

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 12/33/28. The contractual start date was in May 2017. The draft report began editorial review in December 2017 and was accepted for publication in August 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Claire Roberts reports grants, in addition to non-financial support, from Novartis Pharmaceuticals UK Ltd (Frimley, UK), outside the submitted work. Peter Howarth reports part-time employment with GlaxoSmithKline plc (London, UK) as a global medical expert.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Kapoor et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background and rationale
The burden of severe asthma
Epidemiology
Asthma affects > 5.4 million people in the UK, with nearly 500,000 experiencing severe symptoms and frequent exacerbations that are inadequately controlled with available treatments. 1,2 The burden of severe asthma on the NHS is enormous, accounting for 80% of the total asthma cost (£1B3), with frequent exacerbations and expensive medications generating much of this cost. 4 In 2009, there were 1131 deaths caused by asthma,5 with those whose asthma remains poorly controlled facing the greatest risk. 6,7 Patients with severe asthma bear the greatest burden of asthma morbidity. They experience more frequent and severe exacerbations,8 which reduce their quality of life, impair their ability to work and place an enormous burden of anxiety on them and their families. 9 There is also an increased risk of significant depression. 10 One in five asthmatics in the UK report serious concerns that their next asthma attack will be fatal. 1 As highlighted in the 2010 Asthma UK report Fighting for Breath,11 these patients also face discrimination from employers, health-care professionals and society as a whole as a result of their asthma.
The unmet need in severe asthma
Current treatments, including oral corticosteroids (OCSs), ‘steroid-sparing’ immunosuppressants and monoclonal antibody therapies, often have limited efficacy and potentially serious side effects (steroids, immunosuppressive agents) or are prohibitively expensive (monoclonal antibodies). The adverse effects of long-term oral steroids include adrenal suppression, decreased bone mineral density, diabetes mellitus and increased cardiovascular mortality. 12 The anti-immunoglobulin E (IgE) treatment omalizumab (Xolair®; Novartis Pharmaceuticals UK Ltd, Frimley, UK) has been shown to reduce exacerbations by up to 50%13 and improve quality of life in severe allergic asthma, but costs up to £26,640 per year. 14 The National Institute for Health and Care Excellence (NICE)14 reappraised the use of omalizumab in 2012 and, although recognising the grave effects of severe uncontrolled asthma on quality of life for patients and their families, concluded that it is cost-effective within the NHS only when its use is limited to those with severe persistent confirmed allergic IgE-mediated asthma experiencing four or more severe exacerbations in the preceding 12 months. A large number of patients are therefore left with a significant unmet clinical need and a specific requirement for therapies that reduce systemic steroid exposure.
It is also important to acknowledge the often unrecognised role that carers play in looking after patients with poorly controlled severe asthma. The Fighting for Breath11 document highlighted the strain that this can place on the physical and mental health of this vital informal workforce. In addition, having to take time off work, having to work part-time or not being able to work at all as a result of a patient’s care needs places a significant financial burden on carers.
National/international strategies to improve asthma care
The 2011 Department of Health and Social Care report An Outcomes Strategy for COPD and Asthma in England15 recognises the huge burden that poorly controlled asthma places on people’s lives and the NHS. It also describes the political commitment to improve asthma control and reduce asthma-related emergency health-care needs and deaths. 15 The 2011 British Thoracic Society (BTS) and Scottish Intercollegiate Guidelines Network (SIGN) national asthma guideline16 and the 2010 World Health Organization (WHO) consultation on severe asthma8 have highlighted an urgent need for research in severe asthma, acknowledging the limitations of available treatments and the dearth of clinical trials on which to base management recommendations. In its research strategy for 2016–21, Asthma UK17 identified the development of new treatments as a priority for improving clinical outcomes and patient well-being and reducing the cost of treating severe asthma within the NHS. It also identified the need to gain a better understanding of the impact of exposure to substances known to trigger asthma and the impact of strategies that regulate and control this exposure as a key priority.
The significance of allergen exposure and environmental interventions
More than 70% of people with severe asthma are sensitised to common aeroallergens and/or moulds,18 with the level of allergen exposure determining symptom severity; those exposed to high allergen levels are at increased risk of exacerbations and hospital admissions. 19–22 Domestic exposure to allergens is also known to act synergistically with viruses in sensitised patients to increase the risk and severity of exacerbations. 23 Allergen avoidance has been widely recognised as a logical way to treat these patients. 24 In controlled conditions, long-term allergen avoidance in sensitised asthmatics reduces airway inflammation with consequent symptomatic improvement, which is further supported by high-altitude, clean-air studies. 25–27 Unfortunately, effective methods of allergen reduction have proved elusive,28,29 with current measures unable to reduce allergen load sufficiently to yield a consistent clinical improvement, thus leaving a significant gap in the potential strategies for reducing asthma severity through allergen reduction.
Rationale for temperature-controlled laminar airflow therapy
At night, airborne particles are carried by a persistent convection current established by the warm body, transporting allergens from the bedding area to the breathing zone. 30 Proof-of-concept studies have shown that the temperature-controlled laminar airflow (TLA) device reduces the total number of airborne particles of > 0.5 µm in the breathing zone by 3000-fold (p < 0.001) and cat allergen exposure by 30-fold (p = 0.043) and significantly reduces the increase in the number of particles generated when turning in bed for all particle sizes. 31 Compared with a best-in-class traditional air cleaner, TLA is able to reduce exposure to potential allergens by a further 99%. 32 We postulate that this highly significant reduction in nocturnal exposure, targeted to the breathing zone, explains why TLA may succeed when so many other measures, including traditional air cleaners, have failed.
Evidence of benefit with temperature-controlled laminar airflow therapy
Compared with placebo, the TLA device has proven efficacy for asthma-related quality of life and bronchial inflammation (measured by exhaled nitric oxide) in a pan-European, multicentre, 12-month Phase III study33 (n = 282, age range 7–70 years). The greatest benefit was seen in the more severe asthma patients requiring higher intensity treatment [Global INitiative for Asthma (GINA) step 4] and with poorly controlled asthma [7-point Asthma Control Questionnaire (ACQ) score of < 19]. GINA step 4 is consistent with American Thoracic Society (ATS)/European Respiratory Society (ERS) severe asthma guideline definitions34 and BTS/SIGN guideline treatment step 4 [inhaled corticosteroid (ICS) dose of ≥ 1000 µg/day beclomethasone dipropionate (BDP) equivalent plus an additional controller medication such as a long-acting β2-agonist, leukotriene receptor antagonist or sustained-release theophylline]. Although not powered to ascertain an effect on exacerbations, a post hoc analysis of the Phase III study data showed a decreased exacerbation rate in more severe patients treated with TLA compared with placebo, with a trend towards significance (mean TLA 0.23 exacerbations/year, placebo 0.57 exacerbations/year; p = 0.07). 33 Although a cost-effectiveness analysis based on the results from this trial found no significant differences in emergency department (ED) visits, hospitalisation days, medication usage and, therefore, overall costs between the two study groups,35 this may have reflected the fact that the trial was powered to detect a difference in asthma-related quality of life and did not specifically include patients at risk of exacerbations (average annual rate was 0.2 exacerbations per year), a predictor of increased asthma health-care resource use and costs. 36 Despite the lack of a significant reduction in health-care resource use and associated costs, subsequent economic modelling showed that TLA would be cost-effective in Sweden at the current monthly rental price (SEK2000, ≈£167), mainly because of increases in quality of life. 34
Using the results from a very small, 10 + 10-week randomised controlled crossover trial in Sweden,37 a modelling study addressed the potential cost-effectiveness of TLA therapy over a projected 5-year period. 38 Assuming no impact of TLA on health-care resource use, TLA was cost-effective compared with placebo at a device cost of €8200 (≈£6890), at a willingness-to-pay threshold of €35,000 (≈£30,000) per quality-adjusted life-year (QALY) gained. We considered that the greatest potential for cost-effectiveness could occur in severe asthma through reducing exacerbations, which was not addressed in the Swedish trial because of the short observation period and less severe patient population included.
A further pragmatic, patient-centred randomised controlled trial (RCT) of this novel non-pharmacological treatment in severe allergic asthma was thus warranted.
Objectives
The aim of the Laminar Airflow in Severe asthma for Exacerbation Reduction (LASER) trial was to assess whether or not home-based nocturnal TLA treatment can effectively reduce asthma-related morbidity over a 1-year period in a real-life group of patients in the UK with poorly controlled, severe allergic asthma.
Primary objective
The primary objective was to determine whether or not nocturnal TLA treatment reduces the frequency of severe asthma exacerbations (defined as an acute deterioration in asthma requiring treatment with systemic corticosteroids).
Secondary objectives
The secondary objectives were to:
-
assess the impact of nocturnal TLA treatment on asthma control, including:
-
current clinical asthma control, which is the extent to which the clinical manifestations of asthma (i.e. symptoms, reliever use and airway obstruction) have been reduced or removed by treatment
-
the risk of future adverse asthma outcomes, which includes loss of control, exacerbations, accelerated decline in lung function and side effects of treatment
-
-
ascertain the effect of TLA treatment on quality of life in participants with poorly controlled, severe allergic asthma and their carers
-
qualitatively evaluate participants’ perceptions, values and opinions of the device to identify potential modifications to improve participant acceptance and to inform future use of the device within the NHS setting
-
evaluate the impact of TLA treatment on health-care utilisation and related costs and its impact on education/work days lost
-
fully assess the cost-effectiveness, both at 1 year and over the lifetime of the participant, of nocturnal TLA treatment using a cost–utility analysis to determine the incremental cost per QALY gained.
Chapter 2 Methods
The objective of the LASER trial was to assess whether or not home-based nocturnal TLA treatment can effectively reduce asthma-related morbidity over a 1-year period in a real-life group of patients with poorly controlled, severe allergic asthma. The trial was sponsored by Portsmouth Hospitals NHS Trust and funded by the National Institute for Health Research Health Technology Assessment (HTA) programme. The first participant was recruited to the trial in May 2014 and the final participant completed participation during January 2017.
Patient and public involvement
The design of the methods for this trial was supported by patient and public involvement (PPI) members in several ways. First, Mrs Wilsher and Mr Boughton worked with the investigators and co-applicants during the grant submission stage to inform the outcomes and objectives, for example by including carers inthe study design. During the trial, they assisted with the design and participated in the delivery of patient information events and designed study advertisements for use on social media platforms. They also assisted with writing the plain English summary for the ethics application and the patient information sheet and consent form. Further support was provided by Mr Supple, an expert patient member from Asthma UK, who sat on the Trial Management Group (TMG). Representatives from Allergy UK also assisted with the organisation and delivery of patient-facing events and the design and posting of recruitment materials on the Allergy UK website. Mrs Wilsher is further involved in the interpretation of data from the qualitative analyses and is currently contributing to a publication on the importance of the use of social media in recruitment to clinical trials.
Trial design
The LASER trial was designed exclusively to meet the objective previously described. The trial design determined to best meet this objective was a multicentre, randomised, double-blind, placebo comparator, parallel-group trial design, with each individual participant trialling the active or placebo device for 12 months.
A placebo comparator was chosen as other add-on treatments in severe asthma (e.g. omalizumab and bronchial thermoplasty) vary greatly in indication, use and delivery, are not suitable for every patient and would therefore not be able to be used consistently or safely in an ‘active’ control group. Participants were randomised in a 1 : 1 ratio to receive either an active treatment device or a placebo device. Throughout the trial, participants in both treatment arms received standard asthma care in accordance with the national BTS/SIGN guidelines for the management of asthma in adults. 16
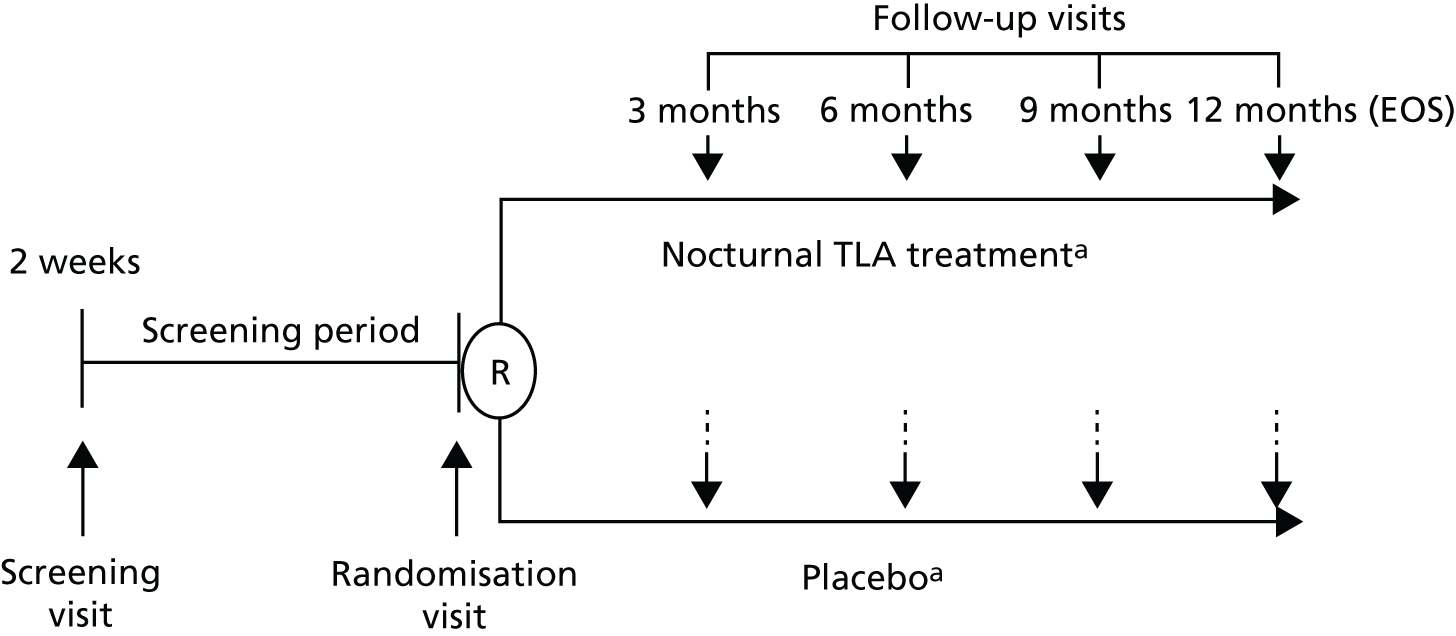
Figure 1 presents a simple overview of the trial design, highlighting the six study visits (1, screening visit; 2, randomisation visit; 3–6, follow-up visits at months 3, 6, 9 and 12 months, respectively). The full trial flow chart and data collected at each of these study visits are summarised in Table 1, presented in full in Table 27 and explained throughout this chapter.
FIGURE 1.
Simple overview of trial design. EOS, end of study; R, randomisation. a, In addition to existing asthma treatments that will not be adjusted during the trial.
| Visit | Data collected |
|---|---|
| Scheduled data | |
| Visit 1: screening | Lung function (spirometry + bronchodilator reversibility) (FeNO) |
| Allergy testing (skin prick tests) (total IgE) (serum-specific IgE) | |
| Questionnaires (ACQ) | |
| Visit 2: baseline | Lung function (spirometry + bronchodilator reversibility) |
| Questionnaires (ACQ, AQLQ, EQ-5D-5L, SNOT-22, AC-QoL) | |
| 2-week diary submissiona | |
| Visits 3–6: treatment period | Lung function (spirometry) (FeNO) |
| Questionnaires (ACQ, AQLQ, ED-5D-5L, SNOT-22, WPAI) | |
| 2-week diary submissiona | |
| TLA diary review of device adherence/health-care usage | |
| Visit 6 only (end of study) | Lung function (bronchodilator reversibility) |
| Additional questionnaires (GETE, AC-QoL) | |
| Telephone call: 1-month telephone call | Telephone review – participant-reported adherence |
| Unscheduled data | |
| Exacerbation history collected throughout study at ‘exacerbation reviews’ | |
Trial protocol
Figure 2 consolidates and summarises the major content of the trial protocol into a simple flow chart. The major activities in the trial are described below, linked to the corresponding sections of this report for full details, for the reader’s convenience. The data collected at these visits are listed in Table 1.
-
Participant recruitment (see Recruitment).
-
Study visits: participants were required to attend the following six study visits to collect the scheduled data –
-
screening visit [see Screening visit (–2 weeks)]: purpose of data collection = screen participant against trial inclusion and exclusion criteria (see Participants)
-
randomisation visit [see Randomisation visit 2 (0 months)]: purpose = assign participant to active or placebo device treatment arm (see Participants and minimisation criteria)
-
3-month follow-up visit
-
6-month follow-up visit
-
9-month follow-up visit
-
12-month follow-up visit: common purpose of study visits 3–6 = secondary outcome data collection (see Secondary outcomes).
-
-
Unscheduled data collection: purpose of data collection = primary outcome data collection (see Primary outcome).
-
Optional focus group sessions: purpose of data collection = capture individuals’ perceptions, expectations and meanings to explore acceptance, level of personal control, motivation and usefulness of the TLA device (see Focus groups).
FIGURE 2.
Trial flow chart. ORTU, Oxford Respiratory Trials Unit; v, visit. a, Telephone review only; b, expected 10% dropout. Light green shading represents the study team with participant. Dark green shading represents the TLA engineer.
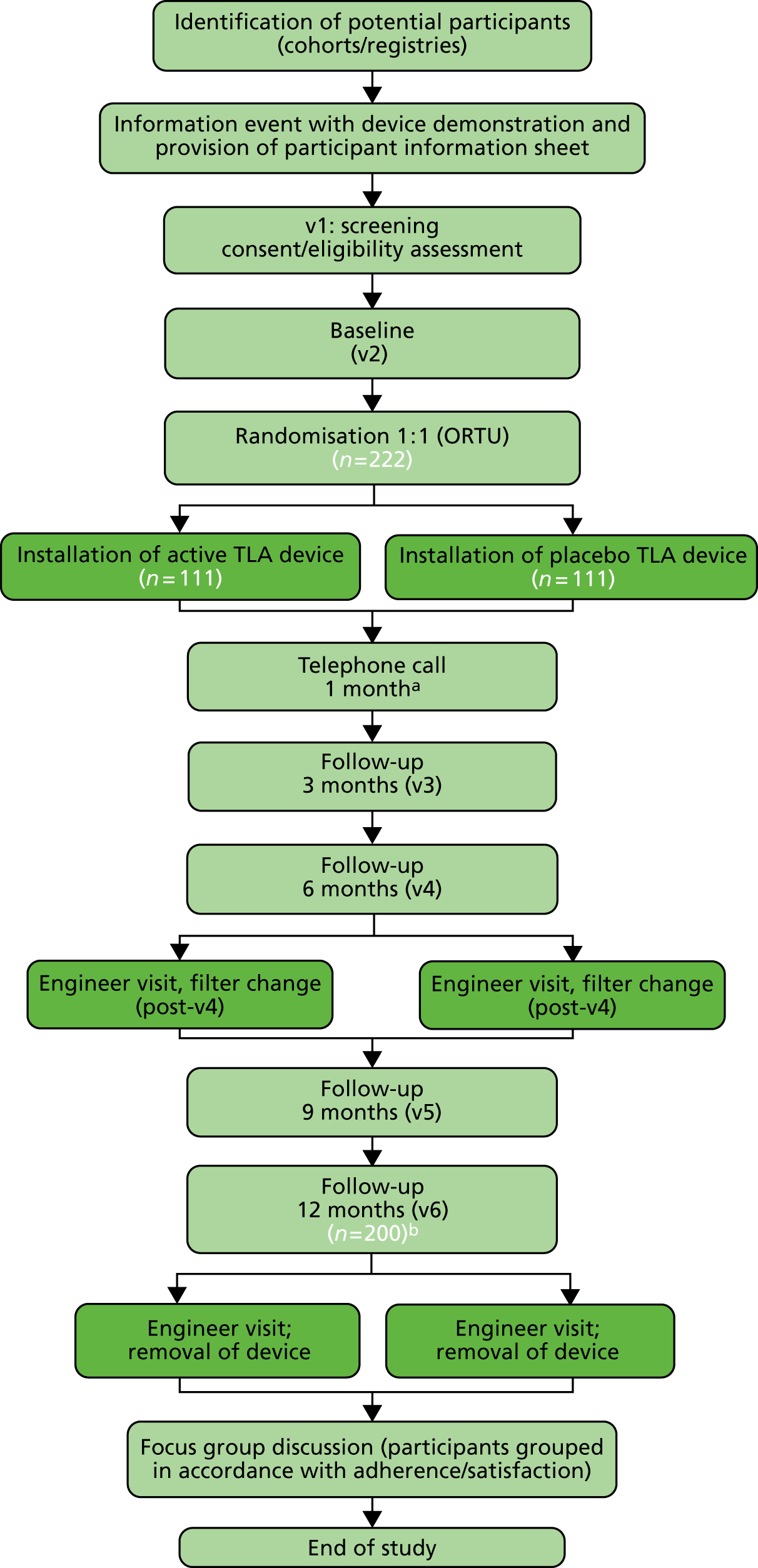
Further quantitative data are detailed in the subsections of Study assessments.
The internal pilot study
A 4-month internal pilot study was used to validate the feasibility of the trial for achieving its objectives. Specifically, the internal pilot study evaluated the following indicators of trial feasibility at the five initial recruiting centres during the first 4 months of recruitment. Amendments were made to the original trial protocol based on the resulting insights, including practical modifications to trial processes that could be implemented to enhance participant and partner experience during the remainder of the trial.
-
Recruitment and retention of participants:
-
participant numbers emerging from the screening, consent and randomisation processes
-
participant experiences of the recruitment process
-
time from randomisation to device installation
-
adherence to follow-up (including the 1-month telephone review).
-
-
Data collection methods and quality: quality and completeness of the following trial outputs –
-
clinical exacerbation reports
-
participant diaries
-
participant case report forms (CRFs)
-
participant questionnaires.
-
-
Participant and partner experiences:
-
potential barriers to trial recruitment
-
challenges to trial adherence, including device acceptability.
-
A trained qualitative researcher elicited the participant and partner experience feedback through semistructured one-to-one telephone interviews during study month 9 (recruitment month 3). Ten trial participants and two participant partners (aged ≥ 18 years) living within the same home and sharing the same bedroom environment were invited to take part in the qualitative interviews. Although it was intended to recruit participants from different study sites, this was not possible within the 3-month deadline because of the initial slow recruitment rate. All telephone interviews were therefore conducted at the main study site, Portsmouth. Participants and their partners shared their views in separate telephone interviews.
Amendments to the trial protocol
Three amendments were made to the trial protocol following trial initiation. Each of these amendments was deemed necessary to improve the efficiency and effectiveness of the trial for meeting its objectives. The amendments were as follows:
-
7 August 2014 (minor): Vitalograph Asma-1 USB device removed. Reason: the international stock levels of the original device were low; switching to similarly licensed devices minimised delays.
-
23 October 2014 (minor): removal of 12-month limit on historical bronchial challenge test results. Reason: several participants failed screening as their bronchial challenge test had fallen just outside the 12-month window. Removing this criterion ensured that more participants with asthma were able to be recruited.
-
28 May 2015 (major): three amendments to the trial inclusion criteria: (i) reduction in the lower age for participation from 18 years to 16 years, (ii) reduction in the length of the pre-screening stability period from 4 weeks to 2 weeks and (iii) reduction in the required ICS dose from > 1000 µg/day of BDP or equivalent to ≥ 1000 µg/day of BDP or equivalent. Reason: all amendments were based on examination of the reasons why participants failed screening. Working with patient advisors, amendments to the protocol were made to ensure that more participants were able to be screened as well as randomised.
All material in this report refers to the version of the protocol in place after these three amendments were made.
Eligibility and inclusion/exclusion criteria
Participants
The selected eligibility criteria reflected a population of participants with severe exacerbation-prone allergic asthma, which was also consistent with previous similar trials in this population (see Table 28).
Inclusion criteria
Potential participants had to meet all of the following inclusion criteria by randomisation visit 2 to be considered eligible for the study:
-
Adults (aged 16–75 years inclusive).
-
A clinical diagnosis of asthma for ≥ 6 months supported by evidence of any one of the following –
-
Airflow variability with a mean diurnal peak expiratory flow (PEF) variability {calculated by [PEF(highest) – PEF(lowest)]/[PEF(mean)]} of > 15% during the baseline 2-week period or a variability in forced expiratory volume in 1 second (FEV1) of > 20% across clinic visits within the preceding 12 months, with concomitant evidence of airflow obstruction (FEV1/FVC ratio < 70%, where FVC is forced vital capacity).
-
Airway reversibility with an improvement in FEV1 of ≥ 12% or 200 ml after inhalation of 400 µg of salbutamol (Ventolin® GlaxoSmithKline UK) via a metered dose inhaler and spacer at the first study visit or within the preceding 12 months.
-
Airway hyper-responsiveness demonstrated by methacholine challenge testing, with a provocative concentration of methacholine (Nova Laboratories, Leicester, UK) causing at least a 20% reduction in FEV1 (PC20) of ≤ 8 mg/ml, or an equivalent test (see Table 29).
-
-
Severe asthma –
-
Requirement for high-dose ICSs (≥ 1000 µg/day of BDP or equivalent; see Table 30) plus a second controller (long-acting β2-agonist or antimuscarinic, theophylline or leukotriene antagonist) and/or systemic corticosteroids.
-
If on maintenance corticosteroids, the maintenance dose must have been stable for 3 months; this excluded any interim need for short-term steroid bursts to treat exacerbations.
-
-
Poorly controlled asthma demonstrated by both –
-
Two or more severe asthma exacerbations requiring systemic corticosteroids of ≥ 30 mg of prednisolone or equivalent daily (or ≥ 50% increase in dose if maintenance dose of ≥ 30 mg prednisolone) for ≥ 3 days during the previous 12 months, despite the use of high-dose ICSs and additional controller medication.
-
ACQ (7-point scale) score of > 1 at screening visit 1 and randomisation visit 2.
-
-
Atopic status –
-
Sensitisation to one or more perennial indoor aeroallergens (including house dust mites, domestic pets or fungi) to which participants are likely to be exposed during the study, demonstrated by a positive skin prick test against Dermatophagoides pteronyssinus (Der p 1) or Dermatophagoides farinae (Der f 1), Aspergillus fumigatus (Asp f 1), Alternaria alternata (Alt a 1) or Cladosporium herbarum (Cla h 1), cat – Felis domesticus (Fel d 1) – or dog – Canis familiaris (Can f 1) (wheal diameter of ≥ 3 mm more than a negative control test) or specific IgE of ≥ 0.35 IU/l determined by blood test.
-
-
Recent medical stability –
-
Exacerbation free and taking stable maintenance asthma medications (not including short-acting bronchodilator or other reliever therapies) for at least 2 weeks prior to screening visit 1 and in the period between screening visit 1 and randomisation visit 2 (the screening period). Participants suffering a severe exacerbation during the screening period were rescreened 2 weeks after returning to their maintenance asthma medications.
-
-
Adherence –
-
Able to use the TLA device during sleep on at least five nights per week (excluding holidays).
-
Able to understand and give written informed consent prior to participation in the trial and able to comply with the trial requirements.
-
Exclusion criteria
Potential participants who met any of the following exclusion criteria were excluded from participating in the study:
-
Current smokers or ex-smokers abstinent for < 6 months.
-
Ex-smokers with a ≥ 15 pack-year smoking history.
-
Partner who is a current smoker and smokes within the bedroom where the TLA device is installed.
-
TLA device cannot be safely installed within the bedroom.
-
Intending to move out of study area within the follow-up period. (Participants moving out of the study area after randomisation were not automatically withdrawn. Every effort was made to continue treatment and trial follow-up.)
-
Documented poor treatment adherence.
-
Occupational asthma with continued exposure to known sensitising agents in the workplace.
-
Previous bronchial thermoplasty within 12 months of randomisation.
-
Treatment with omalizumab (Xolair®, Novartis Pharmaceuticals UK Ltd, Grimley, UK) (anti-IgE) within 120 days of randomisation.
-
Using long-term oxygen, continuous positive airway pressure or non-invasive ventilation routinely overnight, as it is known that this impairs the effect of the TLA device.
-
Uncontrolled symptomatic gastro-oesophageal reflux that may act as a persistent asthma trigger.
-
Presence of clinically significant lung disease other than asthma, including smoking-related chronic obstructive pulmonary disease (COPD), bronchiectasis associated with recurrent bacterial infection, allergic bronchopulmonary aspergillosis (mycosis), pulmonary fibrosis, sleep apnoea, pulmonary hypertension or lung cancer, that, in the opinion of the principal investigator (PI), is likely to be contributing significantly to a participant’s symptoms.
-
Clinically significant comorbidity (including cardiovascular, endocrine, metabolic, gastrointestinal, hepatic, neurological, renal, haematological and malignant conditions) that remains uncontrolled with standard treatment.
-
Currently taking part in other interventional respiratory clinical trials.
Centres and care providers
The feasibility was considered of 25 secondary care providers as recruiting centres for the LASER trial. The 14 sites listed in Table 2 were selected, all with trial teams embedded within respiratory departments. Each of these recruiting centres activated the trial at different dates during the study period and these activation dates are also shown in Table 2.
| Site | Date of activation | Number randomised, n (%) |
|---|---|---|
| Portsmouth (trial lead site) | 7 May 2014 | 74 (31) |
| Liverpool (Aintree) | 17 June 2014 | 13 (5) |
| Birmingham (Heartlands) | 26 June 2014 | 28 (12) |
| Leicester | 09 July 2014 | 14 (6) |
| Southampton | 09 July 2014 | 25 (10) |
| Bradford | 09 September 2014 | 9 (4) |
| Royal Liverpool | 20 January 2015 | 18 (8) |
| London (St George’s) | 22 January 2015 | 11 (5) |
| Chester | 03 February 2015 | 6 (3) |
| Oxford | 26 February 2015 | 7 (3) |
| Hull | 02 April 2015 | 10 (4) |
| Maidstone | 07 July 2015 | 17 (7) |
| Birmingham (Queen Elizabeth) | 30 July 2015 | 5 (2) |
| Belfast | 26 August 2015 | 3 (1) |
The trial was very well supported by the lead site’s local Clinical Research Network (Wessex) as well as by the 13 other recruiting sites. This enabled the trial to facilitate the support of general practice surgeries in the Portsmouth area as participant identification centres (PICs). To ensure swift recruitment at the sites and to avoid protracted contractual negotiations between sites, commissioners and the supplier, in particular with regard to excess treatment costs (ETCs), it was agreed that the lead site and sponsor (Portsmouth Hospitals NHS Trust) would assume responsibility for receipt and distribution of all TLA devices and arrange separate research contracts with the sites. This prevented other sites from having separate contracts with the supplier with the risk that they may not be able to recover the ETCs associated with the active intervention. This risk was particularly acute because the sites and participants would be blinded to the treatment allocation and so any costs recovered for the active intervention could be based on only retrospective invoicing, often carried over into subsequent financial years, which would further complicate financial contracts.
Excess treatment costs
The Department of Health and Social Care agreed to provide a subvention payment to cover the ETCs for those recruited to the active intervention arm, contributing £2088 per participant. The sponsor was able to access this funding on behalf of all sites only once the study had recruited sufficient participants in a year to reach a threshold of £50,000 costs. At this point, the Department of Health and Social Care would contribute £2088 for each additional participant recruited to the active intervention arm of the study. The first year began on the date that a centre recruited its first participant to the study and the second year began on the first anniversary of that date, the third year on the second anniversary, and so on. The maximum subvention available for this trial was £131,768. The NHS was required to meet all other treatment costs associated with the study through local commissioning arrangements. The sponsor was able to invoice the Department of Health and Social Care 6-monthly in arrears for the ETCs incurred above the £50,000-per-annum threshold on behalf of all of the recruitment sites. Centres in Wessex (Portsmouth and Southampton) had made arrangements for the Wessex Clinical Research Network to fund the ETCs up to the £50,000 threshold and the trial and recruitment were set up to maximise that support, which we gratefully acknowledge.
Interventions
Treatment compared with comparator
Active devices
The active TLA device (Airsonett®; Airsonett AB, Ängelholm, Sweden) significantly reduces nocturnal allergen exposure by filtering ambient air through a high-efficiency particulate air filter, slightly cooling the air (0.5–0.8 °C) and ‘showering’ it over the participant during sleep. The reduced temperature allows the filtered air to descend in a laminar stream, displacing allergen-rich air from the breathing zone and thereby reducing allergen exposure without creating draught or dehydration. 39 The device is installed next to the participant’s bed, as shown in Figure 3, and is easy to use with no identified safety concerns in previous trials. The device is CE (Conformité Européenne) marked and licensed for use in the UK for allergic asthma. The device uses the same amount of electricity as a 60-W light bulb and has an anticipated lifespan of 5 years, with filter changes required every 6 months.
FIGURE 3.
The TLA device manufactured by Airsonett. Reproduced with permission from Airsonett AB (Ängelholm, Sweden).

Placebo devices
The placebo devices were adjusted to deliver isothermal air instead of slightly cooled air and holes in the filter effectively allowed the air to bypass it while still maintaining an equivalent sound and airflow level as with the active device. This allowed the placebo device to deliver a laminar flow of non-filtered, non-descending, isothermal air that, when mixed with the warm body convection current, ascended towards the ceiling and thus had no effect on the normal air flow pattern around the breathing zone. There was no difference in the air delivery rate, perceived air movements or sound level between the active and the placebo device. The human body is not able to detect an absolute temperature difference of 0.75°C and, therefore, there is no perceptible temperature difference sleeping beneath an active or a placebo device. Electricity usage was the same as for the active device and the filter was changed at 6-monthly intervals.
Asthma care during the trial
Treatments when stable
All participants were evaluated during the study follow-up visits (study visits 3–6) by clinicians with expertise in severe asthma. These experts were able to identify and exclude alternative or comorbid pathologies contributing to poor asthma control and confirm treatment adherence.
No adjustment or reduction in asthma medications (excluding antihistamines and nasal corticosteroids) was allowed during the trial (unless required for patient safety reasons) because of the significant risk of precipitating severe asthma exacerbations. Any variation in non-asthma medication usage was recorded at each follow-up visit [including the use of over-the-counter (OTC) medications].
Those participants using variable maintenance and adjustable reliever therapy (MART), which combines ICS and bronchodilator therapy in a single inhaler, were converted to a fixed-dose regimen (preferably without changing inhalers) and an alternative short-acting bronchodilator [e.g. salbutamol, terbutaline (Bricany®, AstraZeneca UK Ltd)] by the site team for the duration of the trial. A LASER ‘BDP equivalent’ dose calculator was developed to allow centres to easily calculate the BDP equivalent of their ICS based on the dose and frequency of use and on known pharmacokinetics of all available inhaled therapy.
Participants using a self-management plan (SMP) prior to the trial were allowed to continue and were asked not to change this during the trial treatment period.
Asthma exacerbations
Asthma exacerbations were managed following best clinical practice in the appropriate setting following the national BTS/SIGN guidelines. 16
If participants required urgent medical attention at any time during the follow-up period, they were instructed to call 999 and/or to attend the ED. Participants who did not require urgent medical attention were instructed to follow their normal process for seeking medical attention from their general practitioner (GP), practice nurse or asthma specialist within working hours and to contact their local primary care out-of-hours service at other times.
Participants who self-managed their OCSs were instructed to contact 999 if they required urgent medical attention or to self-manage in the community as directed by their agreed SMP if they did not require urgent medical attention.
Participants reported severe exacerbations to their local site trial team as soon as possible after exacerbation onset.
Clinicians prescribed the process for reducing and ultimately stopping corticosteroid treatment and returning to a normal maintenance dose after each exacerbation, determined by individual patient need.
Intervention standardisation
Prior to shipping, the manufacturer (Airsonett) ensured that all devices were quality checked to CE standard on air temperature regulation, airflow and breathing-zone particle reduction metrics. It also provided all of the required quality control documentation.
Adherence monitoring
To simplify adherence to the intervention, study devices were programmed at installation to automatically turn on for a minimum of 10 hours to cover the participants’ normal sleeping hours. This could be over-ridden by participants should they wish to start the treatment at a different time or turn off the device. Participants were allowed to increase their use of the device, for example for daytime naps. All episodes of use of the device were documented in the daily participant-completed TLA diary, which was collected at the follow-up visits (see Temperature-controlled laminar airflow diary).
Outcomes
The trial used validated, standardised primary and secondary outcomes for clinical asthma trials recommended by the ATS/ERS40 and endorsed by the Core Outcome Measures in Effectiveness Trials (COMET) initiative. 41 Comparison of data at multiple time points was carried out to assess the magnitude and rate of treatment response and variation in level of control.
Primary outcome
There was one primary outcome: severe asthma exacerbations occurring within the 12-month follow-up period.
Severe asthma exacerbations are defined in accordance with ATS/ERS guidelines40 as a worsening of asthma requiring systemic corticosteroids [≥ 30 mg of prednisolone or equivalent daily (or a ≥ 50% increase in dose if maintenance dose is ≥ 30 mg of prednisolone)] for ≥ 3 days. Courses of corticosteroids separated by ≥ 7 days were treated as separate severe exacerbations.
Measurement of primary outcome
Participants were asked to start a participant exacerbation diary (PED) when exceeding the ‘exacerbation dose’ threshold of systemic corticosteroids, individually defined for each participant during randomisation visit 2. The PED included PEF measurements (using the trial electronic PEF device), OCS dose, reliever medication use and nocturnal asthma symptoms. Participants were asked to report severe exacerbations to their local site trial team as soon as possible after onset via a dedicated telephone line or a secure NHS e-mail account. Whenever possible, participants were asked to attend an exacerbation review with their local trial team within 72 hours to corroborate the exacerbation, at which the local trial team completed an exacerbation review form.
If participants were not able to attend an exacerbation review, the PED was collected at the next follow-up visit.
Information about exacerbations was also collected from the participant-completed daily diary in which participants recorded their daily corticosteroid dose (TLA diary) and from follow-up visit forms completed by the clinician delivering each follow-up visit.
Details about how these various sources of exacerbation data were combined to make a useable primary outcome are detailed in Data collection.
Secondary outcomes
Quantitative
The three quantitative secondary outcomes of the LASER trial were:
-
Asthma control – to assess the impact of nocturnal TLA treatment on asthma control.
-
Quality of life – to ascertain the effect of TLA treatment on quality of life in participants with poorly controlled severe allergic asthma and their adult carers.
-
Impact and cost-effectiveness – to evaluate the impact of TLA treatment on health-care utilisation and related costs to fully assess the cost-effectiveness, both at 1 year and over the lifetime of the participant, of nocturnal TLA treatment using a cost–utility analysis to determine the incremental cost per QALY gained, and to evaluate the impact of TLA treatment on education/work days lost.
The data from which these outcomes could be determined were collected as detailed in the following sections. All data collected at the screening or randomisation visit were recorded by the attending clinician on CRFs. All data collected at any of the 3-, 6-, 9- or 12-month follow-up visits were recorded by the attending clinician on follow-up visit forms.
Asthma control
The following indicators of current asthma control were determined and recorded at the randomisation visit (as baseline data) and at each follow-up visit (3, 6, 9 and 12 months during the trial, as intervention data):
-
lung function measures –
-
pre-bronchodilator FEV1
-
mean morning pre-bronchodilator PEF rate over 2 weeks preceding the follow-up visit
-
fraction of exhaled nitric oxide (FeNO)
-
-
ACQ score
-
Asthma Control Diary (ACD) score over 2 weeks preceding the follow-up visits
-
22-outcome Sino-Nasal Outcome Test (SNOT-22) score.
The following indicators of risk of future adverse asthma outcomes were also determined and recorded at these visits:
-
severe exacerbations (see Asthma exacerbations)
-
systemic corticosteroid use over the 12-month follow-up period only
-
post-bronchodilator FEV1 at the 12-month follow-up only.
Quality of life
The following health-related participant quality-of-life scores were determined and recorded at the randomisation visit (as baseline data) and at each follow-up visit (3, 6, 9 and 12 months, as intervention data):
-
Standardised Asthma Quality of Life Questionnaire [AQLQ(S)] score
-
EuroQol-5 Dimensions, five-level version (EQ-5D-5L) score.
The following health-related adult carer quality-of-life score was determined and recorded at the randomisation visit (as baseline data) and at study visit 6 only (12 months, as intervention data), if the carer consented to this:
-
Adult Carers Quality of Life (AC-QoL) questionnaire score.
Impact and cost-effectiveness
The impact and cost-effectiveness measures that were calculated and recorded at follow-up visits were:
-
Work Productivity and Activity Impairment (Asthma) [WPAI(A)] score at the randomisation visit (as baseline data) and at 3, 6, 9 and 12 months (as intervention data)
-
Work Productivity and Activity Impairment (Caregiver) [WPAI(CG)] score at the randomisation visit (as baseline data) and at 12 months only (as intervention data)
-
health-care resource use and costs at 3, 6, 9 and 12 months.
Device usage
The following measures of device usage were determined and recorded at the follow-up visits to identify participants with persistent or recurrent non-adherence to the device usage requirement of five nights per week, excluding holidays:
-
participant-reported device usage at 3, 6, 9 and 12 months
-
engineer-reported device usage at 6 and 12 months only.
Persistent or recurrent non-adherence could lead to a participant being withdrawn from the trial.
Global Evaluation of Treatment Effect questionnaire
The Global Evaluation of Treatment Effect (GETE) questionnaire was completed at 12 months by both participants and physicians as a simple measure of the perceived treatment effectiveness of the TLA device, using five responses ranging from excellent (complete control of asthma) to worsening (deterioration in asthma).
Qualitative
In addition to the quantitative secondary outcomes described (see Quantitative), there was one qualitative secondary outcome:
-
acceptability – to qualitatively evaluate participant and carer perceptions, values and opinions of the trial process and TLA device to identify potential modifications to improve participant acceptance and to inform future implementation of the device within the NHS setting.
Supplementary variable
Indoor Air Quality Questionnaire
It was decided that the air quality of the indoor home environment should be measured as a supplementary variable in the LASER trial to maximise the impact of the investigation into non-intrusive management of people with severe allergic asthma. The statistical analysis of these measurements is not included in this report.
Sample size calculation
Members of the PPI group felt that a reduction of one severe exacerbation per year with a new treatment would be worthwhile to them and have a meaningful impact on their life. Based on an estimated rate of two severe asthma exacerbations per participant over the 12-month period in the placebo group (informed by previous trials; see Table 28), it was calculated that a minimum of 222 participants (111 per group) were required to provide 80% power (at the 5% two-sided significance level) to detect a clinically meaningful 25% reduction in the average exacerbation rate in the group using the TLA device. This sample size was based on a Poisson regression model with the treatment group as the covariate and a 10% overall dropout rate. 42 A review of comparative interventions of proven efficacy in severe asthma gave effect sizes ranging from 21% to 63%, with a mean of 41% (see Table 28). Given that this was a pragmatic trial in which the intervention was expected to be less effective than in an efficacy trial, a more conservative effect size of 25% was chosen deliberately. This represents, on average, one less severe exacerbation every 2 years.
Termination conditions
The Data and Safety Monitoring Committee (DSMC), a committee independent of the sponsor and with the major aims of safeguarding the interests of trial participants, monitoring the main outcome measures, including safety and efficacy, and monitoring data quality and completeness, was granted authority to be supplied with interim data emerging from the trial during the period of recruitment as frequently as was requested. In the light of interim data and other evidence from relevant studies, the DSMC could inform the Trial Steering Committee (TSC) if, in its view, there was proof beyond reasonable doubt that any part of the protocol under investigation was either clearly indicated or contraindicated, either for all participants or for a particular subgroup of trial participants. A difference of at least 3 standard errors (SEs) in the interim analysis of the major end point was indicated as potential justification for halting or modifying the trial prematurely.
Two post hoc analyses of the primary outcome data were requested by the DSMC: (1) using an alternative definition of severe exacerbation and (2) subgroup analysis by allergy to dust mites.
Recruitment
Participants
The trial opened to recruitment at Portsmouth Hospital on 7 May 2014, with the first participant randomised on 25 May 2014 and the last recruited in January 2016. In total, 545 participants were screened against the inclusion/exclusion criteria for the trial during this period: 56 of these were excluded and 489 were approached for consent. A total of 100 participants either refused consent or were excluded, with 389 being consented. Of these, 149 participants failed the pre-screening phase; 240 participants were entered into the trial and randomised.
The overall recruitment rate is shown in Figure 4. It can be seen that the target participant recruitment rate was not met during the first 8 months of the trial. This was partly because of multiple recruiting sites activating their trial sites later than anticipated. Nevertheless, the revised recruitment trajectory was met and the recruitment target was ultimately exceeded (240 participants recruited instead of 222) because the 2-week pre-screening phase meant that new participants had already begun the pre-screening phase once the target had been met. It was felt unethical to exclude these participants on the basis of meeting a target.
FIGURE 4.
Participant recruitment rates.
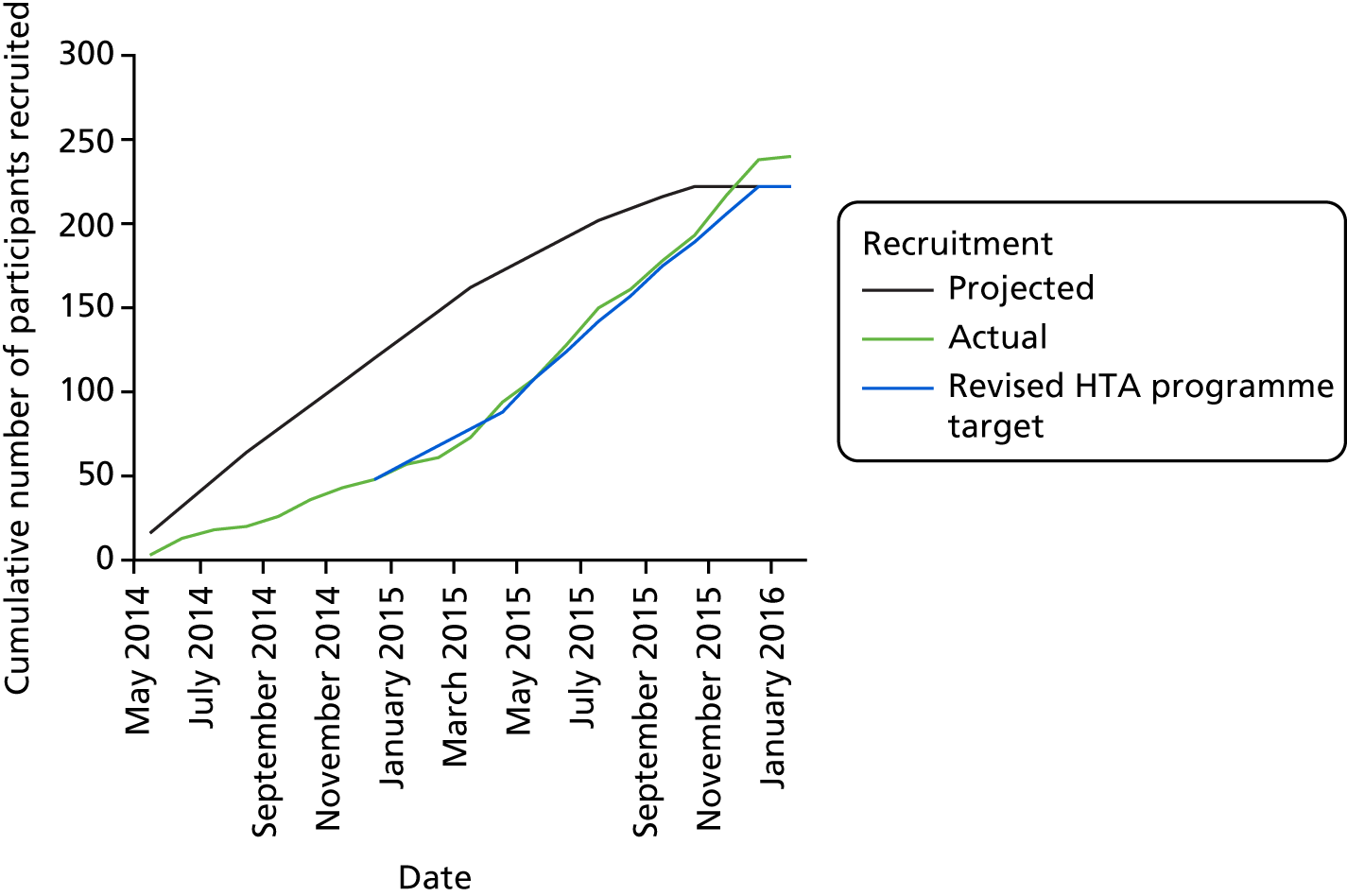
Table 3 lists the sources that referred the 240 recruited participants to the trial. In total, 80% of participants were recruited as existing participants of the 14 health-care provider recruiting sites or PIC general practice surgeries. The majority of the remaining participants were recruited through social media channels, which are described in Social media.
| Referral source | Randomised participants, n (%) |
|---|---|
| Existing clinic patient | 192 (80) |
| Social media (Trialbee) | 27 (11) |
| Social media [Asthma UK/Allergy UK/Tillison Consulting (Waterlooville, UK)] | 13 (5) |
| Newspaper advertisement | 0 |
| Radio advertisement | 1 (< 1) |
| Other | 7 (3) |
| Total | 240 |
Adult carers and partners
Four adult carers consented to participation in the trial for the purposes of collecting quality-of-life and productivity information about these stakeholders; 32 partners gave informed consent for inclusion in the qualitative study.
Website
The trial website (see www.lasertrial.co.uk; accessed 29 November 2018) (see Figure 5 for a screenshot), which was approved by the relevant ethics committee, enabled the public to submit the following screening details if they were interested in participating in the trial: age, postcode, smoking status, allergies, number of exacerbations in the previous 12 months, current asthma treatment and current asthma symptoms. The trial team was notified of those individuals who passed these screening questions. The process was approved by the information governance framework of the sponsor and that of its Caldicott Guardian.
FIGURE 5.
The LASER trial website landing page.

Strategies for increasing trial publicity and channelling more traffic to the LASER website were required to facilitate recruitment to time and target.
Television coverage
In September 2014, Professor Chauhan (Chief Investigator) appeared alongside one of the trial participants in a news feature on British Broadcasting Corporation (BBC) South Today. Following the broadcast, BBC South published the video on its Facebook (Facebook, Inc., Menlo Park, CA, USA; www.facebook.com) and Twitter (Twitter, Inc., San Francisco, CA, USA; www.twitter.com) accounts. The Facebook post included a comment displaying the trial website URL so that potential participants could access more information and register for the trial. The Google Analytics tool (Google Inc., Mountain View, CA, USA) revealed that this social media interaction led to a significant increase in website traffic on the day of the broadcast, with 160 individual website sessions (Figure 6).
FIGURE 6.
BBC South social media content and result. Reproduced with permission from BBC South (Southampton, UK).

Social media
As more evidence is emerging from both commercially driven and academic trials that social media can be a useful tool in recruitment, a social media strategy for the LASER trial was developed. This strategy was considered to be especially appropriate for the trial population, who tend to be younger patients and are therefore likely to be working and have less time to engage with the traditional advertising and recruitment pathways, but who are likely to explore the availability of new treatments and technologies for their condition via the internet and social media platforms. All social media postings were approved by the ethics committee and moderated by the trial co-ordinator and the trial lead research nurse, who were involved in recruiting for the trial.
National charities
First, we partnered with national charities with an interest in the new treatment, Asthma UK and Allergy UK, which publicised the trial on their websites and posted trial information on both Twitter and Facebook that signposted patients to the LASER trial website; see Figure 7 for example content from Asthma UK, which has 29,000 followers on Twitter and has had > 40,000 likes on Facebook. Allergy UK has 5000 followers and has had > 7000 likes on Facebook.
FIGURE 7.
Asthma UK website, Facebook and Twitter content. Reproduced with permission from Asthma UK (London, UK).

Using Google Analytics, a spike in activity on the website was seen when posts/tweets were published.
Facebook and Twitter
Second, we partnered with Tillison Consulting (see https://tillison.co.uk; accessed 29 November 2018) to develop a LASER trial Facebook page and set up business accounts that facilitated trial advertisements on Facebook and Twitter. This business account permits targeted advertising, enabling us to set demographic parameters based on trial inclusion criteria (aged between 18 and 65 years, both men and women, location up to 50 km from a recruiting centre) and also to target people who have a search or following history that indicates an interest in asthma or asthma awareness. The tweets and posts directed people towards the study website and through the screening questions. Techniques such as ‘Google remarketing’ were also used, in which people who had visited the website would be shown ‘branded adverts’ in the following 30-day period, although these were less successful than the social media campaigns.
On Facebook, > 100,000 individuals were reached, there were 1361 clicks through to the website and a large number of patients engaged with the trial via ‘likes’, ‘shares’ and ‘comments’, which would have further increased the trial’s visibility if it led to patients sharing the information with friends or family with the condition. Similarly, on Twitter, > 150,000 people were reached.
Trialbee
Finally, in March 2015 we partnered with the Swedish company Trialbee (Malmö, Sweden), which specialises in social media solutions for clinical trials. On arriving at the Trialbee LASER trial ‘landing page’ (Figure 8), where there is information about the trial, interested patients answered a series of initial screening questions concerning their eligibility and those that passed screening were passed on to the trial team.
FIGURE 8.
Text from the Trialbee LASER trial landing page.

The Trialbee screening questions were answered 14,059 times, with 910 people passing questions. Of these, and following more in-depth screening by our trial team, 57 were eligible for the trial and 27 of these were consented and randomised to participate in the trial. This represented 16% of people recruited during this time period, demonstrating the power of social media for recruiting patients to clinical trials. In addition to being a cost-effective way to recruit patients, another benefit of social media engagement is that it empowers people to approach the research team of their own volition, thus possibly selecting more motivated, engaged research participants.
Adaptation of the recruitment approach
Many reasons for an initial delay in recruitment were encountered, as described in the following sections.
Delays in opening the first four and subsequent sites
There were a number of delays in opening the initial four recruiting sites because of delays in research and development (R&D) department approvals. We therefore identified a number of potential additional recruiting centres.
Change in National Institute for Health and Care Excellence recommendations for omalizumab
After the trial began, NICE reappraised its technology appraisal guidance for omalizumab,14 a drug manufactured by Novartis that is a costly injectable anti-IgE therapy, delivered from centres with asthma specialists, and a treatment option that competes directly with the TLA device tested in this trial. In the 2007 guidance,43 the NICE recommendation included confirmation of IgE-mediated allergy to a perennial allergen by clinical history and allergy skin testing and with either two or more severe exacerbations of asthma requiring hospital admission within the previous year or three or more severe exacerbations of asthma within the previous year, at least one of which required admission to hospital and a further two that required treatment or monitoring in excess of the patient’s usual regimen in an accident and emergency (A&E) unit.
The revised guidance broadened the inclusion criteria to include any patient who needed continuous or frequent treatment with OCSs (defined as at least four courses in the previous year).
This expanded the population of patients who might be eligible for omalizumab (no requirement to attend hospital or EDs and everyone on OCSs for asthma, irrespective of their prior exacerbation history). This had a negative impact on LASER trial recruitment as it dramatically shrunk the anticipated ‘prevalent’ population of severe asthma patients previously identified by recruiting centres when performing the extensive feasibility assessments and recruitment projections.
Omalizumab treatment pathway
This change in the licensed indication for omalizumab led to considerable variation among centres regarding the treatment option offered to patients. Several centres offered omalizumab therapy to patients prior to them being considered for the LASER trial, even though they potentially met the eligibility criteria for both treatments; some specialists were concerned that half of the patients may be randomised to placebo in this trial and therefore could still remain potentially uncontrolled.
The eligibility criteria allowed the recruitment of participants with severe allergic asthma who had failed or responded only partially to omalizumab therapy after a washout period of 120 days. Even then, a ‘trial’ of omalizumab treatment involves 2-weekly injections for 16 weeks before a patient is deemed to have responded or failed treatment. Thus, any potential participant who was first offered omalizumab in preference to the LASER trial device would have received omalizumab for 16 weeks (4 months) and then could not be screened for another 3 months, delaying recruitment to the LASER trial even further.
Competition with other commercial research trials
There has been a plethora of monoclonal antibody treatments for severe asthma in the last 3 years, sponsored by the pharmaceutical industry and facilitated by all local Clinical Research Networks through their commercial portfolio to encourage centres to participate in more commercial trials. The 30-day metrics of the Clinical Research Networks ensures that centres are incentivised and performance managed to recruit patients into all portfolio trials, including those that were competing with the LASER trial. Naturally, nearly all of our centres were approached to participate in such trials, with significant variations between centres in terms of participation; in some cases participants were recruited to other trials in preference to the LASER trial.
NHS England service specifications for severe asthma centres
In 2013, NHS England controversially approved the service specification for a limited number of centres to be designated as specialist asthma centres (SACs) (Birmingham, Brompton, Leicester and Manchester being approved at the time of commencement of the LASER trial). The designation and numbers of SACs were finally confirmed in March 2019. One criterion for specialist status had been the introduction of ‘bronchial thermoplasty’ as a new treatment option in severe asthma, with a requirement that each SAC perform 10 procedures per year to maintain status; those aspiring to become a SAC are required to perform a similar number of procedures over a 12-month period. Given the political incentives, potential participants eligible for the LASER trial were, in some instances, considered for bronchial thermoplasty in preference to the LASER trial in order to meet the politico-clinical targets set by the service specification.
Screen failures prior to randomisation
The number of patients requiring active screening to the point of consenting was not known; consequently, centres had been unaware of the number of potential participants that needed to be screened over any given period of time to ensure a sufficient volume of randomisation. Based on estimates, it was anticipated that two of every three patients screened would progress on to randomisation. Although relatively minor, this guidance allowed centres to manage their recruitment performance.
Resource problems
Despite the trial being on the Clinical Research Network portfolio, there were several instances of Clinical Research Networks not being able to support sites with clinical staff because of lack of funding, which required the transfer of potential participants to other centres. Other centres similarly completed a feasibility assessment but did not open because of staff shortages.
Solutions to boost recruitment
The actions taken to overcome the challenges to recruitment were wide-ranging:
-
increase the number of centres
-
greater engagement and performance management of existing centres
-
website optimisation
-
search engine optimisation
-
use of Google Display
-
Google remarketing
-
use of social media
-
media broadcasts and outputs to facilitate recruitment
-
broaden eligibility criteria (i.e. reduction in ICS dose required, reduction in lower age limit for inclusion and reduction of screening period from 4 to 2 weeks)
-
modification of patient pathway at existing centres to ensure trial visibility (Chief Investigator visited some of the centres)
-
detailed interrogation of primary care for suitable participants.
Participant screening and enrolment
Screening visit (–2 weeks)
At the screening visit, informed consent was sought for participation in the main trial as well as the qualitative focus group sessions. Informed consent preceded any study procedures (including tests to ascertain eligibility for trial inclusion), thus ensuring that individuals had had an opportunity to fully discuss the participant information sheet (PIS) with the research team.
Individuals who met all of the inclusion criteria and did not meet any of the exclusion criteria pertinent to the screening visit (namely the baseline spirometry including the reversibility test, skin prick test, blood test and ACQ score criteria described in Eligibility and inclusion/exclusion criteria) were trained in the use of the electronic PEF meter to measure morning and evening PEF (prior to taking asthma medications) and in completion of the ACD for the 2 weeks prior to randomisation visit 2.
If appropriate, a PIS was given to each participant for his or her adult carer and/or partner to complete, if he or she also wished to participate.
Extension of the screening period
To continue to be deemed potentially eligible for the trial, individuals needed to demonstrate acceptable compliance with the electronic PEF recordings and ACD during the 2-week screening period. However, in the event of electronic PEF device malfunction or if, in the investigator’s opinion, there were significant extenuating circumstances, the screening period was extended by up to a further 2 weeks. Participants experiencing a severe exacerbation during the screening period were no longer eligible but could be rescreened 2 weeks after returning to their maintenance asthma medications.
Randomisation visit 2 (0 months)
The following data collected during the screening period and at the randomisation visit (study visit 2) were used to assess whether or not potential participants fulfilled the following remaining eligibility criteria (see Eligibility and inclusion/exclusion criteria for a full description of the eligibility criteria):
-
demographics, asthma history and asthma review (see Demographics, asthma history and asthma review)
-
review of the ACD, including electronic PEF recordings (see Asthma Control Diary)
-
ACQ score (see Participant questionnaires).
Those individuals who met these remaining eligibility criteria were confirmed as eligible for participation. The data used in the final eligibility assessment were supplemented by the following data from the CRF to act as baseline data (see Secondary outcomes, Quantitative, for descriptions of these measures):
-
SNOT-22, AQLQ(S), EQ-5D-5L, WPAI(A) and Indoor Air Quality Questionnaire scores
-
FeNO
-
baseline spirometry after withholding bronchodilator (pre-bronchodilator FEV1).
If appropriate, informed consent from adult carers of participants to participate in the trial was sought. If a participant’s carer was unable to attend, an additional appointment was arranged. After giving informed consent the carers completed:
-
the AC-QoL questionnaire
-
the WPAI(CG) questionnaire.
If appropriate, informed consent from partners of participants for inclusion in the qualitative study was also sought. Again, if a participant’s partner was unable to attend, an additional appointment was arranged.
Finally, participants were then provided with the materials required to measure and record the primary and secondary trial outcome data (see Outcomes and Secondary outcomes, respectively, for outcome definitions), including a TLA diary for self-reported device usage (see Temperature-controlled laminar airflow diary), a resource use log for health-care utilisation over each 3-month follow-up period (see Resource use log), at least three PEDs (see Participant exacerbation diary and exacerbation review form) and a 2-week ACD (including electronic PEF recordings; see Asthma Control Diary) issued for completion prior to the 3-month follow-up visit.
Randomisation
Devices
The trial statistician generated a list of LASER trial-specific device numbers (L-numbers) coded against a X or a Y (i.e. active or placebo), which they sent in a password-protected electronic file to the following Airsonett personnel only: Chief of Operations, the Director of R&D and the Director of Quality Assurance. This list was generated using the Stata® version 13.1 command RALLOC (StataCorp LP, College Station, TX, USA). A total of 400 codes were generated in blocks of 20 (10 active and 10 placebo) in line with Airsonett manufacturing the devices in blocks of 20.
The Airsonett personnel with access to the list oversaw the manufacture of the active and placebo devices according to the L-numbers. Each device was labelled with both L-number and manufacturing serial number.
Participants and minimisation criteria
Once the eligibility of an individual was confirmed at the randomisation visit, the trial team at the recruiting site contacted the Oxford Respiratory Trials Unit (ORTU; part of the Oxford Clinical Trials Research Unit) to arrange randomisation. Participants were randomised in a 1 : 1 ratio to receive either an active TLA device or a placebo device. Randomisation was undertaken centrally by Sealed Envelope™ Ltd (London, UK) using a validated computer randomisation program including a non-deterministic minimisation algorithm to ensure balanced allocation of participants across the two treatment groups for each clinical site, prevalent compared with incident cases and the following prognostic factors at baseline, which are key indicators of future exacerbation risk: exacerbation frequency in the previous 12 months (two, three or more than three), use of OCSs (yes/no) and pre-bronchodilator FEV1 (> 50% predicted yes/no). In essence, this approach accounted for the characteristics of the participants who had been previously randomised when randomising each new participant. By trial end, 119 of the 240 participants were allocated an active device and 121 were allocated a placebo device.
Participants previously known to the recruiting centre were termed prevalent participants, whereas participants not previously known to the recruiting centre but referred from another centre or through a social media channel were termed incident participants.
Once participant randomisation was complete, Sealed Envelope sent a secure e-mail to the local trial team to confirm randomisation and to provide the information required for implementation, described in the following section. It should be noted that the device allocation was embedded into the Sealed Envelope system.
Implementation
After randomisation of each new participant to the active or placebo treatment group, Sealed Envelope selected which device with the appropriate treatment would be received by each randomised participant. It then sent secure e-mail and text messages to the local independent device distributor (Bishopsgate Specialist Logistics & Installation, Swindon, UK), which specialises in medical devices, with the following details: participant trial number, allocated L-number (without X or Y designation so that the allocated treatment arm remained concealed) and an exclusive link for the engineering team to log in to access participants’ contact details.
Device installation
The Bishopsgate engineering team contacted participants within 72 hours of their randomisation visit to arrange device delivery and installation. Members of the engineering team were trained, with certificates of competency based on completion of a Good Clinical Practice (GCP) training course on trial procedures, and they followed a standard device delivery, filter change and removal protocol developed by the trial team. The agreement with Bishopsgate was that devices should be installed within 10 working days, excluding weekends and bank holidays. Members of the engineering team left written instructions on device operation for participants during the installation visit.
Device maintenance
Devices had a filter change 6 months into their use in the trial, automatically calculated from the date of randomisation. The Trial Manager informed Bishopsgate on a monthly basis of which filter renewals were due.
Troubleshooting
One item discussed during the telephone review conducted by the site research team with participants 1 month into their trial period was troubleshooting on any issue related to the trial, including difficulties with using the device.
Blinding
The methods described above for the manufacture of the active and placebo devices, the allocation of participants to the two treatment arms and the implementation of device installation ensured that all participants, trial teams and members of the installation team were blinded to the trial treatments. This ensured that everyone apart from the statistician who generated the codes for the devices and the programmers at Sealed Envelope was blinded to treatment allocation. Airsonett was the only party to know the differences between the real and the placebo devices but it did not have access to any information related to participants recruited to the trial or device usage once in the UK.
Study assessments
This section describes the quantitative and qualitative data collected at the study visits, including those recorded between study visits:
-
quantitative data –
-
demographics, asthma history and asthma review
-
lung function measures:
-
pre-bronchodilator FEV1
-
reversibility testing
-
FeNO
-
-
allergy testing:
-
skin prick test
-
serum-specific IgE testing
-
measurement of serum total IgE and peripheral blood eosinophil count
-
-
participant questionnaires:
-
ACQ
-
AQLQ(S) (disease-specific quality of life)
-
EQ-5D-5L (generic quality of life)
-
SNOT-22 (rhinosinusitis health status)
-
Indoor Air Quality Questionnaire
-
GETE questionnaire
-
WPAI(A) questionnaire
-
-
carer questionnaires:
-
AC-QoL questionnaire
-
WPAI(CG)
-
-
pre-visit data collection:
-
ACD – PEF rate, symptom and reliever medication use
-
TLA diary – device usage, corticosteroid dose, reliever usage, work/study days lost
-
resource use log: health-care usage
-
PED
-
-
-
qualitative data –
-
focus groups.
-
These quantitative and qualitative data were used for one or more of the following six purposes:
-
to inform eligibility of an individual to participate in the trial against the inclusion and exclusion criteria (see Eligibility and inclusion/exclusion criteria, Participants) (P1)
-
to build the primary outcome data set (see Primary outcome for the primary outcome definition) (P2)
-
to build the secondary outcome data sets (see Secondary outcomes for the secondary outcomes definitions) (P3)
-
to create participant factor data sets used to control the primary and secondary outcome data against associations unrelated to the treatment (P4)
-
to contribute to the baseline outcome data sets (P5)
-
to build the supplementary variable data sets that may be used for future analyses, not all of which are mentioned in Supplementary variable (P6).
The purpose of each type of data mentioned in the following section is indicated by the codes P1–P6.
Quantitative data
Demographics, asthma history and asthma reviews
The following data were recorded about each participant on the CRF at the randomisation visit only:
-
demographics –
-
age (P1, P4)
-
sex (P4)
-
socioeconomic class (P4)
-
ethnicity (P4)
-
-
asthma history –
-
date of asthma diagnosis (P1, P4)
-
history of life-threatening and near-fatal asthma exacerbations [intensive treatment unit (ITU) admissions] (P6)
-
number of severe asthma exacerbations in previous 12 months (P1, P5)
-
history of previous asthma treatment (P1)
-
history of atopy (P1)
-
family history of asthma/atopy (P6)
-
asthma triggers (P1)
-
medical or surgical comorbidities (P1)
-
occupational history (P1)
-
smoking history (P1)
-
height (cm)/weight (kg) for measuring predicted lung function (P3).
-
The following data were collected at each follow-up visit as well as at the randomisation visit and recorded by the attending clinician on the follow-up visit form/CRF:
-
asthma review
-
current asthma symptoms and treatment (P1, P3, P5)
-
current medications (P1)
-
history of severe asthma exacerbations since the previous trial visit and current participant-reported clinical status (still in exacerbation or recovered) (P2)
-
unscheduled asthma-related health-care use (P3)
-
work/study days lost as a result of asthma symptoms (P3).
-
Lung function measures
The following indicators of lung function were collected at the study visits:
-
Pre-bronchodilator FEV1 (P1, P3, P5) – spirometry was conducted at the screening visit, the randomisation visit and the 3-, 6-, 9- and 12-month follow-up visits to collect the following variables:
-
FEV1 (l)
-
FVC (l)
-
FEV1/FVC ratio
-
forced expiratory flow at 25–75% of the pulmonary volume (FEF25–75) (%).
FEV1 and FVC were documented both as absolute values and as a percentage of the predicted value. A spirometer conforming to ATS/ERS standards44 was used as specified by the manufacturer’s instructions.
-
-
Reversibility testing (P1, P3, P5) – post-bronchodilator FEV1 (both percentage change and volume change) was measured at the screening visit and 12-month follow-up visit only. Following ATS/ERS standards,44 post-bronchodilator FEV1 was defined as FEV1 recorded 15 minutes after administration of 400 µg of salbutamol via a metered dose inhaler and spacer device. An improvement in FEV1 post-bronchodilator use of ≥ 12% or 200 ml was considered significant.
-
FeNO (P1, P3, P5) – FeNO was measured before spirometry at the randomisation visit and the 3-, 6-, 9- and 12-month follow-up visits. The measurements were made using a NIOX MINO® device (Aerocrine AB®, Solna, Sweden) as specified by the manufacturer’s instructions and outlined in the ATS/ERS standards. 45
Allergy testing
The following allergy tests were made during the screening visit to determine whether the allergy-related trial inclusion criteria were met:
-
Skin prick testing (P1) – a standard skin prick test procedure using common indoor aeroallergen (Der p 1, Der f 1, Asp f 1, Alt a 1, Cla h 1, Fel d 1 and Can f 1) extracts along with negative (saline) and positive (histamine) controls was performed on all subjects. This occurred during the randomisation visit instead of at the screening visit if antihistamine hold was required (see Appendix 1). Skin prick testing was performed in accordance with the practice parameter released by the American Academy of Allergy, Asthma and Immunology. 46 A positive skin prick test reaction was measured as a wheal of at least 3 mm diameter greater than the negative control.
-
Serum-specific IgE testing (P1) – if skin prick testing was not available, a blood sample was taken to measure serum-specific IgE to common indoor aeroallergens (Der p 1, Der f 1, Asp f 1, Alt a 1, Cla h 1, Fel d 1 and Can f 1). A specific-serum IgE concentration of > 0.35 IU/l was considered to represent allergen sensitisation. Serum-specific IgE testing could also have been used if there was uncertainty about a skin prick test result or if there was a negative skin prick test result in the context of a patient on long-term maintenance systemic corticosteroids.
-
Measurement of serum total IgE and peripheral blood eosinophil count (P1) – a blood sample was collected to measure serum total IgE and peripheral blood eosinophil levels.
Participant questionnaires
-
Asthma Control Questionnaire (P3): the well-validated 7-item ACQ47 was used to assess asthma control over the 7 days leading up to the screening visit, the randomisation visit and the 3-, 6-, 9- and 12-month follow-up visits. It was administered at the same time during each visit, with the participant blind to the results of other tests. The ACQ includes five symptom scores, the amount of daily rescue bronchodilator usage and a measure of airway calibre (FEV1% predicted). Responses are given on a 6-point scale and the overall score is the mean of the responses (0 = totally controlled, 6 = severely uncontrolled). The ACQ has strong evaluative and discriminative properties and has been shown to be very responsive to within-patient changes in asthma control over time. It has a validated minimally important difference of 0.5 to demonstrate clinical significance.
-
Standardised Asthma Quality of Life Questionnaire (P3): asthma-specific quality of life was measured using the AQLQ(S)48 at the randomisation visit and the 3-, 6-, 9- and 12-month follow-up visits. The AQLQ(S) consists of 32 questions within four domains – symptoms, activity limitation, emotional function and environmental stimuli – and it has strong measurement properties and a validated minimally important difference of 0.5. 49 Patients are asked to think about how they have been during the previous 2 weeks and to respond to each of the 32 questions on a 7-point scale (7 = not impaired at all, 1 = severely impaired). The overall AQLQ(S) score is the mean of all 32 responses and the individual domain scores are the means of the items in those domains.
-
EuroQol-5 Dimensions, five-level version (P3): generic health-related quality of life (HRQoL) was measured using the EQ-5D-5L questionnaire50 at the randomisation visit and the 3-, 6-, 9- and 12-month follow-up visits. The EQ-5D-5L is a standardised measure of health providing a simple generic measure of health for clinical and economic appraisal. Patients are asked to think about their health on the day that they are completing the questionnaire and report on any problems (none, slight, moderate, severe and unable/extreme) relating to five attributes (mobility, self-care, usual activities, pain/discomfort and anxiety/depression). Patients are then required to rate their health using a 100-point visual analogue scale (VAS) (0 = worst health you can imagine, 100 = best health you can imagine). The EQ-5D-5L is the most widely used HRQoL measure in adults in the UK and has been shown to be a reliable and valid means of measuring quality of life in asthma patients. 51 In addition to the EQ-5D-5L score, the EuroQol Visual Analogue Scale (EQ-VAS) was also ascertained at each of these visits. The EQ-VAS was determined by asking the participants to indicate their health status at each visit on a 20-cm vertical scale with end points of 0 and 100.
-
22-item Sino-Nasal Outcome Test (P3): the SNOT-22 score is a well-validated and sensitive measure of rhinosinusitis health status52 that was recorded at the randomisation visit and the 3-, 6-, 9- and 12-month visits. The SNOT-22 consists of 22 questions related to symptoms and the social/emotional impact of those symptoms (symptoms are rated on a scale from 0 = no problem to 5 = problem as bad as it can be). Participants were asked to rate the problems according to how they had been over the previous 2 weeks.
-
Indoor Air Quality Questionnaire (P6): the Indoor Air Quality Questionnaire was completed by participants at the randomisation visit to identify key factors affecting air quality within the home environment. This questionnaire is a bespoke domestic indoor air quality assessment tool derived from a combination of the Stockholm53 and Southampton54 Indoor Environment Questionnaires.
-
Global Evaluation of Treatment Effect questionnaire (P3): the GETE questionnaire55 is a simple measure of perceived treatment effectiveness that was completed by participants and clinicians at the 12-month visit. This questionnaire has been used in the evaluation of other treatments in patients with severe allergic (IgE-mediated) asthma. For the purposes of the LASER trial, it required participants and physicians to rate the global treatment effectiveness of the TLA device as excellent (complete control of asthma), good (marked improvement of asthma), moderate (discernible but limited improvement in asthma), poor (no appreciable change in asthma) or worsening (deterioration in asthma).
-
Work Productivity and Activity Impairment (P3): the WPAI(A) version 2 is a validated questionnaire tool for assessing work productivity and activity impairment56 that was completed by participants at the randomisation visit and at the 3-, 6-, 9- and 12-month visits. This questionnaire consists of six questions addressing absenteeism, presenteeism (reduced effectiveness while working), overall work productivity loss (absenteeism and presenteeism) and activity impairment. Participants were asked to count the number of hours that they had missed from work because of problems associated with their asthma and the number of hours missed for other reasons (such as holidays) over the past 7 days. In addition, participants were asked to report the impact of asthma on their work and on the ability to do regular daily activities (on a scale from 1 = asthma had no effect to 7 = asthma completely prevented me). WPAI(A) outcomes are measured as percentages; a higher percentage relates to greater impairment and reduced productivity. A modified WPAI(A) was used for student participants.
Adult carer questionnaires
-
Adult Carer Quality of Life (P3): the AC-QoL questionnaire57 measures the overall quality of life of adult, unpaid carers and was completed by participant carers at the randomisation visit and again at the 12-month visit. The eight domains of this 40-item questionnaire assess support for caring, carer choice, carer stress, money matters, personal growth, sense of value, ability to care and carer satisfaction. Carers are asked to recall their experiences over the previous 2-week period. Scores on the questionnaire have a possible range of 0–120, with higher scores indicating better carer quality of life.
-
Work Productivity and Activity Impairment (P3): a modified WPAI for caregivers [WPAI(CG)] was completed at the randomisation visit and 12-month follow-up visit.
Pre-visit data collection
Asthma Control Diary
Participants were issued with a validated ACD58 at the screening visit, the randomisation visit and the 3-, 6- and 9-month visits to record data for the 2 weeks leading up to each subsequent visit. Towards the end of the trial, a text reminder system was set up to remind participants when to start their ACD and to remind them of the date of their follow-up visit.
Participants recorded the following data on a daily basis for 2 weeks:
-
PEF rate – during the trial, participants performed three morning PEF measurements using a handheld device supplied by the trial team and recorded these measurements in the ACD. During the screening period, participants also performed three evening PEF measurements to assess variability as part of the eligibility assessment. These additional PEF data were stored on the PEF device and downloaded at the randomisation visit.
-
Symptom and reliever medication use – the ACD measures a morning score (two items; 0–6 point scale), a bedtime score including bronchodilator requirement (four items; 0–6 point scale) and a best morning PEF rate measured as percentage of predicted best (0–6 point scale). The overall daily score is the mean of the responses (0 = perfectly controlled, 6 = severely uncontrolled).
-
Device usage – device usage data (displayed on the device screen) documenting the number of hours that the device had been active were collected by:
-
participants at 3, 6, 9 and 12 months
-
the engineering team at 6 and 12 months to coincide with planned filter changes.
These data were recorded in the ACD, for review at the 3-, 6-, 9- and 12-month follow-up visits. Device usage was also one item discussed during the 1-month telephone review, revealing where mitigating steps needed to be put in place to improve adherence early on in the trial.
-
Temperature-controlled laminar airflow diary
Participants were issued with a TLA diary at the randomisation visit to record the following data on a daily basis:
-
use of the TLA device (hours), that is, participant-reported treatment adherence
-
reliever used (number of times)
-
dose of corticosteroids (mg)
-
work/study days lost because of asthma symptoms (hours).
Adherence to completing the TLA diary was another item discussed during the 1-month telephone review, revealing where mitigating steps needed to be put in place to improve adherence early on in the trial.
Resource use log
Participants were issued with a resource use log at the randomisation visit and the 3-, 6- and 9-month follow-up visits to record health-care resource use between study visits. The purpose of these logs was to help participants recollect events as they completed the participant questionnaires administered during the follow-up visits. To keep the questionnaires as simple as possible, they did not distinguish whether the contacts with the health-care service were through the NHS or through private providers, meaning that an underlying assumption of the resource use analyses is that all contacts were financed by the NHS.
The date stamps of the hospitalisation records were correlated with those recorded by health-care providers to avoid double counting.
Participant exacerbation diary and exacerbation review form
Participants were asked to start a PED when exceeding the ‘exacerbation dose’ threshold of systemic corticosteroids individually defined for each participant during randomisation visit 2. The PED recorded the following during exacerbations for a period of 7 days: morning PEF ( l/minute) (recorded using the trial-supplied electronic PEF device), steroid dose (mg), reliever use (number of times) and nocturnal wakening (yes/no).
In addition to completing the PED, participants were asked to report severe exacerbations to their local site trial team as soon as possible after onset via a dedicated telephone line or a secure NHS e-mail account. Whenever possible, participants were asked to attend an exacerbation review session with their local trial team within 72 hours to corroborate the exacerbation, at which the local trial team completed the exacerbation review form. The following information was captured on the exacerbation review form: onset of symptoms, dose of steroid used (mg), corroboration of use of oral steroids of at least 30 mg for 3 days, and change in symptoms and reliever use.
If participants were not able to attend an exacerbation review, a review of the exacerbation occurred by telephone and a exacerbation review form was still completed. If a telephone review was not possible either, an exacerbation review form was completed at the next follow-up visit. The nature of the review (face-to-face or telephone) was recorded on the exacerbation review form and the participant’s PED was collected.
Follow-up visit forms
A regular 3-monthly follow-up visit form was completed by the attending clinician, with participant input at each follow-up visit (at 3, 6, 9 and 12 months), prompting discussion with participants about key issues related to the preceding 3 months. This included number of exacerbations, a review of the TLA diary, medication use, health resource use log, work and study days lost, reported device use, completion of trial questionnaires, FeNO and spirometry.
Qualitative data collection
Sampling and recruitment
Informed consent for participation in the qualitative study was sought at the screening visit. All participants taking part in the LASER trial were contacted towards the end of their 12-month follow-up period with an invitation to attend local focus group sessions.
Not all participants who consented to take part in the focus group sessions were selected: 5–10 participants were selected for each of the focus group sessions on the basis that they best reflected multiple variation (including balance of sex, age and ethnicity).
Focus groups
A focus group is an interview technique in which a group of individuals are encouraged to debate and discuss specific topic areas. The rationale for choosing focus group discussions during the LASER trial was to stimulate alternative views and experiences, evaluate the process and assess the feasibility of the intervention.
In total, 28 participants were invited to attend focus group sessions based on the fact that they had completed at least 9 months of the trial and had consented to attend local focus group sessions when asked at the start of the trial; 20 of these 28 participants agreed to participate and were sent invitation letters to attend one of three focus group sessions hosted by the Portsmouth site during January 2016. The three sessions were held on separate dates at different times of the day, including an evening focus group, in order to provide choice to those invited to attend and maximise attendance. The venue chosen was a non-NHS, non-trial centre, within an easily accessible location with on-site parking, namely the Premier Inn at Port Solent.
Acceptance of the invitations covered consent for the use of verbatim quotations and assurance that the information that a participant provided would be kept strictly in confidence and anonymised.
The focus groups were led by a senior qualitative researcher from the University of Portsmouth, with a senior medical research fellow attending as facilitator and note keeper.
Focus group 1 was held on 21 January 2016 at 18.00. Five participants agreed to attend but only one participant attended on the day. It was agreed to conduct a one-to-one interview with this participant, following the focus group topic guide.
Focus group 2 was held on 26 January 2016 at 10.00. Seven participants agreed to attend but only three participants attended on the day.
Focus group 3 was held on 28 January 2016 at 14.00. Eight participants agreed to attend but only six attended on the day.
In total, 10 participants took part in the focus group sessions, seven females and three males. These 10 participants comprised both satisfied and non-satisfied participants (determined from a combination of device-reported usage and participant-reported device usage).
At the beginning of each focus group discussion, the facilitator checked that participants were aware of the purpose of the discussion, likely topics to be discussed and their right to leave, at any time, without giving a reason for doing so. All participants were asked to respect the views of others and to take turns in speaking to aid the recording of the discussions.
The topics were based on key themes identified during the qualitative telephone interviews conducted during the pilot phase. Free discussion of experiences and ideas was encouraged throughout.
The focus group discussions were audio-recorded and lasted between 60 and 90 minutes. Afterwards, audio-recordings were transcribed verbatim by the Way With Words (London, UK) secure online transcription service. Participants were offered tea or coffee and biscuits, with water available throughout.
Trial conduct
The trial was conducted under the guidance and requirements of the HTA programme. A TSC and a DSMC were appointed with an operational charter; both were independently chaired. The TMG met or teleconferenced weekly during the trial from set-up to the final data collection. Several measures were taken to engage with trial centres to ensure good levels of recruitment. This included three investigator meetings (one in Portsmouth and two in Leicester, at a central location), 3-monthly teleconferences with research nurses and PIs at trial centres and a weekly e-mail that included the trial recruitment numbers at each site (allowing visibility of progress on a weekly basis) and whether or not centres had hit a prespecified recruitment target for that month. These targets were set by the TMG and Chief Investigator. It also included a ‘tip of the week’ that answered some of the frequently asked questions (FAQs) from trial staff. A dedicated LASER trial co-ordinator and LASER trial nurse were appointed to engage with centres more frequently and facilitate recruitment. A dedicated website and Twitter handle ensured that awareness of the trial remained high.
Statistical methods
All statistical analyses were undertaken using a validated statistical package: Stata/IC version 14.2. The results are presented in this report as comparative summary statistics (difference in response rate or means), with 95% confidence intervals (CIs), in accordance with the Consolidated Standards of Reporting Trials (CONSORT) 2010 statement. 59 All of the tests were carried out at a 5% two-sided significance level.
Defining populations for data analysis
Primary statistical analysis
The primary statistical analysis was based on the intention-to-treat (ITT) population, which is defined as all participants included and analysed according to their allocated treatment group irrespective of the treatment received. Note that, in the primary analysis, the five participants who were randomised and withdrew consent to allow data usage were still included, with data set to missing and an assumption that no severe exacerbations were experienced. All other participant data deemed to be ineligible post randomisation or associated with protocol violations were also included in these primary outcome data. Two blinded analyses (without information on treatment allocation added to the clinical database) of the data were undertaken to define the per-protocol population used for the sensitivity analyses (see Defining populations for data analysis) and the minimum data population to identify participants with a threshold minimum number of data:
-
Per-protocol population (as the ITT population but excluding) –
-
participants who did not receive a device
-
participants who withdrew consent for data use.
-
-
Minimum data population – all participants who have at least 90 days of steroid dose information reported (in the TLA diary or PED) or who have reported at least one PED. This requirement included the assumed doses in the TLA diary and PEDs when the dosage was left blank (see Procedure for dealing with missing, unused and spurious data for explanation).
Secondary statistical analysis
Analysis of secondary outcomes was carried out on a variable-by-variable complete case basis, that is, participants were included in each individual secondary outcome analysis only if all data relevant to that specific analysis were available, with no requirement for their remaining (non-relevant) data. Secondary outcomes with insufficient data were not analysed.
Analysis of the primary end point
Definitions
The primary end point definition was rate of severe exacerbations in a 12-month period.
Severe exacerbations were defined in accordance with ATS/ERS guidelines40 as a worsening of asthma requiring systemic corticosteroids, that is, ≥ 30 mg of prednisolone or equivalent daily (or a ≥ 50% increase in dose if maintenance 30 mg of prednisolone or above) for ≥ 3 days.
Courses of corticosteroids separated by ≥ 7 days were treated as separate severe exacerbations.
A post hoc analysis included the worsening of asthma requiring systemic corticosteroids, that is, ≥ 10 mg of prednisolone or equivalent daily for ≥ 3 days, with exacerbations separated by ≥ 7 days treated as separate exacerbations.
Data collection
In an attempt to not miss any exacerbations, the primary outcome was collected from several different sources, which are listed in Table 4, along with a description of the source of these data (patient or trial site). A description of the nature of the data available for each source is also given as some sources provided detailed information including dates and doses of corticosteroids, and others provided only the number of severe exacerbations with no corroboration regarding the severity and duration of the attack in relation to the definition described above (see Temperature-controlled laminar airflow diary, Participant exacerbation diary and exacerbation review form and Follow-up visit forms for full descriptions of the data collected by each of these sources).
| Source | Abbreviation | Timing/description | Source |
|---|---|---|---|
| Dated severe exacerbations | |||
| TLA diary | TLA | Daily | Participant |
| PED | PED | As severe exacerbation happens | Participant |
| Exacerbation review form | REV | After reported severe exacerbation | Site |
| Adverse event/serious adverse event | SAE | Ad hoc | Site |
| Hospitalisations recorded on follow-up visit form | HOSP | Every 3 months, with dates of hospitalisation obtained retrospectively from site | Information provided by participant and date provided by site |
| Primary outcome | Dated severe exacerbations | Combination of all of the above, with duplicates removed | |
| Undated severe exacerbations | |||
| Number of severe exacerbationsa recorded on follow-up visit form | FU | Every 3 months | Information provided by participant and recorded by site |
| Alternative primary outcome | Maximum severe exacerbations | Maximum of dated severe exacerbations (primary outcome) and those determined from the follow-up visit form for each 3-month time period and summed for the full 12-month follow-up period | |
After discussion with the trial team, investigators, TSC and DSMC, it was decided that the most appropriate information to use for the primary outcome was the dated information as this was the most accurate and would enable duplicate reporting to be removed and the definition to be applied. The primary outcome for this study was therefore determined from the dated primary outcome data only. The undated primary outcome data were used for the sensitivity analyses only, which are described in Sensitivity analyses.
Statistical model
The primary efficacy end point, which was the rate of severe exacerbations over the 12-month period, was analysed using the negative binomial model. The only adjustment made to the model was the minimisation factors used in the random assignment. The minimisation factors used in the model were prevalent compared with incident cases (new to the centre), exacerbation frequency in the previous 12 months (two, three, more than three), use of OCSs (yes/no) and pre-bronchodilator FEV1 (> 50% predicted yes/no). The recruiting centre was included in the model.
The negative binomial model was chosen over the originally planned Poisson model because it is more flexible and gave a better fit to the overall blinded data.
In the current data, there are approximately 60 cases in which the participant reported a severe exacerbation in the PED but there was no report or no data in the daily diary. There are also approximately 50 severe exacerbations reported by the research team without corresponding daily diary entries or a PED report. Thus, there are approximately 110 severe exacerbations without documentation of how long the participants were being observed. Therefore, as it could not be ascertained which days participants were actively reporting severe exacerbations if they did not complete the daily diary, it was decided that, for dated severe exacerbations, the observation period would extend from randomisation to 365 days after randomisation for the primary analysis. By taking a full year as the denominator for all participants, we essentially assumed that all days were at risk as we did not have exact days at risk for the model. This method most likely leads to an underestimation of the rate of events; however, it was deemed the best approach to handling the problem of a large number of missing diary data. See the methods section on the project web page (www.journalslibrary.nihr.ac.uk/programmes/hta/123328/#/; accessed 29 November 2018) for more details.
When using undated data from the follow-up visit form, all data up to and including the 12-month follow-up visit were used.
Sensitivity analyses
The following individual sensitivity analyses were carried out to test the robustness of the primary outcome to the missing data.
1. Best and worst case
The primary analysis was repeated but with the following modified and plausible best- and worst-case severe exacerbation substitutions for those participants who did not contribute a minimum number of data (see Defining populations for data analysis):
-
Worst imputed case:
-
placebo arm with missing data – best possible outcome is assumed
-
active arm with missing data – worst possible outcome is assumed.
-
-
Best imputed case:
-
placebo arm with missing data – worst possible outcome is assumed
-
active arm with missing data – best possible outcome is assumed.
-
Usually a best- and worst-case scenario would use the maximum and minimum number of observed exacerbations, respectively. Instead, the 90th and 10th percentiles were used to avoid using extreme values, referring to the best- and worse-case analysis as modified best- and worst-case analysis. This should put a reasonable bound on the assumptions made about missing data and how these affect the outcomes of the trial. It is believed that most other reasonable attempts at estimating the missing data will fall between these two extreme bounds, and this will aid correct interpretation of the results.
The most plausible best and worst outcomes were assumed for the missing data, with limits decided by clinical expertise guided by trends in the accessible recorded data.
2. Participant population definition
The primary analysis was repeated with the ITT participant population replaced by the per-protocol population (see Defining populations for data analysis for descriptions), meaning that participants who did not receive a device and the five who withdrew their consent for their data to be used in the analyses were excluded from the primary analysis. All other protocol deviations were deemed minor.
3. Alternative definitions of the primary outcome
The primary analysis and best- and worst-case analyses were repeated using the following alternative definitions of the primary end point (see Data collection for descriptions of the dated and undated primary outcome sources):
-
3(a) maximum – the maximum of the dated and undated severe exacerbations at each 3-month time point and summed for the year
-
3(b) follow-up visit form data only – the number of severe exacerbations reported on the follow-up visit form only
-
3(c) dichotomised primary outcome – the primary outcome of number of severe exacerbations was replaced by the dichotomised variable (participants experiencing no severe exacerbations vs. participants experiencing one or more severe exacerbations) and analysed according to the method described in Statistical model but using a logistic regression model with the minimisation factors
-
3(d) dichotomised maximum – the primary analysis was conducted on the dichotomised maximum of the dated and undated severe exacerbations.
Data processing
Several sources provided primary end point data. Reports of exacerbations close in date were inspected and adjusted to prevent duplicate counting of events; for example, if the PED said that an event started 1 day later than the diary data suggested, then only one exacerbation was counted.
Thereafter, and following blinded review of the data (blinded to treatment allocation) and discussion with the clinical team, including the Chief Investigator, TSC and DSMC, data processing decisions as given in the statistical analysis plan (SAP) supplement [see the methods section on the project web page (www.journalslibrary.nihr.ac.uk/programmes/hta/123328/#/)] were made.
Analysis of the secondary end point
Complete-case analysis (or, in the event that we were looking over multiple time points, cases with at least one reported value for the specific secondary outcome) was carried out for all secondary outcomes, that is, only available data were analysed with no missing data imputation. At each time point, the number of available outcomes between placebo and the TLA device are reported.
A multilevel mixed-effects linear regression including the treatment term, a time point term, an interaction term of treatment arm and time point, the baseline measure and the minimisation factors, and that was adjusted for repeated observations on participants, was used to analyse the repeatedly collected secondary outcomes. The minimisation factors for the secondary outcomes were prevalent compared with incident cases, exacerbation frequency in the previous 12 months (two, three, more than three), use of OCSs (yes/no) and pre-bronchodilator FEV1 (> 50% predicted yes/no). The linear combination of the treatment term and the treatment time interaction term is reported.
For continuous variables with only baseline and 12-month data available, the minimisation factors and the baseline variable were included in the model.
Procedure for dealing with missing, unused and spurious data
We experienced the problem of missing data in the form of both total non-response post randomisation (e.g. attrition or withdrawal) and item non-response (when some but not all of the required information was collected from the participants, e.g. an intermittently missing end point because of the participant not filling in the diary). Please see the SAP supplement in the methods section on the project web page (www.journalslibrary.nihr.ac.uk/programmes/hta/123328/#/) for a description of the procedures for dealing with data issues.
Qualitative analysis
Focus group discussions were digitally recorded, transcribed verbatim and entered into NVivo 8 (QSR International, Warrington, UK), a qualitative software package for systematic and transparent data management. A pseudonym was assigned to each participant at recruitment. After tape recordings had been transcribed, the pseudonyms were used to refer to individuals and no ‘real’ names were included in any reports to maintain participant anonymity. Care was taken to always ensure that any direct quotes used in study reports or papers to illustrate the findings were not directly attributable to individuals.
We used framework analysis, a three-stage analytical process, to analyse the qualitative data. This analysis method involves identifying initial themes by indexing the content of the data and using these themes to guide the formation of a framework within which transcribed material is synthesised. Key categories are then identified to help describe the data.
The two researchers who had collected the qualitative data independently coded it. They then compared findings and scrutinised the framework matrix to see if there was agreement with the categories generated. In the case of disagreement, a solution was sought to clarify the meaning of a code/theme developed until mutual consent was reached. The aim of this stage was to attempt to enhance the validity of the development of the conceptual framework and to guard against researcher bias. Following analysis of all focus groups, it was observed that data saturation had been achieved as no new themes emerged from the participants.
Finally, the researchers explored patterns of association between the categories and attempted to explain why those patterns occurred.
Subgroup analysis
No subgroup analysis was originally planned for this report but a post hoc analysis was requested by the DSMC to assess the primary outcome for participants who do and do not use dust mite covers.
Chapter 3 Results
Participants
Recruitment
Screening and recruitment for the LASER trial took place between 25 May 2014 and 11 January 2016, resulting in the randomisation of 240 participants. A total of 14 centres randomised participants into the trial.
Baseline characteristics
The baseline characteristics of the 240 participants randomised into the two participant groups are summarised in Table 5. The five participants who withdrew consent to use previously collected data (see Participants with minimum data, deaths and those with withdrawal forms completed for details) are indicated by ‘Missing’. Note that these data were actually collected at three different time points as per the participant flow diagram: at screening, at randomisation and at baseline.
| Factor | Level | Trial group | Total (N = 240) | |
|---|---|---|---|---|
| Placebo (N = 121) | TLA device (N = 119) | |||
| Origin of case, n (%) | ||||
| New to clinic (incident) or existing patient (prevalent)a | Incident | 26 (21) | 24 (20) | 50 (21) |
| Prevalent | 94 (78) | 91 (76) | 185 (77) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Demographics | ||||
| Sex, n (%) | Female | 90 (74) | 82 (69) | 172 (72) |
| Male | 30 (25) | 33 (28) | 63 (26) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Age at randomisation (years), n (%) | 16–17 | 1 (1) | 0 (0) | 1 (< 1) |
| 18–34 | 33 (27) | 26 (22) | 59 (25) | |
| 35–59 | 69 (57) | 70 (59) | 139 (58) | |
| 60–75 | 17 (14) | 19 (16) | 36 (15) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Age at randomisation (years), mean (SD) | 45.3 (13.8), n = 120 | 46.8 (13.8), n = 115 | 46.1 (13.8), n = 235 | |
| Ethnicity, n (%) | White | 103 (85) | 100 (84) | 203 (85) |
| Bangladeshi | 1 (1) | 0 (0) | 1 (< 1) | |
| Black Caribbean | 1 (1) | 2 (2) | 3 (1) | |
| Black other | 1 (1) | 0 (0) | 1 (< 1) | |
| Indian | 5 (4) | 4 (3) | 9 (4) | |
| Mixed white | 2 (2) | 2 (2) | 4 (2) | |
| Other | 1 (1) | 2 (2) | 3 (1) | |
| Pakistani | 5 (4) | 4 (3) | 9 (4) | |
| Unknown | 1 (1) | 1 (1) | 2 (1) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Category of body mass index (kg/m2), n (%) | Severely underweight (15–16) | 1 (1) | 1 (1) | 2 (1) |
| Normal (18.5–25) | 24 (20) | 26 (22) | 50 (21) | |
| Overweight (25–30) | 33 (27) | 38 (32) | 71 (30) | |
| Obese class I (30–35) | 33 (27) | 26 (22) | 59 (25) | |
| Obese class II (35–40) | 9 (7) | 17 (14) | 26 (11) | |
| Obese class III (≥ 40) | 18 (15) | 6 (5) | 24 (10) | |
| Invalid data | 2 (2) | 1 (1) | 3 (1) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Body mass index (kg/m2), mean (SD) | 31.2 (7.2), n = 120 | 29.6 (5.9), n = 115 | 30.4 (6.6), n = 135 | |
| Smoking, n (%) | ||||
| Smoking status | Ex-smoker | 36 (30) | 22 (18) | 58 (24) |
| Never smoked | 84 (69) | 93 (78) | 177 (74) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Does anyone smoke in the bedroom where the TLA will be installed? | No | 120 (99) | 115 (97) | 235 (98) |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Does anyone smoke in the house? | No | 116 (96) | 112 (94) | 228 (95) |
| Yes | 4 (3) | 3 (3) | 7 (3) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Exacerbation history | ||||
| Exacerbation frequency in preceding 12 months: baseline data, n (%) | 0 | 1 (0.8) | 1 (0.8) | 2 (0.8) |
| 1 | 0 (0.0) | 1 (0.8) | 1 (0.4) | |
| 2 | 36 (29.8) | 35 (29.4) | 71 (29.6) | |
| 3 | 25 (20.7) | 33 (27.7) | 58 (24.2) | |
| 4 | 22 (18.2) | 15 (12.6) | 37 (15.4) | |
| 5 | 13 (10.7) | 17 (14.3) | 30 (12.5) | |
| 6 | 6 (5.0) | 5 (4.2) | 11 (4.6) | |
| 7 | 3 (2.5) | 2 (1.7) | 5 (2.1) | |
| 8 | 1 (0.8) | 1 (0.8) | 2 (0.8) | |
| 9 | 0 (0.0) | 1 (0.8) | 1 (0.4) | |
| 10 | 3 (2.5) | 1 (0.8) | 4 (1.7) | |
| 12 | 3 (2.5) | 1 (0.8) | 4 (1.7) | |
| 14 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| 18 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| 20 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| 25 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| Invalid data | 3 (2.5) | 2 (1.7) | 5 (2.1) | |
| Missing | 1 (0.8) | 4 (3.4) | 5 (2.1) | |
| Exacerbation frequency in preceding 12 months, n (%) | 2 | 38 (31) | 37 (31) | 75 (31) |
| 3 | 30 (25) | 27 (23) | 57 (24) | |
| > 3 | 52 (43) | 51 (43) | 103 (43) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Number of exacerbations in preceding 12 months, mean (SD) | 4.3 (3.7), n = 116 | 3.5 (1.8), n = 112 | 3.9 (2.9), n = 228 | |
| Hospital admissions in preceding 12 months, n (%) | 0 | 81 (66.9) | 80 (67.2) | 161 (67.1) |
| 1 | 21 (17.4) | 17 (14.3) | 38 (15.8) | |
| 2 | 8 (6.6) | 11 (9.2) | 19 (7.9) | |
| 3 | 6 (5.0) | 2 (1.7) | 8 (3.3) | |
| 4 | 1 (0.8) | 4 (3.4) | 5 (2.1) | |
| 6 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| 7 | 0 (0.0) | 1 (0.8) | 1 (0.4) | |
| 10 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| 12 | 1 (0.8) | 0 (0.0) | 1 (0.4) | |
| Missing | 1 (0.8) | 4 (3.4) | 5 (2.1) | |
| Previous asthma-related ITU admission, n (%) | No | 96 (79) | 87 (73) | 183 (76) |
| Yes | 24 (20) | 28 (24) | 52 (22) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Physiological measures | ||||
| Pre-bronchodilator FEV1 > 50%,a n (%) | No | 28 (23) | 27 (23) | 55 (23) |
| Yes | 92 (76) | 88 (74) | 180 (75) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Pre-bronchodilator FEV1 (l), mean (SD) | 2.0 (0.8), n = 118 | 2.1 (0.9), n = 115 | 2.1 (0.8), n = 233 | |
| Pre-bronchodilator FEV1 (% predicted), mean (SD) | 68.5 (21.7), n = 118 | 69.9 (22.6), n = 115 | 69.2 (22.1), n = 233 | |
| Pre-bronchodilator FVC (l), mean (SD) | 3.0 (1.0), n = 118 | 6.2 (33.9), n = 115 | 4.6 (23.9), n = 233 | |
| Pre-bronchodilator FVC (% predicted), mean (SD) | 86.4 (19.4), n = 118 | 88.3 (17.5), n = 115 | 87.4 (18.5), n = 233 | |
| FENO (p.p.b.), mean (SD) | 36.3 (36.0), n = 114 | 38.7 (35.7), n = 110 | 37.5 (35.8), n = 224 | |
| Asthma treatments, n (%) | ||||
| Use of maintenance corticosteroids?a,b | No | 91 (75) | 86 (72) | 177 (74) |
| Yes | 29 (24) | 29 (24) | 58 (24) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Category of baseline dose of maintenance steroid (mg/day, prednisolone equivalent) | 1–9 | 10 (8) | 13 (11) | 23 (10) |
| 10–19 | 5 (4) | 12 (10) | 17 (7) | |
| 20–29 | 5 (4) | 0 (0) | 5 (2) | |
| 30 | 1 (1) | 1 (1) | 2 (1) | |
| 40 | 2 (2) | 1 (1) | 3 (1) | |
| None | 97 (80) | 88 (74) | 185 (77) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Steroid-sparing immunosuppressant? | No | 115 (95) | 109 (92) | 224 (93) |
| Yes | 5 (4) | 6 (5) | 11 (5) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Monoclonal antibody therapy? | No | 105 (87) | 106 (89) | 211 (88) |
| Yes | 15 (12) | 9 (8) | 24 (10) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
| Bronchial thermoplasty? | No | 119 (98) | 112 (94) | 231 (96) |
| Yes | 1 (1) | 3 (3) | 4 (2) | |
| Missing | 1 (1) | 4 (3) | 5 (2) | |
These baseline data indicate that the two participant groups were proportionately balanced.
Participant flow
The overall flow of the 240 participants randomised into the trial is shown in Figure 9. This figure shows the compliance of the two participant groups with device usage (measured as days in possession of the device during the 365 days of trial participation), as well as their compliance with reporting of the steroid dose (the number of days that the steroid dose was reported during the 365 days of trial participation). There was a requirement for participants to remain active in the trial for a minimum of 6 months before they became eligible for 4 years’ post-trial provision of the active device.
FIGURE 9.
The LASER trial CONSORT flow diagram: participants randomised (n = 240). ORTU, Oxford Respiratory Trials Unit. a, Did not meet minimum data criteria of at least one reported PED and/or 90 days of reporting the dosage taken. In the main analysis, all participants were included using the available data from all dated sources, verified by dosage information when available. For sensitivity analyses, estimates of the number of exacerbations were substituted for those participants who did not meet the minimum data criteria.

Figure 9 also presents the numbers of participants in each of the primary outcome analysis populations (see Chapter 2, Primary statistical analysis, for descriptions), segmented by active and placebo intervention. All participants were included in the ITT analysis with data retained as missing for those participants who withdrew consent to all use of their data (see Participants with minimum data, deaths and those with a withdrawal form completed for details of these withdrawals). The per-protocol population excluded 11 participants who either did not receive the device or withdrew consent to use all data: three from the placebo group and eight from the active group.
Participants with minimum data, deaths and those with a withdrawal form completed
In total, 59 participants did not have the minimum data, died or had a withdrawal form completed. In some cases, data were provided after the withdrawal date, bringing into question the withdrawal status. In other cases, the withdrawals related to participants who were described as being lost to follow-up. Five participants withdrew consent for data that had been collected previously. Details of the 59 participants are provided in Table 31. All participants were included in the primary analysis (ITT population; see Chapter 2, Defining populations for data analysis for description). Of the 59 participants, 31 were in the active group and 28 were in the placebo group.
Missing data
Steroid dose is the key indicator of the primary outcome; unfortunately, a considerable number of steroid dose data were not collected from the primary outcome data sources (as shown in Chapter 2, Primary outcome).
For both the active and the placebo groups, we calculated the total number of possible study days from the date of randomisation to the 12-month follow-up visit if available; the total number of possible study days was given as 365 days if no 12-month follow-up visit occurred. If participants withdrew earlier than 12 months, the number of study days was calculated from randomisation to the point of withdrawal.
Placebo group
For the placebo group, data for 29,147 (66.1%) of the 44,091 possible study days of daily maintenance steroid use were collected; 6419 (22.0%) of the 29,147 days of maintenance dose information analysed were based on the assumption of a zero dose because this field of the daily diary was left blank but other fields (i.e. any of device use, reliever use or time off work) were completed.
In addition to missing maintenance steroid dose data, exacerbation steroid dose data were also missing. A 30-mg dose was assumed for the 24 days for placebo group cases for which the exacerbation dose was left blank in the PED.
Active group
For the active group, data for 28,753 (66.2%) of the 43,398 possible study days of maintenance steroid dose use were collected; 4819 (16.8%) of the 28,753 days of maintenance dose information analysed were based on the assumption of a zero dose because this field of the daily diary was left blank but other fields were completed.
In addition to missing maintenance steroid dose data, exacerbation steroid dose data were also missing. A 30-mg dose was assumed for the 25 days for active group cases for which the exacerbation dose was left blank in the PED.
The rate at which dose data were available for analysis (either because they were reported or because a zero dose was assumed) over the 1-year duration of the trial is shown in Figure 10. The assumed data (zero dose or PED data) are plotted on a participant-by-participant basis in Figure 11. The voids indicate missing data, which are plotted in Figure 12. Table 6 summarises the missing data assumptions.
FIGURE 10.
Percentage of the 240 randomised participants who had a dose of oral corticosteroid reported (or an assumed zero dose) on the nth study day. Day of randomisation = 0, 1-year post randomisation = 365 days. LOWESS (locally weighted scatterplot smoothing) smoothed average applied with bandwidth 0.02.

FIGURE 11.
Assumed data: zero dose (in green) and 30-mg dose (in blue) by participant and days since randomisation.

FIGURE 12.
Missing dose data by participant and days since randomisation. Long tails are the result of follow-up visits that do not have related TLA diary data. For each participant, at the beginning of the trial, the diary was often not completed prior to delivery of the device. After the device was available, the completion rate was around 70% for the rest of the trial.

| Variable | Trial group | |
|---|---|---|
| Placebo | TLA device | |
| Possible study days: days from randomisation to 12-month follow-up visit (or 365 days, if later), mean (SD) | 373.5 (16.2) | 376.0 (22.2) |
| Days when dose was reported by the participant in the daily diary or severe exacerbation report, mean (SD) | 244.2 (133.7) | 245.7 (137.3) |
| Days when a zero dose was assumed, mean (SD) | 54.1 (89.8) | 41.5 (77.6) |
| Has minimum data recorded, including assumed data,a,b n (%) | ||
| No | 23 (19) | 22 (18) |
| Yes | 98 (81) | 97 (82) |
Outcomes
Primary outcome sources of data
Data pertaining to the primary outcome were reported in several sources, as described in Chapter 2 (see Data collection). The percentage of dated severe asthma exacerbations identified from each of these sources is provided in Table 7. Only 18% of the dated severe exacerbations were reported in all three expected sources: the TLA diary, PED and exacerbation review form. In total, 40 (12%) of the severe exacerbations were reported only on the exacerbation review form, with no other supporting data.
| Source | Frequency | % |
|---|---|---|
| – – – – – HOS | 11 | 3.20 |
| – – – – sAE – | 1 | 0.29 |
| – – – REV – – | 40 | 11.63 |
| – – – REV – HOS | 3 | 0.87 |
| – – T/P REV – HOS | 1 | 0.29 |
| – PED T/P – – | 30 | 8.72 |
| – PED T/P – – HOS | 1 | 0.29 |
| – PED T/P REV – – | 26 | 7.56 |
| – PED T/P REV sAE – | 2 | 0.58 |
| – PED T/P REV sAE HOS | 1 | 0.29 |
| TLA – T/P – – – | 56 | 16.28 |
| TLA – T/P – – HOS | 7 | 2.03 |
| TLA – T/P – sAE – | 2 | 0.58 |
| TLA – T/P REV – – | 31 | 9.01 |
| TLA – T/P REV – HOS | 7 | 2.03 |
| TLA PED T/P – – – | 57 | 16.57 |
| TLA PED T/P – – HOS | 4 | 1.16 |
| TLA PED T/P – sAE – | 1 | 0.29 |
| TLA PED T/P REV – – | 53 | 15.41 |
| TLA PED T/P REV – HOS | 8 | 2.33 |
| TLA PED T/P REV sAE – | 1 | 0.29 |
| TLA PED T/P REV sAE HOS | 1 | 0.29 |
| Total | 344 | 100.00 |
A summary of the distribution of steroid doses consumed by each participant group, spanning both maintenance and exacerbation steroid dose ranges, is provided in Table 8. This summary table groups the individual doses into 10 ranges, chosen to highlight the shape of the distribution. The full breakdown of the dose data is provided in Table 32.
| TLA/PED steroid dose (mg) | Trial group | Total | ||||
|---|---|---|---|---|---|---|
| Placebo | Active | |||||
| Number of study days | % of treatment group | Number of study days | % of treatment group | Number of study days | % of treatment group | |
| Missing | 14,944 | 33.9 | 14,645 | 33.7 | 29,589 | 33.8 |
| 0 | 16,384 | 51.7 | 15,976 | 47.9 | 32,360 | 49.8 |
| 0 A | 6419 | 4819 | 11,238 | |||
| 2–5 | 1221 | 2.8 | 1895 | 4.4 | 3116 | 3.6 |
| 2–5 A | 0 | 1 | 1 | |||
| 6–10 | 2284 | 5.2 | 2684 | 6.2 | 4968 | 5.7 |
| 6–10 A | 4 | 0 | 4 | |||
| 11–20 | 1033 | 2.3 | 1431 | 3.3 | 2464 | 2.8 |
| 11–20 A | 1 | 2 | 3 | |||
| 25–30 | 846 | 2.0 | 825 | 2.0 | 1671 | 2.0 |
| 25–30 A | 22 | 13 | 35 | |||
| 25–30 AP | 15 | 4 | 19 | |||
| 25–30 P | 9 | 21 | 30 | |||
| 31–40 | 750 | 1.7 | 857 | 2.0 | 1607 | 1.9 |
| 31–40 A | 13 | 25 | 38 | |||
| 41–50 | 108 | 0.2 | 31 | 0.08 | 139 | 0.2 |
| 41–50 A | 0 | 5 | 5 | |||
| 51–100 | 33 | 0.07 | 154 | 0.4 | 187 | 0.2 |
| 51–100 A | 0 | 10 | 10 | |||
| 101–200 | 5 | 0.01 | 0 | 0 | 5 | 0.006 |
| 101–200 A | 0 | 0 | 0 | |||
| Total | 44,091 | 100 | 43,398 | 100 | 87,489a | 100 |
The numbers of participants reporting none, one or more dated exacerbations during the 12 months of the trial is shown in Table 9 by treatment group.
| Number of dated exacerbations | Trial group, n (%) | Total (N = 240), n (%) | |
|---|---|---|---|
| Placebo (N = 121) | TLA device (N = 119) | ||
| 0 | 55 (45) | 49 (41) | 104 (43) |
| 1 | 23 (19) | 25 (21) | 48 (20) |
| 2 | 17 (14) | 16 (13) | 33 (14) |
| 3 | 9 (7) | 16 (13) | 25 (10) |
| 4 | 7 (6) | 8 (7) | 15 (6) |
| 5 | 3 (2) | 3 (3) | 6 (3) |
| 6 | 2 (2) | 1 (1) | 3 (1) |
| 7 | 2 (2) | 1 (1) | 3 (1) |
| 8 | 2 (2) | 0 (0) | 2 (1) |
| 10 | 1 (1) | 0 (0) | 1 (< 1) |
Primary outcome
As shown in Table 10, there was no statistical difference in the rates of severe exacerbations between the two participant groups (p = 0.62). In addition, there was no statistical difference in these rates when the outcomes were dichotomised (Table 11).
| Outcome | Trial group | Risk ratioa | SD | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TLA device | |||||||||
| N/events | Mean | SD | N/events | Mean | SD | |||||
| Total dated severe exacerbations within 1 year | 121/179 | 1.48 | 2.03 | 119/165 | 1.39 | 1.57 | 0.92 | 0.15 | 0.66 to 1.27 | 0.6167 |
| Sensitivity analysisa | ||||||||||
| Dated severe exacerbations within 1 year in the per-protocol population (2b) | 118/179 | 1.52 | 2.04 | 111/164 | 1.48 | 1.59 | 0.951 | 0.15 | 0.69 to 1.31 | 0.7575 |
| Maximum severe exacerbations for 3-month periods, summed for year (3ab) | 121/258 | 2.13 | 2.75 | 119/235 | 1.97 | 2.15 | 0.912 | 0.14 | 0.68 to 1.23 | 0.5444 |
| Severe exacerbations reported on the 3-monthly follow-up visit form, corrected for other data (3bb) | 121/234 | 1.93 | 2.64 | 119/209 | 1.76 | 2.01 | 0.892 | 0.14 | 0.65 to 1.22 | 0.4689 |
| Outcomes treated as dichotomiseda | Trial group | Odds ratio (SD) | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | TLA device | ||||||||
| N | Count | Percentage | N | Count | Percentage | ||||
| Dated severe exacerbations within 1 year: yes/no (3ca) | 121 | 66 | 0.55 | 119 | 70 | 0.59 | 1.192 (0.32) | 0.71 to 2.01 | 0.5096 |
| Maximum severe exacerbations reported: yes/no (3da) | 121 | 78 | 0.64 | 119 | 82 | 0.69 | 1.227 (0.35) | 0.70 to 2.14 | 0.4696 |
Sensitivity analyses
The sensitivity analyses designed to test the robustness of the results to the missing data, participant population definition and alternative definitions of the primary outcome (as described in Chapter 2, Sensitivity analyses) show that the primary outcome results in Table 10 are very sensitive to the assumptions made; by varying these assumptions the primary outcome treatment effects could either favour or not favour the TLA device. This conclusion was drawn by investigating the best- and worst-case scenarios for those participants who did not meet the minimum data requirement (see Chapter 2, Asthma Control Diary for definition). The following best- and worst-case scenarios describe the best- and worst-case numbers of severe exacerbations that might have occurred in this population of 45 participants during the 12-month trial period but which were not captured in the primary outcome data set:
-
modified best and worst case –
-
best case = 0
-
worst case = 4
-
-
plausible best and worst case –
-
best case = 0
-
worst case = 2.
-
The results of the sensitivity analyses made using the best- and worst-case scenarios are as follows:
-
The point estimates of the risk ratio ranged from 1.4 (worst for active) to 0.6 (best for active) in the modified best- and worst-case analysis for the dated exacerbations, highlighting the wide range of possible outcomes under different assumptions about the outcomes for those with less than the minimum number of data reported. The plausible best- and worst-case analysis gave point estimates of the risk ratio ranging from 1.1 to 0.7 (sensitivity analysis 2; see Chapter 2, Study assessments, Participant exacerbation diary and exacerbation review form).
-
The best- and worst-case analysis carried out using the maximum number of exacerbations as outcomes resulted in point estimates of 1.5 to 0.6, again highlighting the wide range of possible outcomes under different assumptions about the outcomes for those with less than the minimum number of data reported (sensitivity analysis 3a; see Chapter 2, Study assessments, Participant exacerbation diary and exacerbation review form).
-
The best- and worst-case analysis carried out using the dichotomised dated exacerbations (participants with one or more severe exacerbations vs. no severe exacerbations) resulted in point estimates ranging from 2.6 to 0.5 (sensitivity analysis 3c; see Chapter 2, Study assessments, Participant exacerbation diary and exacerbation review form). The corresponding analysis made using the maximum number of severe exacerbations definition resulted in point estimates ranging from 3.5 to 0.5 (sensitivity analysis 3d; see Chapter 2, Study assessments, Participant exacerbation diary and exacerbation review form).
Table 10 presents the risk ratio point estimates for the different primary outcome sensitivity analyses. None of these risk ratios was statistically significant. There is therefore no evidence that the null hypothesis of no difference between the active and the placebo treatment can be rejected. The number of missing data could mask a difference between the treatments, but this could be in either direction.
Secondary outcomes: quantitative
The asthma control secondary outcome results (lung function, ACQ score, ACD score and SNOT-22 score) are presented in Table 12. The remaining secondary outcome results (quality of life, impact and health economics) are presented in Chapter 4.
| Outcome | Time point | Trial group | Treatment effect | SD | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TLA device | ||||||||||
| n | Mean | SD | n | Mean | SD | ||||||
| Lung function | |||||||||||
| Pre-bronchodilator FEV1 (l) | Screening | 119 | 2.02 | 0.86 | 113 | 2.01 | 0.84 | ||||
| Randomisation | 118 | 2.02 | 0.81 | 115 | 2.09 | 0.86 | |||||
| 3 months | 100 | 2.05 | 0.76 | 98 | 2.14 | 0.89 | |||||
| 6 months | 100 | 2.04 | 0.81 | 94 | 2.16 | 0.86 | |||||
| 9 months | 92 | 1.99 | 0.79 | 88 | 1.98 | 0.78 | |||||
| 12 months | 91 | 2.01 | 0.79 | 85 | 2.01 | 0.8 | |||||
| Pre-bronchodilator FEV1, mixed-model analysis | –0.014 | 0.05 | –0.11 to 0.08 | 0.7607 | |||||||
| FeNO | Screening | 0 | NA | NA | 0 | NA | NA | ||||
| Randomisation | 114 | 36.35 | 35.98 | 110 | 38.74 | 35.74 | |||||
| 3 months | 97 | 35.13 | 34.81 | 95 | 34.06 | 32.03 | |||||
| 6 months | 95 | 32.67 | 35.07 | 91 | 32.71 | 30.67 | |||||
| 9 months | 87 | 30.31 | 29.2 | 81 | 36.45 | 37.33 | |||||
| 12 months | 89 | 33.39 | 33.11 | 82 | 32.56 | 30.19 | |||||
| FeNO, mixed-model analysis (p.p.b.) | –1.843 | 2.58 | –6.91 to 3.22 | 0.4756 | |||||||
| Post-bronchodilator FEV1 | Screening | 115 | 2.32 | 0.85 | 111 | 2.27 | 0.84 | ||||
| Randomisation | 0 | NA | NA | 0 | NA | NA | |||||
| 3 months | 0 | NA | NA | 0 | NA | NA | |||||
| 6 months | 0 | NA | NA | 0 | NA | NA | |||||
| 9 months | 0 | NA | NA | 0 | NA | NA | |||||
| 12 months | 81 | 2.17 | 0.77 | 84 | 2.24 | 0.8 | |||||
| Post-bronchodilator FEV1, regression analysis | 0.04 | 0.05 | –0.07 to 0.15 | 0.4628 | |||||||
| Average daily maximum peak flow (l/minute) | Screening | 91 | 311.22 | 117.84 | 82 | 336.62 | 126.78 | ||||
| Randomisation | 0 | 0 | NA | NA | 0 | NA | |||||
| 3 months | 93 | 321.52 | 118.68 | 87 | 341.24 | 130.94 | |||||
| 6 months | 87 | 319.66 | 124.62 | 91 | 351.11 | 130.95 | |||||
| 9 months | 80 | 307.97 | 112.97 | 80 | 338.3 | 130.52 | |||||
| 12 months | 76 | 319.72 | 121.46 | 74 | 349.83 | 124.5 | |||||
| Average daily maximum peak flow, mixed-model analysis | 14.729 | 7.35 | 0.32 to 29.14 | 0.0452 | |||||||
| Patient-reported outcomes | |||||||||||
| ACQ score | Screening | 107 | 2.98 | 0.99 | 107 | 2.9 | 0.96 | ||||
| Randomisation | 110 | 3.05 | 1.08 | 106 | 2.84 | 1 | |||||
| 3 months | 81 | 2.54 | 1.27 | 84 | 2.26 | 1.02 | |||||
| 6 months | 89 | 2.55 | 1.27 | 84 | 2.24 | 1.03 | |||||
| 9 months | 83 | 2.45 | 1.21 | 78 | 2.38 | 1.18 | |||||
| 12 months | 82 | 2.42 | 1.28 | 79 | 2.31 | 1.16 | |||||
| ACQ score, mixed-model analysis | –0.054 | 0.11 | –0.26 to 0.15 | 0.6061 | |||||||
| ACD scores averaged over available 14 days | Screening | 32 | 2.08 | 1 | 31 | 2.18 | 1.19 | ||||
| Randomisation | 0 | NA | NA | 0 | NA | NA | |||||
| 3 months | 36 | 1.42 | 1.02 | 28 | 1.84 | 1.37 | |||||
| 6 months | 33 | 1.68 | 1.35 | 37 | 1.96 | 1.19 | |||||
| 9 months | 26 | 1.8 | 1.17 | 34 | 2.02 | 1.22 | |||||
| 12 months | 36 | 1.59 | 1.08 | 34 | 1.94 | 1.27 | |||||
| ACD scores averaged over available 14 days, mixed-model analysis | 0.212 | 0.25 | –0.28 to 0.71 | 0.4008 | |||||||
| SNOT-22 score | Screening | 0 | NA | NA | 0 | NA | NA | ||||
| Randomisation | 120 | 41.17 | 21.74 | 114 | 41.78 | 21.32 | |||||
| 3 months | 106 | 38.53 | 21.28 | 100 | 34.95 | 20.95 | |||||
| 6 months | 101 | 39.33 | 22.25 | 97 | 34.16 | 20.7 | |||||
| 9 months | 95 | 37.99 | 22.38 | 89 | 35.24 | 22.12 | |||||
| 12 months | 96 | 36.3 | 23.57 | 90 | 35.69 | 21.18 | |||||
| SNOT-22 score, mixed-model analysis | –3.265 | 1.85 | –6.9 to 0.37 | 0.0781 | |||||||
The only asthma control secondary outcome that was found to be statistically significant was daily maximum peak flow (p = 0.045), based on data from 150 of 240 participants reporting at 12 months. All other aspects of the lung function secondary outcome (pre-bronchodilator FEV1, FeNO and post-bronchodilator FEV1), as well as ACQ, ACD and SNOT-22 scores, were not found to be statistically significant. It should be noted that, because a large number of peak flow data were missing, and there was one significant result compared with six non-significant results (the multiple testing problem), this significant result could have occurred by chance.
Post hoc analyses
The TSC requested two post hoc analyses of the primary outcome: (1) analysis of an alternative definition of a severe exacerbation and (2) subgroup analysis of patients who do and do not use dust mite covers.
Alternative definition of a severe exacerbation
Following a review of the recently reported literature, in which exacerbations have been defined using a less stringent definition, the original definition of the primary end point (see Chapter 2, Primary outcome) was amended so that comparison with these studies could be undertaken. In the new definition, an exacerbation was defined as a 10-mg increase in steroid dose over any maintenance dose for 3 consecutive days. All other parts of the definition remained the same, that is, exacerbations were considered to be separate if participants were on the maintenance dose or on a dose that was lower than the maintenance dose for at least 7 days between them.
The primary outcome results calculated for this new primary end point definition are presented in Table 13. This analysis also supports no rejection of the hypothesis of no difference between the treatment groups and therefore provides the same conclusion for the primary outcome as the analysis performed according to the original definition of the primary end point (see Table 10).
| Post hoc analysis: alternative definition of the primary end point | Trial group | Risk ratio | SE | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TLA device | |||||||||
| N/events | Mean | SD | N/events | Mean | SD | |||||
| Total dated severe exacerbations within 1 year | 21/186 | 1.54 | 2.05 | 119/189 | 1.59 | 1.75 | 1.02 | 0.17 | 0.7 to 1.4 | 0.910 |
Subgroup analysis: allergy to dust mites
A subgroup analysis was performed to investigate the primary outcome only for those participants allergic to dust mites to discriminate those participants with asthma symptoms on exposure to allergen from those with allergy unrelated to their asthma symptoms. Participants allergic to dust mites were identifiable from two questions on the screening form:
-
Those who answered ‘yes’ to the following option in Q10: ‘Does the participant suffer from (wheeze, cough, chest tightness, breathlessness) when carrying out housework such as dusting, vacuuming and changing bedding?’
-
Those who answered ‘yes’ to Q10a: ‘Does the participant routinely use dust mite covers on bedding (pillow/duvet covers)?’
In total, 221 participants answered yes to Q10, meaning that only 14 answered no (excluding the five participants who withdrew consent for their data to be used). The low number of ‘no’ responses prevented the requested subgroup analysis from being performed on these data because of insufficient statistical power. A total of 129 participants answered yes to Q10a, meaning that 106 participants answered no (again excluding the five participants who withdrew consent for their data to be used). These participant numbers were sufficient for a post hoc analysis to test the interaction effect of the treatment and routine use of dust mite covers. The interaction was found to be non-significant (1.2; p = 0.566). The estimates of the effect in each subgroup based on the model using this same interaction are shown in Table 14.
| Post hoc analysis: does the participant routinely use dust mite covers on bedding? | Trial group | Risk ratio | SE | 95% CI | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | TLA device | |||||||||
| N/events | Mean | SD | N/events | Mean | SD | |||||
| No | 51/77 | 1.5 | 1.8 | 55/62 | 1.1 | 1.3 | 0.85 | 0.21 | 0.5 to 1.4 | 0.514 |
| Yes | 69/102 | 1.5 | 2.2 | 60/103 | 1.7 | 1.8 | 1.16 | 0.27 | 0.7 to 1.8 | 0.519 |
Secondary outcomes: qualitative
Pilot phase telephone interviews
In total, 12 telephone interviews were conducted with participants and partners during September and October 2014 (see Chapter 2, The internal pilot study, for a description of these interviews). A full report of the findings from these interviews was prepared and submitted to the funding body (see Report Supplementary Material 1). A summary of the categories of themes emerging from these interviews is provided in the following sections, according to the responses from the 10 participants and those from the two partners. The method by which the content of these interviews was distilled down to eight categories is described in Chapter 2, Qualitative analysis.
Participant experiences
Participants gave a variety of different reasons for taking part in the LASER trial. The most common reason was to help improve asthma control and reduce the risk of exacerbations and hospital admissions. This was seen as beneficial to the trial participants themselves as well as to future users of the device and patients with asthma. Participants also saw the trial as an opportunity to learn more about their asthma. This trial was seen as being different from other clinical trials by virtue of the facts that it was testing a non-pharmaceutical treatment and that this was an additional treatment to be used alongside current asthma treatments.
The information events were well received and seen as a useful educational opportunity. Participants enjoyed the opportunity to meet other patients with asthma and the opportunity to discuss the trial with members of the trial team. Participants who attended an event with other family members felt that this was useful so that everyone was fully informed of what participation in the trial would involve.
All participants found the information sheet easy to read and accessible. They all felt that they had been given adequate time to read the information and ample opportunity to ask questions to come to an informed decision before signing the consent form. A device template was included in the information pack so that participants could be reassured that the device would fit into their bedroom. Participants found the device template to be particularly useful; however, one participant noted that there was no explanation of which orientation the template should be used in.
Some participants found that the initial study visits (which could take up to 90 minutes) were longer than expected.
Device delivery and installation generally went smoothly and delivery was scheduled at a convenient time for the majority of participants. Two participants, however, had problems with the delivery team not being competent to deliver and install the device. All participants were able to accommodate the treatment device in their bedroom but six had to make minor modifications. Three had to move a bedside table and three had to move their bed to accommodate the device. It was decided that future participants would be informed of the possible need for bedroom modification to accommodate the device before device delivery to check that they would be happy with this.
Nine of the 10 participants had used the trial website (shown in Figure 5) at least once but most had used the website only once or at the start of the trial and had not revisited the site. Participants felt that it would be useful for the website to include a forum where they could share their experiences.
Participants made several suggestions about how the device could be modified for future users. These included:
-
device size – reduce the size of the device and make it portable if possible
-
airshower – rotate the neck of the device to make changing the bed easier and allow the device’s ‘airshower’ to be moved out of the way when necessary without having to move the base of the device
-
shelf size – increase the size of the shelf, which was felt to be a useful design feature but too small to accommodate what would otherwise be stored on a bedside table
-
noise – five participants commented on the noise of the device but did not feel that this was a barrier to device use and in three cases participants felt that the noise was helping them sleep
-
smell – one participant mentioned that the device had an unpleasant smell when turned on
-
heat – the same participant who reported the unpleasant smell also reported that the device generated heat.
Two comments were made about the trial processes: one participant requested the addition of reminders between visits to prompt participants to commence the diary and PEF data collection in the 2 weeks before a study visit and one participant requested increased contact between the trial team and participants between study visits.
Partner experiences
Both partners reported that they were happy for the device to be installed in their home because they were keen for anything that might lead to an improvement in their partner’s asthma control.
Both partners attended an information event and received information about the trial. Both felt that they had received adequate information.
One partner reported that the bedroom had to be significantly modified to accommodate the device. The bed had to be completely reorientated so that the device was not too close to a window.
One partner had visited the trial website at the start of the trial but had not returned. The other participant had not had time to visit the website.
Both partners commented on the noise of the device. Both reported that it was louder than they had perceived it to be at the information event. Neither felt that the noise of the device was a barrier to its use.
Table 15 lists the recommendations and modifications made as a result of these participant and partner interview findings.
| Category | Learning points and actions |
|---|---|
| Motivation |
Participants provided a number of reasons for taking part in the trial
|
| Information events |
Positive feedback from attending information events
|
| Participant information packs/PIS |
The orientation of the device template is currently not clearly defined
|
| Trial visits 1 and 2 |
Insufficient information
|
| Device delivery/installation |
Awareness of the need for bedroom modification
|
| Trial website |
The current website is underused
|
| The TLA device |
A number of small, but bothersome, problems have been identified
|
| Trial process |
Limited contact with trial participants between visits
|
Focus groups
A total of 10 participants (without partners) attended the three focus groups conducted by the Portsmouth site during January 2016 (see Chapter 2, Focus groups, for a description of these focus group sessions). Some of the topics discussed at these focus group sessions were prompted by the attending researcher, based on the categories that emerged from the pilot phase telephone interviews (see Pilot phase telephone interviews). The remaining topics of discussion were generated from the attending participants without prompts. All of the topics were consolidated into categories according to the methodology described in Chapter 2 (see Qualitative analysis) and these categories are described in detail in the following sections.
The learning points applicable to this clinical trial and the TLA product specifically and to future clinical trials generally are highlighted at the end of each section. Selected anonymised verbatim quotes are included to best illustrate the theme being described.
It is worth noting that these qualitative findings are based on a small purposive sample of trial participants and it is unlikely that the comments made during the focus group interviews can be generalised to include all participants in the trial and their experiences of using the TLA device. Nevertheless, the responses received do represent a range of participants and describe their experiences of the treatment, the device and the trial.
Category 1: motivation for participation
When asked about their motivation for taking part in the LASER trial, a number of different reasons were given by participants including personal gain and helping others. All of the focus group participants said that they were pleased to have been asked to take part in the trial:
I was over the moon. I was so pleased . . . to be offered the chance of this was really something special to me.
MW
I thought it was brilliant.
GL
Participants also described their desperation for new treatments that might help to improve their asthma control and how they would be willing to try anything to improve their symptoms:
. . . just the fact that somebody was prepared to try something new.
AP
I was keen to try anything that might help.
GJ
Yes, a real positive out of what was feeling a very, very negative all the time, an opportunity to make a difference.
UF
I’m so ill I would’ve tried anything.
SF
Participants recognised that they might not be receiving an active treatment device during the 12-month trial period but this had not deterred them from participating:
Well, it’s just part of the trial, isn’t it and if there’s a trial there’s always the placebo so . . . but as long as we get the right results at the end.
MW
I was just fingers crossed that I got the one that worked, because it was a half and half, wasn’t it, so yeah.
SF
. . . if it doesn’t work it’s not going to cost you anything to try it.
AP
. . . when the results of this get together and it all gets processed . . . in a year or 2 years’ time or whatever and the results feed back to the consultants . . . hopefully I’ll be one of the people that benefit from the findings, whether I’ve got a good machine or not.
IB
A number of participants said that being offered an active TLA device free of charge on completion of the 12-month trial period had been a further motivating factor:
I finished last October so it’s reassuring to have 4 years of continuing process.
SP
You know that eventually you’ll have one that works either way.
AP
. . . we’re going to get a proper one after the end of the year . . . that’s good.
MW
A number of participants recognised the importance of this research to determine whether or not the TLA device is a viable treatment option for asthma patients and the importance of finding new treatment options. Two participants further personalised this, recognising that future family members might benefit:
Well, not necessarily me, but maybe my children or children’s children or whatever . . . Yeah, that any information gathered from my survey plus the results from other people’s, all goes together for the future for scientists to assess to make the world a better place primarily for asthma people.
IB
Two participants spoke about how they liked the fact that this was a device trial rather than a drug trial. Two others were motivated by the fact that the treatment ‘made sense’ as they were allergic to indoor allergens and the device was designed to filter and ‘clean the air’:
If it had been drugs, I would’ve thought longer about whether I’d signed up and possibly not done that.
GJ
I had done some research into quite expensive air filter . . . I stopped looking after I had the trial, but the ones that aren’t just purifiers, and they’re a few hundred pounds and if you’re not sure if that’s going to work it’s a bit of an outlay. It might have even been £1000.
AP
-
Participants were keen to take part in a non-pharmaceutical trial in which a device offers the potential to alleviate personal symptoms and provide improvements for future asthma sufferers.
-
A key motivation to take part in the trial included having access to an expensive device that would be too expensive to purchase privately.
Category 2: device delivery and installation
Participants’ experiences of the device delivery and installation process (see Chapter 2, Device installation, for a description) were generally positive, with most participants reporting that the process was easily arranged and straightforward. Participants were required to take time off work for device delivery and installation as the Bishopsgate engineering team was available for delivery and installation only during normal working hours:
I think it’s just one young chap on his own at first who installed it, it was just quite straightforward . . . [unclear] and then put it to the appropriate height . . . he was very pleasant.
SP
I had to have a day off work to wait for the little man to come, two little men . . . it had to be installed and then we had to be trained up on using it . . . which is fine, but I’ve had a bad year and used up all my holiday . . . and this was just another one of those things; so that’s a downside for me . . . it’s a shame it couldn’t just be ‘here are some instructions, assemble it yourself’.
IB
-
It is important for participant satisfaction with the trial experience that device delivery and implementation can be flexible.
-
It is important for participant confidence in the new technology that the engineers who install the device are trained and knowledgeable.
Category 3: user experience
One of the most important reasons for conducting the focus group interviews was to understand participants’ perceptions of TLA treatment, including tolerability of the device and the impact of TLA treatment. Participants were asked to comment on both positive and negative feelings towards the treatment device.
A number of the focus group participants, but not all, felt that there had been an improvement in their asthma symptoms and frequency of exacerbations over the year that they had received the trial treatment, acknowledging that they did not know if they were on an active or a placebo device:
I’ve gone from having four or five exacerbations a year to none.
DS
. . . my cough wasn’t nearly so bad after I’d been using it for a while.
SP
I’m certainly a lot better than I was before the trial started. Whether it’s to do with the machine or not, I don’t know. But I’m delighted. I have had other problems but certainly my breathing has markedly improved.
GL
I’m not too sure. I’ve had a few exacerbations; I’ve been in hospital, I think, beginning of November and I’m on steroids at the moment now. So I am not too good at the moment. I can breathe a bit better but I’ve been off work this week.
ED
Two participants reported that they felt that their sleep quality had improved, although one of them was unsure whether or not their asthma had also improved. Interestingly, one participant felt that perhaps the TLA treatment might be having a beneficial effect in reducing her husband’s snoring:
I’m finding that I’m sleeping much better.
SP
Once we got over the noise, we slept really well and even my husband said we’ll keep this machine because we sleep better under it.
ED
I have to say, I think he got a bit quieter, I wondered if he was benefiting from it. I don’t . . . you know.
SF
Category 4: bedroom modification
Participants were asked about their experience of having the TLA treatment device in their bedroom. Most of the focus group participants had to make minor or major modifications to their bedroom to accommodate the treatment device. Minor modifications included removal of a bedside table to allow the device to be installed by the bed. In some cases, more significant moving of furniture and bedroom reconfiguration were required. One participant had to sleep with the door open throughout the trial period.
We had to swap sides of the bed, which was a bit odd after 20 years . . . Because it doesn’t fit the other side, so because the way the bedroom’s laid out with the furniture . . . you couldn’t swap it round because the position of the window and so yeah, that was a bit odd.
GJ
We have a small bedroom and we have individual single beds that were pushed together but now they’re apart to get the [device in] . . . If I was a bit more sensitive and perhaps we’d been married less long I would have said it’s come between us . . . But it’s all worthwhile, definitely worthwhile, but we did have to do some major upheaval to get everything right in the bedroom.
MW
It’s so wide that I can’t shut the bedroom door . . . because of where the bed is and the shape of the room I think, I can’t shut the bedroom door, which I don’t really care about. But when we’ve got visitors staying my snoring helps keep them awake as well and if we could shut the door it would be slightly better for them.
IB
Category 5: negative device experience
A number of participants commented on the noise of the device. The device is quoted as having a noise level of 38 dB, equivalent to a quiet whisper or a suburban street, and participants were made aware of this in the PIS. Some participants and their partners found the noise difficult to get used to at first but a number of participants commented on how they got used to the noise and even missed it when they were away from home:
No problems with the noise whatsoever.
DS
I’m ex-Navy and my husband’s Navy and it makes us feel like we’re on a ship, it’s like a [inaudible: laughter] you’ve got that hum in the background.
GJ
Very difficult to get used to the first few nights. Not much sleep at all. Because it’s just constant noise. I was expecting it to be a lot quieter having heard it at the hotel . . . But there is noise and it’s a lot more than I expected. But you do get used to it. And then, actually, you go somewhere else and you haven’t got it and you can’t sleep. So it’s almost like mood music. You get used to it then you can’t live without it.
ED
My wife . . . is a bit of a light sleeper, so she’s getting a bit grumpified [sic] about the bloody machine . . . it keeps her awake . . . the noise when it starts up and the noise when it goes off, it shakes and vibrates a little bit, and the air coming out of the machine.
JC
The 1-month pilot phase telephone interviews identified that some participants had noticed a distinctive smell whenever the device was switched on and one participant complained about the heat generated by the device. Each of the focus groups was therefore asked specifically about the heat and smell. One participant in focus group 2 reported that she had noticed a smell of dust and another participant in the same focus group noticed that some heat was generated by the mechanism of the device:
It does smell, that’s why I wonder whether mine’s not an actual device because sometimes it smells of dust when it first turns on, so I get the slight odour of dust.
GJ
Mine is hot.
GL
A number of participants highlighted some minor health and safety concerns. For example, a number of participants reported that, because of its position above the pillow, there were occasions when they had hit their head or hand on the airshower of the TLA device while getting in or out of bed. The airshower has a rough surface that is engineered to ensure that the descending laminar airflow is kept separate from the ambient room air. No major injuries were sustained but participants questioned why this was such an abrasive surface:
You learn to not sit up too quick.
GL
It’s like pumice stone, scrapes your knuckle . . . If I sit up, my head doesn’t touch it. When I knock my head on it is when we’re trying to make the bed.
UF
A number of participants commented on the weight of the device. This was in the context of trying to move the device to clean the room. The device weighs 23 kg. Participants were asked, as much as possible, not to move the device during the trial. Participants commented that they would like the device to be lighter so that it could be moved more easily and made portable so that it could be taken away from home when on holiday or spending extended periods away from the home:
I would like a bit more movement underneath so small casters or something. I’m always afraid of moving it too much out of position but it would help if it was a bit easier, especially when I’m breathless.
UF
. . . it’s got a quite heavy base. I mean, I’ve put the Hoover edges to it, I do Hoover , I just . . . but I have trouble . . . [it’s not like a chair you can drag and] . . . the base is very heavy.
SF
Participants were asked not to use a fan or an air conditioning unit in the room where the device was installed as this would affect the laminar flow of cooled, filtered air. Participants were also asked not to have windows open if possible. If a window was open, they were asked to ensure that the door was shut to prevent a cross-draught from disturbing the laminar airflow. One participant complained that she had been unable to sleep with a fan on during the trial and had come close to withdrawing from the trial:
You can’t have any air flow through the bedroom. For me, the biggest problem is I wasn’t sleeping . . . I was so hot, I was absolutely dripping, . . . I normally have a fan on me all night, and this thing isn’t blowing any cold air onto me. And I was just so tired because I was absolutely sweltering. No window open . . . I would say nearly switched it off during the summer to have the fan on because I was so hot.
MW
-
Some participants were convinced that they knew whether they had an active or a placebo device based on their perceived characteristics of their device.
-
Noise was the most frequent negative experience of using the device, affecting both participants and partners. This is an important consideration as this may be a reason why some participants might not be able to use the device in the future.
-
The weight of the machine led participants to anticipate future difficulties with regard to maintaining domestic cleaning.
-
The height of the arm and the rough abrasive surface resulted in participants sustaining minor injuries. None of the participants who took part in the focus groups felt that this was a barrier to the use of the treatment device, but these concerns were reported back to the device manufacturer for consideration.
Category 6: design modifications
The most frequent observation was that the Perspex shelf integral to the device and aiming to provide the same functionality as a bedside table was too small to hold what would usually be stored on a bedside table, such as a lamp, medications or a drink. This was relevant as most participants had been required to remove a bedside table in order to accommodate the treatment device. Participants were concerned that the device might malfunction if they were to inadvertently spill liquid onto or into it.
It was suggested that a useful design feature would be an integral light for reading in place of a bedside lamp.
Some participants reported problems with the power cord, which is inserted under the device. Participants reported that this was difficult to locate and, because of the angle of insertion, the cord had a tendency to become dislodged or fall out.
Participants also agreed that it would be helpful if the neck of the device could be modified from a fixed design to one that could swivel, enabling the device user to move it out of the way when getting into and out of bed and when making the bed.
Participants complained that the device was too heavy to move. This was particularly in the context of cleaning the bedroom; they felt that they were unable to clean effectively around the device without being able to move it. Participants felt that a lighter, more portable, machine might be beneficial so that it could be used in other rooms of the house, for instance while watching television:
I wouldn’t have thought it would be that difficult to get it to go behind the bed instead of at the side of the bed, probably redesign the machine to some extent. We can pull the bed out that way but this side is not functional.
GL
A better table attachment, because it replaced the bedside table and there’s not a lot of space, I read a lot of books, there’s a lot of books. It would be helpful to have something you felt confident putting your glass of water on, which I don’t put my glass of water on it . . . If it falls into the device, then what, it’s not going to be ideal.
GJ
I thought it was broken for a while, it seemed dead, but because the power cord goes up into the bottom, I’d actually left the power cord in and it fell out. You switch it on at the wall. But I must have moved it somehow and the power cord just fell out the bottom because it goes in upwards.
AP
An arm that swivels round so you could get up and sit back.
SF
I’ve got no light by my bedside table now. So if I wake up during the night and I need my tablets, I am having to feel for them and end up dropping them on the floor.
MW
-
The suggested design modifications were reported back to the device manufacturer.
Category 7: study procedures
The focus group participants who had attended an information event found it to be useful. They particularly appreciated the opportunity to see the device and found that the demonstration was helpful in deciding whether or not they would be able to accommodate the device in the bedroom and whether or not they felt prepared to have the device in their home for a 12-month trial period. They also found it useful to be told what would be expected of them if they were to take part in the trial. They appreciated the opportunity to meet other asthma sufferers, recognising that they sometimes felt isolated. This helped to encourage participation:
It was very good, we saw the machine on the bed and Professor Chauhan gave an overview of what it was, what we’re trying to achieve, how it worked and you could all have a little lie down and test it, test it out. And we were asked to bring along husbands, wives and partners and they were given the chance to go ‘well, are you OK with this, sleeping under this machine’.
SF
And also to meet other people in the room I thought was really important because you can feel very isolated when you have this going on, so it’s really good to talk to other people and see how they felt about it as well.
UF
I thought absolutely, but coming to a session like that with lots of other people and they all had similar stories, so that just . . . I was already going to say yes, but it kind of just sealed it.
SF
When asked about the written information received, participants felt that there was a lot of information but that this was not unreasonable. The majority felt that the written information was clearly written, although one participant particularly appreciated receiving a follow-up telephone call after the information event, as it provided an opportunity to discuss the trial with a member of the research team:
It may have been a lot in there but in all honesty, probably like a lot of people, you scanned it, read it and signed it . . . it was nothing unreasonable.
AP
I think everything was covered and, yes, there was plenty to read but I think I wanted to have as much information as there was available so I know what it was all about.
UF
But a telephone call, I could pause and ask questions. I could get answers to those, we could move on to the next bit. I could ask more questions. It was quite interactive, which is what I like.
IB
Participants generally spoke positively about the study visits. Some participants said they would not have been able to take part in the LASER trial if the follow-up visits had not been scheduled for the same day as their regular 3-monthly clinic appointments (which all people with severe asthma should receive) as they could not miss that many days of work:
As I explained to [trial co-ordinator] when I started up the thing, I don’t want to do any more visits than I have to.
IB
Some participants found the frequency of trial visits reassuring as they felt that they were being monitored more closely, although one participant was less keen because of the requirement for her to perform lung function tests:
I found sometimes it was a bit gruelling, doing the breathing tests and FeNO was quite . . . I felt quite lightheaded afterwards. But it was . . . I was just so glad to be monitored, to have people and the liaison with the GP and so on.
GJ
Participants said that they found the text reminder system set up towards the end of the trial to remind them when to start their 2-week peak flow diary and to remind them of the date of their follow-up visit useful. One participant commented on the large number of questionnaires that had to be completed at the follow-up visits and the degree of repetition in some of the questions. However, despite the volume of questionnaires, one participant felt that they really made her think about her asthma and that this had been helpful:
You do like paper! I could have shortened it up. But obviously they originated from different people.
GL
Some participants had visited the trial website (see Figure 5) but most had visited the website only at the start of the trial and had not returned. For those who had used the website, one participant found the trial status graphic useful and she visited the site from time to time to look at how many participants had been recruited. A suggested improvement from one participant was that previous research investigating treatment with the TLA device should have been available:
Went on there at the beginning, not back again.
SP
And I can’t remember it was on . . . I found it on the website or not, but if it’s not there, that would be useful for me to have . . . Not to assume that people wouldn’t be interested but give them that choice.
GJ
I’ve been going on to see how many people they’ve now got for it because it’s got a thing that tells you.
MW
-
Comprehensive written information is key to enable potential clinical trial participants to decide whether or not to take part.
-
Giving potential participants an opportunity to speak with a member of the research team so that their remaining questions can be answered is also important.
-
Information events allow the device to be demonstrated and provide another opportunity for potential participants to ask questions.
-
Meeting with other people with asthma at the information events was valued.
-
Multiple study visits and the collection of data can be burdensome, but careful planning to ensure, when possible, that these activities are combined with routine visits can be helpful.
-
A trial website can be useful, particularly at the beginning of a trial. To attract longer-term usage, the website should provide ongoing knowledge support, updates and a social media platform.
Safety, harm and unintended effects
Two participants died during the trial and one died after the primary outcome time point at 12 months. The causes of death (days after randomisation) were (1) cardiac event/secondary sepsis (day 22), (2) unknown cause (day 345) and (3) aortic aneurysm (day 417).
Table 16 summarises the adverse events and serious adverse events reported in the sources of primary outcome data (see Chapter 2, Analysis of the primary end point, for a list of sources) by treatment group. The protocol did not require asthma exacerbations to be reported separately as serious adverse events or adverse events, so these are excluded from the table.
| Event description | Serious adverse events (n) | Adverse events (n) | ||||
|---|---|---|---|---|---|---|
| Active | Placebo | Total | Active | Placebo | Total | |
| Acute coronary syndrome | 1 | 1 | 0 | |||
| Acute pulmonary embolism | 1 | 1 | 0 | |||
| Aortic aneurysm | 1 | 1 | 0 | |||
| Bronchospasm (following lung function tests) | 1 | 1 | 1 | 1 | ||
| Cellulitis | 1 | 1 | 2 | 3 | ||
| Dental disease | 0 | 1 | 1 | |||
| Depression | 0 | 1 | 1 | |||
| Dry eyes | 0 | 1 | ||||
| Dry mouth | 0 | 1 | 1 | |||
| Ear infection | 0 | 1 | 1 | |||
| Exacerbation of eczema | 0 | 1 | 1 | |||
| Exacerbation of multiple sclerosis | 0 | 2 | 2 | |||
| Fall | 0 | 1 | 1 | |||
| Fracture (ankle/leg) | 1 | 1 | 1 | 1 | ||
| Gastroenteritis | 1 | 1 | 0 | |||
| Gynaecological symptoms | 1 | 1 | 1 | 1 | ||
| Headache/migraine | 1 | 1 | 4 | 4 | ||
| Hip fracture | 1 | 1 | 0 | |||
| Hypertension | 0 | 1 | 1 | |||
| Hypokalaemia | 1 | 1 | 0 | |||
| Ischaemic heart disease | 1 | 1 | 0 | |||
| Kidney infection | 1 | 1 | 0 | |||
| Lower respiratory tract infection | 2 | 2 | 4 | 1 | 1 | |
| Musculoskeletal pain | 0 | 2 | 2 | |||
| Pneumonia | 1 | 1 | 0 | |||
| Post-surgical injury | 1 | 1 | 0 | |||
| Sore eye | 0 | 2 | 2 | |||
| Sore throat | 0 | 3 | ||||
| Stroke | 1 | 1 | 0 | |||
| Superficial skin injury | 0 | 1 | 1 | |||
| Trauma to toe | 0 | 1 | 1 | |||
| Upper gastrointestinal bleed | 1 | 1 | 0 | |||
| Upper respiratory tract infection | 0 | 2 | 2 | |||
| Upper respiratory tract symptoms | 0 | 1 | 1 | |||
| Viral meningitis | 1 | 1 | 0 | |||
| Total | 14 | 9 | 23 | 9 | 24 | 29 |
The numbers of adverse events were similar in both groups and none of the serious adverse events was device related following causality assessment. Five adverse events (in four patients) were considered to be probably related to the device, all in the placebo group. These included (1) a sore eye thought to be from a piece of the machine gauze falling into the eye at night, (2) back of the hand grazed against the machine gauze, (3) sneezing when using the device and (4) headaches and a sore throat, which resolved on stopping use of the device.
Chapter 4 Economic evaluation
Study question
Severe asthma accounts for 80% of the total NHS asthma cost (£1B3) and frequent exacerbations generate much of this cost. 4 Treatments available on the NHS to reduce the frequency of exacerbations include steroid and immunosuppressive agents, but these treatments have limited efficacy and potentially serious side effects. Monoclonal antibodies such as the anti-IgE treatment omalizumab have been shown to reduce exacerbations by up to 50% and improve quality of life in patients with severe allergic asthma,13 but these treatments are prohibitively expensive. Omalizumab costs up to £26,640 per year,35 which NICE concluded in its 2012 reappraisal of this drug (with 2013 update) is cost-effective only for those people with severe persistent confirmed allergic IgE-mediated asthma who have experienced four or more severe exacerbations in the preceding 12 months as an add-on to optimised standard therapy. 14,60 This guidance had two caveats: (1) participants would have to have needed continuous or frequent treatment with OCSs and (2) the manufacturer of omalizumab would need to make discounts available.
With an annual rental cost of £2000 a year, the TLA device could present a cost-effective method for treating people with severe allergic asthma. Prior to the LASER trial, the cost-effectiveness of TLA could be estimated only from modelling secondary data using a number of assumptions, which resulted in inconclusive conclusions. 35,38,61 This economic evaluation has instead been carried out using data collected in a pragmatic, multicentre, double-blind, placebo-controlled, parallel-group RCT with an embedded economic evaluation. In addition, using results from the LASER trial and long-term published data, a Markov model was used to model the cost-effectiveness of the TLA device over the lifetime of the participant.
Data sources, context and definitions
The economic evaluation was carried out from the primary perspective of the NHS. Secondary analyses included all major additional resources likely to be relevant from a societal perspective in this population: productivity losses (because of time off work or study) and out-of-pocket expenses on OTC and complementary medicines and supplements. All costs are presented in Great British pounds (£) for the year 2015/16.
Participant data
The following sources of quantitative data were collected during the follow-up visits to assess the subjective effect, impact on quality of life and cost-effectiveness of the intervention:
-
GETE questionnaire
-
AQLQ(S)
-
EQ-5D-5L
-
AC-QoL questionnaire
-
WPAI(A) questionnaire
-
WPAI(CG) questionnaire.
Descriptions of all of these sources of data are provided in Chapter 2, Participant questionnaires.
Resource use
Participants indicated their resource use over the previous 3-month period in a questionnaire provided to them at each follow-up visit, using the resource use log they had completed during that period as an aide memoir. These resource uses were assigned to one of the following health-care resource categories:
-
community care, including visits to a –
-
GP (at the surgery, at home or by the telephone)
-
nurse (at the surgery or at home)
-
physiotherapist
-
occupational therapist
-
psychologist
-
counsellor
-
-
hospital care, including –
-
visits to the A&E department
-
ambulance use
-
length of stay in hospital including stays in an ITU or any procedures while in hospital
-
-
prescribed medications, for both asthma-related and other conditions.
All of this health-care resource use was assumed to be borne by the NHS. Only resource use associated with OTC and complementary medications was assigned to non-NHS sources in this economic evaluation.
Unit costs
NHS
Unit costs for consultations with GPs and nurses were taken from the Personal Social Services Research Unit’s Unit Costs of Health and Social Care publications for 201562 and 2016. 63 For all other NHS contacts, unit costs were derived from the NHS Reference Costs 2015 to 2016. 64 For outpatient visits, we used the weighted average of all consultant-led, non-admitted face-to-face attendances provided by the NHS during a year (including weighting by first or follow-up appointment). The same method was used for visits to physiotherapists, occupational therapists and psychologists. In the absence of specific unit costs for counsellors, we assumed that these would be the same as for a psychologist. For visits to A&E, we used the weighted average of all emergency medicine contacts, excluding dental care and participant dead on arrival. For ambulance transport to A&E, the unit cost of a call to the emergency services was included, as well as that for ambulance transport.
Using the reasons for hospitalisation reported by participants in the resource use questionnaires, we obtained diagnosis and procedure codes. These were then translated into a Healthcare Resource Group (HRG) using the HRG4+ 2015/16 Reference Costs Grouper (https://digital.nhs.uk/services/national-casemix-office/downloads-groupers-and-tools/grouper-and-tools-archive/; accessed 2 April 2019). Each HRG was then linked to a series of elective, non-elective and day case reference costs obtained from the NHS Reference Costs 2015 to 2016. 64
Non-NHS, including productivity
Costs of prescribed and OTC medications were obtained from the British National Formulary (BNF). 65 For complementary medicines or supplements not listed in the BNF, unit costs were derived from commercial suppliers such as Holland & Barrett (Nuneaton, UK), which is one of the largest suppliers of health foods in the UK.
To cost the hours of missed work as a result of asthma, we used the mean hourly earnings for men and women, which are £17.01 and £14.06, respectively. 66
Statistical analysis
As the analysis of the LASER trial data was undertaken on an ITT basis, multiple imputation was used to impute missing cost and utility values for the reference case analysis (i.e. analysis of all participants regardless of whether complete data were available or not). 67,68
Global Evaluation of Treatment Effect questionnaire
Although the trial protocol stated that both participants and trial physicians would complete a GETE questionnaire at the 12-month follow-up visit, for a number of participants only one GETE questionnaire was completed. When only one questionnaire was filled in, in some cases it was not possible to determine whether the participant or physician had filled in the questionnaire. In these cases we assumed, under recommendation from trial staff responsible for participant follow-up, that the participant had filled in the questionnaire. As a result, analysis of the GETE questionnaire data is presented based on responses from trial physicians, confirmed responses from participants and confirmed and possible responses from participants. The percentages with which each of these stakeholder groups rated the global treatment effectiveness of the TLA device as excellent, good, moderate, poor or worsening were compared across the two participant groups using chi-squared tests.
Quality of life
Standardised Asthma Quality of Life Questionnaire: disease-specific quality of life
The overall AQLQ(S) score for each participant at each follow-up visit was calculated as the mean of all 32 responses to the questionnaire. We also evaluated each participant’s score for each domain individually (symptoms, activity limitations, emotional function and environmental stimuli) as the mean of all responses in that domain. The mean differences in the AQLQ(S) scores of the two treatment groups (both across all questions and for each domain individually) were calculated for each follow-up visit with 95% CIs. The statistical significance of these differences was assessed using two-sided t-tests. The differences in the means of the 12-month and randomisation scores were also compared using two-sided t-tests.
EuroQol-5 Dimensions: generic quality of life
As recommended by NICE,69 and for those participants with complete EQ-5D-5L and EQ-VAS data only, the responses to each of the five questions in the EQ-5D-5L were converted into utilities using the validated mapping function to derive utility values for the EQ-5D-5L from those existing for the EQ-5D-3L (EuroQol-5 Dimensions, three-level version). 70 The mean differences in the utility values and EQ-VAS scores between the two treatment groups at each follow-up visit were calculated with 95% CIs. The statistical significance of these differences was assessed using two-sided t-tests. Changes between randomisation and 12-month scores were also compared using two-sided t-tests.
Carer quality of life
An insufficient number of AC-QoL questionnaires were completed by adult carers of the participants for these results to be meaningful.
Quality-adjusted life-years
Survival was estimated using the Kaplan–Meier survival function at 12 months post randomisation. A quality-adjusted survival curve was generated by plotting, against time, the product of the mean utility values of participants living at time t (determined as described in EuroQol-5 Dimensions: generic quality of life) and the probability of surviving to time t, with t being set to 3 months, in order to create four periods of 3 months. The area under this quality-adjusted survival curve then gave the mean quality-adjusted survival in each treatment group. 71 Utility was assumed to change linearly between each follow-up visit (i.e. each 3-month period), rather than changing at the midpoint between follow-up visits or being maintained from one follow-up visit to another.
For each treatment group, QALYs were calculated with 95% CIs determined non-parametrically from 1000 bootstrap differences. Mean QALY differences between the two participant groups were also calculated with 95% CIs estimated using the 1000 bootstrap differences. With this approach, the interpretation of a 95% CI ranging from minus to positive is that there is no statistically significant difference at the traditional level (p = 0.05).
To capture the whole participant sample in the QALY calculations, for those participants who withdrew from the analysis their missing utility estimates were assumed to be the same as the mean for that treatment group. The analyses were conducted on all participant data as well as on complete-case data only, that is, for participants with complete EQ-5D-5L responses from each follow-up visit and who did not die during the trial.
Resource use
NHS resource use
Except for number of days in hospital, statistical differences in NHS resource use between the two participant groups were evaluated assuming a Poisson distribution. Differences in number of days in hospital between groups were assessed using a Student’s two-sided t-test. Resource use was calculated for each 3-month follow-up visit period using the available data, as well as for the full 12-month period using complete data only, that is, from those participants with 3-, 6-, 9- and 12-month resource data recorded.
Non-NHS resource use
The mean differences in the expense borne by participants in the two participant groups over the 12-month trial period for OTC medicines and supplements were calculated with 95% CIs. The statistical significance of these differences was assessed using two-sided t-tests.
Productivity losses
The responses to the six WPAI(A) questions were used to assess productivity losses. For the question ‘Currently employed?’, the percentages for the two participant groups were calculated and the difference in these rates was assessed using the chi-squared test. The differences between the groups in terms of the responses to the remaining questions, which all have numerical responses (‘Hours missed due to asthma’, ‘Hours missed due to other reasons’, ‘Hours actually worked’, ‘Impact of asthma on work productivity’ and ‘Impact of asthma on regular daily activities’), were assessed using a Student’s two-sided t-test.
For participants with missing follow-up visit WPAI(A) responses, we used, when available, data from other questionnaires collected at that follow-up visit that also asked about the number of days of work and/or study missed as a result of asthma. In addition, when WPAI(A) information was missing, we assumed that those participants who reported being retired at baseline would continue to be so at follow-up. The WPAI(A) determined the number of daily work hours missed over a 7-day interval; we multiplied this by 91.25 days to calculate the productivity losses (i.e. work hours missed) over a 3-month period representing the interval between study visits.
An insufficient number of caregiver WPAI questionnaires were completed by adult carers of the participants to power a statistical analysis of these responses.
Costs
Costs of the device
The costs of the TLA device were provided directly by the manufacturer (Airsonett). 72 After consultation with Airsonett, we assumed that the lifetime of the device would be 10 years. We estimated costs, excluding value-added tax (VAT), under three different scenarios:
-
Device rental. Under this scenario, rather than purchase the device, the NHS would rent the device from the manufacturer. Under this rental agreement, the costs of device reconditioning, replacement of spare parts, servicing and filter changes would be included. The costs for the initial month of rental were £690 and for subsequent months were £174. Hence, the annual cost would be £2604 over the first year of rental and £2088 for subsequent years. Averaging the cost of rental over 10 years, the mean annual cost would be £2140.
-
Purchase with no full service agreement. Under this scenario, the NHS would purchase the device for £8780. It would then have to cover the costs of reconditioning, filters (two every year, excluding the first one, which is provided with the device) and any device accessories that would need replacing. Table 17 provides the unit costs of reconditioning, filters and accessories and the number of times that these would be required over the lifetime of the TLA device. We assumed that each accessory would require replacing once because of breakage.
To estimate a mean annual cost, we first estimated the present value cost of device acquisition, reconditioning, filter changes and accessories. For this, we discounted all future costs using an annual rate of 3.5%, assumed that reconditioning and device accessory changes would occur at 5 years and added these costs to the costs of device acquisition and one filter change (i.e. costs incurred during the first year). In present value terms, the cost of acquisition, reconditioning, filter changes and device accessories would be £17,797. To obtain an equivalent annual cost,73 we depreciated these costs over the 10-year lifetime of the device using an annual rate of 3.5%. Therefore, the equivalent annual cost of this scenario to the NHS was £2140.
-
Purchase with full service agreement. Under this scenario, the NHS would purchase the device for £8780. With a full service agreement the NHS would then, after the first year, pay an annual maintenance fee of £1125.60, which would cover the costs of device reconditioning, replacement of spare parts, servicing and filter changes. To estimate a mean annual cost, we first estimated the present value cost of acquisition and service of the device. For this, we discounted the future costs of servicing using an annual rate of 3.5% and added these costs to the acquisition cost. In present value terms, the cost of acquisition and servicing of the device would be £17,643. To obtain an equivalent annual cost, we depreciated these costs over the 10-year lifetime of the device using an annual rate of 3.5%. 74 Therefore, the equivalent annual cost of this scenario to the NHS was £2121.
Given that there was no considerable difference in the annual cost under these three scenarios, we used the costs under scenario 1 (i.e. rental agreement) in the analysis.
Total societal costs
The total societal cost is the sum of the NHS resource use and non-NHS resource use, including costs arising from the productivity losses. The difference between groups in the mean total societal cost at each 3-month follow-up visit for available cases, as well as for the full 12 months where complete data existed, was assessed using Student’s two-sided t-test.
Cost-effectiveness
Complete-case analysis
To estimate the cost-effectiveness of the TLA device compared with placebo, we undertook a complete-case analysis including only participants who had complete cost and EQ-5D-5L utility information. We carried out an incremental analysis, with the mean cost difference between the TLA device and placebo divided by the mean QALY difference to give the incremental cost-effectiveness ratio (ICER). The non-parametric percentile method75 used to calculate the CI around this ratio used 1000 bootstrap estimates of the mean cost and QALY differences. We used the cost-effectiveness acceptability curve to show the probability that the TLA device is cost-effective at 1 year for different values of the NHS’s willingness to pay for an additional QALY. 74 Cost-effectiveness was estimated both from a NHS and from a societal perspective.
Reference case analysis, including imputation of missing data
Descriptive analyses revealed that 79% (380/484) of observations in the placebo group had complete cost and EQ-5D-5L data compared with 76% (360/476) of observations in the treatment arm. Logistic regression models comparing missing indicator baseline variables with experiencing a severe exacerbation showed that having missing cost data was significantly negatively associated with age (p < 0.031) and experiencing a severe exacerbation (p < 0.001). Missing utility data were also negatively associated with age (p < 0.079) and experiencing a severe exacerbation (p < 0.001). This suggests that the data were not missing at random, as is assumed in the complete-case analysis. This may lead to bias in analyses, as participants with complete data may vary from those with missing data, and to a loss of statistical power, as missing data reduce the available sample size. 76
As the analysis of the LASER trial data was undertaken on an ITT basis, multiple imputation was used to impute missing cost and utility values for the reference case analysis (i.e. analysis of all participants regardless of whether complete data were available or not). 67,68 As is recommended best practice, imputation was implemented separately by randomised treatment allocation. 77 Costs were imputed at the most disaggregated level at which the model would converge. However, it was not possible to impute individual costs for ambulance use and hospitalisation costs. As a result, we imputed values for the combined total hospital care costs category (including outpatient, A&E, ambulance and hospitalisation costs). The only exception to this was for the five participants who withdrew from the study following randomisation (and, in addition, who withdrew consent for their data to be used for analysis). As we were unable to use any of their baseline characteristics to impute missing outcome data, we imputed missing cost and utility data for these participants using the average estimates for cost and utility, stratified by treatment arm.
Rather than imputing missing responses for each of the five domains in the EQ-5D-5L, we imputed the overall EQ-5D-5L utility score. 78 The imputation of costs and utilities was conducted using predictive mean matching (i.e. imputation of data from similar participants with complete data) to account for the skewed nature of both cost and utility data. Imputation was conducted using the trial minimisation criteria (i.e. clinical site, age, sex, baseline exacerbation rate, use of OCSs and an indicator of pre-bronchodilator FEV1 of < 50%) as well as number of exacerbations at follow-up. Following the rule of thumb that the number of imputations should be at least equal to the percentage of incomplete cases (originally 21% imputations and 24% incomplete cases),79 we generated 30 replacement values for each missing case, generating 30 imputed data sets.
Using the Stata (version 14) ‘mi estimate’ command, we obtained mean and standard deviation (SD) estimates of costs and utilities across participant groups using the ‘mi estimate: reg’ ordinary least squares regression command.
Long-term cost-effectiveness
Markov modelling was used to extrapolate the long-term cost-effectiveness of TLA therapy for patients with severe asthma from the 12-month LASER trial data and relevant reputable third-party published work.
Markov model
The Markov model was developed in Microsoft Excel® version 16.0 (Microsoft Corporation, Redmond, WA, USA) to follow a hypothetical, homogeneous cohort over 40 years and consists of four distinct health states – (1) daily symptoms, (2) severe exacerbations, (3) severe exacerbation-related death and (4) death by other causes – as shown in Figure 13. Participants begin in the model suffering from daily symptoms with a probability of either experiencing an exacerbation or dying from all other causes. Participants who experience an exacerbation can recover and move back to the daily symptoms state or suffer another exacerbation. They also have a small probability of an exacerbation being fatal or dying from other causes. The model has a cycle length of 3 months, mimicking the follow-up periods in the LASER trial. In the model, the cohort starts at an age of 47 years, which is the mean age of participants in the LASER trial. QALYs and costs incurred after the first year are discounted at an annual rate of 3.5%.
FIGURE 13.
Markov model.
Model parameters
The parameters in the Markov model are described in the following sections.
Transition probabilities and treatment effectiveness
A severe asthma exacerbation is incorporated into the model as a binary variable, indicating the occurrence of one or more exacerbations over a 3-month period. This effectiveness measure was used to reflect the set-up of the Markov model and to minimise the number of health states required in the model. The total number of 3-month periods in which participants had at least one severe asthma exacerbation was 129 (out of 485) in the placebo group. Therefore, the absolute risk of severe exacerbations was 0.27 (129/485) in the placebo group. This estimate was used as the transition probability between daily asthma symptoms and severe exacerbations. As per NICE guidance,14 it was also assumed that this would be the probability of remaining in the severe exacerbation health state.
In the TLA device group, the total number of 3-month periods in which participants had at least one severe asthma exacerbation was 126 (out of 480). The absolute risk of severe exacerbations was therefore 0.26 (126/480) in this group, giving a relative risk of 0.987.
Mortality
The probability of non-asthma-related mortality was constructed using age- and sex-specific mortality rates from life tables for England and Wales from 2015. 80 The probability of non-asthma-related mortality was estimated by subtracting the probability of asthma-related mortality [International Statistical Classification of Diseases and Related Health Problems, 10th revision (ICD-10)81 medical classifications relating to asthma: ICD-10 codes J45 and J46] from the probability of all-cause death. Weighted means according to the sex balance in the LASER trial (73% women) were used.
The LASER trial was not powered to identify asthma-related mortality. Therefore, the probability of death from a severe asthma exacerbation was obtained from previously published estimates. We reviewed the systematic review of asthma-related mortality conducted as part of the NICE guidance,62 which identified two studies. 79,82
As per NICE guidance,62 we obtained the estimate of asthma-exacerbation-related mortality from the study by de Vries et al. 82 This study estimated mortality rates for participants at different asthma treatment steps according to BTS/SIGN guidelines. 16 As the LASER trial population is most likely to reflect participants at step 5 in the BTS/SIGN guidelines, we obtained the mortality rate for this group: 0.4 cases per 100 person-years. This reflects a 3-monthly probability of death of 0.001 (SE 0.0002).
Utility
Utility information required for the model was based on LASER trial data after imputing for missing cases. Regression analyses were performed to assess the relationship between utility and use of the TLA device and asthma exacerbations. Analyses of utility pose challenges because they are negatively skewed and censored at 1. 83 Consequently, a two-part model was used: a logistic regression to assess the predictors of reporting problems in the EQ-5D-5L and, conditional on reporting problems in the EQ-5D-5L, a general gamma linear model with a log identity to determine the predictors of utility loss (i.e. 1 – utility). Using the product of the two-part model, the mean expected utility gain/loss associated with different participant characteristics was estimated.
Costs
All cost parameters required for the Markov model were derived directly from the individual participant-level data obtained from the LASER trial. These included the costs of the TLA device, NHS costs, societal costs and the impact of asthma exacerbations on NHS and societal costs.
We used the rental costs of the TLA device as supplied by Airsonett; hence, for the first cycle of the model (i.e. the first 3 months), we used a cost of £1038 and for all subsequent cycles we used a cost of £522.
Model estimates for NHS costs incurred by participants with asthma and the impact of asthma exacerbations on them were based on trial data after imputing for missing cases. To assess the impact of asthma exacerbations on 3-monthly NHS and societal costs, generalised linear models with a gamma distribution and log link function were used to model the relationship between the variance and the conditional mean, clustered at the participant level.
Sensitivity analyses
The uncertainty around the Markov model parameters was incorporated into the model using probabilistic sensitivity analysis. The uncertainty around costs, utilities and absolute and relative risks was estimated using a non-parametric bootstrap procedure with 1000 resamples from the LASER trial data. The probability of exacerbation-related death was parameterised using the beta distribution. The cost of the device and the probabilities of death from other causes were assumed not to vary.
Results
Global Evaluation of Treatment Effect questionnaire
As seen in Table 33, over half of all participants in both participant groups experienced a perceived improvement in their asthma over the 12 months of the trial. However, regardless of who completed the GETE questionnaire, either participant or trial physician, there were no statistically significant differences in responses between the two treatment groups.
Standardised Asthma Quality of Life Questionnaire
The two participant groups had similar overall AQLQ(S) scores at randomisation (p = 0.213; see Table 34). In both groups there was a statistically significant improvement in overall quality of life over the trial. When quality of life at 12 months was compared with that at randomisation, an improvement in the overall AQLQ(S) score of 0.57 (p < 0.001) in the placebo group and 0.68 (p < 0.001) in those receiving the TLA device was identified. Although the improvement in overall quality of life was higher in participants in the TLA group, this improvement was not statistically significant compared with the improvement in participants in the placebo group (p = 0.543).
Overall, AQLQ(S) scores in the two participant groups were similar at each follow-up visit, except at 6 months, when participants receiving the TLA device had a significantly higher quality of life than those receiving placebo (4.74 vs. 4.30, respectively; p = 0.020; see Table 34). This difference at 6 months was also observed for the following domains: activity limitation (p = 0.022) and environmental stimuli (p = 0.009). In addition, participants in the TLA group also had higher quality of life scores in the domain for environmental stimuli at 9 months (p = 0.038).
EuroQol-5 Dimensions utility
Responses to the EQ-5D-5L questionnaire are presented in Table 38. The utilities that these responses were converted into are shown in Table 18. For participants who completed the EQ-5D-5L at both randomisation and 12 months, there was no statistically significant improvement in overall quality of life over the trial in either participant group (p = 0.983 for the placebo group and p = 0.105 for the TLA device group). In addition, no significant differences in utility values were observed between the two groups at any of the follow-up visits (using data that were complete for each follow-up visit only).
| Measure | Trial group, mean (SD), n | p > |z| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo | TLA device | |||
| EQ-5D-5L utility | ||||
| Randomisation | 0.67 (0.25), 119 | 0.68 (0.26), 115 | 0.633 | 0.02 (–0.05 to 0.08) |
| 3 months | 0.68 (0.27), 104 | 0.73 (0.23), 98 | 0.123 | 0.05 (–0.01 to 0.12) |
| 6 months | 0.67 (0.28), 101 | 0.71 (0.24), 96 | 0.265 | 0.04 (–0.03 to 0.12) |
| 9 months | 0.64 (0.30), 95 | 0.72 (0.22), 90 | 0.052 | 0.08 (–0.00 to 0.15) |
| 12 months | 0.67 (0.30), 96 | 0.74 (0.24), 90 | 0.081 | 0.07 (–0.01 to 0.15) |
| Difference at 12 months | –0.00 (0.17), 96 | 0.03 (0.17), 90 | 0.226 | 0.04 (–0.02 to 0.08) |
| EQ-VAS score | ||||
| Randomisation | 58 (20), 120 | 61 (19), 115 | 0.329 | 2 (–2 to 7) |
| 3 months | 59 (23), 104 | 65 (18), 98 | 0.032 | 6 (1 to 12) |
| 6 months | 63 (21), 102 | 64 (20), 95 | 0.690 | 1 (–5 to 7) |
| 9 months | 62 (21), 94 | 65 (19), 89 | 0.193 | 4 (–2 to 10) |
| 12 months | 65 (20), 95 | 67 (18), 90 | 0.471 | 2 (–4 to 8) |
| Difference at 12 months | 7 (19), 95 | 7 (20), 90 | 0.790 | 1 (–5 to 7) |
Table 18 also shows that there was a statistically significant improvement in overall quality of life over the trial in both participant groups when assessed by EQ-VAS scores. When quality of life at 12 months was compared with that at randomisation, an improvement in VAS score of 7 was observed both for those receiving placebo and for those receiving the TLA device (p = 0.002 and p = 0.001, respectively). At 3 months, participants in the TLA device group had significantly higher VAS scores than those in the placebo group (65 vs. 59; p = 0.032).
Table 39 presents the same results but only for those participants who had complete EQ-5D-5L utility and VAS scores at all four follow-up visits.
Quality-adjusted life-years
Survival data were combined with the EQ-5D-5L utilities to estimate QALYs at 1 year after randomisation. Given that only two participants died during the trial follow-up (one in each group), the life-years gained over the trial period were virtually identical between the two groups (Table 19).
| Survival data | Trial group, mean (95% CI) | Mean difference (95% CI) | |
|---|---|---|---|
| Placebo | TLA device | ||
| Life-years gained (survival) | |||
| Overall sample | 0.999 (0.998 to 1.000) | 0.992 (0.975 to 1.000) | –0.007 (–0.024 to 0.001) |
| Complete cases | 0.999 (0.998 to 1.000) | 0.989 (0.967 to 1.000) | –0.010 (–0.032 to 0.001) |
| QALYs gained | |||
| Overall sample | 0.663 (0.622 to 0.706) | 0.712 (0.679 to 0.746) | 0.049 (–0.006 to 0.103) |
| Complete cases | 0.666 (0.618 to 0.710) | 0.718 (0.679 to 0.753) | 0.052 (–0.008 to 0.110) |
Regardless of whether QALYs were estimated for the whole sample or only for those participants with complete EQ-5D-5L information at all follow-up visits, participants in the TLA device group gained more QALYs than those in the placebo group. However, these differences did not reach statistical significance (determined by the fact that the CIs range from negative to positive numbers in Table 19).
Resource use and costs
NHS perspective
Information on 3-month NHS resource use and unit costs, inpatient health-care unit costs and 3-month NHS costs collected at each follow-up visit is presented in Tables 40–42, respectively. Table 35 presents the mean NHS resource use estimates over the 12 months after randomisation. The only statistically significant reductions in NHS resource use patterns in the treatment group compared with the placebo group over the 12 months after randomisation were in the numbers of general practice consultations (both surgery visits and telephone consultations), occupational therapist visits and psychologist visits; over the course of the year, participants in the TLA device group had 0.94 less general practice surgery consultations (95% CI –1.68 to –0.19; p = 0.014), 0.64 less general practice telephone consultations (95% CI –1.10 to –0.19; p = 0.005), 0.37 less occupational therapist visits (95% CI –0.95 to –0.22; p = 0.002) and 0.43 less psychologist visits (95% CI –0.76 to –0.23; p < 0.001) than participants in the placebo group. Conversely, the TLA device group had statistically significantly more physiotherapy visits on average than the placebo group (0.42, 95% CI 0.04 to 0.80; p = 0.032).
The cost of all this NHS resource use is shown in Table 20. Excluding the costs of the TLA device, over the 12 months after randomisation, participants in the placebo and TLA device groups incurred mean annual NHS costs of £4188 (SD £5004) and £4182 (SD £6091), respectively (p = 0.995). In both groups, the main cost drivers were inpatient stays (£1575 in the placebo group and £1385 in the TLA device group), which accounted for around one-third of all NHS costs in both groups.
| Resource use category | Trial group, mean (SD) (£) | p > |z| | Mean difference (95% CI) (£) | |
|---|---|---|---|---|
| Placebo (n = 81) | TLA device (n = 79) | |||
| General practice consultations | ||||
| Surgery | 221 (222) | 187 (203) | 0.320 | –34 (–100 to 33) |
| Home | 15 (64) | 5 (20) | 0.183 | –10 (–25 to 5) |
| Telephone | 65 (174) | 47 (83) | 0.416 | –18 (–60 to 25) |
| Nurse consultations | ||||
| Surgery | 36 (63) | 34 (105) | 0.862 | –2 (–29 to 25) |
| Home | 9 (53) | 10 (61) | 0.933 | 1 (–17 to 18) |
| Physiotherapist visits | 61 (160) | 82 (365) | 0.648 | 20 (–67 to 108) |
| Occupational therapist visits | 27 (108) | 2 (16) | 0.040 | –25 (–50 to –1) |
| Psychologist visits | 87 (420) | 23 (125) | 0.194 | –64 (–161 to 33) |
| Counsellor visits | 93 (388) | 65 (315) | 0.617 | –28 (–139 to 83) |
| Total community care costs | 614 (975) | 453 (724) | 0.240 | –160 (–429 to 108) |
| Outpatient visits | 389 (499) | 350 (659) | 0.672 | –39 (–221 to 143) |
| A&E visits | 107 (184) | 126 (248) | 0.594 | 18 (–50 to 86) |
| Ambulance use | 93 (256) | 77 (214) | 0.666 | –16 (–90 to 58) |
| Hospitalisations | ||||
| Non-ITU | 1247 (2712) | 1251 (3257) | 0.993 | 4 (–931 to 939) |
| ITU | 328 (2255) | 134 (865) | 0.477 | –193 (–729 to 343) |
| Total hospital care costs | 2163 (4518) | 1938 (4376) | 0.749 | –226 (–1615 to 1164) |
| Asthma medications | 1212 (1255) | 1513 (3047) | 0.413 | 301 (–423 to 1026) |
| Other prescribed medications | 199 (288) | 278 (572) | 0.268 | 79 (–61 to 220) |
| Total prescribed medications | 1411 (1295) | 1791 (3087) | 0.309 | 381 (–355 to 1117) |
| Total | 4188 (5004) | 4182 (6091) | 0.995 | –6 (–1744 to 1733) |
| Total – including costs of TLA device | 4188 (5004) | 6296 (6101) | 0.018 | 2108 (368 to 3849) |
Costs of asthma prescription medications for both groups were also considerable: £1212 (SD £1255) in the placebo group (accounting for 29% of total NHS costs) and £1513 (SD £3047) in the TLA device group (accounting for 36% of total NHS costs). A total of seven participants were prescribed omalizumab (four in the placebo group and three in the TLA device group; p = 0.718).
After taking into account the costs of the TLA device, there was a statistically significant difference in total NHS costs at 12 months. Participants in the TLA device group incurred mean costs of £6296 (SD £6101) whereas those in the placebo group incurred mean costs of £4188 (SD £5004) (mean difference £2108, 95% CI £368 to £3849; p = 0.018).
Productivity losses and other non-NHS costs
As shown in Table 36, half of all participants in each participant group were not working at randomisation. There were no significant differences in employment between the two participant groups at subsequent follow-ups. In terms of self-reported hours missed from work as a result of asthma, responses were non-significantly different between the two participant groups at randomisation and at each of the four follow-ups, except at 12 months. At 12 months, participants in the TLA device group reported having missed 2.1 (SD 7.7) hours during the past 7 days whereas those in the placebo group reported having missed 4.0 (SD 9.3) hours during the past 7 days (p = 0.049). Regardless of this finding, participants rated the impact of asthma on their ability to work and perform their daily activities similarly across all follow-ups.
Three-monthly non-NHS costs (including the costs of non-prescribed medications and productivity losses) are reported per follow-up visit in Table 43. Table 21 presents the means of these non-NHS resource use estimates over the 12 months after randomisation. For both participant groups, the costs of non-prescribed medications were negligible. Mean annual losses because of missed hours of work as a result of asthma were £1615 (SD £4590) in the placebo group and £807 (SD £2331) in the TLA device group (p = 0.152). Hence, overall non-NHS costs incurred over the 12 months after randomisation were £1628 (SD £4593) in the placebo group and £826 (SD £2330) in the TLA device group, which is not significantly different.
| Resource use category | Trial group, mean (SD) (£) | p > |z| | Mean difference (95% CI) (£) | |
|---|---|---|---|---|
| Placebo (n = 85) | TLA device (n = 84) | |||
| Medications | ||||
| OTC | 5 (12) | 11 (44) | 0.264 | 6 (–4 to 15) |
| Supplementary | 8 (23) | 8 (25) | 0.973 | 0 (–7 to 7) |
| Asthma productivity costs | 1615 (4590) | 807 (2331) | 0.152 | –808 (–1915 to 300) |
| Total non-NHS costs | 1628 (4593) | 826 (2330) | 0.155 | –802 (–1910 to 306) |
Cost-effectiveness: complete-case analysis
NHS perspective
A total of 78 participants in the placebo group and 77 participants in the TLA device group had complete follow-up data for both quality of life (QALYs) and NHS costs. As shown in Table 22, the mean total NHS costs for these complete quality of life and NHS cost cases, including the cost of the TLA device, were £4171 (SD £5051) for the placebo group and £6360 (SD £6167) for the TLA device group, a mean difference of £2189 (95% CI £401 to £3977; p = 0.017). The mean QALY gain was 0.68 in the placebo group and 0.72 in the TLA device group, a mean difference of 0.04 (95% CI –0.02 to 0.10). The incremental cost per QALY gained when the TLA device was compared with placebo was therefore £49,685.
| Resource use category | Trial group, mean (SD) | p > |z| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo (n = 78) | TLA device (n = 77) | |||
| NHS costs (£) | ||||
| General practice consultations | ||||
| Surgery | 223 (224) | 188 (206) | 0.308 | –35 (–104 to 33) |
| Home | 9 (41) | 5 (20) | 0.372 | –5 (–15 to 6) |
| Telephone | 66 (177) | 46 (9) | 0.362 | –20 (64 to 24) |
| Nurse consultations | ||||
| Surgery | 36 (64) | 34 (107) | 0.900 | –2 (–30 to 26) |
| Home | 9 (54) | 10 (61) | 0.898 | 1 (–17 to 19) |
| Physiotherapist visits | 63 (163) | 82 (369) | 0.682 | 19 (–71 to 109) |
| Occupational therapist visits | 28 (110) | 2 (16) | 0.039 | –26 (–52 to –1) |
| Psychologist visits | 90 (428) | 23 (127) | 0.189 | –67 (–167 to 33) |
| Counsellor visits | 90 (393) | 66 (319) | 0.675 | –24 (–138 to 90) |
| Total community care costs | 617 (990) | 457 (733) | 0.255 | –160 (–436 to 117) |
| Outpatient visits | 396 (504) | 357 (666) | 0.684 | –39 (–226 to 149) |
| A&E visits | 110 (186) | 127 (251) | 0.622 | 18 (–53 to 88) |
| Ambulance use | 96 (260) | 79 (217) | 0.646 | –18 (–94 to 58) |
| Hospitalisations | ||||
| Non-ITU | 1210 (2725) | 1284 (3293) | 0.878 | 74 (–884 to 1033) |
| ITU | 340 (2297) | 138 (876) | 0.471 | –202 (–755 to 351) |
| Total hospital care costs | 2151 (4581) | 1984 (4424) | 0.818 | –167 (–1596 to 1262) |
| Prescribed medications | ||||
| Asthma medications | 1201 (1268) | 1522 (3084) | 0.396 | 321 (–425 to 1068) |
| Other prescribed medications | 203 (292) | 283 (579) | 0.274 | 81 (–65 to 226) |
| Total prescribed medication costs | 1403 (1311) | 1806 (3124) | 0.296 | 402 (–356 to 1161) |
| Total | 4171 (5051) | 4247 (6156) | 0.933 | 76 (–1710 to 1862) |
| Total – including costs of TLA device | 4171 (5051) | 6360 (6167) | 0.017 | 2189 (401 to 3977) |
| QALYs | Mean (95% CI) | Mean (95% CI) | ||
| QALYs gained | 0.68 (0.63 to 0.72) | 0.72 (0.68 to 0.76) | NA | 0.04 (–0.02 to 0.10) |
| ICER | ||||
| ICER when TLA compared with placebo (£) | 49,685 | |||
| Probability TLA is cost-effective | 0.15 | |||
At the conventional £20,000-per-QALY-gained threshold, the TLA device would have a probability of being cost-effective of 0.15 from a NHS perspective (Figure 14). Only at a willingness-to-pay threshold of £50,000 per QALY gained would the probability of the TLA device being cost-effective be > 0.5 from a NHS perspective.
FIGURE 14.
Cost-effectiveness acceptability curve from a NHS perspective.
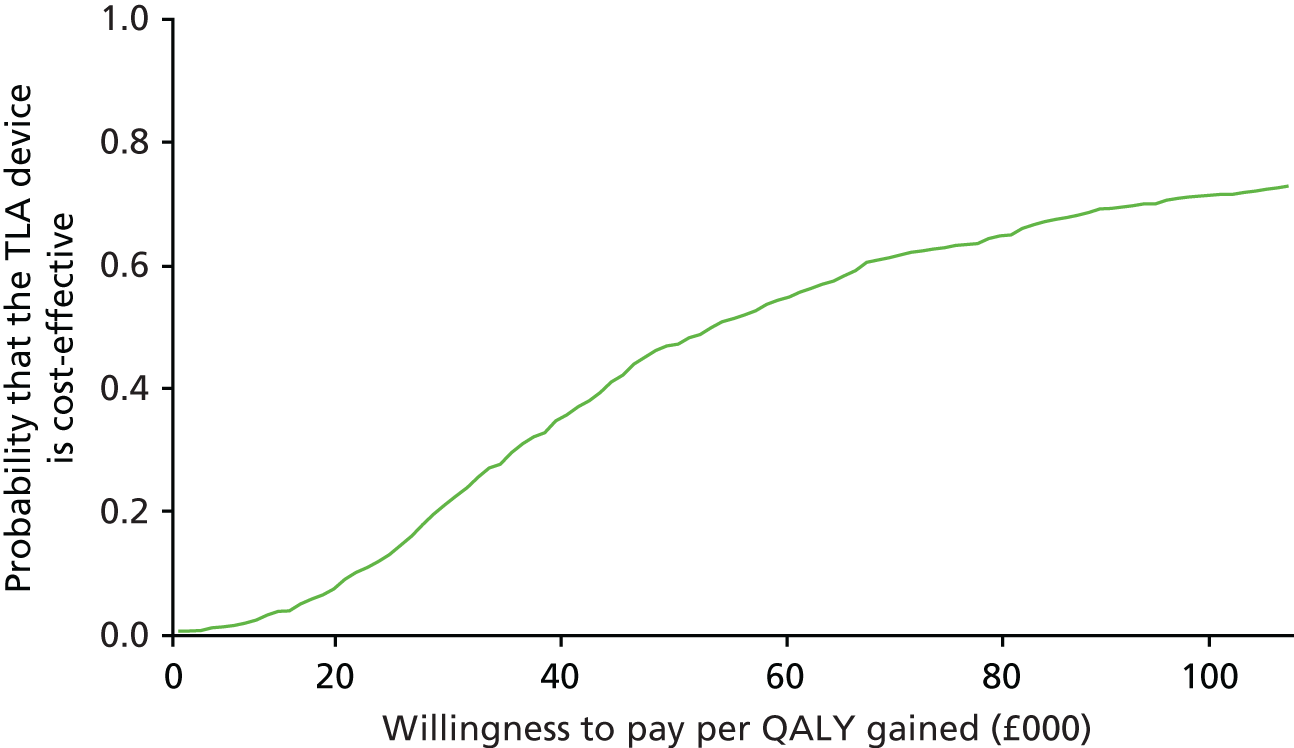
Societal perspective
A total of 74 participants in each participant group had complete follow-up visit data for both quality of life (QALYs) and societal costs (NHS and non-NHS resource use). As shown in Table 23, the difference in mean total societal costs between the groups was not significant: £5706 (SD £7207) in the placebo group and £7278 (SD £6986) in the TLA device group, a mean difference of £1572 (95% CI –£820 to £3966; p = 0.196). The mean QALY gain was 0.67 in the placebo group and 0.72 in the TLA device group, a mean difference of 0.05 (95% CI –0.01 to 0.11).
| Resource use category | Trial group, mean (SD) | p > |z| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo (n = 74) | TLA device (n = 74) | |||
| NHS costs (£) | ||||
| General practice consultations | ||||
| Surgery | 229 (228) | 183 (204) | 0.194 | –46 (–117 to 24) |
| Home | 10 (42) | 5 (21) | 0.363 | –5 (–16 to 6) |
| Telephone | 70 (181) | 47 (84) | 0.335 | –22 (–68 to 23) |
| Nurse consultations | ||||
| Surgery | 37 (65) | 35 (109) | 0.905 | –2 (–31 to 27) |
| Home | 9 (55) | 11 (63) | 0.908 | 1 (–18 to 20) |
| Physiotherapist visits | 66 (166) | 84 (376) | 0.707 | 18 (–76 to 113) |
| Occupational therapist visits | 30 (113) | 2 (16) | 0.037 | –28 (–54 to –2) |
| Psychologist visits | 95 (439) | 24 (129) | 0.184 | –71 (–176 to 34) |
| Counsellor visits | 95 (403) | 69 (325) | 0.662 | –26 (–145 to 93) |
| Total community care costs | 642 (1010) | 460 (746) | 0.215 | –182 (–470 to 107) |
| Outpatient visits | 402 (508) | 358 (675) | 0.655 | –44 (–238 to 150) |
| A&E visits | 114 (190) | 129 (255) | 0.688 | 15 (–58 to 88) |
| Ambulance use | 101 (266) | 79 (220) | 0.575 | –23 (–102 to 57) |
| Hospitalisations | ||||
| Non-ITU | 1269 (2786) | 1336 (3350) | 0.896 | 67 (–934 to 1068) |
| ITU | 341 (2355) | 143 (893) | 0.502 | –197 (–776 to 381) |
| Total hospital care costs | 2227 (4689) | 2045 (4502) | 0.810 | –182 (–1676 to 1311) |
| Prescribed medications | ||||
| Asthma medications | 1147 (1155) | 1549 (3144) | 0.305 | 401 (–368 to 1171) |
| Other prescribed medications | 208 (298) | 273 (575) | 0.385 | 66 (–83 to 214) |
| Total NHS costs | 4224 (5145) | 4327 (6260) | 0.913 | 103 (–1759 to 1964) |
| Medications | ||||
| OTC | 4 (8) | 11 (47) | 0.235 | 7 (–4 to 17) |
| Supplementary | 7 (20) | 7 (25) | 0.907 | 0 (–7 to 8) |
| Asthma productivity costs | 1471 (4532) | 821 (2358) | 0.276 | –650 (–1824 to 524) |
| Total non-NHS costs | 1482 (4531) | 839 (2358) | 0.281 | –643 (–1816 to 531) |
| Societal costs | 5706 (7207) | 5166 (6973) | 0.656 | –540 (–2931 to 1851) |
| Societal costs – including costs of TLA device | 5706 (7207) | 7278 (6986) | 0.196 | 1572 (–820 to 3966) |
| QALYs | Mean (95% CI) | Mean (95% CI) | ||
| QALYs gained | 0.67 (0.62 to 0.72) | 0.72 (0.68 to 0.76) | N/A | 0.05 (–0.01 to 0.11) |
| ICER | ||||
| ICER when TLA compared with placebo (£) | 29,831 | |||
| Probability TLA is cost-effective | 0.36 | |||
From a societal perspective, the incremental cost per QALY gained when the TLA device was compared with placebo was £29,831. At the conventional £20,000-per-QALY-gained threshold, the TLA device would have a probability of 0.36 of being cost-effective (Figure 15). Only at a willingness-to-pay threshold of > £30,000 per QALY gained would the probability of the TLA device being cost-effective be > 0.5 from a societal perspective.
FIGURE 15.
Cost-effectiveness acceptability curve from a societal perspective.

Cost-effectiveness: reference case
After multiple imputation of missing cost and utility data, the ICER for the TLA device compared with placebo was estimated from both a NHS and a societal perspective (Table 24). Mean total NHS costs, excluding costs of the TLA device, were non-significantly different between the two participant groups: £3730 (SD £5143) in the placebo group and £3670 (SD £4483) in the TLA device group, a mean difference of –£60 (95% CI –£1317 to £1200; p = 0.926). Mean total societal costs, excluding the TLA device cost, were also non-significantly different between the two participant groups: £5208 (SD £6798) in the placebo group and £4500 (SD £5839) in the TLA device group, a mean difference of –£708 (95% CI –£2368 to £972; p = 0.401). The mean QALY gain was 0.67 in the placebo group and 0.72 in the TLA device group, a mean difference of 0.05 (95% CI –0.001 to 0.10).
| Resource use category | Trial group, mean (SD) | p > |t| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo (n = 121) | TLA device (n = 119) | |||
| NHS costs (£) | ||||
| General practice consultations | ||||
| Surgery | 197 (200) | 180 (186) | 0.502 | –17 (–67 to 33) |
| Home | 13 (55) | 4 (17) | 0.089 | –9 (–20 to 1) |
| Telephone | 62 (148) | 47 (76) | 0.334 | –15 (–46 to 16) |
| Nurse consultations | ||||
| Surgery | 32 (87) | 25 (54) | 0.620 | –5 (–23 to 14) |
| Home | 12 (58) | 7 (49) | 0.501 | –5 (–19 to 9) |
| Physiotherapist visits | 58 (299) | 47 (151) | 0.737 | –11 (–51 to 72) |
| Occupational therapist visits | 21 (89) | 1 (13) | 0.029 | –20 (–38 to –2) |
| Psychologist visits | 88 (366) | 20 (104) | 0.068 | –68 (–140 to 5) |
| Counsellor visits | 102 (347) | 62 (263) | 0.382 | –40 (–129 to 50) |
| Total hospital care costs | 1925 (3952) | 1706 (3645) | 0.671 | –219 (–1233 to 795) |
| Prescribed medications | ||||
| Asthma medications | 1076 (2528) | 1320 (1116) | 0.338 | 244 (–257 to 744) |
| Other prescribed medications | 186 (479) | 248 (259) | 0.238 | 62 (–41 to 164) |
| Total NHS costs | 3730 (5143) | 3670 (4483) | 0.926 | –60 (–1317 to 1200) |
| Total NHS costs – including costs of TLA device | 3730 (5306) | 5538 (4497) | 0.005 | 1808 (541 to 3074) |
| Medications | ||||
| OTC | 11 (53) | 17 (22) | 0.291 | 6 (–5 to 16) |
| Supplementary | 6 (22) | 7 (19) | 0.738 | 1 (–4 to 6) |
| Asthma productivity costs | 1583 (4297) | 1020 (2759) | 0.253 | –562 (–1528 to 404) |
| Total non-NHS costs | 1594 (4299) | 963 (2400) | 0.185 | –631 (–1566 to 304) |
| Societal costs | 5208 (6798) | 4500 (5839) | 0.401 | –708 (–2368 to 972) |
| Societal costs – including costs of TLA device | 5208 (6819) | 6367 (6002) | 0.172 | 1159 (–507 to 2846) |
| QALYs | Mean (95% CI) | Mean (95% CI) | ||
| QALYs gained | 0.67 (0.62 to 0.69) | 0.72 (0.68 to 0.73) | N/A | 0.05 (–0.001 to 0.10) |
| ICER from a NHS perspective | ||||
| ICER when TLA compared with placebo | 36,219 | |||
| Probability TLA is cost-effective | 0.21 | |||
| ICER from a societal perspective | ||||
| ICER when TLA compared with placebo | 27,321 | |||
| Probability TLA is cost-effective | 0.40 | |||
From a NHS perspective, the incremental cost per QALY gained when the TLA device was compared with placebo was £36,219. At the conventional £20,000-per-QALY-gained threshold, the TLA device would have a probability of 0.21 of being cost-effective (Figure 16). Only at a willingness-to-pay threshold of > £39,000 per QALY gained would the probability of the TLA device being cost-effective be > 0.5 from a NHS perspective.
FIGURE 16.
Reference case: cost-effectiveness acceptability curve from a NHS perspective.

From a societal perspective, the incremental cost per QALY gained when the TLA device was compared with placebo was £27,321. At the conventional £20,000-per-QALY-gained threshold, the TLA device would have a probability of 0.40 of being cost-effective (Figure 17). Only at a willingness-to-pay threshold of > £29,000 per QALY gained would the probability of the TLA device being cost-effective be > 0.5 from a societal perspective.
FIGURE 17.
Reference case: cost-effectiveness acceptability curve from a societal perspective.
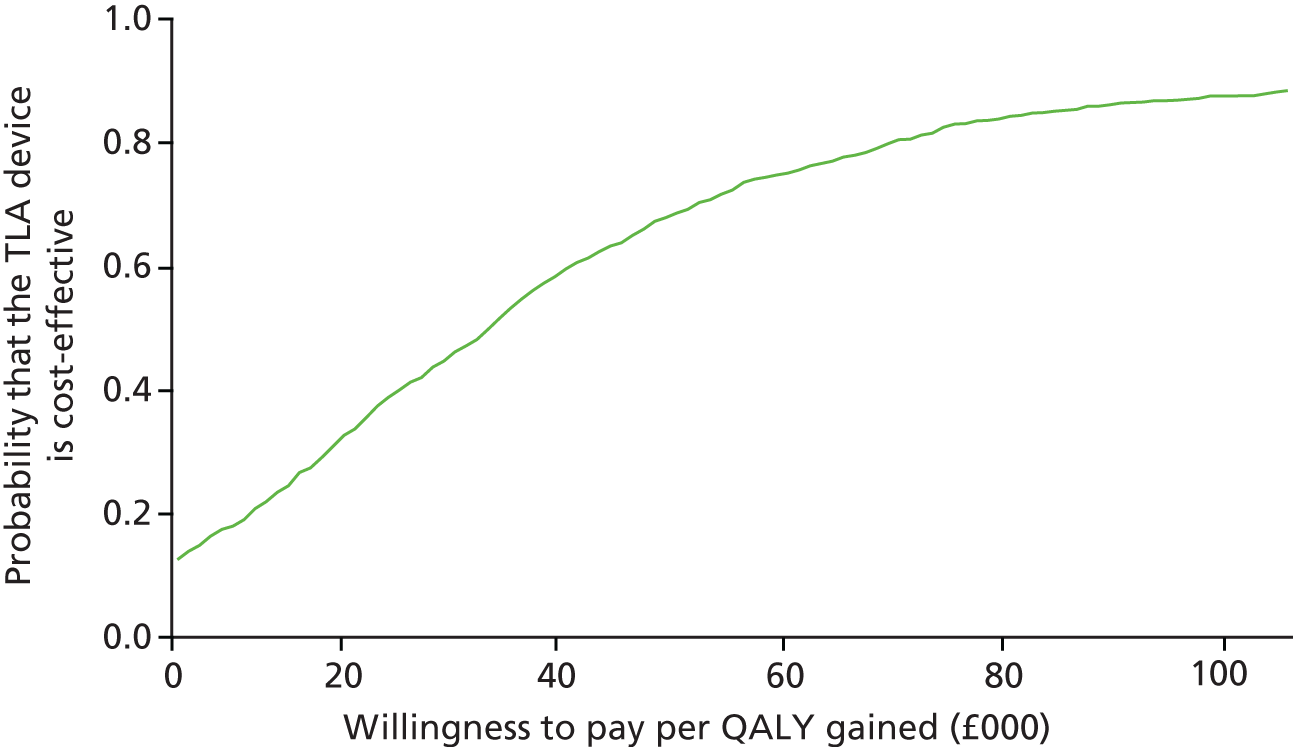
Model-based economic evaluation
The parameters used in the model-based economic evaluation used to infer long-term cost-effectiveness of TLA therapy are shown in Table 37.
When the model was run over the lifetime of the participant (assumed to be 40 years), the incremental cost per QALY gained when the TLA device was compared with placebo was £38,124 from a NHS perspective and £38,062 from a societal perspective (Table 25).
| Cost-effectiveness category | Trial group | Mean difference | |
|---|---|---|---|
| Placebo | TLA device | ||
| Mean per-patient costs (95% CI) (£) | |||
| NHS perspective | 73,940 (60,380 to 87,089) | 102,776 (89,936 to 115,883) | 28,836 (25,442 to 32,188) |
| Societal perspective | 99,732 (80,733 to 117,577) | 128,520 (109,559 to 144,935) | 28,789 (23,805 to 32,833) |
| Mean QALYs gained per patient (95% CI) | 12.40 (11.41 to 13.02) | 13.15 (12.39 to 13.46) | 0.76 (–0.04 to 1.54) |
| ICER from a NHS perspective | |||
| ICER when TLA compared with placebo | 38,124 | ||
| Probability TLA is cost-effective | 0.05 | ||
| ICER from a societal perspective | |||
| ICER when TLA compared with placebo | 38,062 | ||
| Probability TLA is cost-effective | 0.06 | ||
The results of the probabilistic sensitivity analysis indicated that the TLA device would have a 0.05 and 0.06 probability of being cost-effective at the conventional £20,000-per-QALY-gained threshold from a NHS and societal perspective, respectively (Figures 18 and 19, respectively).
FIGURE 18.
Markov model: cost-effectiveness acceptability curve from a NHS perspective.

FIGURE 19.
Markov model: cost-effectiveness acceptability curve from a societal perspective.
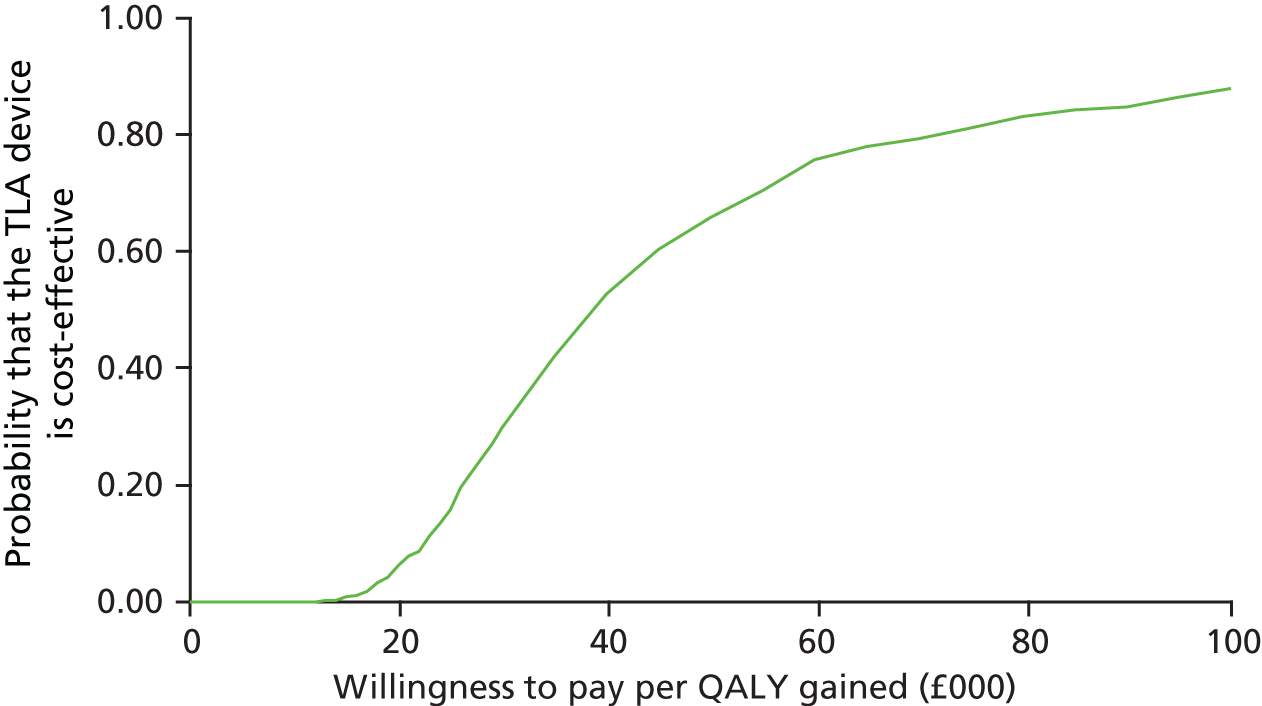
Economic evaluation discussion
Although previous economic evaluations have been undertaken on the use of TLA to treat severe asthma, this is the first evaluation to be based on a pragmatic, multicentre, double-blind, placebo-controlled, parallel-group RCT. These results show that use of the TLA device did not reduce overall health-care resource usage and, therefore, NHS costs. This lack of statistically significant differences in costs between the groups reflected the fact that the LASER trial did not detect any differences in exacerbations – a predictor of increased asthma health-care use and costs. According to the LASER trial data, having at least one severe exacerbation during a 3-month period significantly increases NHS costs by £1506 (95% CI £1192 to £1933).
Previous model-based economic evaluations of TLA have shown more promising results, suggesting that the device might be cost-effective. Subsequent modelling of the uncontrolled severe asthma subgroup in a previous multicentre, double-blind, placebo-controlled, parallel-group RCT33 suggested that TLA would be cost-effective in Sweden because of increases in quality of life. 35 Results from our trial-based economic evaluation did show that the use of the TLA device yielded higher levels of generic (based on the EQ-5D-5L) and disease-specific [based on the AQLQ(S)] HRQoL, with statistically significantly higher quality of life at some intermediary follow-up visits. However, these increases in quality of life were not sufficient to offset the annual costs associated with use of the TLA device, which resulted in an incremental cost per QALY gain that was above the £20,000-per-QALY-gained threshold used by NICE to determine cost-effectiveness.
Another model-based economic evaluation undertaken in the UK61 suggested that use of the TLA device is cost-effective at an incremental cost per QALY gained of £8998, which is well below the £20,000-per-QALY-gained threshold commonly used. This analysis was based on a small before-and-after study in 30 patients with severe difficult-to-control asthma,84 which showed a significant reduction in the number of asthma exacerbations, reduced use of A&E health care and (a non-significant) reduction in inpatient intensive care treatment. The study therefore suggested significant NHS cost savings of £1535 associated with use of the TLA device, which, after inclusion of device costs, yielded an additional cost of £553 per participant using the TLA device. In this study, in which no significant reduction in NHS costs was found, the additional cost of using the TLA device was £1808 per patient.
However, the economic evaluation of the LASER trial also had its limitations. As reported in previous chapters, there was a considerable number of missing data, mainly because of the number of participants withdrawing from the study or contact being lost at the follow-up visits. As a result, of a total of 121 and 119 participants randomised to the placebo and TLA device groups, respectively, complete cost and utility data were obtained for only 78 (64%) and 77 (65%) participants, respectively. We tried to overcome this limitation by imputing missing data for the reference case using currently recommended techniques, which generated similar results as in the complete- and reference-case analyses, but missing data remain an important limitation of this study.
Another limitation of this economic study was the fact that we did not receive Hospital Episode Statistics data collected centrally by the NHS on time, meaning that we could not validate the hospital outpatient, A&E visit and hospital admissions resource use data collected through the use of participant questionnaires. However, we have no reason to believe that any recall biases would have differed between the two participant groups and, as a result, have any impact on the final cost-effectiveness results.
In conclusion, evidence from the LASER trial shows that, at present, nocturnal TLA treatment is unlikely to be cost-effective in the treatment of severe asthma in either the short term or the long term. Although there was some evidence that the TLA device improved participants’ overall quality of life, this was offset in the cost-effectiveness analyses by the additional costs of the device.
Chapter 5 Main discussion
The TLA device has previously been shown to be beneficial in patients with allergic asthma, with improvements in asthma control and some evidence of improvements in quality of life. 31,33 In this pragmatic, multicentre, randomised placebo-controlled trial in patients with severe, exacerbation-prone allergic asthma, we were unable to demonstrate consistent benefits of the device over placebo in terms of reducing exacerbation frequency (despite assessment of exacerbations from a variety of sources). Sensitivity analyses including alternative sources of reported exacerbations (e.g. exacerbations recalled at follow-up visits or a combination of dated reports of exacerbations and exacerbations recalled at a follow-up visit, rather than dated reports only) did not reveal any further differences between treatment groups in the primary outcome.
Although there were no significant differences in the secondary outcomes of spirometric lung function (pre- or post-bronchodilator FEV1), asthma control or eosinophilic airway inflammation, there was a significant improvement in mean daily PEF in favour of the TLA device. However, only 150 out of the 240 participants provided data at the 12-month follow-up visit. In light of a lack of improvement in other parameters of lung function, and the sparse data limitations, this improvement needs cautious interpretation, although the levels of improvement in PEF are of the order of the expected magnitude of reductions in peak flow during exacerbations in this patient group. The results of the economic evaluation showed that use of the TLA device yielded higher levels of generic and disease-specific HRQoL, with statistically significantly higher HRQoL at some intermediary follow-up visits. However, given that TLA use was not associated with any reduction in overall NHS health-care use, these increases in HRQoL were not sufficient to offset the annual costs associated with use of the TLA device, which resulted in an incremental cost per QALY gain that was above the £20,000-per-QALY-gained threshold used by NICE to determine cost-effectiveness.
There were no significant differences in safety concerns between the groups, with adverse events being similar between the groups and none of the serious adverse events being considered to be device related. The TLA device was well received by participants and carers and the qualitative analyses in both the pilot study and at the end of the trial showed high levels of acceptability of the device.
The different outcomes in this trial compared with previous TLA studies and other anti-allergy interventions in this patient group require interpretation, and these are discussed in more detail in the following sections.
Assessment of exacerbations
The chosen primary outcome of severe exacerbations is important as it drives the costs of severe asthma because of the use of unscheduled care. Our definition was accurately described and consistent with international trial end point criteria, although we chose a threshold of ≥ 30 mg of prednisolone as this is consistent with the BTS/SIGN guidance for the treatment of severe acute asthma. Post hoc analyses of a lower threshold of ≥ 10 mg also did not reveal any significant findings. We had sought to ensure that we collected severe exacerbation reports from multiple sources to avoid under-reporting and to provide some clinical, if not data, corroboration that a severe exacerbation had occurred. By attempting to capture more information, we also increased the potential risk of missing data.
We collected data from the PED, which was filled out only during severe exacerbations, the TLA diary, filled out ‘continuously’, and the exacerbation review form, completed by the clinical team when the patient registered an exacerbation so that a clinical decision could be made to determine whether or not the increase in symptoms was a genuine severe exacerbation (or just a moderate exacerbation with increased symptoms and medication use but short of needing oral corticosteroids). Asthma exacerbations requiring hospitalisation were not required to be reported as serious adverse events but some of the centres still reported them as such. Furthermore, to ensure that severe asthma exacerbations requiring hospitalisation were reported if they had not been included in the PED, TLA diary, exacerbation review form or serious adverse event report, at the follow-up visits at 3, 6, 9 and 12 months we asked specifically for the number of hospital admissions and reasons for admission in the preceding 3 months. This included asthma hospitalisations with an estimate of length of stay and ITU admissions when appropriate. As we would expect in a real-world RCT, data completion across all formats was not consistent. Many participants completed their TLA diary but some did not; occasionally, clinical teams advised participants who were unable to complete their TLA diary to at least fill out their PED or come for an exacerbation assessment (exacerbation review form) when undergoing an exacerbation. We thus cleaned and matched all of the different sources of exacerbations by date to avoid double counting, for example courses of OCSs that coincided with a hospitalisation. We also combined the TLA diary and PED by date and dose, as in some the data were incomplete, for example the date of commencement was offset by a few days, in order to create a combined TLA diary/PED source of exacerbation. Transferring 12 exacerbations recorded as hospitalisations on the follow-up visit forms or recorded as serious adverse events resulted in 344 combined ‘dated exacerbations’. We relied on participants completing their TLA diary or PED when well or during exacerbations. As many patients did not complete the potentially onerous daily diary for the 12 months of the trial, there were significant ‘blank spaces’, interpreted either as missing data or that participants did not have an exacerbation if they also left the PED blank. Ultimately, this may potentially have led to severe exacerbations being missed.
Examining the follow-up visit form responses, it was evident that some participants either under-reported or over-reported severe exacerbations compared with their ‘dated exacerbations’, that is, based on the evidence of OCS use. Surprisingly, there was evidence of over-reporting of severe exacerbations even by some participants who filled out their TLA diary fully. It is possible that, by reporting on these exacerbations in the follow-up visit, some participants may have had recall bias. The consequence of all this is that the number of trial end-point-dated severe exacerbations may not be the same as the sum of the number of severe exacerbations reported by the participants at the follow-up visits, as moderate exacerbations may also have been reported at these follow-up visits.
We therefore sought to examine all possible measures of severe exacerbations, using the dated exacerbations in the primary analysis and incorporating other exacerbations into sensitivity analyses. By using dated exacerbations, there was certainty of data and clinical evidence of OCS use to meet our protocol definition; however, when TLA diary or PED data were missing, we assumed that patients did not have any severe exacerbations during these blank spaces. Using either the severe exacerbations reported on the follow-up visit form or the maximum of either the dated or the follow-up visit form exacerbations allowed the inclusion of more exacerbations – potential exacerbations for which the TLA diary was left blank – and also allowed for instances of under- or over-reporting of the dated sources. However, these last two methods used in the sensitivity analyses were both subject to recall bias, as we could not be certain that participants did not include non-protocol or moderate exacerbations. Other disadvantages of this approach were that it did not take account of whether or not the participants met the minimal clinical trial end point criteria, it did not allow for those who under-reported exacerbations on the follow-up visit form when we had evidence of more exacerbations from dated exacerbation sources, it could not ascertain whether or not participants contravened the 7-day rule for separating exacerbations and it potentially allowed for over-reporting by participants (i.e. for one exacerbation to be reported as two). Despite these different methods, we detected no differences in exacerbation frequency between the different arms of the trial.
We also considered the impact of missing TLA diary data on the primary outcome of the rate of severe asthma exacerbation events. The missing data from the TLA diary relate to the failure to record any value (diary entry left blank); the majority of the data were missing because of a failure to record a value of zero when it is probable that no exacerbation occurred. When participants were taking doses of OCSs according to levels in the protocol definition of an exacerbation (≥ 30 mg), the proportion of cases in which a value in the TLA diary had to be assumed to be zero was small; when participants were taking doses of OCSs that were much lower than levels in the protocol definition of an exacerbation (< 30 mg), the proportion of cases in which values had to be assumed to be zero was much larger. This indicated that participants were less likely to record lower doses of OCSs, for example when on maintenance OCSs or when tapering from prolonged courses, or to record a zero when they were not taking any OCSs and so were not in exacerbation (the commonest occurrence). Although in the primary analysis we had sought to determine not the severity and duration of exacerbations but, rather, their frequency, this approach may have led to us underestimating the rate of events, as we did not have available the number of days that participants were ‘at risk’ as the denominator in the model and we had to assume that all days (when there was no value recorded in the TLA diary) were potentially at risk. The number of potential missing data according to the TLA diary was significant and could have caused a type 2 error for the primary outcome, that is, not showing a treatment benefit even if such an effect existed. Ultimately, despite merging all available records to provide an integrated record of the number of exacerbations per participant, we were not able to demonstrate a difference in frequency of exacerbations between groups.
Appropriateness of the patient population and minimisation
There was a remarkable reduction (approximately 50%) in exacerbation frequency in both groups from baseline to completion of the trial, but no difference between groups. Across the two groups, 43% of participants reported no exacerbations during the 12-month trial despite having had at least two exacerbations in the preceding year. The most likely cause of the reduction in number of events is the lack of data. Although potentially attributable to missing data in this trial, this phenomenon of improvement in outcomes under study conditions has been reported for other diseases, as well as previously for asthma, and is not explicable simply by a regression to the mean effect; we would have expected that not to have occurred in a year-long study. Possible reasons other than improved adherence to treatments may include the support gained from regular visits and access to health-care professionals from trial teams, who are often integrated in clinical care teams too, and which may have improved longer-term care (no reduction in medications was allowed unless it was for safety reasons). We also recruited participants who may not have been known to the recruiting site and so the opportunity to treat other comorbidities that may drive asthma control may have been missed prior to enrolment and may have become evident during the trial. It is remarkable that four participants in the placebo group recorded a prior exacerbation frequency of 14, 18, 20 and 25 in the previous year. The impact of this was, however, minimised by including the exacerbation frequency in the previous 12 months (two, three, or more than three) and further analysis of the dated exacerbations during the trial showed no participants with a skewed exacerbation frequency between groups. It is therefore possible that the trial team may have selected participants with easily modifiable factors and this may not have been related to their source of origin (incident or prevalent population) as they were minimised at randomisation and thus balanced between groups. This has to be examined in the context of the challenges to recruitment in this patient population in light of other competing treatments such as new monoclonal antibodies therapies and the change in NICE guidance for omalizumab during the trial. Overall, the number of participants crossing over to alternative treatments during the trial was small (data not presented in this report) and so the statistical effects of crossover are unlikely to be significant. The fact that the majority of withdrawals occurred within the first 3 months reduces the likelihood that they occurred because of a failure of treatment effect (which might cause some participants to change to other alternative treatments).
Although not presented in this report, we measured the number of hours that the device was used by each participant. The majority of participants possessed a device for > 6 months of the trial, making them eligible to benefit from the 4-year post-trial provision period. This incentive may have helped with participant retention but this is not a perfect indicator of adherence as even measured hours is not representative of the time spent under the device. Nevertheless, the TLA diaries, which recorded use of the device, suggested that, when it was recorded, most participants used the device for a sufficient number of hours to include overnight sleep (≥ 8 hours). There were instances when participants were on holiday or abroad and in this pragmatic trial we allowed them to continue participation in the trial as long as this meant that they spent no longer than a week without the device. The number of withdrawals because of dissatisfaction with the device was low and the qualitative analysis confirmed the utility of the device and general acceptability despite its size by the bedside. A list of non-insurmountable modifications to the device emerging from the focus group sessions has been passed on to the manufacturer.
Trial conduct
All trial centres had a face-to-face site initiation by the trial co-ordinator to ensure that all trial procedures were followed. Despite this, 12 incidents of admission to hospital because of an exacerbation were identified from serious adverse event reporting (with or without other acute comorbidities); these were reframed as data contributing to the primary outcome rather than reportable adverse events.
Some trial teams recognised the onerous nature of the TLA diary, and this may explain why some participants were less likely to record values of zero when well and why often only the exacerbation review forms were completed, with the accompanying PED. Earlier data quality checks at sites may have identified this problem earlier.
Despite these issues, overall trial engagement was good and this was maintained at trial centres by sending a weekly e-mail including a ‘Tip of the week’, recruitment FAQs and information about how centres were progressing with meeting their recruitment targets for the coming month. This allowed trial teams to have up-to-date information and weekly visibility of trial progress. A dedicated trial mobile number (available 24 hours) allowed the lead site to address any potential queries immediately; the feedback from trial teams regarding this was very positive. Several monthly and other regular teleconferences with the lead LASER trial nurse allowed good practice to be shared, and this was found to enhance engagement among centres. Further incentivisation with a 6-monthly iPad (Apple Inc., Cupertino, CA, USA) competition for centres that provided the highest data quality, at least in terms of the return of CRFs, follow-up visit form completeness and timely handling of queries, ensured a good rate of return of data.
Comparison with other evidence on the temperature-controlled laminar airflow device
A larger placebo-controlled trial of the TLA device had previously shown an improvement in asthma-related quality of life and a reduction in exhaled nitric oxide, with a greater benefit shown in patients with severe asthma (GINA step 4) and with poor control. 33 However, this trial included a heterogeneous group of asthmatics (age range 7–70 years), randomised 2 : 1 to active treatment and placebo, about half of whom had GINA step 4 severity of asthma, meaning that the event rate of exacerbations was low. Consequently, despite an adequate number of participants, the trial was unable to show a reduction in exacerbation frequency. In the LASER trial, with a higher proportion of people with severe asthma, we were able to demonstrate improvements in both generic and asthma-related quality of life. Nevertheless, despite the more exacerbation-prone population included in our trial, we were unable to demonstrate the cost-effectiveness of the TLA device by an incremental cost per QALY gain below the £20,000 gained threshold used by NICE.
In another smaller, open-label, single-arm study of the TLA device, in which the participant population was more severely exacerbation prone than the population in our trial (33% were on OCSs and 43% were on omalizumab treatment at baseline, i.e. a significant number of BTS step 5 participants), TLA treatment was found to reduce the exacerbation rate from 3.6 per year to 1.3 per year, a 64% reduction. 37 This study differed from ours in that participants were their ‘own control’ rather than there being a placebo group and 50% of participants were children, with an even sex distribution. We report a similar reduction in exacerbation frequency before and after the trial in both groups, but with a predominantly female and obese allergic asthma population reflective of ‘real-world’ severe asthma patients. Thus, we have confirmed the benefit of improved quality of life, in keeping with other evidence, but not a reduction in eosinophilic inflammation or exacerbations. We did not test for an effect of the device on the systemic effects of allergy, although there was an improvement in the sinonasal score in favour of the active device (comparable to the minimally important difference for the SNOT-22 questionnaire), which was not statistically significant in this population (p = 0.07).
Allergic asthma and its link with symptoms
It is plausible that a proportion of severe asthma cases attributable to allergy may be overestimated and that aetiological mechanisms other than allergy may be important in the pathogenesis of severe asthma. For instance, numerous studies have reported a strong association between asthma exacerbations and respiratory viral infections, suggesting a viral-induced mechanism. 85 Rather than being mutually exclusive, viruses and allergens may interact in increasing the risk of severe asthma and exacerbation frequency. 86 It is not possible from the data obtained in the LASER trial to determine the aetiological mechanisms of each individual exacerbation and whether or not other factors were playing a role in the reported exacerbations. A better understanding of the aetiology of specific exacerbations may have demonstrated a different outcome in the trial.
Consistent with European standards,87 allergic sensitisation was defined in the LASER trial as either an allergen-specific serum IgE detected of > 0.35 kU/l or a positive skin prick test to a defined allergen with a mean wheal diameter of ≥ 3 mm. These allergy tests have high sensitivity, but in themselves do not necessarily signify disease. A considerable proportion of non-asthmatic individuals are sensitised to one or more aeroallergens, and a positive test in an asthmatic patient does not always result in clinical response on exposure to that allergen. There is a difference between allergic asthma with asthma symptoms induced by exposure to a defined allergen and asthma in a subject characterised as being sensitised but with no relation between allergen exposure and clinical reaction. In the LASER trial, we attempted to overcome this issue by recording both allergen sensitisation by either skin prick testing or measurement of serum-specific IgE levels as well as documenting evidence of the presence of symptoms on exposure to allergens to which the subject was sensitised. This is imprecise as it relies on subjective reporting of symptoms on exposure to allergens and may have led to the inclusion of participants who did not have purely allergic asthma.
Exacerbations and symptoms often coexist but are not always interlinked. Some patients have significant daily symptoms and require high-intensity treatment to maintain quality of life and prevent deterioration in lung function but do not experience frequent exacerbations. It is also recognised that there is an asthmatic phenotype characterised by development of sudden severe asthma symptoms and exacerbations in otherwise mild or asymptomatic patients, often triggered by exposure to an allergen, a drug, an air pollutant or a volatile organic compound, a viral infection or another unknown trigger. The huge heterogeneity in asthma highlights the importance of characterising specific asthma phenotypes aiming to provide precision therapeutics targeted towards each individual’s disease. Subgroup analysis of trial populations might help to identify specific phenotypes or subgroups of allergic asthmatic patients who might benefit from specific treatments, such as TLA.
Dissociation between asthma symptoms and exacerbations
Previous landmark asthma studies have provided good evidence showing that there is a dissociation between asthma symptoms and exacerbations.
Early studies investigating the safety and efficacy of mepolizumab (Nucala®, GlaxoSmithKline) showed no clinical safety concerns but were unable to demonstrate efficacy in the measured primary outcomes. In a double-blind, placebo-controlled study of 24 mild allergic asthmatic patients,88 researchers were unable to demonstrate any significant improvement in airway hyper-reactivity, peak expiratory flow or FEV1 in the group treated with mepolizumab compared with the placebo group despite seeing a significant reduction in airway and blood eosinophil counts in the mepolizumab-treated group. A further double-blind, placebo-controlled study89 randomised 24 participants to mepolizumab or placebo. Again, researchers were unable to demonstrate any improvement in their clinical endpoints of FEV1, PEF or airway hyper-reactivity despite, again, showing a significant reduction in blood and airway eosinophil counts in the active treatment group. Flood-Page et al. 90 went on to carry out a large multicentre RCT in 362 asthma patients with persistent symptoms. Again, they were unable to demonstrate significant clinical improvements in symptoms, lung function measures or quality of life in the active treatment group.
Phase II and III studies of mepolizumab treatment in carefully selected populations of severe eosinophilic asthmatic patients were later able to demonstrate a significant reduction in the annualised asthma exacerbation rate despite not showing consistent improvements in asthma symptoms or quality-of-life measures. The DREAM (Dose Ranging Efficacy And safety with Mepolizumab) trial91 treated 621 patients with intravenous mepolizumab for 52 weeks. There was a significant reduction in the exacerbation rate in the mepolizumab-treated participants compared with participants in the placebo group. It was noted that the significant reduction in exacerbation risk was only associated with small and non-clinically significant improvement in measures of asthma symptoms, asthma control, lung function or quality of life.
The SIROCCO study group92 investigating the anti-interleukin (IL)-5 antibody, benralizumab (Fasenra™, AstraZeneca), were able to show a similar treatment benefit in reducing the annualised exacerbation rate and were able to show an improvement in FEV1 in the active treatment group but, again, could not demonstrate a consistent improvement in asthma symptoms in the active treatment group.
These studies suggest dissociation between symptoms and risk of exacerbation in patients with severe asthma. This evidence is further supported by studies carried out to reduce eosinophilic airways inflammation. 23 Green et al. 23 were able to show that treatment strategies aimed at normalising sputum eosinophil counts were able to reduce asthma exacerbations and hospital admissions. Despite the significant reduction in exacerbation rate in the sputum management group, the group was unable to demonstrate any improvement in symptom scores, quality of life or lung function measures.
Previous trials of TLA treatment in asthma have demonstrated improvements in asthma quality of life and, recognising the dissociation between symptoms, quality of life and exacerbations in severe asthma, it may be that further studies to demonstrate efficacy of TLA treatment should focus on symptom control and quality of life given the lack of demonstrable improvement in exacerbation reduction seen in the LASER trial.
Device compliance
The majority of participants possessed a device for more than 6 months of the trial, making them eligible to benefit from the 4-year post-trial provision of an active TLA device free of charge. This incentive may have helped with participant retention, but this is not a perfect indicator of adherence. During the LASER trial, participants were asked to commit to using the device on at least five out of seven nights of the week, excluding holidays, recognising that this was a pragmatic trial and that the device was not sufficiently portable to allow participants to take on holiday or away from home. Participants were encouraged to use the device every day if possible and to ensure that the device was switched on when sleeping under it at night.
In order to try to capture a measure of device compliance, participants were asked to record their use of the device in the daily TLA diary including whether the device was used (yes or no) and the number of hours the device was used. Where it was recorded, most participants used the device for a sufficient number of hours to include overnight sleep (8 or more hours). In addition to this, participants were asked to report the ‘device-reported use’. The device automatically recorded the number of hours ‘in use’ when the device was switched on. The device was pre-programmed at the time of installation by the engineer to turn on and switch off at times defined by the individual participant to cover the ‘earliest possible time to bed’ and ‘latest possible time to rise’. Participants were able to override this function and could turn on the device earlier than programmed and switch off the device earlier than programmed. This also gave participants flexibility to use the device during daytime naps if they wished.
Unfortunately, it is not possible to determine from the device-reported use whether or not the participant was sleeping under the device during the time that the device was switched on. Difficulties in measuring device compliance mean that it is not possible to absolutely confirm that each participant used the device for a satisfactory length of time to receive a treatment benefit.
Indeed, it is not known what length of time a participant would be required to sleep under the device to obtain a positive treatment effect. It would be important in future studies of TLA treatment to have a more robust measure of treatment compliance to allow further analysis of treatment outcome determined by compliance.
Novel methods of recruitment including social media
We were able to explore the benefits of using different social media platforms to recruit and randomise participants. Of the 240 randomised participants, approximately 20% were recruited from sources other than clinic registers at participating centres. Furthermore, fewer than 1% of participants were recruited from newspaper and radio advertisement, which are more traditional methods of recruitment, but no less costly. We demonstrated that use of dedicated trial websites, Google remarketing, targeted Facebook advertisements and national charity Facebook and Twitter account use can enhance recruitment. The most successful method of recruitment, however, was the use of Trialbee, a dedicated website used for directing participants after a short screening questionnaire to the relevant local trial sites. We recommend further exploration of the use of digital media for recruitment to large multicentre trials.
Chapter 6 Conclusion
Despite evidence of the efficacy of the TLA device in allergic asthma, in this pragmatic, placebo-controlled RCT we were unable to demonstrate any effect on severe exacerbation frequency and demonstrated an effect on only some of the secondary outcomes. Despite the multiple sources of severe exacerbation data used, it is possible that the numbers of severe exacerbations may have been under-reported, and thus the potential benefits of TLA therapy remain inconclusive. Any further trial would benefit from having a singular, robust measure of severe exacerbations. This is the first trial of the TLA device in severe asthma and, despite the outcomes, the type of patient who may benefit from the TLA device requires further exploration. The reasons for the large decreases in exacerbation frequency in participants in severe asthma trials in both the active and the intervention groups also need to be further explored and accounted for in future trials. Although it was not the primary aim of this trial, we found that the use of social media can enhance trial recruitment and that such recruitment methods are superior to more traditional methods of recruitment in this population of severe allergic asthma patients.
Acknowledgements
We would like to acknowledge the participants and their partners and carers, as well as the participating trial centres, the TMG, the TSC and the DSMC:
Participating trial centres:
-
Belfast – Professor Liam Heaney
-
Birmingham (Heartlands) – Dr Adel Mansur
-
Birmingham (Queen Elizabeth) – Dr Benjamin Sutton
-
Bradford – Dr Dinesh Saralaya
-
Chester – Dr Stephen Scott
-
Hull – Dr Jaymin Morjaria
-
Leicester – Professor Peter Bradding
-
Liverpool (Aintree) – Dr Kevin Mortimer
-
Royal Liverpool – Dr Hassan Burhan
-
London (St Georges) – Professor Emma Baker
-
Maidstone – Dr Syed Hussain
-
Oxford – Professor Ian Pavord
-
Portsmouth – Professor Anoop J Chauhan (Chief Investigator)
-
Southampton – Professor Peter Howarth.
Trial Management Group:
-
Dr William Storrar – Trial Co-ordinator
-
Dr Michael Schlussel, Dr Beverly Shirkey and Sue Dutton – Statisticians
-
Emma Hedley – Trial Manager
-
Lara Balls – Lead Research Nurse
-
David Supple – patient representative
-
Bishopsgate representatives (Imogen Speakman and Kate Kelly)
-
Dr Melissa Kapoor – Research Analyst (trial write-up and business support to the Chief Investigator)
-
Sue Marshall (general trial support and all activities related to the smooth running of the trial)
-
Dr Pedro Aparício – Research Analyst.
Trial Steering Committee:
-
Professor Stephen Durham (Chairperson) – independent
-
Dr William Oldfield (Member) – independent
-
Dr Simon Crowther (Member) – independent
-
Mr David Supple (Public Member) – independent
-
Ms Debby Waddell (Member) – independent
-
Dr Derrick Crump (Member) – independent
-
Dr Jessica Harris (Member) – independent
-
Professor Anoop J Chauhan (Member) – non-independent
-
Dr Will Storrar (Member) – non-independent.
Data and Safety Monitoring Committee:
-
Professor Stephen Spiro (Chairperson) – independent
-
Dr Andrew Boyle (Member) – independent
-
Mr Mike Bradburn (Member) – independent
-
Ms Ellen Lee (Member) – independent.
We would also like to thank Trudi Simmons at the Department of Health and Social Care for guidance on the subvention and excess treatment costs. We also gratefully acknowledge the support of Airsonett for its swift supply and distribution of all TLA devices and training of the Bishopsgate engineering teams. Particular thanks to:
-
Jonas Baghammar
-
Frederik Werner
-
Anders Malm (in memoriam)
-
Magnus Lundberg
-
Krupa Patel
-
Ann-Marie Tindberg
-
Pal Svensson.
Contributions of authors
Melissa Kapoor (Data Scientist, Research Analyst and Chief Operating Officer) contributed to the interpretation of data, drafting and revision of the report and approval of the report.
Will Storrar (Trial Co-ordinator and Consultant in Respiratory Medicine) contributed to the conception and design of the trial, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Lara Balls (Lead Trial Nurse, Research and Innovation) contributed to the conception and design of the trial, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Thomas P Brown (Co-PI in Portsmouth, Consultant in Respiratory Medicine) contributed to the conception and design of the trial, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Adel Mansur (Co-applicant on grant, PI at Heartlands Hospital in Birmingham, Consultant and Senior Lecturer in Allergy and Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Emma Hedley (Clinical Trial Manager) contributed to the interpretation of data, drafting and revision of the report and approval of the report.
Tom Jones (Clinical Research Fellow in Asthma and Airways Disease) contributed to the interpretation of data, drafting and revision of the report and approval of the report.
Claire Roberts (Clinical Research Fellow in Asthma and Airways Disease) contributed to the interpretation of data, drafting and revision of the report and approval of the report.
Beverly Shirkey (Trial Statistician) contributed to the analysis of data, drafting and revision of the report and approval of the report.
Susan Dutton (Senior Statistician) contributed to the analysis of data, drafting and revision of the report and approval of the report and provided statistical oversight throughout the trial.
Ramon Luengo-Fernandez (Co-applicant on grant, Senior Health Economist) contributed to the conception and design of the study, analysis and interpretation of the health economic data, drafting and revision of the report and approval of the report.
Matthew Little (Health Economist) contributed to the analysis and interpretation of the health economic data and revision and approval of the report.
Ann Dewey (Co-applicant on grant, Senior Lecturer in Qualitative Research) contributed to the conception and design of the study, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Sue Marshall (business support at the sponsor site) contributed to the conception and design of the trial, drafting and revision of the report and approval of the report.
Carole Fogg (Co-applicant on grant, Senior Lecturer in Trials Methodology) contributed to the conception and design of the study, interpretation of data, drafting and revision of the report and approval of the report.
Keith Boughton (Co-applicant on grant, patient advisor at Wessex Asthma Network) contributed to the conception and design of the study and revising and approval of the report.
Najib Rahman (Co-applicant on grant, Lead of Oxford Respiratory Trials Unit and Associate Professor of Respiratory Medicine) contributed to the conception and design of the study, safety reporting, analysis and interpretation of data and revision and approval of the report.
Ly-Mee Yu (Co-applicant on grant, Senior Statistician) contributed to the conception and design of the study, analysis and interpretation of data and revision and approval of the report.
Peter Bradding (Co-applicant on grant, PI at Leicester, Professor of Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Peter Howarth (Co-applicant on grant, PI at Southampton, Professor of Respiratory Medicine) contributed to the conception and design of the study, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Anoop J Chauhan (Chief Investigator, lead applicant on grant, Trial Guarantor, PI at Portsmouth, Professor of Respiratory Medicine) contributed to the conception and design of the trial, delivery and management of the trial, analysis and interpretation of data, drafting and revision of the report and approval of the report.
Publications
Storrar W, Brown TP, Balls L, Chauhan AJ, Fogg C. Recruitment to clinical trials – the use of social media. Trials 2015;16(Suppl. 2):O77.
Storrar W, Dewey A, Chauhan AJ, Fogg C, Lanning E, Brown TP, Balls L. Early qualitative analysis to enhance trial processes. Trials 2015;16(Suppl. 2):P73.
Storrar W, Roberts C, Gunatilake S, Jones TL, Brown TP, Chauhan AJ. Educational information events to enhance recruitment to clinical research trials. Eur Resp J 2015;46(Suppl. 59):PA2778.
Storrar W, Fogg C, Brown TP, Dennison P, Yu LM, Dewey A, et al. Temperature-controlled laminar airflow in severe asthma for exacerbation reduction (the LASER trial): study protocol for a randomised controlled trial. Trials 2016;17:15.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Living on a Knife Edge. A Powerful and Moving Account of Living with Serious Symptoms of Asthma. London: Asthma UK; 2004.
- Holgate ST, Polosa R. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet 2006;368:780-93. https://doi.org/10.1016/S0140-6736(06)69288-X.
- Asthma Facts and Statistics. London: Asthma UK; 2017.
- Hoskins G, McCowan C, Neville RG, Thomas GE, Smith B, Silverman S. Risk factors and costs associated with an asthma attack. Thorax 2000;55:19-24. https://doi.org/10.1136/thorax.55.1.19.
- Tough SC, Hessel PA, Ruff M, Green FH, Mitchell I, Butt JC. Features that distinguish those who die from asthma from community controls with asthma. J Asthma 1998;35:657-65. https://doi.org/10.3109/02770909809048968.
- Turner MO, Noertjojo K, Vedal S, Bai T, Crump S, Fitzgerald JM. Risk factors for near-fatal asthma. A case–control study in hospitalized patients with asthma. Am J Respir Crit Care Med 1998;157:1804-9. https://doi.org/10.1164/ajrccm.157.6.9708092.
- Campbell MJ, Holgate ST, Johnston SL. Trends in asthma mortality. BMJ 1997;315. https://doi.org/10.1136/bmj.315.7114.1012.
- Bousquet J, Mantzouranis E, Cruz AA, Aït-Khaled N, Baena-Cagnani CE, Bleecker ER, et al. Uniform definition of asthma severity, control, and exacerbations: document presented for the World Health Organization Consultation on Severe Asthma. J Allergy Clin Immunol 2010;126:926-38. https://doi.org/10.1016/j.jaci.2010.07.019.
- Rodrigo GJ, Rodrigo C, Hall JB. Acute asthma in adults: a review. Chest 2004;125:1081-102. https://doi.org/10.1378/chest.125.3.1081.
- Nejtek VA, Brown ES, Khan DA, Moore JJ, Van Wagner J, Perantie DC. Prevalence of mood disorders and relationship to asthma severity in patients at an inner-city asthma clinic. Ann Allergy Asthma Immunol 2001;87:129-33. https://doi.org/10.1016/S1081-1206(10)62206-5.
- Fighting for Breath. The Hidden Lives of People with Severe Asthma. London: Asthma UK; 2010.
- Manson SC, Brown RE, Cerulli A, Vidaurre CF. The cumulative burden of oral corticosteroid side effects and the economic implications of steroid use. Respir Med 2009;103:975-94. https://doi.org/10.1016/j.rmed.2009.01.003.
- Humbert M, Beasley R, Ayres J, Slavin R, Hébert J, Bousquet J, et al. Benefits of omalizumab as add-on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005;60:309-16. https://doi.org/10.1111/j.1398-9995.2004.00772.x.
- National Institute for Health and Care Excellence . Omalizumab for Treating Severe Persistent Allergic Asthma 2013. www.nice.org.uk/guidance/TA278 (accessed 14 September 2017).
- An Outcomes Strategy for People with Chronic Obstructive Pulmonary Disease (COPD) and Asthma in England. London: Department of Health and Social Care; 2011.
- National Guideline on the Management of Asthma. London: British Thoracic Society; 2011.
- Asthma UK Research Strategy 2016–2021. London: Asthma UK; 2016.
- Heaney LG, Brightling CE, Menzies-Gow A, Stevenson M, Niven RM. British Thoracic Society Difficult Asthma Network . Refractory asthma in the UK: cross-sectional findings from a UK multicentre registry. Thorax 2010;65:787-94. https://doi.org/10.1136/thx.2010.137414.
- Custovic A, Taggart SC, Francis HC, Chapman MD, Woodcock A. Exposure to house dust mite allergens and the clinical activity of asthma. J Allergy Clin Immunol 1996;98:64-72. https://doi.org/10.1016/S0091-6749(96)70227-0.
- Tunnicliffe WS, Fletcher TJ, Hammond K, Roberts K, Custovic A, Simpson A, et al. Sensitivity and exposure to indoor allergens in adults with differing asthma severity. Eur Respir J 1999;13:654-9. https://doi.org/10.1183/09031936.99.13365499.
- Langley SJ, Goldthorpe S, Craven M, Morris J, Woodcock A, Custovic A. Exposure and sensitization to indoor allergens: association with lung function, bronchial reactivity, and exhaled nitric oxide measures in asthma. J Allergy Clin Immunol 2003;112:362-8. https://doi.org/10.1067/mai.2003.1654.
- Rosenstreich DL, Eggleston P, Kattan M, Baker D, Slavin RG, Gergen P, et al. The role of cockroach allergy and exposure to cockroach allergen in causing morbidity among inner-city children with asthma. N Engl J Med 1997;336:1356-63. https://doi.org/10.1056/NEJM199705083361904.
- Green RM, Custovic A, Sanderson G, Hunter J, Johnston SL, Woodcock A. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ 2002;324. https://doi.org/10.1136/bmj.324.7340.763.
- Custovic A, Simpson A, Chapman MD, Woodcock A. Allergen avoidance in the treatment of asthma and atopic disorders. Thorax 1998;53:63-72. https://doi.org/10.1136/thx.53.1.63.
- van Velzen E, van den Bos JW, Benckhuijsen JA, van Essel T, de Bruijn R, Aalbers R. Effect of allergen avoidance at high altitude on direct and indirect bronchial hyperresponsiveness and markers of inflammation in children with allergic asthma. Thorax 1996;51:582-4. https://doi.org/10.1136/thx.51.6.582.
- Peroni DG, Boner AL, Vallone G, Antolini I, Warner JO. Effective allergen avoidance at high altitude reduces allergen-induced bronchial hyperresponsiveness. Am J Respir Crit Care Med 1994;149:1442-6. https://doi.org/10.1164/ajrccm.149.6.8004296.
- Grootendorst DC, Dahlén SE, Van Den Bos JW, Duiverman EJ, Veselic-Charvat M, Vrijlandt EJ, et al. Benefits of high altitude allergen avoidance in atopic adolescents with moderate to severe asthma, over and above treatment with high dose inhaled steroids. Clin Exp Allergy 2001;31:400-8. https://doi.org/10.1046/j.1365-2222.2001.01022.x.
- Gøtzsche PC, Johansen HK. House dust mite control measures for asthma. Cochrane Database Syst Rev 2008;2. https://doi.org/10.1002/14651858.CD001187.pub3.
- Sublett JL. Effectiveness of air filters and air cleaners in allergic respiratory diseases: a review of the recent literature. Curr Allergy Asthma Rep 2011;11:395-402. https://doi.org/10.1007/s11882-011-0208-5.
- Spilak MP, Sigsgaard T, Takai H, Zhang G. A comparison between temperature-controlled laminar airflow device and a room air-cleaner in reducing exposure to particles while asleep. PLOS ONE 2016;11. https://doi.org/10.1371/journal.pone.0166882.
- Gore RB, Boyle RJ, Hanna H, Custovic A, Gore C, Svensson P, et al. Personal allergen exposures are increased by changes in sleep position and improved by temperature controlled laminar airflow. Thorax 2010;65:A87-8. https://doi.org/10.1136/thx.2010.150961.27.
- Sigsgaard T. Temperature Regulated Laminar Airflow (TLA): TLA Principles and Practice n.d.
- Boyle RJ, Pedroletti C, Wickman M, Bjermer L, Valovirta E, Dahl R, et al. Nocturnal temperature controlled laminar airflow for treating atopic asthma: a randomised controlled trial. Thorax 2012;67:215-21. https://doi.org/10.1136/thoraxjnl-2011-200665.
- Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 2014;43:343-73. https://doi.org/10.1183/09031936.00202013.
- Persson U, Hofmarcher T. Consulting Report. Cost Effectiveness Analysis. Lund: Swedish Institute for Health Economics; 2012.
- Vervloet D, Williams AE, Lloyd A, Clark TJH. Costs of managing asthma as defined by a derived Asthma Control Test™ score in seven European countries. Eur Respir Rev 2006;15:17-23. https://doi.org/10.1183/09059180.06.00009803.
- Pedroletti C, Millinger E, Dahlén B, Söderman P, Zetterström O. Clinical effects of purified air administered to the breathing zone in allergic asthma: a double-blind randomized cross-over trial. Respir Med 2009;103:1313-19. https://doi.org/10.1016/j.rmed.2009.03.020.
- Brodtkorb TH, Zetterström O, Tinghög G. Cost-effectiveness of clean air administered to the breathing zone in allergic asthma. Clin Respir J 2010;4:104-10. https://doi.org/10.1111/j.1752-699X.2009.00156.x.
- Gore RB, Hanna H, Custovic A, Gore C, Svensson P, Warner JO. Effect of a novel temperature controlled laminar airflow device on personal breathing zone cat allergen exposure. Allergy 2010;65.
- Reddel HK, Taylor DR, Bateman ED, Boulet LP, Boushey HA, Busse WW, et al. An official American Thoracic Society/European Respiratory Society statement: asthma control and exacerbations: standardizing endpoints for clinical asthma trials and clinical practice. Am J Respir Crit Care Med 2009;180:59-9. https://doi.org/10.1164/rccm.200801-060ST.
- COMET . Core Outcome Measures in Effectiveness Trials n.d. www.comet-initiative.org (accessed 2 April 2019).
- Royston P. Multiple imputation of missing values. Stata J 2004;4:227-41. https://doi.org/10.1177/1536867X0400400301.
- NICE Technology Appraisal Guidance 133: Omalizumab for Severe Persistent Allergic Asthma. London: NICE; 2007.
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. ATS/ERS Task Force: standardisation of lung function testing. Standardisation of spirometry. Eur Respir J 2005;26:319-38. https://doi.org/10.1183/09031936.05.00034805.
- European Respiratory Society . ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide. Am J Respir Crit Care Med 2005;171:912-30. https://doi.org/10.1164/rccm.200406-710ST.
- Bernstein I, Li J, Bernstein D, Hamilton R, Spector S, Tan R, et al. Allergy diagnostic testing: an updated practice parameter. Ann Allergy Asthma Immunol 2008;100:S1-148. https://doi.org/10.1016/S1081-1206(10)60305-5.
- Juniper EF, O’Byrne PM, Guyatt GH, Ferrie PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J 1999;14:902-7. https://doi.org/10.1034/j.1399-3003.1999.14d29.x.
- Juniper EF, Guyatt GH, Ferrie PJ, Griffith LE. Measuring quality of life in asthma. Am Rev Respir Dis 1993;4:832-8. https://doi.org/10.1164/ajrccm/147.4.832.
- Juniper EF, Guyatt GH, Willan A, Griffith LE. Determining a minimal important change in a disease-specific Quality of Life Questionnaire. J Clin Epidemiol 1994;47:81-7. https://doi.org/10.1016/0895-4356(94)90036-1.
- EQ-5D-5L User Guide: Basic Information on How to Use the EQ-5D-5L Instrument. Rotterdam: EuroQ Group; 2011.
- Pickard AS, Wilke C, Jung E, Patel S, Stavem K, Lee TA. Use of a preference-based measure of health (EQ-5D) in COPD and asthma. Respir Med 2008;102:519-36. https://doi.org/10.1016/j.rmed.2007.11.016.
- Hopkins C, Gillett S, Slack R, Lund VJ, Browne JP. Psychometric validity of the 22-item Sinonasal Outcome Test. Clin Otolaryngol 2009;34:447-54. https://doi.org/10.1111/j.1749-4486.2009.01995.x.
- Engvall K, Norrby C, Sandstedt E. The Stockholm Indoor Environment Questionnaire: a sociologically based tool for the assessment of indoor environment and health in dwellings. Indoor Air 2004;14:24-33. https://doi.org/10.1111/j.1600-0668.2004.00204.x.
- de Bilderling G, Chauhan AJ, Jeffs JA, Withers N, Johnston SL, Holgate ST, et al. Gas cooking and smoking habits and the risk of childhood and adolescent wheeze. Am J Epidemiol 2005;162:513-22. https://doi.org/10.1093/aje/kwi238.
- Lloyd A, Turk F, Leighton T, Canonica GW. Psychometric evaluation of Global Evaluation of Treatment Effectiveness: a tool to assess patients with moderate-to-severe allergic asthma. J Med Econ 2007;10:285-96. https://doi.org/10.3111/13696990701478856.
- Reilly MC, Zbrozek AS, Dukes EM. The validity and reproducibility of a work productivity and activity impairment instrument. PharmacoEconomics 1993;4:353-65. https://doi.org/10.2165/00019053-199304050-00006.
- Joseph S, Becker S, Elwick H, Silburn R. Adult Carers Quality of Life questionnaire (AC-QoL): development of an evidence based tool. Mental Health Rev J 2012;17:57-69. https://doi.org/10.1108/13619321211270380.
- Juniper EF, O’Byrne PM, Ferrie PJ, King DR, Roberts JN. Measuring asthma control. Am J Respir Crit Care Med 2000;162:1330-4. https://doi.org/10.1164/ajrccm.162.4.9912138.
- Schulz KF, Altman DG, Moher D. CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. https://doi.org/10.1136/bmj.c332.
- Omalizumab for the Treatment of Severe Persistent Allergic Asthma in Children Aged 6–11. London: NICE; 2010.
- Brazier P, Schauer U, Hamelmann E, Holmes S, Pritchard C, Warner JO. Economic analysis of temperature-controlled laminar airflow (TLA) for the treatment of patients with severe persistent allergic asthma. BMJ Open Respir Res 2016;3. https://doi.org/10.1136/bmjresp-2015-000117.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2015. Canterbury: PSSRU, University of Kent; 2016.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2016. Canterbury: PSSRU, University of Kent; 2017.
- NHS Reference Costs 2015 to 2016. London: Department of Health and Social Care; 2016.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; n.d.
- Earnings and Working Hours. Newport: ONS; 2017.
- Rubin DB. Multiple imputation after 18+ years. JASA 1996;91:473-89. https://doi.org/10.1080/01621459.1996.10476908.
- Schafer JL, Graham JW. Missing data: our view of the state of the art. Psychol Methods 2002;7:147-77. https://doi.org/10.1037/1082-989X.7.2.147.
- Position Statement on Use of the EQ-5D-5L Valuation Set. London: NICE; 2017.
- van Hout B, Janssen MF, Feng YS, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15. https://doi.org/10.1016/j.jval.2012.02.008.
- Billingham L, Abrams K, Jones D. Methods for the analysis of quality-of-life and survival data in health technology assessment. Health Technol Assess 1999;3.
- Båghammar J. GBP Prices and Conditions. Ängelholm: Airsonett AB; 2017.
- Drummond MF, Sculpher MJ, Torrance GW, O’Brien BJ, Stoddart GL. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2005.
- van Hout BA, Al MJ, Gordon GS, Rutten FF. Costs, effects and C/E-ratios alongside a clinical trial. Health Econ 1994;3:309-19. https://doi.org/10.1002/hec.4730030505.
- Briggs AH, Wonderling DE, Mooney CZ. Pulling cost-effectiveness analysis up by its bootstraps: a non-parametric approach to confidence interval estimation. Health Econ 1997;6:327-40. https://doi.org/10.1002/(SICI)1099-1050(199707)6:4<327::AID-HEC282>3.0.CO;2-W.
- Briggs A, Clark T, Wolstenholme J, Clarke P. Missing . . . presumed at random: cost-analysis of incomplete data. Health Econ 2003;12:377-92. https://doi.org/10.1002/hec.766.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99. https://doi.org/10.1002/sim.4067.
- Faria R, Gomes M, Epstein D, White IR. A guide to handling missing data in cost-effectiveness analysis conducted within randomised controlled trials. PharmacoEconomics 2014;32:1157-70. https://doi.org/10.1007/s40273-014-0193-3.
- Watson L, Turk F, James P, Holgate ST. Factors associated with mortality after an asthma admission: a national United Kingdom database analysis. Respir Med 2007;101:1659-64. https://doi.org/10.1016/j.rmed.2007.03.006.
- National Life Tables, UK: 2013–2015. Newport: ONS; 2017.
- International Statistical Classification of Diseases and Related Health Problems: Tenth Revision. Geneva: World Health Organization; 2004.
- de Vries F, Setakis E, Zhang B, van Staa TP. Long-acting β2-agonists in adult asthma and the pattern of risk of death and severe asthma outcomes: a study using the GPRD. Eur Respir J 2010;36:494-502. https://doi.org/10.1183/09031936.00124209.
- Pullenayegum EM, Tarride J-E, Xie F, O’Reilly D. Calculating utility decrements associated with an adverse event. Med Decis Making 2011;31:790-9. https://doi.org/10.1177/0272989X10393284.
- Schauer U, Bergmann KC, Gerstlauer M, Lehmann S, Gappa M, Brenneken A, et al. Improved asthma control in patients with severe, persistent allergic asthma after 12 months of nightly temperature-controlled laminar airflow: an observational study with retrospective comparisons. Eur Clin Respir J 2015;2. https://doi.org/10.3402/ecrj.v2.28531.
- Holt PG, Strickland DH, Sly PD. Virus infection and allergy in the development of asthma: what is the connection?. Curr Opin Allergy Clin Immunol 2012;12:151-7. https://doi.org/10.1097/ACI.0b013e3283520166.
- Green RH, Brightling CE, McKenna S, Hargadon B, Parker D, Bradding P, et al. Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet 2002;360:1715-21. https://doi.org/10.1016/S0140-6736(02)11679-5.
- Heinzerling L, Mari A, Bergmann K-C, Bresciani M, Burbach G, Darsow U, et al. The skin prick test – European standards. Clin Transl Allergy 2013;3. https://doi.org/10.1186/2045-7022-3-3.
- Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, Walls CM, et al. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyperresponsiveness, and the late asthmatic response. Lancet 2000;356:2144-8. https://doi.org/10.1016/S0140-6736(00)03496-6.
- Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil’s role remains uncertain as an interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med 2003;167:199-204. https://doi.org/10.1164/rccm.200208-789OC12406833.
- Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, et al. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med 2007;176:1062-71. https://doi.org/10.1164/rccm.200701-0850C.
- Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 2012;380:651-9. https://doi.org/10.1016/S0140-6736(12)60988-X.
- Bleeker ER, Fitzgerald JM, Chanez P, Papi A, Weinstein SF, Barker P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high dosage inhaled corticosteroids and long acting B2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled Phase 3 trial. Lancet 2016;388:2115-27. https://doi.org/10.1016/S0140-6736(16)31324.
- Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med 2009;360:973-84. https://doi.org/10.1056/NEJMoa0808991.
- Hanania NA, Alpan O, Hamilos DL, Condemi JJ, Reyes-Rivera I, Zhu J, et al. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med 2011;154:573-82. https://doi.org/10.7326/0003-4819-154-9-201105030-00002.
- Castro M, Rubin AS, Laviolette M, Fiterman J, De Andrade Lima M, . Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med 2010;181:116-24.
- Busse WW, Israel E, Nelson HS, Baker JW, Charous BL, Young DY, et al. Daclizumab improves asthma control in patients with moderate to severe persistent asthma: a randomized, controlled trial. Am J Respir Crit Care Med 2008;178:1002-8. https://doi.org/10.1164/rccm.200708-1200OC.
- Pauwels RA, Löfdahl CG, Postma DS, Tattersfield AE, O’Byrne P, Barnes PJ, et al. Effect of inhaled formoterol and budesonide on exacerbations of asthma. Formoterol and Corticosteroids Establishing Therapy (FACET) International Study Group. N Engl J Med 1997;337:1405-11. https://doi.org/10.1056/NEJM199711133372001.
- Crapo RO, Casaburi R, Coates AL, Enright PL, Hankinson JL, Irvin CG, et al. Guidelines for methacholine and exercise challengetesting-1999. Am J Respir Crit Care Med 2000;162:309-29.
- Anderson SD, Brannan J, Spring J, Spalding N, Rodwell LT, Chan KA, et al. A new method for bronchial-provocation testing in asthmatic subjects using a dry powder of mannitol. Am J Respir Crit Care Med 1997;156:758-65. https://doi.org/10.1164/ajrccm.156.3.9701113.
Appendix 1 Interference of medications with skin test reactions
| Drug | Abstinence required before testing |
|---|---|
| Antihistamines | |
| First-generation H1-antihistamines | |
| Hydroxyzine | > 2 days |
| Second-generation H1-antihistamines | |
| Cetirizine | 7 days |
| Loratadine | 3 days |
| Fexofenadine | 2 days |
| H2-blockers | 0 |
| Glucocorticosteroids | |
| Topical | > 1 week (in area being tested) |
| Nasal | 0 |
| Inhaled | 0 |
| Systemica | 0 |
| Other medication | |
| Tricyclic antidepressants | |
| Doxepin | 7 days |
| Desipramine | 3 days |
| SSRIs | |
| Citalopram/fluoxetine/sertraline | 0 |
| Beta-agonists | 0 |
| Anticholinergics | 0 |
| Leukotriene receptor antagonist | 0 |
| Theophylline | 0 |
Appendix 2 Trial visit and data collection schedule
| Trial data | Data collection time point | |||||||
|---|---|---|---|---|---|---|---|---|
| Screening | Randomisation | 1 month | 3, 6, 9, 12 months | Exacerbation | ADE | SADE | Withdrawal | |
| CRFs/follow-up visit forms | ||||||||
| Screening visit | ✗ | |||||||
| Randomisation visit | ✗ | |||||||
| Randomisation form | ✗ | |||||||
| 1-month telephone follow-up | ✗ | |||||||
| 3-month visit | ✗ | |||||||
| Exacerbation review | ✗ | |||||||
| ADE form | ✗ | |||||||
| SADE form | ✗ | |||||||
| Withdrawal form | ✗ | |||||||
| Questionnaires (participant) | ||||||||
| ACQ | ✗ | ✗ | ✗ | |||||
| AQLQ(S) | ✗ | ✗ | ||||||
| EQ-5D-5L | ✗ | ✗ | ||||||
| SNOT-22 | ✗ | ✗ | ||||||
| WPAI(A) | ✗ | ✗ | ||||||
| Indoor air quality | ✗ | |||||||
| Questionnaires (carer) | ||||||||
| AC-QoL | ✗ | |||||||
| WPAI(CG) | ✗ | |||||||
| Diaries | ||||||||
| Asthma control – screening | ✗ | |||||||
| Asthma control – follow-up | ✗ | |||||||
| TLA diary | ✗ | |||||||
| Health-care resource use diary | ✗ | |||||||
| Exacerbation diary | ✗ | |||||||
Appendix 3 Comparative trials summary for sample size and magnitude of effect
| Author and year | Treatment | n | Baseline exacerbation ratea | Placebo group exacerbation rate | Exacerbation reduction: treatment vs. placebo | % reduction | ICS dose | Exacerbation definition |
|---|---|---|---|---|---|---|---|---|
| Pavord 201291 | Mepolizumab | 621 (four groups) | 3.73 (SD 0.8) | 2.40 (SD 0.11) over 52 weeks | 1.24 vs. 2.40 (75 mg) | 48 (75 mg) | 880 µg of fluticasone propionate equivalent per day, with or without maintenance OCSs | Requiring OCSs or an ED visit + objective evidence that asthma had worsened |
| 1.46 vs. 2.40 (250 mg) | 39 (250 mg) | |||||||
| 1.15 vs. 2.40 (750 mg) | 52 (750 mg) | |||||||
| Haldar 200993 | Mepolizumab | 32 | 5 | 3.4 over 12 months | 2.0 vs. 3.4 | 41 | 1000–4000 BDP equivalent, mean 2000 µg | Requiring OCSs |
| Green 200286 | Sputum eosin-guided treatment | 74 | 2.0 (SD 3.0) in placebo group | 2.95 over 12 months | 0.95 vs. 2.95 | 68 | High dose > 1600 µg of BDP | Requiring OCSs or PEF of ≤ 70% |
| Humbert 200513 | Omalizumab | 419 | 2.41 (SD 1.09) in 14 months | 0.91 (95% CI 0.73 to 1.14) over 28 weeks | 0.68 vs. 0.91 (severe 0.24 vs. 0.48) | 50 | > 1000 µg per day of BDP, GINA 2002 step 4 | Requiring OCSs |
| Hanania 201194 | Omalizumab | 850 | 1.9 (SD 1.5) in 12 months | 0.88 over 48 weeks | 0.66 vs. 0.88 | 25 | > 1000 µg per day of fluticasone dipropionate | Requiring OCSs (or an increase in dose if on maintenance dose) |
| Castro 201095 | Bronchial thermoplasty | 288 | Not recorded | 0.70 (SD 0.122) over 12 months | 0.48 vs. 0.70 | 32 | > 1000 µg per day of BDP | Requiring OCSs or doubling of dose of ICS |
| Busse 200896 | Daclizumab | 115 (3 : 1 treatment to placebo) | Not recorded | Not recorded | 25% vs. 47.6% at 252 days | 47 | Moderate to severe | % of participants in each group suffering an exacerbation requiring systemic corticosteroids |
| Pauwels 199797 | Badesonide/formoterol | 852 | Not recorded | 0.91 | 0.34 vs. 0.91 | 63 | Low to moderate | Requiring OCSs |
Appendix 4 Equivalence table for bronchial challenge testing
| Challenge test | Positive result |
|---|---|
| Direct | |
| Methacholine98 | PC20 < 8 mg/ml |
| Histamine | PC20 < 8 mg/ml |
| Indirect | |
| Mannitol99 | PD15 < 635 mga |
| Exercise98 | Fall in FEV1 of ≥ 10% from baselineb |
Appendix 5 Definition of high-dose inhaled corticosteroids
| ICS | Threshold daily dose (µg) considered as high in adults |
|---|---|
| BDP | > 1000 (DPI or CFC MDI); > 500 (HFA MDI) |
| Budesonide | > 800 (MDI or DPI) |
| Ciclesonide | > 320 (HFA MDI) |
| Fluticasone propionate | > 500 (HFA MDI or DPI) |
| Mometasone furoate | > 800 (DPI) |
| Triamcinolone acetonide | > 2000 |
Appendix 6 Participant withdrawals and oral corticosteroid doses reported during the trial
| Description of withdrawal | Withdrawal reason | Reason for withdrawal reason specified | Withdrew consent to use previous data | Had minimum data | Death | Follow-up visits received (months) | Participant group |
|---|---|---|---|---|---|---|---|
| Did not meet minimum data requirement | No | 3 6 9 12 | Placebo | ||||
| Did not meet minimum data requirement | No | 3 6 9 12 | Active | ||||
| Did not meet minimum data requirement | No | 3 6 9 12 | Placebo | ||||
| Did not meet minimum data requirement | No | 3 6 9 12 | Placebo | ||||
| Did not meet minimum data requirement | No | – 6 9 12 | Placebo | ||||
| Did not meet minimum data requirement | No | – 6 9 12 | Placebo | ||||
| Did not meet minimum data requirement | No | 3 6 9 12 | Active | ||||
| Death | Yes | Yes | 3 6 9 – | Placebo | |||
| Data withdrawal | Yes | No | – – – – | Active | |||
| Data withdrawal | Yes | No | – – – – | Placebo | |||
| Data withdrawal | Yes | No | – – – – | Active | |||
| Data withdrawal | Yes | No | – – – – | Active | |||
| Data withdrawal | Yes | No | – – – – | Active | |||
| Withdrawal form completed | Ineligibility | Yes | 3 6 – – | Placebo | |||
| Withdrawal form completed | Ineligibility | Yes | 3 6 – – | Active | |||
| Withdrawal form completed, death | Other | ‘Death, patient’ | No | Yes | – – – – | Active | |
| Withdrawal form completed | Other | Constant sneezing when using TLA device | No | – – – – | Placebo | ||
| Withdrawal form completed | Other | Getting headaches and sore throat from the device | Yes | 3 – – – | Placebo | ||
| Withdrawal form completed | Other | Increased asthma severe exacerbations | No | 3 – – – | Placebo | ||
| Withdrawal form completed | Other | Machine did not fit in room | No | – – – – | Active | ||
| Withdrawal form completed | Other | Mental health problems | Yes | 3 6 – – | Active | ||
| Withdrawal form completed | Other | Moving house again | Yes | 3 6 – – | Placebo | ||
| Withdrawal form completed | Other | Non-compliance with device. Did not attend study visits | Yes | – – – – | Active | ||
| Withdrawal form completed | Other | Not wanting to participate any more | Yes | 3 – – – | Placebo | ||
| Withdrawal form completed | Other | Participant did not attend multiple visits and did not respond to telephone calls | No | – – – – | Placebo | ||
| Withdrawal form completed | Other | Participant left the family home and was unable to use the machine or have it reinstalled elsewhere and wished to withdraw from the study | No | – 6 – – | Active | ||
| Withdrawal form completed | Other | Participant moved out of area and did not want to continue follow-up at another hospital | Yes | 3 – – – | Active | ||
| Withdrawal form completed | Other | Participant became unwell during pregnancy – contributed to decision to withdraw | No | – – – – | Placebo | ||
| Withdrawal form completed | Other | Participant did not attend appointments or respond to telephone contact | No | 3 – – – | Placebo | ||
| Withdrawal form completed | Other | Participant unable to commit to last appointment | No | 3 6 9 – | Placebo | ||
| Withdrawal form completed | Other | Unable to arrange installation of the TLA device | No | – – – – | Placebo | ||
| Withdrawal form completed | Other | Unable to comply with protocol | No | 3 – – – | Active | ||
| Withdrawal form completed | Other | Unable to contact participant | No | – – – – | Active | ||
| Withdrawal form completed | Other | Unable to contact participant | No | 3 – – – | Active | ||
| Withdrawal form completed | Other | Unable to contact participant | No | 3 – – – | Placebo | ||
| Withdrawal form completed | Other | Unable to contact participant | Yes | 3 6 – – | Active | ||
| Withdrawal form completed | Other | Participant moved abroad | No | 3 – – – | Placebo | ||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | 3 – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | 3 – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | 3 – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | 3 6 – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | 3 6 – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | No | – – – – | Placebo | |||
| Withdrawal form completed | Participant withdrew consent | Yes | 3 6 9 – | Active | |||
| Withdrawal form completed | Participant withdrew consent | Yes | 3 6 – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | Yes | 3 6 – – | Active | |||
| Withdrawal form completed | Participant withdrew consent | Yes | 3 6 – – | Active |
| TLA diary/PED steroid dose (mg) | Trial group, n | Total, n | |
|---|---|---|---|
| Placebo | TLA device | ||
| Missing | 14,944 | 14,645 | 29,589 |
| 0 | 16,384 | 15,976 | 32,360 |
| 0 A | 6419 | 4819 | 11,238 |
| 2 | 15 | 0 | 15 |
| 3 | 0 | 8 | 8 |
| 4 | 4 | 6 | 10 |
| 5 | 1202 | 1881 | 3083 |
| 5 A | 0 | 1 | 1 |
| 6 | 1 | 1 | 2 |
| 7 | 2 | 7 | 9 |
| 7 A | 4 | 0 | 4 |
| 7.5 | 368 | 7 | 375 |
| 8 | 5 | 20 | 25 |
| 9 | 0 | 167 | 167 |
| 9.5 | 6 | 0 | 6 |
| 10 | 1902 | 2482 | 4384 |
| 11 | 0 | 117 | 117 |
| 12 | 0 | 174 | 174 |
| 12.5 | 5 | 125 | 130 |
| 13 | 0 | 2 | 2 |
| 14 | 14 | 2 | 16 |
| 15 | 307 | 490 | 797 |
| 20 | 707 | 521 | 1228 |
| 20 A | 1 | 2 | 3 |
| 25 | 229 | 140 | 369 |
| 25 A | 1 | 6 | 7 |
| 30 | 617 | 685 | 1302 |
| 30 A | 21 | 7 | 28 |
| 30 AP | 15 | 4 | 19 |
| 30 P | 9 | 21 | 30 |
| 34 | 1 | 0 | 1 |
| 35 | 84 | 59 | 143 |
| 35 A | 0 | 2 | 2 |
| 37 | 3 | 0 | 3 |
| 40 | 662 | 798 | 1460 |
| 40 A | 13 | 23 | 36 |
| 45 | 34 | 0 | 34 |
| 50 | 74 | 31 | 105 |
| 50 A | 0 | 5 | 5 |
| 55 | 7 | 0 | 7 |
| 60 | 7 | 16 | 23 |
| 80 | 0 | 138 | 138 |
| 100 | 19 | 0 | 19 |
| 100 A | 0 | 10 | 10 |
| 200 | 5 | 0 | 5 |
| Total | 44,091 | 43,398 | 87,489a |
Appendix 7 Tables of health economic analyses
| Response | Trial group, n (%) | p > |z| | |
|---|---|---|---|
| Placebo | TLA device | ||
| Completed by trial physiciana | |||
| Complete control | 1 (3) | 1 (3) | 0.861 |
| Marked improvement | 13 (33) | 13 (42) | |
| Discernible but limited improvement | 13 (33) | 9 (29) | |
| No appreciable change | 11 (28) | 8 (26) | |
| Worsening | 1 | 0 | |
| Completed by participant, confirmed onlyb | |||
| Complete control | 4 (11) | 2 (6) | 0.425 |
| Marked improvement | 15 (39) | 12 (39) | |
| Discernible but limited improvement | 7 (18) | 10 (32) | |
| No appreciable change | 12 (32) | 6 (19) | |
| Worsening | 0 | 1 (3) | |
| Completed by participant, confirmed and possiblec | |||
| Complete control | 7 (8) | 5 (6) | 0.347 |
| Marked improvement | 34 (38) | 33 (38) | |
| Discernible but limited improvement | 22 (24) | 32 (37) | |
| No appreciable change | 25 (28) | 16 (18) | |
| Worsening | 2 (2) | 1 (1) | |
| Time point | Trial group, mean (SD) | p > |z| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo | TLA device | |||
| Overall score, across all four domains | ||||
| Randomisation | 3.87 (1.22), n = 120 | 4.07 (1.20), n = 115 | 0.213 | 0.20 (–0.11 to 0.51) |
| 3 months | 4.39 (1.41), n = 107 | 4.67 (1.25), n = 100 | 0.139 | 0.27 (–0.09 to 0.65) |
| 6 months | 4.30 (1.42), n = 101 | 4.74 (1.22), n = 97 | 0.020 | 0.44 (0.07 to 0.81) |
| 9 months | 4.36 (1.53), n = 95 | 4.65 (1.35), n = 90 | 0.166 | 0.29 (–0.12 to 0.72) |
| 12 months | 4.50 (1.47), n = 96 | 4.76 (1.33), n = 90 | 0.219 | 0.25 (–0.15 to 0.66) |
| Difference at 12 months | 0.57 (1.12), n = 96 | 0.68 (1.24), n = 90 | 0.543 | 0.11 (–0.24 to 0.45) |
| Symptoms domain | ||||
| Randomisation | 3.74 (1.28) | 3.93 (1.21) | 0.233 | 0.19 (–0.13 to 0.51) |
| 3 months | 4.35 (1.51) | 4.59 (1.34) | 0.239 | 0.23 (–0.15 to 0.63) |
| 6 months | 4.25 (1.53) | 4.63 (1.28) | 0.061 | 0.38 (–0.02 to 0.78) |
| 9 months | 4.36 (1.57) | 4.58 (1.46) | 0.319 | 0.22 (–0.22 to 0.66) |
| 12 months | 4.56 (1.54) | 4.62 (1.36) | 0.761 | 0.06 (–0.36 to 0.49) |
| Difference at 12 months | 0.74 (1.29) | 0.69 (1.32) | 0.816 | –0.04 (–0.42 to 0.33) |
| Activity limitation domain | ||||
| Randomisation | 4.02 (1.29) | 4.29 (1.30) | 0.103 | 0.28 (–0.06 to 0.61) |
| 3 months | 4.47 (1.41) | 4.76 (1.34) | 0.141 | 0.28 (–0.09 to 0.66) |
| 6 months | 4.39 (1.43) | 4.84 (1.28) | 0.022 | 0.45 (0.07 to 0.83) |
| 9 months | 4.41 (1.54) | 4.75 (1.43) | 0.121 | 0.34 (–0.09 to 0.77) |
| 12 months | 4.54 (1.49) | 4.92 (1.35) | 0.070 | 0.38 (–0.03 to 0.79) |
| Difference at 12 months | 0.47 (1.09) | 0.62 (1.19) | 0.394 | 0.14 (–0.19 to 0.47) |
| Emotional function domain | ||||
| Randomisation | 3.83 (1.58) | 3.95 (1.47) | 0.572 | 0.11 (–0.28 to 0.51) |
| 3 months | 4.36 (1.73) | 4.72 (1.52) | 0.111 | 0.36 (–0.08 to 0.81) |
| 6 months | 4.21 (1.79) | 4.67 (1.58) | 0.055 | 0.46 (–0.01 to 0.94) |
| 9 months | 4.33 (1.92) | 4.55 (1.59) | 0.399 | 0.22 (–0.29 to 0.73) |
| 12 months | 4.39 (1.82) | 4.68 (1.68) | 0.257 | 0.29 (–0.22 to 0.80) |
| Difference at 12 months | 0.51 (1.36) | 0.76 (1.59) | 0.259 | 0.25 (–0.18 to 0.67) |
| Environmental stimuli domain | ||||
| Randomisation | 3.92 (1.51) | 4.02 (1.52) | 0.599 | 0.10 (–0.29 to 0.49) |
| 3 months | 4.38 (1.56) | 4.62 (1.57) | 0.277 | 0.24 (–0.19 to 0.67) |
| 6 months | 4.32 (1.64) | 4.90 (1.44) | 0.009 | 0.58 (0.14 to 1.01) |
| 9 months | 4.25 (1.63) | 4.73 (1.45) | 0.038 | 0.48 (0.03 to 0.93) |
| 12 months | 4.36 (1.65) | 4.79 (1.59) | 0.070 | 0.43 (–0.04 to 0.90) |
| Difference at 12 months | 0.41 (1.42) | 0.69 (1.53) | 0.205 | 0.28 (–0.15 to 0.70) |
| Resource use | Trial group, mean (SD) | p > |z| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo (n = 81) | TLA device (n = 79) | |||
| General practice consultations | ||||
| Surgery | 6.14 (6.15) | 5.20 (5.65) | 0.014 | –0.94 (–1.68 to –0.19) |
| Home | 0.16 (0.70) | 0.05 (0.22) | 0.070 | –0.12 (–0.26 to 0.01) |
| Telephone | 2.33 (6.28) | 1.70 (3.01) | 0.005 | –0.64 (–1.10 to –0.19) |
| Outpatient visits | 3.11 (3.99) | 2.80 (5.27) | 0.250 | –0.31 (–0.85 to 0.22) |
| A&E visits | 0.78 (1.33) | 0.91 (1.80) | 0.360 | 0.13 (–0.15 to 0.42) |
| Ambulance use | 0.44 (1.22) | 0.37 (1.03) | 0.443 | –0.08 (–0.27 to 0.12) |
| Hospitalisations | 0.65 (1.26) | 0.57 (1.37) | 0.495 | –0.08 (–0.32 to 0.16) |
| Length of stay (days) | 2.20 (5.51) | 2.67 (6.76) | 0.629 | 0.47 (–1.45 to 2.40) |
| Nurse consultations | ||||
| Surgery | 2.49 (4.36) | 2.33 (7.29) | 0.503 | –0.16 (–0.65 to 0.32) |
| Home | 0.22 (1.27) | 0.24 (1.47) | 0.810 | 0.02 (–0.13 to 0.17) |
| Physiotherapist visits | 1.27 (3.29) | 1.68 (7.52) | 0.032 | 0.42 (0.04 to 0.80) |
| Occupational therapist visits | 0.40 (1.57) | 0.03 (0.23) | 0.002 | –0.37 (–0.95 to –0.22) |
| Psychologist visits | 0.58 (2.80) | 0.15 (0.83) | < 0.001 | –0.43 (–0.76 to –0.23) |
| Counsellor visits | 0.62 (2.59) | 0.43 (2.10) | 0.110 | –0.19 (–0.42 to 0.04) |
| Time point | Trial group | p > |z| | |||
|---|---|---|---|---|---|
| Placebo | TLA device | ||||
| Currently employed, n (%) | |||||
| Randomisation | N = 117 | 59 (50) | N = 111 | 59 (53) | 0.681 |
| 3 months | N = 106 | 54 (51) | N = 98 | 56 (57) | 0.375 |
| 6 months | N = 97 | 47 (48) | N = 95 | 56 (59) | 0.145 |
| 9 months | N = 93 | 43 (46) | N = 89 | 49 (55) | 0.234 |
| 12 months | N = 91 | 48 (53) | N = 86 | 49 (57) | 0.572 |
| Hours missed of work during past 7 days because of asthma, mean (SD) | |||||
| Randomisation | N = 55 | 3.6 (8.7) | N = 58 | 2.2 (5.8) | 0.306 |
| 3 months | N = 54 | 3.7 (9.5) | N = 56 | 2.4 (9.2) | 0.484 |
| 6 months | N = 47 | 3.5 (8.9) | N = 55 | 2.1 (7.7) | 0.386 |
| 9 months | N = 43 | 3.1 (9.2) | N = 48 | 2.1 (5.0) | 0.513 |
| 12 months | N = 49 | 4.0 (9.3) | N = 50 | 1.1 (4.1) | 0.049 |
| Hours missed of work during past 7 days for other reasons, mean (SD) | |||||
| Randomisation | N = 54 | 3.4 (13.6) | N = 58 | 2.8 (9.2) | 0.779 |
| 3 months | N = 53 | 4.2 (9.4) | N = 56 | 4.5 (10.5) | 0.867 |
| 6 months | N = 46 | 3.3 (12.2) | N = 55 | 2.8 (7.8) | 0.807 |
| 9 months | N = 43 | 7.0 (27.0) | N = 47 | 3.4 (9.0) | 0.393 |
| 12 months | N = 48 | 1.5 (5.6) | N = 49 | 0.8 (3.5) | 0.436 |
| Hours actually worked during past 7 days, mean (SD) | |||||
| Randomisation | N = 57 | 28.3 (16.4) | N = 59 | 31.8 (17.6) | 0.270 |
| 3 months | N = 54 | 23.9 (18.8) | N = 56 | 28.4 (17.4) | 0.190 |
| 6 months | N = 47 | 26.9 (19.3) | N = 56 | 30.4 (17.7) | 0.350 |
| 9 months | N = 44 | 24.8 (18.0) | N = 49 | 30.9 (20.8) | 0.133 |
| 12 months | N = 48 | 28.8 (18.0) | N = 49 | 31.1 (15.5) | 0.506 |
| Impact of asthma on work productivity,a mean (SD) | |||||
| Randomisation | N = 61 | 4.0 (2.8) | N = 54 | 3.4 (2.8) | 0.285 |
| 3 months | N = 51 | 2.8 (2.8) | N = 53 | 2.8 (2.5) | 0.895 |
| 6 months | N = 50 | 3.0 (3.0) | N = 55 | 2.7 (2.5) | 0.543 |
| 9 months | N = 43 | 2.4 (2.5) | N = 50 | 3.4 (3.0) | 0.083 |
| 12 months | N = 47 | 2.5 (2.7) | N = 54 | 2.7 (2.6) | 0.658 |
| Impact of asthma on regular daily activities,a mean (SD) | |||||
| Randomisation | N = 116 | 5.4 (2.4) | N = 109 | 5.0 (2.5) | 0.266 |
| 3 months | N = 105 | 4.6 (2.9) | N = 95 | 4.1 (2.5) | 0.171 |
| 6 months | N = 93 | 4.6 (2.9) | N = 96 | 3.9 (2.9) | 0.141 |
| 9 months | N = 89 | 4.2 (2.9) | N = 90 | 4.1 (2.7) | 0.807 |
| 12 months | N = 90 | 3.9 (3.0) | N = 87 | 3.9 (2.7) | 0.966 |
| Parameter | NHS | Societal |
|---|---|---|
| Costs (£) | ||
| Cost of one cycle for daily symptoms | 808 | 1057 |
| Cost of one cycle in the severe exacerbation state | 1506 | 2121 |
| Cost of device in the first cycle | 1038 | |
| Cost of device for all subsequent cycles | 522 | |
| Quality-of-life adjustments/utilities | ||
| Daily asthma symptoms – placebo | 0.69 | |
| Daily asthma symptoms – TLA device | 0.75 | |
| Severe asthma exacerbation – placebo | 0.60 | |
| Severe asthma exacerbation – TLA device | 0.66 | |
| Transition probabilities | ||
| Between daily asthma symptoms and exacerbation state | 0.27 | |
| Between exacerbation state and daily asthma symptoms | 0.73 | |
| Dying from a severe exacerbation | 0.001 | |
| Other parameters | ||
| Effectiveness of device in reducing the risk of severe exacerbations | 0.99 | |
| Mean initial age of participants (years) | 47 | |
| Annual discount rate for costs (%) | 3.5 | |
| Annual discount rate for outcomes (%) | 3.5 | |
| Response | Trial group, n (%) | ||||
|---|---|---|---|---|---|
| Randomisation | 3 months | 6 months | 9 months | 12 months | |
| Placebo | |||||
| Mobility | |||||
| No problems in walking about | 38 (32) | 46 (44) | 44 (44) | 33 (35) | 37 (39) |
| Slight problems in walking about | 28 (23) | 14 (13) | 17 (17) | 17 (18) | 22 (23) |
| Moderate problems in walking about | 33 (28) | 29 (28) | 26 (26) | 35 (37) | 24 (25) |
| Severe problems in walking about | 21 (18) | 16 (15) | 13 (13) | 10 (11) | 13 (14) |
| Unable to walk about | 0 | 0 | 1 (1) | 0 | 0 |
| Self-care | |||||
| No problems washing or dressing | 87 (73) | 83 (79) | 72 (72) | 65 (68) | 61 (64) |
| Slight problems washing or dressing | 13 (11) | 11 (10) | 17 (17) | 13 (14) | 22 (23) |
| Moderate problems washing or dressing | 17 (14) | 9 (9) | 8 (8) | 10 (11) | 9 (9) |
| Severe problems washing or dressing | 2 (2) | 2 (2) | 3 (3) | 5 (5) | 4 (4) |
| Unable to wash or dress | 0 | 0 | 1 (1) | 2 (2) | 0 |
| Usual activities | |||||
| No problems doing usual activities | 24 (20) | 32 (31) | 34 (34) | 30 (32) | 31 (32) |
| Slight problems doing usual activities | 38 (32) | 23 (22) | 20 (20) | 20 (21) | 28 (29) |
| Moderate problems doing usual activities | 40 (34) | 33 (32) | 34 (34) | 28 (29) | 25 (26) |
| Severe problems doing usual activities | 17 (14) | 13 (13) | 10 (10) | 15 (16) | 10 (10) |
| Unable to do usual activities | 0 | 3 (3) | 3 (3) | 2 (2) | 2 (2) |
| Pain/discomfort | |||||
| No pain or discomfort | 45 (38) | 40 (38) | 37 (37) | 35 (37) | 37 (39) |
| Slight pain or discomfort | 33 (28) | 29 (28) | 29 (29) | 20 (21) | 25 (26) |
| Moderate pain or discomfort | 28 (23) | 24 (23) | 24 (24) | 26 (27) | 19 (20) |
| Severe pain or discomfort | 12 (10) | 9 (9) | 9 (9) | 12 (13) | 12 (13) |
| Extreme pain or discomfort | 2 (2) | 3 (3) | 2 (2) | 2 (2) | 3 (13) |
| Anxiety/depression | |||||
| Not anxious or depressed | 61 (51) | 57 (54) | 54 (53) | 45 (47) | 54 (56) |
| Slightly anxious or depressed | 29 (24) | 26 (25) | 24 (24) | 24 (25) | 15 (16) |
| Moderately anxious or depressed | 21 (18) | 12 (11) | 8 (8) | 14 (15) | 15 (16) |
| Severely anxious or depressed | 6 (5) | 7 (7) | 14 (14) | 9 (9) | 10 (10) |
| Extremely anxious or depressed | 3 (3) | 3 (3) | 1 (1) | 3 (3) | 2 (2) |
| TLA device | |||||
| Mobility | |||||
| No problems in walking about | 49 (43) | 49 (49) | 43 (44) | 43 (48) | 45 (50) |
| Slight problems in walking about | 24 (21) | 21 (21) | 23 (24) | 16 (18) | 20 (22) |
| Moderate problems in walking about | 31 (27) | 22 (22) | 20 (21) | 23 (26) | 16 (18) |
| Severe problems in walking about | 11 (10) | 7 (7) | 10 (10) | 8 (9) | 9 (10) |
| Unable to walk about | 0 | 0 | 0 | 0 | 0 |
| Self-care | |||||
| No problems washing or dressing | 86 (75) | 78 (80) | 74 (77) | 73 (81) | 71 (79) |
| Slight problems washing or dressing | 14 (12) | 11 (11) | 13 (14) | 8 (9) | 11 (12) |
| Moderate problems washing or dressing | 13 (11) | 6 (6) | 9 (9) | 7 (8) | 5 (6) |
| Severe problems washing or dressing | 2 (2) | 3 (3) | 0 | 2 (2) | 3 (3) |
| Unable to wash or dress | 0 | 0 | 0 | 0 | 0 |
| Usual activities | |||||
| No problems doing usual activities | 34 (30) | 37 (37) | 38 (40) | 38 (42) | 46 (51) |
| Slight problems doing usual activities | 39 (34) | 31 (31) | 27 (28) | 24 (27) | 18 (20) |
| Moderate problems doing usual activities | 29 (25) | 21 (21) | 20 (21) | 17 (19) | 19 (21) |
| Severe problems doing usual activities | 11 (10) | 10 (10) | 9 (9) | 10 (11) | 7 (8) |
| Unable to do usual activities | 2 (2) | 0 | 2 (2) | 1 (1) | 0 |
| Pain/discomfort | |||||
| No pain or discomfort | 42 (37) | 37 (38) | 36 (37) | 34 (38) | 35 (39) |
| Slight pain or discomfort | 33 (29) | 31 (32) | 31 (32) | 30 (33) | 26 (29) |
| Moderate pain or discomfort | 22 (19) | 24 (24) | 22 (23) | 15 (16) | 23 (26) |
| Severe pain or discomfort | 15 (13) | 5 (5) | 4 (4) | 10 (11) | 4 (4) |
| Extreme pain or discomfort | 3 (3) | 1 (1) | 3 (3) | 1 (1) | 2 (2) |
| Anxiety/depression | |||||
| Not anxious or depressed | 64 (56) | 59 (60) | 55 (57) | 52 (58) | 57 (63) |
| Slightly anxious or depressed | 26 (23) | 22 (22) | 26 (27) | 25 (28) | 16 (18) |
| Moderately anxious or depressed | 17 (15) | 12 (12) | 10 (10) | 11 (12) | 12 (13) |
| Severely anxious or depressed | 7 (6) | 3 (3) | 4 (4) | 2 (2) | 4 (4) |
| Extremely anxious or depressed | 1 (1) | 2 (2) | 1 (1) | 0 | 1 (1) |
| Time point | Trial group, mean (SD) | p > |z| | Mean difference (95% CI) | |
|---|---|---|---|---|
| Placebo | TLA device | |||
| EQ-5D-5L utility | ||||
| Number with complete EQ-5D-5L utility | n = 86 | n = 84 | ||
| Randomisation | 0.66 (0.26) | 0.70 (0.23) | 0.259 | 0.04 (–0.03 to –0.11) |
| 3 months | 0.66 (0.27) | 0.74 (0.21) | 0.055 | 0.07 (–0.00 to 0.15) |
| 6 months | 0.67 (0.27) | 0.72 (0.22) | 0.151 | 0.05 (–0.02 to 0.13) |
| 9 months | 0.67 (0.30) | 0.72 (0.22) | 0.159 | 0.06 (–0.02 to 0.14) |
| 12 months | 0.67 (0.30) | 0.73 (0.24) | 0.178 | 0.06 (–0.03 to 0.14) |
| Difference at 12 months | 0.00 (0.17) | 0.02 (0.17) | 0.497 | 0.02 (–0.04 to 0.07) |
| EQ-VAS score | ||||
| Number with complete VAS score | n = 85 | n = 84 | ||
| Randomisation | 59 (19) | 60 (19) | 0.747 | 1 (–5 to 7) |
| 3 months | 59 (22) | 65 (18) | 0.073 | 6 (–1 to 12) |
| 6 months | 63 (22) | 66 (18) | 0.212 | 4 (–2 to 10) |
| 9 months | 62 (21) | 66 (19) | 0.226 | 4 (–2 to 10) |
| 12 months | 65 (21) | 68 (17) | 0.426 | 2 (–3 to 8) |
| Difference at 12 months | 7 (20) | 7 (20) | 0.916 | –0 (–6 to 6) |
| Resource use | Unit cost (£) | Time point (months), mean (SD), n | |||
|---|---|---|---|---|---|
| 3 | 6 | 9 | 12 | ||
| Placebo, mean (SD) | |||||
| General practice consultations | |||||
| Surgery | 3663 | 1.69 (2.04), 104 | 1.28 (1.68), 102 | 1.70 (2.10), 94 | 1.38 (2.21), 97 |
| Home | 9162,63 | 0.04 (0.31), 104 | 0.03 (0.22), 102 | 0.06 (0.52), 94 | 0.03 (0.17), 97 |
| Telephone | 2862,63 | 0.72 (2.49), 104 | 0.58 (1.64), 102 | 0.70 (1.82), 94 | 0.40 (1.36), 95 |
| Outpatient visits | 12564 | 0.74 (1.17), 104 | 0.67 (1.09), 102 | 0.85 (1.34), 94 | 0.68 (1.40), 95 |
| A&E visits | 13864 | 0.31 (0.74), 104 | 0.18 (0.50), 102 | 0.14 (0.40), 94 | 0.18 (0.41), 95 |
| Ambulance use | 20964 | 0.24 (0.78), 104 | 0.12 (0.48), 102 | 0.06 (0.29), 94 | 0.11 (0.31), 95 |
| Hospitalisations | 0.24 (0.62), 104 | 0.18 (0.53), 102 | 0.15 (0.41), 94 | 0.15 (0.39), 97 | |
| Length of stay | 46064 | 0.63 (2.10), 103 | 0.38 (1.51), 100 | 0.57 (2.08), 94 | 0.74 (2.45), 97 |
| Nurse consultations | |||||
| Surgery | 1462 | 0.76 (2.72), 103 | 0.61 (1.57), 102 | 0.49 (0.97), 94 | 0.57 (1.13), 95 |
| Home | 4162 | 0.12 (1.09), 103 | 0.07 (0.38), 102 | 0.03 (0.31), 94 | 0.12 (1.03), 95 |
| Physiotherapist visits | 4964 | 0.20 (0.90), 103 | 0.35 (1.39), 102 | 0.39 (1.54), 94 | 0.37 (1.58), 95 |
| Occupational therapist visits | 6964 | 0.12 (1.09), 103 | 0.08 (0.61), 102 | 0.10 (0.66), 94 | 0.03 (0.18), 95 |
| Psychologist visits | 15064 | 0.19 (1.09), 103 | 0.20 (0.92), 102 | 0.11 (0.56), 94 | 0.12 (0.85), 95 |
| Counsellor visits | 15064 | 019 (1.00), 103 | 0.31 (1.56), 102 | 0.12 (0.72), 94 | 0.05 (0.42), 95 |
| TLA device, mean (SD) | |||||
| General practice consultations | |||||
| Surgery | 3663 | 1.54 (2.22), 101 | 1.31 (1.74), 96 | 1.22 (1.64), 88 | 1.36 (1.93), 89 |
| Home | 9162,63 | 0.02 (0.14), 101 | 0.02 (0.14), 96 | 0 (0), 88 | 0 (0), 89 |
| Telephone | 2862,63 | 0.46 (0.88), 101 | 0.44 (0.88), 96 | 0.45 (0.91), 88 | 0.51 (1.39), 89 |
| Outpatient visits | 12564 | 0.74 (1.80), 101 | 0.86 (1.93), 96 | 0.68 (1.73), 88 | 0.70 (1.56), 89 |
| A&E visits | 13864 | 0.31 (0.78), 101 | 0.21 (0.56), 96 | 0.18 (0.58), 88 | 0.16 (0.42), 89 |
| Ambulance use | 20964 | 0.18 (0.55), 101 | 0.06 (0.35), 96 | 0.11 (0.47), 88 | 0.04 (0.26), 89 |
| Hospitalisations | 0.30 (0.74), 101 | 0.11 (0.43), 96 | 0.09 (0.39), 88 | 0.12 (0.42), 89 | |
| Length of stay | 46064 | 1.14 (3.07), 101 | 0.42 (1.45), 96 | 0.32 (1.73), 88 | 0.93 (3.74), 89 |
| Nurse consultations | |||||
| Surgery | 1462 | 0.72 (2.30), 101 | 0.48 (1.90), 96 | 0.35 (1.55), 88 | 0.55 (1.63), 89 |
| Home | 4162 | 0.12 (1.19), 101 | 0.02 (0.20), 96 | 0.02 (0.21), 88 | 0.03 (0.32), 89 |
| Physiotherapist visits | 4964 | 0.33 (1.43), 101 | 0.47 (2.49), 96 | 0.40 (1.96), 88 | 0.42 (1.57), 89 |
| Occupational therapist visits | 6964 | 0 (0), 101 | 0.01 (0.10), 96 | 0.01 (0.11), 88 | 0 (0), 89 |
| Psychologist visits | 15064 | 0.02 (0.14), 101 | 0.10 (0.73), 96 | 0.02 (0.15), 88 | 0.01 (0.11), 89 |
| Counsellor visits | 15064 | 0.12 (0.60), 101 | 0.15 (0.94), 96 | 0.10 (0.86), 88 | 0.07 (0.64), 89 |
| Reason | Unit cost (£) | Source64 |
|---|---|---|
| Abdominal pain | ||
| LoS = 1 with no intervention | 445 | NES HRG FZ90B |
| Accidental overdose | ||
| LoS = 1 with no intervention | 406 | Weighted average of NES HRGs WH04D and WH04E |
| Anaphylaxis | ||
| LoS = 1 with no intervention | 375 | NES HRG WH05Z |
| LoS > 1 with no intervention | 621 | Weighted average of NEL and EL HRG WH05Z |
| Angina/chest pain | ||
| LoS = 1 with no intervention | 398 | Weighted average of NES HRGs EB12A–EB12C and EB13A–EB13D |
| Asthma | ||
| LoS = 1 with no intervention | 424 | Weighted average of NES HRGs DZ15N and DZ15P-DZ15R |
| LoS = 1 with intervention | 765 | NES HRG DZ15M |
| LoS > 1 with no intervention | 408 | Weighted average of NEL and EL HRG DZ15M |
| LoS > 1 with intervention | 460 | Weighted average of NEL and EL HRGs DZ15N and DZ15P-DZ15R |
| Atrial fibrillation | ||
| LoS = 1 with no intervention | 466 | Weighted average of NES HRGs EB07A–EB07E |
| Back pain | ||
| Los > 1 with no intervention | 571 | Weighted average of NEL and EL HRGs HC32H, HC32K and HC32J |
| Broken leg | ||
| LoS > 1 with intervention | 645 | Weighted average of NEL and EL HRGs HT13A–HT13E |
| Cataracts | ||
| Day case with intervention | 878 | Weighted average of DC HRGs BZ30A, BZ30B, BZ31A, BZ31B, BZ32A, BZ32B and BZ33Z |
| LoS = 1 with intervention | 742 | Weighted average of NES HRGs BZ30A, BZ30B, BZ31A, BZ31B, BZ32A, BZ32B, BZ33Z and BZ34A–BZ34C |
| Chest infection | ||
| LoS > 1 with no intervention | 415 | Weighted average of NEL and EL HRGs DZ22M, DZ22N, DZ22P and DZ22Q |
| Collapse | ||
| LoS = 1 with no intervention | 430 | Weighted average of NES HRGs EB08A–EB08E |
| Colonoscopy | ||
| Day case | 573 | Weighted average of DC HRGs FZ51Z, FZ52Z and FZ53Z |
| Epilepsy | ||
| LoS > 1 with no intervention | 507 | Weighted average of NEL and EL HRGs AA26C–AA26H |
| Food poisoning | ||
| LoS > 1 with no intervention | 440 | Weighted average of NEL and EL HRGs FZ36N, FZ36M, FZ36P and FZ36Q |
| Fundoplication | ||
| LoS > 1 | 1358 | Weighted average of NEL and EL HRGs FZ83C–FZ83H, FZ83J and FZ83K |
| Gastroscopy | ||
| LoS = 1 | 549 | Weighted average of NES HRGs FZ60Z, FZ61Z, FZ62A, FZ62B, FZ63Z, FZ64A, FZ64B and FZ65Z |
| Infected finger | ||
| LoS > 1 with intervention | 868 | Weighted average of NEL and EL HRGs HE42A and HE42B |
| Lumbar decompression | ||
| LoS > 1 | 2290 | Weighted average of NEL and EL HRGs HC64A–HC64C |
| Meningitis | ||
| LoS > 1 with no intervention | 498 | Weighted average of NEL and EL HRGs AA22C–AA22G |
| Miscarriage | ||
| LoS > 1 with intervention | 775 | Weighted average of NEL and EL HRGs MB08A |
| Nasal polypectomy | ||
| LoS = 1 | 1251 | NES HRG CA23Z |
| LoS > 1 | 1944 | Weighted average of NEL and EL HRG CA23Z |
| Nerve release | ||
| LoS = 1 with intervention | 1639 | NES HRG HN55Z |
| Osteoarthritis | ||
| LoS > 1 with intervention | 612 | Weighted average of NEL and EL HRGs HD24D–HD24H |
| Pain management | ||
| LoS = 1 | 443 | Weighted average of NES HRGs WH08A and WH08B |
| Planned surgery | ||
| LoS = 1 with intervention | 742 | Weighted average of NES HRGs BZ30A, BZ30B, BZ31A, BZ31B, BZ32 A, BZ32B, BZ33Z and BZ34A–BZ34C |
| Pleurisy | ||
| LoS = 1 with no intervention | 373 | Weighted average of NES HRGs DZ28A and DZ28B |
| Pneumonia | ||
| LoS > 1 with no intervention | 395 | Weighted average of NEL and EL HRGs DZ23L–DZ23N and DZ11R–DZ11V |
| Post-surgical infection | ||
| LoS > 1 with no intervention | 512 | Weighted average of NEL and EL HRGs WH07E–WH07G |
| Prostate surgery | ||
| LoS > 1 | 1364 | Weighted average of NEL and EL HRGs LB21 A, LB21B, LB22Z, LB25D–LB25F, LB26A and LB26B |
| Pyelonephritis | ||
| LoS > 1 with no intervention | 393 | Weighted average of NEL and EL HRGs LA04N and LA04P-LA04S |
| Shortness of breath | ||
| LoS = 1 with no intervention | 412 | Weighted average of NES HRGs DZ19L–DZ19N |
| LoS = 1 with intervention | 775 | Weighted average of NES HRGs DZ19H, DZ19J and DZ19K |
| Stroke | ||
| LoS > 1 with no intervention | 461 | Weighted average of NEL and EL HRGs AA35A–AA35F |
| Surgical complication | ||
| LoS = 1 with no intervention | 509 | Weighted average of NES HRGs WH07E–WH07G |
| Surgical injury to stoma | ||
| LoS > 1 with intervention | 993 | Weighted average of NEL and EL HRGs FZ74C–FZ74F |
| Surgical removal of ovarian cysts | ||
| LoS = 1 | 1021 | NES HRG MA24Z |
| Tooth extraction | ||
| Day case | 758 | Weighted average of DC HRGs CD04A, CD04B, CD05A, CD05B, CD06A, CD06B, CD07A and CD07B |
| Urinary tract infection | ||
| LoS > 1 with no intervention | 393 | Weighted average of NES HRGs LA04N and LA04P–LA04S |
| Resource use | Time point (months), mean (SD), n | |||
|---|---|---|---|---|
| 3 | 6 | 9 | 12 | |
| Placebo | ||||
| General practice consultations | ||||
| Surgery | 61 (74), 104 | 46 (60), 102 | 61 (75), 94 | 50 (80), 97 |
| Home | 4 (28), 104 | 3 (20), 102 | 6 (48), 94 | 3 (16), 97 |
| Telephone | 20 (69), 104 | 16 (45), 102 | 19 (50), 94 | 11 (38), 95 |
| Outpatient visits | 93 (147), 104 | 83 (136), 102 | 106 (168), 94 | 86 (175), 95 |
| A&E visits | 42 (102), 104 | 24 (68), 102 | 19 (56), 94 | 25 (57), 95 |
| Ambulance use | 50 (163), 104 | 25 (99), 102 | 13 (60), 94 | 22 (64), 95 |
| Hospitalisations | ||||
| LoS (non-ITU) | 292 (895), 103 | 189 (661), 100 | 339 (1242), 94 | 500 (2195), 97 |
| LoS (ITU) | 13 (131), 103 | 66 (663), 100 | 113 (794), 94 | 109 (733), 97 |
| Nurse consultations | ||||
| Surgery | 11 (39), 104 | 9 (23), 102 | 7 (14), 94 | 8 (16), 97 |
| Home | 5 (45), 104 | 3 (16), 102 | 1 (13), 94 | 5 (43), 97 |
| Physiotherapist visits | 10 (44), 104 | 17 (67), 102 | 19 (75), 94 | 18 (77), 97 |
| Occupational therapist visits | 8 (75), 104 | 5 (42), 102 | 7 (45), 94 | 2 (12), 97 |
| Psychologist visits | 29 (163), 104 | 29 (139), 102 | 16 (84), 94 | 17 (127), 97 |
| Counsellor visits | 29 (150), 104 | 47 (234), 102 | 18 (108), 94 | 8 (63), 97 |
| Asthma prescribed medications | 260 (195), 107 | 292 (320), 102 | 310 (409), 94 | 302 (408), 95 |
| Other prescribed medications | 41 (64), 107 | 45 (69), 101 | 58 (82), 94 | 53 (82), 95 |
| TLA device | ||||
| General practice consultations | ||||
| Surgery | 56 (80), 101 | 47 (63), 96 | 44 (59), 88 | 49 (69), 89 |
| Home | 2 (13), 101 | 2 (13), 96 | 0 (0), 88 | 0 (0), 89 |
| Telephone | 13 (24), 101 | 12 (24), 96 | 13 (25), 88 | 14 (39), 89 |
| Outpatient visits | 92 (226), 101 | 108 (241), 96 | 85 (217), 88 | 87 (195), 89 |
| A&E visits | 42 (108), 101 | 29 (77), 96 | 25 (80), 88 | 22 (58), 89 |
| Ambulance use | 37 (116), 101 | 13 (73), 96 | 24 (97), 88 | 9 (54), 89 |
| Hospitalisations | ||||
| LoS (non-ITU) | 535 (1377), 101 | 146 (468), 96 | 130 (707), 88 | 478 (1969), 89 |
| LoS (ITU) | 0 (0), 101 | 104 (729), 96 | 0 (0), 88 | 75 (703), 89 |
| Nurse consultations | ||||
| Surgery | 10 (33), 101 | 7 (28), 96 | 5 (22), 88 | 8 (24), 89 |
| Home | 5 (49), 101 | 1 (8), 96 | 1 (9), 88 | 1 (13), 89 |
| Physiotherapist visits | 16 (69), 101 | 23 (121), 96 | 19 (95), 88 | 20 (76), 89 |
| Occupational therapist visits | 0 (0), 101 | 1 (7), 96 | 1 (7), 88 | 0 (0), 89 |
| Psychologist visits | 3 (21), 101 | 16 (110), 96 | 3 (23), 88 | 2 (16), 89 |
| Counsellor visits | 18 (91), 101 | 22 (141), 96 | 15 (129), 88 | 10 (95), 89 |
| Asthma prescribed medications | 262 (225), 102 | 379 (852), 99 | 398 (901), 91 | 393 (896), 89 |
| Other prescribed medications | 47 (91), 102 | 66 (173), 99 | 69 (155), 91 | 74 (184), 89 |
| Resource use | Time point (months), mean (SD), n | |||
|---|---|---|---|---|
| 3 | 6 | 9 | 12 | |
| Placebo | ||||
| Medications | ||||
| OTC | 1 (4), 106 | 1 (6), 100 | 1 (4), 94 | 1 (4), 97 |
| Supplementary | 2 (7), 106 | 4 (13), 102 | 0 (4), 95 | 1 (5), 97 |
| Asthma productivity costs | 441 (1725), 114 | 403 (1590), 109 | 319 (1481), 103 | 473 (1685), 107 |
| TLA device | ||||
| Medications | ||||
| OTC | 2 (11), 102 | 3 (13), 99 | 2 (11), 90 | 2 (11), 89 |
| Supplementary | 2 (7), 102 | 2 (7), 98 | 2 (8), 91 | 2 (9), 90 |
| Asthma productivity costs | 336 (1712), 106 | 303 (1574), 104 | 255 (892), 98 | 147 (741), 97 |
List of abbreviations
- ACD
- Asthma Control Diary
- ACQ
- 7-Point Asthma Control Questionnaire
- AC-QoL
- Adult Carers Quality of Life
- A&E
- accident and emergency
- AQLQ(S)
- Standardised Asthma Quality of Life Questionnaire
- ATS
- American Thoracic Society
- BBC
- British Broadcasting Corporation
- BDP
- beclomethasone dipropionate
- BNF
- British National Formulary
- BTS
- British Thoracic Society
- CE
- Conformité Européenne
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- DSMC
- Data and Safety Monitoring Committee
- ED
- emergency department
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- EQ-VAS
- EuroQol Visual Analogue Scale
- ERS
- European Respiratory Society
- ETC
- excess treatment cost
- FAQ
- frequently asked question
- FeNO
- fraction of exhaled nitric oxide
- FEV1
- forced expiratory volume in 1 second
- FVC
- forced vital capacity
- GETE
- Global Evaluation of Treatment Effect
- GINA
- Global INitiative for Asthma
- GP
- general practitioner
- HRG
- Healthcare Resource Group
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ICS
- inhaled corticosteroid
- IgE
- immunoglobulin E
- ITT
- intention to treat
- ITU
- intensive treatment unit
- LASER
- Laminar Airflow in Severe asthma for Exacerbation Reduction
- NICE
- National Institute for Health and Care Excellence
- OCS
- oral corticosteroid
- OTC
- over the counter
- PED
- participant exacerbation diary
- PEF
- peak expiratory flow
- PI
- principal investigator
- PIC
- participant identification centre
- PIS
- participant information sheet
- p.p.b.
- parts per billion
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- R&D
- research and development
- SAC
- specialist asthma centre
- SAP
- statistical analysis plan
- SD
- standard deviation
- SE
- standard error
- SIGN
- Scottish Intercollegiate Guidelines Network
- SMP
- self-management plan
- SNOT-22
- 22-item Sino-Nasal Outcome Test
- TLA
- temperature-controlled laminar airflow
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- VAS
- visual analogue scale
- WPAI(A)
- Work Productivity and Activity Impairment (Asthma)
- WPAI(CG)
- Work Productivity and Activity Impairment (Caregiver)