

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 13/04/105. The contractual start date was in December 2015. The draft report began editorial review in July 2017 and was accepted for publication in November 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Mark J Peters reports membership of the National Institute for Health Research (NIHR) Health Technology Assessment General Board since June 2018. Kathryn M Rowan is a member of the NIHR Health Services and Delivery Research Board.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Inwald et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background and rationale
Infectious diseases remain a major cause of mortality and morbidity, accounting for > 25% of deaths in children < 5 years of age in Europe. 1 In the UK, approximately 1000 children present to paediatric intensive care units (PICUs) with severe sepsis each year, of whom ≈8% die. 2 Mortality increases, sometimes up to 17%, when children presenting to emergency departments (EDs) with severe sepsis are included. 3 In addition, serious morbidity in PICU survivors is high. 4
Rapid fluid bolus resuscitation is integral to the UK management of children presenting to EDs with septic shock. In 2009, based on the best available evidence, the American College of Critical Care Medicine – Pediatric Advanced Life Support (ACCM-PALS)’s clinical guideline5 recommended fluid bolus resuscitation with a bolus size of 20 ml/kg, up to a total of 200 ml/kg over the first hour, for the management of children and neonates with septic shock. However, this recommendation is predominantly based on weak evidence from retrospective observational studies involving small numbers of children. 6–8
In Africa, a recent, rigorous, multicentre, randomised controlled trial (RCT), the Fluid Expansion as Supportive Therapy (FEAST) trial,9 compared fluid bolus resuscitation of 20 ml/kg with maintenance fluid in > 3000 children with severe infection. The study reported a 35% increase in mortality associated with fluid bolus resuscitation. 9 Although conducted in a low-income setting, the FEAST9 trial raised considerable uncertainty and highlighted the lack of evidence for fluid bolus resuscitation for children in middle- and high-income settings. 10,11
A recent systematic review of fluid bolus resuscitation in children with septic shock reported that fluid boluses were associated with increased mortality compared with maintenance fluid. 12 However, the only RCT comparing bolus with maintenance fluid identified in this review was the FEAST9 trial. The systematic review identified one additional RCT comparing different fluid volumes. 13 This trial compared an aggressive fluid resuscitation strategy (20–40 ml/kg over 15 minutes) with usual fluid bolus resuscitation (20 ml/kg over 20 minutes up to a maximum of 60 ml/kg over 60 minutes) in 147 children, aged from 1 month to 12 years, presenting with septic shock to a single ED in India. 13 The total volume of fluid administered was higher in the aggressive fluid resuscitation group than in the usual fluid resuscitation group, but there was no difference in mortality or in the other clinical outcomes measured, except for an increase in hepatomegaly at 20 minutes in the aggressive fluid resuscitation group.
To date, no trials have compared a restrictive fluid bolus resuscitation strategy with recommended fluid bolus resuscitation in children with septic shock in the developed world. One single-centre pilot RCT has been completed in Canada. 14 This trial aimed to achieve a ‘fluid-sparing’ resuscitation strategy through earlier initiation of vasoactive medication (following initial fluid bolus resuscitation of ≥ 40 ml/kg within the 2 hours prior to randomisation).
When considering repeating the FEAST9 trial in the UK, the existence and adoption of clinical guidelines (such as the ACCM-PALS guideline)5 and training courses (such as Advanced Paediatric Life Support15) make it very unlikely that clinicians in UK EDs would accept a randomisation allocation to maintenance fluid only. However, with accumulating adult and paediatric data suggesting that excessive fluid administration is associated with worse patient outcomes, and even an increased risk of death,16 the optimal volume of fluid for children presenting with septic shock remains an important unanswered question.
The Fluids in Shock (FiSh) trial proposed to evaluate, in a pragmatic RCT, whether or not a restrictive strategy (fluid bolus resuscitation of 10 ml/kg), compared with the current recommended strategy (fluid bolus resuscitation of 20 ml/kg), is associated with improved outcomes for children presenting to UK EDs with presumed septic shock.
First, it was imperative to establish whether or not a large multicentre RCT of a restrictive fluid bolus resuscitation strategy would be feasible and acceptable in the UK. Clinical trials, such as the proposed FiSh trial, are expensive, and the chances of successful completion are improved if the feasibility and pilot testing of certain key parameters can be clearly demonstrated.
Aim
To explore and test, in a combined feasibility study and pilot trial (conducted as a smaller version of the full trial), important key parameters needed to inform the design and ensure the successful conduct of the FiSh trial and to report a clear recommendation for continuation or not to the full trial.
Objectives
Feasibility study
-
To review and explore, with input from parents/legal representatives, acceptability, potential barriers, information/documentation, decision-making and deferred consenting [or research without prior consent (RWPC)].
-
To review and explore, with input from parents/legal representatives, the potential patient-centred primary, and other important secondary, outcome measures.
Pilot trial with integrated perspectives study
-
To test the willingness of clinicians to screen, recruit and randomise eligible participants.
-
To estimate the recruitment rate.
-
To test, following randomisation, the delivery of and adherence to the intervention and demonstrate separation between the groups.
-
To test acceptability of the deferred consenting procedures and documentation.
-
To test follow-up for the identified, potential patient-centred primary and other important secondary outcome measures and adverse event (AE) reporting.
-
To inform the final selection of a patient-centred primary outcome measure.
-
To estimate the characteristics of the selected patient-centred primary outcome measure to inform sample size estimation.
-
To inform the content of, and time needed for, final data collection.
Progression criteria
The objectives would be reported against the following progression criteria:
-
Formal sign-up of sites (the required number of hub-and-spoke models) to achieve the overall sample size based on the observed recruitment rate, as determined by the pilot trial objectives concerning identification, recruitment rate, acceptability of deferred consenting procedures, selection of the patient-centred primary outcome measure and sample size estimation.
-
The recruitment rate achieved in the pilot trial (taking into account loss of participants as a result of declining deferred consent) being consistent with achieving the overall sample size required – as determined by the objectives concerning selection of the patient-centred primary outcome measure and sample size estimation – in an acceptable time frame from the sites giving formal sign-up in the first feasibility objective.
-
At least 80% of fluid bolus resuscitation in the external pilot study being delivered at the correct volume and timing ± 10%, as determined by the external pilot study objective concerning adherence.
-
The absolute total volume of fluid administered during the first hour and during the first 4 hours being lower (by ≥ 25%) in the more restrictive fluid bolus resuscitation group than in the current recommended fluid bolus resuscitation group, as determined by the external pilot study objective concerning separation.
Chapter 2 Feasibility study
Methods
Study design
The FiSh feasibility study was a qualitative semistructured interview study.
Research governance
The FiSh feasibility study was sponsored by Imperial College Healthcare NHS Trust and co-ordinated by the University of Liverpool and Intensive Care National Audit and Research Centre (ICNARC) Clinical Trials Unit (CTU). An ethics application was made to the North West – Liverpool Central Research Ethics Committee (REC) on 16 October 2015 and a favourable opinion was received on 15 December 2015 (REC reference number: 15/NW/0913). The protocol is available at www.journalslibrary.nihr.ac.uk/programmes/hta/1304105/#/ (accessed 23 May 2016). Local NHS permissions were obtained for the NHS trusts that participated as Participant Identification Centres (PICs) for recruitment routes 1 and 2 (see Recruitment).
Study management
The FiSh feasibility study was led by a qualitative researcher co-investigator (KW) with support from the Study Management Group (SMG), which included the chief investigator (DI) and other co-investigators (AC, DAH, NJ, MDL, PRM, SN, MJP and KMR). An experienced research associate (CBOH) was employed to conduct, transcribe and analyse the interviews.
Network support
To maintain the profile of the study, updates were provided at national meetings, such as the Paediatric Emergency Research in the UK and Ireland (PERUKI) annual meeting and the biannual Paediatric Intensive Care Society Study Group (PICS-SG) meetings.
Patient and public involvement
Two parents (NJ and AC) of children with experience of admission to hospital with severe infection were co-investigators and members of the SMG. They provided valuable input into the design and conduct of the study, including reviewing documents for parent interviews [e.g. draft pilot trial participant information sheets (PISs) and a list of potential outcomes, including outcome descriptions] and informing study recruitment approaches (i.e. identification of social media groups and charities). They were also involved in the review of study progress and findings.
Design and development of the protocol
The design and development of the protocol, including sample estimation, recruitment strategy and interview topic guide, were informed by previous trials conducted in paediatric emergency and critical care in the NHS and earlier research. 17–20 Previous relevant research was used to develop an interview topic guide (see Appendix 1) and draft PIS for the pilot trial (see Appendix 2). 18,19,21 The topic guide contained open-ended questions and prompts to help explore parents’/legal representatives’ views on the acceptability of the FiSh trial, including the draft pilot trial PIS and approach to consent. A separate set of questions was developed for bereaved parents/legal representatives.
Literature review
The literature review for potential outcome measures was conducted prior to the FiSh feasibility study start date (December 2015). The review included:
-
previous systematic reviews of the literature
-
a search through literature referenced in guidelines provided by the International Liaison Committee on Resuscitation
-
a search on the Core Outcome Measures in Effectiveness Trials (COMET) Initiative database
-
a search on the Evidence in Paediatric Intensive Care Collaboration database
-
a search on ClinicalTrials.gov for recent paediatric emergency and critical care studies, including the SQUEEZE14 (Septic Shock Reversal is Quicker in Pediatric Patients Randomized to an Early Goal Directed Fluid-sparing Strategy vs. Usual Care) pilot trial.
A number of studies, trials and systematic reviews were identified. From these, a list of potential outcome measures (see Appendix 3) was compiled to inform discussions with parents/legal representatives during the interview.
Amendments to the study protocol
Following receipt of a favourable opinion of the study protocol from the REC on 15 December 2015, there were no amendments.
Recruitment
Hospitals
Invitations for expressions of interest in participating as a PIC for recruitment routes 1 and 2 were sent to lead clinicians in paediatric emergency and paediatric critical care medicine at a number of UK NHS hospitals.
Eligibility
Parents/legal representatives with a child presenting to an ED or being admitted to a PICU with a severe infection within the previous 3 years were eligible to take part in the study, unless they were unable to speak and read English. In this instance, severe infection was defined as any condition leading to inpatient treatment for sepsis or septic shock, such as meningococcal septicaemia.
Recruitment and sampling procedure
Based on previous studies,19 it was anticipated that 15–25 parents/legal representatives would need to be recruited to reach data saturation. The study procedures for recruitment are summarised in Figure 1.
FIGURE 1.
Summary of study procedures for recruitment of participants.

Recruitment route 1: postal contact
Clinicians used hospital medical records to identify the 15 most recent parents/legal representatives (including up to five bereaved) of children who met the inclusion criteria. Those identified were sent a postal invitation, including a covering letter and FiSh feasibility study PIS describing how to register interest in taking part.
Recruitment route 2: advertising in paediatric intensive care units
Posters and copies of the FiSh feasibility study PIS were placed in family/relative waiting rooms near the PICU.
Recruitment route 3: advertising online, including social media
An advertisement was posted on Twitter (Twitter Inc., San Francisco, CA, USA; www.twitter.com) and Facebook (Facebook Inc., Menlo Park, CA, USA; www.facebook.com), which invited parents/legal representatives to register interest in participating in the study. Relevant charities and parent support groups were asked to place the advertisement on their website and social media.
Interviews
Screening
Parents’/legal representatives’ requests to participate were responded to in sequential order. Once eligibility was confirmed, an interview date and time were scheduled. The draft pilot trial PIS (see Appendix 2) and list of potential outcomes (see Appendix 3) were e-mailed to parents/legal representatives to read before the interview. Interviews aimed for data saturation, that is, the point at which no new major themes are discovered in analysis. 22,23 Screening stopped when data saturation was reached. 22,23
Informed consent
Audio-recorded verbal consent was sought over the telephone before the interview. This involved reading each aspect of the consent form to parents/legal representatives, including consent for audio-recording and to receive a copy of the findings when the study ended. Each box was ticked on the consent form when verbal consent was provided. Informed consent discussions were audio-recorded for auditing purposes.
Conduct of the interview
The telephone interview began with a discussion about the aims of the study, an opportunity for questions and checking that the parent/legal representative had had sufficient time to read the draft pilot trial PIS and list of potential outcomes.
The interview then commenced using the interview topic guide to explore:
-
the acceptability of the proposed trial
-
any potential barriers for participation in the trial and how these could be addressed
-
parents’/legal representatives’ views on the length and content of the draft pilot trial PIS, leaflets and posters
-
parents’/legal representatives’ decision-making in the emergency setting
-
the acceptability of RWPC (deferred consent)
-
the trial design, including the selection of outcome measures.
Example questions are shown in Appendix 1. Respondent validation was used to add unanticipated topics to the topic guide as interviewing and analysis progressed. 24 After the interview, participants were sent a copy of the consent form and a thank-you letter, including a £30 Amazon (Amazon.com, Inc., Bellevue, WA, USA) voucher to thank them for their time.
Transcription
Digital audio-recordings were transcribed verbatim by a professional transcription company (Voicescript Ltd, Bristol, UK) in accordance with the Data Protection Act 1998. 25 Transcripts were anonymised and checked for accuracy. All identifiable information, such as names (e.g. of patients, family members or the hospital that their child was admitted to), was removed.
Data analysis
A psychologist (CBOH) led the analysis with assistance from a sociologist (KW). Analysis was broadly interpretive and iterative (Table 1). 31,32 Informed by the constant comparative approach, the aim was to provide accurate representation of parental views on trial design and acceptability. 29,33–35 NVivo 10 (QSR International, Warrington, UK) software was used to assist in the organisation and coding of data.
| Phase | Description |
|---|---|
| Familiarisation with data and generating initial themes | CBOH read and reread a sample of transcripts, noting down initial ideas around main themes and potential codes |
| Developing the coding framework | Using NVivo 10 software, CBOH created codes for initial main themes (e.g. thoughts on the FiSh trial, thoughts on the draft pilot trial PIS, thoughts on RWPC, decision-making in the emergency setting and misunderstandings and misconceptions) and began developing a coding framework using line-by-line coding, comparing between transcripts as part of a constant comparative approach26–28 |
| Initial coding meeting | CBOH and KW met to discuss early themes and develop the coding framework |
| Second coding (coded by a second person) | KW second coded a sample of transcripts (two bereaved parent interviews, one non-bereaved parent interview – 15% of transcripts) and made notes on any new themes identified and how the framework could be refined |
| Second coding meeting | CBOH and KW met to discuss, reflect and refine the specifics of each theme in the coding framework29 |
| Completion of coding of transcripts | CBOH completed coding of all transcripts in preparation for write-up |
| Write-up and final revision of coding | CBOH and KW developed the manuscript using themes to relate back to the study aims, ensuring key findings and recommendations were relevant to the FiSh trial design and site staff training (i.e. catalytic validity).28,30 Final discussion and development of selected themes occurred during the write-up phase |
Results
Participating NHS trust hospitals
Expressions of interest were received from seven NHS hospitals. Local research and development approval was obtained for four NHS hospitals in January 2016 for recruitment via routes 1 and 2.
Participants
A total of 58 parents registered interest, of whom 29 were screened (Figure 2); three were deemed ineligible and five did not confirm a date for interview. Data saturation was reached when 21 parents from 20 families were interviewed. 22,23,35 No individuals identified themselves as legal representatives and, therefore, this term is not used in the remainder of this chapter.
FIGURE 2.
The FiSh feasibility study recruitment process.
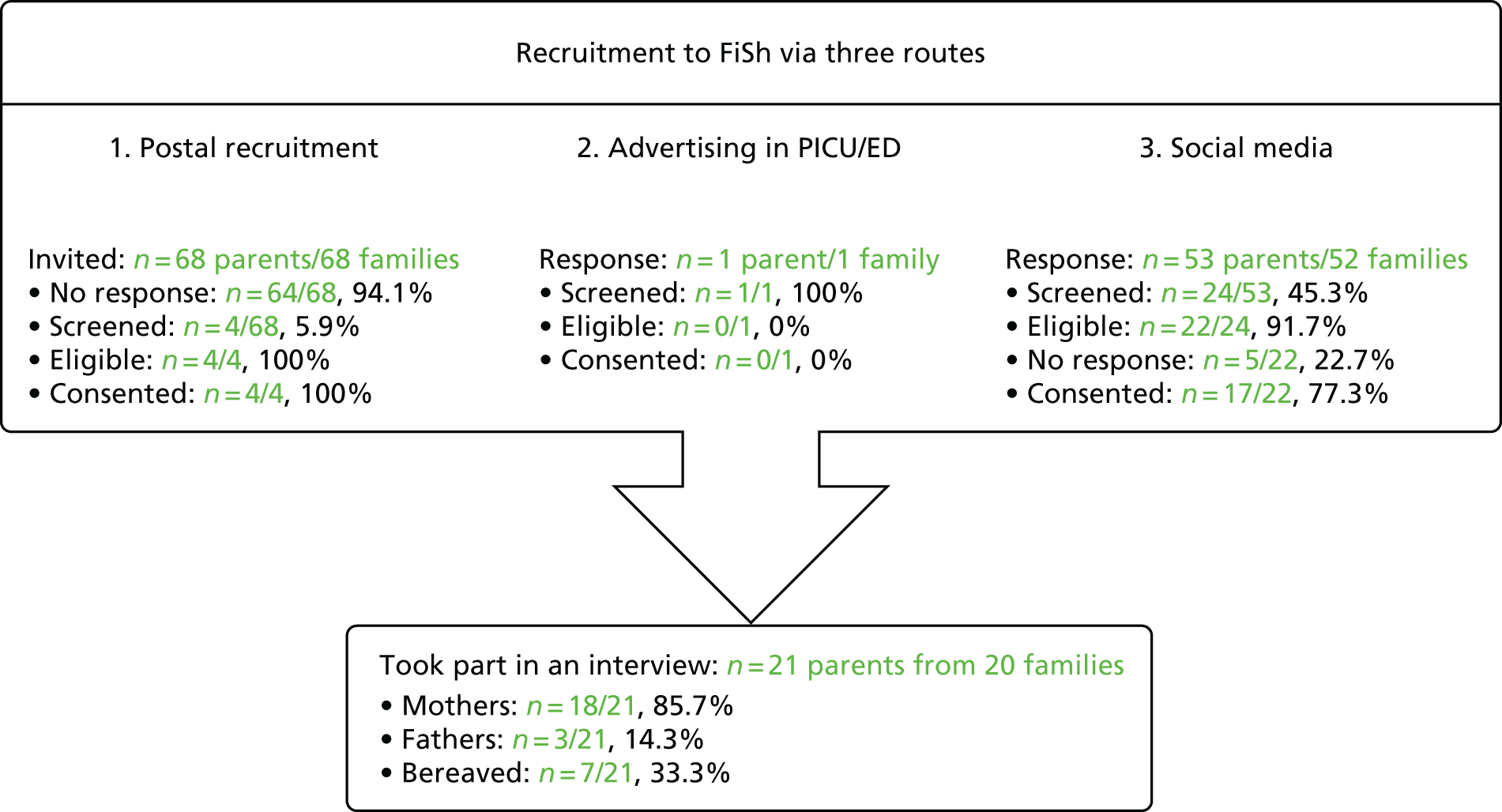
Characteristics
The sample included 18 mothers (five bereaved) and three fathers (two bereaved). Bereaved parents were interviewed a mean of 12.3 months [standard deviation (SD) 10.1 months] since admission (range 3–31 months) and non-bereaved parents a mean of 17.6 months (SD 9.9 months) since admission (range 1–41 months). The mean length of hospital stay for their children was 7.1 days (SD 9.6 days; range: 1–25 days) for bereaved parents and 33.9 days (SD 47.4 days; range 4–140 days) for non-bereaved parents. Eight parents had previously participated in a clinical trial. One mother had experience of RWPC. Interviews took between 30 and 55 minutes.
Parental perspectives
Research without prior consent is acceptable but some initial concerns
A general definition of RWPC was first read to parents (Box 1). Many said that they may be initially surprised to discover that their child had been entered into a trial without their prior consent (see Appendix 4 for quotations by theme). However, concerns subsided once reasons for using RWPC in emergency situations were considered. Parents went on to respond favourably to the concept of RWPC, describing it as a logical solution to enable research in challenging circumstances.
Due to the need to treat a patient in an emergency without delay, or because parents may not always be present when a child needs treatment, it is not always appropriate or possible to obtain consent before a child is entered into a trial. To enable research to be conducted in the emergency setting, many countries (including the UK) allow consent to be sought as soon as possible afterwards. This is for permission to use the data already collected and to continue in the trial. This is research without prior consent (sometimes called deferred consent). Research without prior consent is a relatively new approach to seeking consent in the UK.
Support for the FiSh trial but some concerns and misconceptions
Overall, parents supported the FiSh trial. Many viewed trial participation as a way to help other families and children in the future. Some parents thought that the consent discussion could provide a ‘distraction’ [Participant (P) 9, mother, non-bereaved] but foster ‘a sense of control in a situation where you feel completely out of control’ (P6, mother, non-bereaved).
However, support for the FiSh trial appeared to be dependent on the intervention being successful and ‘how well [their child] was’ (P4, mother, non-bereaved). Parents, including those who were bereaved, said that they would wonder if their child’s participation in the trial was ‘the reason [their child] didn’t survive’ (P19, mother, bereaved), in that eventuality.
Some parents had specific concerns and misconceptions that influenced their views on the acceptability of the FiSh trial. Many were concerned that a change from current practice might jeopardise their child’s chances of survival. This was linked to the misconception that the currently recommended fluid bolus volumes are evidence based. Parents were concerned that restrictive fluid bolus therapy would be insufficient, with the current liberal approach to fluid bolus resuscitation viewed as being more likely to save a child’s life.
In response, parents were directed to the relevant sections of the draft pilot trial PIS and provided tailored explanations, such as the weak evidence base for current recommended practice, how fluid resuscitation is part of a larger treatment package, and monitoring procedures. Such explanations appeared to address parents’ initial concerns and misconceptions about the proposed FiSh trial.
Unclear or missing study information
All parents considered the draft pilot trial PIS to be clear and concise. However, parents raised questions about some aspects of the trial (Box 2), indicating that key information was missing. Importantly, this information was prioritised by parents, affecting their understanding and views about the acceptability of the trial. Many said that they would not ask questions or raise concerns with a member of site staff approaching them for consent.
-
Would the amount of fluid given be corrected if my child was not stabilising?
-
Does the amount of fluid have a direct impact on outcome?
-
What is a fluid bolus?
-
What is the timing of fluid bolus administrations?
-
Would I be able to find out which group my child was randomised to?
-
Does the fluid treatment apply regardless of the child’s age?
Timing of consent
Parents suggested that the discussion about consent should occur after the initial stress had subsided and their child’s condition had stabilised, preferably within 24–48 hours. Parents expressed the need for the research team to gauge appropriate timing of this discussion in consultation with the clinical team.
The researcher asked bereaved parents to consider a scenario in which their child had been entered into the trial before death and a practitioner approached them after death to discuss the trial. They suggested that site staff should be prepared to address concerns about whether or not trial participation was ‘a reason as to why [death] happened’ (P21, mother, bereaved). They emphasised a need for sensitivity and time, particularly if a child had died very quickly and without warning. Parents described their anger in the initial stages of bereavement, which they believed would negatively affect their decision-making abilities and their response to a discussion about consent. Nevertheless, all bereaved parents agreed that it would be acceptable to discuss the trial at a later time, after they had left hospital. Bereaved parents valued medical research and, in general, said that they would have consented to the use of their child’s data as a way to help other children in the future.
Bereaved parents’ views were then sought on the most appropriate way of contacting parents to discuss the trial following the death of a child (Box 3). Several parents thought that an appropriately timed face-to-face discussion with a nurse or consultant would be preferable to a telephone call or letter. The majority, however, supported contact via post at 4 weeks and then at 8 weeks after death as long as the proposed ‘opt-out’ approach provided was emphasised in bold and the letter personalised, ideally by a known member of the clinical team. However, parents described grief as a ‘very personal matter’ (P17, mother, bereaved), making it difficult to develop general recommendations on how best to approach parents in this situation.
Research associate presented several options to consider:
-
Face-to-face discussion with a nurse or doctor.
-
Telephone call from a nurse or doctor.
-
Personalised letter 4 weeks after randomisation, followed by a second letter 8 weeks after randomisation (i.e. if no response is received after sending the initial letter). Letters would explain how to opt out of the study and that there would be no need to respond if they wanted their child’s data to be used in the trial.
Outcomes of importance to parents
Appendix 3 includes the list of outcomes and accompanying descriptive text sent to parents before the telephone interviews. In this section of the interview, a definition of an outcome was first read to parents, including an explanation about why it was important to explore parents’ perspectives about important outcomes (see Appendix 1). Parents were asked to think about their experience of their child being admitted for severe infection and then prompted to share their views on what effects of treatment they hoped for (i.e. what indicators were most important to suggest that their child was getting better).
Many parents described looking for important indicators of improvement as ‘a gradient of seriousness’ (P6, mother, non-bereaved), initially looking for ‘the worst [outcome] and then . . . you sort of progressively aim towards the sort of next hurdle to get over’ (P10, mother, non-bereaved).
Appendix 3 shows the full list of outcomes identified in the analysis of parent descriptions. On average, parents described six outcomes. In order of importance (i.e. defined as how many parents mentioned a particular outcome when asked directly which indicators were most important to them), parents prioritised the following outcomes:
-
long-term effects, for example long-term health and development, disabilities
-
‘getting better’, for example looking and behaving more like their normal selves
-
organ/physiological functioning, for example blood pressure, temperature and heart rate
-
time spent on treatments and machines
-
survival
-
time spent in hospital.
The majority of parents described the provided list of outcome measures as ‘fair’ (P16, father, bereaved) and ‘thorough’ (P20, mother, non-bereaved). No additional outcomes (i.e. unrelated to categories provided on the list) were identified by parents. Bereaved parents found it difficult to consider outcome measures other than survival:
Now that’s difficult, because coming from where I am coming from I would just say that they survive. For me would be the number one criteria.
P19, mother, bereaved
Key findings to inform the pilot trial
This qualitative study provides insight into the acceptability of the FiSh trial to parents by exploring the views of parents with relevant experience. Consistent with the CONNECT (CONseNt methods in paediatric Emergency and urgent Care Trials) study findings and associated guidance on RWPC,18,19,26,27 some parents initially expressed concerns about the concept of RWPC. However, initial concerns subsided when reasons why informed consent could not be sought were offered. As also shown in previous research,17,28 parents thought that they might be unable to provide a rational, informed decision about research in an acute emergency situation and supported alternative approaches to prospective informed consent as a way to enable research in time-critical situations to improve treatments for critically ill children. 17,19,28,30,36,37
The findings highlight specific concerns and misunderstandings that initially influenced parental views on the acceptability of the FiSh trial. 38 Concerns included the proposed change from current clinical practice and its potential effect on a child’s recovery. Although the draft pilot trial PIS included a description of the weak evidence for current practice, many held a misconception that this was the proven optimal treatment. Moreover, several parents were unclear about the nature of fluid bolus therapy and only one understood that 20 ml/kg of fluid would be given to all children before being entered into the trial. Nevertheless, all parents reported that they would have provided consent for the use of their child’s data in the FiSh trial, valuing the opportunity to advance medical research and viewing their child’s participation as a means to help similar families in the future. 19 This is consistent with previous findings that emphasise the need for simple, non-medicalised information to improve parental understanding of trial information and inform their research decisions. 19,21,39 Tailored explanations appeared to address parents’ priorities, concerns and misconceptions. These findings were used to develop the FiSh pilot trial site staff training, which emphasises the need for recruiters to provide opportunities for questions as parents are unlikely to voice potential concerns. 38,40
Consistent with previous studies, which explored approaches to consent in time-critical situations,17,19,20,30,41 the timing of the consent was found to be likely to affect parental responses to the trial. Although many parents said that consultation with the clinical team would help research staff to gauge when to approach families for consent,27 their views on when and how this initial contact should happen differed depending on whether or not their child had survived. Parents of children who had survived a severe infection expressed how they would wish to be approached in hospital, ideally within 24–48 hours of randomisation, once their child’s condition had stabilised. 26 In contrast, bereaved parents emphasised that parents in this situation should not be approached immediately after their child’s death, as this may heighten feelings of grief and anger. It was this perceived burden to vulnerable families that led to a waiver of consent (i.e. no trial discussion) when a child died in the FEAST trial. 17,30,42 However, our findings do not support the model used in the FEAST trial,43 as the bereaved parents we interviewed expressed a wish to discuss the use of their child’s information in the FiSh trial, as long as the timing of this discussion was appropriate. 18,27
The FiSh feasibility study findings thus provide new insight into what should happen if a child dies after being entered into a trial. 26 Although some bereaved parents preferred a face-to-face research discussion, the majority were happy to be contacted by post, with an opportunity to meet with a researcher at a later date. Bereaved parents responded favourably to the opt-out approach proposed. Importantly, parents recommended that the letter should be sent from a practitioner known to the family, with whom they had developed a close and trusting rapport. 26 As septic shock is associated with an 8–17% mortality rate in the ED and PICU,2,3 these findings are particularly important and will help to design a trial that is appropriate for the needs of vulnerable families. The views of bereaved parents also highlight the need for practitioners to prepare to respond to parents who are concerned that trial participation may have resulted in harm.
Finally, parents provided their views about the outcomes that are most important to them in children with severe infection. These qualitative findings were incorporated into the FiSh pilot trial design to ensure the selection and measurement of outcomes that were patient centred.
Chapter 3 Methods for pilot trial with integrated perspectives study
Study design
The FiSh pilot trial was a pragmatic, open, multicentre, parallel-group, pilot RCT with an integrated perspectives study element involving parents/legal representative of trial participants and site staff involved in the RCT. The integrated perspectives study comprised questionnaires and interviews with parents/legal representatives, as well as focus groups and interviews with the site staff towards the end of the pilot trial recruitment period.
Research governance
The pilot trial was sponsored by Imperial College Healthcare NHS Trust and co-ordinated by the ICNARC CTU. An application was submitted to the Health Research Authority (HRA) on 19 April 2016 and received full approval on 22 June 2016 (Integrated Research Application System reference number: 195544; REC reference number: 16/LO/0854). The trial protocol is available at www.journalslibrary.nihr.ac.uk/programmes/hta/1304105/#/.
The pilot trial was registered with the ISRCTN (International Standard Randomised Controlled Trial Number) registry. Registration was confirmed on 11 August 2016 (ISRCTN15244462). The pilot trial was adopted onto the National Institute for Health Research (NIHR) Clinical Research Network (CRN) portfolio on 9 June 2016 and issued the NIHR CRN portfolio number 31037.
Local confirmation of capacity and capability was obtained from each participating NHS trust. A clinical trial site agreement, based on the model agreement for non-commercial research in the health service, was signed by each participating NHS trust and the sponsor (Imperial College Healthcare NHS Trust).
Following guidelines from NIHR,44 a Trial Steering Committee (TSC), with a majority of independent members, was convened to oversee the pilot trial on behalf of the funder (NIHR) and the sponsor (Imperial College Healthcare NHS Trust). The TSC met three times during the pilot trial and comprised an independent chairperson, an independent lay member (representing parent perspectives), independent clinicians (specialising in paediatric emergency and critical care medicine), the chief investigator (DI) and the lead qualitative researcher (KW).
In addition, an independent Data Monitoring and Ethics Committee (DMEC) was convened to monitor pilot trial data and ensure the safety of pilot trial participants. The DMEC met twice during the pilot trial and comprised two expert clinicians specialising in paediatric emergency and critical care medicine (one of whom acted as chairperson) and an experienced statistician.
Study management
The study manager was responsible for the day-to-day management of the pilot trial, with support from the senior trial manager, data manager, study statistician and research assistant. The researcher was responsible for the day-to-day management of the integrated perspectives study elements with support from the qualitative researcher co-investigator. The SMG, chaired by the senior trial manager, was responsible for overseeing day-to-day management of the pilot trial and comprised the chief investigator (DI) and co-investigators (AC, DAH, NJ, PRM, SN, MJP, KMR and KW). The SMG met regularly throughout the pilot trial to ensure adherence to the pilot trial protocol and to monitor its conduct and progress.
Network support
To maintain the profile of the pilot trial, updates were provided at national meetings, such as the PERUKI annual meeting and biannual PICS-SG meetings.
Patient and public involvement
Two parents (NJ and AC) of children with experience of admission to hospital with severe infection were co-investigators and members of the SMG. They provided valuable input into the design and conduct of the study. They were also involved in the review of study progress and findings.
Design and development of the protocol
Using the FEAST trial9 as a starting point, the FiSh pilot trial protocol was originally conceived as a comparison of fluid bolus resuscitation with no fluid bolus resuscitation (i.e. solely maintenance fluid). However, following consultation with clinicians in UK paediatrics, paediatric ED and PICU, it became clear that this approach would not be acceptable, and that the lowest agreed intervention bolus size was 10 ml/kg after an initial bolus of 20 ml/kg. The 10-ml/kg fluid bolus size was then proposed and deemed acceptable, following discussion, at a meeting of would-be collaborators of the FiSh pilot trial in 2012, with the comparator being the currently recommended bolus size of 20 ml/kg. This strategy was then further ratified by the PERUKI research collaborative. Thus, the proposed intervention bolus size of 10 ml/kg represented the current position of equipoise among UK clinicians.
The use of vasopressors was considered during protocol development. However, the clinical consultation indicated that the protocolised use of vasopressors would not be pragmatic in an ED-based study, in which central venous access and advanced monitoring would be variably available, and that the study question should be limited to fluid bolus size.
Following submission of the grant application and approval of funding, the SMG updated their review of the recent literature,45 which resulted in some additional minor changes to the protocol: tachycardia was removed as one of the clinical signs of shock for the pilot trial, as it could be a symptom of many different conditions outside the scope of the pilot trial and could thus lead to inappropriate randomisations. The SMG came to a consensus view that the most pragmatic clinical criteria for shock in the context of suspected infection in childhood were systolic blood pressure below the fifth percentile for age or a CRT of ≥ 3 seconds. Fluid boluses would be delivered within a 15-minute cycle rather than the 10 minutes initially proposed as this was thought to be too quick, and hence difficult to achieve, particularly in older children and adolescent patients. Additional safety features were added to prevent the administration of excessive amounts of fluid. Fluid bolus resuscitation would be withheld if participants showed signs of fluid overload, defined as rales (crackles) on auscultation with a stethoscope, pulmonary oedema (lung) fluid in the endotracheal tube or new or increasing hepatomegaly (liver enlargement), as per the 2009 ACCM-PALS guidance,5 which was current when the protocol was written. The maximum volume of fluid that could be given with the pilot trial protocol would be 120 ml/kg (excluding the original 20 ml/kg given as part of the inclusion criteria) rather than the initially proposed volume of 160 ml/kg.
As described in Chapter 2, the perspectives of parents involved in the FiSh feasibility study were used to develop pilot trial information materials and sections of the protocol related to recruitment and consent seeking, as well as site staff training. These findings were also used to develop parent/legal representative interview and site staff focus group topic and interview guides for the integrated perspectives study elements.
Amendments to the pilot trial protocol
Following receipt of approval of the pilot trial protocol from the HRA on 22 June 2016, three non-substantial amendments were submitted and categorised; there were no substantial amendments.
-
Minor amendment 1 (10 August 2016): the addition of a new research site and change of principal investigator (PI) at one site.
-
Minor amendment 2 (16 November 2016): edits were made to the protocol to clarify the maximum volume of fluid that can be delivered per bolus, as this was not clear in the previous iteration. Following review by the TSC, updates were also made to the PISs for parents/legal representatives to clarify the unknown risks and benefits of 10-ml/kg versus 20-ml/kg fluid boluses. In addition, minor amendments were made to the information sheet for 8- to 10-year-olds to make the description of the two interventions clearer and to update some of the images to make them more relatable to the pilot trial (i.e. a picture of a child in bed with a bag of fluid vs. a child sneezing). The TSC also suggested the addition of two new participant materials, namely a contact details card, with the telephone numbers of parents/legal representatives who agreed to take part in a telephone interview and a reminder when that would occur, to help potentially boost contact and arrangement of interviews, and an additional envelope label, to be added onto the envelope when contacting bereaved parents/legal representatives about the pilot trial, informing parents/legal representatives that the contents were related to their child, providing them the opportunity to open the envelope at their own discretion.
-
Minor amendment 3 (10 February 2017): a 1-month extension to the recruitment period.
NHS support costs
Studies in emergency and critical care are challenging and expensive to conduct. Unlike other areas of health care, such as oncology, recruitment cannot take place solely within usual office hours. Resources are needed to enable screening and recruitment 24 hours per day, 7 days per week. To this end, resources equivalent to 0.12 whole-time equivalent band 6 research nurses were successfully agreed with the North Thames Local Clinical Research Network (LCRN) before the submission of the research grant. Resources were based on an estimated 12 eligible admissions per hospital per year, of whom approximately nine would be recruited and randomised 1 : 1 between 10-ml/kg and 20-ml/kg fluid boluses. Using these recommendations, participating sites, assisted by the SMG, negotiated the resources required locally for the pilot trial with their respective research and development departments and LCRNs.
Patient and public involvement
Engagement with patients was vital to the successful conduct of the pilot trial. Three parents of children who had experienced septic shock and had been admitted to a PICU provided oversight of the pilot trial: two were co-investigators (AC and NJ) and members of the SMG, and one was an independent member of the TSC. They provided input into the conduct of the pilot trial, including reviewing the literature to be given to participants and their families (e.g. PISs and parent/legal representative interview topic guide).
Pilot trial
Sites
Sites were set up under a ‘hub-and-spoke’ model within three geographical regions: Bristol, London (North Thames) and Southampton. The ‘hubs’ were four regional hospitals with integral PICUs (two hospitals covered the same region), three of which also had an integrated ED and paediatric assessment unit [(PAU) or equivalent]. The ‘spokes’ were nine hospitals within the selected regions, which had an ED but did not have an integral PICU. The criteria for inclusion as a study site were:
-
sites meet all responsibilities as stated in the pilot trial clinical trial site agreement
-
sites identify and sign up a local PI
-
sites identify a responsible FiSh trial research nurse (to be funded, or part-funded, centrally)
-
sites agree to incorporate the FiSh trial into routine ED and PAU (or equivalent) activity, in particular, by highlighting the importance of screening at ED and PAU (or equivalent) presentation
-
sites agree to adhere to randomisation allocation and to ensure adherence to the protocol
-
sites agree, when possible, to recruit all eligible patients to the FiSh trial and to maintain a screening log.
The ‘hub’ sites were chosen during the grant application stage. For the nine ‘spoke’ sites, invitations for expressions of interest were sent to lead clinicians in emergency and critical care medicine at NHS hospitals within the three regions. Sites that expressed interest were asked to complete a site feasibility questionnaire. After reviewing the questionnaires, teleconferences were held with all potential hospitals to assess the engagement and support of the clinical team, the integration of the pilot trial into clinical practice (e.g. screening, randomising, delivering the intervention) and any potential challenges/barriers to running the pilot trial at their site. The final sites included in the study are listed in Table 2.
| Site | Name of PI |
|---|---|
| Bristol Royal Hospital for Children (B) | Dr Mark D Lyttle |
| Chelsea and Westminster Hospital (NT) | Dr Hester Yorke |
| Great Ormond Street Hospital for Children (NT) | Professor Mark J Peters |
| Musgrove Park Hospital (B) | Dr Esther Wilson |
| Northwick Park Hospital (NT)a | Dr Sathish Bangalore |
| Queen Alexandra Hospital (S) | Dr Alan Charters |
| Royal Devon and Exeter Hospital (B) | Dr Simon Parke |
| Royal United Hospital (B) | Dr Elizabeth Gilby |
| Salisbury District Hospital (S)a | Dr Nick Brown |
| Southampton General Hospital (S) | Dr Jane Bayreuther |
| St Mary’s Hospital (NT) | Dr David Inwald |
| Watford General Hospital (NT) | Dr Solomon Kamal-Uddin |
| Whittington Hospital (NT) | Dr Kerry Robinson |
Site initiation
Site teams from all participating sites attended a site initiation meeting before the commencement of patient screening. Site initiation meetings were held at each participating site between 13 June 2016 and 23 November 2016. The purpose of these meetings was to present the background and rationale for the FiSh trial, present the FiSh feasibility study findings and discuss delivery of the protocol, including the screening and recruiting of patients, as well as the delivery of the intervention, data collection and validation, and safety monitoring. The operational challenges of conducting the pilot trial at sites were discussed in detail, including strategies for ensuring effective communication within the ED, PAU (or equivalent), retrieval transport services and PICU. The PI from each participating site attended the relevant meeting. A standardised slide set from the site initiation meetings was circulated to facilitate internal training within a participating site.
Investigator site file
An investigator site file was provided to all participating sites. This contained all essential documents for the conduct of the pilot trial and included the approved pilot trial protocol; all relevant approvals (e.g. local confirmation of capacity and capability); a signed copy of the clinical trial site agreement; the delegation of trial duties log; copies of the approved PISs, parent/legal representative consent forms and participant assent form (one for 8- to 10-year-olds and a second for those aged ≥ 11 years); and all standard operating procedures, for example for screening patients, randomising participants, delivering the intervention, obtaining informed consent or assent and collecting and entering data onto the secure, dedicated, electronic case report from (eCRF). The site PI was responsible for maintaining the investigator site file. Responsible staff at sites were authorised to carry out pilot trial duties (e.g. consenting, oversight of the delivery of the intervention) by the site PI on the delegation of trial duties log. This included a confirmation that the individual had been adequately trained to carry out the specific duty.
Communication
The study manager, with support from the data manager and research assistant, maintained close contact with the PI and research team at participating sites by e-mail and telephone throughout the pilot trial. Teleconferences were held every month with research teams at participating sites. The purpose of these was to provide updates on pilot trial progress and to provide a forum for site teams to ask questions, discuss local barriers and challenges to the conduct of the pilot trial and delivery of the intervention, and to share successes and best practice. Notes, including ‘hints and tips’, from the teleconferences were distributed to all participating sites. Teleconferences were also held with individual site teams as required to address site-specific issues in the conduct of the pilot trial.
Site monitoring visits
At least one routine monitoring visit was conducted at all participating sites during the pilot trial. During the site visit, the investigator site file was checked for completeness (i.e. that all essential documents were present); the parent/legal representative consent forms and participant assent forms (if applicable) were checked to ensure that the relevant correctly completed form was present for each participant recruited into the pilot trial (or, if the parents/legal representatives were followed up for consent by post, evidence of the postal consent covering letters for non-responders); and a random sample of participant case report forms (CRFs) were checked against the source data for accuracy and completeness. After the visit, the PI and site team were provided with a report summarising the documents that had been reviewed and any actions required by the site team. The site PI was responsible for addressing the actions and reporting back to the ICNARC CTU. Additional visits were conducted on a risk-based approach using recruitment rates, data quality and adherence to the protocol as central monitoring triggers.
Maintenance and motivation
During the pilot trial, an e-mail was sent each week to site teams with an update on participant recruitment and a newsletter was sent every 2 months. These provided an opportunity to clarify any issues related to the conduct of the pilot trial and to share ideas for maximising recruitment, as well as maintaining motivation and involvement through regular updates on progress. To maintain the profile of the pilot trial at participating sites, inclusion criteria and trial flow posters were displayed in staff areas and at relevant locations within the ED and resuscitation bays, PAU (or equivalent) and PICU, for example, in fluid-bag storage areas. Furthermore, information posters were displayed in family/relative waiting rooms, pocket cards summarising the eligibility and shock criteria were distributed, branded pens were distributed to staff, labels for patient notes (e.g. ‘This is a patient is on a clinical trial’) were provided and certificates were given from PIs to clinical staff in recognition of their contribution to the trial. Additional materials, such as a quiz and biweekly localised newsletter, were provided to boost the profile of the pilot trial going into the last few weeks of recruitment.
Support
A 24 hours per day, 7 days per week, telephone support service was available to site teams, via the CTU, for advice on the screening and recruitment of patients and the delivery of the intervention. This ensured access to clinicians to answer any queries on the eligibility of patients and delivery of the intervention.
Recruitment
The trial procedures for recruitment and follow-up of participants are summarised in Figure 3.
FIGURE 3.
Summary of trial procedures for recruitment and follow-up of participants.
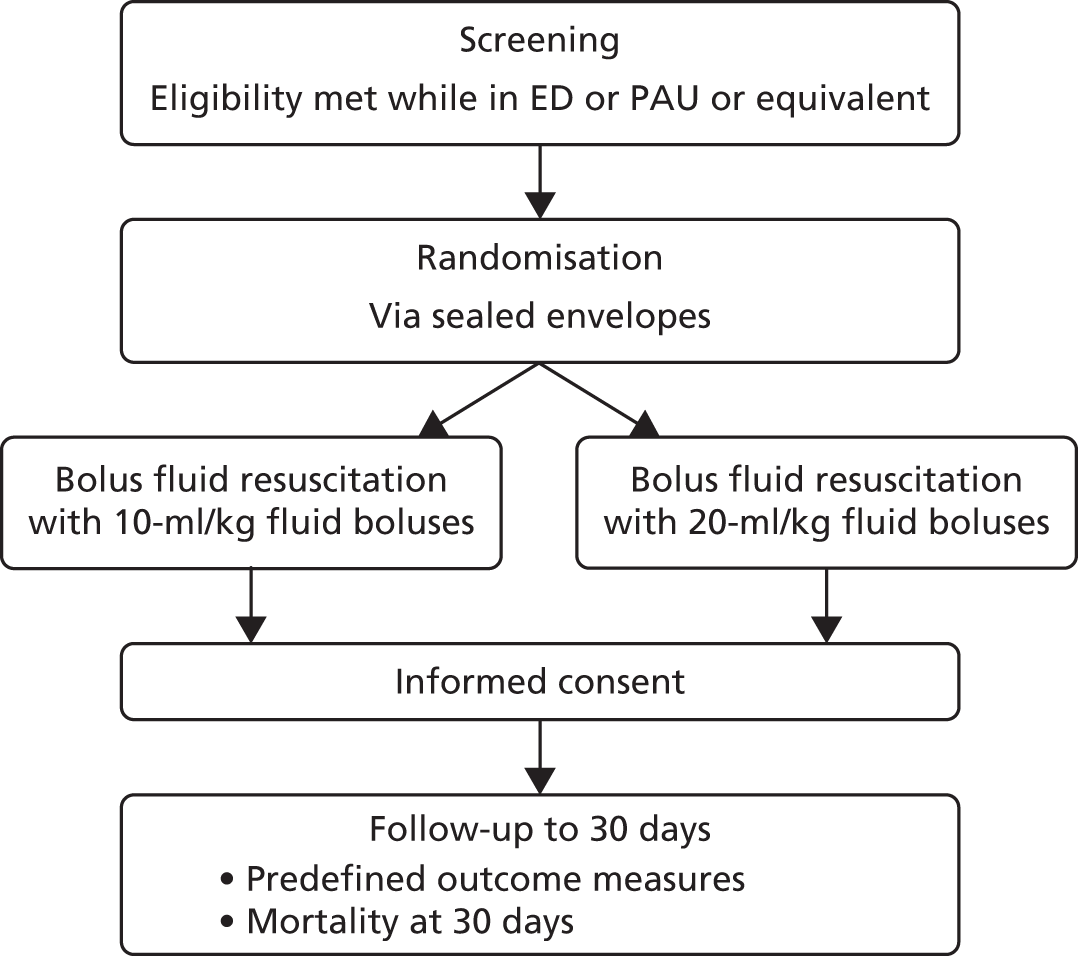
Eligibility criteria
Patients were eligible for inclusion in the pilot trial if they met all of the following criteria:
-
aged ≥ 37 weeks (corrected gestational age) and < 16 years
-
clinical suspicion of infection
-
clinical signs of shock (defined as age-adjusted hypotension or prolonged CRT of ≥ 3 seconds) after receipt of 20 ml/kg of fluid bolus
-
recruitment and randomisation to take place while patient is in an acute assessment area.
Exclusion criteria
Patients were excluded from the pilot trial if they met any of the following criteria:
-
prior receipt of > 20 ml/kg of fluid bolus
-
conditions in which fluid bolus resuscitation should be curtailed
-
full active resuscitation not within current goals of care.
Initiation of screening and recruitment
Following attendance at a site initiation meeting, screening and recruitment was commenced at participating sites once the clinical trial site agreement had been signed and necessary approvals were in place. To promote awareness of the pilot trial and to facilitate recruitment, posters and leaflets providing information about the FiSh trial were displayed in the ED, PAU (or equivalent) and family/relative waiting rooms. Potentially eligible patients were identified, confirmed as eligible by the local research and clinical teams and then randomised before informed consent was obtained, because patients requiring fluid bolus resuscitation as treatment for septic shock would most often need this treatment to be started in a life-threatening emergency, in which any delay in commencing treatment could have been detrimental.
Randomisation and allocation procedure
Following confirmation of eligibility, participants were randomised using sealed randomisation envelopes located at each hospital. Participants were randomly allocated, 1 : 1, to either the 10-ml/kg fluid bolus group or the 20-ml/kg fluid bolus group, from an a priori computer-generated list of randomised permuted blocks (with variable block lengths) stratified by recruiting site. Staff at participating sites were advised to call either their local FiSh trial on-call clinicians or the 24 hours per day, 7 days per week, telephone support service if they needed to address any emergency recruitment/randomisation issues.
Screening log
To enable full and transparent reporting for the pilot trial, brief details of all patients who met eligibility criteria, those who met all inclusion criteria plus one or more of the exclusion criteria and those who did not meet the ‘clinical signs of shock after receipt of 20 ml/kg of fluid bolus’ inclusion criterion were recorded in the screening log. The reasons for eligible patients not being recruited were recorded and included the patient being excluded by the treating clinician and logistical reasons. No patient identifiers were recorded in the screening log.
Consent
Staff members who had received training on the background, rationale and purpose of the FiSh trial and on the principles of the good clinical practice guidelines were authorised by the PI to take informed consent from parents/legal representatives. This method used for the FiSh pilot trial is known as ‘deferred’ or ‘retrospective’ consent, referred to here as RWPC. However, the terms ‘deferred’ and ‘retrospective’ are misnomers, as a child will have already received an intervention as part of the pilot trial before any information is given or consent is sought. Rather, the process should be understood, first, as the provision of information about what has already happened, and, second, as an invitation to consent for future procedures (when appropriate) and permission for the use of any data already collected.
Once notified of the recruitment of a participant to the pilot trial, a member of the site research team approached the parents/legal representatives as soon as practically and appropriately possible after randomisation to discuss consent for the pilot trial.
Information about the pilot trial was provided to the parents/legal representatives and included the purpose of the pilot trial, the reasons why informed consent could not be sought from parents/legal representatives before their child was entered into the pilot trial, what participation in the pilot trial meant (i.e. permission for the use of data already collected and for their child to continue to take part in the pilot trial), participant confidentiality, future availability of the results of the pilot trial and funding of the pilot trial. Information on completing an optional questionnaire and/or taking part in a telephone interview was also provided. This information was provided in the pilot trial PIS, along with the name and contact details of the local PI and research nurse(s), which was given to the parents/legal representatives to read before making their decision for their child’s data to be used, or not, and for their child to continue to take part, or not, in the pilot trial. Once the staff member taking informed consent was satisfied that the parents/legal representatives had read and understood the PIS and all their questions about the pilot trial had been answered, the parents/legal representatives were invited to sign the consent form.
Death prior to consent being sought
The views of bereaved parents who participated in the FiSh feasibility study (see Chapter 2) and guidance on RWPC in emergency and critical care trials were used to inform the approach to consent when a child had died before consent had been sought from parents/legal representatives. 27 In this situation, a researcher would obtain information from colleagues and bereavement counsellors to establish the most appropriate research team member to notify the parents/legal representatives of the pilot trial. Consent could have been sought from parents/legal representatives following the death of their child and before their departure from the hospital; however, it was at the discretion of the site staff to determine if this was appropriate for each individual family. If consent was not sought before the parents’/legal representatives’ departure from the hospital, then the parents/legal representatives were sent a covering letter (personalised by the most appropriate clinical team member) and a copy of the PIS (a version for bereaved parents/legal representatives) and consent form (postal version for bereaved parents/legal representatives) by post 4 weeks after randomisation. The letter explained how to opt in or out of the pilot trial, directed them to the PIS for detailed information on the pilot trial and provided telephone contact details if parents/legal representatives wished to discuss the pilot trial with a member of the site research team. If there was no response 4 weeks after sending the initial letter, a follow-up letter, along with the PIS and consent form, was sent. The second letter provided the same information as the first letter; in addition, this letter also confirmed that, if no consent form was received within 4 weeks of receipt of the letter, then the participant’s data would be included in the pilot trial unless the family notified the site research team otherwise.
Discharge prior to consent being sought
In the situation in which a participant was discharged from hospital before consent had been sought, the most appropriate member of the site research team attempted at least one telephone call to the parents/legal representatives within 5 working days of hospital discharge to inform them of the participant’s involvement in the pilot trial and to provide details of the pilot trial. Following on from the call, as well as if there was no response to the call, the parents/legal representatives were sent a covering letter (personalised by the most appropriate clinical team member) and a copy of the PIS and consent form (postal version) by post. The letter explained how to opt in or out of the pilot trial, directed parents/legal representatives to the PIS for detailed information on the pilot trial and provided telephone contact details if parents/legal representatives wished to discuss the pilot trial with a member of the site research team. If there was no response 4 weeks after sending the initial letter, a follow-up letter, along with the PIS and consent form, was sent. The second letter provided the same information as the first letter; in addition, this letter also confirmed that, if no consent form was received within 4 weeks of receipt of the letter, then the participant’s data would be included in the pilot trial unless the family notified the site research team otherwise.
Assent
Owing to the severity of illness and its effect on the mental state of the target population, it was not considered possible to involve pilot trial participants in the consenting process. Instead, there was a possibility for participants to assent to the trial, before hospital discharge, if the participant was ≥ 8 years old (e.g. old enough to be able to make an informed decision) and their condition allowed (e.g. they regained capacity). Pilot trial participants would have been provided with an age-appropriate PIS and asked to sign an assent form, if appropriate. Parents/legal representatives would have been involved in this discussion. In all other respects, the assenting procedures followed the consenting procedures as described above. If the participant was likely to regain capacity following hospital discharge, then an age-appropriate PIS was provided to parents/legal representatives to discuss with the participant, following recovery.
Treatment groups
Participants were randomly allocated to fluid bolus resuscitation using boluses of two different sizes over a 4-hour period (Figure 4). The period was divided up into 15-minute cycles, with one bolus of either 10 ml/kg or 20 ml/kg to be delivered within each cycle, at a rate left to the discretion of the treating clinician. There was no attempt to blind clinicians to the allocated treatment group as this was not felt to be pragmatic, given the ED setting and the need for clinical staff to prescribe, draw up and administer the fluid boluses. The maximum volume of fluid that could be given per bolus was either 500 ml (for those allocated to 10-ml/kg fluid boluses) or 1000 ml (for those allocated to 20-ml/kg fluid boluses). Other interventions, including referral to PICU, were not protocolised and were left to the discretion of the treating clinician. As a pragmatic pilot trial, FiSh also did not dictate the use of specific fluid products. At the end of each cycle, should the age-adjusted clinical signs of shock [defined as age-adjusted hypotension (Table 3) or prolonged CRT of ≥ 3 seconds] persist, then another bolus of the same size (i.e. according to the randomly allocated group) was given – again, at a rate left to the discretion of the treating clinician but within a 15-minute cycle.
FIGURE 4.
Fluid bolus resuscitation algorithm.

| Age group | SBP (mmHg) |
|---|---|
| 0 days–1 week | < 60 |
| 1 week–1 year | < 70 |
| 1–2 years | < 75 |
| 2–5 years | < 80 |
| 5–12 years | < 85 |
| 12–16 years | < 90 |
The cycles repeated until either the end of the 4-hour resuscitation period or any of the hold criteria occurred. If a participant’s age-adjusted clinical signs of shock resolved or if a participant showed signs of fluid overload (defined as pulmonary oedema – either rales on auscultation or pulmonary oedema fluid in the endotracheal tube – or new or increasing hepatomegaly), the delivery of further fluid boluses was withheld. If, within the 4-hour resuscitation period, fluid boluses were indicated again, that is, age-adjusted clinical signs of shock were present without signs of fluid overload, then the cycles were recommenced with the allocated boluses until the end of the 4-hour resuscitation period. After the 4-hour resuscitation period, any further treatment was at the discretion of the treating clinician.
The maximum volume of fluid that could be given within the pilot trial protocol, regardless of allocation, was 120 ml/kg (excluding the 20-ml/kg fluid bolus pre randomisation). If > 120 ml/kg of fluid was required, then further treatment was at the discretion of the treating clinician.
Outcome measures
The combined objectives were to test whether or not all of the processes could work together, and to inform the design, and ensure the successful conduct, of the FiSh trial (should this be the recommendation).
Objectives 1, 2 and 4 were measured by the:
-
proportion of eligible patients recruited
-
number of participants recruited per site per month
-
proportion of parents/legal representatives refusing consent.
Objective 3 was measured by:
-
the proportion of fluid boluses delivered at the correct volume and time during the intervention period
-
comparing the total volume of fluid received in each arm during the intervention period.
Objectives 5–8 were measured by the:
-
proportion of complete data for each outcome measure
-
characteristics of potential outcome measures
-
observed AEs
-
time taken for data collection and entry
-
proportion of required data that could be linked to routine sources.
Safety monitoring
Participants were monitored for AEs occurring between randomisation and 30 days following randomisation. Specified AEs were as follows: pulmonary oedema, cerebral oedema, extravasation injury, amputation and skin graft. An unspecified AE was defined as an unfavourable symptom or disease temporally associated with the use of the pilot trial treatments (regardless of whether or not it was related to the pilot trial treatment) that was not deemed to be a direct result of the participant’s medical condition and/or standard emergency or critical care treatment. All AEs were recorded in the eCRF and reported, as part of routine reporting throughout the pilot trial, to the DMEC. AEs that were assessed to be serious (i.e. causing prolonged hospitalisation or resulting in persistent or significant disability/incapacity), life-threatening or fatal – collectively termed serious adverse events (SAEs) – were reported to the ICNARC CTU and reviewed by a clinical member of the SMG. SAEs that were unspecified and considered to be possibly, probably or definitely related to the pilot trial treatment were reported to the REC within 15 calendar days of the event being reported.
Data collection
A secure, dedicated eCRF, hosted by ICNARC, was set up to enable pilot trial data to be entered by staff at participating sites. The eCRF was accessible only to authorised users and access was approved centrally by the study manager, data manager or research assistant (after cross-checking the site delegation of study duties log). Each individual was provided with a unique username and password and had access to data only for participants recruited at their site. The data set for the FiSh trial included the minimum data required to confirm patient eligibility, describe the patient population, monitor and describe the delivery of the intervention, assess outcomes and enable linkage to the Paediatric Intensive Care Audit Network [(PICANet) the UK national clinical audit for paediatric intensive care] and the NHS Spine web portal. PICANet data were available only for patients admitted to a PICU and included mechanical ventilation days, length of PICU stay and PICU mortality.
Baseline data
Data were collected to enable the patient to be randomised and included confirmation that the patient met all of the inclusion criteria and none of the exclusion criteria. The following data were also collected at baseline to enable follow-up and describe the patient population:
-
full name and home postcode of the patient
-
date of birth
-
sex
-
raw physiology data to enable calculation of the Paediatric Index of Mortality version 2r (PIM2r) score, the standard severity of illness scoring system in use across UK PICUs, according to the 2016 recalibration46–48
-
severe comorbidities defined according to PIM2r that were present and documented in past medical history.
Intervention data
Data were collected before, during and after each bolus delivery throughout the 4-hour intervention period to monitor adherence to the treatment allocation (10-ml/kg or 20-ml/kg, fluid boluses) and to describe delivery of the 10-ml/kg fluid boluses compared with 20-ml/kg fluid boluses (usual resuscitation). The data collected included fluid bolus delivery, including volume and type of fluid and bolus end time, and assessments, including systolic blood pressure, CRT and shock-confirmed time.
Treatment summary data
Following the intervention period, the following data were collected:
-
other interventions delivered during the intervention period, including mechanical ventilation and vasoactive drugs
-
total fluid bolus and total fluids (including fluid bolus) received from randomisation to the end of both 4 and 24 hours
-
site of infection and causative organism (if available) and type of intravenous antibiotics first administered.
For patients who were admitted to a PICU, daily intervention data were obtained through PICANet.
Outcome data
At the time of discharge from the acute hospital, the following data were collected:
-
the locations of care during the patient’s stay in the acute hospital, for example ED, PAU (or equivalent), PICU or ward
-
date of discharge from, or death in, the acute hospital
-
discharge location, for example home, nursing home or other hospital.
All patients were followed up at 30 days post randomisation for all-cause mortality via the NHS Spine web portal.
Data management
Data management was an ongoing process. Data entered by sites onto the eCRF were monitored and checked throughout the recruitment period to ensure that they were as complete and accurate as possible. Two levels of data validation were incorporated into the eCRF. The first was to prevent obviously erroneous data from being entered, for example entering a date of birth that occurred after the date of randomisation. The second level involved checks for data completeness and any unusual data entered, for example a physiological variable, such as systolic blood pressure, that was outside the predefined range. The data manager could generate data request checks, listing all outstanding data queries, at any time via the eCRF. The site PI was responsible for ensuring that all data queries were resolved. Ongoing data entry and validation at sites was closely monitored by the data manager and any concerns were raised with the site PI.
Adherence to the pilot trial protocol was closely monitored, including adherence to all elements of the fluid bolus resuscitation protocol. Any queries relating to adherence were also generated as data request checks via the eCRF. For each query, the PI and/or site research team were asked to explain the reason for any non-adherence to the protocol. If deemed necessary, a teleconference was arranged with the site to ensure effective plans were put in place to improve future adherence.
Sample size
The pilot trial was set up as a small RCT without a defined primary outcome and, hence, without a usual power calculation to determine sample size. Instead, the sample size had been determined to be adequate to estimate critical parameters to be tested to a necessary degree of precision. With respect to sample size, a frequently quoted ‘rule of thumb’ for the sample size of pilot trials is to recruit 30 participants. 49 However, more recent research has demonstrated that such a sample size will result in an imprecise estimate of the SD of a potential outcome measure, which will frequently lead to definitive studies that are either underpowered (if the imprecision of the estimated SD is not taken into account in the sample size calculation) or inefficient (if it is). 50 Sim and Lewis50 suggest that a sample size of around 60 participants would usually be sufficient to estimate the SD for a continuous outcome measure; however, the authors note that estimating the precision of a binary outcome will require a larger sample size, typically requiring between 98 and 260 participants. Based on available data from PICANet and the PICS-SG severe sepsis audit,3 it was anticipated that each of the EDs of regional hospitals with PICUs in the pilot trial and each of the nine feeder hospital EDs – a total of 12 sites – would recruit approximately one child per month, giving a total of around 108 children.
Interim analysis
Unblinded, comparative data on recruitment, withdrawal, adherence (to the allocated treatment) and AEs were regularly reviewed by an independent DMEC. No formal interim analyses were performed owing to the nature of the pilot trial.
Analysis principles
All analyses were based on the intention-to-treat principle. Participants were analysed in accordance with the treatment group they were randomised to, irrespective of whether or not the allocated treatment was received (i.e. regardless of whether they did or did not adhere to the fluid bolus resuscitation protocol). All tests were two-sided with significance levels set at a p-value of < 0.05 and with no adjustment for multiplicity.
Statistical analysis
Statistical analyses were conducted in accordance with a prespecified statistical analysis plan written before the final analysis. The final analyses were conducted using Stata®/SE version 14.0 (StataCorp LP, College Station, TX, USA).
Screening and randomisation
Recruitment to the pilot trial, treatment allocation and completeness of follow-up were illustrated using a Consolidated Standards of Reporting Trials (CONSORT) flow diagram. All participating sites maintained screening logs of patients who were eligible (fulfilled all of the inclusion criteria and none of the exclusion criteria) but were not randomised, who fulfilled all of the inclusion criteria but met one or more of the exclusion criteria, and who did not fulfil ‘clinical signs of shock after receipt of 20 ml/kg of fluid bolus’ inclusion criterion. Reasons for non-recruitment were categorised and summarised (see Figure 7). Numbers and percentages of screened and randomised infants and children were presented. The following were reported for each treatment group:
-
the number of patients screened
-
the number of patients who received a 20-ml/kg fluid bolus
-
the number of patients with resolved shock after the initial 20-ml/kg fluid bolus
-
the number of patients who were not eligible
-
the number of patients randomised –
-
among those patients randomised, those who did not meet inclusion criteria or met one or more of the exclusion criteria.
-
Recruitment rate
Recruitment to the pilot trial was presented as a rate over the recruitment period and per month, overall and per site. The variation in recruitment rates across sites was presented as a funnel plot (recruitment rate against months open to recruitment).
Refused consent
The number and percentage of participants in each treatment group that had RWPC either refused initially or subsequently withdrawn was reported.
Baseline characteristics
Baseline demographic and clinical data were summarised by treatment group. Statistical tests for differences between the groups were not reported, as these may be misleading. Discrete variables were summarised as numbers and percentages, which were calculated according to the number of participants for whom data were available; when values were missing, the denominator was reported. Continuous variables were summarised by standard measures of central tendency and dispersion, either mean and SD and/or median and interquartile range (IQR) as specified below:
-
age, mean (SD) and median (IQR)
-
sex, n (%)
-
weight, mean (SD) and median (IQR)
-
inclusion criteria –
-
clinical signs of shock, n (%)
-
systolic blood pressure value at which criterion for clinical signs of shock was met, mean (SD)
-
CRT value at which criterion for clinical signs of shock was met, mean (SD)
-
-
-
acute severity of illness –
-
PIM2r score (2016 UK recalibration), median (IQR)
-
-
infection, n (%) –
-
site
-
confirmed organism.
-
Adherence
Non-adherence to the allocated treatment was reported for each treatment group as the number and percentage of participants:
-
who did not receive first bolus
-
for whom for subsequent boluses –
-
bolus was given but shock criteria were not met
-
shock criteria were met but no bolus was given.
-
Delivery of care
Delivery of care was summarised by treatment group but not subjected to statistical testing. As with baseline characteristics, discrete variables were summarised as numbers and percentages. Percentages were calculated according to the number of participants for whom data were available; when values were missing, the denominator was reported. Continuous variables were summarised by both mean (SD) and median (IQR) to maximise information on the distribution of these key parameters of the intervention. Variables reported were:
-
number of boluses delivered
-
total volume of fluid bolus resuscitation at 1 hour and during the intervention period
-
receipt of fluid bolus resuscitation of 10 ml/kg (± 10%)
-
receipt of fluid bolus resuscitation of 20 ml/kg (± 10%)
-
delivery of boluses within 15 minutes (overall and by age group)
-
fluid boluses delivered at correct volume and within 15 minutes during the intervention period.
Overall adherence was reported as the number and percentage of participants in each treatment group with fluid boluses delivered at the correct volume and timing during the intervention period. The distribution of the total volume of fluid received during the intervention period in each treatment group was presented as a histogram. Separation between the treatment groups was assessed by the difference in the mean total volume of fluid, tested with a t-test and reported difference in means with 95% confidence interval (CI).
Outcomes
The following potential outcome measures were reported for each treatment group:
-
hospital mortality
-
mortality within 30 days post randomisation
-
length of hospital stay
-
transferred to a PICU
-
length of stay in a PICU among those admitted
-
days alive and not in a PICU up to 30 days (deaths within 30 days score of 0; infants and children not admitted to PICU score of 30)
-
receipt of mechanical ventilation
-
duration of mechanical ventilation among those ventilated
-
days alive and free of mechanical ventilation up to 30 days (deaths within 30 days score of 0; infants and children not receiving mechanical ventilation score of 30).
Although this was a pilot RCT and not powered to detect differences in outcomes, the potential outcome measures were tested between the groups to gain further understanding of the potential magnitude of any treatment effect from the width of the CIs. Differences in binary outcomes were tested with Fisher’s exact test and presented as the number and percentage in each treatment group, absolute risk difference and relative risk with 95% CIs. Differences in lengths of stay and duration of organ support were tested with the Wilcoxon rank-sum test and presented as median (IQR) in each treatment group. Differences in days alive and not in a PICU and days without mechanical ventilation were tested with the t-test and presented as difference in means with 95% CI.
For all potential outcome measures, the number of participants with complete data in each treatment group was reported. For measures requiring data linkage with routine data sources, the proportion of successfully linked records was reported.
Safety monitoring
The number and percentage of participants experiencing each AE and SAE (occurring between randomisation and 30 days) were reported for each treatment group.
Integrated perspectives study
Participants
Based on previous research and the pilot trial sample size,18 it was estimated that 100–150 parents/legal representatives (including both mothers and fathers of randomised children) would be recruited to the questionnaire element and 15–25 parents/legal representatives would be recruited to the interview element. In addition, three focus groups at the hub sites and up to 10 telephone interviews with staff who could not attend the focus groups were planned.
Eligibility
Parents/legal representatives
Parents/legal representatives who did and did not consent to their child’s participation in the pilot trial were eligible to take part in the questionnaire and interview elements, unless they were unable to speak and read English.
Site staff
Site staff who were involved in screening, recruiting, randomising and consenting parents/legal representatives during the pilot trial were eligible to take part in the focus groups or interviews. There were no exclusion criteria.
Recruitment and consent procedure
Parents/legal representatives
As part of the pilot trial consent discussion, parents/legal representatives were provided with information about the integrated perspectives study elements in the pilot trial PIS. Researchers explained the different aspects of the pilot trial and asked parents/legal representatives if they would like to complete the FiSh trial consent questionnaire (see Appendix 5) and/or provide contact details on the consent form if they wished to take part in a telephone interview. Following the consent discussion, parents/legal representatives (both, if present) were asked to complete the questionnaire and place it in a stamped addressed envelope to be returned, preferably before they left hospital. Parents/legal representatives contacted about the pilot trial by post (see Death prior to consent being sought and Discharge prior to consent being sought) were provided with information on the telephone interview only. A researcher contacted parents/legal representatives to arrange an interview within 1 month of consent. Consent for audio-recording of interviews was checked verbally before the interview began.
Site staff
An invitation to participate in a focus group at one of the three hub sites was e-mailed to site PIs and/or lead research nurses. They were also asked to send the invitation to all staff involved in the FiSh pilot trial at their site. The e-mail included a PIS for staff, which provided information about the purpose of the integrated perspectives study elements; what the focus group/interview would involve; how the integrated perspectives study elements were funded; and the consequences of participating or not. Telephone interviews were provided as an alternative option for staff who could not attend a focus group. An e-mail invitation was also sent to the PI and/or lead research nurse at each spoke site inviting them to take part in a telephone interview. Written consent was sought from staff before the focus group began, and verbal consent was sought before the telephone interview began. Consent for audio-recording of interviews was checked before the focus group or interview began.
Interviews and focus groups
Telephone interviews with parents/legal representatives
The telephone interview began with a discussion about the parent’s/legal representative’s child’s admission to hospital and current well-being, the aims of the interview, an outline of the topics to be covered in the interview and an opportunity for questions. The interview then commenced using the interview topic guide to explore views and experiences about the child’s admission to hospital, including their diagnosis; the pilot trial consent process, including how the pilot trial was communicated by staff and information materials; consent and decision-making, including how pilot trial processes could be improved; child assent; and outcome measures. Example questions are shown in Appendix 6. Respondent validation was used to add unanticipated topics to the topic guide as interviewing and analysis progressed. 24 After the interview was complete, parents/legal representatives were sent a copy of the consent form, a thank-you letter and £30 shopping voucher to thank them for their time.
Focus groups and interviews with site staff
Focus groups and interviews began by asking staff to introduce themselves and their role within the FiSh pilot trial. The focus group/interview aims and topics to be covered were discussed, followed by an opportunity for questions. In the focus groups, a voting system, using TurningPoint software version 5 (Turning Technologies, Youngstown, OH, USA), was used alongside verbally administered questions. This involved some of the questions (see Appendix 7) being presented to the group via a laptop presentation and each participant using a wireless handset to select their answer from those shown on the screen. A test question was used at the beginning of the focus group to help demonstrate how the voting system would work alongside verbal questions. This method was to used enable the collection of data from all participants, as well as to generate statistical data from all sites alongside qualitative data from group discussions. The same topic guide was used for focus groups and interviews, although questions (e.g. voting system questions) were adapted for telephone interviews.
Focus groups and interviews explored staff views on, and experiences of, site training; FiSh pilot trial screening and randomisation processes; protocol deviations; recruitment and consent, including parent/legal representative responses to RWPC; and FiSh pilot trial forms, including CRFs and consent forms. Example questions are shown in Appendix 7. Respondent validation was used to add unanticipated topics to the topic guide as interviewing and analysis progressed. 24 After the focus groups and interviews were complete, all participants were sent a copy of the consent form (if applicable) and thanked for their time.
Transcription
Digital audio-recordings were transcribed verbatim by a professional transcription company (Voicescript Ltd) in accordance with the Data Protection Act 1998. 25 Transcripts were anonymised and checked for accuracy. All identifiable information, such as names (e.g. of patients, family members or the hospital that their child was admitted to), was removed.
Data analysis
The analysis of qualitative data were broadly interpretive and iterative (see Table 19). 31,32 Informed by the constant comparative approach, the aim was to provide accurate representation of parental views on trial design and acceptability. 29,33–35 NVivo 10 software was used to assist in the organisation and coding of data. Data from voting handsets were cleaned and entered into IBM SPSS Statistics version 20.0 (IBM Corporation, Armonk, NY, USA). Descriptive statistics are presented with percentages.
Chapter 4 Results of the pilot trial
Participants: sites
The four ‘hub’ sites were chosen during the grant application stage. For the nine ‘spoke’ sites, expressions of interest were received from 21 NHS hospitals across the three UK regions, with 18 completing a site feasibility questionnaire. After the initial selection of the nine ‘spoke’ sites, two sites withdrew and were replaced:
-
one hospital had a restructuring of their research team that led to a reduction in resources in ED (date of withdrawal – 3 June 2016)
-
one hospital had a lack of resources and support for the study in their paediatric team (including capacity for additional workload) (date of withdrawal – 18 August 2016).
A total of 12 hospitals in England obtained local confirmation of capacity and capability and opened to recruitment between 13 July and 23 November 2016. Seven sites were opened within the first 2 months of the pilot trial opening on 13 July 2016 (Figure 5). The London region included a second hub site (PICU only), which did not recruit patients as it has no ED, but was open to receive patients randomised at the spokes from 9 August 2016.
FIGURE 5.
Sites open to recruitment.
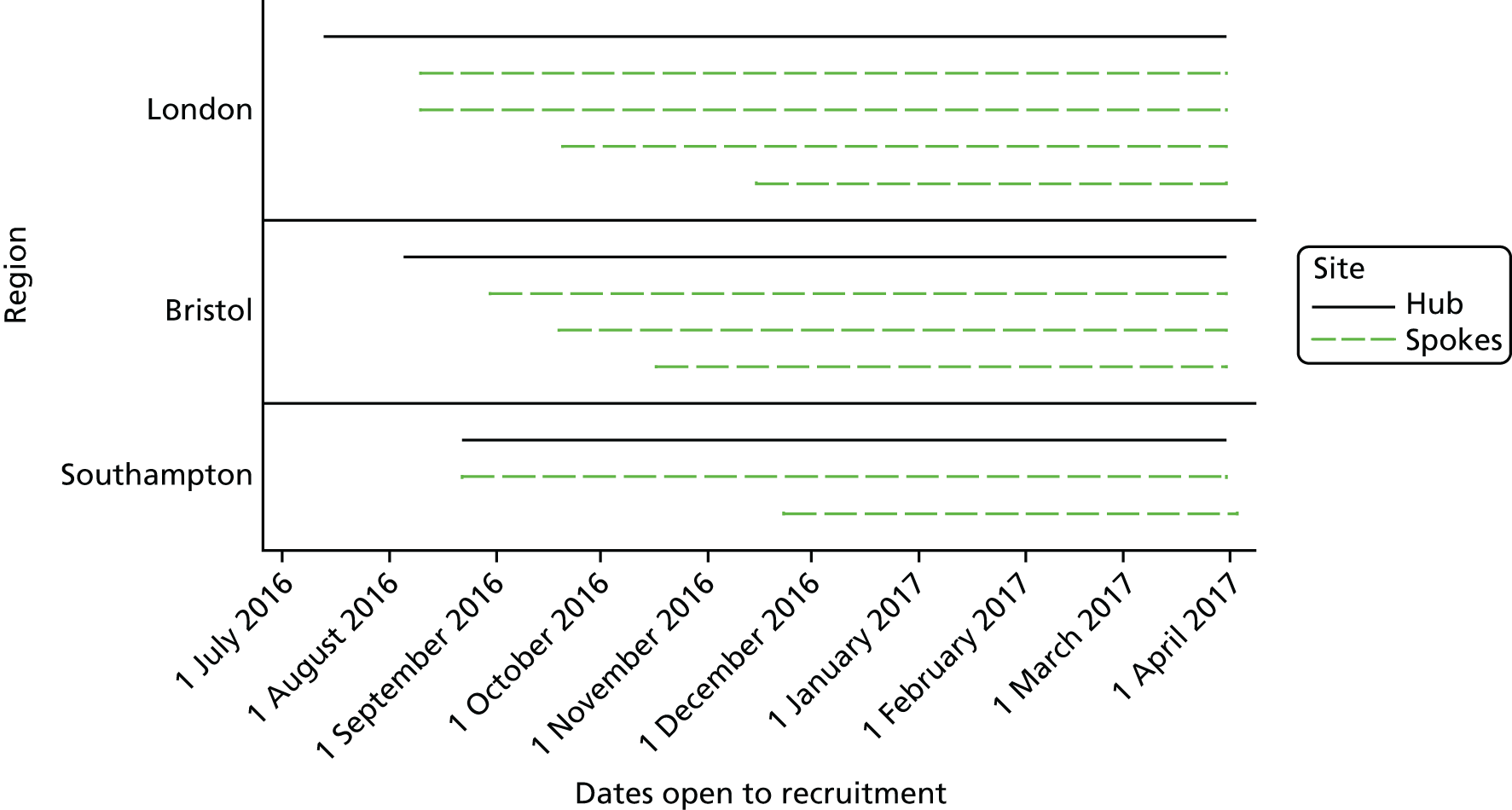
The median time from receiving the local document pack, including the HRA initial assessment letter, to the pilot trial opening at sites (i.e. start of screening) was 95 days (IQR 81–124 days) and to the first participant recruited was 150 days (IQR 124–178 days) (Figure 6). Reasons for delays in opening were as follows:
-
hospital research and development teams transitioning to the new HRA approval
-
delays in contracting with the sponsor
-
time needed to train staff across up to four departments (ED, paediatrics, PICU and retrieval) – this affected not only arranging the initial site initiation visit but the training to be done by the local teams
-
arranging replacement sites for the withdrawn sites
-
a new area of research for some sites – for some of the sites, this was their first foray into paediatric research in the emergency care setting, thus additional time and training was needed.
FIGURE 6.
Duration in days from receiving local document pack to (a) opening to recruitment; and (b) first participant recruited.
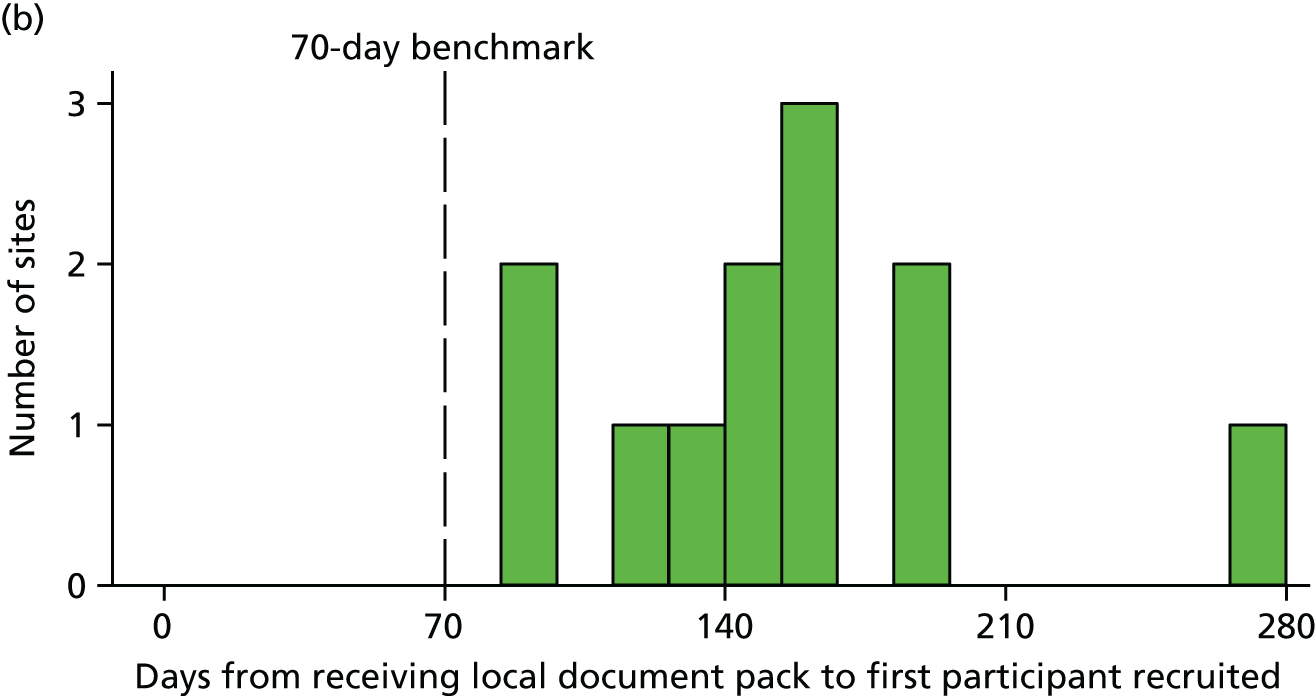
Overall, sites participated in the FiSh pilot trial for a median of 7 months (IQR 6–8 months). All sites remained open until the end of recruitment on 31 March 2017 (see Figure 5). One site recruited an additional participant on 3 April 2017 after the study had closed to recruitment, as not all staff in the site were aware that the study had closed.
Characteristics of participating sites
The characteristics of the 12 EDs recruiting patients to the FiSh pilot trial, compared with all EDs participating in the PERUKI research collaborative, are shown in Table 4. The characteristics were broadly similar. There was a lower percentage of paediatric hospitals participating in the FiSh pilot trial than in PERUKI, and a slightly lower proportion with PICU on site, but these characteristics are likely to be over-represented among PERUKI participants relative to the national picture.
| Characteristics | EDs | |
|---|---|---|
| Participating in FiSh pilot trial | All in PERUKI | |
| Number of EDs (n) | 12 | 52 |
| Tertiary referral hospital, n (%) | 5 (41.7) | 28 (53.8) |
| Type of paediatric ED, n (%) | ||
| Combined adult and paediatric ED | 6 (50.0) | 17 (32.7) |
| Separate paediatric ED in mixed hospital | 5 (41.7) | 24 (46.2) |
| Paediatric hospital ED | 1 (8.3) | 11 (39.3) |
| Annual paediatric ED census (thousands), median (IQR) [n] | 22.5 (17–30) [12] | 27 (20–37) [49] |
| PICU on site, n (%) | 3 (25.0) | 20 (38.5) |
Participants: patients
In total, 297 patients were screened between 13 July 2016 and 3 April 2017 (Table 5 and Figure 7). Of these, 88 (29.6%) patients received a fluid bolus of < 20 ml/kg and did not meet the defined clinical signs of shock. The remaining 209 (70.4%) patients received a fluid bolus of ≥ 20 ml/kg; of these, 101 (48.3%) patients were assessed as requiring further fluid boluses within the ED or PAU (or equivalent). Of these 101 patients, there were 18 (17.8%) patients who met one or more exclusion criteria. Of these, 12 had received an initial bolus of > 20 ml/kg and met no other exclusion criteria and so were potentially eligible patients who were missed. In total, 83 of the patients screened were eligible for the pilot trial. Eight (9.6%) of these were not recruited. As a consequence of clinical staff having insufficient knowledge of the pilot trial, most of the small number of eligible patients were not recruited. Overall, 75 out of the 83 eligible patients (90.4%) were recruited to the trial. The percentage of eligible patients recruited reduces to 78.9% if the 12 patients who received an initial bolus of > 20 ml/kg are included in the denominator.
| Variables | Participants |
|---|---|
| Screened (n) | 297 |
| Received fluid bolus | |
| ≥ 20 ml/kg, n (%) | 209 (70.4) |
| < 20 ml/kg, n (%) | 88 (29.6) |
| N | 297 |
| Still in shock after bolus | |
| Yes, n (%) | 101 (48.3) |
| No, n (%) | 108 (51.7) |
| N | 209 |
| Met exclusion criteria | |
| No, n (%) | 83 (82.2) |
| Yes, n (%) | 18 (17.8) |
| N | 101 |
| Randomised | |
| Yes, n (% of those eligible) | 75 (90.4) |
| Missed, n (% of those eligible) | 6 (7.2) |
| Other, n (% of those eligible) | 2 (2.4) |
| N | 83 |
FIGURE 7.
The CONSORT flow diagram.
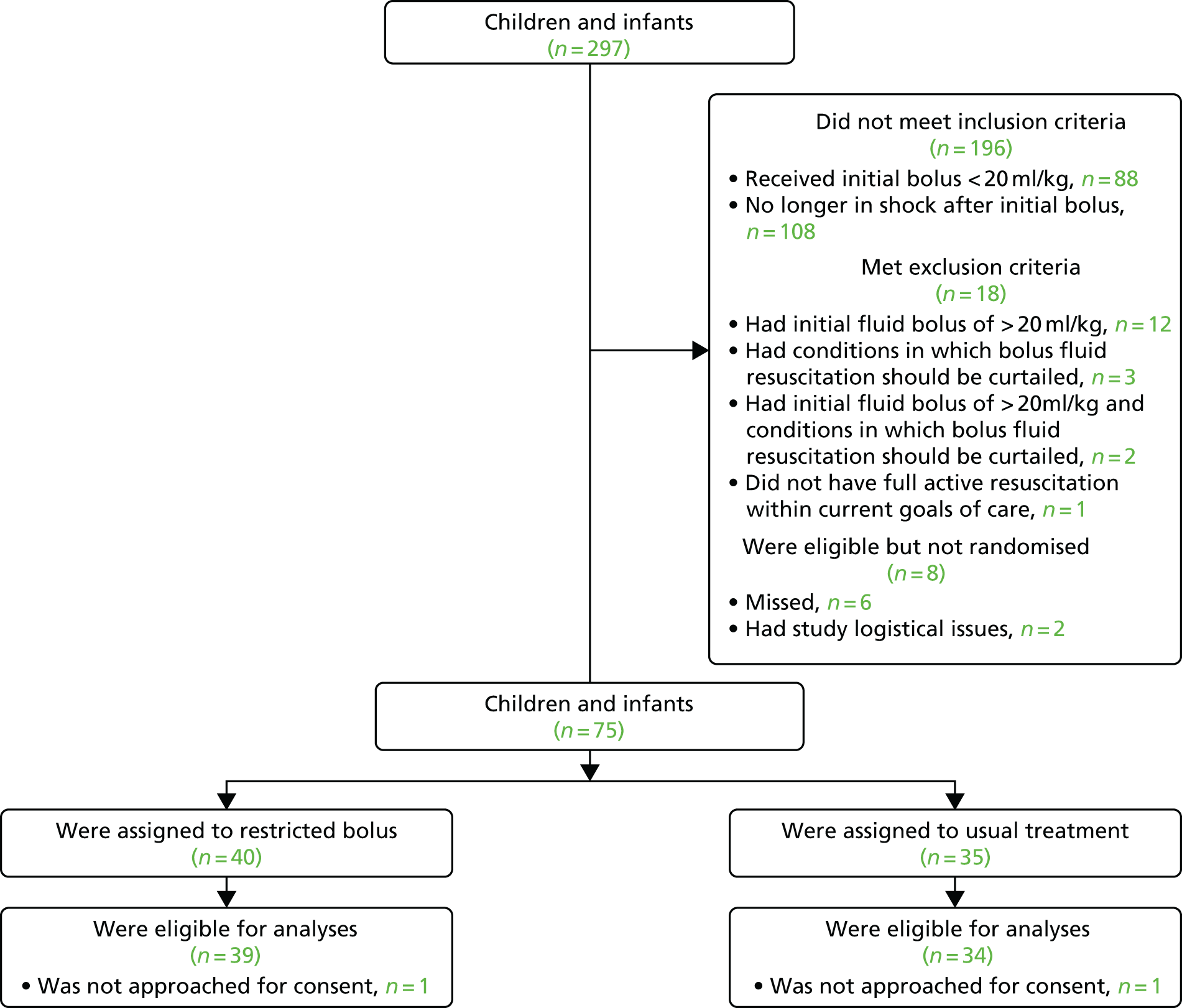
The 75 participants were recruited between 17 August 2016 and 3 April 2017 (Figure 8), with 40 randomised to the 10-ml/kg fluid boluses and 35 randomised to 20-ml/kg fluid boluses (Table 6). Nine participants were subsequently identified as having been randomised in error: seven had not met the inclusion criteria (six met local definitions for shock using lactate or heart rate but did not meet the FiSh trial shock criteria; one was randomised on the ward) and two had met an exclusion criterion. All nine participants randomised in error had been allocated to the 10-ml/kg fluid bolus group.
FIGURE 8.
Cumulative recruitment over time compared with planned (pre-trial) and anticipated (based on actual site opening dates).

| Variables | Treatment group | |
|---|---|---|
| 10 ml/kg | 20 ml/kg | |
| Randomised, n | 40 | 35 |
| Subsequently identified as randomised in error, n (%) | ||
| Did not meet inclusion criteria | 7 (17.5) | 0 (0.0) |
| Met exclusion criteria | 2 (5.0) | 0 (0.0) |
| Consent obtained, n (%) | ||
| Yes – in hospital | 26 (65.0) | 30 (85.7) |
| Yes – post discharge | 8 (20.0) | 1 (2.9) |
| No response (included) | 5 (12.5) | 3 (8.6) |
| Not approached (withdrawn) | 1 (2.5) | 1 (2.9) |
| Included in ITT analysis, n | 39 | 34 |
There was variation across the 12 sites in the rate of recruitment (Figure 9). The overall recruitment rate was 0.9 participants per site per month (95% CI 0.7 to 1.2 participants per site per month), compared with a pre-trial estimate of one participant per site per month. The median recruitment rate across individual sites was 0.6 participants per month (IQR 0.3–1.0 participants per month), and the highest recruitment rate was 3.2 participants per month.
FIGURE 9.
Recruitment rate with 95% CI, overall and by site.

Participants were recruited into the pilot trial throughout all days of the week, but the lowest recruitment was on Friday (Figure 10). Participants were generally recruited after 17.00 up until midnight (Figure 11).
FIGURE 10.
Number of participants, randomised by day of the week.
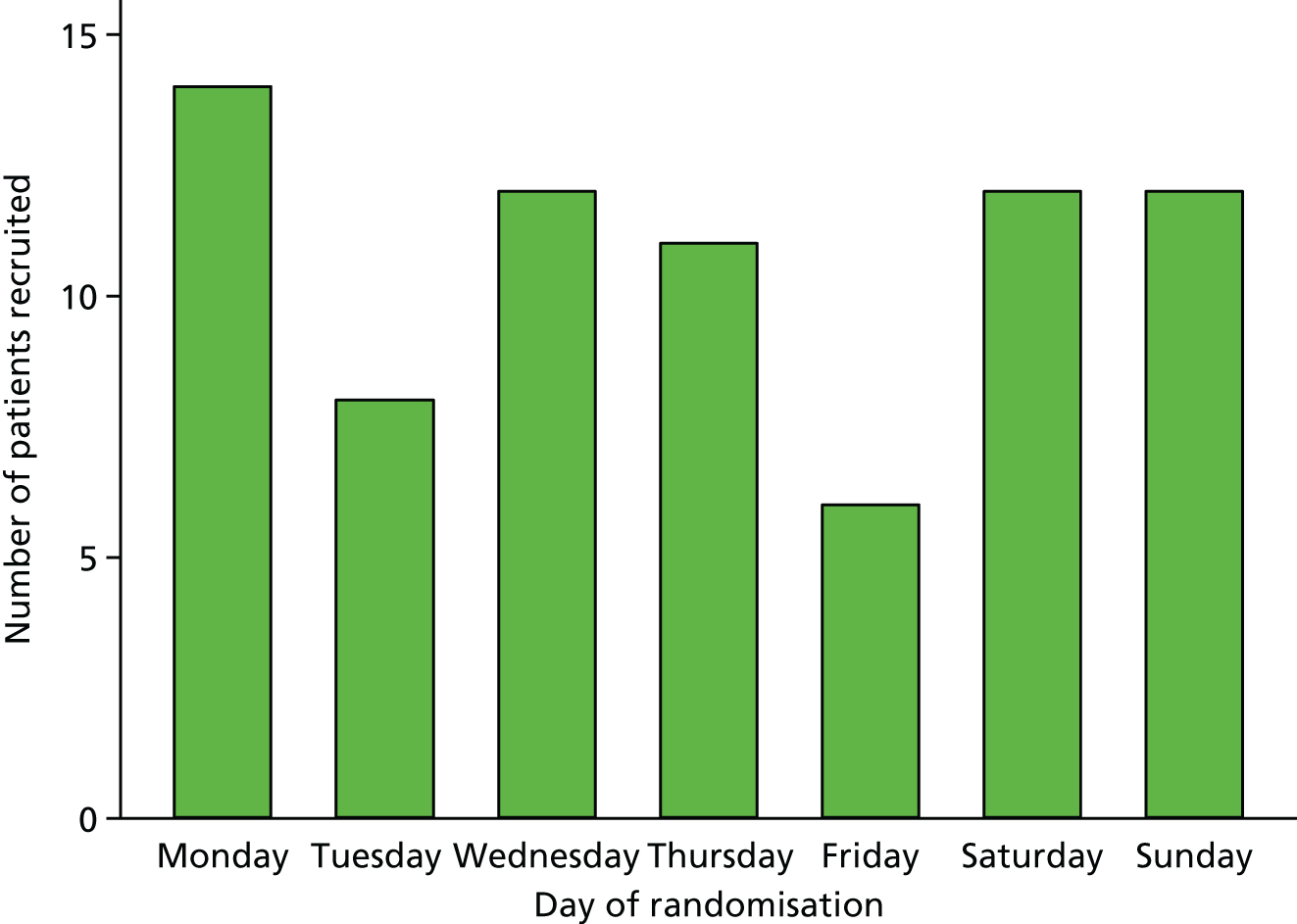
FIGURE 11.
Number of participants, randomised by time of the day.
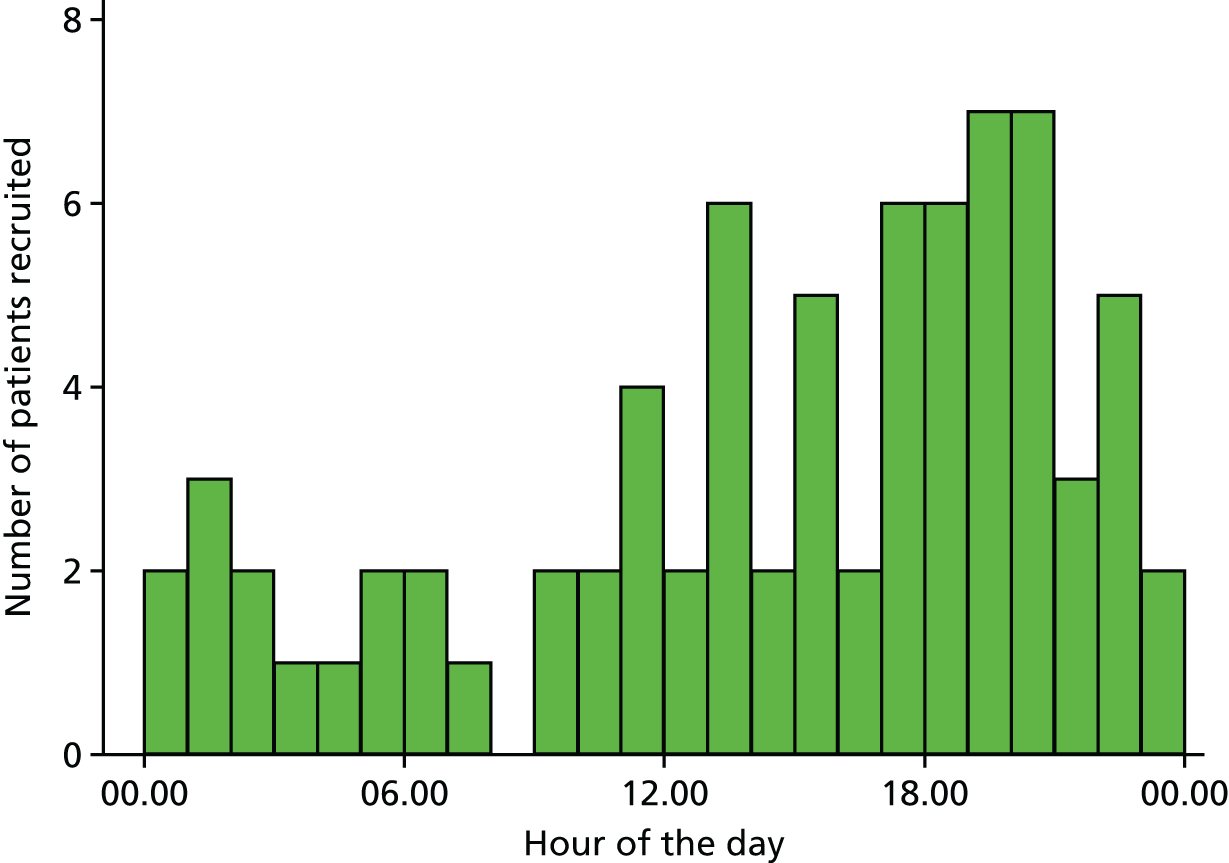
The majority of parents/legal representatives provided informed consent while their child was still in hospital (n = 56, 74.7%; see Table 6). For the remaining children, parents/legal representatives were approached via post, that is, after their child had been discharged and before consent could be sought; a response was received for nine (52.9%) patients. No response was received for eight (47.1%) patients; the follow-up letters, approved by the REC, indicated that, if no response was received, then the patient’s data would be used. Two (2.7%) patients (one from each treatment group) were withdrawn from the pilot trial as the parents/legal representatives could not be approached for informed consent, resulting in data from 73 patients for analysis. With regard to participant assent, although attempts were made to include children and young people in the discussion about the pilot trial, child assent was not taken by any of the participating sites, because children were either too young or too sick before discharge for a discussion about research. Follow-up was completed on 3 May 2017.
Characteristics of patients at baseline
The treatment groups were, on the whole, well matched at baseline (Table 7), with slight differences in age and, consequently, weight. The median age of patients was 11 months for the 10-ml/kg fluid bolus group and 2 months for the 20-ml/kg fluid bolus group (Figure 12). The participants randomised to the 10-ml/kg fluid bolus group had a mean weight at baseline of 13.3 kg, compared with 9.1 kg for the 20-ml/kg fluid bolus group, which is in line with the slightly older age distribution in this group.
| Characteristics | Treatment group | |
|---|---|---|
| 10 ml/kg (n = 39) | 20 ml/kg (n = 34) | |
| Age (months) | ||
| Mean (SD) | 30 (47) | 21 (41) |
| Median (IQR) | 11 (1–35) | 2 (1–17) |
| Gender, n (%) | ||
| Male | 24 (61.5) | 18 (52.9) |
| Female | 15 (38.5) | 16 (47.1) |
| Weight (kg) | ||
| Mean (SD) | 13.3 (15.6) | 9.1 (9.4) |
| Median (IQR) | 9 (5–13) | 5 (4–10) |
| Shock criteria met, n (%) | ||
| CRTa only | 30 (76.9) | 30 (88.2) |
| Age-adjusted hypotension only | 2 (5.1) | 2 (5.9) |
| Both CRTa and age-adjusted hypotension | 1 (2.6) | 2 (5.9) |
| Neither | 6 (15.4) | 0 (0.0) |
| PIM2r (2016) score (%) | ||
| Mean (SD) | 2.4 (1.5) | 2.4 (1.8) |
| Median (IQR) | 2.1 (1.6–2.7) | 2.0 (1.6–2.5) |
| Infection confirmed, n (%) | ||
| No | 19 (48.7) | 17 (50.0) |
| Yes | 20 (51.3) | 17 (50.0) |
FIGURE 12.
Distribution of age, by treatment group. (a) 10-ml/kg fluid bolus group; and (b) 20-ml/kg fluid bolus group.

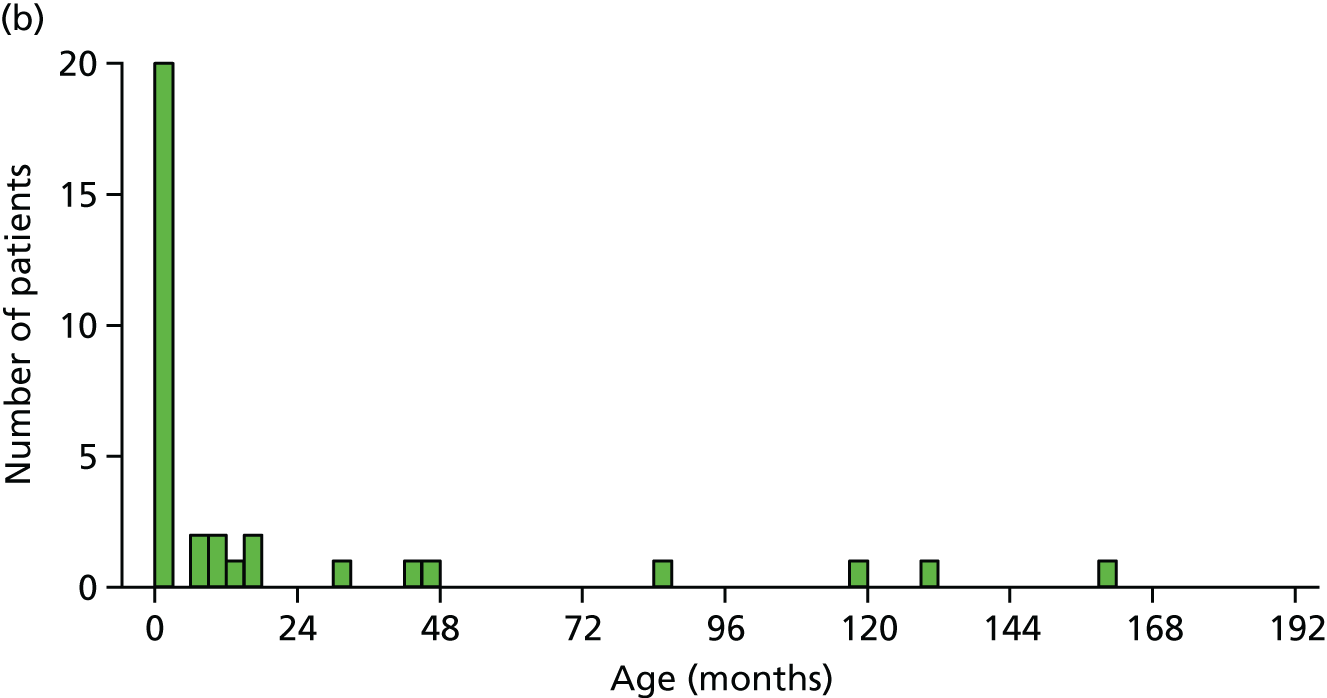
More than half the participants were male (61.5% in the 10-ml/kg fluid bolus group; 52.9% in the 20-ml/kg fluid bolus group). Most participants met the shock criteria because their CRT was ≥ 3 seconds (76.9% in the 10-ml/kg fluid bolus group; 88.2% in the 20-ml/kg fluid bolus group). The PIM2r score was balanced across treatment groups.
There were 37 participants (51.3% in the 10-ml/kg fluid bolus group; 50% in the 20-ml/kg fluid bolus group) recorded as having a confirmed infection. For 32 of these, the organism causing the infection was identified (Table 8). Of these, only 13 were bacterial infections in sterile sites. These included five Escherichia coli infections, two Enterococcus faecalis infections, three Streptococcus (two group A and one group B) infections, one Staphylococcus aureus infection, one Neisseria meningitidis infection and one Gram-positive coccal (unspecified) infection. There were 18 viral infections, 15 of which were respiratory pathogens (eight respiratory syncytial virus, five rhinovirus, one influenza A and one metapneumovirus), two of which were identified as enterovirus on blood polymerase chain reaction (PCR) analysis and one of which was identified as rotavirus in stools. One patient had falciparum malaria.
| Type of infection | Treatment group, n (%) | |
|---|---|---|
| 10 ml/kg (n = 20) | 20 ml/kg (n = 17) | |
| Bacterial | 5 (25.0) | 8 (47.1) |
| Viral | 10 (50.0) | 8 (47.1) |
| Other | 1 (5.0) | 0 (0.0) |
| Confirmed but not identified | 4 (20.0) | 1 (5.9) |
Protocol deviations
All participants randomised to the 20-ml/kg fluid bolus group received their first bolus (Table 9). Three participants randomised to the 10-ml/kg fluid bolus group did not receive their first bolus, although were correctly identified as being in shock, because it was deemed that they no longer required fluid post randomisation. Of the subsequent boluses, one participant in the 20-ml/kg fluid bolus group was administered a bolus despite the shock criteria (age-adjusted hypotension and CRT) not being met. Five participants in total (three from the 10-ml/kg fluid bolus group and two from the 20-ml/kg fluid bolus group) met the shock criteria; however, they were not identified or given a bolus. No participant experienced more than one deviation.
| Variables | Treatment group, n (%) | |
|---|---|---|
| 10 ml/kg | 20 ml/kg | |
| Number of participants | 39 | 34 |
| Did not receive first bolus | 3 (7.7) | 0 (0.0) |
| Of subsequent boluses | ||
| Bolus given, shock criteria not met | ||
| Deviations | 0 (0.0) | 1 (2.9) |
| Shock criteria met, no bolus given | ||
| Deviations | 3 (7.7) | 2 (5.9) |
Treatment delivery by group
The majority of participants in both groups received only one bolus (Table 10). During the total 4-hour intervention period, only four patients received four or more boluses.
| Variables | Treatment group | |
|---|---|---|
| 10 ml/kg | 20 ml/kg | |
| Number of patients | 39 | 34 |
| Number of boluses delivered, n (%) of patients | ||
| 0 | 3 (7.7) | 0 (0.0) |
| 1 | 23 (59.0) | 25 (73.5) |
| 2 | 8 (20.5) | 6 (17.6) |
| 3 | 3 (7.7) | 1 (2.9) |
| ≥ 4 | 2 (5.1) | 2 (5.9) |
| Total volume of fluid received during the first hour (ml) | ||
| Mean (SD) | 173 (254) | 184 (190) |
| Median (IQR) | 100 (50–180) | 121 (76–200) |
| n | 39 | 34 |
| Total volume of fluid received during the first hour (ml/kg) | ||
| Mean (SD) | 13.5 (8.0) | 20.7 (8.3) |
| Median (IQR) | 10 (10–20) | 20 (20–20) |
| n | 39 | 34 |
| Total volume of fluid received during the intervention period (ml) | ||
| Mean (SD) | 188 (325) | 243 (275) |
| Median (IQR) | 100 (50–210) | 124 (76–263) |
| n | 39 | 34 |
| Total volume of fluid received during the intervention period (ml/kg) | ||
| Mean (SD) | 14.5 (11.1) | 25.7 (12.0) |
| Median (IQR) | 10 (10–20) | 20 (20–29) |
| n | 39 | 34 |
| Total number of boluses delivered | 58 | 48 |
| Volume of bolus, n (%) of boluses | ||
| < 10 ml/kga | 2 (3.4) | 3 (6.3) |
| 10 ml/kga | 56 (96.6) | 5 (10.4) |
| 20 ml/kga | 0 (0.0) | 40 (83.3) |
| n | 58 | 48 |
| Delivery of bolus within 15 minutes, n (%) of boluses | ||
| No | 9 (19.1) | 14 (31.8) |
| Yes | 38 (80.9) | 30 (68.2) |
| n | 47 | 44 |
| Delivery of bolus within 15 minutes by age group, n (%) of boluses | ||
| < 1 year | ||
| No | 3 (11.1) | 7 (24.1) |
| Yes | 24 (88.9) | 22 (75.9) |
| n | 27 | 29 |
| 1–< 2 years | ||
| No | 2 (22.2) | 1 (33.3) |
| Yes | 7 (77.8) | 2 (66.7) |
| n | 9 | 3 |
| 2–< 5 years | ||
| No | 1 (14.3) | 2 (50.0) |
| Yes | 6 (85.7) | 2 (50.0) |
| n | 7 | 4 |
| ≥ 5 years | ||
| No | 3 (75.0) | 4 (50.0) |
| Yes | 1 (25.0) | 4 (50.0) |
| n | 4 | 8 |
| Fluid boluses delivered at correct volume and within 15 minutes, n (%) of boluses | ||
| No | 10 (21.3) | 20 (45.5) |
| Yes | 37 (78.7) | 24 (54.5) |
| n | 47 | 44 |
The mean total volume of fluid given during the first hour of the intervention period was 6% lower in the 10-ml/kg fluid bolus group than in the 20-ml/kg fluid bolus group (173 vs. 184 ml). The mean total volume of fluid given during the entire 4-hour intervention period was 23% lower in the 10-ml/kg fluid bolus group than in the 20-ml/kg fluid bolus group (188 vs. 243 ml; Figure 13), but the difference was not statistically significant (mean difference −55 ml, 95% CI −196 to 88 ml, p = 0.448; Table 11).
FIGURE 13.
Distribution of total volume of fluid received during intervention period (ml). (a) The 10-ml/kg fluid bolus group; and (b) the 20-ml/kg fluid bolus group.
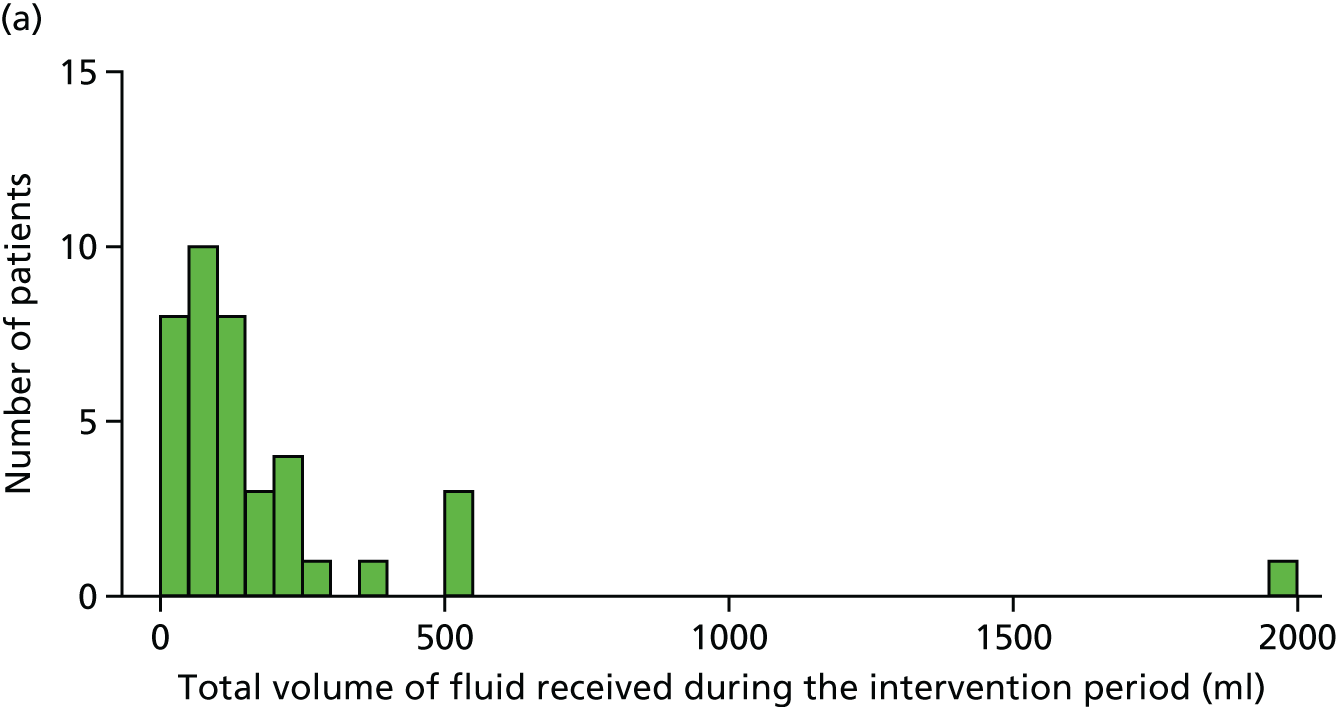

| Variables | Treatment group, mean volume (SD) | Mean difference (95% CI) | p-value | |
|---|---|---|---|---|
| 10 ml/kg (n = 39) | 20 ml/kg (n = 34) | |||
| Total volume of fluid (ml) | 188 (325) | 243 (275) | –54 (–196 to 88) | 0.448 |
| Total volume of fluid (ml/kg) | 14.5 (11.1) | 25.7 (12.0) | –11.2 (–16.6 to –5.8) | < 0.001 |
Owing to the baseline imbalance in weights, separation between the groups was also assessed in ml/kg (not prespecified in the statistical analysis plan). The mean total volume of fluid, in ml/kg, given during the first hour was 35% lower in the 10-ml/kg fluid bolus group than in the 20-ml/kg fluid bolus group (13.5 ml/kg vs. 20.7 ml/kg), and during the entire 4-hour intervention period was 47% lower (14.5 vs. 27.5 ml/kg; Figure 14). At the end of the 4-hour intervention period, this corresponded to a statistically significant mean difference of –11.2 ml/kg (95% CI –16.6 to –5.8 ml/kg; p < 0.001; see Table 11).
FIGURE 14.
Distribution of total volume of fluid received during intervention period (ml/kg). (a) The 10-ml/kg fluid bolus group and (b) the 20-ml/kg fluid bolus group.
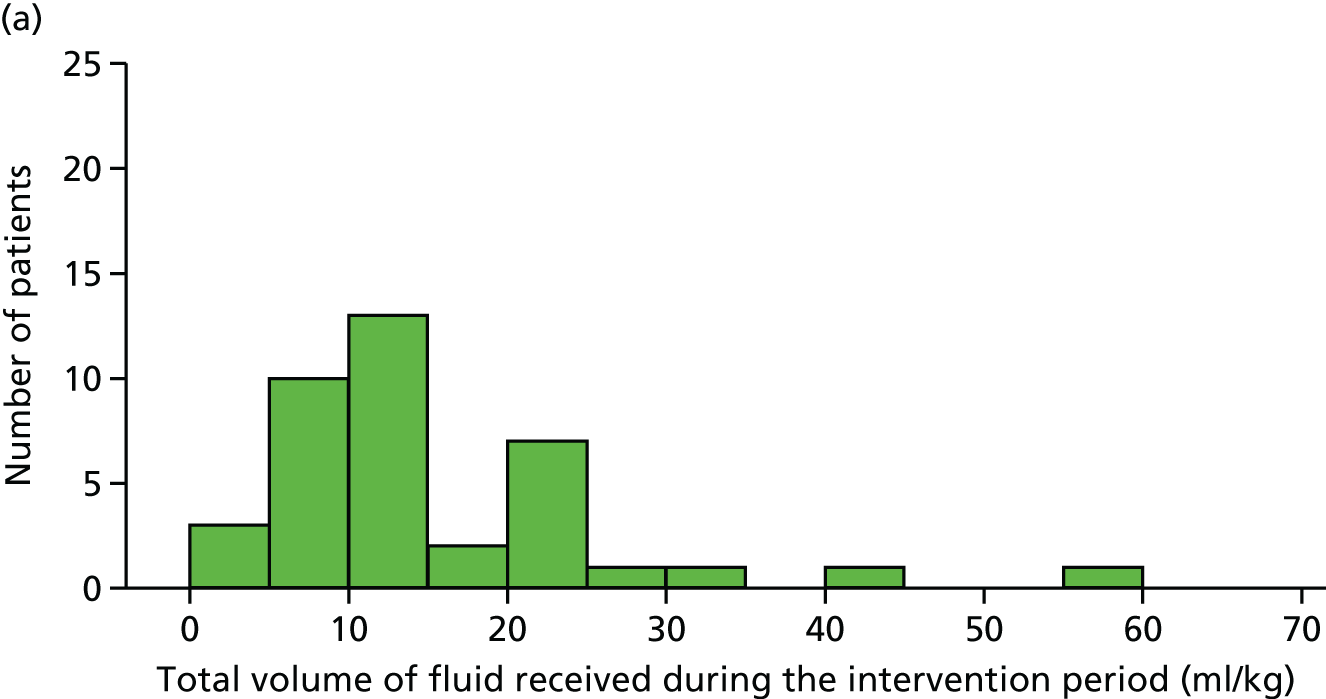
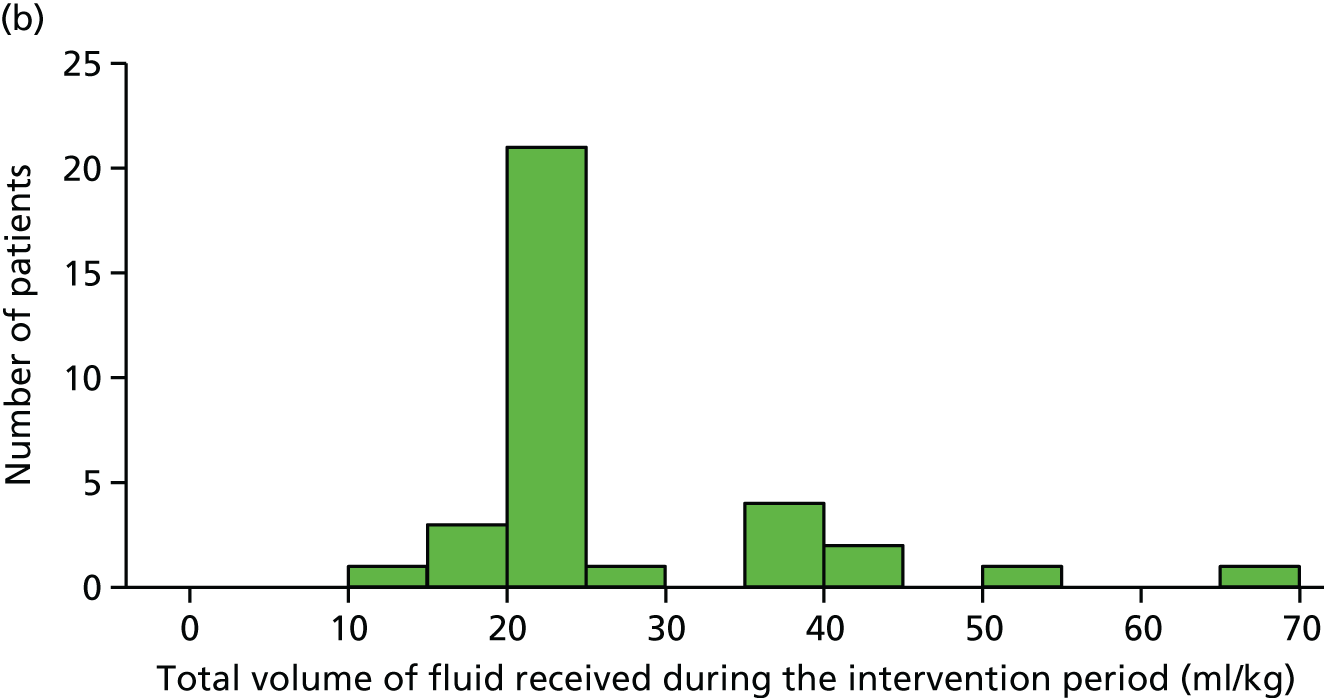
None of the boluses delivered in the 10-ml/kg fluid bolus group was > 10 ml/kg; however, two (3.4%) were < 10 ml/kg (see Table 10). In the 20-ml/kg fluid bolus group, no boluses of > 20 ml/kg were delivered; however, five boluses (10.4%) of 10 ml/kg and three (6.3%) of < 10 ml/kg were delivered.
Out of 106 boluses delivered, there were a total of 91 boluses (85.8%) with complete data to determine the timing of bolus delivery (47 in the 10-ml/kg fluid bolus group and 44 in the 20-ml/kg fluid bolus group). Thirty-eight out of 47 boluses (80.9%) delivered to the 10-ml/kg fluid bolus group were administered within 15 minutes, whereas 30 out of 44 (68.2%) delivered to the 20-ml/kg fluid bolus group were administered within 15 minutes. There were no clear indications of difference when looking at different age groups, owing to the smaller numbers of older children.
Thirty-seven boluses (78.7%) were delivered at the correct volume and within 15 minutes in the 10-ml/kg fluid bolus group, whereas 24 boluses (54.5%) were delivered at the correct volume and within 15 minutes in the 20-ml/kg fluid bolus group.
Potential outcome measures
All participants (n = 16) admitted to a PICU in one of the FiSh trial sites were successfully linked to PICANet data. Five participants were admitted to a non-FiSh-trial PICU and were not able to be linked as research approvals were not in place. Mechanical ventilation was defined as invasive ventilation via endotracheal tube, invasive ventilation via tracheostomy tube, advanced ventilatory support (jet ventilation) or advanced ventilatory support (oscillatory ventilation). All patients were successfully followed up for mortality following hospital discharge using the NHS Spine web portal.
No participants died either in hospital or by 30 days post randomisation in either treatment group (Table 12). As anticipated for a small pilot trial, there were no significant differences between the groups in any of the other outcomes. To assist with powering any future trial, 95% CIs for the outcomes in each treatment group are presented in Table 13.
| Potential outcome measures | Treatment group | Effect estimate (95% CI) | p-value | |
|---|---|---|---|---|
| 10 ml/kg (n = 39) | 20 ml/kg (n = 34) | |||
| Hospital mortality, n/N (%) | 0/39 (0.0) | 0/34 (0.0) | Not estimable | |
| Length of hospital stay (days), median (IQR) [n] | 4 (3–7) [39] | 5 (4–8) [34] | 0.225a | |
| Transferred to PICU, n/N (%) | 10/39 (25.6) | 11/34 (32.4) | –6.7 (–27.6 to 14.1)b | 0.354 |
| 0.79 (0.38 to 1.63)c | ||||
| Length of stay in PICU (hours), median (IQR) [n] | 45 (18–143) [10] | 119 (52–228) [11] | 0.091a | |
| Days alive and free of PICU up to 30 days,d mean (SD) [n] | 28.9 (2.4) [39] | 27.9 (3.6) [34] | 1.0 (–0.4 to 2.4)e | 0.160 |
| Receipt of mechanical ventilation, n/N (%) | 4/36 (11.1) | 8/32 (25.0) | –13.9 (–32.1 to 4.3)b | 0.119 |
| 0.44 (0.15 to 1.34)c | ||||
| Duration of mechanical ventilationf (days), median (IQR) [n] | 6 (4–8) [4] | 5.5 (4–8.5) [8] | 0.797 | |
| Days alive and free of mechanical ventilation up to 30 days,d mean (SD) [n] | 29.3 (2.1) [36] | 28.5 (2.7) [32] | 0.8 (–0.4 to 2.0)e | 0.192 |
| Mortality at 30 days,d n/N (%) | 0/39 (0.0) | 0/34 (0.0) | Not estimable | |
| Potential outcome measures | Treatment group, estimate (95% CI) | |
|---|---|---|
| 10 ml/kg (n = 39) | 20 ml/kg (n = 34) | |
| Hospital mortality (%) | 0 (0.0 to 9.0) | 0 (0.0 to 10.2) |
| Length of hospital stay (days), median | 4 (3 to 5) | 5 (4 to 7) |
| Transferred to PICU (%) | 25.6 (14.6 to 41.1) | 32.4 (19.1 to 49.2) |
| Length of stay in PICU (hours), median | 45 (8 to 143) | 119 (51 to 230) |
| Days alive and free of PICU up to 30 days,a mean | 28.9 (28.2 to 29.7) | 27.9 (26.7 to 29.2) |
| Receipt of mechanical ventilation (%) | 11.1 (4.4 to 25.3) | 25.0 (13.3 to 42.1) |
| Duration of mechanical ventilationb (days), median | 6 (2 to 10) | 5.5 (2 to 9) |
| Days alive and free of mechanical ventilation up to 30 days,a mean | 29.3 (28.6 to 30.0) | 28.5 (27.5 to 29.6) |
| Mortality at 30 daysa (%) | 0 (0.0 to 9.0) | 0 (0.0 to 10.2) |
Safety monitoring
There were no reported SAEs; four AEs in total were reported (Table 14). Three were reported in the 10-ml/kg fluid bolus group. One of these was pulmonary oedema (an expected AE), which was deemed unlikely to be related to the trial intervention, and the other two AEs were not related to the intervention (Table 15). There was one AE in the 20-ml/kg fluid bolus group, which was not related to the trial intervention.
| AEs | Treatment group, n (%) | |
|---|---|---|
| 10 ml/kg (n = 39) | 20 ml/kg (n = 34) | |
| AEs | 3 (5.1) | 1 (2.9) |
| SAEs | 0 (0.0) | 0 (0.0) |
| Treatment group | Event | Expectedness | Severity | Relatedness |
|---|---|---|---|---|
| 10 ml/kg | Pulmonary oedema | Expected | Moderate | Unlikely |
| 10 ml/kg | Recurrent urinary tract infection | Unexpected | Moderate | None |
| 10 ml/kg | Head injury | Unexpected | Mild | None |
| 20 ml/kg | Rash after taking oral antibiotics at home | Unexpected | Mild | None |
Admissions to paediatric intensive care units
Baseline characteristics and treatment delivery for participants admitted to a PICU compared with participants not admitted to a PICU are presented in Table 16. Participants admitted to a PICU had a higher average severity of illness as assessed by the PIM2r (2016) score (mean 3.6% vs. 1.9%), were more likely to have an infection (71% vs. 42%) and received a greater average volume of fluid during the intervention period (mean 24.4 ml/kg vs. 17.8 ml/kg).
| Characteristics | Admission to PICU | |
|---|---|---|
| Admitted (n = 21) | Not admitted (n = 52) | |
| Age (months) | ||
| Mean (SD) | 41 (59) | 19 (35) |
| Median (IQR) | 10 (0–65) | 4 (1–19) |
| Gender, n (%) | ||
| Male | 11 (52.4) | 31 (59.6) |
| Female | 10 (47.6) | 21 (40.4) |
| Weight (kg) | ||
| Mean (SD) | 14.4 (19.4) | 10.1 (9.6) |
| Median (IQR) | 7 (3–18) | 7 (5–12) |
| Shock criteria met, n (%) | ||
| CRTa only | 16 (76.2) | 44 (84.6) |
| Age-adjusted hypotension only | 3 (14.3) | 1 (1.9) |
| Both CRTa and age-adjusted hypotension | 2 (9.5) | 1 (1.9) |
| Neither | 0 (0.0) | 6 (11.5) |
| PIM2r (2016) score (%) | ||
| Mean (SD) | 3.6 (2.6) | 1.9 (0.6) |
| Median (IQR) | 2.6 (2.3–3.8) | 1.8 (1.5–2.1) |
| Infection confirmed, n (%) | ||
| No | 6 (28.6) | 30 (57.7) |
| Yes | 15 (71.4) | 22 (42.3) |
| Number of boluses delivered, n (%) of patients | ||
| 0 | 2 (9.5) | 1 (1.9) |
| 1 | 10 (47.6) | 38 (73.1) |
| 2 | 3 (14.3) | 11 (21.2) |
| 3 | 3 (14.3) | 1 (1.9) |
| ≥ 4 | 3 (14.3) | 1 (1.9) |
| Total volume of fluid received during the intervention period (ml) | ||
| Mean (SD) | 267 (305) | 192 (301) |
| Median (IQR) | 120 (60–360) | 104 (73–185) |
| Total volume of fluid received during the intervention period (ml/kg) | ||
| Mean (SD) | 24.4 (17.9) | 17.8 (9.6) |
| Median (IQR) | 20 (20–30) | 20 (10–20) |
Potential outcome measures for participants admitted to a PICU (pooled across the treatment groups) are presented in Table 17.
| Potential outcome measures | Number of patients | Value | 95% CIa |
|---|---|---|---|
| Length of hospital stay (days), median (IQR) | 21 | 8 (4–12) | 4 to 12 |
| Length of stay in PICU (hours), median (IQR) | 21 | 95 (35–145) | 35 to 145 |
| Days alive and free of PICU up to 30 days,b mean (SD) | 21 | 24.7 (3.5) | 23.1 to 26.3 |
| Receipt of mechanical ventilation, n (%) | 16 | 12 (75.0) | 50.5 to 89.8 |
| Duration of mechanical ventilationc (days), median (IQR) | 12 | 6 (4–8.5) | 4 to 9 |
| Days alive and free of mechanical ventilation up to 30 days,b mean (SD) | 16 | 25.6 (3.5) | 23.7 to 27.4 |
Chapter 5 Results of the integrated perspectives study
Participants: parents/legal representatives
A total of 52 out of 75 (69%) parents of patients enrolled in the pilot trial consented for inclusion in the perspectives study element of the study (i.e. agreed to complete a questionnaire and/or to be interviewed). No participants identified themselves as legal representatives. Of the 52 parents who agreed to be approached for interview, 34 (65%) were purposively selected and invited by telephone or e-mail. Of these, 11 (32%) did not respond, one was deemed ineligible owing to an inability to understand English and two declined to be interviewed on telephone contact. Data saturation was reached following interviews with 20 parents. 22,23 Of the 18 parents not contacted for interview, 10 (19%) were from one high-recruiting site already represented in the interview sample, and so were not contacted to prevent sample bias. A total of eight (15%) parents were not contacted as a result of data saturation22,23 being reached. A questionnaire was received from 45 out of 52 (87%) parents from 44 families. Figure 15 presents the recruitment process.
FIGURE 15.
Parent/legal representative recruitment process and sample characteristics in the integrated perspectives study element of the pilot trial. a, Does not include if more than one parent was asked to participate – data unknown.

Characteristics
Of the 20 parents (19 mothers and one father) interviewed by telephone and 45 parents (34 mothers and 11 fathers) who completed a questionnaire, all had consented to the use of their child’s information in the pilot trial and none was bereaved. Parents interviewed were recruited from 10 out of the 12 (83%) recruiting sites and questionnaires were received from 9 out of the 12 (75%) recruiting sites. Parents were interviewed a median of 26.5 days after admission (range 14–96 days) and the median length of hospital stay for their child was 3 days (range 1–23 days). One parent had previous experience of their child participating in medical research; none had experience of RWPC. Telephone interviews with parents took between 30 and 60 minutes. All 45 parents completed the questionnaire on site, before their child was discharged from hospital.
Participants: site staff
Three focus groups were conducted with 20 site staff from the three recruiting ‘hub’ pilot trial sites (Figure 16). In addition, seven telephone interviews were conducted with 7 out of 20 (35%) invited staff, one of whom was from a ‘hub’ site and six of whom were from a ‘spoke’ site.
FIGURE 16.
Site staff focus group and telephone interview recruitment process and sample characteristics in the integrated perspectives study element of the pilot trial.

Characteristics
Of the 27 participating site staff, 22 (81%) were involved in the clinical care of children [17/20 (85%) of focus group participants and 5/7 (71%) of telephone interview participants]. The staff sample included 14 (52%) nurses and 13 (48%) registrar- or consultant-grade doctors with a modal range of 1–2 years of experience in recruiting to paediatric clinical trials. Telephone interviews took between 30 and 60 minutes and focus groups lasted approximately 90 minutes.
Parental perspectives
Research without prior consent is acceptable
Before any discussion of the pilot trial during interviews, parents were prompted to describe their understanding of RWPC to explore explanations provided to them during the consent discussion, as well as their views on this method of consent. Although no parents had any previous knowledge of RWPC, the majority (n = 15/20, 75%) were able to appropriately describe this approach, explaining and supporting its use in emergency situations:
They just said that in these situations they just obviously stabilise the child as a priority and then discuss it afterwards because then there’s obviously no pressure to be involved and that’s sort of how it was explained. Um, so I hadn’t heard of it, no, but it doesn’t surprise me. In an emergency situation, you do what you’ve got to do, don’t you, and then deal with stuff afterwards.
P25, mother, telephone interview
Following the general discussion of RWPC, the focus shifted towards parents’ views on the use of this method in the pilot trial. Consistent with previous studies18,19,27 and the feasibility study findings described in Chapter 2, several parents (n = 7/20, 35%) were initially shocked or surprised to discover that their child had been enrolled into the pilot trial without prior consent. As shown in Table 18, 13 out of 45 (28%) parents who completed a questionnaire agreed with statement 2: ‘I was initially surprised to find out that my child had already been entered into FiSh’. However, following tailored explanation from site staff about why informed consent was not sought,30 all parents stated that they supported RWPC in this pilot trial:
I was initially surprised that obviously this had happened, because obviously the consent happened afterwards, but I wasn’t sort of concerned or anything, and then they explained everything really clearly, and the information leaflet was very good as well.
P12, mother, telephone interview
| Statement | Responses, n (%) | ||
|---|---|---|---|
| Agree | Neither agree nor disagree | Disagree | |
| 1. The practitioner checked that it was a convenient time to discuss research before discussing FiSh | 41 (91.1) | 2 (4.4) | 2 (4.4) |
| 2. I was initially surprised to find out that my child had already been entered into FiSh | 13 (28.9) | 19 (42.2) | 13 (28.9) |
| 3. The information I received about FiSh was clear and straightforward to understand | 42 (93.3) | 3 (6.7) | 0 (0) |
| 4. I understood why consent for my child’s participation in FiSh was sought after the treatment had been given | 44 (97.8) | 1 (2.2) | 0 (0) |
| 5. I had enough opportunity to ask questions about FiSh | 43 (95.6) | 2 (4.4) | 0 (0) |
| 6. I was satisfied with the deferred consent process for FiSh | 39 (86.7) | 6 (13.3) | 0 (0) |
| 7. It was difficult to take in the information I was given about FiSh | 1 (2.2) | 6 (13.3) | 38 (84.4) |
| 8. It was difficult to make a decision about FiSh | 1 (2.2) | 3 (6.7) | 41 (91.1) |
| 9. I made this decision | 42 (93.3) | 1 (2.2) | 2 (4.4) |
| 10. Someone took this decision away from me | 2 (4.4) | 1 (2.2) | 42 (93.3) |
| 11. I was not in control of this decision | 4 (8.9) | 1 (2.2) | 40 (88.9) |
| 12. The decision about the research was inappropriately influenced by others | 2 (4.4) | 3 (6.7) | 40 (88.9) |
Indeed, the majority (n = 39/45, 86.7%) of parents who completed the questionnaire agreed with statement 6: ‘I was satisfied with the deferred consent process in FiSh’; no parents disagreed with this statement. Parents (n = 44/45, 97.8%) also agreed with statement 4, indicating that they had ‘understood why consent for my child’s participation in FiSh was sought after the treatment had been given’. Moreover, both questionnaire and interview responses indicated that, despite their children being entered into the FiSh pilot trial without prior consent, the majority of parents felt that their consent decision about the use of their child’s data was voluntary18,51,52 [42/45 (93.3%) agreed with statement 9: ‘I made this decision’; 42/45 (93.3%) disagreed with statement 10: ‘Someone took this decision away from me’; 40/45 (88.9%) disagreed with statement 11: ‘I was not in control of this decision’ and 40/45, 88.9% disagreed with statement 12: ‘The decision about research was inappropriately influenced by others’].
Importantly, however, support for RWPC in emergency care trials was felt by some to depend on the context of the specific study:
I think probably my concern was probably a strange concern, insomuch on this study, ‘cause I completely agree with it. It’s more, I think, a concern to give the impression that I think that consent after the fact is a good thing, because I don’t necessarily think it is. I think there are situations where I would strongly disagree with it. That’s really my only concern, that I would be in some way condoning that process as a process in itself, rather than it being a process specifically about, you know, this sort of subject.
P06, mother, telephone interview
More specifically, several parents stated that practitioners explained that rapid fluid bolus resuscitation is the standard treatment for children presenting to UK EDs, despite a lack of evidence for current recommended practice (i.e. 20 ml/kg). 3–5 This explanation, combined with a detailed description of fluid bolus therapy, helped to reassure parents that there were good reasons for RWPC in the FiSh pilot trial and that their child’s safety would not be compromised, as both treatment arms involved carefully titrated administrations of fluid:
I think because he was still getting the same, he was still getting the fluids and essentially still had the same amount but it was just over different sort of periods of time, I personally didn’t feel like it mattered ‘cause he was still getting the same thing.
P12, mother, telephone interview
Obviously the fluid is what he needed so just giving him a small amount wouldn’t have made a difference because they were keeping an eye on him, so it could have been added to.
P26, mother, telephone interview
Similarly, several parents indicated that they would have been concerned about site staff not seeking their prior consent if the trial had involved the administration of a new drug or any other ‘unknown’ clinical procedure, including surgery:
Um, if it’s a trial, I would want to know if it was medicine or drugs or if it was unknown surgery. So, unknown territory. The fluids, they’re going to get fluids anyway so for me, [RWPC] was not even vaguely a concern.
P40, mother, telephone interview
Finally, the majority (15/20, 75%) of parents interviewed said that their child’s survival probably affected their response to the pilot trial and willingness to provide consent. This was consistent with parental survey responses that explored parents’ reasons for consenting to the trial (see Table 19 and subsequent text):
I think that again it’d be down to outcome. If you have a positive outcome, I think the reaction would be completely different than if you had a negative outcome. I think then it could cause a lot of, kind of questioning on the parents’ part, had, you know, he been given a different drug or whatever, would the outcome be different. And actually it could, it could cause an awful lot of ongoing pain, wondering, you know, whether or not that was a factor that tipped the balance, you know.
P06, mother, telephone interview
Nevertheless, the remainder of parents interviewed stated that they would have supported the pilot trial and its method for consent even in less positive circumstances, suggesting that they would not have linked the trial to poor recovery. These views appear to have been influenced not only by the nature of the trial, but also by parental trust in doctors to prioritise their child’s life over research, as well as the sensitive manner of site research staff:
No, I’m pretty sure that if, if they started to do the FiSh and then something went completely wrong, I’m sure rather than stick to the trial, they’d put the patient first and think, right we need to do this, forget looking at the clock, this needs to be done.
P37, mother, telephone interview
The following conversation is from a telephone interview:
Hypothetically speaking, how would you have felt if they approached you about this study and your son had not recovered as well as he did?
Well the approach was really gentle and it wasn’t at all, um, you know, we weren’t being strong-armed into anything. So I don’t think, err, I know it’s a, would be a stressful and awful time anyway but I think it was done so delicately that I don’t think we would have been upset to be asked.
Do you think you would have had any concerns that the trial was linked to poor recovery?
No, I don’t think so. I mean, like I’ve said previously, if it was to do with different drug trials, then yeah 100%, but not for a you know, rehydration solution or whatever the trial is.
However, as the following quotation illustrates, some parents might question whether or not trial participation was to blame for a child’s poor recovery. This finding suggests that in such circumstances site staff should be prepared to address parents’ questions and concerns about their child’s poor recovery in the context of trial participation:
I understand how serious it was and I can’t thank them enough but I think everyone sometimes wants someone to blame and I think if it didn’t go the way that, that it was supposed to, I wouldn’t necessarily be blaming them but I would want an answer to that question.
P36, father, telephone interview
Responses to the pilot trial
Following the discussion of RWPC, the interview conversation shifted to the pilot trial. Parents were prompted for their views on several aspects of the trial, including their understanding of the aims, acceptability of the interventions involved and the reasons for consent (Table 19).
| Reason for consent | Identified as [n (%)] | |
|---|---|---|
| A reason (n = 45) | The main reason (n = 25) | |
| 1. To help my child | 23 (52.3) | 1 (4.0) |
| 2. To help other children in the future | 44 (100) | 19 (76.0) |
| 3. I felt that medical studies like FiSh are important | 33 (75.0) | 3 (12.0) |
| 4. I trusted the doctor or nurse who explained FiSh | 27 (60.0) | 0 (0) |
| 5. The treatment had already been given to my child | 26 (57.8) | 0 (0) |
| 6. My child recovered | 22 (48.9) | 2 (8.0) |
| 7. I did not feel comfortable saying no to the nurse or doctor who explained | 0 (0) | 0 (0) |
| 8. Other | 0 (0) | 0 (0) |
All parents interviewed had provided consent for the pilot trial. The parents cited their child’s trial participation as a way to help advance children’s emergency treatment to help other children in the future:
I kind of thought about other children and thought ‘do you know what, like it’s probably a good idea that they relook at this just to make sure that every child is getting the right treatment’.
P01, mother, telephone interview
The questionnaire included a series of statements to establish the reasons why parents had consented for their child’s information to be used in the pilot trial. Many parents ticked multiple reasons (mode, five reasons; mean, four reasons). In line with the qualitative findings, all parents indicated that a reason they provided consent for the use of their child’s information in the pilot trial was to help other children in the future (see Table 19), with 19 out of 25 (76%) indicating that this was the main reason for consent. A high proportion also indicated that they provided consent owing to a belief that medical research studies like the FiSh trial are important, with 3 out of 25 (12%) indicating that this was their main reason. Interestingly, half of parents in both interviews and questionnaires stated that they had participated because their child had recovered, which supports our qualitative findings that show that parents were happy to consent as their child had recovered by the time of consent.
Also of interest was that half of parents indicated that they consented to help their child, although this was a main reason for consent for only one parent. Only half of the parents interviewed appeared to understand the uncertainty around optimal fluid bolus resuscitation therapy and how the FiSh trial is needed because ‘we don’t know what is best’ (P33, mother, telephone interview). Interviews also revealed a potential lack of equipoise among some parents (7/20, 35%). This related to beliefs that one arm of fluid bolus resuscitation for septic shock was safer than the other, that the volume administered to their child was the correct amount given their recovery, or that both interventions were ‘the same essentially’ (P12, mother, telephone interview):
I really didn’t mind that. Obviously, they’re saying the small amount would be better for the child, so to me, you know, he was fine and he came round quick . . . so if someone had given him the larger amount I would have probably panicked anyway.
P26, mother, telephone interview
The operation was a success, you gave the right amount of fluid. Her, in our daughter’s case, we believe that that was the right amount because she didn’t need any more until the operation and I think that kind of kept her going.
P36, mother, telephone interview
In addition, the subject of the trial (i.e. septic shock) as well as its perceived severity appeared to have a significant impact on some parents. Specifically, 3 out of 20 (15%) mothers who took part in an interview, one of whom also completed a questionnaire, described their upset at finding out that their child had been treated for septic shock during the consent discussion. As the following quotations illustrate, clinical staff had not spoken to these parents using this term before research staff had approached them to discuss the pilot trial:
I had no idea my child had septic shock. When I was told about the study was the first time I found out, which was quite distressing.
P06, mother, questionnaire
We didn’t actually know he was being treated for septic shock . . . until they gave me the letter and I read it, I didn’t actually know that’s what he was being treated for.
P07, mother, telephone interview
I just heard sepsis and then I almost hit the floor.
P39, mother, telephone interview
One mother stated that ‘it was almost awkward for the nurse ‘cause she didn’t realise that she was breaking the news’ (P06, mother, telephone interview).
These parents recommended that, in future research, a member of the clinical team should have a discussion with parents about what has happened and their child’s condition, before a discussion about research occurs:
What could have been improved was to make sure the parents know what actually happened in the situation before speaking about a trial.
P26, mother, telephone interview
Approaching parents for consent
The majority of parents, including 41 out of 45 (91%) of those who completed the questionnaire, indicated that site staff had checked that it was a convenient time to discuss the pilot trial. During interviews, parents stated that they felt that they were approached appropriately within 24–48 hours of their child’s admission, ‘after sort of the initial, um, scary period’ (P01, mother, telephone interview), when the emergency situation had passed. Parents appeared to appreciate the timing of the discussion and that the research nurse had ‘made sure that it was the right time’ (P03, mother, telephone interview) to discuss research.
Two parents who completed a questionnaire indicated that the researcher had not checked the timing of the consent discussion, although this did not appear to affect their ability to understand the information or their satisfaction with the consent process. Although 38 out of 45 (84.4%) disagreed with statement 7 (see Table 18), indicating that they did not find it difficult to take in the pilot trial information, one parent agreed that it had been difficult to take in the information and six neither agreed nor disagreed with the statement. Furthermore, consistent with other studies in this area,38,40 many parents said that they did not ask questions or raise concerns with site staff. A few parents stressed that it was important for staff to tailor the amount of trial information provided to help meet individual needs and preferences:
You’ve got parents with very different levels of interest, education and cultures. I am somebody who likes to be as informed as possible and I’ll ask questions if I don’t understand. But, I think it’s a two-way process. Maybe the researcher has the option of going down the road of explaining to a parent if they want to know but perhaps doesn’t go down that road if the parent chooses to have sufficient information. Err, I think I would have appreciated a little more information in situ.
P38, mother, telephone interview
As per the findings in other critical care studies,19,27 parents stated that they would not wish to be approached for consent during resuscitation, as their child’s life was the priority. As the following quotation illustrates, parents questioned if they would want, or be able to take in, study information when their child was being resuscitated. One-third of parents indicated that they would have felt pressured to provide a decision about the pilot trial if approached by staff during resuscitation. They also suggested that, under such circumstances, parents would be more likely either to refuse to take part or to provide a decision that was uninformed as heightened emotions may affect parental capacity:
My focus had to be on my child. He needed me, right there and right then, it would have been, you know, noise in the background, and I probably would have nodded and not even taken it in.
P06, mother, telephone interview
I mean it’s such a stressful situation, you just need to be, but you just need to be as calm and relaxed as possible. By throwing that on to you I think people would just say no.
P13, mother, telephone interview
Interestingly, 5 out of 20 (25%) parents who were interviewed described how they were spoken to by site staff about the pilot trial during resuscitation. Of these, most parents (4/5, 80%) were unperturbed by this method of approach; however, this appeared to depend on the brevity of the conversation. One mother suggested that being approached by site staff during resuscitation was inappropriate and ‘just not necessary and just not a priority for the parent’ (P38, mother, telephone interview). She stated that she ‘would have been happy for them [site staff] to be there’, provided the explanation provided was simplified and kept brief:
I don’t think it’s necessary in the resus [resuscitation] department unless the parent asks. I think there’s so much going on so unless they ask or they overhear and they say, ‘oh, what’s that?’ Without brushing them off, you know, just a brief explanation is sufficient, which is what I had, which was perfect.
P40, mother, telephone interview
So, I was in A&E [accident and emergency] and there was a lady standing by the door sort of watching everything that was going on in the Resus [Resuscitation] Room. She introduced herself to me as somebody doing research and she said FiSh trial like the FiSh research project. And obviously I, you know, I remember because FiSh is obviously a fairly memorable acronym. And then she described it, but to be honest I remember her talking about fluids and giving probably too much information for me at that point in time.
P38, mother, telephone interview
Finally, during interviews parents were asked to reflect on who (doctor, nurse or researcher) approached them to discuss the pilot trial and whether or not their role affected the trial acceptability and their consent decision. The majority (70%) stated that they would not have a role preference so long as the practitioner ‘believed in the trial and its purpose’ (P13, mother, telephone interview) and was able to provide parents with ‘simple and clear’ (P40, mother, telephone interview) tailored explanations on key trial information. Nevertheless, several parents stated that they would appreciate a consultant’s involvement in the initial stages of consent. More specifically, they suggested that interacting with someone directly involved in their child’s care could promote parental reassurance and foster confidence in the pilot trial:
Well we had, I think the doctor had mentioned it and I think coming from the doctor that’s looking after your child, you feel a bit more confident in what they’re . . . because you’ve got confidence in them, you’re gonna have confidence in what they’re coming to do.
P35, mother, telephone interview
Consistency in who discussed the pilot trial and sensitivity of approach were also valued by parents. These factors appeared to positively affect pilot trial decision-making processes, reducing the likelihood of confusion and helping parents decide to take part:
I don’t remember who it was but it was definitely a different person and that’s a bit confusing in hospital ‘cause just as a consultant changes shift, then a nurse changes shift, so it seemed like the research team changed shift and I think if it had have been the same person it would have been easier to associate that individual with the research.
P38, mother, telephone interview
I would say yeah, their manner did play a part in us going ahead with it. I think if they’d have sort of come in straight faced and very sort of to the point, then we may have been a bit like, ‘oh God, what’s this?’. But because they sort of made it really easy to understand and, um, had a good rapport and sort of made us feel a bit more light-hearted about our actual stay at the hospital, um, it, it sort of made it easier to say yes.
P01, mother, telephone interview
Participant written information and terminology
Parents thought that the PIS was comprehensive: ‘I think it was quite informative from what I can remember . . . it pointed out probably everything that we needed to know at that point’ (P35, mother, telephone interview), and generally clear: ‘It was really, really easy to read . . . very straightforward’ (P13, mother, telephone interview), despite using certain medical terminology throughout: ‘I did think, you know because I’m not a clinical person, that like the language that it was using, it was explained quite well. I did actually understand what it was all about’ (P26, mother, telephone interview). These views were also reflected in questionnaire responses, with 42 out of 45 (93.3%) parents agreeing with statement 3: ‘The information I received about FiSh was clear and straightforward to understand’. No parents disagreed with the statement (see Table 18). The majority of parents (43/45, 95.6%) also indicated that they had enough opportunities to ask questions about the FiSh trial (statement 5, Table 18).
Although two parents reported that further explanation about their child’s treatment in the context of the pilot trial would have been appreciated – ‘Um, explain it more. I don’t really know what happened to my daughter in that sense, I don’t know what’s sort of gone on’ (P19, mother) – the large majority indicated that detailed verbal explanation of the pilot trial had been given by site staff. This discussion, in addition to the written PIS, was valued by parents and appeared to assist their understanding of the pilot trial:
I think the conversation definitely helped. I don’t think I would have fully sort of understood it just from the paperwork, but the doctor sort of went through it and made it, elaborated on some of the points.
P01, mother, telephone interview
Some parents suggested providing a shorter and more basic pamphlet which clearly summarises key aspects of the trial prioritised by parents, including trial safety and aims:
All I needed to know was that it wasn’t detrimental, it wasn’t harmful. You know, my son’s part of a study but actually at the end of the day he’s going to be getting exactly the same as somebody else, you know, not on the study. It’s just that you’re documenting it all and it’s going somewhere. Perhaps that’s, you know, I don’t need any more information than my son is safe.
P40, mother, telephone interview
Finally, as discussed, a minority of parents described significant distress at finding out that their child had been treated for septic shock during the consent process, which included delivery of the pilot trial PIS. In addition to recommending that a member of the clinical team should initially discuss the child’s condition with the child’s parents, parents agreed with suggested changes to PIS sentence structure and terminology specific to septic shock. For example, parents recommended emphasising ‘suspected’ infection or ‘suspected’ sepsis throughout all written information materials alongside a clinical conversation that briefs parents on how some words used (i.e. sepsis or septic shock) may be upsetting:
That leaflet is great but perhaps, like you say, the wording needs to change a little bit with regards [sic] to the septic shock bit.
P34, mother, telephone interview
Certainly, even just the word, rather than, if I’d been told about it right from the beginning that it’s an infection ‘cause you can tell that, you know, the immediate blood results and the high temperature and whatever. But until the blood results come back they don’t necessarily know, it could just be viral, couldn’t it? But I guess from a parental point of view, just knowing that your child is being treated immediately for suspected infection, at the moment we don’t know what it is. But then if that comes back as being more serious than an infection of whatever, this is actually the blood, then you know. I think parents need a build-up of information so it’s not so shocking when it all gets slammed into them.
P40, mother, telephone interview
Outcome measures
As part of an iterative process, findings from the FiSh feasibility study (see Chapter 2) were incorporated into the topic guide as interviewing progressed, in order to further explore parents’ perspectives about important outcome measures. Consistent with methods described in Chapter 2, parents were asked to think about their experience of their child being admitted for severe infection and then prompted to share their views on what effects they hoped the fluid bolus treatment had to help their child (i.e. what indicators were most important to suggest that their child was getting better).
On average, parents discussed seven outcomes. Appendix 3 shows the full list of outcomes identified in the analysis of parent descriptions when asked directly which indicators were most important to them. The top prioritised outcomes, in order of the how commonly they were described as being most important, were as follows:
-
organ/physiological functioning, such as blood pressure, temperature and heart rate
-
looking and behaving more like self, including becoming ‘more alert’ (P25, mother)
-
machines as an indicator of improvement, including ‘the figures’ (P40, mother) and ‘the fewer machines that were operating’ (P38, mother)
-
time spent on treatments
-
long-term effects
-
survival
-
time spent in hospital.
Site staff perspectives
FiSh pilot trial site initiation training
Site staff focus groups and interviews began with questions aimed at exploring staff views and experiences about the FiSh pilot trial site initiation training, including whether or not they felt that the training had sufficiently prepared them for the pilot trial. Over half (17/27, 63%) had been trained at the site initiation meeting, whereas 10 out of 27 (37%) had received training from a member of their own site team, often the PI or research nurse. Three site staff had received both forms of training. Without exception, site staff stated that the training had prepared them for recruitment and consent in the pilot trial. As the following quotation illustrates, site PIs stated that the training had provided them with the confidence and knowledge to disseminate the pilot trial training to the rest of their team:
Yeah, it was absolutely fine . . . it made me feel confident enough to go and do the training for the extended team.
P02, focus group 1
Site staff valued having access to the training slides after the site initiation meeting so that they could refer back to them at a later date. Access to training materials had assisted the dissemination of training more widely across relevant departments and teams:
The mixture of having the information to refer back to, so the protocols that you’ve got along with the consent inform – you’ve got to have the background knowledge, so you’ve got to sort of attend the training to know what you’re doing to fully understand why you’re doing it, but also to have the reminder bits of the bits that are important to explain to the families at the point at which you’re getting consent.
P01, focus group 1
I think having that resource was really good and everything you need to deliver training is in there.
P22, telephone interview
Some staff stated that the training had outlined clear pilot trial procedures, including simple recruitment criteria, which they felt were important in ensuring the engagement of all site staff, particularly those who work in the ED:
Who you recruit, who you randomise into it is . . . they’ve made it so kind of simple . . . it was very clear as to which cohort of patients they were looking for.
P21, telephone interview
Yeah, it was simple, wasn’t it, and that’s why it encourages, in A&E [accident and emergency] for instance, to get involved because it’s not complicated to pick the children out and to put them into the study.
P02, focus group 2
Others stated that, before the training, they had no previous experience of RWPC, which had made them feel anxious about being involved in the pilot trial. However, the inclusion of specific training on this approach to consent, which incorporated parents’ perspectives from the FiSh feasibility study, appeared to help prepare them for consent discussions with families in the pilot trial:
Um, I hadn’t really been involved in retrospective consent before, so it was very useful kind of hearing them kind of talk about the kind of background of retrospective consent and why they were doing it, and all that kind of stuff. Because it’s really helped when I’ve gone and consented people, explaining why we’re doing it now and we didn’t do it yesterday type of thing. Um, so that probably was the more useful bit out of it, if that makes sense.
P21, telephone interview
The training package was very well put together and it had lots of relevance. Um, you know, supporting evidence for the rationale and lots of interesting sort of qualitative stuff about the methodology.
P22, telephone interview
I think with this particular study, what was quite concerning for most was the consenting part, obviously, because everybody was feeling how do we sort of speak to parents when they’re ready, so like we’ve asked them when their child has already been entered into the study in a way. And I think, having had the sort of the initiation day and sort of discussions, I think that gave me sort of a sound understanding of it.
P26, telephone interview
Screening pilot trial patients
Site staff described the methods used to help raise awareness of the pilot trial to ensure that every patient admitted to the ED or PAU (or equivalent) needing a fluid bolus was screened for the pilot trial. This included placing FiSh trial posters, stickers and memos where fluid was stored to remind staff to screen, as well as using computer systems to regularly search for potentially eligible recently admitted patients. In most sites, all clinical staff were described as being involved in screening. In some sites, a bleep system was successfully used, ‘so the A&E [accident and emergency] team know that in the event that there’s a child coming in who’s requiring fluids and sort of fits into the sort of FiSh criteria’ (P26, telephone interview), to inform research nurses that a consent discussion would be needed. Others stated that there were a few designated research nurses responsible for monitoring patients for eligibility to the pilot trial, as well as other studies being run at their site. One site used a rota system to ensure that someone was watching and checking computer systems for potentially eligible patients during the day:
We have a rota, and it’s Monday to Friday. There’s somebody watching from 8 till 6-ish, 8-ish, it depends. Some days it’s later than others.
P13, focus group 2
Site research staff stated that potentially eligible patients were sometimes missed at weekends, and that they would use computer systems to identify them the following week and retrospectively complete screening forms. During focus groups, staff members were asked to use the voting handsets to indicate whether or not the screening process could be improved. A total of 13 out of 20 (65%) clinicians indicated the process could be improved, whereas 7 out of 20 (35%) did not. Staff who took part in a telephone interview also stated that the screening process worked well, with few suggestions for improvement.
Suggestions for improvements to the screening process to help ensure that patients are not missed in the FiSh trial included ensuring 24/7 research cover in the ED, making sure that all staff were trained (including new staff on rotation) and ensuring that study posters are placed in the optimal location:
I mean I think it would be fantastic if the research cover could be a bit integrated into the areas that we’re recruiting because I do think that it raises awareness.
P16, focus group 2
So just in terms of improving it for the future, ‘cause we’ve said that there are multiple clinicians and nurses involved and they change every 6 months. Training the clinicians.
P20, focus group 3
We have got a big poster down there but maybe we need to think . . . of where it is. It’s on the back wall.
P10, focus group 2
Randomising pilot trial patients
The sealed-envelope method of randomisation was viewed as being simple and easy to use. During focus groups, staff members were asked to use the voting handsets to indicate whether or not they felt that the randomisation process could be improved. The majority (14/20, 70%) indicated that the randomisation process could not be improved. This view was shared by three out of seven (42.8%) staff who took part in a telephone interview.
The system is very much in place. I do like in ED the system where we use the sealed envelopes because it’s quick and easy.
P23, telephone interview
Problems identified with the process included an envelope being opened in error when a child did not meet the inclusion criteria or an envelope being opened too early. Staff stated that these issues occurred at the start of the pilot trial when some site members who ‘didn’t really understand the process’ (P25, telephone interview) were not completely familiar with the pilot trial protocol.
In one focus group, site staff stated that the treatment allocation had been misread. They suggested that, for the FiSh trial, the allocation should be in larger, bold text on the case record form, so that there is no doubt which treatment should be given:
They [ED clinician] were very confident they had done the right thing, but from the paperwork, it obviously was indicating they had not done the right thing, but they didn’t realise at all, because they felt, ‘we’ve recruited them to FiSh, isn’t that great?’ And we’ve given the second bolus and then we reassessed them and all of this, and it’s just such a great documentation but just unfortunately the wrong bolus.
P19, focus group 3
One staff member described how their localised protocol for sepsis treatment differed from the pilot protocol, which led to two patients being randomised in error:
I think we’ve been slightly overenthusiastic, if that makes sense. So, our local protocol is currently very heavily weighted towards lactates in sepsis, and if you’ve got high lactates, give fluid. Um, so we’ve had two children that, as per FiSh randomisation criteria, haven’t fitted needing a second fluid bolus. But as per our local sepsis protocol, have fitted being given a second bolus, i.e., their lactate hasn’t improved. And people have I guess kind of rightly in the face of sepsis more than 20 kilo[gram]s let’s randomise them into FiSh. But actually their cap [capillary] refill and their blood pressures have been fine. So I think, that’s kind of local protocol fighting the FiSh protocol. It’s not the fault of FiSh, it’s just it clashing with our local protocol.
P21, telephone interview
Protocol adherence
During the focus groups, staff members were asked to use the voting handsets to indicate whether or not they had experienced any difficulties adhering to the pilot trial protocol. Of these, 6 out of 20 (30%) staff indicated that they had experienced protocol adherence difficulties, whereas 5 out of 20 (25%) had not. However, 9 out of 20 (45%) staff opted not to respond to the question using their handsets, although insight was gained into protocol deviations at these sites through group discussion. Some staff (3/7, 43%) who took part in telephone interviews described some difficulty with protocol adherence. In addition to the randomisation issues described above, protocol deviations described by staff related to a lack of equipoise and difficulties in administering fluid boluses within 15-minute cycles.
Lack of equipoise because of a belief that a restrictive fluid bolus resuscitation is safer
Descriptions of non-adherence to the 20-ml/kg fluid bolus allocated treatment indicated a lack of equipoise among some staff. As the following quotation illustrates, some clinicians did not feel that it was safe to give a patient the 20-ml/kg fluid bolus treatment allocation because of fears about fluid overload:
I know that certainly one of the people who recruited said, ‘I recruited them and then thought, I really hope that it’s not going to be 20 [ml] per kilo’.
P18, focus group 3
The child was allocated to 10 or 20 [ml/kg], I think it was 10, but the doctor who was treating them decided I don’t want them to have 10, I want them to have 5. Um, and it’s just, it’s just trying to get that across where the 10 is safe, 20 is safe but you can’t deviate from what the protocol says.
P06, focus group 1
There was the ICU [intensive care unit] consultant down there and the ED consultant, and there was a problem with equipoise essentially, that they felt that, or they didn’t like the intervention so they just went off, they just went off-piste, gave less.
P02, focus group 2
As the following quotation illustrates, some staff had concerns about the safety of administering a 20-ml/kg fluid bolus allocation to smaller children:
I opened the envelope and it said 20 ml per kilo[gram] but at that time they’ve already decided 10 ml per kilo . . . And I think the child was really quite distressed and was working really hard, the mum was quite distressed so it was a very stressful situation . . . I think they would still have not given the 20 ml because of her size and because of also at that precise moment she was working hard, you know, I think they would have not . . . I don’t think it was clinically viable to give her 20 ml per kilo.
P26, telephone interview
Some staff suggested that smaller children (i.e. those < 3 kg) should be excluded from the FiSh trial:
So the 2.1 kilo[gram]s, I suppose just raises issues with the very small ones, whether they should exclude them in the next study.
P17, focus group 2
But I think having 20 ml per kilo for babies which are like 2 kilos or 3 kilos, I think that itself is quite difficult sometimes and we might probably have a lot more deviations. So that one size fits all, I think personally doesn’t work.
P26, telephone interview
A few staff described the ‘clinical creep’ of a restricted fluid bolus resuscitation without scientific evidence, highlighting the need to conduct the FiSh trial:
It’s interesting the patient we failed to consent at the weekend, the registrar gave 20 ml per kilo and then gave a further 10, rather than giving 20 and 20. I think that’s, you know, so what we’ve got here is examples of clinical creep of lower volume fluid resuscitation in the absence of evidence. And, I think, you know, it just highlighted for me that actually if we don’t do the study now, it’s going to, you know, we’re never going to know. And so it was, and I think I can probably persuade a parent about that, you know, that you just don’t know and we did the best.
P22, telephone interview
Lack of familiarity with the trial protocol caused by low recruitment rates
There was some discussion in two focus groups about there being not enough children randomised for clinical staff to become familiar with the protocol. This may be an additional factor contributing to protocol deviations:
If you don’t get them coming through then people lose familiarity with the processes, however much you know you train them.
P02, focus group 2
Yeah. I mean that’s not trivial, ‘cause actually there’s something about the event rates being high enough for enough staff being familiar with it. I think there were not quite as many as we hoped.
P02, focus group 2
Challenges in administering fluid bolus within specified time frames
Site staff were asked about the timing of the first fluid bolus administration after the randomisation envelope was opened. Most stated that the bolus was administered promptly, often within 15 minutes ‘[be]cause you’re wanting to get that bolus pretty quick’ (P06, focus group 1). Only one participant stated that they had found it easy to adhere to the protocol because ‘the criteria and protocol are straightforward’ (P23, telephone interview). Some spoke of how they had been initially concerned about being able to deliver fluid boluses within the specified time frame and how they ‘changed their practice’ and had ‘gotten faster’ (P01, focus group 1) at delivering the fluid bolus during the pilot trial. The majority of staff interviewed spoke of the challenges that they had experienced in administering the allocated fluid bolus within 15-minute cycles. Staff in one focus group stated that the 15-minute time frame did not intuitively fit with usual clinical practice:
Giving the bolus within 15 minutes of randomisation, I just don’t think that’s feasible. I don’t think I’ve looked at the CRF for a single child who has actually been achieved within the 15-minute window.
P19, focus group 3
Yeah, I mean that’s what I suppose I was referring to that process, trying to follow that protocol.
P20, focus group 3
It’s challenging.
P18, focus group 3
In real time doesn’t actually reflect practice.
P20, focus group 3
I mean you were constantly reassessing right, but maybe not 15 minutely.
P19, focus group 3
I think, yeah, so the rigid protocolisation of fluid therapy, although that was the way you were supposed to, had to, do it for the trial process, doesn’t intuitively fit with clinical practice.
P20, focus group 3
At two sites, staff described issues with the medical devices used to deliver a fluid bolus, such as a slow pump or small cannula:
Also we’ve got an issue at the minute in that our IVACs [IVAC® infusion pump (CareFusion, San Diego, CA, USA)] will only . . . the fastest they go is 600 ml an hour, which isn’t fast enough for anybody’s fluid bolus, whereas it used to go at 999 ml an hour. So that could give you, you know, your bolus every 15 minutes if you wanted. Whereas now the pumps are too slow, so I think people are resorting more to pushing because the pump isn’t an option until they’re reconfigured. Honestly, this pump thing has been a problem.
P08, focus group 1
The calibre of the cannula allows it to be possible but you can’t necessarily push it that quickly in through a cannula of that size in every child, depending on how it’s placed and things like that.
P18, focus group 3
Case report form
Many staff stated that it was difficult to complete the CRF in ‘real time’ while they were focused on resuscitating a child. Many felt that this had led to missing or incomplete CRF data, which were difficult for research nurses to complete retrospectively:
If they’re [ED staff] not recording things at the time that those boxes are wanting, then it makes it difficult for us [research nurses] to know what to put in those boxes.
P24, telephone interview
Many stated that research support in the ED was needed to help complete the CRF as, although the form was ‘really straightforward’ (P24, telephone interview), it was difficult and ‘time-consuming’ (P23, telephone interview) to complete while treating a critically ill child. As one ED clinician described, randomising and treating participants over the weekend without research support was stressful:
Both of our patients were randomised at the weekends, so there was no research input whatsoever it was all done off the back of the nurses that we had trained. Um, and so the only thing that they said is that it’s just a very, very scary experience and that, you know, they’re still trying to concentrate on the child. But they also want to get the trial right, so it’s a very stressful experience.
P25, telephone interview
Some staff suggested that additional training and experience of completing the CRF would help to improve the quality of data entry, whereas others felt that changes should be made to the recording of CRT to reflect a score rather than a number of seconds:
You put your capillary refill time in like point whatever of a second. That isn’t how people measure it. So that immediately put people off as a sort of first hurdle. And when you’ve sort of fallen at the first hurdle, I think people lost trust with it.
P27, telephone interview
Suggestions were made to help improve the format of the CRF, including having specific sections to complete in an emergency situation, as well as a clearer way of recording the time that a fluid bolus was administered:
It looks quite daunting, so maybe if there was a way of trimming it down so that only the bare essentials that are needed from the emergency, and then the rest could be picked up retrospectively from notes, from the research team. Which is kind of how we had to work the first one.
P25, telephone interview
One comment would be just to allow for a time box at the top of each of those sections so that, you know, I know it says 15 minutes but what time was that? Was it actually 15 minutes? And then that would probably help you with the data collecting as well. Um, I don’t, I didn’t feel like a flow from reassessing at the end of the 15 minutes to whether a trial bolus was given or not, I didn’t think that was a natural progression.
P19, focus group 3
Additional issues to consider for the FiSh trial protocol
Indices of shock: capillary refill time
Staff in one focus group questioned the use of CRT in seconds as an indication that a child was still in shock and the quality of CRF data for this element of the pilot trial:
I’m not quite sure how many people would have been using capillary refill [time] to help guide them as to whether a child remained shocked or not, and I don’t know how much good data you’ve got on the capillary refill time for all the children recruited.
P01, focus group 1
No, noticed also our local early warning score observation charts, um when you record the cap refill [time], if it’s recorded, it has a score rather than an actual number of seconds, so if it’s below 3 seconds you score nought, if it’s above 3 seconds, so 3 to 4 seconds, you score one, or two or so forth. So it’s not actually counting the number of seconds, so you don’t actually record um, a cap refill score, you record an early warning score based off of them. Does that make sense?
P02, focus group 1
Communication between teams
When staff members were asked to describe what they thought had been successful in the pilot trial, many referred to communication between hospital teams and units:
I think the fact that we have had really good relationships with ED and with the Paed[iatric]s Team has been really good.
P22, telephone interview
However, communication between hospitals and transfer teams was also one area which staff in one focus group felt could be improved in the FiSh trial. As the following quotations illustrate, there is a need to keep transfer teams informed when new sites open to recruitment:
I think you have the staggered start with some of the sites. I think what’s confusing, the confusion I think that from [transfer team name] end was knowing whether, say for example, [hospital] was live or not live, ‘cause the poster just said these are the four hospitals, if you get a referral from any of these four hospitals and they randomise the patient then you continue whatever is meant to be done . . . But [hospital] wasn’t live for a long time, so teams that got referrals from [hospital] started asking questions, saying, ‘oh have you randomised?’ And they didn’t know anything about it. It caused a bit of confusion. I don’t know what the easy answer is.
P17, focus group 2
Maybe we need to be better at communicating with you, which we probably should have done but we didn’t.
P11, focus group 2
Inclusion criteria
Two staff members suggested that a review of the inclusion criteria for the FiSh trial, if feasible, should consider extending eligibility to include children who deteriorate on a ward rather than restricting eligibility to children being randomised in the ED or PAU (or equivalent):
So I guess in terms of feeding that back, thinking about that for a bigger study I guess what we should think about . . . do we need to push out the window further to capture the patients that are the kind of slow-burner patients that end up going to the ward and then collapsing maybe beyond the first 4 hours. But then they haven’t been eligible to go into the study because they’re either beyond the first 4 hours, they’re not in A&E [accident and emergency] anymore. So we need to think a little bit about that I think, if there’s a bigger study.
P08, focus group 1
Experiences of research without prior consent in the pilot trial
Staff involved in other NIHR-funded critical care research stated that they were ‘less daunted’ by the use of RWPC in the pilot trial, as their previous experiences of such consent discussions with parents ‘had all gone without a hitch’ (P21, telephone interview). Staff at one site stated that they did not have any initial reservations about the use of RWPC in the pilot trial:
It was going to be quite an interesting experience and we were all quite excited about it.
P25, telephone interview
However, the majority of staff involved did not have prior experience of RWPC and described their initial anxieties about this consent method before the pilot trial began. As one PI described, the site team took their time to consider the consent process before agreeing to participate in the pilot trial:
We had to go through the process quite a lot, that was sort of the sticking point for the trial for quite a long time until they understood why it’s working that way.
P25, telephone interview
Many staff were concerned about how parents would react to being told that their child had been entered into the pilot trial without their prior consent, particularly if their child was still ‘really sick’ at the time of the conversation or ‘had died’ (P01, focus group 1). As described previously, the FiSh feasibility study findings, which showed how parents support the use of RWPC in the FiSh trial, appeared to have alleviated some initial staff anxieties:
I think I, along with the majority of people, was quite anxious about the honesty with the parents to begin with, was quite anxious about how they’d react, and I think the biggest thing that helped me was hearing that there’d been views from parents that they actually were happy about it.
P20, focus group 3
However, it was often the experience of conducting the pilot trial that helped to fully address any remaining anxieties about the use of this approach to consent in the pilot trial. Staff stated that parents had responded positively to the consent discussion and appeared to understand the reasons why informed consent could not have been sought prospectively. Furthermore, staff confidence in conducting the pilot trial appeared to increase over time:
And obviously the first deferred consent . . . you know it’s the see one, do one, type thing and then I think you realise, ‘oh, actually it is OK’.
P02, focus group 1
Both of them [parents of two recruited patients] were really, really understanding and recognised why we hadn’t asked them at the time.
P18, focus group 3
But as time has gone on, people have bought into it more and more and more.
P02, focus group 1
All staff supported the use of RWPC in the FiSh trial and thought that seeking prospective informed consent in the ED or PAU (or equivalent) when a child was being resuscitated would not be acceptable to families.
Staff viewed the consent form as simple and easy to use. One focus group participant suggested that the form should be one sided so that ‘nothing could be missed’ (P18, focus group 3). Finally, staff stated that child assent had most often not been sought as children were ‘too young’ (P27, telephone interview), or ‘didn’t have the capacity’ (P19, focus group 3) to be involved in pilot trial consent discussions with parents. Only one site member stated that ‘in retrospect, thinking about it, we should have thought to go back and get assent from that child once she’d been woken up’ (P18, focus group 3).
Summary of parents’ perspectives in the pilot trial
The integrated perspectives study findings add to the feasibility work (see Chapter 2) to show that the trial protocol was feasible and acceptable to parents. Consistent with the feasibility study findings and with associated guidance on RWPC,18,19,26,27 some parents of children randomised to the FiSh trial were initially surprised about the concept of RWPC. However, following tailored explanations from site staff about why prospective consent was not sought, all parents stated that they supported the RWPC approach in the pilot. Again, parents strongly valued the advancement of medical research and stated that they provided consent as a way to help other families and children in the future. 17,19,28,30,36,37
The use of the feasibility study findings to develop site staff initiation training on how to explain the pilot trial, including RWPC, appeared to assist parents’ understanding in the pilot trial. Importantly, concerns and misunderstandings about key aspects of the trial observed in parents who took part in the feasibility study were not observed in the pilot trial sample. Parents of children who took part in the pilot trial recalled site staff explanations about the nature of fluid bolus resuscitation and how this was standard care, not a new or novel treatment for septic shock, and about the lack of evidence for current recommended practice (i.e. 20 ml/kg). These verbal explanations, rather than written trial information, were prioritised by parents and helped to reassure them that there were good reasons for conducting this research without seeking prior informed consent, and that their child’s safety would not be compromised, as both treatment arms involved carefully titrated administrations of fluid. Nevertheless, researchers should bear in mind that parents may struggle to voice their questions or concerns during research discussions. 38,40 In line with previous studies in this area, these findings should be incorporated into staff training to encourage site staff involved in the FiSh trial to use open-ended questions and provide ample opportunities for questions,38,53 in order to identify and respond to parental concerns. 40,54
Importantly, trial acceptability and parents’ willingness to consent to their child’s information being included in the pilot trial appeared to hinge, in part, on the nature of the trial and their child’s recovery. 19,43 Not all children who experience septic shock will make a full recovery; therefore, site staff should be prepared to respond to parental questions or concerns related to the possibility that trial participation may have resulted in harm. 26,27 In line with our previous findings,19 parents viewed this study, involving variations in routinely used fluid bolus resuscitation therapy, as safer than trials of a new drug or surgery, and therefore as an acceptable study in which to use RWPC. Of added interest, several parents indicated that they consented in order to help their child, and some revealed a preference for one fluid bolus volume over another. This may indicate that parents did not understand that the optimal fluid bolus resuscitation volume for septic shock is not known, and, therefore, any direct benefit could not be assured. We recommend highlighting this in staff training programmes and including it as a key message to convey in consent for a full trial, if planned.
The professional background of the member of site staff approaching families for consent in the FiSh trial did not appear to affect significantly trial acceptability and willingness to provide consent – provided that adequate explanation was offered and familiarity was demonstrated with the child’s clinical case. Parental accounts of the pilot trial consent process supports existing guidance for site staff to check appropriate timing with the clinical team27 before approaching parents within 24–48 hours, when the acute situation has resolved. 17,19,30,38,41 Indeed, discussions during resuscitation were, for the most part, viewed as inappropriate unless conversations were kept brief, or parents had explicit trial-related questions.
Importantly, some parents became distressed when first hearing that their child had been treated for septic shock during the pilot trial discussion. We recommend that site staff check that a member of the clinical team has spoken to parents about their child’s condition and treatment before discussing the FiSh trial. To assist understanding and parent–practitioner communication, parents emphasised the need for clear and concise written information materials alongside verbal tailored explanation. 38,40 Similar to the findings in other clinical trials,19,39 parents recommended amending language used in both written and verbal explanations, and provided insight on what parents regard as appropriate language specifically in the context of septic shock. This recommendation may be related to a recent increase in media coverage on sepsis in children, with the term being associated with a high risk of mortality in children with severe infection. As a result, terminology such as ‘suspected’ sepsis or ‘suspected’ septic shock may not have been used sufficiently in the PIS and pilot trial verbal explanations. This small change to patient information could reduce parents’ initial anxieties relating to the severity of their child’s illness.
Finally, parents provided their views about what outcomes are most important in children with severe infection. In order of importance, parents reported that improvement in organ and physiological functioning, looking and behaving more like their normal selves, improvement in clinical figures and less need for machines could be used as indicators that fluid bolus resuscitation therapy was effective. Interestingly, these parents, who were interviewed shortly after their children’s treatment for septic shock, prioritised shorter-term rather than long-term outcomes, which were potentially most salient to them at the time of being interviewed. This is in contrast to the prioritisation of longer-term outcomes, such as disabilities, by parents in the feasibility study who were interviewed approximately 1 year after their children’s hospital admission. Child survival as an outcome was rarely discussed by parents in the pilot trial sample, all of whom had children who had survived. Such findings may suggest that parents’ prioritisation of outcomes may be influenced by their experience of their child’s illness, the survival of their child and the point at which they are asked about outcomes of importance in the course of their child’s illness. These combined qualitative findings should be incorporated into any future priority setting or core outcome set development work to ensure the selection and measurement of outcomes that are patient centred.
Overall, these findings highlight the value of doing pre-trial research to inform approaches to recruitment and consent in challenging trials. 55 Incorporation of these findings into staff initiation training, approaches to consent and information materials for the full FiSh trial, if recommended, will help parents to make informed decisions about the use of their children’s information.
Summary of site staff perspectives in the pilot trial
Overall, findings demonstrated that site staff supported the pilot trial and were willing to screen, recruit and randomise eligible patients to the FiSh trial. The study also provides new evidence in addition to the existing literature18–20,27 that can be used by practitioners involved in the design, ethics approval and conduct of children’s emergency care trials. Staff valued the FiSh pilot trial site initiation training, which sufficiently prepared them for recruitment and approach to consent. Access to site initiation training materials after the site initiation meeting helped PIs disseminate training across relevant departments and teams, a model that can be replicated in the FiSh trial. Similar to previous studies,18,20 confidence in conducting the pilot trial appeared to increase over the recruitment period, when staff became more familiar with the screening, randomisation and consenting processes.
Some staff without prior experience of RWPC were initially anxious about being involved in the pilot trial. Concerns were mainly related to how parents would react to being informed that their child had already been included in the pilot trial without their prior consent, potentially affecting the parent–practitioner relationship. 20 The inclusion of FiSh feasibility study findings in site initiation training, which showed parental support for the FiSh trial and its use of RWPC, helped to prepare staff for pilot trial consent discussions with parents. However, in staff without previous experience of this method, it was often the direct experience of conducting the pilot trial, and observing parents’ positive responses to the consent discussion, that provided staff with full confidence about the acceptability of this approach to consent in the FiSh trial. Consistent with previous findings,18,20 the views of practitioners with prior experience of RWPC should be considered in the design and conduct of future paediatric critical care trials, which should not focus solely on the beliefs of those without prior experience of this method of consent. Furthermore, future research would benefit from audio-recording RWPC discussions20,56 to improve future training by providing insights into how practitioners explain this to parents and to identify parental views and concerns. Suggestions for improvements to the screening process for the FiSh trial included increasing research support in the ED, ensuring that all relevant staff are fully trained when the medical staff rotation occurs every 6 months, and ensuring that posters and study materials are placed in the optimal location.
The pilot trial recruitment process and inclusion criteria were described as being simple to follow in an emergency situation. The majority of staff stated that the pilot trial randomisation process worked well, and, although some protocol deviations had occurred, these often occurred in the first few patients recruited at their site. Suggestions were made to improve the CRF for the FiSh trial. In addition to research support in the ED to help complete the CRF, staff suggested that the form should be reformatted, so that sections to be completed ‘in real time’ during resuscitation are clarified, or separated from sections that could be completed retrospectively from medical notes. As shown in other studies, staff can find recruitment difficult, with too many competing demands on their time,57 and this is particularly pertinent in a paediatric emergency care situation. Additional training would be needed on randomisation and CRF completion in a full FiSh trial, such as a simulation of the screening, randomisation and allocation procedures in the FiSh trial site initiation training. A step-by-step guide, or video, might help to improve the quality of CRF completion and, therefore, the quality of data obtained.
Staff found the administration of fluid bolus within the 15-minute time frame to be challenging. Although some stated that they had changed their practice and had been able to administer fluid boluses more quickly to adhere to the trial protocol, there were issues with medical devices (i.e. slow pumps and small cannulas) used to deliver fluid boluses. Such medical devices would need to be reviewed in preparation for the definitive FiSh trial.
Finally, telephone interviews and focus groups with staff indicated a lack of equipoise among some ED clinicians who had not adhered to the pilot trial protocol when a child had been randomised to a 20-ml/kg fluid bolus allocation because of concerns about fluid overload. Some staff also described a ‘clinical creep’ of a tendency to use restricted volume fluid bolus resuscitation without scientific evidence. Studies have consistently shown that trial recruitment can be negatively influenced by staff perceptions and experiences, including the decision not to recruit eligible participants because of a lack of equipoise with regard to particular treatments or individuals. 58–61 Although it appeared that only a small proportion of FiSh trial site staff were not in clinical equipoise, it has been suggested by other studies that this number would probably increase in the light of increasing use of one’s own preferred method of treatment and in the absence of forthcoming evidence. 61 Moreover, previous findings suggest that the role and importance of equipoise may not always be fully understood by all. 60 Taken together, these findings highlight the crucial need to conduct the definitive FiSh trial, incorporating explicit training on clinical equipoise, to identify the optimal fluid resuscitation volume for children with septic shock, to achieve the best possible outcomes and to inform clinical guidelines and practice in the NHS.
Overall, clear communication and ensuring that participants understand trial information and discussions is a challenge for practitioners in many trials. 62,63 This demonstrates the significant value and impact of our findings for future trials by providing recommendations for the enhancement of staff training in preparation for the conduct of ethically sound research in the paediatric critical care setting.
Strengths and limitations
This study involved parents from 10 of the 12 hospital sites that took part in the FiSh pilot trial, and staff from 9 of the 12 hospital sites that took part. Our sample size was relatively small. However, data saturation was reached,22,23 and we involved parents with direct experience of their child being enrolled in the FiSh pilot trial, as well as staff involved in recruitment and consent procedures. Data on whether or not more than one parent agreed to take part in the questionnaire element of the study were unknown; thus, a comparison of the consent rates for the interview element was not possible. We aimed to maximise diversity within our qualitative sample by selecting for interview bereaved and non-bereaved mothers and fathers who had consented and those who had declined to consent for the FiSh pilot trial, and doctors and nurses, from both high-recruiting and low-recruiting sites. However, opportunities to purposively sample fathers, bereaved parents and parents who declined to consent were limited as a result of high consent rates, particularly among mothers, and no deaths in the trial. Therefore, our understanding of these groups is limited and does not provide further insights beyond those described in the FiSh feasibility study (see Chapter 2) on the acceptability to parents of RWPC when a child has died.
Interestingly, all questionnaires received were completed on-site and all parents interviewed reported that they would be interested in taking part in future research. Therefore, our sample is likely to comprise parents with an interest in research. Although a high proportion of participating parents were from the highest recruiting site, interviews were purposively sampled across pilot sites to help ensure that there was sample variance and to capture different recruitment experiences.
Our insight into the consent process was also limited by the subjective recollections of parents and practitioners. Nevertheless, our qualitative data were strengthened by the integrated perspectives mixed-methods approach, providing a multiperspective understanding of parents’ and site staff opinions and experiences of RWPC, specifically in the context of the pilot trial.
As a final point, although attempts were made to include children in the discussion about the pilot trial, child assent was not taken by any of the participating sites. Practitioners attributed this limitation to the fact that children were often too young or too sick before discharge for a discussion about research. Thus, there is a need to consider how children and young people with capacity might be informed and engaged in consent discussions when they have recovered.
Chapter 6 Discussion and conclusions
Principal findings
Feasibility study
This was a qualitative interview study involving 21 parents (18 mothers and 3 fathers, and 7 of whom were bereaved) with children admitted to a UK ED with presumed septic shock in the previous 3 years. The study provided insight into the acceptability of the FiSh trial to parents. The majority of parents interviewed were unfamiliar with RWPC, yet supported its use in the FiSh trial. Parents were concerned about the change from currently recommended treatment, but were reassured by an explanation of the current evidence base, the rationale for fluid bolus therapy and the monitoring procedures that would be in place. Parents made recommendations about both the timing of consent and the patient information materials. Bereaved parents suggested that site staff should not attempt to obtain consent immediately after a child’s death, but supported a personalised postal ‘opt-out’ approach to RWPC for the FiSh trial. All parents interviewed in this phase of the project would have provided consent for the use of their child’s data in the FiSh trial, citing altruism as their main motive.
Parents also gave views on outcome measures that they considered to be most important in children with severe infection. Parents commonly discussed the following outcomes: long-term effects, including health and development; the child looking and behaving more normally; improvement in organ and physiological function; less need for medications and machines; and survival.
Pilot trial
The pilot trial was conducted in 13 hospitals from July 2016 to April 2017. Inclusion criteria were children presenting to ED with clinical suspicion of infection and shock after receiving 20 ml/kg of fluid. Children were randomised to 10- or 20-ml/kg fluid boluses every 15 minutes for 4 hours if in shock. Exclusion criteria were prior receipt of > 20 ml/kg of fluid, conditions requiring fluid restriction and full active resuscitation not being within the goals of care. A number of potential end points were collected. Seventy-five children were randomised: 40 into the 10-ml/kg fluid bolus group and 35 into the 20-ml/kg fluid bolus group. Two children were withdrawn as their parents were unable to be approached to provide consent, but no parents declined consent. Baseline characteristics were broadly similar, with some imbalance in age and weight. The majority of children (59% in the 10-ml/kg fluid bolus group and 74% in the 20-ml/kg fluid bolus group) required only a single trial bolus before shock resolved. The mean total volume of fluid received (in ml/kg) was significantly lower in the 10-ml/kg fluid bolus group than in the 20-ml/kg fluid bolus group. Fluid boluses were delivered in accordance with the protocol in 79% of children in the 10-ml/kg fluid bolus group and 55% in the 20-ml/kg fluid bolus group. There were no deaths or SAEs. Overall, 29% were admitted to PICU. Length of hospital stay, transfers to a PICU, length of stay in a PICU and days alive and not in a PICU up to 30 days post presentation to an ED did not differ significantly between the groups.
Integrated perspectives study
This was a questionnaire, interview and focus group study involving all hospitals participating in the pilot trial. Forty-five parents completed a questionnaire, 20 families and 7 site staff (3 doctors and 4 nurses) were interviewed and 20 site staff (10 doctors and 10 nurses) participated in three focus groups. One-third of parents were surprised to discover that their child had been entered into the pilot trial without prior consent. Nevertheless, all parents supported the trial because they were reassured by explanations about why prior informed consent could not be sought, the nature of fluid bolus resuscitation and lack of evidence for current recommended practice. Parents suggested improvements to how the trial should be introduced, including how ‘suspected’ sepsis should be emphasised. Parents prioritised shorter-term physiological outcomes, such as reduced heart rate, over longer-term health outcomes, such as disabilities. This is in contrast to the prioritisation of longer-term outcomes by parents in the feasibility study who were interviewed approximately 1 year after their child’s hospital admission. Child survival as an outcome was rarely discussed by parents of children who took part in the pilot study, none of whom was bereaved. Such findings may suggest that parents’ prioritisation of outcomes may be influenced by their child’s illness experience, survival and the point at which they are asked about outcomes of importance in the course of their child’s illness. These combined qualitative findings should be incorporated into any future patient-centred outcome priority-setting exercise or core outcome set development work to ensure that the selection and measurement of outcomes for sepsis research are patient centred. Site staff made some suggestions to improve the protocol and the training package. There was a lack of equipoise among some parents and some clinical staff who favoured a 10-ml/kg fluid bolus volume. However, overall, both clinical and research staff supported the FiSh trial, with their confidence in delivering the protocol increasing over time.
Interpretation
The combined findings on whether or not the FiSh trial is both feasible and acceptable were that, in general, parents whose children had been admitted to hospital with severe infection supported the proposed FiSh trial, including the use of RWPC. The findings from the feasibility study were used to develop the information sheets and consent forms, the pilot trial protocol, site initiation training and outcome measures. Acceptability to families was confirmed by the findings from the pilot trial; although two patients were withdrawn post randomisation, this was as a result of it being inappropriate to approach the parents to obtain consent, and the remainder were consented into the study successfully. The FiSh trial was seen to be acceptable by parents in the integrated perspectives study.
Logistical difficulties, including delays to local approvals, meant that the pilot trial started 6 weeks later than expected, with the first site opening on 13 July 2016 rather than 1 June 2016 as originally planned. Rather than all sites opening simultaneously, sites opened over a period of 4 months, with the final site opening on 23 November 2016. The final two sites that were opened were replacements for two originally planned sites that were subsequently found not to have adequate research infrastructure to support the study. The delay in sites opening was mitigated as much as possible by extending the recruitment period by 1 month; however, total study site months at the conclusion of the trial were 80 rather than 108. This reduced the anticipated recruitment total from 108 to 80. In this challenging context, the trial was successfully delivered, with screening logs showing 83 eligible patients across the study sites over the study period. Out of 83 eligible patients, 75 (90.4%) were randomised. Nine of these randomised participants were deemed to have been randomised in error (coincidentally, all were allocated to the 10-ml/kg fluid bolus group). The main reason for this was one site applying its own criteria, based on lactate, to identify children in shock rather than those specified in the protocol.
The intervention was delivered in accordance with protocol in the majority of participants. However, there were a substantial minority of participants in whom protocol deviations occurred, with more deviations in the 20-ml/kg fluid bolus group than in the 10-ml/kg fluid bolus group. One reason for non-adherence was that some clinicians may lack equipoise, apparently preferring restrictive 10-ml/kg fluid boluses in some scenarios, despite 20 ml/kg being the current recommended practice. There were also difficulties delivering the 20-ml/kg fluid boluses within 15 minutes. Furthermore, the randomised population were also significantly less unwell than expected when the study was designed, with only a minority of patients receiving more than one fluid bolus or being admitted to a PICU.
These findings were further supported by the integrated perspectives study, which indicated that the FiSh trial was acceptable to parents and site staff involved in the pilot trial, although some problems with equipoise were revealed. If a definitive trial was to be delivered, the findings would have been used to improve site initiation training and to refine the study protocol, aiming to increase the confidence of site staff in screening, recruitment and consent, and to help parents to make informed decisions about the use of their child’s information.
Strengths and limitations
Feasibility study
Overall, the findings supported the proposed FiSh pilot trial, including the use of RWPC. Although the sample size was small, data saturation was achieved and, importantly, both bereaved and non-bereaved parents were included. However, as the majority of parents were recruited via social media, our sample may have comprised parents with a pre-existing interest in research and may not be representative of the parents of children who might be recruited in a trial. Additional qualitative research was therefore conducted within the FiSh pilot trial to confirm the findings in the feasibility study and to understand the perspectives of site staff.
Pilot randomised controlled trial
The pilot trial was successfully delivered, with screening logs showing that > 90% of eligible patients were recruited. Twelve patients were ineligible as a result of receiving > 20 ml/kg of fluid; the opportunity may have been missed to randomise these patients earlier. It is also possible that these may have represented a subset of sicker patients. Despite the logistical challenge posed by setting up 13 study sites for a short pilot, recruitment was very close to the target of one child per site per month. The process of RWPC worked smoothly, although it was not tested in the emotional aftermath of a child’s death, as, thankfully, no patients died during the course of the study. The intervention was by and large delivered successfully, with good separation being achieved in terms of volume of fluid delivered. Potential outcomes were successfully collected as part of the trial data collection, with more data coming from the linkage with PICANet. Some participants had missing data from the PICU stay because of referral to a PICU that was not in the pilot trial. It would be vital to have local approvals for all PICUs for a larger trial.
However, there were significant logistical problems in setting up some of the smaller study sites, two of which were discovered during this process to have inadequate research infrastructure to support the study. This necessitated the recruitment of another two sites in the middle of the pilot trial. Although the overall recruitment rate was just under the anticipated recruitment rate of one patient per site per month, with a final rate of 0.9 patients per site per month, recruitment was driven mainly by three study sites (Southampton, Chelsea and Westminster, and Bristol), which between them recruited 38 participants. The remainder of the study sites struggled to recruit more than a handful of participants each.
In the event of a larger study looking at fluid bolus volumes being undertaken in the UK, it would be important to consider those sites that might be best suited for such a study. These would probably be large university hospitals with a children’s ED, an integrated PICU/high-dependency unit and good pre-existing research infrastructure, rather than small district hospitals.
Most importantly, the population was not as unwell as expected. The mean PIM2r score (a PICU severity of illness score giving a population risk of mortality) was 2.1%. There were 51 out of 73 (70%) participants who required only one study fluid bolus and only 8 out of 73 (11%) participants required more than two study boluses. Only 21 out of 73 patients (29%) were transferred to a PICU. All the participants survived. Furthermore, despite many guidelines suggesting that elective intubation and ventilation should be considered when ≥ 40 ml/kg of fluid has been administered in this clinical setting, because of the risk of pulmonary oedema, out of the 46 children who received ≥ 40 ml/kg (±10%) of fluid, including the 20 ml/kg pre randomisation, 16 were transferred to a PICU. Thus, not only were the participants less unwell than expected, but many of those who were apparently unwell enough to merit referral to a PICU, or discussion with a PICU retrieval team, improved and remained under the care of general paediatricians. As the intervention was not blinded, it is possible that potential outcome measures, such as admission to a PICU, may be subject to bias either directly through knowledge of the allocated treatment (performance bias) or through the decision to admit to a PICU being influenced by treatment factors including the volume of fluid received. It was not considered practical to blind clinicians to the allocated treatment in the ED setting.
Overall, the pilot trial was successfully delivered. However, there were more protocol deviations in the 20-ml/kg fluid bolus group than in the 10-ml/kg fluid bolus group and qualitative data implied that some clinicians may lack equipoise. The randomised population was less unwell than expected and only a minority of participants received more than one fluid bolus or ended up in a PICU. Thus, the trial as currently conceived is not feasible.
Possible reasons for the patients being less unwell than expected in this study are explored below.
Integrated perspectives study
This study involved parents from 10 of the 12 hospital sites that randomised participants to the FiSh pilot trial and staff from 9 of the 12 hospitals. Although the sample size was relatively small, data saturation was reached. However, opportunities to sample fathers, bereaved parents and parents who declined to consent were limited, as only one father consented to be interviewed, all children recruited into the pilot trial survived and no parents declined consent. This limits our understanding of these groups and means that firm recommendations cannot be made on how to conduct RWPC when a child has died. However, our qualitative data were strengthened by the integrated perspectives mixed-methods approach, providing an overall understanding by investigating the experiences and opinions of both parents and site staff.
Although attempts were made to include children and young people in the discussion about the pilot trial, child assent was not taken by any of the participating sites, because children were either too young or too sick before discharge for a discussion about research. Thus, children’s views on the FiSh trial remain unknown.
Overall results in context
Parent and site staff perspectives: research without prior consent
Our findings add to the existing literature on RWPC in paediatric emergency and critical care settings: they provide an important contribution and demonstrate the value of using qualitative methods in considering the family-centred parameters needed to inform the design and conduct of challenging trials. 53,64
Other studies looking at fluid bolus therapy
Current guidelines recommend aggressive fluid resuscitation in children with severe sepsis and septic shock. The latest ACCM-PALS guidelines65 recommend rapid fluid bolus resuscitation (of 20 ml/kg, up to a total of 60 ml/kg over the first 15 minutes) during the first hour of emergency assessment and treatment.
The literature supporting the ACCM-PALS recommendations consists of retrospective studies involving small numbers of children. 6–8 There are no data from prospective clinical trials. However, there is now increasing evidence that fluid overload in critical illness is deleterious. In Africa, the recent FEAST9 multicentre RCT reported a 30% increase in mortality associated with fluid bolus resuscitation. A recent systematic review66 identified only one RCT, other than FEAST, that investigated different size fluid bolus therapy in children presenting to EDs in septic shock. 13 This was a single-centre study in India, randomising 147 children to 40 ml/kg of fluid over 15 minutes followed by dopamine, compared with 20 ml/kg of fluid over 20 minutes up to a maximum of 60 ml/kg over 1 hour followed by dopamine. No difference was found in any outcome measure between the groups, although the 40-ml/kg fluid bolus group had a higher incidence of hepatomegaly than the 20-ml/kg fluid bolus group, indicating fluid overload.
Thus, at the time of writing, the optimum strategy for fluid bolus resuscitation in children with septic shock remains unknown in the developed world. One paediatric fluid resuscitation RCT, other than the FiSh trial, is currently in progress. The SQUEEZE14 trial is a Canadian RCT that is comparing fluid with early inotrope therapy. Results are eagerly awaited. In PICU, many observational studies67–70 have recently shown that fluid overload is associated with morbidity and mortality in a variety of clinical settings.
Our data also suggest that the FEAST9 trial, along with the PICU studies, may have already affected clinician equipoise in the UK, with a preference for less generous fluid boluses than those recommended in the current guidelines already entering routine clinical practice. Further work is thus required to confirm equipoise still exists before a large-scale trial of fluid bolus volumes is undertaken in the UK.
The impact of vaccination
Only 32 out of 73 (44%) participants in this study had a confirmed bacterial infection in a sterile site or a viral infection. Of these infections, 18 were viral, the majority of which were respiratory and likely, therefore, to have minimal cardiovascular dysfunction. Thirteen participants had confirmed bacterial infection in sterile sites, only one of which (a single case of meningococcal sepsis) was preventable by a vaccine. Such results would until recently have been impossible to imagine – that, over a 9-month period in the winter in 12 UK EDs, so few children would present with confirmed bacterial sepsis. A few years ago, the incidence of meningococcal and pneumococcal sepsis, in particular, was much higher (about 600 cases of group B meningococcus were reported in 201371). Although more recent absolute figures are not available, the incidence is now much lower, following the introduction of group B meningococcal vaccination in September 2015. Since then, there has been a 50% incidence rate ratio reduction in group B meningococcal cases in vaccine-eligible children. 72 The incidence of group C meningococcal infection was already very low following the introduction of the vaccine in 1999. In relation to pneumococcal disease, in 2006, a seven-valent vaccine, pneumococcal conjugate vaccine (PCV7), was added to routine childhood immunisation, and in 2010 a PCV containing polysaccharide from 13 common capsular types (PCV13) replaced PCV7. Before the introduction of PCV7, the mean annual incidence of invasive pneumococcal disease was 44.4 per 100,000 children < 2 years of age, with a mean case fatality rate of 3.5%. Following the introduction of PCV7 and now PCV13, the mean annual incidence of invasive pneumococcal disease is currently at around 10–12 per 100,000 children. The impact of the comprehensive childhood vaccination now offered in the UK on vaccine-preventable infections cannot, thus, be understated.
Furthermore, there has been a recent trend in many hospitals to implement ‘sepsis bundles’ to help staff recognise and manage early sepsis. In the UK, evidence-based guidelines for recognition and early management of children with sepsis have been produced and disseminated by the National Institute for Health and Care Excellence (NICE). 73 Thus, it is also possible that some children presented early to the ED, were recognised early, treated promptly and responded to treatment. This would reduce the severity of illness in the population as a whole and also reduce the need for fluid bolus therapy. Unfortunately, data that might determine whether or not this was the case were not collected as part of the FiSh feasibility study; hence, this possibility must remain a speculation.
Overall, it is most likely that vaccination, and possibly improved recognition and management of early sepsis, is the cause of both the unexpected low severity of illness of the population of children randomised into the FiSh pilot trial and the difficulties many sites had in recruiting more than a handful of participants.
Measurement against the progression criteria
Progression criterion 1
-
Formal sign-up of sites (the required number of hub-and-spoke models) to achieve the overall sample size based on the observed recruitment rate, as determined by the pilot trial objectives concerning identification, recruitment rate, acceptability of deferred consenting procedures, selection of the patient-centred primary outcome measure and sample size estimation.
-
Response: qualitative work showed that there was buy-in for the FiSh trial, but as a result of the full trial not being deemed feasible there has been no formal sign-up of sites.
Progression criterion 2
-
The recruitment rate achieved in the pilot trial (taking into account the loss of participants as a result of declining deferred consent) was consistent with achieving the overall sample size required (as determined by the objectives concerning selection of the patient-centred primary outcome measure and sample size estimation) in an acceptable time frame from the sites giving formal sign-up in the first feasibility objective.
-
Response: the overall recruitment rate in the pilot trial was very close to anticipated, although there was considerable variation across sites. However, the severity of illness of the participants recruited was much lower than expected and, if the inclusion criteria were to be changed to restrict to a more severely ill cohort, then the number of eligible children would inevitably be reduced and the feasibility of completing recruitment in an acceptable time frame may be brought into question.
Progression criterion 3
-
At least 80% of fluid bolus resuscitation in the external pilot study was delivered at the correct volume and timing, ± 10%, as determined by the external pilot study objective concerning adherence.
-
Response: adherence to both volume and timing was close to achieving the 80% threshold in the 10-ml/kg fluid bolus group but was substantially worse in the 20-ml/kg fluid bolus group, suggesting that, despite current recommendations, rapid infusion of 20-ml/kg, fluid boluses does not represent current usual practice in the UK.
Progression criterion 4
-
The absolute total volume of fluid administered during the first hour and during the first 4 hours is lower (by ≥ 25%) in the more restrictive fluid bolus resuscitation group than in the current recommended fluid bolus resuscitation group, as determined by the external pilot study objective concerning separation.
-
Response: the absolute total volume of fluid administered during the first hour was only 7% lower in the 10-ml/kg fluid bolus group than in the 20-ml/kg fluid bolus group, and by the end of the 4-hour intervention period it was 23% lower. However, once imbalance in children’s weights between the groups was taken into account, this criterion was achieved, with the volume of fluid (in ml/kg) being 35% lower in the first hour and 44% lower over the entire 4-hour period.
Recommendations for research
Recommendation 1: the definitive FiSh trial, with its current design, should not be conducted
The full FiSh trial, with the current design, is not feasible, for the reasons given above. However, the optimal volume for fluid bolus resuscitation in the developed world remains unknown and infection is still an important cause of death in young children, even in the post-vaccine era. Thus, it may be that a different trial, with different inclusion criteria, or a different intervention, would be feasible and would answer this question. Possibilities might include adjusting the inclusion criteria to randomise patients to the trial only after receiving 40-ml/kg fluid bolus resuscitation, to select a more unwell population. Disadvantages of this approach would be the lower number of eligible patients in each centre and the difficulty in achieving separation. Alternatively, an intervention that compared usual management with a fluid-sparing/early inotrope arm, such as that currently being tested in the SQUEEZE trial,14 might have more chance of success.
Recommendation 2: further observational and epidemiological research should be conducted
Further observational and epidemiological research is required, both in EDs and in PICUs, to determine both current management and if there is still a population of children in the UK in whom a study looking at fluid bolus volumes could usefully be conducted. This would require the prospective collection of data on children presenting to EDs with suspected sepsis. In order to succeed, such a project would need a collaborative approach involving paediatric EDs, Acute Paediatric Units and PICUs. In relation to PICUs, it should be possible to perform an audit, linking to – and using – PICANet data collection systems, to determine the number of children on PICUs with severe sepsis and septic shock, where they are being admitted from (ED or ward) and their comorbidities.
Recommendation 3: further understanding of the moving clinical landscape is needed
Further qualitative work is required to determine whether or not clinical equipoise still exists among front-line professionals dealing with acutely ill children in relation to the research question. Our data seem to suggest that the equipoise, which previously existed, may already be lost in favour of clinicians using smaller, more limited, fluid bolus volume resuscitation than that currently ‘officially’ recommended. This may be a consequence of the FEAST9 trial or of smaller observational studies demonstrating a link between fluid bolus administration or fluid overload and poor outcomes in critically ill children.
Acknowledgements
We wish to thank the NIHR Health Technology Assessment programme for funding this trial. We also wish to thank all the patients and staff from all the sites that participated in the trial. We would like to thank all the staff at ICNARC, with special thanks to Dr Sheila Harvey and Amy Andrews.
Research staff at participating sites
We acknowledge that there have been many other individuals who made a contribution within the participating sites. It is impossible to thank everyone personally; however, we would like to thank research staff at the following hospitals:
Bristol Royal Hospital for Children (Pauline Jackson, Anna Laskey, Mark Lyttle, Alice Smith and Peter Davis); Chelsea and Westminster Hospital (Sara Edwards, Victoria Wilson and Hester Yorke); Great Ormond Street Hospital for Children (Lauran O’Neill, Mark Peters and Fran Standing); Musgrove Park Hospital (Debs Heard, Jayne Foot and Esther Wilson); Northwick Park Hospital (Sathish Bangalore and Katie Blundell); Queen Alexandra Hospital (Simon Birch, Alan Charters, Zoe Daly and Kat Ellinor); Royal Devon and Exeter Hospital (Caroline Harrill, Nicola Jones, Simon Parke, Jennie Small and Su Wilkins); Royal United Hospital (Elizabeth Gilby and Anna Wedgwood); Salisbury District Hospital (Jim Baird, Nick Brown, Sarah Diment and Dee Mead); Southampton General Hospital (Jane Bayreuther, Ruth Ensom, Lisa Fairhead and John Pappachan); St Mary’s Hospital (David Inwald, Rikke Joergensen, Farhana Abdulla and Salina Persand); Watford General Hospital (Solomon Kamal-Uddin and Jess Fitzsimmons); and Whittington Hospital (Sheik Pahary and Kerry Robinson).
Study management team
Ruth Canter (study manager), Andrew Fleming (senior data manager), Dr David Harrison (senior statistician), Nick Hudson (data manager), Dr David Inwald (chief investigator), Paul Mouncey (senior researcher), Dr Caitlin O’Hara (research associate), Professor Kathryn Rowan (CTU director), Steven Saunders (research assistant), Dr Kerry Woolfall (co-investigator) and Zohra Zenasni (study statistician).
Study Management Group
Ruth Canter (study manager), Anjali Carter (co-investigator), Dr David Harrison (senior statistician), Dr David Inwald (chief investigator), Dr Nicola Jones (co-investigator), Dr Michael Levin (co-investigator), Dr Mark Lyttle (co-investigator), Paul Mouncey (senior researcher), Dr Simon Nadel (co-investigator), Dr Caitlin O’Hara (research associate), Professor Mark Peters (co-investigator), Professor Kathryn Rowan (CTU director) and Dr Kerry Woolfall (co-investigator).
Trial Steering Committee
Dr Stephen Playfor (independent, chairperson), Emma Bull (independent), Dr David Inwald (chief investigator), Jason Pott (independent), Dr Damian Roland (independent), Dermot Shortt (independent), Dr Claire Snowden (independent) and Dr Kerry Woolfall (co-investigator).
Data Monitoring and Ethics Committee
Dr Colin Powell (chairperson), Dr Duncan Macrae and Rejina Verghis.
Contributions of authors
David Inwald conceived and designed the pilot trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically revised the manuscript.
Ruth R Canter managed the pilot trial, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Kerry Woolfall conceived and designed the qualitative elements of the study, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically revised the manuscript.
Caitlin B O’Hara carried out the qualitative elements of the study, contributed to the acquisition, analysis and interpretation of the data, and drafted and critically revised the manuscript.
Paul R Mouncey contributed to the design of the trial and the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Zohra Zenasni contributed to the analysis and interpretation of the data and drafted and critically reviewed the manuscript.
Nicholas Hudson contributed to the analysis and interpretation of the data and critically reviewed the manuscript.
Steven Saunders contributed to the acquisition, analysis and interpretation of the data and critically reviewed the manuscript.
Anjali Carter provided parental input into the study, contributed to the acquisition, analysis and interpretation of the data, and critically reviewed the manuscript.
Nicola Jones provided parental input into the study, contributed to the acquisition, analysis and interpretation of the data, and critically reviewed the manuscript.
Mark D Lyttle contributed to the design of the trial and the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Simon Nadel contributed to the design of the trial and the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Mark J Peters contributed to the design of the trial and contributed to the acquisition, analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
David A Harrison contributed to the design of the study and the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Kathryn M Rowan designed the study, contributed to acquisition, analysis and interpretation of the data, and drafted and critically revised the manuscript.
Publications
O’Hara CB, Canter RR, Mouncey PR, Carter A, Jones N, Nadel S, et al. A qualitative feasibility study to inform a randomised controlled trial of fluid bolus therapy in septic shock. Arch Dis Child 2018;103:28–32.
Inwald DP, Canter R, Woolfall K, Mouncey P, Zenasni Z, O'Hara C, et al. Restricted fluid bolus volume in early septic shock: results of the Fluids in Shock pilot trial. PERUKI (Paediatric Emergency Research in the UK and Ireland) and PICS SG (Paediatric Intensive Care Society Study Group) [published online ahead of print August 7 2018]. Arch Dis Child 2018.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 2010;375:1969-87. https://doi.org/10.1016/S0140-6736(10)60549-1.
- 2014 Annual Report – Tables and Figures. Leeds and Leicester: University of Leeds and University of Leicester; 2014.
- Inwald DP, Tasker RC, Peters MJ, Nadel S. Paediatric Intensive Care Society Study Group (PICS-SG) . Emergency management of children with severe sepsis in the United Kingdom: the results of the Paediatric Intensive Care Society sepsis audit. Arch Dis Child 2009;94:348-53. https://doi.org/10.1136/adc.2008.153064.
- Vyse A, Anonychuk A, Jäkel A, Wieffer H, Nadel S. The burden and impact of severe and long-term sequelae of meningococcal disease. Expert Rev Anti Infect Ther 2013;11:597-604. https://doi.org/10.1586/eri.13.42.
- Brierley J, Carcillo JA, Choong K, Cornell T, Decaen A, Deymann A, et al. Clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock: 2007 update from the American College of Critical Care Medicine. Crit Care Med 2009;37:666-88. https://doi.org/10.1097/CCM.0b013e31819323c6.
- Carcillo JA, Davis AL, Zaritsky A. Role of early fluid resuscitation in pediatric septic shock. JAMA 1991;266:1242-5. https://doi.org/10.1001/jama.1991.03470090076035.
- Booy R, Habibi P, Nadel S, de Munter C, Britto J, Morrison A, et al. Meningococcal Research Group . Reduction in case fatality rate from meningococcal disease associated with improved healthcare delivery. Arch Dis Child 2001;85:386-90. https://doi.org/10.1136/adc.85.5.386.
- Han YY, Carcillo JA, Dragotta MA, Bills DM, Watson RS, Westerman ME, et al. Early reversal of pediatric-neonatal septic shock by community physicians is associated with improved outcome. Pediatrics 2003;112:793-9. https://doi.org/10.1542/peds.112.4.793.
- Maitland K, Kiguli S, Opoka RO, Engoru C, Olupot-Olupot P, Akech SO, et al. Mortality after fluid bolus in African children with severe infection. N Engl J Med 2011;364:2483-95. https://doi.org/10.1056/NEJMoa1101549.
- Hilton AK, Bellomo R. A critique of fluid bolus resuscitation in severe sepsis. Crit Care 2012;16. https://doi.org/10.1186/cc11154.
- Myburgh J, Finfer S. Causes of death after fluid bolus resuscitation: new insights from FEAST. BMC Med 2013;11. https://doi.org/10.1186/1741-7015-11-67.
- Ford N, Hargreaves S, Shanks L. Mortality after fluid bolus in children with shock due to sepsis or severe infection: a systematic review and meta-analysis. PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0043953.
- Santhanam I, Sangareddi S, Venkataraman S, Kissoon N, Thiruvengadamudayan V, Kasthuri RK. A prospective randomized controlled study of two fluid regimens in the initial management of septic shock in the emergency department. Pediatr Emerg Care 2008;24:647-55. https://doi.org/10.1097/PEC.0b013e31818844cf.
- Parker MJ, Thabane L, Fox-Robichaud A, Liaw P, Choong K. Canadian Critical Care Translational Biology Group . A trial to determine whether septic shock-reversal is quicker in pediatric patients randomized to an early goal-directed fluid-sparing strategy versus usual care (SQUEEZE): study protocol for a pilot randomized controlled trial. Trials 2016;17. https://doi.org/10.1186/s13063-016-1689-2.
- Advanced Paediatric Life Support n.d. www.alsg.org/en/files/PFactsheet.pdf (accessed September 2018).
- Inwald DP, Butt W, Tasker RC. Fluid resuscitation of shock in children: what, whence and whither?. Intensive Care Med 2015;41:1457-9. https://doi.org/10.1007/s00134-015-3905-z.
- Molyneux S, Njue M, Boga M, Akello L, Olupot-Olupot P, Engoru C, et al. ‘The words will pass with the blowing wind’: staff and parent views of the deferred consent process, with prior assent, used in an emergency fluids trial in two African hospitals. PLOS ONE 2013;8. https://doi.org/10.1371/journal.pone.0054894.
- Woolfall K, Frith L, Gamble C, Gilbert R, Mok Q, Young B. CONNECT advisory group . How parents and practitioners experience research without prior consent (deferred consent) for emergency research involving children with life threatening conditions: a mixed method study. BMJ Open 2015;5. https://doi.org/10.1136/bmjopen-2015-008522.
- Woolfall K, Young B, Frith L, Appleton R, Iyer A, Messahel S, et al. Doing challenging research studies in a patient-centred way: a qualitative study to inform a randomised controlled trial in the paediatric emergency care setting. BMJ Open 2014;4. https://doi.org/10.1136/bmjopen-2014-005045.
- Woolfall K, Frith L, Gamble C, Young B. How experience makes a difference: practitioners’ views on the use of deferred consent in paediatric and neonatal emergency care trials. BMC Med Ethics 2013;14. https://doi.org/10.1186/1472-6939-14-45.
- Knapp P, Raynor DK, Silcock J, Parkinson B. Can user testing of a clinical trial patient information sheet make it fit-for-purpose? – a randomized controlled trial. BMC Med 2011;9. https://doi.org/10.1186/1741-7015-9-89.
- Baker SE, Edwards R. How Many Qualitative Interviews is Enough?. Southampton: National Centre for Research Methods; 2012.
- Guest G, Bunce A, Johnson L. How many interviews are enough? An experiment with data saturation and variability. Field Methods 2006;18:59-82. https://doi.org/10.1177/1525822X05279903.
- Mays N, Pope C. Qualitative research in health care. Assessing quality in qualitative research. BMJ 2000;320:50-2. https://doi.org/10.1136/bmj.320.7226.50.
- Data Protection Act 1998. London: The Stationery Office; 1998.
- Woolfall K, Frith L, Dawson A, Gamble C, Lyttle MD, Young B. CONNECT advisory group . Fifteen-minute consultation: an evidence-based approach to research without prior consent (deferred consent) in neonatal and paediatric critical care trials. Arch Dis Child 2015;101:49-53. https://doi.org/10.1136/archdischild-2015-309245.
- Young B, Gamble C, Frith L, Dawson A, Parker M, Breen R, et al. Research Without Prior Consent (Deferred Consent) in Trials Investigating the Emergency Treatment of Critically Ill Children: CONNECT Study Guidance. Liverpool: University of Liverpool; 2015.
- Morris MC, Nadkarni VM, Ward FR, Nelson RM. Exception from informed consent for pediatric resuscitation research: community consultation for a trial of brain cooling after in-hospital cardiac arrest. Pediatrics 2004;114:776-81. https://doi.org/10.1542/peds.2004-0482.
- Strauss A, Corbin J. Basics of Qualitative Research: Techniques and Procedures for Developing Grounded Theory. Thousand Oaks, CA: Sage Publications Ltd; 1998.
- Maitland K, Molyneux S, Boga M, Kiguli S, Lang T. Use of deferred consent for severely ill children in a multi-centre phase III trial. Trials 2011;12. https://doi.org/10.1186/1745-6215-12-90.
- Braun V, Clarke V. What can ‘thematic analysis’ offer health and wellbeing researchers?. Int J Qual Stud Health Well-Being 2014;9. https://doi.org/10.3402/qhw.v9.26152.
- Braun V, Clarke V. Using thematic analysis in psychology. Qualitative Res Psychol 2006;3:77-101. https://doi.org/10.1191/1478088706qp063oa.
- Glaser B. The constant comparative method of qualitative analysis. Social Problems 1965;12:436-45. https://doi.org/10.2307/798843.
- Stiles WB. Evaluating qualitative research. Evid Based Ment Health 1999;2:99-101. https://doi.org/10.1136/ebmh.2.4.99.
- Tong A, Sainsbury P, Craig J. Consolidated criteria for reporting qualitative research (COREQ): a 32-item checklist for interviews and focus groups. Int J Qual Health Care 2007;19:349-57. https://doi.org/10.1093/intqhc/mzm042.
- Brierley J, Larcher V. Emergency research in children: options for ethical recruitment. J Med Ethics 2011;37:429-32. https://doi.org/10.1136/jme.2010.040667.
- Jansen TC, Kompanje EJ, Bakker J. Deferred proxy consent in emergency critical care research: ethically valid and practically feasible. Crit Care Med 2009;37:65-8. https://doi.org/10.1097/CCM.0b013e3181920851.
- Woolfall K, Shilling V, Hickey H, Smyth RL, Sowden E, Williamson PR, et al. Parents’ agendas in paediatric clinical trial recruitment are different from researchers’ and often remain unvoiced: a qualitative study. PLOS ONE 2013;8. https://doi.org/10.1371/journal.pone.0067352.
- Reinert C, Kremmler L, Burock S, Bogdahn U, Wick W, Gleiter CH, et al. Quantitative and qualitative analysis of study-related patient information sheets in randomised neuro-oncology phase III-trials. Eur J Cancer 2014;50:150-8. https://doi.org/10.1016/j.ejca.2013.09.006.
- Gillies K, Entwistle VA. Supporting positive experiences and sustained participation in clinical trials: looking beyond information provision. J Med Ethics 2012;38:751-6. https://doi.org/10.1136/medethics-2011-100059.
- Menon K, Ward R. Canadian Critical Care Trials Group . A study of consent for participation in a non-therapeutic study in the pediatric intensive care population. J Med Ethics 2014;40:123-6. https://doi.org/10.1136/medethics-2012-101075.
- Jansen TC, Kompanje EJ, Druml C, Menon DK, Wiedermann CJ, Bakker J. Deferred consent in emergency intensive care research: what if the patient dies early? Use the data or not?. Intensive Care Med 2007;33:894-900. https://doi.org/10.1007/s00134-007-0580-8.
- Gamble C, Nadel S, Snape D, McKay A, Hickey H, Williamson P, et al. What parents of children who have received emergency care think about deferring consent in randomised trials of emergency treatments: postal survey. PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0035982.
- National Institute for Heath Research . Research Governance Guidelines n.d. www.nihr.ac.uk/funding-and-support/documents/funding-for-research-studies/how-to-apply/NETSCC_Project_Oversight_Groups_Guidance.pdf (accessed September 2018).
- Marlais M, Lyttle MD, Inwald D. Ten concerns about blood pressure measurement and targets in paediatric sepsis. Intensive Care Med 2017;43:433-5. https://doi.org/10.1007/s00134-016-4642-7.
- Slater A, Shann F, Pearson G. PIM2: a revised version of the Paediatric Index of Mortality. Intensive Care Med 2003;29:278-85. https://doi.org/10.1007/s00134-002-1601-2.
- Annual Report of the Paediatric Intensive Care Audit Network: January 2008–December 2010 Summary Report. Leeds and Leicester: University of Leeds and University of Leicester; 2011.
- Annual Report of the Paediatric Intensive Care Audit Network: November 2016. Leeds and Leicester: University of Leeds and University of Leicester; 2016.
- Lancaster GA, Dodd S, Williamson PR. Design and analysis of pilot studies: recommendations for good practice. J Eval Clin Pract 2004;10:307-12. https://doi.org/10.1111/j.2002.384.doc.x.
- Sim J, Lewis M. The size of a pilot study for a clinical trial should be calculated in relation to considerations of precision and efficiency. J Clin Epidemiol 2012;65:301-8. https://doi.org/10.1016/j.jclinepi.2011.07.011.
- Beauchamp T, Childress J. Principles of Biomedical Ethics. Oxford: Oxford University Press; 2001.
- Gill D. Ethics Working Group of the Confederation of European Specialists in Paediatrics . Ethical principles and operational guidelines for good clinical practice in paediatric research. Recommendations of the Ethics Working Group of the Confederation of European Specialists in Paediatrics (CESP). Eur J Pediatr 2004;163:53-7. https://doi.org/10.1007/s00431-003-1378-5.
- Donovan J, Mills N, Smith M, Brindle L, Jacoby A, Peters T, et al. Quality improvement report: improving design and conduct of randomised trials by embedding them in qualitative research: ProtecT (prostate testing for cancer and treatment) study. Commentary: presenting unbiased information to patients can be difficult. BMJ 2002;325:766-70. https://doi.org/10.1136/bmj.325.7367.766.
- Mills N, Donovan JL, Wade J, Hamdy FC, Neal DE, Lane JA. Exploring treatment preferences facilitated recruitment to randomized controlled trials. J Clin Epidemiol 2011;64:1127-36. https://doi.org/10.1016/j.jclinepi.2010.12.017.
- O’Cathain A, Thomas KJ, Drabble SJ, Rudolph A, Goode J, Hewison J. Maximising the value of combining qualitative research and randomised controlled trials in health research: the QUAlitative Research in Trials (QUART) study – a mixed methods study. Health Technol Assess 2014;18. https://doi.org/10.3310/hta18380.
- Boeije H. A purposeful approach to the constant comparative method in the analysis of qualitative interviews. Qual Quant 2002;36:391-409. https://doi.org/10.1023/A:1020909529486.
- Ross S, Grant A, Counsell C, Gillespie W, Russell I, Prescott R. Barriers to participation in randomised controlled trials: a systematic review. J Clin Epidemiol 1999;52:1143-56. https://doi.org/10.1016/S0895-4356(99)00141-9.
- Donovan JL, Paramasivan S, de Salis I, Toerien M. Clear obstacles and hidden challenges: understanding recruiter perspectives in six pragmatic randomised controlled trials. Trials 2014;15. https://doi.org/10.1186/1745-6215-15-5.
- Howard L, de Salis I, Tomlin Z, Thornicroft G, Donovan J. Why is recruitment to trials difficult? An investigation into recruitment difficulties in an RCT of supported employment in patients with severe mental illness. Contemp Clin Trials 2009;30:40-6. https://doi.org/10.1016/j.cct.2008.07.007.
- Ziebland S, Featherstone K, Snowdon C, Barker K, Frost H, Fairbank J. Does it matter if clinicians recruiting for a trial don’t understand what the trial is really about? Qualitative study of surgeons’ experiences of participation in a pragmatic multi-centre RCT. Trials 2007;8. https://doi.org/10.1186/1745-6215-8-4.
- Turnham H, Agbeko RS, Furness J, Pappachan J, Sutcliffe AG, Ramnarayan P. Non-invasive respiratory support for infants with bronchiolitis: a national survey of practice. BMC Pediatr 2017;17. https://doi.org/10.1186/s12887-017-0785-0.
- Dixon-Woods M, Ashcroft RE, Jackson CJ, Tobin MD, Kivits J, Burton PR, et al. Beyond ‘misunderstanding’: written information and decisions about taking part in a genetic epidemiology study. Soc Sci Med 2007;65:2212-22. https://doi.org/10.1016/j.socscimed.2007.08.010.
- Austin JK. Ethical issues related to the increased emphasis on children participating in research. Chronic Illn 2006;2:181-2. https://doi.org/10.1177/17423953060020031101.
- Shilling V, Williamson PR, Hickey H, Sowden E, Smyth RL, Young B. Processes in recruitment to randomised controlled trials (RCTs) of medicines for children (RECRUIT): a qualitative study. Health Technol Assess 2011;15. https://doi.org/10.3310/hta15150.
- Davis AL, Carcillo JA, Aneja RK, Deymann AJ, Lin JC, Nguyen TC, et al. American College of Critical Care Medicine clinical practice parameters for hemodynamic support of pediatric and neonatal septic shock. Crit Care Med 2017;45:1061-93. https://doi.org/10.1097/CCM.0000000000002425.
- Gelbart B, Glassford NJ, Bellomo R. Fluid bolus therapy-based resuscitation for severe sepsis in hospitalized children: a systematic review. Pediatr Crit Care Med 2015;16:e297-307. https://doi.org/10.1097/PCC.0000000000000507.
- Flori HR, Church G, Liu KD, Gildengorin G, Matthay MA. Positive fluid balance is associated with higher mortality and prolonged mechanical ventilation in pediatric patients with acute lung injury. Crit Care Res Pract 2011;2011. https://doi.org/10.1155/2011/854142.
- Valentine SL, Sapru A, Higgerson RA, Spinella PC, Flori HR, Graham DA, et al. Fluid balance in critically ill children with acute lung injury. Crit Care Med 2012;40:2883-9. https://doi.org/10.1097/CCM.0b013e31825bc54d.
- Sinitsky L, Walls D, Nadel S, Inwald DP. Fluid overload at 48 hours is associated with respiratory morbidity but not mortality in a general PICU: retrospective cohort study. Pediatr Crit Care Med 2015;16:205-9. https://doi.org/10.1097/PCC.0000000000000318.
- Bhaskar P, Dhar AV, Thompson M, Quigley R, Modem V. Early fluid accumulation in children with shock and ICU mortality: a matched case-control study. Intensive Care Med 2015;41:1445-53. https://doi.org/10.1007/s00134-015-3851-9.
- Immunisation Against Infectious Disease and Children’s Health. London: PHE; 2013.
- Parikh SR, Andrews NJ, Beebeejaun K, Campbell H, Ribeiro S, Ward C, et al. Effectiveness and impact of a reduced infant schedule of 4CMenB vaccine against group B meningococcal disease in England: a national observational cohort study. Lancet 2016;388:2775-82. https://doi.org/10.1016/S0140-6736(16)31921-3.
- Sepsis: Recognition, Diagnosis and Early Management. London: NICE; 2016.
Appendix 1 Feasibility study example interview topics and questions
| Interview topics | Example questions |
|---|---|
| Knowledge and experience |
|
| Participant information |
|
| Consent decision-making |
|
| Approach to consent | (Following the description of RWPC, given in Chapter 2, Box 1)
|
| Participation |
|
| Outcome measures |
|
| For bereaved parents only |
|
Appendix 2 Draft pilot trial participant information sheet
Appendix 3 List of potential outcome measures
Appendix 4 Selected quotations from parents by theme
| Theme | Subtheme | Example quotations |
|---|---|---|
| RWPC is acceptable, but some initial concerns | Logical solution | At first I was like ‘What? You’d do that without even finding out if it was OK?’ And then I kind of thought about it and actually that does make senseP15, mother, bereavedI had absolutely no problem with it [RWPC]P2, mother, non-bereaved, prior experience of RWPCIt totally makes sense because as a parent that’s been through it, if they had come and asked us lots of different questions it probably would have brought up a lot more questions, which would have resulted in the subsequent delay in treating [child’s name] . . . and at that time there wasn’t a lot of time to kind of faff about telling us things. So yeah, it totally makes senseP15, mother, bereaved |
| Acceptability dependent on nature of the trial | It depends what they’re actually doing . . . When it’s your child, I think as long as it isn’t sort of sold as part of an experimentP6, mother, non-bereavedI mean I don’t have any issues with it. The only concern would be that I would want to know if you’ve done something without my consent has there been an impact on him in a negative way . . . Would it have altered anything? And if you can give me assurances around that, I can’t see that there’s any risk either wayP10, mother, non-bereaved | |
| Support for the FiSh trial, but some concerns and misconceptions | Outcome dependent | I wouldn’t have had a problem at all if someone had come to me a few hours afterwards and said, ‘Can we use the information?’ . . . I’d have been quite happy that she’d have been involved in the trial because I know that obviously it’s worked and she’s betterP4, mother, non-bereavedPersonally, if she had stabilised then yeah, that would be fine . . . If I thought she was touch and go and she was being used as a trial perhaps then I would feel ‘hang on a minute . . .’ Because you want anything you can cling onto. A reason or anything. So I would think what is the reason that she didn’t survive, was it because she was given 10 and not 20?P19, mother, bereaved |
| Misconception: current practice has been proven to be effective | I think you just want the tried and tested, don’t you? It’s the difference between saying well we can put a plaster on this, or not put a plaster on it. I would be like well, what do you normally do? Well, we normally put a plaster on it. Well, put a plaster on it thenP6, mother, non-bereavedObviously we know 20 ml/kg works . . . That’s the thing, isn’t it? It’s like, well why are you changing it if it already works?P21, mother, bereaved | |
| Misconception: more fluid is better | To me it was just give her as much as you can, that’s what was in my mind. They kept saying another bolus, another bolus. In my mind, that was what was saving her lifeP1, mother, non-bereavedUltimately at a rate of doing it 20 ml per kilo, obviously they might stabilise fasterP21, mother, bereaved | |
| Tailored explanation is important to address concerns and misconceptions | That’s quite important, I think . . . reiterating that the fluid treatment is part of a package of things and that it’s not necessarily the be all and end all of the treatmentP17, mother, bereavedI think hearing those two points, the two key points around there being limited or no evidence really that the way we’re doing it is the right way . . . I think that would definitely sway me to take partP2, mother, non-bereavedI’d be quite happy that they’d keep an eye on things and keep monitoring closelyP4, mother, non-bereaved | |
| Unclear or missing study information | I mean it’s changed when you said he would have had the standard treatment anyway and then they would go on to the trial. I suppose if it’s emphasised that everyone gets the same initial treatment, the emergency treatment . . . that would be easierP16, father, bereavedIf he was randomly selected to only have the 10 ml which is, you know, not with what the guidelines go with at the moment, would he have been constantly monitored and would that amount have been increased if they thought it was necessary? Or would they have just rigidly stuck to their guns? That sort of thing I think would have run through my mindP17, mother, bereavedDoes it say anywhere what the bolus actually does? That might be something, ‘cause I was thinking I don’t really know. I clocked that the idea is to reduce it but not actually what it doesP13, mother, non-bereavedI just wondered if the 20 ml has to be given over a certain amount of time?P17, mother, bereaved | |
| Not voicing concerns | I think generally . . . your child’s ill, somebody takes them off you and knows what they’re doing, you just kind of let them deal with it. You don’t get too involved other than just asking how they’re doing, but you don’t necessarily voice concernsP5, mother, non-bereavedI don’t think it would be at the forefront of what’s going on to question the doctorP11, mother, non-bereaved | |
| Approaching non-bereaved parents for a RWPC discussion | Timing | I think it’d be very much dependent on the actual time . . . if I was asked this once my child was stable, I’d know . . . he’s OK . . . before that point I’d have probably found it quite hard to make any decisionsP5, mother, non-bereavedBeing in that stressful situation, I don’t think it would benefit the parent or the staff that are having to ask these questions . . . Once a parent’s a bit calmer, it literally will open up a whole different world to you guysP11, mother, non-bereavedThe real first 24 hours might be too soon possibly, because emotions are running high and you know, there’s a lot of new, strange things happening. It might be a little bit much to ask at that pointP7, father, non-bereaved |
| Gauge on a case-by-case basis | I think that’s probably where it would be down to each individual, you know, case by case . . . it’s always going to be a difficult position but people react completely different to their child being in that sort of environment . . . So I think it would be about the timing, and some degree of judgement on behalf of the medical staff to actually ask, ‘Can this person reasonably given consent at this time?’P10, mother, non-bereaved | |
| Approaching bereaved parents for a RWPC discussion | Prepare for parental concerns about RWPC | I think probably my only concern is, as a bereaved parent, you are always looking for a reason as to why it happened. And it possibly would lay the door open for people to go ‘You did it wrong, you shouldn’t have done that.’ That’s my only concern. It gives you angle – it’s the doctor’s fault because they did this and they didn’t ask meP21, mother, bereaved |
| RWPC is acceptable as a way to help others | If it helps one other family, it’s fine by us. At the end of the day that’s what it’s about. We can’t bring our [child] back and if this helps one other child not go through the suffering, the pain, that’s fine by us. I would just say just get on with it. Just do itP14, father, bereavedTo be honest, I am so desperate to help, or if [child’s name] information can be used in any way . . . If it could help stop it happening to someone else, or help understand why, I would be happy for them to use it for research without confirming it first. For me, I can’t think of any negativesP19, mother, bereaved | |
| Do not approach in hospital | I don’t think you should do it straight away. I don’t think it should be done in hospital. It’s just an overwhelming experience is all I can say. And then to have that, you won’t take it in, you won’t understand. You’ll be more angry that your child is dead and they did this and they didn’t tell youP21, mother, bereavedOn the day, I think you’re more likely to get parents who will want to opt out. And I think that you will get more of an opt-out the earlier that you do itP17, mother, bereaved | |
| Acceptability of postal contact and ‘opt-out’ approach | I think a letter is the right way round but with a lot of sensitivity in the way it’s worded because it will come as a shock to familiesP18, father, bereavedI think the opt-out rather than the opt-in is fair because I think if it’s an opt-in situation, you’re going to lose the data. And I think by opting-out you give the people, if they have strong feelings the other way, you give them that optionP16, father, bereavedCould you make that in like bold or underlined or something, that they will be included? Because it’s quite often that when you receive something like that you’re like, well, if you don’t get in touch then that’s it, you don’t get included. Whereas you’re saying actually if we don’t get in touch with you, you are being included in it. And I think, yeah maybe that little bit could be highlightedP15, mother, bereaved | |
| Importance of parent–practitioner relationships | Depending on your stay, you become quite familiar with certain nurses and you do have a rapport with them. And I think some, from our point of view, some nurses we would definitely have accepted letters from them probably a bit better than from other nursesP15, mother, bereaved | |
| Patient-centred outcomes | (1) ‘Getting better’ (e.g. looking and behaving more like their normal selves) | Actually, when she was in the PICU, one of the things for me, being not medical, was looking more like herself, if that makes sense. To me that was the big measure of she was getting betterP13, mother, non-bereaved |
| (2) Survival | Well I guess for me he was in a critical situation, so it was just about getting him to survive at that stageP10, mother, non-bereaved | |
| (3) Organ/physiological functioning (e.g. blood pressure, temperature and heart rate) | Temperature was always a big one. It showed that he was getting better to meP21, mother, bereaved | |
| (4) Long-term effects (e.g. disabilities, long-term health and development) | The fact there would be no problems in the future because that’s what you worry about, the long-term future things. So, whether there’ve been any other repercussions because of the infection is a big thing I thinkP4, mother non-bereaved | |
| (5) Time spent on machines and medications | By the third or fourth day all her drugs were getting reduced and she was slowly coming off the machines, so sort of we knew then that she was making a recovery. So maybe how long they’re on a machineP3, mother, non-bereaved |
Appendix 5 Parent/legal representative consent questionnaire
Appendix 6 Pilot trial example parent/legal representative interview topics and questions
| Interview topics | Example questions |
|---|---|
| Child’s hospital admission experience |
|
| FiSh pilot trial consent process |
|
| Consent in the emergency setting |
|
| Decision-making |
|
| Decision-making (those who did not consent to participate in the FiSh trial) |
|
| Outcome measures |
|
| Child assent (not for bereaved parents) |
|
| For bereaved parents only | (If received letter in post)
|
Appendix 7 Pilot trial example site staff focus group/interview topics and questions
| Interview topics | Example questions |
|---|---|
| Training |
|
| Screening |
|
| Randomisation |
|
| Protocol deviations |
|
| Recruitment and consent in the FiSh trial |
|
| Completing forms |
|
List of abbreviations
- ACCM-PALS
- American College of Critical Care Medicine – Pediatric Advanced Life Support
- AE
- adverse event
- CI
- confidence interval
- CONNECT
- CONseNt methods in paediatric Emergency and urgent Care Trials
- CRF
- case report form
- CRN
- Clinical Research Network
- CRT
- capillary refill time
- CTU
- clinical trials unit
- DMEC
- Data Monitoring and Ethics Committee
- eCRF
- electronic case report form
- ED
- emergency department
- FEAST
- Fluid Expansion as Supportive Therapy
- FiSh
- Fluids in Shock
- HRA
- Health Research Authority
- ICNARC
- Intensive Care National Audit and Research Centre
- IQR
- interquartile range
- LCRN
- Local Clinical Research Network
- NIHR
- National Institute for Health Research
- P
- participant
- PAU
- paediatric assessment unit
- PCV
- pneumococcal conjugate vaccine
- PERUKI
- Paediatric Emergency Research in the UK and Ireland
- PI
- principal investigator
- PIC
- Participant Identification Centre
- PICANet
- Paediatric Intensive Care Audit Network
- PICS-SG
- Paediatric Intensive Care Society Study Group
- PICU
- paediatric intensive care unit
- PIM2r
- Paediatric Index of Mortality version 2r
- PIS
- participant information sheet
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RWPC
- research without prior consent
- SAE
- serious adverse event
- SD
- standard deviation
- SMG
- Study Management Group
- SQUEEZE
- Septic Shock Reversal is Quicker in Pediatric Patients Randomized to an Early Goal Directed Fluid-sparing Strategy vs. Usual Care
- TSC
- Trial Steering Committee