

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 09/01/48. The contractual start date was in June 2013. The draft report began editorial review in January 2017 and was accepted for publication in November 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Kamlesh Khunti received honoraria and research grants from AstraZeneca plc, Novartis Pharmaceuticals UK Ltd, Novo Nordisk Ltd, Sanofi S.A., Eli Lilly and Company, Merck Sharp & Dohme Corp., Janssen Research and Development, Boehringer Ingelheim Ltd and Roche Diagnostics Ltd. He has acted as an advisor to the NHS Health Check programme, National Institute for Health and Care Excellence (NICE) Guidance and the Diabetes Prevention Programme External Reference Group. M Angelyn Bethel reports non-financial support from Merck Serono during the conduct of the study. Outside the submitted work, she received grants from Merck Sharp & Dohme Corp. and AstraZeneca, acted as an advisory board member for AstraZeneca, Boehringer Ingelheim and Novo Nordisk, as a speaker for Sanofi and received non-financial support from Bayer plc. Rury R Holman reports ACE (Acarbose Cardiovascular Evaluation) trial funding and lecture fees from Bayer, trial funding from AstraZeneca and Merck Sharp & Dohme Corp. and lecture fees from Boehringer Ingelheim, outside the submitted work. He acted as an advisory board member for Merck, Sharp & Dohme, Elcelyx Therapeutics, Inc., Novartis Pharmaceuticals UK, Novo Nordisk and Servier Laboratories Ltd. Melanie Davies has acted as a consultant, advisory board member and speaker for Novo Nordisk, Sanofi, Eli Lilly and Company, Merck Sharp & Dohme Corp., Boehringer Ingelheim, AstraZeneca and Janssen Research and Development, as an advisory board member for Servier Laboratories and as a speaker for Mitsubishi Tanabe Pharma Corporation and Takeda Pharmaceuticals International, Inc. She has received grants in support of investigator and investigator-initiated trials from Novo Nordisk, Sanofi, Eli Lilly and Company, Boehringer Ingelheim and Janssen Research and Development. She has acted as an advisor to the NHS Health Check programme and the NICE Guidance and is Director of a National Institute for Health Research Biomedical Centre and a Clinical Trials Unit. John R Petrie reports personal fees from Novo Nordisk, Eli Lilly and Company, Janssen Research and Development and Boehringer Ingelheim, grants, personal fees and non-financial support from Sanofi, non-financial support from Merck KGaA and Itamar Medical Ltd, grants from the Juvenile Diabetes Research Foundation Ltd and grants and personal fees from Quintiles (now IQVIA), outside the submitted work. Martin Rutter reports grants from Novo Nordisk, outside the submitted work. He owns stocks and shares in GlaxoSmithKline plc and acted as an advisory board member for Roche Diagnostics Ltd. Naveed Sattar reports personal fees from Boehringer Ingelheim, Janssen Research and Development, Novo Nordisk and Eli Lilly and Company and grants and personal fees from AstraZeneca, outside the submitted work. Simon J Griffin reports receiving an honorarium and reimbursement of travel expenses from Eli Lilly and Company, associated with membership of an Independent Data Monitoring Committee for a randomised trial of a medication to lower glucose levels, and an honorarium from Janssen Research and Development for speaking at an educational meeting in 2015.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Griffin et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction (including background and rationale)
Cardiovascular disease (CVD) is a major public health problem in the UK and is associated with substantial premature mortality, morbidity and financial cost. 1 People with established type 2 diabetes (T2D) have an increased risk of CVD, but the degree to which that risk is modulated through glucose control is unclear. Observational studies show a consistent and continuous association between glycaemia and CVD risk, even below the diagnostic threshold for T2D. 2–4 Despite this association, interventional trials in patients with T2D have not consistently demonstrated reductions in CVD risk with intensive glucose control. 5,6 The UK Prospective Diabetes Study (UKPDS), conducted in people with newly diagnosed diabetes, showed a reduced risk of myocardial infarction [relative risk 0.67, 95% confidence interval (CI) 0.51 to 0.89] and all-cause mortality (relative risk 0.73, 95% CI 0.59 to 0.89) with metformin-based treatment. 7 The hypothesis that tight glycaemic control is more important early in the course of T2D or prior to its diagnosis in the setting of non-diabetic hyperglycaemia (NDH), before macrovascular disease has developed, could unify these findings and is supported by subgroup analyses showing a greater CVD effect in those with a shorter duration of T2D. 8–10 Metformin is typically well tolerated and is now widely considered to be the first-line therapy for T2D. However, there are relatively few data from adequately powered randomised trials with long-term follow-up concerning the frequency of adverse effects associated with metformin. Consequently, although metformin is used by millions of people worldwide, it is possible that serious adverse events (SAEs) remain undiscovered.
The effect of glucose lowering on CVD events and mortality in people with NDH is unknown. This is an important gap in the current research base,11 particularly at a time when population-based programmes to identify those at a high risk of CVD and T2D are being undertaken. NDH affects 15–20% of the UK adult population and around 50% of all CVD events attributable to higher-than-normal glucose levels occur in this group. 3 In randomised controlled trials, both intensive lifestyle interventions and metformin have been shown to reduce the risk of progression to diabetes in individuals with NDH. 12,13 However, properly designed and powered studies to evaluate the impact of metformin on CVD outcomes in people with NDH have not been carried out. If a cheap drug, such as metformin, was shown to reduce CVD risk by reductions in glycaemia or by other mechanisms14 it would have important population health and economic benefits.
Hyperglycaemia, insulin resistance and obesity are risk factors for malignancy. 15 Observational data link metformin use to a 37% lower incidence of major common cancers, including bowel, lung and breast cancers,16 and this is supported by a body of evidence describing plausible mechanisms of action in addition to glucose lowering. 17 Among patients with T2D, the use of metformin is associated with a halving of cancer mortality. 18 However, these epidemiological data are limited by treatment indication bias and residual confounding and cannot establish causality. Indeed, a recent meta-analysis does not support the hypothesis that metformin lowers cancer risk by one-third. 19 By randomising a large number of participants to metformin therapy or placebo and following them up over 5 years, we will be able to quantify the effects on the incidence of CVD and cancer as well as the frequency of important adverse effects.
Aims and rationale
The Glucose Lowering In Non-diabetic hyperglycaemia Trial (GLINT) was designed to evaluate the impact of metformin (Glucophage® SR, Merck KGaA, Bedfont Cross, Middlesex, UK) compared with placebo on a composite CVD end point of non-fatal myocardial infarction, non-fatal stroke and CVD death, when added to the usual care of people with NDH and high modelled CVD risk, over approximately 5 years’ follow-up. Secondary objectives included measuring the impact of metformin on the incidence of diabetes, non-melanoma cancer, death from non-melanoma cancer and death from any cause and on functional status, health utility and patient satisfaction, as well as quantifying the cost-effectiveness of metformin treatment. The full study, in which we planned to enrol approximately 13,000 participants aged ≥ 40 years with NDH [glycated haemoglobin (HbA1c) level between 36.6 and 47.5 mmol/mol] and an estimated 10-year CVD risk of ≥ 20%, was powered to detect a 17% risk reduction in the composite CVD end point. We planned to provide participants with a theory-based brochure containing advice about reducing the risk of heart attack and/or diabetes based on previous prevention studies and randomise them on a 1 : 1 basis to either 500 mg of prolonged-release metformin or placebo, up to three tablets a day, as tolerated. Here, we report the results of an initial feasibility study that enrolled 249 participants, with a minimum follow-up period of 6 months.
Feasibility study aims
In this feasibility phase we assessed the different design aspects of the full trial in order to refine the design and protocol. We aimed to ascertain whether or not eligible participants could be recruited and retained, to demonstrate that trial processes were sufficiently robust and to ascertain the acceptability, tolerability and safety of the intervention and trial procedures to participants. We will use data from the feasibility study to determine the most efficient strategies for participant recruitment, retention, monitoring and outcome assessment, to estimate the number of eligible participants required for the full trial and to inform the assessment of the feasibility of the full trial.
Objectives
-
Assess the feasibility of the recruitment of practices and practice consortia to GLINT during a period of reorganisation of health services and establishment of a vascular risk assessment programme (the NHS Health Check programme). 20
-
Assess the feasibility of the recruitment of participants, including those from multiethnic populations, to GLINT at a time when uncertainties about the optimal mode of assessment of diabetes risk and glycaemia within the NHS Health Check programme are being resolved.
-
Examine the feasibility and relative efficiency of three proposed recruitment strategies:
-
recruitment via the NHS Health Check programme
-
recruitment directly from participating general practices
-
recruitment from existing research registers of people with NDH.
-
-
Assess the efficiency of different search strategies, and the availability and accessibility of information, in computerised medical records concerning inclusion and exclusion criteria, such as records of NDH HbA1c and estimates of modelled 10-year CVD risk.
-
Assess if recorded estimates of modelled CVD risk and its constituent variables are accurate compared with those estimated based on data collected at the baseline visit in the feasibility study and hence whether or not CVD risk estimates in medical records could be utilised in recruitment in the main trial.
-
Examine the feasibility and acceptability of the randomisation procedure to patients and practitioners.
-
Describe the characteristics of recruited participants including ethnic diversity and estimate modelled 10-year CVD risk as an input to the revised sample size calculations for the main trial.
-
Examine the feasibility of the delivery mechanism of the investigational medicinal product (IMP).
-
Examine the acceptability of the IMP to patients and practitioners.
-
Estimate adherence to the IMP in both randomised groups.
-
Consider strategies to optimise adherence.
-
Assess change in predefined safety parameters, including renal function and plasma vitamin B12 levels, to inform decisions about the level of safety monitoring required in the full trial.
-
Examine the feasibility of the system of adverse event (AE) reporting.
-
Assess the acceptability and feasibility of collecting end-point data from participants, their general practitioners (GPs) and routine data sources [Office for National Statistics (ONS), Health and Social Care Information Centre (HSCIC) (now NHS Digital), cancer registries and Hospital Episode Statistics (HES)].
-
Examine patient experiences of taking part in the trial by questionnaire/interview.
-
Establish systems for collecting resource use and quality-of-life data for future cost-effectiveness analyses.
-
Finalise the costing for the full trial and negotiate with other funders.
The demonstration of treatment effect was not an objective within the feasibility phase.
Chapter 2 Trial design and methods
Trial design
The Glucose Lowering In Non-diabetic hyperglycaemia Trial is a multicentre, individually randomised, double-blind, parallel-group, pragmatic, primary prevention trial comparing the effect on a macrovascular composite outcome of adding prolonged-release metformin or the matched placebo to the usual care of people with NDH and high CVD risk in the community. We standardised one aspect of usual care by providing all participants with a theory-based brochure containing advice about reducing the risk of heart attack and/or diabetes that was based on previous prevention studies in Leicester (the brochure can be viewed at http://leicesterdiabetescentre.org.uk/GLINT). 21
Here we report the findings of the feasibility study that will inform the design and conduct of the main trial.
Sponsorship, ethics review and research governance
The study was hosted initially by the Cambridge Medical Research Council (MRC) Epidemiology Unit and sponsored by the MRC. When the MRC Epidemiology Unit became a university unit, the University of Cambridge assumed responsibility for sponsorship. Shortly before the planned study initiation meeting in April 2014, sponsorship arrangements were revised in line with a new University of Cambridge School of Medicine policy regarding Clinical Trials of Investigational Medicinal Products (CTIMPs). Investigators were advised that the university would no longer be the sole sponsor of CTIMPs, which henceforth had to be jointly sponsored by the university and a NHS trust. Consequently, following detailed review by the Cambridge Clinical Trials Unit (CCTU) on behalf of Cambridge University Hospitals NHS Foundation Trust, the study was jointly sponsored by the Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge. As per the sponsor requirements, the CCTU undertook regulatory submissions, monitoring of the lead site (Cambridge) and submission of reports concerning SAEs and development safety update reports to the competent authority. The National Research Ethics Service Committee East of England – Cambridge South reviewed the study for ethics approval and gave a favourable opinion on 28 January 2014 (reference number 13/EE/0415). Clinical trials authorisation was granted by the Medicines and Healthcare products Regulatory Agency (MHRA) on 2 January 2014 (MHRA reference 24551/0018/001-0001, EudraCT number 2012-005570-56). The timelines for sponsorship and ethics and governance review are shown in Figures 1 and 2.
FIGURE 1.
Timeline of changes in trial sponsorship. HTA, Health Technology Assessment; REC, Research Ethics Committee.

FIGURE 2.
Timeline of regulatory approvals. REC, Research Ethics Committee.

The project was led by the MRC Epidemiology Unit (University of Cambridge) in collaboration with the Diabetes Trials Unit (DTU) (University of Oxford) and the Diabetes Research Centre (University of Leicester). Sponsor oversight was undertaken by the CCTU on behalf of the Cambridge University Hospitals NHS Foundation Trust and the University of Cambridge.
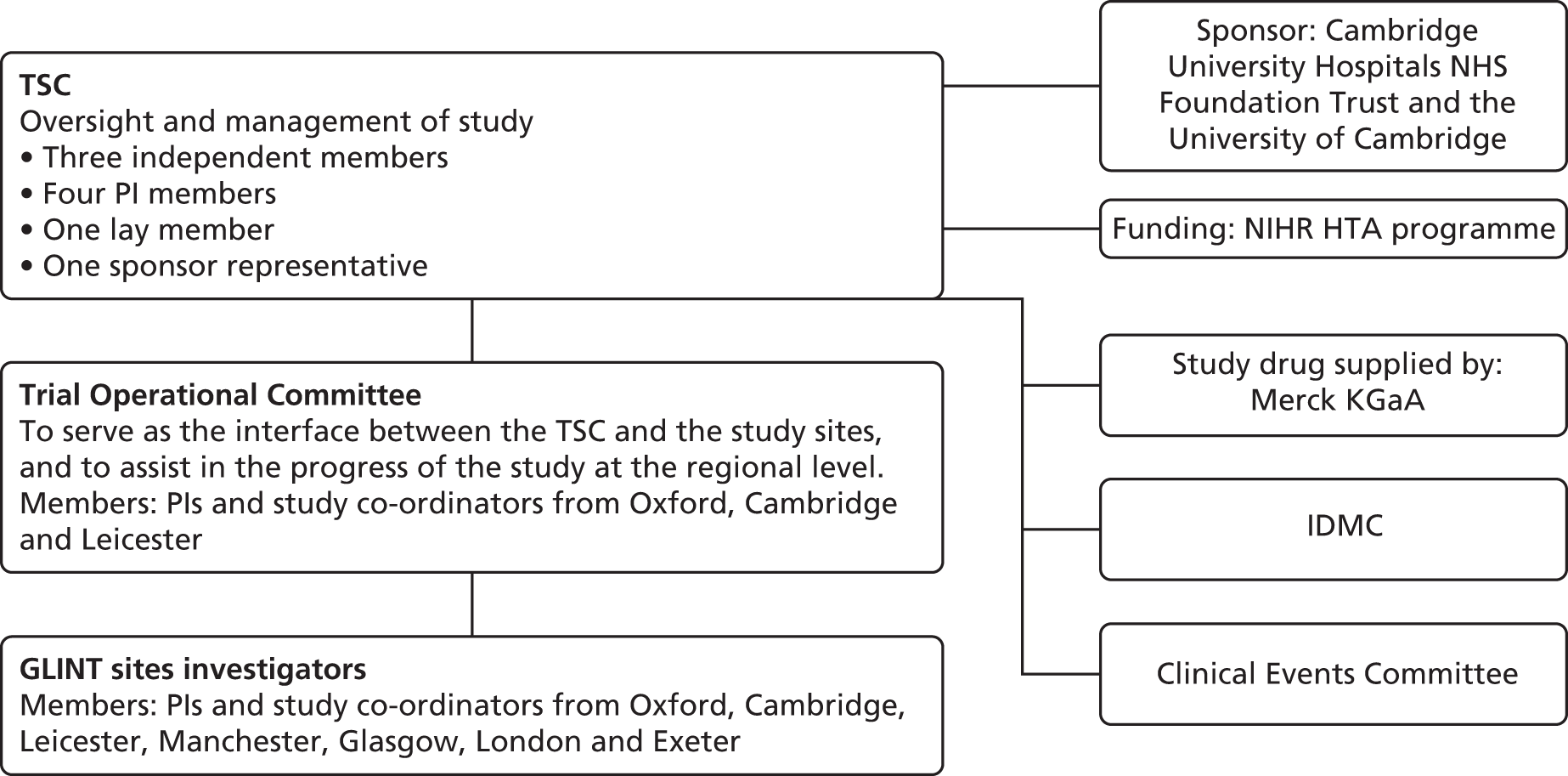
A Trial Steering Committee (TSC), operating under an agreed charter (see Appendix 1 for the list of members and www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation to view the charter), was charged with the oversight of the scientific, professional and operational conduct of the study and was the main decision-making body for GLINT. The TSC comprised nine individuals: three senior independent academic representatives who are experts in their field (one of whom chaired the committee), four GLINT principal investigators, one lay representative and one sponsor representative. When the lay member encountered difficulties with complying with the meeting schedule, an additional lay member was recruited to ensure that there was appropriate patient representation on the committee. All TSC members were approved by the funder. The TSC met four times during the feasibility phase (July 2012, February 2013, October 2013 and November 2015), with two additional informal non-quorate progress updates provided (May 2014 and June 2015).
An Independent Data Monitoring Committee (IDMC) was responsible for reviewing the unblinded safety information from the trial as it accrued. The committee consisted of four senior academic members with experience of conducting randomised trials in the fields of CVD, cancer and diabetes. The independent statistician member was not affiliated with the MRC Epidemiology Unit, the DTU or the sponsor. Members were approved by the funder and are listed in Appendix 2. The IDMC reviewed the protocol and agreed to the charter (www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation) prior to trial initiation (March 2014). The committee then met by teleconference to review unblinded data (including SAEs) after 249 participants had been followed up for 3 months (May 2016).
The Trial Operational Committee was composed of centre leads and was co-chaired by representatives from the MRC Epidemiology Unit and the DTU. Its primary role was to serve as the interface between the TSC and the study sites. Given the small number of events requiring independent adjudication, membership of the Clinical Endpoint Committee (for the charter see www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation) was restricted to one person (Professor Jane Armitage, Clinical Trial Service Unit, University of Oxford) during the feasibility phase.
The overall governance structure of GLINT, including the lines of responsibility and reporting, is shown in Figure 3.
FIGURE 3.
Overview of trial governance. HTA, Health Technology Assessment; NIHR, National Institute for Health Research; PI, principal investigator.
Patient and public involvement
The participant information sheet and consent form were reviewed by a patient and public involvement (PPI) group prior to ethics submission. The suggested changes to the documents were implemented.
Two of the TSC members were patient representatives at different times during the trial. A lay representative has been a member of the GLINT TSC since its inception. He has been involved in the design and management of the research, including commenting on the methods and materials for participant recruitment and follow-up and the means of delivering the study drug to participants. He gave his opinion concerning the acceptability and feasibility of the available options for each aspect of the trial that related to participants.
Feedback about study participation has been sought through questionnaires and we were planning to invite a subset of GLINT participants to take part in focus groups to further inform the development of the protocol for the full trial. The PPI panel at Cambridge University Hospitals NHS Foundation Trust reviewed the lay summary for the report, as well as the summary of results for the trial participants. The panel was also approached to provide feedback on the design of the full GLINT proposal and members reviewed the lay summary for the grant application. Participant and GP recommendations will be implemented in the full GLINT proposal.
The CCTU, on behalf of the sponsor, was responsible for monitoring trial sites according to the schema shown in Figure 4. See Trial recruitment for a description of the trial sites.
FIGURE 4.
Trial site monitoring schema.

Trial setting
The study took place in general practices and clinical research facilities in Cambridgeshire, Norfolk and Leicestershire.
Trial recruitment
A total of 10 general practices in Cambridge acted as sites where participants were identified, consent procedures were undertaken and baseline measurements were collected. A further 21 practices (seven in Cambridgeshire and 14 in Leicestershire) contributed to the identification of potentially eligible participants as participant identification centres (PICs). Participants who were identified by this route were seen at local clinical research facilities to undertake consent procedures and baseline measurements. Practices were recruited with support from Comprehensive Local Research Network (CLRN) staff. The recruitment and initiation of general practice sites and identification of potentially eligible individuals through searches of general practice electronic medical records necessitated several face-to-face meetings between investigators and staff from individual practices or groups of practices.
Four clinical research facilities that were part of the University of Cambridge MRC Epidemiology Unit, in Cambridge, Ely, Wisbech and Norfolk, and one in Leicester (Leicester Diabetes Research Centre) took part in the study. All research procedures were undertaken by a research nurse or suitably qualified research assistant either from within the research team or provided by the National Institute for Health Research (NIHR) Eastern Clinical Research Network. All staff had undergone appropriate training including attendance at a trial initiation meeting.
We intended to recruit participants through three complementary routes, detailed in the following sections.
The NHS Health Check programme
The NHS Health Check programme was implemented across England in April 2009. People aged 40–74 years with no prior diagnosis of CVD, diabetes or kidney disease are invited for a Health Check every 5 years. The NHS Health Check programme was selected to identify potentially eligible GLINT participants because the Health Check measurements include all components of the CVD risk score algorithm, as well as a measure of glycaemia for the subgroup of people at increased risk of developing T2D.
We initially planned to collaborate with Public Health England and local authorities (who are responsible for co-ordinating the NHS Health Check programme) so that, when potentially eligible individuals were identified during a health check, they could be provided with brief information about the study and the study centre contact details. However, those responsible for co-ordinating the NHS Health Check programme were focused on delivering the programme and increasing uptake and were not in a position to embed the initiation of GLINT participant recruitment into their existing routines.
Existing research registers
We searched GLINT investigators’ research registers for potentially eligible participants. The registers include participants from previous population-based observational studies and trials that have included assessments of glycaemia and CVD risk factors. Registered participants have provided consent to be contacted about future research projects.
General practitioner electronic records
In collaboration with GPs and study staff, practice staff in Leicester and Cambridge searched electronic patient medical records for values for fasting plasma glucose/HbA1c and an estimate of modelled 10-year CVD risk. An initial assessment of general practice medical records in Cambridge practices demonstrated that few NHS Health Checks had been undertaken in recruited practices and few records contained both recent values for fasting plasma glucose/HbA1c and an estimate of modelled 10-year CVD risk. Consequently, in the Cambridge practices, we utilised the Cambridge Diabetes Risk Score to identify potentially eligible individuals. The Cambridge Diabetes Risk Score is an algorithm that uses routinely collected non-biochemical parameters to identify those at risk of developing T2D and CVD. 22,23 Parameters include age, body mass index (BMI), prescription of antihypertensive medication, family history of diabetes and history of smoking.
Potentially eligible participants (see Inclusion and exclusion criteria) were sent the brief participant information sheet for the study with a covering letter by post from their general practice. Prior to contacting identified individuals by post, GPs were asked to review the list generated by the medical record search to exclude any patients for whom an invitation would be inappropriate, in their professional judgement, for reasons other than those listed on the exclusion criteria. For potential participants who were identified from the existing research registers, brief information sheets with a covering letter were sent from the study centres. The brief participant information sheet invited individuals to contact the study centre if they were interested in participating in the trial. Individuals who responded were sent the full participant information sheet, including a copy of the consent form. The brief and full participant information sheets and covering letter are available at www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation. Although the initial response rate to the invitation was reasonable (14%), the eligibility rate among those who agreed to take part was lower than expected (39%). We therefore increased the specificity of the general practice record search strategy by inviting participants from the upper end of the practice population distribution of the Cambridge Diabetes Risk Score, which resulted in an increase in the eligibility rate.
People were invited to attend a local clinical research facility or their participating study practice to discuss the study and undergo eligibility assessments. Individuals were recruited to participate in the full trial, with an expected duration of treatment and follow-up of around 5 years.
Inclusion and exclusion criteria
Inclusion criteria
To be included in the study, the participants were required to have met all of the following criteria:
-
understand the study procedures, the alternative treatments available and the risks involved in the study and voluntarily agree to participate by providing written informed consent
-
aged ≥ 40 years
-
HbA1c level of ≥ 36.6 mmol/mol but < 47.5 mmol/mol (≥ 5.5% but < 6.5%) measured within 1 year prior to enrolment
-
estimated 10-year CVD risk of ≥ 20% as assessed by the Framingham Risk Score24 or QRISK225 scores (laboratory values used for the risk calculation must have been collected no more than 1 year prior to enrolment)
-
estimated glomerular filtration rate (eGFR) of ≥ 45 ml/minute/1.73 m2 as determined by the Modification of Diet in Renal Disease (MDRD)-4 method (measured within 6 months prior to enrolment)
-
agree to allow study staff to contact his or her GP and/or consultant to notify them of study participation and to obtain all medical records necessary for complete data ascertainment during the follow-up period (including recording of NHS number and access to HES)
-
agree to be tagged for mortality with the ONS and to be tagged for –
-
time to first non-melanoma cancer diagnosis (supplied by the National Cancer Registry)
-
death attributable to a non-melanoma cancer cause according to the primary/underlying cause of death on the death certificate (supplied by the ONS).
-
Exclusion criteria
The participants could not enter the study if any of the following applied:
-
unable to provide written informed consent
-
prior history of physician-diagnosed T2D (participants with a history of gestational diabetes that resolved after pregnancy were permitted to enrol)
-
prior history of CVD, defined as a myocardial infarction or a surgical or percutaneous coronary revascularisation procedure or a stroke (haemorrhagic or ischaemic) (participants with a prior transient ischaemic attack or unstable angina were not excluded and could be enrolled)
-
planned or anticipated coronary revascularisation procedure within 6 months following enrolment (participants with a previous peripheral revascularisation procedure were not excluded and could be enrolled)
-
breastfeeding or known to be pregnant
-
taking metformin (for any reason) or had taken metformin in the last 3 months
-
history of cirrhosis of the liver or other significant hepatic impairment, as assessed by medical history
-
end-stage renal disease [chronic kidney disease (CKD) stage 3b or worse, eGFR of < 45 ml/minute/1.73 m2].
-
in the investigator’s opinion, a medical history that indicated a life expectancy of < 2 years or that might limit an individual’s ability to take the study treatments for the duration of the study
-
any other significant disease or disorder that, in the opinion of the investigator or the potential participant’s GP, would put the participant at risk because of participation in the study or could influence the results of the study or the participant’s ability to participate in the study
-
enrolled or participated, within 12 weeks prior to enrolment, in another experimental protocol involving the use of an investigational drug or device or an intervention that would interfere with the conduct of the study.
Randomisation
A nominated Oxford DTU statistician (with no other involvement in GLINT) generated a sequence of unique randomisation numbers for each study site using a computer macro. The statistician, in strict confidence, randomly assigned treatment allocation codes for metformin prolonged-release tablets or placebo in a 1 : 1 ratio to each randomisation number, blocked within each site and with a block size of four.
The randomisation numbers and their respective treatment allocation codes were provided in strict confidence to a nominated Oxford DTU senior applications programmer and the IDMC independent statistician only. The nominated programmer stored them in the encrypted Medication & Codebreak Database with access being available only for a single participant at a time and only to staff who were authorised to unblind assigned study medication (the chief investigator and one nominated principal investigator). A secure system automatically produced medication distribution lists detailing which study medication packs should be sent to which participants. This system identified from the Trial Management System Database which participants required titration or maintenance packs. It then utilised the encrypted Medication & Codebreak Database to allocate the next available pack that had a treatment code that matched the treatment code that was allocated to a participant’s randomisation number. Finally, it accessed the encrypted contact database to acquire the participant contact details that were required for the mailing label.
Immediately after database lock, the Oxford DTU informatics group incorporated the treatment allocation codes into the Trial Management System Database, linking participant data with treatment allocation so that unblinded data could be made available for analysis. The randomisation numbers alone (i.e. without the treatment allocation codes) were added to the Trial Management System for allocation to participants who were eligible for randomisation. Randomisation was referred to in the participant information sheets and discussed during the consent process.
Intervention
Participants were allocated, in addition to usual care, to 500 mg of prolonged-release metformin (Glucophage SR) or the matched placebo, up to three tablets per day administered orally. We standardised one aspect of usual care by providing all participants with a theory-based brochure containing advice about reducing the risk of heart attack and/or diabetes that was based on previous prevention studies carried out in Leicester. 21,26,27 Participants were given a card that provided information about the trial and study drug and safety instructions (see Appendix 3).
Dosage
Randomised participants received the IMP packaged in high-density polyethylene bottles with dosing instructions. Having ascertained that the proposed packaging would fit through the majority of letter boxes available in large hardware stores, the IMP was posted directly (by Anderson Brecon Ltd, Hay-on-Wye, Hereford) to the participants’ homes in 16-weekly batches. We telephoned the first 10 participants at each site to confirm receipt of the study drug and to assess acceptability of this mode of delivery. All patient contacts were documented in site files. For the first batch following randomisation, individuals were advised to take one tablet per day (500 mg of prolonged-release metformin or placebo) for 4 weeks, titrating up to two tablets per day for weeks 5–8 and three tablets per day thereafter. We contacted participants by telephone, at approximately week 4 and week 8, to assess tolerance and to further explain the titration process. The bottle labels for the titration and maintenance packs are shown in Appendix 4. We sent letters to participants summarising the information concerning the dose of the study drug in both the titration and the maintenance phases and advice about safety (www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation).
Any participants who could not tolerate 1500 mg per day of the study drug were advised to take the highest tolerated dose. The tolerated dose was determined during the titration telephone calls at week 4 and week 8 and at the 3- and 6-month visits. Participants were asked to continue taking their study medication for the duration of the feasibility study.
Participants with moderate renal impairment (CKD stage 3a, eGFR 45–59 ml/minute/1.73 m2) recorded during their participation in the study were recommended a maximum daily dose of 1000 mg of prolonged-release metformin/placebo. Participants whose eGFR fell to < 45 ml/minute/1.73 m2 were discontinued from taking the study drug but remained in the study for follow-up.
If participants experienced symptoms suggestive of episodes of hypoglycaemia, they were advised to down-titrate their dose. Any participants with continued potential hypoglycaemic episodes were advised to discontinue the study medication.
Legal status of the drug
Merck KGaA (Bedfont Cross, Middlesex, UK) are the marketing authorisation holders of Glucophage SR 500-mg prolonged-release tablets (marketing authorisation number PL 11648/0054). Glucophage SR is licensed for use in the treatment of T2D in adults for whom dietary management and exercise alone does not result in adequate glycaemic control. The study was carried out under a Clinical Trial Authorisation. The IMP dossiers for Glucophage SR and matching placebo are included as additional editorial documentation (www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation).
Concomitant therapy
Concomitant medications were used at the discretion of participants’ usual care providers. Open-label use of metformin was contraindicated during the study except for those participants who developed T2D, in which case up to a maximum of 1 g per day was allowed in conjunction with continued study medication. Participants’ GPs were provided with information about the maximum dose of metformin that they could prescribe according to the dose of study drug that the participant was taking (www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation).
Any prescribed medication other than the study medication was recorded at the baseline visit based on information provided by participants. Information on prescribed medications was updated using data collected from the GP questionnaires that were completed at 4 months, 1 year and the end of the study.
Outcome measures and data collection
Main outcome measures for the feasibility study
The outcomes assessed in the feasibility study consisted of a combination of process and outcome measures:
-
number of general practices recruited
-
numbers of people identified through the different search strategies
-
proportion of participants who responded to the invitation letter
-
availability of qualifying laboratory values in participants’ medical records
-
difference between the qualifying laboratory values obtained from medical records and those obtained at baseline visits
-
proportion of consenting participants who were randomised
-
baseline characteristics of recruited participants by study group.
In the treatment phase of the feasibility study, the following process and outcome measures were assessed as per the schedule of events (see Schedule of events):
-
reliability and acceptability of the delivery of the IMP to participants’ homes
-
proportions of GP and participant questionnaires returned
-
participant medication adherence, assessed using the following sources:
-
the study database (number of people who were no longer being sent study medication)
-
during dose titration telephone calls at approximately 4 and 8 weeks
-
during follow-up clinic visits at 3 and 6 months
-
questionnaire responses
-
-
frequency of daily tablet taking
-
proportions of participants taking one, two and three tablets per day
-
reasons for non-adherence
-
feasibility of (S)AE reporting and collection of data for outcomes, morbidities, quality of life and health service use
-
frequency, nature and severity of adverse effects including SAEs
-
proportions of participants attending follow-up measures at 3 and 6 months and the characteristics of those with missing data
-
change in HbA1c level and renal function (eGFR and creatinine) between baseline, 3 and 6 months
-
change in levels of cholesterol [total, high-density lipoprotein (HDL) and low-density lipoprotein (LDL)] and plasma vitamin B12 between baseline and 6 months
-
changes in laboratory test results between baseline and 6 months among those still taking the study drug at 6 months
-
frequency of study end points
-
values for functional status [Short Form questionnaire-8 items (SF-8)] and health utility [EuroQol-5 Dimensions (EQ-5D)] measures
-
frequency of participant withdrawal from the study.
Baseline data collection
During the face-to-face baseline visit, data were collected directly into the electronic case report form (eCRF) that was created as part of the Trial Management System Database using InferMed MACRO version 3 (InferMed Ltd, London, UK) software developed by DTU staff for the trial. These data included:
-
sociodemographic information – date of birth, sex, ethnicity, smoking status and alcohol consumption
-
medical history (including diabetes, CVD and previous cancer) and any medication use
-
clinical variables – height, weight and blood pressure measured according to standardised operating procedures
-
modelled 10-year cardiovascular risk (QRISK2/Framingham Risk Score)
-
self-reported morbidity (CVD, cancer, T2D), health service use in the past 12 months [adapted Health Economics Research Centre (HERC) questionnaire] and health utility and functional status (EQ-5D and SF-8).
Blood samples were taken and sent to the local NHS clinical biochemistry laboratories at Leicester General Hospital and Addenbrooke’s Hospital for measurement of HbA1c, total cholesterol, HDL cholesterol, triglycerides, vitamin B12, creatinine and alanine aminotransferase (ALT) using standardised protocols. The LDL cholesterol level was estimated using the Friedewald equation. 28 Once test results had been returned from the laboratory, data were entered onto the eCRF. Data were used to estimate the Framingham modelled 10-year CVD risk, which was entered onto the eCRF.
In addition, with consent, additional research samples were collected and stored at –80 °C for genetic and other biomarker studies. All samples were logged into a central database, link anonymised and stored either at access-controlled local sites or in an off-site Human Tissue Authority-licensed facility.
We contacted participants’ general practices to collect additional information, including recorded modelled 10-year CVD risk scores and prescribed medication. In Cambridgeshire, historical qualifying laboratory results were requested from GPs. In Leicestershire, these were available through the hospital iLab Operations Software system (Agilent, Santa Clara, CA, USA).
Follow-up data collection
Follow-up information on adherence to study medication, side effects, treatment satisfaction and events was collected by paper questionnaires, sent to the participants and their GPs. Questionnaires were sent to participants and their GPs in parallel, at 4 months, 1 year (when applicable) and the end of the study (see www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation). Participants were asked how many tablets of study medication they took each day (possible responses: one, two, three and none), how regularly they had taken study medication during the previous year (possible responses: every day, most days, only occasionally and never) and if they were willing to continue taking the study medication (possible responses: yes or no). The presence of side effects was assessed with a binary question and how bothersome side effects were was assessed with a single question with a seven-point Likert-type scale response. Treatment satisfaction was assessed using questions with seven-point Likert-type scale response sets.
Participants and GPs were asked to return the completed questionnaires to the DTU in Freepost envelopes. GPs were instructed to include any prescriptions and information relating to any events. Those who did not respond to the questionnaires were sent reminders at 4 weeks and again at 8 weeks. For the end-of-study questionnaire, a reminder was sent at 4 weeks only. With the exception of the end-of-study questionnaire, participants and GPs were contacted by telephone if they did not respond to these reminders.
In the week commencing 31 May 2016, a brief survey was sent along with a study newsletter to all participants who had not withdrawn from data collection. The survey (available at www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation) asked about the willingness of participants to complete future GLINT questionnaires using an online system and allowed participants to leave open-ended comments in a free-text box about their experience of the study. No reminders were sent to non-respondents.
Table 1 shows the timings and details of each study visit. The recruitment visit (visit 1), visit 2 and visit 3 were clinic visits. All other visits were carried out remotely, either by telephone or by questionnaire.
Schedule of events
| Visit activities | Recruitment | Visit | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | |||||||
| Baseline (day 0) | 4 weeks | 8 weeks | 3-month visit | 4-month mail-out | 6-month visit | Annual GP/participant questionnaire | End-of-study questionnaire | ||
| Demographics and medical history | ✓ | ✓ | |||||||
| Biochemistry: HbA1c, total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides and creatinine levels | ✓ | ✓ | ✓ | ||||||
| Vitamin B12 level, liver function tests, total cholesterol and LDL and HDL cholesterol levels | ✓ | ✓ | |||||||
| Height, weight and blood pressure | ✓ | ||||||||
| Modelled 10-year cardiovascular risk (Framingham Risk Score) | ✓ | ||||||||
| Informed consent and stored blood sample authorisation | ✓ | ||||||||
| Randomisation | ✓ | ||||||||
| Drug dispensation starts | ✓ | ||||||||
| Telephone call to check titration competency and AEs | ✓ | ✓ | |||||||
| Medication count | ✓ | ✓ | |||||||
| Participant questionnaire | |||||||||
| Self-reported morbidity (CVD, cancer and T2D) | ✓ | ✓ | ✓ | ✓ | |||||
| Medication adherence | ✓ | ✓ | ✓ | ||||||
| Health service use in past 12 months (adapted from HERC questionnaire) | ✓ | ✓ | ✓ | ✓ | |||||
| EQ-5D score | ✓ | ✓ | ✓ | ✓ | |||||
| Treatment satisfaction/SF-8 | ✓ | ✓ | ✓ | ||||||
| AEs/side effects | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| GP questionnaire | |||||||||
| Vital status, morbidity (CVD, cancer and T2D) | ✓ | ✓ | ✓ | ||||||
| Current prescribed medication | ✓ | ✓ | ✓ | ||||||
| AEs and reported pregnancies | ✓ | ✓ | ✓ | ||||||
Main trial outcomes
The participant questionnaires included a section for entering study events, divided into three subsections: (1) cardiovascular events, (2) cancer events and (3) other events including diabetes. A table with examples was provided (see www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation). The GP questionnaire was designed to capture the same type of information but it also included a section for reporting a death event. We also collected data concerning functional status (SF-8), health utility (EQ-5D) and health service use (frequency of consultations with a GP, nurse, physiotherapist or other health worker and frequency of consultations in the outpatient or accident and emergency department in the previous 12 months).
Data from the returned questionnaires were entered into the database by DTU staff, in duplicate. All data were manually reviewed and potential events (both AEs and outcome events) were flagged.
Information from medication prescriptions was entered into the database by the local study team in Cambridge.
When a participant or GP indicated that an event or a primary or secondary end point (as per the definition of the main trial end points) had occurred, further information was collected from the participant’s GP by the research teams by telephone or e-mail, if not already provided with the questionnaire. Additional information was then entered into the database by the local study teams.
Safety information
During the face-to-face visits at 3 and 6 months, participants were asked about any AEs that had occurred. Furthermore, the paper questionnaires included a section for entering illnesses, side effects and hospitalisations. Finally, participants were encouraged to report AEs to the study centre using the Freephone number provided. Non-SAEs were reported only if they resulted in cessation of the IMP and the event was considered to be related to the study drug. For this study, the reference safety information is provided in section 4.0 of the summary of product characteristics (SmPC) for Glucophage SR prolonged-release metformin. 29 The following information was recorded: description of the event, date of onset, dosage of the study drug and any required changes, assessment of seriousness, assessment of expectedness, relevant medical history, concomitant medications, causality assessment and resolution/outcome.
Serious adverse events
Serious adverse events were reported when they met the definition in the GLINT protocol, as follows:
A SAE is defined as any untoward event or medical occurrence that, at any dose:
-
resulted in death
-
was life-threatening (note that the term ‘life-threatening’ in the definition of a ‘serious adverse event’ refers to an event in which the participant was at risk of death at the time of the event; it does not refer to an event that hypothetically might have caused death if it were more severe)
-
required inpatient hospitalisation or prolongation of existing hospitalisation
-
resulted in persistent or significant disability/incapacity
-
was a congenital anomaly/birth defect
-
was another important medical event deemed significant by the investigator.
Note that other events that did not result in death, were not life-threatening or did not require hospitalisation were considered to be a SAE when, based on appropriate medical judgement, the event jeopardised the participant and required medical or surgical intervention to prevent one of the outcomes listed above.
Serious adverse events were recorded on the eCRF via one of the two paths discussed in the following sections.
Direct participant contact with the study centre
We instructed participants to contact their study centre if they had any serious illness, had a hypoglycaemic event that required the assistance of another person, were admitted to hospital for any reason or discontinued their drug. When such a study contact occurred, study centre staff conducted a telephone interview to establish the participant’s status and record any relevant clinical and safety data. If the centre staff had any concerns about the participant’s status they instructed the participant to contact his or her GP.
Questionnaires (month 4, year 1 when applicable and at the end of the study)
Safety data were collected using the participant questionnaire, which included questions to capture safety data. The questionnaires sent to the participants’ GPs also included questions relating to safety.
Completed questionnaires were mailed to the DTU along with any relevant supporting documentation. If further data were required in addition to the questionnaires, the study staff contacted participants and/or their GP.
The Cambridge study team received immediate notification by e-mail if a SAE had been recorded on the trial eCRF and ensured that the event was reported to the CCTU for reporting to the MHRA within the appropriate timelines. In the event that a participant contacted the study staff and immediate eCRF data entry was not possible, a SAE form was faxed or e-mailed to the Cambridge study team to ensure that reporting timelines could be met.
Expected events, which may or may not fulfil the criteria for serious, are listed in the Modified Safety Reporting section of the GLINT protocol (www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation). Any events that occurred that met the criteria for serious, but which were not listed in the Modified Safety Reporting procedure, were collected and reported. However, those detailed below were subject to a modified safety reporting plan (see Modified Safety Reporting flow chart at www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation). These events included (1) primary and secondary study end points, (2) other cardiovascular events of interest (that may be associated with the primary cardiovascular end points and are included in the items sent for independent adjudication), (3) other non-cardiovascular events of interest (e.g. incident cancer, incident T2D), and (4) known common toxicities of metformin.
Expected adverse reactions/serious adverse reactions
The common side effects of metformin include gastrointestinal upset and diarrhoea. Hypoglycaemia does occur rarely and lactic acidosis is a very rare but serious AE. Evidence from participants with NDH in the US Diabetes Prevention Program (DPP) suggests that hypoglycaemia is not expected and SAEs are very rare. 12 We advised GLINT participants about the symptoms suggestive of hypoglycaemia (such as tachycardia, palpitations, shakiness, sweating, inability to concentrate, dizziness, hunger, blurred vision, obvious impairment of motor function and confusion or inappropriate behaviour that reverses after intake of carbohydrates) and how to respond. Serious hypoglycaemia (requiring the assistance of another person) was regarded as a SAE triggering the safety reporting mechanism described above. We issued each participant with a card indicating that he/she was participating in a research study (see Appendix 3). This card included the name and telephone number of the investigational site to be contacted in case of an emergency, including any episode of suspected hypoglycaemia requiring the assistance of another person. The card also specified temporary interruption of treatment with study medication if participants underwent general anaesthesia or investigation with iodine-containing radiographic contrast media and if participants were at risk of tissue hypoxia or sudden deterioration of renal function, for example as a result of severe dehydration or infection, shock, sepsis, acute heart or respiratory failure, hepatic impairment or recent myocardial infarction.
An extremely rare but potentially serious side effect of metformin is lactic acidosis. The risk of lactic acidosis is increased among people with CKD stage 3b or worse. Consequently, we excluded individuals with an eGFR of < 45 ml/minute/1.73 m2 and measured the creatinine level at baseline, 3 and 6 months in the feasibility phase. Participants’ GPs were reminded of contraindications to metformin and this rare side effect and the need to discontinue study medication in the aforementioned situations (1) when they were informed that their participant had entered the study, (2) at the 4-month questionnaire phase in the feasibility phase, (3) annually thereafter when receiving their questionnaire, and (4) in the study newsletter. Written reminders detailing the action to be taken by participants in the event of a severe illness, including renal failure, were sent to participants with the 4-month questionnaire and every 12 months when they received their annual questionnaire. Participants who developed CKD (stage 3b or worse, eGFR of < 45 ml/minute/1.73 m2) during the study were not offered further metformin/placebo. The mechanism for reporting and responding to potential AEs is shown in Safety information.
Chronic metformin use has been associated with vitamin B12 deficiency. 30 Severe vitamin B12 deficiency can result in neuropathic symptoms that can be mistaken for diabetic neuropathy. However, the strength of this association has varied in epidemiological studies and the clinical significance of mild vitamin B12 deficiency syndromes is unclear. We measured plasma vitamin B12 levels at the baseline and 6-month visits. If a participant had a vitamin B12 level of < 200 ng/l, the results were reviewed by a study clinician and his or her GP was contacted if necessary. The results of the feasibility study will inform a decision on whether or not it is necessary to continue monitoring vitamin B12 levels in the full trial.
Statistical methods
The analysis of the feasibility phase was descriptive and by randomised group (metformin/placebo), no p-values were calculated, and it includes a presentation of the process and outcome measures listed above separately by randomised group. The analysis followed a statistical analysis plan finalised in March 2016 (see www.journalslibrary.nihr.ac.uk/programmes/hta/090148/#/documentation).
Sample size
Formal sample size estimates were not appropriate for a study assessing feasibility rather than treatment effect and were not calculated. However, it is possible to quantify the precision with which certain parameters can be estimated in a feasibility study. Estimated values for each parameter were extrapolated from other similar studies and the precision of such an estimate (the width of the 95% CI) was then estimated based on the original planned sample size of 500 participants for the feasibility study.
The expected recruitment rate via the NHS Health Check programme, based on data from ADDITION-Leicester,31 was 20%. With 500 individuals, the 95% CI around an estimate of 20% would be from 17% to 24%. However, this study31 required an oral glucose tolerance test, so a recruitment rate of 20% may be an underestimate of what could be expected in GLINT (which does not require fasting or an oral glucose tolerance test). If, instead, the recruitment rate was 50%, the 95% CI around this estimate would be from 46% to 54%.
The expected modelled 10-year CVD risk, based on data from EPIC (European Prospective Investigation of Cancer)-Norfolk,32 was 2.2% per year. With 500 individuals, the 95% CI around an estimate of 2.2% would be from 1.1% to 3.9%.
In relation to the expected adherence to the IMP, based on data from the DPP,12 the proportion of participants who took ≥ 80% of the prescribed dose of the study medication was around 80% at 4 months. With 500 individuals, the 95% CI around an estimate of 80% of participants adhering at 4 months would be from 76% to 83%.
Following the 12-month delay in trial initiation because of changes in sponsorship (described in Table 2), we revised the estimates for a sample size of 250 participants. For medication adherence with 250 participants, the 95% CI around the estimate of adherence (80%) increased and was 75% to 85%. For modelled CVD risk with 250 participants, the 95% CI around the estimate of 2.2% per year was from 0.9% to 5.2%.
| Amendment date | Details of change |
|---|---|
| Amendment 1, dated 4 July 2014 |
|
| Amendment 2, dated 10 November 2014 |
|
| Amendment 3, dated 1 April 2016 |
|
Blinding
The study used a double-blind design: neither the participant and his or her GP nor GLINT personnel were aware of which treatment the participant was allocated to. The placebo had the same visual appearance as the IMP.
Data handling and record keeping
Trial Master File
The Trial Master File was located at the MRC Epidemiology Unit, Cambridge Biomedical Campus, and the other investigator sites had Investigator Site Files. All essential documents to demonstrate the compliance of the study and study personnel with the conditions and principles of good clinical practice and all applicable regulatory requirements were stored in the Trial Master File.
Source data
Study data included data collected as part of the NHS Health Check programme, data held in the existing research registries and data held in the databases of participating general practices. These source data were entered onto the eCRF by the research team at each site. For any data acquired that were entered directly onto the eCRF, the eCRF was considered the source data. Consent forms and any outstanding tests that were required to confirm eligibility were completed at the recruitment site. Data from annual questionnaires that were sent to participants and GPs were entered onto the eCRF by study personnel at the DTU. Further data on potential CVD events were collected by the study co-ordinators from GPs and participants when necessary.
Data protection and patient confidentiality
Local databases were used to identify and recruit potential participants. Thereafter, participants were randomised and all study data were entered into a Trial Management System Database using InferMed MACRO version 3 software and validation procedures. Participants were identified throughout the trial by a study-specific unique participant identification number. A separate encrypted participant contact database was used to store participant names, addresses, NHS numbers and other identifying details required for drug distribution. Access to the participant contact database was restricted to the secure computer system used to facilitate distribution of the study medication and DTU informatics personnel not otherwise involved in GLINT who were able to amend the details when changes were required.
Local sites stored source data for each participant. These were labelled with the participants’ study number, initials and date of birth. Data included the original signed and dated consent form, the results of any laboratory tests or procedures carried out and any additional documentation obtained to support events reported in the annual questionnaires. The eCRF was also considered to be source data for any data obtained from the participant during the baseline visits that were immediately entered onto the eCRF. Appendix 5 lists the data and the sources from which they were obtained. Source documents for other data points were kept in local co-ordinating centres and entered onto the eCRF by study personnel. The DTU sent out the annual questionnaires to the participants and their GPs and entered the data on receipt of the completed documents.
The GLINT staff ensured that the participants’ anonymity was maintained. The participants were identified only by initials and a participant identification number on the eCRF and Trial Management System Database. Consent forms, GP questionnaires and participant questionnaires necessarily contained participant names. However, all documents were stored securely and were accessible only to study staff and authorised personnel.
Feasibility of collection of clinical events
Data on events that potentially constituted the primary end points of the main study were collected as part of the feasibility study. If necessary, further information was obtained from participants or their GPs before the data were independently adjudicated by Professor Jane Armitage (Clinical Trial Service Unit, University of Oxford), who was unaware of study group allocation. The specific end points that were adjudicated were CVD mortality, non-fatal myocardial infarction, non-fatal stroke, all-cause mortality, hospital admission for congestive heart failure, hospitalisation for unstable angina and coronary, cerebrovascular or peripheral revascularisation. Events that were regarded as secondary end points for the main trial were reported but not adjudicated. These events included non-melanoma cancer, death attributable to a non-melanoma cancer cause and physician-diagnosed T2D.
Amendments to the protocol
A summary of all substantial study amendments is presented in Table 2.
Chapter 3 Results
Participant recruitment
In Cambridgeshire, we included 10 GP sites and 7 PICs. Fourteen PICs took part in Leicestershire.
In total, 4129 patients were identified from the GP surgery searches carried out by both GP sites and PICs across Cambridgeshire and Leicestershire and an additional 1122 potentially eligible people were found through searches of existing research databases in Cambridgeshire, Leicestershire and Norwich.
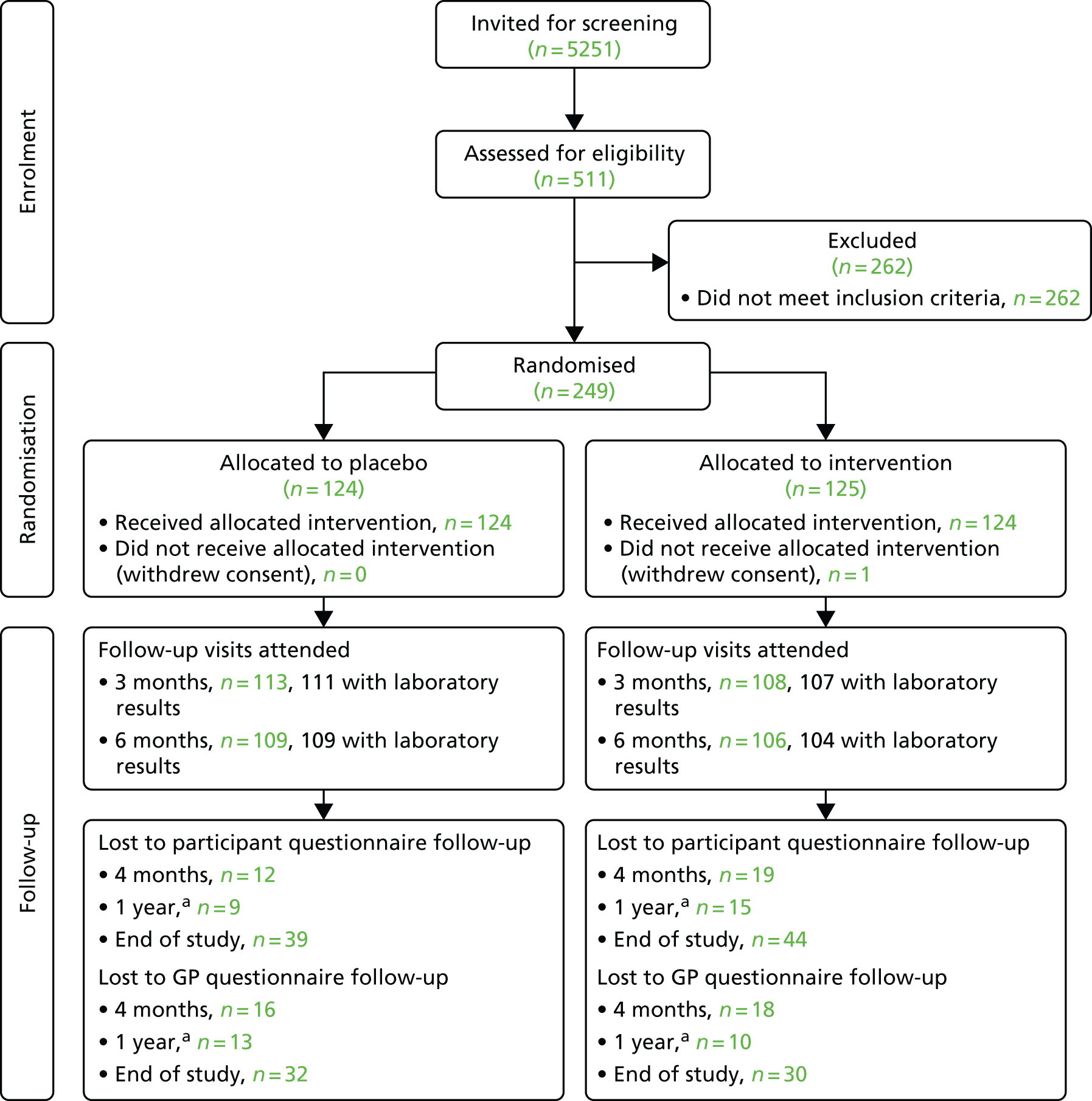
We posted a total of 5251 invitations to potentially eligible people. Of the people who were invited, 511 (9.7%) responded, attended a screening visit and consented to take part in the study. Following assessment against the inclusion and exclusion criteria, 262 people (51.3%) were found to be ineligible: 215 (82.1%) did not meet the CVD risk score criterion of ≥ 20% and 86 (32.8%) had an ineligible HbA1c score, with 46 (17.6%) being ineligible on both CVD risk and HbA1c criteria, and 7 (2.7%) were ineligible based on other criteria. Over a period of 9 months (from March to November 2015), we randomised 249 people (219 men and 30 women) into the study, which represents 4.7% of those originally invited.
Source of recruited participants
The GP sites and PICs were the main sources of eligible and willing participants, with 153 participants recruited by this route. The NHS Health Check programme directly provided only two recruited participants. Ninety-four people were recruited from existing research databases, as shown in Table 3.
| Source of recruitment | Study group, n (%) | |
|---|---|---|
| Placebo | Metformin | |
| NHS Health Check programme | 1 (0.8) | 1 (0.8) |
| Investigator research database | 45 (36.3) | 49 (39.2) |
| General practice electronic record search | 78 (62.9) | 75 (60.0) |
| Total | 124 | 125 |
The percentage of those invited who were randomised was 8.4% for the research database approach, 3.2% for the GP surgery search approach and 4.8% overall.
Weekly rates of recruitment increased as the study progressed, as shown in Figure 5.
FIGURE 5.
Participant recruitment.
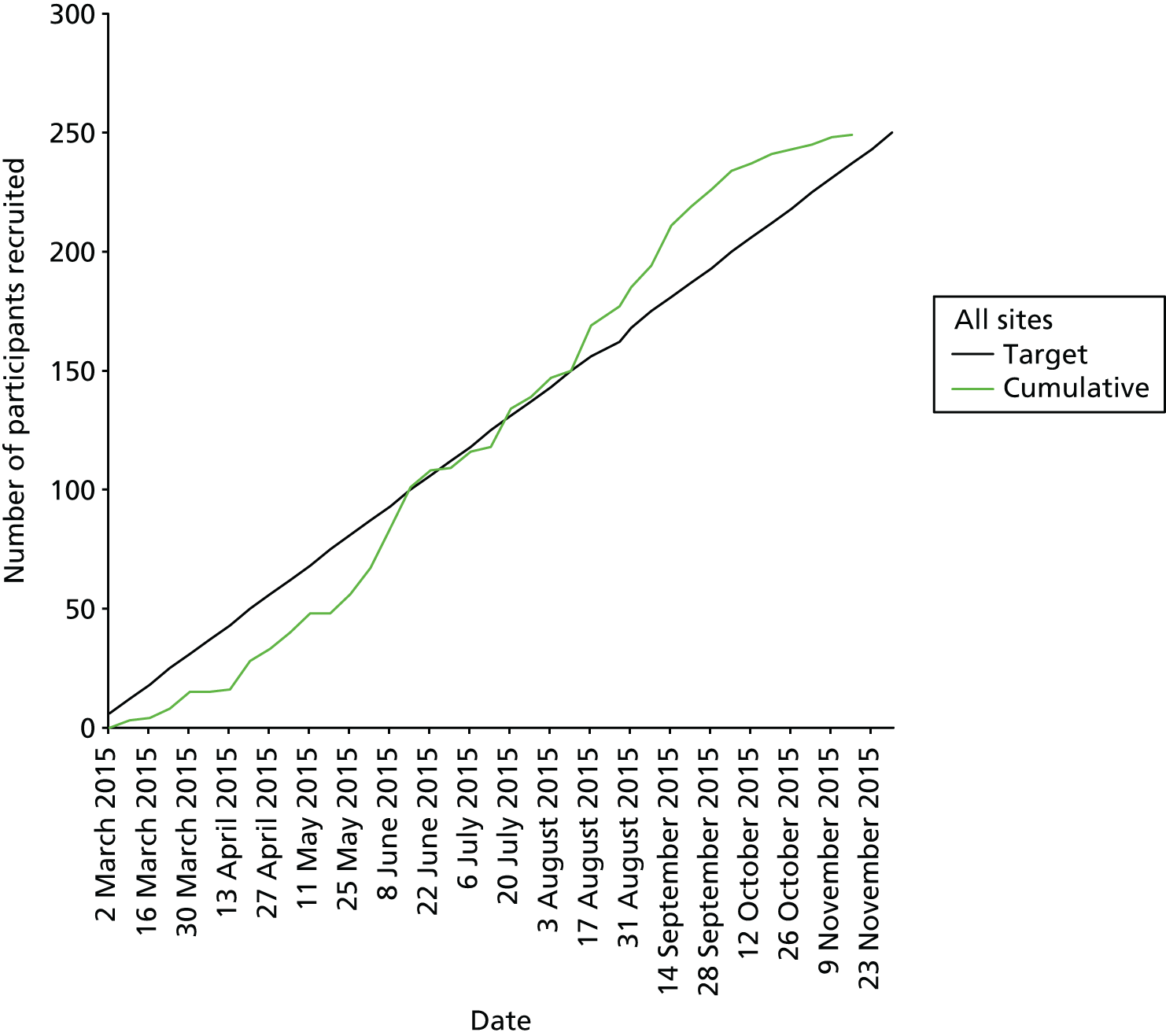
The increased efficiency of recruitment was due, in part, to the refinement of the criteria for searching GP medical records (using the Cambridge Diabetes Risk Score and selecting potential participants from the upper part of the population distribution for the risk score). This led to an increase in the proportion of individuals who consented to take part who met the eligibility criteria, as shown in Figure 6. In the initial search, we used a Cambridge Diabetes Risk Score threshold of > 0.37 in combination with an age of ≥ 40 years and a modelled 10-year CVD risk of ≥ 20% and randomly sampled from the resulting distribution. This resulted in 2.5% of those invited and 17.3% of those consenting being randomised. When we invited individuals from the upper part of the distribution of the Cambridge Diabetes Risk Score, this resulted in 5.2% of those invited and 51.5% of those consenting being randomised.
FIGURE 6.
Mean weekly percentage of participants randomised/number consenting to participate.

Staff working in CLRNs hosting GLINT and staff working in most of the recruited practices were unable to undertake searches of electronic medical records without assistance from study staff. Consequently, to recruit a practice as a PIC, explain the study design and procedures and assist with electronic searches of medical records and preparation of mail merges for invitation letters, study staff were required to visit participating general practices on several occasions.
Participant baseline characteristics
Participant characteristics are shown in Tables 4 and 5. Of the 219 men and 30 women randomised, the majority were of white ethnicity, with only five participants reporting a non-white background. This reflects the demographics of participating practices, which served predominantly white populations in spite of the overall ethnic diversity of the populations of Cambridge and Leicester. We recruited a relatively senior population with a mean age of 70 years. Participants were generally overweight and had elevated blood pressure and a mean modelled 10-year CVD risk of close to 30%. The majority were ex-smokers or current smokers. Over half were prescribed statins. The participants generally had normal liver and renal function tests at baseline. The mean HbA1c level was 41 mmol/mol (5.9%).
| Characteristic | Study group | |
|---|---|---|
| Placebo | Metformin | |
| Demographic variables, n (%) | N = 124 | N = 125 |
| Male | 111 (89.5) | 108 (86.4) |
| Ethnicity | ||
| White | 121 (97.6) | 123 (98.4) |
| Mixed | 0 (0.0) | 0 (0.0) |
| Asian/Asian British | 2 (1.6) | 2 (1.6) |
| Other (Mexican American) | 1 (0.8) | 0 (0.0) |
| Previous cancer | ||
| Yes | 5 (4.0) | 15 (12.0) |
| No | 119 (96.0) | 110 (88.0) |
| Smoking | ||
| Never | 43 (34.7) | 49 (39.2) |
| Ex-smoker | 67 (54.0) | 54 (43.2) |
| Current | 14 (11.3) | 22 (17.6) |
| Clinical variables, mean (SD) | ||
| Age (years) | 70.1 (6.2) | 69.9 (7.2) |
| BMI (kg/m2) | 30.1 (4.5) | 30.1 (4.4) |
| Height (m) | 1.7 (0.1) | 1.7 (0.1) |
| Weight (kg) | 89.8 (15.9) | 89.4 (16.2) |
| SBP (mmHg) | 141.9 (15.2) | 143.2 (14.6) |
| DBP (mmHg) | 80.5 (8.9) | 82.0 (11.1) |
| Self-reported medication, n (%) | N = 110 | N = 111 |
| Antihypertensive drugs | 73 (66.4) | 80 (72.1) |
| ACE inhibitor | 44 (40.0) | 54 (48.6) |
| Beta-blocker | 13 (11.8) | 15 (13.5) |
| Calcium antagonist | 46 (41.8) | 39 (35.1) |
| Diuretic | 19 (17.3) | 27 (24.3) |
| Other | 7 (6.4) | 10 (9.0) |
| Cholesterol-lowering drugs | 61 (55.5) | 65 (58.6) |
| Statins | 60 (54.5) | 58 (52.3) |
| Aspirin | 17 (15.5) | 16 (14.4) |
| Characteristic | Study group | |||
|---|---|---|---|---|
| Placebo | Metformin | |||
| n | Mean (SD) | n | Mean (SD) | |
| Qualifying biochemical variables | ||||
| HbA1c (mmol/mol) | 56 | 40.3 (2.7) | 48 | 40.7 (2.9) |
| eGFR (ml/minute/1.73 m2) | 53 | 91.3 (20.0) | 45 | 82.3 (18.2) |
| Framingham Risk Score | 124 | 29.3 (8.8) | 125 | 30.6 (11.1) |
| Cholesterol (mmol/l) | 67 | 4.8 (0.9) | 61 | 4.8 (1.1) |
| HDL cholesterol (mmol/l) | 59 | 1.3 (0.3) | 55 | 1.3 (0.3) |
| LDL cholesterol (mmol/l) | 56 | 2.7 (0.8) | 51 | 2.8 (0.8) |
| Triglycerides (mmol/l) | 58 | 1.5 (0.6) | 54 | 1.9 (1.4) |
| Creatinine (µmol/l) | 54 | 76.4 (16.9) | 45 | 83.7 (16.4) |
| Feasibility biochemical variables | ||||
| HbA1c (mmol/mol) | 124 | 40.6 (2.7) | 125 | 41.3 (2.7) |
| eGFR (ml/minute/1.73 m2) | 124 | 90.5 (18.2) | 125 | 89.4 (18.8) |
| Framingham Risk Score | 124 | 27.9 (7.6) | 125 | 29.6 (9.2) |
| Cholesterol (mmol/l) | 124 | 4.9 (0.9) | 125 | 5.0 (0.9) |
| HDL cholesterol (mmol/l) | 124 | 1.3 (0.4) | 125 | 1.3 (0.3) |
| LDL cholesterol (mmol/l) | 120 | 2.7 (0.9) | 123 | 2.8 (0.8) |
| Triglycerides (mmol/l) | 124 | 1.9 (1.1) | 125 | 2.0 (1.0) |
| Creatinine (µmol/l) | 124 | 78.7 (13.8) | 125 | 79.5 (16.1) |
| Vitamin B12 (ng/l) | 124 | 338.0 (103.3) | 125 | 353.3 (112.7) |
| ALT (U/l) | 124 | 26.8 (9.9) | 125 | 28.4 (22.3) |
Randomised groups were well matched with small differences in the proportion of participants who had a previous diagnosis of cancer (placebo 4%, metformin 12%), the proportion of current smokers (placebo 11.3%, metformin 17.6%) and the proportion prescribed angiotensin-converting enzyme inhibitors (placebo 40.0%, metformin 48.6%), as shown in Table 4. A large proportion of participants (98.5%) agreed to provide a sample for genetics/biomarker research. Among recruiting practices and participants, no concerns were raised relating to the issue and process of randomisation.
Qualifying laboratory test results
Qualifying laboratory results pre-dating recruitment to the trial were available for fewer than half of the participants (see Table 5). For example, we obtained a creatinine result from general practice or laboratory records (within the preceding 6 months) for 40% of participants and a cholesterol value for 67%. However, when information about test results was available in the GP medical records, it matched well with the data obtained at the baseline visit at the study centre (see Table 3).
Reliability and acceptability of the delivery of the investigational medicinal product to participants’ homes
Postal delivery of the study drug to participants’ homes appeared to be acceptable and worked well. No issues were escalated from the telephone calls to the first 10 participants at each site to indicate a problem with medication delivery. Only 5 out of 360 placebo packs (1.4%) and 4 out of 366 metformin packs (1.1%) required replacement.
Participant progress through the trial
The progress of participants through the trial is shown in the Consolidated Standards of Reporting Trials (CONSORT) flow diagram in Figure 7. One participant withdrew consent for follow-up for the primary outcome using register data.
FIGURE 7.
Participant flow. a, Of those who reached the 1-year follow-up time point prior to the end of the study.
Questionnaire response rates and visit attendance rates
The proportion of questionnaires that were completed and returned remained reasonably high throughout the study, for both GPs and participants, as shown in Table 6. Baseline data were 100% complete. At the end of the study (September 2016, 20 months after the first participant was recruited), the response rate among participants had fallen to 67% of those randomised, which equates to 74% of those who had not declined to receive a questionnaire. Response rates were slightly higher among participants in the placebo group than among those in the metformin group at all time points (75.2% vs. 72.3% of those issued at the end of the study, respectively). At the end of the study, the questionnaire response rate among GPs had fallen to 79% of those issued.
| Time point, recipient | Questionnaires | Reminders, n | ||||
|---|---|---|---|---|---|---|
| Issued, n | Returned, n | Returned, % of participants issued | Returned, % of participants randomised | First | Second | |
| 4 months | ||||||
| Participant | 245 | 218 | 89.0 | 87.6 | 66 | 38 |
| GP | 247 | 215 | 87.0 | 86.3 | 123 | 69 |
| 1 year | ||||||
| Participanta | 133 | 110 | 82.7 | N/A | 31 | 13 |
| GP | 144 | 122 | 84.7 | N/A | 40 | 19 |
| End of study | ||||||
| Participant | 225 | 166 | 73.8 | 66.7 | 79 | N/A |
| GP | 238 | 187 | 78.6 | 75.1 | 87 | N/A |
Adherence to the study drug
Adherence was assessed using the following sources:
-
study database (the number of people who were no longer being sent study medication)
-
dose titration telephone calls at approximately 4 and 8 weeks
-
follow-up clinic visits at 3 and 6 months
-
questionnaire responses.
Adherence varied over time as participants who had discontinued study medication were encouraged to restart, including restarting at a lower dose if the reason for discontinuation had been AEs. The frequency of premature discontinuation of the study drug was just under 30% by 6 months, with no significant difference between the study groups (log-rank test for the difference between groups: p = 0.39), as shown in Figure 8.
FIGURE 8.
Premature discontinuation of the study drug since randomisation.

By the end of follow-up, the probability of premature discontinuation of the study drug was 38.3% (metformin 43.2%, placebo 32.0%). The total person-years of exposure to the study drug were 114.6 in the placebo group and 112.9 in the metformin group. The mean duration of exposure was 0.92 years [standard deviation (SD) 0.46 years] and 0.90 years (SD 0.46 years), respectively. As shown in Table 7, at the 3-month visit, 192 out of the 221 participants who provided data reported that they were still taking the IMP (placebo, n = 97; metformin, n = 95; 87% of respondents). Assuming that all those who did not return a questionnaire were not taking the study drug, only 77% of participants were still taking the study drug after 3 months. Among questionnaire responders, 85.4% reported taking the study drug at 6 months. Assuming that all non-responders were non-adherent, the proportion taking the study drug at 6 months was 73%. The most common reason, in both study groups, for not taking the study drug was ‘refusal’, which related to overall participation in the study rather than necessarily the experience of AEs. Nevertheless, side effects were given as the reason for not taking the study drug by up to 31% of questionnaire respondents in the metformin group and 17% in the placebo group (see Table 7).
| Study drug status | Time point | |||||||
|---|---|---|---|---|---|---|---|---|
| 4 weeks | 8 weeks | 3 months | 6 months | |||||
| N | n (%) | N | n (%) | N | n (%) | N | n (%) | |
| Placebo group | ||||||||
| On study drug | 103 | 106 | 113 | 109 | ||||
| Yes | 91 (88.3) | 91 (85.8) | 97 (85.8) | 92 (84.4) | ||||
| No | 12 (11.7) | 15 (14.2) | 16 (14.2) | 17 (15.6) | ||||
| Reasons for not taking study drug | 12 | 15 | 16 | 17 | ||||
| AE | 0 (0.0) | 0 (0.0) | 1 (6.3) | 1 (5.9) | ||||
| Lost tablets | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||
| Forgot | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||
| Refused | 3 (25.0) | 8 (53.3) | 7 (43.8) | 5 (29.4) | ||||
| Stopped | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||
| Other | 7 (58.3) | 5 (33.3) | 6 (37.5) | 7 (41.2) | ||||
| Side effects | 2 (16.7) | 2 (13.3) | 2 (12.5) | 4 (23.5) | ||||
| Metformin group | ||||||||
| On study drug | 103 | 107 | 108 | 104 | ||||
| Yes | 92 (89.3) | 94 (87.9) | 95 (88.0) | 90 (86.5) | ||||
| No | 11 (10.7) | 13 (12.1) | 13 (12.0) | 14 (13.5) | ||||
| Reasons for not taking study drug | 11 | 13 | 13 | 14 | ||||
| AE | 0 (0.0) | 1 (7.7) | 1 (7.7) | 2 (14.3) | ||||
| Lost tablets | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||
| Forgot | 2 (18.2) | 0 (0.0) | 0 (0.0) | 0 (0.0) | ||||
| Refused | 1 (9.1) | 6 (46.2) | 4 (30.8) | 4 (28.6) | ||||
| Stopped | 0 (0.0) | 0 (0.0) | 1 (7.7) | 0 (0.0) | ||||
| Other | 6 (54.5) | 2 (15.4) | 3 (23.1) | 4 (28.6) | ||||
| Side effects | 2 (18.2) | 4 (30.8) | 4 (30.8) | 4 (28.6) | ||||
The majority of participants were able to titrate up to the maximum dose of the study drug. Among those participants for whom data were available (207/249), 81.5% in the placebo group and 75.6% in the metformin group reported taking three tablets per day at 6 months, as shown in Table 8. Participants reported that they had taken study medication on around 85% of the previous 14 days. This value was consistent over the first 6 months and did not vary between study groups.
| Study drug status | Time point | |||
|---|---|---|---|---|
| 4 weeks | 8 weeks | 3 months | 6 months | |
| Placebo group | ||||
| Tablets per day [N; n (%)] | ||||
| One | 91; 48 (52.7) | 91; 9 (9.9) | 97; 9 (9.3) | 92; 9 (9.8) |
| Two | 91; 41 (45.1) | 91; 46 (50.5) | 97; 16 (16.5) | 92; 8 (8.7) |
| Three | 91; 2 (2.2) | 91; 36 (39.6) | 97; 72 (74.2) | 92; 75 (81.5) |
| % of last 14 days taking any study medication [N; mean (SD)] | 102; 87.0 (29.3) | 105; 84.2 (33.1) | 111; 84.1 (35.2) | 106; 84.4 (34.4) |
| Metformin group | ||||
| Tablets per day [N; n (%)] | ||||
| One | 92; 42 (45.7) | 93; 10 (10.8) | 95; 7 (7.4) | 90; 9 (10.0) |
| Two | 92; 48 (52.2) | 93; 43 (46.2) | 95; 21 (22.1) | 90; 13 (14.4) |
| Three | 92; 2 (2.2) | 93; 40 (43.0) | 95; 67 (70.5) | 90; 68 (75.6) |
| % of last 14 days taking any study medication [N; mean (SD)] | 103; 85.0 (33.0) | 104; 86.3 (31.6) | 107; 83.9 (34.2) | 101; 84.7 (33.6) |
The question that was posed at 4 months concerning the regularity of taking study medication was answered by 85% of participants. Among respondents, 87% of the metformin group and 91% of the placebo group reported taking study medication every day or most days, as shown in Table 9. Assuming that all those who did not return questionnaires were not taking any study drug then the proportions taking the study drug every day or most days at 4 months were 72% in the metformin group and 79% in the placebo group.
| Regularity of taking study medication | Study group, n (%) | |
|---|---|---|
| Placebo (N = 108) | Metformin (N = 103) | |
| Every day | 82 (75.9) | 76 (73.8) |
| Most days | 16 (14.8) | 14 (13.6) |
| Only occasionally | 6 (5.6) | 6 (5.8) |
| Never | 4 (3.7) | 7 (6.8) |
Reported side effects
At 4 months, 48 out of the 201 participants who responded (23.9%) reported having side effects associated with the study medication, with similar proportions in the placebo and metformin groups (23.8% and 24.0%, respectively), as shown in Table 10. All but one of the participants reporting side effects also graded their severity, with 28% in the placebo group compared with 14% in the metformin group rating the side effects as being very bothersome.
| Participant-reported side effects | Study group, n (%) | |
|---|---|---|
| Placebo | Metformin | |
| Side effects from study medication? | N = 105 | N = 96 |
| Yes | 25 (23.8) | 23 (24.0) |
| No | 80 (76.2) | 73 (76.0) |
| How bothersome are the side effects? | N = 25 | N = 22 |
| 0 = Not bothersome at all | 2 (8.0) | 2 (9.1) |
| 1 | 3 (12.0) | 1 (4.5) |
| 2 | 3 (12.0) | 5 (22.7) |
| 3 | 7 (28.0) | 2 (9.1) |
| 4 | 2 (8.0) | 6 (27.3) |
| 5 | 1 (4.0) | 3 (13.6) |
| 6 = Very bothersome | 7 (28.0) | 3 (13.6) |
Adverse events and serious adverse events
Overall, there were 64 reported AEs, serious and non-serious, among 50 participants. All AEs and SAEs are listed by study group in Tables 11 and 12, respectively. In the placebo group, 10 participants (8.06%) experienced non-SAEs. In the metformin group, 18 participants (14.4%) experienced a total of 19 events (one individual having more than one event); event details are given in Table 11. Eight events in the placebo group and 12 events in the metformin group were related to the gastrointestinal system. There were 35 SAEs reported during the trial, 13 in the placebo group and 22 in the metformin group, none of which was deemed to be related to the study medication (see Table 12).
| Placebo group | Metformin group |
|---|---|
| Faecal soiling | Chest pain |
| Nausea | Diarrhoea |
| Nausea and loss of appetite | Constipation and headache |
| Constipation and dry skin | Gastrointestinal upset |
| Stomach upset (no diarrhoea), light-headedness and dizziness | Gastritis reported by GP. Discharge summary indicated pulmonary embolism |
| Tiredness and confusion | Indigestion and belching |
| Tiredness and not feeling right | Diarrhoea |
| Abdominal pain | Abdominal pain |
| Constipation | Light-headedness, breathlessness, tiredness and flatulence |
| Constipation | Loose stools and bloating |
| Loose stools, gastric upset and extreme flatulence | |
| Muscle pain in both arms | |
| Nausea | |
| Nausea and diarrhoea | |
| Taste disturbance and bloated stomach | |
| Thirst and dry mouth | |
| Tiredness and loss of appetite | |
| Other | |
| Other |
| Placebo group | Metformin group |
|---|---|
| Skin abscess, cellulitis | Gallstones, admitted to hospital for ERCP |
| Cellulitis of the right leg | Laparoscopic cholecystectomy |
| Guillain–Barré syndrome | Head injury |
| Laparoscopic cardiomyotomy and fundoplication | ERCP procedure |
| Gallstones and kidney stones | Lymphoma |
| Gall bladder and liver infection | Left lower-lobe pneumonia |
| Chronic atrial fibrillation diagnosed | Hyponatraemia |
| Lumbar decompression surgery | Phimosis |
| Left hip replacement | Hip replacement |
| Prostate operation | Left knee replacement |
| Knee replacement | Gall bladder operation |
| Postural hypotension, diarrhoea | Broken rib that required patient hospitalisation |
| Chest infection and shortness of breath | Viral gastroenteritis and dehydration |
| Alcohol withdrawal | |
| Unintentional overdose | |
| Vascular dementia | |
| Lower respiratory tract infection, pleurisy | |
| Bronchitis | |
| COPD | |
| Knee replacement | |
| Group A streptococcal infection | |
| Total knee replacement |
Change in laboratory test results during follow-up
Over 6 months, in the metformin group, HbA1c level decreased by 1.7 mmol/mol, ALT by 2.5 U/l, total cholesterol by 0.2 mmol/l and LDL cholesterol by 0.1 mmol/l and there was no change in triglyceride levels, as shown in Table 13. In contrast, in the placebo group, there were no changes in total cholesterol, LDL cholesterol or ALT levels, but triglyceride values increased by 0.1 mmol/l and HbA1c levels fell by 0.8 mmol/mol. Concerning renal function, eGFR declined by 4.3 ml/minute/1.73 m2 over 6 months in the metformin group and by 6.9 ml/minute/1.73 m2 in the placebo group. Creatinine levels increased by 3.2 µmol/l over 6 months in the metformin group and by 5.5 µmol/l in the placebo group. One participant in each group developed CKD stage 3b (eGFR of < 45 ml/minute/1.73 m2) over 6 months, leading to discontinuation of study medication. Mean plasma vitamin B12 values increased in both groups between baseline and 6 months (by 26.6 ng/l and 2.1 ng/l in the placebo and metformin groups, respectively). At baseline, eight participants in the placebo group and nine in the metformin group had plasma vitamin B12 levels below 211 ng/l (the lower end of the laboratory reference range). At 6 months, only one participant in each group had a plasma vitamin B12 level below 211 ng/l.
| Laboratory test | Follow-up time point | Change | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 3 months | 6 months | Baseline to 3 months | Baseline to 6 months | ||||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
| Placebo group | ||||||||||
| HbA1c (mmol/mol) | 124 | 40.6 (2.7) | 111 | 40.3 (3.0) | 109 | 39.9 (3.0) | 111 | –0.4 (2.2) | 109 | –0.8 (1.9) |
| eGFR (ml/minute/1.73 m2) | 124 | 90.5 (18.2) | 111 | 85.1 (15.4) | 107 | 83.7 (16.9) | 111 | –5.2 (10.7) | 107 | –6.9 (10.2) |
| Cholesterol (mmol/l) | 124 | 4.9 (0.9) | N/A | N/A | 108 | 5.0 (0.9) | N/A | N/A | 108 | 0.0 (0.6) |
| HDL cholesterol (mmol/l) | 124 | 1.3 (0.4) | N/A | N/A | 108 | 1.3 (0.3) | N/A | N/A | 108 | 0.0 (0.2) |
| LDL cholesterol (mmol/l) | 120 | 2.7 (0.9) | N/A | N/A | 105 | 2.8 (0.9) | N/A | N/A | 104 | 0.0 (0.5) |
| Creatinine (µmol/l) | 124 | 78.7 (13.8) | 111 | 83.1 (13.5) | 107 | 84.4 (14.6) | 111 | 3.9 (8.3) | 107 | 5.5 (7.7) |
| Vitamin B12 (ng/l) | 124 | 338.0 (103.3) | N/A | N/A | 108 | 363.6 (96.7) | N/A | N/A | 108 | 26.6 (68.5) |
| Metformin group | ||||||||||
| HbA1c (mmol/mol) | 125 | 41.3 (2.7) | 107 | 40.6 (3.1) | 104 | 39.5 (3.5) | 107 | –0.6 (1.9) | 104 | –1.7 (2.4) |
| eGFR (ml/minute/1.73 m2) | 125 | 89.4 (18.8) | 107 | 85.9 (17.0) | 106 | 85.0 (16.6) | 107 | –2.7 (9.5) | 106 | –4.3 (10.2) |
| Cholesterol (mmol/l) | 125 | 5.0 (0.9) | N/A | N/A | 106 | 4.8 (0.9) | N/A | N/A | 106 | –0.2 (0.6) |
| HDL cholesterol (mmol/l) | 125 | 1.3 (0.3) | N/A | N/A | 106 | 1.3 (0.3) | N/A | N/A | 106 | 0.0 (0.1) |
| LDL cholesterol (mmol/l) | 123 | 2.8 (0.8) | N/A | N/A | 105 | 2.6 (0.7) | N/A | N/A | 104 | –0.1 (0.5) |
| Creatinine (µmol/l) | 125 | 79.5 (16.1) | 107 | 82.3 (17.4) | 106 | 82.6 (15.8) | 107 | 2.4 (7.5) | 106 | 3.2 (8.3) |
| Vitamin B12 (ng/l) | 125 | 353.3 (112.7) | N/A | N/A | 105 | 364.8 (93.1) | N/A | N/A | 105 | 2.1 (70.0) |
Changes in laboratory test results from baseline to 3 months and from baseline to 6 months in the metformin group relative to the placebo group are shown in Table 14. Compared with placebo, metformin was associated with small improvements in HbA1c, eGFR, total and LDL cholesterol, and creatinine, and a reduction in plasma vitamin B12 levels.
| Laboratory test | Time frame, difference (95% CI) | |||
|---|---|---|---|---|
| Baseline to 3 months | Baseline to 6 months | |||
| HbA1c (mmol/mol) | –0.10 (–0.63 to 0.44) | –0.82 (–1.39 to –0.24) | ||
| eGFR (ml/minute/1.73 m2) | 2.12 (–0.33 to 4.57) | 2.31 (–0.20 to 4.81) | ||
| Cholesterol (mmol/l) | N/A | N/A | N/A | –0.16 (–0.31 to 0.00) |
| HDL cholesterol (mmol/l) | N/A | N/A | N/A | 0.00 (–0.04 to 0.04) |
| LDL cholesterol (mmol/l) | N/A | N/A | N/A | –0.11 (–0.25 to 0.02) |
| Creatinine (µmol/l) | –1.44 (–3.51 to 0.63) | –2.22 (–4.30 to –0.14) | ||
| Vitamin B12 (ng/l) | N/A | N/A | N/A | –16.43 (–32.85 to –0.01) |
| Triglycerides (mmol/l) | N/A | N/A | N/A | –0.11 (–0.29 to 0.07) |
| ALT (U/l) | N/A | N/A | N/A | –1.30 (–3.44 to 0.83) |
Analyses restricted to those participants who were still taking the study drug at 6 months’ follow-up (Table 15) demonstrated broadly similar results to those shown in Table 14. Changes in HbA1c, total and LDL cholesterol, triglycerides, ALT, eGFR and creatinine all favoured the metformin group. The mean changes in plasma vitamin B12 level from baseline to 6 months among those still taking the study drug were –4.6 ng/l and +30.5 ng/l in the metformin and placebo groups, respectively.
| Laboratory test | Follow-up time point | Change | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 3 months | 6 months | Baseline to 3 months | Baseline to 6 months | ||||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
| Placebo group | ||||||||||
| HbA1c (mmol/mol) | 92 | 40.9 (2.8) | 91 | 40.4 (3.0) | 92 | 40.0 (3.0) | 91 | –0.5 (1.9) | 92 | –0.9 (1.8) |
| eGFR (ml/minute/1.73 m2) | 92 | 89.0 (16.7) | 91 | 84.6 (16.2) | 90 | 82.3 (16.6) | 91 | –4.4 (8.9) | 90 | –6.5 (9.3) |
| Cholesterol (mmol/l) | 92 | 5.0 (0.9) | N/A | N/A | 91 | 5.0 (0.9) | N/A | N/A | 91 | 0.0 (0.5) |
| HDL cholesterol (mmol/l) | 92 | 1.3 (0.3) | N/A | N/A | 91 | 1.3 (0.3) | N/A | N/A | 91 | 0.0 (0.2) |
| LDL cholesterol (mmol/l) | 88 | 2.8 (0.9) | N/A | N/A | 88 | 2.8 (0.9) | N/A | N/A | 87 | 0.0 (0.5) |
| Creatinine (µmol/l) | 92 | 79.9 (13.0) | 91 | 83.7 (13.8) | 90 | 85.7 (14.2) | 91 | 3.6 (7.8) | 90 | 5.6 (7.7) |
| Vitamin B12 (ng/l) | 92 | 327.5 (86.4) | N/A | N/A | 91 | 358.5 (90.5) | N/A | N/A | 91 | 30.5 (58.3) |
| Metformin group | ||||||||||
| HbA1c (mmol/mol) | 90 | 41.3 (2.7) | 89 | 40.7 (3.1) | 89 | 39.5 (3.6) | 89 | –0.6 (1.9) | 89 | –1.9 (2.4) |
| eGFR (ml/minute/1.73 m2) | 90 | 88.5 (17.4) | 89 | 85.7 (17.3) | 90 | 84.6 (16.1) | 89 | –2.6 (9.4) | 90 | –3.9 (10.0) |
| Cholesterol (mmol/l) | 90 | 5.0 (0.9) | N/A | N/A | 90 | 4.8 (0.9) | N/A | N/A | 90 | –0.2 (0.6) |
| HDL cholesterol (mmol/l) | 90 | 1.3 (0.3) | N/A | N/A | 90 | 1.3 (0.3) | N/A | N/A | 90 | 0.0 (0.1) |
| LDL cholesterol (mmol/l) | 89 | 2.8 (0.8) | N/A | N/A | 89 | 2.6 (0.7) | N/A | N/A | 88 | –0.1 (0.5) |
| Creatinine (µmol/l) | 90 | 80.0 (16.1) | 89 | 82.5 (17.9) | 90 | 82.9 (15.8) | 89 | 2.4 (7.5) | 90 | 2.9 (8.5) |
| Vitamin B12 (ng/l) | 90 | 362.6 (106.9) | N/A | N/A | 89 | 358.4 (91.2) | N/A | N/A | 89 | –4.6 (64.2) |
End-point data collection
The total duration of participant follow-up was 124.5 person-years in the metformin group (mean 1.00 years, SD 0.30 years) and 122.1 person-years in the placebo group (mean 0.98 years, SD 0.29 years). Assessing the feasibility of using routine national data sources (e.g. HES) to ascertain CVD events was one of the aims of the feasibility study. We obtained participants’ consent to utilise register data linked to their identifiable information, including NHS number, and prepared an application for the HSCIC (now known as NHS Digital). However, following discussion with the TSC, we decided not to pursue the application for several reasons: (1) we were able to identify possible study end points through other routes, (2) the high cost of the application process, (3) the considerable delay experienced by applicants [UK researchers were experiencing delays of up to 12 months in obtaining approvals and data following applications to the HSCIC (now NHS Digital) and the Myocardial Ischaemia National Audit Project at the time of the GLINT feasibility trial], (4) the absence of any cardiovascular events among study participants at the time of the proposed HSCIC application and (5) we had demonstrated the feasibility of the process in a concurrent study (ADDITION) involving GLINT investigators from Cambridge and Leicester. 34 Therefore, it was felt that pursuing the application would not provide additional useful information.
Reported primary and secondary outcomes
A total of 10 events that met the definition of an outcome (either primary or secondary) for the full trial were recorded during the feasibility study: two deaths (one attributable to a strangulated hernia and one attributable to type 1 respiratory failure, pneumonia and sepsis), three CVD events, three physician diagnoses of T2D and two cases of non-melanoma cancer. These represent five primary end points for the full trial and five secondary end points for the full trial. Of the reported events, six were in the placebo group and four were in the metformin group, as shown in Table 16.
| Outcome | Study group, n (%) | |
|---|---|---|
| Placebo (N = 124) | Metformin (N = 125) | |
| Mortality | 1 (0.8) | 1 (0.8) |
| Cardiovascular event | 2 (1.6) | 1 (0.8) |
| Cancer | 1 (0.8) | 1 (0.8) |
| Diabetes | 2 (1.6) | 1 (0.8) |
Health utility and functional status
Among those who responded to the questionnaire, health utility was unaffected by participation in the trial (as measured by the change from baseline) or by allocation to the metformin or placebo group, as shown in Table 17.
| Health utility item | Follow-up time point | Change (baseline to 4 months) | ||||
|---|---|---|---|---|---|---|
| Baseline | 4 months | n | Mean score (SD) | |||
| n | Mean score (SD) | n | Mean score (SD) | |||
| Placebo group | ||||||
| Mobility | 124 | 1.3 (0.5) | 112 | 1.3 (0.5) | 112 | 0.0 (0.3) |
| Self-care | 124 | 1.0 (0.2) | 112 | 1.1 (0.3) | 112 | 0.0 (0.2) |
| Usual activities | 124 | 1.2 (0.4) | 112 | 1.2 (0.4) | 112 | 0.0 (0.4) |
| Pain/discomfort | 124 | 1.5 (0.5) | 112 | 1.4 (0.5) | 112 | –0.1 (0.5) |
| Anxiety/depression | 124 | 1.1 (0.3) | 112 | 1.1 (0.4) | 112 | 0.0 (0.3) |
| Health state | 123 | 82 (13) | 108 | 81 (15) | 107 | –2 (11) |
| Metformin group | ||||||
| Mobility | 125 | 1.2 (0.4) | 105 | 1.2 (0.4) | 105 | 0.0 (0.4) |
| Self-care | 125 | 1.0 (0.1) | 104 | 1.0 (0.2) | 104 | 0.0 (0.1) |
| Usual activities | 125 | 1.2 (0.4) | 102 | 1.2 (0.4) | 102 | 0.0 (0.4) |
| Pain/discomfort | 125 | 1.4 (0.5) | 104 | 1.4 (0.5) | 104 | 0.0 (0.5) |
| Anxiety/depression | 125 | 1.2 (0.4) | 105 | 1.2 (0.4) | 105 | 0.0 (0.4) |
| Health state | 125 | 83 (14) | 106 | 81 (12) | 106 | –2 (11) |
Functional status, assessed at 4 months using the SF-8 measure, did not differ between study groups, as shown in Table 18.
| Functional status item | Study group | |||
|---|---|---|---|---|
| Placebo | Metformin | |||
| n | Mean score (SD) | n | Mean score (SD) | |
| Health (1–6) | 109 | 2.6 (0.9) | 106 | 2.5 (0.9) |
| Limited activity (1–5) | 110 | 1.8 (1.0) | 106 | 1.7 (0.9) |
| Work difficulty (1–5) | 109 | 1.7 (1.0) | 106 | 1.6 (0.8) |
| Body pain (1–6) | 112 | 2.5 (1.2) | 104 | 2.2 (1.1) |
| Energy (1–5) | 112 | 2.2 (0.8) | 105 | 2.2 (0.6) |
| Social activity (1–4) | 112 | 1.5 (0.8) | 105 | 1.5 (0.7) |
| Emotional problems (1–5) | 111 | 1.5 (0.8) | 106 | 1.6 (0.8) |
| Work emotional problems (1–5) | 111 | 1.3 (0.6) | 106 | 1.3 (0.5) |
Participant survey
The survey was sent to all 249 randomised participants, together with the newsletter. It was completed by 150 participants (60%), with 54 completing it online and 96 completing it on paper. Of the 150 respondents, 89 (57%) said that they would be willing to complete future surveys online, 57 (38%) were unwilling to complete future surveys online and four (3%) did not respond to this question. Of those who were unwilling to complete future surveys online, 26 (46%) had limited access to the internet and 17 (30%) were unwilling to enter personal details online. This equates to 17% and 11% of total responders, respectively. A total of 63 comments were reported in the free-text section of the brief survey. The most common problem that people reported in relation to the survey was technical difficulties when using the online systems (n = 24). Several of those people with no internet access or who had difficulty navigating the system wanted more contact with the study team by telephone. With regard to the study medication, 10 participants reported that they were unhappy with the size and texture of the tablets, which they found difficult to swallow. Four participants wanted more contact/check-ups from clinicians during and following the study. Four participants made positive comments about the newsletter, as this made them feel involved in the study and gave them a greater understanding of the trial and the medication. Randomisation was not referred to by any participants in the free-text response section of the questionnaire.
Chapter 4 Discussion and conclusions
In this report, we describe the conduct and findings of the feasibility phase of GLINT, a large-scale clinical outcome trial to quantify the effects of metformin on the incidence of CVD and cancer among people with NDH and high CVD risk. We have demonstrated that most aspects of the trial design and conduct appear to be feasible, but some aspects will require adaptation for the main trial, drawing on the lessons learned and to ensure that there is sufficient power to reliably address the primary end point.
Acceptability of the study to general practitioners and patients
It is clear that primary care practitioners were sufficiently interested in the research question addressed in this study to become involved. Feedback at practice recruitment meetings confirmed that the topic was relevant and that there was uncertainty about management of the growing number of patients who would be eligible for the study, further justifying the study aims. In addition, the limited direct workload for the practices associated with participation was deemed appropriate. However, the delays introduced by sponsorship changes meant that initial enthusiasm had waned somewhat by the time that the initiation of the trial took place. The sponsor requirement that each general practice that recruited ≥ 20 participants would need to undergo a formal monitoring visit is unlikely to be feasible in the full trial.
The trial design, procedures, treatment and duration (individuals were recruited to a trial with an anticipated duration of > 5 years) appeared to be sufficiently acceptable to individuals such that 9.7% of those receiving a letter from their GP about the study attended the baseline assessment, signed a consent form and agreed to take part. This compares favourably with recent similar trials, such as A Study of Cardiovascular Events iN Diabetes (ASCEND),35 involving the distribution of the study drug by post. Once randomised, only one participant withdrew consent for follow-up for end points via their GP and routine data sources.
Efficiency of recruitment methods
Between March and November 2015, we recruited and obtained consent from ≥ 500 people to take part in the trial, 249 of whom were eligible and were randomised. As such, we demonstrated the feasibility of participant recruitment. However, simply scaling up the same recruitment procedures is unlikely to be feasible, as discussed in Recruitment from general practices. There were inefficiencies, in particular duplicate data entry, in using local databases to manage the information and study co-ordination prior to randomisation and using the InferMed MACRO system to host data entry and data storage and co-ordinate all participant contacts post randomisation. For the full trial, all study procedures should be co-ordinated by one central trial management system.
Recruitment from general practices
The majority of participants (61.4%) were recruited from general practices. Although the GLINT feasibility study was dependent on the co-operation of 10 sites and 21 PICs, the level of support required to recruit and manage those practices is not sustainable on a larger scale. Personal visits from the principal investigators to recruit practices, and technical support from the study team to assist with searching of electronic medical records and sending out invitations to potential participants, is not feasible for a UK-wide endeavour. Alternative approaches, such as mass mailshots within federations of practices and research-accredited pharmacies and liaisons with existing screening and prevention programmes, will need to be used.
The availability in electronic medical records of recent information concerning inclusion/exclusion criteria varied but for most variables this was < 50%. Qualifying laboratory test results were available for fewer than half of the participants but, when available, closely corresponded with the information obtained at the baseline visit. Consequently, in the early phase of recruitment, the proportion of individuals agreeing to take part who were subsequently randomised was significantly less than 50%. Following the application of a diabetes risk score search in the practices, and stratification of practice populations using the risk score, this proportion increased to 49% for the whole study (i.e. one out of two individuals who attended the baseline assessment at a study centre was randomised).
Recruitment via the NHS Health Check programme
The NHS Health Check programme did not contribute as many participants as had been expected. First, NHS Health Checks are centrally mandated but implemented by local authorities. Staff undertaking NHS Health Checks in Cambridgeshire and Leicestershire were focused on programme delivery and meeting targets. In 2015, they were not in a position to facilitate opportunistic recruitment to GLINT by referring individuals to the study centre for eligibility assessments or by providing eligible individuals with our brief information sheet and contact details. Fewer NHS Health Checks were conducted in recruited practices than expected. There have been differences in uptake between regions and it was noted that the uptake in Cambridge was lower than in Leicester. Between 2013 and 2015, in Leicestershire, the proportion of eligible people invited for a Health Check was 70%,36 whereas, in Cambridgeshire, only 62% of eligible people were invited during this period. The proportion of people who responded to the invitation for a Health Check was 38% in Cambridgeshire and 40% in Leicestershire. In Cambridgeshire, the NHS Health Check programme has been targeted at more deprived communities whose general practices are less likely to become involved in research. Without these NHS Health Checks, the information required to identify potentially eligible individuals was rarely available in the medical records.
Measurement of glycaemia, a key eligibility criterion in the feasibility phase of GLINT, has also varied because of local decision-making about how NHS Health Checks are delivered. A greater proportion of patients in the Leicestershire practices had a recent value for HbA1c level in their medical records than in the Cambridgeshire practices. The Health Checks diabetes filter is currently under consultation. The new recommendations include the use of existing diabetes risk scores, such as the Cambridge22 and Leicester37 diabetes risk scores, which, as we have shown previously38,39 and in this study, perform reasonably well in identifying individuals at high risk of diabetes and CVD.
Recruitment from research databases
We demonstrated the relative efficiency of recruiting from existing research databases by targeting invitations to those with known values for eligibility criteria; 8.4% of individuals identified by this route were eventually randomised. However, results from tests relating to eligibility criteria that were undertaken several years ago are unlikely to remain valid. Hence, the efficiency of this approach is, to some degree, dependent on how recently the individuals in the research database were assessed.
We demonstrated that information for key eligibility criteria, for example concerning renal function, was available in the general practice electronic medical records for nearly half of the randomised participants and that the values for variables extracted from the medical records were very similar to those obtained at the baseline visit at the study centres.
In conclusion, none of the recruitment strategies assessed would allow sufficient numbers to be recruited rapidly for a large outcome study. Although we will continue to explore links with NHS programmes to facilitate recruitment to GLINT (e.g. the NHS Health Check programme and the NHS Diabetes Prevention programme), we should not depend on NHS referrals to facilitate recruitment. Oxford investigators have experience of other recruitment methods using secondary care electronic health records, which have enabled thousands of patients to be randomised into cardiovascular outcome trials, and these methods will be adopted for recruitment into the main study. In addition, mass participant recruitment to trials from primary care is increasingly being enabled by organisations such as NorthWest Ehealth [http://nweh.co.uk/products/farsite (accessed 4 January 2018)], a not-for-profit partnership between the NHS and the University of Manchester. NorthWest EHealth undertakes searches of electronic medical records across hundreds of practices and then arranges mailshots to potentially eligible individuals, at a fraction of the cost per recruited participant of the feasibility study and requiring limited input from research and practice staff. Furthermore, much of this recruitment activity is eligible for NHS support cost funds. Given the low frequency of recent HbA1c values in the medical records, we would also propose dispensing with this as an inclusion criterion and, instead, identify people mainly on the basis of cardiovascular risk.
The original GLINT proposal was based upon modelled CVD risk using data from the EPIC-Norfolk study. 32 The feasibility study demonstrated that the CVD risk of recruited participants (and, hence, the number of events) was likely to be lower than this. Furthermore, the adherence to metformin was lower than expected and observed in previous studies. Finally, the original sample size calculation was based on an effect size estimate of a 17% reduction in risk of events, which we updated to a 15% risk reduction following the feasibility study. For these reasons, we needed a larger sample size for the full trial than originally estimated. As part of our application for funds to support the full trial, we updated the sample size calculations as below.
Randomisation of at least 20,000 participants with at least 5 years’ follow-up and until at least 2700 unrefuted primary outcomes occur. This calculation is based on (1) an estimated hazard ratio with full compliance of 0.85, (2) approximately 78% compliance overall and (3) 90% power at 2p < 0.05.
One of the key logistical challenges in the feasibility study concerned the co-ordination of baseline assessments, which included obtaining consent. To scale up from 249 participants to around 20,000 participants, we propose to remove the need for face-to-face consultations to obtain consent for the majority of participants, instead utilising the internet, correspondence and telephone calls, as per A Study of Cardiovascular Events iN Diabetes (ASCEND). 35
Randomisation
We have demonstrated the feasibility of the randomisation procedure, which efficiently generated groups that were well matched for the majority of baseline characteristics. We had originally planned to undertake focus groups at the end of the feasibility study in which we would have addressed the acceptability of the study procedures, including randomisation. However, the significant delays that we experienced in relation to changes in sponsorship meant that this was not possible. Among recruiting practices and participants, no concerns were raised relating to the issue and process of randomisation. Furthermore, randomisation was not referred to by any participants in the free-text response section of the questionnaire nor during the regular contacts with study co-ordinators. We have therefore inferred that the randomisation process was acceptable.
Characteristics of recruited participants
Participants were generally elderly (mean age 70 years) and predominantly male and overweight or obese. The majority were ex-smokers or current smokers and were prescribed antihypertensive medication. To some extent this reflects the variables included in the risk score used to identify potentially eligible individuals. Participants were predominantly white. We expected that recruited participants would be representative of the overall population in the areas we recruited from and that a larger proportion would have come from an Asian background. However, the participant demographics reflect those of the populations of recruited practices. In the case of the Leicester site, the surgeries that took part were located around the county of Leicestershire, rather than in the city centre. Recruiting general practices from more-deprived inner-city areas with greater ethnic diversity remains a challenge. The mean modelled 10-year CVD risk based on practice records was 30%, confirming that it is feasible to recruit individuals at high risk of CVD events. However, although CVD risk scores exhibit reasonable discrimination, their calibration is less good and modelled risk tends to overestimate observed risk. Furthermore, just over half of the participants were prescribed statins. Consequently, the expected number of actual CVD events is likely to be lower than estimates based on modelled risk. This has implications for the size of the main study and the characteristics of those who might be invited in order to maximise event rates.
Adherence to study medication
Delivering the study drug by post was feasible and efficient. A very small proportion of medication packs had to be re-sent. Concerns that the medication packs might not fit through some letter boxes did not materialise. However, there was a high level of discontinuation of the study drug within 6 months, although the frequency did not differ between study groups. In the DPP, a similar proportion of participants had discontinued metformin (Glucophage 850 mg, twice daily) at 6 months and this proportion remained stable for a further 2.5 years, at which point it declined to between 65% and 70%. 12 In the DPP, adherence to placebo was consistently higher than adherence to metformin, which contrasts with our study, in which we used a prolonged-release preparation. Reports of AEs associated with the study medication were similar for metformin and placebo, although the side effects experienced by the placebo group were regarded as more ‘bothersome’. There were some anecdotal reports about the large size and unpleasant texture of the study medication. Furthermore, among those participants who discontinued the study medication, a greater proportion in the metformin group than in the control group attributed side effects as the reason for discontinuation. Among participants taking the study drug, most were able to tolerate the full 1500-mg dose per day and had taken their medication on 85% of the preceding 14 days. Nevertheless, the level of discontinuation of the study drug in both groups suggests the need for a pre-randomisation run-in period in the full trial.
Pre-randomisation run-in to improve treatment adherence, participant retention and questionnaire response rates
For the feasibility study, we did not include a pre-randomisation run-in period but will do so for the main study. The main advantages of this are twofold. For interventions that are not universally well tolerated, such as metformin, a run-in period allows all potential participants to take the study drug before being randomised so that only those people who tolerate metformin at the end of the run-in period are randomised. This increases the number of people who need to be screened but has been shown to improve post-randomisation adherence. Second, the run-in period allows the exclusion of those people who are initially enthusiastic about trial participation but who then lose interest and stop complying with the study procedures (e.g. questionnaire completion) and are more likely to drop out post randomisation, resulting in a serious impact on study power.
Follow-up by questionnaire and electronic records/registers
We obtained appropriate consent for the use of routine data and registers for the tracking of outcomes and, although we did not pursue the formal application for HSCIC (now NHS Digital) approvals, we have demonstrated that such an approach is feasible in parallel studies. 31 We also demonstrated the feasibility of collecting sufficient data for independent adjudication concerning potential end points. We demonstrated the feasibility of collecting data on functional status, health utility and health service use. Questionnaire response rates were reasonable at 4 months but fell to 75% by the end of the study, although, for a significant proportion of participants, the end-of-study questionnaire coincided fairly closely with a previous questionnaire. It is clear that using internet-based data collection instruments is not feasible for all participants and hence postal paper questionnaires would still be required. The declining response rate underlines the value of collecting outcome data using routine sources, of including study procedures in a run-in phase in a main trial and of maintaining contact with study participants through newsletters.
Safety monitoring
In the feasibility study, we included study centre visits at 3 and 6 months, mainly for the purpose of collecting blood samples to monitor safety. Attendance levels were high (86% at 6 months). Given the small sample size, the results of analyses of differences between study groups should be interpreted with caution.
Renal function and risk of lactic acidosis
There were small declines in renal function over 6 months, as one might expect in this study population with a mean age of 70 years. However, there were no differences between groups. The concern in relation to metformin has not been that it is associated with renal AEs, but rather that, in the presence of impaired renal function, the risk of lactic acidosis, a rare but serious condition, is increased. This remains unproven but, nevertheless, significantly impaired renal function is a contraindication to the use of metformin. During the course of the feasibility study, the SmPC was changed and stipulated that metformin should not be prescribed if the eGFR is < 45 ml/minute/1.73 m2 and that eGFR should be monitored. Also during the course of the feasibility study, a systematic review40 was published demonstrating the safety of metformin among people with impaired renal function. The review showed that drug levels generally remain within the therapeutic range and lactate concentrations are not substantially increased when used in patients with mild-to-moderate CKD (eGFRs of 30–60 ml/minute/1.73 m2). Furthermore, the overall incidence of lactic acidosis in metformin users (3–10 per 100,000 person-years) was similar to the background rate in the population with diabetes. A recently conducted placebo-controlled trial41 of 173 patients, in which an eGFR of < 45 ml/minute/1.73 m2 was an exclusion criterion, showed no difference in lactate levels over 18 months. A Cochrane Database Systematic Review by Salpeter et al. 42 and a meta-analysis of English and non-English literature by the same authors43 reported that the risk of lactic acidosis is essentially nil in the context of clinical trials of metformin, including those that did not specify kidney disease as an exclusion criterion. It is apparent that patients who develop lactic acidosis while taking metformin typically have an acute supervening illness, for example sepsis, acute kidney or liver failure or cardiovascular collapse, which precipitates the metabolic decompensation causing lactic acidosis. Therefore, even though metformin is renally excreted, and its clearance is impaired in mild-to-moderate CKD, drug levels are still largely maintained within a therapeutic range when the eGFR is > 30 ml/minute/1.73 m2 and do not seem to significantly affect circulating lactate levels. Observational studies40 suggest a potential benefit of metformin on macrovascular outcomes, even in patients with renal contraindications for its use. The recognition of metformin’s safety has led to its common use in patients with CKD.
There has been increasing pressure for regulatory agencies to relax their guidelines regarding metformin prescribing in renal impairment, with an updated joint position statement44 issued by the American Diabetes Association and the European Association for the Study of Diabetes in 2015 suggesting that the renal safety guidelines in the USA may be ‘overly restrictive’. Following a comprehensive review of the medical literature, the US Food and Drug Administration issued updated guidance45 in April 2016 that relaxed the renal threshold for metformin to allow its use in patients with an eGFR of > 30 ml/minute/1.73 m2, with consideration of dose reduction and monitoring of renal function in those patients at highest risk. The European Medicines Agency issued similarly updated guidance46 in October 2016 to allow metformin use in patients with an eGFR of > 30 ml/minute/1.73 m2.
Having considered the data on changes in renal function from the feasibility study and the totality of evidence for the safety of metformin in patients with impaired renal function, we intend to revise the entry criteria for the full trial to exclude only those with severe renal dysfunction, defined as requiring ongoing follow-up in a specialist nephrology clinic, or with an eGFR of < 30 ml/minute/1.73 m2 and we will limit the maximum dose of the study drug to two tablets (equivalent to 1000 mg of metformin) for those participants aged > 80 years or with an eGFR of 30–45 ml/minute/1.73 m2. Interim monitoring in a subset of patients at high risk of progression of renal dysfunction is being considered in the revised trial design.
Vitamin B12
Metformin was not associated with reductions in plasma vitamin B12 levels over 6 months. At baseline, 6.83% of participants had a vitamin B12 level below the laboratory reference range and this had fallen to 0.94% at 6 months. Changes in vitamin B12 levels have been reported among people with insulin-treated T2D receiving a mean dose of 2050 mg per day of metformin. 47 However, it is uncertain whether or not these findings are generalisable to people with NDH who are treated with a lower dose. Previous studies47,48 have shown that metformin is associated with reductions in serum vitamin B12 levels, but these have remained within the normal range and have not been associated with adverse health outcomes such as anaemia, neuropathy or reduced quality of life. Based on these data, it would be premature to introduce vitamin B12 monitoring for all T2D patients receiving metformin48 or for participants in our trial. We have no robust data showing that vitamin B12 monitoring in individuals receiving metformin would yield clinical benefits.
Cardiovascular risk factors, health utility and functional status
Compared with placebo, treatment with metformin was associated with small improvements in the following cardiovascular and metabolic risk factors over 6 months: HbA1c, ALT, total cholesterol, LDL cholesterol and triglycerides. Changes in HbA1c level were consistent with those observed in the DPP12 and the more recent Carotid Atherosclerosis: MEtformin for Insulin ResistAnce Study (CAMERA). 41 Any observed reduction in the risk of CVD and cancer attributable to metformin is unlikely to be solely as a result of its effect on glycaemia. We showed that metformin was not associated with any detrimental effects on health utility or functional status within 4 months. One limitation of the feasibility study is that we did not include follow-up measures of weight and blood pressure, although the small sample size would have constrained interpretation of between-group differences.
Serious adverse events
Systems for collecting information about SAEs and reporting them to the sponsor and IDMC functioned well, although the number of events was small. A greater number of SAEs were reported by participants in the metformin group than by participants in the placebo group; however, none was considered to be related to the study medication.
Ensuring an adequate event rate for study power
The feasibility study was not expected to provide information on cardiovascular event rates but the types of patient recruited suggest that, if limited to a primary prevention population, the cardiovascular event rate is likely to be much lower than the 2% that was originally modelled.
In our original proposal we suggested recruiting some individuals with pre-existing CVD and some without. However, some reviewers and funders strongly recommended that GLINT should be a primary prevention study. We believe that the main study question, quantifying the effects of metformin on the risk of CVD events, can be feasibly answered only by recruiting mainly secondary prevention patients with a prior history of CVD. We intend to revise the GLINT inclusion criteria to recruit a cohort of around 80% secondary CVD prevention participants and 20% primary CVD prevention participants.
People with pre-existing CVD are at higher CVD risk, and are more easily identified in both primary and secondary care and recruited to studies, than individuals with a high modelled risk but no prior history of CVD. The Oxford Clinical Trial Service Unit, with whom we plan to work on the main study, has extensive experience of identifying high-risk patients with CVD from hospital-based electronic records. In the 1990s, it randomised 20,000 patients using these methods in the Heart Protection Study49 and, in subsequent years, 12,000 UK post-myocardial infarction patients were randomised in SEARCH (Study of the Effectiveness of Additional Reductions in Cholesterol and Homocysteine),50 8000 UK patients (out of 25,000 worldwide) were randomised in the HPS2-THRIVE (Treatment of HDL to Reduce the Incidence of Vascular Events) study51 and 8000 (out of 30,000 worldwide) were randomised in the HPS3-REVEAL (HPS3/TIMI55-REVEAL Randomized EValuation of the Effects of Anacetrapib through Lipid-modification) study. 52 We will use similar methods, alongside primary care registers, to recruit approximately 20,000 patients to be randomised cost-effectively into GLINT.
Chapter 5 Future research recommendations
We have demonstrated that a large, simple, pragmatic randomised trial comparing the effects of prolonged-release metformin with placebo on the risk of CVD events is potentially feasible. The study question remains important for the reasons outlined in Chapter 1. However, we have a number of recommendations concerning changes to the design and conduct of the study to make it possible for the trial to be scaled up efficiently. These recommendations include:
-
changing the inclusion criteria to recruit people with and without pre-existing CVD to increase the recruitment and event rates
-
using large primary care and secondary care databases to improve participant identification
-
conducting follow-up remotely to reduce costs and improve efficiency
-
including a run-in period prior to randomisation to optimise adherence to the study procedures and drug.
Acknowledgements
We acknowledge the contribution of all GLINT participants, CLRN nurses and general practices. Without their input, the trial would not have been possible.
Contributions of authors
Simon J Griffin (General Practice – Professor of General Practice, Institute of Public Health, University of Cambridge) was involved in the conception of the original GLINT design; prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
M Angelyn Bethel (Endocrinology – Assistant Professor of Diabetes & Endocrinology, Deputy Director, Diabetes Trials Unit, University of Oxford) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Rury R Holman (Diabetes – Professor of Diabetic Medicine, Diabetes Trials Unit Director, NIHR Senior Investigator, Honorary Consultant Physician, Radcliffe Department of Medicine, University of Oxford) was involved in the conception of the original GLINT design; prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Kamlesh Khunti (General Practice – Head of Department and Professor of Primary Care Diabetes and Vascular Medicine, Diabetes Research Centre, University of Leicester) was involved in the conception of the original GLINT design; prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Nicholas Wareham (Epidemiology – Director MRC Epidemiology Unit, co-Director of the Institute of Metabolic Science, University of Cambridge; Honorary Consultant, Addenbrooke’s Hospital) was involved in the conception of the original GLINT design; prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Gwen Brierley (Clinical Research Manager – MRC Epidemiology Unit, University of Cambridge) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Melanie Davies (Diabetes – Professor of Diabetes Medicine, Diabetes Research Centre/Department of Health Sciences, University of Leicester) was involved in the conception of the original GLINT design; prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Andrew Dymond (Study Co-ordinator – Senior Volunteer Recruitment Co-ordinator, The Cambridge NIHR BioResource, University of Cambridge) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Rose Eichenberger (Programme Manager – MRC Epidemiology Unit, University of Cambridge) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Philip Evans [General Practice – Associate Professor of General Practice and Primary Care, University of Exeter Medical School (Primary Care)] prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Alastair Gray (Health Economics – Health Economics Research Centre, Nuffield Department of Population Health, University of Oxford) prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Colin Greaves [Behavioural Science – Associate Professor in Psychology Applied to Health, University of Exeter Collaboration for Academic Primary Care (APEx)] prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Kyla Harrington (Study Co-ordinator – Programme Manager, University of Leicester) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Graham Hitman (Diabetes – Professor of Molecular Medicine and Diabetes, Blizard Institute, Queen Mary University of London) prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Greg Irving (General Practice – GP and Clinical Lecturer in General Practice, Department of Public Health and Primary Care, University of Cambridge) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Sarah Lessels (Senior Trial Co-ordinator – Scottish Clinical Trials Unit, Edinburgh University) contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Ann Millward (Diabetes – Professor in Clinical Diabetes, Peninsula Medical School, Plymouth University) prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
John R Petrie [Diabetes – Professor of Diabetic Medicine, Institute of Cardiovascular and Medical Sciences, British Heart Foundation (BHF) Glasgow Cardiovascular Research Centre] prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Martin Rutter (Cardiometabolic Medicine – Senior Lecturer and Honorary Consultant Physician, University of Manchester) prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Mike Sampson (Diabetes – Consultant for Diabetes and Endocrinology, Norfolk and Norwich University Hospital, NHS Trust/Norwich Medical School; Honorary Professor of Diabetes, University of East Anglia) contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Naveed Sattar (Metabolic Medicine – Professor/Honorary Consultant, Cardiovascular and Medical Sciences, University of Glasgow) prepared the application for grant funding to support the trial; contributed to the design of the work and the interpretation of data; revised the draft report critically for important intellectual content; and prepared the results for publication.
Stephen Sharp (Statistics – Senior Statistician, MRC Epidemiology Unit, University of Cambridge) prepared the application for grant funding to support the trial; contributed to the design of the work and the acquisition, analysis and interpretation of data; drafted the work and revised it critically for important intellectual content; and prepared the results for publication.
Contributions of others
We are grateful to the independent members of the TSC: Professor Mike Clarke (Chairperson), Professor Paul Little [Programme Director of Programme Grants for Applied Research (PGfAR), Editor-in-Chief for the PGfAR journal and member of the NIHR Journals Library Board], Professor Des Johnston, Mr Roger Hughes, Mr Kirit Mistry and Dr Sabine Kläger.
We acknowledge the helpful input from the IDMC: Professor Bryan Williams, Professor David Dearnaley, Professor Simon Heller, Professor Stuart Pocock and Mr Tom Godec.
We are grateful to Professor Jane Armitage for carrying out the adjudication for all GLINT events.
We are grateful to Joanne Keenan and Zoë Doran, former employees of the DTU, for their contribution to the development and initiation of GLINT.
Aside from the authors, the Cambridge GLINT team has included Clare Boothby, Debora Lucarelli, Rebecca Simmons, Matt Sims, James Sylvester, Jaimie Taylor, the MRC Epidemiology Unit field team, the study co-ordination team, the laboratory team and the data management team.
Aside from the authors, the Leicester GLINT team has included Emer Brady, Carla Christie, Carolyn Currie, Adrian Manise, Danielle McKenna, Pramodini Radia, Francesco Zaccardi and the Leicester Diabetes Centre clinical and nursing team.
Aside from the authors, the Oxford GLINT team has included Zoe Doran, Anne Gilligan, Vanessa Gregory, Joanne Keenan, Ian Kennedy, Steven Richards (all provided assistance with database design and management), Ada Tse (assistance with operational delivery), Ruth Coleman (independent statistician) and Rishi Patel (manuscript review).
We thank the CCTU for its contribution to the sponsor oversight of GLINT.
We thank the Cambridge University Hospitals NHS Foundation Trust Department of Clinical Biochemistry and the NIHR Cambridge Biomedical Research Centre Core Biochemical Assay Laboratory for carrying out the biochemical assays.
We acknowledge Merck KGaA for providing metformin and matching placebo.
Initial development of GLINT was supported by funding from a Diabetes Research Network writing group award.
Data sharing statement
All available data can be obtained from the corresponding author.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Luengo-Fernandez R, Leal J, Gray A, Petersen S, Rayner M. Cost of cardiovascular diseases in the United Kingdom. Heart 2006;92:1384-9. https://doi.org/10.1136/hrt.2005.072173.
- Levitan EB, Song Y, Ford ES, Liu S. Is nondiabetic hyperglycemia a risk factor for cardiovascular disease? A meta-analysis of prospective studies. Arch Intern Med 2004;164:2147-55. https://doi.org/10.1001/archinte.164.19.2147.
- Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med 2004;141:413-20. https://doi.org/10.7326/0003-4819-141-6-200409210-00006.
- Coutinho M, Gerstein HC, Wang Y, Yusuf S. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care 1999;22:233-40. https://doi.org/10.2337/diacare.22.2.233.
- Ray KK, Seshasai SR, Wijesuriya S, Sivakumaran R, Nethercott S, Preiss D, et al. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trials. Lancet 2009;373:1765-72. https://doi.org/10.1016/S0140-6736(09)60697-8.
- Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, Duckworth WC, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia 2009;52:2288-98. https://doi.org/10.1007/s00125-009-1470-0.
- Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577-89. https://doi.org/10.1056/NEJMoa0806470.
- ACCORD study group . Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008;358:2545-59. https://doi.org/10.1056/NEJMoa0802743.
- Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008;358:2560-72. https://doi.org/10.1056/NEJMoa0802987.
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009;360:129-39. https://doi.org/10.1056/NEJMoa0808431.
- Deedwania P, Kosiborod M, Barrett E, Ceriello A, Isley W, Mazzone T, et al. Hyperglycemia and acute coronary syndrome: a scientific statement from the American Heart Association Diabetes Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 2008;117:1610-19. https://doi.org/10.1161/CIRCULATIONAHA.107.188629.
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393-40. https://doi.org/10.1056/NEJMoa012512.
- Ramachandran A, Snehalatha C, Mary S, Mukesh B, Bhaskar AD, Vijay V. Indian Diabetes Prevention Programme (IDPP) . The Indian Diabetes Prevention Programme shows that lifestyle modification and metformin prevent type 2 diabetes in Asian Indian subjects with impaired glucose tolerance (IDPP-1). Diabetologia 2006;49:289-97. https://doi.org/10.1007/s00125-005-0097-z.
- Boyle JG, Salt IP, McKay GA. Metformin action on AMP-activated protein kinase: a translational research approach to understanding a potential new therapeutic target. Diabet Med 2010;27:1097-106. https://doi.org/10.1111/j.1464-5491.2010.03098.x.
- Becker S, Dossus L, Kaaks R. Obesity related hyperinsulinaemia and hyperglycaemia and cancer development. Arch Physiol Biochem 2009;115:86-9. https://doi.org/10.1080/13813450902878054.
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 2009;32:1620-5. https://doi.org/10.2337/dc08-2175.
- Duncan BB, Schmidt MI. Metformin, cancer, alphabet soup, and the role of epidemiology in etiologic research. Diabetes Care 2009;32:1748-50. https://doi.org/10.2337/dc09-1183.
- Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 2010;33:322-6. https://doi.org/10.2337/dc09-1380.
- Stevens RJ, Ali R, Bankhead CR, Bethel MA, Cairns BJ, Camisasca RP, et al. Cancer outcomes and all-cause mortality in adults allocated to metformin: systematic review and collaborative meta-analysis of randomised clinical trials. Diabetologia 2012;55:2593-603. https://doi.org/10.1007/s00125-012-2653-7.
- Robson J, Dostal I, Sheikh A, Eldridge S, Madurassinghe V, Griffiths C, et al. The NHS Health Check in England: an evaluation of the first 4 years. BMJ Open 2016;6. https://doi.org/10.1136/bmjopen-2015-008840.
- Are You at Risk of Developing Cardiovascular Disease? Information Booklet. Leicester: University of Leicester; 2013.
- Rahman M, Simmons RK, Harding AH, Wareham NJ, Griffin SJ. A simple risk score identifies individuals at high risk of developing type 2 diabetes: a prospective cohort study. Fam Pract 2008;25:191-6. https://doi.org/10.1093/fampra/cmn024.
- Chamnan P, Simmons RK, Hori H, Sharp S, Khaw KT, Wareham NJ, et al. A simple risk score using routine data for predicting cardiovascular disease in primary care. Br J Gen Pract 2010;60:e327-34. https://doi.org/10.3399/bjgp10X515098.
- D’Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation 2008;117:743-53. https://doi.org/10.1161/CIRCULATIONAHA.107.699579.
- Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, Minhas R, Sheikh A, et al. Predicting cardiovascular risk in England and Wales: prospective derivation and validation of QRISK2. BMJ 2008;336:1475-82. https://doi.org/10.1136/bmj.39609.449676.25.
- Yates T, Davies M, Gorely T, Bull F, Khunti K. Effectiveness of a pragmatic education program designed to promote walking activity in individuals with impaired glucose tolerance: a randomized controlled trial. Diabetes Care 2009;32:1404-10. https://doi.org/10.2337/dc09-0130.
- Khunti K, Gray LJ, Skinner T, Carey ME, Realf K, Dallosso H, et al. Effectiveness of a diabetes education and self management programme (DESMOND) for people with newly diagnosed type 2 diabetes mellitus: three year follow-up of a cluster randomised controlled trial in primary care. BMJ 2012;344. https://doi.org/10.1136/bmj.e2333.
- Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972;18:499-502.
- Merck Serono . Glucophage SR 500mg, 750mg and 1000mg Prolonged Release Tablets. Summary of Product Characteristics. Updated 4 November 2015 n.d. www.medicines.org.uk/EMC/medicine/20952/SPC (accessed 7 January 2016).
- Sahin M, Tutuncu NB, Ertugrul D, Tanaci N, Guvener ND. Effects of metformin or rosiglitazone on serum concentrations of homocysteine, folate, and vitamin B12 in patients with type 2 diabetes mellitus. J Diabetes Complicat 2007;21:118-23. https://doi.org/10.1016/j.jdiacomp.2005.10.005.
- Webb DR, Khunti K, Gray LJ, Srinivasan BT, Farooqi A, Wareham N, et al. Intensive multifactorial intervention improves modelled five year coronary heart disease risk in screen-detected type 2 diabetes mellitus: a cluster randomized controlled trial. Diabetic Med 2012;29:531-40. https://doi.org/10.1111/j.1464-5491.2011.03441.x.
- Day N, Oakes S, Luben R, Khaw KT, Bingham S, Welch A, et al. EPIC-Norfolk: study design and characteristics of the cohort. European Prospective Investigation of Cancer. Br J Cancer 1999;80:95-103.
- Cambridge University Hospitals NHS Foundation Trust . Standard Operating Procedure R&Amp;D SOP001 n.d. www.cuh.nhs.uk/sites/default/files/research/RD_SOP001_CTIMP%20Delegation%20Roles%20and%20Responsibilities%20%282%29_0.pdf.
- Griffin SJ, Borch-Johnsen K, Davies MJ, Khunti K, Rutten GE, Sandbæk A, et al. Effect of early intensive multifactorial therapy on 5-year cardiovascular outcomes in individuals with type 2 diabetes detected by screening (ADDITION-Europe): a cluster-randomised trial. Lancet 2011;378:156-67. https://doi.org/10.1016/S0140-6736(11)60698-3.
- Aung T, Haynes R, Barton J, Cox J, Murawska A, Murphy K, et al. Cost-effective recruitment methods for a large randomised trial in people with diabetes: A Study of Cardiovascular Events iN Diabetes (ASCEND). Trials 2016;17. https://doi.org/10.1186/s13063-016-1354-9.
- NHS Health Check . NHS Health Check Data n.d. www.healthcheck.nhs.uk/commissioners_and_providers/data/ (accessed 11 May 2016).
- Gray LJ, Taub NA, Khunti K, Gardiner E, Hiles S, Webb DR, et al. The Leicester Risk Assessment score for detecting undiagnosed type 2 diabetes and impaired glucose regulation for use in a multiethnic UK setting. Diabet Med 2010;27:887-95. https://doi.org/10.1111/j.1464-5491.2010.03037.x.
- Chamnan P, Simmons RK, Khaw K-T, Wareham NJ, Griffin SJ. Estimating the population impact of screening strategies for identifying and treating people at high risk of cardiovascular disease: modelling study. BMJ 2010;340. https://doi.org/10.1136/bmj.c1693.
- Chamnan P, Simmons RK, Hori H, Khaw KT, Wareham NJ, Griffin SJ. A simple risk score using routine data for predicting cardiovascular disease in primary care. Br J Gen Pract 2010;60:590-6. https://doi.org/10.3399/bjgp10X515098.
- Inzucchi SE, Lipska KJ, Mayo H, Bailey CJ, McGuire DK. Metformin in patients with type 2 diabetes and kidney disease: a systematic review. JAMA 2014;312:2668-75. https://doi.org/10.1001/jama.2014.15298.
- Preiss D, Lloyd SM, Ford I, McMurray JJ, Holman RR, Welsh P, et al. Metformin for non-diabetic patients with coronary heart disease (the CAMERA study): a randomised controlled trial. Lancet Diabetes Endocrinol 2014;2:116-24. https://doi.org/10.1016/S2213-8587(13)70152-9.
- Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst Rev 2010;4. https://doi.org/10.1002/14651858.CD002967.pub3.
- Salpeter SR, Buckley NS, Kahn JA, Salpeter EE. Meta-analysis: metformin treatment in persons at risk for diabetes mellitus. Am J Med 2008;121:149-57. https://doi.org/10.1016/j.amjmed.2007.09.016.
- Inzucchi SE, Bergenstal RM, Buse JB, Diamant M, Ferrannini E, Nauck M, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015;38:140-9. https://doi.org/10.2337/dc14-2441.
- US Food and Drug Administration . FDA Revises Warnings Regarding Use of the Diabetes Medicine Metformin in Certain Patients With Reduced Kidney Function. Safety Announcement [4-8-2016] n.d. www.fda.gov/downloads/Drugs/DrugSafety/UCM494140.pdf (accessed 28 November 2016).
- European Medicines Agency . Use of Metformin to Treat Diabetes Now Expanded to Patients With Moderately Reduced Kidney Function. Recommendations For Patients With Kidney Impairment Updated in Product Information 2016.
- de Jager J, Kooy A, Lehert P, Wulffelé MG, van der Kolk J, Bets D, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B12 deficiency: randomised placebo controlled trial. BMJ 2010;340. https://doi.org/10.1136/bmj.c2181.
- Vidal-Alaball J, Butler CC. Reduced serum vitamin B12 in patients taking metformin. BMJ 2010;340. https://doi.org/10.1136/bmj.c2198.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R. Heart Protection Study Collaborative Group . MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet 2003;361:2005-16. https://doi.org/10.1016/S0140-6736(03)13636-7.
- Armitage J, Bowman L, Wallendszus K, Bulbulia R, Rahimi K, Haynes R, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet 2010;376:1658-69. https://doi.org/10.1016/S0140-6736(10)60310-8.
- The HPS2-THRIVE Collaborative Group . Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 2014;371:203-12. https://doi.org/10.1056/NEJMoa1300955.
- University of Oxford Clinical Trial Service Unit . HPS3/TIMI55-REVEAL/Trial n.d. www.revealtrial.org/ (accessed 17 January 2017).
Appendix 1 Trial Steering Committee membership
Independent
Professor Mike Clarke, chairperson (Queen’s University Belfast).
Professor Paul Little (University of Southampton).
Professor Des Johnston (Imperial College London, University of London).
Mr Roger Hughes (PPI).
Mr Kirit Mistry (PPI).
Dr Sabine Kläger (Sponsor Representative).
Non-independent (four principal investigators; the remainder are observers according to our charter)
Professor Simon J Griffin.
Professor Nicholas Wareham.
Dr Greg Irving.
Mr Andrew Dymond.
Professor Rury R Holman.
Professor M Angelyn Bethel.
Mrs Sarah Lessels.
Professor Kamlesh Khunti.
Professor Melanie Davies.
Mrs Kyla Harrington.
Appendix 2 Independent Data Monitoring Committee and Ethics Committee membership
Professor Bryan Williams, chairperson (University College London).
Professor Stuart Pocock (London School of Hygiene & Tropical Medicine).
Professor David Dearnaley (Institute of Cancer Research).
Professor Simon Heller (University of Sheffield).
Mr Tom Godec, responsible for unblinded data analysis (London School of Hygiene & Tropical Medicine).
Appendix 3 Participant study identification card
Appendix 4 Investigational medicinal product packaging label
Appendix 5 Data sources
| Data | Source |
|---|---|
| Consent | Consent form |
| Medical screening | eCRF |
| Sociodemographic | eCRF |
| Height | eCRF |
| Weight | eCRF |
| BMI | eCRF |
| Blood pressure | eCRF |
| Qualifying laboratory results | Source database |
| Laboratory results | Biochemistry output (varying at different sites) |
| Drug accountability | eCRF/questionnaire (depending on time point in study) |
| Drug satisfaction | Participant questionnaire |
| Participant-reported cardiovascular events | Participant questionnaire |
| Participant-reported cancer | Participant questionnaire |
| Participant-reported diabetes | Participant questionnaire |
| EQ-5D | Participant questionnaire |
| SF-8 | Participant questionnaire |
| HERC questionnaire | Participant questionnaire |
| GP-reported events | GP questionnaire |
| Pregnancy | GP questionnaire |
| Medication list | eCRF |
| AE/SAE form | eCRF or paper case report form |
List of abbreviations
- AE
- adverse event
- ALT
- alanine aminotransferase
- BMI
- body mass index
- CCTU
- Cambridge Clinical Trials Unit
- CI
- confidence interval
- CKD
- chronic kidney disease
- CLRN
- Comprehensive Local Research Network
- CTIMP
- Clinical Trial of an Investigational Medicinal Product
- CVD
- cardiovascular disease
- DPP
- Diabetes Prevention Program
- DTU
- Diabetes Trials Unit
- eCRF
- electronic case report form
- eGFR
- estimated glomerular filtration rate
- EQ-5D
- EuroQol-5 Dimensions
- GLINT
- Glucose Lowering In Non-diabetic hyperglycaemia Trial
- GP
- general practitioner
- HbA1c
- glycated haemoglobin
- HDL
- high-density lipoprotein
- HERC
- Health Economics Research Centre
- HES
- Hospital Episode Statistics
- HSCIC
- Health and Social Care Information Centre
- IDMC
- Independent Data Monitoring Committee
- IMP
- investigational medicinal product
- LDL
- low-density lipoprotein
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRC
- Medical Research Council
- NDH
- non-diabetic hyperglycaemia
- NIHR
- National Institute for Health Research
- ONS
- Office for National Statistics
- PIC
- participant identification centre
- PPI
- patient and public involvement
- SAE
- serious adverse event
- SD
- standard deviation
- SF-8
- Short Form questionnaire-8 items
- SmPC
- summary of product characteristics
- T2D
- type 2 diabetes
- TSC
- Trial Steering Committee