

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 11/136/83. The contractual start date was in January 2013. The draft report began editorial review in July 2019 and was accepted for publication in June 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2021. This work was produced by Primrose et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2021 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
International guidelines recommend that after completion of treatment for primary colorectal cancer with curative intent, some form of patient follow-up is instated with the aim of detecting treatable recurrence. 1–4 Indeed, there is abundant evidence that circumscribed recurrence is amenable to further potentially curative treatment. 5,6 However, the optimal methodology of follow-up is not known. A Cochrane review7 was performed to evaluate this but included studies in which follow-up was not linked to any treatment that could improve survival. Therefore, the results are difficult to interpret. Of the trials published in the past decade, GILDA,8 Follow-up After Colorectal Surgery (FACS) trial9 and COLOFOL10 are important to consider.
The GILDA8 trial mainly utilised ultrasound-based imaging, which is insufficiently sensitive to detect recurrence and is not applicable to the current management of colorectal cancer. Furthermore, all of the patients had tumour marker [carcinoembryonic antigen (CEA)] analysis performed and, therefore, the GILDA trial did not evaluate follow-up strategies that did not involve regular blood tests. It was a negative trial. By contrast, COLOFOL10 examined two intensive schedules of follow-up, both including computerised tomography (CT) imaging and CEA blood tests but with different frequencies. Crucially, it did not include stage I cancers. This was also a negative trial.
The FACS trial9 evaluated the use of CEA and CT imaging in a 2 × 2 trial design. This included a minimum surveillance arm in which patients had purely symptomatic follow-up with the possible addition of a single CT scan at 12–18 months. This trial had an end point of identification of recurrent disease that was treatable with curative intent. The trial was positive by this primary end point. All three intensive arms (i.e. CEA only, CT only, and CEA and CT combined) identified more treatable recurrences than the minimum follow-up arm. Importantly, it showed that there was no benefit in having both regular CEA measurements and CT imaging.
The FACS trial included patients with stage I–III colorectal cancer. Although predictably, the incidence of relapse was higher among those with higher-stage cancer, patients with all stages of cancer benefited equally from follow-up. This is because a greater proportion of the patients with recurrence from early-stage cancers were able to undergo further treatment with curative intent. 5,9 Therefore, the benefit of follow-up is not stage dependent.
In recent years, the influence of the immune system in cancer has come to the fore and immunotherapy has become mainstream in the treatment of certain cancers. In colorectal cancer, the observation that a lymphocytic infiltrate in the tumour and enlargement of draining lymph nodes correlates with a better outcome was made a long time ago. 11,12 A seminal study in 200613–18 refined this further, reporting that density of infiltration of colorectal cancer with T lymphocytes [cluster of differentiation (CD)3+] and memory T cells (CD45RO+) correlates better with outcome than conventional tumour node metastasis (TNM) staging. Although subsequent studies have not shown the same strength of correlation with outcome as the initial paper,13 there is a consistent relationship between immune cell infiltration and cancer recurrence.
It is important to consider why the initial reports demonstrated the density T-cell infiltrate to be such a strikingly strong prognostic marker. Careful evaluation of the survival curves reveals frequency of recurrence to be far higher than one would expect by stage. By contrast, the recurrence rate in the FACS trial was lower than many anticipated for a cohort of stage I–III colorectal cancer, but it is in fact in keeping with other modern studies. This is most likely explained by the robust staging that patients underwent prior to being eligible for recruitment to FACS (CT imaging of the chest/abdomen and a CEA level of ≤ 10 µg/l on completion of treatment) meaning that a large number with residual disease were excluded from the study. It is therefore surmised that the cohorts of patients utilised for some studies of immune cell infiltrates may have used understaged patients.
It is therefore crucial that the prognostic relevance of T-cell infiltrates are assessed in a well-staged cohort of patients managed in the modern era of imaging, surgery and adjuvant treatments. The FACS cohort provides such a population and allows us to determine whether or not there is a group of patients in whom the risk of relapse is so low that follow-up is not required.
The work by Galon et al. 13 generally uses full-face tissue block sections. This is not suitable for routine use because of the level of difficulty and the cost involved. If widespread application is to be introduced then any process has to be suitable for routine use in histopathology laboratories. Therefore, it was decided to use tissue microarrays (TMAs). This is a system whereby multiple cores of tissue can be examined in a single section, greatly decreasing the time and cost of processing because material from multiple patients can be managed together. The disadvantage is that only six cores, in general, will be sampled from each tumour; therefore, less of the tumour is assessed when using TMAs than when using full-face tissue block sections.
Aim and objectives
The aim is to determine if immunophenotypic analysis of T lymphocytes within the primary tumour can identify:
-
the risk of relapse
-
a subset of patients in whom follow-up is not necessary.
Chapter 2 Methods
Design
This is a translational observational study utilising tissue and clinicopathological data from the completed FACS trial. 9 The intention is to correlate the incidence of recurrence and the recurrence that is treatable with curative intent with the immune cell milieu in the primary tumour.
Participants
Participants were recruited from the centre at which they received their primary treatment for colorectal cancer, spanning 39 NHS hospitals across all regions of England. To be eligible for the study, participants had to have undergone curative surgery for primary colorectal cancer and, after extensive testing (histology and imaging, and a CEA level of ≤ 10 µg/l), were confirmed to have no residual disease. Further detail regarding the study setting, participant inclusion and exclusion criteria, and randomisation procedures can be found in the original publication. 9
Patient and public involvement
The original FACS study had significant patient and public involvement. This substudy did not.
Tissue collection
The original FACS proposal did not include funding to obtain tissue from the primary tumours of the patients in the study but the ethics permissions and patient consent allowed for this. The present grant funding enabled the collection of tissue blocks from the centres. This work was undertaken by the Southampton Clinical Trials Unit and tissue stored in the Human Tissue Authority-registered Southampton Tissue Bank until used.
Tissue processing
The tissue blocks were sectioned and stained with routine haematoxylin and eosin (H&E) on an automated Dako CoverStainer (Agilents Diagnostics, Santa Clara, CA, USA) (see Appendices 2 and 3). The sections were examined by a pathologist and the sites for the six cores to be taken were marked. Three cores were taken from the centre of the tumour and three were taken from the invasive margin. TMAs were produced on a MiniCore® machine (ALPHELYS, Plaisir, France) (see Appendix 4) and recorded on the map shown (Figure 1). Sections from the TMA blocks were stained with H&E (see Appendix 2) and then for the T-cell markers CD3+ (all T cells) and CD45RO+ (memory T cells). CD3+ and CD45RO+ immunohistochemistry staining was performed on an automated Dako Autostainer 48 Link (Agilents Diagnostics) (see Appendix 3). The CD3+ antibody (Dako, Agilents Diagnostics) is ready to use and the CD45RO+ antibody (Dako, Agilents Diagnostics) is concentrated and was used at a dilution of 1 : 2500. The methodology is detailed in Appendices 5 and 6.
FIGURE 1.
Tissue acquisition and processing.
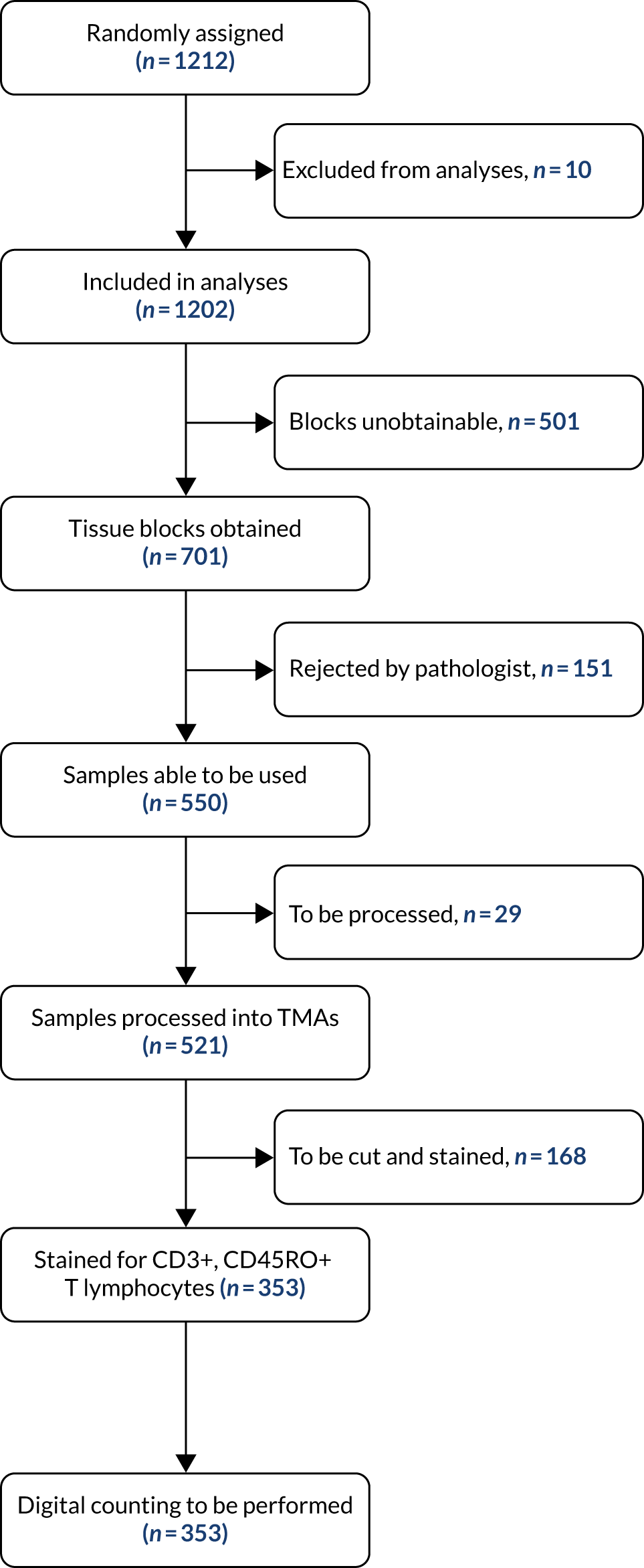
Chapter 3 Results
Figure 1 illustrates the tumour tissue available for analysis of patients in the FACS trial. Of the 1202 randomised patients, tissue blocks were obtained for 701 patients. In cases where the initial block sent contained no cancer, another block was requested. Samples from a total of 151 patients were judged to be inadequate by the study pathologists (i.e. they contained no cancer). As a result, tumour tissue blocks were processed for 550 of the 1202 randomised patients. This is an acceptable proportion for a retrospective tissue collection when the collection was not built into the original protocol.
Cell counting
The initial intention was to manually count cells using a standard microscope. Although this proved possible, rapidly became clear that the time required for the project was not feasible. Our calculation was that counting 17 TMAs for two antibodies only would take around 480 hours or 80 6-hour days. Consideration was given to using a ‘citizen science’-type programme,19 but there were no resources to organise such a complex system. In 2015, it became clear that automated analysis was coming to the fore, and in 2016 a Digital Pathology Accelerator award to Belfast and Southampton enabled appropriate hardware and software to be installed. Attempts were made to scan the TMAs on a Zeiss Axio Scan.Z120 (ZEISS, Oberkochen, Germany) whole-slide scanner at resolution equivalent to ×20 magnification. Initial image analysis was carried out using QuPath (developed at the University of Edinburgh; originally created at the Centre for Cancer Research & Cell Biology at Queen’s University Belfast as part of research projects funded by Invest Northern Ireland and Cancer Research UK). 21 Unfortunately, neither the hardware nor the software functioned correctly.
Numerous problems occurred, including the misregistration of the cores (the number assigned by the machine being at variance with the actual core number) in the software (Figure 2) requiring laborious manual reassignment. These problems were compounded by the loss of key science staff following the 2016 referendum, a situation only remedied in 2019. Some preliminary data were obtained (for the abstract, see Appendix 1). For each tumour region, high (Hi) and low (Lo) CD3+ and CD45RO+ densities were determined according to the median of the cohort. To investigate the combined effect of CD3+ and CD45RO+ densities in both tumour regions, patients were divided into three groups: Lo (low density of both markers in both tumour regions), Hi (high density of both markers in both tumour regions) and Het (heterogeneous, i.e. high density in some regions and low in others). Examples of the stained TMAs are shown in Figures 2–5. The data indicated a potential survival benefit associated with tumours exhibiting a high infiltrate of CD3- and CD45RO-positive immune cells, but these results are preliminary and the reproducibility has not yet been established.
FIGURE 2.
Example of a TMA map showing misregistration by the analysis software.

FIGURE 3.
Example of a TMA map stained with H&E.

FIGURE 4.
A TMA map showing staining for CD3+ lymphocytes (staining brown).

FIGURE 5.
A TMA map showing staining for CD45RO+ lymphocytes (staining brown).

This benefit appeared to be limited to those with left-sided tumours. At present, an attempt is being made to analyse the entire cohort with different software (Definiens Developer XD 2.7; Definiens AG, Munich, Germany).
Chapter 4 Discussion
This study aimed to build on the clinicopathological data acquired during the FACS trial of colorectal cancer follow-up, which is a unique resource. It provides a population of accurately staged patients and, unlike trials of adjuvant therapies, includes patients with early-stage cancers. Although the original grant application was written many years ago, it, fortunately, had the foresight to include consent to collect tumour tissue from these patients. Utilising the resource obtained in a subsequent grant has meant that is has been possible to assess the tumour tissue for approximately half of the patients in the trial.
Traditional prognostic tumour features, such as stage at presentation and differentiation, are predictive of relapse. However, the overall relapse rate of completely resected cancers is relatively low. These features are insufficient to identify a cohort that would not require follow-up. 9
It is clear that the immune system is critical to the outcome in cancer cases and it is clear that the measurement of immune cells in the primary tumour has prognostic significance. In the original paper by Galon et al.,13 the predictive value was dramatic. However, these patients were almost certainly not staged with the rigour that would be normal now, as carried out in the FACS trial, and it is likely that many of the supposedly early-stage patients would actually have had occult metastatic disease. More recent studies give a more reliable assessment in all probability, but the predictive value remains. 14–18
The tissue blocks obtained from the patients in the FACS trial provide a resource for future research. We were able to use TMA technology to examine immune cells in multiple patients in one block, hence producing a method that is economical and could be adapted for routine clinical use. Unfortunately, we were not able to perfect machine counting of immune cells within the time frame of this study. We were able to make the system work on a small subset of the specimens, but these results are unvalidated and not suitable for publication at the present time.
The failure to achieve manual counting was not predictable as pathology experts advised that it was reasonable. We had anticipated using undergraduates during their special study module to perform this counting with the expectation of a publication. However, this proved unfeasible.
We have demonstrated that machine counting is possible. This possibility first became available in 2016 when the technology first became widespread. The Zeiss Axio Scan.Z120 was introduced to Belfast and Southampton as part of an Accelerator Award in Digital Pathology. There were significant problems with integration of both the hardware and the software (QuPath was used initially). 21 The scanner in Belfast was replaced as it was considered unfit for purpose, but the same scanner remains in Southampton. Scans were originally obtained in Belfast, but problems with the software made the results unable to be used. In 2017, images were acquired in Southampton from a subset of the cohort. As detailed, the misregistration of the cores by the software made any analysis very challenging. Enough data were acquired for an abstract but there has not been the opportunity to confirm the reproducibility of these results or complete the analysis on the whole cohort.
Scientists with the capability to use the hardware and the software are rare. After the 2016 referendum, the scientist involved in this work relocated to outside the UK. It proved impossible to replace this skill set until 2019. There is the intention to continue and develop this work further. The initial approach was to use a limited but validated range of markers (i.e. CD3+, CD45RO+) as proof of principle. However, any publishable work would at least include CD8+ (cytotoxic T cells) and FOXP3+ as a minimum. 14–18 It is also clear that the lymphocytic infiltrate is correlated with the more immunogenic microsatellite unstable [microsatellite instable (MSI) high] tumours, and this can also be assessed on the TMAs. 18
In summary, despite the technical challenges, machine counting of tumour-infiltrating lymphocytes in TMAs that are formed from the tumours of patients with colorectal cancer is potentially feasible and this might be undertaken as a routine service in a regional pathology laboratory with an appropriate digital scanner and easily available software. However, high-level operator expertise is needed at present. It remains to be seen whether or not these biomarkers are sufficiently discriminating to have an impact on patient management.
Acknowledgements
The authors would like to acknowledge the key role of the other FACS trial investigators in providing the data analysed in this substudy.
University of Southampton, UK
Louisa Little, Andrea Corkhill (Clinical Trial Manager), Scott Regan and Jane Mellor (Clinical Trial Co-ordinators).
University of Oxford, UK
Alice Fuller Helen Campbell (Research Fellow in Health Economics).
Oxford University hospitals Trust, UK
Helen Bungay (Clinical Radiologist).
Participating NHS hospitals
Birmingham Heartlands Hospital (Mr Gamal Barsoum); Castle Hill Hospital, Hull (Mr John Hartley); Charing Cross Hospital (Mr Peter Dawson); Cumberland Infirmary (Dr Jonathan Nicoll); Darent Valley Hospital (Mr Mike Parker); Derriford Hospital, Plymouth (Mr Mark Coleman); Grantham and District Hospital (Mr Dilip Mathur); Harrogate District Hospital (Mr Jon Harrison); Hillingdon Hospital (Mr Yasser Mohsen); Hinchingbrooke Hospital (Dr Litee Tan); King’s Mill Hospital (Mr Mukul Dube); Leeds St James (Mr Simon Ambrose); Leeds General Infirmary (Mr Paul Finan); Leighton General Hospital (Mr Arif Khan); Maidstone Hospital (Dr Mark Hill); Mayday Hospital (Croydon University Hospital) (Mr Muti Abulafi); Newham University Hospital (Mr Roger Le Fur); Oxford Radcliffe Hospitals (Professor Neil Mortensen); Queen Alexandra/Portsmouth (Mr Daniel O’Leary); Queen Elizabeth Hospital, Birmingham (Dr Neil Steven); Queen’s Hospital Burton-on-Trent (Mr Stelios Vakis); Queen’s Medical Centre, Nottingham (Professor John Scholefield); Royal Cornwall Hospital (Mr Ponnandai Arumugam); Royal Derby Hospital (Mr Jonathan Lund); Royal Shrewsbury (Mr Trevor Hunt); Russels Hall Hospital (Professor David Ferry); Scarborough Hospital (Dr Ian Renwick); Southampton General Hospital (Professor John Primrose); St Mark’s Hospital, Harrow (Professor John Northover and Dr Arun Gupta); St Peter’s Hospital, Chertsey (Mr Philip Bearn); St Richard’s Hospital, Chichester (Mr Neil Cripps); Taunton and Somerset (Dr Mary Tighe); Torbay Hospital (Mr Rupert Pullan); Manor Hospital, Walsall (Mr Jonathan Stewart); Warrington Hospital (Mr Barry Taylor); West Middlesex Hospital (Mr Subramanian Ramesh); Wexham Park Hospital (Dr H Wasan); Worcester Royal Hospital (Mr Stephen Lake); and Wycombe General Hospital (Dr Andrew Weaver).
Data Monitoring and Ethics Committee
The Data Monitoring and Ethics Committee was Jack Hardcastle, Emeritus Professor of Surgery, University of Nottingham; Michael Campbell, Professor of Statistics, University of Sheffield; and David Whynes, Professor of Health Economics, University of Nottingham.
We also acknowledge the invaluable contribution of the local National Institute for Health Research (NIHR) cancer research networks, of the NHS trusts and of the patients who agreed to participate in this trial.
Funding and conduct of main trial
The main FACS project was funded by the UK NIHR Health Technology Assessment (HTA) programme (project number 99/10/99). 22 The trial was conducted in accordance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use – Good Clinical Practice in Clinical Trials (2001/20/EC) and the NHS NIHR Governance Framework.
Role of funding agency
The funding agency (NIHR HTA) had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; nor any decision to submit the manuscript for publication.
Contributions of authors
John N Primrose (https://orcid.org/0000-0002-2069-7605) (Professor of Surgery) was co-principal investigator on the main FACS trial, obtained the funding for the substudy, advised on the clinical interpretation of the findings, and was principal author of the report.
Siân A Pugh (https://orcid.org/0000-0002-1866-8338) (Specialist Registrar in Oncology) provided clinical support for the data analysis and helped draft the manuscript at each stage.
Gareth Thomas (https://orcid.org/0000-0003-3832-7335) (Professor of Cancer Pathology) provided the pathological expertise required for the study.
Matthew Ellis (https://orcid.org/0000-0002-5264-0531) (Senior Fellow, Cancer Sciences Division) undertook the image acquisition from the TMA and undertook the analysis of the later samples.
Karwan Moutasim (https://orcid.org/0000-0002-3460-596X) (Consultant Pathologist) provided pathological expertise and marked the full-face slides.
David Mant (Emeritus Professor of General Practice) was co-principal investigator on the main FACS trial, obtained the funding for the substudy, helped Bethany Shinkins design and conduct the main analysis, and drafted the manuscript.
All authors commented on more than one draft of the manuscript and approved the final draft.
Data-sharing statement
The data reported here are a subset extracted from the main FACS trial data set. Requests to access anonymised data from the FACS trial for the purposes of non-commercial research for patient benefit should be addressed to Professor Primrose (j.n.primrose@soton.ac.uk). At present, there are no data from this substudy that can be shared. It is likely that data will become available in due course and after a full publication will be made available as above.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Bastiaenen VP, Hovdenak Jakobsen I, Labianca R, Martling A, Morton DG, Primrose JN, et al. Consensus and controversies regarding follow-up after treatment with curative intent of nonmetastatic colorectal cancer: a synopsis of guidelines used in countries represented in the European Society of Coloproctology. Colorectal Dis 2019;21:392-416. https://doi.org/10.1111/codi.14503.
- National Institute for Health and Care Excellence (NICE) . Colorectal Cancer: Diagnosis and Management. Clinical Guideline 131 2011. nice.org.uk/guidance/cg131 (accessed July 2020).
- Glimelius B, Tiret E, Cervantes A, Arnold D. ESMO Guidelines Working Group . Rectal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013;24:vi81-8. https://doi.org/10.1093/annonc/mdt240.
- Labianca R, Nordlinger B, Beretta GD, Mosconi S, Mandalà M, Cervantes A, et al. ESMO Guidelines Working Group . Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2013;24:vi64-72. https://doi.org/10.1093/annonc/mdt354.
- Pugh SA, Shinkins B, Fuller A, Mellor J, Mant D, Primrose JN. Site and stage of colorectal cancer influence the likelihood and distribution of disease recurrence and postrecurrence survival: data from the FACS randomized controlled trial. Ann Surg 2016;263:1143-7. https://doi.org/10.1097/SLA.0000000000001351.
- Kanas GP, Taylor A, Primrose JN, Langeberg WJ, Kelsh MA, Mowat FS, et al. Survival after liver resection in metastatic colorectal cancer: review and meta-analysis of prognostic factors. Clin Epidemiol 2012;4:283-301. https://doi.org/10.2147/CLEP.S34285.
- Jeffery M, Hickey BE, Hider PN. Follow-up strategies for patients treated for non-metastatic colorectal cancer. Cochrane Database Syst Rev 2007;1. https://doi.org/10.1002/14651858.CD002200.pub2.
- Rosati G, Ambrosini G, Barni S, Andreoni B, Corradini G, Luchena G, et al. A randomized trial of intensive versus minimal surveillance of patients with resected Dukes B2-C colorectal carcinoma. Ann Oncol 2016;27:274-80. https://doi.org/10.1093/annonc/mdv541.
- Primrose JN, Perera R, Gray A, Rose P, Fuller A, Corkhill A, et al. Effect of 3 to 5 years of scheduled CEA and CT follow-up to detect recurrence of colorectal cancer: the FACS randomized clinical trial. JAMA 2014;311:263-70. https://doi.org/10.1001/jama.2013.285718.
- Wille-Jørgensen P, Syk I, Smedh K, Laurberg S, Nielsen DT, Petersen SH, et al. Effect of more vs less frequent follow-up testing on overall and colorectal cancer-specific mortality in patients with stage II or III colorectal cancer: the COLOFOL randomized clinical trial. JAMA 2018;319:2095-103. https://doi.org/10.1001/jama.2018.5623.
- Jass JR, Ajioka Y, Allen JP, Chan YF, Cohen RJ, Nixon JM, et al. Assessment of invasive growth pattern and lymphocytic infiltration in colorectal cancer. Histopathology 1996;28:543-8. https://doi.org/10.1046/j.1365-2559.1996.d01-467.x.
- George S, Primrose J, Talbot R, Smith J, Mullee M, Bailey D, et al. Will Rogers revisited: prospective observational study of survival of 3592 patients with colorectal cancer according to number of nodes examined by pathologists. Br J Cancer 2006;95:841-7. https://doi.org/10.1038/sj.bjc.6603352.
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006;313:1960-4. https://doi.org/10.1126/science.1129139.
- Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J Clin Oncol 2009;27:5944-51. https://doi.org/10.1200/JCO.2008.19.6147.
- Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol 2014;232:199-20. https://doi.org/10.1002/path.4287.
- Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The link between the multiverse of immune microenvironments in metastases and the survival of colorectal cancer patients. Cancer Cell 2018;34:1012-26. https://doi.org/10.1016/j.ccell.2018.11.003.
- Pagès F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018;391:2128-39. https://doi.org/10.1016/S0140-6736(18)30789-X.
- Maby P, Tougeron D, Hamieh M, Mlecnik B, Kora H, Bindea G, et al. Correlation between density of CD8+ T-cell infiltrate in microsatellite unstable colorectal cancers and frameshift mutations: a rationale for personalized immunotherapy. Cancer Res 2015;75:3446-55. https://doi.org/10.1158/0008-5472.CAN-14-3051.
- BBC . Citizen n.d. www.bbc.co.uk/programmes/articles/4BZZdHm64S051q2lnZ1Nr7p/citizen-science (accessed June 2019).
- Carl Zeiss n.d. www.zeiss.com/microscopy/int/products/imaging-systems/axio-scan-z1.html (accessed June 2019).
- Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7. https://doi.org/10.1038/s41598-017-17204-5.
- Mant D, Gray A, Pugh S, Campbell H, George S, Fuller A, et al. A randomised controlled trial to assess the cost-effectiveness of intensive versus no scheduled follow-up in patients who have undergone resection for colorectal cancer with curative intent. Health Technol Assess 2017;21. https://doi.org/10.3310/hta21320.
- Pugh SA, Moutasim K, Jenkins NM, Shinkins B, Thomas G, Mant D, et al. Association between density of tumor infiltrating lymphocytes and disease-free survival (DFS) in patients with resected stage I-III colorectal cancer in the FACS randomized trial. J Clin Oncol 2018;36.
Appendix 1 Abstract from American Society of Clinical Oncology meeting 2018
This abstract has been reprinted with permission. © 2018 American Society of Clinical Oncology. All rights reserved. Pugh SA, Moutasim K, Jenkins NM, Shinkins B, Thomas G, Mant D, et al. Association between density of tumor infiltrating lymphocytes and disease-free survival (DFS) in patients with resected stage I-III colorectal cancer in the FACS randomized trial. J Clin Oncol vol. 36, iss. 15, p. 3573. 23
Background
Accumulating evidence demonstrates an association between density of tumor infiltrating lymphocytes and outcome in colorectal cancer (CRC). This study sought to assess the prognostic utility using an accurately staged cohort of patients followed up in a clinical trial.
Methods
Observational analysis of data from the FACS (follow-up after CRC surgery) trial after 5 years of follow-up. All patients had undergone treatment with curative intent for stage I–III primary CRC, with microscopically clear margins, no evidence of metastases on axial imaging and CEA < 10 µg/l following completion of treatment. Immune cell densities were quantified in the centre (CT) and invasive margin (IM) of all tumors for both CD3 and CD45RO. For each tumor region high (Hi) and low (Lo) CD3 and CD45RO densities were determined according to the median of the cohort to investigate association with disease-free survival (DFS).
Results
Tumor samples have been analysed from 297 patients to date for which the combined 5-year DFS is 83% (left sided CRC 81%, right sided CRC 85%). High densities of CD3 and CD45RO positive cells in both tumor regions were associated with a superior outcome: CD3CTIMHiCD45ROCTIMHi 94% 5-year DFS vs CD3CTIMLoCD45ROCTIMLo 81%, HR 0.36 95% CI 0.15–0.89 p = 0.04. This difference was most notable in left sided CRC: CD3CTIMHiCD45ROCTIMHi 96% 5yr DFS vs CD3CTIMLoCD45ROCTIMLo 78%, HR 0.13 95% CI 0.04–0.41 p = 0.02. In right sided CRC the difference was not significant: CD3CTIMHiCD45ROCTIMHi 91% 5-year DFS vs CD3CTIMLoCD45ROCTIMLo 83%, HR 0.70 95% CI 0.17–2.89 p = 0.62.
Conclusions
In a well characterised and followed up cohort of CRC patients within a clinical trial we have demonstrated that CD3CTIMHiCD45ROCTIMHi left sided tumors have a significantly better outlook. The potential for these data to impact on the need for clinical follow up in patients with left sided CRC should be examined in a prospective study. Clinical trial information: 61091474.
Appendix 2 The H&E protocol on Dako CoverStainer
| Station | Reagent | Time (minutes) |
|---|---|---|
| 1 | Xylene | 3.00 |
| 2 | Xylene | 3.00 |
| 3 | 95% IDA | 0.10 |
| 4 | 95% IDA | 2.00 |
| 5 | 70% IDA | 2.00 |
| 6 | Tap water | 1.00 |
| 7 | Haematoxylin | 0.45 |
| 8 | Distilled water | 1.00 |
| 9 | Blueing buffer | 1.00 |
| 10 | Tap water | 1.00 |
| 11 | 70% IDA | 1.00 |
| 12 | Eosin Phlox B | 1.30 |
| 13 | 95% IPA | 1.00 |
| 14 | 100% IPA | 1.00 |
| 15 | 100% IPA | 1.00 |
| 16 | 100% IPA | 1.00 |
| 17 | Xylene | 1.00 |
| 18 | Xylene | Exit |
Appendix 3 Immunohistochemistry procedures using the Dako PT Link and Dako Autostainer 48S Link platform
Appendix 4 Tissue microarray procedure
-
1. Gather all of the blocks based on the marked H&E-stained slides for TMA.
-
2. Go to TMA designer options to design your TMA.
-
2a. Choose your recipient block (the size of the block you are using).
-
2b. Make a new tissue array template design (this will include the punch size that you are going to use for making your TMA and the spacing of the spots; it will give you exact numbers of the spots you are going to create). The punch size we usually use is 1000 μm and a spacing between 800 µm and 1000 µm. This will also give you the total number of spots by row and by column, and you can also mark your orientation or even edit the spots you want by deleting the spots you will not be going to use. In addition, you can save this for your TMA project.
-
2c. List of tissue types options (you can save here the tissue types that you are going to use, i.e. orientation, colorectal central and colorectal invasive, and assign a colour code for this) and initial of the one making the TMA project.
-
2d. Making of Tissue Array Project design. Name of the project is the first step followed by downloading the list of your donor blocks that you created in an Excel file (Microsoft Excel®, Microsoft Corporation, Redmond, WA, USA) (this includes the histology number of the block and name of the patient; you may include the sex, hospital number, date of birth, date of diagnosis and site). It will also ask you to download the tissue types that you wanted and apply how many replicate numbers of cores you wanted for each block. Afterwards, the computer will fill the tissue array and will ask you to save the TMA project you created.
-
3. Click on the MiniCore icon and select the name of the project you saved, and the machine will tell you to load the blocks that you require and unload the blocks afterwards. The TMA removes a wax core from the recipient TMA block to allow space to insert a tissue core. The marked slide will be put on top of the donor block. The inbuilt camera can take a picture of this and the marked points on the slide are selected by clicking on the appropriate areas. This puts a marker where the donor block is going to get the specimen that it will transfer into a recipient block. It will do this repeatedly on each of the blocks depending on how many replicates (usually three per block) you created until it finishes the whole project.
-
4. Put the TMA block created in a 40-degree oven overnight to assist in the wax adhering to the tissue cores. This helps to reduce the number of cores that you lose when cutting sections. Sections are cut and stained with H&E and/or any other immunohistochemical stain that you want for the project.
Appendix 5 The CD3+ protocol on the Dako Autostainer 48 Link
| Station | Reagent | Time (minutes) |
|---|---|---|
| 1 | FLEX buffer | 0.00 |
| 2 | Endogenous peroxidase block | 5.00 |
| 3 | FLEX buffer | 0.00 |
| 4 | CD3+ RTU | 15.00 |
| 5 | FLEX buffer | 0.00 |
| 6 | FLEX-labelled polymer/HRP | 20.00 |
| 7 | FLEX buffer | 0.00 |
| 8 | FLEX buffer | 5.00 |
| 9 | FLEX DAB + chromogen | 5.00 |
| 10 | FLEX DAB + chromogen | 5.00 |
| 11 | FLEX buffer | 0.00 |
| 12 | Haematoxylin | 5.00 |
| 13 | Distilled water | 0.00 |
| 14 | FLEX buffer | 5.00 |
| 15 | Distilled water | 0.00 |
Appendix 6 The CD45RO+ protocol on the Dako Autostainer 48 Link
| Station | Reagent | Time (minutes) |
|---|---|---|
| 1 | FLEX buffer | 0.00 |
| 2 | Endogenous peroxidase block | 5.00 |
| 3 | FLEX buffer | 0.00 |
| 4 | CD45RO+ 1 : 2500 | 20.00 |
| 5 | FLEX buffer | 0.00 |
| 6 | FLEX mouse linker | 15.00 |
| 7 | FLEX buffer | 0.00 |
| 8 | FLEX-labelled polymer/HRP | 20.00 |
| 9 | FLEX buffer | 0.00 |
| 10 | FLEX buffer | 5.00 |
| 11 | FLEX DAB + chromogen | 5.00 |
| 12 | FLEX DAB + chromogen | 5.00 |
| 13 | FLEX buffer | 0.00 |
| 14 | Haematoxylin | 5.00 |
| 15 | Distilled water | 0.00 |
| 16 | FLEX buffer | 5.00 |
| 17 | Distilled water | 0.00 |
List of abbreviations
- CD
- cluster of differentiation
- CEA
- carcinoembryonic antigen
- CRC
- colorectal cancer
- CT
- computerised tomography
- DFS
- disease-free survival
- FACS
- Follow-up After Colorectal Surgery
- H&E
- haematoxylin and eosin
- HTA
- Health Technology Assessment
- IDA
- industrial denatured alcohol
- NIHR
- National Institute for Health Research
- TMA
- tissue microarray