

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 11/36/29. The contractual start date was in March 2013. The draft report began editorial review in May 2016 and was accepted for publication in December 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Adrian Taylor reports personal fees from Zimmer Inc., Corin Group and DePuy Synthes Companies outside the submitted work. Martin McNally reports personal fees from Bonesupport AB outside the submitted work. R Andrew Seaton reports personal fees from previous consultancy and funding for speaking at educational meetings (Novartis Pharma) and consultancy for Merck Sharp & Dohme Corp. (MSD) outside the submitted work. Harriet Hughes reports other competing interests from Gilead Sciences Inc., MSD, Biocomposites, and personal fees from Biocomposites and Cubist Pharmaceuticals outside the submitted work. Jennifer Bostock was a member of the Health Services and Delivery Research Commissioned Panel Members during this project.
Disclaimer
This report contains transcripts of interviews conducted in the course of the research and contains language that may offend some readers.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2019. This work was produced by Scarborough et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2019 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Some of the material in this chapter has previously been published in our description of the trial, reproduced from Li et al. 1 This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Scientific background
Infections involving bones and joints are increasingly common. In the NHS in the UK, approximately 250,000 orthopaedic operations are performed annually, including 160,000 hip and knee replacements. 2 Around 1% of these are reported to have been complicated by postoperative infection. 3 In addition, there are around 5000 diabetic foot infections with associated osteomyelitis and a smaller number of infections of the axial skeleton. Treatment costs are estimated to be between £20,000 and £40,000 per patient. 4–6 Many consider a prolonged course (4–6 weeks) of intravenous (IV) antibiotics to be the ‘gold standard’ during the early phase of treatment for bone and joint infections. 7–9 However, such practice derives from an era prior to properly embedded pharmacokinetic principles, during which a widely held view was established that IV therapy is ‘stronger’ than PO therapy. As a result, IV antibiotic therapy is often preferred to oral (PO) therapy and has become an accepted standard of care even for many non-acute infections. The evidence base supporting this practice is, however, limited and there is a growing body of literature from randomised controlled trials (RCTs) and pharmacokinetic studies that suggest that an early switch to PO antibiotics is as effective as continued IV antibiotics. These studies have included patients with pneumonia,10 urinary tract infections,11 low-risk neutropenic sepsis,12 skin and soft tissue infections13 and endocarditis caused by Staphylococcus aureus. 14 There are no large RCTs of PO versus IV antibiotics for bone and joint infection but, provided that agents are carefully chosen with respect to bioavailability and tissue penetration, there is no biologically plausible reason to believe that bone and joint infections should be any different. A Cochrane review of five small trials involving a total 180 participants with bone or joint infection showed no benefit of IV as compared with PO therapy. 14,15 The largest single trial in this meta-analysis comprised 59 patients and the authors concluded that there is currently insufficient evidence to inform a widespread change in practice. Subsequent to this meta-analysis, a further trial involving 42 patients with S. aureus osteomyelitis, who were randomised to either IV cloxacillin or PO combination therapy with co-trimoxazole and rifampicin, showed similar results. 16 Observational studies have reported high success rates for prosthetic joint infection managed by two-stage revision and a shortened course of IV antibiotics or use of antibiotic cement spacers,17,18 but observational comparisons are prone to confounding by indication whereby, for example, only those patients with a better underlying prognosis are switched early to PO antibiotics.
Prolonged IV antibiotic therapy mandates placement of an IV vascular access device, which carries a risk of complications such as catheter-related infection and thromboembolic disease. 6,19 PO antibiotic therapy mitigates such risks,20,21 is more convenient for the patient and is less costly. On the other hand, PO therapy carries a greater risk of poor adherence, gastrointestinal intolerance and variable serum levels related to drug bioavailability.
Nonetheless, for the majority of bone and joint infections, clinicians are able to identify an appropriate PO antibiotic regimen with high PO bioavailability and good tissue penetration. This strategy, however, has not yet been compared with IV treatment in a large clinical trial. Therefore, we set out to address this issue.
Initially, we conducted a single-centre pilot study that concluded in March 2013. 1 The results were reviewed by an independent Data Monitoring Committee (DMC), which advised that it was safe and appropriate to extend the trial. Thereafter, we broadened recruitment to multiple centres and transferred the data from the 228 participants in the pilot study to the database for a multicentre trial, the findings of which are reported here.
The Oral versus IntraVenous Antibiotics (OVIVA) trial was funded by the Health Technology Assessment (HTA) programme. The trial was in full compliance with the Helsinki Declaration22 and has ethics approval (Research Ethics Committee reference number 09/H0604/109 for the single-centre pilot study and Research Ethics Committee reference number 13/SC/0016 for the multicentre trial) from the NHS health research authority.
Explanation of rationale
The objective of the study was to compare the efficacy and safety of IV versus PO antibiotic therapy for patients with bone and joint infection. Six weeks of IV therapy is the current standard of care for some or all of the patients in the hospital trusts that took part in this study. Antibiotics commonly used for IV therapy are often not suitable for oral use (because they are not absorbed) and PO antibiotics are often not suitable for IV use (either because an IV preparation is not available or because they require more frequent dosing than is logistically practical with outpatient IV therapy). It would not, therefore, have been possible simply to randomise the route of administration without this affecting the choice of antibiotic. The choice of antibiotic was subject to patient factors, the organisms identified and the site of infection, and the preferred antibiotic may have changed during treatment as laboratory results were returned or in response to drug reactions. Thus, it was not feasible to develop a protocol specifying particular antibiotics to cover each eventuality for either IV or PO antibiotic choice. In this study, therefore, we randomised participants to an PO or IV ‘strategy’. The choice of individual antibiotics within the randomised strategy was made by a clinician specialised in managing clinical infection and was based on bioavailability, side effect profile, spectrum of activity and, while waiting for culture results, patient risk factors for resistant organisms.
Health economic rationale
The objective of the health economics analysis was to explore the cost-effectiveness of IV antibiotics compared with PO antibiotics. Cost-effectiveness is judged using incremental costs per health outcome. Two analyses were planned in the economic evaluation: a cost–utility analysis (CUA) using quality-adjusted life-years (QALYs) as the health outcome (cost per QALY gained) and a cost-effectiveness analysis (CEA) using definitive failures as an outcome (cost per definitive failure averted). As PO antibiotics were found to be non-inferior to IV antibiotics, the CEA was not carried out based on the assumption that there would be no difference by more than the predefined non-inferiority margin in the number of definitive treatment failures. A CEA would not be informative under these conditions. The health economic analyses focus on the CUA, analysing differences in health-related quality of life (QALYs) and differences in costs between treatment arms. The planned primary health economic analysis was within trial and had a time horizon of 12 months. The intention-to-treat (ITT) population was used. The planned secondary analysis was an extrapolation of trial results beyond the 12 months’ follow-up. However, as there was no difference in failure rate between PO and IV antibiotics, extrapolation was not necessary, therefore, only the primary health economic analysis is presented.
Chapter 2 Methods
Some of the material in this chapter has previously been published in our description of the trial. 1 Reproduced from Li et al. 1 This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Trial design
The trial was a parallel-group, unblinded, non-inferiority multicentre (1 : 1) RCT.
Trial participants
Participants were recruited from the following 26 secondary care centres, all of which are NHS hospitals in England and Scotland:
-
Birmingham Heartlands Hospitals
-
Bristol Royal Infirmary University Hospitals
-
Cambridge University Hospitals
-
Gartnavel General Hospital, Greater Glasgow and Clyde
-
Guy’s and St Thomas’ Hospitals, London
-
Hull and East Yorkshire NHS Trust
-
Leeds Teaching Hospitals
-
Newcastle upon Tyne Hospitals
-
NHS Lothian Hospitals, Edinburgh
-
Oxford University Hospitals
-
Royal Free Hospital, London
-
Royal National Orthopaedic Hospital, Stanmore
-
Royal Hallamshire Hospital, Sheffield
-
Royal Liverpool and Broadgreen University Hospitals
-
NHS Tayside, Dundee
-
Tunbridge Wells Hospital, Kent
-
Brighton and Sussex University Hospitals
-
Wansbeck Hospital, Northumbria
-
Medway Maritime Hospital, Kent
-
Norfolk and Norwich Hospitals
-
Queen Elizabeth Hospital, King’s Lynn
-
Blackpool Teaching Hospitals
-
Northwick Park Hospital, London
-
Northampton General Hospital
-
University Hospitals of North Midlands, Stoke on Trent
-
Whittington Hospital, London.
All sites routinely used 6 weeks of IV antibiotic therapy as their standard initial treatment for some or all categories of bone and joint infection, and all were able to deliver IV antibiotics to patients after discharge from hospital.
Participants were considered for inclusion when an infection specialist reviewed a patient with a bone or joint infection that was considered to require at least 6 weeks of antibiotic therapy. The contact was triggered through the routine care pathway, for example following referral by a surgical team, a referral from primary care direct to infectious disease services or by following up a laboratory result. Informed consent was obtained from each participant by research staff, trained in good clinical practice, after assessing their understanding of the patient information sheet (PIS). Eligibility was determined by the following inclusion and exclusion criteria.
Inclusion criteria
The participant had to meet each of the following criteria:
-
a clinical syndrome comprising any of the following – (1) localised pain, (2) localised erythema, (3) temperature > 38.0 °C or (4) a discharging sinus or wound
-
willing and able to give informed consent
-
aged ≥ 18 years
-
had received ≤ 7 days of IV therapy after an appropriate surgical intervention to treat bone or joint infection (regardless of presurgical antibiotics) or, if no surgical intervention was required, the patient had received ≤ 7 days of IV therapy after the start of planned curative treatment for the relevant clinical episode
-
life expectancy of > 1 year
-
bone and joint infection in one of the following categories –
-
native osteomyelitis (i.e. bone infection without metalwork) including haematogenous or contiguous osteomyelitis
-
native joint sepsis treated by excision arthroplasty
-
prosthetic joint infection treated by debridement and retention, by one-stage revision or by excision of the prosthetic joint (with or without planned reimplantation)
-
orthopaedic device or bone-graft infection treated by debridement and retention, or by debridement and removal
-
spinal infection, including discitis, osteomyelitis or epidural abscess.
-
Exclusion criteria
The participant was ineligible if he or she met any one of the following criteria:
-
S. aureus bacteraemia on presentation or within the previous month
-
bacterial endocarditis, either on presentation or within the previous month (note: there were no study mandated investigations, so participants were not required to have echocardiograms, blood cultures or any other investigations to exclude endocarditis in the absence of a clinical indication)
-
any other concomitant infection that, in the opinion of the clinician responsible for the patient, required a prolonged course of IV antibiotic therapy (e.g. mediastinal infection or central nervous system infection)
-
mild osteomyelitis, defined as bone infection that, in the opinion of the physician, would not usually require a 6-week course of IV antibiotic therapy
-
an infection for which there were no suitable antibiotic choices to permit randomisation between the two arms of the trial (e.g. when organisms were only sensitive to IV antibiotics)
-
prior enrolment in the trial
-
septic shock or systemic features requiring IV antibiotic therapy in the opinion of the treating clinician (the patient could be re-evaluated if these features resolved)
-
unlikely to comply with trial requirements following randomisation in the opinion of the investigator
-
clinical, histological or microbiological evidence of mycobacterial, fungal, parasitic or viral aetiology of the infection
-
receiving an investigational medical product as part of another clinical trial.
The use of antibiotic-loaded cement in spacers, bone substitutes or beads at the site of infection was not an exclusion criterion, but was recorded. Pregnancy, renal failure and liver failure were not exclusion criteria, provided that suitable antibiotic options could be identified for both IV and PO therapy prior to randomisation.
Randomisation
An electronic randomisation service, with telephone back-up if necessary, was provided through a clinical trials unit (CTU). After confirming the patient’s eligibility, the randomisation service assigned a sequentially allocated study number and informed the investigator of the treatment allocation in real time and by confirmatory e-mail. Randomisation was stratified by site to take account of variation in clinical practice between centres.
The local clinician or study nurse was responsible for documenting participants’ enrolment in their clinical notes and for informing the participant’s general practitioner (GP).
Interventions
Eligible patients were randomised (1 : 1) to complete the first 6 weeks of antibiotic therapy with either IV or PO antibiotic therapy. The selection of individual antibiotics within the allocated strategy (i.e. PO or IV antibiotics) was the responsibility of the infection specialist caring for the patient, based on microbiological assessments, the side effect profile, patient preferences and epidemiological factors suggesting the likelihood of antibiotic resistance. In the event of a culture-negative bone or joint infection (or when there was a delay in availability of culture results), the infection specialist selected the most appropriate empiric therapy. When new information became available, the infection specialist was permitted to alter the choice of antibiotic agent according to clinical need. If the participant remained within the allocated administration strategy, they remained within protocol; if this was not possible, the participant was deemed to have met a secondary end point (i.e. early termination of the randomised strategy).
Patients randomised to the IV strategy were expected to complete 6 weeks of IV antibiotics. When necessary for optimal care, clinicians were permitted to use adjunctive PO agents in patients treated with IV therapy (e.g. PO rifampicin as adjunctive therapy for biofilm-related staphylococcal infection). Patients randomised to PO therapy were expected to commence their randomised strategy as soon as possible but were permitted to remain on IV therapy for up to 7 days from the start of the treatment episode, which was most commonly the date of surgical intervention, without being considered an end point. This provided an opportunity for complete recovery from anaesthesia and for antibiotic selection based on culture and susceptibility testing after surgery. If a participant who was randomised to PO therapy required IV antibiotic therapy for an unrelated intercurrent illness during the initial 6 weeks of treatment, or experienced vomiting, inability to swallow or other concern about absorption of PO medication, then IV antibiotic therapy could be substituted for up to 5 days. If IV antibiotic prescribing exceeded the limits set in the PO strategy, the patient was deemed to have met a secondary end point but still contributed to the ‘ITT’ analysis, and study follow-up therefore continued.
If at any point the randomised strategy (IV or PO) was no longer compatible with good clinical care, then the study participant was withdrawn from their randomised treatment arm and an end point was recorded. Appropriate reasons for discontinuing the allocated treatment were, for example, no suitable medication was available within the allocated strategy because of adverse reactions, contraindications and susceptibility testing results. Failure to maintain IV access was considered a legitimate reason for discontinuing IV antibiotics and switching to PO antibiotics to complete the first 6 weeks. In such cases, the event was recorded as a secondary end point, which was most commonly an early exit from allocated treatment strategy. However, a wound discharge, superficial erythema or other clinical sign related to infection or resolution of infection was not an appropriate indication for changing PO to IV, or vice versa, as there was equipoise regarding efficacy.
For any patient who was withdrawn from their randomised strategy, each case was discussed with the study chief investigator or delegate of the chief investigator beforehand. Changing the antibiotic while remaining within the allocated strategy did not need discussion, but such decisions were made by a clinician with appropriate training in managing infection. Patients who were withdrawn from the allocated strategy for any reason continued to be followed up according to the trial protocol (unless they specifically declined this) and were included in ‘ITT’ analysis of efficacy, but not in the ‘according-to-protocol’ analysis (unless they had completed at least 4 weeks within their randomised strategy).
Dose adjustments based on renal or hepatic function, drug interactions or other factors were permitted in accordance with drug labelling information: the British National Formulary (BNF) and local pharmacy guidelines.
Follow-on antibiotic treatment after the initial 6 weeks was allowed in either arm of the trial, but the choice of agent, duration and route of administration were not governed by the trial protocol.
All systemic antibiotics used (including dose, route of administration and duration) were recorded in the case report form (CRF) from the date of randomisation to final follow-up at 1 year. Local antibiotic use in cement or bone fillers was recorded but topical antibiotic use for superficial wounds was not.
There were no formal withdrawal criteria in this study other than at the request of a participant. All patients were free to withdraw their consent at any time; if they elected to withdraw from the allocated treatment strategy during the randomised treatment phase, they were deemed to have met a secondary end point but were still followed up and included in the analysis, provided that appropriate consent had been obtained.
Assessments
Data on inclusion criteria, patient characteristics, operative details and comorbidities were collected at the baseline/enrolment visit and entered onto the web-based database by the trial sites.
While an inpatient, study clinician or research nurse maintained contact with the clinical team to identify potential end points, and to ensure implementation of the randomised antibiotic strategy. Following hospital discharge, participants were seen according to clinically determined follow-up plans. Trial-specific clinical data were obtained from either face-to-face contact with the participants or from the relevant case records at 6 weeks (range day 21 to day 63), 4 months (range day 70 to day 180) and 1 year (range day 250 to day 420). Research staff at the recruiting centre were responsible for entering the data from clinical reviews. If the patient did not attend clinic within the specified date ranges, the investigator arranged a telephone review with the participant or the participant’s GP to identify potential end points or serious adverse events (SAEs). If, based on the telephone discussion, a further clinical review was indicated, the investigator facilitated this and advised the patient accordingly.
A study clinician reviewed the source documents from routine care visits when completing investigator reviews. They recorded:
-
microbiology and histology results and date of discharge (first review only)
-
outpatient visits since randomisation
-
SAEs
-
readmissions for inpatient care
-
type of IV catheter (line) used and any line-related complications
-
episodes of Clostridium difficile-associated diarrhoea
-
antibiotic use, including dosage, route and model of care (e.g., district nurse, self-administered or daily clinic visits)
-
presence or absence of any potential end points
-
reasons for not completing the planned antibiotic course (if applicable).
The EuroQol-5 Dimensions, three-level version (EQ-5D-3L) questionnaires to assess quality of life were requested at baseline, day 14, day 42, day 120 and day 365. The baseline EuroQol-5 Dimensions (EQ-5D) data were entered by the researcher and then filed in the site file. Subsequent EQ-5D questionnaires were handed to participants with prepaid envelopes to the central CTU in Oxford for data entry and filing. EQ-5D data were not routinely collected during the single-centre pilot study.
Oxford Hip Score (OHS) or Oxford Knee Score (OKS) questionnaires were given to patients with an infection in the hip or knee. Returns were requested at baseline, day 120 and day 365. Baseline data were entered at trial sites, but subsequent returns were sent directly to Oxford for data entry and filing.
A subset of participants was monitored through a Medication Event Monitoring System (MEMS). These consist of tablet containers with a cap that records every opening with a date and time stamp, which subsequently can be downloaded for analysis permitting monitoring of medication adherence.
Objectives
The primary objective was to determine whether or not PO antibiotics are non-inferior to IV antibiotics for serious bone and joint infection, as judged by the proportion of patients experiencing definitive treatment failure during 1-year of follow-up.
Secondary objectives were to compare the following end points according to treatment allocation:
-
SAEs, including death (i.e. all cause) according to treatment allocation
-
line complications (i.e. infection, thrombosis or other events requiring early removal or replacement of the line)
-
C. difficile-associated diarrhoea
-
‘probable’ and ‘possible’ treatment failure as composites with definitive treatment failure
-
early termination of the planned 6-week period of PO or IV antibiotics because of adverse events, patient preference or any other reason
-
resource allocation using (1) length of inpatient hospital stay, (2) frequency of outpatient visits and (3) antibiotic prescribing costs
-
quality of life, as evaluated by EQ-5D questionnaire
-
OHS and OKS (when infection was in the hip or knee)
-
adherence, as indicated by MEMS in a subset of participants. [In a subset of sites (i.e. Oxford University Hospitals, Guy’s and St Thomas’ Hospitals Trust, Royal Free Hospital Trust and Royal National Orthopaedic Hospital), PO antibiotics were dispensed to patients in pill containers with MEMS.]
Outcomes
Potential primary end points were identified through post-randomisation prospective surveillance, and reviewed by an end-point committee blind to the treatment group. The primary end point was failure of infection treatment, for which definite failure was indicated by one or more of the following:
-
isolating bacteria from two or more samples of bone/spine/periprosthetic tissue, when the bacteria were phenotypically indistinguishable
-
a pathogenic organism (e.g. S. aureus but not S. epidermidis) on a single, closed aspirate or biopsy of native bone or spine
-
diagnostic histology on bone/periprosthetic tissue
-
formation of a draining sinus tract arising from bone/prosthesis
-
recurrence of frank pus adjacent to the bone/prosthesis.
Secondary end points were:
-
SAEs, including death (i.e. all cause) according to treatment allocation
-
line complications (i.e. infection, thrombosis or other events requiring early removal or replacement of the line)
-
‘probable’ or ‘possible’ treatment failure as composites with definitive treatment failure – these were determined by a blinded end-point committee review and determined according to the following criteria:
-
loosening of a prosthesis, confirmed radiologically, or
-
non-union of a fracture after 6 months, confirmed radiologically, or
-
superficial spreading erythema, treated as cellulitis with an antibiotic for > 1 week, when results from deep tissue samples did not meet the primary end point as described above
when appropriate, deep tissue samples were sent for microbiology and the results of culture were negative, and either (a), (b) or (c) were met, the end point was regarded as ‘possible’. On the other hand, when deep tissue samples were not sent for microbiology, and either (a), (b) or (c) were met, then the end point was regarded as ‘probable’
-
-
early termination of the planned 6-week period of PO or IV antibiotics because of adverse events, patient preference or any other reason
-
resource allocation determined by (1) length of inpatient hospital stay, (2) frequency of outpatient visits and (3) antibiotic prescribing costs
-
quality of life evaluated by EQ-5D questionnaire
-
OHS and OKS (when infection was in the hip or knee)
-
adherence to PO medication.
The study clinicians determined secondary end points 1, 2, 4 and 5. The blinded end-point review committee determined primary end points and secondary end point 3, by reviewing relevant clinical notes redacted for personal details and any information that might have betrayed the treatment allocation. Participant questionnaires determined secondary end points 6 and 7. Secondary end point 8 was determined by MEMS at four sentinel sites.
Adherence and Medication Event Monitoring Systems
Patient adherence to antibiotic therapy may directly influence the outcome of treatment. In order to avoid intrusion and to minimise undue influence on patient behaviour, participants did not receive any direct antibiotic adherence support (such as text message reminders or telephone monitoring), but the importance of adherence was explained at the time of recruitment and reinforced at the time of discharge. The PIS included information written by the patient representatives explaining the importance and underlying rationale of medication adherence.
In order to validate adherence, selected sites (i.e. Oxford University Hospitals, Guy’s and St Thomas’ Hospitals, The Royal National Orthopaedic Hospital and The Royal Free Hospital, London), dispensed PO antibiotics in pill containers with MEMS. 23,24 Sensors in the pill container lids (caps) detected opening and closing and recorded these events with a time and date stamp. The sensor data were downloaded and read at a later date to assess whether or not patients had opened their bottles at times consistent with their prescription. MEMS were used only with specific consent from participants. If more than one antibiotic was prescribed, MEMS sensors were used for the more frequently dosed antibiotic.
Safety
As the OVIVA trial did not involve randomisation to a specific therapy, it was not a ‘Clinical Trial of an Investigational Medicinal Product’, as defined by the European Union directive 2001/20/EC. 25 Safety reporting therefore referred to the trial sponsor and the DMC. All SAEs identified within a year of randomisation were recorded.
If an investigator became aware of an unexpected SAE during the trial, he or she contacted the chief investigator who clarified clinical details and reported the SAE to the sponsor. If, in the opinion of the chief investigator or the sponsor, an unexpected SAE might have been relevant to participant safety, a detailed report including an assessment of causality and severity was forwarded to the DMC. In turn, the DMC made a recommendation to the Trial Steering Committee regarding the safety of the trial in the light of this report.
Expected SAEs that did not undergo expedited reporting were defined as:
-
complications of bone/joint surgery
-
complications of the bone/joint infection for which the patient was undergoing treatment (including potential end points)
-
drug reactions as detailed in the product literature [i.e. the summary of product characteristics (SmPC) or BNF]
-
drug reactions for concurrent medications given for routine clinical care as detailed in the product literature (i.e. SmPC or BNF)
-
intercurrent illness causally related to the comorbid conditions that the investigator believed were likely diagnoses, given the patient’s history, age and other factors.
The investigators used their judgement, such that SAEs that technically met the definitions above for expectedness, but that seemed unexpected in terms of severity, duration or other factors, may have been reported as unexpected.
Statistical methods
Full details of the statistical methods used are detailed in a statistical analysis plan (see Appendix 1), which was agreed and signed off prior to locking the database.
Sample size
An initial sample size estimation of 1050 was based on an expected overall failure rate of 5% and a non-inferiority margin of 5% (or a relative increase of 100%), with a one-sided alpha = 0.05, 90% power and 10% loss to follow-up. This was derived from short-term follow-up in the single-centre pilot study in which 10 participants experienced a primary end point in the first 197 randomisations.
Pooled data from a planned interim analysis during the multicentre study demonstrated that the true event rate was likely to be closer to 12.5%. To account for this, we adjusted the non-inferiority margin to 7.5% (or a relative increase of 60%). As the final control group failure rate remained unknown, recruitment continued as planned until October 2015 to achieve the largest possible sample size within the original target, and to optimise the potential utility of subgroup analyses. The DMC and ethics committee approved this as an amendment to the protocol.
Primary analysis
The proportions of participants experiencing a primary end point at 1-year follow-up (definitive treatment failure as adjudicated by a blinded end-point review committee) were tabulated by randomised strategy (i.e. PO vs. IV therapy). Non-inferiority was defined as the absolute, upper 90% confidence interval (CI) around the unadjusted difference (PO vs. IV) being < 7.5%. The primary analysis was based on the ITT population, whereby all participants were included based on their randomised strategy (PO vs. IV strategy). Missing end-point data were handled by multiple imputation. Supporting analysis included a complete-case analysis, a per-protocol (PP) analysis and an analysis whereby those with missing end points were assumed not to have experienced a definitive treatment failure. Sensitivity analyses explored the impact of informatively missing data.
Secondary analyses
Secondary analyses focused on consistency of point estimates and 95% CI, rather than formal comparisons with the 7.5% non-inferiority margin. Adjusted quantile regression models or rank sum tests were used to compare continuous secondary outcomes, and proportions of participants with secondary end points were presented (including chi-squared tests). Interaction tests were used to determine the consistency of treatment effects by prespecified subgroups, including the type of baseline surgical procedure, infecting pathogen and the clinician’s specified antibiotic intentions, as recorded prior to randomisation, and whether or not this planned antibiotic regimen included rifampicin.
Deviations from the statistical analysis plan
Additional post hoc subgroup analyses performed were:
-
metal retained versus no metal retained in baseline surgical procedure
-
known pathogen versus pathogen unknown
-
participants with and without peripheral vascular disease.
Software employed
Analyses were undertaken using Stata® (version 14SE, StataCorp LP, College Station, TX, USA).
Randomisation
A randomisation list, stratified by site and prepared by a statistician, was held securely by a CTU. The randomisation sequence was created using Stata 12IC statistical software and was stratified by centre with a 1 : 1 allocation using random blocks of size 8–14. Allocation concealment was achieved through the use of sequentially allocated study numbers. After confirming a patient’s eligibility, the study clinician contacted the CTU via a website link (with telephone back-up if required) to be provided with a study number and the associated randomised treatment allocation (PO vs. IV for the first 6 weeks of antibiotics). An automated e-mail confirming these data was then forwarded to the clinician randomising the patient. All participants were randomised after confirmation of eligibility but within 7 days of the start of their treatment episode.
Blinding
The study was open label. Blinding was not possible, as we considered that giving prolonged IV placebo would pose an unnecessary risk to participants and, therefore, would be unethical. Because an open-label study is at risk of bias, we appointed an end-point review committee. The end-point review committee was composed of three independent clinicians (two infectious diseases specialists and one orthopaedic surgeon) with expertise in the management of orthopaedic infections.
All relevant notes relating to a potential end point were reviewed and redacted for both personal identifiable information and specifics of antibiotic treatment or IV line insertion, which could have indicated the route of administration of antibiotics. The end-point review committee was therefore blind to treatment allocation. The redacted notes were forwarded to the end-point review committee, which examined them against objective criteria, to determine whether or not an end point had been met, either by consensus or by a vote called by the chairperson if consensus could not be reached.
The end-point review committee was only required to review definite or potential treatment failures. All other end points were determined directly by the local study clinicians.
Summary of changes to the project protocol
-
Adjustment of non-inferiority margin as described under Sample size.
-
Extension of recruitment period at no additional cost.
Health economic analysis methods
The economic evaluation is based on health-care resource use and quality-of-life data collected during the trial. All costs and health outcomes were measured and collected within 1 year, so that no discount rate was applied.
Resource use
Costs were measured from a NHS and Personal Social Services perspective. Resource use included antibiotic medication, IV administration and complications, and inpatient stays. These were completed for the time periods of 42, 120 and 365 days following randomisation. Costs for medication were obtained from the BNF. 26 Inpatient stays were valued using NHS reference costs27 and IV administration resources and costs were taken from the literature28,29 and adjusted for inflation using the Hospital and Community Health Index. 30 Costs were reported for 2015 in Great British pounds. Antibiotic resource use includes all antibiotics prescribed to each participant in the 12-month follow-up period. Inpatient stays are per bed-day, and IV administration includes the cost of IV line insertion and removal for each IV episode per participant, cost of line complications when a new line is needed and the cost of the outpatient parenteral antimicrobial therapy (OPAT) team, if applicable.
Total costs per participant were calculated by assigning unit costs to within-trial resource use for each participant. 29 Unit costs and their sources are presented in Table 1.
| Resource | Unit cost | Source |
|---|---|---|
| Antibiotic | Various | BNF26 |
| Inpatient stay | £295.80 per overnight stay | NHS Reference Costs 2014 to 2015 27 |
| IV administration | ||
| Insertion: PICCa | £190 | Expert opinion |
| Removal | £34.12 | Expert opinion |
| OPAT type | ||
| District nurse | £58 per hour | NHS Reference Costs 2014 to 2015 27 |
| Infusion centre attendance | £109 per hour | NHS Reference Costs 2014 to 2015 27 |
Health outcomes
Outcomes are measured using QALYs. QALYs are a combination of both quality and length of life. Quality-of-life data were collected using the EQ-5D-3L,31 administered at baseline, at 14 days, 42 days, 120 days and 365 days and if an end point or SAE occurred. The EQ-5D data collected when a SAE occurred were subsequently not used, as the available data were insufficient to provide additional information for the analysis. The EQ-5D-3L is a generic quality-of-life measure comprising five questions and a visual analogue scale (VAS). The questions cover five domains: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. There are three levels of severity for each domain (‘no pain’, ‘moderate pain’ and ‘extreme pain’). The EQ-5D instrument provides 243 predefined health states. Responses are pooled into a three-digit number labelling the respondent’s health state (from ‘111’, meaning no health-related problems, to ‘333’, meaning extreme health-related problems in all five domains). 32
The EQ-5D-3L responses were converted to utility measures using the tariff developed for the UK general population. 33 This utility is combined with the length of time the person is in each health state using standard area-under-the-curve methods to calculate QALYs. Patient-specific QALYs were estimated using utility values from each follow-up point and weighting each time interval by the patient’s utility during that period. A utility score of 1 is equivalent to full health and 0 is equivalent to death. It is possible to have a negative utility score, for which the patient’s health state is worse than death. Discrete changes in utility values between follow-up time points were assumed to be linear.
Analysis
The total cost per participant in each intervention was summed and divided by the number of participants in each arm to calculate the mean cost per participant in each arm, along with the difference in means and 95% CI.
The mean QALY per participant for each intervention was calculated by summing all participants’ QALYs and dividing by the number of participants in that intervention arm. The difference in the means was calculated along with 95% CIs.
The analysis was carried out in Stata version 14.0. Complete cases were analysed initially and multiple imputation was used to explore the effect of missing data on the analysis.
Missing data
The nature of the missing data was analysed and an appropriate method to replace missing data utilised. 34,35 Missing data for resource use and EQ-5D-3L were handled using multiple imputation, which requires less strong assumptions than complete-case analysis. Multiple imputation requires a more relaxed assumption that data are missing at random. The probability of having missing data is independent of unobserved values and the missing data may depend on observed data. 36 Missing resource and quality-of-life data were imputed using multiple imputation by chained equation. 37
The regression analyses used to impute missing data included information on ‘baseline surgical procedures’.
The following assumptions were made:
-
the cost of a line insertion and removal was applied to the initial 6-week period of the intervention. 29 In addition, it was assumed that an IV episode with a gap of ≤ 2 days between IV drugs did not require a new line to be inserted and a cost was not applied for insertion/removal; if the gap between episodes was > 2 days, it was assumed that a new line had to be inserted and the old line was removed, and a cost was assigned accordingly
-
the OPAT type recorded at the 42-day follow-up visit was used for each participant for all IV episodes in the 12-month follow-up period29
-
any durations of antibiotics, IV episodes and inpatient stays per participant were truncated at 365 days, as the follow-up period is 365 days
-
OPAT costs were applied at 1 hour per day, if applicable
-
participants with an OPAT type of ‘infusion centre’ had a weighted cost of two out of five to self-administrating and three out of five to district nurse applied to the length of IV episode following discharge from hospital; this was the proportion of district nurse to self-administering OPAT witnessed in the trial
-
for participants with missing data for OPAT type, a weighted average cost of two out of five toself-administrating and three out of five to district nurse was applied to the length of IV episode
-
for participants with missing data for IV line type, the cost of a peripherally inserted central catheter line was used as this was used by the majority of participants during the trial.
Sensitivity analysis
Instead of using the above weighted average for participants with missing OPAT type, two scenarios were explored: applying solely the cost of a district nurse, and applying solely the cost of self-administration.
To explore the uncertainty around the cost and QALY differences and the resulting incremental cost-effectiveness ratio (ICER), a non-parametric bootstrapping technique was employed with 1000 iterations. Results are presented using a cost-effectiveness plane, showing all 1000 cost-effectiveness pairs.
Long-term outcomes
Owing to the non-inferiority margin being met in the trial, the extrapolation of failure rates was not carried out as there would have been no difference in rates extrapolated forward.
Chapter 3 Results
Recruitment
The timeline of recruitment into the study was as follows:
-
date of start of recruitment – 26 March 2013, start of main study; 3 June 2010, start of internal pilot
-
date of end of recruitment – 31 October 2015
-
date of end follow-up – 31 October 2016
-
date of final analysis – 1 November 2016–20 January 2017
-
target number of subjects – 1054 (527 per arm) including the pilot.
Originally, recruitment to the OVIVA study was to conclude at the end of October 2014. Owing to the initial recruitment being lower than expected, the trial was granted an extension without additional funding. The above presented timelines take into account this extension.
Study participants
Information on screening, eligibility, randomisations and follow-up is shown in the Consolidated Standards of Reporting Trials (CONSORT) flow diagram in Figure 1.
FIGURE 1.
The OVIVA trial CONSORT flow diagram. a, An additional seven deaths were reported within the acceptable range for the day 365 follow-up. The final follow-up for these participants is not considered missing. These deaths are reported in Serious adverse events.

Screening logs were only available in the multicentre study and not all sites completed screening logs adequately. Therefore, the CONSORT flow diagram may overestimate conversion rates from screened to eligible and eligible to randomised participants.
Participants were excluded from the PP population if they received < 4 weeks of their allocated strategy for reasons other than possible or probable recurrence of infection and/or had missing data for the primary end point.
Data quality
Data collection and compliance
Data on inclusion criteria, patient characteristics, operative details and comorbidities were collected at the baseline/enrolment visit and entered onto the web-based database by the trial sites.
Three clinical reviews were performed for each participant during the follow-up:
-
day 42 (accepted range day 21–63)
-
day 120 (accepted range day 70–180)
-
day 365 (accepted range day 250–420).
Clinical assessment compliance
Table 2 shows the data completeness for clinical assessments at three follow-up points. The number of missing baseline and follow-up CRFs may not coincide with the number of participants withdrawn or lost to follow-up at the relevant assessment time point. This is because CRFs could be completed to indicate participant withdrawal and loss to follow-up, as well as to record relevant clinical data up to the time of withdrawal/loss to follow-up.
| Assessment | Antibiotics | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IV | PO | Total | |||||||
| Complete | Expected | % | Complete | Expected | % | Complete | Expected | % | |
| Baseline | 527 | 527 | 100.00 | 527 | 527 | 100.00 | 1054 | 1054 | 100.00 |
| Day 42 | 523 | 527 | 99.24 | 523 | 527 | 99.24 | 1046 | 1054 | 99.24 |
| Day 120 | 517 | 527 | 98.10 | 521 | 527 | 98.86 | 1038 | 1054 | 98.48 |
| Day 365 | 514 | 527 | 97.53 | 517 | 527 | 98.10 | 1031 | 1054 | 97.82 |
Questionnaire compliance
Some potential participants were willing to take part in the trial, but were unwilling to complete quality-of-life data. This contributed to the low compliance rate for the questionnaires (Table 3).
| Assessment | Antibiotics | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IV | PO | Total | |||||||
| Complete | Expected | % | Complete | Expected | % | Complete | Expected | % | |
| Baseline | 386 | 414 | 93.24 | 388 | 412 | 94.17 | 774 | 826 | 93.70 |
| Day 14 | 307 | 414 | 74.15 | 308 | 412 | 74.76 | 615 | 826 | 74.46 |
| Day 42 | 326 | 414 | 78.74 | 336 | 412 | 81.55 | 662 | 826 | 80.15 |
| Day 120 | 295 | 414 | 71.26 | 286 | 412 | 69.42 | 581 | 826 | 70.34 |
| Day 365 | 285 | 414 | 68.84 | 276 | 412 | 66.99 | 561 | 826 | 67.92 |
Although not routinely collected from 228 participants in pilot phase of the OVIVA study, 122 questionnaires were received for these participants (1 at day 14, 74 at day 42, 32 at day 120 and 15 at day 365). These data were included in the analysis of the EQ-5D-3L.
Table 4 displays the number of participants for whom the OHS and OKS could be calculated at the relevant time points. The OHS and OKS could be calculated when up to two items are missing. A low number of additional questionnaires were received for which the calculation of the OHS was not possible owing to missing items (4 at baseline, 2 at day 120 and 2 at day 365). A low number of additional questionnaires were received for which the calculation of the OKS was not possible owing to missing items (3 at day 120 and 2 at day 365). These questionnaires were not classed as complete and are not included any of the subsequent summaries and analyses.
| Assessment | Antibiotics | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IV | PO | Total | |||||||
| Complete | Expected | % | Complete | Expected | % | Complete | Expected | % | |
| OHS | |||||||||
| Baseline | 74 | 87 | 85.06 | 71 | 81 | 87.65 | 145 | 168 | 86.31 |
| Day 120 | 64 | 87 | 73.56 | 59 | 81 | 72.84 | 123 | 168 | 73.21 |
| Day 365 | 60 | 87 | 68.97 | 57 | 81 | 70.37 | 117 | 168 | 69.64 |
| OKS | |||||||||
| Baseline | 99 | 111 | 89.19 | 88 | 98 | 89.80 | 187 | 209 | 89.47 |
| Day 120 | 75 | 111 | 67.57 | 69 | 98 | 70.41 | 144 | 209 | 68.90 |
| Day 365 | 75 | 111 | 67.57 | 67 | 98 | 68.37 | 142 | 209 | 67.94 |
Baseline characteristics
Trial site was the only stratification factor for randomisation in this trial. Tables 5 and 6 provide an overview of the baseline data. As these data were collected on different CRFs, not all data were available for all randomised participants.
| Trial site and characteristicsa | Antibiotic, n (%) | Total (N = 1054), n (%) | |
|---|---|---|---|
| IV (N = 527) | PO (N = 527) | ||
| Oxford University Hospitals | 256 (48.58) | 256 (48.58) | 512 (48.58) |
| Bristol Royal Infirmary | 3 (0.57) | 3 (0.57) | 6 (0.57) |
| Western General Hospital Edinburgh | 5 (0.95) | 3 (0.57) | 8 (0.76) |
| Guy’s and St Thomas London | 19 (3.61) | 17 (3.23) | 36 (3.42) |
| Royal Free London | 23 (4.36) | 22 (4.17) | 45 (4.27) |
| Queen Elizabeth Hospital King’s Lynn | 0 (0.00) | 1 (0.19) | 1 (0.09) |
| Royal Liverpool University Hospital | 36 (6.83) | 34 (6.45) | 70 (6.64) |
| Addenbrookes Hospital Cambridge | 25 (4.74) | 24 (4.55) | 49 (4.65) |
| Royal Hallamshire Hospital Sheffield | 2 (0.38) | 0 (0.00) | 2 (0.19) |
| Royal Victoria Infirmary Newcastle | 1 (0.19) | 1 (0.19) | 2 (0.19) |
| Ninewells Hospital Dundee | 7 (1.33) | 7 (1.33) | 14 (1.33) |
| Gartnaval Hospital Glasgow | 23 (4.36) | 21 (3.98) | 44 (4.17) |
| Birmingham Heartlands | 23 (4.36) | 25 (4.74) | 48 (4.55) |
| Royal National Orthopaedic Hospital Stanmore | 63 (11.95) | 63 (11.95) | 126 (11.95) |
| Hull Royal Infirmary | 5 (0.95) | 6 (1.14) | 11 (1.04) |
| Medway Hospital | 0 (0.00) | 2 (0.38) | 2 (0.19) |
| University Hospital of North Staffordshire | 0 (0.00) | 1 (0.19) | 1 (0.09) |
| Leeds General Infirmary | 14 (2.66) | 14 (2.66) | 28 (2.66) |
| Northampton General Hospital | 4 (0.76) | 1 (0.19) | 5 (0.47) |
| Maidstone and Tunbridge Wells | 1 (0.19) | 3 (0.57) | 4 (0.38) |
| Royal Sussex County Hospital Brighton | 2 (0.38) | 5 (0.95) | 7 (0.66) |
| Northumbria NHS Trust | 5 (0.95) | 8 (1.52) | 13 (1.23) |
| Norfolk and Norwich University Hospital | 4 (0.76) | 2 (0.38) | 6 (0.57) |
| Blackpool Teaching Hospitals | 2 (0.38) | 4 (0.76) | 6 (0.57) |
| Northwick Park London | 3 (0.57) | 3 (0.57) | 6 (0.57) |
| Whittington Hospital London | 1 (0.19) | 1 (0.19) | 2 (0.19) |
| Gendera | |||
| Male | 320 (60.72) | 358 (67.93) | 678 (64.33) |
| Female | 207 (39.28) | 169 (32.07) | 376 (35.67) |
| Age (years)b | 61 (49–70) (18–92) | 60 (49–70) (18–91) | 60 (49–70) (18–92) |
| Clinical variable | Antibiotic | Total (N = 1054a), n (%) | |
|---|---|---|---|
| IV (N = 527a), n (%) | PO (N = 527a), n (%) | ||
| Information on inclusion criteriab | |||
| Localised pain | 397 (75) | 403 (76) | 800 (76) |
| Localised erythema | 226 (43) | 207 (39) | 433 (41) |
| Temperature > 38.0 °C | 62 (12) | 62 (12) | 124 (12) |
| Discharging sinus/wound | 296 (56) | 285 (54) | 581 (55) |
| Information on the baseline surgical procedure | |||
| Chronic osteomyelitis debrided, no current implant or device | 153 (29) | 169 (32) | 322 (31) |
| Chronic osteomyelitis as above, but not debrided | 25 (4.7) | 29 (5.5) | 54 (5.1) |
| Implant or device present and retained | 124 (24) | 123 (23) | 247 (23) |
| Removal of orthopaedic device for infection | 89 (17) | 78 (15) | 167 (16) |
| Prosthetic joint implant removed | 68 (13) | 67 (13) | 135 (13) |
| Prosthetic joint implant, one-stage revision | 47 (8.9) | 43 (8.2) | 90 (8.5) |
| Discitis/spinal osteomyelitis/epidural abscess debrided | 8 (1.5) | 5 (1.0) | 13 (1.2) |
| Discitis/spinal osteomyelitis/epidural abscess but not debrided | 13 (2.5) | 13 (2.5) | 26 (2.5) |
| Information on anatomical site affected by the infection | |||
| Left | 225 (43) | 240 (46) | 465 (44) |
| Right | 252 (48) | 241 (46) | 493 (47) |
| Bilateralc | 50 (9.5) | 46 (8.7) | 96 (9.1) |
| Further information on anatomical sited | |||
| Spinal infection | 37 (7.0) | 35 (6.6) | 72 (6.8) |
| Upper limb infection | 43 (8.2) | 59 (11) | 102 (9.7) |
| Lower limb infection | 436 (83) | 419 (80) | 855 (81) |
| Other area of infection | 12 (2.3) | 14 (2.7) | 26 (2.5) |
| Details on lower limb infectionse | n = 436 | n = 418 | n = 854 |
| Hip | 110 (25) | 104 (25) | 214 (25) |
| Knee | 133 (31) | 115 (27) | 248 (29) |
| Foot | 89 (20) | 86 (21) | 175 (20) |
| Other area of lower limb infection | 105 (24) | 113 (27) | 218 (26) |
| Operative findings | |||
| Draining sinus arising from bone/prosthesis | 177 (34) | 142 (27) | 319 (30) |
| Frank pus adjacent to bone/prosthesis | 179 (34) | 186 (35) | 365 (35) |
| Information on local antibiotics used during the operation | |||
| No | 360 (68) | 348 (66) | 708 (67) |
| Cement | 129 (24) | 109 (21) | 238 (23) |
| Beads | 36 (6.8) | 69 (13) | 105 (10) |
| Missingf | 2 (0.38) | 1 (0.19) | 3 (0.28) |
| Antibiotics added to the cement during the operation | n = 165 | n = 178 | n = 343 |
| Gentamicin | 86 (52) | 99 (56) | 185 (54) |
| Vancomycin | 29 (18) | 31 (17) | 60 (17) |
| Tobramycin | 5 (3.0) | 12 (6.7) | 17 (5.0) |
| Otherg | 34 (21) | 30 (17) | 64 (19) |
| Missingh | 11 (6.7) | 6 (3.4) | 17 (5.0) |
| Comorbiditiesb,i | |||
| Diabetes | 107 (20) | 98 (19) | 205 (19) |
| Renal failure | 11 (2.1) | 11 (2.1) | 22 (2.1) |
| Ischaemic heart disease | 43 (8.2) | 45 (8.5) | 88 (8.4) |
| Peripheral vascular disease | 31 (5.9) | 32 (6.1) | 63 (6.0) |
| Previous stroke or TIA | 19 (3.6) | 22 (4.2) | 41 (3.9) |
| Dementia | 1 (0.19) | 1 (0.19) | 2 (0.19) |
| Immunosuppressing medication | 28 (5.3) | 17 (3.2) | 45 (4.3) |
| HIV infection (if tested for) | 1 (0.19) | 3 (0.57) | 4 (0.38) |
| Rheumatoid arthritis or systemic autoimmune disease | 47 (8.9) | 38 (7.2) | 85 (8.1) |
| Current smoker | 61 (12) | 79 (15) | 140 (13) |
| Malignancy (curent or diagnosed within the last 2 years) | 17 (3.2) | 17 (3.2) | 34 (3.2) |
Table 7 shows information on histology and microbiology results and the diagnostic certainty of infection as determined by baseline criteria. Although these samples were taken at trial entry, data were collected at the day 42 CRF. Therefore, data for eight participants were missing, as this form was received for only 1046 participants. One further participant, who was withdrawn soon after randomisation, also has missing data for their histology and microbiology. This accounts for nine missing data points in this table. For those participants who did not fulfil the predefined definition for definite infection, an independent blinded committee reviewed the case records to assign categorisation.
| Symptom or sign | Antibiotic | Total (n = 1054)a | |
|---|---|---|---|
| IV (n = 527)a | PO (n = 527)a | ||
| Deep tissue histology resultb | |||
| Infected | 266 (50.47) | 277 (52.56) | 543 (51.52) |
| Equivocal | 13 (2.47) | 17 (3.23) | 30 (2.85) |
| Uninfected | 31 (5.88) | 32 (6.07) | 63 (5.98) |
| Not done | 212 (40.23) | 197 (37.38) | 409 (38.80) |
| Missingc | 5 (0.95) | 4 (0.76) | 9 (0.85) |
| Deep tissue microbiology resultb | |||
| ≥ 2 samples positive with the same organism | 357 (67.74) | 338 (64.14) | 695 (65.94) |
| ≥ 2 samples taken but only 1 sample positive with a given pathogenic organism | 20 (3.80) | 32 (6.07) | 52 (4.93) |
| Only 1 sample taken which is positive for a pathogenic organism via closed biopsy | 25 (4.74) | 30 (5.69) | 55 (5.22) |
| Culture negative | 77 (14.61) | 78 (14.80) | 155 (14.71) |
| ≥ 2 samples taken but only 1 sample positive with a given non-pathogenic organism | 21 (3.98) | 25 (4.74) | 46 (4.36) |
| Not doned | 22 (4.17) | 20 (3.80) | 42 (3.98) |
| Missingc | 5 (0.95) | 4 (0.76) | 9 (0.85) |
| Results from the deep tissue microbiology (when available) | (n = 500) | (n = 503) | (n = 1003) |
| S. aureus presentb | 196 (39.20) | 182 (36.18) | 378 (37.69) |
| Coagulase-negative Staphylococcus presentb | 137 (27.40) | 135 (26.84) | 272 (27.12) |
| Streptococcus species presentb | 72 (14.40) | 73 (14.51) | 145 (14.46) |
| Pseudomonas species presentb | 28 (5.60) | 23 (4.57) | 51 (5.08) |
| Other Gram-negative organism(s) presentb | 84 (16.80) | 84 (16.70) | 168 (16.75) |
| Infection status at presentb | |||
| Definite infection | 478 (90.70) | 476 (90.32) | 954 (90.51) |
| Probable infection | 13 (2.47) | 10 (1.90) | 23 (2.18) |
| Possible infection | 30 (5.69) | 27 (5.12) | 57 (5.41) |
| Infection status unclear | 6 (1.14) | 13 (2.47) | 19 (1.80) |
| Missinge | 0 (0.00) | 1 (0.19) | 1 (0.09) |
Note that there were participants who did not fulfil the definition of infection at baseline in accordance with the protocol but who were treated for infection on clinical grounds. These participants are summarised under ‘infection status unclear’. A decision was made by the trial team to include these participants into the ‘possible infection’ category in all subsequent summaries.
Numbers analysed
The following patient populations were utilised in the analysis:
Intention to treat
All randomised participants were analysed according to their allocated intervention.
Modified intention-to-treat analysis
All randomised participants with both baseline and at least one post-baseline assessment for patient reported outcomes. For all other outcomes, randomised participants with at least one post-baseline assessment. Participants were excluded from this analysis if relevant baseline covariates (when relevant) were not available. In other words, the modified intention-to-treat analysis (MITT) population was the complete-cases subset of the ITT population.
Per protocol
The PP population was defined as all participants who received at least 4 weeks of their randomised strategy and, if in the PO group, did not exceed the limits set for the use of IV antibiotics (i.e. 5 continuous days at any one time). Participants who were recorded to have exited early from their randomised strategy owing to possible or probable recurrence of infection were also included in the PP population. Participants were included in the PP analyses if sufficient outcome and baseline data (when relevant) were available.
Compliance
Treatment compliance
Compliance with the randomised strategy, including early exit, are secondary end points and are summarised in the results section.
Withdrawals and protocol violations
Withdrawals and losses to follow-up
Out of the 1054 randomised participants, 42 (3.98%) were reported as withdrawn or lost to follow-up. Follow-up for these participants ceased for the following reasons:
-
participant withdrew from study, n = 14
-
participant lost/did not attend scheduled clinic visits and was no longer contactable, n = 12
-
patient had died, n = 16.
An additional seven deaths were reported within the acceptable range for the day 365 follow-up. The final follow-up for these participants is not considered missing. These deaths are reported in Serious adverse events.
Additional information on withdrawals and losses to follow-up by treatment arm can be found in the CONSORT statement. End-point data are available for three of these participants.
Protocol violations/deviations
The trial team are not aware of any protocol violations to date. The following 19 protocol deviations occurred:
-
One participant who lacked capacity to provide personal informed consent was recruited in error. The participant was immediately withdrawn from further study related activity and all subsequent data were recorded as missing.
-
One participant was recruited despite having had staphylococcal bacteraemia within the 30 days prior to randomisation. The participant had completed the course of therapy for bacteraemia by the time he was recruited to the trial. He was retained in the trial despite this deviation from the protocol.
-
One participant was randomised on two separate occasions, once in the pilot study and once in the multicentre study. This patient was withdrawn from further study-related activity following realisation of the error and all subsequent data were recorded as missing.
-
Eight participants (four in each arm) were discontinued early from their randomised strategy without an appropriate explanation. In all cases, a change to the prescription arose either as a result of an administrative error or on the advice of a clinician who was not involved with the OVIVA trial.
-
Seven patients randomised to PO therapy switched to their randomised strategy beyond the 7 days allowed from start of treatment episode. The median delay in IV to PO switch from the start of the treatment episode in these patients was 12 days (range 10–19 days).
-
One participant randomised to IV therapy started their IV treatment 8 days after the start of the treatment episode.
Blinding
Blinding was not applicable to the study; participants, clinical staff and the trial team were not blinded to the randomised intervention.
The independent end-point review committee was blinded: end points were assessed based on patient notes provided by trial sites, which were subsequently redacted by the trial staff at Oxford. Only one incident of unblinding was reported (OV1053). This unblinding was accidental and occurred as a result of inadequate redaction of notes. No other issues were reported by the blinded reviews.
Primary analyses
Analysis using multiple imputation utilising all randomised participants
The frequency and proportions of participants experiencing primary end points (i.e. definitive treatment failures as identified by the independent end-point review committee), as well as those for whom the end-point data were missing because of participants withdrawing from the trial or being lost to follow-up prior to the 1-year post randomisation assessment, are shown in Table 8. This summary includes all randomised participants.
| Analysis | Number (rate) of definitive treatment failures, n (%) | Risk difference (90% CI) | |
|---|---|---|---|
| IV antibiotic | PO antibiotic | ||
| ITT population (all randomised participants, N = 1054)a | 74 (14.04) [Missing:b 21 (3.98)] | 67 (12.71) [Missing:b 18 (3.42)] | –1.38% (–4.94% to 2.19%)c |
| MITT subset (all participants with available outcome data, N = 1015)c | 74 (14.62) | 67 (13.16) | –1.46% (–5.03% to 2.11%) |
| All randomised participants, assuming no definite treatment failures for those with missing outcome data (N = 1054)d | 74 (14.04) | 67 (12.71) | –1.33% (–4.78% to 2.12%) |
| PP population (N = 909)e | 69 (15.58) | 61 (13.09) | –2.49% (–6.31% to 1.34%) |
The results from the primary analysis and the supporting analyses are displayed graphically in Figure 2. The non-inferiority margin of 7.5% is indicated by the dashed line.
FIGURE 2.
Forest plot of risk differences (95% CI) by analyses performed (PO vs. IV).

Adjusted logistic regression model
The model uses the occurrence of definite treatment failure as adjudicated by the blinded end-point review committee as the outcome and adjusts for randomised strategy, age, comorbidity (when sufficient observations are available), infecting pathogen and baseline surgical procedure.
The baseline surgical procedures have been categorised as follows:
-
chronic osteomyelitis debrided, no current implant or device
-
discitis/spinal osteomyelitis/epidural abscess debrided
-
chronic osteomyelitis as above but not debrided, or discitis/spinal osteomyelitis/epidural abscess but not debrided
-
implant or device present and retained [i.e. debridement, antibiotics and implant retention (DAIR)]
-
removal of orthopaedic device for infection
-
prosthetic joint implant removed
-
prosthetic joint implant, one-stage revision
-
the OVIVA trial infection criteria not met.
When participants fall into more than one category, they were assigned to the lowest numeric category in the above list. Categories with very low counts were combined with the next (lower) category.
All randomised participants are included in this model by using multiple imputation for missing outcome data. Insufficient incidence of comorbidities were observed for dementia and HIV (human immunodeficiency virus) infection. Infecting pathogen and baseline surgical procedure were categorised as defined in the section on primary end points.
There was no evidence of effect of the randomised strategy on the odds of experiencing a definitive treatment failure during the trial follow-up. The quantile regression models are adjusted for covariates. The covariate adjustment aims to separate the effect of the randomised intervention from other factors that may also have an influence on the odds of participants experiencing a definitive treatment failure during the trial follow-up. Low numbers may have been included in some levels of the categorical explanatory variables; the coefficients therefore have low power and should be interpreted cautiously.
Diagnostic checks demonstrated that the model has limited predictive ability (pseudo-R2 = 4%). However, the main purpose of the model was to obtain an average treatment effect, rather than to obtain accurate predictions for individual participants, and adequate goodness of fit was demonstrated when comparing the average predicted and observed probabilities of treatment failures in either arm. Lowess plots demonstrated linear relationships between the independent variables and the predictors. Investigation of the residuals showed some departure from normality; the majority of the residuals, except those at either end of the range of linear predictions, seemed independent from predicted values.
Time-to-event modelling
To assess any potential bias in the post-randomisation surveillance, which would present as a delay in time to meeting an end point in one randomised group or loss to follow-up or death without an event, a time-to-event analysis was performed.
This analysis focused on the timing of definitive treatment failures and was not adjusted for baseline characteristics. Six participants were withdrawn immediately after randomisation. They are therefore not included in the following summaries.
The data entered under the ‘date of review’ for the day 365 assessment were used as the date at which the follow-up was censored for participants who did not experience a definitive treatment failure and who were not lost to follow-up. In some instances, these reviews were performed retrospectively, and the data entered reflect the timing of the review instead of the date at which data for the relevant participants were reviewed (i.e. a date within the follow-up window for the day 365 visit). Therefore, the time from randomisation to the final assessment was capped at 420 days.
There was no evidence to suggest that the hazard ratio between the treatment arms was statistically significantly different from 1. This suggests that there was no post-randomisation surveillance bias between the trial arms.
The Kaplan Meier curves in Figure 3 show the definitive treatment failure-free time-to-event rates. Again, there does not seem to be any evidence to suggest that the time to definitive treatment failure differed between trial arms.
FIGURE 3.
Kaplan–Meier curves for time to treatment failure by randomised strategy.

The test for the proportional hazards assumption, as well as log-log plots, indicate that the proportional hazards assumption is met (for the majority of the plot, i.e. the time where the majority of treatment failures occur).
Adjustment of p-values for multiple testing
There was no multiple testing, as only a single primary outcome was considered. All additional analyses were undertaken with an intention to further inform the results from the primary analysis. Therefore, significance levels used were 0.05, and 95% CIs were reported.
The DMC reviewed interim summaries and a formal interim analysis; however, it was expected that the DMC would only recommend early stopping if there was a very significantly worse outcome in the PO antibiotic group compared with the IV group (i.e. guided by the Haybittle–Peto stopping boundary). Therefore, the significance level used to determine early termination of the trial is very low (i.e. 0.001) and no formal adjustment of the p-value for the final analysis was considered necessary.
Missing data
Missing data were taken into account in the primary analysis, based on the ITT population, using multiple imputation. This was described in detail in the statistical analysis plan (see Appendix 1).
The multiple imputation and the MITT (complete-cases ITT) analyses make the assumption that data are missing at random. The sensitivity analysis looks at the impact of informatively missing data, assuming that data are missing not at random.
In addition to the assumptions made in the above supporting analyses (i.e. assuming no definitive failures for all participants with missing end-point data), this sensitivity analysis considers two extreme missing not at random assumptions (best-case/worst-case scenarios).
The first missing not at random sensitivity analysis makes the assumption that all participants with missing end-point data in the PO arm had a definitive treatment failure, while those with missing end-point data in the IV arm did not have a definitive treatment failure.
The second missing not at random sensitivity analysis makes the assumption that all participants with missing end-point data in the PO arm had no definitive treatment failure, while those with missing end-point data in the IV arm had a definitive treatment failure.
The sensitivity analyses did not alter the results from the primary trial analysis and, therefore, did not change the overall conclusions of the trial (i.e. that the non-inferiority criteria were met). Therefore, the trial results are robust to missing data.
Prespecified subgroup analysis
All subgroup analyses are based on the MITT population. Subgroup analyses for definite/probable/possible infection at baseline are repeated for the PP population.
Odds ratios were obtained from logistic regression models using definitive treatment failure as the dependent variable, and treatment allocation, the relevant subgroup as well as the interaction term as the only covariates.
The number of treatment failures by treatment arm observed in some of the subgroups were low. Therefore, some of the interaction effects may not be very robust (as indicated by wide CIs) or cannot be included in the plots.
Figure 4 summarises all subgroup analyses, showing the point estimates of the odds ratios, the 95% CIs and the numbers included in the analyses.
FIGURE 4.
Forest plot of OR (95% CI) for subgroup analyses (PO vs. IV). a, Baseline surgical procedure 1 (chronic osteomyelitis debrided, no current implant or device or discitis/spinal osteomyelitis/epidural abscess debrided), baseline surgical procedure 2 (chronic osteomyelitis as above, but not debrided or discitis/spinal osteomyelitis/epidural abscess but not debrided), baseline surgical procedure 3 [implant or device present and retained (i.e. DAIR)], baseline surgical procedure 4 (removal of orthopaedic device for infection or prosthetic joint implant removed) and baseline surgical procedure 5 (prosthetic joint implant, one-stage revision); b, ad hoc subgroup analyses. F/N, Failure/no failure.

Based on the analysis, there was no evidence to suggest a statistically significant difference in the odds of treatment failure between the treatment arms. Odds ratios of > 1 favour IV therapy (i.e. indicate that the odds of experiencing a treatment failure in the PO arm were higher than the odds in the IV arm), whereas odds ratios of < 1 favour PO therapy.
Prespecified subgroup analysis considering infection subgroups at randomisation
Subgroup analysis of definite versus probable/possible infection at baseline
Modified intention-to-treat analysis analysis (complete-cases intention to treat)
A total of 1015 participants were included in this subgroup analysis.
The odds ratio of definitive treatment failures (PO vs. IV) in those with definitive infection at baseline was approximately 0.91, and the odds ratio for those with probable or possible infection was 0.56. Figure 4 shows that the CIs for both odds ratios cross 1. The results for the probable/possible infection subgroup shows a lot of uncertainty owing to the small numbers included into this analysis.
The overall interaction heterogeneity p-value is 0.531, indicating that there is no statistically significant difference in the treatment effect between the subgroups.
Per-protocol analysis
A total of 909 participants were included in this subgroup analysis.
The odds ratio of definitive treatment failures (PO vs. IV) in those with definitive infection at baseline was approximately 0.84, and the odds ratio for those with probable or possible infection was 0.56. CIs for both odds ratios cross 1. The results for the probable/possible infection subgroup shows a lot of uncertainty owing to the small numbers included into this analysis.
The overall interaction heterogeneity p-value is 0.612, indicating that there is no evidence that the interaction between randomised treatment and the subgroups is statistically significantly different from 1.
Subgroup analysis of definite/probable versus possible infection at baseline
Modified intention-to-treat analysis analysis (complete-cases intention to treat)
Data for 1015 participants are included in this subgroup analysis.
The odds ratio of definitive treatment failures (PO vs. IV) in those with definitive or probable infection at baseline was approximately 0.89, and the odds ratio for those with possible infection was 0.94. CIs for both odds ratios cross 1. The results for the probable/possible infection subgroup shows a lot of uncertainty owing to the small numbers included into this analysis.
The overall interaction heterogeneity p-value is 0.955, indicating that there is no evidence that the interaction between randomised treatment and the subgroups is statistically significantly different from 1.
Per-protocol analysis
Data for 909 participants are included in this subgroup analysis.
The odds ratio of definitive treatment failures (PO vs. IV) in those with definitive or probable infection at baseline was approximately 0.80, and the odds ratio for those with possible infection was 1.08. Results for the probable/possible infection subgroup shows a lot of uncertainty owing to the small numbers included into this analysis.
The overall interaction p-value is 0.782, indicating that there is no evidence that the interaction between randomised treatment and the subgroups is statistically significantly different from 1.
Prespecified subgroup analysis considering the baseline surgical procedure
Subgroup analysis was used to determine the consistency of treatment effects by the baseline surgical procedure. Information on the type of infection was collected at the enrolment of trial participants and categorised as follows:
-
baseline surgical procedure 1 – chronic osteomyelitis debrided, no current implant or device or discitis/spinal osteomyelitis/epidural abscess debrided
-
baseline surgical procedure 2 – chronic osteomyelitis as above, but not debrided or discitis/spinal osteomyelitis/epidural abscess but not debrided
-
baseline surgical procedure 3 – implant or device present and retained (i.e. DAIR)
-
baseline surgical procedure 4 – removal of orthopaedic device for infection or prosthetic joint implant removed
-
baseline surgical procedure 5 – prosthetic joint implant, one-stage revision.
Results from a logistic regression model (ITT population) with the occurrence of the primary end point (i.e. definite treatment failure as adjudicated by the blinded end-point review committee) as the outcome, and the randomised treatment as well as the baseline surgical procedure (as a five-level categorical variable) and the interaction between randomised treatment and baseline surgical procedure as explanatory variables are presented.
The interaction model does not indicate that any of the treatment/baseline surgical procedure interactions are likely to be significant.
The overall interaction heterogeneity p-value is 0.263, indicating that there is no evidence that the interaction between randomised treatment and the subgroups was statistically significantly different from 1.
In an additional post hoc subgroup analysis, restricted to metal retained versus not retained, we included participants from the following baseline surgical procedure categories:
-
No metal retained –
-
chronic osteomyelitis debrided, no current implant or device
-
removal of orthopaedic device for infection
-
prosthetic joint implant removed.
-
-
Metal retained –
-
implant or device present and retained (i.e. DAIR)
-
prosthetic joint implant, one-stage revision.
-
A total of 928 participants were included in this subgroup analysis.
The overall interaction heterogeneity p-value is 0.120, indicating that there is no evidence that the interaction between randomised treatment and the subgroups was statistically significantly different from 1.
Prespecified subgroup analysis considering the infecting pathogen
Subgroup analysis was used to determine the consistency of treatment effects by infecting pathogen.
Information on the following five infecting pathogens, and if there was no pathogen, was collected:
-
S. aureus
-
Pseudomonas species
-
Gram-negative organism(s) (other than Pseudomonas)
-
Streptococcus species
-
coagulase negative Staphylococcus
-
no infecting pathogen identified.
When evidence for more than one of the above pathogens was present on the deep tissue microbiology results taken prior to randomisation, they were assigned to the lowest numeric category in the above list. The infecting pathogen was therefore a single variable with six levels.
The above categories for the infecting pathogens were chosen as part of a pragmatic approach and included the main causative organism categories. It was felt that insufficient numbers of patients would be available for other infecting pathogens to enable meaningful statistical subgroup analysis.
A total of 1015 participants (i.e. all participants with valid end-point data) were included in this summary. Participants without an identified infecting pathogen were categorised as no infecting pathogen identified. As no failures occurred in the PO arm of the Pseudomonas spp. subgroup, an odds ratio could not be calculated for this group.
The overall interaction heterogeneity p-value was 0.295, indicating that there is no evidence that the interaction between randomised treatment and the subgroups was statistically significantly different from 1.
In the prespecified subgroup analysis, the point estimate suggests that IV therapy may confer an advantage for patients in whom no infecting pathogen was identified.
Therefore, an additional post hoc analysis was performed to investigate the odds of definitive treatment failure by treatment arm in participants with any pathogen compared with no pathogen identified.
The overall interaction heterogeneity p-value is 0.069, indicating that there is no evidence of an interaction between randomised treatment and whether or not the pathogen was known.
Table 9 summarises the use of antibiotics according to whether the pathogen was known or unknown. These data were available for 1011 participants.
| Antibiotics used | Antibiotics, n (%) | Total (N = 1011), n (%) | |||
|---|---|---|---|---|---|
| IV | PO | ||||
| Known pathogen (N = 391) | Unknown pathogen (N = 113) | Known pathogen (N = 396) | Unknown pathogen (N = 111) | ||
| Glycopeptides (IV) used | 141 (36.06) | 65 (57.52) | 17 (4.29) | 4 (3.60) | 227 (22.45) |
| Penicillins (IV) used | 27 (6.91) | 7 (6.19) | 5 (1.26) | 3 (2.70) | 42 (4.15) |
| Cephalosporins (IV) used | 144 (36.83) | 26 (23.01) | 4 (1.01) | 2 (1.80) | 176 (17.41) |
| Carbapenems (IV) used | 37 (9.46) | 3 (2.65) | 5 (1.26) | 0 (0.00) | 45 (4.45) |
| Other single IV antibiotic used | 28 (7.16) | 7 (6.19) | 1 (0.25) | 1 (0.90) | 37 (3.66) |
| Combination IV antibiotics used | 29 (7.42) | 6 (5.31) | 5 (1.26) | 1 (0.90) | 41 (4.06) |
| Penicillins (PO) used | 7 (1.79) | 1 (0.88) | 61 (15.40) | 20 (18.02) | 89 (8.80) |
| Quinolones (PO) used | 24 (6.14) | 7 (6.19) | 146 (36.87) | 39 (35.14) | 216 (21.36) |
| Tetracyclines (PO) used | 1 (0.26) | 3 (2.65) | 44 (11.11) | 11 (9.91) | 59 (5.84) |
| Macrolides/lincosamide (PO) used | 7 (1.79) | 3 (2.65) | 51 (12.88) | 15 (13.51) | 76 (7.52) |
| Other single PO antibiotic (PO) used | 5 (1.28) | 5 (4.42) | 42 (10.61) | 11 (9.91) | 63 (6.23) |
| Combination PO antibiotics (PO) used | 6 (1.53) | 5 (4.42) | 65 (16.41) | 19 (17.12) | 95 (9.40) |
This exploratory analysis demonstrates that in the IV arm, glycopeptides were the antibiotic category of choice when the infecting pathogen was not identified.
Prespecified subgroup analysis considering the intended and actual antibiotic choice
A subgroup analysis considered the clinician’s specific antibiotic intentions, as recorded prior to randomisation, as a categorical variable. The antibiotic intentions were categorised into the following groups based on the intended drug (Table 10). The rationale for this was to ensure that participants should have options for both IV therapy and PO therapy, thus demonstrating true equipoise from the infection specialist over the effectiveness of the two trial arms at the point of randomisation.
| Treatments | |
|---|---|
| Planned IV | Planned PO |
|
|
The results of the logistic regression models using the occurrence of the primary end points (i.e. definite treatment failure as adjudicated by the blinded end-point review committee) as the outcome, and the randomised treatment as well as the subcategory of the antibiotic intention and the interaction between the two variables are displayed below. Separate analyses are shown for the planned IV and planned PO treatments.
Intravenous and PO intentions were not documented for all participants, as it was not a requirement in the initial single-centre pilot study. Only those participants of the MITT population with available IV and PO plans were included into these analyses (913 participants).
Planned intravenous treatments
Of the 917 participants for whom relevant data were available, 380 were planned to receive glycopeptides if randomised to IV therapy. Of these, 216 were subsequently randomised to IV therapy and 164 were randomised to PO therapy. The asymmetry between the arms for missing data might suggest that these data fields were not reliably completed prior to randomisation.
The overall interaction heterogeneity p-value is 0.416, indicating that there is no evidence that the interaction between randomised treatment and the subgroups was statistically significantly different from 1.
Planned oral treatments
Of the 945 participants for whom relevant data were available, 131 were planned to receive penicillins if randomised to PO therapy. Of these, 57 were subsequently randomised to IV therapy and 74 were randomised to PO therapy. The asymmetry between the arms for missing data might suggest that these data fields were not reliably completed prior to randomisation.
The overall interaction p-value is 0.800, indicating that there is no evidence that the interaction between randomised treatment and the subgroups is statistically significantly different from 1.
Inclusion of rifampicin into the planned intravenous and oral choices
According to the available data, adjunctive rifampicin was included in the planned IV regimen in 142 partipants. Of these, 73 were subsequently randomised to IV therapy and 69 were randomised to PO therapy. The overall interaction heterogeneity p-value is 0.876, indicating that there was no evidence that the interaction between randomised treatment and the subgroups is statistically significantly different from 1.
Rifampicin was included in the planned PO regimen in 487 participants. Of these, 228 were subsequently randomised to IV therapy and 259 were randomised to PO therapy.
The overall interaction heterogeneity p-value is 0.122, indicating that there is no evidence that the interaction between randomised treatment and the subgroups was statistically significantly different from 1.
Treatment by peripheral vascular disease interaction
A final post hoc subgroup analysis looked at the effect of peripheral vascular disease as recorded at randomisation. This factor was identified as being associated with the outcome in the adjusted logistic regression analysis performed as part of the supporting analyses.
A total of 1015 participants (i.e. all participants with valid end-point data) were included in this summary.
The overall interaction heterogeneity p-value is 0.467, indicating that there was no evidence of an interaction between randomised treatment and whether or not peripheral vascular disease was present.
Adverse events and complications
Clostridium difficile
Summaries for episodes of C. difficile include all participants for whom at least one follow-up assessment was entered onto the database (Table 11).
| Episode | Antibiotic | Total (n = 1046) | |
|---|---|---|---|
| IV (n = 523) | PO (n = 523) | ||
| Episode of C. difficilea | 9 (1.72) | 5 (0.96) | 14 (1.34) |
Information on C. difficile was collected on the day 42 and day 120 CRFs. Day 42 forms were received for 1046 participants; all were included in this analysis. The day 120 form was received for a subset of these participants.
C. difficile data were missing for three participants: two in the IV arm and one in the PO arm. Two of these participants were withdrawn prior to their day 42 follow-up, and one died; therefore, the relevant information was not available for these participants (risk difference –0.8%, 95% CI –2.2% to 0.6%).
Using all participants with non-missing data (MITT population, n = 1043), there was no evidence of an association between randomised strategy and the occurrence of episodes of C. difficile (p-value = 0.298, using Fisher’s exact test).
Serious adverse events
All reported and confirmed SAEs were included in the summaries reported in Tables 12 and 13.
| SAEs | Antibiotic | Total (n = 1046) | |
|---|---|---|---|
| IV (n = 527) | PO (n = 527) | ||
| SAE reporteda | 146 (27.70) | 138 (26.19) | 284 (26.94) |
| Number of SAEs reporteda | |||
| 0 | 381 (72.30) | 389 (73.81) | 770 (73.06) |
| 1 | 109 (20.68) | 89 (16.89) | 198 (18.79) |
| 2 | 20 (3.80) | 29 (5.50) | 49 (4.65) |
| 3 | 9 (1.71) | 7 (1.33) | 16 (1.52) |
| 4 | 4 (0.76) | 10 (1.90) | 14 (1.33) |
| 5 | 1 (0.19) | 2 (0.38) | 3 (0.28) |
| 6 | 2 (0.38) | 1 (0.19) | 3 (0.28) |
| 11 | 1 (0.19) | 0 (0.00) | 1 (0.09) |
| SAE information | Antibiotic | Total (n = 444) | |
|---|---|---|---|
| IV (n = 220) | PO (n = 224) | ||
| Timing of SAE onset from randomisation (in weeks)a | 18 (4, 36), (0, 57) | 16 (4, 35), (0, 56) | 17 (4, 35), (0, 57) |
| SAE expecteda | |||
| Yesb | 220 (100.00%) | 224 (100.00%) | 444 (100.00%) |
| SAE related to randomised interventiona | |||
| No | 218 (99.09%) | 220 (98.21%) | 438 (98.65%) |
| Yes | 2 (0.91%) | 4 (1.79%) | 6 (1.35%) |
| SAE outcomea | |||
| Resolved | 154 (70.00%) | 172 (76.79%) | 326 (73.42%) |
| Ongoingc | 10 (4.55%) | 19 (8.48%) | 29 (6.53%) |
| Resolved with sequelae | 39 (17.73%) | 27 (12.05%) | 66 (14.86%) |
| Death | 17 (7.73%) | 6 (2.68%) | 23 (5.18%) |
| SAE severitya | |||
| Mild | 56 (25.45%) | 43 (19.20%) | 99 (22.30%) |
| Moderate | 119 (54.09%) | 123 (54.91%) | 242 (54.50%) |
| Severe | 45 (20.45%) | 58 (25.89%) | 103 (23.20%) |
Table 14 shows details for the SAEs reported in relation to randomisation.
| ID number | Randomised treatment | Date of | SAE description as documented on the trial database | Outcome | Timing (weeks) | |
|---|---|---|---|---|---|---|
| Randomisation | SAE onset | |||||
| 2113 | PO | 22 October 2013 | 30 October 2013 | Admitted with nausea and vomiting. Had brief break from PO ciprofloxacin, which was then restarted | Resolved | 1 |
| 2386 | PO | 1 July 2014 | 6 July 2014 | Diarrhoea, nausea, vomiting and exhaustion associated with (liquid) PO antibiotics. Admitted for symptomatic therapy and switched to IV ceftriaxone | Resolved | 0 |
| 2664a | PO | 1 May 2015 | 24 June 2015 | Left arm swelling investigated for midline infection; no infection or line complication found | Resolved | 7 |
| 2694b | IV | 11 June 2015 | 16 August 2015 | Reaction to doxycycline: burning lower leg and blisters on feet. Widespread pruritus | Resolved with sequelae | 9 |
| 2701c | IV | 17 June 2015 | 26 June 2015 | Severe oesophagitis thought to be attributable to PO antibiotics | Resolved | 1 |
| 2796 | PO | 18 September 2015 | 22 September 2015 | Participant had an unplanned admission as a result of intolerance of PO antibiotics. Symptoms were loose stools and a reduction in the effectiveness of the participant’s methadone | Resolved | 0 |
The frequency of line complications
The following summaries refer primarily to participants randomised to the IV strategy; therefore, no statistical tests were performed (Table 15).
| Line complications | Antibiotic | Total (n = 1046a) | |
|---|---|---|---|
| IV (n = 523) | PO (n = 523) | ||
| IV line detailsb | |||
| Not present | 64 (12.24) | 489 (93.50) | 553 (52.87) |
| PICC | 450 (86.04) | 30 (5.74)c | 480 (45.89) |
| Hickman | 5 (0.96) | 1 (0.19) | 6 (0.57) |
| Otherd | 2 (0.38) | 2 (0.38) | 4 (0.38) |
| Missing | 2 (0.38) | 1 (0.19) | 3 (0.29) |
| Line complicationsb | 49 (9.37) | 5 (0.96) | 54 (5.16) |
| Nature of line complicationsb | (n = 49) | (n = 5e) | (n = 54) |
| Mechanical failure | 24 (48.98) | 3 (60.00) | 27 (50.00) |
| Thrombophlebitis/thrombosis | 13 (26.53) | 1 (20.00) | 14 (25.93) |
| Infection | 12 (24.49) | 1 (20.00) | 13 (24.07) |
| Line removed as result of line complicationsb | (n = 49) | (n = 5e) | (n = 54) |
| Yes | 42 (85.71) | 4 (80.00) | 46 (85.19) |
| Replacement of line after removal | (n = 42) | (n = 4) | (n = 46) |
| Yes | 18 (42.86) | 4 (100.00) | 22 (47.83) |
Information on line complications was collected on the day 42 and day 120 CRFs. Day 42 forms were received for 1046 participants and these are all included in Table 15. The day 120 form was received for a subset of these participants.
Information on line complications was missing for three participants: two in the IV arm and one in the PO arm. Two of these participants were withdrawn prior to their day 42 follow-up, and one died; therefore, the relevant information is not available for these participants.
Five line complications were reported in the PO arm of the trial. Of these, four related to participants who exited early from their allocated treatment strategy and were treated with IV therapy. One further line complication in the PO arm arose in relation to a planned second stage procedure, which took place after the completion of the 6 weeks’ randomised strategy.
Early termination of the planned 6-week strategy
Information on early termination from the allocated treatment strategy was collected on the day 42 and day 120 CRFs.
A total of 1046 participants had at least one of these forms available, and they are included in the following summaries (Table 16).
| Reasons for early exit from allocated treatment strategya | Antibiotic | Total [166/1046 (15.87%)] | |
|---|---|---|---|
| IV [99/523 (18.93%)] | PO [67/523 (12.81%)] | ||
| Intolerance | 26 (26.26) | 23 (34.33) | 49 (29.52) |
| Patient preference | 19 (19.19) | 5 (7.46) | 24 (14.46) |
| Difficulties with IV access or administration | 41 (41.41) | 0 (0.00) | 41 (24.70) |
| Intercurrent illness | 2 (2.02) | 8 (11.94) | 10 (6.02) |
| Due to possible or probable recurrenceb | 1 (1.01) | 15 (22.39) | 16 (9.64) |
| Good clinical response | 1 (1.01) | 0 (0.00) | 1 (0.60) |
| Otherc | 9 (9.09) | 15 (22.39) | 24 (14.46) |
| Reason not available | 0 (0.00) | 1 (1.49) | 1 (0.60) |
Information on early exits was missing for three participants. Two of these were withdrawn prior to their day 42 follow-up and one died; therefore, the relevant information was not available for these participants.
Pearson’s chi-squared test (H0: no association between treatment and early exit) suggests that there was evidence of an association between randomised treatment arm and early exit from the allocated strategy (p = 0.006).
Quality of life evaluated by the EuroQol-5 Dimensions, three-level version questionnaire
The EQ-5D-3L index ranges from –0.594 to 1, with higher values indicating better health states and zero indicating a health state equivalent to death.
The EQ-5D VAS ranges from 0 to 100, with higher values indicating better health states.
The results for the quantile regression on the EQ-5D-3L index and VAS showed no evidence of an effect of randomised strategy on the median outcome at any of the follow-up time points.
The quantile regression models were adjusted for a number of covariates, as outlined in the statistical analysis plan. The covariate adjustment aimed to separate the effect of the randomised intervention from other factors which may also have an influence on the EQ-5D-3L and EQ-5D VAS at follow-up.
Similarly, there was no evidence of an effect of randomised strategy on the median OHS outcomes at any follow-up time point. However, there was evidence to suggest that the randomised strategy has a statistically significant effect on the median outcome of the OKS at both the day 120 and day 365 follow-up in favour of PO therapy.
The quantile regression models were adjusted for a number of covariates, as outlined in the statistical analysis plan. The covariate adjustment aimed to separate the effect of the randomised intervention from other factors that may also have had an influence on the OHS and OKS at follow-up.
Adherence to oral medication
The MEMS was used in a subset of sites (Oxford University Hospitals, Guy’s and St. Thomas’ Hospitals, Royal Free London and Royal National Orthopaedic Hospital). MEMS caps were returned by 63 participants allocated to the PO arm. Compliance with PO therapy at 42 days according to MEMS data ranged from 45% to 100%.
Antibacterial agents used for treatment
This section presents data on the antibiotic regimens received by participants during the first 42 days of the trial.
Data for 1044 participants were available for these summaries (Table 17). No antibiotic data were available for five trial participants and, for an additional five participants, insufficient antibiotic information was available to categorise their antibiotic regimen.
| Antibiotic regimena | Antibiotic | Total (n = 1044) | |
|---|---|---|---|
| IV (n = 521) | PO (n = 523) | ||
| Glycopeptides (IV) used | 214 (41.07) | 22 (4.21) | 236 (22.61) |
| Penicillins (IV) used | 38 (7.29) | 11 (2.10) | 49 (4.69) |
| Cephalosporins (IV) used | 173 (33.21) | 8 (1.53) | 181 (17.34) |
| Carbapenems (IV) used | 41 (7.87) | 5 (0.96) | 46 (4.41) |
| Other single IV antibiotic used | 35 (6.72) | 2 (0.38) | 37 (3.54) |
| Combination IV antibiotics used | 35 (6.72) | 6 (1.15) | 41 (3.93) |
| Penicillins (PO) used | 8 (1.54) | 83 (15.87) | 91 (8.72) |
| Quinolones (PO) used | 33 (6.33) | 191 (36.52) | 224 (21.46) |
| Tetracyclines (PO) used | 4 (0.77) | 57 (10.90) | 61 (5.84) |
| Macrolides/lincosamide (PO) used | 10 (1.92) | 68 (13.00) | 78 (7.47) |
| Other single PO antibiotic used | 10 (1.92) | 54 (10.33) | 64 (6.13) |
| Combination PO antibiotics used | 13 (2.50) | 87 (16.63) | 100 (9.58) |
The categories in this table were not mutually exclusive; participants could fall into more than one category. A total of 145 participants fell into two antibiotic categories, three participants fell into three categories and one participant fell into four categories.
All participants in the PO arm who received IV antibiotics were early exits from their randomised strategy. All participants in the IV arm who received PO antibiotics were early exits from their randomised strategy or were on adjunctive PO therapy.
Figure 5 shows the proportion of participants on IV antibiotic therapy on each day from the start of treatment episode through to day 60, by treatment arm. As expected according to the trial protocol, the figure shows a marked decline in IV use around day 7 in the PO arm and around day 42 in the IV arm. Participants who were randomised to PO therapy but were receiving IV therapy after day 7 represent either early exits from strategy, permissible short-term IV therapy for concomittant illness or, in seven cases, protocol deviation. Two further apparent protocol deviations were likely to be a result of data entry error.
FIGURE 5.
Proportion of participants on IV antibiotics from start of treatment episode through to day 60.
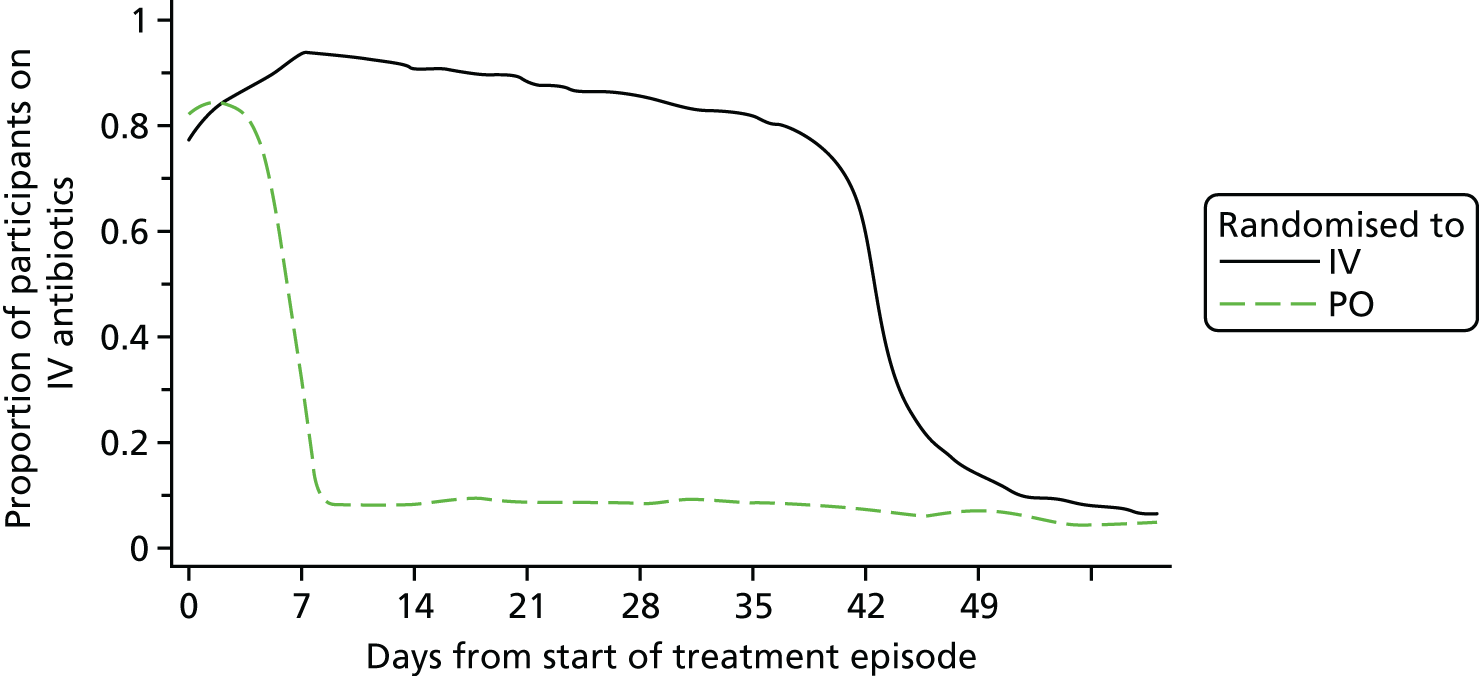
The number of patients continuing long-term antibiotic treatment (after 6 weeks) and time to permanent discontinuation of all antibiotic treatment (defined as the first day when antibiotics are not taken for the next 14 days) are displayed in Table 18 and Figure 6. Antibiotic use was capped at 400 days when use was recoded beyond that period.
| Long-term use | Antibiotic | Total (n = 1049) | |
|---|---|---|---|
| IV (n = 523) | PO (n = 526) | ||
| Antibiotic treatment continued beyond 6 weeksa | |||
| No | 139 (26.58) | 125 (23.76) | 264 (25.17) |
| Yes | 384 (73.42) | 401 (76.24) | 785 (74.83) |
| Duration of antibiotic useb | 78 (42–99) (1–400) | 71 (43–94) (2–400) | 76 (42–96) (1–400) |
FIGURE 6.
Time to permanent discontinuation of antibiotics.
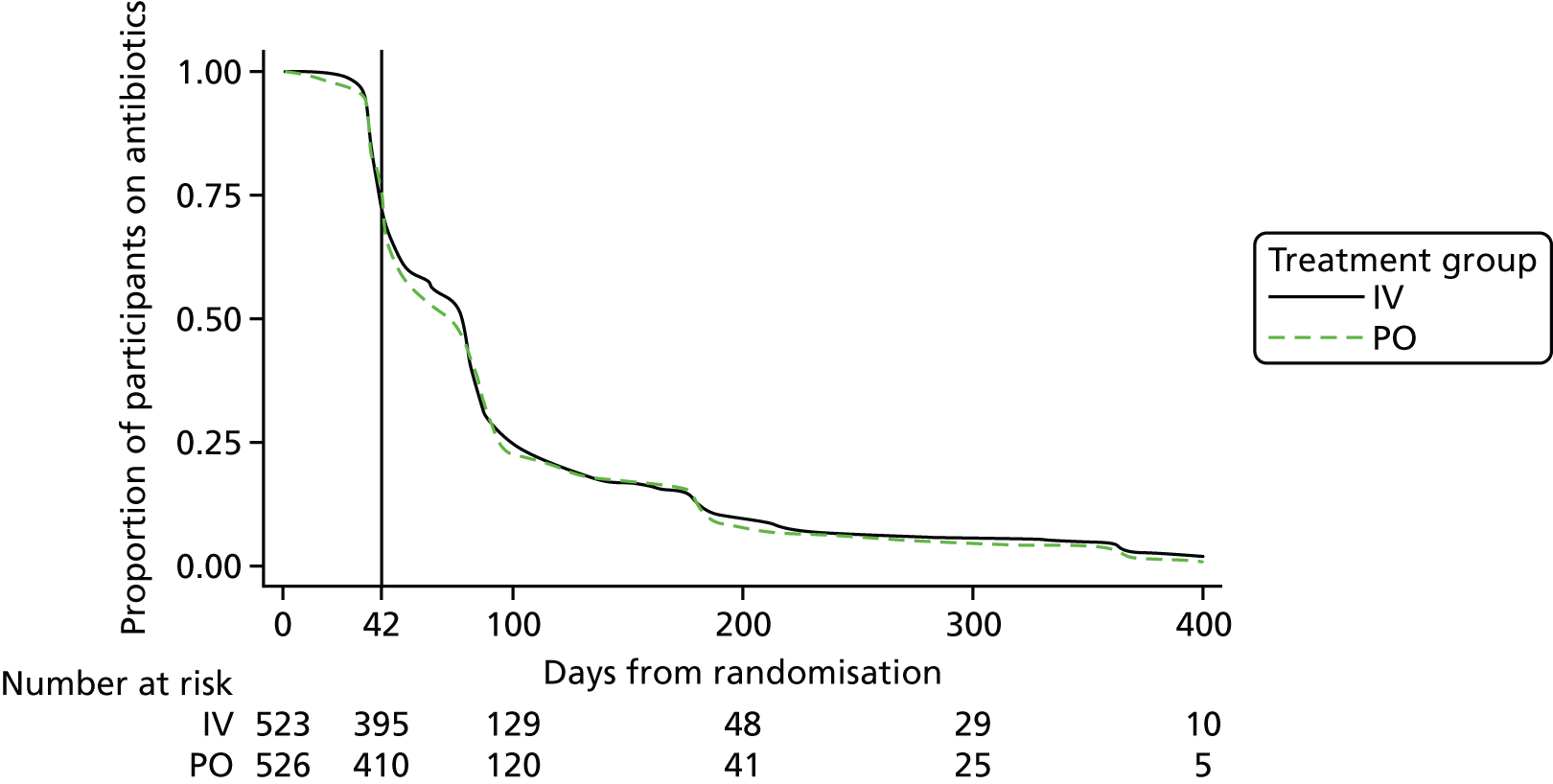
Using the Wilcoxon rank-sum test, there was no evidence of effect of randomised strategy on the median long-term use of antibiotics between the treatment arms (p = 0.628).
Agreement between intended and received antibiotics
This section presents agreements between the planned PO and IV antibiotics as stated prior to randomisation and actual antibiotic administered. Included in the summaries are participants for whom both the intended and actual antibiotic choices were available (Table 19). For one participant, the information provided was insufficient for categorisation.
| Compliancea | Antibiotic | Total (n = 1044) | |
|---|---|---|---|
| IV (n = 521) | PO (n = 523) | ||
| Complete match | 370 (71.02) | 374 (71.51) | 744 (71.26) |
| Partial match | 68 (13.05) | 90 (17.21) | 158 (15.13) |
| No match | 83 (15.93) | 58 (11.09) | 141 (13.51) |
| Missingb | 0 (0.00) | 1 (0.19) | 1 (0.10) |
Summaries are categorised as follows.
-
Full match: received their randomised strategy and remained within the intended antibiotic group.
-
Partial match: received their randomised strategy but deviated from the intended antibiotic group.
-
No match: received < 50% of planned therapy within randomised strategy.
Note that the definition for the ‘no match’ category is different from that in the approved statistical analysis plan (version 2.0). Originally, this category was defined as ‘early exit from randomised strategy’. The updated definition was felt to be more accurate and clinically relevant.
Duration of primary hospital stay
Time from randomisation to discharge is summarised in Figure 7. This summary excludes participants who were treated as outpatients (length of stay of zero) and those who died during their initial hospital stay. Note that readmission post discharge was recorded as a SAE and represented a secondary end point.
FIGURE 7.
Time from randomisation to discharge.
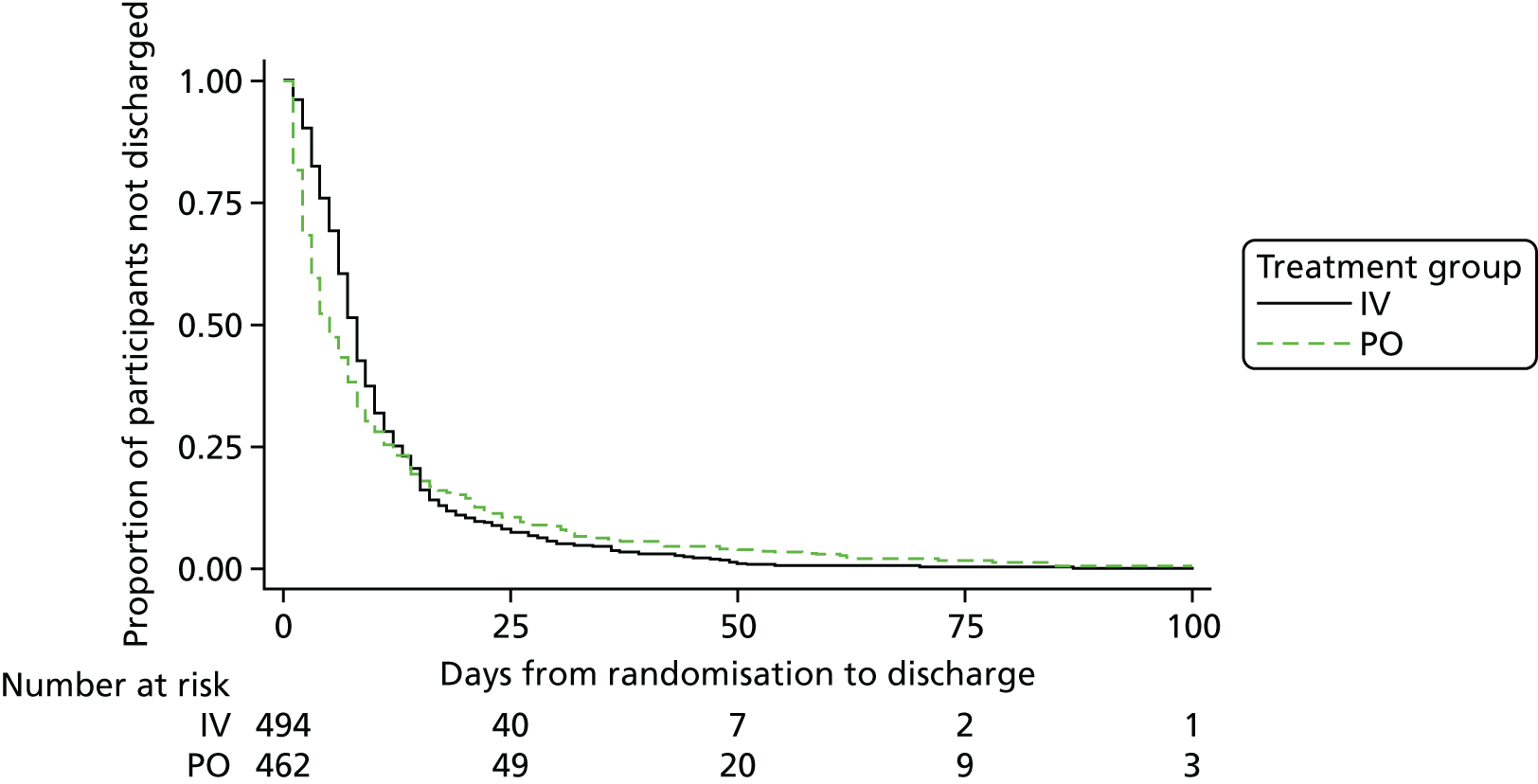
Using the Wilcoxon rank-sum test, there is sufficient evidence to suggest a difference in the median time from randomisation to discharge between the treatment arms (p < 0.001).
Results of health economics analysis
Results from the trial indicate that PO antibiotics are non-inferior to IV antibiotics. Results are presented for complete cases as well as using the ITT population, for which missing values were replaced using imputation methods.
Missing data
The effects of missing data were explored using both mean and multiple imputation. Missing cost values were replaced at the aggregate total cost level using both mean imputation and multiple imputation. Missing quality-of-life data were replaced at utility score level at each EQ-5D-3L follow-up point using multiple imputation.
Resource use
Only a small proportion of patients had missing resource use data (IV arm, n = 12; PO arm, n = 14). Table 20 shows data, for any indication, from incident admission through to 1 year of follow-up for the mean number of antibiotic prescriptions, antibiotic duration in days, mean number of inpatient admissions, the mean length of stay as inpatient and the total number of days that a patient received IV therapy. The antibiotic duration sums the duration of all antibiotic use, including simultaneous use. The IV duration includes the length of IV episodes for which an IV line was needed to administer IV antibiotics (including more than one IV antibiotic taken at the same time as another).
| Resource type | Antibiotic | Difference | 95% CI | |||
|---|---|---|---|---|---|---|
| IV | PO | |||||
| Mean (SD) | n (%) | Mean (SD) | n (%) | |||
| Number of antibiotic prescriptions | 6.70 (3.74) | 515 (97.7) | 6.43 (3.93) | 513 (97.3) | 0.276 | –0.19 to 0.74 |
| Antibiotic duration (days) | 189.8 (177.5) | 515 (97.7) | 185.6 (156.3) | 513 (97.3) | 4.18 | –16.29 to 24.65 |
| Number of inpatient admissions | 1.83 (1.15) | 515 (97.7) | 1.82 (1.11) | 513 (97.3) | 0.01 | –0.12 to 0.14 |
| Inpatient duration (days) | 26.22 (24.28) | 515 (97.7) | 26.35 (28.47) | 513 (97.3) | –0.125 | –3.36 to 3.11 |
| Total number of days IV therapy was received | 52.58 (40.37) | 515 (97.7) | 17.96 (33.52) | 513 (97.3) | 34.62 | 30.08 to 39.16 |
There were no statistically significant differences between arms for antibiotic prescriptions and duration, number of inpatient stays or total inpatient duration over 1 year. However, there was a statistically significant difference in the mean number of days’ IV therapy was received. On average, the total number of days that IV therapy was received was 34.62 days longer in the IV arm than in the PO arm. Table 21 presents the mean costs in both arms for unadjusted complete cases.
| Cost category | Antibiotics | Difference | 95% CI | p-value | |||
|---|---|---|---|---|---|---|---|
| IV | PO | ||||||
| Mean (SD) | n (%) | Mean (SD) | n (%) | ||||
| Antibiotics | £1992 (£2545) | 515 (97.7) | £1207 (£2043) | 513 (97.3) | £785 | £502 to £1067 | < 0.01 |
| Inpatient stays | £7756 (£7183) | 515 (97.7) | £7793 (£8420) | 513 (97.3) | –£37 | –£995 to 920 | 0.94 |
| IV costs | £3527 (£2920) | 515 (97.7) | £1548 (£1618) | 513 (97.3) | £1979 | £1690 to £2268 | < 0.01 |
| Total costs (excluding surgical costs) | £13,275 (£10,113) | 515 (97.7) | £10,549 (10,371) | 513 (97.3) | £2727 | £1473 to £3980 | < 0.01 |
For unadjusted complete cases, the total mean non-surgical cost was £13,275 in the IV arm compared with £10,549 in the PO arm. The observed difference in mean total cost between arms was £2727, a statistically significant result. The mean cost differences for antibiotics and IV costs were also statistically significant, but there was no statistically significant difference in inpatient costs between the IV and PO arm over 1 year.
To explore the difference in costs between IV and PO antibiotics for a 42-day course, trial results were used to calculate the mean daily cost of all antibiotics in each arm. The mean cost of a 42-day (6-week) course of antibiotics (antibiotics only) was £997 [standard deviation (SD) £873] for IV antibiotics and £188 (SD £648) for PO antibiotics.
Health outcomes: quality-adjusted life-years
The proportion of available data for each EQ-5D-3L questionnaire is presented in Table 22. These values include zero utility scores after death for deceased participants.
| Time point | Antibiotic | |
|---|---|---|
| IV (%) | PO (%) | |
| Baseline | 73.2 | 73.6 |
| 14 days | 58.4 | 58.6 |
| 6 weeks | 69.4 | 71.0 |
| 4 months | 59.2 | 58.1 |
| 12 months | 57.1 | 54.3 |
The complete-case EQ-5D-3L questionnaire results at dimension level showed that, in all domains, there was a lower proportion of participants in levels 2 and 3 at the 365-day follow-up than earlier follow-ups. This indicated that there was improvement in all aspects of the EQ-5D-3L from mobility through to anxiety, and this was seen in both the IV and PO arms.
The data for mean EQ-5D-3L utilities at baseline and at 14, 42, 120 and 365 days, along with mean QALYs, showed that there were no statistically significant differences in mean utilities at any follow-up point or in mean QALYs. Results consider a zero utility score for patients who died during the trial. 29 The utilities in both arms improved at each follow-up point compared with the previous follow-up point. Available data percentages ranged from 73.6% at baseline to 54.3% at the 365-day follow-up.
Imputation results
Mean and multiple imputation was carried out for total costs and are presented in Table 23.
| Total costs | Antibiotic (mean costs) | Difference (SE) | 95% CI | |
|---|---|---|---|---|
| IV | PO | |||
| Mean imputation results | ||||
| Total costs | £13,141 (SD £10,036) | £10,406 (SD £10,269) | £2735 (£625) | £1508 to £3963 |
| Multiple imputation results | ||||
| Total costs | £13,274 (SE £446) | £10,534 (SE £453) | £2740 (£638) | £1488 to £3992 |
| Total QALYs | 0.537 (SE 0.013) | 0.545 (SE 0.015) | –0.007 (0.019) | –0.05 to 0.03 |
Results for both mean and multiple imputation were consistent with the results from the base-case complete-case analysis; the mean cost differences for mean and multiple imputations were £2735 and £2740, respectively, compared with £2727 for complete-case analysis. All of these results showed a statistically significant difference between arms. The results of the multiple imputation for QALYs show a difference of –0.007 between arms, compared with 0.023 for complete cases. Neither of these results were statistically significant.
Sensitivity analysis
The results of the sensitivity analysis were similar to the base-case results, with a difference in total costs between arms of £2617–2887, compared with £2727 in the complete-case results.
Cost-effectiveness
Mean costs were observed to be lower in the PO arm and mean QALYs were higher in the PO arm than the IV arm, suggesting that the strategy of treating bone and joint infections with PO antibiotics is a dominant strategy. However, there is uncertainty surrounding this result, which is explored further in the next section.
Uncertainty
Although the difference in costs between strategies was found to be statistically significant, there is uncertainty around the magnitude of this difference and we can be 95% confident that this difference is between £1488 and £3992. The difference between QALYs is not statistically significant and results suggest that we can be 95% confident that the real difference in total QALYs between treatment arms is between –0.045 and 0.031 and, therefore, favours neither strategy.
Figure 8 shows the cost-effectiveness plane with 1000 bootstrap samples of the ICER. It also shows a point estimate giving the mean differences in costs and QALYs between treatment arms. This figure also includes a line showing the £30,000 threshold currently used to assess cost-effectiveness by National Institute for Health and Care Excellence (NICE), and the lower and upper 95% CI from the bootstrap samples. 35 All bootstrap samples had a lower cost in the PO arm than the IV arm, and most (82.8%) of cost-effectiveness pairs are in the south-east quadrant. In this quadrant, lower costs and higher QALYs can be observed for the PO arm as compared with the IV arm, which makes a PO intervention dominant for these samples. 29 However, some samples fall into the south-west quadrant of the plane, where patients in the PO arm have fewer QALYs than patients in the IV arm. Similar to observed cost-effectiveness results, non-parametric bootstrapping also resulted in a negative ICER of –£108,500, indicating that the PO strategy was dominant, with the 95% CI ranging from –£1,592,000 to £1,126,000. The mean cost difference was £2924 (standard error £1031) in favour of the PO strategy, and mean QALYs were 0.027 (standard error 0.031) in favour of the PO strategy. The lower 95% CI line indicates that there is a possibility that the true difference in QALYs may be in the south-west quadrant. When the IV intervention results in higher QALYs than the PO intervention, there is uncertainty that the PO arm dominates; 17.2% of the bootstrap samples fall in the south-west quadrant, where the IV strategy is less costly but also results in higher QALYs. The strategy of treating bone and joint infections with PO antibiotics is dominant in the lower limit of the 95% CI, and it is also dominant in the point estimate of the ICER and has an upper limit of £1,126,000 per QALY gained.
FIGURE 8.
Cost-effectiveness plane. Reproduced with permission from McMeekin et al. 29 © 2019 McMeekin et al. This is an open access article distributed under the terms of the Creative Commons Attribution Licence, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
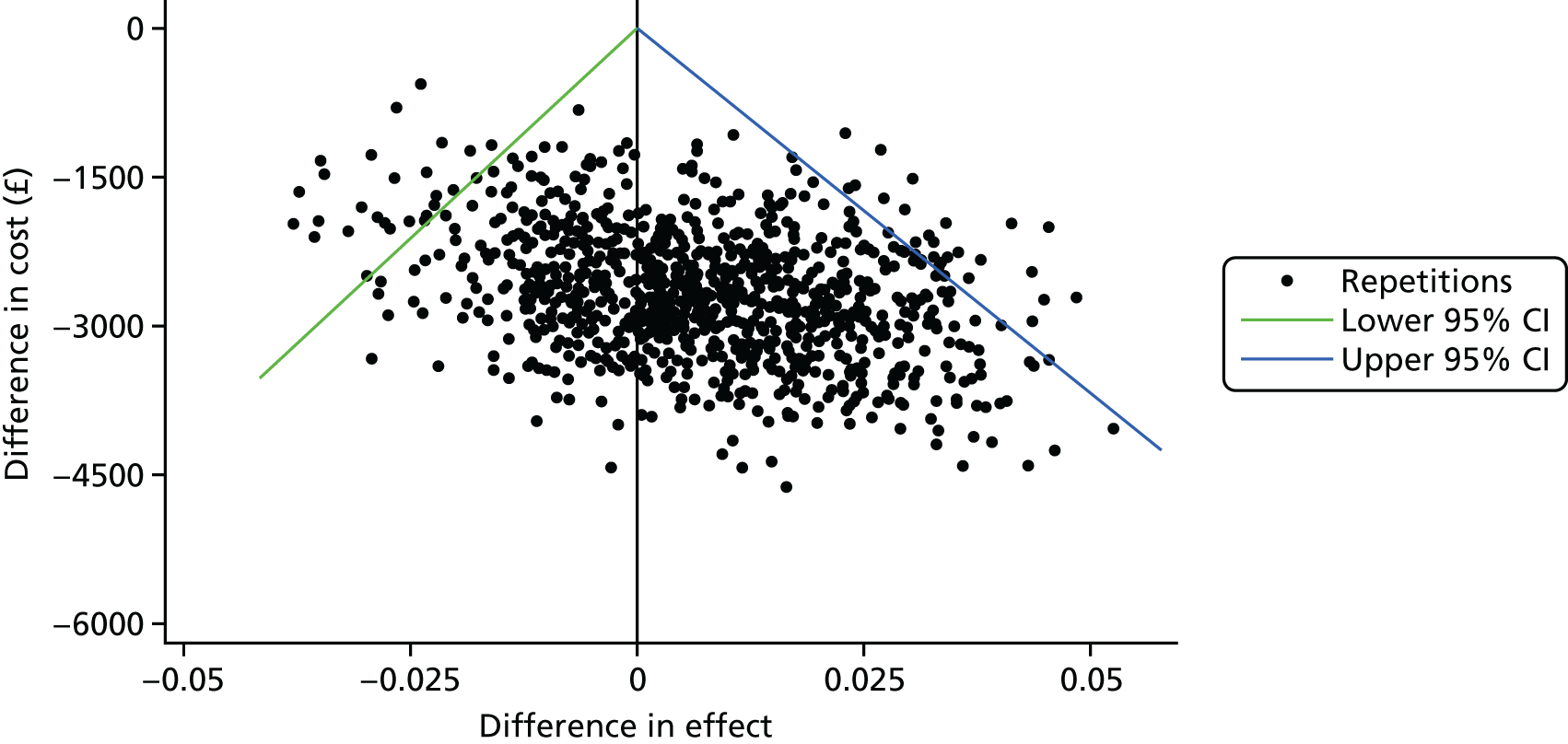
Chapter 4 Discussion
Some of the material in this chapter has previously been published in our description of the trial, reproduced from Li et al. 1 This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Despite a widely held view that the successful management of bone and joint infection requires IV rather than PO antibiotics,38–40 there is no evidence to suggest that PO antibiotic therapy results in worse outcomes. Nonetheless, there is significant variation in practice, with some centres advocating prolonged courses of IV therapy, some using short courses of IV therapy and others relying primarily on locally administered antibiotic agents. 41,42 Such lack of consensus demonstrates that the current trial addresses an important question and that the results are likely to influence practice. This is reflected in a study43 in which 500 infectious diseases physicians were asked to prioritise more than 100 research questions relating to infection. Four out of the top five responses related to route of administration of antibiotics and the top two concerned orthopaedic infection specifically.
The aim of the trial was to determine whether or not PO antibiotic therapy is non-inferior to IV antibiotic therapy when used for the first 6 weeks in the treatment of bone and joint infection. All recruiting centres routinely used a 6-week course of IV therapy for some or all bone and joint infections as their standard care pathway.
The results of the OVIVA trial demonstrate that PO therapy, when used during the initial 6 weeks in the treatment in bone and joint infection, is non-inferior to IV therapy. This finding held true for the ITT analysis, the complete-case analysis (which excluded participants for whom no valid end-point data were available), the PP analysis (defined by participants who received at least 4 weeks of their allocated treatment strategy or who exited early owing to potential treatment failure) and for sensitivity analyses (which substituted missing primary end points with the most extreme possible outcomes).
A secondary analysis, which included 16 possible and probable treatment failures as composites with definitive treatment failure, also demonstrated non-inferiority of PO therapy as compared with IV therapy.
Predefined subgroup analyses focusing on diagnostic certainty at baseline, surgical procedure, bacterial pathogen, trial site and planned IV or PO antibiotic regimen at the time of randomisation showed no evidence of a differential effect of either treatment arm. Similarly, post hoc analyses relating to retention of metalware, peripheral vascular disease and culture-negative infection demonstrated no advantage of IV over PO therapy. For a pragmatic and unselective trial, with significant heterogeneity in the population under study, the findings from these subgroup analyses are reassuring and, overall, suggest that the results from this trial can reasonably be assumed to be broadly generalisable. However, none was sufficiently powered formally to compare outcomes following IV and PO therapy and, although we believe that we selected the most important subgroups for analysis, we cannot exclude the possibility that there are otherwise unidentified subgroups for whom IV (or PO) therapy may be superior.
Adverse events and complications
For the purposes of this trial, C. difficile diarrhoea and complications relating to IV access devices (or lines) were analysed as secondary end points rather than SAEs.
As expected, line complications were significantly more common among participants randomised to IV therapy. Although the incidence was relatively modest, the mortality associated with line infection in particular has been reported at 12–25%. 44 Elimination of this risk by using PO therapy may therefore offer considerable advantage and, extrapolating from the presumed incidence data that we used to inform the design of the OVIVA trial, could avert 19–40 deaths annually in the UK.
The incidence of C. difficle diarrhoea and SAEs did not differ significantly between the treatment arms. A total of 23 patients died during the conduct of the study but none was considered related to the randomised strategy.
Patient-reported outcome measures
Data collection for the EQ-5D-3L was suboptimal. Although we cannot be certain that the missing data arose randomly, the trial showed no evidence that randomised strategy had any effect on the EQ-5D index at any time point.
The OHS and OKS were originally designed as assessment tools to indicate whether or not patients may have reached a threshold for joint replacement. 45,46 Although not necessarily directly applicable to the population included in this trial, they have been validated as research tools. We therefore collected the data as part of an assessment of functional outcome. For the OHS, the unadjusted scores indicated a difference between the trial arms at 120 and 365 days in favour of IV therapy, but in an adjusted quantile regression model there was insufficient evidence to suggest that the randomised strategy had a statistically significant effect on the median outcome at any time point. For the OKS, unadjusted scores indicated a difference between the trial arms at 120 and 365 days in favour of PO therapy, and the adjusted quantile regression model amplified this difference, which was statistically significant at both time points. A plausible biological explanation for this is lacking but it is possible that the difference between the adjusted and unadjusted estimates is attributable to the fact that only a small subgroup of the population is included in either the OHS or OKS sample.
There was clear evidence of improvement over time in all elements of the EQ-5D, and in the OHS and OKS, in both arms of the trial. This suggests that patients’ mobility, self-care, activity level, pain, psychological status and joint function generally improved progressively following the start of treatment for their incident bone or joint infection.
Adherence to medication
At the outset of this trial, we were concerned that if PO therapy proved inferior to IV therapy, this might have arisen as a result of ‘failure of compliance with oral therapy’ rather than ‘therapeutic failure of oral antibiotics’. To address this concern, we issued very clear guidance on the importance of adherence, both verbally and in writing, at the time of randomisation. We did not subsequently provide direct adherence support, such as text reminders, as this would be difficult to translate into routine practice.
Follow-on antibiotic therapy
The total duration of treatment, including follow-on therapy after the initial 6 weeks of treatment, is usually determined by a combination of factors such as the presence or absence of metalware, the organism isolated, the certainty with which all non-vital tissue has been excised and the availability of options for further surgical intervention should an infection recur. Previous studies have shown that timing of recurrence of infection is commonly related to the cessation of antibiotic therapy. 47 In this open-label trial, we were concerned that clinicians might inadvertently (or deliberately) extend follow-on therapy for participants who had been randomised to the PO arm. Therefore, we analysed the total duration of therapy in all participants. The results demonstrated no evidence of prolongation of follow-on therapy in one or other arm of the trial. There were clear indicators that the proportion of participants remaining on therapy fell markedly at 6 weeks, 3 months, 6 months and 1 year. Although these time points reflect clinical practice, they probably represent digit preference rather than an evidence base governing total duration of therapy. It suggests that significant redundancy may be built into our current practice; if so, there could be considerable gains in terms of cost and antibiotic minimisation if optimal duration of therapy could be more clearly defined prospectively.
Health economics
As the results of the trial indicate that PO antibiotics are non-inferior to IV antibiotics, there is no possibility of incremental benefit in outcomes of one treatment over the other. Therefore, it was not considered necessary or useful to carry out a full economic evaluation. The results of the EQ-5D-3L questionnaires reflected the main trial outcome of definitive failures; there was no statistically significant difference in the QALYs between arms. 29 This was reinforced by post hoc regression of QALYs on ‘definite failure’, for which the indicator variable for failure was found to be statistically significant, confirming that the EQ-5D measure is sensitive to the end point. However, the end point was found not to differ between arms. The difference in costs between arms was £2740 using multiple imputation, indicating that the use of PO antibiotics to treat a bone or joint infection was significantly cheaper (when taking into account the cost of antibiotics, IV administration and inpatient stays over the course of 1 year) than the use of IV antibiotics. With PO antibiotics being non-inferior to IV administration, and the costs in the PO arm being significantly less than the IV arm during the trial, the results suggest that the PO arm was a dominant strategy.
However, there was uncertainty around these results. Although there was no statistical difference in the QALYs, and PO antibiotics were found to be non-inferior to IV antibiotics using the primary outcome, the uncertainty around the economic results was explored further. These results suggested that, although in 82.8% of the bootstrap sample the PO strategy is dominant, 17.2% of the samples indicated that the IV strategy would result in higher QALYs than the PO strategy and still at a higher cost. However, at the NICE willingness-to-pay threshold of £30,000 there was a 100% probability of the PO intervention being more cost-effective than the IV intervention.
Despite the economic burden of bone and joint infection, economic studies in this area are rare. 48 A cost-effectiveness study that compared exchange arthroplasty with debridement and prosthetic retention for infected total hip arthroplasty in the elderly found that retention and debridement improved quality-adjusted life expectancy and also increased costs in 65- and 80-year-old men and women over a lifetime. 49 The ICER ranged from US$500 for frail 80-year-old men to US$21,800 for 65-year-old women. Kapadia et al. 50 conducted an economic evaluation in which they explored using chlorhexidine cloths before total knee arthroplasty and reported that for 1000 patients having total knee arthroplasty, a net saving of US$2.1M occurred. 29 This study assumed an estimated cost of US$130,000 per revision owing to infection, with 22 patients in a cohort of 1000 without use of the cloth becoming infected and six infections in the cohort using the cloth. Two studies estimated revision costs for infected prosthetics: for infected hip arthroplasties, estimated costs were £22,00051 and for infected knees, estimated costs were £30,000. 52 These costs included the revision surgery and subsequent inpatient stay. 29 A 2013 review summarised the economic literature in the treatment of periprosthetic joint infections, looking at prevention, treatment and surgical options. 53 Unlike the OVIVA trial, the treatment costs included the surgical costs of revision based on an estimated average cost of US$50,000 to US$60,000 per patient with an infected total hip arthroplasty. 48 None of these studies compared treatment costs of IV with PO antibiotics. The OVIVA trial estimated non-surgical costs over the year to be £13,274 for those treated with IV antibiotics and £10,534 for those treated with PO antibiotics.
After imputation of missing values for resource use and health outcomes (QALYs), results remained consistent with those obtained from complete-case analysis. QALYs reflect the primary outcome of non-inferiority. Results from sensitivity analyses were also consistent with the complete-case and imputation results.
Strengths
The OVIVA trial was pragmatic in that it was fully embedded into usual care and, as far as possible, reflected standard practice in all respects other than randomisation of treatment strategy and data collection. No additional diagnostic investigations, trial-specific clinic visits or blood tests were required of the participants. This had the advantage of reducing the influence of possible differential observer effects by treatment arm.
The OVIVA trial was a large trial and clinically evaluable primary end-point data were available for 1015 participants representing 96.3% of those randomised. This was well within the 10% allowance in the sample size calculation for loss to follow-up.
Of the 39 participants lost to follow-up, only 14 (seven in each arm) withdrew consent to further involvement with the trial. This suggests that the study design and patient pathway were generally acceptable.
The primary end point was hard in that it was predefined by clinically relevant criteria as used in daily practice. When these criteria were not clearly fulfilled, a blinded end-point committee assessed potential treatment failures from clinical records redacted for personal identifiers and for any indication of the randomisation arm. The latter included redaction for all specifically named antibiotics, any reference to IV lines and any reference to line complications, side effects and use of MEMS containers.
Recruitment criteria were highly inclusive. The hypothesis behind the trail was based strictly on the pharmacokinetic principle that carefully selected PO antibiotics are as likely as IV antibiotics to achieve sufficiently high concentrations to effect eradication of infection. Such a principle is unlikely to be influenced by factors such as site of infection, retention of metal or causative organism. A more restrictive recruitment strategy, for example selecting only participants with S. aureus infection or primary arthroplasty, would have made the trial prohibitively long and would have limited the utility of the results. Although the inclusive recruitment criteria resulted in an heterogeneous study population, we believe that the advantages of generalisability outweigh the disadvantages of heterogeneity.
Weaknesses
The OVIVA trial was an open-label study. The decision to use this design was based on the two principles. First, exposure of patients to a placebo IV therapy for a period of up to 6 weeks would have posed unnecessary risks associated with the use of an intravascular access device and would, therefore, have been unethical. Second, owing to the number of different antibiotics required to provide optimal care for all patients randomised, it was not feasible to provide a matched placebo in every case. Although an open-label design leaves the trial open to bias, the primary end points were determined according to predefined criteria and an independent committee that was blinded to treatment allocation. This was achieved through redaction from case notes of any information that might have betrayed the treatment allocation (e.g. reference to IV access, OPAT, drug names, therapeutic drug monitoring). Primary end points were defined by objective clinical and microbiological criteria, assessment of which required attendance at, or admission to, hospital. Therefore, they were hard end points, the interpretation of which was unlikely to have been influenced by treatment allocation or other confounding factors.
The OVIVA trial was designed as a pragmatic trial that relied on routine care records as the primary source of data. There were no research-specific clinic visits and no research-specific investigations. When possible, and provided that the timings were commensurate with follow-up requirements of the trial, follow-up data were collected through direct patient contact at their routine clinical reviews. When this was not possible, the protocol allowed for telephone follow-up with the patient and the GP. Although primary end-point data were available for all except 39 participants, it is possible that some potential treatment failures were not identified. Although we think this unlikely, there is no reason to believe that unidentified losses influenced one arm more than the other.
Eligibility for recruitment to the trial was based on clinical criteria rather than diagnostic laboratory results. There are several reasons why we did not include histological or microbiological results as part of the inclusion criteria. First, approximately 15% of bone and joint infections diagnosed clinically are not confirmed microbiologically, for example, as a result of prior exposure to antibiotics or sampling error. 54 Nonetheless, such patients are commonly treated for infection diagnosed on clinical criteria alone. Second, the results of laboratory tests, particularly the histology results, are not always available within 7 days of sampling; had we relied on laboratory results as part of the inclusion criteria, many patients would have had to be excluded from this trial on account of this delay. Third, the pragmatic design of this trial gave due autonomy over clinical management to the surgeon or physician responsible for the patient. If, according to a research definition, infection was deemed not present, the trial could potentially have undermined a clinician’s decision to treat an infection based on clinical criteria alone. Finally, in order to account for the possibility that uninfected patients were included, every case that failed to meet a strict prospective definition of infection at baseline was reviewed by an independent committee for a consensus decision on their infection status at the time of recruitment.
There are three circumstances in which an apparent deviation from the allocated treatment arm might have arisen.
First, participants were permitted IV therapy for up to 7 days following the start of planned curative therapy, regardless of their randomised strategy (in most instances, the ‘start of planned curative therapy’ was the date of definitive surgical intervention). The rationale for this was to allow patients to recover from the effects of anaesthesia before starting PO therapy and to allow sufficient time for microbiological results to inform the optimal choice of antimicrobial agent. The availability of microbiological results was not a requirement prior to randomisation but, because standard practice usually includes initial broad-spectrum empiric IV antibiotics while waiting for microbiology results, we felt that a requirement for immediate postoperative use of PO therapy might undermine clinical decision-making. Furthermore, operative findings were an important component of eligibility and, as a result, most patients were randomised in the postoperative period.
Second, for participants randomised to IV therapy, the use of adjunctive PO agents was permitted. This may at first seem counterintuitive in a study that aimed to compare IV with PO therapy, but is based on common practice outside the context of the trial. Examples include PO rifampicin, which is routinely used alongside IV therapy in the management of biofilm-associated staphylococcal disease and metronidazole, which is commonly used in polymicrobial osteomyelitis. To exclude patients allocated to the IV arm but who require adjunctive PO therapy would likely incur a bias in favour of PO therapy.
Third, participants randomised to the PO arm were allowed up to 5 days of IV therapy to allow for treatment of intercurrent illness or for short periods when, for unrelated reasons, PO therapy was not appropriate. It was not designed to be used as a rescue treatment for the bone infection under therapy and the protocol made this very clear. To withdraw participants on the grounds that they had an unrelated concomitant illness which, in the opinion of a physician independent of the trial, required IV therapy would have been considered unethical or discriminatory by some readers. Given that all patients had to have been prescribed at least 6 weeks of therapy for the incident bone infection, we believe that a short course of IV therapy in a small minority of patients is unlikely significantly to have influenced the results. To ensure transparency around both of these circumstances, all antibiotic use (including dose, route of administration and duration) was recorded from the day of randomisation through to 1-year follow-up.
There was considerable variation in the number of participants recruited at each site, with over half being recruited at just two centres. Both were tertiary referral specialist units, which consequently have high volumes of orthopaedic surgery and its associated complications. Therefore, it is unsurprising that these centres accrued a higher number of eligible patients than other centres. In addition, the single-centre pilot study contributed 228 participants at one of these sites. The asymmetry of recruiting between sites is unlikely to influence interpretation of the results and, in the 11 sites for which subgroup analysis was possible, there was no evidence of an interaction between randomised treatment strategy and study site.
Follow-up in this trial was for 1 year with the facility to obtain final clinical review data up to 420 days. The data points were made deliberately wide because the trial relied on routine clinic attendance to capture end points and adverse events. Had we included trial-specific clinics at more tightly defined time points, the trial may have been open to a greater influence of observer bias. Because follow-up was limited to 1 year, we cannot, of course, assume from our results that very late recurrences will be equally distributed between those who were randomised to IV and PO therapy. However, there is no biologically plausible reason to suggest otherwise and, given that the median total duration of therapy was around 11 weeks, we believe that there was unlikely to be an advantage of following up participants for longer than 1 year.
There are some important caveats relating to antibiotic therapy in orthopaedic surgery and are detailed below.
First, the effective management of bone and joint infection is critically dependent on effective surgical management. This may include careful and complete surgical debridement and excision, a meticulous sampling framework to optimise diagnostics in the microbiology and histopathology laboratories, and early vital soft tissue cover. Regardless of the route of administration, antibiotic therapy is likely to be ineffective without appropriate and adequate surgical intervention. Researchers involved in this trial were self-selected and are therefore likely to represent centres with surgical expertise specifically in the management of bone and joint infection.
Second, the antimicrobial agents used in this trial were chosen by specialists in clinical infection, with reference to bioavailability, tissue penetration and likely effectiveness against the known or presumed pathogen. It cannot be assumed that an antibiotic will be effective based simply on the reported susceptibility of the target organisms. This is particularly true for PO antibiotics for which a wide range of agents is available and for which oral bioavailability and dose are critical for efficacy.
Third, because adherence to therapy is plausibly better with supervised IV therapy than with self-administered PO therapy, participants in this trial were provided with written information explaining the importance of adherence. It is critical that, if the findings of this study are used to support a change in practice from routine use of IV to PO therapy, due consideration should be given to mechanisms to promote adherence.
Fourth, patients managed with long courses of IV therapy, commonly through an OPAT service, are likely to be more closely supervised than those on PO therapy during the first 6 weeks of treatment. Although the trial did not demonstrate any difference in time to treatment failure between the two arms, it may be that, because of their involvement in the trial, participants on PO therapy were more closely followed up than they might have been outside the context of a trial. Extrapolation of the results into routine practice should therefore take account of the need for adequate monitoring of patients after discharge.
Comparison with previous studies
There have been a limited number of studies on this topic. A Cochrane review,15 which included data from five trials involving a total of 180 patients with chronic osteomyelitis, demonstrated no benefit of IV over PO antibiotic therapy, although the authors concluded that there was insufficient evidence to inform clinical practice. We believe that the OVIVA trial provides sufficient support for these findings to allow widespread adoption of PO therapy in this setting. Our results concur with The Infectious Diseases Society of America guideline on the management of prosthetic joint infections,55 and a review by Fraimow,56 which suggest that the use of highly bioavailable PO agents may be an appropriate alternative to IV therapy, provided that patient factors do not limit the drug’s pharmacokinetic properties.
Implications for practice
Patient pathways
There is a clear professional mandate to ensure patient-centred treatment, including, when possible, limitation of hospital attendances, promotion of an independent life style and greater patient choice over their own treatment. 57 Such gains are less achievable with IV therapy than they are with PO therapy for the following reasons:
-
Use of IV therapy results in a delay in discharge from hospital, most commonly while awaiting insertion of an IV access device and setting up OPAT. The median delay, quantified prospectively as part of the OVIVA trial, was 3 days for patients managed with IV therapy.
-
Clinic visits, although not quantified in this trial, would almost certainly have been more numerous among those randomised to IV therapy than to PO therapy on account of therapeutic drug monitoring and line checks/manipulation.
-
Patients on IV therapy after discharge from hospital, unless self-administering, may find it inconvenient to either arrange access in their own homes for a visiting nurse or to attend the hospital infusion centre on a regular basis, usually daily. Furthermore, the presence of a line may restrict social and sporting activities such as swimming, which patients may regard as important for their rehabilitation. Although we did not specifically collect qualitative data to assess patient preference or satisfaction, we contributed to a separate trial over the same period. 58 This study suggested that, for prolonged IV therapy, self-administration yielded significant cost savings, although patient preference was for home visits by a specialist nurse. The study did not compare preference for IV therapy with preference for PO therapy.
-
Conversations with our patient and public involvement (PPI) representatives revealed that a potential further advantage of oral administration is the sense of control that patients feel over their own treatment, particularly on account of the ease, familiarity and portability of tablets, that they would not have had with IV therapy.
Patient safety
The use of IV lines pose risks that are not directly relevant to patients on PO therapy. For the purposes of this trial, line complications were recorded as secondary end points (rather than SAEs) and included line fracture or blockage, bleeding, thromboembolic events and line-related infection. Line-related infection carries with it a crude mortality estimate of up to 25%. 59 Around 10% of evaluable patients who were randomised to IV therapy for the OVIVA study suffered a line-related complication. This figure is either lower than or similar to other reports, although the populations involved are not directly comparable. 60,61 Nonetheless, we believe that almost all of these complications could have been avoided in participants managed with PO therapy.
Although we would not have expected line-related complications among those randomised to PO therapy, five such instances were recorded. In four of these participants, the line was placed following early exit from the randomised strategy (i.e. the participant was switched from PO to IV therapy during the intervention period) and, in one participant, the line complication occurred after completion of the intervention period.
Cost
The acquisition cost of IV antibiotics is generally greater than that of PO therapy. In the OVIVA trial, the mean total drug cost for those randomised to IV therapy was almost £800 greater than PO therapy. IV therapy incurs additional costs relating to drug administration and prolonged hospitalisation. The mean non-surgical treatment costs over the year for patients randomised to IV therapy was approximately £2700 greater than that of patients randomised to PO therapy. Extrapolation based on the most conservative estimates used to inform the design of the OVIVA trial suggests a potential saving to the NHS of over £30M if PO rather than IV therapy becomes the standard of care in the early treatment of bone and joint infection across the UK.
Discussion with service users and PPI representatives suggested that the socioeconomic costs associated with IV therapy represent a significant burden. Time absent from work, child care and travel to hospital clinics were considered important. Additional hidden costs relating to delayed discharge, worry about complications, lack of social engagement, work absence and effects on family/social environment were all thought to be higher in those managed with IV therapy.
Antibiotic resistance
Antimicrobial stewardship activity has become a major priority in health care. PO therapy provides an opportunity for a reduction in use of broad-spectrum antibiotics. The global threat of antimicrobial resistance has been highlighted by the UK Chief Medical Officer as a very major risk to the NHS. Progress in limiting the overall exposure to broad-spectrum antibiotics will reduce this threat and limit the risks of health care associated infections such as C. difficile, methicillin-resistant S. aureus and carbapenemase-producing Enterobacteriaceae. This is of particular relevance to the treatment of orthopaedic infections, which commonly mandate an extended course of antibiotic therapy. Although not all PO antibiotics are narrow spectrum, it is easier to select targeted therapy with PO agents than it is with IV agents. The effective use of PO therapy in orthopaedic infection will help preserve the broad-spectrum IV antibiotic use for serious infections, particularly when therapeutic options are limited.
Implications and suggestions for research
-
Given the advantages of PO over IV therapy discussed, the findings of this trial suggest that it would be value to prospectively investigate its use in other conditions in which prolonged courses of IV therapy are thought to be necessary for optimal outcome. These include bacteraemia, endocarditis and meningitis. Although the incidence of these conditions is lower than that of bone and joint infection, there is still considerable potential for patient benefit, cost reduction and improved antimicrobial stewardship.
-
To further support patient safety, cost improvement and antimicrobial stewardship, additional work to define the optimal total duration of antibiotic therapy in bone and joint infection is necessary. Currently, there is considerable variation between centres and between clinicians, which suggests that there may be significant redundancy in antibiotic use. This almost certainly contributes to the risk of emerging resistance to antimicrobials, an issue that is high on the agenda of the Department of Health and Social Care and the medical community globally.
-
Effective antibiotic therapy requires the presence of therapeutic drug levels at the site of the infected tissue. This depends on both bioavailability and tissue penetration. Optimising antibiotic choice will require a programme of work that may include techniques such as microdialysis of tissue fluid at the site of deep surgical infection.
-
There is currently growing interest in the literature around environmental factors that might influence SSI rates. To limit the number of bone and joint infections arising as a complication of surgery, there is considerable scope to investigate simple perioperative interventions that could reduce the risk of surgical site infection in orthopaedics. Examples might include the influence of different patient-warming technologies during surgery, the effect of preoperative transfusion and choice of postoperative dressings.
Chapter 5 Conclusions
Despite its limitations, this trial is the largest study of its type addressing the question of route of administration of antibiotic therapy in bone and joint infection. Currently, the majority of centres that manage complex bone and joint infections routinely use a prolonged course of IV antibiotics in the early phase of therapy. The results of this trial suggest that this strategy has no advantage over PO therapy. In addition, use of PO therapy will allow clinicians to mitigate the risks associated with IV access devices normally used for long-term IV therapy. There was no significance difference in the incidence of C. difficile diarrhoea or SAEs.
For patients, PO therapy provides an opportunity for earlier discharge from hospital, autonomy and independence in managing their medications, and limitation of the risks associated with prolonged use of IV access devices. These advantages have to be weighed against the risk of poor adherence with unsupervised PO therapy. Although this did not appear to influence clinical outcome in the trial, adherence monitoring during the trial may have influenced the behaviour of a subset of participants.
The demonstration of non-inferiority of PO therapy provides an important opportunity for antimicrobial stewardship. Because the choice of PO agents is generally greater than that of IV agents, it seems likely that the results of this trial will facilitate much greater capacity for individualisation of therapy, thereby ensuring that the use of broad-spectrum agents can be better restricted to cases in which no alternatives exist. It provides an important opportunity to support the global fight against emerging antimicrobial resistance.
The results from the OVIVA trial could provide an opportunity for considerable cost savings. These arise primarily from the shorter period of hospitalisation, the drug costs and the resources associated with their administration. The health economic analyses in the OVIVA trial suggested that, on average, the non-surgical treatment costs over 1 year for patients randomised to PO therapy were approximately £2700 less than those of IV therapy.
Translation of the results from the OVIVA trial into clinical practice is likely to have important implications for patients, health-care practitioners in the field of orthopaedic infection and the health economy.
Acknowledgements
The OVIVA trial was funded by the National Institute for Health Research (HTA project number 11/36/29).
The Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences (NDORMS) co-ordinated the study via the Surgical Interventions Trials Unit and the trial sponsor was Oxford University Hospitals NHS Trust.
The authors greatly appreciate the considerable contributions of Neil French, Martin Llewelyn and Colette Smith as DMC members, and of David Beard and Cushla Cooper for their support through the CTU. The authors also acknowledge the valued input from Fraser Old (public patient representative), Alice Harin, Lorrayne Jefferies, Louise Spoors, Patrick Julier and all the members of the infectious diseases and orthopaedic departments at Oxford University Hospitals NHS Trust.
Our gratitude also to Brian Angus, Andrew Woodhouse, Tony Berendt, Parham Sendi, Ivor Byren and Hemant Pandit for their substantial intellectual input and advice throughout.
The authors would like to thank members of the United Kingdom Clinical Infection Research Group (UKCIRG), principal investigators and research staff across the 26 contributing sites. These included Gavin Barlow, Oliver Koch, Catherine Sargent, Caroline Barker, Jim Buckley, Aula Abbara, Tumena Corrah, Martin Williams, Rashmi Sharma, Achyut Guleri, Bijayendra Singh, Uli Schwabb, Jonathon Campion, Nick Nicolaou, Sentil Velayudhum, Michael Kelsey, Parvez Moondi, Ian Dos Remedios, Neena Bodasing, Julia Grieg, Stephen Mapham, Sophie Collier, Conor Bowman, Fiona Fitzgerald, Kim Cox, Jocelyn Marshall, Karen Bisnauthsing, Antonio Querol-Rubiera, Sam Stafford, Helen Reynolds, Marie Claire Hoyle, Parizade Raymode, Kareen Darnley, Angela Dunne, Claire Vallance, Deborah Bunne, Chris Herriott, James Calderwood, Elizabeth Saunders, Sue Brown, Natalia Waddington, Rebecca Houlihan, Louise Flintoff and Helen Sankey.
We are extremely grateful to the funding agency and to all patients who participated.
Contributions of authors
Matthew Scarborough (Consultant in Clinical Infection) was the chief investigator and was responsible for design, recruitment, data capture, analysis, interpretation and manuscript preparation.
Ho Kwong Li (Research Fellow in Infectious Diseases) was the trial physician and was responsible for design, recruitment, data capture, analysis, interpretation and manuscript preparation.
Ines Rombach (Trial Statistician) was responsible for design, data analysis, interpretation and manuscript preparation.
Rhea Zambellas (Trial Co-ordinator) was responsible for data capture, analysis and manuscript preparation.
A Sarah Walker (Professor of Medical Statistics) was the senior statistician and was responsible for trial design, analysis and manuscript preparation.
Martin McNally (Consultant Orthopaedic Surgeon) was responsible for recruitment, data capture, interpretation and manuscript revision.
Bridget Atkins (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Michelle Kümin (Post-doctoral Scientist) was the research co-ordinator and was responsible for analysis and manuscript preparation.
Benjamin A Lipsky (Professor of Infectious Diseases) was the end-point review committee chairperson and was responsible for trial design and manuscript preparation.
Harriet Hughes (Consultant in Infectious Diseases and Microbiology) was on the end-point review committee, and was responsible for trial design and manuscript revision.
Deepa Bose (Consultant Orthopaedic Surgeon) was on the end-point review committee and was responsible for trial design and manuscript revision.
Simon Warren (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Damien Mack (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Jonathan Folb (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Elinor Moore (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Neil Jenkins (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Susan Hopkins (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
R Andrew Seaton (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Carolyn Hemsley (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Jonathan Sandoe (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Ila Aggarwal (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Simon Ellis (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Rebecca Sutherland (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Claudia Geue (Health Economist) was responsible for data analysis and interpretation and manuscript preparation.
Nicola McMeekin (Health Economist) was responsible for data analysis and interpretation and manuscript preparation.
Claire Scarborough (Research Fellow in Infectious Diseases) was the trial physician and was responsible for data capture, analysis and manuscript preparation.
John Paul (Consultant in Clinical Infection) was on the Trial Steering Committee and was responsible for trial design, data interpretation and manuscript review.
Graham Cooke (Consultant in Clinical Infection) was on the Trial Steering Committee and was responsible for trial design, data interpretation and manuscript review.
Jennifer Bostock (PPI Representative) was on the Trial Steering Committee and was responsible for trial design and manuscript preparation.
Elham Khatamzas (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Nick Wong (Research Fellow in Infectious Diseases) was responsible for recruitment, data capture and manuscript revision.
Andrew Brent (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Jose Lomas (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Philippa Matthews (Consultant in Clinical Infection) was responsible for recruitment, data capture and manuscript revision.
Tri Wangrangsimakul (Research Fellow in Infectious Diseases) was responsible for recruitment, data capture and manuscript revision.
Roger Gundle (Consultant Orthopaedic Surgeon) was responsible for recruitment, data capture and manuscript revision.
Mark Rogers (Consultant Orthopaedic Surgeon) was responsible for recruitment, data capture and manuscript revision.
Adrian Taylor (Consultant Orthopaedic Surgeon) was responsible for recruitment, data capture and manuscript revision.
Guy E Thwaites (Professor of Infectious diseases) was responsible for trial design, public engagement and manuscript review.
Philip Bejon (Professor of Infectious Diseases) identified the research question and was responsible for trial design, recruitment, data capture, analysis, interpretation and manuscript preparation.
Publications
Li HK, Scarborough M, Zambellas R, Cooper C, Rombach I, Walker AS, et al. Oral versus intravenous antibiotic treatment for bone and joint infections (OVIVA): study protocol for a randomised controlled trial. Trials 2015;16:583.
Li HK, Rombach I, Zambellas R, Walker AS, McNally MA, Atkins BL, et al. Oral versus intravenous antibiotics for bone and joint infection. N Engl J Med 2019;380:425–36.
McMeekin N, Geue C, Briggs A, Rombach I, Li HK, Bejon P, et al. Cost-effectiveness of oral versus intravenous antibiotics (OVIVA) in patients with bone and joint infection: evidence from a non-inferiority trial [version 1; peer review: awaiting peer review]. Wellcome Open Res 2019;4:108.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Please note exclusive use will be retained until the publication of major outputs. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Li HK, Scarborough M, Zambellas R, Cooper C, Rombach I, Walker AS, et al. Oral versus intravenous antibiotic treatment for bone and joint infections (OVIVA): study protocol for a randomised controlled trial. Trials 2015;16. https://doi.org/10.1186/s13063-015-1098-y.
- Lew DP, Waldvogel FA. Osteomyelitis. Lancet 2004;364:369-79. https://doi.org/10.1016/S0140-6736(04)16727-5.
- Zimmerli W, Ochsner PE. Management of infection associated with prosthetic joints. Infection 2003;31:99-108. https://doi.org/10.1007/s15010-002-3079-9.
- Legrand E, Flipo RM, Guggenbuhl P, Masson C, Maillefert JF, Soubrier M, et al. Management of nontuberculous infectious discitis. Treatments used in 110 patients admitted to 12 teaching hospitals in France. Joint Bone Spine 2001;68:504-9. https://doi.org/10.1016/S1297-319X(01)00315-3.
- Berman SJ, Johnson EW. Out-patient parenteral antibiotic therapy (OPAT): clinical outcomes and adverse events. Hawaii Med J 2001;60:31-3.
- Matthews PC, Conlon CP, Berendt AR, Kayley J, Jefferies L, Atkins BL, et al. Outpatient parenteral antimicrobial therapy (OPAT): is it safe for selected patients to self-administer at home? A retrospective analysis of a large cohort over 13 years. J Antimicrob Chemother 2007;60:356-62. https://doi.org/10.1093/jac/dkm210.
- Tice A. Outpatient parenteral antimicrobial therapy (OPAT): a global perspective. Introduction. Chemotherapy 2001;47:1-4. https://doi.org/10.1159/000048562.
- Yong C, Fisher DA, Sklar GE, Li SC. A cost analysis of Outpatient Parenteral Antibiotic Therapy (OPAT): an Asian perspective. Int J Antimicrob Agents 2009;33:46-51. https://doi.org/10.1016/j.ijantimicag.2008.07.016.
- Tice AD, Marsh PK, Craven PC, McEniry DW. Home intravenous antibiotic therapy. Am J Med 1993;94:114-15. https://doi.org/10.1016/0002-9343(93)90134-B.
- Oosterheert JJ, Bonten MJ, Schneider MM, Buskens E, Lammers JW, Hustinx WM, et al. Effectiveness of early switch from intravenous to oral antibiotics in severe community acquired pneumonia: multicentre randomised trial. BMJ 2006;333. https://doi.org/10.1136/bmj.38993.560984.BE.
- Montini G, Toffolo A, Zucchetta P, Dall’Amico R, Gobber D, Calderan A, et al. Antibiotic treatment for pyelonephritis in children: multicentre randomised controlled non-inferiority trial. BMJ 2007;335. https://doi.org/10.1136/bmj.39244.692442.55.
- Shenep JL, Flynn PM, Baker DK, Hetherington SV, Hudson MM, Hughes WT, et al. Oral cefixime is similar to continued intravenous antibiotics in the empirical treatment of febrile neutropenic children with cancer. Clin Infect Dis 2001;32:36-43. https://doi.org/10.1086/317552.
- Stevens DL, Herr D, Lampiris H, Hunt JL, Batts DH, Hafkin B. Linezolid versus vancomycin for the treatment of methicillin-resistant Staphylococcus aureus infections. Clin Infect Dis 2002;34:1481-90. https://doi.org/10.1086/340353.
- Heldman AW, Hartert TV, Ray SC, Daoud EG, Kowalski TE, Pompili VJ, et al. Oral antibiotic treatment of right-sided staphylococcal endocarditis in injection drug users: prospective randomised comparison with parenteral therapy. Am J Med 1996;101:68-76. https://doi.org/10.1016/S0002-9343(96)00070-8.
- Conterno LO, da Silva Filho CR. Antibiotics for treating chronic osteomyelitis in adults. Cochrane Database Syst Rev 2009;3. https://doi.org/10.1002/14651858.CD004439.pub2.
- Euba G, Murillo O, Fernández-Sabé N, Mascaró J, Cabo J, Pérez A, et al. Long-term follow-up trial of oral rifampin-cotrimoxazole combination versus intravenous cloxacillin in treatment of chronic staphylococcal osteomyelitis. Antimicrob Agents Chemother 2009;53:2672-6. https://doi.org/10.1128/AAC.01504-08.
- Whittaker JP, Warren RE, Jones RS, Gregson PA. Is prolonged systemic antibiotic treatment essential in two-stage revision hip replacement for chronic Gram-positive infection?. J Bone Joint Surg Br 2009;91:44-51. https://doi.org/10.1302/0301-620X.91B1.20930.
- Hart WJ, Jones RS. Two-stage revision of infected total knee replacements using articulating cement spacers and short-term antibiotic therapy. J Bone Joint Surg Br 2006;88:1011-15. https://doi.org/10.1302/0301-620X.88B8.17445.
- Ceroni D, Regusci M, Pazos JM, Saunders CT, Kaelin A. Risks and complications of prolonged parenteral antibiotic treatment in children with acute osteoarticular infections. Acta Orthop Belg 2003;69:400-4.
- Nguyen S, Pasquet A, Legout L, Beltrand E, Dubreuil L, Migaud H, et al. Efficacy and tolerance of rifampicin-linezolid compared with rifampicin-cotrimoxazole combinations in prolonged oral therapy for bone and joint infections. Clin Microbiol Infect 2009;15:1163-9. https://doi.org/10.1111/j.1469-0691.2009.02761.x.
- Daver NG, Shelburne SA, Atmar RL, Giordano TP, Stager CE, Reitman CA, et al. Oral step-down therapy is comparable to intravenous therapy for Staphylococcus aureus osteomyelitis. J Infect 2007;54:539-44. https://doi.org/10.1016/j.jinf.2006.11.011.
- World Medical Association . Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 2013;310:2191-4. https://doi.org/10.1001/jama.2013.281053.
- Diaz E, Levine HB, Sullivan MC, Sernyak MJ, Hawkins KA, Cramer JA, et al. Use of the Medication Event Monitoring System to estimate medication compliance in patients with schizophrenia. J Psychiatry Neurosci 2001;26:325-9.
- Farley J, Hines S, Musk A, Ferrus S, Tepper V. Assessment of adherence to antiviral therapy in HIV-infected children using the Medication Event Monitoring System, pharmacy refill, provider assessment, caregiver self-report, and appointment keeping. J Acquir Immune Defic Syndr 2003;33:211-18. https://doi.org/10.1097/00126334-200306010-00016.
- European Commission . Directive 2001 20 EC of the European Parliament and of the Council of 4 April 2001 2001.
- Joint Formulary Committee . British National Formulary n.d. www.medicinescomplete.com (accessed 12 December 2016).
- Department of Health and Social Care . NHS Reference Costs 2014 to 2015 2016.
- Wu O, Boyd K, Paul J, McCartney E, Ritchie M, Mellon D, et al. Hickman catheter and implantable port devices for the delivery of chemotherapy: a phase II randomised controlled trial and economic evaluation. Br J Cancer 2016;114:979-85. https://doi.org/10.1038/bjc.2016.76.
- McMeekin N, Geue C, Briggs A, Rombach I, Li HK, Bejon P, et al. Cost-effectiveness of oral versus intravenous antibiotics (OVIVA) in patients with bone and joint infection: evidence from a non-inferiority trial [version 1; peer review: awaiting peer review]. Wellcome Open Res 2019;4. https://doi.org/10.12688/wellcomeopenres.15314.1.
- Curtis L. Unit Costs of Health and Social Care 2015. Canterbury: PSSRU, University of Kent; 2015.
- EuroQol G . EuroQol – a new facility for the measurement of health-related quality of life. Health Pol 1990;16:199-208. https://doi.org/10.1016/0168-8510(90)90421-9.
- EuroQol . EuroQol – About EQ-5D 2017. https://euroqol.org (accessed 13 September 2017).
- Dolan P. Modeling valuations for EuroQol health states. Med Care 1997;35:1095-108. https://doi.org/10.1097/00005650-199711000-00002.
- Ramsey SD, Willke RJ, Glick H, Reed SD, Augustovski F, Jonsson B, et al. Cost-effectiveness analysis alongside clinical trials II-An ISPOR Good Research Practices Task Force report. Value Health 2015;18:161-72. https://doi.org/10.1016/j.jval.2015.02.001.
- NICE . Guide to the Methods of Technology Appraisal: 2013 n.d. www.nice.org.uk/article/pmg9/chapter/foreword (accessed 13 September 2017).
- Briggs A, Clark T, Wolstenholme J, Clarke P. Missing. presumed at random: cost-analysis of incomplete data. Health Econ 2003;12:377-92. https://doi.org/10.1002/hec.766.
- Faria R, Gomes M, Epstein D, White IR. A guide to handling missing data in cost-effectiveness analysis conducted within randomised controlled trials. PharmacoEconomics 2014;32:1157-70. https://doi.org/10.1007/s40273-014-0193-3.
- Waldvogel FA, Medoff G, Swartz MN. Osteomyelitis: a review of clinical features, therapeutic considerations and unusual aspects. N Engl J Med 1970;282:198-206. https://doi.org/10.1056/NEJM197001222820406.
- Berendt A, McNally M, Warell DA, Cox TM, Firth JD. Oxford Textbook of Medicine. Oxford: Oxford University Press; 2010.
- Mader JT, Calhoun J, Mandell GL, Bennett JE, Dolin R. Principles and Practice of Infectious Diseases. London: Churchill Livingstone; 1995.
- Johannsson B, Taylor J, Clark CR, Shamsuddin H, Beekmann SE, Polgreen P. Infectious Diseases Society of America Emerging Infections Network . Treatment approaches to prosthetic joint infections: results of an Emerging Infections Network survey. Diagn Microbiol Infect Dis 2010;66:16-23. https://doi.org/10.1016/j.diagmicrobio.2009.08.016.
- Stengel D, Bauwens K, Sehouli J, Ekkernkamp A, Porzsolt F. Systematic review and meta-analysis of antibiotic therapy for bone and joint infections. Lancet Infect Dis 2001;1:175-88. https://doi.org/10.1016/S1473-3099(01)00094-9.
- Paterson DL. Steering Group of the Australasian Society for Infectious Diseases Clinical Research Network . Determining research priorities for clinician-initiated trials in infectious diseases. Med J Aust 2013;198:270-2. https://doi.org/10.5694/mja12.11703.
- Soufir L, Timsit JF, Mahe C, Carlet J, Regnier B, Chevret S. Attributable morbidity and mortality of catheter-related septicemia in critically ill patients: a matched, risk-adjusted, cohort study. Infect Control Hosp Epidemiol 1999;20:396-401. https://doi.org/10.1086/501639.
- Dawson J, Fitzpatrick R, Carr A, Murray D. Questionnaire on the perceptions of patients about total hip replacement. J Bone Joint Surg Br 1996;78:185-90. https://doi.org/10.1302/0301-620X.78B2.0780185.
- Dawson J, Fitzpatrick R, Murray D, Carr A. Questionnaire on the perceptions of patients about total knee replacement. J Bone Joint Surg Br 1998;80:63-9. https://doi.org/10.1302/0301-620X.80B1.7859.
- Byren I, Bejon P, Atkins BL, Angus B, Masters S, McLardy-Smith P, et al. One hundred and twelve infected arthroplasties treated with ‘DAIR’ (debridement, antibiotics and implant retention): antibiotic duration and outcome. J Antimicrob Chemother 2009;63:1264-71. https://doi.org/10.1093/jac/dkp107.
- Fernandez-Fairen M, Torres A, Menzie A, Hernandez-Vaquero D, Fernandez-Carreira JM, Murcia-Mazon A, et al. Economical analysis on prophylaxis, diagnosis, and treatment of periprosthetic infections. Open Orthop J 2013;7:227-42. https://doi.org/10.2174/1874325001307010227.
- Fisman DN, Reilly DT, Karchmer AW, Goldie SJ. Clinical effectiveness and cost-effectiveness of 2 management strategies for infected total hip arthroplasty in the elderly. Clin Infect Dis 2001;32:419-30. https://doi.org/10.1086/318502.
- Kapadia BH, Johnson AJ, Issa K, Mont MA. Economic evaluation of chlorhexidine cloths on healthcare costs due to surgical site infections following total knee arthroplasty. J Arthroplasty 2013;28:1061-5. https://doi.org/10.1016/j.arth.2013.02.026.
- Vanhegan IS, Malik AK, Jayakumar P, Ul Islam S, Haddad FS. A financial analysis of revision hip arthroplasty: the economic burden in relation to the national tariff. J Bone Joint Surg Br 2012;94:619-23. https://doi.org/10.1302/0301-620X.94B5.27073.
- Kallala RF, Vanhegan IS, Ibrahim MS, Sarmah S, Haddad FS. Financial analysis of revision knee surgery based on NHS tariffs and hospital costs: does it pay to provide a revision service?. Bone Joint J 2015;97–B:197-201. https://doi.org/10.1302/0301-620X.97B2.33707.
- Hernández-Vaquero D, Fernández-Fairen M, Torres A, Menzie AM, Fernández-Carreira JM, Murcia-Mazon A, et al. Treatment of periprosthetic infections: an economic analysis. Sci World J 2013;2013. https://doi.org/10.1155/2013/821650.
- Berbari EF, Hanssen AD, Duffy MC, Steckelberg JM, Ilstrup DM, Harmsen WS, et al. Risk factors for prosthetic joint infection: case-control study. Clin Infect Dis 1998;27:1247-54. https://doi.org/10.1086/514991.
- Osmon DR, Berbari EF, Berendt AR, Lew D, Zimmerli W, Steckelberg JM, et al. Executive summary: diagnosis and management of prosthetic joint infection: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis 2013;56:1-10. https://doi.org/10.1093/cid/cis966.
- Fraimow HS. Systemic antimicrobial therapy in osteomyelitis. Semin Plast Surg 2009;23:90-9. https://doi.org/10.1055/s-0029-1214161.
- Hackett M. Homecare Medicines ‘Towards a Vision for the Future’ n.d. https://www.media.dh.gov.uk/111w1-Homecare-medicines-towards-a-vision-for-the-future2 (accessed 11 May 2018).
- Minton J, Murray CC, Meads D, Hess S, Vargas-Palacios A, Mitchell E, et al. The Community IntraVenous Antibiotic Study (CIVAS): A Mixed-methods Evaluation of Patient Preferences for and Cost-effectiveness of Different Service Models for Delivering Outpatient Parenteral Antimicrobial Therapy. Southampton: NIHR Journals Library; 2017.
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 2004;39:309-17. https://doi.org/10.1086/421946.
- Kadam PD, Chuan HH. Erratum to: Rectocutaneous fistula with transmigration of the suture: a rare delayed complication of vault fixation with the sacrospinous ligament. Int Urogynecol J 2016;27. https://doi.org/10.1007/s00192-016-2952-5.
- Noonan P, Petersen T, Hanson S, Simpson P, Dasgupta M. 1081: comparing complication rates in central lines versus peripherally inserted central catheters. Crit Care Med 2016;44. https://doi.org/10.1097/01.ccm.0000509756.02057.eb.
Appendix 1 Statistical analysis plan
List of abbreviations
- BNF
- British National Formulary
- CEA
- cost-effectiveness analysis
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CTU
- clinical trials unit
- CUA
- cost–utility analysis
- DAIR
- debridement, antibiotics and implant retention
- DMC
- Data Monitoring Committee
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-3L
- EuroQol-5 Dimensions, three-level version
- GP
- general practitioner
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ITT
- intention to treat
- IV
- intravenous
- MEMS
- Medication Event Monitoring System
- MITT
- modified intention-to-treat analysis
- NICE
- National Institute for Health and Care Excellence
- OHS
- Oxford Hip Score
- OKS
- Oxford Knee Score
- OPAT
- outpatient parenteral antimicrobial therapy
- OVIVA
- Oral Versus IntraVenous Antibiotics
- PIS
- patient information sheet
- PO
- oral
- PP
- per protocol
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SD
- standard deviation
- SmPC
- summary of product characteristics
- VAS
- visual analogue scale