

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 13/04/30. The contractual start date was in October 2014. The draft report began editorial review in May 2019 and was accepted for publication in November 2019. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Martin Dennis, Maree Hackett, Graeme J Hankey, Gillian Mead and Erik Lundström report grants from the National Health and Medical Research Council (Australia) and funding from the Swedish Research Council Framework grant in clinical therapy research during the conduct of the study. Maree Hackett also reports grants from The Stroke Association (London, UK), grants from the National Institute for Health Research (NIHR) Stroke Research Network and a grant in clinical therapy research during the conduct of the study, and grants from the National Heart Foundation of Australia outside the submitted work. She also held a National Health and Medical Research Council (Australia) Career Development Fellowship, level 2 (reference APP1141328) (2018–21). Stephanie Lewis reports being a member of the NIHR Health Technology Assessment General Committee (2016 to present). Peter Sandercock reports lecture fees from Bayer AG (Leverkusen, Germany) paid to his department, outside the submitted work.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2020. This work was produced by Dennis et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2020 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
The burden of stroke
Approximately 130,000 people have a stroke each year in the UK and, even with acute treatments, about 50% of survivors will have long-term residual disability. 1 This places a huge burden on health and social services and informal carers. Although more can be done to implement treatments that we know are effective (e.g. the more widespread provision of thrombolysis and thrombectomy and more rapid access to stroke units), there is still an urgent need to identify new treatments that might reduce neurological impairments, disability and dependency after stroke. One promising intervention that needs to be tested is a widely used antidepressant drug, fluoxetine, which is a selective serotonin reuptake inhibitor (SSRI).
Serotonin reuptake inhibitors in animal models
In animals, SSRIs have several potentially beneficial effects on both normal and diseased brains. First, they have a neurotrophic effect. Neurotrophins are involved in embryogenesis and organogenesis, control neural plasticity in adults, regulate synaptic activity and neurotransmitter synthesis and are essential for the regeneration of nerves. 2 Adult neurogenesis is generally restricted to the subependymal cells of the ventricular system and the subgranular zone of the dentate gyrus in the hippocampus. 3 SSRI antidepressants increase neurogenesis and expression of neurotrophic/growth factors in the adult hippocampus,4 which is likely to account for the behavioural benefits of antidepressants in animals. 5 Importantly, several studies have shown that migration of new neurones to damaged areas of brain may occur,6 and that neurogenesis may also occur in areas of damaged brain in patients who have had ischaemic stroke. 7 Second, fluoxetine may have a neuroprotective effect associated with its anti-inflammatory effect (e.g. repression of microglia activation)8 and enhancement of specific protein expression (e.g. hypoxia-inducible factor-1 alpha and heme oxygenase-1). 9 Third, SSRIs can indirectly affect the adrenergic system through the upregulation of beta-1 receptors. 10
Selective serotonin reuptake inhibitors and motor function in humans
In healthy humans, functional magnetic resonance imaging studies have demonstrated that fluoxetine can modulate cerebral motor activity. 11 In eight patients who had a pure motor stroke who were given fluoxetine, there was hyperactivation in the ipsilesional primary motor cortex during a motor task; moreover, fluoxetine significantly improved motor skills in the affected side. 12 In a small-scale randomised trial of patients who had a unilateral stroke, the administration of citalopram, another SSRI, was associated with a significant improvement in neurological status, as measured with the National Institutes of Health Stroke Scale (NIHSS),13 and a decrease of motor excitability over the unaffected hemisphere, as measured by transmagnetic stimulation. 14 Zittel et al. 15 investigated the effects of a single dose of 40 mg of citalopram in eight chronic stroke patients; dexterity was significantly improved. In a trial of 52 hemiplegic patients who were randomly allocated to receive one of three treatments (20 mg/day of fluoxetine vs. 150 mg/day of maprotiline vs. placebo) for 3 months against a background of physical therapy, those allocated to receive fluoxetine demonstrated the greatest recovery from disability. 16
The FLuoxetine for motor recovery After acute ischaeMic strokE (FLAME) trial17 evaluated the effects of SSRIs on motor recovery after stroke. This double-blind, placebo-controlled, multicentre trial randomised 118 patients who had an ischaemic stroke and unilateral motor weakness to receive either 20 mg of fluoxetine daily or placebo for 3 months. At day 90, the improvement in the Fugl-Meyer Assessment Motor Score from baseline was significantly greater in the fluoxetine group [57 patients, adjusted mean 34.0, 95% confidence interval (CI) 29.7 to 38.4] than in the placebo group (56 patients, adjusted mean 24.3, 95% CI 19.9 to 28.7) (p = 0.003). In a post hoc analysis, the frequency of independent patients [modified Rankin Scale (mRS) of 0–2]18 was significantly higher in the fluoxetine group than in the placebo group (26.3% vs. 8.9%; p = 0.015), although there were no significant differences at other cut-off points. The small sample size limits the study’s generalisability. All patients also received physiotherapy (of unknown intensity), so we do not know whether or not fluoxetine on its own, or with less intense physiotherapy, would also be effective. Importantly, we also do not know whether or not any benefits of fluoxetine persist beyond the treatment period and whether or not fluoxetine might improve outcome in stroke patients without motor deficits. Nevertheless, these promising but inconclusive results clearly justify further larger trials in patients who have motor deficits.
Might selective serotonin reuptake inhibitors be of benefit in recovery of non-motor aspects of stroke?
Several small studies have suggested that fluoxetine might have other neurological benefits (e.g. increased activation of agonist and antagonist muscles in paretic arms after stroke,19 and improvements in executive function after stroke20). We do not know whether or not these beneficial effects of antidepressants are independent of their antidepressant effect. 21
In people with depression, SSRIs modulate the hyperactivity of the hypothalamic pituitary axis (HPA). 22 After stroke, activation of the HPA occurs, resulting in hypercortisolism. Hypercortisolism is associated with the development of delirium after stroke and also predicts worse long-term outcome. 23 Thus, SSRIs might, by attenuating the hypercortisolism that is present after stroke, improve outcomes, including cognition.
Systematic review of effects of fluoxetine on post-stroke outcomes
In 2011, when the Fluoxetine Or Control Under Supervision (FOCUS) trial was being planned, a recent systematic review of randomised controlled trials (RCTs) testing whether or not a course of treatment with fluoxetine started shortly after stroke onset might improve function and prevent post stroke depression identified six RCTs published before December 2009, which together randomised 385 patients. 24 Meta-analysis demonstrated that fluoxetine helped recovery in neurological function (weighted mean difference –4.72, 95% CI –8.31 to –1.13), improved independence in activities of daily living (weighted mean difference –8.04, 95% CI –13.40 to –2.68) and reduced the incidence of post-stroke depression [odds ratio (OR) 0.25, 95% CI 0.11 to 0.56]. A Cochrane review of selective serotonin receptor antagonists in stroke25 subsequently identified 56 trials comparing SSRIs with a control intervention (e.g. usual care or placebo), which were given in the first year after stroke. Fifty-two trials (4059 participants) reported data that could be included in the meta-analyses. Of these 52 trials, 28 used fluoxetine and 31 recruited patients within 3 months of stroke onset. The meta-analyses demonstrated beneficial effects of SSRIs on dependency, disability, neurological deficit, depression and anxiety at the end of treatment. There were benefits even in patients without depression at recruitment. However, there was substantial heterogeneity in the estimates of effect sizes; sensitivity analyses suggested that methodological limitations of many of the included trials may have led to overestimation of effect sizes and there was an excess of gastrointestinal side effects in patients receiving a SSRI. 25 Furthermore, most trials excluded people with cognitive impairment and aphasia, and only eight trials followed patients up after treatment had been discontinued.
Why choose fluoxetine?
There are many SSRI antidepressant medications available. We chose to evaluate fluoxetine because it is one of the most widely studied. Its safety profile is very well established, and the drug is well tolerated in long-term use, even in older patients. There was more evidence for its effectiveness in stroke than for that of alternatives, such as citalopram. 25 A number of manufacturers produce the drug and the price was low, which makes it particularly attractive to health services that are under severe cost pressures. Finally, of all the SSRIs, it has the longest half-life; therefore, gradual reduction in dose is not required when withdrawing the drug (which is inevitable in a trial), which is typically carried out to avoid the possibility of a SSRI-withdrawal syndrome. 26
Potential concerns of using fluoxetine in stroke patients
There are potential risks associated with giving fluoxetine to a wide range of stroke patients. Its reported interaction with antiplatelet and anticoagulant medication might increase bleeding risk, although this is usually minor and limited to bruising. Like other antidepressants, fluoxetine may lower the seizure threshold and, therefore, could increase the frequency of post-stroke seizures. In our Cochrane review, there was a non-significant excess of seizures in patients who were allocated SSRIs. 25 Therefore, we excluded from the FOCUS trial patients who had a history of epileptic seizures. An adverse effect on glycaemic control in diabetic patients has been recorded. Hyponatraemia is a recognised adverse effect and may prove to be more common among stroke patients who may be taking concomitant angiotensin-converting enzyme (ACE) inhibitors, diuretics and proton pump inhibitors. Observational studies have suggested that bone fractures are more common in those taking SSRIs, and this has variably been attributed to an increased risk of fractures in depression, increased risk of falling while taking SSRIs (possibly owing to drowsiness, increased activity or motor effects) and direct effects of SSRIs on bone strength. 27,28 Furthermore, there are already concerns that stroke patients have a greater risk of falls owing to their neurological and functional deficits or concurrent medications (e.g. antihypertensive medication), and greater risk of fractures owing to osteoporosis affecting hemiplegic limbs. 29,30
Nevertheless, fluoxetine has been very commonly prescribed for several years for selected patients who have had a stroke to treat depression and emotionalism without major problems emerging.
Patients who are commenced on psychotropic drugs, including fluoxetine, are encouraged to monitor the effects on their psychomotor function before resuming driving. However, stroke patients in the UK are advised not to drive for at least 1 month after a stroke, which should provide ample time in the trial for any potentially important adverse effects that would affect their driving ability to become apparent.
Rationale for the study
The need for large randomised trials of fluoxetine in stroke
Given these encouraging data, which suggested that fluoxetine might have substantial benefits for a wide range of stroke patients, there was an urgent need to carry out RCTs with adequate power to reliably detect clinically important benefits. Given that fluoxetine is inexpensive (approximately £2.50 per patient per month in the UK), simple to administer and generally well tolerated, if it had an effect that was a fraction of that seen in the FLAME trial17 it would be a very worthwhile treatment option for patients, their carers and health and social services.
The need to identify the patients who might particularly benefit from treatment
Although fluoxetine might improve outcome for a range of stroke patients, it is also plausible, given its diverse pharmacological effects, that the balance of risk and benefit may vary in patients who have had different types of stroke. For instance, pre-clinical work had suggested that motor recovery may be specifically enhanced. In addition, fluoxetine influences bleeding risk, particularly in those taking antithrombotic medication, so there could be differences in effectiveness between patients who have had an ischaemic stroke (who are taking antithrombotics) and patients who have had a haemorrhagic stroke. Patients who have had a severe stroke associated with cognitive and communication problems may be at greater risk of adverse effects because they are unable to report early problems, but they might also have more to gain from a treatment that enhances recovery. In addition, patients who have had a severe stroke are normally at greater risk of post-stroke depression (which may be associated with stroke severity); however, as a consequence of their deficits, these patients are at greater risk that their post-stroke depression is not recognised and, thus, not treated.
The FOCUS trial collaboration (see Appendix 1 for membership) aimed to robustly address several research questions.
Primary research question:
-
Does the routine early administration of fluoxetine [20 mg once per day (o.d.)] for 6 months after an acute stroke improve patients’ functional outcome?
Secondary research questions:
-
If fluoxetine improves functional outcome, does any functional improvement persist after treatment is stopped?
-
Among patients who have had an acute stroke –
-
If there is motor impairment, does fluoxetine improve patients’ motor function and does any improvement persist after treatment is stopped?
-
If there is communication impairment, does fluoxetine improve patients’ communication function and does any improvement persist after treatment is stopped?
-
If there are impairments that preclude the formal assessment of post-stroke mood, does fluoxetine improve patients’ functional outcomes?
-
Does fluoxetine improve patients’ outcome with respect to mood, fatigue, cognition, health-related quality of life (HRQoL) or participation and does any improvement persist after treatment is stopped?
-
Does fluoxetine reduce the cost of health care over the first year?
-
Does fluoxetine increase the risk of serious adverse events?
-
Chapter 2 Methods
The full trial protocol is available at www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#.
Design overview
The FOCUS trial was a pragmatic, investigator-led, multicentre, parallel-group, double-blind, placebo-controlled trial with broad entry criteria and follow-up to ascertain the primary and secondary outcomes at 6 and 12 months.
Setting
The FOCUS trial was carried out in hospital-based stroke services in the UK.
Participant inclusion/exclusion criteria
Inclusion criteria
-
Aged ≥ 18 years.
-
Brain imaging was compatible with intracerebral haemorrhage or ischaemic stroke.
-
Randomisation could be undertaken between 2 and 15 days after stroke onset.
-
Persisting focal neurological deficit was present at the time of randomisation. This needed to be severe enough to warrant 6 months’ treatment with the FOCUS trial medication from the patient’s or carer’s perspective.
Exclusion criteria
-
Subarachnoid haemorrhage (except where secondary to a primary intracerebral haemorrhage).
-
Unlikely to be available for follow-up for the next 12 months (e.g. had no fixed home address).
-
Unable to speak English and had no close family member available to help with follow-up forms.
-
Other life-threatening illness (e.g. advanced cancer) that would have made 12-month survival unlikely.
-
History of epileptic seizures.
-
History of allergy to fluoxetine.
-
Contraindications to fluoxetine, including:
-
hepatic impairment (alanine aminotransferase level more than 3 times the upper normal limit).
-
renal impairment (creatinine level of > 180 µmol/l).
-
-
Pregnancy or breastfeeding, and women of childbearing age not taking contraception. Minimum contraception was an oral contraceptive.
-
Previous drug overdose or attempted suicide.
-
Already enrolled into a clinical trial of an investigational medicinal product (IMP).
-
Current or recent (within the previous month) depression requiring treatment with a SSRI antidepressant.
-
Current medications that have a serious interaction with fluoxetine, including:
-
Use of a monoamine oxidase inhibitor during the previous 5 weeks [e.g. phenelzine (Nardil®, Kyowa Kirin Ltd, Tokyo, Japan), isocarboxacid, tranylcypromine, moclobemide (Manerix®, Mylan, Canonsburg, PA, USA), selegiline (Eldepryl®, Orion Pharma UK, Newbury, UK) and rasagiline (Azilect®, Teva UK Ltd, Castleford, UK)].
-
Pimozide (Orap®, Eumedica Pharmaceuticals, Basel, Switzerland).
-
Metoprolol for heart failure [introduced late in 2016 after a change to the summary of product characteristics (SmPC)] (see Report Supplementary Material 1).
-
Consent
The investigator was responsible for ensuring that informed consent was obtained and the consent form was completed, signed and dated by all parties before any protocol-specific procedures were carried out.
Participant information booklets (PIBs) and informed consent forms (ICFs) were provided (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#; accessed 7 May 2020). Separate versions were available for patients with capacity; proxies were used for patients without capacity. We developed easy-access versions for patients or proxies with cognitive or communication difficulties (see Appendix 2). The verbal explanation to the participant was provided by the investigator or designated person, and aimed to cover all the elements specified in the PIB/ICF. The participants were given every opportunity to clarify any points that they did not understand and, if necessary, ask for more information. Participants could withdraw their consent to participate at any time without loss of benefits to which they would otherwise be entitled.
The participants consented to their medical records being inspected by regulatory authorities and representatives of the sponsor(s) and agreed that the information held and maintained by NHS Digital and other central UK NHS bodies could be shared with us and may be used to help contact them or provide information about their health status.
Written informed consent from the patient was always sought where possible. If this was not possible because the patient could not write, the randomising clinician or nurse could gain witnessed verbal consent. Laws governing consent procedures, and in particular those governing incapacitated adults and their involvement in research, were followed.
The patient or personal legal representative received a folder including a copy of the relevant version of the PIB, a copy of the completed ICF and a patient diary that contained contact details for the trial co-ordinating centre and prompted the recording and reporting of safety outcomes and adverse events, etc. The original ICF and PIB were filed in the site file with the randomisation form (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#; accessed 7 May 2020). The completed ICF was also scanned and uploaded onto the secure trial website or e-mailed, or faxed, to the trial office, before randomisation. The trial management system prompted the research team to do so via e-mail and/or fax until the consent form had been received.
Randomisation
Having obtained consent, the randomising person collected the baseline data necessary to complete a randomisation form (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#; accessed 7 May 2020) and entered the patient’s baseline data into our computerised central randomisation service by means of a secure 24 hours per day/7 days per week (24/7) web interface. After the computer program checked these baseline data for completeness and consistency, it allocated that patient a unique study identification number and a treatment pack number that corresponded to either fluoxetine or placebo. The system applied a minimisation program to achieve balance between the treatment groups for four factors:
-
delay since stroke onset (2–8 vs. 9–15 days)
-
predicted 6-month outcome (based on the six simple variable model)31
-
presence of a motor deficit (based on NIHSS)13
-
presence of aphasia (based on NIHSS).
The six simple variable model is a statistical model that predicts survival and functional outcome after stroke. 31 The variables are (1) the patient’s age, (2) whether or not the patient was independent prior to the stroke, (3) whether or not they lived alone, (4) whether or not after the stroke the patient could lift both arms off the bed, (5) walking without help of another person and (6) talking without being confused (i.e. normal on the verbal component of the Glasgow Coma Scale32).
The minimisation algorithm randomly allocated the first patient to a treatment, but allocated each subsequent patient to the treatment that minimised the imbalance between the treatment groups with respect to the prognostic factors. 33 It was designed to allocate equal numbers to each of the two treatment groups (i.e. a 1 : 1 ratio). To ensure that we retained a random element to treatment allocation, patients were allocated to the group that minimised differences between groups with a probability of 0.8. The system contained a list of treatment codes for each centre and that matched the stocks held at that centre. At the end of the session, each patient was allocated a treatment code that corresponded to an active (20 mg of fluoxetine o.d.) or placebo treatment pack that contained a 6-month supply of capsules held at that centre.
The randomisation system took account of the drug stocks that were held locally to (1) ensure that the allocated treatment was available and (2) minimise wastage. The randomisation system automatically generated an e-mail/fax to the centre co-ordinator and the local research pharmacist to ensure that the allocated treatment was prescribed. The pharmacist or co-ordinator could access treatment codes to replace lost study medication through a secure website by entering the patient’s study ID number and date of birth.
To facilitate drug reconciliation and stock control, the pharmacist or local co-ordinator removed an adhesive treatment number label (flag) from the medication bottle, stuck it onto the confirmation of allocation fax and faxed it back to the trial co-ordinating centre. The trial management system prompted them to do so via e-mail and/or fax until the fax was received.
Following randomisation, the trial co-ordinating centre sent a letter to inform the general practitioner (GP) of the patient’s enrolment in the trial, including a copy of the consent form and the follow-up schedule (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#; accessed 7 May 2020).
The interventions
The interventions were 20 mg of fluoxetine o.d. or placebo for 6 months. The study medication (active and placebo) was manufactured by Unichem (Mumbai, India), imported by Niche Generics Ltd (Hitchin, UK), purchased from Discovery Pharmaceuticals Ltd (Castle Donington, UK) and quality assured, packaged, labelled and distributed by Sharp Clinical Services (Tredegar, UK). Patients were supplied with 186 capsules and were prescribed the study medication (20-mg capsules of fluoxetine or placebo) to be taken daily. If the patient was unable to swallow capsules and had an enteral feeding tube in place, the capsules were broken open and the contents put down the tube.
We measured adherence to the study medication in several ways: recording the date of first and last dose taken, number of missed doses while in hospital, capsule counts when unused capsules were returned and estimated adherence at 6-month follow-up. We recorded the reasons for stopping the study medication early. Our primary measure of adherence was the best estimate of the interval between the first and the last dose based on all of the information available. Therefore, for a particular participant, a capsule count might lead us to modify the estimate of the timing of the last dose (see Chapter 3 for more detail).
Blinding
The patient, their families, the health-care team including the pharmacist, the staff in the co-ordinating centre and anyone involved in outcome assessments were blinded to the treatment allocation by using a placebo capsule that was visually identical to the fluoxetine capsules, even when broken open to allow the administration of the trial medication down an enteral feeding tube.
An emergency unblinding system was available. If a clinician thought that they needed to know the allocated treatment for a patient, they were asked to telephone a 24/7 helpline that was manned by staff from a co-ordinating centre, and provided access, directly or indirectly, to one of our chief investigators. The case for unblinding was discussed and, if agreed, the clinician was given a unique code (based on a simple arithmetic manipulation of the date) to unlock the web-based unblinding system. The clinician could then enter the patient’s details, along with the reason(s) for unblinding, and they were provided with the treatment allocation. This was designed so that those in the co-ordinating centre and those conducting follow-up remained blind to the treatment allocation. Our information technology system logged any attempts to unblind.
Primary outcome
The primary outcome was the mRS (based on ordinal analysis to maximise power and to avoid the problem of including patients with a mRS of > 2 prior to their stroke) at 6 months after randomisation. 18,34 We also collected data on mRS at 12 months (one of our secondary objectives). Patients who died were attributed a score of 6 for this analysis.
The mRS is a simple, time-efficient measure with well-studied reliability that is used to categorise levels of functional outcome (see Table 10). It has been used extensively in large, multicentre stroke trials.
Any misclassification of patients into an inappropriate mRS category may reduce the power of the trial. To minimise misclassification and intermodality differences, we used the simplified modified Rankin Scale questionnaire (smRSq) described by Bruno et al. 18,35,36 This can be delivered via telephone and postal questionnaires and has been completed by patients and proxies. 36,37
Secondary outcomes
To answer our secondary objectives, we collected the following outcome measures:
-
Deaths from all causes by 6 and 12 months.
-
The EuroQol-5 Dimensions, five-level version (EQ-5D-5L) to provide an overall measure of HRQoL and to allow a health economic analysis based on quality-adjusted life-years (QALYs). 38
-
The Mental Health Inventory – 5 questions (MHI-5), which is derived from the Short Form questionnaire-36 items (SF-36) and provided a measure of depression and anxiety symptoms. This brief measure performs well, compared with longer questionnaires (e.g. Mental Health Inventory – 18 questions, General Health Questionnaire –12 questions and General Health Questionnaire –30 questions), in the detection of depression and anxiety symptoms. 39–41
-
The vitality subscale of the SF-36 was used to assess patients’ levels of fatigue. 42,43
-
The Stroke Impact Scale (SIS) provided an overall assessment of patient outcome as well as allowing us to assess the effect of treatment on specific outcomes of importance to the patients. The SIS is a stroke-specific, comprehensive health status measure. The scale was developed with input from patients and caregivers and comprises eight domains (strength, hand function, activities of daily living/instrumental activities of daily living, mobility, communication, emotion, memory and thinking, and participation) from across the full impairment–participation continuum. 44–46 It also provided an overall assessment of recovery with a visual analogue scale. The scale has been validated for use by proxy respondents and has been delivered via telephone and postal questionnaires. 45,47,48
Safety outcomes
-
New diagnosis of depression since randomisation. This was collected at 6- and 12-month follow-up with GPs and participants. We recorded who had made the diagnosis, whether or not any treatment, and specifically any treatment with antidepressant medication, was initiated and whether or not there was any attempt at suicide or self-harm. Sometimes patients were started on an antidepressant without a clear prior diagnosis; in these cases, we made an individual judgement based on all available information and whether the antidepressant had been started for new depression (or low mood) or for another indication, such as neuropathic pain, anxiety or emotionalism without depression.
-
Other adverse events, including further strokes (ischaemic or haemorrhagic), acute coronary events, upper gastrointestinal haemorrhage, falls resulting in injury, new bone fractures, epileptic seizures, symptomatic hypoglycaemia (< 3 mmol/l), hyperglycaemia (> 22 mmol/l) and hyponatraemia (Na+ < 125 mmol/l). Retrospectively, we also categorised patients with other serious bleeds (e.g. lower gastrointestinal, renal tract and subdural) and thrombotic events (deep-vein thrombosis, pulmonary embolism, mesenteric thrombosis, ischaemic limbs) that led to hospital admission. Information on these events were collected via centres, GPs and participants at discharge and 6 months, although we became aware of some events occurring later because they led to hospital admissions that we recorded at the 12-month follow-up.
Follow-up
The principal investigator (PI) and researchers at each site collected the local data listed in the schedule of study assessments below. The chief investigators and the research team in the central co-ordinating office collected the central data (Table 1).
| Assessment | Days | Weeks | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2–15 | 4–6 | 12 | 24 | 26 | 30 | 50 | 52 | 54 | |
| Local | |||||||||
| Screen of eligibility | ✓ | ||||||||
| Check results of post-stroke bloods | ✓ | ||||||||
| Give PIB to patient and/or carer | ✓ | ||||||||
| Consent | ✓ | ||||||||
| Collect baseline data | ✓ | ||||||||
| Randomise | ✓ | ||||||||
| Record treatment code/study number | ✓ | ||||||||
| Prescribe study medication | ✓ | ||||||||
| Dispense 6 months’ worth of treatment | ✓ | ||||||||
| Fax treatment code | ✓ | ||||||||
| Complete discharge form, including: | + | ||||||||
| Adverse events | + | ||||||||
| All medications | + | ||||||||
| Adherence | + | ||||||||
| Updated contact details | + | ||||||||
| Central (postal or telephone) | |||||||||
| E-mail/fax notification of allocation | ✓ | ||||||||
| Letter informing GP of participation | ✓ | ||||||||
| 1-month follow-up for outpatients | o | ||||||||
| Send fax alert following discharge to GP of patient participation | ✓ | ||||||||
| Courtesy call to participant | ✓ | ||||||||
| 3-month prompt to patients | ✓ | ||||||||
| GP questionnaire | ✓ | ||||||||
| New depression | ✓ | ✓ | |||||||
| Other adverse events | o | ✓ | |||||||
| Follow-up on previous adverse events | ✓ | ✓ | |||||||
| All medications | o | ✓ | ✓ | ||||||
| Adherence | o | ✓ | |||||||
| Resource use | ✓ | ✓ | |||||||
| Patient follow-up | |||||||||
| Safety outcomes and adverse events | o | ✓ | |||||||
| Follow-up on previous adverse events | ✓ | ✓ | |||||||
| Adherence | o | ✓ | |||||||
| mRS | ✓ | ✓ | |||||||
| SIS | ✓ | ✓ | |||||||
| MHI-5 | ✓ | ✓ | |||||||
| EQ-5D-5L (HRQoL) | ✓ | ✓ | |||||||
| SF-36 vitality subscale | ✓ | ✓ | |||||||
| Resource use | ✓ | ✓ | |||||||
| Retrieve residual capsules (pill count, reconciliation and destruction) | ✓ | ||||||||
Study safety assessments
Our monitoring system was primarily aimed at identifying suspected unexpected serious adverse reactions (SUSARs), but also at identifying whether or not the frequency of serious adverse reactions was greater than in other populations given fluoxetine and sufficiently common to offset any benefits. We did not aim to detect the occurrence of the very many adverse events that occur in stroke patients and that were very unlikely to be related to participation in the trial or the medication.
The trial materials given to the patient and/or their carer contained details of the known adverse reactions to fluoxetine (based on the SmPC) and the adverse events that commonly occur after stroke. They received a diary in which they were encouraged to record the date and nature of any adverse events.
Patients who were enrolled while they were an inpatient had a hospital discharge form completed by the local co-ordinator at the time of discharge from the recruiting centre or shortly after. The data collected were entered on a secure web-based form or faxed to the co-ordinating centre to ensure that we were alerted to any important adverse reactions. We regularly prompted centres to complete discharge forms for patients with incomplete data.
Patients who were enrolled while they were an outpatient had a central follow-up at 1 month after recruitment to detect safety outcomes and adverse reactions.
At 12 weeks after randomisation, the trial co-ordinating centre staff posted a reminder to the patients to report any adverse events or difficulties with the trial medication, but this was not followed up unless a response was received.
All surviving patients who had not withdrawn consent or indicated that they did not want to be contacted directly were followed up at 6 and 12 months after randomisation, whether or not they adhered to their allocated treatment. At each follow-up, the GP was asked about safety outcomes and other adverse events. In order to detect adverse reactions between the scheduled follow-ups, patients, their carers or their GPs could report any adverse reactions to us via:
-
post – a Freepost envelope and adverse events form to return to us with details of any adverse reactions that the patient had experienced
-
a helpline – a telephone number that allowed the patients or their doctors to leave a message (if non-urgent) or to access a trial doctor (if urgent).
About 2 weeks before any central follow-up was due, the trial co-ordinating centre staff contacted the GPs (or hospital co-ordinators if no discharge form had been received) to check that the patient was alive and that they may be approached for follow-up. The GP was asked (and paid a fee of £56.00) to provide a list of non-IMPs and to complete a questionnaire including information regarding the patient’s adherence to the IMP, details of any safety outcomes or adverse events, hospital admissions and up-to-date contact details for the patient.
If appropriate, the trial co-ordinating centre then posted a questionnaire to the patient at 4 weeks (only for those recruited as outpatients), 26 weeks and 52 weeks. If the patient did not respond to the postal questionnaire, they were telephoned by the co-chief investigators at 6 months and by a trained member of the team at 12 months. The questionnaire at 26 and 52 weeks aimed to capture the primary and secondary outcomes and included the outcome of any adverse events that have been reported earlier in the follow-up. If the patient had incapacity, the next of kin (proxy) was asked to complete and return the forms. If the patient was unable to speak English, we asked that their carer supported them in filling out the forms. If the follow-up information could not be obtained by the postal or telephone questionnaire, we asked the local research team to arrange a face-to-face follow-up at a clinic or home visit.
Data linkage and extract to determine outcome and long-term survival
We collected data from our participating hospitals, the patients and their GPs about hospital admissions during the first 12 months. However, we also planned to obtain information about the health status and resource use of participants to determine outcomes beyond the end of the trial from the Health and Social Care Information Centre. This function has been devolved to NHS Digital in England and Wales and the eData Research and Innovation Service (eDRIS) in Scotland. No centrally held data are available for Northern Ireland.
Management of depression in the trial
Our hypothesis was that new episodes of depression would be less commonly diagnosed and treated in the group allocated to fluoxetine. We ascertained cases of depression by:
-
asking about a diagnosis or initiation of an antidepressant during hospital admission or during the first month – this was recorded on the locally completed discharge form or the 1-month central follow-up form
-
asking the GP at 6 months and 12 months
-
asking the patient (or their proxy) at 6 months and 12 months.
Because the primary question addressed by the FOCUS trial was whether or not a SSRI (20 mg of fluoxetine o.d.) enhanced recovery from stroke, it would be an advantage if the control group were kept free from any SSRIs, including fluoxetine. However, it would be unethical to deny patients in the trial access to effective antidepressant treatment. We therefore asked collaborating clinicians and the patients’ GPs to adhere to the following treatment guideline.
If a patient in the FOCUS trial was diagnosed as having depression (or pathological emotionalism) that the responsible clinician judged to be severe enough to justify treatment with antidepressant drugs, we recommended that, if possible, they should avoid any SSRIs and prescribe either mirtazapine or trazodone. Both drugs are compatible with fluoxetine (there are no common or important interactions), although because mirtazapine has some serotonergic activity there is likely to be a slightly greater risk of precipitating a serotonergic syndrome. Both drugs were recommended by the National Institute for Health and Care Excellence (NICE) for treatment of depression in patients with physical illness. 26 The clinician might alternatively use a tricyclic antidepressant of their choice. We advised that patients taking the trial drug and another antidepressant should be monitored carefully (e.g. check plasma sodium levels to exclude hyponatraemia) to identify any potential interactions.
Sample size
We planned to enrol at least 3000 patients in the main phase of the FOCUS trial. This aimed to provide 90% power with a two-sided 5% level of significance to detect a 5.6% absolute increase in percentage with mRS 0–2 from 27.0% to 32.6% based on an ordinal analysis, which is statistically more efficient than an analysis that dichotomises the mRS. 34
In arriving at this sample size, we took account of the effect sizes seen in the FLAME trial17 alongside the effects that we judged clinicians and their patients would find interesting. Because fluoxetine is safe and inexpensive, the FOCUS trial sought to reliably detect a moderate, but nonetheless clinically important, benefit that might be associated with widespread use of fluoxetine in this population. However, we also took account of the feasibility of enrolling large numbers of patients into the FOCUS trial.
We based our expected outcomes for our placebo group on the distribution of the mRS score measured at 6 months after randomisation in the CLOTS trials,49,50 which evaluated graduated compression stockings.
We used the ordered categorical data method described by Machin et al. 51
The Trial Steering Committee (TSC) reviewed the target sample size and could adjust this based on:
-
advice from the Data Monitoring Committee (DMC)
-
accruing data on –
-
the enrolment into specific prespecified subgroups
-
completeness of follow-up
-
distribution of mRS categories in the population of enrolled subjects (i.e. both treatment groups combined), overall and in specific patient categories (e.g. those with motor deficits or aphasia).
-
For example, if the distribution of mRS was different from that anticipated, then the sample size could be increased. This approach had the advantage that such sample size adjustments could be made without reference to the accumulating unblinded data, and avoided the need for conditional power calculations, which could be unreliable.
Statistical analyses
Our statistical analysis plan was published prior to completion of data collection. 52 We summarise the plan here but, rather than reproduce the whole plan in detail, we have indicated in Chapters 3–7 which analyses were not specified in the published plan. For all analyses, unless otherwise specified, we retained participants in the treatment group to which they were originally assigned, irrespective of the treatment they actually received (i.e. an intention-to-treat analysis). A statistical significance level of p < 0.05 (two tailed) was applied to all analyses. The final prespecified analyses were performed on the data set after any ‘cleaning’ that was required had been completed and the database was locked. The treatment allocation was only then unblinded. Certain post hoc analyses presented in this report included data that had been further cleaned after the main analyses had been carried out and published to correct minor anomalies detected during the analyses.
Primary analysis
This aimed to address our primary research question: does the routine early administration of fluoxetine (20 mg o.d.) for 6 months after an acute stroke improve patients’ functional status at 6 months? To minimise missing data, our analyses of the primary outcome included a mRS score obtained between 90 days and 1 year after randomisation, taking the value measured closest to the 6-month time point.
The primary analysis used an ordinal logistic regression adjusted for factors in the baseline minimisation but also reported in an unadjusted manner. This approach is recommended by the Medicines and Healthcare products Regulatory Agency (UK). The ordinal analysis of mRS was conducted by treatment allocation, under the assumption of proportional odds in the model. This assumption was tested using the score test for proportional odds assumption.
All of the analyses were programmed by our trial statistician (CG), but the primary analysis was also independently programmed by a second statistician and the results were compared; any inconsistencies were identified and resolved by discussion.
Secondary analyses
Analyses of secondary outcomes and analysis of our primary outcome (ordinal mRS) in predefined subgroups were carried out to address the other research questions. Where the outcome of interest was binary, comparison by treatment group was examined using a binary logistic regression and adjusted for factors used in the minimisation algorithm.
Where the outcome of interest was continuous, descriptive statistics are presented [n, mean, standard deviation (SD), minimum, maximum, median, Q1, Q3] and were categorised by allocated treatment. Owing to the nature of the distribution of these measures in this population, a simple unadjusted analysis was performed comparing the two treatment groups using a Mann–Whitney U-test (i.e. not adjusted for variables in the minimisation algorithm).
These analyses were conducted for the following outcomes at 6 and 12 months:
-
fatigue measured by the vitality subscale of the SF-36
-
individual SIS domain scores, a ‘motor score’ derived from averaging scores across three domains (arm, hand, leg and foot strength; hand function; and mobility), a ‘physical function score’ derived by averaging across four domains (arm, hand, leg and foot strength; hand function; mobility; and daily activities) and recovery based on the visual analogue scale
-
quality of life as measured by the EQ-5D-5L.
Our analyses aimed to answer the following questions:
-
If fluoxetine improves functional status (mRS) at 6 months, does any improvement in functional status persist after treatment is stopped? To answer this question, we used ordinal logistic regression to compare functional status (mRS scores) at the 12-month follow-up, as for our primary analysis.
-
Does fluoxetine influence the secondary outcome measures (living circumstances, quality of life, fatigue, stroke impact and mood) at 6 months and/or 12 months? The binary outcomes are living at home or with relative versus care home, hospital or long-term care; mRS at 6 months and 12 months (mRS 0–2 vs. mRS 3–6); and new diagnosis of depression corroborated by the GP or hospital after randomisation by 6 months and 12 months. The continuous outcomes are EQ-5D-5L, vitality subscale of SF-36, SIS and MHI-5.
-
Does fluoxetine increase the risk of serious adverse events? We compared the proportion of patients having any of the following adverse events (all binary outcomes) between randomisation and cessation of the trial medication (i.e. IMP), based on treatment received rather than intention to treat:
-
any recurrent stroke
-
ischaemic stroke [not transient ischaemic attacks (TIAs)]
-
haemorrhagic stroke
-
acute coronary syndromes
-
epileptic seizure
-
episode of hyponatraemia (Na+ < 125 mmol/l)
-
upper gastrointestinal bleeding
-
other major bleeds (lower gastrointestinal, extracranial, urinary or intracranial but extracerebral)
-
poorly controlled diabetes including hyperglycaemia (> 22 mmol/l) or symptomatic hypoglycaemia
-
falls resulting in injury
-
new bone fractures
-
attempted suicide/self-harm.
-
-
If fluoxetine is clinically effective, is it also cost-effective? We carried out a within-trial economic analysis of direct resource costs and health outcomes on an intention-to-treat basis. A health service perspective was adopted for measuring and valuing health service use over a 12-month time horizon. The methods are described in Chapter 6.
-
Is fluoxetine associated with longer survival? Functional outcome at 6 months post stroke is strongly associated with long-term survival; therefore, we wished to determine whether or not any benefits to functional outcome would translate into longer-term survival. 53 We used Cox proportional hazards regression to analyse the effect of treatment on survival to 12 months. We adjusted for the variables included in our minimisation algorithm. We present this analysis graphically [cumulative hazard of death (%) vs. time], providing a hazard ratio (HR) with 95% CIs and a p-value. This analysis will be repeated if survival data for a more prolonged period become available and sufficient resources are available to perform and report the analyses.
-
Does the presence or absence of any of the following factors materially alter the effect of fluoxetine on our primary outcome?
-
Stroke pathology (ischaemic vs. haemorrhagic vs. uncertain pathological type).
-
Age (≤ 70 or > 70 years).
-
Stroke severity [i.e. baseline probability of a good outcome on mRS calculated with the six simple variable model31 to see if effects remain constant across the range of stroke severities (≤ 0.15 vs. > 0.15–1)].
-
Patients who were unable to consent for themselves, as this subgroup will allow us to address the question of whether or not routine use of fluoxetine is likely to benefit patients in whom a formal assessment of mood is impossible because of communication and cognitive problems.
-
Inability to assess mood because of communication or cognitive problems [NIHSS Q1b > 0 or Q1c > 0 or Q9 > 1 or unable to answer Patient Health Questionnaire 2 (PHQ2)54 at randomisation]. Defining the patients’ ability to have their baseline mood assessed based on NIHSS and PHQ2 is likely to be more meaningful than based on patient or proxy consent (see d above), especially because no proxy consent is allowed in Efficacy oF Fluoxetine – a randomisEd Controlled Trial in Stroke (EFFECTS) (Sweden).
-
Patients with and without depression at baseline because our systematic review suggested that the effects of SSRIs were greater in those who were depressed. 25 Depression at baseline was defined as an affirmative response to our baseline question of whether or not the patient has current depression or to both questions in the PHQ2 at baseline.
-
The functional status (mRS) at 6 months was compared with ordinal logistic regression in these mutually exclusive subgroups by entering a treatment-by-subgroup interaction into the regression model.
-
-
In patients with motor deficits at randomisation, does fluoxetine improve motor function? Patients with motor deficits were defined as those with a motor deficit affecting the face/arm or leg (based on NIHSS Q5–9 of > 0). For this subgroup analysis, in addition to comparing their overall functional outcome based on the ordinal analysis of mRS, we compared the motor score with the physical function scores based on the SIS domains described above.
-
In patients with aphasia at randomisation, does fluoxetine improve communication? Patients with aphasia were defined as those with an NIHSS Q9 of > 0. For this subgroup analysis, in addition to comparing their overall functional outcome based on mRS based on ordinal analysis, we compared with the SIS communication subscale.
-
For questions 7 and 8, because patients may have a combination of neurological deficits, individual patients may appear in more than one subgroup.
-
Is there a relationship between functional status at 6 months and mood and is this relationship affected by fluoxetine? We performed exploratory analyses of potential mediating factors (e.g. the role of depression).
Missing data
Our randomisation systems did not allow investigators to proceed to treatment allocation without entering complete baseline data. The mRS, our primary outcome, includes death; therefore, the number of participants with missing mRS at follow-up was small. Anyone with a missing mRS was not included in any analysis requiring mRS (complete-case analysis).
For secondary outcomes [e.g. SIS, MHI-5, vitality subscale of the SF-36 and EQ-5D-5L] for which missing data were expected because data were not available for patients who did not survive, we presented results for those who were alive at follow-up and any discrepancies in death rates between groups were taken into account in the interpretation. Missing data for single questions within scores were handled as detailed by each scoring method. Where responses to all questions within a scale or subscale were missing, that patient was not included in that part of the analysis.
Protocol deviations, adherence and blinding
Inclusion/exclusion violations: we reported the number and percentage of participants randomised who did not meet the entry criteria (e.g. non-strokes), with exclusion criteria. However, they were included in the primary analysis. A secondary analysis excluded ineligible patients (see below).
Unblinding: we reported the number of patients who required unbinding of study medication during the trial by treatment group and, where available, present the reasons for unblinding.
Adherence: each participant was issued with a 6-month supply of trial medication (186 capsules). At 6 months, they were asked if they had completed the course and taken all of the capsules and how often they took capsules on average. They were asked the reasons for stopping, as well as the date of stopping. Where possible, we retrieved and counted the unused trial medication. Before unblinding, we derived an estimated date on which the patient was thought to have taken their last dose of trial medication and used the interval (days) from first dose to that date as our main measure of adherence. This was based on all of the available information. We used a combination of the following to define several types of non-adherence to the protocol (see 1–8 below):
-
inclusion/exclusion violations
-
the answers to the adherence questions (see above)
-
number, percentage and duration of any open-label SSRI intake before the 6-month follow-up
-
the reasons for stopping trial medication.
A so-called intention-to-treat analysis, in which patients’ outcomes are analysed in the groups that they were randomised to regardless of treatment received, provides the least biased and most robust evidence of the effect of treatment. However, the observed treatment effect may be reduced if a large number of patients are included who are unlikely to benefit because they did not have a stroke or more likely where a large proportion of patients do not receive the allocated treatment or actually received the alternative treatment (i.e. cross-overs).
Where the primary analysis does not demonstrate an improvement of functional outcome (mRS) at the 6-month follow-up, the question arises of whether or not this is this likely to be a result of poor adherence to the protocol and/or trial medication? This is important because we would not wish to abandon a potentially useful treatment simply because of poor adherence to trial protocols or trial medication. These might be improved in any future trials. We undertook further per-protocol analyses to reassure the clinical community that the trials have not underestimated any treatment effect to an extent that would alter future clinical practice, or more likely the need for further randomised trials of SSRI in stroke.
Inevitably, analyses that try to take account of adherence introduce a degree of patient selection and, thus, are likely to introduce bias.
These prespecified exploratory sensitivity analyses to account for non-adherence included all of the analyses of the primary outcome and selected secondary outcomes:
-
living at home or with relative versus care home, hospital or long-term hospital care
-
mRS at 6 months and 12 months (mRS 0–2 vs. mRS 3–6)
-
new diagnosis of depression between randomisation and 6 months and 12 months
-
SIS domain scores
-
averaged score over all SIS domains
-
SF-36 vitality subscale score
-
utility based on EQ-5D-5L and population preferences.
These analyses do not include any analysis of subgroups defined on the basis of baseline variables. The following groups were sequentially added to the group excluded from the analyses:
-
Patients who did not meet the entry criteria for the trial.
-
Patients who did not receive any trial medication.
-
Patients who received < 90 days of trial medication because of failures in trial procedures, for example failures to transfer trial medication with patients during moves between hospitals, care homes and home. The 90-day cut-off point was chosen because previous trials have tested this duration of treatment with apparent benefit. 17,25
-
Patients who received < 90 days of trial medication because of patient or relative concerns but not because of suspected adverse reactions.
-
Patients who received < 90 days of trial medication because they experienced symptoms that were attributed to the trial medication.
-
Patients who had been allocated to placebo who received a SSRI (fluoxetine or other) within the first 90 days and the SSRI was not known to have been stopped within 10 days of starting.
-
Patients who had been allocated to fluoxetine who received a SSRI (fluoxetine or other) within the first 90 days and the SSRI was not known to have been stopped within 10 days of starting.
-
Patients who did not complete at least 150 days of treatment. We chose this cut-off point because patients sometimes received the questionnaires shortly before 6 months, and some stopped the trial medication at that point, whereas others finished the 186 capsules. We regarded both as fully adherent.
Research governance
The trial was co-ordinated by a Project Management Group: Professor Martin Dennis, Professor Gillian Mead (the joint chief investigators and PIs for two participating sites), Karen Innes (trial manager) and Catriona Graham (trial statistician).
Trial co-ordinating centre
The trial co-ordinating centre was responsible for all aspects of the management of the FOCUS trial and was based at the Centre for Clinical Brain Sciences at the University of Edinburgh. Responsibilities included regulatory submissions and compliance; financial management; monitoring of sites; training; patient information and communication; end-point assessment; data collection systems and data management; IMP management; statistical analysis; reports and publications; and archiving of the trial master file in accordance with funder and sponsor requirements.
Trial Steering Committee
A TSC was established, including a stroke survivor and a carer, to oversee the conduct and progress of the trial. The terms of reference of the TSC, the draft template for reporting and the names and contact details were agreed at its first meeting (see Report Supplementary Material 1).
Data Monitoring Committee
An independent DMC was established to oversee the safety of participants in the trial. During the period of recruitment into the study, interim analyses of the baseline and follow-up data were supplied, in strict confidence, to the chairperson of the DMC, along with any other analyses that the committee requested. In the light of these analyses, the DMC could advise the chairperson of the TSC if, in their view, the randomised comparisons had provided (1) ‘proof beyond reasonable doubt’ that for all, or some, the treatment is clearly indicated or clearly contraindicated and (2) evidence that might reasonably be expected to materially influence future patient management. Appropriate criteria of proof beyond reasonable doubt were not specified precisely, but the DMC worked on the principle that a difference of at least three standard errors in an interim analysis of a major outcome event (e.g. death from all causes or independent survival at 6 months) would be needed to justify halting, or modifying, the study before the planned completed recruitment. This criterion has the practical advantage that the exact number of interim analyses would be of little importance, and so no fixed schedule was proposed. Following a report from the DMC, the TSC decided whether to modify entry to the study (or seek extra data).
The terms of reference of the DMC, the DMC charter and the names and contact details were agreed at the first meeting of the DMC (see Report Supplementary Material 2).
Patient and public involvement
In December 2010, service users overseeing a study of post-stroke fatigue commented on our plans for the FOCUS trial. We met with the group in May 2011. We included their suggestions about patient information booklets (i.e. explaining the rationale for using fluoxetine in people without depression, listing all side effects), and agreed that all trial participants would be sent a summary of the trial results if they wished. A group of stroke survivors with aphasia developed the easy-access version of the patient information booklet, guided by Professor Marian Brady (see Appendix 2). A stroke survivor, Judith Williamson, from the National Institute for Health Research (NIHR) Stroke Research Network (SRN), and Zena Jones (manager of the NIHR SRN patient and public involvement group) attended our first investigator meeting (in June 2011). In March 2013, the SRN patient and public involvement group advised us how to enhance the proportion of eligible patients consenting, and how to facilitate follow-up. This advice featured in our Autumn 2013 newsletter to sites. In November 2013, Ms Jones and Ms Williamson endorsed our plans to telephone patients immediately after hospital discharge. They edited the script that would guide the telephone call. They commented on a draft of the funding application to NIHR. Ms Williamson and a carer were on our TSC, offering advice and comments throughout the trial. They were particularly influential in discussion of how we would disseminate the results of the trial to participants and their families. They commented on the final newsletter, which was sent out on the day the results were presented at conference, and published in The Lancet. 55 They have also had input into the drafting of this report, specifically the Plain English summary.
Chapter 3 Results 1: conduct
The main results of the FOCUS trial have been published. 55 In this chapter, and following chapters, we present the main results and additional information.
Recruitment
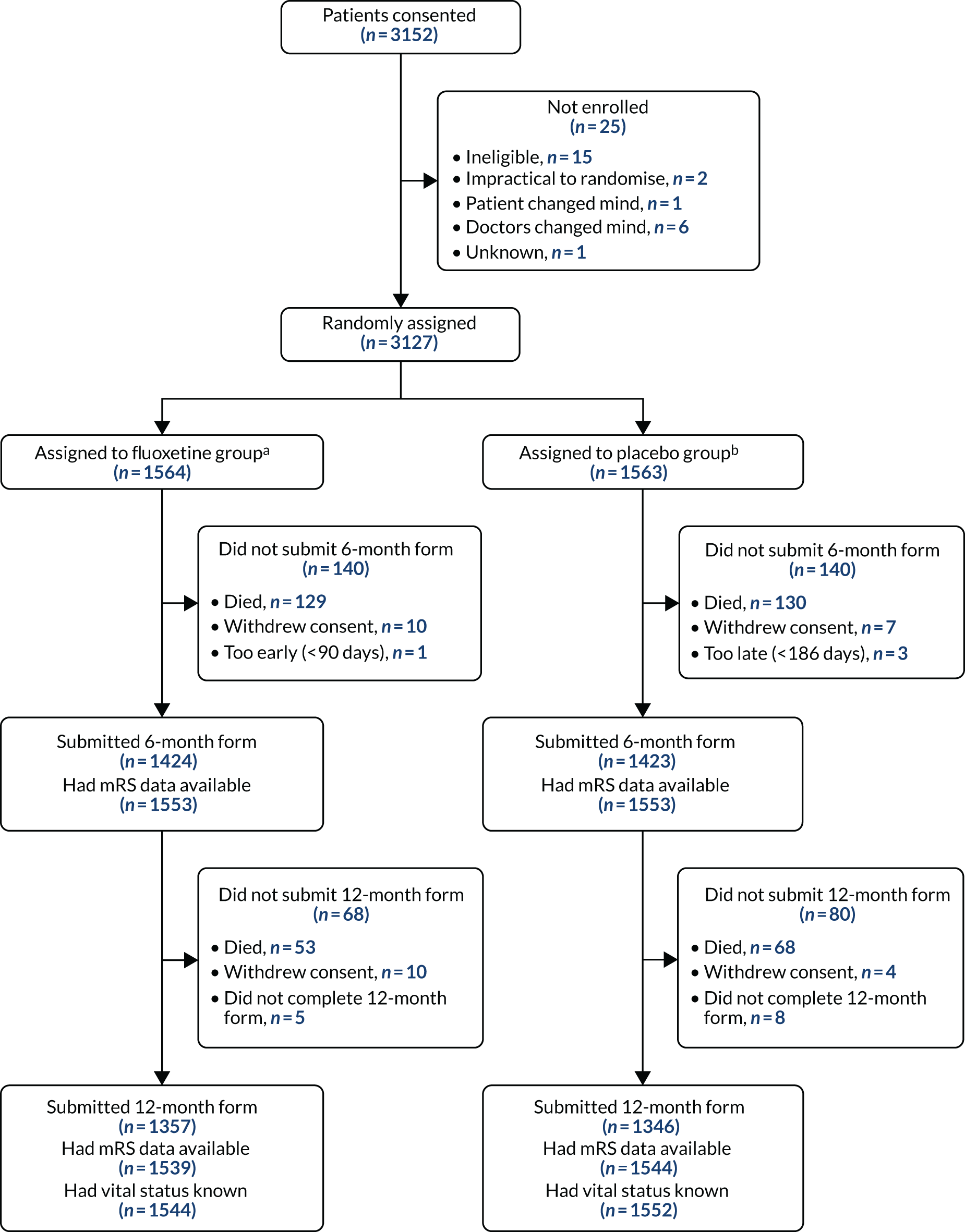
Between 10 September 2012 and 31 March 2017, 103 UK hospitals consented 3152 patients and enrolled 3127 patients. Recruitment was stopped after we had exceeded our minimum target of 3000 patients to account for recruitment of ineligible patients and withdrawals (Figure 1).
FIGURE 1.
Recruitment graph showing planned vs. actual recruitment.

The network of centres was built on the networks that we had established to carry out previous trials, including the CLOTS49,50 and IST343 trials. We enrolled new centres throughout the trial period. The trial co-ordinating centre worked closely with the research teams at prospective centres, and the majority of site initiation visits were carried out remotely using telephone and video conferencing. The trial was facilitated by the NIHR-funded research networks, which provided funding for local research nurses. Centres also received a £300 pharmacy start-up fee, and approximately £46 per patient recruited.
Thirty-one patients were identified as ineligible between obtaining consent and randomisation; in other cases, the patients, their proxy or their treating clinician changed their mind about participation in the trial. Of the 3127 patients who were enrolled, 1564 were allocated to the fluoxetine group and 1563 were allocated to the placebo group. Eleven of these patients did not meet our eligibility criteria: two in each group had a final diagnosis other than stroke and seven others were identified as meeting exclusion criteria after randomisation (e.g. a history of epilepsy, self-harm or some other contraindication to fluoxetine). The ineligible patients were retained in our intention-to-treat analyses. The patients’ progress through the trial is shown in Figure 2.
FIGURE 2.
Participant flow. a, 1544 inpatients with discharge form; 20 recruited as outpatients; b, 1536 inpatients with discharge form; 27 recruited as outpatients. Reproduced from the FOCUS trial collaboration. 55 © 2018 The Author(s). Published by Elsevier Ltd. This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Baseline characteristics of recruited patients
The baseline characteristics of the two treatment groups were well balanced with respect to all measured variables (Tables 2–6). 55
| Characteristics of patients randomised | Allocated treatment | |
|---|---|---|
| Fluoxetine (N = 1564) | Placebo (N = 1563) | |
| Sex, n (%) | ||
| Female | 589 (37.66) | 616 (39.41) |
| Male | 975 (62.34) | 947 (60.59) |
| Age, n (%) | ||
| ≤ 70 years | 666 (42.58) | 664 (42.48) |
| > 70 years | 898 (57.42) | 899 (57.52) |
| Age (years), mean (SD) | 71.24 (12.35) | 71.48 (12.06) |
| Ethnicity, n (%) | ||
| Asian | 30 (1.92) | 31 (1.98) |
| Black | 35 (2.24) | 29 (1.86) |
| Chinese | 0 (0.00) | 1 (0.06) |
| Other | 4 (0.26) | 9 (0.58) |
| White | 1495 (95.59) | 1493 (95.52) |
| Marital status, n (%) | ||
| Married | 879 (56.20) | 846 (54.13) |
| Partner | 93 (5.95) | 91 (5.82) |
| Divorced/separated | 109 (6.97) | 100 (6.40) |
| Widowed | 337 (21.55) | 354 (22.65) |
| Single | 124 (7.93) | 150 (9.60) |
| Other | 22 (1.41) | 22 (1.41) |
| Living arrangement, n (%) | ||
| Living with someone else | 1057 (67.58) | 1034 (66.15) |
| Living alone | 485 (31.01) | 516 (33.01) |
| Living in an institution | 10 (0.64) | 4 (0.26) |
| Other living arrangement | 12 (0.77) | 9 (0.58) |
| Employment, n (%) | ||
| Full-time employment | 287 (18.35) | 258 (16.51) |
| Part-time employment | 76 (4.86) | 70 (4.48) |
| Retired | 1122 (71.74) | 1134 (72.55) |
| Unemployed/disabled | 53 (3.39) | 60 (3.84) |
| Other employment | 26 (1.66) | 41 (2.62) |
| Characteristics of patients randomised | Allocated treatment, n (%) | |
|---|---|---|
| Fluoxetine (N = 1564) | Placebo (N = 1563) | |
| Coronary heart disease | 281 (17.97) | 300 (19.19) |
| Ischaemic stroke/TIA | 274 (17.52) | 294 (18.81) |
| Diabetes | 337 (21.55) | 303 (19.39) |
| Hyponatraemia | 19 (1.21) | 26 (1.66) |
| Intracranial bleed | 27 (1.73) | 23 (1.47) |
| Upper gastrointestinal bleed | 25 (1.60) | 26 (1.66) |
| Bone fractures | 241 (15.41) | 256 (16.38) |
| Depression | 130 (8.31) | 123 (7.87) |
| Characteristics of patients randomised | Allocated treatment, n (%) | |
|---|---|---|
| Fluoxetine (N = 1564) | Placebo (N = 1563) | |
| Stroke diagnosis | ||
| Non-stroke (final diagnosis) | 2 (0.13) | 2 (0.13) |
| Ischaemic stroke | 1410 (90.15) | 1406 (89.96) |
| Intracerebral haemorrhage | 154 (9.85) | 157 (10.04) |
| OCSP classification of ischaemic strokes | ||
| Total anterior circulation infarct | 318 (20.33) | 317 (20.28) |
| Partial anterior circulation infarct | 561 (35.87) | 553 (35.38) |
| Lacunar infarct | 307 (19.63) | 283 (18.11) |
| Posterior circulation infarct | 191 (12.21) | 230 (14.72) |
| Uncertain | 33 (2.11) | 23 (1.47) |
| Cause of stroke: modified TOAST classification | ||
| Large artery disease | 278 (17.77) | 234 (14.97) |
| Small vessel disease | 252 (16.11) | 218 (13.95) |
| Embolism from heart | 377 (24.10) | 411 (26.30) |
| Another cause | 38 (2.43) | 35 (2.24) |
| Unknown/uncertain | 465 (29.73) | 508 (32.50) |
| Characteristics of patients randomised | Allocated treatment | |
|---|---|---|
| Fluoxetine (N = 1564) | Placebo (N = 1563) | |
| SSV | ||
| Age (years), mean (SD) | 71.24 (12.35) | 71.48 (12.06) |
| Independent before stroke, n (%) | 1431 (91.50) | 1435 (91.81) |
| Living alone, n (%) | 485 (31.01) | 516 (33.01) |
| Able to lift both arms off bed, n (%) | 924 (59.08) | 935 (59.82) |
| Able to talk and not confused, n (%) | 1166 (74.55) | 1164 (74.47) |
| Able to walk without help from another person, n (%) | 435 (27.81) | 412 (26.36) |
| Probability that alive and independent, median (IQR) (derived from SSV) | 0.28 (0.07–0.63) | 0.26 (0.07–0.63) |
| 0 to ≤ 0.15, n (%) | 592 (37.85) | 591 (37.81) |
| > 0.15 to 1, n (%) | 972 (62.15) | 972 (62.19) |
| NIHSS, median (IQR) | 6 (3–11) | 6 (3–11) |
| Presence of a motor deficit, n (%) | 1361 (87.02) | 1361 (87.08) |
| Presence of aphasia, n (%) | 457 (29.22) | 449 (28.73) |
| Current diagnosis of depression (patient/proxy reported), n (%) | 26 (1.66) | 18 (1.15) |
| Taking a non-SSRI antidepressant, n (%) | 65 (4.16) | 77 (4.93) |
| Current mood: PHQ2,54 n (%) | ||
| 2 yes responses | 81 (5.18) | 60 (3.84) |
| 1 yes response | 136 (8.70) | 130 (8.32) |
| 0 yes responses | 1347 (86.13) | 1373 (87.84) |
| Characteristics of patients randomised | Allocated treatment | |
|---|---|---|
| Fluoxetine (N = 1564) | Placebo (N = 1563) | |
| Delay (days) since stroke onset at randomisation | ||
| Delay, mean (SD) | 6.93 (3.64) | 6.98 (3.64) |
| 2–8, n (%) | 1070 (68.41) | 1072 (68.59) |
| 9–15, n (%) | 494 (31.59) | 491 (31.41) |
| Enrolled as a hospital inpatient (not outpatient clinic), n (%) | 1544 (98.72) | 1536 (98.27) |
| Patient consented, n (%) | 1136 (72.63) | 1118 (71.53) |
| Proxy consented, n (%) | 428 (27.37) | 445 (28.47) |
Withdrawal
The term withdrawal is used widely in RCTs but it is important to be precise about what is meant. In the FOCUS trial, we separately categorised patients into the following groups:
-
patients who had stopped their trial medication, but were content to be followed up
-
patients who may or may not have stopped their medication but no longer wanted to be contacted directly for follow-up information, but were content for us to obtain follow-up information from family, friends, GPs or routine data sources
-
patients or their proxies who withdrew consent and wanted the patient and their data to be taken out of the study from that point on, so that we used data collected up to that point only.
When we were informed that the patients wished to withdraw, we clarified which of the above applied. Only 31 patients withdrew consent; the timing and treatment allocations are shown in the participant flow diagram (see Figure 2) and treatment allocations were fairly well balanced.
Discharge forms
A discharge form (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#) was completed for all 3040 patients enrolled as inpatients.
The 6- and 12-month follow-ups
We completed the 12-month follow-up in the trial in June 2018. The participant flow diagram (see Figure 2) shows the completeness of follow-up with respect to our primary outcome and vital status. Table 7 shows the methods of follow-up to obtain these data. Forty-nine per cent of 6-month follow-up assessments were obtained by postal questionnaire (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#). The remainder required a telephone reminder or were completed by telephone interview. The telephone follow-ups for participants not returning postal 6-month questionnaires were carried out by the two co-chief investigators (MD and GM) who were trained and certified in the use of the mRS and had conducted an independent validation of the smRSq. 18,35,36 Those conducting the 12-month telephone follow-ups had received training in their application.
| Method of follow-up | Allocated treatment, n (%) | |
|---|---|---|
| Fluoxetine | Placebo | |
| 6 months | ||
| Completed 6-month postal questionnaire without telephone prompting | 693 (48.6) | 700 (49.1) |
| Required prompting or clarification by telephone to complete 6-month questionnaire | 312 (21.9) | 276 (19.4) |
| Completed 6-month questionnaire by telephone | 420 (29.5) | 450 (31.6) |
| Total completing 6-month questionnaire | 1425 (100.0) | 1426 (100.0) |
| 12 months | ||
| Completed 12-month postal questionnaire without telephone prompting | 745 (54.9) | 743 (55.2) |
| Required prompting or clarification by telephone to complete 12-month questionnaire | 195 (14.4) | 179 (13.4) |
| Completed 12-month questionnaire by telephone | 417 (30.7) | 424 (31.5) |
| Total completing 12-month questionnaire | 1357 (100.0) | 1346 (100.0) |
Based on our previous trials, we had expected 80% of follow-up to be returned by postal questionnaire, rather than 50%. This placed a much greater burden on the central co-ordinating team than expected. In addition, in the FOCUS trial, patients were followed up at 6 and 12 months, doubling the number of follow-ups. Because missing outcome data would reduce the power of the trial, and can more importantly introduce bias because it is rarely missing randomly, the central team allocated far more time and resources to follow-up than had been planned.
Reasons for the low response rate
We did not formally assess the reasons for the low rate of response to the postal questionnaires, but the following issues contributed:
-
Centres providing incomplete or inaccurate postal addresses for participants. Increasingly, stroke services admit patients acutely to one hospital and then transfer the patients to another hospital, or community rehabilitation centre, for ongoing care. We collected the discharge address on our discharge form, and this was often completed when patients moved from the centre in which they were recruited. Downstream facilities often did not inform us that the patient had then been discharged to an address other than their original home address. Our team spent a lot of time tracking down these patients to obtain their current address.
Anecdotally, patients had often not opened our postal questionnaires because they believed them to be circulars. Any indication of the content of the letter on the outside of the envelope (e.g. contains FOCUS trial questionnaires) could have an impact on patient confidentiality and was, therefore, not used.
-
Our 6- and 12-month follow-up questionnaires included the SIS. This is long, containing 59 items in several domains. In one domain, the Likert scaling for three of the nine items is reversed (i.e. good outcomes have low instead of high scores in the 1–5 range). Patients found this confusing, and they often entered internally inconsistent information, which we then queried by telephone.
-
The burden of carrying out telephone follow-ups to clarify or complete information received by post, or to complete the whole follow-up by telephone, was considerable, and was increased as a result of several factors, including:
-
Patients changing telephones from landlines to mobiles, which meant that our co-ordinating team spent a lot of time communicating with centres, downstream health-care facilities, patients’ GPs and proxies to obtain up-to-date contact information.
-
Patients and proxies increasingly not answering calls from unknown numbers, which are often assumed to be marketing calls or scams. In many cases, patients or their proxies were telephoned at different times of the day and week, often on multiple occasions, before contact was actually made. To overcome this barrier, we often texted the recipient in advance of a phone call to increase the likelihood that they would answer. In future trials, it would be useful at the time of recruitment to enter the trial co-ordinating centre’s number into the patients’ and/or proxies’ mobile phones as a contact so they could make a more informed decision about whether or not to answer a telephone call from the trial centre.
-
-
General practitioners and their staff varied greatly in the assistance they would provide in completing the GP questionnaires and in helping us contact patients. Most were very helpful but:
-
Some refused to provide information because they were unaware that the patient had provided written consent for them to do so. We had routinely sent GPs a copy of the patient information and the completed consent form with a covering letter at the time of recruitment. It appears that this was often filed and, therefore, it was not obvious to the staff when they received a request 6 or 12 months later. Some patients had moved house and changed GPs, which meant that the original trial documentation was not necessarily available to the new GP.
-
Some GPs reported that they were simply too busy to help.
-
Some felt that the fee of £54 negotiated with the primary care network was insufficient, and that they required a much larger sum to complete a follow-up questionnaire. We occasionally paid a little more, but usually we obtained the data via an alternative route.
-
Unblinding
The emergency unblinding procedure was carried out for only three patients. One was on the request of a coroner after the patient died, a second was on the request of the sponsor, for a suspected unexpected serious adverse reaction, and the third was because the responsible clinician felt that knowledge of the treatment would significantly alter their management of the patient. All of the patients had been allocated fluoxetine.
Adherence
The primary measure of adherence was the estimated duration of study medication (interval in days from first to last dose of study medication) based on all available data. Capsule counts were available in 398 (25.6%) of those allocated fluoxetine and 410 (26.4%) of those allocated placebo. The patients returned a median of 32 [interquartile range (IQR) 10–135] capsules in the fluoxetine group and 33 (IQR 11–139) capsules in the placebo group. Our primary measure of adherence was available in 1417 (91%) patients in each group. The median duration of treatment was 185 (IQR 149–186) days in the fluoxetine group and 183 (IQR 136–186) days in the placebo group. The median delay between randomisation and first dose was 0 (IQR 0–1) days in both treatment groups. A total of 1519 (97%) participants in the fluoxetine group and 1494 (96%) participants in the placebo group received their first dose by day 2 after randomisation (Table 8). Table 9 shows the number and proportion of patients meeting our eligibility criteria and different levels of adherence to the study medication. Patients stopping the trial medication because of perceived adverse effects within the first 90 days was only marginally more common in the fluoxetine group (n = 143, 9.1%) than in the placebo group (n = 122, 7.8%). About two-thirds of patients took the study medication for at least 150 days.
| Interval to first dose | Allocated treatment, n (%) | Total, n (%) | |
|---|---|---|---|
| Fluoxetine | Placebo | ||
| 0 days | 817 (52) | 816 (52) | 1633 (52) |
| 1 day | 658 (42) | 619 (40) | 1277 (41) |
| 2 days | 44 (3) | 59 (4) | 103 (3) |
| 3 days | 17 (1) | 17 (1) | 34 (1) |
| ≥ 4 days | 16 (1) | 30 (2) | 46 (1) |
| Missing | 8 (1) | 11 (1) | 19 (1) |
| Total | 1564 (100) | 1563 (100) | 3127 (100) |
| Groups cumulatively excluded | Number meeting each exclusion criterion | Cumulative number removed from analysis | Number remaining in fluoxetine group | Number remaining in placebo group |
|---|---|---|---|---|
| None: as per intention-to-treat analysis | 0 | 0 | 1553 | 1553 |
| Ineligible: did not meet all inclusion criteria | 11 | 11 | 1548 | 1547 |
| Received no study medication after randomisation | 17 | 26 | 1540 | 1540 |
| Received < 90 days of study medication owing to failure to follow trial procedures | 128 | 152 | 1480 | 1474 |
| Received < 90 days of study medication owing to patient/carer/doctor choice | 208 | 342 | 1405 | 1359 |
| Received < 90 days of study medication owing to suspected adverse reaction | 265 | 607 | 1262 | 1237 |
| Allocated placebo but received SSRI for > 10 days within 90 days | 84 | 628 | 1262 | 1216 |
| Allocated fluoxetine and received SSRI for > 10 days within 90 days | 52 | 651 | 1239 | 1216 |
| Received < 150 days of study medication unless died earlier still taking study medication | 847 | 892 | 1122 | 1092 |
| Received < 150 days of study medication for any reason including death | 975 | 1016 | 1051 | 1039 |
Problems with adherence
We identified some reasons for non-adherence to the trial medication. These included:
-
Recruiters not checking that they had someone named on the delegation log who could sign the initial prescription. Usually this just led to a short delay in starting the medication, but was more complicated if the patient was discharged or moved hospital prior to receiving the first dose.
-
Delays in obtaining the trial medication from pharmacies, especially when the patient was randomised later in the day.
-
In discharge letters to GPs, it was sometimes unclear that patients were in a placebo-controlled trial and that patient had been given their trial medication. This sometimes led GPs to prescribe fluoxetine or nursing home staff to ask for fluoxetine to be prescribed on admission or when a patient’s supply was running low.
-
Patients, or their families or nursing home staff, often wanted the trial medication in a blister pack, in a dosette box provided by pharmacists or in liquid form and so they would then request that the GPs prescribe fluoxetine – GPs would comply with this. Patients and relatives could put their trial medication into a dosette box, but we were unable to identify any means of getting pharmacies to put the trial medication into dosette boxes or blister packs. Thus, some patients allocated to the placebo group were inadvertently given fluoxetine by their GP.
We introduced a safety alert letter (see www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#) after patient 1000 was recruited. This was automatically generated when we received the discharge form (see www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#). The introduction of the safety fax had no effect on the frequency of inappropriate prescribing of antidepressants, and fluoxetine in particular, in the randomised patients.
The 24/7 helpline was very helpful in reducing these problems; in many cases, patients, their families or health-care staff rang the helpline to clarify the situation and we were able to reduce the non-adherence by talking with all of these groups.
Confirmation of safety outcome events and data cleaning
This was carried out by the data manager and co-chief investigator (MD) prior to unblinding of the treatment code. We did not have an event or outcome adjudication committee because there is evidence that these do not significantly improve data quality. 58 We aimed to obtain information from patients, their relatives, GPs and hospitals (usually via the research co-ordinators) to confirm the event, its nature and its date. We established some rules that were applied so that assumptions were made consistently.
Monitoring
We monitored the quality and integrity of the accumulating clinical data in accordance with a protocol agreed with the study sponsors [the Academic and Clinical Central Office for Research and Development (ACCORD) representing the University of Edinburgh and NHS Lothian], which involved central statistical monitoring, supplemented by on-site monitoring and detailed source data verification in the co-ordinating centre and triggered visits when patterns in the data at a centre seemed anomalous. All baseline data and in-hospital and 6- and 12-month outcome data were subject to verification checks built into the randomisation and data management system. In practice, almost all monitoring visits to centres were triggered by the occurrence of protocol deviations and violations, rather than any concerns about data quality.
Closeout
All trial monitoring activities, including those for trial closure, were carried out in compliance with the agreed FOCUS monitoring plan and in accordance with the trial sponsor’s standard operating procedures to ensure that all study-related activities were reconciled, recorded and reported at the end of the trial in accordance with the trial protocol and all applicable regulatory requirements and that they complied with good clinical practice.
Remote closeouts were conducted by the FOCUS central team, using a combination of sponsor-approved checklists (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#; accessed 7 May 2020) and trial-specific instructions and documents for reconciliation of the investigator site file for completeness. A completed closeout checklist was available for all centres. Bespoke per-site reports were provided to each site to reconcile all essential documentation required for the investigator site file in preparation for archiving. The site files included site delegation logs, documentation of staff qualifications and training (good clinical practice, curricula vitae), IMP management and reconciliation, consent confirmation, randomisation records, documentation of violations, deviations, serious adverse events and SUSARs. A final closeout visit to the co-ordinating centre and trial master file review was conducted by the trial sponsor in preparation for final closure and archiving of all essential trial master file documents.
Chapter 4 Results 2: patient outcomes and events
The main results of the trial have been published. 55
The number and percentage of patients in each mRS category by treatment group is shown in Table 10.
| Primary outcome | Allocated treatment, n (%) | |
|---|---|---|
| Fluoxetine | Placebo | |
| Disability on the mRS at 6 months | ||
| 0: no symptoms | 114 (7.34) | 124 (7.98) |
| 1: no clinically significant disability despite symptoms | 302 (19.45) | 309 (19.90) |
| 2: slight disability – unable to do everything | 156 (10.05) | 155 (9.98) |
| 3: moderate disability – unable to live independently but can walk | 518 (33.35) | 510 (32.84) |
| 4: moderately severe disability and unable to walk without help from another person | 121 (7.79) | 122 (7.86) |
| 5: severe disability – unable to sit up | 213 (13.72) | 203 (13.07) |
| 6: dead | 129 (8.31) | 130 (8.37) |
| Total number of patients with mRS | 1553 (100.00) | 1553 (100.00) |
| Number of patients with missing mRS | 11 | 10 |
| Total number of patients randomised | 1564 | 1563 |
The primary outcome at 6 months in the two treatment groups is compared in Figure 3. An ordinal comparison of the distribution of patients across the mRS at 6 months, adjusted for variables included in the minimisation algorithm, was similar in the two groups [common odds ratio (COR) 0.951, 95% CI 0.839 to 1.079; p = 0.439], where a COR in favour of placebo is < 1.0. The unadjusted analysis provided similar results (COR 0.961, 95% CI 0.848 to 1.089; p = 0.531). The ordinal analysis of mRS has been conducted by treatment allocation, under the assumption of proportional odds in the model. This assumption was found to hold using the score test for proportional odds assumption (p = 0.9947). Comparing the mRS dichotomised into 0–2 and 3–6, there was similarly little difference between the groups (adjusted OR 0.955, 95% CI 0.812 to 1.123; p = 0.576; unadjusted OR 0.957, 95% CI 0.827 to 1.107; p = 0.352).
FIGURE 3.
Comparison of the distribution of patients across the seven categories of the mRS in the two allocated treatments. Reproduced from the FOCUS trial collaboration. 55 © 2018 The Author(s). Published by Elsevier Ltd. This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
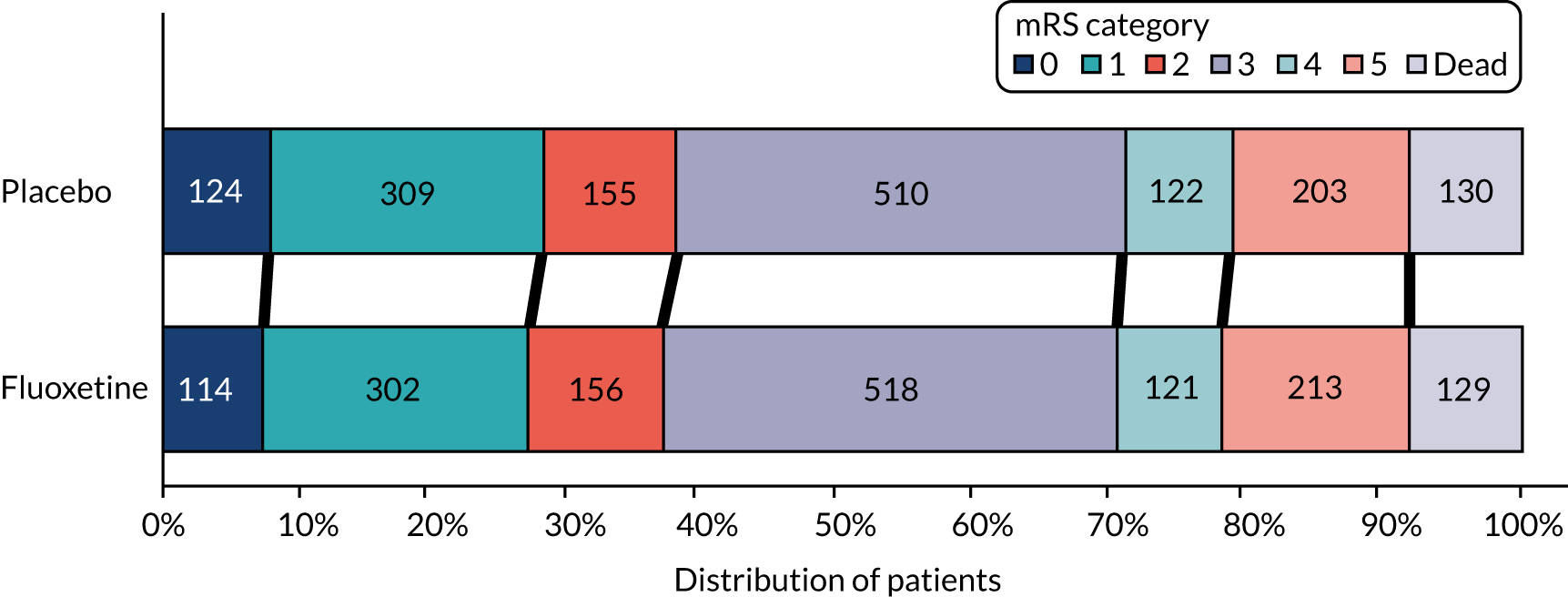
The results of our prespecified subgroup analyses are shown in Table 11. There were no statistically significant interactions between the prespecified subgroups and the effect of treatment on the primary outcome.
| 6 months, adjusted analysis | Allocated treatment (n) | COR | 95% CI | p-value | |
|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||
| Overall | 1553 | 1553 | 0.952 | 0.840 to 1.079 | 0.439 |
| Variables used in the minimisation | |||||
| Probability of being alive and independent at 6 months | |||||
| 0 to ≤ 0.15 | 590 | 586 | 1.026 | 0.836 to 1.258 | 0.326 |
| 0.15 to 1 | 963 | 967 | 0.906 | 0.771 to 1.063 | |
| Delay from onset to randomisation (days) | |||||
| 2–8 | 1061 | 1067 | 0.957 | 0.822 to 1.114 | 0.951 |
| 9–15 | 492 | 486 | 0.940 | 0.750 to 1.178 | |
| Motor deficit | |||||
| No | 203 | 201 | 1.207 | 0.847 to 1.721 | 0.153 |
| Yes | 1350 | 1352 | 0.919 | 0.803 to 1.052 | |
| Aphasia | |||||
| No | 1099 | 1108 | 0.894 | 0.770 to 1.038 | 0.123 |
| Yes | 454 | 445 | 1.107 | 0.874 to 1.403 | |
| Other prespecified subgroup analyses | |||||
| Stroke type | |||||
| Haemorrhagic | 153 | 156 | 0.816 | 0.546 to 1.221 | 0.427 |
| Ischaemic | 1400 | 1397 | 0.969 | 0.848 to 1.107 | |
| Age group (years) | |||||
| ≤ 70 | 661 | 661 | 0.947 | 0.780 to 1.151 | 0.944 |
| > 70 | 892 | 892 | 0.952 | 0.806 to 1.124 | |
| Who gave consent | |||||
| Proxy | 427 | 443 | 0.944 | 0.741 to 1.204 | 0.899 |
| Patient | 1126 | 1110 | 0.940 | 0.810 to 1.092 | |
| Inability to assess mood | |||||
| No | 1167 | 1165 | 0.891 | 0.770 to 1.031 | 0.089 |
| Yes | 386 | 388 | 1.125 | 0.871 to 1.452 | |
| Baseline depression | |||||
| No | 1457 | 1483 | 0.952 | 0.836 to 1.084 | 0.805 |
| Yes | 96 | 70 | 1.030 | 0.586 to 1.798 | |
Table 12 shows the effect of fluoxetine on our primary outcome in subgroups defined by their meeting the eligibility criteria and being adherent to the study medication to different degrees (see Table 9). These are a series of prespecified per-protocol analyses that sequentially exclude subgroups of patients who either did not meet our eligibility criteria or had incomplete adherence to the study medication. There was no trend towards greater benefit in those with greater adherence.
| Groups cumulatively excluded | Number remaining in fluoxetine group | Number remaining in placebo group | COR for mRS | 95% CI | p-value |
|---|---|---|---|---|---|
| None: as per intention-to-treat analysis | 1553 | 1553 | 0.951 | 0.839 to 1.079 | 0.439 |
| Ineligible: did not meet all inclusion criteria | 1548 | 1547 | 0.949 | 0.837 to 1.077 | 0.418 |
| Received no study medication after randomisation | 1540 | 1540 | 0.948 | 0.835 to 1.076 | 0.406 |
| Received < 90 days of study medication owing to failure to follow trial procedures | 1480 | 1474 | 0.958 | 0.842 to 1.090 | 0.514 |
| Received < 90 days of study medication owing to patient/carer/doctor choice | 1405 | 1359 | 0.912 | 0.797 to 1.042 | 0.175 |
| Received < 90 days of study medication owing to suspected adverse reaction | 1262 | 1237 | 0.936 | 0.813 to 1.078 | 0.360 |
| Allocated placebo but received SSRI for > 10 days within 90 days | 1262 | 1216 | 0.923 | 0.801 to 1.064 | 0.268 |
| Allocated fluoxetine and received SSRI for > 10 days within 90 days | 1239 | 1216 | 0.927 | 0.804 to 1.068 | 0.294 |
| Received < 150 days of study medication unless died earlier still taking study medication | 1122 | 1092 | 0.888 | 0.765 to 1.032 | 0.121 |
| Received < 150 days of study medication for any reason including death | 1051 | 1039 | 0.921 | 0.788 to 1.075 | 0.296 |
Table 13 shows the safety outcomes at 6 months. Those allocated fluoxetine were less likely to be diagnosed with a new episode of depression [n = 210 (13.0%) vs. n = 269 (16.9%), difference in proportion –3.78%, 95% CI –1.26% to –6.30%; p = 0.003]. Those allocated fluoxetine had an increased risk of fractures [n = 45 (2.9%) vs. n = 23 (1.5%), difference in proportion 1.41%, 95% CI 0.38% to 2.43%; p = 0.007]. There were no statistically significant differences in other safety outcomes, although there were expected trends towards more events in the fluoxetine group for adverse effects listed in the SmPC for fluoxetine [epileptic seizures, falls, hyponatraemia (Na+ < 125 mmol/l – reported by treating clinician) and markers of poor diabetic control] (see the project web page: www.journalslibrary.nihr.ac.uk/programmes/hta/130430/#; accessed 7 May 2020). There were only small differences in the rates of thrombotic and bleeding events (Table 14) despite concerns that fluoxetine might affect platelet function and interact with antithrombotic medications.
| Adverse events by 6 months | Allocated treatment, n (%) | Difference (%) | 95% CI (%) | p-value | |
|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||
| Epileptic seizures | 58 (3.71) | 40 (2.56) | 1.15 | –0.07 to 2.37 | 0.065 |
| Fall with injury | 120 (7.67) | 94 (6.01) | 1.66 | –0.11 to 3.43 | 0.066 |
| Fractured bone | 45 (2.88) | 23 (1.47) | 1.41 | 0.38 to 2.43 | 0.007 |
| Hyponatraemia Na+ < 125 mmol/l | 22 (1.41) | 14 (0.90) | 0.51 | –0.24 to 1.26 | 0.181 |
| Hyperglycaemia | 23 (1.47) | 16 (1.02) | 0.45 | –0.33 to 1.22 | 0.260 |
| Symptomatic hypoglycaemia | 23 (1.47) | 13 (0.83) | 0.64 | –0.11 to 1.39 | 0.094 |
| New depression | 210 (13.43) | 269 (17.21) | –3.78 | –6.30 to –1.26 | 0.003 |
| New antidepressant prescription | 280 (17.90) | 357 (22.84) | –4.94 | –7.76 to –2.12 | 0.001 |
| Attempted/actual suicide | 3 (0.19) | 2 (0.13) | 0.06 | –0.02 to 0.34 | 0.655 |
| Adverse events by 6 months | Allocated treatment, n (%) | Difference (%) | 95% CI (%) | p-value | |
|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||
| Any stroke | 56 (3.58) | 64 (4.09) | –0.51 | –1.86 to 0.83 | 0.454 |
| All thrombotic events | 78 (4.99) | 92 (5.89) | –0.90 | –2.49 to 0.69 | 0.268 |
| Ischaemic stroke | 43 (2.75) | 45 (2.88) | –0.13 | –1.29 to 1.03 | 0.826 |
| Other thrombotic events | 20 (1.28) | 27 (1.73) | –0.45 | –1.30 to 0.40 | 0.303 |
| Acute coronary events | 15 (0.96) | 23 (1.47) | –0.51 | –1.28 to 0.26 | 0.191 |
| All bleeding events | 41 (2.62) | 38 (2.43) | 0.19 | –0.91 to 1.29 | 0.735 |
| Haemorrhagic stroke | 7 (0.45) | 9 (0.58) | –0.13 | –0.60 to 0.37 | 0.615 |
| Upper gastrointestinal bleed | 21 (1.34) | 16 (1.02) | 0.32 | –0.44 to 1.08 | 0.409 |
| Other major bleeds | 13 (0.83) | 14 (0.90) | –0.06 | –0.71 to 0.58 | 0.845 |
We also carried out a safety analysis, which analysed patients according to the treatment they received rather than the treatment they were allocated. These data are provided in Report Supplementary Material 3. The other secondary outcomes (fatigue, mood, HRQoL and SIS) are compared in Tables 15 and 16. Those treated with fluoxetine had better mood measured on the MHI-5 at the 6-month follow-up than those allocated placebo [median 76 (IQR 60–88) vs. 72 (IQR 56–88); p = 0.010]. This is consistent with the lower rate of new episodes of depression that we observed. There were no statistically significant differences in any other secondary outcomes at 6 months, including any of the nine domains of the SIS, the vitality subscale of the SF-36 and the EQ-5D-5L.
| Outcome | Allocated treatment | p-value (Mann–Whitney U-test) | |||||
|---|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||||
| Missing (n) | Median | IQR | Missing (n) | Median | IQR | ||
| Vitality subscale of SF-36 | 19 | 56.25 | 37.5–75.00 | 21 | 56.25 | 43.75–56.25 | 0.673 |
| MHI-5 | 26 | 76.00 | 60.00–88.00 | 22 | 72.00 | 56.00–88.00 | 0.010 |
| EQ-5D-5L | 12 | 0.56 | 0.21–0.74 | 4 | 0.56 | 0.19–0.75 | 0.587 |
| SIS domain | Allocated treatment | p-value (Mann–Whitney U-test) | |||||
|---|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||||
| Missing (n) | Median | IQR | Missing (n) | Median | IQR | ||
| Strength | 13 | 56.25 | 31.25–81.25 | 14 | 62.50 | 37.50–81.25 | 0.701 |
| Hand ability | 14 | 45.00 | 0.00–90.0 | 18 | 50.00 | 0.00–90.00 | 0.482 |
| Mobility | 9 | 63.89 | 36.11–86.11 | 7 | 63.89 | 33.33–88.89 | 0.549 |
| Motor | 12 | 54.86 | 27.31–83.33 | 13 | 56.78 | 28.75–82.64 | 0.513 |
| Daily activities | 11 | 62.50 | 37.50–90.00 | 13 | 65.00 | 35.00–90.00 | 0.624 |
| Physical function | 11 | 56.77 | 30.38–84.31 | 12 | 58.82 | 30.56–84.10 | 0.515 |
| Memory | 23 | 82.14 | 57.14–96.43 | 18 | 82.14 | 57.14–96.43 | 0.307 |
| Communication | 12 | 89.29 | 67.86–100 | 11 | 92.86 | 71.43–100.0 | 0.192 |
| Emotion | 42 | 75.00 | 58.33–88.89 | 29 | 75.00 | 58.33–88.89 | 0.469 |
| Participation | 12 | 62.50 | 37.50–87.50 | 15 | 65.63 | 40.63–87.50 | 0.260 |
| Recovery (VAS) | 16 | 60.00 | 40.00–80.00 | 9 | 60.00 | 40.00–80.00 | 0.982 |
The number and percentage of patients in each mRS category at the 12-month follow-up by treatment group is shown in Table 17. An ordinal comparison of the distribution of patients across the mRS at 12 months, adjusted for variables included in the minimisation algorithm, was similar in the two groups (COR 1.015, 95% CI 0.894 to 1.151; p = 0.820), where a COR in favour of placebo is < 1.0. The unadjusted analysis provided similar results (COR 1.011, 95% CI 0.892 to 1.145; p = 0.866). When comparing the mRS dichotomised into 0–2 and 3–6, there was similarly little difference between the groups (adjusted OR 1.033, 95% CI 0.879 to 1.215; p = 0.691; unadjusted OR 1.019, 95% CI 0.880 to 1.181; p = 0.799).
| mRS at 12 months | Allocated treatment, n (%) | |
|---|---|---|
| Fluoxetine | Placebo | |
| 0 | 133 (8.64) | 145 (9.39) |
| 1 | 251 (16.31) | 237 (15.35) |
| 2 | 178 (11.57) | 175 (11.33) |
| 3 | 494 (32.10) | 505 (32.71) |
| 4 | 90 (5.85) | 81 (5.25) |
| 5 | 211 (13.71) | 203 (13.15) |
| 6 (dead) | 182 (11.83) | 198 (12.82) |
| Missing mRS | 25 | 19 |
| Total number of patients randomised | 1564 | 1563 |
The survival of patients in the two treatment groups is compared in Figure 4. Anyone who died after 1 year was considered to be alive and censored at 365 days and any participant who withdrew consent was considered to be alive and was censored at the time of withdrawal. There was no statistically significant difference in the hazards of death over the first 12 months after randomisation (HR 0.929, 95% CI 0.756 to 1.141; p = 0.482) adjusted for baseline variables.
FIGURE 4.
The Kaplan–Meier survival curves for the two allocated treatments with number of subjects at risk. Times have been capped at 365 days. Reproduced from the FOCUS trial collaboration. 55 © 2018 The Author(s). Published by Elsevier Ltd. This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.

Table 18 shows the number of patients with a new episode of depression or who had started an antidepressant between randomisation and 12 months. The difference in the cumulative number of patients diagnosed with a new episode of depression over the 12 months between the two treatment groups was no longer statistically significant. More patients had been started on antidepressants in the placebo group than in the fluoxetine group, but some were started on antidepressants for indications other than depression.
| Outcome by 12 months | Allocated treatment, n (%) | Difference (%) | 95% CI (%) | p-value | |
|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||
| New depression | 292 (18.67) | 327 (20.92) | –2.25 | –5.04 to 0.54 | 0.114 |
| New antidepressant prescription | 358 (22.89) | 410 (26.23) | –3.34 | –6.36 to –0.33 | 0.030 |
The difference in MHI-5 scores between the groups at 6 months was not sustained at 12 months (Table 19), and there were no statistically significant differences between treatment groups in vitality or HRQoL at 12 months.
| Outcome | Allocated treatment | p-value (Mann–Whitney U-test) | |||||
|---|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||||
| Missing (n) | Median | IQR | Missing (n) | Median | IQR | ||
| Vitality subscale of SF-36 | 31 | 50.00 | 37.50–75.00 | 35 | 50.00 | 37.50–75.00 | 0.904 |
| MHI-5 | 34 | 72.00 | 56.00–88.00 | 36 | 76.00 | 56.00–88.00 | 0.711 |
| EQ-5D-5L excluding dead patients | 18 | 0.59 | 0.24–0.75 | 13 | 0.59 | 0.27–0.77 | 0.309 |
The SIS scored in the two treatment groups at 12 months is shown in Table 20. There was little difference between the treatment groups on any of the domains.
| SIS domain | Allocated treatment | p-value (Mann–Whitney U-test) | |||||
|---|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | ||||||
| Missing (n) | Median | IQR | Missing (n) | Median | IQR | ||
| Strength | 35 | 56.25 | 31.25–75.00 | 31 | 56.25 | 37.50–75.00 | 0.384 |
| Hand ability | 34 | 50.00 | 0.00–90.00 | 33 | 50.00 | 5.00–90.00 | 0.281 |
| Mobility | 28 | 66.67 | 36.11–88.89 | 29 | 66.67 | 38.89–88.89 | 0.543 |
| Motor | 33 | 55.56 | 28.80–83.33 | 31 | 58.61 | 31.20–83.70 | 0.329 |
| Daily activities | 30 | 67.50 | 40.00–90.00 | 33 | 67.50 | 40.00–90.00 | 0.581 |
| Physical function | 30 | 57.81 | 32.81–84.24 | 31 | 60.10 | 33.54–85.28 | 0.372 |
| Memory | 33 | 78.57 | 60.71–96.43 | 32 | 82.14 | 57.14–96.43 | 0.425 |
| Communication | 32 | 89.29 | 67.86–100.0 | 29 | 89.29 | 71.43–100.0 | 0.314 |
| Emotion | 46 | 72.22 | 58.33–86.11 | 44 | 73.61 | 58.33–88.89 | 0.744 |
| Participation | 32 | 65.63 | 40.63–90.63 | 33 | 65.63 | 40.63–90.63 | 0.930 |
| SIS recovery (VAS) | 24 | 60.00 | 40.00–80.00 | 24 | 60.00 | 40.00–80.00 | 0.933 |
We assessed the effect of treatment among the subgroup with motor deficit at baseline (n = 2722) and among those who had a mRS at 6 months (n = 2702) but found no evidence of an effect on the mRS (p = 0.2172). Of the 2722 participants who had a motor deficit at baseline, 2438 had a motor score outcome [fluoxetine median 48.43 (IQR 24.98–78.84) vs. placebo median 52.66 (IQR 25.28–77.22); p = 0.471]. In addition, in the 906 patients with aphasia at baseline, 899 had a mRS at 6 months and 794 had a SIS communication domain score at 6 months. There was little difference in the mRS (see Table 11) or SIS communication scores between the treatment groups [fluoxetine median 64.29 (IQR 32.14–89.29) vs. placebo 64.29 (IQR 35.71–89.29); p = 0.497].
Chapter 5 Results 3: post hoc analyses to better understand the observed effect of fluoxetine on the risk of bone fractures
Introduction
The statistically significant excess of bone fractures (see Table 13) that we observed is potentially important for several reasons. First, many observational studies have demonstrated an association between SSRI use and fractures. 27,28 However, the question of whether or not this association is causal has remained. RCTs provide stronger evidence of causality than any other research method. Second, if fractures affect patients’ functional outcomes, then this excess might have offset some beneficial effects of fluoxetine on functional outcomes. Third, if SSRIs, and fluoxetine specifically, cause fractures, the following question arises: what are the mechanisms by which they have this effect? Is it owing to an increased risk of falling, which could be through several possible mechanisms, or the proposed effects of SSRIs on bone density, or both, or neither?
We have, therefore, carried out some post hoc analyses in an attempt to answer the following questions:
-
What sort of fractures occurred after stroke and were they likely to have had an impact on patients’ function?
-
Might the increased risk of fractures, along with their associated loss of function, have offset the beneficial effects of fluoxetine on neurological recovery?
-
What baseline factors were associated with fracture risk?
-
Does the temporal pattern of fractures in the FOCUS trial provide any useful information about the relative importance of the potential mechanisms of SSRI-induced fractures?
Methods
We have published a brief account of further analyses and results that attempts to address these questions. 59 We extracted further information from our trial database while remaining blind to treatment allocation. Here we present the post hoc analyses in more detail. Having identified the excess risk of bone fractures in our prespecified analyses,52 we also prespecified further analyses to address our additional research questions about fractures. We coded baseline medications to distinguish non-SSRI antidepressants, blood pressure lowering, bone density reducing (e.g. glucocorticoids) or bone density increasing (e.g. calcium, vitamin D and bisphosphonates) medications and those that might increase risk of falls (e.g. tranquillisers). In addition, we extracted available data on the fracture site and any associated falls or seizures. We had not systematically collected data that indicated the side of the body where fracture(s) occurred. We compared the number of fractures occurring in those with and without specific characteristics but formally tested each variable by plotting Kaplan–Meier survival curves in those with and without each characteristic and compared these with the log-rank statistic. We did not formally test differences in fracture risk where numbers were small (i.e. fewer than six). We included all variables with a p-value of < 0.1 into a Cox proportional hazards model to identify independent predictors of fracture risk. We repeated these analyses focusing on fractures only at sites typically associated with low bone density (i.e. neck of femur, wrist and vertebrae).
Results
Type of fractures
In our original published analysis of fractures,55 we included 68 patients with at least one fracture each (see Table 13), where the fracture had been diagnosed on radiography after randomisation. However, in three patients, on reviewing all of the evidence, the fracture had probably occurred before randomisation. We excluded these patients (two from the fluoxetine group and one from the placebo group) from further analyses. Sixty-five of the 3127 (2.1%) patients who were enrolled had 67 definite new fractures (two patients sustained more than one fracture simultaneously) within 6 months of randomisation. The differences in fracture risk between the fluoxetine and the placebo group remained statistically significant (in a univariate analysis) having removed the three patients [43 (2.75%) vs. 22 (1.41%); difference 1.34%, 95% CI 0.34% to 2.34%; p = 0.009]. Among the 65 patients with fractures, 59 (90.8%) fractures resulted from a fall. The sites of the fractures are shown in Table 21; 26 patients (40%) had a neck of femur fracture, which was quite likely to have had an impact on patients’ functional outcome.
| Fracture-related outcome | Number | % |
|---|---|---|
| Number of patients sustaining a fracture | 65 | |
| Number of fractures sustained | 67 | 100.0 |
| Site of fracture | ||
| Neck of femur | 26 | 40.0 |
| Vertebral | 10 | 15.4 |
| Any long bone | 10 | 15.4 |
| Wrist | 7 | 10.8 |
| Rib | 4 | 6.2 |
| Pelvis | 3 | 4.6 |
| Clavicle | 1 | 1.5 |
| Other (e.g. skull and patella) | 6 | 9.2 |
| Site associated with osteoporosis | 40 | 61.5 |
| Associated with a fall | 59 | 90.8 |
| Associated with an epileptic seizure | 1 | 1.5 |
Effect of removing the patients with fracture from estimates of effect on modified Rankin Scale
Removing the 65 patients with at least one fracture during follow-up from the primary analysis (ordinal imbalance) did not significantly alter the estimate of effect of fluoxetine on the mRS (COR including those with fractures 0.951, 95% CI 0.839 to 1.079; p = 0.439; COR for those without fractures 0.961, 95% CI 0.847 to 1.093; p = 0.545). 59
Risk factors for fractures
Those who had a fracture within 6 months were older [mean age 76 (SD 12.2) vs. 71 (SD 11.6) years, difference 4.9 years, 95% CI for difference 2.0 to 7.9 years; p = 0.001] and had slightly more severe strokes [median NIHSS 7.0 (IQR 4–11) vs. 6.0 (IQR 3–11); p = 0.407]. The numbers (%) of patients with specific baseline characteristics divided into those with and without subsequent fractures are shown in Table 22, along with the p-value for the difference based on the log-rank analysis of the Kaplan–Meier curves. The differences were statistically significant only between those aged > 70 years and younger patients, between women and men and between those allocated to fluoxetine and those allocated to placebo. Medical history including prior fractures, stroke pathology, NIHSS, type of deficit and medications of different types had no statistically significant associations with fracture risk. Previous depression and being able to walk at the time of the stroke did not reach the 5% level of significance; however, they were included in our Cox proportional hazards model as they were approaching significance (p < 0.1).
| Characteristic | Fracture by 6 months, n (%) | Log-rank statistic | |
|---|---|---|---|
| No | Yes | ||
| Total number of patients randomised | 3062 (100.0) | 65 (100.0) | |
| Randomised treatment | |||
| Fluoxetine | 1521 (49.7) | 43 (66.2) | 0.008 |
| Placebo | 1541 (50.3) | 22 (33.9) | |
| Sex | |||
| Female | 1167 (38.1) | 38 (58.5) | 0.001 |
| Male | 1895 (61.9) | 27 (41.5) | |
| Age group (years) | |||
| ≤ 70 | 1313 (42.9) | 17 (26.2) | 0.003 |
| > 70 | 1749 (57.1) | 48 (73.9) | |
| Before the stroke | |||
| Dependent in ADL | 253 (8.3) | 8 (12.3) | 0.176 |
| Ischaemic stroke/TIA | 557 (18.2) | 11 (16.9) | 0.882 |
| Diabetes | 628 (20.5) | 12 (18.5) | 0.760 |
| Bone fractures | 486 (15.9) | 11 (16.9) | 0.829 |
| Depression | 244 (8.0) | 9 (13.9) | 0.091 |
| Stroke type | |||
| Intracerebral haemorrhage | 301 (9.8) | 10 (15.4) | 0.132 |
| Stroke deficits at baseline | |||
| Unable to walk | 2227 (72.7) | 53 (81.5) | 0.085 |
| Unable to lift both arms | 1243 (40.6) | 25 (38.5) | 0.870 |
| Unable to talk | 779 (25.4) | 18 (27.7) | 0.520 |
| Motor deficit on NIHSS | 2665 (87.0) | 57 (87.7) | 0.797 |
| Visual field deficit on NIHSS | 844 (27.6) | 14 (21.5) | 0.326 |
| Limb ataxia on NIHSS | 753 (24.6) | 17 (26.2) | 0.832 |
| Baseline medications | |||
| Non-SSRI antidepressant | 137 (4.5) | 5 (7.7) | |
| Treatments for osteoporosis | 287 (9.4) | 5 (7.7) | |
| Major or minor tranquillisers | 121 (4.0) | 3 (4.6) | |
| Parkinson’s disease medication | 14 (0.5) | 2 (3.1) | |
| BP-lowering medication | 2178 (71.1) | 52 (80.0) | 0.100 |
| Treatments for vertigo | 129 (4.2) | 5 (7.7) | |
| Any of these drugs of interest | 2349 (76.7) | 55 (84.6) | 0.116 |
The Cox proportional hazards model with all those variables reaching and approaching statistical significance is shown in Table 23.
| Parameter | Value | Probability > χ2 | HR | HR 95% CI |
|---|---|---|---|---|
| Sex | Female | 0.008 | 1.978 | 1.193 to 3.280 |
| Age group | > 70 years | 0.018 | 1.973 | 1.123 to 3.467 |
| Previous depression | No/unknown | 0.135 | 0.581 | 0.285 to 1.184 |
| Able to walk | No | 0.240 | 1.462 | 0.776 to 2.755 |
| Randomised treatment | Fluoxetine | 0.009 | 1.992 | 1.191 to 3.330 |
When those variables that do not reach statistical significance at the 5% level are sequentially removed, the resulting Cox model (Table 24) contained just three variables: sex, age group and randomised treatment.
| Parameter | Value | Probability > χ2 | HR | HR 95% CI |
|---|---|---|---|---|
| Sex | Female | 0.003 | 2.131 | 1.294 to 3.511 |
| Age group | > 70 years | 0.017 | 1.972 | 1.127 to 3.451 |
| Randomised treatment | Fluoxetine | 0.008 | 2.000 | 1.196 to 3.344 |
The Cox proportional hazards model (see Table 23) showed that only age of > 70 years (HR 1.97, 95% CI 1.13 to 3.45; p = 0.017), female sex (HR 2.131. 95% CI 1.294 to 3.511; p = 0.003) and fluoxetine treatment (HR 2.00, 95% CI 1.196 to 3.344; p = 0.008) were independent predictors of fracture. The model was almost identical when only those with fractures at sites likely to be affected by osteoporosis (i.e. neck of femur, vertebral and wrist) were included.
Temporal pattern of fractures
The Kaplan–Meier curve comparing fracture risk in the two treatment groups is shown in Figure 5. The risks appear to diverge early after randomisation, which might suggest that an increased risk of falling associated with taking fluoxetine contributed to the excess of fractures. A delayed divergence might have suggested that the excess was mainly owing to effects on bone density, which presumably would not have occurred immediately.
FIGURE 5.
Kaplan–Meier curves to 6 months, with number of subjects at risk, comparing the risk of fracture in those allocated fluoxetine and placebo where patients dying or being lost to follow-up were censored. Reproduced from Dennis et al. 59 © American Heart Association, Inc.

Chapter 6 Results 4: health economic evaluation
Introduction
We undertook a within-trial economic analysis to estimate the cost-effectiveness of fluoxetine on an intention-to-treat basis. The primary treatment effect in the economic analysis was estimated using an individual-level regression model for average (mean) incremental costs and incremental survival times over 12 months after randomisation.
Methods
Resource use and costs
The number and duration of hospital stays and other secondary care contacts were recorded using information obtained from the case report form. We had planned to use data from NHS Digital in England and Wales, and from eDRIS for patients recruited in Scotland, but we were unable to obtain these data in time to produce analyses that could be included in this report.
Unit costs and analysis
We converted length-of-stay distributions into cost estimates based on a per-diem hospital cost. Resource use was valued from the perspective of the NHS using the 2017/18 and 2018/19 national tariffs with currencies and prices for 2018. 60 Per-diem hospital costs were derived using tariff information for Healthcare Resource Group codes AA22C to AA22G (Cerebrovascular Accident, Nervous System Infections or Encephalopathy) with varying levels of complexity and complications. 60 Our base case simulated unit costs using a gamma distribution around a mean of £515 per day with higher unit costs for the first 2 days to allow for conditional payments (best-practice tariffs) for acute stroke care (i.e. direct admissions to an acute stroke unit, initial brain imaging and thrombolysis assessment). We also considered other cost distributions, including the application of trim points for lengths of hospital stay beyond 63 days with lower unit costs of £240 per day. The results from the sensitivity analysis were quantitatively and qualitatively similar to those of the base-case analysis, so are not reported. Other secondary care contacts were costed using corresponding currencies and prices from the national tariff. The NHS indicative price for 20-mg capsules of fluoxetine was £2.27 for 30 capsules. 61 No discounting of resource costs was conducted as the time horizon was limited to 12 months for within-trial analysis.
Health outcomes
Survival times for the first 12 months after randomisation were measured in days, with anyone alive at 365 days being censored. Self-reported HRQoL at 6 and 12 months of follow-up was measured using the EQ-5D-5L preference-based scale. The EQ-5D-5L index values were calculated using English value sets. We also validated the EQ-5D-5L by checking the concordance with the mRS. We planned to use a standard multiplicative model to estimate quality-adjusted survival (QALYs) calculated by the area under linear interpolation of the EQ-5D-5L index value trajectory for each individual with survival times, the EQ-5D-5L utility index value at 6 and 12 months and a modelled baseline EQ-5D-5L utility index value. If no group differences in the EQ-5D-5L emerged, QALYs would not be estimated and the analysis would focus on any difference in survival days.
Cost-effectiveness model specification
The primary treatment effect in the economic analysis was estimated using an individual-level regression model for average (mean) incremental costs and incremental survival times over 12 months after randomisation. The model accounts for the joint distribution of costs and survival times using a general specification that allowed for different parametric and conditional distributions. Incremental cost-effectiveness ratios (ICERs) are reported for the difference in mean total costs between treatment groups divided by the difference in mean number of survival days between treatment groups. All analyses were based on cases with complete information on resource use and survival times.
Sensitivity analysis
Mean costs and survival times and differences between intervention treatment groups in costs and survival times were based on a bootstrapped analysis using 1000 (and 10,000) replicates. Uncertainty in the ICER was visually represented as a cost-effectiveness plane. Incremental net benefits and cost-effectiveness acceptability curves were not calculated if the ICER was centred on zero (i.e. no difference in mean costs and survival times). We conducted a companion analysis of cost-effectiveness in which we truncated the cumulative cost distribution at 6 months and estimated the incremental costs in relation to incremental differences in the primary outcome (mRS at 6 months).
Long-run economic analysis and assessment of treatment effect heterogeneity
Longer-run modelling, estimating the distribution of costs and quality-adjusted survival times calculated over the expected patient lifetimes, was planned if differences in the primary outcome emerged between treatment groups at 6 months. Our intention was to use a microsimulation model calibrated using information gained from the within-trial analysis of cost-effectiveness combined with additional data from (1) Assessment oF FluoxetINe In sTroke recoverY (AFFINITY) and EFFECTS,62 (2) trials and observational studies reporting longer-term costs, survival and HRQoL following stroke, and (3) expert beliefs on the distributions of parameters where information was less readily available.
Secondary analyses are planned to address heterogeneous treatment effects using the pooled individual-level data from FOCUS, AFFINITY and EFFECTS. 62 Subpopulations with different average treatment effects will be identified using ‘regression tree’ or ‘recursive partitioning’ methods. These data-driven analyses complement prespecified subgroup analyses examining individual and group covariates of substantive interest, such as stroke severity (NIHSS) and the six simple variable model for prognosis. 31
Results
Resource use and cost analysis
Hospital inpatient use and attendances and duration of fluoxetine therapy are reported in Table 25. At 12 months, the average number of inpatient hospital days was just over 30 days for both treatment groups, with little difference in the average number of hospital attendances. Fluoxetine therapy lasted 143 days on average, with a median of 6 months. The distributions of cumulative total costs (Table 26) were similar between the groups, with mean total costs of £18,784 (median £11,150) for patients allocated to fluoxetine and £18,297 (median £10,871) for patients allocated to placebo.
| Resource use | Allocated treatment | |||
|---|---|---|---|---|
| Fluoxetine (n = 1556) | Placebo (n = 1553) | |||
| Mean (SD) | Median (IQR) | Mean (SD) | Median (IQR) | |
| Hospital inpatient days | 32 (39) | 16 (5–46) | 31 (39) | 15 (4–43) |
| Hospital attendances | 0.343 (0.582) | 0 (0–1) | 0.338 (0.605) | 0 (0–1) |
| Fluoxetine therapy (days) | 143 (64) | 182 (101–186) | 0 | 0 |
| Cost (£) | Allocated treatment | |||
|---|---|---|---|---|
| Fluoxetine (n = 1548) | Placebo (n = 1553) | |||
| Mean (SD) | Median (IQR) | Mean (SD) | Median (IQR) | |
| Hospital inpatient days | 18,561 (20,502) | 10,870 (5113–26,195) | 18,137 (20,214) | 10,632 (4805–24,855) |
| Hospital attendances | 162 (277) | 0 (0–405) | 159 (289) | 0 (0–397) |
| Fluoxetine therapy | 11 (5) | 14 (8–14) | 0 | 0 |
| Total costs | 18,784 (20,504) | 11,150 (5231–26,400) | 18,297 (20,201) | 10,871 (5129–24,855) |
Health outcomes
Mean survival times up to 12 months were 334 days (11 months) for both treatment groups. The pattern and distribution of EQ-5D-5L dimensions and levels were similar at 6 months, with little change at 12 months (Table 27). The frequency of ‘no problems’ (i.e. level 1) and ‘problems’ (i.e. levels 2 to 5) across the five dimensions at 6 months revealed a slight advantage in mobility and usual activity for the placebo group and less anxiety and depression in the fluoxetine group. These differences diminished by 12 months. The 10 most frequent EQ-5D-5L profiles accounted for around one-fifth of profiles and there was a high degree of concordance between treatment groups at 6 and 12 months (Table 28). EQ-5D-5L index values averaged around 0.47 at 6 months with no significant difference between the treatment groups; at 12 months, there was a slight increase in the mean index value to 0.50 but no sign of difference between patients allocated to fluoxetine or placebo (Table 29). Given the similar survival trajectories and EQ-5D-5L index values up to 12 months, there is no evidence of a difference in quality-adjusted survival times.
| Dimension | Level | 6 months (%) | 12 months (%) | ||||
|---|---|---|---|---|---|---|---|
| Allocated treatment | Total (N = 2835) | Allocated treatment | Total (N = 2672) | ||||
| Fluoxetine (n = 1413) | Placebo (n = 1422) | Fluoxetine (n = 1339) | Placebo (n = 1333) | ||||
| Mobility | No problems | 17.8 | 21.4 | 19.6 | 19.5 | 21.5 | 20.5 |
| Slight problems | 23.4 | 22.9 | 23.1 | 24.5 | 22.7 | 23.6 | |
| Moderate problems | 24.6 | 23.0 | 23.8 | 25.7 | 26.0 | 25.9 | |
| Severe problems | 17.5 | 17.5 | 17.5 | 16.1 | 15.8 | 15.9 | |
| Unable to | 16.8 | 15.2 | 16.0 | 14.3 | 13.9 | 14.1 | |
| Self-care | No problems | 37.1 | 37.1 | 37.1 | 37.9 | 39.4 | 38.7 |
| Slight problems | 19.4 | 19.8 | 19.6 | 21.4 | 20.5 | 20.9 | |
| Moderate problems | 19.3 | 18.0 | 18.7 | 18.8 | 16.1 | 17.4 | |
| Severe problems | 8.5 | 8.2 | 8.4 | 7.5 | 9.1 | 8.3 | |
| Unable to | 15.7 | 16.8 | 16.3 | 14.4 | 15.0 | 14.7 | |
| Usual activity | No problems | 14.8 | 17.3 | 16.1 | 17.9 | 17.5 | 17.7 |
| Slight problems | 22.1 | 21.6 | 21.8 | 21.1 | 25.2 | 23.2 | |
| Moderate problems | 20.2 | 18.5 | 19.4 | 22.8 | 19.7 | 21.3 | |
| Severe problems | 13.4 | 14.5 | 13.9 | 13.8 | 13.7 | 13.7 | |
| Unable to | 29.5 | 28.1 | 28.8 | 24.4 | 23.9 | 24.1 | |
| Pain/discomfort | No pain | 36.4 | 37.2 | 36.8 | 35.8 | 36.7 | 36.2 |
| Slight pain | 29.3 | 28.3 | 28.8 | 30.8 | 33.3 | 32.1 | |
| Moderate pain | 24.6 | 24.1 | 24.3 | 23.1 | 21.5 | 22.3 | |
| Severe pain | 7.9 | 8.0 | 7.9 | 7.5 | 6.8 | 7.2 | |
| Extreme pain | 1.9 | 2.3 | 2.1 | 2.8 | 1.7 | 2.2 | |
| Anxiety/depression | Not anxious | 53.4 | 49.0 | 51.2 | 45.6 | 47.6 | 46.6 |
| Slightly anxious | 25.3 | 26.9 | 26.1 | 34.6 | 31.7 | 33.1 | |
| Moderately anxious | 16.0 | 17.7 | 16.8 | 14.4 | 15.8 | 15.1 | |
| Severely anxious | 3.9 | 4.7 | 4.3 | 3.9 | 3.7 | 3.8 | |
| Extremely anxious | 1.4 | 1.8 | 1.6 | 1.6 | 1.4 | 1.5 | |
| Rank | Allocated treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | |||||||
| EQ-5D-5L profile | n | % | % (cumulative) | EQ-5D-5L profile | n | % | % (cumulative) | |
| Analysis at 6 months | ||||||||
| 1 | 1 1 1 1 1 | 90 | 6.3 | 6.3 | 1 1 1 1 1 | 105 | 7.4 | 7.4 |
| 2 | 2 1 2 1 1 | 34 | 2.4 | 8.7 | 1 1 2 1 1 | 33 | 2.3 | 9.7 |
| 3 | 2 1 2 2 1 | 30 | 2.1 | 10.8 | 2 1 2 1 1 | 31 | 2.2 | 11.9 |
| 4 | 5 5 5 1 1 | 29 | 2.0 | 12.8 | 1 1 1 2 1 | 27 | 1.9 | 13.7 |
| 5 | 1 1 2 1 1 | 27 | 1.9 | 14.7 | 2 1 2 2 1 | 23 | 1.6 | 15.4 |
| 6 | 2 1 1 1 1 | 25 | 1.8 | 16.5 | 5 5 5 1 1 | 21 | 1.5 | 16.8 |
| 7 | 1 1 1 2 1 | 17 | 1.2 | 17.7 | 2 2 2 1 1 | 19 | 1.3 | 18.2 |
| 8 | 5 5 5 2 1 | 16 | 1.1 | 18.8 | 5 5 5 2 1 | 18 | 1.3 | 19.4 |
| 9 | 5 5 5 2 2 | 16 | 1.1 | 19.9 | 1 1 1 1 2 | 16 | 1.1 | 20.6 |
| 10 | 2 2 2 2 1 | 14 | 1.0 | 20.9 | 2 1 1 1 1 | 16 | 1.1 | 21.7 |
| Total | 1425 | 100.0 | 1426 | 100.0 | ||||
| Analysis at 12 months | ||||||||
| 1 | 1 1 1 1 1 | 105 | 7.7 | 7.7 | 1 1 1 1 1 | 102 | 7.6 | 7.6 |
| 2 | 1 1 1 2 1 | 29 | 2.1 | 9.9 | 1 1 2 1 1 | 35 | 2.6 | 10.2 |
| 3 | 2 1 1 1 1 | 25 | 1.8 | 11.7 | 2 1 2 2 1 | 32 | 2.4 | 12.6 |
| 4 | 1 1 2 1 1 | 21 | 1.6 | 13.3 | 1 1 1 2 1 | 27 | 2.0 | 14.6 |
| 5 | 5 5 5 2 2 | 21 | 1.6 | 14.8 | 2 1 2 1 1 | 26 | 1.9 | 16.5 |
| 6 | 2 1 2 2 1 | 20 | 1.5 | 16.3 | 5 5 5 2 2 | 22 | 1.6 | 18.1 |
| 7 | 5 5 5 3 3 | 20 | 1.5 | 17.8 | 1 1 2 1 2 | 20 | 1.5 | 19.6 |
| 8 | 2 1 2 1 1 | 19 | 1.4 | 19.2 | 2 1 1 1 1 | 20 | 1.5 | 21.1 |
| 9 | 2 2 2 2 2 | 19 | 1.4 | 20.6 | 2 1 1 2 1 | 17 | 1.3 | 22.4 |
| 10 | 2 1 2 2 2 | 18 | 1.3 | 21.9 | 1 1 2 2 1 | 16 | 1.2 | 23.6 |
| Total | 1357 | 100.0 | 1346 | 100.0 | ||||
| EQ-5D-5L index | Allocated treatment | Mean difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| 6 months | 1413 | 0.470 (0.358) | 1422 | 0.475 (0.360) | –0.005 (–0.031 to 0.022) | 0.716 |
| 12 months | 1339 | 0.491 (0.357) | 1333 | 0.505 (0.352) | –0.013 (–0.040 to 0.014) | 0.330 |
Cost-effectiveness
Table 30 and Figure 6 present the cost-effectiveness results at 12 months. Although there is a slight difference of around £500 in the mean total costs and a very small increase (< 1 day) in survival times for the fluoxetine group, neither of these differences are significant. The ICER of £2609 per day is not significantly different from zero, as illustrated in the cost-effectiveness plane (see Figure 6). The joint distribution of incremental costs and incremental survival days is essentially centred on zero.
| Outcome | Allocated treatment | Mean difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Fluoxetine | Placebo | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| Total costs (£) | 1548 | 18,784 (20,504) | 1553 | 18,297 (20,201) | 487 (–947 to 1920) | 0.506 |
| EQ-5D-5L index | 1339 | 0.491 (0.357) | 1333 | 0.505 (0.352) | –0.013 (–0.040 to 0.014) | 0.330 |
| Survival days | 1560 | 334 (84) | 1561 | 334 (85) | 0.19 (–5.75 to 6.13) | 0.951 |
| ICERa | 2609 (–11,931 to 17,149) | 0.725 | ||||
FIGURE 6.
Cost-effectiveness plane with means centred.

Finally, given the statistically significant increase in fracture risk at 6 months in the fluoxetine group, exploratory analysis suggests that this is likely to account for around 37% of the difference in mean total costs reported above as patients who sustained a fracture reported a £13,330 (95% CI £8453 to £18,207) increase in mean total costs at 12 months.
Chapter 7 Results 5: an updated systematic review of randomised controlled trials of fluoxetine in stroke patients
This chapter is reproduced from Mead et al. 63 International Journal of Stroke. Copyright © 2019 by SAGE Publications. Reprinted by permission of SAGE Publications, Ltd.
Introduction
A 2012 Cochrane systematic review of SSRIs for stroke recovery25 suggested that SSRIs, including fluoxetine, reduced disability in stroke patients even if they did not have depression, but poor methodological quality of the trials probably introduced bias. In general, systematic reviews and meta-analyses should be updated as soon as there are new studies that might change the conclusions of the review. Thus, we, in collaboration with many other individuals (see Appendix 3), conducted an updated meta-analysis focusing specifically on the role of fluoxetine for stroke recovery, rather than on all SSRIs. We have included the results of the FOCUS trial and any other RCTs completed since the earlier review. 25 We sought to determine whether or not fluoxetine, at any dose, given within the first year after stroke to patients who did not have to have mood disorders at randomisation, compared with usual care or placebo, reduced disability and dependency at the end of treatment, reduced neurological deficits and fatigue, and improved motor function, mood and cognition at the end of treatment and follow-up, with the same number of or fewer adverse effects. The methods and results of this updated systematic review have been published63 and are included here to put the FOCUS trial results into context.
Methods
Protocol and registration
We followed PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analysis) to complete and report this study. We did not register the current review on PROSPERO as we used the same methods as the 2012 Cochrane review,25 except for (1) including fluoxetine trials only, (2) excluding trials that require patients to have mood disorders at randomisation, (3) simplifying our sensitivity analyses by excluding trials at high or unclear risk of bias in at least one domain rather than considering each domain individually, (4) excluding trials comparing fluoxetine plus another ‘active treatment’ with the ‘active treatment’ and (5) defining incomplete outcome data reporting as systematic differences in withdrawals between groups rather than a total of > 5%. These five criteria were agreed prior to study selection and data extraction.
After study selection and data extraction, but prior to analyses, we decided to report the proportion of participants who were independent (mRS 0–2) rather than the proportion of participants who were dependent (mRS 3–5).
Random-effects models were used in the 2012 Cochrane review25 because we assumed that the included studies would represent a random sample of the effect sizes that could be observed. Given that the large FOCUS trial55 had systematically different results from the smaller trials, a random-effects model would have given disproportionate weight to smaller studies. 64 Therefore, we report fixed-effects models. We undertook sensitivity analyses using random-effects models and report any major differences between the two.
Eligibility criteria
-
Participants: stroke in the previous year. Stroke was defined as sudden-onset focal neurological disturbance, assumed to be vascular in origin and lasting > 24 hours. 65 We excluded trials requiring patients to have a mood disorder at randomisation.
-
Types of intervention: any dose of fluoxetine, any mode of delivery, given for any duration.
-
Comparator group: usual care or a placebo. We excluded studies comparing fluoxetine plus another ‘active treatment’ with ‘active treatment’ alone, because of possible interactions.
-
Outcomes: we prespecified two co-primary outcomes – independence or disability at the end of treatment (using any measure) – rather than the single primary outcome of independence because changes in disability (performance of activities of daily living) could be of importance to patients even without a change in overall dependence. Independence (or not) was defined as a dichotomous variable; we expected to find this commonly reported using the mRS. We anticipated disability being measured using a number of different continuous outcome measures, which we planned to combine using standardised mean differences (SMDs).
-
Secondary outcomes: independence or disability at the end of follow-up. Neurological score, new depression during the trial but not based on standardised criteria, anxiety, cognition, quality of life, fatigue, health-care costs, death, motor scores, adverse events (at the end of treatment and/or at the end of follow-up), ‘leaving the trial before scheduled follow-up’, which included any reason other than death for missing outcome data.
-
Report characteristics: we included all reports irrespective of year of publication, language and publication status. Where necessary, we sought unpublished data from authors.
Information sources
Our information sources and search strategy are described in Appendix 2. We screened reference lists from review articles and included papers. We contacted experts to identify additional studies.
Study selection
Duplicate references were removed using Covidence software (Melbourne, VIC, Australia; www.covidence.org). Titles and abstracts were scrutinised by two authors. Obviously irrelevant articles were excluded. Full texts of potentially relevant articles were retrieved and inclusion criteria applied by two authors. A third author was involved if there was disagreement. We included studies meeting our criteria.
Data collection process
Two reviewers independently extracted data from the new trials using Covidence. We contacted the authors if data were missing or required in a different format.
Data items
Continuous and dichotomous data were extracted. If trials reported the same number of patients at the beginning and the end, we assumed that there had been no deaths. If there was no description of how adverse events were recorded, we included any available data on adverse events, but did not assume the absence of serious adverse events unless the authors had explicitly reported this. If there was a different number of patients at the end of the trial, we extracted data on deaths and dropouts for other reasons. The denominator was the number of patients for whom a particular outcome was available.
Risk of bias of individual studies
Two authors applied the same criteria as previously. 25 We included allocation (selection bias), blinding (performance bias and detection bias), incomplete outcome data (systematic differences between groups in withdrawals from a study), selective reporting and other potential sources of bias.
Prespecified sensitivity analyses
Sensitivity analyses explored the influence of bias by excluding studies with unclear or high risk of bias across at least one key domain. 64
Summary measures and synthesis of results
Risk ratios (RRs) were used for dichotomous data and for ordinal scales with an established cut-off point. SMDs were used for continuous data and ordinal scales with no standard cut-off point. We prespecified our interpretation of SMD: 0.2 represents a small effect, 0.5 represents a moderate effect and 0.8 represents a large effect. 64 Unpublished data kindly provided by the authors of one trial66 reported medians, IQRs and ranges (Dr Juan Marquez Romero, Mexican Institute of Social Security, Departmento de Neurologia Aguascalientes, Mexico, 20 September 2018, personal communication.). We estimated the mean and SD using the best available method. 67
Risk of bias across studies
A funnel plot was used to investigate publication bias. When available, we scrutinised protocols to investigate selective reporting.
Subgroup analyses
Given that fluoxetine may be more effective when given earlier after stroke, we aimed to explore the influence of time since stroke at recruitment on our primary outcome by categorising studies as < 3 months, 3 to 6 months, 6 to 9 months and 9 to 12 months since the stroke.
Results
From the database searches, we identified 3412 references, removed 426 duplicates, screened 2988 references and assessed 499 full texts for eligibility (Figure 7). Three published papers had the same grant number,68–70 very similar inclusion criteria and recruited patients from the same hospital during overlapping time periods; one appeared to be the 3-year follow-up data70 from one of the earlier publications. 69 Thus, we included the publication with the largest number of patients reporting our prespecified outcomes69 and categorised the other two68,70 as ‘awaiting assessment’ pending further information. We identified three further new eligible trials from the database searches66,71,72 and one by contact with experts. 73 We also included the FOCUS trial. 55
FIGURE 7.
Flow diagram showing selection of studies. Reproduced from Mead et al. 63 International Journal of Stroke. Copyright © 2019 by SAGE Publications. Reprinted by permission of SAGE Publications, Ltd.
These six new trials (n = 3710)55,66,69,71–73 were added to seven eligible trials12,16,17,74–78 (n = 435) identified in the 2012 Cochrane review25 (total of 13 completed trials, n = 4145, Table 31). One further registered trial was withdrawn because it recruited no patients. 79
| Study (first author and year) | Country | Participants (pathological type and time since stroke) | Number recruited | Number included at end of treatment | Dose and duration of fluoxetine | Control | Outcomes reported by the trial authors | Follow-up period |
|---|---|---|---|---|---|---|---|---|
| Birchenall 201972 | France | Stroke or brain haemorrhage, day 3 to day 15 | 6 (study terminated early) | 6 | 20 mg daily for 3 months | Placebo | Several clinical and TMS measurements, death | End of treatment and at month 6 |
| Chollet 201117 | France | Ischaemic stroke, 5–10 days | 118 | 113 | 20 mg daily for 3 months | Matching placebo | Primary outcome: FMMS. Secondary endpoints: NIHSS, mRS and MADRS at 0, 30 and 90 days. AEs | End of treatment |
| Dam 199616 | Italy | Ischaemic stroke, 1–6 months | 35 | 33 | 20 mg daily for 12 weeks | Matching placebo | HDRS, HSS (total, gait and motor scores), BI, death, AEs | End of treatment |
| FOCUS collaboration 201855 | UK | Any stroke, 2–15 days | 3127 | 3106 | 20 mg daily for 6 months | Matching placebo |
Primary: mRS Secondary: SIS, depression, MHI5, fatigue, EQ-5D-5L, health care costs |
End of treatment and then 6 months later |
| He 200474 | China | First ever stroke, all pathological types; mean time 3.1 days in fluoxetine and 3.5 days in control | 84 | 71 | 20 mg daily for 8 weeks | Usual stroke care | HAMD, SSS, AEs | End of treatment |
| He 201669 | China | Ischaemic stroke, within 1 week | 374 | 350 | 20 mg daily for 90 days | Usual care | NIHSS, BI, AEs | End of treatment and at day 180 |
| Kong 200775 | China | Any pathological type, within 7 days | 90 | 73 | 20 mg daily for 8 weeks | Matching placebo |
HAMD, BI, NIHSS Somatic side effects and hyponatraemia |
End of treatment |
| Li 200476 | China | Any pathological type; mean time to recruitment was 2 days | 67 | 67 | 20 mg daily for 4 weeks | Routine stroke care | HAMD, CSS; AEs in fluoxetine group | End of treatment |
| Marquez-Romero 201366 | Mexico | Intracerebral haemorrhage within 10 days | 32 | 30 | 20 mg daily for 90 days | Matching placebo |
Primary: FMMS, mRS Secondary: NIHSS, BI, AEs |
End of treatment |
| Pariente 200112 | France | Lacunar ischaemic stroke | 8 | 8 | Single 20-mg dose | Placebo | Finger tapping and clinical scales presented only as graphs. fMRI activation location | Post treatment |
| Robinson 200077 (follow-up reported in Mikami 201178) | USA and Argentina | All pathological types, within 6 months | 33 | 28 | Dose increased over 3 weeks from 10 mg to 30 mg daily; total 12 weeks | Matching placebo | HDRS, mRS, FIM, MMSE, JHFI, death, AEs | End of treatment |
| Shah 201673 | India | Haemorrhagic stroke, 5–10 days after onset | 89 | 84 | 10 mg for 1 week; increased to 20 mg after 1 week; total 3 months | Inert capsule ‘similar’ to fluoxetine |
Primary outcome: FMMS mRS and AEs |
End of treatment |
| Zhao 201171 | China | Stroke with aphasia, ‘early treatment with fluoxetine', precise time not stated | 82 | 71 | 20 mg daily for 12 weeks | Standard care | MESSS, ADL | End of treatment |
Several ongoing RCTs together aim to recruit about 3775 patients.
Risk of bias
There were four high-quality trials (n = 3283) with a low risk of bias across important quality criteria17,55,66,72 (Figure 8). One terminated early having recruited six patients, and reported no deaths. 72
FIGURE 8.
Risk of bias. +, low risk of bias; ?, unclear risk of bias; –, high risk of bias. Reproduced from Mead et al. 63 International Journal of Stroke. Copyright © 2019 by SAGE Publications. Reprinted by permission of SAGE Publications, Ltd.

Results of studies and synthesis of results
Co-primary outcomes: independence (Figure 9) and disability at end of treatment (Figure 10)
FIGURE 9.
Forest plot: mRS (0–2) at the end of treatment. df, degrees of freedom; M–H, Mantel–Haenszel. Reproduced from Mead et al. 63 International Journal of Stroke. Copyright © 2019 by SAGE Publications. Reprinted by permission of SAGE Publications, Ltd.

FIGURE 10.
Forest plot: disability at the end of treatment. df, degrees of freedom. IV, inverse variance. Reproduced from Mead et al. 63 International Journal of Stroke. Copyright © 2019 by SAGE Publications. Reprinted by permission of SAGE Publications, Ltd.
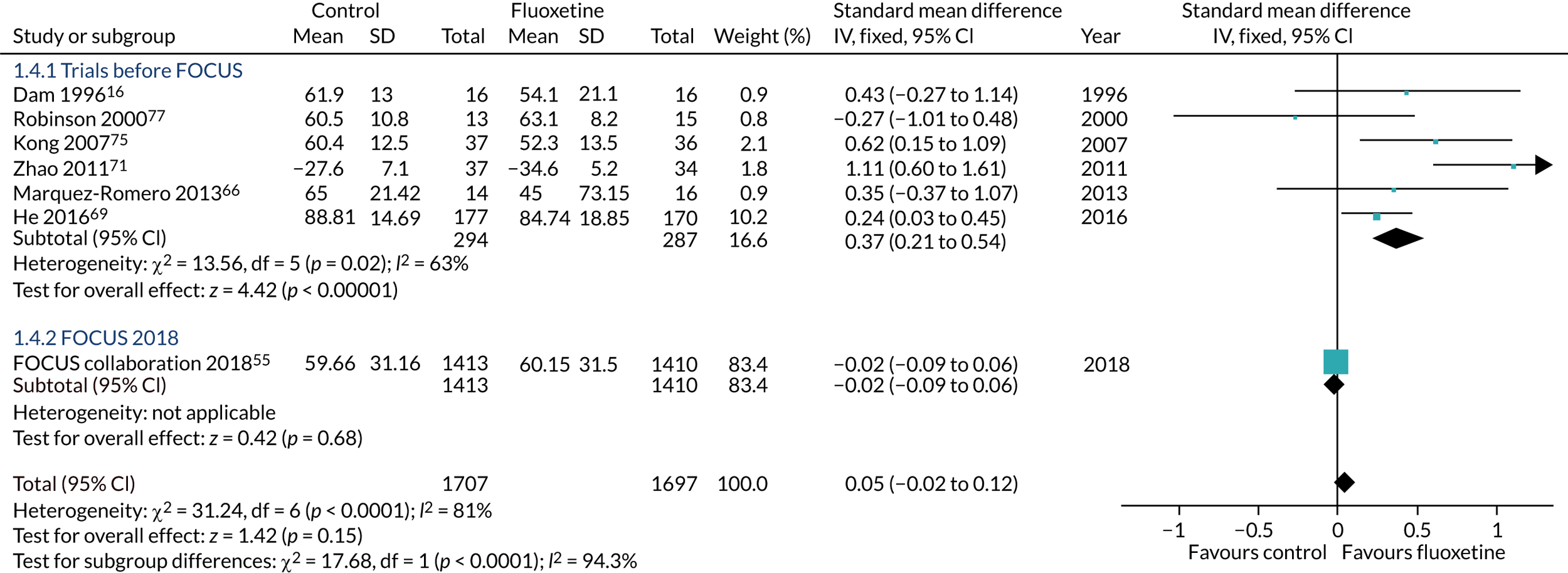
Three trials (n = 3249) reported independence. 17,55,66 Fixed-effects meta-analysis found no difference in the proportion who were independent (36.6% fluoxetine vs. 36.7% control, RR 1.00, 95% CI 0.91 to 1.09; p = 0.99; I2 = 78%) and no difference in disability (seven trials, n = 3404, SMD 0.05, 95% CI –0.02 to 0.12; p = 0.15; I2 = 81%).
Two other trials73,78 reported improvements in mRS in the fluoxetine group but the data were in a format that could not be used in the meta-analysis and the authors did not respond to our requests for clarification.
Random-effects models demonstrated a small but statistically significant benefit of fluoxetine for disability (SMD 0.34, 95% CI 0.04 to 0.64; p = 0.03; I2 = 81%), and a higher RR (RR 1.87, 95% CI 0.74 to 4.56; p = 0.19; I2 = 78%) of being independent than the fixed-effects models because of the greater weight given to smaller positive trials.
Secondary outcomes at the end of treatment: summary effect sizes (Table 32)
| Outcome | Number of trials (number of participants) contributing to the meta-analysis | Effect size (RR or SMD) and 95% CI | p-value | I 2 |
|---|---|---|---|---|
| Independent (mRS 0–2) | 3 (n = 3249) | RR 1.00 (0.91 to 1.09) | 0.99 | 78% |
| Disability | 7 (n = 3404) | SMD 0.05 (–0.02 to 0.12) | 0.15 | 81% |
| Neurological deficit score | 8 (n = 803) | SMD –0.28 (–0.42 to –0.14) | < 0.0001 | 77% |
| Depression-continuous data | 6 (n = 3113) | SMD –0.16 (–0.23 to –0.09) | < 0.0001 | 92% |
| Depression-dichotomous | 2 (n = 3194) | RR 0.77 (0.65 to 0.90) | 0.001 | 53% |
| Motor score | 5 (n = 3079) | SMD 0.06 (–0.02 to 0.13) | 0.12 | 95% |
| Cognition | 2 (n = 2834) | SMD –0.04 (–0.11 to 0.03) | 0.32 | 0% |
| Death | 11 (n = 3824) | RR 1.0 (0.79 to 1.26) | 1.00 | 0% |
| Seizures | 7 (n = 3815) | RR 1.49 (1.05 to 2.11) | 0.03 | 0% |
| Gastrointestinal symptoms (nausea, diarrhoea, abdominal pain) | 7 (n = 688) | RR 1.38 (0.99 to 1.94) | 0.06 | 8% |
| Serious bleeding | 2 (n = 3477) | RR 1.10 (0.72 to 1.62) | 0.67 | 0% |
| Leaving before the end of first follow-up | 11 (n = 3972) | RR 0.92 (0.61 to 1.40) | 0.71 | 0% |
Fluoxetine was associated with better neurological scores (eight trials, n = 803, SMD –0.28, 95% CI –0.42 to –0.14; p < 0.001; I2 = 77%), lower (fewer depressive symptoms) depression scores (six trials, n = 3113, SMD –0.16, 95% CI –0.23 to –0.09; p < 0.0001; I2 = 92%), fewer diagnoses of depression (two trials, n = 3194, RR 0.77, 95% CI 0.65 to 0.90; p = 0.001; I2 = 53%) but more seizures (seven trials, n = 3815, 3.9% vs. 2.6%, RR 1.49, 95% CI 1.05 to 2.11; p = 0.03; I2 = 0). Random-effects models gave broadly similar results. The FOCUS trial identified a small excess of bone fractures in the fluoxetine group, which was statistically significant (see Table 13). No other trial reported fractures.
End of follow-up
Two trials (n = 2924) reported disability at the end of follow-up (SMD 0.11, 95% CI –0.17 to 0.40; p = 0.45; I2 = 85%; fixed effects). Only one trial (FOCUS) reported independence at the end of follow-up; there was no difference between groups. 55
Sensitivity analyses: high-quality trials at low risk of bias only (Table 33) (fixed effects)
| Outcome | Number of trials and participants contributing to the meta-analysis | Effect size (RR or SMD) and 95% CI | p-value | I 2 |
|---|---|---|---|---|
| Independent (mRS 0–2) | 3 (n = 3269) | RR 1.00 (0.91 to 1.09) | 0.99 | 78% |
| Disability | 2 (n = 2853) | SMD –0.01 (–0.09 to 0.06) | 0.75 | 0% |
| Neurological deficit score | 2 (n = 142) | SMD –0.30 (–0.63 to 0.04) | 0.08 | 0% |
| Depression (continuous data) | 2 (n = 2861) | SMD –0.11 (–0.19 to –0.04) | 0.002 | 69% |
| Motor score | 3 (n = 2936) | SMD 0.02 (–0.05 to 0.09) | 0.58 | 88% |
| Death | 4 (n = 3260) | RR 0.99 (0.79 to 1.25) | 0.95 | 0% |
| Gastrointestinal symptoms | 2 (n = 148) | RR 2.19 (1.0 to 4.76) | 0.05 | 0% |
| Leaving the trial before first follow-up | 4 (n = 3283) | RR 1.01 (0.48 to 2.10) | 0.98 | 0% |
| Seizures | 3 (n = 3275) | RR 1.47 (0.99 to 2.18) | 0.06 | 0% |
The fixed-effects models found a small but statistically significant effect on depression scores at the end of treatment (two trials, n = 2861, SMD –0.11, 95% CI –0.19 to –0.04; p = 0.002; I2 = 69%). Random-effects models found a slightly larger effect size for depression, which was not statistically significant (SMD –0.23, 95% CI –0.56 to 0.10; p = 0.07; I2 = 61%).
Subgroup analyses
We did not conduct subgroup analyses because all trials except two (n = 68) recruited patients within 3 months of stroke onset.
Chapter 8 Discussion
In this section, we first discuss the main results of the FOCUS trial,55 and the possible explanations of why we did not confirm the very encouraging results of the earlier RCTs included in our 2012 systematic review. 25 We then discuss the results of our post hoc analyses relating to fractures and, finally, the results of our updated systematic review, which aims to put the FOCUS trial results into the context of all completed similar trials.
The FOCUS trial, along with its sister trials in Australasia/Vietnam (AFFINITY) and Sweden (EFFECTS),62 which are ongoing at the time of this report, was established to answer several questions. Our discussion will first focus on whether or not we have addressed these questions.
Primary question: does the routine, early administration of fluoxetine (20 mg o.d.) for 6 months after an acute stroke improve patients’ functional outcome?
Our results do not support the hypothesis that a course of fluoxetine improves patients’ outcomes. We have considerable confidence that this is the correct conclusion because of the trial’s methodological strengths.
The strengths of the study, supporting the internal validity of the results, are that bias was minimised by central randomisation without any prospect of foreknowledge; blinding of patients, carers and outcome assessment (with only three episodes of unblinding); very few losses to follow-up (< 1%); and published prespecified intention-to-treat analyses. The small difference in the number of patients stopping the trial medication for perceived adverse effects suggests that unblinding owing to adverse effects was unlikely to have had a significant effect on our results. In any case, expectation bias would normally be expected to bias the result in favour of the active treatment. Random error was also minimised by randomising a large number of patients and high rates of follow-up, providing much greater statistical power than previous similar trials.
However, it is important to consider whether or not methodological factors could have caused us to miss an improvement in outcome. Possible explanations might include:
-
Inadequate power to detect a small but clinically significant improvement in functional outcome. Our sample size estimate indicated that we had 90% probability of detecting an effect equivalent to a COR of 1.23 at the 5% significance level. The planned total sample size for the FOCUS, AFFINITY and EFFECTS trials,62 6000 patients, provides 90% power to detect a smaller effect size equivalent to a COR of 1.16. Our estimate of effect in the FOCUS trial was a COR of 0.951 (95% CI 0.839 to 1.079; p = 0.439; these 95% CIs, which include neither 1.16 nor 1.23, make it very unlikely that we have overlooked a treatment effect of the size that we had considered likely or of clinical importance).
-
Inclusion of the wrong type of stroke patients. For instance, those included in the FLAME trial,17 on average, had more severe strokes and all had motor deficits. Our prespecified subgroup analyses (see Table 11) did not identify any interaction between our overall effect and any subgroup. We specifically explored whether or not there was a benefit for patients who had a motor deficit at baseline; there was not.
The external validity of the results, at least for the UK stroke population, is supported by the large number of participating hospitals throughout the UK. Compared with unselected stroke patients admitted to UK hospitals (Table 34), there were a few differences to the subjects enrolled in the trial. 80,81 The patients enrolled in the FOCUS trial had slightly more severe strokes (NIHSS 6 vs. 4), which probably reflected the inclusion criteria that required patients to have a neurological deficit persisting at the time of enrolment. Also, 60% were male, compared with a UK average of 50%: an unexplained but common observation in stroke trials. 82 Patients enrolled were slightly younger (mean 71.4 years) than unselected patients who were admitted to hospital with stroke in the UK, which might partly explain the male preponderance, with older women being under-represented. Many of the studies included in the previously published systematic review of RCTs of fluoxetine were from China; non-white patients made up < 5% of those recruited in the FOCUS trial. The ongoing AFFINITY trial is recruiting in Vietnam and will include a much larger proportion of Asian patients. 62
| Characteristics of patients randomised | Allocated treatment | Comparative data from UK national audits | ||
|---|---|---|---|---|
| Fluoxetine | Placebo | Sentinel Stroke National Audit Programme (N = 74,307) | Scottish Stroke Care Audit (N = 9345) | |
| All patients, n (%) | 1564 (100.00) | 1563 (100.00) | ||
| Female, n (%) | 589 (37.66) | 616 (39.41) | 50% | 49% |
| Male, n (%) | 975 (62.34) | 947 (60.59) | 50% | 51% |
| Age (years), mean (SD) | 71.24 (12.35) | 71.48 (12.06) | 77 | 73 |
| Lives alone, n (%) | 485 (31.01) | 516 (33.01) | 38% | |
| Independent before stroke, n (%) | 1431 (91.50) | 1435 (91.81) | 81% | 82% |
| Past history, n (%) | ||||
| Prior ischaemic stroke/TIA | 274 (17.52) | 294 (18.81) | 27% | |
| Known diabetes | 337 (21.55) | 303 (19.39) | 19% | |
| Stroke type, n (%) | ||||
| Ischaemic stroke | 1410 (90.15) | 1406 (89.96) | 88% | 87% |
| Intracerebral haemorrhage | 154 (9.85) | 157 (10.04) | 11% | 13% |
| Stroke severity | ||||
| Able to walk at enrolment, n (%) | 435 (27.81) | 412 (26.36) | 48% | |
| Able to lift both arms off bed, n (%) | 924 (59.08) | 935 (59.82) | 63% | |
| Able to talk and not confused, n (%) | 1166 (74.55) | 1164 (74.47) | 66% | |
| NIHSS, median (IQR) | 6 (3–11) | 6 (3–11) | 4 (2–10) | |
| Enrolled as a hospital inpatient, n (%) | 1544 (98.72) | 1536 (98.27) | 100% | |
The primary outcome measure, the modified Rankin Scale, was too insensitive
Some might criticise our use of the mRS, and specifically the smRSq, because it is too simple and not a directly observed assessment of function, but is patient or proxy reported. However, the smRSq is a valid, reliable measure of functional outcome, which is patient centred, thus ensuring that our results are relevant for patients and their families. 18,35–37 We cannot definitely exclude an effect of fluoxetine on a directly measured neurological deficit, such as the Fugl-Meyer Motor Score, which was measured in the FLAME trial. 17 A structured neurological examination during follow-up would have been impractical to include in this large pragmatic multicentre trial. In addition, local face-to-face assessments of outcomes are likely to be more prone to unblinding owing to patients reporting adverse effects of trial medication to those assessing outcome, than would occur through postal and telephone follow-up. However, we have shown that even if fluoxetine improves neurological deficits, a resulting improvement in functional status measured with the mRS or SIS is unlikely.
Non-adherence might have diluted any benefit
The main limitation of the FOCUS trial was the degree of non-adherence to the trial medication, which might have led us to underestimate any treatment effect. However, adherence measured in the FOCUS trial was superior to that reported in routine clinical practice and not very different between the treatment groups in the trial. 83 Where reduced adherence resulted from patients experiencing possible adverse reactions or a perceived change (or lack of change) in their condition, then this might be more likely to differ between the fluoxetine and placebo groups. We repeated the analysis of our primary outcome, having sequentially excluded patients with different reasons for, and different degrees of, adherence. Such per-protocol analyses may increase the risk of bias, usually in favour of the active treatment. Our analyses (see Table 12) did not show any trend towards greater benefit from fluoxetine in those with greater adherence.
Excess of fractures may have offset the functional benefits
Removing the 65 patients with one or more fractures during follow-up from the primary analysis (ordinal analysis of the mRS) did not significantly alter the estimate of effect of fluoxetine on mRS (COR including those with fractures 0.951, 95% CI 0.839 to 1.079; p = 0.439; COR for those without fractures 0.961, 95% CI 0.847 to 1.093; p = 0.545).
Patients recruited received lower background rehabilitation intensity than in FLAME
The proponents of fluoxetine as a medication that helps functional recovery claim that it amplifies the effects of physical therapy by enhancing neuroplasticity. 17 We had no measure of the intensity of rehabilitation or delivery of physical therapies during follow-up. This is likely to have varied considerably between centres and patients. Our results are very likely to apply to the effects of fluoxetine in patients admitted to UK hospitals with a stroke. The EFFECTS trial is being carried out in Sweden where the impression is that patients receive higher doses of physiotherapy and other physical rehabilitation than in the UK. The EFFECTS trial is also estimating the intensity of physiotherapy treatment. The planned individual patient data meta-analysis will be able to determine if there are differences in the effect of fluoxetine on functional outcome between the UK and Sweden, and perhaps explore any interaction with intensity of physiotherapy.
Secondary questions
If fluoxetine improves functional outcome, does any improvement persist after treatment is stopped?
We demonstrated no effect at 6 months, and the data at 12 months do not suggest any improvement in any measure of functional outcome in the fluoxetine group compared with the placebo group.
Does the routine early administration of fluoxetine after acute stroke causing motor impairment improve patients’ motor function and does any improvement persist after treatment is stopped?
There was no evidence of benefit from fluoxetine on motor outcomes (as defined by relevant domains of the SIS) at 6 or 12 months in those patients with motor dysfunction at baseline based on the NIHSS. Similarly, in those with communication difficulties at baseline, there was no evidence of benefit from fluoxetine on the communication domain of the SIS at 6 or 12 months in those patients with communication dysfunction at baseline based on the NIHSS.
In those patients with impairments that preclude the formal assessment of post-stroke mood, does fluoxetine improve outcomes?
A prespecified subgroup analysis did not indicate any interaction between the patients’ ability or not to have their mood assessed at baseline and a treatment effect (see Table 11).
Does fluoxetine improve patients’ outcome with respect to mood, fatigue, cognition, health-related quality of life or participation and does any improvement persist after treatment is stopped?
The only improvement in patients’ outcome at 6 months was a small improvement in mood, measured with the MHI-5 (see Table 15). This was consistent with our observation that new episodes of depression were less commonly diagnosed and thus treated within the first 6 months in the group allocated fluoxetine (see Table 13). The difference in MHI-5 between groups and the difference in rates of new depression by 12 months were no longer statistically significant (see Tables 19 and 20). No other secondary measure of outcome was significantly improved.
A previous systematic review included five RCTs, including FLAME,17 in non-depressed stroke patients, which tested whether or not SSRIs (two fluoxetine, two sertraline and one escitalopram) prevented the development of post-stroke depression. 84 In a pooled analysis, 23 out of 248 (9.3%) patients treated with a SSRI developed post-stroke depression, compared with 59 out of 242 (24.4%) patients treated with a placebo; the authors reported an OR of 0.37 (95% CI 0.22 to 0.61; p = 0.001). 84 The rate of depression in the placebo arms of these trials was much higher than that in the FOCUS trial, which may have reflected the characteristics of the patients, who tended to have had more severe strokes than those in the FOCUS trial, or perhaps different methods of diagnosing depression. This is consistent with our findings in the direction, if not the magnitude, of the treatment effect.
Does fluoxetine reduce the cost of health care over the first year?
No; there was no evidence from our health economic analyses that fluoxetine was associated with a reduced cost of health care over the first year (see Tables 25 and 26).
Does fluoxetine increase the risk of serious adverse events?
Our data showed a statistically significant increase in the risk of bone fractures in the group allocated fluoxetine. The observed 1.4% absolute excess of bone fractures at 6 months with fluoxetine in the FOCUS trial is consistent with previous reports from large case–control and cohort studies. 28–30 The size of the increased risk in the previous observational studies tended to be greater than in the FOCUS trial, but this might be attributable to the inherent confounding by treatment indication in observational studies.
The rates of other serious adverse reactions referred to in the SmPC of fluoxetine, which we included as secondary outcomes in this trial (e.g. epileptic seizures, falls, hyponatraemia, uncontrolled diabetes and upper gastrointestinal bleeding), were higher in the fluoxetine group than in the placebo group, but the absolute differences were small and were not statistically significant (see Table 13). It seems likely that there is an excess of these recognised adverse effects, but we had an insufficient number of patients to confirm this reliably. Despite concerns about fluoxetine’s effect on platelet function and interactions with antiplatelet and anticoagulant medications, we observed no effect on bleeding or thrombotic adverse events (see Table 14). The trial was not powered to detect increases in these recognised adverse effects of fluoxetine.
The AFFINITY and EFFECTS trials, which are of similar design to the FOCUS trial, but with smaller recruitment targets, are in progress. 62 These should allow us to confirm the effects on post-stroke depression and bone fractures and provide more precise estimates of the benefits and harms of early fluoxetine to guide its use in stroke patients and perhaps other elderly people with comorbidities.
Summary
In summary, the FOCUS trial has shown that 20 mg of fluoxetine started early and given daily for 6 months after an acute stroke did not influence patients’ functional outcome, but did decrease the occurrence of depression and increase bone fractures. These results do not support the routine use of fluoxetine for the prevention of post-stroke depression or to promote recovery of function. Ongoing trials and a planned individual patient data meta-analysis are needed to confirm or refute a modest benefit of fluoxetine for functional outcome, either overall or in particular subgroups, and to provide more precise estimates of any harms.
Discussion of the post hoc analyses relating to fractures occurring during the 6-month treatment period
The most common site of fractures among patients with stroke enrolled in the FOCUS trial and assigned fluoxetine or placebo for 6 months was the neck of femur, and most fractures were in sites associated with osteoporosis; almost all resulted from a fall. Greater age, female sex and allocation to fluoxetine were independent predictors of subsequent fractures. An increased risk of falling is likely to explain much of the excess risk of fractures because most fractures were associated with a fall. Falls with injury were more common in the fluoxetine group than in the placebo group [n = 120 (7.67%) vs. n = 94 (6.01%); p = 0.0663], although the difference did not quite reach statistical significance and the risks in the two treatment groups diverged early after randomisation. No other baseline factors analysed had statistically significant associations with fracture risk.
These findings in an adequately powered prospective randomised trial confirm that the association between SSRI and fracture risk noted in observational studies is real and is not due to methodological biases, and that SSRI medication is likely to be causative. Our finding that greater age and female sex are associated with a greater fracture risk confirms the findings of previous observation studies in stroke. 28–30 The risk of fractures appears to increase soon after randomisation (see Figure 5) and most fractures were related to falls (see Table 21), suggesting that the predominant mechanism explaining the excess of fractures is likely to be the non-significant excess of falls seen in the FOCUS trial compared with the control group (see Table 13). This might be due to effects on cognition, co-ordination, balance or activity levels. However, we cannot exclude a contribution from fluoxetine’s possible effect on bone density.
These predefined secondary analyses have several limitations. The number of fractures in the first 6 months was modest, which means that we were only able to identify powerful predictors of fracture risk. Our only baseline indicators of bone density were previous fractures and the use of medications to reduce bone density loss at baseline. We had no direct measures of bone density. In addition, the effect of fluoxetine, and other medications, may have been diluted by non-adherence or changes to medication after randomisation. We did not collect data on current medication at the time of the fracture. Also, we did not systematically collect fractures beyond 6 months so cannot determine whether or not the effect of fluoxetine on fracture risk persists, as it might if it causes osteoporosis, or whether or not the risk subsides after stopping if it caused falls by affecting balance, etc., or increased activity by reducing depression. We did not systematically collect data on the side of the fracture so we could not reliably confirm previous findings that fractures most often affect the side of any weakness. 30
The cost-effectiveness of fluoxetine
There was no evidence that fluoxetine improves health outcomes, and it did not have a significant effect on the costs of health care, even though it reduced the frequency of new depression and increased the risk of fractures. Treatment with fluoxetine is cheap, but treatment is not justified by the clinical or economic outcomes.
The FOCUS results in the context of all similar randomised controlled trials
The updated systematic review of fluoxetine for stroke recovery identified 13 trials that recruited > 4000 patients, of which four trials (n = 3283) were of high methodological quality.
There were no differences between groups for the co-primary outcomes of dependency and disability. Fluoxetine was associated with better neurological scores at the end of treatment, lower depression scores (fewer symptoms of depression) and fewer diagnoses of depression, although the effect sizes were all small and there was substantial heterogeneity. There was a higher risk of seizures with fluoxetine. However, when only high-quality trials were considered, the only statistically significant difference between groups was lower depression scores (fewer symptoms) at the end of treatment.
We used fixed-effects models as these give appropriate weight to larger trials. The sensitivity analysis using random-effects models found a large benefit of fluoxetine on independence (RR 1.87) because of the disproportionate weight given to smaller trials. Fixed- and random-effects models produced only slightly different effect sizes for depression scores.
Previous meta-analyses suggested that fluoxetine might reduce dependency and disability if given early after stroke. 24,25,85 This meta-analysis, which includes many more patients than previous reviews, has not confirmed these promising effects. Although one of the reviews85 strongly recommended fluoxetine to promote neurological recovery, this recommendation was based on the results of just four reports,17,68,74,75 only one of which was high quality. 17
Thus, these data do not support the routine prescription of fluoxetine early after stroke in order to reduce dependency and disability. Clinicians and patients may wish to consider the routine use of fluoxetine early after stroke for its small effects on reducing the incidence of new depression, but this would need to be weighed up against the excess of seizures and bone fractures. The most important depression-related outcome after stroke is suicide, which is about double what it is in the non-stroke population; however, it is rare, and for that reason the RCTs shed no light on whether or not antidepressant treatment reduces the risk.
There are some limitations at the study and outcome level: only four trials were of high methodological quality, and not all had been registered prospectively or they reported the same outcomes. Furthermore, different scales were used for the same outcome; although we used SMD to combine data, the interpretation of SMD is not intuitive, and clinicians prefer to know the effect size on a familiar scale (e.g. Functional Independence Measure). Two large ongoing trials (AFFINITY and EFFECTS62) are using the same measures as those used in the FOCUS trial. A future meta-analysis will report mean differences for continuous data.
We did not register the review in PROSPERO, but we used almost the same methods as the 2012 Cochrane review, which had a prospectively published protocol. 25 We used sensitive searches developed by Cochrane Stroke,86 and there was complete retrieval of identified research, no language restrictions and inclusion of unpublished data.
About three-quarters of the patients were from the FOCUS trial. There was quite marked heterogeneity, even for the high-quality trials (see Table 33); this might be explained by the different types of patients and health-care settings. Five of the low-quality trials were from China; the three reporting disability all found favourable effects of fluoxetine. As the evidence base increases, it may be possible to undertake meta-regression analyses to determine the factors (e.g. country, health-care setting and trial quality) associated with good outcome.
Conclusions
The results of the FOCUS trial show that, in the context of the UK NHS, routine use of fluoxetine does not improve functional outcomes in patients with a recent stroke. There was evidence that its use reduces the risk of developing new episodes of depression after stroke, but that this is at the expense of an increased risk of adverse effects, including bone fractures.
Ongoing trials (AFFINITY and EFFECTS62), which used almost identical methods to those of the FOCUS trial, and the investigators with whom we have closely collaborated, will report in 2020. An individual patient data meta-analysis including data from FOCUS, AFFINITY and EFFECTS is planned. This will indicate whether or not the findings of the FOCUS trial are generalisable to other countries, ethnic groups and health-care systems. They will also help to provide more precise, and generalisable, estimates of the risks associated with several months of fluoxetine treatment in a predominantly elderly population. Such data are likely to be relevant to the use of fluoxetine, and perhaps other SSRIs, in similar populations (i.e. elderly patients with comorbidities). This knowledge could be of use when counselling patients about the pros and cons of starting treatment with fluoxetine.
Most previous RCTs of fluoxetine, and other SSRIs, have been carried out in patients with depression, who are often much younger than those recruited into stroke trials, and have only tested short courses with the purpose of demonstrating an improvement in mood. 87 There is a good case for studying the effects of prolonged courses of fluoxetine, not just those on mood, in older patients who are likely to be more susceptible to adverse effects. Such information would be useful in balancing the risks against the perceived benefits of using SSRIs for a variety of indications.
In advance of the FOCUS trial results becoming available, we carried out systematic reviews to determine whether or not SSRIs might be of benefit in conditions other than stroke through the same mechanisms that we thought might underpin the hypothesised benefit in stroke. All the reviews suggested promising effects of SSRIs. 88–90 However, before embarking on large trials in these other conditions, the relevance of the neutral results of the FOCUS trial needs to be considered carefully.
Acknowledgements
We would like to acknowledge all the patients and their families who participated in the FOCUS trial, the nursing staff who assisted at collaborating sites, the Scottish Stroke Research Network staff and NIHR research staff, without whom the trial would not have been possible.
Contributions of authors
Professor Martin Dennis (https://orcid.org/0000-0003-1148-8972) (Stroke Physician, University of Edinburgh, UK) was co-chief investigator; participated in the TSC; was involved in the design of the trial; collected, verified and analysed data; and drafted this report.
Professor John Forbes (https://orcid.org/0000-0002-8255-3762) (Health Economist, University of Limerick, Ireland) participated in the TSC, was involved in the design of the trial and analysed health economic data.
Ms Catriona Graham (https://orcid.org/0000-0003-1889-712X) (Unblinded Statistician, University of Edinburgh, UK) participated in the TSC, was involved in the design of the trial, wrote the first draft of the statistical analysis plan and verified and analysed data.
Professor Maree Hackett (https://orcid.org/0000-0003-1211-9087) (Psychologist, University of Sydney, Australia) was involved in the trial design and helped carry out relevant systematic reviews.
Professor Graeme J Hankey (https://orcid.org/0000-0002-6044-7328) (Neurologist, University of Western Australia, Australia) was involved in the trial design and helped carry out relevant systematic reviews.
Professor Allan House (https://orcid.org/0000-0001-8721-8026) (Psychiatrist, University of Leeds, UK) was involved in the trial design and advised on the management of depression within the trial.
Professor Stephanie Lewis (https://orcid.org/0000-0003-1210-2314) (Blinded Statistician, University of Edinburgh, UK) was involved in the trial design and advised on the statistical analysis plan.
Professor Erik Lundström (https://orcid.org/0000-0002-5313-9052) (Neurologist, Karolinska Institutet, and Uppsala University, Sweden) was involved in the design of the trial.
Professor Peter Sandercock (https://orcid.org/0000-0001-8484-0135) (Neurologist, University of Edinburgh, UK) chaired the TSC of the initial phase.
Professor Gillian Mead (https://orcid.org/0000-0001-7494-2023) (Stroke Physician/Geriatrician, University of Edinburgh, UK) was co-chief investigator, participated in the TSC, was involved in the design of the trial and data collection, and co-ordinated the systematic review of the RCTs.
All members of the writing committee listed here have commented on the analyses and drafts of this report and have seen and approved the final version of the report.
Publications
Mead G, Hackett ML, Lundstrom E, Murray V, Hankey GJ, Dennis M. The FOCUS, AFFINITY and EFFECTS trials studying the effect(s) of fluoxetine in patients with a recent stroke: a study protocol for three multicentre randomised controlled trials. Trials 2015;16:369.
Graham C, Lewis S, Forbes J, Mead G, Hackett ML, Hankey GJ, et al. The FOCUS, AFFINITY and EFFECTS trials studying the effect(s) of fluoxetine in patients with a recent stroke: statistical and health economic analysis plan for the trials and for the individual patient data meta-analysis. Trials 2017;18:627.
Dennis M, Forbes J, Graham C, Hackett ML, Hankey GJ, House A, et al. Fluoxetine and fractures after stroke: exploratory analyses from the FOCUS trial. Stroke 2019;50:11.
FOCUS Trial Collaboration. Effects of fluoxetine on functional outcomes after acute stroke (FOCUS): a pragmatic, double-blind, randomised, controlled trial. Lancet 2019;393:265–74.
Mead GE, Legg L, Tilney R, Hsieh CF, Wu S, Lundström E, Hankey GJ. Fluoxetine for stroke recovery: meta-analysis of randomized controlled trials [published online ahead of print October 17 2019]. Int J Stroke 2019.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review and appropriate agreements being in place.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- GBD 2016 Stroke Collaborators . Global, regional, and national burden of stroke, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2019;18:439-58. https://doi.org/10.1016/S1474-4422(19)30034-1.
- Lang UE, Jockers-Scherübl MC, Hellweg R. State of the art of the neurotrophin hypothesis in psychiatric disorders: implications and limitations. J Neural Transm 2004;111:387-411. https://doi.org/10.1007/s00702-003-0100-0.
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annu Rev Neurosci 2005;28:223-50. https://doi.org/10.1146/annurev.neuro.28.051804.101459.
- Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol 2007;18:391-418. https://doi.org/10.1097/FBP.0b013e3282ee2aa8.
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003;301:805-8. https://doi.org/10.1126/science.1083328.
- Wiltrout C, Lang B, Yan Y, Dempsey RJ, Vemuganti R. Repairing brain after stroke: a review on post-ischaemic neurogenesis. Neurochem Int 2007;50:1028-41. https://doi.org/10.1016/j.neuint.2007.04.011.
- Taupin P. Stroke-induced neurogenesis: physiopathology and mechanisms. Curr Neurovasc Res 2006;3:67-72. https://doi.org/10.2174/156720206775541769.
- Lim C, Kim S, Park J, Kim C, Yoon S, Lee J. Fluoxetine affords robust neuroprotection in the post ischemic brain via its anti-inflammatory effect. J Neurosci Res 2009;87:1037-45. https://doi.org/10.1002/jnr.21899.
- Shin KT, Kang MS, Lee HY, Seo MS, Kim SF, Kim CD, et al. Fluoxetine and sertraline attenuate postischemic brain injury in mice. Korean J Physiol Pharmacol 2009;13:257-63. https://doi.org/10.4196/kjpp.2009.13.3.257.
- Palvimaki E, Laakso A, Kuoppamaki M, Syvalahti E, Hietala J. Up-regulation of l-adrenergic receptors in rat brain after chronic citalopram and fluoxetine treatments. Psychopharmacology 1994;115:543-6. https://doi.org/10.1007/BF02245579.
- Loubinoux I, Boulanouar K, Ranjeva J-F, Carel C, Berry I, Rascol O, et al. Cerebral functional magnetic resonance imaging activation modulated by a single dose of the monoamine neurotransmission enhancers fluoxetine and fenozolone during hand sensorimotor tasks. J Cereb Blood Flow Metab 1999;19:1365-75. https://doi.org/10.1097/00004647-199912000-00010.
- Pariente J, Loubinoux I, Carel C, Albucher J-F, Leger A, Manelfe C, et al. Fluoxetine modulates motor performance and cerebral activation of patients recovering from stroke. Ann Neurol 2001;50:718-29. https://doi.org/10.1002/ana.1257.
- The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group . Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 1995;333:1581-7. https://doi.org/10.1056/NEJM199512143332401.
- Acler M, Robol E, Fiaschi A, Manganotti P. A double blind placebo RCT to investigate the effects of serotonergic modulation on brain excitability and motor recovery in stroke patients. J Neurol 2009;256:1152-8. https://doi.org/10.1007/s00415-009-5093-7.
- Zittel S, Weiller C, Liepert J. Citalopram improves dexterity in chronic stroke patients. Neurorehabil Neural Repair 2008;22:311-14. https://doi.org/10.1177/1545968307312173.
- Dam M, Tonin P, De Boni A, Pizzolato G, Casson S, Ermani M, et al. Effects of fluoxetine and maprotiline on functional recovery in post stroke hemiplegic patients undergoing rehabilitation therapy. Stroke 1996;27:1211-14. https://doi.org/10.1161/01.STR.27.7.1211.
- Chollet F, Tardy J, Albucher JF, Thalamas C, Berard E, Lamy C, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo-controlled trial. Lancet Neurol 2011;10:123-30. https://doi.org/10.1016/S1474-4422(10)70314-8.
- Bruno A, Shah N, Lin C, Close B, Hess DC, Davis K, et al. Improving modified Rankin Scale assessment with a simplified questionnaire. Stroke 2010;41:1048-50. https://doi.org/10.1161/STROKEAHA.109.571562.
- Berends HI, Nijlant J, van Putten M, Movig KL, IJzerman MJ. Single dose of fluoxetine increases muscle activation in chronic stroke patients. Clin Neuropharmacol 2009;32:1-5.
- Narushima K, Paradiso S, Moser DJ, Jorge R, Robinson RG. Effect of antidepressant therapy on executive function after stroke. Br J Psychiatry 2007;190:260-5. https://doi.org/10.1192/bjp.bp.106.025064.
- Tallelli P, Werring DJ. Pharmacological augmentation of motor recovery after stroke: antidepressants for non-depressed patients?. J Neurol 2009;256:1159-60. https://doi.org/10.1007/s00415-009-5070-1.
- Nikisch G, Mathé AA, Czernik A, Thiele J, Bohner J, Eap CB, et al. Long-term citalopram administration reduces responsiveness of HPA axis in patients with major depression: relationship with S-citalopram concentrations in plasma and cerebrospinal fluid (CSF) and clinical response. Psychopharmacology 2005;181:751-60. https://doi.org/10.1007/s00213-005-0034-3.
- Barugh AJ, Grey P, Shenkin SD, MacLullich AM, Mead GE. Cortisol levels and the severity and outcomes of acute stroke: a systematic review. J Neurol 2014;261:533-45. https://doi.org/10.1007/s00415-013-7231-5.
- Yi ZM, Liu F, Zhai SD. Fluoxetine for the prophylaxis of poststroke depression in patients with stroke: a meta-analysis. Int J Clin Pract 2010;64:1310-17. https://doi.org/10.1111/j.1742-1241.2010.02437.x.
- Mead GE, Hsieh CF, Lee R, Kutlubaev MA, Claxton A, Hankey GJ, et al. Selective serotonin reuptake inhibitors (SSRIs) for stroke recovery. Cochrane Database Syst Rev 2012;11. https://doi.org/10.1002/14651858.CD009286.pub2.
- National Institute for Health and Care Excellence (NICE) . Depression in Adults With a Chronic Physical Health Problem Treatment and Management 2009. http://guidance.nice.org.uk/CG91/QuickRefGuide/pdf/English (accessed 27 August 2019).
- Wadhwa R, Kumar M, Talegaonkar S, Vohora D. Serotonin reuptake inhibitors and bone health: a review of clinical studies and plausible mechanisms. Osteoporos Sarcopenia 2017;3:75-81. https://doi.org/10.1016/j.afos.2017.05.002.
- Coupland C, Dhiman P, Morriss R, Arthur A, Barton G, Hippisley-Cox J. Antidepressant use and risk of adverse outcomes in older people: population based cohort study. BMJ 2011;343. https://doi.org/10.1136/bmj.d4551.
- Dennis MS, Lo KM, McDowall M, West T. Fractures after stroke: frequency, types, and associations. Stroke 2002;33:728-34. https://doi.org/10.1161/hs0302.103621.
- Myint PK, Poole KE, Warburton EA. Hip fractures after stroke and their prevention. QJM 2007;100:539-45. https://doi.org/10.1093/qjmed/hcm067.
- Counsell C, Dennis M, McDowall M, Warlow C. Predicting outcome after acute and subacute stroke: development and validation of new prognostic models. Stroke 2002;33:1041-7. https://doi.org/10.1161/hs0402.105909.
- Teasdale G, Jennett B. Assessment of coma and impaired consciousness. The Lancet 1974;304:81-4.
- Altman DG, Bland JM. Treatment allocation by minimisation. BMJ 2005;330. https://doi.org/10.1136/bmj.330.7495.843.
- The Optimising Analysis of Stroke Trials (OAST) Collaboration . Can we improve the statistical analysis of stroke trials? Statistical reanalysis of functional outcomes in stroke trials. Stroke 2007;38:1911-15. https://doi.org/10.1161/STROKEAHA.106.474080.
- Bruno A, Close B, Switzer JA, Hess DC, Gross H, Nichols FT, et al. Simplified modified Rankin Scale questionnaire correlates with stroke severity. Clin Rehabil 2013;27:724-7. https://doi.org/10.1177/0269215512470674.
- Bruno A, Akinwuntan AE, Lin C, Close B, Davis K, Baute V, et al. Simplified modified rankin scale questionnaire: reproducibility over the telephone and validation with quality of life. Stroke 2011;42:2276-9. https://doi.org/10.1161/STROKEAHA.111.613273.
- Dennis M, Mead G, Doubal F, Graham C. Determining the modified Rankin score after stroke by postal and telephone questionnaires. Stroke 2012;43:851-3. https://doi.org/10.1161/STROKEAHA.111.639708.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Berwick DM, Murphy JM, Goldman PA, Ware JE, Barsky AJ, Weinstein MC. Performance of a five-item mental health screening test. Med Care 1991;29:169-76. https://doi.org/10.1097/00005650-199102000-00008.
- McCabe CJ, Thomas KJ, Brazier JE, Coleman P. Measuring the mental health status of a population: a comparison of the GHQ-12 and the SF-36 (MHI-5). Br J Psychiatry 1996;169:516-21. https://doi.org/10.1192/bjp.169.4.516.
- Hoeymans N, Garssen AA, Westert GP, Verhaak PF. Measuring mental health of the Dutch population: a comparison of the GHQ-12 and the MHI-5. Health Qual Life Outcomes 2004;2. https://doi.org/10.1186/1477-7525-2-23.
- Mead G, Lynch J, Greig C, Young A, Lewis S, Sharpe M. Evaluation of fatigue scales in stroke patients. Stroke 2007;38:2090-5. https://doi.org/10.1161/STROKEAHA.106.478941.
- Mead GE, Graham C, Dorman P, Bruins SK, Lewis SC, Dennis MS, et al. UK Collaborators of IST . Fatigue after stroke: baseline predictors and influence on survival. Analysis of data from UK patients recruited in the International Stroke Trial. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0016988.
- Duncan PW, Bode RK, Min Lai S, Perera S. Glycine Antagonist in Neuroprotection Americans Investigators . Rasch analysis of a new stroke-specific outcome scale: the Stroke Impact Scale. Arch Phys Med Rehabil 2003;84:950-63. https://doi.org/10.1016/S0003-9993(03)00035-2.
- Duncan PW, Reker DM, Horner RD, Samsa GP, Hoenig H, LaClair BJ, et al. Performance of a mail-administered version of a stroke-specific outcome measure, the Stroke Impact Scale. Clin Rehabil 2002;16:493-505. https://doi.org/10.1191/0269215502cr510oa.
- Duncan PW, Wallace D, Lai SM, Johnson D, Embretson S, Laster LJ. The Stroke Impact Scale version 2.0. Evaluation of reliability, validity, and sensitivity to change. Stroke 1999;30:2131-40. https://doi.org/10.1161/01.str.30.10.2131.
- Duncan P, Reker D, Kwon S, Lai SM, Studenski S, Perera S, et al. Measuring stroke impact with the stroke impact scale: telephone versus mail administration in veterans with stroke. Med Care 2005;43:507-15. https://doi.org/10.1097/01.mlr.0000160421.42858.de.
- Kwon S, Duncan P, Studenski S, Perera S, Lai SM, Reker D. Measuring stroke impact with SIS: construct validity of SIS telephone administration. Qual Life Res 2006;15:367-76. https://doi.org/10.1007/s11136-005-2292-2.
- Dennis M, Sandercock PA, Reid J, Graham C, Murray G, Venables G, et al. Effectiveness of thigh-length graduated compression stockings to reduce the risk of deep vein thrombosis after stroke (CLOTS trial 1): a multicentre, randomised controlled trial. Lancet 2009;373:1958-65. https://doi.org/10.1016/S0140-6736(09)60941-7.
- CLOTS (Clots in Legs Or sTockings after Stroke) Trial Collaboration . Thigh-length versus below-knee stockings for deep thrombosis prophylaxis after stroke: a randomized trial. Ann Intern Med 2010;153:553-62.
- Machin D, Campbell MJ, Tan SB. Sample Size Tables for Clinical Studies. Chichester: John Wiley & Sons; 2008.
- Graham C, Lewis S, Forbes J, Mead G, Hackett ML, Hankey GJ, et al. The FOCUS, AFFINITY and EFFECTS trials studying the effect(s) of fluoxetine in patients with a recent stroke: statistical and health economic analysis plan for the trials and for the individual patient data meta-analysis. Trials 2017;18. https://doi.org/10.1186/s13063-017-2385-6.
- Slot KB, Berge E, Dorman P, Lewis S, Dennis M, Sandercock P. Oxfordshire Community Stroke Project, the International Stroke Trial (UK) . Impact of functional status at six months on long term survival in patients with ischaemic stroke: prospective cohort studies. BMJ 2008;336:376-9. https://doi.org/10.1136/bmj.39456.688333.BE.
- Whooley MA, Avins AL, Miranda J, Browner WS. Case-finding instruments for depression. Two questions are as good as many. J Gen Intern Med 1997;12:439-45. https://doi.org/10.1046/j.1525-1497.1997.00076.x.
- FOCUS Trial Collaboration . Effects of fluoxetine on functional outcomes after acute stroke (FOCUS): a pragmatic, double-blind, randomised, controlled trial. Lancet 2018;393:265-74.
- Bamford J, Sandercock P, Dennis M, Burn J, Warlow C. Classification and natural history of clinically identifiable subtypes of cerebral infarction. Lancet 1991;337:1521-6. https://doi.org/10.1016/0140-6736(91)93206-O.
- Adams HP, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35-41. https://doi.org/10.1161/01.str.24.1.35.
- Pogue J, Walter SD, Yusuf S. Evaluating the benefit of event adjudication of cardiovascular outcomes in large simple RCTs. Clin Trials 2009;6:239-51. https://doi.org/10.1177/1740774509105223.
- Dennis M, Forbes J, Graham C, Hackett ML, Hankey GJ, House A, et al. Fluoxetine and fractures after stroke: exploratory analyses from the FOCUS trial. Stroke 2019;50:3280-2. https://doi.org/10.1161/STROKEAHA.119.026639.
- NHS Improvement . Proposed National Tariff Prices: Planning for 2017 18 and 2018 19 n.d. https://improvement.nhs.uk/resources/proposed-national-tariff-prices-1718-1819/ (accessed 13 April 2020).
- National Institute for Health and Care Excellence and British National Formulary . Fluoxetine n.d. https://bnf.nice.org.uk/medicinal-forms/fluoxetine.html (accessed 13 April 2020).
- Mead G, Hackett ML, Lundström E, Murray V, Hankey GJ, Dennis M. The FOCUS, AFFINITY and EFFECTS trials studying the effect(s) of fluoxetine in patients with a recent stroke: a study protocol for three multicentre randomised controlled trials. Trials 2015;16. https://doi.org/10.1186/s13063-015-0864-1.
- Mead GE, Legg L, Tilney R, Hsieh CF, Wu S, Lundström E, et al. Fluoxetine for stroke recovery: meta-analysis of randomised controlled trials. Int J Stroke 2019. https://doi.org/10.1177/1747493019879655.
- Higgins JPT, Green S. Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration; 2011.
- Hatano S. Experience from a multicentre stroke register: a preliminary report. Bull World Health Organ 1976;54:541-53.
- Marquez-Romero JM, Arauz A, Ruiz-Sandoval JL, Cruz-Estrada Ede L, Huerta-Franco MR, Aguayo-Leytte G, et al. Fluoxetine for motor recovery after acute intracerebral hemorrhage (FMRICH): study protocol for a randomized, double-blind, placebo-controlled, multicenter trial. Trials 2013;14. https://doi.org/10.1186/1745-6215-14-77.
- Wan X, Wang W, Liu J, Tong T. Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Med Res Methodol 2014;14. https://doi.org/10.1186/1471-2288-14-135.
- Guo Y, He Y, Tang B, Ma K, Cai Z, Zeng S, et al. Effect of using fluoxetine at different time windows on neurological functional prognosis after ischemic stroke. Restor Neurol Neurosci 2016;34:177-87. https://doi.org/10.3233/RNN-150535.
- He YT, Tang BS, Cai ZL, Zeng SL, Jiang X, Guo Y. Effects of fluoxetine on neural functional prognosis after ischemic stroke: a randomized controlled study in China. J Stroke Cerebrovasc Dis 2016;25:761-70. https://doi.org/10.1016/j.jstrokecerebrovasdis.2015.11.035.
- He Y, Cai Z, Zeng S, Chen S, Tang B, Liang Y, et al. Effect of fluoxetine on three-year recurrence in acute ischemic stroke: a randomized controlled clinical study. Clin Neurol Neurosurg 2018;168:1-6. https://doi.org/10.1016/j.clineuro.2018.02.029.
- Zhao P, Wang JP. Effects of antidepressants on neurofunctional recovery of post-stroke patients with aphasia. J Dalian Med Univ 2011;33:55-7.
- Birchenall J, Térémetz M, Roca P, Lamy JC, Oppenheim C, Maier MA, et al. Individual recovery profiles of manual dexterity, and relation to corticospinal lesion load and excitability after stroke -a longitudinal pilot study. Neurophysiol Clin 2019;49:149-64. https://doi.org/10.1016/j.neucli.2018.10.065.
- Shah IA, Asimi RP, Kawoos Y, Wani MA, Wani MA, Dar MA. Effect of fluoxetine on motor recovery after acute haemorrhagic stroke: a randomized trial. J Neurol Neurophysiol 2016;7.
- He P. Randomized controlled observation on the effect of early application of fluoxetine in preventing depression after stroke. Chin Clin Rehabil 2004;8:6016-17.
- Kong Y. Fluoxetine for poststroke depression: a randomized placebo controlled clinical trial. Neural Regeneration Research 2007;2:162-5. https://doi.org/10.1016/S1673-5374(07)60036-X.
- Li J, He Q-Y, Han M-F. Recent effect of fluoxetine in improving neurologic impairment and preventing post-stroke depression in the early stage. Chin Postgrad Med 2004;8:1208-9.
- Robinson RG, Schultz SK, Castillo C, Kopel T, Kosier JT, Newman RM, et al. Nortriptyline versus fluoxetine in the treatment of depression and in short-term recovery after stroke: a placebo-controlled, double-blind study. Am J Psychiatry 2000;157:351-9. https://doi.org/10.1176/appi.ajp.157.3.351.
- Mikami K, Jorge RE, Adams HP, Davis PH, Leira EC, Jang M, et al. Effect of antidepressants on the course of disability following stroke. Am J Geriatr Psychiatry 2011;19:1007-15. https://doi.org/10.1097/JGP.0b013e31821181b0.
- Black-Schaffer R. Fluoxetine for Motor, Aphasia, and Neglect Recovery After Ischemic Stroke (flAN) 2012. https://clinicaltrials.gov/ct2/show/NCT01674868 (accessed 25 September 2018).
- Bray BD, Cloud GC, James MA, Hemingway H, Paley L, Stewart K, et al. Weekly variation in health-care quality by day and time of admission: a nationwide, registry-based, prospective cohort study of acute stroke care. Lancet 2016;388:170-7. https://doi.org/10.1016/S0140-6736(16)30443-3.
- NHS National Services Scotland . Scottish Stroke Improvement Programme. 2017 Report n.d. www.strokeaudit.scot.nhs.uk/Downloads/docs/2017-07-11-SCCA-Report.pdf (accessed 19 July 2018).
- Tsivgoulis G, Katsanos AH, Caso V. Under-representation of women in stroke randomized controlled trials: inadvertent selection bias leading to suboptimal conclusions. Ther Adv Neurol Disord 2017;10:241-4. https://doi.org/10.1177/1756285617699588.
- Sansone RA, Sansone LA. Antidepressant adherence: are patients taking their medications?. Innov Clin Neurosci 2012;9:41-6.
- Salter KL, Foley NC, Zhu L, Jutai JW, Teasell RW. Prevention of poststroke depression: does prophylactic pharmacotherapy work?. J Stroke Cerebrovasc Dis 2013;22:1243-51. https://doi.org/10.1016/j.jstrokecerebrovasdis.2012.03.013.
- Gu SC, Wang CD. Early selective serotonin reuptake inhibitors for recovery after stroke: a meta-analysis and trial sequential analysis. J Stroke Cerebrovasc Dis 2018;27:1178-89. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.11.031.
- Cochrane Stroke n.d. https://stroke.cochrane.org/ (accessed 13 April 2020).
- Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet 2018;391:1357-66. https://doi.org/10.1016/S0140-6736(17)32802-7.
- Jones HE, Joshi A, Shenkin S, Mead GE. The effect of treatment with selective serotonin reuptake inhibitors in comparison to placebo in the progression of dementia: a systematic review and meta-analysis. Age Ageing 2016;45:448-56. https://doi.org/10.1093/ageing/afw053.
- Foley P, Lawler A, Chandran S, Mead GE. Potential disease-modifying effects of selective serotonin reuptake inhibitors in multiple sclerosis: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2014;85:709-10. https://doi.org/10.1136/jnnp-2013-306829.
- Dixon O, Mead G. Selective serotonin reuptake inhibitors for mild cognitive impairment: a systematic review. J Neurol Disord Stroke 2013;1.
- Townend E, Brady M, McLaughlan K. A systematic evaluation of the adaptation of depression diagnostic methods for stroke survivors who have aphasia. Stroke 2007;38:3076-83. https://doi.org/10.1161/STROKEAHA.107.484238.
- Brady MC, Stott DJ, Norrie J, Chalmers C, St George B, Sweeney PM, et al. Developing and evaluating the implementation of a complex intervention: using mixed methods to inform the design of a randomised controlled trial of an oral healthcare intervention after stroke. Trials 2011;12. https://doi.org/10.1186/1745-6215-12-168.
- Brady MC, Fredrick A, Williams B. People with aphasia: capacity to consent, research participation and intervention inequalities. Int J Stroke 2013;8:193-6. https://doi.org/10.1111/j.1747-4949.2012.00900.x.
- Collaboration of Aphasia Triallists . Enabling People With Aphasia to Participate in Research: Resources for Stroke Researchers n.d. www.aphasiatrials.org/clinicians-and-researchers/ (accessed 16 September 2019).
- University of Sheffield . Big CACTUS n.d. www.sheffield.ac.uk/scharr/sections/dts/ctru/bigcactus (accessed 27 August 2019).
- University of Central Lancashire . ICONS II n.d. www.uclan.ac.uk/research/explore/projects/icons-two-trial.php (accessed 27 August 2019).
- Thomalla G, Boutitie F, Fiebach JB, Simonsen CZ, Nighoghossian N, Pedraza S, et al. Effect of informed consent on patient characteristics in a stroke thrombolysis trial. Neurology 2017;89:1400-7. https://doi.org/10.1212/WNL.0000000000004414.
Appendix 1 Membership of the FOCUS Trial Collaboration
Co-chief investigators
Martin Dennis and Gillian Mead.
Writing group
Martin Dennis (chairperson), John Forbes, Catriona Graham, Maree Hackett, Graeme J Hankey, Allan House, Stephanie Lewis, Erik Lundström, Peter Sandercock and Gillian Mead.
FOCUS Trial Co-ordinating Centre
Rosemary Anderson, David Buchanan, Ann Deary, Jonathan Drever, Ruth Fraser, Catriona Graham, Karen Innes, Connor McGill, Aileen McGrath, David Perry, Pauli Walker and Carol Williams.
Telephone 6- and 12-month follow-up (numbers conducted)
Martin Dennis (1394), Carol Williams (227), Gillian Mead (220), Rosemary Anderson (173), Yvonne Chun (60), Lynn Dinsmore (50), Emma Maschauer (46), Wellcome Trust Clinical Research Facility (Greig Fraser, Katherine Lawrence and Alison Shaw) (27), Amanda Barugh (15), Shadia Mikhail (8), Gordon Blair (6), Ingrid Hoeritzauer (6) and Maggie Scott (4).
Trial Steering Committee
Stroke Association funded phase: Peter Sandercock (chairperson), Steff Lewis (statistician), Judith Williamson (patient involvement), Martin Dennis (co-chief investigator), Gillian Mead (co-chief investigator), John Forbes (health economist), Graeme J Hankey (AFFINITY), Maree Hackett (AFFINITY), Veronica Murray (EFFECTS) (deceased), Karen Innes (trial manager) and Ray French (sponsor representative).
NIHR funded phase: David Stott (independent chairperson), David Burgess (lay member), Jonathan Emberson (independent statistician), Graham Ellis, Pippa Tyrrell, Judith Williamson (patient involvement), Martin Dennis (co-chief investigator), Gillian Mead (co-chief investigator), Karen Innes (trial manager) and Ray French (sponsor representative).
Co-applicants on funding applications
NIHR Stroke Research Network Portfolio Development Application
Gillian Mead (co-PI and convener of the group), Martin Dennis (co-PI), Marian Brady, John Forbes, Maree Hackett, Allan House, Steff Lewis, Malcolm MacLeod, Hazel Milligan, David Perry, Peter Sandercock, Frank Sullivan, Frederike van Wijck and Caroline Watkins.
Stroke Association
Gillian Mead, Martin Dennis, Peter Sandercock, Malcolm MacLeod, Stephanie Lewis, Frank Sullivan, Allan House, John Forbes, Maree Hackett, Craig Anderson and Graeme J Hankey.
NIHR Health Technology Assessment
Gillian Mead, Martin Dennis, Peter Sandercock, Malcolm MacLeod, Stephanie Lewis, D Morales, Allan House, John Forbes, Maree Hackett, Craig Anderson and Graeme J Hankey.
Independent Data Monitoring Committee
Peter Langhorne, Fiona Reid and Helen Rodgers.
Other activities
Marian Brady (Glasgow Caledonian University) helped produce aphasia-friendly versions of the patient information leaflect and consent form (see Appendix 2). Dan Morales reviewed information sent to GPs.
Investigational medicinal product
Manufactured by Unichem (Mumbai, India).
Sourced through Niche Generics Ltd (Hitchin, UK) and Discovery Pharmaceuticals Ltd (Castle Donington, UK).
Packaged and distributed by Bilcare Ltd (Pune, India), then Sharp Clinical Services (Tredegar, UK).
Participating centres
We have listed each hospital with the total number of patients recruited [n], followed by the names of the local PI(s) and other significant contributors in that centre. The hospitals are ordered depending on the numbers recruited.
Recruiting centres
Royal Infirmary Edinburgh, Edinburgh [141] [G Mead (PI), N Hunter, R Parakramawansha, A Fazal, P Taylor, W Rutherford, K McCormick, R Buchan, A MacRaild, Y Chun, R Paulton, S Burgess, D McGowan, J Skwarski, F Proudfoot, R Murphy, A Barugh, J Perry]; Leeds General Infirmary, Leeds [123] [J Bamford (PI), C Bedford, D Waugh, E Veraque, M Kambafwile, L Makawa, P Smalley, M Randall, L Idrovo, A Hassan, T Thirugnana-Chandran, R Vowden, J Jackson]; St Thomas’ Hospital, London [115] [A Bhalla (PI), C Tam, Professor A Rudd, C Gibbs, J Birns, L Lee Carbon, E Cattermole, A Cape, L Hurley, K Marks, S Kullane]; Royal Hampshire County Hospital, Winchester [110] [N Smyth (PI), E Giallombardo, C Eglinton, J Wilson, D Dellafera, P Reidy, M Pitt, L Sykes, A Frith, V Croome, J Duffy, D Cooke, M Hancevic, L Kerwood, C Narh, C Merritt, J Willson]; Royal Hallamshire Hospital, Sheffield [107] [A Ali (PI), S Bell, T Jackson, H Bowler, C Kamara, A Naqvi, J Howe, K Stocks, G Dunn, K Endean, F Claydon, S Duty, C Doyle, K Harkness, E Richards, M Meegada, A Maatouk, L Barron, K Dakin, R Lindert, Professor A Majid]; Calderdale Royal Hospital, Halifax [81] [P Rana (PI), A Nair, C Brighouse-Johnson, J Greig, M Kyu, S Prasad, M Robinson, B Mclean, I Alam, L Greenhalgh, Z Ahmed]; University Hospitals of North Midlands NHS Trust, Stoke-on-Trent [80] [Professor C Roffe (PI), S Brammer, A Barry, C Beardmore, K Finney, H Maguire, P Hollinshead, J Grocott, I Natarajan, J Chembala, R Sanyal, S Lijko, N Abano, A Remegoso, P Ferdinand, S Stevens, C Stephen, P Whitmore, A Butler, C Causley, R Varquez, G Muddegowda, R Carpio, J Hiden, H Denic]; Royal Devon and Exeter Hospital, Exeter [73] [J Sword (PI), F Hall, J Cageao, S Keenan, R Curwen, M James, P Mudd, C Roughan, H Kingwell, A Hemsley, C Lohan, S Davenport, T Chapter, A Bowring, M Hough, D Strain, K Gupwell, K Miller, A Goff, E Cusack, S Todd, R Partridge, G Jennings, K Thorpe, J Stephenson, K Littlewood]; Monklands Hospital, Airdrie [70] [M Barber (PI), F Brodie, S Marshall, D Esson, C McInnes, I Coburn, F Ross, V Withers, E Bowie, H Barcroft, L Miller]; York Hospital, York [69] [P Willcoxson (PI), M Keeling, M Donninson, R Evans, D Daniel, J Coyle, M Elliott, P Wanklyn, J Wightman, E Iveson, A Porteous, N Dyer, M Haritakis, M Ward, L Wright, J Bell, C Emms, P Wood, P Cottrell, L Doughty, L Carr, C Anazodo, M O Neill, J Westmoreland, R Rodriguez, R Mir, C Donne, E Bamford, P Clark Brown]; Pinderfields Hospital, Wakefield [67] [A Stanners (PI), I Ghouri, A Needle, M Eastwood, M Carpenter, P Datta, R Davey, F Razik, G Bateman, J Archer, V Balasubramanian, L Jackson, L Benton, J Ball, R Bowers, J Ellam, K Norton]; Southend University Hospital NHS Foundation Trust, Essex [64] [P Guyler (PI), S Tysoe, P Harman, A Kundu, T Dowling, S Chandler, O Omodunbi, T Loganathan, S Noor, S Kunhunny, D Sinha, A Siddiqui, A Siddiqui, M Sheppard, S Shah, S Kelavkar, K Ng, L Wilson, A Ropun, L Kamuriwo, R Orath Prabakaran, E France, S Rashmi]; Pilgrim Hospital, Boston [63] [D Mangion (PI), C Constantin, S Markova, A Hardwick, J Borley, L De Michele Hock, T Lawrence, J Fletcher, K Netherton, R Spencer, H Palmer]; Lincoln County Hospital, Lincoln [60] [M Soliman (PI), S Leach, Professor J Sharma, R Brown, C Taylor, I Wahishi, S Arif, S Bell, A Fields, S Butler, J Hindle, E Watson, J Borley, C Hewitt]; University Hospital Aintree, Liverpool [60] [C Cullen (PI), D Hamill, Z Mellor, T Fluskey, V Hankin, A Keeling, R Durairaj, D Wood, J Peters, D Shackcloth, R Tangney, T Hlaing, V Sutton, M Harrison, S Stevenson, J Ewing]; Bradford Royal Infirmary, Bradford [59] [C Patterson (PI), J Price, H Wilson, H Ramadan, S Maguire, S Khan, R Bellfield, U Hamid, M Hooley, R Ghulam, L Masters, W Gaba, O Quinn]; Luton and Dunstable University Hospital, Luton [56] [L Sekaran (PI), M Tate, N Mohammed, S Sethuraman, L Alwis, R Robinson, K Bharaj, R Pattni, F Justin, C Tam, M Chauhan, L Eldridge, S Mintias, J Palmones]; Bristol Royal Infirmary, Bristol [54] [C Holmes (PI), L Guthrie, P Murphy, N Devitt, J Leonard, M Osborn, L Ball, A Steele, E Dodd, A Holloway, P Baker, R Patel, I Penwarden, S Caine, S Clarke, L Dow, S Williams, R Wynn-Williams]; John Radcliffe Hospital, Oxford [51] [J Kennedy (PI), A DeVeciana, P Mathieson, I Reckless, R Teal, U Schulz, Professor G Ford, P Mccann]; St George’s Healthcare NHS Trust, London [47] [G Cluckie (PI), G Howell, J Ayer, B Moynihan, R Ghatala, B Clarke, G Cloud, B Patel, U Khan, N Al-Samarrai, F Watson, T Adedoyin, S Trippier, N Chopra, L Zhang, L Choy, K Kennedy, R Williams, V Jones, N Clarke, A Dainty, A Blight]; South Glasgow University Hospital, Glasgow [45] [J Selvarajah (PI), W Smith, F Moreton, A Welch, D Kalladka, B Cheripelli, E Douglas, A Lush, X Huang, S El Tawil, N Day, K Montgomery, H Hamilton, D Ritchie, S Ramachandra, K McLeish]; Northwick Park Hospital, Harrow [44] [D Cohen (PI), B Badiani, M Abdul-Saheb, A Chamberlain, M Mpelembue, R Bathula, M Lang, J Devine, L Alwis, L Southworth, L Burgess, N Epie, A David, E Owoyele, F Guo, A Oshodi, V Sudkeo]; Royal Bournemouth Hospital, Bournemouth [44] [K Thavanesan (PI), D Tiwari, J Bell, C Ovington, E Rogers, R Bower, G Hann, B Longland, O David, A Hogan, S Loganathan, J Roberts, C Cox, S Orr, M Keltos]; Yeovil District Hospital, Yeovil [41] [K Rashed (PI), D Wood, B Williams-Yesson, J Board, S De Bruijn, C Buckley, C Vickers, S Board, J Allison, E Keeling, T Duckett, D Donaldson, C Barron, L Balian, A Edwards, J Wilson]; Royal Derby Hospital, Derby [40] [T England (PI), A Hedstrom, E Bedford, M Harper, E Melikyan, W Abbott, K Subramanian, M Goldsworthy]; The Princess Royal Hospital, Telford [40] [M Srinivasan (PI), I Mukherjee, U Ghani, A Yeomans, D Donaldson, F Hurford, R Chapman, S Shahzad, O David, N Motherwell, L Tonks, R Young]; Gloucestershire Royal Hospital, Gloucester [39] [D Dutta (PI), P Brown, F Davis, D Ward, J Turfrey, M Obaid, B Cartwright, B Topia, J Spurway, C Hughes, L Hill, S OConnell, K Collins, R Bakawala]; Countess of Chester Hospital, Chester [38] [K Chatterjee (PI), T Webster, S Haider, P Rushworth, F Macleod, C Perkins, A Nallasivan, E Burns, S Leason, T Carter, S Seagrave]; Airedale General Hospital, Keighley [37] [E Sami (PI), S Parkinson, M Hassan, S Naqvi, L Armstrong, S Mawer, G Darnbrook, C Booth, B Hairsine, M Smith, S Williamson, F Farquhar]; Queen Elizabeth Hospital, Gateshead [36] [B Esisi (PI), T Cassidy, B McClelland, G Mankin, M Bokhari, D Sproates]; Walsall Manor Hospital, Walsall [35] [E Epstein (PI), R Blackburn, S Hurdowar, N Sukhdeep, S Razak, N Upton, A Hashmi, K Osman]; New Cross Hospital, Wolverhampton [34] [K Fotherby (PI), A Willberry, D Morgan, G Sahota, K Jennings-Preece, D Butler, S Das, A Stevens, N Ahmad, K Kauldhar]; Royal Cornwall Hospital, Truro [34] [F Harrington (PI), A Mate, J Skewes, K Adie, K Bond, G Courtauld, C Schofield, L Lucas, A James, S Ellis, B Maund, L Allsop, C Brodie, M Johnson, E Driver, K Harris, M Drake, K Moore, E Thomas]; Wycombe Hospital, High Wycombe [34] [M Burn (PI), A Hamilton, S Mahalingam, A Benford, D Hilton, F Reid, A Misra, L Hazell, K Ofori, M Mathew, A Thomas, S Dayal, I Burn]; University Hospital North Durham, Durham [32] [D Bruce (PI), M Naeem, R Burnip, R Hayman, P Earnshaw, E Brown, S Clayton, P Gamble, S Dima, M Dhakal, G Rogers, L Stephenson, R Nendick, Y Pai, K Nyo]; Victoria Hospital, Kirkcaldy [32] [V Cvoro (PI), M Couser, M Simpson, A Tachtatzis, K Ullah, K McCormick, R Cain, N Chapman, S Pound, S McAuley]; William Harvey Hospital, Ashford [32] [D Hargroves (PI), B Ransom, K Mears, K Griffiths, L Cowie, T Hammond, T Webb, I Balogun, H Rudenko, A Thomson, D Ceccarelli, A Gillian, E Beranova, A Verrion, N Chattha, N Schumacher, A Bahk, S Walker]; Queen Elizabeth Hospital, Birmingham [31] [D Sims (PI), R Jones, J Smith, R Tongue, M Willmot, C Sutton, E Littleton, J Khaira, S Maiden, J Cunningham, Y Chin, C Green, M Bates, K Ahlquist]; Royal Sussex County Hospital, Brighton [31] [I Kane (PI), J Breeds, T Sargent, L Latter, A Pitt Ford, T Levett, N Gainsborough, P Thompson, A Dunne, E Barbon, S Hervey]; Poole Hospital, Poole [30] [S Ragab (PI), T Sandell, C Dickson, S Power, J Dube, N Evans, B Wadams, S Elitova, B Aubrey, T Garcia]; Victoria Hospital, Blackpool [29] [J Mcllmoyle (PI), A Ahmed, C Dickinson, C Jeffs, S Dhar, K Jones, J Howard, C Armer, J Frudd, S Kumar, A Potter, S Donaldson]; Watford General Hospital, Watford [29] [D Collas (PI), S Sundayi, L Denham, D Oza, E Walker, J Cunningham, M Bhandari]; Sandwell General Hospital, Birmingham [28] [S Ispoglou (PI), R Evans, K Sharobeem, A Hayes, J Howard-Brown, E Walton, S Shanu, S Billingham]; Southampton General Hospital, Southampton [27] [N Weir (PI), G Howard, E Wood, L Sykes, V Pressly, P Crawford, H Burton, A Walters, J Marigold, R Said, C Allen, S Evans, S Egerton, J Hakkak, J Andrews, R Lampard, S Smith, C Cox, S Tsang, R Creeden, I Gartrell, F Smith]; The County Hospital, Hereford [26] [C Jenkins (PI), F Price, J Pryor, A Hedges, L Moseley, L Mercer, C Hughes]; Addenbrooke’s Hospital, Cambridge [25] [E Warburton (PI), D Handley, S Finlay, N Hannon, A Espanol, S Kelly, J Mcgee, Professor H Markus, D Chandrasena, J Sesay, D Hayden, H Hayhoe, J Macdonald, M Bolton, J Mitchell, C Farron, E Amis, D Day, A Culbert, L Whitehead, S Crisp, J Francis]; Sunderland Royal Hospital, Sunderland [25] [J OConnell (PI), E Osborne, R Beard, P Corrigan, A Smith, M Edwards, L Mokoena, N Sattar, M Myint, R Krishnamurthy]; West Suffolk Hospital, Bury St Edmonds [25] [A Azim (PI), S Whitworth, A Nicolson, S Alam, J White, M Krasinska-Chavez, J Imam, S Chaplin, D Singh, J Curtis, L Wood]; Western General Hospital, Edinburgh [25] [Professor M Dennis (PI), R Buchan, W Rutherford, J Skwarski, D McGowan]; Forth Valley Royal Hospital, Larbert [24] [A Byrne (PI), C McGhee, A Smart, Professor M MacLeod, F Donaldson, J Blackburn, C Copeland, J Wilson, R Scott]; Royal Liverpool University Hospital, Liverpool [24] [P Fitzsimmons (PI), G Fletcher, A Manoj, P Cox, L Trainor, P Lopez, M Wilkinson, L Denny, K Kavanagh, H Allsop]; Queen Alexandra Hospital, Portsmouth [23] [U Sukys (PI), S Valentine, D Jarrett, K Dodsworth, M Wands, C Watkinson, W Golding, N Khan, J Tandy, R Butler, K Yip, C James, Y Davies, M Williams, A Suttling]; Royal Berkshire NHS Foundation Trust, Reading [23] [K Nagaratnam (PI), N Mannava, N Haque, N Shields, K Preston, G Mason, K Short, G Uitenbosch, G Lumsdale]; Royal Preston Hospital, Preston [23] [H Emsley (PI), S Sultan, B Walmsley, S Ahmed, D Doyle, A McLoughlin, L Hough, B Gregary, S Raj]; Wythenshawe Hospital, Manchester [22] [A Maney (PI), S Blane, G Gamble, A Hague, B Charles, B Duran, C Lambert, K Stagg]; Musgrove Park Hospital, Taunton [21] [R Whiting (PI), S Brown, M Hussain, M Harvey, J Homan, L Foote, L Graham, C Lane, L Kemp, J Rowe, H Durman, L Brotherton, N Hunt, J Foot, A Whitcher, C Pawley]; Norfolk and Norwich University Hospital, Norwich [21] [P Sutton (PI), S Mcdonald, D Pak, A Wiltshire, J Balami, C Self, J Jagger, A Metcalf, G Healey, M Crofts, A Chakrabarti, C Hmu, J Keshet-Price, G Ravenhill, C Grimmer, T Soe, I Potter, P Tam, M Langley]; Aberdeen Royal Infirmary, Aberdeen [20] [M MacLeod (PI), P Cooper, M Christie, J Irvine, A Joyson, F Annison, D Christie, C Meneses, A Johnson, S Nelson, V Taylor, J Furnace, H Gow, J Reid, R Clarke]; East Surrey Hospital, Redhill [19] [Y Abousleiman (PI), S Bloom, S Goshawk, J Purcell, T Beadling, S Collins, S Jones, S Sangaralingham, E Munuswamy Vaiyapuri, M Landicho, Y Begum, S Mutton, J Allen, J Lowe, M Hughes]; The Royal Victoria Hospital, Belfast [19] [I Wiggam (PI), S Tauro, S Cuddy, B Wells]; Derriford Hospital, Plymouth [17] [A Mohd Nor (PI), C Eglinton, N Persad, M Kalita, M Weinling, S Weatherby, D Lashley, A Pace, C Brown, A Mucha, A Shah, J Baker, M Marner, J Westcott, N Wilmshurst, R Cowan, D Waugh]; Doncaster Royal Infirmary, Doncaster [17] [D Chadha (PI), M Fairweather, D Walstow, R Fong]; Morriston Hospital, Swansea [17] [M Krishnan (PI), H Thompson Jones, C Lynda, C Hughes, C Clements, R Williams, T Anjum, S Storton, D Lynne, L Thomas, S Tucker, D Colwill, P Jones]; The Hillingdon Hospital NHS Foundation Trust, Uxbridge [17] [E Vasileiadis (PI), A Parry, C Mason, M Holden, K Petrides, T Nishiyama, H Mehta, S Mumani]; Perth Royal Infirmary, Perth [16] [S Johnston (PI), C Almadenboyle, S Carson, S Ross, P Nair, M Stirling, E Tenbruck]; James Cook University Hospital, Cleveland [15] [D Broughton (PI), A Annamalai, J Wong, D Tryambake, L Dixon, A Skotnicka, J Thompson, A Sigsworth, S Whitehouse, J Pagan]; Lister Hospital, Stevenage [15] [A Pusalkar (PI), H Beadle, K Chan, P Dangri, A Asokanathan, A Rana, S Gohil, K Crabtree, A Cook, M Massyn, P Aruldoss, S Dabbagh]; Salisbury District Hospital, Salisbury [15] [T Black (PI), C Clarke, R Fennelly, L Nardone, V DiMartino, A Anthony, D Mead, M Tribbeck]; St Peter’s Hospital, Chertsey [14] [B Affley (PI), C Sunderland, E Young, L Goldenberg, A Khan, P Wilkinson, L Abbott, R Nari, S Lock, J Stewart, A Shakhon, R Pereira, M DSouza, S Dunn, N Cron, A Mckenna]; Colchester General Hospital, Colchester [13] [R Sivakumar (PI), S Cook, A Wright, J Ngeh, R Saksena, J Ketley-O’Donel, R Needle, E Chinery]; University College London Hospitals NHS Foundation Trust, London [13] [R Greenwood (PI), L Howaniec, C Watchurst, K Patel, R Erande, M Brezitski, N Passeron, E Elliott, N Oji, D Austin, A Banaras, C Hogan, T Corbett, R Shah]; Warrington Hospital, Warrington [13] [M Kidd (PI), G Hull, J Simpson, S Punekar, J Nevinson, H Penney, J Ward, W Wareing, N Hayes, K Bunworth, L Connell, K Mahawish, G Drummond]; Worthing Hospital, Worthing [13] [N Sengupta (PI), M Metiu, C Gonzalez, J Margalef, S Funnell, G Peters, I Chadbourn]; Dorset County Hospital, Dorchester [12] [H Proeschel (PI), P Ashcroft, S Sharpe, S Jones, P Cook, D Jenkinson, D Kelly, H Bray]; Queen Elizabeth The Queen Mother Hospital, Margate [12] [G Gunathilagan (PI), K Griffiths, K Mears, A Gillian, S Jones, S Tilbey, S Abubakar, E Beranova]; King’s Mill Hospital, Mansfield [11] [M Cooper (PI), A Rajapakse, A Nasar, J Janbieh, L Wade, L Otter, I Wynter, S Haigh, R Boulton, J Burgoyne, A Boulton]; Stepping Hill Hospital, Stockport [11] [J Vassallo (PI), A Hasan, L Orrell, A Khan, S Qamar, S Graham, D Leonard, E Hewitt, M Haque, J Awolesi, E Bradshaw, A Kent]; Bronglais General Hospital, Aberystwyth [10] [P Jones (PI), C Duggan, A Hynes, E Nurse, S Raza, U Pallikona, B Edwards, G Morgan, H Tench, R Loosley, K Dennett, T Trugeon-Smith, R Jones, S Jones, R Williams, D Robson]; Hull Royal Infirmary, Hull [10] [R Rayessa (PI), A Abdul-Hamid, V Lowthorpe, K Mitchelson, E Clarkson, H Rhian, A Fleming]; Broomfield Hospital, Chelmsford [9] [R Kirthivasan (PI), J Topliffe, R Keskeys, S Williams, F McNeela, E Bohannan, L Cooper, S Shah, G Zachariah, F Cairns, T James, L Fergey, S Smolen, A Lyle, E Cannon, S Omer]; Whiston Hospital, Prescot [9] [S Mavinamane (PI), S Meenakshisundaram, L Ranga, J Bate, A Hill, M Hargreaves, T Smith, S Dealing, L Harrison]; Frimley Park Hospital, Frimley [8] [S Amlani (PI), G Gulli, M Hawkes-Blackburn, N Hunter, S Levy, L Francis, S Holland, A Peacocke, J Amero, M Burova, O Speirs]; Harrogate Hospital, Harrogate [7] [S Brotheridge (PI), S Al Hussayni, H Lyon, C Hare, S Jackson, L Stephenson, J Featherstone, A Bwalya]; Royal Blackburn Hospital, Blackburn [7] [A Singh (PI), M Goorah, J Walford, A Bell, C Kelly, D Rusk, D Sutton, F Patel, S Duberley, K Hayes, L Hunt]; Scarborough Hospital, Scarborough [7] [E Ahmed El Nour (PI), S Dyer, L Brown, K Elliott, E Temlett, J Paterson, P Wood, M Haritakis, S Honour, C Box, P Cottrell, J Westmoreland, S Young, R Furness]; West Cumberland Hospital, Whitehaven [7] [E Orugun (PI) (deceased), H Crowther, R Glover, C Brewer, S Thornthwaite]; Macclesfield District General Hospital, Macclesfield [6] [M Sein (PI), K Haque, L Bailey, S Wong, E Gibson, K Burton, L Brookes, K Rotchell, K Waltho, C Lindley, A Murray, D Leonard, M Holland]; Royal Lancaster Infirmary, Lancaster [6] [P Kumar (PI), M Khan, P Harlekar, C Culmsee, L Booth, J Drew, J Ritchie, N Mackenzie, C Thomas, J Barker]; Weston General Hospital, Weston Super Mare [6] [M Haley (PI), D Cotterill, L Lane, D Simmons, R Warinton, G Saunders, H Dymond, S Kidd, C Little, Y Neves-Silva]; Basildon and Thurrock University Hospital, Basildon [5] [B Nevajda (PI), M Villaruel, S Patel, U Umasankar, A Man, N Gadi, N Christmas, R Ladner, R Rangasamy, G Butt, W Alvares]; Ulster Hospital, Belfast [5] [M Power (PI), S Hagan, K Dynan, D Wilson, S Crothers, C Leonard, B Wroath, G Douris]; Antrim Area Hospital, Antrim [4] [D Vahidassr (PI), B Gallen, S McKenna, A Thompson, C Edwards, C McGoldrick, M Bhattad]; Epsom General Hospital, Epsom [3] [J Putteril (PI), R Gallifent, E Makanju, M Lepore, C McRedmond, L Arundell, A Goulding]; Fairfield General Hospital, Bury [3] [K Kawafi (PI), P Jacob, L Turner, N Saravanan, L Johnson, D Morse, R Namushi, S Humphrey, R Patel, J McLaughlin]; Leighton Hospital, Crewe [3] [M Salehin (PI), S Tinsley, T Jones, D Bailey, L Garcia-Alen, L Kalathil, R Miller, N Gautam, J Horton, J Meir, E Margerum, A Ritchings, A Jones, K Amor]; Royal Free Hospital, London [3] [V Nadarajan (PI), J Laurence, S Fung Lo, S Melander, P Nicholas, E Woodford, G McKenzie, V Le, J Crause]; St Helier Hospital, Carshalton [3] [P OMahony (PI), C Orefo, C McDonald, V Jones, E Osikominu, T Khan, G Appiatse, E Makanju, A Wardale, M Augustin, H Stone]; North Middlesex University Hospital NHS Trust, London [2] [R Luder (PI), M Bhargava, G Bhome, V Johnson, R Shah, D Chesser, H Bridger, E Murali]; South Tyneside General Hospital, South Shields [2] [J Scott (PI), S Morrison, A Burns, J Graham, M Duffy]; Princess Royal Hospital, Haywards Heath [1] [K Ali (PI), T Sargent, E Pitcher, J Gaylard, J Newman]; Rotherham Hospital, Rotherham [1] [S Punnoose (PI), M Khan, S Oakley, V Murray, C Bent, R Walker, K Purohit, A Rees, M Davy, S Besley, O Chohan]; Royal London Hospital (Barts Health), London [1] [L Argandona (PI), L Cuenoud, H Hassan, E Erumere, A OCallaghan, L Howaniec, O Redjep, G Auld, P Gompertz, A Song, R Hungwe, H Kabash, T Tarkas]; and Royal Surrey County Hospital, Guildford [1] [A Blight (PI), S Jones, G Livingstone, F Butler, S Bradfield, L Gordon, J Schmit, A Wijewardane, C Medcalf, T Edmunds, R Wills, C Peixoto].
Appendix 2 Development of easy-access versions of patient information and consent forms by Professor Marian Brady, Research Group Lead for Living with Stroke, Glasgow Caledonian University
Aim
Development of accessible versions of participant information sheets and consent forms for the FOCUS trial.
Methods
Initial drafts of accessible versions of the standard FOCUS information sheets and consent forms were prepared for review by people with aphasia based on established methodologies91,92 and sound ethical principles93 by an experienced speech and language therapist and stroke rehabilitation researcher (Marian Brady).
Accessible versions of the standard materials were drafted:
-
participant information sheets
-
participant consent forms.
Two meetings were planned with Speakeasy (Bury, UK) members in Dundee on 28 October and again on 4 November 2011. Version 1 of the accessible materials were sent in advance of the first meeting to members of the Speakeasy group (peer support group for people with aphasia following stroke) based in Dundee. These were sent in advance to provide sufficient time for the Speakeasy members with language impairment to review and prepare in advance of the first meeting. The process of drafting and review with comments was scheduled to continue to contribute to the iterative development of accessible materials until the Speakeasy group felt that they were suitable to be used in the trial with people with aphasia.
Results
The members reviewed version 1 of the proposed accessible materials in advance of the meeting. Eight members attended. The materials were reviewed on a statement-by-statement basis with reference (where necessary) to the standard materials. In this way, we ensured that the essence of the statement contained in the standard format was retained in the accessible version, and made comments.
Version 2 of the accessible materials was prepared in response to the group’s comments. Version 2 was reviewed by the FOCUS trial team at the University of Edinburgh, including the PIs, trial manager and other trial staff. They raised some feasibility issues regarding the version 2, which was in colour and on A4 sheets. They queried whether printing the accessible versions in black and white (considerably cheaper than the colour) would be acceptable to the group and to people with aphasia. They also highlighted that printing in black and white would allow sites to easily photocopy the materials, which would support implementation of the trial at sites. They also asked whether or not the A4 size of the materials could be reduced to an A5 booklet, again to support implementation.
Version 3 of the materials was shared with the Speakeasy group (black and white and A5 version), with a specific response requested in relation to the above issues via the post to members who had participated in the first meeting. The Speakeasy members responded that although colour printing would be preferable, for cost reasons the black-and-white versions were acceptable. They also felt that although larger A4 was preferable, the A5 version would also be acceptable. All other changes were accepted, and no further changes were requested. A second scheduled face-to-face meeting was agreed to be unnecessary and was cancelled.
The ethics committee responded positively to the accessible versions of information sheets and consent forms and agreed that accessible versions could be made available to all potential trial participants as deemed required by trial staff. The ethics committee’s response to the materials was fed back to the Speakeasy members.
Discussion and conclusions
In addition, and beyond the FOCUS trial, the accessible materials used in this trial contributed to the NIHR template materials supporting the involvement of people with aphasia in stroke research. 94
Reflections/critical perspective
Involvement of people with aphasia supported the development of accessible materials to support the ethical inclusion of people with aphasia in the FOCUS trial. The perspective of people with a language impairment is vital to the development of such materials as it is not possible for people with intact language skills to perceive all the challenges experienced with a language impairment.
Preparation of accessible versions of materials in stroke research is increasingly standard practice (Big CACTUS95 and ICONS II96). There is some suggestion that reliance on proxy consent may contribute to selection bias in the participant recruitment. 97
Appendix 3 Contributors to the updated systematic review, search strategy and references
Contributors to updated systematic review
Gillian E Mead, MD, Fellow of Royal College of Physicians of Edinburgh, Professor of Stroke and Elderly Care Medicine, University of Edinburgh, Edinburgh, UK.
Lynn Legg, Doctor of Philosophy (PhD), Senior Researcher, Department of Medicine for the Elderly, Royal Alexandra Hospital, Paisley, UK.
Russel Tilney, Doctor of medicine (MD), Department of Medicine, Mater Dei Hospital, Malta.
Cheng Fang Hsieh, MD, Division of Geriatrics and Gerontology, Department of Internal Medicine and Department of Neurology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan.
Simiao Wu, MD, PhD, Department of Neurology, West China Hospital, Sichuan University, Chengdu, China.
Erik Lundström, Associate Professor of Neurology, Karolinska Institutet, Department of Clinical Neuroscience; and Adjunct Senior Lecturer at Department of Neuroscience, Neurology, Uppsala University, Uppsala, Sweden.
Ann Sofie Rudberg, MD, Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden.
Mansur Kutlubaev, MD, PhD, Department of Neurology, G.G. Kuvatov Republican Clinical Hospital; Department of Neurology, Bashkir State Medical University, Ufa, Russia.
Martin S Dennis, Professor of Stroke Medicine, University of Edinburgh, Edinburgh, UK.
Babak Soleimani, MB, Bachelor of Medicine and Bachelor of Surgery, Clinical Fellow, NHS Lothian, UK.
Amanda Barugh, PhD, Consultant Stroke Physician, NHS Lothian, UK.
Maree L Hackett, PhD, Programme Head Mental Health, Associate Professor, Faculty of Medicine, University of New South Wales, Sydney; Professor of Epidemiology, The University of Central Lancashire, UK; The George Institute for Global Health, Australia.
Graeme J Hankey, MD, Fellow of the Royal Australasian College of Physicians, Professor of Neurology, Medical School, The University of Western Australia, WA, Australia.
Acknowledgements
Joshua Cheyne of Cochrane Stroke ran the literature searches.
Maureen Harding obtained articles for full-text review.
Dr Juan Marquez Romero provided unpublished data from the fluoxetine for motor recovery after acute intracerebral hemorrhage (FMRICH) trial.
Professor Chollet responded to requests for information about other new trials.
Dr Jan Bembenek and Professor Anna Czlonkowska provided further information about FOCUS-Poland.
Professor Peter Sandercock commented on the manuscript.
Contributions
Gillian Mead conceived the study, screened references, extracted data, assessed risk of bias, carried out the analyses and wrote the first draft.
Lynn Legg searched for selected studies for inclusion, collected data, assessed risk of bias, managed studies through the review process and contributed to the final version.
Russel Tilney, Cheng Fang Hsieh, Simiao Wu, Erik Lundström, Ann-Sofie Rudberg, Mansur Kutlubaev, Babak Soleimani and Amanda Barugh screened citations, retrieved potentially relevant papers and screened their eligibility for the systematic review, assisted with data extraction and drafted the manuscript for submission.
Maree Hackett extracted data and edited the final manuscript.
Graeme J Hankey and Martin Dennis conceived the study, provided expertise in relation to analysis methods and edited the draft paper.
Funding
This review was not specifically funded. Maree L Hackett held a National Health and Medical Research Council (Australia) Career Development Fellowship, level 2 (reference APP1141328) (2018–21).
Search strategy for updated systematic review
The following searches were carried out (Cochrane Stroke Group Trials Register; date searched: 17 July 2018):
-
Cochrane Central Register of Controlled Trials (CENTRAL) (via The Cochrane Library) (date range searched: 2018, issue 6).
-
MEDLINE (via Ovid) (date range searched: 1948 to 17 July 2018).
-
EMBASE (via Ovid) (date range searched: 1980 to 17 July 2018).
-
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (via EBSCOhost) (date range searched: 1982 to 17 July 2018).
-
Allied and Complementary Medicine (AMED) (via Ovid) (date range searched: 1985 to 17 July 2018)
-
PsycINFO (via Ovid) (date range searched: 1967 to 17 July 2018).
-
US National Institutes of Health Ongoing Trials Register [via ClinicalTrials.gov (www.clinicaltrials.gov)].
-
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch) (date searched: 26 June 2018).
Cochrane Central Register of Controlled Trials (CENTRAL) (via The Cochrane Library) (2018, issue 6) search strategy
-
#1. MeSH descriptor Cerebrovascular Disorders explode all trees
-
#2. (stroke in Title, Abstract or Keywords or poststroke in Title, Abstract or Keywords or post-stroke in Title, Abstract or Keywords or cerebrovasc* in Title, Abstract or Keywords or (brain in Title, Abstract or Keywords and vasc* in Title, Abstract or Keywords) or (cerebral in Title, Abstract or Keywords and vasc* in Title, Abstract or Keywords) or cva* in Title, Abstract or Keywords or apoplex* in Title, Abstract or Keywords or SAH in Title, Abstract or Keywords)
-
#3. ((brain* in Title, Abstract or Keywords or cerebr* in Title, Abstract or Keywords or cerebell* in Title, Abstract or Keywords or intracran* in Title, Abstract or Keywords or intracerebral in Title, Abstract or Keywords) and (ischemi* in Title, Abstract or Keywords or ischaemi* in Title, Abstract or Keywords or infarct* in Title, Abstract or Keywords or thrombo* in Title, Abstract or Keywords or emboli* in Title, Abstract or Keywords or occlus* in Title, Abstract or Keywords))
-
#4. ((brain* in Title, Abstract or Keywords or cerebr* in Title, Abstract or Keywords or cerebell* in Title, Abstract or Keywords or intracerebral in Title, Abstract or Keywords or intracranial in Title, Abstract or Keywords or subarachnoid in Title, Abstract or Keywords) and (haemorrhage* in Title, Abstract or Keywords or hemorrhage* in Title, Abstract or Keywords or haematoma* in Title, Abstract or Keywords or hematoma* in Title, Abstract or Keywords or bleed* in Title, Abstract or Keywords))
-
#5. MeSH descriptor hemiplegia this term only
-
#6. MeSH descriptor paresis explode all trees
-
#7. MeSH descriptor Gait Disorders, Neurologic explode all trees
-
#8. (hemipleg* in Title, Abstract or Keywords or hemipar* in Title, Abstract or Keywords or paresis in Title, Abstract or Keywords or paretic in Title, Abstract or Keywords)
-
#9. (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8)
-
#10. MeSH descriptor Serotonin Uptake Inhibitors explode all trees
-
#11. (serotonin in Title, Abstract or Keywords or 5-HT in Title, Abstract or Keywords or “5 HT” in Title, Abstract or Keywords or 5-hydroxytryptamine in Title, Abstract or Keywords or “5 hydroxytryptamine” in Title, Abstract or Keywords)
-
#12. (uptake in Title, Abstract or Keywords or reuptake in Title, Abstract or Keywords or re-uptake in Title, Abstract or Keywords)
-
#13. inhib* in Title, Abstract or Keywords
-
#14. (#11 and #12 and #13)
-
#15. SSRI* in Title, Abstract or Keywords
-
#16. (alaproclat* in Title, Abstract or Keywords or cericlamin* in Title, Abstract or Keywords or citalopram in Title, Abstract or Keywords or dapoxetin* in Title, Abstract or Keywords or escitalopram in Title, Abstract or Keywords or femoxetin* in Title, Abstract or Keywords or fluoxetin* in Title, Abstract or Keywords or fluvoxamin* in Title, Abstract or Keywords or paroxetin* in Title, Abstract or Keywords or sertralin* in Title, Abstract or Keywords or trazodone in Title, Abstract or Keywords or vilazodone in Title, Abstract or Keywords or zimelidine in Title, Abstract or Keywords)
-
#17. (Celexa in Title, Abstract or Keywords or Cipramil in Title, Abstract or Keywords or Cipram in Title, Abstract or Keywords or Recital in Title, Abstract or Keywords or Emocal in Title, Abstract or Keywords or Dalsan in Title, Abstract or Keywords or Sepram in Title, Abstract or Keywords or Seropram in Title, Abstract or Keywords or Citox in Title, Abstract or Keywords or Priligy in Title, Abstract or Keywords or Lexapro in Title, Abstract or Keywords or Cipralex in Title, Abstract or Keywords or Seroplex in Title, Abstract or Keywords or Esertia in Title, Abstract or Keywords or Prozac in Title, Abstract or Keywords or Fontex in Title, Abstract or Keywords or Seromex in Title, Abstract or Keywords or Seronil in Title, Abstract or Keywords or Sarafem in Title, Abstract or Keywords or Ladose in Title, Abstract or Keywords or Motivest in Title, Abstract or Keywords or Fluctin in Title, Abstract or Keywords or fluox in Title, Abstract or Keywords or Lovan in Title, Abstract or Keywords or Luvox in Title, Abstract or Keywords or Fevarin in Title, Abstract or Keywords or Faverin in Title, Abstract or Keywords or Favoxil in Title, Abstract or Keywords or Movox in Title, Abstract or Keywords or Paxil in Title, Abstract or Keywords or Seroxat in Title, Abstract or Keywords or Sereupin in Title, Abstract or Keywords or Aropax in Title, Abstract or Keywords or Deroxat in Title, Abstract or Keywords or Divarius in Title, Abstract or Keywords or Rexetin in Title, Abstract or Keywords or Xetanor in Title, Abstract or Keywords or Paroxat in Title, Abstract or Keywords or Loxamine in Title, Abstract or Keywords or Zoloft in Title, Abstract or Keywords or Lustral in Title, Abstract or Keywords or Serlain in Title, Abstract or Keywords or Asentra in Title, Abstract or Keywords)
-
#18. (#10 or #14 or #15 or #16 or #17)
-
#19. (#9 and #18).
MEDLINE (via Ovid) search strategy
-
1. cerebrovascular disorders/or exp basal ganglia cerebrovascular disease/or exp brain ischemia/or exp carotid artery diseases/or exp intracranial arterial diseases/or exp “intracranial embolism and thrombosis”/or exp intracranial hemorrhages/or stroke/or exp brain infarction/or vertebral artery dissection/
-
2. (stroke or poststroke or post-stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
-
3. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
-
4. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
-
5. hemiplegia/or exp paresis/
-
6. (hemipleg$ or hemipar$ or paresis or paretic).tw.
-
7. exp Gait Disorders, Neurologic/
-
8. or/1-7
-
9. exp Serotonin Uptake Inhibitors/
-
10. ((serotonin or 5-HT or 5 HT or 5-hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re-uptake) adj5 inhib$).tw.
-
11. SSRI$1.tw.
-
12. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw,nm.
-
13. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw,nm.
-
14. 9 or 10 or 11 or 12 or 13
-
15. 8 and 14
-
16. exp animals/not humans.sh.
-
17. 15 not 16
-
18. Randomized Controlled Trials as Topic/
-
19. random allocation/
-
20. Controlled Clinical Trials as Topic/
-
21. control groups/
-
22. clinical trials as topic/or clinical trials, phase i as topic/or clinical trials, phase ii as topic/or clinical trials, phase iii as topic/or clinical trials, phase iv as topic/
-
23. Clinical Trials Data Monitoring Committees/
-
24. double-blind method/
-
25. single-blind method/
-
26. Placebos/
-
27. placebo effect/
-
28. cross-over studies/
-
29. Multicenter Studies as Topic/
-
30. Therapies, Investigational/
-
31. Drug Evaluation/
-
32. Research Design/
-
33. Program Evaluation/
-
34. evaluation studies as topic/
-
35. randomized controlled trial.pt.
-
36. controlled clinical trial.pt.
-
37. (clinical trial or clinical trial phase i or clinical trial phase ii or clinical trial phase iii or clinical trial phase iv).pt.
-
38. multicenter study.pt.
-
39. (evaluation studies or comparative study).pt.
-
40. meta analysis.pt.
-
41. meta-analysis as topic/
-
42. random$.tw.
-
43. (controlled adj5 (trial$ or stud$)).tw.
-
44. (clinical$ adj5 trial$).tw.
-
45. ((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
-
46. (quasi-random$ or quasi random$ or pseudo-random$ or pseudo random$).tw.
-
47. ((multicenter or multicentre or therapeutic) adj5 (trial$ or stud$)).tw.
-
48. ((control or experiment$ or conservative) adj5 (treatment or therapy or procedure or manage$)).tw.
-
49. ((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
-
50. (coin adj5 (flip or flipped or toss$)).tw.
-
51. latin square.tw.
-
52. versus.tw.
-
53. (cross-over or cross over or crossover).tw.
-
54. placebo$.tw.
-
55. sham.tw.
-
56. (assign$ or alternate or allocat$ or counterbalance$ or multiple baseline).tw.
-
57. controls.tw.
-
58. (treatment$ adj6 order).tw.
-
59. (meta-analy$ or metaanaly$ or meta analy$ or systematic review or systematic overview).tw.
-
60. or/18-59
-
61. 17 and 60
EMBASE (via Ovid) search strategy
-
1. cerebrovascular disease/or basal ganglion hemorrhage/or exp brain hematoma/or exp brain hemorrhage/or exp brain infarction/or exp brain ischemia/or exp carotid artery disease/or cerebral artery disease/or cerebrovascular accident/or exp intracranial aneurysm/or exp occlusive cerebrovascular disease/or stroke/
-
2. stroke unit/or stroke patient/
-
3. (stroke or poststroke or post-stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
-
4. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
-
5. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
-
6. hemiparesis/or hemiplegia/or paresis/
-
7. (hemipleg$ or hemipar$ or paresis or paretic).tw.
-
8. or/1-7
-
9. exp serotonin uptake inhibitor/
-
10. ((serotonin or 5-HT or 5 HT or 5-hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re-uptake) adj5 inhib$).tw.
-
11. SSRI$1.tw.
-
12. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw.
-
13. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw,tn.
-
14. 9 or 10 or 11 or 12 or 13
-
15. 8 and 14
-
16. limit 15 to human
-
17. Randomized Controlled Trial/
-
18. Randomization/
-
19. Controlled Study/
-
20. control group/
-
21. clinical trial/or phase 1 clinical trial/or phase 2 clinical trial/or phase 3 clinical trial/or phase 4 clinical trial/or controlled clinical trial/
-
22. Double Blind Procedure/
-
23. Single Blind Procedure/or triple blind procedure/
-
24. placebo/
-
25. “types of study”/
-
26. research subject/
-
27. random$.tw.
-
28. (controlled adj5 (trial$ or stud$)).tw.
-
29. (clinical$ adj5 trial$).tw.
-
30. ((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw.
-
31. (quasi-random$ or quasi random$ or pseudo-random$ or pseudo random$).tw.
-
32. ((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw.
-
33. (coin adj5 (flip or flipped or toss$)).tw.
-
34. versus.tw.
-
35. placebo$.tw.
-
36. controls.tw.
-
37. or/17-36
-
38. 16 and 37
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (via EBSCOhost) search strategy
-
S23. S12 and S22
-
S22. S13 or S17 or S18 or S19 or S20 or S21
-
S21. AB Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra
-
S20. TI Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra
-
S19. TI (alaproclat* or cericlamin* or citalopram or dapoxetin* or escitalopram or femoxetin* or fluoxetin* or fluvoxamin* or paroxetin* or sertralin* or trazodone or vilazodone or zimelidine) OR AB (alaproclat* or cericlamin* or citalopram or dapoxetin* or escitalopram or femoxetin* or fluoxetin* or fluvoxamin* or paroxetin* or sertralin* or trazodone or vilazodone or zimelidine)
-
S18. TI SSRI* OR AB SSRI*
-
S17. S14 and S15 and S16
-
S16. TI inhib* OR AB inhib*
-
S15. TI (uptake or reuptake or re-uptake) OR AB (uptake or reuptake or re-uptake)
-
S14. TI (serotonin or 5-HT or 5 HT or 5-hydroxytryptamine or 5 hydroxytryptamine) OR AB (serotonin or 5-HT or 5 HT or 5-hydroxytryptamine or 5 hydroxytryptamine)
-
S13. (MH “Serotonin Uptake Inhibitors+”)
-
S12. S1 or S2 or S3 or S6 or S9 or S10 or S11
-
S11. TI (hemipleg* or hemipar* or paresis or paretic) or AB (hemipleg* or hemipar* or paresis or paretic)
-
S10. (MH “Hemiplegia”)
-
S9. S7 and S8
-
S8. TI (haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed*) or AB (haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed*)
-
S7. TI (brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid) or AB (brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid)
-
S6. S4 and S5
-
S5. TI (ischemi* or ischaemi* or infarct* or thrombo* or emboli* or occlus*) or AB (ischemi* or ischaemi* or infarct* or thrombo* or emboli* or occlus*)
-
S4. TI (brain* or cerebr* or cerebell* or intracran* or intracerebral) or AB (brain* or cerebr* or cerebell* or intracran* or intracerebral)
-
S3. TI (stroke or poststroke or post-stroke or cerebrovasc* or brain vasc* or cerebral vasc or cva or apoplex or SAH) or AB (stroke or poststroke or post-stroke or cerebrovasc* or brain vasc* or cerebral vasc or cva or apoplex or SAH)
-
S2. (MH “Stroke Patients”) OR (MH “Stroke Units”)
-
S1. (MH “Cerebrovascular Disorders”) OR (MH “Basal Ganglia Cerebrovascular Disease+”) OR (MH “Carotid Artery Diseases+”) OR (MH “Cerebral Ischemia+”) OR (MH “Cerebral Vasospasm”) OR (MH “Intracranial Arterial Diseases+”) OR (MH “Intracranial Embolism and Thrombosis”) OR (MH “Intracranial Hemorrhage+”) OR (MH “Stroke”) OR (MH “Vertebral Artery Dissections”)
Allied and Complementary Medicine (AMED) (via Ovid) search strategy
-
1. cerebrovascular disorders/or cerebral hemorrhage/or cerebral infarction/or cerebral ischemia/or cerebrovascular accident/or stroke/
-
2. (stroke or poststroke or post-stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
-
3. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
-
4. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
-
5. hemiplegia/
-
6. (hemipleg$ or hemipar$ or paresis or paretic).tw.
-
7. or/1-6
-
8. antidepressive agents/
-
9. ((serotonin or 5-HT or 5 HT or 5-hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re-uptake) adj5 inhib$).tw.
-
10. SSRI$1.tw.
-
11. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw.
-
12. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw.
-
13. 8 or 9 or 10 or 11 or 12
-
14. 7 and 13
PsycINFO (via Ovid) search strategy
-
1. cerebrovascular disorders/or cerebral hemorrhage/or exp cerebral ischemia/or cerebral small vessel disease/or cerebrovascular accidents/or subarachnoid hemorrhage/
-
2. (stroke or poststroke or post-stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw.
-
3. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw.
-
4. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw.
-
5. (hemipleg$ or hemipar$ or paresis or paretic).tw.
-
6. hemiparesis/or hemiplegia/
-
7. or/1-6
-
8. exp serotonin reuptake inhibitors/
-
9. ((serotonin or 5-HT or 5 HT or 5-hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re-uptake) adj5 inhib$).tw.
-
10. SSRI$1.tw.
-
11. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw.
-
12. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw.
-
13. 8 or 9 or 10 or 11 or 12
-
14. 7 and 13
Search strategy for the trial registers
-
Patient: stroke.
-
Intervention: alaproclate OR cericlamineOR citalopram OR clomipramine OR dapoxetine OR etoperidone OR femoxetine OR fenfluramine OR fluoxetine OR fluvoxamine OR norfenfluramine OR paroxetine OR sertraline OR trazodone OR vilazodone OR zimelidine.
-
Comparison: placebo.
-
Trial status: ongoing OR Recruiting OR Not yet recruiting OR Active.
-
Age: adult OR older adult.
-
Methods: Randomised Controlled Study.
List of abbreviations
- AFFINITY
- Assessment oF FluoxetINe In sTroke recoverY
- CI
- confidence interval
- COR
- common odds ratio
- DMC
- Data Monitoring Committee
- eDRIS
- eData Research and Innovation Service
- EFFECTS
- Efficacy oF Fluoxetine – a randomisEd Controlled Trial in Stroke
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- FLAME
- FLuoxetine for motor recovery After acute ischaeMic strokE
- FOCUS
- Fluoxetine Or Control Under Supervision
- GP
- general practitioner
- HPA
- hypothalamic pituitary axis
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- ICER
- incremental cost-effectiveness ratio
- ICF
- informed consent form
- IMP
- investigational medicinal product
- IQR
- interquartile range
- MD
- doctor of medicine
- MHI-5
- Mental Health Inventory – 5 questions
- mRS
- modified Rankin Scale
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NIHSS
- National Institutes of Health Stroke Scale
- o.d.
- once per day
- OR
- odds ratio
- PhD
- Doctor of Philosophy
- PHQ2
- Patient Health Questionnaire 2
- PI
- principal investigator
- PIB
- participant information booklet
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RR
- risk ratio
- SD
- standard deviation
- SF-36
- Short Form questionnaire-36 items
- SIS
- Stroke Impact Scale
- SMD
- standardised mean difference
- SmPC
- summary of product characteristics
- smRSq
- simplified modified Rankin Scale questionnaire
- SRN
- Stroke Research Network
- SSRI
- selective serotonin reuptake inhibitor
- SUSAR
- suspected unexpected serious adverse reaction
- TIA
- transient ischaemic attack
- TSC
- Trial Steering Committee
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/hta24220).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.