
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 10/104/20. The contractual start date was in October 2013. The draft report began editorial review in October 2016 and was accepted for publication in February 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Michael Kerr reports personal fees from Johnson & Johnson outside the submitted work. Paul Deslandes and the Cardiff and Vale University Health Board department he previously worked for received an honorarium from Janssen-Cilag Ltd for a speaking engagement in 2014 and funding for conference registration fees in 2015. Kerry Hood is a member of the National Institute for Health Research Clinical Trials Unit Standing Advisory Committee and a member of the Health Technology Assessment General Board.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by McNamara et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Prevalence of learning disability and antipsychotic prescribing
The age-specific rate of registered learning disability (LD) in people aged ≥ 16 years in Wales is 0.47% (Local Government Data Unit 2011)1 and the rate of adult users of LD services in England is also estimated to be 0.47% of the adult population. 2 In the two countries combined, this results in an estimated 200,000 adults with LDs.
An audit of adults with LDs in primary care in Wales (n = 9947) found that 29% were prescribed antipsychotic medication. 3 An earlier and smaller primary care study in England4 found that 21% of 357 adults with LDs were prescribed antipsychotic medication. Applying the average of the two estimates to the number of people above suggests that there are 50,000 adults with LDs in England and Wales who are prescribed antipsychotic drugs. A comparison of the Perry et al. 3 and Molyneux et al. 4 rates of antipsychotic prescribing for adults with LDs in primary care gives no reason to think that the prescription of antipsychotics is declining. Indeed, a more recent North American survey5 found that 45% of 4069 individuals with a LD who lived in the community and received services from the New York State Office for People with Developmental Disabilities received antipsychotic medication, with 39% receiving atypical drugs and 6% being prescribed typical drugs.
The rate of prescription of antipsychotic medication in this population far exceeds the estimated prevalence of psychosis (3–4%). 6,7 The discrepancy may be accounted for by the use of antipsychotic medications for the treatment of behavioural problems, which is the most common reason that they are prescribed. 4,8–10 Rates of prescription among samples of people with LDs with challenging behaviour cluster around 50%11,12 and may be as high as 80–95% among those in specially designated services. 13,14 After taking account of prescription of antipsychotics for the treatment of psychosis, about 42,000 of the estimated 50,000 adults with LDs in England and Wales may be receiving these medications to treat or control challenging behaviour. In a recent study of primary care prescribing commissioned by Public Health England,15 it was estimated that between 30,000 and 35,000 adults with a LD were being prescribed an antipsychotic, an antidepressant or both, despite having no recorded diagnosis of psychosis or affective disorder. This estimate equates to 16.2% of adults in England registered as having a LD by their general practitioner (GP).
Clinical effectiveness and cost-effectiveness of antipsychotics for challenging behaviour
The clinical effectiveness of antipsychotic medications in treating or controlling challenging behaviour has not been demonstrated. 16 Their use may, therefore, be considered mistreatment in some cases. 10 A Cochrane review failed to find evidence to support such treatment17 and a review of 56 treatment trials found that the great majority lacked scientific rigour and the remainder found conflicting results. 18 A more recent review19 found evidence to suggest that a number of atypical antipsychotics are effective in treating challenging behaviour in adults with LDs and autism, although the authors acknowledge that tolerability and the balance of benefit versus adverse effects is unclear in this population. A recent cohort study of mental illness, challenging behaviour and psychotropic drug use in LD also concluded that more evidence with regard to the safety and efficacy of these medications is needed when prescribed for challenging behaviour without a corresponding diagnosis of severe mental illness. A double-blind randomised controlled trial (RCT) exploring the impact on aggression of haloperidol (a typical antipsychotic), risperidone (an atypical antipsychotic) and a placebo found that patients who were given the placebo showed no evidence of worse response than patients assigned to either of the antipsychotic drugs at any time point. 20 An accompanying economic evaluation concluded that the treatment of challenging behaviour among people with LDs by antipsychotic medication is not a cost-effective option. 21
Perry et al. 3 reported that there were 4714 prescriptions among the 2891 people who were prescribed antipsychotic medication, of which 2008 (43%) were for atypical medications. Romeo et al. 21 reported mean half-year medication costs for groups enrolled in a trial of risperidone and haloperidol as £127 and £8, respectively. Using these as estimates for the cost of all atypical and typical medications prior to the start of this trial produced an estimate of the full-year treatment costs for the 2891 people in the Perry et al. 3 audit of £553,328. Extrapolated to the 42,000 figure estimated above gave an annual total cost of £8M for England and Wales without including GP consultation or other NHS costs. However, medication costs are subject to change over time and the cost of risperidone has decreased since these calculations were made.
Known side effects of antipsychotic medications
Apart from a lack of therapeutic and cost-effectiveness for the treatment of challenging behaviour, concern about the high use of antipsychotic medication for this purpose is related to the common occurrence of a range of possible adverse medication side effects in LD populations. 22 These include possible adverse cardiovascular side effects, including thromboembolism, and central and autonomic nervous system and endocrine function side effects, including extrapyramidal side effects, akathisia and other muscle or movement disorders, which may in the case of tardive dyskinesia or tardive akathisia become permanent. 23 Moreover, certain atypical medications are associated with an increased risk of obesity and diabetes mellitus. 24,25 Mahan et al. 26 found that individuals taking psychotropic medication had significantly higher scores on the Matson Evaluation of Drug Side-effects scale in four domains: skin/ allergies/temperature, central nervous system: general, central nervous system: Parkinsonism/dyskinesia and central nervous system: behavioural/akathisia.
These side effects are consistent with those in other populations, such as in adults with schizophrenia,27 in whom movement disorder (typical antipsychotics in particular), weight gain (atypical antipsychotics) and sedation (typical and atypical) are commonly reported, and in adults with dementia, in whom the use of antipsychotics has been associated with stroke and increased mortality. 28–30
Prescribing guidelines for the management of challenging behaviour
The recent National Institute for Health and Care Excellence guidance31 acknowledges the limited and low-quality evidence supporting the use of antipsychotics for the management of challenging behaviour in adults with a LD. The guidance also states that antipsychotics should be prescribed only if (1) psychological or other interventions have been unsuccessful in managing behaviour, (2) treatment for any comorbid conditions (physical or psychological) has not resulted in improved behaviour and (3) risk to the individual or others is significant, owing to aggression, violence or self-injury. Further recommendations include the use of medication only in conjunction with behavioural/other interventions, ensuring that appropriate strategies are in place to review prescribing and any benefits or adverse effects and ensuring that there is a plan in place to stop medication, particularly when prescribing is transferred to community or primary care.
In 2014, NHS England commissioned the Winterbourne Medicines Programme32 to review use of antipsychotic prescribing in adults with LDs. The work has identified high levels of inappropriate psychotropic drug prescribing in primary care and in adults with LDs who are detained under the Mental Health Act 2007,33 medication, in some instances, appearing to have been prescribed for challenging behaviour rather than for the underlying mental health condition. As a result of this work, a number of recommendations have been made, including greater involvement of people with LDs, their families and carers in decision-making, provision of active care pathways for challenging behaviours and instigation of a collaborative ‘call to action’34 approach, bringing together patients, families, carers, health professionals and improvement experts to agree the actions required to reduce inappropriate use of antipsychotics. This approach has been used successfully in reducing antipsychotic prescribing in dementia. 28 As a result of the work undertaken by NHS England, The Royal College of Psychiatrists35 has issued a recent report on psychotropic drug prescribing in this population, recommending regular (preferably 3-monthly) review of treatment response and side effects. The report also includes a foreword from Dr Paul Lelliott, Deputy Chief Inspector Hospitals: Mental Health, Care Quality Commission, who states:
There is compelling evidence that a significant number of people with intellectual disabilities are prescribed psychotropic medication that, at best, is not helping them. In particular, there is a risk that doctors are prescribing medication to treat behaviour that is an expression of distress or a mode of communication rather than a mental disorder.
Reproduced with permission from the Royal College of Psychiatrists35 © 2016 The Royal College of Psychiatrists
Withdrawal of antipsychotic medication
A number of drug withdrawal studies have investigated predictors of successful withdrawal from antipsychotic medication,36,37 but these are limited by being retrospective, non-randomised, uncontrolled or inadequately rigorous in measurement. A retrospective clinical audit investigating change from thioridazine for safety reasons among 119 adults with LDs reported poor clinical outcomes. Most adults with LDs were given alternative antipsychotics and a few withdrew. Significant minorities experienced onset or deterioration of adverse effects with the introduction of new drugs or of challenging behaviour or mental ill health, and costs to the specialist psychiatric service rose. 38 However, a randomised controlled withdrawal study reported more positive results. Ahmed et al. 39 conducted a trial in which 56 participants were randomised to an experimental group (n = 36) or a control group (n = 20). In the experimental group drug dose was to be reduced over a 6-month period, in four stages, approximately 1 month apart, between baseline and post-intervention evaluation. Overall, full withdrawal was achieved in 33% of the this group and a reduction of at least 50% was achieved in a further 19%; in 48%, medication was reinstated to baseline levels after partial to full withdrawal. Drug reduction was not associated with higher levels of challenging behaviour and drug reinstatement was not associated with either staff-reported or directly observed measures of challenging behaviour. A recent systematic review40 of reduction or withdrawal of antipsychotics concluded that these drugs can be reduced in adults with a LD, although the authors noted that some participants in some studies did experience adverse effects. However, the authors also note that much of the evidence to date is from relatively small and biased samples and that interventions and comparators are generally inadequately described. It is not possible from the available evidence to identify individual characteristics that could predict a poor response to withdrawal.
A large controlled and blinded randomised trial of the impact of planned withdrawal on resulting drug dosage, behaviour, psychiatric symptoms, safety and the consequent costs of treatment is therefore required.
The ANDREA-LD trial
The initial purpose of this study was to conduct a sufficiently large blinded RCT to investigate whether or not antipsychotic medication prescribed to adults with LDs for the treatment of challenging behaviour could be reduced or withdrawn entirely without adversely affecting their behaviour or mental health or causing a corresponding increase in financial costs. We proposed to limit recruitment to patients receiving risperidone or haloperidol in order to increase the feasibility of blinding while including, within the trial, both atypical and typical medications, specifically those found to be ineffective for challenging behaviour by Tyrer et al. 20 Moreover, as Ahmed et al. 39 found in their open study that reinstatement of medication occurred for almost half of the sample, despite being unrelated to reported or directly observed changes in the level of challenging behaviour, this study was designed to compare the extent of medication change between blinded and unblinded conditions and explore the perceptions of clinicians and carers about medication usage. However, as Chapter 2 describes, poor recruitment led to modified aims for the study.
Chapter 2 Methods
The community-led ANtipsychotic Drug REduction for Adults with Learning Disabilities (ANDREA-LD) trial was originally designed as a large-scale non-inferiority trial of an antipsychotic withdrawal programme in primary care. The main outcome was the level of reported aggression at 9 months post randomisation. With slower than expected recruitment in primary care, the study was expanded to recruit via community learning disability teams (CLDTs). However, because of significant challenges to various elements of set-up and recruitment, the trial closed early and is therefore reported as an exploratory pilot study (as defined by the National Institute for Health Research Health Technology Assessment programme). Sections Design, Objectives, Site selection, Participants, Recruitment process, Interventions, Procedures, Outcomes and Statistical methods detail the trial as it was originally designed, and Alterations to study design describes the changes made in response to lower than expected recruitment rates. Exploratory pilot study design details the final presentation of the trial following the decision to close early.
Design
The ANDREA-LD trial was a two-arm randomised (1 : 1) double-blind placebo-controlled non-inferiority withdrawal trial. Those randomised to the intervention arm progressed through a dose reduction regime, while those in the control arm received their treatment as usual. After the baseline assessment, follow-up assessments were planned for 6, 9 and 12 months. The aim was to recruit 310 adults with LDs without psychosis and who were currently receiving one of two antipsychotics (risperidone or haloperidol) for the treatment of challenging behaviour. Ethics approval was given by Wales Research Ethics Committee 3.
Objectives
Primary objectives
The primary objective was to evaluate the impact of a blinded antipsychotic medication withdrawal programme for adults with LDs without psychosis compared with treatment as usual. More specifically, we wanted to determine whether or not reduction or withdrawal of antipsychotic medication prescribed for challenging behaviour without psychosis could be safely achieved without a corresponding increase in aggression, as indicated in previous non-blinded studies. The primary outcome (aggression) was to be assessed at baseline and 9 months (blinded) with levels of aggression compared between the intervention (reduced medication) and control (standard treatment) arms.
Secondary objectives
A secondary objective was to explore the potential non-efficacy-based barriers to drug reduction in clinical practice. We aimed to complete qualitative telephone interviews with principal investigators (PIs), carers and participants to explore their perceptions of involvement in the trial and medication usage, in addition to a final assessment of medication dosage at 12 months.
Site selection
Sites were originally intended to be general practices. Research-active general practices across four health boards in south Wales (Cardiff and Vale University, Cwm Taf, Abertawe Bro Morgannwg University and Aneurin Bevan health boards) were approached to participate in the trial. One GP at each site would be recruited to act as PI and at least one practice nurse was recruited to the task of taking delivery of and handing out study medication to participants.
Participants
Inclusion criteria
Adults with a LD were eligible for the trial if they met all of the inclusion criteria and none of the exclusion criteria. Inclusion criteria were that the patient:
-
was aged ≥ 18 years
-
had a recognised LD as judged by administrative classification (e.g. on LD register, in receipt of an annual LD health check or in receipt of LD services)
-
was currently prescribed risperidone or haloperidol for the treatment of challenging behaviour.
Exclusion criteria
Patients were excluded if:
-
they had a current diagnosis of psychosis
-
they had had a known recurrence of psychosis following previous drug reduction in the past 3 years
-
the clinician primarily responsible for their care judged for any other reason that participation in a drug reduction programme may be contraindicated
-
the research team were unable to identify an appropriate individual to complete outcome assessments.
Recruitment process
Participating sites were asked to identify all patients in their records who had a LD and were receiving either risperidone or haloperidol. PIs then examined the list and excluded any patient who they felt met the exclusion criteria. It was then up to the PI to approach the remaining individuals (or their carers if appropriate) with information about the trial and details of how to indicate a willingness to be approached to participate. This could have been either via completion of an expression of interest form returned in a pre-paid envelope to the study team or by the PI handing contact details directly to the study team, with the individual’s permission.
Once an expression of interest was received, the study team made contact with the patient (or their carer) to discuss the study in more detail, identify key personnel (to provide consent if necessary and complete outcome assessments) and arrange a screening assessment. The screening assessment would be carried out in order to assess the potential participant’s capacity, to gain informed consent and to ensure that inclusion criteria were fully met. Approximately 2 weeks after the screening assessment had taken place, a baseline assessment was carried out. The participant was then randomised by a member of the study team to either experimental reduction or to control treatment as usual (i.e. maintenance of current medication level).
Informed consent
It was expected that although some participants would have the capacity to give informed consent, there would also be a proportion judged by researchers to lack capacity. In such cases, consent from a personal legal representative was sought instead (failing that, from a professional legal representative). Assessment of capacity was made by members of the trial team or the research network, who were professionals with considerable experience in assessing capacity in this population. It was permitted for assessments of capacity and consent to be undertaken by the PI at site if necessary. Criteria for consent included presumption of capacity, an assessment of the individual’s understanding of the risks or benefits of taking part in the trial, their ability to retain this information and their ability to communicate their decision-making. Potential participants were given a plain language and pictorial participant information sheet at least 24 hours in advance of their meeting with the trial team that they could go through with a carer or legal representative (as appropriate) in their own time and at their own speed.
Potential participants with capacity
Upon meeting with the researcher, the trial and potential risks and benefits were explained verbally in simple terms. The researcher checked frequently that the potential participant understood the explanation. Once all questions had been answered individuals who indicated that they were happy to take part in the trial were asked to tick or initial each statement on the consent form as a means of indicating their consent and to sign the form. This process was witnessed and signed off by a carer who was independent of the research team. Participants could decide to withdraw their consent at any stage. A small sample of participants with capacity were also invited to take part in a qualitative interview at the end of the study. Capacity was assessed again at this time.
Potential participants who lacked capacity
When capacity was judged by an experienced researcher to be lacking, a similarly straightforward explanation of the trial and its potential risks and benefits was given verbally to a personal legal representative or, failing that, to a professional legal representative. Neither the personal nor the professional legal representative was connected with the conduct of the trial (e.g. the PI). That individual was asked to give consent on the participant’s behalf. Again, consent could be withdrawn at any stage. Legal representatives were kept informed of all material changes to the trial or participant’s condition to enable them to exercise their right of reviewing the person’s participation in the trial.
Carers of potential participants/principal investigators
The participant’s main carer was also asked to give separate consent to complete assessments designed to be completed by a third party and to consent to taking part in the qualitative interviews at the end of the trial if selected. PIs were also asked to consent to participate in the qualitative interviews.
Risks and expected benefits
Risk was considered against the recognised risks of long-term antipsychotic medication and, therefore, the potential benefits of withdrawal. Benefits would include reduction of cardiovascular risk, in particular stroke, reduction of musculoskeletal risk from tardive dyskinesia and other extrapyramidal side effects, reduction in acute life-threatening risk of malignant neuroleptic syndrome and a broad spectrum of psychosocial benefits from reduction of sedation, associated alertness and concentration and learning. Societal benefits would include increased contribution from adults with LDs who are not constrained by unnecessary medication and reduced expenditure/resource use on unnecessary treatment and medical complications of long-term antipsychotic medication use. However, withdrawal may be associated with the following risks.
-
The emergence of tardive dyskinesia. Advice for PIs on the recognition, assessment and management approaches was included in the detailed treatment and safety package prepared by the trial team.
-
Emergence of unrecognised psychiatric illness. There remained a slight possibility that, especially in the case of those on very long-term antipsychotics, the drugs masked an underlying mental illness. This, if present, was most likely to be an anxiety disorder. Advice for PIs on the recognition and assessment of psychiatric symptoms was included in the detailed treatment and safety package prepared by the trial team. A clinical algorithm (described in Interventions) was developed to support the primary care team to follow the appropriate treatment and care pathways. Clear guidance was available for predicted scenarios in which unblinding may be necessary, such as the emergence of psychotic symptoms.
-
Deterioration in behaviour. A previous study39 showed that measurable behavioural deterioration was uncommon following drug reduction, but other studies have shown greater deterioration and that carer concern can be high. Advice on assessing a meaningful behaviour change was provided in the PI support package. As behavioural signs and psychiatric symptoms for this population are intertwined, the clinical algorithm referred to above also dealt with behaviour change.
Supporting secondary care services
In the case of individuals recruited through general practice and who had involvement with LD services, contact was made with these teams regarding confirmation of eligibility. At this time, a description was given of the study protocol, the PI support package (see Interventions) and the procedure for accessing the code break.
It was not expected that the study would have a considerable impact on the current well-developed specialist LD services. These services would most probably already be aware of many of the individuals involved in the study, and we estimated that the chance of severe deterioration would be small and would be distributed across at least six health boards. It was possible that the study might have increased the referrals to LD services because of a greater awareness of the issue of antipsychotic drug prescribing across primary care. Such referrals would be a positive outcome; LD teams are skilled in drug assessment, and regular review is a key component of good clinical care.
Interventions
The intervention group progressed through up to four approximately equal reduction stages to full withdrawal over a 6-month period while the control group maintained baseline treatment. The following rules were used to decide each participant’s Investigational Medicinal Product (IMP) regime:
-
Participants in both arms stayed on the same number of tablets throughout the study when feasible.
-
For those in the intervention arm:
-
Reductions from stage to stage were as equal as possible, but when this was not possible larger reductions were made first.
-
Reductions were made in such a way that there was only one tablet in each encapsulation.
-
If a participant was on multiple doses per day, preference was given to reducing the middle of the day doses, later doses and then earlier doses.
-
Schedules were reviewed by clinicians and could be changed if there was a valid clinical reason.
-
Drugs were supplied to ensure blinding, but treatment was led by PIs, and, although blinded to whether or not medication was being reduced, the PI retained discretion to delay progression to the next step (i.e. to maintain current medication level).
Sites were supported by a detailed treatment and safety package showing clear clinical contact and decision-making to support drug reduction. The chief investigator and co-applicants produced this guidance, focusing on how to respond to participant and carer queries, including those concerned with behavioural deterioration, emergent features of tardive dyskinesia or psychiatric symptomatology. The guidance started with a ‘management flow chart’ and this was followed by more detail on elements such as history taking, examination, consultation with the research team, making appropriate referrals and information on the code-breaking practice. The flow chart (Figure 1) was designed as an easy-access decision-making tool. Each box in the flow diagram pertains to specific issues that were addressed in more detail over the following pages of the support package. PIs were given training in how to use the manual and its content at the point of site initiation and were provided with contact details of the study’s chief investigator and clinical reviewer. As part of their training, PIs were also requested to add labels to participants’ medical notes in order to flag their participation in the trial.
FIGURE 1.
ANDREA-LD clinicians ‘management flow diagram’. Note that the cross-references within the figure refer to those within the support package.

Treatment achieved at 6 months was maintained for a further 3 months under blind conditions. At 9 months following collection of follow-up data, the blinding was broken. PIs were informed of the participant’s treatment allocation and current medication dosage. It was the responsibility of the PIs to then reveal the allocation to the participant and their carer, to handle any further prescribing and to communicate with the participant’s wider care team.
Supply of blinded medication
In order to achieve effective blinding, medication was encapsulated. Risperidone and haloperidol tablets of varying doses were encapsulated based on estimates of the likely numbers of participants recruited on each medication at the common doses. Encapsulated placebo medication that was identical in appearance to active medications was also produced. All participants experienced a change in the supply of their antipsychotic medication at the outset of the study. Although individuals started the trial on their usual dose of medication, it was important to ensure that the number of tablets that they took daily remained constant over the blinded period and that the effective dose could be reduced across dose reduction steps. In order to allow participants to familiarise themselves with their new medication, a run-in period was built into the programme for all participants, regardless of allocation and prior to any reduction.
Manufacturing estimates assumed that all participants would achieve at least a 50% reduction. In reality, the number of reduction steps achieved was likely to be much more variable, although this assumption allowed for a reasonable degree of flexibility. Manufacturing estimates included provision of medications to all participants up to 9 months, when the blind was lifted.
Investigational Medicinal Products were manufactured by St Mary’s Pharmaceutical Unit (SMPU) under its Manufacturer’s Authorisations for IMPs licence and dispensed using Nomad® Clear 2 trays (Omnicell Ltd, Manchester, UK) in accordance with participant-specific prescriptions. SMPU was to do this under section 37 of the Medicines for Human Use (Clinical Trials) Regulations (2004)41 as ‘post (Qualified Person) certification labelling for safety purposes’. Nomad Clear 2 trays are disposable trays with separate compartments for days of the week as well as times of day – morning, midday, evening and bedtime. IMP was dispensed monthly for 9 months. Although participants were to take 28 days of medication at each stage, enough IMP was provided for 33 days (to allow for a +5-day window around the planned 28-day time frame between medication review visits) in case of any delays or issues in getting the prescription.
Participant-specific prescriptions were issued to SMPU by the research team following consent and randomisation. SMPU then dispensed and dispatched these directly to site, where they were formally received and kept secure by a practice nurse (or designated individual). Accountability documentation was completed on receipt of the IMP and returned to the study team, thus evidencing the ownership of the IMP. Prescriptions were handed out only to the patient or their carer, legal representative or researcher by authorised site staff. Again, accountability documentation was completed and returned to the study team at this time. This process was repeated for the IMP of each new month following the PI’s decision to allow the participant to progress through the trial.
When any new prescriptions of study drug were collected from site, unused IMP from previous stages had to be returned by the participant or their carer. Sites were then responsible for the destruction of any unused study medication in accordance with local procedure and following authorisation from the trial manager. Completion of accountability documentation at various time points allowed the study team to evidence the location of IMP throughout the trial.
Toxicity was not expected and use of all pro re nata (PRN) medication was permitted and recorded in study diaries during the trial. Drug reduction was unlikely to cause interaction with other drugs; however, it was recommended that participants taking warfarin underwent more frequent international normalised ratio tests. All concomitant medication was permitted and details of any medications that had been taken were collected by the research team. IMP was stored at ambient temperatures at site; therefore, no temperature monitoring was undertaken.
Procedures
Piloting
Once the trial was open for recruitment, arrangements were piloted in primary care for 6 months in order to test the assumptions and practicalities of trial processes and recruitment. At the end of this period, any adjustments would be made as necessary and the study was to then continue until full recruitment.
Principal investigator visits/contact
Participants in both trial arms (intervention and control) had five appointments with the PI in total. The first four took place in the 2 weeks preceding the release of each new batch of blinded medication and were approximately 28 calendar days apart. The purpose of the appointments was for the PI to make an assessment of whether or not there were any concerns about the participant’s progression to the next stage of the trial. When face-to-face appointments could not be held, the PI could consult over the telephone. It was the responsibility of participating PIs to provide participants and the study team with details of each of these appointments. The PI provided appointment cards to the participant or their carer and were responsible for reminding participants of the appointment nearer the time of the visit to ensure attendance. The site was also responsible for rearranging any appointments as necessary. The appointment card contained the contact details of the PI, an emergency number for participants or carers to use should they need to and a reminder of the amount of medication the participant had been taking when they started the study. It was important that the PI was the first point of contact for participants or carers if they had any concerns. The fifth PI visit took place after the 9-month assessment and was for the PI to unblind the participant and their carer and reveal the treatment allocation. It was also the point at which a discussion would take place regarding the participant’s care from there on.
Practice nurse visits
Participants (or their carer/representative) in both trial arms (intervention and control) collected their prescribed study medication from the practice nurse monthly until the blind was broken at 9 months. At each of these visits, the practice nurse took receipt of any unused medication from the previous prescription before distributing any new medication. The practice nurse would then complete accountability paperwork before destroying any unused medication on confirmation from the trial manager.
Assessments and follow-up
Eligibility data were collected at screening. Full data were to be collected at baseline and post intervention, approximately 9 months from randomisation (Table 1). Data on medication and psychopathology [Modified Overt Aggression Scale (MOAS), Aberrant Behaviour Checklist (ABC), Psychiatric Assessment Schedule for Adults with Developmental Disability (PAS-ADD) checklist] and costs [Client Service Receipt Inventory (CSRI)] were to be obtained at 6 months and 12 months. All data collection was face-to-face, either at site or during home visits.
| Assessment time points | Measures and data collection | Participant involved | Estimated time to complete appointment (hours) |
|---|---|---|---|
| Screening | Age, gender, current medication, ABS, Mini PAS-ADD checklist | Carer | 1.5 |
| Baseline | Medication, MOAS, ABC, PAS-ADD checklist, CSRI | Carer | 1.5 |
| ASC, DISCUS | Participant/carer | ||
| 6 months | MOAS, ABC, PAS-ADD checklist, CSRI | Carer | 1.5 |
| 9 months | MOAS, ABC, PAS-ADD checklist, DISCUS, ASC, CSRI | Carer | 1.5 |
| 12 months | Medication, MOAS, ABC, PAS-ADD checklist, CSRI | Carer | 1.5 |
Details of outcomes and follow-up time points can be seen in Table 2 and were the same for both experimental and control groups.
| Outcomes | Measure | Time point | Estimated time to complete assessment (minutes) |
|---|---|---|---|
| Adaptive behaviour | ABS | Screening | 40 |
| Mental health | PAS-ADD checklist | Screening,a baseline, 6 months, 9 months, 12 months | 30 |
| Adverse effects of psychotropic medication | ASC | Baseline, 9 months | 15 |
| Movement disorders | DISCUS | Baseline, 9 months | 7 |
| Aggression | MOAS | Baseline, 6 months, 9 months, 12 months | 5 |
| Other challenging behaviour | ABC | Baseline, 6 months, 9 months, 12 months | 10 |
| Costs | CSRI (modified) | Baseline, 6 months, 9 months, 12 months | 10 |
Outcomes
Screening measure
Information collected included age, gender, current medication and psychiatric history. In addition, adaptive behaviour was assessed using the Adaptive Behaviour Scale (ABS)42 as a means also to estimate intelligence quotient (IQ). 43 Current mental health status was assessed using the PAS-ADD interviews. 44 The data gathered were used to confirm inclusion and exclusion criteria. If required, clinical review was undertaken for those exceeding thresholds for the ABS (a score that converts to an estimated IQ of > 70 using the method described by Moss and Hogg43) and/or the Mini PAS-ADD checklist (a score for section M, potentially indicative of psychotic disorder, of > 2).
Primary outcome measure
The primary outcome measure was aggression and was evaluated using the MOAS. 45 The MOAS rates four categories of aggression (verbal aggression, destruction of property, self-mutilation and physical aggression to others) each on a scale of 0–4 but then weighted by an ascending index of seriousness. The measurement to be used here was a non-inferiority comparison; therefore, a score difference of ≤ 3 was to be taken as clinically non-significant.
Secondary outcome measures
Secondary outcome measures at baseline, 6-month, 9-month and 12-month assessments were as follows.
-
The ABC46 comprising 58 behaviours, each relating to one of five subscales, to assess other challenging behaviour.
-
The PAS-ADD checklist47 to monitor mental health. The PAS-ADD checklist is a 25-item questionnaire designed for use primarily with care staff and families. The scoring system includes threshold scores, which, if exceeded, indicate the presence of a potential psychiatric problem in the scale’s three diagnostic domains (affective or neurotic disorder, possible organic condition and psychotic disorder). The proportions of people reaching threshold scores for possible mental ill health were to be compared.
-
The Antipsychotic Side-effect Checklist (ASC)48 was used to measure adverse effects of psychotropic medication. The ASC comprises a list of the more common or clinically important side effects of antipsychotic treatment.
-
The Dyskinesia Identification System Condensed User Scale (DISCUS)49 was used to assess movement disorders. A psychometrically derived DISCUS threshold of 5 was to be used.
-
The CSRI was modified for use in those with LDs and has been used previously in LD research. 21,50 The CSRI was used to collect data on a comprehensive range of services used and support received by each individual in the study. The collection of these data facilitated the calculation of the cost of medication, health, social care and unpaid carer inputs incurred by trial participants because of challenging behaviour or mental ill health.
The primary and secondary outcomes relating to challenging behaviour and mental health were to be analysed for non-inferiority with other secondary outcomes, such as medication usage and adverse effects, to be analysed for difference. A description of all scales, the range of their possible values and their interpretation is given in Appendix 4.
Statistical methods
Randomisation and unblinding
The offline password-protected randomisation programme was designed by the trial statistician and based on the method of minimisation. Allocations were balanced with respect to medication type (risperidone/haloperidol) and dose: low (< 4 mg for risperidone and < 5 mg for haloperidol) and high (at least 4 mg for risperidone and at least 5 mg for haloperidol). A random component, set at 80%, was used alongside the minimisation procedure to increase the integrity of the minimisation process (i.e. there was an 80% chance that the allocation would minimise the imbalance with respect to the aforementioned balancing variables).
Following consent and baseline assessments, participants were randomised to either the intervention arm (gradual reduction) or the control arm (treatment as usual) in a 1 : 1 ratio by a member of the study team. Any unblinding was performed only after authorisation from the chief investigator or (if not available) an authorised clinical reviewer who was an appropriately qualified clinician and member of the trial management team. In the event of an emergency, the treating clinician would have access to details of the participant’s baseline dose (i.e. the dose at which they entered the study) and so could treat accordingly.
Sample size
We originally aimed to randomise 310 participants (155 per group) in total, which would have provided 90% power to fit a one-sided 95% confidence interval (CI) around the between-group difference in mean MOAS scores at 9 months post randomisation. A sample size of 310 assumed a non-inferiority margin of 3 and a standard deviation (SD) of 8 (i.e. an effect size of 0.375) and had been adjusted to allow for 20% attrition.
Main analysis
The original proposed primary analysis focused on a comparison of MOAS scores at 9-month follow-up between the two trial arms. An analysis of covariance model, with baseline MOAS score and variables balanced on/stratified by at randomisation (medication type, dosage and recruitment source) controlled for as covariates, would have been fitted. Using the estimates from this model, a one-sided 95% CI of the adjusted mean difference in MOAS scores at 9-month follow-up (intervention – control) would be calculated. Non-inferiority would have been concluded if the limit of the CI was < 3 in all study populations [complete case (CC); full intention to treat (ITT), with multiple imputation used to impute missing outcome data; and a per protocol (PP), which would include participants who had outcome data available, had not withdrawn from trial treatment and, if they were allocated to the intervention group, had experienced at least one reduction].
A complier average causal effect analysis would have been performed as a secondary analysis of the primary outcome, to obtain an ITT estimate in the treatment adherent. If non-inferiority was concluded, a superiority analysis of the difference in MOAS scores between trial arms was planned in the CC and ITT populations, using a two-sided 90% CI.
All secondary analyses (antipsychotic medication use, other challenging behaviour, mental health, adverse effects, movement disorders) would have been conducted using the CC population, with those secondary outcomes assessed for non-inferiority (challenging behaviour and mental health) and adverse effects also being analysed using the PP population.
Potential moderators of the effect of the intervention on MOAS score (e.g. age, gender, medication type or adherence to intervention) would have been explored in multivariable analyses using interaction terms. It was also originally proposed to model aggression levels using mixed models to explore changes over time.
Cost-effectiveness analysis
The original proposed main cost-effectiveness analysis focused on the comparison of the two trial arms through the calculation of incremental cost-effectiveness ratios, defined as the difference between trial arms in mean costs divided by the difference in mean outcome (MOAS score) over 9 months.
It was proposed to conduct the main cost-effectiveness analyses from health and social care agencies and a wider societal perspective to include health and social care agencies and unpaid carers. To inform the cost-effectiveness analyses from these two perspectives, it was proposed that comprehensive data on health, social care and other services used by individuals were included in the study, using a tailored version of the CSRI. To estimate component costs, service use data were due to be combined with the unit costs for each service using long-run marginal opportunity cost principles. For services in which national figures were not available or not suitable, we proposed to calculate best estimates of long-run marginal cost; values and time spent by friends or relatives providing support were due to be estimated using the unit costs of a local authority care worker. Three main categories of costs due to be analysed were (1) medication costs; (2) medication costs, aggregated health and social care costs, consisting of inpatient admissions, outpatient appointments, accident and emergency department contacts and community-based health and social care contacts; and (3) medication costs, aggregated health and social care costs and cost of time spent caregiving by relatives and friends.
Costs were proposed to cover the period from baseline to 6 months (the end of the full treatment withdrawal period) and 6–9 months (3 months following the full treatment withdrawal period). The MOAS score was to be used as the primary measures of effectiveness in a series of cost-effectiveness analyses. As cost data are likely to be skewed, and to explore if unobserved difference in service use at baseline between the allocation groups may result in differences in cost between treatment groups, regression analysis using bootstrapping was proposed, adjusting for baseline covariates (MOAS score, baseline costs and variables balanced on/stratified by at randomisation: medication type, dosage and recruitment source).
A series of cost-effectiveness analyses was to be conducted by combining outcomes with costs from health and social care agencies and unpaid carers in turn. In the event that the experimental reduction group had lower costs and better outcomes than its comparator, it would have been interpreted as the dominant treatment, and if the experimental reduction group had higher costs and worse outcomes than the comparator treatment, the experimental reduction group would have been dominated by the comparator. If the experimental reduction group was both more effective and more costly than its comparator, the nature of the trade-offs to be made would have been made using cost-effectiveness acceptability curves (CEACs). To generate the CEAC and non-parametric bootstrapping of the costs and effectiveness, data would have been used to generate the joint distribution of incremental mean costs and incremental effects. The CEAC shows the likelihood of one treatment arm being seen as cost-effective relative to another treatment arm given different (implicit monetary) values placed on incremental outcome improvements.
We originally planned to use one-way sensitivity analyses to examine robustness of the findings to (1) changes in the unit costs of informal support, (2) analyses based on all randomised participants whose 9-month follow-up MOAS score is known (CC population) and (3) analyses based on all randomised participants (ITT population).
Qualitative study
We undertook qualitative telephone interviews with a proportion of carers, PIs and participants who took part in the trial. One of the main purposes of these interviews was to gain insight into the non-efficacy-based barriers to drug reduction in clinical practice, as well as attributions of behavioural changes in relation to potential reduction of medication. The interviews were scheduled to take place during the unblinded phase of the trial between the 9- and 12-month time points and were to ascertain (1) views about participating in the study, (2) reasons for any partial or full reinstatement of medication after unblinding and (3) views about antipsychotic medication use to treat or control challenging behaviour for the participant, in particular, and the patient group in general. PI interviews also focused on PI views of the support package and views about how the patient and carer(s) managed during the trial period. Interviews were expected to take up to 30 minutes.
We aimed to interview up to 60 carers and the corresponding PI. It was hoped that both parties would agree to take part in these paired interviews, but we accepted that this was not guaranteed. The sample was to be selected, purposefully incorporating participants from both trial arms and from across the geographical recruitment areas.
We also hoped to interview a proportion of participants of the ANDREA-LD trial. Those taking part would be required to have the capacity to provide consent for a face-to-face interview. Interview topics for participants focused on (1) the reasons of participants for participating in the trial, (2) how they felt they managed during the trial period and (3) their views about taking medicines to help with their behaviour.
Carers and participants who agreed to take part in an interview were offered a £10 high street shopping voucher to thank them for their time and considered views. PIs who participated in interviews were offered £50. With the participants’ consent, all interviews were audio-recorded, transcribed and anonymised.
Qualitative analysis
It was proposed that data from the transcribed anonymised telephone interviews would be subject to thematic analysis as described by Braun and Clarke. 51 Thematic analysis allows researchers to take an initial inductive approach towards the data set. Following familiarisation with the data, researchers index data according to a priori and emerging themes. A priori themes are informed by the research literature on the topic of antipsychotic medication for people with LDs. Analysis is facilitated by use of the computer-assisted qualitative data analysis software package NVivo version 10 (QSR International, Warrington, UK). Data from each data set (participants, carers and PIs) would be analysed separately and then comparisons made across data sets.
Alterations to study design
Recruitment via community learning disability teams
Despite expansion of recruitment in primary care to areas in England, it was apparent that targets would not be achieved in the predicted time frames by relying on this route. We gained approval from the funders to expand recruitment to CLDTs, with LD psychiatrists acting as PIs and hospital-based pharmacies taking on the role of the practice nurses and dispensing trial medication.
Twenty LD psychiatrists from six trusts in Wales and England were then recruited to act as sites in the trial. An additional 13 hospital pharmacies were recruited in order to dispense trial medication.
Other alterations
Evidence from screening logs showed that the number of potential participants receiving haloperidol was much lower than expected. For this reason, the decision was taken not to manufacture blinded haloperidol medication but to continue to recruit only those taking risperidone. With these changes in place, the randomisation programme was also altered so that allocations were stratified by recruitment source (general practice/CLDT) and balanced with respect to medication dose only: low (< 4 mg for risperidone) and high (at least 4 mg for risperidone).
Exploratory pilot study design
Exploratory pilot study methods
In November 2015, the decision was taken to close the trial to recruitment because of the difficulties described. At this point, 22 participants had been recruited into the trial. The study team submitted a close-down plan to the funders and it was agreed that all randomised participants would continue to receive the intervention and follow-up to 9 months. This meant that the trial would be complete by the end of June 2016 and would be reported as an exploratory pilot study (as defined by the Health Technology Assessment programme). As such, there were a few key alterations to the methods previously described, which are detailed in Table 3.
| Change | Study component | Changes to design |
|---|---|---|
| 1 | Sample size, recruitment and retention | No specified sample size; 22 recruited |
| 2 | Length of follow-up | Reduced from 12 months to 9 months post randomisation |
| 3 | Intervention | No change |
| 4 | Analysis of primary and secondary outcomes | Still pre-planned, but primarily focusing on outcomes related to conducting antipsychotic drug withdrawal trials in a LD population (see Exploratory study analysis for more details) |
| 5 | Qualitative analysis | Interviews brought forward to 4- to 6-month time point. Focus shifted to feedback on involvement in the trial and sample reduced |
| 6 | Economic evaluation | Not reported |
Exploratory study analysis
As the required sample size would not be achieved, we planned to focus on estimating feasibility outcomes. With a particular interest in recruitment and retention, we planned to estimate the following:
-
the number and proportion of primary care practices/CLDTs that progressed through the various stages from initial approach to recruitment of participants
-
the number and proportion of recruited participants who progressed through the various stages of the study.
We also compared trial arms regarding the following clinical outcomes:
-
MOAS at 6 and 9 months post randomisation
-
level of psychotropic medication use, assessed at the 6-month and 9-month post-randomisation assessments
-
ABC at 6 and 9 months post randomisation
-
PAS-ADD checklist at 6 and 9 months post randomisation
-
ASC at 9 months post randomisation
-
DISCUS at 9 months post randomisation
-
use of PRN medication over the study period
-
use of other interventions to manage challenging behaviour at 9 months post randomisation, including:
-
physical intervention/restraint
-
seclusion
-
PRN medication
-
-
costs and service utilisation at 6 and 9 months post randomisation.
The analysis of recruitment and retention outcomes was descriptive, with frequencies and percentages reported both overall and split by recruitment route (primary care or CLDTs). Pre-randomisation variables, including those related to the participant, their recruitment route and their starting medication, were used to explore the association between participant characteristics and their progression through the study post randomisation (to stage 4 of the intervention).
Clinical outcomes were compared between arms using appropriate regression models (linear or logistic, depending on type of variable). Data transformations were made, when required, to fulfil regression assumptions. The analyses were also adjusted for the corresponding clinical score at baseline (if measured), as well as variables that were balanced on at randomisation [dose of antipsychotic medication (< 4 mg or ≥ 4 mg) and recruitment route (primary care or secondary care)].
Three analysis sets were considered:
-
The ITT population, which comprised all randomised participants. When participants had either withdrawn from the study intervention (but remained in the trial for follow-up assessments) or not provided follow-up assessments at 6 or 9 months post randomisation, it was assumed that they returned to their original starting dose.
-
The modified intention-to-treat population (MITT), which comprised all randomised participants whose follow-up data were known.
-
The PP population, which comprised all randomised participants who had progressed to stage 4 of the intervention (regardless of the trial arm to which the participant was randomised).
The analysis of the level of psychotropic medication at 6 and 9 months was conducted in the ITT, MITT and PP populations. The remaining clinical outcomes analyses were conducted in the MITT and PP populations only.
As it was the original primary outcome, further exploratory analyses were performed with the MOAS score:
-
The MOAS score at 9 months post randomisation was fitted with a two-sided 90% CI in order to reflect the original primary analysis intended for this study.
-
Individual trajectories for the MOAS scores at baseline, 6 months and 9 months post randomisation were plotted and described, with particular attention paid to individuals whose MOAS scores changed by at least 4 points (a change considered to be clinically meaningful).
Economic analysis
Owing to the small sample size of the exploratory pilot, economic analysis was not reported as planned.
Chapter 3 Setting up and delivering a drug reduction trial for adults with learning disabilities: challenges and lessons learned
The unique nature of this trial meant that there were particular challenges faced that had not been experienced by other research in this area. These have been summarised under the following headings.
Recruitment
Recruitment of sites
Research-active general practices in south Wales and south-west England were recruited into the study in the first instance. Initially, the number of practices interested was quite low; therefore, we expanded our approach to include practices that were not known to be research active. In total, approaches were made to 351 practices in four health boards in south Wales and 127 in eight Clinical Commissioning Groups in south-west England using a variety of methods, including e-mails, mailshots, articles in GP magazines/circulars and follow-up phone calls. The result was an active decline response from 204 practices across Wales and 17 practices in England and no response from the rest.
When recruitment later moved to CLDTs, we approached 30 LD psychiatrists also in south Wales and south-west England. While the decision to participate in the research could be made at a practice level by GPs, this decision required buy-in not only from the whole health board or trust at secondary care level but also from the LD services and teams. In one area, meetings were held with a member of the trial team and representatives from the whole LD directorate before a decision was made to participate. Concerns centred mainly around the buy-in from wider care teams and how any escalations in behaviour would be handled and communicated between relevant parties. Of the 30 LD psychiatrists approached, 20 across six health boards/trusts agreed to take part in the trial.
In primary care, a site was considered to be the general practice, and in secondary care a site referred to the LD directorate of each health board or trust. Owing to the changing nature of the NHS, establishing which was the appropriate body to obtain approval from was more complex than expected. In one area of England, provision of LD services was provided on behalf of the NHS by a community interest company. Research was not something commonly dealt with by the company; therefore, the permissions process was unclear and took time to establish. This had the knock-on effect of delaying the recruitment of investigators and, thus, of participants.
Gaining permissions to use community LD services in south Wales was also challenging. The Abertawe Bro Morgannwg University Health Board LD directorate acts as the provider of LD services across Bridgend, Cardiff, Merthyr, Neath Port Talbot, Rhondda Cynon Taf and Swansea. This meant that permission to recruit participants through the LD clinics in these areas was gained through the Abertawe Bro Morgannwg University Health Board but the permission to use hospital pharmacies that were local to those clinics was gained through each individual health board.
Recruitment of participants depended on ensuring that approvals were in place for an investigator and a local hospital pharmacy at the same time. This had an impact on recruitment in one health board, as approval had been granted to recruit through a LD clinic by one health board but the health board in charge of the local hospital pharmacy delayed granting permission for some months while the board reviewed the request. Securing NHS costs added to the delay in gaining permission to recruit in certain areas, as the allocation process had become more complex with this change in study recruitment.
Recruitment of investigators
Along with permission from research and development boards, it was necessary for investigators to have undertaken Good Clinical Practice (GCP) training before undertaking research activities. This proved to be an obstacle for many, particularly in secondary care settings in which clinician involvement in research was less common. GCP training is specific to clinical trials and typically takes up to 3 hours to complete – a time commitment that was not always easy for clinicians to accommodate. Despite having 16 investigators based in primary care sites and 20 in CLDTs, only 14 of these recruited any participants into the trial: three GPs and 11 community LD psychiatrists. Low recruitment rates on the part of PIs are explored in more detail through interviews that are reported in Chapter 6.
Of the 11 psychiatrists who recruited participants, three were specialist registrars in LDs. The specialist registrars were able to provide invaluable support for the trial in that they took on the role of investigator, obtained GCP training and were able to see a number of participants on behalf of other clinicians who might not have had the time to get involved in research. Specialist registrars worked on rotation, however, and if they moved to different health boards or trusts there were difficulties in maintaining care of participants as part of the trial. Fortunately, we were able to overcome this in the current trial and arrange for other clinicians to take on the role of investigator.
Recruitment of participants
Details of the eligibility criteria were clearly laid out in the trial protocol, and practices and LD teams were able to identify sufficient numbers of potentially eligible patients. However, the number of potentially eligible patients who were actually approached with details about the trial was fairly low, particularly among primary care clinicians.
The study team drafted an audit tool that provided search terms and Read codes for general practices to help them identify those who might be eligible to take part. Once patients had been identified, GPs appeared reluctant to directly approach these patients or their carers about the study and to invite them to take part. It is not clear what the reasons for this were, as we did not gain any feedback through interviews with primary care clinicians (this is discussed in more detail in Chapter 6). Anecdotal feedback suggests that it may have been in part because of the nature of the consent procedure as well as concern about taking on decisions for care that are normally the domain of secondary care LD clinicians. However, following the change in recruitment from primary care to CLDTs, primary care practices became participant ID (identification) centres, rather than full sites, in an attempt to mitigate this issue. When clinicians had identified patients who might potentially be eligible, it was then possible to refer them to the CLDT clinicians who would be able to discuss the study in more detail. This potential was not subsequently utilised by GPs and did not result in participants coming from leads in primary care.
Consent procedure
There is understandable anxiety over the capacity of individuals with intellectual impairment to participate in clinical trials. Within the drafting of the Mental Capacity Act 200552 specific provision was made relating to the care, treatment and decisions on behalf of people who lack capacity, including participation in clinical trials. Further, important information is contained in the Medicines for Human Use (Clinical Trials) Regulations (2004). 41 As a Clinical Trial of an Investigational Medicinal Product (CTIMP), the ANDREA-LD trial was required to adhere to the latter regulations (which supersede the Mental Capacity Act 2005), meaning that, in the case of those who lacked capacity, consent would need to be given by a personal or professional legal representative of the participant. Although clinicians (particularly CLDT psychiatrists) are well versed in the Mental Capacity Act 2005,52 a lack of experience in research means that many are not familiar with differences in the consent procedure as specified under the Medicines for Human Use (Clinical Trials) Regulations (2004). 41
It was expected that many potential participants in the ANDREA-LD trial would probably lack capacity to give informed consent, and so clear study-specific guidance explaining the Regulations was drafted. The guidance specified that the following factors needed to be taken into consideration.
-
First, no patient would be treated as unable to make a decision unless all practical steps to help them to do so had been taken without success.
-
Second, even if an individual lacked capacity, their opinion would still be taken into consideration.
-
Third, should they lack capacity, the research team would, in accordance with due legal process, consult with relevant parties to clarify whether or not their participation was in the patient’s best interest.
Even with this guidance and reassurance, it became apparent that the consent process for the trial would be more challenging than expected.
Not only were clinicians apprehensive regarding the consent regulations, but wider care teams, carers and support staff were as well. The impact of this meant that it took much longer than hoped to explain the consent procedure to individuals and also to identify someone willing to act as a legal representative when the participant lacked capacity. On average, anecdotal evidence suggests it was taking between 2 and 3 weeks just to complete phone calls and arrange a meeting in order to complete the consent procedure. This was especially the case in residential settings, where carers, who would be well placed to provide consent, were required to refer this decision to managers and seniors, whose involvement often meant that a best interests meeting was called with wider care teams. With added layers of referral, it was often difficult to identify an individual who would be willing to provide any necessary consent.
Intervention delivery
Dosing
After extensive discussions, the decision was made by the trial team to include patients on any dose of risperidone, provided it was not being prescribed for psychosis and that the participant showed no evidence of psychosis. The rationale for this was that, if the withdrawal programme was to be shown to be effective and safe, it needed to work for individuals who would be on varying doses of medication. If the patient was deemed clinically eligible to enter the trial on all other criteria, their dose of risperidone should not be a restricting factor. By not limiting the entry criteria in this way, the pool of potentially eligible participants was also increased. We hypothesised, based on clinical experience, that doses of risperidone would be relatively low in the target population given that it was being prescribed for challenging behaviour as opposed to psychosis, for which larger doses are clinically indicated. The trial team, therefore, felt that there would probably be a limited number of dose combinations to cover as part of the trial. Careful consideration was given to the practicalities of how participants would receive their individually tailored medication regime.
Blinding medication
Blinding clinicians, carers and participants to treatment allocation presented a practical challenge. How could varying medication strengths be potentially tapered off without revealing which arm the participant had been randomised to? The decision was made to overencapsulate all trial medication using size 0 Swedish orange hypromellose capsules so that all of the strengths of risperidone and the placebo looked the same. The number of pills taken each day would also need to remain the same throughout the trial. To achieve this, the trial statistician created an algorithm that was used at baseline to create a unique dosing schedule for each participant (Table 4). Based on the participants’ prescription upon entering the trial, the algorithm calculated how many capsules each individual would need to take on a daily basis to ensure that the blind would remain intact. As a result of manufacturing constraints, only specific tablet strengths of risperidone were used in the trial (0.5 mg, 1 mg, 2 mg). The algorithm aimed to make the reductions as equal as possible using the minimum number of capsules as possible, based on up to four possible drug reduction stages within a 6-month period. (Note: it was not possible to reduce medication in four stages for participants on very low doses of risperidone, e.g. 0.5 mg daily.) If the participant had been allocated to the reduction arm, as the dose of active medication was reduced, a placebo capsule was introduced into the treatment regime. This allowed a constant number of capsules to be taken throughout the trial but with a variation in dose as necessary to accommodate the reducing regime.
| Time slot | Dose (mg) | Number of tablets |
|---|---|---|
| Baseline IMP regime: total daily dose 1.5 mg of risperidone for 33 days. To be taken as follows: | ||
| Morning | 1.0 | 1 |
| Midday | ||
| Bedtime | 0.5 | 1 |
| Stage 1 IMP regime: total daily dose 1.0 mg of risperidone for 33 days. To be taken as follows: | ||
| Morning | 1.0 | 1 |
| Midday | ||
| Bedtime | 0.0 | 1 |
| Stage 2 IMP regime: total daily dose 0.5 mg of risperidone for 33 days. To be taken as follows: | ||
| Morning | 0.5 | 1 |
| Midday | ||
| Bedtime | 0.0 | 1 |
| Stage 3 IMP regime: total daily dose 0.0 mg of risperidone for 33 days. To be taken as follows: | ||
| Morning | 0.0 | 1 |
| Midday | ||
| Bedtime | 0.0 | 1 |
| Stage 4 IMP regime: total daily dose 0.0 mg of risperidone for 33 days. To be taken as follows: | ||
| Morning | 0.0 | 1 |
| Midday | ||
| Bedtime | 0.0 | 1 |
Using the Nomad trays with separate compartments for days of the week as well as times of day meant that participant-specific doses of IMP could be safely dispensed and delivered. The trial team was able to specify which tablets (and thus which strength) should be taken at which time while maintaining the blind.
Because of the change in appearance of participants’ normal medication, a run-in period was implemented, which allowed individuals to get accustomed to using the Nomad trays and taking the slightly larger than normal capsules. For the vast majority, the change in the appearance of the medication was not a problem. Only one participant had difficulty taking the trial medication and had to be excluded. It was also important for carers to become accustomed to the new medication during this period, as they would be responsible for ensuring that participants took their medication as prescribed. Written and verbal information was provided to carers on how to handle study medication and the importance of using the Nomad trays correctly. It was important to ensure that carers fully understood how we were using the capsules and trays to blind the medication and for them to be clear on how to raise any concerns they had. In the case of participants residing in a staffed house, it was particularly important that everyone involved in providing that person with their medication knew about the trial.
A number of individuals required more tailored IMP deliveries, which included aligning IMP dispensing with that of other prescriptions the participant may be taking in order that medication administration record sheets could be completed more easily. These are sheets that serve as a record of the drugs administered to a patient at a residential setting. Although Nomad trays were used along with European Union Good Manufacturing Practice annex 13-compliant labelling, two residential settings delayed the progression of participants onto study medication by insisting that an extra label, signed off by the clinician, was included. Individual requests such as these became extremely time-consuming and difficult for the study team to manage.
Progression through the trial
Participant safety and careful monitoring of behaviour was of utmost importance in the trial design. The programme of reducing medication was devised to allow PIs to have regular contact with participants and to provide the opportunity to delay any potential reductions at any point if there was concern about the individual. This meant that trial medication could only be given out on a monthly basis once the PI had confirmed how the participant was to progress (Figure 2). To accommodate this, monthly prescriptions were dispensed and delivered to site within a 10 working day time frame. PIs therefore made their monthly contact with participants 2 weeks from the start of a new medication stage to allow adequate time for a new batch of trial medication to be dispensed and delivered. The PI’s decision regarding progression to the next study stage was translated into an IMP order by the study team and sent through to the pharmaceutical unit for dispensing. This pattern of working required investigators to be prompt in their communication with the trial team and to see participants within the specified time frame. When this was done, the system worked well; however, it also meant that the trial team needed to provide constant oversight to investigators on a real-time basis, which increased the burden on the study team’s workload and monitoring.
FIGURE 2.
Flow diagram for PI visits and IMP collection.

Dispensing medication
Another challenge was to ensure that instructions for taking medication while maintaining the blind were clear. To do this, the study team chose to deliver medication using the Nomad dosing system. Some participants and carers would have been used to receiving medication in this type of dosing tray, which is designed to make it more straightforward for individuals to know how much medication to take and when.
Once IMP had been manufactured at SMPU in Cardiff, it then had to be dispensed into the Nomad trays in accordance with the participant-specific prescriptions. The trial team made extensive investigations into who would be able to carry out the dispensing and then how to get the IMP to participants in different geographical areas throughout south Wales and south-west England. After discussions with various parties, the option chosen was to use SMPU not only to manufacture the IMP but to also supply to the patient-specific orders in a Nomad system under section 37 of the Medicines for Human Use (Clinical Trials) Regulations (2004)41 as the process of ‘post (Qualified Person) certification labelling for safety purposes’. SMPU was then able to dispatch IMP orders as necessary directly to specified sites or pharmacies via courier to be received by a designated member of staff.
When recruiting in primary care settings, each practice nurse’s role was to take receipt of participants’ medication and ensure that it was handed out according to the protocol. They also completed accountability records to evidence the whereabouts of the IMP. Enough IMP for 33 days was delivered at each stage, which meant that there was usually unused IMP that needed to be returned and destroyed. Any unused IMP from a previous stage could have been at a higher dose for participants in the reduction arm. It was important that the correct blinded medication was used each time.
Once recruitment in CLDTs began, it was apparent that the set-up used in primary care would not translate to CLDTs, as LD psychiatry clinics were not equipped to handle, store and dispose of medication as required by GCP. To resolve this, alternative solutions were examined, including the use of community pharmacies and delivering IMP via the post. The priority was to minimise the disruption experienced by the participant or their carer in obtaining medication on a frequent basis. This had, of course, to be balanced with what would be practical for the study team to set up and maintain and what would fit with regulatory requirements. The benefit of using community pharmacies would mean an IMP pick-up location local to each participant. However, the drawback was that it was not feasible for the study team to set up and maintain a potentially very large number of pharmacies in this way (we would not know which pharmacy would be most appropriate to use until the participant had been recruited). It would not be possible to ensure that agreements, GCP training and initiation were all in place prior to issuing study medication. The use of postal services to supply IMP was also not feasible because of regulatory issues and ensuring that any unused IMP was returned.
The approach taken was to use hospital-based pharmacies. Larger hospitals with ready GCP-trained clinical trials pharmacies were used to take receipt of IMP orders from SMPU and to issue medication to participants or their carers. Participants often had other health-care reasons to visit local hospitals; therefore, picking up trial medication from these locations was not overly burdensome for participants or their carers. For those who did find this a challenge, the study team was able to co-ordinate medication delivery in person by various members of the research team and supporting research network. However, this was possible for only a small number of participants. Researchers did not have the capacity to do this for all, as it involved special journeys from the office to the hospital pharmacy, on to the participant’s home, back to the pharmacy and then back to the office.
Summary
Trial procedures as laid out in the protocol were designed to allow the study to operate efficiently and in such a way as to accommodate individual requirements when possible. However, a number of factors outside the design of the trial had varying impacts on the delivery of the trial.
The main premise of the original trial design had been that it would be possible for clinicians in primary care to implement a drug reduction programme for adults with LDs who were being prescribed antipsychotic medication in the absence of psychosis. It became apparent fairly early on into the trial that there were a number of factors that were inhibiting this. First and foremost, the reluctance of GPs to be involved in the trial was a major stumbling block. Added to this, the requirement for investigators to have GCP training meant considerable delays in site recruitment. Second, the consent procedure for entering participants into the trial was new territory for many clinicians and delays were created as GPs referred to more specialist secondary care clinicians, who were also involved in the patient’s care, for advice.
The trial then moved to recruiting participants through CLDTs with LD psychiatrists acting as PIs. Obtaining appropriate permissions to recruit from secondary care services was not always straightforward and the complexity meant it took longer than expected. Obtaining GCP was also a delaying factor for secondary care clinicians, many of whom had little or no clinical trials experience.
Although confidence over consent procedures for participants who lacked capacity seemed to be an issue for primary care clinicians, secondary care investigators appeared more comfortable with the process, even though it was different from their normal practice. However, when it came to carers who were asked to provide consent for a participant who lacked capacity, many had knowledge only of the Mental Capacity Act 2005,52 and thus it took longer than expected and required more engagement with wider care teams to explain the procedure as outlined under Medicines for Human Use (Clinical Trials) Regulations (2004). 41 It would often take up to 2–3 weeks to ensure that an appropriate person was able to provide consent for an individual who lacked capacity to take part in the trial.
Once the procedure for maintaining the blind and delivering IMP to participants had been established, the system ran relatively smoothly. Use of the Nomad system worked well, and the majority of investigators and carers had no problem in completing trial procedures. There did, however, emerge a small number of individual tailoring requirements to certain aspects of the trial that were requested by some carers. As the trial only had a small number of recruits, the team could accommodate these; however, had recruitment been higher, it would have been more of a problem for the team to keep track of these. A limited number of investigators also required closer monitoring to ensure that procedures were being undertaken correctly and on time, which again put a time and workload burden on the trial team.
Initially, securing sites to recruit participants took a considerable amount of time but, of the challenges listed here, the elements that created the biggest ongoing impact on the trial team in terms of time and resource use were the tailored processes requested by some individuals and the monitoring required to ensure that study documentation was being completed on time by some investigators. Study processes were very time dependent, and it was crucial that all parties involved completed tasks as required. When this did not happen, the study team had to work quickly to ensure the progression of the participant in the trial.
As explained in the qualitative results, the analyst was blinded to the allocation of each participant during the analysis and drafting of this report. At the time of interview, nine carers were blinded to the allocation and seven had been unblinded. Participants 7, 17, 36 and 58 were all in the intervention arm of the study, and all were withdrawn because of the concerns of carers or the participants themselves about behaviour. Although some of these behaviours had been ongoing before the ANDREA-LD study, some of these concerns seemed to be very real for the carers. Participants 6, 35 and 53 were in the control arm and were withdrawn from the study. The carer of participant 6 reported that the participant had been withdrawn because of concerns about hallucinations. However, the carer also signalled that a history of allergic reactions to medications may have also been a reason for the clinician’s cautious approach in withdrawing the participant. Unfortunately, interviews with the carers of participants 35 and 53 were not conducted, possibly because the carer had wished to withdraw him/herself and the participant from the study completely, and so we have no qualitative data on the reasons for their withdrawal. In addition, of note, although perhaps not surprising, is the lack of equipoise towards the study arm by the carers and (to a lesser extent) clinicians. This is seen in discussion about their reasons for participating (seen as an individual benefit in terms of supported withdrawal rather than more altruistic reasons for benefit to the LD population in general). This lack of equipoise may account for some expressions of hope, reported in Chapter 5, Speculation about the arm of the trial, the unblinding phase and future management of the participant, that the participant had been successfully withdrawn from medication.
Chapter 4 Statistical analysis
Recruitment
Approximately 500 potential sites were contacted in total: 470 general practices and 30 CLDTs in south Wales and south-west England. Of those contacted, 79 expressed an interest and took part in the trial (16%), which comprised the majority of CLDTs contacted (20/30, 67%) and a minority from primary care (59/470, 13%). The majority of sites expressing interest in taking part identified potentially eligible participants (n = 61, or 77%) and became sites to recruit participants (n = 36, or 59%). From those sites, 18 provided a participant who was screened (37%), with a higher percentage of CLDTs providing participants for screening (50% vs. 28%), and 10 provided a participant who was randomised (20%), again with a higher yield from CLDTs (30% vs. 14%) (Table 5).
| Stage | General practices | CLDTs | Overall |
|---|---|---|---|
| Contacted | 470 | 30 | 500 |
| Expressed interest | 59 | 20 | 79 |
| Identified potentially eligible participants | 41 | 20 | 61 |
| Became sites to recruit participants | 16 | 20 | 36 |
| Provided a participant who was screened | 8 | 10 | 18 |
| Provided a participant who was randomised | 4 | 6 | 10 |
Thirty-six participants were screened in total: five participants screened in primary care and 31 from CLDTs. Of those 36 screened, 23 went on to complete a baseline assessment (63.9%): three coming from primary care (60.0%) and 20 from CLDTs (64.5%).
In total, 22 participants were then randomised following a baseline assessment (61.1% of those screened). This can be broken down as follows: for primary care, 80% of those screened and 100% of those who completed a baseline assessment were randomised; for CLDT, 61.3% of those screened and 95% of those who completed a baseline assessment were randomised (Table 6 and Figure 3).
| Stage | Primary care | CLDT | Overall |
|---|---|---|---|
| Screened | 5 | 31 | 36 |
| Completed baseline | 3 | 20 | 23 |
| Randomised | 3 | 19 | 22 |
FIGURE 3.
Consolidated Standards of Reporting Trials diagram. a, Participant was ‘held’ at the same level of medication but continued with the trial.
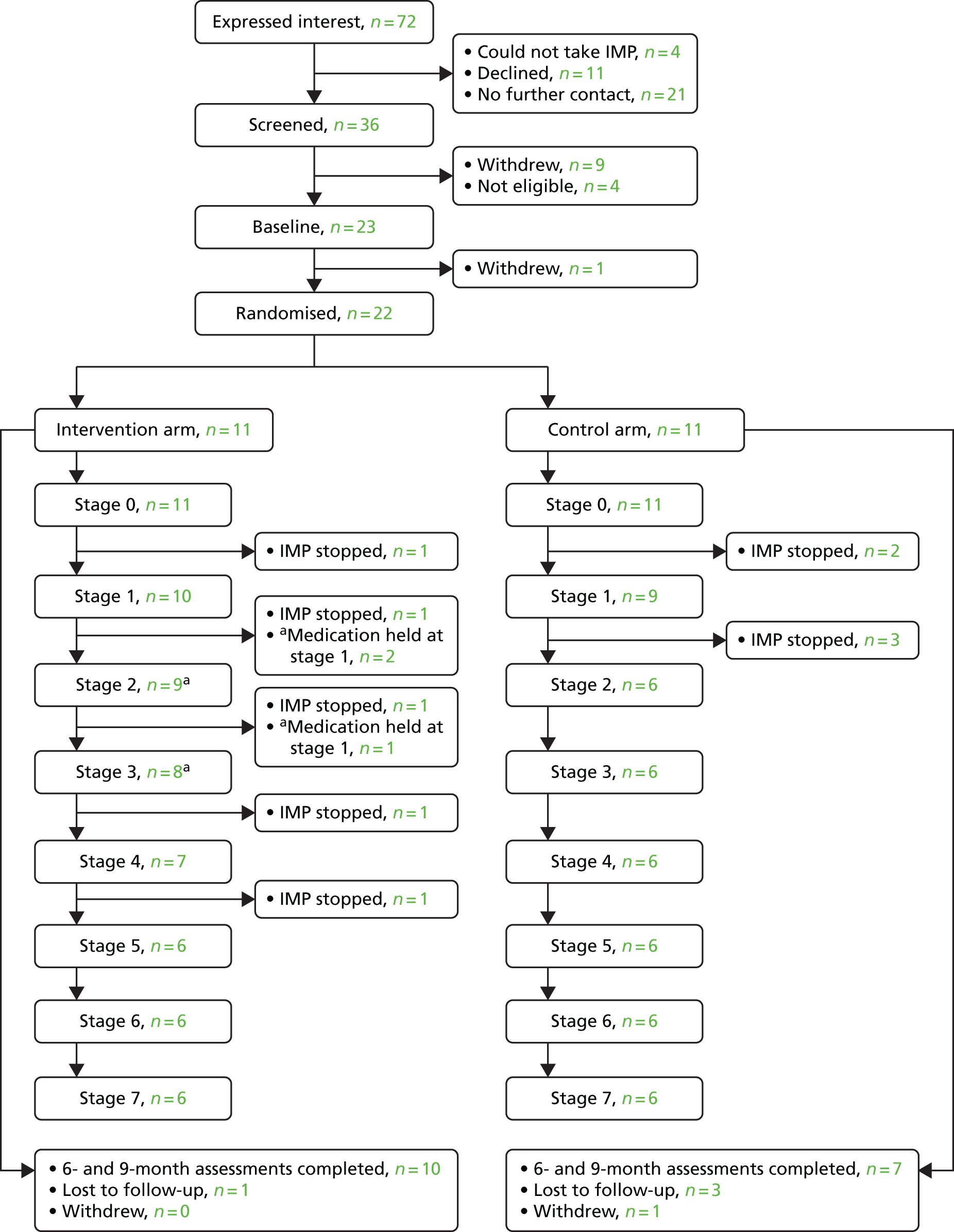
Baseline data
Pre-randomisation characteristics of study participants
Participants were well balanced with respect to variables collected pre randomisation (including at screening and during baseline visits). Fifteen participants were male (68%) and the mean age was 43 years (SD 13.4 years). There were no self-referrals. The majority of participants were consented into the study by a legal representative (n = 18, or 82% of those randomised). Ten of the randomised participants had a diagnosis of autistic spectrum disorder (45%) and none had ever been diagnosed with attention deficit hyperactivity disorder (ADHD). Ten had been diagnosed with epilepsy (45%), with all 10 currently on medication for epilepsy, four having had a seizure in the last year and seven having had a seizure in the past 5 years.
Regarding strategies used by carers to manage challenging behaviour, three participants were managed using physical intervention (14%), six were managed using seclusion (29%) and nine using PRN medication (43%). Most participants were in regular contact with their LD team (n = 21, or 95%), and when confidence handling challenging behaviour was self-rated (by carer) on a scale of 1 to 10 (1 being least confident and 10 being most confident), the median score was 10 [interquartile range (IQR) 8–10] (Table 7).
| Pre-randomisation characteristic | Trial arm | Overall (N = 22) | |
|---|---|---|---|
| Control (N = 11) | Intervention (N = 11) | ||
| Recruitment route | |||
| Primary care | 2 (18) | 2 (18) | 4 (18) |
| CLDT | 9 (82) | 9 (82) | 18 (82) |
| Self-referral | |||
| No | 11 (100) | 11 (100) | 22 (100) |
| Yes | 0 (0) | 0 (0) | 0 (0) |
| Gender | |||
| Male | 7 (64) | 8 (73) | 15 (68) |
| Female | 4 (36) | 3 (27) | 7 (32) |
| Mean age (years) | 42 | 44 | 43 |
| SD | 10.7 | 16.1 | 13.4 |
| Minimum, maximum | 24, 61 | 21, 68 | 21, 68 |
| Consent | |||
| Participant | 1 (9) | 3 (27) | 4 (18) |
| Legal representative | 10 (91) | 8 (73) | 18 (82) |
| Diagnosis of autistic spectrum disorder | |||
| No | 5 (45) | 7 (64) | 12 (55) |
| Yes | 6 (55) | 4 (36) | 10 (45) |
| Current diagnosis of ADHD | |||
| No | 11 (100) | 11 (100) | 22 (100) |
| Yes | 0 (0) | 0 (0) | 0 (0) |
| Ever been diagnosed with ADHD | |||
| No | 10 (91) | 11 (100) | 21 (95) |
| Yes | 1 (9) | 0 (0) | 1 (5) |
| Diagnosed with epilepsy | |||
| No | 7 (64) | 5 (45) | 12 (55) |
| Yes | 4 (36) | 6 (55) | 10 (45) |
| Currently on medication for epilepsy | |||
| No | 5 (56) | 5 (45) | 10 (50) |
| Yes | 4 (44) | 6 (55) | 10 (50) |
| Seizure in the last year | |||
| No | 8 (80) | 9 (82) | 17 (81) |
| Yes | 2 (20) | 2 (18) | 4 (19) |
| Seizure in the past 5 years | |||
| No | 8 (80) | 6 (55) | 14 (67) |
| Yes | 2 (20) | 5 (45) | 7 (33) |
| Use of a physical intervention to manage challenging behaviour | |||
| No | 11 (100) | 8 (73) | 19 (86) |
| Yes | 0 (0) | 3 (27) | 3 (14) |
| Training in physical interventions (if answered Yes to having used a physical intervention) | |||
| No | N/A | 0 (0) | 0 (0) |
| Yes | N/A | 3 (100) | 3 (100) |
| Length of time since physical intervention training last received (if answered Yes to having received physical intervention training) | |||
| < 6 months ago | N/A | 2 (67) | 2 (67) |
| 6 months to 1 year ago | N/A | 1 (33) | 1 (33) |
| 1 to 2 years ago | N/A | 0 (0) | 0 (0) |
| > 2 years ago | N/A | 0 (0) | 0 (0) |
| Use of seclusion to manage challenging behaviour | |||
| No | 5 (50) | 10 (91) | 15 (71) |
| Yes | 5 (50) | 1 (9) | 6 (29) |
| Use of PRN medication to manage challenging behaviour | |||
| No | 6 (60) | 6 (55) | 12 (57) |
| Yes | 4 (40) | 5 (45) | 9 (43) |
| In regular contact with the LD team | |||
| No | 0 (0) | 1 (9) | 1 (5) |
| Yes | 11 (100) | 10 (91) | 21 (95) |
| Median confidence handling challenging behaviour score | 10 | 10 | 10 |
| IQR | 8.0–10.0 | 8.5–10.0 | 8.0–10.0 |
| Minimum, maximum | 7, 10 | 7, 10 | 7, 10 |
Participants were on a median risperidone dose of 1.5 mg prior to randomisation (IQR 1.0–2.0 mg), with two participants on a dose of 4 mg or higher (9%). The majority of participants were given risperidone twice a day (n = 17, or 77% of those randomised), and among those whose data were known/available 20 participants had been on risperidone for more than 1 year (Table 8).
| Pre-randomisation risperidone medication characteristic | Trial arm | Overall | |
|---|---|---|---|
| Control | Intervention | ||
| Median total daily dose in mg | 1.5 | 1.0 | 1.5 |
| IQR | 1.0–2.0 | 1.0–2.0 | 1.0–2.0 |
| Minimum, maximum | 0.5, 4 | 0.5, 8 | 0.5, 8 |
| Total daily dose less than 4 mg | 10 (91) | 10 (91) | 20 (91) |
| Total daily dose at least 4 mg | 1 (9) | 1 (9) | 2 (9) |
| Frequency (times per day) | |||
| Once | 2 (18) | 3 (27) | 5 (23) |
| Twice | 9 (82) | 8 (73) | 17 (77) |
| Length of time on risperidone | |||
| Not known | 0 (0) | 1 (9) | 1 (5) |
| > 1 year | 10 (100) | 10 (91) | 20 (95) |
The mean ABS total score was 162 (SD 61.7), with no participants having an ABS-derived IQ that was 70 or higher. Median scores were 0 on all domains of the Mini PAS-ADD interview, for both 4-week and 2-year recall periods, and mental health thresholds were rarely triggered. One participant triggered the threshold for having a depressive disorder in the last 2 years, one for an anxiety disorder in the last 2 years, and three for psychosis in the last 2 years. No thresholds were triggered for disorders in the past 4 weeks (Table 9).
| Outcome measure | Trial arm | Overall | |
|---|---|---|---|
| Control | Intervention | ||
| Adaptive behaviour (ABS) | |||
| Total raw score (mean) | 145 | 180 | 162 |
| SD | 54.0 | 66.8 | 61.7 |
| Minimum, maximum | 58, 237 | 78, 296 | 58, 296 |
| Derived IQ score (mean) | 28 | 27 | 27 |
| SD | 9.5 | 13.7 | 11.4 |
| Minimum, maximum | 18, 44 | 11, 55 | 11, 55 |
| IQ less than 70, n (%) | 10 (100) | 10 (100) | 20 (100) |
| IQ at least 70, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Mental health (Mini PAS-ADD interview) | |||
| Depressive disorder subscale | |||
| Score in the past 4 weeks (median) | 0 | 0 | 0 |
| IQR | 0–2 | 0–1 | 0–1 |
| Minimum, maximum | 0, 3 | 0, 2 | 0, 3 |
| Threshold in the past 4 weeks, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Score in the past 2 years (median) | 0 | 0 | 0 |
| IQR | 0–0 | 0–1 | 0–0 |
| Minimum, maximum | 0, 0 | 0, 12 | 0, 12 |
| Threshold in the past 2 years, n (%) | 0 (0) | 1 (13) | 1 (7) |
| Anxiety disorder subscale | |||
| Score in the past 4 weeks (median) | 0 | 0 | 0 |
| IQR | 0–2 | 0–0 | 0–0 |
| Minimum, maximum | 0, 4 | 0, 0 | 0, 4 |
| Threshold in the past 4 weeks, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Score in the past 2 years (median) | 0 | 0 | 0 |
| IQR | 0–0 | 0–0 | 0–0 |
| Minimum, maximum | 0, 0 | 0, 13 | 0, 13 |
| Threshold in the past 2 years, n (%) | 0 (0) | 1 (20) | 1 (13) |
| Hypomania/mania (expansive mood) subscale | |||
| Score in the past 4 weeks (median) | 0 | 0 | 0 |
| IQR | 0–2 | 0–0 | 0–1 |
| Minimum, maximum | 0, 2 | 0, 1 | 0, 2 |
| Threshold in the past 4 weeks, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Score in the past 2 years (median) | 0 | 0 | 0 |
| IQR | 0–1 | 0–1 | 0–1 |
| Minimum, maximum | 0, 1 | 0, 1 | 0, 1 |
| Threshold in the past 2 years, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Obsessive–compulsive subscale | |||
| Score in the past 4 weeks (median) | 0 | 0 | 0 |
| IQR | 0–1 | 0–1 | 0–1 |
| Minimum, maximum | 0, 2 | 0, 2 | 0, 2 |
| Threshold in the past 4 weeks, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Score in the past 2 years (median) | 0 | 0 | 0 |
| IQR | 0–0 | 0–0 | 0–0 |
| Minimum, maximum | 0, 0 | 0, 0 | 0, 0 |
| Threshold in the past 2 years, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Psychosis subscale | |||
| Score in the past 4 weeks (median) | 0 | 0 | 0 |
| IQR | 0–0 | 0–0 | 0–0 |
| Minimum, maximum | 0, 0 | 0, 0 | 0, 0 |
| Threshold in the past 4 weeks, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Score in the past 2 years (median) | 0 | 0 | 0 |
| IQR | 0–0 | 0–0 | 0–0 |
| Minimum, maximum | 0, 3 | 0, 2 | 0, 3 |
| Threshold in the past 2 years, n (%) | 1 (9) | 2 (18) | 3 (14) |
| Unspecified disorder subscale | |||
| Score in the past 4 weeks (median) | 0 | 0 | 0 |
| IQR | 0–2 | 0–0 | 0–0 |
| Minimum, maximum | 0, 2 | 0, 0 | 0, 2 |
| Threshold in the past 4 weeks, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Score in the past 2 years (median) | 0 | 0 | 0 |
| IQR | 0–0 | 0–0 | 0–0 |
| Minimum, maximum | 0, 0 | 0, 5 | 0, 5 |
| Threshold in the past 2 years, n (%) | 0 (0) | 0 (0) | 0 (0) |
Clinical scores were generally low at baseline. The median MOAS total score was 1 (IQR 0–2). Median scores on the ABC were highest for the irritability (median 4, IQR 1–8) and hyperactivity subscales (median 5, IQR 2–13). The median scores on the PAS-ADD checklist were 0, and no participants met thresholds for any conditions/disorders. At least one antipsychotic side effect was reported by 16 participants (73%), with a median total number of side effects of 1 (IQR 0–3). No participants met the threshold for a possible movement disorder prior to randomisation (Table 10).
| Outcome measure | Trial arm | Overall | |
|---|---|---|---|
| Control | Intervention | ||
| Aggression (MOAS) | |||
| Total score | 1 (0–2, 0, 34) | 1 (0–3, 0, 9) | 1 (0–2, 0, 34) |
| Verbal aggression subscale | 0 (0–1, 0, 30) | 0 (0–1, 0, 7) | 0 (0–1, 0, 30) |
| Physical aggression against objects subscale | 0 (0–0, 0, 0) | 0 (0–0, 0, 2) | 0 (0–0, 0, 2) |
| Physical aggression against self subscale | 0 (0–2, 0, 2) | 0 (0–0, 0, 3) | 0 (0–1, 0, 3) |
| Physical aggression against others subscale | 0 (0–0, 0, 2) | 0 (0–0, 0, 1) | 0 (0–0, 0, 2) |
| Other challenging behaviour (ABC) | |||
| Irritability subscale | 4 (2–6, 0, 23) | 5 (1–9, 0, 31) | 4 (1–8, 0, 31) |
| Lethargy subscale | 2 (1–10, 0, 16) | 1 (0–9, 0, 16) | 2 (0–9, 0, 16) |
| Stereotypy subscale | 0 (0–2, 0, 12) | 0 (0–1, 0, 8) | 0 (0–1, 0, 12) |
| Hyperactivity/non-compliance subscale | 5 (3–14, 0, 21) | 5 (2–10, 0, 26) | 5 (2–13, 0, 26) |
| Inappropriate speech subscale | 0 (0–4, 0, 8) | 1 (1–5, 0, 11) | 1 (0–5, 0, 11) |
| Mental health (PAS-ADD checklist) | |||
| Possible organic condition total score | 0 (0–0, 0, 1) | 0 (0–0, 0, 4) | 0 (0–0, 0, 4) |
| Meets threshold for possible organic condition, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Affective or neurotic disorder total score | 0 (0–0, 0, 1) | 0 (0–0, 0, 0) | 0 (0–0, 0, 1) |
| Meets threshold for affective or neurotic disorder, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Psychotic disorder total score | 0 (0–0, 0, 0) | 0 (0–0, 0, 1) | 0 (0–0, 0, 1) |
| Meets threshold for psychotic disorder, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Adverse effects of psychotropic medication (ASC) | |||
| No side effects present, n (%) | 3 (27) | 3 (27) | 6 (27) |
| At least one side effect present, n (%) | 8 (73) | 8 (73) | 16 (73) |
| Total number of side effects present | 2 (1–4, 0, 10) | 1 (1–2, 0, 7) | 1 (0–3, 0, 10) |
| Movement disorders (DISCUS) | |||
| Total score | 0 (0–0, 0, 4) | 0 (0–1, 0, 2) | 0 (0–0, 0, 4) |
| Threshold met, n (%) | 0 (0) | 0 (0) | 0 (0) |
Randomisation characteristics
Twenty-two participants were randomised in total, with 11 allocated to each arm. As previously described, the majority of participants were on a total daily dose of risperidone of < 4 mg, and the majority were recruited from CLDTs. The arms were well balanced with respect to these key variables (Table 11).
| Total daily dose | Trial arm | Overall | |
|---|---|---|---|
| Control | Intervention | ||
| Primary care | |||
| < 4 mg | 2 | 1 | 3 |
| At least 4 mg | 0 | 1 | 1 |
| Overall | 2 | 2 | 4 |
| CLDT | |||
| < 4 mg | 8 | 9 | 17 |
| At least 4 mg | 1 | 0 | 1 |
| Overall | 9 | 9 | 18 |
| Overall | |||
| < 4 mg | 10 | 10 | 20 |
| At least 4 mg | 1 | 1 | 2 |
| Overall | 11 | 11 | 22 |
Participant retention outcomes
Of the 22 participants randomised, progression from stage 0 (run-in stage) to stage 1 (first reduction stage) was achieved by 19 (86%), with three participants withdrawing from trial medication prior to progression. Progression from stage 1 to stage 2 was achieved by 13 participants (59%), with a further four participants withdrawing from trial medication and two having their progression delayed. Thirteen participants continued progression through stage 3 and stage 4, with one participant withdrawing from trial medication prior to each progression point. Therefore, progression through all four stages of reduction (potential reduction if allocated to the control arm) was achieved by 13 participants (59%), with six of these participants being from the control arm and seven being from the intervention. An additional one intervention participant withdrew from trial treatment following progression to stage 4.
Follow-up data at 6 and 9 months post randomisation were obtained for 17 participants (77% of those randomised), with 10 intervention participants and seven control participants followed up (Table 12).
| Stage | Trial arm | Overall | |
|---|---|---|---|
| Control | Intervention | ||
| Total randomised | 11 (100) | 11 (100) | 22 (100) |
| Intervention receipt | |||
| Stage 0 to stage 1 | |||
| Withdrew before progressing to stage 1 | 2 (18) | 1 (9) | 3 (14) |
| Progressed from stage 0 to stage 1 | 9 (82) | 10 (91) | 19 (86) |
| Stage 1 to stage 2 | |||
| Withdrew between stage 1 and stage 2 | 3 (27) | 1 (9) | 4 (36) |
| Delayed progression between stage 1 and stage 2 | 0 (0) | 2 (18) | 2 (9) |
| Progressed from stage 1 to stage 2 | 6 (55) | 7 (64) | 13 (59) |
| Stage 2 to stage 3 | |||
| Withdrew between stage 2 and stage 3 | 0 (0) | 1 (9) | 1 (5) |
| Delayed progression between stage 2 and stage 3 | 0 (0) | 1 (9) | 1 (5) |
| Progressed from stage 2 to stage 3 | 6 (55) | 7 (64) | 13 (59) |
| Stage 3 to stage 4 | |||
| Withdrew between stage 3 and stage 4 | 0 (0) | 1 (9) | 1 (5) |
| Progressed from stage 3 to stage 4 | 6 (55) | 7 (64) | 13 (59) |
| Stage 4 to stage 4 (repeat 1) | |||
| Withdrew between stage 4 and stage 4 (repeat 1) | 0 (0) | 1 (9) | 1 (5) |
| Progressed from stage 4 to stage 4 (repeat 1) | 6 (55) | 6 (55) | 12 (55) |
| Stage 4 to stage 4 (repeat 2) | |||
| Progressed from stage 4 to stage 4 (repeat 2) | 6 (55) | 6 (55) | 12 (55) |
| Stage 4 to stage 4 (repeat 3) | |||
| Progressed from stage 4 to stage 4 (repeat 3) | 6 (55) | 6 (55) | 12 (55) |
| Participant follow-up | |||
| Completed 6-month follow-up | 7 (64) | 10 (91) | 17 (77) |
| Completed 9-month follow-up | 7 (64) | 10 (91) | 17 (77) |
Participants who progressed to stage 4 tended to be older [the mean age of those who progressed was 47 years (SD 12.3 years), compared with 37 years (SD 13.2 years) for those who did not progress] and to have higher MOAS total score, ABC-lethargy and ABC-hyperactivity scores at baseline and were more likely to have their challenging behaviour managed using PRN medication prior to randomisation (62% of those who progressed and 13% of those who did not progress). Participants who progressed were less likely to have a diagnosis of autism spectrum disorder (31% for those who progressed and 67% for those who did not), and were less likely to have consented themselves (8% for those who progressed and 33% for those who did not) (Table 13).
| Variable | Did not progress to stage 4 (n = 9) | Progressed to stage 4 (n = 13) | p-valueb |
|---|---|---|---|
| Participant characteristics | |||
| Mean age (years) (SD) | 37 (13.2) | 47 (12) | 0.080 |
| Gender (male) | 5 (56) | 10 (77) | 0.290 |
| Consent provided by participant | 3 (33) | 1 (8) | 0.125 |
| Diagnosis of autism spectrum disorder | 6 (67) | 4 (31) | 0.096 |
| Diagnosed with epilepsy | 5 (56) | 5 (39) | 0.429 |
| Mean ABS raw score (SD) | 176.9 (66.49) | 152.5 (59.22) | 0.401 |
| Challenging behaviour characteristics | |||
| Median MOAS total score (IQR) | 0 (0–1) | 1 (1–4) | 0.055 |
| Median ABC (IQR) | |||
| Irritability score | 3 (0–5) | 5 (4–10) | 0.213 |
| Lethargy score | 0 (0–1) | 9 (2–10) | 0.011 |
| Stereotypy score | 0 (0–1) | 0 (0–3) | 0.430 |
| Hyperactivity score | 3 (1–5) | 8 (5–14) | 0.087 |
| Inappropriate speech | 0 (0–1) | 2 (0–5) | 0.154 |
| ASC | |||
| Presence of antipsychotic medication side effects | 6 (67) | 10 (77) | 0.595 |
| Number of side effects present, median (IQR) | 1 (0–2) | 2 (1–3) | 0.356 |
| Use of physical intervention to manage CB | 1 (11) | 2 (15) | 0.774 |
| Use of seclusion to manage CB | 2 (25) | 4 (31) | 0.776 |
| Use of PRN medication to manage CB | 1 (13) | 8 (62) | 0.027 |
| Median confidence managing CB (IQR) | 10 (9–10) | 10 (8–10) | 0.797 |
| Antipsychotic medication characteristics | |||
| Median starting dose of risperidone (IQR) | 2 (1–2.5) | 1.5 (1–1.5) | 0.357 |
| Dosing frequency | |||
| Once daily | 2 (22) | 3 (23) | 0.962 |
| Twice daily | 7 (78) | 10 (77) | |
Clinical outcomes
Aggression (Modified Overt Aggression Scale total score)
At 6 months post randomisation, MOAS total scores remained low but were higher in those randomised to the intervention arm, with differences more discernible for the MITT population {control mean 3.0 [standard error (SE) 1.86], intervention mean 4.5 (SE 4.5)} than for the PP population [control mean 3.5 (SE 2.13), intervention mean 3.6 (SE 1.85)]. The adjusted mean differences for both populations indicated higher MOAS total scores for those randomised to the intervention arm, although 95% CIs were wide and included zero.
Modified Overt Aggression Scale total scores were higher at 9 months post randomisation than they were at 6 months, remaining higher in those randomised to the intervention arm in both MITT [control, mean 3.7 (SE 3.55); intervention, mean 7.7 (SE 3.51)] and PP [control, mean 4.3 (SE 4.14); intervention, mean 9.3 (SE 4.94)] populations. The adjusted mean differences both indicated higher MOAS total scores for those randomised to the intervention arm, although as for data at 6 months, 95% CIs were wide and included zero (Table 14).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | Adjusted mean differencea,b | 95% CI of adjusted mean difference | |
|---|---|---|---|---|---|
| Control | Intervention | ||||
| 6 months | |||||
| MITT (n = 17) | 3.0 (1.86) | 4.5 (1.62) | 1.5 (2.49) | 0.47 | –0.55 to 1.48 |
| PP (n = 13) | 3.5 (2.13) | 3.6 (1.85) | 0.1 (2.80) | 0.05 | –1.03 to 1.13 |
| 9 months | |||||
| MITT (n = 17) | 3.7 (3.55) | 7.7 (3.51) | 4.0 (5.15) | 0.90 | –0.42 to 2.22 |
| PP (n = 13) | 4.3 (4.14) | 9.3 (4.94) | 5.0 (6.58) | 0.99 | –0.90 to 2.88 |
Reflecting on our originally planned primary analysis (i.e. between-group comparison of the MOAS total score at 9 months post randomisation, with a two-sided 90% CI fitted and the upper limit of the CI inspected for non-inferiority), Table 15 and Figure 4 demonstrate that the upper limit of the CI in both the MITT and the PP populations (and for unadjusted and adjusted analyses) includes the stated non-inferiority margin. Therefore, we were unable to conclude non-inferiority on the basis of this study.
| Unadjusted/adjusted | Mean differencea | 90% CI of mean difference |
|---|---|---|
| MITT (n = 17) | ||
| Unadjusted | 0.89 | –0.19 to 1.98 |
| Adjustedb | 0.90 | –0.18 to 1.98 |
| PP (n = 13) | ||
| Unadjusted | 0.80 | –0.63 to 2.22 |
| Adjustedb | 0.99 | –0.54 to 2.52 |
FIGURE 4.
Forest plot illustrating between-group mean differences on the MOAS at 9 months post randomisation, with two-sided 90% CIs. Estimates/CIs are on the ln [x + 1] scale. Original non-inferiority margin of 3 has been translated onto this scale (1.39).

Figure 5 plots MOAS total scores for each individual at each time point, separating participants by their trial arm and whether or not they progressed onto stage 4. A change in the MOAS total score of 4 was deemed clinically important. 20 For the majority of participants, change in MOAS total scores over time was slight. However, five participants experienced a change from baseline in MOAS total score of at least 4. Two of these participants had been allocated to the intervention arm and had progressed to stage 4, one had been allocated to the control arm and had progressed to stage 4 and two had been allocated to the intervention arm and had not progressed to stage 4 (with both of these participants experiencing an increase in their MOAS total score at 6 months and then a decrease between 6 and 9 months).
FIGURE 5.
Individual MOAS total scores over time by trial arm and whether or not the participant progressed onto stage 4 of the intervention. (a) Control, did not progress to stage 4; (b) intervention, did not progress to stage 4; (c) control, did progress to stage 4; and (d) intervention, did progress to stage 4.
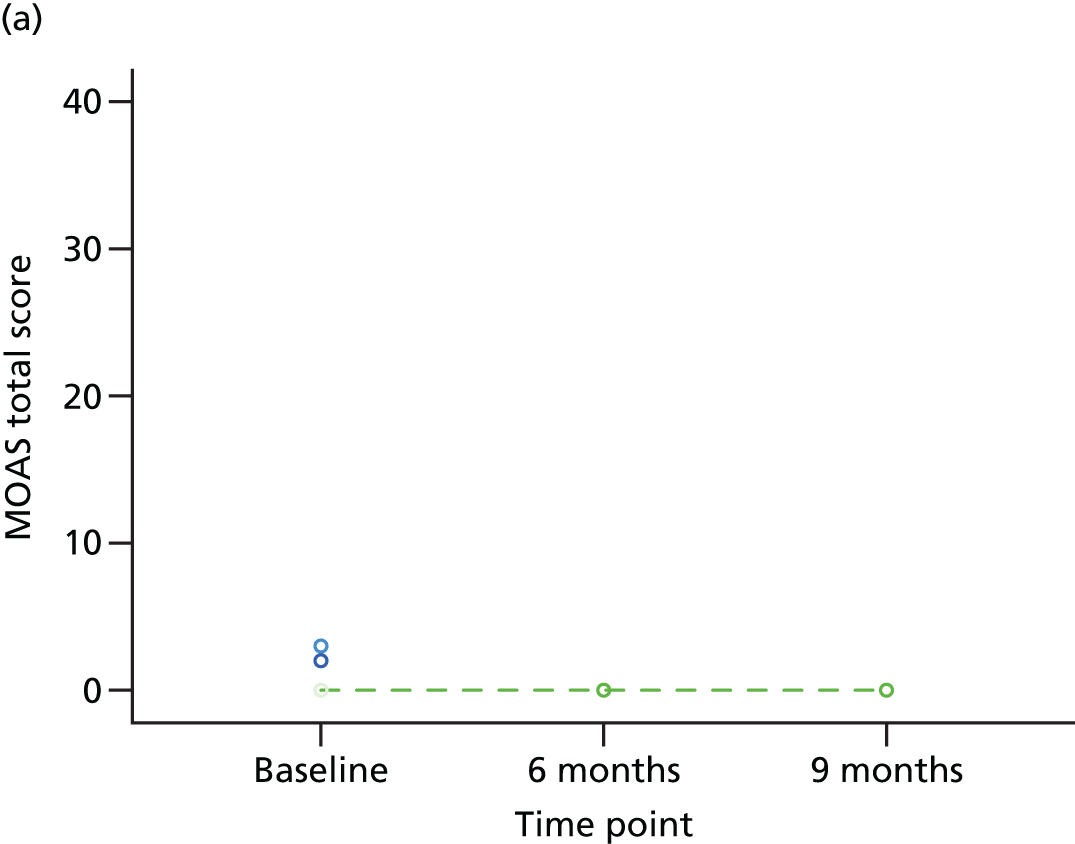
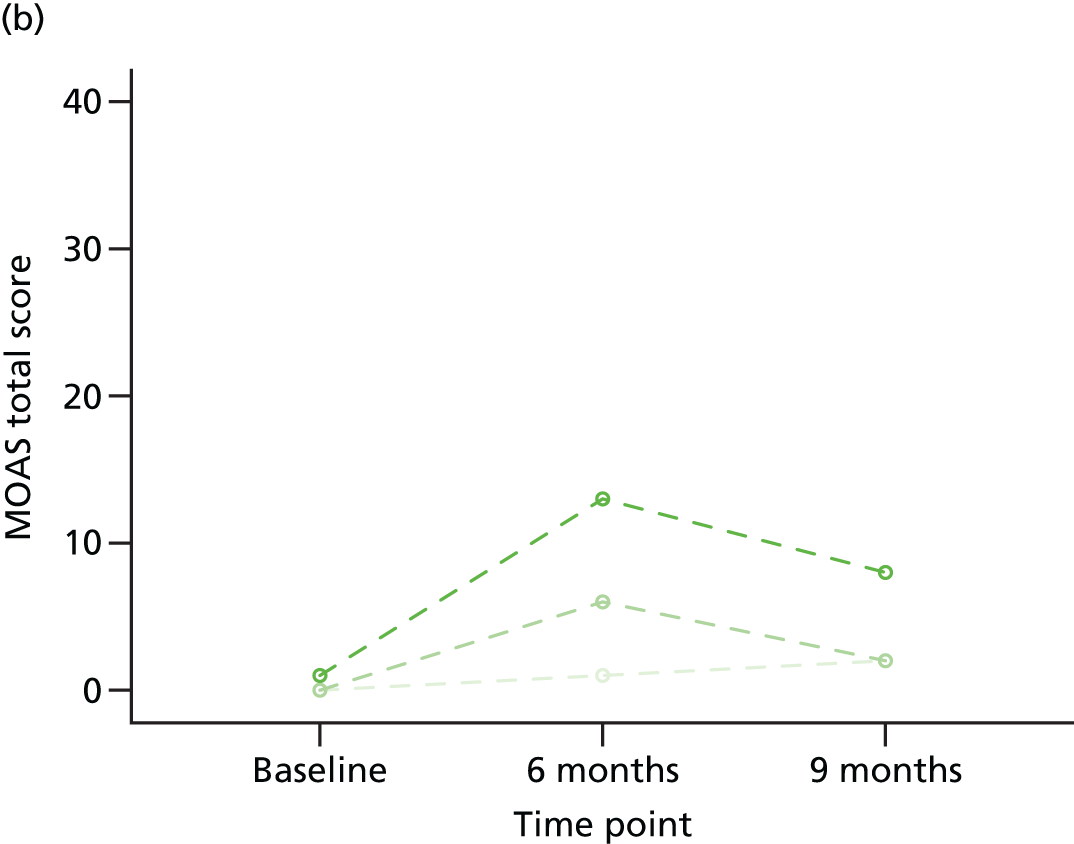

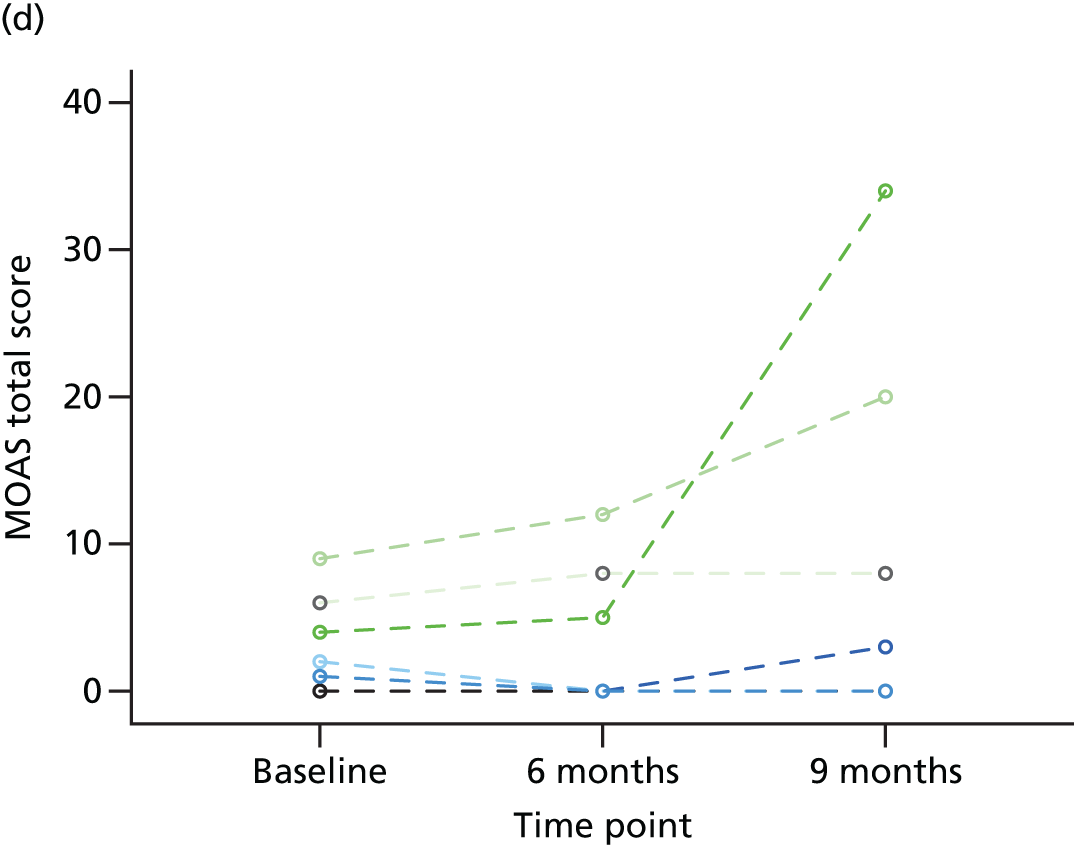
Psychotropic medication use
The average total daily dose of risperidone was lower in those randomised to the intervention arm than in those randomised to the control group at both 6 and 9 months post randomisation and in both ITT and MITT populations. In the MITT population (i.e. in those for whom this outcome was available), the mean was 1.6 mg lower in the intervention arm (SE 0.24 mg), whereas in the ITT population (i.e. assuming risperidone use had returned to baseline levels in those for whom this outcome was not available) the mean was 0.6 mg lower (SE 0.78 mg). As one participant withdrew from trial treatment soon after progressing to stage 4 (i.e. there were no additional withdrawals before the 6- and 9-month time points), the differences in level of psychotropic medication use at 6 and 9 months were identical (Table 16).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | |
|---|---|---|---|
| Control | Intervention | ||
| 6 months | |||
| ITT (n = 22) | 1.7 (0.29) | 1.1 (0.73) | –0.6 (0.78) |
| MITT (n = 12) | 1.6 (0.24) | 0.0 (0.00) | –1.6 (0.24) |
| 9 months | |||
| ITT (n = 22) | 1.7 (0.29) | 1.1 (0.73) | –0.6 (0.78) |
| MITT (n = 12) | 1.6 (0.24) | 0.0 (0.00) | –1.6 (0.24) |
Other challenging behaviour (Aberrant Behaviour Checklist subscales)
At 6 months post randomisation, mean ABC subscale scores were higher in those randomised to the intervention arm, with differences generally more discernible for the PP population than for the MITT population. While descriptively the means were consistently higher for the intervention arm, the adjusted mean difference was negative (indicating a lower adjusted mean in the intervention arm) for the irritability subscale (PP population) and inappropriate speech subscale (MITT and PP populations). However, other than for the stereotypy subscale (both MITT and PP populations), all 95% CIs included zero.
Although mean scores on the irritability and stereotypy subscales remained higher in the intervention arm at 9 months post randomisation, mean lethargy, hyperactivity and inappropriate speech scores were lower in the intervention arm. The 95% CIs of the adjusted mean differences for all ABC subscales included zero at 9 months (Table 17).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | Adjusted mean differencea,b | 95% CI of adjusted mean difference | |
|---|---|---|---|---|---|
| Control | Intervention | ||||
| 6 months | |||||
| Irritability subscale | |||||
| MITT (n = 17) | 7.1 (3.16) | 11.7 (4.00) | 4.6 (5.48) | 0.60 | –4.82 to 6.02 |
| PP (n = 13) | 7.7 (3.69) | 13.1 (5.68) | 5.5 (7.05) | –2.30 | –9.33 to 4.74 |
| Lethargy subscale | |||||
| MITT (n = 17) | 4.6 (2.03) | 7.5 (3.39) | 3.0 (4.42) | 4.00 | –3.73 to 11.73 |
| PP (n = 13) | 5.2 (2.30) | 8.4 (4.83) | 3.3 (5.66) | 2.77 | –7.92 to 13.46 |
| Stereotypy subscale | |||||
| MITT (n = 17) | 2.1 (1.53) | 4.2 (1.33) | 2.1 (2.05) | 2.88 | 0.36 to 5.34 |
| PP (n = 13) | 2.5 (1.77) | 5.0 (1.77) | 2.5 (2.52) | 3.97 | 0.96 to 6.98 |
| Hyperactivity/non-compliance subscale | |||||
| MITT (n = 17) | 8.3 (2.90) | 11.0 (3.23) | 2.7 (4.57) | 2.20 | –4.52 to 8.91 |
| PP (n = 13) | 9.4 (3.19) | 11.9 (4.65) | 2.5 (5.84) | 1.30 | –8.06 to 10.66 |
| Inappropriate speech subscale | |||||
| MITT (n = 17) | 3.0 (1.07) | 3.7 (1.32) | 0.7 (1.82) | –0.55 | –2.35 to 1.26 |
| PP (n = 13) | 3.2 (1.25) | 4.9 (1.84) | 1.3 (2.30) | –1.03 | –3.47 to 1.42 |
| 9 months | |||||
| Irritability subscale | |||||
| MITT (n = 17) | 5.5 (1.65) | 8.4 (3.15) | 2.9 (4.04) | 0.36 | –5.34 to 6.05 |
| PP (n = 13) | 5.4 (2.00) | 9.7 (4.34) | 4.3 (5.05) | 0.27 | –8.47 to 9.02 |
| Lethargy subscale | |||||
| MITT (n = 17) | 4.3 (1.21) | 4.0 (1.02) | –0.3 (1.59) | –0.29 | –3.76 to 3.18 |
| PP (n = 13) | 4.7 (1.36) | 3.7 (0.99) | –1.0 (1.65) | –0.76 | –4.69 to 3.17 |
| Stereotypy subscale | |||||
| MITT (n = 17) | 2.1 (1.28) | 2.5 (0.99) | 0.4 (1.60) | 0.95 | –1.34 to 3.24 |
| PP (n = 13) | 2.5 (1.46) | 2.6 (1.34) | 0.1 (1.98) | 0.98 | –2.05 to 4.01 |
| Hyperactivity/non-compliance subscale | |||||
| MITT (n = 17) | 6.9 (1.84) | 5.6 (1.71) | –1.3 (2.56) | –1.53 | –6.24 to 3.19 |
| PP (n = 13) | 7.7 (1.96) | 5.9 (2.24) | –1.8 (3.03) | –1.84 | –8.51 to 4.83 |
| Inappropriate speech subscale | |||||
| MITT (n = 17) | 2.0 (0.66) | 2.4 (0.85) | 0.4 (1.12) | –0.30 | –1.91 to 1.31 |
| PP (n = 13) | 2.0 (0.78) | 2.7 (1.17) | 0.7 (1.46) | –0.55 | –2.79 to 1.70 |
Mental health (Psychiatric Assessment Schedule for Adults with Developmental Disability checklist)
Mean scores on the subscales of the PAS-ADD checklist were higher in the intervention group at both 6 and 9 months post randomisation and for both MITT and PP populations. Owing to a high frequency of zero scores from participants, linear regression analyses were not possible for all outcomes. Similar to the analysis of ABC subscales, although descriptively the mean scores were consistently higher for the intervention arm, for two analyses (affective/neurotic disorder at 9 months for the PP population and psychotic disorder at 9 months in the MITT population) the adjusted mean difference (when an adjustment was made for the recruitment route and corresponding score at baseline) indicated that these were lower in the intervention arm. However, all 95% CIs of the adjusted mean differences included zero (Table 18).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | Adjusted mean differencea,b | 95% CI of adjusted mean difference | |
|---|---|---|---|---|---|
| Control | Intervention | ||||
| 6 months | |||||
| Possible organic disorder | |||||
| MITT (n = 17) | 0.1 (0.14) | 1.0 (0.60) | 0.9 (0.73) | 0.26 | –0.28 to 0.79 |
| PP (n = 13) | 0.2 (0.17) | 1.4 (0.81) | 1.3 (0.83) | ||
| Affective or neurotic disorder | |||||
| MITT (n = 17) | 0.3 (0.30) | 0.8 (0.51) | 0.5 (0.67) | ||
| PP (n = 13) | 0.4 (0.35) | 0.9 (0.71) | 0.5 (0.83) | ||
| Psychotic disorder | |||||
| MITT (n = 16) | 0.0 (0.00) | 0.2 (0.13) | 0.2 (0.13) | ||
| PP (n = 12) | 0.0 (0.00) | 0.3 (0.18) | 0.3 (0.22) | ||
| 9 months | |||||
| Possible organic disorder | |||||
| MITT (n = 17) | 0.1 (0.14) | 0.9 (0.46) | 0.8 (0.48) | ||
| PP (n = 13) | 0.2 (0.17) | 1.3 (0.61) | 1.1 (0.63) | 0.52 | –0.23 to 1.26 |
| Affective or neurotic disorder | |||||
| MITT (n = 17) | 0.3 (0.18) | 0.8 (0.59) | 0.5 (0.73) | ||
| PP (n = 13) | 0.2 (0.17) | 1.1 (0.83) | 1.0 (0.91) | –0.27 | –0.70 to 0.16 |
| Psychotic disorder | |||||
| MITT (n = 16) | 0.3 (0.33) | 0.4 (0.31) | 0.1 (0.47) | –0.29 | –0.95 to 0.62 |
| PP (n = 12) | 0.0 (0.00) | 0.6 (0.43) | 0.6 (0.43) | ||
At 6 months post randomisation, the threshold for a possible organic disorder was triggered by one participant in the intervention arm. At 9 months post randomisation, the threshold for an affective or neurotic disorder was triggered by one participant in the intervention arm and the threshold for a psychotic disorder was triggered by two participants (one per arm) (Table 19).
| Participant population | Trial arm, n (%) | |
|---|---|---|
| Control | Intervention | |
| 6 months | ||
| Possible organic disorder | ||
| MITT (n = 17) | 0 (0) | 1 (10) |
| PP (n = 13) | 0 (0) | 1 (14) |
| Affective or neurotic disorder | ||
| MITT (n = 17) | 0 (0) | 0 (0) |
| PP (n = 13) | 0 (0) | 0 (0) |
| Psychotic disorder | ||
| MITT (n = 16) | 0 (0) | 0 (0) |
| PP (n = 12) | 0 (0) | 0 (0) |
| 9 months | ||
| Possible organic disorder | ||
| MITT (n = 17) | 0 (0) | 0 (0) |
| PP (n = 13) | 0 (0) | 0 (0) |
| Affective or neurotic disorder | ||
| MITT (n = 17) | 0 (0) | 1 (10) |
| PP (n = 13) | 0 (0) | 1 (14) |
| Psychotic disorder | ||
| MITT (n = 16) | 1 (17) | 1 (10) |
| PP (n = 12) | 0 (0) | 1 (14) |
Adverse effects of psychotropic medication (Antipsychotic Side-effect Checklist)
In the MITT population, at least one side effect of psychotropic medication was reported by seven control participants (100%) and eight intervention participants (80%) when they were asked at 9 months post randomisation. In the PP population (i.e. in those who had progressed to stage 4 and for whom outcome data were available), all participants reported at least one side effect (Table 20).
| Participant population | Trial arm, n (%) | |
|---|---|---|
| Control | Intervention | |
| 9 months | ||
| At least one side effect present | ||
| MITT (n = 17) | 7 (100) | 8 (80) |
| PP (n = 13) | 6 (100) | 7 (100) |
The mean number of psychotropic medication side effects reported at 9 months post randomisation was higher in the control arm [MITT, mean 2.6 (SE 0.72)] than the intervention arm [MITT, mean 1.4 (SE 0.31)], with mean numbers similar for the PP population. However, the 95% CI of the adjusted mean difference included zero for both analyses (Table 21).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | Adjusted mean differencea,b | 95% CI of adjusted mean difference | |
|---|---|---|---|---|---|
| Control | Intervention | ||||
| 9 months | |||||
| Total number of side effects reported | |||||
| MITT (n = 17) | 2.6 (0.72) | 1.4 (0.31) | –1.2 (0.70) | –1.04 | –2.52 to 0.44 |
| PP (n = 13) | 2.7 (0.84) | 1.7 (0.29) | –1.0 (0.89) | –0.74 | –2.71 to 1.23 |
Movement disorders (Dyskinesia Identification System Condensed User Scale)
At 9 months post randomisation, the mean total DISCUS score was higher in the intervention arm [MITT, mean 2.1 (SE 1.67)] than in the control arm [MITT, mean 0.0 (SE 0.00)], while the mean for intervention participants who progressed to stage 4 was higher again [PP, mean 3.0 (SE 2.35)] (Table 22). One participant in the intervention arm met the threshold for a possible movement disorder (Table 23).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | |
|---|---|---|---|
| Control | Intervention | ||
| 9 months | |||
| Total DISCUS score | |||
| MITT (n = 17) | 0.0 (0.0) | 2.1 (1.67) | 2.1 (1.67) |
| PP (n = 13) | 0.0 (0.0) | 3.0 (2.35) | 3.0 (2.35) |
| Participant population | Trial arm, n (%) | |
|---|---|---|
| Control | Intervention | |
| 9 months | ||
| DISCUS threshold met | ||
| MITT (n = 17) | 0 (0) | 1 (10) |
| PP (n = 13) | 0 (0) | 1 (14) |
Managing challenging behaviour
At 9 months post randomisation, carers reported using physical intervention to manage challenging behaviour for two participants (both allocated to the intervention arm), although only one of these had progressed through to stage 4. They reported using seclusion for three participants (both allocated to the control arm) and PRN medication for nine participants (four allocated to the control arm and five to the intervention arm), although for the intervention arm only six of these participants had progressed to stage 4 (three per arm) (Table 24).
| Participant population | Trial arm, n (%) | |
|---|---|---|
| Control | Intervention | |
| 9 months | ||
| Use of physical intervention to manage challenging behaviour | ||
| MITT (n = 17) | 0 (0) | 2 (20) |
| PP (n = 13) | 0 (0) | 1 (14) |
| Use of seclusion to manage challenging behaviour | ||
| MITT (n = 17) | 3 (43) | 0 (0) |
| PP (n = 13) | 3 (50) | 0 (0) |
| Use of PRN medication to manage challenging behaviour | ||
| MITT (n = 17) | 4 (57) | 5 (50) |
| PP (n = 13) | 3 (50) | 3 (43) |
The mean score on the scale measuring confidence handling challenging behaviour was higher for the intervention arm [MITT, mean 9.3 (SE 0.26)] than for the control arm [MITT, mean 8.7 (SE 0.36)], with scores similar for the PP population. However, the 95% CIs of the adjusted mean differences included zero (Table 25).
| Participant population | Trial arm, mean (SE) | Unadjusted mean difference (SE)a | Adjusted mean differencea,b | 95% CI of adjusted mean difference | |
|---|---|---|---|---|---|
| Control | Intervention | ||||
| 9 months | |||||
| Confidence handling challenging behaviour | |||||
| MITT (n = 17) | 8.7 (0.36) | 9.3 (0.26) | 0.6 (0.43) | 0.59 | –0.28 to 1.45 |
| PP (n = 13) | 8.8 (0.40) | 9.6 (0.20) | 0.7 (0.45) | 0.67 | –0.26 to 1.60 |
Use of pro re nata medication
Use of PRN medication post randomisation was captured using carer-reported diaries. However, as indicated by Table 26, the majority of participants did not return these diaries. For those who did, it would appear that PRN use was generally higher in those allocated to the intervention arm and appeared to increase over time. This is also illustrated in Figure 6.
| Time period (weeks) | n | Trial arma | Overalla | |
|---|---|---|---|---|
| Control | Intervention | |||
| 1–5 | 10 | 1 (25) | 2 (33) | 3 (30) |
| 6–10 | 11 | 1 (20) | 3 (50) | 4 (36) |
| 11–15 | 14 | 2 (33) | 3 (38) | 5 (36) |
| 16–20 | 12 | 2 (40) | 4 (57) | 6 (50) |
| 21–25 | 10 | 1 (17) | 3 (75) | 4 (40) |
| 26–30 | 6 | 0 (0) | 1 (50) | 1 (17) |
| 31–35 | 6 | 0 (0) | 3 (100) | 3 (50) |
FIGURE 6.
Pro re nata use following randomisation.
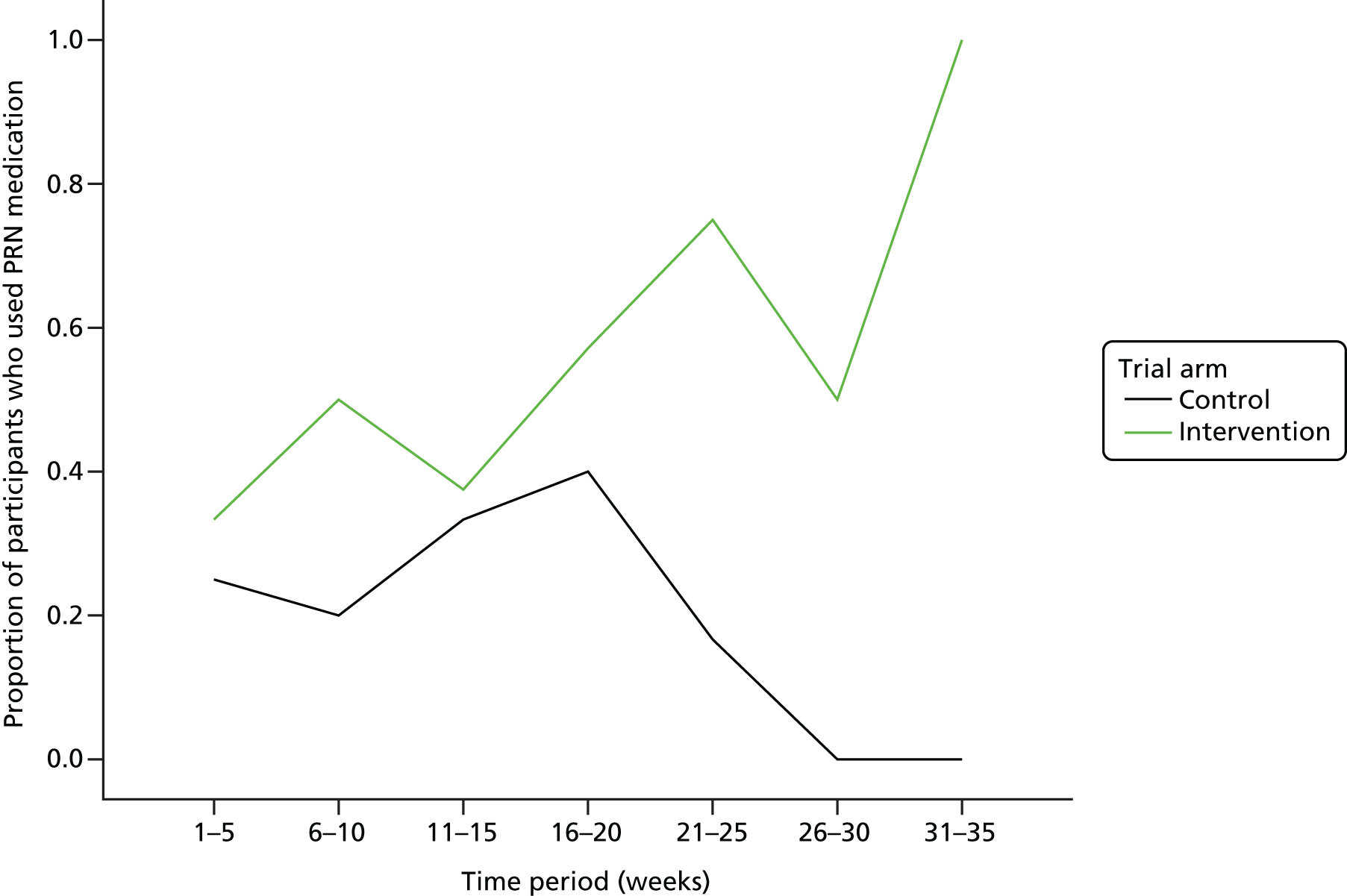
Safety reporting
During the course of the trial, there were four adverse events reported and one serious adverse event (SAE). The SAE was categorised as ‘an event which required intervention to prevent outcomes such as hospitalisation’. There was reported deterioration in the participant’s mental health with increased agitation, tearfulness and depressive thoughts as well as reports of hearing a voice. Deterioration was such that crisis intervention was required to avoid hospital admission. It was suspected that these symptoms were not a side effect of the trial medication but a recurrence of symptoms masked by the risperidone and, hence, a serious adverse reaction to the withdrawal (i.e. the intervention being trialled during the study) as opposed to the medication itself. This participant withdrew from the trial intervention and the blind was broken. On entry into the trial, the participant had been taking 2.5 mg of risperidone daily and progressed through three stages of reduction so that, when the blind was broken at the time of the SAE, they were taking 0.5 mg daily.
Chapter 5 Qualitative study: exploring carer, participant and clinician experiences of the ANDREA-LD trial and of medication reduction
Rationale for qualitative substudy
As specified in the original protocol, we planned to undertake qualitative telephone interviews with a proportion of carers, participants and recruiting clinicians who took part in the trial. One of the main purposes of these interviews was to gain insight on barriers to drug reduction in clinical practice, as well as attributions of behavioural changes in relation to potential reduction of medication. The interviews had originally been scheduled to take place during the unblinded phase of the trial between the 9- and the 12-month time points, but because of the need to close trial recruitment early, and the timetable of the subsequent close-down plan, the interviews were brought forward to be conducted between 4 and 6 months after randomisation.
With awareness among the ANDREA-LD team that recruitment issues were becoming increasingly apparent, the qualitative interviews about participants’ views of the trial took on greater importance. The team hypothesised that the ANDREA-LD study was challenging to clinicians, carers and participants because of issues relating to capacity to consent, views about the necessity of antipsychotic medication, observed changes in behaviour and the challenges of conducting research with participants who were in residential homes.
Aim
The qualitative study was therefore set up to explore the experiences and challenges of the study with study participants, their carers, and clinicians using qualitative methods.
Objectives
The objectives of the qualitative interviews were to ascertain:
-
views about their reasons for participating in the study and concerns about participating both before and after randomisation
-
experiences of taking part in the study including the support received from the study team
-
whether or not the participant’s behaviour had changed during the trial and why those changes may have occurred, and reasons for any partial or full reinstatement to usual care medication during the study period
-
views on the study medication (size, colour, taste, etc.) and study processes including consenting arrangements and completion of assessments
-
views about the use of antipsychotic medication to treat behavioural problems for the participant in particular and also within the LD population in general
-
views about what should happen to participants once the trial finished, including thoughts about whether or not any participants should continue to be withdrawn from antipsychotic medication or should remain on the usual dose.
Study design
We used qualitative research methods because this allowed us to explore respondents’ views and experiences in depth, including topics that we were unable to predict in advance. We thought it important to understand the views of a range of stakeholders; therefore, our respondents included participants, carers (including family and professional carers) and recruiting clinicians. As the study had been altered to focus on recruitment in secondary care, only clinicians in these services were included in the interviews. Data were collected through a combination of face-to-face interviews with participants, either face-to-face or telephone interviews with carers, and telephone interviews with clinicians. All data were audio-recorded with the respondent’s consent.
Recruitment
Our aim was to interview as many patients, carers and recruiting clinicians as feasibly possible within the time frame and from the participants of the trial.
Interviews with carers
In total, 16 carers (five parents and 11 professional carers) were invited to be interviewed by ANDREA-LD research staff. Interviews were conducted at the carer’s home, in a private room within the residential care home or by telephone.
Interviews with patient participants
With the support of residential care staff in three cases and a parent carer in the remaining case, a research assistant approached ANDREA-LD patient participants who had been identified as having mental capacity to consent for themselves during the screening phase of the study. Consent to participate in the qualitative study was undertaken. Interviews were conducted in a private room (an office or lounge) within the residential setting in three cases and in an office within a day centre in the remaining case. All interviews were conducted by a research assistant. A residential carer was present during three of the interviews; a member of day service staff was present during the fourth interview. Carers were asked to support the participants, but to refrain from answering on behalf of the participants.
Telephone interviews with clinicians
Eleven secondary care clinicians were contacted to take part in a telephone interview.
Ethics requirements
Ethics approval for the qualitative study was given as part of the main ANDREA-LD ethics application. All respondents consented to be approached with information about the qualitative study at the point of consent into the main trial. At the point at which the interviews took place, they were provided with an information sheet about the purpose of the qualitative study and what was being asked of them. All respondents signed a consent form prior to the interview and, in the case of telephone interviews, this was returned in the post. Participating clinicians received a £50 voucher as a ‘thank you payment’ for participating in an interview, patient participants and carers received a £10 voucher.
Data collection
An interview topic guide defined the main topics while allowing flexibility to pursue issues in more depth as they emerged from the interviews. The semistructured interview schedules for each of the three groups of participants are included in Appendix 2. All data were transcribed verbatim and anonymised on transcription.
Data analysis
Data were analysed using thematic analysis with an abductive approach (incorporating themes that had been identified in advance and themes that were derived from the data). 53 This approach involves systematically coding data according to a thematic framework, which is developed iteratively. The thematic framework was applied to the data using the coding software package NVivo version 10. Interpretations were discussed between members of the ANDREA-LD team. All quotations presented have been anonymised and respondents have been given ID numbers. In order to associate the responses from carers with the participant they care for, carer and participant interviews share a common numeric ID number, so that carer 7 is the carer for participant 7. However, it was not possible to use this formula for the clinicians, as some clinicians had more than one participant recruited in ANDREA-LD. The clinician ID numbers therefore do not relate to the carer or participant ID numbers.
It is also worth noting that the analysis and report writing for this chapter were conducted blind to which arm each participant was in. It was only when the report had been drafted and commented on by other authors that the lead analyst was unblinded to the randomisation of participants and this information was then added to the data extracts. At this point, a further section was added to the discussion section to reflect on participants’ responses in the context of their trial arm allocation.
Results
Interviews with carers
All 16 (five parent and 11 professional) carers who had been invited to participate in the study agreed to take part. The five parents interviewed had given advice as legal representative that their son or daughter could participate in the ANDREA-LD study. Interviews lasted between 12 and 31 minutes with an average time of 19 minutes.
Reasons for participating in the study
The majority of carers had positive reasons for participation, such as a desire to reduce the participant’s medication if possible. This was seen as desirable, as it was felt that medication such as antipsychotics (but also antidepressants) were modifying the participant’s personality. Other staff members felt that the ANDREA-LD study presented an opportunity to review a participant’s medication within a supported environment. As such, reasons of personal benefit rather than population-level altruism seemed to be the main motivators:
She wanted to try reducing her risperidone um, and she’d been talking with the psychiatrist about it, because she’d managed to come off of her citalopram [antidepressant] which she’d been on for about 7 years and um, [participant 7] is um, highly emotionally reliant on taking things. She sees it as a fix-all, doctors fix everything and pills fix everything and um, we’ve been trying to kind of get her to be more reliant on stabilising her own mood, and having, kind of, safety nets in place for her to check and balance her own mood, rather than relying on lots of medication that made her a zombie really.
Carer 7, staff (participant in intervention arm and was withdrawn during stage 3)
The opportunity to participate was particularly welcomed if the participant had been experiencing side effects of being on antipsychotic medication:
. . . they asked, they asked my permission, and I said ‘if it’s going to do good’ because he was putting on so much weight and when he was on this, this trial, he, he did lose a lot of weight.
Carer 17, parent (participant in intervention arm and was withdrawn at the end of stage 3)
Can you tell me your reasons for participating in the trial in the first place?
Yeah because [participant 22’s] blood test had shown that he needed to come off the risperidone anyway.
Carer 22, parent (participant in control arm and interview conducted at stage 7)
There was one parent who felt that their negative views about antipsychotic medication had not been taken seriously by the health-care professionals to date and that this was their chance to be proactive about medication reduction:
. . . because I didn’t believe that, um, that the medication was right for [participant 8], it wasn’t, I didn’t think it was doing anything for him really. I weighed it up I thought ‘well, if I don’t take part, well I, I couldn’t say anything could I?’
Carer 8, parent (participant in control arm and interview conducted at stage 6)
In contrast, there were professional carers who were eager to use the ANDREA-LD study as a mechanism to persuade family members that reduction of medication could be a positive management strategy within the confines of a supported trial and gradual medication reductions without issues of bias clouding the carers’ judgement about whether or not medication reduction was affecting behaviour:
[Psychiatrist] thought that he’d be a perfect candidate for the study. He’s on risperidone um, she wanted to take him off it, and the study came up at the time and she thought that might be a quick way to reduce them. And then also it would be blind, well double blind, so no one would really know whether he was coming off it or not. We’ve had some issues with his family not wanting him to come off it, because of past behaviours that they’d experienced, um so we thought that it would be a good way to, if obviously he took the reduction route, then it would be a good way for him to come off without having those um, maybe false um . . .
OK.
What they called? Um behaviours.
Carer 66, staff (participant in intervention arm and interview conducted at stage 5)
However, a small minority of carers had rather passive reasons for participation. When asked why they had consented involvement with the ANDREA-LD trial, some carers did not appear to have thought critically about their participation. Instead, they had followed the lead of the clinician:
Well um I uh suppose uh it was just that um [doctor’s name] said about it.
OK.
Then um who am I to object? You know?
Carer 6, parent (participant in control arm and was withdrawn at stage 0)
Can you tell me what the reasons were for [name] participating in the trial?
Um, it was well, it was suggested by her psychiatrist.
OK.
Um. Because it was thought that she could, that she’d probably be coming off them anyway, so to do the trial first.
Carer 28, staff (participant in control arm and interview conducted at stage 5)
In some cases the carers reported that, ultimately, it had been the participant’s decision whether or not to participate in the trial:
There was a discussion between [psychiatrist] and [team leader] and [participant 52] about kind of like the benefits of going ahead with it.
OK.
And they had an open discussion and [participant 52] ultimately decided that she’d go for it.
OK, so the ultimate decision was left with [participant 52]?
Yeah, yeah, yeah, she consented to it, she um, she was all positive about it the whole time, um yeah I can remember [team leader] was enthusiastic about it but ultimately it was [participant 52’s] decision.
Carer 52, staff (participant in intervention arm and interview conducted at stage 7)
Views about the trial
Concerns about the trial were surprisingly rare given that, for most carers, the ANDREA-LD study was the first large trial or CTIMP that they had experienced. For some, the uncertainty about whether or not the participant was in the withdrawal or treatment-as-normal arm was a slight cause of concern, but they understood that this was necessary as part of trial procedures:
Were there any concerns that you, or um, maybe her father had about her participating in the trial?
Um, only just, um, the fact that, because she was coming off the risperidone, just, well, we didn’t know what, if she was going to be coming off or, or still on it, so um, but the fact that the psychiatrist was thinking of taking her off anyway it was no different.
OK, any concerns you have now that the, kind of, trial is going through?
No.
Carer 28, parent (participant in control arm and interview conducted at stage 5)
Even when carers reported that the participant in their care had not responded well to the trial in terms of changes in their behaviour, they still seemed to support the principle of the trial and were positive about the way in which it had been conducted:
Overall I’d say it was really quite positive, you know. Fair enough, [participant 7] didn’t react very well to reducing medication that rapidly but that’s not to say it might not be fine for someone else. I suppose it’s all dependant on how people react to a reduction really. But the support was there, you know, um everyone explained stuff really quite well, um I think yeah, I haven’t got any kind of major issues with it.
Carer 7, staff (participant in intervention arm and was withdrawn at stage 3)
For a complex CTIMP in which many participants lived in residential homes, there were surprisingly no real issues with study procedures, including the delivery of the medication:
Consent forms and the trial sheets and the diaries and things like that, how did you, kind of get on with all of those?
Yeah, they were good.
OK, and what about the assessments, because I know there were quite a few assessments we asked, um you to do, so how did you find those?
Yeah, they were fine as well.
Carer 28, staff (participant in control arm and interview conducted at stage 5)
It was very well organised basically we received delivery the day before the um new meds step box would be completed um so it was delivered in advance of at least 4 days um and yes I was able to contact them if I needed to make rearrangements but again my concerns again at the beginning with the [CLDT] um [psychiatrist] and the GP gave us the constant that they’re there at the end of the phone. So yes I feel that the project was very well managed and very supportive.
Carer 9, staff (participant in intervention arm and interview conducted at stage 4)
There was also a sense from some carers that it had exposed them to research procedures and ideas that had stretched and interested them in a way that was beneficial to themselves and the people they care for:
I’ve really enjoyed it because I think it is opened up to you sort of look at other people, ‘Should they be on this? Should they be on that? Can we reduce that?’ It does, because sometime you can plod along, and yes they have their yearly review but actually do we really look at their medication?
Carer 24, parent (participant in intervention arm and interview conducted at stage 3)
There was one notable exception to the views of carers from the mother of participant 17, who, although she had no complaints of the conduct of the study, felt that the experience had been traumatic for herself and the participant. She was clear that this was a research experience that she did not want to repeat:
So what was your, your general views or your experiences of being involved in the trial, so kind of, how much impact do you think that the study had on, on um you and [participant]?
Well [participant] was, well he was like a zombie, he was no good for anything. He wasn’t eating and like I was saying about his clothes and that, and, and I was so upset.
Yes.
You know, I wouldn’t want him to do any trials like that again.
Carer 17, parent (participant in intervention arm and was withdrawn at stage 3)
When respondents were pressed about how the study could be done differently, the main comment concerned the study medication, which was larger than the normal medication, and, for a minority of participants, was difficult to swallow or possibly caused issues to participants who were wary of obvious (in terms of size, shape and colour) changes to their medication:
Maybe the tablet could be a bit smaller because it is quite a bit of a horse pill to take.
Carer 57, staff (participant in intervention arm and interview conducted at stage 4)
Mindful of the problems ANDREA-LD had experienced in participant recruitment, respondents were also asked about how the study might be promoted more among professionals and the public. Suggestions were made about promoting it in professional journals, making GPs aware, including it in newsletters or making charities such as the National Autistic Society aware of the study.
Reasons for withdrawal from the ANDREA-LD study
Ten participants were withdrawn from the ANDREA-LD study before the 9-month study completion period and unblinding of the trial arm. During interviews, carers reported that reasons for withdrawal were a result of changes in behaviour and changes in health and well-being, including increases in the number of seizures, more violent behaviour, hallucinations, low mood or depression, distress, tetchy/irritable/grumpy behaviours, restlessness, self-harming behaviours, obsessive behaviours, such as repeatedly dressing and undressing, an intensification of autistic behaviours, memory lapses, a lack of interest in personal hygiene and appearance, developing a ‘zombie-like’ personality and a loss of appetite.
For some carers, the decision to withdraw the participant from the study was not an easy one and could sometimes cause friction or disappointment among team members as they debated the pros and cons of study withdrawal:
So what were the reasons for like, the withdrawal of medication?
I think it was because, I don’t know whether he’d had an increase in seizures or, because he’s had these episodes that were so um, like I say, with downstairs staff, you know, that they felt he, that he was becoming more violent or whatever, that maybe that was why, it was like ‘oh no, we’re not happy, let’s stick him back on the old . . .’
Ok, um, and so when he withdraw from the study, did he just go back to normal?
Yeah, just went back straight on. It was perhaps a couple of weeks and you know ‘oh this isn’t working as it’s supposed to’. Well, well we knew there was going to be, you know, pros and cons or whatever, you know to do with his behaviour. But I did think perhaps sort of try and stick at it for a little bit longer [sighs] I don’t know.
Carer 36, staff (participant in intervention arm and was withdrawn at stage 0)
In some cases, it was the relatives who had pushed for a study withdrawal even though the residential carers were more willing to continue the participant on the study medication:
Well we have been having our concerns we weren’t ready to jump in straight away ourselves but um [participant 58’s] mum she was concerned because she had noticed quite a deterioration when she was with him because when she visited she didn’t like any staff around and [participant 58] didn’t either. He tended to play up a bit more. And yeah she was saying he had been pushing things a lot more um he had actually attacked the stepdad.
Carer 58, staff (participant in intervention arm and was withdrawn at stage 2)
There was also a feeling that for some participants the withdrawal of medication had been too rapid, particularly if they had recently been experiencing other medication changes:
I think for [participant 7] it was too quick, I think you know, for some people its fine to go up and down a milligram or two or three but she dropped two-thirds of her medication overnight and that had a pretty profound effect, I mean the reduction in her citalopram was painfully slow, um but it’s what [participant 7] needed because it was a, a very slow reduction in her security blanket, while she was figuring out what else she could rely on.
Carer 7, staff (participant in intervention arm and was withdrawn stage 3)
One carer reported that her daughter had been withdrawn from the study by her doctor as she had been experiencing hallucinations (although there may also have been concerns about allergic reactions to medications); however, withdrawing from the study did not stop her experiencing hallucinations:
So what were the reasons for [participant 6’s] withdrawal from the study?
Well as far as I know it was these hallucinations he thought that it that was what the cause of it was she’s still having them you see. [Doctor’s name] he took her off.
So he thought the trial medication was causing the hallucinations?
As I said she has a history of being allergic to medications.
OK, so when he took them off did the hallucinations stop?
No, no.
OK um, so when they stopped the trial medication do you know what happened then with her medication? Did she just go back to . . . ?
Yeah, what she had before.
Carer 6, parent (participant in control arm and was withdrawn at stage 0)
Positive changes in behaviour
Although some carers noted a deterioration in behaviour, it is worth noting that a few carers during interviews did report positive changes in behaviour during the trial study period. These included increased energy levels/increased motivation, less anxiety and less self-harming behaviour. Other carers reported no notable changes in behaviour. Interestingly, if this was the case, they tended to assume that the participant had been on the placebo (maintaining normal dose) arm of the study:
Any thoughts about um the behaviour changes for [participant 22]? Were they better or worse while they been on the trial?
We haven’t seen any. That’s what makes me think he’s still on the same dose.
Carer 22, parent (participant in control arm and interview conducted at stage 7)
Changes in carers’ behaviour as a consequence of the trial
The interviewer also raised this issue of whether or not the carer thought that they had changed their own attitudes and behaviour towards the participant during the trial period. This was particularly considered in relation to whether or not they might be more observant of the participants’ behavioural changes. Generally, the carers understood that this higher level of observation was an important part of the trial, and some appeared to be aware that this was controlled for by the randomisation process and double blinding of the study medication. There was some acknowledgement that, because of the need to complete the study forms, they were bound to be observant of behaviours:
Yes I am more aware, I was more aware, of [participant 8’s] behaviour. Just extra vigilant.
Carer 8, parent (participant in control arm and interview conducted at stage 6)
So everyone was more aware so it could be awareness of you know if we see any anxiety building up then this is what we do to help him to calm. Possibly a different approach to managing the behaviour.
Carer 9, staff (participant in intervention arm and interview conducted at stage 4)
Speculation about the arm of the trial, the unblinding phase and future management of the participant
Because of the need to close the study early, we had to interview participants before they had been told which arm of the trial they had been randomised to. However, during interviews many carers speculated about the arm of the trial the participant had been in. For some carers who had observed no significant worsening of behaviours, there was the hope (usually tinged with doubt) that the participant had been in the reduction arm of the trial:
It will be fantastic if he’s off them but if he’s not . . .
Carer 22, parent (participant in control arm and interview conducted at stage 7)
For many carers there was also a sense of impending excitement about the unblinding stage:
So we’re putting bets on whether he’s taking it or not.
Yes you’ve not far to go, I think you’re nearly at the end.
It will be so interesting.
Carer 8, staff (participant in control arm and interview conducted at stage 6)
In relation to what happens after the trial, many carers seemed to think that this was primarily a decision for the leading clinician to make:
. . . what happens in the future with [participant] in regards to her medication?
Well um I don’t um I mean it’s up to [clinician].
Carer 6, parent (participant in control arm and interview conducted at stage 0)
There was hope among the carers that the trial might result in some of the participants being withdrawn from the study medication after the trial. However, this seemed to be an option only for participants who had been in the medication withdrawal (intervention) arm of the trial:
Do you have any kind of inkling as to which arm he might have been in or, have you thought about what, what’s going to happen at the end?
Because of his behaviour change we are thinking of all scenarios, so um, I mean I, I hope that it has worked and he’s no longer on it, because I think, you know, sometimes to have unnecessary medication, I think you know come off it, but I hope that, that his behaviour change hasn’t been because of this and I hope he has been on the one that’s been reduced off a bit and he doesn’t no longer take it. Because I think, you know, yeah I do so, because I think the less tablets you take in life, the better really.
Carer 24, parent (participant in intervention arm and interview conducted at stage 3)
Views about antipsychotic medication for the participant and the learning disability population in general
There was a feeling among most carers that antipsychotic medication is not ideal for anyone with a LD and that non-medical therapies and that interventions are generally preferable. Carers were aware of the controversy that surrounds this issue. At times words such as ‘chemical restraint’ and ‘relics of the past’ were used to describe antipsychotic medication:
We’ve a psychologist that works here and we try to be on the, kind of, forefront of kind of um, new ideas and kind of seeing if, you know, and obviously we were quite aware there’s quite a lot of controversy between, kind of like what drugs are um, um are used traditionally with LD and personality disorder that don’t necessarily need to, in, in that modern age don’t need to be happening so obviously we are quite forthcoming with, like, new ideas like that.
Carer 47, staff (participant in control arm and interview conducted at stage 4)
There was also acceptance that, in some cases, medication was necessary to address imbalances in the brain that could not be repaired by other therapeutic interventions:
I think obviously antipsychotic medication is to deal with chemical imbalances in the brain, whereas kind of learnt behaviours is not necessarily going to be corrected by that, so I’m, like what we was talking earlier, I’m aware that the kind of like, there’s a bit of controversy on, its, you know people like you guys are trying to challenge that and try and move it forward so it’s more centred around kind of, um support and talking therapies and stuff like that rather than um, medication which is not really justified.
Carer 52, staff (participant in intervention arm and interview conducted at stage 7)
Other members of staff were more accepting of the need for medication and compared it to more benign personal needs, such as coffee or company:
I think everybody’s got a threshold, as long as they stay on that plateau that if it takes antipsychotic medication to keep them on that level I don’t have any issues with that whatsoever. There are certain things that I need throughout the day to keep me on an even keel, as we all do some people it’s coke some people it’s coffee, and other people it’s just company, everybody has got their own needs when it comes to keeping a level head.
Carer 58, staff (participant in intervention arm and interview conducted at stage 2)
Parent carers were fairly pragmatic about the need for antipsychotic medication within this population, and no very strong views were expressed. Most felt that if antipsychotic drugs were needed then that was an acceptable intervention:
Well, I, I can’t speak about other people, but if I’ve got to give medication, as the last resort, I will. But um, if there’s any other form, you know, of anything, you know like taking him out of the situation, distracting him, I would prefer that to just pop a pill down him like.
Carer 8, parent (participant in control arm and interview conducted at stage 6)
One parent had a fairly positive view about the need for antipsychotic medication to the point that she thought it was worth giving just to benefit from the reassurance of having a medication to rely on:
What are your views more generally on um, using antipsychotic medication for the treatment of challenging behaviour?
I think it’s, I think it’s good.
You think it’s good?
Yeah because I do think it’s almost like it’s that satisfaction, she thinks it will help her calm down, yeah so, I think in some instances I think it’s that placebo kind of effect, it’s quite good and I think it works out whether you actually need that medication or not.
Carer 24, parent (participant in intervention arm and interview conducted at stage 3)
Interviews with patient participants
Four patient participants consented to take part in an interview, all of whom had participated in the ANDREA-LD study, although two had withdrawn at some point during the study. Interviews lasted between 6 minutes and 18 minutes with an average of 11 minutes, although the study team had expected that they might be longer than this.
Reasons for participating in the study
Of the four participants interviewed, one (participant 6) could not remember the study or why she had been involved. One (participant 33) appeared happy to be on the trial but could not express why, instead reiterating that he liked to be on the new red tablets and did not want to come off them. Participant 52 said she wanted to take part because she wanted to see if she could ‘change her life’ by controlling her mood swings. One participant (participant 7) was very motivated to participate in the trial, thinking that she wanted to see how would she would do without the medication and later described in the interview that she had been ‘brave’ to do it:
OK, can you tell me then, so thinking right back, what were your reasons for taking part in the study?
I think one of the reasons why I decided to take part in the study was because I wanted to see, like, how, I would do, with, with um the medication and that sort of thing, um, but then it went terribly wrong and I was very ill.
Participant 7 (participant in intervention arm and was withdrawn stage 3)
At least two of these participants (7 and 52) said that they had been actively involved in the decision-making process of taking part and that their views had been sought:
So was it [clinician] who mentioned it to you, like just at an appointment?
Yeah, yeah, yeah, yeah.
And then um, so did you decide yes or no, did you speak to other people about it?
I decided it, yeah.
OK, did [clinician] give you like information about the project? Like some sheets and . . . ?
Yeah, she did, yeah.
Yeah? OK, did you have to talk to anybody about taking part or did you decide yourself?
I have to talk to some people about it.
Participant 52 (participant in intervention arm and interview conducted at stage 7)
Experience of the trial
All four participants interviewed felt that the study processes had been fine. They seemed fairly unphased by the regularity and nature of the assessments:
I think there was a lady, was it [name] I think who was coming to do the assessments.
I think she did yeah.
How did they go?
They were alright.
They were alright? No problems?
No, no problems.
Participant 52 (participant in intervention arm and interview conducted at stage 7)
The participant interviewed who had been most motivated to take part in the ANDREA-LD study was upset about how she felt during the trial period. This was expressed not as anger towards the study but rather as disappointment that the outcome was not what she had hoped for and that the study had taken a large emotional toll on her:
So positive or negative, please say exactly what you think, overall what do you think about taking part in the study?
I don’t think I want to do it again.
No? I don’t blame you.
I really don’t want to do it again.
OK, so the study had quite a big effect on you.
On me mentally.
Yeah.
And emotionally.
Participant 7 (participant in intervention arm and was withdrawn stage 3)
The other three participants were more positive about their experiences on the trial:
So do you think that if there was another study, which was looking at changing medication again, do you think you would take part again?
I’d take part again.
Is there anything you think we would, you would like us to change?
Stay on the red tablets for good.
Participant 33 (participant in control arm and interview conducted at stage 4)
Changes in behaviour and reasons for withdrawal from the trial
Participant 6 reported that she had been experiencing sleep disturbances, possibly related to paranoia or hearing unusual talking at night. Participant 7 reported that she had been experiencing stress and had been very upset and losing her temper with staff a lot. This had resulted in her ending her participation in the trial early. Participant 33 was not able to acknowledge any changes in behaviour but appeared to be happy continuing on his medication. Participant 52 reported that she had felt a positive change in her behaviour since being on the study:
I feel quite, um, I’ve had a bit of changes, yeah.
OK. In what way?
Um . . . I don’t necessarily want to hurt myself so much.
OK, so that’s a good change?
Yeah.
Participant 52 (participant in intervention arm and interview conducted at stage 7)
Views of antipsychotic medication
Despite the willingness to be involved in the ANDREA-LD trial, all of the four participants felt that antipsychotic medication was helpful to them. None of the participants was able to reflect on whether or not medication was a suitable treatment more generally for the population of people with LDs.
Interviews with clinicians
We approached 11 clinicians, but one had moved jobs leaving no forwarding details, one was on maternity leave and one was unavailable for interview because of other commitments. In total, eight secondary care clinicians took part in a telephone interview, although in one interview the recording device did not record well enough to enable transcription; therefore, we are including seven interviews in our analysis. Interviews lasted between 9 and 27 minutes with an average interview length of 16 minutes.
Reasons for participating in the trial
Clinicians discussed both personal and professional reasons for their interest in the study. Some clinicians talked about an ongoing interest in the study question of whether or not antipsychotics could be safely withdrawn for some patients. Most talked about their involvement in the study with colleagues and the wider team. Some clinicians had not participated in research studies or CTIMPs before and said that experience was part of the attraction of involvement. There were thought to be benefits to the wider team in terms of learning about research procedures and awareness raising about prescribing of antipsychotics:
Research is something that has always been part of my career and I think really interesting, in fact I found that in the last maybe, probably, 8–10 years I’ve been a bit out of touch with proper research and this gave me a chance to just be back, even if it was from a, within the background not as a main, not as a main, one of the main researchers, but that gave me a chance to get back in touch with research which I have always been very interested.
Clinician 4
There was also the attraction of being able to contribute to an important clinical question:
Obviously it’s a contentious area and we need a definitive answer one way or the other in terms of whether um, people with challenging behaviour do benefit from longer term use of antipsychotic medication, um I’m very aware of people becoming put on antipsychotic medication because of bad episodes, remain on it for many years later without it being adequately reviewed. I wanted to support high-quality research, firm believer that we need applied research, and jobbing psychiatrists being part of research, not just um, people with, with academic roles.
Clinician 6
Concerns about participating in the trial
Most concerns related to issues of recruitment and consenting arrangements. In relation to recruitment, the clinicians reported that many carers had been reluctant to be involved in the study or that carers had concerns about the rapidity of the withdrawal:
I guess really the uptake from some patients or their carers, yeah uh I think people generally get anxious when they think they’re going to take some medication away or may take some medications away that sort of . . . yeah it’s challenging.
So was there a little bit of figuring out how to manage that?
Yeah how to manage it and reassure people but I think that has affected the uptake.
Clinician 3
There were also concerns about the exclusion and inclusion criteria of the study. Although some clinicians seemed to consider that any patient of theirs on either risperidone or haloperidol was a potential candidate for the study, other clinicians appeared to apply the criteria more selectively and approached only participants and carers who they thought were ready to appropriately withdraw:
I didn’t have any real concerns for the patients we selected either because I think they were quite carefully selected and chosen the people who were on a medication that we thought perhaps they could do without.
Clinician 7
A couple of clinicians (6 and 7) also raised the issue that they thought people with autism were a distinct population who might not be suitable for the trial:
I think the population we looked at was correct, I think the only difficulty that I saw was, we do have patients with autism and sometimes they’re on small doses of antipsychotics and trying to, although it’s not for a psychosis, it’s for other reasons, so I think they may have been included. And when they’re taken off, even a small dose of antipsychotics they had difficulties I think, so that maybe something, that they’re a slightly separate population to people without autism.
Clinician 7
In relation to capacity, there were some concerns about whether or not participants with some capacity really had the opportunity to make an informed choice about their participation and contribute to the study. Furthermore, clinicians reported that obtaining consent from carers (personal legal representatives) had been challenging:
We did spend quite a lot of time trying to find out who was the proper person to discuss informed consent and talk about informed consent, you know in the paperwork it was very clear that the carers could give full consent, but the reality was that the carers were not happy, were quite uneasy with that.
Clinician 3
Experience of the study
Some clinicians who had no prior experience of being involved in CTIMPs reported that the study set-up stage had been time-intensive and, in their opinion, unnecessarily complicated:
I think what initially before actually doing something there was a whole I think there was just so much paper work and so many versions so actually that was a bit confusing um but I think once we’d done the first one and actually walked the process so to speak um it just became clearer. But the paperwork actually didn’t help, there was quite a lot.
Clinician 1
Sometimes problems about documentation seemed to spill into the main study period:
I’ll tell you what was a bit of a faff was having to fax over the sheets to the study centre once they were filled in because the fax never seemed to get there.
Clinician 2
However, generally, once the study was running, study processes had gone relatively smoothly and clinicians were generally very grateful of the support they received from the research team:
Yes once recruited the person I felt really supported and in fact I felt that, you know, in a way it was very straightforward just to do the follow-ups yes that was absolutely fine, yeah there were no problems at all there.
Clinician 4
Only one clinician felt that contact with and support from the study team had been rather lacking:
Maybe looking back, if there had been more frequent, for example telecontact, around the study itself. For other studies we have kind of given um, better kind of um, feedback on the study as well.
Clinician 5
Changes in behaviour and reasons for withdrawal from the study
Clinicians were not as forthcoming with comments about behaviour changes as the carers had been during interviews. One clinician stated that the clinicians relied on carers to report changes in the behaviour of study participants to them. Mainly, they thought that behaviours were stable and, when there had been changes, these were generally explained by other influences within the participant’s life such as an acute infection, admission to hospital or changes with other medication. As with the carer interviews, some positive changes in behaviour were reported as well, including regaining personality.
Some clinicians also expressed frustration that some of their patients had been withdrawn from the ANDREA-LD study because of concerns about behaviour changes from carers only to find that the participant was on the full dose of medication:
I did rely a lot on carers or family members to tell me about the behaviour. I always told the carers and the person ‘please do let me know if you see any significant changes, anything that will concern’. I made a point every single time, um but sometimes with the carers whether they could read more into something or not, because there was one person who unfortunately pulled out, they, they pulled out because they saw a change of behaviour and the person was still on there, I think it was still on their first month, therefore he wasn’t yet on the active medication study, he was still just on the same dose but with the placebo tablet.
Clinician 4
Clinicians also reported instances of patients who had withdrawn from the study, but perhaps for good reason. Even so, there was still some expressed regret that the patient had to do it before they finished the trial:
The second one is the one we had to break the blind, and that’s where um, in retrospect the family and the carers absolutely accurately um, guessed when he came off his final dose of medication. So they knew. They could see, and again that’s a young autistic man, going back to this autism business, they could see when that last dose went, he had that background agitation returned and of course he was aggressive or whatever, um unfortunately it was, the blind was broken.
Clinician 6 (participant in intervention arm and interview conducted at stage 2)
On other occasions clinicians reported that unblinding and withdrawal from the study was completely necessary, and there was no question that they wanted the participant withdrawn from the trial:
[Participant 17] we had to unblind, so there were concerns about significant change in behaviour that seemed to correlate with his involvement in the trial and potential reduction of his medication.
Clinician 7 (participant in intervention arm and was withdrawn at stage 3)
Management of participants after the completion of the study
How participants were managed, or were expected to be managed, after their ANDREA-LD involvement differed depending on the participant’s and carer’s experience of behaviours during the study period. For some, this meant continued and complete withdrawal from the study medication, but for others it meant a return to a small dose of the medication, primarily to abate concerns from family members or carers about observed changes in the participant’s behaviour:
One of your participants has finished in the study, any changes in um, prescribing medication since they finished?
No, I mean if they had been withdrawn, they have withdrawn, we have not put them back, but those who have been on the, who have not been on the placebo arm and they want back on the medication we went back on the medication for a short period to try and start again the process of withdrawing the medication.
Clinician 5
When the blind was broken, um for the one participant, um if you can, reflecting on how care kind of continued from there.
Yeah, he went back on the meds.
Yeah, and that was, that was just . . .
Small dose. That was at the request of his parents, well he’s in supported accommodation, the staff would’ve hung on in there, but the parents were saying, you know, ‘we noticed a difference, he’s less settled, he’s hit us, which he hasn’t done for months, um he’s not how he was’ and the carers said ‘look, you know we’ve noticed it as well’ so it was a bit of a no brainer in that he hadn’t had any side effects from the medication, we just put him on, back on the lowest possible dose and sure enough it took the edge off him again.
Clinician 7 (participant in intervention arm and was withdrawn stage 3)
Views about the use of antipsychotic medication in the wider learning disability population
There was a general view that antipsychotic medication has its uses in managing challenging behaviour within this population but is probably overused. There was also an awareness that some patients were prescribed these medications initially for a short period, or had their dose increased initially for a short period, but a lack of review meant that the patient remained on the medication for unnecessarily long periods or at unnecessary strengths:
I truly believe that when antipsychotics are prescribed for challenging behaviour, if they were prescribed on low dosages, sometime it can make a really, really important difference because that is what you see in real practice. But I think that we need to be able to evidence that really, really well and look at what are the side effects for the person.
Clinician 4
I wouldn’t prescribe medication just purely for behaviour problems, I would always definitely make sure that is part of the behaviour management protocol, so there are steps in place.
Clinician 5
There was also a feeling that the clinicians who were participating in the ANDREA-LD study were perhaps more consistent with nationally agreed standards in relation to antipsychotic prescribing than clinicians working on the boundaries of evidence-based care:
We’ve done audit on it, you know, it is an area that’s been looked at time and time again, which goes back to the fundamental problem with ascertainment, that we’ve primarily gone to the good services who have already been doing this and perhaps weren’t able to get so rapidly that the areas where practice might not have been so um, in keeping with national standards.
Clinician 6
There was also a sense of disappointment that, as a result of recruitment problems, the ANDREA-LD study would not be able to deliver an answer to the question of whether or not these medications can be safely withdrawn in people with LDs:
I think this is a really important subject, I think there has been some research in the past but obviously there has not really been a trial like this where you’re looking at um, a placebo control double-blind trial, because um, prospective, I think it’s a really good, and I think unfortunately we didn’t get that many people engaged in it which has caused a bit of an issue.
Clinician 7
Limitations of the qualitative study
Although the qualitative study reported here incorporates views from a wide range of stakeholders, we acknowledge that our data may be biased. For example, we were able to interview only a very small proportion of participants in the ANDREA-LD trial. We found that even the participants who had been deemed to have capacity to be interviewed about their views still struggled cognitively with some of the interview content. The need for the interviewer to rephrase questions to make it more accessible to the participants may have resulted in more leading questions, which leads to bias, as this is a population who have a tendency to agree with statements. The interview data for the four participants should, therefore, be treated with some caution. For the interviews with carers and clinicians, there may be other methodological problems such as response bias. Carers who felt that the trial had been a particularly bad experience or clinicians who felt that trial procedures had been particularly tricky to handle may have been more motivated to respond to the invitation to an interview and therefore may not be representative of the all carers and clinicians. Owing to the need to complete the interviews in a relatively short time, within the study close-down period, interviews were conducted when participants were between 4 and 6 months post randomisation. Many participants, therefore, were not fully through the trial procedures and, consequently, some of them would not have been fully withdrawn from medication.
Conclusion
This qualitative study about stakeholders’ views of the ANDREA-LD study found that carers, participants and clinicians who took part in the ANDREA-LD study were generally in agreement that it was an important research question, that study procedures, such as delivery of medication and assessments, were acceptable and that study support from the research team was good. Generally, there was a feeling that this study should be supported by the LD community but also an awareness that it was a challenging study. Issues that caused more concern included consenting arrangements (particularly carers’ concerns about acting as a personal legal representative), whether or not the study inclusion and exclusion criteria were appropriate (particularly surrounding issues of whether or not to include participants with autism) and the size of the study medication. In addition, carers in particular reported that participants experienced a number of negative behaviours during the study period, but these were not always attributed to drug reduction and many of the behaviours were not new within the study period. Despite the number of adverse behaviours reported in the qualitative interviews, only one SAE was reported to the trial team, and many behaviours were ongoing regardless of the trial. It is notable, however, that none of the stakeholders felt that such a trial was not possible or that research involving people in a residential home had been a particular barrier to recruitment or retention.
Chapter 6 Discussion
The purpose of this study was to conduct a blinded RCT to investigate whether or not antipsychotic medication prescribed to adults with LDs for the treatment of challenging behaviour could be reduced or withdrawn entirely without adversely affecting their behaviour or mental health or causing a corresponding increase in financial costs. In addition, we hoped to further understand the process of drug reduction from the point of view of people with a LD, carers and professionals and to further our knowledge of the process of running high-quality RCTs in people with a LD.
Unfortunately, recruitment was largely unsuccessful, particularly from primary care, and a revised strategy with a focus on recruiting from community psychiatric services did not demonstrate a significant improvement within the required time frame. The study therefore closed early to recruitment and is reported as a pilot study. We believe that, despite the difficulties in recruitment, the study has provided invaluable information in the following three domains:
-
the impact of antipsychotic drug reduction on patients, carers and services
-
the nature of future studies of antipsychotic drug reduction
-
the nature of future RCTs in people with a LD.
Main findings
Before addressing these areas, we will summarise the main findings from our pilot study.
-
Recruitment from primary care was unsuccessful, with very poor uptake and only rare successful participant completion.
-
Recruitment from community psychiatric services was possible. The numbers were slow and considerable start-up time was needed before recruitment built up.
-
There was considerable attrition from approach to randomisation.
-
Progression through the trial was independent of whether the participant was in the treatment group or the control group.
-
Reasons for participant withdrawal appear independent of trial arm.
-
Participants demonstrated relatively low levels of expressed aggression at baseline.
-
Participants experienced high levels of side effects at baseline.
-
Prediction of successful progression (through drug withdrawal or equivalent control phases) is very tentative but the presence of a diagnosis of autism, the use of PRN medication and individuals with capacity to consent may predict poor progression.
-
Clinically significant changes in aggression, challenging behaviour and mental health did occur for a minority of participants, although it is difficult to assess the significance of these changes given the very low overall sample size.
-
Such symptomatology appears to be managed by the use of PRN medication.
-
A considerable reduction in antipsychotic drug load is achieved through drug reduction.
-
Those undergoing drug reduction have a reduction in side effects but previously masked movement disorder may appear.
-
Participants, carers and clinicians who took part were in agreement over the need for drug reduction and found the trial process acceptable.
-
Difficulties were faced in the consenting process for some people, especially in relation to the role of legal representative.
-
Carers rarely reported significant side effects and behaviours experienced were generally in keeping with the individual’s habitual behaviours.
Study challenges
The initial recruitment through primary care was chosen because the future intervention was deemed most likely to occur at a primary care level. However, this was wholly unsuccessful and the delay incurred, with associated use of resource, led in a large part to the overall failure of recruitment. For our chosen population, numbers per practice were small, invitations to participate were often not responded to and concerns over the need for support from psychiatric services were raised, despite the inclusion of a comprehensive GP support package. Although GPs were not interviewed, there is some anecdotal evidence to suggest that GPs were concerned about safety of participants, their own expertise in this area and the level of workload associated with the study (i.e. number of study-specific consultations required per patient).
Recruitment in community psychiatric services was more successful but it was significantly negatively impacted by a lack of GCP training among professionals, delays in set-up and then difficulties in identifying patients, as many were reported as having previously failed drug reduction and, therefore, they were felt to be ineligible by the clinician. Difficulty in identifying appropriate persons to provide consent was an issue in both general practice and CLDTs. Carer beliefs about the efficacy of these medications to manage behaviour and fears about the consequences of reduction were also reported as reasons for declining participation. Carer and clinician concerns about safety were also likely to have been exacerbated by the limited availability of alternative (behavioural) interventions for challenging behaviour, and in particular the fact that no alternative to drug reduction was offered as part of the current trial. Although the rationale for this was based on previous research that suggested alternative interventions are not likely to be needed for those managed in community settings, provision of an alternative intervention may have improved recruitment. Although negative consequences of withdrawal in the current sample were infrequent and relatively minor, a low-intensity behavioural alternative may offset such changes in a larger sample.
Our findings may relate to a section of the population who already receive relatively low-dose antipsychotic drugs but who are not currently displaying high-level challenging behaviour (on formal scales). Aggression, behaviour and psychological ill health were generally low at baseline. This needs further exploration. One suggestion would be that some people with a LD on antipsychotics are not in fact displaying challenging behaviour. However, PRN usage and use of restraint over time in the control group suggests that behaviour is still perceived as an issue, with concerns relating to behaviour raised by carers. How drug reduction applies to those on high levels of antipsychotic medication with continuing high-level challenging behaviour may need further exploration.
The impact of antipsychotic drug reduction on patients, carers and services
Our study has shown pilot data that we believe identify that drug reduction is possible and safe. However, the findings indicate a signal that reduction is not without change. Changes were observed in behavioural and mental health measures and in the development of movement disorder in some participants. This is important because previous open studies have reported changes although concerns were raised about biased reporting.
These changes offer an opportunity to provide focused support and tailored behavioural interventions to patients undergoing drug reduction in the future. They will aid clinicians in advising individuals and carers on the impact of reduction and predicting areas of individual need.
Data from carer interviews highlighted that concerns about drug side effects and efficacy were key reasons for participation in the study. The responses reflected an ambiguity when considering the effect of antipsychotics and revealed that people were not convinced of their efficacy.
A key finding from the study was the ability of carers to identify perceived symptoms that were both positive and negative. Negative perceived symptoms, such as reports of low mood or depression, distress, restlessness and intensification of autistic behaviours, were reasons for discontinuation throughout the trial. It is worth noting, however, that these were not always attributed to drug reduction, and many of the behaviours did not newly arise during the study period. These were countered by positive perceived symptoms, such as reports of increased energy levels, increased motivation, decreased anxiety and fewer self-harming behaviours. Some carers reported no notable changes in behaviour, which, interestingly, they tended to attribute to the participant being in the ‘maintaining normal dose’ arm of the trial. We believe that this shows the importance of a greater need to support carers in their understanding of the likely symptomatology during drug withdrawal.
National Institute for Health and Care Excellence guidance on the use of antipsychotics54 provides no detail on how to manage drug reduction. Guidance on the use of antipsychotics from the Royal College of Psychiatrists suggests that clinicians should ‘taper off the drug based on its effectiveness’. 55 We recommend that antipsychotic drug withdrawal is seen not as a passive process but as one that has predicted needs, and for this reason guidance is needed for practitioners, carers and patients.
Recommendation
We would like to see the Royal College of Psychiatrists, potentially working with the Royal College of General Practitioners, establish a working group with relevant stakeholders to provide guidance on antipsychotic drug withdrawal. This guidance should provide an auditable standard for drug withdrawal including recommendations for levels of behavioural and other service support and accessible information for people with a LD and their carers. Development of guidance for carers on this issue would also be beneficial. As Sheehan and Hassiotis note in their recent systematic review,40 a systems approach to this complex issue is required.
The nature of future studies of antipsychotic drug reduction
As discussed in this report, the study has provided important insights into the experiences of people taking part in drug reduction studies that should influence future trial development. First, it seems that, despite the many barriers faced in recruitment, these were not reflected in the experience of carers, professionals or people with a LD.
The practical running of the study was well received, and relatively complex issues, such as ethics, blinding and overwrapping of medication, were not particularly problematic.
The problems we faced were largely related, first, to recruitment in primary care and, second, to the preparedness of services dealing with people with LDs and antipsychotics to take part in research. In particular, there is clearly no functioning network of LD specialists who can respond rapidly to research requests. We believe this is an issue with the current trial, which addressed an area of concern for carers and professionals but not necessarily for those receiving the medication.
One of the key challenges in a drug reduction study lies within carer and professional concern that medication is being reduced and that this is causing change. If this is the case, it is something that could be relatively easily reversible. Our study showed that, in fact, incidents for which it was necessary to remove the blind were equal between groups. It would appear that this concern is a powerful drive independent of intervention.
Recommendation
Future studies are needed to explore interventions that could reduce unnecessary antipsychotic drug use in people with a LD. They should involve interventions that, in addition to the process of reduction, address the concerns of professionals and carers by having extra support or alternative (i.e. non-pharmacological) interventions available for perceived or actual deterioration. It may be that reduction should be non-blinded and these alternative support mechanisms should instead be compared with treatment as usual.
The nature of future randomised controlled trials in people with a learning disability
This pilot study has provided information of value to those who want to conduct further high-quality interventional RCTs in people with a LD.
The study has shown that carers and people with a LD were not overly troubled by even quite complex trial processes involving over-encapsulated medication and data collection at multiple time points. Carer and participant concerns can, therefore, be overcome when they are adequately informed of the process.
This study suggests that, although there is a clear need, primary care services are not currently well equipped to deliver this type of intervention. This is important for other studies, which should explore the clinical competencies needed and how these apply to primary care if that is where the target population predominantly receives health care.
In the case of further studies in drug withdrawal, it is clear that services and professionals are not currently well prepared for recruiting into interventional pharmaceutical studies. This has also been noted in other recent studies. A recent pilot study of the use of statins to slow cognitive decline in people with Down syndrome showed similar challenges in recruitment. In that study, 21 out of a target of 181 participants were recruited, 13 of whom completed the study. 56 This is a particular concern in people with a LD, who are often excluded from participation in RCTs and therefore the potential benefits of high-quality interventions.
Recommendation
We would like to see measures put in place to improve recruitment to studies in people with a LD and for consideration to be given to how adults with a LD might also be included in general population trials. These measures may include a commitment from the Royal College of Psychiatrists for GCP training (including covering recruitment with reduced or no capacity) to be mandatory for its members and for Clinical Commissioning Groups and other health groups, and for performance-related measures linked to recruitment to be considered for LD studies.
Conclusion
Despite increasing guidance on the use of antipsychotic medication, no guidance exists for reducing the medication. This pilot study has provided valuable insights into the development of such guidance and beyond this to support improved access to trials for people with a LD.
Acknowledgements
We would like to thank Jackie Swain and Christian Barlow for their help with administering the study and Rhys Williams-Thomas for his artwork and help preparing the final report for submission.
We would like to acknowledge Health and Care Research Wales staff in Wales and network staff in England for their help with recruitment and follow-up. We are also grateful to the general practices and CLDTs that helped us to recruit the participants and deliver the intervention along with the hospital pharmacies. We would also like to thank all those who kindly gave up their time to participate in the trial.
Finally, we would also like to thank the members of our Trial Steering Committee [Sally-Ann Cooper (chairperson), Eric Emerson, Alan Montgomery, Irwin Nazareth, Wayne Crocker, Jonathan Richards, Joe Powell and Pauline Young] and Data Monitoring Ethics Committee [Angela Hassiotis (chairperson), Umesh Chauhan and Alan Watkins] for their help and guidance.
Contributions of authors
Rachel McNamara (Senior Research Fellow, Psychology) contributed to study and intervention design. She also provided oversight to trial management and led the writing of Chapters 1 and 6.
Elizabeth Randell (Research Associate, Psychology) contributed to study and intervention design. She also managed the trial and led the writing of Chapters 2 and 3.
David Gillespie (Research Associate, Statistics) contributed to study and intervention design. He conducted the quantitative analyses and led the writing of Chapter 4, as well as contributing to drafting and revising the report.
Fiona Wood (Senior Lecturer, Qualitative Methods) contributed to study and intervention design. She led the qualitative analyses of the interviews and led the writing of Chapter 5, as well as contributing to drafting and revising the report.
David Felce (Professor, Learning Disabilities) contributed to the design of the trial and the intervention, as well as overall implementation of the study. He contributed to drafting and revising the report.
Renee Romeo (Senior Lecturer, Health Economics) contributed to study and intervention design, led the health economics analyses and contributed to drafting and revising the report.
Lianna Angel (Research Assistant, Data Management) was a Data Manager and contributed to drafting and revising the report.
Aude Espinasse (Research Associate, Trial and Data Management) was a Data Manager and contributed to interpretation of the results. She also contributed to revising the report.
Kerry Hood (Professor, Statistics) oversaw the quantitative analyses, was involved in the design of the trial and contributed to drafting and revising the report.
Amy Davies (Assistant Psychologist, Learning Disabilities) was a Research Assistant and contributed to data collection. She also contributed to revising the report.
Andrea Meek (Research Assistant, Learning Disabilities) was a Research Assistant and contributed to data collection and conducting qualitative interviews. She also contributed to drafting and revising the report.
Katy Addison (Research Assistant, Data Management) was a Data Manager and contributed to revising the report.
Glyn Jones (Consultant Psychiatrist, Learning Disabilities) contributed to study and intervention design. He contributed to revising the report.
Paul Deslandes (Senior Lecturer, Pharmacy) contributed to study and intervention implementation and to revising the report.
David Allen (Professor, Learning Disabilities) contributed to study and intervention design and to revising the report.
Martin Knapp (Professor, Health Economics) contributed to study and intervention design and contributed to the Health Economics.
Ajay Thapar (Research Fellow, General Practice) contributed to study and intervention design and to revising the report.
Michael Kerr (Professor, Learning Disabilities) led the design of the trial and the intervention as well as overall implementation of the study. He contributed to drafting and revising the report.
All authors approved the final version of the report.
Data sharing statement
We shall make data available to the scientific community with as few restrictions as feasible, while retaining exclusive use until the publication of major outputs. Anonymised data can be obtained by contacting the corresponding author.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- InfoBaseCymru . Local Government Data Unit 2011 n.d. www.infobasecymru.net/IAS/themes/2011census (accessed July 2016).
- Emerson E, Hatton C. CEDR Research Report 2008 (1): People with Learning Disabilities in England. Lancaster: Lancaster University; 2008.
- Perry JL, Kerr M, Felce D, Bartley S, Tomlinson J. Monitoring the Public Health Impact of Health Checks for Adults with a Learning Disability in Wales. Cardiff: Public Health Wales; 2010.
- Molyneux P, Emerson E, Caine A. Prescription of psychotropic medication to people with intellectual disabilities in primary health-care settings. J Appl Res Intellect Disabil 1999;12:46-57. https://doi.org/10.1111/j.1468-3148.1999.tb00049.x.
- Tsiouris JA, Kim SY, Brown WT, Pettinger J, Cohen IL. Prevalence of psychotropic drug use in adults with intellectual disability: positive and negative findings from a large scale study. J Autism Dev Disord 2013;43:719-31. http://dx.doi.org/10.1007/s10803-012-1617-6.
- Cooper SA, Smiley E, Morrison J, Allan L, Williamson A, Finlayson J, et al. Psychosis and adults with intellectual disabilities. Prevalence, incidence, and related factors. Soc Psychiatry Psychiatr Epidemiol 2007;42:530-6. http://dx.doi.org/10.1007/s00127-007-0197-9.
- Sheehan R, Hassiotis A, Walters K, Osborn D, Strydom A, Horsfall L. Mental illness, challenging behaviour, and psychotropic drug prescribing in people with intellectual disability: UK population based cohort study. BMJ 2015;351. https://doi.org/10.1136/bmj.h4326.
- Clarke DJ, Kelley S, Thinn K, Corbett J. Psychotropic drugs and mental retardation: 1. Disabilities and the prescription of drugs for behaviour and for epilepsy in three residential settings. J Intellect Disabil Res 1990;34:385-95. https://doi.org/10.1111/j.1365-2788.1990.tb01549.x.
- Robertson J, Emerson E, Gregory N, Hatton C, Kessissoglou S, Hallam A. Receipt of psychotropic medication by people with intellectual disability in residential settings. J Intellect Disabil Res 2000;44:666-76. https://doi.org/10.1046/j.1365-2788.2000.00307.x.
- Tsiouris JA. Pharmacotherapy for aggressive behaviours in persons with intellectual disabilities: treatment or mistreatment?. J Intellect Disabil Res 2010;54:1-16. http://dx.doi.org/10.1111/j.1365-2788.2009.01232.x.
- Emerson E, Alborz A, Kiernan C, Mason H, Reeves D, Swarbrick R, et al. The HARC Challenging Behaviour Project. Report 5: Treatment, Management and Service Utilisation. Manchester: Hester Adrian Research Centre, University of Manchester; 1997.
- Kiernan C, Reeves D, Alborz A. The use of anti-psychotic drugs with adults with learning disabilities and challenging behaviour. J Intellect Disabil Res 1995;39:263-74. https://doi.org/10.1111/j.1365-2788.1995.tb00518.x.
- Perry J, Felce D, Allen D, Meek A. Resettlement outcomes for people with severe challenging behaviour moving from institutional to community living. J Appl Res Intellect Disabil 2011;24:1-17. https://doi.org/10.1111/j.1468-3148.2010.00567.x.
- Robertson J, Emerson E, Pinkney L, Caesar E, Felce D, Meek A, et al. Treatment and management of challenging behaviours in congregate and noncongregate community-based supported accommodation. J Intellect Disabil Res 2005;49:63-72. https://doi.org/10.1111/j.1365-2788.2005.00663.x.
- Glover G, Williams A, Branford D, Avery R, Chauhan U, Hoghton M, et al. Prescribing of Psychotropic Drugs to People with Learning Disabilities andor Autism by General Practitioners in England. London: Public Health England; 2015.
- Scheifes A, Stolker JJ, Egberts ACG, Nijman HLI, Heerdink ER. Representation of people with intellectual disabilities in randomised controlled trials on antipsychotic treatment for behavioural problems. J Intellect Disabil Res 2011;55:650-64. https://doi.org/10.1111/j.1365-2788.2010.01353.x.
- Brylewski J, Duggan L. Antipsychotic medication for challenging behaviour in people with intellectual disability: a systematic review of randomized controlled trials. J Intellect Disabil Res 1999;43:360-71. http://dx.doi.org/10.1046/j.1365-2788.1999.043005360.x.
- Matson JL, Neal D. Psychotropic medication use for challenging behaviors in persons with intellectual disabilities: an overview. Res Dev Disabil 2009;30:572-86. http://dx.doi.org/10.1016/j.ridd.2008.08.007.
- Sawyer A, Lake JK, Lunsky Y, Liu SK, Desarkar P. Psychopharmacological treatment of challenging behaviours in adults with autism and intellectual disabilities: a systematic review. Res Autism Spectr Disord 2014;8:803-13. https://doi.org/10.1016/j.rasd.2014.03.021.
- Tyrer P, Oliver-Africano PC, Ahmed Z, Bouras N, Cooray S, Deb S, et al. Risperidone, haloperidol, and placebo in the treatment of aggressive challenging behaviour in patients with intellectual disability: a randomised controlled trial. Lancet 2008;371:57-63. https://doi.org/10.1016/S0140-6736(08)60072-0.
- Romeo R, Knapp M, Tyrer P, Crawford M, Oliver-Africano P. The treatment of challenging behaviour in intellectual disabilities: cost-effectiveness analysis. J Intellect Disabil Res 2009;53:633-43. https://doi.org/10.1111/j.1365-2788.2009.01180.x.
- de Leon J, Greenlee B, Barber J, Sabaawi M, Singh NN. Practical guidelines for the use of new generation antipsychotic drugs (except clozapine) in adult individuals with intellectual disabilities. Res Dev Disabil 2009;30:613-69. https://doi.org/10.1016/j.ridd.2008.10.010.
- Baumeister AA, Sevin JA, King BH, Reiss S, Aman MG. Psychotropic Medication and Developmental Disabilities: The Internatinal Consensus Handbook. Columbus, OH: Ohio State University; 1998.
- Frighi V, Stephenson MT, Morovat A, Jolley IE, Trivella M, Dudley CA, et al. Safety of antipsychotics in people with intellectual disability. Br J Psychiatry 2011;199:289-95. http://dx.doi.org/10.1192/bjp.bp.110.085670.
- Newcomer JW. Second-generation (atypical) antipsychotics and metabolic effects: a comprehensive literature review. CNS Drugs 2005;19:1-93. https://doi.org/10.2165/00023210-200519001-00001.
- Mahan S, Holloway J, Bamburg JW, Hess JA, Fodstad JC, Matson JL. An examination of psychotropic medication side effects: does taking a greater number of psychotropic medications from different classes affect presentation of side effects in adults with ID?. Res Dev Disabil 2010;31:1561-9. https://doi.org/10.1016/j.ridd.2010.05.006.
- Bagnall AM, Jones L, Ginnelly L, Lewis R, Glanville J, Gilbody S, et al. A systematic review of atypical antipsychotic drugs in schizophrenia. Health Technol Assess 2003;7. https://doi.org/10.3310/hta7130.
- Banerjee S. The Use of Antipsychotic Medication for People with Dementia: Time for Action. London: Department of Health; 2009.
- Douglas IJ, Smeeth L. Exposure to antipsychotics and risk of stroke: self controlled case series study. BMJ 2008;337. http://dx.doi.org/10.1136/bmj.a1227.
- Ballard C, Hanney ML, Theodoulou M, Douglas S, McShane R, Kossakowski K, et al. The dementia antipsychotic withdrawal trial (DART-AD): long-term follow-up of a randomised placebo-controlled trial. Lancet 2009;8:151-7. https://doi.org/10.1016/S1474-4422(08)70295-3.
- National Institute for Health and Care Excellence . Challenging Behaviour and Learning Disabilities: Prevention and Interventions for People With Learning Disabilities Whose Behaviour Challenges 2015.
- NHS England . Winterbourne Medicines Programme: Improving the Use of Medicines in People With Learning Disabilities – NHS Improving Quality Report Apr2014–Apr2015 2015. www.nhsiq.nhs.uk/media/2671659/nhsiq_winterbourne_medicines.pdf (accessed July 2016).
- Mental Health Act 2007. London: The Stationery Office; 2005.
- NHS England . Urgent Action Pledged on Over-Medication of People With Learning Disabilities n.d. www.england.nhs.uk/2015/07/urgent-pledge (accessed July 2016).
- Faculty of Psychiatry of Intellectual Disability . Psychotropic Drug Prescribing for People With Intellectual Disability, Mental Health Problems and Or Behaviours That Challenge: Practice Guidelines. Faculty Report FR ID 09 2016. www.rcpsych.ac.uk/pdf/FR_ID_09_for_website.pdf (accessed July 2016).
- Branford D. Factors associated with the successful or unsuccessful withdrawal of antipsychotic drug therapy prescribed for people with learning disabilities. J Intellect Disabil Res 1996;40:322-9. https://doi.org/10.1111/j.1365-2788.1996.tb00637.x.
- Luchins DJ, Dojka DM, Hanrahan P. Factors associated with reduction in antipsychotic medication dosage in adults with mental retardation. Am J Ment Retard 1993;98:165-72.
- Stevenson C, Rajan L, Reid G, Melville C, McGilp R, Cooper S-A. Withdrawal of antipsychotic drugs from adults with intellectual disabilities. Ir J Psychol Med 2004;21:85-90. https://doi.org/10.1017/S0790966700008417.
- Ahmed Z, Fraser W, Kerr MP, Kiernan C, Emerson E, Robertson J, et al. Reducing antipsychotic medication in people with a learning disability. Br J Psychiatry 2000;176:42-6. https://doi.org/10.1192/bjp.176.1.42.
- Sheehan R, Hassiotis A. Reduction or discontinuation of antipsychotics for challenging behaviour in adults with intellectual disability: a systematic review. Lancet Psychiatry 2017;4:238-56.
- Medicines for Human Use (Clinical Trials) Regulations. London: The Stationery Office; 2004.
- Nihira K, Lambert NM, Leland H. Adaptive Behavior Scale: Residential and Community. Examiner’s Manual. Austin, TX: PRO-ED, Inc.; 1993.
- Moss S, Hogg J. Estimating IQ from adaptive behaviour information in people with moderate or severe intellectual disability. J Appl Res Intellect Disabil 1997;10:61-6. https://doi.org/10.1111/j.1468-3148.1997.tb00007.x.
- Prosser H, Moss S, Costello H, Simpson N, Patel P, Rowe S. Reliability and validity of the Mini PAS-ADD for assessing psychiatric disorders in adults with intellectual disability. J Intellect Disabil Res 1998;42:264-72. https://doi.org/10.1046/j.1365-2788.1998.00146.x.
- Oliver PC, Crawford MJ, Rao B, Reece B, Tyrer P. Modified Overt Aggression Scale (MOAS) for people with intellectual disability and aggressive challenging behaviour: a reliability study. J Appl Res Intellect Disabil 2007;20:368-72. https://doi.org/10.1111/j.1468-3148.2006.00346.x.
- Aman MG, Singh NN. The Aberrant Behaviour Checklist. New York, NY: Slosson Educational Publications; 1986.
- Moss S, Prosser H, Costello H, Simpson N, Patel P, Rowe S, et al. Reliability and validity of the PAS-ADD checklist for detecting psychiatric disorders in adults with intellectual disability. J Intellect Disabil Res 1998;42:173-83. https://doi.org/10.1046/j.1365-2788.1998.00116.x.
- Dott SG, Weiden P, Hopwood P, Awad AG, Hellewell JS, Knesevich J, et al. An innovative approach to clinical communication in schizophrenia: the approaches to schizophrenia communication checklists. CNS Spectr 2001;6:333-8. https://doi.org/10.1017/S1092852900022045.
- Kalachnik JE, Sprague RL. The Dyskinesia Identification System Condensed User Scale (DISCUS): reliability, validity, and a total score cutoff for mentally-ill and mentally-retarded populations. J Clin Psychol 1993;49:177-89.
- Beecham J, Knapp MRJ, Thornicroft GJ, Brewin CR, Wing JK. Measuring Mential Health Needs. London: Gaskell; 1992.
- Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol 2006;3:77-101. https://doi.org/10.1191/1478088706qp063oa.
- Mental Capacity Act 2005. London: The Stationery Office; 2005.
- Green J, Thorogood N. Qualitative Methods for Health Research. London: Sage; 2004.
- Challenging Behaviour and Learning Disabilities: Prevention and Interventions for People with Learning Disabilities Whose Behaviour Challenges. London: NICE; 2015.
- Psychotropic Drug Prescribing for People with Intellectual Disability, Mental Health Problems andor Behaviours that Challenge: Practice Guidelines. Cardiff: Royal College of Psychiatrists; 2016.
- Cooper SA, Ademola T, Caslake M, Douglas E, Evans J, Greenlaw N, et al. Towards onset prevention of cognition decline in adults with Down syndrome (The TOP-COG study): a pilot randomised controlled trial. Trials 2016;17. http://dx.doi.org/10.1186/s13063-016-1370-9.
Appendix 1 ANDREA-LD principal investigator support package
Part 1
FIGURE 7.
ANDREA-LD PI management flow chart. Note that the cross-references within the figure refer to those in Part 2 of the support package.
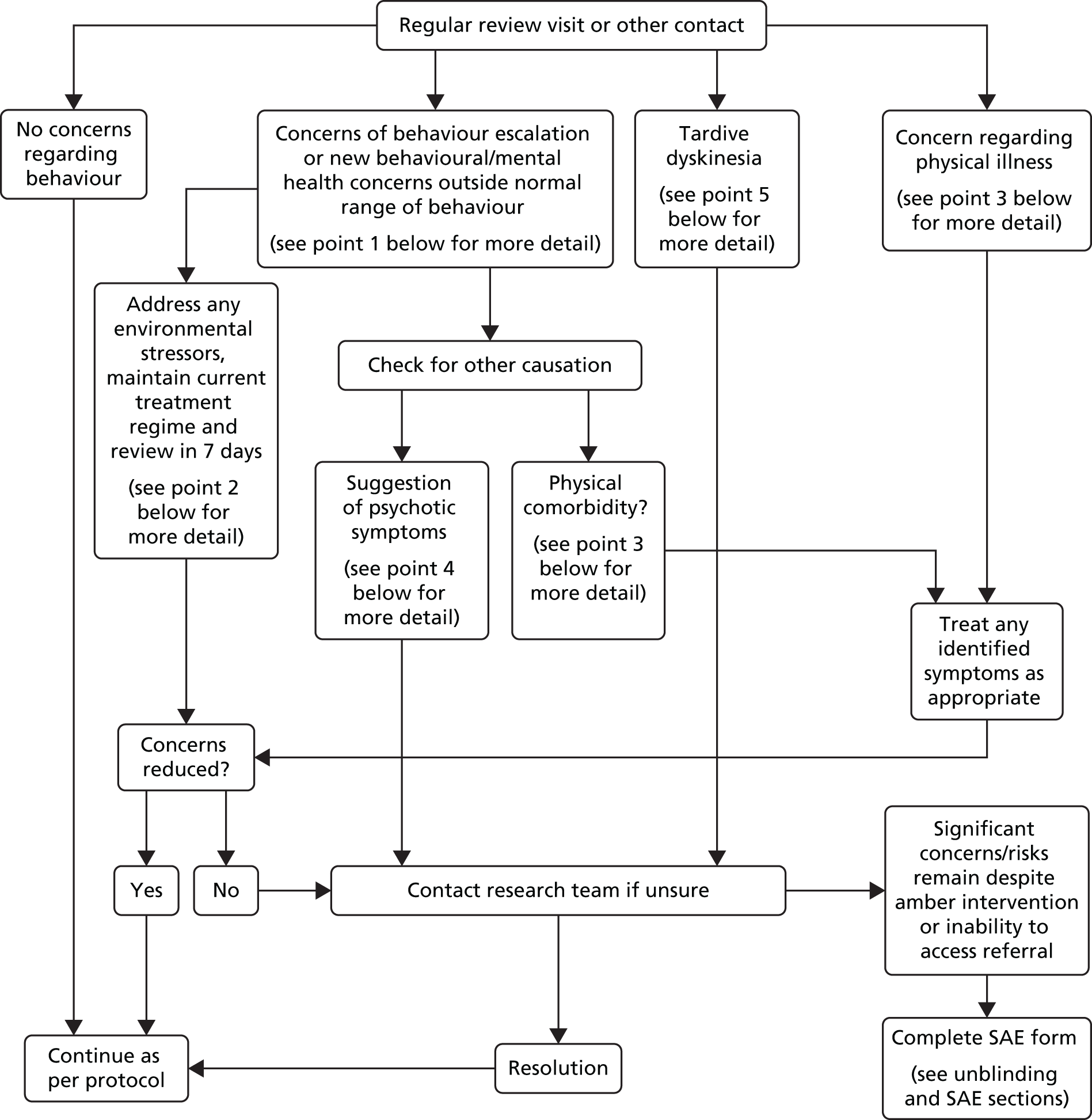
Part 2
1. Concerns of behaviour escalation or new behavioural/mental health concerns outside normal range of behaviour
It is quite likely that carers or families will raise concerns relating to the individual for whom they care in relation to alteration of behaviour. In this first stage of assessment you can ask a few simple questions to signpost the next step in the flow chart:
Does this seem to be a new condition rather than worsening of usual behaviour?
If it is not worsening of usual behaviour, it is worth deciding if there is an underlying physical or psychiatric condition. Completely new patterns of behaviour are unusual so detailed questioning will ensure that it is definitely a new behaviour. If it does seem a new behaviour, then it is best to follow the advice on physical illness (likely to be common) or psychotic symptoms (likely to be rarer) in the flow chart. You may be directed to which step by the nature of the behaviour and other symptomatology.
If it is the usual pattern of behaviour then the next step is to assess its severity.
If this is worsening of usual behaviour – has the individual displayed behaviour as concerning as this before starting in the study?
If the answer is ‘yes’, then it is likely that this is simply a fluctuation in normal patterns as people with challenging behaviour tend to have ups and downs in the normal course of their condition.
In such situations it is unlikely to be a direct result of the study. Such fluctuations are best approached by watchful waiting.
If the behaviour is of a greater degree or the current degree causes great concern, then the most likely short-term reason for deterioration is environmental stressors-so please follow the advice in the environmental stressor box. If there is no such stressor then checking for a physical condition or new psychiatric symptoms would be worthwhile.
2. Environmental stressors
If carers do report a change in the level of a person’s challenging behaviour, it’s important to try and identify what the reasons for this might be. More often than not, these will be linked with some kind of change in the person’s environment rather than any change in the person themselves. Some good questions to put to carers include:
-
Has there been any change in the person’s routine (e.g. have they been provided with their normal pattern of day activity, been able to go out as often as they normally do, been able to see people that they normally see)?
-
Has there been any change in the people working with the person? (For example have there been a lot of relief staff on duty who don’t know the person well and not fully briefed regarding key aspects of their care?; has a particular carer of whom the person is particularly found been away from for some reason?; have the person’s parents been away on holiday and unable to visit?; has the service manager been off on leave?)
-
Are all carers currently implementing agreed support plans (particularly behavioural support plans)? How do carers check that this is the case?
-
Have there been any reductions in levels of support to the person that impact on the ability to implement these plans?
-
Are the reported increases well-known ones that happen at a particular time of year? (Christmas and birthdays are often difficult times for people.)
-
Has the person been exposed to any recent trigger (e.g. having to go to particular places that they don’t like or asked to do things that they’re not keen on; have they been left alone for lengthy periods)?
This is not an exhaustive list – but it gives you an idea of things to probe for. Carers will often not be aware that these sorts of issues may in themselves explain why the person may have been a little more difficult lately. The solution here is clearly to prompt the carers to address the environmental issues identified rather than to treat the person. Allowing some time for this to occur and the behaviour to settle is the best strategy here therefore.
Escalation of challenging behaviour and non-psychotic psychiatric symptoms
Escalation of challenging behaviour is especially linked to anxiety symptoms and may particularly be associated with environmental stressors. Addressing these stressors, as outlined above, may resolve the problem. If this does not resolve the issue please refer to the What to do if Challenging Behaviour Escalates section below.
3. Physical illnesses
Individuals with a LD are especially prone to developing physical illness but, given communication difficulties, behavioural difficulties may be the presenting feature. Sources of pain [such as ear infections, headaches, abdominal pain (because of urinary tract infection, constipation)] may be especially difficult to pick up and should be actively sought out.
4. Suggestion of psychotic symptoms
The participants in this study will have been screened for the presence of serious mental illness so, in theory, few if any should experience a relapse of psychosis or evolution of a new illness. However, the presence of mental illness in people with LD can be masked by a variety of factors including intellectual function and communication barriers. Therefore, some people whose medication was thought to have been originally prescribed for ‘challenging behaviours’ may have in fact had an undiagnosed psychotic illness, such as schizophrenia or a bipolar disorder.
If a patient in the study starts to develop an unusual pattern of speech or behaviour, it is important to remain vigilant for potential psychosis. In people with mild LD, the presentation would usually be similar to that seen in the general population, that is, abnormal beliefs out of keeping with their social situation which are not amenable to reason or abnormal perceptions which are not explained by obvious environmental stimuli. However, when the person lacks a level of intellectual function or emotional development commensurate with a chronological age, their ability to express their symptoms coherently may be severely diminished. Here, a carer’s account of unusual behaviours or communication which is out of keeping with their pre-morbid presentation may be critical in highlighting the evolution of serious mental illness.
In these circumstances it is appropriate to contact your local CLDT to ask for an urgent psychiatric assessment. They may be able to reassure you, or could, if necessary, ask for the medication code to be broken to identify the current prescription and withdrawal of the patient from the study.
5. Tardive dyskinesia
Tardive dyskinesia is characterised by chewing or sucking movements, grimacing and choreoathetoid movements particularly affecting the face, but also potentially the limbs and, most importantly, the muscles responsible for swallowing and respiration. While this syndrome can occasionally be seen in drug-naïve patients, it is more common in those who have taken antipsychotic medication for many years. However, neither dose nor duration of treatment is the sole determinant, and it is also more common in women, the elderly and patients with diffuse brain pathology. Almost half of the cases arise when drugs are being reduced or discontinued.
The cause is uncertain but could be super sensitivity to dopamine, resulting from prolonged dopaminergic blockade, and arises with many antipsychotic agents. Many treatments have been tried, but none is universally effective, hence the need to avoid prolonged unnecessary prescription of antipsychotic medication, especially at higher doses.
Should any patient develop symptoms suggestive of tardive dyskinesia during the study, it is recommended that you contact your local learning disability team for further psychiatric evaluation, including, if necessary a breaking of the code to identify the current prescription and withdrawal of the patient from the study. This advice does not preclude you from seeking other expert opinion, such as a consultant neurologist if you feel this to be a more appropriate course of action.
What to do if challenging behaviour escalates
If a patient’s challenging behaviour escalates during the study there are a number of options. Firstly, it is important to rule out any treatable physical or remediable environmental causes. It is unlikely, but not impossible, that an escalation in challenging behaviour may be related to some previously unrecognised psychosis in which case referral to your local Psychiatric or Learning Disability Service is warranted.
By far the most likely scenario is that this is a simple fluctuation in the patient’s normal pattern of challenging behaviour which should be addressed through the usual range of support strategies. However, if you feel that adjustment in medication is necessary you may wish to prescribe a short course of Benzodiazepine (agent and dose will be dependent upon individual patient characteristics) or, if appropriate, an ‘as required’ option to be administered as necessary when alternative calming strategies have proved unsuccessful.
You may, if simple interventions prove unsuccessful, contact the study team with a view to delaying the next incremental reduction in medication or in an extreme situation withdraw from the study.
Adverse events
Definitions and reporting procedures
It is the responsibility of the Investigator to report all adverse events to South East Wales Trials Unit within 24 hours of becoming aware of it. Any queries concerning adverse event reporting should be directed to the Trial Manager in the first instance.
How to report
Complete an ANDREA-LD SAE form as follows and fax to South East Wales Trials Unit on XXXX.
-
The first report should be marked as ‘Initial’. Any other types of report (‘follow-up’ or ‘final’ will be prompted by the study team.
-
Complete the ‘Report Date’ and ‘Details of Subject affected by Event’.
-
Complete ‘Details of Event’. Note, an adverse event is considered serious if it:
-
results in death
-
is life-threatening (Note: the term ‘life-threatening’ in the definition of serious refers to an event in which the participant was at risk of death at the time of the event; it does not refer to an event which hypothetically might have caused death if it were more severe.)
-
required hospitalisation or prolongation of existing hospitalisation (Note: Hospitalisation is defined as an inpatient admission, regardless of the length of stay, even if the hospitalisation is a precautionary measure, for continued observation. Pre-planned hospitalisation, e.g. for pre-existing conditions which have not worsened or elective procedures does not constitute an adverse event.)
-
results in persistent or significant disability or incapacity
-
consists of a congenital anomaly or birth defect
-
any other medically important condition (Note: other events that may not result in death are not life-threatening, or do not require hospitalisation may be considered as a SAE when, based upon appropriate medical judgement, the event may jeopardise the participant and may require medical or surgical intervention to prevent one of the outcomes listed above).
-
-
Complete ‘Details of Investigational Medicinal Product(s)’ section with as much detail as possible. Note, for the question ‘Is the SAE related to the IMP?’, consider the following:
-
Most adverse events and drug reactions that occur in this trial, whether they are serious or not, may be due to drug reduction. They will not be toxicity related effects. The assignment of the causality should be made using the definitions in the table below.
-
| Relationship | Description |
|---|---|
| Unrelated | No evidence of any causal relationship with the trial/intervention |
| Unlikely | Little evidence to suggest a casual relationship with the trial/intervention (e.g. the event did not occur within a reasonable time after administration of the trial medication). There is another reasonable explanation for the event (e.g. the participant’s clinical condition, other concomitant treatment) |
| Possible some evidence | Possible some evidence to suggest a causal relationship with the trial/intervention (e.g. because the event occurs within a reasonable time after administration of the trial medication). However, the influence of other factors may have contributed to the event(e.g. the participant’s clinical condition, other concomitant treatments) |
| Probable evidence | Probable evidence to suggest a causal relationship. The influence of other factors is unlikely |
| Definite clear evidence | Definite clear evidence to suggest a causal relationship and other possible contributing factors can be ruled out |
| Not assessable | Insufficient or incomplete evidence to make a clinical judgement of the causal relationship |
-
Complete ‘Details of other treatment’ and ‘Further information relevant to assessment’ sections with as much detail as possible.
-
Sign the form and send a copy as described at the end of the form. Place the original in the Trial Site File and add a copy to the patient’s medical notes.
Reporting summary
FIGURE 8.
Reporting summary. CRF, case report form; MHRA, Medicines and Healthcare products Regulatory Agency; REC, Research Ethics Committee; SAR, serious adverse reaction; SUSAR, suspected unexpected serious adverse reaction.
Unblinding
Emergency
If, having worked through the management flow chart above, you still have significant concerns or feel that risks remain despite amber intervention or there was an inability to access referral, you may feel it appropriate to request emergency unblinding of treatment allocation.
In such a situation, you must report an adverse event as described above by completing a Serious Adverse Event (SAE) Form (this can be found in your Site File) and submitting it to the Trial Manager. The Chief Investigator or Clinical Reviewer will then contact you to discuss the issues surrounding the situation and, if necessary, confirm that the participants treatment allocation needs to be unblinded.
Routine
Breaking the code (blind) at the 9-month visit.
When each patient reaches their 9-month visit, it will be time for the blind to be broken, thus revealing whether they were in the reduction arm or were receiving treatment as normal.
The ANDREA-LD team will reveal the allocation to you along with details of the dose of medication the patient is currently taking. It will be your responsibility to have a discussion with the patient and their carer (or representative) in order to relay this information.
For those still receiving medication, any ongoing treatment and dosage could be based on the dose the patient finished on depending on the clinical judgement of their clinician and the patient’s needs at the time.
Once the blind has been broken and the patient’s treatment allocation and drug dosage is known, we would like you to share this information with any secondary services involved in their care. Knowledge of how the patient progressed in the trial could be of importance in future decision-making regarding treatment.
Contact details
ADDRESS
South East Wales Trial Unit
7th Floor Neuadd Meirionnydd
Cardiff University
Heath Park
Cardiff
CF14 4YS
Fax
FAO ANDREA-LD Trial
XXXX
Telephone
Trial Manager – XXXX
Data Manager – XXXX
Trial Administrator – XXXX
E-mail – XXXX
Clinical support
Professor Michael Kerr – XXXX
E-mail – XXXX
Dr Glyn Jones – XXXX
E-mail – XXXX
Secretary – Sandra Jones, XXXX
Appendix 2 ANDREA-LD interview schedules
Carer interviews
Question 1. Reasons for participating in the trial
Can you tell me about why you were interested in [the person you care for] participating in the study? (Prompts: Where did they hear about it?, Who did they discuss it with? Have they been involved in other research projects? Who was involved in the decision?)
Question 2. Concerns about participating in the trial
Did you have any concerns about participating in the trial? (Prompts: Did you have any best interest meetings? Who did you discuss it with? What were your concerns? Are you still concerned?)
Question 3. Views about why others might or might not want to participate in the trial
Do you think that other participants or other carers might have concerns about participating in the trial? (Prompts: What might those concerns be?)
Question 4. Thoughts about any behaviour changes of person with LD (for better, for worse, or just different) while they have been on the trial and their ideas for why behaviour might have changed
How do you think you (the person you care for) has been generally while they have been in the trial? (Prompts: Has their physical or mental health improved or declined? Behaviour changes?)
Question 5. Thoughts about carers own behaviour during the trial
Do you think you respond or behave differently to [the person in the study]?
Question 6. Views on the study medication
Do you think the medication was difficult for [the participant] to take? (Prompts: size, colour, texture, etc.)
Question 7. General experiences of being involved in the trial
Overall what was your experience of the trial? How much impact did the study have on you? (Prompts: trial procedures e.g. consent forms, info sheets, trial questionnaires and measures, being present at the assessments, appointment times, arrangements for collecting the medication, difficulties taking the medication, possible side effects.)
Question 8. Views about the use of antipsychotic medication to treat or control challenging behaviour for the participant and for people with LD in general
Have you had any thoughts about how antipsychotic medication is used to treat or control challenging behaviour for people with LD? (Prompts: read anything in newspapers or magazines or websites, talked to people about this?)
Question 9. Reasons for any partial or full reinstatement of medication after unblinding or reasons for withdrawal from the study
(If the participant withdrew) what were your reasons for coming off the study and do you know what medication [the participant] went back to after you withdrew?
Question 10. Thoughts about how the trial could be promoted should it run again as a full RCT
Can you think how we might raise awareness of the study should it be repeated?
Question 11. Thoughts about what might happen after the trial
The ANDREA-LD study was about seeing if we can safely reduce antipsychotic medication for people with LDs. When the trial has finished, what do you think should happen to [the participant’s] medication? (Prompts: do you have concerns about this?)
Secondary care clinician interviews
Question 1. Can you tell me about why you were interested in participating in the stud? (Prompts: Who did they hear about it from? Who did they discuss it with? Have they been involved in other research projects? Who was involved in the decision? Are there any benefits?)
Question 2. Do you or your team have any concerns about participating in the trial (for themselves and for the patient population)? (Prompts: time involved, risk to patients and their carers.)
Question 3. Do you think the trial is suitable for certain types of patients? (Prompts: those with milder disabilities, those who live at home or those who live in a home.)
How did you decide which participants to approach for the study?
Question 4. Can you tell me what you thought about some of the study processes (screening, support package, consent processes, patient follow-up)?
Question 5. Do you have any views about how [the participant’s] behaviour may have changed (for better, for worse, or just different) while they have been on the trial and their ideas for why behaviour might have changed?
Question 6. How do you think the patient and their carer managed while during the trial period? (Prompts: Were they more anxious? Were you more anxious about them?)
Question 7. You may be aware of the debate about the value of prescribing antipsychotic medication to people with LDs to treat or control challenging behaviour. Do you have any views about this? What kind of evidence would you like to see that you think would be useful to help psychiatrists make good prescribing decisions?
Question 8. What would be your solution to the problem of overprescribing antipsychotic medication to this patient population? [Prompts: substitute other drugs (benzodiazepenes), substitute other (non-pharmacological) intervention.]
Question 9. What were your reasons for any partial or full reinstatement of medication after unblinding or reasons for withdrawal from the study? (Prompts: concerns about the patients behaviour, carers or patients concerns about their behaviour, other general health problems or life event.)
Participant interviews
The schedule will loosely follow the carer schedule, but with changes between interviews depending on what the participants could cope with, their level of language, capacity, etc.
Appendix 3 ANDREA-LD patient and public involvement
The coapplicants for this trial have experience of involving people with LDs in the inception, conduct and dissemination of research. An experienced member of All Wales People First (Jonathan Richards) and Pauline Young, a parent whose child has a LD, joined the Trial Steering Committee. They were able to provide feedback on the trial protocol and study information as well as advice on the best ways available to introduce the research to potential participants.
Appendix 4 Description of the scales used during the ANDREA-LD study
| Scale | Scale range | Interpretation |
|---|---|---|
| MOAS | 0–120 | Higher scores imply higher levels of aggression |
| ABC | ||
| Irritability subscale | 0–45 | Higher scores imply higher levels of irritability |
| Lethargy subscale | 0–48 | Higher scores imply higher levels of lethargy |
| Stereotypy subscale | 0–21 | Higher scores imply higher levels of stereotypy |
| Hyperactivity/non-compliance subscale | 0–48 | Higher scores imply higher levels of hyperactivity/non-compliance |
| Inappropriate speech subscale | 0–12 | Higher scores imply higher levels of inappropriate speech |
| PAS-ADD checklist | ||
| Possible organic disorder subscale | 0–8 | Higher scores imply higher severity of a possible organic disorder |
| Affective or neurotic disorder subscale | 0–25 | Higher scores imply higher severity of an affective or neurotic disorder |
| Psychotic disorder subscale | 0–4 | Higher scores imply higher severity of a psychotic disorder |
| Mini PAS-ADD interview | ||
| Depressive disorder subscale | 0–30 | Higher scores imply higher severity of a depressive disorder |
| Anxiety disorder subscale | 0–22 | Higher scores imply higher severity of an anxiety disorder |
| Hypomania/mania (expansive mood) subscale | 0–19 | Higher scores imply higher severity of hypomania/mania |
| Obsessive–compulsive subscale | 0–10 | Higher scores imply higher severity of an obsessive compulsive disorder |
| Psychosis subscale | 0–13 | Higher scores imply higher severity of psychosis |
| Unspecified disorder subscale | 0–11 | Higher scores imply higher severity of an unspecified disorder |
| DISCUS | ||
| Total score | 0–60 | Higher scores imply more severe movement disorders |
| Confidence handling challenging behaviour | 0–10 | Higher scores imply more confidence handling challenging behaviour |
List of abbreviations
- ABC
- Aberrant Behaviour Checklist
- ABS
- Adaptive Behaviour Scale
- ADHD
- attention deficit hyperactivity disorder
- ANDREA-LD
- Community-led ANtipsychotic Drug REduction for Adults with Learning Disabilities
- ASC
- Antipsychotic Side-effect Checklist
- CC
- complete case
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CLDT
- community learning disability team
- CSRI
- Client Service Receipt Inventory
- CTIMP
- Clinical Trial of an Investigational Medicinal Product
- DISCUS
- Dyskinesia Identification System Condensed User Scale
- GCP
- Good Clinical Practice
- GP
- general practitioner
- ID
- identification
- IMP
- investigational medicinal product
- IQ
- intelligence quotient
- IQR
- interquartile range
- ITT
- intention to treat
- LD
- learning disability
- MITT
- modified intention to treat
- MOAS
- Modified Overt Aggression Scale
- NICE
- National Institute for Health and Care Excellence
- PAS-ADD
- Psychiatric Assessment Schedule for Adults with Developmental Disability
- PI
- principal investigator
- PP
- per protocol
- PRN
- pro re nata
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SD
- standard deviation
- SE
- standard error
- SMPU
- St Mary’s Pharmaceutical Unit