
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 14/27/02. The contractual start date was in June 2015. The draft report began editorial review in November 2016 and was accepted for publication in February 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Hanna Bergman worked for Enhance Reviews Ltd during the preparation of this report and during the preparation of Cochrane reviews related to this report, and was paid for her contribution in doing so. Enhance Reviews Ltd is a private company that performs systematic reviews of literature and currently does not take commissions from industry. Hanna Bergman works for Cochrane Response, an evidence consultancy that takes commissions from health-care guideline developers and policy-makers. Adriani Nikolalopoulou was paid for contributing to the statistical analysis for this report. Karla Soares-Weiser was the managing director of Enhance Reviews Ltd. Karla Soares-Weiser has since moved to work for Cochrane, has not drawn a salary from this project, and had limited involvement in co-ordinating the activities of this project.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Bergman et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background
Since the 1950s, antipsychotic medication has been used extensively to control psychotic symptoms and to reduce the harm caused by the symptoms of chronic mental illness, including schizophrenia, bipolar disorder and dementia. Other illnesses that necessitate long-term antipsychotic treatment include autism, Tourette syndrome and other behavioural disturbances. Antipsychotic drugs are associated with a wide range of adverse effects, including tardive dyskinesia (TD), the late onset of involuntary, repetitive body movements, often involving the face and tongue. Critical problems associated with severe TD include difficulty swallowing, locomotion difficulties, involvement of respiratory muscles and speech being rendered unintelligible. TD can be extremely disfiguring, compounds stigma and is associated with poor compliance to treatment. 1
Tardive dyskinesia occurs in > 20% of people who use first-generation antipsychotic (FGA) drugs continually for > 3 months,1 and every year 4–5% of those who continually use these drugs begin to show signs of TD. 1 When second-generation antipsychotic (SGA) drugs were introduced in the 1990s, many hoped that they would not cause TD. 2,3 Although the risks of developing TD with SGA drugs do seem to be reduced, they have not been eliminated. 1,3 There is some evidence to indicate that rates of TD do not differ at all between first- and second-generation antipsychotic drugs, making the distinction between the two ‘generations’ of drugs increasingly redundant. 2 Recent assessments of the incidence and prevalence of TD range from 5% to 60% of patients taking antipsychotic medication for long periods. 4 For example, one recent, well-conducted survey from the Netherlands found that, of 209 people with chronic severe mental illness receiving antipsychotic medication, 28% had TD (yearly incidence rate of TD 19.6%). 5,6 Furthermore, the study reconfirmed that TD was positively associated with age [hazard ratio per year exposure 1.04, 95% confidence interval (CI) 1.02 to 1.06]. 5,6
The large, definitive US randomised trial of antipsychotic treatments for schizophrenia [Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE)], with a 4-year period of follow-up, obtained an incidence rate of TD of around 17% and found no significant difference in rates between first- and second-generation (olanzapine, quetiapine, risperidone, ziprasidone) antipsychotics. 7 A prospective cohort study of 352 psychiatric outpatients confirmed this,8 but a meta-analysis of nine other studies carried out by the same study authors showed that the yearly TD incidence rate for FGAs was significantly higher than for SGAs; however, many of these studies were not predesigned to detect TD. 8 Another, later, prospective cohort study found no significant difference in TD incidence rates between risperidone and olanzapine in 207 elderly psychiatric in- and outpatients. 9
As a result of widespread use of SGA drugs, increased off-label use and an ageing population, the frequency of TD is likely to be higher than thought,10,11 and increasing. The problem will be considerably greater for people in countries in which the use of newer drugs is less prevalent. 12,13
Given this high incidence and prevalence, the need for prevention or treatment is clear; unfortunately, there has been sparse evidence to guide clinicians. 14,15 Although many treatments have been tested, no one intervention has been shown clearly to be effective.
Although antipsychotic reduction and/or cessation would seem to be a logical first step in the management of antipsychotic-induced TD, this is not always possible in the clinical setting because of the over-riding need to manage current psychotic symptoms and/or reduce the risk of relapse. Changes in several antipsychotic medications have been produced in the last few decades that claim to cause less or no TD. 16 These claims may or may not be true, and certainly evidence does point to the fact that thoughtful use of older-generation drugs is not associated with more TD than with newer treatments. 17 In the search for ways to manage antipsychotic-induced TD, certain antipsychotic medications have themselves been proposed as specific treatments for the condition. 18 The usual rationale for such trials relates to variations in the receptor-blocking profile that distinguishes the compound of interest from antipsychotics in general. As for TD, treatment options for other movement disorders also include antipsychotic dose reduction or the switch to a newer antipsychotic. 19–21 Tetrabenazine is the only Food and Drug Administration-approved drug to specifically treat a movement disorder, Huntington’s chorea;20,22 consequently, and because of the lack of viable treatment options for TD, tetrabenazine has been suggested as a treatment for TD as well. 23
Drugs that reduce the activity of the cholinergic cells (anticholinergic drugs) are widely used to help treat other antipsychotic-induced movement disorders, such as Parkinsonism and dystonia. It is hypothesised that alterations in striatal cholinergic neurons could serve as pathophysiological basis for TD24 and, therefore, patients would benefit from cholinergic drugs. Benzodiazepines, the most widely used gamma-aminobutyric acid (GABA) agonists, have also been suggested as potential interventions for TD. Chronic blockade of dopamine receptors in TD leads to inactivity in another set of cells that employ GABA. 25 Also, there is evidence from animal experiments suggesting that GABA dysfunction may be associated with movement disorders. 26 Benzodiazepines have been included as a candidate treatment for TD in several practice guidelines27–29 and are also used to treat other movement disorders. 19,21,30
Vitamin E (tocopherol) is a lipid-soluble antioxidant that acts as a free radical scavenger and has also been proposed as a treatment for antipsychotic-induced TD. 31 There has been some suggestion that the chronic use of antipsychotics may cause abnormal production of highly active atoms and chemical groups (cytotoxic free radicals), which may damage specific cells in the brain. This, in turn, could be responsible for the appearance of TD. 32 Vitamin E may assist in minimising damage caused by cytotoxic free radical overproduction, and may prevent or decrease the severity of TD, particularly among those in whom onset occurred in the preceding 5 years. 33,34
Another agent under investigation for treatment of TD is buspirone, an anxiolytic drug acting as a partial agonist for the serotonin 5-HT1A (5-hydroxytryptamine subtype 1A) receptors, with additional low affinity as an antagonist for the dopamine D2 autoreceptors. A number of studies on TD animal models have found that buspirone ameliorated symptoms. 35,36
Other, non-pharmacological, treatments should also be examined in the context of TD. ‘Mind–body’ interventions, including both relaxation techniques and hypnosis, are reported to benefit patients with a number of neurological disorders. 37 The use of different relaxation techniques38,39 and hypnosis40 has also been examined in tic disorders and in Parkinson’s disease, with some positive preliminary findings; however, their effectiveness in movement disorders and TD specifically has yet to be systematically investigated.
We are aware that TD is not exclusive to people with schizophrenia, but, to illustrate the point regarding the disparate nature of evidence, a comprehensive database with more than 500 controlled trials comparing 101 different interventions used to improve or prevent deterioration of symptoms of antipsychotic-induced TD in schizophrenia was published in 1996. 41,42 The studies in this database were, largely, very small and poorly reported. 41,42 After categorisation according to treatment groups, nine Cochrane reviews were performed (first published in 1995–6 and periodically updated since). 18,23,43–49 An overview of all published Cochrane reviews was published in 1999. 50 These reviews reported a lack of information on the efficacy of most interventions, in particular the logical – but often impractical – step of stopping antipsychotic treatment. 18 Many with TD are faced with a lifetime of suffering from this disfiguring adverse effect.
This is a good time to revisit this difficult area for several reasons:
-
The research community has recognised that TD is not a problem of the past and may be an increasing problem of the future.
-
Widening the inclusion criteria to well beyond people with schizophrenia may lead to a broader appreciation of the research landscape, with opportunities for cross-fertilisation of ideas for prevention/treatment.
-
New approaches have been tested. 51
-
Methods in systematic reviewing have become considerably more sophisticated, with new techniques to employ evidence from, for example, network meta-analysis (NMA). 52
-
Dissemination of information is warranted, and methods for dissemination are much wider than has previously been the case, potentially generating further impact for this neglected area of research.
There may not be definitive answers available for the best way to prevent or treat TD, but this work will use all the best available evidence, highlight if there is good evidence for a specific treatment path, and provide high-quality evidence for choice of treatments and techniques for future testing.
Chapter 2 Hypotheses tested in the review (research questions)
To summarise evidence from clinical trials and observational studies of interventions used for treating or preventing deterioration of symptoms of antipsychotic-induced TD by performing an overview of systematic reviews, including updating Cochrane reviews, and NMA.
Specific objectives
-
To identify all relevant evaluative studies.
-
To produce a broad-brush overview of the evaluative research in this area and prioritise the top 10 candidate treatments for head-to-head comparisons.
-
To extract all relevant useful quantitative data on evaluations of the treatments, and to ensure that the source of these data is entirely transparent and made available for future researchers.
-
To produce reviews by:
-
updating nine existing relevant Cochrane reviews for different groups of interventions comparing TD with placebo
-
adding head-to-head comparisons reporting for the treatment and prevention of deterioration of symptoms of antipsychotic-induced TD to all Cochrane reviews in:
-
– adults with schizophrenia
-
– adults with dementia
-
-
ranking identified interventions according to relevance for the NHS and selecting the potentially relevant ones for NMA
-
performing a NMA.
-
-
To work collaboratively to tailor this evidence to clinical, research and public needs using dissemination techniques appropriate for all three.
Chapter 3 Methods
Part A: methods for patient and public involvement
This project brought together expertise from a range of fields to plan and deliver the review. The main part was review work. In order to assess if current research met the needs of people with experience of TD, a small consultation was planned, taking results from the reviews and exploring whether or not the assessed outcomes matched service user priorities for managing TD. The consultation was advertised by e-mail via the McPin Foundation’s large circulation list of people who are interested in being involved. It was also advertised on their website. Interested people were asked to contact the McPin Foundation to book a place to attend. Reimbursement for time and out-of-pocket expenses was offered.
A lay overview of the previously published version of a Cochrane review evaluating the effects of vitamin E in TD47 gave the foundation for the discussions. All of the researchers involved in the consultation were extremely experienced in involving patients and the public. The session was planned to provide time to reflect on current research on TD and to consider gaps in knowledge.
The discussion was audio-taped and the service users were invited to write comments on Post-it® (3M, Bracknell, UK) notes and paper tablecloths, which were then collected and reviewed. The researchers listened to the recordings after the session and noted any points relevant to the above-mentioned questions that would have an impact on the funded systematic review. Full transcription and formal analyses were not appropriate in this case, as the consultation was not a piece of empirical qualitative work. Furthermore, two of the consultation facilitators had extensive experience in involving patients and the public in research and expert knowledge in this paradigm, including hosting focus groups (or, in this case, a consultation).
Informed by the results of the consultation, we updated outcomes for the summary-of-findings table for the systematic reviews. See Appendix 1 for the full report.
Part B: methods for systematic review
Please see Appendix 2 for differences between the project protocol and the review.
Interventions being assessed
We aimed to evaluate any intervention used for treating or preventing deterioration of symptoms of antipsychotic-induced TD. There is a vast array of strategies to deal with TD – one review identified over 100. 50 Based on our experience with Cochrane reviews in this research area, we grouped the interventions as follows:
-
vitamins
-
GABA agonists
-
benzodiazepines
-
anticholinergics
-
cholinergics
-
calcium channel blockers
-
non-antipsychotic dopaminergics and noradrenergics
-
specific antipsychotic drugs
-
antipsychotic reduction or cessation including intermittent therapy
-
other interventions, including botulin toxin, insulin or lithium, among others.
We compared interventions with other interventions used to treat or prevent deterioration of symptoms of antipsychotic-induced TD of relevance to people in the NHS, placebo or no intervention.
Prioritisation of interventions for the NHS
From the included studies we listed all interventions, regardless of the primary condition, in order to map research activity. From this mapping, we chose to target, for this report, the top 10 interventions that seem to have demonstrated some efficacy and that are relevant for clinical practice and the NHS.
Measurement of outcomes
The following outcomes were included in the overview:
-
clinical improvement of TD symptoms
-
deterioration of TD symptoms
-
adverse events – extrapyramidal symptoms
-
adverse events – all
-
mental state
-
acceptability of the treatment – leaving the study early
-
social confidence, social inclusion, social networks, or personalised quality-of-life measures [this outcome was designated as important to patients, informed by the results of the patient and public involvement (PPI) consultation].
The Cochrane reviews included several more outcomes.
Design and theoretical/conceptual framework
We included randomised or quasi-randomised controlled trials containing data related to antipsychotic-induced TD, irrespective of language or place of publication. We also considered observational studies for inclusion with the following designs: (1) non-randomised controlled trials, (2) prospective cohort studies with a control group and (3) case–control studies. The systematic reviews and the overview of reviews follow Cochrane design and methodology. 53
Target population
We included studies of adults with a diagnosis of antipsychotic-induced TD (according to any criteria), regardless of the primary condition.
Inclusion/exclusion criteria
We excluded studies in which participants had used antipsychotic drugs for < 3 months or in which the antipsychotic doses had not been stable for at least 1 month4 (except in analyses of antipsychotic switch, withdrawal or reduction). In addition, we excluded studies evaluating children and adolescents, or studies evaluating interventions that are not relevant to the NHS.
We also excluded studies that were > 10 years old that otherwise qualified for inclusion, but reported no useable data and in which:
-
we contacted study authors requesting data, but received no reply
-
we were unable to contact any of the study authors.
Setting/context
Participants may be receiving treatment in any setting, any country or any health-care system.
Search strategy
We attempted to identify all relevant studies regardless of language or publication status (published, unpublished, in press and in progress).
We searched Cochrane Schizophrenia Group’s Study-Based Register of Trials on 16 July 2015 using the following string:
*Tardive Dyskinesia* in Healthcare Condition Field of Study.
In such a study-based register, searching the major concept retrieves all the synonym keywords and relevant studies because all the studies have already been organised based on their interventions and linked to the relevant topics. The Cochrane Schizophrenia Group’s Register of Trials is compiled by systematic searches of major resources [including Allied and Complementary Medicine Database (AMED), Bioscience Information Service, Cumulative Index to Nursing and Allied Health Literature (CINAHL), EMBASE, MEDLINE, PsycINFO and PubMed, and registries of clinical trials including CT.Gov, International Standard Randomised Controlled Trial Number (ISRCTN) and the World Health Organization’s International Clinical Trials Registry Platform registries] and their monthly updates, hand-searches, grey literature and conference proceedings (see Group’s Module: http://onlinelibrary.wiley.com/o/cochrane/clabout/articles/SCHIZ/frame.html). There are no language, date, document type or publication status limitations for inclusion of records into the register.
We also searched the Cochrane Dementia and Cognitive Improvement Group’s Register of Trials via the Cochrane Register of Studies Online (CRSO; http://crso.cochrane.org/) on 21 July 2015 using the following string:
DEMENTIA:CC AND (*Tardive* OR *Dyskinesia*):TI,AB,KY.
For more information about this register, see the register’s page (www.medicine.ox.ac.uk/alois/content/about-alois).
Finally, we searched EMBASE, MEDLINE, and PsycINFO for observational studies on 9 January 2017, and details of the search strategy can be found in Appendix 3.
We inspected references of all identified studies for further relevant studies.
As some of the Cochrane reviews have not been updated during the past decade, and systematic reviews methods have changed considerably during this period of time, we also cross-checked all included, awaiting assessment, ongoing and excluded studies in the suite of nine Cochrane reviews on antipsychotic-induced TD.
Selection of studies
We uploaded search results into a web-based system (DistillerSR®, Evidence Partners, Ottawa, ON, Canada; www.systematic-review.ca). At least two reviewers (out of Antonio Grande, Rosie Asher, Hanna Bergman and Karla Soares-Weiser) independently screened all citations and abstracts identified by the search. Two reviewers (Hanna Bergman and Karla Soares-Weiser) inspected all studies from the nine Cochrane reviews on TD. We obtained full reports for potentially eligible studies and these were independently screened by two review authors (Antonio Grande and Rosie Asher). Disagreements were resolved through discussion with reviewers (Hanna Bergman and Karla Soares-Weiser). We documented justifications for excluding studies from the review.
Data extraction and management
Reviewer Rosie Asher extracted data from all included studies. These were cross-checked by Antonio Grande, and further validated by Hanna Bergman. Any disagreements about data extraction were documented and resolved by consensus. Any potential differences or data entry problems were discussed and decisions documented.
If more than one publication was identified reporting data from the same participants, the main publication was considered as the one with more information or with longer-term outcomes; all others were considered companion publications and data were only collected from these if they had not been provided in the main publication.
We attempted to contact authors in order to obtain missing information or for clarification whenever necessary.
We extracted data into tabular format, with an ‘address’ to each point in the document from which each data element had been taken. This allows future researchers to verify extraction and avoid duplication of effort. All data extracted in this way are fully available to researchers. 54
We extracted data from graphs in GetData Graph Digitizer software version 2.26 (GetData Graph Digitizer, S Federov, Moscow, Russia).
Some specific outcomes
No clinically important improvement in tardive dyskinesia
‘No clinically important improvement’ was defined as < 50% improvement on any scale measuring TD, or as defined by triallists of the individual studies. For this outcome we assumed that participants with missing data did not improve.
We have shown details of the scales that provided usable data below.
Brief Psychiatric Rating Scale
The Brief Psychiatric Rating Scale (BPRS) is an 18-item scale measuring positive symptoms, general psychopathology and affective symptoms. 55 The original scale has 16 items, although a revised 18-item scale is commonly used. Total scores can range from 0 to 126. Each item is rated on a seven-point scale, with high scores indicating more severe symptoms.
Extrapyramidal Symptom Rating Scale
The Extrapyramidal Symptom Rating Scale (ESRS) was developed to assess four types of drug-induced movement disorders: Parkinsonism, akathisia, dystonia and TD. 56 The score for TD, ranging from 0 to 42, is based on the sum of all seven items in the TD objective examination.
Simpson–Angus Scale
The Simpson–Angus Scale (SAS)57 is a 10-item scale, with a scoring system of 0–4 for each item, measuring drug-induced Parkinsonism, a short-term drug-induced movement disorder. A low score indicates low levels of Parkinsonism.
Udvalg for Kliniske Undersøgelser Side-Effect Rating Scale
The Udvalg for Kliniske Undersøgelser (UKU) was developed to provide a comprehensive side-effect rating scale with well-defined and operationalised items to assess the side effects of psychopharmacological medications. 58 The scoring sheet includes 48 items, with higher scores indicating more side effects.
Assessment of risk of bias of the included studies
Rosie Asher classified and Hanna Bergman cross-checked studies as being at low, unclear or high risk of bias, based on domain-specific assessments of risk of bias done using the Cochrane Collaboration’s existing risk-of-bias tool. 53 If the raters disagreed, we made the final rating by consensus, with the involvement of another member of the review group. Where inadequate details of randomisation and other characteristics of trials were provided, we contacted authors of the studies in order to obtain further information.
We incorporated these judgements in assessing limitations in study design for outcomes in the summary-of-findings table (see Table 2).
Risk-of-bias assessment for observational studies was performed by a senior systematic reviewer (Artemisia Kakourou) using a tool that is currently being tested by Cochrane. 59 The following domains were assessed: (1) confounding and selection bias (including confounders measured and addressed, use of matching and methods of adjustment), (2) performance bias (including any considerations of co-intervention), (3) missing data, (4) detection (for cohort studies) or recall bias (for case–control studies) and (5) selective reporting bias.
Data analysis
Analyses of single studies
Dichotomous data
For each study, the risk ratio (RR) and 95% CI were derived for people receiving the intervention compared with the control.
Continuous data
We included continuous data from rating scales only if:
-
the psychometric properties of the measuring instrument had been described in a peer-reviewed journal60
-
the measuring instrument was not written or modified by only one of the authors of the particular study from which the data were taken, but had also received independent validation.
For each study, the mean difference (MD) between groups and 95% CIs were estimated.
We also produced descriptive tables summarising information about study design, risk of bias and results of all included studies. Data were presented by each specific intervention according to the main diagnosis (schizophrenia or dementia).
Crossover trials
A major concern of crossover trials is the carry-over effect. This occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state, despite a washout phase. For the same reason, crossover trials are not appropriate if the condition of interest is unstable. 61 As both effects are very likely in severe mental illness, we used only data of the first phase of crossover studies.
Meta-analyses
Where studies were considered substantively similar enough for meta-analysis to be appropriate, we carried out fixed-effects analyses using the RevMan software version 5.3.5 (The Cochrane Collaboration, The Nordic Cochrane Centre, Copenhagen, Denmark).
We understand that there is no closed argument for preference for use of fixed- or random-effects models. The random-effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This often seems to be true to us and the random-effects model takes into account differences between studies, even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random-effects model. It puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect, these studies can either inflate or deflate the effect size. We chose the fixed-effects model for all analyses.
Observational studies
We provided an overview of evidence from observational studies. Study characteristics, results and conclusions were tabulated and summarised.
Variation in efficacy according to characteristics of individuals and studies
Visual inspection of the forest plots was used to evaluate the potential statistical heterogeneity (differences between the true intervention effects in the different studies). Heterogeneity was quantified by estimating the between-study variance τ2- and the I2-statistics,62,63 which measures the percentage of observed variation that can be attributed to true differences between the studies. 62 In forest plots and meta-analyses, τ2 was estimated using the restricted maximum likelihood estimator,64 whereas its 95% CIs were estimated by the Q-profile method. 65
Summarising and interpreting results
We used the Grading of Recommendations, Assessment Development and Evaluation (GRADE) approach66–68 to assess the evidence of the various interventions. For all NHS-prioritised interventions and outcomes, we have presented a summary-of-findings table (see Table 2) based on the GRADE results.
Investigation of heterogeneity
We considered a degree of heterogeneity inevitable, and hence we planned to explore only important heterogeneity (I2 ≥ 75%) using metaregression or subgroup analyses for the effect modifiers: (1) risk of bias in the different study designs; (2) length of antipsychotic use; (3) underlying disease (dementia or schizophrenia); (4) sex/age; (4) type of treatment use, specifically first- or second-generation antipsychotics; and (5) whether or not other concomitant drug interventions were used. Analyses were homogeneous with no important heterogeneity (I2 ≥ 75%).
Sensitivity analyses
To ensure that our imputations did not bias our results, we planned to restrict the analyses to studies considered to be at low, and low or unclear risk of selection and detection bias. However, all studies were at unclear risk of selection and detection bias, and we did not carry out this restricted analysis.
Planning of future studies
To judge the sufficiency of the evidence for the comparison of switching to any FGAs versus any SGAs, we calculated the conditional power of an updated meta-analysis for the particular comparison as described in Sutton et al. 69 We further investigated whether or not hypothetical future studies are likely to alter the meta-analysis results using extended funnel plots. 70 Given the small number of studies available, a fixed-effect inverse-variance meta-analysis model was assumed for this analysis.
Power of an updated meta-analysis based on simulations of new studies
We estimated the power of an updated meta-analysis through the simulation of (sufficiently similar) hypothetical ‘new’ studies and calculating the proportion of times that the meta-analysis result would be statistically significant. 69 The event rate was assumed to be equal to that observed, and the number of simulations on which we estimated power was 1000.
Extended funnel plots
We further assessed whether or not future studies are likely to alter the meta-analysis result via extended funnel plots. 70 A colour code appended in conventional funnel plots illustrates where the result of an updated meta-analysis would lie, depending on the effect estimate and the standard error of a hypothetical new study to be added to the evidence base.
Part C: methods for network meta-analysis
In order to facilitate clinical decision-making and a plan of future research, we planned to conduct a NMA as we expected that few studies reported trials with head-to-head comparisons of different interventions.
We carried out an exploratory NMA, and the results are presented in Appendix 4. The main reasons for the decision of only presenting the results in the appendix are (1) there were few data, (2) there was a median of one study per comparison, ranging up to 11 for cholinergic drugs and 13 for vitamin E, (3) there were no differences between pairwise meta-analyses and NMA and (4) there were no sufficiently connected networks.
Chapter 4 Part A: results of patient and public involvement
Dawn-Marie Walker worked with the McPin Foundation to organise an event to which a group of service users (n = 6) were invited and at which there was the opportunity to discuss the review’s results. All of the service users had TD or were at risk of developing it. All attendees recognised TD as a serious condition: ‘TD can be as debilitating as the psychosis itself’. They recognised that TD could increase stigma, as one could not hide it, which in turn would have a negative impact on one’s self-esteem. Indeed, there were suggestions for a therapeutic intervention to help people with TD learn coping mechanisms. The attendees argued that prevention was better than cure, and wondered how much psychiatrists knew about TD and, in turn, how much patients knew prior to taking a medication. With regard to the outcomes of the trial, they thought that the review placed too much emphasis on pharmaceutical interventions and were concerned that an adverse effect of medication was being treated by other medications. Owing to the lack of definite findings about a treatment for TD, one commented: ‘I’m appalled by the poverty of this evidence base given how debilitating TD is’ (Figure 1).
FIGURE 1.
Message from one of the participants of the PPI consultation of service user perspectives on TD research.

One of the questions participants posed was whether or not research could be done to try to identify those who are at risk of TD. There was also some debate about the similarities in presentation between Tourette syndrome and TD, with a number of public awareness campaigns helping reduce the stigma of Tourette syndrome, and some participants asked if a similar approach would work for TD. When the outcome measures cited in the review were discussed, the attendees thought all of them were important; however, they felt that some relating to empowerment and autonomy, such as knowledge of TD (health-care practitioner, patient and public) or a social integration scale (see Appendix 1), were missing.
Chapter 5 Part B: results of systematic reviews
Search and screening
The update search retrieved 704 references from the Cochrane Schizophrenia Group’s Register and 29 references from the Cochrane Dementia and Cognitive Improvement Group’s Register. Four duplicate reports included in both these registers were removed. In addition, as we aimed to code all studies, we independently re-extracted the data of all included and excluded studies in the published TD Cochrane reviews and cross-checked all references; 222 additional records were found in the reference lists of previously published Cochrane reviews. In total, we screened 947 records. After excluding irrelevant references when screening the titles and abstracts, we identified 565 potentially relevant full-text articles that were assessed for eligibility. We excluded 398 full-text articles (grouped into 329 studies) with documented reasons for exclusion (see Appendix 5). We included 112 studies (167 references) in the nine Cochrane reviews (see Appendix 6), including two studies awaiting classification and 11 ongoing studies.
We did not identify any included studies for people with dementia and antipsychotic-induced TD. See Figure 2 for the screening and study selection process.
FIGURE 2.
The Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram.

Studies were assessed in Chinese, Danish, French, German, Japanese, Korean, Persian, Portuguese, Spanish and English. There were 10 included studies in Chinese,71–80 three in German,81–83 three in Japanese,84–86 and one each in Persian87 and in Portuguese. 88
The observational studies search retrieved 3312 references. After de-duplication, 2702 references were screened. A total of 2261 titles and abstracts were excluded, and 41 full texts were retrieved and screened. Thirty studies (31 references) were excluded and eight studies (10 references) were included [see Figure 11 in Appendix 3 for the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram].
Prioritisation of interventions
In consultation with a NHS consultant psychiatrist (Clive E Adams), we identified the 10 interventions that are mostly relevant for the NHS, and these interventions (30 unique studies) were included in the current report. The 10 were chosen for ‘local’ accessibility, breadth of approach and practicality. We realise that opinions could differ on which choice should have been made, but it was directed by having available trials and also being accessible in the UK’s NHS. The 10 interventions prioritised as the most relevant for the NHS were anticholinergics, antipsychotics, antipsychotic reduction, antipsychotic withdrawal, benzodiazepines, buspirone, hypnosis and relaxation, placebo, treatment as usual (TAU) and vitamin E. These 10 interventions are included in the pairwise comparisons of this report and in the NMA.
Box 1 lists all interventions from eligible randomised trials included in the Cochrane reviews, and the interventions prioritised and reported in this overview are highlighted in bold. The full Cochrane reviews should be the point of reference for details of every study and outcome (see Appendix 6). This report represents a summary.
-
Anticholinergic: procyclidinea.
-
Anticholinergic continuation: biperiden.
-
Anticholinergic withdrawal: biperiden.
-
Antipsychotic continuation.
-
Antipsychotic reduction.
-
Antipsychotic withdrawal (with placebo).
-
Benzodiazepine: clonazepam.
-
Benzodiazepine: diazepam.
-
Calcium channel blocker: diltiazem hydrochloride.
-
Calcium channel blocker: diltiazem hydrochloride.
-
Calcium channel blocker: nifedipine.
-
Cholinergic medication: deanol.
-
Cholinergic medication high dose: deanol, 2 g.
-
Cholinergic medication low dose: deanol, 1 g.
-
Cholinergic medication: galantamine.
-
Cholinergic medication: lecithin.
-
Cholinergic medication: meclofenoxate hydrochloride.
-
Cholinergic medication: rivastigmine.
-
GABA agonist: baclofen.
-
GABA agonist: GABA.
-
GABA agonist: progabide.
-
GABA agonist: sodium valproate.
-
GABA agonist: THIP.
-
Miscellaneous: L-stepholidine.
-
Miscellaneous: branched-chain amino acids.
-
Miscellaneous: buspirone.
-
Miscellaneous: ceruletide.
-
Miscellaneous: cyproheptadine.
-
Miscellaneous: dihydrogenated ergot alkaloids/co-dergocrine mesylate.
-
Miscellaneous: oestrogen.
-
Miscellaneous: gamma-linolenic acid supplementation (oil of evening primrose).
-
Miscellaneous: Ginkgo biloba standardised extract (EGb-761).
-
Miscellaneous: hypnosis or relaxation.
-
Miscellaneous: insulin.
-
Miscellaneous: levetiracetam.
-
Miscellaneous: lithium.
-
Miscellaneous: MAO inhibitors (isocarboxazid, selegiline).
-
Miscellaneous: melatonin.
-
Miscellaneous: omega-3 fatty acid (ethyl-eicosapentaenoic acid).
-
Miscellaneous: papaverine.
-
Miscellaneous: pemoline.
-
Miscellaneous: phenylalanine.
-
Miscellaneous: piracetam.
-
Miscellaneous: promethazine.
-
Miscellaneous: ritanserin.
-
Miscellaneous: VMAT2 inhibitor (NBI-98854).
-
Non-neuroleptic catecholaminergic: amantadine.
-
Non-neuroleptic catecholaminergic: bromocriptine.
-
Non-neuroleptic catecholaminergic: carbidodopa/levodopa.
-
Non-neuroleptic catecholaminergic: l-DOPA.
-
Non-neuroleptic catecholaminergic: oxypertine.
-
Non-neuroleptic catecholaminergic: reserpine.
-
Non-neuroleptic catecholaminergic: tiapride.
-
Non-neuroleptic catecholaminergic: tetrabenazine.
-
Non-neuroleptic catecholaminergic: celiprolol.
-
Non-neuroleptic catecholaminergic: methyldopa.
-
Phenobarbital (as active placebo).
-
Placebo.
-
Switch to a different FGA.
-
Switch to a different FGA (not specified).
-
Switch to a different FGA (haloperidol).
-
Switch to a different FGA [molindone (Moban®; Endo Pharmaceuticals Inc., Malvern, PA, USA)]b.
-
Switch to a different FGA (thiopropazate)b.
-
Switch to a different FGA (zuclopentixol)b.
-
Switch to SGA.
-
Switch to SGA (amisulpride).
-
Switch to SGA (clozapine).
-
Switch to SGA (olanzapine).
-
Switch to SGA (quetiapine).
-
Switch to SGA (risperidone).
-
Switch to SGA (ziprasidone).
-
TAU.
-
Vitamin B6 (pyridoxal 5′-phosphate).
-
Vitamin E.
l-DOPA, l-3,4-dihydroxyphenylalanine; MAO, monoamine oxidase; THIP, 4,5,6,7-tetrahydroisoxazolo[5,4-c]pyridin-3-ol; VMAT2, vesicular monoamine transporter 2.
Not used (in a head-to-head comparison with isocarboxazid).
Not used (in a head-to-head comparison with another FGA).
Accessible data
Because of copyright it is not possible to share the full text of original papers, but all data have been extracted and tabulated, and the exact location of every piece of data is recorded in these tables. Pairing these tables with the original report allows tracking of data back to full text. These extracted data are freely available on Cochrane Schizophrenia Group’s website via ResearchGate (ResearchGate GmBH, Berlin, Germany). 54 Also, the extracted data beside the linked full-text reports are available to be used for research purposes in Cochrane Schizophrenia Group’s Study-Based Register of Trials.
Description of studies
Studies included in overview
Randomised controlled trials
We included 30 unique clinical trials (54 articles published between 1973 and 201175,78,89–139) reporting results for the effects of the prioritised interventions on clinical improvement and deterioration of TD symptoms, mental state, adverse events and acceptability of treatment. None of the included studies reported on quality of life. All studies were described as being randomised controlled. Seven of the 30 studies used a crossover design with two periods89–95 and, as planned, we used only data from before the first crossover (see Appendix 2, Unit of analysis issues). Studies were conducted in North America (15 studies89,92,93,96,97,101,104,117,120,121,123,128,129,137,139), Asia (10 studies75,78,90,91,94,108,112,115,127,138), Europe (four studies95,98,119,130) and Africa (one study110), with a total of 1255 participants included. Studies included both men and women of mostly wide age ranges, but participants were mainly men in their fifties, with mean ages ranging from 32 to 68 years.
An overview of characteristics of the included studies contributing data for this report are presented in Table 1 and full details of study characteristics are available in Appendix 7.
| Included studies (first author and year of publication) | Study characteristic | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Methods | Participants | Interventions | Outcomes | ||||||||||||||||
| Randomised | Double blind | Design | Duration (weeks) | Diagnosis | n | Age (years) | Sex | Group 1 intervention | Dose | Group 2 intervention | Dose | Other groups | Other medications allowed | TD symptoms | Study discontinued | QoL measures | Mental state | Adverse events | |
| Antipsychotic drugs | |||||||||||||||||||
| Kazamatsuri et al., 197396 | ✗ | ✗ | Parallel | 24 | Chronic SCZ and TD | 13 | Mean 55.8 | F and M | Haloperidol (after 4-week washout) | 4 mg b.i.d. (weeks 15–24 16 mg/day) | Tetrabenazine (after 4-week washout) | 50 mg b.i.d. (weeks 15–24 200 mg/day) | Antidiabetics and anticonvulsants | ✗ | ✗ | ||||
| Kane et al., 198397 | ✗ | ✗ | Parallel | 48 | SCZ/schizoaffective and TD | 8 | 17–60 | Unknown | Fluphenazine | Low dose (1.25–5 mg every 2 weeks) | Fluphenazine maintenance | Standard dose 12.5–50 mg/2 weeks) | Procyclidine/flurazepam/diazepam | ✗ | ✗ | ||||
| Cookson, 198798 | ✗ | ✗ | Parallel | 44 | SCZ | 18 | Mean 44.5 | F and M | Flupentixol decanoate | 50% reduction from original dose | Flupentixol maintenance | Standard dose | Procyclidine/haloperidol/zuclopentixol decanoate/amitriptyline | ✗ | ✗ | ||||
| Chouinard and Arnott, 1992,99,100 1993;102 Chouinard et al., 1993;103 Chouinard, 1995101 | ✗ | ✗ | Parallel | 8 | SCZ | 135 | Mean 39 | F and M | Risperidone | 2 mg per day (n = 8), 6 mg per day (n = 6), 10 mg per day (n = 6), 16 mg per day (n = 11) | Haloperidol | 20 mg per day | Placebo | Benzodiazepines/biperiden or procyclidine | ✗ | ||||
| Tamminga et al., 1994104 | ✗ | ✗ | Parallel | 52 | SCZ and TD | 32 | Mean 35.57 | F and M | Clozapine and placebo | 293.8 mg per day | Haloperidol and benztropine | 28.5 mg/day | N/A | ✗ | |||||
| Bai et al., 2002,105 2003,108 2005;106 Pai et al., 2002,107 2001109 | ✗ | ✗ | Parallel | 12 | SCZ and TD | 42 | Mean 50.2 | F and M | Risperidone (after 2-week washout) | 2 mg per day increased to 6 mg per day (6 weeks) and maintenance (12 weeks) | Placebo | 2 mg per day increased to 6 mg per day (6 weeks) and maintenance (12 weeks) | Benzodiazepines/antiparkinson medications | ✗ | ✗ | ✗ | |||
| Emsley et al., 2004110,111 | ✗ | Parallel | 50 | SCZ and TD | 45 | Mean 49.2 | F and M | Quetiapine (after 2-week washout) | 100 mg per day increased to 400 mg per day | Haloperidol (after 2-week washout) | 5 mg per day increased to 10 mg per day | Benzodiazepines/anticholinergic agents | ✗ | ✗ | ✗ | ||||
| Bai et al., 2005112–114 | ✗ | Parallel | 24 | SCZ and TD | 80 | Mean 50.2 | F and M | Olanzapine | Unknown | Amisulpride | Unknown | FGA (unknown dose) | N/A | ✗ | ✗ | ✗ | ✗ | ||
| Chan et al., 2010115,116 | ✗ | Parallel | 24 | SCZ/schizoaffective and TD | 60 | Mean 45.3 | F and M | Risperidone (after 3–7 days washout) | 1.9 mg per day increased to 4.1 mg per day | Olanzapine | 8.1 mg per day increased to 12.6 mg per day | N/A | ✗ | ✗ | ✗ | ||||
| Caroff et al., 2011;117 Miller et al., 2005118 | ✗ | ✗ | Parallel | 78 | SCZ and TD | 200 | Mean 47.2 | F and M | Olanzapine | 7.5 mg q.d./b.i.d./t.i.d./q.i.d. | Quetiapine | 200 mg/q.d./b.i.d./t.i.d./q.i.d. | Risperidone 1.5 mg/q.d./b.i.d./t.i.d./q.i.d. or ziprasidone 40 mg/q.d./b.i.d./t.i.d./q.i.d. | N/A | ✗ | ||||
| Anticholinergic drugs | |||||||||||||||||||
| Greil et al., 1984119 | ✗ | ✗ | Parallel | 7 | SCZ and TD | 10 | Mean 56.6 | F and M | Biperiden | Dose stopped after 4 weeks and placebo was then given for 3 weeks | Biperiden | Dose stopped after 1 week and placebo given for 6 weeks | Antipsychotic medications | ✗ | |||||
| Benzodiazepines | |||||||||||||||||||
| Bobruff et al., 1981120 | ✗ | ✗ | Parallel | 2 | Psychiatry patients and TD | 21 | Mean 51.6 | F and M | Clonazepam | 3.9 mg per day | Phenobarbital (as active placebo) | 88.6 mg per day | Antipsychotics | ✗ | ✗ | ✗ | |||
| Weber et al., 198389 | ✗ | Cross over | 24 | SCZ/brain syndrome/unknown and TD | 15 | Mean 57.4 | F and M | Standard care and diazepam | 6–25 mg per day | Standard care | Unknown | Antipsychotic and anticholinergic medications | ✗ | ✗ | ✗ | ||||
| Csernansky et al., 1988121,122 | ✗ | ✗ | Parallel | 5–6 | SCZ and TD | 17 | Unknown | Unknown | Diazepam | 7.2 mg per day | Placebo | 48.3 mg per day | Alprazolam | Anticholinergics | ✗ | ✗ | |||
| Xiang and Zhen, 199775 | ✗ | ✗ | Parallel | 8 | SCZ and TD | 24 | Mean 39.4 | F and M | Standard care and clonazepam | 4–6 mg per day | Standard care and placebo | Unknown | Antipsychotic and anticholinergic medications | ✗ | ✗ | ||||
| Vitamin E | |||||||||||||||||||
| Elkashef et al., 199093 | ✗ | ✗ | Cross over | 10 | SCZ/schizoaffective and TD | 10 | Mean 56.6 | F and M | Vitamin E | 400 IU per day (1 week), 400 IU b.i.d. (1 week), 400 IU t.i.d. (2 weeks) | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Schmidt et al., 199195 | ✗ | ✗ | Cross over | 4 | SCZ/schizoaffective/depression and TD | 23 | Mean 45 | F and M | Vitamin E | 1200 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Egan et al., 199292 | ✗ | ✗ | Cross over | 12 | SCZ/schizoaffective/depression/BD and TD | 21 | Mean 43.9 | F and M | Vitamin E | Week 1: 400 IU per day; week 2: 800 IU per day; week 3: 1200 IU per day; weeks 4–6: 1600 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Adler et al., 1992,124 1993,125,126 1998123 | ✗ | ✗ | Parallel | 36 | SCZ/depression and TD | 40 | Mean 58 | F and M | Vitamin E | Dose increasing to 1600 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ||||
| Akhtar et al., 1993127 | ✗ | ✗ | Parallel | 4 | Psychiatry patients and TD | 32 | Mean 53 | F and M | Vitamin E | 600 mg per day increased to 1200 mg per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | ✗ | ||
| Dabiri et al., 1994128 | ✗ | ✗ | Parallel | 12 | Psychiatry patients and TD | 12 | Mean 51 | F and M | Vitamin E | Week 1: 400 IU per day; week 2: 800 IU per day; weeks 3–12: 1200 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Lam et al., 199494 | ✗ | ✗ | Cross over | 16 | SCZ and TD | 16 | Mean 61.8 | F and M | Vitamin E | Week 1: 400 IU per day; week 2: 400 IU b.i.d.; weeks 3–6: 400 IU t.i.d. | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ||||
| Lohr and Calgiuri, 1996129 | ✗ | ✗ | Parallel | 8 | SCZ/depression/BD and TD | 55 | Mean 48.9 | F and M | Vitamin E | 1600 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Dorevitch et al., 199791 | ✗ | ✗ | Cross over | 20 | SCZ and TD | 10 | Mean 63.1 | F and M | Vitamin E | Dose increasing to 1600 IU per day | Placebo | Unknown | Chlorpromazine | ✗ | ✗ | ||||
| Dorevitch et al., 199790 | ✗ | ✗ | Cross over | 20 | SCZ/schizoaffective and TD | 40 | Mean 64.4 | F and M | Vitamin E | Week 1: 400 IU per day; week 2: 800 IU per day; week 3: 1200 IU per day; weeks 4–8: 1600 IU | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Sajjad, 1998130 | ✗ | ✗ | Parallel | 28 | TD | 20 | Mean 68 | F and M | Vitamin E | 400 mg per day increased to 1600 mg per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | |||
| Tracy et al., 1997;131 Lohr and Lavori, 1998;132 Edson et al., 1997;133 Caligiuri et al., 1997;134 Adler et al., 1994,135 1999;137 Brindler, 2001136 | ✗ | ✗ | Parallel | 52 | SCZ/schizoaffective and TD | 158 | Mean 50 | F and M | Vitamin E | 1600 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ✗ | ✗ | ||
| Zhang et al., 2004138 | ✗ | ✗ | Parallel | 12 | SCZ and TD | 41 | Mean 54.5 | F and M | Vitamin E | Week 1: 800 IU per day; weeks 2–12: 1200 IU per day | Placebo | Unknown | Antipsychotics | ✗ | ✗ | ||||
| Buspirone | |||||||||||||||||||
| Zeng, 199578 | ✗ | ✗ | Parallel | 6 | TD | 42 | Mean 32.5 | F and M | Buspirone | Dose management (1–12 capsules per day) | Placebo | Dose management (1–12 capsules per day) | Antipsychotic and anticholinergic medications | ✗ | ✗ | ||||
| Hypnosis and relaxation | |||||||||||||||||||
| Glover, 1980139 | ✗ | Parallel | 8 sessions | SCZ and TD | 15 | Mean 34.9 | F and M | Hypnosis or relaxation | 8 sessions | TAU | 8 sessions | Psychotropics | ✗ | ||||||
In addition to the included studies:
-
We have requested details on participants from study authors to determine the eligibility for one study comparing dexetimide and benzhexol. 140
-
One study described as a double-blind, randomised study on vitamin E could not be identified after exploring numerous sources. 141
-
The full text of a randomised controlled trial (RCT), published in 1992, comparing buspirone and placebo could not be identified. 142
-
The full text of a RCT described in a trial registry comparing quetiapine with risperidone could not be identified143
-
One study comparing cannabidiol extract with vitamin E is ongoing. 144
Full details of characteristics for ongoing trials and studies awaiting classification are available in Appendix 8.
Observational studies
We included eight unique observational studies (10 articles published between 1983 and 2016145–154) reporting results for the effects of the prioritised interventions on clinical improvement and deterioration of TD symptoms and mental state. None of the included studies reported on quality of life, adverse events or acceptability of the intervention. Two studies (three references) were described as non-randomised controlled145–147 and six (seven references) were described as prospective cohorts. 148–154 Studies were conducted in North America (four studies145,149,151,153), Asia (two studies150,152) and Europe (two studies146,148). A total of 200 participants were included. Studies included adults, both men and women of mostly wide age ranges, with mean ages ranging from 26 to 84 years.
An overview of characteristics of the included observational studies contributing data to this report is presented in Appendix 3 (see Table 4).
Studies excluded from this review
Randomised controlled trials
Sixty-nine studies (99 articles) did not investigate prioritised comparisons and were not included in this report. These studies investigated calcium channel blockers (three studies), cholinergic medication (14 studies), GABA antagonists (11 studies), non-antipsychotic dopaminergic or noradrenergic medication (nine studies), FGAs versus other FGAs (three studies), anticholinergic versus monoamine oxidase (MAO) inhibitors (one study) and various miscellaneous, experimental treatments, such as lithium, melatonin and insulin (28 studies). Full details of these studies and results of comparisons are available in the Cochrane reviews and an overview is available in Appendix 9.
Observational studies
Please see Appendix 3 (see Table 5) for details of references excluded at full-text screening. In addition, one of the included observational studies was not prioritised for this report because it investigated deep-brain stimulation, not one of the NHS-relevant interventions. 146,147
Risk-of-bias assessments
Randomised controlled trials
Detailed risk-of-bias assessments of all included studies are in Appendix 7.
Overall risk of bias for the included studies was rated as being high to unclear. It is astonishing to note that only one of the studies was rated as being free from risk of selection bias. 137 The remaining trials reported inadequately on randomisation and allocation concealment. Furthermore, seven studies were rated as being at high risk of performance bias and 13 were rated as being of unclear risk. This was mainly a result of trials being open label, or poor reporting of blinding. One study was rated as being at high risk of detection bias and 18 were rated as being of unclear risk; this is mainly because of poor reporting. Ten studies were rated as being at high risk of attrition bias (because of high or imbalanced dropout rates) and two at unclear risk. Thirteen studies were rated as being at high risk of reporting bias as a result of selective reporting of outcome measures, and 12 were rated at an unclear risk. We sought information from study authors where risk of bias was rated as being unclear.
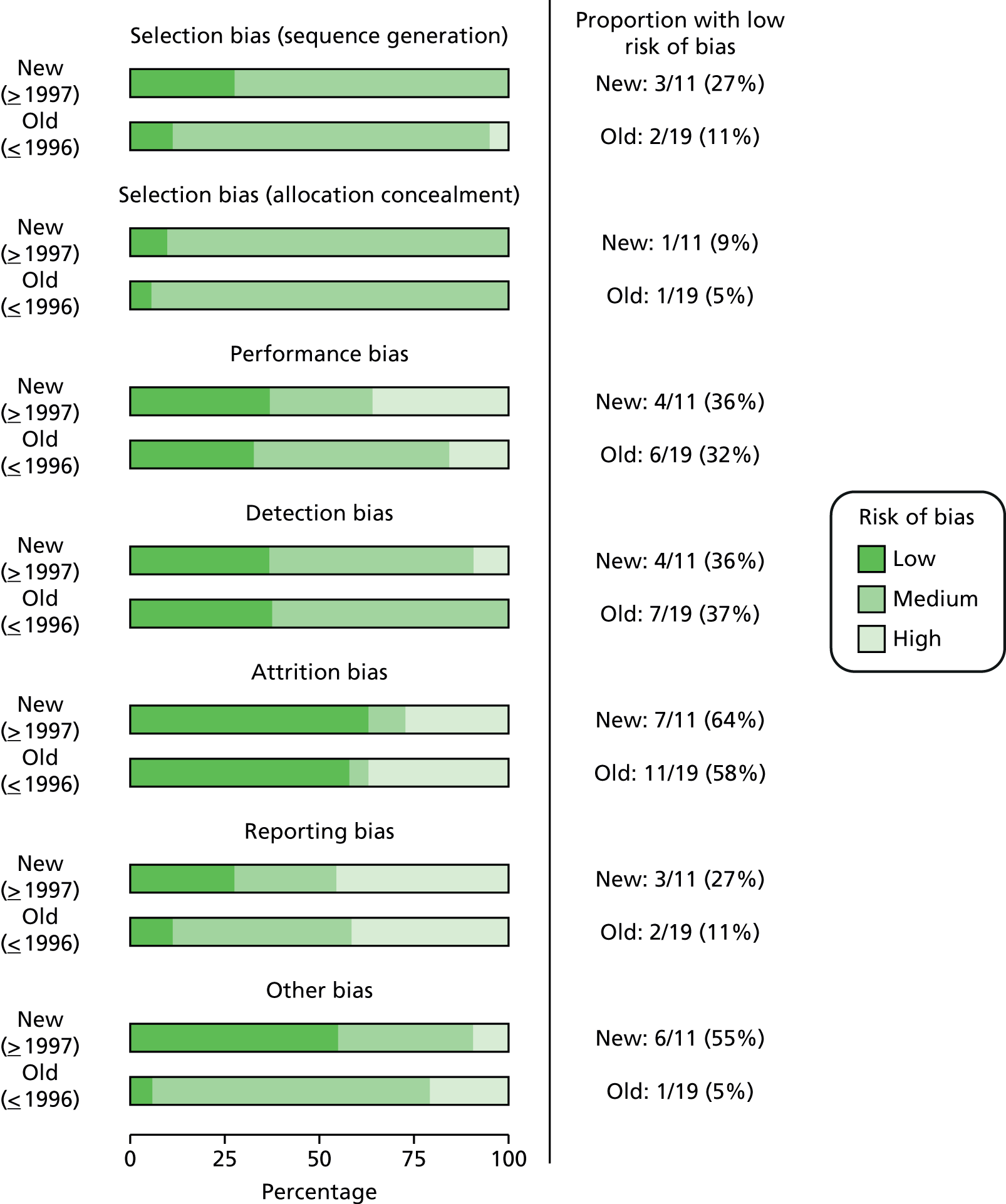
As a post hoc comparison, we evaluated risk of bias in studies published within the past 20 years (1997–2011) compared with older studies published until 1996 (Figure 3). We found that methodological quality had improved only marginally over time on most risk-of-bias categories (selection, performance, attrition and reporting biases). There was no change for detection bias, and other bias had improved over time.
FIGURE 3.
Old (1973–96) vs. new (1997–2011) studies risk of bias.
Observational studies
Detailed risk-of-bias assessments of all included studies are in Appendix 3 (see Table 4).
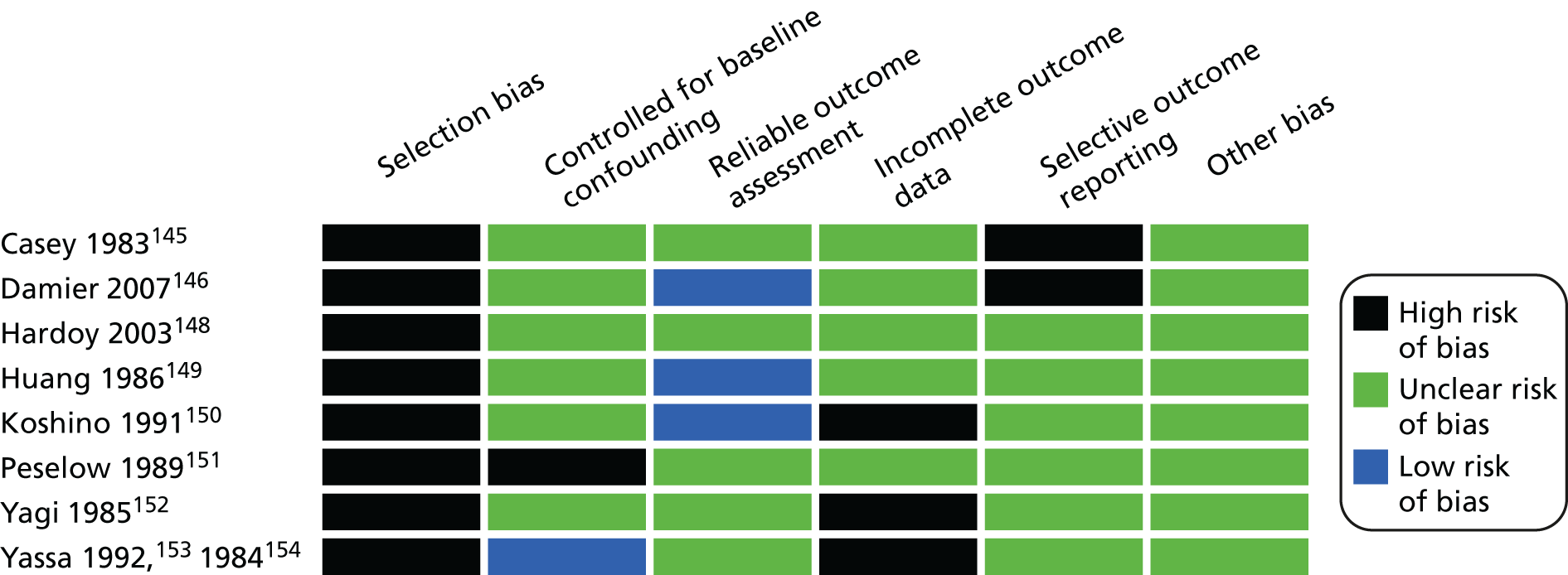
Overall risk of bias for the included observational studies was rated as being high to unclear. None of the observational studies was free from risk of selection bias, one study reported controlling for baseline confounding, and three studies reported a reliable outcome assessment. For the domains of incomplete outcome data and selective outcome reporting, none of the studies reported mechanisms to avoid bias (Figure 4).
FIGURE 4.
Overview of included observational studies risk of bias.
Effects of interventions
Table 2 summarises the results from RCTs for all comparisons. Forest plots for all analyses from RCTs are in Appendix 10. An overview of results from observational studies is in Appendix 3 (see Table 4).
| Intervention | Comparison | Outcome (follow-up) | Effect estimate (95% CI) | n | Quality of the evidence (GRADE) | Rationale for GRADE |
|---|---|---|---|---|---|---|
| Antipsychotic drugs | ||||||
| Reduced dose of antipsychotics | Continuing antipsychotics | TD: no improvement (44–48 weeks) | RR 0.42 (0.17 to 1.04) | 17 (two RCTs)97,98 | + – – – (very low) (R1, R2) |
|
| TD: deterioration (44–48 weeks) | RR 0.61 (0.11 to 3.31) | 17 (two RCTs)97,98 | + – – – (very low) (R1, R2) | |||
| Mental state: relapse (44–48 weeks) | RR 3.00 (0.16 to 57.36) | 8 (one RCT)97 | + – – – (very low) (R2, R3) | |||
| Leaving the study early (44–48 weeks) | RR 0.33 (0.06 to 1.99) | 8 (one RCT)97 | + – – – (very low) (R2, R3, R4) | |||
| Switch to different antipsychotic (risperidone/haloperidol) | Antipsychotic withdrawal (placebo) | TD: no improvement (12 weeks) | RR 0.45 (0.23 to 0.89) | 42 (one RCT)105–109 | + + – – (low) (R1, R2) |
|
| General mental state (12 weeks) | MD –4.3 (–10.48 to 1.88) | 42 (one RCT)105–109 | + – – – (very low) (R1, R3) | |||
| Adverse effects (8–12 weeks) | RR 2.08 (0.74 to 5.86) | 48 (one RCT)99–103 | + – – – (very low) (R1, R3) | |||
| Leaving the study early (12 weeks) | RR 0.60 (0.16 to 2.25) | 50 (one RCT)105–109 | + – – – (very low) (R1, R3, R5) | |||
| Switch to SGA (amisulpride/clozapine/olanzapine/risperidone/quetiapine) | Switch to different FGA | TD: no improvement (6 months) | RR 0.80 (0.52 to 1.22) | 45 (one RCT)110,111 | + – – – (very low) (R1, R2) |
|
| General mental state (1 year) | RR 1.83 (0.62 to 5.39) | 45 (one RCT)110,111 | + – – – (very low) (R1, R5) | |||
| Adverse effects (6 months) | RR 0.52 (0.31 to 0.89) | 82 (two RCTs)99–103,110,111 | + + – – (low) (R3, R4) | |||
| Leaving the study early (24–52 weeks) | RR 1.41 (0.74 to 2.67) | 168 (three RCTs)104,110–114 | + – – – (very low) (R6, R7, R8) | |||
| Olanzapine | Amisulpride | Adverse effects (6 months) | MD –0.35 (–2.44 to 1.74) | 54 (one RCT)112–114 | + – – – (very low) (R1, R2) |
|
| General mental state (6 months) | MD 1.32 (–1.94 to 4.58) | 54 (one RCT)112–114 | + – – – (very low) (R1, R2) | |||
| Leaving the study early (6 months) | RR 1.93 (0.19 to 20.12) | 57 (one RCT)112–114 | + – – – (very low) (R1, R2, R3) | |||
| Olanzapine | Risperidone | TD: no improvement (6 months) | RR 1.25 (0.82 to 1.90) | 60 (one RCT)115,116 | + – – – (very low) (R1, R2) |
|
| Adverse effects (6 months) | MD –0.70 (–1.33 to –0.07) | 60 (one RCT)115,116 | + + – – (low) (R1, R3) | |||
| General mental state (6 months) | RR 1.00 (0.15 to 6.64) | 60 (one RCT)115,116 | + – – – (very low) (R1, R2) | |||
| Leaving the study early (6–18 months) | RR 0.73 (0.57 to 0.95) | 170 (two RCTs)115–118 | + – – – (very low) (R3, R4, R5) | |||
| Olanzapine | Quetiapine | Leaving the study early (18 months) | RR 0.70 (0.54 to 0.90) | 116 one RCT)117,118 | + – – – (very low) (R1, R2, R4) |
|
| Olanzapine | Ziprasidone | Leaving the study early (18 months) | RR 0.77 (0.56 to 1.05) | 82 (one RCT)117,118 | + – – – (very low) (R1, R3, R4) | |
| Quetiapine | Risperidone | Leaving the study early (18 months) | RR 1.05 (0.88 to 1.25) | 118 (one RCT)117,118 | + – – – (very low) (R1, R3, R4) | |
| Quetiapine | Ziprasidone | Leaving the study early (18 months) | RR 1.10 (0.86 to 1.40) | 90 (one RCT)117,118 | + – – – (very low) (R1, R3, R4) | |
| Ziprasidone | Risperidone | Leaving the study early (18 months) | RR 0.95 (0.74 to 1.23) | 84 (one RCT)117,118 | + – – – (very low) (R1, R2, R4) | |
| Haloperidol | Tetrabenazine | TD: no improvement (18 weeks) | RR 1.07 (0.51 to 2.23) | 13 (one RCT)96 | + – – – (very low) (R1, R2) |
|
| TD: deterioration (18 weeks) | RR 0.86 (0.07 to 10.96) | 13 (one RCT)96 | + – – – (very low) (R1, R2) | |||
| Leaving the study early (18 weeks) | RR 4.38 (0.25 to 76.54) | 13 (one RCT)96 | + – – – (very low) (R1, R2, R3) | |||
| Anticholinergic drugs | ||||||
| Withdrawal of biperiden (stopping after 1 week) and AP continuation | Continuation of biperiden (stopping after 4 weeks) and AP continuation | Leaving the study early (7 weeks) | RR 2.14 (0.11 to 42.52) | 10 (one RCT)119 | + – – – (very low) (R1, R2, R3) |
|
| Benzodiazepines | ||||||
| Benzodiazepines (clonazepam, diazepam) and AP continuation | AP continuation with/without placebo | TD: no improvement (5–10 weeks) | RR 1.12 (0.60 to 2.09) | 32 (two RCTs)89,121,122 | + – – – (very low) (R1, R2) |
|
| TD: deterioration (5–10 weeks) | RR 1.48 (0.22 to 9.82) | 30 (two RCTs)89,121,122 | + – – – (very low) (R1, R2) | |||
| Leaving the study early (5–10 weeks) | RR 2.73 (0.15 to 48.04) | 56 (three RCTs)75,89,121,122 | + – – – (very low) (R1, R2, R3) | |||
| Clonazepam and AP continuation | Phenobarbital (as active placebo) and AP continuation | TD: no improvement (2 weeks) | RR 0.44 (0.20 to 0.96) | 21 (one RCT)120 | + – – – (very low) (R4, R5) | |
| Adverse effects (2 weeks) | RR 1.53 (0.97 to 2.41) | 21 (one RCT)120 | + – – – (very low) (R4, R5) | |||
| Leaving the study early (2 weeks) | N/E: no reported events | 21 (one RCT)120 | + – – – (very low) (R3, R4, R5) | |||
| Vitamin E | ||||||
| Vitamin E and AP continuation | Placebo and AP continuation | TD: no improvement (up to 1 year) | RR 0.95 (0.89 to 1.01) | 264 (six RCTs)93–95,123–126,130–137 | + + – – (low) (R1, R2) |
|
| TD: deterioration (up to 1 year) | RR 0.23 (0.07 to 0.76) | 85 (five RCTs)93–95,123–126,130 | + + – – (low) (R1, R2) | |||
| Adverse effects (up to 1 year) | RR 1.21 (0.35 to 4.15) | 205 (nine RCTs)90–93,95,123–128,130 | + – – – (very low) (R3, R4) | |||
| Leaving the study early (up to 1 year) | RR 1.07 (0.64 to 1.80) | 232 (eight RCTs)90–92,94,123–126,128,129,138 | + – – – (very low) (R2, R3, R5) | |||
| Miscellaneous treatments | ||||||
| Buspirone and AP continuation | Placebo and AP continuation | TD: no improvement (6 weeks) | RR 0.53 (0.33 to 0.84) | 42 (one RCT)139 | + + – – (low) (R1, R2) |
|
| Leaving the study early (6 weeks) | N/E: no reported events | 42 (one RCT)139 | – | |||
| Hypnosis/relaxation and AP continuation | TAU (AP continuation) | TD: no improvement (eight sessions) | RR 0.45 (0.21 to 0.94) | 15 (one RCT)78 | + – – – (very low) (R1, R2) |
|
| TD: deterioration (eight sessions) | RR 0.18 (0.01 to 3.81) | 15 (one RCT)78 | + – – – (very low) (R1, R3) | |||
| Leaving the study early (eight sessions) | N/E: no reported events | 15 (one RCT)78 | – | |||
Comparison 1: reduced dose of antipsychotics versus continuing antipsychotics
Two very small randomised trials97,98 conducted with schizophrenia or schizoaffective disorder inpatients and outpatients in the UK and USA reported on reduced doses compared with standard doses of flupentixol and fluphenazine. Evidence was of very low quality (see Table 2); therefore, we are uncertain of the results:
-
TD symptoms improved to a clinically important extent for significantly more people allocated to antipsychotic reduction than antipsychotic continuation after 44–48 weeks (very low-quality evidence, two RCTs,97,98 17 people; RR 0.42, 95% CI 0.17 to 1.04; I2 = 0%).
-
There was no significant difference in deterioration of TD symptoms at 44–48 weeks (very low-quality evidence, two RCTs,97,98 17 people; RR 0.61, 95% CI 0.11 to 3.31; I2 = 33%).
-
The number relapsing was not significantly different in the antipsychotic reduction group (1/4) and the antipsychotic maintenance group (0/4) at 44–48 weeks (one RCT,97 eight people; RR 3.00, 95% CI 0.16 to 57.36).
-
The number of people leaving the study early was not significantly different in the antipsychotic reduction group (1/4) and the antipsychotic maintenance group (3/4) (very low-quality evidence, one RCT,97 eight people; RR 0.33, 95% CI 0.06 to 1.99).
For this comparison there were no studies that reported on adverse events or social confidence, social inclusion, social networks or personalised quality of life.
Observational studies
First-generation antipsychotics: dose discontinuation versus decrease versus increase
Three small observational studies reported on discontinuing antipsychotics compared with a decrease or increase of the antipsychotic doses. 145,150,153,154 The studies were conducted in patients with a serious mental illness, mainly schizophrenia, in Canada, Japan and the USA. Evidence was rated as being of low to very low quality; therefore, we are uncertain of the results:
-
Casey and Toenniessen,145 a small prospective cohort study (n = 27), found that psychiatric patients with TD whose antipsychotic medication was reduced or discontinued showed greater improvement in TD symptoms after 5 years of follow-up than patients whose dosage of antipsychotic medication was increased (55–65% vs. 35%). Other outcomes were not reported.
-
Koshino et al. ,150 a small prospective cohort study (n = 28), found that the severity of TD was unchanged in 39.3% of the patients, improved in 17.9%, fluctuated in 21.4% and worsened in 21.4% at 11 years’ follow-up. The outcome was not associated with discontinuation, increase or decrease in the dosage of antipsychotics.
-
Yassa et al. ,153,154 also a small prospective cohort study (n = 44), reported that 50% of patients had no change in their TD severity, 20% had an improvement and 30% had a worsening of their TD. Little difference was noted in those patients whose medication was decreased (33% had no change in TD severity, 42% had increased TD severity and 25% had decreased TD severity) and those whose medication remained unchanged (56% had no change in TD severity, 25% had increased TD severity and 19% had decreased TD severity) at 10 years’ follow-up.
Comparison 2: switch to a different antipsychotic versus antipsychotic withdrawal (with placebo)
Two small randomised trials101,108 conducted with schizophrenic inpatients in Canada and Taiwan reported on switching to risperidone or haloperidol compared with placebo and withdrawing antipsychotics. Evidence was rated as being of low to very low quality (see Table 2); therefore, we are uncertain of the results:
-
TD symptoms improved to a clinically important extent for significantly more people allocated to antipsychotic switch to risperidone than those allocated to placebo at 12 weeks (low-quality evidence, one RCT,105–109 42 people; RR 0.45, CI 0.23 to 0.89).
-
There was no significant difference in the use of antiparkinsonism drugs between switching to risperidone or haloperidol compared with placebo at 8–12 weeks (two comparisons from one RCT,99–103 48 people; RR 2.08, CI 0.74 to 5.86; I2 = 0%).
-
General mental state was measured using the continuous BPRS scale (see Some specific outcomes). There was no significant difference between switching to risperidone compared with placebo on the average end-point score of the BPRS at 12 weeks (one RCT,105–109 42 people; MD –4.30, CI –10.48 to 1.88).
-
Using antipsychotics did not significantly increase the chances of a person leaving the study early at 12 weeks (very low-quality evidence, one RCT,105–109 50 people; RR 0.60, CI 0.16 to 2.25).
For this comparison there were no studies that reported on deterioration of TD symptoms or social confidence, social inclusion, social networks or personalised quality of life.
Observational studies
First-generation antipsychotics: dose discontinuation versus maintenance
Three small observational studies reported on discontinuing antipsychotics compared with maintenance of the standard doses. 149,151,152 The studies were conducted in patients with a serious mental illness, mainly schizophrenia, in the USA and Japan. Evidence was rated as being of low to very low quality; therefore, we are uncertain of the results:
-
Huang,149 a very small prospective cohort study (n = 10), found that psychiatric patients with TD whose antipsychotic medication was reduced or discontinued showed a greater improvement in TD symptoms after 4 years of follow-up than patients whose dosage of antipsychotic medication remained unchanged (60% vs. 21%). Other outcomes were not reported.
-
Peselow et al.,151 a small prospective cohort study (n = 31), reported a statistically significant decrease in abnormal movements at 1 year of follow-up; this improvement was offset by the fact that 15 of the 21 (71.4%) patients discontinued from antipsychotic treatment relapsed.
-
Yagi and Itoh,152 also a small prospective cohort study (n = 20), reported that, at 10 years’ follow-up, 64% (9/14) of patients in whom antipsychotics were discontinued or decreased after the occurrence of TD presented a clinically important improvement in symptoms; this also occurred in 75% (3/4) of those for whom the antipsychotic dose had been maintained. The authors suggested that the outcome of TD was determined by the patient’s age at onset rather than by the course of antipsychotic treatment.
Comparison 3a: switch to one antipsychotic versus switch to a different antipsychotic
Six small randomised trials101,104,110,112,115,117 of inpatients and outpatients with schizophrenia and schizoaffective disorder conducted in in Canada, South Africa, Taiwan and the USA reported on switching to a SGA (amisulpride, clozapine, olanzapine, risperidone, quetiapine, ziprasidone) compared with switching to a different antipsychotic, either a FGA (haloperidol, unspecified FGA) or another SGA. Evidence was rated as being of low to very low quality (see Table 2); therefore, we are uncertain of the results:
-
There were no significant differences on clinically important improvement in TD symptoms at 6 months between quetiapine and haloperidol (low-quality evidence, one RCT,110,111 45 people; RR 0.80, 95% CI 0.52 to 1.22) or between olanzapine and risperidone (very low-quality evidence, one RCT,115,116 60 people; RR 1.25, 95% CI 0.82 to 1.90).
-
The number of people in need of antiparkinsonism drugs was significantly lower in the group allocated to quetiapine than in the group allocated to haloperidol (one RCT,110,111 45 people; RR 0.45, 95% CI 0.21 to 0.96), but there was no significant difference between the groups allocated to risperidone or haloperidol (one RCT,99–103 37 people; RR 0.68, 95% CI 0.34 to 1.35).
-
Extrapyramidal symptoms at 6 months, as measured by the ESRS, were lower among participants on olanzapine than in those on risperidone (one RCT,115,116 60 people; MD –0.70, 95% CI –1.33 to –0.07), but there was no significant difference in extrapyramidal symptoms at 6 months, as measured by on SAS, at 6 months between participants on olanzapine and those receiving amisulpride (one RCT,112–114 54 people; MD –0.35, 95% CI –2.44 to 1.74).
-
There were no significant differences in general adverse events at 6 months, as measured on the UKU scale, between patients on olanzapine (one RCT,112–114 53 people; MD 0.08, 95% CI –1.85 to 2.01) or amisulpride (one RCT,112–114 53 people; MD –0.55, 95% CI –2.33 to 1.23) and thos receiving an unspecified FGA, or between those on olanzapine and those on amisulpride (one RCT,112–114 54 people; MD 0.63, 95% CI –0.93 to 2.19).
-
There were no significant differences in deterioration of mental state at 1 year between patients on quetiapine and those on haloperidol (one RCT,110,111 45 people; RR 1.83, 95% CI 0.62 to 5.39), or at 6 months between patients on olanzapine and those on risperidone (one RCT,115,116 60 people; RR 1.00, 95% CI 0.15 to 6.64) or at 6 months, measured on the BPRS, between patients on olanzapine and those on amisulpride (one RCT,112–114 54 people; MD 1.32, 95% CI –1.94 to 4.58).
-
People allocated to olanzapine were less likely to leave the study early, that is after 6–18 months, than those allocated to risperidone (two RCTs,115–118 170 people; RR 0.73, 95% CI 0.57 to 0.95; I2 = 0%) or quetiapine (one RCT,117,118 116 people; RR 0.70, 95% CI 0.54 to 0.90).
-
There were no significant differences at 6 months to 1 year in acceptability of treatment, defined as not leaving the study early, between patients receiving olanzapine or amisulpride and those receiving an unspecified FGA,112–114 or between those receiving clozapine or quetiapine and those receiving haloperidol,104,110,111 or between patients receiving olanzapine and those receiving amisulpride112–114 or ziprasidone,117,118 or between those on quetiapine and those on risperidone or ziprasidone,117,118 or between patients on ziprasidone and those on risperidone. 117,118
For this comparison there were no studies that reported on deterioration of TD symptoms or social confidence, social inclusion, social networks or personalised quality of life.
Observational studies
First-generation antipsychotics and gabapentin versus second-generation antipsychotics and gabapentin
One small observational study compared first-generation antipsychotics with gabapentin to second-generation antipsychotics with gabapentin in patients with serious mental illness (schizoaffective, bipolar I disorder and schizophrenic patients) and TD, in Italy. 148 This prospective cohort study (n = 30) reported that gabapentin treatment reduced TD symptoms with a mean percentage improvement on the Abnormal Involuntary Movement Scale (AIMS) of 47.5% (standard deviation ±18.2%) among all treated patients regardless of the antipsychotic used. Those on SGAs (mean 11.2 patients, standard deviation 4.8 patients; n = 18) reported that symptoms improved slightly more than those on FGAs (mean 18.2 patients, standard deviation 5.5 patients; n = 4).
Comparison 3b: specific antipsychotic versus other drug – haloperidol versus tetrabenazine
A very small randomised trial96 conducted with psychiatric inpatients in the USA compared haloperidol with tetrabenazine. The evidence was rated as being of very low quality (see Table 2); therefore, we are uncertain of the results:
-
There was no significant difference in clinically important improvement in TD symptoms at 18 weeks between patients receiving haloperidol and those receiving tetrabenazine (very low-quality evidence, one RCT,96 13 people; RR 1.07, 95% CI 0.51 to 2.23).
-
There was no significant difference in deterioration of TD symptoms at 18 weeks between patients receiving haloperidol and those receiving tetrabenazine (very low-quality evidence, one RCT,96 13 people; RR 0.86, 95% CI 0.07 to 10.96).
-
At 18 weeks there was no significant difference in the proportion of participants who had left the study early between the haloperidol (2/7 participants) and tetrabenazine groups (0/6 participants) (very low-quality evidence, one RCT,96 13 people; RR 4.38, 95% CI 0.25 to 76.54).
For this comparison there were no studies that reported on adverse events, mental state or on social confidence, social inclusion, social networks or personalised quality of life.
Comparison 4: withdrawal of anticholinergics versus continuation of anticholinergics
A very small randomised trial119 conducted in schizophrenia patients in Germany compared stopping biperiden after 1 week or after 4 weeks. The evidence was rated as being of very low quality (see Table 2); therefore, we are uncertain of the results:
-
There was no significant difference at 7 weeks in the proportion of people leaving the study early between those withdrawn from anticholinergic therapy (1/6 participants) and those who continues (0/4 participants) (very low-quality evidence, one RCT,119 10 people; RR 2.14, 95% CI 0.11 to 42.52).
For this comparison there were no studies with useable data on clinically important improvement or deterioration of TD symptoms, adverse events, mental state or on social confidence, social inclusion, social networks or personalised quality of life.
Comparison 5: benzodiazepines versus placebo, treatment as usual or active placebo (with antipsychotic management)
Four small randomised trials75,89,120,122 conducted with psychiatric inpatients and outpatients in China and the USA compared diazepam or clonazepam and antipsychotic continuation with placebo, TAU or phenobarbital as active placebo and antipsychotic continuation. The evidence was rated as being of very low quality (see Table 2); therefore, we are uncertain of the results:
-
There was no significant difference in ‘no clinically important improvement of TD symptoms’ at 5–10 weeks between patients on benzodiazepines and those receiving placebo or no treatment (very low-quality evidence, two RCTs,89,121,122 32 people; RR 1.12, 95% CI 0.60 to 2.09; I2 = 14%). One trial found that clonazepam was more beneficial than phenobarbital (as active placebo) at 2 weeks (very low-quality evidence, one RCT,120 21 people; RR 0.44, 95% CI 0.20 to 0.96).
-
There was no significant difference in deterioration of TD symptoms at 5–10 weeks (very low-quality evidence, two RCTs,89,121,122 30 people; RR 1.48, 95% CI 0.22 to 9.82; I2 = 19%).
-
One study reported on mental state average end-point scores using the BPRS scale and noted no difference between the diazepam and TAU groups at 10 weeks (one RCT,89 11 people; MD –0.50, 95% CI –13.83 to 12.83).
-
One trial found no significant difference in the number of participants experiencing adverse events after 2 weeks’ treatment with clonazepam or phenobarbital (as active placebo) (very low-quality evidence, one RCT,120 21 people; RR 1.53, 95% CI 0.97 to 2.41). All participants allocated to clozapine (10) and 7 out of 11 participants allocated to phenobarbital experienced an adverse event.
-
Three studies reported that no participants left the study early. 75,120–122 One study reported that 2 out of 33 participants allocated to diazepam, but none (out of 23) allocated to TAU, left the study early and, subsequently, found no significant difference between the two groups at 10 weeks (very low-quality evidence, one RCT,89 56 people; RR 2.73, 95% CI 0.15 to 48.04).
For this comparison there were no studies that reported on social confidence, social inclusion, social networks or personalised quality of life.
Comparison 6: vitamin E versus placebo (with antipsychotic management)
Thirteen randomised trials90–95,123,127–130,137,138 in psychiatric inpatients and outpatients in China (one study138), Hong Kong (one study94), Israel (two studies90,91), India (one study127), Switzerland (one study95), the UK (one study130) and the USA (six studies92,93,123–126,128,129,131–137) reported on vitamin E (gamma-tocopherol) and antipsychotic continuation compared with placebo and antipsychotic continuation. The evidence was rated as being of low to very low quality (see Table 2); therefore, we are uncertain of the results. After up to 1 year:
-
There was no significant difference between the vitamin E and placebo groups in the numbers of patients experiencing no clinically important improvement in TD symptoms (low-quality evidence, six RCTs,93–95,123–126,130–137 264 people; RR 0.95, 95% CI 0.89 to 1.01; I2 = 0%).
-
The number of participants who showed deterioration of TD symptoms was significantly lower in the vitamin E group than in the placebo group (low-quality evidence, five RCTs,93–95,123–126,130 85 people; RR 0.23, 95% CI 0.07 to 0.76; I2 = 0%)
-
One study131–137 measured adverse events (extrapyramidal symptoms) using the SAS and found no significant difference between the vitamin E and placebo groups (very low-quality evidence, 104 people; MD 1.10, 95% CI –1.02 to 3.22).
-
There was no significant difference in the incidence of any adverse event (very low-quality evidence, nine RCTs,90–93,95,123–128,130 205 people; RR 1.21, 95% CI 0.35 to 4.15; I2 = 0%).
-
There was no significant difference in mental state, as measured by the BPRS, between vitamin E and placebo groups (three RCTs,127,129,131–137 165 people; MD –0.20, 95% CI –3.21 to 2.82; I2 = 38%).
-
There was no significant difference in acceptability of treatment (leaving the study early) [very low-quality evidence, medium term (overall ≈20% loss to follow-up), eight RCTs,90–92,94,123–126,128,129,138 232 people; RR 1.07, 95% CI 0.64 to 1.80; I2 = 0%].
For this comparison there were no studies that reported on social inclusion, social networks or personalised quality of life.
Comparison 7: buspirone versus placebo (with antipsychotic management)
One small randomised trial,78 conducted with psychiatric inpatients in China, reported on buspirone and antipsychotic continuation compared with placebo and antipsychotic continuation. Evidence was rated as being of low quality (see Table 2); therefore, we are uncertain of the results:
-
The number of participants reporting clinically important improvement in TD symptoms after 6 weeks was signficicantly higher in the buspirone group than in the placebo group (low-quality evidence, one RCT,78 42 people; RR 0.53, 95% CI 0.33 to 0.84).
-
Acceptability of treatment, measured by the number of participants leaving the study early, could not be estimated, as the included study did not report any events.
For this comparison there were no studies that reported on deterioration of TD symptoms, adverse events, mental state or on social confidence, social inclusion, social networks or personalised quality of life.
Comparison 8: hypnosis and relaxation versus treatment as usual (with antipsychotic management)
One very small randomised trial,139 conducted with psychiatric inpatients in the USA, reported on hypnosis or relaxation and antipsychotic continuation compared with TAU and antipsychotic continuation. The evidence was rated as being of very low quality (see Table 2); therefore, we are uncertain of the results:
-
Clinically important improvement in TD symptoms after eight sessions was reported by significantly more participants in the hypnosis or relaxation group than in the TAU group (very low-quality evidence, one RCT,139 15 people; RR 0.45, 95% CI 0.21 to 0.94).
-
There was no significant difference in deterioration of TD symptoms after eight sessions between the hypnosis or relaxation group and the TAU group (very low-quality evidence, one RCT,139 15 people; RR 0.18, 95% CI 0.01 to 3.81).
-
Acceptability of treatment (leaving the study early) could not be estimated, as the included study reported no events.
For this comparison there were no studies that reported on adverse events, mental state or on social confidence, social inclusion, social networks or personalised quality of life.
Analysis of the robustness of the results (sensitivity analyses)
Risk of bias
We planned to restrict the analyses to studies considered to be at low, and low or unclear, risk of selection and detection bias. None of the included studies was rated as being at a low risk of both selection and detection bias. Studies were rated as being either at an unclear risk of bias or at a low and unclear risk (see Appendix 7, Table 13), except Glover,139 which was the only study rated as being at high risk of selection bias. Glover139 was the only study that investigated hypnosis and relaxation.
Imputed values
We would have undertaken a sensitivity analysis to assess the effects of including data from cluster randomised trials in which we used imputed values for the intracluster correlation coefficient in calculating the design effect. However, we identified no cluster randomised trials for inclusion.
Planning future studies
No clinical improvement of tardive dyskinesia symptoms
Only one study110 comparing ‘switch to FGA’ with ‘switch to SGA’ reported the outcome ‘no clinical improvement’. The odds ratio (OR) comparing these two treatments was 1.96 (95% CI 0.56 to 6.92), indicating an insignificant advantage of ‘switch to SGA’ compared with ‘switch to FGA’. The wide CI surrounding the effect estimate suggests that the existing evidence might not be adequate to conclude which of the two interventions is more effective. The power curve in Figure 5 shows the power of an updated meta-analysis considering that a new study with sample size indicated in the horizontal axis is added to the evidence base. The power of a meta-analysis including a new study with a small sample size would remain low (e.g. we would achieve a power of < 40% randomising 100 more patients). To achieve a power of 80% for the meta-analysis, a new study with a total sample size of 800 patients would need to be designed and included in the meta-analysis model. The extended funnel plot could not be drawn given the availability of a single study.
FIGURE 5.
Power curves with 95% CIs for the outcome ‘no clinical improvement of TD symptoms’ for the comparison ‘switch to FGA’ vs. ‘switch to SGA’.
Total discontinuation rates
Three studies comparing ‘switch to FGA’ to ‘switch to SGA’ and reporting ‘total discontinuation rates’ were available. The resulting OR was 0.54 (95% CI 0.21 to 1.42) in favour of a ‘switch to FGA’ using the fixed-effect inverse-variance meta-analysis model. For a new study to make an important contribution to the existing evidence by rendering the power of the meta-analysis 80%, it would have to have a total sample size of ≥ 1000 patients (Figure 6). The implications of including a hypothetical new study in the meta-analysis are illustrated in the extended funnel plot of Figure 7. The inclusion of an additional study lying in the left-hand light-green region of Figure 7 would result in the updated meta-analysis showing a significant result in favour of a ‘switch to FGA’. As none of the existing studies lies in this region, it is considered unlikely that a new trial will change meta-analysis conclusions. The possibility that a meta-analysis would change the inference in favour of a ‘switch to SGA’ is even smaller, as it would require the inclusion of a study with a very small standard error (smaller than 0.1) demonstrating a favoured outcome for the particular treatment. Thus, despite the fact that meta-analysis is inconclusive, it is not likely that a new study would change its conclusions given that its sample size is not substantially large.
FIGURE 6.
Power curves with 95% CIs for the outcome ‘total discontinuation rates’ for the comparison ‘switch to FGA’ vs. ‘switch to SGA’.
FIGURE 7.
Extended funnel plot for the outcome ‘total discontinuation rates’ for the comparison ‘switch to FGA’ vs. ‘switch to SGA’: contours for impact of a new study.

Chapter 6 Part C: results of the network meta-analysis
We intended to synthesise available evidence from treatment options of interest using a NMA model. 155–157 However, the sparseness of the existing evidence imposed important barriers in the analysis rendering the presentation of NMA results as our main analysis impractical. In particular, comparisons were typically informed by very few studies, and many studies had few or even zero events. Analysing and interpreting few data can be particularly challenging, and simulation studies have shown that many of the most commonly used meta-analytic methods produce biased estimates and misleading conclusions when events are rare. 158,159 Challenges in the analysis of few data include the difficulty of justifying the use of distributional approximations to statistics of interest and the potential risk of small studies including unrepresentative populations. 159,160
Use of NMA can benefit the evidence synthesis of few data by borrowing strength across treatment comparisons and gaining information through the contribution of indirect evidence. Moreover, sharing parameters across the entire network can provide information on their inference; here, we assumed a common heterogeneity parameter across all treatment comparisons. Although the assumption of a common heterogeneity is expected to hold in this setting, formal investigation of between-study variations is limited by the sparseness of the data. Despite efforts to strengthen the evidence body and sharing parameters across networks, analysing and interpreting NMA results under sparseness was challenging; results of NMA for the outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’ are presented in Appendix 4. Network effects were almost identical to their pairwise meta-analysis counterparts when direct evidence existed; any differences are attributed to the estimation of heterogeneity. When direct evidence was absent, indirect estimates were highly imprecise, failing to produce useful summaries on the relative effectiveness of the interventions of interest and consequently to provide interpretable results to be used for decision-making. Moreover, no closed loops of evidence were formed in the network for the primary outcome and only one existed for total discontinuation rates, making it impossible to evaluate the validity of the consistency assumption. The interventions of interest that were set to be on the priority list did not form a connected network that could be analysed at once; this further limited the value of performing NMA and precluded us from presenting it as our main analysis.
Despite the barriers that lack of sufficient research data may impose, decisions often need to rely on few data. Thus, exploration of possible ways in which inferences could be made based on a limited evidence base would be useful. Use of external evidence, both eliciting expert opinions and using observational data, has been considered elsewhere. 160 The presence of few data, along with the associated highly imprecise NMA effects, highlights the uncertainty surrounding the relative effectiveness between alternative treatment options for TD and underlines the need for further research to be conducted. Future studies should be planned (see Chapter 8, Recommendations for research) to enrich the existing evidence base and, by making the synthesis of data in a NMA model sensible, to enlighten the relative effectiveness between available treatment options.
Several methods, tailored to outcomes with very low frequency, have been developed. 161–163 Rücker et al. 161 proposed the arcsine difference as an alternative effect size measure that enables such studies to be included in a meta-analysis. Despite its advantages, the arcsine method provides an effect size that is difficult to interpret and is poorly understood by clinicians.
Chapter 7 Discussion
Summary of main results
The search
This area of research does not seem to be active. We have identified additional data, but most trials pre-date the year 2000, with only six studies (of prioritised interventions) published between 2000 and 2011. Possible explanations for this include lack of concern with TD in the research community, discouragement regarding the possibility of identifying effective treatments, or, more positively, decreased emergence of the problem in research-active communities because of more thoughtful use of antipsychotic drugs.
In addition to RCTs, we identified eight small prospective cohort studies that reported on efficacy of interventions (mostly antipsychotics) for the treatment of TD.
Few data
The great majority of studies testing treatments for people with TD are short and very small. This whole review of many comparisons shows that only hundreds, not thousands, of people have been randomised, and no one with dementia and TD. Any effect of treatment is likely to be subtle and so substantial sample sizes are needed to show differences with acceptable confidence. This also applies to observational studies, in which eight prospective studies reported on 200 patients with TD.
Many outcomes were not measured at all by included studies. We may have been overambitious in hoping for some of these outcomes in TD trials, but simple reporting of social impact and quality of life does not seem unreasonable, and is of particular interest to patients and carers.
Outcomes
Tardive dyskinesia symptoms
We found low-quality evidence of clinically important improvement in TD symptoms after 12 weeks for switching antipsychotic to risperidone compared with withdrawing antipsychotics (with placebo) (one study, 42 people; RR 0.45, 95% CI 0.23 to 0.89), and after 6 weeks for buspirone compared with placebo while continuing antipsychotics as usual (one study, 42 people; RR 0.53, 95% CI 0.33 to 0.84). We also found low-quality evidence that use of vitamin E could prevent deterioration of TD symptoms compared with placebo while continuing antipsychotics as usual after 1 year (five studies, 85 people; RR 0.23, 95% CI 0.07 to 0.76). Because the quality of evidence is low, we have limited confidence in the effect estimates and CIs; the true effects may be substantially different.
Furthermore, we found very low-quality evidence of clinically important improvement in TD symptoms after 1 year for antipsychotic reduction compared with antipsychotic continuation (two studies, 17 people; RR 0.42, 95% CI 0.17 to 1.04), after 2 weeks for clonazepam compared with phenobarbital as active placebo while continuing antipsychotics as usual (one study, 21 people; RR 0.44, 95% CI 0.20 to 0.96) or for hypnosis or relaxation compared with placebo while continuing antipsychotics as usual for eight sessions (one study, 15 people; RR 0.45, 95% CI 0.21 to 0.94). Because the quality of evidence is very low, we have very little confidence in the effect estimates and CIs; the true effects are likely to be substantially different.
There was very low-quality evidence from observational studies of an improvement in TD symptoms when antipsychotics were discontinued or decreased; on average, these studies were very small, had an unbalanced number of participants in each group and selective outcome reporting bias.
For the remaining comparisons we found low- to very low-quality evidence of little or no difference between groups, but, again, our confidence in these results is limited.
Adverse effects
There was low-quality evidence that fewer people taking SGAs than taking FGAs needed antiparkinsonism medication because of extrapyramidal side effects after 1 year (two studies, 82 people; RR 0.52, 95% CI 0.31 to 0.89). There was also low-quality evidence that after 6 months extrapyramidal symptoms, as measured on the ESRS, were less common in the olanzapine group than in the risperidone group (one study, 60 people; MD –0.70, 95% CI –1.33 to –0.07). Finally, there was very low-quality evidence that after 2 weeks fewer people on phenobarbital as an active placebo than on clonazepam had experienced any adverse events (one study, 21 people; RR 1.53, 95% CI 0.97 to 2.41).
None of the observational studies reported on adverse events for the interventions.
As a result of the low to very low quality of this evidence, our confidence in these results is limited.
For the remaining comparisons, we found low- to very low-quality evidence of little or no difference between groups, but, again, our confidence in these results is limited.
Mental state
We found low- to very low-quality evidence of little or no difference between groups of all comparisons, but, again, our confidence in these results is limited.
Acceptability of treatment: leaving the study early
It is always unclear what leaving a study early means for the participant. It could be related to the participant rejecting treatment for a series of reasons, or attributable to participants finding the trial intolerable. It also could be a function of a trial design in which participants, although willing to continue, are asked to leave because of some degree of protocol violation. In any event, for most of the interventions the numbers of participants leaving the study early were not different for those allocated to either group. Fewer participants allocated to olanzapine than to risperidone (two studies, 170 people; RR 0.73, 95% CI 0.57 to 0.95) or to quetiapine (one study, 116 people; RR 0.70, 95% CI 0.54 to 0.90) left the study early after 6–18 months. Evidence was of very low quality for both comparisons; therefore, we have very little confidence in the effect estimates and CIs; the true effects are likely to be substantially different.
Social confidence, social inclusion, social networks or personalised quality of life
This group of outcomes was selected as being of importance to patients for the 2016 review update following a service user consultation. No studies were identified that reported on any of these outcomes.
Overall completeness and applicability of evidence
Completeness
We excluded 22 studies of prioritised interventions published between 1971 and 2004 because they did not report data that could be used in the review. We contacted the study authors wherever possible, but no further information was available.
As part of this work, the service user consultation participants highlighted their preferred outcomes (Box 2). These largely correlated with the perspectives of the clinicians and reviewers – listing clear, clinically meaningful effects on TD, adverse effects or leaving the study early – as being of importance. The consultation added the outcome of some measure of social confidence/inclusion/networks and/or quality of life. There were no data for the measure of social confidence/inclusion/networks and/or quality of life, but in reality all others were incomplete – perhaps with the exception of vitamin E. The large trials – or enough small trials on the same topic – have just not been undertaken. The difficulty of carrying out randomised studies in this area should not be underestimated. However, time and time again pioneering triallists have proved that it is possible.
1.1 Improved to a clinically important extent.
1.2 Deteriorated.
2. Mental state 3. Adverse effects3.1 Any adverse event.
3.2 Adverse effects: no clinically significant extrapyramidal adverse effects.
4. Acceptability of treatment4.1 Leaving the study early.
5. Social confidence, social inclusion, social networks or personalised quality-of-life measures5.1 No significant change in social confidence, social inclusion, social networks or personalised quality-of-life measures for either recipients of care or caregiver.
Another problem is that there seems to be little evidence of collaboration; no two trials are the same. With collaborative effort we could have enough people randomised across time to have answers to some practical issues. Currently, we cannot even be confident that dose reduction really helps. Of course, researchers will always be attracted to try the next compound, but this overview illustrates that there are enough ‘loose ends’ in the past work regarding entirely practical interventions to encourage some large collaborative efforts in randomisation.
This overview – and the clear incompleteness of the data on this old, well-recognised condition – also, we think, serves to encourage some consideration about trial design. Past work does not serve people with TD particularly well. In the 30 years of, largely, pilot studies, trial methodology within mental health has evolved, with larger pragmatic trials becoming more prevalent. The service user consultation has provided outcomes fitting with a pragmatic randomised trial design (see Box 2). This trial, which need not be that expensive, could be undertaken wherever TD is a concern and need not be constrained to the somewhat fragmented services often seen in ‘Western’ medicine.
Applicability
Most trials in this review were hospital based, but nevertheless featured the type of patients likely to be encountered in everyday care. Many of the interventions are readily accessible. The outcomes pose a greater problem of applicability. Scale-derived findings may be applicable, but even the original measures do not really describe how findings are relevant to day-to-day care. Whenever possible, we have extracted outcomes such as ‘improved/not improved to a clinically important extent’. For the degree of importance of the change, we have to trust the judgement of triallists from a wide variety of backgrounds and care cultures.
Quality of the evidence
Overall, the quality of the evidence is low to very low. This means that we have limited to very little confidence in the effect estimates, and the true effect may be, or is likely to be, substantially different from the estimate of the effect. The main reasons for our low confidence in the evidence were:
-
poor study methodology and reporting of methods, resulting in downgrading evidence for risk of bias
-
very small sample sizes, resulting in downgrading evidence for imprecision
-
wide CIs (often attributable to low event rates) that included appreciable benefit or harm for the intervention as well as no effect, resulting in downgrading evidence for imprecision.
Please see Table 2 for full details.
Potential biases in the review process
Missing studies
We have made every effort to identify relevant trials. However, these studies are all small and it is likely that we have failed to identify other studies of limited power. It is likely that such studies would also not be in favour of the intervention investigated; if they had been so, it is more likely that they would have been published in accessible literature. We do not, however, think it likely that we have failed to identify large relevant studies.
Introducing bias
We have tried to be balanced in our appraisal of the evidence, but could have inadvertently introduced bias. We have tried to intentionally add bias towards treatments useful within the NHS, but have found no other innovations that really hold promise. We welcome comments or criticisms. We tried to ensure that searches for trials were wide-ranging, covering as many data sources as possible, but we still could easily have missed studies. We think it unlikely, however, that we would have missed large trials with important outcomes.
It is an unavoidable fact that many of the authors were familiar with this literature for many years before undertaking this full overview. However, the PPI exercise was undertaken, largely, blind to the results of the Cochrane reviews and in time to pre-date (and therefore direct) the construction of the summary-of-findings tables.
Agreements and disagreements with other studies or reviews
The only other relevant quantitative review on this topic we know of is the previous Cochrane review. 50 This update expands and improves this review, but does not substantially change the findings or the conclusions.
Chapter 8 Conclusions
Implications for health care
Clinicians, policy-makers and people with/at risk of TD are little better informed on this issue than they were decades ago. Underpowered randomised trials and observational studies of limited quality have repeatedly failed to provide answers.
Although it seems prudent to use the lowest effective dosage of antipsychotic drug possible (within the licensed range) for individual patients, there is no evidence that antipsychotic discontinuation will improve TD symptoms.
Current treatments for TD are prescribed in hopes that they will have an impact on TD, but none have a strong base in evidence. It could be argued that these treatments are only ethical within well-designed pragmatic trials aimed at informing clinical practice in people with this debilitating problem.
Recommendations for research
Tardive dyskinesia reviews have data from current trials extracted, tabulated and traceable to source. 54 TD reviews, whether or not those within Cochrane, could use this resource to save time and money. These are reliably extracted data for sharing.
The NMA highlights one context in which support for this technique is ill advised. Where studies are short, small, have similar results and are of poor quality, NMA is not indicated.
All relevant trials, even if not primarily addressing the issue of TD, should report appropriate binary outcomes on groups of people with this problem.
Our public consultation recognised the importance of TD, and participants reacted to the poor quality of research evidence and lack of progress in addressing TD over time. People attending felt that the current outcomes could be enhanced by addressing core concerns of service users such as social networks, quality of life and employment. Ideas for further research included prevalence studies, addressing social stigma, understanding causal mechanisms, developing psychological therapies to address TD specifically and looking at the role of peer support in managing TD. The full details are reported in Appendix 1.
The recommendations of the public consultation for focusing on specific key outcomes in our work were implemented directly into the summary-of-findings tables presented in this work and in the Cochrane reviews. In turn, these form the basis of the outcome list.
This review summarises more than three decades of pioneering work, but also highlights a systemic failure to properly address the ongoing issue of TD for clinicians or patients.
More thoughtful use of antipsychotic medication may reduce its prevalence, but TD nevertheless remains a problem. 5 Most people needing antipsychotic medication live in low- and middle-income countries, where the highest potency antipsychotic drugs may be the only ones available. TD is with us from treatments of the past, and continues to emerge from treatment practices of the present.
We realise that we are applying pragmatic clinical demands on studies that may never have been designed to provide them. Largely, the studies we have identified for inclusion were of short duration and grossly underpowered. The studies used proxy outcomes, often out of necessity, as sensitive scales may show effects even if they are not pragmatic clinical outcomes. However, even in the syntheses we have been able to do, combining the power of similar studies on any outcome seems unlikely to provide sufficient power to illustrate real effects. We feel that the overview, Cochrane reviews and NMA reported here illustrate the need for not only more well-designed, -conducted and -reported pilot studies, but also much larger pragmatic studies reporting outcomes familiar to clinicians and patients.
Pioneering researchers will probably continue to undertake pilot randomised studies. All such studies should make all data available, including those on outcomes suggested by the public consultation, even if underpowered, to highlight clear differences. Randomised trials of treatments for people with established TD are indicated, with the most obvious recommended outcome for a large study being dose reduction. Such trials should be large (> 800 participants), perhaps with accrual supported through accurate local/national registers. The studies should be of adequate duration (1 year minimum), with test interventions that are acceptable and record outcomes relevant to everyone. Such trials could open opportunities for research in places that may be less well funded but carry the burden of care.
Public consultation in the UK has provided a list of simple, and, we think, universally relevant, practical outcomes for the large trials. These, along with any other routinely collected data, include outcomes that can be used for risk–benefit analyses and economic considerations.
These large trials should take place before another three decades pass.
There are many small, short trials investigating interventions for people with schizophrenia and TD but none for those with dementia and TD. Public consultation highlighted the need for updated prevalence studies of TD in groups of people with schizophrenia, those exposed to antipsychotic medication and, finally, patients with dementia.
Use of crossover design
Triallists find it difficult to identify people with both TD and schizophrenia to participate in trials. 95 Randomised crossover designs are used in the hope of improving the power of the study to find outcomes of interest. In this design, participants are initially randomised to one of the experimental interventions and then, at a prespecified time, cross over to the treatment that they did not receive at first. Conditions with a more stable time course than TD are better suited for crossover studies. 164
The carry-over effect introduces additional difficulties. Many substances used to treat TD may well persist in the body for long periods after discontinuation; unless crossover studies include a mid-study washout period (which ensure that the participant is free from the inital treatment before starting the next arm of the study), any effect of treatment may continue into the second, placebo, arm of the trial – the ‘carry-over effect’. In addition, carry-over may involve the regrowth or retreat of neuroreceptors. This slow rebalancing, if started, could continue long after all traces of intervention drugs are gone, so the physiological half-life of the experimental treatment may not be the only variable to consider when thinking through the issues of carry-over. TD is also an unstable condition, and people with TD may not remain compliant with medication. All these factors make the arguments for not using crossover methodology strong, despite the initial attraction. 164–166
Planning of future studies
The relative effectiveness and safety of a ‘switch to FGA’ compared with a ‘switch to SGA’ is considered to be of great importance in terms of deterioration of symptoms of antipsychotic-induced TD. However, only a handful of studies examined that particular comparison – one and three studies for the outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’ were available, respectively. NMA did not offer any additional advantage or further insight on the ‘switch to FGA versus switch to SGA’ comparison; no indirect evidence feeding this comparison existed and, thus, the network estimates were identical to their pairwise meta-analysis counterparts (see Appendix 4).
Figures 6 and 7 imply that, although the meta-analysis can be considered reasonably robust to the addition of new studies with a small sample size, conclusions might change if large studies are added. If further studies are to be designed and conducted, a total sample size of 1000 patients would give a good prospect of reaching a conclusive result for both outcomes. Decisions on whether or not new studies are to be conducted should take into account the feasibility of such a sample size. In any case, informed and evidence-based decisions would require the systematic assessment of existing evidence before embarking into new research. 167,168
Acknowledgements
People
Rosie Asher (Clinical Research Associate) screened references and full texts, extracted and source-coded data, and assessed and data-extracted studies in Hebrew for Cochrane reviews. Antonio Jose Grande (Research Associate) screened references and full texts, cross-checked data, helped organise references and analyses, and assessed and data-extracted studies in Spanish and Portuguese for Cochrane reviews. Farhad Shokraneh (Information Specialist) conducted the search, made the traceable data available, and assessed and data-extracted studies in Persian for Cochrane reviews. Ben Grey (Senior Peer Researcher, the McPin Foundation) advised on PPI and wrote plain language summaries for Cochrane reviews. Vanessa Pinfold (Research Director, McPin Foundation) advised on PPI. Ruth Sayers (Peer Researcher, McPin Foundation) and Megan Rees (Public Involvement in Research Co-ordinator, McPin Foundation) conducted the PPI consultation together with author Dawn-Marie Walker. Artemisia Kakourou (Medical Doctor, Systematic Reviewer) assessed and data-extracted observational studies and studies in French for Cochrane reviews. Loukia Spineli (Research Associate, Statistician) helped with data extraction, data cross-checking and organising references for Cochrane reviews. Nicholas Henschke (Systematic reviewer) helped with report writing for Cochrane reviews. Nancy Owens (Senior Communications Manager) assisted with proofreading. Molly Grimes (Clinical Psychologist) assisted with copy-editing. Linda Levi (Psychiatry Research Co-ordinator) helped with creating tables for the National Institute for Health Research (NIHR) report and updating background sections for Cochrane reviews. Daphna Fenchel (Psychiatry Research Associate) helped with background for the NIHR report. Sai Zhao assessed and data-extracted studies in Chinese for Cochrane reviews. Stefan Leucht and Johannes Schneider-Thoma assessed and data-extracted studies in German for Cochrane reviews. Yusuke Ogawa assessed and data-extracted studies in Japanese for Cochrane reviews. Lisa Korsbek assessed studies in Danish for Cochrane reviews. Suyoung Kim assessed studies in Korean for Cochrane reviews
Funding
This report was funded by the UK’s NIHR Health Technology Assessment programme (NIHR HTA 14/27/02) and without this our work for this report would have been impossible. The funding has built on the volunteer input, which will continue after the end of the funding period. The funders have had no influence on the content of the reviews or final report.
Contributions of authors
Hanna Bergman (Systematic Reviewer, systematic review methods) co-ordinated updates of the nine Cochrane reviews on which this report is based, co-ordinated traceable data coding, selected studies, extracted, analysed and interpreted data, created summary-of-findings tables and wrote the final report.
Dawn-Marie Walker (Associate Professor, PPI) was one of the researchers who was awarded the grant with Karla Soares-Weiser and Clive E Adams, helped to design the project, oversaw the patient involvement and discussed the findings from the review with them, helped write the PPI section and reviewed the document through iterative drafts.
Adriani Nikolakopoulou (Doctor of Philosophy Student in Biostatistics, evidence synthesis methods) planned and conducted the NMA, and wrote the NMA sections of the report.
Karla Soares-Weiser (Deputy Editor in Chief for Cochrane, until September 2015 was the Managing Director of Enhance Reviews, psychiatry, evidence synthesis) was actively involved in the preparation of the original reviews, helped write the proposal, helped supervise the search and selection, co-ordinated the overall process and wrote the final report.
Clive E Adams (Chairperson of Mental Health Services Research, systematic reviewing, schizophrenia) helped do original reviews, helped supervise the search and selection, co-ordinated the overall process, and helped assimilate and write the final report.
Publications
Currently, only this report is published, but nine Cochrane reviews (see Appendix 6) are updated and are going through to full publication.
Soares-Weiser K, Mobsy C, Holliday E. Anticholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 1997;2:CD000204.
Tammenmaa IA, McGrath JJ, Sailas E, Soares-Weiser K. Cholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2002;3:CD000207.
Soares-Weiser K, Irving Claire B, Rathbone J. Miscellaneous treatments for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2003;2:CD000208.
Bhoopathi PS, Soares-Weiser K. Benzodiazepines for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2006;3:CD000205. http://dx.doi.org/10.1002/14651858.CD000205.pub2
El-Sayeh HG, Lyra da Silva JP, Rathbone J, Soares-Weiser K. Non-neuroleptic catecholaminergic drugs for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2006;1:CD000458.
Soares-Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database Syst Rev 2006;1:CD000459.
Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma-aminobutyric acid agonists for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;4:CD000203. http://dx.doi.org/10.1002/14651858.CD000203.pub3
Essali A, Deirawan H, Soares-Weiser K, Adams CE. Calcium channel blockers for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;11:CD000206. http://dx.doi.org/10.1002/14651858.CD000206.pub3
Soares-Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;2:CD000209. http://dx.doi.org/10.1002/14651858.CD000209.pub2
Data sharing statement
Extracted data are freely available on Cochrane Schizophrenia Group’s website via ResearchGate (http://dx.doi.org/10.13140/RG.2.2.28907.95529).
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Tarsy D, Lungu C, Baldessarini RJ. Epidemiology of tardive dyskinesia before and during the era of modern antipsychotic drugs. Handb Clin Neurol 2011;100:601-16. http://dx.doi.org/10.1016/B978-0-444-52014-2.00043-4.
- Rosenheck RA. Evaluating the cost-effectiveness of reduced tardive dyskinesia with second-generation antipsychotics. Br J Psychiatry 2007;191:238-45. https://doi.org/10.1192/bjp.bp.106.035063.
- Miller DD, Eudicone JM, Pikalov A, Kim E. Comparative assessment of the incidence and severity of tardive dyskinesia in patients receiving aripiprazole or haloperidol for the treatment of schizophrenia: a post hoc analysis. J Clin Psychiatry 2007;68:1901-6. https://doi.org/10.4088/JCP.v68n1210.
- Kane JM, Bloom FE, Kupfer DJ. Psychopharmacology: 4th Generation of Progress. New York, NY: Raven Press; 1995.
- Bakker PR, de Groot IW, van Os J, van Harten PN. Predicting the incidence of antipsychotic-induced movement disorders in long-stay patients: a prospective study. Epidemiol Psychiatr Sci 2013;22:375-9. http://dx.doi.org/10.1017/S204579601300019X.
- Bakker PR, de Groot IW, van Os J, van Harten PN. Antipsychotic-induced movement disorders in long-stay psychiatric patients: a prospective study. Schizophr Res 2014;153:88-9. https://doi.org/10.1016/S0920-9964(14)70282-8.
- Miller DD, Caroff SN, Davis SM, Rosenheck RA, McEvoy JP, Saltz BL, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry 2008;193:279-88. http://dx.doi.org/10.1192/bjp.bp.108.050088.
- Woods SW, Morgenstern H, Saksa JR, Walsh BC, Sullivan MC, Money R, et al. Incidence of tardive dyskinesia with atypical versus conventional antipsychotic medications: a prospective cohort study. J Clin Psychiatry 2010;71:463-74. https://doi.org/10.4088/JCP.07m03890yel.
- Woerner MG, Correll CU, Alvir JM, Greenwald B, Delman H, Kane JM. Incidence of tardive dyskinesia with risperidone or olanzapine in the elderly: results from a 2-year, prospective study in antipsychotic-naïve patients. Neuropsychopharmacology 2011;36:1738-46. http://dx.doi.org/10.1038/npp.2011.55.
- Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics 2014;11:166-76. https://doi.org/10.1007/s13311-013-0222-5.
- Maher AR, Theodore G. Summary of the comparative effectiveness review on off-label use of atypical antipsychotics. J Manag Care Pharm 2012;18:1-20. https://doi.org/10.18553/jmcp.2012.18.s5-b.1.
- Ballesteros J, Gonzalez-Pinto A, Bulbena A. Tardive dyskinesia associated with higher mortality in psychiatric patients: results of a meta-analysis of seven independent studies. J Clin Psychopharmacol 2000;20:188-94. https://doi.org/10.1097/00004714-200004000-00011.
- Martins ES, Rosso A, Coutinho E, Adams C, Huf G. Prevalence of tardive dyskinesia and all-cause mortality amongst patients in a large psychiatric institute in Rio de Janeiro. Rev Psiquiatr Clin 2011;38.
- National Institute for Health and Care Excellence . Psychosis and Schizophrenia in Adults: Treatment and Management. NICE Clinical Guideline 178 2014. www.nice.org.uk/guidance/cg178 (accessed 20 January 2017).
- Taylor D, Paton C, Kapur S. The Maudsley Prescribing Guidelines. London: Informa Healthcare; 2009.
- Lieberman JA, Fleishhacker W. Introduction. Br J Psychiatry 1996;168:7-8. https://doi.org/10.1142/9789812819796_0001.
- Chouinard G, Chouinard V-A. Atypical antipsychotics: CATIE study, drug-induced movement disorder and resulting iatrogenic psychiatric-like symptoms, supersensitivity rebound psychosis and withdrawal discontinuation syndromes. Psychother Psychosom 2008;77:69-77. https://doi.org/10.1159/000112883.
- Soares-Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database Syst Rev 2006;1. http://dx.doi.org/10.1002/14651858.CD000459.pub2.
- Bratti IM, Kane JM, Marder SR. Chronic restlessness with antipsychotics. Am J Psychiatry 2007;164:1648-54. https://doi.org/10.1176/appi.ajp.2007.07071150.
- Killoran A, Biglan KM. Current therapeutic options for Huntington’s disease: good clinical practice versus evidence-based approaches?. Mov Disord 2014;29:1404-13. http://dx.doi.org/10.1002/mds.26014.
- van Harten PN, Hoek HW, Kahn RS. Acute dystonia induced by drug treatment. BMJ 1999;319:623-6. https://doi.org/10.1136/bmj.319.7210.623.
- Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for symptomatic treatment in Huntington’s disease. Cochrane Database Syst Rev 2009;3. https://doi.org/10.1002/14651858.cd006456.pub2.
- El-Sayeh HG, Lyra da Silva JP, Rathbone J, Soares-Weiser K. Non-neuroleptic catecholaminergic drugs for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2006;1. https://doi.org/10.1002/14651858.cd000458.pub2.
- Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias, neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Bio Psychiatry 1993;34:713-38. https://doi.org/10.1016/0006-3223(93)90044-E.
- Barnes TRE, Edwards JG, Barnes TRE. Antipsychotic Drugs and their Side-Effects. London: Academic Press/Harcourt Brace & Company; 1993.
- Gunne LM, Häggström JE, Sjöquist B. Association with persistent neuroleptic-induced dyskinesia of regional changes in brain GABA synthesis. Nature 1984;309:347-9. https://doi.org/10.1038/309347a0.
- Gardos G, Cole JO, Bloom FE, DJ K. Psychopharmacology The Fourth Generation of Progress. New York, NY: Raven Press; 1994.
- Tardive Dyskinesia: A Task Force Report of the American Psychiatric Association. Washington, DC: American Psychiatric Association; 1992.
- Jeste DV, Lohr JB, Clark K, Wyatt RJ. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol 1988;8:38-4. https://doi.org/10.1097/00004714-198808001-00008.
- Lima AR, Soares-Weiser K, Bacaltchuk J, Barnes TR. Benzodiazepines for neuroleptic-induced acute akathisia. Cochrane Database Syst Rev 2002;1.
- Rotrosen J, Adler L, Lohr J, Edson R, Lavori P. Antioxidant treatment of tardive dyskinesia. Prostaglandins Leukot Essent Fatty Acids 1996;55:77-81. https://doi.org/10.1016/S0952-3278(96)90149-0.
- Cadet JL, Lohr JB. Possible involvement of free radicals in neuroleptic-induced movement disorders. Evidence from treatment of tardive dyskinesia with vitamin E. Ann N Y Acad Sci 1989;570:176-85. https://doi.org/10.1111/j.1749-6632.1989.tb14918.x.
- Feltner DE, Hertzman M. Progress in the treatment of tardive dyskinesia: theory and practice. Hosp Community Psychiatry 1993;44:25-34.
- Jeste DV, Caligiuri MP. Tardive dyskinesia. Schizophr Bull 1993;19:303-15. https://doi.org/10.1093/schbul/19.2.303.
- Queiroz CM, Frussa-Filho R. Effects of buspirone on an animal model of tardive dyskinesia. Prog Neuropsychopharmacol Biol Psychiatry 1999;23:1405-18. https://doi.org/10.1016/S0278-5846(99)00074-3.
- Haleem DJ, Samad N, Haleem MA. Reversal of haloperidol-induced tardive vacuous chewing movements and supersensitive somatodendritic serotonergic response by buspirone in rats. Pharmacol Biochem Behav 2007;87:115-21. https://doi.org/10.1016/j.pbb.2007.04.007.
- Wahbeh H, Elsas SM, Oken BS. Mind-body interventions: applications in neurology. Neurology 2008;70:2321-8. http://dx.doi.org/10.1212/01.wnl.0000314667.16386.5e.
- Ajimsha MS, Majeed NA, Chinnavan E, Thulasyammal RP. Effectiveness of autogenic training in improving motor performances in Parkinson’s disease. Complement Ther Med 2014;22:419-25. http://dx.doi.org/10.1016/j.ctim.2014.03.013.
- Franklin SA, Walther MR, Woods DW. Behavioral interventions for tic disorders. Psychiatr Clin North Am 2010;33:641-55. http://dx.doi.org/10.1016/j.psc.2010.04.013.
- Elkins G, Sliwinski J, Bowers J, Encarnacion E. Feasibility of clinical hypnosis for the treatment of Parkinson’s disease: a case study. Int J Clin Exp Hypn 2013;61:172-82. http://dx.doi.org/10.1080/00207144.2013.753829.
- McGrath J, Davies G, Soares K. Writing to authors of systematic reviews elicited further data in 17% of cases. BMJ 1998;316. https://doi.org/10.1136/bmj.316.7131.631a.
- Soares K, McGrath J, Adams C. Evidence and tardive dyskinesia. Lancet 1996;347:1696-7. https://doi.org/10.1016/S0140-6736(96)91525-1.
- Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma-aminobutyric acid agonists for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;4. http://dx.doi.org/10.1002/14651858.CD000203.pub3.
- Bhoopathi PS, Soares-Weiser K. Benzodiazepines for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2006;3. http://dx.doi.org/10.1002/14651858.CD000205.pub2.
- Essali A, Deirawan H, Soares-Weiser K, Adams CE. Calcium channel blockers for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;11. http://dx.doi.org/10.1002/14651858.CD000206.pub3.
- Soares-Weiser K, Irving Claire B, Rathbone J. Miscellaneous treatments for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2003;2. https://doi.org/10.1002/14651858.cd000208.
- Soares-Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;2. http://dx.doi.org/10.1002/14651858.CD000209.pub2.
- Soares-Weiser K, Mobsy C, Holliday E. Anticholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 1997;2. https://doi.org/10.1002/14651858.cd000204.
- Tammenmaa IA, McGrath JJ, Sailas E, Soares-Weiser K. Cholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2002;3. https://doi.org/10.1002/14651858.cd000207.
- Soares KV, McGrath JJ. The treatment of tardive dyskinesia – a systematic review and meta-analysis. Schizophr Res 1999;39:1-16. https://doi.org/10.1016/S0920-9964(99)00021-3.
- Rana AQ, Chaudry ZM, Blanchet PJ. New and emerging treatments for symptomatic tardive dyskinesia. Drug Des Devel Ther 2013;7:1329-40. http://dx.doi.org/10.2147/DDDT.S32328.
- Cipriani A, Higgins JP, Geddes JR, Salanti G. Conceptual and technical challenges in network meta-analysis. Ann Intern Med 2013;159:130-7. http://dx.doi.org/10.7326/0003-4819-159-2-201307160-00008.
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 2011. www.cochrane-handbook.org (accessed 11 April 2017).
- Adamd CE, Walker D-M, Gray B, Shokraneh F. Appendix: Traceable Extracted Data from Included Studies of Tardive Dyskinesia Reviews 2016. www.researchgate.net/publication/308698005_Appendix_Traceable_Extracted_Data_from_Included_Studies_of_Tardive_Dyskinesia_Reviews?channel=doi&linkId=57ebe1c508ae92a5dbd051c1&showFulltext=true (accessed 13 June 2017).
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychol Rep 1962;10:799-812. https://doi.org/10.2466/pr0.1962.10.3.799.
- Chouinard G, Ross-Chouinard A, Annable L, Jones BD. The Extrapyramidal Symptom Rating Scale. Can J Neurol Sci 1980;7.
- Simpson GM, Angus JW. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl 1970;212:11-9. https://doi.org/10.1111/j.1600-0447.1970.tb02066.x.
- Lingjaerde O, Ahlfors UG, Bech P, Dencker SJ, Elgen K. The UKU side effect rating scale. A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl 1987;334:1-100. https://doi.org/10.1111/j.1600-0447.1987.tb10566.x.
- Higgins JPT, Ramsay C, Reeves BC, Deeks JJ, Shea B, Valentine JC, et al. Issues relating to study design and risk of bias when including non-randomized studies in systematic reviews on the effects of interventions. Res Synth Methods 2013;4:12-25. https://doi.org/10.1002/jrsm.1056.
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. Br J Psychiatry 2000;176:249-52. https://doi.org/10.1192/bjp.176.3.249.
- Elbourne DR, Altman DG, Higgins JP, Curtin F, Worthington HV, Vail A. Meta-analyses involving cross-over trials: methodological issues. Int J Epidemiol 2002;31:140-9. https://doi.org/10.1093/ije/31.1.140.
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60. http://dx.doi.org/10.1136/bmj.327.7414.557.
- Higgins JP, Whitehead A. Borrowing strength from external trials in a meta-analysis. Stat Med 1996;15:2733-49. https://doi.org/10.1002/(SICI)1097-0258(19961230)15:24%3C2733::AID-SIM562%3E3.0.CO;2-0.
- Raudenbush SW, Cooper H, Hedges LV, Valentine C. The Handbook of Research Synthesis and Meta-Analysis. New York, NY: Russel Sage Foundation; 2009.
- Viechtbauer W. Confidence intervals for the amount of heterogeneity in meta-analysis. Stat Med 2007;26:37-52. http://dx.doi.org/10.1002/sim.2514.
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables-binary outcomes. J Clin Epidemiol 2013;66:158-72. http://dx.doi.org/10.1016/j.jclinepi.2012.01.012.
- Guyatt GH, Oxman AD, Sultan S, Glasziou P, Akl EA, Alonso-Coello P, et al. GRADE guidelines: 9. Rating up the quality of evidence. J Clin Epidemiol 2011;64:1311-16. http://dx.doi.org/10.1016/j.jclinepi.2011.06.004.
- Mustafa RA, Santesso N, Brozek J, Akl EA, Walter SD, Norman G, et al. The GRADE approach is reproducible in assessing the quality of evidence of quantitative evidence syntheses. J Clin Epidemiol 2013;66:736-42. http://dx.doi.org/10.1016/j.jclinepi.2013.02.004.
- Sutton AJ, Cooper NJ, Jones DR, Lambert PC, Thompson JR, Abrams KR. Evidence-based sample size calculations based upon updated meta-analysis. Stat Med 2007;26:2479-500. http://dx.doi.org/10.1002/sim.2704.
- Langan D, Higgins JP, Gregory W, Sutton AJ. Graphical augmentations to the funnel plot assess the impact of additional evidence on a meta-analysis. J Clin Epidemiol 2012;65:511-19. http://dx.doi.org/10.1016/j.jclinepi.2011.10.009.
- Cai N. A controlled study on the treatment of tardive dyskinesia using 1-stepholidine. Zhonghua Shen Jing Jing Shen Ke Za Zhi 1988;21:281-3.
- Chen J, Zhong X, Cao Z. A double-blind auto-control study on the effect of bromocriptine on tardive dyskinesia. Chin J Pharmacoepidemiol 1995;4:203-5.
- Mei H, Zhu Q. γ-aminobutyric acid in the treatment of 20 cases of tardive dyskinesia. Herald Med 2008;27:304-5.
- Shi X, Zhu F, Zhang X, Zhang JX, Zhang XM, Wei LH, et al. Melatonin in treatment of schizophrenia with tardive dyskinesia: a comparison study of cognitive function. Linchuang Jingshen Yixue Zazhi 2009;19:391-3.
- Xiang H, Zhen C. Clonazepam therapy of tardive dyskinesia: a double-blind trial. West China Med J 1997;12:17-8.
- Yang X, Meng F, Cui Y. Promethazine treatment of tardive dyskinesia: a double blind placebo controlled study. Chin Ment Health J 1999;13:365-7.
- Yin XR, Xie BQ, Jiang L. A double-blind comparative study of sodium valproate in treating of TD. J Clin Psychol Med 2004;14:92-3.
- Zeng ZX. Treatment of tardive dyskinesia with buspirone. Med J Chinese Civil Admin 1995;7:202-3.
- Zeng ZX. Pemoline in the treatment of tardive dyskinesia. Chin J New Drugs Clin Remedies 1996;15:240-1.
- Zeng ZX, Li ZC, Yu XW, Zhang YD, Cao ZC. A double-blind trial of flunarizine therapy for tardive dyskinesia. Chin J Pharmaco Epidemiol 1994;3:183-4.
- Hebenstreit GF, Hoffmann H, Hoffmann W, Pittner H. Beta blockade with celiprolol in tardive dyskinesia patients treated with neuroleptics. Wien Klin Wochenschr 1986;98:388-92.
- Kocher R, Hobi V, Linder M, Studer K. Treatment with dimethylaminoethanol (deanol) in neuroleptic induced tardive dyskinesia. Schweiz Arch Neurol Neurochir Psychiatr 1980;126:103-9.
- Lucius G. Uber Die Therapeutische Wirksamkeit Von Dimethylaminoaethanol Bei Neuroleptikainduzierten Späthyperkinesen 1978.
- Koshino Y, Hiramatsu H, Isaki K, Yamaguchi N. A double-blind clinical trial of dihydrogenated ergot alkaloids in antipsychotic-induced tardive dyskinesia. Clin Psychiatry 1983;25:627-35.
- Koshino Y, Kurata K, Hosokawa K, Yamaguchi N. Double-blind trial of cyproheptadine on neuroleptic induced tardive dyskinesia. Clin Psychiatry 1979;21:421-6.
- Yagi G, Kamishima K, Miura S. Meclofenoxate hydrochloride (Lucidril) in tardive dyskinesia – a double-blind placebo-controlled study. Rinsho Hyoka 1990;18:455-79.
- Jahanian AA, Rezaei O, Fadai F, Yaraghchi A. The effectiveness of rivastigmine in reducing tardive dyskinesia symptoms in patients with schizophrenia. Iran J Psychiatry Clin Psychol 2014;20:29-34.
- Karniol IG, Giampietro AC, Moura DS, Vilela WA, Oliveira MA, Zuardi AW. A double-blind study of the effect of L-dopa in psychotic patients with tardive dyskinesia. Acta Psiquiatr Psicol Am Lat 1983;29:261-6.
- Weber SS, Dufresne RL, Becker RE, Mastrati P. Diazepam in tardive dyskinesia. Drug Intell Clin Pharm 1983;17:523-7. https://doi.org/10.1177/106002808301700705.
- Dorevitch A, Kalian M, Shlafman M, Lerner V. Treatment of long-term tardive dyskinesia with vitamin E. Biol Psychiatry 1997;41:114-16. https://doi.org/10.1016/S0006-3223(96)00367-8.
- Dorevitch A, Lerner V, Shalfman M, Kalian M. Lack of effect of vitamin E on serum creatine phosphokinase in patients with long-term tardive dyskinesia. Int Clin Psychopharmacol 1997;12:171-3. https://doi.org/10.1097/00004850-199705000-00008.
- Egan MF, Hyde TM, Albers GW, Elkashef A, Alexander RC, Reeve A, et al. Treatment of tardive dyskinesia with vitamin E. Am J Psychiatry 1992;149:773-7. http://dx.doi.org/10.1176/ajp.149.6.773.
- Elkashef AM, Ruskin PE, Bacher N, Barrett D. Vitamin E in the treatment of tardive dyskinesia. Am J Psychiatry 1990;147:505-6. http://dx.doi.org/10.1176/ajp.147.4.505.
- Lam LC, Chiu HF, Hung SF. Vitamin E in the treatment of tardive dyskinesia: a replication study. J Nerv Ment Dis 1994;182:113-14. https://doi.org/10.1097/00005053-199402000-00009.
- Schmidt M, Meister P, Baumann P. Treatment of tardive dyskinesias with vitamin E. Eur Psychiatry 1991;6:201-7.
- Kazamatsuri H, Chien CP, Cole JO. Long-term treatment of tardive dyskinesia with haloperidol and tetrabenazine. Am J Psychiatry 1973;130:479-83. http://dx.doi.org/10.1176/ajp.130.4.479.
- Kane JM, Rifkin A, Woerner M, Reardon G, Sarantakos S, Schiebel D, et al. Low-dose neuroleptic treatment of outpatient schizophrenics. I. Preliminary results for relapse rates. Arch Gen Psychiatry 1983;40:893-6. https://doi.org/10.1001/archpsyc.1983.01790070083010.
- Cookson IB. The effects of a 50% reduction of cis(z)-flupenthixol decanoate in chronic schizophrenic patients maintained on a high dose regime. Int Clin Psychopharmacol 1987;2:141-9. https://doi.org/10.1097/00004850-198704000-00008.
- Chouinard G, Arnott W. The effect of risperidone on extrapyramidal symptoms in chronic schizophrenic patients. Biol Psychiatry 1992;31. https://doi.org/10.1016/0006-3223(92)90579-O.
- Chouinard G, Arnott W. Antidyskinetic effect of risperidone in chronic schizophrenic patients. Clin Neuropharmacol 1992;15. https://doi.org/10.1097/00002826-199202001-00514.
- Chouinard G. Effects of risperidone in tardive dyskinesia: an analysis of the Canadian multicenter risperidone study. J Clin Psychopharmacol 1995;15:36-44. https://doi.org/10.1097/00004714-199502001-00007.
- Chouinard G, Arnott W. An Antidyskinetic Effect of Risperidone n.d.
- Chouinard G, Jones B, Remington G, Bloom D, Addington D, MacEwan GW, et al. A Canadian multicenter placebo-controlled study of fixed doses of risperidone and haloperidol in the treatment of chronic schizophrenic patients. J Clin Psychopharmacol 1993;13:25-40. https://doi.org/10.1097/00004714-199302000-00004.
- Tamminga CA, Thaker GK, Moran M, Kakigi T, Gao XM. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry 1994;55:102-6.
- Bai YM, Lin CC, Yu SC. Risperidone for severe tardive dyskinesia: one year follow up study. Int J Neuropsychopharmacol 2002;5.
- Bai YM, Yu SC, Chen JY, Lin CY, Chou P, Lin CC. Risperidone for pre-existing severe tardive dyskinesia: a 48-week prospective follow-up study. Int Clin Psychopharmacol 2005;20:79-85. https://doi.org/10.1097/00004850-200503000-00003.
- Pai YM, Yu SC, Lin CC. Risperidone in Reducing Tardive Dyskinesia: A Double-Blind, Placebo-Controlled Study n.d.
- Bai YM, Yu SC, Lin CC. Risperidone for severe tardive dyskinesia: a 12-week randomized, double-blind, placebo-controlled study. J Clin Psychiatry 2003;64:1342-8. https://doi.org/10.4088/JCP.v64n1110.
- Pai YM, Yu SC, Lin CC. Risperidone in Reducing Tardive Dyskinesia: A Double-Blind, Placebo-Controlled Study n.d.
- Emsley R, Turner HJ, Schronen J, Botha K, Smit R, Oosthuizen PP. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry 2004;65:696-701. https://doi.org/10.4088/JCP.v65n0516.
- Emsley RA, Turner J, Schronen J, Botha K, Smit R, Oosthuizen PP. Quetiapine: Greater Improvements in Tardive Dyskinesia Versus Haloperidol n.d.
- Bai YM, Ping LY, Lin CC, Wang YC, Liou YL, Wu BJ, et al. Comparative effects of atypical antipsychotic on tardive dyskinesia and neurocognition: a 24-week randomized, single-blind, controlled study. Eur Neuropsychopharmacol 2005;15. https://doi.org/10.1016/S0924-977X(05)80979-4.
- Bai YM, Ping LY, Lin CC, Wang YC, Liou YL, Wu BJ, et al. Comparative Effects of Atypical Antipsychotic on Tardive Dyskinesia and Neurocognition: A 24-Week Randomized, Single-Blind, Controlled Study n.d.
- Bai YM. Tardive Dyskinesia and Cognitive Function 2008. https://clinicaltrials.gov/ct2/show/NCT00926965 (accessed 20 January 2017).
- Chan HY, Chiang SC, Chang CJ, Gau SS, Chen JJ, Chen CH, et al. A randomized controlled trial of risperidone and olanzapine for schizophrenic patients with neuroleptic-induced tardive dyskinesia. J Clin Psychiatry 2010;71:1226-33. https://doi.org/10.4088/JCP.09m05155yel.
- NCT00621998 . Risperidone and Olanzapine for the Schizophrenic Patients With Neuroleptic-Induced Tardive Dyskinesia 2008. https://clinicaltrials.gov/ct2/show/NCT00621998 (accessed 20 January 2017).
- Caroff SN, Davis VG, Miller DD, Davis SM, Rosenheck RA, McEvoy JP, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry 2011;72:295-303. http://dx.doi.org/10.4088/JCP.09m05793yel.
- Miller DD, McEvoy JP, Davis SM, Caroff SN, Saltz BL, Chakos MH, et al. Clinical correlates of tardive dyskinesia in schizophrenia: baseline data from the CATIE schizophrenia trial. Schizophr Res 2005;80:33-4. https://doi.org/10.1016/j.schres.2005.07.034.
- Greil W, Haag H, Rossnagl G, Rüther E. Effect of anticholinergics on tardive dyskinesia. A controlled discontinuation study. Br J Psychiatry 1984;145:304-10. https://doi.org/10.1192/bjp.145.3.304.
- Bobruff A, Gardos G, Tarsy D, Rapkin RM, Cole JO, Moore P. Clonazepam and phenobarbital in tardive dyskinesia. Am J Psychiatry 1981;138:189-93. http://dx.doi.org/10.1176/ajp.138.2.189.
- Csernansky JG, Riney SJ, Lombrozo L, Overall JE, Hollister LE. Double-blind comparison of alprazolam, diazepam, and placebo for the treatment of negative schizophrenic symptoms. Arch Gen Psychiatry 1988;45:655-9. https://doi.org/10.1001/archpsyc.1988.01800310063008.
- Csernansky JG, Tacke U, Rusen D, Hollister LE. The effect of benzodiazepines on tardive dyskinesia symptoms. J Clin Psychopharmacol 1988;8:154-5. https://doi.org/10.1097/00004714-198804000-00028.
- Adler LA, Edson R, Lavori P, Peselow E, Duncan E, Rosenthal M, et al. Long-term treatment effects of vitamin E for tardive dyskinesia. Biol Psychiatry 1998;43:868-72. https://doi.org/10.1016/S0006-3223(97)00027-9.
- Adler LA, Peselow E, Angrist B, Duncan E, Lee M, Rosenthal M, et al. Vitamin E in Tardive Dyskinesia: Effects of Longer Term Treatment n.d.
- Adler LA, Peselow E, Duncan E, Rosenthal M, Angrist B. Vitamin E in tardive dyskinesia: time course of effect after placebo substitution. Psychopharmacol Bull 1993;29:371-4.
- Adler LA, Peselow E, Rotrosen J, Duncan E, Lee M, Rosenthal M, et al. Vitamin E treatment of tardive dyskinesia. Am J Psychiatry 1993;150:1405-7. http://dx.doi.org/10.1176/ajp.150.9.1405.
- Akhtar S, Jajor TR, Kumar S. Vitamin E in the treatment of tardive dyskinesia. J Postgrad Med 1993;39:124-6.
- Dabiri LM, Pasta D, Darby JK, Mosbacher D. Effectiveness of vitamin E for treatment of long-term tardive dyskinesia. Am J Psychiatry 1994;151:925-6. http://dx.doi.org/10.1176/ajp.151.6.925.
- Lohr JB, Caligiuri MP. A double-blind placebo-controlled study of vitamin E treatment of tardive dyskinesia. J Clin Psychiatry 1996;57:167-73.
- Sajjad SH. Vitamin E in the treatment of tardive dyskinesia: a preliminary study over 7 months at different doses. Int Clin Psychopharmacol 1998;13:147-55. https://doi.org/10.1097/00004850-199807000-00001.
- Tracy K, Adler LA, Rotrosen J, Edson R, Lavori P. Interrater reliability issues in multicentric trials, part I: theoretical concepts and operational procedures used in Department of Veterans Affairs Cooperative Study 394. Psychopharmacol Bull 1997;33:53-7.
- Lohr JB, Lavori P. Whither vitamin E and tardive dyskinesia?. Biol Psychiatry 1998;43:861-2.
- Edson R, Lavori P, Tracy K, Adler LA, Rotrosen J. Interrater reliability issues in multicentric trials, part II: statistical procedures used in Department of Veterans Affairs Cooperative Study #394. Psychopharmacol Bull 1997;33:59-67.
- Caligiuri MP, Lohr JB, Rotrosen J, Adler L, Lavori P, Edson R, et al. Reliability of an instrumental assessment of tardive dyskinesia: results from VA Cooperative Study #394. Psychopharmacology 1997;132:61-6. https://doi.org/10.1007/s002130050320.
- Adler LA, Rotrosen J, Lavori P, Edson R. Vitamin E treatment of TD: development of a VA cooperative study. Biol Psychiatry 1994;35:730-1. https://doi.org/10.1016/0006-3223(94)91073-1.
- Bridler R. Vitamin E is ineffective in treatment of late dyskinesias. Praxis 2001;90:809-10.
- Adler LA, Rotrosen J, Edson R, Lavori P, Lohr J, Hitzemann R, et al. Vitamin E treatment for tardive dyskinesia. Veterans Affairs Cooperative Study #394 Study Group. Arch Gen Psychiatry 1999;56:836-41. https://doi.org/10.1001/archpsyc.56.9.836.
- Zhang XY, Zhou DF, Cao LY, Xu CQ, Chen DC, Wu GY. The effect of vitamin E treatment on tardive dyskinesia and blood superoxide dismutase: a double-blind placebo-controlled trial. J Clin Psychopharmacol 2004;24:83-6. http://dx.doi.org/10.1097/01.jcp.0000104912.75206.2b.
- Glover O. Alternative Treatment Modalities for Drug Induced Psychomotor Dysfunctions 1980.
- Zeng ZX, Fenglian C, Lin L. A clinical research of dexetimide and benzhexol for treatment of drug-induced tremor. Herald Med 1996;15:130-1.
- Kar-Ahmadi M. Vitamin E in the management of drug induced tardive dyskinesia: a double-blind randomized clinical trial. J Res Med Sci 2002;4:311-20.
- Garcia G, Crismon ML. Double-blind placebo controlled study using buspirone in the treatment of tardive dyskinesia. ASHP Midyear Clin Meet 1992;27.
- Reynolds C. A Six Month, Rater Blind Comparison of Quetiapine and Risperidone in the Treatment of Tardive Dyskinesia in Patients with Schizophrenia. Leeds: National Research Register; 2002.
- Kajero J. Investigation of the Potential Beneficial Effects of Cannabidiol in the Treatment of Tardive Dyskinesia 2015. www.isrctn.com/ISRCTN14688109 (accessed 20 January 2017).
- Casey DE, Toenniessen LM. Neuroleptic treatment in tardive dyskinesia: can it be developed into a clinical strategy for long-term treatment?. Mod Probl Pharmacopsychiatry 1983;21:65-79. https://doi.org/10.1159/000408484.
- Damier P, Thobois S, Witjas T, Cuny E, Derost P, Raoul S, et al. Bilateral deep brain stimulation of the globus pallidus to treat tardive dyskinesia. Arch Gen Psychiatry 2007;64:170-6. https://doi.org/10.1001/archpsyc.64.2.170.
- Pouclet-Courtemanche H, Rouaud T, Thobois S, Nguyen JM, Brefel-Courbon C, Chereau I, et al. Long-term efficacy and tolerability of bilateral pallidal stimulation to treat tardive dyskinesia. Neurology 2016;86:651-9. http://dx.doi.org/10.1212/WNL.0000000000002370.
- Hardoy MC, Carta MG, Carpiniello B, Cianchetti C, Congia S, D’Errico I, et al. Gabapentin in antipsychotic-induced tardive dyskinesia: results of 1-year follow-up. J Affect Disord 2003;75:125-30. https://doi.org/10.1016/S0165-0327(02)00043-5.
- Huang CC. Comparison of two groups of tardive dyskinesia patients. Psychiatry Res 1986;19:335-6. https://doi.org/10.1016/0165-1781(86)90128-9.
- Koshino Y, Wada Y, Isaki K, Kurata K. A long-term outcome study of tardive dyskinesia in patients on antipsychotic medication. Clin Neuropharmacol 1991;14:537-46. https://doi.org/10.1097/00002826-199112000-00006.
- Peselow ED, Angrist BM, Rotrosen J. Changes in tardive dyskinesia after fluphenazine decanoate discontinuation. Ann Clin Psychiatry 1989;1:187-91. https://doi.org/10.3109/10401238909149978.
- Yagi G, Itoh H. A 10-year follow-up study of tardive dyskinesia – with special reference to the influence of neuroleptic administration on the long-term prognosis. Keio J Med 1985;34:211-19. https://doi.org/10.2302/kjm.34.211.
- Yassa R, Nair NP. A 10-year follow-up study of tardive dyskinesia. Acta Psychiatr Scand 1992;86:262-6. https://doi.org/10.1111/j.1600-0447.1992.tb03264.x.
- Yassa R, Nair V, Schwartz G. Tardive dyskinesia: a two-year follow-up study. Psychosomatics 1984;25:852-5. https://doi.org/10.1016/S0033-3182(84)72946-X.
- Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res Synth Methods 2012;3:80-97. https://doi.org/10.1002/jrsm.1037.
- White IR, Barrett JK, Jackson D, Higgins JP. Consistency and inconsistency in network meta-analysis: model estimation using multivariate meta-regression. Res Synth Methods 2012;3:111-25. http://dx.doi.org/10.1002/jrsm.1045.
- Lu G, Welton NJ, Higgins JP, White IR, Ades AE. Linear inference for mixed treatment comparison meta-analysis: A two-stage approach. Res Synth Methods 2011;2:43-60. https://doi.org/10.1002/jrsm.34.
- Bradburn MJ, Deeks JJ, Berlin JA, Russell Localio A. Much ado about nothing: a comparison of the performance of meta-analytical methods with rare events. Stat Med 2007;26:53-77. http://dx.doi.org/10.1002/sim.2528.
- Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta-analysis of sparse data. Stat Med 2004;23:1351-75. http://dx.doi.org/10.1002/sim.1761.
- Soares MO, Dumville JC, Ades AE, Welton NJ. Treatment comparisons for decision making: facing the problems of sparse and few data. J R Stat Soc Ser A 2014;177:259-79. https://doi.org/10.1111/rssa.12010.
- Rücker G, Schwarzer G, Carpenter J, Olkin I. Why add anything to nothing? The arcsine difference as a measure of treatment effect in meta-analysis with zero cells. Stat Med 2009;28:721-38. http://dx.doi.org/10.1002/sim.3511.
- Kuss O. Statistical methods for meta-analyses including information from studies without any events-add nothing to nothing and succeed nevertheless. Stat Med 2015;34:1097-116. https://doi.org/10.1002/sim.6383.
- Warren FC, Abrams KR, Golder S, Sutton AJ. Systematic review of methods used in meta-analyses where a primary outcome is an adverse or unintended event. BMC Med Res Methodol 2012;12. http://dx.doi.org/10.1186/1471-2288-12-64.
- Fleiss JL. The Crossover Study. The Design and Analysis of Clinical Experiments. Chichester: John Wiley & Sons; 1984.
- Armitage P. Should we cross off the crossover?. Br J Clin Pharmacol 1991;32:1-2. https://doi.org/10.1111/j.1365-2125.1991.tb05604.x.
- Pocock SJ. Crossover Trials. Clinical Trials A Practical Approach. Chichester: John Wiley & Sons; 1983.
- Ferreira ML, Herbert RD, Crowther MJ, Verhagen A, Sutton AJ. When is a further clinical trial justified?. BMJ 2012;345. http://dx.doi.org/10.1136/bmj.e5913.
- Clarke M. Doing new research? Don’t forget the old. PLOS Med 2004;1. http://dx.doi.org/10.1371/journal.pmed.0010035.
- Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. II. Short-term efficacy of dopamine-blocking agents haloperidol and thiopropazate. Arch Gen Psychiatry 1972;27:100-3. https://doi.org/10.1001/archpsyc.1972.01750250086012.
- Borenstein M, Hedges LV, Higgins JPT. Introduction to Meta-Analysis. Chichester: John Wiley & Sons; 2011.
- Salanti G, Ades AE, Ioannidis JP. Graphical methods and numerical summaries for presenting results from multiple-treatment meta-analysis: an overview and tutorial. J Clin Epidemiol 2011;64:163-71. http://dx.doi.org/10.1016/j.jclinepi.2010.03.016.
- Rücker G, Schwarzer G. Ranking treatments in frequentist network meta-analysis works without resampling methods. BMC Med Res Methodol 2015;15. http://dx.doi.org/10.1186/s12874-015-0060-8.
- Higgins JP, Green S, Scholten RJ, Higgins JP, Green S. Cochrane Handbook for Systematic Reviews of Interventions. Chichester: John Wiley & Sons, Ltd; 2008.
- Jansen JP, Naci H. Is network meta-analysis as valid as standard pairwise meta-analysis? It all depends on the distribution of effect modifiers. BMC Med 2013;11. http://dx.doi.org/10.1186/1741-7015-11-159.
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177-88. https://doi.org/10.1016/0197-2456(86)90046-2.
- White IR. Network meta-analysis. Stata J 2015;15:951-85.
- Rücker G. Network meta-analysis, electrical networks and graph theory. Res Synth Methods 2012;3:312-24. http://dx.doi.org/10.1002/jrsm.1058.
- Rücker G, Schwarzer G. Reduce dimension or reduce weights? Comparing two approaches to multi-arm studies in network meta-analysis. Stat Med 2014;33:4353-69. http://dx.doi.org/10.1002/sim.6236.
- R Development Core Team . R: A Language and Environment for Statistical Computing 2008. www.r-project.org/ (accessed 11 April 2017).
- Chaimani A, Higgins JP, Mavridis D, Spyridonos P, Salanti G. Graphical tools for network meta-analysis in STATA. PLOS ONE 2013;8. http://dx.doi.org/10.1371/journal.pone.0076654.
- Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JP. Predicting the extent of heterogeneity in meta-analysis, using empirical data from the Cochrane Database of Systematic Reviews. Int J Epidemiol 2012;41:818-27. https://doi.org/10.1093/ije/dys041.
- Rhodes KM, Turner RM, Higgins JP. Predictive distributions were developed for the extent of heterogeneity in meta-analyses of continuous outcome data. J Clin Epidemiol 2015;68:52-60. http://dx.doi.org/10.1016/j.jclinepi.2014.08.012.
- Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta-analysis of randomized controlled trials. J Clin Epidemiol 1997;50:683-91. https://doi.org/10.1016/S0895-4356(97)00049-8.
- Higgins JP, Jackson D, Barrett JK, Lu G, Ades AE, White IR. Consistency and inconsistency in network meta-analysis: concepts and models for multi-arm studies. Res Synth Methods 2012;3:98-110. http://dx.doi.org/10.1002/jrsm.1044.
- Jackson D, Barrett JK, Rice S, White IR, Higgins JP. A design-by-treatment interaction model for network meta-analysis with random inconsistency effects. Stat Med 2014;33:3639-54. http://dx.doi.org/10.1002/sim.6188.
- Hempel S, Miles JN, Booth MJ, Wang Z, Morton SC, Shekelle PG. Risk of bias: a simulation study of power to detect study-level moderator effects in meta-analysis. Syst Rev 2013;2. http://dx.doi.org/10.1186/2046-4053-2-107.
- Chaimani A, Salanti G. Using network meta-analysis to evaluate the existence of small-study effects in a network of interventions. Res Synth Methods 2012;3:161-76. http://dx.doi.org/10.1002/jrsm.57.
- Lublin H, Gerlach J, Hagert U, Meidahl B, Mølbjerg C, Pedersen V, et al. Zuclopenthixol, a combined dopamine D1/D2 antagonist, versus haloperidol, a dopamine D2 antagonist, in tardive dyskinesia. Eur Neuropsychopharmacol 1991;1:541-8. https://doi.org/10.1016/0924-977X(91)90008-I.
- Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res 1990;3:315-20. https://doi.org/10.1016/0920-9964(90)90016-Z.
- Glazer WM, Hafez HM, Benarroche CL. Molindone and haloperidol in tardive dyskinesia. J Clin Psychiatry 1985;46:4-7.
- Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. Washington, DC: American Psychiatric Association; 2000.
- Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry 1982;39:486-7. https://doi.org/10.1001/archpsyc.1982.04290040080014.
- DSM-IIl-R: Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Association; 1987.
- National Center for Health Statistics . Classification of Diseases and Injuries 2002. ftp://ftp.cdc.gov/pub/Health_Statistics/NCHS/Publications/ICD-9/ucod.txt (accessed on 6 June 2017).
- Chinese Classification of Mental Disorders, Second Edition, Revised (CCMD-2-R). Nanjing: Dong Nan University Press; 1995.
- World Health Organization . The Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines n.d. www.who.int/classifications/icd/en/bluebook.pdf (accessed 20 July 2017).
- Bucci L. The dyskenesias: a new therapeutic approach. Dis Nerv Syst 1971;32:324-7.
- Loonen AJM, Verwey HA, Roels PR, van Bavel LP, Doorschot CH. Is diltiazem effective in treating the symptoms of (tardive) dyskinesia in chronic psychiatric inpatients? A negative, double-blind, placebo-controlled trial. J Clin Psychopharmacol 1992;12:39-42. https://doi.org/10.1097/00004714-199202000-00007.
- Schwartz B, McCarthy MF, Kendrick K, Rosse R, Deutsch S. Effect of nifedipine on motor skill learning in schizophrenia. Schizophr Res 1997;24. https://doi.org/10.1016/S0920-9964(97)82352-3.
- Schwartz BL, Fay-McCarthy M, Kendrick K, Rosse RB, Deutsch SI. Effects of nifedipine, a calcium channel antagonist, on cognitive function in schizophrenic patients with tardive dyskinesia. Clin Neuropharmacol 1997;20:364-70. https://doi.org/10.1097/00002826-199708000-00009.
- Beckham BJ. Lecithin Therapy for Tardive Dyskinesia 1981.
- Caroff SN, Walker P, Campbell C, Lorry A, Petro C, Lynch K, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry 2007;68:410-15.
- Caroff SN. Treatment of Tardive Dyskinesia With Galantamine 2005. https://clinicaltrials.gov/ct2/show/NCT00164242 (accessed 20 January 2017).
- de Montigny C, Chouinard G, Annable L. Ineffectiveness of deanol in tardive dyskinesia: a placebo controlled study. Psychopharmacology 1979;65:219-23. https://doi.org/10.1007/BF00492207.
- Gelenberg AJ, Dorer DJ, Wojcik JD, Falk WE, Brotman AW, Leahy L. A crossover study of lecithin treatment of tardive dyskinesia. J Clin Psychiatry 1990;51:149-53.
- George J, Pridmore S, Aldous D. Double blind controlled trial of deanol in tardive dyskinesia. Aust N Z J Psychiatry 1981;15:68-71. https://doi.org/10.3109/00048678109159413.
- Jackson IV. Cholinergic enhancement in tardive dyskinesia. Curr Ther Res Clin Exp 1978;24:725-33.
- Jackson IV, Davis LG, Cohen RK, Nuttall EA. Lecithin administration in tardive dyskinesia: clinical and biomedical correlates. Biol Psychiatry 1981;16:85-90.
- Jackson IV, Nuttall EA, Ibe IO, Perez-Cruet J. Treatment of tardive dyskinesia with lecithin. Am J Psychiatry 1979;136:1458-60. http://dx.doi.org/10.1176/ajp.136.11.1458.
- Bockenheimer S, Lucius G. Deanol in tardive dyskinesia: a double-blind study. Arch Psychiatr Nervenkr 1976;222:69-75. https://doi.org/10.1007/BF00369796.
- Ogunmefun A, Hasnain M, Alam A, Osuala T, Regenold WT. Effect of donepezil on tardive dyskinesia. J Clin Psychopharmacol 2009;29:102-4. http://dx.doi.org/10.1097/JCP.0b013e3181934475.
- Price LA. Lecithin Treatment for Tardive Dyskinesia: A Clinical Evaluation 1982.
- Tarsy D, Bralower M. Deanol acetamidobenzoate treatment in choreiform movement disorders. Arch Neurol 1977;34:756-8. https://doi.org/10.1001/archneur.1977.00500240044007.
- Ojima Y, Tsubaki M, Yagi G, Kamishima K, Miura S. Experimental design and analysis for determination of improvement rating by video imaging – a double-blind placebo-controlled study for Meclofenoxate hydrochloride (Lucidril) in tardive dyskinesia. Rinsho Hyoka 1991;19:267-76.
- Yagi G, Kamizima K, Miura S. Meclofenoxate (Lucidril) in Tardive Dyskinesia – A Double-Blind Placebo-Controlled Study n.d.
- Ananth J, Djenderedjian A, Beshay M, Kamal M, Kodjian A, Barriga C. Baclofen in the treatment of tardive dyskinesia. Curr Ther Res Clin Exp 1987;42:111-14.
- Burner M, Giroux C, L’Heritier C, Garreau M, Morselli PL. Preliminary observations on the therapeutic action of progabide in tardive dyskinesia. Brain Dysfunct 1989;2:289-96.
- Fisk GG, York SM. The effect of sodium valproate on tardive dyskinesia – revisited. Br J Psychiatry 1987;150:542-6. https://doi.org/10.1192/bjp.150.4.542.
- Gerlach J. The relationship between parkinsonism and tardive dyskinesia. Am J Psychiatry 1977;134:781-4. http://dx.doi.org/10.1176/ajp.134.7.781.
- Gerlach J, Rye T, Kristjansen P. Effect of baclofen on tardive dyskinesia. Psychopharmacology 1978;56:145-51. https://doi.org/10.1007/BF00431840.
- Glazer WM, Moore DC, Bowers MB, Bunney BS, Roffman M. The treatment of tardive dyskinesia with baclofen. Psychopharmacology 1985;87:480-3. https://doi.org/10.1007/BF00432517.
- Linnoila M, Viukari M, Kietala O. Effect of sodium valproate on tardive dyskinesia. Br J Psychiatry 1976;129:114-19. https://doi.org/10.1192/bjp.129.2.114.
- Nair NP, Yassa R, Ruiz-Navarro J, Schwartz G. Baclofen in the treatment of tardive dyskinesia. Am J Psychiatry 1978;135:1562-3. http://dx.doi.org/10.1176/ajp.135.12.1562.
- Nair NP, Lal S, Schwartz G, Thavundayil JX. Effect of sodium valproate and baclofen in tardive dyskinesia: clinical and neuroendocrine studies. Adv Biochem Psychopharmacol 1980;24:437-41.
- Stewart RM, Rollins J, Beckham B, Roffman M. Baclofen in tardive dyskinesia patients maintained on neuroleptics. Clin Neuropharmacol 1982;5:365-73. https://doi.org/10.1097/00002826-198212000-00004.
- Stewart RM, Rollins J, Beckam B, Roffman M. Baclofen for tardive dyskinesia: a double-blind placebo-controlled trial. Neurology 1982;32.
- Thaker GK, Tamminga CA, Alphs LD, Lafferman J, Ferraro TN, Hare TA. Brain gamma-aminobutyric acid abnormality in tardive dyskinesia. Reduction in cerebrospinal fluid GABA levels and therapeutic response to GABA agonist treatment. Arch Gen Psychiatry 1987;44:522-9. https://doi.org/10.1001/archpsyc.1987.01800180032006.
- Castro F, Carrizo E, Prieto de Rincón D, Rincón CA, Asián T, Medina-Leendertz S, et al. Effectiveness of melatonin in tardive dyskinesia. Invest Clin 2011;52:252-60.
- Emsley R, Niehaus DJ, Koen L, Oosthuizen PP, Turner HJ, Carey P, et al. The effects of eicosapentaenoic acid in tardive dyskinesia: a randomized, placebo-controlled trial. Schizophr Res 2006;84:112-20. https://doi.org/10.1016/j.schres.2006.03.023.
- Elmsley R, Oosthuizen PP. Double-Blind, Randomized, Parallel-Group Comparison of Ethyl-eicosapentaenoic Acid (ethyl-EP A) versus Placebo as add-on Medication in 84 Patients with Established Tardive Dyskinesia. Muscatine, IA: Stanley Foundation Research Programs; 2002.
- Emsley R. Ethyl Eicosapentanoic Acid for Tardive Dyskinesia. Muscatine, IA: Stanley Foundation Research Programs; 2009.
- NCT00114595 . A Double-Blind, Randomised, Parallel-Group Comparison of Ethyl-Eicosapentaenoic Acid (ethyl-Epa) Versus Placebo As Add-on Medication in Patients With Established Tardive Dyskinesia 2005. https://clinicaltrials.gov/ct2/show/NCT00114595 (accessed 20 January 2017).
- Emsley R, Niehaus DJ, Oosthuizen PP, Koen L, Ascott-Evans B, Chiliza B, et al. Safety of the omega-3 fatty acid, eicosapentaenoic acid (EPA) in psychiatric patients: results from a randomized, placebo-controlled trial. Psychiatry Res 2008;161:284-91. http://dx.doi.org/10.1016/j.psychres.2007.06.029.
- Gardos G, Granacher RP, Cole JO, Sniffin C. The effects of papaverine in tardive dyskinesia. Prog Neuropsychopharmacol 1979;3:543-50. https://doi.org/10.1016/0364-7722(79)90008-0.
- Glazer WM, Naftolin F, Morgenstern H, Barnea ER, MacLusky NJ, Brenner LM. Estrogen replacement and tardive dyskinesia. Psychoneuroendocrinology 1985;10:345-50. https://doi.org/10.1016/0306-4530(85)90011-3.
- Goff DC, Renshaw PF, Sarid-Segal O, Dreyfuss DA, Amico ET, Ciraulo DA. A placebo-controlled trial of selegiline (L-deprenyl) in the treatment of tardive dyskinesia. Biol Psychiatry 1993;33:700-6. https://doi.org/10.1016/0006-3223(93)90119-X.
- Hajioff J, Wallace M. Effect of co-dergocrine mesylate on tardive dyskinesia. A preliminary report. Psychopharmacology 1983;79:1-3. https://doi.org/10.1007/BF00433006.
- Kojima T, Yamauchi T, Miyasaka M, Isaki K, Nakane Y, Takahashi R, et al. Treatment of tardive dyskinesia with ceruletide: a double-blind placebo-controlled study. Saishin-Igaku 1989;44:2177-88.
- Kojima T, Yamauchi T, Miyasaka M, Koshino Y, Nakane Y, Takahashi R, et al. Treatment of tardive dyskinesia with ceruletide: a double-blind, placebo-controlled study. Psychiatry Res 1992;43:129-36. https://doi.org/10.1016/0165-1781(92)90127-O.
- Lerner V. Piracetam for Tardive Dyskinesia. Muscatine, IA: Stanley Foundation Research Programs; 2009.
- Libov I, Miodownik C, Bersudsky Y, Dwolatzky T, Lerner V. Efficacy of piracetam in the treatment of tardive dyskinesia in schizophrenic patients: a randomized, double-blind, placebo-controlled crossover study. J Clin Psychiatry 2007;68:1031-7. https://doi.org/10.4088/JCP.v68n0709.
- Anon . Piracetam reduces TD symptoms tardive dyskinesia. Brown Uni Psychopharmacol Update 2007;18:3-4.
- NCT00190008 . Therapeutic Use of Piracetam for Treatment of Patients Suffering from Tardive Dyskinesia – A Double Blind, Placebo-Controlled Crossover Study 2005. https://clinicaltrials.gov/ct2/show/NCT00190008 (accessed 20 January 2017).
- MacKay AV, Sheppard GP, Saha BK, Motley B, Johnson AL, Marsden CD. Failure of lithium treatment in established tardive dyskinesia. Psychol Med 1980;10:583-7. https://doi.org/10.1017/S0033291700047498.
- Matsunaga T, Ohyama S, Takehara S, Kabashima K, Moriyama S, Tsuzuki J, et al. The effect of ceruletide on tardive dyskinesia: a double-blind placebo-controlled study. Prog Neuropsychopharmacol Biol Psychiatry 1988;12:533-9. https://doi.org/10.1016/0278-5846(88)90112-1.
- Meco G, Bedini L, Bonifati V, Sonsini U. Ritanserin in tardive dyskinesia: a double-blind crossover study versus placebo. Curr Ther Res Clin Exp 1989;46:884-94.
- Mosnik DM, Spring B, Rogers K, Baruah S, Waziri R. Phenylalanine loading effects on tardive dyskinesia severity in schizophrenics. Schizophr Res 1995;15. https://doi.org/10.1016/0920-9964(95)95640-U.
- Mosnik DM. Phenylalanine Loading Effects on Tardive Dyskinesia Severity in Schizophrenics 1994.
- Mosnik DM, Spring B, Rogers K, Baruah S. Tardive dyskinesia exacerbated after ingestion of phenylalanine by schizophrenic patients. Neuropsychopharmacology 1997;16:136-46. https://doi.org/10.1016/S0893-133X(96)00054-1.
- Mouret J, Khomais M, Lemoine P, Sebert P. Low doses of insulin as a treatment of tardive dyskinesia: conjuncture or conjecture?. Eur Neurol 1991;31:199-203. https://doi.org/10.1159/000116678.
- O’Brien CF, Jimenez R, Hauser RA, Factor SA, Mandri DF, Castro-Gyol JC. Kinect 2: NBI-98854 Treatment of Moderate to Severe Tardive Dyskinesia n.d.
- NCT01733121 . Nbi-98854 Dose Titration Study for the Treatment of Tardive Dyskinesia 2012. http://ClinicalTrials.gov/show/NCT01733121 (accessed 20 January 2017).
- Rastogi SC, Blowers AJ, Gibson AC. Co-dergocrine (hydergine) in the treatment of tardive dyskinesia. Psychol Med 1982;12:427-9. https://doi.org/10.1017/S0033291700046778.
- Richardson MA, Bevans ML, Read LL, Chao HM, Clelland JD, Suckow RF, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry 2003;160:1117-24. http://dx.doi.org/10.1176/appi.ajp.160.6.1117.
- Shamir E, Barak Y, Plopsky I, Zisapel N, Elizur A, Weizman A. Is melatonin treatment effective for tardive dyskinesia?. J Clin Psychiatry 2000;61:556-8. https://doi.org/10.4088/JCP.v61n0803.
- Shamir E, Barak Y, Shalman I, Laudon M, Zisapel N, Tarrasch R, et al. Melatonin treatment for tardive dyskinesia: a double-blind, placebo-controlled, crossover study. Arch Gen Psychiatry 2001;58:1049-52. https://doi.org/10.1001/archpsyc.58.11.1049.
- Shamir EZ, Barak F, Shalman I, . Melatonin Treatment for Tardive Dyskinesia: A Double-Blind, Placebo-Controlled, Cross-Over Study n.d.
- NCT00175955 . An 8-Week Exploratory, Double-Blind, Placebo Controlled, Randomized Trial – Evaluation of the Efficacy and Safety of Levetiracetam up to 3000 Mg Day (250-500 Mg Oral Tablets in Bid Administration) on Neuroleptic-Induced Tardive Dyskinesia in Subjects With Stable Axis I Psychiatric Disorder, Aged from at Least 18 Years to 80 Years 2005. https://clinicaltrials.gov/ct2/show/NCT00175955 (accessed 20 January 2017).
- Wolkin A, Jordan B, Peselow E, Rubinstein M, Rotrosen J. Essential fatty acid supplementation in tardive dyskinesia. Am J Psychiatry 1986;143:912-14. http://dx.doi.org/10.1176/ajp.143.7.912.
- Woods SW, Saksa JR, Baker CB, Cohen SJ, Tek C. Effects of levetiracetam on tardive dyskinesia: a randomized, double-blind, placebo-controlled study. J Clin Psychiatry 2008;69:546-54. https://doi.org/10.4088/JCP.v69n0405.
- NCT00291213 . Levetiracetam Treatment of Tardive Dyskinesia 2006. https://clinicaltrials.gov/ct2/show/NCT00291213 (accessed 20 January 2017).
- Zhang WF, Tan YL, Zhang XY, Chan RC, Wu HR, Zhou DF. Extract of Ginkgo biloba treatment for tardive dyskinesia in schizophrenia: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2011;72:615-21. http://dx.doi.org/10.4088/JCP.09m05125yel.
- Zhang F, Bigos K, Weinberger D. Genome-Wide Analysis of Antipsychotic Drug Response in Schizophrenia n.d.
- Tan Y. Extract of Ginkgo Biloba and Tardive Dyskinesia 2008. https://clinicaltrials.gov/ct2/show/NCT00672373 (accessed 20 January 2017).
- Buruma OJ, Roos RA, Bruyn GW, Kemp B, van der Velde EA. Tiapride in the treatment of tardive dyskinesia. Acta Neurol Scand 1982;65:38-44. https://doi.org/10.1111/j.1600-0404.1982.tb03059.x.
- Roos RAC, Buruma OJS, Bruyn GW, Kemp B, van der Velde EA, Zelvelder WG. Tiapride in Huntington’s chorea and tardive dyskinesia. A double-blind, placebo controlled crossover clinical trial. J Drug Res 1982;7:1234-9.
- Huang CC, Wang RI, Hasegawa A, Alverno L. Evaluation of reserpine and alpha-methyldopa in the treatment of tardive dyskinesia. Psychopharmacol Bull 1980;16:41-3.
- Huang CC, Wang RI, Hasegawa A, Alverno L. Reserpine and alpha-methyldopa in the treatment of tardive dyskinesia. Psychopharmacology 1981;73:359-62. https://doi.org/10.1007/BF00426466.
- Pappa S, Tsouli S, Apostolou G, Mavreas V, Konitsiotis S. Effects of amantadine on tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Clin Neuropharmacol 2010;33:271-5. https://doi.org/10.1097/WNF.0b013e3181ffde32.
- Pappa S, Tsouli S, Apostolou G, Mavreas V, Konitsiotis S. Efficacy of amantadine in the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Biol Psychiatry 2009.
- Pappa S, Tzouli S, Mavreas V, Konitsiotis S. Efficacy of an NMDA receptor antagonist in the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Schizophr Res 2012;136. https://doi.org/10.1016/S0920-9964(12)71045-9.
- Rust M. Tiapride treatment of tardive dyskinesia due to long-term neuroleptic treatment. Sem Hop 1984;60:2195-6.
- Simpson GM, Yadalam KG, Stephanos MJ. Double-blind carbidopa/levodopa and placebo study in tardive dyskinesia. J Clin Psychopharmacol 1988;8:49-51. https://doi.org/10.1097/00004714-198808001-00009.
- Soni SD, Freeman HL, Bamrah JS, Sampath G. Oxypertine in tardive dyskinesia: a long-term controlled study. Acta Psychiatr Scand 1986;74:446-50. https://doi.org/10.1111/j.1600-0447.1986.tb06267.x.
Appendix 1 Patient and public involvement report: tardive dyskinesia – adding perspectives from personal experience to the research agenda
Introduction
On 15 April 2016, the McPin Foundation hosted a consultation group to gather feedback from people with a lived experience of TD. This endeavour was undertaken by the Cochrane Schizophrenia Group at the University of Nottingham in an effort to inform our systematic review. The consultation was commissioned by a group of researchers who have completed a NIHR-funded systematic review to ascertain effective interventions to treat TD. An integral part of any health research is to gain the service user perspective; therefore, the results of the review were discussed. Another aim of the session was to elicit what people with lived experience thought would be a good research project in this area.
Methods
The consultation was planned to enable the voices of people with personal experience of TD to be heard. The consultation was advertised by e-mail via the McPin Foundation’s large circulation list of people who have an expressed interest in being involved, as well as on their website. Interested people were asked to contact the McPin Foundation to book a place to attend. Prior to the meeting, two documents were circulated to attendees: a lay report providing an overview of the review and one of the individual systematic reviews that had been included. These documents gave the foundation for the discussions of the day.
The consultation was held at the McPin Foundation offices in London, UK. Reimbursement for time and out-of-pocket expenses was offered. The consultation was facilitated by Ruth Sayers (Peer Researcher at the McPin Foundation), with support from Megan Rees (Public Involvement in Research Co-ordinator at the McPin Foundation) and Dr Dawn-Marie Walker (Associate Professor at the University of Southampton). All of these researchers have extensive experience in involving patients and the public in research consultation. Furthermore, although this collaboration is not empirical qualitative research per se, both Ruth and Dawn-Marie have expert knowledge in this paradigm, including hosting focus groups (or in this case a collaboration). The session was planned to provide time to reflect on current research on TD and to consider gaps in knowledge.
Following an introduction to the consultation by Ruth Sayers, Dr Dawn-Marie Walker gave an oral overview of the review and the findings.
The group was then shown a video clip from YouTube (YouTube, LLC, San Bruno, CA, USA) showing people with TD. The primary purpose of showing the clip was to give attendees an overview of the effects of TD and to provide a common starting point for the discussion. The YouTube clip shown towards the beginning of the consultation was entitled ‘Tardive Dyskinesia’. Uploaded on 12 June 2016, the clip is a training digital versatile disc (DVD) that presents the AIMS exam by showing a range of abnormal involuntary movement-associated conditions in patients, including scoring by an expert medical panel.
The clip can be found at www.youtube.com/watch?v=FUr8ltXh1Pc (accessed 13 June 2017).
Attendees were then asked to consider:
-
What is important to people who have experience of managing TD alongside living with severe mental illness?
-
Are the outcomes used in current TD research, as reflected in the Cochrane reviews, appropriate from a lived experience perspective?
-
What other outcomes might be important to service users and carers for research into TD?
-
Ideas for future research in the area.
The consultation included open group discussions and prioritisation of ideas. All discussions were audio-recorded, while the attendees were asked to write down their ideas throughout the day on paper tablecloths and Post-it notes to help keep an accurate record of discussion and in order to encourage everyone to participate (see Figures 1, 8 and 9). The researchers listened to the recordings after the session and noted any points relevant to the above mentioned questions that would have impact on the funded systematic review. Full transcription and formal analyses were not appropriate in this case, as the consultation was not a piece of empirical qualitative work.
FIGURE 8.
Some comments from the consultation.

FIGURE 9.
A key concern.

Group demographics
A total of six people attended the consultation, excluding facilitators. All collaborators were mental health service users and one was a carer. All service users were taking, or had previously taken, antipsychotics. The researchers acknowledge that a larger, diverse group may have presented a wider range of perspectives on the review; however, for the type of involvement we anticipated, a more formal method for recruitment (e.g. purposive sampling) would not have been appropriate.
| Category | Participants’ details |
|---|---|
| Sex | Male, n = 0; female, n = 6 |
| Age group (years) | 25–34, n = 2; 35–44, n = 1; 45–54, n = 1; 55–64, n = 1; ≥ 65, n = 1 |
| Ethnic group | White British, n = 4; other, n = 2 |
| Service user/carer | Service user, n = 5; carer, n = 1 |
| Antipsychotic use | Taken in past: olanzapine, quetiapine, thioridazine, haloperidol, risperidone olanzapine, sulpiride, quetiapine, haloperidol Currently taking antipsychotics: olanzapine, Depakote® (AbbVie Inc., North Chicago, IL, USA), venlafaxine |
Findings
Within the relatively open format of the consultation, the group were asked to bear in mind the four consultation questions. A number of attendees, including facilitators, were disturbed by the YouTube clip shown at the session, particularly its sole emphasis on identifying the physical symptoms of TD.
That’s how others see me! Mad old woman from a 1950s asylum.
The group went on to discuss the debilitating nature of TD. One attendee noted that, unlike symptoms of psychosis such as hearing voices and hallucinations, people with TD are unable to conceal the effects of TD when they are out in public. This, in turn, can have a very negative impact on a person’s self-esteem and ability to maintain social networks.
TD can be as debilitating as the psychosis itself.
From group discussions, a key theme that emerged was informed consent and the extent to which service users are made aware of the adverse effects of antipsychotic medication. There was a consensus that, on the whole, people are not given enough information about the adverse effects of antipsychotic medication. This lack of information makes it impossible for people to weigh the pros and cons of taking medications prior to beginning treatment. Informed consent is not only a key principle of treatment, but it also leads to higher levels of ‘treatment adherence’ and treatment satisfaction. Attendees felt that informed consent was important in both inpatient and outpatient settings.
I think psychiatrists presume that patients are stupid and can’t make an informed choice.
Although attendees acknowledged that increasing the level of information provided to people would not directly lead to a lower incidence of TD, it would probably lead to people feeling more empowered and better able to accept the consequences of any treatment. Although we acknowledge that published evidence suggests that clinical efficacy is more important to patients than the side-effect profile of antipsychotics, a clear message that emerged from this consultation was the need for full informed consent obtained by outlining adverse effects in a patient-centred consultation. Only one of the collaborators had heard of TD before, although all had taken antipsychotics at some time.
Key recommendation for research outcomes in TD: measure the extent to which people feel informed about their treatment and the possibility of adverse effects such as TD.
Participants also noted the importance of people having access to quality, evidence-based information about TD. This would make service users less reliant on clinicians for information, and support full informed consent.
Key recommendation for research outcomes in TD: measure service users’ access to quality information about TD.
Discussions about informed consent led into a discussion about accountability. Attendees highlighted service users’ feelings of anger and impotence that result from experiencing the distressing adverse effects of medication, particularly in cases in which people have not previously been provided with adequate information. In many cases, people have no way of holding the medical profession to account because adverse effects of medication are often similar to defined symptoms of mental illness and, thus, it is difficult for people to prove a direct link with medication. This is not the case with TD, as there is a general consensus that TD results solely from medication consumption. Accountability was an important outcome, particularly for people who have developed lifelong TD as a result of taking medication.
Key recommendation for research outcomes in TD: for people who have developed lifelong TD as a result of taking medication, to what extent do organisations/individuals take responsibility? Are people supported or encouraged to seek accountability?
Prevention was another key theme in the discussion. Attendees were concerned that adverse effects of medication are often treated with more medication and that the research included in the Cochrane review placed an over-reliance on pharmaceutical interventions to treat TD. They wondered, ‘Why are all of the approaches pharmacological?’.
Furthermore, in light of the Cochrane review’s findings, attendees were not confident that reducing or stopping taking antipsychotic drugs reduces instances of TD.
I’m appalled by the poverty of this evidence base given how debilitating tardive dyskinesia is.
Attendees suggested other avenues that may be worth exploring, including attempting to understand the causal mechanisms behind TD through brain imaging.
Key recommendation for future research in TD: understanding the causal mechanisms that result in TD as well as developing methods to assess individuals’ risk of developing TD as a result of medication consumption.
As the group discussed ideas for future research into TD, the issue of prevalence was raised. Is TD a diminishing problem? Prevalence was not addressed in the research compiled by the Cochrane review and the group were not aware of any substantive data to suggest that the prevalence of TD is decreasing. A number of recommendations were made in relation to prevalence.
Key recommendation for future research in TD: understanding the prevalence of medication-related TD.
Key recommendation for research outcomes in TD: measuring clinician awareness of TD as a side effect of psychiatric medications.
Key recommendation for research outcomes in TD: measuring the level of reporting with regard to incidences of TD.
Following the discussion about prevention and prevalence, the group considered the best ways of supporting those already living with TD and the role that research can play. None of the research that has taken place thus far has explored the effectiveness of psychological therapies, peer support and social interventions to help people to cope with the symptoms of TD. Coping mechanisms are very important in the absence of effective treatments, particularly for those who experience these adverse effects long term. Attendees noted that some of the most debilitating aspects of living with TD stem from social stigma and the negative impacts of TD on an individual’s confidence:
Look at that lady!
People point at me, particularly children.
Tardive dyskinesia makes you feel vulnerable because it’s so obvious.
The group made a number of suggestions relating to managing the symptoms of TD, as well as measuring the effectiveness of particular treatments in relation to service users’ confidence, social inclusion and quality of life.
Key recommendation for future research in TD: what psychological therapies are effective in managing the symptoms of TD?
Key recommendation for future research in TD: is peer support effective in managing the symptoms of TD?
Key recommendation for research outcomes in TD: social confidence, social inclusion, social networks, personalised quality-of-life measures and employment.
The group discussed the parallels between Tourette syndrome and TD. A number of public awareness campaigns have been successful in informing the public about Tourette syndrome, and this in turn has reduced social stigma. The group suggested that similar campaigns would probably be effective in reducing the stigma associated with TD.
Key recommendation for future research in TD: measuring public awareness of TD.
Finally, attendees were asked to review the outcomes that have been used in TD research to date to assess their relevance. As illustrated in the Cochrane review, the outcomes used in research relating to TD are as follows:
-
improvement in TD
-
level of functioning
-
improvement/reduction in psychiatric symptoms
-
deterioration
-
relapse
-
mental state changes
-
acceptability of treatment
-
quality of life
-
satisfaction with care
-
adverse effects
-
hospital admission
-
death
-
dropped out of trial/left the study early.
There was consensus within the group that all of the outcomes used to date have their merits and that their relevance would depend on a large number of factors including the type of treatment being assessed and trial design. However, the list of outcomes included in the Cochrane review has some notable omissions. Outcomes and areas of research that have thus far been underexplored are listed below.
List of key recommendations for outcomes and research in to tardive dyskinesia
Outcomes
-
Measure the extent to which service users feel informed about their treatment and the possibility of adverse effects such as TD.
-
Measure patients’ access to quality information about TD.
-
For people who have developed lifelong TD as a result of taking medication, to what extent do organisations/individuals take responsibility? Are service users supported or encouraged to seek accountability?
-
Measuring clinician awareness of TD as a side effect of psychiatric medications.
-
Measuring the level of reporting with regard to incidences of TD.
-
Measuring social confidence, social inclusion, social networks, personalised quality-of-life measures and employment.
-
Measuring public awareness of TD (Figure 10).
FIGURE 10.
Key outcomes of interest.

Future research
-
Understanding the causal mechanisms that result in TD as well as developing methods to assess individuals’ risk of developing TD as a result of medication consumption.
-
Understanding the prevalence of medication-related TD.
-
What psychological therapies are effective in managing the symptoms of TD?
-
Is peer support effective in managing the symptoms of TD?
It is important to note that the above list of recommendations reflects the context within which they were suggested, either as additional outcomes to be considered within future TD research or as future research projects.
However, it was clear that almost all of the recommendations relating to ‘outcomes’ could equally be important areas of interest for future research in and of themselves. Moreover, some studies that are not solely focused on ascertaining the prevalence of medication-related TD may be improved by including an outcome measure to understand the prevalence of TD among their participant group.
Reflections of the facilitating team
Megan Rees
I really enjoyed the session and given I had little prior experience of working in the field of TD, I found the group’s discussions very enlightening.
When it came to the most important outcomes for research, attendees unanimously supported the goals of research included in the Cochrane review. Preventing and treating the symptoms of TD were, for obvious reasons, a key concern of service users. However, attendees were quick to highlight important outcomes that appeared to be missing from the research. One such ‘missing’ outcome referred to as ‘informed prescribing’ particularly struck me. After watching a rather graphic video of the effects of TD, there was a palpable sense of injustice. A number of attendees wondered how many people who are prescribed antipsychotics are made aware of such severe side effects and expressed how important it is that service users are given the opportunity to make an informed choice before taking medication. If, as the review found, we are unable to effectively prevent or treat this particular side effect, some emphasis must be placed on giving service users enough information that they are able to essentially own their decisions when it comes to medication. This would at least mitigate against the feeling of powerlessness and subjugation that many people feel when they experience medication side effects that they were not initially made aware of.
The group made a number of highly insightful suggestions throughout the day but it was their focus on outcomes relating to empowerment and autonomy that were so striking given that these outcomes were conspicuous by their absence in the research that has taken place so far.
Dawn Marie-Walker
I really enjoyed the session, and was reassured by the passionate responses from the service users that this research is really worthwhile.
Since being part of this work, one of my PhD [doctor of philosophy] students from Saudi Arabia has had a nephew with severe mental health difficulties. His nephew has been given vast amounts of medication, including anti psychotics, and what has resulted, from the description of my student, as TD.
Although initially my colleagues and I thought TD was a declining problem (due to having far more knowledge about it and medication regimes), it appears that it is still a grave problem internationally. Also in dementia, where antipsychotics are prescribed off licence, it may also be more of a problem.
Ruth Sayers
I appreciated the openness and engagement of the people who attended the workshop. Individual accounts of experiencing TD differed considerably, but all showed clearly the level of distress, vulnerability and stigmatisation that can be associated with tardive dyskinesia. Lack of awareness of TD was compared with the growing awareness of Tourette’s, and the efforts being made to de-stigmatise that condition, especially with young people.
Several felt angry that they had not been given sufficient information at the time of prescribing about side effects of antipsychotics to make an informed choice – to enable them to balance the risks for themselves. There were many questions raised about how much was known, and how much doctors know, or reported, about TD, and therefore whether the actual prevalence is known, in the UK or elsewhere. Suggestions about what might help people included greater knowledge and an opportunity to avoid TD, and personal and social support to cope with the stigmatising condition. I hope that the workshop raised some important issues for further exploration.
Conclusion and next steps
It is clear that service users and carers from the consultation thought that research into TD to date has been limited and that further exploration is required. They supported the outcomes used in Cochrane schizophrenia review work on TD, but would recommend that the field is broadened. In addition, a formal recommendation was to put information on the prevalence of TD into the public domain. If data on prevalence do not currently exist, service users and carers recommend that this be sought out urgently. There was acknowledgement that data might include under-reporting, but this was felt to be an important benchmark for understanding.
The ultimate goal of research is to improve service user outcomes. The consultation group felt that there were some key issues that needed to be addressed. First, it was felt that better information about TD was needed, so that service users and their carers can make informed choices about medication. Second, strategies for coping with TD were identified as essential. A greater emphasis needs to be placed on psychological and social interventions for managing the symptoms of TD. For people already living with persistent symptoms of TD, supporting people in the management of the numerous impacts of TD was very important. Third, the consultation group felt that social stigma needed to be addressed as public reactions to people living with TD can be as hard to cope with as the symptoms of underlying mental health problems themselves, such as schizophrenia.
Appendix 2 Differences between protocol and review
| Details of difference | Comments |
|---|---|
| We planned to include evidence from crossover trials. We only included evidence from the first phase of crossover trials | A major concern of crossover trials is the carry-over effect. This occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a washout phase. For the same reason, crossover trials are not appropriate if the condition of interest is unstable.61 As both effects are very likely in severe mental illness, we used only data of the first phase of crossover studies |
| The planned outcomes list was reviewed and updated | As a consequence of the PPI session, outcome measures for the review were reviewed to also reflect outcomes important to patients |
| We planned to rely on evidence from the NMA. We decided not to rely on evidence from the NMA | The complete NMA was performed and it is available in Appendix 4. We have very little confidence in the results of the NMA because of (1) few data, (2) few studies in each comparison, (3) no differences between pairwise meta-analyses and NMA, and (4) not sufficiently connected networks. Therefore, we only used the results of the NMA to support planning future studies in this area |
| We carried out a different search from the protocol-specified search | As the Cochrane Schizophrenia Group maintains a good register that is regularly updated with a variety of databases and grey literature, we believed it was more appropriate to run the searches for all potential RCT TD references in their register. We also searched included and excluded studies of published Cochrane reviews |
Appendix 3 Observational studies: additional methods and results
Search strategy and results
See Figure 11 for the PRISMA diagram of observational study screening and study selection process.
FIGURE 11.
The PRISMA diagram of observational study screening and study selection process.

The search strategy and results per database are presented below.
EMBASE
Date searched: 9 January 2017.
Date range searched: 1974 to 2017 week 2.
Number of results: 696.
Search strategy
-
exp cohort analysis/ or exp longitudinal study/ or exp prospective study/ or exp case control study/ or exp follow up/ or cohort$.tw. or (case$ and control$).tw.
-
tardive dyskinesia/ or ‘tardive dyskinesia?’.mp.
-
1 and 2
-
Limit 3 to human
Ovid MEDLINE In-Process & Other Non-Indexed Citations and Ovid MEDLINE
Date searched: 9 January 2017.
Date range searched: 1946 to 9 January 2017.
Number of results: 2072.
Search strategy
-
exp cohort studies/ or epidemiologic methods/ or exp case-control studies/ or (case$ and control$).tw. or cohort$.tw.
-
tardive dyskinesia/ or ‘tardive dyskinesia?’.mp.
-
1 and 2
-
Limit 3 to humans
PubMed
Date searched: 9 January 2016.
Date range searched: up to 9 January 2017.
Number of results: 377.
Search strategy
-
Therapy/Broad[filter] AND (‘observational study’[Publication Type] OR ‘observational studies as topic’[MeSH Terms] OR ‘observational studies’[All Fields]).
-
tardive dyskinesia/ or ‘tardive dyskinesia?’.mp.
-
1 and 2
-
Limit 3 to humans
PsycINFO
Date searched: 9 January 2017.
Date range searched: 1806 to January week 1 2017.
Number of results: 167.
Search strategy
-
cohort analysis/ or followup studies/ or exp longitudinal studies/ or (case$ and control$).tw. or cohort$.tw.
-
tardive dyskinesia/ or ‘tardive dyskinesia?’.mp.
-
1 and 2
-
Limit 3 to human
Results
Included studies
| Study characteristics | Outcomes | Results | Conclusion; risk of bias | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Selection bias | Controlled for baseline confounding | Reliable outcome assessment | Incomplete outcome data (attrition bias) | Selective outcome reporting | Other bias | |||||
| Casey and Toenniessen, 1983145 5-year NRCT (n = 27) of 30- to 77-year-old F and M inpatients with various mental disorders and TD in the USA Comedications: lithium |
Discontinuation of FGAs | Decrease of FGAs | Increase of FGAs | A small NRCT found that psychiatric patients with TD whose antipsychotic medication was reduced or discontinued showed greater improvement in TD symptoms (even resolution of symptoms) after 5 years of follow-up, compared with patients whose dosage of antipsychotic medication was increased or remained unchanged | ||||||
| Mean (%) improvement in TD symptoms (AIMS) | 55 | 65 | 35 | |||||||
| Mental state (relapse) (n/N) | 4/10 | 8/10 | 7/7 | High | UC | UC | UC | High | UC | |
| Damier et al., 2007146 6-month Phase II NRCT (n = 10) of 26- to 69-year-old F and M participants with various mental disorders and TD in France Comedications: benzodiazepine, mianserin and amitriptyline |
Mean (%) improvement in TD symptoms (ESRS) | There was a 50% improvement (range 30–66%) (p = 0.002) with bilateral globus pallidus deep-brain stimulation compared with no brain stimulation | A very small NRCT found that bilateral globus pallidus deep-brain stimulation seems to offer a greater benefit (50%) in decreasing TD symptoms at 6 months’ follow-up compared with no stimulation | |||||||
| High | UC | Low | UC | High | UC | |||||
| Hardoy et al., 2003148 1-year prospective cohort study (n = 30) of F and M outpatients with various mental disorders and TD in Italy Comedications: antipsycotics and mood stabilisers |
TD symptoms scale scores (AIMS mean end point) (low = less severe) | Gabapentin ± typical antipsychotics: 18.2 (SD 5.5); n = 4 | Gabapentin ± atypical antipsychotics: 11.2 (SD 4.8); n = 18 | A small prospective cohort study found that gabapentin treatment reduced TD symptoms in schizoaffective, bipolar I disorder and schizophrenic patients with a mean percentage of improvement at AIMS of 47.5% (SD ± 18.2%) in all treated patients. A trend towards improvement was revealed in both the participants taking concurrently atypical antipsychotics and those concurrently on traditional antipsychotics, with those on atypical antipsychotics (mean AIMS score 11.2, SD 4.8; n = 18) doing a little better than those on traditional antipsychotics (mean 18.2, SD 5.5; n = 4) | ||||||
| Dosage: gabapentin commenced at 300 mg/day, increased after 2 days to 600 mg/day and reached 900–1200 mg/day during the first week. Mean dosage administered: 1170 ± 278 mg/day | High | UC | UC | UC | UC | UC | ||||
| Huang, 1986149 4-year prospective cohort study (n = 10) of 50- to 68-year-old F and M inpatients with various mental disorders and TD in the USA Comedications: benztropine |
Discontinuation or reduced dose of FGAs | No change in dose of FGAs | A very small prospective cohort study found that psychiatric patients with TD whose antipsychotic medication was reduced or discontinued showed greater improvement in TD symptoms after 4 years of follow-up, compared with patients whose dosage of antipsychotic medication remained unchanged at 4 years’ follow-up | |||||||
| TD symptoms scale scores: mean end point (Kazamatsuri et al.169) (low = less severe) | 1.9; n = 5 | 3.3; n = 5 | ||||||||
| Mean improvement in TD symptoms | 60%; n = 5 | 21%; n = 5 | High | UC | Low | UC | UC | UC | ||
| Koshino et al., 1991150 11-year prospective cohort study (n = 28) of 37- to 77-year-old F and M participants with various mental disorders and TD in Japan Comedications: not reported |
Decreased dose of FGAs | Increased dose or no change of FGAs | A small prospective cohort study found that the severity of TD was unchanged in 39.3% of the patients, improved in 17.9% of patients, fluctuated in 21.4% of patients and worsened in 21.4% of patients at 11 years’ follow-up. The outcome was not associated with patient sex, age, duration of primary illness, dosage of antipsychotics or changes in dosage | |||||||
| Improvement in TD symptoms (n/N) | 2/13 | 3/15 | ||||||||
| No change in TD symptoms (n/N) | 4/13 | 7/15 | ||||||||
| Worsening of TD symptoms (n/N) | 3/13 | 3/15 | ||||||||
| Fluctuation of TD symptoms (n/N) | 4/13 | 2/15 | ||||||||
| Dosage: the mean daily dose of FGAs was 221.4 mg (SD 153.7 mg) of CPZE (average for all groups) | High | UC | Low | High | UC | UC | ||||
| Peselow et al., 1989151 1-year prospective cohort study (n = 31) of F and M inpatients with schizophrenia and TD in the USA Comedications: not reported |
Discontinuation of fluphenazine decanoate | Maintenance of fluphenazine decanoate | A small prospective cohort study found that, although there was a statistically significant decrease in abnormal movements at 1-year follow-up, this improvement was offset by the fact that 15 of the 21 (71.4%) patients discontinued from antipsychotic treatment relapsed | |||||||
| No clinically important improvement in TD symptoms (n/N) | 14/21 | 9/10 | ||||||||
| TD symptoms scale scores: mean end-point AIMS score | 5.76; n = 21 | 7.8; n = 10 | ||||||||
| Mental state (relapse) (n/N) | 15/21 | 1/10 | High | High | UC | UC | UC | UC | ||
| Dosage: average 41.93 mg (SD ± 21.9 mg) biweekly | ||||||||||
| Yagi and Itoh, 1985152 10-year prospective cohort study (n = 20) of 35- to 84-year-old F and M participants with various mental disorders and TD in Japan Comedications: antipsychotics |
Discontinuation or decreased dose of antipsychotics | Antipsychotic maintenance | A small prospective cohort study found that the long-term outcome (10 years) of TD is determined by the patient’s age at onset rather than by the course of antipsychotic treatment (discontinuation, maintenance or decreased dose) after the occurrence of TD | |||||||
| No clinically important improvement in TD symptoms (n/N) | 5/14 | 1/4 | ||||||||
| Disappearance of TD (n/N) | 6/14 | 2/4 | ||||||||
| Mental state (relapse) (n/N) | 3/14 | 1/4 | High | UC | UC | High | UC | UC | ||
| Yassa et al., 1992153,154 10-year prospective cohort study (n = 44) of 42- to 83-year-old F and M inpatients and outpatients with various mental disorders and TD in Canada Comedications: anticholinergic medication, lithium carbonate, antidepressant |
No change in antipsychotic dose (dosage: 357 mg/dl) | Decrease in antipsychotic dose (dosage: 312 mg/dl) | A small prospective cohort study found that the majority (50%) had no change in their TD severity, 20% had an improvement and 30% had a worsening of their TD. Little difference was noted in those patients whose medication was decreased (33% had no change in TD severity, 42% had increased TD severity and 25% had decreased TD severity) and those whose medication remained unchanged (56% had no change in TD severity, 25% had increased TD severity and 19% had decreased TD severity) at 10 years’ follow-up | |||||||
| TD symptoms scale scores: mean endpoint AIMS score | 4.8 (SD 3.8); n = 32 | 6.6 (SD 4.7); n = 12 | ||||||||
| No change in TD severity (n/N) | 18/32 | 4/12 | ||||||||
| Increase in TD severity (n/N) | 8/32 | 5/12 | ||||||||
| Decrease in TD severity (n/N) | 6/32 | 3/12 | High | Low | UC | High | UC | UC | ||
Description of excluded studies
Thirty studies (31 references) were excluded at full-text screening. Reasons for exclusion were: not an observational study (seven studies), observational study with no control group (19 studies), study only measuring prevalence (three studies) or no treatment was provided (one study). Table 5 shows full references and reasons for exclusion per study.
| Study | Reason for exclusion |
|---|---|
| Ascher-Svanum H, Zhu B, Faries D, Peng X, Kinon BJ, Tohen M. Tardive dyskinesia and the 3-year course of schizophrenia: results from a large, prospective, naturalistic study. J Clin Psychiatry 2008;69:1580–8 | No treatment provided |
| Bai YM, Yu SC, Chen JY, Lin CY, Chou P, Lin CC. Risperidone for pre-existing severe tardive dyskinesia: a 48-week prospective follow-up study. Int Clin Psychopharmacol 2005;20:79–85 | 48-week open-label follow-up of RCT (12 weeks: risperidone × placebo) with all receiving risperidone |
| Barron ET, McCreadie RG. One year follow-up of tardive dyskinesia. Br J Psychiatry 1983;143:423–4 | TD prevalence only |
| Caine ED, Polinsky RJ, Kartzinel R, Ebert MH. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry 1979;136:317–20 | Already excluded RCT: Tourette syndrome, Huntington disease and drug-induced atypical dyskinesia, no TD symptoms at baseline |
| Chaplin RH. Risperidone, tardive dyskinesia, and the elderly. Am J Psychiatry 2001;158:1336–7 | Review/commentary/editorial |
| Chen PH, Liu HC. Rapid improvement of neuroleptic-induced tardive dyskinesia with levetiracetam in an interictal psychotic patient. J Clin Psychopharmacol 2010;30:205–7 | Case series/case report |
| Chouinard G, Annable L, Mercier P, Ross-Chouinard A. A five year follow-up study of tardive dyskinesia. Psychopharmacol Bull 1986;22:259–63 | TD prevalence only |
| Cortese L, Caligiuri MP, Williams R, Schieldrop P, Manchanda R, Malla A, Harricharan R. Reduction in neuroleptic-induced movement disorders after a switch to quetiapine in patients with schizophrenia. J Clin Psychopharmacol 2008;28:69–73 | Already excluded RCT; people with schizophrenia, no TD symptoms at baseline |
| Factor SA. Propranolol therapy for tardive dyskinesia revisited. Mov Disord 2012;27:1703 | Case series/case report |
| Glazer WM, Moore DC, Schooler NR, Brenner LM, Morgenstern H. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry 1984;41:623–7 | No comparison group. Reported probabilities based on regression analyses |
| Glazer WM, Morgenstern H, Schooler N, Berkman CS, Moore DC. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry 1990;157:585–92 | No comparison group. Reported probabilities based on regression analyses |
| Hatcher-Martin JM, Armstrong KA, Scorr LM, Factor SA. Propranolol therapy for tardive dyskinesia: a retrospective examination. Parkinsonism Relat Disord 2016;32:124–6 | Observational study without a control group (mentioned tetrabenazine as treatment of choice) |
| Heimburger RF. Dentatectomy in the treatment of dyskinetic disorders. Confin Neurol 1967;29:101–6 | Case series/case report |
| Kantrowitz JT, Srihari VH, Tek C. Resolution of tardive dyskinesia after addition of aripiprazole to haloperidol depot. J Clin Psychopharmacol 2007;27:525–6 | Case series/case report |
| Kucerová H. Olanzapine and improvement of tardive dyskinesia. Eur Psychiatry 2002;17:421–4 | Case series/case report |
| Lee JG, Shin BS, Lee YC, Park SW, Kim YH. Clinical effectiveness of the Kampo medicine kamishoyosan for adjunctive treatment of tardive dyskinesia in patients with schizophrenia: a 16-week open trial. Psych Clin Neurosci 2007;61:509–14 | Observational study without a control group |
| Louzã MR, Bassitt DP. Maintenance treatment of severe tardive dyskinesia with clozapine: 5 years’ follow-up. J Clin Psychopharmacol 2005;25:180–2 | Case series/case report |
| Mendhekar D, Aggarwal A. Olanzapine and trihexyphenidyl-induced tardive dyskinesia. Indian J Pharmacol 2005;37:263 | Case series/case report |
| Michael N, Sourgens H, Arolt V, Erfurth A. Severe tardive dyskinesia in affective disorders: treatment with vitamin E and C. Neuropsychobiology 2002;46(Suppl. 1):28–30 | Case series/case report |
| Morgenstern H, Glazer WM, Woods SW. Risperidone and tardive dyskinesia. Int J Geriatr Psychiatry 2001;16:541–2 | Review/commentary/editorial |
| Naber D, Leppig M, Grohmann R, Hippius H. Efficacy and adverse effects of clozapine in the treatment of schizophrenia and tardive dyskinesia – a retrospective study of 387 patients. Psychopharmacology 1989;99:S73–6 | Retrospective case series |
| O’Brien CF, Jimenez R, Hauser RA, Factor SA, Burke J, Mandri D, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomised, double-blind, placebo-controlled study. Mov Disord 2015;30:1681–7 | RCT – included in Cochrane review |
| Pi EH, Simpson GM. Atypical neuroleptics: clozapine and the benzamides in the prevention and treatment of tardive dyskinesia. Mod Probl Pharmacopsychiatry 1983;21:80–6 | Review/commentary/editorial |
| Rajarethinam R, Dziuba J, Manji S, Pizzuti A, Lachover L, Keshavan M. Use of aripiprazole in tardive dyskinesia: an open label study of six cases. World J Biol Psychiatry 2009;10:416–19 | Case series/case report |
| Saltz BL, Kane JM, Woerner MG, Lieberman JA, Alvir JM, Blank K, et al. Prospective study of tardive dyskinesia in the elderly. Psychopharmacol Bull 1989;25:52–6 | Only TD prevalence |
| Sharma A, Ramaswamy S, Dewan VK. Resolution of ziprasidone-related tardive dyskinesia with a switch to aripiprazole. Prim Care Companion J Clin Psychiatry 2005;7:36 | Case series/case report |
| Singh MM, Becker RE, Pitman RK, Nasrallah HA, Lal H. Sustained improvement in tardive dyskinesia with diazepam: indirect evidence for corticolimbic involvement. Brain Res Bull 1983;11:179–85 | Before-and-after study, irrelevant study design |
| Thara R. Use of antipsychotics and tardive dyskinesia. J Postgrad Med 2004;50:172 | Review/commentary/editorial |
| van Harten PN, Hoek HW, Matroos GE, van Os J. Evidence that lithium protects against tardive dyskinesia: the Curaçao Extrapyramidal syndromes study VI. Eur Neuropsychopharmacol 2008;18:152–5 | Observational study without a control group |
| Viallet F, Gayraud D, Gombert C, Renie L, Martinez-Almoyna L, Di Legge S, et al. Utility of tetrabenazine for managing L-Dopa induced dyskinesias in advanced Parkinson’s disease: a retrospective observational study on 10 patients. Mov Disord 2014;29:S149 | Observational study without a control group |
| Yasui-Furukori N, Kikuchi A, Katagai H, Kaneko S. The effects of electroconvulsive therapy on tardive dystonia or dyskinesia induced by psychotropic medication: a retrospective study. Neuropsychiatr Dis Treat 2014;10:1209–12 | Case series/case report |
Appendix 4 Network meta-analysis on comparative safety and clinical effectiveness of interventions for antipsychotic-induced tardive dyskinesia: methods and results
Objectives
We aimed to compare the safety and clinical effectiveness of interventions for deterioration of symptoms of antipsychotic-induced TD. We also aimed to generate a clinically meaningful hierarchy of the eligible interventions according to their efficacy and safety.
Methods
Criteria for considering studies for this review
Types of interventions
We included interventions used to treat or prevent deterioration of symptoms of antipsychotic-induced TD of relevance for people in the NHS, indicated as priority interventions: ‘switch to SGA (including switch to amisulpride, clozapine, olanzapine, quetiapine, risperidone, ziprasidone)’, ‘antipsychotic (AP) reduction’, ‘antipsychotic maintenance/TAU (including AP)’, ‘antipsychotic withdrawal (with placebo)’, ‘FGA (any)’, ‘anticholinergic and AP continuation’, ‘anticholinergic withdrawal and AP continuation’, ‘benzodiazepines and AP continuation’, ‘buspirone and AP continuation’, ‘hypnosis or relaxation and AP continuation’, ‘vitamin E and AP continuation’ and ‘placebo (with AP continuation)’.
We assumed that any patient who met the inclusion criteria was, in principle, equally likely to be randomised to any of the interventions and, thus, the transitivity assumption was likely to hold on the onset.
Types of outcome measures
The following outcomes were measured:
-
primary outcome – no clinical improvement of TD symptoms (< 50% improvement on scales)
-
secondary outcome – total discontinuation rates.
We intended to analyse all planned outcomes described in the main paper but we were unable to do so because of the limited data available. We estimated the relative ranking of the competing interventions according to both of the above outcomes.
Data collection and analysis
Measures of treatment effect
Relative treatment effects
Odds ratios were employed for dichotomous outcomes. When continuous outcomes were measured, we analysed them using the MD if all studies used the same measure to assess the same outcome. Standardised mean difference, Hedge’s adjusted g, was used when a different measure was used across studies to assess a common continuous outcome. 170
Relative treatment ranking
We estimated p-scores, which are the most frequent analogues of surface under the cumulative ranking curves (SUCRAs), to obtain a hierarchy of the competing interventions. 171,172
-
Assessment of clinical and methodological heterogeneity within treatment comparisons.
We assessed the presence of clinical and methodological heterogeneity within each pairwise comparison by comparing trial and study population characteristics across all eligible trials. Considerable differentiation in synthesised studies in terms of patient, study and intervention characteristics might lead to a lack of usefulness of obtained results. 173
-
Assessment of transitivity across treatment comparisons
The assumption underlying NMA implies that one can learn about the relative effectiveness of ‘A versus B’ via a common comparator, for instance C. 155,174 We were unable to compare the distribution of effect modifiers across comparisons because of the limited data, but we compared the particular study characteristics qualitatively. Moreover, we assessed if the indication of the included interventions varied according to the alternative it is compared against.
Data synthesis
Methods for direct treatment comparisons
Initially, standard pairwise meta-analysis was performed for all pairwise comparisons with at least two studies using the random-effects inverse variance model in Stata. 175
Methods for indirect and mixed comparisons
Network meta-analysis integrates direct and indirect evidence for each pairwise comparison to derive relative treatment effects between all competing treatments. We intended to perform NMA using the methodology of multivariate meta-analysis in which different treatment comparisons are handled as different outcomes using the ‘network’ package (which includes the ‘mvmeta’ command) in Stata. 156,176 As a result of the substantial number of treatment nodes and the version of Stata available, however, analysis using the ‘network’ package was not feasible and we performed NMA using graph theoretical methods as described in Rücker. 177,178 To this aim, we used the ‘netmeta’ package in R. 179 We also used available Stata routines to present the evidence base and to illustrate the results. 180 We produced a plot to present jointly the relative ranking of treatments for ‘no clinical improvement’ and ‘total discontinuation rates’, and we used a hierarchical cluster analysis to group interventions in meaningful subsets. 180
Assessment of statistical heterogeneity
Assumptions when estimating the heterogeneity
In pairwise meta-analysis we assumed different heterogeneity variances for each comparison. In NMA, we assumed a common heterogeneity variance across all treatment comparisons in the network.
Measures and tests for heterogeneity
Between-study variance τ2 was estimated in both pairwise and NMA using the DerSimonian and Laird estimator. 175 We assessed statistical heterogeneity based on the magnitude of the estimated parameter. We also compared the magnitude of τ2 with empirical distributions derived in Turner et al. 181 and Rhodes et al. 182
Assessment of statistical inconsistency
Network meta-analysis assumes consistency between various sources of evidence; that means that direct and indirect evidence is expected to be in agreement. However, it might be that the assumption of consistency is violated either in certain parts or in the entire network. We intended to evaluate statistical inconsistency using both local and global methods. In particular, we intended to evaluate the consistency assumption using the loop-specific approach. 183 Employing this method, we would estimate the disagreement between direct and indirect evidence in each closed loop (inconsistency factors).
Moreover, we intended to evaluate inconsistency in the entire network using the design-by-treatment interaction model. 156,184,185 However, there was only one closed loop in the network for the ‘total discontinuation rates’ outcome and, thus, we only judged on inconsistency for this loop using the loop-specific approach.
Investigation of heterogeneity and inconsistency
Several metaregression and subgroup analyses were planned in order to assess the impact of potential effect modifiers on the treatment effects. Our intention was to explore the impact of study and population characteristics fitting network metaregression models in a Bayesian environment using the WinBUGS software version 1.4.3 (MRC Biostatistics Unit, Cambridge, UK) and considering vague prior distributions for the covariates. As these analyses are known to have low power,186,187 their presentation would be of questionable usefulness in the case of very few data.
Sensitivity analysis
We planned to perform the following four sensitivity analyses to ensure the robustness of the NMA results:
-
analysis restricted to studies rated as being at low risk of selection bias
-
analysis restricted to studies rated as being at low or unclear risk of selection bias
-
analysis restricted to studies rated as being at low risk of detection bias
-
analysis restricted to studies rated as being at low or unclear risk of detection bias.
Results
Summary
The primary outcome (no clinical improvement of TD symptoms) was reported in 46 studies (one three-arm study and 45 two-arm studies), including 1560 patients. Total discontinuation rates were reported in 78 studies (one four-arm study, one three-arm study and 76 two-arm studies) with 2965 patients. The number of studies and the number of participants per comparison with available direct data are given in Table 6.
| Comparisons | No clinical improvement of TD symptoms | Total discontinuation rates | ||
|---|---|---|---|---|
| Number of studies | Number of participants | Number of studies | Number of participants | |
| Placebo (with AP continuation) vs.: | ||||
| Benzodiazepine (clonazepam, diazepam) and AP continuation | 1 | 17 | 2 | 41 |
| Branched-chain amino acids and AP continuation | 1 | 52 | 1 | 52 |
| Buspirone and AP continuation | 1 | 42 | 1 | 42 |
| Ceruletide and AP continuation | – | – | 1 | 85 |
| Cholinergic medication (deanol, galantamine, lecithin, meclofenoxate hydrochloride) and AP continuation | 3 | 17 | 11 | 278 |
| Cyproheptadine and AP continuation | – | – | 1 | 42 |
| Dihydrogenated ergot alkaloids/co-dergocrine mesylate and AP continuation | 1 | 28 | 2 | 48 |
| Dopaminergic (amantadine, bromocriptine, carbidopa/levodopa, oxypertine, reserpine, tiapride) and AP continuation | 1 | 20 | 6 | 163 |
| GABA agonist (baclofen, GABA, progabide, sodium valproate, THIP) and AP continuation | 6 | 258 | 6 | 218 |
| Ginkgo biloba standardised extract (EGb-761) and AP continuation | 1 | 157 | 1 | 157 |
| Insulin and AP continuation | 1 | 20 | 1 | 20 |
| Levetiracetam and AP continuation | – | – | 2 | 119 |
| Lithium and AP continuation | 1 | 11 | 1 | 11 |
| MAO inhibitor (isocarboxazid, selegiline) and AP continuation | 1 | 33 | 1 | 33 |
| Melatonin and AP continuation | 2 | 32 | 3 | 54 |
| Noradrenergic (celiprolol, methyldopa) and AP continuation | 1 | 20 | 1 | 35 |
| Oestrogen and AP continuation | 1 | 12 | 1 | 12 |
| Oil of evening primrose and AP continuation | 1 | 16 | 1 | 16 |
| Omega-3 fatty acid and AP continuation | – | – | 1 | 84 |
| Pemoline and AP continuation | 1 | 46 | 1 | 46 |
| Phenylalanine and AP continuation | – | – | 1 | 18 |
| Piracetam and AP continuation | – | – | 1 | 40 |
| Promethazine and AP continuation | 1 | 34 | 1 | 34 |
| Ritanserin and AP continuation | 1 | 10 | 1 | 10 |
| VMAT2 inhibitor (NBI-98854) and AP continuation | 1 | 88 | 1 | 88 |
| Vitamin B6 and AP continuation | 1 | 45 | – | – |
| Vitamin E and AP continuation | 6 | 264 | 13 | 475 |
| 1-Stepholidine and AP continuation | 1 | 57 | 1 | 57 |
| MAO inhibitor AP vs. anticholinergic (biperiden, procyclidine) and AP continuation | 1 | 20 | 1 | 20 |
| Antipsychotic maintenance/TAU (including AP) vs.: | ||||
| Benzodiazepine (clonazepam, diazepam) and AP continuation | 1 | 15 | 1 | 15 |
| Hypnosis or relaxation and AP continuation | 1 | 15 | – | – |
| Antipsychotic reduction (reduced dose FGA) | 2 | 17 | 1 | 8 |
| Active placebo (phenobarbital) and AP continuation vs. benzodiazepine (clonazepam, diazepam) and AP continuation | 1 | 21 | 1 | 21 |
| Switch to haloperidol/unspecified FGA vs.: | ||||
| Dopaminergic (tetrabenazine) and AP withdrawal | 1 | 13 | – | – |
| Dopaminergic (amantadine, bromocriptine, carbidopa/levodopa, oxypertine, reserpine, tiapride) and AP continuation | 1 | 13 | 1 | 13 |
| Switch to amisulpride | – | – | 1 | 55 |
| Switch to clozapine | – | – | 1 | 39 |
| Switch to molindone (FGA) | – | – | 1 | 18 |
| Switch to olanzapine | – | – | 1 | 56 |
| Switch to quetiapine | 1 | 45 | 1 | 45 |
| Switch to thiopropazate (FGA) | 1 | 20 | 1 | 20 |
| Switch to zuclopentixol | 1 | 15 | – | – |
| Dopaminergic (amantadine, bromocriptine, carbidopa/levodopa, oxypertine, reserpine, tiapride) and AP continuation vs. noradrenergic (celiprolol, methyldopa) and AP continuation | 1 | 20 | – | – |
| Switch to risperidone vs.: | ||||
| Switch to olanzapine | 1 | 60 | 2 | 170 |
| Switch to ziprasidone | – | – | 1 | 84 |
| Switch to quetiapine | – | – | 1 | 118 |
| Switch to ziprasidone vs.: | ||||
| Switch to olanzapine | – | – | 1 | 82 |
| Switch to quetiapine | – | – | 1 | 90 |
| Switch to amisulpride vs. switch to olanzapine | – | – | 1 | 57 |
| Switch to quetiapine vs. switch to olanzapine | – | – | 1 | 116 |
| Antipsychotic withdrawal (placebo) vs. switch to risperidone | 1 | 50 | 1 | 50 |
| Anticholinergic withdrawal (biperiden stopped after 1 week) and AP continuation vs. anticholinergic AP | – | – | 1 | 10 |
Pairwise meta-analysis results
From the available comparisons with direct data described in Table 6, we kept data only for those that compared interventions described in Chapter 5, Prioritisation of interventions. Table 7 and Figures 12 and 13 show the available direct estimates for outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’ for comparisons including interventions of priority with at least two studies available. Direct evidence suggests that ‘switch to olanzapine’ appears to be associated with lower discontinuation rates than ‘switch to risperidone’, whereas no important differences were detected between ‘vitamin E and AP continuation’ and ‘placebo with AP continuation’ for the outcome ‘total discontinuation rates’. In terms of no clinical improvement of TD symptoms, ‘vitamin E and AP continuation’ has an insignificant advantage over ‘placebo with AP continuation’. The comparison of ‘antipsychotic maintenance/TAU (including AP)’ versus ‘antipsychotic reduction (reduced dose FGA)’ is not statistically significant, but the overall treatment effect estimate does not rule out a beneficial effect of the second intervention.
| Comparisons | No clinical improvement of TD symptoms | Total discontinuation rates | ||
|---|---|---|---|---|
| OR (95% CI) | τ | OR (95% CI) | τ | |
| Placebo (with AP continuation) vs.: | ||||
| Benzodiazepine (clonazepam, diazepam) and AP continuation | – | – | Excluded | Excluded |
| Vitamin E and AP continuation | 2.28 (0.76 to 6.88) | 0 | 1.02 (0.64 to 1.62) | 0 |
| Antipsychotic maintenance/TAU (including AP) vs. antipsychotic reduction (reduced dose FGA) | 8.41 (0.91 to 77.72) | 0 | – | – |
| Switch to risperidone vs. switch to olanzapine | – | – | 2.17 (1.10 to 4.26) | 0 |
FIGURE 12.
Pairwise meta-analysis results for active treatments vs. placebo (with AP continuation) for outcome ‘no clinical improvement of TD symptoms’ (comparisons with more than two studies, random-effects model, different heterogeneity parameters across comparisons). Heterogeneity was estimated using the method-of-moments estimator.
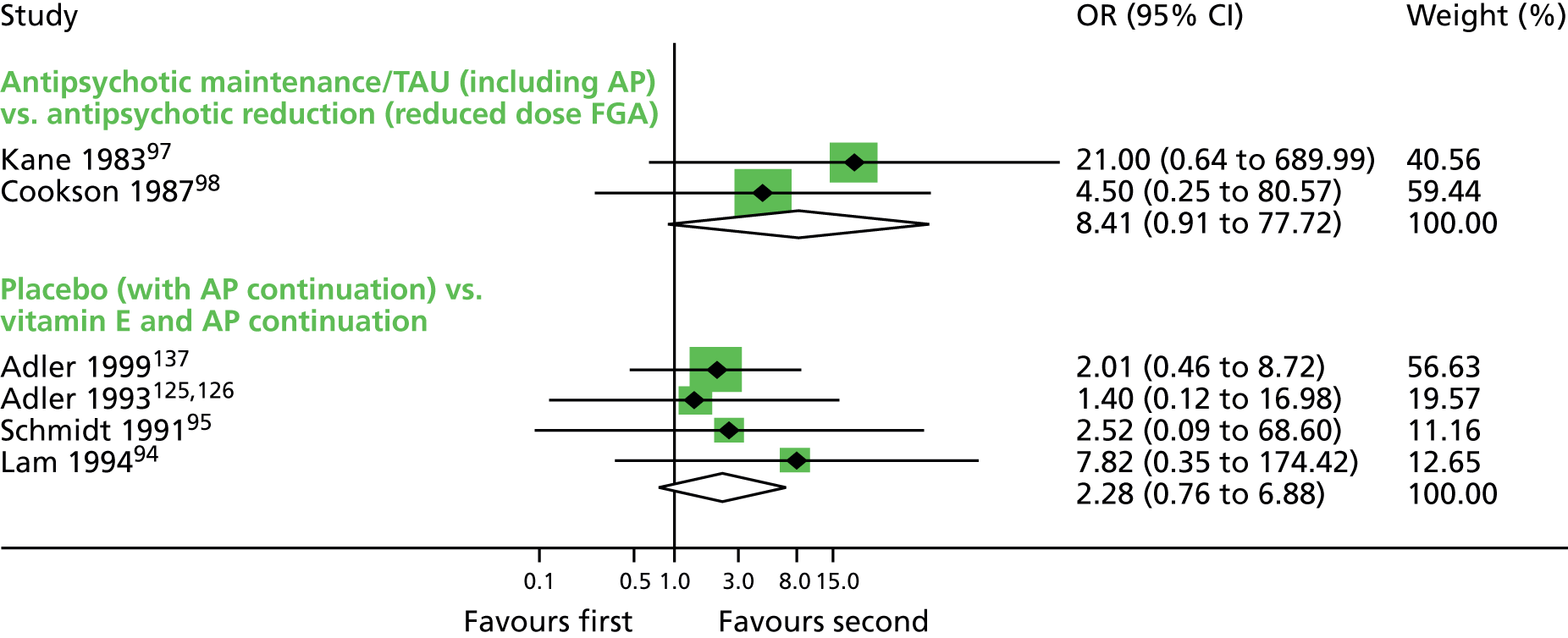
FIGURE 13.
Pairwise meta-analysis results for active treatments vs. placebo (with AP continuation) for outcome ‘total discontinuation rates’ (comparisons with more than two studies, random-effects model, different heterogeneity parameters across comparisons). Heterogeneity was estimated using the method-of-moments estimator.
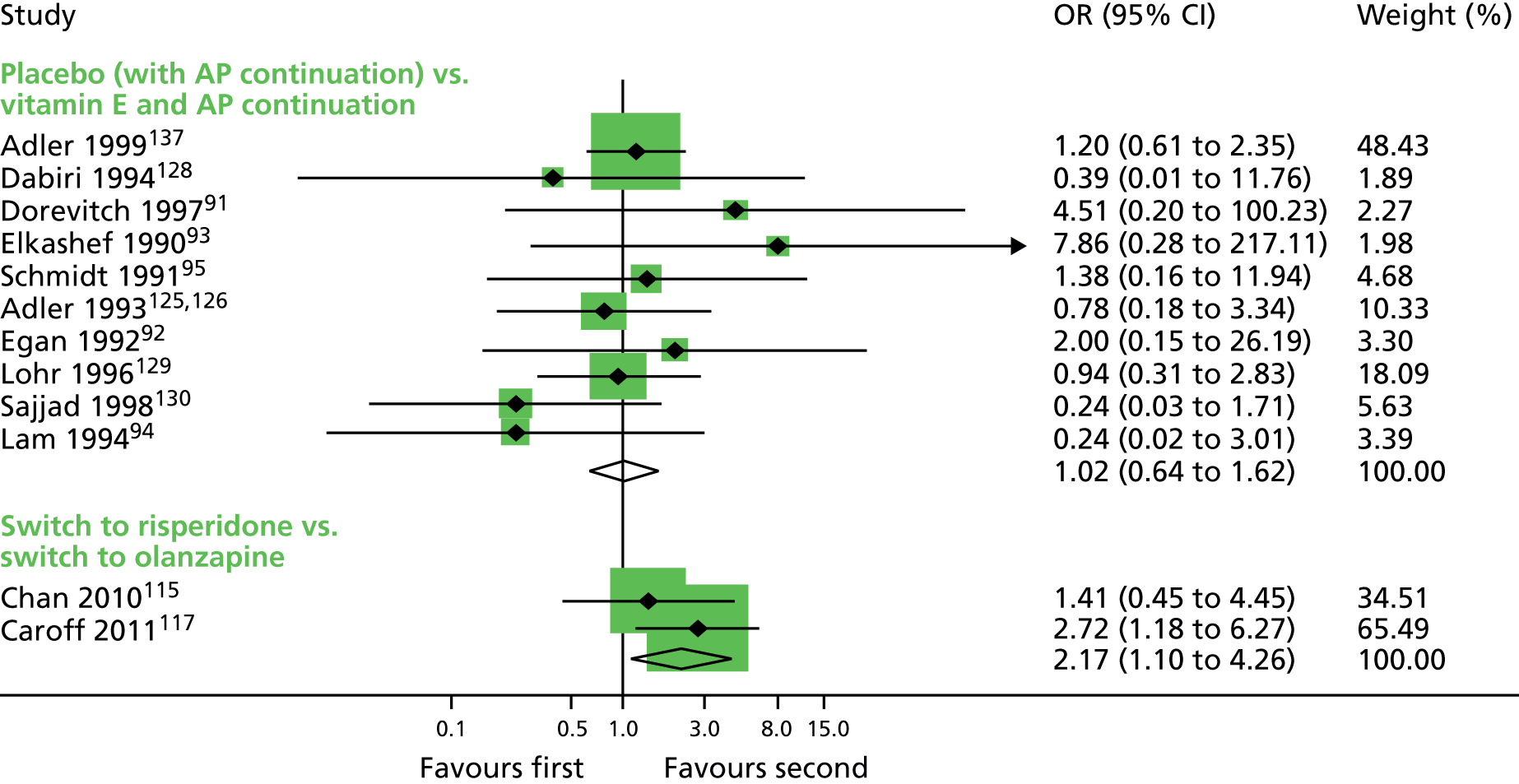
Network meta-analysis results
No clinical improvement of tardive dyskinesia symptoms
Evidence for the outcome ‘no clinical improvement of TD symptoms’ formed two disconnected networks that were analysed separately using NMA. The two formed networks for the outcome ‘no clinical improvement of TD symptoms’ are illustrated in Figure 14 [included treatments: ‘benzodiazepine (clonazepam, diazepam) and AP continuation’, ‘buspirone and AP continuation’, ‘MAO inhibitor (isocarboxazid, selengiline) and AP continuation’, ‘vitamin E and AP continuation’, ‘anticholinergic (biperiden, procyclidine) and AP continuation’, ‘antipsychotic maintenance/TAU (including AP)’, ‘hypnosis or relaxation and AP continuation’, ‘antipsychotic reduction (reduced dose FGA)’] and Figure 15 (included treatments: ‘switch to haloperidol’, ‘switch to thiopropazate’, ‘switch to quetiapine’). Nodes represent available treatments and edges represent available comparisons. Nodes and edges are weighted according to the number of studies involved in each treatment. Two studies105–109,115,116 compared treatments that were connected to neither of the two networks and, thus, were excluded from the NMA. ‘MAO inhibitor (isocarboxazid, selengiline) and AP continuation’ is included in the first subnetwork of Figure 14 despite the fact that it is not in the list of priority interventions as it connects ‘placebo (with AP continuation)’ to ‘anticholinergic (biperiden, procyclidine) and AP continuation’, the relative effectiveness of which is of interest.
FIGURE 14.
Network plot for the first subnetwork for the outcome ‘no clinical improvement of TD symptoms’.

FIGURE 15.
Network plot for the second subnetwork for the outcome ‘no clinical improvement of TD symptoms’.
Table 8 shows the NMA results for the network illustrated in Figure 14 for the outcome ‘no clinical improvement of TD symptoms’. Studies in which all participants were classified as events or non-events in both groups were excluded. The forest plot in Figure 16 shows the ORs of all treatments versus ‘placebo (with AP continuation)’ derived from the NMA. According to Table 8 and Figure 16, the NMA suggests that ‘hypnosis or relaxation and AP continuation’ has the greatest benefit over ‘placebo (with AP continuation)’, whereas ‘buspirone and AP continuation’ and ‘antipsychotic reduction (reduced dose FGA)’ are also more effective than ‘placebo (with AP continuation)’. ‘Anticholinergic (biperiden and procyclidine) and AP continuation’ appears to be less effective than ‘placebo (with AP continuation)’. The results are consistent with the corresponding effect estimates derived from pairwise meta-analysis. It should be noted, however, that any judgements on the relative effectiveness of the treatments are mitigated by the high imprecision associated with most network estimates.
| Intervention | OR (95% CI) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Anticholinergic (biperiden, procyclidine) and AP continuation | Benzodiazepine (clonazepam, diazepam) and AP continuation | Buspirone and AP continuation | Hypnosis or relaxation and AP continuation | MAO inhibitor (isocarboxazid, selengiline) and AP continuation | Antipsychotic maintenance/TAU (including AP) | Antipsychotic reduction (reduced dose FGA) | Placebo (with AP continuation) | Vitamin E and AP continuation | |
| Anticholinergic (biperiden, procyclidine) and AP continuation | – | 0 (0 to 0.09) | 0 (0 to 0.01) | 0 (0 to 0) | 0.01 (0 to 0.33) | 0 (0 to 0.03) | 0 (0 to 0.01) | 0 (0 to 0.01) | 0 (0 to 0.05) |
| Benzodiazepine (clonazepam, diazepam) and AP continuation | 908.73 (11.22 to 73,567.47) | – | 0.17 (0.01 to 2.33) | 0.01 (0 to 0.67) | 12.73 (0.61 to 267.28) | 0.17 (0.01 to 2.56) | 0.02 (0 to 0.67) | 1.75 (0.23 to 13.16) | 0.85 (0.09 to 8.22) |
| Buspirone and AP continuation | 5426.4 (77.13 to 381,770.62) | 5.97 (0.43 to 83) | – | 0.06 (0 to 8.6) | 76 (4.45 to 1298.45) | 1 (0.02 to 44.23) | 0.12 (0 to 9.62) | 10.45 (1.93 to 56.64) | 5.09 (0.7 to 36.88) |
| Hypnosis or relaxation and AP continuation | 86,632 (204.27 to 36,740,218.23) | 95.33 (1.49 to 6100.75) | 15.96 (0.12 to 2190.69) | – | 1213.33 (7.01 to 210,075.5) | 15.89 (0.69 to 365.14) | 1.89 (0.04 to 88.25) | 166.38 (1.64 to 16,971.54) | 81.34 (0.71 to 9268.58) |
| MAO inhibitor (isocarboxazid, selengiline) and AP continuation | 71.4 (3 to 1696.74) | 0.08 (0 to 1.65) | 0.01 (0 to 0.22) | 0 (0 to 0.14) | – | 0.01 (0 to 0.78) | 0 (0 to 0.16) | 0.14 (0.01 to 1.34) | 0.07 (0.01 to 0.82) |
| Antipsychotic maintenance/TAU (including AP) | 5452.36 (30.85 to 963,526.55) | 6 (0.39 to 92.28) | 1 (0.02 to 44.66) | 0.06 (0 to 1.45) | 76.36 (1.28 to 4567.86) | – | 0.12 (0.01 to 1.1) | 10.5 (0.35 to 313.68) | 5.12 (0.15 to 178.19) |
| Antipsychotic reduction (reduced dose FGA) | 45,832.92 (164.1 to 12,801,390.75) | 50.44 (1.49 to 1710.27) | 8.45 (0.1 to 686.61) | 0.53 (0.01 to 24.7) | 641.92 (6.1 to 67,591.57) | 8.41 (0.91 to 77.72) | – | 88.26 (1.52 to 5118.87) | 43.03 (0.65 to 2838.5) |
| Placebo (with AP continuation) | 519.27 (10.48 to 25,740.14) | 0.57 (0.08 to 4.3) | 0.1 (0.02 to 0.52) | 0.01 (0 to 0.61) | 7.27 (0.74 to 71.11) | 0.1 (0 to 2.85) | 0.01 (0 to 0.66) | – | 0.49 (0.17 to 1.37) |
| Vitamin E and AP continuation | 1065.09 (18.8 to 60,350.44) | 1.17 (0.12 to 11.29) | 0.2 (0.03 to 1.42) | 0.01 (0 to 1.4) | 14.92 (1.22 to 182.13) | 0.2 (0.01 to 6.8) | 0.02 (0 to 1.53) | 2.05 (0.73 to 5.75) | – |
FIGURE 16.
Network meta-analysis results for comparisons ‘placebo (with AP continuation) vs. active treatments for outcome ‘no clinical improvement of TD symptoms’ (random-effects model, common heterogeneity parameter across comparisons). Heterogeneity was estimated using the methods-of-moment estimator.

The subnetwork corresponding to Figure 15 is formed by two studies only; a third study that was connected to the network188 was excluded as all participants were classified as events. Thus, we do not present indirect estimates for the particular network as the value of drawing inferences would be doubtful because of the substantially limited data availability. The only study that compared ‘switch to FGA’ with ‘switch to SGA’ for the outcome ‘no clinical improvement’ was Emsley et al. ,110,111 in which an OR of 1.96 (95% CI 0.56 to 6.92) in favour of ‘switch to SGA’ was calculated. This comparison does not benefit from the NMA as it is not connected with the largest subnetwork of Figure 14 and there is no indirect evidence that can be used to strengthen evidence on the relative effectiveness of the two interventions.
Total discontinuation rates
Evidence for the outcome ‘total discontinuation rates’ formed two disconnected networks that were analysed separately using NMA, and are illustrated in Figures 17 and 18. Nodes represent available treatments and edges represent available comparisons. Nodes and edges are weighted according to the number of studies involved in each treatment. ‘MAO inhibitor (isocarboxazid, selengiline) and AP continuation’ is included in the subnetwork of Figure 17 despite the fact that it is not in the list of priority interventions as it connects ‘placebo (with AP continuation)’ to ‘anticholinergic (biperiden, procyclidine) and AP continuation’.
FIGURE 17.
Network plot for the first subnetwork for the outcome ‘total discontinuation rates’.

FIGURE 18.
Network plot for the second subnetwork for the outcome ‘total discontinuation rates’.
Studies in which all participants were classified as events or non-events in both groups were excluded. The forest plot in Figure 19 shows the ORs of all treatments versus ‘placebo (with AP continuation)’ derived from the NMA corresponding to the network plot of Figure 17. Tables 9 and 10 summarise the network estimates corresponding to the networks of Figures 17 and 18, respectively. As is shown in Tables 9 and 10 and Figure 19, most network estimates are highly imprecise (with rather wide CIs), rendering any conclusions on relative treatment effectiveness impractical. No statistically significant differences occur for any treatment versus ‘placebo (with AP continuation)’ in terms of discontinuation rates.
FIGURE 19.
Network meta-analysis results for the comparisons of active treatments vs. placebo for the outcome ‘total discontinuation rates’ (using a random-effects model and using a common heterogeneity parameter across comparisons) corresponding to the subnetwork of Figure 15. Heterogeneity was estimated using the methods-of-moment estimator.
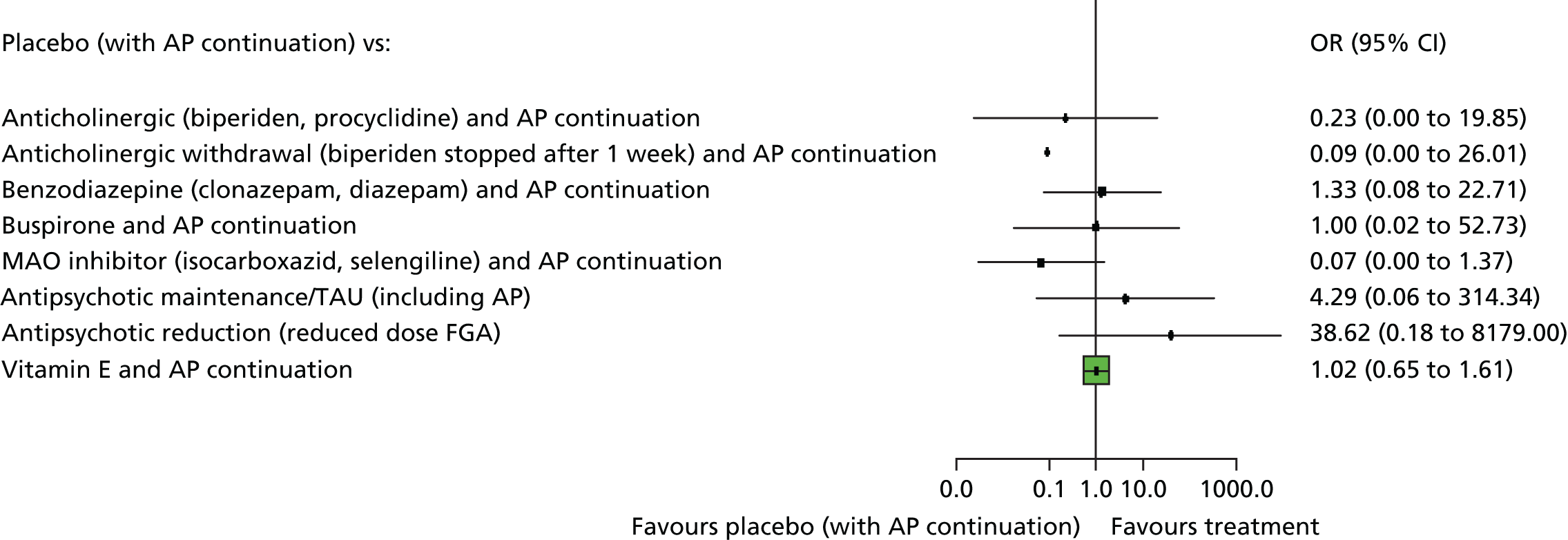
| Intervention | OR (95% CI) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Anticholinergic (biperiden, procyclidine) and AP continuation | Anticholinergic withdrawal (biperiden stopped after 1 week) and AP continuation | Benzodiazepine (clonazepam, diazepam) and AP continuation | Buspirone and AP continuation | MAO inhibitor (isocarboxazid, selengiline) and AP continuation | Antipsychotic maintenance/TAU (including AP) | Antipsychotic reduction (reduced dose FGA) | Placebo (with AP continuation) | Vitamin E and AP continuation | |
| Anticholinergic (biperiden, procyclidine) and AP continuation | – | 0.41 (0.01 to 12.64) | 5.81 (0.03 to 1154.15) | 4.38 (0.01 to 1716.99) | 0.3 (0.01 to 8.33) | 18.79 (0.04 to 9209.95) | 169.14 (0.16 to 180,458.34) | 4.83 (0.05 to 380.6) | 4.47 (0.05 to 397.87) |
| Anticholinergic withdrawal (biperiden stopped after 1 week) and AP continuation | 2.45 (0.08 to 76.13) | – | 14.26 (0.03 to 7831.6) | 10.75 (0.01 to 10,546.15) | 0.74 (0.01 to 87.82) | 46.13 (0.04 to 54,962.09) | 415.15 (0.17 to 985,782.71) | 10.75 (0.04 to 3004.46) | 10.98 (0.04 to 3125.61) |
| Benzodiazepine (clonazepam, diazepam) and AP continuation | 0.17 (0 to 34.21) | 0.07 (0 to 38.53) | – | 0.75 (0.01 to 98.99) | 0.05 (0 to 3.2) | 3.24 (0.13 to 80.99) | 29.12 (0.31 to 2728.71) | 0.75 (0.04 to 12.91) | 0.77 (0.04 to 13.67) |
| Buspirone and AP continuation | 0.23 (0 to 89.54) | 0.09 (0 to 91.28) | 1.33 (0.01 to 174.17) | – | 0.07 (0 to 9.86) | 4.29 (0.01 to 1482.25) | 38.62 (0.05 to 30,257.79) | 1 (0.02 to 52.73) | 1.02 (0.02 to 55.29) |
| MAO inhibitor (isocarboxazid, selengiline) and AP continuation | 3.32 (0.12 to 91.6) | 1.35 (0.01 to 160.25) | 19.26 (0.31 to 1187.7) | 14.52 (0.1 to 2079.47) | – | 62.31 (0.33 to 11,645.88) | 560.82 (1.22 to 258,196.52) | 14.52 (0.73 to 287.84) | 14.83 (0.72 to 304.38) |
| Antipsychotic maintenance/TAU (including AP) | 0.05 (0 to 26.08) | 0.02 (0 to 25.83) | 0.31 (0.01 to 7074) | 0.23 (0 to 80.48) | 0.02 (0 to 3) | – | 9 (0.37 to 220.93) | 0.23 (0 to 17.07) | 0.24 (0 to 17.86) |
| Antipsychotic reduction (reduced dose FGA) | 0.01 (0 to 6.31) | 0 (0 to 5.72) | 0.03 (0 to 3.22) | 0.03 (0 to 20.28) | 0 (0 to 0.82) | 0.11 (0 to 2.73) | – | 0.03 (0 to 5.48) | 0.03 (0 to 5.71) |
| Placebo (with AP continuation) | 0.23 (0 to 19.85) | 0.09 (0 to 26.01) | 1.33 (0.08 to 22.71) | 1 (0.02 to 52.73) | 0.07 (0 to 1.37) | 4.29 (0.06 to 314.34) | 38.62 (0.18 to 8197) | – | 1.02 (0.65 to 1.61) |
| Vitamin E and AP continuation | 0.22 (0 to 19.9) | 0.09 (0 to 25.94) | 1.3 (0.07 to 23.07) | 0.98 (0.02 to 53.02) | 0.07 (0 to 1.38) | 4.2 (0.06 to 315.42) | 37.82 (0.18 to 8167.79) | 0.98 (0.62 to 1.55) | – |
| Intervention | OR (95% CI) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Antipsychotic withdrawal (placebo) | Switch to amisulpride | Switch to clozapine | Switch to haloperidol/unspecified FGA | Switch to molindone | Switch to olanzapine | Switch to quetiapine | Switch to risperidone | Switch to thiopropazate | Switch to ziprasidone | |
| Antipsychotic withdrawal (placebo) | – | 5.48 (0.31 to 97.6) | 0.66 (0.03 to 13.64) | 2.73 (0.36 to 20.93) | 2.73 (0.03 to 247.59) | 3.85 (0.71 to 20.94) | 1.38 (0.23 to 8.08) | 1.83 (0.39 to 8.67) | 13.63 (0.32 to 588.99) | 1.98 (0.31 to 12.77) |
| Switch to amisulpride | 0.18 (0.01 to 3.26) | – | 0.12 (0 to 3.35) | 0.5 (0.04 to 5.8) | 0.5 (0 to 55.39) | 0.7 (0.07 to 7.38) | 0.25 (0.02 to 2.79) | 0.33 (0.03 to 3.79) | 2.49 (0.05 to 136.84) | 0.36 (0.03 to 4.52) |
| Switch to clozapine | 1.51 (0.07 to 30.91) | 8.24 (0.3 to 227.37) | – | 4.11 (0.44 to 38.23) | 4.11 (0.04 to 408.14) | 5.8 (0.45 to 74.88) | 2.07 (0.17 to 24.8) | 2.76 (0.21 to 36.85) | 20.53 (0.43 to 987.99) | 2.98 (0.21 to 43.25) |
| Switch to haloperidol/unspecified FGA | 0.37 (0.05 to 2.81) | 2.01 (0.17 to 23.37) | 0.24 (0.03 to 2.27) | – | 1 (0.02 to 55.8) | 1.41 (0.4 to 4.94) | 0.5 (0.17 to 1.5) | 0.67 (0.18 to 2.51) | 5 (0.21 to 118.65) | 0.73 (0.17 to 3.17) |
| Switch to molindone | 0.37 (0 to 33.29) | 2.01 (0.02 to 223.34) | 0.24 (0 to 24.22) | 1 (0.02 to 55.8) | – | 1.41 (0.02 to 95.3) | 0.5 (0.01 to 32.53) | 0.67 (0.01 to 46.3) | 5 (0.03 to 835.73) | 0.73 (0.01 to 52.66) |
| Switch to olanzapine | 0.26 (0.05 to 1.41) | 1.42 (0.14 to 14.92) | 0.17 (0.01 to 2.23) | 0.71 (0.2 to 2.48) | 0.71 (0.01 to 47.8) | – | 0.36 (0.16 to 0.78) | 0.48 (0.24 to 0.93) | 3.54 (0.12 to 106.65) | 0.51 (0.19 to 1.38) |
| Switch to quetiapine | 0.73 (0.12 to 4.27) | 3.98 (0.36 to 44.28) | 0.48 (0.04 to 5.78) | 1.98 (0.67 to 5.89) | 1.98 (0.03 to 127.88) | 2.8 (1.28 to 6.14) | – | 1.33 (0.57 to 3.12) | 9.91 (0.35 to 282.22) | 1.44 (0.5 to 4.18) |
| Switch to risperidone | 0.55 (0.12 to 2.58) | 2.99 (0.26 to 33.77) | 0.36 (0.03 to 4.84) | 1.49 (0.4 to 5.56) | 1.49 (0.02 to 102.44) | 2.1 (1.07 to 4.12) | 0.75 (0.32 to 1.75) | – | 7.44 (0.24 to 229.66) | 1.08 (0.39 to 3.02) |
| Switch to thiopropazate | 0.07 (0 to 3.17) | 0.4 (0.01 to 22.07) | 0.05 (0 to 2.34) | 0.2 (0.01 to 4.75) | 0.2 (0 to 33.43) | 0.28 (0.01 to 8.51) | 0.1 (0 to 2.87) | 0.13 (0 to 4.15) | – | 0.15 (0 to 4.78) |
| Switch to ziprasidone | 0.5 (0.08 to 3.25) | 2.76 (0.22 to 34.5) | 0.34 (0.02 to 4.86) | 1.38 (0.32 to 6.01) | 1.38 (0.02 to 99.73) | 1.94 (0.72 to 5.21) | 0.69 (0.24 to 2.01) | 0.93 (0.33 to 2.59) | 6.88 (0.21 to 226.2) | – |
Sensitivity analysis merging switch to antipsychotics
As a sensitivity analysis, we further conducted a NMA for the subnetwork of Figure 18 in which all switches to SGAs were merged into a ‘switch to SGA (any)’ treatment node, and all switches to FGAs were merged into a ‘switch to FGA (any)’ treatment node. The Caroff et al. ,117,118 Chan et al. ,115,116 Glazer et al. 189,190 and Kazamatzuri et al. 169 studies were excluded from this analysis as they examined either second- or first-generation antipsychotics only, and thus were representing a single treatment node. The network plot for this analysis is represented in Figure 20. Nodes and edges are weighted according to the number of studies involved in each treatment.
FIGURE 20.
Network plot for the second subnetwork of Figure 18 for the outcome ‘total discontinuation rates’, in which switch to first- and second-generation antipsychotics have been merged to ‘switch to FGA (any)’ and ‘switch to SGA (any)’ treatment nodes, respectively.

As the network presented in Figure 20 comprised only four trials, we did not perform NMA as the validity of the results of such an analysis would be questionable. The comparison ‘switch to FGA (any) versus switch to SGA (any)’ was informed by three studies, resulting in a pairwise meta-analysis OR of 0.54 (95% CI 0.21 to 1.42) in favour of ‘switch to FGA’. There is no indirect evidence to enrich the available information for this comparison and, thus, the use of NMA does not contribute to the knowledge regarding the relative effectiveness of the two interventions.
Comparison of heterogeneity parameters with empirical distributions
For a binary mental health outcome and a ‘non-pharmacological versus any’ comparison type, a median value of 0.13 is suggested for τ. 181 The specific value is greater than our estimation of heterogeneity (0) for both outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’.
Evaluation of inconsistency
We intended to evaluate the consistency assumption using the loop-specific approach in Stata using a common heterogeneity within each loop (but different across loops). 180 We also intended to further assess the assumption of consistency in the entire network simultaneously using the design-by-treatment interaction model in Stata. 156,176 However, for the outcome ‘no clinical improvement of TD symptoms’ all loops were formed by multiarm studies only (consistent by definition) and, thus, consistency could not be evaluated. For the outcome ‘total discontinuation rates’ only one loop was formed for the subnetwork illustrated in Figure 18, ‘switch to olanzapine – switch to quetiapine – switch to haloperidol’; the inconsistency factor using the loop-specific approach was estimated at 1.45, with a (truncated) CI (0 to 4.51) indicating a lack of evidence of inconsistency.
Relative ranking of treatments
Table 11 shows the p-scores of the treatments involved in the outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’ (networks of Figures 14 and 17), which are frequent analogues of SUCRAs. 171,172
| Treatment | No clinical improvement of TD symptoms | Total discontinuation rates |
|---|---|---|
| Hypnosis or relaxation and AP continuation | 0.89 | – |
| Antipsychotic reduction (reduced dose FGA) | 0.85 | 0.90 |
| Buspirone and AP continuation | 0.66 | 0.56 |
| Antipsychotic maintenance/TAU (including AP) | 0.62 | 0.74 |
| Vitamin E and AP continuation | 0.36 | 0.59 |
| Benzodiazepine (clonazepam, diazepam) and AP continuation | 0.35 | 0.61 |
| Placebo (with AP continuation) | 0.24 | 0.58 |
| MAO inhibitor (isocarboxazid, selengiline) and AP continuation | 0.10 | 0.19 |
| Anticholinergic (biperiden, procyclidine) and AP continuation | 0.01 | 0.38 |
| Anticholinergic withdrawal (biperiden stopped after 1 week) and AP continuation | – | 0.29 |
No clinical improvement of tardive dyskinesia symptoms
The p-score value of ‘hypnosis or relaxation and AP continuation’ is 89%, indicating that it is 89% as effective as a treatment that would be ranked always first without uncertainty. ‘Anticholinergic (biperiden, procyclidine) and AP continuation’ appears to be the worst treatment in terms of ‘no clinical improvement of TD symptoms’ as it has a p-score close to 0. These findings are in agreement with the network effect estimates presented in Table 8 and Figure 16.
Total discontinuation rates
‘Antipsychotic reduction (reduced dose FGA)’ has the greatest p-score (90%) in terms of total discontinuation rates. Uncertainty in treatment effects escalates in uncertainty in treatment ranking resulting in many p-scores around 50%.
Clustered ranking plot for the outcomes ‘no clinical improvement of tardive dyskinesia symptoms’ and ‘total discontinuation rates’
In Figure 21 we have ranked treatments according to the outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’. Hierarchical cluster analysis is performed to group the competing treatments. Different colours represent different groups of treatments considering jointly their relative ranking for two outcomes. Treatments that belong to the same group may be considered as being of comparable performance with respect to both outcomes. According to Figure 21, ‘antipsychotic reduction (reduced dose FGA)’ has the highest performance on both outcomes in terms of ranking for the two considered outcomes. ‘Anticholinergic (biperiden, procyclidine) and AP continuation’ and ‘MAO inhibitor (isocarboxazid, selengiline) and AP continuation’ can be considered as the treatments having the worst joint performance for the outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’.
FIGURE 21.
Clustered ranking based on p-scores for the outcomes ‘no clinical improvement of TD symptoms’ and ‘total discontinuation rates’. Hierarchical cluster analysis is performed to group the competing treatments.
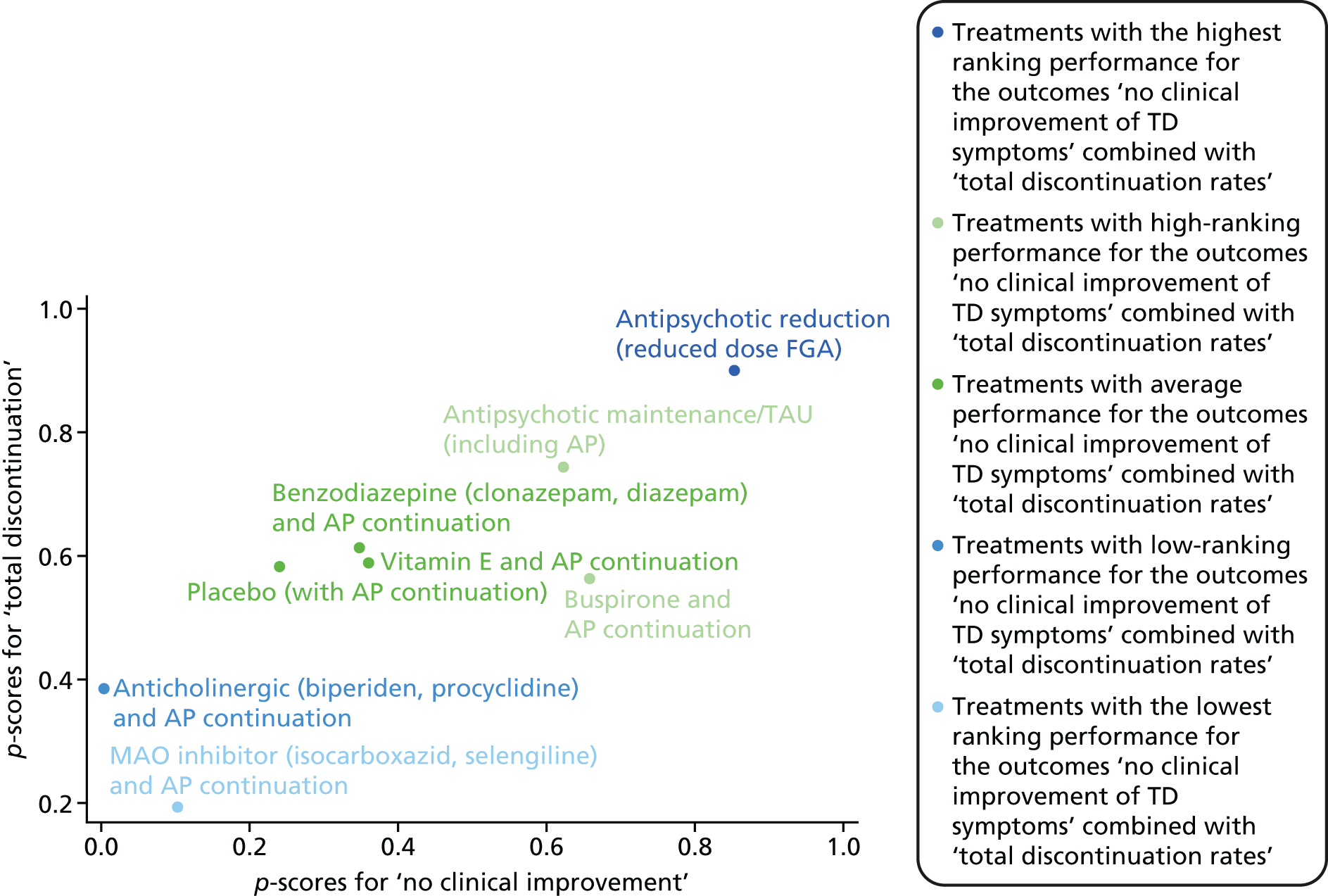
Appendix 5 Studies excluded from the search: reasons for exclusion
Summary
Table 12 summarises the number of studies and references excluded from the review with reasons for exclusion.
| Reason for exclusion | Number of studies (number of references from which studies found) |
|---|---|
| Not RCT or randomised comparison | 170 (201) |
| Randomised but not TD | 88 (103) |
| Randomised, TD, but not stabilised on antipsychotics | 5 (6) |
| Randomised, TD, no usable data reported – authors contacted to confirm lack of data | 15 (19) |
| Randomised, TD, but no usable data reported – no author contact details, study > 20 years old | 8 (12) |
| Randomised, TD, but no separate data reported on minority with TD – authors contacted to confirm lack of data | 3 (3) |
| Randomised, TD, but crossover trial with no separate data reported for phase before crossing over to second treatment – authors contacted to confirm lack of data | 26 (36) |
| Randomised, TD, but crossover trial with no separate data reported for phase before crossing over to second treatment – no author contact details, study > 20 years old | 14 (18) |
| Total | 329 (398) |
References for Appendix 5 and reasons for exclusion
Not randomised controlled trial or randomised comparison
Adler L, Duncan E, Reiter S, Angrist B, Peselow E, Rotrosen J. Effects of calcium-channel antagonists on tardive dyskinesia and psychosis. Psychopharmacol Bull 1988;24:421–5.
Albus M, Naber D, Muller-Spahn F, Douillet P, Reinertshofer T, Ackenheil M. Tardive dyskinesia: relation to computer-tomographic, endocrine, and psychopathological variables. Biol Psychiatry 1985;20:1082–9.
Alexander PE, Van Kammen DP, Bunney WE. Serum calcium and magnesium in schizophrenia: relationship to clinical phenomena and neuroleptic treatment. Br J Psychiatry 1978;133:143–9.
Alexander PE, Van Kammen DP, Bunney WE Jr. Serum calcium and magnesium levels in schizophrenia. II. Possible relationship to extrapyramidal symptoms. Arch Gen Psychiatry 1979;36:1372–7.
Ananth JV, Ban TA, Lehmann HE. An uncontrolled study with thiopropazate in the treatment of persistent dyskinesia. Psychopharmacol Bull 1977;13:9.
Anderson BG, Reker D, Ristich M, Friedman E, Banay-Schwartz M, Volavka J. Lecithin treatment of tardive dyskinesia – a progress report. Psychopharmacol Bull 1982;18:87–8.
Yackulic CF, Anderson BG, Reker D, Webb E, Volavka J. The safety of lecithin diet supplementation in schizophrenic patients. Biol Psychiatry 1982;17:1445–8.
Andersson U, Häggström JE, Nilsson MI, Widerlöv E. Remoxipride, a selective dopamine D2 receptor antagonist, in tardive dyskinesia. Psychopharmacology 1988;94:167–71.
Asher SW, Aminoff MJ. Tetrabenazine and movement disorders. Neurology 1981;31:1051–4.
Asnis GM, Sachar EJ, Langer G, Halpern FS, Fink M. Normal prolactin responses in tardive dyskinesia. Psychopharmacology 1979;66:247–50.
Athanassenas G, Papadopoulos E, Kourkoubas A, Tsitourides S, Gabriel J, Hoïdas S, Frangos E. Serum calcium and magnesium levels in chronic schizophrenics. J Clin Psychopharmacol 1983;3:212–16.
Bartels M, Mezger G, Schmalzing G, Schonle PW. Long-term Treatment of Tardive Dyskinesia with Lecithin. Proceedings of the Symposium der Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie, Nuremberg, Germany, 1981.
Bjorndal N, Casey DE, Gerlach J. Enkephalin, morphine, and naloxone in tardive dyskinesia. Psychopharmacology 1980;69:133–6.
Blaha L. [A strategy to reduce the frequency of early dyskinesias under high-dosed haloperidol treatment.] Arzneimittel Forschung 1980;30:1208.
Blum I, Munitz H, Shalev A, Roberts E. Naloxone may be beneficial in the treatment of tardive dyskinesia. Clin Neuropharmacol 1984;7:265–7.
Blum I, Nisipeanu PF, Roberts E. Naloxone in tardive dyskinesia. Psychopharmacology 1987;93:538.
Bowers MB, Moore D, Tarsy D. Tardive dyskinesia: a clinical test of the supersensitivity hypothesis. Psychopharmacology 1979;61:137–41.
Brambilla F, Gessa GL, Sciascia A, Latina A, Maggioni M, Perna GP, et al. Treatment of drug-resistant chronic schizophrenics with an association of neuroleptics and the calcium antagonist nimodipine. Eur Psychiatry 1992;7:177–82.
Branchey MH, Branchey LB, Bark NM, Richardson MA. Lecithin in the treatment of tardive dyskinesia. Commun Psychopharmacol 1979;3:303–7.
Branchey MH, Branchey LB, Richardson MA. Effects of neuroleptic adjustment on clinical condition and tardive dyskinesia in schizophrenic patients. Am J Psychiatry 1981;138:608–12.
Branchey MH, Branchey LB, Richardson MA. Effects of gradual decrease and discontinuation of neuroleptics on clinical condition and tardive dyskinesia. Psychopharmacol Bull 1981;17:118–20.
Buchanan RW, Kirkpatrick B, Summerfelt A, Hanlon TE, Levine J, Carpenter TW Jr. Clinical predictors of relapse following neuroleptic withdrawal. Biol Psychiatry 1992;32:72–8.
Calne DB, Claveria LE, Teychenne PF, Haskayne L, Lodge-Patch IC. Pimozide in tardive dyskinesia. Trans Am Neurol Assoc 1974;99:166–70.
Campbell M, Adams P, Perry R, Spencer EK, Overall JE. Tardive and withdrawal dyskinesia in autistic children: a prospective study. Psychopharmacol Bull 1988;24:251–5.
Armenteros, JL, Adams PB, Campbell M, Eisenberg ZW. Haloperidol-related dyskinesias and pre- and perinatal complications in autistic children. Psychopharmacol Bull 1995;31:363–9.
Campbell M, Armenteros JL, Malone RP, Adams PB, Eisenberg ZW, Overall JE. Neuroleptic-related dyskinesias in autistic children: a prospective longitudinal study. J Am Acad Child Adolesc Psychiatry 1997;36:835–43.
Caroff SN, Campbell EC, Havey J, Sullivan KA, Mann SC, Gallop R. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry 2001;62:772–5.
Caroff SN, Campbell EC, Havey JC, Sullivan KA, Katz IR, Mann SC. Treatment of tardive dyskinesia with donepezil. J Clin Psychiatry 2001;62:128–9.
Carpenter WT, Rey AC, Stephens JH. Covert dyskinesia in ambulatory schizophrenia. Lancet 1980;2:212–13.
Casey DE, Denny D. Deanol in the treatment of tardive dyskinesia. Am J Psychiatry 1975;132:864–7.
Casey DE, Denney D. Pharmacological characterization of tardive dyskinesia. Psychopharmacology 1977;54:1–8.
Casey DE. Mood alterations during deanol therapy. Psychopharmacology 1979;62:187–91.
Casey DE, Hammerstad JP. Sodium valproate in tardive dyskinesia. J Clin Psychiatry 1979;40:483–5.
Casey DE, Gerlach J, Simmelsgaard H. Sulpiride in tardive dyskinesia. Psychopharmacology 1979;66:73–7.
Casey DE, Gerlach J, Magelund G, Christensen TR. Gamma-acetylenic GABA in tardive dyskinesia. Arch Gen Psychiatry 1980;37:1376–9.
Casey DE, Gerlach J, Magelund G, Christensen TR. Gamma-acetylenic GABA in tardive dyskinesia. Adv Biochem Psychopharmacol 1980;24:577–80.
Casey DE, Korsgaard S, Gerlach J, Jørgensen A, Simmelsgaard H. Effect of des-tyrosine-gamma-endorphin in tardive dyskinesia. Arch Gen Psychiatry 1981;38:158–60.
Casey DE, Korsgaard S, Gerlach J. Des-tyr-gamma-endorphin in Tardive Dyskinesia. Proceedings of the World Congress of Biological Psychiatry, Stockholm, Sweden, 28 June–3 July 1981.
Blum I, Nisipeanu PF, Roberts E, Casey DE, Korsgaard S, Gerlach J. Des-tyr-gamma-endorphin in Tardive Dyskinesia. Proceedings of the 3rd World Congress of Biological Psychiatry, Stockholm, Sweden, 28 June–3 July 1981.
Casey DE. Tardive dyskinesia: what is the natural history? Int Drug Ther Newsl 1983;18:13–16.
Cassady SL, Thaker GK, Tamminga CA. Pharmacologic relationship of anti saccade and dyskinesia in schizophrenic patients. Psychopharmacol Bull 1993;29:236–40.
Chouza C, Romero S, Lorenzo J, Camano JL, Fontana AP, Alterwain P, et al. [Clinical trial of tiapride in patients with dyskinesia (author’s transl).] Sem Hop 1982;58:725–33.
Claveria LE, Teychenne PF, Calne DB, Haskayne L, Petrie A, Pallis CA, Lodge-Patch IC. Tardive dyskinesia treated with pimozide. J Neurol Sci 1975;24:393–401.
Cowen MA, Green M, Bertollo DN, Abbott K. A treatment for tardive dyskinesia and some other extrapyramidal symptoms. J Clin Psychopharmacol 1997;17:190–3.
Crane GE. Tardive dyskinesia in schizophrenic patients treated with psychotropic drugs. Agressologie 1968;9:209–18.
Crane GE, Ruiz P, Kernohan WJ, Wilson W, Royalty N. Effects of drug withdrawal on tardive dyskinesia. Act Nerv Super 1969;11:30–5.
Crane GE. Deanol for tardive dyskinesia. N Engl J Med 1975;292:926.
Curran JP. Management of tardive dyskinesia with thiopropazate. Am J Psychiatry 1973;130:925–7.
Curran DJ, Nagaswami S, Mohan KJ. Treatment of phenothiazine induced bulbar persistent dyskinesia with deanol acetamidobenzoate. Dis Nerv Syst 1975;36:71–3.
Danion JM, Singer L, Tell G, Schechter P. [Gamma-vinyl GABA and tardive dyskinesia: a single-blind study versus placebo.] Ann Med Psychol 1984;142:101–10.
Davis KL, Berger PA, Hollister LE. Letter: Choline for tardive dyskinesia. N Engl J Med 1975;293:152. http://dx.doi.org/10.1056/NEJM197507172930317
Davis KL, Hollister LE, Barchas JD, Berger PA. Choline in tardive dyskinesia and Huntington’s disease. Life Sci 1976;19:1507–15.
Davis KL, Berger PA, Hollister LE. Deanol in tardive dyskinesia. Am J Psychiatry 1977;134:807. http://dx.doi.org/10.1176/ajp.134.7.807
Davis KL, Berger PA. Pharmacological investigations of the cholinergic imbalance hypotheses of movement disorders and psychosis. Biol Psychiatry 1978;13:23–49.
De Silva L, Huang CY. Deanol in tardive dyskinesia. Br Med J 1975;3:466.
Delwaide PJ, Hurlet A. Bromocriptine and buccolinguofacial dyskineasias in patients with senile dementia. A quantitative study. Arch Neurol 1980;37:441–3.
Diamond BI, Borison RL. Basic and clinical studies of neuroleptic-induced supersensitivity psychosis and dyskinesia. Psychopharmacol Bull 1986;22:900–5.
Dixon L, Thaker G, Conley R, Ross D, Cascella N, Tamminga C. Changes in psychopathology and dyskinesia after neuroleptic withdrawal in a double-blind design. Schizophrenia Res 1993;10:267–71.
Duncan E, Adler L, Angrist B, Rotrosen J. Nifedipine in the treatment of tardive dyskinesia. J Clin Psychopharmacol 1990;10:414–16.
Escobar JI, Kemp KF. Letter: Dimethylaminoethanol for tardive dyskinesia. N Engl J Med 1975;292:317–18. http://dx.doi.org/10.1056/NEJM197502062920617
Fahn S. Long Term Treatment of Tardive Dyskinesia with Presynaptically Acting Dopamine-Depleting Agents. In Fahn S, Calne DB, Shoalson I, eds. Advances in Neurology: Experimental Therapeutics of Movement Disorders. New York, NY: Raven Press; 1983. pp. 267–7.
Fahn S. A therapeutic approach to tardive dyskinesia. J Clin Psychiatry 1985;46:19–24.
Falk WE, Wojick JD, Gelenberg AJ. Diltiazem for tardive dyskinesia and tardive dystonia. Lancet 1988;1:824–5.
Fann WE, Lake CR, Gerber CJ, McKenzie GM. Cholinergic suppression of tardive dyskinesia. Psychopharmacologia 1974;37:101–7.
Fann WE, Sullivan JL, Miller RD, McKenzie GM. Deanol in tardive dyskinesia: a preliminary report. Psychopharmacologia 1975;42:135–7.
Fann WE, Stafford JR, Thornby JI, Richman BW. Chronic deanol administration in tardive dyskinesia. Clin Pharmacol Therapeu 1976;19:106.
Ferrari P, Robotti E, Nardini M. [Experimental design of a pilot study on amantadine in the extrapyramidal syndrome induced by neuroleptic drugs.] Boll Chim Farm 1972;111:610–15.
Freeman H, Soni SD. Oxypertine for tardive dyskinesia. Br J Psychiatry 1980;136:522–3.
Freeman HL, Soni SD, Carpenter L. A controlled trial of oxypertine in tardive dyskinesia. International Pharmacopsychiatry 1980;15:281–91.
Freeman HL, Soni SD. Treatment of Tardive Dyskinesia with Oxypertine. Supplement to Progress in Neuropsychopharmacology. Proceedings of the 12th Congress of Collegium Internationale Neuro-psychopharmacologicum, 1980.
Gardos G, Cole JO. Pilot study of cyproheptadine (Periactin) in tardive dyskinesia. Psychopharmacol Bull 1978;14:18–20.
Gardos G, Cole JO, Rapkin RM, LaBrie RA, Baquelod E, Moore P, et al. Anticholinergic challenge and neuroleptic withdrawal. Changes in dyskinesia and symptom measures. Arch Gen Psychiatry 1984;41:1030–5.
Gelenberg AJ, Doller-Wojcik JC, Growdon JH. Choline and lecithin in the treatment of tardive dyskinesia: preliminary results from a pilot study. Am J Psychiatry 1979;136:772–6.
Gerlach J, Thorsen K. The movement pattern of oral tardive dyskinesia in relation to anticholinergic and antidopaminergic treatment. Int Pharmacopsychiatry 1976;11:1–7.
Gerlach J. The relationship between parkinsonism and tardive dyskinesia. Am J Psychiatry 1977;134:781–4.
Gerlach J, Simmelsgaard H. Tardive dyskinesia during and following treatment with haloperidol, haloperidol + biperiden, thioridazine, and clozapine. Psychopharmacology 1978;59:105–12.
Gerlach J, Casey DE. Sulpiride in tardive dyskinesia. Acta Psychiatr Scand Suppl 1984;311:93–102.
Gerlach J, Casey DE. [Dogmatil in delayed dyskinesia.] Sem Hop 1985;61:1369–75.
Bjørndal N, Casey D, Gerlach J. Dopamine antagonist and agonist treatment of tardive dyskinesia. Adv Biochem Psychopharmacol 1980;24:541–5.
Gerlach J, Korsgaard S, Noring U. Primary (Initial) and Secondary (Tardive) Dyskinesia: Effect of Fluperlapine, a New Atypical Neuroleptic Drug. In Usdin E, ed. Catecholamines: Neuropharmacology and Central Nervous System – Therapeutic Aspects. New York, NY: Liss; 1984. pp. 73–8.
Korsgaard S, Noring U, Gerlach J. Fluperlapine in tardive dyskinesia and parkinsonism. Psychopharmacology 1984;84:76–9.
Gibson AC. Sodium valproate and tardive dyskinesia. Br J Psychiatry 1978;133:82.
Gibson AC. Depot Fluphenazine and Tardive Dyskinesia in an Outpatient Population. In Fann WE, Smith RC, Davis JM, Domino EF, eds. Tardive Dyskinesia: Research and Treatment. New York, NY: Spectrum Publications; 1980. pp. 315–24.
Gibson AC. Effect of drug holidays on tardive dyskinesia. In Usdine E, Forrest IS, eds. Phenothiazines and Structurally Related Drugs: Basic and Clinical Studies. New York, NY: Elsevier-North Holland; 1980. pp. 333–6.
Ginsberg DL. Gabapentin reduces neuroleptic-induced tardive dyskinesia. Prim Psychiatry 2003;10:31–2.
Glazer WM, Morre DC, Schooler NR, Brenner LM, Morgenstern H. Tardive dyskinesia: a discontinuation study. Arch Gen Psychiatry 1984;41:623–7.
Glazer WM, Bowers MB, Charney DS, Heninger GR. The effect of neuroleptic discontinuation on psychopathology, involuntary movements, and biochemical measures in patients with persistent tardive dyskinesia. Biol Psychiatry 1989;26:224–33.
Granacher RP, Baldessarini RJ, Cole JO. Deanol for tardive dyskinesia. N Engl J Med 1975;292:926–7.
Grebb JA, Shelton RC, Taylor EH, Bigelow LB. A negative, double-blind, placebo-controlled, clinical trial of verapamil in chronic schizophrenia. Biol Psychiatry 1986;21:691–4.
Growdon JH, Hirsch MJ, Wurtman RJ, Wiener W. Oral choline administration to patients with tardive dyskinesia. N Engl J Med 1977;297:524–7. http://dx.doi.org/10.1056/NEJM197709082971002
Growdon JH. Effects of choline on tardive dyskinesia and other movement disorders. Psychopharmacol Bull 1978;14:55–6.
Wurtman RJ, Growdon JH. Dietary enhancement of CNS neurotransmitters. Hosp Pract 1978;13:71–7.
Haggstrom J. Sulpride in tardive dyskinesia. Curr Therap Res 1980;27:164–9.
Hanus H, Tůma I, Fusek J, Patocka J. [Treatment of tardive dyskinesias with 7-methoxycrine. II.] Sb Ved Pr Lek Fak Karlovy Univerzity Hradci Kralove Suppl 1993;36:47–53.
Heresco-Levy U, Greenberg D, Lerer B, Dasberg H, Brown WA. Trial of maintenance neuroleptic dose reduction in schizophrenic outpatients: two-year outcome. J Clin Psychiatry 1993;54:59–62.
Heresco-Levy U, Greenberg D, Lerer B, Dasberg H, Brown WA. Two-year trial of maintenance neuroleptic dose reduction in schizophrenic out-patients: predictors of relapse. Isr J Psychiatry Relat Sci 1995;32:268–75.
Hogarty GE, Ulrich RF, Mussare F, Aristigueta N. Drug discontinuation among long term, successfully maintained schizophrenic outpatients. Dis Nerv Syst 1976;37:494–500.
Inada T, Beasley CM, JR, Tanaka Y, Walker DJ. Extrapyramidal symptom profiles assessed with the Drug-Induced Extrapyramidal Symptom Scale: comparison with Western scales in the clinical double-blind studies of schizophrenic patients treated with either olanzapine or haloperidol. Int Clin Psychopharmacol 2003;18:39–48.
Inderbitzin LB, Lewine RRJ, SchellerGilkey G, Swofford CD, Egan GJ, Gloersen BA, et al. A double-blind dose-reduction trial of fluphenazine decanoate for chronic, unstable schizophrenic patients. Am J Psychiatry 1994;151:1753–9.
Ingram NA, Newgreen DB. The use of tacrine for tardive dyskinesia. Am J Psychiatry 1983;140:1629–31.
Izumi K, Tominaga H, Koja T, Nomoto M, Shimizu T, Sonoda H, et al. Meclofenoxate therapy in tardive dyskinesia: a preliminary report. Biol Psychiatry 1986;21:151–60.
Jeste DV, Olgiati SG, Ghali AY. Masking of tardive dyskinesia with four times-a-day administration of chlorpromazine. Dis Nerv Syst 1977;38:755–8.
Jeste DV, Potkin SG, Sinha S, Feder S, Wyatt RJ. Tardive dyskinesia – reversible and persistent. Arch Gen Psychiatry 1979;36:585–90.
Johnson DAW, Pasterski G, Ludlow JM, Street K, Taylor RDW. The discontinuance of maintenance neuroleptic therapy in chronic schizophrenic patients: drug and social consequences. Acta Psychiatr Scand 1983;67:339–52.
Jus K, Jus A, Gautier J, Villeneuve A, Pires P, Pineau R, Villeneuve R. Studies on the action of certain pharmacological agents on tardive dykinesia and on the rabbit syndrome. Int J Clin Pharmacol 1974;9:138–45.
Jus A, Jus K, Fontaine P. Long term treatment of tardive dyskinesia. J Clin Psychiatry 1979;40:72–7.
Kalachnik JE, Harder SR, Kidd-Nielsen P, Errickson E, Doebler M, Sprague RL. Persistent tardive dyskinesia in randomly assigned neuroleptic reduction, neuroleptic nonreduction, and no-neuroleptic history groups: preliminary results. Psychopharmacol Bull 1984;20:27–32.
Kane JM, Woerner MG, Pollack S, Safferman AZ, Lieberman JA. Does clozapine cause tardive dyskinesia? J Clin Psychiatry 1993;54:327–30.
Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry 1972;27:95–9.
Kinon BJ, Jeste DV, Kollack-Walker S, Stauffer V, Liu-Seifert H. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry 2004;28:985–96.
Kinon BJ, Stauffer VL, Wang L, Thi K, Niewoehner J, Kollack-Walker S. Olanzapine Improves Dyskinesia in Patients with Schizophrenia. International Congress on Schizophrenia Research, Colorado Springs, CO, USA, 29 March–2 April 2003.
Kinon B, Stauffer VL, Wang L, Thi KT, Kollack-Walker S. Olanzapine improves tardive dyskinesia in patients with schizophrenia in a controlled prospective study. Int J Neuropsychopharmacol 2002;5(Suppl. 1):165.
Kinon BJ, Stauffer VL, Wang L, Thi KT. Olanzapine improves tardive dyskinesia in patients with schizophrenia. Schizophren Res 2002;53(Suppl. 1):191.
Kinon BJ, Stauffer VL, Wang L, Thi KT. Olanzapine Improves Tardive Dyskinesia in Patients with Schizophrenia. 155th Annual Meeting of the American Psychiatric Association, Philadelphia, PA, USA, 18–23 May 2002.
Kirch D, Hattox S, Bell J, Murphy R, Freedman R. Plasma homovanillic acid and tardive dyskinesia during neuroleptic maintenance and withdrawal. Psychiatry Res 1983;9:217–23.
Klawans HL, Rubovits R. Effect of cholinergic and anticholinergic agents on tardive dyskinesia. J Neurol Neurosurg Psychiatr 1974;37:941–7.
Kollack-Walker S, Kinon BJ, Stauffer VL, Wang L, Thi KT. Olanzapine improves tardive dyskinesia in patients with schizophrenia. Schizophren Res 2003;60:359.
König P, Chwatal K, Havelec L, Riedl F, Schubert H, Schultes H. Amantadine versus biperiden: a double-blind study of treatment efficacy in neuroleptic extrapyramidal movement disorders. Neuropsychobiology 1996;33:80–4.
Korsgaard S, Casey DE, Damgaard Pedersen NE, Jørgensen A, Gerlach J. Vasopressin in anergic schizophrenia. A cross-over study with lysine-8-vasopressin and placebo. Psychopharmacology 1981;74:379–82.
Korsgaard S, Casey DE, Gerlach J, Hetmar O, Kaldan B, Mikkelsen LB. The effect of tetrahydroisoxazolopyridinol (THIP) in tardive dyskinesia: a new gamma-aminobutyric acid agonist. Arch Gen Psychiatry 1982;39:1017–21.
Korsgaard S, Casey DE, Gerlach J. Effect of gamma-vinyl GABA in tardive dyskinesia. Psychiatry Res 1983;8:261–9.
Korsgaard S, Casey D, Gerlach J. GABA Agonist Treatment of Tardive Dyskinesia. Proceedings of the 13th Congress of Collegium Internationale Neuropsychopharmacologicum, Jerusalem, Israel, 20–25 June 1982.
Kumar BB. Treatment of tardive dyskinesia with deanol. Am J Psychiatry 1976;133:978.
Lambert PA, Cantiniaux P, Chabannes JP, Tell GP, Schechter PJ, Koch-Weser J. [Therapeutic trial of gamma-vinyl GABA, an inhibitor of GABA transaminase, in tardive dyskinesias induced by neuroleptics.] Encephale 1982;8:371–6.
Laterre EC, Fortemps E. Letter: Deanol in spontaneous and induced dyskinesias. Lancet 1975;1:1301.
Leblanc G, Cormier H, Gagné MA, Vaillancourt S. Effects of neuroleptic reduction in schizophrenic outpatients receiving high doses. Can J Psychiatry 1994;39:223–9.
Leblhuber F. Treatment of permanent tardive dyskinesia with tiapride, a selective D2-receptor blocking agent. Clin Neuropharmacol 1987;10:458–61.
Lejoyeux M, Gorwood P, Stalla-Bourdillon A, Adès J. [Translation and application of the Simpson and Angus Scale of Extrapyramidal Symptoms.] Encephale 1993;19:17–21.
Levy MI, Davis BM, Mohs RC, Kendler KS, Mathé AA, Trigos G, et al. Apomorphine and schizophrenia. Treatment, CSF, and neuroendocrine responses. Arch Gen Psychiatry 1984;41:520–4.
Lieberman JA, Saltz BL, Johns CA, Pollack S, Kane JM. Clozapine effects on tardive dyskinesia. Psychopharmacol Bull 1989;25:57–62.
Lieberman JA, Saltz BL, Johns CA, Pollack S, Borenstein M, Kane J. The effects of clozapine on tardive dyskinesia. Br J Psychiatry 1991;158:503–10.
Lin CC, Chen JY, Bai YM, Chen TT, Wang YC, Liou YJ, Lin WK. Remission of tardive dyskinesia following the switch from clozapine to zotepine: 2-year follow-up. Eur Neuropsychopharmacol 2006;16(Suppl. 4):410.
Littrell K, Magill AM. The effect of clozapine on preexisting tardive dyskinesia. J Psychosoc Nurs Ment Health Serv 1993;31:14–18.
Lonowski DJ, Sterling FE, King HA. Electromyographic assessment of dimethylaminoethanol (deanol) in treatment of tardive dyskinesia. Psychol Rep 1979;45:415–19.
MacKay AV, Sheppard GP, Saha BK, Motley B, Johnson AL, Marsden CD. Failure of lithium treatment in established tardive dyskinesia. Psychol Med 1980;10:583–7.
Marsalek M, Filip V, Praskova H, Karen P. An open trial with 7-methoxytacrine in tardive dyskinesia. Eur Neuropsychopharmacol 1994;4:369.
Meco G, Bedini L, Bonifati V, Sonsini U. Risperidone in the treatment of chronic schizophrenia with tardive dyskinesia. A single-blind crossover study versus placebo. Curr Therap Res 1989;46:876–83.
Mehta D, Mehta S, Mathew P. Failure of deanol in treating tardive dyskinesia. Am J Psychiatry 1976;133:1467.
Miller DD, Flaum M, Arndt S, Fleming F, Andreasen NC. Effect of antipsychotic withdrawal on negative symptoms in schizophrenia. Neuropsychopharmacology 1994;11:11–20.
Monteleone P, Maj M, Ariano MG, Iovino M, Fiorenza L, Steardo L. Prolactin response to sodium valproate in schizophrenics with and without tardive dyskinesia. Psychopharmacology 1988;96:223–6.
Moore DC, Bowers MB. Identification of a subgroup of tardive dyskinesia patients by pharmacologic probes. Am J Psychiatry 1980;137:1202–5.
Morselli PL, Fournier V, Bossi L, Musch B. Clinical activity of GABA agonists in neuroleptic- and L-dopa-induced dyskinesia. Psychopharmacology Suppl 1985;2:128–36.
Nagao T, Ohshimo T, Mitsunobu K, Sato M, Otsuki S. Cerebrospinal fluid monoamine metabolites and cyclic nucleotides in chronic schizophrenic patients with tardive dyskinesia or drug-induced tremor. Biol Psychiatry 1979;14:509–23.
Nasrallah HA, Dunner FJ, Smith RE, McCalley-Whitters M, Sherman AD. Variable clinical response to choline in tardive dyskinesia. Psychol Med 1984;14:697–700.
Nasrallah HA, Dunner FJ, McCalley-Whitters M. A placebo-controlled trial of valproate in tardive dyskinesia. Biol Psychiatry 1985;20:205–8.
Nechifor M, Vaideanu C, Palamaru I, Borza C, Mindreci I. The influence of some antipsychotics on erythrocyte magnesium and plasma magnesium, calcium, copper and zinc in patients with paranoid schizophrenia. J Am Coll Nutr 2004;23:549S–51S.
Noring U, Povlsen UJ, Casey DE, Gerlach J. Effect of a cholinomimetic drug (RS 86) in tardive dyskinesia and drug-related parkinsonism. Psychopharmacology 1984;84:569–71.
Pai YM, Yu SC, Lin CC. Risperidone in Reducing Tardive Dyskinesia: A Double-Blind, Placebo-Controlled Study. Annual Meeting of the American Psychiatric Association, New Orleans, LA, USA, 5–10 May 2001.
Pai Y-M, Yu S-C, Lin C-C. Risperidone in Reducing Tardive Dyskinesia: A Double-Blind, Placebo-Controlled Study. 155th Annual Meeting of the American Psychiatric Association, Philadelphia, PA, USA, 18–23 May 2002.
Paulson GW, Rizvi CA, Crane GE. Tardive dyskinesia as a possible sequel of long-term therapy with phenothiazines. Clin Pediatr 1975;14:953–5.
Peacock L, Solgaard T, Lublin H, Gerlach J. Clozapine versus typical antipsychotics. A retro- and prospective study of extrapyramidal side effects. Psychopharmacology 1996;124:188–96.
Peet M, Laugharne J, Rangarajan N, Reynolds GP. Tardive dyskinesia, lipid peroxidation and sustained amelioration with vitamin E treatment. Int Clin Psychopharmacol 1993;8:151–3.
Peet M, Laugharne J, Rangarajan N, Reynolds G. Tardive dyskinesia. Hosp Community Psychiatry 1993;44:795.
Prange A, Wilson JC, Morris CE, Hall CD. Preliminary experience with tryptophan and lithium in the treatment of tardive dyskinesia. Psychopharmacol Bull 1973;9:36–7.
Price WA, Pascarzi GA. Use of verapamil to treat negative symptoms in schizophrenia. J Clin Psychopharmacol 1987;7:357.
Pyke J, Seeman MV. Neuroleptic-free intervals in the treatment of schizophrenia. Am J Psychiatry 1981;138:1620–1.
Quitkin F, Rifkin A, Gochfeld L, Klein DF. Tardive dyskinesia: are first signs reversible? Am J Psychiatry 1977;134:84–7.
Rapoport J, Kumra S, Jacobsen LK. The Spectrum of Extrapyramidal Symptoms in Children and Young Adults. 150th Annual Meeting of the American Psychiatric Association, San Diego, CA, USA, 17–22 May 1997.
Ray R, Ramakrishnan N, Rao BS. Oral choline in tardive dyskinesia. Indian J Med Res 1982;76:628–31.
Reda FA, Scanlan JM, Kemp K, Escobar JI. Letter: Treatment of tardive dyskinesia with lithium carbonate. N Engl J Med 1974;291:850.
Reda FA, Escobar JI, Scanlan JM. Lithium carbonate in the treatment of tardive dyskinesia. Am J Psychiatry 1975;132:560–2.
Reker D, Anderson B, Yackulic C, Cooper TB, Banay-Schwartz M, Leon C, Volavka J. Naloxone, tardive dyskinesia, and endogenous beta-endorphin. Psychiatry Res 1982;7:321–4.
Volavka J, Anderson B, Koz G. Naloxone and naltrexone in mental illness and tardive dyskinesia. Ann N Y Acad Sci 1982;398:97–102.
Rektor J. [The cholinergic system in the pathophysiology of tardive dyskinesia.] Cesk Psychiatr 1988;84:289–96.
Ringwald E. [Dopamine-receptor stimulators and neuroleptic-induced dyskinesia (author’s transl).] Pharmakopsychiatr Neuropsychopharmakol 1978;11:294–8.
Rondot P, Bathien N. Movement disorders in patients with coexistent neuroleptic-induced tremor and tardive dyskinesia: EMG and pharmacological study. Adv Neurol 1987;45:361–6.
Bathien N, Sevestre P, Rondot P, Morselli PL, Van Landeghem V. The Effect of Progabide, a Specific GABAergic Agonist, on Neuroleptic-Induced Tardive Dyskinesia – A Result of a Pilot Study. Proceedings of the 13th Collegium Internationale Neuropsychopharmacologicum Congress, Jerusalem, Israel, 20–25 June 1982.
Ross JL, Mackenzie TB, Hanson DR, Charles CR. Diltiazem for tardive dyskinesia. Lancet 1987;1:268.
Roxburgh PA. Treatment of persistent phenothiazine-induced oral dyskinesia. Br J Psychiatry 1970;116:277–80.
Sarbulescu A, Alexandrescu L, Georgescu M. Comparative study of two benzodiazepines (‘diazepam’ versus ‘nitrazepam’) in the treatment of postneuroleptic tardive dyskinesia. Rev Roum Neurol Psychiatr 1986;24:189–93.
Schultz SK, Miller DD, Arndt S, Ziebell S, Gupta S, Andreasen NC. Withdrawal-emergent dyskinesia in patients with schizophrenia during antipsychotic discontinuation. Biol Psychiatry 1995;38:713–19.
Seeman MV. Tardive dyskinesia: two-year recovery. Compr Psychiatry 1981;22:189–92.
Simpson GM, Branchey MH, Lee JH, Voitashevsky A, Zoubok B. Lithium in tardive dyskinesia. Pharmakopsychiatr Neuropsychopharmakol 1976;9:76–80.
Simpson GM, Zoubok B, Lee JH. An early clinical and toxicity trial of EX 11-582A in chronic schizophrenia. Curr Ther Res Clin Exp 1976;19:87–98.
Simpson GM, Lee JH, Shrivastava RK, Branchey MH. Baclofen in the treatment of tardive dyskinesia and schizophrenia. Psychopharmacol Bull 1978;14:16–18.
Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology 1978;56:75–80.
Singh MM, Nasrallah HA, Lal H, Pitman RK, Becker RE, Kucharski T, et al. Treatment of tardive dyskinesia with diazepam: indirect evidence for the involvement of limbic, possibly GABA-ergic mechanisms. Brain Res Bull 1980;5(Suppl. 2):673–80.
Singh MM, Becker RE, Pitman RK, Nasrallah HA, Lal H, Dufresne RL, et al. Diazepam-induced changes in tardive dyskinesia: suggestions for a new conceptual model. Biol Psychiatry 1982;17:729–42.
Singh MM, Becker RE, Pitman RK, Nasrallah HA, Lal H. Sustained improvement in tardive dyskinesia with diazepam: indirect evidence for corticolimbic involvement. Brain Res Bull 1983;11:179–85.
Small JG, Milstein V, Marhenke JD, Hall DD, Kellams JJ. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry 1987;48:263–7.
Smith RC, Tamminga CA, Haraszti J, Pandey GN, Davis JM. Effects of dopamine agonists in tardive dyskinesia. Am J Psychiatry 1977;134:763–8.
Smith JS, Kiloh LG. Six month evaluation of thiopropazate hydrochloride in tardive dyskinesia. J Neurol Neurosurg Psychiatr 1979;42:576–9.
Soni SD, Freeman HL, Hussein EM. Oxypertine in tardive dyskinesia: an 8-week controlled study. Br J Psychiatry 1984;144:48–52.
Spivak B, Schwartz B, Radwan M, Weizman A. alpha-Tocopherol treatment for tardive dyskinesia. J Nerv Ment Dis 1992;180:400–1.
Spivak B, Mester R, Abesgaus J, Wittenberg N, Adlersberg S, Gonen N, Weizman A. Clozapine treatment for neuroleptic-induced tardive dyskinesia, parkinsonism, and chronic akathisia in schizophrenic patients. J Clin Psychiatry 1997;58:318–22.
Stahl SM, Thornton JE, Simpson ML, Berger PA, Napoliello MJ. Gamma-vinyl-GABA treatment of tardive dyskinesia and other movement disorders. Biol Psychiatry 1985;20:888–93.
Tamminga CA, Smith RC, Ericksen SE, Chang S, Davis JM. Cholinergic influences in tardive dyskinesia. Am J Psychiatry 1977;134:769–74.
Tell GP, Schechter PJ, Koch-Weser J, Cantiniaux P, Chabannes JP, Lambert PA. Effects of gamma-vinyl GABA. N Engl J Med 1981;305:581–2.
Turek I, Kurland AA, Hanlon TE, Bohm M. Tardive dyskinesia: its relation to neuroleptic and antiparkinson drugs. Br J Psychiatry 1972;121:605–12.
Villeneuve A, Böszörményi Z. Treatment of drug-induced dyskinesias. Lancet 1970;1:353–4.
Villeneuve A, Cazejust T, Côté M. Estrogens in tardive dyskinesia in male psychiatric patients. Neuropsychobiology 1980;6:145–51.
Volavka J, O’Donnell J, Muragali R, Anderson BG, Gaztanaga P, Boggiano W, et al. Lithium and lecithin in tardive dyskinesia: an update. Psychiatry Res 1986;19:101–4.
Wang D, Xie F, Gao Z. Persistent tardive dyskinesia treated with clonazepam. Chin J Pharmacoepidemiol 2002;11:284–6.
Wirshing WC, Freidenberg DL, Cummings JL, Bartzokis G. Effect of anticholinergic agents on patients with tardive dyskinesia and concomitant drug-induced parkinsonism. J Clin Psychopharmacol 1989;9:407–11.
Wolf MA, Bailly L, Diener JM, Martinet JP, Peretti S, Garneau Y. Sevrage neuroleptique complet chez des patients schizophrenes presentant une symptomatologie intense et resistant au traitement. Encephale 1991;17:255–61.
Wright P, Tollefson GD, Beasley CM, Tamura RN, Tran PV, Potvin JII. A blinded, controlled, long-term study of the comparative incidence of treatment-emergent tardive dyskinesia with olanzapine or haloperidol. Schizophren Res 1998;29:206.
Zander KJ, Fischer B, Zimmer R, Ackenheil M. Long-term neuroleptic treatment of chronic schizophrenic patients: clinical and biochemical effects of withdrawal. Psychopharmacology 1981;73:43–7.
Zapletálek M, Hanus H, Fusek J, Hrdina V. First experience with the application of 7-methoxytacrine to psychiatric patients. Act Nerv Super 1989;31:305–6.
Zarebinski JM, Royds JN. Sulpiride in tardive dyskinesia. S Afr Med J 1990;78:374–5.
Zwanikken GJ, Oei TT, Kimya S, Amery W. Safety and efficacy of prolonged treatment with Tremblex (dexetimide), an antiparkinsonian agent. A controlled study. Acta Psychiatr Belg 1976;76:467–9.
Randomised but not tardive dyskinesia
Adler LA, Angrist B, Rotrosen J. Metoprolol versus propranolol. Biol Psychiatry 1990;27:673–5.
Apseloff G, Mullet D, Wilner KD, Anziano RJ, Tensfeldt TG, Pelletier SM, Gerber N. The effects of ziprasidone on steady-state lithium levels and renal clearance of lithium. Br J Clin Pharmacol 2000;49(Suppl. 1):61–4.
Barnes T. A Prospective Study of Tardive Dyskinesia in the Elderly; Single-Blind Comparison of Amisulpride and Risperidone. Leeds: National Research Register; 2002.
Bisol LW, Brunstein MG, Ottoni GL, Ramos FLP, Borba DL, Daltio CS, et al. Is flunarizine a long-acting oral atypical antipsychotic? A randomized clinical trial versus haloperidol for the treatment of schizophrenia. J Clin Psychiatry 2008;69:1572–9.
Bitter I, Slabber M, Pretorius J, Bartko GY, Danics Z, Dossenbach M, et al. Olanzapine versus clozapine in patients non responsive or intolerant to standard acceptable treatment of schizophrenia. Int J Neuropsychopharmacol 2000;3(Suppl. 1):141.
Brecher M, Okamoto A, Napolitano J, Kane JM, Risperidone Study Group. Low frequency of tardive dyskinesia in elderly patients with dementia exposed to risperidone for up to one year. Am J Geriatr Psychiatry 1999;7:53–4.
Brecher M, Okamoto A, Napolitano J, Kane JM, The Risperidone Study Group. Low frequency of tardive dyskinesia in elderly patients with dementia exposed to risperidone for up to one year. Schizophren Res 1999;1–3:362.
Brecher M. Follow-up Study of Risperidone in the Treatment of Patients with Dementia: Interim Results on Tardive Dyskinesia and Dyskinesia Severity. Proceedings of the 11th European College of Neuropsychopharmacology Congress, Paris, France, 31 October–4 November 1998.
Caine ED, Polinsky RJ, Kartzinel R, Ebert MH. The trial use of clozapine for abnormal involuntary movement disorders. Am J Psychiatry 1979;136:317–20.
Carman JS, Wyatt RJ. Calcium: pacesetting the periodic psychoses. Am J Psychiatry 1979;136:1035–9.
Chaplin R, Timehin C. Informing patients about tardive dyskinesia: four-year follow up of a trial of patient education. Aust N Z J Psychiatry 2002;36:99–103.
Chaplin R, Zipursky RB. An educational session on tardive dyskinesia increased patients’ knowledge at 6 months without affecting compliance or clinical stability. Evidence-Based Med 1998;3:153.
Chaplin R, Kent A. Informing patients about tardive dyskinesia. Controlled trial of patient education. Br J Psychiatry 1998;172:78–81.
Chiu SS. A Placebo-Controlled Cross Study of Panax ginseng in Augmentation of Antipsychotics in 60 Partially Treatment Responsive Patients with Schizophrenia. 2006. URL: https://clinicaltrials.gov/ct2/show/NCT00401089 (accessed 6 June 2017).
Chouinard G, Annable L, Kropsky M. A double-blind controlled study of pipothiazine palmitate in the maintenance treatment of schizophrenic outpatients. J Clin Pharmacol 1978;18:148–54.
Chouinard G, Annable L, Ross-Chouinard A, Kropsky ML. Ethopropazine and benztropine in neuroleptic-induced parkinsonism. J Clin Psychiatry 1979;40:147–52.
Chouinard G, Annable L, Campbell W. A randomized clinical trial of haloperidol decanoate and fluphenazine decanoate in the outpatient treatment of schizophrenia. J Clin Psychopharmacol 1989;9:247–53.
Chouinard G, Vainer JL, Belanger MC, Turnier L, Beaudry P, Roy JY, et al. Risperidone and clozapine in the treatment of drug-resistant schizophrenia and neuroleptic-induced supersensitivity psychosis. Progr Neuropsychopharmacol Biol Psychiatry 1994;18:1129–41.
Cookson JC. Side effects during long-term treatment with depot antipsychotic medication. Clin Neuropharmacol 1991;14(Suppl. 2):24–30.
Cortese L, Caligiuri MP, Williams R, Schieldrop P, Manchanda R, Malla A, et al. Reduction in neuroleptic-induced movement disorders after a switch to quetiapine in patients with schizophrenia. J Clin Psychopharmacol 2008;28:69–73.
Crane GE. High doses of trifluperazine and tardive dyskinesia. Arch Neurol 1970;22:176–80.
Curson DA, Barnes TR, Bamber RW, Platt SD, Hirsch SR, Duffy JC. Long-term depot maintenance of chronic schizophrenic out-patients: the seven year follow-up of the Medical Research Council fluphenazine/placebo trial. II. The incidence of compliance problems, side-effects, neurotic symptoms and depression. Br J Psychiatry 1985;146:469–74.
Davidson M, Harvey PD, Vervarcke J, Gagiano CA, De Hooge JD, Bray G, et al. A long-term, multicenter, open-label study of risperidone in elderly patients with psychosis. On behalf of the Risperidone Working Group. Int J Geriatr Psychiatry 2000;15:506–14.
DiMascio A, Bernardo DL, Greenblatt DJ, Marder JE. A controlled trial of amantadine in drug-induced extrapyramidal disorders. Arch Gen Psychiatry 1976;33:599–602.
DiMascio A, Bernardo DL, Greenblatt DJ, Marder JE. A controlled trial of amantadine in drug-induced extrapyramidal disorders. Psychopharmacol Bull 1977;13:31–3.
Dorfman-Etrog P, Hermesh H, Prilipko L, Weizman A, Munitz H. The effect of vitamin E addition to acute neuroleptic treatment on the emergence of extrapyramidal side effects in schizophrenic patients: an open label study. Eur Neuropsychopharmacol 1999;9:475–7.
Dose M. [The significance of calcium antagonists and anticonvulsants for the pharmacotherapy of psychoses.] Habilitationsschrift der Technischen Universitat Munchen 1991;1991:11–48.
Double DB, Warren GC, Evans M, Rowlands RP. Efficacy of maintenance use of anticholinergic agents. Acta Psychiatr Scand 1993;88:381–4.
Egan MF, Zhao X, Smith A, Troyer MD, Uebele VN, Pidkorytov V, et al. Randomized controlled study of the T-type calcium channel antagonist MK-8998 for the treatment of acute psychosis in patients with schizophrenia. Hum Psychopharmacol 2013;28:124–33.
Ehrenreich H, Hinze-Selch D, Stawicki S, Aust C, Knolle-Veentjer S, Wilms S, et al. Improvement of cognitive functions in chronic schizophrenic patients by recombinant human erythropoietin. Mol Psychiatry 2007;12:206–20.
Elie R, Morin L, Tétreault L. [Effects of ethopropazine and trihexyphenidyl on several parameters of the neuroleptic syndrome.] Encephale 1972;61:32–52.
Emsley R, Myburgh C, Oosthuizen P, van Rensburg SJ. Randomized, placebo-controlled study of ethyl-eicosapentaenoic acid as supplemental treatment in schizophrenia. Am J Psychiatry 2002;159:1596–8.
Fann WE, Lake CR. Amantadine versus trihexyphenidyl in the treatment of neuroleptic-induced parkinsonism. Am J Psychiatry 1976;133:940–3.
Fay-McCarthy M, Kendrick KA, Rosse RB, Schwartz BL, Peace T, Wyatt RJ, Deutsch SI. The effect of nifedipine on akathisia and agitation in patients with movement disorders. Schizophren Res 1997;24:208.
Fay-McCarthy M, Kendrick KA, Rosse RB, Schwartz BL, Peace T, Wyatt RJ, Deutsch SI. The effect of nifedipine on tardive dyskinesia: a double blind study in eighteen patients. Schizophren Res 1997;24:271.
Gerlach J, Thorsen K, Fog R. Extrapyramidal reactions and amine metabolites in cerebrospinal fluid during haloperidol and clozapine treatment of schizophrenic patients. Psychopharmacology 1975;40:341–50.
Goetz CG, Stebbins GT, Chung KA, Hauser RA, Miyasaki JM, Nicholas AP, et al. No PD dyskinesia scale protects against placebo responses: a comparison of seven scales. Move Disord 2013;28:S115–16.
Goetz C, Stebbins G, Chung K, Hauser R, Miyasaki J, Nicholas A, et al. No PD dyskinesia scale protects against placebo responses: a comparison of multiple scales. Neurology 2013;80(Suppl. 7):P04.187.
Goldberg SC, Shenoy RS, Sadler A, Hamer R, Ross B. The effects of a drug holiday on relapse and tardive dyskinesia in chronic schizophrenics [proceedings.] Psychopharmacol Bull 1981;17:116–17.
Gulmann NC, Bahr B, Andersen B, Eliassen HM. A double-blind trial of baclofen against placebo in the treatment of schizophrenia. Acta Psychiatr Scand 1976;54:287–93.
Gutierrez M, Alpert M, Guimon J, Friedhoff AJ, Veramendi V. [Controlled study on the possibilities of L-dopa in the residual extrapyramidal syndrome caused by neuroleptics.] Actas Esp Psiquiatr 1979;7:181–8.
Hershon HI, Kennedy PF, McGuire RJ. Persistence of extra-pyramidal disorders and psychiatric relapse after withdrawal of long-term phenothiazine therapy. Br J Psychiatry 1972;120:41–50.
Hogarty GE, McEvoy JP, Munetz M, DiBarry AL, Bartone P, Cather R, et al. Dose of fluphenazine, familial expressed emotion, and outcome in schizophrenia – results of a two-year controlled study. Arch Gen Psychiatry 1988;45:797–805.
Huang J-L, Fu Z-C, Sun Q-Q. Trilafon in combination with nimodipine in the treatment of 30 senile patients with first-episode schizophrenia. Herald Med 2004;23:152–3.
Janicak PG, Sharma RP, Pandey G, Davis JM. Verapamil for the treatment of acute mania: a double-blind, placebo-controlled trial. Am J Psychiatry 1998;155:972–3.
Jean-Noel B, Wood AJ, Kiesler GM, Birkett M, Tollefson GD. Olanzapine vs. Clozapine: an International Double Blind Study in the Treatment of Resistant Schizophrenia. Proceedings of the 11th World Congress of Psychiatry, Hamburg, Germany, 6–11 August 1999.
Jolley AG, Hirsch SR, McRink A, Manchanda R. Trial of brief intermittent neuroleptic prophylaxis for selected schizophrenic outpatients: clinical outcome at one year. BMJ 1989;298:985–90.
Jolley AG, Hirsch SR, Morrison E, McRink A, Wilson L. Trial of brief intermittent neuroleptic prophylaxis for selected schizophrenic outpatients: clinical and social outcome at two years. BMJ 1990;301:837–42.
Kopala L, Rabinowitz J, Emsley R, Mcgorry P. Extra-pyramidal signs and symptoms (EPS) in recent onset schizophrenia: a comparison of risperidone and haloperidol. Schizophren Res 2004;67:187.
Krupitsky E, Burakov A, Romanova T, Vegso S, Krystal J. Nimodipine attenuates psychotigenic effects of ketamine in humans. Eur Neuropsychopharmacol 1999;9:S347.
Lara D. Flunarizine for Schizophrenia. Muscatine, IA: Stanley Foundation Research Programs; 2009.
Levine J, Schooler NR, Severe J, Escobar J, Gelenberg A, Mandel M, et al. Discontinuation of oral and depot fluphenazine in schizophrenic patients after one year of continuous medication: a controlled study. Adv Biochem Psychopharmacol 1980;24:483–93.
Lieberman JA, Kane JM, Sarantakos S, Gadaleta D, Woerner M, Alvir J, Ramos-Lorenzi J. Prediction of relapse in schizophrenia. Arch Gen Psychiatry 1987;44:597–603.
Lieberman JA, Alvir J, Geisler S, Ramos-Lorenzi J, Woerner M, Novacenko H, et al. Methylphenidate response, psychopathology and tardive dyskinesia as predictors of relapse in schizophrenia. Neuropsychopharmacology 1994;11:107–18.
Liebman HM, Woo W. The addition of tiagabine to antipsychotic medication in the treatment of recent-onset schizophrenia by modification of developmental pruning of prefrontal circuitry. Schizophren Res 2010;117:380–1.
Marder SR, Van Putten T, Mintz J, Lebell M, McKenzie J, May PR. Low- and conventional-dose maintenance therapy with fluphenazine decanoate. Two-year outcome. Arch Gen Psychiatry 1987;44:518–21.
Marder SR, Van Putten T, Mintz J, McKenzie J, Lebell M, Faltico G, May PR. Costs and benefits of two doses of fluphenazine. Arch Gen Psychiatry 1984;41:1025–9.
McCreadie RG, Dingwall JM, Wiles DH, Heykants JJ. Intermittent pimozide versus fluphenazine decanoate as maintenance therapy in chronic schizophrenia. Br J Psychiatry 1980;137:510–17.
NCT00310661. A Dual-Centre, Double-Blind, Randomized, Placebo-Controlled, Parallel-Group Study to Determine the Effects of Various Adjunctive Doses of Sarizotan in the Treatment of Patients with neuroleptic-Induced Tardive Dyskinesia. 2006. URL: https://clinicaltrials.gov/ct2/show/NCT00310661 (accessed 29 May 2016).
NCT00425815. A Placebo-Controlled Trial of org 24448 (Ampakine) Added to Atypical Antipsychotics in Patients with Schizophrenia. 2007. URL: https://clinicaltrials.gov/ct2/show/NCT00425815?term=NCT00425815&rank=1 (accessed 6 June 2017).
NCT00469664. A Double Blind Placebo Controlled Study of Guanfacine Adjunctive Treatment to Atypical Antipsychotics for Cognitive Dysfunction in Schizophrenia. 2007. URL: https://clinicaltrials.gov/ct2/show/NCT00469664 (accessed 6 June 2017).
NCT00512070. Melatonin Metabolism Abnormality in Patients with Schizophrenia or Schizoaffective Disorder Treated with Olanzapine and Melatonin Dose Finding for the Correction of the Metabolic Abnormality. 2007. URL: https://clinicaltrials.gov/ct2/show/NCT00512070?term=NCT00512070&rank=1 (accessed 6 June 2017).
NCT00845000. Acute Effects of SCH 420814 on Dyskinesia and Parkinsonism in Levodopa Treated Patients. 2009. URL: https://clinicaltrials.gov/ct2/show/NCT00845000?term=NCT00845000&rank=1 (accessed 29 May 2016).
Newcomer JW, Riney SJ, Vinogradov S, Csernansky JG. Plasma prolactin and homovanillic acid as markers for psychopathology and abnormal movements after neuroleptic dose decrease. Psychopharmacol Bull 1992;28:101–7.
Newton JE, Cannon DJ, Couch L, Fody EP, McMillan DE, Metzer WS, et al. Effects of repeated drug holidays on serum haloperidol concentrations, psychiatric symptoms, and movement disorders in schizophrenic patients. J Clin Psychiatry 1989;50:132–5.
Odejide OA, Aderounmu AF. Double-blind placebo substitution: withdrawal of fluphenazine decanoate in schizophrenic patients. J Clin Psychiatry 1982;43:195–6.
O’Suilleabhain P, Dewey RB. A randomized trial of amantadine in Huntington disease. Arch Neurol 2003;60:996–8.
Peluso MJ, Lewis SW, Barnes TR, Jones PB. Extrapyramidal motor side-effects of first- and second-generation antipsychotic drugs. Br J Psychiatry 2012;200:387–92.
Perry R, Campbell M, Green WH, Small AM, Die Trill ML, Meiselas K, et al. Neuroleptic-related dyskinesias in autistic children: a prospective study. Psychopharmacol Bull 1985;21:140–3.
Perry R, Campbell M, Adams P, Lynch N, Spencer EK, Curren EL, Overall JE. Long-term efficacy of haloperidol in autistic children: continuous versus discontinuous drug administration. J Am Acad Child Adolesc Psychiatry 1989;28:87–92.
Petit P, Bottai T, Pujalte D, Hue B, Blayac JP, Pouget J. Clonazepam in neuroleptic-induced akathisia: efficacy and dose-response relationship. Fundamental Clin Pharmacol 1994;8:287.
Pickar D, Wolkowitz OM, Doran AR, Labarca R, Roy A, Breier A, Narang PK. Clinical and biochemical effects of verapamil administration to schizophrenic patients. Arch Gen Psychiatry 1987;44:113–18.
Popov MIu. [The use of nifedipine as a corrector of extrapyramidal side-effects of classical neuroleptics.] Zh Nevrol Psikhiatr Im S S Korsakova 2008;108:28–33.
Price W, Giannini AJ, Loiselle R. Antischizophrenia effects of verapamil. Int J Neurosci 1986;31:577–8.
Price WA. Antipsychotic effects of verapamil in schizophrenia. Hillside J Clin Psychiatry 1987;9:225–30.
Raptis C, Garcia-Borreguero D, Weber MM, Dose M, Bremer D, Emrich HM. Anticonvulsants as adjuncts for the neuroleptic treatment of schizophrenic psychoses: a clinical study with beclamide. Acta Psychiatr Scand 1990;81:162–7.
Rosenheck R, Perlick D, Bingham S, Liu-Mares W, Collins J, Warren S, et al. Effectiveness and cost of olanzapine and haloperidol in the treatment of schizophrenia: a randomized controlled trial. JAMA 2003;290:2693–702.
Sachdev P, Loneragan C. Intravenous benztropine and propranolol challenges in tardive akathisia. Psychopharmacology 1993;113:119–22.
Salmasi FB, Jazayeri M, Ghaeli P, Hashemian F, Akhondzadeh S, Raisi F, et al. Comparing the effects of high-dose vitamin E with those of placebo on insulin resistance in patients with schizophrenia treated with olanzapine. J Clin Psychopharmacol 2009;29:182–3.
Shah BB, Connolly B, Mestre TA, Prashanth LK, Miyasaki JM, Steeves T, et al. Famotidine for the Treatment of Levodopa-Induced Dyskinesia: An Ongoing ‘N-of-1’ Study. 26th Annual Symposium on Etiology, Pathogenesis and Treatment of Movement Disorders, Irving, TX, USA, 11 May 2012.
Shenoy RS, Sadler AG, Goldberg SC, Hamer RM, Ross B. Effects of a six-week drug holiday on symptom status, relapse, and tardive dyskinesia in chronic schizophrenics. J Clin Psychopharmacol 1981;1:141–5.
Sikich L, Williamson K, Malekpour A, Bashford RA, Hooper S, Sheitman B, Lieberman JA. Interim Results of a Randomized Controlled Trial of Haloperidol, Risperidone, and Olanzapine in Psychotic Youth. Proceedings of the 38th Annual Meeting of the American College of Neuropsychopharmacology, Acapulco, Mexico, 12–16 December 1999.
Sikich L, Horrigan JP, Lieberman JA, Barnhill LJ, Sheitman BB, Courvoisie HE. Comparative Use of Olanzapine and Risperidone in Psychotic Youth. Proceedings of the 154th Annual Meeting of the American Psychiatric Association, New Orleans, LA, USA, 5–10 May 2001.
Sikich L. Critical Decisions in the Treatment of Adolescent and Pediatric Psychosis. Proceedings of the 154th Annual Meeting of the American Psychiatric Association, New Orleans, LA, USA, 5–10 May 2001.
Sikich L. Critical Decisions in the Treatment of Adolescent and Pediatric Psychosis. Proceedings of the 155th Annual Meeting of the American Psychiatric Association, Philadelphia, PA, USA, 18–23 May 2002.
Singh H, Hunt JI, Vitiello B, Simpson GM. Neuroleptic withdrawal in patients meeting criteria for supersensitivity psychosis. J Clin Psychiatry 1990;51:319–21.
Speller JC, Barnes TRE, Curson DA, Pantelis C, Alberts JL. One-year, low-dose neuroleptic study of in-patients with chronic schizophrenia characterised by persistent negative symptoms. Amisulpride v. haloperidol. Br J Psychiatry 1997;171:564–8.
Barnes TRE, Speller JC, Curson DA, Pantelis C, Alberts JL. A one-year dose reduction study in chronic schizophrenic inpatients: amisulpride vs haloperidol. Schizophren Res 1992;6:107.
Suh GH, Son HG, Ju YS, Jcho KH, Yeon BK, Shin Y, et al. A randomized, double-blind, crossover comparison of risperidone and haloperidol in Korean dementia patients with behavioral disturbances. Am J Geriatr Psychiatry 2004;12:509–16.
Thapa PB, Meador KG, Gideon P, Fought RL, Ray WA. Effects of antipsychotic withdrawal in elderly nursing home residents. J Am Geriatr Soc 1994;42:280–6.
Tollefson GD, Beasley CM, Tamura RN, Tran PV, Potvin JH. Blind, controlled, long-term study of the comparative incidence of treatment-emergent tardive dyskinesia with olanzapine or haloperidol. Am J Psychiatry 1997;154:1248–54.
Tamura RN, Tollefson GD, Dellva MA, Beasley CM, Glazer WM, Morgenstern H. What is the differential risk of tardive dyskinesia with the novel antipsychotic olanzapine? Schizophren Res 1998;29:176.
Beasley CM, Dellva MA, Tamura RN, Morgenstern H, Glazer WM, Ferguson K, Tollefson GD. Randomised double-blind comparison of the incidence of tardive dyskinesia in patients with schizophrenia during long-term treatment with olanzapine or haloperidol. Br J Psychiatry 1999;174:23–30.
Tran PV, Dellva MA, Tollefson GD, Beasley CM, Potvin JH, Kiesler GM. Extrapyramidal symptoms and tolerability of olanzapine versus haloperidol in the acute treatment of schizophrenia. J Clin Psychiatry 1997;58:205–11.
Vaddadi KS, Soosai E, Chiu E, Dingjan P. A randomised, placebo-controlled, double blind study of treatment of Huntington’s disease with unsaturated fatty acids. Neuroreport 2002;13:29–33.
Wang Q, Wei X. Propranolol and verapamil therapy antipsychotics due to sinus tachycardia control study. Central Plains Med J 1995;22:26–7.
Wang F, Lu Y. The control study of clonazepam and artane in akathisia. J Clin Psychosom Dis 2000;6:19–20.
Wei X-Y, Zhu A, Li N, Chen Z. Propranolol vs verapamil in treating drug-induced sinus tachycardia. Chin J N Drugs Clin Remedies 1995;14:336–7.
Wei P. Efficacy of nimodipine combined sulpiride treatment of the negative symptoms of schizophrenia. Int Med Health Guidance News 2008;14:54–6.
Williamson DJ, Tran PV, Beasley CM, Tollefson GD, Sanger T, Satterlee WG. Clinical efficacy and safety of olanzapine, a new atypical antipsychotic agent. J Psychopharmacol 1995;9:A47.
Wirshing DA, Marshall BD, Green MF, Mintz J, Marder SR, Wirshing WC. Risperidone in treatment-refractory schizophrenia. Am J Psychiatry 1999;156:1374–9.
Wistedt B, Wiles D, Jørgensen A. A depot neuroleptic withdrawal study neurological effects. Psychopharmacology 1983;80:101–5.
Yaryura-Tobias JA, Krumholz WV, Wolpert A, White L, Merlis S. Calcium as a proposed treatment for drug-induced extrapyramidal symptoms. Psychopharmacol Bull 1968;4:36–7.
Randomised, tardive dyskinesia, but not stabilised on antipsychotics
Cassady SL, Thaker GK, Moran M, Birt A, Tamminga CA. GABA agonist-induced changes in motor, oculomotor, and attention measures correlate in schizophrenics with tardive dyskinesia. Biol Psychiatry 1992;32:302–11.
Jankovic J. Treatment of hyperkinetic movement disorders with tetrabenazine: a double-blind crossover study. Ann Neurol 1982;11:41–7.
Lieberman JA, Alvir J, Mukherjee S, Kane JM. Treatment of tardive dyskinesia with bromocriptine. A test of the receptor modification strategy. Arch Gen Psychiatry 1989;46:908–13.
Perovich RM, Lieberman JA, Fleischhacker WW, Alvir J. The behavioral toxicity of bromocriptine in patients with psychiatric illness. J Clin Psychopharmacol 1989;9:417–22.
Simpson GM, Voitashevsky A, Young MA, Lee JH. Deanol in the treatment of tardive dyskinesia. Psychopharmacology 1977;52:257–61.
Tamminga CA, Crayton JW, Chase TN. Improvement in tardive dyskinesia after muscimol therapy. Arch Gen Psychiatry 1979;36:595–8.
Randomised, tardive dyskinesia, no usable data reported – authors contacted to confirm lack of data
Alpert M, Friedhoff AJ, Diamond F. Use of dopamine receptor agonists to reduce dopamine receptor number as treatment for tardive dyskinesia. Adv Neurol 1983;37:253–8.
Andia I, Zumarraga M, Zabalo MJ, Bulbena A, Davila R. Differential effect of haloperidol and clozapine on plasma homovanillic acid in elderly schizophrenic patients with or without tardive dyskinesia. Biol Psychiatry 1998;43:20–3.
Diehl A, Braus DF, Büchel C, Krumm B, Medori R, Gattaz WF. [Tardive dyskinesia: pergolid, a possible therapeutic option.] Psychiatr Prax 2003;30:333–7. http://dx.doi.org/10.1055/s-2003-42166
Diehl A, Dittmann RW, Gattaz W, Rubin M, Hundemer HP. Low Dose Pergolide in the Treatment of Tardive Dyskinesia (TD): A Double Blind, Placebo Controlled Randomised Cross Over Trial. 11th World Congress of Psychiatry, Hamburg, Germany, 6–11 August 1999.
Diehl A, Hundemer HP, Rubin M, Dittmann RW, Gattaz W. Low-dose pergolide in the treatment of tardive dyskinesia (TD): a double-blind, placebo-controlled randomized cross-over trial. J Eur Coll Neuropsychopharmacol 1999;9:S359.
Frangos E, Athanasenas E. Lioresal in the Treatment of Neuroleptic-Induced Tardive Dyskinesia. Proceedings of the 12th CINP Congress, Gothenburg, Sweden, 22–26 June 1980.
Fudge RC, Thailer SA, Alpert M, Intrator J, Sison CE. The effects of electromyographic feedback training on suppression of the oral-lingual movements associated with tardive dyskinesia. Biofeedb Self-Regulat 1991;16:117–29.
Herz MI, Glazer WM, Mostert MA, Sheard MA, Szymanski HV, Hafez H, et al. Intermittent vs maintenance medication in schizophrenia. Two-year results. Arch Gen Psychiatry 1991;48:333–9.
Johnson DA, Ludlow JM, Street K, Taylor RD. Double-blind comparison of half-dose and standard-dose flupenthixol decanoate in the maintenance treatment of stabilised out-patients with schizophrenia. Br J Psychiatry 1987;151:634–8.
Junker D, Steigleider P, Gattaz WF. Alpha-tocopherol in the treatment of tardive dyskinesia. Schizophren Res 1992;6:122–3.
Koller WC, Barr A, Biary N. Estrogen treatment of dyskinetic disorders. Neurology 1982;32:547–9.
Leys D, Vermersch P, Danel T, Comayras S, Goudemand M, Caron J, Petit H. Diltiazem for tardive dyskinesia. Lancet 1988;1:250–1.
Quinn N, Marsden CD. A double blind trial of sulpiride in Huntington’s disease and tardive dyskinesia. J Neurol Neurosurg Psychiatr 1984;47:844–7.
Quinn N, Marsden, CD. [Double blind trial of dogmatil in Huntington chorea and tardive dyskinesia.] Sem Hop1985;61:1376–80.
Sommer BR, Cohen BM, Satlin A, Cole JO, Jandorf L, Dorsey F. Changes in tardive dyskinesia symptoms in elderly patients treated with ganglioside GM1 or placebo. J Geriatr Psychiatry Neurol 1994;7:234–7.
Peselow ED, Irons S, Rotrosen J, Alonso MT, Dorsey F. GM1 ganglioside as a potential treatment in tardive dyskinesia. Psychopharmacol Bull 1989;25:277–80.
Spohn HE, Coyne L, Spray J. The effect of neuroleptics and tardive dyskinesia on smooth-pursuit eye movement in chronic schizophrenics. Arch Gen Psychiatry 1988;45:833–40.
Spohn HE, Coyne L. The effect of attention/information processing impairment of tardive dyskinesia and neuroleptics in chronic schizophrenics. Special Issue: Tardive dyskinesia and cognitive dysfunction. Brain Cogn 1993;23:28–39.
Thaker GK, Nguyen JA, Strauss ME, Jacobson R, Kaup BA, Tamminga CA. Clonazepam treatment of tardive dyskinesia: a practical GABAmimetic strategy. Am J Psychiatry 1990;147:445–51.
Randomised, tardive dyskinesia, but no usable data reported – no author contact details, study > 20 years old
Borison RL, Shah C, White TH, Diamond BI. Atypical and typical neuroleptics and tardive dyskinesia. Psychopharmacol Bull 1987;23:218–20.
Greendyke RM, Webster JC, Kim J, Kim H. Lack of efficacy of pindolol in tardive dyskinesia. Am J Psychiatry 1988;145:1318–19.
Kabes J, Sikora J, Pisvejc J, Hanzlicek L, Skondia V. Effect of piracetam on extrapyramidal side effects induced by neuroleptic drugs. Int Pharmacopsychiatry 1982;17:185–92.
Kabes J, Sikora J, Pisvejc J, Skoudia V. Piracetam Action in Neuroleptic Induced Extrapyramidal Side-Effects. In Proceedings of the 12th CINP Congress, Supplement to Progress in Neuropsychopharmacology. Oxford: Pergamon Press; 1980.
Sikora J, Kabes J, Pisvejc J. [Management of neuroleptic side-effects with piracetam (author’s translation).] Cesk Psychiatr 1981;77:137–42.
Kabes J, Sikora J, Stary O, Piisvejc J, Hanzlicek L. Piracetam effectivity in tardive dyskinesia. A double-blind placebo-controlled cross-over trial. Cesk Psychiatr 1983;79:339–45.
Kabes J, Sikora J, Stary O, Pisvejc J. Dose-dependent effect of piracetam in tardive-dyskinesia – double-blind placebo controlled trial. Activitas Nervosa Superior 1985;27:64–6.
Ludatscher JI. Stable remission of tardive dyskinesia by L-dopa. J Clin Psychopharmacol 1989;9:39–41.
Marsalek M, Filip V, Petrovsky M, Klar I, Filipova M, Klaschka J. 7-MEOTA in the treatment of tardive dyskinesia. Double-blind placebo controlled study. Homeostasis 1997;38:7.
Rzewuska M, Soucka K. Therapeutic Effect of Diltiazem in Tardive Dyskinesia. Proceedings of the 8th European College of Neuropsychopharmacology Congress, Venice, Italy, 30 September–4 October 1995.
Rzewuska M, Sobucka K. Therapeutic effect of diltiazem in tardive dyskinesia. Eur Neuropsychopharmacol 1995;5:390–1.
Sikora J, Kabes J, Pisvejc J. [Management of neuroleptic side-effects with piracetam (author’s translation).] Cesk Psychiatr 1981;77:137–42.
Randomised, tardive dyskinesia, but no separate data reported on minority with tardive dyskinesia – authors contacted to confirm lack of data
de Jesus Mari J, Lima MS, Costa AN, Alexandrino N, Rodrigues-Filho S, de Oliveira IR, Tollefson GD. The prevalence of tardive dyskinesia after a nine month naturalistic randomized trial comparing olanzapine with conventional treatment for schizophrenia and related disorders. Eur Arch Psychiatry Clin Neurosci 2004;254:356–61.
Klett CJ, Caffey E. Evaluating the long-term need for antiparkinson drugs by chronic schizophrenics. Arch Gen Psychiatry 1972;26:374–9.
Suddath RL, Straw GM, Freed WJ, Bigelow LB, Kirch DG, Wyatt RJ. A clinical trial of nifedipine in schizophrenia and tardive dyskinesia. Pharmacol Biochem Behav 1991;39:743–5.
Randomised, tardive dyskinesia, but crossover trial with no separate data reported for phase before crossing over to second treatment – authors contacted to confirm lack of data
Domino EF, May WW, Demetriou S, Mathews B, Tait S, Kovacic B. Lack of clinically significant improvement of patients with tardive dyskinesia following phosphatidylcholine therapy. Biol Psychiatry 1985;20:1189–96.
Doongaji DR, Jeste DV, Jape NM, Sheth AS, Apte JS, Vahia VN, et al. Effects of intravenous metoclopramide in 81 patients with tardive dyskinesia. J Clin Psychopharmacol 1982;2:376–9.
Fann WE, Davis JM, Wilson IC. Methylphenidate in tardive dyskinesia. Am J Psychiatry 1973;130:922–4. http://dx.doi.org/10.1176/ajp.130.8.922
Friis T, Rosted-Christensen T, Gerlach J. Sodium valproate and biperiden in neuroleptic-induced akathisia, parkinsonism and hyperkinesia: a double-blind crossover study with placebo. Acta Psychiatr Scand 1983;67:178–87.
Gelenberg AJ, Wojcik J, Falk WE, Bellinghausen B, Joseph AB. CDP-choline for the treatment of tardive dyskinesia: a small negative series. Compr Psychiatry 1989;30:1–4.
Hemnani TJ, Dashputra PG, Sarda RN. Metoclopramide in tardive dyskinesia. Ind J Pharmacol 1982;14:309–12.
Hemanani TJ, Dashputra PG, Sarda RN. Metoclopramide in tardive dyskinesia. Ind J Psychiatry 1983;25:134–7.
Jeste DV, Cutler NR, Kaufmann CA, Karoum F. Low-dose apomorphine and bromocriptine in neuroleptic-induced movement disorders. Biol Psychiatry 1983;18:1085–91.
Joe SH, Suh KY, Lee BY. Effect of lecithin on tardive dyskinesia. Korea Uni Med J 1985;22:197–206.
Jus A, Villeneuve A, Gautier J, Jus K, Villeneuve C, Pires P, Villeneuve R. Deanol, lithium and placebo in the treatment of tardive dyskinesia. A double-blind crossover study. Neuropsychobiology 1978;4:140–9.
Korsgaard S. Baclofen (Lioresal) in the treatment of neuroleptic-induced tardive dyskinesia. Acta Psychiatr Scand 1976;54:17–24.
Lal S, Ettigi P. Comparison of thiopropazate and trifluoperazine on oral dyskinesia – a double blind study. Curr Ther Res Clin Exp 1974;16:990–7.
Lieberman J, Pollack S, Lesser M, Kane J. Pharmacologic characterization of tardive dyskinesia. J Clin Psychopharmacol 1988;8:254–60.
Lindenmayer JP, Gardner E, Goldberg E, Opler LA, Kay SR, van Praag HM, et al. High-dose naloxone in tardive dyskinesia. Psychiatry Res 1988;26:19–28.
Lohr JB, Cadet JL, Lohr MA, Larson L, Wasli E, Wade L, et al. Vitamin E in the treatment of tardive dyskinesia: the possible involvement of free radical mechanisms. Schizophr Bull 1988;14:291–6.
Lohr JB, Cadet JL, Lohr MA, Jeste DV, Wyatt RJ. Alpha-tocopherol in tardive dyskinesia. Lancet 1987;1:913–14.
Cadet JL, Lohr JB. Possible involvement of free radicals in neuroleptic-induced movement disorders. Evidence from treatment of tardive dyskinesia with vitamin E. Ann N Y Acad Sci 1989;570:176–85.
Nasrallah HA, Dunner FJ, McCalley-Whitters M, Smith RE. Pharmacologic probes of neurotransmitter systems in tardive dyskinesia: implications for clinical management. J Clin Psychiatry 1986;47:56–9.
Nordic Dyskinesia Study Group. Effect of different neuroleptics in tardive dyskinesia and parkinsonism. A video-controlled multicenter study with chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden. Psychopharmacology 1986;90:423–9.
Gerlach J, Ahfors UG, Amthor KF. Effect of different neuroleptics in tardive dyskinesia and parkinsonism. A video-controlled multicenter study with chlorprothixene, perphenazine, haloperidol and haloperidol and biperiden. Psychopharmacology 1986;90:423.
Gerlach J. Tardive dyskinesia. Pathophysiological mechanisms and clinical trials. Encephale 1988;14:227–32.
Povlsen UJ, Noring U, Meidahl B, Korsgaard S, Waehrens J, Gerlach J. [The effects of neuroleptics on tardive dyskinesias. A video-controlled, randomized study of chlorprothixene, perphenazine, haloperidol and haloperidol + biperiden.] Ugeskr Laeg 1987;149:1682–5.
Penovich P, Morgan JP, Kerzner B, Karch F, Goldblatt D. Double-blind evaluation of deanol in tardive dyskinesia. JAMA 1978;239:1997–8.
Perez-Cruet J, Menendez I, Alvarez-Ghersi J, Falcon JR, Valderrabano O, Castro-Urrutia EC, et al. Double-blind study of lecithin in the treatment of persistent tardive dyskinesia. Boletin Asociacion Medica Puerto Rico 1981;73:531–7.
Ricketts RW, Singh NN, Ellis CR, Chambers S, Singh YN, Carmanico SJ, et al. Calcium channel blockers and vitamin E for tardive dyskinesia in adults with mental retardation. J Develop Phys Disabil 1995;7:161–74.
Shriqui CL, Bradwejn J, Annable L, Jones BD. Vitamin E in the treatment of tardive dyskinesia: a double-blind placebo-controlled study. Am J Psychiatry 1992;149:391–3.
Stearns AI, Sambunaris A, Elkashef AM, Issa F, Egan MF, Wyatt RJ. Selegiline for Negative Symptoms and Tardive Dyskinesia. Proceedings of the 149th Annual Meeting of the American Psychiatric Association, New York, NY, USA, 4–9 May 1996.
Tamminga CA, Chase TN. Bromocriptine and CF 25-397 in the treatment of tardive dyskinesia. Arch Neurol 1980;37:204–5.
Tamminga CA, Thaker GK, Ferraro TN, Hare TA. GABA agonist treatment improves tardive dyskinesia. Lancet 1983;2:97–8.
Tamminga CA, Thaker GK, Goldberg ST. Tardive Dyskinesia: GABA Agonist Treatment. In Usdin E, Carlsson A, Dahlstrom A, Engel J, editor(s). Catecholamines: Neuropharmacology and Central Nervous System – Therapeutic Aspects. New York, NY: Alan R Liss; 1984. pp. 69–72.
Thaker GK, Hare TA, Tamminga CA. GABA system: clinical research and treatment of tardive dyskinesia. Mod Probl Pharmacopsychiatry 1983;21:155–67.
Nguyen JA, Thaker GK, Tamminga CA. Gamma-aminobutyric-acid (GABA) pathways in tardive dyskinesia. Psychiatr Ann 1989;19:302–9.
Vaddadi KS, Courtney P, Gilleard CJ, Manku MS, Horrobin DF. A double-blind trial of essential fatty acid supplementation in patients with tardive dyskinesia. Psychiatry Res 1989;27:313–23.
Vaddadi KS, Gileard CJ. Essential fatty acids, tardive dyskinesia, and schizophrenia. In Horrobin DF, ed. Omega-6 Essential Fatty Acids: Pathophysiology and Roles in Clinical Medicine. Hoboken, NJ: Wiley-Liss; 1990. pp. 333–43.
Wonodi I, Adami H, Sherr J, Avila M, Hong LE, Thaker GK. Naltrexone treatment of tardive dyskinesia in patients with schizophrenia. J Clin Psychopharmacol 2004;24:441–5.
Yamada K, Kanba S, Ashikari I, Ohnishi K, Yagi G, Asai M. Nilvadipine is effective for chronic schizophrenia in a double-blind placebo-controlled study. J Clin Psychopharmacol 1996;16:437–9.
Randomised, tardive dyskinesia, but crossover trial with no separate data reported for phase before crossing over to second treatment – no author contact details, study > 20 years old
Angus S, Sugars J, Boltezar R, Koskewich S, Schneider NM. A controlled trial of amantadine hydrochloride and neuroleptics in the treatment of tardive dyskinesia. J Clin Psychopharmacol 1997;17:88–91.
Auberger S, Greil W, Ruther E. Tiapride in the treatment of tardive dyskinesia. A double-blind study. Pharmacopsychiatry 1985;18:61–2.
Greil W, Auberger S, Haag H, Ruther E. Tiapride: effects on tardive dyskinesia and on prolactin plasma concentrations. Neuropsychobiology 1985;14:17–22.
Bateman DN, Dutta DK, McClelland HA, Rawlins MD. The effect of metoclopramide and haloperidol on tardive dyskinesia [proceedings.] Br J Pharmacol 1979;66:475P–476P.
Bateman DN, Dutta DK, McClelland HA, Rawlins MD. Metoclopramide and haloperidol in tardive dyskinesia. Br J Psychiatry 1979;135:505–8.
Braun A, Mouradian MM, Mohr E, Fabbrini G, Chase TN. Selective D-1 dopamine receptor agonist effects in hyperkinetic extrapyramidal disorders. J Neurol Neurosurg Psychiatr 1989;52:631–5.
Browne J, Silver H, Martin R, Hart R, Mergener M, Williams P. The use of clonidine in the treatment of neuroleptic-induced tardive dyskinesia. J Clin Psychopharmacol 1986;6:88–92.
Chien C, Jung K, Ross-Townsend A. Efficacies of agents related to GABA, dopamine, and acetylcholine in the treatment of tardive dyskinesia [proceedings.] Psychopharmacol Bull 1978;14:20–2.
Delwaide PJ, Desseilles M. [Controlled therapeutic study of spontaneous bucco-linguo-facial dyskinesias.] Sem Hop 1979;55:1585–9.
Gardos G, Granacher RP, Cole JO, Sniffin C. The effects of papaverine in tardive dyskinesia. Prog Neuropsychopharmacol 1979;3:543–50.
Gerlach J, Thorsen K, Munkvad I. Effect of lithium on neuroleptic-induced tardive dyskinesia compared with placebo in a double-blind cross-over trial. Pharmakopsychiatrie Neuro-Psychopharmakologie 1975;8:51–6.
Godwin-Austen RB, Clarke T. Persistant phenothiazine dyskinesia treated with tetrabenazine. Br Med J 1971;4:25–6.
Schwartz M, Moguillansky L, Lanyi G, Sharf B. Sulpiride in tardive dyskinesia. J Neurol Neurosurg Psychiatr 1990;53:800–2.
Silver H, Geraisy N, Schwartz M. No difference in the effect of biperiden and amantadine on parkinsonian- and tardive dyskinesia-type involuntary movements: a double-blind crossover, placebo-controlled study in medicated chronic schizophrenic patients. J Clin Psychiatry 1995;56:167–70.
Silver H, Geraisy N, Schwartz M. No difference in the effect of biperiden and amantadine on parkinsonian- and tardive dyskinesia-type involuntary movements: a double-blind crossover, placebo-controlled study in medicated chronic schizophrenic patients. J Clin Psychiatry 1995;56:435.
Singer K, Cheng MN. Thiopropazate hydrochloride in persistent dyskinesia. Br Med J 1971;4:22–5.
Singer K, Cheng MN. Thiopropazate Hydrochloride (Dartalan) in Persistent Dyskinesia with Phenothiazine Therapy. 5th World Congress of Psychiatry, Ciudad de Mexico, Mexico, 28 November–4 December 1971.
Viukari M, Linnoila M. Effect of methyldopa on tardive dyskinesia in psychogeriatric patients. Curr Ther Res Clin Exp 1975;18:417–24.
Appendix 6 Cochrane reviews on antipsychotic-induced tardive dyskinesia
Published Cochrane reviews on TD are listed below. They can be accessed through The Cochrane Library. All these reviews have been updated and are in the pre-publication process at the time of writing.
Soares-Weiser K, Mobsy C, Holliday E. Anticholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 1997;2:CD000204.
Tammenmaa I, McGrath J, Sailas E, Soares-Weiser K. Cholinergic medication for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2002;3:CD000207.
Soares-Weiser K, Joy C. Miscellaneous treatments for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2003;2:CD000208.
Bhoopathi PS, Soares-Weiser K. Benzodiazepines for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2006;3:CD000205.
El-Sayeh HG, Lyra da Silva JP, Rathbone J, Soares-Weiser K. Non-neuroleptic catecholaminergic drugs for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2006;1:CD000458.
Soares-Weiser K, Rathbone J. Neuroleptic reduction and/or cessation and neuroleptics as specific treatments for tardive dyskinesia. Cochrane Database Syst Rev 2006;1:CD000459.
Alabed S, Latifeh Y, Mohammad HA, Rifai A. Gamma-aminobutyric acid agonists for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;4:CD000203.
Essali A, Deirawan H, Soares-Weiser K, Adams CE. Calcium channel blockers for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;11:CD000206.
Soares-Weiser K, Maayan N, McGrath J. Vitamin E for neuroleptic-induced tardive dyskinesia. Cochrane Database Syst Rev 2011;2:CD000209.
Appendix 7 Detailed study characteristics and risk-of-bias assessments
FIGURE 22.
Summary of risk-of-bias assessments for included studies. –, high risk of bias; +, low risk of bias; ?, unclear risk of bias.


Antipsychotic drugs
| Included study | Description | |
|---|---|---|
| Bai et al., 2003108 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions | After a 4-week washout period with all original conventional antipsychotics discontinued:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: supported by Janssen-Cilag Taiwan, Johnson & Johnson Taiwan Ltd | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . subjects were randomly assigned to the risperidone or placebo groups’, further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | . . . double-blind . . . A placebo with an identical appearance to the risperidone dose was prescribed for the placebo group using the same dose schedule |
| Blinding of outcome assessment (detection bias) | Low risk | The TD condition was evaluated blindly by a psychiatrist with the Abnormal Involuntary Movement Scale (AIMS) every 2 weeks |
| Incomplete outcome data (attrition bias) | Low risk | Seven of 49 participants withdrew:Four subjects dropped out due to psychotic symptom exacerbation (2 subjects during the washout period: 1 subject in the placebo group and 1 subject in the risperidone group). Another 3 subjects withdrew due to a medical condition (infectious disease, heart condition, and lung carcinoma) |
| Selective reporting (reporting bias) | Unclear risk | Unclear if all predefined outcomes have been reported. A protocol is not available for verification |
| Other bias | Low risk | The study seems to be free of other sources of bias |
| Bai et al., 2005112,113 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions | No washout period on the discontinuation of all conventional antipsychotics was reported:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: the study was supported by grants from National Science Council, Taiwan | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘The subjects were randomized to three groups’, further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | . . . single-blind and controlled study |
| Blinding of outcome assessment (detection bias) | Unclear risk | ‘. . . single-blind and controlled study’. Blinding details of outcome assessors not reported |
| Incomplete outcome data (attrition bias) | Low risk | Finally 76 cases (95%) completed the 24-week study, 2 cases in the olanzapine groups withdrew due to impaired liver function, 1 case in the amisulpride group due to infectious disease, and 1 case in the FGA controlled groups withdrew due to unstable psychiatric conditionIntention-to-treat analyses with last-observation-carried-forward method applied |
| Selective reporting (reporting bias) | Low risk | All outcomes appear to have been reported |
| Other bias | Low risk | The study seems to have been free of other sources of bias |
| Caroff et al., 2011117 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions | Overlap in administration of the antipsychotic drugs that patients received before study entry was permitted for the first 4 weeks after randomisation to allow a gradual transition to study medication:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: supported by the Clinical Antipsychotic Trials of Intervention Effectiveness project, National Institute of Mental Health. This article was based on results from the Clinical Antipsychotic Trials of Intervention Effectiveness project, supported by the National Institute of Mental Health. Astra Zeneca Pharmaceuticals LP, Bristol-Myers Squibb Company, Forest Pharmaceuticals Inc., Janssen Pharmaceutical Products LP, Eli Lilly and Company, Otsuka Pharmaceutical Co. Ltd, Pfizer Inc. and Zenith Goldline Pharmaceuticals Inc. provided medications for the studies. This material is based on work also supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research Development, with resources and the use of facilities at the Philadelphia Veterans Affairs Medical Center | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Patients were initially randomly assigned’, further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | . . ., double-blind conditions, . . . Identical-appearing capsules contained olanzapine (7.5 mg), quetiapine (200 mg), risperidone (1.5 mg), perphenazine (8 mg), or ziprasidone (40 mg) |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of outcome assessors not reported |
| Incomplete outcome data (attrition bias) | High risk | The primary clinical outcome measure was time to all-cause treatment discontinuation. Total population (n = 200): 74% discontinuation. Olanzapine: 31/54 (57%); quetiapine: 51/62 (82%); risperidone: 44/56 (79%); ziprasidone: 21/28 (75%). Reasons for withdrawal reported |
| Selective reporting (reporting bias) | High risk | Original CATIE study:The primary clinical outcome measure was time to all-cause treatment discontinuation. Secondary outcomes included discontinuations for intolerability, inefficacy, and patient decision; rates of discontinuations; mean modal dose; and change from baseline in the PANSS and neurocognitive composite scoresAll outcomes not fully reported for the TD population |
| Other bias | High risk | Post hoc analysis; modified diagnostic criteria for TD were applied at baseline and a 3-month history of antipsychotic exposure was not required |
| Chan et al., 2010115 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions | Following a washout period of 3–7 days:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: supported by research grant from the Taoyuan Mental Hospital and from the Department of Health, Executive Yuan, Taiwan | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | . . . randomly assigned to receive either olanzapine or risperidone with a 1-to-1 ratio by coin method with a 6-block design |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | . . . primary care physicians and patients were not blinded |
| Blinding of outcome assessment (detection bias) | Low risk | Two investigators (C.-H.C. and J.-J.C.) served as blinded raters . . . The BPRS, CGI-S, AIMS and global impression of ESRS were performed at baseline and at weeks 1, 2, 3, 4, 8, 12, 16, 20, and 24 or at end point visit by blinded-rater |
| Incomplete outcome data (attrition bias) | Low risk | Nine out of 30 in the risperidone and 7 out of 30 in the olanzapine groups dropped out from the study; reasons reportedAll patients who were randomly assigned and had at least 1 post-baseline assessment were included in the intent-to-treat (ITT) analysis. If the ITT subjects withdrew from the study earlier than scheduled, then the last observation carried forward method was employed to extend the end point scores |
| Selective reporting (reporting bias) | Low risk | Data for all outcomes in the trial registry, NCT00621998, have been reported |
| Other bias | Low risk | The study seems to be free of other sources of bias |
| Chouinard et al., 1993102,103 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | Diagnosis: chronic schizophrenia (DSM-III-R criteria193), n = 135 Age: mean 39 years, range 19–60 years Sex: 34 male and 14 female History: duration TD not reported; the most common pre-study medications were haloperidol, procyclidine, lorazepam, benztropine and chlorpromazine; the most commonly used depot antipsychotic agents were haloperidol decanoate, fluphenazine decanoate, flupentixol decanoate and pipothiazine palmitate |
|
| Interventions | Mean duration of washout phase 6 days:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: not reporteda | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . randomly assigned’, details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | . . . identical tablets |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of raters not reported |
| Incomplete outcome data (attrition bias) | Low risk | 33% of participants terminated the study early because of an insufficient therapeutic response. All early terminations were included in the intention-to-treat analysis |
| Selective reporting (reporting bias) | High risk | Outcomes not fully reported |
| Other bias | High risk | Subgroup with TD |
| Cookson, 198798 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions | No washout period before study entry:
|
|
| Outcomes |
|
|
| Notes | Dr Cookson kindly provided additional information | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | ‘. . . randomised in blocks of 4 and stratified by neuroleptic dose and gender’, implies adequate random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No allocation concealment details |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double blind’, no further details |
| Blinding of outcome assessment (detection bias) | Unclear risk | ‘. . . double blind’, no further details |
| Incomplete outcome data (attrition bias) | Low risk | All patients seem to have completed the study |
| Selective reporting (reporting bias) | Unclear risk | All outcomes proposed in the methods were reported, but some were not presented adequately. No protocol available to check as well |
| Other bias | High risk | The randomised allocation of the small number of patients in the pilot study results in inequalities between the 2 groups at entry and confounded comparisons of group mean values during the study |
| Emsley et al., 2004110 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | Diagnosis: schizophrenia (DSM-IV191), TD (Schooler and Kane criteria192), n = 45 Age: 49.2 (SD 14.5) years, range 18–65 years Sex: 16 male and 29 female History: duration of TD not reported; at least 3 months antipsychotic exposure; patients with established psychiatric disorder who do not receive clozapine |
|
| Interventions | After an initial screening visit, subjects were tapered from all psychotropic medication over a 2-week period:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: supported in part by the Medical Research Council of South Africa, Cape Town and the University of Stellenbosch. Trial medication and monitoring of the study were provided by AstraZeneca, Wilmington, DE, USA | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Subjects were then randomly assigned’, further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | ‘. . . investigator-blinded’, further blinding details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | ‘. . . investigator-blinded’, further blinding details not reported |
| Incomplete outcome data (attrition bias) | High risk | 43% dropouts (including the two subjects excluded in the early stages). 10/22 (45%) patients in the quetiapine group and 8/23 (35%) haloperidol patients dropped out |
| Selective reporting (reporting bias) | High risk | Adverse effects: extrapyramidal symptoms (other than dyskinesia) not fully reported |
| Other bias | Low risk | The study seems to be free of other sources of bias. Baseline characteristics are balanced in the compared groups |
| Kane et al., 198397 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes | TD (‘no clinical improvement’; ‘not any improvement’; ‘deterioration’), reported as adverse effects:
|
|
| Notes | Sponsorship source: this investigation was supported in part by grants from the National Institute of Mental Health. Dr Woerner kindly provided unpublished data for one site of the main study and only these are used in this review; the sex ratios are not available. If people in this study developed TD, participation was stopped and they were classified as leaving the study early | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised using random numbers table |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind’. Details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | ‘. . . double-blind’. Details not reported |
| Incomplete outcome data (attrition bias) | High risk | 4/8 participants left the study early |
| Selective reporting (reporting bias) | High risk | Not all data were reported |
| Other bias | High risk | Only subsample with TD from one site included in this review |
| Kazamatsuri et al., 197396 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions | 4-week washout from antiparkinsonian and antipsychotic medication (all replaced by placebo), then:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: Supported in part by Public Health Service grant from the National institute of Mental Health. Tetrabenazine and placebo tablets were provided by Hoffman-La Roche | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The 13 patients were divided randomly into two groupsFurther details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | Blinding of participants and personnel not reported |
| Blinding of outcome assessment (detection bias) | Low risk | A frequency count of mouth movements (18), done by a psychiatrist blind to the study design was used to assess oral dyskinesia |
| Incomplete outcome data (attrition bias) | High risk | Two out of seven (29%) subjects dropped out from the haloperidol group. There were no dropouts from the tetrabenazine group. The dropouts were not entered in the analysis (data reported for all subject up until week 16, inclusive) |
| Selective reporting (reporting bias) | High risk | TD scale scores and extrapyramidal symptoms scale scores not fully reported |
| Other bias | Unclear risk | Insufficient information to make a judgement |
| Tamminga et al., 1994104 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions | After the stabilisation period, each patient was withdrawn from antipsychotic treatment for 4 weeks to allow a antipsychotic-free assessment of their dyskinetic symptoms. Then:
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: sponsorship source not reported. Authors were contacted for updated data but at the time of preparing this review no more information had been received | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Subjects were then blindly randomised to two different drug groupsFurther details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Staff, patients, and all raters were blind to the drug group; one non rating physician and one nurse were non blind to dispense medication and monitor safetyNo further details are provided |
| Blinding of outcome assessment (detection bias) | Low risk | Staff, patients, and all raters were blind to the drug group; one non rating physician and one nurse were non blind to dispense medication and monitor safety’No further details are provided |
| Incomplete outcome data (attrition bias) | High risk | Of 43 enrolled participants, four did not complete the study and seven were withdrawnOne subject from each treatment group was dropped for leukopenia. The other 5 clozapine subjects were dropped for noncompliance (1 patient), decompensation (1 patient), seizure (1 patient), hypotension (1 patient), and ECG [electrocardiogram] changes (1 patient)Data has been reported for completers only |
| Selective reporting (reporting bias) | Unclear risk | Unclear if all predefined outcomes have been reported. Efficacy data reported in graphs as means only. A study protocol is needed for firm conclusions |
| Other bias | Unclear risk | Preliminary results as four subjects had not completed the study |
Anticholinergic drugs
| Included study | Description | |
|---|---|---|
| Greil et al., 1984119 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . randomly assigned’; further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Double-blind . . . investigators were not informed about the study design |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of raters was not mentioned |
| Incomplete outcome data (attrition bias) | Low risk | Nine patients completed the trial. One patient dropped out one week after biperiden withdrawal because of severe parkinsonism; in this patient, only one rating could be carried out while on the placebo |
| Selective reporting (reporting bias) | High risk | TD symptoms data were not reported per randomised group, but before biperiden removal vs. after biperiden removal |
| Other bias | Unclear risk | Insufficient information to make a judgement |
Benzodiazepines
| Included study | Description | |
|---|---|---|
| Bobruff et al., 1981120 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | Diagnosis: psychiatric patients (details not reported). Obvious TD (at least three scores of mild or one score of moderate on AIMS), n = 21 Duration of TD: not reported Age: mean 51.6 years; range 36–63 years Sex: 16 male and five female |
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: supported in part by NIMH grant. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Patients were randomly assigned’; further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind’. Details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | ‘. . . double-blind’. Details not reported |
| Incomplete outcome data (attrition bias) | Low risk | Although not clearly reported, it seems that all subjects completed the double-blind phase (data reported for all 21 subjects) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes seem to have been reported but not as mean (SD). Also, as protocol is not available, it is not possible to verify that all predefined outcomes were reported |
| Other bias | Unclear risk | Insufficient information to make a judgement |
| Csernansky et al., 1988121,122 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: supported by a Public Health Service grant and a grant from the National Institute of Mental Health, a VA Career Development Award to the first author, a grant from the Upjohn Company and the Research Service of the VA. Participants were extracted post hoc from a larger study examining benzodiazepines for the treatment of the negative symptoms of schizophrenia. Data on age, sex, baseline medication doses, side effects and dropout rate for the initial cohort are provided in the parent study | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were randomly assigned to the treatment with either Alprozalam, Diadepam, or placebo . . .Further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Patients were randomly assigned to the treatment with either alprozalam, diadepam, or placebo under double-blind conditions. Identical capsules contained either 1 mg of alprozalam, 10 mg of diazepam, or the drug carrier as placebo |
| Blinding of outcome assessment (detection bias) | Low risk | Two independent raters |
| Incomplete outcome data (attrition bias) | Unclear risk | Fifty-five RDC schizophrenic outpatients were rated using the Gerlach Dyskinesia Scale (GDS) before, and at weekly intervals during, treatment . . . 17 patients were identified with rateable TD symptoms at baseline . . .All 17 subjects were entered to analysis. However, as 72 subjects were enrolled in the original study, it is unclear if relevant data for any of the 17 out of 72 subjects that dropped out are missing |
| Selective reporting (reporting bias) | Unclear risk | All outcomes for the main study seem to have been reported. A protocol is not available for verification. Although mental state and adverse effects have not reported separately for subjects with TD symptoms, TD was not an inclusion criterion and thus does not seem to affect biasSince TD was not a criterion for inclusion into or exclusion from the trial, it was only by chance that we identified 17 patients with TD symptoms |
| Other bias | High risk | Participants with TD at baseline were extracted post hoc from a larger study examining benzodiazepines for the treatment of the negative symptoms of schizophrenia |
| Weber et al., 198389 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Each patient was assigned randomly . . .’; further details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | As one of the groups received an intervention and the second standard care, blinding of participants and personnel could not have been possible |
| Blinding of outcome assessment (detection bias) | Low risk | . . . rater-blind . . . The rating scales were administered by trained observers who did not know which patients received diazepam |
| Incomplete outcome data (attrition bias) | Low risk | 13% dropout rateFifteen patients began the study. Two failed to complete the entire protocol (one because she continued to receive diazepam throughout the study and the other because she was discharged from the hospital) |
| Selective reporting (reporting bias) | Unclear risk | The outcomes seem to have been reported. However, a protocol is not available for verification |
| Other bias | Unclear risk | Change in medication for two participants may have had a confounding effect; however, both substitutions occurred 4 weeks into the second phase of the study |
| Xiang and Zhen, 199775 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: sponsorship source not reported. Participants with stable or aggravating symptoms of TD after suspending antipsychotics for 2 weeks were excluded | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . randomised controlled trial’. The author did not state the method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | . . . double blind . . . The two drugs were contained in capsules with same appearanceBlinding of participants and key study personnel ensured |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of outcome assessment not reported |
| Incomplete outcome data (attrition bias) | Low risk | All participants competed the study |
| Selective reporting (reporting bias) | Low risk | The author reported all measured outcomes |
| Other bias | Low risk | Free from other bias |
Vitamin E
| Included study | Description | |
|---|---|---|
| Adler et al., 1993125,126 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions | Stable antipsychotic medication: dose average (CPZE) vitamin E 536 mg/day (SD 642 mg/day); placebo 921 mg/day (SD 1026 mg/day). Compliance assessed by pill counts | |
| Outcomes |
|
|
| Notes |
|
|
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were randomly assigned to treatment with Vitamin E, 400 IU, or one matching placebo capsule, by mouth, b.i.d.No further details provided |
| Allocation concealment (selection bias) | Unclear risk | We used a randomisation of 3 : 2 (vitamin E to placebo) to maximise the number of patients receiving active treatment while maintaining the blindNo further details provided |
| Blinding of participants and personnel (performance bias) | Unclear risk | Both rater and patient were blind to the patient’s drug assignmentNo further details provided |
| Blinding of outcome assessment (detection bias) | Unclear risk | Both rater and patient were blind to the patient’s drug assignment’No further details provided |
| Incomplete outcome data (attrition bias) | Low risk | One patient dropped out after 2 weeks due to non-compliance . . . Two patients developed significant medical illnesses . . . unrelated to study treatment . . . By prior design, treatment for the first 8 patients was terminated after 8 weeks |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported |
| Other bias | Unclear risk | Baseline AIMS scores were somewhat higher in the vitamin E group than in the placebo group; however, this difference was not statistically significant. Small sample size |
| Adler et al., 1999137 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
Compliance assessed by pill counts |
|
| Outcomes |
|
|
| Notes | Source of funding: Cooperative Studies Program of the Department of Veterans Affairs, Veterans Affairs Headquarters, Washington, DC, USA. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation co-ordinated centrally |
| Allocation concealment (selection bias) | Low risk | Allocation with ‘biased coin’ method, stratified by site, age and baseline TD |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double blind: no further details |
| Blinding of outcome assessment (detection bias) | Unclear risk | Double blind: no further details |
| Incomplete outcome data (attrition bias) | Low risk | Of the 51 subjects who did not complete 1 year, most changed their minds about participating (n = 18), moved too far away from a site to continue in the study (n = 11), or were classified as ‘whereabouts unknown’ (n = 8) . . . Per protocol, we analysed the data according to the intention-to-treat principle |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported |
| Other bias | Unclear risk | No significant differences between groups’ baseline characteristics. Small sample size |
| Akhtar et al., 1993127 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | Diagnosis: psychiatric disorder (Spitzer criteria) and antipsychotic-induced TD (Schooler and Kane criteria192), n = 32 Sex: 14 female and 18 male Age: vitamin E, mean 53.06 years (SD 13.39 years); placebo, mean 56.87 years (SD 11.13 years) |
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Authors contacted but did not reply. Source of funding: not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The patients were then randomly assignedDetails not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Low risk | . . . double blind manner to receive either one capsule of 600 mg vitamin E or an identical placebo |
| Blinding of outcome assessment (detection bias) | Low risk | Both, the investigators and raters were blind to the nature of therapy . . . active drug or placebo till the completion of analysis |
| Incomplete outcome data (attrition bias) | Low risk | The study results seem to include all participants and there seem to be no dropouts from the study |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported |
| Other bias | Unclear risk | There was no significant difference in the demographic profile of the two groups. Small sample size |
| Dabiri et al., 1994128 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Authors contacted but did not reply. Source of funding: not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . random allocation’, no further details |
| Allocation concealment (selection bias) | Low risk | . . . patients were randomly divided into treatment and placebo groups by a non-clinical staff member |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind study’, details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Each patient was rated blindly by one of us (L.M.D.) before and after treatment using the Abnormal Involuntary Movement Scale (AIMS)Blinding details not reported |
| Incomplete outcome data (attrition bias) | Low risk | One patient who was taking vitamin E stopped treatment after 2 weeks because of diarrhoea, leaving five patients taking placebo and six vitamin E |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported, but there is no study protocol to confirm that all planned outcomes were reported |
| Other bias | Unclear risk | No statistically significant differences in AIMS baseline scores between groups. Very small sample size |
| Dorevitch et al., 199791 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Source of funding: not reported. Teva Pharmaceuticals supplied the vitamin E and placebo for this study. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . randomised’. Details not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind’. Blinding details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | ‘. . . double-blind’. Blinding details not reported |
| Incomplete outcome data (attrition bias) | Low risk | The study results seem to include all participants and there seem to be no dropouts from the study |
| Selective reporting (reporting bias) | High risk | TD symptoms (AIMS) were assessed but not reported |
| Other bias | Unclear risk | Baseline characteristics not reported. Very small sample size |
| Dorevitch et al., 199790 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Source of funding: not reported. Teva Pharmaceuticals supplied the vitamin E and placebo for this study. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Randomised’ – no further details |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind’; blinding details were not reported |
| Blinding of outcome assessment (detection bias) | Low risk | Two senior psychiatrists served as blinded raters |
| Incomplete outcome data (attrition bias) | Unclear risk | Two patients did not complete the study. Both patients were from the placebo phase of the placebo-vitamin E sequence group. One died while choking on food and the second as the result of a traffic accident |
| Selective reporting (reporting bias) | High risk | Addition of vitamin E or placebo did not adversely affect patient mental status as measured by brief psychiatric rating scale (BPRS)BPRS data not fully reported |
| Other bias | Unclear risk | Baseline characteristics not reported. Small sample size |
| Egan et al., 199292 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Three patients were not included in the data analysis: one dropped out and two had inconsistent vitamin E blood levels. Source of funding: not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were assigned randomlyDetails not reported |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind.’ Details not reported |
| Blinding of outcome assessment (detection bias) | Low risk | All raters were blind to treatment with either placebo or vitamin E |
| Incomplete outcome data (attrition bias) | High risk | Not ITT analysis:Eighteen patients who demonstrated high blood levels of vitamin E were included in the data analysisThree patients were excluded from the analysis |
| Selective reporting (reporting bias) | High risk | Data for mental state (PSAS and NSAS) not reported |
| Other bias | Unclear risk | Baseline characteristics not reported. Very small sample size |
| Elkashef et al., 199093 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Source of funding: not reported. Hollman-La Roche Inc., supplied the drug and placebo for this study. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The subjects were then assigned in a random, double-blind manner . . .No further details |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | Double blind: no further details |
| Blinding of outcome assessment (detection bias) | Unclear risk | The subjects were evaluated biweekly by a blind trained rater using the AIMS and the Brief Psychiatric Rating Scale (BPRS)Details of blinding not reported |
| Incomplete outcome data (attrition bias) | High risk | 2/5 participants in the placebo group dropped out, whereas none in the vitamin E group dropped out:Two patients did not complete the study, one because of noncompliance and the other experienced substantial side effects (nausea) while taking placebo |
| Selective reporting (reporting bias) | High risk | AIMS data partially reported and BPRS evaluated but not reported |
| Other bias | Unclear risk | The baseline severity of TD was closely matched in the two groups. Very small sample size |
| Lam et al., 199494 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | Diagnosis: schizophrenia (DSM-III-R193) and antipsychotic-induced TD (Schooler and Kane criteria192), n = 16 Sex: seven female and five malec Age: average 61.8 years (SD 12.8 years)c History: no history of chronicity of TD |
|
| Interventions | Stable antipsychotic medication. For those taking antipsychotic medication, the average daily dose was 365 mg CPZE | |
| Outcomes | TD symptoms: AIMS Leaving study early (assuming equal randomisation into the two groups) Unable to use: mental state – BPRS (no mean or SD reported), adverse effects |
|
| Notes | Four people left study early (no information about allocation), the reasons being death, deterioration of symptoms of schizophrenia, bacillary dysentery (all stated not to be related to treatment) and poor compliance. Authors contacted and replied, no more information available. Source of funding: not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Subjects were then selected randomlyNo further details |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind’. Details not reported |
| Blinding of outcome assessment (detection bias) | Low risk | Subjects were evaluated weekly with the AIMS . . . and Brief Psychiatric Rating Scale . . ., respectively, by two independent blind raters at the initial stabilisation period, and the last 2 weeks of each test period |
| Incomplete outcome data (attrition bias) | High risk | Twelve subjects completed the trial. One patient died of unrelated medical illness, one contracted bacillary dysentery and was dropped from the trial, and one had poor compliance and refused to continue medication. It was not reported which groups these participants were allocated to |
| Selective reporting (reporting bias) | High risk | TD symptoms data not reported as mean (SD); BPRS data not reported per period. Adverse effects not reported per group |
| Other bias | Unclear risk | Baseline characteristics not reported. Very small sample size |
| Lohr et al., 1996129 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions | Stable psychotropic medication for at least 1 month prior to entry into study. Antipsychotic dose average (CPZE) vitamin E 706 mg/day (SD 680 mg/day); placebo 376 mg/day (SD 242 mg/day) | |
| Outcomes |
|
|
| Notes | Source of funding: Partial funding by a VA Merit Review grant and United States Public Health Service grants. Vitamin E and placebo supplied by Hoffmann-La Roche Inc. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Patients were randomly assigned to receive either active vitamin E or sesame oil placebo gel caps |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment details not reported |
| Blinding of participants and personnel (performance bias) | Low risk | Patients were randomly assigned to receive either active vitamin E or sesame oil placebo gel caps, which were indistinguishable from the active gel caps |
| Blinding of outcome assessment (detection bias) | Unclear risk | Insufficient information to make a judgement |
| Incomplete outcome data (attrition bias) | High risk | Dropout rate of 36% (20/55 patients) but not reported per study group:2 developed manic symptoms necessitating medical changes, and 18 were non-compliant with either the vitamin E or the psychotropic medication. These 20 patients, who did not differ significantly from the remaining 35 patients in terms of age, gender, or diagnosis, were dropped from the study |
| Selective reporting (reporting bias) | High risk | Adverse effects: extrapyramidal side effects (parkinsonism) – data not reported |
| Other bias | Unclear risk | There were no significant differences in baseline characteristics between the two study groups. Small sample size |
| Sajjad, 1998130 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Source of funding: not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | . . . the patients were randomly divided into two groups using . . . a computer statistic programme |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | As an active group was compared with TAU, the study could not be double blinded. The only person blinded seems to have been the doctor. . . the dose increased by another doctor not involved in the ratings and who, therefore, was blind as to whether or not the patient was receiving a-tocopherol for the first month of the trial |
| Blinding of outcome assessment (detection bias) | High risk | Rater initially blind. However, after 1 month, the rater performed statistical tests and, hence, blindness was not maintained |
| Incomplete outcome data (attrition bias) | High risk | 40% dropout rate (12/20 participants completed the study): 6 out of 11 subjects in the intervention and 2 out of 9 subjects in the control group did not complete the trial. By the fourth month there were 12 patients left in the trial: five in the treatment group and seven in the control group. Patients excluded at this stage included those whose dose of antipsychotic medication was changed |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported |
| Other bias | Unclear risk | Mean AIMS scores and age were similar between groups at baseline. Very small sample size |
| Schmidt et al., 199195 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | Diagnosis: schizophrenia, depression, schizoaffective psychoses (no criteria) and antipsychotic-induced TD (no criteria), n = 23 Sex: 12 female and 11 male Age: average 45 years, range 21–88 years |
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | It was observed that two of the patients who benefited from the vitamin E therapy continued taking it: after stopping vitamin E medication, one of them experienced an increase in TD, whereas in the other the beneficial effect was still observed even 3 months later. Source of funding: not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . randomised pattern’, no further details |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | Unclear risk | ‘. . . double-blind’. Details not reported |
| Blinding of outcome assessment (detection bias) | Unclear risk | Details not reported |
| Incomplete outcome data (attrition bias) | Low risk | Of the 13 patients initially randomised to vitamin E, two left before the end of the study (one died and the other withdrew). Of the 10 patients initially randomised to placebo, two left before the end of the study (one died and the other had his treatment modified) |
| Selective reporting (reporting bias) | Unclear risk | All expected outcomes have been reported but there is no study protocol to confirm that all planned outcomes were reported |
| Other bias | Unclear risk | Baseline characteristics similar between study groups. Very small sample size, crossover design |
| Zhang et al., 2004138 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants | ||
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Source of funding: Not reported. Declarations of interest: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘Eligible patients were randomly assigned’; no further details |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment method not reported |
| Blinding of participants and personnel (performance bias) | Low risk | . . . either capsulized vitamin E (n = 22) or identically capsulized placebo (n = 19) using a double-blind fashion |
| Blinding of outcome assessment (detection bias) | Low risk | TD and psychotherapy were assess by blinded investigators |
| Incomplete outcome data (attrition bias) | Low risk | All randomised subjects seem to have completed the study |
| Selective reporting (reporting bias) | High risk | Outcome data were not reported for mental symptoms (PANSS) |
| Other bias | Low risk | No significant differences in demographic data were observed between vitamin E and placebo groups |
Buspirone
| Included study | Description | |
|---|---|---|
| Zeng, 199578 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes | Clinical response TD: AIMS Adverse events: dizziness, headache, nausea, vomiting Unable to use: blood routine examination, urine routine test and liver function test, electrocardiography, electroencephalography (the author only stated results of these tests were normal, but did not report the data) |
|
| Notes | Funding source: not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | ‘. . . randomly assigned . . .’, the author did not state the method of randomisation |
| Allocation concealment (selection bias) | Unclear risk | The author did not state the method of allocation concealment |
| Blinding of participants and personnel (performance bias) | Low risk | . . . double blind study, the interventions were coded as intervention A or B by the researcher in pharmacy . . . Participants and personnel did not know the allocation result. The two drugs were contained in capsules with same appearanceBlinding of participants and key study personnel ensured |
| Blinding of outcome assessment (detection bias) | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) | Low risk | All participants completed the study |
| Selective reporting (reporting bias) | Low risk | The author reported all measured outcomes |
| Other bias | Low risk | None obvious |
Hypnosis and relaxation
| Included study | Description | |
|---|---|---|
| Glover, 1980139 | ||
| Study characteristics | ||
| Characteristic | Description | |
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes | Sponsorship source: sponsorship source not reported | |
| Risk of bias | ||
| Bias | Authors’ judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Quasi-randomised. Assigned to the three groups in order of approaching the clinic |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment not reported |
| Blinding of participants and personnel (performance bias) | High risk | As subjects in group 1 received hypnosis, those in group 2 received relaxation training and those in group 3 received TAU without any other treatment, blinding could not be achieved |
| Blinding of outcome assessment (detection bias) | Unclear risk | Blinding of outcome assessors not reported |
| Incomplete outcome data (attrition bias) | Low risk | There were no refusals, or drop-outs among the referrals |
| Selective reporting (reporting bias) | Low risk | It seems that all outcomes have been reported. However, data is not usable |
| Other bias | Unclear risk | Baseline characteristics were similar but sample sizes very small |
Appendix 8 Characteristics of studies awaiting classification and ongoing
| Kar-Ahmadi, 2002141 | |
|---|---|
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Notes | A copy of this study was not available in The British Library |
| Zeng et al., 1996140 | |
| Methods | RCT |
| Participants | Schizophrenia with drug-induced tremor, n = 68 |
| Interventions |
|
| Outcomes |
|
| Notes | In Chinese, assessed by Sai Zhao. Study authors have been contacted to find out if participants were diagnosed with TD |
| Garcia and Crismon, 1992142 | |
|---|---|
| Study name | Double-blind placebo controlled study using buspirone in the treatment of tardive dyskinesia |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes | AIMS score |
| Notes | Abstract of a study protocol, there are no data to be extracted |
| Kajero, 2015144 | |
| Study name | Investigation of the potential beneficial effects of cannabidiol in the treatment of tardive dyskinesia |
| Methods | Randomised, double-blind, placebo-controlled study |
| Participants | Target number of participants: 28 per group Adults aged > 18 years who currently meet the ICD-10196 diagnosis of a psychotic disorder, verified with the Mini International Neuropsychiatric Interview questionnaire and who currently meet the clinical diagnosis of TD confirmed with the AIMS. Patients should currently be receiving treatment for a psychotic disorder and should be on either atypical or conventional antipsychotics |
| Interventions |
|
| Outcomes |
|
| Starting date | 1 December 2015 |
| Notes | Source of funding: Federal Neuropsychiatric Hospital, Nigeria. Trial is part of a Stellenbosch University PhD. Intention to publish date: 1 January 2018 |
| Reynolds, 2002143 | |
| Methods |
|
| Participants | Schizophrenic patients with TD |
| Interventions |
|
| Outcomes | Prevalence and severity of abnormal involuntary movements |
| Notes | Very limited information from two trial registries. We were unable to locate author contact details |
Appendix 9 Non-prioritised comparisons: results overview
| Study; setting | Participant characteristics | Interventions | Outcome | Effect estimate (95% CI); n | Risk of bias | |||
|---|---|---|---|---|---|---|---|---|
| Selection | Performance | Detection | Attrition | |||||
| Anticholinergics | ||||||||
| Bucci, 1971;197 outpatients in the USA |
|
|
TD: No clinical improvement | RR 4.20 (1.40 to 12.58); 20 | UC | High | High | Low |
| Adverse events: any | RR 0.33 (0.02 to 7.32); 20 | UC | High | High | Low | |||
| Leaving the study early | RR 0.33 (0.02 to 7.32); 20 | UC | Low | Low | Low | |||
| Calcium channel blockers | ||||||||
| Loonen et al., 1992;198 inpatients in the Netherlands |
|
|
Mental state: deterioration | Not estimable;b 18 | UC | Low | UC | High |
| Schwartz et al., 1997;199,200 setting not reported, the USA |
|
|
This study did not report on any of the selected outcomes | Not estimable; 15 | UC | UC | UC | High |
| Zeng et al., 1994;80 inpatients in China |
|
|
Adverse events: any | Not estimable;b 20 | UC | Low | UC | Low |
| Cholinergic medication | ||||||||
| Beckham, 1981;201 inpatients and outpatients in the USA |
|
|
Mental state: deterioration | RR 0.33 (0.01 to 7.81); 50 | UC | Low | Low | High |
| Leaving the study early | RR 0.50 (0.17 to 1.45); 50 | UC | Low | Low | High | |||
| Caroff et al., 2007;202,203 inpatients in the USA |
|
|
Leaving the study early | RR 3.00 (0.96 to 9.39); 38 | UC | Low | Low | UC |
| de Montigny et al., 1979;204 inpatients in Canada |
|
|
Leaving the study early | Not estimable;b 20 | UC | Low | Low | Low |
| Gelenberg et al., 1990;205 outpatients in the USA |
|
|
This study did not report on any of the selected outcomes | Not estimable; 14 | UC | UC | UC | High |
| George et al., 1981;206 inpatients in Australia |
|
|
Leaving the study early | Not estimable;b 33 | UC | Low | Low | Low |
| Jackson, 1978;207 inpatients in the USA |
|
|
TD: no clinical improvement | RR 0.84 (0.39 to 1.81); 6 | UC | UC | Low | UC |
| TD: deterioration | RR 0.36 (0.09 to 1.51); 6 | UC | UC | Low | UC | |||
| Mental state: deterioration | Not estimable;b 6 | UC | UC | Low | UC | |||
| Leaving the study early | Not estimable;b 6 | UC | Low | Low | UC | |||
| Jackson et al., 1979;208,209 inpatients in the USA |
|
|
TD: no clinical improvement | RR 0.71 (0.31 to 1.66); 6 | UC | Low | Low | UC |
| TD: deterioration | RR 0.33 (0.02 to 5.97); 6 | UC | Low | Low | UC | |||
| Mental state: deterioration | Not estimable;b 6 | UC | Low | Low | UC | |||
| Leaving the study early | Not estimable;b 6 | UC | Low | Low | UC | |||
| Jahanian et al., 2014;87 inpatients in Iran |
|
|
This study did not report on any of the selected outcomes | Not estimable; 40 | UC | UC | UC | UC |
| Kocher et al., 1980;82 inpatients in Switzerland |
|
|
TD: deterioration | RR 1.00 (0.17 to 5.77); 20 | UC | UC | Low | Low |
| Leaving the study early | Not estimable;b 20 | UC | Low | Low | Low | |||
| Lucius, 1978;83,210 inpatients in Germany |
|
|
TD: deterioration | RR 3.00 (0.45 to 19.93); 10 | UC | UC | Low | UC |
| Mental state: deterioration | RR 0.33 (0.02 to 6.65); 10 | UC | UC | Low | UC | |||
| Leaving the study early | RR 0.33 (0.02 to 6.65); 10 | UC | Low | Low | UC | |||
| Ogunmefun et al., 2009;211 setting and country not reported |
|
|
TD: no clinical improvement | RR 1.00 (0.70 to 1.43); 10 | UC | UC | Low | UC |
| TD: deterioration | RR 0.67 (0.06 to 7.85); 10 | UC | UC | Low | UC | |||
| Leaving the study early | Not estimable;b 10 | UC | Low | Low | UC | |||
| Price, 1982;212 inpatients in the USA |
|
|
TD: deterioration | RR 3.00 (0.13 to 68.26); 30 | UC | Low | Low | Low |
| Leaving the study early | Not estimable;b 30 | UC | Low | Low | Low | |||
| Tarsy and Bralower, 1977;213 inpatients and outpatients in the USA |
|
|
TD: no clinical improvement | RR 1.00 (0.43 to 2.34); 5 | UC | UC | UC | Low |
| TD: deterioration | RR 2.00 (0.16 to 25.75); 5 | UC | UC | UC | Low | |||
| Mental state: deterioration | RR 1.20 (0.08 to 18.75); 5 | UC | UC | UC | Low | |||
| Leaving the study early | RR 1.20 (0.08 to 18.75); 5 | UC | Low | Low | Low | |||
| Yagi, 1990;86,214,215 inpatients in Japan |
|
|
TD: deterioration | RR 1.87 (0.18 to 19.55); 60 | UC | UC | UC | Low |
| Adverse events: any | RR 0.56 (0.15 to 2.14); 60 | UC | UC | UC | Low | |||
| Leaving the study early | Not estimable;b 60 | UC | Low | Low | Low | |||
| GABA agonists | ||||||||
| Ananth et al., 1987;216 inpatients in Canada |
|
|
TD: deterioration | Not estimable;b 10 | UC | Low | UC | Low |
| Mental state: deterioration | Not estimable;b 10 | UC | Low | UC | Low | |||
| Adverse events: any | Not estimable;b 10 | UC | Low | UC | Low | |||
| Leaving the study early | Not estimable;b 10 | UC | Low | Low | Low | |||
| Burner et al., 1989;217 setting and country not reported |
|
|
TD: no clinical improvement | RR 0.68 (0.36 to 1.25); 13 | UC | Low | UC | Low |
| Mental state: deterioration | RR 1.82 (0.11 to 30.27); 13 | UC | Low | UC | Low | |||
| Leaving the study early | RR 1.09 (0.05 to 21.67); 13 | UC | Low | Low | Low | |||
| Fisk and York, 1987;218 inpatients and outpatients in the UK |
|
|
TD: no clinical improvement | RR 0.94 (0.80 to 1.11); 62 | Low | Low | Low | High |
| TD: deterioration | RR 3.41 (0.77 to 15.19); 47 | Low | Low | Low | High | |||
| Mental state: deterioration | RR 2.27 (0.22 to 23.38); 47 | Low | Low | Low | High | |||
| Leaving the study early | RR 2.42 (0.86 to 6.77); 62 | Low | Low | Low | High | |||
| Gerlach, 1977;219,220 inpatients in Denmark |
|
|
TD: deterioration | RR 2.45 (0.11 to 53.25); 18 | UC | UC | UC | UC |
| Mental state: deterioration | RR 4.09 (0.22 to 74.78); 18 | UC | UC | UC | UC | |||
| Leaving the study early | RR 0.20 (0.01 to 3.70); 20 | UC | Low | Low | UC | |||
| Glazer et al., 1985;221 inpatients in the USA |
|
|
TD: no clinical improvement | RR 0.87 (0.66 to 1.14); 31 | UC | UC | UC | High |
| TD: deterioration | RR 0.94 (0.06 to 13.68); 31 | UC | UC | UC | High | |||
| Mental state: deterioration | Not estimable;b 31 | UC | UC | UC | High | |||
| Linnoila et al., 1976;222 inpatients in Finland |
|
|
This study did not report on any of the selected outcomes | Not estimable; 32 | UC | Low | UC | Low |
| Mei and Zhu, 2008;73 inpatients in China |
|
|
TD: no clinical improvement | RR 0.67 (0.45 to 0.98); 40 | UC | Low | Low | Low |
| Mental state: average end-point score | MD 0.03 (–3.29 to 3.35); 40 | UC | Low | Low | Low | |||
| Nair et al., 1978;223,224 inpatients in the USA |
|
|
This study did not report on any of the selected outcomes | Not estimable; 10 | UC | UC | Low | UC |
| Stewart et al., 1982;225,226 setting and country not reported |
|
|
TD: no clinical improvement | RR 0.90 (0.60 to 1.36); 33 | UC | UC | UC | UC |
| TD: deterioration | RR 0.65 (0.07 to 6.45); 30 | UC | UC | UC | UC | |||
| Leaving the study early | RR 0.68 (0.07 to 6.76); 33 | UC | Low | Low | UC | |||
| Thaker et al., 1987;227 inpatients in the USA |
|
|
Mental state: deterioration | RR 3.00 (0.24 to 37.67); 2 | UC | UC | Low | Low |
| Yin et al., 2004;77 inpatients in China |
|
|
TD: no clinical improvement | RR 0.80 (0.68 to 0.94); 79 | UC | Low | UC | Low |
| Leaving the study early | RR 3.00 (0.13 to 71.51); 80 | UC | Low | Low | Low | |||
| Miscellaneous treatments | ||||||||
| Cai, 1988;71 setting and country not reported |
|
|
TD: no clinical improvement | RR 0.54 (0.35 to 0.82); 57 | UC | Low | Low | Low |
| Mental state: average end-point score | MD –4.50 (–7.60 to –1.40); 20 | UC | Low | Low | Low | |||
| Adverse events: any | Not estimable;b 57 | UC | Low | Low | Low | |||
| Leaving the study early | Not estimable;b 57 | UC | Low | Low | Low | |||
| Castro et al., 2011;228 inpatients and outpatients in Venezuela |
|
|
TD: no clinical improvement | RR 0.74 (0.44 to 1.23); 13 | UC | Low | UC | Low |
| Mental state: deterioration | Not estimable;b 13 | UC | Low | UC | Low | |||
| Adverse events: any | Not estimable;b 13 | UC | Low | UC | Low | |||
| Leaving the study early | Not estimable;b 13 | UC | Low | Low | Low | |||
| Emsley et al., 2006;229–233 inpatients and outpatients in South Africa |
|
|
Mental state: deterioration | RR 0.49 (0.05 to 5.14); 75 | UC | Low | UC | Low |
| Adverse events: EPS | MD 0.30 (–1.17 to 1.77); 75 | UC | Low | UC | Low | |||
| Leaving the study early | RR 0.57 (0.27 to 1.22); 84 | UC | Low | Low | Low | |||
| Gardos et al., 1979;234 inpatients in the USA |
|
|
This study did not report on any of the selected outcomes | Not estimable; 22 | UC | High | Low | UC |
| Glazer et al., 1985;235 outpatients in the USA |
|
|
TD: no clinical improvement | RR 1.18 (0.76 to 1.83); 12 | UC | UC | UC | Low |
| TD: deterioration | RR 0.20 (0.01 to 3.35); 11 | UC | UC | UC | Low | |||
| Adverse events: any | RR 0.33 (0.02 to 6.86); 12 | UC | UC | UC | Low | |||
| Leaving the study early | RR 1.00 (0.08 to 12.56); 12 | UC | Low | Low | Low | |||
| Goff et al., 1993;236 outpatients in the USA |
|
|
TD: no clinical improvement | RR 1.37 (0.96 to 1.94); 33 | UC | Low | Low | High |
| Leaving the study early | RR 10.39 (0.62 to 173.97); 33 | UC | Low | Low | High | |||
| Hajioff and Wallace, 1983;237 inpatients in the UK |
|
|
Leaving the study early | RR 0.33 (0.02 to 7.32); 20 | UC | Low | Low | Low |
| Kojima et al., 1992;238,239 inpatients and outpatients in Japan |
|
|
TD: deterioration | RR 0.33 (0.01 to 7.90); 66 | UC | UC | UC | High |
| Adverse events: any | RR 1.13 (0.61 to 2.07); 85 | UC | UC | UC | High | |||
| Leaving the study early | RR 1.09 (0.49 to 2.40); 85 | UC | Low | Low | High | |||
| Koshino et al., 1979;85 inpatients in Japan |
|
|
TD: deterioration | RR 0.33 (0.01 to 7.74); 42 | UC | UC | UC | Low |
| Adverse events: any | RR 0.33 (0.04 to 2.95); 42 | UC | UC | UC | Low | |||
| Leaving the study early | RR 0.33 (0.01 to 7.74); 42 | UC | Low | Low | Low | |||
| Koshino et al., 1983;84 inpatients in Japan |
|
|
TD: no clinical improvement | RR 0.45 (0.21 to 0.97); 28 | UC | UC | UC | Low |
| TD: deterioration | RR 0.33 (0.01 to 7.55); 28 | UC | UC | UC | Low | |||
| Mental state: deterioration | RR 0.50 (0.05 to 4.90); 28 | UC | UC | UC | Low | |||
| Adverse events: any | RR 2.33 (0.75 to 7.23); 28 | UC | UC | UC | Low | |||
| Leaving the study early | Not estimable;b 28 | UC | Low | Low | Low | |||
| Libov et al., 2007;240–243 inpatients in Israel |
|
|
Adverse events: EPS | MD 2.50 (–4.73 to 9.73); 35 | UC | UC | Low | High |
| Leaving the study early | RR 0.23 (0.03 to 1.85); 40 | UC | Low | Low | High | |||
| MacKay et al., 1980;244 inpatients in the UK |
|
|
TD: no clinical improvement | RR 1.59 (0.79 to 3.23); 11 | UC | Low | Low | Low |
| TD: deterioration | RR 4.29 (0.25 to 72.90); 11 | UC | Low | Low | Low | |||
| Adverse events: any | RR 6.00 (0.38 to 94.35); 11 | UC | Low | Low | Low | |||
| Leaving the study early | RR 2.57 (0.13 to 52.12); 11 | UC | Low | Low | Low | |||
| Matsunaga et al., 1988;245 inpatients in Japan |
|
|
TD: deterioration | RR 2.85 (0.12 to 65.74); 37 | UC | UC | UC | High |
| Adverse events: any | RR 3.79 (0.47 to 30.77); 37 | UC | UC | UC | High | |||
| Meco et al., 1989;246 inpatients in Italy |
|
|
TD: no clinical improvement | RR 1.00 (0.70 to 1.43); 10 | UC | UC | UC | Low |
| TD: Deterioration | RR 0.47 (0.02 to 9.26); 10 | UC | UC | UC | Low | |||
| Mental state: deterioration | RR 0.47 (0.02 to 9.26); 10 | UC | UC | UC | Low | |||
| Mosnik et al., 1995;247–249 inpatients and outpatients in the USA |
|
|
Leaving the study early | RR 2.45 (0.11 to 53.25); 18 | UC | Low | Low | Low |
| Mouret et al., 1991;250 inpatients in Morocco |
|
|
TD: no clinical improvement | RR 0.52 (0.29 to 0.96); 20 | UC | UC | UC | UC |
| TD: deterioration | RR 0.14 (0.01 to 2.45); 20 | UC | UC | UC | UC | |||
| Leaving the study early | Not estimable;b 20 | UC | Low | Low | UC | |||
| O’Brien et al., 2014;251,252 inpatients and outpatients in the USA |
|
|
TD: no clinical improvement | RR 0.58 (0.41 to 0.82); 88 | UC | UC | Low | UC |
| Adverse events: any | RR 1.88 (0.73 to 4.84); 88 | UC | UC | Low | UC | |||
| Leaving the study early | RR 1.26 (0.39 to 4.03); 88 | UC | Low | Low | UC | |||
| Rastogi et al., 1982;253 inpatients in the UK |
|
|
This study did not report on any of the selected outcomes | Not estimable; 40 | UC | Low | UC | UC |
| Richardson et al., 2003;254 inpatients and outpatients in the USA |
|
|
TD: no clinical improvement | RR 0.79 (0.63 to 1.00); 52 | UC | UC | Low | UC |
| TD: deterioration | RR 0.29 (0.07 to 1.19); 36 | UC | UC | Low | UC | |||
| Leaving the study early | RR 0.84 (0.37 to 1.92); 52 | UC | Low | Low | UC | |||
| Shamir et al., 2000;255 inpatients in Israel |
|
|
TD: no clinical improvement | RR 1.00 (0.83 to 1.21); 19 | Low | Low | Low | Low |
| TD: deterioration | RR 0.22 (0.01 to 4.05); 19 | Low | Low | Low | Low | |||
| Adverse events: any | Not estimable;b 19 | Low | Low | Low | Low | |||
| Leaving the study early | Not estimable;b 19 | Low | Low | Low | Low | |||
| Shamir et al., 2001;256,257 inpatients in Israel |
|
|
Adverse events: any | Not estimable;b 22 | Low | Low | UC | Low |
| Leaving the study early | Not estimable;b 22 | Low | Low | Low | Low | |||
| Shi et al., 2009;74 inpatients in China |
|
|
This study did not report on any of the selected outcomes | Not estimable; 76 | UC | High | UC | Low |
| UCB Pharma, 2005;258 inpatients in Belgium and Bulgaria |
|
|
Adverse events: any | RR 0.51 (0.25 to 1.04); 69 | UC | UC | UC | High |
| Leaving the study early | RR 0.21 (0.03 to 1.67); 69 | UC | Low | Low | High | |||
| Wolkin et al., 1986;259 inpatients and outpatients in the USA |
|
|
TD: no clinical improvement | RR 1.00 (0.69 to 1.45); 16 | UC | UC | UC | Low |
| TD: deterioration | RR 1.50 (0.34 to 6.70); 16 | UC | UC | UC | Low | |||
| Mental state: average end-point score | MD –6.00 (–15.99 to 3.99); 10 | UC | UC | UC | Low | |||
| Leaving the study early | Not estimable;b 16 | UC | Low | Low | Low | |||
| Woods et al., 2008;260,261 outpatients in the USA |
|
|
Mental state: deterioration | RR 0.67 (0.12 to 3.65); 50 | UC | UC | UC | Low |
| Leaving the study early | RR 1.80 (0.70 to 4.62); 50 | UC | Low | Low | Low | |||
| Yang et al., 1999;76 inpatients in China |
|
|
TD: no clinical improvement | RR 0.24 (0.11 to 0.55); 34 | Low | UC | Low | Low |
| Mental state: average end-point score | MD 0.70 (–3.77 to 5.17); 34 | Low | UC | Low | Low | |||
| Adverse events: any | MD –0.10 (–0.53 to 0.33); 34 | Low | UC | Low | Low | |||
| Adverse events: EPS | MD –0.50 (–1.36 to 0.36); 34 | Low | UC | Low | Low | |||
| Zeng, 1996;79 inpatients in China |
|
|
TD: no clinical improvement | RR 0.48 (0.29 to 0.77); 46 | UC | Low | UC | Low |
| Leaving the study early | Not estimable;b 46 | UC | Low | Low | Low | |||
| Zhang et al., 2011;262–264 inpatients in China |
|
|
TD: no clinical improvement | RR 0.88 (0.81 to 0.96); 157 | Low | Low | Low | Low |
| Mental state: deterioration | RR 0.34 (0.01 to 8.16); 157 | Low | Low | Low | Low | |||
| Leaving the study early | RR 0.25 (0.03 to 2.22); 157 | Low | Low | Low | Low | |||
| AP reduction and/or cessation and APs | ||||||||
| Glazer and Hafez, 1990;189,190 outpatients in the USA |
|
|
Leaving the study early | Not estimable;b 18 | UC | Low | Low | Low |
| Kazamatsuri et al., 1972;169 inpatients in the USA |
|
|
TD: no clinical improvement | RR 1.53 (0.58 to 4.05); 20 | UC | UC | Low | Low |
| TD: deterioration | RR 1.22 (0.09 to 16.92); 20 | UC | UC | Low | Low | |||
| Leaving the study early | RR 0.24 (0.01 to 4.44); 20 | UC | Low | Low | Low | |||
| Lublin et al., 1991;188 inpatients in Denmark and Finland |
|
|
TD: no clinical improvement | RR 1.00 (0.79 to 1.27); 15 | UC | High | Low | Low |
| TD: deterioration | RR 0.88 (0.16 to 4.68); 15 | UC | High | Low | Low | |||
| Adverse events: EPS | MD –4.81 (–12.15 to 2.53); 15 | UC | High | Low | Low | |||
| Non-AP catecholaminergic drugs | ||||||||
| Buruma et al., 1982;265,266 inpatients in the Netherlands |
|
|
Leaving the study early | Not estimable;b 12 | UC | Low | Low | Low |
| Chen et al., 1995;72 inpatients in China |
|
|
Leaving the study early | Not estimable;b 20 | UC | Low | Low | Low |
| Hebenstreit et al., 1986;81 inpatients in Austria |
|
|
Quality of life: no improvement | RR 0.87 (0.68 to 1.12); 35 | UC | Low | UC | UC |
| Leaving the study early | RR 5.28 (0.27 to 102.58); 35 | UC | Low | Low | UC | |||
| Huang et al., 1980;267,268 inpatients in the USA |
|
|
TD: no clinical improvement | RR 0.33 (0.14 to 0.80); 20 | UC | Low | UC | UC |
| TD: deterioration | RR 0.33 (0.02 to 7.32); 20 | UC | Low | UC | UC | |||
|
TD: no clinical improvement | RR 0.60 (0.19 to 1.86); 20 | UC | Low | UC | UC | ||
| TD: deterioration | Not estimable;b 20 | UC | Low | UC | UC | |||
|
TD: no clinical improvement | RR 0.52 (0.29 to 0.96); 20 | UC | Low | UC | UC | ||
| TD: deterioration | RR 0.33 (0.02 to 7.32); 20 | UC | Low | UC | UC | |||
| Karniol et al., 1983;88 inpatients in Brazil |
|
|
This study did not report on any of the selected outcomes | Not estimable; 20 | UC | Low | UC | Low |
| Pappa et al., 2010;269–271 outpatients in Greece |
|
|
Leaving the study early | Not estimable;b 22 | UC | Low | Low | Low |
| Rust, 1984;272 inpatients in France |
|
|
Leaving the study early | Not estimable;b 50 | UC | Low | Low | Low |
| Simpson et al., 1988;273 inpatients in the USA |
|
|
TD: deterioration | RR 1.78 (0.44 to 7.25); 17 | UC | Low | UC | High |
| Leaving the study early | RR 0.18 (0.01 to 3.27); 17 | UC | Low | Low | High | |||
| Soni et al., 1986;274 inpatients in the UK |
|
|
Mental state: deterioration | RR 2.20 (0.22 to 22.45); 42 | UC | UC | Low | High |
| Leaving the study early | RR 1.73 (0.83 to 3.58); 42 | UC | Low | Low | High | |||
Appendix 10 Analyses: forest plots for prioritised comparisons
Antipsychotic reduction versus continuation
Reduced versus standard dose of flupentixol decanoate98 or fluphenazine decanoate97
Figures 23–26 present forest plots of outcome analyses for antipsychotic reduction versus continuation.
FIGURE 23.
Antipsychotic reduction vs. continuation: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 44–48 weeks). df, degrees of freedom; M–H, Mantel–Haenszel.
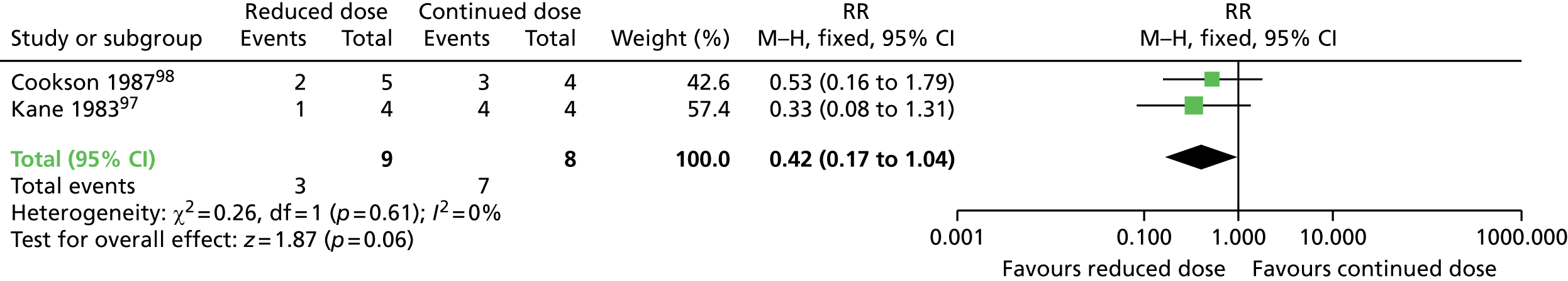
FIGURE 24.
Antipsychotic reduction vs. continuation: forest plot for the outcome ‘TD – deterioration’ (follow-up 44–48 weeks). df, degrees of freedom; M–H, Mantel–Haenszel.

FIGURE 25.
Antipsychotic reduction vs. continuation: forest plot for the outcome ‘mental state – relapse’ (follow-up 44–48 weeks). M–H, Mantel–Haenszel.
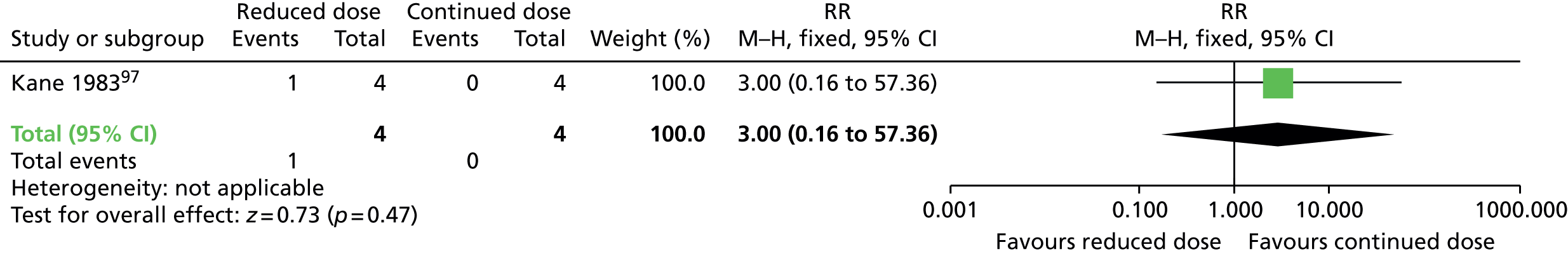
FIGURE 26.
Antipsychotic reduction vs. continuation: forest plot for the outcome ‘leaving the study early’ (follow-up 44–48 weeks). M–H, Mantel–Haenszel.

Antipsychotic switch versus withdrawal (with placebo)
Figures 27–30 present forest plots of outcome analyses for antipsychotic switch versus antipsychotic withdrawal.
FIGURE 27.
Antipsychotic switch vs. withdrawal: forest plot for the outcome ‘TD: no clinically important improvement’ (follow-up 12 weeks). M–H, Mantel–Haenszel.
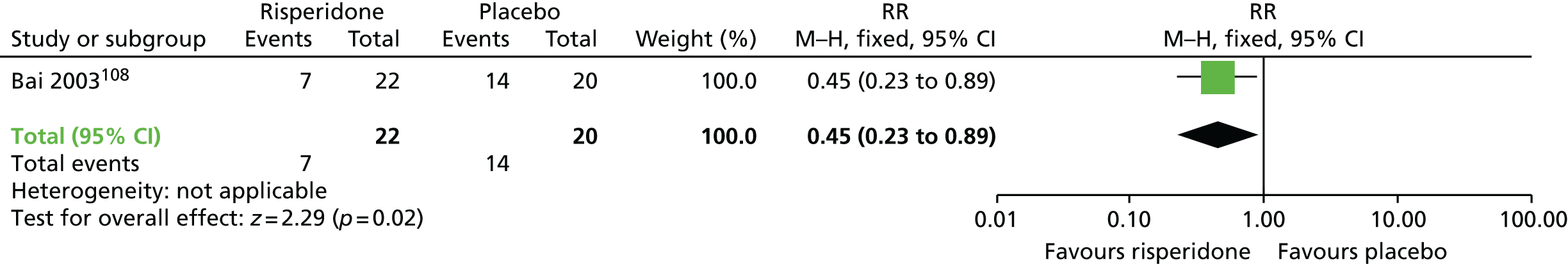
FIGURE 28.
Antipsychotic switch vs. withdrawal: forest plot for the outcome ‘general mental state – average end-point score (BPRS, high score means worse outcome)’ (follow-up 12 weeks). IV, inverse variance.

FIGURE 29.
Antipsychotic switch vs. withdrawal: forest plot for the outcome ‘adverse events – need of antiparkinsonism drugs’ (follow-up 8–12 weeks). df, degrees of freedom; IV, inverse variance. a, Split placebo group.
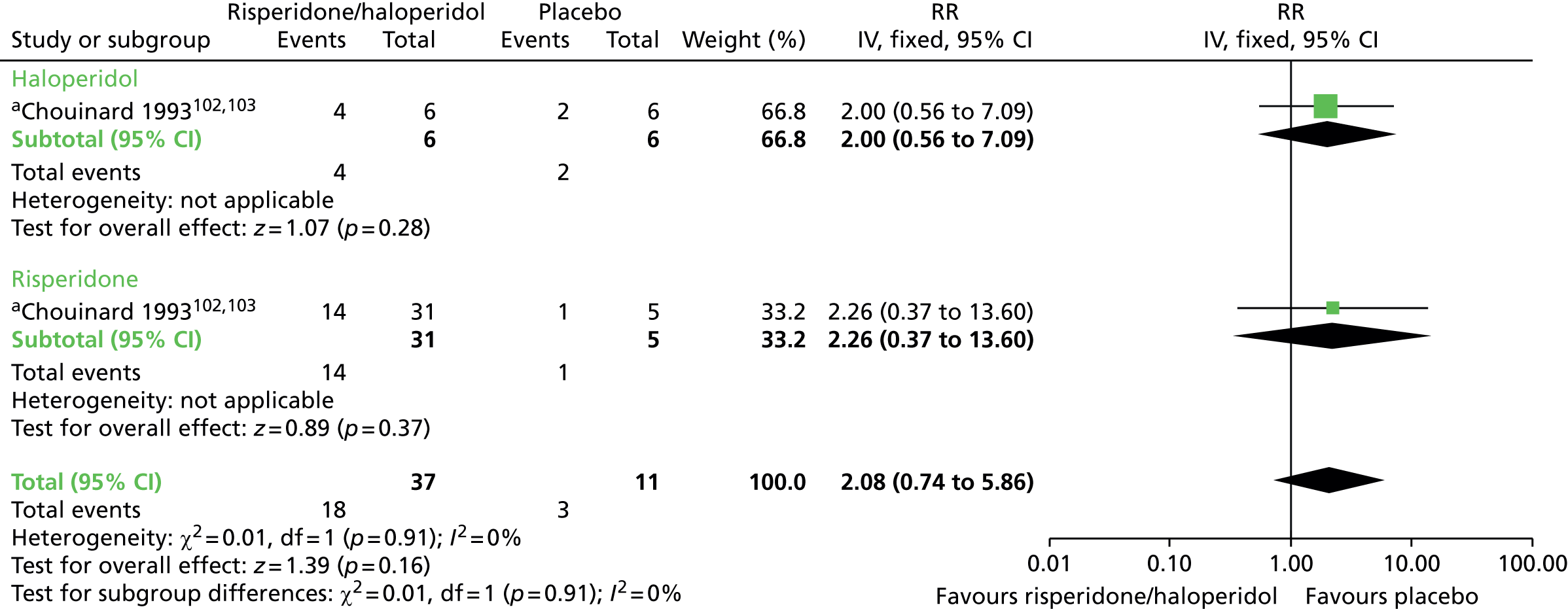
FIGURE 30.
Antipsychotic switch vs. withdrawal: forest plot for the outcome ‘leaving the study early’ (follow-up 12 weeks). IV, inverse variance.

Switch to second-generation antipsychotic versus switch to first-generation antipsychotic
Figures 31–35 present forest plots of outcome analyses for switch to SGA versus switch to FGA.
FIGURE 31.
Switch to SGA vs. switch to FGA: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 6 months). M–H, Mantel–Haenszel.

FIGURE 32.
Switch to SGA vs. switch to FGA: forest plot for the outcome ‘adverse events – need of antiparkinsonism drugs’ (follow-up 1 year). df, degrees of freedom; M–H, Mantel–Haenszel.
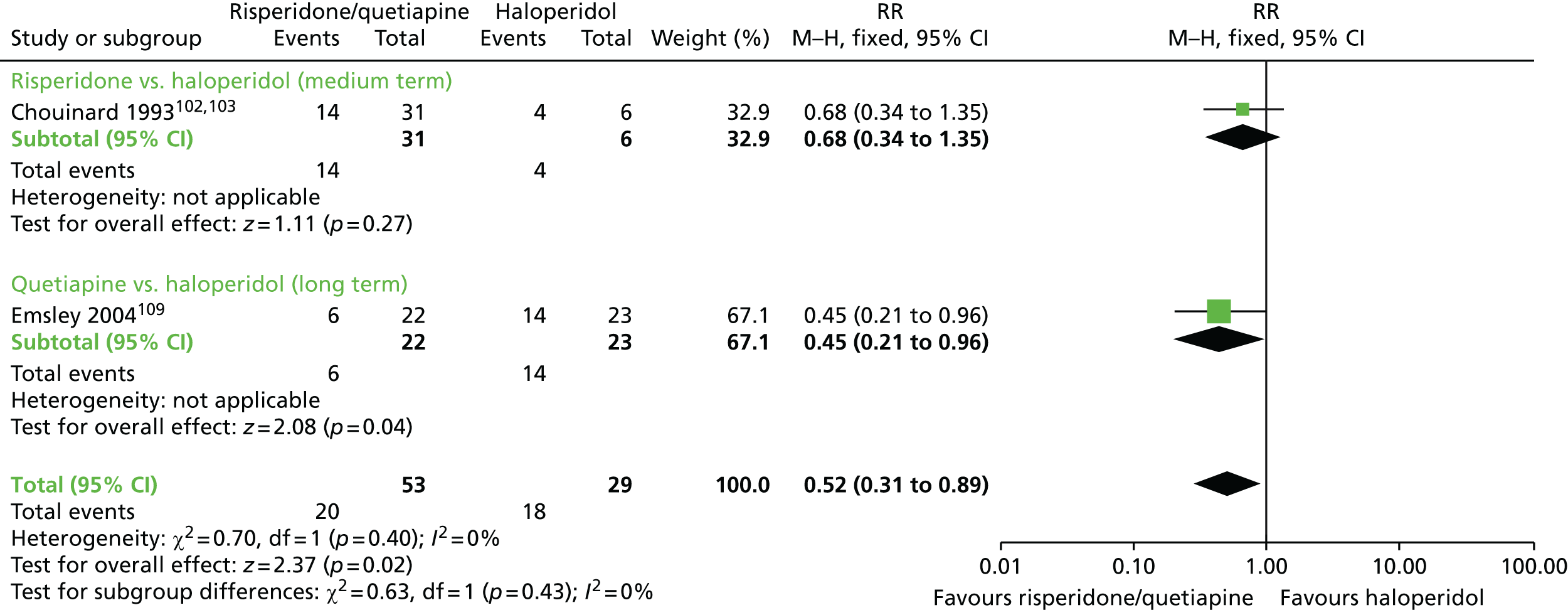
FIGURE 33.
Switch to SGA vs. switch to FGA: forest plot for the outcome ‘adverse events: general – average change scores (UKU, high score means worse outcome)’ (follow-up 6 months). ASP, amisulpride; IV, inverse variance; OLZ, olanzapine; SD, standard deviation.
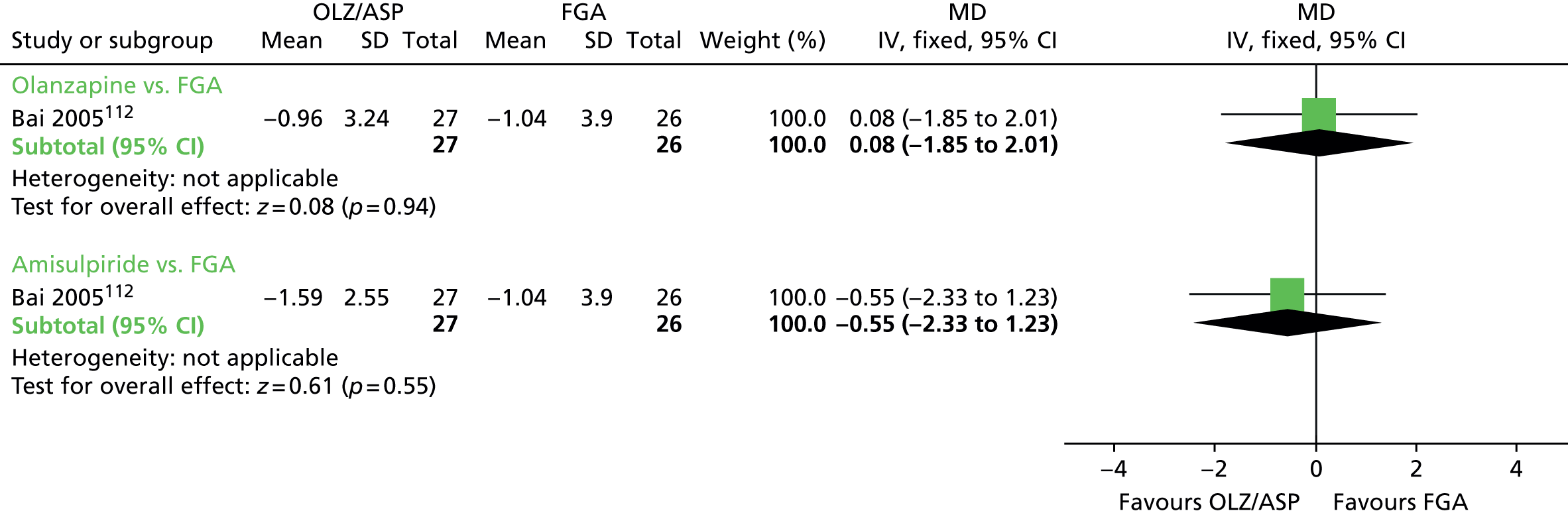
FIGURE 34.
Switch to SGA vs. switch to FGA: forest plot for the outcome ‘mental state – deterioration’ (follow-up 1 year). M–H, Mantel–Haenszel.

FIGURE 35.
Switch to SGA vs. switch to FGA: forest plot for the outcome ‘leaving the study early’ – medium term (follow-up 24–52 weeks). df, degrees of freedom; IV, inverse variance. a, FGA group split.

Switch to second-generation antipsychotic versus switch to another second-generation antipsychotic
Figures 36–48 present forest plots of outcome analyses for switch to SGA versus switch to another SGA.
Olanzapine versus amisulpride
FIGURE 36.
Olanzapine vs. amisulpride: forest plot for the outcome ‘general mental state – average change score (BPRS, high score means worse outcome)’ (follow-up 6 months). IV, inverse variance; SD, standard deviation.

FIGURE 37.
Olanzapine vs. amisulpride: forest plot for the outcome ‘adverse events: parkinsonism – average change score (SAS, high score means worse outcome)’ (follow-up 6 months). IV, inverse variance; SD, standard deviation.

FIGURE 38.
Olanzapine vs. amisulpride: forest plot for the outcome ‘adverse events: general – average change scores (UKU, high score means worse outcome)’ (follow-up 6 months). IV, inverse variance; SD, standard deviation.

FIGURE 39.
Olanzapine vs. amisulpride: forest plot for the outcome ‘leaving the study early’ (follow-up 6 months). IV, inverse variance.

Olanzapine versus risperidone
FIGURE 40.
Olanzapine vs. risperidone: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 6 months). M–H, Mantel–Haenszel.
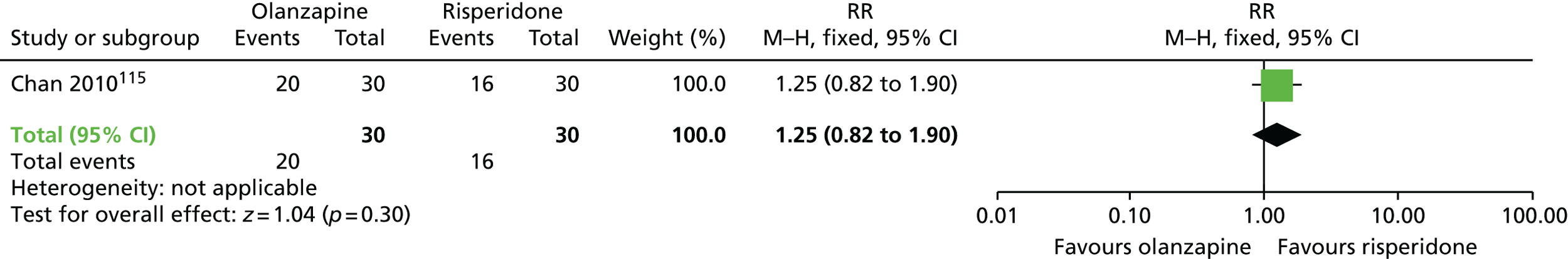
FIGURE 41.
Olanzapine vs. risperidone: forest plot for the outcome ‘mental state – deterioration’ (follow-up 6 months). M–H, Mantel–Haenszel.

FIGURE 42.
Olanzapine vs. risperidone: forest plot for the outcome ‘adverse effects: parkinsonism – average change score (ESRS, high score means worse outcome)’ (follow-up 6 months). IV, inverse variance; SD, standard deviation.

FIGURE 43.
Olanzapine vs. risperidone: forest plot for the outcome ‘leaving the study early’ (follow-up 6–18 months). df, degrees of freedom; IV, inverse variance.

Olanzapine versus quetiapine
FIGURE 44.
Olanzapine vs. quetiapine: forest plot for the outcome ‘leaving the study early’ (follow-up 18 months). IV, inverse variance.

Olanzapine versus ziprasidone
FIGURE 45.
Olanzapine vs. ziprasidone: forest plot for the outcome ‘leaving the study early’ (follow-up 18 months). IV, inverse variance.

Quetiapine versus risperidone
FIGURE 46.
Quetiapine vs. risperidone: forest plot for the outcome ‘leaving the study early’ (follow-up 18 months). IV, inverse variance.

Quetiapine versus ziprasidone
FIGURE 47.
Quetiapine vs. ziprasidone: forest plot for the outcome ‘leaving the study early’ (follow-up 18 months). IV, inverse variance.

Ziprasidone versus risperidone
FIGURE 48.
Ziprasidone vs. risperidone: forest plot for the outcome ‘leaving the study early’ (follow-up 18 months). IV, inverse variance.

Antipsychotic versus other drug
Haloperidol versus tetrabenazine
Figures 49–51 present forest plots of outcome analyses for haloperidol versus tetrabenazine.
FIGURE 49.
Haloperidol vs. tetrabenazine: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 18 weeks). IV, inverse variance.

FIGURE 50.
Haloperidol vs. tetrabenazine: forest plot for the outcome ‘TD – deterioration’ (follow-up 18 weeks). IV, inverse variance.
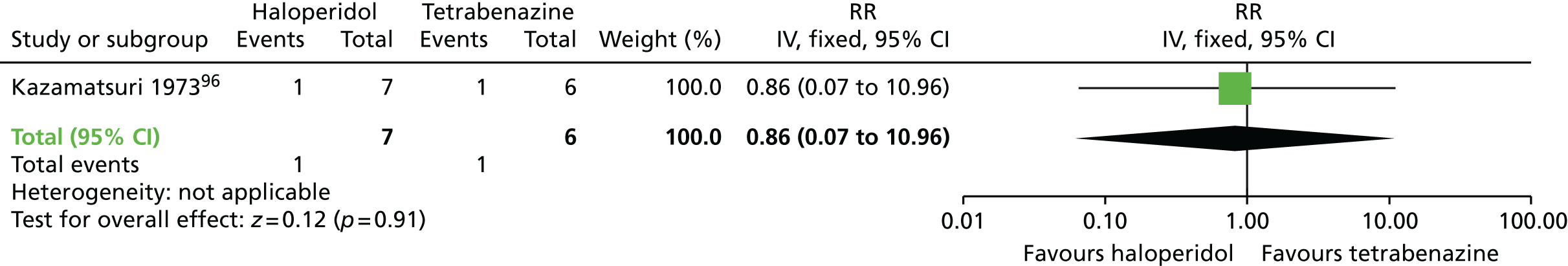
FIGURE 51.
Haloperidol vs. tetrabenazine: forest plot for the outcome ‘leaving the study early’ (follow-up 18 weeks). M–H, Mantel–Haenszel.

Anticholinergic withdrawal versus continuation
Figure 52 presents a forest plot of outcome analysis for anticholinergic withdrawal versus continuation.
FIGURE 52.
Anticholinergic withdrawal vs. continuation: forest plot for the outcome ‘leaving the study early’ (follow-up 7 weeks). IV, inverse variance.

Benzodiazepines versus placebo/treatment as usual
Figures 53–56 present forest plots of outcome analyses for benzodiazepines versus placebo or TAU.
FIGURE 53.
Benzodiazepines vs. placebo/TAU: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 5–10 weeks). df, degrees of freedom; M–H, Mantel–Haenszel.

FIGURE 54.
Benzodiazepines vs. placebo/TAU: forest plot for the outcome ‘TD – deterioration’ (follow-up 5–10 weeks). df, degrees of freedom; IV, inverse variance.

FIGURE 55.
Benzodiazepines vs. placebo/TAU: forest plot for the outcome ‘mental state – average end-point score (BPRS, high score means worse outcome)’ (follow-up 5–10 weeks). IV, inverse variance; SD, standard deviation.

FIGURE 56.
Benzodiazepines vs. placebo/TAU: forest plot for the outcome ‘leaving the study early’ (follow-up 5–10 weeks). M–H, Mantel–Haenszel.

Benzodiazepines versus phenobarbital (as active placebo)
Figures 57–59 present forest plots of outcome analyses for benzodiazepines versus phenobarbital (as active placebo).
FIGURE 57.
Benzodiazepines vs. phenobarbital: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 2 weeks). IV, inverse variance.

FIGURE 58.
Benzodiazepines vs. phenobarbital: forest plot for the outcome ‘adverse events – short term’ (follow-up 2 weeks). IV, inverse variance.

FIGURE 59.
Benzodiazepines vs. phenobarbital: forest plot for the outcome ‘leaving the study early’ (follow-up 2 weeks). IV, inverse variance.
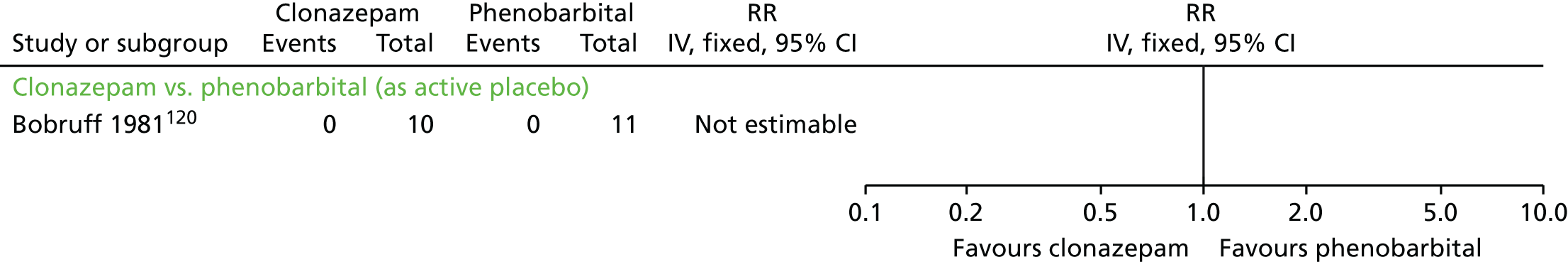
Vitamin E versus placebo
Figures 60–65 present forest plots of outcome analyses for vitamin E versus placebo.
FIGURE 60.
Vitamin E vs. placebo: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up up to 1 year). df, degrees of freedom; M–H, Mantel–Haenszel.
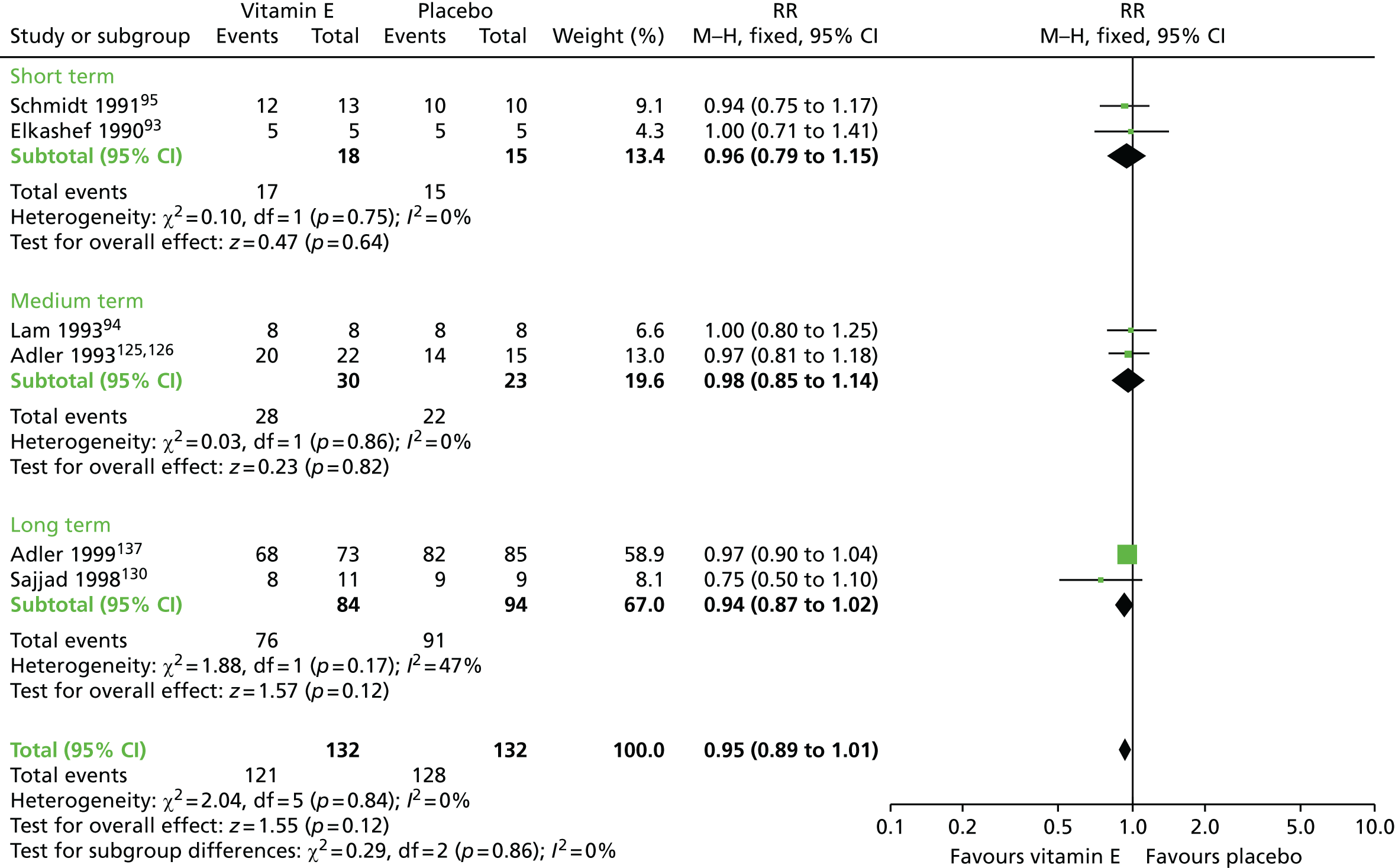
FIGURE 61.
Vitamin E vs. placebo: forest plot for the outcome ‘TD – deterioration of symptoms’ (follow-up up to 1 year). df, degrees of freedom; M–H, Mantel–Haenszel.

FIGURE 62.
Vitamin E vs. placebo: forest plot for the outcome ‘adverse events: extrapyramidal adverse events – long term (SAS, high score means worse outcome)’ (follow-up up to 1 year). IV, inverse variance; SD, standard deviation.

FIGURE 63.
Vitamin E vs. placebo: forest plot for the outcome ‘any adverse effect’ (follow-up up to 1 year). df, degrees of freedom; M–H, Mantel–Haenszel.
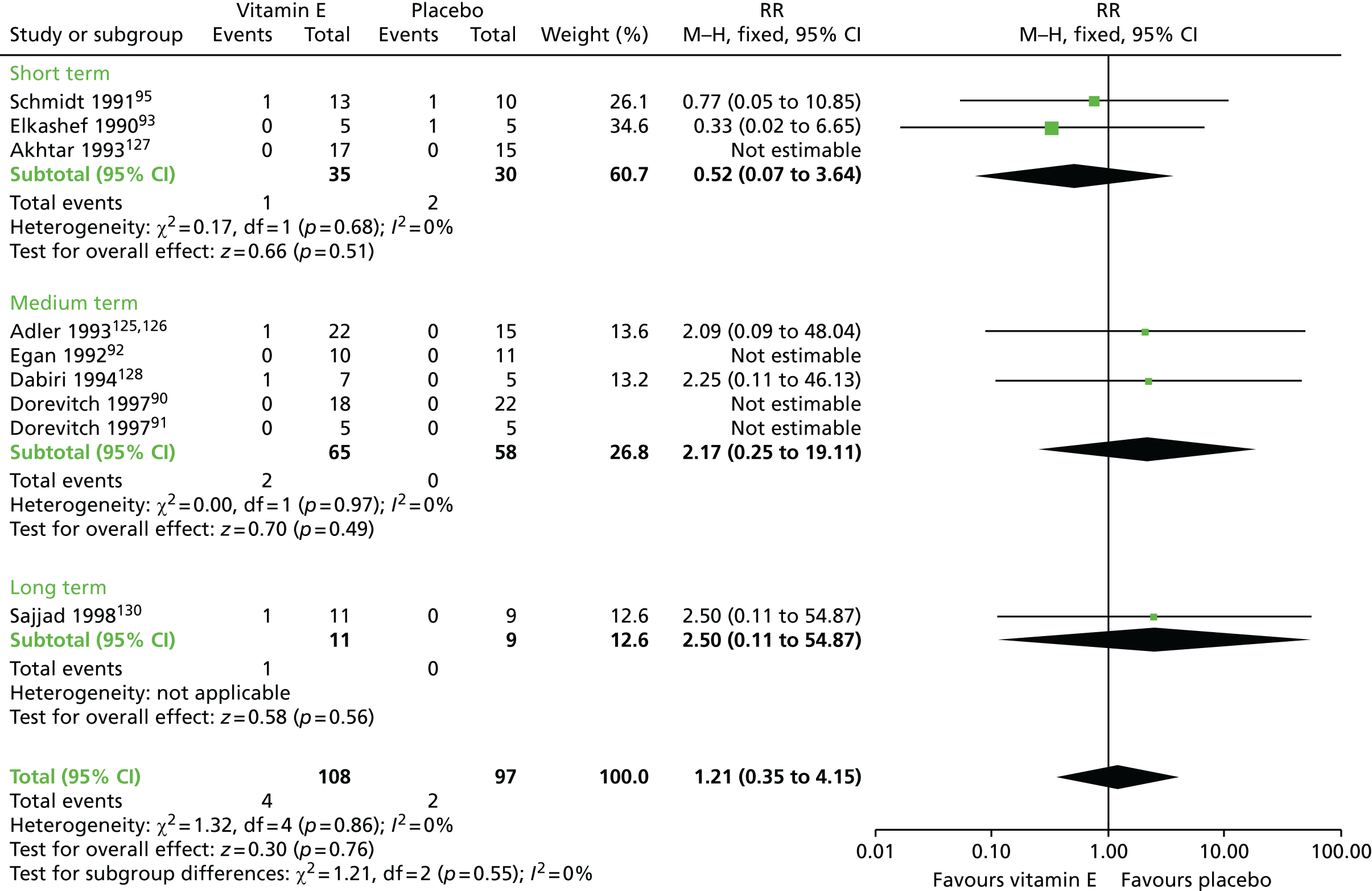
FIGURE 64.
Vitamin E vs. placebo: forest plot for the outcome ‘mental state – Average score (BPRS, high score means worse outcome)’ (follow-up up to 1 year). df, degrees of freedom; IV, inverse variance; SD, standard deviation.
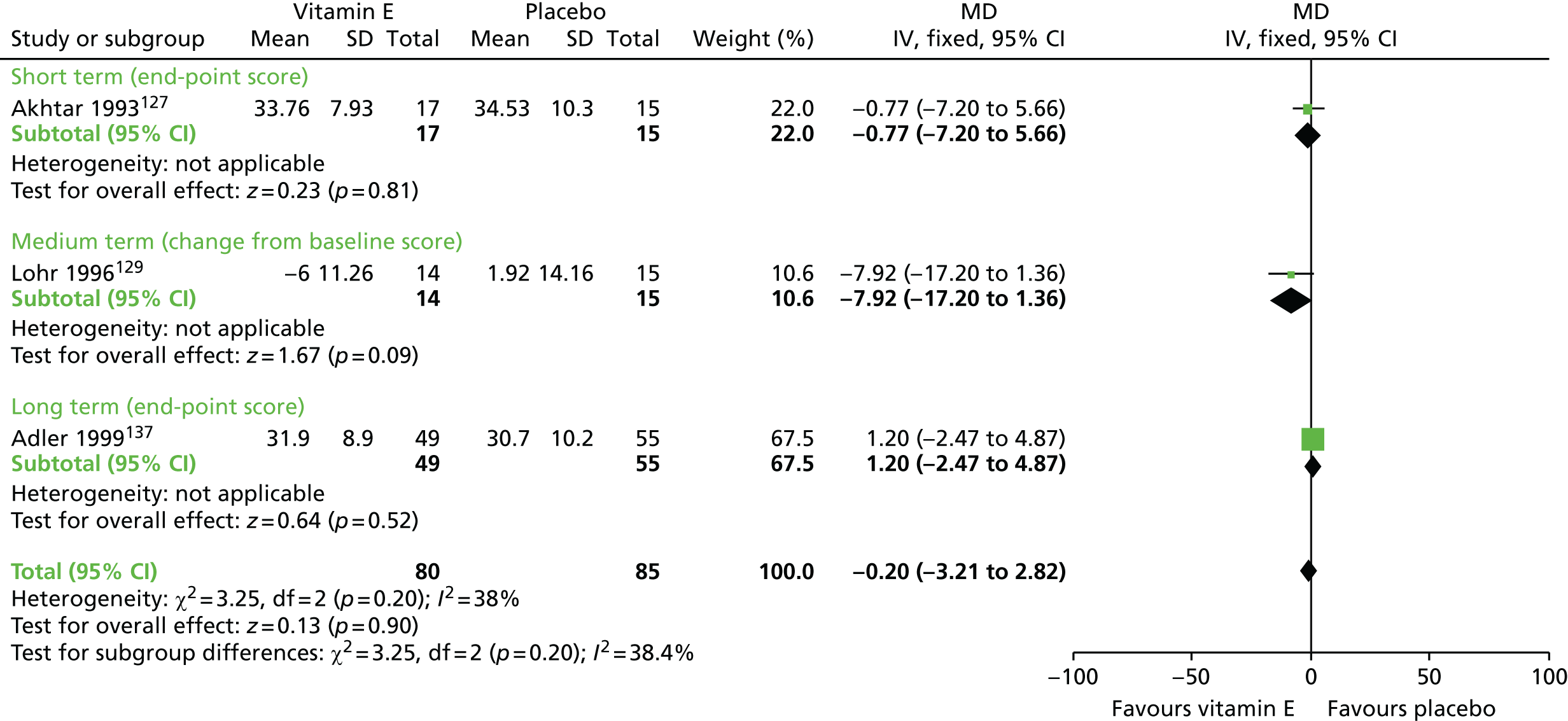
FIGURE 65.
Vitamin E vs. placebo: forest plot for the outcome ‘leaving the study early’ (follow-up up to 1 year). df, degrees of freedom; M–H, Mantel–Haenszel.
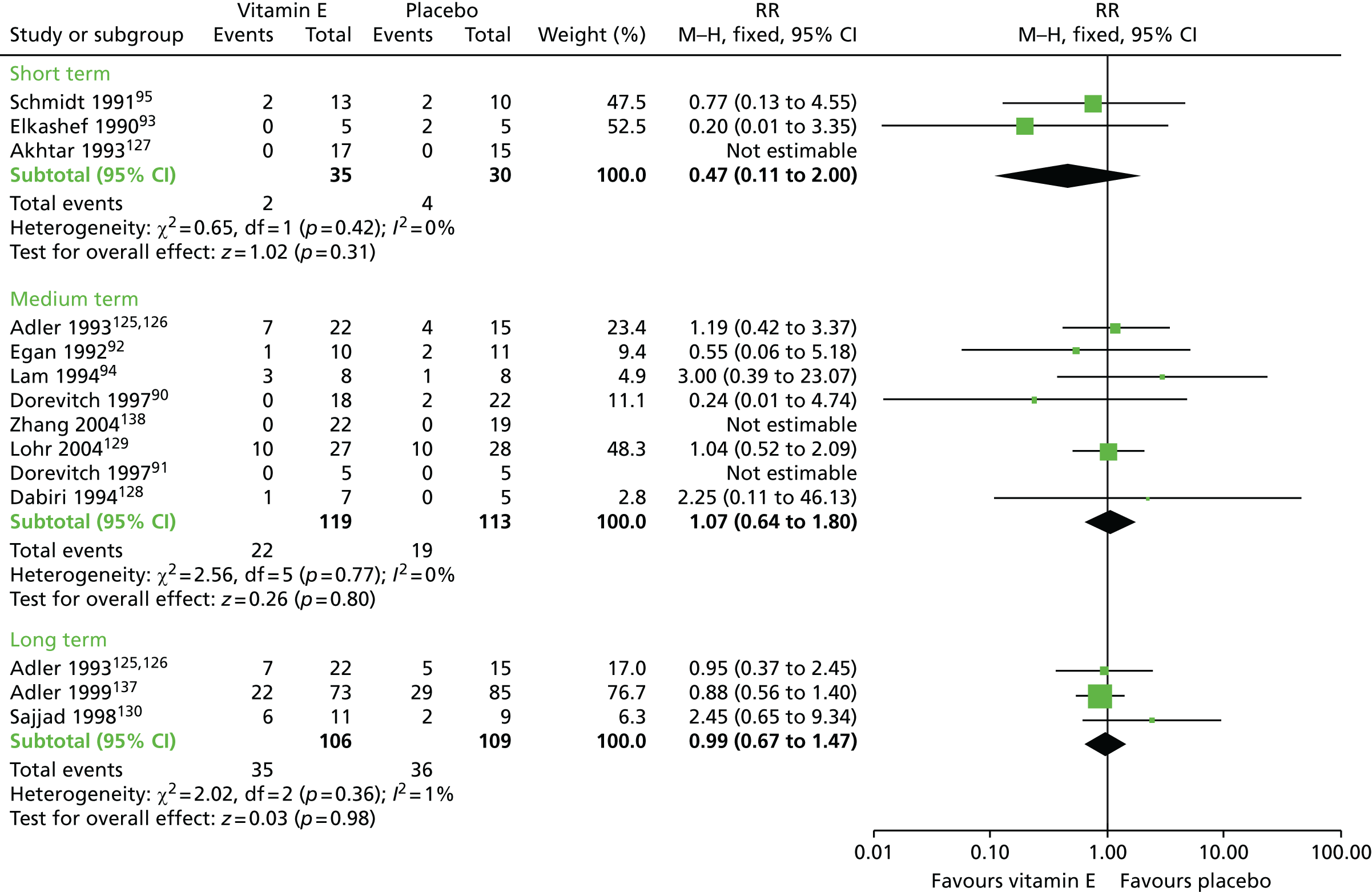
Buspirone versus placebo
Figures 66 and 67 present forest plots of outcome analyses for buspirone versus placebo.
FIGURE 66.
Buspirone vs. placebo: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up 6 weeks). M–H, Mantel–Haenszel.

FIGURE 67.
Buspirone vs. placebo: forest plot for the outcome ‘leaving the study early’ (follow-up 6 weeks). M–H, Mantel–Haenszel.
Hypnosis or relaxation versus treatment as usual
Figures 68–70 present forest plots of outcome analyses for hypnosis or relaxation versus TAU.
FIGURE 68.
Hypnosis or relaxation vs. TAU: forest plot for the outcome ‘TD – no clinically important improvement’ (follow-up eight sessions). M–H, Mantel–Haenszel.

FIGURE 69.
Hypnosis or relaxation vs. TAU: forest plot for the outcome ‘TD – deterioration’ (follow-up eight sessions). M–H, Mantel–Haenszel.
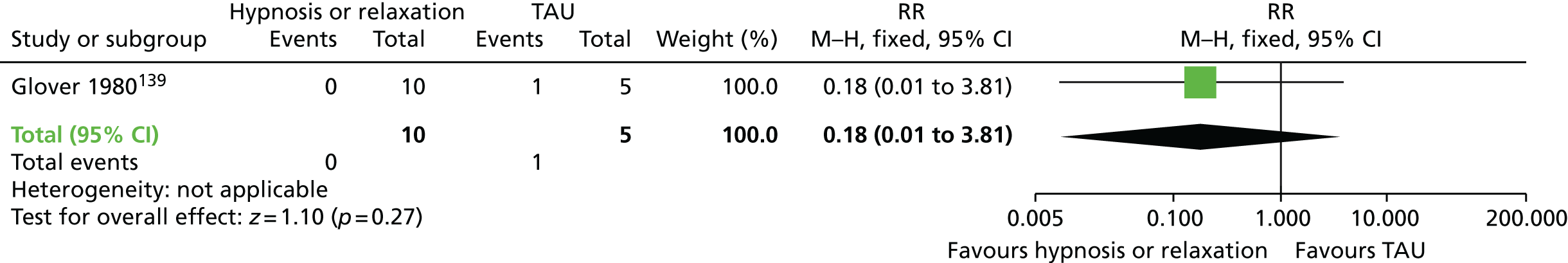
FIGURE 70.
Hypnosis or relaxation vs. TAU: forest plot for the outcome ‘leaving the study early’ (follow-up eight sessions). M–H, Mantel–Haenszel.

List of abbreviations
- AIMS
- Abnormal Involuntary Movement Scale
- BPRS
- Brief Psychiatric Rating Scale
- CI
- confidence interval
- ESRS
- Extrapyramidal Symptom Rating Scale
- FGA
- first-generation antipsychotic
- GABA
- gamma-aminobutyric acid
- GRADE
- Grading of Recommendations, Assessment Development and Evaluation
- MAO
- monoamine oxidase
- MD
- mean difference
- NIHR
- National Institute for Health Research
- NMA
- network meta-analysis
- OR
- odds ratio
- PPI
- patient and public involvement
- PRISMA
- Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- RCT
- randomised controlled trial
- RR
- risk ratio
- SAS
- Simpson–Angus Scale
- SGA
- second-generation antipsychotic
- TAU
- treatment as usual
- TD
- tardive dyskinesia
- UKU
- Udvalg for Kliniske Undersøgelser