
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 14/151/06. The contractual start date was in July 2015. The draft report began editorial review in January 2016 and was accepted for publication in June 2016. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Hettle et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction and aims
The term ‘regenerative medicine’ refers to a field of research and clinical applications dealing with the process of replacing or regenerating human cells, tissues or organs to restore or establish normal function. 1 Regenerative medicine is not a new field of medicine, as it encompasses bone marrow or organ transplants. However, the development of newer types of regenerative medicine such as cell-based therapies (often using stem cells or progenitor cells to produce tissues), gene therapy and tissue engineering has raised the possibility that diseases that are currently deemed chronic or fatal may be curable. Most regenerative medicines will be classed by the European Medicines Agency (EMA) as ‘advanced-therapy medicinal products’ (ATMPs), which are essentially treatments based on engineered cells or tissues. Although regenerative medicines may offer great potential, the route to this new era of medicine might not be straightforward. Product development and production to commercially viable levels may involve many challenges. Furthermore, efficacy and safety evaluations of regenerative medicines may be difficult compared with efficacy and safety evaluations of conventional pharmaceutical treatments. For example, although when pharmaceuticals are discontinued their adverse effects are likely to improve, some regenerative medicines may cause prolonged toxicities, especially when cells persist long term; such adverse effects might also not become evident for years.
An inquiry by the House of Lords Science and Technology Committee into regenerative medicine was set up to pinpoint the UK’s strengths in this area, to identify barriers to translation (applying findings from basic research to a clinical setting) and commercialisation (primarily delivering treatments in the health-care market) and to recommend solutions. The report – published in July 20132 – concluded that, although the UK has a great potential resource in the NHS, which could make it an attractive place for investment, it is currently underprepared to realise the full potential of regenerative medicine. One of the report’s recommendations was that the Department of Health should establish a regenerative medicine expert working group to develop a NHS regenerative medicine delivery readiness strategy and action plan, which was to report back to the Secretary of State for Health by December 2014. 2 In response to this, the Regenerative Medicine Expert Group (RMEG) was convened and was given the remit to monitor progress on the government’s response to the House of Lords inquiry and to develop, in partnership with other stakeholders, a strategy for regenerative medicine in the NHS and provide an action plan. One of the major discussion areas for the RMEG was that, even when therapies have real potential, this may not be known with a high level of certainty at the time that an ATMP first comes to market, as the available evidence base is often limited. In its report (p. 6)3 the RMEG stated that:
In order for NHS patients to benefit from regenerative medicines, robust and effective product evaluation has to be made to inform commissioning decisions. National Institute for Health and Care Excellence (NICE) guidance is essential in speeding up the adoption and spread of high value regenerative medicines in healthcare. However, applying the Institute’s appraisal methodology, based on cost utility analysis, to products whose true value may not be known for many years can be challenging, due to the inherent uncertainty of estimating long-term benefit from evidence derived from short-term studies.
Contains public sector information licensed under the Open Government Licence v3.0. © Crown Copyright 2015
The assessment of the cost-effectiveness of regenerative medicines may raise particular challenges compared with the assessment of the cost-effectiveness of other types of technologies. Important challenges may include the potential curative nature and claims of long-term/lifetime benefits; the potentially rapid changes that may arise in product characteristics over time; potential longer-term patient safety issues because of persistence; organisational and scaling issues; and the potentially significant upfront costs that may arise. Whether the conceptual differences between regenerative medicines and other types of technologies (e.g. pharmaceuticals and medical devices) mean that a different approach to the assessment of cost-effectiveness is required needs to be investigated.
The RMEG Evaluation and Commissioning Subgroup proposed that the National Institute for Health and Care Excellence (NICE) commission a ‘mock technology appraisal’ (TA) on an exemplar regenerative medicine product and develop an outline plan for such a study. This proposal was reflected in the final report and recommendations of the RMEG (p. 6),3 which stated further that:
We encourage the Institute to consider the findings from these studies with a view to assessing whether changes to its methods and processes are needed. Evaluation and commissioning, as with all steps of the product development pathway, need to be supported by clear, up-to-date and accessible advice and guidance.
Contains public sector information licensed under the Open Government Licence v3.0. © Crown Copyright 2015
Through RMEG Evaluation and Commissioning Subgroup discussions and further input from the Cell Therapy Catapult, it was concluded that undertaking a study involving a real commercial product was not feasible for a number of reasons: there would be significant commercial sensitivities; products undergoing regulatory review would be candidates for a real appraisal; and using a product at an earlier stage in clinical development is not helpful as the evidence base would be even less mature and, therefore, it would not have the attributes of an ‘exemplar’ product. It was therefore proposed to undertake the evaluation of a hypothetical product: chimeric antigen receptor (CAR) T-cell therapies (see Chapter 3, Clinical efficacy and safety issues arising from European Medicines Agency, National Institute for Health and Care Excellence and Food and Drug Administration assessments of licensed regenerative medicines). This decision was made on the basis that CAR T-cell therapies are quite a new product class – none is currently licensed – for which there is emerging evidence of clinical benefit. An evaluation of these therapies might also appropriately exemplify some of the main challenges faced by new regenerative medicines. The Cell Therapy Catapult has knowledge of and experience with gene-modified T-cells and therefore worked with others on the advisory group to develop the basis of the target product profile (TPP).
The objectives of this study were to:
-
test the application of NICE appraisal methodology to regenerative medicines, identifying challenges and any areas where methods research and/or adaptation of methodology would be appropriate
-
identify specific issues related to the appraisal of regenerative medicines using the current NICE appraisal process and decision framework
-
develop a framework for those developing regenerative medicines to facilitate understanding of how NICE evaluates clinical effectiveness and cost-effectiveness and to identify the most important evidence areas to develop before cost-effectiveness can be reasonably estimated.
Chapter 2 Background
Issues identified by the European Medicines Agency as being specific to advanced therapy medicinal products
Most of the new, innovative regenerative medicines that are evaluated by the EMA are likely to be categorised as ATMPs. The Committee for Advanced Therapies (CAT) is the EMA committee responsible for assessing the quality, safety and efficacy of ATMPs (and for following scientific developments in the field). The EMA and CAT have issued a range of documents providing guidance regarding the development of ATMPs; one of these [issued in 2008, based on the requirements of a European Union (EU) regulation] is a guideline for post-authorisation follow-up entitled Guideline on Safety and Efficacy Follow-up – Risk Management of Advanced Therapy Medicinal Products. 4 Such rules were needed because of the ‘novelty, complexity, and technical specificity’ of ATMPs (p. 7). 4
In this guideline the concerns about risks relate to:
-
living donors (where applicable)
-
quality characteristics (e.g. origin and characteristics of cells and vectors; quality assurance issues)
-
storage and distribution of products (e.g. stability, preservation, thawing)
-
administration and readministration procedures (e.g. immune reactions)
-
interaction of the product and the patient (e.g. immunogenicity, malignancy)
-
scaffolds, matrices and biomaterials (e.g. biodegradation)
-
product persistence (e.g. availability of rescue procedures or antidotes)
-
health-care professionals, caregivers or other close contacts with the product.
Concerns about the efficacy of ATMPs relate mainly to the uncertainty about how effective they may be in ‘real-life’ settings in the long term and include the following:
-
Possible temporal changes in the characteristics of the living material in ATMPs may affect efficacy.
-
The time required for new tissue to be fully functional may be several years (use of surrogate end points needed for marketing authorisation, but confirmation with clinical end points needed in post-authorisation phase).
-
Some ATMPs may be a once in a lifetime treatment and long-term follow-up is needed to demonstrate the sustainability of efficacy.
-
Efficacy may be highly dependent on the quality of the administration procedure (e.g. patient conditioning, surgery). This may differ between clinical trial and normal health-care settings.
-
Cell therapy products with a limited lifetime may require an efficacy follow-up system that monitors the dynamics of efficacy (this will help to determine need and timing of reapplication).
The issues highlighted in the guideline for the design of the studies needed to monitor long-term safety and efficacy include careful consideration of:
-
sample size (high potential for dropouts over many years of follow-up)
-
the dynamics of the disease and the effects of the product (different approaches needed for detecting early vs. late complications)
-
the use of usual clinical practice for follow-up whenever possible to limit additional procedures and interventions
-
the appropriate duration of follow-up of living donors (when applicable)
-
the feasibility of follow-up of close contacts and offspring (when applicable).
Both the safety and efficacy follow-up systems are defined as any systematic collection and collation of data that is designed in a way that enables learning about safety and/or efficacy of an ATMP. This may include passive or active surveillance, observational studies or clinical trials. The guideline stresses that both the efficacy and the safety follow-up systems are not a substitute for the need for adequate data to be available at the time of authorisation to enable proper benefit–risk evaluation. 4
Overview of wider regulatory evidence requirement issues and the evolving pathways for approval
The ethics, feasibility and reliability of small randomised controlled trials
Although the randomised controlled trial (RCT) is the expected level of evidence needed for regulatory assessments, it is recognised that for some indications such expectations are unrealistic. Conducting RCTs in populations with severe or advanced disease may be problematic for a variety of reasons. Such populations may be very small and, consequently, recruitment into an adequately sized trial would require a large number of centres and would be very expensive and take a very long time. In addition, when no alternative treatments exist, patients with life-threatening diseases or severe morbidity typically need, and desire, accelerated access to innovative new therapies. Patients with more severe or advanced disease may be more willing to accept the risks of an experimental therapy. In such situations randomisation to a control treatment may be ethically problematic (because of an absence of clinical equipoise).
A health technology assessment (HTA) review of ethics issues in the design and conduct of RCTs described numerous situations in which alternative non-randomised designs are morally or practicably preferable. These included when large differences between treatments are expected; when a disease, if left untreated, is lethal and for which there is no known effective treatment (i.e. unmet need); and when a disease is rare and recruitment is slow. 5 Additionally, when trial populations are small, it may be difficult to differentiate a true treatment effect from a chance effect. Important chance imbalances in relevant prognostic factors between groups at baseline are more likely in small trials. The HTA review highlighted the problem of underpowered RCTs, which were described as ‘necessarily unethical’ as they were unlikely to produce clear-cut answers. 5 This argument was supported by 15 articles that stipulated the statistical necessity for random errors in measured effects of treatments to be small in comparison with the size of the therapeutic effect sought. Other articles in the review discussed the ethics of stopping RCTs early when there is some evidence of efficacy and the subsequent problems that this may cause, with reduced statistical precision, clinicians not being persuaded by results and secondary trial aims being compromised being some of the key problems.
A related HTA review discussed further the ethics issues that may arise when early-phase (e.g. single-arm) trials produce very encouraging results: it may be unethical to conduct a further trial if the intervention is apparently effective in a small number of patients. 6 In such a situation, the argument for a trial rests on demonstrating a grey area between a reasonable hope that the intervention is effective in a few patients and a rational and justified belief that it is effective for the studied patient population more generally (i.e. the evidence to date has sufficient external validity).
Possible alternatives to the randomised controlled trial
More recently, a framework for using unfamiliar trial designs when rare diseases are studied has outlined several possible alternative approaches. 7 The framework aims to facilitate research when populations are small. Two of the ‘adaptive’ designs outlined may be particularly relevant for regenerative medicines, for which treatment intentions may be curative. The first is responsive–adaptive randomisation, which maximises allocation to the most effective treatment and minimises the required sample size. Outcomes for previous participants affect the subsequent treatment allocation probabilities. This ‘play the winner’ rule has the potential to reduce the number of patients who are allocated to less effective treatments and can therefore reduce the ethical concerns associated with randomisation. However, this design is limited to studies that assess rapidly available outcomes (as results from previous patients are needed to influence future allocations). Modified designs have also been outlined to counter the criticism that comparisons may be obtained in which only one patient has received conventional treatment. The second adaptive design that may also be useful for studying regenerative medicines is the internal pilot design. This design eliminates the loss of scarce eligible participants because of participation in a prior pilot study. Once the pilot phase is finished, a sample size is recalculated with the study continuing until this number is recruited; patients from the pilot phase are included in the final analysis.
A EMA reflection paper on methodological issues associated with adaptive designs suggested that such designs ‘would be best utilised as a tool for planning clinical trials in areas where it is necessary to cope with difficult experimental situations’ (p. 3). 8 Cited examples of such situations included ‘small populations or orphan diseases with constraints to the maximum amount of evidence that can be provided’ and when there are ‘ethical constraints to experimentation’ (p. 10). However, the US Food and Drug Administration (FDA)9 raised two principal issues with regard to adaptive design methods more broadly:
-
whether the adaptation process has led to design, analysis or conduct flaws that have introduced bias which increases the chance of a false conclusion that the treatment is effective (a type I error)
-
whether the adaptation process has led to positive study results that are difficult to interpret, irrespective of having control of type I error.
This draft FDA guidance document also noted that, for some of the more recently developed adaptive methods (including adaptive randomisation methods), the magnitude of the risk of bias and the size of the potential bias, and how to eliminate these effects, are not yet well understood.
Although adaptive designs may be useful in some situations, it is still likely that single-arm trials will form the basis of many submissions for the regulatory approval of regenerative medicines (because of the nature of the target populations). Nevertheless, a study that reviewed 31 oncology drugs or biologics approved by the FDA (between 1973 and 2006) without a randomised trial that incorporated a comparator treatment, supportive care or placebo arm concluded that such drugs have a reassuring record of long-term safety and efficacy despite the fact that nearly all of the evidence studies were single-arm Phase II trials. 10 The median number of patients studied per approval was 79 (range 40–413); response rate was the primary end point for most drugs, and the median objective response rate was 33%. At the time of publication (2009) all but one of the drugs were still approved; marketing authorisation for gefitinib (Iressa®; AstraZeneca, Cambridge, UK) was rescinded after a RCT showed no survival improvement. Nineteen drugs have additional uses, with formal FDA approvals obtained for 11.
Evolving regulatory pathways
Since the late 1980s and early 1990s, regulators and HTA bodies/payers around the world have produced new approaches to provide patients with timely access to new medicines. 11 These new regulatory pathways can also improve competitiveness; shortened product development times prior to licensing can be very beneficial and more appealing to emerging small and medium-sized enterprises.
An overview of the relevant EMA regulatory accelerated access pathways is presented in Table 1. The main mechanism for accelerated access of these pathways is the reduced development time.
| Designation (year of introduction) | Use | Notes |
|---|---|---|
| Approval under exceptional circumstances (1993) | Medicines with urgent public health need for which comprehensive data cannot be provided | Justifications for not being able to provide comprehensive data include rarity of the condition, lack of scientific knowledge (e.g. diagnostic tools) and contrary to medical ethics. Post-authorisation data collection required, which usually includes an identified programme of studies, the results of which form the basis of an annual reassessment of the benefit–risk profile |
| Accelerated assessment (2005) | Medicines of major interest to public health, particularly those representing a therapeutic innovation | Review time shortened to 150 days compared with the standard of 210 days. This pathway has very rarely been used |
| Conditional marketing authorisation (2005) | Seriously debilitating and life-threatening conditions, medicinal product for emergency use or orphan medicinal products; must address unmet medical need | Authorised for 1 year with the option to renew as long as benefit–risk profile remains positive. The condition is that the manufacturer will initiate or, preferably, continue studies to reduce uncertainty about benefits and risks to enable conversion to full authorisation. A periodic safety update report is required at 6-month intervals |
| Parallel scientific advice between EMA and FDA (2009) | Important medicinal oncology, vaccine, orphan, paediatric, nanotechnology, advanced therapy, pharmacogenomics or blood products. Products usually have fast-track designation in the USA | Expected advantages are increased dialogue between the two agencies and sponsors from the beginning of the life cycle of a new product, a deeper understanding of the bases of regulatory decisions and the opportunity to optimise product development and avoid unnecessary testing replication or unnecessary diverse testing methodologies. Scheduling of parallel scientific advice can be challenging |
| Adaptive licensing (2014 pilot) | Medicines to treat an unmet medical need for a serious condition, especially when no alternative therapies exist | Open to interventions in the early stages of development (during or prior to phase II). Multi-stakeholder participation desirable. Enhanced monitoring of drug safety and drug utilisation controls required after initial authorisation |
The EMA’s most recent development in this area is the adaptive licensing pilot programme, which was launched in 2014. The programme utilises the regulatory processes within the existing EU legal framework and is defined as being a prospectively planned adaptive approach to bringing drugs to market. It is more of a staggered iterative system than previous approval pathways. Such a ‘life-cycle approach’ to acquiring and (re)assessing evidence will consider the basis of decision-making in the following stages of a product’s life cycle: development, licensing, reimbursement, monitoring/post-licence evidence and drug utilisation. 11 Importantly, the approach encompasses both the authorised indication and the potential further therapeutic uses of the medicine. The EMA changed the name of the pilot project from ‘adaptive licensing’ to ‘adaptive pathways’ to better reflect the idea of a lifespan approach.
The pilot project aims to examine whether or not this kind of approach to medicine development and authorisation will offer advantages in terms of achieving the best balance between the need for timely patient access and the importance of providing adequate, evolving information on benefits and risks. In so doing it is expected to develop thinking in the following areas:12
-
encourage developers of medicines to consider all regulatory tools and flexibilities within the existing EU legal framework when planning the life cycle of medicine development
-
explore the extent to which regulatory demands for the generation of evidence around efficacy and safety are compatible with demands around evidence generation from other stakeholders (e.g. HTA bodies, payers, patient organisations)
-
investigate in a timely manner the hurdles that exist in realising the most efficient medicine development pathways, including the role and limitations of real-world data.
Ideas for refining and improving this lifespan approach are developing at pace. For example, with MAPPs (Medicines Adaptive Pathways to Patients) the development plan across target populations and indications will be agreed upfront with the EMA, which distinguishes the MAPPs process from the conventional indication expansion approach. The MAPPs plan may include a range of studies, such as RCTs, single-arm studies, pragmatic trials and other forms of real-world study. 13 A newly formed public–private project called ADAPT SMART (Accelerated Development of Appropriate Patient Therapies: a Sustainable, Multi-stakeholder Approach from Research to Treatment-outcomes), which is funded by the EU Innovative Medicines Initiative, aims to facilitate and accelerate the availability of MAPPs. 14 NICE is one of the 32 international partners that together represent regulators, patients, academia and industry. The challenge for ADAPT SMART is to develop a MAPPs model that aligns the needs of all stakeholders, including patients, member state payers, regulators, medical practitioners and industry. A major task will be the identification of opportunities and obstacles, and providing a framework for MAPPs that will overcome the latter and seize the former. ADAPT SMART will address the challenges to the broad implementation of MAPPs by exploring new concepts to align the various stakeholders and create a consensus on what evidence will be required, how multiple sources of available data can be best used to facilitate MAPPs and which scientific challenges related to MAPPs need to be addressed. 14
In the UK there is another initiative that may facilitate the pathway to market: the Medicines and Healthcare products Regulatory Agency (MHRA) operates an early access to medicines scheme (EAMS), which was launched in April 2014. This voluntary scheme (which does not replace the normal licensing procedures) is aimed at unlicensed or off-label treatments deemed by the MHRA to be ‘promising innovative medicines’ (PIMs) for treating life-threatening or seriously debilitating conditions for which there is unmet need. Once a PIM designation is obtained (stage 1 of the process), the MHRA can then provide benefit and risk information (stage 2 – scientific opinion) to doctors who may wish to prescribe the unlicensed medicine under their own responsibility. However, it appears somewhat unclear how the EAMS assessment output may impact on ongoing or forthcoming EMA assessments of the same therapy (e.g. in terms of speeding up processes or reducing repetition of information). Further uncertainty around EAMS exists regarding how therapies with this regulatory status can be funded. As EAMS is not accompanied by any funding arrangements, meeting the costs of the therapy is currently the responsibility of the manufacturer; this can act as a barrier to adoption, especially for high-cost therapies produced by small enterprises.
Regenerative medicines in the new regulatory environment
The experience gleaned from the EMA adaptive licensing pilot so far appears to be quite limited with respect to regenerative medicines: as of May 2015 one of only three candidate ATMPs had been selected for a ‘stage II’ proposal. 15 By far the most accommodating regulatory environment for developing regenerative medicines is currently Japan, where, under the new 2014 legislation, regenerative medicines can receive accelerated conditional approval after a single clinical study, provided the trial has demonstrated the therapy to be safe, with evidence of a probable therapeutic benefit. This approach aims to dramatically accelerate patient access and meaningfully shorten clinical development times, thus promoting investment (as faster, cheaper development, coupled with accelerated commercialisation, would shift the risk–reward ratio favourably from an investment perspective). 16 However, there is concern that this approach may leave Japan with regenerative medicines that are unrecognised by other countries because of efficacy concerns: the lack of an explicit plan for determining efficacy during the conditional approval period points to a strong underlying assumption that regenerative medicines will ultimately prove efficacious, whereas experience from other areas of clinical research suggests that such optimism may be misplaced. 17 The initial demonstration of safety based on only Phase I trial data is an additional major concern.
The concern raised about the limited evidence that will probably be presented when a product is submitted for regulatory approval is by no means limited to the Japanese regenerative medicine experience. As many regenerative medicines will be developed with the initial aim of treating small patient populations in which there is unmet need, it is likely that they will be evaluated via a regulatory pathway that offers patients accelerated access to the new treatment. A consequence of this is that many of the studies submitted will be early-phase, small single-arm trials. Nevertheless, the Japanese regulations excepted, the newer regulatory pathways being developed across the world do not focus specifically on facilitating the licensing of regenerative medicines. The newer pathways are primarily aimed at addressing unmet need in serious conditions for which no alternatives exist, regardless of the type of technology. However, much of the focus and expectation for success in this area seems to have been directed at regenerative medicines, possibly because they may evolve over time and may therefore, ultimately, not be restricted and limited by having single modes of action. The submission of evidence that is based on single-arm studies appears to be less to do with regenerative medicines being a ‘special case’ category of interventions, but rather a consequence of the seriously ill, very small populations of patients with unmet medical needs who are often the initial target of new regenerative medicines.
Chapter 3 Technology appraisal methodology issues that may be particularly relevant to regenerative medicines
Clinical efficacy and safety issues arising from European Medicines Agency, National Institute for Health and Care Excellence and Food and Drug Administration assessments of licensed regenerative medicines
Methods
From the regenerative medicine literature and experts in the field we sought to identify regenerative medicines that have been granted marketing authorisation in the EU. In addition to EMA assessment documents, we also sought any NICE or FDA documents. We extracted key details from these reports, with a primary focus on identifying issues that might be unique, or particular, to regenerative medicines.
Results
We identified six regenerative medicines that are (or have been) licensed in the EU: ChondroCelect® (TiGenix, Leuven, Belgium), matrix-applied characterised autologous cultured chondrocyte implant (MACI) (Sanofi, Gentilly, France), Glybera® (alipogene tiparvovec; uniQure, Amsterdam, the Netherlands), Holoclar® (Holostem Advanced Therapies, Modena, Italy), PROVENGE® (sipuleucel-T; Dendreon Corporation, Seattle, WA, USA) and ReCell® (Avita Medical, London, UK). No allogeneic therapies were identified – all were autologous. Summary details are presented in Table 2; more comprehensive details can be found in Appendix 1 (see Table 42).
| Summary | Glybera18 | MACI19–21 | ChondroCelect20,22 | Holoclar23 | PROVENGE24–26 | ReCell27 |
|---|---|---|---|---|---|---|
| Year of EMA MA | 2012 | 2013a | 2009 | 2014 | 2013b | 2005c |
| Type of RM | Gene therapy | Autologous cells seeded on porcine collagen membrane | Suspension of autologous cells | Autologous tissue-engineered product (includes stem cells) | Autologous active cellular immunotherapy | Stand-alone autologous cell-harvesting device (for immediate delivery to wound surface) |
| Indication | Adults with familial lipoprotein lipase deficiency (LPLD; confirmed by genetic testing), detectable levels of LPL protein and suffering from at least one pancreatitis episode despite dietary fat restriction | Skeletally mature patients for the repair of symptomatic cartilage defects of the knee | Repair of single symptomatic cartilaginous defects of the femoral condyle of the knee in adults | Corneal lesions with associated (limbal) stem cell deficiency because of ocular burns | Asymptomatic or minimally symptomatic metastatic (non-visceral) hormone-relapsed prostate cancer in men for whom chemotherapy is not yet clinically indicated | Adults or children with (1) partial-thickness burns including scalds caused by hot water where mesh grafting is not required, (2) large-area burns; full-thickness or deep partial-thickness burns including where mesh grafting is required |
| Orphan status? | Yes | No | No | Yes | No | No |
| Claiming to meet unmet medical need? | Yes | No | No | Yes | No | No |
| Trial design | Three single-arm studies | One RCT (multicentre) | One RCT (multicentre) | Three retrospective case series (multicentre) | Three RCTs (multicentre) | Three RCTs (single centre) and eight observational studies |
| Trial size | Combined total n = 27 | n = 144 | n = 118 | Combined total n = 148 | Main RCT n = 512 | Main RCT n = 82 |
| Length of follow-up | 12–18 weeks | 2 years | 5 years | 1 year | 3 years | 6 months |
| Comparator | Two observational studies (combined n = 40) of patients receiving only diet reduction and no active treatment | RCT had a control arm of patients receiving microfracture | RCT had a control arm of patients receiving microfracture | Patients acted as their own controls – outcomes were compared with baseline data | Placebo group of RCT: one-third of the patient’s cells were reinfused but were not activated with the fusion protein | RCT had a control arm of patients receiving split-thickness skin grafting |
| Adverse events | No obvious serious adverse events seemingly related to Glybera | Most were surgery related rather than product related | Most were surgery related rather than product related | Out of a total of 11 serious adverse events, three were judged to be related to Holoclar | Main risks were infusion reactions and (catheter-related infections) | None reported |
| Surrogate outcome? | Yes – levels of fasting triglycerides | Yes – magnetic resonance imaging or histology scoring of structural and functional repair | Yes – structural repair (histology) | Yes – corneal epithelial integrity and absence of significant neovascularisation | Yes – time to progression, antigen response | No |
| Real clinical outcome? | Yes – pancreatitis events | Yes – Knee Injury and Osteoarthritis Outcome Score | Yes – Knee Injury and Osteoarthritis Outcome score | Yes – visual acuity | Yes – overall survival | Yes – several wound-healing outcomes |
| Estimate of HRQoL | SF-36 for earlier time points | Absence of reliable quality-of-life data20 | ‘Lack of good quality of life data’20 | Not assessed | Not assessed | Not reported |
ChondroCelect and MACI are both therapies for treating knee cartilage defects. ChondroCelect was the first ATMP to receive marketing authorisation, in 2009. The marketing authorisation for MACI was suspended in September 2014 as an authorised manufacturing site no longer existed. Holoclar is a therapy used for treating corneal lesions resulting from burns to the eye. In 2014 it became the first stem cell-based ATMP to gain regulatory approval (conditional marketing authorisation was granted). PROVENGE is an active cellular immunotherapy for asymptomatic or minimally symptomatic metastatic hormone-relapsed prostate cancer when chemotherapy is not yet indicated; this therapy purportedly helps the immune system to selectively attack cancer cells (rather than directly attacking tumour cells, as happens with CAR T-cell therapies). EMA marketing authorisation was granted in June 2013 but withdrawn in May 2015 at the request of the manufacturer, for commercial reasons. Glybera is used to treat familial lipoprotein lipase deficiency (a rare genetic disorder) with associated pancreatitis. Its mechanism of action is viral vector delivery of a therapeutic gene to muscle cells. In 2012 it became the first gene therapy to be approved in Europe or the USA. The ReCell spray-on skin system is a regenerative medicine device. It harvests a small amount of a patient’s skin cells, which are then processed to produce a mixed cell population for immediate delivery onto burn wound surfaces. ReCell can be given rapidly as there is no need for proliferation of the harvested skin cells. A Conformité Européere (CE) mark was granted in 2005 (under Medical Devices Directive 93/42/EEC28).
Study designs
Randomised trial evidence formed the basis of the regulatory submissions for four of these six regenerative medicines. This would be expected as, for the four therapies in question (ChondroCelect, MACI, ReCell and PROVENGE), the disorders being treated were not rare and alternative therapies existed. However, for PROVENGE, both the European Public Assessment Report (EPAR)24 and NICE Evidence Review Group (ERG) report26 commented on the lack of blinding and the use of crossover, which allowed placebo patients to receive active treatment following disease progression, making interpretation of the post-progression overall survival (OS) results difficult. Nevertheless, there were no design issues for the other three therapies (ChondroCelect, MACI and ReCell), demonstrating that ATMP/regenerative medicine status in itself may not necessarily be a barrier to submitting randomised trial evidence (as discussed at the end of Chapter 2).
Holoclar and Glybera were not studied in RCTs. Both had orphan designations and indications for when there is unmet medical need; randomised trials were therefore not viable. A single-group study design was therefore deemed acceptable in both EMA assessments. 18,23 However, whereas for Holoclar the CAT accepted that the condition (eye burns) would not improve spontaneously (making it more plausible that observed benefits resulted from treatment), for Glybera there were concerns that the reduction of pancreatitis events may possibly be due to temporal rarity and inherent variability of events over time (i.e. the resulting apparent benefit may have resulted from chance). Perhaps it is for this reason that these two therapies took very different routes to approval. Whereas conditional marketing authorisation was achieved for Holoclar without any prior negative Committee for Medicinal Products for Human Use (CHMP) decisions, Glybera had a much more difficult route to acquiring marketing authorisation. Negative CAT and CHMP opinions on Glybera were issued in June 2011. Following a request for re-examination, the CAT recommended the granting of marketing authorisation under exceptional circumstances in October 2011, but the CHMP did not recommend approval. Glybera was finally granted approval in July 2012 with a more restricted licence (the approval being for patients with lipoprotein lipase deficiency and severe or multiple pancreatitis attacks). It appears that EMA concerns about the efficacy of Glybera remain, prompting Germany’s G-BA [Gemeinsamer Bundesausschuss (Federal Joint Committee for healthcare regulation)] (which makes reimbursement decisions) to suspend its assessment of Glybera. 29
The issue that this comparison of Glybera with Holoclar raises (the likelihood of cure or improvement without experimental treatment) could be an important consideration for both the design and the interpretation of future regenerative medicine trials. It is for conditions for which spontaneous cure or improvement is unlikely that so much is expected of regenerative medicines; the extent of the problems perceived to result from single-arm trial evidence may well depend on the ‘game-changing’ possibilities of the therapy being assessed.
Persistence and adverse events
The requirement for, and implications of, long-term persistence of the six licensed therapies in treated patients varied. For ChondroCelect, MACI, Holoclar and ReCell the aim is for therapeutic cells to become integrated in recipients for as long as possible and to ultimately produce new cells. Long-term data are needed for evaluations of true therapeutic success in this respect, and adverse effects associated with longer-term persistence seem unlikely. Unknown long-term durability was highlighted in the ChondroCelect22 and MACI19 EPARs. Although the negative persistence effects of Glybera were thought to be minimal [the risk of cancer by integration of viral vector deoxyribonucleic acid (DNA) was thought to be low], the EMA’s conclusions on efficacy noted that the proposed single treatment was insufficient to provide a durable and measurable effect on triglycerides, suggesting that the therapy did not persist in recipients for long enough. 18 Little information could be found on the implications of the long-term persistence of PROVENGE within patients. 24 However, prior to infusion into patients, PROVENGE is associated with a very short shelf life. An overview of the manufacturing and scale-up issues that may be encountered with regenerative medicines can be found in this report’s discussion.
The only other adverse events that were noteworthy in terms of informing evaluations of future regenerative medicine studies were immune reactions. For patients receiving Glybera, the use of immunosuppression did not result in a reduction of unwanted immunogenicity. 18 Acute infusion reactions were identified as a risk in patients who had received PROVENGE and the risk of autoimmune reactions in non-prostatic tissues could not be ruled out. 24
Use of surrogate outcomes
Both surrogate and real clinical outcomes were evaluated for five of the six regenerative medicines; the ReCell studies did not need to use surrogates, with all outcomes having clear clinical importance. 27 The use of surrogate outcomes was most problematic in the assessment of PROVENGE, as the OS results were not supported by the progression-free survival (PFS) or the time-to-progression (TTP) results. 24 Many members of the CHMP felt strongly that, in light of these seemingly contradictory results, the efficacy evidence should be convincing and ideally corroborated by other secondary end points, which was not the case. The NICE ERG report also highlighted the lack of consistency between the surrogate outcomes and OS. Surrogate outcomes are discussed more broadly in Review of the use of surrogate end points as primary outcome measures in definitive effectiveness trials of new therapeutic agents.
Evolving therapies
A key difference between regenerative medicines and conventional medicines is the likelihood that specific treatments may change or evolve over time. The only example of this issue in the reports identified in this section related to the cartilage cell (chondrocytes) treatments for cartilage defects of the knee (MACI and ChondroCelect). 19–22 When both were assessed by NICE they were third-generation products. The ERG report noted the ‘general problem when long-term results are needed but the technology continues to evolve’ (p. 148),20 the implication being that, by the time long-term trials results become available, the therapy may well have been superseded by a (apparently superior) next-generation treatment.
Summary
The key issues arising from the reports of licensed regenerative medicines, that is, the issues that may be beneficial to consider when appraising future regenerative medicines, were:
-
the importance of considering the likelihood of cure or improvement without experimental treatment when evaluating the results of single-arm studies
-
the positive and negative implications of long-term persistence of therapies within patients
-
the use of reliable surrogate outcomes (i.e. the need for validation of the relationship between surrogates and real clinical outcomes)
-
the problems of long-term evaluations when therapies evolve over time
-
none of the six regenerative medicines approved for use in the EU to date was an allogeneic therapy.
Study biases: an overview of their importance and methods to quantify and adjust for their impact
Regenerative medical technologies will often seek (and receive) EMA/FDA approval with limited or no data from randomised experiments. In such cases, estimates of effectiveness will be based on observational data and single-arm experimental studies. Recent examples include Holoclar, which received EMA authorisation based on retrospective case series (combined n = 148),23 and Glybera, which was licensed based on single-arm studies (combined n = 27). 18
The focus of this section is therefore on making comparisons using historical controls and non-randomised evidence more generally, as this is likely to represent the typical way in which single-arm studies will be used in any future regenerative medicine submissions when evidence from randomised trials is unavailable. This section will provide an overview of the reliability of using observational data and data from single-arm trials and current methods used to minimise potential confounding bias. Most manufacturer submissions to NICE are likely to be based on efficacy evidence from randomised trials; this overview is therefore important as it may highlight areas in which NICE might consider that methods development research is needed to enhance the TA programme. Specifically, this section seeks to address the following three questions.
-
To what extent do estimates of effectiveness obtained from non-randomised studies (NRSs) agree with those obtained from randomised trials? (i.e. the quantification of bias)
-
What techniques are available to adjust for confounding bias in NRSs and how reliable are they?
-
What are the specific challenges of using single-arm studies to estimate treatment effectiveness?
Methods
Pragmatic surveys of the literature were carried out to address these research questions. One review addressed the reliability of obtaining treatment effectiveness estimates in comparative NRSs. Two further separate reviews were carried out with respect to the second research question: one focusing on methods to adjust for bias in the evidence synthesis process and a second on methods of analysing individual patient data (IPD) from NRSs. A final review explored the literature relating specifically to single-arm studies. For each review a number of key articles were identified using unstructured searches of MEDLINE and studies known to the team. Based on these key studies snowballing techniques were then applied in which citation searches were carried out and references checked for relevant studies. Citations and references of any additional studies identified were then also checked until no further relevant studies were identified.
Records identified in both the searches of MEDLINE and citation searches were screened by a single reviewer and the full texts of those deemed potentially relevant were obtained and also screened by a single reviewer.
Results
Quantification of bias in observational studies
A total of 14 studies were identified as relevant to the first research question (quantification of bias in observational studies). 30–43 A summary of the methods and findings of each the 14 identified studies is presented in Appendix 2 (see Table 42).
All 14 studies relevant to the quantification of bias in observational studies sought to quantify the extent of bias in NRSs by comparing the results of RCTs with those of NRSs. In six of the studies,33–35,37,38,41 data were sourced from published meta-analyses that included both RCTs and NRSs. Five other studies30,32,39,40,42 took a different approach and searched for NRSs that compared treatment effects and then carried out a further search to locate relevant RCTs. Beynon et al. 31 took a similar sampling approach, randomly selecting RCTs from the Cochrane Central Register of Controlled Trials database and then conducting searches for NRSs that had addressed the same topic.
The method of analysis in the majority of studies involved pooling the evidence from randomised and non-randomised sources separately. The resulting summary effects from the randomised and non-randomised evidence were then compared. Despite these similarities in approach, a considerable range of methods was used to compare summary estimates of effect, with multiple outcome measures often being employed. Common outcomes included:
-
assessment of direction of effect
-
subjective assessment of overlap of confidence intervals (CIs) and proximity of summary estimates
-
tests of statistical difference in summary estimates of effect obtained from randomised and non-randomised evidence
-
the calculation of ratios of odds or risk ratios.
The lack of a common method of comparison is problematic as it presents a significant barrier to making comparisons across studies and indicates a lack of consensus around how to measure the degree of concordance between results obtained from randomised studies and those obtained from NRSs. Furthermore, the employment of multiple methods of comparison in many studies can be considered a potential source of bias, as no attempt was made to adjust comparisons for multiple testing.
Of the 14 included studies, seven30,35,37,38,40–42 concluded that there were no systematic differences in either the size or the direction of effect estimates obtained from NRSs compared with those from RCTs. Five studies31,32,34,39,43 concluded that effect estimates obtained from NRSs were systematically larger than those obtained from RCTs. This included the largest study by Ioannidis et al. ,34 which contained RCTs and NRSs from 45 topic areas. The authors of the other two studies33,36 felt unable to draw any meaningful conclusions about the comparability of estimates obtained from RCTs and NRSs.
Study design and study quality were investigated in a number of the studies and were discussed in nearly all of the studies included in this review. Study design was identified as a likely factor in determining the reliability of estimates of clinical effectiveness obtained from NRSs.
Two studies excluded NRSs that used historical control groups. 37,42 Concato et al. 37 justified this exclusion based on previous evidence presented in Sacks et al. ,39 who reported that 79% of interventions tested were considered effective in trials with historical controls, whereas only 20% were considered effective in RCTs. Further empirical evidence of the potential for bias in studies using historical controls is also presented in Ioannidis et al. ,34 Algra and Rothwell40 and Golder et al. ,41 who all found that there were fewer discrepancies between the results of RCTs and NRSs when studies with historical controls were excluded. Ioannidis et al. 34 also found that results from prospective NRSs contained fewer discrepancies compared with effect estimates from randomised studies than did retrospective studies, either with current or historical controls. Investigations into broader measures of quality have also revealed similar results. MacLehose et al. 35 classified NRSs as being of either high or low quality and observed that comparisons between randomised evidence and high-quality NRSs tended to show much smaller discrepancies than comparisons between randomised studies and low-quality NRSs.
Adjustment for bias in non-randomised studies
A total of 28 studies44–71 were identified as relevant to the second research question on the techniques available to adjust for confounding bias in NRSs (details of these reviews are presented in Appendix 2).
A key factor in the reliability of estimates of effectiveness based on observational data is the statistical analysis used; a large number of studies have sought to develop and evaluate methods for adjusting and eliminating bias resulting from confounding. A summary of the studies that have looked at methods of adjustment for confounding bias in NRSs and how reliable they are62–70 is presented in Appendix 3. Overall, it is unclear which methods are most appropriate in certain circumstances and further research is needed. Furthermore, adjusting for bias when comparing single-arm trials with historical controls requires IPD; this can be difficult to access, although approaches for recreating IPD data have been developed, such as the algorithm by Guyot et al. 72 Consequently, results generated from NRSs will be subject to an unknown degree of uncertainty, even after adjustment for confounding.
Challenges of using single-arm trials to estimate effectiveness
A total of 10 articles were identified as being relevant to the issue of using single-arm trials to estimate effectiveness. 73–82 One of these was a recent review paper73 that discusses both the opportunities and the challenges involved in using studies without a control group. Single-arm designs have the advantage of requiring fewer patients, all of whom receive the experimental treatment, thereby reducing the cost of trials in terms of patients, funding and effort. This section discusses the issues of making comparisons using single-arm studies and how comparable results from single-arm studies and comparative randomised studies are.
Making comparisons using single-arm studies
Without a direct, concurrent comparator in single-group studies, both explicit and implicit comparisons are frequently made. 73 Implicit comparisons are made when the expected outcomes in the absence of the intervention of interest are believed to be well known and the expected effect size from the intervention is large. Explicit comparisons are made when the investigators compare the single group of subjects before and after an intervention or when the investigators choose to incorporate a historical comparator in the analysis (e.g. historical data from the research institution or from an external cohort or existing database). Each of these alternative study designs has particular challenges and advantages. The particular challenges are discussed in the following sections.
Implicit comparison is acceptable when the natural history of the disease is known with (near) certainty, the study participants are representative of the broader patient population in terms of disease severity and prognosis (in the absence of treatment) and the outcomes in untreated patients are well known, with a large observed effect in the study group. 73 Examples can be seen in the recent TAs of new drugs for hepatitis C by NICE, for which, because of the objective outcome and large treatment benefit, regulatory approval had been granted based on short-term single-arm trials. 83 However, even for diseases with an apparently uniform prognosis, there may be subtle yet clinically relevant differences between patients who are enrolled in the single-arm trial and those who do not qualify and also between those in the trial and the historical controls. Careful review of the study population and eligibility criteria is needed to make an assessment concerning external validity. 73
When considering clinical effectiveness based on single-arm trials, the comparison is often made implicitly: a survey found that roughly half of Phase II studies did not cite the source of their historical response rates. 74 This is never sufficient for the purposes of a cost-effectiveness analysis, in which it is essential to have some reasonable estimate of the treatment’s effectiveness relative to a control. This requirement has the implication that such implicit comparisons are likely to be rarely of relevance to submissions to NICE, which will by necessity always contain an economic component.
Studies that use before-and-after designs (sometimes referred to as pre–post designs) assess the difference in response before and after the administration of an intervention in a single group of patients. Patients therefore serve as their own controls. For before-and-after designs to provide unbiased estimates of effectiveness it is necessary to eliminate all alternative explanations for observed treatment effects. It is therefore necessary to eliminate the possibility of improvement as a result of adjunctive therapies administered concurrently or carryover effects from therapies administered before the intervention of interest should be considered. Furthermore, natural recovery presents another potential explanation for an observed before–after improvement in a health outcome in a single-group comparison. Drawing valid and meaningful inferences about treatment effect using single-group observational studies is therefore problematic when evaluating conditions that are fluctuating or intermittent and this limits their applicability. Further to the above, before-and-after designs can be subject to the effects of regression to the mean, which can simulate improvements in disease outcomes but which result from the elective sampling of patients at a peak severity in the natural history of disease, which has a tendency to return to average severity levels over time regardless of interventions administered. 75 Before-and-after designs are therefore most appropriate for chronic conditions in which disease status is stable over time or in which the natural history of the disease is certain, such that any variation in disease status/progression is likely to result from the intervention. Before-and-after designs consequently are most commonly used for the evaluation of surgical interventions and other irreversible interventions. Before-and-after studies can also be useful when a disease is rare (as fewer patients need be recruited) or when ethical issues mean that using a control group would be inappropriate, such as in end-of-life (EoL) care and childhood diseases. In these cases, however, the weaknesses highlighted above are likely to remain, but can be mitigated by the inability to carry out comparative studies.
Comparative estimates of effectiveness can be generated from single-arm data sets by comparing results with historical data obtained either from the same research institution or from an external cohort or database. The interpretation of single-group studies with historical controls is, however, complicated by specific challenges to the validity of historical comparisons resulting from differences between patients selected as historical controls and those recruited to the single-arm studies. Differences between the patient populations of a single treated group and historical controls can arise for a variety of reasons, including differences among accrual sites or over time in patient characteristics (e.g. age, performance status or other prognostic factors). For example, more recently diagnosed patients may have milder manifestations of a condition because of improved (and therefore commonly increased) diagnostic sensitivity. Treatment effects may also be attributable to secular trends in clinical care (e.g. changes in diagnostic methods, classification criteria or outcome ascertainment).
There are many additional reasons why patients in a single-arm Phase II study may not be comparable to those in some hypothetical historical group. 76 Phase II trials involving new agents are typically undertaken in large academic medical centres, where the patient population may vary in many ways from that in a subsequent Phase III trial (e.g. the population may be more mobile or more heavily pretreated or have a better socioeconomic status or receive better supportive care). For new agents there is a natural enthusiasm among the investigators for the new agent and a desire for it to ‘look good’. This enthusiasm may manifest itself in various ways, such as setting the historical response rate at a low value74 or enrolling only patients who look in some sense ‘promising’. These aspects cause problems in an uncontrolled Phase II study, but not in a randomised Phase II study.
However, if historical data are available from previous randomised Phase III trials, the historical estimate of the response rate for the standard treatment may be more accurate than the estimate obtained from the control arm of a randomised Phase II trial, which is based on a smaller sample size. 76
To address the problem of reliable historical benchmarks for single-arm Phase II trials, efforts have been made to amass historical databases and derive historical control data for future trials in specific disease sites. Examples include stage IV melanoma77 and advanced pancreatic cancer. 78 The availability of these kinds of data is extremely important for better evaluation and analysis of data from single-arm trials and is essential to generate the estimates of relative effectiveness needed in economic models for the assessment of cost-effectiveness.
Comparability of results from single-arm studies and randomised designs
There is a growing body of literature on whether Phase II trials should be single arm or randomised (which is now the more common approach), with the focus on which design is most efficiently associated with success in Phase III RCTs, particularly in the context of cancer drug development. From one perspective this appears not to be directly relevant to the issue of the product development of regenerative medicines, for which the issue is not which design best helps companies decide which drugs to take on to a Phase III trial, but rather how companies and regulators can manage development when the long-established expectations for pivotal evidence are unlikely to be met. This body of literature, however, includes a number of studies that have sought to evaluate the reliability of estimates of effectiveness from single-arm studies and their relative performance compared with those from randomised trials.
A simulation study79 investigated the difference between randomised Phase II trials and single-arm Phase II trials under realistic statistical parameters and with a historical control success rate of 20% and a target success rate of 40%. The study found that both designs produced similar results when there was no variation in historical control success rate but that even a modest variation in historical control success rate inflated the false-positive rate in single-arm trials. Furthermore, increasing the size of the single-arm trial inflated the false positive rate. Another simulation study80 aimed to quantify the impact of a policy of all single-arm Phase II trials compared with randomised Phase II trials on the number of Phase III trials conducted using active agents. The parameters modelled in this study included between-institution variability in the standard care response rate, treatment effect and estimate of historical control rate; the presence of historical bias (over- or under-estimation of the response rate in the historical controls as a result of changing care); and the proportion of Phase II trials conducted using active agents. The study found that single-arm trials resulted in a higher percentage of Phase III trials conducted using active agents when there was a minimal standard of care activity (i.e. high unmet need) or when the historical control rate was overestimated (with a high control rate a randomised trial was less likely to identify a treatment benefit). Randomised Phase II trials performed better when the historical control rate was underestimated or when it was highly variable. These results reflect those of Tang et al. 79 in demonstrating that historical bias has a large impact on the reliability of results from single-arm trials. Similar findings were reported when a Bayesian approach was used to compare single-arm and randomised studies, based on a binary response variable, in terms of their abilities to reach the correct decision about a new treatment. 81 The study found that the accuracy of the estimate of the success rate for the standard agent, obtained from historical data, has a crucial role: when the response rate for the standard agent is correctly estimated, the single-arm studies are preferred but, as the magnitude of the misspecification increases or as the total number of patients accrued get larger, two-arm studies tend to be preferred.
A more recent publication investigated the superiority of randomised Phase II trials over single-arm Phase II trials to predict success at Phase III for oncology drugs. 82 In this study, published Phase III trials testing systemic cancer therapy were identified through a MEDLINE search. Statistical analysis was performed using the generalised estimating equation method, correlating Phase II features with Phase III outcome. The results found that of 189 eligible Phase III trials the primary outcome was positive in 79 (41.8%) (success) and these were supported by 336 Phase II trials, including 66 randomised Phase II trials; positive Phase II outcome, randomised or not, correlated with positive Phase III outcome (p = 0.03). Randomised Phase II trials were not superior to single-arm Phase II trials at predicting Phase III study success. The authors concluded that, given the added resources required to conduct randomised Phase II trials, further research into Phase II trial designs is required.
In summary, these studies confirm that results from single-arm trials can be considered as reliable indicators of treatment benefit only when the disease natural history is very well known, the patient population is homogeneous and the control (standard care) treatment has little impact on outcomes. It is interesting that increasing the size of single-arm trials is not helpful.
Effect estimates from single-centre compared with multicentre trials
Single-centre trials may produce significantly larger effect estimates than multicentre trials. Although no publications were found examining this effect in NRSs, there are relevant publications for RCTs. Overestimation of treatment effect in single-centre RCTs has been discussed and quantified in critical care medicine;84,85 a relative overestimation of 36% was found in a study that compared 41 single-centre studies (median n = 40) with 41 multicentre studies (median n = 223). 84 Trial- or review-specific examples of this effect have also been reported in neonatology. 86
Possible reasons for the larger effect estimates may be that single-centre studies:
-
are more prone to bias than multicentre studies84
-
recruit fewer patients than multicentre studies (smaller studies tend to report larger effects)
-
may have treatment effect magnitudes that are affected by the high levels of centre expertise
-
may recruit populations that are unduly homogeneous.
These factors may limit the reliability or the external validity (generalisability) of single-arm trial results.
Relevance to future regenerative medicine submissions
Although RCTs continue to be the dominant method for evaluating treatment effectiveness, a large number of studies has been conducted devoted to establishing the relatability of evidence from NRSs. This sizable literature demonstrates both the value and the challenges of using observational data. Although the evidence is mixed regarding the reliability of observational data for evaluating treatment effectiveness, the existing studies do seem to indicate that, in some cases at least, confounding is a potential issue and will impact on treatment effectiveness estimates. Furthermore, the current evidence suggests that retrospective studies and, in particular, historical control studies are more likely to result in biased estimates of effect. As observed in Clinical efficacy and safety issues arising from European Medicines Agency, National Institute for Health and Care Excellence and Food and Drug Administration assessments of licensed regenerative medicines, many recent regenerative medicine submissions have been based on data from single-arm studies, which have been compared with historical controls. The findings of this review therefore suggest that a degree of caution is necessary in interpreting estimates from these comparisons, as bias in estimates of effectiveness from historical comparisons will add additional uncertainty not accounted for in the CIs/credible intervals presented. A key factor in the reliability of estimates of effectiveness based on observational data is the statistical analysis used, and a large number of studies have similarly sought to develop and evaluate methods for adjusting and eliminating bias resulting from confounding. Despite this, it is unclear which methods are most appropriate in certain circumstances and further research is needed. Consequently, results generated from NRSs will be subject to an unknown degree of uncertainty, even after adjustment for confounding. Single-arm trials are reliable indicators of treatment benefit only when the natural history of the disease is very well known, the patient population is homogeneous and the control treatment has little impact on outcomes. It is interesting that increasing the size of single-arm trials is not always helpful.
If regenerative medicines continue to be targeted at tightly defined conditions, with a narrow population to minimise heterogeneity, when patients have little or no chance of recovery/improvement otherwise, the use of NRSs and, in particular, single-arm studies may be adequate. To complement the data from such trials, robust accurate evidence of the outcomes achieved with standard care must be provided. When appropriate, methods to adjust for confounding should be employed, with the selection of the method used being explicit and based on sound reasoning. Confidence in estimates of effect may also increase by utilising multiple methods of adjustment, although care should be taken to ensure that methods are appropriate to the decision problem in question. However, many regenerative medicines may require highly skilled and specialised facilities for optimum delivery. Consequently, the evidence on their efficacy and safety may be derived from only small, single-centre studies, which (more often than not) might overestimate effect estimates or which might lack the external validity needed to support more widespread uptake of the intervention.
In terms of NICE methods and processes, methods research may be considered to inform guidance both for manufacturers (e.g. minimum reporting requirements for analysis methods for comparing single-arm trial data with historical control data) and for ERGs (e.g. checklists for appraising how historical control data were identified and analysed by manufacturers).
Review of the use of surrogate end points as primary outcome measures in definitive effectiveness trials of new therapeutic agents
Introduction
As discussed earlier (see Clinical efficacy and safety issues arising from European Medicines Agency, National Institute for Health and Care Excellence and Food and Drug Administration assessments of licensed regenerative medicines), it can be anticipated that almost all of the pivotal trials of regenerative medicines submitted for assessment for marketing authorisation will utilise a surrogate or intermediate outcome (or end point). A surrogate may be either a laboratory or a physiological measure of the patients’ experience that could be used to predict or provide an early measure of therapeutic effect. This section presents an overview of surrogate outcome measures and their use in clinical research and highlights issues pertinent to the development and appraisal of regenerative medicines.
Methods
To describe the use of surrogate end points as primary outcome measures in trials of new therapeutic agents a review of the most relevant and up-to-date literature was performed. The review was not systematic but was designed more as a pragmatic rapid review to assimilate current information and opinion on the use and suitability of surrogates in therapeutic trials. The review began with a search of key guidelines on the use of surrogate end points produced by the FDA, NICE DSU (Decision Support Unit) (University of Sheffield) and European Network for Health Technology Assessment (EUnetHTA) and survey results produced by the National Institute for Health Research (NIHR) HTA programme on the cost-effective use of surrogate outcomes. Citation and reference searches followed, which produced a library of relevant peer-reviewed publications and statistical reports on evidence for the use of surrogate end points in medicine. All relevant studies identified are presented in Appendix 4 (see Table 44).
Definition and examples of surrogate outcomes
Ideally, it is expected that the relative effectiveness of drugs and treatments will be based on final clinical end points,87 that is, an outcome that the patient, the clinician and other stakeholders hope to avoid such as morbidity, impaired quality of life and/or death. 88 RCTs with large sample sizes and extended follow-up periods are often required to capture the statistical significance of a treatment’s or an intervention’s impact on a patient-relevant outcome. 87 However, the requirements of RCTs are often impractical when considered alongside pressures of time for products to go to market and in particular the urgent need for new treatments for patients with chronic but life-threatening diseases. The principal rationale for the use of a surrogate outcome is a more rapid assimilation of data without the need for large and lengthy trials in patients for whom mortality rates are high or treatment options are few. 89
For example, OS is considered the gold standard to measure benefit in many clinical trials as it provides a precise and statistically and clinically meaningful end point. However, mature OS data are difficult to achieve because of the length of time needed and the number of deaths required for appropriate statistical analyses. Furthermore, OS as a measure of therapeutic success becomes less useful as the course and duration of diseases such as cancer move from being acute to more chronic; longitudinal effects of chronic disease such as comorbidities and additional ongoing treatments add further limitations to OS as an outcome. 90,91 As a solution, there has recently been a steady move (by regulatory bodies) away from OS as a clinical end point measure and towards more short-term surrogate measures.
A generally accepted definition of a surrogate has followed that of Temple (p. 4):92 ‘a laboratory measurement or physical sign used as a substitute for a clinically meaningful endpoint that measures directly how a patient feels, functions or survives’. However, chronic disease programmes and patient-reported outcomes have meant that a broader definition is now needed to better fit the HTA perspective. 93,94 Although the term ‘intermediate end point’ is sometimes used synonymously with surrogate end point,95 it is often used to refer to more patient-relevant outcomes than those typically thought of as surrogates. However, for the purposes of this report, the term ‘surrogate outcome’ will be used in its broadest sense.
Examples of approved drugs based on the use of validated surrogate end points include antihypertensives and blood pressure in stroke research, cholesterol-lowering agents and serum cholesterol and treatments for glaucoma and intraocular pressure;96 CD4 count for acquired immunodeficiency syndrome (AIDS) or death in human immunodeficiency virus (HIV) infection;97 and bone density for bone fracture in osteoporosis. 89 However, occasionally such approvals have to be revised when long-term data become available. The drug gefitinib was approved in the USA in 2003 for patients with non-small-cell lung cancer based on tumour response rate, a surrogate end point. When, in 2005, the results from later studies showed no significant benefit in terms of survival, the FDA withdrew approval for its use in new patients. Therefore, although surrogate end points offer the potential of real benefit – in providing patients with faster access to treatments and saving triallists time and resources – they may also have important drawbacks. Most notably (as the gefitinib example demonstrates), there may be uncertainty about the relationship between surrogate and real clinical end points and this may result in treatment efficacies being overestimated. A meta-epidemiological study that compared 84 trials that used surrogate outcomes with 101 trials that used patient-relevant outcomes showed that trials reporting surrogate end points had larger treatment effects: on average, trials using surrogate outcomes reported treatment effects that were 28–48% higher than those of trials using final patient-relevant outcomes and this result was consistent across sensitivity and secondary analyses. 98 The study characteristics of trials using surrogate outcomes and those of trials using patient-relevant outcomes were well balanced except for median sample size (371 vs. 741) and single-centre status (23% vs. 9%). Their risks of bias did not differ. This finding illustrates the importance of surrogate end points being appropriately validated and of quantifying the level of certainty of association of treatment effect between the surrogate and patient-relevant final outcomes. 98
Validation
Surrogate outcomes can be unreliable without sufficient validation; for example, two major antiarrhythmic drugs, encanaide and flecanaide, reduced arrhythmia but caused a more than threefold increase in overall mortality99 and cardiac inotropes improved short-term cardiac haemodynamic function but can increase mortality. 100 Such examples may fuel uncertainty about the validity of surrogates. The results of a questionnaire study of 74 stakeholders in the drug development of cardio-renal disease indicated that, although the use of surrogates is not opposed, most are not considered valid. 101 Out of the four surrogate outcomes suggested as an end point for trials – blood pressure, glycated haemoglobin (HbA1c), albuminuria or C-reactive protein (CRP) – only use of blood pressure was considered moderately accurate. Questionnaire responders from industry valued the accuracy of surrogates consistently higher than academic and regulatory responders.
General principles of validation
For a surrogate to be a reliable outcome measure it is generally accepted that the measure must be on the ‘causal pathway’ from the intervention to the clinical outcome. 89 The possible reasons for treatment or trial failure associated with surrogate end points have been discussed by Fleming and DeMets102 and more recently by Taylor and Elston:89
-
the surrogate is not on the causal pathway of the disease process
-
of several causal pathways of disease, the intervention affects only the pathway mediated by the surrogate
-
the surrogate is not on the pathway of the intervention’s effect or is insensitive to its effect
-
the intervention has mechanisms of action independent of the disease process (and so its effect will not be captured by a surrogate outcome).
A number of guidelines have been proposed for assessing the validity of surrogate end points87,89,100,102 and further work has also been published on scoring schemas for the value of surrogates. 103
As a result of a review, Elston and Taylor88 recommended that, before a surrogate outcome is accepted, a systematic review of the evidence for the validation of the surrogate/final outcome relationship should be conducted. Furthermore, the evidence on surrogate validation should be presented according to an explicit hierarchy, such as:
-
level 1 – evidence demonstrating that treatment effects on the surrogate correspond to effects on the patient-related outcome (from clinical trials)
-
level 2 – evidence demonstrating a consistent association between surrogate outcome and final patient-related outcome (from epidemiological/observational studies)
-
level 3 – evidence of biological plausibility of the relationship between surrogate and final patient-related outcome (from pathophysiologic studies and/or understanding of the disease process).
Methods for the statistical validation of surrogates as outcome measures have also developed. 99,104,105
Validation of specific surrogate outcomes
Surrogate outcomes in oncology
A recently published systematic review of trial-level meta-analyses of randomised trials quantifying the association between surrogate and final outcomes in cancer included 36 studies. 106 The review found that all validation studies used only a subset of the available trials and that the evidence supporting the use of surrogate outcomes in cancer trials is limited. The results are summarised in Table 3.
| Surrogate and clinical outcome | Number of studies | Range of correlation coefficients | Level of correlation (low, medium or high) |
|---|---|---|---|
| Pathological complete response for event-free survival | 2 | 0.17–0.28 | Low |
| Pathological complete response for OS | 2 | 0.30–0.49 | Low |
| Response rate for OS | 11 | 0.32–0.68 | Low to medium |
| Locoregional control for OS | 2 | 0.52–0.84 | Medium to high |
| Event-free survival for OS | 3 | 0.79–0.86 | High |
| Disease-free survival for OS | 7 | 0.62–0.98 | High |
| PFS for OS | 30 | 0.29–0.99 | Low to high |
| TTP for OS | 3 | 0.54–0.69 | Medium |
The results of the review indicate that little research effort has been invested in validating tumour response as a surrogate for clinical outcomes; the available evidence suggests that better tumour-level surrogate outcomes are required. The clinical outcome surrogates (intermediate outcomes) for OS, particularly PFS, have been better studied and appear to perform better. However, the range of results for PFS indicates that the validation of a surrogate in one disease and setting cannot be assumed to hold for other diseases and settings.
Progression-free survival or time to progression
The suitability of PFS or TTP as an appropriate surrogate measure in advanced or metastatic cancer research has been reviewed. 94 The review identified 19 papers covering eight different tumour types. Data sets included the relationship between the measures within aggregated trial data and the effect on individuals within IPD. The studies employed a variety of different data sets and statistical techniques, but the lack of standardisation across the studies made it very difficult for the review to identify any consistent relationship between the surrogate and the overall outcome measure.
In a recent review of current statistical approaches to surrogate end point validation based on meta-analysis in various advanced-tumour settings,107 the suitability of PFS and TTP was assessed using three validation frameworks: Elston and Taylor’s framework, the German Institute of Quality and Efficiency in Health Care’s (IQWiG) framework and the Biomarker Surrogacy Evaluation Schema (BSES3). The findings suggested that the strength of the association between the two surrogates and OS was generally low. The level of evidence (observation level vs. treatment level) available varied considerably by cancer type and evaluation tools and was not always consistent, even within one specific cancer type. This study emphasises the challenges of surrogate end point validation and the importance of building consensus on the development of evaluation frameworks.
A recently published study analysed the degree of difference in treatment effects between surrogate end points and OS in RCTs of pharmacological therapies in advanced colorectal cancer. 108 Univariate and multivariate random-effects meta-analyses were used to estimate pooled summary treatment effects. The ratio of hazard ratios (HRs) to odds ratios (ORs) and differences in medians were used to quantify the degree of difference in treatment effects between the surrogate end points and OS. The study found a larger treatment effect for the surrogates than for OS. The authors suggested that previous surrogacy relationships observed between PFS/TTP and OS in selected settings may not apply across other classes or lines of therapy. 108
Minimal residual disease
Minimal residual disease (MRD) is a surrogate outcome that has been accepted by a regulatory agency, the FDA. With current intensive treatments, many acute leukaemia patients will enter morphological complete remission (CR). This is typically defined as patients having < 5% blasts (abnormal, immature cells) in the bone marrow. If no further therapy is given after entering CR, most patients will relapse, demonstrating that microscopy-based evaluations are incapable of detecting all tumour cells. However, diagnostic techniques can now quantify and monitor MRD, which is invisible to the trained eye, in patients in CR. The ability to quantitatively measure the amount of MRD at various times after achieving CR can guide subsequent treatment. 109 Studies have shown that MRD before stem cell transplantation is a strong independent predictor of subsequent relapse in children with high-risk or very high-risk acute lymphocytic (lymphoblastic) leukaemia (ALL). 110,111
Threshold levels for MRD may vary depending on the population being considered. For children receiving first-line chemotherapy for ALL, leukaemia cell concentrations of 0.01% (1 in 10,000) have been described as optimal for identifying higher-risk patients for potential intervention. 112 For children with ALL who have had a previous relapse, the best MRD threshold for predicting disease-free survival (DFS) at 10 years has been reported as 0.001%. 113 The FDA has concluded that the evidence base to indicate that early MRD status is the strongest predictor of long-term event-free survival (EFS) in ALL is unequivocal. 114 It added that the magnitude of the importance of its critical role in risk stratification for treatment decisions has furthered the consideration of its potential as a surrogate end point for clinical trials of investigational therapeutic interventions. However, results from the UKALL R3 trial, which compared different chemotherapy treatments for children in first relapse, showed that the longer-term outcome of having MRD-negative status in patients who have already had one relapse may well vary according to how the status was achieved. 115 There is, therefore, some uncertainty in how MRD negativity correlates to long-term outcomes in relapsed populations.
Current issues for health technology assessment and cost-effectiveness models
Regulatory bodies find it acceptable for trials to be shorter, to have fewer participants and to use surrogate outcomes when populations are rare and there is a high unmet clinical need. However, a commitment to ongoing research is mandatory to receive longer-term approval; if research is not continued or if it is continued but efforts to validate the surrogate fail, the approval will be withdrawn. 96 From a regulatory and HTA perspective, the absence of data on clinical end points might be acceptable when a clinical end point is difficult or impossible to study. The EUnetHTA summarised its findings into eight recommendations for end points used in the relative effectiveness assessment (REA) of pharmaceuticals:87
-
Efficacy assessments of pharmaceuticals should be based whenever possible on final patient-relevant clinical end points (e.g. morbidity, overall mortality).
-
Biomarkers and intermediate end points will be considered as surrogate end points if they can reliably substitute for a clinical end point and predict its clinical benefit.
-
Surrogate end points should be adequately validated and this must have been demonstrated based on biological plausibility and empirical evidence.
-
Validation of a surrogate is normally undertaken in a specific population and for a specific drug intervention. Demonstration of surrogate validation both within and across drug classes should be thoroughly justified.
-
The availability of a sufficiently large safety database is particularly important and evidence on safety outcomes should always be reported.
-
The absence of data on clinical end points might be acceptable when a clinical end point is difficult or impossible to study (very rare or delayed) or the target population is too small to obtain meaningful results on relevant clinical end points even after very long follow-up (very slowly progressive and/or rare diseases). However, these exceptions need to be carefully argued and agreed in advance.
-
Reassessment requirements for further data should be clearly defined when an assessment has been previously made based on surrogate end points.
-
Further methodological research on the use of surrogate outcomes is needed to inform future REA approaches for the handling of surrogates.
Similarly, Elston and Taylor88 recommended that a HTA or cost-effectiveness model based on a surrogate outcome should be undertaken only when it is not possible to base the assessment of clinical effectiveness and cost-effectiveness on final patient-related outcomes [i.e. mortality, important clinical events and health-related quality of life (HRQoL)]. In such cases, a systematic review of the evidence for the validation of the surrogate/final outcome relationship should be performed and the evidence on surrogate validation should be presented according to an explicit hierarchy.
Given the difficulty in validating surrogate outcomes, which conflicts with the need to use such outcomes in clinical research, Ciani and Taylor93 commented on the requirement to recognise the need for pragmatic high-level evidence, preferably from meta-analyses and regression modelling using both surrogate and final outcomes for HTA. This is demonstrated by a study conducted to illustrate the potential to reduce uncertainty around the clinical outcome by estimating it from a multivariate meta-analysis. 116 Bayesian multivariate meta-analysis was used to synthesise data on correlated outcomes in rheumatoid arthritis. Estimates from the Health Assessment Questionnaire (HAQ) were mapped onto the HRQoL measure, the EuroQol-5 Dimensions (EQ-5D) questionnaire, and the effect was compared with mapping the HAQ obtained from the univariate approach. The results showed that use of multivariate meta-analysis can lead to reduced uncertainty around the effectiveness parameter. By allowing all of the relevant data to be incorporated in estimating clinical effectiveness outcomes, including data from surrogate outcomes, multivariate meta-analysis can improve the estimation of health utilities through mapping methods.
In their review of HTA and cost-effectiveness models, Taylor and Elston89 found that only one of the four reports undertook a systematic review to specifically seek the evidence base for the association between surrogate and final outcomes. Furthermore, this was the only report to provide level 1 surrogate–final outcome validation evidence (i.e. RCT data) showing a strong association between the change in surrogate outcome (biopsy-confirmed acute rejection) and the change in final outcome (graft survival) at an individual patient level. The outcome of the review was to make recommendations for the evaluation of surrogate end points in a HTA (these are listed in Appendix 4, Table 44).
Taylor and Elston’s89 HTA publication has been key to providing insight into the use of surrogates within the HTA and cost-effectiveness models framework and presents the range of approaches. This includes HR calculation, transition probabilities within a model of natural history and predictive risk equations, used by researchers to quantify the relationship between surrogate and clinical end points. 88
In addition to calls for the validation of commonly used surrogate outcomes, there is a need for novel, more appropriate, more valid outcomes. An editorial in the Journal of Clinical Oncology117 commented on the large number of novel anti-tumour agents currently being tested in ever smaller groups of patients with increasingly specific tumour characteristics. Cancer types will continue to be divided into many subentities that differ from each other in terms of genetic make-up, natural course and sensitivity to systemic treatments. This, together with the limited number of patients who are available for clinical studies, means that a new approach to oncology research is needed. The editorial called for more intensive efforts at the preclinical stage to better understand the mode of action of potential new agents and for this information to be used to select more precisely the target population and appropriate and valid surrogate outcomes. By so doing it should be possible to achieve a higher success rate in Phase III studies, with smaller numbers of patients needed.
Summary
-
Studies looking at surrogates for OS demonstrate how difficult it is to validate even commonly used surrogates.
-
On average, it seems that trials using surrogate outcomes report larger treatment effects (28–48%) than trials using final patient-relevant outcomes.
-
However, a desire to get regenerative medicines to market quickly means that manufacturer submissions are likely to be supported by short-term trials reporting primary outcomes that are surrogates.
-
Regulatory agencies may accept evidence based on surrogate outcomes; for example, the FDA accepts that MRD is the strongest predictor of long-term EFS in ALL, although there is considerable uncertainty about its value in relapsed populations.
-
The choice of surrogate outcomes must be researched, explicit and justified. Ideally, a systematic review of the evidence for the validation of the surrogate/final outcome relationship should be performed and the evidence on surrogate validation should be presented according to an explicit hierarchy.
-
Analyses, at whatever stage of development and maturity of data, should include all available outcome data to minimise uncertainty.
Scoping review of potential cost-effectiveness issues
The assessment of the cost-effectiveness of regenerative medicines and cell therapies may raise additional challenges compared with the assessment of the cost-effectiveness of other types of technologies. A focused scoping review was undertaken to help to identify potential conceptual differences between regenerative medicines/cell therapy products and other more conventional technologies. The objective of the scoping review was to identify possible characteristics that could make any assessment of cost-effectiveness, uncertainty and the value of further evidence different from that for other technologies. These characteristics also provided a basis for subsequent exploratory work to assess the appropriateness of existing decision frameworks for health technologies. A related objective was to identify areas in which additional methodological development may be required.
The scoping review was based on completed and ongoing NICE TAs for regenerative medicines and broader literature that has attempted to identify potential challenges.
Previous regenerative medicine evaluations evaluated within the National Institute for Health and Care Excellence technology appraisal process
Methods
A review of previous NICE TAs of regenerative medicines and cell therapy products was conducted. The primary aim of the review was to identify any common themes and potential analytical challenges relating to the assessment of the cost-effectiveness of regenerative medicines and cell therapy products.
Results
The National Institute for Health and Care Excellence has previously evaluated two regenerative medicines within the existing TA process: autologous chondrocyte implantation (ACI) for the treatment of cartilage defects in the knee joints118–120 and sipuleucel-T (PROVENGE) for treating asymptomatic or minimally symptomatic metastatic hormone-relapsed prostate cancer. 121
Autologous chondrocyte implantation
Autologous chondrocyte implantation has now been appraised on three separate occasions by NICE: originally in 2000 (TA16)119 and as separate re-reviews in 2005 (TA89)120 and 2015 (as part of an ongoing review). 118 The original guidance in TA16 has since been replaced by TA89 and documentation from the initial appraisal has been removed from NICE’s website. Hence, our review focused on the separate re-reviews. However, it was reported in the final appraisal determination (FAD) for TA89 that, when the original guidance was produced in 2000, data from completed RCTs for ACI were not available.
For the re-review in 2005,120 four controlled trials were subsequently considered, two comparing ACI with microplasty (n = 40 and n = 100) and two comparing ACI with microfracture (n = 80 and n = 66). Follow-up across the trials varied between 1 and 2 years. Three publications relating to a Swedish longer-term case series for ACI were also identified, describing outcomes for up to 11 years after surgery.
In reviewing the various documents for TA89 there appears to be no specific reference made to any distinct challenges of evaluating ACI based on its classification as a regenerative medicine or any specific discussion related to possible innovation. However, the lack of medium- to long-term outcomes associated with ACI and their durability was highlighted as a key limitation in the FAD. The committee also noted concerns that the comparative trial evidence had a follow-up of only 1–2 years and longer-term case series data appeared to show similar benefits for most treatment modalities.
Although uncertainties surrounding long-term outcomes are clearly not unique to regenerative medicines, the Assessment Group (AG) concluded in TA89 that there was insufficient evidence for ACI to produce a robust cost per quality-adjusted life-year (QALY) estimate for ACI. Instead, the AG undertook ‘illustrative modelling’ of the cost-effectiveness of ACI in three ‘increasingly speculative’ stages, incorporating alternative assumptions relating to the short term (2 years), medium term (10 years) and long term (up to 50 years). The conventional NICE reference case for cost-effectiveness was applied,122 although deterministic approaches (i.e. point estimates were assumed for input parameters) were applied. A discount rate of 1.5% for health benefits and 6% for costs were applied in line with the recommended rates at the time of the appraisal. 123
The cost-effectiveness results were sensitive to the time horizon and the assumptions employed within the chosen periods. In both the short-term and medium-term analyses, ACI was reported to be dominated by the current standard of care (microfracture/mosaicplasty). In the long-term analyses, the possible avoidance of knee replacements was taken into account and the incremental cost-effectiveness ratio (ICER) of ACI compared with microfracture was reported to be between £3200 and £3650 per QALY.
Autologous chondrocyte implantation was subsequently not recommended in TA89120 for routine use, being given an only in research (OIR) recommendation.
As of 23 November 2016 this re-review is still ongoing. 118 The rapid evolvement of ACI over time was highlighted in the appraisal consultation document (ACD) and the branded MACI product being appraised was now classified as a third-generation ACI.
The AG undertook a ‘review of reviews’ comparing the effectiveness of ACI (any generation) with that of microfracture. In total, 12 systematic reviews were identified. Studies within the reviews were reported by the AG to be heterogeneous, with follow-up of between 6.5 months and 7.5 years. The AG considered the results of the reviews to be inconclusive on the effectiveness of ACI compared with microfracture.
The National Institute for Health and Care Excellence received separate submissions for ChondroCelect (Swedish Orphan Biovitrum, Stockholm, Sweden), MACI (Aastrom, now called Vericel Corporation, Cambridge, MA, USA) and OsCell (Robert Jones and Agnes Hunt Orthopaedic Hospital, Oswestry, UK). 118 Although both ChondroCelect and MACI had EMA approval, the submission by OsCell was based on a product provided via the hospital exemption licence, which allows provision for OsCell to supply chondrocytes for use in ACI under the professional responsibility of a medical practitioner. There was a marked difference in list prices (excluding value-added tax) between the products: £18,301 for ChondroCelect, £16,226 for MACI and £4135 for OsCell. However, costs were also noted to vary in different settings because of negotiated procurement discounts.
The manufacturer submission supporting ChondroCelect provided evidence of clinical effectiveness from four new sources not considered in TA89: a RCT (n = 118) with up to 5 years’ follow-up; a ‘compassionate use’ case series (n = 370); an ongoing registry-based cohort study (n = 308) with up to 3 years’ follow-up; and data from a Belgian reimbursement scheme (254 procedures undertaken over a 3-year period).
The submission supporting MACI described new clinical evidence from two RCTs (n = 144 and n = 60, both with up to 2 years’ follow-up) and a subsequent ongoing extension study (up to 3 years’ additional follow-up, with interim data reported for the first year).
The OsCell submission reported interim (up to 5 years’ follow-up) clinical effectiveness evidence from a UK RCT [ACTIVE (Autologous Chondrocyte Transplantation/Implantation Versus Existing treatments) trial (n = 390), including first-, second- and third-generation ACI] and evidence from a separate cohort study (n = 366) with up to 3 years’ follow-up.
Separate cost-effectiveness analyses were presented by the manufacturers of ChondroCelect and OsCell and the AG. A discount rate of 3.5% for both health benefits and costs was applied in line with the recommended rates at the time of the appraisal. 122 The base-case ICERs for ACI compared with microfracture in the manufacturers’ and AG models were approximately £6000–7000 and £16,000 per QALY gained, respectively.
In the ACD, the committee concluded that, although there was more clinical effectiveness data than at the time of the previous NICE TA guidance, the evidence base for the technology is still emerging and no comparative clinical effectiveness data had been reported beyond 5 years. Innovation was formally considered and the committee agreed that ACI, albeit not new, is technically innovative. However, the committee concluded that the uncertainties in clinical effectiveness were such that the technologies could not be considered innovative in the context of a NICE appraisal. 118
In relation to the cost-effectiveness evidence, the committee considered that OsCell had underestimated its cell costs and that the true cost may approach that of MACI and ChondroCelect. The committee concluded that, although the cost to the NHS of providing the cells for ACI was somewhat uncertain, the cost estimate used within the AG and the ChondroCelect model were reasonable for the purposes of decision-making.
The committee concluded that a lifetime horizon was preferable because it captured all of the costs and consequences of treatment, but the lack of long-term data with which to populate a model generated large uncertainties. The committee concluded that there was no ICER available that included the assumptions that it considered plausible. Furthermore, it was not persuaded that ACI was proven to be a cost-effective treatment and neither did it consider that the available data robustly supported ACI being better than other treatments.
The committee therefore issued a provisional OIR recommendation because the clinical effectiveness and cost-effectiveness of ACI remains uncertain. Hence, ACI was not recommended for routine use in the NHS unless it is part of existing or new clinical studies. It was stated that ‘these studies should generate robust outcome data and include both interventional and observational studies’ (p. 45 of the ACD). 118
Sipuleucel-T
The National Institute for Health and Care Excellence issued guidance for sipuleucel-T in February 2015. 121 The appraisal was subsequently withdrawn in May 2015 following the withdrawal of the marketing authorisation for sipuleucel-T. However, prior to this NICE had conducted a full appraisal of the technology, rejecting it because of the cost-effectiveness estimates exceeding the threshold considered to represent value for money to the NHS. 122
Clinical effectiveness evidence was based on three Phase III, double-blind, multicentre RCTs conducted in the USA and Canada that compared sipuleucel-T with placebo (n = 512, n = 127 and n = 98). The primary end point for the pivotal [Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT)] trial was OS and the median follow-up was 34 months. The main secondary end point was TTP. The risk of death was reported to be statistically significantly lower in the sipuleucel-T group than in the placebo group (HR 0.78, 95% CI 0.61 to 0.98). The trial also demonstrated that patients randomised to sipuleucel-T survived for longer (median 25.8 months) than patients randomised to placebo (median 21.7 months), with a difference of 4.1 months.
Subgroup analysis suggested important clinical differences based on baseline prostate-specific antigen (PSA) concentration, with a difference of 13 months (HR 0.51, 95% CI 0.31 to 0.85) in median survival for the quartile of patients with the lowest baseline PSA concentration compared with 2.8 months (HR 0.84, 95% CI 0.55 to 1.29) for the quartile with the highest PSA concentration. The manufacturer suggested that sipuleucel-T has a delayed onset of action because it is an immunotherapy, so giving it early in the course of disease progression (as indicated by a low PSA) could provide patients with more time to benefit from sipuleucel-T. However, the ERG cautioned that the subgroup of patients in the IMPACT trial with the lowest quartile baseline PSA level had been identified in a post hoc analysis, with no clinical significance attached to the specific PSA concentration in this group.
A conventional Markov (partitioned-survival) model was submitted by the manufacturer to inform cost-effectiveness with a lifetime time horizon (10 years). Parametric survival analyses were used to extrapolate the trial data to the lifetime horizon. Conventional discount rates (3.5% for costs and benefits) were applied.
In the manufacturer’s base-case analysis, the ICER for sipuleucel-T compared with best supportive care was £124,875 per QALY gained. In the subgroup with the lowest quartile baseline PSA level, the ICER for sipuleucel-T compared with best supportive care was £48,672 per QALY gained. The manufacturer also conducted sensitivity analyses with an alternative comparator (abiraterone rather than best supportive care; Zytiga®, Janssen Biotech Inc., Horsham, PA, USA) and applied assumed discounts to the price of abiraterone of ≥ 30%; these analyses resulted in ICERs for sipuleucel-T compared with abiraterone of at least £511,663 per QALY gained.
The ERG noted uncertainty surrounding the extrapolation of survival data and chose an alternative survival distribution for OS in its exploratory analyses alongside other proposed amendments. In the ERG’s base case, the ICER for sipuleucel-T compared with BSC was £111,417 per QALY gained. The ERG’s analysis for the low PSA concentration subgroup resulted in an ICER of £61,381 per QALY gained for sipuleucel-T compared with BSC.
In considering the cost data and assumptions within the manufacturer’s submission, it was noted that the acquisition cost of sipuleucel-T included the costs of leukapheresis, patient tests associated with leukapheresis, transportation of white blood cells and the manufacture and transportation of sipuleucel-T. However, given the complex administration of sipuleucel-T and the lack of experience in the UK of using the treatment, the committee was unsure whether the NHS would incur additional costs of using sipuleucel-T that were not included in the economic model. The committee also considered that there may be patient travel costs associated with sipuleucel-T treatment because of its provision within specialist centres, which had not been included in the model. These issues were considered to add uncertainty to the estimates of cost-effectiveness.
In considering the clinical relevance of the subgroups, the committee heard that the clinical experts were unable to identify a single PSA value that was currently used for guiding treatment decisions. The committee considered that registry data could have been used to assess whether outcomes after treatment with sipuleucel-T in clinical practice were similar to those in the IMPACT trial for patients with low baseline PSA concentrations, but they were not presented with this information by the manufacturer. The manufacturer reported that such a registry had been established (PROCEED – A Registry of Sipuleucel-T Therapy in Men With Advanced Prostate Cancer) but that data were considered too immature to inform OS.
In relation to potential innovation, the committee reported that it was aware that sipuleucel-T is an autologous cellular immunotherapy and is the first treatment for this indication that is not cytotoxic or based on hormone therapy. However, the committee concluded that no evidence had been presented for any specific additional benefits that were not already captured in the QALY estimates. 121
The committee concluded that there were areas of considerable uncertainty in the results generated by the model and noted that all of the ICERs estimated by the manufacturer and the ERG fell substantially above the range normally considered cost-effective (£20,000–30,000 per QALY gained). 121
Issues and common themes
The existing NICE TAs raise a number of issues and several common themes emerge. The innovative nature of ACI (most recent ACD only) and sipuleucel-T were acknowledged by both committees. However, these considerations appear to relate more to an appreciation of the technical nature of the innovations than to any specific attributes of the innovations that might lead to a distinct benefit that may not be appropriately reflected in the reference case measure of QALYs. Importantly, no evidence was presented in either appraisal that led the committee to consider that these specific attributes of innovation were relevant.
The high levels of uncertainty surrounding the cost-effectiveness results were highlighted in both appraisals. In the most recent appraisal of ACI,118 this led the committee to conclude that there was no ICER available that included the assumptions that it considered plausible; neither was it persuaded that ACI was ‘proven’ to be a cost-effective treatment. The committee appraising sipuleucel-T concluded that, despite the considerable uncertainty in the results generated by the model, all of the ICERs estimated by the manufacturer and the ERG appeared to be substantially above the range normally considered cost-effective. 121 The difference in the committee’s subsequent recommendations (i.e. OIR for ACI and reject for sipuleucel-T) suggests that the committees may have reached different conclusions on the potential of both products to be cost-effective despite the inherent uncertainties.
The rapidly changing nature of regenerative medicines and challenges raised by this is evident across the series of appraisals for ACI. Over the 15-year period that the separate appraisals have been undertaken, the initial first-generation ACI products (ACI-C) have been superseded by second- and third-generation products. This has resulted in potential challenges in relation to quantifying the long-term uncertainties as newer generations emerge – during the time over which longer-term evidence has emerged, newer generations of ACI have also arrived. The generalisability of the longer-term evidence to the newer generations has raised additional issues and challenges. For example, the AG in the most recent ACI appraisal excluded longer-term evidence available from the first-generation of ACI products on the basis that these products had now been superseded by newer generations. 118 This approach effectively assumes that existing evidence cannot be generalised across different generations of products. If such a position were routinely taken, this may pose a potential challenge to manufacturers in terms of providing data that are considered sufficiently robust within a time frame that permits sufficient commercial return to warrant their research and development (R&D) expenditure. The extent to which evidence can be generalised or transferred between generations remains an important consideration.
Similar uncertainties arise for more conventional technologies in relation to the constant evolution of knowledge over time and subsequent challenges for HTA and cost-effectiveness assessments. The challenge of determining when evidence is sufficiently ‘robust’ within a technology’s overall life cycle to undertake a HTA/cost-effectiveness assessment is summarised by what has been termed ‘Buxton’s law’ (i.e. it is always too early until, unfortunately, it’s suddenly too late). 124 These challenges have led to an increased appreciation of the importance of employing an iterative approach to cost-effectiveness assessments, such that, as new evidence emerges, progressively more certain estimates are derived and earlier policy decisions can be subsequently reconsidered. 125,126 However, as highlighted by the ACI appraisal, more specific challenges may arise for appraising newer generations of products in relation to the extent to which evidence is considered generalisable or transferable across different product generations. Furthermore, the potential high upfront costs and the scale of any irrecoverable costs, as discussed in more detail in later sections, may be important additional considerations within these iterative assessments.
Additional uncertainties were also identified across both appraisals in relation to the costs that would be incurred by the NHS. Within the ACI appraisal, uncertainties were identified surrounding the acquisition costs of the technologies themselves (i.e. because of local price negotiations and concerns regarding the proposed cost of the product provided under hospital exemption), as well as the appropriate cost or tariff to apply to other elements of the overall procedure. 118 The complexity of provision and lack of experience in the UK of using the product was also identified as an issue within the appraisal of sipuleucel-T. 121 Uncertainties arising from this, alongside the proposed provision within specialist centres and the possible impact on travel costs for patients, led the committee to conclude that additional uncertainties existed surrounding whether all relevant costs had been appropriately included within the model.
Importantly, the RCTs that informed the basis of the regulatory submissions for ChondroCelect, MACI and sipuleucel-T were also central to the subsequent submissions to NICE and the economic models developed to support these. 121 Follow-up ranged from between 2 and 5 years for ChondroCelect and MACI and additional evidence was also submitted from ongoing extension studies and other registries. In the case of sipuleucel-T, the pivotal (IMPACT) trial was powered on OS with a median follow-up time of 34 months. Consequently, neither appraisal provides an indication of any additional challenges that may be raised for regenerative medicines or cell therapies that have received regulatory approval based on uncontrolled studies or employing surrogate outcomes. However, it seems reasonable to conclude that the uncertainties expressed in relation to cost-effectiveness within the existing appraisals are likely to be magnified in this eventuality.
Broader consideration of potential conceptual differences and possible methodological challenges for cost-effectiveness analyses
A separate review of known references and key citations of these was undertaken to identify other potential conceptual differences between regenerative medicine and cell therapies and more conventional medicines to identify potential methodological challenges for cost-effectiveness assessments.
During the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) conference held in November 2014, a workshop was held to discuss potential HTA and reimbursement challenges for regenerative medicines and curative treatments. As part of the workshop, Towse127 argued that many of the challenges for curative therapies appear similar to those for disease-modifying therapies for chronic diseases, sharing several associated problems, most notably (1) the use of short-term trials using surrogate outcomes that may not produce relevant clinical outcomes, (2) the use of outcomes that may not be sustained over time and (3) safety problems that may emerge over time.
In considering how these uncertainties might be formally incorporated within policy decisions regarding reimbursement, Towse127 acknowledged that value of information approaches provide a potential analytical framework. This framework formally evaluates the potential trade-off between the net health benefits to current patients from early access to the technology and those to future patients from withholding access to the technology until additional research has been conducted. The framework can then be used to help guide more appropriate policy choices between (1) adopt and reimburse now, (2) delay adoption/reimbursement and undertake further research (i.e. OIR) and (3) adopt/reimburse now and undertake further research [akin to coverage with evidence development (CED) or approve with research (AWR)]. 128 Towse127 also acknowledged the importance of risk-sharing approaches and particularly how these could enhance the value of CED/AWR.
As discussed in Clinical efficacy and safety issues arising from European Medicines Agency, National Institute for Health and Care Excellence and Food and Drug Administration assessments of licensed regenerative medicines, assessments of the clinical effectiveness and cost-effectiveness of regenerative medicines and cell-based therapies, particularly early in their life cycle, may be less extensive and lower in quantity than evidence for more conventional pharmaceuticals. Under these circumstances it may become even more critical to consider conditional reimbursement and possible risk-sharing agreements between the manufacturer and the payer.
Towse127 concluded that the main reimbursement challenges for regenerative medicines relate more to their financing than to the methodology of HTA and cost-effectiveness. In particular, concern was raised by Towse regarding whether health-care systems could cope with the potential high upfront costs of a curative treatment that appeared cost-effective using conventional thresholds, thereby presenting a potential barrier to adoption.
In the same workshop, Faulkner129 explored differences between regenerative medicines/cell therapies and conventional biologics. Specific reference was made to the more limited understanding by physicians and payers, leading to potentially greater requirements being made for longer-term data collection to more robustly demonstrate value. The multiple procedural steps required for some therapies were identified as another potential challenge, as these may be subject to different regulatory and reimbursement processes. Similar concerns were raised by another speaker, Husereau,130 who highlighted that regenerative medicines can be considered as both a biologic and a device/procedure, with many countries also having different reimbursement procedures, often based on cost minimisation for the funding of procedures because of fixed Healthcare Resource Group (HRG) pricing. In common with the arguments made by Towse, both Husereau and Faulkner also highlighted the challenge of applying a single financing approach for reimbursement.
The potential to enable a disease cure or prolonged therapeutic effect was also identified as a relevant characteristic by both Faulkner129 and Husereau. 130 However, Husereau130 identified a specific challenge surrounding the classification of a cure and its distinction from a prolonged therapeutic effect. To be considered curative, a therapy needed to demonstrate ‘no chance of re-entering suboptimal health state from same disease’. In reality, it seems unlikely that a new therapy can be definitively classified as curative prospectively, as many of the required elements cannot be demonstrated until a full lifetime of a cohort of treated patient has passed.
Husereau130 also raised the question of whether there is anything specific regarding curative therapies relative to standard treatments that could be perceived as providing additional benefits to patients beyond the current QALY framework. This was further considered in a subsequent publication. 131 Husereau reported that, although there was limited direct empirical evidence to address this specific question, important insights could be generated from the large literature exploring valuation issues for treatment (which he noted was often labelled as ‘cure’ within these studies) compared with prevention. Husereau reported a potential disconnect between existing literature reporting individual preferences and that reporting societal preferences. When given a choice between prevention and treatment, individuals appear to state a preference for prevention. However, similar preferences are not apparent when (societal) willingness to pay is considered. This disconnect was attributed to separate psychological factors including time, certainty of individual decisions and the valuation of identifiable compared with statistical lives. Husereau concluded that if a similar disconnect exists for curative therapies relative to standard treatments this could lead to considerable public debate and that further research was required.
Importantly, several of these issues and challenges highlighted in the workshop do not appear to be unique to regenerative medicines and cell-based therapies and similar challenges are often present when appraising more conventional biologics, companion diagnostics and devices more generally. However, it appears reasonable to conclude that these issues will be more commonly faced within evaluations concerning regenerative medicines and cell therapies. Furthermore, as many regenerative medicines and cell therapies will be considered as both a biologic and a device/procedure, manufacturers may have to address the specific regulatory and reimbursement challenges faced by both pharmaceutical and device manufacturers internationally.
Because of the personalised nature of regenerative medicines, the manufacturing and production process is typically more complex than that for traditional drug therapies. Current pharmaceutical manufacture is largely based around drugs being prepared, tested and manufactured in bulk at a consistent quality in advance of need, using automated processes. In contrast, many regenerative medicines require significant personalisation at the point of need. The complexity of the process, and the high level of personalisation required, may result in significantly higher marginal costs of production than for conventional pharmaceuticals or biologics. Inevitably, these additional complexities are likely to lead to higher upfront costs to health-care systems. However, high-upfront-cost therapies are not limited to regenerative medicine. A recent example is Sovaldi® (sofosbuvir; Gilead Sciences, Foster City, CA, USA) for hepatitis C, which was recommended by NICE in some genotypes of the disease despite its high initial cost. 132
This complexity and personalisation is likely to be coupled with a requirement for the provision of additional health-care services within the overall process. The additional demands raise issues around the impact on the wider health-care setting, both at a marginal cost and wider infrastructure level. These demands may differ according to the extent of the services provided by the manufacturer and those requiring separate funding by the health-care system. In the sipuleucel-T appraisal,121 the lack of experience in the UK of using the treatment raised additional uncertainties about whether the NHS would incur additional costs that were not reflected in any of the scenarios evaluated, raising an additional source of uncertainty.
As the provision of regenerative medicines and cell-based therapies often entails multiple procedural steps (e.g. cell extraction, processing and administration) and may be undertaken alongside additional procedures (e.g. leukapheresis in the case of sipuleucel-T and arthrotomy for ACI), additional uncertainties are likely to be raised concerning the generalisability and transferability of evidence between different settings. That is, separating out the specific effect of the regenerative medicine or cell therapy from the effect of broader health provision, which itself may be subject to significant variation across different health-care systems, represents an important challenge to HTAs and cost-effectiveness assessments.
Many regenerative medicines and cell therapies also appear likely to share similar ‘unique’ characteristics to those reported for medical devices. 133 For example, particular parts of the procedural process may change significantly over time, experiencing incremental or step changes as new processes and infrastructure develops. Additionally, the requirement for highly specialised infrastructure and staff indicates the potential for a learning curve over time both for manufacturers and for health-care providers. Although increased automation methods and the ‘scaling out’ of services may subsequently reduce the need for highly specialised staff (and lower the marginal costs of production), the infrastructure requirements and implications for possible learning curve effects are likely to be an important consideration when assessing the cost-effectiveness of regenerative medicines and cell therapies.
In addition to issues related to uncertainty, issues of irrecoverable costs may pose an additional challenge. Irrecoverable costs are those costs that once committed cannot be recouped if changes occur at a later time, most commonly thought of as investment costs associated with the capital expenditure on equipment, new facilities or training and learning costs. These are likely to be most significant when the introduction of a new regenerative medicine or cell therapy imposes additional infrastructure requirements on the health system. Within economic evaluations, these costs are commonly annuitised and allocated as ‘per-patient’ costs by spreading the cost over the number of patients likely to be treated during the lifetime of the equipment. However, if reimbursement decisions about the technology change before the end of the lifetime of the equipment (e.g. approval is withdrawn), then these costs may not be recovered and hence need to be explicitly considered.
The risks of these more conventional types of irrecoverable costs to the health system may be more limited if the manufacturer provides the necessary infrastructure and associated training. However, irrecoverable costs also potentially exist at the patient level. Regenerative and cell therapies are developed, by design, to have a significant (if not permanent) period of effect, during which they may be neither removable nor reversible. The irreversibility of these therapies implies that any uncertainty associated with the long-term efficacy and adverse event profile has a greater potential significance than for treatments that can easily be reversed or switched.
Potential approaches to addressing health technology assessment challenges
Many of the issues associated with regenerative medicines will inevitably impact on the level of uncertainty associated with the cost-effectiveness of the technology when introduced into clinical practice. Even when products have a significant potential to confer important clinical advances over current therapies, this may not be known with a high level of certainty at the time at which a regenerative medicine is licensed. Inevitably, a new technology’s cost-effectiveness may be more difficult to determine in these circumstances and schemes that allow the development of further evidence or entail a risk-sharing component may be required.
Managed entry agreements (MEAs) are an increasingly common policy response to dealing with uncertainty in the evidence base of new health technologies entering the market. MEAs are also commonly referred to as performance-based risk-sharing agreements and patient access schemes (PASs). A taxonomy provided by Walker et al. 134 makes an important distinction between MEAs based on reductions in the effective price (e.g. akin to the majority of PASs implemented currently alongside NICE guidance) and those associated with evidence generation (e.g. OIR, CED). Both approaches have the aim of reducing risk and decision uncertainty to the health-care system, albeit through separate mechanisms. Walker et al. 134 concluded that reimbursement decisions and the possible use of MEAs should be based not only on the expected value of a technology but also on the value of further research, the anticipated effect of coverage on further research and the costs associated with reversing the decision (i.e. irrecoverable costs).
Similar conclusions are reflected in NICE’s current Guide to the Methods of Technology Appraisal (pp. 69–70),122 which outlines several factors that need to be considered (e.g. the need for and value of further research as well as the feasibility of obtaining trials) when clinical effectiveness is considered limited or weak.
How to determine when efficiency is sufficiently weak or uncertain, such that MEAs are appropriate policy responses, remains a key methodological issue that has important implications both for policy making and research investments made by the regenerative medicine industry. A more formal framework has recently been proposed that has established the key principles of the assessments needed. 128 NICE’s DSU has also recently published a report outlining potential methods for assessing MEAs within the TA programme. 135
Concerns surrounding the potential high upfront costs of regenerative medicines and their affordability to health-care systems have also received particular attention in the literature, leading several authors to conclude that alternative financing approaches may also have to be considered. For example, Towse127 outlined three alternative financing routes: (1) pay for performance, (2) amortisation and (3) innovative financing schemes. The potential issues and challenges related to alternative financing approaches in the context of regenerative medicine and cell-based therapies are discussed in the following sections.
Fixed price schemes
The simplest and most common approach to reimbursement is the payment of a fixed price at the time of treatment, potentially subject to discounts agreed via a PAS. This approach has the benefit of being relatively manageable and low cost to implement. Furthermore, subject to uncertainties concerning the eligible patient population size and subsequent uptake rates, the budget impact is also largely predictable. However, if the therapy is expensive and/or the patient population is large, the total budget impact to the funder may raise issues concerning system ‘affordability’ (i.e. the ability to displace sufficient activities elsewhere in the health-care system to provide the additional funds necessary to provide the new treatment). This may have implications for subsequent implementation.
Although ‘affordability’ is not explicitly considered by NICE, the committee is requested to take ‘account of how the incremental cost-effectiveness of the technology being appraised relates to other interventions or technologies currently or potentially applied in the NHS’ (p. 65). 122 When significant uncertainty exists surrounding future outcomes, a fixed price scheme exposes the funder to the risk of overpayment for outcomes that may not be realised. A fixed pricing mechanism may be potentially optimal in situations in which there is little uncertainty about the long-term outcomes; there are high costs of patient follow-up; and/or the resulting budget impact is likely to represent a marginal change to overall NHS spend.
Amortisation
Amortisation has been raised as an alternative financing approach for curative treatments, particularly for chronic diseases. 127,136 Gottlieb and Carino136 identified that most health-care finance systems are not currently structured to be able to pay to rapidly cure everyone of a chronic disease using a treatment that may be priced much higher than the existing chronic therapies. Payment models were therefore advocated that could more easily spread the potentially high upfront costs of a curative treatment and be more closely aligned to the time period over which health and economic benefits are realised. Such financing schemes are common for the payment of medical devices and other capital equipment. However, for a regenerative medicine or cell therapy, instead of spreading out payment over the lifetime of the capital equipment, amortisation would spread payment over the expected duration of benefits.
Gottlieb and Carino136 highlighted several issues and challenges relating to operationalising such a scheme. These included the potential need for another financial intermediary to act as a third party to the transactions, the need to alter accounting standards and potential conflict with the manufacturer’s desire to secure immediate revenue to maximise return on its investment. The magnitude of the initial R&D costs compared with the ongoing marginal costs of production might also influence the interest rate that the funder would have to offer to sufficiently incentivise both existing and future manufacturers.
Pay for performance
Although the use of an amortisation approach to financing might address the constraints imposed within current financial structures, this approach does not reduce the risk to health-care systems of uncertain future health benefits that may not be realised in routine clinical practice.
In contrast, a pay-for-performance-type mechanism ensures that the total price paid is more directly related to the performance of the therapy in clinical practice. This mechanism requires agreement between the funder and the manufacturer on the measure of performance (e.g. response, relapse or mortality), the programme of data collection and analyses required to monitor performance and the payment mechanism itself (e.g. fixed price at time of treatment with rebate, retrospective reimbursement for treatment ‘successes’ or amortised payments directly linked to performance over time).
As with amortisation, the potential to spread repayments over the longer term reduces the short-term budget impact. This financing approach also potentially addresses the uncertainties surrounding the potential health benefits and the risk of overpayment (i.e. when the opportunity costs are subsequently revealed to be greater than the acquisition cost) by the funder. Inevitably, such a mechanism is likely to be both more complex and more expensive to implement than a simple PAS or amortisation approach. However, there are examples of existing PASs within the NICE TA programme that already incorporate performance assessments, and discontinuation rules are commonly applied within NICE appraisals to ‘optimise’ cost-effectiveness and reduce decision uncertainty.
A pay-for-performance mechanism is potentially optimal if there is large uncertainty about long-term outcomes, a relatively low cost of patient follow-up and monitoring of the outcome(s) of interest (relative to the level of uncertainty) and a large total budget impact that, as with amortisation, can be spread over time. The potential challenges concern how performance in clinical practice would be monitored and evaluated and whether a simple assessment of continued treatment ‘success’ is feasible or not.
A recent paper by Edlin et al. 137 proposed a leasing approach for innovative technologies as an alternative payment strategy combining elements of amortisation with pay-for-performance approaches. The advantages of this approach is that it both addresses the funding constraints caused by existing finance structures and ensures that the risks associated with uncertain future health benefits are more appropriately shared between the funder and the manufacturer.
Edlin et al. 137 proposed that, having established the price at which the technology is expected to be cost-effective, the ‘lease’ payment due for each period of health delivered could be established by calculating a stream of payments over the expected lifetime of the technology that has the same expected net present value as the agreed price. The subsequent leasing scheme would work by paying the manufacturer for each period of time that health is delivered at the individual patient level, that is, if the observed effectiveness in clinical practice was equal to the expected effectiveness, the manufacturer would receive the full value of the technology over the agreed period. However, if observed effectiveness was less than expected, payment would stop and the risk to the health system of overpayment would be limited. Furthermore, manufacturers would be rewarded for technologies that resulted in better health outcomes than expected by receiving additional payments over extended periods of time.
Using trastuzumab (Herceptin®, Genetech, San Francisco, CA, USA) in early breast cancer as an exemplar, and linking the lease to relapse-free survival (RFS), Edlin et al. 137 demonstrated that the scheme not only reduced the total budgetary impact but also resulted in a more appropriate share of risk between the manufacturer and the funder, while simultaneously reducing the value of further research. Edlin et al. 137 concluded that such a scheme could help to promote the rapid adoption of innovative technologies into routine clinical practice.
Innovative financing
Several authors have argued for even more innovative approaches to pricing to be considered, seeking inspiration from the wider financial world, for example innovative licensing and the issuing of bonds, by which third-party payers cover the costs of treatment, benefiting from the respective interest rate paid by the health-care funder. 131 Such mechanisms have had some success in the provision of vaccination programmes in developing nations (through the International Finance Facility for Immunisation scheme), appealing to investors seeking ethical investments. An alternative mechanism, considerably closer to a pay-for-performance mechanism, is the Health Impact Fund, in which manufacturers distribute innovations at cost but are rewarded with performance-based bonuses.
Possible implications for National Institute for Health and Care Excellence methods and processes
In considering the potential characteristics of regenerative medicines and cell-based therapies and associated challenges for HTA, NICE will need to consider whether changes to its current processes and methods are required or not. Importantly, some of the potential challenges highlighted are already considered within the existing methods guide. 122 For example, the committee is already requested to recognise that the evidence base will necessarily be weaker for some technologies, such as those used to treat patients with very rare diseases. If considered appropriate, this could be extended to include regenerative and cell therapies. Similarly, although the magnitude of the budget impact is stated not to determine the appraisal committee’s decision, the existing methods guide indicates that the committee may require more robust evidence on the effectiveness and cost-effectiveness of technologies that are expected to have a large impact on NHS resources. However, a potential conflict may arise between the certainty required for interventions with a large budget impact and subsequent deliberations regarding the acceptability of ‘weaker’ evidence.
The National Institute for Health and Care Excellence’s existing processes also make separate provision for specific disease and technology characteristics, which may be relevant to many regenerative medicines and cell therapies. NICE’s current EoL criteria allow the committee, when considering the overall health benefits, to explore a QALY weighting that is different from that of the reference case, assuming that all of the stated criteria are met. The methods guide also states that this approach can be used in other circumstances when instructed by the NICE board. Further research may be warranted to determine whether a similar weighting approach might be appropriate for regenerative medicines and cell therapies. However, there remains an issue regarding whether such a weight should be based on product-specific characteristics or patient-specific characteristics (i.e. not confined to product type).
Within the provisions and regulations of the Health and Social Care Act 2012138 relating to NICE, due regard is also required concerning ‘the desirability of promoting innovation in providing health services or social care in England’ (see Chapter 6). 122 This is currently incorporated within the committee’s deliberative process for situations in which the most plausible ICER exceeds £20,000 per QALY gained. In this situation, the innovative nature of the technology, specifically if it adds ‘demonstrable and distinctive benefits of a substantial nature which may not have been adequately captured in the reference case QALY measure’ (p. 68) is accounted for. 122 Importantly, neither of the previous regenerative medicines appraised by NICE to date were considered to demonstrate such benefits. 118,121
The NICE methods guide also permits separate provision to be made via the specific discount rate that it applies. 122 Within the NICE reference case for cost-effectiveness analysis, the same annual discount rate is required to be used for both costs and benefits (currently 3.5%). However, the use of a non-reference case discount of 1.5% for both costs and health effects is permitted in cases in which treatment restores people who would otherwise die or have a very severely impaired life to full or near full health and when this is sustained over a very long period (normally at least 30 years) and is highly likely to be achieved. Hence, certain regenerative medicines and cell therapies may be considered to meet these criteria. However, uncertainties remain regarding how the likelihood of achieving these long-term health benefits will be considered by the committee, particularly in the context of the uncertainties outlined in this section. Furthermore, the stipulation that the committee will also need to be satisfied that the introduction of the technology does not commit the NHS to significant irrecoverable costs raises additional issues about whether these criteria will be met.
Issues of discounting have been widely considered in the economic literature in relation to preventative treatments and, particularly, vaccination programmes. The appropriateness of employing different discount rates and/or different rates over time is an area that requires further consideration, particularly for potentially curative regenerative medicines and cell-based therapies. Westra et al. 139 explored the impact of employing alternative discount rate approaches for human papillomavirus vaccination based on different time-varying methods: a stepwise approach (a constant rate is applied for a set period and lowered in subsequent periods), a hyperbolic approach (the discount rate declines over time) and a time-shifted approach. A recent review by Jit and Mibei140 also noted that the UK Treasury currently recommends stepwise discounting to all public sector bodies, but at a very slowly declining rate (3.5% for the first 30 years, declining to 3.0% from year 31, with further declines from year 76). Although the use of discounting seeks to incorporate social preferences rather than to alleviate uncertainty, further consideration could be given to its application to regenerative medicines and cell therapies.
Another approach commonly employed to better characterise the uncertainties surrounding longer-term benefits and to inform the committee’s deliberations relates to the time horizon of the analysis and the methods of extrapolation. Within NICE’s current methods guide,122 alternative scenarios are requested to be routinely considered to compare the implications of different methods for the extrapolation of the results. Several other country-specific guidelines for cost-effectiveness analyses request the presentation of alternative scenarios based on different time horizons and comparing within-trial results with extrapolated results. 141–143
Alteration of the time horizon of the evaluation, away from the lifetime analysis recommended by NICE when appropriate, acts to reduce the uncertainty by excluding the impact of costs and outcomes that occur in the long term, when uncertainty is likely to be greatest. Although such analyses are potentially informative in terms of understanding how influential particular assumptions are over the period of extrapolation, restricting the horizon risks omitting important costs and outcomes related to a particular technology and simply shifts the risk associated with particular uncertainties from the health system to the manufacturer.
Similarly, separating the within-trial results from the extrapolated results (or considering alternative scenarios for extrapolation) has been argued to allow separation of the uncertainty associated with downstream consequences from other sources of uncertainty. Mortimer144 suggested that this approach could enable decision-makers to assign a weight to the results of the extrapolation to take account of various uncertainties. However, it was also acknowledged that such a comparison is not always explicitly made and that implicit comparisons were often problematic, as the relationship between the within-trial and extrapolation period may not be predictable. Issues of predictability may be a particular challenge for regenerative medicines and cell therapies. Although it is commonly argued that a within-trial analysis is conservative with respect to cost-effectiveness estimates, the author identified situations in which this may not be true, for example when long-term adverse effects offset any initial gains or when increased survival is associated with additional costs related to the disease and/or other unrelated diseases. Mortimer144 also highlighted that the relativity in results between within-trial analysis and the results of extrapolation was made even more problematic when uncertainty as a result of future technological change was introduced. Several key factors were highlighted as affecting the relativity of results between within-trial and extrapolated analysis, including the timing of potential technological advances, the proportion of patients who could benefit when the new technology becomes available and the effectiveness of the new technology.
Conclusions
The review has identified a number of common themes and potential challenges in relation to HTAs and assessments of cost-effectiveness for regenerative medicines and cell therapies. Some of the challenges identified do not appear to be unique to these types of therapies and are also faced by manufacturers of more conventional pharmaceuticals, biologics and devices. However, it seems likely that these challenges may be faced more routinely for regenerative medicines and cell therapies.
There is already provision within NICE’s methods guide to accommodate some of these aspects,122 although potential challenges may arise in ensuring that this is consistently applied between committees and understood by manufacturers. NICE will also need to consider whether further amendments to its processes and methods are required. Broader consideration will also need to be given to approaches that may extend beyond NICE’s existing remit, for example alternative funding approaches. Consequently, other bodies and manufacturers themselves may also have an important role in identifying more innovative approaches to seeking reimbursement that recognise the inherent uncertainties and lead to a more efficient sharing of associated risk.
Chapter 4 Exemplar technology appraisal of a regenerative medicine
Selection of exemplar
Following RMEG subgroup discussions and further input from the Cell Therapy Catapult, it was decided that undertaking an exemplar appraisal involving a real commercial product was not feasible for a number of reasons: there would be significant commercial sensitivities; products undergoing regulatory review would be candidates for a real appraisal; and using a product at an earlier stage in clinical development would not be helpful as the evidence base would be even less mature and therefore would not have the attributes of an ‘exemplar’ product. It was therefore proposed to undertake the evaluation of a hypothetical product.
As a result of both RMEG subgroup discussions and technical meeting discussions, the type of regenerative medicine chosen as the hypothetical product was CAR T-cell therapy specific to the antigen CD19 (cluster of differentiation 19). The chosen indication was relapsed or refractory ALL. This specific combination was selected based on the existence of relatively mature data sets in the context that none of the currently available CAR T-cell products is licensed.
About chimeric antigen receptor T-cell therapies
Chimeric antigen receptor T-cell therapies have been under development for around 20 years. The specific CAR T-cell therapies considered in this appraisal consist of autologous (i.e. the treated individual’s) T-cells that are genetically modified to redirect the target of the T-cell receptors. These receptors target specific proteins found on the surface of leukaemia cells, in this case the protein CD19, which is present on B-cell leukaemias as well as on healthy B cells, but which is not found on haematopoietic stem cells (which are situated in the bone marrow) or on other tissues. 145 The activated T-cells can then attack and destroy the leukaemia B cells. Persistence of a given CAR T-cell therapy within the body is linked to the properties of the T-cell from which the cells were derived as well as the immune environment into which they are infused. CAR T-cell therapies have already begun to evolve, with second-generation therapies currently being trialled in Phase II studies. Research efforts at developing future generations are focused on addressing the key challenges of T-cell target specificity, persistence and ability to exert the desired anti-tumour effects as well as identifying new target antigens. 146 CAR T-cell therapies have recently emerged as regenerative medicines with promising potential to treat haematological cancers. In July 2014 the FDA granted ‘breakthrough therapy’ status to the CAR T-cell therapy CTL019 (manufactured by Novartis, Basel, Switzerland) for the treatment of adult and paediatric relapsed or refractory ALL. 147
Although CAR T-cell therapies may offer relapsed or refractory B-cell acute lymphocytic (lymphoblastic) leukaemia (B-ALL) patients a ‘bridge’ to stem cell transplantation, or possibly even a cure for B-ALL, it is likely that patients will need to be monitored for some key adverse effects that are often reported. These include cytokine release syndrome (CRS) and B-cell aplasia (an absence of B cells). CRS occurs as a result of cytokines being released from the successfully targeted cancer cells and can result in various symptoms such as fever, headache, nausea and a rash. The severity of CRS appears to be proportional to the tumour burden. Although CRS is an adverse effect of CAR T-cell therapy, there may be a correlation between the development of CRS and response to therapy; patients who do not develop CRS may be less likely to benefit from CAR T-cells, whereas those who develop CRS often respond to the therapy. Although there may be some correlation between developing CRS and efficacy, there does not appear to be a strong correlation between the degree of CRS and response to therapy. 148
B-cell aplasia is an expected adverse effect of successful CAR T-cell therapies, which eliminate normal mature and precursor B cells. As long as CAR T-cells persist, B-cell aplasia continues (which provides what appears to be a highly accurate pharmacodynamic marker of CAR function). 148 B-cell aplasia is a manageable disorder; patients may be treated with intravenous immunoglobulin (IVIG), although this is an expensive treatment. Persistent B-cell aplasia could result in an increased risk of infection even with replacement therapy. 149
Overview of disease
B-cell acute lymphoblastic leukaemia is a subtype of ALL. ALL is a cancer that starts from the immature lymphocytes in the bone marrow and then invades the blood fairly quickly, spreading to other parts of the body including the lymph nodes, liver, spleen, central nervous system (brain and spinal cord) and testicles (in males). The term ‘acute’ means that the leukaemia can progress quickly and, if not treated, is probably fatal within a few months. The American Cancer Society has estimated that, including both children and adults, in 2015 there were about 6250 new cases of ALL (3100 in males and 3150 in females) and around 1450 deaths from ALL (800 in males and 650 in females). 150 The risk for developing ALL is highest in children aged < 5 years. Although most cases of ALL occur in children, most deaths from ALL (about four out of five) occur in adults. 150
UK statistics presents a similar picture. Statistics for the incidence of ALL in the UK (2009–11) are provided by Cancer Research UK,151 based on data sourced from the UK Office for National Statistics (ONS), Information Services Division Scotland, the Welsh Cancer Intelligence and Surveillence Unit and the Northern Ireland Cancer Registry. Across all ages in 2011 there were 654 new cases reported in the UK (377 in males and 277 in females), with crude incidence rates of 1.2 for males and 0.9 for females (per 100,000). Incidence is strongly related to age, but ALL is unusual as it does not follow the pattern of increasing incidence with age seen for most cancers; instead, the highest incidence rates are in children, teenagers and young adults. In the UK between 2009 and 2011, an average of 65% of cases were diagnosed in people aged < 25 years and only 6% of cases were diagnosed in those aged ≥ 75 years. Age-specific incidence rates are highest in infants aged 0–4 years and drop sharply through childhood, adolescence and young adulthood, reaching their lowest point at age 35–39 years and increasing slightly thereafter. Incidence rates are similar between males and females in all age groups except for those aged 15–19 years, when age-specific rates are significantly higher in males (male-to-female ratio of around 22 : 10). Averaged across all patients aged < 30 years the mean number of cases of ALL per year is 462.
There are subtypes of ALL based on the type of lymphocyte (B cell or T cell) and how mature these leukaemia cells are. 150 About 80–85% of ALL cases are B-ALL. There are several subtypes of B-ALL: early precursor B-ALL (early pre-B-ALL, also called pro-B-ALL); common ALL; pre-B-ALL; and mature B-ALL (also called Burkitt leukaemia). This last type is rare, accounting for only about 2–3% of childhood ALL; it is essentially the same as Burkitt lymphoma and is treated differently from most leukaemias. T-cell acute lymphocytic (lymphoblastic) leukaemia (T-ALL) constitutes about 15–20% of cases of ALL. This type of leukaemia affects males more than females and generally affects older children more than does B-ALL. It often causes an enlarged thymus (a small organ in front of the windpipe), which can sometimes cause breathing problems. It may also spread to the cerebrospinal fluid (the fluid that surrounds the brain and spinal cord) early in the course of the disease.
Based on an estimate of 82.5% of cases of ALL being B-ALL, there will be 540 cases of B-ALL in the UK per year, of which 381 will be in those aged < 30 years. Approximately 20% of these cases will be refractory to treatment or relapse,152 giving an estimate for young people with relapsed/refractory B-ALL in the UK of 76.
Overview of current practice
Although the management of ALL in adults and children is similar, the prognosis is different, with that in adults (aged > 30 years) being much poorer than that in the younger age group. Hence, they are generally considered as two distinct clinical groups.
With stepwise improvements in risk-adapted chemotherapy and supportive care over the past five decades, current overall cure rates of newly diagnosed ALL are approaching 90% in the developed world in children and around 50% in adults. 153 Treatment involves induction with combination chemotherapy for the attainment of CR (both clinical and haematological) followed by post-remission maintenance therapy with or without haematopoietic stem cell transplantation (HSCT) (which enhances relapse prevention, particularly in patients aged < 35 years). However, because of the morbidity and mortality risks associated with transplant, HSCT is usually reserved for high-risk patients.
US National Comprehensive Cancer Network guidelines for first-line treatment are based on risk stratification and age, as follows:154
-
Philadelphia chromosome-positive (Ph+) ALL (adolescents and young adults) – chemotherapy and tyrosine kinase inhibitor (TKI), followed by allogeneic stem cell transplantation (ASCT) if an appropriate donor is available; if transplantation is not feasible, continue multiagent chemotherapy and a TKI
-
Ph+ ALL (adults aged < 65 years) – chemotherapy and TKI; consider ASCT if an appropriate donor is available and the patient has a good performance status and no or limited comorbidities; if transplantation is not feasible, continue multiagent chemotherapy and a TKI
-
Ph+ ALL (adults aged ≥ 65 years or with substantial comorbidities) – TKI and corticosteroids or TKI and chemotherapy (evaluate end-organ reserve, end-organ dysfunction and performance status)
-
Philadelphia chromosome-negative (Ph–) ALL (adolescents and young adults) – paediatric-style multiagent chemotherapy
-
Ph– ALL (adults aged < 65 years) – multiagent chemotherapy
-
Ph– ALL (adults aged ≥ 65 years or with substantial comorbidities) – multiagent chemotherapy or corticosteroids (evaluate end-organ reserve and end-organ dysfunction).
However, little progress has been made in the treatment of relapsed ALL. Following initial induction and maintenance therapy most adults will relapse and long-term leukaemia-free survival is achieved in only 20–30% of cases; following relapse, response rates to further chemotherapy are low at around 20–30% and long-term OS rates of 3–24% have been reported. 155 From a UK study of 608 adult patients, OS at 5 years in newly diagnosed patients was 38% (95% CI 36% to 41%) but after relapse was only 7% (95% CI 4% to 9%). 156
Relapse is less common in paediatric ALL but accounts for the highest proportion of cancer deaths in children. 157 Studies of Nordic and Austrian data found that, of children with ALL, 25% had a first relapse, 8% had a second relapse158,159 and 2% had a third relapse. 159 Around 50% of relapsed ALL in children does not respond to salvage therapy and for these patients survival rates are < 10%. 159 In children, age and white blood cell count at primary diagnosis of ALL are the most important prognostic factors for relapse: age < 1 year or ≥ 10 years is associated with the worst prognosis. In addition, site of relapse and duration of first remission are the major criteria for the classification of patients after first relapse. 157
Therapy after relapsed ALL consists of re-induction chemotherapy followed by consolidation chemotherapy and/or HSCT. Time to relapse (length of first remission), site of relapse and ALL immunophenotype are established factors that are prognostic at first relapse and can be used to determine further treatment. 157 B-ALL has a better prognosis than T-ALL. Various regimens have been investigated and re-induction remission rates of 71–95% have been reported; the higher rates are generally associated with later first relapse. Patients who are refractory to re-induction therapy or who have a further relapse have a poor prognosis, with survival rates of < 10%. 159 Failure to achieve CR after late re-induction chemotherapy is associated with previous failures to achieve CR or short remission. The proportion of patients achieving CR has been shown to reduce with subsequent relapses: in a study of 225 patients with ALL (including 195 patients with B-ALL), the mean (standard error) CR rates were 83% (4%) for early first marrow relapse, 93% (3%) for late first marrow relapse, 44% (5%) for second marrow relapse and 27% (6%) for third marrow relapse. 152 Five-year DFS rates in second CR and third CR were 27% (4%) and 15% (7%), respectively. Although some therapies with curative intent are capable of inducing a second remission in patients refractory to previous therapy, these are often associated with high treatment-related morbidity, mortality and minimal survival. 160 Such patients are eligible for innovative therapies in Phase I or Phase II trials. Therapies for relapsed B-ALL that have been licensed by the EMA or the FDA are discussed in Review of licensed treatments for relapsed/refractory B-cell acute lymphoblastic leukaemia. In particular, clofarabine (Evoltra®; Genzyme Europe, Naarden, the Netherlands), a purine nucleoside anti-metabolite, (which affects DNA elongation, synthesis and repair), was granted EMA marketing authorisation in 2006 for use in children and young adults with a second or greater relapse (or refractory patients). The pivotal trial of clofarabine (n = 61 with second or greater relapse) reported an overall remission rate of 20% (12/61 patients), with 16% (10/61) of patients going on to receive HSCT. 161 Clofarabine had been studied only in single-arm trials and marketing authorisation was granted ‘under exceptional circumstances’.
As discussed in Chapter 3 (see Review of the use of surrogate end points as primary outcome measures in definitive effectiveness trials of new therapeutic agents), the use of CR as an outcome is not specific at predicting which patients might subsequently relapse. In recent years, evaluation of response to therapy in B-ALL patients has become more precise with the development of methods to detect MRD. Although the FDA has concluded that the evidence base to indicate that early MRD status is the strongest predictor of long-term EFS in ALL is unequivocal, there is some uncertainty surrounding how MRD correlates with long-term outcomes in relapsed populations. The use of MRD as a surrogate end point is discussed further in Chapter 3.
Haematopoietic stem cell transplantation
Allogeneic HSCT is a potentially curative treatment option and a number of studies have demonstrated improved outcomes compared with chemotherapy. However, there are difficulties in the interpretation of the findings of many such trials, with issues such as patient selection bias, the specific nature of the HSCT, the source of the donor cells and the adverse effects associated with various specific transplant therapies. 157
The risks associated with transplant include graft rejection, delayed immune reconstitution, graft versus host disease and vulnerability to infections. In addition, there is significant toxicity associated with the chemoradiotherapy conditioning required before transplant. 157 The adverse effects of HSCT are not limited to those occurring in the short term. One study investigated long-term survival and late deaths among 1458 ALL patients who were disease free 2 years after allogeneic HSCT; the median follow-up was around 80 months. 162 Of the 167 deaths, new cancers accounted for around 10% of the primary causes of death, with graft versus host disease accounting for 23% of deaths.
One study examined the impact of MRD status in 157 patients with ALL in morphological remission undergoing allogeneic HSCT following a myeloablative conditioning regimen (12 patients were post two or more relapses). 163 The 3-year OS for those who were MRD negative pre transplant was 68% and for those who were MRD positive was 40%, whereas the probabilities of relapse were 16% and 33% for the two groups, respectively. The trend towards increased relapse in those with MRD was seen in patients with B-ALL (HR 2.87) and in those with T-ALL (HR 7.07); the small number of T-ALL patients (n = 24) meant that it could not be determined if the effect was statistically significantly larger in T-ALL patients. Post transplant, among those with any sample positive for MRD, the risk of subsequent relapse was higher (HR 3.21) as was the risk for overall mortality (HR 2.54). Similar findings were reported in a study that included a slightly larger sample of those post second or later relapse (n = 18). 110 Based on these and other studies, there is the suggestion of benefits through MRD-directed therapies, although controlled trials are needed to define their value. 109
Immunotherapies are showing promise and are being investigated. These therapies are targeted at specific surface antigens expressed on the target cells: naked and unconjugated antibodies; immunoconjugates and immunotoxins; bi-specific T-cell-engaging (BiTE) therapy [Blincyto® (blinatumomab; Amgen, Thousand Oaks, CA, USA)]; and CAR T-cells. The last two target CD19. 155
Decision problem
The key aspects of the decision problem to be addressed were identified and agreed at a meeting between the academic group and a subgroup of the project advisory group (topic experts). These were based on discussion of the Phase I/II CAR T-cell trials in B-ALL,164–166 which had been identified by literature searches performed by Cell Therapy Catapult. The agreed components of the decision problem were as follows:
-
Intervention – CD19 CAR T-cell therapies.
-
Indication – patients with B-ALL who have relapsed (with no further planned curative chemotherapy or HSCT) or who are refractory to standard chemotherapy. As described in Overview of current practice, the treatment pathways and prognosis for patients aged < 30 years and > 30 years are very different. Consequently, the indication considered in this assessment has focused on those aged < 30 years.
-
Subgroups – sources of heterogeneity such as relapsed/refractory status, previous HSCT, CAR design, dose, conditioning chemotherapy, tumour burden at the time of therapy or age of the patients may be explored.
-
Comparators – best supportive care (e.g. salvage chemotherapy).
-
Efficacy outcomes – response criteria such as CR, partial response/remission (PR) and MRD; OS; progression and/or EFS; persistence of CAR T-cells; HRQoL; and rates of HSCT.
-
Adverse event outcomes – CRS, B-cell aplasia, febrile neutropenia and neurological effects.
Review of evidence of clinical effectiveness: chimeric antigen receptor T-cell therapies
Methods
Initially, studies of CD19 CAR T-cells in B-ALL were identified by staff at Cell Therapy Catapult (who are part of the project advisory group, being experts in cell-based regenerative medicines). PubMed was searched using the search terms ‘CD19’, ‘chimeric antigen receptor’, ‘CAR’ and ‘CD19 CAR’ with a cut-off date of 21 October 2014. The ClinicalTrials.gov trials registry and relevant published reviews of CAR T-cell therapies were also searched.
To identify any further relevant clinical trials we performed update searches in MEDLINE and EMBASE up to May 2015, using the same search terms as those previously described. One reviewer performed an initial screen of the abstracts and those deemed potentially relevant were then screened by a second reviewer. Google (Google Inc., Mountain View, CA, USA) searches to identify further data on already-identified trials were also undertaken. Our clinical advisor was also contacted regarding any relevant 2015 conferences where new data may have been presented. To further inform the study design details of the hypothetical data sets, the ClinicalTrials.gov trials registry was searched for ongoing trials that had commercial involvement: the focus was on trials designed with the likely aim of acquiring marketing authorisation.
Overview of studies
Three published papers were identified from the Cell Therapy Catapult searches. 164–166 No further studies were identified from the MEDLINE and EMBASE update searches. Two conference abstracts167,168 (relating to two of the published studies) and one conference video169 (relating to one of the published studies) with more up-to-date data were identified from the Google searches.
Of the planned or (other) ongoing CAR T-cell studies identified on ClinicalTrials.gov, seven had commercial involvement: three were Phase II trials, all with estimated enrolments of 67 patients; one was a Phase I/II trial with an estimated enrolment of 80 patients; and three were Phase I trials (Table 4). All were single-arm studies. Two of the Phase II trials were multicentre studies (i.e. they listed more than one centre for recruitment in the contacts and locations field) and three had a primary outcome that assessed response or remission; the time frames stated for these outcomes ranged from 9 weeks to 1 year. Only one trial reported the collection of longer-term survival data, with a stated time frame of 5 years for OS, EFS and RFS.
| ClinicalTrials.gov identifier, start date and estimated end date | Study name | Phase and design | Estimated enrolment | Primary outcome(s) and time frame | Secondary outcomes and time frame |
|---|---|---|---|---|---|
| NCT02228096, August 2014–November 2017 | Study of efficacy and safety of CTL019 in pediatric ALL patients (relapsed/refractory) | II; single arm, open label, multicentre | 67 | Overall response rate, which includes CR and CRi, as determined by assessments of peripheral blood, bone marrow and central nervous system symptoms, physical examination and cerebrospinal fluid (1 year) | Adverse events and laboratory abnormalities (type, frequency and severity) (1 year) |
| NCT02167360, June 2016–June 2017 | Study of efficacy and safety of CTL019 in adult ALL patients (relapsed/refractory) | II; single arm, open label, multicentre | 67 | Safety (1 year) | None reported |
| NCT02435849, April 2015–April 2021 | Determine efficacy and safety of CTL019 in paediatric patients with relapsed and refractory B-ALL | II; single arm, open label, multicentre | 67 | Overall remission rate, which includes CR and CRi, as determined by independent review committee assessment (6 months) | Percentage of patients who achieve CR or CRi at month 6 without stem cell transplant between CTL019 infusion and the month 6 response assessment; percentage of patients who achieve CR or CRi and proceed to stem cell transplant while in remission before the month 6 response assessment; duration of remission (60 months); percentage of patients who achieve CR or CRi with MRD-negative bone marrow (60 months); RFS (60 months); EFS (60 months); OS (60 months); in vivo cellular pharmcokinetic profile (levels, persistence, trafficking) of CTL019 cells (60 months); prevalence/incidence of immunogenicity to CTL019 (60 months) |
| NCT01840566, April 2013–April 2016 | High dose therapy and autologous stem cell transplantation followed by infusion of chimeric antigen receptor (CAR) modified T-cells directed against CD19+ B-cells for relapsed and refractory aggressive B cell non-Hodgkin lymphoma | I; single arm, open label | 18 | Maximum tolerated dose; safety (2 years) | 2-year PFS; OS (2 years) |
| NCT01430390, September 2011–September 2016 | In vitro expanded allogeneic Epstein–Barr virus specific cytotoxic T-lymphocytes (EBV-CTLs) genetically targeted to the CD19 antigen in B-cell malignancies | I; single arm, open label | 26 | Safety (3 years); persistence of escalating doses (3 years) | Assess the effects of the adoptively transferred CD19-specific T-cells on the progression of leukaemia (3 years); quantitate the number of CAR-positive T-cells in the blood at defined intervals post infusion to determine their survival and proliferation in the host (3 years); assess the long-term status of treated patients (15 years) |
| NCT01683279 (PLAT-01), December 2012–January 2016 | A pediatric trial of genetically modified autologous T cells directed against CD19 for relapsed CD19+ acute lymphoblastic leukemia | I; single arm, open label | 18 | Number of participants with adverse events (42 days) | Persistence of the CD19 CAR+ T-cells (42 days); determine whether there is anti-leukaemic activity of the CD19 CAR+ T-cells (42 days) |
| NCT02028455 (PLAT-02), January 2014–January 2017 | A pediatric and young adult trial of genetically modified T-cells directed against CD19 for relapsed/refractory CD19+ leukemia | I/II; single arm, open label | 80 | Safety (30 days); MRD-negative CR (63 days); releasable cell product generated (28 days) | Persistence of the CD19 CAR+ T-cells (63 days); number of participants with recrudescence or development of acute graft versus host disease (63 days); number of participants who have T-cells ablated with cetuximab (3 years) |
Efficacy results
Details of the three trials identified from the Cell Therapy Catapult searches are presented in Table 5. Two of the studies were Phase I trials164,166 and one was categorised as a Phase I/IIA trial165 (on ClinicalTrials.gov); safety was the primary outcome in all trials (one study also had maximum tolerated dose as a co-primary outcome164). All trials had recruited < 40 patients, although two were ongoing. 165,166
| Study details | Study (authors, year, product and sponsor) | |||||
|---|---|---|---|---|---|---|
| Lee et al., 2015,164 CD19 CAR T-cells, NIH funded | Maude et al., 2014,165 CTL019, Novartis | Grupp et al., 2014167 (abstract) | Grupp 2014169 (video) | Davila et al., 2014,166 19–28z CAR T-cells, Juno Therapeutics | Park et al., 2015168 (abstract) | |
| Design | ||||||
| ClinicalTrials.gov identifier | NCT01593696 | NCT01626495 (children, young adults), NCT01029366 (adults) | NCT01044069 | |||
| Primary outcome(s) | SAEs, maximum tolerated dose | SAEs | SAEs | |||
| Study design | Phase I feasibility/dose escalation | Phase I/IIA | Phase I | |||
| No. of centres | 1 | 2 (adult n = 1, children n = 1) | 1 | |||
| Planned duration of follow-up | 5 years (from ClinicalTrials.gov) | Unclear (but appears to be 1 year for most outcomes) | 2 years (from ClinicalTrials.gov) | |||
| Sample size | 21 | 30 | 30 | 39 | 16 | 33 (32 evaluable) |
| Duration of follow-up (to date) | Median 10 months | 6 months (median 7 months) | 6 months | 6 months | NR | Median 5.1 months |
| Population | ||||||
| Relapsed/refractory | 14/7 | 27/3 | NR | NR | All 16 relapsed | 14 patients had three or more previous lines of therapy |
| No. of previous relapses | One: n = 6 patients; two or more: n = 8 patients | One: n = 5 patients; two or more: n = 22 patients | NR | NR | Appears to be one | NR |
| Age | 14 children, 7 adults: range 5–27 years | 25 children: median 11 years (range 5–22 years); 5 adults: median 47 years (range 26–60 years) | All children or young adults: median age 10 years (range 5–22 years) | All children or young adults | Adults: median 50 years (range 18–60+ years) | Median 54 years (range 22–74 years) |
| No. of B-ALL patients | 20/21a | 29/30b | NR | NR | 16/16 | NR |
| MRD threshold | < 0.01% by flow cytometry | Unclear, but measured by flow cytometry | NR | NR | Unclear, but measured by flow cytometry | NR |
| MRD negative at baseline | 0 | 5/25 children | 5/25 | NR | 2/15 | NR |
| Previous HSCT | 8 | 18/25 children | NR | NR | 4/16 | 11 |
| Intervention | ||||||
| CAR T-cell dose (per kg) | Mostly 1 × 106 (15/21) or 3 × 106 (4/21) | 1–10 × 107 or 5–50 × 108 (if > 50 kg) | 107–108 | 3 × 106 | 1–3 × 106 | |
| Conditioning regimen | Fludarabine and cyclophosphamide | 15 patients had fludarabine and cyclophosphamide | NR | NR | Cyclophosphamide | NR |
| Type of dosing regimen | Single dose; three patients had second infusion | Given over 1–3 days | Split dose (days 1 and 2) | |||
| Efficacy outcomes | ||||||
| CR | Day 28: 14/21 (66.7%, 95% CI 43.0% to 85.4%); 70% (95% CI 45.7% to 88.1%) in 20 patients with B-ALL | 1 month: 27/30 (90%) | 27/30 (90%) | Day 28: 36/39 (92%); probability at 6 months 76% (95% CI 61% to 94%) | 10/16 (63%); time point unclear (median time to CR/CRi 24.5 days) | 29/32 (91%) |
| PR | NR | NR | NR | NR | NR | |
| MRD negative | Day 28: 12/20 (60%, 95% CI 36.1% to 80.9%) in 20 patients with B-ALL | 22/30 (at perhaps 1 month – time point unclear) | 23/30 (77%) | NR | 12/16 (75%); time point unclear | 23/28 MRD-evaluable patients (82%) |
| OS | Probability at 9.7 months: 51.6% (median 10 months) | Probability at 6 months: 78% (95% CI 65% to 95%) | Probability at 6 months: 78% (95% CI 63% to 95%) | NR | NR | 6 months: 58% (95% CI 36% to 74%) |
| PFS | 78.8% in the 12 patients achieving MRD-negative status (beginning at 4.8 months) | NR | NR | NR | NR | NR |
| EFS | NR | Probability at 6 months: 67% (95% CI 51% to 88%) | Probability at 6 months: 63% (95% CI 47% to 84%) | Probability at 6 months: 70% | NR | NR |
| Persistence of CAR T-cells | 18/21 had detectable CAR T-cells with peak expansion occurring around day 14. No CAR T-cells detected after day 68 in any patient | Probability at 6 months: 68% (95% CI 50% to 92%) | 6 months: 68% (95% CI 50% to 92%) | NR | Peak of CAR T-cells within 1–2 weeks, with decreases to low or undetectable levels by 2–3 months | NR |
| HSCT | 10 | 3 children withdrew from study following treatment with CTL019 to have HSCT | NR | 3 | 7 out of 10 eligible patients; of the remaining 6, 3 were contraindicated, 2 declined and 1 was being evaluated | 11 |
| Quality of life | NR | NR | NR | NR | NR | NR |
| Safety outcomes | ||||||
| CRS | Grade 3: 3; grade 4: 3 | All patients had CRS; mild to moderate in 22/30 patients, severe in 8/30 patients (needed ICU treatment); 9 patients received tocilizumab and all patients recovered fully | All responding patients developed grade 1–4 CRS | 7/16 had severe CRS (definition of severe provided); patients received steroids and/or tocilizumab | 7 had severe CRS | |
| B-cell aplasia | Prolonged B-cell aplasia did not occur | Occurred in all patients who had a response and persisted for up to 1 year after CTL019 cells were no longer detectable; these patients received immunoglobulin replacement | 6 months: 73% (95% CI 57% to 94%) | NR | NR | NR |
| Febrile neutropenia | Grade 3: 7 | 22/30 required hospitalisation | NR | NR | Grade 3: 11 | NR |
| Neurological effects | Hallucinations: 5; dysphasia: 1 | 13 had neurological effects; 6 had delayed encephalopathy | NR | NR | Grade 3 altered mental status: 5; grade 3 altered mental status: 1 | NR |
| Notes | ||||||
| IPD data available in published trial report | Age, sex, no. of relapses, percentage marrow blasts, previous treatment, CR, MRD, HSCT | Lymphodepleting chemotherapy, CR, severe CRS, persistence, B-cell aplasia | Age, salvage chemotherapy, MRD, HSCT, CRS | |||
| CR definition | < 5% marrow blasts, absence of circulating blasts and no extramedullary sites of disease with an absolute neutrophil count of ≥ 1000/µl and a platelet count of ≥ 100,000/µl | Morphological assessment of the bone marrow as M1 (< 5% leukaemic blasts) with no evidence of extramedullary disease | Restoration of normal haematopoiesis with a neutrophil count of > 1000 × 106/l, a platelet count of > 100,000 × 106/l and a haemoglobin level of > 10 g/dl. Blasts should be < 5% in a post-treatment bone marrow differential. No clinical evidence of leukaemia for a minimum of 4 weeks | |||
| Other notes | All toxicities fully reversible; 15 years’ follow-up for delayed adverse events (FDA guidance) | Reasons for not having HSCT after CTL019: lack of suitable donor, previous HSCT, family choice | CAR T-cells given as bridge to HSCT; 2 patients who achieved CR after CAR T-cells had a CR prior to CAR T-cell infusion | |||
As can be seen from Table 5, notable clinical heterogeneity was evident both within and across trials. One study was of children and adults165 and one studied children and young adults. 164 The remaining study included only adults166 and so ultimately was not of further use for the assessment. Most patients had had a previous relapse following remission; a small proportion of patients were refractory to previous treatments. In two studies the CAR T-cell treatments were mostly used to enable patients to receive HSCT (i.e. used as a bridging therapy). 164,166 The remaining study appeared to recruit a more difficult-to-treat population, with most patients having two or more relapses and previous HSCT; here, the treatment intention may possibly have been curative. 165 CAR T-cell design also varied, with either the CD28 or the 4-1BB co-stimulatory domains being used, a difference that might explain the more prolonged persistence of circulating CAR T-cells seen in one of the studies. 165 Persistence of CAR T-cell therapies in the body can result in benefit and risk, depending on the duration of persistence.
Two surrogate end points were reported in all three trials: CR and MRD. Rates of CR ranged from around 70% to 90%. As would be expected, the rates of achieving a status of MRD were lower, ranging from around 60% to 80%, although only one trial stated the MRD threshold used, which was 0.01% (i.e. one cancer cell in 10,000 normal cells). 164 All three trials reported OS data. In one trial the probability of OS was 52% at 9.7 months. 164 The other two trials reported probabilities of OS at 6 months, which were 58%168 and 78%, respectively. 165
Adverse events
The key adverse events noted in the trials were CRS, B-cell aplasia, febrile neutropenia and various neurological effects. In two studies most patients had mild to moderate CRS,164,165 although a greater incidence of severe CRS was evident in the trial in adults. 166 Affected patients were treated with steroids or tocilizumab. Two of the three studies reported the incidence of B-cell aplasia. In one study prolonged B-cell aplasia did not occur164 and in the other B-cell aplasia occurred in all patients who had a response and persisted for up to 1 year after CAR T-cells were no longer detectable. 165 Significant proportions of patients had febrile neutropenia or neurological adverse effects such as hallucinations or altered mental status (see Table 5).
Summary issues for the target product profile and hypothetical data sets
-
The B-ALL population is narrowly defined with extremely poor prognosis and limited alternative therapy options. This is likely to be typical of regenerative medicines.
-
Therapy potentially offers a ‘cure’.
-
There are potentially serious adverse effects of therapy.
-
Limited data are available (single-arm studies).
-
Appropriate comparator and control data need to be identified/generated.
Review of licensed treatments for relapsed/refractory B-cell acute lymphoblastic leukaemia
A pragmatic review of the other treatments for relapsed/refractory B-ALL that have been licensed by the EMA or the FDA was undertaken. This was carried out to further inform decisions to help construct the CAR T-cell therapy hypothetical data sets and to help to put them in context. Three treatments were quickly identified from the B-ALL literature and EMA/FDA websites: Evoltra (clofarabine), Blincyto (blinatumomab) and Marqibo® (vincristine sulphate liposome injection; Talon Therapeutics, San Francisco, CA, USA). This number of treatments was deemed sufficient for the purposes of this exercise.
Evoltra – known as Clolar® in the USA – was granted EMA marketing authorisation under exceptional circumstances in 2006. It is a purine nucleoside anti-metabolite (which affects DNA elongation, synthesis and repair). Blincyto and Marqibo were both licensed by the FDA under the accelerated approval programme (in 2014 and 2012, respectively); in this programme, drugs for serious conditions that fill an unmet medical need may be approved based on a surrogate end point. Blincyto, which has also been granted a ‘breakthrough therapy’ designation, is a monoclonal antibody designed to specifically attach to CD19 proteins on leukaemia cells. Marqibo provides targeted delivery of vinicristine, which involves encapsulation of vinicristine in nanoparticle liposomes.
Marqibo and Blincyto are licensed for use in adults and Evoltra for use in children and young adults. All three treatments have an orphan product designation, all claim to meet unmet medical need and the submissions for all were primarily based on data from Phase II single-arm trials. 170–173 However, whereas Marqibo and Evoltra are licensed for patients with a second or greater relapse (or refractory patients), the approval for Blincyto is broader, covering patients with Ph– relapsed or refractory B-cell precursor ALL; over half of the patients in the pivotal Blincyto study172 had had only one relapse. A consequence of these different populations can be seen in the pivotal trial sample sizes, with smaller study sample sizes for Marqibo (n = 65)173 and Evoltra (n = 61)170,171 and a larger sample size for Blincyto (n = 189). 172
Primary outcomes for all three trials were based on remission status (CR and/or overall remission). All studies also reported OS. For the second or greater relapse populations, treatment with Evoltra resulted in an overall remission rate of 20% (12/61 patients), with 16% (10/61) of patients going on to receive HSCT;170,171 treatment with Marqibo resulted in a CR rate of 15% (10/65 patients) (although the figure was 12% based on the FDA’s assessment), with 18% (12/65) receiving HSCT. 173 However, most of these patients did not achieve CR with Marqibo. As would be expected, a higher rate of CR was seen in the Blincyto trial (42%)172 than in the Marqibo173 and Evoltra170,171 trials, as most patients were at first relapse. Further results and other assessment details are presented in Appendix 5.
The EMA review of Evoltra stated that, given the efficacy seen early on in the clinical programme, studies using a placebo comparator were considered clinically unethical. 170 Active comparator studies were also not deemed to be appropriate as there were no other recognised therapeutic options available: ‘The indication is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive data on clinical efficacy and safety’ (p. 35). 170 Marketing authorisation was therefore granted ‘under exceptional circumstances’. 170 The All Wales Medicines Strategy Group (AWMSG) recommended Evoltra only if the intended use was as a bridge to HSCT (and recommended that it should not be used with palliative intent). 171 The FDA approval of Marqibo seemed less straightforward; committee members consistently stated that the proposed Phase III trial was critical in assessing the benefit of Marqibo. 173 Some members indicated that the trial should be completed before approval, whereas several indicated that accelerated approval may be appropriate but with the expectation that this approval would be withdrawn if the Phase III trial failed to confirm clinical benefit. The post-approval study was a multicentre Phase III randomised trial comparing standard vinicristine with Marqibo in older adults with newly diagnosed untreated Ph– ALL; the proposed sample size was 348.
For Blincyto, a confirmatory Phase III RCT was required to compare Blincyto with standard care chemotherapy in relapsed/refractory adults; this was ongoing at the time of submission to the FDA. 172 A 2 : 1 randomisation ratio was used, with more patients receiving blinatumomab. Around 400 patients were expected to be enrolled and the primary end point is OS.
Single-arm B-cell acute lymphoblastic leukaemia trials: identifying appropriate control data
As discussed in Chapter 3 (see Study biases: an overview of their importance and methods to quantify and adjust for their impact), although the results of single-arm trials can be compared with historical control data, the results of such comparisons can be considered as reliable indicators of treatment benefit only when the disease natural history is very well known, the patient population is homogeneous and the standard of care treatment has little impact on outcomes. For Evoltra, Blincyto and Marqibo, different approaches were used to devise a control data set.
For Blincyto a weighted analysis of patient-level data from 694 retrospective controls (1990–2014) was performed. 174 This study, which was of relapsed/refractory adults treated with standard of care therapy, utilised databases in several EU countries and the USA. The manufacturer of Blincyto (Amgen) also conducted a model-based meta-analysis of clinical study data to project the effect of Blincyto relative to existing therapies. For its ongoing trial in children, Amgen cited both a key paper on prognostic factors in B-ALL152 and the CR rates seen with Clolar (clofarabine), stating that the primary efficacy end point would be met if the CR + CRh (complete remission with incomplete haematological recovery) rate was at least 22.5% (suggesting efficacy similar to or greater than that for clofarabine). 172 For the Marqibo FDA submission, literature searches were performed to identify response rates in relevant patients. 173 The study reporting the best historical comparison data still had some key differences with regard to the Marqibo trial population, most notably in terms of line of treatment, eligibility for transplant and site of adjudication. 175 Comparisons were made with the closest matched subgroup of patients in the historical study: patients who received third-line single-agent treatment. 173 Clofarabine was assessed in 2006 and thus the aforementioned O’Brien study175 was not available. Instead, data were obtained from German and Dutch cancer registries; simple comparisons of median survival results were presented to the EMA. 170
Summary
-
Studies that form the basis of regulatory submissions for treatments for patients with second or greater relapsed/refractory B-ALL will be small (around 65 patients), Phase II, single-arm trials.
-
Primary end points will be surrogate end points such as CR.
-
Confirmatory randomised trials may be appropriate and viable in related larger populations for whom other treatment options exist.
-
For very small patient populations it is likely to be difficult to identify published prognostic studies that have suitable historical control data. Other strategies may therefore be needed, such as seeking access to national patient databases (in order to perform new studies).
Chapter 5 The target product profile and hypothetical data sets
Summary of issues to consider to inform the creation of the target product profiles and hypothetical data sets
The innovative nature of regenerative medicines, together with the indications that many of them will be expected to target initially (populations with high levels of unmet medical need), means that there is a collective desire to expedite their approval and appraisal. This ambition may run counter to the need for additional vigilance relating to robust evidence and long-term outcomes. Regulatory bodies must therefore endeavour to balance urgency of patient need with the requirement for robust evidence on efficacy and safety. This can be managed through a combination of regulatory approval based on limited – although promising – data, combined with post-approval requirements for continued data collection. From the perspective of NICE appraisals, this means that the evidence base available at the time of product approval may be highly uncertain; the cost of this uncertainty has to be a key part of the decision-making process.
The reviews have identified several broad issues relevant to uncertainty around the clinical evidence for the creation of the TPPs and hypothetical data sets for the exemplar.
-
It is not universally the case that regenerative medicines (or ATMPs) will be tested using NRS designs. Rather, submitted pivotal studies may well in fact be randomised, notably when levels of unmet need are low and diseases/conditions are not rare; in such cases, the maturity of data (which would be available at the time of a NICE appraisal) has been up to 5 years’ duration.
-
When single-arm trials, or case series, do form the basis of a regulatory submission, a key consideration when judging uncertainty should be the likelihood of cure or improvement without experimental treatment. However, it may be very difficult to identify published prognostic studies that have suitable historical control data. Other strategies for obtaining historical data may well be needed, such as seeking access to national patient databases.
-
When single-arm trial data are compared with historical data and appropriate methods to adjust for confounding are employed, the selection of the method used must be explicit and based on sound reasoning; despite advances in statistical techniques, clear challenges remain in generating accurate unbiased estimates of effect from non-randomised data.
-
Results from single-arm trials can be considered as reliable indicators of treatment benefit only when the disease natural history is very well known, the patient population is homogeneous and the control (standard of care) treatment has little impact on outcomes.
-
Although more mature evidence, such as confirmatory RCTs, may sometimes be viable in the specific population, it might also be expected only in larger, similar populations (e.g. B-ALL patients in first relapse). This raises the possibility of incorporating indirectly relevant but more reliable (and possibly more mature) data into the analysis, to reduce uncertainty.
-
The high technology status of regenerative medicines may imply greater potential for variation in response across both individuals and centres. This is likely to have implications in terms of the generalisability of efficacy and safety estimates obtained from small single-centre (probably expert centre), single-arm studies; in the absence of larger or more varied trials, this might be addressed only by access to IPD so that potential predictors of response or effect modifiers may be investigated.
-
Another key issue is that pivotal trials in regulatory submissions are likely to report primary end points that are surrogates for real clinical end points. On average, trials using surrogates report larger treatment effects than trials using final patient-relevant outcomes. This has implications for effect estimate uncertainty, especially when only surrogate end points are reported; the choice of surrogate outcomes used should be researched, explicit and justified. Nevertheless, to maximise the use of all available data, and to reduce overall uncertainty, multivariate meta-analysis methods to analyse data should be considered, whatever the maturity of the evidence base.
-
Related to the issue of surrogates as primary outcomes is that of duration of follow-up: use of intermediate shorter-term outcomes avoids the need for long follow-up. The consequence of this is that, even when OS data are recorded, these data are immature at the point of regulatory approval.
-
Regenerative medicines are by their nature innovative products and may be subject to continuing development, with new generations having improved efficacy. This may pose problems when evaluating long-term efficacy and safety, for example when determining to what extent the long-term safety data from a first-generation product can be used to inform the long-term safety of a related newly licensed second-generation product. This may mean that, as well as bioavailability-type studies, key trials conducted earlier in the development process may have to be replicated or adjustments may have to be made in the analyses of trial data to account for their indirectness.
For the specific purpose of deciding what to include in the exemplar hypothetical data sets, the best information to begin with comes from the published and ongoing trials for CAR T-cells, together with the EMA-/FDA-licensed non-regenerative medicines for relapsed B-ALL. These indicate that a minimum data set would consist of a small (around 65 patients), Phase II, single-arm trial, with a surrogate end point such as CR as the primary end point. MRD, the surrogate end point that is the strongest predictor of long-term EFS in ALL, is also likely to be reported, although there is considerable uncertainty about its value in relapsed populations.
Historical control data must be identified, which should reflect the treatment that B-ALL patients would receive in the absence of CAR T-cell therapies being available. This is necessary to utilise the hypothetical trial evidence within the economic analyses. A key challenge for constructing the historical control group would therefore be identifying the population included within the single-arm studies and selecting an appropriate control group: any selected control group is unlikely to exactly match the tiny population included in the single-arm studies and so comparisons would therefore be subject to confounding. To mitigate the effects of any such bias, a second challenge would be to identify and apply the most appropriate methods to adjust for confounding.
The small sample sizes available from the trials of CAR T-cell therapies in relapsed/refractory B-ALL imply that estimates of effect are likely to be inexact and imprecise and this should be considered when creating the more mature data sets.
More mature data sets would be expected to have larger (tending towards appropriately powered) sample sizes to reduce the width of the CI around any effect estimate. It should be noted that this would not influence the magnitude of any potential bias and may lead to increased confidence in an incorrect estimate of effect. Increasing the sample size may, however, also allow for a wider range of statistical methods to mitigate the effects of confounding and therefore have an indirect effect on reducing any bias in the effect estimate.
A RCT could be included in a more mature data set or the availability of data from a RCT in B-ALL patients in first relapse could be proposed. This latter possibility raises methodological questions of how the results of confirmatory RCTs in an indirect population might be used to re-evaluate the uncertainty of the direct evidence base.
Background to developing the target product profiles and evidence sets
Data from the three published trials for CAR T-cell therapies164–166 were discussed at a meeting of the project advisory group on 24 June 2015. Based on these discussions it was decided that, for the purposes of the exemplar, the population would comprise children and young adults who had experienced two or more relapses or who were refractory to treatment (with older adults excluded). It was further decided that the exemplar would explore both of the therapeutic goals of the CAR T-cell therapy, encompassing bridging and remission/curative intents.
Two TPPs were subsequently developed to be considered as part of the exemplar appraisal:
-
CAR T-cell therapy used as a bridge to HSCT, in which the primary goal of treatment is to induce short-term remission of disease to maximise the opportunity for successful HSCT
-
CAR T-cell therapy used with curative intent, in which the primary goal of CAR T-cell treatment is long-term remission/cure of disease (with or without HSCT).
These two approaches to treatment with CAR T-cell therapy imply two potentially different contexts in which therapy may be appraised. Consequently, there are separate implications arising from the different applications, which require their consideration as two distinct scenarios.
In the bridge to HSCT scenario, the survival benefits of treatment are determined primarily by the subsequent receipt of HSCT and the associated benefits that stem from this. As such, the health benefits of CAR T-cell therapy are closely linked to the HSCT status of the cohort in the immediate period following CAR T-cell therapy. From a regulatory and reimbursement standpoint, the primary determinant of treatment efficacy is likely to include short-term end points such as remission and, potentially, MRD status. These data may also be supported by data on the outcomes of HSCT after CAR T-cell therapy. Marketing approval may therefore be achieved through demonstrating clinical benefit in terms of remission, MRD status (potentially) and subsequent rates of HSCT.
In the curative intent scenario, the survival benefit of treatment is considered to be as a direct result of CAR T-cell therapy itself. In this context, there is no separate surrogate treatment or process (i.e. HSCT) that determines the long-term benefits of therapy. From a regulatory standpoint, the primary determinant of the efficacy of treatment in this scenario is likely to include longer-term clinical end points such as EFS and OS, and increased levels of data maturity may be required.
New technologies are submitted to licensing agencies to seek regulatory approval and are subject to NICE appraisal at various stages of development of the supporting evidence base. To explore the impact of different levels of precision and maturity in the evidence base, three hypothetical data sets were constructed for each TPP:
-
the minimum set – the minimum data considered potentially sufficient for CAR T-cell therapy to be granted conditional regulatory approval
-
the intermediate set – a variant of the minimum set in which the efficacy and safety of CAR T-cell therapy have been assessed over a longer follow-up period
-
the mature set – a variant of the intermediate set in which the efficacy and safety of CAR T-cell therapy have been assessed in a larger clinical study but with a similar follow-up period as in the intermediate set.
In developing the TPPs, it was not our intention to directly compare the separate scenarios or to use these to infer differences between the alternative CAR T-cell therapies currently being developed. Neither were the different evidence sets intended to be prescriptive regarding the sufficiency of evidence for the purposes of regulatory or reimbursement processes. Instead, the hypothetical TPPs were developed to provide an exploration of potential issues and challenges associated with varying levels of precision and maturity in the underlying evidence base and the potential impact that these might have on subsequent assessments of cost-effectiveness and associated decision uncertainty.
In total, six evidence sets were developed spanning the separate TPPs (three sets for bridge to HSCT and three sets for curative intent). Each of the six evidence sets included hypothetical efficacy and safety data for CAR T-cell therapy and for a historical control. The efficacy and safety estimates in the bridge to HSCT and curative intent TPPs were derived from data from Lee et al. 164 and Maude et al. ,165 respectively, reflecting the clinical heterogeneity and the potentially different treatment intentions reported in Chapter 4 (see Review of evidence of clinical effectiveness: chimeric antigen receptor T-cell therapies).
Defining a historical control
The lack of control data within existing CAR T-cell studies necessitated the selection of a historical control from existing published literature to inform the TPPs and economic model. As discussed in Chapter 3 (see Study biases: an overview of their importance and methods to quantify and adjust for their impact), the use of a historical control introduces potential bias as observed or unobserved confounders other than the treatments may impact the outcomes of interest. As such, a direct comparison of the CAR T-cell results and historical control data may be subject to bias.
Observable sources of confounding can be potentially adjusted for in a number of ways, either at a single-study level or through the synthesis of evidence from a number of studies (as discussed in Appendix 2). A key source of potential observable confounding relates to differences in patient characteristics, which are known to be related to subsequent prognosis. To identify prognostic factors that might provide a basis for adjusting a historical control to account for potential prognostic imbalance, a pragmatic search was conducted using Google and Google Scholar to identify previously published multivariate prognostic models for patients with ALL.
The search identified 12 potentially relevant studies. However, five of these did not report sufficient detail on the results to be considered further. 159,160,175–177 A summary of the patient characteristics in the remaining seven studies is provided in Appendix 6. None of the prognostic models specifically focused on the population of interest. In addition, there appeared to be little consistency across the prognostic factors selected for inclusion in the multivariate analyses, with no single factor considered across all models and only a few of which could be applied to the patient characteristics reported within existing CAR T-cell studies. Hence, although formal adjustment for potential bias is desirable, the lack of access to IPD meant that this was not considered feasible within the exemplar.
A separate pragmatic search was subsequently conducted using Google and Google Scholar to identify possible historical control studies that might be more generalisable to the population of interest (i.e. based on age and previous history of relapse) and which might minimise the potential for bias in the absence of a formal adjustment for confounding. This search identified two studies considered to be potentially generalisable to the population considered within the exemplar evaluation. 160,178 Jeha et al. 178 reported on a Phase II, open-label study of clofarabine in paediatric patients with refractory or relapsed ALL. Von Stackelberg et al. 160 conducted a retrospective analysis of outcomes in children and adolescents with ALL who had not responded to salvage therapy and evaluated the OS of the patient population given different treatment modalities (curative, palliative and no therapy).
For consistency across TPPs and evidence sets, the same historical control intervention and informative data were implemented in both scenarios. Clofarabine and the study by Jeha et al. 178 were subsequently selected to act as the control treatment and source of historical control data for the following reasons:
-
Clofarabine is considered a standard of care chemotherapy for B-cell relapsed refractory ALL, alongside other chemotherapies such as fludarabine, cytarabine, granulocyte colony-stimulating factor and idarubicin (Zavedos, Pfizer, New York City, NY, USA) (FLAG-IDA).
-
It is the only EMA-licensed treatment available for ALL in paediatric patients who have relapsed or who are refractory after receiving at least two prior regimens and for whom there is no other treatment option anticipated to result in a durable response.
-
Although clofarabine has not been appraised by NICE for this indication, clofarabine is currently funded through the Cancer Drugs Fund (CDF). At the time of writing, clofarabine was the only treatment for relapsed and refractory B-ALL approved by the CDF.
-
The Phase II study population was also considered to be broadly consistent with populations enrolled in the studies by Maude et al. 165 and Lee et al. 164
Developing the evidence sets and target product profiles
The first step in developing the hypothetical evidence was to define the sample size and maturity of the evidence (i.e. follow-up) for the minimum, intermediate and mature evidence sets. The second step involved generating the efficacy and safety data conditional on the sample and maturity levels specified for each of the evidence sets. This involved the synthesis of data reported in the existing CAR T-cell historical control studies and simulation modelling.
Defining the sample size and maturity of evidence in the evidence sets studies
Current evidence on the efficacy of CAR T-cell therapies is limited to early Phase I/II studies with patient numbers of between 16 and 30. 164,165,179 It is anticipated that larger clinical studies will be needed to meet the minimum requirements for positive regulatory approval. This evidence is expected to come from a series of Phase II/III studies. Several previous pharmaceutical treatments for ALL have been granted regulatory approval based on efficacy and safety data from single-arm Phase II studies with sample sizes ranging from 61 to 189. This appears to be reflected in the design of planned and ongoing CAR T-cell studies in ALL. According to the ClinicalTrials.gov trials registry, there are currently three registered Phase II trials investigating the efficacy and safety of CAR T-cell therapies. The planned sample size of these trials is 67 patients (see Table 4). There is also one Phase I/II trial with an estimated enrolment of 80 patients.
The expected minimum data requirement for regulatory approval of CAR T-cell therapy was therefore set in the region of 60–80 patients. This sample size was used in both the minimum and the intermediate evidence sets. The mature evidence set was assumed to be based on trial evidence derived from a larger sample of patients than in the minimum and intermediate evidence sets. This evidence set was designed to reflect a scenario in which the evidence base for CAR T-cell therapy could include data from a more conventional RCT (or alternatively a larger uncontrolled study) with sufficient duration of follow-up to determine the longer-term efficacy for key clinical end points including OS. In practice, the sample size and maturity for such a study would be determined by a number of factors, including conventional statistical power calculations, likely accrual rates, the competitive landscape and overall study costs. In the time available, it was not feasible to formally consider these elements in estimating the anticipated sample size for this study. Instead, the sample size was based on the planned enrolment size of an ongoing Phase III trial of blinatumomab in adult ALL, identified from previous hand-searching of ClinicalTrials.gov. 180 In this study, the planned enrolment was for 400 patients to be randomised to blinatumomab or standard of care at a ratio of 2 : 1. As such, of the total 400 randomised patients, 133 would be randomised to the control arm and 267 would be randomised to blinatumomab. For the mature set, the study sample size was therefore set in the region of 120–140 patients per treated group (240–280 in total).
In using this specific sample size for the mature evidence set, we recognise that there are differences between the patients recruited into the blinatumomab trial and the specific population being considered here both in terms of age and previous history of relapse. However, for the purposes of a hypothetical exemplar this was considered to provide a reasonable basis for investigating the potential impact of increased precision.
The intermediate and mature evidence sets were also assumed to be based on trial evidence, with longer efficacy follow-up durations than the minimum set. For the minimum set, trial follow-up was based on a similar duration to that reported within existing CAR T-cell studies, with a median follow-up of approximately 10 months. For the intermediate and mature sets, trial follow-up was based on the maximum planned study duration for all Phase II CAR T-cell trials registered on the ClinicalTrials.gov trials registry. Across these studies, the longest planned follow-up period was 5 years.
A summary of the targeted sample size and level of evidence maturity considered across each of the evidence sets is provided in Table 6.
| Variable | Minimum data set | Intermediate data set | Mature data set |
|---|---|---|---|
| Sample size | 60–80 | 60–80 | 120–140 |
| Study follow-up (months) | 10 (median) | 60 (maximum) | 60 (maximum) |
Estimating the efficacy of chimeric antigen receptor T-cell and comparator treatments in the evidence sets
For all dichotomous outcomes, including response, remission and use of HSCT, parameter estimates were extracted directly from the existing CAR T-cell and clofarabine publications. The effect of increased sample size on the variance parameter for each dichotomous outcome was modelled using a beta distribution. As these outcomes tend to be measured during the first few months of a study, it is expected that longer follow-up would not directly impact these parameter estimates.
For the OS end point, parameter estimates were derived by digitising the Kaplan–Meier (KM) curves reported in the main study publications and using the algorithm by Guyot et al. 72 to impute the patient-level time-to-event and event-type (censored or event) data. These data were then analysed using conventional semiparametric survival modelling techniques using the statistical programming platform R (version 3.0.2; The R Foundation for Statistical Computing, Vienna, Austria). 181 This included assessments of landmark survival probabilities at 6, 12 and 60 months and derivation of the HR for CAR T-cell therapy compared with standard of care therapies and restricted mean survival times (i.e. in the event of non-proportional hazards, formally tested using the Schoenfeld residual test).
Using the studies by Maude et al. 165 and Lee et al. ,164 it was possible to generate samples of between 21 and 30 patients treated with CAR T-cell therapy. However, the expected minimum sample size for regulatory approval may be in the region of 60–80 patients. Therefore, it was necessary to increase the size of the imputed data sets from between 21 and 30 to between 60 and 80. This was achieved by replicating each of the imputed data sets until the total sample size was between 60 and 80 patients for the minimum/intermediate evidence sets and between 120 and 140 patients for the mature evidence set. By creating the pooled sets in this way, the mean survival probabilities and KM plots for OS remained consistent across evidence sets, whereas the variance around those estimates was allowed to vary in line with the sample size.
For the mature and intermediate data sets it was also necessary to simulate an increase in the duration of study follow-up to account for a scenario in which CAR T-cell studies had a longer follow-up duration, up to a maximum of 5-years. This adjustment was made by adding survival time to the imputed patient records, which were censored after the last recorded event in each study. These patients were subsequently assumed to be re-censored at their new survival time. The same approach was applied in both the CAR T-cell and the clofarabine arms. By extending the study time of patients who were censored after the last recorded event, the following assumptions were made:
-
patients who were alive and censored at the end of the studies were likely to be ‘cured’ of ALL, such that they would also be alive and censored at the end of the longer follow-up period
-
patients who were censored prior to the last event were assumed to have been lost to follow-up, such that additional trial follow-up would not lead to further information on the timing of death (or re-censoring) in those individual patients.
This approach ensured that the KM curves remained consistent across the evidence sets. The net result of this adjustment is that the more mature evidence sets contain more information on the long-term survival of those alive and censored at the end of the current studies. An illustration of this approach is provided in Figure 1.
FIGURE 1.
Illustration of adjustment to the data sets to account for additional study maturity.

It is important to highlight that, in practice, additional follow-up time points in trials would be likely to result in changes to the KM curves and estimates of survival benefit. To predict these changes would require access to IPD to elicit the characteristics of patients who are censored at shorter follow-up times and to then predict the unobserved event time for the censored patients, conditional on their characteristics. With the imputed data it is not possible to identify the characteristics of those who are censored and there are too few data to develop a prediction equation for the unobserved event times.
Finalised target product profiles
Bridge to haematopoietic stem cell transplantation target product profiles
Data on the evidence sets assumed for the clinical efficacy of CAR T-cell therapy as a bridge to HSCT are reported in Table 7 (OS) and Table 8 (dichotomous end points, adverse events). The associated KM plots are reported in Figure 2.
| End point | Minimum data set | Intermediate data set | Mature data set | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean estimate (uncertainty) (95% CI) | Comparative efficacy (95% CI) | Mean estimate and uncertainty (95% CI) | Comparative efficacy (95% CI) | Mean estimate (uncertainty) (95% CI) | Comparative efficacy (95% CI) | ||||
| CAR T-cell therapy | Standard of care | CAR T-cell therapy | Standard of care | CAR T-cell therapy | Standard of care | ||||
| Sample size | 63 | 61 | 124 | 63 | 61 | 124 | 126 | 122 | 248 |
| Median time to censoring (follow-up; OS end point) (months) | – | – | 11.3 (9.9 to14.1) | – | – | 53.6 (14.1 to 54.2) | – | – | 53.6 (52.9 to 54.2) |
| OS | |||||||||
| Landmark survival probability at 6 months (%) | 66.8 (52.4 to 77.8) | 32.0 (20.6 to 43.9) | – | 66.8 (52.4 to 77.8) | 32.0 (20.6 to 43.9) | – | 66.8 (57.0 to 74.9) | 32.0 (23.8 to 40.4) | – |
| Landmark survival probability at 12 months (%) | 51.6 (36.1 to 65.0) | 20.7 (11.4 to 32.1) | – | 51.6 (36.1 to 65.0) | 20.7 (11.4 to 32.1) | – | 51.6 (40.8 to 61.3) | 20.7 (13.8 to 28.7) | – |
| Landmark survival probability at 60 months (%) | NA | NA | NA | 51.6 (36.1 to 65.0) | 20.7 (11.4 to 32.1) | – | 51.6 (40.8 to 61.3) | 20.7 (13.8 to 28.7) | – |
| HR (95% CI) | – | – | 0.331 (0.203 to 0.539) | – | – | 0.309 (0.190 to 0.503) | – | – | 0.307 (0.218 to 0.434) |
| Test for proportionality (p-value < 0.05 indicates non-proportional hazards) | – | – | 0.101 | – | – | 0.0368 | – | – | 0.0211 |
| Restricted mean survival time analysis (months) | 11.37 | 5.98 | 5.388 (3.175 to 7.601) | 33.02 | 10.96 | 22.06 (12.868 to 31.254) | 33.02 | 10.96 | 22.06 (15.56 to 28.57) |
| End point | Minimum and intermediate data sets | Mature data set | ||||
|---|---|---|---|---|---|---|
| Mean estimate (uncertainty) (95% CI) (%) | OR (95% CI) | Mean estimate (uncertainty) (95% CI) (%) | Comparative efficacy (95% CI) | |||
| CAR T-cell therapy | Standard of care | CAR T-cell therapy | Standard of care | |||
| Sample size | 63 | 61 | 124 | 126 | 122 | 248 |
| CR | 64.9 (52.6 to 76.5) | 12.5 (6.1 to 21.5) | 15.6 (5.5 to 34.1) | 65.7 (57.7 to 73.6) | 12.0 (6.9 to 18.1) | 15.4 (7.5 to 28.9) |
| MRD negative | 54.7 (42.8 to 67.7) | 2.7 (0.4 to 7.5) | 88.5 (14.3 to 394.2) | 56.0 (47.6 to 64.7) | 2.3 (0.5 to 5.4) | 82.7 (21.2 to 243.9) |
| Probability of HSCT | 47.3 (35.6 to 59.6) | 16.4 (8.5 to 26.6) | 5.2 (2.2 to 11.2) | 47.5 (39.1 to 56.8) | 15.6 (10.1 to 22.3) | 5.2 (2.9 to 9.3) |
| CRS | 28.9 (19.0 to 40.7) | 1.5 (0.0 to 5.3) | – | 28.2 (20.8 to 36.1) | 0.9 (0.0 to 3.3) | – |
| Encephalopathy | 6.3 (2.0 to 13.3) | 1.6 (0.0 to 5.8) | 5.7 (2.4 to 10.7) | 0.8 (0.0 to 2.9) | ||
| Hypotension | 22.7 (13.7 to 34.2) | 19.2 (10.7 to 29.6) | 22.5 (15.8 to 30.4) | 18.6 (12.4 to 25.8) | ||
| Febrile neutropenia | 33.8 (22.8 to 45.0) | 48.6 (36.7 to 61.0) | 33.2 (24.8 to 41.6) | 49.1 (40.5 to 58.2) | ||
| Neutropenia (neutrophil count decreased) | 87.7 (78.8 to 94.6) | 16.2 (8.0 to 25.9) | 88.4 (82.2 to 93.2) | 15.5 (9.8 to 22.1) | ||
| Anaemia | 67.4 (55.9 to 78.0) | 1.6 (0.0 to 5.8) | 67.8 (59.8 to 75.4) | 0.8 (0.0 to 2.9) | ||
| Thrombocytopenia (platelet count decreased) | 52.9 (41.2 to 64.7) | 1.6 (0.1 to 5.7) | 52.7 (44.3 to 61.7) | 0.8 (0.0 to 2.9) | ||
| Leukopenia (white cell count decreased) | 88.0 (79.0 to 94.5) | 1.6 (0.0 to 6.2) | 88.4 (82.0 to 93.4) | 0.8 (0.0 to 2.7) | ||
| Hypokalaemia | 46.9 (34.6 to 58.9) | 1.8 (0.0 to 6.1) | 47.3 (39.3 to 55.5) | 0.8 (0.0 to 3.0) | ||
| Hypophosphataemia | 42. (30.6 to 54.1) | 1.6 (0.0 to 5.9) | 42.1 (33.7 to 50.7) | 0.8 (0.0 to 3.0) | ||
FIGURE 2.
Kaplan–Meier plots of time from randomisation to death for CAR T-cell and standard of care therapies in the different scenarios: (a) Bridge to HSCT – current trial follow-up; (b) curative intent – current trial follow-up; (c) bridge to HSCT – mature trial follow-up; and (d) curative intent – mature trial follow-up.


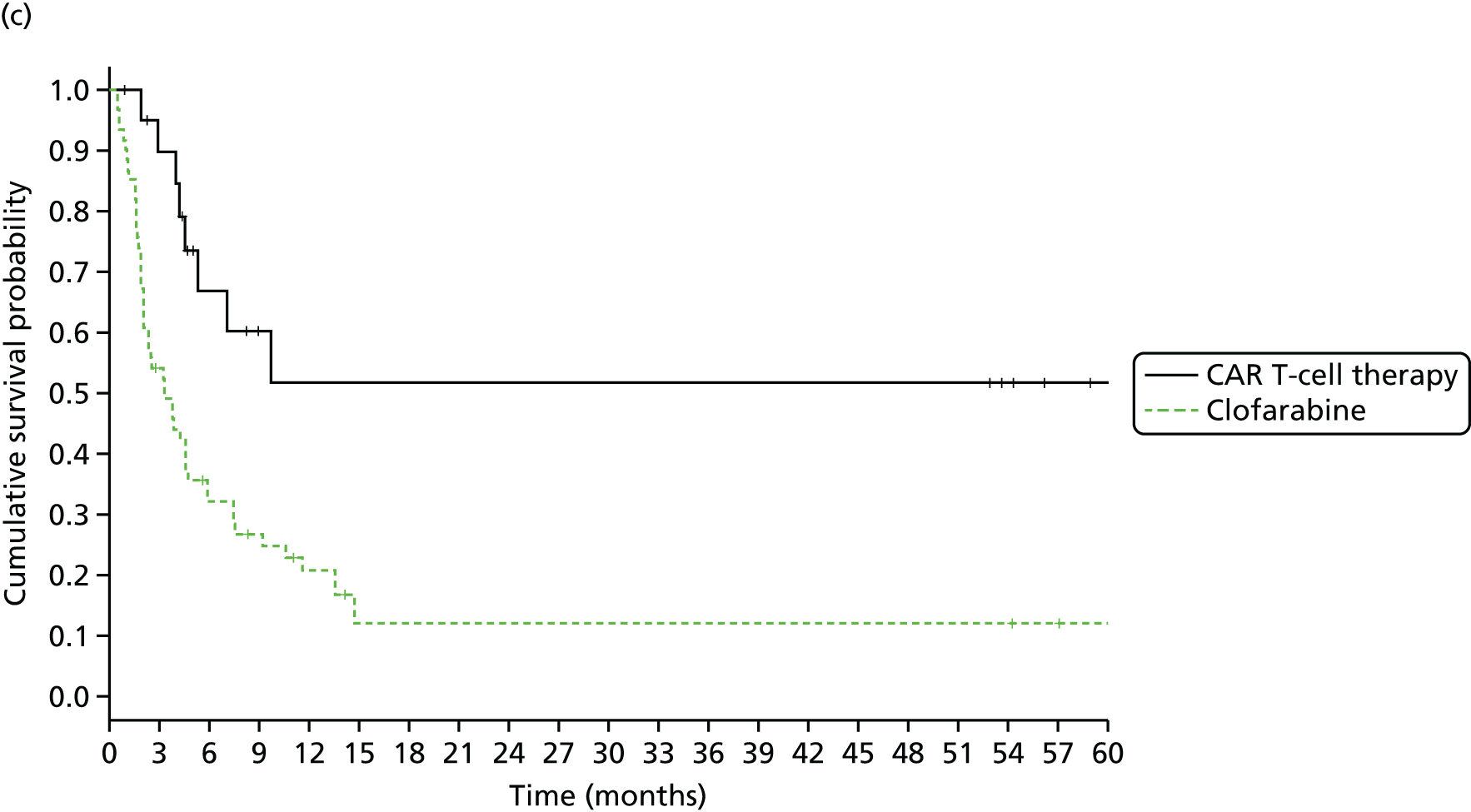
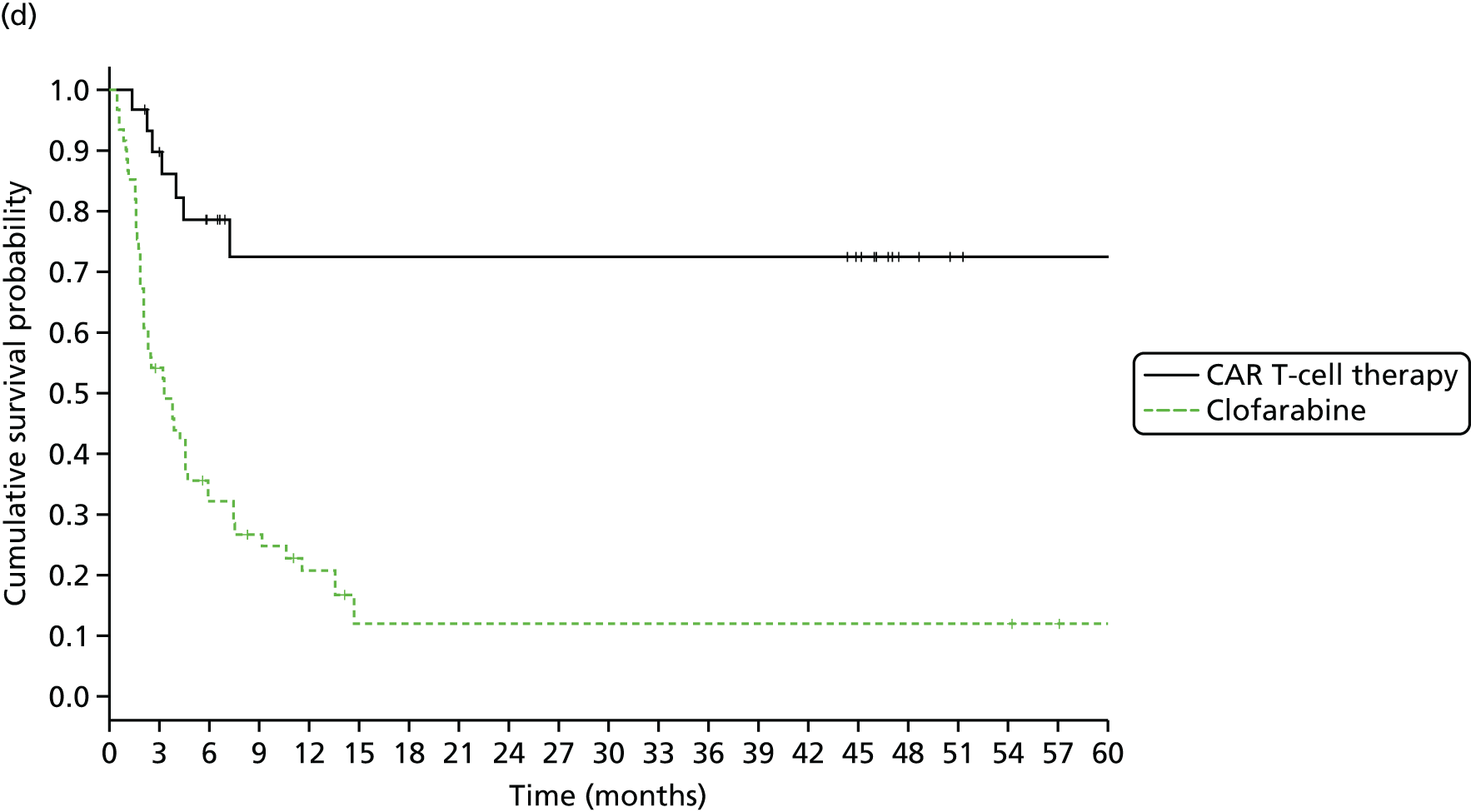
Minimum data set
In terms of OS, CAR T-cell therapy was assumed to be associated with improved probabilities of survival at months 6 (66.8%) and 12 (51.6%) compared with standard of care therapy (32.0% at 6 months and 20.7% at 12 months). Treatment with CAR T-cell therapy was associated with a statistically significant improvement in the time to death, with a HR of 0.33 (95% CI 0.203 to 0.539).
In a restricted mean survival time analysis, treatment with CAR T-cell therapy was associated with a mean extension to life expectancy of 5.39 months (95% CI 3.18 to 7.60 months) compared with standard of care therapy. The median follow-up in the minimum set was 11.3 months.
Intermediate data set
Given the consistency in the assumptions and the KM data assumed across the evidence sets, similar results were observed in the intermediate set as were reported in the minimum set. However, evidence was now also assumed to be reported on the survival benefits up to 5 years, with assumed 5-year landmark survival probabilities of 51.6% and 20.7% for CAR T-cell and standard of care therapy, respectively. Treatment with CAR T-cell therapy was associated with a statistically significant improvement in the time to death, with a HR of 0.31 (95% CI 0.19 to 0.50).
With increased data maturity compared with the minimum set, there was a greater trend towards non-proportional hazards. In the restricted mean survival time analysis, treatment with CAR T-cell therapy was associated with a mean improvement in life expectancy of 22.06 months (95% CI 12.87 to 31.25 months) compared with standard of care therapy. The median follow-up in the intermediate data set was 53.6 months.
Mature data set
With increased precision and data maturity compared with the minimum set, treatment with CAR T-cell therapy was associated with a statistically significant improvement in the time to death, with a HR of 0.31 (95% CI 0.22 to 0.43). In the restricted mean survival time analysis, treatment with CAR T-cell therapy was associated with a mean improvement in life expectancy of 22.06 months (95% CI 15.56 to 28.57 months) compared with standard of care therapy. The increased sample size is reflected in a more precise estimate of the mean life expectancy in this evidence set compared with the intermediate set, evidenced by the tighter CIs reported. The median follow-up in the mature data set was 53.6 months.
Curative intent target product profiles
Data on the evidence sets assumed for the clinical efficacy of CAR T-cell therapy as a curative treatment option are reported in Table 9 (OS) and Table 10 (dichotomous end points, adverse events). The associated KM plots are reported in Figure 2.
| End point | Minimum data set | Intermediate data set | Mature data set | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean estimate (uncertainty) (95% CI) | Comparative efficacy (95% CI) | Mean estimate (uncertainty) (95% CI) | Comparative efficacy (95% CI) | Mean estimate (uncertainty) (95% CI) | Comparative efficacy (95% CI) | ||||
| CAR T-cell therapy | Standard of care | CAR T-cell therapy | Standard of care | CAR T-cell therapy | Standard of care | ||||
| Sample size | 60 | 61 | 121 | 60 | 61 | 121 | 120 | 122 | 242 |
| Median time to censoring (follow-up; OS end point) (months) | – | – | 10.03 (8.33 to 11.41) | – | – | 45.2 (14.1 to 46.8) | – | – | 45.2 (44.3 to 46.0) |
| OS | |||||||||
| Landmark survival probability at 6 months (%) | 78.5 (65.3 to 87.2) | 32.0 (20.6 to 43.9) | – | 78.5 (65.3 to 87.2) | 32.0 (20.6 to 43.9) | – | 78.5 (69.7 to 85.1) | 32.0 (23.8 to 40.4) | – |
| Landmark survival probability at 12 months (%) | 72.5 (57.3 to 83.1) | 20.7 (11.4 to 32.1) | – | 72.5 (57.3 to 83.1) | 20.7 (11.4 to 32.1) | – | 72.5 (62.2 to 80.4) | 20.7 (13.8 to 28.7) | – |
| Landmark survival probability at 60 months (%) | NA | NA | NA | 72.5 (57.3 to 83.1) | 20.7 (11.4 to 32.1) | – | 72.5 (62.2 to 80.4) | 20.7 (13.8 to 28.7) | – |
| HR (95% CI) | – | – | 0.204 (0.113 to 0.370) | – | – | 0.180 (0.099 to 0.327) | – | – | 0.179 (0.117 to 0.272) |
| Test for proportionality (p-value < 0.05 indicates non-proportional hazards) | – | – | 0.699 | – | – | 0.784 | – | – | 0.678 |
| Restricted mean survival time analysis (months) | 17.04 | 6.57 | 10.47 (7.59 to 13.34) | 43.86 | 10.96 | 32.94 (24.38 to 41.43) | 10.96 | 32.94 (26.87 to 38.93) | 43.86 |
| End point | Minimum and intermediate data sets | Mature data set | ||||
|---|---|---|---|---|---|---|
| Mean estimate (uncertainty) (95% CI) (%) | OR (95% CI) | Mean estimate (uncertainty) (95% CI) (%) | Comparative efficacy (95% CI) | |||
| CAR T-cell therapy | Standard of care | CAR T-cell therapy | Standard of care | |||
| Sample size | 60 | 61 | 121 | 120 | 122 | 242 |
| CR | 90.0 (81.3 to 96.2) | 11.5 (4.7 to 20.6) | 97.25 (25.9 to 284.0) | 90.0 (84.0 to 94.7) | 11.5 (6.5 to 17.8) | 81.42 (33.1 to 177.1) |
| MRD negative | 73.4 (61.5 to 83.7) | 1.6 (0.0 to 6.0) | 1719.0 (39.26 to 7169.0) | 73.4 (65.2, 80.8) | 1.6 (0.0, 4.5) | 344.4 (54.42 to 1462.0) |
| Probability of HSCT | 10.0 (3.8 to 18.7) | 14.8 (7.1 to 24.7) | 0.738 (0.193 to 1.913) | 10.0 (5.3 to 16.0) | 14.8 (9.0 to 21.6) | 0.686 (0.283 to 1.379) |
| CRS | 27.0 (16.6 to 38.9) | 1.5 (0.0 to 5.3) | – | 27.0 (19.5 to 35.3) | 0.9 (0.0 to 3.3) | – |
| Encephalopathy | 20.0 (11.0 to 31.0 | 1.6 (0.0 to 5.8) | 20.0 (13.4 to 27.6) | 0.8 (0.0 to 2.9) | ||
| Hypotension | 27.1 (16.7 to 38.9) | 19.2 (10.7 to 29.6) | 27.0 (19.5 to 35.2) | 18.6 (12.4 to 25.8) | ||
| Febrile neutropenia | 73.0 (61.2 to 83.3) | 48.6 (36.7 to 61.0) | 73.0 (64.8 to 80.5) | 49.1 (40.5 to 58.2) | ||
| Neutropenia (neutrophil count decreased) | 1.6 (0.0 to 5.8) | 16.2 (8.0 to 25.9) | 0.8 (0.0 to 2.9) | 15.5 (9.8 to 22.1) | ||
| Anaemia | 1.6 (0.0 to 5.8) | 1.6 (0.0 to 5.8) | 0.8 (0.0 to 2.7) | 0.8 (0.0 to 2.9) | ||
| Thrombocytopenia (platelet count decreased) | 1.6 (0.0 to 5.8) | 1.6 (0.1 to 5.7) | 0.8 (0.0 to 2.9) | 0.8 (0.0 to 2.9) | ||
| Leukopenia (white cell decreased) | 1.6 (0.0 to 5.8) | 1.6 (0.0 to 6.2) | 0.8 (0.0 to 2.9) | 0.8 (0.0 to 2.7) | ||
| Hypokalaemia | 1.6 (0.0 to 6.2) | 1.5 (0.0 to 5.3) | 0.8 (0.0 to 3.0) | 0.8 (0.0 to 3.0) | ||
| Hypophosphataemia | 1.6 (0.0 to 5.8) | 1.6 (0.0 to 5.8) | 0.8 (0.0 to 3.0) | 0.8 (0.0 to 3.0) | ||
Minimum data set
Chimeric antigen receptor T-cell therapy was assumed to be associated with improved probabilities of survival at months 6 (78.5%) and 12 (72.5%) compared with standard of care therapy (32.0% and 20.7% at 6 and 12 months, respectively). Treatment with CAR T-cell therapy was associated with a statistically significant improvement in the time to death, with a HR of 0.20 (95% CI 0.11 to 0.37).
In a restricted mean survival time analysis, treatment with CAR T-cell therapy was associated with a mean extension to life expectancy of 10.47 months (95% CI 7.59 to 13.34 months) compared with standard of care therapy. The median follow-up in the minimum set was 10.03 months.
Intermediate data set
Five-year landmark survival probabilities of 72.5% and 20.7% for CAR T-cell therapy and standard of care therapy were assumed, respectively. Treatment with CAR T-cell therapy was associated with a statistically significant improvement in the time to death, with a HR of 0.18 (95% CI 0.10 to 0.33).
In contrast to the bridge to HSCT intermediate data set, there was no apparent trend towards non-proportional hazards over time. In the restricted mean survival time analysis, CAR T-cell therapy was associated with a mean improvement in life expectancy of 32.94 months (95% CI 24.38 to 41.43 months) compared with standard of care therapy. The median follow-up in the intermediate data set was 45.2 months.
Mature data set
Treatment with CAR T-cell therapy was associated with a statistically significant improvement and a more precise estimated HR than in the minimum and intermediate data sets (HR 0.179, 95% CI 0.117 to 0.272). In the restricted mean survival time analysis, treatment with CAR T-cell therapy was associated with a similar mean extension to life expectancy as in the intermediate set but with increased precision. The estimate of the improvement in life expectancy was 32.94 months (95% CI 26.87 to 38.93 months) compared with standard of care therapy. The median follow-up in the mature data set was 45.2 months.
A summary of the six evidence sets across the two separate TPPs is provided in Table 11.
| Attribute | Minimum data set | Intermediate data set | Mature data set |
|---|---|---|---|
| TPP: bridge to HSCT | |||
| Median time to censoring (follow-up) (months) | 11.3 | 53.6 | 53.6 |
| OS: HR (95% CI); difference in restricted mean survival time (95% CI) (months) | 0.331 (0.203 to 0.539); 5.4 (3.2 to 7.6) | 0.309 (0.190 to 0.503); 22.1 (12.9 to 31.3) | 0.307 (0.218 to 0.434); 22.1 (15.6 to 28.6) |
| CR (95% CI) | |||
| CAR T-cell therapy | 64.9 (52.6 to 76.5) | 64.9 (52.6 to 76.5) | 65.7 (57.7 to 73.6) |
| Standard of care – clofarabine | 12.5 (6.1 to 21.5) | 12.5 (6.1 to 21.5) | 12.0 (6.9 to 18.1) |
| MRD negative (95% CI) (%) | |||
| CAR T-cell therapy | 54.7 (42.8 to 67.7) | 54.7 (42.8 to 67.7) | 56.0 (47.6 to 64.7) |
| Standard of care – clofarabine | 2.7 (0.4 to 7.5) | 2.7 (0.4 to 7.5) | 2.3 (0.5 to 5.4) |
| CRS (95% CI) (%) | |||
| CAR T-cell therapy | 28.9 (19.0 to 40.7) | 28.9 (19.0 to 40.7) | 28.2 (20.8 to 36.1) |
| Standard of care – clofarabine | 1.5 (0.0 to 5.3) | 1.5 (0.0 to 5.3) | 0.9 (0.0 to 3.3) |
| Febrile neutropenia (95% CI) (%) | |||
| CAR T-cell therapy | 33.8 (22.8 to 45.0) | 33.8 (22.8 to 45.0) | 33.2 (24.8 to 41.6) |
| Standard of care – clofarabine | 48.6 (36.7 to 61.0) | 48.6 (36.7 to 61.0) | 49.1 (40.5 to 58.2) |
| TPP: curative intent | |||
| Median time to censoring (follow-up) (months) | 10.03 | 45.2 | 45.2 |
| OS: HR (95% CI); difference in restricted mean survival time (95% CI) (months) | 0.204 (0.113 to 0.370); 10.5 (7.6 to 13.3) | 0.180 (0.099 to 0.327); 32.9 (24.4 to 41.4) | 0.179 (0.117 to 0.272); 32.9 (26.9 to 38.9) |
| CR (95% CI) (%) | |||
| CAR T-cell therapy | 90.0 (81.3 to 96.2) | 90.0 (81.3 to 96.2) | 90.0 (84.0 to 94.7) |
| Standard of care – clofarabine | 11.5 (4.7 to 20.6) | 11.5 (4.7 to 20.6) | 11.5 (6.5 to 17.8) |
| MRD negative (95% CI) (%) | |||
| CAR T-cell therapy | 73.4 (61.5 to 83.7) | 73.4 (61.5 to 83.7) | 73.4 (65.2 to 80.8) |
| Standard of care – clofarabine | 1.6 (0.0 to 6.0) | 1.6 (0.0 to 6.0) | 1.6 (0.0 to 4.5) |
| CRS (95% CI) (%) | |||
| CAR T-cell therapy | 27.0 (16.6 to 38.9) | 27.0 (16.6 to 38.9) | 27.0 (19.5 to 35.3) |
| Standard of care – clofarabine | 1.5 (0.0 to 5.3) | 1.5 (0.0 to 5.3) | 0.9 (0.0 to 3.3) |
| Febrile neutropenia (95% CI) (%) | |||
| CAR T-cell therapy | 73.0 (61.2 to 83.3) | 73.0 (61.2 to 83.3) | 73.0 (64.8 to 80.5) |
| Standard of care – clofarabine | 48.6 (36.7 to 61.0) | 48.6 (36.7 to 61.0) | 49.1 (40.5 to 58.2) |
Chapter 6 Review of cost-effectiveness evidence for chimeric antigen receptor T-cell therapy and other interventions for acute lymphocytic leukaemia
Methods
No previously published studies on the potential cost-effectiveness of CAR T-cell therapy for ALL were identified in our searches (see Appendix 7). To inform the conceptualisation and development of the economic model, a separate review of published studies evaluating the cost-effectiveness of other treatments for ALL was conducted. The primary aim of the review was to inform key structural assumptions and potential parameter sources required for the model. Hence, the review focused on the main methodological approaches taken in the studies identified, rather than the specific results reported.
A two-part approach was taken, consisting of a systematic review and a more pragmatic search. Details of the search strategy employed to inform the systematic review are provided in Appendix 8 (see Table 47). The pragmatic search searched for any publicly available reports considering the cost-effectiveness of any intervention in ALL using Google and Google Scholar; in addition, the relevant websites for NICE and AWMSG were searched to identify previous appraisals for ALL.
Results
The systematic search identified 489 records, 11 of which were deemed potentially relevant after a review of their titles and abstracts. However, after obtaining the full articles, none of these studies was found to be a full economic evaluation and hence these studies were not subsequently considered within the model conceptualisation stage. The pragmatic search using Google and Google Scholar found two papers deemed relevant to the primary aim of the review. 182,183
Costa et al. 182 conducted a cost-effectiveness evaluation of unrelated stem cell transplantation for adults with acute leukaemia (ALL and acute myeloid leukaemia) structured around a 20-year Markov model. The study concluded that the two forms of transplantation considered (cord blood and bone marrow/peripheral blood stem cells) were cost-effective compared with no transplantation. The study found that, despite the high initial cost and short-term mortality associated with the transplantation procedures, the resulting life-year gains achieved by surviving patients were significant.
Lis et al. 183 considered the cost-effectiveness of clofarabine combined with chemotherapy in children and adolescents with ALL who have failed at least two previous therapies compared with nelarabine (Atriance®, GlaxoSmithKline, Brentford, UK) and FLAG-IDA, through the use of a lifetime Markov model. After the initial treatments, a proportion of patients was assumed to subsequently receive HSCT; this proportion varied given the response to initial treatment (complete, partial, complete without platelet recovery or no response) and the treatment arm. A patient who survived for 2 years post HSCT was assumed to be cured of ALL; no cure was possible without HSCT. The authors found clofarabine to be cost-effective compared with both comparators. The result was driven by the success of a therapy in achieving a bridge to HSCT and thus a potential cure. As clofarabine was associated with a greater proportion of patients experiencing an initial CR, it had the greatest proportion of patients undergoing HSCT and thus cured patients.
The search of NICE and AWMSG appraisals found that the only appraisal by NICE for ALL (dasatinib, ID386)184 was discontinued in 2008 because of the low number of patients anticipated to be treated. By contrast, the AWMSG provided details of four separate appraisals in ALL, although one of these (imatinib, no. 2014) did not receive a formal submission by the manufacturer. 171,185–187 Of the remaining AWMSG appraisals, only the final appraisal recommendations (FARs) are made publicly available, limiting the detail available on the evaluative approaches. Only two of the appraisals (clofarabine171 and nelarabine185) provided sufficient detail to review.
Clofarabine171 was recommended by the AWMSG for children and adolescents with ALL who are relapsed or refractory after at least two previous regimens and for whom no other treatment is anticipated. Within the FAR, an important restriction was placed on the recommendation such that clofarabine should be given only to patients in whom there is an intention to proceed to HSCT. This recommendation was based on the findings that clofarabine did not appear to be cost-effective for patients who did not subsequently receive HSCT. In the submission, clofarabine was compared with palliative care alone. Palliative care was assumed to be associated with a very short median survival time (9–10 weeks) based on historical control data.
Although limited details of the modelling approach are reported, it is evident that the primary structural driver within the model is the bridging role of clofarabine to HSCT, with potentially significant gains in life-years assumed for patients who subsequently receive HSCT. The manufacturer assumed that the success of HSCT in achieving long-term remission (and cure) was driven by the achievement of remission (complete, with platelet involvement or partial) at the time of transplantation. Hence, improved rates of remission achieved with clofarabine compared with palliative care directly equate to long-term survival. The model submitted assumed that patients who received HSCT and survived for 1 year were cured, returning to the mortality risks and utilities of the general population.
Nelarabine185 was recommended by the AWMSG for the treatment of patients with T-ALL and T-cell lymphoblastic leukaemia whose disease has not responded to, or has relapsed, following treatment with at least two chemotherapy regimens. Best supportive care was used as the main comparator and clofarabine was considered in a separate scenario based on indirect comparisons. In common with the restriction previously applied within its recommendations for clofarabine in ALL, the AWMSG also restricted treatment to patients for whom there is an intention to proceed to HSCT. This restriction was based on a similar finding that the cost-effectiveness of nelarabine was closely related to the assumed increase in the proportion of patients subsequently receiving HSCT (and their related long-term health gains). The base-case analysis presented survival based on within-trial estimates with no extrapolation conducted. This was considered to be an extremely conservative estimate. Separate scenarios were presented considering the long-term survival of post-HSCT patients and were found to have a major impact on the results. The base-case ICER of £102,281 per QALY gained was subsequently reduced to £51,169 if post-HSCT survival was assumed to be 2 years and to £25,523 if normal life expectancy was assumed in patients who survived for > 1 year (i.e. cure at 1 year).
Implications for model conceptualisation
The systematic and pragmatic searches highlighted a number of potential implications for our evaluation. Within existing studies, it is clear that the main benefit of existing treatments has been related to their ability to provide a ‘bridge’ to HSCT. The primary factor determining cost-effectiveness in the reviewed literature was the increased likelihood of receiving HSCT with a new treatment and the associated assumptions made regarding subsequent health gains associated with transplantation. Only a limited survival gain was attributed to patients who did not subsequently receive HSCT, such that none of the treatments reviewed appeared to be cost-effective as a palliative option.
The key structural assumptions employed within these studies were the potentially curative effect of HSCT and the short life expectancy assumed for the comparator treatments (best supportive care/palliative treatment alone) derived from historical controls. The majority of studies assumed a ‘cure point’ associated with HSCT, although the timing was different across studies. The ‘cure point’ was assumed to represent the time at which patients are assumed to no longer be at risk of disease relapse. The study by Costa et al. 182 assumed that at 5 years post transplantation the patient will be free of any procedural mortality risk or any risk of disease recurrence. In Lis et al. 183 and the AWMSG appraisal of nelarabine185 this cure point was assumed to be 2 years after HSCT, whereas in the AWMSG appraisal of clofarabine171 the cure point was assumed to be 1 year after HSCT.
The studies also differed in the assumptions made concerning subsequent survival after the ‘cure point’. Costa et al. 182 acknowledged that long-term ALL survivors are likely to be subject to significant comorbidities over their remaining lifetime despite being leukaemia free. To account for the impact of comorbidities, an assumption was made that the long-term survival of ALL patients would be 50% less than that in the general population. The authors acknowledge that this was an arbitrary adjustment because of the lack of data on the long-term mortality rate in long-term survivors of ALL reported at the time. In contrast, the study reported by Lis et al. 183 and the clofarabine submission171 effectively assumed no additional comorbidities (i.e. beyond those experienced by the general population) beyond the ‘cure point’. Hence, patients were subsequently assumed to return to the age-adjusted mortality risk and utility of the general population. The AWMSG raised concerns that, not only was this assumption insufficiently justified but, also, the model was very sensitive to changes in the long-term survival probability.
In the absence of RCT data, each model incorporated historical control data as the basis to inform outcomes associated with the comparator (best supportive care, palliative care and clofarabine within a scenario for the submission for nelarabine). 171,185–187 However, insufficient details were reported regarding the source of the historical control data used and whether attempts were made to identify possible biases or to formally account for potential confounding.
The existing cost-effectiveness literature is limited in ALL. No completed NICE appraisals of licensed treatments for ALL were identified. Furthermore, of the studies published, none was reported in sufficient detail to provide a suitable basis for informing the exemplar application. In the absence of previous NICE appraisals or sufficient reporting within existing publications, the development of a de novo model to inform the exemplar application was considered necessary. Full details of this are reported in the next chapter.
Chapter 7 The exemplar economic model
Overview
There are several distinct issues and challenges relating to the modelling of costs and outcomes that arise from the separate TPPs:
-
In the bridge to HSCT TPP, the primary health benefits of treatment are gained by enabling more patients to successfully undergo HSCT, an established intervention that has known curative potential. For economic modelling purposes it may therefore be desirable to introduce a structural link between HSCT and overall treatment benefit (i.e. survival) in the model. The introduction of a link between a potential established surrogate outcome or process and final health benefits also enables the use of evidence external to the CAR T-cell evidence sets (i.e. survival post HSCT). This structural link may also provide decision-makers with greater confidence surrounding the modelled health benefits of treatment for survival, given that model projections would depend largely on the established benefits of HSCT. In terms of decision uncertainty, this approach would also mean that the uncertainty surrounding the cost-effectiveness of CAR T-cell therapy is partly determined by the maturity and sample size of the evidence sets and partly by the maturity, sample size and acceptability of external evidence obtained from other sources.
-
In the curative intent TPP, the case for introducing a structural link between final health benefits and a surrogate outcome or process such as HSCT is more limited than in the bridge to HSCT case, given that it is primarily CAR T-cell therapy itself that is expected to provide the curative benefits. In this context, it may be more appropriate to model long-term outcomes through the direct extrapolation of EFS and OS data from the CAR T-cell trial evidence sets, as opposed to modelling long-term outcomes through a separate surrogate process. In this case, the decision uncertainty surrounding the cost-effectiveness of treatment would be determined solely by the maturity and sample size of data from the evidence sets.
Patient population
In this evaluation we assessed the cost-effectiveness of CAR T-cell therapy in the treatment of children and young adults with two or more relapses or refractory ALL. The baseline demographic characteristics of this patient group are summarised in Table 12.
Comparator
The comparator treatment to CAR T-cell therapy was defined as standard of care. In the base case, the standard of care treatment was assumed to be clofarabine. The mean cost for a course of clofarabine treatment is approximately £43,200 per patient. 171
As part of a separate sensitivity analysis, the standard of care treatment was assumed to be FLAG-IDA. The mean cost for a course of FLAG-IDA treatment is approximately £3803 per patient (see Administration and monitoring costs).
Model development
The approaches to modelling the cost-effectiveness of CAR T-cell therapy varied between the separate scenarios. Therefore, two de novo decision models were developed and used to assess the cost-effectiveness of CAR T-cell therapy across the two scenarios:
-
bridge to HSCT model – based on a landmark responder model consisting of two related decision models:
-
a short-term decision tree to predict the remission and transplant status of the population in the immediate period following CAR T-cell or comparator therapy
-
a series of partitioned survival (or area under the curve) models to predict the longer-term survival of patients conditional on remission and transplant status
-
-
curative intent model – based on a simple three-state (alive and event free, alive post event, dead) partitioned survival model.
The two models share a number of common features, which are outlined in Table 13.
| Factor | Chosen values | Justification |
|---|---|---|
| Time horizon | Lifetime horizon (up to a maximum age of 100 years) | Necessary to capture the potential lifetime impacts of short-term and potentially ongoing mortality benefit |
| Cycle length | 1 month | Sufficient length to capture relevant transitions in the model |
| Mid- or half-cycle correction | Mid-cycle correction employed | To guard against over- or under-predicting state occupancy in the model |
| Measure of health effects | QALYs | In accordance with the current NICE reference case for cost-effectiveness.122 Necessary to quality short-term and potentially ongoing mortality benefits and associated adverse events |
| Discounting | 3.5% for costs and health effects over the lifetime horizon | In accordance with the current NICE reference case. Alternative discounting rates explored using sensitivity analysis |
| Perspective | NHS/PSS | In accordance with the current NICE reference case |
Further details specific to each of the two model structures are reported in the following sections.
Bridge to haematopoietic stem cell transplantation scenario
Key structural assumptions
The bridge to HSCT model consists of a decision tree model (days 0–56) and a series of partitioned survival models (day 56 to lifetime) that, when combined, provide an estimate of the lifetime costs and effectiveness of treatment in ALL. An illustration of the structure of the model is provided in Figure 3.
FIGURE 3.
Illustration of the bridge to HSCT model.

The short-term decision tree component of the model consists of three chance nodes that represent a series of clinically relevant events that may occur during the first 56 days (2 months) of treatment:
-
remission status at day 28 (remission, no remission or death)
-
MRD status at day 28 (negative or positive)
-
transplantation status at day 56 (HSCT or no HSCT).
These events are considered to be prognostic of the duration and quality of life of patients with ALL and were therefore included in the model to link short-term measures of trial efficacy to longer-term health outcomes. The three nodes of the decision tree are sequenced in the order of remission, MRD status and transplantation status.
At the first chance node, the hypothetical cohort are distributed across three states: remission, no remission or death. In the model, remission is defined using the criteria applied in the CAR T-cell and clofarabine clinical trials. 164,178 CR is defined as < 5% marrow blasts by flow cytometry, an absence of circulating blasts and no extramedullary sites of disease with an absolute neutrophil count of ≥ 1000/µl and a platelets count of ≥ 100,000/µl. In accordance with both the Lee et al. 164 study and the Jeha et al. 178 study, remission status is determined at day 28 (month 1) of the simulation.
At the second chance node, patients with remission are reassigned to one of two states: remission and MRD negative or remission and MRD positive. A MRD-negative status is defined as < 0.01% marrow blasts and a MRD-positive status is determined by marrow blasts of between 0.01% and 5% (at > 5% patients are no longer in remission).
At the third and final node (day 56), all patients are assigned to states corresponding to the use of HSCT (HSCT vs. no HSCT). The final determination of health status (remission – MRD – HSCT) was assumed to occur at day 56 (month 2) of the simulation. This time period was chosen based on the mean time from CAR T-cell therapy to HSCT estimated from data reported in the study by Lee et al. 164 (mean 54 days, 95% CI 45 to 77 days).
At the end of the decision tree phase, the cohort is assigned to six mutually exclusive states (presented in order of best prognosis):
-
HSCT – remission and MRD negative
-
HSCT – remission and MRD positive
-
HSCT – no remission
-
no HSCT – remission
-
no HSCT – no remission
-
death.
After day 56, the long-term survival of the cohort is modelled through a series of related partitioned survival models (see Figure 3) that are used to model the long-term outcomes of treatment (day 56 to lifetime). The model includes four distinct partitioned survival models that are used to evaluate survival in the following groups:
-
HSCT – remission and MRD negative
-
HSCT – remission and MRD positive
-
HSCT – no remission
-
no HSCT.
Haematopoietic stem cell transplantation recipients with MRD-negative status prior to transplantation are assumed to have the best prognosis in terms of long-term survival. An increasing level of marrow blasts is assumed to be associated with a lower probability of long-term survival, such that HSCT recipients with MRD-positive status have (on average) a worse survival prognosis than MRD-negative patients. HSCT recipients who fail to achieve remission prior to transplantation are assumed to have the poorest prognosis of all HSCT patients.
For patients who did not receive HSCT, the probability of OS was significantly lower than for HSCT patients. It was assumed that CR was not associated with improved probabilities of survival in non-HSCT patients. This assumption was made on the basis that in the bridging scenario it is through HSCT (and not remission in the absence of HSCT) that meaningful gains in survival can be achieved. The impact of this assumption on the results of the evaluation was tested in the one-way sensitivity analyses, in which it was assumed that non-remission non-HSCT patients had an inferior survival prognosis to remission non-HSCT patients.
At year 5 of the simulation, those who were alive were subsequently assumed to be long-term survivors of ALL. From this point forward, the cohort was considered to be effectively ‘cured’ of ALL and experienced the mortality risk profile consistent with that of a long-term survivor of ALL. The mortality risks after year 5 were therefore modelled based on general population age- and sex-adjusted all-cause risks of mortality adjusted for excess morbidity and mortality reported in cohorts of long-term survivors of ALL. 188 The approach is more formally described in Model development.
The model also included treatment-related adverse events. These include events such as CRS, encephalopathy, hypotension, febrile neutropenia, neutropenia, anaemia, thrombocytopenia, leukopenia, hypokalaemia and hypophosphataemia. The costs and consequences of these events were assumed to occur at the start of the evaluation. As prolonged B-cell aplasia did not occur in the Lee et al. 164 study, the costs and consequences of this were not included within the bridge to HSCT scenario. The key structural assumptions applied in the model are outlined in Table 14.
| Input | Assumption |
|---|---|
| Surrogate relationship between MRD status and HSCT | A lower marrow blast status prior to transplantation (as captured through MRD status) is associated with a higher probability of experiencing sustained remission and long-term survival benefits in ALL |
| HSCT | All HSCT events were assumed to occur at day 56 of the simulated time horizon. No further HSCT events were permitted after this point |
| Survival during the first 5 years of the evaluation time horizon | Survival post HSCT was modelled based on the constant transition probability. There is no difference in survival between remission non-HSCT patients and non-remission non-HSCT patients |
| Survival after the first 5 years of the evaluation time horizon | All patients alive at 5 years post HSCT are considered to be long-term survivors of the disease. Long-term survivors of ALL experience excess morbidity and mortality compared with the general population |
| Treatment/retreatment | In the base case it was assumed that each patient would receive a single full course of therapy. Retreatment with CAR T-cell therapy or standard of care therapy was not permitted in the base case but was considered in the sensitivity analysis |
| Treatment effect | Compared with standard of care therapy, CAR T-cell therapy improves the probability of remission, the probability of a MRD-negative status and the probability of successful HSCT. The clinical parameter estimates used to inform the models and TPPs can be generalised to the UK NHS |
| Patient follow-up | After HSCT, patients receive ongoing care and rehabilitation up to 2 years post HSCT. Patients who do not receive HSCT are assumed to require hospitalisation prior to death |
| Adverse events | Treatment-related adverse events were considered in the evaluation and included events such as CRS, the incidence of which is expected to increase with the use of CAR T-cell therapy. The costs and health consequences of adverse events were accrued at the start of the evaluation |
| HRQoL | Patients who achieve remission status are assigned a higher utility weight than patients who do not achieve remission. Transplantation is associated with a one-off decrement to HRQoL |
Clinical justification for the structure of the model
The conceptual structure of the bridge to HSCT model is based on an assumed relationship between HSCT use and final clinical benefits and the assumption that the effectiveness of HSCT is dependent on MRD status prior to transplantation.
Allogeneic HSCT is a potentially curative treatment option in patients with ALL. However, the long-term benefits of HSCT are uncertain, with some patients experiencing long-term benefits, including the effective cure/suppression of ALL, and other patients experiencing relapse and/or mortality shortly after transplantation. In this evaluation, survival benefits are established through remission and MRD status prior to HSCT.
Several studies have investigated the relationship between remission/MRD status prior to HSCT and the long-term outcomes of HSCT therapy. 110,111,189 These studies have shown, to varying degrees, that MRD status prior to HSCT appears to be an important prognostic determinant of long-term RFS and OS, with MRD-negative (< 0.01% marrow blasts) patients experiencing superior survival compared with MRD-positive (> 0.01% to 5% marrow blasts) patients, including within studies of children with relapsed ALL. These data support the assumption of a continuous relationship between MRD level prior to HSCT and 5-year survival probability.
For patients who do not receive HSCT, the long-term outcomes of treatment are generally poor. In the study by von Stackelberg et al. ,160 the median survival in refractory patients who failed to respond to induction therapy and who went on to receive palliative care was 89 days (equivalent to 3.17 months).
In the model, it was assumed that all patients who did not receive HSCT (including remission and non-remission patients) went on to receive palliative care, having exhausted all treatment strategies that may be curative. Remission status was not considered to be prognostic of survival in the non-HSCT population, such that non-HSCT patients who achieved remission were assumed to be at the same risk of mortality as non-HSCT patients who failed to achieve remission. However, as discussed later in Model inputs: utilities, all patients with remission were assigned an improved health utility compared with those who failed to achieve remission. These benefits were, however, assumed not to extend to improved life expectancy.
In previous economic evaluations in ALL (reviewed in Chapter 6), it had been assumed that survivors of ALL experience the same mortality risk profile as that of the general population. This assumption implies that there is no excess mortality or morbidity risk associated with their previous illness. This assumption is not supported by the published literature, which generally reports excess mortality and morbidity among the long-term ALL survivor population compared with match-adjusted individuals without ALL (i.e. siblings). 190,191 In the model, the risk of mortality assigned to survivors of ALL was set equal to the general population background all-cause mortality risk profile, with an adjustment for an increased mortality risk among survivors of ALL.
The point at which patients were assumed to be long-term survivors of ALL (5 years) was based on the definition used in a number of published studies reporting long-term survival data in ALL. None of these studies provides an explicit rationale for selecting 5 years as the cure point and, to our knowledge, there appears to have been no published attempts to empirically justify the widespread use of the 5-year cure point. However, across a number of studies, the KM curves for post-HSCT survival appear to stabilise within the 5-year time frame, such that the curve becomes flat and the incidence of death reduces to near zero.
Efficacy parameter estimates
In the decision tree component of the model, the data for remission, MRD and HSCT status of the modelled cohort were derived from the separate evidence sets reported in Chapter 5 (see Bridge to haematopoietic stem cell transplantation target product profiles). The key assumptions required to generate the estimates for the evidence sets are outlined in Table 15. A continuity correction was applied to particular estimates to take account of the low numbers (e.g. when 0 events were recorded) in the probabilistic analysis.
| Node parameter 1 | Node parameter 2 | Node parameter 3 | Estimate (%) | Probabilistic distribution | Minimum data set, n | Intermediate data set, n | Mature data set, n | Key assumptions |
|---|---|---|---|---|---|---|---|---|
| CAR T-cell therapy | ||||||||
| Remission | 66.7 | Dirichlet distribution | 63 | 63 | 126 | Remission probability based on 14 of 21 patients achieving remission in Lee et al.164 By day 56, one of 21 patients had died (5%). All remaining patients (28.6%) were assumed to have non-remission | ||
| No remission | 28.3 | |||||||
| Death (day 0–56) | 5.0 | |||||||
| Remission | MRD negative | 85.7 | Beta distribution | 12 patients in Lee et al.164 had a MRD-negative status. In total, 14 patients were in remission. Thus, 12 of 14 patients were MRD negative and in remission | ||||
| MRD positive | 14.3 | |||||||
| Remission | MRD negative | HSCT | 83.3 | Beta distribution | 12 patients were MRD negative in Lee et al.,164 of whom 10 underwent HSCT | |||
| No HSCT | 16.7 | |||||||
| Remission | MRD positive | HSCT | 0 | Beta distribution | In Lee et al.,164 no MRD-positive patients received HSCT | |||
| No HSCT | 100 | |||||||
| No remission | HSCT | 0 | Beta distribution | In Lee et al.,164 none of the patients who failed to achieve remission received HSCT | ||||
| No HSCT | 100 | |||||||
| Clofarabine | ||||||||
| Remission | 11.5 | Dirichlet distribution | 61 | 61 | 122 | Remission based on seven of 61 patients achieving CR in Jeha et al.178 By day 56, 32.7% of patients had died (based on digitisation of the published KM curve). All remaining patients (55.8%) were assumed not to be in remission | ||
| No remission | 55.8 | |||||||
| Death (day 0–56) | 32.7 | |||||||
| Remission | MRD negative | 14.3 | Beta distribution | One patient with remission had undergone HSCT and was considered to be in long-term remission (> 200 days alive). This patient was assumed to have a MRD-negative status (MRD status not reported in Jeha et al.178). Thus, one of seven remission patients was MRD negative | ||||
| MRD positive | 85.7 | |||||||
| Remission | MRD negative | HSCT | 100.0 | Beta distribution | Assumption that all patients who were MRD negative went on to receive HSCT | |||
| No HSCT | 0.0 | |||||||
| Remission | MRD positive | HSCT | 16.7 | Beta distribution | Seven patients were in remission, of whom one was assumed to be MRD negative and had undergone HSCT. Of the remaining six patients (MRD positive), a further one patient had undergone HSCT. Thus, one of six MRD-positive patients had undergone HSCT178 | |||
| No HSCT | 83.3 | |||||||
| No remission | HSCT | 20.6 | Beta distribution | In total, nine patients underwent HSCT in Jeha et al.178 Two HSCT patients were in remission, with the remaining seven HSCT patients not in remission. During the initial 56 days, an estimated 34 (55.8%) patients were not in remission. Thus, seven of 34 no-remission patients underwent HSCT | ||||
| No HSCT | 79.4 | |||||||
Figure 4 presents the proportion of patients occupying each state at the end of the decision tree model. The model predicts that 48% of patients receiving CAR T-cell therapy and 15% of patients receiving standard of care treatment will receive HSCT. All patients who underwent HSCT following CAR T-cell therapy were assumed to have a MRD-negative status. In contrast, most patients who underwent HSCT after receiving clofarabine had not achieved remission (11.5%) prior to transplantation, with only a small proportion of patients receiving HSCT after CR (1.6% MRD negative, 1.6% MRD positive).
FIGURE 4.
Proportion of patients occupying each state at the end of the decision tree model (day 56) by CAR T-cell and comparator group: bridge to HSCT scenario.

With the structural link included within the model, it was necessary to use external data rather than the evidence sets themselves for the purposes of extrapolation and estimating lifetime mortality. This was necessary because the existing survival data for CAR T-cell therapy were not reported in terms of being conditional on remission, MRD or HSCT status. Hence, the parameter estimates for the partitioned survival analyses were sourced from two external studies: Leung et al. 111 for the post-HSCT survival probabilities and von Stackelberg et al. 160 for the non-HSCT survival probabilities. A summary of the survival rates is provided in Table 16.
| Treatment status | Status prior to treatment | Exponential rate parameter (standard error) | Sampling distribution used in probabilistic analysis | Proportion alive and considered ‘effectively’ cured at year 5 (%) | Mean time to death following HSCT (years)a | Notes | Source |
|---|---|---|---|---|---|---|---|
| HSCT | MRD negative (< 0.01% bone marrow blasts) | – | NA | 99.0 | 43.70 | Based on all-cause mortality, with adjustment for excess mortality in ALL survivors | Assumption |
| MRD positive (> 0.01% to 5% bone marrow blasts) | 0.0121 (0.0232) | Log-normal (applied to rate) | 48.5 | 22.43 | Leung et al.111 report a 5-year post-HSCT survival probability of 48.5% in patients with a MRD-positive status. The 5-year cumulative probability was converted to a monthly rate using the equation [–(1/60) × log(0.485)] | Leung et al.111 | |
| No remission (> 5% to 25% bone marrow blasts) | 0.0175 (0.0232b) | Log-normal (applied to rate) | 35.1 | 16.74 | 5-year post-HSCT survival probability estimated by fitting a linear regression model to the survival data by MRD status, reported in Leung et al.111 The independent variable in the regression was the cumulative hazard rate at year 5 and the dependent variable was the mid-point of each MRD category (on the log to base 10 scale). To predict the cumulative hazard at year 5 for patients without remission prior to HSCT the mid-point MRD level of 15% was used | ||
| No HSCT | All patients | 0.2425 (0.2085) | Log-normal (applied to rate) | 0 | 0.35 | See main text | Von Stackelberg et al.160 |
In the base case it was assumed that all transplant recipients in remission and with a MRD-negative status prior to HSCT reverted to the same mortality rates as seen in long-term survivors of ALL, from the time point of HSCT. Employing this assumption, as opposed to using the data reported in Leung et al. 111 for this population, provided a more consistent prediction of survival data from Lee et al. ,164 in which it was reported that all 10 HSCT recipients with a MRD-negative status were leukaemia free and alive at the end of study follow-up.
Transplant recipients in remission and with a MRD-positive status were assumed to have an inferior long-term survival prognosis compared with those who were MRD negative. Similarly, recipients who failed to respond to therapy were assumed to have an inferior long-term prognosis compared with those who responded (including MRD-positive and MRD-negative recipients). Parameter estimates were obtained from Leung et al. 111 and were modelled assuming an exponential distribution for time to death.
The Leung et al. 111 data were used in the base-case analysis as this was the only study identified in the literature review that reported post-HSCT survival in patients who failed to achieve remission (marrow blasts > 5.0%). The parameter estimate for no-remission HSCT patients forms an important part of predicting the long-term survival benefits of standard of care therapy, as approximately 11% of the standard of care population underwent HSCT, despite having failed to achieve CR (vs. 0% of the CAR T-cell trial population).
For patients who do not receive HSCT (including remission and non-remission patients), long-term survival was modelled using data from von Stackelberg et al. 160 A series of parametric survival functions was fitted to estimates of patient-level data generated from the published KM curve. According to goodness-of-fit statistics, the best-fitting distribution was the log-normal. However, when the function was applied in the model, the predicted OS for the total CAR T-cell and standard of care populations became visibly disjointed, with the risk of mortality in the decision tree phase being significantly greater than the risk being applied at the start of the partition survival phase. Consequently, there was an uncharacteristic ‘plateau’ in the modelled survival curve between day 56 (month 2) and day 84 (month 3). This plateau effect was caused by an initially low probability of death predicted from the von Stackelberg et al. 160 data.
Because of the implausible nature of the survival curve, an alternative survival distribution for von Stackelberg et al. 160 was selected in the base case. To be consistent with the approach used in modelling post-HSCT survival,111 the exponential distribution was chosen for the base-case analysis. The mean time to death with the exponential function was 0.35 years, which is consistent with the mean time to death estimated using the log-normal distribution (0.34 years).
The validation of the responder model in predicting the outcomes of the Lee et al. 164 and Jeha et al. 178 studies was assessed by comparing the predicted survival probabilities from the model with the KM data extracted from these studies. As shown in Figure 5, the final model appears to provide an accurate prediction of reported survival for both CAR T-cell therapy and the comparator.
FIGURE 5.
Model prediction (lines) compared with reported KM curves of OS: bridge to HSCT scenario.

The background all-cause mortality risks were obtained from the interim life tables published by the UK ONS. 188 The ONS data report annual all-cause mortality rates by sex and age (yearly increments from 0 to 100 years). A sex-averaged mortality risk was derived based on a cohort that was 33.3% female (n = 7/21). 164 An adjustment factor for excess mortality in ALL survivors was also incorporated and modelled using data from MacArthur et al. 191 [standardised mortality ratio (SMR) 9.1, 95% CI 7.8 to 10.5]. These data were combined using the following equation:
where TP(x) is the monthly transition probability for the cohort with average age x, MR(y, x) is the ONS all-cause mortality rate for sex y and average age x, P(y) is the proportion of the cohort with sex y, and SMR is the SMR for long-term ALL survivors compared with the general population. The factor of 1/12 was included to convert the annualised mortality rates from the ONS to monthly rates, and probabilities, for use in the model. The mortality risk was assumed to remain constant within each year of the cohort’s age.
Curative intent model
Structural assumptions
A simple three-state partitioned survival model was developed to assess the cost-effectiveness of CAR T-cell therapy used with curative intent. The three health states included in the model were alive and event free, alive with relapsed disease and death. An illustration of the structure of the model is provided in Figure 6.
FIGURE 6.
Illustration of the partitioned survival model structure. (a) Illustration of the state structure; and (b) illustration of partitioned survival.


The health state of alive and event free included all patients who either had stable disease or had responded to therapy. The health state of alive with relapsed disease included patients who had failed induction therapy, had relapsed after previously responding to treatment or had developed second malignancies. This definition is based on the criteria used in the UK ALL study. 115
State occupancy in the model was derived using the partitioned survival technique. This involved the direct extrapolation of EFS and OS curves, which were then used to estimate the proportion of patients occupying each of the three states using the following equations:
where P(event,t) is the cumulative survival probability for the event at time t.
Data on EFS were not available from either the CAR T-cell164 or the clofarabine178 studies. In the absence of data, the EFS curve was derived from the available OS data, through assuming a proportional relationship between EFS and OS. This relationship is justified on the basis that EFS is highly correlated with OS as it includes death prior to recurrence.
In the short term it was assumed that the cumulative hazard function for EFS would be proportional to the cumulative hazard function for OS. This was modelled based on data from the UK ALL study. 115 The proportional relationship between EFS and OS is not expected to continue indefinitely, given the potential for cure of disease and the expectation that after a finite period of time all patients alive in the simulation would also be free of relapsed disease (EFS = OS). This is equivalent to saying that, at some point in time, all patients who are alive are long-term survivors of ALL. Therefore, in the model, the proportional relationship between EFS and OS was assumed to continue up to year 5 of the simulation (the assumed point of ‘effective’ cure in ALL). After year 5, the cumulative survival probabilities for EFS were assumed to be flat up to the point at which EFS is equal to OS. In all cases, EFS was always assumed to be less than or equal to OS to avoid a negative number of patients being assigned to the relapsed disease state.
In common with the bridge to HSCT scenario, at year 5 of the simulation, those who were alive in the curative intent model were also subsequently assumed to be long-term survivors of ALL. From this point forward, the cohort was considered to be effectively ‘cured’ of ALL and experienced the mortality risk profile consistent with a long-term survivor of ALL. The mortality risks after year 5 were also modelled based on general population age- and sex-adjusted all-cause risks of mortality adjusted for excess morbidity and mortality reported in cohorts of long-term survivors of ALL.
The model evaluation also included the costs and consequences of treatment-related adverse events, which included CRS and B-cell aplasia, whose occurrence is specifically associated with CAR T-cell therapy. Other events captured in the model include encephalopathy, hypotension, febrile neutropenia, neutropenia, anaemia, thrombocytopenia, leukopenia, hypokalaemia and hypophosphataemia. All events, with the exception of B-cell aplasia, were assumed to occur at the time of treatment initiation and to resolve within the first year of therapy. The cost–consequences of these events were therefore captured at the start of the evaluation.
The occurrence of B-cell aplasia in patients treated with CAR T-cells is an expected consequence of CAR T-cell therapy and is linked to the proliferation of CAR T-cells and the associated durability of the clinical effect. Consequently, for some patients, treatment of B-cell aplasia is expected to persist beyond the first year post CAR T-cell therapy. To capture this in the model, a series of survival models was fitted to data on the time to CD19 positivity or relapse reported in Maude et al. 165 and used to predict the proportion of patients requiring treatment for B-cell aplasia. The primary assumptions made in the curative intent model scenario are summarised in Table 17.
| Input | Assumption |
|---|---|
| Survival during the first 5 years | Survival was modelled based on a weighted average survival distribution |
| Survival after the first 5 years | All patients alive at 5 years are considered to be long-term survivors of the disease. Long-term survivors of ALL experience excess morbidity and mortality compared with the general population |
| Treatment/retreatment | In the base case it was assumed that all patients received a single full course of therapy. Retreatment with CAR T-cell therapy or standard of care therapy was not permitted in the base case, but was considered in the sensitivity analysis |
| Treatment effect | Treatment with CAR T-cell therapy is assumed to lead to an increase in the number of patients achieving a sustained cure for ALL and therefore extend the life expectancy of patients with ALL. The clinical parameter estimates used to inform the models and TPPs can be generalised to the UK NHS |
| Adverse events | Treatment-related adverse events were considered in the evaluation and included CRS and B-cell aplasia. The costs and health consequences of all adverse events except B-cell aplasia were accrued at the start of the evaluation. The costs of B-cell aplasia were modelled by estimating the probability of patients having B-cell aplasia over time, using data from Maude et al.165 |
| HRQoL | Patients who are event free are assigned a higher utility weight than patients who have relapsed disease. Transplantation is associated with a one-off adjustment to utilities |
Efficacy parameter estimates: partitioned survival model
The primary data sources for OS in the curative intent model were the same imputed patient data used to derive the evidence sets reported in Chapter 5 (see Curative intent target product profiles). Each separate evidence set was then analysed using parametric survival modelling to inform the 5-year survival estimates and projections applied within the cost-effectiveness analyses. The parametric analyses were undertaken using the FlexSurv package in the statistical programming platform R (version 3.0.2).
A series of survival distributions was considered in the analysis, including exponential, log-normal, Weibull and Gompertz. Because of the potential curative nature of CAR T-cell therapy (and therefore the potential for an unconventional hazard function), a series of flexible cubic spline models was also considered in the analysis. The cubic spline models were based on those developed by Royston and Parmar. 192 Cubic spline models expressed on the proportional odds scale were used as they appeared to converge to an optimised solution more frequently than the proportional hazards or probit variants of the cubic spline model. A series of one-, two-, three- and four-knot spline models was considered. The knots were evenly distributed across the time scale of the study, as per the default settings for the FlexSurv package in R.
Separate curves were fitted to the hypothetical CAR T-cell data and the comparator data to allow both the shape and the scale of the distribution to vary between these. Alternative options include fitting proportional hazards models to a data set containing both treatments and including a covariate in the regression for treatment assignment. This alternative approach was not considered here, given that an earlier assessment of the validity of the proportional hazards assumption illustrated that this assumption may not consistently hold across all evidence sets.
Within cost-effectiveness studies it is common practice to use a single survival distribution in the base-case analysis. This is chosen based on goodness-of-fit statistics, the fit of each distribution to the KM curves and the clinical plausibility of subsequent model projections over the full time horizon. However, it is unlikely that a single survival distribution can adequately characterise uncertainties over the longer-term extrapolation period. The robustness of the ICER estimates to alternative distributions can be considered within separate sensitivity analyses or scenarios. However, transparency concerns may exist regarding this approach if their weighting is not explicitly specified in subsequent policy decisions.
To more formally account for the uncertainty surrounding choice of survival distribution, a model-averaging approach was adopted using the methods outlined in Jackson et al. 193 This technique involves the parameterisation of uncertainty surrounding the choice of distribution through including all plausible survival functions as part of a weighted distribution and sampling both the parametric uncertainty associated within each distribution and the uncertainty (or weights) surrounding the choice of preferred method. Through the probabilistic analysis, it is therefore possible to estimate the joint distribution of uncertainty around the parameter estimates and the choice of survival function.
Each model is assigned a weight that represents the adequacy of that distribution in predicting the lifetime survival of the modelled cohort in comparison to all other distributions considered in the model. There are a number of measures of model adequacy that can be considered. Examples include statistical adequacy measures such as the Akaike information criterion (AIC) and Bayesian information criterion and expert judgement. The weights considered in this evaluation were based on AIC scores. As outlined in Jackson et al. ,193 the AIC value reported from each survival distribution was converted to a probability weight (wk) using the following equations:
The weighted distribution was then applied in the base-case analysis. Different model weights and parameter estimates were considered across the three different data sets, as outlined in the following sections.
Minimum data set
A summary of the goodness-of-fit statistics for each distribution fitted to the imputed survival data across each of the evidence sets is provided in Table 18.
| Distribution | CAR T-cell therapy | Standard of care | ||
|---|---|---|---|---|
| AIC | AIC-based weight (%) | AIC | AIC-based weight (%) | |
| Exponential | 127.91 | 1.9 | 302.15 | 0.1 |
| Weibull | 129.88 | 0.7 | 303.31 | 0.0 |
| Gamma | 129.91 | 0.7 | 304.09 | 0.0 |
| Gompertz | 139.21 | 0.0 | 303.01 | 0.0 |
| Log-normal | 128.70 | 1.3 | 291.00 | 13.8 |
| Spline with a single knot | 121.02 | 60.1 | 288.65 | 44.6 |
| Spline with two knots | 122.97 | 22.7 | 290.41 | 18.5 |
| Spline with three knots | 124.93 | 8.5 | 291.32 | 11.7 |
| Spline with four knots | 126.44 | 4.0 | 291.42 | 11.2 |
According to the AIC statistic, the distribution with the best goodness of fit to the CAR T-cell data was the spline model with a single knot (AIC = 121.02), followed by the spline model with two knots (AIC = 122.97). The spline model with a single knot was assigned the highest single weight of 60.1% and was followed by the two-knot (22.7%), three-knot (8.5%) and four-knot (4.0%) spline configurations. A visual comparison of the survival data based on the weighted distribution and several single distributions is reported in Figure 7.
FIGURE 7.
Plot comparing fit of weighted distribution and key single distributions to CAR T-cell data: curative intent scenario.
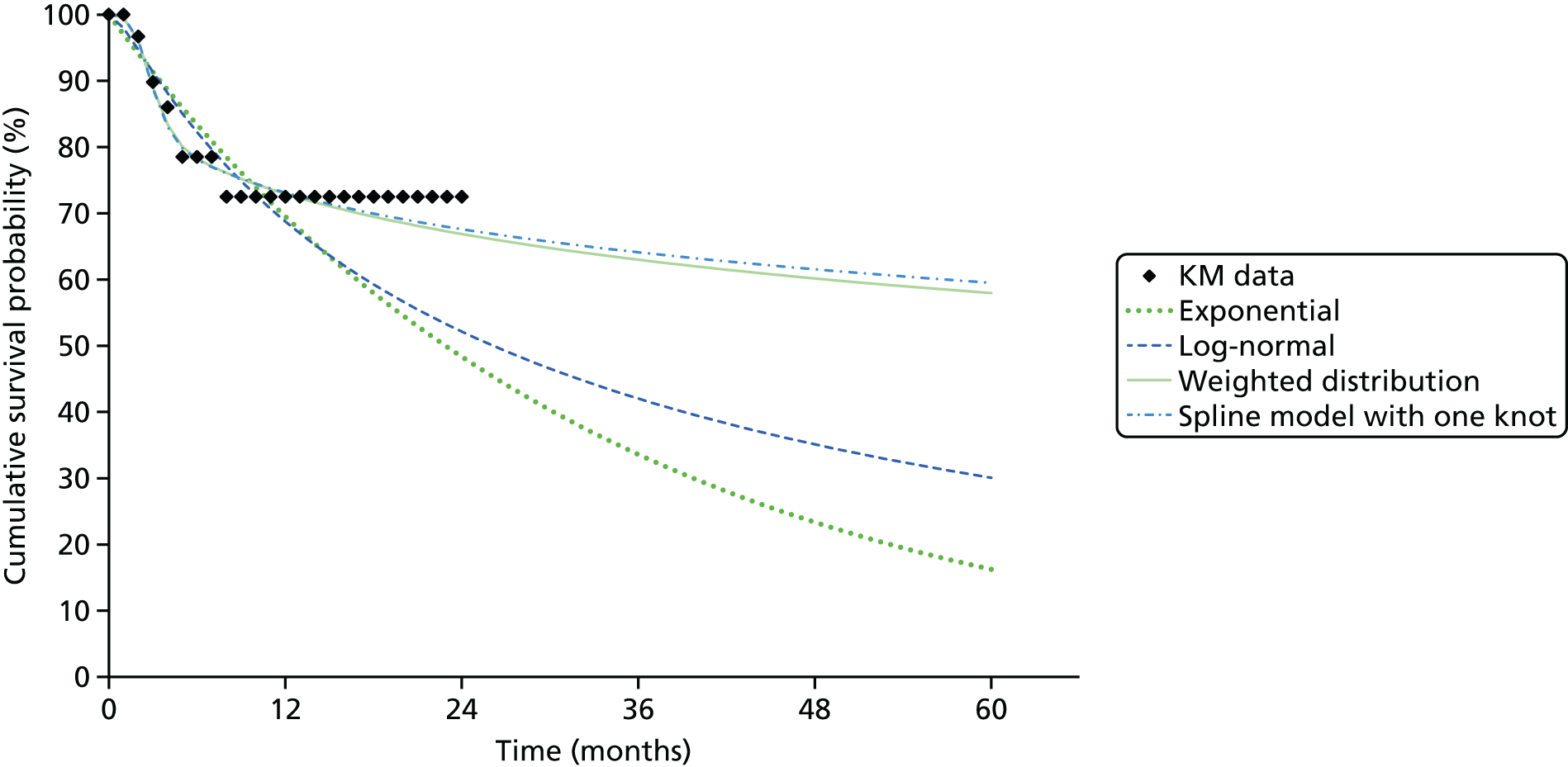
Because of the limited maturity in the minimum data set, there was considerable variation in the predicted long-term survival of the modelled cohort, as shown by the spread of survival trajectories in Figure 7. Although the ‘best-fitting’ spline models appeared to generate a robust fit to the data over the first 3 months of the study, the functions were not able to accurately predict the tail of the distribution. In this case, the ‘best-fitting’ model underestimated the KM probabilities from month 18 of the simulated time horizon. The weak fit of the model to the tail of the KM curve is partly driven by the limited data available to support the continued flattening of the curve. As shown in the following section, with additional data maturity, the parametric models tend to provide a better prediction of the tail of the KM curve as there are more data to support the long-term flattening of the survival curve.
In the standard of care group, the distribution with the optimal predictive validity as judged using the AIC was also the spline model with a single knot (AIC = 288.65). A weight of 44.6% was assigned to the spline model with a single knot, followed by weights of 18.5% for the spline model with two knots and 13.8% for the log-normal model. A visual comparison of the survival data based on the weighted distribution and several single distributions is reported in Figure 8.
FIGURE 8.
Plot comparing fit of weighted distribution and key single distributions to standard of care data: curative intent scenario.
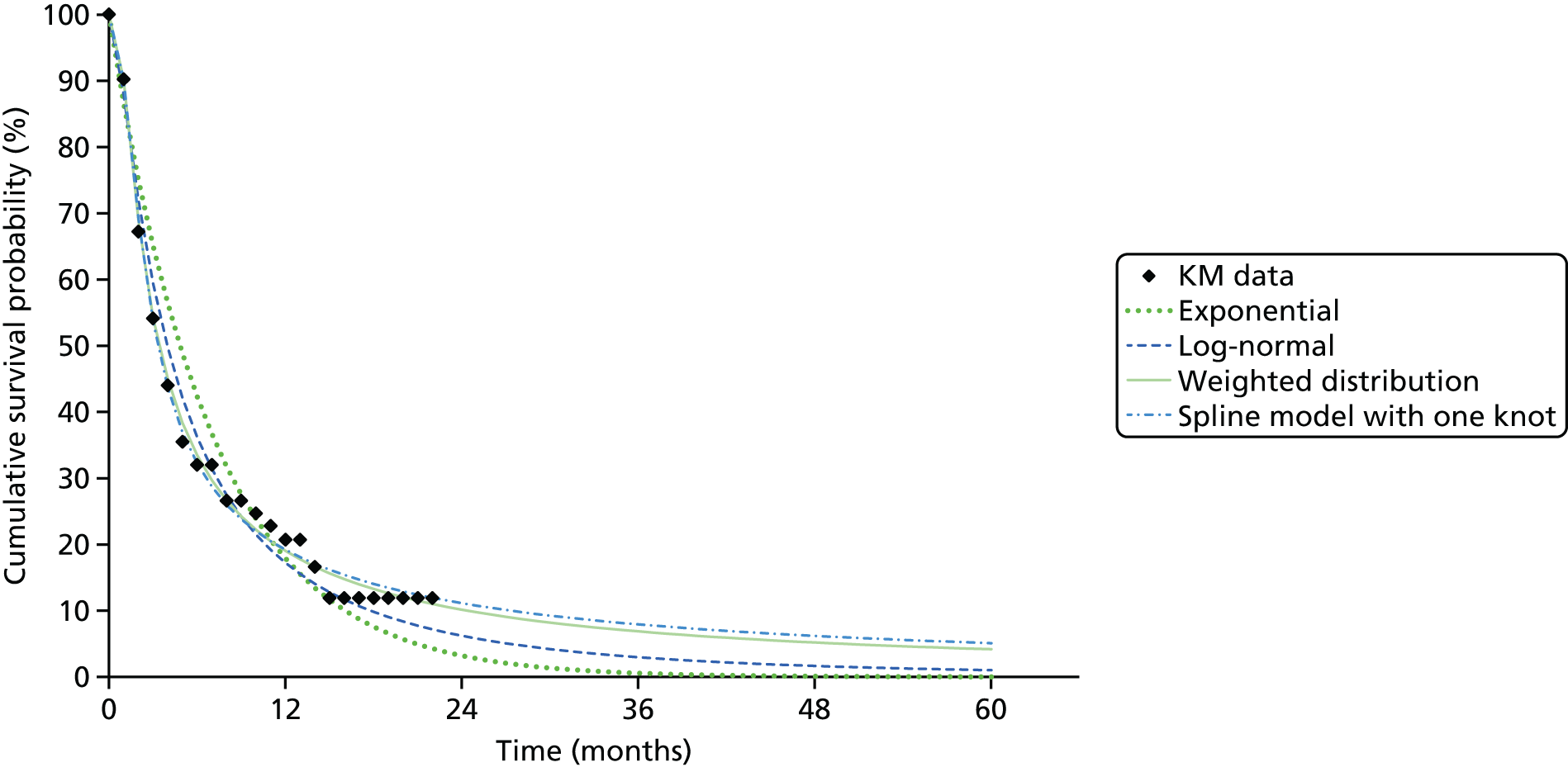
Intermediate and mature data sets
Summaries of the goodness-of-fit statistics and weights are reported for the intermediate and mature evidence sets in Tables 19 and 20, respectively.
| Distribution | CAR T-cell therapy | Standard of care | ||
|---|---|---|---|---|
| AIC | AIC-based weight (%) | AIC | AIC-based weight (%) | |
| Exponential | 157.43 | 0.0 | 345.45 | 0.0 |
| Weibull | 147.51 | 0.0 | 329.54 | 0.0 |
| Gamma | 148.64 | 0.0 | 337.70 | 0.0 |
| Gompertz | 161.81 | 0.0 | 343.38 | 0.0 |
| Log-normal | 143.41 | 0.0 | 308.08 | 0.1 |
| Spline with a single knot | 125.18 | 61.2 | 296.00 | 56.0 |
| Spline with two knots | 126.95 | 25.3 | 298.08 | 19.7 |
| Spline with three knots | 128.84 | 9.8 | 299.58 | 9.4 |
| Spline with four knots | 130.84 | 3.6 | 298.66 | 14.8 |
| Distribution | CAR T-cell therapy | Standard of care | ||
|---|---|---|---|---|
| AIC | AIC-based weight (%) | AIC | AIC-based weight (%) | |
| Exponential | 312.85 | 0.0 | 688.89 | 0.0 |
| Weibull | 291.02 | 0.0 | 655.07 | 0.0 |
| Gamma | 293.28 | 0.0 | 671.39 | 0.0 |
| Gompertz | 339.75 | 0.0 | 644.99 | 0.0 |
| Log-normal | 282.81 | 0.0 | 612.15 | 0.0 |
| Spline with a single knot | 244.36 | 60.3 | 586.00 | 34.0 |
| Spline with two knots | 245.89 | 28.0 | 588.17 | 11.5 |
| Spline with three knots | 247.65 | 11.7 | 589.17 | 7.0 |
| Spline with four knots | NA | NA | 585.32 | 47.6 |
The additional maturity of the data in these evidence sets and the superior AIC statistics associated with the flexible spline models resulted in none of the standard distributions being assigned a weight > 0.1%. The different levels of precision resulted in small differences in the weights assigned to the spline models across the intermediate and mature evidence sets.
Visual comparisons of the survival data based on the weighted distribution and several single distributions are reported in Figure 9.
FIGURE 9.
Plot comparing fit of weighted distribution and key single distributions to KM curves across the data sets: curative intent scenario. (a) CAR T-cell therapy minimum data set; (b) standard of care minimum data set; (c) CAR T-cell therapy intermediate/mature data set; and (d) standard of care intermediate/mature data set.

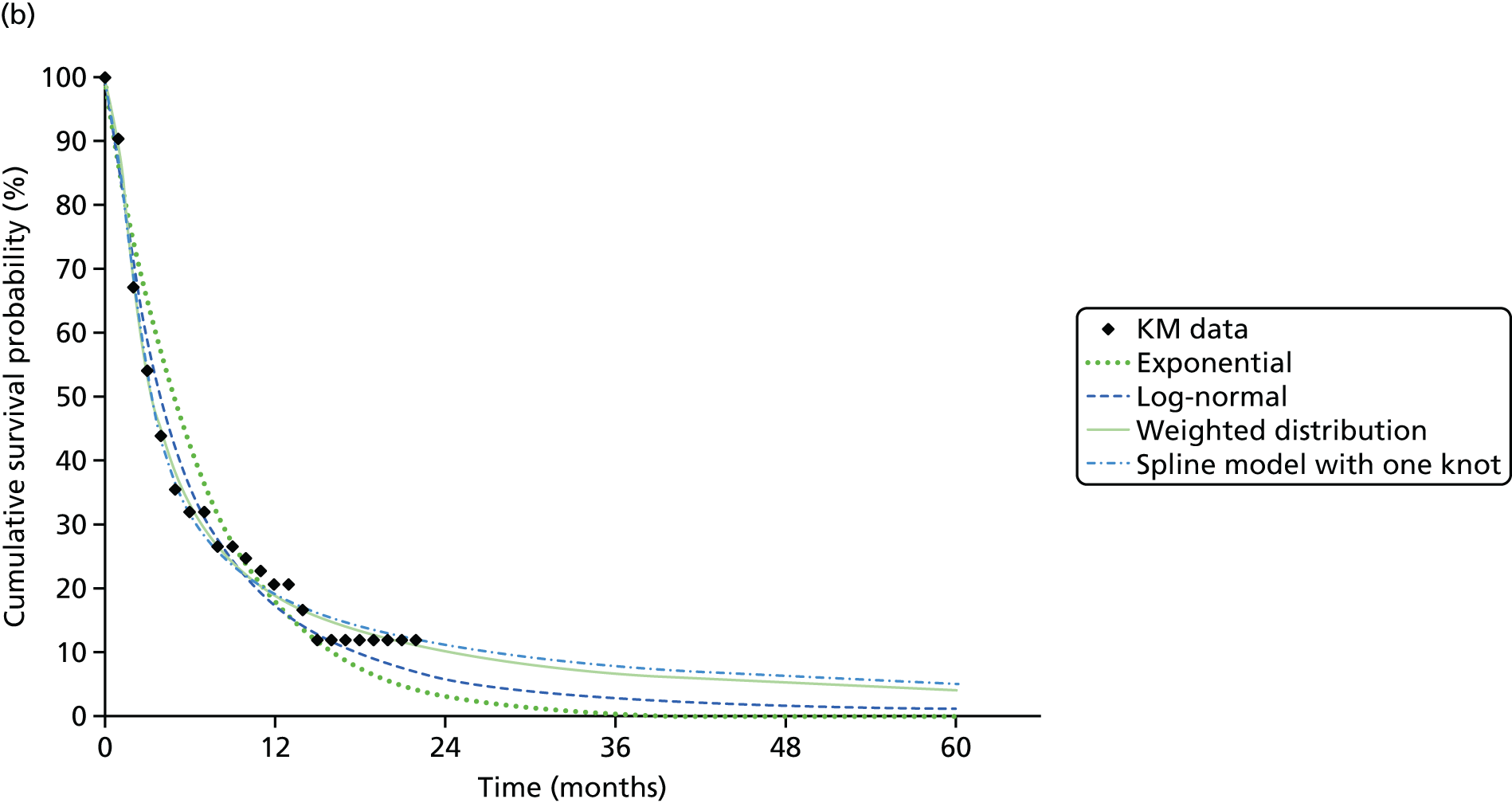
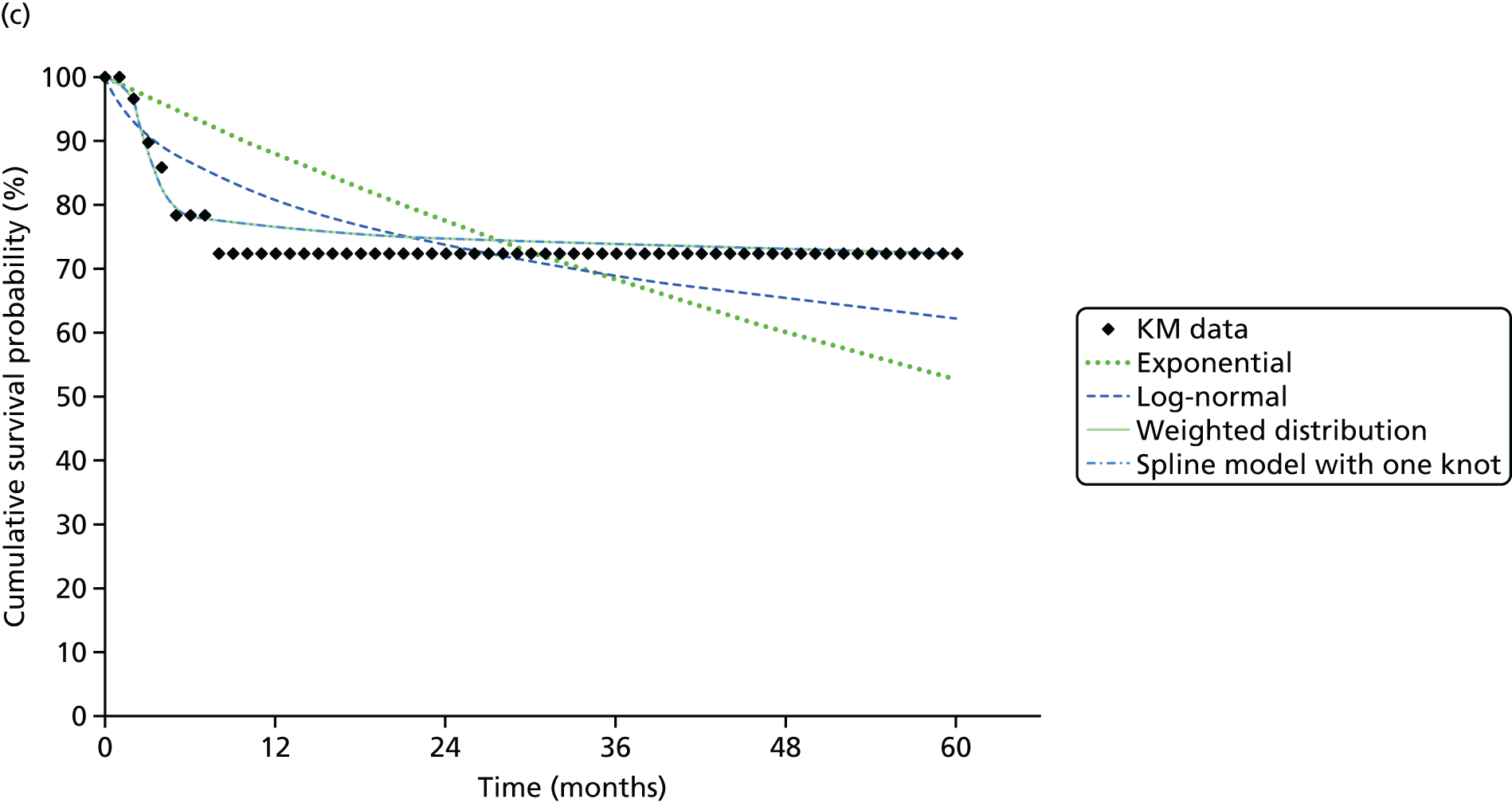
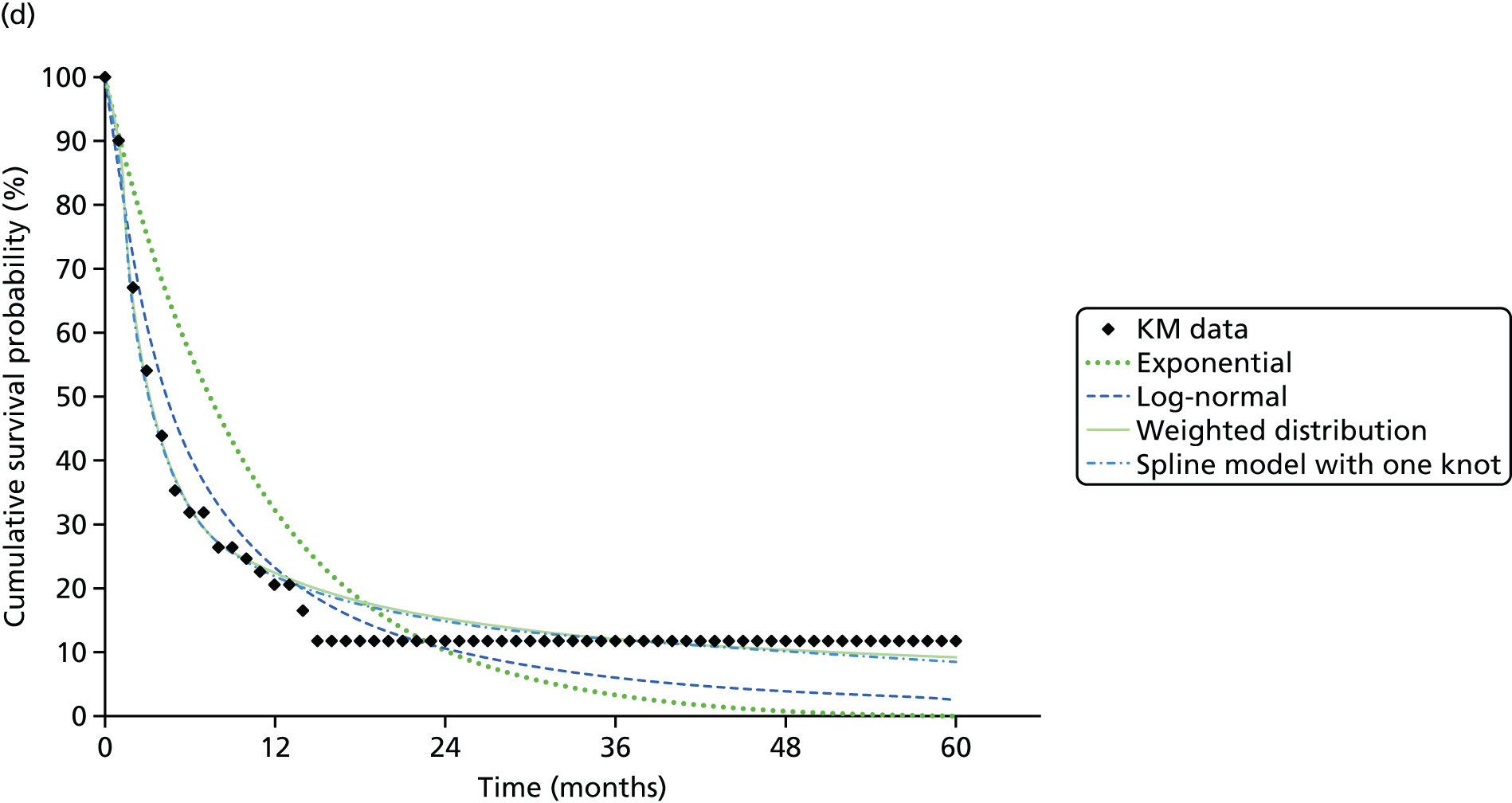
In comparing across evidence sets, the survival models fitted to the intermediate and mature evidence sets appear to have a shallower slope than those fitted to the minimum evidence set, resulting in a longer tail to the predicted survival curves. This is driven by the assumption that in the more mature evidence sets there is greater certainty over the ‘curative’ benefit of treatment because of additional evidence on patient survival up to month 60 of the hypothetical evidence set (vs. maximum survival of approximately 24 months in the minimum data set). This is broadly equivalent to saying that, in the intermediate and mature evidence sets, there is greater certainty over the flattening of the KM curve.
When comparing across competing survival models, the intermediate and mature evidence sets are also associated with a more consistent set of survival projections than the minimum data set. This leads to a narrower range of potential survival probabilities being predicted at later time points in both the intermediate data set and the mature data set. Therefore, unlike the bridge to HSCT model, additional evidence maturity in the curative model leads to a different projection of survival benefit, as well as impacting on the parametric uncertainty surrounding model extrapolations.
There are slight differences in the survival curves predicted from the intermediate and mature evidence sets because of differences in the weights applied to different functions. These differences cannot be clearly seen on the plots as the difference in weights is marginal. The key difference between these evidence sets is the additional sample size assigned to the mature data set, which primarily impacts on the uncertainty/precision surrounding survival estimates, which is not shown on these plots.
Adverse events: B-cell aplasia
A series of survival models was fitted to data on the time to CD19 positivity or relapse reported in Maude et al. 165 and used to predict the proportion of patients requiring treatment for B-cell aplasia. The best-fitting distribution was the Weibull distribution.
The accuracy of the partitioned survival model in predicting the outcomes of the Maude et al. 165 and Jeha et al. 178 studies was assessed by comparing the predicted survival probabilities from the model with the KM data. As shown in Figure 10, the final models appear to provide an accurate prediction of the extracted KM curve for OS in both studies.
FIGURE 10.
Model prediction (lines) vs. extracted KM curves (markers) of OS in all patients across the three evidence sets: (a) minimum data set; (b) intermediate data set; and (c) mature data set.


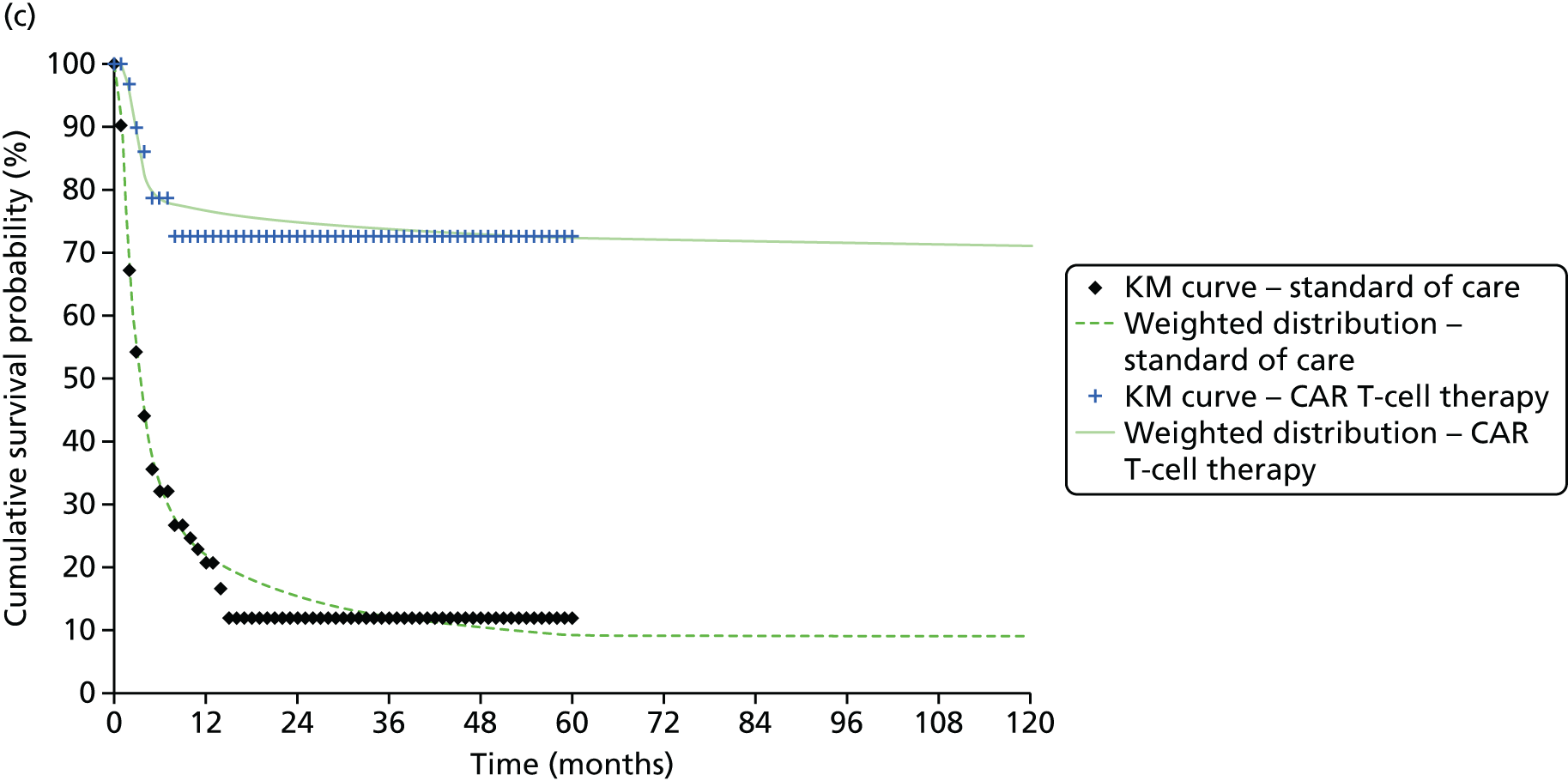
Resource use and costs: bridge to haematopoietic stem cell transplantation and curative models
The resource use and costs incorporated within each separate model were based on the following components:
-
treatment acquisition costs
-
administration and monitoring costs
-
adverse events
-
HSCT
-
long-term costs.
Treatment acquisition costs
Chimeric antigen receptor T-cell therapy
The complex nature of regenerative medicines and the treatment pathway makes it necessary to disentangle the separate procedural elements of the CAR T-cell treatment process and to make assumptions concerning those elements that would be included within the acquisition cost of the therapy itself and those that might represent additional procedural costs that would need to be separately provided and funded by the NHS itself.
Levine et al. 145 summarises the CAR T-cell process as follows: separating the processes of leukapheresis, conditioning chemotherapy and infusion from the transduction and expansion. We assumed the same separation to represent those components of care that would be provided (and funded separately) by the NHS and those that would be undertaken by the manufacturer and included within the acquisition cost of CAR T-cell therapy. Hence, we assumed that the acquisition cost of CAR T-cell therapy would not include the cost to the NHS of providing leukapheresis, conditioning chemotherapy or cell infusion and that these are assumed to represent additional costs to the NHS.
In the absence of licensed products being available, there are currently no commercially available estimates of the acquisition cost of CAR T-cell therapy. Informal sources have indicated that future acquisition costs may be in the region of US$150,000–500,000. 194 Within the exemplar, we have assumed that the manufacturer would employ a value-based approach to pricing, such that the acquisition cost would be set at a level such that the resulting cost-effectiveness (ICER) estimates would be close to NICE’s current threshold range. In the context of the specific population considered, we have assumed that this would be in line with the £50,000 per QALY estimate based on NICE’s current approach to treatments at the EoL. 195 We subsequently explored the impact of alternative prices and payment schemes using separate scenarios. Full details of the hypothetical prices assumed across the separate scenario are reported in Chapter 8.
The acquisition cost of conditioning therapy (£329.86) was estimated from the regimen used in the study by Lee et al. ,164 which was 25 mg/m2 per day of fludarabine on days –4, –3 and –2 and 900 mg/m2 per day of cyclophosphamide on day –2.
The acquisition cost associated with clofarabine was derived from the AWMSG appraisal of clofarabine,171 which reported a cost of £43,200 per patient treated based on the average costs of the drug volumes used in the CLO-212 study178 (based on 1.8 cycles of treatment, a patient body surface area of 1.2 m2 and the licensed dose of 52 mg/m2/5-day treatment cycle).
The acquisition costs of FLAG-IDA were considered as part of a separate sensitivity analysis and were estimated by applying unit costs from the British National Formulary196 to a dosing guide published by the Royal Surrey NHS Trust. 197 Assuming an average body surface area of 1.2 m2 and an average of 1.76 cycles of treatment198 gives the estimate of £3809 per patient.
Administration and monitoring costs
In addition to the acquisition costs, it is important to consider the resource use and costs associated with administration and subsequent monitoring. All patients regardless of subsequent treatment are assumed to require an initial non-elective hospitalisation. For clofarabine and FLAG-IDA it is assumed that the costs of this hospitalisation also include all costs associated with the monitoring and administration of treatment. For CAR T-cell therapy, the same initial hospitalisation is assumed to occur for the administration of the conditioning therapy. However, because of the additional production period required to manufacture the CAR T-cells (in the region of 11 days currently), an additional elective hospitalisation is also assumed during which CAR T-cells are subsequently administered and the patient monitored. The cost of a single leukapheresis procedure is also applied to CAR T-cell patients.
Table 21 reports these per-patient costs and the sources and associated assumptions.
| Parameter | Cost | Source/assumption |
|---|---|---|
| 1. Acquisition costs | ||
| 1a. CAR T-cell therapy | ||
| Acquisition cost of CAR T-cell therapy | Threshold analysis | Threshold price analysis based on three approaches detailed in the accompanying text |
| Conditioning therapy | £329.86 per patient | Acquisition costed directly from Lee et al.164 assuming full use of 2 × 50-mg fludarabine vials and 1 × 500-mg and 1 × 1-g vials of cyclophosphamide and a body surface area of 1.2 m2;198 infusion costs assumed included in CAR T-cell therapy administration costs |
| 1b. Clofarabine | ||
| Acquisition cost of clofarabine | £43,200 per patient | Cost presented in AWMSG FAR for clofarabine,171 excluding costs of administration |
| 1c. FLAG-IDA | ||
| Acquisition cost of FLAG-IDA | £3808.57 per patient | Cost per cycle estimated from the Royal Surrey NHS Trust guide,197 average body surface area of 1.2 m2 and the average number of cycles of FLAG-IDA of 1.76198 |
| 2. Administration and monitoring costs | ||
| 2a. CAR T-cell therapy | ||
| Leukapheresis | £1627 per patient | Weighted average of HRGs for stem cell and bone marrow harvest199 |
| Initial hospitalisation for conditioning | £7179.99 | HRG paediatric ALL admissions weighted average non-elective long stay199 |
| Additional hospitalisation for CAR T-cell treatment | £5831.72 | HRG paediatric ALL admissions weighted average elective inpatient199 |
| 2b. Clofarabine | ||
| Hospitalisation over treatment period | £7179.99 | HRG paediatric ALL admissions weighted average non-elective long stay199 |
| 2c. FLAG-IDA | ||
| Hospitalisation over treatment period | £7179.99 | HRG paediatric ALL admissions weighted average non-elective long stay199 |
| 3. Adverse events | ||
| CRS | £2857.99 per patient per grade 4 or severe CRS event | Combination of the drug cost (£1193 HRG for cytokine inhibitor drugs) plus cost of ICU hospitalisation (£1664.99 HRG weighted ALL advanced critical care paediatric ICUs)199 |
| B-cell aplasia | £1075 per month per patient for the first 3 months | Dose of 0.5 g/kg of IVIG every 4 weeks until the patient is no longer in need of treatment (i.e. CD19 positivity, relapse or death)196 |
| Febrile neutropenia | £0 | Assumed included in CRS costs |
| Encephalopathy | £539.24 per patient per adverse event | HRG paediatric ALL admissions weighted excess bed-day non-elective inpatient stay199 |
| Hypotension | ||
| Neutropenia | ||
| Anaemia | ||
| Thrombocytopenia | ||
| Leukopenia | ||
| Hypokalaemia | ||
| Hypophosphataemia | ||
| 4. HSCT | ||
| Transplantation | £89,879.15 per patient | Weighted average of paediatric transplant HRGs, elective inpatients only199 |
| Follow-up costs | £61,965 per living patient | Sum of follow-up costs from UK Stem Cell Strategy Oversight Committee report200 (< 6 months = £28,390, 6–12 months = £19,502, 12–24 months = £14,073). In the model these will be included as time and OS dependent |
| 5. Long-term costs | ||
| Post non-HSCT population | £7179.77 at point of death | HRG paediatric ALL admissions weighted average non-elective long stay199 |
| Curative model population | £7179.77 at point of recurrence | HRG paediatric ALL admissions weighted average non-elective long stay199 |
Adverse events
The individual costing of each adverse event for the alternative treatments could entail double counting, as some aspects of these may already be included in the hospitalisation costs used for the administration and monitoring costs of each treatment. Therefore, an assumption is made that all grade 3 and 4 adverse events (except CRS and B-cell aplasia, as discussed below) require an extension of hospitalisation by 1 day, with a cost based on the excess bed-day HRG cost as shown in Table 21.
For CRS, a combination of the acquisition cost of cytokine inhibitor drugs and the cost of an admission to a paediatric intensive care unit is assumed for all cases of grade 4 or severe CRS.
B-cell aplasia is assumed to be treated with a regimen of IVIG, given at a dose of 0.5 g/kg every 4 weeks until the patient is no longer in need of treatment (i.e. CD19 positivity, relapse or death). We assumed the population treated to have an average weight of 49.5 kg. Rounding down of each dose to the nearest vial in line with national prescribing recommendations201 (i.e. 20-g vial per dose), the cost per vial is estimated as £850. In addition, an administration cost of £225 per dose is assumed.
Haematopoietic stem cell transplantation
Three potential sources of cost estimates of HSCT were identified and considered:
-
NHS reference costs. 199 This provides estimates of completed HRG activity and unit costs across six different paediatric allogeneic transplantation categories. Although intuitively appealing because of the relevance to our population and UK context, concerns have been raised200 that these do not capture the full cost of HSCT because of their focus on a single admission period.
-
London Specialised Commissioning Group report. 202 This report estimated a national tariff for adult blood and bone marrow transplants based on the phases of transplantation from decision to transplant to 100 day post-transplantation follow-up care. However, no details are given on how the estimate was derived. In addition, the estimate considers only an adult population.
-
UK Stem Cell Strategy Oversight Committee report. 200 This report used the results from a Dutch study published in 2002 that reported the cost of allogeneic adult unrelated bone marrow transplantation. This estimate includes all initial costs of the transplantation as well as follow-up costs for up to 2 years after the transplantation. The inclusion of the longer-term follow-up costs addresses the primary concern around existing NHS reference costs. However, there is uncertainty about the generalisability of the cost to the specific population considered here.
To take account of the limitations around each of the three data sources, the model combines estimates from both the NHS reference costs and the UK Stem Cell Strategy Oversight Committee report. The London Specialised Commissioning Group report was discounted because of a lack of detail on how the estimate was derived.
The cost of HSCT is considered in two parts: (1) the cost of the procedure and associated hospitalisation and (2) the cost of long-term care. Although all three sources provide an estimate of the cost of the procedure, both the London Specialised Commissioning Group and the UK Stem Cell Strategy Oversight Committee focus on adult populations. Existing HRG costs report a higher cost of the procedure for paediatric patients, with paediatric HRG costs of between £21,622 and £74,434 more than the equivalent adult HRG costs across the four different forms of allogeneic transplantation reported. 199 Therefore, the cost of the procedure has been estimated as the weighted average (by frequency of HRG) of all paediatric allogeneic transplantations from the HRG costs.
As previously noted, the HRG costs include only the costs accrued during the admission in which the transplantation occurred. Hence, any longer-term costs will not be included. To estimate the longer-term costs, an estimate of post-transplantation costs from the UK Stem Cell Strategy Oversight Committee report was used. 200 No further adjustment was made to the estimate. In using this estimate the same assumptions were made about the appropriateness of the original source of the costs. 203 It was additionally assumed that, unlike the cost of the procedure, long-term costs are independent of type of transplantation and age of patient at the time of transplantation.
Model inputs: utilities
Literature review
A pragmatic approach was taken to identify potentially relevant sources for health utilities. Google and Google Scholar were used to search for publicly available utility estimates, alongside a search of known economic evaluations and HTA appraisals in ALL (see Appendix 7). The search focused on utility estimates for children with ALL, regardless of treatment provided. Two systematic reviews of utility studies in paediatric ALL were identified. 204,205
Van Litsenburg et al. 205 reviewed the measurement of HRQoL (used synonymously with utilities) in paediatric patients with ALL using the Health Utilities Index (HUI). The study identified 15 studies reporting utilities in this population using both HUI2 and HUI3. The van Listenburg et al. 205 review has several issues that limit its relevance to our model. First, no attempt was made to meta-analyse the results, with the review summarising only the individual utility estimates from each study. In addition, the results were reported by phase of care, often focusing on specific time points in the treatment pathway rather than on specific health states relevant to our modelling. Given the time constraints in our work, a more detailed consideration of each study was not considered feasible.
Kelly et al. 204 undertook a decision analysis of cranial radiation therapy for paediatric T-ALL patients, including a systematic review of utility studies to inform this. Although the study focused on T-ALL, the review of utilities did not stipulate type of ALL and hence included all forms of ALL. The study used existing mapping functions to convert generic HRQoL measures [Short Form questionnaire-36 items (SF-36) and Child Health Rating Inventories (CHRIs)] to preference-based utility estimates (HUI2 and EQ-5D). Of particular relevance to our model were the states of ‘in the state of relapse’ and ‘cured after relapse’, with mean utility estimates of 0.75 (range 0.44–1) and 0.91 (0.87–0.95), respectively.
In addition, the pragmatic search also identified a number of published economic evaluations that had used utility estimates. 171,182,183,185
Of the three AWMSG FARs related to ALL, one did not report any utility results from the manufacturer’s submission (dasatinib). 186 The clofarabine FAR171 reported that all patients who survived post 1 year after HSCT were assumed to have the utility of the general population. All other states modelled were varied between 0.2 and 1 as scenario analyses to demonstrate that the results were not sensitive to the utility values of those who do not survive long term. The nelarabine FAR185 reported that non-responders and untreated patients were assumed to have a utility of 0.64. This value was referenced from Health Outcomes Data Repository data from patients with lymphoid leukaemia and, as such, represents patients in secondary care. In addition, all patients who undergo successful transplantation were assumed to have a utility of 0.92 based on the study by Sung et al. 206
The study by Sung et al. 206 considers physician-elicited estimates of utility for acute myeloid leukaemia patients who have survived without recurrent disease post transplantation. Sung et al. 206 additionally present estimates of disutility (i.e. decrement associated with an event) associated with treatment with chemotherapy and transplantation, estimated as 0.42 (plausible range 0.16–0.83) and 0.57 (plausible range 0.31–0.87), respectively. No estimates of the duration of these disutilities are presented.
Similar to the study by Sung et al. ,206 the economic evaluation of clofarabine for paediatric ALL conducted by Lis et al. 183 included an elicitation exercise involving physicians because of a lack of relevant utility estimates available at the time. Lis et al. 183 reported utility estimates for treatment with palliative care (0.26), clofarabine without HSCT (0.34) and clofarabine with HSCT but surviving for < 1 year (0.48), as well as for survival post HSCT for 1 year (0.80), 2 years (0.85) and beyond (0.88).
Although these values appear to be generally consistent with the results reported within the systematic reviews, the magnitude of the treatment disutilities appears higher. It is plausible that this discrepancy may be the result of the use of physician rather than patient utility elicitation.
Informing the model states
All model utility inputs applied in the model are summarised in Table 22.
| Parameter | Utility (95% CI) | Source/assumption |
|---|---|---|
| Treatment disutilities | ||
| HSCT disutility | 0.57 for 1 year (0.33 to 0.87) | Sung et al.:206 ‘disutility of undergoing BMT’ expert VAS elicitation |
| Adverse events | ||
| CRS | 0 for 1 week | Assume severity of ICU hospitalisation associated with utility of 0 |
| Short-term utility | ||
| Relapse | 0.75 (0.44 to 1) | Kelly et al.:204 ‘in the state of relapse’ mapped value from CHRIs to EQ-5D |
| Remission | 0.91 (0.87 to 0.95) | Kelly et al.:204 ‘cured after relapse-all relapsed patients treated with CRT’ mapped value from SF-36 to HUI2; need to assume no long-term disutility adverse events from CRT |
| Long-term utility | ||
| Long-term disutility | Remission utility (0.91) with age-adjusted decrement | To reflect ageing of cohort |
Treatment disutilities
Because of a lack of literature on the short-term impacts on health utility associated with both chemotherapy and HSCT, we based our estimates on the study by Sung et al. 206 A decrement in utility of 0.57 for HSCT and 0.42 for all forms of chemotherapy was assumed. Both estimates were assumed to incorporate all short-term adverse events associated with both treatments. However, Sung et al. 206 failed to report any estimate of duration associated with the estimated disutility for either treatment. Therefore, we assumed that disutilities apply for 1 year post treatment initiation. As the disutility estimate for all forms of chemotherapy is the same in both treatment arms, the impact will cancel out and was therefore excluded from our model.
Adverse events
As discussed in the previous section, all HSCT and chemotherapy adverse events are assumed to be incorporated in the treatment disutility estimates applied. The only additional adverse events to consider are those specifically associated with CAR T-cell therapy. As discussed in the cost section, only CRS and B-cell aplasia are expected to be associated with a potential additional burden not considered elsewhere in the model. The pragmatic literature review was unable to find any specific estimates of disutility or duration associated with either adverse event.
For severe (grade 4) CRS it was assumed that, because of the severity of initial onset of the event and associated intensive care admission, a utility of 0 is incurred for 1 week. For B-cell aplasia, although there is a large cost burden associated with its management, there is little evidence of any significant impact on patient utility. In existing CAR T-cell studies, B-cell aplasia appears to be either well managed or short-lived, with no reported cases of associated intensive care hospitalisation. Therefore, no disutility was assumed for cases of B-cell aplasia.
Short-term health-related quality of life
The model considers the short-term response as either relapse or remission. The utility estimates to inform these states were derived from the study by Kelly et al. ,204 with a utility of 0.75 assigned to the relapse state and 0.91 to the remission state.
Long-term health-related quality of life
Patients with the severe form of ALL considered in the model are likely to experience long-term comorbidities associated with the disease and associated disutility. As such, the utility score estimated for the state of remission was applied with an additional age-related decrement.
Conclusions
Two de novo decision models were developed to assess the cost-effectiveness of CAR T-cell therapy within the two separate TPPs (bridge to HSCT and curative intent) across each of the separate evidence sets. Although a number of common inputs and assumptions were employed across both models, the two models had important structural differences that led to differences both in the underlying modelling approach and in the use of external evidence.
In the bridge to HSCT scenario, the primary health benefits of treatment with CAR T-cell therapy were assumed to be driven by an increase in the proportion of patients receiving HSCT and the subsequent success of HSCT itself (based on remission and MRD status). The introduction of an epidemiological ‘link’ between a potential established surrogate outcome and/or process (i.e. MRD and HSCT status) and final health benefits (i.e. OS and QALYs) also enabled the use of external evidence to be utilised alongside the separate hypothetical evidence sets generated. A landmark response model was developed utilising evidence from the hypothetical evidence sets to inform short-term outcomes of remission, HSCT and MRD status and external evidence to estimate OS conditional on these shorter-term outcomes. Hence, the key assumption employed within this scenario is that external evidence substantiating the relationship between MRD and HSCT status in studies in which CAR T-cells have not been used can be generalised to patients in whom CAR T-cells have been used. Importantly, the results of our validation work appear to demonstrate that, with minor calibration and adjustment, the combination of trial-reported evidence on short-term outcomes (remission, HSCT and MRD status) and external evidence on their relationship to OS appeared to closely match the OS estimates directly reported within the studies used to generate the evidence sets for CAR T-cell therapy and the comparator (clofarabine).
In the curative intent model a different assumption was employed, specifically that the CAR T-cell therapy itself potentially confers longer-term and potentially curative benefits without the need to bridge to HSCT. In this context, the case for use of a structural link between final health benefits and a surrogate outcome or process such as HSCT appears more limited. Instead, a simple three-state partitioned survival model was developed to model long-term outcomes through the direct extrapolation of OS data from the evidence sets. An important consideration within this model was whether or not the use of conventional parametric survival functions (e.g. exponential, Weibull, log-normal) would adequately capture the potential for a less conventional hazard function that might be observed for a curative treatment and how this might be affected by different levels of precision and maturity of evidence. Consequently, our work considered the goodness of fit of conventional survival functions and more flexible survival models (e.g. spline-based models developed by Royston and Parmar192). A key finding was that the more flexible survival models appeared to more closely approximate the observed hazard function across each of the evidence sets. To our knowledge, although the use of these more flexible survival models is briefly discussed within existing NICE technical support documents,207 we are not aware of any examples of their use to date by manufacturers or AGs within the NICE TA process. Consequently, further research may be required to more formally consider the appropriateness of alternative survival modelling approaches to regenerative medicines and cell-based therapies, including more flexible models and cure fraction models. 208
The importance of the level of data maturity in deriving robust survival projections for the economic model was evident in our results. Although the ‘best-fitting’ spline models appeared to generate a robust fit to the data over the first 3 months of the KM estimate used in the minimum data set, the functions were not able to accurately predict the tail of the distribution. Furthermore, considerable variation was evident in the predicted long-term survival of the modelled cohort, with a significant spread in the projected survival trajectories employing different parametric functions. Consequently, we concluded that it was unlikely that a single survival distribution could adequately characterise uncertainties over the longer-term extrapolation period. Although the robustness of the ICER estimates to alternative distributions can be explored in separate sensitivity analyses or scenarios, concerns may exist regarding the transparency of subsequent decisions if the weighting of these is not explicitly specified in subsequent policy decisions.
To more formally account for the uncertainty surrounding choice of survival distribution, a model-averaging approach was adopted. This technique involves the parameterisation of uncertainty surrounding the choice of distribution, combining results from a series of alternative survival functions as part of a weighted distribution. This approach samples both the parametric uncertainty associated within each distribution and the uncertainty (or weights) surrounding the choice of preferred method. Through the probabilistic analysis, it is therefore possible to estimate the joint distribution of uncertainty around the parameter estimates and the choice of survival function.
In contrast to the minimum set, the additional data maturity in the intermediate and mature evidence sets results in greater certainty over the long-term survival benefits of treatment. This leads to reduced variability in the potential trajectories for the survival benefits of treatment. In addition, with more mature evidence, the fitted survival models are better able to predict the tail of the KM curve. Therefore, unlike the bridge to HSCT model, additional evidence maturity in the curative model leads to different projections of survival benefit, as well as impacting on the parametric uncertainty surrounding model extrapolations. The weights in the exemplar model were based on standard measures of statistical fit. However, these weights could also be informed by clinical judgement and the committee’s deliberations.
Given the inevitable uncertainties that are likely to exist regarding the longer-term benefits of regenerative medicines and cell-based therapies and their implications for the robustness of subsequent cost-effectiveness estimates, further methodological research could be usefully undertaken to help inform how these uncertainties might be appropriately quantified in a transparent manner to inform subsequent decisions. A key consideration here would be the extent to which these weights can be defined prior to the committee’s deliberations or whether they should be more directly informed by them. Given the potential complexity in both undertaking these analyses and communicating the results, more efforts should be made to ensure that informal judgements can be more explicitly incorporated in a timely and transparent manner. 209
A key assumption employed within both models is that from year 5 onwards all patients who remained alive were assumed to experience a similar mortality risk profile as that of a long-term survivor of ALL. Hence, the mortality risks assumed in both models after year 5 were based on matched general population estimates of the all-cause risk of mortality adjusted for excess morbidity and mortality reported in cohorts of long-term survivors of ALL. As data were not assumed to be available beyond 5 years, it is not possible to determine the possible direction and/or magnitude of any possible bias that this approach might introduce. However, this period is consistently utilised within existing studies of ALL and appears clinically to represent an important time point for patients to reach without subsequent relapse. Hence, for the purposes of extrapolation and the exemplar, it was considered a reasonable basis for informing subsequent longer-term extrapolations. This assumption also impacted on reducing some of the longer-term uncertainties that would inevitably arise from the extrapolation of the data beyond the maximum reported follow-up across the evidence sets considered for CAR T-cell therapies. Clearly, if additional follow-up data were available, then the validity of such an approach could be more formally considered and any claims of longer-term benefits could be more robustly substantiated.
Our searches to inform other model parameters identified other important uncertainties. The existing HRQoL data on ALL were limited and several assumptions were required. Importantly, no existing CAR T-cell study had incorporated measures of HRQoL that could be considered directly in the model. In the absence of these data, assumptions were made based on external studies to account for the possible magnitude of HRQoL benefits of achieving remission, alongside any negative impacts resulting from the model of therapy (i.e. HSCT, chemotherapy) and other specific adverse events. Our model focused specifically on the impact of CRS and B-cell aplasia. Importantly, no studies were identified on the potential HRQoL impact of these specific events, which are likely to be associated with CAR T-cell therapy, necessitating the use of potentially arbitrary assumptions. Further research to generate more robust estimates of HRQoL appropriate for cost-effectiveness analysis is clearly required, together with more specific research that more formally demonstrates the impact of specific therapeutic modalities (including CAR T-cell therapy).
Finally, our research also identified important uncertainties regarding both the likely acquisition costs of CAR T-cells and the other key elements of the process (e.g. costs of leukapheresis, costs of conditioning therapies, level of hospitalisation required for different aspects such as conditioning, subsequent administration and monitoring costs). Furthermore, no account was taken of the potential costs incurred by patients and their families. Based on previous NICE TA appraisals, additional evidence would need to be provided by manufacturers to more robustly determine the potential costs to the NHS to avoid these uncertainties regarding the costing assumptions being raised. An important uncertainty identified related to the costs of HSCT and any additional costs that may arise from longer-term management of patients. A variety of possible sources were identified in our review and important differences were observed across these. Further studies would be useful to more formally cost the short-term and longer-term implications of HSCT in paediatric populations and also to determine the generalisability of studies reporting estimates from outside the UK.
Although the existence of possible learning curves was identified as an important issue in the conceptual review, these were not directly considered within the exemplar. Some aspects of these may become more apparent as larger studies report, particularly those involving centres with different levels of expertise. Hence, some aspects of learning may be reflected within the results from larger studies and/or specific factors may become more apparent in terms of how these might be incorporated within cost-effectiveness assessments. For example, as experience with using CAR T-cell therapies develops, this may have important implications for both the identification and the management of potential adverse events, as well as the provision of the therapy itself. An assumption is made in the exemplar model that the different stages of CAR T-cell therapy would require separate hospitalisations (i.e. for the initial conditioning therapy and later for the subsequent administration of the CAR T-cells and ensuring monitoring). However, as experience and knowledge continues to develop, aspects of the process may evolve over time such that the subsequent administration and monitoring may be undertaken in a less resource-intensive setting. Although the existence of learning curves has received significant attention in the clinical literature, to date, their implications for and application within cost-effectiveness analysis remain limited and warrant further investigation. 133
Finally, an important assumption made within the exemplar relates to the acquisition cost of CAR T-cell therapy itself. In the absence of a commercially available product and published price, an assumption was made that the manufacturer would employ a value-based approach to its decision such that the resulting cost-effectiveness (ICER) estimate was close to NICE’s cost-effectiveness threshold. In the context of the exemplar, this was assumed to be based on the maximum range of the threshold considered by NICE, assuming that the existing EoL criteria are met. Importantly, this price is not considered to be indicative of the final acquisition cost that might be set when commercially available products are available. Neither are we making the assumption that NICE’s current EoL criteria would apply. Rather, the basis for setting the price using the existing cost-effectiveness threshold was to enable different interested parties to better understand the potential impact of other uncertainties (e.g. precision and maturity of evidence) within NICE’s current decision-making process, identifying potential trade-offs that may exist and illustrating how these uncertainties might be more explicitly addressed within different MEAs (i.e. evidence generation and/or pricing schemes). Although it is clearly possible to examine a range of different possible prices for the CAR T-cell therapy within the exemplar, it was considered that this approach may result in the subsequent panel decision process becoming unmanageable (i.e. multiple pricing scenarios) and would lessen the generalisability learning that the exemplar was developed to highlight.
Chapter 8 Assessment of cost-effectiveness, uncertainty and the value of alternative policy options
Overview
The exemplar in Chapter 7 was developed to highlight some of the specific challenges that may present themselves to manufacturers and AGs in terms of developing and populating a cost-effectiveness model. Consideration is now given to how such estimates could be presented and communicated to the committee. In doing this, we consider the analyses routinely requested within NICE’s existing methods guide122 but also consider whether further analyses may provide useful additional insights to help inform subsequent committee deliberations.
Based on the scoping review reported in Chapter 3 (see Scoping review of potential cost-effectiveness issues), we also considered analyses relating to some of the broader issues and approaches identified previously (e.g. alternative payment mechanisms), which, although potentially outside the existing remit of NICE, may provide additional insights to other interested bodies and manufacturers.
Importantly, the use of non-reference case approaches and additional analyses beyond those requested within NICE’s existing process and methods guide is not intended to be prescriptive. Neither are they comprehensive, given the multiplicity of issues and challenges raised. Instead, they have been provided to help explore whether or not additional information and analyses may be helpful in informing the committee’s deliberations and the nature of such analyses.
Consideration will subsequently be given to whether particular analyses helped to inform particular considerations within NICE’s deliberations within the exemplar appraisal and to identify areas in which further methodological and applied work may be required.
Acquisition costs of chimeric antigen receptor T-cell therapies
As noted in Chapter 7 (see Chimeric antigen receptor T-cell therapy), the acquisition cost for CAR T-cell therapy in the exemplar was assumed to be based on a value-based approach from the manufacturer, such that it would be priced at a level so that the ICER for CAR T-cell therapy would be close to the upper limit of NICE’s EoL threshold range (circa £50,000 per QALY gained). Because of differences in the projected survival benefits of treatment across the separate TPPs, the subsequent cost of CAR T-cell therapy varied across the TPPs, with one-off acquisition costs of £356,100 assumed in the bridge to HSCT scenario and £528,600 in the curative intent scenario.
A full summary of the CAR T-cell therapy acquisition costs assumed across the separate pricing scenarios described in subsequent sections (one-off fixed cost, monthly leasing price, discounted list price via PASs) is provided in Table 23.
| Scenario | One-off acquisition cost per patient (£) | Monthly leasing price (£) | Discounted list price (10%) per patient (£) |
|---|---|---|---|
| Bridge to HSCT | 356,100 | 2756.27 | 320,490 |
| Curative intent | 528,600 | 3282.66 | 475,740 |
Bridge to haematopoietic stem cell transplantation target product profile
Per-patient analyses: minimum evidence set
The sequence of assessments starts with a conventional assessment of cost-effectiveness at the patient level based on the minimum evidence set reported in Chapter 5. Disaggregated costs and outcomes are presented in Table 24.
| Outcome | CAR T-cell therapy | Standard of care | Increment |
|---|---|---|---|
| Costs (£) | |||
| Course of treatment (including conditioning) | 358,057 | 43,200 | 314,857 |
| Hospitalisation for treatment | 13,012 | 7180 | 5832 |
| Adverse event costs | 2750 | 442 | 2308 |
| HSCT and related follow-up costs | 71,918 | 21,380 | 50,538 |
| Non-HSCT follow-up costs | 3391 | 3759 | –368 |
| Total costs | 449,128 | 75,962 | 373,166 |
| QALYs | |||
| Decision tree | 0.14 | 0.11 | 0.03 |
| Post HSCT MRD negative | 8.82 | 0.30 | 8.52 |
| Post HSCT MRD positive | 0.00 | 0.16 | –0.16 |
| Post HSCT no remission | 0.00 | 0.72 | –0.72 |
| No HSCT remission | 0.06 | 0.03 | 0.03 |
| No HSCT no remission | 0.07 | 0.11 | –0.04 |
| QALY loss from HSCT | –0.27 | –0.08 | –0.19 |
| Total QALYs | 8.82 | 1.36 | 7.46 |
| Total life-years | 10.60 | 1.77 | 8.83 |
| Proportion of patients receiving HSCT (undiscounted) | 48% | 15% | 33% |
The mean incremental cost of CAR T-cell therapy compared with standard of care over a patient’s lifetime was estimated to be £373,166, with CAR T-cell therapy resulting in an additional 7.46 QALYs. The incremental cost per QALY gained over a lifetime horizon was £49,995 (Table 25), which can be compared against the cost-effectiveness threshold. This can also be expressed as the per-patient net health effect (NHE), including benefits, harms and NHS/Personal Social Services costs. The NHE is the difference between any health gained with the intervention and the health forgone elsewhere in the health-care system and can be expressed in both monetary and QALY terms. With an ICER of approximately £50,000 per QALY, the incremental NHE at a threshold of £50,000 is close to zero (i.e. 0.001 QALYs or £41 per patient), that is, the additional health gained with the intervention is almost exactly offset by health forgone elsewhere.
| Per-patient level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||
|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) |
| CAR T-cell therapy | 449,128 | 8.82 | 49,995 | –0.158 (–7919) | 0.001 (41) |
| Standard of care | 75,962 | 1.36 | –0.159 (–7960) | – | |
Given the uncertainties surrounding longer-term outcomes, it may also be informative to consider how incremental NHEs accumulate over time or the ‘investment profile’ with CAR T-cell therapy, shown in Figure 11. The initial per-patient cost for CAR T-cell patients is attributable to the additional acquisition and administration costs of the CAR T-cells and associated HSCT costs. The ‘kink’ in the curve that appears early on represents the associated HSCT costs, which are assumed to be incurred at day 28. These negative NHEs are gradually offset by positive NHEs in later periods resulting from the ongoing mortality benefits assumed from successfully bridging to HSCT. However, it is only after 60 years that the initial losses are sufficiently compensated for by later gains, that is, CAR T-cell therapy appears to be close to breaking even (i.e. NHE ≥ 0).
FIGURE 11.
Investment profile: bridge to HSCT TPP.
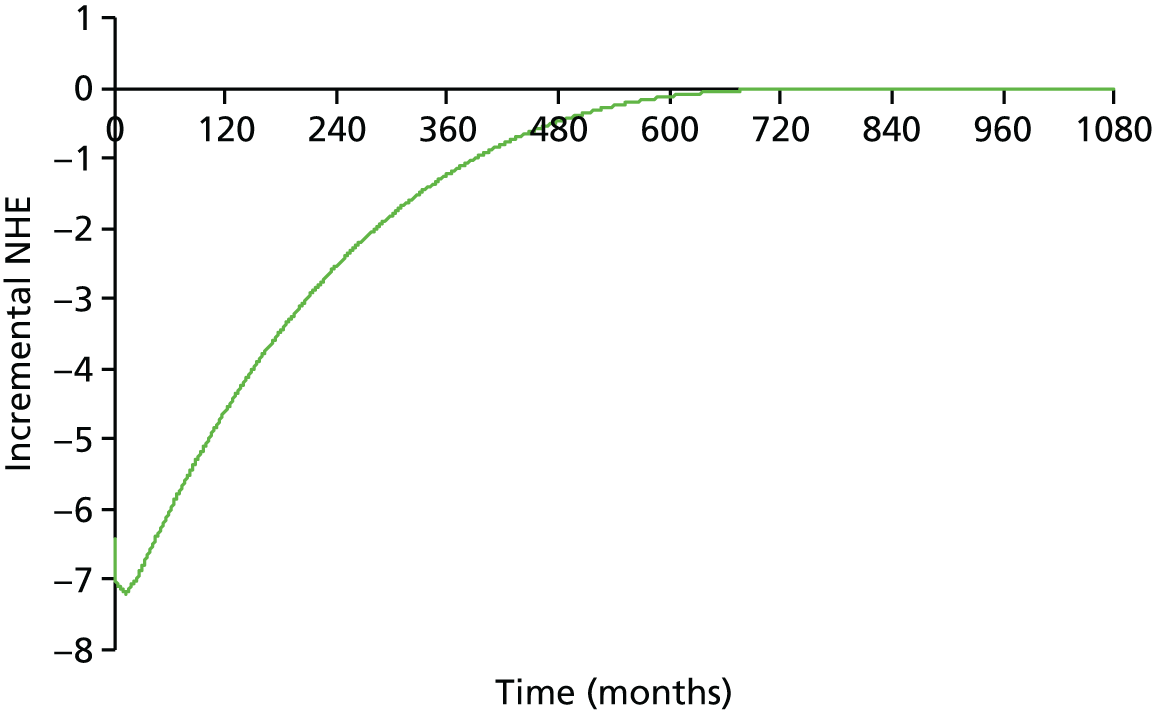
Population-level analyses: minimum evidence set
Net health effects can also be presented for a population of patients over time. Although the presentation of population NHEs is not formally requested within the existing NICE methods guide,122 population-based analyses are requested to be submitted to assess population impacts within Section 5.12 (Impact on the NHS). In addition, Section 6.4.1. states that in situations in which the evidence of clinical effectiveness is ‘absent, weak or uncertain’, the committee is requested to ‘balance the potential net benefits to current NHS patients of a recommendation not restricted to research with the potential net benefits to both current and future NHS patients of being able to produce guidance and base clinical practice on a more secure evidence base’ (p. 69-71). 122
Analyses of population NHEs may therefore provide additional information to help inform the committee’s deliberations regarding possible research recommendations (Section 6.4 of the current methods guide122). Analyses of population NHEs require information about the prevalence and future incidence of the target population and a judgement about the time horizon over which the technology will be used in clinical practice. As outlined in Appendix 8, the expected incidence of eligible cases for the exemplar was estimated to be approximately 38 per annum. The technology time horizon was set to 10 years in the base case.
Table 26 reports the population NHE for CAR T-cell therapy over the 10-year technology time horizon. Over this period, the use of CAR T-cell therapy is estimated to generate an additional 2356 QALYs (discounted values) within the population considered compared with the current standard of care. However, as the additional lifetime cost of £117.78M (£141.75M – £23.97M; discounted values) requires other treatments to be displaced and health to be forgone by other patients in the NHS, overall the additional QALYs are exactly offset by health forgone elsewhere. Hence, the incremental population NHE at a £50,000 per QALY threshold is 0.26 QALYs (£12,813).
| Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||
|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) |
| CAR T-cell therapy | 141,751,559 | 2785.04 | 49,995 | –49.99 (–2,499,490) | 0.26 (12,813) |
| Standard of care | 23,974,719 | 429.25 | –50.25 (–2,512,303) | ||
A series of one-way sensitivity analyses was undertaken to assess the sensitivity of the model results to changes in assumptions. The results of these sensitivity analyses are presented in Table 27.
| Scenario | Incremental cost (£) | Incremental QALYs | ICER (£) | Incremental NHE at willingness to pay of £50,000, QALYs (£) |
|---|---|---|---|---|
| Base case (0% repeat CAR T-cell treatment) | 117,776,840 | 2355.79 | 49,995 | 0.26 (12,813) |
| Repeat CAR T-cell treatment – monthly probability of 0.5% | 193,649,693 | 2355.79 | 82,201 | –1517.20 (–75,860,040) |
| Repeat CAR T-cell treatment – monthly probability of 0.1% | 132,951,410 | 2355.79 | 56,436 | –303.24 (–15,161,757) |
| Discounting – 0% costs and health effects | 117,863,631 | 4608.43 | 25,576 | 2251.16 (112,557,826) |
| Discounting – 6% costs and health effects | 117,718,706 | 1662.50 | 70,808 | –691.87 (–34,593,729) |
| Discounting – 0% costs and 6% health effects | 117,863,631 | 1662.50 | 70,895 | –694.77 (–34,738,654) |
| Discounting – 6% costs and 0% health effects | 117,718,706 | 4608.43 | 25,544 | 2254.06 (112,702,751) |
| Discounting – 1.5% costs and 1.5% health effects | 117,825,625 | 3350.89 | 35,162 | 994.38 (49,718,835) |
| Discounting – UK Treasury-recommended step discounting of 3.5% up to year 30, 3% thereafter (both costs and health effects) | 117,776,840 | 2374.47 | 49,601 | 18.94 (946,799) |
| Standard of care costs based on FLAG-IDA | 130,211,131 | 2355.79 | 55,273 | –248.43 (–12,421,478) |
| Hazard rate for death in non-remission no-HSCT patients increased from 0.2425 (mean time to death 0.34 years) to 0.6075 (mean time to death 0.14 years) | 117,775,723 | 2363.47 | 49,832 | 7.95 (397,705) |
The one-way sensitivity analyses indicate that the results of the evaluation are sensitive to assumptions regarding the potential for retreatment with CAR T-cell therapy and the assumed discounting rate for health effects in the model. The results of the evaluation are less sensitive to assumptions about the discounting rate for costs, to the impact of remission status on survival in non-HSCT patients and to reducing the cost of standard of care treatment to values consistent with treatment using FLAG-IDA (assuming similar efficacy to that of clofarabine).
If the committee was to consider the criteria met for applying the non-reference case discount rate of 1.5% for both costs and health effects (i.e. when treatment restores people who would otherwise die or have a very severely impaired life to full or near full health and when this is sustained over a very long period, normally at least 30 years), then the ICER would reduce to £35,162 per QALY and CAR T-cell therapy would be associated with an additional population NHE equivalent to 994 QALYs (£49.72M) in comparison to health forgone elsewhere.
Employing the stepwise discounting recommended by the UK Treasury210 to all public sector bodies makes only a small difference to the ICER (£49,601 vs. £49,995), with the incremental population NHE increasing to 18.94 QALYs (£946,799).
Although the results of the evaluation appear to be sensitive to assumptions about the potential for retreatment with CAR T-cell therapy, this was not considered to represent such a challenge in this TPP. CAR T-cell therapy was assumed to be used as a one-off therapy to induce remission and to improve the likelihood and outcomes of HSCT. It was assumed that patients would not receive a repeat treatment in the event of not achieving remission; nor would patients who were successfully treated with HSCT receive further treatments with CAR T-cell therapy.
Probabilistic analysis
The results of the probabilistic analysis are shown in Table 28.
| Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) |
| CAR T-cell therapy | 141,556,652 | 2716.4 | 55,090 | –114.8 (–5,738,274) | –215.9 (–10,794,902) | 26.1 | 56.3 (2,813,197) |
| Standard of care | 24,728,297 | 595.7 | 101.1 (5,056,627) | ||||
The probabilistic ICER increased to £55,090 because of the model non-linearities. Consequently, the population NHE is now negative, with an overall loss to the health system of 215.9 QALYs (£10.79M). At a £50,000 cost-effectiveness threshold, the probability that CAR T-cell therapy is the most cost-effective option is 26.1%.
The cost-effectiveness acceptability planes and curves are presented in Figures 12 and 13, respectively.
FIGURE 12.
Cost-effectiveness acceptability plane: bridge to HSCT TPP – minimum evidence set. WTP, willingness to pay.
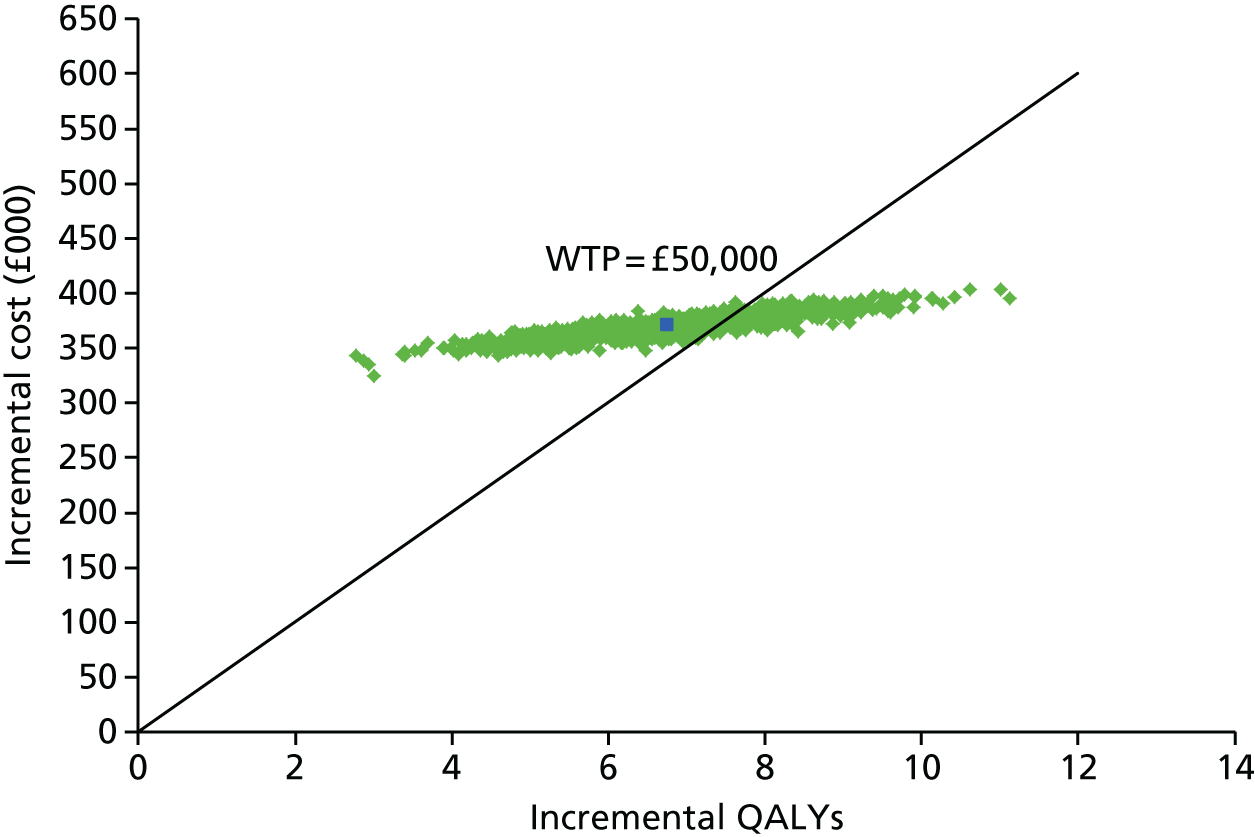
FIGURE 13.
Cost-effectiveness acceptability curve: bridge to HSCT TPP – minimum evidence set.

In addition to considering how uncertain a decision is to approve or reject a technology based on expected cost-effectiveness, an assessment of the scale of the likely consequences may also be potentially informative to the committee, particularly in deliberations related to possible research recommendations. An assessment of the potential consequences of uncertainty is important because it indicates the scale of the population NHEs that could be gained if uncertainty surrounding this decision could be resolved immediately. 128 This estimate also represents an expected upper bound to the benefits of more research. This may help to inform subsequent research recommendations. For example, if the maximum potential benefits of further research are considered unlikely to sufficiently justify the research costs, then it may not be worthwhile to issue further research recommendations.
These same consequences are referred to using the term ‘payer uncertainty burden’ (PUB) in the DSU report on managed access. 211 Elsewhere in the literature, these have been defined as the expected value of perfect information (EVPI) and the overall expected opportunity loss. Within the DSU report they are further defined as the value of the risk of making a particular decision because of uncertainty (expressed in either monetary or health units), combining two key concepts: first, the probability that the strategy with the highest expected NHE may not be the optimal strategy (i.e.1 – probability that the intervention is cost-effective based on the probabilistic results) and, second, the consequences of a ‘wrong’ decision in terms of QALYs and NHS costs that could have been saved if the truly optimal strategy had been selected instead.
Assuming a 10-year technology horizon, the consequences of decision uncertainty in the minimum data set are estimated to be 56.3 QALYs (£2.83M). Figure 14 shows how the scale of the consequences of decision uncertainty varies across different cost-effectiveness thresholds, reaching a peak at a £55,000 threshold.
FIGURE 14.
Consequences of decision uncertainty: bridge to HSCT TPP – minimum evidence set.

A summary of the population-level incremental NHEs, net monetary benefits, probability of cost-effectiveness and consequences of decision uncertainty across a range of willingness-to-pay thresholds is presented in Table 29.
| Cost-effectiveness threshold (£) | Incremental NHE, QALYs | Incremental NMB (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) |
|---|---|---|---|---|
| 20,000 | –3720.75 | –74,414,973 | 0 | 0 (0) |
| 30,000 | –1773.61 | –53,208,283 | 0 | 0 (0) |
| 50,000 | –215.90 | –10,794,902 | 26.1 | 56.3 (2,813,197) |
| 75,000 | 562.96 | 42,221,825 | 94.1 | 9.5 (710,894) |
| 100,000 | 952.39 | 95,238,551 | 99.3 | 0.6 (63,592) |
At conventional cost-effectiveness thresholds between £20,000 and £30,000 per QALY gained, the probability that CAR T-cell therapy is cost-effective compared with standard of care is 0%. Consequently, because of the high certainty that CAR T-cell therapy is not cost-effective at conventional thresholds (i.e. assuming that EoL criteria do not apply), there are no consequences of decision uncertainty. At a cost-effectiveness threshold of £50,000 per QALY gained, the probability that CAR T-cell therapy is cost-effective compared with standard of care is 26.1%. In this case, the expected population health consequence of decision uncertainty is 56.3 QALYs. These expected consequences can be interpreted as an estimate of the population NHE that could be gained if the uncertainty surrounding this decision could be resolved immediately and provide an expected upper bound on the benefits of more research. The corresponding expected monetary cost of decision uncertainty is approximately £2.8M. At thresholds of £75,000 and £100,000 per QALY gained, the probability that CAR T-cell therapy is cost-effective increases to > 94%. Because there is now high certainty that CAR T-cell therapy is cost-effective at these thresholds, the corresponding consequences of decision uncertainty reduce to < 10 QALYs (or < £1M in monetary terms).
Alternative pricing scenarios probabilistic analysis
A series of alternative pricing schemes has been generated to explore their potential impact on cost-effectiveness and decision uncertainty. These schemes were selected to reflect the possible approaches that have been suggested to address the potential HTA challenges, as highlighted previously in Chapter 3 (see Methods). The schemes considered were:
-
A leasing scheme approach based on the approach outlined by Edlin et al. 137 In this scenario, the technology is assumed to be leased from the company. The monthly ‘lease’ payment was established by calculating a stream of payments over the expected survival duration of the patients that has the same expected net present value as the agreed price. Hence, payment was assumed to continue on a monthly basis while a patient remained alive.
-
A pay for performance scheme in which payment is made retrospectively only for patients who achieve remission (CR) within a specified period (e.g. 28 days). Alternatively, an initial upfront payment could be made for all, with a separate ‘clawback’ agreed for patients who do not achieve remission.
-
A more conventional PAS providing a fixed percentage discount (e.g. 10%).
The probabilistic results based on alternative hypothetical pricing scenarios are shown in Table 30. The scatterplots showing each iteration of incremental costs and incremental effects considered in the probabilistic sensitivity analysis are provided in Figure 15.
| Pricing scenario | Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) | |
| Base case | CAR T-cell therapy | 141,556,652 | 2716.4 | 55,090 | –114.77 (–5,738,274) | –215.9 (–10,794,902) | 26.1 | 56.3 (2,813,197) |
| Standard of care | 24,728,297 | 595.7 | 101.13 (5,056,627) | |||||
| Leasing method | CAR T-cell therapy | 140,082,600 | 2727.04 | 54,227 | –74.61 (–3,730,478) | –179.94 (–8,997,139) | 22.1 | 22.5 (1,123,900) |
| Standard of care | 24,678,802 | 598.91 | 105.33 (5,266,662) | |||||
| Payment for remission patients only (70% on average) | CAR T-cell therapy | 102,099,708 | 2708.82 | 36,430 | 666.83 (33,341,351) | 577.2 (–28,861,808) | 96.8 | 3.9 (195,152) |
| Standard of care | 24,614,048 | 581.87 | 89.59 (4,479,543) | |||||
| Fixed pricing discount (10%) | CAR T-cell therapy | 130,229,928 | 2707.12 | 49,857 | 102.52 (5,125,971) | 6.05 (302,586) | 51.8 | 131.2 (6,558,209) |
| Standard of care | 24,818,199 | 592.83 | 96.47 (4,823,385) | |||||
FIGURE 15.
Scatterplots of incremental costs and incremental effectiveness across the four pricing scenarios: bridge to HSCT TPP. (a) Base case; (b) leasing method; (c) payment for remission only; and (d) fixed discount. Blue box = mean of the probabilistic sensitivity analysis values. WTP, willingness to pay.
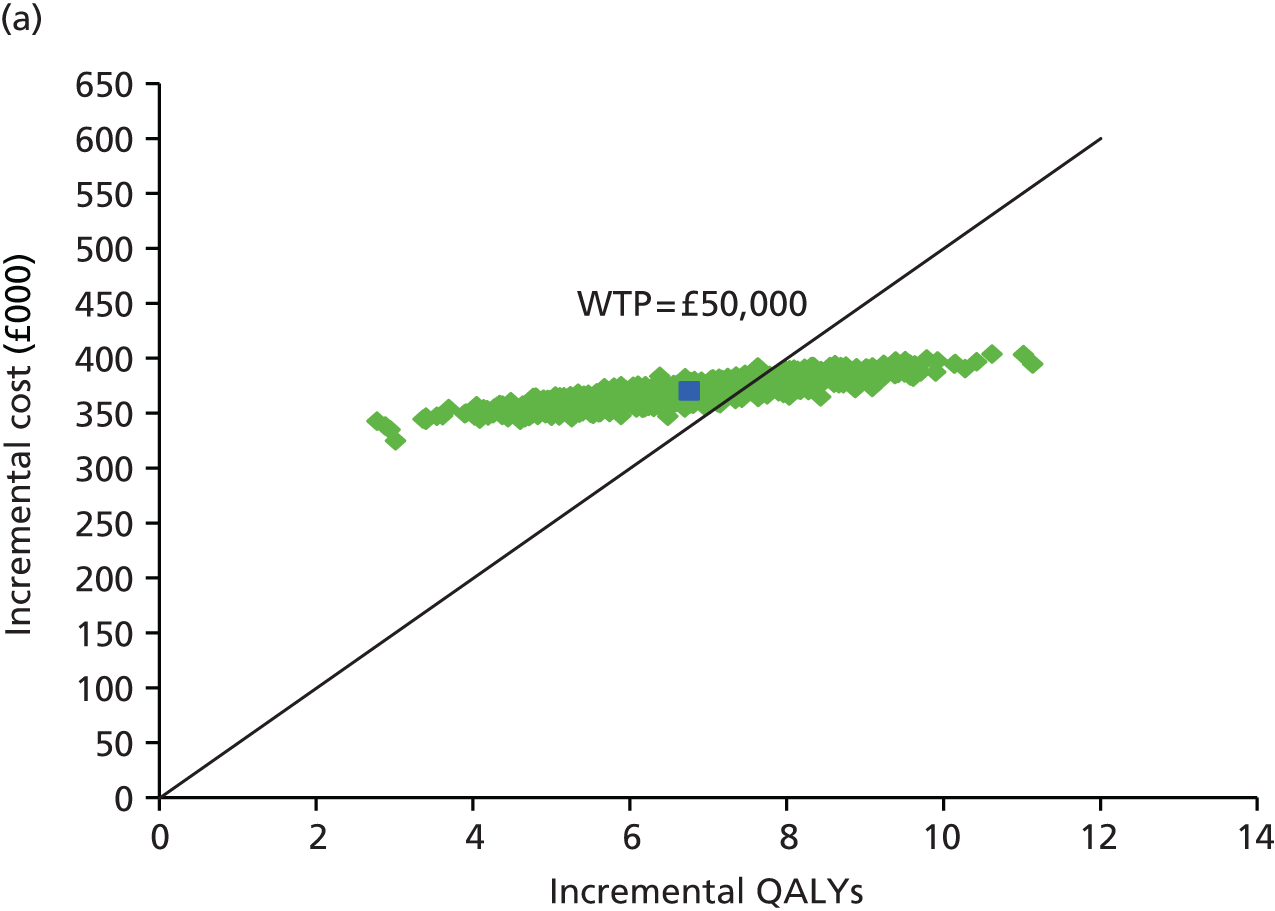
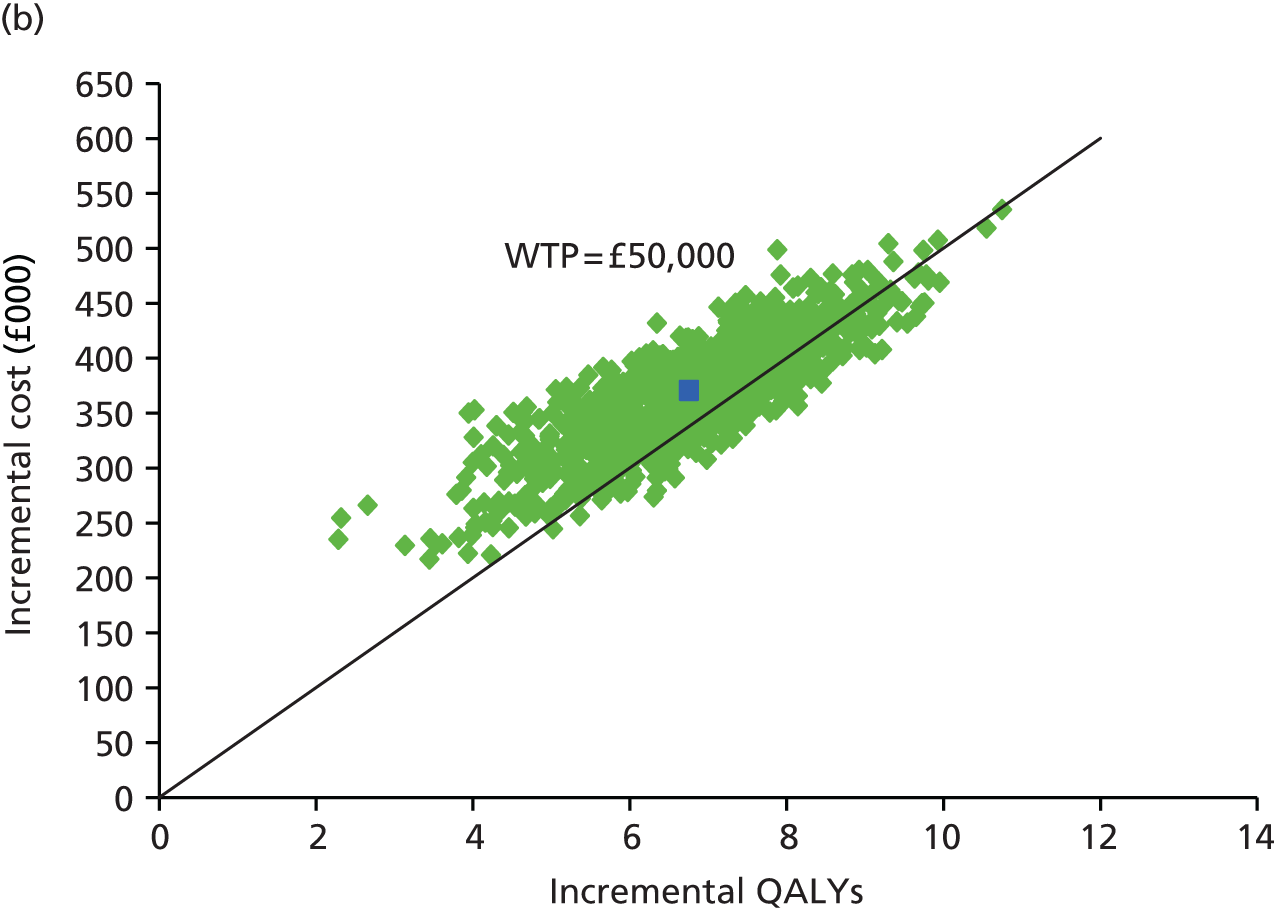


The impact of the different pricing schemes on the sampled outputs of the probabilistic sensitivity analysis is shown graphically in Figure 15. In the base case (fixed cost for CAR T-cell therapy), the cloud of simulated outcomes from the probabilistic sensitivity analysis is flat, such that there is considerable variability around the QALY gains of treatment, but little relative variability around the incremental costs. By introducing a leasing method, the cost of CAR T-cell therapy becomes more closely linked to the effectiveness of treatment, such that the cloud of simulated outcomes from the probabilistic sensitivity analysis is reoriented around the willingness-to-pay threshold. With both the remission and discounted schemes, the cost-effectiveness of treatment is improved and the cloud of simulated outcomes is shifted downwards on the chart.
A comparison plot of the consequences of decision uncertainty across the alternative pricing scenarios for different cost-effectiveness thresholds is shown in Figure 16.
FIGURE 16.
Comparison plot of the consequences of decision uncertainty across alternative pricing schemes: bridge to HSCT TPP – minimum evidence set.
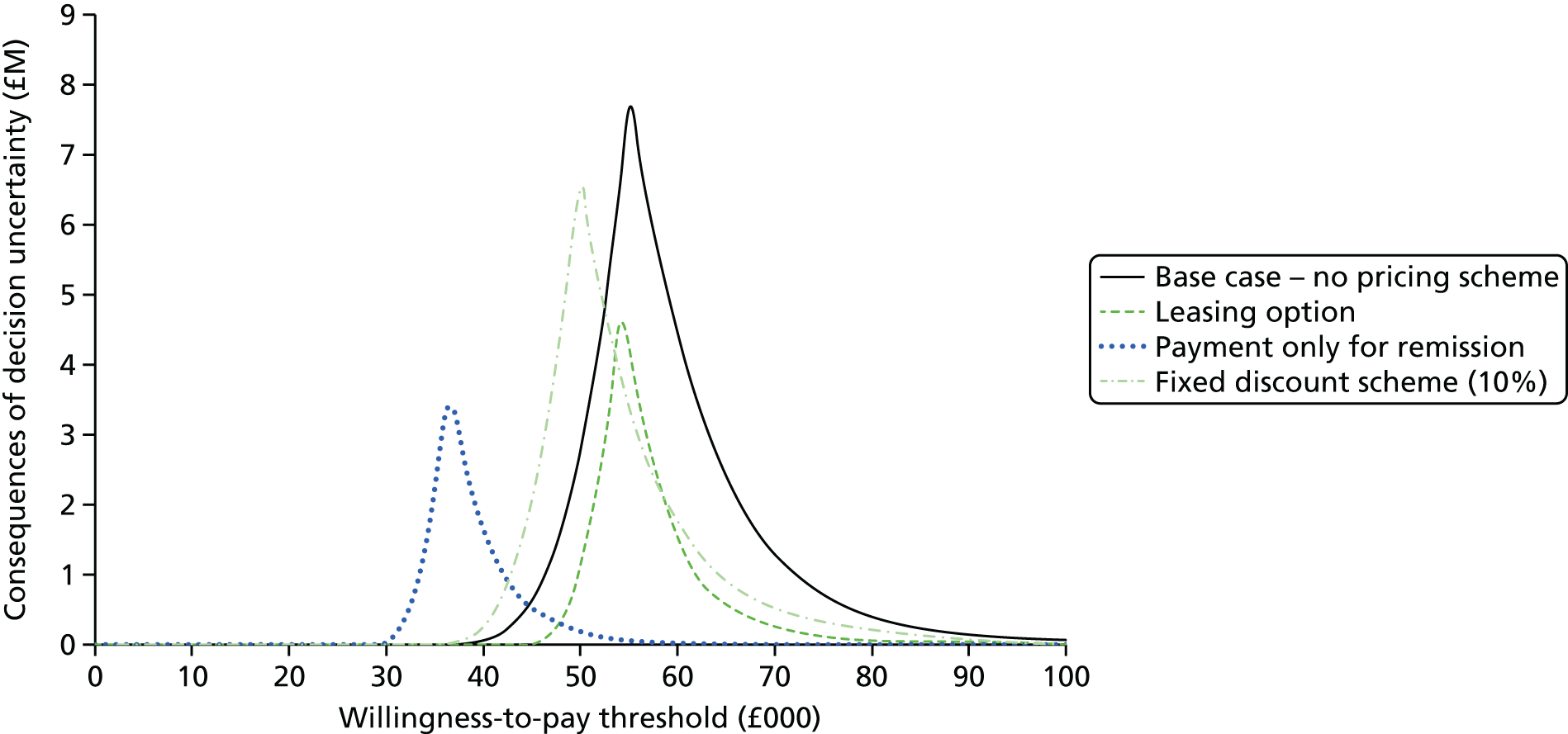
Under a fixed one-off acquisition cost approach, assumed in the main analyses, the NHS bears all of the risks associated with uncertainty surrounding whether the expected benefits of therapy will be realised in routine clinical practice. Hence, the consequences of decision uncertainty to the NHS appear highest with this scheme (56.3 QALYs, £2.81M). The alternative schemes result in reductions in decision uncertainty and associated consequences to the NHS. However, the impact of this and the mechanism by which it is achieved differ across the separate approaches.
The leasing approach results in only a minor difference in the ICER. Similar levels of decision uncertainty also remain (i.e. the probability that the intervention is cost-effective is similar to that under a fixed one-off acquisition cost approach). However, the scale of the consequences of the uncertainty to the NHS is significantly reduced under this scheme. This scheme limits the risk to the NHS of overpaying for a technology that does not achieve the expected outcomes, significantly lowering the consequences of decision uncertainty to 22.5 QALYs (£1.12M).
The use of a pay for performance scheme improves the expected cost-effectiveness and, as a result, reduces both the level of decision uncertainty and the scale of the consequences of this uncertainty. Restricting payment to only patients who achieve remission improves expected cost-effectiveness (£36,430 per QALY), leading to a higher probability of being cost-effective (96.8%), thereby reducing the consequences of uncertainty to 3.9 QALYs (£195,000). The use of a more conventional PAS, based on an assumed 10% reduction in the acquisition cost, works in a similar manner by improving both expected cost-effectiveness (£49,857 per QALY) and the likelihood that the treatment is cost-effective (51.8%). However, as the ICER now lies closer to the threshold in absolute terms, the consequences are increased to 131.2 QALYs (£6.56M).
The comparison plot more clearly shows the impact of the alternative pricing schemes. The alternative schemes affect both the shape of the distribution of the consequences across the separate cost-effectiveness thresholds as well as their position.
Alternative evidence sets probabilistic analysis
All of the analyses reported previously have been based on the minimum evidence set. The impact of the alternative evidence sets on expected cost-effectiveness, the level of decision uncertainty and the scale of the consequences is reported in Table 31 and Figure 17.
| Evidence set | Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) | |
| Minimum (base case) | CAR T-cell therapy | 141,556,652 | 2716.4 | 55,090 | –114.77 (–5,738,274) | –215.9 (–10,794,902) | 26.1 | 56.3 (2,813,197) |
| Standard of care | 24,728,297 | 595.7 | 101.13 (5,056,627) | |||||
| Intermediate | CAR T-cell therapy | 141,556,652 | 2716.4 | 55,090 | –114.77 (–5,738,274) | –215.9 (–10,794,902) | 26.1 | 56.3 (2,813,197) |
| Standard of care | 24,728,297 | 595.7 | 101.13 (5,056,627) | |||||
| Mature | CAR T-cell therapy | 141,680,276 | 2764.76 | 53,462 | –68.84 (–3,442,181) | –151.92 (–7,595,782) | 28.1 | 48.1 (2,406,886) |
| Standard of care | 24,375,545 | 570.58 | 83.07 (4,153,600) | |||||
FIGURE 17.
Scatterplots of incremental costs and incremental effectiveness across evidence sets: bridge to HSCT TPP. (a) Minimum and intermediate evidence sets; and (b) mature evidence set. Blue box = mean of the probabilistic sensitivity analysis values. WTP, willingness to pay.


As highlighted in Chapter 5, the use of a separate structural/surrogate link within the bridge to HSCT TPP was employed to allow the incorporation of external evidence on the relationship between remission MRD and HSCT status. A limitation of our analysis is that the same external evidence is then used across each of the separate evidence sets. This means that the additional follow-up assumed in both the intermediate and the mature evidence sets is not adequately reflected in the results. Consequently, the ICER and associated decision uncertainty are identical across the minimum and intermediate data sets. Furthermore, the differences in results between these evidence sets and the mature evidence set is driven entirely by the increased precision (i.e. because of higher patient numbers) in the short-term remission, MRD and HSCT rates, as opposed to the additional maturity of follow-up data that may be available.
In practice, the additional follow-up reported in more mature studies could either replace the existing surrogate relationship employed here or be synthesised and combined with the external evidence. Hence, the value that the additional follow-up brings in terms of either confirming an assumed surrogate relationship or increasing the precision around this relationship is not adequately captured in these analyses.
Despite these limitations, the separate evidence sets may still provide an important comparison for the committee to consider, specifically in relation to how its deliberations might be affected in situations in which the same ICER and decision uncertainty are reported but under different circumstances, that is, situations in which the results are based entirely on external surrogate relationships compared with situations in which the results are based on actual observed data from a longer-term trial or follow-up.
As expected, the health consequence of decision uncertainty in the mature evidence set (48.1 QALYs, £2.41M) is lower than that reported in the minimum set (56.2 QALYs, £2.81M) at a threshold of £50,000 per QALY gained. These consequences are reduced by the increased precision associated with the larger sample in terms of the short-term remission, MRD and HSCT rates.
A comparison plot of the consequences of decision uncertainty between the minimum/intermediate evidence sets and the mature evidence set across a range of cost-effectiveness thresholds is provided in Figure 18.
FIGURE 18.
Comparison plot of the consequences of decision uncertainty across alternative evidence sets: bridge to HSCT TPP.

Presentation of the scale of consequences using population NHEs allows some important comparisons to be made across the separate pricing approaches and the different evidence sets. Specifically, these comparisons could provide a more explicit basis for considering the value of direct price reductions that might be realised through a conventional PAS (or less conventional schemes that work by indirectly lowering the effective price) compared with the provision of additional evidence (both precision and maturity) in terms of reducing decision uncertainty and its consequences.
In the bridge to HSCT TPP, significant reductions in the level and scale of the consequences of decision uncertainty (i.e. the risk faced by the NHS) appear to be achieved by more innovative pricing approaches such as pay for performance and leasing approaches than those that might be realised by the provision of further evidence. Such information might provide an important basis for discussions between manufacturers and NICE in terms of how the existing uncertainties that exist might be appropriately managed, ensuring that risks and benefits are more appropriately shared.
Curative intent target product profile
A similar sequence of assessments and analyses was conducted based on the curative intent TPP. In contrast to the bridge to HSCT TPP, differences in the results across the evidence sets are more evident, as the results are directly informed by the data assumed within these rather than by employing evidence from external sources.
Per-patient analyses: minimum evidence set
Again, the sequence of assessments starts with a conventional assessment of cost-effectiveness at the patient level based on the minimum evidence set. Disaggregated costs and outcomes are presented in Table 32. The mean incremental cost of CAR T-cell therapy over an individual patient’s lifetime was estimated to be £503,256 and CAR T-cell therapy resulted in an additional 10.07 QALYs.
| Outcome | CAR T-cell therapy | Standard of care | Increment |
|---|---|---|---|
| Costs (£) | |||
| Course of treatment (including conditioning) | 530,557 | 43,200 | 487,357 |
| Hospitalisation for treatment | 13,012 | 7180 | 5832 |
| Adverse event costs | 20,513 | 442 | 20,070 |
| HSCT and related follow-up costs | 15,092 | 22,267 | –7175 |
| Non-HSCT follow-up costs | 4189 | 7016 | –2827 |
| Total costs | 583,362 | 80,106 | 503,256 |
| QALYs | |||
| Event free | 10.62 | 0.83 | 9.79 |
| Recurrent disease | 0.62 | 0.37 | 0.25 |
| Adverse events | 0.00 | 0.00 | 0.00 |
| QALY loss from HSCT | –0.06 | –0.08 | 0.03 |
| Total QALYs | 11.18 | 1.11 | 10.07 |
| Total life-years | 13.42 | 1.47 | 11.95 |
The expected cost-effectiveness of CAR T-cell therapy and the per-patient NHEs are shown in Table 33. In common with the previous TPP, the acquisition cost was set such that the ICER (£49,994) was close to the upper limit of NICE’s EoL threshold range (circa £50,000 per QALY gained).
| Patient level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||
|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) |
| CAR T-cell therapy | 583,362 | 11.18 | 49,994 | –0.49 (–24,509) | 0.001 (61) |
| Standard of care | 80,106 | 1.11 | –0.49 (–24,570) | – | |
The accumulation of NHEs over time, or, equivalently, the ‘investment profile’ per patient, is shown in Figure 19.
FIGURE 19.
Investment profile: curative intent TPP.

At the start of the time horizon, the initial high costs of treatment are far in excess of the immediate health benefits of treatment, leading to a negative NHE. Over time, the initial negative NHEs are gradually offset by the accrual of the residual health benefits of treatment (i.e. cure). In common with the bridge to HSCT TPP, it is only after approximately 60 years that the initial losses are sufficiently compensated for by later gains such that CAR T-cell therapy appears to be close to breaking even (i.e. NHE ≥ 0).
The shape of the investment profile differs slightly across the separate TPPs, with the early kink that was shown in the bridge to HSCT TPP not evident here. The lack of the kink is due to the small number of patients who are assumed to receive HSCT in the curative intent TPP. Hence, the resulting investment profile is smoother, although higher initial negative NHEs are reported because of the higher acquisition cost assumed within this TPP.
Population-level analyses: minimum evidence set
The expected per-patient effects of treatment were also extended to a population level based on similar assumptions concerning incidence (approximately 38 patients per annum) and the technology time horizon (10 years).
Table 34 reports the population NHEs for CAR T-cell therapy over the 10-year technology time horizon. Over this period, the use of CAR T-cell therapy is estimated to result in an additional 3177 QALYs (discounted values) within the population considered compared with the current standard of care. However, as the additional lifetime costs of £158.84M (discounted values) require other treatments to be displaced and health to be forgone by other patients, overall the additional QALYs are almost exactly offset by health forgone elsewhere. The resulting incremental population NHE is 0.39 QALYs expressed in health terms and £19,269 expressed in monetary terms.
| Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||
|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) |
| CAR T-cell therapy | 184,117,952 | 3527.65 | 49,994 | –154.71 (–7,735,298) | 0.39 (19,269) |
| Standard of care | 25,282,579 | 350.56 | –155.09 (–7,754,567) | ||
A series of one-way sensitivity analyses was conducted to assess the sensitivity of the model results to changes in assumptions or model settings. The results of these sensitivity analyses are presented in Table 35.
| Scenario | Incremental cost (£) | Incremental QALYs | ICER (£) | Incremental NHE, QALYs (£) |
|---|---|---|---|---|
| Base case (0% repeat CAR T-cell treatment) | 158,835,372 | 3177.09 | 49,994 | 0.39 (19,269) |
| Repeat CAR T-cell treatment – monthly probability of 1% | 429,511,483 | 3177.09 | 135,190 | –5413.14 (–270,656,842) |
| Repeat CAR T-cell treatment – monthly probability of 0.5% | 294,173,428 | 3177.09 | 92,592 | –2706.38 (–135,318,786) |
| Repeat CAR T-cell treatment – monthly probability of 0.1% | 185,902,983 | 3177.09 | 58,514 | –540.97 (–27,048,342) |
| Discounting – 0% costs and health effects | 160,095,703 | 6127.15 | 26,129 | 2925.24 (146,261,823) |
| Discounting – 6% costs and health effects | 158,456,968 | 2272.68 | 69,723 | –896.46 (–44,823,167) |
| Discounting – 0% costs and 6% health effects | 160,095,703 | 2272.68 | 70,444 | –929.24 (–46,461,901) |
| Discounting – 6% costs and 0% health effects | 158,456,968 | 6127.15 | 25,861 | 2958.01 (147,900,557) |
| Discounting – 1.5% costs and 1.5% health effects | 159,368,09 | 4478.47 | 35,585 | 1291.1 (64,555,226) |
| Discounting – UK Treasury-recommended step discounting of 3.5% up to year 30, 3% thereafter (both costs and health effects) | 158,853,044 | 3202.28 | 49,606 | 25.22 (1,260,898) |
| Standard of care costs based on FLAG-IDA | 171,269,663 | 3177.09 | 53,908 | –248.3 (–12,415,022) |
Again, the results of the one-way sensitivity analyses indicate that the results of the evaluation in the curative intent TPP are sensitive to assumptions about the potential for retreatment with CAR T-cell therapy and the assumed discounting rate for health effects in the model. The results of the evaluation are relatively insensitive to assumptions about the discounting rate for costs, the use of a stepped discounting rate (vs. constant discounting rates) and a reduction in the cost of standard of care treatment to values consistent with treatment using FLAG-IDA (keeping the same efficacy).
If the committee was to consider the criteria met for applying the non-reference case discount rate of 1.5% for both costs and health effects, then the ICER would reduce to £35,585 per QALY, and CAR T-cell therapy would be associated with an additional population NHE equivalent to 1291 QALYs (£64.56M) in comparison to health forgone elsewhere.
Employing the stepwise discounting recommended by the UK Treasury again makes only a small difference to the ICER results (£49,606 vs. £49,994), with an incremental population NHE of 25 QALYs (£1.26M) in comparison to health forgone elsewhere.
The sensitivity of the results to assumptions about the potential for retreatment with CAR T-cell therapy was considered to represent a more important issue within this TPP, that is, the longer-term survival benefits are directly linked to the curative potential of the CAR T-cells themselves rather than to an intermediate treatment such as HSCT. Consequently, the potential need to re-administer CAR T-cell therapy over a longer period represents an important additional source of uncertainty within this TPP, particularly for the minimum data set with a relatively short follow-up period.
Probabilistic analysis
The results of the probabilistic analysis are shown in Table 36. The probabilistic ICER increased to £50,906 because of the model non-linearities. Consequently, the population NHE is now negative, with an overall loss to the health system of 56 QALYs (£2.82M). At a £50,000 cost-effectiveness threshold, the probability that CAR T-cell therapy is the most cost-effective option is 50.7%.
| Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|
| Treatment | E[Costs] (£) | E[QALYs] | ICER (£) | E[NHE], QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) |
| CAR T-cell therapy | 183,931,590 | 3501.50 | 50,906 | –177.13 (–8,856,695) | –56.4 (–2,823,943) | 50.7 | 304.6 (15,229,876) |
| Standard of care | 25,270,727 | 384.76 | –120.66 (–6,032,752) | ||||
The cost-effectiveness acceptability planes and curves are presented in Figures 20 and 21, respectively.
FIGURE 20.
Cost-effectiveness acceptability plane: curative intent TPP – minimum evidence set. WTP, willingness to pay.
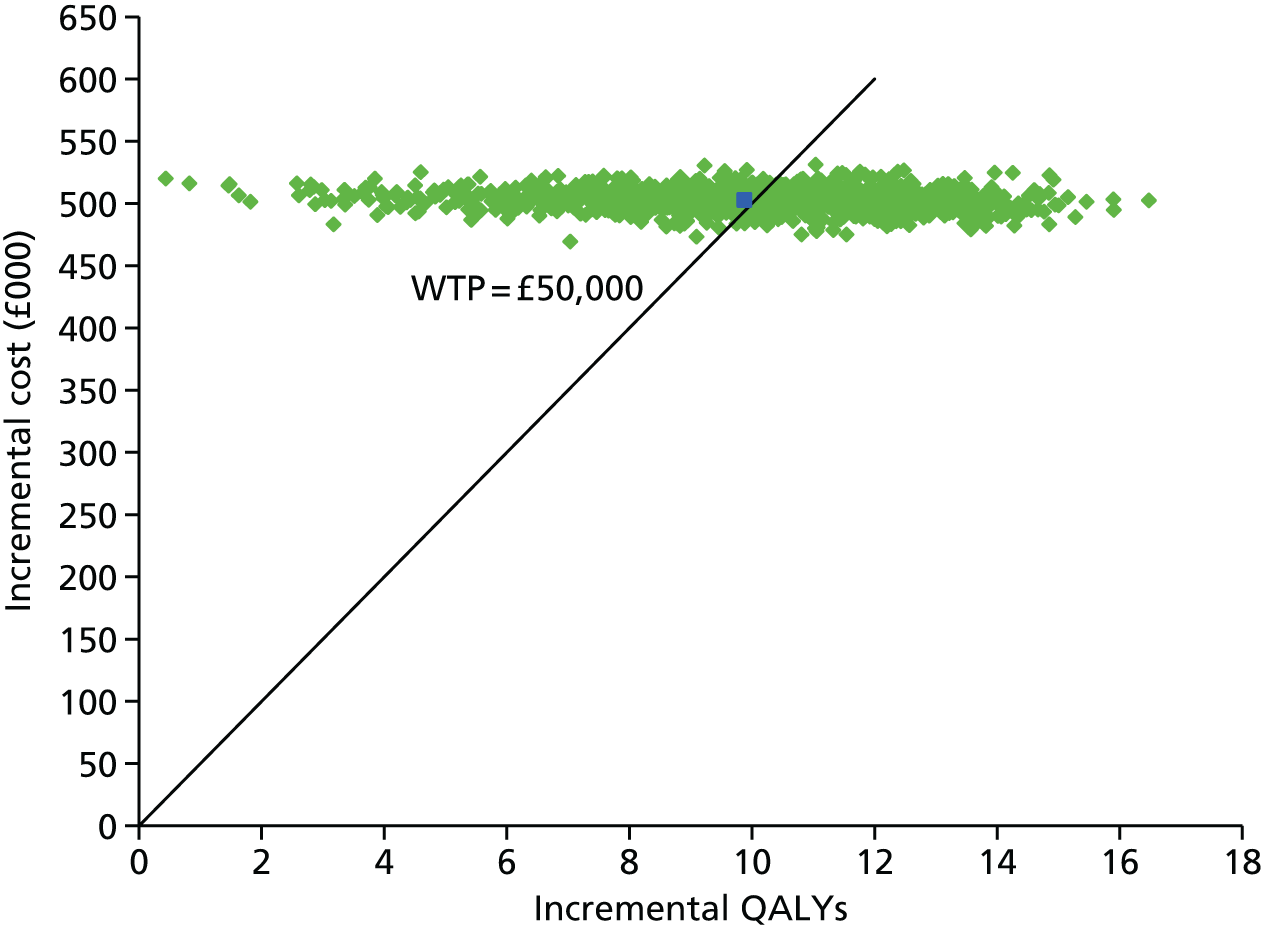
FIGURE 21.
Cost-effectiveness acceptability curve: curative intent TPP – minimum evidence set.
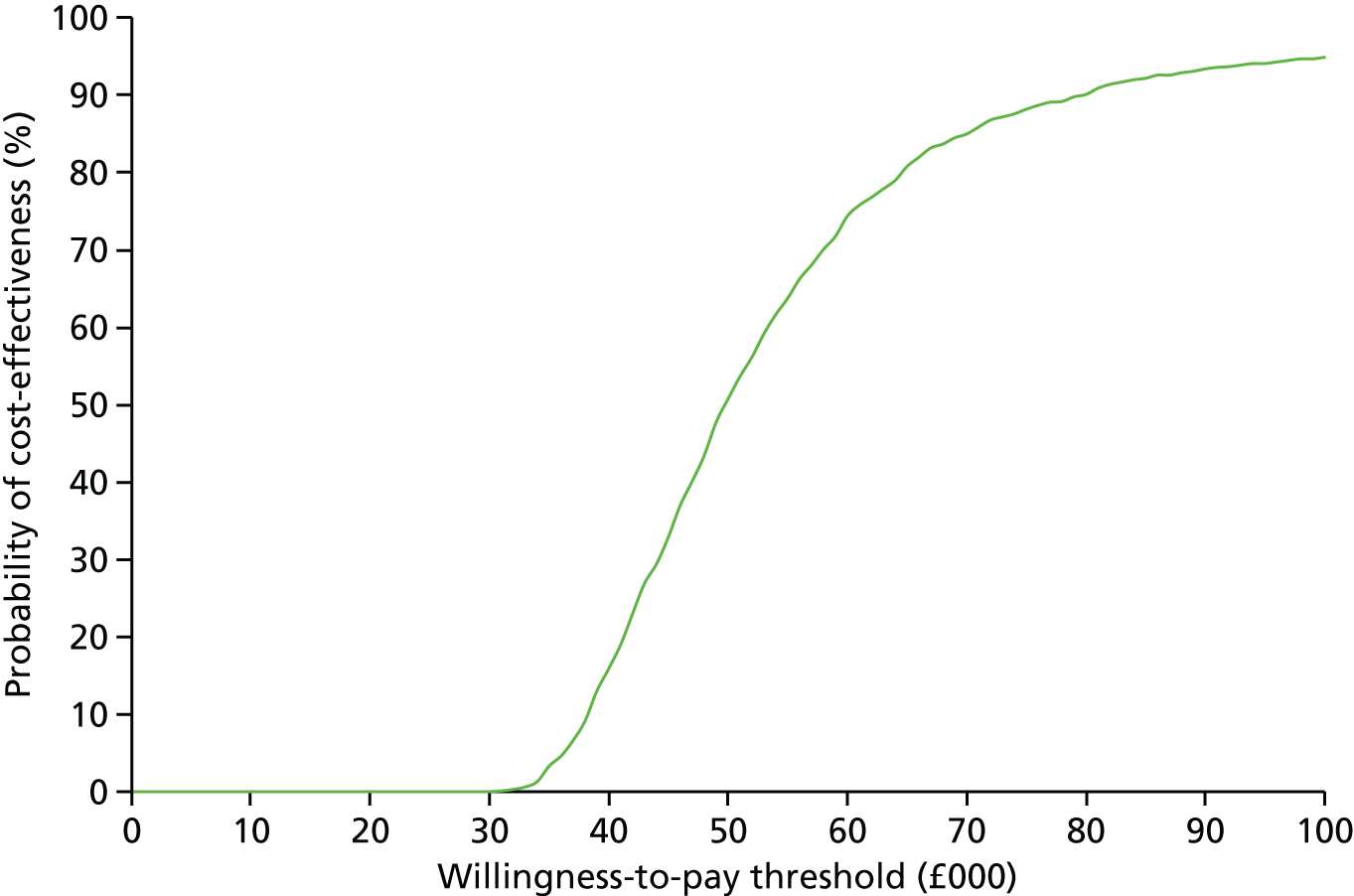
The consequences of decision uncertainty in the minimum evidence set are estimated to be 304.6 QALYs (£15.23M). Figure 22 shows how the scale of the consequences of decision uncertainty varies across different cost-effectiveness thresholds, reaching a peak at the £50,000 threshold.
FIGURE 22.
Consequences of decision uncertainty: curative intent TPP – minimum evidence set.
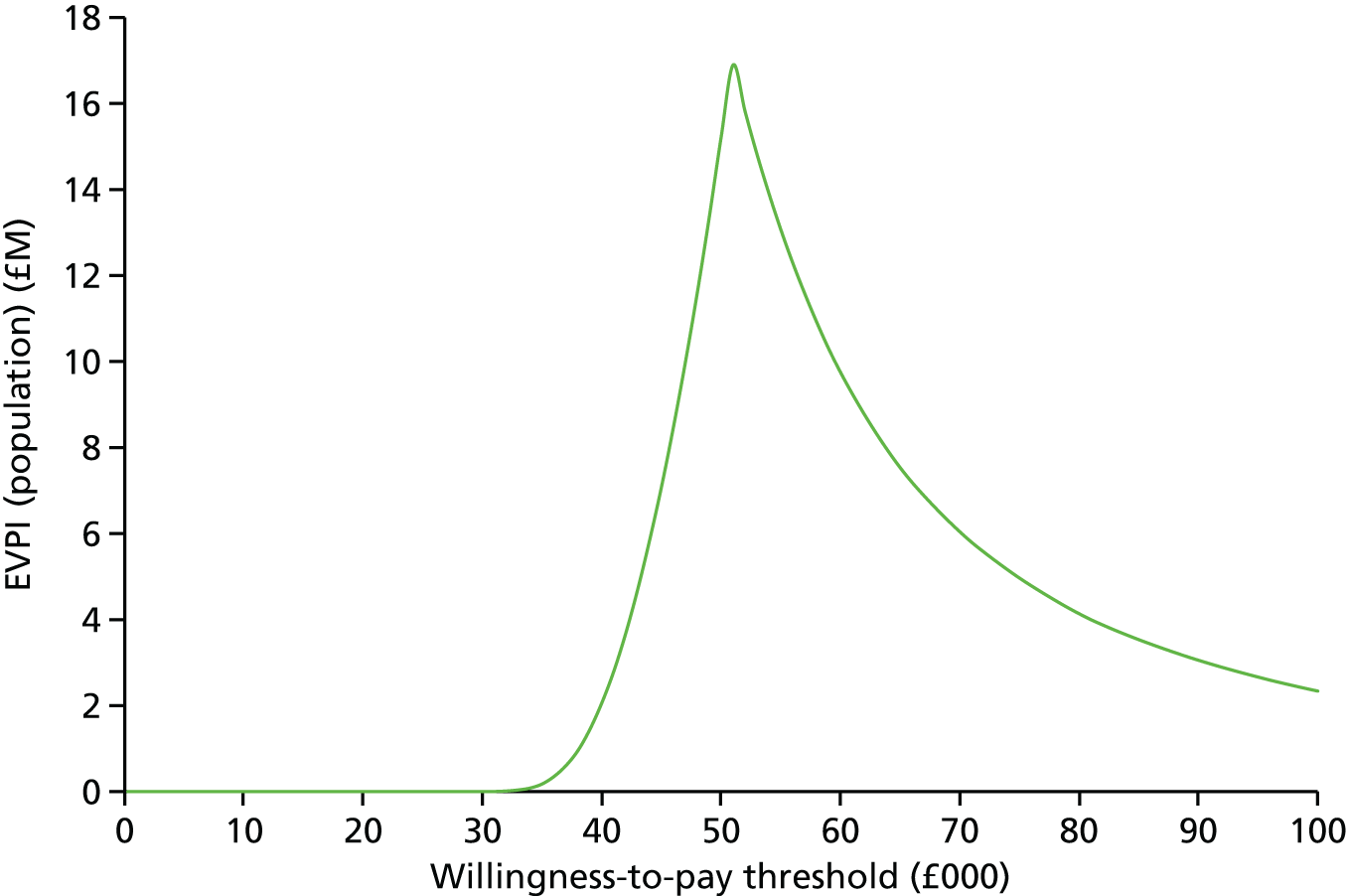
A summary of the population-level incremental NHEs, net monetary benefits, probability of cost-effectiveness and consequences of decision uncertainty across a range of cost-effectiveness thresholds is presented in Table 37.
| Cost-effectiveness threshold (£) | Incremental NHE, QALYs | Incremental NMB (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) |
|---|---|---|---|---|
| 20,000 | –4816.30 | –96,326,095 | 0.0 | 0 (0) |
| 30,000 | –2171.96 | –65,158,711 | 0.0 | 0 (0) |
| 50,000 | –56.48 | –2,823,943 | 50.7 | 304.6 (15,229,876) |
| 75,000 | 1001.26 | 75,094,517 | 88.1 | 66.2 (4,963,418) |
| 100,000 | 1530.13 | 153,012,977 | 94.8 | 23.4 (2,336,731) |
At conventional thresholds of between £20,000 and £30,000 per QALY gained, the probability that CAR T-cell therapy is cost-effective compared with standard of care is 0%. Consequently, there are no consequences of decision uncertainty at these threshold values. At a willingness-to-pay threshold of £50,000 per QALY gained, the probability that CAR T-cell therapy is cost-effective compared with standard of care is 50.7%. In this case, the expected population health consequence of decision uncertainty is 305 QALYs (10 years). The corresponding expected monetary cost of decision uncertainty is approximately £15.23M. At thresholds of £75,000 and £100,000 per QALY gained, the probability that CAR T-cell therapy is cost-effective increases to > 88%. Despite there being high certainty that CAR T-cell therapy is cost-effective at these thresholds, the corresponding consequences of decision uncertainty remain relatively high at 66 QALYs (£4.96M in monetary terms) and 23 QALYs (£2.34M), respectively.
Alternative pricing scenarios probabilistic analysis
The probabilistic results based on alternative hypothetical pricing scenarios are shown in Table 38. The scatterplots showing the iterations of incremental costs and incremental effects for the four pricing scenarios considered in the probabilistic sensitivity analysis are provided in Figure 23. A comparison plot of the consequences of decision uncertainty across the alternative pricing scenarios is shown in Figure 24.
| Pricing scenario | Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (£) | Consequences of decision uncertainty, QALYs (£) | |
| Base case | CAR T-cell therapy | 183,931,590 | 3501.50 | 50,906 | –177.13 (–8,935,381) | –56.48 (–2,902,629) | 50.7 | 304.6 (15,229,876) |
| Standard of care | 25,270,727 | 384.76 | –120.66 (–6,032,752) | |||||
| Leasing method | CAR T-cell therapy | 181,832,300 | 3488.85 | 50,618 | –147.79 (–7,389,708) | –38.21 (–1,910,653) | 49.2 | 65.6 (3,277,969) |
| Standard of care | 25,317,596 | 396.77 | –109.58 (–5,479,055) | |||||
| Payment for remission patients only (90% on average) | CAR T-cell therapy | 167,127,512 | 3510.80 | 45,708 | 168.25 (8,412,636) | 266.50 (13,325,042) | 63.9 | 236.1 (11,803,131) |
| Standard of care | 25,219,827 | 406.15 | –98.25 (–4,912,407) | |||||
| Fixed pricing discount (10%) | CAR T-cell therapy | 167,054,363 | 3535.49 | 45,131 | 194.40 (9,719,974) | 305.88 (15,293,860) | 64.2 | 209.1 (10,456,541) |
| Standard of care | 25,301,914 | 394.56 | –111.48 (–5,573,886) | |||||
FIGURE 23.
Scatterplots of incremental costs and incremental effectiveness across the four pricing scenarios: curative intent TPP. (a) Base case; (b) leasing method; (c) payment for remission only; and (d) fixed discount. Blue box = mean of the probabilistic sensitivity analysis values. WTP, willingness to pay.

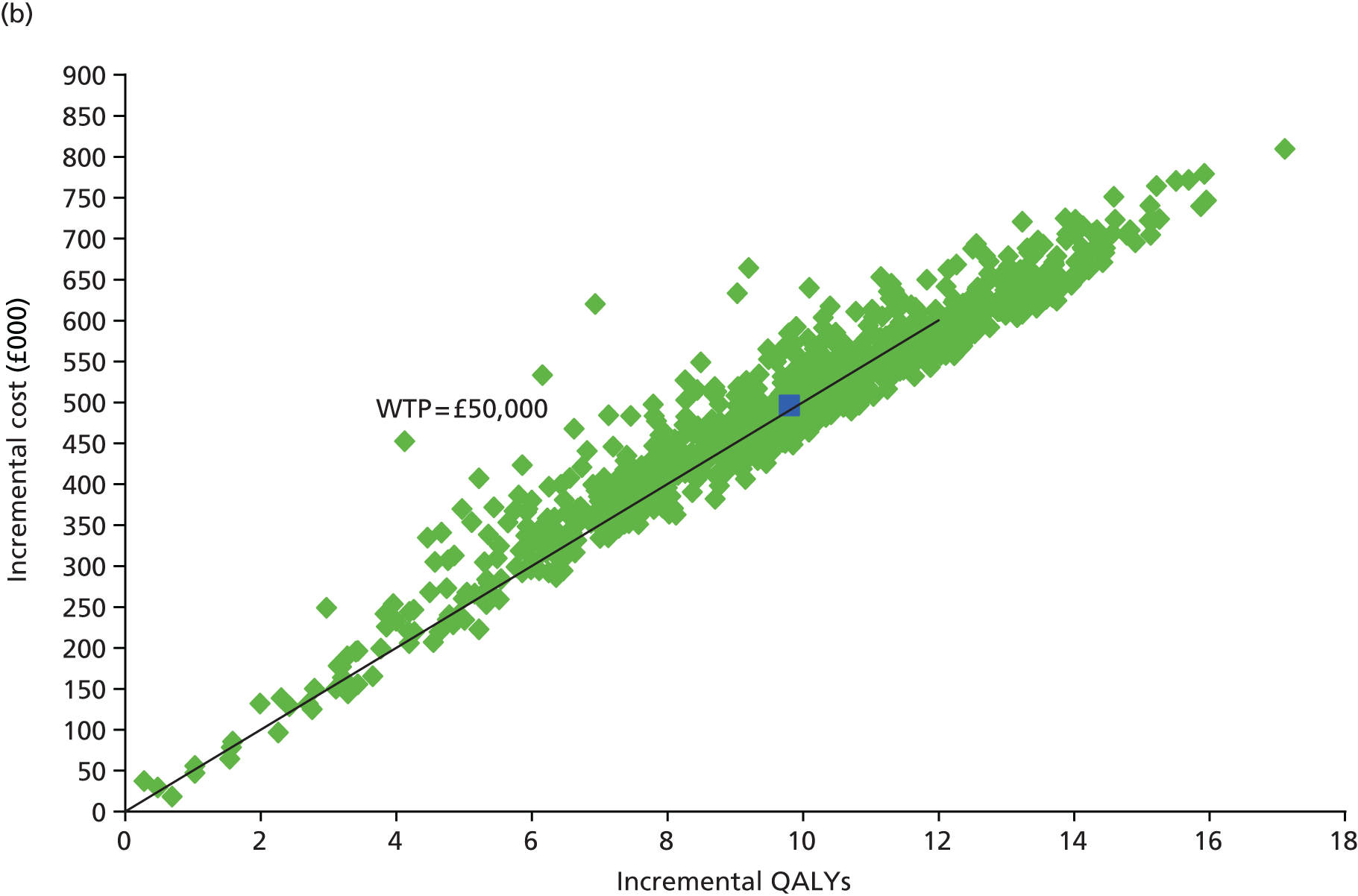

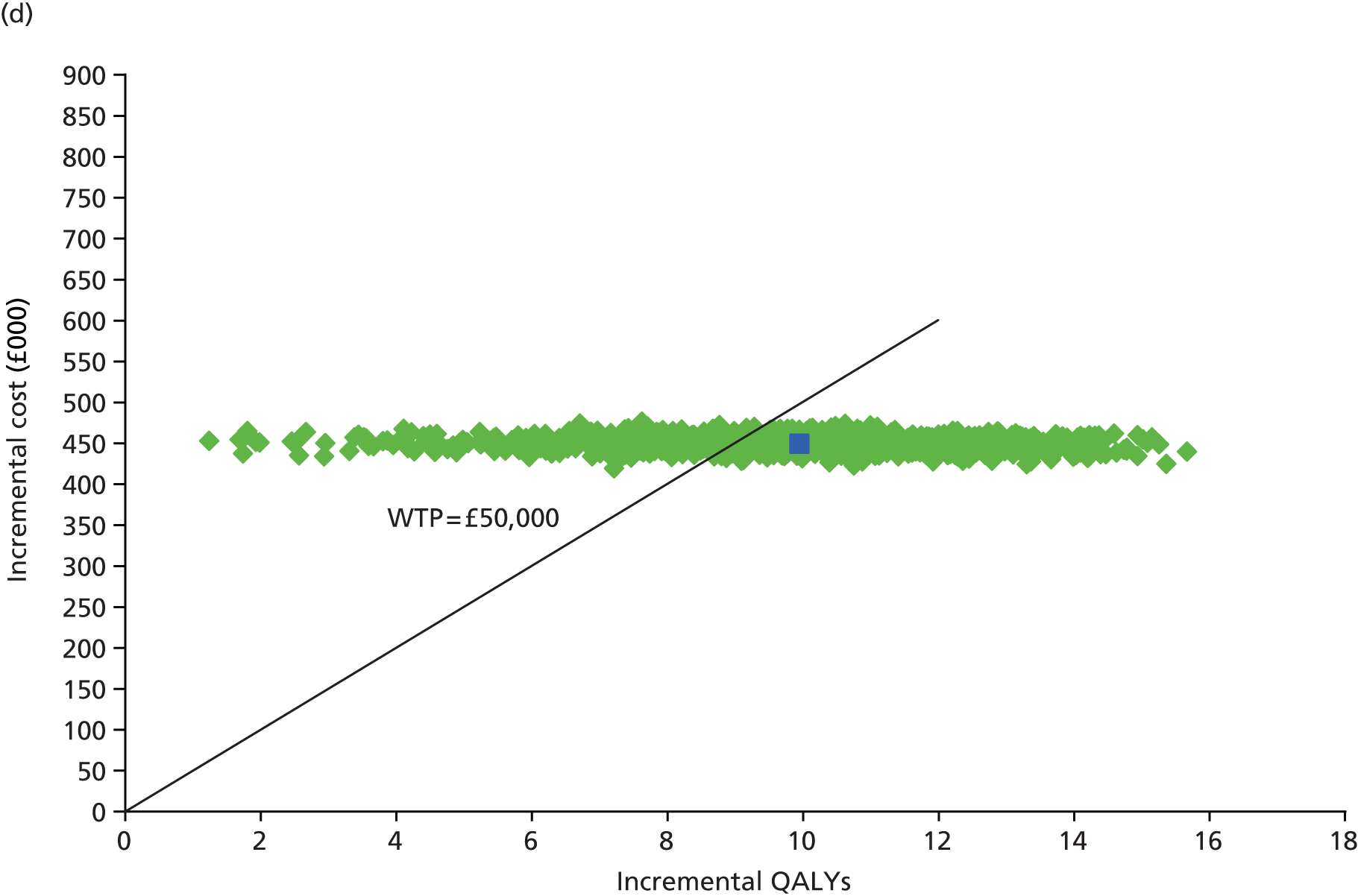
FIGURE 24.
Comparison plot of consequences of decision uncertainty across alternative pricing schemes: curative intent TPP – minimum evidence set.
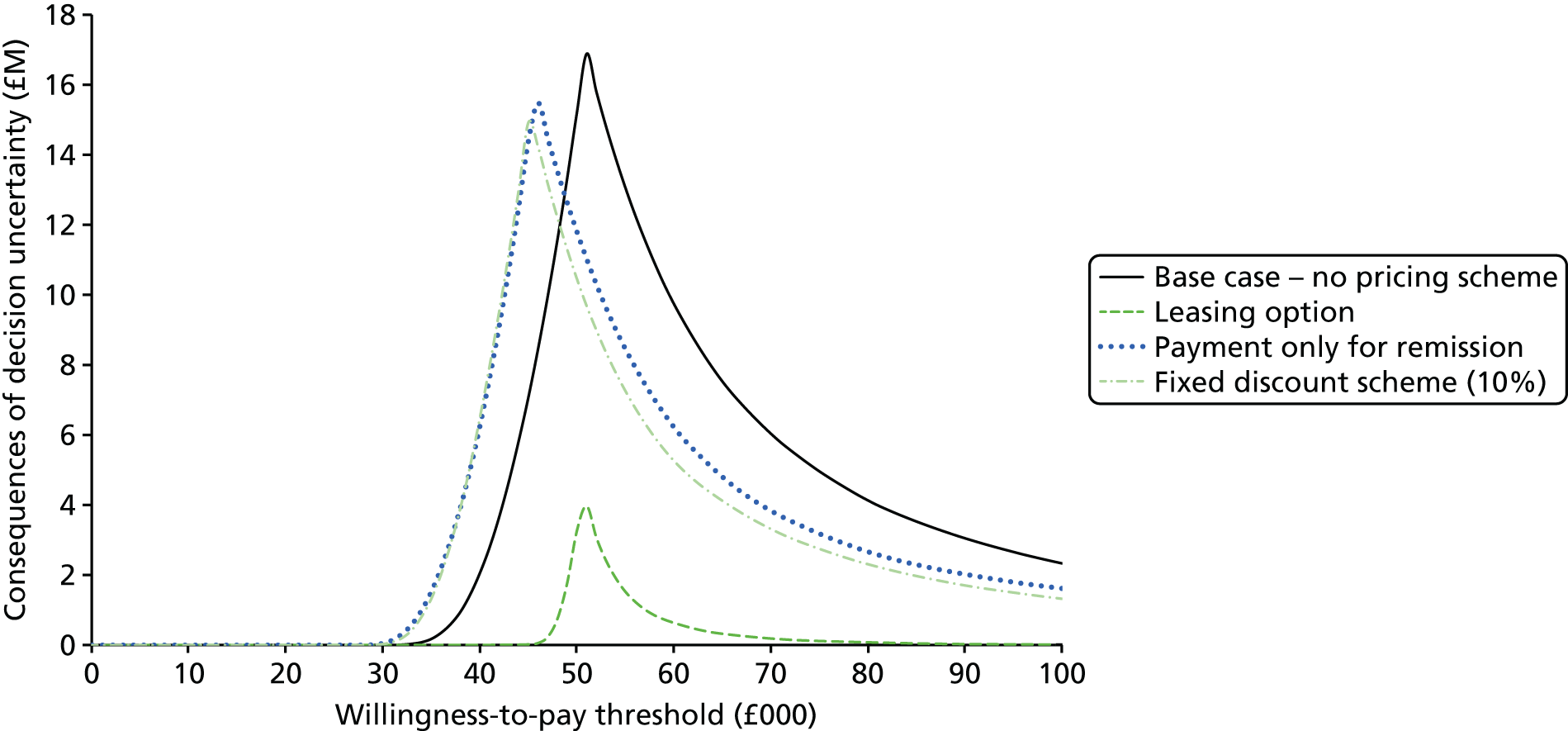
As observed in the previous analysis, the fixed one-off acquisition cost approach is associated with the highest potential consequences because of decision uncertainty (304.6 QALYs, £15.23M). As before, the alternative schemes result in reductions in decision uncertainty and associated consequences to the NHS. However, the impact of these reductions and the mechanisms by which they are achieved differ across the separate approaches.
The leasing approach results in only a minor difference in the ICER. Similar levels of decision uncertainty also remain (i.e. the probability that the intervention is cost-effective is similar to that under a fixed one-off acquisition cost approach). However, the scale of the consequences of the uncertainty to the NHS is significantly reduced with this scheme. This scheme limits the risk to the NHS of overpaying for a technology that does not achieve the expected outcomes, significantly lowering the consequences of decision uncertainty from > 300 QALYs in the base case to 65.6 QALYs (£3.28M) with the leasing approach.
Alternative evidence sets probabilistic analysis
The results of the probabilistic analysis are shown in Table 39 and Figure 25. A comparison plot of the consequences of decision uncertainty across the evidence sets is shown in Figure 26.
| Evidence set | Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | E[Costs] (£) | E[QALYs] | ICER (£) | E[NHE], QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) | |
| Minimum | CAR T-cell therapy | 183,931,590 | 3501.50 | 50,906 | –177.13 (–8,935,381) | –56.48 (–2,902,629) | 50.7 | 304.6 (15,229,876) |
| Standard of care | 25,270,727 | 384.76 | –120.66 (–6,032,752) | |||||
| Intermediate | CAR T-cell therapy | 183,586,917 | 4296.77 | 43,344 | 625.03 (31,251,488) | 486.22 (24,311,227) | 85.9 | 40.6 (2,031,623) |
| Standard of care | 25,264,818 | 644.10 | 138.81 (6,940,262) | |||||
| Mature | CAR T-cell therapy | 183,560,268 | 4307.12 | 43,252 | 635.91 (31,795,547) | 494.47 (24,723,328) | 91.5 | 14.1 (707,136) |
| Standard of care | 25,103,273 | 643.51 | 141.44 (7,072,220) | |||||
FIGURE 25.
Comparison plot of the consequences of decision uncertainty across alternative evidence sets: curative intent TPP.

FIGURE 26.
Scatterplots of incremental costs and incremental effectiveness across evidence sets: curative intent TPP. (a) Base case; (b) intermediate evidence set; and (c) mature evidence set. Blue box = mean of the probabilistic sensitivity analysis values.

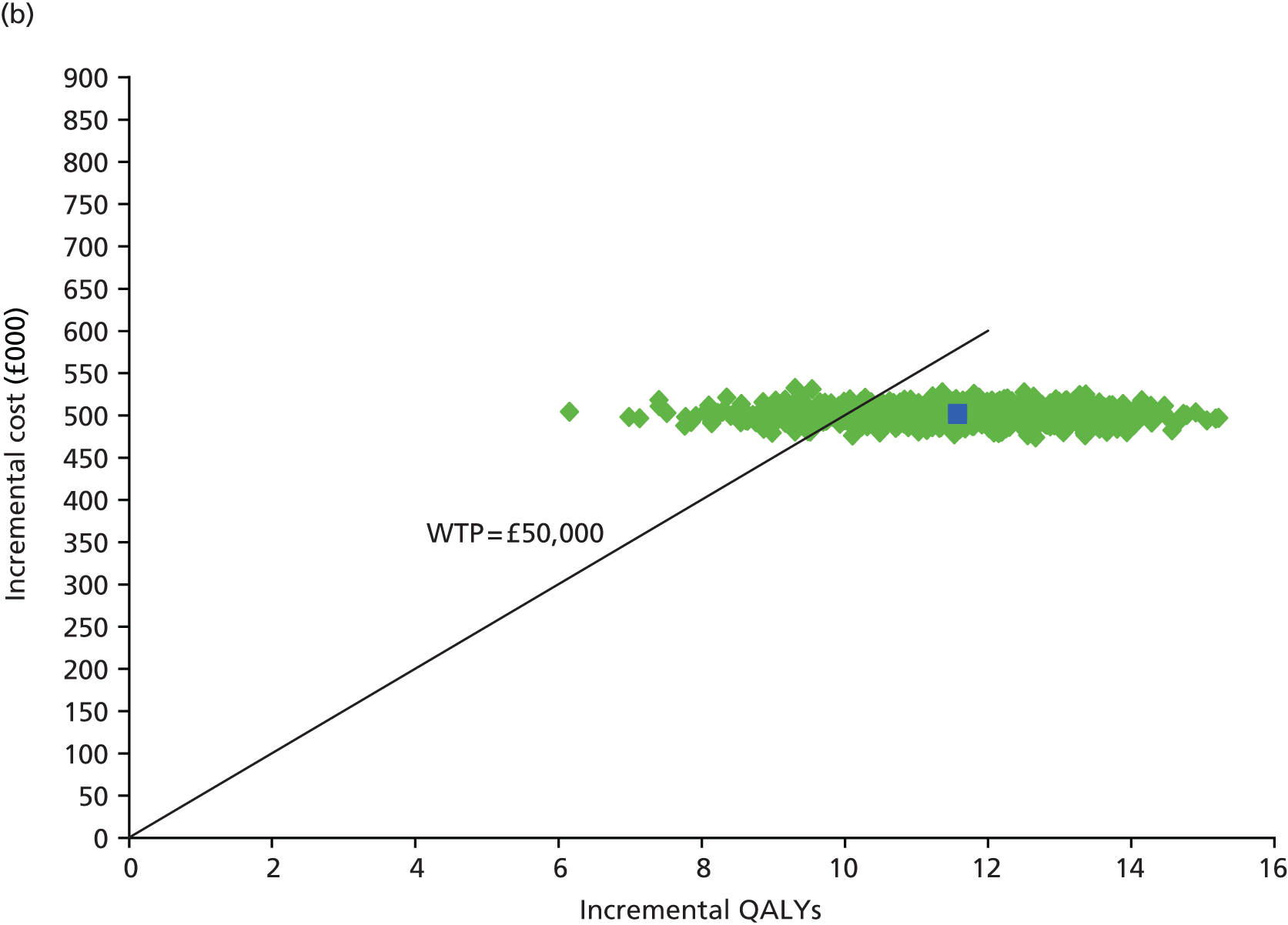
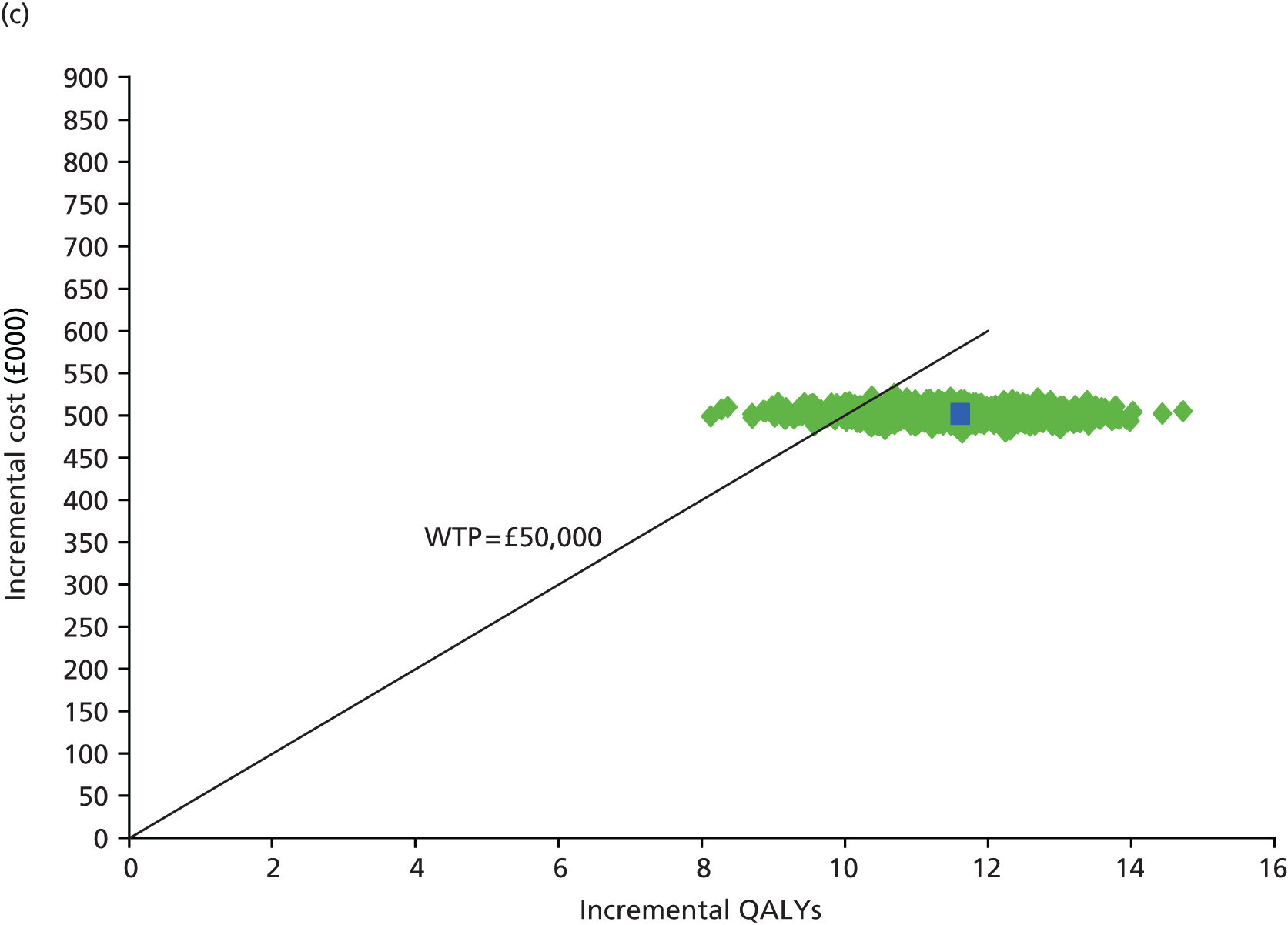
As expected, the health consequences of decision uncertainty in the mature evidence set (14.1 QALYs, £707,000) are lower than those reported in the minimum evidence set (304.6 QALYs, £15.23M) at a threshold of £50,000 per QALY gained. These consequences are reduced by the increased certainty surrounding the trajectory of the parametric survival curves and the effect of increased maturity on improving the cost-effectiveness of CAR T-cell therapy. As is evident from Figure 26, the increased certainty over the longer-term survival benefits of treatment (represented by the longer follow-up assumed in the intermediate and mature evidence sets) has a proportionately greater effect in reducing decision uncertainties within the minimum evidence set than the increased precision of greater patient numbers (i.e. reflected only in the mature evidence set).
Conclusions
The primary purpose of this chapter was to report the potential cost-effectiveness of CAR T-cell therapy within the separate scenarios considered and to highlight key uncertainties surrounding these results. An important aspect of this work was also to consider how these estimates could be presented and communicated to the committee to inform its deliberations. In doing this, we presented analyses based on approaches routinely requested within NICE’s existing methods guide. 122 We also undertook additional analyses that may provide useful additional insights to help inform subsequent committee deliberations and the potential nature of such analyses.
The sequence of assessments presented started with a conventional assessment of cost-effectiveness at the patient level based on the minimum evidence set. Disaggregated estimates of the costs and outcomes were presented, together with resulting cost-effectiveness estimates based on the ICER. These results were also expressed using NHEs, representing the difference between any health gained with the intervention and health forgone elsewhere in the health-care system, expressed either in monetary or in QALY terms. The impact of uncertainties was explored using conventional one-way sensitivity analyses (i.e. varying individual parameters or specific assumptions) and probabilistic approaches (i.e. exploring the impact of joint uncertainty across all parameters). Conventional scatterplots and acceptability curves were utilised to graphically show the impact of parameter uncertainties and other more methodological uncertainties (e.g. the appropriate discount rate). The analyses also explored the potential impact if the committee considered the criteria met for applying the non-reference case discount rate of 1.5% for costs and health effects.
In addition to the analyses undertaken using the conventional reference case approaches, a series of more exploratory analyses was also undertaken. In particular, the per-patient assessments were subsequently scaled up to population assessments, requiring an estimate of the number of potentially eligible patients (assumed to be approximately 38 patients per annum) and an assessment of the ‘technology time horizon’, that is, the period over which the therapy might be utilised within clinical practice (assumed to be 10 years in the exemplar). Although the population-level analyses are not formally requested within the existing NICE methods guide for reporting cost-effectiveness results,122 an assessment of population impact is required within section 5.12 (Impact on the NHS). Hence, these exploratory analyses were considered to be consistent with the requirement to consider the population impact and the specific requests within section 6.4.1 (Research recommendations) for the committee to balance the potential NHEs of current and future NHS patients when considering making research recommendations.
The results of the population-based analyses were summarised in terms of incremental NHEs (both in terms of QALYs and equivalent monetary value) together with an assessment of the probability that CAR T-cell therapy was cost-effective. Alongside these more conventional assessments, an assessment of the scale of the likely consequences was considered to be potentially informative to the committee, particularly in deliberations related to possible research recommendations. An estimate of the consequences of existing decision uncertainty was subsequently derived, reflecting the possible scale of NHEs that could be gained if uncertainty surrounding this decision could be resolved.
Using the different analyses, the impact of alternative pricing scenarios was explored, including conventional PASs (i.e. equivalent to a fixed price reduction) as well as more sophisticated schemes based on pay for performance and leasing approaches. Similarly, the impact of the alternative evidence sets was explored to establish the implications of increased precision and maturity assumed in the intermediate and mature evidence sets.
An important consideration within this work is the extent to which current NICE methods and processes are likely to appropriately quantify the potential uncertainties surrounding regenerative medicines and cell-based therapies to ensure that appropriate policy decisions are made regarding the adoption and spread of potentially promising technologies. Our findings show that the conventional assessments requested within the current TA process may not be sufficient. Estimates of the ICER and associated uncertainty (e.g. probability that a technology is cost-effective) were shown to be similar in one of the TPPs despite being based on three different evidence sets with varying levels of precision and maturity. Consequently, it is unclear how these differences would be reflected within the current deliberative process. Although it is acknowledged that different conclusions might be reached based on informal judgements, the importance of ensuring transparency in subsequent decisions remains a key principle of NICE and appears to be critical for manufacturers in developing appropriate R&D and pricing strategies.
The presentation of the scale of the consequences using population NHEs provided a clearer distinction between the different evidence sets and the impact of alternative pricing schemes. Consequently, their more routine application within the TA process for regenerative and cell-based therapies may be an important consideration for NICE. Furthermore, such comparisons could also provide a more transparent and explicit basis for considering the value of direct price reductions that might be realised using a conventional PAS (or less conventional schemes that work by indirectly lowering the effective price) compared with the provision of additional evidence (both precision and maturity) in terms of reducing decision uncertainty and its consequences. Such information might provide an important basis for discussions between manufacturers, NICE and other relevant parties in terms of how the existing uncertainties that exist might be appropriately managed, ensuring that risks and benefits are more appropriately shared.
Chapter 9 Issues arising from the National Institute for Health and Care Excellence panel meeting
Aseparate panel and meeting were convened by NICE to discuss the findings from Chapters 1–8 of the report. The panel included clinical experts and current and past NICE committee members and was chaired by Professor Andrew Stevens (current chairperson of the NICE Technology Appraisal Committee). A full list of panel members is provided in Appendix 9. The objective of the panel meeting was to assess the clinical effectiveness and cost-effectiveness evidence informing the separate TPPs and to identify potential issues and challenges for the NICE TA appraisal process and methods.
A summary of the clinical effectiveness and cost-effectiveness evidence was presented to the panel, together with an overview of key technical and process issues for consideration. The panel was then presented with a series of separate decision scenarios reflecting the two separate TPPs (bridge to HSCT and curative intent), the three evidence sets (minimum, intermediate and mature) and the impact of different pricing approaches based on the minimum evidence set. The panel was requested to deliberate on the scenarios and to provide ‘hypothetical’ decisions and outline the main considerations for these. The panel was requested to focus particularly on the role of uncertainty (clinical effectiveness and cost-effectiveness) to (1) identify key areas of uncertainty, (2) understand the nature of assessments/analyses that could help to inform deliberations and (3) explore the impact of different pricing approaches and different evidence sets.
The main clinical effectiveness and cost-effectiveness issues discussed by the panel are summarised in the following sections. This is followed by a summary of the panel discussions related to the separate scenarios.
Clinical issues
When asked for their thoughts, following a presentation of the clinical effectiveness and safety issues, the panel clinical experts commented that, although the data for CAR T-cell therapies were limited, the results nevertheless appeared to be very encouraging when T-cell therapy was compared with the best available alternative (clofarabine). They added that manufacturers nevertheless need guidance on how to account for the uncertainty of trial results, given the availability of only short-term data and the potential long-term effects. It is likely that future cell therapies will be aimed at larger populations (which ties in with the EMA’s adaptive pathways approach; see Chapter 2), and the panel members also highlighted that clarity was needed around how data requirements might change according to the size of the population.
The clinical experts stated that knowledge is improving about which patients will have side effects from CAR T-cell therapies. Knowledge on predictors of response (effect modifiers) was less developed, although the panel thought that the possibility of ERGs having access to IPD during any assessment could be an important step to help identify possible effect modifiers and to assess the reliability of submitted evidence. For this assessment, it was suggested that there might be interest in whether relapsed patients responded better than refractory patients.
The clinical experts were also asked about the potential variability in the efficacy and safety profiles of these types of interventions as a result of manufacturing variability and heterogeneity in patient response. It was considered that any differences in efficacy and safety because of variability in the manufacturing process are likely to be largest early on, but will be optimised with time. Variability of efficacy and safety as a result of individual patient heterogeneity is, however, likely to remain.
In response to a question from the panel about the success rates when manufacturing individual treatments, the clinicians said that, although the success rates for ‘expanding’ CAR T-cells is high for B-ALL patients, it is difficult to tell which patients’ (cells) can be successfully expanded (i.e. successful manufacture of the bespoke treatment). They stated that patients may die before the cell-based therapies can be produced and administered. It was noted that it will, therefore, be very important that trials report data relating to the full intention-to-treat population, including those patients for whom CAR T-cell expansion was not successful. If any patients required retreatment this should also be clearly reported.
There were serious concerns from the panel regarding the level of uncertainty in the evidence base, in particular that it was based on single-arm trials with possibly large unknown biases. There were concerns from the panel that certain efficacy estimates, particularly for the minimum data set, might be too optimistic, and questions were asked about whether any such biases could be quantified and adjusted for; it would be useful to see the impact of more pessimistic efficacy estimates on the cost-effectiveness results. There were concerns around the long-term benefit of the therapy and whether or not the estimate of OS in the minimum data set really could be carried into the mature data set.
Another issue that was raised was the panel being provided with knowledge about what further research had been mandated by the EMA (e.g. for conditional approvals). Understanding this may be key to knowing how much present uncertainty, and at what cost, can be accepted. The difficulties of decommissioning services once treatments are approved were also raised as potential problems.
Cost-effectiveness issues
A key issue regarding the cost-effectiveness results and implications for the ‘hypothetical’ decisions was whether the panel considered that existing criteria considered within the TA process in relation to EoL and 1.5% discounting (applied to costs and health outcomes) could be applied. The panel accepted that, based on the patient numbers, current prognosis and the likely treatment benefit, CAR T-cell therapy for relapsed/refractory ALL would be likely to meet existing criteria for EoL. However, the panel also noted that the existing criteria might need to be reconsidered more generally for therapies with curative potential. It was argued by one panel member that the EoL criteria were developed to cover scenarios in which people with conditions such as cancer with a short life expectancy were given some extension but whose life expectancy was still short. It was suggested that different QALY weights might need to be considered over a longer period of projected survival benefits for therapies that have curative potential.
The use of the 1.5% discounting was also discussed by the panel. Although it was noted that the existing criteria had been developed in response to a similar decision context, the panel was also aware that the criteria had been applied in only one previous appraisal (the TA for which it was developed212). The lack of precedent was noted and the panel concluded that its application could generate significant debate in future appraisals. Hence, no conclusion was reached during the panel meeting about its application to CAR T-cell therapy. The use of stepped discounting recommended by the UK Treasury was discussed by the panel but was considered to be more relevant for interventions that might have important intragenerational impacts (e.g. immunisation) as opposed to longer-term intergenerational effects.
In addition to the concerns it noted in relation to the possible biases and additional uncertainty arising from comparisons based on single-arm trials, the panel raised questions about whether or not there were wider structural uncertainties relevant to regenerative medicines and cell-based therapies that were not fully captured within the analyses presented. The panel concluded that identifying sources of potential bias and appropriately reflecting structural sources of uncertainty would be important considerations in future appraisals, and manufacturers would need to clearly report how these had been addressed within their submissions.
The panel discussed the sequence of assessments presented in the cost-effectiveness section and the exploratory approaches to quantifying decision uncertainty based on an assessment of the scale of the consequences associated with each decision using population NHEs. The panel agreed that these exploratory approaches provided a clearer and potentially important distinction between the different evidence sets and the impact of alternative pricing schemes. The panel also acknowledged that such assessments provided important information that could help to inform its deliberations. However, the panel further noted that, although such assessments were helpful and represented a useful starting point for deliberations, they were not necessarily sufficient to inform its final decisions. In particular, the panel expressed difficulty in determining how to interpret the numbers presented without a formal reference point to establish whether the consequences were sufficiently high to impact on their decisions and/or potential research recommendations.
The panel acknowledged that the estimates of the consequences represented a theoretical upper bound to the value of further research. However, the panel concluded that it would be important to explore these consequences further, in terms of both the underlying distribution (as opposed to the expected mean value of the consequences) and needing to decompose the overall estimate in relation to specific sources of uncertainty. This latter aspect was considered particularly important in determining the extent to which particular sources of uncertainty could be resolved by additional research, the type of research that might be most appropriate and, finally, whether this research would be feasible following a positive approval. The panel was also aware of the relevance of existing published work128 and work by the NICE DSU211 that would be important to consider in any review of potential processes or methods.
Prior to a more detailed discussion of the specific decision scenarios, the panel outlined a number of more general considerations related to the cost-effectiveness evidence and results:
-
In discussing the appropriate cost-effectiveness threshold for the purpose of NICE decision-making, the panel was clear that £50,000 per QALY (assuming that the EoL criteria applied) represented an absolute upper bound to the range that NICE would consider acceptable. The panel concluded that other considerations (e.g. innovation) would not be applied in conjunction with the higher threshold considered in an EoL appraisal. Furthermore, the panel also considered that the upper end of the range was unlikely to be considered appropriate in the presence of significant evidential uncertainties.
-
The panel concluded that, if the hypothetical price of CAR T-cell therapy had been set using the conventional cost-effectiveness threshold range (£20,000–30,000 per QALY), this could have mitigated some of these uncertainties, increasing the likelihood of a positive recommendation.
-
The panel appreciated that there was a difference between the deterministic estimates of the ICER and the probabilistic estimates of the ICER because of the non-linearity between the parameter inputs and the model outputs (i.e. mean costs, QALYs and ICER). The panel also noted that, for some analyses, these differences resulted in ICER estimates that could have a material impact on its decisions (i.e. situations in which the deterministic and probabilistic estimates lie on either side of the cost-effectiveness threshold). The panel concluded that the probabilistic estimates were the more appropriate basis for informing its decisions.
-
The panel raised issues regarding the possible nature and magnitude of any irrecoverable costs that might be incurred by the NHS and the implications for its decisions. The panel concluded that an ‘exit strategy’ for the NHS would be a key consideration for interventions that appear highly promising but for which significant uncertainties and irrecoverable costs may exist.
-
The panel acknowledged that the different pricing schemes had important impacts both in terms of the ICER and in terms of the allocation of any risk between the NHS and manufacturers. The concept of the ‘leasing approach’ was identified as a potentially important option and there was consensus among the panel that this warranted further exploration by NICE and manufacturers (e.g. logistics, costs and overall feasibility).
-
The panel recognised the various issues and challenges likely to be faced by the manufacturers of regenerative medicines and cell-based therapies. The panel also noted that many of the issues and implications identified did not appear to be specific to these types of therapies but were also apparent in appraisals of more conventional products. However, the panel acknowledged that the challenges may be faced more routinely by manufacturers of regenerative medicines and cell-based therapies and that the resulting levels of uncertainty (and the potential scale of the consequences) may exceed those that existing committees might conclude could be appropriately dealt with by existing processes and the current methods guide.
Panel discussion of the different scenarios
Following a general discussion of clinical effectiveness and cost-effectiveness issues, the panel was presented with a series of ‘decision scenarios’ based on the results reported in Chapter 8. For each TPP, the scenarios started with the minimum evidence set and a fixed acquisition cost for CAR T-cell therapy (scenario 1). Scenario 2 explored the impact of alternative pricing approaches based on the same minimum evidence set. Scenarios 3 and 4 were based on the results from the intermediate and mature evidence sets, respectively, assuming a fixed acquisition cost.
For each scenario, the panel was presented with a summary of the deterministic ICER and the probabilistic, population-level results, including an estimate of the ICER, an estimate of the incremental NHE (expressed in monetary and QALY terms), the probability of CAR T-cell therapy being cost-effective and an assessment of the scale of the consequences of decision uncertainty (again expressed in monetary and QALY terms).
A summary of the panel considerations is provided below:
-
For scenario 1, the panel understood that the deterministic estimate of the ICER for CAR T-cell therapy was close to the £50,000 upper bound of the ICER range considered acceptable currently when the EoL criteria are met. However, the panel concluded that the probabilistic estimates of the ICER were more appropriate given the model non-linearity. As the probabilistic ICER in the base case for both TPPs exceeded the upper bound of the ICER range, the panel concluded that CAR T-cell therapy would be unlikely to represent an efficient use of NHS resources in scenario 1. The panel concluded that, although other aspects of innovation were discussed that were important considerations for CAR T-cell therapy, additional weight should not be incorporated over and above that which had already been permitted when applying the EoL criteria.
-
For scenario 2, the panel acknowledged the different impacts of the alternative pricing schemes on the ICER, the scale of the consequences of decision uncertainty and the apportionment of any risk between the NHS and a manufacturer. The panel noted that the lifetime leasing scheme resulted in a significant reduction in the scale of decision uncertainty compared with a fixed acquisition cost. The panel also acknowledged that the leasing scheme could also provide an important exit strategy for the NHS given the high uncertainties that were evident. There was consensus among panel members that innovative financing schemes could be an important consideration in future appraisals.
-
The panel also noted that there were important differences in the scale of the consequences of decision uncertainty across the separate TPPs, with significantly higher consequences reported in the curative intent TPP. The panel understood that the use of an external surrogate relationship between MRD, HSCT and remission status in the bridge to HSCT TPP had an important impact on reducing the scale of the decision consequences over the modelled time horizon.
-
The panel found it difficult to determine the policy significance of the estimates reported for decision uncertainty without further analyses and an appropriate reference point. However, the panel also acknowledged in the bridge to HSCT TPP that the magnitude of the incremental NHE (i.e. the NHE that might be gained from immediate approval) significantly exceeded the scale of the consequences of decision uncertainty for the pricing schemes formed on a pay for performance approach based on achieving remission. The panel understood that the higher incremental NHE reported in these scenarios (and reduction in the consequences of decision uncertainty) was driven by the lower ICER as a result of the direct or indirect impact on the acquisition cost of CAR T-cell therapy and that this had an important impact on the scale of the consequences of decision uncertainty.
-
Faced with the high levels of uncertainty, the panel concluded that schemes that reduced the ICER to significantly below the upper bound of the EoL range and closer to the more conventional ICER range (£20,000–30,000 per QALY) would increase the likelihood of approval.
-
The panel also acknowledged that the reduction in the consequences of decision uncertainty in the leasing and pay for performance schemes arose because of the risks being shared between the NHS and manufacturers. Although the ICER of the lifetime leasing method exceeded the upper bound of the EoL range, the panel concluded that it may have looked more favourably on a combined scheme involving a fixed price discount and a leasing element in the bridge to HSCT TPP. However, in the absence of being provided with a formal assessment of this scheme, the panel felt that it was not possible to make a clear recommendation.
-
The panel was less clear on potential recommendations across the different pricing schemes for the curative intent TPP. Although the panel acknowledged that the consequences of decision uncertainty were reduced by each of the alternative pricing approaches, it remained concerned at the scale of the consequences that remained. Again, the panel concluded that it may have looked more favourably on a combined scheme involving a fixed price discount and a leasing element but noted that it had not been presented with results from such a scenario.
-
The panel was also aware that different prices were assumed across the separate TPPs, reflecting the different effectiveness estimates reported in the different studies used in each TPP. The panel indicated that, if the same price that was used in the bridge to HSCT TPP had been applied in the curative intent TPP, this would have potentially significantly improved the ICER and lowered the consequences of decision uncertainty.
-
Faced with higher consequences in the curative intent TPP, the panel concluded that the combination of using the same price as in the bridge to HSCT TPP and a leasing scheme would potentially improve the ICER and lower the consequences of decision uncertainty to a level that could potentially be acceptable. Again, in the absence of being provided with a formal assessment of such a scheme, the panel felt that it was not possible to make a clear recommendation.
-
The panel discussed the additional evidence sets that had been generated for each TPP (scenarios 3 and 4). The panel acknowledged that these estimates were generated using a series of assumptions and hence remained subject to various additional uncertainties. However, the panel understood the principles that were being considered and that there were important differences across the evidence sets for the separate TPPs. The panel understood that the difference across the TPPs was primarily the use of an external surrogate relationship in the bridge to HSCT TPP. The panel acknowledged that greater uncertainty could arise in situations in which a robust surrogate relationship had not been demonstrated and that ensuring that evidence is sufficiently robust (i.e. in terms of precision and/or maturity) for decision-making would be an important consideration. The panel noted that the consequences of decision uncertainty in the intermediate evidence set for the curative intent TPP were significantly reduced compared with those in the minimum evidence set and were closer to the scale of those reported for the minimum evidence set for the bridge to HSCT TPP, for which a surrogate relationship had been assumed.
-
The panel understood that the scale of the consequences was further reduced in the mature evidence sets because of increased precision (compared with the intermediate data set) and maturity (compared with the minimum evidence set) and that this was most evident in the curative intent TPP because additional surrogate evidence had not been included.
-
The panel acknowledged the challenges and difficulties of generating mature evidence at the point at which a product is launched. However, the panel considered that the principles outlined through the different assessments would be important in informing future deliberations. In particular, the panel noted that a comparison of the magnitude of the incremental NHE and the consequences of decision uncertainty provided an important starting point for deliberations regarding the scale of the NHE that could be achieved by immediate approval and that which might be achieved by further research.
-
The panel noted that further assessments could be helpful to inform (1) whether a positive approval decision might alter incentives to undertake the type of research necessary to resolve the main sources of uncertainty and (2) the full opportunity costs of approval and rejection decisions. The panel concluded that further information concerning the distribution of the consequences and further exploration of the main sources of these consequences would provide important additional information.
Additional exploratory analyses undertaken after the panel discussion
Following the panel meeting, a series of additional exploratory analyses was undertaken to capture some of the specific requests and considerations that were identified during the panel discussions. These analyses are not intended to be comprehensive but rather to reflect on some of the main points raised and to consider any further implications.
Information on the distribution of consequences is shown in Figure 27 based on the minimum evidence set of the curative intent TPP. The most common outcome (50.7%) is for CAR T-cell therapy to be cost-effective at a willingness-to-pay threshold of £50,000. Consequently, there are no negative consequences to the NHS in these instances. However, in 49.3% of iterations (1 – probability of CAR T-cell therapy being cost-effective), the decision to recommend CAR T-cell therapy may be incorrect (at a willingness-to-pay threshold of £50,000). The consequences of making an incorrect decision are expressed in terms of the NHEs forgone. In this analysis, most of the negative consequences are < 1000 QALYs (probability of 36.4%). The probability that the negative consequences exceed 1000 and 3000 QALYs is 12.9% and 0.1%, respectively.
FIGURE 27.
Distribution of the consequences of decision uncertainty.

The panel was also interested in exploring the impact of a number of alternative pricing schemes on the cost-effectiveness of CAR T-cell therapy and associated decision uncertainty. These schemes included the application of the bridge to HSCT fixed acquisition cost to the curative intent TPP, as well as a lifetime leasing approach with or without an additional 10% price discount.
Applying both a lifetime leasing method and a 10% discount to the cost of CAR T-cell therapy to the minimum evidence set analysis for the curative intent TPP improved the ICER (£45,502 per QALY), resulting in a large decrease in the consequences of decision uncertainty and an increase in the probability of cost-effectiveness, as shown in Table 40.
| Pricing scenario | Population level | Cost-effectiveness threshold of £50,000 per QALY gained | ||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Cost (£) | QALYs | ICER (£) | NHE, QALYs (£) | Incremental NHE, QALYs (£) | Probability cost-effective (%) | Consequences of decision uncertainty, QALYs (£) | |
| Base case | CAR T-cell therapy | 183,931,590 | 3501.50 | 50,906 | –177.13 (–8,935,381) | –56.48 (–2,902,629) | 50.7 | 304.6 (15,229,876) |
| Standard of care | 25,270,727 | 384.76 | –120.66 (–6,032,752) | |||||
| Lifetime leasing and 10% discount (£2955 per month) | CAR T-cell therapy | 164,420,596 | 3458.93 | 45,502 | 170.52 (8,525,896) | 275.00 (13,750,033) | 87.2 | 27.2 (1,358,584) |
| Standard of care | 25,321,756 | 401.95 | –104.48 (–5,224,137) | |||||
| Same pricing as bridge to HSCT TPP (fixed cost of £356,100) | CAR T-cell therapy | 129,435,001 | 3446.20 | 34,337 | 857.50 (42,874,913) | 951.11 (47,555,583) | 85.6 | 73.1 (3,655,992) |
| Standard of care | 25,178,368 | 409.95 | –93.61 (–4,680,670) | |||||
| Same total cost as bridge to HSCT TPP with lifetime leasing (£2211.42 per month) | CAR T-cell therapy | 129,689,785 | 3532.92 | 33,277 | 939.12 (46,956,030) | 1050.02 (52,500,851) | 99.4 | 2.3 (112,597) |
| Standard of care | 25,219,874 | 393.50 | –110.90 (–5,544,821) | |||||
| Same total cost as bridge to HSCT TPP with lifetime leasing and 10% discount (£1990.28 per month) | CAR T-cell therapy | 117,750,114 | 3509.04 | 29,713 | 1154.04 (57,701,888) | 1262.40 (63,120,093) | 100.0 | 0 (0) |
| Standard of care | 25,302,238 | 397.68 | –108.36 (–5,418,205) | |||||
Applying the bridge to HSCT fixed acquisition cost of CAR T-cell therapy (£356,100) to the minimum evidence set analysis for the curative intent TPP significantly improved the cost-effectiveness of curative CAR T-cell therapy, resulting in an ICER of £34,337 per QALY (see Table 40). With improved cost-effectiveness, the expected consequences of decision uncertainty are also improved, decreasing from 304 QALYs (£15.23M) in the base case to 73.1 QALYs (£3.66M). Applying a lifetime leasing method resulted in further reductions in the consequences of decision uncertainty to 2.3 QALYs (£0.11M). By applying an additional 10% discount alongside the leasing and bridge to HSCT acquisition cost, it was possible to eliminate the potential consequences of decision uncertainty (at a willingness-to-pay threshold of £50,000).
These additional analyses further reinforce the importance of considering the implications of decision uncertainty for the ICER as well as the scale of the consequences. A key finding from these additional analyses is that the consequences of decision uncertainty in the minimum evidence set can be significantly lowered by reductions in price or the application of alternative pricing schemes. Indeed, the additional exploratory analyses reveal that the scale of the consequences might be reduced to a similar or even lower magnitude than that which could be resolved through the provision of further evidence alone. Furthermore, by reducing the opportunity costs of early approval, increased flexibility in pricing and pricing approaches would allow more patients to receive early access to potentially innovative regenerative medicines and cell-based therapies.
Chapter 10 Discussion
Implications for the National Institute for Health and Care Excellence technology appraisal process
Modifications (which may sometimes be informed by methods research) might be considered to update the methods guidance provided to manufacturers and ERGs in the following areas.
Use of surrogate end points
The choice of surrogate end points used by manufacturers in their submissions must be researched, explicit and justified. Ideally, a systematic review should be performed to evaluate the strength of the association between the surrogate and the patient-relevant outcome, and the evidence on surrogate validation should be presented according to an explicit hierarchy.
Pivotal study design and the use of historical control data sets
For manufacturer submissions, consideration should be given to the benefits of producing recommendations and/or minimum reporting requirements in terms of the methods used to obtain and analyse single-arm trial data when they are compared with historical control data. When single-arm trial data form the main basis of an assessment, a clear rationale should be given for the type of comparisons made (implicit or explicit) and for the choice of the historical control data that were selected. For example, the gold standard for historical data might be matched data obtained from a patient database (rather than relying on published studies, which might not fit the trial population being studied well enough). When designing trials, manufacturers should bear in mind that multicentre trials are likely to produce more reliable and generalisable results than single-centre trials.
Evidence Review Groups might benefit from using checklists when appraising how historical control data were identified and analysed by manufacturers.
Efficacy estimates
Submission of IPD might be beneficial for ERGs, especially when data sets are small. Use of multivariate meta-analysis can lead to reduced uncertainty around the effectiveness parameter. By allowing all of the relevant data to be incorporated in estimating clinical effectiveness outcomes – including data from surrogate outcomes – multivariate meta-analysis can improve the estimation of health utilities through mapping methods.
Manufacturers should report the data for the full trial population, that is, all eligible patients, including patients who died before they could receive treatment and patients for whom a bespoke (autologous) treatment could not be produced.
The role of any further mandatory trial evidence
Manufacturers should provide details of mandated further studies (e.g. those related to conditional approvals or approvals made through the EMA’s adaptive pathways approach). Future reports from the ADAPT SMART project should provide details about how the use of development plans across target populations agreed upfront with the EMA is working. Guidance may be needed regarding methodological approaches to utilising ‘confirmatory’ trial data in a related indication to update the decision that NICE made for the original indication.
Consideration is also needed regarding the precise role that NICE will have in EMA adaptive pathways processes. For example, what will be the mechanisms by which the EMA updates NICE with new efficacy and safety data for conditionally approved ATMPs (in a timely way) and how will NICE deal with the new data (process wise)?
Extrapolation approaches
Given the inevitable uncertainties that are likely to exist regarding the longer-term benefits of regenerative medicines and cell-based therapies, further methodological research could be usefully undertaken to help inform how these uncertainties might be appropriately quantified in a transparent manner to inform cost-effectiveness analyses. Further research may be particularly helpful to determine the appropriateness of alternative survival modelling approaches to regenerative medicines and cell-based therapies, including more flexible survival models and cure fraction models.
The level of data maturity is an important factor in deriving robust survival projections that are required for cost-effectiveness assessments. When follow-up is immature, a single ‘best-fitting’ survival distribution may not adequately characterise uncertainties over the longer-term extrapolation period. Although the robustness of the ICER estimates to alternative distributions can be explored through separate sensitivity analyses or scenarios, the transparency of the process may be impacted if the weighting of these is not explicitly considered in subsequent policy decisions. The feasibility and appropriateness of model-averaging approaches may also need to be considered more formally. The advantage of these approaches is that the parametric uncertainty associated within each distribution and the uncertainty (or weights) surrounding the choice of preferred method can be more explicitly characterised. However, given the potential complexity in both undertaking these analyses and communicating the results, efforts will need to be made to ensure that models are developed so that informal judgements can be explicitly incorporated in a timely and transparent manner.
Irrecoverable costs and possible learning curve effects
Given the complexity of the treatment pathways that may be required for regenerative medicines and cell-based therapies, manufacturers will need to report clearly the resource and cost assumptions of the different processes required to determine whether the full costs to the NHS have been included and any aspects for which uncertainties may exist. Issues of irrecoverable costs may need to be considered more formally, particularly if a new technology could impose additional infrastructure requirements on the health system. If reimbursement decisions about the technology change before the end of the lifetime of the equipment (e.g. approval is withdrawn), then these costs may not be recovered and hence need to be explicitly considered.
The existence and possible impact of learning curves may also be an important issue for clinical effectiveness and cost-effectiveness assessments. Although the existence of learning curves has received attention in the clinical literature, the relevance of recent work in this area in the context of assessing the cost-effectiveness of medical devices should be considered. 133
Quantification of decision uncertainty
Presentation of the scale of the consequences of decision uncertainty using population NHEs may provide an important additional approach to quantifying decision uncertainty to the assessments already routinely specified within the existing TA methods guide. 122 The implications of existing research128 and research by the DSU211 will also need to be considered by NICE to determine whether further changes to its processes or methods may be helpful for informing the nature of any additional assessments that may be required.
Such assessments could provide a more transparent and explicit basis for discussions between manufacturers, NICE and other relevant stakeholders in terms of how the existing uncertainties might be appropriately managed, ensuring that risks and benefits are more appropriately shared. Broader consideration will also need to be given to approaches that may extend beyond NICE’s existing remit, for example alternative payment schemes. Consequently, other bodies and manufacturers themselves may also have an important role in identifying more innovative approaches to seeking reimbursement that recognise the inherent uncertainties and lead to a more efficient sharing of associated risk.
Existing criteria
The National Institute for Health and Care Excellence’s existing processes also make separate provision for specific disease and technology characteristics that may be relevant to many regenerative medicines and cell-based therapies. Although NICE’s current EoL criteria allow the committee to explore a QALY weighting that is different from that of the reference case, the appropriateness of these criteria may need to be considered in relation to treatments that have curative potential. Further methodological research may also be important to determine whether an alternative weighting approach might be more appropriate for curative therapies. Existing research has identified a potential disconnect between individual and societal preferences concerning valuation of treatment compared with preventative interventions. Further research that is more specifically focused on the concept of cure may provide important additional insights.
Although the NICE methods guide permits the use of a non-reference case discount to be applied in specific contexts, it remains unclear whether regenerative medicines and cell-based therapies would meet the existing criteria (e.g. uncertainties over the projected benefits and/or potentially significant irrecoverable costs). Consequently, NICE may need to provide additional guidance to ensure that manufacturers understand the likelihood of meeting these criteria.
Acknowledgements
We acknowledge and thank Professor Robert Hawkins (Cancer Research UK Professor of Medical Oncology, University of Manchester and Christie Hospital) and Dr Beki James (Consultant Paediatric Haematologist, Leeds Teaching Hospitals NHS Trust) for their advice on clinical issues; Natalie Mount and Panos Kefalas from Cell Therapy Catapult for their advice and their comments on the report; and Kath Wright for her help in searching for studies and managing references.
The views expressed in this report are those of the authors and not necessarily those of the NIHR HTA programme. Any errors are the responsibility of the authors.
Contributions of authors
Robert Hettle contributed to the project protocol, conducted the analysis and modelling for the exemplar and wrote sections of the report.
Mark Corbett contributed to the project protocol, was lead reviewer for the clinical issues sections and the clinical review of the exemplar and wrote sections of the report.
Sebastian Hinde contributed to the protocol, conducted the review of issues relating to cost-effectiveness, contributed to the analysis and modelling for the exemplar and wrote sections of the report.
Robert Hodgson contributed to the protocol and prepared the clinical issues section on study biases.
Julie Jones-Diette prepared the clinical issues section on surrogate outcomes.
Nerys Woolacott had overall responsibility for the clinical sections of the report. She contributed to the protocol, the preparation of all clinical sections and the writing of the report.
Stephen Palmer had overall responsibility for the cost-effectiveness sections. He contributed to the protocol and to all aspects of the cost-effectiveness work, including the writing of the report.
Data sharing statement
This report is based on an assessment of a hypothetical case study and therefore the data generated are not suitable for sharing beyond what are contained within the report. Further information can be obtained from the corresponding author.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Mason C, Dunnill P. A brief definition of regenerative medicine. Regen Med 2008;3:1-5. https://doi.org/10.2217/17460751.3.1.1.
- Regenerative Medicine Report. London: The Stationery Office; 2013.
- Building on Your Own Potential: A UK Pathway for Regenerative Medicine. London: Department of Health; 2015.
- Guideline on Safety and Efficacy Follow-Up – Risk Management of Advanced Therapy Medicinal Products. London: EMA; 2008.
- Edwards SJ, Lilford RJ, Braunholtz DA, Jackson JC, Hewison J, Thornton J. Ethical issues in the design and conduct of randomised controlled trials: a review. Health Technol Assess 1999;2.
- Ashcroft RE, Chadwick DW, Clark SR, Edwards RH, Frith L, Hutton JL. Implications of socio-cultural contexts for the ethics of clinical trials. Health Technol Assess 1997;1.
- Gupta S, Faughnan ME, Tomlinson GA, Bayoumi AM. A framework for applying unfamiliar trial designs in studies of rare diseases. J Clin Epidemiol 2011;64:1085-94. http://dx.doi.org/10.1016/j.jclinepi.2010.12.019.
- Reflection Paper on Methodological Issues in Confirmatory Clinical Trials Planned with an Adaptive Design. London: EMA; 2007.
- Adaptive Design Clinical Trials for Drugs and Biologics. Draft Guidance. Silver Spring, MD: FDA; 2010.
- Tsimberidou AM, Braiteh F, Stewart DJ, Kurzrock R. Ultimate fate of oncology drugs approved by the US food and drug administration without a randomized trial. J Clin Oncol 2009;27:6243-50. http://dx.doi.org/10.1200/JCO.2009.23.6018.
- Baird LG, Banken R, Eichler HG, Kristensen FB, Lee DK, Lim JC, et al. Accelerated access to innovative medicines for patients in need. Clin Pharmacol Ther 2014;96:559-71. http://dx.doi.org/10.1038/clpt.2014.145.
- Questions and Answers Following the Initial Experience of the Adaptive Licensing Pilot Project. London: EMA; 2014.
- Lucas F. Medicines Adaptive Pathways to Patients (MAPPs) – opportunities and challenges in Europe. Value &Amp; Outcomes Spotlight 2015:11-3.
- ADAPTSMART . Accelerated Development of Appropriate Patient Therapies: A Sustainable, Multi-Stakeholder Approach from Research to Treatment-Outcomes n.d. http://adaptsmart.eu (accessed 11 September 2015).
- European Commission Expert Group on Safe and Timely Access to Medicines for Patients (STAMP) . Regulatory Tools for Early Access: Accelerated Assessment Procedure n.d.
- Van Bokkelen G, Morsy M, Kobayashi T-H. Demographic transition, health care challenges, and the impact of emerging international regulatory trends with relevance to regenerative medicine. Curr Stem Cell Rep 2015;1:102-9. https://doi.org/10.1007/s40778-015-0013-5.
- Sipp D. Conditional approval: Japan lowers the bar for regenerative medicine products. Cell Stem Cell 2015;16:353-6. http://dx.doi.org/10.1016/j.stem.2015.03.013.
- European Medicines Agency . Glybera (Alipogene Tiparvovec): EMEA H C 002145 2012. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002145/WC500135476.pdf (accessed 17 January 2017).
- European Medicines Agency . MACI (Matrix Applied Characterised Autologous Cultured Chondrocytes): EMEA H C 002522 0000 2013. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002522/WC500145888.pdf (accessed 17 January 2017).
- NICE . Autologous Chondrocyte Implantation for Repairing Symptomatic Articular Cartilage Defects of the Knee (Including a Review of TA89) 2014. www.nice.org.uk/guidance/GID-TAG446/documents/knee-cartilage-defects-autologous-chondrocyte-implantation-id686-assessment-report2 (accessed 17 January 2017).
- US Food and Drug Administration . Autologous Cultured Chondrocytes: Summary for Basis of Approval n.d. www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM109341.pdf (accessed 17 January 2017).
- European Medicines Agency . ChondroCelect (Characterised Viable Autologous Cartilage Cells Expanded Ex Vivo Expressing Specific Marker Proteins): EMEA H C 000878 2009. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/000878/WC500026035.pdf (accessed 17 January 2017).
- European Medicines Agency . Holoclar (Ex Vivo Expanded Autologous Human Corneal Epithelial Cells Containing Stem Cells): EMEA H C 002450 0000 2014. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002450/WC500183405.pdf (accessed 17 January 2017).
- European Medicines Agency . Provenge (Autologous Peripheral Blood Mononuclear Cells Activated With PAP-GM-CSF (Sipuleucel-T)): EMEA H C 002513 0000 2013. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002513/WC500151101.pdf (accessed 17 January 2017).
- US Food and Drug Administration . Approval History, Letters, Reviews, and Related Documents – Provenge n.d. www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/ucm213554.htm (accessed 17 January 2017).
- NICE . Final Appraisal Determination: Sipuleucel-T for Treating Asymptomatic or Minimally Symptomatic Metastatic Hormone-Relapsed Prostate Cancer 2015. www.nice.org.uk/guidance/ta332/documents/prostate-cancer-metastatic-hormone-relapsed-sipuleucelt-1st-line-id573-final-appraisal-determination-document2 (accessed 17 January 2017).
- NICE . The ReCell Spray-On Skin System for Treating Skin Loss, Scarring and Depigmentation After Burn Injury 2014. www.nice.org.uk/guidance/mtg21 (accessed 17 January 2017).
- Council of the European Communities . Council Directive 93 42 EEC of 14 June 1993 Concerning Medical Devices 1993. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:31993L0042:EN:HTML (accessed 17 January 2017).
- Reuters . German Regulator Puts UniQure Gene Therapy Appraisal on Hold 2015. www.reuters.com/article/2015/04/17/health-genetherapy-germany-idUSL5N0XE0X620150417 (accessed 17 November 2016).
- Benson K, Hartz AJ. A comparison of observational studies and randomized, controlled trials. N Engl J Med 2000;342:1878-86. https://doi.org/10.1056/NEJM200006223422506.
- Beynon R, Savovic J, Harris R, Altman D, Sterne J, Deeks J. Quantification of bias in the results of non-randomised studies compared with randomised studies. Z Evid Fortbild Qual Gesundhwes 2008;102:6-69.
- Dahabreh IJ, Sheldrick RC, Paulus JK, Chung M, Varvarigou V, Jafri H, et al. Do observational studies using propensity score methods agree with randomized trials? A systematic comparison of studies on acute coronary syndromes. Eur Heart J 2012;33:1893-901. http://dx.doi.org/10.1093/eurheartj/ehs114.
- Hartz A, Bentler S, Charlton M, Lanska D, Butani Y, Soomro GM, et al. Assessing observational studies of medical treatments. Emerg Themes Epidemiol 2005;2. https://doi.org/10.1186/1742-7622-2-8.
- Ioannidis JP, Haidich AB, Pappa M, Pantazis N, Kokori SI, Tektonidou MG, et al. Comparison of evidence of treatment effects in randomized and nonrandomized studies. JAMA 2001;286:821-30. https://doi.org/10.1001/jama.286.7.821.
- MacLehose RR, Reeves BC, Harvey IM, Sheldon TA, Russell IT, Black AM. A systematic review of comparisons of effect sizes derived from randomised and non-randomised studies. Health Technol Assess 2000;4.
- Shepherd J, Bagnall A-M, Colquitt J, Jacqueline D, Duffy S, Sowden A. Sometimes Similar, Sometimes Different: A Systematic Review of Meta-Analyses of Randomised and Non-Randomised Policy Intervention Studies. York: University of York; 2006.
- Concato J, Shah N, Horwitz RI. Randomized, controlled trials, observational studies, and the hierarchy of research designs. N Engl J Med 2000;342:1887-92. https://doi.org/10.1056/NEJM200006223422507.
- Britton A, McKee M, Black N, McPherson K, Sanderson C, Bain C. Choosing between randomised and non-randomised studies: a systematic review. Health Technol Assess 1998;2.
- Sacks H, Chalmers TC, Smith H. Randomized versus historical controls for clinical trials. Am J Med 1982;72:233-40. https://doi.org/10.1016/0002-9343(82)90815-4.
- Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol 2012;13:518-27. http://dx.doi.org/10.1016/S1470-2045(12)70112-2.
- Golder S, Loke YK, Bland M. Comparison of pooled risk estimates for adverse effects from different observational study designs: methodological overview. PLOS ONE 2013;8. http://dx.doi.org/10.1371/journal.pone.0071813.
- Lonjon G, Boutron I, Trinquart L, Ahmad N, Aim F, Nizard R, et al. Comparison of treatment effect estimates from prospective nonrandomized studies with propensity score analysis and randomized controlled trials of surgical procedures. Ann Surg 2014;259:18-25. http://dx.doi.org/10.1097/SLA.0000000000000256.
- Abraham NS, Durairaj R, Young JM, Young CJ, Solomon MJ. How does an historic control study of a surgical procedure compare with the ‘gold standard’?. Dis Colon Rectum 2006;49:1141-8. https://doi.org/10.1007/s10350-006-0614-2.
- Verde PE, Ohmann C. Combining randomized and non-randomized evidence in clinical research: a review of methods and applications. Res Synth Methods 2015;6:45-62. http://dx.doi.org/10.1002/jrsm.1122.
- Doi SA. Evidence synthesis for medical decision making and the appropriate use of quality scores. Clin Med Res 2014;12:40-6. http://dx.doi.org/10.3121/cmr.2013.1188.
- Droitcour J, Silberman G, Chelimsky E. A new form of meta-analysis for combining results from randomized clinical trials and medical-practice databases. Int J Technol Assess Health Care 1993;9:440-9. https://doi.org/10.1017/S0266462300004694.
- Jackson C, Best N, Richardson S. Improving ecological inference using individual-level data. Stat Med 2006;25:2136-59. https://doi.org/10.1002/sim.2370.
- Jackson C, Best N, Richardson S. Hierarchical related regression for combining aggregate and individual data in studies of socio-economic disease risk factors. J R Stat Soc Ser A Stat Soc 2008;171:159-78.
- Prevost TC, Abrams KR, Jones DR. Hierarchical models in generalized synthesis of evidence: an example based on studies of breast cancer screening. Stat Med 2000;19:3359-76. https://doi.org/10.1002/1097-0258(20001230)19:24%3C3359::AID-SIM710%3E3.0.CO;2-N.
- Welton NJ, Ades AE, Carlin JB, Altman DG, Sterne JAC. Models for potentially biased evidence in meta-analysis using empirically based priors. J R Stat Soc Ser A Stat Soc 2009;172:119-36. https://doi.org/10.1111/j.1467-985X.2008.00548.x.
- Soares MO, Dumville JC, Ades AE, Welton NJ. Treatment comparisons for decision making: facing the problems of sparse and few data. J R Stat Soc Ser A Stat Soc 2014;177:259-79. https://doi.org/10.1111/rssa.12010.
- Spiegelhalter DJ, Best NG. Bayesian approaches to multiple sources of evidence and uncertainty in complex cost-effectiveness modelling. Stat Med 2003;22:3687-709. https://doi.org/10.1002/sim.1586.
- Turner RM, Spiegelhalter DJ, Smith GC, Thompson SG. Bias modelling in evidence synthesis. J R Stat Soc Ser A Stat Soc 2009;172:21-47. https://doi.org/10.1111/j.1467-985X.2008.00547.x.
- McNamee R. Regression modelling and other methods to control confounding. Occup Environ Med 2005;62:500-6. https://doi.org/10.1136/oem.2002.001115.
- Harrell FE. Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis. New York, NY: Springer; 2001.
- Bland JM, Altman DG. Matching. BMJ 1994;309. https://doi.org/10.1136/bmj.309.6962.1128.
- Austin PC. An Introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res 2011;46:399-424. https://doi.org/10.1080/00273171.2011.568786.
- Austin PC. Type I error rates, coverage of confidence intervals, and variance estimation in propensity-score matched analyses. Int J Biostat 2009;5. http://dx.doi.org/10.2202/1557-4679.1146.
- Austin PC, Grootendorst P, Anderson GM. A comparison of the ability of different propensity score models to balance measured variables between treated and untreated subjects: a Monte Carlo study. Stat Med 2007;26:734-53. https://doi.org/10.1002/sim.2580.
- Austin PC, Mamdani MM. A comparison of propensity score methods: a case-study estimating the effectiveness of post-AMI statin use. Stat Med 2006;25:2084-106. https://doi.org/10.1002/sim.2328.
- Lunceford JK, Davidian M. Stratification and weighting via the propensity score in estimation of causal treatment effects: a comparative study. Stat Med 2004;23:2937-60. https://doi.org/10.1002/sim.1903.
- Cepeda MS, Boston R, Farrar JT, Strom BL. Comparison of logistic regression versus propensity score when the number of events is low and there are multiple confounders. Am J Epidemiol 2003;158:280-7. https://doi.org/10.1093/aje/kwg115.
- Crosby DA, Dowsett CJ, Gennetian LA, Huston AC. A tale of two methods: comparing regression and instrumental variables estimates of the effects of preschool child care type on the subsequent externalizing behavior of children in low-income families. Dev Psychol 2010;46:1030-48. http://dx.doi.org/10.1037/a0020384.
- Kurth T, Walker AM, Glynn RJ, Chan KA, Gaziano JM, Berger K, et al. Results of multivariable logistic regression, propensity matching, propensity adjustment, and propensity-based weighting under conditions of nonuniform effect. Am J Epidemiol 2006;163:262-70. https://doi.org/10.1093/aje/kwj047.
- Martens EP, Pestman WR, de Boer A, Belitser SV, Klungel OH. Systematic differences in treatment effect estimates between propensity score methods and logistic regression. Int J Epidemiol 2008;37:1142-7. http://dx.doi.org/10.1093/ije/dyn079.
- Shah BR, Laupacis A, Hux JE, Austin PC. Propensity score methods gave similar results to traditional regression modeling in observational studies: a systematic review. J Clin Epidemiol 2005;58:550-9. https://doi.org/10.1016/j.jclinepi.2004.10.016.
- Stukel TA, Fisher ES, Wennberg DE, Alter DA, Gottlieb DJ, Vermeulen MJ. Analysis of observational studies in the presence of treatment selection bias: effects of invasive cardiac management on AMI survival using propensity score and instrumental variable methods. JAMA 2007;297:278-85. https://doi.org/10.1001/jama.297.3.278.
- Sturmer T, Joshi M, Glynn RJ, Avorn J, Rothman KJ, Schneeweiss S. A review of the application of propensity score methods yielded increasing use, advantages in specific settings, but not substantially different estimates compared with conventional multivariable methods. J Clin Epidemiol 2006;59:437-47. https://doi.org/10.1016/j.jclinepi.2005.07.004.
- Laborde-Castérot H, Agrinier N, Thilly N. Performing both propensity score and instrumental variable analyses in observational studies often leads to discrepant results: a systematic review. J Clin Epidemiol 2015;68:1232-40. http://dx.doi.org/10.1016/j.jclinepi.2015.04.003.
- Biondi-Zoccai G, Romagnoli E, Agostoni P, Capodanno D, Castagno D, D’Ascenzo F, et al. Are propensity scores really superior to standard multivariable analysis?. Contemp Clin Trials 2011;32:731-40. http://dx.doi.org/10.1016/j.cct.2011.05.006.
- Hamre HJ, Glockmann A, Kienle GS, Kiene H. Combined bias suppression in single-arm therapy studies. J Eval Clin Pract 2008;14:923-9. http://dx.doi.org/10.1111/j.1365-2753.2007.00903.x.
- Guyot P, Ades AE, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan–Meier survival curves. BMC Med Res Methodol 2012;12. http://dx.doi.org/10.1186/1471-2288-12-9.
- Paulus JK, Dahabreh IJ, Balk EM, Avendano EE, Lau J, Ip S. Opportunities and challenges in using studies without a control group in comparative effectiveness reviews. Res Synth Methods 2014;5:152-61. http://dx.doi.org/10.1002/jrsm.1101.
- Vickers AJ, Ballen V, Scher HI. Setting the bar in Phase II trials: the use of historical data for determining ‘go/no go’ decision for definitive Phase III testing. Clin Cancer Res 2007;13:972-6. https://doi.org/10.1158/1078-0432.CCR-06-0909.
- Torgerson DJ, Torgerson C. Designing Randomised Trials in Health, Education and the Social Sciences: An Introduction. Basingstoke: Palgrave Macmillan; 2008.
- Sargent DJ, Taylor JM. Current issues in oncology drug development, with a focus on Phase II trials. J Biopharm Stat 2009;19:556-62. http://dx.doi.org/10.1080/10543400902802474.
- Korn EL, Liu PY, Lee SJ, Chapman JA, Niedzwiecki D, Suman VJ, et al. Meta-analysis of Phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future Phase II trials. J Clin Oncol 2008;26:527-34. http://dx.doi.org/10.1200/JCO.2007.12.7837.
- Philip PA, Chansky K, LeBlanc M, Rubinstein L, Seymour L, Ivy SP, et al. Historical controls for metastatic pancreatic cancer: benchmarks for planning and analyzing single-arm Phase II trials. Clin Cancer Res 2014;20:4176-85. http://dx.doi.org/10.1158/1078-0432.CCR-13-2024.
- Tang H, Foster NR, Grothey A, Ansell SM, Sargent DJ. Excessive false-positive errors in single-arm Phase II trails: a simulation-based analysis. J Clin Oncol 2009;25.
- Pond GR, Abbasi S. Quantitative evaluation of single-arm versus randomized Phase II cancer clinical trials. Clin Trials 2011;8:260-9. http://dx.doi.org/10.1177/1740774511401764.
- Sambucini V. Comparison of single-arm vs. randomized Phase II clinical trials: a Bayesian approach. J Biopharm Stat 2015;25:474-89. http://dx.doi.org/10.1080/10543406.2014.920856.
- Monzon JG, Hay AE, McDonald GT, Pater JL, Meyer RM, Chen E, et al. Correlation of single arm versus randomised Phase 2 oncology trial characteristics with Phase 3 outcome. Eur J Cancer 2015;51:2501-7. http://dx.doi.org/10.1016/j.ejca.2015.08.004.
- NICE . Hepatitis C: Guidance and Advice List n.d. www.nice.org.uk/guidance/published?type=ta&title=hepatitis%20C (accessed 17 January 2017).
- Unverzagt S, Prondzinsky R, Peinemann F. Single-center trials tend to provide larger treatment effects than multicenter trials: a systematic review. J Clin Epidemiol 2013;66:1271-80. http://dx.doi.org/10.1016/j.jclinepi.2013.05.016.
- Bellomo R, Warrillow SJ, Reade MC. Why we should be wary of single-center trials. Crit Care Med 2009;37:3114-19. http://dx.doi.org/10.1097/CCM.0b013e3181bc7bd5.
- Sinha IP, Sinha SK. Single-center trials in neonatology: issues to consider. Semin Fetal Neonatal Med 2015;20:384-8. http://dx.doi.org/10.1016/j.siny.2015.08.003.
- EUnetHTA . Endpoints Used in Relative Effectiveness Assessment of Pharmaceuticals: Surrogate Endpoints 2013. https://eunethta.fedimbo.belgium.be/sites/5026.fedimbo.belgium.be/files/Surrogate%20Endpoints.pdf (accessed 21 November 2016).
- Elston J, Taylor RS. Use of surrogate outcomes in cost-effectiveness models: a review of United Kingdom health technology assessment reports. Int J Technol Assess Health Care 2009;25:6-13. http://dx.doi.org/10.1017/S0266462309090023.
- Taylor RS, Elston J. The use of surrogate outcomes in model-based cost-effectiveness analyses: a survey of UK Health Technology Assessment reports. Health Technol Assess 2009;13. http://dx.doi.org/10.3310/hta13080.
- Wilson MK, Karakasis K, Oza AM. Outcomes and endpoints in trials of cancer treatment: the past, present, and future. Lancet Oncol 2015;16:e32-42. http://dx.doi.org/10.1016/S1470-2045(14)70375-4.
- Aziz A, Kempkensteffen C, May M, Lebentrau S, Burger M, Chun FK, et al. Prognostic, predictive and potential surrogate markers in castration-resistant prostate cancer. Expert Rev Anticancer Ther 2015;15:649-66. http://dx.doi.org/10.1586/14737140.2015.1038247.
- Temple R, Nimmo WS, Tucker GT. Clinical Measurement in Drug Evaluation. New York, NY: John Wiley; 1995.
- Ciani O, Taylor RS. Surrogate, friend or foe? The need for case studies of the use of surrogate outcomes in cost-effectiveness analyses. Health Econ 2013;22:251-2. http://dx.doi.org/10.1002/hec.2826.
- Davis S, Tappenden P, Cantrell A. A Review of Studies Examining the Relationship between Progression-Free Survival and Overall Survival in Advanced or Metastatic Cancer. Sheffield: Decision Support Unit, School of Health and Related Research, University of Sheffield; 2012.
- Gøtzsche PC, Liberati A, Torri V, Rossetti L. Beware of surrogate outcome measures. Int J Technol Assess Health Care 1996;12:238-46. https://doi.org/10.1017/S0266462300009594.
- Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx 2004;1:189-95. https://doi.org/10.1602/neurorx.1.2.189.
- Lerche la Cour J, Brok J, Gøtzsche PC. Inconsistent reporting of surrogate outcomes in randomised clinical trials: cohort study. BMJ 2010;341. http://dx.doi.org/10.1136/bmj.c3653.
- Ciani O, Buyse M, Garside R, Pavey T, Stein K, Sterne JA, et al. Comparison of treatment effect sizes associated with surrogate and final patient relevant outcomes in randomised controlled trials: meta-epidemiological study. BMJ 2013;346. http://dx.doi.org/10.1136/bmj.f457.
- Buyse M, Molenberghs G. Criteria for the validation of surrogate endpoints in randomized experiments. Biometrics 1998;54:1014-29. https://doi.org/10.2307/2533853.
- Bucher HC, Guyatt GH, Cook DJ, Holbrook A, McAlister FA. Users’ guides to the medical literature: XIX. Applying clinical trial results. A. How to use an article measuring the effect of an intervention on surrogate end points. Evidence-Based Medicine Working Group. JAMA 1999;282:771-8. https://doi.org/10.1001/jama.282.8.771.
- Schievink B, Lambers Heerspink H, Leufkens H, De Zeeuw D, Hoekman J. The use of surrogate endpoints in regulating medicines for cardio-renal disease: opinions of stakeholders. PLOS ONE 2014;9. http://dx.doi.org/10.1371/journal.pone.0108722.
- Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled?. Ann Intern Med 1996;125:605-13. https://doi.org/10.7326/0003-4819-125-7-199610010-00011.
- Lassere MN, Johnson KR, Boers M, Tugwell P, Brooks P, Simon L, et al. Definitions and validation criteria for biomarkers and surrogate endpoints: development and testing of a quantitative hierarchical levels of evidence schema. J Rheumatol 2007;34:607-15.
- Berger VW. Does the Prentice criterion validate surrogate endpoints?. Stat Med 2004;23:1571-8. https://doi.org/10.1002/sim.1780.
- Baker SG, Kramer BS. Surrogate endpoint analysis: an exercise in extrapolation. J Natl Cancer Inst 2013;105:316-20. http://dx.doi.org/10.1093/jnci/djs527.
- Prasad V, Kim C, Burotto M, Vandross A. The strength of association between surrogate end points and survival in oncology: a systematic review of trial-level meta-analyses. JAMA Intern Med 2015;175:1389-98. http://dx.doi.org/10.1001/jamainternmed.2015.2829.
- Ciani O, Davis S, Tappenden P, Garside R, Stein K, Cantrell A, et al. Validation of surrogate endpoints in advanced solid tumors: systematic review of statistical methods, results, and implications for policy makers. Int J Technol Assess Health Care 2014;30:312-24. http://dx.doi.org/10.1017/S0266462314000300.
- Ciani O, Buyse M, Garside R, Peters J, Saad ED, Stein K, et al. Meta-analyses of randomized controlled trials show suboptimal validity of surrogate outcomes for overall survival in advanced colorectal cancer. J Clin Epidemiol 2015;68:833-42. http://dx.doi.org/10.1016/j.jclinepi.2015.02.016.
- Buckley SA, Appelbaum FR, Walter RB. Prognostic and therapeutic implications of minimal residual disease at the time of transplantation in acute leukemia. Bone Marrow Transplant 2013;48:630-41. http://dx.doi.org/10.1038/bmt.2012.139.
- Elorza I, Palacio C, Dapena JL, Gallur L, Sánchez de Toledo J, Díaz de Heredia C. Relationship between minimal residual disease measured by multiparametric flow cytometry prior to allogeneic hematopoietic stem cell transplantation and outcome in children with acute lymphoblastic leukemia. Haematologica 2010;95:936-41. http://dx.doi.org/10.3324/haematol.2009.010843.
- Leung W, Pui CH, Coustan-Smith E, Yang J, Pei D, Gan K, et al. Detectable minimal residual disease before hematopoietic cell transplantation is prognostic but does not preclude cure for children with very-high-risk leukemia. Blood 2012;120:468-72. http://dx.doi.org/10.1182/blood-2012-02-409813.
- Borowitz MJ, Devidas M, Hunger SP, Bowman WP, Carroll AJ, Carroll WL, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood 2008;111:5477-85. https://doi.org/10.1182/blood-2008-01-132837.
- Eckert C, Hagedorn N, Sramkova L, Mann G, Panzer-Grümayer R, Peters C, et al. Monitoring minimal residual disease in children with high-risk relapses of acute lymphoblastic leukemia: prognostic relevance of early and late assessment. Leukemia 2015;29:1648-55. http://dx.doi.org/10.1038/leu.2015.59.
- Minimal Residual Disease (MRD) as a Surrogate Endpoint in Acute Lymphoblastic Leukemia (ALL) Workshop. FDA Briefing Document. Silver Spring, MD: FDA; 2012.
- Parker C, Waters R, Leighton C, Hancock J, Sutton R, Moorman AV, et al. Effect of mitoxantrone on outcome of children with first relapse of acute lymphoblastic leukaemia (ALL R3): an open-label randomised trial. Lancet 2010;376:2009-17. http://dx.doi.org/10.1016/S0140-6736(10)62002-8.
- Bujkiewicz S, Thompson JR, Sutton AJ, Cooper NJ, Harrison MJ, Symmons DP, et al. Use of Bayesian multivariate meta-analysis to estimate the HAQ for mapping onto the EQ-5D questionnaire in rheumatoid arthritis. Value Health 2014;17:109-15. http://dx.doi.org/10.1016/j.jval.2013.11.005.
- Sleijfer S, Wagner AJ. The challenge of choosing appropriate end points in single-arm Phase II studies of rare diseases. J Clin Oncol 2012;30:896-8. http://dx.doi.org/10.1200/JCO.2011.40.6942.
- Knee Cartilage Defects – Autologous Chondrocyte Implantation [ID686]. London: NICE; TBC; n.d.
- Autologous Cartilage Transplantation for Full Thickness Cartilage Defects in Knee Joints. London: NICE; 2000.
- The Use of Autologous Chondrocyte Implantation for the Treatment of Cartilage Defects in the Knee Joints. London: NICE; 2005.
- Sipuleucel-T for Treating Asymptomatic or Minimally Symptomatic Metastatic Hormone-Relapsed Prostate Cancer. London: NICE; 2015.
- Guide to the Methods of Technology Appraisal. London: NICE; 2013.
- Guide to the Methods of Technology Appraisal. London: NICE; 2004.
- Mooney GH. Economics, Medicine and Health Care. Harlow: Pearson Education; 2003.
- Sculpher M, Drummond M, Buxton M. The iterative use of economic evaluation as part of the process of health technology assessment. J Health Serv Res Policy 1997;2:26-30.
- Fenwick E, Claxton K, Sculpher M, Briggs A. Improving the Efficiency and Relevance of Health Technology Assessment: the Role of Iterative Decision Analytic Modelling. York: University of York, Centre for Health Economics; 2000.
- Towse A. Regenerative Medicine: A European HTA Perspective n.d.
- Claxton K, Palmer S, Longworth L, Bojke L, Griffin S, McKenna C, et al. Informing a decision framework for when NICE should recommend the use of health technologies only in the context of an appropriately designed programme of evidence development. Health Technol Assess 2012;16. http://dx.doi.org/10.3310/hta16460.
- Faulkner E. What Value Do We Place on a Cure? Value Demonstration Challenges Associated With Innovator and Regenerative Therapies in the EU, North America and Asia n.d.
- Husereau D. What Value Do We Place on Cure? n.d.
- Husereau D. How do we value a cure?. Expert Rev Pharmacoecon Outcomes Res 2015;15:551-5. http://dx.doi.org/10.1586/14737167.2015.1039519.
- Sofosbuvir for Treating Chronic Hepatitis C. London: NICE; 2015.
- McKenna C, Claxton K, Palmer S, Sculpher M, Epstein D, Iglesias C, et al. WP5: uncertainty and value of information analysis for medical devices. Deliverable 5.1: evaluation of uncertainty and value of further research for devices – key methodological issues. Milan: MedtecHTA, Bocconi University; 2014.
- Walker S, Sculpher M, Claxton K, Palmer S. Coverage with evidence development, only in research, risk sharing, or patient access scheme? A framework for coverage decisions. Value Health 2012;15:570-9. http://dx.doi.org/10.1016/j.jval.2011.12.013.
- Grimm S, Strong M, Brennan A, Wailoo A. Framework for Analysing Risk in Health Technology Assessments and Its Application to Managed Entry Agreements. Report by the Decision Support Unit 2016. www.nicedsu.org.uk/DSU%20Managed%20Access%20report%20FINAL.pdf (accessed 17 January 2017).
- Gottlieb S, Carino T. Establishing new payment provisions for the high cost of curing disease. AEI Research 2014. www.aei.org/wp-content/uploads/2014/07/-establishing-new-payment-provisions-for-the-high-cost-of-curing-disease_154058134931.pdf (accessed 17 November 2016).
- Edlin R, Hall P, Wallner K, McCabe C. Sharing risk between payer and provider by leasing health technologies: an affordable and effective reimbursement strategy for innovative technologies?. Value Health 2014;17:438-44. http://dx.doi.org/10.1016/j.jval.2014.01.010.
- Health and Social Care Act 2012. London: The Stationery Office; 2012.
- Westra TA, Parouty M, Brouwer WB, Beutels PH, Rogoza RM, Rozenbaum MH, et al. On discounting of health gains from human papillomavirus vaccination: effects of different approaches. Value Health 2012;15:562-7. http://dx.doi.org/10.1016/j.jval.2012.01.005.
- Jit M, Mibei W. Discounting in the evaluation of the cost-effectiveness of a vaccination programme: a critical review. Vaccine 2015;33:3788-94. http://dx.doi.org/10.1016/j.vaccine.2015.06.084.
- Canadian Agency for Drugs and Technologies in Health . Guidelines for the Economic Evaluation of Health Technologies: Canada 2006. www.cadth.ca/sites/default/files/pdf/186_EconomicGuidelines_e.pdf (accessed 17 January 2017).
- Australian Government, Department of Health . Guidelines for Preparing a Submission to the Pharmaceutical Benefits Advisory Committee 2016. https://pbac.pbs.gov.au/content/information/files/pbac-guidelines-version-5.pdf (accessed 17 January 2017).
- Guidance to Manufacturers for Completion of New Product Assessment Form (NPAF). Glasgow: SMC; 2013.
- Mortimer D. Modelling downstream effects in the presence of technological change. Pharmacoeconomics 2008;26:991-1003. http://dx.doi.org/10.2165/0019053-200826120-00003.
- Levine BL. Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther 2015;22:79-84. http://dx.doi.org/10.1038/cgt.2015.5.
- Catapult Cell Therapy . Genetically Modified T Cell Therapies for Cancer – Basic Facts n.d. https://ct.catapult.org.uk/sites/default/files/publication/T-Cell-Therapies-for-Cancer-Basic-Facts-14-Feb-2014-9.pdf (accessed 17 November 2015).
- Hedden J. CAR-T Cell Therapy CTL019 Gets FDA Breakthrough Therapy Status 2014. www.datamonitorhealthcare.com/car-t-cell-therapy-ctl019-gets-fda-breakthrough-therapy-status/ (accessed 17 November 2016).
- Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Blood 2015;25:4017-23. https://doi.org/10.1182/blood-2014-12-580068.
- Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 2014;123:2625-35. http://dx.doi.org/10.1182/blood-2013-11-492231.
- American Cancer Society . How Is Childhood Leukemia Classified? 2016. www.cancer.org/cancer/leukemiainchildren/detailedguide/childhood-leukemia-how-classified (accessed 7 September 2015).
- Cancer Research UK . Acute Lymphoblastic Leukaemia (ALL) Statistics 2014. www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/leukaemia-all (accessed 7 September 2015).
- Ko RH, Ji L, Barnette P, Bostrom B, Hutchinson R, Raetz E, et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: a Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol 2010;28:648-54. http://dx.doi.org/10.1200/JCO.2009.22.2950.
- Bassan R, Spinelli O. Minimal residual disease monitoring in adult ALL to determine therapy. Curr Hematol Malig Rep 2015;10:86-95. http://dx.doi.org/10.1007/s11899-015-0252-7.
- Seiter K, Besa EC. Acute Lymphoblastic Leukemia (ALL) Guidelines 2014. http://emedicine.medscape.com/article/2249076-overview (accessed 9 December 2016).
- Kebriaei P, Poon ML. Future of therapy in acute lymphoblastic leukemia (ALL) – potential role of immune-based therapies. Curr Hematol Malig Rep 2015;10:76-85. http://dx.doi.org/10.1007/s11899-015-0251-8.
- Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 2007;109:944-50. https://doi.org/10.1182/blood-2006-05-018192.
- Fuster JL. Current approach to relapsed acute lymphoblastic leukemia in children. World J Hematol 2014;3:49-70. https://doi.org/10.5315/wjh.v3.i3.49.
- Saarinen-Pihkala UM, Heilmann C, Winiarski J, Glomstein A, Abrahamsson J, Arvidson J, et al. Pathways through relapses and deaths of children with acute lymphoblastic leukemia: role of allogeneic stem-cell transplantation in Nordic data. J Clin Oncol 2006;24:5750-62. https://doi.org/10.1200/JCO.2006.07.1225.
- Reismuller B, Peters C, Dworzak MN, Potschger U, Urban C, Meister B, et al. Outcome of children and adolescents with a second or third relapse of acute lymphoblastic leukemia (ALL): a population-based analysis of the Austrian ALL-BFM (Berlin-Frankfurt-Munster) study group. J Pediatr Hematol Oncol 2013;35:e200-4. https://doi.org/10.1097/MPH.0b013e318290c3d6.
- von Stackelberg A, Völzke E, Kühl JS, Seeger K, Schrauder A, Escherich G, et al. Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM study group. Eur J Cancer 2011;47:90-7. http://dx.doi.org/10.1016/j.ejca.2010.09.020.
- European Medicines Agency (EMA) . Scientific Discussion: Evoltra, INN-Clofarabine 2013. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000613/WC500031194.pdf (accessed 6 February 2017).
- Socié G, Stone JV, Wingard JR, Weisdorf D, Henslee-Downey PJ, Bredeson C, et al. Long-term survival and late deaths after allogeneic bone marrow transplantation. Late Effects Working Committee of the International Bone Marrow Transplant Registry. N Engl J Med 1999;341:14-21. https://doi.org/10.1056/NEJM199907013410103.
- Bar M, Wood BL, Radich JP, Doney KC, Woolfrey AE, Delaney C, et al. Impact of minimal residual disease, detected by flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute lymphoblastic leukemia. Leuk Res Treatment 2014;2014. http://dx.doi.org/10.1155/2014/421723.
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a Phase 1 dose-escalation trial. Lancet 2015;385:517-28. https://doi.org/10.1016/S0140-6736(14)61403-3.
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507-17. http://dx.doi.org/10.1056/NEJMoa1407222.
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014;6. https://doi.org/10.1126/scitranslmed.3008226.
- Grupp SA, Maude SL, Shaw P, Aplenc R, Barrett DM, Callahan C, et al. 380 T Cells Engineered With a Chimeric Antigen Receptor (CAR) Targeting CD19 (CTL019) Have Long Term Persistence and Induce Durable Remissions in Children With Relapsed, Refractory ALL n.d.
- Park JH, Riviere I, Wang X, Bernal Y, Purdon T, Halton E, et al. Efficacy and Safety of CD19-Targeted 19-28z CAR Modified T Cells in Adult Patients With Relapsed or Refractory B-ALL n.d.
- Grupp SA. CAR Targeting CTL019 in Children With Relapsed, Refractory ALL 2014. www.youtube.com/watch?v=F5XAhsir4kY (accessed 9 December 2016).
- European Medicines Agency . Evoltra (Clofarabine): EMEA H C 000613 Article 46–035 2014. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/000613/WC500164760.pdf (accessed 17 January 2016).
- All Wales Medicines Strategy Group . Final Appraisal Report: Clofarabine (Evoltra®) 2007. www.awmsg.org/awmsgonline/app/appraisalinfo/92 (accessed 9 December 2016).
- Blinatumomab (AMG 103). Background Information for the Pediatric Subcommittee of the Oncologic Drugs Advisory Committee Meeting, 04 December 2012. Thousand Oaks, CA: Amgen Inc.; 2012.
- Marqibo® (Vincristine Sulfate Liposomes Injection) for the Treatment of Advanced Relapsed andor Refractory Philadelphia Chromosome Negative (Ph-) Adult Acute Lymphoblastic Leukemia. Silver Spring, MD: FDA; 2012.
- ClinicalTrials.gov . Historical Data Analysis of Hematological Remission and Survival in Adults With R R Acute Lymphoblastic Leukemia n.d. https://clinicaltrials.gov/ct2/show/NCT02003612 (accessed 17 November 2016).
- O’Brien S, Thomas D, Ravandi F, Faderl S, Cortes J, Borthakur G, et al. Outcome of adults with acute lymphocytic leukemia after second salvage therapy. Cancer 2008;113:3186-91. http://dx.doi.org/10.1002/cncr.23919.
- Goekbuget N, Topp MS, Zugmaier G, Viardot A, Stelljes M, Neumann S, et al. Anti-CD19 BiTE blinatumomab induces high complete remission rate in adult patients with relapsed B-precursor ALL: updated results of an ongoing Phase II trial. Ann Oncol 2012;23:i20-1.
- Kantarjian HM, Thomas D, Ravandi F, Faderl S, Jabbour E, Garcia-Manero G, et al. Defining the course and prognosis of adults with acute lymphocytic leukemia in first salvage after induction failure or short first remission duration. Cancer 2010;116:5568-74. http://dx.doi.org/10.1002/cncr.25354.
- Jeha S, Gaynon PS, Razzouk BI, Franklin J, Kadota R, Shen V, et al. Phase II study of clofarabine in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. J Clin Oncol 2006;24:1917-23. https://doi.org/10.1200/JCO.2005.03.8554.
- Davila ML, Bouhassira DC, Park JH, Curran KJ, Smith EL, Pegram HJ, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int J Hematol 2014;99:361-71. http://dx.doi.org/10.1007/s12185-013-1479-5.
- ClinicalTrials.gov . Ph 3 Trial of Blinatumomab Vs Investigator’s Choice of Chemotherapy in Patients With Relapsed or Refractory ALL 2016. https://clinicaltrials.gov/ct2/show/NCT02013167?term=blincyto&type=Intr&phase=2&rank=1 (accessed 17 January 2017).
- R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2013.
- Costa V, McGregor M, Laneuville P, Brophy JM. The cost-effectiveness of stem cell transplantations from unrelated donors in adult patients with acute leukemia. Value Health 2007;10:247-55. https://doi.org/10.1111/j.1524-4733.2007.00180.x.
- Lis J, Kawalec P, Glasek M. Economic evaluation of acute lymphoblastic leukaemia treatment with clofarabine (Evoltra®) combined with chemotherapy for children and adolescents in Poland. J Health Policy Outcomes Res 2012;2:20-33.
- Leukaemia (Acute Lymphoblastic) – Dasatinib (ID386). London: NICE; 2008.
- All Wales Medicines Strategy Group . Final Appraisal Report: Nelarabine (Atriance®) 2009. www.awmsg.org/awmsgonline/app/appraisalinfo/216 (accessed 9 December 2016).
- All Wales Medicines Strategy Group (AWMSG) . Final Appraisal Report: Dasatinib (Sprycel®). Advice no.: 0407 – December 2007 2007.
- All Wales Medicines Strategy Group (AWMSG) . Final Appraisal Report: Mercaptopurine (Xaluprine®). Advice no.: 2412 – September 2012 2012.
- National Life Tables, England & Wales, 1980–82 to 2011–13. Newport: ONS; 2014.
- Campana D, Leung W. Clinical significance of minimal residual disease in patients with acute leukaemia undergoing haematopoietic stem cell transplantation. Br J Haematol 2013;162:147-61. http://dx.doi.org/10.1111/bjh.12358.
- Mody R, Li S, Dover DC, Sallan S, Leisenring W, Oeffinger KC, et al. Twenty-five-year follow-up among survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Blood 2008;111:5515-23. http://dx.doi.org/10.1182/blood-2007-10-117150.
- MacArthur AC, Spinelli JJ, Rogers PC, Goddard KJ, Abanto ZU, McBride ML. Mortality among 5-year survivors of cancer diagnosed during childhood or adolescence in British Columbia, Canada. Pediatr Blood Cancer 2007;48:460-7. https://doi.org/10.1002/pbc.20922.
- Royston P, Parmar MK. Flexible parametric proportional-hazards and proportional-odds models for censored survival data, with application to prognostic modelling and estimation of treatment effects. Stat Med 2002;21:2175-97. https://doi.org/10.1002/sim.1203.
- Jackson CH, Thompson SG, Sharples LD. Accounting for uncertainty in health economic decision models by using model averaging. J R Stat Soc Ser A Stat Soc 2009;172:383-404. https://doi.org/10.1111/j.1467-985X.2008.00573.x.
- Reaves A. Can CAR-T Therapy Live up to the Biotech IPO Hype? 2015. http://news.investors.com/technology/012615-736198-cancer-car-t-therapy-drives-hot-biotech-ipos.htm (accessed 17 November 2016).
- Appraising Life-Extending, End of Life Treatments. London: NICE; 2009.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; n.d.
- FLAG-IDA Dosing Guide. Guildford: Royal Surrey County Hospital NHS Foundation Trust; 2015.
- Tavil B, Aytac S, Balci YI, Unal S, Kuskonmaz B, Yetgin S, et al. Fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin (FLAG-IDA) for the treatment of children with poor-prognosis acute leukemia: the Hacettepe experience. Pediatr Hematol Oncol 2010;27:517-28. http://dx.doi.org/10.3109/08880018.2010.493578.
- NHS Reference Costs 2013 to 2014. London: Department of Health; 2015.
- UK Stem Cell Strategy Oversight Committee . Unrelated Donor Stem Cell Transplantation in the UK: Effective Affordable Sustainable 2014. www.nhsbt.nhs.uk/download/unrelated_donor_stem_cell_transplantation_in_the_uk.pdf (accessed 17 November 2016).
- Good Medical Practice. London: GMC; 2013.
- Adult Blood and Bone Marrow Transplant Services: Costing Model Guidelines. London: London Specialised Commissioning Group; 2009.
- van Agthoven M, Groot MT, Verdonck LF, Lowenberg B, Schattenberg AV, Oudshoorn M, et al. Cost analysis of HLA-identical sibling and voluntary unrelated allogeneic bone marrow and peripheral blood stem cell transplantation in adults with acute myelocytic leukaemia or acute lymphoblastic leukaemia. Bone Marrow Transplant 2002;30:243-51. https://doi.org/10.1038/sj.bmt.1703641.
- Kelly MJ, Pauker SG, Parsons SK. Using nonrandomized studies to inform complex clinical decisions: the thorny issue of cranial radiation therapy for T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer 2015;62:790-7. http://dx.doi.org/10.1002/pbc.25451.
- van Litsenburg RR, Kunst A, Huisman J, Ket JC, Kaspers GJ, Gemke RJ. Health status utilities in pediatrics: a systematic review of acute lymphoblastic leukemia. Med Decis Making 2014;34:21-32. http://dx.doi.org/10.1177/0272989X13497263.
- Sung L, Buckstein R, Doyle JJ, Crump M, Detsky AS. Treatment options for patients with acute myeloid leukemia with a matched sibling donor: a decision analysis. Cancer 2003;97:592-600. https://doi.org/10.1002/cncr.11098.
- NICE DSU Technical Support Document 14: Survival Analysis For Economic Evaluations Alongside Clinical Trials – Extrapolation With Patient-Level Data. Sheffield: School of Health and Related Research, University of Sheffield; 2011.
- Lambert PC, Thompson JR, Weston CL, Dickman PW. Estimating and modeling the cure fraction in population-based cancer survival analysis. Biostatistics 2007;8:576-94. https://doi.org/10.1093/biostatistics/kxl030.
- Bujkiewicz S, Jones HE, Lai MC, Cooper NJ, Hawkins N, Squires H, et al. Development of a transparent interactive decision interrogator to facilitate the decision-making process in health care. Value Health 2011;14:768-76. http://dx.doi.org/10.1016/j.jval.2010.12.002.
- The Green Book: Appraisal and Evaluation in Central Government. London: The Stationery Office; 2011.
- NICE Decision Support Unit . Framework for Analysing Risk in Health Technology Assessments and Its Application to Managed Entry Agreements 2016.
- Mifamurtide for the Treatment of Osteosarcoma. London: NICE; 2011.
- Gravante G, Di Fede MC, Araco A, Grimaldi M, De Angelis B, Arpino A, et al. A randomized trial comparing ReCell system of epidermal cells delivery versus classic skin grafts for the treatment of deep partial thickness burns. Burns 2007;33:966-72. https://doi.org/10.1016/j.burns.2007.04.011.
- Park JH, Heggie KM, Edgar DW, Bulsara MK, Wood FM. Does the type of skin replacement surgery influence the rate of infection in acute burn injured patients?. Burns 2013;39:1386-90. https://doi.org/10.1016/j.burns.2013.03.015.
- Buyse M, Burzykowski T, Carroll K, Michiels S, Sargent DJ, Miller LL, et al. Progression-free survival is a surrogate for survival in advanced colorectal cancer. J Clin Oncol 2007;25:5218-24.
- Hawkins N, Richardson G, Sutton AJ, Cooper NJ, Griffiths C, Rogers A, et al. Surrogates, meta-analysis and cost-effectiveness modelling: a combined analytic approach. Health Econ 2012;21:742-56. http://dx.doi.org/10.1002/hec.1741.
- Oriana C, Martin H, Toby P, Chris C, Ruth G, Claudius R, et al. Complete cytogenetic response and major molecular response as surrogate outcomes for overall survival in first-line treatment of chronic myelogenous leukemia: a case study for technology appraisal on the basis of surrogate outcomes evidence. Value Health 2013;16:1081-90. http://dx.doi.org/10.1016/j.jval.2013.07.004.
- De Gruttola V, Fleming T, Lin DY, Coombs R. Perspective: validating surrogate markers – are we being naive?. J Infect Dis 1997;175:237-46. https://doi.org/10.1093/infdis/175.2.237.
- Ellenberg S, Hamilton JM. Surrogate endpoints in clinical trials: cancer. Stat Med 1989;8:405-13. https://doi.org/10.1002/sim.4780080404.
- Fleming TR, Prentice RL, Pepe MS, Glidden D. Surrogate and auxiliary endpoints in clinical trials, with potential applications in cancer and AIDS research. Stat Med 1994;13:955-68. https://doi.org/10.1002/sim.4780130906.
- Freedman LS, Graubard BI, Schatzkin A. Statistical validation of intermediate endpoints for chronic diseases. Stat Med 1992;11:167-78. https://doi.org/10.1002/sim.4780110204.
- Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med 1989;8:431-40. https://doi.org/10.1002/sim.4780080407.
- Herson J. The use of surrogate endpoints in clinical trials (an introduction to a series of four papers). Stat Med 1989;8:403-4. https://doi.org/10.1002/sim.4780080403.
- Holloway RG, Dick AW. Clinical trial end points: on the road to nowhere?. Neurology 2002;58:679-86. https://doi.org/10.1212/WNL.58.5.679.
- Zee J, Xie SX. Alzheimer’s Disease Neuroimaging Initiative . Assessing treatment effects with surrogate survival outcomes using an internal validation subsample. Clin Trials 2015;12:333-41. http://dx.doi.org/10.1177/1740774515583488.
- US Food and Drug Administration . Drug Approval Package: Clolar (Clofarabine) n.d. www.accessdata.fda.gov/drugsatfda_docs/nda/2004/21-673_Clolar.cfm (accessed 18 January 2017).
- Center for Drug Evaluation and Research . Secondary Medical Review: Blinatumomab Blincyto 2014. www.accessdata.fda.gov/drugsatfda_docs/nda/2014/125557Orig1s000MedR.pdf (accessed 6 February 2017).
- Nguyen K, Devidas M, Cheng SC, La M, Raetz EA, Carroll WL, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia 2008;22:2142-50. https://doi.org/10.1038/leu.2008.251.
- Tavernier E, Boiron JM, Huguet F, Bradstock K, Vey N, Kovacsovics T, et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia 2007;21:1907-14. https://doi.org/10.1038/sj.leu.2404824.
- Oriol A, Vives S, Hernández-Rivas JM, Tormo M, Heras I, Rivas C, et al. Outcome after relapse of acute lymphoblastic leukemia in adult patients included in four consecutive risk-adapted trials by the PETHEMA study group. Haematologica 2010;95:589-96. https://doi.org/10.3324/haematol.2009.014274.
- Schrappe M, Hunger SP, Pui CH, Saha V, Gaynon PS, Baruchel A, et al. Outcomes after induction failure in childhood acute lymphoblastic leukemia. N Engl J Med 2012;366:1371-81. https://doi.org/10.1056/NEJMoa1110169.
- Thomas DA, Kantarjian H, Smith TL, Koller C, Cortes J, O’Brien S, et al. Primary refractory and relapsed adult acute lymphoblastic leukemia: characteristics, treatment results, and prognosis with salvage therapy. Cancer 1999;86:1216-30. https://doi.org/10.1002/(SICI)1097-0142(19991001)86:7<1216::AID-CNCR17>3.0.CO;2-O.
- Office for National Statistics . Cancer Registration Statistics, England, 2013 2015. www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/cancerregistrationstatisticsengland/2015-07-10 (accessed 9 December 2016).
- American Cancer Society . What Are the Key Statistics about Acute Lymphocytic Leukemia? 2015. www.cancer.org/cancer/leukemia-acutelymphocyticallinadults/detailedguide/leukemia-acute-lymphocytic-key-statistics (accessed 21 September 2015).
Appendix 1 Regenerative medicines licensed by the European Medicines Agency
| Glybera (alipogene tiparvovec): EMA assessment (CAT and CHMP) 201218 | |||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EMA marketing authorisation under exceptional circumstances | |||||||||||||||||||||||||||||||||||||||||||
| Nature of the disease | |||||||||||||||||||||||||||||||||||||||||||
| Indication | The indication initially applied for was (p. 11):18Glybera is indicated for the long term correction of lipoprotein lipase deficiency, to control or abolish symptoms and prevent complications in adult patients clinically diagnosed with lipoprotein lipase deficiency (LPLD)The indication for which a licence was granted is more restricted (p. 98):18Glybera is indicated for adult patients diagnosed with familial lipoprotein lipase deficiency (LPLD) and suffering from at least one pancreatitis episode despite dietary fat restriction. The diagnosis of LPLD has to be confirmed by genetic testing. The indication is restricted to patients with detectable levels of LPL protein | ||||||||||||||||||||||||||||||||||||||||||
| Orphan status? | Yes | ||||||||||||||||||||||||||||||||||||||||||
| Is this a rare condition? | The calculated prevalence of this condition was reported to be 0.02 per 10,000 | ||||||||||||||||||||||||||||||||||||||||||
| What is the natural history of the disease without this treatment/with current treatment? | LPLD is a rare autosomal recessive inherited condition caused by homozygosity or compound heterozygosity for mutations in the LPL gene. The condition may become evident only after several episodes of pancreatitis in adolescence or adulthood. Laboratory investigation reveals genuine lactescent plasma (lipaemia) because of the increased chylomicron concentrations. The symptom severity is proportional to the degree of chylomicronaemia and the most severe complication associated with LPLD is pancreatitis. Pancreatitis in a LPLD subject may lead to admission to an intensive care unit. In severe cases, patients may eventually develop chronic pancreatitis, ultimately resulting in endocrine and exocrine pancreatic insufficiency. Treatment of LPLD patients currently consists of severe reductions in dietary fat to < 20% of caloric intake. Compliance with this dietary regimen is very difficult and, even with good compliance, the diet is often ineffective at reducing chylomicronaemia and triglyceride levels. Currently, no triglyceride-lowering drug is available. Enzyme replacement therapy is not expected to be effective because of the short intravascular half-life of the LPL protein | ||||||||||||||||||||||||||||||||||||||||||
| Nature of the medicine | |||||||||||||||||||||||||||||||||||||||||||
| How does it work? | Glybera is a replication-deficient adeno-associated viral vector designed to deliver and express the human LPL gene variant LPLS447X. Transduction of part of the skeletal muscle mass is expected to restore a level of LPL activity that is sufficient to hydrolyse the triglyceride-rich lipoproteins and influence lipid homoeostasis and thus lead to clinical improvement or stabilisation | ||||||||||||||||||||||||||||||||||||||||||
| Is it claiming to meet an otherwise unmet need? | Yes, the therapeutic aim of Glybera was to control symptoms of LPLD and prevent complications in adult patients clinically diagnosed with LPLD | ||||||||||||||||||||||||||||||||||||||||||
| How is it given? | A sterile solution for injection presented as single-use vials. Each vial contains 3 × 1012 genomic copies (gcs) of alipogene tiparvovec (AAV1-LPLS447X) in 1 ml of a phosphate-based formulation buffer containing 5% sucrose. Glybera is to be administered once at multiple sites intramuscularly at a dose of 1 × 1012 gcs/kg of body weight. Note that Glybera is intended as a single procedure but with multiple injections (up to 60 injection sites) administered under regional or spinal anaesthesia. All 27 patients reported adverse events related to the injection procedure | ||||||||||||||||||||||||||||||||||||||||||
| Are there any comparator treatments? | Reducing chylomicronaemia and triglyceride levels by reducing dietary fat to < 20% of caloric intake | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of the intervention evolving over time? | The applicant uses two different company codes to differentiate between the current production system (AMT-011) and the previous production system (AMT-010). There were changes during the development phases but the CHMP felt that issues relating to these had been resolved and ‘consistency of product quality throughout development has been shown’ (p. 14)18 | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of persistence of the treatment within the patient? | Negative effects of persistence – it is considered that, although recombinant adeno-associated virus has a potential integration risk, the risk of a consequent cancer is minimal. In the context of treating patients with this disease, these data suggest an acceptable safety profile.18 Overall, the CAT and CHMP agreed that the data do not substantiate a concern for tumourigenicity | ||||||||||||||||||||||||||||||||||||||||||
| Positive effects of persistence – the post-treatment observation period was insufficient for conclusions to be made about a rate of change of pancreatitis events long term. The totality of evidence derived from all studies combined suggested that AMT-011 may temporarily reduce mean fasting triglyceride levels but the proposed single treatment was insufficient to provide a durable and measurable effect | |||||||||||||||||||||||||||||||||||||||||||
| Trial design | |||||||||||||||||||||||||||||||||||||||||||
| Trial description | Study numberDose (gc/kg)Number of patientsDuration of monitoringDuration of follow-upStatusPREPARATION-01None1813–78 weeks–CompletedCT-AMT-010–011 × 1011412 weeks5 yearsActive phase completed, follow-up ongoing3 × 10114PREPARATION-02None222–83 weeks–CompletedCT-AMT-011–013 × 1011612 weeks5 yearActive phase completed, follow-up ongoing1 × 10128CT-AMT-011–021 × 1012518 weeks (including 4 weeks’ run-in)1 yearCompletedTwo observational preparation studies were carried out to collect baseline data (no treatment control) Glybera was studied in three uncontrolled, open-label interventional studies (CT-AMT-010–01, CT-AMT-011–01 and CT-AMT-011–02) with a combined total sample size of 27. Three different dose regimens were evaluated in CT-AMT-010–01, CT-AMT-011–01 and CT-AMT-011–02. CT-AMT-011–02 was a safety and efficacy trial; initially planned as a controlled study it was subsequently amended to an uncontrolled study because of difficulties in identifying patients with a high baseline risk of pancreatitis. It should be noted that a different Glybera product was used in the AMT-010 and AMT-011 trials because of a change in the manufacturing process. The first cohort in the AMT-011 trials (n = 2 subjects) was administered 3 × 1011 gc/kg of AMT-011 to serve as a bridging arm to gauge the similarity in the safety and efficacy of AMT-011 relative to AMT-010. CT-AMT-011–01 and CT-AMT-011–02 included an immunosuppressive regimen. CT-AMT-011–01 included a combination of ciclosporin A (3 mg/kg/day) and mycophenolate mofetil (2 g/day) given over 12 weeks. The regimen in CT-AMT-011–02 was the same as that in CT-AMT-011–01 but also included a single bolus of methylprednisolone (single intravenous bolus of 1 mg/kg) given half an hour before administration of ciclosporin A and mycophenolate mofetil. Efficacy was assessed over 12 weeks, with long-term follow up planned for 5 years. The analysis of pancreatitis events was attempted post hoc by examining the number of events or admissions to an intensive care unit retrospectively as this was not a prespecified analysis |
Study number | Dose (gc/kg) | Number of patients | Duration of monitoring | Duration of follow-up | Status | PREPARATION-01 | None | 18 | 13–78 weeks | – | Completed | CT-AMT-010–01 | 1 × 1011 | 4 | 12 weeks | 5 years | Active phase completed, follow-up ongoing | 3 × 1011 | 4 | PREPARATION-02 | None | 22 | 2–83 weeks | – | Completed | CT-AMT-011–01 | 3 × 1011 | 6 | 12 weeks | 5 year | Active phase completed, follow-up ongoing | 1 × 1012 | 8 | CT-AMT-011–02 | 1 × 1012 | 5 | 18 weeks (including 4 weeks’ run-in) | 1 year | Completed | ||
| Study number | Dose (gc/kg) | Number of patients | Duration of monitoring | Duration of follow-up | Status | ||||||||||||||||||||||||||||||||||||||
| PREPARATION-01 | None | 18 | 13–78 weeks | – | Completed | ||||||||||||||||||||||||||||||||||||||
| CT-AMT-010–01 | 1 × 1011 | 4 | 12 weeks | 5 years | Active phase completed, follow-up ongoing | ||||||||||||||||||||||||||||||||||||||
| 3 × 1011 | 4 | ||||||||||||||||||||||||||||||||||||||||||
| PREPARATION-02 | None | 22 | 2–83 weeks | – | Completed | ||||||||||||||||||||||||||||||||||||||
| CT-AMT-011–01 | 3 × 1011 | 6 | 12 weeks | 5 year | Active phase completed, follow-up ongoing | ||||||||||||||||||||||||||||||||||||||
| 1 × 1012 | 8 | ||||||||||||||||||||||||||||||||||||||||||
| CT-AMT-011–02 | 1 × 1012 | 5 | 18 weeks (including 4 weeks’ run-in) | 1 year | Completed | ||||||||||||||||||||||||||||||||||||||
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | PREPARATION-01: 18 LPL-deficient patients aged ≥ 18 years with type I hyperchylomicronaemia, post-heparin LPL activity < 25% of the normal level and plasma concentration of triglycerides > 95th percentile for age and sex. Seventeen subjects completed the study; one subject died of a cardiac arrest | ||||||||||||||||||||||||||||||||||||||||||
| CT-AMT-010–01: 8/18 patients from the PREPARATION-01 cohort with confirmed homozygotic and compound heterozygotic LPL gene mutations | |||||||||||||||||||||||||||||||||||||||||||
| PREPARATION-02: 22 subjects with LPLD, LPL activity ≤ 20% of normal, LPL mass > 5% of normal and fasting plasma triglyceride concentration > 10 mmol/l. Twenty subjects completed the study; two subjects withdrew | |||||||||||||||||||||||||||||||||||||||||||
| CT-AMT-011–01: 15/22 subjects from PREPARATION-02 cohort; one subject was withdrawn and thus 14 subjects entered the study long term. Follow-up extended up to 5 years | |||||||||||||||||||||||||||||||||||||||||||
| CT-AMT-011–02: five patients enrolled to examine postprandial chylomicron metabolism, fasting triglyceride level, serum LPL activity and pancreatitis; only one patient provided data | |||||||||||||||||||||||||||||||||||||||||||
| Trial size/total trial population | Combined total n = 27 | ||||||||||||||||||||||||||||||||||||||||||
| Length of follow-up | See table above | ||||||||||||||||||||||||||||||||||||||||||
| Control/comparator used | The two observational studies (PREPARATION-01 and PREPARATION-02), which included patients receiving only diet reduction and no active treatment, acted as the control for the active treatment studies. Note: some patients (not all) from the PREPARATION-01 and PREPARATION-02 studies took part in the active treatment studies | ||||||||||||||||||||||||||||||||||||||||||
| How is the control/comparator constructed? | See previous section | ||||||||||||||||||||||||||||||||||||||||||
| Outcomes | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 1 | PREPARATION studies – fasting plasma triglyceride levels and disease complications in LPL-deficient subjects on a low-fat diet | ||||||||||||||||||||||||||||||||||||||||||
| Active treatment studies – across the three studies a measure of the reduction in fasting plasma triglyceride levels to < 10 mmol/l or to 40% of the starting level was either a primary or a secondary outcome | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 2 | PREPARATION studies – to record the incidence of pancreatic events in the context of the safety evaluation | ||||||||||||||||||||||||||||||||||||||||||
| Active treatment studies – a reduction in frequency and/or severity of clinical signs and symptoms related to LPL deficiency (i.e. eruptive xanthomas, lipaemia retinalis, pancreatitis, episodes of abdominal pain, plasma lactescence, lack of energy/fatigue and quality of life and diabetes management). The incidence of pancreatitis was the most clinically meaningful end point | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 3 | Other measures of the effect of active treatment [e.g. clearance of chylomicrons and other determinants of the biological activity of the LPL (LPLS447X) transgene product] | ||||||||||||||||||||||||||||||||||||||||||
| Adverse events | Overall, Glybera was well tolerated by all patients during the initial 12-week observational period and during the long-term phase of observation (up to 3 years with CT-AMT-010–01). All reactions were self-limiting and mild in nature. There were no obvious serious adverse events seemingly related to Glybera | ||||||||||||||||||||||||||||||||||||||||||
| Surrogate or intermediate clinical outcome? | Yes – the effect on lipid profiles, such as a reduction in fasting triglycerides to < 10 mmol/l or a > 40% reduction in fasting triglycerides, is a surrogate marker of LPL activity-related clinical benefit. A reduction in post-prandial chylomicronaemia has been proposed as an alternative surrogate marker and, subject to clinical validation, a reduction in post-prandial chylomicronaemia could be accepted as a surrogate marker for efficacy | ||||||||||||||||||||||||||||||||||||||||||
| Real clinical outcome? | Yes (a reduction in pancreatitis events was suggested using retrospective data) | ||||||||||||||||||||||||||||||||||||||||||
| Summary of efficacy evidence | |||||||||||||||||||||||||||||||||||||||||||
| Overall evidence base provided | CT-AMT-011–02 was the only study yielding data allowing the possibility of making a link between surrogate and clinical end points (pp-CM metabolism, fasting triglyceride levels, serum LPL activity, pancreatitis). Only one patient out of five responded to the treatment The presented data set in relation to the restricted indication included 12 out of 27 patients treated with Glybera, who were aged 40–70 years of age and were diagnosed with LPLD relatively late in life The reduction in post-prandial chylomicronaemia as an alternative surrogate marker for efficacy, although not validated at present, was considered biologically plausible and acceptable. The data on pancreatitis remain very limited and include a very small number of patients (n = 12) with limitations acknowledged in the statistical analysis In summary, the evidence generated by the reduction in pancreatitis events and severity of attacks, although hampered by statistical limitations and by fluctuations in the occurrence of pancreatitis, suggested that Glybera leads to a clinically relevant reduction in pancreatitis risk, at least in some patients. This is also supported by the reduction in hospital admissions and intensive care unit stays. Of particular note is the fact that, although about half of the 17 patients required an intensive care unit stay because of pancreatitis events before treatment, no intensive care unit stay was recorded in the same patients after treatment, compared with non-treated patients |
||||||||||||||||||||||||||||||||||||||||||
| Estimate of effect on HRQoL | The reduction in SF-36 scores (from both the physical functioning and the mental domains) in three out of five patients in the CT-AMT-011–02 study at week 14 following treatment was of major concern. The applicant explained the quality-of-life reduction by adverse events and immunosuppression. However, the data on quality of life from later time points (up to week 52) and from all other studies conducted with Glybera are not available | ||||||||||||||||||||||||||||||||||||||||||
| Other issues | |||||||||||||||||||||||||||||||||||||||||||
| Any issues of scale-up for the product? |
|
||||||||||||||||||||||||||||||||||||||||||
| Is further evidence requested for approval? |
|
||||||||||||||||||||||||||||||||||||||||||
| Notes | Given the rarity of LPLD (prevalence in the EU of 2 in 1,000,000), the uncontrolled study design applied in all three clinical trials using subjects as their own control was accepted and in line with the scientific advice given. Development of studies was hampered by difficulties in the recruitment of sufficient numbers of patients The Scientific Advisory Group considered that it is not possible to exclude completely the hypothesis that the reduction in the incidence of pancreatitis in some patients is due to the inherent temporal rarity of pancreatitis events. Issues inherent to retrospective data assessment in comparison to prospective data assessment were highlighted by the CAT Across the three active treatment trials the primary and secondary outcomes were not the same |
||||||||||||||||||||||||||||||||||||||||||
| MACI: EMA assessment (CHMP and CAT) 2013,19 NICE multiple technology appraisal 201420 | |||||||||||||||||||||||||||||||||||||||||||
| EMA marketing authorisation in April 2013, which was subsequently suspended in September 2014 (an authorised manufacturing site no longer existed) | |||||||||||||||||||||||||||||||||||||||||||
| Nature of the disease | |||||||||||||||||||||||||||||||||||||||||||
| Indication | MACI is to be used in skeletally mature patients for the repair of symptomatic cartilage defects of the knee (grades III and IV of the arthroscopic staging of osteochondral lesions as described by the modified Outerbridge scale) | ||||||||||||||||||||||||||||||||||||||||||
| Orphan status? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is this a rare condition? | No. Cartilage injuries were observed in 5–11% of diagnostic knee arthroscopies in predominantly young adult populations with knee pain | ||||||||||||||||||||||||||||||||||||||||||
| What is the natural history of the disease without this treatment/with current treatment? | Cartilage defects of the knee occur along a spectrum of disease and severity. Larger, more chronic lesions are often symptomatic, may contribute to joint misalignment and can cause disabling symptoms such as pain, catching, locking and swelling. Focal chondral lesions that are left untreated may progress to debilitating joint pain, dysfunction and degenerative arthritis | ||||||||||||||||||||||||||||||||||||||||||
| Nature of the medicine | |||||||||||||||||||||||||||||||||||||||||||
| How does it work? | It is the first advanced-therapy medicine to be combined with a medical device – in this case the cells are embedded in a biodegradable matrix. It attempts to generate hyaline or hyaline-like cartilage. ACI requires two surgical procedures, first to harvest autologous chondrocytes, which are then grown extracorporeally, and then to transplant the cultivated cells back into the lesions. The benefit of ACI over other restoration techniques is that larger lesions can be treated | ||||||||||||||||||||||||||||||||||||||||||
| Is it claiming to meet an otherwise unmet need? | No, other treatment options (such as microfracture) exist and clinical practice varies | ||||||||||||||||||||||||||||||||||||||||||
| How is it given? | Autologous chondrocytes are seeded onto a collagen membrane of porcine origin, which is secured into the lesion with fibrin glue. At implantation, the membrane is trimmed to the correct size and shape and implanted cell-side down into the base of the defect; the implant is secured in place using fibrin sealant. The recommended dose of MACI implant is 500,000–1,000,000 cells/cm2 of defect. The dose is the same for all patients, regardless of age | ||||||||||||||||||||||||||||||||||||||||||
| Are there any comparator treatments? | Repair techniques such as microfracture aim at marrow stimulation and induce the formation of fibrocartilage repair tissue to treat patients with focal chondral defects in the knee. These techniques penetrate the subchondral bone and cause release of marrow components into the defect site. The reparative response produced from these procedures is one that may generate primarily fibrocartilage. Single-stage restoration techniques such as osteochondral autograft, mosaicplasty and osteochondral allograft attempt to replace the cartilage defect with host or donor articular cartilage | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of the intervention evolving over time? | Yes – MACI is a third-generation ACI product. ERG report (for NICE) stated that ‘There is a general problem when long-term results are needed but the technology continues to evolve’ (p. 148)20 | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of persistence of the treatment within the patient? | In concept, the MACI implant would contribute to the repair of articular cartilage defects through proliferation of seeded chondrocytes, resulting in synthesis of hyaline-like repair tissue | ||||||||||||||||||||||||||||||||||||||||||
| Trial design | |||||||||||||||||||||||||||||||||||||||||||
| Trial description | The clinical data consist of the pivotal Superiority of MACI Versus Microfracture Treatment (SUMMIT) multicentre, randomised, open-label parallel-group trial (MACI00206) supported by several clinical studies reported in the literature.19 The internal study reports were small-scale, non-randomised prospective studies. The aim of the SUMMIT trial was to demonstrate the superiority of the MACI implant compared with arthroscopic microfracture for the treatment of symptomatic articular cartilage defects of the femoral condyle, including the trochlea | ||||||||||||||||||||||||||||||||||||||||||
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Male and female patients aged between 18 and 55 years (inclusive) with at least one symptomatic Outerbridge grade III or IV focal cartilage defect on the medial femoral condyle, lateral femoral condyle and/or trochlea (defect size ≥ 3.0 cm2 irrespective of location) | ||||||||||||||||||||||||||||||||||||||||||
| Trial size/total trial population | 144 patients: 72 patients MACI, 72 patients microfracture | ||||||||||||||||||||||||||||||||||||||||||
| Length of follow-up | 2-year follow-up data already collected from the MACI00206 study (5-year follow-up planned) | ||||||||||||||||||||||||||||||||||||||||||
| Control/comparator used | Microfracture treatment | ||||||||||||||||||||||||||||||||||||||||||
| How is the control/comparator constructed? | RCT | ||||||||||||||||||||||||||||||||||||||||||
| Outcomes | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 1 | Co-primary end point of Knee Injury and Osteoarthritis Outcome Score (KOOS) for pain and function (sports and recreational activities) | ||||||||||||||||||||||||||||||||||||||||||
| Response outcome 2 | Secondary end point: histology of cartilage forming (histological evaluation of structural repair of evaluable biopsies harvested from the core of the index lesion during arthroscopy) | ||||||||||||||||||||||||||||||||||||||||||
| Response outcome 3 | Magnetic resonance imaging (MRI) of cartilage – MRI assessments of structural repair parameters | ||||||||||||||||||||||||||||||||||||||||||
| Adverse events | Most adverse events were thought to be surgery related rather than product related | ||||||||||||||||||||||||||||||||||||||||||
| Surrogate or intermediate clinical outcome? | Yes – structural and functional repair of cartilage defects as measured by MRI or histology scoring | ||||||||||||||||||||||||||||||||||||||||||
| Real clinical outcome? | Yes – KOOS | ||||||||||||||||||||||||||||||||||||||||||
| Summary of evidence | |||||||||||||||||||||||||||||||||||||||||||
| Overall evidence base provided | A clinically and statistically significant difference in improvement from baseline to week 104 was seen for the co-primary end point of KOOS for pain and function in patients treated with MACI over the comparator (p = 0.001). Significantly more patients treated with MACI (87.50%) met the responder analysis criteria than patients treated with microfracture (68.06%), which is considered clinically relevant. The primary efficacy end point was corroborated by several other patient-reported outcome measures and a responder analysis of the primary efficacy measures demonstrated superior clinical efficacy for patients treated with MACI compared with microfracture | ||||||||||||||||||||||||||||||||||||||||||
| Estimate of HRQoL | Knee-related quality of life is one of the five key dimensions of KOOS, although the NICE report highlighted the ‘lack of good quality of life data’ (p. 60)20 | ||||||||||||||||||||||||||||||||||||||||||
| Other issues | |||||||||||||||||||||||||||||||||||||||||||
| Any issues of scale-up for the product? | The manufacture of the product is patient specific (autologous). Production will be centralised at one site | ||||||||||||||||||||||||||||||||||||||||||
| Is further evidence requested for EMA/FDA approval? |
|
||||||||||||||||||||||||||||||||||||||||||
| Any additional information provided? |
|
||||||||||||||||||||||||||||||||||||||||||
| ChondroCelect – characterised viable autologous cartilage cells expanded ex vivo expressing specific marker proteins: EMA assessment 2009,22 NICE multiple technology appraisal 201420 | |||||||||||||||||||||||||||||||||||||||||||
| EMA marketing authorisation | |||||||||||||||||||||||||||||||||||||||||||
| Nature of the disease | |||||||||||||||||||||||||||||||||||||||||||
| Indication | The indication for ChondroCelect is repair of single symptomatic cartilaginous defects of the femoral condyle of the knee [International Cartilage Repair Society (ICRS) grade III or IV] in adults | ||||||||||||||||||||||||||||||||||||||||||
| Orphan status? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is this a rare condition? | No | ||||||||||||||||||||||||||||||||||||||||||
| What is the natural history of the disease without this treatment/with current treatment? | The healing capacity of articular cartilage is poor and damaged articular cartilage is thought to be a precursor to the development of osteoarthritis. Damaged articular cartilage can result in pain, loss of joint function and disability. An early intervention on symptomatic cartilage lesions may prevent or delay irreversible changes in the joint surface. Currently, there is no uniform approach to managing significant knee cartilage defects | ||||||||||||||||||||||||||||||||||||||||||
| Nature of the medicine | |||||||||||||||||||||||||||||||||||||||||||
| How does it work? | ChondroCelect is a suspension of approximately 10,000 autologous cartilage cells per microlitre of medium for autologous use. The cells have been obtained by ex vivo expansion of chondrocytes isolated from a biopsy of the articular cartilage from the patient’s knee. The active substance is a centrifuged pellet of 4–12 million cells that were expanded ex vivo, harvested and washed. The expansion process is designed to preserve the integrity and function of the cells and particularly to maintain the cells’ ability to produce hyaline cartilage | ||||||||||||||||||||||||||||||||||||||||||
| Is it claiming to meet an otherwise unmet need? | No. Other treatment options exist and clinical practice varies | ||||||||||||||||||||||||||||||||||||||||||
| How is it given? | In the first step a cartilage biopsy is obtained arthroscopically from healthy articular cartilage from a lesser weight-bearing area of the patient’s knee, approximately 4 weeks prior to implantation. Chondrocytes are isolated from the biopsy by enzymatic digestion, expanded in vitro, characterised and delivered as a suspension of 1 × 104 cells/µl for implantation in the same patient. During the second step of the procedure the expanded chondrocyte suspension is implanted during open-knee surgery | ||||||||||||||||||||||||||||||||||||||||||
| Are there any comparator treatments? | Repair techniques such as microfracture aim at marrow stimulation and induce the formation of fibrocartilage repair tissue to treat patients with focal chondral defects in the knee. These techniques penetrate the subchondral bone and cause release of marrow components into the defect site. The reparative response produced from these procedures is one that may generate primarily fibrocartilage. Single-stage restoration techniques such as osteochondral autograft, mosaicplasty and osteochondral allograft attempt to replace the cartilage defect with host or donor articular cartilage | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of the intervention evolving over time? | Yes – ChondroCelect is a third-generation ACI product | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of persistence of the treatment within the patient? | Implanted cells become a structural part of newly formed cartilage | ||||||||||||||||||||||||||||||||||||||||||
| Trial design | |||||||||||||||||||||||||||||||||||||||||||
| Trial description | Study TIG/ACT/01/2000 is a Phase III, multicentre RCT to compare ChondroCelect with microfracture in the repair of symptomatic single cartilaginous lesions of the femoral condyles of the knee. Supportive study: prospective, long-term follow-up study of patients in the Belgian Armed Forces treated with ChondroCelect (TIG/ACT/02)22 | ||||||||||||||||||||||||||||||||||||||||||
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | TIG/ACT/01/2000: patients aged between 18 and 50 years who had a single symptomatic cartilage lesion of between 1 and 5 cm2 of the femoral condyles met the inclusion criteria | ||||||||||||||||||||||||||||||||||||||||||
| TIG/ACT/02: this study is a prospective, non-comparative, open-label study of 2–5 years’ duration in 20 patients with single and multiple symptomatic cartilage defects, in any location of the knee, who underwent chondrocyte implantaton using ChondroCelect | |||||||||||||||||||||||||||||||||||||||||||
| Trial size/total trial population | TIG/ACT/01/2000: 118 participants: n = 57 ChondroCelect, n = 61 microfracture | ||||||||||||||||||||||||||||||||||||||||||
| TIG/ACT/02: Of all reported lesions, 80% were reported to be of ICRS Grade III or IV. Of 24 femoral lesions reported in 19 patients, 21 were treated with CCI | |||||||||||||||||||||||||||||||||||||||||||
| Length of follow-up | TIG/ACT/01/2000: 12 months, extended to 36 months for adverse events | ||||||||||||||||||||||||||||||||||||||||||
| TIG/ACT/02: 5 years | |||||||||||||||||||||||||||||||||||||||||||
| Control/comparator used | Microfracture is considered an effective standard treatment for smaller femoral cartilage lesions according to currently available literature and is an acceptable control therapy | ||||||||||||||||||||||||||||||||||||||||||
| How is the control/comparator constructed? Source of comparative data? Confounding? | RCT | ||||||||||||||||||||||||||||||||||||||||||
| Outcomes | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 1 | Knee Injury and Osteoarthritis Outcome Score (KOOS) | ||||||||||||||||||||||||||||||||||||||||||
| Response outcome 2 | Structural repair | ||||||||||||||||||||||||||||||||||||||||||
| Adverse events | The overall safety summary showed that the main difference in treatment-related adverse events between ChondroCelect and microfracture was related to the open knee surgery (arthrotomy), which caused an increase in joint swelling and possible joint effusion. Cartilage hypertrophy can be reduced by using a biomembrane to cover the lesion and will therefore not pose a major safety concern in future applications of ChondroCelect. However, a higher number of patients in the microfracture arm have a treatment failure and require a subsequent surgical intervention. Therefore, the short- and long-term complication rate is not higher for ChondroCelect than for microfracture | ||||||||||||||||||||||||||||||||||||||||||
| Surrogate or intermediate clinical outcome? | Yes – structural repair (histological analysis) | ||||||||||||||||||||||||||||||||||||||||||
| Real clinical outcome? | Yes – KOOS | ||||||||||||||||||||||||||||||||||||||||||
| Summary of efficacy evidence | |||||||||||||||||||||||||||||||||||||||||||
| Overall evidence base provided | The mean change in overall KOOS from baseline to the average of 12–18 months was slightly higher for patients in the ChondroCelect group than for patients in the microfracture group. The results fulfil the predefined criteria for non-inferiority and changes are clinically relevant. The results of the histological analysis of structural repair at 12 months favoured ChondroCelect and the difference was statistically significant for both qualitative and quantitative analyses. It was, however, acknowledged that this end point was not in compliance with good clinical practice as it was developed during the conduct of the study as the original a priori determined primary efficacy point was considered invalid | ||||||||||||||||||||||||||||||||||||||||||
| Estimate of HRQoL | Knee-related quality of life is one of the five key dimensions of KOOS, although the NICE report highlighted the ‘lack of good quality of life data’ (p. 60)20 | ||||||||||||||||||||||||||||||||||||||||||
| Other issues | |||||||||||||||||||||||||||||||||||||||||||
| Any issues of scale-up for the product? | The manufacture of the product is patient specific (autologous). Production will be centralised at one site | ||||||||||||||||||||||||||||||||||||||||||
| Is further evidence requested for approval? | The good clinical practice inspection highlighted the number of missing data on the structural end point and the change to the ICRS II readout in the pivotal study as major concerns. The CAT considered the following particular causes for concern:
|
||||||||||||||||||||||||||||||||||||||||||
| Holoclar – ex vivo expanded autologous human corneal epithelial cells containing stem cells: EMA assessment (CHMP, CAT, Committee for Orphan Medicinal Products) 201423 | |||||||||||||||||||||||||||||||||||||||||||
| EMA conditional marketing authorisation | |||||||||||||||||||||||||||||||||||||||||||
| Nature of the disease | |||||||||||||||||||||||||||||||||||||||||||
| Indication | Corneal lesions, with associated (limbal) stem cell deficiency (LSCD), as a result of ocular burns. The clinical spectrum of LSCD includes pain, photophobia, inflammation, corneal neovascularisation and eventually the reduction or complete loss of visual acuity | ||||||||||||||||||||||||||||||||||||||||||
| Orphan status? | Designated as an orphan medicinal product (2008) in the following indications: corneal lesions with associated LSCD as a result of ocular burns | ||||||||||||||||||||||||||||||||||||||||||
| Is this a rare condition? | The condition is considered to be rare, with an estimated prevalence of 0.34 per 10,000 | ||||||||||||||||||||||||||||||||||||||||||
| What is the natural history of the disease without this treatment/with current treatment? | If left untreated, the condition may progress to a stage whereby persistent epithelial defects present with an associated high risk for the development of bacterial keratitis, corneal perforation and blindness | ||||||||||||||||||||||||||||||||||||||||||
| Nature of the medicine | |||||||||||||||||||||||||||||||||||||||||||
| How does it work? | Holoclar is specifically ex vivo expanded autologous human corneal epithelial cells containing stem cells and replaces damaged corneal epithelium cells and creates a reservoir of limbic stem cells (LSCs) in LSC-deficient areas of the cornea for continuous regeneration. It consists of a transparent circular sheet of living tissue containing autologous human corneal epithelial cells, limbal stem cells and derived transient amplifying cells | ||||||||||||||||||||||||||||||||||||||||||
| Is it claiming to meet an otherwise unmet need? | Yes, the product claims to respond to an unmet medical need by providing a new active substance to treat patients with irreversible and extensive damage as a result of an ocular burn. At the time of application, no medicinal products had been approved in the EU/European Economic Area (EEA) for this indication and there was no gold standard treatment | ||||||||||||||||||||||||||||||||||||||||||
| How is it given? | Single topical placement without systemic effect | ||||||||||||||||||||||||||||||||||||||||||
| Are there any comparator treatments? | Limbal allografts, which have an associated risk of rejection and which require long-term systemic immunosuppression. Non-expanded limbal autografts from the healthy fellow eye, which may lead to iatrogenic induction of LSCD in the donor eye | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of the intervention evolving over time? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of persistence of the treatment within the patient? | Negative effects of persistence – possible risks include systemic distribution of cells derived from Holoclar that are tumour forming, accelerated immune response or transmission of adventitious agents. The cells are not expected to migrate beyond the ocular surface or to produce systemic effects. Tumourigenicity was investigated in vivo and results suggested a low risk | ||||||||||||||||||||||||||||||||||||||||||
| Positive effects of persistence – some information on the potential for biodistribution was derived from a historical data set: data from a histological and morphological evaluation of corneal material collected from 26 patients who had undergone perforating keratoplasty post LSC transplantation with Holoclar. Available long-term follow-up data up to 10 years after autologous cultivated limbal stem cells transplantation, although limited, supported persistence of treatment success beyond 12 months. Additional long-term efficacy data will be collected in the margins of a post-authorisation safety study to confirm this outcome | |||||||||||||||||||||||||||||||||||||||||||
| Trial design | |||||||||||||||||||||||||||||||||||||||||||
| Trial description | Multicentre retrospective observational case series Primary efficacy/safety study and supportive study |
||||||||||||||||||||||||||||||||||||||||||
| HLSTM01 (1998–2007) | HLSTM02 (1998–2007) | HLSTM04 (2008–2013) | |||||||||||||||||||||||||||||||||||||||||
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Male or female with moderate/severe LSCD; median age 49 years, mostly adults | Male or female with moderate/severe LSCD; median age 43.5 years, mostly adults | 15 patients treated from 2008 (from additional centres not originally provided as part of HLSTM01) | ||||||||||||||||||||||||||||||||||||||||
| Trial size/total trial population | 104 patients with moderate/severe LSCD; two centres | 29 patients with moderate/severe LSCD; seven centres | 15 patients with moderate/severe LSCD; three centres | ||||||||||||||||||||||||||||||||||||||||
| Length of follow-up | 12 months post-intervention assessment, max. 10 years’ follow-up: 28% of patients 1–2 years, 22% of patients 2–3 years, 12% of patients ≥ 5 years to a maximum of 10 years post transplantation After year 5, only five patients had long-term follow-up data, of whom four were reported to have continued treatment success |
||||||||||||||||||||||||||||||||||||||||||
| Control/comparator used | Patients acted as their own controls – outcomes were compared with baseline data | ||||||||||||||||||||||||||||||||||||||||||
| How is the control/comparator constructed? Source of comparative data? Confounding? | See above. The assumption that the condition would not heal was accepted, so any healing could be ascribed to Holoclar | ||||||||||||||||||||||||||||||||||||||||||
| Outcome | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 1 | Successful transplant at 12 months based on the co-presence of clinical signs: (1) a superficial corneal neovascularisation classified as ‘none’ or ‘mild’ and (2) epithelial defects classified as ‘none’ or ‘trace’ | ||||||||||||||||||||||||||||||||||||||||||
| Response outcome 2 | Symptomatic relief (pain, burning, photophobia) | ||||||||||||||||||||||||||||||||||||||||||
| Response outcome 3 | Improvement in visual acuity or visual stabilisation at month 12 compared with baseline | ||||||||||||||||||||||||||||||||||||||||||
| Surrogate or intermediate clinical outcome? | Yes – corneal epithelial integrity and absence of significant corneal neovascularisation | ||||||||||||||||||||||||||||||||||||||||||
| Real clinical outcome? | Yes – improved visual acuity | ||||||||||||||||||||||||||||||||||||||||||
| Adverse events | Eye-related disorders were the most commonly observed adverse events, occurring in 57% of the safety population. Overall, the rate of serious adverse events was low. Out of a total of 11 serious adverse events, three were judged as related to the administration of Holoclar | ||||||||||||||||||||||||||||||||||||||||||
| Summary of efficacy evidence | |||||||||||||||||||||||||||||||||||||||||||
| Overall evidence base provided |
|
||||||||||||||||||||||||||||||||||||||||||
| Estimate of HRQoL | Not assessed | ||||||||||||||||||||||||||||||||||||||||||
| Other issues | |||||||||||||||||||||||||||||||||||||||||||
| Any issues of scale-up for the product? | Clinical success depends on factors unique to cell therapies, including manufacturing procedures, clinical and pharmacological standardisation of protocols and regulation. Manufacture of the active substance is patient specific and the manufacturing process is state of the art and highly complex. As such, the applicant implemented a training programme for surgeons to ensure collection of seed material and a structured approach to manufacturing consisting of many monitored stages and substage in-process controls. The applicant was also required to provide further evidence on the stability of the product (integrity and viability) and transport information | ||||||||||||||||||||||||||||||||||||||||||
| Is further evidence requested for EMA/FDA approval? | A multinational, multicentre, prospective, open-label, uncontrolled interventional study to assess the efficacy and safety of autologous cultivated LSC grafting for restoration of corneal epithelium in patients with LSCD caused by ocular burns was required by December 2020. A major objection was raised with regard to the proliferation of irradiated cells and further validation was requested. Evidence was provided in the form of a demonstration of several methods to show that the irradiated cells do not proliferate. Paediatric application was deferred at the time of submission pending further measures | ||||||||||||||||||||||||||||||||||||||||||
| Notes | At the time of the application > 200 patients had already been treated with Holoclar in clinical practice (since 1998); however, many clinics declined the request to provide data. The assessors considered that this may introduce bias but felt that the supporting literature and the similarity of the findings reported to those in the published articles provided some confidence in the numbers and, therefore, they were happy to allow the data. Supportive data from published articles were also considered by the CAT and this appears to have had a strong influence on the decision-making process, although only supportive information was provided. As the condition was considered to have a low incidence the small sample size was considered to be acceptable. The CAT noted that at baseline the majority of patients already presented with no or only trace epithelial defects and as such already presented with a successful treatment outcome. However, they considered that LSCD is a condition with impaired ability to maintain or restore an intact corneal epithelium and so defects over the follow-up period were considered clinically relevant. The fact that the studies were uncontrolled and not randomised further added to the uncertainties of the validity of the data set, but this was considered inevitable because of the lack of a suitable comparator, considering that there is neither an approved treatment for LSCD nor a ubiquitous accepted standard of care. As this condition would not heal spontaneously, the single-arm, uncontrolled design was considered acceptable by the CAT | ||||||||||||||||||||||||||||||||||||||||||
| PROVENGE (sipuleucel-T/autologous peripheral blood mononuclear cells activated with prostatic acid phosphatase–granulocyte-macrophage colony-stimulating factor): EMA assessment 2013 (CAT and CHMP),24 NICE STA 2014,26 FDA assessment 200925 | |||||||||||||||||||||||||||||||||||||||||||
| EMA marketing authorisation in June 2013, which was withdrawn in May 2015 at the request of the manufacturer for commercial reasons | |||||||||||||||||||||||||||||||||||||||||||
| Nature of the disease | |||||||||||||||||||||||||||||||||||||||||||
| Indication | Asymptomatic or minimally symptomatic metastatic (non-visceral) hormone-relapsed prostate cancer in men for whom chemotherapy is not yet clinically indicated | ||||||||||||||||||||||||||||||||||||||||||
| Orphan status? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is this a rare condition? | Hormone-refractory metastatic prostate cancer affects around 5000 patients per year in the UK | ||||||||||||||||||||||||||||||||||||||||||
| What is the natural history of the disease without this treatment/with current treatment? | Asymptomatic patients have a median OS of 18–24 months. Patients with symptomatic disease have a median OS of 9–16 months | ||||||||||||||||||||||||||||||||||||||||||
| Nature of the medicine | |||||||||||||||||||||||||||||||||||||||||||
| How does it work? | Sipuleucel-T is an autologous active cellular immunotherapy product designed to stimulate an antigen (CD59) immune response to prostate cancer. Patients’ peripheral blood mononuclear cells are incubated with a recombinant fusion protein, the prostate protein prostatic acid phosphatase | ||||||||||||||||||||||||||||||||||||||||||
| Is it claiming to meet an otherwise unmet need? | No | ||||||||||||||||||||||||||||||||||||||||||
| How is it given? | Following blood sampling leukapheresis is performed (day 1) after which sipuleucel-T is manufactured at a central facility (days 2–3) and then infused into the patient (day 3 or 4). This process happens three times, at approximately 2-week intervals | ||||||||||||||||||||||||||||||||||||||||||
| Are there any comparator treatments? | Best supportive care (radiotherapy, bisphosphonates, steroids, analgesics, active surveillance), abiraterone acetate | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of the intervention evolving over time? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of persistence of the treatment within the patient? | No. The achievement and maintenance of the antigen response was assessed – maximum duration tested was 26 weeks in one trial. There was no clear indication of whether or not persistence was required for benefit. No adverse effects related to persistence of antigen response were mentioned | ||||||||||||||||||||||||||||||||||||||||||
| Trial design | |||||||||||||||||||||||||||||||||||||||||||
| Trial description | D9902B (IMPACT)24 | D9902A24 | D990124 | ||||||||||||||||||||||||||||||||||||||||
| Multicentre RCT (with crossover allowed after progression) using a 2 : 1 ratio (favouring allocation to sipuleucel-T) | As for IMPACT trial | As for IMPACT trial | |||||||||||||||||||||||||||||||||||||||||
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Asymptomatic or minimally symptomatic metastatic hormone-relapsed prostate cancer | Asymptomatic metastatic hormone-relapsed prostate cancer | Asymptomatic metastatic hormone-relapsed prostate cancer | ||||||||||||||||||||||||||||||||||||||||
| Trial size/total trial population | 512 | 98 | 127 | ||||||||||||||||||||||||||||||||||||||||
| Length of follow up | 3 years [follow-up was planned to continue until the number of events (deaths) reached that required by the analysis plan] | 3 years | 3 years | ||||||||||||||||||||||||||||||||||||||||
| Control/comparator used | Placebo, consisting of one-third of the patient’s cells being reinfused, but the cells had not been activated with the fusion protein; the remaining two-thirds were cryopreserved | As for IMPACT study | As for IMPACT study | ||||||||||||||||||||||||||||||||||||||||
| How is the control/comparator constructed? Source of comparative data? Confounding? | Following confirmation of disease progression, placebo patients could receive activated cells (i.e. very similar to sipuleucel-T) derived from their cryopreserved cells. Open-label phase | As for IMPACT study | As for IMPACT study | ||||||||||||||||||||||||||||||||||||||||
| Outcomes | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 1 | OS | Time to disease progression | Time to disease progression | ||||||||||||||||||||||||||||||||||||||||
| Response outcome 2 | Time to objective disease progression | OS | Time to onset of disease-related pain | ||||||||||||||||||||||||||||||||||||||||
| Response outcome 3 | Safety | Time to objective disease progression | Grade 3 adverse events | ||||||||||||||||||||||||||||||||||||||||
| Surrogate or intermediate clinical outcome? | TTP. Antigen response was also measured. Note: this did not correlate with the OS results | ||||||||||||||||||||||||||||||||||||||||||
| Real clinical outcome? | OS | ||||||||||||||||||||||||||||||||||||||||||
| Adverse events | Overall, the leukapheresis procedure and PROVENGE infusions were well tolerated. The main risks identified were acute infusion reactions, toxicities (e.g. citrate toxicity) associated with the leukapheresis procedure and infections (principally associated with catheters). Treatment with PROVENGE may lead to unwanted long-term immunological effects in the body system. This potential risk is adequately addressed in the risk management plan. Additional data will become available to further characterise the long-term safety profile of PROVENGE through registries | ||||||||||||||||||||||||||||||||||||||||||
| Summary of efficacy evidence | |||||||||||||||||||||||||||||||||||||||||||
| Overall evidence base provided | For the IMPACT trial, OS was significantly improved with sipuleucel-T (HR 0.8, 95% CI 0.61 to 0.98; p = 0.03) but there was no difference in time to objective disease progression (HR 0.95, 95% CI 0.77 to 1.17; p = 0.63). Two trials reported a significant advantage in terms of OS favouring sipuleucel-T, although no significant differences in time to disease progression were seen in any of the three trials. The RCTs had a low risk of bias, but only up to the point of disease progression, after which crossover from placebo to active intervention was permitted. No analyses were performed to adjust for crossover. In addition, the lack of significant effect on PFS may also have been because of a delay in effect | ||||||||||||||||||||||||||||||||||||||||||
| Estimate of HRQoL | Not assessed | ||||||||||||||||||||||||||||||||||||||||||
| Other issues | |||||||||||||||||||||||||||||||||||||||||||
| Any issues of scale-up for the product? | Yes, patients’ cells must be transferred from their local hospital to a central manufacturing facility and then back again to the local hospital. The final product has a short shelf life | ||||||||||||||||||||||||||||||||||||||||||
| Is further evidence requested for approval? | Periodic safety update reports | ||||||||||||||||||||||||||||||||||||||||||
| Notes | Thirteen members of the CHMP did not agree with the CHMP’s recommendation and the granting of a marketing authorisation. The objections were based around whether the differences in OS resulted from a true and clinically relevant effect of sipuleucel-T. The effect was not supported by either the PFS results or the TTP results. Importantly, in case of disagreement between these outcomes the efficacy evidence should be particularly convincing and ideally corroborated by other secondary end points, which was not the case. A lower proportion of patients were treated with docetaxel in the placebo group and also underwent delayed treatment with docetaxel in the placebo group (in the pivotal trial), which may have had an effect on OS. Confounding may also have been caused by the crossover from placebo to the active treatment (sipuleucel-T prepared from cryopreserved cells); as stated above, no analyses were performed to adjust for crossover and, therefore, the treatment effect may have been underestimated ‘Lack of consistency’ between TTP and OS, and possible confounding of the OS results by non-randomised post-progression post-blinding treatment, was also noted in the ERG report for NICE (appraisal now withdrawn). A possible reason for the lack of association between OS and TTP was that current clinical metrics of progression assessed in bone are inadequate. In addition, immune responses may require time to develop and the lack of difference in progression could result from such a delayed anti-tumour response FDA analyses of docetaxel treatment following randomisation did not provide evidence that the survival difference between the two arms was attributable to docetaxel. The FDA statistician based these analyses on the following assumption, which was thought very likely to be true: more patients with good prognosis were in the placebo arm than in the sipuleucel-T arm in the subgroup receiving docetaxel. This implies that more patients with poor prognosis were in the placebo arm in the sipuleucel-T arm in which patients did not receive docetaxel, as overall the two treatments were comparable In May 2015 the EU marketing authorisation for PROVENGE was withdrawn at the request of the manufacturer (Dendreon) for commercial reasons |
||||||||||||||||||||||||||||||||||||||||||
| ReCell Spray-On Skin system: NICE Medical Technologies Evaluation Programme (MTEP) 201427 | |||||||||||||||||||||||||||||||||||||||||||
| Authorisation granted in 2005 under Medical Devices Directive 93/42/EEC28 | |||||||||||||||||||||||||||||||||||||||||||
| Nature of the disease | |||||||||||||||||||||||||||||||||||||||||||
| Indication | Adults or children treated in burns units or centres for (1) partial-thickness burns including scalds caused by hot water for which mesh grafting is not required and (2) large-area burns – full-thickness or deep partial-thickness burns including where mesh grafting is required | ||||||||||||||||||||||||||||||||||||||||||
| Orphan status? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is this a rare condition? | No | ||||||||||||||||||||||||||||||||||||||||||
| What is the natural history of the disease without this treatment/with current treatment | The treatment of burns can be considered in two phases: acute and reconstructive. The acute phase is the initial management of the injury with the intention that burn wound healing will occur with minimal scarring and physical limitation. The reconstructive phase aims to improve the functional or visual impact of scarring, usually by surgical means, and may be carried out months or years after the initial injury. Full-thickness burns of > 1 cm in diameter need skin grafts because the regenerative components of the skin have been lost. Healing can occur only from the edges of the wound; without a graft the skin contracts, leading to a poor cosmetic outcome and reduced mobility. Deep dermal burns are unlikely to heal within 3 weeks and will therefore often need grafting | ||||||||||||||||||||||||||||||||||||||||||
| Nature of the medicine | |||||||||||||||||||||||||||||||||||||||||||
| How does it work? | ReCell is a stand-alone autologous cell harvesting device that enables a thin split-thickness skin biopsy to be processed to produce a mixed cell population for immediate delivery onto a prepared wound surface | ||||||||||||||||||||||||||||||||||||||||||
| Is it claiming to meet an otherwise unmet need? | No | ||||||||||||||||||||||||||||||||||||||||||
| How is it given? | The ReCell device allows a small, thin split-thickness shave biopsy to be physically and enzymatically broken down, yielding a viable suspension of mixed keratinocytes, fibroblasts and melanocytes that can be immediately sprayed or dripped on to the de-epithelialised area. The process is rapid – around 30 minutes – and does not require specialist skills or facilities to carry out. A cell suspension derived from a 1-cm2 biopsy is sufficient to treat an area of around 80 cm2, making it particularly valuable for patients with limited available healthy donor sites | ||||||||||||||||||||||||||||||||||||||||||
| Are there any comparator treatments? | Partial-thickness burns: biosynthetic dressings or standard dressings; large-area burns: skin mesh graft alone or skin mesh graft plus biosynthetic dressing | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of the intervention evolving over time? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is there any mention of persistence of the treatment within the patient? | No | ||||||||||||||||||||||||||||||||||||||||||
| Trial design | |||||||||||||||||||||||||||||||||||||||||||
| Trial description | Eleven studies were included in the submission to NICE:27 three RCTs and eight observational studies. Two of the RCTs were pilot studies with very small sample sizes (n = 13 and 14). All but one of the observational studies were also small (range 5–40 patients) case series. The two main studies are summarised below | ||||||||||||||||||||||||||||||||||||||||||
| Gravante et al.213 | Park et al.214 | ||||||||||||||||||||||||||||||||||||||||||
| Single-centre RCT | Retrospective cohort study (three groups) | ||||||||||||||||||||||||||||||||||||||||||
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Adults with deep partial-thickness burns (< 320 cm2) | Burns treated with skin grafting or replacement; all ages | |||||||||||||||||||||||||||||||||||||||||
| Trial size/total trial population? | 82 | 767 | |||||||||||||||||||||||||||||||||||||||||
| Length of follow-up | 6 months | Not reported | |||||||||||||||||||||||||||||||||||||||||
| Control/comparator used | Split-thickness skin grafting | ReCell Spray-On Skin system plus standard skin graft and standard skin graft alone | |||||||||||||||||||||||||||||||||||||||||
| How is the control/comparator constructed? Source of comparative data? Confounding? | RCT | Both the intervention and the two comparators used historical data. Multiple regression was used, although sex and type of burn agent were not included in the model input variables. Burn depth was greater in patients treated with a standard skin graft than in patients treated with ReCell alone, although burn depth was controlled for in the multiple regression | |||||||||||||||||||||||||||||||||||||||||
| Outcomes | |||||||||||||||||||||||||||||||||||||||||||
| Response outcome 1 | Time to complete epithelialisation | Wound infection | |||||||||||||||||||||||||||||||||||||||||
| Response outcome 2 | Aesthetic and functional quality of the scar | Graft loss | |||||||||||||||||||||||||||||||||||||||||
| Response outcome 3 | Wound infections | ||||||||||||||||||||||||||||||||||||||||||
| Response outcome 4 | Postoperative pain | ||||||||||||||||||||||||||||||||||||||||||
| Surrogate or intermediate clinical outcome? | No | No | |||||||||||||||||||||||||||||||||||||||||
| Real clinical outcome? | Yes | Yes | |||||||||||||||||||||||||||||||||||||||||
| Adverse events | None reported | None reported | |||||||||||||||||||||||||||||||||||||||||
| Summary of efficacy evidence | |||||||||||||||||||||||||||||||||||||||||||
| Overall evidence base provided | The RCT found ReCell and standard skin graft to be comparable in terms of wound healing time and long-term aesthetics, but ReCell was significantly less painful and the mean size of the donor site was significantly smaller. These results were reflected in the large cohort study, which also found no difference in terms of wound infection. The remaining evidence was supportive, indicating a range of patients who can be treated with ReCell. The External Assessment Centre concluded that ReCell may be a clinically suitable alternative to the use of split-thickness skin grafts in mid-deep partial-thickness burns. There was no clinical evidence examining the use of ReCell in partial-thickness burns, which are considered not to require skin grafting. There was also no evidence demonstrating improved outcomes for the use of ReCell plus split-thickness skin grafts compared with split-thickness skin grafts alone | ||||||||||||||||||||||||||||||||||||||||||
| Estimate of effect HRQoL | Not reported | ||||||||||||||||||||||||||||||||||||||||||
| Other issues | |||||||||||||||||||||||||||||||||||||||||||
| Any issues of scale-up for the product? | No | ||||||||||||||||||||||||||||||||||||||||||
| Is further evidence requested for approval? | NICE concluded that despite the potential benefits of the ReCeII Spray-On Skin system in terms of helping improve healing of acute burns, there was limited evidence regarding its use in clinical practice to curently support routine use in the NHS.27 NICE recommended research to address uncertainties about the claimed patient and system benefits of the ReCell Spray-On Skin system. Clinical outcomes should include time to 95% healing, length of hospital stay, cosmetic appearance of the scar and function of the burned area with ReCell compared with standard care | ||||||||||||||||||||||||||||||||||||||||||
| Notes | Note: within the NICE assessment the claimed benefits of ReCell in the case for adoption presented by the sponsor were:
|
||||||||||||||||||||||||||||||||||||||||||
Appendix 2 Adjustment for bias in non-randomised studies
Methods developed to adjust effect estimates obtained from NRSs for potential biases have taken two broad approaches: adjust at the study level or adjust as part of the process of evidence synthesis. These approaches are discussed separately in the following sections.
Adjusting for bias in the evidence synthesis process
The review of the literature on methods to adjust for bias in the evidence synthesis process identified 10 relevant studies. 44–53 These articles included two comprehensive reviews44,45 as well as individual articles, all of which were identified in the review articles. Many of the techniques described in Verde and Ohmann44 and Doi,45 however, have limited applicability to regenerative medicine (i.e. when only limited evidence from a small number of studies is available), as they require significant numbers of studies or data from RCTs to be applied. A small number of these techniques can, however, be applied when only a single study or a small number of studies is available. These methods are outlined in the following sections.
Adjusting using external data
Welton et al. 50 present a Bayesian hierarchical model to model bias in RCTs that are at high risk of bias. The authors developed a mixed-effects model in which treatment effects are considered as fixed and bias effects are considered as random. Estimates of bias in any given meta-analysis were given as a function of the prior distribution, which was estimated from published meta-analyses of RCTs and data from the current meta-analysis. When a meta-analysis contained no information about the size and magnitude of the bias, that is, when there were only high-risk studies, the estimate of bias was based on the prior distribution alone. This method allows treatment effect estimates to include information from the high-risk studies, accounting for the uncertainty in the magnitude of the bias in any particular meta-analysis. This technique was designed with adjustment of RCTs in mind but is extendable to the adjustment of NRSs whereby RCTs represent the low-risk studies and NRSs represent the high-risk studies. An appropriate library of meta-analyses combining data from RCTs and NRSs would, however, be necessary to apply this technique.
Elicitation
Turner et al. ,53 recognising the practical limitations of basing adjustment on external empirical data, proposed an alternative approach in which the direction and magnitude of biases are elicited by reviewers. This method can deal with multiple sources and types of bias including both internal validity bias and external validity bias. In brief, Turner et al. 53 proposed that authors design an idealised study aimed at answering the specific question in mind. This study may not be feasible to carry out and is simply a tool for exploring bias in the completed studies. To identify the potential biases, the completed studies are compared with the idealised study considering a number of potential sources of bias. For each form of bias identified, assessors then elicit the likely magnitude and variance of the bias. These estimates of the magnitude and variance of the potential biases can then be used to adjust treatment effect estimates accounting for both the magnitude and the uncertainty of any potential bias identified. External empirical evidence of bias can be included in the analysis rather than relying on eliciting values, but it is assumed that these data will be largely unavailable.
Adjusting for bias at the study level
There are a number of established statistical methods for analysing NRSs that attempt to minimise the potential bias from confounding. Each of these methods is briefly described in the following sections and is followed by a brief review of the literature discussing the efficacy of these methods.
Regression analysis
Confounding bias occurs in the context of estimating clinical effectiveness when individual patient characteristics, such as age, sex and disease duration, that influence efficacy outcomes are also correlated with treatment received. Regression analysis seeks to directly adjust for these potential confounding variables by building a statistical model54 of the form:
Regression models therefore allow the estimation of the treatment effect conditional on these confounding variables. There are many types of regression model. The choice of any particular model depends on the characteristics of the outcome variable (i.e. continuous or categorical) and on the way that it is mathematically related to the explanatory variables. Typically, for dichotomous outcomes, a logistic regression model is used. For continuous outcomes a linear regression model is used and for time-to-event data a proportional hazards regression (Cox regression) model is used.
Theoretically, regression models can be used to entirely eliminate bias attributable to confounding as long as the appropriate parameters are included with a regression equation. However, in reality, either confounding factors will be unobserved, preventing their inclusion in the regression model, or a lack of understanding of the disease process will mean that we do not know to include them in the regression model. When such unobserved confounders are not included in the regression model, confounding bias can persist. Regression techniques can be used in conjunction with other methods for adjusting of confounding, including propensity scoring and instrumental variables. 54 Regression models also require a minimum number of participants per additional explanatory variable, with a useful rule of thumb being at least 10 observations per explanatory variable. 55 This requirement may limit their application to regenerative medicine where effectiveness estimates can be based on relatively small studies.
Stratification
Stratification involves the division of participants into subgroups with respect to categorical (or categorised quantitative) prognostic factors, for example classifying age into decades or weight into quartiles. The intervention effect is then estimated in each stratum and a pooled estimate is calculated across strata. This procedure can be interpreted as a meta-analysis at the level of an individual study. Major limitations are that it is feasible and meaningful only when effects are consistent across strata and that it can usually be employed for only few variables, as strata increase exponentially in keeping with the number of stratification factors. 54 As such, stratification can only minimise rather than completely remove the bias resulting from confounding.
Matching
Matching involves selecting participants with similar values for important prognostic factors to make the control and treatment groups more similar so that any differences between the treatment group and the control group cannot be a result of differences in the matched variables. Matching can be carried out both prospectively or retrospectively. Matching prospectively can, however, cause significant recruitment problems. Matching retrospectively can also cause problems as it is not always possible to match individuals. In large studies it is often easier to use an unmatched control group and to use regression analysis to adjust for what would have been matched on. 56 Matching can, however, be useful in small studies when there are insufficient participants to adjust for multiple variables at once. 56 As such, matching may be a potentially useful technique for controlling for confounding when using historical controls with the small single-arm studies that have typified regenerative medicine clinical evidence. Although matching can be used to reduce confounding bias, it is unlikely to completely account for all difference because of unobserved confounding.
Instrumental variables analysis
Instrumental variables techniques attempt to approximate the experimental approach by using an instrument variable or variables. A parameter is considered a valid instrument if it meets the following two conditions:
-
the instrument must be correlated with the receiving of treatment (or exposure)
-
the instrumental variable must be independent of (uncorrelated with) unobserved confounders.
When a valid instrument exists the instrumental variables approach leads to unbiased estimates equivalent to those from a randomised study. Indeed, randomisation can be thought of as the perfect instrumental variable as it is by definition perfectly associated with treatment allocation and independent of unobserved heterogeneity. The problem with the instrumental variables approach, however, is that identification of a valid instrument is often difficult. Furthermore, although the first requirement of a valid instrument is easily tested, the second requirement is essentially untestable and therefore we can never be certain that an instrument is valid. The application of an instrumental variables approach also leads to significant reduction in the power to detect a difference, particularly when the instrumental variable is poorly correlated with treatment allocation. This issue may be particular problematic in regenerative medicine where studies are often small with a low power to detect differences between alternative treatments.
Propensity scoring
Propensity scoring, rather than being a single method, is a suite of methods that considers confounding bias as a form of selection bias in which treatment allocation is acknowledged to be non-random and which considers that treatment selection is often influenced by a patient’s characteristics. 57 All propensity scoring methods seek to model this process of treatment selection and estimate the propensity to receive treatment based on baseline patient characteristics. Conditional on the propensity score the distribution of baseline characteristics will be similar in both the treatment group and the control group. Therefore, in patients with similar propensity scores patient characteristics will be the same, independent of whether treatment was received. The propensity is typically estimated using a logistic regression model, although other methods have been applied. The estimated propensity score can be used to remove the effects of confounding in four different ways. 57 These are described very briefly below:
-
Matching – the propensity score is used to match participants in the treatment and control groups who have similar values of the propensity score.
-
Stratification – subjects are ranked on the propensity score and stratified into groups, typically quintiles. Stratum-specific mean differences are then calculated and these differences are effectively meta-analysed to estimate an overall difference in means.
-
Inverse probability of treatment weighting (IPTW) – this involves using the propensity score as a weight such that an individual participant’s weight is equal to the inverse of the probability of receiving the treatment.
-
Covariate – the propensity score is added as a covariate within a regression equation. The propensity score can be added either with or without additional explanatory variables.
A number of studies have sought to compare propensity scoring methods to ascertain which is the most effective at removing confounding bias. 57–61 These studies have shown matching and IPTW to be more effective than stratification or covariate adjustment. 58–61 The principal advantage of propensity scoring over other adjustment methods such as regression analysis is that it can be used even with small sample sizes and therefore may be particularly relevant to regenerative medicine. Propensity scoring also has a number of disadvantages. First, propensity scoring controls only for differences in observed variables and does nothing to remove bias resulting from unobserved characteristics. Second, including variables that affect whether a treatment is received but not the outcome of interest increases the variance of the estimated treatment effect without a concomitant reduction in bias. This is problematic as sometimes it can be difficult to establish which variables will impact only on which treatment is received. 57
Effectiveness of adjustment methods
Our review identified a total of nine studies: eight studies62–69 compared the results of regression analysis, instrumental variables and propensity scoring and a further study70 discussed the relative merits of the alternative methods of adjustment. Two of these studies were systematic reviews: Shah et al. 66 reviewed comparisons between propensity scoring and regression methods and Shah et al. 66 and Laborde-Castérot et al. 69 reviewed comparisons between propensity scoring and instrumental variable analyses.
Propensity scoring compared with regression analysis
Six studies62,64–68 compared propensity scoring with regression analysis. The conclusions from these studies were inconsistent. Two studies66,68 concluded that estimates obtained from regression analysis are similar to those obtained using propensity scoring. However, two studies64,67 also came to the opposite conclusion, that estimates obtained from regression analysis and propensity scoring differ significantly. One simulation study65 comparing the two methods considered propensity scoring to be the superior method, whereas another study62 found that propensity scoring is superior when the number of events per confounder is low. The disparate results of these studies means that it is difficult to draw conclusions regarding the relative performance of the different approaches, but Kurth et al. 64 make an important observation that potentially explains these different results. Kurth et al. 64 note that each method of adjustment answers subtly different questions as it makes different assumptions. This inevitably means that different methods of adjustment will yield different results. Kurth et al. 64 advise that researchers need to consider carefully the population for which an overall treatment estimate is most appropriate.
Regression analysis compared with instrumental variables analysis
Only two studies compared regression analysis with instrumental variable methods. 63,67 Crosby et al. 63 found that results from regression analysis and instrumental variable methods differed somewhat and suggested that instrumental variable methods are potentially superior. Stukel et al. 67 compared all three methods of adjustment (regression analysis, instrumental variables and propensity scoring) and concluded that instrumental variables may lead to less biased estimates of treatment effects. Although the evidence on instrumental variables is limited it nevertheless suggests that instrumental variables may offer advantages over other methods and may produce the least biased estimates.
Propensity scoring compared with instrumental variables analysis
A recently published systematic review found 55 comparisons (37 studies) between propensity scoring and instrumental variables analysis. 69 The review found there to be a slight/fair agreement between the methods (Cohen’s kappa coefficient = 0.21, 95% CI 0.00 to 0.41). In 23 cases (42%) the results were non-significant using one method but significant using the other method; using instrumental variables methods the results were non-significant in most cases (87%). The study authors recommended caution when interpreting the results of these analyses and that further research is needed to clarify the roles of these methods.
In addition to the seven empirical studies identified,62–67,69 a discussion paper by Biondi-Zoccai et al. 70 provides a useful overview of the alternative methods of adjustment. Biondi-Zoccai et al. 70 concluded that there is no clearly superior method, noting that ‘both standard multivariable methods and propensity scores have key limitations, and none is able to take into account unknown confounders’ (p. 736). Biondi-Zoccai et al. ,70 however, go on to suggest that propensity scoring methods may have advantages over regression methods when the sample size is small and that, although instrumental variables methods are not without their limitations, they are the only methods that allow for unobserved confounding to be adjusted for.
Adjustment methods applied specifically to single-arm trials
Hamre et al. 71 carried out a study aimed at improving methods to minimise bias in single-arm studies. Four bias factors were suppressed stepwise: attrition bias (by replacing missing values with the baseline value carried forward), bias from natural recovery (by sample restriction to patients with disease duration of 12 months), regression to the mean because of symptom-driven self-selection (by replacing baseline scores with scores 3 months before enrolment) and bias from adjunctive therapies (by sample restriction to patients not using adjunctive therapies). In the cohort analysed, these four bias factors could together explain a maximum of 37% of the 0- to 6-month improvement in disease score. However, this method has not been widely tested on other cohorts.
Quantifying bias in observational studies
| Study name | Sampling methods | Selection criteria | Number of studies included | Outcomes | Conclusions |
|---|---|---|---|---|---|
| Abraham et al., 200643 | A case–control study and RCT of the effectiveness of laproscopic surgery were carried out and the results compared | NA | One RCT and one NRS. No topic areas were in oncology | Direction of measured effects, statistical significance of effects, magnitude of measured effects | The results of a surgical historical control trial compared favourably with those of a RCT conducted under similar circumstances in determining the direction of measured effects but tended to yield larger estimates of effect magnitudes |
| Algra and Rothwell 201240 | PubMed and the National Library of Medicine were searched | Papers were eligible for inclusion if they reported results of case–control and cohort studies of the use of aspirin or non-steroidal anti-inflammatory drugs and risk of cancer | 12 oncology areas; six RCTs and 195 NRSs | Subjective assessment of similarity, correlation between estimated effect sizes | Results of methodologically rigorous NRSs are consistent with those obtained from RCTs, but sensitivity is particularly dependent on appropriately detailed recording and analysis of aspirin use |
| Benson and Hartz 200030 | NRSs published between 1985 and 1998 were searched for in MEDLINE and the Cochrane Database of Systematic Reviews. These were matched to RCTs investigating the same interventions by searching MEDLINE | NRSs were included if they met the following criteria: did not use an experimental design, included a control group, treatment was provided by a physician and assessed the difference between two treatments. No restrictions were applied to included RCTs other than that they were relevant to one of the included NRSs | 19 topic areas were included with one topic area in oncology; 53 RCTs and 83 NRSs | Overlap in CIs, subjective assessment of similarity of ORs | There was little evidence that effect estimates differed systematically between NRSs and RCTs. The authors noted that there may be clinically important differences and that their data set was relatively small |
| Beynon et al., 200831 | Randomly selected RCTs from the Cochrane Central Register of Controlled Trials, followed by searches for NRSs addressing the same topics | RCTs or NRSs reporting all-cause mortality | Six topic areas were included (not reported whether topic areas included oncology); 54 RCTs and 27 NRSs | Ratio of ORs | Suggested that NRSs overestimated treatment effects by 10% on average, compared with RCTs. However, these are only preliminary results |
| Britton et al., 199838 | Searched for studies comparing results from NRSs and RCTs in four areas: coronary artery bypass grafting, calcium antagonists, stroke units and malaria vaccines | The results of a RCT must be compared with those of a NRS or the results of several RCTs combined must be compared with those of several NRSs combined; the intervention must be the same and carried out in similar settings; the control arms of the studies must receive similar therapy; there must be comparable outcome measures, preferably valid and reliable | Three topic areas were included, none in oncology; 29 RCTs and five NRSs | Subjective assessment of differences | No evidence from stroke units or calcium antagonists to support using adjustment of observational data to close the gap on RCT data. Differences are probably due to patient characteristics |
| Concato et al., 200037 | Searched five major journals in MEDLINE between 1991 and 1995 | Meta-analyses of RCTs or NRSs. Excluded studies with historical controls and those that did not report point estimates | Five topic areas, one in oncology; 55 RCTs and 44 NRSs | Subjective comparison of point estimates and range of estimates obtained from the different study types | The results of NRSs are not systematically larger than those obtained from RCTs |
| Dahabreh et al., 201232 | MEDLINE search for NRSs and other studies of acute coronary syndromes. The search was limited to the top eight journals in cardiac and cardiovascular systems and the top four in medicine (general and internal) as defined by Thompson Reuters. RCTs were identified using searches of MEDLINE, the Cochrane Database of Systematic Reviews and relevant guidelines | Any NRS that used propensity scoring to estimate the treatment efficacy of therapeutic interventions administered to patients with acute coronary syndromes. RCTs were matched on the basis of interventions, patient populations and type of mortality outcomes investigated in the NRSs | 17 topic areas were included; 63 RCTs and 21 NRSs | Proportion of studies in which the ratio of NRS and RCT treatment effects was < 0.70 or > 1.43; number of comparisons in which the difference between RCTs and NRSs was statistically significantly different; how often the direction of the treatment effect estimated from the NRS and RCT evidence was the same | For the treatment of acute coronary syndromes, observational studies using propensity scoring methods produce treatment effect estimates that are of a more extreme magnitude than those from RCTs, although the differences are rarely statistically significant |
| Golder et al., 201341 | Searched multiple databases including the Cochrane Methodology Register, Database of Abstracts of Reviews of Effects and Web of Knowledge for methodological studies relating to the incorporation of adverse effects into systematic reviews | Any meta-analysis including RCTs and NRSs aimed at quantifying the relative adverse effects of a health-care intervention | 58 meta-analyses in 19 topic areas; 311 RCTs and 222 NRSs | Descriptive summary of overlap in CIs, direction of results and statistical significance of results | Empirical evidence from this overview indicates that there is no difference on average in the risk estimate of the adverse effects of an intervention derived from meta-analyses of RCTs and meta-analyses of observational studies. Some indication that case–control studies gave higher estimates of harm than RCTs |
| Hartz et al., 200533 | Used data obtained from previous studies | Included meta-analyses from two previous comparisons of RCTs and NRSs that included at least four observational studies | 10 topic areas, with none on oncology; 62 RCTs and 113 NRSs | Number of comparisons in which the difference between RCTs and NRSs was statistically significantly different, comparison of failure rates in the intervention and control groups, reporting of characteristics and efforts to address confounding in observational studies | Poor methodological reporting in NRSs prevents conclusions about relative size of effect estimates from being drawn |
| Ioannidis et al., 200134 | Searched MEDLINE and The Cochrane Library as well as previous studies and personal data | Meta-analyses of RCTs and NRSs in which one of the primary outcomes was of binary form and was analysed in the meta-analysis | 45 topic areas, with five topic areas in oncology; 240 RCTs and 168 NRSs | Correlation of summary effects obtained from randomised and non-randomised evidence, proportion of summary effects obtained from NRSs that were larger than those obtained from RCTs, number of comparisons in which the differences between RCTs and NRSs were statistically significant | Despite good correlation between estimates obtained from RCTs and those obtained from NRSs, NRSs on average tended to produce larger estimates of effectiveness |
| Lonjon et al., 201442 | A systematic search of MEDLINE and PubMed for NRSs. Sensitive searches of PubMed were then carried out to identify relevant RCTs. Searches for RCTs were limited to 5 years before and after the oldest and most recent NRSs were published | Prospective NRSs using propensity scoring to evaluate a surgical procedure | Evidence evaluating 31 clinical questions was included; 94 RCTs and 70 NRSs | Ratio of ORs | There was no statistically significant difference in treatment effect between NRSs using propensity scoring analysis and RCTs. Prospective NRSs with suitable and careful propensity scoring analysis can be relied on as evidence when RCTs are not possible |
| MacLehose et al., 200035 | The Cochrane Library, Database of Abstracts of Reviews of Effects and the Science Citation Index were searched. Additionally, references of relevant papers identified were searched and experts were consulted | Studies reporting estimates of effect from RCTs and NRSs. This could be from a single study or a pooled analysis from multiple studies | 14 topic areas, with five topic areas in oncology; number of RCTs and NRSs not reported | Ratio of relative risks, ratio of risk differences, comparison of number of events in the intervention and control groups | Concluded that when the quality of NRSs is high the disparity between outcomes between RCTs and NRSs is small. However, the authors advised caution about the generalisability of the findings |
| Sacks et al., 198239 | MEDLINE was searched for RCTs and NRSs addressing the same topic | Studies reporting estimates of effect from RCTs and NRSs | Not reported whether topic areas included oncology; 56 RCTs and 50 NRSs | Magnitude of differences, performance of control group | The data suggest that biases in patient selection may irretrievably weight the outcome of case–control studies in favour of new therapies |
| Shepherd et al., 200636 | A ‘comprehensive’ search of a number of databases was completed. No further information was provided | Systematic reviews published between 1999 and 2004; evaluated a policy intervention; included both RCTs and NRSs; and included quantitatively synthesised evidence | 16 meta-analyses from one topic area were included (not in oncology); number of RCTs and NRSs included was not reported | Proportion of reviews in which authors graded the results of RCTs and NRSs ‘similar’, ‘not similar’ or ‘mixed’ | Suggested that there may be some evidence of differences in results between RCTs and NRSs. However, noted that the lack of consistent criteria to evaluate such differences and lack of exploration into possible other explanations for any differences mean that it is not possible to draw any strong conclusions |
Appendix 3 Studies comparing bias adjustment methods
| Study | Objective | Methods compared | Summary of findings |
|---|---|---|---|
| Biondi-Zoccai et al., 201170 | Discussion piece comparing relative merits of alternative methods of adjusting for confounding bias in NRSs | Regression analysis, propensity scoring and instrumental variables | Propensity scoring may have advantages over other methods of adjustment, but all methods have important limitations |
| Cepeda et al., 200362 | Simulations study comparing logistic regression with propensity scoring in terms of bias, precision, empirical coverage probability, empirical power and robustness | Propensity scoring and logistic regression | Logistic regression is superior to propensity scoring when the number of events is greater than eight per confounder |
| Crosby et al., 201063 | To assess the potential usefulness of instrumental variables and ordinary least squares regression for addressing biases that can confound causal inferences in child-care research | Regression analysis and instrumental variables | Some discrepancies in results obtained using regression analysis and instrumental variables. Suggested that instrumental variables may be superior to regression analysis as a method of accounting for confounding bias |
| Kurth et al., 200664 | To assess the utility of different techniques to adjust for confounding | Propensity scoring and logistic regression | Different methods to control for confounding yielded extremely different treatment effect estimates. This disparity is suggested to be a result of each analysis answering a different question implicit or explicit to that method of adjustment |
| Laborde-Castérot et al., 201569 | Systematic review of studies comparing the performance of propensity scoring with that of instrumental variables analysis | Propensity scoring and instrumental variables analysis | There was slight/fair agreement between the methods (Cohen’s kappa coefficient = 0.21, 95% CI 0.00 to 0.41). In 42% of cases the results were non-significant using one method but significant using the other method; using instrumental variables methods the results were non-significant in 87% of cases |
| Martens et al., 200865 | Simulation study comparing the treatment effect estimates from propensity scoring and logistic regression | Propensity scoring and logistic regression | On average, estimates from propensity scoring are closer to the true marginal treatment effects than those generated by logistic regression |
| Shah et al., 200566 | Systematic review: to determine whether adjusting for confounder bias in observational studies using propensity scores gives different results from traditional regression modelling | Propensity scoring and standard regression analysis | Observational studies had similar results whether using traditional regression or propensity scores to adjust for confounding. Propensity scoring produced modestly more conservative estimates of effect on average |
| Stukel et al., 200767 | To compare four analytical methods for removing the effects of selection bias in observational studies | Regression analysis, propensity scoring (via matching and covariate adjustment) and instrumental variables | Estimates of the observational association of cardiac catheterisation with long-term acute myocardial infarction mortality are highly sensitive to analytical method used. Compared with standard modelling, instrumental variables analysis may produce less biased estimates of treatment effects |
| Sturmer et al., 200668 | To examine the use of propensity scoring methods and whether results obtained using propensity scoring differ substantially from those obtained using standard regression techniques | Propensity scoring and standard regression analysis | Little evidence that propensity scoring yielded substantially different estimates from those obtained using conventional multivariable methods |
Appendix 4 Studies on surrogate end points
| Study | Description/aim | Summary/findings |
|---|---|---|
| Use of surrogate measures as clinical end points | ||
| Davis et al., 201294 | The aim of the review was to examine the evidence available on the relationship between PFS/TTP and OS to support surrogate end points in advanced cancer | PFS and TTP are sometimes regarded as valid surrogate outcomes in the absence of a mature data set, but an estimate of OS is still needed within the economic analysis. The relationship between surrogate and OS can be used to populate the economic model. Unfortunately, when comparing studies, the lack of a standardised methodology or approach made it difficult for the authors to establish a relationship. The findings support Taylor and Elston89 in that any cost-effectiveness analysis based on a surrogate relationship between PFS and OS should be supported with a transparent explanation of how the relationship is quantified and presented alongside appropriate validation analysis and supporting literature |
| Katz 200496 | An article presenting the FDA regulatory context in relation to problems of interpretation of data from clinical trials in which unvalidated surrogate markers are used as primary outcomes | From a regulatory standpoint, the use of biomarkers and surrogate outcomes is supported when used in the appropriate context and they can be ‘shown to confer a clinical benefit to patients’ (p. 190). In research in which there are few if any available alternative treatment options, accelerated approval on the basis of the drug product having an effect on a surrogate end point may be granted by the FDA. The surrogate end point is expected to be based on research evidence other than survival or irreversible morbidity. Important points to note with regard to the regulation of surrogate outcome use in research include (p. 190):
|
| Bucher et al., 1999100 | The JAMA (Journal of the American Medical Association) Evidence-Based Medicine Working Group’s thoughts on the validity of surrogate outcome measures | For a surrogate to be valid there must be ‘no important effects of that intervention on the outcome of interest that are not mediated through, or captured by, the surrogate’ (p. 772). ‘Reliance on surrogate end points may be beneficial or harmful’ (p. 771) and the clinician needs to assess more than a single study to be confident that a surrogate end point is an adequate measure of outcome |
| Fleming and DeMets, 1996102 | The most commonly used guidance on the validity of surrogate end points | For the surrogate to be a reliable outcome measure it must be on the causal pathway from the intervention to the clinical outcome; this is the ‘setting that provides the greatest potential for the surrogate end point to be valid’ (p. 606). Reasons for failure when using surrogate outcomes include (p. 606):
|
| Health technology assessment and regulation | ||
| Elston and Taylor, 200988 | Paper published prior to the following HTA programme report by Taylor and Elston89 that specifically discusses the use of surrogate outcomes in cost-effectiveness models | This key discussion paper on the role of surrogate outcomes in cost-effectiveness models is often cited as such and includes the following recommendations (pp. 12–13): When it is not possible to base the clinical effectiveness and cost-effectiveness of the health technology on final patient outcome data and there is a requirement to use a surrogate outcome, the following should be undertaken:
|
| EUnetHTA 201387 | Recommendations for end points used in REAs of pharmaceuticals. EUnetHTA summarised its findings into eight recommendations for end points used in REAs of pharmaceuticals (pp. 6–7) | EUnetHTA summarised its findings into eight recommendations for end points used in REAs of pharmaceuticals (p. 6):
|
| Taylor and Elston, 200989 | Full HTA programme report to explore the use of surrogate outcomes in HTA and provide a basis for guidance for their future use, validation and reporting | The focus of this report was the use of surrogate outcomes in cost-effectiveness models within UK HTA programme reports. HTA programme reports were selected to examine the role of surrogate outcomes on cost-effectiveness models within the UK between 2005 and 2006. Selection was based on treatment effectiveness/efficacy, a cost-effectiveness model being included in the report and the cost-effectiveness model being primarily based on a surrogate outcome. Only one HTA reported the results of a systematic review that presented the evidence base for the association between surrogate and final outcomes. A key output from this work was the design of a schema used to evaluate the cost-effectiveness ratio of a surrogate for use in HTA reports |
| Key publications | ||
| Aziz et al., 201591 | A review of the current evidence for the treatment of metastatic castration-resistant prostate cancer and the advantages of using prognostic and/or predictive markers as surrogate end points in clinical trials | This review aimed to address the prospects for the future application and clinical use of biomarkers in the field of metastatic castration-resistant prostate cancer, including highlighting possible obstacles and solutions: ‘Suitable parameters serving as surrogates for intermediate and long-term endpoints and reflecting individual benefit, respectively, need to be identified and proven’ (p. 649) |
| Bujkiewicz et al., 2014116 | A study to examine the possibility of reducing the uncertainty around the clinical utility using multivariate meta-analysis | In the areas of highest priority in health care, decisions are required to be made on a short time scale. Therefore, alternative clinical outcomes, including surrogate end points, are increasingly being considered for use in evidence synthesis as part of economic evaluationp. 109The results of this research suggest that multivariate meta-analysis can improve the estimation of health utilities through mapping methods; however, more research is needed to determine the circumstances under which uncertainty is reduced |
| Buyse et al., 2007215 | In this study the authors examine the relationship between PFS and OS in a set of historical trials of colorectal cancer | The end point for trials assessing chemotherapy for advanced cancer was OS. This study examined the relevance of PFS as a surrogate end point in studies with prolonged follow-up periodsThe rank correlation coefficient between PFS and OS was equal to 0.82 (95% CI, 0.82 to 0.83). The correlation coefficient between treatment effects on PFS and on OS ranged from 0.99 (95% CI, 0.94 to 1.04) when all trials were considered to 0.74 (95% CI, 0.44 to 1.04)p. 5218Data are presented that suggest that additional measures are required to validate PFS as a surrogate for OS in colorectal cancer studies. Recommendations include a comparison between the effects of treatment on the true end point and the effects of treatment on the surrogate at the population level as well as quantifying the association between the true and the surrogate end points after taking treatment into account at the individual level |
| Ciani et al., 201398 | The aim of this research was to quantify and compare the treatment effect and risk of bias of trials reporting biomarkers or intermediate outcomes (surrogate outcomes) vs. trials using final patient-relevant primary outcomes | This meta-epidemiological study examined 84 trials using surrogate outcomes and 101 using patient-relevant outcomes:The preliminary results suggest trials reporting surrogate primary outcomes are more likely to report larger treatment effects than trials reporting final patient relevant primary outcomesp. 346As the study characteristics of the two trial types (surrogate outcomes and those reporting patient-relevant outcome) were well balanced and there were no differences in risk of bias, the authors conclude that the findings could not be explained by these factors |
| Ciani and Taylor, 201393 | Letter to the editor commenting on analytical approaches discussed by Hawkins et al.216 with regard to the use of surrogates in HTA and cost-effectiveness models | Presents opinion on the three main issues raised by Hawkins et al.216 with regard to best practice for the use of surrogate outcomes in HTA and cost-effectiveness models:
|
| Oviana et al., 2013217 | This case study aimed to illustrate the validation of complete cytogenic response (CCyR) and major molecular response (MMR) as surrogate outcomes for OS in chronic myelogenous leukaemia (CML) and how this evidence was used to inform NICE’s recommendation on the public funding of first-line treatments | A case study to provide insight into the use of surrogate outcome evidence in HTA. It is often a requirement that surrogate outcome data be based on the findings of RCTs and that the link between the treatment effects be apparent for both the surrogate outcome and the final outcome. The findings from this case study suggest that a lower level of evidence (i.e. observational association) may be acceptable |
| Ciani et al., 2014107 | The authors state that it is essential that candidate surrogate end points be properly validated but that there is no consensus on statistical methods for such validation. This study proposed a method for validation | A review of three statistical approaches to surrogate–end point validation (Elston and Taylor’s88 framework, the IQWiG framework and BSES3) was performed. The findings suggest that the strength of the association between two surrogates, PFS and TTP, and OS was generally low. The authors discuss the challenges of surrogate–end point validation and emphasise the importance of building consensus on the development of evaluation frameworks |
| Ciani et al., 2015108 | To examine the treatment effects on three surrogate end points vs. OS based on a meta-analysis of RCTs of drug interventions in advanced colorectal cancer (aCRC) | Univariate and multivariate random-effects meta-analyses were used to estimate pooled summary treatment effects reported in RCTs of pharmacological therapies in aCRC over a 10-year period. The treatment effects on PFS, TTP and tumour response rate were all compared with those for OS. The authors found larger treatment effects for the surrogate end points than for OS, with differences in median PFS/TTP higher than differences in OS by an average of 0.5 months. The authors conclude that the surrogacy relationships observed between PFS and TTP vs. OS in selected settings may not apply across other clinical classes or lines of therapy |
| De Gruttola et al., 1997218 | In this study the authors consider why surrogate end points can be unreliable and illustrate the importance of variability in evaluating the reliability of surrogates, with specific focus on HIV/AIDS treatment | In order for a marker to be a valid surrogate by the ‘Prentice’ definition (Prentice 1989), it must capture all of a treatment's beneficial and harmful effects[Although] partial surrogate markers that capture some of a treatment’s effect may provide insight into biologic mechanisms, analyses of the degree of surrogacy must be regarded with cautionp. 243 |
| Ellenberg and Hamilton 1989219 | A review of surrogate end points in clinical trials with a special focus on cancer | The authors argue that a surrogate end point is usually proposed on the basis of a biological rationale. Overall, the paper describes how, in cancer studies with survival time as the primary end point, the surrogate outcomes often used are tumour response, TTP and time to reappearance of disease, as these events occur earlier |
| Fleming et al., 1994220 | In this paper the authors discuss the applicability of surrogate end point criteria, with an emphasis on cancer and AIDS research settings | The authors conclude that using biological markers as axillary end points appears to provide an improvement in efficiency when assessing treatment effect, although only a modest improvement:There is potential for data on pertinent intermediate endpoints to play an auxiliary role in strengthening true endpoint analysesThe gains will be particularly evident when sufficient follow-up occurs to observe both auxiliary and true endpointsp. 965 |
| aFleming and DeMets, 1996102 | This paper provides examples to illustrate how surrogate end points may provide misleading assessments of actual effects of treatment on the health of patients | In theory, for a surrogate end point to be an effective substitute for the clinical outcome, effects of the intervention on the surrogate must reliably predict the overall effect on the clinical outcomep. 605The authors argue that in reality this is not always the case and believe that, although surrogate end points have value in guiding decisions about whether the intervention is promising enough to justify a large definitive trial with clinically meaningful outcomes, in definitive Phase III trials, unless the surrogate end point has already been rigorously established, the primary end point should be the clinical outcome used |
| Freedman et al., 1992221 | In this paper the authors expand on the work of Prentice222 with respect to the criteria for validation of intermediate variables or surrogate end points, by describing and discussing the statistical implementation of these criteria and by using the example of serum cholesterol as an intermediate end point for coronary heart disease | The authors state that a major obstacle in the study of the aetiology of chronic diseases and the development of effective prevention is the long latent period between the initiation of the disease and its diagnosis. Intermediate end points or surrogate end points are of interest in the study of several diseases as they can usually be observed prior to the clinical appearance of disease. In this paper an attempt is made to clarify the criteria that may be used to validate an intermediate end point. The authors found that the original general criteria were difficult to test in practice and as such found that the validation analysis would require some aspect of statistical modelling |
| Gøtzsche et al., 199695 | In this paper the authors review the justification for the use of surrogates and conclude that ‘reliance on them may be harmful’ | Surrogate outcomes can be any measurable event or value related to the disease and true outcome of interest. A surrogate in one trial may be the true outcome in another, depending on the purpose of the study. The authors discuss the risk of making assumptions based on surrogate outcome measures using a bone mineral density study as an example. They report that, although clinically relevant interventions would be expected to improve clinical outcomes, measures made on surrogate outcomes may be unreliable as a true measure of a positive effect |
| Herson 1989223 | An introduction to a series of papers that were presented at a meeting in 1989 to address the interest and controversy around surrogate end points | Long completion times are not only a component of overall cost, but also frequently result in the intervention under investigation being rendered obsolete by the time the trial terminates . . . The use of surrogate endpoints constitutes an effort to control the cost and completion time for clinical trialsp. 403 |
| Holloway and Dick, 2002224 | The authors hypothesise that therapeutic uncertainty in certain areas of clinical research exists because of the use of surrogate outcome measures in clinical trials | many of the clinical trial end points have been surrogate outcome measures rather than end points with clear and convincing value to patientsConsequences of using surrogate outcomes that have not been validated include ambiguous evidence and wasted resources as well as patient harm and missed opportunitiesp. 679 |
| Lerche la Cour et al., 201097 | The authors assessed RCT reports to evaluate the authors’ use of surrogate outcome measures | Of 626 published RCTs, 109 used a surrogate as a primary outcome. Of these trials, 62 clearly reported that the primary outcome was a surrogate. Only 38 also discussed the validity of the surrogate. The authors discuss the ‘shortcomings of surrogates’ in research and the use of such surrogates as primary outcomes in about one-fifth of published RCTs |
| Lessere et al., 2007103 | Review of biomarkers and surrogates to systematically evaluate the surrogacy status of such measures | The result of this research was a recommendation for a new quantitative surrogate validation level of evidence schema designed to evaluate the criteria for biomarkers and surrogate end points:Scores derived from 3 domains – the Target that the marker is being substituted for, the Design of the (best) evidence, and the Statistical strength – are additive. Penalties are then applied if there is serious counterevidenceMost stakeholders agreed that this operationalization of the National Institutes of Health definitions of biomarker, surrogate endpoint, and clinical endpoint was usefulp. 607 |
| Prentice 1989222 | The operational criteria for using surrogate end points in clinical trials are discussed | A criterion for surrogate use is proposed:In order that treatment comparisons based on a surrogate response variable have a meaningful implication for the corresponding true endpoint treatment comparison, a rather restrictive criterion is proposed for use of the adjective ‘surrogate’p. 431Operationally, it is suggested that the surrogate ‘capture’ any relationship between the clinical intervention and the true end point |
| Schievink et al., 2014101 | The authors consider the opinions of different stakeholders concerning the use of surrogate end points in the regulation of medicines | Online questionnaire of various stakeholder groups and medical specialties to inquire under what conditions a surrogate end point should be used:out of four proposed surrogates (blood pressure (BP), HbA1c, albuminuria, CRP) for cardiovascular outcomes or end-stage renal disease, only use of BP for cardiovascular outcomes was deemed moderately accurate (mean: 3.6, SD: 1.1). Specialists in cardiology or nephrology tended to be more positive about the use of surrogate endpointsp. 1 |
| Wilson et al., 201590 | Review of the benefits and limitations of ‘alternative’ trial end points in use for cancer research | Considers the issue of who defines what is a clinically meaningful outcome in cancer treatment: patients, clinicians or regulatory bodies and also highlights the variation in opinion between these groups |
| Zee et al., 2015225 | A study to assess and validate treatment effects with surrogate survival outcomes | The authors conclude that the resulting method is able to account for the uncertainty of surrogate outcomes. The proposed estimator is thought to outperform standard semiparametric survival analysis methods, saving on trial costs and leading to improvements in detecting treatment effects |
Appendix 5 Licensed treatments for relapsed/refractory B-cell acute lymphoblastic leukaemia
| Clofarabine: EMA,170 AWMSG – Evoltra;171 FDA – Clolar226 | |
|---|---|
| Nature of the disease and medicine | |
| Indication(s) | Relapsed or refractory paediatric ALL patients after receiving at least two previous regimens and when there is no other treatment option that is anticipated to result in a durable response |
| How does it work? | Clofarabine is a purine nucleoside antimetabolite (affects DNA elongation, synthesis and repair) |
| Is it claiming to meet an otherwise unmet need? | Yes (indicated in patients in whom no other durable treatment options exist) |
| How is it given? | Intravenous infusion for 5 consecutive days every 2–6 weeks. Dose for paediatrics is 52 mg/m2 over 2 hours |
| Are there any comparator treatments? | Not at the time of evaluation (other than palliative care) |
| Is there any mention of the intervention evolving over time? | No (not a regenerative medicine) |
| Is there any mention of persistence of the treatment within the patient? | No (not a regenerative medicine) |
| Trial design | Only a single efficacy trial available |
| Trial description | CLO 212: multicentre single-arm Phase II trial170 |
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Paediatric patients aged from ≥ 1 to ≤ 21 years |
| Trial size/total trial population | 61 patients |
| Length of follow-up | Data cut-off point was 2 years after the start of recruitment |
| Control/comparator used | Results for clofarabine were compared with rates expected by expert clinical evaluation. No suitable published studies were available to provide appropriate comparator data |
| How is the control/comparator constructed? Source of comparative data? Confounding? | Median survival of 9–10 weeks was estimated (using German and Dutch cancer registries) |
| Outcomes | |
| Response outcome 1 | Overall remission rate (incorporates CR and CR without platelet recovery |
| Response outcome 2 | PR |
| Response outcome 3 | Duration of remission |
| Response outcome 4 | OS |
| Adverse events | Nausea and vomiting in around two-thirds of patients and febrile neutropenia in around one-third. Two patients stopped treatment because of a serious adverse event, although four deaths were considered to be related to clofarabine |
| Surrogate or intermediate clinical outcome? | Remission outcomes |
| Real clinical outcome? | OS |
| Summary of evidence | The overall remission rate was 12/61 (20%). In total, 10/61 patients (16%) went on to receive HSCT. Median survival (all patients) was 17.7 weeks. For the seven patients achieving a CR, the median OS was 66.6 weeks (95% CI 53.7 to 89.4 weeks). The effect in terms of remission and facilitating HSCT was considered to be clinically significant and may have a significant impact on long-term treatment outcome; 8/18 responders received HSCT |
| Overall evidence base provided: trial result summary | |
| Estimate of HRQoL | No information reported |
| Product information and registration | |
| Any issues of scale-up for the product? | No – not a regenerative medicine |
| Is further evidence requested for EMA/FDA approval? | Specific risk-minimisation activities were required. Prescribers were also encouraged to participate in a voluntary adverse event reporting system. In particular, monitoring of systemic inflammatory response syndrome was important |
| Any additional information provided? | The EMA review stated that, given the efficacy seen early on in the clinical programme, studies using a placebo comparator were considered to be clinically unethical. Active comparator studies were not appropriate as there were no other recognised therapeutic options available. ‘The indication is encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive data on clinical efficacy and safety’ (p. 35).170 Marketing authorisation was therefore granted ‘under exceptional circumstances’. The AWMSG recommended use only if the intended use was as a bridge to HSCT (should not be used with palliative intent) |
| Blincyto (blinatumomab): FDA assessment227 | |
| Nature of the disease and medicine | |
| Indication(s) | Ph– relapsed or refractory B-cell precursor ALL |
| How does it work? | Blinatumomab is a monoclonal antibody (a type of protein) that has been designed to specifically recognise and attach to CD19 proteins and to the T-cell receptor/CD3 complex, which is responsible for the activation of some cells of the immune system (the body’s natural defences) called T-cells. By attaching to the cancer cells and the T-cell receptor/CD3 complex blinatumomab is expected to stimulate the T-cells to kill the cancer cells |
| Is it claiming to meet an otherwise unmet need? | Clofarabine and Marqibo already exist as current treatments, although blinatumomab might be a significant alternative because it works in a different way from existing treatments |
| How is it given? | Intravenous infusion over 4 weeks, with 2-week interval between each treatment cycle |
| Are there any comparator treatments? | Yes, clofarabine and Marqibo have been granted accelerated approval by the FDA for a similar indication prior to blinatumomab |
| Is there an issue of the intervention evolving over time? | No (not a regenerative medicine) |
| Is there an issue of persistence? | No (not a regenerative medicine) |
| Trial design | Trial MT103–211 (with supporting data from MT103–206)227 |
| Trial description | Single-arm pivotal Phase II trial (MT103–211) |
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Adults, mean age 39 years |
| Trial size/total trial population | n = 189 (MT103–211) + 36 (MT103–206) |
| Length of follow-up | 24 months |
| Control/comparator used | Historical controls |
| How is the control/comparator constructed? Source of comparative data? Confounding? | Analysis of patient-level data from 694 historical controls: the CR + CRh rate was 24% |
| Outcomes | Trial MT103–211 (with supporting data from MT103–206) |
| Response outcome 1 | Rate of CR + CRh |
| Response outcome 2 | RFS |
| Response outcome 3 | OS |
| Response outcome 4 | HSCT |
| Adverse events | Boxed warning for CRS and neurological toxicities (including seizures) |
| Surrogate or intermediate clinical outcome | CR + CRh, RFS |
| Real clinical outcome | OS |
| Summary of evidence | |
| Overall evidence base provided | CR + CRh rate was 42% (95% CI 34% to 49%). Median RFS was 6.7 months (95% CI < 0.1 to 16.5 months) |
| HRQoL measure | No data |
| Product information and registration | |
| Any issues of scale-up for the product? | No (not a regenerative medicine) |
| Is further evidence requested for EMA/FDA approval? | A confirmatory Phase III RCT comparing blinatumomab with standard care chemotherapy in the same population was ongoing at the time of submission.180 Randomisation method used will ensure a 2 : 1 treatment ratio (i.e. more patients will receive blinatumomab than will receive standard care). OS is the primary end point. In addition, there are four post-marketing commitments to test the stability of the product once stored |
| Any additional information provided? | |
| Marqibo (vincristine sulphate liposomes injection): FDA assessment173 | |
| Nature of the disease and medicine | |
| Indication(s) | Adult ALL patients with Ph– second or greater relapse or who are refractory to treatment |
| How does it work? | Targeted delivery of vincristine is achieved through encapsulating it in nanoparticle liposomes. This allows increased vincristine doses to be achieved without the associated increases in toxicity (dose-limiting neuropathy) |
| Is it claiming to meet an otherwise unmet need? | Yes (no other durable treatment options existed at the time for this indication) |
| How is it given? | Intravenously for 1 hour every week. Four doses = one course of treatment |
| Are there any comparator treatments? | Not at the time of evaluation (other than palliative care) |
| Is there any mention of the intervention evolving over time? | No |
| Is there any mention of persistence of the treatment within the patient? | No |
| Trial design | Only one trial using the correct dose (HBS407). Supporting evidence from a Phase I/II, multicentre, dose-escalation study (VSLI-06) was also submitted173 |
| Trial description | HBS407: multicentre, single-arm, Phase II trial (minimax two-stage design used for sample size) |
| Trial population (adults/children/all); any further specifics of disease not covered in ‘Indication’ | Adults only. All patients had previously been treated with standard vincristine |
| Trial size/total trial population | 65 patients |
| Length of follow-up | Up to 5 years (planned) |
| Control/comparator used | Data from relevant patients included in a retrospective study were identified and used as a historical control group. Median OS was < 3 months |
| How is the control/comparator constructed? Source of comparative data? Confounding? | |
| Outcomes | |
| Response outcome 1 | Rate of CR + CR with incomplete blood count recovery (CRi) |
| Response outcome 2 | Duration of CR + Cri |
| Response outcome 3 | OS |
| Adverse events | Most frequent were constipation (57%) and nausea (52%). Around one-third of patients had a neuropathy adverse event ≥ grade 3 |
| Surrogate or intermediate clinical outcome | CR, Cri |
| Real clinical outcome | OS |
| Summary of evidence | |
| Overall evidence base provided: trial result summary | 10/65 patients (15%) achieved CR or CRi. Five of the eight FDA-confirmed CR + CRi patients had a duration of response of < 1 month (median duration of response for these eight patients was 28 days). Five patients who lived for ≥ 1 year were considered potential long-term survivors; two of the five did not respond to Marqibo. In total, 12 patients received a stem cell transplant; seven patients did not achieve CR or CRi with Marqibo but six of these received other chemotherapy and had a subsequent stem cell transplant |
| Estimate/measure of effect (HRQoL) | No information |
| Product information and registration | |
| Any issues of scale-up for the product? | No |
| Is further evidence requested for EMA/FDA approval? | Post-approval confirmatory commitment study: a multicentre, open-label RCT of standard vincristine vs. Marqibo in adults aged > 60 years with newly diagnosed, untreated Ph– ALL. Proposed sample size of 348 |
| Any additional information provided? | ‘Accelerated approval’ regulations were used. Final vote was ‘yes’, n = 7; ‘no’, n = 4; ‘abstain’, n = 2. Members discussed the liposomal formulation of the product and its possible impact on the effectiveness of the drug; they consistently stated that the proposed Phase III trial was critical in assessing the benefit of Marqibo. Some members indicated that the trial should be completed before approval, whereas several indicated that accelerated approval may be appropriate but with the expectation that this approval would be withdrawn if the Phase III trial failed to confirm clinical benefit. One member stated that the ‘yes’ vote was more an indictment of the lack of other options than enthusiasm about Marqibo |
Appendix 6 Summary of patient characteristics in previously published multivariate prognostic models of acute lymphocytic (lymphoblastic) leukaemia
| Study | Trial | T or B cell | Sample size of interest (for prognostic model) | Duration of follow-up (months), median | Sex (% female) | Children/adults | CNS disease (%) | Proportion with prior transplantation | Number of relapses |
|---|---|---|---|---|---|---|---|---|---|
| Fielding et al., 2007156 | UKALL 12/ECOG 2993 | Both | 609 | 49 | 37 | Adults (15–60 years) | 9 | 0 | 1 |
| Ko et al., 2010152 | Therapeutic Advances in Childhood Leukemia Consortium (TACL) T2005-002 | Both | 225 | NR | 41.30 | Children (0–21 years) | 8.30 | NR | 1 |
| Nguyen et al., 2008228 | Children’s Oncology Group clinical trials (10 trials) | Both | 1961 | 51.7 | 44 | Children | > 20.9 | NR | 1 |
| Tavernier et al., 2007229 | Leucémie Aiguës Lymphoblastique de l’Adulte (LALA)-94 trial | Both | 421 | 51.6 | 33 | Adults (15–55 years) | 15 | NR | 1 |
| Oriol et al., 2009230 | Four PETHEMA (Programa Español de Tratamiento en Hematologia) trials | Both | 263 | NR | 43 | Adults (15–70 years) | < 7 | NR | 1 |
| Schrappe et al., 2012231 | Case based (14 co-operative study groups) | Both | 1041 | 99.6 | 39 | Children (0–18 years) | 6 | NR | Induction failure |
| Thomas et al., 1999232 | MCACC (MD Anderson Cancer Center) cases | Both | 314 | NR | 39 | Adults | 15 | NR | 1 |
Appendix 7 Review of previous economic evaluations in acute lymphocytic (lymphoblastic) leukaemia
Database
Ovid MEDLINE In-Process & Other Non-Indexed Citations and Ovid MEDLINE (1946 to present).
Search strategy
-
acute lymphoblastic leukaemia.ti,ab. (4080)
-
acute lymphoblastic leukaemia.ti,ab. (19,141)
-
Leukaemia, Lymphocytic, Chronic, B-Cell/ (12,539)
-
1 or 2 or 3 (35,395)
-
‘ALL R3’.ti,ab. (7)
-
ALLR3.ti,ab. (2)
-
‘ALL R2’.ti,ab. (31)
-
ALLR2.ti,ab. (0)
-
5 or 6 or 7 or 8 (39)
-
4 or 9 (35,431)
-
economics/ (26,627)
-
exp ‘costs and cost analysis’/or Cost Allocation/or Cost-Benefit Analysis/or Cost Control/or Cost of Illness/or Cost Sharing/or Health Care Costs/or Health Expenditures/ (188,408)
-
economics, dental/ (1861)
-
exp ‘economics, hospital’/or Hospital Charges/or Hospital Costs/ (20,315)
-
economics, medical/ (8619)
-
economics, nursing/ (3916)
-
economics, pharmaceutical/ (2575)
-
(economic$or cost$or price or prices or pricing or pharmacoeconomic$).tw. (536,282)
-
(expenditure$not energy).tw. (20,070)
-
(value adj1 money).tw. (27)
-
budget$.tw. (20,416)
-
11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 (665,721)
-
((energy or oxygen) adj cost).ti,ab. (3059)
-
(metabolic adj cost).ti,ab. (925)
-
((energy or oxygen) adj expenditure).ti,ab. (18,354)
-
or/23–25 (21,563)
-
22 not 26 (660,845)
-
letter.pt. (882,177)
-
editorial.pt. (379,418)
-
historical article.pt. (317,175)
-
28 or 29 or 30 (1,563,299)
-
27 not 31 (630,599)
-
exp animals/not humans/ (4,056,152)
-
32 not 33 (586,163)
-
10 and 34 (489)
Appendix 8 Incidence of relevant population estimate
To estimate the budget impact associated with CAR T-cell therapy it is necessary to estimate the incident population eligible for treatment per year. No observed estimates were available because of the small numbers of patients involved and the late stage of treatment; therefore, an estimate was constructed based on a three-step calculation:
-
estimate of new ALL diagnoses per year in the UK in the age range of interest
-
adjustment for B-ALL
-
adjustment for patients who have relapsed (with no further planned curative chemotherapy or HSCT) or who are refractory to standard chemotherapy.
The ONS publishes registrations of newly diagnosed cases of cancer, shown for ALL in England in Table 47. 233 The population of relevance is assumed to be those aged from birth to 30 years inclusive, consistent with the definition of children and young adults in the study by Lee et al. ,164 giving an annual incidence estimate of 460.
| Sex | Age (years) | Total | ||||||
|---|---|---|---|---|---|---|---|---|
| < 1 | 1–4 | 5–9 | 10–14 | 15–19 | 20–24 | 25–29 | ||
| Male | 6 | 87 | 60 | 36 | 33 | 10 | 14 | 246 |
| Female | 9 | 102 | 48 | 22 | 18 | 6 | 9 | 214 |
| Total | 15 | 189 | 108 | 58 | 51 | 16 | 23 | 460 |
Of these ALL incident cases, an estimated 80–85% are B-ALL. 234 For simplicity, we assumed that 82.5% are B-ALL, giving a B-ALL incidence of 379.5. Finally, Fuster157 estimated that 20% of children experience relapse after current frontline therapy. In addition, Fuster157 finds that, of this population, 50% will not respond to salvage therapy or will suffer a second relapse, giving a population incidence of relevance of 37.95 per annum in England.
Appendix 9 Full list of advisory group and National Institute for Health and Care Excellence panel members
| Andrew Stevens (chairperson) | Professor of Public Health, University of Birmingham |
| Natalie Mount | Chief Clinical Officer, Cell Therapy Catapult |
| Ian McKay | Senior Scientific Officer, Genomics Science and Emerging Therapies, Department of Health |
| Nick Crabb | Programme Director Scientific Affairs, NICE |
| Robert Hawkins | Professor of Medical Oncology, University of Manchester |
| Panos Kefalas | Head of Health Economics and Market Access, Cell Therapy Catapult |
| Matthew Taylor | Director of York Health Economics Consortium, University of York |
| Philip Newsome | Professor of Experimental Hepatology, University of Birmingham |
| Chris Mason | Professor of Regenerative Medicine Bioprocessing, UCL |
| Angela Blake | Head of Health and Value, Pfizer UK |
| Andrew Webster | Director of the Science and Technology Studies Unit, University of York |
| Paul Catchpole | Director of Value and Access, ABPI |
| Michael Hunt | Chief Financial Officer, ReNeuron |
| Siobhan Connor | Clinical Effectiveness Executive, BUPA |
| Holger Mueller | Senior Vice President, Commercial Operations, Cell Medica |
| Ahmed Syed | NHS England |
| Claude Schmitt | Head of Market Access, Rare Diseases, GSK |
| Angela Crossman | Global Market Access Director, Gene Therapy, GSK |
| Helen Tayton-Martin | Chief Operating Officer, Adaptimmune |
| Matthew Durdy | Chief Business Officer, Cell Therapy Catapult |
| Andrew Stevens (chairperson of the panel) | Chair of Appraisal Committee C, Professor of Public Health, University of Birmingham |
| Peter Jackson | Consultant Physician and Honorary Reader in Clinical Pharmacology and Therapeutics, Sheffield Teaching Hospitals NHS Foundation Trust |
| Gary McVeigh | Professor of Cardiovascular Medicine, Queen’s University Belfast, and Consultant Physician, Belfast Health and Social Care Trust |
| Peter Selby | Consultant Physician, Central Manchester University Hospitals NHS Foundation Trust |
| Jonathan Michaels | Honorary Professor of Clinical Decision Science, University of Sheffield |
| Mark Sculpher | Professor of Health Economics, University of York |
| Allan Wailoo | Professor of Health Economics and Director of NICE DSU, University of Sheffield |
| John Cairns | Professor of Health Economics, Public Health and Policy, London School of Hygiene and Tropical Medicine |
| Norman Waugh | Professor of Public Health, Warwick Medical School |
| Paul Miller | Director, Payer Evidence, AstraZeneca |
| Chris O’Regan | Head of Health Technology and Outcomes Research, Merck Sharp & Dohme |
| Danielle Preedy | Assistant Director, NIHR Evaluation, Trials and Studies Coordinating Centre |
| David Chandler | Chief Executive, nominated by Psoriasis and Psoriatic Arthritis Alliance |
Glossary
- Adverse effect
- An abnormal or harmful effect caused by and attributable to exposure to a drug, which is indicated by some result such as death, a physical symptom or visible illness. An effect may be classed as adverse if it causes functional or anatomical damage, causes irreversible change in the homeostasis of the organism or increases the susceptibility of the organism to other chemical or biological stress.
- Antigen CD19 (cluster of differentiation 19)
- A protein present on B-cell leukaemias (as well as on healthy B cells).
- Aplasia
- The failure of an organ or tissue to develop or to function normally.
- Autologous
- Derived from an individual’s own cells.
- Between-study variance
- A measure of statistical heterogeneity that depends on the scale of the outcome measured. It represents the variation in reported study effects over and above the variation expected given the within-study variation.
- Biologic therapy (biological)
- Medical preparation derived from a living organism. Includes anti-tumour necrosis factor drugs and other new drugs that target pathologically active T-cells.
- Consolidation chemotherapy
- Chemotherapy given once a remission is achieved, to sustain a remission.
- Cost–benefit analysis
- An economic analysis that converts the effects or consequences of interventions into the same monetary terms as the costs and compares them using a measure of net benefit or a cost–benefit ratio.
- Cost-effectiveness analysis
- An economic analysis that expresses the effects or consequences of interventions on a single dimension. This would normally be expressed in ‘natural’ units (e.g. cases cured, life-years gained, additional strokes prevented). The difference between interventions in terms of costs and effects is typically expressed as an incremental cost-effectiveness ratio (e.g. the incremental cost per life-year gained).
- Cost–utility analysis
- The same as a cost-effectiveness analysis but the effects or consequences of interventions are expressed in generic units of health gain, usually quality-adjusted life-years.
- Credible interval
- In Bayesian statistics, a credible interval is a posterior probability interval estimation that incorporates problem-specific contextual information from the prior distribution. Credible intervals are used for purposes similar to those that confidence intervals are used for in frequentist statistics.
- Fixed-effect model
- A statistical model that stipulates that the units under analysis (e.g. people in a trial or study in a meta-analysis) are the ones of interest and thus constitute the entire population of units. Only within-study variation is taken to influence the uncertainty of the results (as reflected in the confidence interval) of a meta-analysis using a fixed-effect model.
- Graft rejection
- The rejection of transplanted organs as a result of humoral and cell-mediated responses by the recipient to specific antigens present in the donor tissue.
- Haematological cancers
- Cancer of blood cells, which can be subdivided into three main diseases: leukaemia, lymphoma and myeloma.
- Heterogeneity
- In systematic reviews heterogeneity refers to variability or differences between studies in the estimates of effects. A distinction is sometimes made between ‘statistical heterogeneity’ (differences in the reported effects), ‘methodological heterogeneity’ (differences in study design) and ‘clinical heterogeneity’ (differences between studies in key characteristics of the participants, interventions or outcome measures).
- Immune reconstitution
- A condition in which the patient’s immune system begins to recover after treatment.
- Immune reconstitution inflammatory syndrome
- A condition in which the patient’s immune system begins to recover after treatment but then reacts later with an overwhelming inflammatory response.
- Immunoconjugate
- An antibody joined to a second molecule, usually a toxin, radioisotope or label, for use in immunotherapy.
- Immunophenotype
- The protein type expressed by cells.
- Immunotherapy
- A treatment designed to boost the body’s natural defences to fight cancer by utilising material either from the body or produced in vitro to improve, target or restore immune system function.
- Immunotoxin
- A protein that consists of a targeting portion linked to a toxin, which will bind to a cell and cause endocytosis, allowing the toxin to kill the cell.
- Intention to treat
- An intention-to-treat analysis is one in which all of the participants in a trial are analysed according to the intervention to which they were allocated, whether they received it or not.
- I-squared (I2)
- A measure of ‘statistical heterogeneity’ (differences in the reported effects). It varies between 0 and 1, where 0 indicates that the differences in reported effects are entirely consistent with the within-study uncertainty, and 1 indicates that the differences in reported effects are entirely explained by study characteristics that vary across studies.
- Medical Devices Directive
- The Medical Devices Directive is a directive relating to the safety and performance of medical devices, which were harmonised in the European Union in the 1990s.
- Monoclonal antibody
- An antibody produced in a laboratory from a single clone that recognises only one antigen.
- Open-label study
- A type of study in which both participants and researchers know which treatment is being administered.
- Orphan designation/status
- Based on the European Medicines Agency criteria, a medicine can qualify for orphan status if it is intended for the treatment, prevention or diagnosis of a disease that is life-threatening or chronically debilitating; the prevalence of the condition in the European Union is not more than 5 in 10,000 or it must be unlikely that marketing of the medicine would generate sufficient returns to justify the investment needed for its development; and no satisfactory method of diagnosis, prevention or treatment of the condition concerned can be authorised or, if such a method exists, the medicine must be of significant benefit to those affected by the condition.
- Persistence
- In treatment intended for direct in vivo administration, persistence may describe how long the product is effective in treating a targeted disease. It may also be used to refer to the persistence of the product, for example gene expression or any permanent changes, within the patient as a result of treatment with the product.
- Pharmacodynamic effects
- The study of how a drug behaves in the body.
- Pharmacokinetic effects
- The study of the effect that the body has on a drug.
- Phase I study
- Researchers test a new drug or treatment in a small group of people for the first time to evaluate its safety, determine a safe dosage range and identify side effects.
- Phase II study
- The drug or treatment is given to a larger group of people to see if it is effective and to further evaluate its safety.
- Phase III study
- The drug or treatment is given to large groups of people to confirm its effectiveness, monitor side effects, compare it with commonly used treatments and collect information that will allow the drug or treatment to be used safely.
- Phase IV study
- Study carried out after the drug or treatment has been marketed to gather information on the drug’s effect in various populations and any side effects associated with long-term use.
- Placebo
- An inactive substance or procedure administered to a patient, usually to compare its effects with those of a real drug or other intervention, but sometimes for the psychological benefit to the patient through a belief that he or she is receiving treatment.
- Quality-adjusted life-year
- An index of health gain in which survival duration is weighted or adjusted by the patient’s quality of life during the survival period. Quality-adjusted life-years have the advantage of incorporating changes in both quantity (mortality) and quality (morbidity) of life.
- Quality of life
- A concept incorporating all of the factors that might impact on an individual’s life, including factors such as the absence of disease or infirmity as well as other factors that might affect their physical, mental and social well-being.
- Random-effects model
- A statistical model sometimes used in meta-analysis in which both within-study sampling error (variance) and between-study variation are included in the assessment of the uncertainty (confidence interval) of the results of a meta-analysis.
- Randomised controlled trial
- An experiment in which investigators randomly allocate eligible people into intervention groups to receive or not receive one or more interventions that are being compared.
- Refractory
- A disease that does not respond to attempted forms of treatment.
- Regenerative medicine
- A field of research and clinical applications dealing with the process of replacing or regenerating human cells, tissues or organs to restore or establish normal function.
- Relative risk (synonym: risk ratio)
- The ratio of risk in the intervention group to risk in the control group. The risk (proportion, probability or rate) is the ratio of people with an event in a group to the total number in the group. A relative risk of 1 indicates no difference between comparison groups. For undesirable outcomes, a relative risk that is < 1 indicates that the intervention was effective in reducing the risk of that outcome.
- Salvage chemotherapy
- Chemotherapy given to a patient when all other treatment options are exhausted.
- Sensitivity analysis
- An analysis used to determine how sensitive the results of a study or systematic review are to changes in how it was carried out. Sensitivity analyses are used to assess how robust the results are to uncertain decisions or assumptions about the data and the methods that were used.
- Time to relapse
- Length of first remission.
- Weighted mean difference (in meta-analysis)
- A method of meta-analysis used to combine measures on continuous scales when the mean, standard deviation and sample size in each group are known. The weight given to each study is determined by the precision of its estimate of effect and is equal to the inverse of the variance. This method assumes that all of the trials have measured the outcome on the same scale.
List of abbreviations
- ACD
- appraisal consultation document
- ACI
- autologous chondrocyte implantation
- ADAPT SMART
- Accelerated Development of Appropriate Patient Therapies: a Sustainable, Multi-stakeholder Approach from Research to Treatment-outcomes
- AG
- Assessment Group
- AIC
- Akaike information criterion
- AIDS
- acquired immunodeficiency syndrome
- ALL
- acute lymphocytic (lymphoblastic) leukaemia
- ASCT
- allogeneic stem cell transplantation
- ATMP
- advanced-therapy medicinal product
- AWMSG
- All Wales Medicines Strategy Group
- AWR
- approve with research
- B-ALL
- B-cell acute lymphocytic (lymphoblastic) leukaemia
- BSES3
- Biomarker Surrogacy Evaluation Schema
- CAR
- chimeric antigen receptor
- CAT
- Committee for Advanced Therapies
- CD
- cluster of differentiation
- CDF
- Cancer Drugs Fund
- CED
- coverage with evidence development
- CHMP
- Committee for Medicinal Products for Human Use
- CHRI
- Child Health Rating Inventory
- CI
- confidence interval
- CR
- complete remission (or response)
- CRh
- complete remission (or response) with incomplete haematological recovery
- CRP
- C-reactive protein
- CRS
- cytokine release syndrome
- DFS
- disease-free survival
- DNA
- deoxyribonucleic acid
- DSU
- Decision Support Unit
- EAMS
- early access to medicines scheme
- EFS
- event-free survival
- EMA
- European Medicines Agency
- EoL
- end of life
- EPAR
- European Public Assessment Report
- EQ-5D
- EuroQol-5 Dimensions
- ERG
- Evidence Review Group
- EU
- European Union
- EUnetHTA
- European Network for Health Technology Assessment
- EVPI
- expected value of perfect information
- FAD
- final appraisal determination
- FAR
- final appraisal recommendation
- FDA
- Food and Drug Administration (US)
- FLAG-IDA
- fludarabine, cytarabine, granulocyte colony-stimulating factor and idarubicin
- HAQ
- Health Assessment Questionnaire
- HbA1c
- glycated haemoglobin
- HIV
- human immunodeficiency virus
- HR
- hazard ratio
- HRG
- Healthcare Resource Group
- HRQoL
- health-related quality of life
- HSCT
- haematopoietic stem cell transplantation
- HTA
- health technology assessment
- HUI
- Health Utilities Index
- ICER
- incremental cost-effectiveness ratio
- IMPACT
- Immunotherapy for Prostate Adenocarcinoma Treatment
- IPD
- individual patient data
- IPTW
- inverse probability of treatment weighting
- IQWiG
- German Institute of Quality and Efficiency in Health Care
- ISPOR
- International Society for Pharmacoeconomics and Outcomes Research
- IVIG
- intravenous immunoglobulin
- KM
- Kaplan–Meier
- MACI
- matrix-applied characterised autologous cultured chondrocyte implant
- MAPPs
- Medicines Adaptive Pathways to Patients
- MEA
- managed entry agreement
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRD
- minimal residual disease
- NHE
- net health effect
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NRS
- non-randomised study
- OIR
- only in research
- ONS
- Office for National Statistics
- OR
- odds ratio
- OS
- overall survival
- PAS
- patient access scheme
- PFS
- progression-free survival
- Ph+
- Philadelphia chromosome positive
- Ph–
- Philadelphia chromosome negative
- PIM
- promising innovative medicine
- PR
- partial response/remission
- PSA
- prostate-specific antigen
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RCT
- randomised controlled trial
- REA
- relative effectiveness assessment
- RFS
- relapse-free survival
- RMEG
- Regenerative Medicine Expert Group
- SF-36
- Short Form questionnaire-36 items
- SMR
- standardised mortality ratio
- TA
- technology appraisal
- T-ALL
- T-cell acute lymphocytic (lymphoblastic) leukaemia
- TKI
- tyrosine kinase inhibitor
- TPP
- target product profile
- TTP
- time to progression