
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 11/129/148. The contractual start date was in June 2014. The draft report began editorial review in October 2016 and was accepted for publication in February 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Intravenous immunoglobulin was provided by Biotest AG, Germany, and, should any commercial opportunity arise, the industrial partner has an option for an exclusive licence from the sponsor (Guy’s and St Thomas’ NHS Foundation Trust) and the potential for a revenue-sharing arrangement. Michael Absoud serves on the data safety monitoring board for a study sponsored by Neurim Pharmaceuticals, has received consultation fees from Novartis and is on the editorial advisory board for the International Journal of Language & Communication Disorders. Peter Brex has received fees for speaking and consulting from Biogen Idec, Roche, Sanofi Genzyme, Teva Pharmaceuticals Industries Ltd and Merck Serono. Olga Ciccarelli serves as consultant for Novartis, Biogen Inc. and GE Healthcare and is an Associate Editor of Neurology. Gavin Giovannoni has received consultation and speaking fees from Biogen Idec, GlaxoSmithKline, Merck Serono, Novartis, Sanofi Genzyme and Synthon BV and is on the steering committee for studies sponsored by AbbVie, Biogen Idec, Novartis, Teva Pharmaceuticals Industries Ltd and Roche. Jackie Palace serves on the scientific advisory board for the Charcot Foundation, has performed advisory work for Biogen Idec, Merck Serono, Bayer Schering Pharma, Novartis, Teva Pharmaceuticals Industries Ltd, Gilenya, Ono Pharmaceutical Co., Primary i-Research, Alexion Pharmaceuticals Inc. and Chugai Pharma Europe, receives research support from Merck Serono, Bayer Schering Pharma, Biogen Idec and Teva Pharmaceuticals Industries Ltd and has received conference expenses from Novartis, Merck Serono and Biogen Idec. Michael Pike has received a meeting support grant from EUROIMMUN. Anu Jacob is supported by the NHS National Specialised Commissioning Group for Neuromyelitis Optica, has been a consultant for Shire, Alexion Pharmaceuticals Inc. and Chugai Pharmaceutical Co. Ltd and has received research funding from Biogen Inc. and Alexion Pharmaceuticals Inc. Ming Lim has received consultation fees from CSL Behring and travel grants from Merck Serono and has been awarded educational grants to organise meetings by Novartis, Biogen Idec, Merck Serono and Bayer.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Absoud et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Background
Transverse myelitis (TM) is an immune-mediated disorder of the spinal cord affecting children and adults and is usually characterised by a rapid onset of paraplegia or tetraplegia, loss of sensation and sphincter disturbance. 1 Histologically, TM is characterised by spinal cord immune cellular infiltration, with pathogenesis mediated by a variety of immunological mechanisms. 1 Attacks usually develop over 24 hours and, in some cases, can progress rapidly to a potentially devastating and life-threatening condition. The severity of symptoms depends on the spinal cord level affected, with patients with high cervical lesions often requiring intensive care support to maintain respiratory function. Patients can recover fully from TM but a large number are left with significant disabilities. Recovery occurs within weeks of the onset of symptoms and is most rapid during the first 3–6 months, although further improvement may be seen for up to 2–4 years. 2
Diagnostic criteria for TM were established by the Transverse Myelitis Consortium Working Group in 2002. 3 A proportion of patients initially diagnosed with TM will subsequently relapse, often with the involvement of other parts of the central nervous system (CNS), and may then be diagnosed with either multiple sclerosis (MS) or neuromyelitis optica (NMO). NMO is a relapsing subset of TM, caused by antibodies to aquaporin 4 (AQP4). 4 Clinically, patients have predominantly recurrent episodes of myelitis and optic neuritis. Neurodisability accrues with progressive relapses. Initial presentation may be with myelitis alone, making it clinically and radiologically indistinguishable from TM, and patients are thus subjected to the same acute therapeutic strategies.
The precise number of patients who make a full recovery from TM remains unclear. Studies carried out before the development of the Transverse Myelitis Consortium Working Group criteria may have included patients with a wider range of myelopathies, such as spinal cord infarction,5 or may reflect the greater severity of cases seen at tertiary referral centres such as the Johns Hopkins Transverse Myelitis Centre,6 where up to 20% of patients are reported to make a good recovery. Currently, the only report to reliably inform on the outcome of adult-onset TM is a retrospective French multicentre study applying Transverse Myelitis Consortium Working Group criteria, with 36% of patients with TM having a poor prognosis, defined by death or non-ambulating. 7 Approximately half of children with TM make a good recovery. 8 Hence, the majority of adults and children presenting with TM have either a fair outcome (functional and ambulatory but with varying degrees of spasticity, urgency and/or constipation and some sensory signs) or a poor outcome (remaining completely or largely unable to walk, having at best partial sphincter control and being left with severe sensory deficits). 2 These outcomes have a huge burden on patients and their carers. With conservative estimates of the incidence of TM in the UK being 350 cases per year (based on an incidence of three to seven cases per million),9,10 this clearly imposes a significant cumulative demand on health resources in the UK. Moreover, many patients are affected when they are in the prime of their working life, thus resulting in a loss of productivity and imposing a further financial impact on the country.
Importantly, strategies to reduce disability in patients are urgently required, but there have been no robust randomised controlled trials (RCTs) carried out in children or adults to inform on the optimal treatment of TM. The current clinical consensus is derived from data that are extrapolated mainly from class IV evidence from case series or clinical trials on the treatment of exacerbations of adult MS. 9–12 In adults, this evidence suggests that treatment of relapses with intravenous methylprednisolone (IVMP) shortens relapse duration and speeds up recovery. It is on this evidence that the current standard therapy for TM has been based, with both children and adults treated with high-dose intravenous (IV) steroids for 3–7 days to reduce inflammation, hasten recovery and restore neurological function. 13
Although IV steroids are now the most common treatment for TM, there are other interventions that have proved effective in aiding recovery, but which are not routinely applied. In a retrospective analysis of 122 adults with TM, acute therapies given at one centre between 2001 and 2005 were evaluated, with the finding that some patients benefited from the addition of plasma exchange (PLEX) to IVMP. 11 The efficacy of PLEX was also demonstrated in a small RCT in adults with acute CNS demyelination (including four patients with TM), in whom steroids had failed to induce a remission of symptoms. 14 However, administering PLEX is technically difficult and costly, making it challenging to deliver within the NHS and resulting in it not being universally available.
Treatment with intravenous immunoglobulin (IVIG) is also used increasingly in the management of a range of neurological conditions and its efficacy has been clearly established in RCTs for a handful of these conditions. 15 In adults and children with CNS demyelination who do not respond to steroids, IVIG is often used, although supporting data are limited to small case series and single case reports. 16,17 The most relevant actions of IVIG in the therapy of neurological diseases include (1) the inhibition of complement binding, (2) the neutralisation of pathogenic cytokines, (3) the down-regulation of antibody production and (4) the modulation of Fc-receptor-mediated phagocytosis. Additional actions include the modulation of T-cell function and the enhancement of remyelination. 18 The majority of these factors are common across inflammatory disorders of the CNS, including TM,19 providing a strong rationale for the use of IVIG in the management of TM. In addition, IVIG is cost-effective compared with PLEX and more readily accessible.
We aimed to conduct a multicentre, single-blind, parallel-group RCT to generate evidence to inform clinical and health economic decisions with regard to IVIG use in adults and children with TM.
Risks and benefits
Risks
This study included adults and children. As treatments in both arms of the trial are already used in current clinical practice, those participating faced almost no additional risk beyond what they would experience in treatment outside the trial.
Benefits
Interventions to reduce disability in patients are urgently required. The current management recommendation is largely based on expert opinion,13 as there have been no robust RCTs on the treatment of TM in children or adults to inform on the optimal treatment of TM. This trial aimed to evaluate whether or not IVIG would be beneficial in the management of TM.
Study aim
The randomiSed controlled TRial of IntraVEnous immunoglobulin compared with standard therapy for the treatment of transverse myelitis in adults and children (STRIVE) study was a multicentre, single-blind, parallel-group RCT aimed at generating evidence to inform clinical and health economic decisions regarding IVIG use in adults and children with TM.
The primary objective was to evaluate whether or not additional, and early, treatment with IVIG is of extra benefit in TM compared with current standard therapy with IV steroids.
The secondary objectives were to investigate the following potential benefits:
-
The clinical and para-clinical data collected from patients will provide a robust resource and platform for other clinical studies, including the identification of early predictors of poor outcome.
-
Biobanked samples from patients recruited to the study will be collected and used for carefully designed biological studies by a consortium of established basic science researchers in the field.
Chapter 2 Methods and materials
Trial design
This was a UK, multicentre, single-blind, parallel-group RCT.
Patients randomised to the control arm were prescribed IVMP in line with local clinical practice. Recommended dosages were as follows:
-
paediatric patients: 30 mg/kg or 500 mg/m2 capped to a maximum dose of 1 g/day for 5 days
-
adult patients: 1 g/day for 5 days.
Patients in the intervention arm received the above standard therapy plus additional IVIG:
-
adults: 2 g/kg administered in five divided doses
-
children > 41.2 kg: 2 g/kg administered as for adults
-
children ≤ 41.2 kg: 2 g/kg administered in two divided doses.
Although IVIG dosing does not need to be administered over consecutive days, it must be administered in accordance with the dosing schedule (see Appendix 1).
Patients could be recruited and randomised up to 5 days from the date of first commencing steroid therapy or up to 21 days from the onset of symptoms (if definitely known).
In patients who did not respond to standard IVMP treatment or adjunctive treatment with IVIG, rescue therapy, such as PLEX, would be instituted.
If PLEX was administered it might attenuate the treatment effect of IVIG and, indeed, might have a treatment effect of its own. Guidance parameters were set out to define and standardise the PLEX regime as follows:
-
treatment failure would be considered if no improvement was seen or if deterioration occurred after 14 days from presentation or 5 days after completion of either treatment arm
-
complete PLEX treatment would consist of at least five cycles, with at least 75% of plasma volume exchanged in each cycle, with a 24- to 48-hour interval between each cycle
-
an extra course of IVMP may be given by physicians, often during the lag phase from the decision to proceed with rescue therapy to its initiation (usually 5–7 days).
End-point measures
Primary end-point measure
The primary outcome measure was the change in American Spinal Injury Association (ASIA) Impairment Scale score20 (categories range from A to E, with category E describing normal function and category A describing complete lack of motor and sensory function below the level of injury) from baseline (just prior to randomisation) to 6 months post randomisation, with an improvement of two grades being defined as a good outcome.
Secondary end-point measures
-
ASIA motor scale (0–100) and sensory scale (0–112) scores21 at 3, 6 and 12 months post randomisation.
-
Kurtzke Expanded Disability Status Scale (EDSS) score,22 measured by Neurostatus scoring, at 3, 6 and 12 months.
-
EuroQol-5 Dimensions youth version (EQ-5D-Y) score23 at 3, 6 and 12 months for patients aged 8–12 years (at presentation).
-
EQ-5D five-level version (EQ-5D-5L) score24 at 3, 6 and 12 months for patients aged ≥ 13 years (at presentation).
-
International Spinal Cord Injury Quality of Life (SCI-QOL) Basic Data Set25 at 3, 6 and 12 months for patients aged ≥ 13 years (at presentation).
-
Client Service Receipt Inventory (CSRI)26 at 3, 6 and 12 months.
Tertiary end-point measures
-
International SCI-QOL, Pain, Bladder and Bowel Function Basic Data Sets27,28 at 6 and 12 months post randomisation for patients aged ≥ 13 years (at presentation).
-
Paediatric Quality of Life Inventory (PedsQL™) Parent Report for Toddlers29 at 6 and 12 months for patients aged 2–4 years (at presentation).
-
PedsQL (Parent Report for Young Children30) at 6 and 12 months for patients aged 5–7 years of age (at presentation).
-
International SCI-QOL Pain Basic Data Set31 at 6 and 12 months for patients aged ≥ 13 years (at presentation).
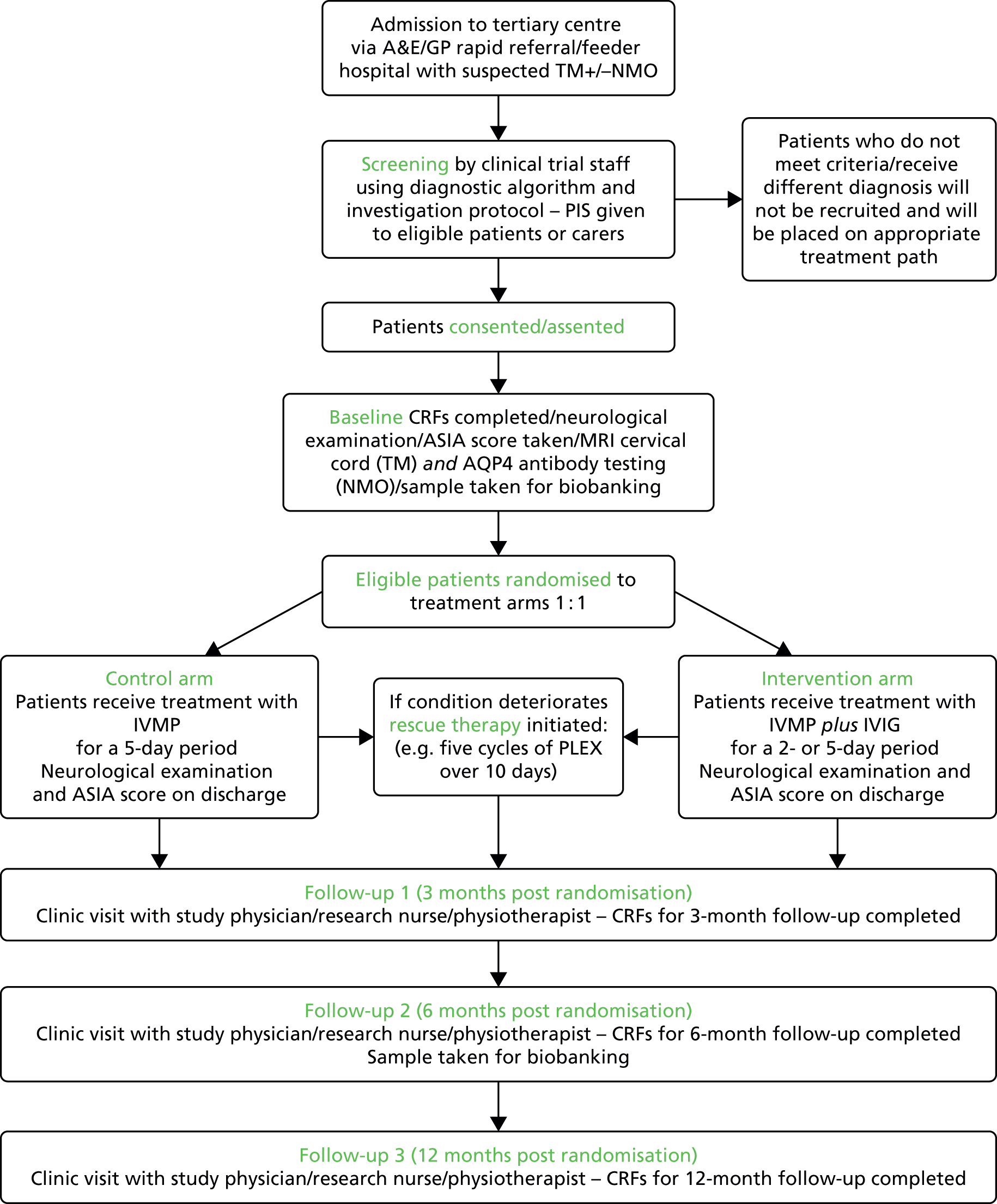
The overall flow of study participants from admission through to randomisation and the final visit is summarised in Figure 1.
FIGURE 1.
Participant flow chart. A&E, accident and emergency; CRF, case report form; GP, general practitioner; MRI, magnetic resonance imaging; PIS, patient information sheet.
Study subjects
Participants were individuals who met the eligibility criteria and diagnostic algorithm (see Appendix 2) and who lived in the catchment area of participating tertiary neurology centres (although neurologists could also recruit patients at district general hospitals or from rapid general practitioner referrals). Eligibility for the study was determined using the following criteria.
Inclusion criteria
Patients were eligible for inclusion in the trial if, on presentation, they:
-
Were aged ≥ 1 year.
-
Had been diagnosed with either acute first-onset TM or a first presentation of NMO. [The Transverse Myelitis Consortium Working Group 2002 criteria for probable TM were used. 3 Hence, following clinical and radiological exclusion of a compressive myelopathy, a patient will be diagnosed with TM if he or she meets all of the following criteria: sensory, motor or autonomic dysfunction attributable to spinal cord disease; bilateral signs and/or symptoms (not necessarily symmetrical); clearly defined sensory level (except in young children aged < 5 years when this is difficult to evaluate); lack of brain magnetic resonance imaging (MRI) criteria consistent with MS (McDonald 2010 space criteria); and progression to nadir between 4 hours and 21 days. Patients with definite modified NMO should meet the following criteria:32 absolute criteria – optic neuritis and acute myelitis (both) – plus two out of three supportive criteria – brain MRI not meeting criteria for MS at disease onset, spinal cord MRI with contiguous T2-weighted signal abnormality extending over three or more vertebral segments, indicating a relatively large lesion in the spinal cord, and AQP4 seropositive status.]
-
Had an ASIA impairment score of A–C.
-
Had commenced steroid treatment but would be randomised no later than 5 days after commencing treatment and, if definitely known, no later than 21 days from the onset of symptoms.
-
Gave assent (< 16 years)/consent to participate in the trial.
Exclusion criteria
Patients were excluded if they showed evidence of one or more of the following:
-
contraindication to IVIG as stated in the product Summary of Product Characteristics (SmPC) or receiving IVIG for other reasons
-
previously known systemic autoimmune disease (e.g. systemic lupus erythematosus) or any evidence of systemic inflammation during the current presentation
-
direct infectious aetiology (e.g. varicella zoster)
-
previous episode of CNS inflammatory demyelination
-
acute disseminated encephalomyelitis
-
other causes of myelopathy not thought to be caused by myelitis (e.g. nutritional, ischaemic, tumour)
-
other disease that would interfere with the assessment of the outcome measures
-
known pregnancy
-
circumstances that would prevent follow-up for 12 months.
Spinal cord inflammation demonstrated by cerebrospinal fluid (CSF) pleocytosis or an elevated immunoglobulin G (IgG) index or gadolinium-enhanced MRI is supportive of an inflammatory aetiology but was not essential for inclusion/exclusion. Individuals with myelitis underwent testing for AQP4 antibodies, as NMO can present as isolated TM. In addition, patients received investigations that were clinically indicated to identify specific non-inflammatory aetiologies.
Sample size
In recognition of TM as a rare condition, the power analysis took into account the inclusion of a futility analysis to be undertaken after recruitment of one-third of the target sample. We assumed that the proportion of participants showing a two-grade improvement (or greater) on the ASIA Impairment scale would be approximately 0.5 (50%) in the control arm and a minimum of 0.75 (75%) in the intervention arm. The sample size calculation was based on the conservative assumption of no correlation between repeated measures.
Randomised 1 : 1, the primary intention-to-treat (ITT) analyses would compare 76 treatment and 76 control patients on the ASIA classification scale at 6 months post randomisation. Based on comparing the difference in the number of successes among the treatment patients and the control patients, the SAS program sample size chi procedure (version 9.22; SAS Institute Inc., Cary, NC, USA) examines all 772 possible trial outcomes under the null and alternative hypotheses. The possible outcomes are then arranged in descending order and cumulative probabilities for every possible value from 76 to –76 are computed. Using a critical value that maintains the tail probability at 0.02355 under the null hypothesis, the probability under the alternative hypothesis is 0.9034. The study thus had 90% power for a two-tailed test with α = 0.05.
The sample size was inflated for attrition, based on our experience and the study design; to minimise any loss to follow-up, we estimated 10% attrition. This would require recruiting a sample size of 152/0.90 = 170 (85 participants per arm).
The ASIA total motor score (0–100) was a secondary outcome. There is little evidence in acute TM to summarise this in terms of variance, mean and correlation. The Stata® function sampsi (version 12; StataCorp LP, College Station, TX, USA) indicated that, using analysis of covariance, with a baseline to end-point correlation of 0.6, the study would have 87% power to detect a difference between the control arm and the treatment arm of a medium to large effect size of 0.4. Such a difference would be of clinical significance.
Randomisation
Treatment allocation was to be stratified at randomisation by service type (adult or child), using stratified block randomisation; the block would randomly vary in size. Participants were randomised 1 : 1 to treatment.
Withdrawal
Patients (or their parents/guardians) had the right to withdraw from the study at any time for any reason. The investigators also had the right to withdraw patients from the study drug in the event of intercurrent illness, adverse events (AEs), serious adverse events (SAEs) and suspected unexpected serious adverse reactions, subsequent evidence of a different aetiology, protocol violations, cure or for administrative or other reasons. Participants who either wished to or had to discontinue study medication would be returned to standard care through their supervising physician, but would continue to provide study-specific data at follow-up visits at 3, 6 and 12 months. It was understood that an excessive rate of withdrawals could jeopardise randomisation outcomes and render the study results uninterpretable; therefore, unnecessary withdrawal of patients was to be avoided. If a patient had decided to withdraw from the study, all efforts would have been made to report the reason for withdrawal as thoroughly as possible.
Study data
The study data were managed as previously described in the published trial protocol. 33
Data management
Data were managed using the InferMed MACRO database system (version 4; Elsevier, Amsterdam, the Netherlands). This system is regulatory compliant. 34–36 An electronic case report form (eCRF) was created using the InferMed MACRO system in collaboration with the trial statisticians and the chief investigator and was maintained by the King’s Clinical Trials Unit (KCTU). It was hosted on a secure dedicated server within King’s College London and source data were entered by authorised staff onto the eCRF with a full audit trail.
Database passwords
Database access was strictly restricted through the use of passwords provided to authorised members of the research team. The chief investigator or site delegate requested access from the KCTU. If a new staff member joined the study, a personalised username and password were requested through the chief investigator or site delegate.
Identifiable data
All participant contact information was stored within the recruiting NHS site, with restricted access from password-protected computers. Accrual data uploaded to the UK Clinical Research Network portfolio database were anonymised and collated by the chief investigator or trial manager to the Local Clinical Research Network. No identifiable data were entered on the eCRF or transferred to the KCTU. Participants were identified on the study database using unique codes and initials. Each investigator maintained accurate patient records detailing observations of each patient enrolled.
Investigational medicinal product
The investigational medicinal product (IMP) was provided as human normal immunoglobulin (Intratect®) [100 g/l solution for infusion in a single 5-g (50-ml) or 10-g (100-ml) glass vial] by Biotest AG, Dreieich, Germany, the marketing authorisation holder of the commercially available Intratect.
An Annex 13 clinical trial labelling exemption37 was in place and was approved by the Medicines and Healthcare products Regulatory Agency (MHRA) for this trial. A standard pharmacy dispensing label was applied to the IMP at the point of dispensing by the pharmacy at each investigator site.
The IMP was stored in a secure area with limited access. Site pharmacies were responsible for the safe and appropriate storage of the IMP at their site in accordance with the manufacturer’s instructions:
-
do not store above 25°C
-
do not freeze
-
keep the vial in the outer carton, to protect from light.
Dosing regimen
Intravenous methylprednisolone (as sodium succinate) was administered in accordance with local clinical guidelines. A single daily dose of 30 mg/kg or 500 mg/m2 (maximum 1 g/day) for 5 days can be used in paediatric patients. Adult patients can receive 1 g/day for 5 days.
Patients randomised to the control arm received no additional treatment, whereas patients randomised to the treatment arm received the above treatment plus 2 g/kg of IVIG in divided doses (see Appendix 1).
Note that IVMP (as sodium succinate) was classed as a non-IMP in this trial and was dispensed by hospital pharmacies in accordance with their standard clinical practice.
Risks
The current risks associated with the Intratect immunoglobulin are detailed in the SmPC for Intratect [see www.medicines.org.uk/emc/medicine/23175/SPC/intratect/ (accessed 21 March 2017)]. A summary of these risks is provided in Appendix 3.
Compliance
Treatment with the IMP was administered under the supervision of the investigator and in a controlled clinical environment. Therefore, full patient compliance with treatment was anticipated.
Concomitant medication
Only relevant immunomodulatory medications were recorded throughout the study.
In patients not responding to control treatment or adjunctive treatment with IVIG, rescue therapy was to be instituted in accordance with local guidelines. In most cases, the rescue therapy of choice was PLEX therapy. This was also to be recorded as a concomitant medication.
Confidentiality
The study staff ensured that participants’ anonymity was maintained, identifying patients by their PIN (personal identification number) and initials only. The study complied with the Data Protection Act 1998,38 which requires data to be anonymised as soon as it is practicable to do so.
Trial sites and study duration
The study was conducted across 15 clinical sites within the UK, with an additional site acting as a participant information centre. The first of the 15 sites was opened for recruitment on 4 March 2015, whereas the final site received its approval to commence recruiting on 8 February 2016. Recruitment was terminated early across all 15 recruiting sites and the single participant identification centre on 11 March 2016.
Trial procedure
Table 1 provides a summary of the trial visit schedule and the associated assessments to be carried out. Some of the questionnaires were specific to particular age groups, with age on presentation used as the criterion.
| Schedule | Time point | ||||||
|---|---|---|---|---|---|---|---|
| T0 (screening, baseline and prediagnosis tests) | T1 (treatment and discharge) | T2 (3 months) | T3 (6 months) | T4 (12 months) | Ongoing | ||
| Rescue therapya | Discharge | ||||||
| Screening with the diagnostic algorithm and core investigations including physical examination | ✗ | ||||||
| Patient information and informed consent | ✗ | ||||||
| Eligibility form | ✗ | ||||||
| Registration form | ✗ | ||||||
| Prediagnosis tests (e.g. MRI and AQP4 testing) | ✗ | ||||||
| Randomisation | ✗ | ||||||
| Biobank samples | ✗ | ✗ | |||||
| ASIA impairment score (A–E) | ✗ | ✗ | ✗ | ✗ | P | ✗ | |
| ASIA motor and sensory scale score | ✗ | ✗ | ✗ | ✗ | S | ✗ | |
| Neurostatus scoring (Kurtzke functional systems and EDSS) | ✗ | ✗ | ✗ | S | ✗ | ||
| EQ-5D-Y questionnaire (8–12 years) | ✗ | ✗ | S | ✗ | |||
| EQ-5D-5L questionnaire (≥ 13 years) | ✗ | ✗ | S | ✗ | |||
| CSRIb | ✗ | S | ✗ | ||||
| SCI-QOL Basic Data Set (≥ 13 years)b | ✗ | S | ✗ | ||||
| SCI Bladder Function Basic Data Set (≥ 13 years)b | T | ✗ | |||||
| SCI Bowel Function Basic Data Set (≥ 13 years)b | T | ✗ | |||||
| SCI-QOL Pain Basic Data Set (≥ 13 years)b | T | ✗ | |||||
| PedsQL (5–7 years)b | T | ✗ | |||||
| PedsQL (2–4 years)b | T | ✗ | |||||
| Treatment form | ✗ | ||||||
| Concomitant medications | ✗ | ||||||
| Discharge form | ✗ | ||||||
| Rescue therapy form (if needed)a | ✗ | ||||||
| Relapse form (at any time point if needed)a | ✗ | ✗ | ✗ | ✗ | |||
| AEs | ✗ | ||||||
| Study status form | ✗ | ✗ | ✗ | ||||
| Withdrawal form (at any time point)a | ✗ | ||||||
Blinding
Because of the technical challenges of masking IVIG from saline, the need for rapid recruitment and the fact that follow-up would take place many months after the event using objective well-defined clinical end points, treatment was not blinded (no placebo). The trial manager, pharmacies and those administering treatment were not blinded to the intervention, whereas staff carrying out primary outcome assessments at follow-up and statistical analyses were blinded.
Screening, baseline and discharge assessments were carried out in the tertiary centres by a study physician/research nurse. Following discharge from treatment in hospital, all primary outcome assessments at follow-up in clinic at the tertiary centre or appropriate neurology centre were carried out by a study physician/research nurse/physiotherapist who had been blinded to treatment. For consistency, whenever possible, the same blinded assessor was required to carry out the assessments at each time point. Although not mandatory, secondary and tertiary outcome assessments at follow-up were to be performed by a blinded member of staff whenever possible.
Laboratory tests
It was planned that all consenting patients would have samples taken for clinical investigations and biobanking at baseline and at the 6-month follow-up. In cases in which samples for clinical investigations had been taken prior to consent, any leftover material would be used for biobanking. No additional samples would be collected unless there was a clinical indication to do so. Samples for biobanking would consist of CSF via lumbar puncture and blood taken by venepuncture for serum, plasma, deoxyribonucleic acid, peripheral blood mononuclear cells and ribonucleic acid (site dependent). Samples were to be stored in one of the two biobanks (London or Cardiff). These samples would not form part of this trial but were intended for further hypothesis-driven biological research, directed by Neil Robertson and Gavin Giovannoni (adult medicine) and Ming Lim (paediatric medicine).
Magnetic resonance imaging sequences
As part of the routine diagnostic process for TM/NMO, brain and spinal cord MRI sequences were acquired when possible, the results of which were in the study’s diagnostic algorithm at screening and recorded as study data if patients entered the trial. Local protocols were in place for the acquisition of MRI sequences, which would usually include gadolinium-enhanced sequences in the event of suspected TM/NMO.
To facilitate the systematic accrual of neuroimaging information it was recommended that reports included:
-
the location of the lesion (which spinal cord level)
-
the size of the lesion (in terms of how many vertebral segments were involved)
-
whether gadolinium injection was used and, if so, whether enhancement was seen.
During the trial period, the study team was able to request anonymised patient scans provided on a compact disc to resolve potential clinical and radiological uncertainties.
Analysis
A comprehensive statistical analysis plan was developed and a descriptive analysis (e.g. summary statistics and plots) was to be performed to investigate the distribution of the primary outcome, that is, the ASIA impairment score, across participants.
Statistical analysis
All analyses were to be pragmatic and follow the ITT principle, that is, patients would be analysed in the group to which they were randomised, irrespective of treatment received, utilising all available follow-up data from all randomised patients. Sensitivity analyses were to be used to assess the robustness of the conclusions to missing outcome data and to departures from randomised treatment.
An interim futility analysis was scheduled to be conducted after 52 patients had provided a response (26 in each treatment arm), with the end point being a change in the ASIA impairment score of two or more grades 6 months after randomisation; the results were to be assessed by the Data Monitoring Committee (DMC).
The final analysis of effectiveness was scheduled to be conducted once the trial database had closed, if the study continued to full recruitment. The DMC was to collate effectiveness and safety data during the trial to inform its recommendations to the Trial Steering Committee (TSC). It was planned to summarise the main effects by intervention arm and assessment time point, with associated 95% confidence intervals.
Primary and secondary outcome analysis
The main objective of the statistical analysis was to assess the effect of IVIG on the primary outcome, the ASIA Impairment Scale (A–E) score, at 6 months post randomisation.
The secondary clinical assessments (EDSS, continuous ASIA motor and sensory scales, SCI-QOL, Pain, Bladder and Bowel Basic Data Sets, PedsQL, EQ-5D and CSRI) with repeated measurements were to be analysed within a linear mixed-model framework, with generalisations of the linear mixed model utilised to allow for outcomes with non-normal data if necessary. Those measures with one follow-up assessment were to be evaluated with a general linear model. The statistical modelling was designed to feature the outcome measure(s) as the dependent variable, with corresponding baseline measure(s) (if applicable), stratification factors and treatment group featuring as covariates.
Recruitment rate, consent rate, loss to follow-up, departures from randomised treatment and the prevalence of SAEs (specifying deaths and intensive therapy unit admissions) were to be reported as descriptive analyses at 3, 6 and 12 months post randomisation and summarised by treatment arm over the course of the study. The chi-squared test (Fisher’s exact test) was to be used for categorical outcomes (e.g. SAEs and mortality).
All analyses were scheduled to be repeated considering age status (adult or child) and putative biological markers as moderators by interaction with treatment group (control or intervention), allowing estimates of treatment effect in the subpopulations to be summarised.
Explanatory analyses to assess the efficacy of the treatment within NMO or idiopathic TM diagnosis by allowing for an interaction with treatment arm were to be carried out. The intracluster correlation coefficient of the sites would have been explored by allowing for site as a random effect in the statistical modelling.
With regard to missing data for post-treatment outcomes, which would have arisen as participants discontinued treatment or were lost to follow-up, regression analysis based on maximum likelihood and resulting inferences would have been valid provided that the missing data-generating mechanism was missing at random (MAR), that is, ‘missingness’ was predicted only by variables that were included in the model, including earlier values of the outcome variables. We aimed to empirically assess whether or not any baseline variables predict missingness and, if this was the case, we planned to condition on such variables by including them in the statistical model. Sensitivity analyses would have been used to assess the robustness of the conclusions to missing outcome data and to departures from randomised treatment, as previously described by White et al. 39
Futility analysis
As previously mentioned, an interim futility analysis was scheduled to be conducted after 52 patients had provided a response, 26 in each treatment arm, with the end point being a change in the ASIA Impairment Scale score of two or more grades at 6 months. The trial would have been terminated with the conclusion that the new treatment was no better than the standard treatment if, based on these 52 patients, the test statistic was less than zero. If the sample sizes were equal, this would occur if the successes under the new treatment were fewer than those under the standard treatment. Otherwise, the trial would proceed to the full sample size of 170 patients. The SAS program two-stage – interim – chi evaluates the design-deleting outcomes that would correspond to futility. The tail probabilities under the null and alternative hypotheses were 0.0228 and 0.8946, respectively. The inclusion of the futility analysis therefore represented a very small loss of power.
The SAS program two-stage – stage 1 – chi evaluates the properties of the first stage of the design. It showed that the probability of abandoning the study at the interim analysis was 0.4449 under the null hypothesis and 0.0201 under the alternative hypothesis. Therefore, there was a good chance of stopping for futility when the treatments were equivalent and a very small chance of stopping for futility when the desired treatment effect was present (see Appendix 4 for the futility analysis plan).
Health economics
The study team aimed to develop a health economic model structure and complete a write-up of its economic analysis within the 12 months following the end of patient data collection (i.e. 42–54 months after the start date).
Economic analysis
Drug pricing data and primary care, secondary care and social care costs were to be calculated as described previously. 40 Costs were to be combined with the primary outcome measure in the form of a cost-effectiveness analysis. If IVIG resulted in higher costs and a better outcome, then an incremental cost-effectiveness ratio would be generated to show the extra cost incurred to achieve an extra unit of improvement. Because of the uncertainty around results, it was planned that cost-effectiveness planes and cost-effectiveness acceptability curves would be used, with bootstrapping of skewed results.
Long-term cost-effectiveness over 5-year and 10-year periods was to be calculated using a Markov model. Response to treatment was to be classified, and transition probabilities between groups were to be derived from 6-month and 12-month follow-up data. Costs and quality-adjusted life-years (QALYs) for each category were to be derived from the trial data. As limited data would be available on long-term costs, we planned to conduct both deterministic and probabilistic sensitivity analyses.
All causes of withdrawal from randomised treatment were to be reported. The chi-squared test (Fisher’s exact test) was to be used for categorical outcomes (e.g. SAEs and mortality).
There would have been missing data for post-treatment outcomes as participants discontinued treatment or were lost to follow-up. Inferences would have been valid provided that the missing data-generating mechanism was MAR and was not predicted by any variables in the model, that is, missingness was predicted only by variables that were included in the model.
Economic evaluation
It was planned to record the use of IVIG, IVMP, additional treatments and rescue PLEX throughout the follow-up period, with costing carried out using drug pricing data from the British National Formulary and the Department of Health. It was planned to record the use of primary care, secondary care and social care at the 3-month, 6-month and 12-month follow-ups using the CSRI, with costs calculated to determine the total cost for the control and treatment arms.
A cost-effectiveness analysis was to be performed using the primary and secondary outcome measures of improvement in ASIA scores and the secondary outcome of QALYs with EQ-5D-Y, EQ-5D-5L and CSRI. If IVIG results in higher costs and a better outcome, then an incremental cost-effectiveness ratio will be generated to show the extra cost incurred to achieve an extra unit of improvement.
Ethics and approvals
This study is registered as EudraCT 2014–002335–34, ClinicalTrials.gov NCT02398994 and Current Controlled Trials ISRCTN12127581. Research Ethics Committee approval was obtained (South Central – Berkshire B; reference number 14/SC/1329) alongside MHRA notification.
Chapter 3 Trial outcome and results
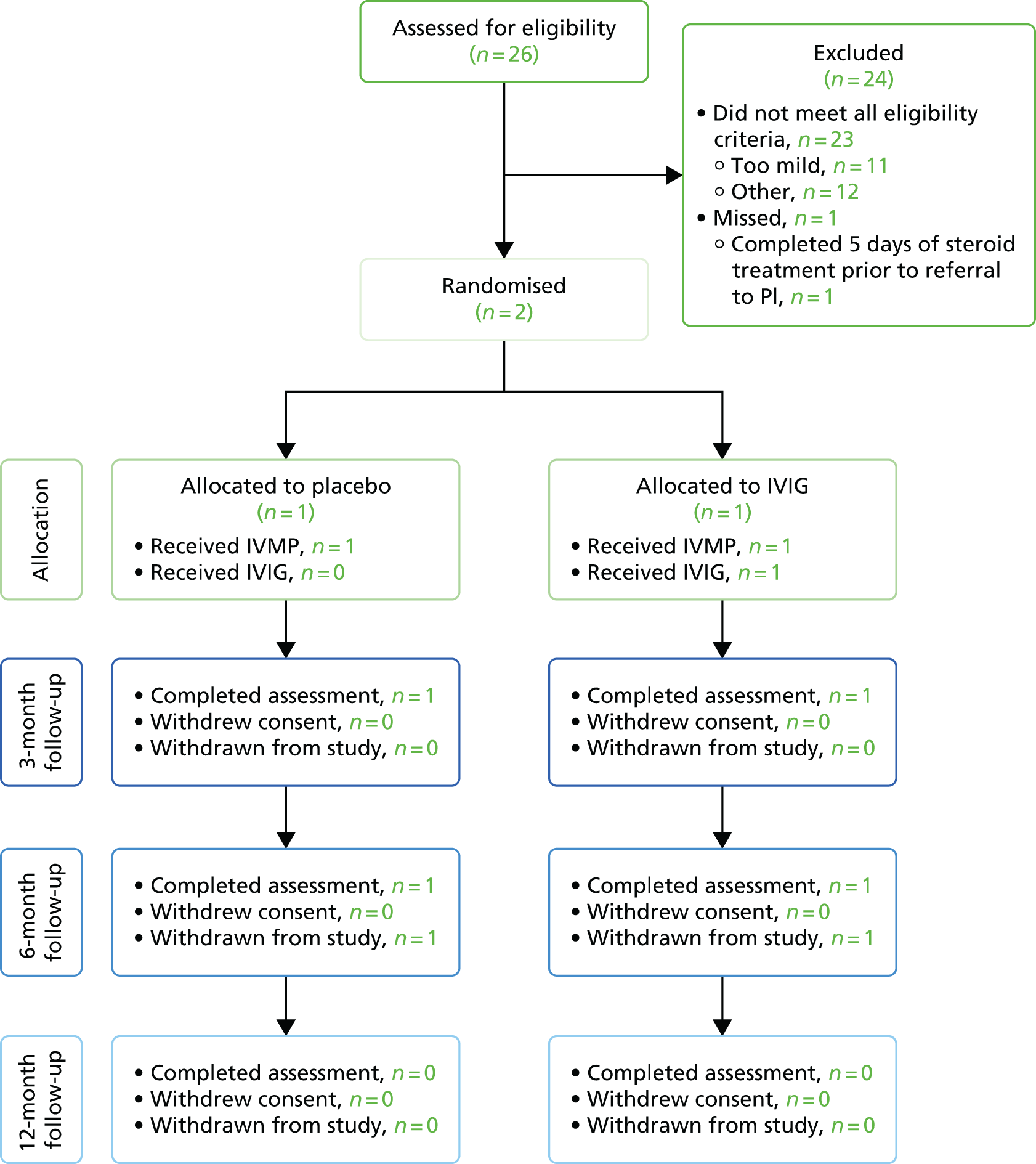
Over the 53 weeks of recruitment, 26 potential participants were screened for eligibility across all 15 sites. Of these 26 participants, 24 were not randomised, with 23 being ineligible [48% of these patients (n = 11/23) had symptoms considered ‘too mild’ for inclusion and the remaining 52% (n = 12/23) were ineligible as they did not meet other inclusion criteria or met the exclusion criteria) and one patient having a delayed referral to the primary investigator. Two participants were therefore recruited into the study (Figure 2), one to each arm of the trial, with each participant followed up for 6 months post randomisation prior to withdrawal from the study. The number of participants randomised into the study was significantly below the target sample size, thus precluding any form of data analysis or detection of a significant difference between the control arm and the treatment arm of the study.
FIGURE 2.
The Consolidated Standards of Reporting Trials (CONSORT) flow diagram. PI, primary investigator.
Baseline and 3-month data summary
Descriptive summarisation of both participants’ data was carried out following a lock of the study data; the trial statistical team remained masked to treatment allocation up to this point. One patient was a white female child aged between 10 and 15 years (student), with an unknown body mass index (BMI). This participant had an ASIA impairment score of C prior to randomisation, meaning that motor function was preserved below the neurological level and more than half of the key muscles below this level had a muscle grade of < 3. At 3 months post randomisation this participant had improved to an ASIA impairment score of D, meaning that at least half of the key muscle functions below the neurological level had a muscle grade of ≥ 3. The other participant was a white adult male aged between 60 and 65 years in full-time employment and with a BMI of 30 kg/m2. This participant had an ASIA impairment score of A prior to randomisation, meaning that no motor or sensory function was preserved in sacral segments S4–S5. At 3 months post randomisation, this participant had not improved. Neither participant suffered a relapse during the trial. The adult participant suffered from two AEs during the trial, a chest infection and pressure sores, which were not related to the trial; the paediatric participant suffered no AEs. Because of the very small sample size it is not possible to comment on whether there is any relationship between the trial arm and any change in ASIA impairment scores.
Withdrawal
Both randomised patients remained in the trial until their second follow-up visit (6 months post randomisation), at which point they were withdrawn from the study by the study team.
The rationale behind the termination and subsequent withdrawal was explained to both participants, both of whom had recourse to further information.
Health economics analysis
As the trial did not recruit a sufficient number of participants, it was not possible to conduct the health economic analysis.
Investigator (opinion leader) feedback
Following the termination of the study, the study team developed a survey that sought to both gain feedback on the study and develop a better understanding of the reasons, in the opinion of the investigators, the study did not recruit to target. The survey questions were sent to 13 of the 15 recruiting sites, as the remaining two sites were those of the chief investigator and the co-investigator/project co-ordinator, both of whom were involved in the design of the survey. Of the 13 investigators contacted, 11 responded. A summary of these responses is provided in Table 2.
| Question | Investigator’s response (n = 11) | ||
|---|---|---|---|
| Agree | Neither agree nor disagree | Disagree | |
| 1. The aim and overall objective of the study was clear | 11 | 0 | 0 |
| 2. The study design was simple enough to understand | 10 | 1 | 0 |
| 3. All of the study procedures (including study visits at 3, 6 and 12 months post treatment) were in line with our standard care for TM/NMO patients | 6 | 3 | 2 |
| 4a. The ASIA score was an appropriate measure to use | 3 | 4 | 4 |
| 4b. What alternative would you have considered if any? | Motor component of ASIA assessment, n = 1; EDSS, n = 2; MSFC, n = 1; assessment of mobility, n = 1; a bespoke assessment score validated in advance, n = 1 | ||
| 5. The trial offered good treatment options for TM/NMO | 7 | 3 | 0 |
| 6. The trial was sufficiently pragmatic | 6 | 2 | 1 |
| 7. The site was an appropriate choice for the trial | 11 | 0 | 0 |
| Investigator | |||
| 1. I think addressing the study question is highly important and should be a priority | 10 | 1 | 0 |
| 2. The outcome of this study would have changed how I treat my TM patients | 9 | 1 | 1 |
| 3. Within my daily workload, I believe I had enough time to allocate to activities that the study required (both clinical and administrative) | 2 | 2 | 7 |
| 4. I generally find it difficult to participate in all kinds of clinical trials nowadays because of a lack of time | 7 | 3 | 1 |
| 5. A study visit by a participant had (or would have had) an undesirable impact on my clinic (e.g. significantly increasing the clinic time or time for administration of the clinic) | 5 | 3 | 3 |
| 6. I generally find it difficult to find time for all study visits nowadays | 4 | 4 | 3 |
| 7. What in your opinion was the single most important reason this trial failed? | ‘Strict inclusion criteria’, n = 4; ‘rarity of cases’, n = 3; ‘choice of outcome measure’, n = 2; ‘early recognition of cases’, n = 3; and ‘finding time to recruit patients’, n = 1 | ||
| 8. What would you have changed about the study? | Broader eligibility criteria, n = 3; inclusion of less severe patients, n = 5; use of different outcome assessment, n = 5; improved awareness among all neurologists and engage all sites earlier, n = 3 | ||
| Study set-up and recruitment | |||
| 1. I was actively involved in the site feasibility discussion before the study opened at my site | 8 | 2 | 0 |
| 2. Set-up of the study felt complicated at times | 5 | 1 | 4 |
| 3. All things considered, I think funding for sites for this study was not really adequate | 7 | 2 | 2 |
| 4. I found my interaction with our R&D department useful during the set-up and administration of this study (if applicable) | 7 | 3 | 1 |
| 5. I was unable to recruit because (choose as many as applicable) | Strict inclusion criteria, n = 9; no cases came to my attention, n = 5; came to know late of case, n = 7; did not have time, n = 0; other, n = 1 | ||
| Other | |||
| 1. I received as much assistance as needed from the study co-ordinating team (trial manager, sponsors, etc.) | 10 | 1 | 0 |
| 2. The study co-ordinating team could have done more to increase awareness of the study among other clinicians or the potential patient population | 4 | 1 | 6 |
| 3. In future, I would be interested in collaborating with this team in designing and running another study | 9 | 2 | 0 |
Chapter 4 Discussion: lessons learned from early closure of the trial
This study aimed to elucidate the added benefit in terms of clinical efficacy and health economics associated with additional and early treatment of TM with IVIG compared with the current standard therapy of IV steroids. However, because the study under-recruited, we were unable to determine whether or not there were any differences between the trial arms. The barriers encountered during the course of the trial are discussed in this chapter. Other researchers planning a similar study would benefit from being aware of these barriers and their potential impact.
Overview of the trial evolution
The first trial recruiting site received approval to start recruiting on 4 March 2015, whereas the final site to receive its approval did so on 8 February 2016. At the point at which the study was suspended (11 March 2016), 15 sites were open to recruitment, with two sites each recruiting one subject. The TSC met on 11 March 2106, when strategies to further improve recruitment into the study were extensively discussed. Following this meeting, the TSC and Trial Management Group (TMG) jointly decided that further recruitment into the study should be suspended with immediate effect. The TSC and TMG came to this decision because the study, on its current trajectory, was unlikely to recruit to sufficient numbers to allow scientifically valid deductions to be made. The study co-ordinating team consulted the National Institute for Health Research (NIHR) monitoring team and, following further discussion with the respective programme directors, early termination of the study was agreed.
The need for better understanding of transverse myelitis epidemiology
A lower than expected frequency of patient encounters is believed to be the main factor that led to the low levels of recruitment observed in this study. The challenges associated with recruitment to rare disease trials are not restricted to this study alone. A large-scale comparison of interventional trials (24,088 trials), of which 2759 (11.5%) were classified as rare disease trials and 21,329 (88.5%) as non-rare disease trials, found that rare disease trials are more likely to be terminated early (13.7%) than non-rare disease trials (6.3%). 41 The same study found that, on average, fewer patients were recruited into rare disease trials (70.1%) than into trials of non-rare diseases (81.6%) than initially estimated. It is perhaps intuitively understandable that, because of the uncommon nature of the disorders under study, one of the most frequent problems faced during the conduct of rare disease trials is recruitment of the requisite number of study subjects. 41,42
The trial team were aware of the challenges associated with recruitment into rare disease trials and a prior feasibility assessment suggested a higher frequency of patient encounters than was eventually observed. With the yearly incidence of TM being approximately 350 cases (109 cases proportionately across the 15 open sites, based on a study team projection that 24 sites would capture 50% of cases of TM in the UK), the relatively low rate of patient encounters reported by all sites was unexpected. Furthermore, based on our estimated 35% recruitment rate, approximately 38 subjects could theoretically have been recruited by the open sites per year, rather than the two actually recruited. Interestingly, although numerous evidence (screening logs, informal discussions with recruiting site personnel and the investigator survey responses) pointed towards a low rate of patient encounters, anonymised data provided by the TM patient group, the Transverse Myelitis Society (TMS), showed that 61 registered members of the society reported having been diagnosed with TM in 2015 (of whom 57 had a confirmed TM diagnosis) (Barbara Babcock, TMS chairperson, May 2016, personal communication) (see Appendix 5). These data suggest that the incidence of the disease may not have been overestimated by the study team, especially as the TMS data are likely to be an underestimate of the true incidence of TM (reflecting only the individuals who actually took time to register with the society). However, an analysis of the geographical distribution of these subjects indicated that more than half resided > 25 miles away from a recruiting site (see Appendix 5). This observation points towards a level of geographical disconnect between the potential patients and the trial sites. Additionally, although local TM cases were often referred to, or had their treatment discussed with, the investigators involved in the trial, it is likely that some potentially eligible subjects were seen and treated exclusively by their local physician. The study team did take steps to ensure wide dissemination of the study among the neurology network by inclusion of the study details in the Association of British Neurologists newsletter and conferences, with an e-mail sent to members, presentation of the study at the TMS annual general meeting and conference, distribution of ward leaflets and admission unit posters at participating sites and the provision of study flyers for use in district general hospitals.
The low frequency of patient encounters was further complicated by the fact that a significant proportion of subjects who were considered for the trial were not eligible for inclusion into the study. One reason for this may be that the investigators may not have wholly appreciated the strictness of the eligibility criteria during the feasibility assessment. Indeed, ‘strict inclusion criteria’ was cited by 82% (n = 9/11) of respondents in the post-trial survey as a key factor that they believed had inhibited recruitment into the study. Notably, these inclusion criteria were mandated by the primary end point of the study, with recruited patients needing to have at least an ASIA impairment score of C to observe at least a two-grade improvement in this scale (see Considerations of the design of the study). This highlights the absolute necessity for further studies to establish both the incidence and, equally as important, the severity of TM cases that are observed.
Navigating operational barriers in the set-up and running of a rare disease trial
Although the low frequency of patient encounters primarily led to the study under-recruiting, other barriers played an important role in complicating the study set-up and the running of the study and ultimately the recruitment of subjects into the study. Hurdles in setting up the trial within the NHS sites, including contract and cost negotiations, local governance and a lack of capacity of research staff (research nurses’ and/or investigators’ competing priorities, i.e. clinical and trial commitments) led to the staggered opening of target sites. The overall impact of this was that, at the time of the study closure, only 62.5% (n = 15/24) of target sites were opened and recruiting. As recruitment logs were not available for sites that failed to open (n = 9), we are not able to say what the frequency of patient encounters would have been. The impact of this is best understood in light of the fact that the 24 originally targeted sites were assumed to represent the neurology services that catered for approximately half of the UK population. With only 15 of these target sites open, approximately 31% of new TM/NMO cases were geographically covered.
Lack of research capacity, described as the unavailability of site research staff or co-investigators or competing clinical priorities and commitments to other trials, played a significant role in five of the 24 target sites failing to open. Unsurprisingly, in the survey of investigators who took part in this study, 63.6% (n = 7/11) found it difficult to run all kinds of clinical trials at the present time because of a lack of time; 27.3% (n = 3/11) found it neither difficult nor easy; and one investigator (9.1%) did not currently find it difficult to run clinical trials because of a lack of time (see Appendix 6 for details).
The protracted contract negotiations and cost allocations highlight a wider issue related to differences in health service provision within different regions, in particular the interpretation of standard of care across different (neurology) services. For instance, although the premise of the study (which informed the funding structure) was that most of the study activities were found within standard patient care, some of these activities, such as the ASIA assessment, were evidently not routinely used within some neurological services. These differences impacted not only on the ability of target sites to be opened, but also on how and when sites could be authorised to commence patient recruitment. As site activation was predicated on at least one researcher within a site being ASIA assessment trained, sites that did not routinely use this assessment tool required a member of the research team (preferentially the principal investigator) to undertake the training and gain competence in ASIA assessment. Moreover, competency in ASIA assessment required online training that could take up to 6 hours. At least one site remained unopened at the point of the study closure despite having all other approvals in place and having already had their site initiation visit, as the primary investigator was unable to complete the ASIA assessment training.
It is not immediately apparent how best to overcome the barriers associated with research capacity. Investigators, especially research-active ones, are likely to routinely balance clinical activities with existing and new clinical trials, whereas research and development (R&D) departments balance the low cost-effectiveness associated with initiating rare disease trials with the cost-effectiveness of high throughput or commercial studies. Ultimately, fiscal remunerations for adopting very rare disease trials are likely to be required to facilitate the running of such trials with their associated barriers.
Considerations of the design of the study
Although the paucity of patient encounters and other operational barriers played a significant role in the study under-recruiting, the contributory effect of the study design (in terms of eligibility criteria) to the under-recruitment of the study cannot be overlooked. An analysis of site screening logs and feedback from investigators indicated that the exclusion of ‘less severe’ patients (i.e. patients with an ASIA impairment score of D) had some impact on the recruitment of patients into the study. Approximately 48% (n = 11/23) of screened patients who did not meet the eligibility criteria did not do so as a result of their symptoms being ‘mild’ or being assessed as having an ASIA impairment score of D. The study team recognised this trend while the study was still active and considered amending the protocol to allow the inclusion of patients assessed as having an ASIA impairment score of D or those whose symptoms were considered to be ‘mild’ at the time of presentation. However, the primary end point of the study required patients to have at least an ASIA impairment score of C to enable a two-grade improvement in this scale to be observed.
The short window for recruitment (5 days from the date of first commencement of steroid therapy) was the second factor that contributed to the low recruitment rate. Although it would have been easy to remove this restriction, the early treatment paradigm was key to the study question and so was retained.
Achievements of the trial
The barriers described individually in the previous section and summarised in the timeline in Figure 3 collectively contributed to a formidable barrier that prevented the study from maintaining steady momentum and recruiting to target. Nevertheless, despite the early termination of the study, the efforts involved in trying to run this trial have resulted in two major hidden rewards. First, the study team has established a network of investigators, including both adult and paediatric neurologists, who are ready to collaborate in future studies and clinical trials in TM and other neuroinflammatory conditions. Second, set-up of the study resulted in the provision of important training to local investigators with regard to evaluating patients with TM. As such, we now have 15 paediatric and adult neurology centres that are more equipped to manage adults and children with TM.
FIGURE 3.
A summary of some of the barriers encountered during the conduct of the trial. CRF, case report form.

Conclusion and recommendations
With 91% (n = 10/11) of the trial investigators (opinion leaders) surveyed agreeing that the study question is important and should be a priority and 82% (n = 9/11) noting that the outcome of the study would have changed how they treat TM patients, it is clear that our study question remains pertinent.
However, a future TM intervention trial would benefit from having robust, up-to-date information on the incidence of TM and the distribution of the severity of patients presenting with TM. Multisource incidence studies in both adults and children within the intended intervention study population would negate the need to extrapolate data. Such a trial must also accommodate the evidence accrued on the spectrum of the severity of TM within patients and thus an alternative outcome measure is required beyond the ASIA Impairment Scale. Here, prospective studies recruiting patients to carefully evaluate the utility of other available measures, such as the ASIA motor score, will be required. Importantly, the lessons learned from this study are easily applicable to other very rare disorders.
Patient and public involvement
Patient and public representatives were actively involved in the design of the research, the management of the research and the development of the participant information resources.
The TMS, Multiple Sclerosis Society and Guthy-Jackson Charitable Foundation were involved in the design of the research. Two members from the TMS served on the TSC. Their involvement was crucial both in finalising the research protocol and patient information sheet and in the overall direction that the study took. Additionally, they provided a lay perspective on the probable public perceptions of the study and offered key advice on how to maximise recruitment.
Acknowledgements
We would like to thank members of the TSC (R Hughes, M Lim, A Jacob, C Lundy, B Babcock, L Gray, M Kappler and M Sanders) and DMC (J Zajicek, S Cotterill and A Parker); the TMS; the Guthy-Jackson Charitable Foundation and UK Children’s Neurological Research Campaign (UKCNRC) for early support of the trial design and subsequent grant applications; and Carla Rush and Rosemary Howe for supporting the trial group on various aspects of the application. This study was also supported by the UK Clinical Research Collaboration-registered KCTU at King’s Health Partners, which is part funded by the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London and by the NIHR Evaluation, Trials and Studies Coordinating Centre. We also acknowledge Professor John Whitehead and the Medical Research Council’s network advice on trials methodology [Methodology Advisory Service for Trials (MAST)] for the design of the futility analysis.
Collaborators
Collaborating principal investigators
M Absoud (co-author), PA Brex (co-author), M Duddy, K Forrest, C Hemingway, A Jacob (co-author), S Jacob, R Kneen, M Lim (chief investigator), K Murray, J Palace (co-author), N Robertson (co-author), K Vijayakumar, E Wassmer, S West and V Williams.
Recruiting centres
Children’s Neurosciences, Evelina London Children’s Hospital at Guy’s and St Thomas’ NHS Foundation Trust, London (ML, MA); Department of Neurology, Guy’s and St Thomas’ NHS Foundation Trust, London (VW); Department of Neurology, King’s College Hospital NHS Foundation Trust, London (PB); Department of Paediatric Neurology, Great Ormond Street Hospital for Children NHS Foundation Trust, London (CH); Department of Paediatric Neurology, Alder Hey Children’s Hospital NHS Foundation Trust, Liverpool (RK); The Walton Centre, Walton Centre NHS Foundation Trust, Liverpool (AJ); Department of Neurology, John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford (JP); Department of Paediatric Neurology, Birmingham Children’s Hospital NHS Foundation Trust, Birmingham (EW); Department of Neurology, University Hospitals Birmingham NHS Foundation Trust, Birmingham (SJ); Department of Neurology, University Hospital of Wales, Cardiff and Vale University Health Board, Cardiff (NR); Department of Paediatric Neurology, North Bristol NHS Trust, Bristol (KV); Department of Paediatric Neurology, Royal Manchester Children’s Hospital, Central Manchester University Hospitals NHS Foundation Trust, Manchester (SW); Department of Paediatric Neurology, University Hospital Southampton NHS Foundation Trust, Southampton (KF); Department of Neurology, Newcastle Hospitals NHS Foundation Trust, Newcastle (MD); Department of Neurology, University of Edinburgh, NHS Lothian (KM).
Contributions of authors
Michael Absoud (Consultant and Research Fellow, Paediatric Neurodisability) contributed to the study and to the protocol conceptualisation and design.
Peter Brex (Consultant Neurologist, Adult Clinical Neurology) provided methodological input and clinical expertise.
Olga Ciccarelli (Reader in Neurology, Neuroimaging) provided methodological input and clinical expertise.
Onyinye Diribe (Clinical Trial Manager) contributed to study management and drafting of the final report.
Gavin Giovannoni (Professor of Neurology, Immunobiology) provided methodological input and clinical trial expertise.
Jennifer Hellier (Statistician) was the primary statistician and provided methodological input.
Rosemary Howe (Clinical Trial Manager) contributed to study management and to initial drafts of aspects of the final report.
Rachel Holland (Statistician) was the primary statistician and provided methodological input.
Joanna Kelly (Data Management Strategic Lead, Trial Data Management) contributed to the trial methodology.
Paul McCrone (Professor of Health Economics) provided methodological input.
Caroline Murphy (Operational Director of KCTU) provided methodological input.
Jackie Palace (Consultant Neurologist, Adult Clinical Neurology) provided methodological input, clinical input and clinical trial expertise.
Andrew Pickles (Professor of Biostatistics) contributed to the statistical analysis.
Michael Pike (Consultant Neurologist, Paediatric Neurology) provided methodological input and clinical expertise.
Neil Robertson (Professor of Neurology, Adult Clinical Neurology) provided methodological input and clinical expertise.
Anu Jacob (Consultant Neurologist, Adult Neurology) contributed to the study and to the protocol conceptualisation and design.
Ming Lim (Consultant and Reader in Neurology, Paediatric Neurologist) contributed to the study and protocol conceptualisation and design and to the drafting of the final report.
All authors reviewed and approved the final version of the report.
Publications
Absoud M, Gadian J, Hellier J, Brex PA, Ciccarelli O, Giovannoni G, et al. Protocol for a multicentre randomiSed controlled TRial of IntraVEnous immunoglobulin versus standard therapy for the treatment of transverse myelitis in adults and children (STRIVE). BMJ Open 2015;5:e008312. http://dx.doi.org/10.1136/bmjopen-2015-008312
Data sharing statement
All key data are included in appendices to the report and any further data desired but not included here can be obtained from the corresponding author.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Kerr DA, Ayetey H. Immunopathogenesis of acute transverse myelitis. Curr Opin Neurol 2002;15:339-47. https://doi.org/10.1097/00019052-200206000-00019.
- Borchers AT, Gershwin ME. Transverse myelitis. Autoimmun Rev 2012;11:231-48. http://dx.doi.org/10.1016/j.autrev.2011.05.018.
- Transverse Myelitis Consortium Working Group . Proposed diagnostic criteria and nosology of acute transverse myelitis. Neurology 2002;59:499-505. https://doi.org/10.1212/WNL.59.4.499.
- Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatr 2013;84:922-30. http://dx.doi.org/10.1136/jnnp-2012-302310.
- Altrocchi PH. Acute transverse myelopathy. Arch Neurol 1963;9:111-19. https://doi.org/10.1001/archneur.1963.00460080021002.
- Kaplin AI, Krishnan C, Deshpande DM, Pardo CA, Kerr DA. Diagnosis and management of acute myelopathies. Neurologist 2005;11:2-18. https://doi.org/10.1097/01.nrl.0000149975.39201.0b.
- de Seze J, Lanctin C, Lebrun C, Malikova I, Papeix C, Wiertlewski S, et al. Idiopathic acute transverse myelitis: application of the recent diagnostic criteria. Neurology 2005;65:1950-3. https://doi.org/10.1212/01.wnl.0000188896.48308.26.
- Absoud M, Lim M, Wassmer E, Pike Michael. Disorders of the Spinal Cord in Children. London: Mac Keith Press; 2013.
- Young J, Quinn S, Hurrell M, Taylor B. Clinically isolated acute transverse myelitis: prognostic features and incidence. Mult Scler 2009;15:1295-302. https://doi.org/10.1177/1352458509345906.
- Absoud M, Lim MJ, Chong WK, De Goede CG, Foster K, Gunny R, et al. Paediatric acquired demyelinating syndromes: incidence, clinical and magnetic resonance imaging features. Mult Scler 2013;19:76-8. https://doi.org/10.1177/1352458512445944.
- Greenberg BM, Thomas KP, Krishnan C, Kaplin AI, Calabresi PA, Kerr DA. Idiopathic transverse myelitis: corticosteroids, plasma exchange, or cyclophosphamide. Neurology 2007;68:1614-17. https://doi.org/10.1212/01.wnl.0000260970.63493.c8.
- Frohman EM, Wingerchuk DM. Clinical practice. Transverse myelitis. N Engl J Med 2010;363:564-72. http://dx.doi.org/10.1056/NEJMcp1001112.
- Scott TF, Frohman EM, De Seze J, Gronseth GS, Weinshenker BG. Therapeutics and Technology Assessment Subcommittee of American Academy of Neurology . Evidence-based guideline: clinical evaluation and treatment of transverse myelitis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2011;77:2128-34. https://doi.org/10.1212/WNL.0b013e31823dc535.
- Weinshenker BG, O’Brien PC, Petterson TM, Noseworthy JH, Lucchinetti CF, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 1999;46:878-86. https://doi.org/10.1002/1531-8249(199912)46:6<878::AID-ANA10>3.0.CO;2-Q.
- Hughes RA, Dalakas MC, Cornblath DR, Latov N, Weksler ME, Relkin N. Clinical applications of intravenous immunoglobulins in neurology. Clin Exp Immunol 2009;158:34-42. http://dx.doi.org/10.1111/j.1365-2249.2009.04025.x.
- Banwell B, Ghezzi A, Bar-Or A, Mikaeloff Y, Tardieu M. Multiple sclerosis in children: clinical diagnosis, therapeutic strategies, and future directions. Lancet Neurol 2007;6:887-902. https://doi.org/10.1016/S1474-4422(07)70242-9.
- Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler 2014;20:501-4. https://doi.org/10.1177/1352458513495938.
- Dalakas MC. Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology 1998;51:S2-8. https://doi.org/10.1212/WNL.51.6_Suppl_5.S2.
- Awad A, Stuve O. Idiopathic transverse myelitis and neuromyelitis optica: clinical profiles, pathophysiology and therapeutic choices. Curr Neuropharmacol 2011;9:417-28. https://doi.org/10.2174/157015911796557948.
- Steeves JD, Lammertse D, Curt A, Fawcett JW, Tuszynski MH, Ditunno JF, et al. Guidelines for the conduct of clinical trials for spinal cord injury (SCI) as developed by the ICCP panel: clinical trial outcome measures. Spinal Cord 2007;45:206-21. https://doi.org/10.1038/sj.sc.3102008.
- Savic G, Bergstrom EM, Frankel HL, Jamous MA, Jones PW. Inter-rater reliability of motor and sensory examinations performed according to American Spinal Injury Association standards. Spinal Cord 2007;45:444-51. https://doi.org/10.1038/sj.sc.3102044.
- Sepülveda M, Blanco Y, Rovira A, Rio J, Mendibe M, Llufriu S, et al. Analysis of prognostic factors associated with longitudinally extensive transverse myelitis. Mult Scler 2013;19:742-8. https://doi.org/10.1177/1352458512461968.
- Scott D, Ferguson GD, Jelsma J. The use of the EQ-5D-Y health related quality of life outcome measure in children in the Western Cape, South Africa: psychometric properties, feasibility and usefulness – a longitudinal, analytical study. Health Qual Life Outcomes 2017;15. https://doi.org/10.1186/s12955-017-0590-3.
- Whitehurst DG, Mittmann N, Noonan VK, Dvorak MF, Bryan S. Health state descriptions, valuations and individuals’ capacity to walk: a comparative evaluation of preference-based instruments in the context of spinal cord injury. Qual Life Res 2016;25:2481-96. https://doi.org/10.1007/s11136-016-1297-3.
- Biering-Sørensen F, Charlifue S, DeVivo M, Noonan V, Post M, Stripling T, et al. International Spinal Cord Injury Data Sets. Spinal Cord 2006;44:530-4. http://dx.doi.org/10.1038/sj.sc.3101930.
- Mahdi-Rogers M, McCrone P, Hughes RA. Economic costs and quality of life in chronic inflammatory neuropathies in southeast England. Eur J Neurol 2014;21:34-9. https://doi.org/10.1111/ene.12245.
- Juul T, Bazzocchi G, Coggrave M, Johannesen IL, Thiyagarajan CC, Poletti E, et al. Reliability of the International Spinal Cord Injury Bowel Function Basic and Extended Data Sets. Spinal Cord 2011;49:886-91. https://doi.org/10.1038/sc.2011.23.
- Goetz LL, Cardenas DD, Kennelly M, Bonne Lee BS, Linsenmeyer T, Moser C, et al. International Spinal Cord Injury Urinary Tract Infection Basic Data Set. Spinal Cord 2013;51:700-4. https://doi.org/10.1038/sc.2013.72.
- Buck D. The PedsQL as a measure of parent-rated quality of life in healthy UK toddlers: psychometric properties and cross-cultural comparisons. J Child Health Care 2012;16:331-8. https://doi.org/10.1177/1367493512448127.
- Oladeji O, Johnston TE, Smith BT, Mulcahey MJ, Betz RR, Lauer RT. Quality of life in children with spinal cord injury. Pediatr Phys Ther 2007;19:296-300. https://doi.org/10.1097/PEP.0b013e31815a12ef.
- Mahnig S, Landmann G, Stockinger L, Opsommer E. Pain assessment according to the International Spinal Cord Injury Pain classification in patients with spinal cord injury referred to a multidisciplinary pain center. Spinal Cord 2016;54:809-15. https://doi.org/10.1038/sc.2015.219.
- Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485-9. https://doi.org/10.1212/01.wnl.0000216139.44259.74.
- Absoud M, Gadian J, Hellier J, Brex PA, Ciccarelli O, Giovannoni G, et al. Protocol for a multicentre randomiSed controlled TRial of IntraVEnous immunoglobulin versus standard therapy for the treatment of transverse myelitis in adults and children (STRIVE). BMJ Open 2015;5.
- Medicines and Healthcare products Regulatory Agency . Good Clinical Practice for Clinical Trials 2014. www.gov.uk/guidance/good-clinical-practice-for-clinical-trials (accessed 30 March 2017).
- US Food and Drug Administration . CFR – Code of Federal Regulations Title 21 n.d. www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=11&showFR=1 (accessed 30 March 2017).
- European Commission . Medicinal Products for Human Use n.d. https://ec.europa.eu/health/human-use/clinical-trials_en (accessed 30 March 2017).
- Great Britain . Statutory Instruments: 2004 No. 1031 – Medicines. The Medicines for Human Use (Clinical Trials) Regulations 2004 – Regulation 46(2) 2004. www.legislation.gov.uk/uksi/2004/1031/pdfs/uksi_20041031_en.pdf (accessed 30 March 2017).
- Data Protection Act 1998. London: The Stationery Office; 1998.
- White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.d40.
- McCrone P, Chisholm D, Knapp M, Hughes R, Comi G, Dalakas MC, et al. Cost–utility analysis of intravenous immunoglobulin and prednisolone for chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol 2003;10:687-94. https://doi.org/10.1046/j.1351-5101.2003.00701.x.
- Bell SA, Tudur Smith C. A comparison of interventional clinical trials in rare versus non-rare diseases: an analysis of ClinicalTrials.gov. Orphanet J Rare Dis 2014;9. http://dx.doi.org/10.1186/s13023-014-0170-0.
- Griggs RC, Batshaw M, Dunkle M, Gopal-Srivastava R, Kaye E, Krischer J, et al. Clinical research for rare disease: opportunities, challenges, and solutions. Mol Genet Metab 2009;96:20-6. http://dx.doi.org/10.1016/j.ymgme.2008.10.003.
Appendix 1 Dosing table for intravenous immunoglobulin administration
Appendix 2 Clinicoradiological diagnostic algorithm
FIGURE 4.
Clinicoradiological diagnostic algorithm. ACE, angiotensin-converting enzyme; ADEM, acute disseminated encephalomyelitis; CMV, cytomegalovirus; EBV, Epstein–Barr virus; HHV, human herpesvirus; HIV, human immunodeficiency virus; HSV, herpes simplex virus; HTLV-1, human T-lymphotrophic virus 1; MCTD, mixed connective tissue disease; SLE, systemic lupus erythematosus; TB, tuberculosis; VZV, varicella zoster virus.

Appendix 3 Common side effects associated with Intratect
Intratect can cause the following AEs, which may occur occasionally: chills, headache, fever, vomiting, allergic reactions, nausea, arthralgia, low blood pressure and mild back pain.
Rarely, human normal immunoglobulins may cause a sudden fall in blood pressure and, in isolated cases, anaphylactic shock, even when the patient has shown no hypersensitivity to previous administration.
Cases of reversible aseptic meningitis, isolated cases of reversible haemolytic anaemia/haemolysis and rare cases of transient cutaneous reactions have also been observed with human normal immunoglobulin, as well as an increase in serum creatinine level and/or acute renal failure.
Very rarely, thromboembolic reactions such as myocardial infarction, stroke, pulmonary embolism and deep-vein thrombosis have been observed.
Other reported adverse reactions are:
-
cardiac disorders – angina pectoris (very rare)
-
general disorders and administration site conditions – rigors (very rare)
-
immune system disorders – anaphylactic shock (very rare), hypersensitivity (very rare)
-
investigations – blood pressure decreased (very rare)
-
musculoskeletal and connective tissue disorders – back pain (very rare)
-
respiratory, thoracic and mediastinal disorders – dyspnoea not otherwise specified (very rare)
-
vascular disorders – shock (very rare).
The AEs reported above are expected in the sense that they are possible known side effects of the study medication, but all reported instances of both SAEs and non-SAEs would have been reported in this study. For a more detailed list of all reactions, see the Intratect SmPC [www.medicines.org.uk/emc/medicine/23175/SPC/intratect/ (accessed 22 March 2017)].
Appendix 4 Futility analysis plan
Proposal for an interim futility analysis
Introduction
Patients suffering from TM will be randomised equally between IVIG (the experimental arm, E) and steroids (the control arm, C). The primary analysis will concern response to treatment, defined as an improvement of two grades on a paralysis assessment scale over a 6-month period following treatment. It is anticipated that the success rate on C will be pC = 0.5. The trial is to have 90% power to achieve significance at the 0.05 level (two-sided) if the success rate on E is pE = 0.75.
The final analysis of the study can be conducted in terms of the statistic χ2 = Σ(O – E)2/E, which can be shown to be equal to Z2/V, where:
where nC and nE denote the numbers of patients and SC and SE denote the numbers of successes on C and E, respectively, n = nC + nE, S = SC + SE, F = n – S, p^C=SC/nC p^E=SE/nE and p¯=12(pC+pE).
In fact, it will be concluded that E is significantly superior to C if C = Z/√V exceeds a suitable critical value k.
For equal randomisation, we have nC = nE = 0.5n and:(2)Z=12(SE−SC) and V=SF4n.
Sample size calculation
The SAS program sample size chi concerns a trial in which 152 patients are randomised, 76 to C and 76 to E. The probability that SC = i and SE = j is found for all i, j = 0,. . .,76. Thus, the probability of all 772 possible trial outcomes is found. The probability is found assuming that pC = pE = 0.5 and assuming that pC = 0.5 and pE = 0.75. The possible outcomes are then arranged in descending order according to T (denoting chi-squared test statistic), and cumulative probabilities of T being larger than or equal to every possible value from 76 to –76 are computed. Reading the last row of the output for which χ = 1.95441 shows that P(χ ≥ 1.95441) is equal to 0.023555 when pC = pE = 0.5 and 0.90338 when pC = 0.5 and pE = 0.75. Thus, the appropriate value for the critical value k is 1.95441. No suitable critical value can be found for n = 150 and so the sample size should be n = 152.
This exact sample size calculation depends on the control success rate being precisely 0.5, although the SAS program can be used to evaluate the decision rule – reject H0 if χ ≥ 1.95441 – under any other pair of success rates. The sample size found is close to that obtained using Stata. Once the data are available, the analysis will be based on Z, allowing for any departures from the intended sample size of 76 in each arm. Additional patients to allow for potential dropouts can be added later.
An interim futility analysis
Suppose that an interim futility analysis is conducted after 52 patients have provided a response (26 in each treatment arm). The trial is then terminated with the conclusion that E is no better than C if, based on these 52 patients, χ < 0. If the sample sizes are equal, this occurs if SE < SC. Otherwise, the trial proceeds to the full sample size of 152, with 76 patients in each treatment arm, and the null hypothesis is rejected if χ ≥ 1.95441.
The SAS program two-stage chi concerns such a design. The probability that SC1 = i1, SC2 = i2, SE1 = j1 and SE2 = j2 is found for all i1, j1 = 0,. . .,26 and all i2, j2 = 0,. . .,50, where SCr and SEr are the success totals in the rth stage of the trial, r = 1, 2. Thus, the probability of every possible combination of outcomes in the two stages of the trial is found. These are ordered by the final value of C and the results for which P(χ ≥ k) = 0.025 under the null hypothesis and ≥ 0.90 under the alternative hypothesis are printed out. This checking program takes a while to run and produces a lot of output. Line 5031 of the output confirms that P(χ ≥ 1.95441) = 0.023555 when pC = pE = 0.5 and 0.90338 when pC = 0.5 and pE = 0.75.
The SAS program two-stage – interim – chi evaluates the design, but this time outcomes in which i1 > j1 are deleted. This corresponds to stopping corresponding trials for futility. In this case, P(χ ≥ 1.95441) = 0.022795 when pC = pE = 0.5 and 0.89462 when pC = 0.5 and pE = 0.75. This represents a very small loss of power.
The SAS program two-stage – stage 1 – chi evaluates the properties of the first stage of the design. It shows that the probability of abandoning the study at the interim analysis is 0.44494 when pC = pE = 0.5 and 0.020060 when pC = 0.5 and pE = 0.75. Thus, there is a good chance of stopping for futility when the treatments are equivalent and a very small chance when the desired treatment effect is present.
Discussion
The calculations described in the previous sections indicate that a futility analysis conducted when approximately one-third of the observations are available would be worthwhile and would have minimal effect on the power. A final analysis conducted ignoring the interim analysis would be slightly conservative in the sense of underestimating the advantage of E over C and reporting a p-value that was bigger (and thus less significant) than any properly adjusted p-value. It would not appear to be worth making such an adjustment.
If 152 patients are recruited over 2 years, then 52 patients would be recruited after 8.2 months. The interim analysis would take place at 14.2 months, by which time a further 38 patients would have been recruited. If the analysis was instant, there would be the potential to reduce the sample size by 62 patients, although this saving would be reduced because of continued recruitment during the analysis period. If recruitment was to stretch beyond 2 years, the benefits of early stopping would increase.
The calculations performed are qualitative as the actual trial might depart from the model investigated here in various small ways. Here, we declare E to be superior to C if χ ≥ 1.95441, although in practice the more conventional criterion of χ ≥ 1.960 would probably be used. The calculations made here are exact, but only for the null hypothesis pC = pE = 0.5 and not for the more general null hypothesis pC = pE. Calculations could be rerun for the criterion χ ≥ 1.960; a slight increase in sample size might be needed to preserve power. In practice, the sample sizes at the interim and final analyses might not be exactly 26 and 76 in each group and they might not be equal in the two groups. The more general formula for C would then be used and this is another reason for retaining the conventional cut-off value of 1.960.
Variations to the procedure, with different sample sizes at the interim and the null, can be evaluated and properties under different pairs of values of pC and pE can be found. It would also be simple to investigate a more stringent futility criterion, requiring C to exceed a value such as 0.5 or 1 to continue. This would make the loss of power more substantial and open up the question of whether it should be compensated for by an increase in sample size.
Notice that no opportunity for stopping at the interim analysis because of strong evidence of efficacy is allowed. If that were allowed, then the properties of the method would need substantial re-evaluation and conventional analyses would no longer be conservative.
Appendix 5 Geographical distribution of those diagnosed with transverse myelitis in 2015
FIGURE 5.
Geographical distribution of those diagnosed with TM in 2015 (as per TMS data) in relation to the trial sites (n = 51 of the 61 registered patients whose postal code allowed for mapping of their residence to local and regional services). Each numbered black circle represents the number of individuals within the region on the map reported (to the TMS) to have been diagnosed with TM in 2015. Each green circle represents a 25-mile radius around a trial recruiting site and each red circle represents a 25-mile radius around a trial site that was targeted for opening before the study was terminated.
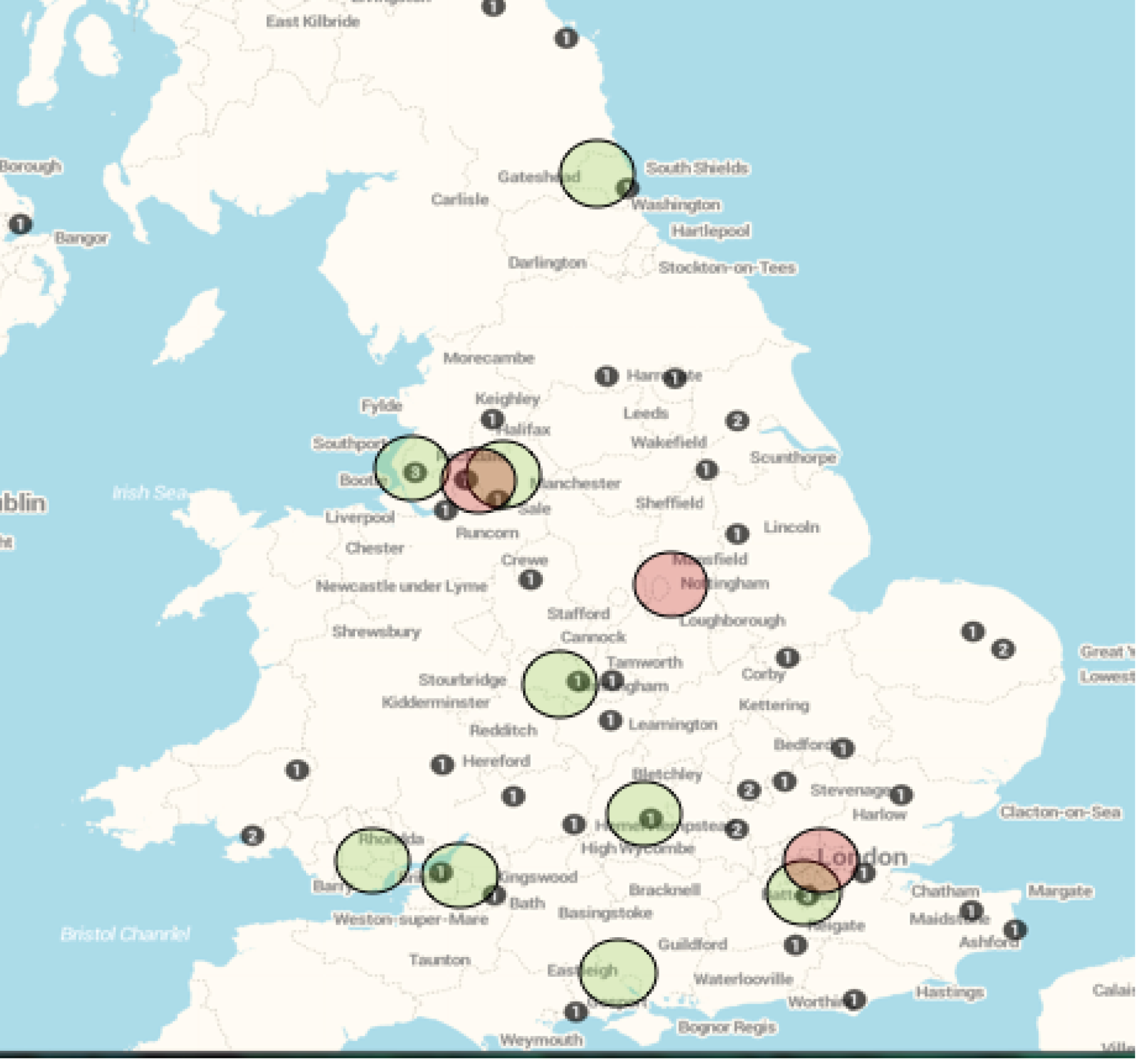
Appendix 6 Investigator responses to the post-trial survey
1. The aim and overall objective of the study was clear (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
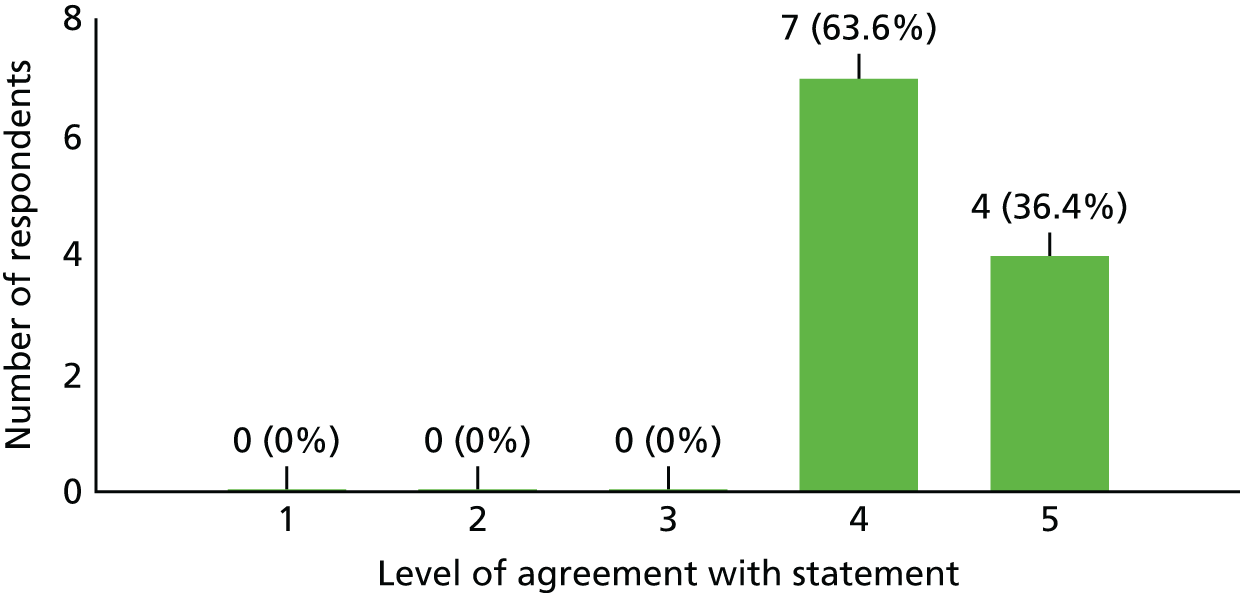
2. The study design was simple enough to understand (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
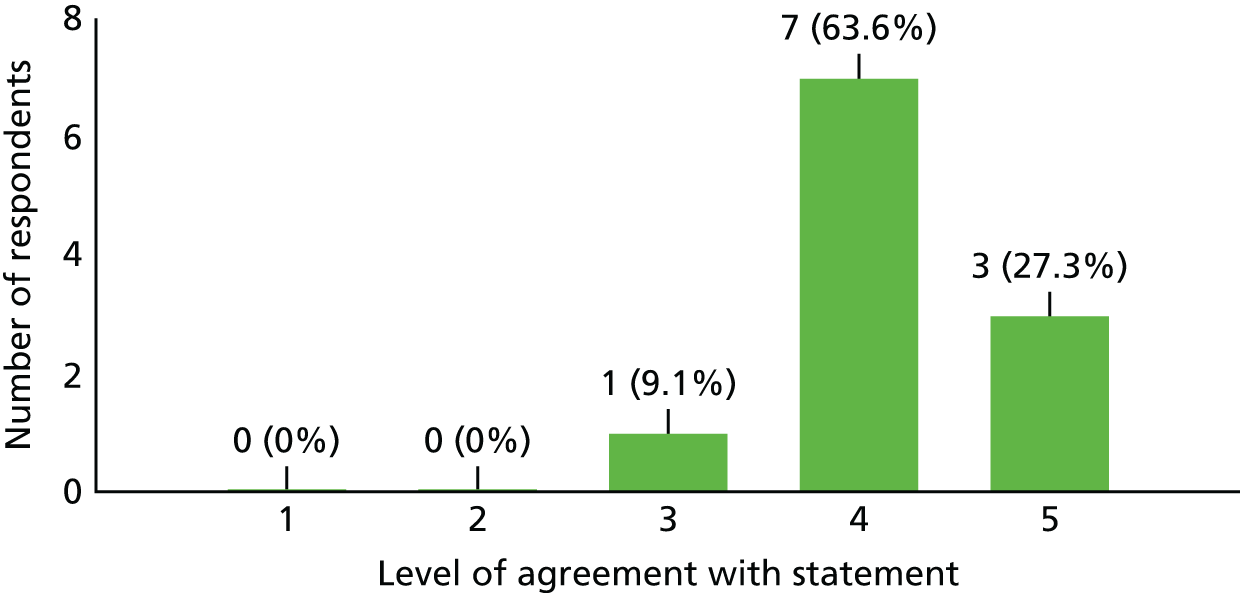
3. All of the study procedures (including study visits at 3, 6 and 12 months post treatment) were in line with our standard care for TM/NMO patients (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

4a. The ASIA score was an appropriate measure to use (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

4b. What alternative would you have considered if any? (four responses)

5. The trial offered good treatment options for TM/NMO (10 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

6. The trial was sufficiently pragmatic (nine responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
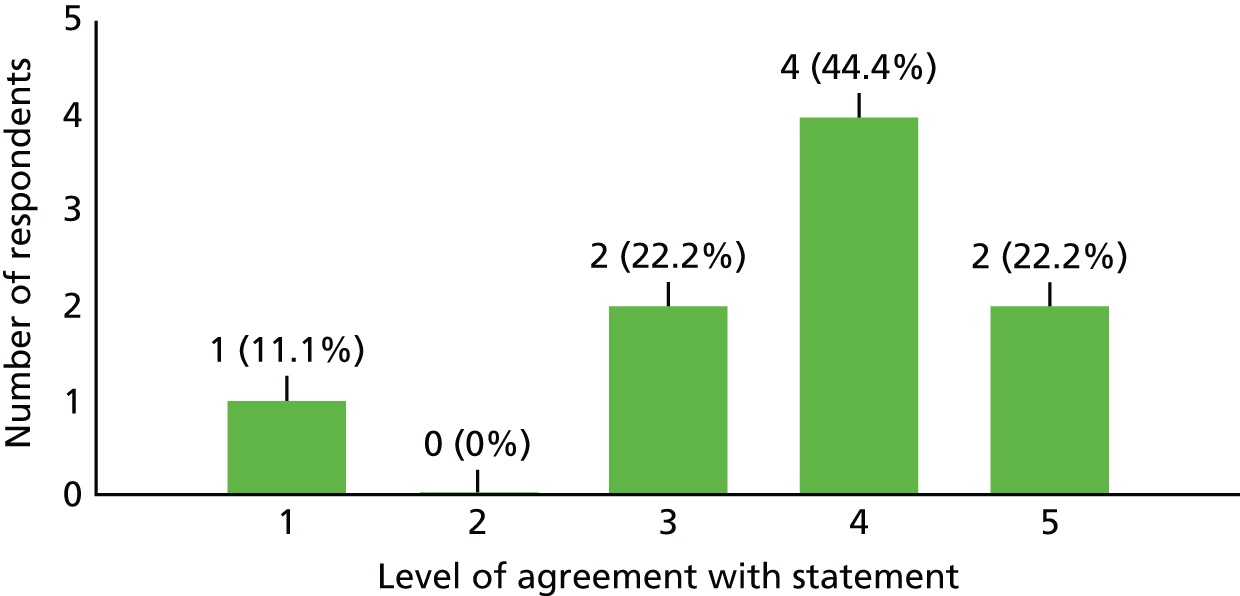
7. The site was an appropriate choice for the trial (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
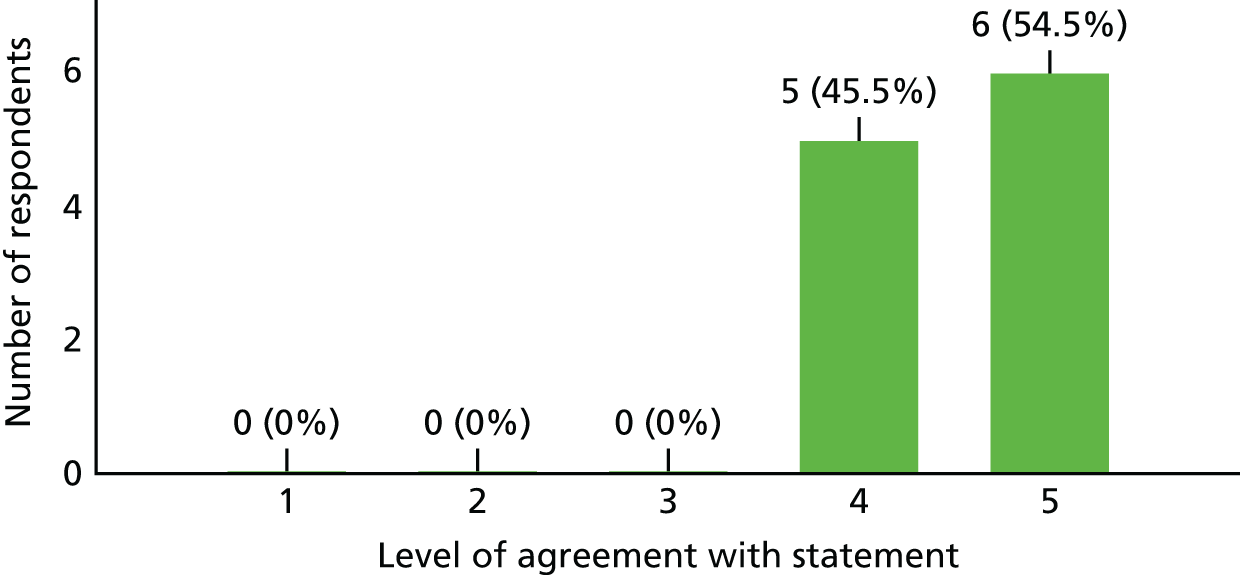
Investigator
1. I think addressing the study question is highly important and should be a priority (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
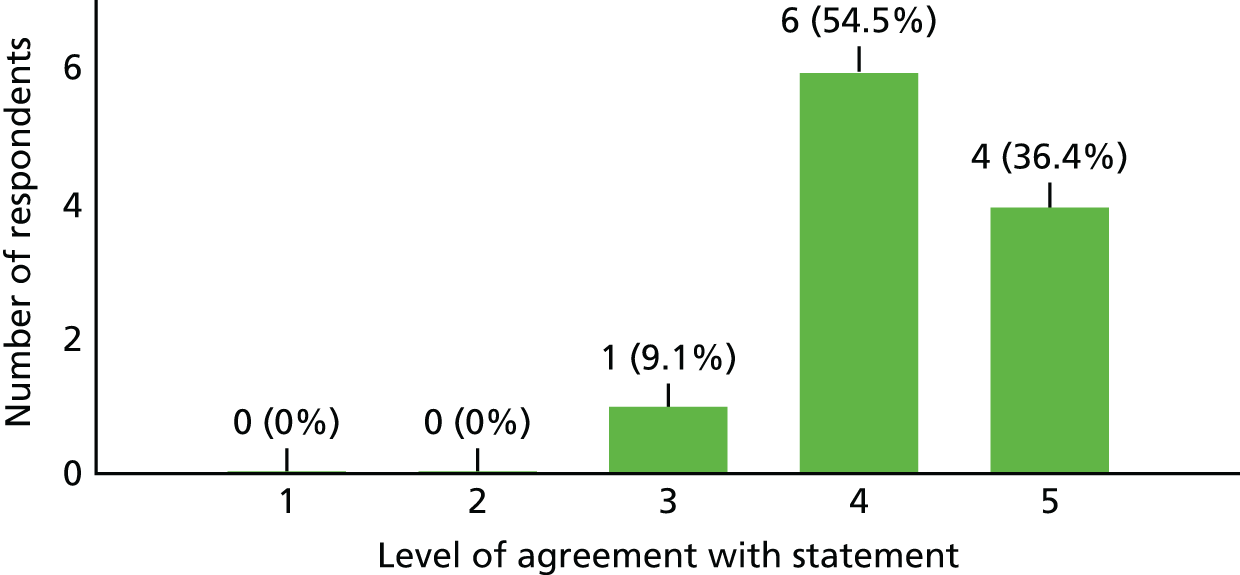
2. The outcome of this study would have changed how I treat my TM patients (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
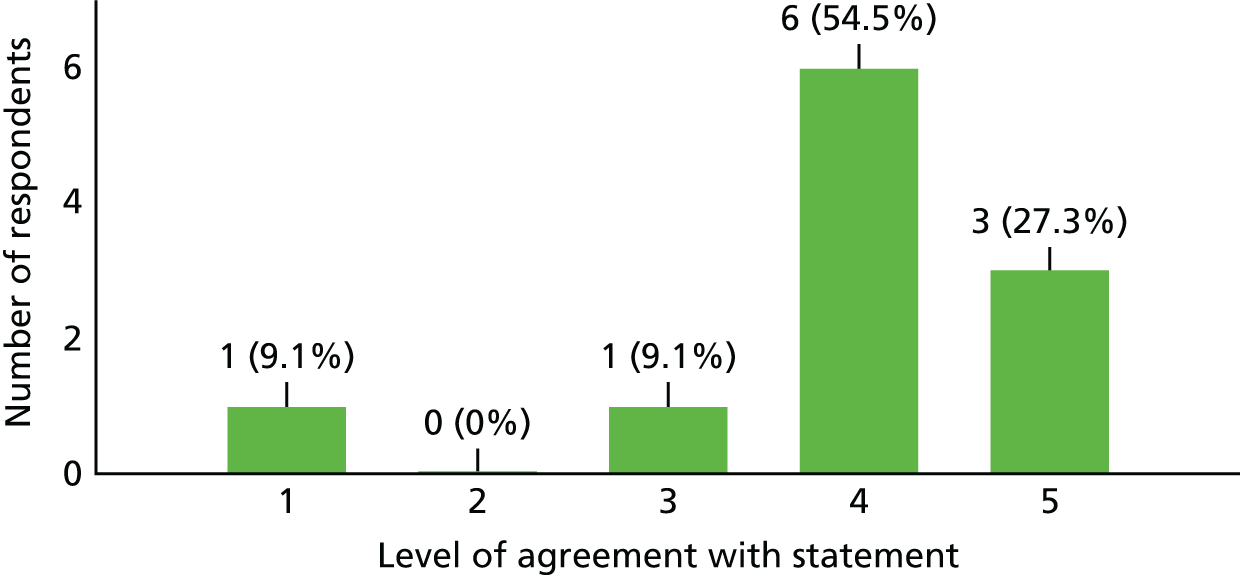
3. Within my daily workload, I believe I had enough time to allocate to activities that the study required (both clinical and administrative) (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
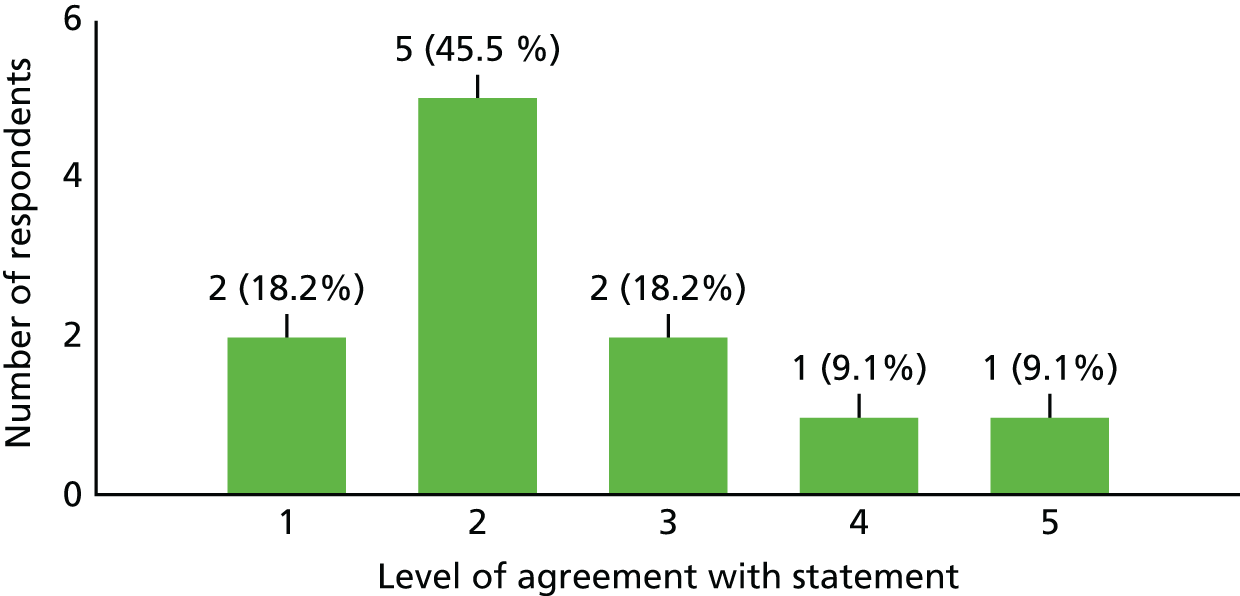
4. I generally find it difficult to participate in all kinds of clinical trials nowadays because of a lack of time (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
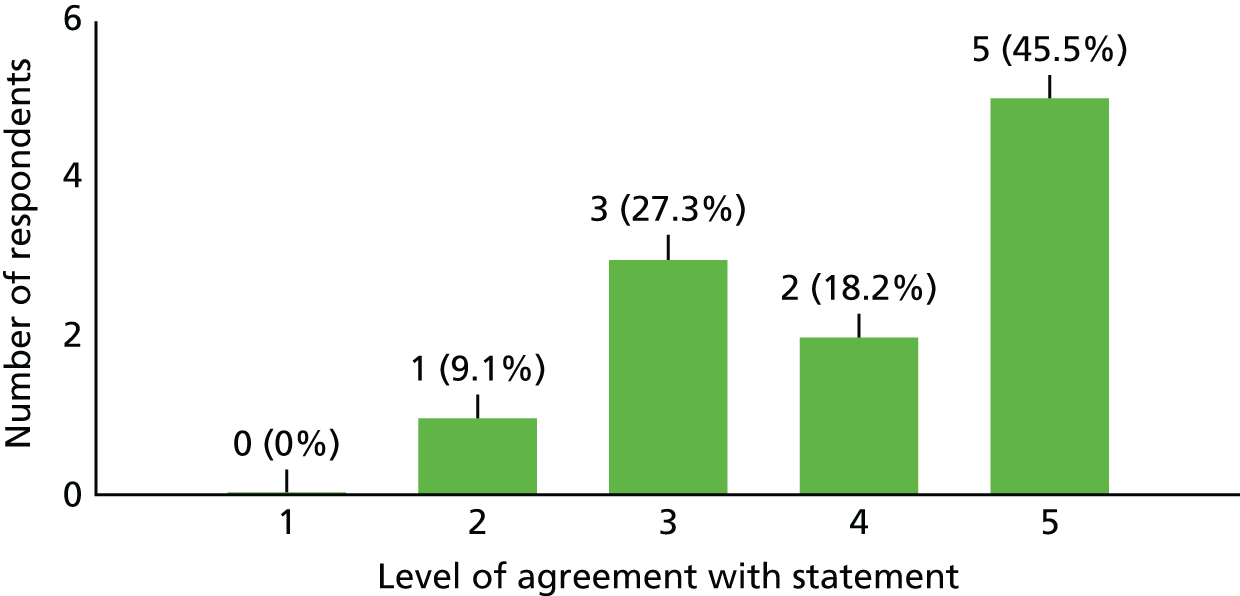
5. A study visit by a participant had (or would have had) an undesirable impact on my clinic (e.g. significantly increasing the clinic time or time for administration of the clinic) (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
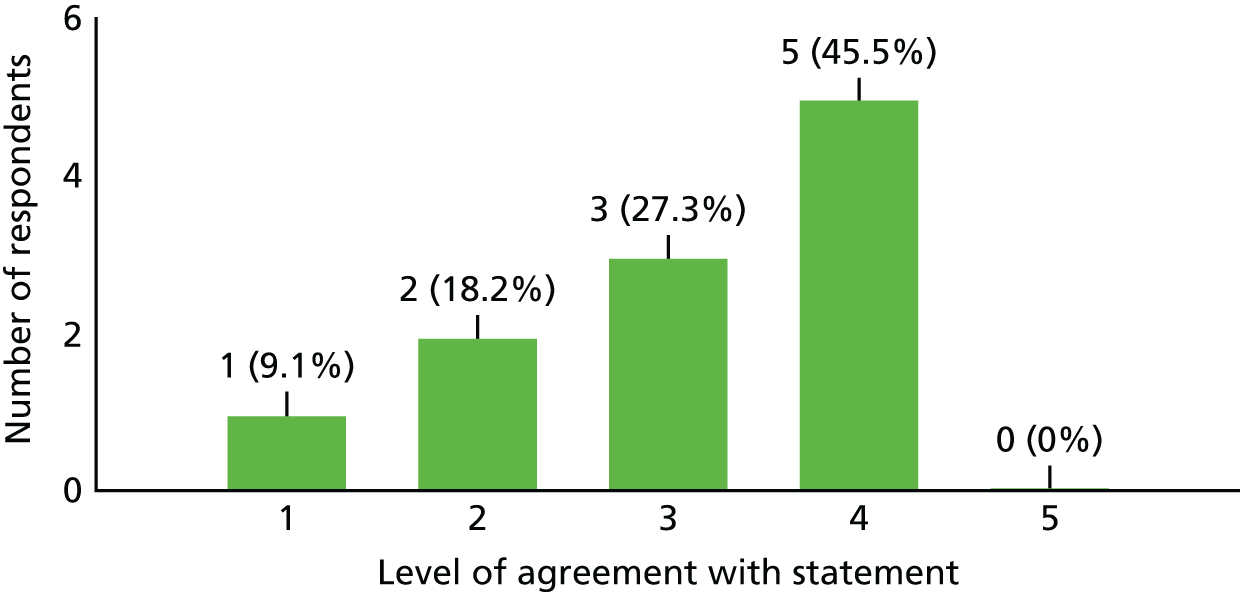
6. I generally find it difficult to find time for all study visits nowadays (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

7. What in your opinion was the single most important reason this trial failed (important, please answer) (11 responses)

8. What would you have changed about the study (important, please answer) (11 responses)

Study set-up and recruitment
1. I was actively involved in the site feasibility discussion before the study opened at my site (10 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

2. Set-up of the study felt complicated at times (10 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

3. All things considered, I think funding for sites for this study was not really adequate (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).

4. I found my interaction with our R&D department useful during the set-up and administration of this study (if applicable) (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
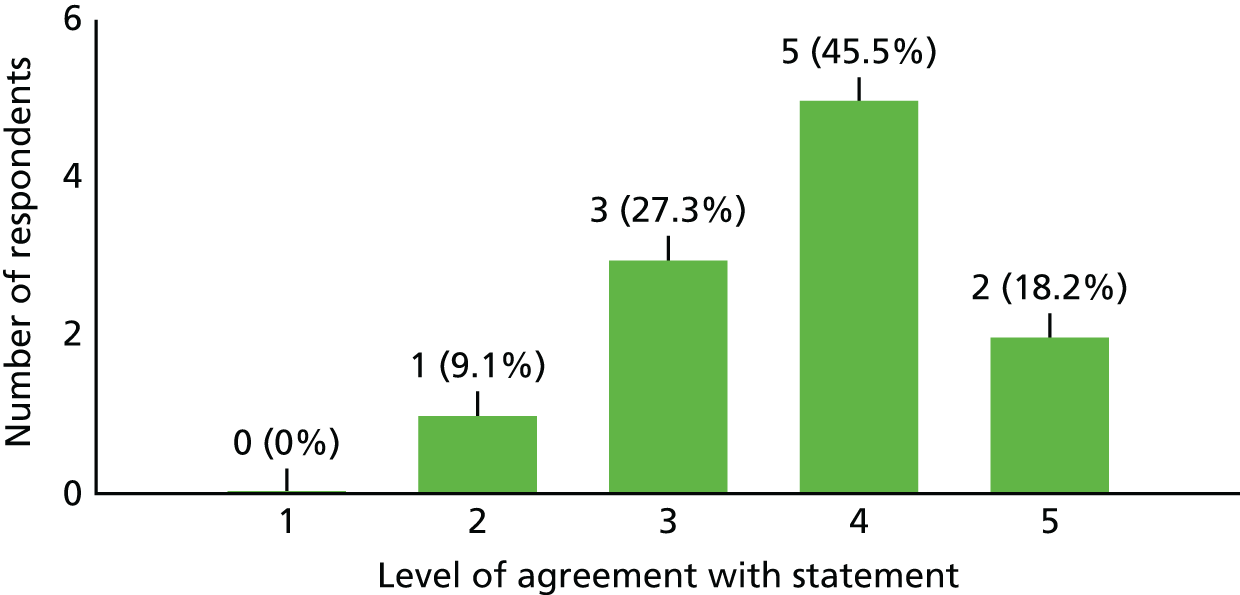
5. I was unable to recruit because (chose as many as applicable) (11 responses)
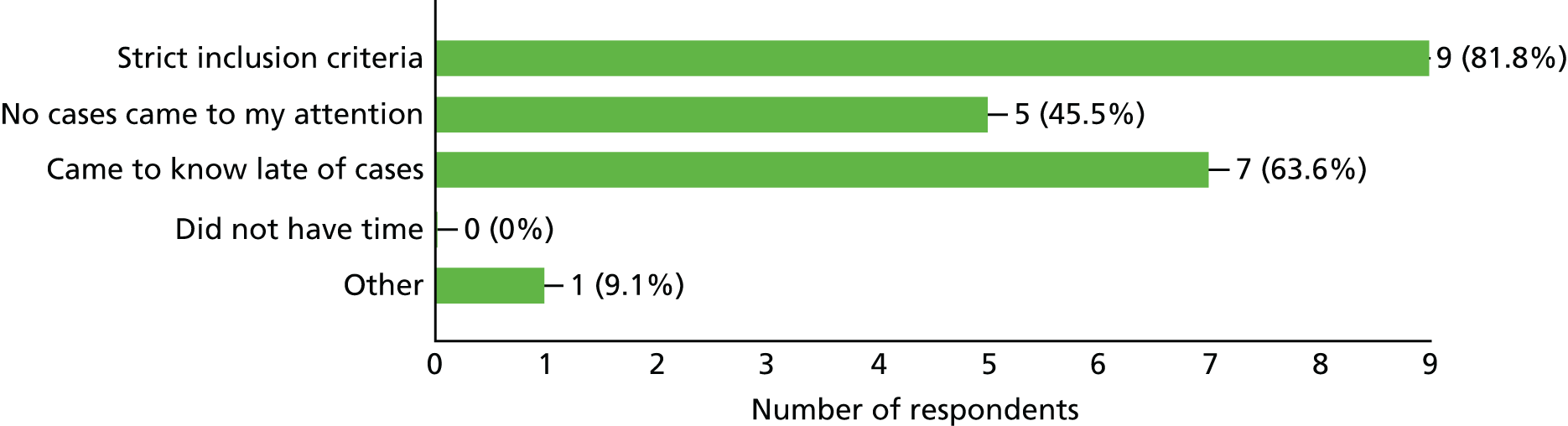
Other
1. I received as much assistance as needed from the study co-ordinating team (trial manager, sponsors, etc.) (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
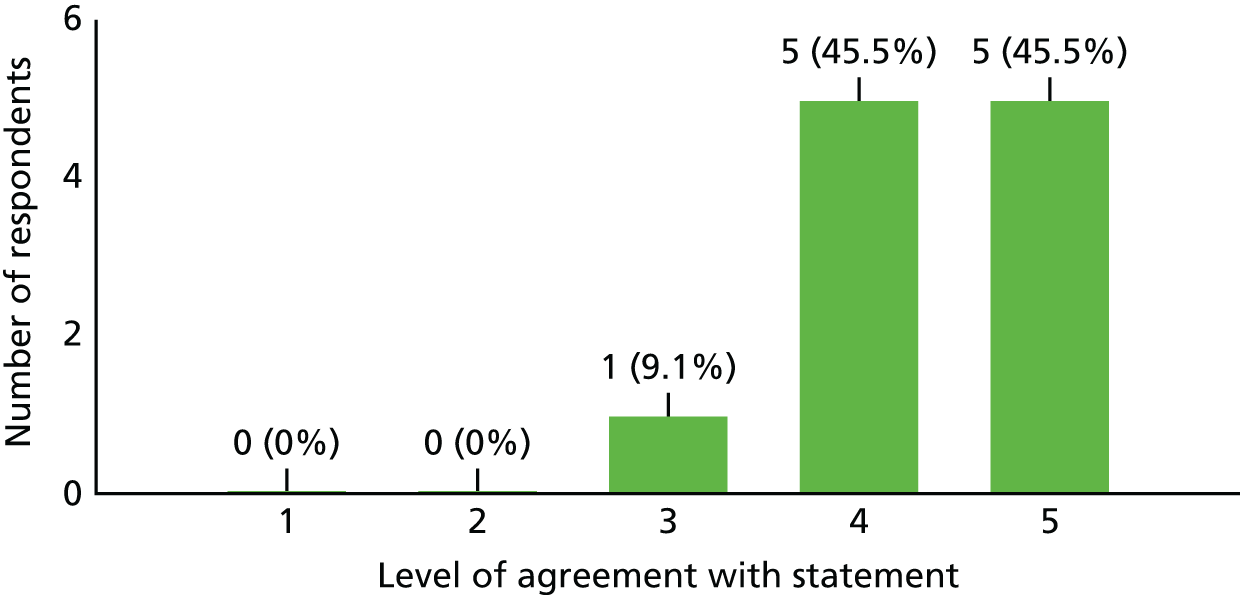
2. The study co-ordinating team could have done more to increase awareness of the study among other clinicians or the potential patient population (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
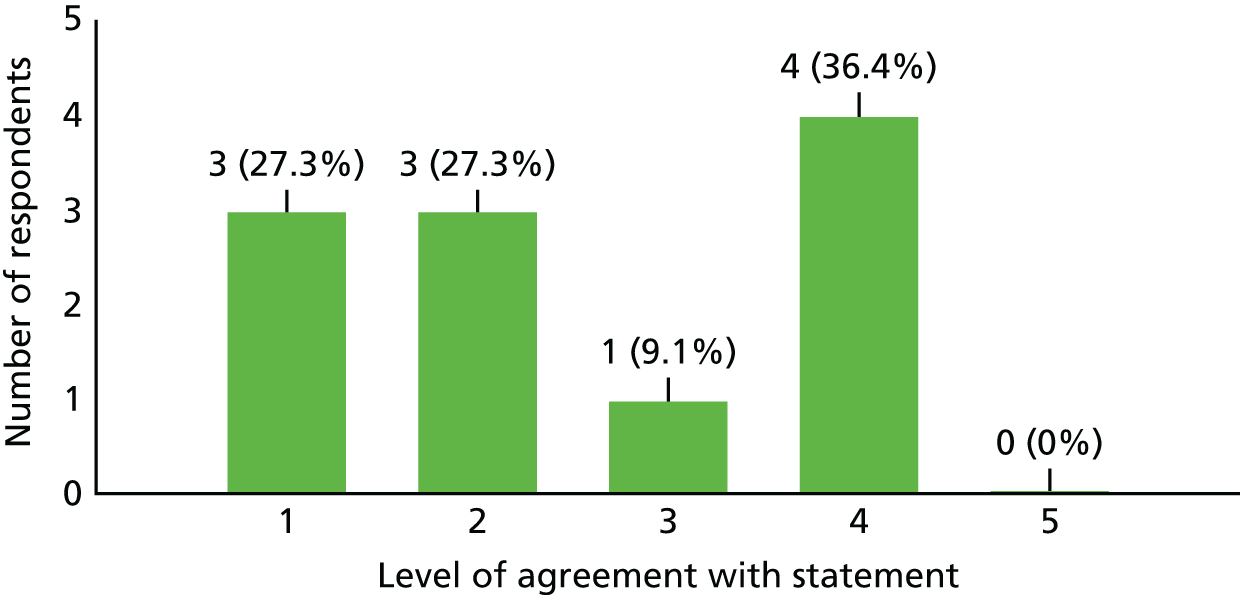
3. In future, I would be interested in collaborating with this team in designing and running another study (11 responses) (1 = strongly disagree, 2 = disagree, 3 = neither agree nor disagree, 4 = agree, 5 = strongly agree).
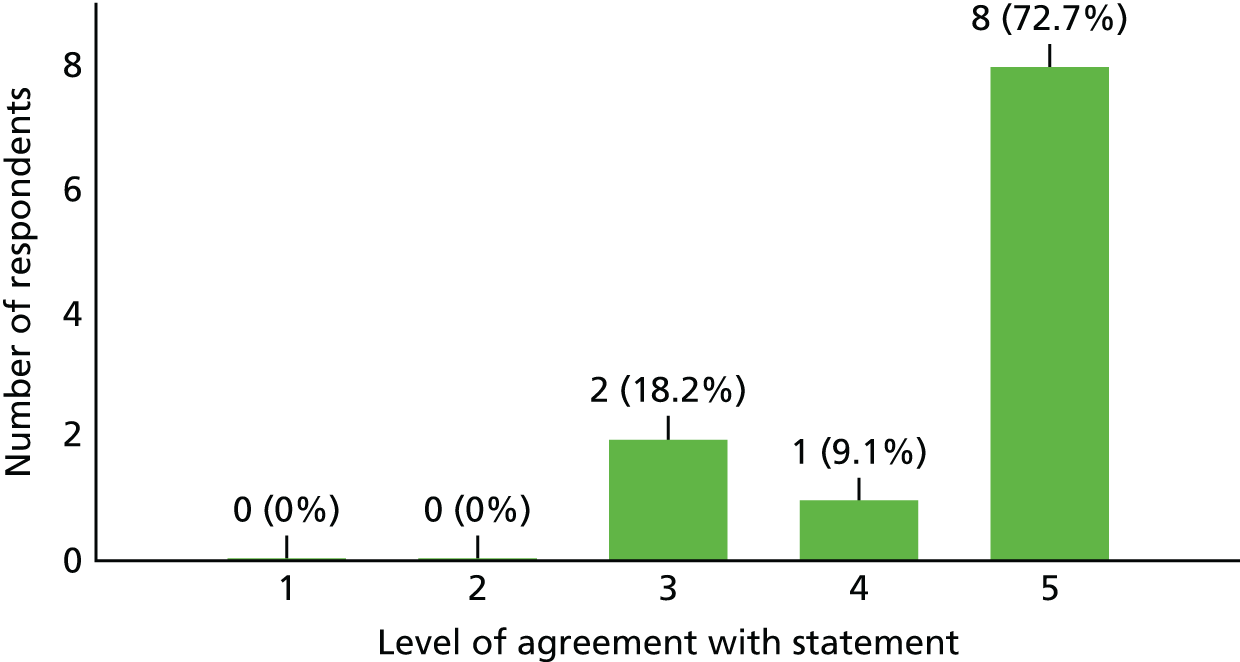
4. Any other comments? Please feel free to be critical, as we would like to learn from any mistakes made in this study (five responses).
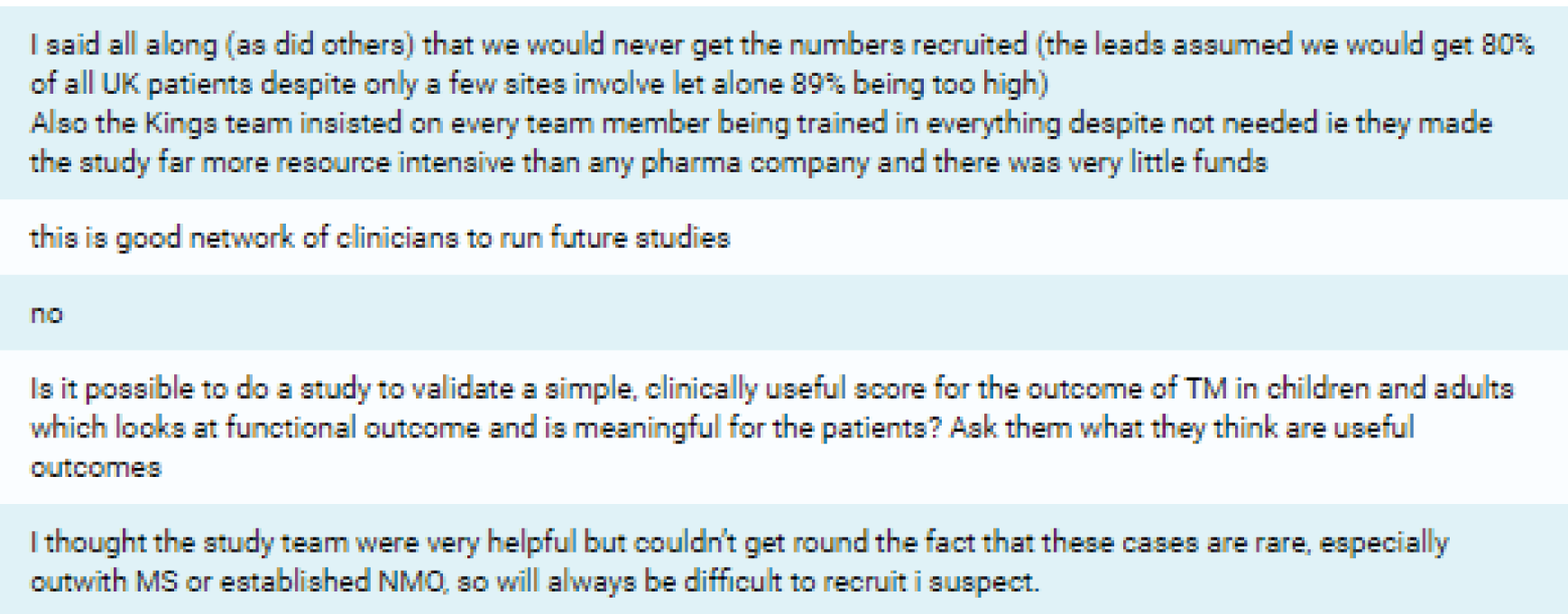
List of abbreviations
- AE
- adverse event
- AQP4
- aquaporin 4
- ASIA
- American Spinal Injury Association
- BMI
- body mass index
- CNS
- central nervous system
- CSF
- cerebrospinal fluid
- CSRI
- Client Service Receipt Inventory
- DMC
- Data Monitoring Committee
- eCRF
- electronic case report form
- EDSS
- Expanded Disability Status Scale
- EQ-5D-5L
- EuroQol-5 Dimensions five-level version
- EQ-5D-Y
- EuroQol-5 Dimensions youth version
- IgG
- immunoglobulin G
- IMP
- investigational medicinal product
- ITT
- intention to treat
- IV
- intravenous
- IVIG
- intravenous immunoglobulin
- IVMP
- intravenous methylprednisolone
- KCTU
- King’s Clinical Trials Unit
- MAR
- missing at random
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRI
- magnetic resonance imaging
- MS
- multiple sclerosis
- NIHR
- National Institute for Health Research
- NMO
- neuromyelitis optica
- PedsQL
- Paediatric Quality of Life Inventory
- PLEX
- plasma exchange
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SCI-QOL
- Spinal Cord Injury Quality of Life
- SmPC
- Summary of Product Characteristics
- TM
- transverse myelitis
- TMG
- Trial Management Group
- TMS
- Transverse Myelitus Society
- TSC
- Trial Steering Committee