | 13 |
| Chest infection | 5 | 2 |
| ALS | 6 | 4 |
| Hypothermia | 1 | 0 |
Physiological measurements
In the light of the early stopping, the TSC requested additional respiratory function data to be collected to augment that which was already collected at baseline (and for DP, immediately pre surgery). Specifically, we wished to assess (1) whether or not the groups were comparable at baseline; (2) whether or not decline appeared more pronounced in the post-surgery period among the NIV plus DPS group than in the NIV group; and (3) the trajectory across time, in general, to see if it offered any other clues by which to explain the findings. We were able to obtain data for patients at some of the sites for FVC, arterial carbon dioxide and ALSFRS-R.
The purpose of the analysis was not to assess efficacy but to investigate possible treatment harm and, consequently, we analysed the group based on actual exposure (i.e. implanted with the pacing device) as opposed to the more conventional intention-to-treat analyses. Measurements taken more than 45 days prior to randomisation or > 1 month after the end of scheduled follow-up were excluded. A generalised least squares regression model was fitted in which the treatment received, time since randomisation and their interaction were the covariates. No attempt was made to impute missing data.
The figures show some evidence that FVC did indeed decline faster in participants who were implanted than those who were not, with the average rate of decline per month being 2.1% steeper among implanted participants than in those without implantation (95% CI –3.4% to –0.8%; p = 0.001). It is not easy to distinguish whether or not the decline is more pronounced in the postoperative period than the preoperative period (and, hence, directly attributable to operation), as the latter is a short period of time. (Figures 23 and 24).
FIGURE 23.
Forced vital capacity against time for implanted participants. The triangle denotes date of procedure and the cross denotes date of death.










FIGURE 24.
Forced vital capacity against time for non-implanted participants. The cross denotes date of death.

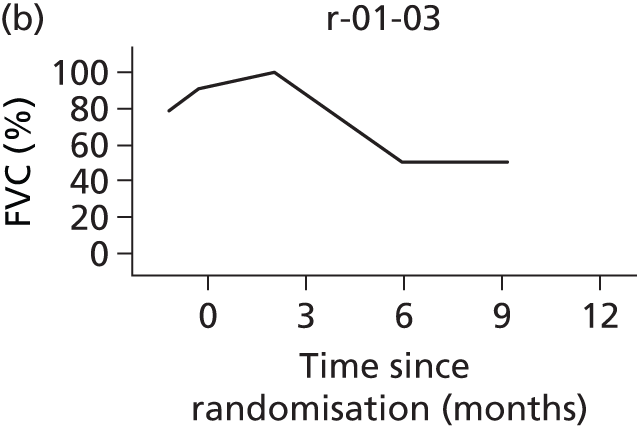
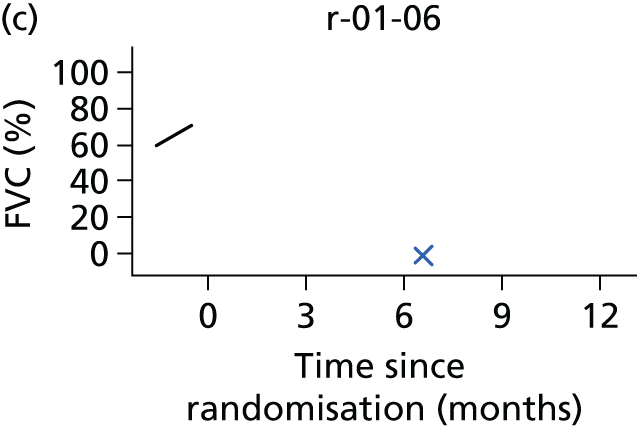

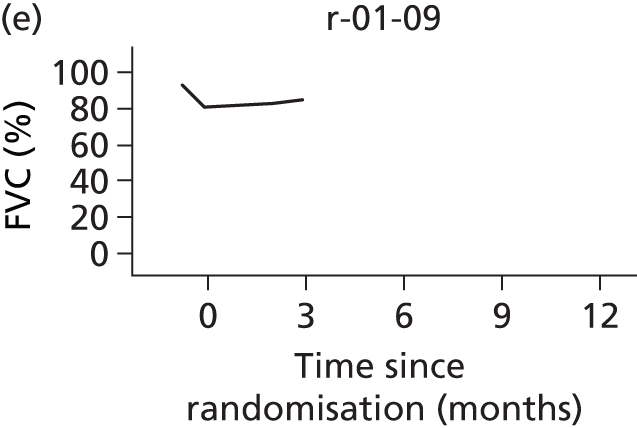

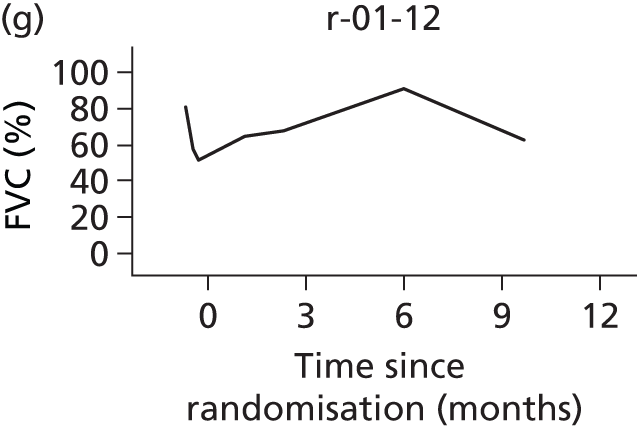


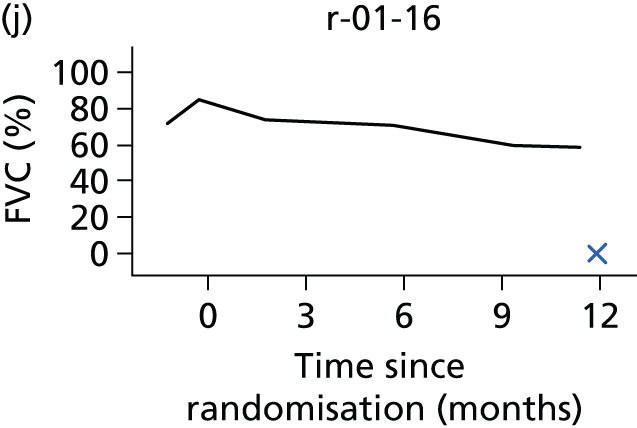


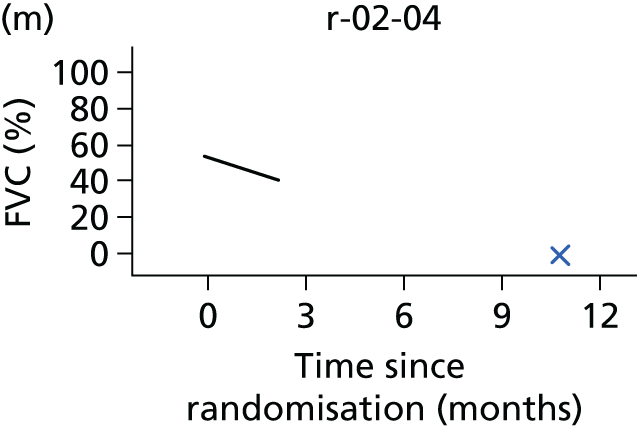
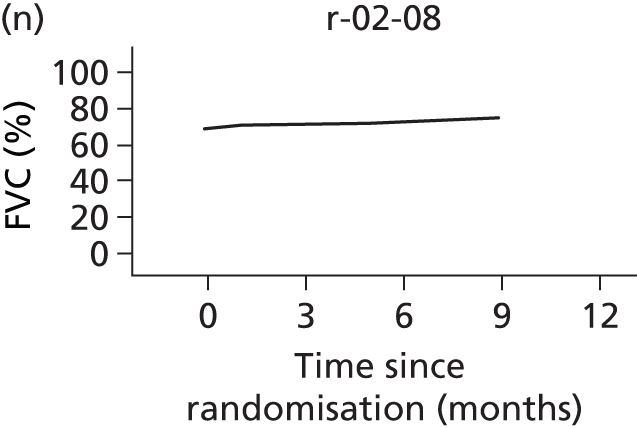




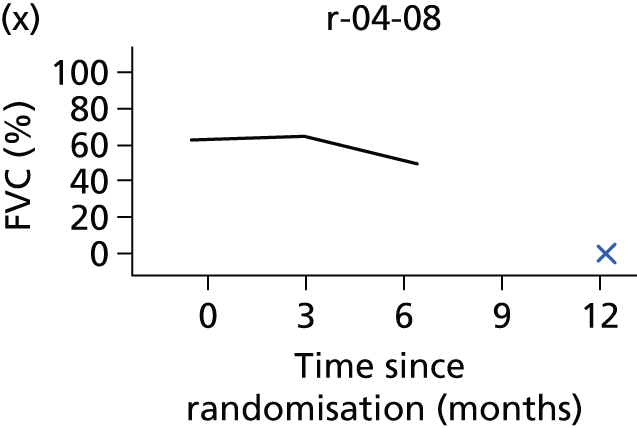
The trajectory for PaCO2 is shown in Figures 25 and 26. In contrast to FVC, the change in PaCO2 is more modest, with no obvious change in levels in either group.
FIGURE 25.
The PaCO2 against time for implanted participants. The triangle denotes date of procedure, the cross denotes date of death.


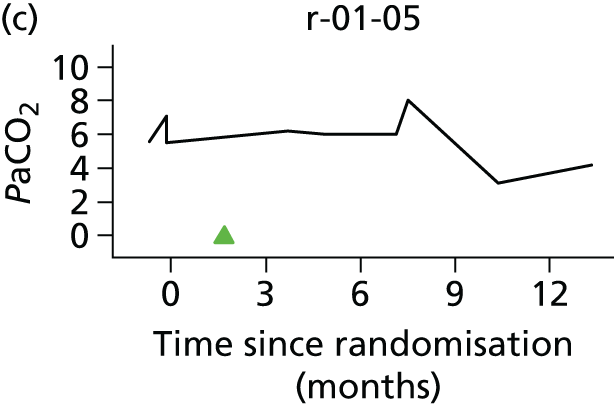

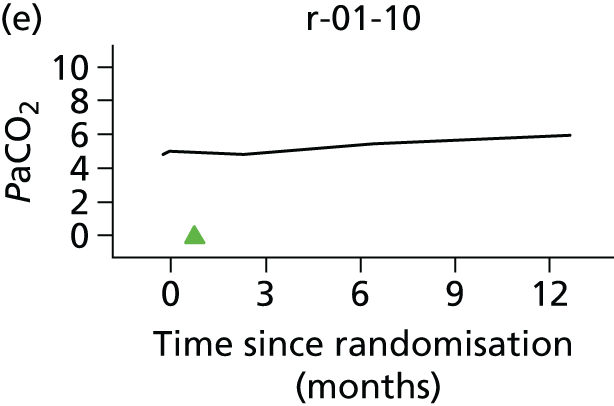
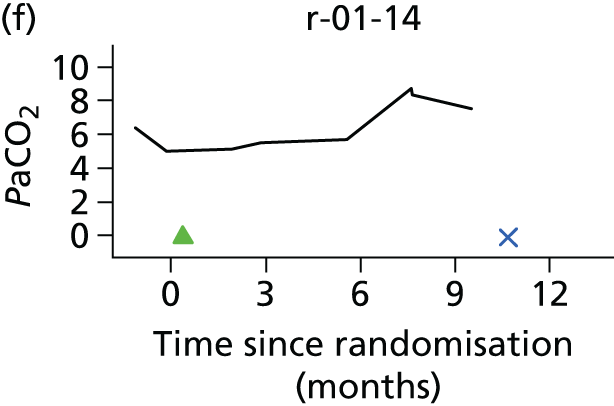
FIGURE 26.
The PaCO2 against time for non-implanted participants. The cross denotes date of death.








Finally the trajectory is shown for ALSFRS-R, a measure of ALS symptomology based on a 12-item questionnaire and ranging from 0 points (most severe) to 48 points (least severe). There was substantial decline in the control group (on average 1.1 point per month), but the rate of decline was greater still among participants who underwent implantation (mean difference 0.4 points per month, 95% CI –0.7 to 0.0 points per month; p = 0.035) (Figures 27 and 28).
FIGURE 27.
The ALSRFS-R score against time for implanted participants. The triangle denotes date of procedure and the cross denotes date of death.


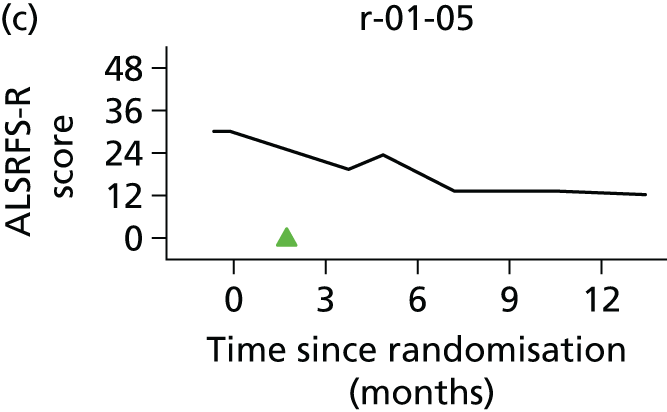
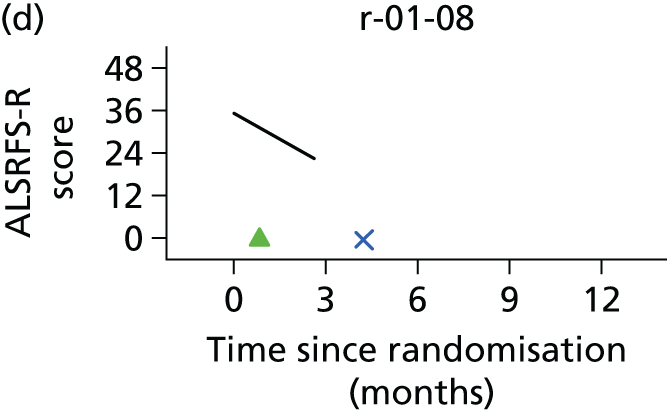

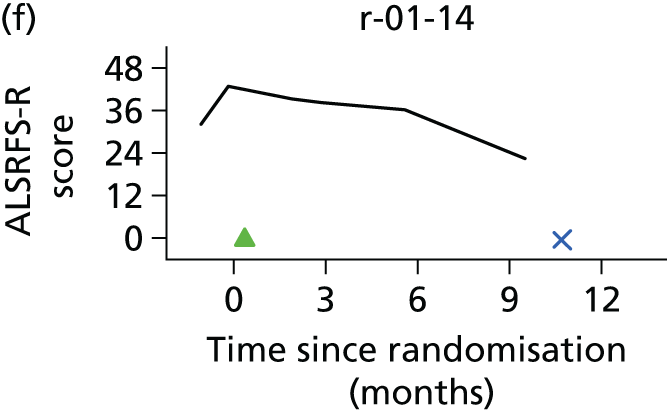


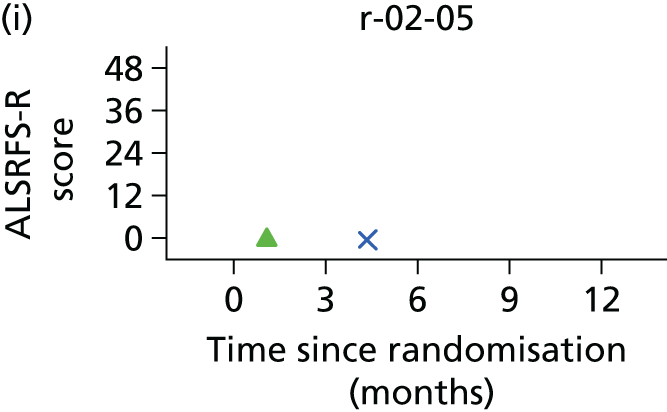












FIGURE 28.
The ALSRFS-R score against time for non-implanted participants. The cross denotes date of death.







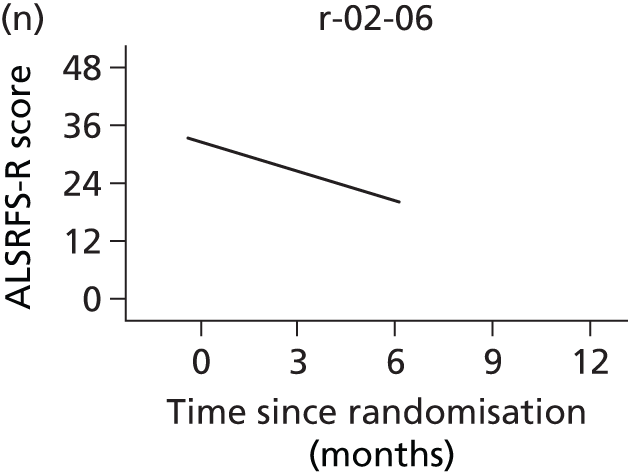
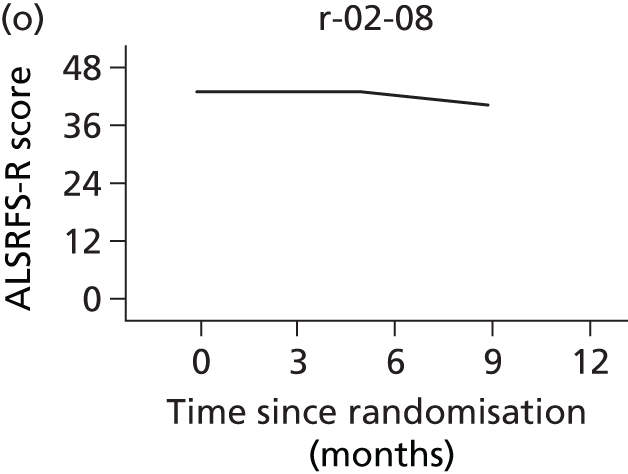
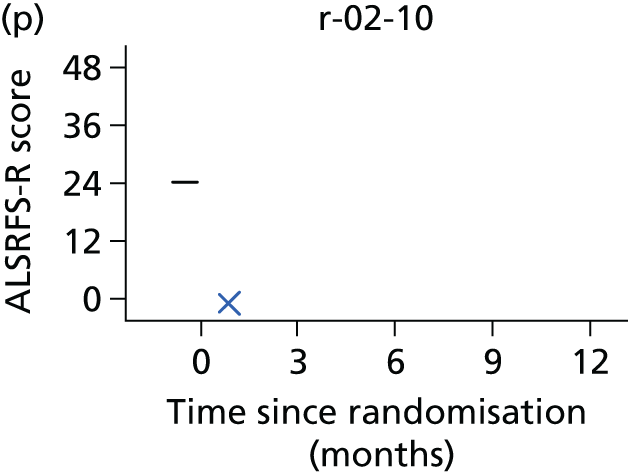
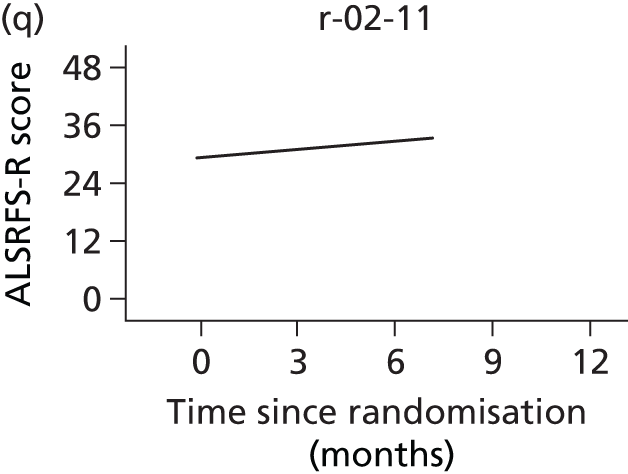
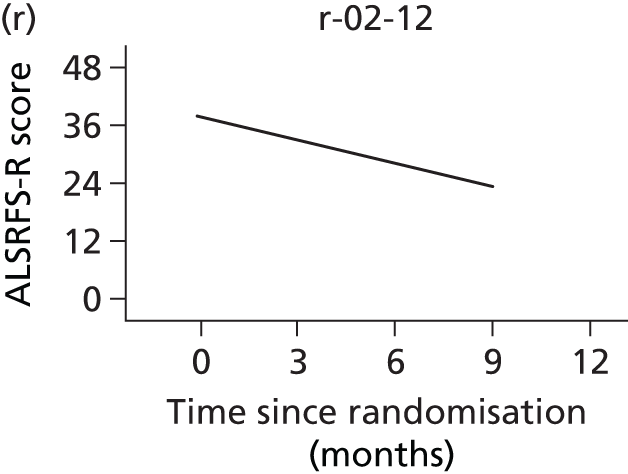
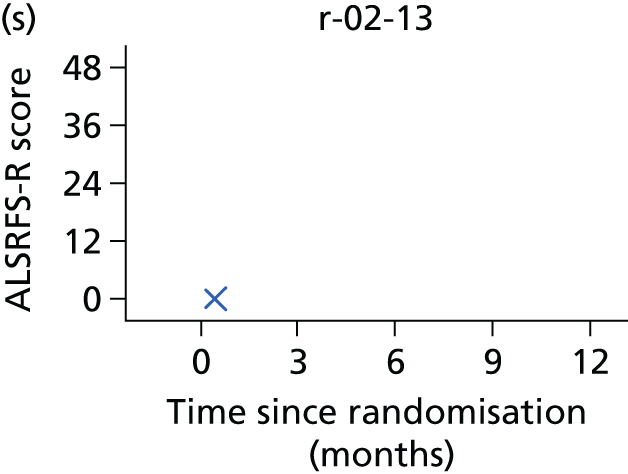


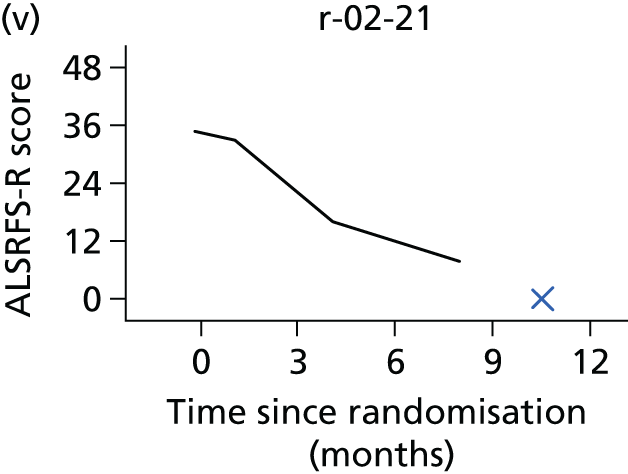



Chapter 5 Qualitative substudy
A qualitative longitudinal study formed a subelement of the trial design. This was an essential part of the DiPALS trial, as it is important not only to investigate the efficacy of DP but also to ensure that any extension of life was not to the detriment of QoL. The qualitative substudy therefore was designed to complement the data collected via the SF-36 and SAQLI by providing additional depth of insight into patient and carer perceptions and experiences of the DPS intervention.
The aim of the qualitative component was to evaluate the acceptability and perceived impact of the intervention on patients with ALS and their family.
Outcomes
The primary end point was the perceptions of patients and carers regarding acceptability and impact of the device.
Qualitative substudy methods
Sampling
We purposively selected participants for the qualitative element of the study from those randomised to the pacing intervention arm of the trial. We intended that our sample would include diversity in terms of patient sex, age, ALS type, and across the different ALS centres taking part in the trial.
Potential participants for the qualitative substudy were identified following randomisation. They were approached by study leads at the research centres at the first follow-up appointment after surgery. The qualitative study component was outlined to potential participants, and an information sheet related to this component was also provided (see Appendices 7 and 8). If they were willing to take part, contact details were provided to a qualitative researcher who was independent of the main trial team.
It was expected that ALS patients would not be able to tolerate an interview in addition to other trial assessments when they attended follow-up visits at local sites. Therefore, an experienced research fellow (SB) conducted semistructured interviews at a time and location that was convenient for participants.
Data collection
Qualitative interviews with patients and carers were carried out at two time points: 1 month following initiation of the pacing intervention and, when possible, 6 months later. Interviews were conducted in patients’ homes lasting 45–60 minutes and were based around a predefined interview schedule (see Appendix 18).
Prior to the interview taking place the researcher checked the contents of the patient information sheet with participants, answered any questions, and a consent form was signed. Participating carers were interviewed together with patients or separately, as preferred by respondents. During the interviews a total communication approach was used for participants with impaired speech, whereby use of gesture, writing, communication aids and forced alternative responses were used to facilitate responses.
Interviews were audio-recorded. The first interview was carried out 1 month following implantation, and focused on expectations and experiences of the surgery, learning to operate the system, and perceptions regarding any impact on life. The second interview, undertaken when possible approximately 6 months post implantation, was considered an appropriate time to allow patients using DPS (and caregivers) to become familiar with the intervention and to have gained an understanding of potential impacts. Therefore, the second interview explored experiences of the system, views regarding DP versus NIV, perceptions of any effects, and reflections in hindsight on taking part in the trial.
Data analysis methods
Interviews were transcribed verbatim and transcript data were analysed using techniques of thematic analysis, where similar concepts or ideas across the interviews are brought together and assigned a code. Relationships between codes are then examined to develop a network of themes and subthemes recurring across the data. Systematic coding and retrieval of data was supported by the ATLAS.ti software (version 7, ATLAS.ti Scientific Software Development GmbH, Berlin, Germany). Data were shared and discussed at several qualitative team meetings to establish consensus for the coding network.
Participant characteristics
Fourteen patients and eight carers took part in the qualitative substudy. Eight patients were male and six were female; 10 were aged in their sixties, and four were aged in their fifties. On the ALS FRS-R at entry into the study, 11 patients were in the mild impairment range for bulbar function and three were in the moderate bulbar impairment range. In terms of limb function, one patient scored > 9, 12 scored 5–8 (moderate impairment), and one scored < 5. Patients were under the care of five different ALS centres. Nine participants were interviewed at both time points, two patients had died in the interval period and two patients preferred not to be followed up because of disease progression. One further person was not using the system at initial visit, or subsequently, and declined follow-up.
Usage of the diaphragm pacing system
At first visit, all but one of the participants had the DPS in use (although one system had technical issues with a broken wire and had not been used for a few days). Ten participants were using DPS during the daytime. Two preferred to use it overnight, and another used it during both day and night. Nine patients had it in operation for 1.5–2 hours per day (in a number of half-hour sessions), with one person reporting 5 hours, and three people estimating at least 6–8 hours’ daily use. Seven patients were also using NIV overnight, and one used NIV in the daytime for short periods. Five participants were struggling to use, or were unable to use, NIV, and one person was awaiting initiation on NIV.
At the 6-month follow-up, use of DPS among this sample of participants during the daytime ranged from 20–24 hours (two patients) to 2–5 hours (five patients). Five patients used NIV overnight, two used it for 20–24 hours; two participants used it for 7–9 hours overnight, one occasionally and another rarely.
Results
Patient and carer experiences
Participants described the process from initial approach to be involved in the study, to ongoing usage of the pacer and perceived outcomes. The recurring themes and subthemes are presented in Figure 29. Owing to the extensive volume of material we focus on the themes most frequently found and include a number of verbatim quotes to illustrate key points.
FIGURE 29.
The patient journey trialling DP.
The surgery
Experiences of the surgery
The prospect of surgery (in particular having a general anaesthetic) was described as a concern for some:
I was more concerned about being put to sleep rather than the surgery.
I was a bit concerned about having the op, yeah; any operation you have, as an ALS patient there is going to be some side effect.
There was obviously the consideration I might not go under and come out properly.
He did have a few worries about it.
She’s bad, she always has been with gas.
Whereas other patients reported little apprehension:
I would say the operation seems quite straightforward.
As far as I’m concerned it’s just a minor operation.
Recovery
Several patients mentioned their expectation that they would stay in hospital only overnight following the surgery. All but two, however, had stayed longer:
Coming home it took about four days I think.
It might’ve been a week and a day.
A day, two days . . . but it turned out to be longer than that.
There were reports of postoperative pain for around half of patients:
My right shoulder, when I came out from anaesthetic, you know, that was killing. I couldn’t move.
I got pain in my shoulders, which they said was referred pain.
He did have painkillers in the first few days, he did have a lot of . . . I think there was quite a bit of pain.
I had a lot of pain, yeah; had this terrible pain on this side.
Impact of surgery
Four participants mentioned that the surgery had impacted adversely on their ALS symptoms:
It took him a while to seem to pick up after it, which I didn’t expect.
It wasn’t bad; the only thing it did do . . . sent my ALS out of kilter.
Since coming out of hospital I’ve definitely felt weaker.
If you have any kind of procedure you are going to take a few steps backwards and you’re going to have to recover again.
Operating the equipment
Expectations versus reality
Participants described their reaction on first seeing the system. All had seen either the equipment or pictures of it at their consultation prior to agreeing to take part and, for most, expectations were not different from reality:
It was what I expected really; I just expected what I’d got.
However, some patients described how they had struggled to visualise exactly what it would look like:
They kept saying well it’s like a pacemaker . . . and we didn’t know really to be honest.
I mean when you wake up and see it on you, it’s a bit of a shock.
I didn’t know what I was expecting.
A lot more (incisions) than what I expected . . . they’d said keyhole I think I just got an idea of a cut here and a cut here.
I don’t know what to expect, basically; I wasn’t sure what it would, cos even looking at a picture you’re not sure what it’s going to look like on you.
The main area of surprise seemed to be regarding the narrow width of the wires:
You kind of think oh wires out your tummy you just expect something that you plug into a computer.
And the wires, cos they’re more like threads . . . you always think wires are thicker don’t you.
There were no instances of patients expressing concerns regarding having technology within and, attached to, their bodies. This perhaps is to be expected from individuals who had agreed to take part in the trial. Participants, although recognising it could be a little strange, mostly seemed to have taken having the system fitted in their stride:
Really weird to have a plug into you; it’s a bit science fiction really.
I’ve got all these bits of wires in me now.
They’re (family) fascinated with it, I’m actually quite OK with it.
It’s not the nicest thing hanging from your diaphragm but . . .
I said yeah, you know, no problem; something to get your head around.
There is something there still physically on your, attached to you.
He (patient) loves the technology.
Ease of operation
There was consensus among participants that the system was easy and trouble-free to operate:
It was a piece of cake, no problems.
It’s easy to manage, there’s nothing to do other than make sure the cable’s connected properly.
It’s so easy to manage; it is a very simple device to use.
That’s easy, yeah, just press two buttons, even I can do that.
It’s really simple.
A small number of patients with severe limb impairment needed assistance to switch the device on and off.
Plugging and unplugging
Plugging the external lead on to the receiver on the body was described as being potentially awkward:
The actual fitting of the pacer on to this is very awkward for somebody with ALS. It’s very small and it’s very hard to get in exactly the right place.
You put it in, it doesn’t click, so you keep trying, trying, trying, you know, so it is a bit annoying actually.
She had a job trying to fit it because it was the other way round, it took her some time.
Sometimes it goes in really easily.
It is a bit of a struggle to get it in.
The difficulty in attaching the lead to the connector was described as being made more challenging owing to the siting of the connector:
If it were a little bit lower down maybe they could do it themselves.
Because where it is it’s quite hard to see, especially for a lady.
If it were a bit lower down it’d just go in.
You can’t see a thing cos it’s under my bust.
I think the location of the wire is such a way that I can’t even look at it properly.
Sensation of the pacer working
Patient reports of the sensation of the pacer working varied considerably. The majority of participants described it as a noticeable, but not painful, feeling:
If I’m watching something, if I’m talking to somebody, you forget about it.
It’s hard to describe; it’s quite strong and almost like beats.
It’s a bit like a needle scratching down your stomach.
It’s like really, really strong butterflies.
It just like your heart ticking away, you know it’s there but you don’t know it.
Just a little electric pulse.
Like a flutter, fluttering feeling.
It’s not painful; within a day you don’t really notice it.
I would not say it’s pleasant, I mean it is definitely unpleasant, but it is not unbearable.
You do feel something but it’s not uncomfortable; he initially used to say ouch, but now he’s stopped doing that.
Others, in contrast, experienced higher levels of discomfort:
It’s like getting shocked with an electric fence.
it affects the left shoulder; it is a bit painful when you inhale.
The most excruciating pain she’s ever experienced in her life, no exaggeration.
Because of the discomfort and pain that he’s had, he hasn’t been using it as he should be.
It can be very uncomfortable; this is very painful.
For three participants, the level of pain experienced limited the length of time that they were able to use the system. At follow-up interview, these individuals had continued to attempt to use the pacer despite their discomfort. One person had found that if it was much more tolerable when lying down at night .
Impact on life
The system was not reported to have an adverse impact on life (apart from those experiencing significant pain):
My life just goes on normally, I work around it.
No it doesn’t have any effect; I can just sit and have it on.
We just fit it round our routine.
You just sit back and watch something else and do something and suddenly half an hour is gone.
Only one patient highlighted that making time for several interventions impacted on their day:
It has now structured our whole day . . . cos we can’t do this at night with three hours on that, two hours on this, feeds. The entire day is structured out by these things now.
Another patient reported that the system set off security alarms in shops, so this affected where they went.
Portability
Several participants mentioned that they had been surprised at the size of the external battery unit, and some that they struggled to put it in a clothing pocket:
It was bigger than I was expecting; well I’d seen it on my [tablet] but of course on there it looks quite small.
It is a little bit on big side, for what’s in it; I would think they could have made that like half size of that.
I wouldn’t like it in my pocket; it might come out.
A bit big even for our pockets.
I’m not sure where box is going to go.
It needs a bag provided, you know.
It needs either a waist strap or a shoulder strap or just something to carry it around in.
The solution that most adopted was to use a waist bag or ‘bum bag’:
We just altered a bum bag, didn’t we, to use.
I’d certainly say well try a bum bag.
My wife thought of that, because it was getting a bit awkward, what do you do with this thing and this box, so she said OK, we can use this bag so we took it out and then she put, put a hole in the side.
Fragility of the system
The system was perceived to require careful handling, with worries regarding pulling or catching and then breaking the external wires and connector, or dropping the external unit. Patients were anxious that if the wires broke that they would need to undergo further surgery, and that if they pulled on the wires that the internal mechanism would be damaged:
Those wires so minute you know you could look at them and they break.
I’ll have to make it really secure so I can’t catch it and pull it.
Because I’m liable to drop it, the flex might just drop and I don’t want it to pull on it.
Well I don’t know how strong the wires are, are they strong enough, I mean if I pull it will it come out.
You’re careful with it, cos obviously it’s quite delicate.
It could slip off, you could knock it.
I think the only thing that is a concern is just if you pulled on the wires.
But if ever I pull it it’s like pulling wires out of your flesh isn’t it really.
The wires could catch on clothing or catch in fingers:
You can catch it quite easily; when you bend over they catch.
I have to be a bit careful I caught it my pyjamas over the thread the other day.
Night-time was a particular concern in terms of pulling the wires:
She was worried if she turned over and pulled it.
I’m a bit worried about sleeping with it cos I turn quite a bit at night.
These worries were not unfounded, with two patients in the sample having to suspend use of the pacer shortly after initiation because of broken wires:
One of the wires is broken.
The reason the wire came out was I was sitting on the bed and the phone rang and I had on it and I forgot so it went to the floor.
Washing and showering
There was variation in level of patient and carer concern regarding getting the system wet. For most individuals, showering with the connectors exposed was not of concern after the incisions had healed:
Not too worried because the wires have to be kind of watered because it’s in your body and there’s fluids there all the time.
I don’t think about it.
She’s been in the bath of couple of times it’s no bother.
It’s fine now; I just shower like you would normally.
Three participants were more cautious and, at follow-up interview, had continued to ensure that the connector was kept dry:
They say there’s no problem if you wet it but he knows that electricity and water don’t mix.
Even now I put a piece of plastic on it.
Some people say don’t get it wet, other people say it doesn’t matter; my advice was just cover it when you shower.
Perceived outcomes
Perceived effect
At first interview, 4 of the 14 patients reported that they had noticed some improvement to their breathing or had a stronger voice, changes that they attributed to using the pacer. Of the nine who could be followed up, two perceived that the pacer had been of benefit to their breathing. The most frequent responses to questions regarding any perceived benefit were uncertainty, and hope that benefit may be in the longer term:
There’s very little I can attribute to this.
I can’t actually tell if it’s helping me to breathe easier or not.
I don’t know.
You don’t know if it’s working or not.
Hopefully the diaphragm will benefit from that exercise and stand me in good stead later on in life that’s the only way I’m looking at it.
The progressive nature of the disease meant that it was challenging for patients and carers to ascertain where a patient would have been if they had not trialled the pacing:
I don’t know how to tell if the having pacer has and will make a difference.
We don’t know how quickly deterioration would be anyway.
He hasn’t seemed to have improved any but then again he could have been worse than this if he’d not had the machine on so it’s really hard to compare isn’t it.
I don’t know whether it’s because of diaphragmatic pacing or whether they think that it would be the same.
It’s hard to tell because we, we feel he’s deteriorated quickly anyway.
Preference for pacing versus non-invasive ventilation
Although there was very limited evidence of perceived benefit, patients generally reported that the pacer was preferable to NIV:
I much prefer this [pacer], I have a problem with the other one [NIV].
If that works is it possible I won’t have to have the NIV.
If they could just replace the mask with that.
Definitely the pacer, because the pacer that is brilliant, the mask is a nightmare.
If I had a choice it’d be this one [points to pacer].
There was a recognition, however, that benefits were more easily attributable to using NIV:
I can’t say that if I’m battling to breathe and I put this thing (pacer) on I get instant relief whereas if I put the NIV on, the minute I put it on I can feel it taking over.
I don’t have enough confidence in this [pacer] to tell me it’s replacing that [NIV].
I think I’d go for the pacer rather than the mask but I feel that the mask does more good.
I’d have the breathing mask because it makes me sleep and I feel better in the morning cos I’ve had a good night’s sleep but I’d pick the pacer if it did the same thing as the breathing mask.
Some patients emphasised that the two interventions were operating differently:
When you get up in the morning you can feel fresh in a way, this is a completely different device which agitates your diaphragm.
To be perfectly honest, with this disease I think you need what you need; they’re not doing the same thing; there’s good things and bad things about both.
Reflections in hindsight
Participants were asked what they would say to another patient with ALS who was considering having the pacing system, and their reflections on taking part in the study. None of the patients at the first interview reported that they regretted having the pacer fitted:
So far we have no regrets.
If you’ve got a chance of it go for it.
With this disease, there are so few things that anybody can do for you that, to be perfectly honest, if you get chance or something then I’d grab it with open arms.
Well I’d say go for it, you’ve got nothing to lose.
At second interview these views were unchanged:
Well tell ‘em to have it, I mean everything that helps to keeping your breathing is obviously good, the only thing is you don’t know if it is helping you or not.
They should go for it.
You don’t have any regrets do you . . . absolutely none; like everything it’s got to be an individual decision because with ALS whatever you do you know that there’s a finite about what you can do but in this instance I would say have a go.
Chapter 6 Discussion
Main findings
The main finding was that DP had a deleterious effect on overall survival. In the DP plus NIV arm compared with the NIV alone arm, the observed reduction in overall survival was 11.5 months and the reduction in survival from symptom onset was 17 months.
The impact on QoL was at best minimal, with no statistically significant differences observed between the arms in patient or carer preplanned QoL measures SF-36, EQ-5D, SAQLI and CBI. The lack of benefit on the SAQLI is particularly noteworthy, as the one RCT of NIV demonstrated improved QoL and respiratory symptoms using this inventory. 1 A planned cost–utility analysis did not go ahead following the lack of efficacy. However, we analysed the EQ-5D-3L scores, as this is a valid QoL tool in its own right. The patient health utility (EQ-5D-3L) was slightly lower in the NIV plus pacing group than in the NIV alone group, especially when a score of 0 was imputed to EQ-5D-3L following death. Differences were modest at any individual time point, but longitudinal analysis demonstrated statistically and clinically significant differences on all patient EQ-5D-3L questionnaires, indicating that at least on one measure, the addition of DP, was associated with a reduction the QoL of patients.
There were more AEs in the NIV plus DPS arm, and many of these were related to the surgery, for example postoperative pain. The type and frequency of the AEs are consistent with those published for ALS, and device-related AEs (e.g. discomfort on pacing or wire failure) were less common than observed in the SSPB, as submitted to the FDA. 11 There are three immediate plausible explanations for the latter finding: (1) improvements in using the device with time, (2) patient under-reporting or (3) the fact that AEs were reported over a longer follow-up (in our study, AEs were reported up to death or 1 year post randomisation, whichever came first). Nevertheless, the low rate of pacing-related AEs support our assertion that the implantation procedures and management of the NeuRX RA/4 DPS was conducted in accordance with previous reports and the manufacturer’s recommendations.
More deaths were observed in the NIV plus pacing arm; however, the causes of death were similar across the arms and consistent with expected causes of death in ALS. This observation suggests that the NeuRX RA/4 DPS is not modifying the disease phenotype of ALS leading to death through some unusual mechanism. The causes of death would support the hypothesis that the disease course is accelerated by the intervention. It is perhaps important to emphasise that postoperative mortality was not encountered, with only one participant death within 3 months of implantation (and this was 45 days after the procedure). This rules out the direct effects of the implantation procedure itself as the cause of the observed harm in the NIV plus pacing arm.
A full health economic analysis of the cost-effectiveness of the NeuRX RA/4 DPS was planned. Given the lack of efficacy and apparent harm, this analysis did not go ahead. We have, however, compared the health-care resource use between the two arms. This is helpful in exploring if there were differences in other treatments or resources that could have impacted on the results. We found no difference in the way participants accessed health care. Similarly, access to other respiratory interventions, such as cough assist, breath stacking and suction, was comparable. There was a tendency for patients in the NIV arm to use cough assist and suction more frequently, whereas those in the NIV plus pacing arm used breath stacking more. The impact of these differences cannot be quantified, but they are unlikely to explain the differences in survival we observe.
Survival in relation to non-invasive ventilation and diaphragm pacing use and compliance
One concern was that perhaps patients in the NIV plus pacing arm would reduce NIV use in favour of DP use. If this occurred, then the negative impact of not using NIV, a proven intervention to prolong life, may have caused the survival difference between arms. Somewhat paradoxically, NIV use was similar in both arms, and, when modelled, NIV use did not influence survival, although this is likely to reflect increasing use of ventilation as prognosis deteriorates. We explored whether or not bulbar function at the time of randomisation and whether or not NIV tolerance influenced survival. In the RCT,1 NIV individuals with poor bulbar function did not experience the survival benefits that those with good bulbar function obtained. Bulbar dysfunction is one obstacle to successful NIV use, but there are others, and indeed, some patients with significant bulbar dysfunction can manage NIV. Therefore, looking at NIV tolerance is likely to be the most informative. The impact of bulbar function at the time of randomisation was negligible among the subgroup of patients who were intolerant of NIV and had a trend towards worse survival in the NIV plus pacing arm than in the NIV arm. Although the numbers are small, this would suggest that DP should not be considered an alternative in patients not able to tolerate NIV. This is particularly disappointing, as despite all efforts there is a consistent 20% or so of individuals with ALS who cannot get on with NIV and would benefit from an alternative means of respiratory support. 2,3
Diaphragm pacing was well tolerated, with the majority of patients reaching the set targets for pacing. We explored whether or not the amount of DP use influenced survival and did not find an association. One might have expected, given the overall findings, that the more an individual used DP, the worse the outcome would be. We did not observe such a dosing effect. The study was not designed to look for a dosing effect and one needs to be cautious in interpreting observational data. However, this observation would lend soft support to the hypothesis that the indirect effects of surgery itself may have been the major contributor to the harm.
Qualitative study
The inclusion of qualitative data relating to patient and carer experiences involved in trials is important, as it ensures that research does not focus only on intervention effectiveness, but also considers elements of implementation and acceptability to participants. The use of a qualitative substudy is a significant strength of this trial, as it provides a comprehensive analysis and interpretation of patient perspectives.
The qualitative data provide valuable insights into the views and experiences of patients and carers following their decision to take part in the trial of DP. For most participants there were few factors adversely impacting on their use of the intervention, although some patients in contrast described severe pain or discomfort.
The qualitative data confirm previous reports of pain experienced by some users of DP and highlight the wide variation in experiences: a longer than expected hospital stay; the perception among some that the surgery had negatively impacted on their functioning; the perceived ease of operation; issues of portability; and concern regarding fragility of the device.
In general, the participants found the device to be acceptable in terms of ease of operation and limited impact on QoL. The majority of users described hoped-for rather than perceived benefit. Although the majority of participants were uncertain regarding any benefit obtained, none appeared to regret their decision to trial the device. They described the limited options available to people with ALS and their willingness to try any available interventions.
Although the data offer important insights into patient and carer views and experiences of DP, the qualitative data may be limited by the small sample size and by the fact that these participants had been invited and agreed to participate in a clinical trial. We were successful in achieving a sample relatively balanced by participant sex; however, other differences between our qualitative substudy sample and wider groups of patients with motor neurone disease may need consideration.
Understanding the results
There has been a presumption that DP offers probable benefit; however, the mechanism by which such benefit is conferred is unclear and unproven. One potential mechanism put forward is that DP reverses an effect on the diaphragm muscle fibres whereby a shift from a predominance of efficient type I fibres to inefficient type IIb fibres occurs. 12 This hypothesis is extrapolated from observations in a population receiving closed-system invasive ventilation support and it is not known whether or not NIV has a similar effect on the diaphragm muscle fibres. 28 A further hypothesis is that DP leads to conditioning of the diaphragm muscle (i.e. increase in muscle mass). There are a few case reports of this, although the functional consequence of this is unknown should it occur. 29 Improved compliance of the lungs and prevention of basal collapse; restoration of the co-ordination of breathing, lost as a result of upper motor neurone dysfunction; and increased tidal volumes are other potential as yet unproven mechanisms. Unhelpfully, we can be no more certain as to why DP may be harmful. The possibilities are a direct effect of pacing (i.e. electrical stimulation) on either the muscle or the phrenic motor neurones. The physiological effects of pacing have not been studied in humans. In canines and rodents, it is clear that neuromuscular damage can be induced depending on the parameters of pacing and that the effects observed differ between healthy and disease models. 8,9 A simpler explanation may be that pacing causes excessive muscle fatigue or that asynchrony between pacing induced diaphragm contraction, patient- and/or NIV-triggered breaths is an issue.
Undertaking surgery in patients with ALS and respiratory failure does present some challenges and angst among health-care professionals. Our study confirmed the finding of others that general anaesthesia can be undertaken safely in terms of perioperative mortality, with no deaths within 30 days of the implantation procedure and only one within the first 3 postoperative months. 7 However, there is evidence that the consequences of surgery and general anaesthesia may be less immediate and not apparent initially. A retrospective review of ALS patients undergoing surgery for any reason demonstrated an apparent acceleration of ALS disease progression post surgery, suggesting a potential disease-modifying effect, albeit one that is not well understood. 10 Direct effects of anaesthetic agents and surgical stress, with the release of systemic pro-inflammatory cytokines, are postulated to contribute to the observed effect. It is possible that such indirect effects of surgery are contributing to the survival differences in the DiPALS trial; however, this is far from certain. In an attempt to minimise patient burden and focus on survival and cost-effectiveness, during the grant peer-review process secondary outcome measures detailing progression of ALS (ALSFRS-R score) and respiratory function (FVC) were removed from the protocol. When the negative outcome was identified, the TSC requested that an attempt was made to retrospectively collect any functional data on participants during the follow-up period. Clearly, this is an unplanned and retrospective analysis on incomplete data and conclusions based on this analysis need to be cautious. The baseline data at randomisation demonstrate the two arms to be balanced in terms of respiratory function and ALSFRS-R score. However, it does appear that there was a change in the rate of decline of FVC and ALSFRS-R score following implantation. This finding, however, would be consistent with either an indirect effect of surgery or a direct effect of pacing, as described in the Discussion.
The results of the Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis trial in context
Our findings are at odds with the FDA summary of SSPB, which reported a survival advantage for DP of 16.1 months from symptom onset and 9 months from the point of NIV initiation, compared with NIV alone, in a historical cohort. 11,30 This compares with our findings of a reduction in survival in the NIV plus DPS group by 17 months from symptom onset and 11.8 months from randomisation. The median survival from symptom onset in DiPALS was 45 months in the NIV arm, 28 months in the NIV plus pacing arm and 56 months in the SSPB pacing study.
This raises several possibilities, which include that there are differences between the populations in the DiPALS trial and in previous cohort studies; the DP intervention has been delivered differently; or other treatments that impact on survival are different between the DiPALS trial and previous studies. We will discuss these three possibilities next.
The Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis trial population compared with other studies of diaphragm pacing
The population in the DiPALS trial other than having a male preponderance is relatively typical of an ALS population with age, proportion of limb to bulbar onset, rate of decline of ALSFRS-R score, all as would be expected. 31 The complete data set, including baseline study population characteristics, from the uncontrolled multicentre cohort study which led to FDA approval on humanitarian grounds of the NeuRX RA/4 DPS (HDE), has to date not yet been published. Therefore, it is not possible to compare the populations and, therefore, it is challenging to fully understand the differences in the reported outcomes. The broad inclusion criteria for the pilot and pivotal phases of the cohort study were evidence of residual bilateral phrenic nerve function and a FVC of < 85% at screening and > 45% at DPS implantation, but specific details, such as the mean age, FVC and bulbar function of the recruited population, are not publicly available. 11 The FVC inclusion is higher than the 75% used in the DiPALS trial and if a significant number of patients lay in the 75–85% range for FVC, then this would contribute to some of the difference observed. More immediately, however, it is noteworthy that of the 144 patients enrolled only 106 were implanted and 84 included in the analysis of overall survival. The reasons for these exclusions are not provided, but we speculate that these may, again in part, explain the disparity. The cohort study contained a lead-in phase of 3 months before implantation during which time patients were monitored. The details of 38 patients who did not undergo implantation are not reported. It seems likely that some have been excluded because they rapidly deteriorated, but this is not clear and there may have been other criteria used. Of the 106 implanted, the data of only 84 patients contribute to the SSPB report, two patients having been lost to follow-up and 20 not meeting the HUD criteria. This cohort is therefore a highly selected group and may not be generalisable to the wider ALS population. In contrast, our trial used an intention-to-treat approach in which all consenting participants were analysed, including patients who subsequently declined rapidly in either group. Therefore, we suspect baseline difference in the study populations may be a major cause of observed survival differences.
Outside ALS, DP is licensed for patients with spinal cord injury in several territories including the USA and Europe. As with ALS, the marketing licence was granted on humanitarian grounds and on the basis of a small (n = 50) cohort study of patients with spinal cord injury resulting in a poorly controlled diaphragm and requiring continuous mechanical ventilation. 11,30 Forty-eight of the 50 had achieved a successful outcome, defined as a continuous period of at least 4 hours without the assistance of a mechanical ventilator; one participant was unable to achieve diaphragmatic pacing. The average follow-up was 1.4 years, during which four deaths were reported, none of which was considered to be related to the device. 11 Thus, as with the ALS cohort data, there is limited evidence to support or refute the safety of DP.
Delivery of diaphragm pacing in the Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis trial compared with other studies
The device manufacturer (Synapse Biomedical) provided training for surgeons and site staff in all aspects of the intervention, as is standard practice when it supplies the NeuRX RA/4 DPS within a service. For example, an experienced surgeon from Synapse Biomedical attended the implantation procedure and proctored the local site surgeon until he or she was competent to insert the devices. At all surgeries Synapse Biomedical staff were present to provide standard technical support. It is likely that the implantation procedures, which followed the manufacturer’s recommendations, were comparable to those undertaken during the earlier cohort study. Supporting this is the fact that the number of surgical complications was lower than previously reported and there was no mortality within the 30-day perioperative period. All diaphragms were stimulatable at surgery supporting our combined clinical and ultrasound assessment of residual phrenic nerve function. No surgeries needed to be abandoned, as planned in the protocol, if on intraoperative mapping of the diaphragm stimulation was not achievable. Following implantation the majority of patients became regular users (mean daily use 6.2 hours) and titrated their use over the course of the study, in line with the protocol and the manufacturer’s recommendations. There were six non-users in the NIV plus pacing arm: five of these did not undergo surgery and one stopped pacing after pulling the wires out after 1 month. The overall survival analysis in the DiPALS trial was by intention to treat; however, if these non-users were excluded, then the HR remained significant (HR 2.71, 95% CI 1.39 to 5.27).
Other confounders
Interventions proven to influence survival in ALS are riluzole, multidisciplinary team care and NIV. 1,4–6 Riluzole use was equal across groups, as this was one of the inclusion criteria. NIV use was comparable across both arms. All participating centres deliver multidisciplinary team care and were specialist centres for treating patients with ALS and respiratory failure. The heath-care resource use indicated patients accessed care in a similar manner, regardless of treatment group.
Limitations
Given that patients allocated DP underwent surgical intervention, it was unavoidable that participants were unblinded as to the intervention. The study assessors were also unblinded to the intervention. The trial statistician was unblinded, but withheld accumulating data from the study team. As the primary outcome measure was objective (overall survival), the risk of bias is small, but there is an unavoidable risk of bias in the subjective patient-reported secondary outcome measures. We considered inserting the DP devices in the control group but not connecting them (sham pacing), to reduce the risk of bias and also to be able to offer DP to control subjects at the end of the 12-month follow-up period, but concluded that implanting control subjects appears less reasonable if one considerd a possible negative outcome. The effect of DP on the ongoing use of NIV was a concern and we asked the question of whether or not patients stopped using the NIV system, which has established survival benefit, in favour of the DP system. However, our data show that this was not the case, with similar daily periods of NIV use across both groups.
There are imbalances between the treatment arms in our study, the most notable of which is that the NIV plus pacing arm was slightly older. We have adjusted the HRs for this (and other) covariates, but also propose that it is unlikely that such a small age difference would have a large impact on ALS survival. Older age is negatively associated with survival, but less so within the ALS population, in which prognosis is poor across all demographics; certainly, we are unaware of any previous work that has identified age as having an impact that could explain a difference of this magnitude. Similar numbers of patients across each group are reported to receive additional respiratory interventions. However, there are differences in the frequency of cough assist use and breath stacking use, among those given devices, across the treatment groups. The effects of these differences are unknown but, again, are unlikely to explain the observed poor survival in the NIV plus pacing group.
Developments since the completion of the Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis trial
Recently, a second randomised trial [Early Stage Amyotrophic Lateral Sclerosis Phrenic Stimulation (RespiStimALS); NCT01583088] has also stopped early citing ‘absence of benefit [and] a statistically significant excess mortality in the group of patients receiving active stimulation’. 32,33 There are two notable differences between this study and the DiPALS trial. First, DP implantation was carried out prior to respiratory failure, with the primary end point being the time to requiring NIV. Second, all participants underwent surgical procedure, but with the control group having a ‘sham’ procedure in which pacing was not instigated until after NIV had started. The fact that an excess mortality was observed in the intervention arm offers compelling evidence that pacing is actively harmful as opposed to surgery. Subsequently, a third randomised study of DPS (NCT01938495) in the USA has suspended enrolment pending further follow-up data from patients already randomised in the study.
Chapter 7 Conclusions
Meaning of the study and implications for clinicians or policy-makers
Diaphragmatic pacing should not be used as a routine treatment for all patients with ALS in respiratory failure. We cannot exclude the possibility that it is beneficial in a subgroup of patients; however, this should not be assumed. Our findings demonstrate that insertion of the NeuRX RA/4 DPS for pacing is harmful when instigated at the point at which an individual with ALS develops respiratory failure. These findings are further supported by a second RCT in which implanting was performed earlier in the disease trajectory, which also stopped early because of excess mortality in the pacing arm (NCT01583088). 32
A poor prognosis and the absence of curative therapy understandably encourage a ‘nothing to lose’ approach among patients and some clinicians alike, with an attendant lowering of the standards of evidence required to adopt a new intervention. Our trial demonstrates the potential for harm that can arise from adopting this approach.
Recommendations for future research
The findings of our trial, and also those of Gonzalez-Bermejo,32 are at odds with the promising survival data presented in a previous cohort11 and, anecdotally, a small number of individual cases using diaphragmatic pacing in our own clinics. Before any further study of DP in ALS is undertaken in the patient population, we would encourage a meta-analysis of all current trial data on DPS in ALS. This may help to identify whether or not there are any specific circumstances in which a patient may benefit from DP, such as those with predominant upper motor neuron disease. In addition, the emerging findings within ALS should prompt some consideration of the evidence for DP use among patients with spinal cord injury. If any further studies take place in the future, they should include measures to understand the mechanism by which harm or benefit occurs as a result of DP.
Acknowledgements
The Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis Research Team
Writing group
Christopher J McDermott, Mike J Bradburn, Wendy O Baird, Susan K Baxter, Cindy L Cooper, Chin Maguire, Judith Cohen and Hannah Cantrill (University of Sheffield); Roger Ackroyd (Sheffield Teaching Hospitals NHS Foundation Trust); Simon Baudouin (Newcastle upon Tyne Hospitals NHS Foundation Trust and the University of Newcastle); Andrew Bentley (University Hospital of South Manchester NHS Foundation Trust); Richard Berrisford (Plymouth Hospitals NHS Trust); Stephen Bianchi (Sheffield Teaching Hospitals NHS Foundation Trust); Stephen C Bourke (Northumbria Healthcare NHS Foundation Trust and Newcastle University); Simon Dixon (University of Sheffield); John Ealing (Salford Royal Hospitals NHS Foundation Trust); Mark Elliott and Abeezar Sarela (Leeds Teaching Hospitals NHS Trust); Simon Galloway (University Hospital of South Manchester NHS Foundation Trust); C Oliver Hanemann (Plymouth University); Philip Hughes (Plymouth Hospitals NHS Trust); Nick Maynard, Kevin Talbot, John Stradling (Oxford University Hospitals NHS Trust); Richard W Orrell (Royal Free London NHS Foundation Trust); Pamela J Shaw (University of Sheffield); and Tim Williams and Dayalan Karat, (Newcastle Hospitals NHS Foundation Trust).
Trial Management Group
Christopher J McDermott, Chin Maguire, Mike J Bradburn, Wendy O Baird, Cindy L Cooper, Simon Dixon, Pamela J Shaw and Hannah Cantrill (University of Sheffield); Helen Woolf, Alison Proctor, Roger Ackroyd and Stephen Bianchi (Sheffield Teaching Hospitals NHS Foundation Trust); Kevin Talbot, Rachael Marsden, Nick Maynard and John Stradling, (Oxford University Hospitals NHS Trust); Tim Williams, Steven Dodds and Simon Baudouin (Newcastle upon Tyne Hospitals NHS Foundation Trust); Andrew Bentley, Katie Lynch and Simon Galloway (University Hospital of South Manchester NHS Foundation Trust); John Ealing (Salford Royal Hospitals NHS Foundation Trust); Philip Hughes, Steve Angus, Richard Berrisford and Agam Jung (Plymouth Hospitals NHS Trust); C Oliver Hanemann (Plymouth University); Mark Elliott, Craig Armstrong, Clair Favager and Abeezar Sarela (Leeds Teaching Hospitals NHS Trust); Stephen Bourke (Northumbria Healthcare NHS Foundation Trust and Newcastle University); and Richard W Orrell, Mark Baker and Concetta Brugaletta (Royal Free London NHS Foundation Trust).
Trial Steering Committee
Carolyn Young (Independent Chairperson, Walton Centre for Neurology and Neurosurgery NHS Foundation Trust), Anita Simmonds (Independent Clinician, Royal Brompton & Harefield NHS Foundation Trust), Lyn Taylor (Independent Statistician, PAREXEL International Corporation, Sheffield), Roger Leek (Independent Patient Representative), Roy Darlison (Independent Patient Representative), Christopher J McDermott (Chief Investigator, University of Sheffield), Mike Bradburn (Study Statistician, University of Sheffield) and Chin Maguire (Trial Manager, University of Sheffield).
Data Monitoring and Ethics Committee
Closed group
Nigel Leigh (Independent Chairperson, Brighton and Sussex Medical School), Michael Dewey (Independent Statistician, University of London) and Aleksander Radunovic (Independent Clinician, Royal London Hospital).
Members of the Open group
Christopher J McDermott (Chief Investigator, University of Sheffield), Mike Bradburn (Study Statistician, University of Sheffield) and Chin Maguire (Trial Manager, University of Sheffield).
Co-applicants
Christopher J McDermott, Cindy L Cooper, Simon Dixon, Pamela Shaw, Wendy O Baird, Mike Bradburn and Patrick Fitzgerald (University of Sheffield); Tim Williams, Simon Baudouin and Dayalan Karat (Newcastle upon Tyne Hospitals NHS Foundation Trust); Kevin Talbot, John Stradling and Nick Maynard (Oxford University Hospitals NHS Trust); Stephen Bianchi and Roger Ackroyd (Sheffield Teaching Hospitals NHS Foundation Trust); Stephen C Bourke (Northumbria Healthcare NHS Foundation Trust and Newcastle University); John Ealing (Salford Royal Hospitals NHS Foundation Trust); and Andrew Bentley and Simon Galloway (University Hospital of South Manchester NHS Foundation Trust).
Local investigators
Christopher J McDermott (University of Sheffield), Tim Williams (Newcastle upon Tyne Hospitals NHS Foundation Trust), Kevin Talbot (Oxford University Hospitals NHS Trust), John Ealing (Salford Royal Hospitals NHS Foundation Trust), Andrew Bentley (University Hospital of South Manchester NHS Foundation Trust), Mark Elliott (Leeds Teaching Hospitals NHS Trust), Philip Hughes (Plymouth Hospitals NHS Trust) and Richard Orrell (Royal Free London NHS Foundation Trust.
We gratefully acknowledge the hard work, support and advice from the following:
-
Rachael Marsden and Hania Piotrowska (Oxford University Hospitals NHS Trust); Katie Lynch (University Hospital of South Manchester NHS Foundation Trust); Steve Dodds (Newcastle upon Tyne Hospitals NHS Foundation Trust); Helen Wollff, Alison Proctor, Theresa Walsh and Gail Mills (Sheffield Teaching Hospitals NHS Foundation Trust); Steve Angus and Jonathan Palmer (Plymouth Hospitals NHS Trust); Craig Armstrong and Clair Favager (Leeds Teaching Hospitals NHS Trust); and, Mark Baker and Concetta Brugaletta (Royal Free London NHS Foundation Trust) for participant screening and data collection
-
Lian Lee (Oxford University Hospitals NHS Trust) and Brian Aspin Dodds (Newcastle upon Tyne Hospitals NHS Foundation Trust) for processing the intervention
-
Katie Biggs (University of Sheffield CTRU) for relief study management
-
Hannah Cantrill (University of Sheffield CTRU) for study monitoring
-
Amanda Loban and Chris Ellis (University of Sheffield CTRU) for support on data management concerns
-
Kylie Cross (University of Sheffield CTRU) and Rebecca Brown (The Sheffield Institute for Translational Neuroscience, University of Sheffield) for administrative and clerical support
-
Ramila Patel (Research Co-ordinator) and Dipak Patel (Research Manager, Research and Development Department, Sheffield Teaching Hospitals NHS Foundation Trust) for their support on ethical and governance issues.
We offer special thanks to the members of our two oversight committees: the TSC and DMEC.
Trial Steering Committee
Carolyn Young (Independent Chairperson, Walton Centre for Neurology & Neurosurgery NHS Foundation Trust, Liverpool), Anita K Simonds (Independent Clinician, National Institute for Health Research Respiratory Biomedical Research Unit, Royal Brompton & Harefield NHS Foundation Trust), Lyn Taylor (Independent Statistician, PAREXEL International Corporation, Sheffield), Roger Leek (Independent Lay Representative, Motor Neurone Disease Association Birmingham and Solihull Group), Roy Darlison (Independent Patient Representative), Christopher J McDermott (Chief Investigator, University of Sheffield), Mike J Bradburn (Statistician, University of Sheffield) and Chin Maguire (Trial Manager, University of Sheffield).
Data Monitoring and Ethics Committee
Nigel P Leigh (Independent Chairperson, Brighton and Sussex Medical School), Michael Dewey (Statistician, University of London), Aleksandar Radunovic (Independent Expert Member, Royal London Hospital), Christopher J McDermott (Chief Investigator, University of Sheffield), Mike J Bradburn (Statistician, University of Sheffield) and Chin Maguire (Trial Manager, University of Sheffield).
Contributions of authors
The following produced the first draft of the report: Christopher J McDermott (Consultant Neurologist), Mike J Bradburn (Statistician), Chin Maguire (Trial Manager), Cindy L Cooper (Professor of Health Services Research and Clinical Trials), Wendy O Baird (Professor of Health Services Research), Susan K Baxter (Qualitative Researcher), Judith Cohen (Research Fellow) and Hannah Cantrill (Research Assistant).
The following conceived of or designed the work: Christopher J McDermott (Consultant neurologist), Mike J Bradburn (Statistician), Cindy L Cooper (Professor of Health Services Research and Clinical Trials), Wendy O Baird (Professor of Health Services Research), Simon Dixon (Professor of Health Economics), Roger Ackroyd (Consultant Surgeon), Simon Baudouin (Senior Lecturer in in Critical Care), Andrew Bentley (Consultant in Respiratory and Intensive Care), Stephen Bianchi (Consultant in Respiratory Medicine), Stephen C Bourke (Consultant Physician in Respiratory and General Medicine), John Ealing (Consultant Neurologist), Patrick Fitzgerald (Health Economist), Simon Galloway (General and Upper Gastrointestinal Surgeon), Dayalan Karat (Consultant Surgeon), Nick Maynard (Consultant Surgeon, Upper Gastrointestinal Surgery), John Stradling (Professor of Respiratory Medicine), Kevin Talbot (Professor of Motor Neuron Biology), Tim Williams (Consultant Neurologist) and Pamela J Shaw (Professor of Neurology).
THE following were involved in the acquisition of data for the work: Christopher J McDermott (Consultant Neurologist), Roger Ackroyd (Consultant Surgeon), Simon Baudouin (Senior lecturer in Critical Care), Andrew Bentley (Consultant in Respiratory and Intensive Care), Richard Berrisford (Surgeon), Stephen Bianchi (Consultant in Respiratory Medicine), Stephen C Bourke (Consultant Physician in Respiratory and General Medicine), John Ealing (Consultant Neurologist), Mark Elliott (Consultant Respiratory Physician), Simon Galloway (General and Upper Gastrointestinal Surgeon), Hisham Hamdalla (Consultant Neurologist), C Oliver Hanemann (Consultant in Neurology), Philip Hughes (Consultant in General and Respiratory Medicine), Ibrahim Imam (Consultant Neurologist), Dayalan Karat (Consultant Surgeon), Nick Maynard (Consultant Surgeon, Upper Gastrointestinal Surgery), Richard W Orrell (Senior Lecturer and Consultant Neurologist), Abeezar Sarela (Consultant in Upper Gastrointestinal Surgical Oncology, Bariatric and Metabolic Surgery and Minimally Invasive Surgery), John Stradling (Professor of Respiratory Medicine), Kevin Talbot (Professor of Motor Neurone Biology), Martin Turner (Senior Clinician Scientist), Tim Williams (Consultant Neurologist) and Wisia Wedzicha (Consultant Respiratory Physician).
The following were involved in the analysis of data: Christopher J McDermott (Consultant Neurologist), Mike J Bradburn (Statistician) and Stephen C Bourke (Consultant Physician in Respiratory and General Medicine).
The following were involved in the interpretation of data for the work: Christopher J McDermott (Consultant Neurologist), Chin Maguire (Trial Manager), Judith Cohen (Research Fellow), Roger Ackroyd (Consultant Surgeon), Simon Baudouin (Senior Lecturer in in Critical Care), Andrew Bentley (Consultant in Respiratory and Intensive Care), Richard Berrisford (Surgeon), Stephen Bianchi (Consultant in Respiratory Medicine), Stephen C Bourke (Consultant Physician in Respiratory and General Medicine), Roy Darlison (TSC PPI representative), John Ealing (Consultant Neurologist), Mark Elliott (Consultant Respiratory Physician), Simon Galloway (General and Upper Gastrointestinal Surgeon), Hisham Hamdalla (Consultant Neurologist), C Oliver Hanemann (Consultant in Neurology), Philip Hughes (Consultant in General and Respiratory Medicine), Ibrahim Imam (Consultant Neurologist), Dayalan Karat (Consultant Surgeon), Roger Leek (TSC lay representative), Nick Maynard (Consultant Surgeon, Upper Gastrointestinal Surgery), Richard W Orrell (Senior Lecturer and Consultant Neurologist), Abeezar Sarela (Consultant in Upper Gastrointestinal Surgical Oncology, Bariatric and Metabolic Surgery and Minimally Invasive Surgery), John Stradling (Professor of Respiratory Medicine), Kevin Talbot (Professor of Motor Neuron Biology), Lyn Taylor (Principal Biostatistician), Anita K Simonds (Professor of Respiratory and Sleep Medicine), Tim Williams (Consultant Neurologist), Wisia Wedzicha (Consultant in Respiratory Physician) and Carolyn Young (Consultant Neurologist and Honorary Professor of Neurology).
The following drafted the monograph: Christopher J McDermott (Consultant Neurologist), Mike J Bradburn (Statistician), Chin Maguire (Trial Manager), Wendy O Baird (Professor of Health Services Research), Susan K Baxter (Qualitative Researcher) and Judith Cohen (Research Fellow).
The following revised the work critically for important intellectual content: Christopher J McDermott (Consultant Neurologist), Mike J Bradburn (Statistician), Chin Maguire (Trial Manager), Cindy L Cooper (Professor of Health Services Research and Clinical Trials), Wendy O Baird (Professor of Health Services Research), Susan K Baxter (Qualitative Researcher), Judith Cohen (Research Fellow), Simon Dixon (Professor of Health Economics), Andrew Bentley (Consultant in Respiratory and Intensive Care), Mark Elliott (Consultant Respiratory Physician), Philip Hughes (Consultant, General and Respiratory Medicine), Richard W Orrell (Senior Lecturer and Consultant Neurologist), Kevin Talbot (Professor of Motor Neurone Biology) and Tim Williams (Consultant Neurologist).
The following were involved in the final approval of the version to be published: Christopher J McDermott (Consultant Neurologist), Mike J Bradburn (Statistician), Andrew Bentley (Consultant in Respiratory and Intensive Care), Mark Elliott (Consultant Respiratory Physician), Philip Hughes (Consultant in General and Respiratory Medicine), Richard W Orrell (Senior Lecturer and Consultant Neurologist), Kevin Talbot (Professor of Motor Neurone Biology) and Tim Williams (Consultant Neurologist).
All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Publications
McDermott CJ, Maguire C, Cooper CL, Ackroyd R, Baird WO, Baudouin S, et al. Protocol for diaphragm pacing in patients with respiratory muscle weakness due to motor neurone disease (DiPALS): a randomised controlled trial. BMC Neurol 2012:12:74.
McDermott CJ, Bradburn MJ, Maguire C, Cooper CL, Baird WO, Baxter SK, et al. Safety and efficacy of diaphragm pacing in patients with respiratory insufficiency due to amyotrophic lateral sclerosis (DiPALS): a multicentre, open-label, randomised controlled trial. Lancet Neurol 2015;14:883–92.
Data sharing statement
Please direct any requests to ctru@sheffield.ac.uk.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol 2006;5:140-7. http://dx.doi.org/10.1016/S1474-4422(05)70326-4.
- Gruis KL, Brown DL, Schoennemann A, Zebarah VA, Feldman EL. Predictors of noninvasive ventilation tolerance in patients with amyotrophic lateral sclerosis. Muscle Nerve 2005;32:808-11. http://dx.doi.org/10.1002/mus.20415.
- O’Neill CL, Williams TL, Peel ET, McDermott CJ, Shaw PJ, Gibson GJ, et al. Non-invasive ventilation in motor neuron disease: an update of current UK practice. J Neurol Neurosurg Psychiatry 2012;83:371-6. http://dx.doi.org/10.1136/jnnp-2011-300480.
- Aridegbe T, Kandler R, Walters SJ, Walsh T, Shaw PJ, McDermott CJ. The natural history of motor neuron disease: assessing the impact of specialist care. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:13-9. http://dx.doi.org/10.3109/17482968.2012.690419.
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585-91. http://dx.doi.org/10.1056/NEJM199403033300901.
- Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996;347:1425-31. http://dx.doi.org/10.1016/S0140-6736(96)91680-3.
- Onders RP, Carlin AM, Elmo M, Sivashankaran S, Katirji B, Schilz R. Amyotrophic lateral sclerosis: the Midwestern surgical experience with the diaphragm pacing stimulation system shows that general anesthesia can be safely performed. Am J Surg 2009;197:386-90. http://dx.doi.org/10.1016/j.amjsurg.2008.11.008.
- Amirjani N, Kiernan MC, McKenzie DK, Butler JE, Gandevia SC. Is there a case for diaphragm pacing for amyotrophic lateral sclerosis patients?. Amyotroph Lateral Scler 2012;13:521-7. http://dx.doi.org/10.3109/17482968.2012.673169.
- Lepore AC, Tolmie C, O’Donnell J, Wright MC, Dejea C, Rauck B, et al. Peripheral hyperstimulation alters site of disease onset and course in SOD1 rats. Neurobiol Dis 2010;39:252-64. http://dx.doi.org/10.1016/j.nbd.2010.03.021.
- Pinto S, Swash M, de Carvalho M. Does surgery accelerate progression of amyotrophic lateral sclerosis?. J Neurol Neurosurg Psychiatry 2014;85:643-6. http://dx.doi.org/10.1136/jnnp-2013-305770.
- Food and Drug Administration . Summary of Safety and Probable Benefit (SSPB) 2011. www.accessdata.fda.gov/cdrh_docs/pdf10/H100006b.pdf (accessed 11 April 2011).
- Onders RP, Elmo M, Khansarinia S, Bowman B, Yee J, Road J, et al. Complete worldwide operative experience in laparoscopic diaphragm pacing: results and differences in spinal cord injuries patients and amyotrophic lateral sclerosis patients. Surg Endosc 2009;23:1433-40. http://dx.doi.org/10.1007/s00464-008-0223-3.
- Kleopa KA, Sherman M, Neal B, Romano GJ, Heiman-Patterson T. Bipap improves survival and rate of pulmonary function decline in patients with ALS. J Neurol Sci 1999;164:82-8. http://dx.doi.org/10.1016/S0022-510X(99)00045-3.
- Aboussouan LS, Khan SU, Meeker DP, Stelmach K, Mitsumoto H. Effect of noninvasive positive-pressure ventilation on survival in amyotrophic lateral sclerosis. Ann Intern Med 1997;127:450-3. http://dx.doi.org/10.7326/0003-4819-127-6-199709150-00006.
- McDermott CJ, Maguire C, Cooper CL, Ackroyd R, Baird WO, Baudouin S, et al. Protocol for diaphragm pacing in patients with respiratory muscle weakness due to motor neurone disease (DiPALS): a randomised controlled trial. BMC Neurol 2012;12. http://dx.doi.org/10.1186/1471-2377-12-74.
- Moher D, Hopewell S, Schulz KF, Montori V, Gotzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c869.
- Hoffmann TC, Glasziou PP, Boutron I, Milne R, Perera R, Moher D, et al. Better reporting of interventions: template for intervention description and replication (TIDieR) checklist and guide. BMJ 2014;348. http://dx.doi.org/10.1136/bmj.g1687.
- Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial ‘clinical limits of amyotrophic lateral sclerosis’ workshop contributors. J Neurol Sci 1994;124:S96-107. http://dx.doi.org/10.1016/0022-510X(94)90191-0.
- Schoenfeld DA. Sample-size formula for the proportional-hazards regression model. Biometrics 1983;39:499-503. http://dx.doi.org/10.2307/2531021.
- Therneau T, Grambsch P. Modeling Survival Data: Extended the Cox Model. New York, NY: Springer-Verlag; 2000.
- Bradburn MJ, Clark TG, Love SB, Altman DG. Survival analysis part III: multivariate data analysis – choosing a model and assessing its adequacy and fit. Br J Cancer 2003;89:605-11. http://dx.doi.org/10.1038/sj.bjc.6601120.
- Royston P, Parmar MKB. The use of restricted mean survival time to estimate the treatment effect in randomized clinical trials when the proportional hazards assumption is in doubt. Stat Med 2011;30:2409-21. http://dx.doi.org/10.1002/sim.4274.
- van Buuren S. Multiple imputation of discrete and continuous data by fully conditional specification. Stat Methods Med Res 2007;16:219-42. http://dx.doi.org/10.1177/0962280206074463.
- Royston P, Altman DG. Regression using fractional polynomials of continuous covariates: parsimonious parametric modelling. J R Stat Soc Ser C 1994;43:429-68. http://dx.doi.org/10.2307/2986270.
- Scott NW, McPherson GC, Ramsay CR, Campbell MK. The method of minimization for allocation to clinical trials: a review. Control Clin Trials 2002;23:662-74. http://dx.doi.org/10.1016/S0197-2456(02)00242-8.
- Payment for Involvement: A Guide for Making Payments to Members of the Public Actively Involved in NHS, Public Health and Social Care Research. Eastleigh: INVOLVE; 2010.
- Bourke SC, Bullock RE, Williams TL, Shaw PJ, Gibson GJ. Noninvasive ventilation in ALS: indications and effect on quality of life. Neurology 2003;61:171-7. http://dx.doi.org/10.1212/01.WNL.0000076182.13137.38.
- Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, et al. Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 2008;358:1327-35. http://dx.doi.org/10.1056/NEJMoa070447.
- Onders RP, Elmo M, Kaplan C, Katirji B, Schilz R. Final analysis of the pilot trial of diaphragm pacing in amyotrophic lateral sclerosis with long-term follow-up: diaphragm pacing positively affects diaphragm respiration. Am J Surg 2014;207:393-7. http://dx.doi.org/10.1016/j.amjsurg.2013.08.039.
- Lechtzin N, Scott Y, Busse AM, Clawson LL, Kimball R, Wiener CM. Early use of non-invasive ventilation prolongs survival in subjects with ALS. Amyotroph Lateral Scler 2007;8:185-8. http://dx.doi.org/10.1080/17482960701262392.
- Hoppitt T, Pall H, Calvert M, Gill P, Yao G, Ramsay J, et al. A systematic review of the incidence and prevalence of long-term neurological conditions in the UK. Neuroepidemiology 2011;36:19-28. http://dx.doi.org/10.1159/000321712.
- Gonzalez-Bermejo J. Early Stage Amyotrophic Lateral Sclerosis Phrenic Stimulation (RespiStimALS) 2015. https://clinicaltrials.gov/ct2/show/record/NCT01583088 (accessed 12 October 2015).
- Assistance Publique – Hôpitaux de Paris . Assistance Publique – Hôpitaux De Paris Stops the ‘Respistimals’ Clinical Trial (‘Early Stage Amyotrophic Lateral Sclerosis Phrenic Stimulation’, NCT01583088) 2015. www.aphp.fr/contenu/sclerose-laterale-amyotrophique-sla-maladie-de-charcot-lassistance-publique-hopitaux-de (accessed 25 October 2015).
Appendix 1 Changes to protocol
| Changes to protocol | Progress report | Date | Approved by |
|---|---|---|---|
| Version 1 | |||
| Version 1 (original submitted to REC) was amended to version 2, on 3 August 2011, following comments from the REC to incorporate the following:Each site has experience in conducting research in patients with ALS and their carers. These healthcare professionals are trained in counselling patients and carers at various stages of the disease. Carers who experience any distress at any time will be dealt with effectively | 1 (dated 23 January 2012) | 17 August 2011 | NRES Committee East of England – Cambridge Central |
| Version 2 | |||
| Version 2 was amended to version 3, on 24 August 2011, as part of a substantial amendment (approved by REC on 20 September 2011), which required some modification to the eligibility criteria. There was also clarity required to ensure that the Ionising Radiation section of the Integrated Research Application System form was completed, as participants require an ultrasound at screening and a radiography postoperatively | 1 (dated 23 January 2012) | 20 September 2011 | NRES Committee East of England – Cambridge Central |
| Version 3 | |||
| Version 3 was amended to version 4, on 10 January 2012, as part of a minor amendment (approved by the sponsors on 12 January 2012). This was an update to clarify that NIV and DP patients both receive their diaries on NIV initiation. As this was just a minor change to the order of giving the diary out, it was reviewed and approved by the sponsor | 2 (dated 13 July 2012) | 20 January 2012 | NRES Committee East of England – Cambridge Central |
| Version 4 | |||
Version 4 was amended to version 5, on 30 October 2012, with the following key changes:
|
3 (dated 14 January 2013) | 4 December 2012 | NRES Committee East of England – Cambridge Central |
| Version 5 | |||
Version 5 was amended to version 6, on 21 May 2013, with the following changes:
|
5 (dated 31 January 2014) | 8 July 2013 | NRES Committee East of England – Cambridge Central |
| Version 6 | |||
| Version 6 was amended to version 7 on 7 October 2013. The protocol was amended to allow respiratory tests up to 2 weeks pre consent, as was standard practice | 5 (dated 31 January 2014) | 29 October 2013 | NRES Committee East of England – Cambridge Central |
| Version 7 | |||
| Version 7 was amended to version 7.1 on 5 November 2013. The data collection table was amended to include data that would be collected from NIV machines and diaries | 5 (dated 31 January 2014) | 15 November 2013 | NRES Committee East of England – Cambridge Central |
| Version 8 | |||
Version 7.1 was amended to version 8, on 16 May 2014, with the following changes:
|
6 (dated 21 July 2014) | 25 June 2014 | NRES Committee East of England – Cambridge Central |
Appendix 2 Trial summary
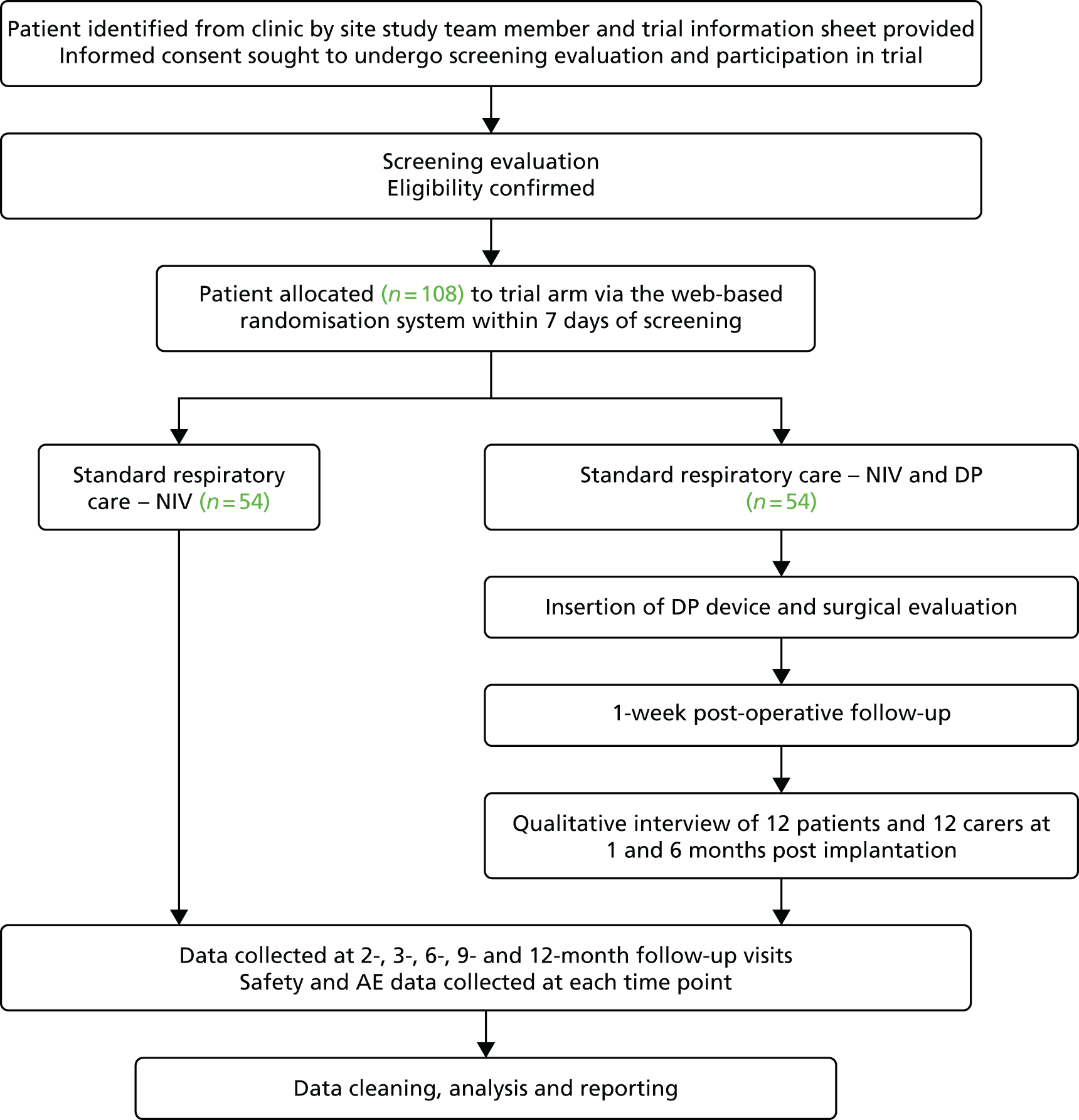
Appendix 3 Participant flow in each trial arm
The carer will be asked to complete the EQ-5D-3L, the CBI and the qualitative interviews. Note that NIV initiation can occur at any point in the screening phase after consent has been obtained.

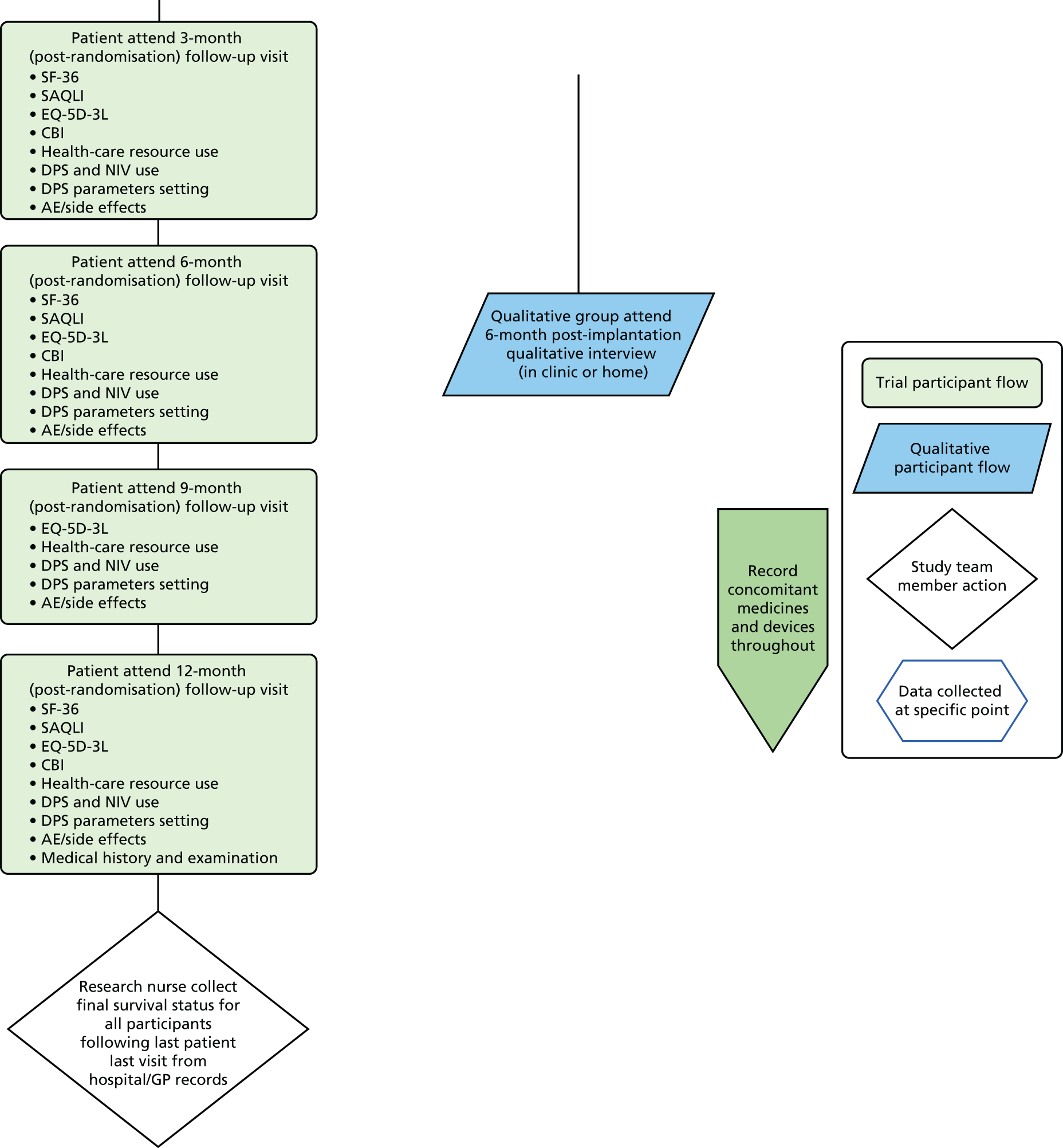
Appendix 4 Screening and randomisation
The carer will be asked to complete the EQ-5D-3L, the CBI and the qualitative interviews. Note that NIV initiation can occur at any point in the screening phase after consent has been obtained. APTT, activated partial thromboplastin time; Ca, calcium; CK, creatine kinase; ECG, electrocardiogram; FBC, full blood count; LFT, liver function test; PT, prothrombin time; U and E, urea and electrolytes; VC, vital capacity.
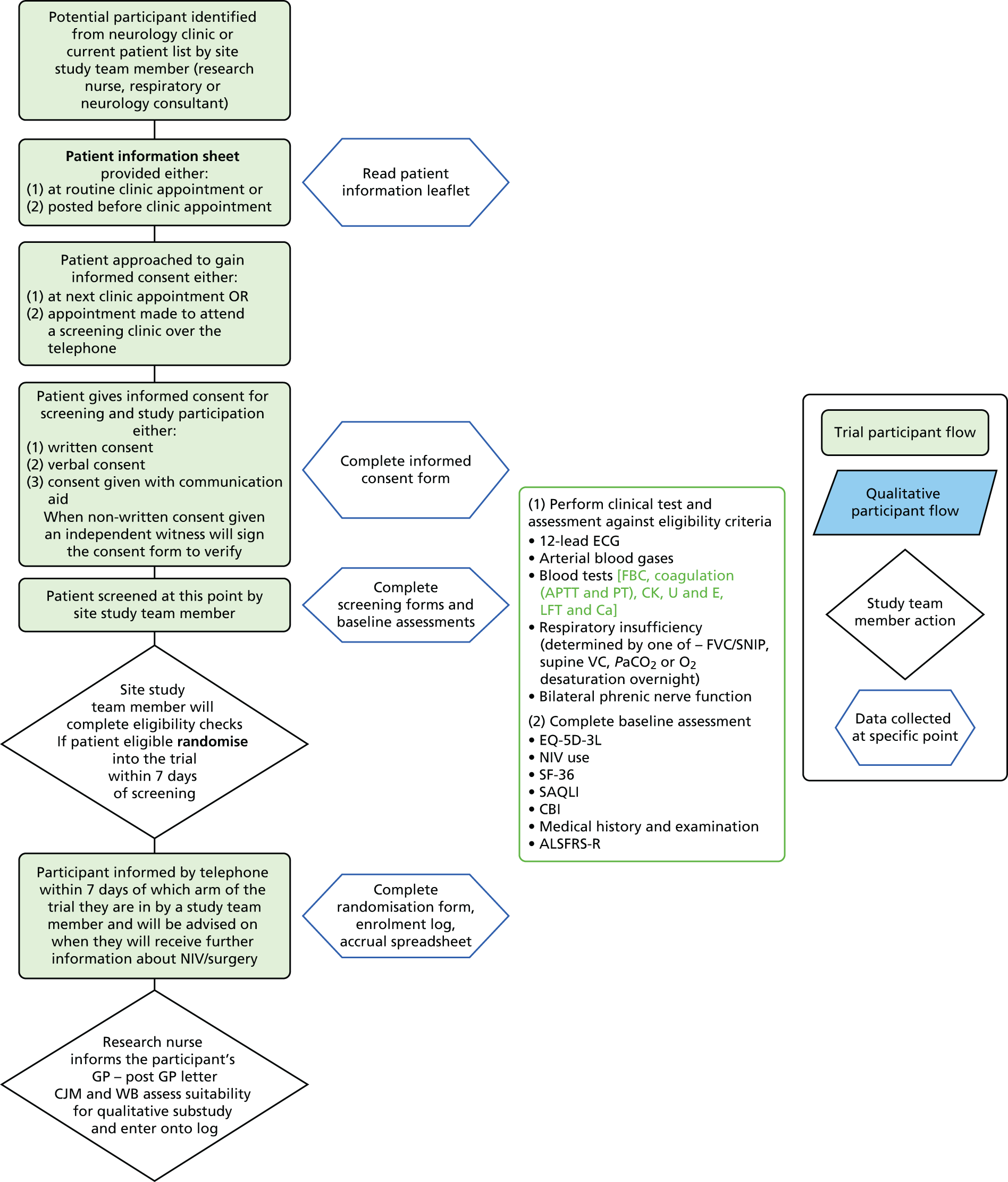
Appendix 5 Patient information sheet: patient
Appendix 6 Patient information sheet: carer
Appendix 7 Patient information sheet: patient qualitative
Appendix 8 Patient information sheet: carer qualitative
Appendix 9 Consent form: patient
Appendix 10 Consent form: carer
Appendix 11 Consent form: patient qualitative
Appendix 12 Consent form: carer qualitative
Appendix 13 Statistical analysis plan
Appendix 14 Stop pacing patient letter (diaphragm pacing arm)
Appendix 15 Stop pacing patient letter (non-invasive ventilation arm)
Appendix 16 Stop pacing general practitioner cover letter (diaphragm pacing arm)
Appendix 17 Pacing discontinuation standard operating procedure
Appendix 18 Qualitative interview topic guide
List of abbreviations
- AE
- adverse event
- ALS
- amyotrophic lateral sclerosis
- ALSFRS-R
- Amyotrophic Lateral Sclerosis Functional Rating Scale – Revised
- CBI
- Caregiver Burden Inventory
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CTRU
- Clinical Trials Research Unit
- DiPALS
- Diaphragm Pacing in patients with Amyotrophic Lateral Sclerosis trial
- DMEC
- Data Monitoring and Ethics Committee
- DP
- diaphragm pacing
- DPS
- diaphragm pacing system
- EPG
- external pulse generator
- EQ-5D-3L
- European Quality of Life-5 Dimensions, three levels
- FDA
- Food and Drug Administration
- FVC
- forced vital capacity
- GP
- general practitioner
- HDE
- humanitarian device exemption
- HR
- hazard ratio
- HUD
- humanitarian-use device
- IQR
- interquartile range
- NIV
- non-invasive ventilation
- PaCO2
- partial pressure of carbon dioxide
- PI
- principal investigator
- PPI
- patient and public involvement
- QoL
- quality of life
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SAQLI
- Sleep Apnoea Quality of Life Index questionnaire
- SF-36
- Short Form questionnaire-36 items
- SMNDRAG
- Sheffield Motor Neurone Disease Research Advisory Group
- SNIP
- Sniff Nasal Inspiratory Pressure
- SOP
- standard operating procedure
- SSPB
- summary of safety and probable benefit document
- TFS
- tracheostomy-free survival
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee