

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 11/107/01. The contractual start date was in April 2014. The draft report began editorial review in September 2016 and was accepted for publication in September 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Soomro et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2017 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Cancer of the kidney accounts for 3% of new cancers and 2% of cancer-related deaths, making it the eighth most common cancer in the UK. In the UK, in 2008, 8757 new cases of kidney cancers were diagnosed (approximately two-thirds of these were < 4 cm in size) and 3848 patients died of kidney cancer. 1–4 Surveillance, Epidemiology, and End Results data5 show that 65,150 patients were diagnosed with, and 13,680 patients died of, renal cancer in the USA in 2013.
The treatment of small renal masses (SRMs) of < 4 cm in size (80% of these are malignant) is evolving. The standard treatment in the past was radical nephrectomy. It is now accepted that the nephron-sparing techniques have similar oncological outcomes but have an additional benefit of preserving kidney function. 6 Among these techniques, partial nephrectomy has emerged as the preferred treatment of SRMs, as it effectively treats the cancer while broadly preserving the kidney function (dependent on ischaemic time) and has good long-term oncological safety. 7 However, it is associated with postoperative morbidity and long hospital stay and recovery. For these and other reasons, such as lack of surgical skills and patient comorbidities, it remains underutilised. Ablative techniques such as radiofrequency ablation (RFA) and cryoablation (CRYO) are also being increasingly used in patients with SRMs. These are particularly attractive as the mean age at diagnosis of renal cancer is 64 years and the procedure incurs significantly fewer complications and inpatient bed stay, and is associated with early recovery. 8
However, ablation still represents an invasive procedure with consumables costs, particularly in the case of CRYO. There are also concerns with ablative techniques regarding the possible persistence of microscopic cancer and a slightly higher chance of persistence of tumour, perhaps necessitating secondary treatment. This leads to increased patient anxiety and additional cost for the providers. In view of these factors, and because current evidence is based mainly on single-centre series and a few meta-analyses, there is uncertainty about the best treatment of SRMs. 9–12 Minimally invasive ablation SRMs (of < 4 cm in size) clearly make available a new treatment option, but robust data comparing relative clinical effectiveness and cost-effectiveness of active surveillance with ablative techniques (RFA/CRYO) are currently not available. A randomised controlled trial (RCT) to answer this question has been identified as a priority by the renal cancer subgroup of the National Cancer Research Institute (NCRI) and has the support of the British Association of Urological Surgeons (BAUS) section of oncology and local National Institute for Health Research (NIHR) clinical research networks. As there remains uncertainty as to the willingness of patients and surgeons to agree to randomisation to this trial and whether or not recruitment and retention would be adequate, a rehearsal pilot addressing the feasibility of a definitive RCT is required.
The natural history of SRMs remains unclear. Almost 66% of newly diagnosed renal cancers are < 4 cm in size. 13 A meta-analysis14 has shown that the majority of small lesions have a slow growth rate (mean rate of 0.28 cm per year) and they rarely metastasise while under active surveillance. Partial nephrectomy became accepted as a standard of care for SRMs when, in a series of 485 patients with renal tumours < 4 cm, followed for over 10 years, cancer-free survival rates at 5 years and 10 years were found to be 96% and 90%, respectively. 15 The local tumour recurrence was 3.5%. Similar results were reported in a meta-analysis16 looking at a series of patients undergoing open partial nephrectomy (OPN) from 1980 to 2000. A study that compared 100 cases of laparoscopic partial nephrectomy (LPN) with OPN concluded that OPN remains the standard of care for SRM. 17 LPN was associated with longer ischaemic time, major intraoperative complications and increased postoperative urological complications. 17 However, in experienced hands, LPN has a comparable oncological efficacy and complication profile. 18 Despite this clear evidence favouring partial nephrectomy, BAUS cancer registry data19 showed that only 721 partial nephrectomies were performed in England and Wales in 2007/8, whereas approximately two-thirds of patients with renal cancer masses of < 4 cm in size underwent radical nephrectomy. This practice alone is contributing to the net population burden of significantly impaired renal function in a population that may already have other comorbidities such as obesity, hypertension and diabetes mellitus.
In RFA, radiofrequency probes are applied into the renal tissue percutaneously using ultrasonography (US), computerised tomography (CT) or magnetic resonance imaging (MRI). There has been some concern that the thermal RFA zone might not be homogeneous and there may be persistence of viable tumour that is not evident on routine radiological surveillance. 20,21 This may be due to the method of tissue heating in RFA, which is considerably reliant on conductive heating. A multi-institutional meta-analysis22 of 1375 renal lesions treated by RFA and CRYO, applied both percutaneously and laparoscopicaly, detailed 600 RFA outcomes at a mean follow-up duration of 15.8 months. Mean patient age was 67 years and mean tumour size was 2.69 cm. This analysis22 yielded a combined subtotal treatment rate and (unexpected) local tumour progression rate of 12.9%, with 8.5% undergoing repeat ablation for treatment completion. It was suggested that true disease persistence might be determined only by delayed post-ablation biopsy. The National Institute for Health and Care Excellence (NICE) has accepted the broad efficacy of RFA but centres are still advised to audit results and outcomes carefully. 23
Cryoablation: CRYO can be performed percutaneously under image guidance or laparoscopicaly by direct visualisation. Again, the meta-analysis by Kunkle and Uzzo22 reviewed multi-institutional outcomes from CRYO, with the majority performed in North American practice under laparoscopy. 24,25 A total of 775 renal lesions were treated; mean tumour size was 2.58 mm and mean patient age was 66 years. This yielded a combined subtotal and (unexpected) local tumour recurrence rate of 5.2%, with only 1.3% undergoing repeat ablation, largely because of the difficulties of a repeat LPN procedure. In this meta-analysis,22 the rate of progression to metastatic disease was similar to that of nephron-sparing surgery, CRYO and RFA. However, in these series, ablative techniques were selected in older patients with small tumours, whereas partial nephrectomy was undertaken in younger patients with larger tumours and had longer post-treatment surveillance. NICE26,27 has accepted the broad efficacy of CRYO but centres are still advised to audit results and outcomes carefully.
Active surveillance studies13,14,28–33 have reported on a small series of patients, showing varying growth rates ranging from 0.09 cm per year to 0.86 cm per year, with most concluding that SRMs grow slowly with a low rate of progression. The rate of metastatic disease is low, between 1% and 7%, with varying lengths of follow-up. 14,34,35 In most cases with metastatic disease, the primary tumour had grown to > 4 cm in diameter. 34 It has also been demonstrated that larger renal cell cancers (RCCs) are significantly associated with higher histological grade, advanced stage and distant metastases, with the significant size cut-off point between 3 and 5 cm. 36,37 This has resulted in the current opinion that small RCCs may grow slowly but then become more aggressive at a size threshold of approximately 4 cm.
However, some small RCCs metastasise when they are < 4 cm in size, and this has led some authors35,38 to question the safety of an active surveillance approach. At present, it is not possible to identify these aggressive tumours on standard radiological characteristics alone.
Currently, a strategy of active surveillance, or watchful waiting, is adopted in cases where the perioperative risks are deemed too high, or when an informed choice is made after balancing the potential risks and benefits of surgery.
Many small RCCs have a slow or immeasurable growth rate; as such, these cancers may not lead to symptoms or metastatic disease within the lifetime of the patient.
Although many small RCCs are indolent, there is significant uncertainty as to which small tumours will behave in a benign fashion and which are more likely to progress and metastasise. A reliable means to predict the behaviour of these small RCCs might enable early definitive treatment for those that are likely to progress or metastasise early and avoid unnecessary procedures, along with the associated morbidity and costs, for those patients with slow-growing or non-growing RCCs that are unlikely to progress within the lifetime of the patient.
At present, the main prognostic factor available is tumour size. This is most commonly measured on a CT scan, with follow-up CT performed to identify an increase in tumour size. A systematic schedule of serial CT scans allows growth, and any acceleration in growth, to be identified, which might suggest tumour progression and likely metastasis.
Most of the masses will be discovered incidentally on CT and US scans. Critically, the technique for follow-up must be able to detect significant increases in renal mass size and provide minimal inter-observer and intraobserver variability. US cannot provide reliable measurements sequentially, so either CT or MRI is ideally required.
The literature to date from one randomised controlled study13 and several retrospective studies suggests that active surveillance may be an initial option for the management of SRMs in healthy individuals with careful follow-up. 13,14,28–34 Size progression of the tumour (> 0.5 cm per year or a maximum diameter of > 3.5 cm; approximately 25% of patients) while on active surveillance may identify a more precise cohort who will actually require intervention.
This approach may produce substantial benefits in terms of reduced morbidity, reduced overall mortality and long-term quality of life (QoL) and these may outweigh the small risk of metastatic disease in patients aged ≥ 70 years. However, this is predicated on the relative morbidity of ablation and surgery.
Chapter 2 Pre-feasibility study phase
Objectives
The objectives of the pre-feasibility study were to determine clinician (radiologists and urologists) equipoise around a trial comparing ablation with active surveillance and to assess whether or not they would participate and recruit patients to such a trial. This was because the threshold for advising patients to consider active surveillance was dependent on the size of the tumour weighed against age and comorbidities of the patient. The second objective was to develop patient information for the feasibility study with the input of patients to ensure that it is clear and comprehensive. In addition, we undertook a survey of the potential participating centres and selected 8 out of the 15 centres approached. This was based on their research capabilities, expertise in offering these interventions and previous experience of undertaking clinical trials.
Survey: clinician equipoise in relation to a trial of ablation or active surveillance in patients with small cell renal tumour
Introduction
Clinician equipoise is assumed to have a negative impact on recruitment to clinical trials. 39 Others40 have demonstrated that equipoise and opinions on a particular trial or design can be explored using a questionnaire survey. A survey of the relevant clinical community was included in the initial funding application, to be conducted in the stage 1 pre-feasibility phase, to determine equipoise and views on a trial comparing ablation with active surveillance in the management of patients with small cell kidney cancer. Ethics approval was sought from the Newcastle University Faculty of Medical Sciences Ethics Committee (00774/2014). This section reports the response to the survey and the survey findings.
Objectives
To determine the point of equipoise for clinicians (in the use of ablation or active surveillance in the management of small cell kidney tumours) to inform the pilot feasibility study, we conducted a survey of radiologists and urologists across the UK. Willingness to recruit patients to a trial comparing these two management options was also explored.
Study subjects
The target sample of clinicians for the survey was interventional radiologists and urologists based in the UK. The British Society of Interventional Radiology (BSIR) kindly circulated a covering e-mail and a link to the questionnaire on our behalf. The BAUS supplied the SURAB (a two-stage randomised feasibility study of SURveillance versus ABlation in the management of incidentally diagnosed SRMs) team with a list of urologists.
Methods
This was a questionnaire survey. The questionnaire was developed within the team, pilot tested with a small number of interventional radiologists and urologists and amended accordingly. The questionnaire included a mix of open and closed questions, multiple-choice questions and vignettes. The content of the questionnaire covered available options for the management of small cell kidney tumours and questions exploring the willingness to randomise to a trial of ablation versus active surveillance in this patient group. A series of vignettes were used to explore which patient clinicians would be happy to recruit to a trial (Table 1). The questionnaire was available online, with the option of a paper version if requested. Completion of the questionnaire was anonymous.
| Would you approach to participate in a trial of active surveillance versus ablation? | |||
|---|---|---|---|
| Aged 50 years | Tumour size (cm) | Charlson score of comorbidity | |
| Patient A | 2.0 | 0 |
Charlson score (points) 0 = no comorbidity |
| Patient B | 2.5 | 0 | |
| Patient C | 3.0 | 0 | |
| Patient D | 3.5 | 0 | |
| Patient E | 2.0 | 2 |
Charlson score (points) 2 = two comorbid conditions (e.g. myocardial infarction and dementia) or a single, more severe condition (e.g. hemiplegia, moderate/severe renal disease) |
| Patient F | 2.5 | 2 | |
| Patient G | 3.0 | 2 | |
| Patient H | 3.5 | 2 | |
A covering e-mail explained the purpose of the survey and a link to the survey was circulated by the BSIR on 4 August 2014. The study team circulated the e-mail and a link to a list of members eligible for the survey provided by the BAUS on 21/22 August 2014, with a second request (owing to a low initial response) 2 weeks later. Members of the study team also circulated both the e-mail and link to their network of clinical colleagues.
Results
A total of 29 out of 666 radiologists and 22 out of 50 urologists responded to the survey. The number of interventional radiologists for whom this survey was relevant is small and the view within the study team was that 29 respondents was an excellent response. In the following sections, we will report the responses to the survey but, because of the small sample size, statistical analysis is not valid and the results are presented descriptively.
To provide a picture of the management options for small cell kidney tumour, respondents were asked about what was available for this patient group in their centre from a choice of active surveillance, RFA and CRYO. Active surveillance was offered by the majority (95%) of centres, with 58% offering RFA and 51% offering CRYO to patients with small cell kidney tumour. Eight respondents skipped this question, but it is not clear whether or not this is because they did not offer any of the options listed.
Attitudes to a trial of active surveillance versus ablation
A total of 79% of the respondents said they would take part in a trial of ablation versus active surveillance in this patient group. Some of those who would not, or were unsure, gave their reasons in the open questions (Box 1).
I’m not sure if it’s reasonable to randomise ‘fit’ patients unless there’s an age cut-off.
I would be unsure due to the ethics of randomising a group we have been treating for many years, admittedly without any level of evidence.
I think there would be problems getting patients to agree to participate. There may be a reluctance to have no treatment. The trial may involve more frequent imaging and that could be an inconvenience.
Almost half (47.5%) of respondents anticipated problems in patients agreeing to a trial comparing ablation with active surveillance, 27.5% did not anticipate problems and 25% were unsure. The reasons given by those who believed it would be problematic to recruit were centred around patient preference for one particular option and patients not being at ease with active surveillance: ‘Some patients are uncomfortable with knowing they have a “tumour” and not having it “dealt with” ’. More than half (55%) of respondents did not envisage any other problems with a trial of this kind. Of the remaining 45%, the other problems given were related to the patients’ perspective, covered in the previous question, but the next most commonly mentioned problem was the lack of a surgical arm and what this would mean for patients who were eligible for surgery or ablation. There were comments that this is a much needed trial but consideration should be given to the potential problems within the clinical community.
Factors that influence which patients are approached
Age was explored as a factor that may have an impact on which patients are approached for the trial. Respondents were asked what age of patients they would feel comfortable recruiting to such a trial from an age range of 45 years to ≥ 80 years. The majority (80%) would be happy to recruit patients aged 75 years, but this dropped to 65% for patients aged 70–89 years and to 25% for patients aged ≥ 80 years; 47% would recruit patients aged 65 years and 15% would recruit patients aged 45–55 years.
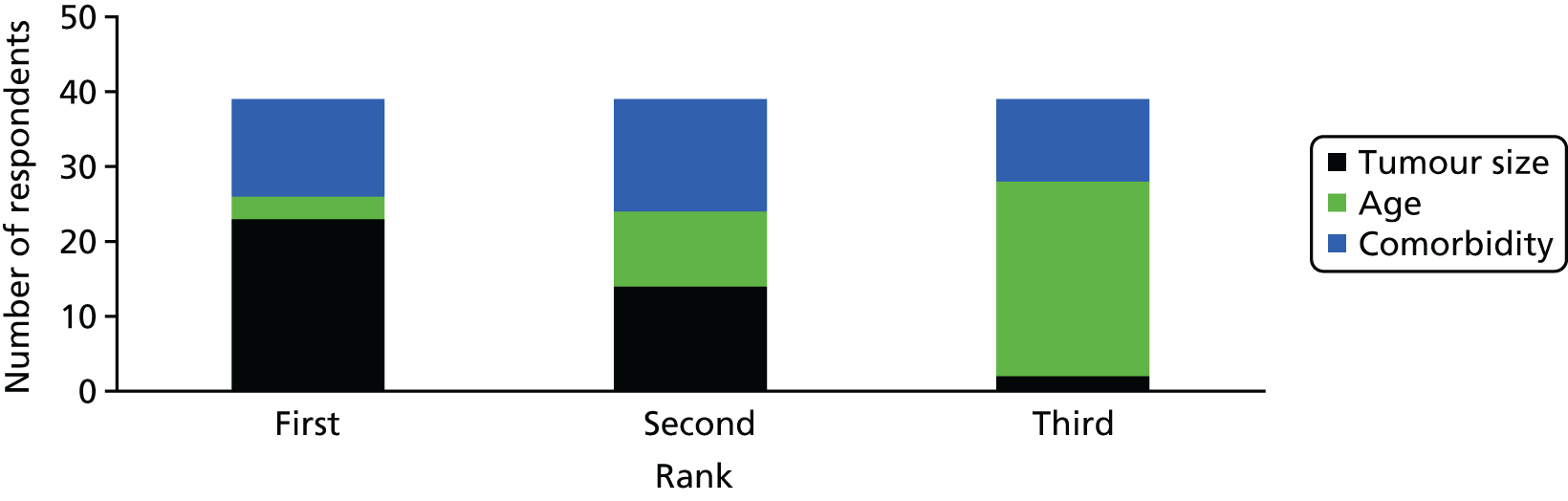
When asked to rank the potential influential factors in the identification of patients for the trial of tumour size, age and presence of comorbidities, tumour size was ranked as the most important for 59% of respondents (Figure 1). The ranking of comorbidity was interesting, with a similar number of respondents ranking it first (n = 13), second (n = 15) or third (n = 12), indicating, perhaps, that opinions on the importance of comorbidities were divided.
FIGURE 1.
Factors ranked as most influential in identifying patients for the trial.
Vignettes
Respondents were asked to consider a series of vignettes (see Table 1, for example) with a range of ages (50–80 years), tumour sizes (2–3.5 cm) and level of comorbidity (Charlson score of 0 or 2 points) in relation to willingness to recruit to the trial.
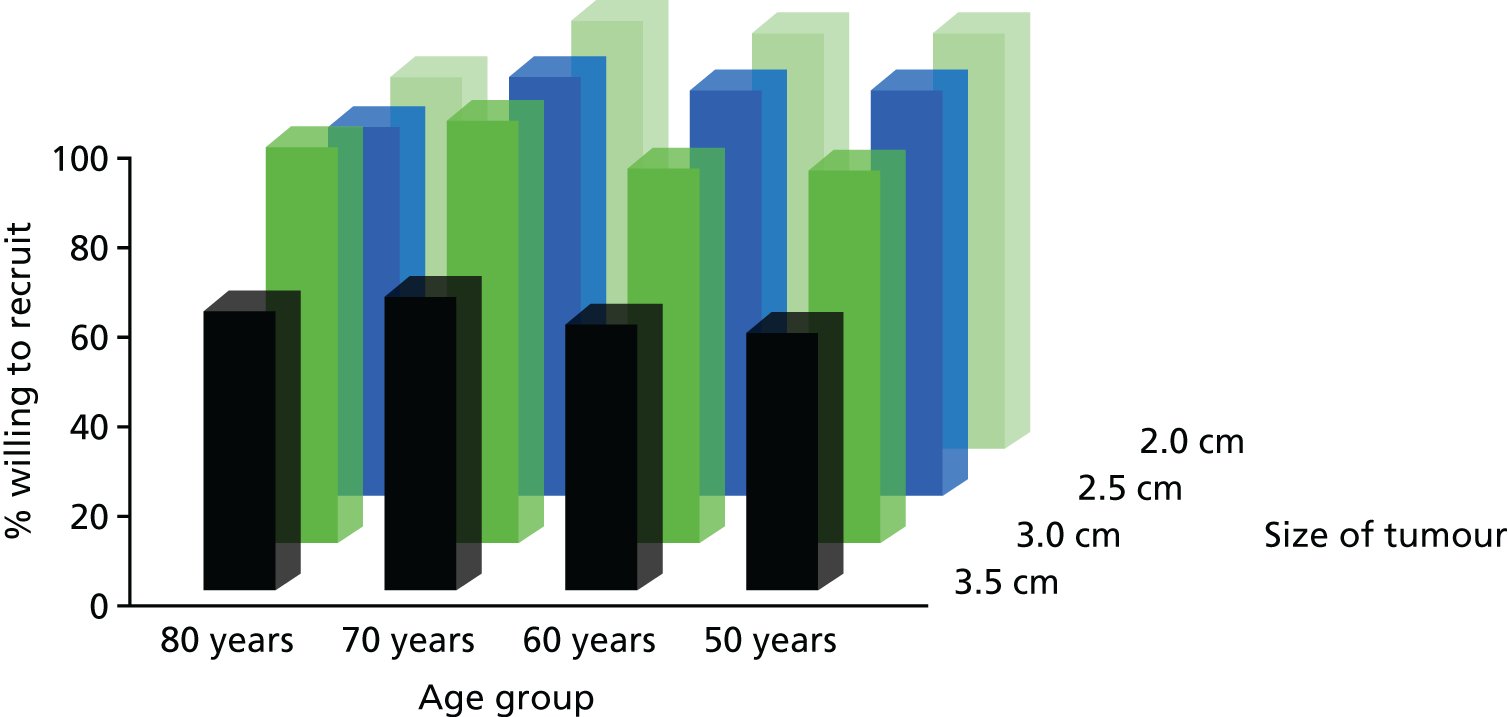
Less than half (45%) of respondents would be willing to recruit patients in the 50–59 years and 60–69 years age groups with tumour size of 2–2.5 cm and no comorbidity (Figure 2). As tumour size increased in this age group, willingness decreased to 23% for tumour size of 3 cm and to 15% for tumour size of 3.5 cm. There was a marked difference in the views of urologists: 47% agreed to approach when tumour size was 2 cm, but figure this dropped to 8% for a tumour size of 3.5 cm (Figure 3). For radiologists this figure was 44% and 22%, respectively (see Figure 4).
FIGURE 2.
Willingness to recruit by age and tumour size and no comorbidity.

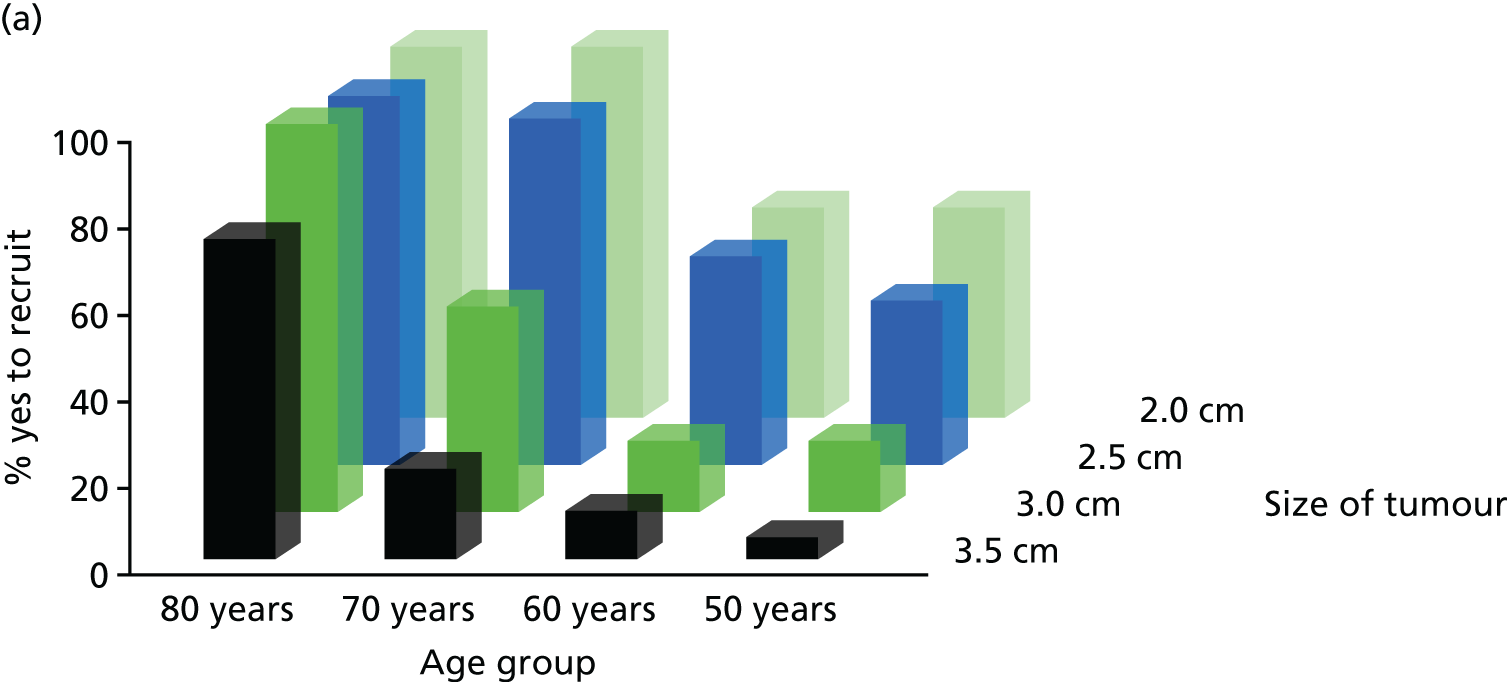
FIGURE 3.
Willingness to recruit by age and tumour size with comorbidity.
For patients aged 70–80 years, the majority (86%) of respondents would recruit if tumour size was of 2 cm and there was no comorbidity. This dropped substantially in the 70–79 years age group with increased tumour size: 48% at 3 cm and 30% at 3.5 cm. Similarly, for the 80–89 years age group, fewer (64%) would recruit if the tumour size was of 3.5 cm than of 3 cm (89%).
The presence of comorbidity (Charlson score of 2 points) had an influence. The majority (86%) of respondents would recruit patients aged 50–69 years with a tumour size of 2–2.5 cm or 3 cm (81%), but this figure dropped to 58% for tumour size of 3.5 cm (Figure 3). There was little or no difference in urologists’ willingness to recruit patients aged 50–79 years with tumours from 2 cm to 3 cm (89–100%) and comorbidity, but this proportion fell to 74% (aged 50–59 years) and 79% (aged 60–69 years) for tumours of 3.5 cm (Figure 4). For radiologists, this dropped from 83% for tumours of size 2–2.5 cm to 72% and 39%, respectively, for tumours of 3 cm and 3.5 cm in size (see Figure 4).
FIGURE 4.
Willingness to recruit by clinician type, age, tumour size and comorbidity. (a) Urologists’ willingness to recruit – no comorbidity; (b) urologists’ willingness to recruit – comorbidity (Charlson score of 2 points); (c) radiologists’ willingness to recruit – no comorbidity; and (d) radiologists’ willingness to recruit – comorbidity (Charlson score of 2 points).



There was an increase in the proportion of respondents who would recruit patients with comorbidity in the 70–79 years age group, to 91% for a tumour size of 3 cm, but this dropped to 64% for a tumour size of 3.5 cm (see Figure 3). For the 80–89 years age group with comorbidity, the percentages dip for a tumour size of 2–2.5 cm (77%), increase for a tumour size of 3 cm (86%) and drop for a tumour size of 3.5 cm (62%). The main difference between radiologists and urologists was in the percentage who would recruit patients aged 80–89 years with comorbidity and a tumour size of 2–2.5 cm (89% of radiologists, compared with 66% of urologists; see Figure 4).
Discussion
The results of this survey demonstrate that clinicians are more likely to recruit to a trial of ablation versus active surveillance patients who have some level of comorbidity and a tumour size of no greater than 3 cm. Willingness to recruit is lower for patients, in all age groups, with larger tumours,. In the case of patients without comorbidity, the majority of clinicians were willing to recruit patients in the 70–79 years age group with tumours of up to 2.5 cm and patients in the 80–89 years age group with tumours of up to 3.5 cm.
There was a similar pattern among radiologists and urologists in willingness to recruit patients without comorbidity. A clear cluster of patients that respondents would recruit were those in the 70–79 years and 80–89 years age groups with a tumour of size 2–2.5 cm and patients in 80–89 years age group only with a tumour size of 3 cm. Considering the upper end of the tumour size, a slightly higher proportion of radiologists than urologists were willing to recruit patients with a tumour size of 3–3.5 cm in the 50–70 years age group.
The presence of comorbidity led to a marked increase in the proportion of both radiologists and urologists who would recruit patients in the 50–59 years age group with a tumour size of 2–3 cm. For some reason, urologists were slightly less willing to recruit patients in the 80–89 years age group with a tumour size of 2–2.5 cm. Radiologists remained cautious about recruiting patients with a tumour size of 3.5 cm, but the proportion willing to recruit such patients was higher than the proportion willing to recruit patients with tumours of the same size but without comorbidity.
Although clinicians who responded to this survey ranked tumour size as the most important factor in recruitment to a trial of ablation versus active surveillance, they considered tumour size less important in the case of patients with comorbidity. Uologists considered ablation or active surveillance to be viable treatment/management options for all age groups and with tumours of all sizes (suggested in the survey) when comorbidity is present. The same can be said for radiologists with the exception of patients with tumours of a size of 3.5 cm.
Limitations
The response rate from urologists was disappointing and was a limitation of this survey as we were able to canvass the views of only half of this group of clinicians. The BAUS, unlike the BSIR, did not circulate the request on our behalf, which might have raised the profile of the survey and resulted in a better response rate. However, the number of interventional radiologists in the UK who conduct ablation is small, and we believe that we were able to capture the views of the majority of this group.
Participatory design study: development and testing of the SURAB trial patient information
Introduction
Recruiting to time and target in clinical trials is a longstanding problem. 41 The reasons for this may be multifactorial, but research conducted to explore patient factors in cancer trials have identified issues such as a dislike of randomisation and the inclusion of a placebo or a no-treatment arm. 42 As the SURAB feasibility study included both randomisation and a no-treatment arm, it was imperative that the rationale for, and importance of, both was fully addressed in the patient information. To do so, it was clear that patient involvement in producing user-friendly and comprehensive information for the SURAB feasibility study was needed. Consumer involvement has also been shown to improve the readability and relevance of patient information. 43 A plan was included in the initial funding application to develop patient information materials, in the stage 1, pre-feasibility, phase, for the patients approached to take the part in the subsequent pilot feasibility study. This stage of the research had ethics approval from the Newcastle University Faculty of Medical Sciences Ethics Committee (00774/2014). The process of recruiting patients for (and conducting) the patient and public involvement (PPI) activity is described in this section.
Objectives
The aim was to produce patient trial recruitment materials that were clear and comprehensive and included all of the information required for the recipients of the trial information. The plan was to develop and test information with patients who have been newly diagnosed with a SRM and those who have recently received treatment (including those under active surveillance). At the same time, their views on the proposed trial and trial processes were also explored.
Methods
As we were working on patient information developed for another trial and the need for the information to meet regulatory requirements, we opted for a participatory design. This means that all stakeholders (in this instance, patients, clinicians and the clinical trial unit team) are actively involved in the process and the end result is information that meets the needs of the patients that the SURAB trial is targeting. We also hoped that the information sheet would facilitate the recruitment process and be something that research nurses in particular could work with when discussing the trial with potential recruits.
It was envisaged that around eight patients would be involved in the design of the information, in either group or one-to-one sessions (whichever was preferable to those involved) with the trial team researcher (JL). We considered that spouses could be included as decisions around research participation are often made in consultation with the patient’s partner. In the group or one-to-one sessions, the pilot feasibility trial was explained (the two interventions being compared, the risks and benefits, what is required of participants in the trial, etc.). We then worked through the information and explored the appropriate content and amount of trial information, and how best to convey the information, including conveying risks and benefits. In addition, patients’ views were sought on the acceptability of approaching participants after they have declined to participate or dropped out of the trial (for the parallel qualitative study) and how best to phrase this in the trial patient information. It was also explained that the patient information had to comply with NHS Research Ethics Committee (REC) guidelines and there were certain elements that had to be included.
The next stage was to test the revised version of the trial information with a new group of patients using cognitive interviews and refine the information accordingly. The two main methods of cognitive interviewing used were (1) thinking aloud and (2) probing. Based on our personal experience with this more tightly focused work, a saturation point is normally reached after 8–10 interviews, when no new comments or issues are raised. We believed that up to eight cognitive interviews would be sufficient. The development work and cognitive interviews were recorded digitally and notes and observations made throughout with the permission of those involved.
Initially, it was hoped that the materials would be developed with patients from scratch. However, it was agreed that this would be a much lengthier process and would require greater buy-in and time from the patients involved. The Newcastle Clinical Trials Unit (NCTU), overseeing the conduct of the trial, had already submitted patient information materials to the NHS REC with a caveat that a revised version would be submitted at a later date after PPI. The information materials the NCTU submitted were based on those used previously in a similar trial and amended accordingly. It was decided that these materials would be the starting point for the participatory design process and the basis of the information for the SURAB trial.
Contact information for activity: getting it right
Designing the information sheet about involvement in the development of the trial information was not straightforward. In order to explain the purpose of, and rationale for, this activity, it was necessary to mention the trial, and there was a concern that patients may think that they were being approached to be trial participants. We needed patient involvement in the design of our information sheet, particularly to ensure it was clear that those approached were not being asked to take part in a trial. However, after contacting two sources (PPI leads from local and national NIHR Clinical Research Networks and a local cancer patient group), support was not forthcoming. To avoid running into difficulties with the timetable for this activity, we had to accept that the best we could do was to rely on our SURAB lay coapplicant and a clinical colleague from a local trust, who commented on the content of the information sheet and suggested some changes.
Identification and contact of patients for the activity
It was considered important to involve patients who had been diagnosed with small cell renal cancer and successfully treated within the last 12 months. They would remember when they received their cancer diagnosis and understand how it would feel being approached to take part in a trial, particularly one that has a no-treatment arm. One hospital site had agreed to send out a letter from the clinician who had cared for the patient, with an information sheet to explain why they were being contacted, a reply sheet they could complete and return (to the researcher) if they were happy to be contacted and a pre-paid envelope. These patients were then contacted by the qualitative researcher who answered any questions they had and determined if they were willing to help. As this was PPI, consent was not required.
Results
Response rate
In the first tranche, 20 letters were sent to patients in one hospital site. Unfortunately, none of those who returned the slip wished to be contacted. We then had to contact another hospital site to request help. One of the research nurses and the database manager from that site helped to identify eligible patients. A second tranche of 20 letters was sent, and 13 patients (nine men and four women) responded to say they were willing to be contacted. Telephone contact was made to answer any questions about the activity and to ascertain whether they had a preference for a group or one-to-one session. Only one person had misunderstood and thought they were being asked to take part in a trial. After learning more about what was required, one person declined any further involvement. Of the remaining 12, three expressed a preference for working in a group session and seven for a one-to-one session; two expressed no preference.
Stage 1: group session
Over several telephone calls with the five people willing to attend a group session, a convenient date and time were arranged. We asked if they would be able to attend for 3 hours. As we had no information other than their name and telephone number from the form they returned, we requested their home address (so we could send out an invitation letter and details of venue) and asked how they would travel to the venue (to organise parking spaces and permits), whether or not they wished to bring a partner and if they had any health or mobility problems we should be aware of. We also sent a copy of the information materials we would be working with should they have the time to look through them before the group met.
Three women and two men attended the group session. No one brought a partner. Ages ranged from 54 years to 77 years. Based on postcode and past or present occupation, four of the five group members’ socioeconomic status was categorised as A/B, which is described as professional and qualified to a high level.
The task began with the researcher (JL) briefly explaining the trial and the purpose of the activity. The participants gave some background on their experience of kidney cancer and what treatment they had received. The patient information materials were handed out and we began to go through each section of the patient information sheet (PIS) in detail.
Several general points were made: the sheer length was off-putting; it was repetitive; it had inconsistencies; and a higher than average level of literacy would be required to fully understand it. The key condition was described in different ways throughout the PIS as ‘renal tumour’, ‘kidney tumour’, ‘kidney cancer’ and ‘mass’. Similarly, the no-treatment arm was described as ‘active surveillance’ or ‘careful observation’. Some parts were considered superfluous or repetitive and some caused alarm. In response to the sentence ‘The tumour normally grows very slowly and can often be safely observed for a period of time’, the group wanted to know ‘how often?’ and ‘for what period of time?’. They thought that the positive aspects about being involved in the trial should be brought to the fore. It was suggested that bullet points of key information be given on the first page, then patients could make a decision if they want to read on. Alternatively, they thought that a short information sheet could be provided, with a longer, more detailed sheet given to patients who wish to know more and who agree to participate. A smaller booklet format was discussed, and it was thought that this might be less daunting. The bullet points should emphasise that patients would not have been approached if they were at risk, and the slow growth rate of this tumour, and should describe what action would be taken if patients in the active surveillance group patients experience any change in their condition. It was felt to be unnecessary to have the information sheet divided into parts 1 and 2. The group did not believe that there was a need to explain why patients are randomised. It was felt that there was too much detail about assessments and it was suggested that a flow chart be included. It was also suggested that information about the qualitative interviews could be reduced and simplified, and the need to call them ‘qualitative’ was queried.
The exercise took around 2 hours and attendees preferred to take refreshments (water, tea, coffee and biscuits) while they worked. Reimbursement was offered for travel and a gift voucher was given to each attendee as a gesture of thanks for their time.
Stage 2: one-to-one session
Amendments were made to the PIS in the light of the comments and suggestions of the group. The major changes were that bullet points were added to the front of the PIS and some of the information around the process for patient participants was replaced with a flow chart. As a result, the number of pages was reduced from 11 to nine.
The seven people who expressed a preference for one-to-one sessions were contacted again and a time was arranged to speak to them face to face. In six cases, the session took place in the participant’s home, and one session, at the request of the individual, was held at the university where the study team was based. The dates and times were confirmed in a letter, which was accompanied by the second version of the PIS so that participants could look through it beforehand.
Six of the sessions went ahead; one patient cancelled due to illness. Five participants were male and one female. This group spanned a broader range of socioeconomic status, categorised as C1/2 to D/E, which ranges from lower qualified professionals (e.g. nurses) to unskilled and casual workers.
As with the group exercise, the background to the study and the purpose of the visit was explained. Two people were slightly perplexed and bemused by the idea of PPI in research and some of the allotted time was taken to explain this in greater depth. The PIS was introduced and, as before, we went through it in detail, exploring each section using cognitive interviewing techniques. Most preferred the researcher to read each sentence and then explore the understanding and identify anything that was not clear.
Individuals tended to remark on similar points in the PIS. The main comments about the bullet points were to tighten up and simplify the text and give further reassurance about the active surveillance arm (that these tumours are slow growing). Comments on the ‘What is the purpose of the study?’ question can be summarised as ‘cut to the chase’ and include only the key points. This section was also perceived to indicate a great deal of uncertainty. Potential trial participants were mainly concerned about the risk of the cancer spreading if untreated and wanted to know how slowly the tumour normally grows, how long it can be observed for, how often these tumours can be observed and what the criteria for observation are. They also thought that the two interventions should be explained. There was a dislike of the term ‘randomisation’ and a preference for ‘random allocation’ or ‘allocated at random’. Some individuals did not understand the rationale for randomisation and thought this should be included. Patients liked the flow chart, and the only comment was about the term ‘randomisation’.
It was considered insensitive to mention in the risk information the minor risk of cancer associated with CT scans when the target population has just been diagnosed with cancer. It was also thought tha, in the same section, the text ‘some people may feel anxious about not receiving treatment and the possible risk of their cancer growing’ may cause concern in patients who had not considered this. One individual was participating in a cohort study and the study information was in a small (A5 size) booklet format that he found easy to read.
Stage 3: Newcastle Clinical Trials Unit and clinical team
The NCTU team reviewed the amended PIS following the group and one-to-one sessions to ensure that the PIS met the regulatory standards. The team members, in general, were very happy with the revised version but, as there is a requirement to inform potential trial participants of alternative treatments, requested that the section ‘What are the alternative treatments?’ be reinstated. The group had found this confusing (as two of the alternative treatments were part of the trial) and thought that this was already covered in the section on the purpose of the study. A compromise was to ensure that details of all potential treatment and management options were listed in the purpose of the study section at the front of the PIS. The clinical team was satisfied that the information around the condition and treatments and the rationale for the SURAB trial were fully explained. The amended PIS was submitted to the NHS REC, which accepted the changes without question.
Discussion
A number of issues were raised and lessons learned from conducting this work to develop patient-relevant materials for a clinical trial. First, securing help to ensure that written communication materials clearly conveyed the message that patients were being approached in a PPI capacity, and were not being asked to participate in a clinical trial, turned out to be rather perplexing. The response for help with this was disappointing but, ultimately, we appear to have managed, apart from in one case, to convey our meaning satisfactorily. Second, we underestimated the amount of time required to liaise with the hospital site that agreed to identify patients and contact them about the study. Furthermore, we had not envisaged the need for a contingency plan, a second hospital site, in the event that none of the patients contacted agreed to help. Fortunately, in the second hospital site, the staff pulled out the stops to identify patients and send our information to them. Third, and by chance, we managed to include patients from a range of socioeconomic groups in our activity, possibly by offering two forms of engagement, namely the group and the one-to-one work. Finally, the patients involved completely transformed the PIS, and the suggestion of a flow chart to replace the rather lengthy and difficult-to-follow patient route through the trial was inspired. The other stakeholders, such as the NCTU team and the trial team, were happy with the majority of changes and were willing to work to come to a resolution that respected the views of the patients involved in developing the materials and also adhered to regulatory and governance requirements.
Chapter 3 Feasibility study
Aims and objectives
The overall aim of this study was to determine whether or not a RCT comparing ablation with active surveillance in the management of SRMs is feasible in terms of recruitment and retention before embarking on a definitive trial. This was a multicentre study with two arms to evaluate the relative clinical effectiveness of ablation and active surveillance in patients with SRMs (< 4 cm). As this was a feasibility study, it was not expected to produce definitive results but would provide a basis for planning a larger definitive, fully powered trial.
The objectives were to:
-
test patient information to gauge comprehensibility and ensure that information perceived to be important for study participants is included
-
quantify the number of eligible patients
-
test patient identification systems and randomisation
-
test appropriateness and feasibility of outcome measures and time scales for collection
-
assess factors that promote or inhibit recruitment and retention in the trial
-
assess potential bias in recruitment and retention, systematic differences between those eligible to be randomised and those eligible but unwilling either by the clinician or the patient
-
examine the mechanism of data collection and assess the completion rates of data collection instruments to inform the full trial.
Trial interventions
In this study, patients who consented to participate were randomised to either ablation or active surveillance. All patients undergo routine biopsy of the renal tumour to confirm that it is a cancerous growth. At centres for which this biopsy is not routine, the participant must consent to the biopsy and the study prior to the biopsy being performed.
Patients randomised to the ablation arm undergo a second compulsory biopsy 6 months after treatment, conducted under US or CT guidance using local analgesia.
The ablation methods used depended on what expertise was available at the study sites. Sites offered only one form of ablation. The permitted ablation methods included RFA, CRYO and microwave ablation (MWA). The paragraphs below provide more detail about the ablative procedures and active surveillance.
Radiofrequency ablation
Radiofrequency probes are carefully positioned into the renal mass lesion percutaneously by image guidance (usually using CT) and laparoscopicaly by direct vision. Radiofrequency probes deliver localised monopolar currents at ‘radiofrequency’ (400–500 kHz) to generate frictional heating in the adjacent tissue. Through direct and conductive heating, this achieves temperatures of up to 105 °C. Tissue destruction occurs by protein denaturation, cell destruction and coagulative necrosis in a sometimes ill-defined sphere around the probe tip. This ablation zone can be compromised by tissue perfusion-mediated cooling and larger adjacent flowing vessels but can usually achieve ablation zones of up to 4–5 cm in diameter. Sometimes probes may be repositioned to achieve the required ablation volume. The procedure is well tolerated but now more usually performed under general anaesthesia to achieve optimal probe positioning and outcomes. When necessary, adjacent bowel or other structures are displaced by contrast-tinted 5% dextrose for retroperitoneal hydrodissection; however, carbon dioxide can also be used. Therapeutic outcomes are confirmed by contrast-enhanced CT or MRI within 3 months post ablation. 44
Cryoablation
Cryoprobes are applied laparoscopicaly or percutaneously by image guidance. Localised tip temperatures of –150 °C and lower can be achieved by utilising the phase change of compressed argon gas delivered through multiple closed-needle applicators, arranged in a pattern to create a confluent ‘therapeutic’ ice ball. Within the induced ice ball, a range of tissue-lethal temperatures are achieved. At the –30 °C isotherm, a double freeze–thaw cycle is believed to yield uniform cell death. Tissue destruction is achieved through disruptive cell necrosis and microvascular injury.
Microwave ablation
Microwave ablation is very similar to RFA. A similar-sized needle/probe is inserted into the lesion under imaging guidance exactly as for RFA. The microwave probe causes heating of the tissue by heating the water molecules within it achieving similar temperatures to RFA, causing cell destruction and coagulative necrosis.
Active surveillance
Patients randomised to active surveillance were asked to adhere to the following schedule – urea and electrolytes including estimated glomerular filtration rate, modification of diet in renal disease (MDRD) equation if performed clinically (there is no need to report this to the study team) and 6-month CT of the abdomen. Participants will undergo aCT of the abdomen (phasing and sequencing details were determined by site as per their local practice).
In case of progression of the growth rate or the size of the tumour in patients on the active surveillance arm, ablation or partial nephrectomy was offered depending on the facilities available in the participating centre.
Progression was considered to have occurred if:
-
growth rate exceeded 2.5 mm per 6 months or
-
tumour volume doubled within 6 months.
Ablation treatment
Our expectation was that ablative treatment would ideally be provided within 1 month of randomisation (SD 14 days) or as per standard national NHS protocols.
The following data were copied into the study electronic case report forms (eCRFs) from NHS medical records in relation to the provision of ablative treatment:
-
type of ablative treatment provided and date/time
-
whether or not the treatment was provided as per the randomisation allocation
-
any reason(s) for not providing treatment as per the randomisation allocation
-
any alternative treatments provided (e.g. surgical excision as an alternative)
-
complications of treatment and adverse events (AEs).
The ablation methods used depended on what expertise was available at study sites. Sites offered only one form of ablation. The permitted ablation methods included RFA, CRYO and MWA. More details about the ablative procedures and active surveillance are given below:
-
Patients recruited to the feasibility study who agreed to be randomised to either ablation or active surveillance underwent routine biopsy of the renal tumour to confirm that it was a cancerous growth. At centres where this biopsy is not routine, the participant must consent to the biopsy and to the study before the biopsy can be performed.
-
Patients randomised to the ablation arm undergo a second compulsory biopsy 6 months after treatment. This was conducted under US or CT guidance using local analgesia.
Methods
This was a multicentre RCT to establish the feasibility of ablation compared with active surveillance in patients with SRMs (< 4 cm). The study was conducted over 30 months.
The components were:
-
a feasibility multicentre RCT comparing ablation with active surveillance in patients with SRMs (< 4 cm) randomising on a 1 : 1 basis
-
a qualitative process evaluation to inform future trial design by exploring patient, clinician and delivery staff experiences of the trial and barriers to participation and recruitment (see Parallel qualitative study)
-
a rehearsal of health economic data collection (see Economic analysis).
Target population
Participants were patients with renal cancer masses < 4 cm.
Inclusion criteria
-
Adults with a renal tumour < 4 cm (confirmation by radiology or by biopsy) in size.
-
American Society of Anesthesiologists (ASA) physical status classification system grades 1 or 2.
-
Aged ≥ 18 years.
-
A CT/MRI scan of abdomen/chest with no evidence of metastases.
Exclusion criteria
-
Patient’s clinician does not feel they would be suitable for the trial (e.g. because of concomitant disease).
-
Multiple SRMs in one kidney.
-
Coagulopathy that cannot be corrected.
-
Previous participation in this study.
-
Inability to give informed consent (carer/proxy consent will not be allowed in this study).
Setting
The trial recruited from eight tertiary NHS centres offering kidney cancer treatment: one centre was in Scotland, two in the north of England and five in the south and south-west of England. These centres were identified from respondents to a national survey implemented by the NCTU.
Outcome measures
Primary
The primary outcome of the trial is feasibility defined as:
-
willingness of patients to be randomised, assessed by reviews of patient screening logs and defined as:
-
number of patients consenting to be randomised as a proportion of all eligible patients approached about the trial, with reasons for non-consent
-
qualitative assessment of barriers to, and facilitators of, recruitment
-
-
willingness of clinicians to randomise patients, assessed via qualitative interviews
-
assessment of retention and dropout rates, defined as:
-
number of patients who started randomised treatment as a proportion of the number randomised, with reasons for early dropout
-
number of patients who completed randomised treatment as a proportion of the number randomised, with reasons for early dropout (including death)
-
qualitative assessment of barriers to, and facilitators of, data collection and participant retention.
-
Secondary
Compliance with interventions and trial processes, defined as the number of patients who completed patient-reported outcomes at each time point as a proportion of the number randomised, including baseline, with reasons for non-compliance. Outcome measures for a definitive trial will be tested in the feasibility study and applied before treatment, at baseline and 3 months and 6 months after treatment.
The following measures were collected:
-
The Short Form questionnaire-36 items (SF-36) score. The SF-36 is a 36-item questionnaire that measures QoL across eight domains, which are both physically and emotionally based. A single item is also included that identifies perceived change in health, making the SF-36 a useful indicator for change in QoL over time and treatment. A summary of physical QoL [physical component summary (PCS)] and emotional QoL [mental component summary (MCS)] is produced. The percentage scores range from 0% (lowest or worst possible level of functioning) to 100% (highest or best possible level of functioning).
-
The Functional Assessment of Cancer Therapy – General (FACT-G) score. The FACT-G is a 27-item compilation of general questions divided into four primary QoL domains: physical well-being, social/family well-being, emotional well-being and functional well-being. It is considered appropriate for use with patients with any form of cancer.
-
The State–Trait Anxiety Inventory (STAI) score. The STAI is a measure of the severity of current symptoms of anxiety. There are two subscales: (1) state evaluates the current state of anxiety and subjective feelings of apprehension, tension, nervousness and worry; and (2) trait evaluates relatively stable aspects of ‘anxiety proneness,’ including general states of calmness, confidence and security. The range of scores for each subtest is 20–80 points, with a higher score indicating greater anxiety.
We also developed and tested health economic data collection tools in the form of a participant costs questionnaire (PCQ). The PCQ has two parts: (1) part A, to be administered at 3- and 6-month follow-ups, and (2) part B, to be administered at 6 months only.
The end of study was the last participant’s final study contact at their 6-month post-treatment follow-up.
Clinical trial screening, recruitment and consent
Identification and screening of patients for the feasibility study and parallel qualitative component
The trial flow chart (Figure 5) illustrates this process as well as data collection time points.
FIGURE 5.
Trial flow chart.

Potential participants were identified in the renal cancer clinics at participating sites. Site principal investigators (PIs) and/or clinical colleagues with documented delegated responsibilities for patient identification and screening performed this task.
An eligibility screening log was completed by the investigator to document participants’ fulfilment of the entry criteria for all patients considered for the study and parallel qualitative component and, subsequently, included or excluded. This information was anonymised and transcribed into screening logs on an ongoing basis via a secure online database.
Patients who agreed or declined to take part in the feasibility study were given an information sheet about the qualitative study and their permission to pass on their contact details to the qualitative researcher was requested. These patients were then contacted by the qualitative researcher, who answered any questions they had and, if they were happy to participate, arranged a convenient time to conduct an interview.
Clinical trial consent
Eligible patients were contacted by the centre PI/nurse lead/nurse specialist to invite them to participate in the trial and parallel qualitative component.
Informed consent was undertaken by appropriate site staff, as per the site delegation log. The delegated staff member (usually the centre PI/nurse lead/nurse specialist) explained the trial and parallel qualitative component to the patient, gave them the information leaflet and answered any questions they had.
Patients were encouraged to take the information leaflet home to discuss it with family and friends and asked to arrange a suitable time for a second meeting (allowing at least 24 hours for this). However, if participants had travelled a long distance to the hospital and would not be returning until an intervention visit, and if returning to hospital for the consent process would be a burden, consent was taken on the same day as information provision. In this case, a member of the local study team phoned the participant 48 hours later to confirm that they still wished to take part. This conversation was documented in the patient’s medical notes.
If a patient declined to participate in the trial and/or qualitative component, the study team (with permission from the patient) documented any reasons available for non-participation in the eligibility screening form and transferred this to the anonymised site screening log. The screening forms and logs ensured that potential participants were approached only once.
After ensuring that the patient had understood the information provided, the research team member delegated to take consent asked the patient to sign and date the consent form agreeing to participate in the feasibility study and/or the parallel qualitative component. Consent by the patient was witnessed and dated by the delegated research team member taking consent.
Written informed consent was always performed before randomisation or any other study-specific procedures/investigations.
The original signed consent form was retained in the investigator site file, with a copy in the clinical notes, a copy faxed to the NCTU (for centralised monitoring) and a copy provided to the participant.
For patients who agreed to take part in the qualitative study, a different consent form was completed at the time of the interview. Owing to the small subject population, the information sheet and consent forms were available only in English.
Clinical trial randomisation and blinding
Randomisation
When all eligibility checks had been made and written informed consent had been obtained, participants in the feasibility study were randomised in a 1 : 1 ratio (stratified by centre) to either active surveillance or ablative treatment (RFA, CRYO or MWA as per centre practice). Randomisation was undertaken using the central web-based randomisation service provided by the NCTU.
The PI at the site or the individual with delegated authority accessed the web-based randomisation system. Patient screening ID, initials and centre (the stratifying variable) were entered into the web-based system, which then returned the allocation status (successful randomisation was followed up by an automated confirmatory e-mail to the site and relevant staff).
Participants were then informed of their allocated treatment group by the site PI or delegated individual following randomisation.
Following allocation, the site organised:
-
the procedure date for those allocated for ablative treatment
-
the active surveillance protocol for those allocated to the active surveillance arm.
Blinding
This was a feasibility study with the primary outcome defined as recruitment and retention rates and qualitative exploration of the patients’ experiences and understanding of the randomisation process and treatment options. Randomised treatments could not be blinded owing to the type of interventions being researched. For this reason, staff were not blinded for the follow-up assessments.
The baseline data capture assessments were to be completed before randomisation in order to reduce any bias in terms of patient attitude to allocated treatment affecting baseline data.
Data collection time points
Baseline
Following written informed consent, a routine, standard care biopsy was performed post consent as part of standard care prior to randomisation in study participants who had not already received this clinically. In sites that did not perform this procedure as standard care, the participant consented to the procedure and the study prior to the biopsy. A compulsory renal biopsy at the core of the ablated lesion was to be performed on all patients who had received ablative treatment. There were three possible biopsy outcomes based on the histology and immunohistochemistry interpretation:
-
failed biopsy
-
no tissue or non-renal tissue
-
normal renal tissue
-
-
fibrosis
-
suggestive of ablation site (inflammation, haemorrhage or haemosiderin)
-
not suggestive of ablation site (old fibrosis, no evidence of recent inflammation or haemorrhage).
-
inconclusive
-
-
renal tumour (classification and grade)
-
tumour cells with Ki67 reactivity
-
tumour cells without Ki67 reactivity
-
The baseline (pre-randomisation) visit involved collection and retrospective collation of the following data:
-
Demographics (e.g. age, sex).
-
Medical history.
-
Blood tests and urinalysis (taken as per local policy, with no requirement to report these to the study team or record them on the study eCRF).
-
SF-36.
-
STAI.
-
FACT-G.
-
Biopsy results were recorded in the baseline eCRF to document relevant details and presence of cancerous growth. If a routine diagnostic biopsy was not standard care at site, consent for the biopsy and the study was given prior to the biopsy procedure.
-
Tumour size and volume were recorded from routine CT/MRI scans already obtained during diagnosis of the tumour. Size was recorded in three planes of measurement (volume = 0.5326 × diameter 1 × diameter 2 × diameter 3). The volume calculation was not to be completed by site.
-
Confirmation was recorded in the eCRF that study inclusion criteria was fulfilled and no exclusion criteria applied.
Patients were randomised only after the baseline visit, review of biopsy results and checking of inclusion/exclusion criteria. Patients were not randomised if the biopsy showed a non-cancerous result.
Three-month follow-up (for all participants)
It was planned that the 3-month follow-up of patients in the ablation arm would take place 3 months (SD 14 days) after the treatment date. In the case of patients in the active surveillance arm, it was planned that this should take place 3 months post randomisation (SD 14 days).
At the 3-month follow-up, the following information was requested for all patients:
-
results of blood tests and urinalysis (taken as per local policy, with no requirement to report these to the study team or record them on the study eCRF)
-
SF-36 score
-
STAI score
-
FACT-G score.
Imaging results were obtained in order to document any changes in tumour progression as follows:
-
For patients receiving ablation (using any of the methods), data were to be captured from a routine CT/MRI of the abdomen performed within 3 months of the ablative procedure.
-
For patients receiving active surveillance, a 3-month post-randomisation scan of the abdomen was optional depending on routine practice at site.
The imaging should allow capture of the following data on the eCRF:
-
Tumour size and volume. Size was recorded in three planes of measurement (volume = 0.5326 × diameter 1 × diameter 2 × diameter 3). The volume calculation was not to be completed by site.
-
Confirmation of any progression of the tumour.
The following additional data were requested for all patients:
-
any changes in treatment plan (e.g. switching from active surveillance to ablation or surgical excision) with dates, times and types of procedures recorded and reasons for a switch in treatment (i.e. reason for crossover)
-
complications of treatment and AEs
-
results of the administration of the health economics questionnaire: PCQ (part A).
Six-month follow-up (for all participants)
It was planned that the 6-month follow-up of patients in the ablation arm would take place 6 months (SD 14 days) after the treatment date. In the case of patients in the active surveillance arm, it was planned that this should take place 6 months post randomisation (SD 14 days).
At the 6-month follow-up the following was requested for all patients:
-
results of blood tests and urinalysis (taken as per local policy, with no requirement to report these to the study team or record them on the study eCRF)
-
SF-36 score
-
STAI score
-
FACT-G score.
For patients who received an ablative procedure, data were captured from a routine follow-up CT/MRI scan of the abdomen performed at 6 months after the procedure in order to document any changes in tumour progression.
For patients receiving active surveillance, data were captured from a routine follow-up CT/MRI scan of the abdomen performed at 6 months after randomisation in order to document any changes in tumour progression.
The imaging allowed capture of the following data on the eCRF:
-
Tumour size and volume. Size was recorded in three planes of measurement (volume = 0.5326 × diameter 1 × diameter 2 × diameter 3). The volume calculation was not to be completed by site.
-
Confirmation of any progression of the tumour.
Tumour volume was calculated from the three-dimensional diameters using the formula to calculate an ellipsoid volume:
If there was progression of the growth rate or the size of the tumour in patients on the active surveillance arm, ablation or partial nephrectomy would be offered depending on the facilities available in the participating centre.
Progression was to be considered to have occurred if growth rate exceeded 2.5 mm per 6 months or tumour volume had doubled by 6 months.
The following additional data were captured for all patients:
-
any changes in treatment plan (e.g. switching from active surveillance to ablation or surgical excision) with dates, times and types of procedures recorded and reasons for a switch in treatment (i.e. reason for crossover)
-
complications of treatment and AEs
-
results of the administration of the health economics questionnaire: PCQ (parts A and B).
Sample size
The target recruitment was a total of 60 patients (30 randomised to the active surveillance arm and 30 to the ablation arm). This figure was based on a recommendation by Lancaster et al. 45 with respect to the number of patients required to yield meaningful estimates of parameters of interest in feasibility studies. With eight participating centres, it was estimated up to 120 patients could be approached; assuming a successful recruitment rate of no less than 50%, this would provide the 60 patients required.
Progression criteria to main trial
Recommendation to move to a definitive, fully powered, multicentre trial is based on the following calculations:
-
The upper 90% confidence interval for the proportion of patients recruited should exceed 50%. The trial should recruit at least 49 patients from 120 approached. This was based on a requirement that the underlying recruitment rate should be at least 50%. The exact 90% confidence interval corresponding to 49 successes from 120 Bernoulli trials is from 32.0% to 50.2% (with 48 successes from 120 trials, the upper interval drops to below 50%). Should recruitment be < 49 patients, this would provide evidence that the recruitment rate is too low and the feasibility of a RCT would be questionable. The upper 90% confidence interval for the retention rate (the proportion of patients recruited who have been followed up) should be ≥ 80%.
-
Economic assessment should suggest that further research likely to be worthwhile.
Adverse events and serious adverse events
The procedure for recording and handling of AEs and serious adverse events (SAEs) was set out in the study protocol.
Adverse events
All non-serious AEs during study participation were to be reported on the study eCRF and sent to the trial manager within 1 month of the form being due. Severity of AEs was graded on a three-point scale (mild, moderate or severe). Relation (causality) and seriousness of the AE to the treatment was to be assessed by the investigator at site in the first instance. The individual investigator at each site was responsible for managing all AEs according to local protocols.
Serious adverse events
All SAEs during study participation were to be reported to the chief investigator (CI) within 24 hours of the site learning of their occurrence. The initial report could be made by telephone or fax. In the case of incomplete information at the time of initial reporting, all appropriate information was to be provided at follow-up as soon as it became available. The relationship of the SAE to study procedures was to be assessed by the investigator at site, as was the expected or unexpected nature of the AE.
Statistical considerations
As a feasibility study, the aim was to provide the foundations for future research in this area and to ensure that a larger-scale research project was feasible and acceptable. We therefore decided to estimate (1) subject availability; (2) the willingness of clinicians to randomise subjects to the trial, and of subjects to be randomised; (3) the proportion of randomised subjects who would complete the trial treatments and data collection; and (4) variability in the data necessary to inform sample size calculations for a future definitive Phase III trial. Our primary focus was, therefore, on descriptive statistics rather than hypothesis testing.
Statistical analyses
The primary purpose of this feasibility study was to assess recruitment and retention. This feasibility study was not designed to make an assessment of treatment efficacy and sample sizes will be too small to make an interim assessment of efficacy. We anticipate that, even if the initial rates are disappointing, the qualitative research might suggest improvements that could be made to recruitment procedures; hence, we have not defined a stopping rule for futility. This trial does not involve the use of drugs and hence issues of toxicity are not expected to be a concern. Complications and AEs are recorded as is usual.
Interval estimates (using 95% confidence intervals) of key parameters of interest will be determined including:
-
the number of patients who agree to be randomised as a proportion of eligible patients identified
-
the number of patients receiving ablation who experience perioperative complications, as a proportion of those randomised to ablation
-
the number of patients for whom we can collect outcomes at 3 and 6 months post treatment, as a proportion of those randomised
-
an estimate of variability (standard deviation) of the QoL measures that will be used in the Phase III trial.
Summary of changes to the project protocol
Changes to the protocol that were made and submitted to the REC are as follows.
Substantial amendment 1: 13 November 2014 (protocol version 2.0)
Changes were made to allow more flexibility for CT/MRI scanning at sites. Post-ablation CT/MRI would be undertaken at 3 and 6 months after treatment (ablation) and at only 6 months in the active surveillance arm. However, the participating centres could carry out additional scanning as per clinical policy of the centre. The scans at 3 and 6 months should be abdominal (we had originally stated chest and abdominal) but the scan phase used should be determined by standard practice at site. Sites can perform other scans as they deem clinically necessary. This change was based on feedback from participating sites. It was also decided that urine and blood values were not required for a feasibility study and there was no requirement for sites to report these results to the study team.
Substantial amendment 2: 16 December 2014 (protocol version 3.0)
Information on how to report SAEs was added.
Updates were also made regarding the baseline biopsy for participants, which resulted in changes to the inclusion and exclusion criteria.
Substantial amendment 3: 26 February 2015 (protocol version 4.0)
The requirement for patients to have a physical status classified as ASA grade 1 or 2 was removed. This was because many patients who would be suitable for randomisation would be elderly and may have other comorbidities that may place them in ASA 2/3 grade. Although patients being considered for surgery should be classified as ASA grade 1/2, it may not be necessary for patients who are being randomised to either active surveillance or ablation to meet this criterion and imposing this restriction might have adversely affected recruitment into the trial.
Following the first independent Data Monitoring Committee meeting, the protocol was altered with regard to statistical analysis. Sites were also allowed to tell participants who were unable to complete the follow-up questionnaires at the clinic appointment that these could be returned by post.
Substantial amendment 4: 12 February 2016 (protocol version 5.0)
Following the Health Technology Assessment (HTA) programme’s request to close the study early to recruitment in November 2015, a close-down plan was prepared and agreed by the HTA, which involved removing the collection of the 6-month follow-up data.
The following protocol changes were made:
-
Obtain verbal consent for the qualitative interviews that are carried out over the telephone.
-
Interview patients who agree to take part in the main study at two time points.
-
Reflect that all study data will be kept for 5 years, as per the Newcastle upon Tyne Hospitals NHS Foundation Trust Archiving Clinical Research Documents standard operating procedure. 46
-
Inform patients about their contribution to the study at the end of the trial in a newsletter, which will include a lay summary of results.
-
Interview all health professionals that are involved in the recruitment process as well as clinicians.
Results
Trial approval and procedures
Ethics consideration
Ethics approval was sought and granted by the Newcastle and North Tyneside 2 Committee, NHS Health Research Authority (reference 14/NE/0155).
Clinical trial assessments/data collection
Pre-screening and screening
-
PIS provided.
-
Eligibility criteria checked.
Recruitment and randomisation
Eight sites screened a total of 154 patients as part of the trial recruitment, of whom 119 (77%) were deemed not eligible, including one patient found to be ineligible post consent (Table 2 and Figure 6).
| Site | Total number of patients screened | Total number of patients randomised |
|---|---|---|
| Newcastle | 69 | 1a |
| Southampton | 33 | 0 |
| St George’s | 23 | 3 |
| Glasgow | 10 | 2 |
| Bristol | 8 | 0 |
| UCLH/Royal Free | 6 | 0 |
| Stevenage | 5 | 0 |
| Leeds | 0 | 0 |
| Oxford | Never opened as a site | Never opened as a site |
FIGURE 6.
Consolidated Standards of Reporting Trials (CONSORT) diagram. a, A list of reasons for patients not being approached and other comorbidities can be found in Table 4, and reasons for declining in Table 5. b, Some sites did not have biopsy as part of standard care and therefore the patients needed to consent to the trial to confirm if their biopsy was cancerous.

The trial recruited the first patient on 11 June 2015 and its last patient on 12 November 2015. Three sites recruited six patients in total before the trial was closed to recruitment. Patients were followed up for a minimum of 6 months and the database was frozen on 23 May 2016.
A total of 81 (68%) of 119 patients were not approached about the trial, the reasons for which are tabulated in Table 3. The majority of patients were not approached because they were deemed to be suitable for surgery (40%) or because of early termination of the trial (33%) and 14% were not approached owing to suitability for active surveillance.
| Reason | Total number of patients |
|---|---|
| Clinician feels patient is more suitable for surgery | 32 |
| Trial closed early to recruitment prior to site approaching the patient | 27 |
| Clinician feels patient is more suitable for active surveillance | 11 |
| Size of the tumour | 3 |
| Clinician feels there is too much to consider in relation to clinical diagnosis | 2 |
| Patient not suitable for surgery | 2 |
| Renal biopsy prior to management plan | 1 |
| Clinical decision to ablate | 1 |
| Referral to local hospital for treatment | 1 |
| Location of tumour | 1 |
| Total | 81 |
A total of 21 (18%) of 119 ineligible patients had other comorbidities as listed in Table 4.
| Number | Information provided by site regarding other comorbidities |
|---|---|
| 1 | Previous lung cancer, cardiac complications, respiratory problems – COPD |
| 2 | Previous colon cancer and radical surgery (April 2015), lung and liver lesions, only for surveillance |
| 3 | Castrate-resistant prostate cancer, heart complications, COPD, diabetes mellitus, renal mass not clinically significant in view of health problems |
| 4 | 90 years, has rectal cancer and hypertension |
| 5 | Previous deep-vein thrombosis/leg amputation, best supportive/palliative care |
| 6 | Alzheimer’s disease, chronic kidney disease, hypertension, metastatic lesion in spine |
| 7 | Non-small cell lung cancer treating with chemotherapy, lifelong smoker |
| 8 | Large abdominal aortic aneurysm needs intervention first |
| 9 | Lung cancer receiving chemotherapy |
| 10 | Renal transplant work-up |
| 11 | Bilateral renal lesion |
| 12 | 84 years, has pleural plaques and shortness of breath, aortic stenosis |
| 13 | Breast carcinoma |
| 14 | Chronic lymphocytic leukaemia, COPD, pulmonary embolism, the left renal mass is probably benign |
| 15 | 89 years, heart disease plus previous coronary artery bypass graft, malignant neoplasm of stomach, previous cerebral vascular attack in 2014 |
| 16 | Patient has history of coronary artery bypass surgery with non-ST elevated myocardial infraction in 2004 and 60% ejection fraction. Patient has mild aortic stenosis, hypercholestraemia and hypertension |
| 17 | CT showed paratracheal mass that needs further investigation |
| 18 | Oncocytoma |
| 19 | Atrial fibrillation, non-alcoholic liver disease and hypotension |
| 20 | Symptomatic ventricular tachycardia (2012) with an intracardiac device implanted, ischaemic cardiomyopathy, atrial fibrillation, osteoarthritis, benin prostatic hyperplasia, gout and chronic kidney disease |
| 21 | Has previous cerebrovascular accident, significant lower body lymphoedema, lower limb cellulitis, grossly obese |
A total of 36 (23%) screened patients were initially eligible for the trial (including one found ineligible post biopsy), of whom 29 (81%) declined participation, for the reasons tabulated in Table 5. The majority of patients [16 (55%)] wanted ablation and a further 10 (34%) wanted active surveillance.
| Reason | Total number of patients |
|---|---|
| Wanted ablation | 16 |
| Wanted active surveillance | 10 |
| Patient did not want active surveillance | 1 |
| Patient not comfortable with randomisation | 1 |
| Patient refused to travel for study | 1 |
| Total | 29 |
After all eligibility checks had been made and written informed consent had been obtained, participants were randomised in a 1 : 1 ratio (stratified by centre) to either active surveillance or ablative treatment (RFA, CRYO or MWA dependent on centre). Randomisation was undertaken using the central web-based randomisation service provided by the NCTU. Eight sites screened potential patients; three sites randomised six patients before the trial was closed to recruitment (Table 6).
| Site | Total number of patients screened | Total number of patients randomised |
|---|---|---|
| Newcastle | 69 | 1a |
| Southampton | 33 | 0 |
| St George’s | 23 | 3 |
| Glasgow | 10 | 2 |
| Bristol | 8 | 0 |
| UCLH/Royal Free | 6 | 0 |
| Stevenage | 5 | 0 |
| Leeds | 0 | 0 |
| Oxford | Never opened as a site | Never opened as a site |
Randomisation by randomising site and randomised treatment arm is tabulated in Table 7, demonstrating the six patients to be randomised in equal numbers to the two research arms.
| Site | Active surveillance (n = 3) | Ablative treatment (n = 3) | Total (n = 6) |
|---|---|---|---|
| Glasgow | 1 | 1 | 2 |
| Newcastle | 1 | 0 | 1 |
| St George’s | 1 | 2 | 3 |
Study population
Demographic and clinical baseline characteristics at randomisation are compared descriptively across treatment groups (see Tables 8 and 9).
Table 8 describes demographic characteristics for comparison across treatment groups, including age, sex, medical history and three QoL scores: a general health questionnaire (SF-36),47 a cancer-specific health status and QoL (FACT-G),48 and the STAI. 49 Owing to the randomised nature of the trial, no significance testing was carried out.
| Patient demographics | Active surveillance (n = 3) | Ablative treatment (n = 3) | All patients (n = 6) |
|---|---|---|---|
| Female | 0 | 0 | 0 |
| Male | 3 | 3 | 6 |
| Age (years) | 59, 65, 69 | 69, 71, 77 | 59–77 |
| Number of patients reporting other medical conditions | 2 | 1 | 3 |
| QoL scores | |||
| SF-36 (PCS) | 49.06, 52.73, 59.23 | 25.61, 46.00, 53.13 | 25.61–59.23 |
| SF-36 (MCS) | 55.18, 60.16, 61.27 | 29.21, 51.22, 55.96 | 29.21–61.27 |
| FACT-G | 46.00, 90.83, 98.33 | 70.67, 96.83, 101.50 | 46.00–101.50 |
| STAI (trait) | 22, 28, 34 | 29, 44, 55 | 22–55 |
| STAI (state) | 22, 26, 32 | 32, 34, 52 | 22–52 |
All six randomised patients were male and aged between 59 and 77 years, with three of them reporting other significant medical conditions.
For the SF-36, baseline measures for PCS ranged from 26% to 59% and for MCS ranged from 29% to 61%. FACT-G scores ranged from 46.0 to 101.5 points across all six patients. STAI scores ranged from 22 to 55 points independent of trait or state scores.
Tumour-related characteristics at baseline, including maximum diameter, are described in Table 9. Maximum tumour diameter ranged from 1.2 cm to 3.3 cm across the six patients. All were identified by CT. Three tumours were pathologically described as clear cell carcinoma, two were papillary cell carcinoma (one type 1, one type 2) and in one case pathology could not be determined. All biopsies were described as lesional tissue with viable renal carcinoma tissue present. None had presence of inflammation, sclerosis or necrotic tumour.
| Tumour characteristics | Active surveillance (n = 3) | Ablative treatment (n = 3) | All patients (n = 6) |
|---|---|---|---|
| Maximum tumour diameter (cm) | 1.2, 2.4, 2.9 | 1.4, 2.6, 3.3 | 1.2–3.3 |
| Scan type | |||
| MRI | 0 | 0 | 0 |
| CT | 3 | 3 | 6 |
| Renal carcinoma type | |||
| Clear cell carcinoma | 1 | 2 | 3 |
| Papillary cell carcinoma type 1 | 0 | 1 | 1 |
| Papillary cell carcinoma type 2 | 1 | 0 | 1 |
| Chromoplate carcinoma | 0 | 0 | 0 |
| Oncocytoma | 0 | 0 | 0 |
| Cannot be determined | 1 | 0 | 1 |
| Other tumour characteristics | |||
| Lesional tissue | 3 | 3 | 6 |
| Viable renal carcinoma tissue present | 3 | 3 | 6 |
| Presence of inflammation and/or sclerosis | 0 | 0 | 0 |
| Necrotic tumour present | 0 | 0 | 0 |
Treatment received
Three patients were randomised to receive ablation. The time from randomisation to first treatment is reported in Table 10, along with ablation type. Compliance with randomised intervention is thus 100% in this very small sample. Two out of three patients received CRYO (at 35 and 41 days post randomisation), one received RFA (at 47 days post randomisation) and no patients received MWA. Ablative treatment should ideally be carried out within 1 month of randomisation (SD 14 days). Both CRYO patients were treated within this timescale, but this target timescale was not achieved in the case of the RFA patient.
| Site | Number of patients randomised to ablation | Number of patients who received ablation | Number of days from randomisation to ablation | Ablation type received | ||
|---|---|---|---|---|---|---|
| RFA | CRYO | MWA | ||||
| Glasgow | 1 | 1 | 47 | 1 | 0 | 0 |
| St George’s | 2 | 2 | 35, 41 | 0 | 2 | 0 |
Safety analysis
No AEs or adverse reactions were reported.
Analysis
A total of 154 patients were screened as part of the trial. Of these, 36 were eligible to be entered into the trial and were provided with study details. Of these patients, seven agreed to be randomised; however, one patient was found ineligible following biopsy results. Six patients (17% of those eligible) were randomised, three patients received ablation and no SAEs were recorded in relation to the trial. Three-month patient-reported outcome data were collected for four (67%) of the six patients, from all three patients in the active surveillance arm and from one patient in the ablation arm. Six-month patient-reported outcome data were collected for three (50%) of the six patients; two of three patients reported (completely) from the active surveillance arm and one patient reported from the ablation arm. Patient-reported outcome measures are shown in Table 11. It was originally planned to determine interval estimates (using 95% confidence intervals) of key parameters of interest. However, owing to very low levels of recruitment, no confidence intervals have been calculated.
| Treatment arms | Patient | SF-36 (PCS) | SF-36 (MCS) | STAI (trait) | STAI (state) | FACT-G | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 3 months | 6 months | Baseline | 3 months | 6 months | Baseline | 3 months | 6 months | Baseline | 3 months | 6 months | Baseline | 3 months | 6 months | ||
| Active surveillance (n = 3) | 1 | 59.23 | 59.33 | 59.6 | 55.18 | 57.11 | 51.97 | 28 | 23 | 28 | 26 | 25 | 39 | 90.83 | 101.00 | 96.00 |
| 2 | 52.73 | 51.49 | 50.41 | 61.27 | 61.72 | 53.36 | 22 | 27 | 26 | 22 | 29 | 28 | 46.00 | 102.00 | 94.83 | |
| 3 | 49.06 | 51 | 51.7 | 60.16 | 56.42 | 48.14 | 34 | 35 | 43 | 32 | 39 | 38 | 98.33 | 88.17 | – | |
| Ablation (n = 3) | 4 | 25.61 | 24.48 | 22.23 | 29.21 | 28.23 | 31.43 | 55 | 59 | 56 | 52 | 54 | 54 | 70.67 | 58.00 | 59.83 |
| 5 | 53.13 | – | – | 55.96 | – | – | 29 | – | – | 32 | – | – | 101.50 | – | – | |
| 6 | 46 | – | – | 51.22 | – | – | 44 | – | – | 34 | – | – | 96.83 | – | – | |
Progression criteria for continuation to a definitive trial (as stated in Statistical considerations) state:
-
The underlying recruitment rate should be at least 50%. At 17%, this recruitment rate was not achieved.
-
Recruitment should be at least 49 patients. With six patients, this recruitment was not achieved.
-
The upper 90% confidence interval for the retention rate (the proportion of patients recruited with complete followed up) should be ≥ 80%. Owing to very low levels of recruitment, confidence intervals have not been calculated. The point estimates for retention are 67% at 3 months and 50% at 6 months and are below what was anticipated.
Given the information above and in the light of the predetermined progression criteria for feasibility, the overall conclusion from the results of this feasibility study is that it is not feasible to move to a definitive Phase III trial with this design.
Economic analysis
Aim
The economic component of the feasibility study aimed to develop and test the health economics data collection tool and assess the ease of data collection required for health economic analysis to inform the definitive trial.
Outcomes data collection
Health-related quality of life (HRQoL) was used as the health economic outcome and was measured by the Short Form questionnaire-6 Dimensions (SF-6D) based on responses from SF-36 questionnaires, which were completed by participants at baseline and at 3 and 6 months’ follow-up. The response rates of the questionnaire were assessed and a summary of utility scores was calculated from the SF-6D.
Health resource use data collection
We took the perspective of both the NHS and the patients, by collecting information on NHS costs that included the costs of intervention and the use of primary and secondary health services, as well as patients’ out-of-pocket expenses relating to the condition.
Intervention resource use
The main cost driver of treating patients with SRM was expected to originate from the intervention (ablation treatments): CRYO, MWA and RFA. The key cost components included staff, consumables, capital and overheads. Data on these costs will be obtained from participating centres. The following information to be used to derive resource use was recorded in the case report form (CRF) for each participant in the ablation arms of the trial:
-
type of anaesthetic used (general or local)
-
grade of anaesthetist present
-
grade of radiologist present
-
grade of assistant staff present
-
number of nursing and assistant staff present
-
time of patient entry and exit from CT suite
-
time of patient entry and exit from recovery room
-
date of admission
-
date of discharge
-
post-treatment complications (Clavien–Dindo grade, if applicable).
Resource use and patients’ out-of-pocket expenses during follow-up
A purposely designed PCQ was developed and tested in the pilot study. The PCQ was used to collect information on patients’ health services use and out-of-pocket expenses during the follow-up period. The PCQ had two parts. Part A collected information on patients’ use of primary and secondary care as a result of problems related to having SRMs, and included hospital outpatient and inpatient visits, general practitioner (GP) visits, nurse visits and other specialist visits during the trial follow-up period. Part A also collected information on any private health insurance or informal care that participants might have in relation to their condition. Part B gathered information on patients’ time and travel costs of attending each of the above-mentioned health services. In terms of time costs, participants were asked about the time spent travelling to, and the time spent at, each health service and what activity they would have been undertaking during that time if not attending the health services. Participants were asked to provide the same information for any relative or carer who accompanied them to each service. In terms of travel costs, participants were asked about their mode of transport and the costs associated with travelling to each health service. Participants were also asked to provide travel cost information for any relative or carer who accompanied them.
In order to reduce recall bias, participants were asked to complete part A of the PCQ at 3-month intervals (at the 3- and 6-month follow-ups). As part B of the PCQ collected information on participants’ latest visit to each of the listed services and would be used to produce unit cost at an aggregated level for each arm, it was administered only once, at the 6-month follow-up.
Response rates of PCQ at different points were calculated and responses to PCQ were examined.
Results
Owing to the early termination of the feasibility trial, only six patients were recruited: three in the control arm and three in the intervention arm.
Resource use
Intervention resource use
Based on data collected on the CRF, of the three patients allocated to the intervention arm, one patient had RFA treatment and two had CRYO treatment. There was no further information apart from the allocated treatment of one of the patients allocated to the intervention arm. Both of the remaining two patients in the intervention arm were given a general anaesthetic; one was attended by a consultant anaesthetist and in the other case the role of the attending physician was unclear. Both patients were treated by a consultant radiologist. Information on any nursing and assisting staff present at the treatment was unavailable, and information on the time of patients entering and exiting theatre and recovery room was incomplete. Both patients had one overnight stay in the hospital. Neither of the patients had post-treatment complications.
Resource use and patients’ out-of-pocket expenses during follow-up
The rates of response to the PCQ are presented in Table 12. Although the response rate is higher in the control arm than in the treatment arm, this comparison is not meaningful given the very small sample size. The loss to follow-up of two patients in the intervention arm was due to a staffing issue at one of the participating sites, which led to patients not being followed up by the research team.
| Follow-up time points | Control arm (n = 3) | Intervention arm (n = 3) |
|---|---|---|
| 3 months (part A) | 100% (3/3) | 33% (1/3) |
| 6 months (parts A and B) | 100% (3/3) | 33% (1/3) |
| Average | 100% | 33% |
In a definitive study, data collected on PCQs would be presented as average costs for each item. These costs would be estimated by multiplying the number of times each individual used a particular service by the number of individuals that gave that response, then summing them across individuals, which would then be divided by the total number of respondents contributing data (examples are given in the footnote of Table 13). However, for this study, the data (see Tables 13 and 14) are presented in their raw form for easier interpretation of responses. Furthermore, owing to the small sample size, any calculation of average costs would not be meaningful.
Table 13 summarises participants’ use of different health services collected from part A of the PCQ. The most common use of health services during the trial’s follow-up period appeared to be outpatient visits. Informal care was given by a relative or friend for the participant in the treatment arm. Planned inpatient stay and GP telephone consultation were also observed for one patient each. However, given the extremely small sample, no meaningful conclusion can be drawn.
| 3 months | 6 months | |||
|---|---|---|---|---|
| Control | Intervention | Control | Intervention | |
| Total number of participants | 3 | 3 | 3 | 3 |
| Questionnaires received | 3 | 1 | 3 | 1 |
| Emergency admission | 0 | 0 | 0 | 0 |
| A&E visit | 0 | 0 | 0 | 0 |
| Planned inpatient stay | (1) × 1 | 0 | 0 | 0 |
| Outpatient visit | (1) × 1 | (1) × 1 | (1) × 1, (2) × 1 | (3) × 1 |
| GP visit | 0 | 0 | 0 | 0 |
| GP home visit | 0 | 0 | 0 | 0 |
| Practice nurse visit | 0 | 0 | 0 | 0 |
| Practice nurse home visit | 0 | 0 | 0 | 0 |
| Telephone consultation | 0 | 0 | (1 × hospital doctor) × 1 | 0 |
| Out-of-hours consultation | 0 | 0 | 0 | 0 |
| Informal care received | 0 | Personal care (6 hours by a relative) × 1, medical care (2 hours by a relative) | 0 | Personal care (4 hours by a relative, 2 hours by a friend), medical care (2 hours by a relative) |
| Private care | 0 | 0 | 0 | 0 |
Table 14 presents patients’ time and travel information on attending each type of health service recorded on part B of the PCQ. With the information recorded on part B of the PCQ, the average costs for an individual to attend each type of the health services could be calculated, which would then be combined with information collected on part A to derive a total costs for each individual’s out-of-pocket expenses.
| 6 months’ follow-up | Control | Intervention |
|---|---|---|
| Total number of participants | 3 | 3 |
| Questionnaires received | 3 | 1 |
| Hospital admission | ||
| Form of transport | (Public transport) × 1, (car) × 1 | (Car) × 1 |
| Miles by car one way | (10) × 1 | (3) × 1 |
| Parking fee (£) | (2.40) × 1 | (0) × 1 |
| Cost of fares (£) | (1.50 public transport) × 1 | – |
| Activity would be doing | (Paid work) × 1, (leisure) × 1 | (Leisure) × 1 |
| Accompanied by another adult | 1 | 1 |
| Activity would be doing by companion | (Paid work) × 1, (house work) × 1 | – |
| Time spent by companion (hours) | (1–2) × 1 | (2–3) × 1 |
| Outpatient visits | ||
| Form of transport | (Public transport) × 1, (car) × 2 | (Car) × 1 |
| Miles by car one way | (6) × 1, (10) × 1 | (3) × 1 |
| Parking fee (£) | (1.20) × 1, (0) × 1 | – |
| Cost of fares (£) | (1.50 public transport) × 1 | – |
| Activity would be doing | (Paid work) × 1, (housework) × 1, (leisure) × 1 | (Leisure) × 1 |
| Time spent in hospital (hours) | (0.5–1) × 1, (2–3) × 1, (3–4) × 1 | (2–3) × 1 |
| Accompanied by another adult | 2 | 1 |
| Activity would be doing by companion | (Paid work) × 1, (leisure) × 1 | (Leisure) × 1 |
| GP/nurse visits | ||
| Form of transport | (Walk) × 2 | – |
| Miles by car one way | – | – |
| Parking fee (£) | – | – |
| Cost of fares (£) | – | – |
| Activity would be doing | (Housework) × 1, (leisure) × 1 | – |
| Time spent in practice (hours) | (0.5–1) × 1, (1–2) × 1 | – |
| Accompanied by another adult | – | – |
| Activity would be doing by companion | – | – |
| Personal care | ||
| Received personal care | – | 1 |
| Form of transport | – | (Walk) × 1 |
| Miles by car one way | – | – |
| Parking fee (£) | – | – |
| Cost of fares (£) | – | – |
| Work affected | ||
| Employment status | (Retired) × 2, (full-time employed) × 1 | (Retired) × 1 |
| Days off work | – | – |
There seem to be some inconsistencies between responses to parts A and B of the PCQ. Only one patient in the control group reported an inpatient stay in part A. However, in part B, a patient from the treatment arm also filled in their travel information for an inpatient stay. No GP/nurse visits were reported in part A, whereas two patients reported travel information for such visits in part B. This type of inconsistency may occasionally occur as a result of a reporting error or problems in the questionnaire design. One of the aims of a feasibility study is to examine and improve processes before conducting a definitive trial. When a larger sample is achieved, such inconsistencies are likely to have an insignificant effect on the results. In this study, we do not have an adequate sample size and the inconsistencies highlight how unreliable it would be to conduct analysis based on such data.
The aim of the PCQ used in the feasibility study was to gather information on the pattern of patients’ use of health services to help determine how the relevant data should be collected in the definitive trial. However, the small sample size did not allow this to be done.
Outcome measured by utility
The response rates of SF-36 are presented in Table 15. Two participants in the intervention arm were lost to follow-up because of a staff issue at one of the participating sites.
| Follow-up time points | Control arm (n = 3) | Intervention arm (n = 3) |
|---|---|---|
| Baseline | 100% (3/3) | 100% (3/3) |
| 3 months | 100% (3/3) | 33% (1/3) |
| 6 months | 100% (3/3) | 33% (1/3) |
| Average | 100% | 55% |
Table 16 presents the utility scores calculated from the SF-6D derived from responses to the SF-36. Average utility at each of the time points (baseline, 3 months and 6 months) and quality-adjusted life-year (QALY) gain over the 6 months period were presented. QALYs were calculated using the area under the curve method (1) with three time points of baseline, 3 months and 6 months. The area under the curve method calculates QALYs by multiplying the QoL with the duration of the life defined by the time points that form the curve. As with the case of resource use data, the sample size for calculating the utility score is too small to draw any meaningful conclusion or make any comparison between trial arms.
| Time points | Control (n = 3) | Intervention (n = 1) |
|---|---|---|
| Baseline | 0.84 | 0.39 |
| 3 months | 0.82 | 0.45 |
| 6 months | 0.70 | 0.38 |
| QALY | 0.40 | 0.21 |
Discussion
The health economic component of the study developed and tested the health economic data collection tool, the PCQ. We also collected information on resource use of the intervention from CRF and on patients’ HRQoL from the administration of the SF-36. The aim was to examine the completeness and ease of collection of the above data, assess feasibility and inform the design of a future definitive trial. Owing to the early termination of the trial, only six patients were recruited and we obtained analysable information on only four out of the six patients. As a result, the above aim could not be achieved owing to the inadequate sample size. However, it is not an indication of the feasibility of the health economics component, as the early termination of the study was due to other reasons that led to recruitment issues. Although we have not been able to assess feasibility of the health economics component, we have developed a workable health economic data collection tool and, based on the data we have been able to collect, it is reasonable to assume that relevant health economic data could be collected should the trial be able to recruit participants following any critical lessons learned from this study.
Parallel qualitative study
Introduction
The purpose of the parallel qualitative study within the SURAB trial was to explore the barriers to, and facilitators of, participation in the trial. Embedded qualitative studies are often a standard component within trials, in particular in feasibility and pilot trials. They can shed light on often inexplicable findings/experience that quantitative data cannot. 50 To assess the feasibility of a definitive trial, we planned to interview a mix of patients in order to address and find solutions to issues around recruitment and retention. We aimed to conduct in-depth face-to-face interviews with patients who:
-
consented (five in the ablation group and five in the active surveillance group)
-
declined (five)
-
withdrew following randomisation (five).
There was also a plan to interview recruiting staff about their views on recruiting to the trial, the trial patient information and processes, any cases in which they were unwilling or unhappy to recruit eligible patients, and explore why and whether or not they considered that there had been a change in their clinical equipoise from first agreeing to be part of the trial.
Aims and objectives
To explore the barriers to, and facilitators of, participation in, and conduct of, the trial.
Objectives
To explore patients’:
-
views on aspects of the informed consent process including method of approach and time to make a decision
-
views on the content and amount of written and verbal trial information and their understanding of it, what they believe to be the purpose of the trial, what the two ‘treatment’ arms involve, what are the risks and benefits and what is required of them
-
reasons why they declined/agreed to participate and under what circumstances would they participate/not participate
-
experiences of participating in the trial, including their views on the arm they were randomised to, whether or not they underwent any further treatment and completion of the outcome measures (SF-36, STAI, FACT-G)
-
reasons for not continuing in the trial until final follow-up.
To explore recruiting staff’s views on:
-
participating in, and recruiting to, the trial
-
the trial patient information
-
cases in which they were unwilling or unhappy to recruit certain eligible patients to the trial.
In this section, we outline the findings of the qualitative parallel study: the barriers to, and facilitators of, the conduct of SURAB; specific facilitators raised by interviewees; interviewees’ views on the early closure of SURAB; and the key issues to be addressed in a future full trial. In Discussion, the key barriers to the trial are summarised.
Participants and recruitment
Potential interviewees for the qualitative study were patients approached to participate in the SURAB feasibility study and recruiting clinicians from the nine participating sites. The sites were Newcastle, Leeds, Bristol, Glasgow, Southampton, Oxford, St George’s University Hospital London, University College London and Stevenage.
Patients
When patients were approached to participate in the trial, they were also asked if they would be willing to be contacted by a member of the research team (JL) to discuss taking part in an interview. This invitation included those who declined to participate in the main SURAB trial. At that point, they gave consent to be contacted by a member of the research team about taking part in an interview. The names and contact numbers of patients were passed via the clinical trials unit to the qualitative researcher (JL). Patients were contacted by telephone to further explain the study, answer any questions they may have and set up a time to conduct the interview if they were happy to proceed. Consent to participate in an interview would be obtained at the time the interview was conducted.
Recruiting clinicians
At the study launch and during site initiation visits, clinicians and research nurses responsible for approaching and recruiting patients to the SURAB trial were informed that they would be asked to participate in an interview to explore their views around trial feasibility. A list of recruiting clinicians and research nurses was obtained from the NCTU that was managing the trial. At the end of the trial, the clinicians and research nurses from this list were approached via e-mail to request that they participate in a face-to-face (Newcastle-based staff only) or telephone interview and agree a date and time for the interview.
Methods
Qualitative methods were used, namely in-depth one-to-one interviews. Logistically, it was more appropriate to conduct telephone interviews with patients and recruiting clinicians outside of the North East region.
Data collection
Interviews were digitally recorded with the permission of the interviewees and transcribed verbatim. A topic guide for the interviews was developed from discussions with the wider team and from literature around trial participation. Although a topic guide was used, interviewees were encouraged to speak freely about any other issues relating to the feasibility study. The guide was revised as new issues emerged in consecutive interviews.
Analysis
Transcript data were managed using NVivo, version 10 (QSR International, Warrington, UK). A thematic framework was derived from the data through a process of data familiarisation to look for emergent themes. The framework was tested and refined and data were coded using the final framework. Data were analysed using a form of constant comparison method. 51
Findings: patient participants
In one site, apparently none of the patients approached for the trial agreed to be contacted about the qualitative substudy. At two other sites, a total of 10 patients agreed to be contacted. Unfortunately, no telephone contact information was provided by the recruiting site for two patients and it was not possible to obtain these details. The remaining eight were contacted and took part in an interview (Table 17), all but one by telephone. Four had declined to take part in the trial and, of the four who agreed to take part in the trial, three were randomised to active surveillance and one to ablation. The four interviewees randomised to active surveillance or ablation remained in the trial until it closed.
| Declined | Agreed | |
|---|---|---|
| Randomised to active surveillance | Randomised to ablation | |
| 4 | 3 | 1 |
Approach to participate in the SURAB trial
The initial approach to take part in the trial was made by a consultant urologist. Those who agreed then met with a research nurse. All of the patients who were interviewed were happy with the approach to take part in the trial and, when applicable, the consent process. The timing of approach about the trial was said to be appropriate. Even considering the fact that the discovery of the tumour was incidental, no one thought it unreasonable to be asked about the trial at the time, or soon after, they received the diagnosis. They all felt they had been given sufficient time to make a decision; in some cases, the discussion about the trial had spanned three outpatient appointments. In a number of cases, the patient had a spouse or family member with them.
Interviewees were positive about both verbal and written trial information, finding both clear and easy to understand. They appeared to understand that they could withdraw from the trial at any point. The patient information booklet was thought to be helpful, and some had referred to it throughout the trial:
If you go to the middle section, it’s more or less a summary that explains it all. So now and again I have a look at that.
Pt03
When asked what they understood to be the purpose of the trial, no one mentioned it was to determine the feasibility of a full trial. Their focus tended to be on the treatment and management of the tumour with comments such as ‘no one knows the best way to treat it’. Only one interviewee misunderstood the purpose of the trial:
It’s to see if it’s frozen if it keeps growing.
Pt01
Randomisation is often said to be difficult for potential trial participants to comprehend, but all interviewees were clear that, if they agreed to take part, the group they were allocated to would be determined at random, by a ‘toss of a coin’. The trial was perceived to be of low risk and a few patients mentioned that the only risk they were aware of was related to the follow-up biopsy and a small chance of bleeding from the biopsy site. There was no perceived benefit, apart from the view that being part of a trial meant ‘better treatment’.
It did explain that there were two options, being left alone and monitored or the ablation and it probably wouldn’t benefit me as such but would benefit others in the future.
Pt02
Interviewees who agreed to the trial understood what was required of them in terms of treatment/management and follow-up visits related to the trial. There were no suggestions for changes or areas of improvement to the approach and consent process.
Decision to participate in the SURAB trial
All interviewees had discussed participation with family and, in one case, their GP. In most cases, their families were important in the decision-making process:
To be honest I was undecided at the time . . . one surgeon told me that I could live for ages with it and I could die with . . . the tumour because it was slow-growing which is encouraging . . . but my family they had spoken to people who had had the operation [ablation] and had been successful and they encouraged me to have the operation.
Pt04
One interviewee, based on a conversation with another patient who had undergone a total nephrectomy, decided that he would prefer ablation. He agreed to the trial and was randomised into the ablation group, but he said that he would have seen the trial through to the end had he been randomised to the active surveillance group. Another said he knew of people who had been on active surveillance for many years without any detrimental effect and this was a factor contributing to his decision to participate.
Attitudes to clinical research generally were explored and, although none of the interviewees had taken part in research previously, they were clearly positive about it and its importance. There was a perception that being part of a trial meant a slightly better standard of treatment:
My father had a heart thing and he took part in a trial and you know people have said if you take part in a trial you get . . . very good treatment . . . if I hadn’t had the [comorbid condition] I would have 100% gone down the trial road.
Pt02
For those who agreed to participate in the trial, the main reasons were altruistic, to help others in the future or because they thought highly of the consultant who approached them about the trial:
Unfortunately I lost my wife last year through cancer so . . . I thought that I should take part in this because if it helped me it would help other people in the future.
Pt02 surveillance
They say it’s for the future and I’ve got four great-grandkids, I’ve got five grandkids you know so if it helps somebody in the future, fair enough.
Pt01
This was often coupled with other factors such as the opportunity to have better treatment or a higher level of monitoring. Also mentioned were the fact that it was a low-risk trial and concerns about the alternative interventions such as surgery and risks of infection:
To be perfectly honest because I know at the end of the day they’re not going to say ‘Alright that’s it’ . . . [the information] explains that if you need any treatment of any sort after the study is finished you get it. It’s like Big Brother in a way.
Pt03
Reasons for declining to participate in the trial varied. One person, mentioned earlier, would have been happy to take part and be randomised but his family persuaded him to request ablation. Another person who had a comorbid condition that warranted continuous treatment thought that the additional visits would be too onerous:
I said . . . I would want to get rid of the cancer if possible rather than be monitored and be backwards and forwards.
Pt02
Only one person stated that they did not feel comfortable with the risk of being randomised to the active surveillance arm and that informed their decision to decline the trial. They were not happy to continue without treatment knowing that they had a cancerous tumour on their kidney and wanted to have the tumour removed immediately.
Views on the arm randomised
Of those who agreed to participate in the trial, none was disappointed with the arm they were randomised to. Prior to randomisation, they did not have a preferred arm and said that they would have seen the trial to its conclusion if they had been in the alternative arm. They were confident that they would be well cared for in either arm. One person also cited as a reason for agreeing the very slow growth of tumour mentioned in the trial information and the fact that the trial was voluntary and participants could withdraw at any point and undergo treatment if in the active surveillance arm.
Trial follow-up and outcome measures
The follow-up visits at 3 and 6 months after random allocation in the active surveillance arm and after treatment in the ablation arm were considered reasonable and not onerous. One interviewee commented that after agreeing to participate in the trial he had thought ‘Oh God, what have I let myself in for’, but, in effect, felt that it was an ‘easy’ trial to take part in and there was ‘not much involved’ (PT08).
Trial participants were required to complete three questionnaires at baseline – (1) a general health survey, (2) health states and QoL and (3) level of anxiety – and the same again at 3 and 6 months with the addition of a PCQ. Interviewees did not find these problematic, but one person commented that the level of anxiety measure was difficult to complete as they could not determine if the stress they were feeling was induced by their work situation or the presence of the tumour.
Findings: recruiting staff
Response to request for interviews
Nine PIs and 16 research nurses (sometimes more than one per site) were e-mailed from a list provided by the NCTU (Table 18).
| Site | Bristol | Glasgow | Leeds | Newcastle | Oxford | St George’s | Southampton | Stevenage | UCL |
|---|---|---|---|---|---|---|---|---|---|
| PI | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ |
| Research nurse | ✓ | ✗ | ✗ | ✓ | ✗ | ✓ | ✗ | ✗ | ✓ |
The site PI and research nurse in Southampton did not respond to requests to participate in an interview; therefore, their views on the feasibility of the SURAB trial in their site are unknown. In Oxford, the research nurse responded to say that the study had not progressed beyond the site initiation visit and, therefore, she felt unable to comment; no response was received from the PI. In Leeds, Glasgow and Stevenage, the research nurses did not respond to e-mails and, therefore, only the opinions of the PIs are represented. In University College London, the PI did not respond to e-mails and, therefore, only the views of the research nurses are represented.
A total of six PIs and five research nurses representing seven sites were interviewed.
Themes and subthemes
The aim of the parallel qualitative study was specifically to explore the barriers to, and facilitators of, the conduct of the SURAB trial. Interviewees’ opinions on the essential attributes of a future trial in terms of design and process were also explored.
What were barriers to the conduct of the trial for some recruiters were not barriers (or were even sometimes facilitators) for others. Therefore, as a constant comparative method of analysis was used, in which similar as well as contrasting ‘deviant’ cases are explored, the barriers and alternative views have been reported together. When interviewees have explicitly voiced what they considered to be facilitators in the conduct of the trial, these have been summarised in a separate section.
A number of themes, categorised broadly under barriers and facilitators, were identified from the interview data. Themes identified as barriers were linked to trial processes, trial design and ‘selling’ (a term used by a few of the interviewees) the trial. Subthemes were identified within trial processes, trial design and ‘selling’ the trial. The themes and subthemes for barriers to, facilitators of and features of a future trial are mapped in Figure 7. The arrows illustrate where the themes and subthemes relate to the key elements to address in the design and conduct of a future full trial.
FIGURE 7.
Themes and subthemes for barriers to, facilitators of and features of a future trial.
Barriers and facilitators: trial processes
Issues around delays or difficulties that had an impact on when recruitment started or the effectiveness of recruitment were raised. These were linked to processual factors such as completion of documentation at sites, sites and trial pathway and management issues linked to the clinical trials unit and trial team.
Bureaucracy
Commencing the identification, screening and recruitment of patients for the SURAB trial had been delayed in four sites. In two sites, this was due to contractual issues and in the other two sites to processing the paperwork. However, in one site, the team had recovered the lost time and reached their projected recruitment target by the time the trial was closed. Another site also had a change of PI, which caused more delays such that there were ‘only 2 weeks to identify and approach patients’ before the trial closed.
Oxford did not proceed beyond the site initiation visit but, as it did not take part in the qualitative study, the reasons for that are unknown.
Sites and trial pathway
The patient route into the trial was a major factor in the ease or otherwise of recruitment to SURAB. Two of the recruiting sites formed the hub of a ‘hub and spoke model’, accepting patient referrals from hospitals around the region. In one of these sites, when assessing patient throughput and potential recruits at the design stage, these patients were believed to be eligible as they were referred to, and discussed, in the hub multidisciplinary team (MDT) meetings. From a governance perspective, it was considered a breach of protocol to approach patients from these ‘spoke’ hospitals as they had not been set up as patient identification centres (PICs). It was not until the trial was under way that the research nurses realised that without this group they would struggle to find the required number of patients in the allotted time:
It maybe could have been better constructed in terms of this role of spoke hospitals in all of this. A really decent proportion of people with small renal tumours come from spoke hospitals . . . maybe, they should’ve been set up as PIC sites from the very beginning.
RS01
In the other site, the PI felt that it was too far for patients identified in the spoke hospitals to travel to the hub site to discuss the trial in person:
. . . half-way through we tried to set up these PICs where that could be done at the referring hospitals, but that never actually happened, it was about to be set up and then the trial got pulled, so we never actually got to a point where patients could be counselled at our referring hospitals.
RS03
One other issue that was related to the hospital site and the care pathway was patients who were identified and recruited in one hospital but who, if they agreed to participate and were randomised into the ablation arm, received their treatment at another site. The interviewee expressed this as a ‘logistical nightmare trying to co-ordinate the two sites’ and difficult in the set-up stage when clinicians responsible for completing documentation were based at different hospitals.
Trial team and trial management
Interviewees were generally positive – or rather non-committal (‘They are what they are’) – about the site initiation visits conducted by the NCTU. There were no suggestions as to how these might have been improved or any changes that could be made. Similarly, interviewees were, in the main, happy with the NCTU’s management of SURAB. Comments were that they were efficient and ‘very supportive and helpful’ and queries were ‘always responded to really quickly’ (RS08). However, at some sites, there was a feeling that the trial had, at times, been managed too stringently. One example was when the nominated PI for the site changed, the trials unit had requested a substantial amount of paperwork to be redone and this delayed the start of recruitment:
Normally if we change PI that PI will just say ‘I’m happy with whatever the old PI did’ but we had to do new file logs and we had to get a new histopathologist. All this stuff takes time and it delayed us . . . I understand that there is some administrative stuff that does need doing when you change PI but it just seemed to be that everything needed redoing.
RS05
Another view was that the experience of the recruiting team was not taken into account by the trials unit and they were not given credit that they could do their job and use their initiative. When the trial had been briefly mentioned at the initial consultation with the patient but their case had yet to be discussed at the MDT, one team would ring patients after that discussion to request that they come back to clinic and ask if they could post out the trial information in the meantime. This was stopped by the trials unit as the trial did not have NHS REC approval to ring patients:
I think it’s probably a bit of a straitjacketed way of doing things, because they can’t possibly tell you every single circumstance of what they’d like you to do . . . To me, the safety issue should be paramount, rather than maybe fretting that you’ve telephoned someone. Or at the very least, make that absolutely stone clear in your protocol to the point where you say, ‘And, yes, do this, and we also do not want you to do this,’ if that’s the case. [Emphasis indicated by interviewee.]
RS01
There were criticisms about the design of the screening log, which was considered to be ‘not fit for purpose in a clinical setting’. This was because of the inability to quickly determine, for example, how many patients had been seen and the reasons as to why they were not eligible:
If a doctor comes in and says, ‘How many have we screened?’ I’m not going to start sorting through 30 A4 pages. . . . we had to make our own Excel screening log . . . And so you can quickly, at a glance, look at them. I’ve given them each a code, my own one to four code, you can sort . . . They’re a requirement, to go to [Clinical Trials Unit] so we do complete them.
RS02
One person also commented that they suspected that not all centres were completing the screening logs in the same way. They had viewed the numbers across the recruiting sites and thought that some were not logging every patient with a tumour size of < 4 cm.
During the course of the trial, there were three changes in senior trial managers and two changes in trial manager and assistant trial manager. Some interviewees alluded to problems early in the trial with delays in replacing these staff, which resulted in ‘a period of inertia’ but that, once this was resolved, things were ‘excellent’ (RS06).
Few interviewees raised the issue of the role of the CI within the trial. Most appeared to consider the problems around recruitment to be something that required a group solution, for example sharing best practice across sites, or fundamental changes to the design. However, one interviewee commented that the CI could have been more ‘hands on’ and had a greater presence across the trial sites:
. . . you have to enthuse people. You have to engage them. You have to cheer them on. You’re the leader on it. You cannot become an invisible person in the background, and expect the trial managers to run it for you. It doesn’t work.
RS07
Barriers and facilitators: trial design
As might be expected, not all of those interviewed were completely happy with the design of the trial. Concerns around trial design related to issues initially with the trial protocol; inclusion and inclusion criteria, namely the need for biopsy and clinician views on whether or not the patient was suitable for the trial; and approach to participate, which includes identification and selection for the trial.
Trial protocol
There were issues around the trial protocol for a number of interviewees. It appeared that some, but not all, PIs had an opportunity to contribute to the protocol; some said they had not been party to discussions or had been asked when it was too late:
We had really good feedback from things that we wanted changed, for instance I think the ASA grades right at the beginning we thought excluded too many patients and that got changed.
RS03
Oh, I think the only issue was that it felt like the protocol had been set in stone before we were asked our opinion on it . . . It can be difficult to change it.
RS04
This lack of early engagement with some of the PIs was said to be the reason the issues around the radiation doses in the documentation submitted to NHS REC were not identified sooner. There was also the problematic issue of biopsy which, for some PIs, was never really resolved, even though they began recruiting:
The protocol involving a biopsy was something that several of us raised concerns about early in the process, but it seem those concerns were not being listened to.
RS04
One other issue was about the amount of what was considered unnecessary work documenting every patient with a tumour size of < 4 cm, even those ruled out as eligible on other criteria:
Recording every lesion that is under 4 cm that is flagged up in the MDT is a lot of work. Even with the ones that the surgeons feel are totally inappropriate, so they would want to give surgery straight off, you would then have to find out what’s happened to the patient. It’s more work than I thought it would be . . . I only did it for a couple of weeks, so the other people must have found it hard.
RS05
Inclusion and exclusion criteria
The major issues around eligibility for the trial were (1) the need for a biopsy to confirm the presence of renal cancer and (2) patients whose clinician did not consider them suitable for the trial. These are discussed in the following subsections.
Less frequently mentioned was size of tumour. For patients to be eligible for the trial, the tumour size had to be < 4 cm. One interviewee expressed frustration that they had a number of patients whose tumour was just over the size threshold [‘We missed a few just by the size. You’d have a 4.2 or something’ (RS08)], and another had been concerned about approaching patients when the size was at the ‘absolute limit’ (RS02), particularly as, if it were any bigger, it would be unlikely that they would have been offered active surveillance. One other interviewee considered the inclusion only of patients with new presentations of small cell kidney cancer problematic. They felt that, had this been extended to include current patients on active surveillance whose tumour had grown, the pool of potential recruits would have been much larger.
Biopsy
One of the inclusion criteria was a confirmed cancerous tumour, which necessitated a biopsy before patients could be randomised to SURAB. Not all of the participating sites routinely biopsied patients with a small cell kidney tumour, which meant a change of practice for the purposes of the trial. At these centres, consent to the SURAB trial was obtained before biopsy but, if it demonstrated that the growth was non-cancerous, then the participant was withdrawn from the study and returned to standard care. Waiting for the result of the biopsy complicated the process of recruitment and made it lengthier. Tracking when the biopsy results were available and when the patient was attending again (so they had the opportunity to speak to them about the trial) was problematic and may have resulted in eligible patients being missed:
It was quite lengthy and quite difficult . . . It was a case of watching for when the biopsy results came out, when their clinical appointments happened, and the timing of it.
RS08
Biopsies were sometimes carried out in a different department by colleagues who were not always core to the MDT, and this meant that ‘it was not a very smooth pathway’ (RS04). In addition, biopsies carry a risk of a bleed, and this was of concern when patients were taking anticoagulants. A bleed can also have a major impact on the management options:
. . . they had a bleed because of the biopsy and that immediately affects the visibility of the tumour . . . you can’t offer cryotherapy and the next thing is nephrectomy. So going from a situation where you could have just watched the patient you actually have now got to take out the whole kidney.
RS06
Although most of the comments were about the biopsy to confirm a diagnosis of cancer, one interviewee was unhappy with the requirement, as part of the trial protocol, for a biopsy at 6 months for patients randomised to the ablation arm. The comment was made that this concern was listened to but ‘not listened to as much’. The need for the 6-month biopsy in the ablation arm patients did not become an issue due to the early closure of the trial:
There was some concern about biopsying again after the cryotherapy to check that the disease had gone. We don’t do that, we rely on CT scans to see if disease has gone, so our radiologists . . . didn’t ethically feel that they wanted to risk biopsying . . . after ablation because it’s not that easy to biopsy that tissue . . . and also you would never know whether you were biopsying the right bit to see whether disease was active anyway.
RS03
Patient’s clinician does not feel would be suitable for trial
Patients were excluded if their physician considered that they would not be a suitable candidate for the trial. On the eligibility form, the example reason given was ‘due to concomitant disease’. This was also interpreted by the sites recruiting to the SURAB trial as excluding those patients who, under non-trial conditions, would not be deemed suitable for active surveillance and ablation. This was the most common reason why patients screened for the trial were deemed ineligible (45/92; Figure 8), followed by the presence of comorbidities (21/92; see Figure 8). Twenty-seven out of the 119 ‘ineligible’ patients in the Consolidated Standards of Reporting Trials (CONSORT) diagram were actually not approached because the trial closed early.
FIGURE 8.
Reasons why patients were deemed ineligible for SURAB.
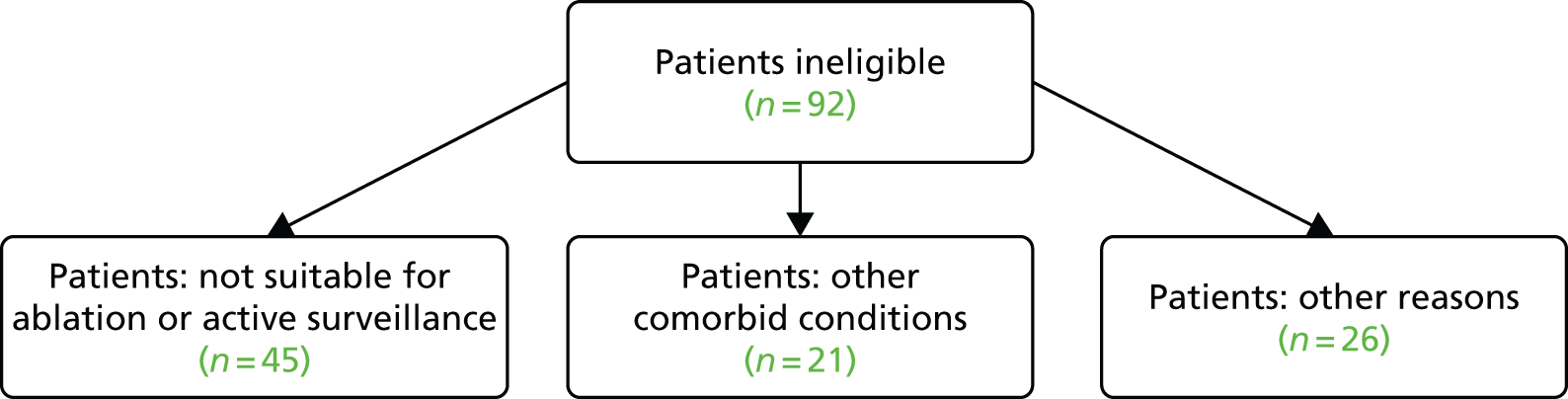
Despite the inclusion criterion of age ≥ 18 years, recruiting clinicians were not comfortable offering the trial to relatively young patients. One PI stated that these patients want ‘closure’; they either want to be discharged from hospital if the tumour is found to be benign or have it quickly removed and ‘they don’t tend to want to be followed up for a very long period of time’ (RS03). In the MDT meetings, it was often decided that it would not be best practice to put younger patients on active surveillance, and, therefore, they would not be offered the trial:
You say ‘Yes, it’s kidney cancer and we’re going to watch it’, that’s fine if you’re 68 or 78 because there’s a very real chance that it will not cause any problems during your lifetime . . . in a 40 year old . . . that’s going to catch up with you at some point and it’s going to need some treatment at some point . . . we’re going to have to do CTs every 6 months for the next 30 years. Is that realistic? No probably not. Is it ever going to go away? No. ‘Am I always going to have this cancer in my kidney doctor?’, ‘Yes’. Can you assure me that it isn’t going to spread?’, ‘No’.
RS09
As it was generally believed to be wrong to recruit younger or fitter patients to the trial and offer random allocation to ablation or active surveillance, the SURAB trial was felt to be a trial primarily for a much older population: ‘definitely a large cohort that we see who are 70 plus, those two options are fine’. However, some clinicians would prefer not to offer ablation to older, less fit people:
When I see an old person who has got co-morbidities and I’m thinking ‘surveillance’, I’m hoping I’m never going to have to treat them.
RS06
One interviewee explained that the patients who present with renal cancer typically fall into two main groups: elderly patients who would not be considered for treatment and would normally be offered active surveillance and people fit for surgery ‘who automatically wanted surgery’ (RS06). There was a view that, particularly in older patients with comorbidities, clinicians would hope to never have to treat them:
Were there any patients that you were unwilling or unhappy to recruit?
Yes . . . we had patients who were too old and frail we had patients who were too young and fit. We would offer typically the younger patients partial nephrectomy and the older patients we wouldn’t be happy to treat them at all.
Despite the mention of a large group of patients in their seventies who would be ideal for SURAB, most interviewees believed, at the time of agreeing to recruit to the trial, that they had underestimated the potential numbers who would be eligible:
If you look at the numbers of tumours that we see less than 4 cm, whatever centimetres, then yes, on paper you think ‘Well even if we pick up 25% of them we’re there. We’re laughing. But for each particular one, it’s quite a big sell for various reasons.
RS09
Although there was no shortage of patients with renal masses (three to five per week was the number quoted by one interviewee), very few (one per fortnight) were in the older age group that clinicians consider suitable for active surveillance. There was a recognition that, in reality, the pool of potential recruits is quite small, if the trial is deemed to be suitable only for older, fitter patients, and this was something not thought of before the trial began:
So we find that patients, if they’re fit enough, they want to have a treatment, if they’re not fit enough, they don’t want anything and the number that are in between that could be randomised is quite a small number.
RS03
Patient identification and approach to participate
At the time of applying for funding, it was proposed that eligible patients would be identified in the MDT meetings. This appears to have happened in most of the sites. This route to identify patients for the trial worked particularly well in one site as patients who were discovered by the radiology department incidentally to have a SRM were referred directly to the MDT rather than to a clinic:
So we would go to an MDT, say with our anticipated 20 patients or something and there’d be another five or more that the X-ray department have picked up.
RS06
The MDT was considered key because of its composition of specialists and the opportunity to fully discuss each patient. However, one interviewee mentioned that, without someone at the MDT championing the SURAB trial, there may sometimes have been a lapse in identifying eligible patients:
I think it’s fair to say that when I turned up to the MDT then SURAB had a better chance of being mentioned, and I suspect that if I didn’t turn up to the MDT then it would not have been on the radar.
RS04
In one site, it was mentioned that the MDT in general is ‘quite pro-treatment’ and there was a reluctance to observe patients with a kidney tumour ‘unless they’re fairly frail and they’ve got lots of co-morbidities’ (RS03). Comments were made about the problem of selection bias, particularly with surgeons recruiting to a trial without a surgical arm. A few PIs questioned whether or not other recruiting clinicians were in equipoise about the trial and if there was a ‘genuine belief that we don’t know if we’re over-treating these lesions’ (RS10):
If you truly believe that we don’t know the right treatment, then you believe in the trial.
RS10
It was considered to be beneficial to the patients who are being approached about the trial for the recruiting clinician to be in equipoise and truly not know, and to be comfortable admitting they do not know, the best way to treat these tumours:
Having faith in the person that you’re hearing the information from makes a huge difference as well. So yes, perhaps we don’t know quite what the answer really is, whether it’s better to ablate it, or whether it’s best to watch it. But I think when you hear that from somebody who gives you the information with good authority and that you have confidence in.
RS11
Following discussion in the MDT meeting, the plan was for these patients to be contacted by the centre nurse lead or nurse specialist, who would invite them to participate in the trial. The same person would then explain the trial to the patient, provide them with the trial information leaflet and answer any questions they may have. The patient would be encouraged to take the information leaflet home and discuss it with family and friends and arrange a suitable time for the second meeting. The centre lead nurse specialist would ask the patient to sign the consent form agreeing to participate in the trial.
In reality, although potentially eligible patients were identified and discussed at the MDT meetings, the initial approach was made by the recruiting clinician, and, in some centres, the patient will have already consulted a urologist. Afterwards, or in some cases at the same time, patients would meet with the research nurse to discuss the trial further and give their consent if they were willing to participate. The ‘best’ system was said to be when the clinician sees the patient and explains the trial and then hands over to the research nurse at the end of the consultation to answer any further questions and obtain consent if possible:
I think just sometimes, with all the best will in the world, you try to make sure you’re at clinics where the patients are going to be, but occasionally, mishaps will happen, and you maybe miss the patient in clinic. The clinician will discuss, and you’ve maybe missed a point at which you could have seen them. But that happens with every trial. It’s just pressure of work, pressure on the systems.
RS01
The presence of the research nurse at the clinic at that time was also thought to be a prompt and reminder for the clinician to mention the trial as they may occasionally forget. The involvement of the research nurse was considered to be important, and some interviewees wanted them to be present at the clinics and to be more proactive in promoting the trial and approaching patients about participation:
[Name] was just very reticent to get in there aggressively, and say, ‘Here, talk to me about this trial’ . . . I feel like we missed the opportunity to come up with a simple way of saying, ‘All right, patient identified, here’s the pathway’ and the way we’re going to do it, sort of acted out ahead of time, say, ‘All right, what I’m going to say is this, and then you say that.’ And just have a bit of coaching, so that we have a better chance of persuading people.
RS04
At one site, the recruiting team cited ‘good teamwork’ and a comprehensive system to identify patients for the trial and monitor them through to the point of consent. This relied not only on those patients identified through the MDT but also those attending the outpatient clinics. As a result of this system, the recruiting team felt that they had ‘complete capture of the small renal masses’ (RS06). The research nurse in this site was present when the PI explained the trial and would speak to them afterwards to answer any further questions:
We would comb through all the patients that are coming into clinic . . . to see, did they meet any of the eligibility criteria? . . . Then, on the day of the clinic itself, . . . I would put a note on the front, to flag up that this patient is to see this particular consultant. I would speak to him beforehand . . . so that he would have that in his mind as well.
RS11
The team would then track the patients’ follow-up appointments so they could be approached again when they returned to clinic.
Barriers and facilitators: ‘selling’ the trial
Four factors were identified that had an impact on recruitment to the trial. These were (1) patient route into the trial, (2) clinician as recruiter, (3) patient preferences and (4) trial intervention options.
Patient route into the trial
The patient route into the trial clearly had a bearing on the ease or otherwise of explaining the trial to patients. At one site, patients had often seen a urologist and discussed treatment before they were referred to the MDT. If they were deemed eligible for the SURAB trial then they met with the clinician recruiting to the trial. The fact that the patient had already had that discussion about their management made it very difficult for the recruiting clinician to offer the trial and convey the uncertainty around ablation and active surveillance. This required the recruiting clinicians, in the face of the patients having already been told what is the ‘best’ treatment, to admit that they do not know what the best treatment is. This resulted in the recruiting clinician feeling that patients would perceive him as an incompetent doctor:
They’ve been told that they’re going to have [treatment]. To then tell them ‘Maybe you don’t need it’, you can just see in their eyes . . . ’What’s he talking about? . . . people don’t like that . . . They want that doctor before who says ‘This is the right treatment!’ This guy, here, he doesn’t seem to know what the best thing is!
RS09
When the patient had not consulted with another clinician before speaking to the PI, the process appeared to be much easier. Although patients may themselves have explored treatment options, the PI did not have to appear as if they were going against the suggested treatment of another clinician:
We have quite a centralised service . . . in that we don’t have other surgeons feeding in if people happen to want ablation. It’s all done out of one centralised clinic, where there’s a surgeon, a radiologist and a nurse specialist all working together.
RS10
Clinician as recruiter
There is an assumption that clinicians will naturally be good recruiters. However, this may depend on the complexity of the trial and how it fits with the usual clinical pathway. Some interviewees questioned the suitability of surgeons recruiting to SURAB:
I think it’s fair to say that SURAB is not a trial that captures surgeons’ imaginations . . . we surgeons get interested and excited about patients who might need surgery [laughs], that’s just our nature. So if the patient’s not being offered surgery then we don’t really see it as core to our role to be involved in recruiting to a trial.
RS04
It was clear that some PIs struggled to deal with a conflict between the role of recruiter and role of clinician; the role of recruiter was often confounded by their clinician role of doing ‘what they do best’:
The problem is, once the patient gets into a room with me, they will ask me, ‘Well, what should I do doctor?’ . . . it’s easy for me to offer treatment, because that’s what I do and that’s what people sort of expect . . .
RS04
Dealing with patients’ expectations that the clinician will advise them on the most appropriate treatment led to a difficulty for some in conveying the uncertainty around ablation versus active surveillance:
Now you could say ‘OK, well we’re just going to observe it for a few months then you can decide to change it’. Yes, but they are coming to you as the source of the knowledge, ‘You’re the expert, tell me what I should do’.
RS09
In this study, there was no training or script provided for recruiters to explain the trial. One interviewee said they had written a script for the purposes of the SURAB trial but one that had been designed for use across all sites would have been helpful. One interviewee commented that there was a greater emphasis on the trial paperwork and less support and training in what is a critical component of a clinical trial – recruitment:
We tend, at the beginning of a trial, to focus on the details of the protocol, . . . that everybody knows what forms to sign off, . . . and actually don’t do the sales. And I think you need to work on a sales pitch, to try to get people involved.
RS04
Some felt that their recruiting skills improved as time went on and there was a definite ‘learning curve as how to do it, and you do get better’:
Everyone was saying ‘You do get better as time goes on’. That means there is a technique or a style that you need to learn, and none of us knew what that was.
RS06
The recruiting staff had telemeetings a few times over the duration of the trial period. This was thought to be a perfect opportunity to discuss how the trial was going but because these were not face to face this was seen as a barrier as ‘if you are all in a room you can express yourself openly’. It was suggested that this time could have been used for the recruiting staff to meet face to face and spend time in role play to highlight ‘what the obstacles are’ (RS06). Role play with a script was mentioned by a few interviewees as something that would have been helpful and a more proactive way of managing the problem: ‘We just seemed to be very passive about the whole thing, instead of taking charge’ (RS04).
One positive outcome of the telemeetings, albeit unfortunately halfway through the recruitment process, was a PI mentioning that the key selling point of the trial was the short period of active surveillance they were asking patients to consider. This had led one site to change its approach to patients about the trial:
. . . that certainly changed the way we counselled patients. I don’t think this was emphasised enough at the beginning . . . because I think if we’d pushed a bit harder with patients that they were going to be observed for six months and then after that they could come out of the trial and go for treatment . . . that might have been something that would have been a bit more attractive for recruitment.
RS3
At a later point, there was a move to improve recruitment by trying to implement methods used in the more successful sites, but at that point it was believed to be too late:
We only shared at the end of the trial period you might say ‘best practice’ for recruitment and it would have been good to talk about that at the beginning.
RS09
Patient preference
It was stated to be a common occurrence for patients who attend the clinic to have a clear idea of what they want in terms of treatment or management of the tumour. A number of patients declined to take part in the trial as they already had decided on treatment. Out of 36 patients provided with study information, 26 declined as they had a preference for ablation (n = 16) or active surveillance (n = 10). In situations in which patients had decided on ablation, bringing in the possibility of active surveillance and then the existence of the SURAB trial was particularly difficult:
. . . if they’ve said ‘I want to have the radio-frequency ablation’ then you can mention ‘OK, well radio-frequency ablation is an option in these circumstance and another option . . .’ . . . you have got to introduce the option of observation and then you’ve got to jump from that to get them to agree to be randomised between the two. So it’s quite a big ask.
RS09
Some patients, even if they had not specifically decided on a particular treatment, wanted something done, which, again, made it difficult to convey the trial with an active surveillance arm as a viable option:
I’m not good at sales, apart from convincing people that I’m a surgeon they can trust and I think filling this trial, it was a difficult sell, . . . they come wanting treatment for their cancer . . . So I think the observation was a difficult sell.
RS04
Further linked to the fact that the SURAB trial was offering treatment versus no treatment, one interviewee said that patients clearly fell into one or the other group with few open to either option:
Even when you had the discussion . . . there are some people who definitely want to avoid any form of intervention and there are some people who definitely want something done no matter what you say to them . . . those people obviously weren’t consented into the main part of the trial.
RS10
One interviewee commented that the population are more informed, can access information via the internet and have a greater awareness of what treatment is available. The information provided as part of the trial gives ‘patients more information than they’ve ever had’ (RS02), and more than they receive routinely.
Intervention options
The intervention options were problematic for some interviewees as they believed there was an imbalance between the two arms of ablation and active surveillance. It was also believed to be difficult to ask patients to agree to be randomised when one of the arms was ‘to do nothing’:
One could imagine a trial where you are comparing two different types of ablation and you say ‘We don’t really know which of these two is the best’ . . . with this one you’re saying ‘Look, I don’t know whether you’re going to get just watched, or we’re going to treat you’, which is quite a big leap between those two things.
RS09
However, other interviewees felt that if the emphasis was placed on the fact that the active surveillance was only for 6 months after which time patients would return to normal care and have treatment if they wished, then the trial could potentially be more appealing:
I think had we pushed a bit harder that they were just going to be observed for 6 months and then after that they could come out of the trial and go for a treatment. I think that might have been something that would have been a bit more attractive for recruitment actually.
RS03
A number of the SURAB sites had participated in a previous trial: CONSERVE [A Feasibility Study for a Multicentre Randomised Controlled Trial to Compare Surgery With Needle Ablation Techniques in People With Small (4 cm) Renal Masses. Soomro N, Newcastle Hospitals NHS Foundation Trust, and Wood J, CTU Newcastle University, 2014]. 52 CONSERVE was mentioned by a number of interviewees in comparison with the SURAB trial in terms of its challenges. On one hand, CONSERVE was believed to be ‘more attractive to patients and maybe clinicians because at least you had two active elements’. On the other hand, it was mentioned as being difficult to recruit to because of the risks with partial nephrectomy; in the light of this, the SURAB trial was initially thought to be an easier trial because the eligible patients would be ‘an older population’ but:
. . . people either come with a preconceived idea . . . or when you tell them what the options are they really like that option. If they really like that option then why would you consider going into something that might not give you that option . . . but might give you an option that you’re really not desperately keen on.
RS09
The alternative view was that there was not a problem with the two arms of ablation and active surveillance but that the target group for such a trial would be older, less fit patients. The short period of active surveillance (6 months) was believed to be an attraction as it meant delaying treatment for a relatively short period:
I couldn’t understand why people were struggling because the two options of surveillance versus cryotherapy and particularly with only a six month mandatory window, it was very easy, I thought, to . . . offer people the two choices. If it was ‘You’re going to be under surveillance until we felt it was necessary to treat’ and that could be years later then I’m not sure patients would have been that easy to randomise.
RS06
There was also the opinion that patients are more interested in trials in which they have a chance of receiving a new intervention that is not available as part of standard care. It was questioned why patients would bother taking part in the trial when they could choose whichever option outside the trial:
What I’ve experienced over all my years in research is once something is available outside the setting of a clinical trial your uptake will be, it used to be about 20% agree to be on the study, I think it’s lower now.
RS02
Facilitators
The key facilitators in response to a question around ‘What worked well in SURAB?’ were patient route into the trial, systems of patient identification and approach and treatment options. It appeared to be ideal if the first contact eligible patients had about their condition was with the recruiting clinician. Interviewees mentioned the benefits of ‘the blank canvas’: patients who, at the point of the trial being discussed, do not have preconceived ideas about which management option they would prefer. To maximise the likelihood of patients not being advised about management by another urologist, a centralised referral system and a ‘totally consultant-led clinic’ with the consultant being the sole clinician recruiting to the trial appeared to be the answer. Linked to this was a keen research nurse and a system to ensure eligible and potential eligible patients do not slip through the net:
There was complete capture from MDT through to [research nurse] who would tell me, ‘Three patients are coming this afternoon, look out for them.’ I knew their names, I would then speak to them, [research nurse] would come and help and we’d have a chat and that was it, you know.
RS06
The other facilitator mentioned was that the SURAB trial was a simple trial with clear inclusion and exclusion criteria, and the treatment options, particularly with only a 6-month active surveillance arm, were attractive.
Response to the early closure of SURAB
The majority of those interviewed expressed disappointment that the trial had closed early. This was the case whether they had just began to recruit, recruitment had picked up or the interviewees said they were doing well:
I have to say I felt hugely deflated because it was completely out of the blue . . . When it’s going well and the plug gets pulled it’s . . . hugely disappointing. It does make you feel that you’ve wasted a lot of time because it was quite labour intensive. I enjoyed doing it but was left with this feeling that it had been a complete waste of time.
RS1
Some interviewees were also puzzled by the early closure, considering that the aim of the SURAB trial was to determine if a future trial of ablation versus active surveillance was feasible:
Isn’t that the whole point of a feasibility study, to see does it work, does it not work? If it’s difficult recruiting to and you don’t get the information you want then the answer is no it doesn’t work.
RS11
Not seeing the trial through to the end was considered to be a missed opportunity; it was an important trial to conduct and a number of unanswered questions remained:
I don’t know where we go from here because I think this is a very, very important question that’s going to get more and more difficult to answer.
RS10
I still feel that there is a place for it . . . I was really disappointed that they pulled the plug . . . I think it is a good study and it could answer a lot of questions.
RS08
Opinions were voiced that recruitment would have improved across all sites had the study been allowed to run until the original end date. Most of the interviewees believed that a trial like that of the SURAB trial is feasible:
I’m sure it’s true that if this trial had been open for another year, the other places would have picked up. They would have started realising how to do it and I bet you, you could have done the study.
RS06
Future trial
With experience in the set-up and patient recruitment in the SURAB trial, interviewees were asked what the key design features of a future full-scale trial should be and what, if anything, they would change. The main points were about the intervention options of ablation versus active surveillance, the role of biopsy and other inclusion criteria, and practical points such as the patient pathway, timing and standardisation of approach to patients about the trial.
One interviewee suggested a system whereby clinicians could see the benefits of any financial reward for recruiting to a trial, or have some control over what the money was used for, rather than the money disappear into the trust ‘pot’. It was felt that this would be a greater in incentive to recruit. It was unclear whether or not a lack of motivation on the part of clinicians was an issue in the SURAB trial as this was not mentioned by any other interviewees. However, in a future trial this could be explored as an incentive.
Treatment options
Interviewees were divided on the value of a trial of ablation versus active surveillance. Opinions ranged from the view that ‘it’s an incredibly important question that we need to answer’ (RS10) to the view that CONSERVE was a ‘much more valid question’ (RS03). The value to the clinical community of observing patients for only 6 months was also raised:
I’m still not sure what the point of that was, if you’re only going to watch them for 6 months, these things are so slow-growing what benefit are you going to really get to show that patients didn’t come to any harm by waiting 6 months? You know, you need to know they don’t come to any harm by waiting 10 years doing nothing.
RS03
However, one other interviewee commented that asking patients to take part in a trial in which they may be randomised to a longer period of observation may have made it more difficult to recruit patients.
It was also believed that, since the inception of the SURAB trial, things have moved on in the treatment of small cell kidney tumours, particularly with the adoption of robotic partial nephrectomy:
The treatment has evolved over time to really suit a different set of patients . . . So you’re almost trying to organise a study and you’re randomising people when we’ve already sort of already boxed them into three different sets of treatment . . . CP-cryo is a really good treatment for those patients that are maybe less suitable for having a proper op, and the reason we have surveillance programmes is . . . quite a lot of tumours in the elderly it’s a waste of time treating them. So when you then try and randomise them it’s bizarre because, you know, we’ve already boxed them into three different groups already.
RS03
A trial tailored to the patient was suggested, in which certain patients would be approached to be randomised to surgery or ablation and other patients to ablation or active surveillance. It was thought that this would be a more attractive option to patients. However, this may not address the issue raised by one interviewee that these trials are not novel enough to attract patients to participate when they can receive the trial options as part of usual care.
One interviewee raised the point that a key question that needs to be addressed is whether or not active surveillance is safe, and they quoted an Italian study under way exploring this. 53 They believe that, once this is determined, it may be easier to recruit patients to a trial of active treatment versus active surveillance.
Trial processes and design
The model of the patient clinical pathway had a major impact on the conduct of the trial in a number of ways. Clearly, it would be difficult for the trial clinician to approach a patient who had already discussed treatment options with another urologist. It could also be problematic for patients to receive conflicting information on what is considered the most appropriate treatment:
It wasn’t just the investigators doing all of this, it was the urologists that were in front of the patients when the small renal mass was being discovered. Once you see a patient with a small renal mass and start this discussion going, you know they start getting ideas in their head at that very early stage.
RS03
It’s to do with capture in the first instance. I think if you have a system in various hospitals where they come from all over the show and . . . and there are a lot of doctors seeing them, those are the ways in which it breaks down.
RS06
One solution would be to have a pool of urologists who are trained to approach patients about the trial. However, concerns were raised about the quality of the approach and ensuring that patients are given full and accurate information.
There is also the issue of the patients from the spoke hospital not being able to be approached about the trial despite their care being delivered by the hub hospital. This is something that should be considered and dealt with before sites begin recruiting. Possible solutions would be to set up the spoke hospitals as PICs or to agree that any patient who is referred to the hub MDT then becomes a hub patient.
Inclusion criteria
As might be expected, there were comments about changes to the inclusion criteria for a future trial. One suggested change was to offer those already on active surveillance the opportunity to take part in the trial if their tumour increased in size. This was considered to be a sizeable group:
If the lesions start growing then patients get twitchy and so we will often biopsy them at that point and if they’re a malignancy then they’re going to more than likely go for a treatment, but some of them will be persuaded to have active surveillance.
RS03
There was a conflict in views on the target population for this trial. Some interviewees suggested broadening the inclusion criteria. Others thought that the specific target population should have been patients whom clinicians considered suitable for ablation, rather than everyone with a malignant tumour < 4 cm:
I just think we could have widened the net, because it was so specific. There seemed to be just a certain age, a certain type and a certain time.
RS08
It’s just delayed treatment versus immediate treatment, with a minimally invasive – a least invasive option in people who can’t really afford to be subjected to too much surgery.
RS06
Similarly, there was some variation in views on what the age range should be. In the main interviewees, it was felt that the trial was more appropriate for those aged > 60, and ideally, > 70 years. The alternative view was that there may be younger people who would be eligible for the trial, albeit rarely:
You might find a 40 year old who has got lots of comorbidity, a really bad chest and their life expectancy is not that great . . . it might be that that renal tumour is not going to cause any problems during their lifetime; therefore, it would be not unreasonable to put them in the trial. But most 40-year-olds, it isn’t really suitable for them.
RS09
Biopsy
Biopsy was a contentious issue for some interviewees, particularly in those sites where it is not routinely carried out. It was suggested that in the planning of any future trial greater consideration should be given to the particular role of biopsy in a trial of ablation versus active surveillance:
I think we need to have a rational discussion about, you know, in hospitals where you don’t normally biopsy, maybe you should only biopsy in those who have been randomised to treatment. The surveillance can just continue to be surveyed on the scan.
RS06
Identification and approach
The issue of equipoise on the part of the recruiting clinician was raised. Standardisation of approach and also removing the clinician from the recruitment equation was suggested as a means of overcoming surgeons’ selection bias:
I wonder if it could’ve been improved by trying to remove some of that surgeon-held bias, and having patients counselled either by [nurse], or by the person not immediately involved in treating them, a bit like they did in Protect. I think that, perhaps, would have removed some of this selection bias that, inevitably, is built into all surgeons when they’re trying to recruit patients.
RS10
The need to standardise the way in which potential participants are approached about taking part, to ensure that all recruiters are saying essentially the same thing, came across strongly. A digital versatile disc (DVD) about the trial was mentioned, but one interviewee said they preferred the personal touch and thought the use of DVD to be ‘quite sterile’. The use of a script appeared to be a more popular option. Linked to this was the suggestion of training for PIs and for research nurses, particularly for the former if they were inexperienced in recruiting to trials and for the latter if they were working in a different clinical field.
A few interviewees mentioned that they would have preferred it if patients had been approached sooner than they were in the SURAB feasibility study. One reason was to alert patients to the trial before they had seen a non-trial urologist and discussed treatment, and the other reason was because it was thought that some patients were missed and not approached about the trial:
If it was done . . . when they’re seen by the consultant . . . ‘By the way, would you like to read this?’ I don’t know how it could be phrased. ‘Should it prove to be cancer, might you consider this?’ I just think if we got to them a little bit earlier, it might help.
RS08
The first point is also linked to the patient route into the trial. In a setting with hub and spoke hospital system, rather than a more centralised service in which the clinician recruiting to the trial would be the first urologist that eligible patients would see, patients from spoke hospitals need to be informed early in the care pathway of the existence of a trial for which they may be a suitable participant.
Discussion
The parallel qualitative study explored the views of patients approached to take part in, and staff recruiting to, the SURAB trial in seven hospital sites. Unfortunately, owing to the low number of patients deemed eligible for the SURAB trial and a poor response rate in one site to the request for patients to take part in an interview, only eight patients were interviewed. One key issue in trials involving patients with cancer is the impact of a no-treatment arm. Before beginning the trial, it was expected that patients would not be comfortable with the no-treatment arm and living with a cancerous tumour on their kidney even for the period of the trial. This was the case for only one person, and one other person declined to participate because of the monitoring required in the active surveillance arm rather than the fact that it involved no treatment. However, the number of interviews was low and it is impossible to say whether or not other patients who declined the trial did so because they did not want to be in the active surveillance arm. Patient views of recruiting staff, and of all aspects of the approach to take part in the trial, were positive and there was little else of note from the patient interview data to inform a future full trial.
From the recruiting staff interview data, the two key factors that had an impact on the ability to recruit the required target sample were the lower than expected number of eligible patients and ‘selling’ the trial. The clinical pathway and patient route into the trial had an impact on the numbers who could be approached for the trial and on the ease of explaining the trial to potential participants. With the hub and spoke model of care, there appears to have been a disagreement or misunderstanding around the approvals: the research nurses believed that it was appropriate to approach patients referred to the hub MDT whereas the NCTU and sponsor responsible for the conduct of the trial did not. The opinion of the Clinical Trials Unit (CTU) and sponsor appears to be that, in this and in another site, the spoke hospitals should be set up as PICs and it was not appropriate to approach patients at those sites. The trial manager began the process for applying for these spoke hospitals to be PICs in October 2015. However, the trial was closed early to recruitment in November 2015. The question remains as to why this was not identified before the trial commenced in these two sites.
The second issue was related to patient route in the trial. This made it difficult for clinicians to convey uncertainty about the treatment when the patient had discussed the most appropriate treatment with a different urologist. Some clinicians may have felt comfortable going against the views of another urologist, but this clearly presented difficulties in SURAB. It could also have been confusing for the patient to have potentially conflicting information on the management of the kidney tumour.
Most of the PIs interviewed said that, when they first signed up to SURAB, they had underestimated the number of patients who would be eligible to participate. It is fair to say that were the SURAB trial to be conducted now, more intensive feasibility work to determine approximate numbers of eligible patients would be carried out by the CTU before accepting a hospital as a site. However, it is doubtful that such feasibility work would reveal whether or not potential PIs were in equipoise about active surveillance and ablation. A lack of equipoise and a bias towards surgery may have resulted in fewer patients being considered eligible for SURAB. Clinician bias could have had an impact on the selection of patients in the MDT meetings and also on the approach to patients in clinic. Without having observational data of the MDT meetings, it is difficult to determine not only who was present but also whether or not there was any element of bias in the discussion about a patient’s eligibility for the SURAB trial. Similarly, in the discussions with patients about the trial in clinic, we do not know if clinicians may have, possibly inadvertently, articulated the trial interventions in a negative way. Patient preference, which was a subtheme within selling the trial, can be due to a lack of equipoise on the part of the recruiter. If recruiting staff truly believe that they do not know the best option between active surveillance and ablation, and are confident to convey that, this can help patients to understand and be in equipoise themselves.
The question of who is the best person to approach patients about a trial like the SURAB trial did emerge in the interviews. Some PIs felt a conflict between their role as surgeon and recruiter, and a number of interviewees mentioned a steep learning curve in the practice of recruitment. Possibly some training or role play in, and a standardised script for, approaching patients may have been beneficial and enhanced recruitment. An alternative option would be for someone else to explain the trial, and this was also raised in the interviews. PIs appeared to value sharing their experience in the periodic telemeetings with other site Pis, and this could have been enhanced as a forum for training. It was also suggested that training or sharing expertise in systems for tracking patients who may be eligible for the SURAB trial would have been beneficial.
Three overarching factors were identified that should be addressed in a future full trial: (1) the treatment options, (2) trial process/design and (3) patient identification/approach. Some, relating to trial process and patient identification, warrant practical solutions and greater engagement of staff involved in recruitment at the planning stage. Others, around treatment options and inclusion criteria, require a full discussion with both clinical and trial design expert input.
Limitations
Although valuable and useful data have been obtained from recruiting staff who agreed to be interviewed, the response to the qualitative study was disappointing from some sites, particularly in the two sites at which neither the PI nor the research nurse agreed to be interviewed. We have the perspectives of both PI and research nurse in only three of the nine SURAB sites. Not having the views of any recruiting staff in two sites and only the PI or research nurse in four sites is a definite limitation of this qualitative study. However, we were able to capture the views of recruiting staff with a range of experience in relation to SURAB: from sites that failed to recruit any patients, from sites that managed to recruit a small number of patients and from sites that achieved their target recruitment rate.
Patient and public involvement
The study protocol was designed with patient and public input from Pat Hanlon, who agreed to be a coapplicant on the funding application. Pat is a trustee of Kidney Cancer UK and has been involved in the charity since 2005. He has experience of involvement in other trials and attends the NCRI Renal Studies Group.
Pat was instrumental in determining the most appropriate time to interview patients, whether this should be very soon after diagnosis or later when they are being followed up. He was supportive of the plan to explore with patients their motives for enrolling in the trial, and, should they drop out, their reasons for doing so, and thought there was much to be learned from this.
Once the trial was under way, he participated in a telemeeting with the qualitative researcher to discuss the content of the topic guide for the interviews with patients. His advice was also sought on the content and design of the letter to patients asking for their help in the development of trial materials (see Chapter 2). In addition, he provided continued support from the launch of the SURAB trial, through attendance at the Trial Steering Committees.
When the team were notified that the study was to be closed down, Pat was consulted on the proposed plan. He also reviewed the first draft of the report and gave the following comments:
The number of patients finally enrolled on the study is disappointing, but perhaps not entirely surprising. A very large proportion (77%) of screened patients was ruled out as ineligible, with the result that only 36 patients were actually approached. This might suggest that to get a workably large sample the net has to be cast widely across sites. Of patients approached only six were finally randomised, the most frequently cited reason for declining being that the patient wanted to be sure of getting ablation. As often shown elsewhere, many patients prefer the certainty of getting at least some treatment to just a 50/50 chance of it.
Although the overall conclusion is that the trial does not meet the criteria for progression to a definitive phrase III trial, there is much in the final report which is of value for future developments in this field. Particularly interesting is the suggested possibility of a MAMS [multiarm multistage] trial which would include some patients with co-morbidities. The qualitative sub-study makes for some interesting reading and produces a number of important insights into patients’ experiences and their understanding of trial processes.
All in all, despite not leading to a phase III trial, the SURAB study can be seen as a most worthwhile exercise from the patient’s point of view.
Reproduced with permission from Pat Hanlon, University of Birmingham, 2017, personal communication2017Pat Hanlon
Chapter 4 Discussion and conclusions
Small kidney cancers are managed by radiologists and urologists in a multidisciplinary setting. Their management varies among kidney centres, which are dependent on the expertise of clinicians and resources available. The NCRI renal cancer group has identified the management of SRMs as an important research question as there is no consensus or high-quality research on the best way to manage these. As the CONSERVE trial, which was supported by Cancer Research UK and looked at the feasibility of surgery versus ablation in small kidney tumours, did not succeed in recruitment, the SURAB trial was planned with a robust qualitative arm. This was to enable identification of barriers to recruitment and to perform a detailed assessment of qualitative issues surrounding recruitment into a surgical trial in kidney cancer in a multispecialty setting.
The main aim of the SURAB trial was to explore the feasibility of conducting a RCT to compare active surveillance with ablation for the management of patients with small kidney tumours with a specific focus on capturing patient willingness to be randomised and clinician willingness to approach eligible patients. The success of this feasibility trial was judged on the basis of three factors: (1) a recruitment rate of at least 50%, (2) a high retention rate and (3) economic criteria that suggest further research is worthwhile. The trial results indicate that these criteria were not met and the overall conclusion is that it is not feasible to move this design to a definitive Phase III trial.
Pre-feasibility study clinician survey
In order maximise recruitment for the trial, the NCTU conducted a survey to identify which centres had the infrastructure to recruit patients. Another survey focusing on surgeons and radiologists was conducted to find which criteria among tumour size, patient’s age and comorbidity would influence their decision in favour of recruitment. In addition to this, focused patient groups were surveyed to optimise patient information literature for the trial. It was found that this period was very useful in preparing for the trial.
Feasibility study
Although the SURAB trial did not achieve its intended recruitment rate, the data from the qualitative substudy have identified issues that have not been previously explored as barriers to surgical studies. Greater engagement with the relevant clinicians and research nurses in the very early stages of the SURAB trial may have facilitated the commencement of recruitment when planned and identified problems in the patient pathway to the trial. In addition, this study has identified the need for support in terms of training and systems sharing of best practice for recruiting staff, and a frank discussion about the potential for bias in the selection and recruitment of patients and how this could be overcome. The lessons learned from SURAB’s qualitative substudy will be applicable to a wide range of surgical studies that intend to recruit from such a complex pathway.
Although we have not been able to assess feasibility of the health economics component, we have developed a workable health economic data collection tool and, based on the data we have been able to collect, it is reasonable to assume that relevant health economic data could be collected should the trial be able to recruit participants following any critical lessons learned from this study.
From a clinical perspective, variation in pre-treatment renal biopsy across the country was one of the points which was highlighted in the feedback from the sites and indicated that relaxation of the criteria might have helped recruitment. The other issue was that partial nephrectomy, which is standard of care for small kidney tumours, was not included in either arm of the trial. This omission deterred clinicians in advising patients for recruitment in the trial. Change in management protocols during the course of the study and delays in getting research and development approval for the participating centres also played a part in the reduction in the overall pool of patients available for recruiting into the research. Health-care professionals continue to regard this as an important question for a SRM, as practice varies around the world, with no widely accepted criteria for deciding whether patients receive an intervention or active surveillance. Currently, this is dictated by local practices and logistics rather than patient benefit or an evidence base of superiority.
Our data suggest that, even if clinical, organisational and operational processes are optimised, a recruitment rate of 50% may be difficult to achieve unless the trial design were based on a three-arm design to directly compare surgery with both ablation and active surveillance. Such a design would ideally be adaptive, possibly a multiarm (MAMS) umbrella trial that would capture the wider patient group including those with comorbidities. 54
Overall, the SURAB trial had a good safety record, with a low incidence of AEs in both arms of the study and no SAEs recorded in relation to the trial. Statistical analysis of patient-reported and clinician-rated outcomes was not possible owing to the recruitment rate.
Research question
The findings from this SURAB trial suggest that a main randomised trial is not feasible. The reason for this failure to recruit was a lack of equipoise on the part of clinicians and an unwillingness of patients to be randomised. This has highlighted the inadequacy of conventional randomised trial methodology to capture all patients when the treatment options are multiple, which include active surveillance, ablation and surgical excision. This was further complicated by the fact that the clinician’s decision-making was affected not only by the size of the tumour but also by the age and comorbidities of the patient.
Although the question of how to best manage SRMs remains important, owing to above-mentioned complexities, the authors would suggest that a MAMS trial would probably be best suited to deliver a successful outcome and provide answers in these questions.
Acknowledgements
We would like to acknowledge the support of the following: the BSIR in circulating the clinician survey on our behalf and for all the clinicians who completed the questionnaire; the staff at hospital trusts in identifying and contacting patients for the participatory design study; the patients who took part in the group and one-to-one work in developing trial materials; the recruiting staff and patients for taking part in the qualitative study; and the PIs at each of the eight sites.
Contributions of authors
Professor Naeem Soomro (Honorary Professor of Urology, CI) developed the protocol and successfully applied for the HTA programme commissioned call. Worked with clinical trials unit in the conduct of the trial. Chaired Trial Management Group meetings. Contributed to and edited the end-of-study report.
Ms Jan Lecouturier (Senior Research Associate – mixed methodologist and specialist in qualitative research) contributed to the study design and production of the funding application. Conducted and analysed the parallel qualitative study and pre-feasibility survey (including design and pilot testing of questionnaire). Developed trial information with patients. Prepared the results from all three for publication.
Dr Deborah D Stocken (Senior Trial Statistician and NCTU Deputy Director) supervised statistical aspects of study design and analysis and responsible for associated statistical documentation including statistical analysis plan, end-of-study report and publication. Responsible for drafting the methods and results sections of this report in relation to the clinical trial component.
Dr Jing Shen (Senior Research Associate, Health Economics) prepared the health economics results for publication.
Dr Ann Marie Hynes (Trial Manager) assisted with the formatting of the final report and the clinical trial results for publication.
Dr Holly F Ainsworth (Research Associate, Statistics) assisted Dr Deborah D Stocken with the preparation of the clinical trial results for publication.
Dr David Breen (Consultant Radiologist) contributed to the initial grant application.
Mr Grenville Oades (Consultant Urological Surgeon) contributed to the initial grant application and trial recruitment.
Mr David Rix (Consultant Urological Surgeon) contributed to the initial grant application and trial recruitment.
Mr Michael Aitchison (Consultant Urological Surgeon) contributed to the initial grant application.
Data sharing statement
All data can be obtained from the corresponding author.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Office for National Statistics . Registrations of Cancer Diagnosed in 2004, England n.d. http://ons.gov.uk/ons/rel/vsob1/cancer-registrations-in-england/2004/index.html (accessed 7 November 2017).
- Northern Ireland Cancer Registry . Registrations of Cancer Diagnosed in 2004, Northern Ireland n.d. www.qub.ac.uk/research-centres/nicr/FileStore/PDF/NIrelandReports/Filetoupload,532181,en.pdf (accessed 14 December 2017).
- Information and Statistics Division . Scotland Registrations of Cancer Diagnosed in 2004, Scotland 2004. www.gov.scot/Publications/2004/12/20257/46697 (accessed 7 November 2017).
- Welsh Cancer Intelligence and Surveillance Unit . Registrations of Cancer Diagnosed in 2004, Wales n.d. www.wcisu.wales.nhs.uk/home (accessed 7 November 2017).
- SEER Stat Fact Sheets: Kidney and Renal Pelvis Cancer. Bethesda, MD: SEER; 2014.
- Van Poppel H, Da Pozzo L, Albrecht W, Matveev V, Bono A, Borkowski A, et al. A prospective randomized EORTC intergroup phase 3 study comparing the complications of elective nephron-sparing surgery and radical nephrectomy for low stage renal cell carcinoma. Eur Urol 2007;51:1606-15. https://doi.org/10.1016/j.eururo.2006.11.013.
- Fergany AF, Hafez KS, Novick AC. Long-term results of nephron sparing surgery for localized renal cell carcinoma: 10-year followup. J Urol 2000;163:442-5. https://doi.org/10.1016/S0022-5347(05)67896-2.
- Hui GC, Tuncali K, Tatli S, Morrison PR, Silverman SG. Comparison of percutaneous and surgical approaches to renal tumor ablation: metaanalysis of effectiveness and complication rates. J Vasc Interv Radiol 2008;19:1311-20. https://doi.org/10.1016/j.jvir.2008.05.014.
- Breen DJ, Rutherford EE, Stedman B, Roy-Choudhury SH, Cast JE, Hayes MC, et al. Management of renal tumors by image-guided radiofrequency ablation: experience in 105 tumors. Cardiovasc Intervent Radiol 2007;30:936-42. https://doi.org/10.1007/s00270-007-9090-x.
- Zagoria RJ, Traver MA, Werle DM, Perini M, Hayasaka S, Clark PE. Oncologic efficacy of CT-guided percutaneous radiofrequency ablation of renal cell carcinomas. AJR Am J Roentgenol 2007;189:429-36. https://doi.org/10.2214/AJR.07.2258.
- Matin SF, Ahrar K, Cadeddu JA, Gervais DA, McGovern FJ, Zagoria RJ, et al. Residual and recurrent disease following renal energy ablative therapy: a multi-institutional study. J Urol 2006;176:1973-7. https://doi.org/10.1016/j.juro.2006.07.016.
- Kunkle DA, Egleston BL, Uzzo RG. Excise, ablate or observe: the small renal mass dilemma – a meta-analysis and review. J Urol 2008;179:1227-33. https://doi.org/10.1016/j.juro.2007.11.047.
- Volpe A, Panzarella T, Rendon RA, Haider MA, Kondylis FI, Jewett MA. The natural history of incidentally detected small renal masses. Cancer 2004;100:738-45. https://doi.org/10.1002/cncr.20025.
- Chawla SN, Crispen PL, Hanlon AL, Greenberg RE, Chen DY, Uzzo RG. The natural history of observed enhancing renal masses: meta-analysis and review of the world literature. J Urol 2006;175:425-31. https://doi.org/10.1016/S0022-5347(05)00148-5.
- Hafez KS, Fergany AF, Novick AC. Nephron sparing surgery for localized renal cell carcinoma: impact of tumor size on patient survival, tumor recurrence and TNM staging. J Urol 1999;162:1930-3. https://doi.org/10.1016/S0022-5347(05)68071-8.
- Uzzo RG, Novick AC. Nephron sparing surgery for renal tumors: indications, techniques and outcomes. J Urol 2001;166:6-18. https://doi.org/10.1016/S0022-5347(05)66066-1.
- Gill IS, Matin SF, Desai MM, Kaouk JH, Steinberg A, Mascha E, et al. Comparative analysis of laparoscopic versus open partial nephrectomy for renal tumors in 200 patients. J Urol 2003;170:64-8. https://doi.org/10.1097/01.ju.0000072272.02322.ff.
- Moinzadeh A, Gill IS, Finelli A, Kaouk J, Desai M. Laparoscopic partial nephrectomy: 3-year followup. J Urol 2006;175:459-62. https://doi.org/10.1016/S0022-5347(05)00147-3.
- BAUS . Cancer Registry 2008 n.d. www.baus.org.uk/_userfiles/pages/files/publications/audit/2008finalanalyses.pdf (accessed 7 November 2017).
- Klingler HC, Marberger M, Mauermann J, Remzi M, Susani M. ‘Skipping’ is still a problem with radiofrequency ablation of small renal tumours. BJU Int 2007;99:998-1001. https://doi.org/10.1111/j.1464-410X.2007.06769.x.
- Weight CJ, Kaouk JH, Hegarty NJ, Remer EM, O’Malley CM, Lane BR, et al. Correlation of radiographic imaging and histopathology following cryoablation and radio frequency ablation for renal tumors. J Urol 2008;179:1277-81. https://doi.org/10.1016/j.juro.2007.11.075.
- Kunkle DA, Uzzo RG. Cryoablation or radiofrequency ablation of the small renal mass: a meta-analysis. Cancer 2008;113:2671-80. https://doi.org/10.1002/cncr.23896.
- Percutaneous Radiofrequency Ablation for Renal Cancer. IPG353. London: NICE; 2010.
- Atwell TD, Farrell MA, Leibovich BC, Callstrom MR, Chow GK, Blute ML, et al. Percutaneous renal cryoablation: experience treating 115 tumors. J Urol 2008;179:2136-40. https://doi.org/10.1016/j.juro.2008.01.144.
- Gill IS, Remer EM, Hasan WA, Strzempkowski B, Spaliviero M, Steinberg AP, et al. Renal cryoablation: outcome at 3 years. J Urol 2005;173:1903-7. https://doi.org/10.1097/01.ju.0000158154.28845.c9.
- Laproscopic Cryotherapy for Renal Cancer. IPG405. London: NICE; 2011.
- Percutaneous Cryotherapy for Renal Cancer. IPG402. London: NICE; 2011.
- Bosniak MA. Observation of small incidentally detected renal masses. Semin Urol Oncol 1995;13:267-72.
- Kato M, Suzuki T, Suzuki Y, Terasawa Y, Sasano H, Arai Y. Natural history of small renal cell carcinoma: evaluation of growth rate, histological grade, cell proliferation and apoptosis. J Urol 2004;172:863-6. https://doi.org/10.1097/01.ju.0000136315.80057.99.
- Kassouf W, Aprikian AG, Laplante M, Tanguay S. Natural history of renal masses followed expectantly. J Urol 2004;171:111-13. https://doi.org/10.1097/01.ju.0000102409.69570.f5.
- Sowery RD, Siemens DR. Growth characteristics of renal cortical tumors in patients managed by watchful waiting. Can J Urol 2004;11:2407-10.
- Kunkle DA, Crispen PL, Chen DY, Greenberg RE, Uzzo RG. Enhancing renal masses with zero net growth during active surveillance. J Urol 2007;177:849-53. https://doi.org/10.1016/j.juro.2006.10.073.
- Jewett MA, Mattar K, Basiuk J, Morash CG, Pautler SE, Siemens DR, et al. Active surveillance of small renal masses: progression patterns of early stage kidney cancer. Eur Urol 2011;60:39-44. https://doi.org/10.1016/j.eururo.2011.03.030.
- Abou Youssif T, Kassouf W, Steinberg J, Aprikian AG, Laplante MP, Tanguay S. Active surveillance for selected patients with renal masses: updated results with long-term follow-up. Cancer 2007;110:1010-14. https://doi.org/10.1002/cncr.22871.
- Guðmundsson E, Hellborg H, Lundstam S, Erikson S, Ljungberg B. Swedish Kidney Cancer Quality Register Group . Metastatic potential in renal cell carcinomas ≤ 7 cm: Swedish Kidney Cancer Quality Register data. Eur Urol 2011;60:975-82. https://doi.org/10.1016/j.eururo.2011.06.029.
- Remzi M, Ozsoy M, Klingler HC, Susani M, Waldert M, Seitz C, et al. Are small renal tumors harmless? Analysis of histopathological features according to tumors 4cm of less in diameter. J Urol 2006;176:896-9. https://doi.org/10.1016/j.juro.2006.04.047.
- Hsu RM, Chan DY, Siegelman SS. Small renal cell carcinomas: correlation of size with tumor stage, nuclear grade, and histologic subtype. AJR Am J Roentgenol 2004;182:551-7. https://doi.org/10.2214/ajr.182.3.1820551.
- Van Poppel H, Joniau S. Is surveillance an option for the treatment of small renal masses?. Eur Urol 2007;52:1323-30. https://doi.org/10.1016/j.eururo.2007.07.025.
- Chard JA, Lilford RJ. The use of equipoise in clinical trials. Soc Sci Med 1998;47:891-8. https://doi.org/10.1016/S0277-9536(98)00153-1.
- Hilton P, Bryant A, Howel D, McColl E, Buckley BS, Lucas M, et al. Assessing professional equipoise and views about a future clinical trial of invasive urodynamics prior to surgery for stress urinary incontinence in women: a survey within a mixed methods feasibility study. Neurourol Urodyn 2012;31:1223-30. https://doi.org/10.1002/nau.22328.
- Sully BG, Julious SA, Nicholl J. A reinvestigation of recruitment to randomised, controlled, multicenter trials: a review of trials funded by two UK funding agencies. Trials 2013;14. https://doi.org/10.1186/1745-6215-14-166.
- Mills EJ, Seely D, Rachlis B, Griffith L, Wu P, Wilson K, et al. Barriers to participation in clinical trials of cancer: a meta-analysis and systematic review of patient-reported factors. Lancet Oncol 2006;7:141-8. https://doi.org/10.1016/S1470-2045(06)70576-9.
- Nilsen E, Myrhaug H, Johansen M, Oliver S, Oxman A. Methods of consumer involvement in developing healthcare policy and research, clinical practice guidelines and patient information material. Cochrane Database Syst Rev 2006;3. https://doi.org/10.1002/14651858.CD004563.pub2.
- Rutherford EE, Cast JE, Breen DJ. Immediate and long-term CT appearances following radiofrequency ablation of renal tumours. Clin Radiol 2008;63:220-30. https://doi.org/10.1016/j.crad.2007.07.002.
- Lancaster GA, Dodd S, Williamson PR. Design and analysis of pilot studies: recommendations for good practice. J Eval Clin Pract 2004;10:307-12. https://doi.org/10.1111/j.2002.384.doc.x.
- Stevenson V, Ridge S. Standard Operating Procedure n.d. https://microsites.ncl.ac.uk/njro/files/2013/02/JRO-12-Archiving-Clinical-Research-Documents-V2.pdf (accessed 7 November 2017).
- Ware JE, Sherbourne CD. The MOS 36-Item Short-Form Health Survey (SF-36): I. Conceptual framework and item selection. Med Care 1992;30:473-83. https://doi.org/10.1097/00005650-199206000-00002.
- Cella DF, Tulsky DS, Gray G, Sarafian B, Linn E, Bonomi A, et al. The Functional Assessment of Cancer Therapy Scale: development and validation of the general measure. J Clin Oncol 1993;11:570-9. https://doi.org/10.1200/JCO.1993.11.3.570.
- Spielberger CD. Manual for the State-Trait Anxiety Inventory (STAI). Palo Alto, CA: Consulting Psychologists Press; 1983.
- Donovan J, Mills N, Smith M, Brindle L, Jacoby A, Peters T, et al. Improving design and conduct of randomised trials by embedding them in qualitative research: ProtecT (prostate testing for cancer and treatment) Study. BMJ 2002;325:766-70. https://doi.org/10.1136/bmj.325.7367.766.
- Glaser BG. The constant comparative method of qualitative analysis. Soc Probl 1965;12:436-45. https://doi.org/10.2307/798843.
- Cancer Research UK . A Study Looking at 3 Different Treatments for Kidney Cancer (CONSERVE) n.d. www.cancerresearchuk.org/about-cancer/find-a-clinical-trial/a-study-looking-3-different-treatments-for-kidney-cancer-conserve (accessed 7 November 2017).
- Oncology Times . New Guidelines for Active Surveillance in Renal Cell Cancer n.d. http://journals.lww.com/oncology-times/fulltext/2017/04250/New_Guidelines_for_Active_Surveillance_in_Renal.16.aspx (accessed 7 November 2017).
- Parmar MK, Carpenter J, Sydes MR. More multiarm randomised trials of superiority are needed. Lancet 2014;384:283-4. https://doi.org/10.1016/S0140-6736(14)61122-3.
Glossary
- SURAB
- A two-stage randomised feasibility study of SURveillance versus ABlation in the management of incidentally diagnosed small renal masses.
List of abbreviations
- AE
- adverse event
- ASA
- American Society of Anesthesiologists
- BAUS
- British Association of Urological Surgeons
- BSIR
- British Society of Interventional Radiology
- CI
- chief investigator
- CRF
- case report form
- CRYO
- cryoablation
- CT
- computerised tomography
- CTU
- Clinical Trials Unit
- DVD
- digital versatile disc
- eCRF
- electronic case report form
- FACT-G
- Functional Assessment of Cancer Therapy – General
- GP
- general practitioner
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- LPN
- laparoscopic partial nephrectomy
- MAMS
- multiarm multistage
- MCS
- mental component summary
- MDT
- multidisciplinary team
- MRI
- magnetic resonance imaging
- MWA
- microwave ablation
- NCRI
- National Cancer Research Institute
- NCTU
- Newcastle Clinical Trials Unit
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- OPN
- open partial nephrectomy
- PCQ
- participant costs questionnaire
- PCS
- physical component summary
- PI
- principal investigator
- PIC
- patient identification centre
- PIS
- patient information sheet
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCC
- renal cell cancer
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RFA
- radiofrequency ablation
- SAE
- serious adverse event
- SF-36
- Short Form questionnaire-36 items
- SF-6D
- Short Form questionnaire-6 Dimensions
- SRM
- small renal mass
- STAI
- State–Trait Anxiety Inventory
- SURAB
- SURveillance versus ABlation
- US
- ultrasonography