

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 11/92/03. The contractual start date was in June 2014. The draft report began editorial review in August 2019 and was accepted for publication in June 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2021. This work was produced by Hykin et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2021 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Retinal vein occlusion (RVO) is the second most common retinal vascular disorder,1,2 after diabetic retinopathy, and comprises branch RVO, hemiretinal vein occlusion and central retinal vein occlusion (CRVO). CRVO has a prevalence of 0.08–0.41%3–5 and a 15-year cumulative incidence rate of 0.5%. 6,7 Approximately 6860 people develop CRVO every year in England and Wales, of these, 5150 develop visual impairment due to macula oedema (MO), which is unlikely to improve spontaneously8–11 and is therefore potentially eligible for treatment, according to the National Institute for Health and Care Excellence (NICE). 12,13
Central retinal vein occlusion is characterised by retinal haemorrhages, venous dilatation and tortuosity in all four quadrants of the retina. 1,7 An increase in hydrostatic pressure at the venous end of the retinal capillary network reduces retinal perfusion, upregulating the production of vascular endothelial growth factor (VEGF), which, in turn, increases retinal capillary permeability and is probably the major cause of MO,14 although the raised hydrostatic pressure per se probably plays a part. 7 VEGF promotes iris and retinal neovascularisation in severe cases. The characteristic presentation of CRVO is sudden painless unilateral decrease in vision due to MO. 8 In severe cases, vision is affected by macular ischaemia or the development of iris neovascularisation and, subsequently, neovascular glaucoma with elevated intraocular pressure, pain, redness and visual loss if the condition is left untreated. CRVO may be bilateral in 5% of cases, and the risk of developing RVO in the contralateral eye within 12 months is approximately 5%. 7,8
Central retinal vein occlusion has two distinct clinical subtypes. 7,8 Non-ischaemic CRVO is characterised by a visual acuity of ≥ 6/30, no relative afferent pupillary defect (RAPD), mild to moderate retinal venous dilatation and tortuosity, and intraretinal haemorrhage and MO. Ischaemic CRVO is characterised by a visual acuity of ≤ 6/36, the presence of a RAPD, and intraretinal haemorrhage with venous dilatation and tortuosity greater than the Central Vein Occlusion Study (CVOS) standard photograph,15 with complications that include MO, macular ischaemia, retinal ischaemia, iris and retinal neovascularisation and neovascular glaucoma. 16 Optical coherence tomography (OCT) confirms and characterises the MO, and fundus fluorescein angiography (FFA) confirms and characterises the extent of macular and retinal ischaemia and the presence of retinal neovascularisation; both investigations guide management. 7,8 Novel morphological OCT biomarkers for CRVO have been identified that may provide important diagnostic and prognostic information, although, to our knowledge, none has been utilised in a large prospective clinical trial to date. 17–19 Conventional seven-field FFA is semiquantitative and, if the total area of angiographic non-perfusion is at least 10 disc areas in size, the prognosis is less good than for the non-ischaemic subtype. 20,21 More recently, wide-angled FFA has allowed a greater proportion of the peripheral retina to be imaged, although the exact amount and distribution of non-perfusion that characterises the subtypes of CRVO have not been well defined. 22,23 Eyes with larger areas of retinal ischaemia on conventional FFA are more prone to neovascular complications. 20 Approximately 15–20% of cases present with ischaemic CRVO, and in 25–34% of cases non-ischaemic CRVO converts to the ischaemic subtype within 3 years. 20,24 Neovascular complications such as iris neovascularisation are typically managed using a combination of retinal laser therapy and anti-VEGF therapy. 7,8
In non-inferiority ophthalmology clinical trials, the primary outcome has typically been a visual acuity difference of –5 Early Treatment Diabetic Retinopathy Study (ETDRS) letters. This is thought to represent a meaningful difference between two treatments, based on the following:
-
Most patients in a busy clinic setting can reliably distinguish an 8-letter (1.5-line) difference on an ETDRS visual acuity chart, but they may perform better than this in a clinical trial setting. 25
-
A 5-letter (1-line) improvement in mean visual acuity in retinal studies typically results in a 50% increase in the number of patients gaining 15-letter (3-line) improvement in visual acuity, suggesting that this is a meaningful difference. 26
-
The choice of a 5-letter margin was 32% higher than the available estimated 12-month placebo-controlled effect of 6.6 letters for ranibizumab (0.5 mg/0.05 ml Lucentis®; Novartis International AG, Basel, Switzerland), the standard (comparator) treatment in this multicentre, double-masked, randomised controlled non-inferiority trial comparing the clinical effectiveness and cost-effectiveness of intravitreal therapy with ranibizumab (Lucentis) versus aflibercept (Eylea) versus bevacizumab (Avastin) for macular oedema due to central retinal Vein Occlusion (LEAVO). This margin choice was, therefore, consistent with maintaining assay sensitivity sufficiently to be able to declare non-inferiority [see www.journalslibrary.nihr.ac.uk/programmes/hta/119203/#/documentation (accessed 14 July 2020)].
-
This margin was accepted by the funder.
Although a 4-letter change has been used as a non-inferiority margin, this was not common practice at the time LEAVO was designed, and we wanted to ensure that LEAVO would be as similar as possible to alternative comparable studies of anti-VEGF therapy in CRVO [e.g. Study of Comparative Treatments for Retinal Vein Occlusion 2 (SCORE2)27].
Central retinal vein occlusion-related macular oedema and antivascular endothelial growth factor therapy
Visual impairment in CRVO is primarily due to MO; it is typically significant, resolution is likely to occur in only the mildest non-ischaemic cases24 and the anatomical improvement of MO may not result in a corresponding improvement in visual acuity. 8 Presenting visual acuity is typically a good predictor of final visual outcome: patients who present with an initial visual acuity of ≥ 6/12 will probably retain good vision, whereas 80% of those who present with a visual acuity of ≤ 6/60 do not improve to > 6/60. 20 The natural history arm of the CVOS showed no change in mean baseline visual acuity over 3 years;20 this finding is supported by the sham arms in the Ranibizumab for the Treatment of Macular Edema after Central Retinal Vein OcclUsIon Study: Evaluation of Efficacy and Safety (CRUISE),9 GALILEO28–30 and COPERNICUS10,28,30,31 licensing trials for ranibizumab and aflibercept (2 mg/0.05 ml Eylea®; Bayer AG, Leverkusen, Germany), in which patients who were initiated on treatment 6 months after randomisation to sham did not achieve as large visual gains as participants randomised to prompt therapy. Therefore, prompt treatment is typically recommended to maximise visual outcomes.
First-line therapy for MO is repeated intravitreal injections of anti-VEGF agents to block the action of VEGF, thereby reducing capillary permeability. 9,32–38 Early studies excluded patients with ischaemic CRVO33,39 as it was questionable whether or not a significant improvement in vision would result from anti-VEGF therapy. More recent (2017) studies40 did not exclude such patients, and this is the approach we adopted in LEAVO to ensure that our study population fully reflected a general UK population likely to present for treatment.
To date, three anti-VEGF agents have been used in the treatment of MO due to CRVO:
-
Ranibizumab is a humanised, affinity-matured VEGF antibody fragment that binds to and neutralises all isoforms of VEGF-A. Ranibizumab was the first anti-VEGF therapy to demonstrate improved visual outcomes in patients with neovascular age-related macular degeneration (nvAMD),41,42 and in 2012 it was licensed by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for MO due to CRVO. This was based on the CRUISE data9 that showed that monthly intraocular ranibizumab therapy improved the mean best corrected visual acuity (BCVA) by +15 ETDRS letters at 6 months and a pro re nata regimen with monthly monitoring improved the mean BCVA by +14 ETDRS letters by 12 months. 9 In an open-label extension [An Open-Label, Multicentre Extension Study to Evaluate the Safety and Tolerability of Ranibizumab in Subjects with Choroidal Neovascularization Secondary to Age-Related Macular Degeneration or Macular Oedema Secondary to Retinal Vein Occlusion Who Have Completed a Genentech-Sponsored Ranibizumab study (HORIZON)] from months 12 to 24, the mean visual acuity in CRVO only patients reduced by 4.1 letters with an average of 3.5 injections in 12 months. Ranibizumab was well tolerated: 6.5% of patients had some degree of cataract after 2 years and < 1% had a rise in intraocular pressure. 38
-
Aflibercept is a fusion protein of the key domains of VEGF receptors 1 and 2 and human IgG Fc that blocks all VEGF-A isoforms and placental growth factor. In 2014, it was licensed by the FDA and the EMA for CRVO based on the GALILEO and COPERNICUS studies, which showed a mean gain of +16.2 letters in BVCA at 12 months and a mean gain of +13.0 letters in BCVA at 24 months, with 60% gaining ≥ 15 letters at 12 months and 49.1% gaining ≥ 15 letters at 24 months. 29–31 Despite these results, and the fact that it was non-inferior to ranibizumab when given every 8 weeks after a loading phase in nvAMD, suggesting improved cost-effectiveness,43 no clinical trial had been undertaken to directly compare aflibercept with ranibizumab or bevacizumab (1.25 mg/0.05 ml Avastin®; F. Hoffmann-La Roche AG, Basel, Switzerland), even though NICE recommended aflibercept for MO due to CRVO [NICE technology appraisal (TA) guidance 30512]. Cumulative safety data have not, to date, shown an increased risk of any ocular or systemic adverse events (AEs) with aflibercept compared with other drugs used for these indications.
-
Bevacizumab is a monoclonal antibody that also inhibits VEGF; it is licensed by the EMA for the treatment of cancer but is used off-label for treatment in the eye. However, it was crucial to fully assess bevacizumab’s suitability for intraocular use because (1) it is substantially cheaper than ranibizumb and aflibercept when divided by a compounding pharmacy into multiple doses from a single 4-ml vial; (2) it was found by the Decision Support Unit44 to be used in NHS trusts across the UK for nvAMD, diabetic macula oedema (DMO), RVO and other less common indications, such as choroidal neovascularisation due to myopia and retinal dystrophies; (3) it is widely used in UK private practice; and (4) there have been concerns about possible systemic side effects following intraocular injection of bevacizumab. 45 Bevacizumab was found to be non-inferior to ranibizumab in terms of macular dysfunction and final visual acuity over 2 years in two large clinical trials: the Inhibit VEGF in Age-related choroidal Neovascularisation (IVAN)45 trial and the Comparison of Age-related macular degeneration Treatments Trials (CATT). 46 These trials also found no increased risk of local or systemic side effects with bevacizumab compared with ranibizumab; although more patients receiving bevacizumab were hospitalised due to serious adverse events (SAEs), the investigators thought that these SAEs were unrelated to bevacizumab. 47
Two independent reviews48,49 had previously suggested an increase in bevacizumab-related side effects, increasing the need to compare the safety of bevacizumab directly with that of ranibizumab. NICE Technology appraisal (TA) 28313 (on ranibizumab) and NICE TA30512 (on aflibercept) for MO secondary to CRVO recommended that additional head-to-head trials including bevacizumab were needed for RVO to carefully examine clinical effectiveness and cost-effectiveness. Therefore, it was proposed that LEAVO be conducted in MO due to CRVO to (1) compare the clinical effectiveness of ranibizumab, aflibercept and bevacizumab in a pragmatic trial over 24 months that followed up patients over the natural history of the disease, (2) compare the cost-effectiveness of the agents in a trial that closely resembled clinical practice and (3) describe the safety profile of each agent in terms of ocular and systemic AEs over 24 months.
Evidence update post LEAVO initiation
Ranibizumab, aflibercept and bevacizumab continue to be used in many countries for multiple retinal diseases, with bevacizumab the most frequently given anti-VEGF agent worldwide, as the licensed alternatives remain too costly. Despite convincing case series and early trials employing bevacizumab, full-scale randomised controlled trials (RCTs) were commissioned and completed by the UK National Institute for Health Research (NIHR) and the US National Institutes of Health to compare bevacizumab with ranibizumab in nvAMD45,46 prior to the licensing of aflibercept. To our knowledge, no RCTs have compared all three agents for nvAMD. Nevertheless, after a review of all the available evidence, the NICE Guideline Committee reported that all three agents were of equivalent efficacy and had similar side effects,50 and systematic reviews found no differences in the risk of vision-threatening complications or systemic AEs. 51,52
Despite this, bevacizumab has not achieved widespread use in the UK. The reasons for this include no clear position on the issue from NHS England or the Medicines and Healthcare products Regulatory Agency (MHRA); likely conflicts of interest among key stakeholders, including physicians; and the belief in some quarters that bevacizumab is an unlicensed medication, rather than a licensed medication being used in an off-label indication. Most recently, a UK judicial review (the Whipple judgement, September 2018), brought by the manufacturers of aflibercept and ranibizumab against north of England Clinical Commissioning Groups that had adopted a policy that off-label bevacizumab should be the preferred option for the treatment of nvAMD, ruled that this was lawful. 53 However, this outcome is now subject to appeal by the manufacturers and the uncertainty continues, which is frustrating as the economic case for bevacizumab is overwhelming. The only retinal condition for which the three anti-VEGF agents have been compared is DMO. The visual gains at 2 years in eyes with moderate and severe visual loss (visual acuity of ≤ 20/50) occurred earlier and were greater in eyes receiving aflibercept therapy. However, among patients with mild initial visual impairment, visual gains were similar across treatment arms, suggesting that bevacizumab could be used for these patients. 54
Robust data remain lacking on long-term comparisons of outcomes with anti-VEGF agents for MO due to CRVO. After the initiation of LEAVO, the secondary outcomes of the randomised, double-masked, Phase III licensing trials of aflibercept for CRVO, the COPERNICUS and GALILEO studies, became available. These showed that the visual and anatomic improvements after fixed monthly dosing through to week 24 and continued pro re nata dosing with monthly monitoring from week 24 to week 52 were largely maintained up to 100 weeks if monitored every 8 weeks, and diminished if monitored quarterly from week 52 to week 100. 29–31 The 12-month single-arm study of an individualised dosing regimen of ranibizumab driven by stabilisation criteria in 357 patients with CRVO also resulted in significant gain in visual acuity (CRYSTAL). 55 The mean number of injections by 12 months was 8.8, with better outcomes in eyes with CRVO of < 3 months’ duration and lower baseline visual acuity. The visual outcomes were similar in eyes with and eyes without baseline macular ischaemia. The study also showed that visual acuity could be stabilised with visual acuity-guided re-treatment criteria up to 100 weeks. 56
Although these trials compared each anti-VEGF agent with sham treatment for MO due to CRVO, RCTs comparing these agents over a longer term have been limited. A RCT comparing aflibercept and ranibizumab on a treat-and-extend regimen over 18 months showed that the frequency of injections was significantly lower in the aflibercept arm than in the ranibizumab arm. 57 The SCORE2 study group randomised 362 patients with MO due to CRVO or hemiretinal vein occlusion 1 : 1 to receive monthly aflibercept or bevacizumab for 6 months, and reported that intravitreal bevacizumab was non-inferior to aflibercept with respect to visual acuity. 27 The participants who responded well to monthly aflibercept and those who responded well to bevacizumab for 6 months in SCORE2 were further randomised to receive either monthly injections or treat-and-extend regimens of aflibercept (for those who responded well to aflibercept) and bevacizumab (for those who responded well to bevacizumab). The 12-month outcome showed that the treat-and-extend arm of each anti-VEGF agent required up to two fewer injections from 6 to 12 months than the monthly mandated treatment arms, although the difference in visual outcomes showed significant variability. 58 A RCT comparing aflibercept and bevacizumab on a one plus pro re nata basis found that those in the aflibercept arm required fewer injections at 12 months. 59
The COMRADE-C trial was a Phase IIIb, multicentre, double-masked, randomised clinical trial that compared a ranibizumab loading phase followed by pro re nata dosing with 0.7 mg of dexamethasone, given only at baseline, for MO due to CRVO, and showed a favourable outcome with ranibizumab. 60 A 2019 systematic review61 evaluating the effectiveness and adverse effects of ranibizumab, aflibercept and bevacizumab in three common retinal conditions, including RVO, reported that none of the 17 included studies showed a clinically important difference (i.e. ≥ 5 letters) in visual acuity gains between agents. There was insufficient evidence to compare bevacizumab and ranibizumab in RVO. Overall, the authors reported that no agent had a clear advantage over another in effectiveness or safety, but in two trials61 both aflibercept and ranibizumab were significantly less cost-effective than repackaged bevacizumab. 61
Another systematic review and network analysis of 11 RCTs of the three anti-VEGF agents for RVO found no statistically significant differences in the proportion of patients who gained at least 15 letters in BCVA, in the mean change from baseline in BCVA, or in the mean change from baseline in central macular thickness at 6 months. 62 However, to date, no RCTs have compared all three anti-VEGF agents for treating this condition over the at least 2-year duration of the disease.
To our knowledge, the LEAVO trial is the first RCT evaluating the comparative clinical effectiveness, cost-effectiveness and relative safety of these three anti-VEGF agents for CRVO-related MO over 100 weeks. In summary, if bevacizumab was shown in LEAVO to be non-inferior to ranibizumab, and aflibercept was non-inferior to ranibizumab, with no new safety concerns, it could be considered for NHS use in MO due to CRVO. In addition, this would provide evidence of its equivalence to the licensed medications in multiple indications and lend substantial support to the case for using bevacizumab in the treatment of nvAMD and other retinal diseases.
Clinical trial objective
The objective of the trial was to compare the relative clinical effectiveness and cost-effectiveness of the anti-VEGF agents bevacizumab (investigational treatment), aflibercept (investigational treatment) and ranibizumab (standard care) in MO due to CRVO over 100 weeks. The trial was intended to determine if bevacizumab or aflibercept was as effective as ranibizumab in reducing visual loss from MO due to CRVO, whether or not they had an equivalent side-effect profile and whether or not either could be considered or recommended for NHS treatment based on non-inferior clinical effectiveness and superior cost-effectiveness.
Primary objectives
-
To determine whether or not bevacizumab is non-inferior to ranibizumab in treating visual loss due to MO secondary to CRVO.
-
To determine whether or not aflibercept is non-inferior to ranibizumab in treating visual loss due to MO secondary to CRVO.
Secondary objectives
-
To determine the difference between arms in terms of mean change in BCVA at 52 weeks.
-
To determine the difference between arms in the proportion of participants with ≥ 15 ETDRS letter improvement (appreciable visual gain), ≥ 10-letter improvement, < 15-letter loss and ≥ 30-letter loss (severe visual loss) at 52 and 100 weeks.
-
To determine the difference between arms in the proportion of participants with ≥ 73 ETDRS letters or > 6/12 Snellen equivalent (i.e. approximate driving visual acuity), ≤ 58 ETDRS letters (≤ 6/24) and ≤ 19 letters (≤ 3/60) [Certificate of Vision Impairment (CVI) partial and severe visual impairment] at 52 and 100 weeks.
-
To determine the difference between arms in the mean change in OCT central subfield thickness (CST) and macular volume at 52 and 100 weeks.
-
To determine the difference between arms in the proportion of participants with an OCT CST of < 320 µm [as measured with the Spectralis® (Heidelberg Engineering, Inc., Franklin, MA, USA) or equivalent] at 52 and 100 weeks (key guide to subsequent NHS clinical practice).
-
To determine the differences between arms in the mean number of intravitreal injections given to each participant at 100 weeks.
-
To determine any differences in the relative effectiveness of the investigational treatments and comparator on quality of life and resource use, reported as incremental cost-effectiveness ratios (ICERs), at 52 and 100 weeks.
-
To detect any differences in the prevalence of local and systemic side effects at 100 weeks.
-
To determine the differences between arms at 100 weeks in the proportion of (1) persistent non-responders who develop a change in retinal non-perfusion, compared with screening, and (2) participants who develop anterior and posterior segment neovascularisation.
-
To determine the differences between arms in terms of mean change in BCVA at 100 weeks.
-
To determine the differences between arms in changes in area of non-perfusion at 100 weeks and OCT anatomical features from baseline to 100 weeks.
Chapter 2 Methods
Trial design
LEAVO was a Phase III, randomised, controlled, double-masked, non-inferiority clinical trial conducted to evaluate the relative clinical effectiveness and cost-effectiveness of intravitreal bevacizumab and aflibercept, compared with ranibizumab, for MO due to CRVO. The intention was to randomise 459 participants with MO due to CRVO in at least one eye in a ratio of 1 : 1 : 1 to ranibizumab (0.5 mg/0.05 ml), aflibercept (2.0 mg/0.05 ml) and bevacizumab (1.25 mg/0.05 ml), all of which would be administered by repeated intravitreal injection, and to follow up these participants for 100 weeks. The study was conducted in the UK NHS across 44 ophthalmology centres that had staff with expertise in retinal disorders and a proven track record of effectiveness research. 2
Participants
The trial population, from which the trial sample was drawn, was adults aged ≥ 18 years with MO secondary to CRVO of < 12 months’ duration who attended one of the 44 NHS ophthalmology centres. The complete inclusion and exclusion criteria are listed in the following sections.
Selection of participants
Inclusion criteria
-
Subjects of either sex aged ≥ 18 years.
-
Clinical diagnosis of centre-involving MO due to CRVO.
-
Central retinal vein occlusion of ≤ 12 months’ duration.
-
Best corrected visual acuity ETDRS letter score in the trial eye of between 78 (approximate Snellen equivalent: 20/32) and 19 (approximate Snellen equivalent: 20/400).
-
Optical coherence tomography CST of > 320 µm (as measured with the Spectralis) (or equivalent with an alternative OCT device) predominantly due to MO secondary to CRVO in the trial eye.
-
Media clarity, pupillary dilatation and subject co-operation sufficient for adequate fundus imaging of the trial eye.
-
Best corrected visual acuity ETDRS letter score in the non-trial eye of ≥ 14 (approximate Snellen equivalent: 20/600).
Exclusion criteria
The following applied to the trial eye only, unless specifically stated otherwise:
-
Macular oedema considered to be caused by a condition other than CRVO (e.g. DMO, Irvine–Gass syndrome).
-
An ocular condition present that, in the opinion of the investigator, might have affected MO or altered visual acuity during the trial (e.g. vitreomacular traction).
-
Any diabetic retinopathy or DMO on baseline clinical examination of the trial eye.
-
Moderate or severe non-proliferative diabetic retinopathy or quiescent, treated or active proliferative diabetic retinopathy or MO in the non-trial eye. Note that mild non-proliferative diabetic retinopathy only was permissible in the non-trial eye.
-
History of treatment for MO due to CRVO in the previous 90 days with intravitreal or peribulbar corticosteroids or in the previous 60 days with anti-VEGF drugs or more than six prior anti-VEGF treatments in the previous 12 months.
-
Active iris or angle neovascularisation, neovascular glaucoma, untreated neovascularisation disc (NVD), neovascularisation elsewhere (NVE) and vitreous haemorrhage or treatment for these conditions in the previous month.
-
Uncontrolled glaucoma (i.e. eye pressure of > 30 mmHg) either untreated or being treated with antiglaucoma medication at screening.
-
Any active periocular or intraocular infection or inflammation (e.g. conjunctivitis, keratitis, scleritis, uveitis, endophthalmitis).
Systemic exclusion criteria
-
Uncontrolled blood pressure, defined as a systolic value of > 170 mmHg and a diastolic value of > 110 mmHg.
-
Myocardial infarction, stroke, transient ischaemic attack, acute congestive cardiac failure or any acute coronary event < 3 months before randomisation.
-
Women of childbearing potential, unless they were using an effective method of contraception during the trial and for 6 months after their last injection for the trial. Effective contraception was defined as one of the following:63
-
Barrier method – condoms or occlusive cap with spermicides.
-
True abstinence – when in line with the preferred and usual lifestyle of the subject. Periodic abstinence (e.g. calendar, ovulation, symptothermal, post-ovulation methods) and withdrawal were not acceptable methods of contraception.
-
Tubal ligation or bilateral oophorectomy (with or without hysterectomy).
-
Male partner sterilisation. The vasectomised male partner should be the only partner of the female participant.
-
Use of established oral, injected or implanted hormonal methods of contraception and intrauterine device.
-
-
Pregnant or lactating women.
-
Men who did not agree to an effective form of contraception for the duration of the trial and for 6 months after their last injection for the trial.
-
Hypersensitivity to the active ingredients of aflibercept, bevacizumab or ranibizumab, or to any of the excipients of these drugs.
-
Hypersensitivity to Chinese hamster ovary cell products or other recombinant human or humanised antibodies.
-
A condition that, in the opinion of the investigator, would preclude participation in the trial.
-
Participation in an investigational trial involving an investigational medicinal product within 90 days of randomisation.
Rescreening of patients2
Patients could be rescreened in the following circumstances:
-
Patients who did not meet the BCVA or OCT CST inclusion criteria could be rescreened a minimum of 4 weeks after their last screening visit if they were thought to meet the eligibility criteria.
-
Individuals who did not meet other modifiable inclusion criteria, for example blood pressure, could be rescreened a minimum of 2 weeks after the last screening visit.
All assessments performed at the initial screening visit were repeated during the rescreening visit, except FFA if the rescreening visit was within 10 weeks of the original screening visit. If a patient was found to be eligible on rescreening and was randomised, their initial entry on the electronic case report form (eCRF) system was updated, rather a ‘new’ patient being created on the system. This avoided such patients being incorrectly counted twice in the Consolidated Standards of Reporting Trials (CONSORT) diagram.
Recruitment
The trial recruited participants from 44 UK ophthalmology centres over 24 months. Recruitment was competitive; however, each site was allocated a minimum target number of participants to recruit and was encouraged to exceed this if possible. Sites were set up strategically: larger sites with greater capacity were initiated first to maximise early recruitment and to ensure that the recruitment period was fully utilised. Eligible patients were invited to participate via their local clinics, or in an invitation letter. At each site, participants were identified from subspecialty retinal, general and eye casualty clinics. Once identified, potential participants underwent a clinical examination, followed by discussion of the trial with an experienced trial clinician, and were provided with the patient information sheet. 2
Trial procedures
Informed consent procedure
The principal investigator or designated subinvestigator was responsible for ensuring that a patient was fully consented after being provided with an adequate explanation of the aims, methods, anticipated benefits and potential hazards of the trial. Patients were advised that any data collected would be held and used in accordance with the Data Protection Act 1998. 64 Patients were given at least 24 hours after receiving the patient information sheet to consider taking part. The principal investigator or designee recorded in the medical notes the date when the patient information sheet was given to the patient and the facts that patients were under no obligation to enter the trial and that they could withdraw at any time without giving a reason. No clinical trial procedures were conducted before consent was taken from a participant; consent denoted enrolment in the trial. A copy of the signed informed consent form was given to the participant. The original signed form was retained at the trial site and a copy was placed in the medical notes. If new safety information resulted in significant changes in the risk/benefit assessment, or if there were significant changes to the protocol or patient information sheet, participants were consented again as appropriate.
Randomisation
Only one eye of each participant was randomised to the trial. In 95% of cases, one eye was affected by CRVO and so was the ‘worse-seeing eye’ and was randomised. On rare occasions, participants had bilateral CRVO that met the eligibility criteria. In these cases, the worse-seeing eye was randomised unless the participant opted for randomisation of the ‘better-seeing eye’. The plan was to recruit 459 adult participants with MO due to CRVO and to randomise them 1 : 1 : 1 at the level of the individual using the method of minimisation incorporating a random element. The three stratifying factors were (1) visual acuity, stratified by screening BCVA letter score [of ≤ 38 (approximate Snellen equivalent: ≤ 6/60), 39–58 (approximate Snellen equivalent: 6/48 to 6/24) or ≥ 59 (approximate Snellen equivalent: ≥ 6/18)]; (2) duration of disease, from date of CRVO diagnosis to commencement of therapy (< 3, 3–6 or > 6 months); and (3) treatment naive versus previous treatment. Each participant was randomised to one of three arms: bevacizumab, aflibercept or ranibizumab. 2
A patient identification number (PIN) was generated by registering a patient on the MACRO eCRF system (InferMed Macro; Elsevier Ltd, Amsterdam, the Netherlands), after consent had been obtained. Randomisation was carried out in a bespoke web-based randomisation system hosted at the King’s Clinical Trials Unit (KCTU). A unique PIN was generated in the MACRO program; this was recorded on all source data worksheets and was used to identify a participant throughout the trial. 2,63 The trial manager allocated all authorised site staff a username and password for the randomisation system. All authorised staff members, who were typically the principal investigator or designee, logged in to the randomisation system and entered a participant’s details, including the unique PIN. Once a participant had been randomised, the system automatically generated e-mails to key staff in the trial. Unmasked e-mails sent to site pharmacies alerted them to a participant’s treatment arm: ranibizumab, aflibercept or bevacizumab. The pharmacy department used the e-mail to cross-check the trial prescription to ensure that the correct medication was dispensed for the correct participant. Additional masked e-mails were generated from the randomisation system and sent to key trial site staff,63 and unmasked e-mails were sent to the emergency unmasking service (ESMS Global Ltd, London, UK) and unmasked trial management staff. 2
Masking of treatment allocation
In randomisation process, only the pharmacy at a local trial site was informed by e-mail of a subject’s treatment allocation; a copy of the e-mail was sent to the emergency unmasking service (ESMS Global Ltd) and to unmasked trial management staff. The trial drug that a participant received was transferred to the dedicated injection room in an opaque masking bag designed to securely and safely transport medication. A unique seal was attached to the bag before it left the pharmacy. The bag had a safe zipped compartment containing a printed form detailing a participant’s unique PIN, their date of birth, the date the drug was dispensed and the injection batch number. Before a participant entered the injection room, the unmasked injector broke the seal and took the drug out of the masking bag. Bevacizumab was provided in a prefilled syringe, but ranibizumab and aflibercept were provided in a vial and drawn into a syringe by the unmasked injector. The syringe was placed on the injection trolley out of view of the participant, who was then invited into the room and asked to lie on the bed, and then received the injection. During the trial the manufacturer of ranibizumab began to provide the drug, in a unique prefilled syringe and vials ceased to be available. In this situation, the unmasked injector took care not to allow the participant to see the syringe either before or after the injection had been given. This was achieved by administering the injection while the participant was lying down and the injection was given via the pars plana in any quadrant of the eye, with the syringe brought to and taken away from the injection site via a participant’s inferotemporal field of vision so that it did not pass across their line of sight. The unmasked injector signed the source notes to the effect that the treatment in the masked bag had been administered to the participant, without specifying the treatment, and also signed the printed form that was in the masking bag. The empty syringe with needle and vial were disposed of in the injection room. The masking bag and completed printed form were returned to the pharmacy. The outer packaging of the drug was disposed of in the injection room. 2
The clinical assessment team, including the site principal investigator, optometrist (i.e. assessor of the primary outcome), site trial co-ordinator, clinical investigator, clinical assessment trial nurse and ophthalmic technician, remained masked throughout the trial, as there was no record of a participant’s treatment arm in the source notes or the case report form (CRF). Similarly, co-ordinators or administrators completing questionnaires with participants in person (or, in extreme circumstances, only over the telephone at specific time points) had details of a participant’s PIN only. If, at any time, information regarding treatment allocation was shared with the outcome assessors, this was recorded in the trial master file, and the person(s) involved met with the site principal investigator to ensure that no repetition occurred and undertook not to convey this information either to the participant or to others involved in the project. Certain secondary outcomes (e.g. interpretation of FFA) occurred at the remote Network of Ophthalmic Reading Centres (NetwORC) UK (Belfast, UK), where the assessors were masked to the treatment allocation. These masking procedures avoided both performance and detection bias. We have described the completeness of outcome data for each outcome, including any unmasking in error, reasons for attrition and exclusions from the analysis. 2 The trial statisticians had access to the accumulating outcome data that were required for reporting to the Data Monitoring and Ethics Committee (DMEC). Both trial statisticians attended both the open and the closed DMEC meetings.
Screening and baseline assessment
A patient had to receive the patient information sheet not later than 24 hours before the screening assessment. The screening and baseline visits could be undertaken on the same day, provided that all test results were available. A patient could return within 10 days of screening for the baseline assessment, at which point the screening procedures were still valid and were not repeated at baseline (see Appendix 3, Table 29).
Milestone and non-milestone visits
Trial milestone assessments, when key research data were collected, occurred at baseline and at weeks 12, 24, 52, 76 and 100. These visits, as well as treatment visits at weeks 4 and 8, were calculated and agreed with a participant prior to randomisation (with flexibility of 0 to 14 days for weeks 4, 8 and 12, and of –14 to 14 days for weeks 24, 52, 76 and 100, from the date of randomisation). It was mandatory for all participants to attend all milestone visits, even if a milestone visit fell < 4 weeks after a treatment visit or if a participant was being followed up every 8 weeks and the next milestone visit fell during the 8-week interval. The intervening trial treatment visits were deliberately flexible to allow normal clinical practice treatment follow-up to be accommodated. All data from the trial milestone visits were entered into the eCRF. For regular treatment visits, only the following information was entered into the eCRF: BCVA; OCT CST; whether or not an injection was given; and, if no injection was given, the reason why. At milestone visits, refracted visual acuity was tested and health economic questionnaires were completed; colour photography was undertaken at baseline and at weeks 52 and 100; and FFA was undertaken at baseline and at week 100, in addition to the clinical examination and OCT tests performed at all other trial visits (see Appendix 3, Table 29).
Trial assessments and methods
Participant demographics, medical and ophthalmic history
This information was retrieved from the participant, from hospital medical records or from a general practitioner. Data included age, sex and ethnic background. Data were also collected on clinically relevant medical history and management in the preceding 24 months, and on any ocular history and treatment. 2
Visual acuity tests
Visual acuity tests were performed by a certified optometrist in a certified visual acuity testing lane using validated ETDRS vision charts and standard operating procedures. 65,66 Refracted visual acuity was carried out in both eyes at screening,63 at weeks 12, 24, 52, 76 and 100, and at the point of withdrawal. For all other visits, the visual acuity was tested with the previous most recent protocol refraction. Visual acuity examiners were masked to the treatment. The visual acuity scores were recorded in the eCRF2 (see Appendix 4).
Standard ophthalmic examination
A standard ophthalmic examination using slit-lamp biomicroscopy included an undilated examination for neovascularisation of the iris (NVI), RAPD and tonometry in both eyes at all visits. Dilated fundus examination was performed in both eyes at all milestone visits (i.e. at screening, at baseline, at weeks 12, 24, 52, 76 and 100, and at the point of withdrawal). At all other visits, dilated fundus examination was performed in the trial eye and, at the discretion of the investigator, in the non-trial eye. Gonioscopy, if indicated, was carried out prior to dilatation at any visit. 2
Spectral-domain optical coherence tomography
The CST and total macular volume in both eyes were recorded in the eCRF from the spectral-domain optical coherence tomography (SD-OCT) thickness map at every visit, and, if applicable, at the point of withdrawal. 63 Any SD-OCT machine could be used for the trial, but the same model of SD-OCT machine had to be used for each individual throughout the period of the trial. SD-OCT images at screening and at weeks 52 and 100 only were transferred to and read by masked graders at the independent NetwORC UK. NetwORC UK provided each site with a trial imaging protocol on how to acquire SD-OCT images, colour fundus photographs and fundus fluorescein angiographs and how to transfer these to NetwORC UK to them. Initial grading of all OCT images at baseline and at weeks 52 and 100 was performed by NetwORC UK. The grading took into account intraretinal oedema, classified as diffuse, cystic or mixed; determined subretinal fluid as being present or absent; and determined vitreoretinal interface abnormalities as being present (as either an epiretinal membrane or vitreomacular traction) or absent. Following the contract variation, additional grading parameters were assessed at NetwORC UK in collaboration with specialised retinal graders at Moorfields Eye Hospital, utilising additional definitions and analyses that had been developed while the trial was in progress. 1,67,68 Only images captured using a Spectralis OCT machine had sufficient detail to support the enhanced grading definitions. Retinal morphology was assessed using the Spectralis® Heidelberg Macular Raster OCT device (Heidelberg Engineering, Inc.) of 31 line scans, 30 × 25 mm in size, at an interscan distance of 240 µm or the equivalent for alternative devices. MO was graded using the entire line-scan series and the central 1500 µm, that is seven scans were employed for vitreomacular interface abnormality and subretinal detachment or equivalent. The remaining parameters were graded using the central 1000 µm only, that is central five-line scans only. A magnification of 300% was used to assess the ellipsoid zone (EZ), disorganisation of the retinal inner layers (DRIL)67,68 and hyperreflective foci (HRF),69,70 with 100% magnification for the remaining parameters. HRF, external limiting membrane (ELM), EZ and cone outer segment tips (COSTs) were graded as positive only if the foveal line showed involvement of the foveal depression such that it was distorted, lessened or absent. 2 For the grading of normal and abnormal individual morphological features, see Appendix 5, Specific grading of individual morphological optical coherence tomography features, and Figures 22 and 23.
Colour fundus photography
Non-stereo, seven-field conventional or wide-angle colour fundus photography (CFP) was performed at screening and at weeks 52 and 100 in the trial eye. CFP confirmed the diagnosis of CRVO and assisted interpretation of features identified by FFA, for example to differentiate between non-perfusion and masking due to haemorrhage. If applicable, CFP was also performed at the point of withdrawal, and at any other trial visit, as per investigator discretion. Colour fundus photographs were transferred to and read by masked graders at the independent NetwORC UK. Either a colour camera capable of taking seven-field colour fundus photographs or a wide-angle system was used, but the same model of camera was used for each individual throughout the trial. The colour photographs were graded by the NetwORC UK. 2
Fundus fluorescein angiography
Non-stereo, seven-field conventional or wide-angle FFA was performed at screening and at week 100 in the trial eye. Any FFA system capable of taking seven-field FFA pictures or a wide-angle system was allowed, but the same system had to be used in the same individual throughout the trial. 2 FFA was used to quantify the degree of retinal ischaemia and for identification of retinal neovascularisation (see Appendix 5, Fundus fluorescein angiography grading). Pseudo-anonymised FFA images were transferred to NetwORC UK, where the standard NetwORC UK 13-sector grid (see Appendix 5, Figure 24) was applied over the wide-angle or montaged seven-field angiography pictures at baseline and at 100 weeks. The first 100 gradings were double-graded. Discrepancies were adjudicated. Subsequently, one in every eight gradings was double-graded. Kappa values for key fields (e.g. detection of new vessels on the disc and new vessels elsewhere) were required to be > 0.8. Any graders who did not achieve this were required to undergo additional training. Each sector in the grid was semiquantified in terms of percentage of non-perfusion (nil, 1–25%, 26–50%, 51–75% and 76–100%), and all available sets of images were analysed to identify how many participants in each arm had experienced a two-step increase (e.g. zero to 26–50%, or 26–50% to 76–100%) in one to five or more sectors (see Appendix 5, Figure 24). This technique was used in preference to the ischaemic index, which estimates the ratio of ischaemic to total retinal area but is very susceptible to image quality and is applicable to wide-angled images only. 22 Therefore, during the trial we used the concentric rings method, which displays superimposed concentric circles, centred on the fovea. 23,71,72 The innermost circle was 1 disc diameter (DD) in size, and is not graded as it represents the foveal avascular zone. The second circle, representing the macular ring (ring M), has a radius of 2.5 DD. Each of the subsequent rings (rings 1, 2, 3 and 4) is placed at increments of 2.5 DD in radius from the foveal centre. Each of these rings is subdivided into 12 equal segments. 23 To calculate the size of the concentric rings required, we assumed that the mean axial length was 24 mm, and excluded 2 mm from this to account for the cornea and part of the anterior chamber. In the model eye, the radius was 11 mm (diameter 22 mm); therefore, the full circumference would have been 69.1 mm (π = 3.142). The wide-angled imaging system (Optos®; Optos, Inc., Marlborough, MA, USA) was able to image 200 degrees of the retina; we used this to calculate the average diameter of retina obtained in a single central image. This was calculated to be 38.4 mm. Using the DD of 1.8 mm, this meant that the diameter of the image was 21.3 DD. A diameter of 21.3 DD resulted in the need for a macular ring plus three/four further rings. 23 Based on our validation study, we identified that ring 4 was gradable, but the superior and inferior segments of rings 3 and 4 were ungradable because the ultra-wide field image had better clarity in the horizontal meridian. For details of this method, see Appendix 5, Figure 25.
Health economic questionnaires
The following quality-of-life and resource use questionnaires were administered at baseline, at 12, 24, 52, 76 and 100 weeks, and at the point of withdrawal: the National Eye Institute Visual Function Questionnaire-25 items (VFQ-25), EuroQol-5 Dimensions (EQ-5D), EuroQol-5 Dimensions with vision bolt-on (EQ-5D-V), and a bespoke resource use questionnaire [see www.journalslibrary.nihr.ac.uk/programmes/hta/119203/#/documentation (accessed 14 July 2020)].
Treatment allocation guess form
Participants and masked optometrists were asked to complete a treatment allocation guess form at week 100, or at the point of withdrawal, to assess how well participant and assessor masking worked in the trial. 2
Definition of the end of the trial
Participants were enrolled in the trial for approximately 100 weeks from the point of randomisation. The end of the trial was defined as the last participant’s last trial visit.
Treatment procedures
Treatment schedule
After mandated administration in all three trial arms at baseline and at 4, 8 and 12 weeks, further pro re nata intervention was administered at weeks 16 and 20 if re-treatment criteria were met and if visual acuity was ≤ 83 letters.
Regardless of whether a treatment was given, the participant was reviewed in 4 weeks. From weeks 24 to 96, the interval was initially 4 weeks (with a visit window of –14 to 14 days), with the potential for the interval to increase to 8 weeks (with a visit window of –14 to 14 days) if criteria for ‘stability’ were achieved. ‘Stability’ was defined as three successive visits from week 16 onwards at which treatment criteria were not met, and so the first time at which treatment could be deferred for 8 weeks was week 24.
Similarly, ‘success’ was defined as an ETDRS letter score of > 83 letters, and if this was present at any re-treatment visit from week 16 onwards, treatment was not given at that visit and the participant was reviewed subsequently. The review occurred 4 weeks later if the initial visit was at 16 weeks, 20 weeks or any other visit if treatment had been given at this or the preceding visit. If no treatment had been given at these two visits, the participant was reviewed 8 weeks later. If, at any subsequent visit, re-treatment criteria were met and BCVA was ≤ 83 ETDRS letters, then re-treatment was commenced (Figure 1). At each visit between weeks 24 and 96 inclusive, ‘non-responder treatment suspension’ criteria could be met. If so, the principal investigator, or their designee, at their discretion, could suspend treatment to prevent therapy in a participant who had not responded to at least their last three injections. If the criteria for restarting therapy after ‘non-responder treatment suspension’ were met, then the participant had to resume therapy. If re-treatment criteria were met at one of the visits that took place every 8 weeks or at an unscheduled visit, then visits every 4 weeks were resumed. Treatment could be ‘deferred’ in certain circumstances, but the participant was asked to still attend the milestone visits.
FIGURE 1.
Re-treatment algorithm for weeks 24–96 of the trial.

Re-treatment criteria
Criteria were met if one or more of the following was present:
-
a decrease in visual acuity of ≥ 6 letters between the current and most recent visit, attributed to an increase in OCT CST
-
an increase in visual acuity of ≥ 6 letters between the current and most recent visit
-
OCT CST of > 320 µm (on Spectralis, or of > 300 µm on other machines) because of intraretinal or subretinal fluid
-
an OCT CST increase of > 50 µm from the lowest previous measurement.
Investigational medicinal products
Comparator: ranibizumab (0.5 mg/0.05 ml)
Ranibizumab is a humanised recombinant monoclonal antibody fragment that binds to VEGF A, preventing receptor interaction and blocking downstream action of VEGF, that is increased vascular permeability, leading to MO in CRVO. It is licensed by the EMA, and NICE has recommended it for use in the treatment of nvAMD, DMO and RVO. NICE TA28313 for MO due to RVO was issued in May 2013. Ranibizumab has been the mainstay of routine clinical care for this condition since the third quarter of 2013 and was the comparator in this trial. It was supplied to each site hospital pharmacy directly from the manufacturer as a part of routine hospital stock. 2
Intervention: aflibercept (2.0 mg/0.05 ml)
Aflibercept is a fusion protein that includes the key binding domains of human VEGF receptors 1 and 2 with human IgG Fc and acts as a dummy receptor for all VEGF isoforms and placental growth factor, preventing increased permeability and MO in CRVO. At initiation of this trial, it was licensed by the EMA, and NICE has recommended it for nvAMD. NICE TA30512 was published in February 2014; NICE recommends this drug as first-line use for CRVO-related MO. Aflibercept was supplied in a glass vial to each site hospital pharmacy directly from the manufacturer as part of routine hospital stock. 2
Intervention: bevacizumab (1.25 mg/0.05 ml)
Bevacizumab is a full-length humanised monoclonal antibody that binds to VEGF A, forming a protein complex incapable of binding to the VEGF receptor, thus blocking downstream VEGF action. In this trial, bevacizumab was supplied in a prefilled plastic syringe in a sealed package to each trial site pharmacy from the Liverpool and Broadgreen Pharmacy Aseptic Unit, Royal Liverpool University Hospital, Liverpool, UK. 2
Site pharmacy storage, ordering and handling procedures of investigational medicinal products
A trial medication dispensing and return log was maintained by the trial site pharmacies. Administration records from these sites were retained by the pharmacy and monitored by the trial manager to ensure that accurate CRF data were recorded. The randomisation system was linked to the investigational medicinal product (IMP) supply. Each site pharmacy was also responsible for appropriate storage, dispensing, disposal, and recall and destruction logs, in accordance with good manufacturing practice73 and good clinical practice,74 and the site hospital pharmacy’s approved policies for IMP accountability and management. Furthermore, each site pharmacy maintained a record of trial drug administration, based on the pre-printed form signed by the unmasked investigator that was returned to the pharmacy at each centre. 2
Investigational medicinal product accountability
Used and unused trial study medication and study medication accountability
Each masking bag contained a pre-printed form that listed the details of the participant’s unique PIN, date of birth, date the drug was dispensed and injection batch number. After performing the intravitreal injection, the unmasked injector signed this form to confirm that the drug had been given to the allocated patient, and they then returned it in the masking bag to the pharmacy. All used drug vials and syringes were disposed of in the injection room and not returned to the pharmacy. Pharmacies in each site maintained a trial medication dispensing log, including date dispensed, batch number, expiry date and return log. The return log was compiled from the form signed by the unmasked injector. In addition, the trial-specific prescriptions were maintained in the pharmacy file for audit purposes. Any administration errors were reported to the chief investigator and trial statistician. In the event that an injection was not given as scheduled, the reason was documented in the participant’s notes and the CRF. The trial monitor checked the pharmacy records against the eCRF. All records were reconciled with the investigator site file at the end of the trial. 2
Description and justification of route of administration and dosage of investigational medicinal product
The approved route of administration (i.e. by intravitreal injection through the pars plana of the eye) was used in all cases under sterile conditions in a designated treatment area in accordance with the guidelines75 for intravitreal injection of the Royal College of Ophthalmologists and any approved procedures at the individual site hospital. The injection could be performed by the unmasked injector(s) only, who was (were) on the hospital site LEAVO delegation log and was (were) experienced in intravitreal injection procedures. The dosages of ranibizumab (0.5 mg/0.05 ml) and aflibercept (2.0 mg/0.05 ml) used in this trial were approved by the EMA, and NICE recommends these doses of these agents for intraocular use. 12,13 The dosage of bevacizumab (1.25 mg/0.05 ml) was the dosage used in the IVAN clinical trial and the CATT of treating wet age-related macular degeneration (AMD), and the standard dose used in clinical practice. Post-injection checks were conducted in accordance with local hospital policy and included a visual acuity, intraocular pressure or optic nerve head perfusion check, or a combination of these. The interval between two doses of all three drugs was not recommended to be less than 4 weeks. 2
Management of complications
Complications, such as the development of ischaemic CRVO, neovascularisation of the angle, NVI, neovascular glaucoma (NVG), NVE and NVD, in the trial eye were recorded as AEs. The diagnosis and management of these complications of CRVO in the trial were at investigator discretion and based on local practice. Laser therapy formed the mainstay of therapy and was recorded as a concomitant procedure. 7,8
Recording and reporting of adverse events and reactions
Routine reporting
The MHRA definitions of AEs and SAEs were adopted for this trial. AEs were reported by the site in the AEs log in the eCRF. All SAEs, serious adverse reactions and suspected unexpected serious adverse reactions (SUSARs) were recorded and reported on the SAE form to the chief investigator/delegate within 24 hours of learning of their occurrence. A record of this notification (including date of notification) was clearly documented to provide an audit trail. In the case of incomplete information at the time of initial reporting, a follow-up report was provided as soon as the information became available. The sites responded promptly to any queries raised by the chief investigator/delegate. The principal investigator/delegate, who had to be a clinician at the site, assessed the relationship of the SAE to any of the trial interventions. The chief investigator was responsible for assessing the expected or unexpected nature of all serious adverse reactions. The chief investigator/delegate, with the support of the KCTU, ensured that Moorfields Eye Hospital, as sponsor, was made aware of any SUSARs and serious adverse reactions that occurred. The chief investigator/delegate, in conjunction with the sponsor, was responsible for reporting all SUSARs to the MHRA and relevant ethics committee within the appropriate time frame.
All principal investigators were informed of all SAEs that were assessed as fulfilling the criteria for a SUSAR (i.e. possibly, probably or definitely related to any trial intervention, and unexpected as per the summary of product characteristics or the protocol). 2
Planned ‘hospitalisations’, non-emergency procedures and adverse event reporting
Some AEs met the definition of serious but did not need to be reported on a SAE report form. Common ophthalmology- and non-ophthalmology-related events that resulted in planned, non-emergency hospital admissions for the investigation or treatment of those events and that were not possibly, probably or definitely related to the IMPs did not need to be reported on a SAE report form. These events were recorded on the AE form and the investigation and treatment of ophthalmology-related events were recorded on the ophthalmology-related concomitant procedure forms. All concomitant medications were recorded on the concomitant medication form. These forms were updated following each trial visit to ensure that the independent DMEC received accurate reports of the occurrence and treatment of AEs. 2
Pregnancy
In the event that a female participant became pregnant, this was reported to the KCTU on a pregnancy form sent by fax or e-mail as soon as the investigator became aware of it. The pregnancy was monitored to determine outcome. Any information related to the pregnancy following the initial report was reported on a follow-up pregnancy form. 2
Data management
Confidentiality
Data were handled, computerised and stored in accordance with the Data Protection Act 1998. 64 Participants were identified via a unique PIN, their date of birth and their initials. Identifiable information was stored in the eCRF and did not leave the site. Any participant contact information was stored in the site on password-protected computers or in secured locations with restricted access.
Data collection tools and source document identification
Written informed consent was obtained before screening and other trial-specific procedures were performed. SAE data were collected on paper SAE report forms and e-mailed or faxed to the KCTU. Summary details of SAEs were transcribed to the AE section of the eCRF. For all other data collected, source data worksheets were used for each patient and data were entered onto the eCRF database. Source data worksheets were reconciled at the end of the trial with a patient’s NHS medical notes in the recruiting site. During the trial, critical clinical information was written in the medical notes to ensure that informed medical decisions could be made in the absence of the trial team. Trial-related clinical letters were copied to the medical notes during the trial. It was the responsibility of the principal investigator and his/her team to ensure that the accuracy of all data entered in the worksheets and the eCRF was in accordance with good clinical practice. The delegation log identified all those personnel with responsibilities for data collection and handling, including those who had access to the trial database. The principal investigator was responsible for ensuring that source data worksheets were filed in a suitably secure location so that source data verification could be undertaken throughout the trial. 2
Data handling and analysis
All trial data and site files were kept on site in a secure location with restricted access.
The trial used an eCRF created using the InferMed MACRO database system. Data were managed using this system. The eCRF was created in collaboration with the trial statistician and the chief investigator and maintained by the KCTU. It was hosted on a dedicated secure server in King’s College London. This system is regulatory compliant; has a full audit trail, data discrepancy functionality and database lock functionality; and supports real-time data cleaning and reporting. The trial manager was responsible for providing usernames and passwords to permitted local trial personnel. Only those authorised by the trial manager were able to use the system. 2,63
Quality assurance
The trial incorporated a range of data management quality assurance functions. The eCRF system contained a range of validations defined by the trial team that alerted sites to inconsistencies in the data being entered, which were monitored by the trial manager. The trial manager provided trial training and ongoing trial support, and conducted regular monitoring visits at each site, checking source data for transcription errors. Any necessary alterations to entered data were date- and time-stamped in the eCRF. A detailed monitoring plan and data management plan was developed and updated as the trial progressed, detailing the quality control and quality assurance checks to be undertaken. 2
Database lock and record-keeping
Prior to database lock, the trial manager reviewed any outstanding warnings on the eCRF and resolved or closed these, as appropriate. Local trial personnel resolved any queries that arose. Once all queries were resolved, no further changes were made to the database unless specifically requested by the trial office in response to the statistician’s data checks. The trial principal investigator reviewed all of the data for each participant and provided e-mail sign-off to verify that all data were complete and correct. At this point, all data were formally locked for analysis. At the end of the trial, each site was supplied with a CD-ROM containing the eCRF data for their site. This was filed locally for any future regulatory inspection or internal audit. The chief investigator is the custodian for the data generated from the trial and is responsible for archiving the original data. All data will be archived for at least 5 years from the end of the trial and will be archived in accordance with sponsor’s and regulatory requirements. Principal investigators were responsible for securely archiving local data generated, essential documents and source data in accordance with local requirements, but for at least 5 years from the end of the trial. 2
Statistical considerations
Sample size calculation
Bevacizumab and aflibercept were hypothesised to be substantially inferior to ranibizumab if, in each case, the mean of the primary outcome (i.e. change in BCVA ETDRS letter score) was worse by a margin of 5 letters, a previously used non-inferiority margin,26,76 representing the minimum visual acuity change that a patient may distinguish. A similar CRVO population9 reported a standard deviation (SD) of 14.3 letters in the ranibizumab (0.5 mg) arm; the 12-month rate of those lost to follow-up was 8.4% in the ranibizumab arms (0.5 mg and 0.3 mg). In the absence of 24-month data, we assumed a comparable SD of 14.3 letters at 100 weeks, and allowed for 15% dropout. The two null hypotheses, that bevacizumab was substantially inferior to ranibizumab, and that aflibercept was substantially inferior to ranibizumab, were each planned to be rejected if the estimated 95% confidence interval (CI) for the difference in treatment means was wholly above the 5-letter margin in each case. Assuming equal efficacy, there was 80% power to reject each null hypothesis and to declare non-inferiority, with 130 followed-up patients analysed per arm. Allowing for 15% missing data at 100 weeks, 459 patients were planned to be randomised to the three arms (equal allocation ratio of 153 participants per arm). Sample size calculations were performed using nQuery Advisor version 4.0 (Statistical Solutions, Saugus, MA, USA). The primary method of analysis was a linear mixed-effects (LME) model with adjustment for baseline, which was expected, other things being equal, to increase the power to detect non-inferiority. The primary method of analysis included all available refracted data of the primary outcome up to and including 100 weeks, including data from the 15% of participants who we anticipated could miss the 100-week primary outcome end point. 2
Statistical considerations
The trial statisticians were responsible for all statistical aspects of the trial, from design through to analysis and dissemination. 2 A detailed statistical analysis plan was completed before the start of the trial; it was commented on by the DMEC and approved by the Trial Steering Committee (TSC). The plan was accompanied by a health economics analysis plan, and was updated and re-approved by the TSC when the protocol was amended.
Target population
The target population, to which inferences from the end of this trial were intended to generalise, was adult patients with MO due to CRVO.
Trial population
The trial population, from which the trial sample was drawn, was further defined to be adults aged ≥ 18 years, with visual impairment due to CRVO-related MO of < 12 months’ duration, who attended one of the 44 ophthalmology centres in the UK that had staff with expertise in retinal disorders and a proven track record of effective research. Only one eye per participant was included in the trial.
Hypotheses
The hypotheses refer to the populations of relevant patients, rather than to trial subjects:
-
The working hypothesis – the so-called ‘working hypothesis’ was the hypothesis that motivated the trial, which the trial results may or may not support. It was that the change in BCVA is non-inferior in patients treated with either aflibercept or bevacizumab, compared with patients treated with ranibizumab.
-
The statistical null hypothesis 1 – bevacizumab is inferior to ranibizumab in eyes with MO due to CRVO at 100 weeks.
-
The statistical null hypothesis 2 – aflibercept is inferior to ranibizumab in eyes with MO due to CRVO at 100 weeks.
-
Statistical alternative hypothesis 1 – bevacizumab is non-inferior to ranibizumab in eyes with MO due to CRVO at 100 weeks.
-
Statistical alternative hypothesis 2 – aflibercept is non-inferior to ranibizumab in eyes with MO due to CRVO at 100 weeks.
Treatment arms
The trial was randomised with equal allocation of participants in a 1 : 1 : 1 ratio to the three arms (see Chapter 2, Randomisation).
Trial samples
Intention to treat
The achieved trial sample comprised those patients who consented to participate and were actually randomised to the trial. 63 These patients were the trial subjects. This randomised trial sample was also the trial intention-to-treat (ITT) population. The ITT principle states that every subject will be analysed according to the treatment group to which they are randomised. In this trial, subjects’ data were analysed according to the ITT strategy,77 under which at least one analysis is recommended to be based on the ITT population. The trial ITT population comprised all randomised participants, regardless of whether there was an error in their eligibility (inclusion/exclusion), whether they had withdrawn post randomisation and whether the correct trial treatments or other interventions were received. 63
Per protocol
A per-protocol set of subjects was also included. These were defined as the subset found to be eligible at entry and who had minimal sufficient exposure to the treatment regimen, defined as four treatments correctly assessed and received during the first six visits up to week 20. For each of the first four visits, a correct treatment was defined as receiving the injection. For the fifth and sixth visits, a correctly assessed and received treatment was defined to be the receipt of an injection if this was indicated to be required by the re-treatment criteria, or the non-receipt of an injection if this was indicated by the re-treatment criteria.
The main reason for having a per-protocol set was that this was a non-inferiority trial, and so the use of the full analysis set would not generally be conservative [see the International Conference on Harmonisation (ICH) guidance, E9, section 5.2.378]. As Lesaffre79 states, ‘dropouts and a poor conduct of the study might direct the results of the two arms towards each other’. Although this can be interpreted as an indication that the per-protocol analysis is the conservative choice for non-inferiority studies, Garrett80 states that ‘The perceived conservative nature of the PP [per protocol] population appears to be much more a reflection of reduced patient numbers than the presence of bias, while bias can be in either direction depending on the pattern of violations’. Moreover, with two active treatments, it may be more likely that any bias affecting both treatments is reduced, in comparison with a placebo-controlled trial. 63
Prominence
Non-inferiority was declared only if both the ITT and the per-protocol analyses supported a non-inferiority conclusion. The Committee on Proprietary Medical Products Points-to-Consider and several other papers support this. 63 The requirement to declare non-inferiority in both the ITT and the per-protocol analyses emphasised the adherence to treatment protocol and the minimisation of exclusions, maintaining power.
Outcomes
Primary outcome
The primary outcome was BCVA in the trial eye, measured in ETDRS letter score at 4 m at 100 weeks. Measurements of BCVA at milestone visits were included in the analysis of the primary outcome. Any BCVA measurement was excluded from the analysis if it is was > 3 SDs below the mean at that time point (including all measurements) and taken within 3 months of the occurrence of a vitreous haemorrhage, or was from another cause unrelated to maculopathy secondary to CRVO (e.g. NVG).
Secondary outcomes
The secondary efficacy outcome measures are listed in the following sections according to their type of variable. They were formally analysed at 52 and 100 weeks, but also measured at other time points.
Continuous outcome variables
-
Visual acuity and clinical outcomes:
-
change from baseline in ETDRS letter score measured at 4 m at 52 weeks
-
change from baseline in mean OCT CST at 52 and 100 weeks
-
change from baseline in macular volume at 52 and 100 weeks
-
number of injections performed in the trial eye by 100 weeks
-
change in retinal non-perfusion as assessed by mean disc area of non-perfusion at 100 weeks.
-
-
Patient-reported outcomes:
-
National Eye Institute VFQ-25 composite score, distance and near subscales at 52 and 100 weeks.
-
quality of life (measured using the EQ-5D and the EQ-5D-V) at 52 and 100 weeks.
-
-
Economic reported outcomes (detailed in the health economics analysis plan):
-
quality-of-life scales (measured using the VFQ-25, the EQ-5D and the EQ-5D-V) at 0, 12, 24, 52, 76 and 100 weeks.
-
resource use at 0, 12, 24, 52, 76 and 100 weeks.
-
Categorical outcome variables
-
Visual acuity and clinical outcomes:
-
participants with ≥ 15 ETDRS letter improvement (appreciable visual gain), ≥ 10-letter improvement, < 15-letter loss and ≥ 30-letter loss (severe visual loss) at 52 and 100 weeks
-
participants with ≥ 73 ETDRS letters, or > 6/12 Snellen equivalent (i.e. approximate driving visual acuity), ≤ 58 letters (≤ 6/24 Snellen equivalent) and ≤ 19 letters (≤ 3/60 Snellen equivalent) (CVI partial and severe visual impairment) at 52 and 100 weeks
-
participants with OCT CST of < 320 µm (on the Spectralis, or of < 300 µm on other machines) at 52 and 100 weeks (key guide to subsequent NHS clinical practice)
-
participants with the anatomical OCT features of diffuse intraretinal oedema, intraretinal cystic change, subretinal fluid or vitreomacular interface abnormality (either vitreomacular traction or epiretinal membrane) over time and at 100 weeks
-
participants with a change in retinal non-perfusion at 100 weeks.
-
-
Safety and tolerability:
-
prevalence of local and systemic side effects at 100 weeks
-
participants who were persistent non-responders and who developed anterior and posterior segment neovascularisation at 100 weeks.
-
Subgroup variables
Three subgroup variables were considered: (1) baseline visual acuity (low, moderate and high: ≤ 38 letters, 39–58 letters and 59–78 letters, respectively), (2) disease duration (< 3 months or ≥ 3 months) and (3) ischaemic compared with non-ischaemic. These variables were based on the fact that the visual gain in the worse-vision group may be higher than that achieved by the better-vision group, and this effect may differ between arms. The shorter the duration of disease, the better the visual acuity outcomes, and this may have varied between treatment arms.
Outcomes requiring derivation
The VFQ-25 is a validated tool for assessing vision-related quality of life. It consists of a base set of 25 vision-targeted questions, representing 11 vision-related subscales, plus an additional single-item question rating general health. The overall composite score is computed as the simple average of the vision-targeted subscale scores, excluding the general health rating question. The overall score can range from 0 (worst possible score) to 100 (best possible score).
The EQ-5D and the EQ-5D-V
The EQ-5D is a generic instrument for describing and valuing health. It is based on a descriptive system that defines health in terms of five dimensions (mobility, self-care, usual activities, pain/discomfort and anxiety/depression). Each dimension [in the EuroQol-5 Dimensions, five-level version (EQ-5D-5L)] has five response categories corresponding to ‘no problems’, ‘slight problems’, ‘moderate problems’, ‘severe problems’ and ‘unable to/extreme problems’. A preference-based score ranges from states worse than dead (< 0) to 1 (full health), anchoring dead at 0. In addition, the EQ-5D includes a visual analogue scale, which records a respondent’s self-rated health on a vertical scale where the end points are labelled ‘best imaginable health state’ (marked as 100) and ‘worst imaginable health state’ (marked as 0). The EQ-5D-V is similar to the EQ-5D-5L, but with another dimension (vision) added to overcome perceived inadequacies in a particular population.
More information is given in Chapter 4, Health-related quality-of-life measures.
Defining outliers
Outliers are observations that have extreme values relative to other observations under the same conditions. An outlier was defined as a data point at least 4 SDs from the mean of its distribution of values observed across other participants. A ‘bivariate outlier’ for checking was defined as a pair of successive serial data points of the same measure for a participant whose difference was at least 4 SDs from the mean of all participants’ such differences. Simple plots of successive pairs of serial measures were used throughout the 24-month period to help identify outliers for data-checking. 63
Handling outliers
Outliers were identified for further investigation by looking at the distributions of the data using histograms, scatterplots or box plots. Univariate tests for the compatibility of the distribution with a normal distribution were not undertaken because they can be too sensitive to departures that are often not relevant to the comparison of means (central limit theorem).
Once an outlier was found, a masked member of the team with sufficient clinical experience was involved in the decision about whether a data value was impossible or implausible or plausible. If an outlier was impossible, then it was set to missing. If an outlier was clinically plausible, then the outlier remained. If an outlier was clinically implausible (but possible), then it was not ignored or deleted, but was retained for the ITT analysis. If outliers remained in the distribution of a variable, then data transformations or non-parametric methods of analysis were considered. A sensitivity analysis was undertaken to check whether or not the outlier was influential by obtaining results with and then without the inclusion of the outlier. If the conclusions changed, then this was noted. 63
Baseline comparability of randomised groups
Baseline descriptions of participants by treatment and overall were summarised. No significance testing was carried out as any differences found might have been chance-generated and not for hypothesised reasons. Continuous variables, such as OCT CST values and VFQ-25 scores, were summarised using means and SDs and/or medians and interquartile ranges (IQRs) for variables presenting a skewed distribution. Categorical variables, such as the proportion of participants gaining ≥ 15 BCVA letters or participants with OCT CST of < 320 µm, were described using numbers and percentages.
Comparison of rates of adherence and follow-up
High compliance and low attrition rates were anticipated for this trial based on previous clinical trial experience. In CRUISE (a study on CRVO), 91.6% of participants completed the active treatment arms at 12 months, and withdrawals were mainly due to physician and patient decisions. 8 A cumulative dropout of approximately 15% by year 2 was predicted for LEAVO and this was reflected in the sample size calculations. Nevertheless, compliance rates and attrition rates were compared and reported by arm using Fisher’s exact test.
Analysis covariates
The ICH E9 guideline78 recommends that consideration be given to accounting for randomisation stratifiers by adjusting for them as covariates in the linear model. This tends to improve the precision of estimated treatment effects. Therefore, for continuous outcomes, the analysis included adjustment for the randomisation stratifiers of screening BCVA letter score (three levels) and disease duration (two levels). This excluded the third stratifier of previous treatment (eye treatment naive vs. had received previous treatment), because the numbers of participants who had received previous treatment was very small; this was approved in the statistical analysis plan [see www.journalslibrary.nihr.ac.uk/programmes/hta/119203/#/documentation (accessed 14 July 2020)] by the TSC.
Baseline
The corresponding baseline measure for a continuous outcome is also often predictive of the outcome at follow-up. Therefore ‘baseline’ (if a baseline measure was collected) was included as an additional covariate when modelling continuous outcomes. 63 This was the case for visual acuity and CST.
Statistical model
The following description of the statistical analysis was applied to obtain results for each of the two investigational treatments, bevacizumab and aflibercept, compared with the standard treatment, ranibizumab.
Primary outcome analysis
Part of this section is reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. The text includes minor additions and formatting changes to the original.
The primary efficacy measure was the change from baseline in refracted BCVA in the trial eye, using the ETDRS letter score, at 100 weeks. The continuous primary outcome was a participant’s longitudinal change in BCVA from baseline to 100 weeks. As more fully described later, this baseline is adjusted for as a continuous covariate. This analysis approach gives results equivalent to those of an approach in which the primary outcome is instead defined to be the cross-sectional 100-week measurement in the same participants. Of these two equivalent approaches, we chose to analyse BCVA and other continuous outcomes at the cross-sectional measurement point. This is convenient, because it means that, if a baseline measurement is missing in a participant with a 100-week outcome, the end point is not considered to be missing. The primary outcome may, therefore, be referred to later as the 100-week visual acuity, rather than as the change in BCVA from baseline to 100 weeks.
The primary outcome was analysed using a LME model incorporating the five post-baseline measurements of the refracted BCVA outcome (at 12, 24, 52, 76 and 100 weeks). This mixed model was, by definition, a mix of random- and fixed-effect terms. The random effect in the model was the participant, represented as a random intercept at each follow-up time point, with allowance for within-participant correlation in the adjusted post-baseline outcomes. The fixed effects in the model were the main effect terms for arm; the two stratifiers, visual acuity and disease duration; ‘time’; and the baseline of the outcome and its missing indicator required for the missing indicator method. The other fixed effects in the model were the interactions between ‘time’ and each of the other fixed effects in the model. This model allowed the treatment effect to be formally tested at 52 weeks and at the primary time point of 100 weeks, and estimated at 24 and 76 weeks. 63
Intention-to-treat strategy
Outcome data were valid and included if the BCVA measure was refracted. All randomised subjects who provided at least one post-baseline valid measurement were included. 63
Per-protocol analysis
For the analysis of the primary outcome, the mixed-effects model was refitted in a reduced per-protocol population, as described in Chapter 2, Per protocol. 63 Only valid (refracted) measurements were included, and so the per-protocol analysis was a subset of the outcome measurements in the 52- and 100-week ITT analysis LME model.
Concluding non-inferiority
Non-inferiority was concluded only if this had been declared by both the ITT analysis and the per-protocol analysis at 100 weeks. Non-inferiority was also assessed secondarily in the ITT and per-protocol populations at 52 weeks from the same models. Non-inferiority was declared if the estimated 95% CI for the difference in means lay wholly above the margin of –5 letters in both the ITT and per-protocol analysis models, primarily at 100 weeks and secondarily at 52 weeks.
Superiority
If non-inferiority was concluded, superiority was assessed from the ITT LME model by reporting the p-value from the two-sided test of the hypothesis of a zero difference in population means using a 5% significance level without the need for correction for multiple testing. 63 In addition, it was planned that, if both investigative treatments were considered non-inferior to the standard treatment at 100 weeks, the investigative treatments would be assessed for superiority relative to each other.
Subgroup analysis
The two subgroup variables were assessed by extending the primary outcome model to include an interaction between arm and each categorical subgroup variable. 63 Subgroup variables with more than two categories that were ordinal were entered as linear in the interaction. The treatment effects were presented in each subgroup category with a 95% CI.
Sensitivity to missing data
An expert missing-data group concluded that, rather than statisticians reacting to missing data at the end of a trial, there should be comprehensive, proactive planning for handling missing data at the stage of designing trials. The group recommended that there should be consideration of missing-data mechanisms (e.g. missing at random), and, if the missing data may be informative, that appropriate sensitivity analyses be undertaken to investigate the robustness of the inferences to the different assumptions made by the main analysis. It has also been recommended that analyses allowing for non-response and low intervention uptake (or compliance) are best specified in advance and included in the analysis plan. As it is expected that compliance will be high from the fear of loss of sight, and as non-inferiority is concluded only when declared in both a compliant per-protocol population and a less compliant ITT population, the focus was the handling of missing data. 63
A sensitivity analysis was undertaken to assess the possibility of alternative plausible values of treatment effect arising from potential mishandling of missing data in the primary analysis model.
The LME model for the primary outcome analysis described above was the first of a two-part approach called the ITT strategy, in which a second analysis examined the sensitivity of the results to missing data in the full randomised, ITT population. This met the ideal of ITT. The approach to missing data taken in the trial followed the published implementation paper81 of the ITT strategy. This was then also applied again to the per-protocol population so that the non-inferiority conclusion could be reassessed under the sensitivity analysis. 63
For the sensitivity analysis, we prespecified a range for best visual acuity from –20 letters to 20 letters, over which the mean of the unobserved outcome data might depart (or be different) from the mean of the observed outcome data. 82 In other words, this range could be thought of as the extent to which a typical subject for whom data are missing may, on average, have had a different estimated treatment effect compared with the corresponding subject for whom outcome data were observed (given the same baseline covariates and follow-up data in the LME model). The range (–20 to 20 letters) was chosen to represent both negative and positive departures that could potentially arise as the ‘net effect’ of alternative reasons that may be unknown, such as dropout as a result of no anticipated further improvement, or dropout as a result of no improvement so far, together with no anticipated achievable improvement. 63
This range of 40 letters (from –20 to 20) was generously wide for exploring the sensitivity of the main results to departures from the missing-at-random assumption, because 20 letters (as the maximum departure in either direction) is larger than the detectable between-arm treatment effect of 3 lines (15 letters) seen in superiority trials (difference in means), which is a sizeable shift in the mean of the distribution for dropouts, compared with completers.
At the end of the trial, the fractions of individuals for whom data were missing for visual acuity at 100 weeks were available in each arm: fi (for intervention) and fc (for control). The parameter representing excess visual acuity in those missing, compared with those observed, δ, will take values by passing across the range –20 to 20. Three scenarios were undertaken in the sensitivity analysis. 77,81,82 These reflected whether or not departures from the missing-at-random assumption applied in the intervention arms only (aflibercept and bevacizumab), in the control arm only (ranibizumab), or in both arms equally and in the same direction (thereby potentially cancelling out across the sensitivity range, if the dropout rate were to be the same in both arms):63
-
Scenario 1 – the treatment effect from the LME model will be increased by fiδ.
-
Scenario 2 – the treatment effect from the LME model will be increased by –fcδ.
-
Scenario 3 – the treatment effect from the LME model will be increased by (fi – fc)δ.
Sensitivity analysis to use of concomitant treatments
The use of concomitant treatments was monitored by the DMEC. 63 It was planned that, if necessary, a sensitivity analysis would be undertaken to examine the robustness of the 100-week per-protocol analysis to the use of concomitant treatments.
Secondary outcome analysis
Secondary outcome analyses (see Appendix 3, Table 28) were on an ITT basis only. All tests were two-sided at the 5% significance level and were interpreted cautiously, with a focus on interpreting effect sizes with 95% CIs. Safety outcomes were reported as unadjusted patient proportions and as rates within and between arms, with 95% CIs, using exact methods when appropriate. Significance tests were used sparingly and were restricted, when possible, to addressing stated hypotheses.
Analysis of continuous outcomes
As for the primary outcome, the analyses of continuous secondary outcomes were compared between arms at 100 weeks using the LME model. The baseline was adjusted for as a covariate, for outcomes for which this was collected at baseline. The missing indicator method was used when there were missing data at baseline. The remaining stratifiers were adjusted for in their categorical form. Time was represented as categorical contrasts in main effect form and in interaction with all other fixed effects. For skewed outcomes, 95% CIs were obtained using the non-parametric bootstrap percentile method. 63
Analysis of binary outcomes
For the binary outcomes, such as the proportion of participants with a ≥ 15 ETDRS letter improvement, differences between two proportions with 95% CIs have been used. Safety outcomes have been reported as unadjusted patient proportions and as rates within and between arms, with 95% CIs, using exact methods when appropriate. 63
Safety meta-analysis
It was not possible to perform a safety meta-analysis because of the lack of comparative outcome data for anti-VEGF therapy in CRVO. Two other comparative studies were completed during LEAVO: the multicentre SCORE227 clinical trial, which compared aflibercept and bevacizumab, given by mandated monthly injection over 6 months, and a small comparative study59 of aflibercept versus bevacizumab in 50 patients with MO due to CRVO who were followed up for 12 months. The latter trial did not publish any tabulated AE data and was discounted. A direct comparison was made with the SCORE2 safety data by comparing them with the first 6 months of LEAVO safety data; this information is presented in the results (see Chapter 3, Comparison with SCORE2 safety data).
Patient and public involvement
As a result of consulting the user involvement officer from the Research Design Service London prior to trial start-up, we (1) consulted the Diabetes Research Network online lay member panel, (2) met with the Central and East London Comprehensive Local Research Network (CLRN) lay member group and (3) formed a service user advisory group of RVO patients. They were asked to comment on the non-expert summary, asked to comment on a brief Microsoft PowerPoint® (Microsoft Corporation, Redmond, WA, USA) overview of the project, asked specific questions and asked to give comments. Overall, they were very supportive; felt that the trial was of benefit to patients; and said that they would definitely participate, although they felt that the dexamethasone intravitreal implant (Ozurdex®; Allergan plc, Dublin, Ireland), originally intended to be a trial arm, should be excluded because of its limited efficacy and side-effect profile. In addition, they thought that aflibercept should be included as it may reduce the frequency of visits and invasive procedures (e.g. dilating and checking the non-trial eye at each visit, which should be avoided when possible), and that they would wish to help in the development of the patient information sheet. This feedback led to us removing Ozurdex from the project; including aflibercept as a third trial arm; minimising trial research visits to six in 2 years; and not dilating the non-trial eye at each visit, to help participants work and commute after their trial visit and to enhance our participant retention activities. The UK RVO service user group helped in the development of the patient information sheet and consent form, reviewing and refining these to make them more accessible and easily understood by all potential participants. One member of the patient group became a member of the TSC, attending every meeting and actively contributing to each.
Once the LEAVO clinical and health economic outcomes were available, the members of the CRVO service user group at Moorfields, additional RVO patients, members of the renamed Barts Health/Queen Mary University of London (QMUL) lay panel and patients with a history of eye disease from the Barts Health/QMUL extended users group were sent a cover letter and questionnaire regarding the trial, which had been reviewed and agreed with the Barts Health/QMUL lay panel chairperson and Moorfields Eye Hospital Biomedical Research Centre patient and public involvement lead. See Chapter 3, Patient and public involvement, for the results. A member of the Royal National Institute of Blind People served as a member of the TSC.
Trial committees
Trial Steering Committee
The TSC was the committee responsible for monitoring the overall integrity, conduct and safety of the trial. It monitored trial progress, investigated any SAEs, and took account of regular reports from the DMEC and communication from the Trial Management Group (TMG). Ultimate responsibility for any decision required on the trial’s continuation lay with the TSC. The TSC comprised an independent chairperson, a professor of statistics, an independent ophthalmologist and general physician, a consultant in public health, a senior Department of Health and Social Care policy-maker and two patient representatives. TSC meetings were held at least annually and arranged by the chief investigator and the trial manager in conjunction with the chairperson. For a list of committee members, see Appendix 2. A representative of Moorfields Eye Hospital (the sponsor) was invited to each meeting. 2
Data Monitoring and Ethics Committee
An independent DMEC of three individuals, one professor of statistics and two retina specialists, met regularly to safeguard the interests of trial participants, assess the safety and efficacy of the interventions during the trial, and monitor the overall conduct of the clinical trial (see Appendix 2). Its terms of reference were to receive and review the progress and accruing data of the trial and to provide the TSC with advice and recommendations on trial conduct. The trial would have been discontinued on the basis of new safety information, or for other reasons given by the DMEC and/or TSC, sponsor, regulatory authority or Research Ethics Committee concerned. All data reviewed by the DMEC determined safety issues. All serious adverse reactions were reported to the KCTU within 24 hours of learning of their occurrence. 2
Trial Management Group and site monitoring
The TMG was responsible for monitoring the delivery of the trial on a day-to-day basis, and was supported and managed via the KCTU. The TMG membership consisted of the chief investigator, the co-lead, the trial manager, the data manager, the lead and trial statisticians and senior members of the KCTU. Other members of the wider research team were also invited on a meeting-by-meeting basis, depending on the scope covered. Trial conduct and data collected were monitored by a combination of central review and site monitoring visits to ensure that these were in accordance with good clinical practice. Trial site monitoring was undertaken by the trial manager, the assistant trial manager and an experienced KCTU trial monitor. The main areas of focus were consent, SAEs and essential documents in trial site files.
Site monitoring included:
-
reviewing all consent forms in the site file and medical notes
-
source data verifying SAEs against medical records
-
source data verifying a proportion of the primary outcome measure against medical records
-
checking essential documents in the investigator site file and trial files.
Central reviews included:
-
ensuring accuracy and completeness of all applications for trial authorisations and submissions of progress/safety reports, prior to submission
-
ensuring that all documentation essential for trial initiation was in place prior to site authorisation
-
reporting and following up all monitoring findings with the appropriate persons in a timely manner.
The investigators and institutions also permitted trial-related monitoring, audits, Research Ethics Committee review and regulatory inspections, providing direct access to source data/documents. Trial participants were informed of this during the informed consent discussion. Participants consented to provide access to their medical notes.
Approvals, reporting and compliance
The trial was approved by the National Research Ethics Committee Service London South East (reference number 14/LO/1043); Clinical Trials Authorisation was given by the MHRA (number 11412/0220/001-0005), and the European Union Drug Regulating Authorities Clinical Trials (EudraCT) number was 2013-003272-12. The trial was run using the standard operating procedures of the sponsor, Moorfields Eye Hospital NHS Foundation Trust. The sponsor provided the oversight of the trial, and the KCTU collaborated with the sponsor to ensure efficient trial delivery. The trial was reported in accordance with the Consolidated Standards of Reporting Trials statement.
Summary of changes made to protocol
After initial substantial amendments [substantial amendment (SA) 1 to SA3] at commencement of the trial clarified the handling of several key issues (e.g. pregnancy, contraception and nurse injectors), subsequent substantial amendments mainly dealt with the addition of sites or a change in principal investigator (see Appendix 3, Table 30). SA6, approved by the Research Ethics Committee on 11 February 2016, included changes to the protocol, in particular the eligibility criteria, to increase recruitment to the trial. The key change requested by the trial team was to increase the upper limit of permissible visual acuity at screening from 73 (Snellen equivalent 6/12) to 78 (Snellen equivalent 6/9) letters. This was to increase recruitment across all trial sites because, as the protocol stood, patients in clinical practice with a visual acuity of 6/9 may have been excluded from the trial, as their visual acuity was too good, and go on to receive treatment in the NHS, and be lost to the trial. This change to the upper limit would potentially allow patients with a visual acuity of 6/9 to enrol in the trial. However, the DMEC and TSC statisticians were concerned that this could introduce a ceiling effect if an abnormally large number of patients with good visual acuity and limited potential to improve were randomised, and could even lead to the trial erroneously declaring non-inferiority. Thus, the statisticians stated that they could not agree to this change unless additional data from other studies was obtained by the chief investigator to determine whether or not a significant ceiling effect was likely to occur. After consultation with the relevant trial sponsors and/or chief investigators, the LEAVO chief investigator and co-lead were able to provide the DMEC and TSC with unpublished results from recent clinical trials (the CRYSTAL55 study of RVO and the US DRCR.net Protocol T sudy54 of DMO) that showed no significant ceiling effect and that a large proportion of such cases gained significant visual acuity. Based on this new information, the TSC and DMEC allowed the protocol change. Additional changes to the eligibility criteria were approved, including an increase (from three to six) in the number of anti-VEGF injections a participant could have received prior to randomisation. The rescreening interval was reduced from 4 to 2 weeks because a number of participants who failed initial screening sought treatment elsewhere before rescreening was possible.
Chapter 3 Clinical results
Participant flow
The original contract commenced on 1 May 2014, with recruitment to start on 1 November 2014. An early contract variation was requested by the LEAVO team, and approved by NIHR, for the contract to commence on 1 June 2014 and for recruitment to start on 1 December 2014. Recruitment was predicted to take 18 months and, therefore, was to finish on 31 May 2016, with last participant, last visit to take place by 31 May 2018, and the trial to close on 31 October 2018. The first participant was randomised on 12 December 2014, but the last was randomised on 16 December 2016, almost exactly 24 months later. As a result, a contract variation was sought to extend the trial by 6 months so that the last participant, last visit would occur by 30 November 2018, and the trial would close on 30 April 2019. The last participant, last visit was actually on 21 November 2018.
Therefore, between December 2014 and December 2016, 586 patients were assessed across 44 UK NHS hospitals (see Appendix 1, Table 27) for eligibility. Of these patients, 123 were excluded: 117 were ineligible, one withdrew consent and five did not proceed for other reasons. Therefore, 463 were managed on protocol (see Appendix 3, Table 29), randomly assigned to receive ranibizumab (n = 155), intravitreal aflibercept (n = 154) or bevacizumab (n = 154), and constituted the ITT population Randomisation was balanced across treatment groups, across hospital sites and within baseline visual acuity strata. The per-protocol population consisted of 145 participants in the ranibizumab arm, 146 in the aflibercept arm and 152 in the bevacizumab arm. Among the ITT population, the 100-week visit was completed by 135 participants in the ranibizumab arm, 133 in the aflibercept arm and 139 in the bevacizumab arm; among the per-protocol population, the same visit was completed by 133 participants in the ranibizumab arm, 128 in the aflibercept arm and 139 in the bevacizumab arm (Figure 2).
FIGURE 2.
The LEAVO CONSORT diagram. a, Models include all participants who have had at least one follow-up visit. PP, per protocol. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.

Recruitment
Overview
NIHR acknowledges the need for experienced trial management and recommends the involvement of a specialised clinical trials unit to conduct trials. We were fortunate to have the multidisciplinary team from the KCTU participate in the trial. As a LEAVO collaborator, the team provided a trial manager, deputy trial manager and experienced monitors, in addition to a senior and a junior statistician, and the expertise of their core team, including the Clinical Trials Unit operations manager, senior data manager and trial methodologist. All these members attended TMG, TSC and DMEC meetings, when appropriate. In addition, the KCTU team members were all available for advice and guidance on a daily basis; working in conjunction with the trial manager, the KCTU was, ultimately, the cornerstone of the trial. 63 It recognised the need to open as many sites as quickly as possible and its senior team spent many hours with the trial manager, ensuring that she was fully familiar with the trial and was able to begin site initiations before recruitment commenced on 1 December 2014. The largest and most experienced sites (e.g. Moorfields and Leeds) were initiated first. Unfortunately, a few weeks before the initiation of the first site, the original trial manager was absent on sick leave and she announced her resignation at the beginning of December 2014. Not unexpectedly, this had a significant impact on site initiation and could have led to very prolonged trial delays. Fortunately, an experienced assistant trial manager had just been appointed and agreed to step up to the trial manager position within a few days of starting. Quite understandably, he took time to familiarise himself with the trial protocol and procedures; therefore, the trial fell significantly behind with site initiations and recruitment. The low point was 39 participants recruited by the end of May 2015, against a predicted target of 76 (51%). However, the new trial manager began to recover the situation in the second quarter of 2015, and the number of site initiations increased: we initiated only eight sites in the first 4 months of recruitment, compared with 13 sites in the succeeding 2 months. As a result, actual recruitment kept pace with predicted recruitment in October, November and December 2015. By November 2015, that is after 12 months of recruitment, we had opened 38 sites, against a target of 40, and recruited 176 participants, against a target of 268 (66%). An additional eight sites were subsequently initiated, to give 46 greenlighted sites open in the first quarter of 2016. By 31 May 2016, when recruitment should have been completed, we had recruited 320 participants, against a target of 459 (70%); by December 2018, we had completed recruitment almost exactly 6 months behind schedule (Figure 3). Table 1 shows the number of participants recruited each month by site, and Table 2 shows the number of participants whom each site recruited per trial arm.
FIGURE 3.
Actual vs. projected recruitment per month.

| Site | Participants (n) | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2014 | 2015 | 2016 | Total | |||||||||||||||||||||||
| December | January | February | March | April | May | June | July | August | September | October | November | December | January | February | March | April | May | June | July | August | September | October | November | December | ||
| Moorfields Eye Hospital | 2 | 3 | 1 | 5 | 3 | 5 | 4 | 2 | 4 | 6 | 3 | 4 | 4 | 3 | 2 | 2 | 3 | 0 | 4 | 4 | 3 | 2 | 5 | 3 | 77 | |
| King’s College Hospital | 1 | 1 | 2 | 1 | 1 | 6 | ||||||||||||||||||||
| Wolverhampton Eye Infirmary, New Cross Hospital | 4 | 2 | 2 | 4 | 3 | 2 | 2 | 1 | 1 | 21 | ||||||||||||||||
| St Paul’s Eye Unit, Royal Liverpool University Hospital | 2 | 2 | 2 | 1 | 2 | 2 | 2 | 13 | ||||||||||||||||||
| University Hospital Southampton | 2 | 4 | 3 | 2 | 1 | 1 | 1 | 14 | ||||||||||||||||||
| Royal Victoria Hospital, Belfast | 1 | 3 | 4 | 1 | 1 | 1 | 1 | 1 | 1 | 14 | ||||||||||||||||
| Royal Blackburn Hospital | 1 | 1 | 1 | 1 | 4 | |||||||||||||||||||||
| Bradford Royal Infirmary | 1 | 3 | 2 | 3 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 2 | 18 | |||||||||||||
| Sussex Eye Hospital | 1 | 4 | 1 | 1 | 2 | 1 | 1 | 11 | ||||||||||||||||||
| Bristol Eye Hospital | 2 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 13 | |||||||||||||||
| West Suffolk Hospital | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 1 | 11 | ||||||||||||||||
| Torbay Hospital | 1 | 1 | 2 | 2 | 1 | 7 | ||||||||||||||||||||
| Essex County Hospital | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 11 | |||||||||||||||
| Hospital of St Cross, Rugby | 1 | 2 | 1 | 1 | 5 | |||||||||||||||||||||
| Birmingham and Midlands Eye Centre | 2 | 1 | 4 | 3 | 1 | 1 | 4 | 16 | ||||||||||||||||||
| Kent and Canterbury Hospital | 1 | 1 | 1 | 1 | 4 | |||||||||||||||||||||
| Frimley Park Hospital | 2 | 1 | 1 | 1 | 1 | 2 | 3 | 1 | 1 | 1 | 1 | 15 | ||||||||||||||
| Whipps Cross University Hospital | 1 | 1 | ||||||||||||||||||||||||
| James Paget University Hospital | 1 | 1 | 2 | 1 | 1 | 1 | 7 | |||||||||||||||||||
| Royal Surrey County Hospital | 3 | 1 | 4 | |||||||||||||||||||||||
| Harrogate District Hospital | 1 | 1 | 2 | |||||||||||||||||||||||
| York Teaching Hospital | 1 | 1 | 1 | 1 | 1 | 5 | ||||||||||||||||||||
| Darlington Memorial Hospital | 1 | 1 | 1 | 1 | 4 | |||||||||||||||||||||
| St James’s University Hospital, Leeds | 1 | 2 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 14 | |||||||||||||
| Hillingdon Hospital | 1 | 1 | 2 | 2 | 1 | 7 | ||||||||||||||||||||
| Eye, Ear and Mouth Unit, Maidstone Hospital | 2 | 1 | 2 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | 14 | |||||||||||||||
| Manchester Royal Eye Infirmary | 1 | 1 | 1 | 1 | 2 | 2 | 1 | 9 | ||||||||||||||||||
| Royal Victoria Infirmary, Newcastle upon Tyne | 2 | 2 | 3 | 1 | 2 | 1 | 1 | 12 | ||||||||||||||||||
| Luton and Dunstable University Hospital | 1 | 1 | 1 | 1 | 1 | 5 | ||||||||||||||||||||
| Cardiff Eye Unit, University Hospital of Wales | 1 | 1 | 1 | 2 | 5 | |||||||||||||||||||||
| Sunderland Eye Infirmary | 1 | 1 | 2 | 1 | 3 | 3 | 1 | 3 | 2 | 2 | 2 | 21 | ||||||||||||||
| Royal Glamorgan Hospital | 1 | 2 | 1 | 1 | 1 | 1 | 2 | 3 | 1 | 1 | 14 | |||||||||||||||
| Royal Hallamshire Hospital | 1 | 2 | 1 | 1 | 3 | 1 | 2 | 1 | 1 | 13 | ||||||||||||||||
| Addenbrooke’s Hospital | 1 | 1 | 2 | 2 | 1 | 1 | 3 | 11 | ||||||||||||||||||
| Gartnavel General Hospital | 1 | 1 | 1 | 1 | 1 | 5 | ||||||||||||||||||||
| Royal Bolton Hospital | 1 | 1 | 1 | 2 | 1 | 6 | ||||||||||||||||||||
| Calderdale Royal Hospital | 1 | 1 | 1 | 1 | 2 | 6 | ||||||||||||||||||||
| Leicester Royal Infirmary | 1 | 1 | 1 | 1 | 1 | 5 | ||||||||||||||||||||
| Norfolk and Norwich Hospital | 2 | 1 | 1 | 4 | ||||||||||||||||||||||
| Cheltenham General Hospital | 1 | 1 | 1 | 1 | 1 | 1 | 6 | |||||||||||||||||||
| Hull Royal Infirmary | 3 | 1 | 1 | 1 | 6 | |||||||||||||||||||||
| Western Eye Hospital | 3 | 1 | 1 | 1 | 2 | 2 | 10 | |||||||||||||||||||
| James Cook University Hospital | 1 | 2 | 3 | |||||||||||||||||||||||
| Princess Alexandra Hospital, Harlow | 1 | 1 | 2 | 4 | ||||||||||||||||||||||
| Total per month | 2 | 4 | 5 | 8 | 11 | 9 | 22 | 14 | 22 | 19 | 30 | 30 | 29 | 21 | 23 | 28 | 24 | 19 | 34 | 24 | 17 | 14 | 25 | 24 | 5 | 463 |
| Cumulative total | 2 | 6 | 11 | 19 | 30 | 39 | 61 | 75 | 97 | 116 | 146 | 176 | 205 | 226 | 249 | 277 | 301 | 320 | 354 | 378 | 395 | 409 | 434 | 458 | 463 | 463 |
| Site | Participants (n) | |||
|---|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | Total | |
| Moorfields Eye Hospital | 25 | 24 | 28 | 77 |
| King’s College Hospital | 3 | 2 | 1 | 6 |
| Wolverhampton Eye Infirmary, New Cross Hospital | 8 | 6 | 7 | 21 |
| St Paul’s Eye Unit, Royal Liverpool University Hospital | 5 | 6 | 2 | 13 |
| University Hospital Southampton | 3 | 6 | 5 | 14 |
| Royal Victoria Hospital, Belfast | 6 | 3 | 5 | 14 |
| Royal Blackburn Hospital | 0 | 1 | 3 | 4 |
| Bradford Royal Infirmary | 3 | 7 | 8 | 18 |
| Sussex Eye Hospital | 6 | 1 | 4 | 11 |
| Bristol Eye Hospital | 5 | 2 | 6 | 13 |
| West Suffolk Hospital | 6 | 4 | 1 | 11 |
| Torbay Hospital | 3 | 3 | 1 | 7 |
| Essex County Hospital | 3 | 2 | 6 | 11 |
| Hospital of St. Cross, Rugby | 1 | 1 | 3 | 5 |
| Birmingham and Midlands Eye Centre | 5 | 5 | 6 | 16 |
| Kent and Canterbury Hospital | 2 | 2 | 0 | 4 |
| Frimley Park Hospital | 5 | 5 | 5 | 15 |
| Whipps Cross University Hospital | 0 | 1 | 0 | 1 |
| James Paget University Hospital | 4 | 3 | 0 | 7 |
| Royal Surrey County Hospital | 0 | 1 | 3 | 4 |
| Harrogate District Hospital | 0 | 1 | 1 | 2 |
| York Teaching Hospital | 0 | 4 | 1 | 5 |
| Darlington Memorial Hospital | 4 | 0 | 0 | 4 |
| St James’s University Hospital, Leeds | 6 | 4 | 4 | 14 |
| Hillingdon Hospital | 2 | 2 | 3 | 7 |
| Eye, Ear and Mouth Unit, Maidstone Hospital | 5 | 5 | 4 | 14 |
| Manchester Royal Eye Infirmary | 2 | 4 | 3 | 9 |
| Royal Victoria Infirmary, Newcastle upon Tyne | 5 | 3 | 4 | 12 |
| Luton and Dunstable University Hospital | 1 | 2 | 2 | 5 |
| Cardiff Eye Unit, University Hospital of Wales | 3 | 1 | 1 | 5 |
| Sunderland Eye Infirmary | 8 | 7 | 6 | 21 |
| Royal Glamorgan Hospital | 5 | 6 | 3 | 14 |
| Royal Hallamshire Hospital | 3 | 4 | 6 | 13 |
| Addenbrooke’s Hospital | 2 | 5 | 4 | 11 |
| Gartnavel General Hospital | 0 | 3 | 2 | 5 |
| Royal Bolton Hospital | 3 | 2 | 1 | 6 |
| Calderdale Royal Hospital | 2 | 3 | 1 | 6 |
| Leicester Royal Infirmary | 2 | 1 | 2 | 5 |
| Norfolk and Norwich Hospital | 1 | 2 | 1 | 4 |
| Hull Royal Infirmary | 0 | 2 | 4 | 6 |
| Cheltenham General Hospital | 4 | 2 | 0 | 6 |
| Western Eye Hospital | 1 | 4 | 5 | 10 |
| James Cook University Hospital | 2 | 1 | 0 | 3 |
| Princess Alexandra Hospital, Harlow | 1 | 1 | 2 | 4 |
| Total | 155 | 154 | 154 | 463 |
Barriers to recruitment and corrective strategies
The following barriers to recruitment were identified:
-
Availability of trial staff, for example masked injectors and trial co-ordinators. Despite fulfilling our initial trial site requirements, several sites were unable to provide sufficient clinician unmasked injector cover (e.g. Rugby) as a result of limited staff availability, or sufficient research co-ordinator time for the trial (e.g. Addenbrooke’s and Hillingdon), the latter in some cases because NHS support costs attributable to LEAVO were not available to the local trial team. We largely resolved the former issue in a substantial amendment that allowed nurses and optometrists who were certified intravitreal injectors in standard NHS clinics to provide unmasked injector cover for LEAVO. We also approached a number of local ophthalmology CLRNs to provide additional co-ordinator time for the trial based on CLRN support costs, and received very helpful support from Rupert Bourne, CLRN National Lead for Ophthalmology, in this regard.
-
Difficulties with the protocol. The following changes were made to the protocol (see Appendix 3, Table 30) –
-
The upper limit of the visual acuity eligibility at baseline was increased from 73 (Snellen equivalent: ≈ 6/12) to 78 letters (Snellen equivalent: ≈ 6/9). Patients in clinical practice with a visual acuity of 6/9 had previously been excluded from the trial as their visual acuity was too good, and they were receiving NHS treatment instead. The change allowed patients with a visual acuity of 6/9 to enrol in the trial.
-
The inclusion criterion for diabetic retinopathy in the trial eye was changed from ‘any previously documented diabetic retinopathy or diabetic macular oedema in the study eye’ to ‘any diabetic retinopathy or diabetic macular oedema at baseline clinical examination of the study eye’. 2 This was to prevent patients being excluded from the trial who presented with a documented history of diabetic retinopathy, which may not have been reliable, rather than clinical evidence based on the trial screening examination.
-
The allowed number of previous anti-VEGF injections was increased from three to six in order to allow patients who had had longer-term treatment for MO due to CRVO (i.e. six injections) to be considered for the trial.
-
Patients who had had recent pan-retinal photocoagulation for NVE, NVD or NVI were considered eligible for the trial within 1 month of treatment rather than within 3 months, as treatment within 1 month would not have had an adverse outcome on anti-VEGF therapy 1 month later.
-
The protocol was altered to change the rescreening interval to 2 weeks, except for visual acuity eligibility, which remained at 4 weeks. Several patients had not enrolled in LEAVO because, for example, they had forgotten to take blood pressure medication, leading to high blood pressure and a screen fail. If they needed to wait 4 weeks before rescreening, as the protocol originally stated, then they typically opted for NHS treatment in the interim; being able to rescreen after 2 weeks prevented them being lost to NHS care.
-
-
Number of sites. Although we planned for 40 sites initially, four withdrew before being initiated, and so we took an early decision to add additional sites. Initially, we planned for a further 12 sites, which would have taken the total to 48 active sites. However, two of these withdrew, and 10 were greenlighted, although one failed to recruit any participants. Nevertheless, these additional sites made a very significant contribution to the last 6 months of recruitment.
-
Site equipment. Several sites had issues with equipment, in particular with wide-angled fluorescein angiography imaging devices and information technology support that allowed communication with the KCTU randomisation software and MACRO trial database, and also allowed data export to the reading centre. We worked with the sites and providers of equipment (e.g. Optos wide-angled imaging) to overcome these issues as quickly as possible.
-
Although we had held an investigator meeting prior to the trial start, a number of optometrists had not been able to attend this and required certification before a site could be greenlighted to recruit patients. To minimise certification delays, we arranged for prompt visits by either lead trial optometrist to any site to undertake optometry certification.
-
Other measures that were used to try to maximise recruitment included the following –
-
A monthly newsletter to every site detailing progress63 and acknowledging each site that had recruited one or more participants in the previous month.
-
An e-mail from the chief investigator to each site team every 2 months encouraging further recruitment.
-
A thank-you e-mail to each site from the chief investigator after each participant was recruited.
-
Reward vouchers each month to the site recruiting the most participants and ‘best site of the month’.
-
Very prompt replies to any site that had queries on any aspect of the trial. We think that this point was critical in keeping sites focused on recruitment and willing to recruit over and above their target, which was something we specifically asked large sites to do.
-
Withdrawals
Appendix 3, Table 31, shows the number of participants who did not complete the week 100 visit in the three arms, and the week of their last visit. Appendix 3, Table 32, shows the number of weeks all withdrawal participants participated in the trial and the reasons for withdrawal. Withdrawals were balanced across treatment arms; overall, more participants completed their week 100 visit [87.9% (407/463)] than had been predicted in the sample size calculation (85%).
Baseline data
Baseline characteristics were well balanced between groups for age, sex and eye involved (Table 3). In the ranibizumab, aflibercept and bevacizumab arms, the mean baseline BCVA was 53.6 (SD 15.1), 54.1 (SD 15.3) and 54.4 (SD 14.2) ETDRS letters, respectively. The numbers recruited to the three stratifier subgroups for visual acuity were equal across arms. The median duration of CRVO in each treatment group was < 1 month; the numbers of participants in the duration of CRVO subgroups of 3–6 months and > 6 months were small and so these groups were combined for analysis purposes, a change that was approved in the final version of the statistical analysis plan. Similarly, the number of participants receiving prior treatment was so small that this stratifier was not analysed. OCT CST was 731.3 µm (SD 227.6 µm), 673.2 µm (SD 189.4 µm) and 676.1 µm (SD 207.0 µm) for the ranibizumab, aflibercept and bevacizumab arms, respectively, with the apparent difference between ranibizumab and the other two groups being approximately 0.5 of a SD, and likely to be attributable to chance.
| Characteristic | Total (N = 463) | Ranibizumab (N = 155) | Aflibercept (N = 154) | Bevacizumab (N = 154) |
|---|---|---|---|---|
| Age (years), mean (SD) | 69.1 (13.0) | 69.2 (13.0) | 68.7 (13.2) | 69.3 (12.8) |
| Female, n (%) | 198 (42.8) | 70 (45.2) | 60 (39.0) | 68 (44.2) |
| Right eye was trial eye, n (%) | 226 (48.8) | 81 (52.3) | 67 (43.5) | 78 (50.6) |
| Mean (SD) BCVA letter score in the trial eyea,b | 54.1 (14.8) | 53.6 (15.1) | 54.1 (15.3) | 54.4 (14.2) |
| BCVA letter score in trial eye, n (%) | ||||
| 19–38 | 85 (18.4) | 31 (20.0) | 27 (17.5) | 27 (17.5) |
| 39–58 | 166 (35.9) | 56 (36.1) | 55 (35.7) | 55 (35.7) |
| 59–78 | 212 (45.8) | 68 (43.9) | 72 (46.8) | 72 (46.8) |
| Median (IQR) duration of CRVO (months)a | 0.9 (0.4–1.7) | 0.9 (0.5–1.8) | 0.9 (0.4–1.7) | 0.9 (0.4–1.7) |
| Duration of trial eye CRVO, n (%) | ||||
| < 3 months | 401 (86.6) | 134 (86.5) | 129 (83.8) | 138 (89.6) |
| 3–6 months | 38 (8.2) | 11 (7.1) | 19 (12.3) | 8 (5.2) |
| > 6 months | 24 (5.2) | 10 (6.5) | 6 (3.9) | 8 (5.2) |
| Previous treatment in trial eye, n (%)a | ||||
| Nil | 446 (96.5) | 148 (96.1) | 149 (96.8) | 149 (96.8) |
| Anti-VEGF therapy | 16 (3.5) | 6 (3.9) | 5 (3.2) | 5 (3.2) |
| CRVO ischaemic status at baseline (trial eye), n (%)a | ||||
| Non-ischaemic | 406 (87.9) | 137 (89.0) | 135 (87.7) | 134 (87.0) |
| Ischaemic | 56 (12.1) | 17 (11.0) | 19 (12.3) | 20 (13.0) |
| OCT (trial eye),a,c mean (SD) | ||||
| CST (µm) | 693.6 (209.8) | 731.3 (227.6) | 673.2 (189.4) | 676.1 (207.0) |
| Total volume (mm3) | 12.7 (2.8) | 13 (2.9) | 12.3 (2.6) | 12.8 (2.9) |
| Lens status (trial eye), n (%) | ||||
| Cataract | 131 (28.4) | 41 (26.6) | 44 (28.6) | 46 (29.9) |
| Pseudophakia | 68 (14.7) | 29 (18.8) | 20 (13) | 19 (12.3) |
| Blood pressure (mmHg),a mean (SD) | ||||
| Systolic | 143.0 (16.8) | 143.1 (17.6) | 142.6 (17.0) | 143.1 (15.7) |
| Diastolic | 79.7 (10.4) | 80.1 (10.2) | 79.1 (10.6) | 79.9 (10.6) |
Derivation of the intention-to-treat model and per-protocol populations
Participants included in the prespecified ITT LME model were derived as follows:
-
The BCVA data were available for 407 of 463 randomly assigned participants (ranibizumab, n = 135; aflibercept, n = 133; and bevacizumab, n = 139) at 100 weeks. Table 4 shows the available BCVA data at 12, 24, 52, 76 and 100 weeks by arm. The model included all participants who had at least one of these follow-up visits; therefore, those without follow-up data did not contribute to the analysis.
-
Only the 76-week measurement in one bevacizumab participant was excluded because of the presence of retinal detachment within 3 months of BCVA recordings, and BCVA was > 3 SDs below the mean at that time point (including all measurements).
-
Therefore, no participants were removed on this basis from the LME model analysis and the ITT and per-protocol populations were not modified by this.
-
A total of 20 participants did not meet the per-protocol definition, so 443 participants constituted the per-protocol population (see Figure 2). 63
| Visit | Mean (SD) BCVA letter score; n participants | |||
|---|---|---|---|---|
| Total (N = 463) | Ranibizumab (N = 155) | Aflibercept (N = 154) | Bevacizumab (N = 154) | |
| Screening | 54.1 (14.8); 459 | 53.6 (15.1); 153 | 54.1 (15.3); 153 | 54.4 (14.2); 153 |
| 12 weeks | 68.4 (15.8); 443 | 67.5 (16.5); 146 | 70.4 (15.1); 148 | 67.3 (15.8); 149 |
| 24 weeks | 65.8 (17.9); 432 | 65 (19.1); 145 | 67.3 (16.9); 146 | 64.9 (17.7); 141 |
| 52 weeks | 66.3 (18.4); 413 | 65.4 (19.4); 139 | 67.2 (17.6); 139 | 66.4 (18.3); 135 |
| 76 weeks | 65.9 (19.0); 397 | 65.7 (19.4); 136 | 66.2 (18.1); 128 | 65.9 (19.6); 133 |
| 100 weeks | 66.2 (19.6); 407 | 65.6 (19.9); 135 | 68.4 (17.9); 133 | 64.6 (20.8); 139 |
Outcomes and estimations
Primary outcome
The mean gain in BCVA letter score was 12.5 with ranibizumab (SD 21.1), 15.1 with aflibercept (SD 18.7) and 9.8 with bevacizumab (SD 21.4) at 100 weeks (Figure 4). First, the primary outcome at 100 weeks was unable to show that bevacizumab was non-inferior in terms of BCVA in both the ITT and per-protocol populations (Table 5). The 95% CI for the adjusted difference between arms at 100 weeks lay below the prespecified acceptable margin of –5 letters (Figure 5). Second, aflibercept was non-inferior, but not superior, to ranibizumab in terms of BCVA in both the ITT and the per-protocol populations (see Table 5 and Figure 5). The 95% CI for the adjusted difference between arms at 100 weeks lay above the prespecified acceptable margin of –5 letters (see Figure 5). The mean BCVA letter score at 24 weeks had decreased by approximately 3 letters across groups following pro re nata injections at weeks 16 and 20, when fewer injections were given (ranibizumab injections, n = 123; aflibercept, n = 76; and bevacizumab, n = 121), but increased gradually thereafter across groups to week 100, during which period participants were seen at least every 8 weeks and received injections promptly if re-treatment criteria were met (see Figure 4). Such peak-and-trough changes in visual acuity were closely mirrored by OCT trough and peak CST results over the 2-year period.
FIGURE 4.
Adjusted mean BCVA letter score across groups to 100 weeks. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.

| Mean (SE) BCVA at screening | Mean (SE) BCVA at 100 weeks; n participants | Adjusted difference between groups (95% CI) at 100 weeks | p-value for non-inferiority (p < 0.025 is significant) | p-value for superiority (p < 0.05 is significant) | ||
|---|---|---|---|---|---|---|
| Aflibercept | Ranibizumab | Aflibercept | Ranibizumab | |||
| Aflibercept vs. ranibizumab ITT | ||||||
| 54.1 (1.2) | 53.6 (1.2) | 68.4 (1.6); 133 | 65.6 (1.7); 135 | 2.23 (–2.17 to 6.63)a,b | 0.0006 | 0.32 |
| Aflibercept vs. ranibizumab per protocol | ||||||
| 55.0 (1.2) | 53.6 (1.3) | 69.5 (1.5); 128 | 65.7 (1.7); 133 | 3.49 (–0.91 to 7.88)a,c | < 0.0001 | 0.12 |
| Bevacizumab | Ranibizumab | Bevacizumab | Ranibizumab | |||
| Bevacizumab vs. ranibizumab ITT | ||||||
| 54.4 (1.1)a | 53.6 (1.2) | 64.6 (1.8); 139 | 65.6 (1.7); 135 | –1.73 (–6.12 to 2.67)b | 0.071 | 0.44 |
| Bevacizumab vs. ranibizumab per protocol | ||||||
| 54.4 (1.2) | 53.6 (1.3) | 64.6 (1.8); 139 | 65.7 (1.7); 133 | –1.67 (–6.02 to 2.68)c | 0.066 | 0.45 |
FIGURE 5.
Forest plot of the primary outcome at 100 weeks. PP, per protocol. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.

The principled sensitivity analysis for missing data supported the primary outcome results (Figures 6 and 7). The sensitivity analysis for outliers was not conducted, as there were no outliers in the ITT and per-protocol populations [see www.journalslibrary.nihr.ac.uk/programmes/hta/119203/#/documentation (accessed 14 July 2020)]. The sensitivity analysis for concomitant treatments taken by one participant in the trial supported the primary outcome results.
FIGURE 6.
Sensitivity analysis for the missing-at-random assumption in the primary outcome analysis assessing non-inferiority of aflibercept. (a) ITT; and (b) per protocol. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.
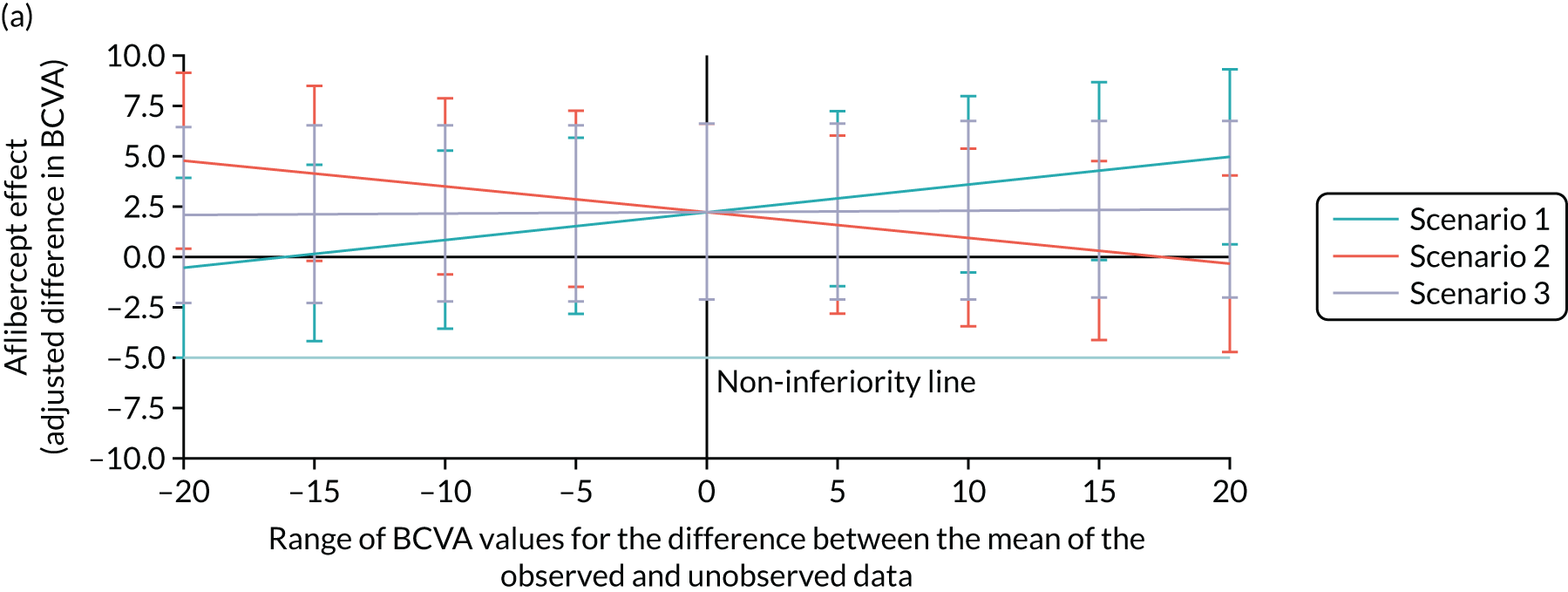
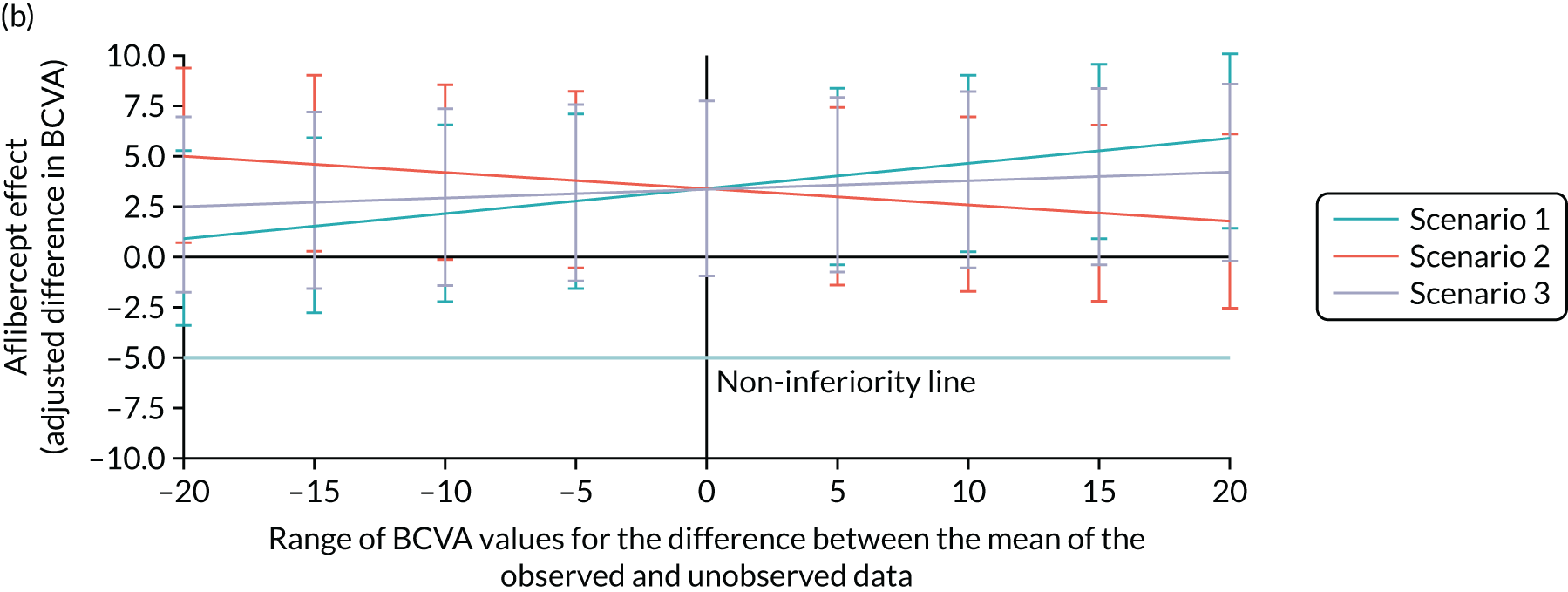
FIGURE 7.
Sensitivity analysis for the missing-at-random assumption in the primary outcome analysis assessing non-inferiority of bevacizumab. (a) ITT; and (b) per protocol. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.
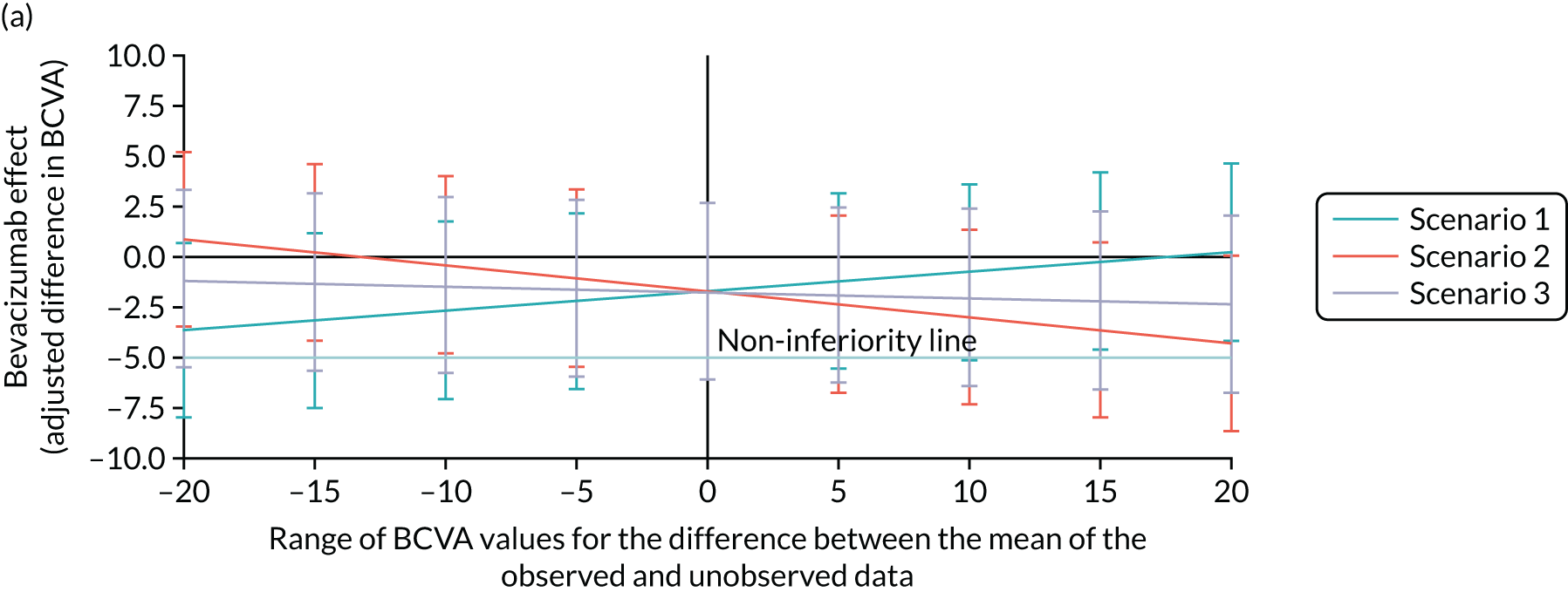
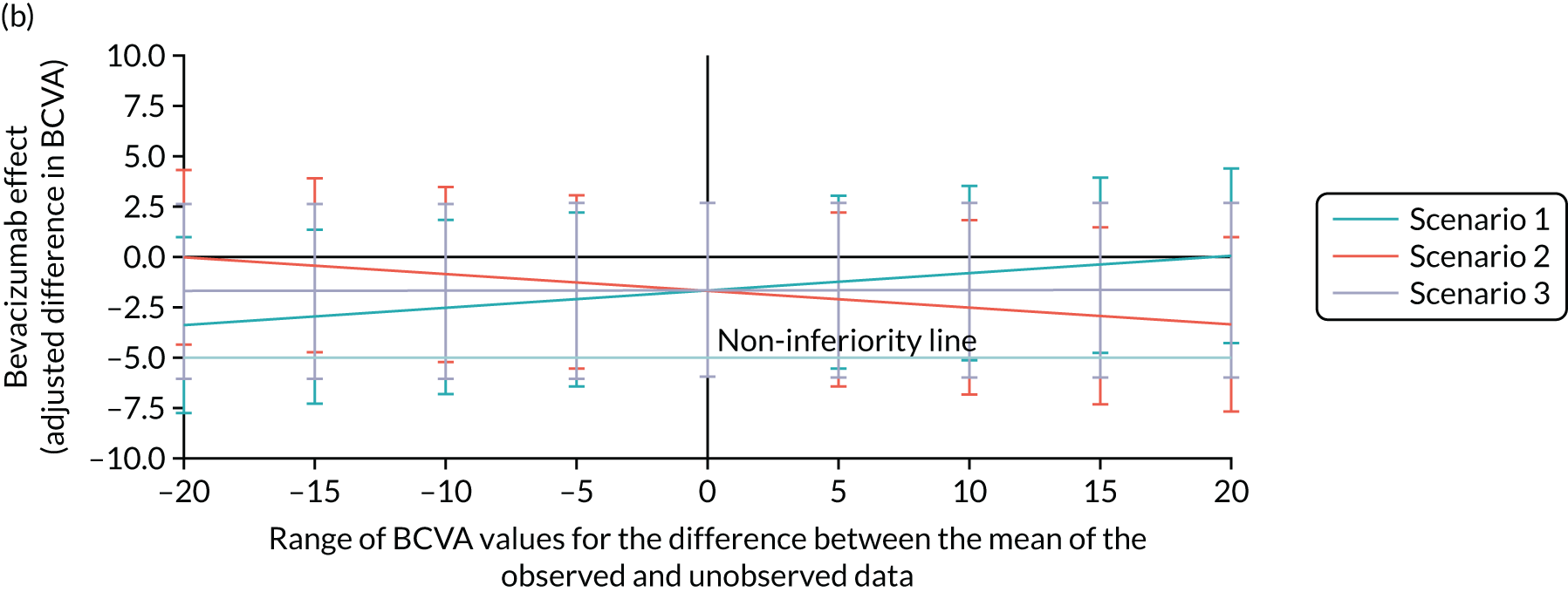
The sensitivity analysis assessed the potential impact on the treatment effect from including participants with unobserved BCVA 24-month primary outcome data in the adjusted primary outcome model. In this analysis, participants with unobserved data were, on average, specified to be able to have score ranging from –20 to 20 BCVA letters away from the scores of their counterparts who did have outcome data observed. This was applied to participants in three scenarios. Scenario 1 involved applying this to participants in the investigative treatment arm (aflibercept) only. Scenario 2 involved applying this to those in the comparator arm (ranibizumab) only. Scenario 3 involved applying this to participants in both arms equally. The x-axis in Figures 6 and 7 represents the range of –20 to 20 BCVA letter scores that those participants with unobserved data were, on average, specified to be able to have relative to the scores of their counterparts who did have data outcome observed. This analysis follows previously described methods. 81 The treatment effect in the main analysis is shown at zero. Vertical bars are 95% CIs for the treatment effect. The 95% CI bars all lie above the non-inferiority margin of –5 letters, supporting the non-inferiority of aflibercept in both the ITT (see Figure 6a) and the per-protocol (see Figure 6b) populations.
For scenario 3, and within most of the ranges of scenarios 1 and 2, the lower CI limit lay below the non-inferiority margin of –5 letters, supporting the main analysis conclusion that bevacizumab lacked non-inferiority. The difference in the mean between those with unobserved BCVA data and those with observed BCVA data would need to be assumed to be 12 letters higher for bevacizumab than for ranibizumab in scenario 1 (or 12.4 letters higher in scenario 2) in order to change the main analysis conclusion of a lack of non-inferiority in both the ITT (see Figure 7a) and the per-protocol (see Figure 7b) populations.
Secondary visual acuity outcomes
Both aflibercept and bevacizumab were non-inferior to ranibizumab at 52 weeks (Table 6). The 95% CI for the adjusted difference in BCVA between arms lay above the prespecified acceptable non-inferiority margin of –5 letters at 52 weeks for both aflibercept and bevacizumab.
| Mean (SE) BCVA at screening | Mean (SE) BCVA at 52 weeks | Adjusted difference between groups (95% CI) at 52 weeks | p-value for non-inferiority (p < 0.025 is significant) | p-value for superiority (p < 0.05 is significant) | ||
|---|---|---|---|---|---|---|
| Aflibercept | Ranibizumab | Aflibercept | Ranibizumab | |||
| Aflibercept vs. ranibizumab ITT | ||||||
| 54.1 (1.2) | 53.6 (1.2) | 67.2 (1.5) (n = 139) | 65.4 (1.6) (n = 139) | 1.33 (–2.62 to 5.28)a,b | 0.0008 | 0.51 |
| Aflibercept vs. ranibizumab per protocol | ||||||
| 55.0 (1.2) | 53.6 (1.3) | 68.4 (1.4) (n = 133) | 65.5 (1.7) (n = 137) | 2.15 (–1.81 to 6.1)a,c | 0.0002 | 0.29 |
| Bevacizumab | Ranibizumab | Bevacizumab | Ranibizumab | |||
| Bevacizumab vs. ranibizumab ITT | ||||||
| 54.4 (1.1) | 53.6 (1.2) | 66.4 (1.6) (n = 135) | 65.4 (1.6) (n = 139) | –0.02 (–3.97 to 3.94)a,b | 0.0067 | 0.99 |
| Bevacizumab vs. ranibizumab per protocol | ||||||
| 54.4 (1.2) | 53.6 (1.3) | 66.4 (1.6) (n = 135) | 65.5 (1.7) (n = 137) | 0.05 (–3.88 to 3.98)a,c | 0.0058 | 0.98 |
The proportions of participants with a ≥ 15-letter gain were 47%, 52% and 45% (Figure 8) in the ranibizumab, aflibercept and bevacizumab arms, respectively, with 63%, 68% and 63%, respectively, gaining ≥ 10 letters at 100 weeks (Figure 9).
FIGURE 8.
Percentage of participants in each group with BCVA improvement of ≥ 15 ETDRS letters at 52 and 100 weeks. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.
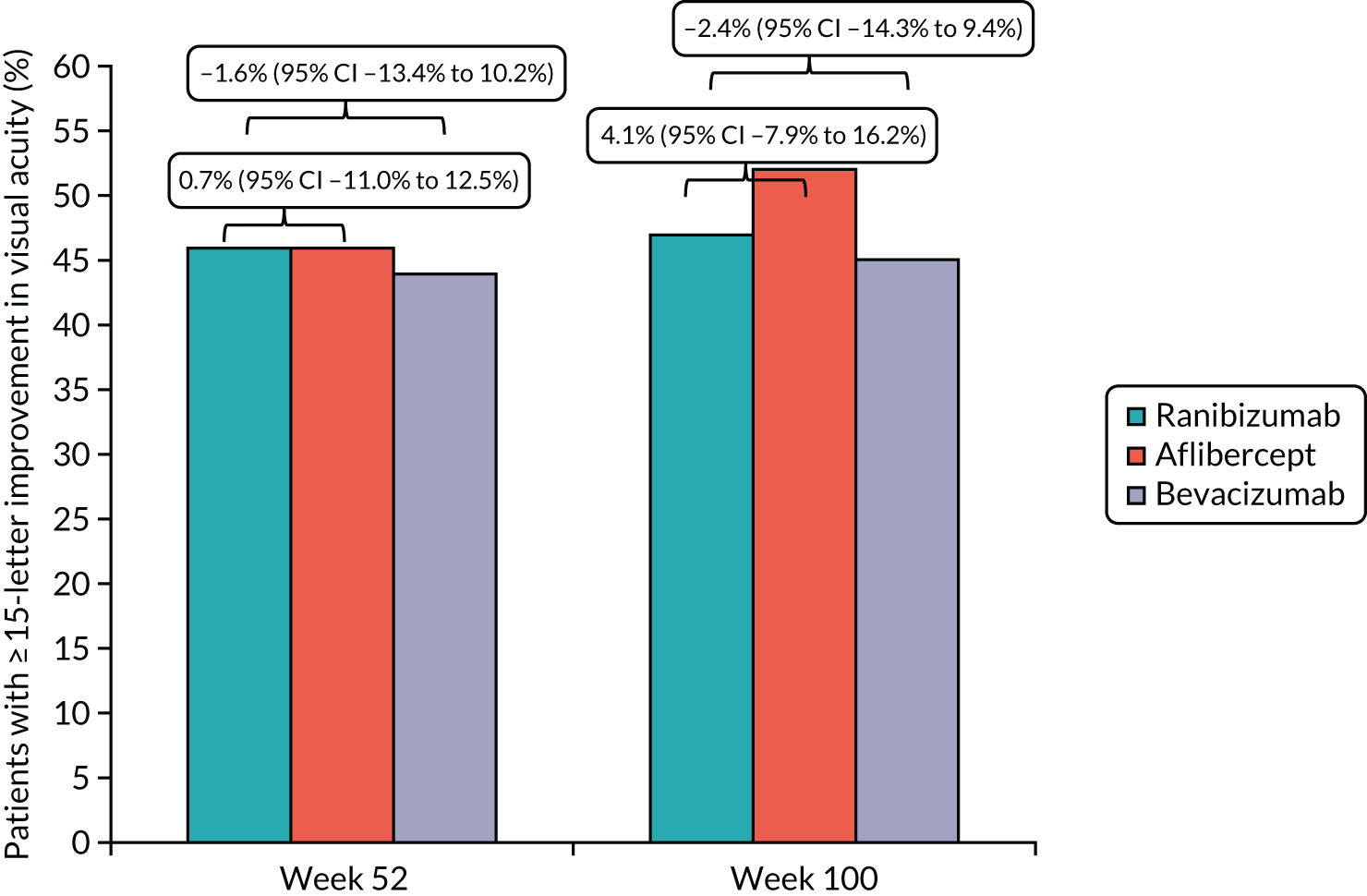
FIGURE 9.
Percentage of participants in each group with improvement of ≥ 10 ETDRS letters at 52 and 100 weeks.

The proportions of participants with a < 15-letter loss were 90%, 93% and 90% in the ranibizumab, aflibercept and bevacizumab arms, respectively (Figure 10), and the proportion of participants with a ≥ 30-letter loss in BCVA was < 6% in each group (Figure 11).
FIGURE 10.
Percentage of participants in each group with loss of < 15 ETDRS letters at 52 and 100 weeks.
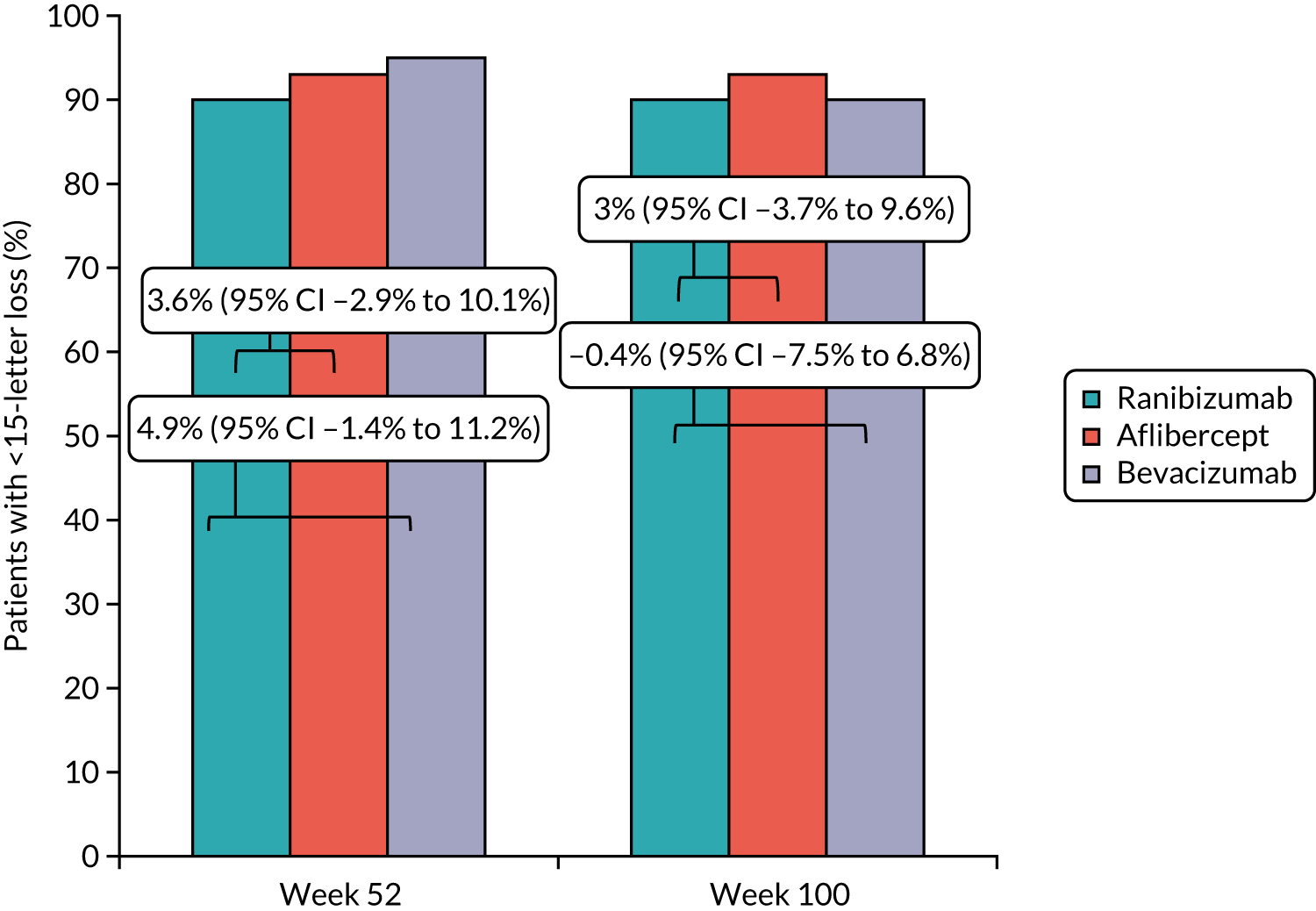
FIGURE 11.
Percentage of participants per group with loss of ≥ 30 ETDRS letters at 52 and 100 weeks.

There were no meaningful differences in the proportion of participants in each group who had prespecified categorical outcomes, for example a final visual acuity of < 19 letters (i.e. eligible for blind registration) (Table 7). Furthermore, there were no subgroup differences in the final visual acuity outcome by baseline stratifiers (Tables 8–10).
| Outcome | Trial group, % (n/N) | Difference in proportions (95% CI) | |||
|---|---|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | Aflibercept vs. ranibizumab | Bevacizumab vs. ranibizumab | |
| Participants with ≥ 73 ETDRS letters (> 6/12 Snellen equivalent) at 100 weeks | 47 (63/135) | 44 (59/133) | 41 (57/139) | –2.3 (–14.2 to 9.6) | –5.7 (–17.4 to 6.1) |
| Participants with ≤ 58 ETDRS letters (≤ 6/24 Snellen equivalent) at 100 weeks | 29 (39/135) | 20 (26/133) | 30 (42/139) | –9.3 (–19.5 to 0.9) | 1.3 (–9.5 to 12.1) |
| Participants with < 19 ETDRS letters (< 3/60 Snellen equivalent) at 100 weeks | 3 (4/135) | 2 (2/133) | 4 (6/139) | –1.5 (–5.0 to 2.1) | 1.4 (–3.1 to 5.8) |
| Participants with ≥ 73 ETDRS letters (> 6/12 Snellen equivalent) at 52 weeks | 42 (59/139) | 42 (59/139) | 39 (53/135) | 0 (–11.6 to 11.6) | –3.2 (–14.8 to 8.4) |
| Participants with ≤ 58 ETDRS letters (≤ 6/24 Snellen equivalent) at 52 weeks | 28 (39/139) | 25 (35/139) | 24 (32/135) | –2.9 (–13.3 to 7.5) | –4.4 (–14.7 to 6.0) |
| Participants with < 19 ETDRS letters (< 3/60 Snellen equivalent) at 52 weeks | 4 (5/139) | 1 (2/139) | 4 (5/135) | –2.2 (–5.8 to 1.5) | 0.1 (–4.3 to 4.5) |
| Visual acuity | Mean (SE) at screening | Mean (SE) at 100 weeks | Adjusted difference between groups (95% CI) | ||
|---|---|---|---|---|---|
| Aflibercept | Ranibizumab | Aflibercept | Ranibizumab | ||
| Aflibercept vs. ranibizumab ITTa | p = 0.91b | ||||
| BCVA≤ 38 letters | 27.3 (1.2) | 27.9 (1.1) | 59.4 (4.2) (n = 25) | 55.1 (3.9) (n = 30) | 3.3 (–6.8 to 13.4) |
| BCVA 39–58 letters | 51.2 (0.8) | 51.3 (0.7) | 65.8 (2.6) (n = 48) | 65.2 (2.8) (n = 45) | –0.5 (–8.0 to 7.0) |
| BCVA 59–78 letters | 66.4 (0.6) | 66.5 (0.5) | 74.2 (1.8) (n = 60) | 71.2 (2.3) (n = 60) | 4.2 (–2.4 to 10.7) |
| Aflibercept vs. ranibizumab per protocolc | p = 0.97b | ||||
| BCVA ≤ 38 letters | 28.7 (1.0) | 27.9 (1.1) | 61.7 (4.3) (n = 22) | 54.9 (4.1) (n = 29) | 5.3 (–5.1 to 15.7) |
| BCVA 39–58 letters | 51.1 (0.8) | 51.5 (0.7) | 67.2 (2.6) (n = 46) | 65.2 (2.8) (n = 45) | 2.0 (–5.4 to 9.5) |
| BCVA 59–78 letters | 66.4 (0.6) | 66.6 (0.6) | 74.2 (1.8) (n = 60) | 71.5 (2.4) (n = 59) | 4.0 (–2.5 to 10.4) |
| Bevacizumab | Ranibizumab | Bevacizumab | Ranibizumab | ||
| Bevacizumab vs. ranibizumab ITTa | p = 0.81b | ||||
| BCVA ≤ 38 letters | 28.8 (1.1) | 27.9 (1.1) | 53.8 (4.7) (n = 23) | 55.1 (3.9) (n = 30) | –2.8 (–12.9 to 7.3) |
| BCVA 39–58 letters | 52.5 (0.7) | 51.3 (0.7) | 64.9 (2.3) (n = 50) | 65.2 (2.8) (n = 45) | –2.3 (–9.7 to 5.2) |
| BCVA 59–78 letters | 65.5 (0.6) | 66.5 (0.5) | 68.2 (2.7) (n = 66) | 71.2 (2.3) (n = 60) | –1.0 (–7.5 to 5.5) |
| Bevacizumab vs. ranibizumab per protocolc | p = 0.82b | ||||
| BCVA ≤ 38 letters | 28.8 (1.1) | 27.9 (1.1) | 53.8 (4.7) (n = 23) | 54.9 (4.1) (n = 29) | –2.6 (–12.6, 7.3) |
| BCVA 39–58 letters | 52.5 (0.7) | 51.5 (0.7) | 64.9 (2.3) (n = 50) | 65.2 (2.8) (n = 45) | –2.2 (–9.6, 5.2) |
| BCVA 59–78 letters | 65.6 (0.6) | 66.6 (0.6) | 68.2 (2.7) (n = 66) | 71.5 (2.4) (n = 59) | –1.1 (–7.5, 5.4) |
| Disease duration | Mean (SE) at screening | Mean (SE) at 100 weeks | Adjusted difference between groups (95% CI) | ||
|---|---|---|---|---|---|
| Aflibercept | Ranibizumab | Aflibercept | Ranibizumab | ||
| Aflibercept vs. ranibizumab ITTa | p = 0.14b | ||||
| CRVO < 3 months | 54.4 (1.4) | 53.9 (1.3) | 68.2 (1.7) (n = 113) | 66.5 (1.9) (n = 116) | 0.8 (–3.9 to 5.6) |
| CRVO ≥ 3 months | 52.6 (2.5) | 51.5 (3.3) | 69.3 (3.2) (n = 20) | 60.6 (3.9) (n = 19) | 10 (–1.3 to 21.4) |
| Aflibercept vs. ranibizumab per protocolc | p = 0.21b | ||||
| CRVO < 3 months | 55.5 (1.3) | 54.0 (1.4) | 69.5 (1.7) (n = 108) | 66.6 (1.9) (n = 114) | 2.2 (–2.5 to 7.0) |
| CRVO ≥ 3 months | 52.6 (2.5) | 51.5 (3.3) | 69.3 (3.2) (n = 20) | 60.6 (3.9) (n = 19) | 10.0 (–1.1 to 21.2) |
| Bevacizumab | Ranibizumab | Bevacizumab | Ranibizumab | ||
| Bevacizumab vs. ranibizumab ITTa | p = 0.33b | ||||
| CRVO < 3 months | 55.0 (1.2) | 53.9 (1.3) | 65.5 (1.8) (n = 127) | 66.5 (1.9) (n = 116) | –1.2 (–5.8 to 3.5) |
| CRVO ≥ 3 months | 49.5 (4) | 51.5 (3.3) | 54.9 (5.2) (n = 12) | 60.6 (3.9) (n = 19) | –7.9 (–20.8 to 5) |
| Bevacizumab vs. ranibizumab per protocolc | p = 0.32b | ||||
| CRVO < 3 months | 55.0 (1.2) | 54 (1.4) | 65.5 (1.8) (n = 127) | 66.6 (1.9) (n = 114) | –1.1 (–5.7 to 3.6) |
| CRVO ≥ 3 months | 49.5 (4.2) | 51.5 (3.3) | 54.9 (5.2) (n = 12) | 60.6 (3.9) (n = 19) | –7.9 (–20.7 to 4.8) |
| Ischaemic or non-ischaemic CRVO | Mean (SE) at screening | Mean (SE) at 100 weeks | Adjusted difference between groups (95% CI) | ||
|---|---|---|---|---|---|
| Aflibercept | Ranibizumab | Aflibercept | Ranibizumab | ||
| Aflibercept vs. ranibizumab ITTa | p = 0.15b | ||||
| Non-ischaemic CRVO | 55.9 (1.2) | 55.1 (1.2) | 68.5 (1.7) (n = 115) | 66.3 (1.8) (n = 122) | 1.1 (–3.6 to 5.9) |
| Ischaemic CRVO | 41.3 (3.8) | 41.6 (4.1) | 67.3 (3.6) (n = 18) | 59.3 (6.5) (n = 13) | 11.2 (–1.9 to 24.3) |
| Aflibercept vs. ranibizumab per protocolc | p = 0.25b | ||||
| Non-ischaemic CRVO | 56.8 (1.2) | 55.2 (1.3) | 69.8 (1.6) (n = 111) | 66.4 (1.8) (n = 120) | 2.7 (–2.0 to 7.4) |
| Ischaemic CRVO | 42.7 (3.7) | 40.8 (4.3) | 67.4 (3.8) (n = 17) | 59.3 (6.5) (n = 13) | 10.8 (–2.2 to 23.8) |
| Bevacizumab | Ranibizumab | Bevacizumab | Ranibizumab | ||
| Bevacizumab vs. ranibizumab ITTa | p = 0.85b | ||||
| Non-ischaemic CRVO | 55.5 (1.2) | 55.1 (1.2) | 65.3 (1.8) (n = 121) | 66.3 (1.8) (n = 122) | –1.7 (–6.4 to 3.0) |
| Ischaemic CRVO | 47.2 (3.7) | 41.6 (4.1) | 60.2 (5.9) (n = 18) | 59.3 (6.5) (n = 13) | –0.4 (–13.4 to 12.7) |
| Bevacizumab vs. ranibizumab per protocolc | p = 0.73b | ||||
| Non-ischaemic CRVO | 55.6 (1.2) | 55.2 (1.3) | 65.3 (1.8) (n = 121) | 66.4 (1.8) (n = 120) | –1.8 (–6.4 to 2.9) |
| Ischaemic CRVO | 46.5 (3.8) | 40.8 (4.3) | 60.2 (5.9) (n = 18) | 59.3 (6.5) (n = 13) | 0.6 (–12.3 to 13.6) |
The were no differences between subgroups in the treatment effects on final visual acuity for any of the three baseline stratifiers.
Optical coherence tomography outcomes
The mean reductions in OCT CST from baseline to 100 weeks were –405 µm for ranibizumab (95% CI –450 µm to 360 µm), –378 µm for aflibercept (95% CI –412 µm to –343 µm), and –334 µm for bevacizumab (95% CI –374 µm to –293 µm). There were no clinically relevant differences across treatment groups for the adjusted difference in CST at 100 weeks: aflibercept versus ranibizumab was –29.3 µm (95% CI –60.9 µm to 2.3 µm); and bevacizumab versus ranibizumab was 21.9 µm (95% CI –9.7 µm to 53.4 µm). The adjusted mean OCT CST across groups increased by approximately 50 µm following pro re nata visits at weeks 16 and 20, closely mirroring the visual acuity data, and decreased gradually thereafter to week 100 (Figure 12). There was no difference in mean macular volume in each trial group at 100 weeks (see Appendix 3, Table 33).
FIGURE 12.
Adjusted mean OCT CST across groups to 100 weeks. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.

The proportion of participants with an OCT CT of < 320 µm at 52 weeks was significantly higher in the aflibercept group (76%) than in the ranibizumab group (63%), a difference of 12.4% (95% CI 1.7% to 23.1%). A similar difference was found at 100 weeks [aflibercept group (81%) and ranibizumab group (66%), a 15.3% difference (95% CI 4.9% to 25.7%)], but a difference between the bevacizumab and ranibizumab groups was found only at week 24 (–18.7%, 95% CI –30.1% to –7.4%) (Figure 13).
FIGURE 13.
Percentage of participants with OCT CST of < 320 µm at 24, 52 and 100 weeks. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.

Injection number
Part of this section is reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This text includes minor additions and formatting changes to the original.
By 100 weeks, ranibizumab group participants had received a mean of 11.8 injections, compared with 10.0 injections for the aflibercept group and 11.5 injections for the bevacizumab group. The difference between the aflibercept and ranibizumab groups was meaningful as early as week 24 [mean difference: –0.4 at week 24 (95% CI –0.6 to –0.2); –1.1 at week 52 (95% CI –1.6 to –0.5); and –1.9 at week 100 (95% CI –2.9 to –0.8)] (Figure 14).
FIGURE 14.
Mean number of injections across treatment groups by weeks 24, 52 and 100. Reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This includes minor additions and formatting changes to the original.

Post hoc bevacizumab versus aflibercept analysis
After being approved by the DMEC, a post hoc analysis was unable to demonstrate that bevacizumab was non-inferior to aflibercept in the ITT analysis at 52 weeks (adjusted mean difference –1.35 letters, 95% CI –5.29 to 2.59 letters) or at 100 weeks (adjusted mean BCVA difference was –3.96 letters, 95% CI –8.34 to 0.42 letters; p = 0.32). The results of the per-protocol analysis were similar. At 100 weeks, there was a significant difference of 1.6 (95% CI 0.5 to 2.7) between the mean number of injections received by participants randomised to bevacizumab and the mean number received by those randomised to aflibercept.
Retinal imaging
Optical coherence tomography imaging
The OCT morphological grading for MO, subretinal detachment and vitreomacular interface abnormality was available for 456 (98.4%) and 396 (85.5%) participants at baseline and week 100, respectively, and showed no difference for any parameter across treatment arms in prevalence or change with time. Across all subgroups, the percentage of participants with any MO and subretinal detachment at baseline had decreased significantly by week 52, and by 75% at week 100 (Table 11).
| Outcome | All | Ranibizumab | Aflibercept | Bevacizumab |
|---|---|---|---|---|
| MO | ||||
| Baseline | ||||
| Null (n) | 7 | 3 | 1 | 3 |
| No evidence, n (%) | 5 (1) | 2 (1) | 2 (1) | 1 (1) |
| Diffuse, n (%) | 19 (4) | 8 (5) | 8 (5) | 3 (2) |
| Cystic, n (%) | 90 (20) | 25 (16) | 33 (22) | 32 (21) |
| Mixed, n (%) | 342 (75) | 117 (77) | 110 (72) | 115 (76) |
| Week 52 | ||||
| Null (n) | 53 | 21 | 13 | 19 |
| No evidence, n (%) | 147 (36) | 56 (42) | 62 (44) | 29 (21) |
| Diffuse, n (%) | 64 (16) | 18 (13) | 24 (17) | 22 (16) |
| Cystic, n (%) | 103 (25) | 27 (20) | 35 (25) | 41 (30) |
| Mixed, n (%) | 96 (23) | 33 (25) | 20 (14) | 43 (32) |
| Week 100 | ||||
| Null (n) | 67 | 22 | 24 | 21 |
| No evidence, n (%) | 150 (38) | 55 (41) | 59 (45) | 36 (27) |
| Diffuse, n (%) | 55 (14) | 17 (13) | 19 (15) | 19 (14) |
| Cystic, n (%) | 87 (22) | 26 (20) | 29 (22) | 32 (24) |
| Mixed, n (%) | 104 (26) | 35 (26) | 23 (18) | 46 (35) |
| Subretinal detachment | ||||
| Baseline | ||||
| Null (n) | 26 | 9 | 8 | 9 |
| No evidence, n (%) | 126 (29) | 39 (27) | 45 (31) | 42 (29) |
| Questionable, n (%) | 9 (2) | 6 (4) | 1 (1) | 2 (1) |
| Definite, n (%) | 196 (43) | 62 (41) | 63 (41) | 71 (48) |
| Week 52 | ||||
| Null (n) | 55 | 22 | 13 | 20 |
| No evidence, n (%) | 352 (86) | 113 (85) | 124 (88) | 115 (86) |
| Questionable, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Definite, n (%) | 56 (14) | 20 (15) | 17 (12) | 19 (14) |
| Week 100 | ||||
| Null (n) | 67 | 22 | 24 | 21 |
| No evidence, n (%) | 342 (86) | 118 (89) | 111 (85) | 113 (85) |
| Questionable, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Definite, n (%) | 54 (14) | 15 (11) | 19 (15) | 20 (15) |
| Vitreomacular interface abnormality | ||||
| Baseline | ||||
| Null (n) | 9 | 3 | 1 | 5 |
| No evidence, n (%) | 250 (55) | 87 (57) | 88 (58) | 75 (50) |
| Questionable, n (%) | 8 (2) | 3 (2) | 2 (1) | 3 (2) |
| Definite, n (%) | 196 (43) | 62 (41) | 63 (41) | 71 (48) |
| Week 52 | ||||
| Null (n) | 53 | 21 | 13 | 19 |
| No evidence, n (%) | 221 (54) | 73 (54) | 76 (54) | 72 (53) |
| Questionable, n (%) | 4 (1) | 0 (0) | 3 (2) | 1 (1) |
| Definite, n (%) | 185 (45) | 61 (46) | 62 (44) | 62 (46) |
| Week 100 | ||||
| Null (n) | 67 | 22 | 24 | 21 |
| No evidence, n (%) | 219 (55) | 74 (56) | 77 (59) | 68 (51) |
| Questionable, n (%) | 9 (2) | 4 (3) | 4 (3) | 1 (1) |
| Definite, n (%) | 168 (42) | 55 (41) | 49 (38) | 64 (48) |
Spectral-domain OCT (Spectralis) image grading was undertaken for additional parameters, including DRIL, COST visibility loss, EZ disruption, loss of ELM integrity and presence of intraretinal HRF. Of 463 participants, 337 were enrolled at sites where Spectralis OCT was available; of these participants, 267 had gradable images at baseline and at weeks 52 and 100 (Table 12). There was no difference in the prevalence of any parameter across treatment groups at any time point. In all treatment groups, DRIL was observed to decrease, and the ELM, EZ and COST retinal layers became better defined with time. This may have represented better visualisation with time, as MO decreased, rather than a specific reconstitution of the parameter. Further investigation and correlation of these findings with visual outcomes will be the subject of a further publication.
| All (N = 267), n (%) | Trial group, n (%) | |||
|---|---|---|---|---|
| Ranibizumab (N = 92) | Aflibercept (N = 89) | Bevacizumab (N = 86) | ||
| DRIL | ||||
| Baseline | ||||
| Absent | 86 (32) | 30 (33) | 31 (35) | 25 (29) |
| Present | 149 (56) | 51 (55) | 48 (54) | 50 (58) |
| Ungradable | 32 (12) | 11 (12) | 10 (11) | 11 (13) |
| Week 52 | ||||
| Absent | 189 (71) | 71 (76) | 60 (67) | 58 (68) |
| Present | 61 (23) | 18 (19) | 21 (24) | 22 (26) |
| Ungradable | 17 (6) | 4 (4) | 8 (9) | 5 (6) |
| Week 100 | ||||
| Absent | 178 (67) | 60 (65) | 61 (69) | 57 (66) |
| Present | 68 (25) | 23 (25) | 24 (27) | 21 (24) |
| Ungradable | 21 (8) | 9 (10) | 4 (4) | 8 (9) |
| HRF | ||||
| Baseline | ||||
| Absent | 62 (23) | 24 (26) | 20 (22) | 18 (21) |
| Present | 204 (76) | 68 (74) | 68 (76) | 68 (79) |
| Ungradable | 1 (0) | 0 (0) | 1 (1) | 0 (0) |
| Week 52 | ||||
| Absent | 132 (49) | 49 (53) | 42 (47) | 41 (48) |
| Present | 135 (51) | 44 (47) | 47 (53) | 44 (52) |
| Ungradable | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Week 100 | ||||
| Absent | 96 (36) | 30 (33) | 39 (44) | 27 (31) |
| Present | 168 (63) | 62 (67) | 48 (54) | 58 (67) |
| Ungradable | 3 (1) | 0 (0) | 2 (2) | 1 (1) |
| ELM | ||||
| Baseline | ||||
| Intact | 66 (25) | 20 (22) | 24 (27) | 22 (26) |
| Not intact | 44 (16) | 17 (18) | 18 (20) | 9 (10) |
| Ungradable | 157 (59) | 55 (60) | 47 (53) | 55 (64) |
| Week 52 | ||||
| Intact | 198 (74) | 71 (76) | 62 (70) | 65 (76) |
| Not intact | 50 (19) | 18 (19) | 20 (22) | 12 (14) |
| Ungradable | 19 (7) | 4 (4) | 7 (8) | 8 (9) |
| Week 100 | ||||
| Intact | 200 (75) | 69 (75) | 67 (75) | 64 (74) |
| Not intact | 49 (18) | 19 (21) | 16 (18) | 14 (16) |
| Ungradable | 18 (7) | 4 (4) | 6 (7) | 8 (9) |
| EZ | ||||
| Baseline | ||||
| Intact | 46 (17) | 15 (16) | 18 (20) | 13 (15) |
| Not intact | 61 (23) | 21 (23) | 21 (24) | 19 (22) |
| Ungradable | 160 (60) | 56 (61) | 50 (56) | 54 (63) |
| Week 52 | ||||
| Intact | 174 (65) | 64 (69) | 54 (61) | 56 (66) |
| Not intact | 75 (28) | 25 (27) | 29 (33) | 21 (25) |
| Ungradable | 18 (7) | 4 (4) | 6 (7) | 8 (9) |
| Week 100 | ||||
| Intact | 172 (64) | 57 (62) | 61 (69) | 54 (63) |
| Not intact | 75 (28) | 30 (33) | 22 (25) | 23 (27) |
| Ungradable | 20 (7) | 5 (5) | 6 (7) | 9 (10) |
| COSTs | ||||
| Baseline | ||||
| Intact | 16 (6) | 8 (9) | 5 (6) | 3 (3) |
| Not intact | 78 (29) | 23 (25) | 31 (35) | 24 (28) |
| Ungradable | 173 (65) | 61 (66) | 53 (60) | 59 (69) |
| Week 52 | ||||
| Intact | 54 (20) | 13 (14) | 25 (28) | 16 (19) |
| Not intact | 170 (64) | 64 (69) | 53 (60) | 53 (62) |
| Ungradable | 43 (16) | 16 (17) | 11 (12) | 16 (19) |
| Week 100 | ||||
| Intact | 65 (24) | 17 (18) | 25 (28) | 23 (27) |
| Not intact | 169 (63) | 66 (72) | 53 (60) | 50 (58) |
| Ungradable | 33 (12) | 9 (10) | 11 (12) | 13 (15) |
Fundus fluorescein angiography image analysis
Of 463 participants at baseline, 461 underwent FFA. At 100 weeks, 407 completed the ITT analysis; 377 underwent FFA, and 30 did not because they declined or had experienced an adverse reaction to the dye at baseline or there were intravenous cannulation/technical difficulties. Of the 377 participants who underwent FFA, 53 could not be graded for other reasons (e.g. the participant had received panretinal photocoagulation before or during the trial), and for 14 participants all images were ungradable, leaving 310 participants with gradable images (Table 13). The percentages of participants in each arm with two-step, or more, worsening in one or more quadrants appeared more frequent in the aflibercept group than in the bevacizumab group, but, as the number of affected quadrants increased, the result across groups tended to converge. Overall, the data showed no meaningful difference between treatment groups in terms of the number of participants with at least two-step worsening of non-perfusion in one or more quadrants.
| Sectors with two-step, or more, capillary non-perfusion worsening (n) | Trial group, n (%) | ||
|---|---|---|---|
| Ranibizumab (N = 105) | Aflibercept (N = 96) | Bevacizumab (N = 109) | |
| 0 | 73 (70) | 62 (65) | 86 (79) |
| 1 | 11 (10) | 18 (19) | 9 (8) |
| 2 | 8 (8) | 6 (6) | 4 (4) |
| 3 | 5 (5) | 4 (4) | 3 (3) |
| 4 | 4 (4) | 1 (1) | 1 (3) |
| 5 | 1 (1) | 1 (1) | 0 (0) |
| ≥ 6 | 3 (3) | 4 (4) | 6 (6) |
The novel concentric ring method for analysing non-perfusion in disc areas, developed by the LEAVO team during the trial, was applicable to 235 of 463 participants randomised who underwent wide-angled Optos FFA. Of these, 184 had images successfully performed at both entry and exit; among these, 31 eyes were poor-quality images either at baseline or at exit. This left 153 gradable images that were converted into disc areas of non-perfusion and form the basis of the comparison between trial groups (Tables 14 and 15).
| Retinal area | Amount of retinal non-perfusion, median (IQR) | |||||||
|---|---|---|---|---|---|---|---|---|
| All (N = 153) | Ranibizumab (N = 57) | Aflibercepta (N = 48) | Bevacizumab (N = 48) | |||||
| Cells | Disc areas | Cells | Disc areas | Cells | Disc areas | Cells | Disc areas | |
| Baseline | ||||||||
| Total area | 3 (1 to 5) | 28.6 (10.4 to 47.4) | 2.5 (1 to 5.3) | 24.6 (8.0 to 49.6) | 3.3 (1.6 to 5) | 30.2 (16.4 to 48.7) | 3 (0.9 to 4.5) | 28.9 (8.6 to 44.4) |
| Posterior (M + R1) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 0) |
| % (n) subjects with posterior > 0 | 14 (21) | 14 (21) | 19 (11) | 19 (11) | 19 (9) | 19 (9) | 2 (1) | 2 (1) |
| Peripheral (R2–R4) | 3 (1 to 5) | 28.4 (10.4 to 47.4) | 2.5 (0.8 to 5) | 24.6 (7.3 to 49.1) | 3 (1.6 to 5) | 30.2 (16.2 to 48.7) | 3 (0.9 to 4.5) | 28.9 (8.6 to 44.4) |
| Week 100 | ||||||||
| Total area | 3 (1.5 to 6) | 30.2 (15.5 to 55.1) | 2.5 (1.3 to 7) | 25.0 (13.0 to 61.4) | 3.8 (2 to 9.4) | 37.0 (20.7 to 72.9) | 3 (1 to 4.5) | 30.2 (10.4 to 44.4) |
| Posterior (M + R1) | 0 (0 to 0) | 0 (0 to 0) | 0 (0 to 1) | 0 (0 to 4.4) | 0 (0 to 1.9) | 0 (0 to 5.5) | 0 (0 to 0) | 0 (0 to 0) |
| % (n) subjects with posterior > 0 | 22 (33) | 22 (33) | 26 (15) | 26 (15) | 29 (14) | 29 (14) | 8 (4) | 8 (4) |
| Peripheral (R2–R4) | 3 (1.5 to 5.5) | 30.2 (15.1 to 51.1) | 2.5 (1.3 to 6.3) | 25.0 (11.9 to 59.1) | 3.5 (2 to 7.5) | 35.2 (20.7 to 69.6) | 3 (1 to 4.5) | 30.2 (10.4 to 44.4) |
| Change in total area | 0.0 (–1.0 to 2.0) | 0.0 (–5.4 to 16.0) | 0.0 (–1.0 to 002.0) | 0.0 (–9.1 to 15.6) | 1.0 (–1.0 to 3.0) | 4.7 (–2.0 to 24.3) | 0.0 (–1.0 to 2.0) | 0.0 (–9.3 to 15.9) |
| Change in posterior | 0.0 (0.0 to 0.0) | 0.0 (0.0 to 0.0) | 0.0 (0.0 to 0.0) | 0.0 (0.0 to 0.0) | 0.0 (0.0 to 0.8) | 0.0 (0.0 to 1.5) | 0.0 (0.0 to 0.0) | 0.0 (0.0 to 0.0) |
| % (n) subjects with an increase in posterior | 17 (26) | 18 (27) | 19 (11) | 19 (11) | 25 (12) | 27 (13) | 6 (3) | 6 (3) |
| Change in peripheral | 0.0 (–1.0 to 2.0) | 0.0 (–7.1 to 15.8) | 0.0 (–1.0 to 2.0) | 0.0 (–9.9 to 15.8) | 1.0 (1.0 to 2.0) | 4.7 (0.0 to 18.0) | 0.0 (–1.0 to 2.0) | 0.0 (–9.3 to 15.9) |
| Change from baseline | Difference in medians (95% CI) | |||
|---|---|---|---|---|
| Aflibercept – ranibizumab | Bevacizumab – ranibizumab | |||
| Cells | Disc areas | Cells | Disc areas | |
| Change in total area | 1.0 (0 to 2.0) | 5.6 (0 to 14.1) | 0.0 (–1.0 to 1.0) | 0.0 (–7.8 to 6.0) |
| Participants with an increase in posterior (%) | 6.0 (–9.7 to 22.0) | 8.0 (–8.0 to 24.1) | –13.0 (–25.6 to 0.3) | –13.0 (–25.6 to 0.3) |
| Change in peripheral | 1.0 (0 to 2.0) | 5.6 (0 to 13.4) | 0.0 (–1.0 to 1.0) | 0.0 (–7.8 to 6.5) |
The median value of baseline non-perfusion for all participants was 28.6 disc areas (IQR 10.4–47.4 disc areas), mostly in the peripheral retina. There was more non-perfusion in the periphery and, notably, in the posterior pole in the ranibizumab (19%) and aflibercept (19%) groups than in the bevacizumab (2%) group. This baseline imbalance between groups was seen at week 100, particularly in the percentage of participants showing an increase in posterior non-perfusion, which may simply reflect higher baseline non-perfusion and, therefore, greater likelihood of progressing. A detailed appraisal of these data is currently being undertaken and will form the basis of a further report. 83
Treatment allocation guess form
The optometrists assessing primary outcomes provided a response on the treatment allocation guess form for 409 of their 463 participants: for 356, they said they did not know; for 53, they made a guess, and were correct in 18 instances, which is consistent with chance. Of the 409 participants, 406 provided a response: 386 did not know and 20 made a guess, of whom eight [i.e. 2% (8/406)] guessed correctly, which is consistent with chance.
Safety outcomes
Part of this section is reproduced from Hykin et al. 2 This is an open access article distributed under the terms of the CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium. This text includes minor additions and formatting changes to the original.
There was one case of infectious endophthalmitis in a trial eye, which followed trabeculectomy bleb infection rather than intravitreal injection. The frequencies of all ocular AEs and Antiplatelet Trialists’ Collaboration (APTC)-defined events were similar between trial arms (Table 16). At 52 weeks, the proportions of participants who were persistent non-responders (defined as not more than a 5-letter gain in visual acuity and an OCT CST decrease of < 50 µm after 24 weeks) were 1/139 for ranibizumab, 5/133 for aflibercept and 5/135 for bevacizumab; at 100 weeks, only one participant, in the bevacizumab group, was a non-responder. During the trial, 25 (5.4%) eyes developed an ischaemic CRVO, 13 (2.8%) developed anterior segment neovascularisation and 6 (1.3%) developed retinal neovascularisation, with no difference across arms (see Table 16).
| AE | Total (N = 463), n (%) | Trial arm, n (%) | Difference (95% CI) (%) | |||
|---|---|---|---|---|---|---|
| Ranibizumab (N = 155) | Aflibercept (N = 154) | Bevacizumab (N = 154) | Aflibercept vs. ranibizumab | Bevacizumab vs. ranibizumab | ||
| Ocular AEs | ||||||
| Infectious endophthalmitis | 1 (0.2) | 0 (0) | 0 (0) | 1 (0.6) | 0.0 (–2.4 to 2.4) | –0.6 (–3.6 to 1.8) |
| Traumatic cataract | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0.0 (–2.4 to 2.4) | 0.0 (–2.4 to 2.4) |
| Retinal tear | 1 (0.2) | 1 (0.6) | 0 (0) | 0 (0) | –0.6 (–3.6 to 1.9) | –0.6 (–3.6 to 1.9) |
| Retinal detachment | 3 (0.6) | 0 (0) | 1 (0.6) | 2 (1.3) | 0.6 (–1.8 to 3.6) | 1.3 (–1.3 to 4.6) |
| Conversion to ischaemic CRVO | 25 (5.4) | 8 (5.2) | 10 (6.5) | 7 (4.5) | 1.3 (–4.2 to 7.0) | –0.6 (–5.9 to 4.6) |
| Anterior segment neovascularisation | 13 (2.8) | 5 (3.2) | 5 (3.2) | 3 (1.9) | 0.0 (–4.5 to 4.5) | –1.3 (–5.6 to 2.8) |
| Retinal neovascularisation | 6 (1.3) | 1 (0.6) | 4 (2.6) | 1 (0.6) | 2.0 (–1.4 to 5.9) | 0.0 (–3.0 to 3.0) |
| Vitreous haemorrhage | 6 (1.3) | 0 (0) | 2 (1.3) | 4 (2.6) | 1.3 (–1.3 to 4.6) | 2.6 (–0.2 to 6.5) |
| IOP elevation | 27 (5.8) | 13 (8.4) | 9 (5.8) | 5 (3.2) | –2.5 (–8.6 to 3.4) | –5.1 (–10.9 to 0.2) |
| Systemic APTC events | ||||||
| Cardiovascular – vascular deaths | 5 (1.1) | 2 (1.3) | 2 (1.3) | 1 (0.6) | 0.0 (–3.4 to 3.4) | –0.6 (–4.0 to 2.4) |
| Cardiovascular – non-fatal MI | 2 (0.4) | 0 (0) | 0 (0) | 2 (1.3) | 0.0 (–2.4 to 2.4) | 1.3 (–1.3 to 4.6) |
| Cardiovascular – non-fatal stroke | 6 (1.3) | 2 (1.3) | 4 (2.6) | 0 (0) | 1.3 (–2.4 to 5.3) | –1.3 (–4.6 to 1.3) |
Eight ranibizumab, seven aflibercept and eight bevacizumab arm participants required panretinal photocoagulation. Two pregnancies were reported during the trial: one in a participant and one in the spouse of a participant. Both of these were followed to term with the delivery of normal neonates.
Systemic serious AEs occurred with an expected and similar frequency between groups (see Table 16), and there were no meaningful differences between groups in the frequency of AEs in the same body system (Table 17).
| Body system | Total (N = 463), n (%) | Trial arm, n (%) | ||
|---|---|---|---|---|
| Ranibizumab (N = 155) | Aflibercept (N = 154) | Bevacizumab (N = 154) | ||
| Cardiovascular – other | 31 (6.7) | 8 (5.2) | 14 (9.1) | 9 (5.8) |
| Respiratory | 20 (4.3) | 4 (2.6) | 6 (3.9) | 10 (6.5) |
| Hepatic | 1 (0.2) | 1 (0.6) | 0 (0) | 0 (0) |
| Gastrointestinal | 19 (4.1) | 8 (5.2) | 8 (5.2) | 3 (1.9) |
| Genitourinary | 13 (2.8) | 2 (1.3) | 7 (4.5) | 4 (2.6) |
| Endocrinal | 1 (0.2) | 0 (0) | 0 (0) | 1 (0.6) |
| Haematological | 1 (0.2) | 0 (0) | 1 (0.6) | 0 (0) |
| Musculoskeletal | 10 (2.2) | 1 (0.6) | 4 (2.6) | 5 (3.2) |
| Neoplastic | 4 (0.9) | 0 (0) | 1 (0.6) | 3 (1.9) |
| Neurological | 6 (1.3) | 1 (0.6) | 3 (1.9) | 2 (1.3) |
| Psychiatric | 2 (0.4) | 1 (0.6) | 0 (0) | 1 (0.6) |
| Immunological | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Dermatological | 2 (0.4) | 0 (0) | 2 (1.3) | 0 (0) |
| Allergies | 1 (0.2) | 0 (0) | 1 (0.6) | 0 (0) |
| Ophthalmological | 9 (1.9) | 3 (1.9) | 3 (1.9) | 3 (1.9) |
| Ear, nose and throat | 1 (0.2) | 0 (0) | 0 (0) | 1 (0.6) |
| Other | 9 (1.9) | 3 (1.9) | 3 (1.9) | 3 (1.9) |
Comparison with Study of Comparative Treatments for Retinal Vein Occlusion 2 safety data
Although it was not possible to perform a safety meta-analysis because of the lack of comparative outcome data in CRVO, as described in Chapter 2, the data from SCORE2 during the initial comparative 6 months were compared with the safety data from the first 6 months of LEAVO (Table 18). A larger number of conversions to ischaemic CRVO were recorded in LEAVO than in SCORE2. This may have been because in LEAVO conversion to ischaemic CRVO was recorded as a direct question in each trial visit sheet; because of early enrolment in LEAVO compared with SCORE 2; and because of the treatment-naive status of most LEAVO participants at randomisation. There were more vitreous haemorrhages recorded in LEAVO than in SCORE2, and more vascular deaths recorded in SCORE2 than in LEAVO. The prevalence of these events was low and it was not thought that there were any meaningful differences between the two studies in the number or type of AEs.
| Event | LEAVO (n) | SCORE2 (n) | |||
|---|---|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | Aflibercept | Bevacizumab | |
| Trial eye | |||||
| Infectious endophthalmitis | 0 | 0 | 1 | – | – |
| Non-infectious endophthalmitis | 0 | 0 | 0 | 0 | 1 |
| Neovascular glaucoma | 1 | 1 | 0 | 1 | 0 |
| Conversion to ischaemic CRVO | 8 | 6 | 6 | 1 | 0 |
| Retinal detachment | 0 | 1 | 1 | 0 | 1 |
| Vitreous haemorrhage | 0 | 2 | 4 | 0 | 1 |
| APTC events | |||||
| Non-fatal MI | 0 | 0 | 0 | 1 | 2 |
| Non-fatal stroke | 0 | 1 | 0 | 1 | 0 |
| Vascular death | 0 | 0 | 0 | 3 | 2 |
| Excluding vascular death | |||||
| Death from any other cause | 1 | 1 | 0 | 1 | 1 |
| Ocular and systemic AEs, not limited to trial eye | |||||
| Participants with any AE | 108 | 99 | 115 | 82 | 98 |
| Total number of all AEs | 301 | 337 | 323 | 184 | 263 |
| Participants with any SAE | 19 | 7 | 14 | 14 | 14 |
| Total number of all SAEs | 20 | 10 | 14 | 18 | 25 |
Patient and public involvement
The lay panel members co-developed the contents and wording of the questions in the following questionnaire.
-
Cost of the medication: if the cheaper medication Avastin was as good as Eylea and Lucentis in improving your vision, and as safe, would you be happy to be given Avastin for your affected eye?
-
Licensed medications: if the cheaper medication Avastin was as good as Eylea and Lucentis in improving your vision, and bearing in mind Avastin is as safe as the other two (see above), would you be concerned about taking Avastin because it had not been licensed by the UK MHRA (i.e. the UK regulatory body that approves new drugs for use in the UK)?
-
Effect of the medications: if the cheaper unlicensed medication Avastin was slightly better at improving vision in your affected eye than the licensed medications Eylea and Lucentis (e.g. an improvement of 2 letters on a visual acuity chart. There are 5 letters on each line, so the difference would be just less than half a line), under this circumstance, would you be happy to be given Avastin? If no, what would be the reason?
-
Effect of the medications: if the cheaper unlicensed medication Avastin was slightly less good at improving vision in your affected eye than the licensed medications Eylea and Lucentis (e.g. a loss of 2 letters on a visual acuity chart. There are 5 letters on each line, so the difference would be just less than half a line), would you be happy to be given Avastin?
-
Effect of the medications: if the cheaper unlicensed medication Avastin was slightly less good (i.e. if you closed your good eye you noticed a slight central blur in the affected eye when reading, but not when looking in the distance and not when using both eyes together), but you were still able to do all regular activities, such as drive, read books and magazines, work machinery, use power tools, would you be happy to be given Avastin?
-
Effect of the medications: if you were asked to commence treatment with Avastin, would you be more likely to agree to this if a licensed alternative (e.g. Eylea) was available that you could change over to if your response to the Avastin was less than expected?
The feedback regarding the final questionnaire content was positive (i.e. it was an important trial to have done, the text was easy to follow and the questions were clear). The results of the trial were under embargo pending publication at the time the questionnaire was sent to patients [n = 22: seven with a history of RVO (but not LEAVO participants), 15 with a history of diabetic eye disease and three regular lay panel members]. The results of the patient and public involvement LEAVO questionnaire are given in Table 19.
| Answer | Question, n (%) responses | |||||
|---|---|---|---|---|---|---|
| 1 | 2a | 3 | 4 | 5 | 6 | |
| Yes | 22 (100) | 6 (27) | 20 (91) | 11 (50) | 15 (68) | 22 (100) |
| Maybe | 0 (0) | 2 (9) | 2 (9) | 3 (14) | 1 (5) | 0 (0) |
| No | 0 (0) | 13 (59) | 0 (0) | 8 (36) | 6 (27) | 0 (0) |
Chapter 4 Health economic evaluation
Introduction
Economic evaluation forms an important part of health technology assessments by evaluating the cost-effectiveness of interventions to determine whether or not they represent value for money. In England, NICE evaluates interventions through its technology appraisal and guidelines programmes. Each programme has a methods guide, which describes a reference case that should be used in cost-effectiveness analyses. 84,85 Our analyses use NICE’s preferred methods in conjunction with other good practice guidelines86,87 to evaluate the cost-effectiveness of intravitreal ranibizumab, aflibercept and bevacizumab for MO due to CRVO.
NICE’s preferred method for cost-effectiveness analysis of interventions delivered in an NHS setting is cost–utility analysis (CUA). 84,85 CUA allows comparisons to be made between disease areas by using a common measure of outcome: cost per quality-adjusted life-year (QALY). QALYs combine morbidity and mortality by using a ‘utility’ to measure health-related quality of life (HRQoL). Utilities are anchored between 0 and 1, where 1 represents perfect health and 0 represents death (utilities of < 0 are permitted, reflecting health states considered to be worse than death).
A CUA is used to compare two or more interventions, using incremental analysis. The outcome of a CUA is an ICER, calculated by dividing the incremental (additional) costs by the incremental QALYs associated with the intervention. The incremental costs are calculated as the difference between the total costs for the intervention and the total costs for the comparator. The incremental QALYs are calculated as the difference between the total QALYs for the intervention and the total QALYs for the comparator.
The results of a CUA can be used in decision-making to determine whether or not interventions represent good value for money. The simplest decisions concern dominance. An intervention is said to ‘dominate’ the comparator (and the comparator is ‘dominated’) when the intervention leads to lower costs and more QALYs than the comparator. In this case, the decision to use the intervention instead of the comparator is clear, as it reduces costs and improves outcomes. In the situation in which an intervention is more costly and leads to more QALYs than the comparator, a decision rule is required to determine whether or not the gain in QALYs is worth the additional cost. In this case, the ICER can be compared with a threshold representing the maximum the funder is willing to pay for each additional QALY.
NICE does not have a specific threshold, but considers a range of maximum acceptable ICERs when deciding if an intervention is cost-effective. Interventions with ICERs of < £20,000 per QALY are generally considered to be cost-effective, whereas decisions regarding interventions with ICERs of between £20,000 and £30,000 per QALY will need to consider additional factors such as uncertainty, innovation, whether or not the HRQoL benefits have been adequately captured, whether or not the treatment meets specific criteria for life-extending treatments at the end of life, and the non-health objectives of the NHS. At > £30,000 per QALY, a stronger case is required for NICE to consider an intervention to be cost-effective. 84
Overview of health economic evaluation methods
Interventions
A full health economic evaluation was conducted, comparing three interventions for MO due to CRVO using data collected as part of LEAVO. The interventions were as follows:
-
Interventions (investigational treatments) –
-
Arm A, treatment: an intravitreal injection of aflibercept (2.0 mg/0.05 ml) administered at baseline and at 4, 8 and 12 weeks as a mandated injection. From week 16 to week 96, treatment was given if one or more re-treatment criteria were met, as specified in the trial protocol. 88 Beyond the 100-week trial period, injections were given based on treatment continuation rules.
-
Arm B, treatment: an intravitreal injection of bevacizumab (1.25 mg/0.05 ml) administered at baseline and at 4, 8 and 12 weeks as a mandated injection. From week 16 to week 96, treatment was given if one or more re-treatment criteria were met. Beyond the 100-week trial period, injections were given based on treatment continuation rules.
-
-
Comparator (standard care) –
-
Arm C, control: an intravitreal injection of ranibizumab (0.5 mg/0.05 ml) administered at baseline and at 4, 8 and 12 weeks as a mandated injection. From week 16 to week 96, treatment was given if one or more re-treatment criteria were met. Beyond the 100-week trial period, injections were given based on treatment continuation rules.
-
Method of economic evaluation
The economic evaluation comprises two parts: a model-based analysis (the primary analysis) and a within-trial analysis (the secondary analysis). The model-based analysis evaluates the three interventions over participants’ lifetimes, extrapolating clinical outcomes beyond the trial period and relating these to costs and QALYs. The within-trial analysis evaluates the three interventions during the trial period using the individual patient-level cost and HRQoL data collected during the trial. The economic evaluation uses CUA. The methods for the economic evaluation were prespecified in health economic and decision-modelling analysis plan (and associated addendum) documents, prior to database lock. 89
The within-trial analysis provides the short-term cost-effectiveness evidence using individual patient-level data on quality of life and costs; therefore, it avoids extrapolation uncertainty. The model-based analysis provides the long-term cost-effectiveness evidence (extrapolating outcomes and costs beyond the trial period); this is the preferred approach for resource allocation decision-making (in line with NICE’s Guide to Methods of Technology Appraisal 201384). To support the development of the economic model, a systematic literature review was undertaken to identify evidence to inform inputs and assumptions.
Settings
Perspective
The economic evaluation uses the NHS and Personal Social Services perspective, consistent with the NICE methods guides. 84,85 Included costs are those incurred by the NHS and Personal Social Services, and so include costs for health-care resource use and interventions. Societal costs, lost productivity and a patient’s personal expenditure (e.g. travel costs) are excluded.
Discounting
Future costs and health outcomes are discounted to reflect time preference. The discount rate for both costs and QALYs is 3.5% per year, consistent with the NICE methods guides. 84,85
Time horizon
The model-based analysis uses a lifetime horizon, calculating costs and QALYs until all modelled patients have died. The within-trial analysis uses the 100-week trial time horizon. NICE states that the time horizon should be ‘long enough to reflect all important differences in costs or outcomes between the technologies being compared’84 (© NICE 2013 Guide to Methods of Technology Appraisal 2013. Available from www.nice.org.uk/process/pmg9/. All rights reserved. Subject to Notice of rights). Using a lifetime horizon reflects the long-term differences between the interventions in terms of effectiveness, time on treatment/discontinuation, and safety outcomes.
Presentation of results
Incremental and pairwise analyses
The economic evaluation reports fully incremental analyses, consistent with the NICE methods guides,84,85 and pairwise analyses, to allow the comparison of each pair of interventions. For the model-based analysis, the fully incremental analysis is presented in Results: model-based analysis, and the pairwise comparisons are presented in Appendix 6, Tables 59–61. For the within-trial analysis, pairwise comparisons are presented in Results: within-trial analysis, and the fully incremental analysis is presented in Appendix 6, Table 66.
Characterisation of uncertainty
The model-based and within-trial analyses each present a base-case analysis and scenario analyses using alternative settings. The base-case and scenario analyses from the model-based analysis use probabilistic sensitivity analysis to incorporate parameter uncertainty (see Methods: within-trial analysis). For deterministic results, see Appendix 6, Table 58.
The base-case and scenario analyses from the within-trial analysis use seemingly unrelated regression to consider the correlation between total costs and QALYs (see Methods: within-trial analysis), and the probabilistic sensitivity analysis is presented as an additional scenario using base-case settings (see Results: within-trial analysis).
Quality assurance
The model was developed by two economic modellers. When one economic modeller added coding or inputs to the model, the other modeller checked these to identify and resolve any errors. The model was debugged by following simulated patients throughout the model, and verifying that the model was picking up the correct inputs and that calculations were being performed as intended. Simulated patient histories for a sample of patients were reviewed to ensure face validity. Results were compared with those from previous models and the within-trial analysis to ensure external validity. The within-trial analysis was checked for face validity, and coding was checked for errors by a second health economist.
Overview of systematic literature review
Objectives
A systematic literature review was undertaken in line with current recommendations. 90,91 The aim of this review was to identify evidence to inform inputs and assumptions for the long-term (> 2 years) economic model of LEAVO. Data requirements for patients with MO secondary to CRVO who were treated with intravitreal injections of ranibizumab (0.5 mg/0.05 ml), aflibercept (2.0 mg/0.05 ml) and bevacizumab (1.25 mg/0.05 ml) comprised:
-
relative clinical effectiveness and safety (including withdrawals and mortality)
-
HRQoL estimates
-
resource use and costs related to treatment, clinic visits, staffing and equipment
-
presence of ischaemic CRVO at baseline
-
prior treatment for CRVO at baseline
-
trial eye OCT CST
-
trial eye BCVA
-
non-trial eye OCT CST
-
non-trial eye BCVA
-
new-onset MO
-
injection frequency.
Methods
Literature searching
Eight electronic databases were searched: MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, The Cochrane Library, the Health Technology Assessment (HTA) database, NHS Economic Evaluation Database (NHS EED), EMBASE, Cumulative Index to Nursing and Allied Health Literature (CINAHL) and Web of Science. Searches were conducted on 28 June 2018, and the databases were searched from the date of inception to 28 June 2018. Additional searches included checking reference lists of relevant studies, grey literature searching and contacting authors.
Free-text terms and subject headings relating to the condition and interventions of interest were used to develop a search strategy (see Appendix 6, Systematic literature review to support the long-term health economic model). To identify systematic reviews, randomised trials, observational studies and economic studies (including quality-of-life studies), appropriate search filters were applied in selected databases.
Study selection, data extraction, critical appraisal and synthesis
Study selection was completed using a two-stage process based on prespecified eligibility criteria (see Appendix 6, Table 35). The titles and abstracts of retrieved records were screened. Potentially relevant full-text articles were then retrieved for detailed examination. Studies were considered for inclusion if they reported on patients with MO secondary to CRVO treated with selected anti-VEGFs [ranibizumab (Lucentis) (0.5 mg/0.05 ml), aflibercept (Eylea) (2.0 mg/0.05 ml) or bevacizumab (Avastin) (1.25 mg/0.05 ml) as a monotherapy, vs. a control, i.e. another active treatment or sham injection]. Prospective uncontrolled before-and-after studies were also reviewed for inclusion. Studies reporting the natural history of CRVO were also sought for inclusion.
Data relating to study characteristics, population characteristics, interventions administered and reported outcomes of interest were extracted into summary tables. After applying the rating of hierarchies of evidence of data sources for economic models92 in study selection (see Appendix 6, Table 55), the most appropriate methodological quality checklist endorsed by the Critical Appraisal Skills Programme (CASP)93 was used for quality assessment of the included studies. The methodological quality of individual studies was considered in study selection. Study selection, data extraction and critical appraisal were undertaken by one reviewer and checked by a second reviewer. Disagreements were resolved by discussion.
Tabular and narrative syntheses were completed because the clinical and methodological heterogeneity of included studies precluded meta-analysis of available evidence.
Results
A total of 1338 unique records were retrieved through literature searches and supplementary searching. Of these, three articles24,34,94 provided evidence of limited relevance for informing or validating the LEAVO economic model (see Appendix 6, Figure 26). A summary of included studies is presented in Table 20. For a list of excluded studies with reasons, see Appendix 6, Table 56. 95
| Characteristic | Campochiaro et al.34 | Novartis94 | McIntosh et al.24 | ||
|---|---|---|---|---|---|
| Study characteristics | |||||
| Sample size (CRVO): patients/eyes | 32 patients | 1048 patients
|
3271 eyes | ||
| Intervention(s) | IVR | IVR | N/A | ||
| Treatment schedule | TER | Not reported | N/A | ||
| Study name | RETAIN | LUMINOUS (NCT01318941) | N/A | ||
| Study design; setting | Non-RCT (open-label extension of CRUISE); USA | Non-RCT (observational, non-interventional, multicentre, open-label, single-arm study); 43 countries, 494 centres | Systematic review of various study types | ||
| Funding | Genentech, Inc., San Francisco, CA, USA | Novartis International AG | Allergan plc | ||
| Duration of follow-up (years) | 4 | 5 | 3 | ||
| Baseline characteristics | |||||
| Mean age (years) | 66.9 (SD not reported) | 69.7 (SD 12.32) | N/A | ||
| % female | Not reported | 41.5% | N/A | ||
| Duration of CRVO at baseline (months) | 4.6 | 12.6 (SD 20.2) | N/A | ||
| BCVA (letters) | 50 | 44.7 (SD 23.88) | N/A | ||
| SD-OCT (µm), mean (SD) | 639 | 463.5 (212.5)a | N/A | ||
| % of patients with ischaemic CRVO | Not reported | Not reported | N/A | ||
| VFQ-25 composite score | Not reported | 73.0 (SD 20.62) | N/A | ||
| Previous ocular history | Not reported |
|
N/A | ||
| Medical history | Not reported | Cardiovascular risk factor,b 4–61.3% | N/A | ||
| Outcomes | |||||
| Primary study outcomes |
|
|
|
||
| Secondary outcomes |
|
|
Not reported | ||
| Quality assessment of included studies | |||||
| Evidence rating (Coyle and Lee92) | 4c | 4;c 2 to 3;d 1e | 3d | ||
| Methodological quality (CASP) | Unclear quality | Unclear quality | Good quality | ||
| Results | |||||
| Visual acuity | |||||
| Mean BCVA from baseline | Mean follow-up: 51.4 months | Month 24 | Month 36 | Month 48 | |
| % of participants with BVCA of ≥ 20/40 | 43.8 | Not reported | Not reported | Not reported | Not reported |
| % of participants who gained ≥ 15 letters | 53.1 | 28.1 | Not reported | Not reported | |
| MO | |||||
| Mean retinal thickness | Month 48 (CFT) | Month 24 (CRT) | Month 36 (CRT) | Month 48 (CRT) | |
|
|
|
|
Not reported | |
| % of participants with CFT of ≤ 250 µm | All participants: 43.8% (14/32) | Not reported | Not reported | ||
| Resource use | |||||
| Mean number of injections per participant (ranibizumab), by time point | |||||
| Month 24 | 4.5 |
|
Not reported | ||
| Month 36 | 3.6 |
|
|||
| Month 48 | 3.3 |
|
|||
| Total number of injections (ranibizumab) | |||||
| All participants | 19.2 (n = 32 participants) | 6224 (n = 1048 participants); approximately 3% were treated in both eyes | |||
| Participants with resolved MO | 28.5 | Not reported | |||
| Participants with unresolved MO | 8.7 | ||||
| Mean duration between consecutive injections (weeks) | Not reported |
|
|||
| Number of visits | Not reported | 11.6 visits by month 48 | |||
| Concomitant treatments | Scatter photocoagulation (n = 2) | 37.1% (CRVO primary treated eye set) received ocular concomitant medications and significant non-drug therapies (not specified) | |||
| 70.8% (CRVO safety set) received concurrent systemic medications and significant non-drug therapies (not specified) | |||||
| AEs | |||||
| Ocular events | Four severe ocular AEs were reported (BRVO and CRVO patients) | All ocular AEs: 10.4% (109/1048) | Not reported | ||
| Ocular SAEs: 0.95% (10/1048) | |||||
| Ocular severe AEs: 1.05% (11/1050) | |||||
| Infectious endophthalmitis: not reported | |||||
| Retinal detachment: not reported | |||||
| Retinal (pigment epithelium) tear: not reported | |||||
| Anterior chamber reaction:f not reported | |||||
| Conjunctival haemorrhage: 0.57% (6/1048) | |||||
| Vitreous haemorrhage: 0.38% (4/1048) | |||||
| Cataract: 1.91% (20/1048) | |||||
| Glaucoma: 0.95% (10/1048) | |||||
| Ocular hypertension (raised intraocular pressure of > 21 mmHg): 0.57% (6/1048) | |||||
| Increased intraocular pressure: 0.86% (9/1048) | |||||
| Visual loss: 0.57% (6/1048) | |||||
| Retinal ischaemia: 0.19% (2/1048) | |||||
| Retinal neovascularisation: 0.19% (2/1048) | |||||
| MO: 0.57% (6/1048) | |||||
| Systemic AEs | 13 severe systemic AEs were reported, including two deaths. Lack of clarity about the incidence of remaining systemic events in patients with CRVO | All systemic AEs: 10.69% (112/1048) | Not reported | ||
| Serious systemic AEs: 6.01% (63/1048) | |||||
| Severe systemic AEs: 3.82% (40/1048) | |||||
| Death: 1.53% (16/1048) | |||||
| Hospitalisation: not reported | |||||
| Non-ocular haemorrhage (gastrointestinal, pulmonary, other non-ocular bleeds): not reported | |||||
| Arterial thromboembolism: not reported | |||||
| Hypertension: 0.76% (8/1048) | |||||
| Myocardial infarction: not reported | |||||
| Cerebrovascular accident (stroke): 0.29% (3/1048) | |||||
| Transient ischaemic attack: 0.29% (3/1048) | |||||
| Systemic AEs, possibly related to ranibizumab and/or ocular injection: 0.29% (3/1048) | |||||
| HRQoL | |||||
| Change in HRQoL (VFQ-25 composite score) from baseline | Not reported |
|
Not reported | ||
| Natural history | |||||
| Baseline visual acuity | 50.0 ETDRS letters | 44.7 ETDRS letters | Initial visual acuity generally poor < (20/40) in all patients. Patients with ischaemic CRVO tend to have lower mean initial visual acuity (20/200) | ||
| Generally, initial visual acuity decreases over time. Ischaemic CRVO is associated with lower subsequent visual acuity over time | |||||
| Development and resolution of MO | Not reported | Not reported | MO resolution occurs in approximately 30–31% of non-ischaemic CRVO eyes by 15 months post occlusion | ||
| MO resolves in up to 73% of ischaemic CRVO eyes by 15 months post occlusion | |||||
| Development of NV | Not reported | Not reported | 33% of non-ischaemic CRVO eyes develop NV by 12–15 months post occlusion | ||
| Up to 20% of ischaemic CRVO eyes develop NV by 8 or 9 months post occlusion | |||||
| Development of NVG | Not reported | Not reported | NVG develops in 23–60% of ischaemic CRVO eyes by 12–15 months post occlusion | ||
| Development of VH | Not reported | Not reported | VH develops in 10% of CRVO eyes by 9 months post occlusion | ||
| Conversion from non-ischaemic CRVO to ischaemic CRVO | Not reported | Not reported | Up to 27% of non-ischaemic CRVO eyes convert to ischaemic CRVO within 10 weeks–13 months post occlusion | ||
| Fellow eye involvement | Not reported | Not reported | Bilateral RVO is present in 0.4–43% of CRVO cases at presentation | ||
| Within 3 years, 1.4% of patients with CRVO develop a CRVO in the fellow eye | |||||
| Within 30 months, 5% of patients with CRVO develop a BRVO in the fellow eye | |||||
| Within 1 year 5% of patients with CRVO develop any RVO in the fellow eye | |||||
Included studies
None of the studies provided a head-to-head comparison of the clinical effectiveness outcomes of interest. Two non-randomised studies, the extended follow-up of patients with macular edema due to bRanch rETinal vein occlusion or centrAl retinal veIn occlusioN previously treated with intravitreal ranibizumab (RETAIN) study (n = 32 participants)34 and the LUMINOUS study (n = 1048 participants),94 provided long-term clinical effectiveness data for ranibizumab only. Participants in the RETAIN study had previously completed two pivotal multicentre US-based Phase III RCTs [CRUISE, for patients with CRVO, and RanibizumaB for the treatment of macular edema following bRAnch Retinal Vein Occlusion (BRAVO), for patients with BRVO]9,33,96 and a subsequent follow-up trial. 38 The mean follow-up period of the RETAIN study was 49.7 months (with a maximum follow-up of 60 months). 34 The LUMINOUS study was a 5-year international multicentre post-authorisation study (n = 43 countries, 494 centres) that evaluated the long-term effectiveness and safety of ranibizumab for all its indications in the real-world setting. Participants with CRVO made up 3.5% (n = 1048) of the entire study population of LUMINOUS (n = 30,153 patients). Evidence relating to the natural history for CRVO was obtained from a systematic review (n = 31 studies, 3271 eyes). 24
Summary of findings
No clinical effectiveness evidence was identified relating to the long-term (i.e. > 2 years’ follow-up) head-to-head comparison of intravitreal injections of ranibizumab (0.5 mg/0.05 ml), aflibercept (2.0 mg/0.05 ml) and bevacizumab (1.25 mg/0.05 ml) in patients with MO secondary to CRVO. There was extensive variation in the reporting and assessment of outcomes of interest.
Long-term visual outcomes were influenced by treatment schedules, CRVO subtype and MO resolution. 34,94 Monthly injections with ranibizumab provided an initial improvement in BCVA and MO resolution. However, this effect was reduced when treatment schedules were on an ‘as-needed’ basis or when follow-up intervals were less frequent. 34 Improved long-term outcomes were observed in patients with early MO resolution [resolved MO vs. unresolved MO at year 4: mean BCVA 73.2 ETDRS letters (20/32) vs. 56.1 ETDRS letters (20/80); mean central foveal thickness (CFT), 171.30 µm vs. 263.40 µm, respectively]. 34 Less than 5% (30/1048) of patients provided relevant data for visual acuity outcomes beyond 2 years of follow-up in the LUMINOUS study. Therefore, the observed general trends in improved vision (gain of 10 or 15 letters in visual acuity, n = 2–8 patients; gain of > 10 letters or a final BCVA of ≥ 73 letters, n = 1 patient, at 48 months) and MO resolution [mean change from baseline –257.1 µm (SD 179.91 µm), n = 7 patients, at 36 months] need to be interpreted with caution. 34 No data were available for mean change from baseline visual acuity according to ETDRS letter categories (LUMINOUS) beyond month 24 for the entire population with CRVO.
Evidence relating to the risk of systemic and ocular AEs following long-term ranibizumab use was mixed because of inadequate sample sizes and inconsistent definitions and reporting. The review also found that most patients with CRVO present with MO. 24 Of the 32 patients enrolled in the RETAIN study, 14 experienced MO resolution (43.8%). 34 A statistically significant difference in change in CFT was noted between patients with resolved MO and those with unresolved MO (263.4 µm vs. 220.6 µm; p = 0.01). The authors reported that ‘more than half still required an average of 6 injections during year 4 to control oedema, and only 25% of those patients had BCVA of 20/40 or better’. 34
The mean number of injections of 0.5 mg of ranibizumab administered in RETAIN was 19.2 over 54 months of follow-up (n = 28 patients). 34 The mean number of injections per patient administered in years 2, 3 and 4 of the study was 4.5, 3.6 and 3.3, respectively. By contrast, the mean number of injections per patient was 5.9 in LUMINOUS, by month 48. 94 A total of 6224 ranibizumab injections were received by patients with CRVO. 94 Although the majority of patients received treatment in only one eye, an estimated 3% were treated in both eyes. 94 Differences in prior intravitreal treatment status did not influence the number of injections received between subgroups.
Available evidence suggests that, after 3 years of treatment, patients receiving ranibizumab tend to experience improved quality of life [VFQ-25 composite score change from baseline of 3.6 (SD 10.70)].
The LUMINOUS study94 reflected real-world management to a greater degree than the RETAIN study. 34 This could explain the higher rate of withdrawals observed. For detailed results of the systematic review, see Appendix 6, Tables 36–54 and Figure 27.
Conclusion
Overall, there was limited evidence to adequately compare the long-term clinical effectiveness and cost-effectiveness of anti-VEGFs used in the management of MO secondary to CRVO. Comparative long-term studies of available vascular therapies for patients with MO secondary to CVRO are needed to inform treatment choices. For a detailed report of this systematic review, see Appendix 6, Systematic literature review to support the long-term health economic model.
Methods: model-based analysis
Model design
A discrete event simulation is used for the health economic model. Discrete event simulations are structured around a set of mutually exclusive events and model the pathway of individual patients through those events according to the time at which each event happens. Each individual patient has specific characteristics that may influence which events happen and when. A patient’s history through the model is recorded and can influence if and when future events happen. Events can occur at any time. Discrete event simulations are so named because they model a discrete sequence of events, but they operate in continuous time (rather than in discrete time intervals).
A discrete event simulation model has five key advantages in this application:
-
Health states are not required – each individual patient’s visual acuity can be tracked over time on a continuous scale.
-
The trial eye and non-trial eye can be modelled separately using data on the change in visual acuity over time.
-
Each patient’s history (previous visits and visual acuity) can be tracked, so the treatment continuation rule (see Chapter 2, Treatment schedule) from LEAVO can be used.
-
The follow-up visit times can be modelled by fixing the time to milestone visits and using the treatment continuation rule from LEAVO to determine other visit times.
-
Individual patients can have different baseline characteristics, to incorporate heterogeneity.
The model diagram is shown in Figure 15. The model was built and all analyses were run in Simul8 Professional Edition (SIMUL8 Corporation, Boston, MA, USA). Once a patient is simulated and has baseline characteristics and an intervention assigned, their times to events are set; these times may be fixed or sampled from a distribution. For times to each event, see Model inputs. The event with the shortest time is the next event that the patient experiences, at which point their characteristics, QALYs and costs are updated. The patient then waits until the next event. The model ends when either the patient has died or the model time horizon is reached. The process is repeated for a large number of patients, and the total costs and QALYs are calculated. The same patients are then simulated through the model again, but with a different intervention. The total costs and QALYs are compared for each intervention to calculate cost-effectiveness results.
FIGURE 15.
Health economic model structure. Reproduced with permission from Pennington et al. 95 This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. The figure includes minor additions and formatting changes to the original figure.

At each model event, costs, utilities, total costs and QALYs are updated. Each model event (i.e. visit to ophthalmologist, ocular AE, withdrawal, new-onset MO in the non-trial eye, annual change in visual acuity, death, model end) is explained in more detail in the following subsections.
Model events
Visit to ophthalmologist
When a modelled patient visits the ophthalmologist, their visual acuity (measured using BCVA letter score) and CST are updated in both eyes, and decision rules are used to determine whether or not the patient receives an anti-VEGF injection, and the time to their next visit.
Within the 100-week trial period, the model uses the same treatment continuation rules as specified in the LEAVO protocol:88
-
All patients (except those who have withdrawn) attend visits and have a mandated injection at baseline and at 4, 8 and 12 weeks.
-
All patients attend visits at weeks 16 and 20, but have an anti-VEGF injection only if their BCVA is > 83 letters and they meet one of the following the re-treatment criteria:
-
decrease in visual acuity of ≥ 6 letters between the previous and the current visit, attributed to an increase in OCT CST
-
increase in visual acuity of ≥ 6 letters between the previous and the current visit
-
an OCT CST of > 320 µm
-
an OCT CST increase of > 50 µm from the lowest previous visit.
-
-
From weeks 24 to 96, the same re-treatment criteria as at weeks 16–20 are applied. If the patient does not meet the re-treatment criteria and was not treated at either of the two previous visits, the time to their next visit is increased from 4 weeks to 8 weeks.
Beyond the trial period, the treatment continuation rules are informed by advice from five clinicians involved in LEAVO (PH, SS, AL, YY and Michael Williams) and by guidance from the Royal College of Ophthalmologists. 8 The following rules are applied:
-
If the patient has not had an injection since year 1, they do not receive an injection and do not visit the ophthalmologist again.
-
Within the first 5 years, the same re-treatment criteria as used in LEAVO are applied to determine whether or not a patient has an injection, but the time to the next visit is increased to 12 weeks. If the patient does not meet the re-treatment criteria and was not treated at either of the two previous visits, they do not receive an injection and do not visit the ophthalmologist again.
-
After 5 years, patients no longer receive injections. They have three further follow-up visits with the ophthalmologist, 12 weeks apart.
For patients who will not visit the ophthalmologist again, the time to visit is set to infinity, and the time to annual change in BCVA score is set at 1 year.
Ocular adverse event
Patients who have an ocular AE are assumed to incur a cost for treating the AE, and remain on treatment. As patients may have more than one AE, the time to AE is resampled from the same distribution.
Withdrawal
Patients who withdraw are assumed to immediately discontinue their assigned intervention and receive no treatment. They no longer visit their ophthalmologist to be assessed for or to receive treatment. As patients cannot withdraw more than once, the time to withdrawal is set at infinity.
New-onset macular oedema in the non-trial eye
Patients may develop MO in the non-trial eye. When this occurs, to reflect the associated change in visual acuity and CST associated with MO, the patient is assigned a new BCVA and CST measurement for the non-trial eye. This is sampled from the baseline characteristics of the trial eye for patients of the same sex and similar age.
Patients who develop MO in the non-trial eye are assumed to receive the same intervention in their non-trial eye as was assigned to their trial eye. Patients who are still on their assigned treatment (and who have not discontinued because of treatment continuation rules or withdrawal) will receive treatment in both eyes, whereas patients who have discontinued or withdrawn from treatment in their trial eye will receive treatment in the non-trial eye only. If a patient is still receiving treatment in their trial eye, their initial visit for the non-trial eye will occur at the same time as the next visit for the trial eye. After this point, the same treatment continuation rule is applied to each eye to determine when the next visit for each eye occurs. If a patient is not still receiving treatment in their trial eye, the patient immediately has a visit for the non-trial eye and follows treatment continuation rules for that eye only.
As patients cannot redevelop MO in the non-trial eye, the time to new-onset MO is then set at infinity.
Annual change in visual acuity
Visual acuity is used to predict utility and resource use. While patients are still visiting the ophthalmologist, their visual acuity is updated at each visit. Once the patient no longer receives injections or no longer has follow-up visits, their visual acuity is tracked using an annual change event. After each annual change, the time to the next annual change is set at 1 year.
Death
When a modelled patient dies, they move immediately to the model end event.
Model end
Once a modelled patient reaches the model end, their costs and QALYs are reported.
Model inputs
Baseline characteristics
The model uses the baseline characteristics of LEAVO participants to preserve the relationship between characteristics. Each modelled patient has the baseline age, sex, trial and non-trial eye BCVA and CST of one of 452 LEAVO participants for whom all of these variables were available at baseline. This approach is consistent with other simulation models in ophthalmology. 97
Central subfield thickness and visual acuity
The re-treatment algorithm assesses both OCT CST and BCVA, so both must be modelled for treated eyes. BCVA in both eyes is important for predicting HRQoL, so BCVA is modelled for both eyes.
Treated eyes
Growth models (longitudinal analyses to estimate growth trajectories over a period of time) are fitted to CST and BCVA from the LEAVO data. In these models, CST (or BCVA) at weeks 12, 24, 52, 76 and 100 are estimated as a function of time, baseline CST (or BCVA), age at baseline, intervention, number of injections and time since last injection. Sex is found not be a significant predictor of CST or BCVA, so it is excluded. Intervention is not a significant predictor of CST or BCVA, but it is included to reflect numerical differences between the interventions.
The equation for yit, the BCVA score for patient i at time t, is as follows:
where
and
where tn2 = 1 for aflibercept and 0 otherwise, and tn3 = 1 for bevacizumab and 0 otherwise, and ξ is an error term.
(The equation for CST follows the same structure, but uses CST at baseline/100 instead of BCVA at baseline/10.)
Whereas η, α and β (age at baseline, CST or BCVA at baseline and intervention) are time-invariant covariates, γ1 and γ2 (number of injections and time since last injection) are time-variant covariates, with values available at 12, 24, 52 and 76 weeks only. To estimate CST and BCVA in the economic model, these covariates are used at the week 12, 24, 52, 76 and 100 visits. For other visits, the following approaches are used:
-
Weeks 4 and 8 – CST and BCVA are calculated at week 12, and linear interpolation is used to estimate CST and BCVA at the week 4 and week 8 visits.
-
Visits from week 16 to week 100, excluding weeks 24 and 52 – CST and BCVA are calculated for the closest milestone visits before and after the non-milestone visits, and interpolation is used to estimate BCVA at the non-milestone visits.
-
Visits beyond week 76 – the time-varying covariates appear similar towards the end of LEAVO, and so models that restricted these covariates to be the same at weeks 76 and 100 were compared with unrestricted models. Log-likelihood tests indicated that the null hypothesis that the restricted models were true should not be rejected. The restricted models suggest that the effect of the number of injections and time since last injection flatten towards the end of LEAVO and can, therefore, be used to extrapolate beyond 100 weeks.
Untreated eyes
Untreated eyes are considered to be eyes that never received treatment or eyes whose treatment has ended or been withdrawn. The same assumption is used for treated eyes whose most recent injection was at least 1 year ago.
Central subfield thickness is not modelled for the non-trial eye unless the patient develops MO in the non-trial eye, in which case CST and BCVA for the non-trial eye are modelled using the same approach as for the trial eye.
Best corrected visual acuity is modelled for untreated eyes using natural history data. The Beaver Dam Eye Study98 was a large population-based study that recorded BCVA in patients over 5 years. This study98 reported the letters gained or lost in the left and right eyes for people aged < 55, 55–64, 65–74 and ≥ 75 years, and has been used in previous CRVO economic models. 51 Combining the right and left eye data, the annual average decrease in BCVA is –0.02 letters [standard error (SE) 0.04 letters] for those aged 55–64 years, 0.26 letters (SE 0.04 letters) for those aged 65–74 years and 0.76 letters (SE 0.06 letters) for those aged ≥ 75 years. There is no change for people aged < 55 years. These data appear consistent with a study of the natural history in CRVO,99 which reports that increasing age was positively associated with visual acuity deterioration, and over 2–5 years in eyes with non-ischaemic CRVO MO, 14% improved, 47% stayed the same and 39% worsened.
Ocular adverse events
The model considers the same ocular AEs as reported in the safety analysis in LEAVO: infectious endophthalmitis, traumatic cataract, retinal tear, retinal detachment, conversion to ischaemic CRVO, anterior segment neovascularisation, retinal neovascularisation, and vitreous haemorrhage and intraocular pressure elevation. As relatively few patients experienced these AEs in LEAVO (e.g. only one patient had infectious endophthalmitis), modelling the time to specific events would be highly uncertain and, in some cases, impossible. Therefore, the model considers the time to any ocular AE, using the data for all ocular AEs (and applying a cost per average AE; see Adverse event costs). When the date of the AE was missing, multiple imputation was used to impute the date based on the trial arm and whether or not an AE occurred.
Survival analysis was used to fit parametric models to extrapolate time to event beyond the trial period. The log-rank test found no statistically significant difference between the time to first AE and time to subsequent AEs (p = 0.128), and the number of subsequent AEs was small, so the time to first AE is used as the time to first or subsequent AEs in the model.
Although the time to AE is not statistically significantly different between the interventions (p = 0.683), they are modelled separately to reflect numerical differences in the deterministic analysis. The probabilistic analysis considers the uncertainty around point estimates, reflecting that the interventions are not significantly different. According to the Akaike information criterion (AIC) and Bayesian information criterion (BIC), the Weibull was the best-fitting parametric model. As no data are available on the ocular AE rates for any of the three interventions beyond the trial period (see Overview of systematic literature review), external validation is not possible. The Weibull is therefore used to model the time to AEs. All three interventions have the same shape parameter of 0.745, demonstrating that the probability of having an ocular AE decreases over time.
Withdrawal
Survival analysis was used to fit parametric models to extrapolate time to withdrawal beyond the trial period. The three interventions are modelled separately to reflect numerical differences, despite non-statistically significant differences in the data (p = 0.572). The AIC and BIC are similar between parametric models, and no external validation was possible because of a lack of data. The Weibull distribution is used to model time to withdrawal event, with shape parameter of 1.385, demonstrating that the probability of withdrawing increases over time.
New-onset macular oedema
Eight of 463 patients in LEAVO either had new-onset MO recorded as an AE or received an anti-VEGF injection in the non-trial eye. This is a small number of observations to fit parametric models to using survival analysis; instead, it is assumed that the occurrence of new-onset MO follows an exponential distribution. The rate of new-onset MO is calculated as 0.009 per year. 100
Mortality
As only 13 LEAVO participants died, the data are not sufficiently mature to be analysed and included in the model. Instead, the model applies an age- and sex-standardised mortality ratio to the probability of death101 for the general UK population102 in order to represent the increased mortality associated with CRVO.
Number of simulated patients
A drawback of individual-level simulation approach is introducing first-order uncertainty (also known as stochastic uncertainty), whereby the mean cost and benefit outcomes may vary between different model runs even if the same input parameters for a given individual (patient) are used. 103 To reduce this type of uncertainty, 7000 patients are simulated for each model run. This ensured that a sufficient number of combinations of different patient characteristics are achieved, and that first-order uncertainty is accounted for by allowing a uniform coverage of a random number seed. Appendix 6, Figure 28, shows that total costs and QALYs are stable when ≥ 7000 patients are sampled.
Health-related quality of life
The model considers patients’ BCVA over their lifetimes. To include patients’ utilities over time, the model predicts utility from BCVA and other demographic variables. This prediction is termed a ‘mapping’ or ‘crosswalk’ and may be used in economic evaluation to convert clinical measures to health utilities when either utility data are not directly available or there is a need to relate clinical outcomes to health utilities in the long term. Developing a mapping requires a data set that contains both the clinical measure and the utility measure. LEAVO provided this data set for BCVA and three measures of utility.
Health-related quality-of-life measures
Three HRQoL questionnaires were used to collect health utility data in the trial:
As specified in the health economic analysis plan and the trial protocol, the VFQ-25 was chosen as the primary measure, with the EQ-5D and EQ-5D-V used in secondary analyses. 9,12 The EQ-5D has been shown to perform poorly in eye disorders, including AMD. 107 Although the VFQ-25 may not meet the NICE reference case,84 non-EQ-5D utility values have been used in economic evaluations in many cases, including eye conditions. 108
Each HRQoL questionnaire was collected at the six milestone visits of LEAVO: baseline and weeks 12, 24, 52, 76 and 100. Utility scores from the VFQ-25 were calculated using the Visual Function Questionnaire-Utility Index (VFQ-UI) for each patient. 109 This tariff uses six items (questions 6, 11, 14, 18, 20 and 25), representing six of the VFQ-25 subscales. 110 Using the crosswalk, the EQ-5D health states were converted to the three-level scale, as this is preferred by NICE. 111,112 Utility scores for the EQ-5D-V were calculated by first taking the EuroQol-5 Dimensions, three-level version (EQ-5D-3L), score and then subtracting the EQ-5D-V score as a utility decrement applied to the individual patient-level data. 106
Mapping from best corrected visual acuity to utility
Data from all milestone visits were combined to maximise the number of observations using a complete-case analysis. At each observation, variables were generated for the visual acuity in the better-seeing eye (BSE) and the worse-seeing eye (WSE), according to whether BCVA was greater in the trial eye or the non-trial eye.
Standard statistical models are often a poor fit to the distribution of utility data113 (particularly EQ-5D data), so adjusted limited dependent variable mixture models (ALDVMMs) are used. Mixture models can be used to represent latent classes (discrete variables that are inferred rather than directly observed) within an overall population, or to provide a very flexible semiparametric framework for modelling distributions with unusual shapes. Limited dependent variables are those whose range of possible values are restricted. ALDVMMs, therefore, represent a flexible framework for developing models to reflect the distribution of utility data.
The ALDVMMs were estimated with one to four components (classes). 114 Models were fitted for the three utility measures. The independent variables used to predict utility in the components were age, sex, BSE BCVA and WSE BCVA. The interaction between the BSE and the WSE was considered as a variable, but its inclusion worsened model fit; therefore, it was excluded from the model specification. BSE BCVA and WSE BCVA are used to determine the probability of a patient belonging to the different components. Intervention is not included as an independent variable, as its impact on utility is expected to be through changing BCVA and not through a treatment-specific effect.
To determine the number of components that should be used for each utility measure, model fit was compared using the mean error, mean absolute error (MAE), root-mean-square error (RMSE), AIC, BIC and visual inspection. In each utility measure, the mean error, MAE and RMSE were generally similar for models with two, three and four components for which component membership was predicted by the BCVA score of the BSE and the WSE. The AIC and BIC, which penalise models with more parameters to reduce overfitting, indicated that the best-fitting model for VFQ-UI has three components, whereas the best-fitting models for the EQ-5D and the EQ-5D-V have two components. Appendix 3, Table 34, shows the model parameters.
The utility in each component is calculated as follows:
-
A temporary variable u is calculated by multiplying the within-component coefficients by the individual patient’s characteristics (as per a regression equation).
-
Parameter a is calculated as –(4)a=uu−uσ.
-
Parameter b is calculated as –(5)b=ul−uσ.
-
Parameter c is calculated as –(6)c=φ(a)−φ(b).
-
Parameter d is calculated as –(7)d=Φ(a)−Φ(b).
-
If parameter c is between –0.00000001 and 0.00000001 parameters, c is set to 0 and d is set to 1.
-
The expected utility within the component is calculated as –(8)(1–Φ(a)+(Φ(a)−Φ(b)))×(u+ (σ×cd))+(ul×Φ(b)),
where uu is the highest feasible utility next to 1, l is the lowest feasible utility, σ is the variance of the component, φ is the probability density function for the normal distribution with mean 0 (SD 1), and Φ is the cumulative distribution function for the normal distribution with mean 0 (SD 1).
The probability of belonging to each component is calculated by the exponentiation of the product of the between-components by an individual patient’s characteristics. For the last component, this will equal 1. The probabilities are then normalised by dividing by the sum of all probabilities.
The expected utility within each component is multiplied by the probability of belonging to each component. The sum of these gives a patient’s utility. The relationship between visual acuity and utility for the BSE and the WSE is provided in Figure 16. A Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA) tool that calculates a patient’s utility score (VFQ-UI, EQ-5D, EQ-5D-V) based on our mappings is provided. 115
FIGURE 16.
The VFQ-UI mapping comparison of observed and predicted scores: (a) VFQ-UI score vs. BSE BCVA score; (b) VFQ-UI score vs. WSE BCVA score; (c) VFQ-UI score vs. BSE ETDRS score; and (d) VFQ-UI score vs. WSE ETDRS score.


Resource use and costs
Costs were calculated using Great British pounds for 2017/18; when costs were not available for this year, they were inflated using the Hospital and Community Health Service (HCHS) index. 116
Intervention costs
The list price is £551.00 for the ranibizumab injection and £816.00 for the aflibercept injection. 117 These prices are used in the base-case analysis. A discount is applied to these costs in scenario analyses to explore the impact of confidential Patient Access Schemes (PASs). The list price of bevacizumab is £243.00; however, this is the cost of a large infusion vial of the drug. 117 As discussed in Chapter 2, Intervention: bevacizumab (1.25 mg/0.05 ml), during LEAVO, the injections of bevacizumab were separated from the larger bottle into pre-filled syringes by the Liverpool and Broadgreen Pharmacy Aseptic Unit. 88 This compounding of the drug was deemed to be legal in a judicial review in 2018, which cited the price per injection as £28. 55 It is assumed that this price includes any costs associated with compounding the drug, such as staff time and storage costs. This price is used in the base-case analysis. Patients incur an injection cost for each eye that is treated.
Visit costs
When a simulated patient visits the ophthalmologist to be assessed against re-treatment criteria, and possibly treated with an anti-VEGF injection, costs are incurred for the visit itself, the OCT examination (if performed) and the drug cost of the injection.
The cost of the initial visit is £140.04: a first multiprofessional consultant-led outpatient ophthalmology visit. Subsequent visits cost £105.19: a follow-up multiprofessional consultant-led outpatient ophthalmology visit. 118 Patients who are receiving treatment in both eyes incur 1.5 times the visit costs, representing clinician advice that approximately half of all patients would have both eyes treated in a single visit, and the other half would require two separate visits.
The cost of the OCT examination is £108.21: a minor vitreous retinal outpatient ophthalmology procedure. 118 This is incurred for each eye for which re-treatment criteria are assessed.
Disease management costs
A bespoke resource use questionnaire was developed to capture resource use relating to a participant’s eye condition during the 100-week trial. Participants were asked to complete the questionnaire at baseline and at weeks 12, 24, 52, 76 and 100. Although resource use questionnaires can be vulnerable to recall bias, the questionnaire captured 9 of the 10 questions recommended as core items in standardised resource measures,119 collecting information relating to hospital admissions, health-care contacts and continuous care and support of patients.
The model includes resource use for the following:
-
visits to the eye consultant, general practitioner (GP), general practice nurse, accident and emergency (A&E), eye A&E and optometrist
-
low-vision appointments
-
telephone calls to the eye hospital helpline, ophthalmologist and GP.
Resource use data were analysed from the resource use questionnaire in LEAVO, with completed measures combined for all patients across the trial period to maximise the number of observations. Resources with < 10 observations were excluded. Participants who developed new-onset MO or who had an AE were excluded from the analysis to prevent double-counting of the resource use associated with these events.
Ordinary least squares regression was performed to estimate the relationship between WSE BCVA and resource use per 3-month period (a higher BCVA score predicted less resource use). When WSE BCVA was not a statistically significant predictor of resource use, the model used the mean resource use for all patients.
The resource use questionnaire asked patients to indicate the number of events, such as the number of visits to eye casualty, number of telephone calls with health-care professionals or number of hours of care received, over the previous 3 or 6 months. However, the duration of each visit was not recorded; therefore, average estimates were used based on the NHS Reference Costs 2017/18118 or the Unit Costs of Health and Social Care 2018,116 when relevant.
When a patient reported a hospital admission, if an associated procedure was named as the reason for the admission, average resource costs associated with the procedure were used based on the NHS reference costs. The number of bed-days reported by a participant was then used to adjust the cost by adding or subtracting the difference between the number of bed-days reported by the participant and the number expected for the procedure, multiplied by the cost of an excess bed-day. If the same concomitant procedure was also reported for this participant, costs were only counted once using the information provided by the participant relating to length of stay. If no reason was recorded for the admission, then the cost of a non-elective excess bed-day was used. 118
The costs and parameters for the resource use regressions are shown in Appendix 3, Table 34.
Adverse event costs
As the model considers any ocular AE, modelled patients who experience ocular AEs incur the average cost for an ocular AE, based on the proportion of participants in LEAVO experiencing each ocular AE. This is calculated by multiplying the number of each type of ocular AE by the cost for treating that ocular AE, and dividing the total by the number of participants in LEAVO who experienced ocular AEs. The cost per ocular AE is the same for the three interventions at £317.96. Costs for each ocular AE are from NHS Reference Costs 2017/18118 or the British National Formulary,117 and are shown in Appendix 3, Table 34.
Blindness costs
Modelled patients may become blind when the BCVA score of both eyes is at ≤ 35 letters, consistent with the definition of severely sight-impaired from the Royal National Institute of Blind People120 and previous models in MO. 51,112 Blindness was tracked at visit and annual BCVA change, both of which are events at which BCVA scores can change. BCVA scores can fluctuate throughout a patient’s lifetime, meaning that a patient can experience more than one blindness episode in their life.
Two sets of costs associated with blindness are defined from literature: one-off costs and recurrent costs. 112,121 When a patient becomes blind for the first time, one-off costs associated with blindness are incurred, including blind registration, low-vision aids and low-vision rehabilitation. As long as a patient remains blind, they incur recurrent costs, including community care, residential care, treatment for depression and hip replacement.
The costs of blindness registration, daily community care and weekly residential care are £60.50, £27.64 and £115.40, respectively. 116 Low-vision rehabilitation and hip replacement unit costs are estimated at £153 and £4170, respectively. 118 The costs of low-vision aids and the annual costs of depression are estimated from Meads and Hyde122 and TA460,123 respectively, both of which are inflated to 2018 values using the HCHS indices. 116
The proportion of blind patients receiving each service is taken from Colquitt et al. 121
The costs and proportion of patients incurring these costs are shown in Appendix 3, Table 34.
Addressing uncertainty
The base-case analysis uses the VFQ-UI, and scenario analyses consider the EQ-5D and EQ-5D-V. Scenario analyses consider shorter time horizons and a cost of £243 for bevacizumab.
Patient Access Schemes are in place for ranibizumab and aflibercept, offering a discount on the list price. 13,51 However, the level of discount is confidential and so is unknown. Therefore, we consider threshold analyses to determine the level of discount that would be needed to change the decision about the most cost-effective intervention.
Results are presented for the base-case, EQ-5D, EQ-5D-V and 100-week scenarios using probabilistic sensitivity analysis to incorporate parameter uncertainty. Whereas deterministic analysis (see Appendix 6, Model-based analysis results: additional data) uses point estimate (mean) inputs, probabilistic sensitivity analysis simultaneously samples all uncertain inputs from their associated distributions. Microsoft Excel was used to sample uncertain parameters from their distributions and to maintain relationships between related parameters. Mean total costs and QALYs are calculated for the modelled patient cohort for each simulation. 95% CIs around the mean and total costs and QALYs are presented using the SE to reflect the uncertainty around the mean. The mean of the mean total costs and QALYs for each intervention are calculated from all of the simulations and used to calculate mean probabilistic ICERs. The uncertainty around the mean probabilistic ICER is calculated using the incremental net monetary benefit (INMB) approach to avoid the mathematical limitations of interpreting uncertainty around a ratio. 124
Running probabilistic sensitivity analysis on a discrete event simulation model is computationally expensive, but it is vital that a sufficient number of simulations are performed so that the model results converge. The number of simulations required for the results to converge can be calculated by comparing the upper and lower bounds of the INMB with zero for a defined cost-per-QALY threshold. 124 Using the tutorial provided by Hatswell et al. 124 and a threshold of £30,000 per QALY, very few probabilistic simulations are required for the analyses. This is because the ICERs are so far away from the threshold of £30,000 per QALY that there is very little uncertainty associated with the decision about which intervention is most cost-effective (the INMB CIs exclude zero). Probabilistic sensitivity analysis is presented using 500 simulations for all scenarios. This is sufficient to ensure that the INMBs have converged for each comparison of two interventions.
Methods: within-trial analysis
Method of economic evaluation
The methods for the within-trial analysis were prespecified in a health economic analysis plan prior to database lock. 89 The primary outcome of the within-trial health economic analysis was to establish the short-term cost-effectiveness of:
-
aflibercept compared with ranibizumab
-
bevacizumab compared with ranibizumab
-
aflibercept compared with bevacizumab.
A fully incremental analysis (ranking the alternative treatment options by total costs and ruling out dominated and extendedly dominated options from the comparison) was also performed. The economic analysis used individual patient-level data collected as part of LEAVO. The total costs and QALYs over the 100-week follow-up period of the trial were used to calculate the incremental cost per QALY gained.
An ITT population was used, including all of the participants randomised to each treatment group. Analyses were conducted using Stata® version 15 (StataCorp LP, College Station, TX, USA) and R (The R Foundation for Statistical Computing, Vienna, Austria).
Health-related quality of life
The individual patient-level QALYs were calculated from the utility scores for each HRQoL questionnaire at baseline and at subsequent follow-up time points using linear interpolation.
Resource use
The costing approach included identification of resource use, measurement and valuation. 125 The resource use associated with delivery of the intervention, hospital admissions, health-care contacts, continuous care and support and concomitant medications and procedures, and the costs associated with blindness, were measured.
Identification of resource use
The within-trial analysis included costs related to a participant’s eye condition as collected using the resource use questionnaire (see Disease management costs); the delivery of the intervention; and concomitant medications.
Information relating to concomitant medications was collected by health-care professionals. Resource use relating to the delivery of the intervention was captured at each visit, and included drug costs, outpatient appointment costs and the costs of any tests commonly conducted at these appointments.
Ocular AEs were captured using the resource use questionnaire and from data relating to concomitant procedures and medications. To capture the relevant costs associated with blindness, the costs of blind registration and low-vision aids were applied to participants who became partially or severely sighted during the trial. A participant was deemed to be partially sighted or severely sight-impaired if their BCVA score was ≤ 58 letters in both eyes. 120 These costs were applied once during the course of the trial, namely at the first time a participant met this criterion, as low-vision aids are thought to be incurred biannually. 50 It is assumed that the same proportion of participants who can register as severely sighted also register as partially sighted and the same costs are incurred for low-vision aids to give a conservative estimate of the cost of blindness. This analysis differs from the model-based analysis to include cost associated with blindness for partially sighted patients.
Measurement of resources
The costs of medications were costed according to standard NHS sources, when available. 117
Valuation of resources
Unit costs, summarised in Appendix 6, Table 57, were applied to each resource use event at the individual patient level to calculate their total cost of resource use over the 100-week trial. Intervention costs used in the within-trial analysis are similar to those used in the model-based analysis (see Intervention costs).
Analytical methods
The base-case CUA was based on multiple imputation using chained equations to account for missing data. The VFQ-UI was used to calculate QALYs. The ICER was estimated comparing bevacizumab and aflibercept with ranibizumab, and aflibercept with bevacizumab. If applicable, the ICER was then compared with the NICE cost-effectiveness threshold range of £20,000–30,000 per QALY gained.
Missing data
Missing data can give misleading estimates in a within-trial cost-effectiveness analysis. A complete-case analysis uses only patients with no missing data on the key cost and benefit outcomes. This is undesirable because it reduces the sample size and affects the power of the study. 126 The following assumptions were made:
-
When a patient died, their utility scores at all subsequent milestone visits were set at zero. Their costs at the next milestone visit were then assumed to be half the costs recorded at the previous visit, unless their next visit was at 52 weeks, in which case the costs were assumed to be the same as the week 24 costs.
-
When a participant withdrew from the trial, if a withdrawal appointment was carried out, cost and utility data were assigned to the nearest milestone visit and all subsequent costs were set at zero and utilities were recorded as missing.
Once the assumptions had been applied to the data, patterns of missing data were assessed using the following descriptive analyses:
-
proportion of missing data by treatment arm, at each follow-up period, to assess whether or not missing data differed by arm
-
missing data patterns to determine whether or not data were missing for all items or individual items of utility scores and resource use items over the trial follow-up.
The multiple imputation chained equation method with predictive mean matching was used to impute missing values of costs, QALYs and baseline covariates. The year 1 QALY imputation model included the following covariates: age, sex, ethnicity, previous treatment, baseline utility, time since diagnosis, baseline BCVA in the trial eye and baseline BCVA in the non-trial eye. The year 2 QALY imputation model also included the imputed year 1 QALY data, as per recommendations by Faria et al. 126 The year 1 cost imputation model included the same covariates as the year 1 QALY model, as well as baseline resource use and site. The year 2 cost imputation model also included the imputed year 1 costs. The number of imputations was based on the highest percentage of missing data for the variables of interest (baseline utility, QALYs and total cost). The imputation was performed per randomisation arm for all imputed variables except baseline covariates for which data were missing, for which imputation was performed across all observations.
Seemingly unrelated regression
A seemingly unrelated regression (SUR) model was used to estimate the difference in mean total costs and QALYs between treatment arms, taking into account the correlation between total costs and QALYs. 127 The SUR model estimated the full variance–covariance matrix, which was further used to address uncertainty. 128 The regression equation for total costs included the randomisation arm. The regression equation for QALY included the randomisation arm and baseline utility in order to control for imbalances in baseline utility between treatment arms. 127,129 The model assumed a normal distribution for both costs and QALYs. 128 Marginal effects in each treatment arm were calculated using the SUR, without adjusting for baseline utility.
Addressing uncertainty
A parametric approach was used to address the uncertainty around the CUA estimates using the following key parameters estimated from the SUR output:
-
difference in mean QALYs
-
SE of mean differential QALYs
-
difference in mean total costs
-
SE of mean differential total costs
-
covariance between total costs and QALYs.
To illustrate uncertainty, cost-effectiveness confidence ellipses and net monetary benefit (NMB) lines with CIs were produced for each pairwise comparison of treatments. In addition, a cost-effectiveness acceptability curve (CEAC) was constructed, illustrating the probability that each treatment was the most cost-effective compared with all alternative treatments at a range of threshold values that varied from £0 to £400,000. To calculate the probability that a treatment was the most cost-effective option, costs and QALYs for each treatment were sampled using a bivariate normal distribution.
Scenario analyses were calculated using SUR output as in the base-case analyses. The scenario analyses were as follows:
-
QALYs estimated using the EQ-5D, using imputed data
-
QALYs estimated using the EQ-5D-V, using imputed data
-
drug price discounts – the CUA carried out using imputed data and applying a 30% and 50% discount to the drug prices of ranibizumab and aflibercept, reflecting possible confidential PASs offered by pharmaceutical companies to the NHS
-
list price for bevacizumab – the CUA carried out using the list price for bevacizumab taken from the British National Formulary (£243)
-
complete-case analysis – the CUA carried out using complete-case data from LEAVO only
-
52-week analysis – the CUA carried out using imputed data from LEAVO up to the week 52 milestone visit.
Results: model-based analysis
Base-case analysis
The results are presented in Table 21. In the base-case analysis, bevacizumab generates the most QALYs, followed by ranibizumab and aflibercept. Aflibercept generates the highest costs, followed by ranibizumab and bevacizumab. The CIs for the incremental costs and QALYS do not contain zero, demonstrating that there is a difference in both costs and effects for the three interventions (however, for QALYs this is numerically small). Bevacizumab dominates (i.e. it is more effective and less costly than) both ranibizumab and aflibercept. The 95% CIs for the NMB at £30,000 per QALY do not contain zero. At a threshold of £20,000–30,000 per QALY, bevacizumab is the most cost-effective intervention (ranibizumab dominates aflibercept).
| Analysis | Total (95% CI) | Incremental (95% CI) | ICER (£) (95% CI) | ||
|---|---|---|---|---|---|
| Costs (£) | QALYs | Costs (£) | QALYs | ||
| Base-case analysis | |||||
| Bevacizumab | 18,353 (17,782 to 18,925) | 9.678 (9.572 to 9.785) | |||
| Ranibizumab | 30,226 (29,386 to 31,066) | 9.635 (9.512 to 9.757) | 11,873 (11,458 to 12,288) | –0.044 (–0.074 to –0.013) | Dominated (INMB:a –14,316 to –12,067) |
| Aflibercept | 35,026 (33,990 to 36,062) | 9.569 (9.429 to 9.710) | 16,673 (16,036 to 17,310) | –0.109 (–0.161 to –0.057) | Dominated (INMB:a –21,864 to –18,040) |
| Scenario analysis: EQ-5D for utilities | |||||
| Bevacizumab | 18,353 (17,782 to 18,925) | 8.782 (8.740 to 8.823) | |||
| Ranibizumab | 30,226 (29,386 to 31,066) | 8.795 (8.754 to 8.836) | 11,873 (11,458 to 12,288) | 0.013 (0.008 to 0.018) | 908,532 (659,881 to 1,476,254) |
| Aflibercept | 35,026 (33,990 to 36,062) | 8.832 (8.790 to 8.874) | 4800 (4445 to 5154) | 0.037 (0.032 to 0.043) | 128,513 (110,116 to 152,663) |
| Scenario analysis: EQ-5D-V for utilities | |||||
| Bevacizumab | 18,353 (17,782 to 18,925) | 8.346 (8.282 to 8.410) | |||
| Ranibizumab | 30,226 (29,386 to 31,066) | 8.351 (8.283 to 8.419) | 12,791 (12,148 to 13,434) | 0.005 (–0.007 to 0.017) | 2,491,676 (INMB:a –12,327 to –11,155) |
| Aflibercept | 35,026 (33,990 to 36,062) | 8.369 (8.289 to 8.449) | 4800 (4445 to 5154) | 0.018 (0.000 to 0.035) | 268,963 (INMB:a –4930 to –3602) |
| Scenario analysis: 100-week time horizon | |||||
| Bevacizumab | 6349 (6293 to 6405) | 1.641 (1.631 to 1.651) | |||
| Ranibizumab | 15,254 (14,962 to 15,545) | 1.641 (1.631 to 1.651) | 8905 (8650 to 9161) | 0.000 (0.000 to 0.001) | 34,067,841 (217,070 to 10,420,696) |
| Aflibercept | 18,844 (18,438 to 19,249) | 1.646 (1.636 to 1.655) | 3590 (3400 to 3780) | 0.005 (0.004 to 0.005) | 793,348 (688,418 to 926,352) |
| Scenario analysis: bevacizumab list price from the BNF117 (£243) | |||||
| Bevacizumab | 23,530 (22,884 to 24,176) | 9.678 (9.572 to 9.785) | |||
| Ranibizumab | 30,226 (29,386 to 31,066) | 9.635 (9.512 to 9.757) | 6696 (6400 to 6992) | –0.044 (–0.074 to –0.013) | Dominated (INMB:a –9084 to –6937) |
| Aflibercept | 35,026 (33,990 to 36,062) | 9.569 (9.429 to 9.710) | 11,496 (10,961 to 12,030) | –0.109 (–0.161 to –0.057) | Dominated (INMB:a –16,636 to –12,905) |
The cost-effectiveness scatterplots (Figure 17) display the variation in the incremental costs and QALYs in the probabilistic samples. These are akin to presenting the SD; although they display the dispersion of the set of values, they do not present the uncertainty around the mean. The 95% CIs using the SE present the uncertainty around the mean, and show a difference in incremental QALYs for the three comparisons.
FIGURE 17.
Model-based analysis: cost-effectiveness scatterplots. (a) Ranibizumab vs. bevacizumab; (b) aflibercept vs. ranibizumab; and (c) aflibercept vs bevacizumab.
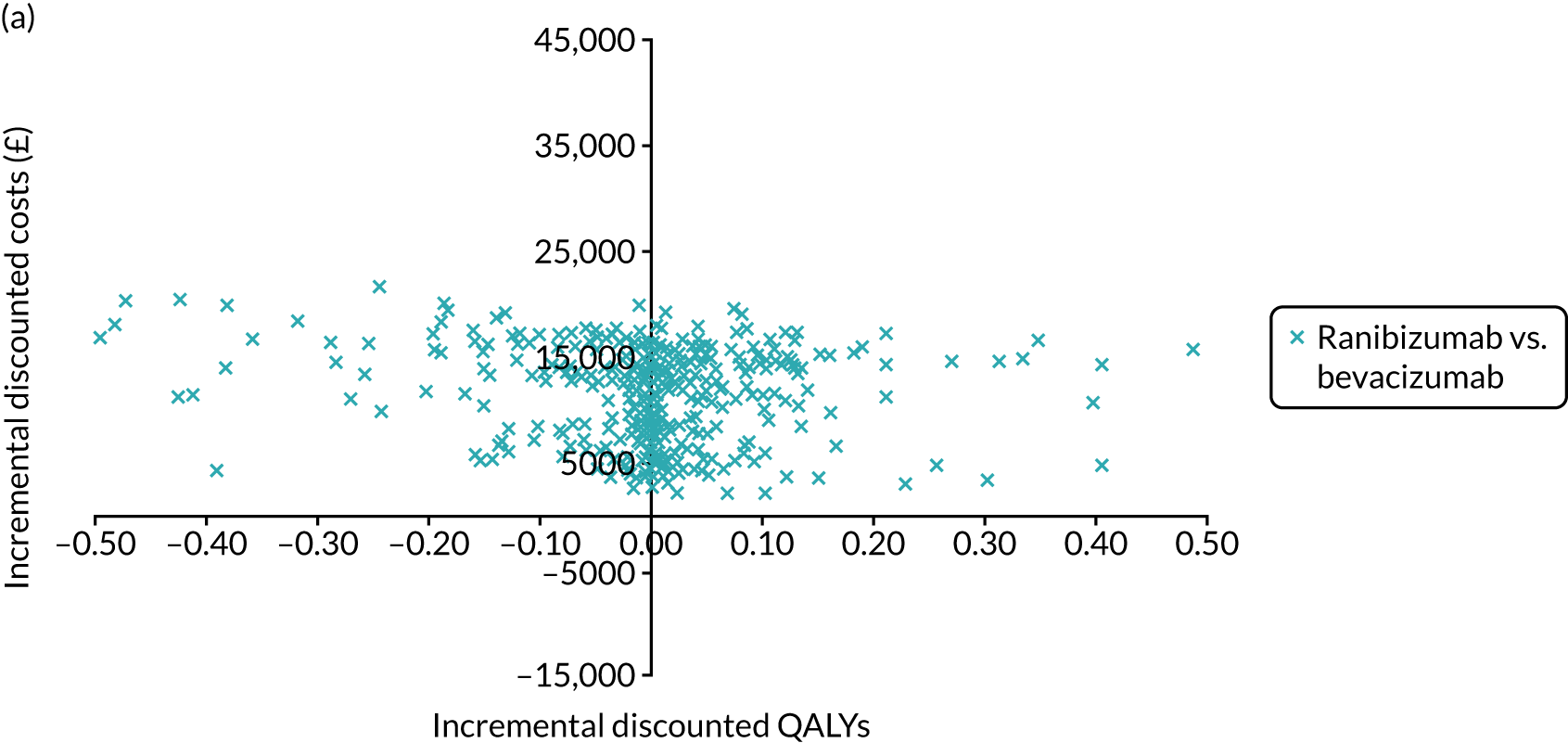
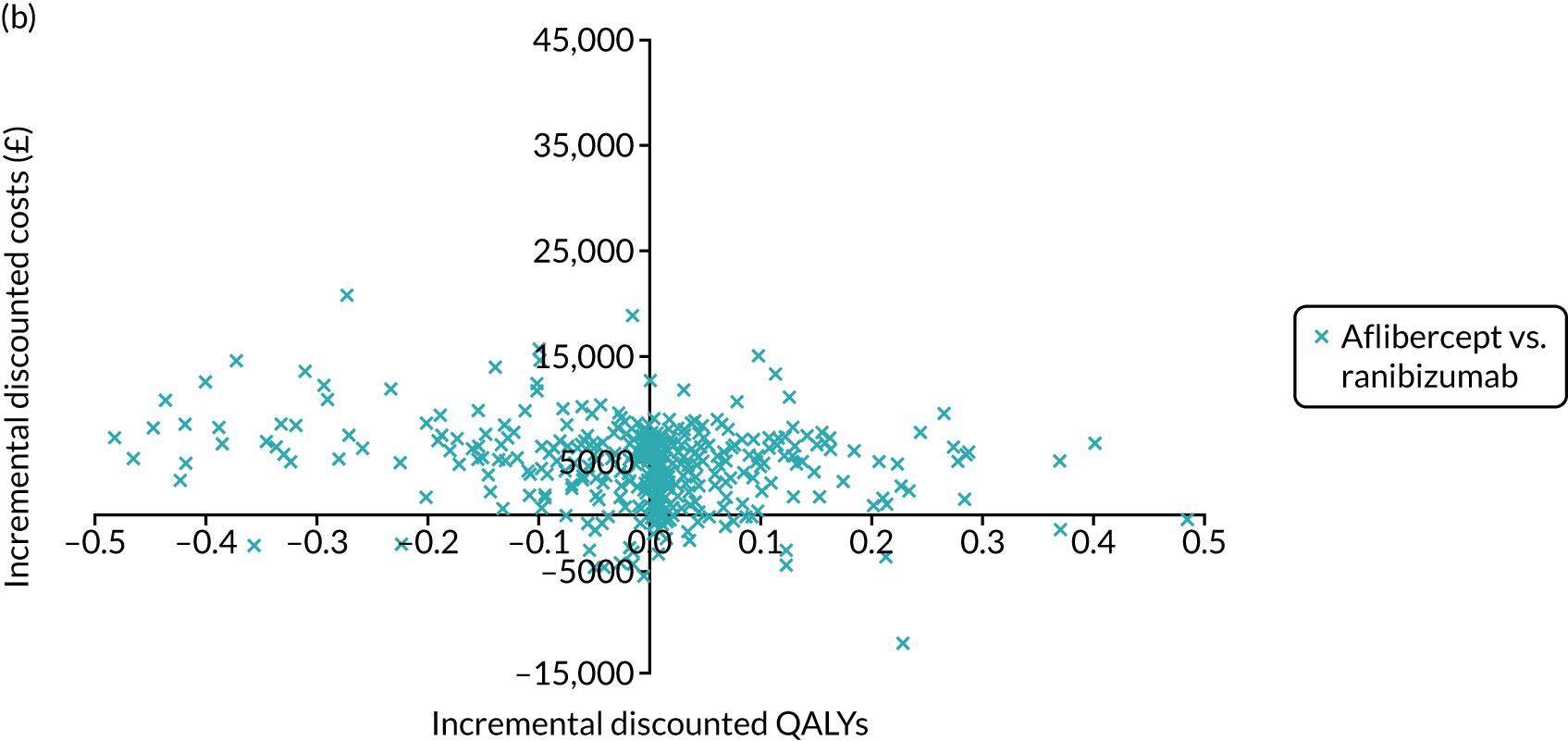
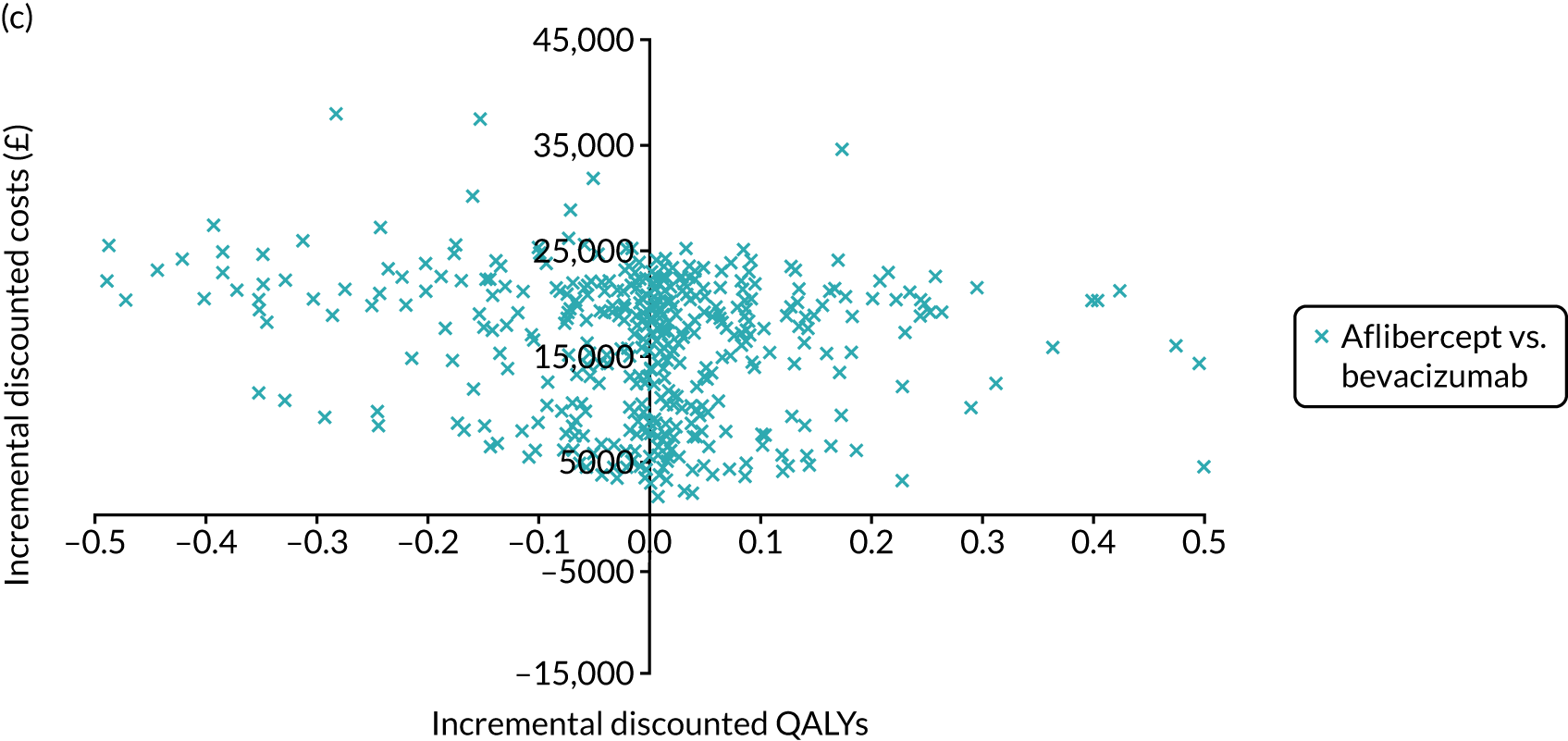
The CEAC (Figure 18) shows that, at £20,000 and at £30,000 per QALY, bevacizumab has the highest probability of being cost-effective (99.6% and 98.4%, respectively). Even at a threshold of £100,000 per QALY, bevacizumab has the highest probability of being cost-effective (92.8%). The probabilistic results demonstrate that bevacizumab is the most cost-effective intervention.
FIGURE 18.
Model-based analysis: CEAC.

The difference in QALYs is due to the difference in the effectiveness of the three interventions (see Table 5) and the relationship between visual acuity and utility. The difference in costs is due to the difference in the cost of the intravitreal anti-VEGF injections (intervention costs), as demonstrated by the cost breakdown in Table 22. The trial eye CST and visit costs are higher for bevacizumab than for aflibercept and ranibizumab because patients require more injections of bevacizumab. However, the drug costs are lower for bevacizumab because the cost of the injection is much lower.
| Type of cost | Costs (£) (95% CI) | ||
|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | |
| Treatment costs | |||
| Trial eye intervention costs | 11,785 (11,387 to 12,184) | 17,156 (16,582 to 17,730) | 634 (614 to 654) |
| Trial eye CST and visit costs | 5427 (5351 to 5503) | 5372 (5299 to 5444) | 5622 (5542 to 5701) |
| Non-trial eye drug costs | 771 (750 to 792) | 1051 (1021 to 1081) | 40 (39 to 41) |
| Non-trial eye CST and visit costs | 268 (262 to 274) | 249 (242 to 255) | 276 (270 to 282) |
| Disease management costs | 9588 (9049 to 10,127) | 10,058 (9435 to 10,681) | 9283 (8807 to 9759) |
| Ocular AEs costs | 1322 (1238 to 1405) | 109 (101 to 117) | 1392 (1301 to 1483) |
| Blindness costs | 1065 (918 to 1212) | 1031 (886 to 1176) | 1107 (957 to 1257) |
| Total costs | 30,226 (29,386 to 31,066) | 35,026 (33,990 to 36,062) | 18,353 (17,782 to 18,925) |
Scenario analyses
For the results of scenario analysis, see Table 21. In the scenarios using the EQ-5D and EQ-5D-V, the costs are unchanged from the base case using VFQ-UI, and the total QALYs for the three interventions are similar. Using the EQ-5D, aflibercept generates the most QALYs, followed by ranibizumab. This is different from the findings for the VFQ-UI base case because the relationship between visual acuity and utility differs for the three utility measures. In these scenarios, although ranibizumab and aflibercept are slightly more effective than bevacizumab, they are not cost-effective because they are much more expensive. The ICER for ranibizumab versus bevacizumab is £908,532 (95% CI £659,881 to £1,476,254) and for aflibercept versus ranibizumab is £128,513 (95% CI £110,116 to £152,663). Using the EQ-5D-V, the results indicate the same trends, but the CI for the incremental effectiveness of ranibizumab compared with bevacizumab contains zero, indicating that the difference is not statistically significant. The CI around the INMB is presented for this comparison, as the ICER may contain dominated results when ranibizumab is less effective than bevacizumab. Aflibercept is more effective than bevacizumab and ranibizumab, but it is not cost-effective.
Using a 100-week time horizon, as in LEAVO, bevacizumab is slightly less effective than ranibizumab and aflibercept, but the ICERs for ranibizumab versus bevacizumab and for aflibercept versus bevacizumab are £34,067,841 and £2,610,554 per QALY, respectively (see Appendix 6, Table 62). However, in this analysis, the 95% CIs for the incremental QALYs for ranibizumab versus bevacizumab contain zero, demonstrating that ranibizumab is not statistically significantly better. Bevacizumab remains the most cost-effective intervention in this scenario.
In the scenario using the list price of £243 per vial of bevacizumab, the costs for ranibizumab and aflibercept and the QALYS for the three interventions are unchanged from the base-case analysis, but the cost of bevacizumab has increased (see Appendix 6, Table 63). However, bevacizumab remains significantly cheaper than ranibizumab and aflibercept, demonstrated by the fact that the CIs for the incremental costs do not contain zero. Bevacizumab continues to dominate ranibizumab and aflibercept.
In deterministic analysis using a 5- and 10-year time horizon, bevacizumab remains the most cost-effective intervention at £20,000–30,000 per QALY (see Appendix 6, Table 58).
In deterministic analysis, to have comparable costs to bevacizumab, at £28 per injection, the PAS discounts for aflibercept and ranibizumab would need to be at least 95%.
Results: within-trial analysis
A total of 462 patients were included in the health economic analysis, with one patient excluded because they had been randomised in error. Thirteen people died and 42 participants withdrew or were lost to follow-up during the trial, and their subsequent costs and QALYs were adjusted, as described in Method of economic evaluation.
Data completeness
Over the 100-week data collection period, data were missing for some participants for baseline utility, QALY parameters for the three quality-of-life measures and total costs. For the extent of these missing data, see Appendix 6, Table 64. The highest proportion of missing data (56%) was recorded for total costs. There were only small differences in the proportion of missing data between the treatment arms. Appendix 6, Figures 29–32, explores the patterns of missing data for cost and HRQoL outcomes. The plots suggest that costs and utilities can be combined at each time period without any major loss of information. The plots suggested that the data were non-monotonic and missing at random; therefore, it was appropriate to use multiple imputation. 126
Utilities
Figure 19 summarises the mean VFQ-UI utility score at each milestone visit, with 95% CIs. There is overlap between the intervals at each of the time points, suggesting no statistical differences between the three arms at all follow-up time points. The mean utility scores from the EQ-5D and the EQ-5D-V are provided (see Appendix 6, Figures 34 and 35, and Table 65).
FIGURE 19.
Within-trial analysis: mean utility scores calculated using the VFQ-UI over 100 weeks. Table shows number of observations at each time point. Reproduced with permission from Pennington et al. 95 This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. The figure includes minor additions and formatting changes to the original figure.

Costs
Table 23 gives a breakdown of the total costs for each of the three treatment arms. Complete-case data were used for estimating the mean costs for each item, and the mean total costs were calculated from imputed data. The mean total costs in each arm are driven by the intervention costs, accounting for 84%, 87% and 76% of the total costs for ranibizumab, aflibercept and bevacizumab, respectively.
| Cost | Cost per patient (£) | |||||
|---|---|---|---|---|---|---|
| Mean (SD); n | Mean (95% CI) | |||||
| Ranibizumab | Aflibercept | Bevacizumab | Aflibercept vs. ranibizumab | Bevacizumab vs. ranibizumab | Aflibercept vs. bevacizumab | |
| Blindness | 1.94 (15.28); 125 | 4.70 (23.51); 129 | 2.96 (18.79); 123 | 2.76 (–2.05 to 7.57) | 1.02 (–3.85 to 5.88) | –1.74 (–6.57 to 3.08) |
| Concomitant medications | 69.03 (342.27); 154 | 22.86 (26.40); 154 | 124.37 (907.96); 154 | –46.17 (–171.35 to 79.01) | 55.34 (–69.84 to 180.52) | 101.51 ( –23.67 to 226.69) |
| Concomitant procedures | 173.23 (567.30); 154 | 222.60 (749.14); 154 | 217.57 (880.10); 154 | 49.37 (–116.66 to 215.4) | 44.34 (–121.69 to 210.37) | –5.03 (–171.06 to 161) |
| Continuous care and support | 7.11 (54.99); 99 | 38.76 (172.27); 88 | 10.43 (82.93); 90 | 31.66 (–0.75 to 64.07) | 3.32 (–28.89 to 35.54) | –28.33 (–61.5 to 4.83) |
| Health-care contacts | 729.36 (815.88); 91 | 710.46 (920.25); 92 | 740.14 (1065.62); 81 | –18.89 (–289.62 to 251.84) | 10.78 (–268.94 to 290.51) | 29.68 (–249.33 to 308.68) |
| Hospital admissions | 54.17 (479.35); 149 | 34.08 (239.58); 149 | 89.32 (689.04); 148 | –20.10 (–134.43 to 94.23) | 35.15 (–79.37 to 149.67) | –55.24 (–169.76 to 59.28) |
| Intervention | 10,991.74 (3973.57); 154 | 12,445.31 (4231.59); 154 | 4784.99 (1247.34); 154 | 1453.57 (687.9 to 2219.23) | –6206.74 (–6972.41 to –5441.08) | 7660.31 (6894.65 to 8425.98) |
| Total costs | 13,014 (3605); 154 | 14,328 (3773); 154 | 6292 (3371); 154 | 1245 (421 to 2070) | –6760 (–7546 to –5973) | 7984 (7209 to 8759) |
Appendix 6, Figure 33, shows histograms of the distribution of total costs by treatment arm. In each arm, few patients incurred extreme high costs, resulting in little skewedness in the cost data.
Base-case analysis
In the base-case analysis (Table 24), the difference in mean total costs between aflibercept and ranibizumab was £1245 (95% CI £421 to £2070); between bevacizumab and ranibizumab arms, the difference was –£6760 (95% CI –£7546 to –£5973); and between aflibercept and bevacizumab, it was £7984 (95% CI £7209 to £8759). Bevacizumab dominated (less costly and with no difference in benefit) ranibizumab, with a probability of cost-effectiveness of 1.00 at the threshold of £20,000 per QALY.
| Outcome | Intervention, mean (SD); n | Comparator, mean (SD); n | Difference,a mean (95% CI) | Probability of being cost-effective at £20,000 per QALY (at £30,000 per QALY) |
|---|---|---|---|---|
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 14,328 (3773); 154 | 13,014 (3605); 154 | 1245 (421 to 2070) | – |
| QALY | 1.651 (0.2374); 154 | 1.627 (0.2471); 154 | 0.004 (–0.0430 to 0.0518) | – |
| ICER | £283,595 | 0.04 (0.10) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 6292 (3371); 154 | 13,014 (3605); 154 | –6760 (–7546 to –5973) | – |
| QALY | 1.666 (0.2426); 154 | 1.627 (0.2471); 154 | 0.018 (–0.0282 to 0.0648) | – |
| ICER | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 14,328 (3773); 154 | 6292 (3371); 154 | 7984 (7209 to 8759) | – |
| QALY | 1.651 (0.2374); 154 | 1.666 (0.2426); 154 | –0.015 (–0.0618 to 0.0322) | – |
| ICER | Aflibercept is dominated | 0.00 (0.00) | ||
Aflibercept was more costly, with a mean QALY difference of 0.004 (95% CI –0.0430 to 0.0518), than ranibizumab, with an ICER of £283,595 per QALY gained and a probability of cost-effectiveness of 0.04 at the threshold of £20,000 per QALY. Aflibercept was dominated by bevacizumab [more costly, with a mean QALY difference of –0.015 (95% CI –0.0618 to 0.0322)], with a probability of cost-effectiveness of 0.00 at the thresholds of £20,000 and £30,000 per QALY.
Uncertainty analysis
The CEAC generated from the parametric analysis, in the base-case analysis, is presented in Figure 20. The CEAC illustrates the probability that each treatment is the most cost-effective, compared with alternative treatments, at a range of threshold values. Bevacizumab has the highest probability of being the most cost-effective of the three treatments at all thresholds considered.
FIGURE 20.
Within-trial analysis: CEAC.

The confidence ellipses graphs (see Appendix 3, Figure 21a–c) represent the point estimate of the ICER in the cost-effectiveness plane, with 50%, 75% and 95% CIs around the point estimate. The ICER for bevacizumab compared with ranibizumab falls in the south-east quadrant of the cost-effectiveness plane, with the 95% confidence ellipse wholly under the horizontal axis but spanning the vertical axis, suggesting certainty around the difference in costs but uncertainty around the difference in QALYs between the two interventions. The ICER for aflibercept compared with ranibizumab falls in the north-east quadrant, again with the 95% confidence ellipse wholly above the horizontal axis but spanning the vertical axis, suggesting certainty in the difference in costs but uncertainty in the difference in QALYs. Uncertainty is also illustrated (see Appendix 6, Figure 36) using the INMB.
Scenario analysis
The results from secondary analyses using the EQ-5D and the EQ-5D-V to estimate QALYs are summarised in Table 25. Although the three HRQoL measures (VFQ, EQ-5D and EQ-5D-V) generated slightly different results, the differences between the three interventions in terms of QALYs were small and uncertain in each analysis. The overall conclusion regarding the most cost-effective treatment is unchanged. Bevacizumab consistently dominates ranibizumab, and, although aflibercept might be slightly more clinically effective than bevacizumab and ranibizumab, it is more costly, resulting in a low probability of cost-effectiveness for both treatments at the £20,000 per QALY threshold.
| Outcome | Mean (SD); n | Difference,a mean (95% CI) | Probability of being cost-effective at £20,000 per QALY (at £30,000 per QALY) | |
|---|---|---|---|---|
| Intervention | Comparator | |||
| EQ-5D | ||||
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 14,271 (3857); 154 | 13,068 (3636); 154 | 1196 (406 to 1986) | – |
| QALY | 1.560 (0.3801); 154 | 1.513 (0.3744); 154 | 0.0184 (–0.0412 to 0.0779) | – |
| ICER | £65,023 | 0.13 (0.26) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 6273 (3384); 154 | 13,068 (3636); 154 | –6783 (–7575 to –5990) | – |
| QALY | 1.535 (0.3759); 154 | 1.513 (0.3744); 154 | 0.0098 (–0.0493 to 0.0690) | – |
| ICER (£) | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 14,271 (3857); 154 | 6273 (3384); 154 | 8035 (7246 to 8824) | |
| QALY | 1.560 (0.3801); 154 | 1.535 (0.3759); 154 | 0.008 (–0.0529 to 0.0683) | |
| ICER | £1,041,476 | 0.00 (0.00) | ||
| EQ-5D-V | ||||
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 14,273 (3720); 154 | 13,000 (3661); 154 | 1325 (499 to 2151) | |
| QALY | 1.516 (0.3856); 154 | 1.472 (0.3666); 154 | 0.0433 (–0.0404 to 0.1269) | |
| ICER | £30,624 | 0.32 (0.49) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 6268 (3368); 154 | 13,000 (3661); 154 | –6713 (–7499 to –5926) | – |
| QALY | 1.500 (0.3757); 154 | 1.472 (0.3666); 154 | 0.0340 (–0.0471 to 0.1151) | – |
| ICER | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 14,273 (3720); 154 | 6268 (3368); 154 | 8012 (7232 to 8793) | – |
| QALY | 1.516 (0.3856); 154 | 1.500 (0.3757); 154 | 0.0032 (–0.0837 to 0.0902) | – |
| ICER | £2,483,943 | 0.00 (0.00) | ||
The results from the fully incremental analysis show that bevacizumab dominates all alternative treatment options (i.e. it is less costly and more effective); therefore, ranibizumab and aflibercept are ruled out by dominance (see Appendix 6, Table 66).
The results from scenario analyses when discount rates of 30% and 50% are applied to the drug costs of ranibizumab and aflibercept are shown in Table 26. These findings suggest that the within-trial cost–utility base-case analysis results are not sensitive to these discount rates. Although the probability of aflibercept being cost-effective, compared with ranibizumab, increased to 11% and 24% at the £20,000 per QALY threshold for the 30% and 50% discounts, respectively, this was still a low probability. Bevacizumab was still cheaper and more effective than ranibizumab, and aflibercept was more costly and less effective than bevacizumab.
| Outcome | Mean (SD); n | Difference,a mean (95% CI) | Probability of being cost-effective at £20,000 per QALY (at £30,000 per QALY) | |
|---|---|---|---|---|
| Intervention | Comparator | |||
| Discount of 30% applied to aflibercept and ranibizumab drug costs | ||||
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 11,727 (2900); 154 | 10,893 (2848); 154 | 833 (203 to 1464) | – |
| QALY | 1.651 (0.2426); 154 | 1.627 (0.2471); 154 | 0.004 (–0.0430 to 0.0518) | – |
| ICER | £189,133 | 0.11 (0.19) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 6227 (2700); 154 | 10,893 (2848); 154 | –4656 (–5280 to –4033) | – |
| QALY | 1.666 (0.2374); 154 | 1.627 (0.2471); 154 | 0.018 (–0.0282 to 0.0649) | – |
| ICER | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 11,727 (2900); 154 | 6227 (2700); 154 | 5476 (4837 to 6116) | |
| QALY | 1.651 (0.2426); 154 | 1.627 (0.2471); 154 | –0.015 (–0.0618 to 0.0322) | |
| ICER | Aflibercept is dominated | 0.00 (0.00) | ||
| Discount of 50% applied to aflibercept and ranibizumab drug costs | ||||
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 10,042 (2553); 154 | 9499 (2538); 154 | 497 (–71 to 1053) | – |
| QALY | 1.651 (0.2426); 154 | 1.627 (0.2471); 154 | 0.004 (–0.0430 to 0.0518) | – |
| ICER | £111,622 | 0.24 (0.32) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 6201 (2419); 154 | 9499 (2538); 154 | –3288 (–3842 to –2734) | – |
| QALY | 1.666 (0.2374); 154 | 1.627 (0.2471); 154 | 0.018 (–0.0282 to 0.0649) | – |
| ICER | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 10,042 (2553); 154 | 6201 (2419); 154 | 3809 (3252 to 4365) | – |
| QALY | 1.651 (0.2426); 154 | 1.666 (0.2374); 154 | –0.015 (–0.0618 to 0.0322) | – |
| ICER | Aflibercept is dominated | 0.00 (0.00) | ||
Results from scenario analyses using the list price of £243 for bevacizumab, complete-case data and a 52-week time horizon are summarised in Appendix 6, Tables 67–69. The same conclusions can be drawn from these analyses as from the base-case analysis, with bevacizumab remaining the most cost-effective treatment option.
Summary of findings from the economic evaluation
Main findings from the model-based analysis
The model-based analysis found that bevacizumab is consistently the most cost-effective intervention at a threshold of £20,000–30,000 per QALY. Bevacizumab, aflibercept and ranibizumab generate very similar QALYs, but bevacizumab leads to substantial cost savings, even when it is assumed that bevacizumab vials cannot be split, which would incur a higher cost per injection. The cost savings associated with bevacizumab are due to the much lower drug cost. For aflibercept and ranibizumab to have comparable costs to bevacizumab, and therefore have a chance of being cost-effective, the PAS discounts on the two licensed products would need to be at least 95%.
The findings were robust to sensitivity analyses, but the use of different utility measures led to differences in the absolute QALYs and ordering of each intervention. This indicates that the estimates of the differences in HRQoL are uncertain, but were consistently small across instruments.
Main findings from the within-trial analysis
The within-trial health economic analysis found strong evidence that bevacizumab is considerably cheaper than ranibizumab and aflibercept, even when potential discount rates are applied to the two licensed products. There was no evidence to suggest a difference in HRQoL between the three treatments regardless of the HRQoL questionnaire used to measure utility; however, the estimates of QALY difference are uncertain. Bevacizumab is more cost-effective option than ranibizumab and aflibercept. Aflibercept is highly unlikely to be cost-effective in the short term (100 weeks), compared with ranibizumab or bevacizumab, using the commonly used cost-effectiveness threshold of £20,000–30,000 per QALY. The cost-effectiveness results are driven by the higher intervention cost for aflibercept, with no additional benefit in terms of QALYs.
Comparison of model-based and within-trial findings
The model-based and within-trial analyses both concluded that bevacizumab is the most cost-effective intervention for treating MO due to CRVO. Both analyses found small differences in QALYs between the three treatments, and substantial cost savings for bevacizumab. Despite the different approaches used for estimating utilities in the model- and trial-based analyses, the cost-effectiveness conclusion remained the same, indicating the robustness of the economic evaluation results.
The total QALYs for each intervention were similar for bevacizumab (model, 1.641; trial, 1.666), aflibercept (model, 1.646; trial, 1.651) and ranibizumab (model, 1.641; trial, 1.627). The total costs for each intervention were also similar for bevacizumab (model, £6349; trial, £6292), aflibercept (model, £18,844; trial, £14,328) and ranibizumab (model, £15,254; trial, £13,014). The similarities between the model- and trial-based costs and QALYs can be viewed as validation of the model-based analysis.
However, there are some differences between the model- and trial-based results. The model-based analysis leads to higher costs for each intervention, despite the exclusion of concomitant medications and procedures (although these make up < £250 per intervention in the trial-based analysis). The differences in costs may be explained by higher intervention drug and administration costs in the model. The within-trial analysis uses information recorded in LEAVO on whether or not a participant had an injection at each visit, whereas the model uses data from LEAVO in combination with the LEAVO re-treatment criteria to allow extrapolation beyond the trial period. The difference between the analyses indicates that some modelled patients receive the intervention when they did not in LEAVO.
The model results follow the same trend as the trial, in that the number of injections was smaller for aflibercept than for bevacizumab or ranibizumab, but the absolute number of injections in each arm is larger. The re-treatment criteria in the model dictate that patients will be re-treated if their CST is > 320 µm, and the CST data used in the model suggest that, on average, bevacizumab and aflibercept patients have a CST above this threshold throughout the trial. Variation between individual patients may have meant that a greater proportion of patients in the trial than in the model had a CST below the threshold. Alternatively, the difference may have arisen because the re-treatment criteria in the trial stipulated that patients should have a CST of > 320 µm due to intraretinal or subretinal fluid, and the model does not consider the reason for CST values. There may have been patients in the trial who had a CST of > 320 µm for other reasons who were not treated, but who would be assumed to be treated in the model. In addition, participants in LEAVO might have missed appointments, which would lead to reduced injection frequency.
There are also differences between the QALYs in the model-based and within-trial analyses. The model- and trial-based analyses both find no significant difference between bevacizumab and ranibizumab, but the model finds that aflibercept generates significantly more QALYs than the other two interventions. This is because the model-based analysis uses BCVA in both eyes (as well as age and sex) to predict utility (and utility is higher for patients with better visual acuity), but the within-trial analysis uses utility data directly. The trial utility data will capture other factors relating to patients’ utility that may not relate to their BCVA, thus adding noise to the data. The relationship between visual acuity and utility is complex, non-linear and, in the observed LEAVO data for WSE, non-monotonic at times (see Figure 16). The mapping used ALDVMMs to try to capture the complex relationship and the distribution of utility data, but found some unusual features: typically ALDVMMs for EQ-5D contain at least three components, with one component representing patients with utility at or below zero. However, in this case, BCVA in the BSE or the WSE did not correlate with EQ-5D scores below zero, and so the model does not contain these separate components, as the covariates cannot predict membership of it.
Some of the QALY differences may also be due to differences in mortality. The within-trial analysis uses mortality data directly, and so includes the deaths of three participants in the ranibizumab group, six in the aflibercept group and four in the bevacizumab group. The model instead links mortality to baseline age, sex and the presence of CRVO; because these are the same for the modelled patients on each treatment, there is no mortality difference between the treatments.
The model-based analysis does not include blind registration and low-vision aid costs for patients who are partially sighted, consistent with previous analyses. 13,51 The within-trial analysis captures these costs and includes blind registration and low-vision aid costs using the same estimates as for patients with severe sight impairment. As cost of blindness was a small proportion of the total costs in both the within-trial and model-based analyses, this difference does not influence the results.
Chapter 5 Discussion
Summary and interpretation of findings
Clinical effectiveness and side-effect profile
The results of this prospective multicentre Phase III RCT demonstrate that repeated intravitreal injections of the three anti-VEGF agents markedly improves BCVA in patients with MO secondary to CRVO over 100 weeks. Aflibercept was non-inferior to ranibizumab in the management of CRVO-related MO at 100 weeks, but it was not superior. The trial was unable to demonstrate that bevacizumab was non-inferior to ranibizumab, as the lower 95% CI extended beyond the non-inferiority margin of –5 letters. The results were consistent in that both the ITT and the per-protocol analyses gave similar results for both comparisons. Furthermore, subsequent sensitivity analyses supported the reliability of the two non-inferiority comparisons. Although post hoc analyses should be interpreted with caution, a comparison of bevacizumab with aflibercept could not demonstrate that bevacizumab was non-inferior to aflibercept.
In clinical terms, the result confirms aflibercept, as well as ranibizumab, use for MO due to CRVO, which was important to demonstrate as both are used widely in UK clinical practice but previously had not been compared directly for this condition. Bevacizumab, on the other hand, could be worse than ranibizumab and aflibercept, or it could be no worse. Practically, this means that, if a patient was being advised on treatments for MO due to CRVO with anti-VEGF therapy, the three agents could not be presented to the patient as being completely equivalent. Clinicians would have a low level of confidence in recommending that a patient who was receiving ranibizumab or aflibercept switch to bevacizumab therapy.
Other visual outcome results across the three groups were similar, with no meaningful differences between ranibizumab, aflibercept and bevacizumab in the number of participants in each group achieving key secondary end points, such as a gain of ≥ 15 BCVA letters or remaining stable (i.e. a < 15-letter loss of visual acuity). The former means that, for patients commencing therapy, there is a 45–50% chance of achieving a three-line improvement in visual acuity. Patients can easily comprehend this by reference to a visual acuity chart when discussing the likely benefits of therapy with their clinician. All patients can be advised that, with regular attendance and adherence to treatment recommendations, there is at least a 90% chance that visual acuity will not deteriorate further. It is reassuring, from a patient perspective, to note that < 4% of participants in the bevacizumab arm experienced a significant loss of vision of ≥ 30 letters, in keeping with data pertaining to ranibizumab and aflibercept in this and in previous studies. 29–31,33
As anticipated, visual acuity improved rapidly during the initial monthly mandated injection phase, but a small mean decrease in visual acuity occurred across all three arms of the trial between weeks 16 and 24, which coincided with the pro re nata injection phase at week 16. Previous studies employed a protocol of six mandated monthly injections from week 0 to week 24. 9,21,27,29,31 During the trial design, we reviewed the available data and concluded that four mandated injections would be sufficient because in CRUISE the increase in visual acuity had reached a plateau by 4 months. 9 This may have been because the study enrolled a carefully selected population of people with non-ischaemic CRVO, who were likely to respond well to therapy. However, we now recognise that subsequent studies27,29,31,58 that introduced broader and more generalisable eligibility criteria indicate that the initial gain in visual acuity takes longer to maximise. Thus, our findings suggest that the loading phase should be extended to 6 months. Had we employed the longer loading phase, it is possible that the gain in visual acuity achieved by the LEAVO participants at week 24 could have been some ≥ 3 letters higher, and more in keeping with gains at 6 months in other studies. 27,29,31
It is also worth noting that SCORE227 and other studies, for example COPERNICUS,29,31 did not maintain such early gains through 1 and 2 years, most likely because follow-up in year 2 was too infrequent to identify and treat those patients who needed regular medication. Notably, the final gain in visual acuity at week 100, compared with baseline, is higher in LEAVO than in any other previously reported study on this condition, and could possibly have been even higher.
We believe that this reflects the importance of timely monitoring in the second year of the study, which should initially be every 4 or 8 weeks, in keeping with the LEAVO protocol. Longer intervals of follow-up in other studies probably led to loss of initial visual gains. 31,34,38 It is possible that follow-ups every 4 or 8 weeks could be extended for selected patients, but this approach was not tested in LEAVO. The adjusted mean visual acuity gains at each time point after baseline had a consistent hierarchy throughout the trial, in that aflibercept group values were higher than ranibizumab group values, which, in turn, were higher than bevacizumab gains. Even at week 76, when the differences between the groups were small, this hierarchy was maintained.
As expected, the three anti-VEGF agents caused a significant and immediate reduction in adjusted OCT CST during mandated injection phase of baseline to 12 weeks. However, the CST increased by approximately 50 µm over the next three visits, as the number of injections administered reduced markedly. This was because intense treatment during the mandated phase meant that re-treatment criteria were frequently not met at the visits at 16 and 20 weeks, leading to a rebound increase in CST by week 24, which closely mirrored the decrease in visual acuity. However, as participants entered the remaining 18 months of the trial, their visits were regularly scheduled every 4 or 8 weeks, resulting in patients who met criteria for re-treatment being treated promptly. This meant that OCT values gradually decreased through to week 100, mirrored by a gradual increase in visual acuity during the same time period, in contrast to other studies in which OCT data did not closely reflect visual acuity changes. 27 A previously unreported finding was that a significantly greater percentage of participants in the aflibercept arm than in the ranibizumab arm had an OCT CST of < 320 µm at weeks 52 and 100. This suggests that aflibercept is more effective than ranibizumab at resolving MO in the longer term, a finding previously reported in exudative AMD and DMO. 43,54 Interestingly, bevacizumab was no less effective than ranibizumab in this regard, unlike in other retinal disorders. 54
Fewer injections were required for aflibercept than for ranibizumab over 100 weeks, a difference that has been reported previously only in a treat-and-extend protocol. 57 The difference was significant as early as 24 weeks, and gradually increased by approximately 0.5 of an injection every 6 months. The post hoc analysis also found that fewer aflibercept than bevacizumab injections were required. This probably reflects the higher binding affinity of aflibercept to the VEGF molecule and a prolonged duration of action. This, coupled with a greater visual acuity gain and more patients achieving a normal thickness OCT CST at 2 years, would be a potential advantage of aflibercept over ranibizumab for MO due to CRVO.
No meaningful differences were seen between groups in OCT morphological grading at baseline or at 100 weeks.
Fundus fluorescein angiography did not detect differences across groups at baseline or exit, but, when the whole cohort is considered, there was overall change in distribution of non-perfusion at week 100, which we are further investigating.
No new safety concerns were identified in LEAVO to suggest any discrepancies in the overall safety profile of the three anti-VEGF agents, which is in keeping with previous reports. The chance of severe visual loss while undergoing anti-VEGF therapy remains low (i.e. in the order of 5% over 2 years) and has been noted in all previous studies. 9,27,29–31,57 This is typically due to development of an ischaemic CRVO, that is an increase in severity of the original occlusion to a point at which retinal blood inflow leads to compromised macular perfusion and possible neovascular complications. Patients were promptly treated with panretinal photocoagulation in such cases, and anti-VEGF therapy for MO may have coincidentally limited the risk of neovascularisation.
When this trial was conceived, it was thought that small amounts of anti-VEGF agents were absorbed into the systemic circulation from an intraocular injection, resulting in a reduction in circulating VEGF concentrations and, possibly, in an increased risk of APTC events, although this cause–effect relationship has not been established. Hence, we planned, in the grant application, to perform a meta-analysis of all comparative anti-VEGF safety data from CRVO studies that we anticipated would be carried out simultaneously with LEAVO. In practice, only the comparative US SCORE2 study27 and a small aflibercept versus bevacizumab trial have been conducted. 59 In addition, the SCORE2 investigators re-randomised their patients at 6 months, depending on whether or not the patients met predefined criteria of being good or poor responders. 58 Thus, a comparison was not possible beyond 6 months, and the comparative prevalence of AEs of anti-VEGF agents used in the two studies up to 6 months showed no difference. No study to date in multiple conditions, including nvAMD, DMO,61 branch and central retinal condition and less common conditions such as pathological myopia, has shown an increased risk of APTC events with bevacizumab, and we do not believe that this issue would be a barrier to the use of this drug in the NHS. The recent judicial review by Mrs Justice Whipple53 emphasised this point and she commented that ensuring that enough compounding pharmacies were available to ensure the large-scale safe production of significant amounts of the drug remained a key issue.
After the trial results were made available, we formulated a questionnaire to gather patient feedback, and received responses from members of the LEAVO CRVO users group that was formed prior to trial initiation, additional patients with RVO, the Barts Health/QMUL lay panel and Barts Heath diabetic patients who had a history of eye disease. We found that two-thirds of patients would consider bevacizumab treatment if the outcome could be worse than licensed alternatives but with such a small difference that it would be very unlikely to prevent them from carrying out their regular daily activities. All said that they would be more likely to agree to this if a licensed alternative was available should they not respond as expected to bevacizumab; provision would probably need to be made for this.
Limitations
The interpretation of the results should be considered in the context of patient eligibility and the trial treatment protocol. It is possible that the trial enrolled people whose eyes had limited potential for visual improvement because of a severe CRVO and compromised retinal perfusion and eyes with good visual acuity that had limited potential to improve because of a ceiling effect. Findings from secondary analyses were supportive but should be interpreted with caution as there was no adjustment for multiple testing. Aflibercept was considered an investigative agent because it was unlicensed when the trial commenced; therefore, all comparisons with bevacizumab were post hoc.
Generalisability (external validity)
The trial was undertaken in a wide range of UK ophthalmic centres. The trial eligibility criteria were purposely as broad as possible to ensure that a population would be recruited who represented patients presenting for NHS standard care. Unlike previous studies, patients with visual acuity of < 6/60 or with a relative afferent pupillary defect were not excluded. The protocol was amended to extend the upper limit of visual acuity from 74 (6/12) to 78 (6/9) letters to allow patients with MO but relatively good vision to enrol in the trial and not opt for NHS standard care. Patients with predisposing conditions (e.g. hypertension and glaucoma) were included and there was no restriction on concomitant medications or procedures during the trial; for example, a participant could undergo cataract surgery if his/her clinician deemed this necessary. The 4- to 8-week follow-up regimen in the second year ensured that the first-year visual acuity gains were maintained; we feel that this was an important part of the trial protocol for NHS centres to replicate. The centres involved in the trial ranged from small NHS departments through secondary referral centres to specialised ophthalmic-only tertiary referral units. All centres and ophthalmologists were able to deliver the trial, and no special expertise or equipment beyond subspecialty retinal expertise was necessary. We believe that the trial is potentially applicable to all UK and overseas ophthalmic centres. We do not believe that there are any related outcomes that the trial did not assess that may affect applicability, and we believe that the 2-year primary outcome and follow-up intervals were appropriate. The concentrations of anti-VEGF therapy in the plasma after 4 weeks are immeasurably low and, because patients did not receive injections after week 96, we would not anticipate any harms occurring beyond week 100 relevant to the trial. The only exception to this might be pregnancy, but both the participant and the spouse of a participant in the trial who were pregnant carried to term with the delivery of normal neonates. Clearly, not all patients in clinical care will respond in the same manner as those in the trial cohort, but we would expect discrepancies only in magnitude rather than in direction, and mostly related to non-adherence to a robust treatment protocol. Overall, the patient feedback from the trial was very positive and we have no reason to believe that any subgroup of patients would decline to receive anti-VEGF therapy in a similar way to the trial protocol.
Overall evidence
Comparative clinical data
The only previous well-powered comparison of anti-VEGF drugs for MO secondary to CRVO prior to LEAVO was SCORE2,27 which randomised 361 patients to aflibercept or bevacizumab, and treated them monthly from baseline to month 5 (six injections). The primary outcome was at 6 months. This differed from LEAVO, in which participants received monthly injections from baseline to month 3 (four injections) followed by pro re nata treatment at mandated visits at weeks 16 and 20, with milestone visual acuity assessments at 6 months. Greater mean BCVA letter gains were achieved in the first 6 months of SCORE2 than in LEAVO [aflibercept: SCORE2, mean 18.9 letters; LEAVO, mean 13.4 letters (SD 16.4 letters)]. This may be explained by the longer initial period of mandated monthly injections in SCORE227 or by differences in eligibility criteria. The baseline BCVA and case mix were dissimilar in these trials, with SCORE2 including patients with hemiretinal vein occlusion and LEAVO including patients with a baseline upper BCVA letter score of 78 (6/9), compared with 74 (6/12) in SCORE2. It is unknown whether or not the initial BCVA gains in SCORE2 could have been maintained through 2 years as the initial patient cohort was re-randomised at 6 months, depending on good and poor response to initial therapy. 58 The CRYSTAL study was a prospective single-arm study of ranibizumab therapy in CRVO with MO that followed up patients for 2 years, with a review at least every 8 weeks in year 2. Although this was a non-comparative study, the follow-up regimen was effective in maintaining first-year visual acuity gains in the second year, even though the number of injections in year 2 averaged only 3.3. This suggests that regular follow-up, with the targeting of patients in need of treatment, is of key importance. 56 Therefore, LEAVO is, to our knowledge, the only large clinical trial of MO due to CRVO to report comparative three-drug outcome data beyond 6 months with sustained visual acuity gains through 100 weeks across treatment arms.
Health economics analysis
The cost-effectiveness analysis found that bevacizumab was the most cost-effective intervention when compared with licensed agents (ranibizumab and aflibercept). In the treatment of MO due to CRVO, the model-based and within-trial analyses found small differences between the QALYs generated by aflibercept, ranibizumab and bevacizumab, but found that bevacizumab led to substantially lower costs. The finding that bevacizumab was the most cost-effective intervention was robust to scenario analyses that varied assumptions and data inputs. If bevacizumab was the standard of care and aflibercept or ranibizumab were new interventions being appraised by NICE, it is highly unlikely that they would be recommended as a cost-effective use of NHS resources.
Treatment with bevacizumab saves £5560 per year when compared with aflibercept, or £4546 per year when compared with ranibizumab (see Appendix 6, Table 69). If the estimated 5700 people diagnosed with MO due to CRVO each year in England and Wales8 were treated with bevacizumab instead of aflibercept, the NHS would save approximately £32M in 1 year (approximately £26M if the patients treated with bevacizumab instead of ranibizumab). Because the cost savings are due to a difference in intervention costs, this result would hold across other health-care systems, as long as the cost per injection for bevacizumab is lower than for aflibercept and ranibizumab.
This trial provides evidence of the cost-effectiveness of anti-VEGF treatment in MO due to CRVO, for which evidence is currently limited. A recent systematic review of the three interventions across retinal conditions did not identify any cost-effectiveness evidence in RVO. 58 This review identified two large US trials that provided evidence that ranibizumab and aflibercept are not cost-effective compared with bevacizumab in other retinal conditions (nvAMD and DMO). The cost-effectiveness findings for MO in the LEAVO trial are consistent with these findings.
The analyses adhered to good practice guidelines,84,86,87,125,126 and had the strengths of being based on data from a well-conducted multicentre randomised trial and having good retention rates over a 100-week follow-up. A key strength of the economic evaluation is the use of three different HRQoL outcome measures, including both disease-specific (VFQ-UI and EQ-5D-V) and generic (EQ-5D) measures. A range of scenario analyses have also been performed providing evidence based on a range of discounted prices for the alternative medications. In the health economics literature, there is always a debate over the relative merits of condition-specific versus generic preference-based measures (in this case VFQ-UI vs. EQ-5D). The argument is that generic measures are not sensitive to particular disease-specific improvements; therefore, the VFQ-UI was seen as a better alternative for the LEAVO population. In addition, bolt-ons to generic measures, such as the EQ-5D-V, were proposed as an alternative approach to retain comparability across different disease areas while improving sensitivity. In this trial, we used the three alternative approaches; we found that the VFQ-UI generated more QALYs for each of the three interventions. However, the incremental QALYs were similar across the three quality-of-life measures.
The strengths of the model-based analysis lie in the model design and the data inputs. A discrete event simulation facilitates the use of a continuous BCVA scale, and avoids arbitrarily grouping patients. This enables the detection of small differences in visual acuity, which are linked to utility and costs, to ensure that the differences between the three treatments are reflected. The model structure further enables consideration of both eyes, and their relationship to utility. The utility mappings follow best practice guidelines113 and up-to-date statistical methods to capture the distributions of utility. The inclusion of age and sex as variables in the utility mappings improved the model fit. In this trial population, quality of life is more likely to be affected by BCVA in both eyes (WSE and BSE). Therefore, our mappings were used to predict utility for each modelled patient using three quality-of-life measures (VFQ-UI, EQ-5D and EQ-5D-V) as a function of age, sex and BCVA in both eyes. Analysing resource use data from LEAVO allowed this to be linked to visual acuity to reflect the changing resource use associated with improvements or deterioration, which has not previously been captured in economic models for MO. 12,13 The use of growth models fitted to longitudinal BCVA and CST data allowed the extrapolation of these inputs over time, and avoided the need to make assumptions regarding effectiveness and injection frequency beyond the trial, as in previous models. 12,13
There are large numbers of missing data in the health economic analysis, but the multiple imputation model for the trial-based analysis suggests that the results are robust. Resource use questionnaires are vulnerable to recall bias. However, in LEAVO, the resource use questionnaire was designed especially for the trial. Resource use is also a small proportion of the overall total cost in each arm, so any changes are unlikely to change the health economic conclusions. Furthermore, results from the complete-case analysis provided similar conclusions, and bevacizumab remained the most cost-effective option.
The primary outcome in LEAVO concerned visual acuity in the trial eye. The model-based analysis considered both the trial and the non-trial eyes and their relationship to utility. However, consideration should be given to the relationship between these outcomes and the reality for patients; although clinical measures assess visual acuity in each eye separately, patients’ overall sight is determined by their visual acuity in both eyes together. Patients’ day-to-day functioning and quality of life may therefore not relate closely to the assessment of visual acuity in the trial eye, and this may explain why the differences in the utilities and QALYs between arms were not significant in the economic evaluation. The mapping from BCVA to utility used a robust estimator of the variance used in the statistical model. A limitation of this was the inclusion of repeated observations of the same patients to increase the number of observations available. A cluster-robust estimator of the SEs could have been used that is robust in the presence of correlation between observations for each individual. This does not change the estimated coefficients from the ALDVMM; it affects only the SEs used in the probabilistic sensitivity analyses.
Chapter 6 Conclusions
Implications for health care
LEAVO was unable to demonstrate that bevacizumab is non-inferior, that is it may be worse or may not be worse than ranibizumab and aflibercept in the management of MO due to CRVO. Clinicians would have a low level of confidence in recommending that bevacizumab is equivalent in clinical effectiveness to the licensed medications for the management of this condition. No differences were detected in the side-effect profile of the treatments in this trial, in keeping with previous trials in this indication. Patients’ quality of life was not significantly different between treatment arms. This suggests that the clinical differences between the treatments were not sufficiently great to affect participants’ regular daily activities, as appraised in this trial. However, it is possible that, in certain situations, patients may undertake or would wish to undertake a visual task in which a difference in visual acuity in one eye may be noticeable to them. It is also important to note that CRVO is typically a unilateral condition, and the vision-related quality of life is dependent on the BSE. Therefore, this finding is not applicable to other retinal conditions such as nvAMD and DMO, as a result of which a larger proportion of patients have bilateral visual impairment.
Compared with aflibercept and ranibizumab, bevacizumab was the most cost-effective treatment for MO due to CRVO. If aflibercept and ranibizumab were to be appraised by NICE in a multitechnology appraisal with bevacizumab, it is highly unlikely that they would be considered cost-effective. Treating patients with bevacizumab would certainly lead to cost savings to the NHS and other health-care systems. However, because the trial could not demonstrate that bevacizumab was non-inferior to the licensed medication, the trial results would need to be discussed in detail with patients, their representatives and funders before the treatment proceeded. The post-trial patient questionnaire responses suggest that approximately two-thirds of patients may be amenable to this approach, assuming that the licensed medications were available in reserve.
Recommendations for research
Additional patient involvement in this area would be required to help quantify more exact numbers of patients willing to consider bevacizumab therapy for MO due to CRVO, and the key factors that would dissuade other patients and whether or not these could be mitigated. This would probably require the full involvement of patients, patient advocate groups and funders to determine if bevacizumab could be introduced in this way. Further larger-scale clinical trials may also be justified for this condition.
Acknowledgements
Support
This project was funded by the NIHR HTA Clinical Evaluation and Trials programme (number 11/92/03). The trial was sponsored by the NIHR Moorfields Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust. The research was supported by the NIHR Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and the University College London Institute of Ophthalmology, the UK Clinical Research Collaboration-registered KCTU at King’s Health Partners, which is part-funded by the NIHR Maudsley Biomedical Research Centre, at South London and Maudsley NHS Foundation Trust, and King’s College London.
We would like to thank Blair McLennan, Aleksandra Kata, Evangelos Georgiou, Beverley White-Alao, Janice Jimenez and Gill Lambert, who formed the trial management of the KCTU; Shakeel Herwitzer of the Liverpool and Broadgreen Pharmacy Aseptic Unit; and Tunde Peto, Vittorio Silvestri, Clare Newell, Michelle McGaughey, Pauline Lenfestey, Karleigh Kelso and Barbara Hamill of the independent NetwORC UK in Belfast. We also thank Luke Nicholson and Piyali Sen, Medical Retina Service; Catherine Grigg and Katie Binstead, Research Optometry Department, Moorfields Eye Hospital; and Bhogal Bhogal, Research Optometrist, Wolverhampton Eye Infirmary. We thank Anthea Sutton, School of Health and Related Research, University of Sheffield, for her support on literature searching for the systematic review work.
We would also like to thank all the patients who participated in this trial; Andi Skilton, patient and public involvement lead at Moorfields Eye Hospital; Gillian Hood, manager of the Diabetes Research Lay Panel Group, QMUL/Barts Health; and the patient and public involvement group in Moorfields Eye Hospital. We are indebted to Barry Hounsome, LEAVO Trial Manager, who sadly passed away during the trial. Finally, we also thank Ellie Fairbank, Research Manager (Monitoring), Simon Bevan and Samantha Low of the NIHR Evaluation Trials and Studies Coordinating Centre and Rupert Bourne, Ophthalmology Lead, NIHR, for their invaluable support and advice throughout the study.
Contributions of authors
Philip Hykin (https://orcid.org/0000-0003-0459-0055) (Consultant Ophthalmic Surgeon) was chief investigator and was responsible for obtaining funding; the trial concept and design; protocol development; data collection, analysis and interpretation; and critical revision of this manuscript for important intellectual content.
A Toby Prevost (https://orcid.org/0000-0003-1723-0796) (Professor, Medical Statistics and Clinical Trials) was a co-applicant and was responsible for trial design, data analysis and interpretation, statistical analysis and critical revision of this manuscript for important intellectual content.
Sobha Sivaprasad (https://orcid.org/0000-0001-8952-0659) (Professor and Consultant Ophthalmologist) was the co-lead investigator and a co-applicant, and was responsible for obtaining funding; trial concept and design; protocol development; data collection, analysis and interpretation; and critical revision of this manuscript for important intellectual content.
Joana C Vasconcelos (https://orcid.org/0000-0001-7709-4058) (Research Fellow, Medical Statistics) was a co-applicant and was responsible for data analysis and interpretation, statistical analysis and critical revision of this manuscript for important intellectual content.
Caroline Murphy (https://orcid.org/0000-0001-7547-8998) (Operations Manager, KCTU) was a co-applicant and was involved in trial design; protocol development; trial management; data collection, analysis and interpretation; and critical revision of this manuscript for important intellectual content.
Joanna Kelly (https://orcid.org/0000-0002-4389-5284) (Senior Data Analyst, KCTU) was a co-applicant and was involved in the trial design and data collection, management, analysis and interpretation.
Jayashree Ramu (https://orcid.org/0000-0002-7411-1729) (Research Fellow, Moorfields Eye Hospital) was involved in the trial design; protocol development; trial recruitment; and data collection, analysis and interpretation. Jayashree was also responsible for critical revision of this manuscript for important intellectual content.
Abualbishr Alshreef (https://orcid.org/0000-0003-2737-1365) (Research Fellow, Health Economics) was involved in the management, analysis and interpretation of the health economics part of the trial, and was responsible for critical revision of this manuscript for important intellectual content.
Laura Flight (https://orcid.org/0000-0002-9569-8290) (Research Associate, Health Economics) was involved in the analysis and interpretation of the health economics part of the trial, and was responsible for critical revision of this manuscript for important intellectual content.
Rebekah Pennington (https://orcid.org/0000-0002-1002-022X) (Research Fellow, Health Economics) was involved in the analysis and interpretation of the health economics part of the trial, and was responsible for critical revision of this manuscript for important intellectual content.
Barry Hounsome (Trial Manager) (deceased) was involved in all aspects of trial management and monitoring.
Ellen Lever (https://orcid.org/0000-0001-8329-6764) (Trial Manager) was involved in all aspects of trial management and monitoring.
Andrew Metry (https://orcid.org/0000-0001-7412-6093) (Research Associate, Health Economics) was involved in the analysis and interpretation of the health economics part of the trial, and was responsible for critical revision of this manuscript for important intellectual content.
Edith Poku (https://orcid.org/0000-0001-6549-5081) (Research Fellow, Systematic Reviewing) was involved in the analysis and interpretation of the health economics part of the trial, and was responsible for critical revision of this manuscript for important intellectual content.
Yit Yang (https://orcid.org/0000-0002-5392-5133) (Professor, Ophthalmology) was responsible for trial design, protocol development, data collection and interpretation, and critical revision of this manuscript for important intellectual content.
Simon P Harding (https://orcid.org/0000-0003-4676-1158) (Professor, Ophthalmology) was responsible for trial design, protocol development, data interpretation, and critical revision of this manuscript for important intellectual content.
Andrew Lotery (https://orcid.org/0000-0001-5541-4305) (Professor, Ophthalmology) was responsible for trial design, protocol development, data collection and interpretation, and critical revision of this manuscript for important intellectual content.
Usha Chakravarthy (https://orcid.org/0000-0002-2606-3734) (Professor, Ophthalmology) was responsible for trial design, protocol development, data interpretation, and critical revision of this manuscript for important intellectual content.
John Brazier (https://orcid.org/0000-0001-8645-4780) (Professor, Health Economics) was a co-applicant and was involved in the design, management, analysis and interpretation of the health economics part of the trial, and was responsible for critical revision of this manuscript for important intellectual content.
All authors reviewed, revised and approved the final version of the manuscript.
Publications
Hykin P, Prevost AT, Vasconcelos JC, Murphy C, Kelly J, Ramu J, et al. Clinical effectiveness of intravitreal therapy with ranibizumab vs. aflibercept vs. bevacizumab for macular edema secondary to central retinal vein occlusion: a randomized clinical trial. JAMA Ophthalmol 2019;137:1256–64.
Hykin P, Sivaprasad S, Prevost AT, Vasconcelos JC, Murphy C, Kelly J, et al. Protocol 14PRT/06545: A Multicentre Phase 3 Double-masked Randomised Controlled Non-Inferiority Trial Comparing the Clinical and Cost Effectiveness of Intravitreal Therapy with Ranibizumab (Lucentis) vs. Aflibercept (Eylea) vs. Bevacizumab (Avastin) for Macular Oedema due to Central Retinal Vein Occlusion (LEAVO trial). URL: www.thelancet.com/protocol-reviews/14PRT-06545 (25 May 2020).
Pennington BM, Hernández-Alava M, Hykin P, Sivaprasad S, Flight L, Alshreef A, Brazier J. Mapping from visual acuity to EQ-5D, EQ-5D with vision bolt-on and VFQ-UI in patients with macular edema in the LEAVO trial. Value Health 2020;23:928–35.
Nicholson L, Vazquez-Alfageme C, Sen P, Patrao N, Peto T, Yang Y, et al. The clinical relevance of ultra-widefield angiography findings in patients with central retinal vein occlusion and macular oedema receiving anti-VEGF therapy [published online ahead of print May 25 2021]. Eye (Lond) 2021.
Pennington R, Alshreef A, Flight A, Metry A, Poku E, Hykin PG, et al. Cost-effectiveness of ranibizumab vs. aflibercept vs. bevacizumab for the treatment of macular oedema due to central retinal vein occlusion: the LEAVO study [published online ahead of print April 26 2021]. PharmacoEconomics 2021.
Data-sharing statement
Consent was not obtained for data-sharing with a third party. The presented data are anonymised and risk of identification is low. All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review. Three years after this report has been published, the data will be deposited with the research and development department in Moorfields Eye Hospital.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Laouri M, Chen E, Looman M, Gallagher M. The burden of disease of retinal vein occlusion: review of the literature. Eye 2011;25:981-8. https://doi.org/10.1038/eye.2011.92.
- Hykin P, Prevost AT, Vasconcelos JC, Murphy C, Kelly J, Ramu J, et al. Clinical effectiveness of intravitreal therapy with ranibizumab vs. aflibercept vs. bevacizumab for macular edema secondary to central retinal vein occlusion: a randomized clinical trial. JAMA Ophthalmol 2019;137:1256-64. https://doi.org/10.1001/jamaophthalmol.2019.3305.
- Rogers S, McIntosh RL, Cheung N, Lim L, Wang JJ, Mitchell P, et al. The prevalence of retinal vein occlusion: pooled data from population studies from the United States, Europe, Asia, and Australia. Ophthalmology 2010;117:313-19.e1. https://doi.org/10.1016/j.ophtha.2009.07.017.
- Ponto KA, Elbaz H, Peto T, Laubert-Reh D, Binder H, Wild PS, et al. Prevalence and risk factors of retinal vein occlusion: the Gutenberg Health Study. J Thromb Haemost 2015;13:1254-63. https://doi.org/10.1111/jth.12982.
- Mitchell P, Smith W, Chang A. Prevalence and associations of retinal vein occlusion in Australia. The Blue Mountains Eye Study. Arch Ophthalmol 1996;114:1243-7. https://doi.org/10.1001/archopht.1996.01100140443012.
- Klein R, Klein BE, Moss SE, Meuer SM. The epidemiology of retinal vein occlusion: the Beaver Dam Eye Study. Trans Am Ophthalmol Soc 2000;98:133-41.
- Williamson TH. Central retinal vein occlusion: what’s the story?. Br J Ophthalmol 1997;81:698-704. https://doi.org/10.1136/bjo.81.8.698.
- Royal College of Ophthalmologists . Retinal Vein Occlusion (RVO) Guidelines 2015. www.rcophth.ac.uk/wp-content/uploads/2015/07/Retinal-Vein-Occlusion-RVO-Guidelines-July-2015.pdf (accessed 17 September 2018).
- Campochiaro PA, Brown DM, Awh CC, Lee SY, Gray S, Saroj N, et al. Sustained benefits from ranibizumab for macular edema following central retinal vein occlusion: twelve-month outcomes of a phase III study. Ophthalmology 2011;118:2041-9. https://doi.org/10.1016/j.ophtha.2011.02.038.
- Brown DM, Heier JS, Clark WL, Boyer DS, Vitti R, Berliner AJ, et al. Intravitreal aflibercept injection for macular edema secondary to central retinal vein occlusion: 1-year results from the phase 3 COPERNICUS study. Am J Ophthalmol 2013;155:429-37. https://doi.org/10.1016/j.ajo.2012.09.026.
- Korobelnik JF, Holz FG, Roider J, Ogura Y, Simader C, Schmidt-Erfurth U, et al. Intravitreal aflibercept injection for macular edema resulting from central retinal vein occlusion: one-year results of the phase 3 GALILEO study. Ophthalmology 2014;121:202-8. https://doi.org/10.1016/j.ophtha.2013.08.012.
- National Institute for Health and Care Excellence . Aflibercept for Treating Visual Impairment Caused by Macular Oedema Secondary to Central Retinal Vein Occlusion 2014. www.nice.org.uk/guidance/ta305 (accessed 18 June 2019).
- National Institute for Health and Care Excellence . Ranibizumab for Treating Visual Impairment Caused by Macular Oedema Secondary to Retinal Vein Occlusion 2013. www.nice.org.uk/guidance/ta283 (accessed 18 June 2019).
- Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF Trap, ranibizumab and bevacizumab. Angiogenesis 2012;15:171-85. https://doi.org/10.1007/s10456-011-9249-6.
- The Central Vein Occlusion Study Group . Baseline and early natural history report. The Central Vein Occlusion Study. Arch Ophthalmol 1993;111:1087-95. https://doi.org/10.1001/archopht.1993.01090080083022.
- Sivaprasad S, Amoaku WM, Hykin P. RVO Guideline Group . The Royal College of Ophthalmologists Guidelines on retinal vein occlusions: executive summary. Eye 2015;29:1633-8. https://doi.org/10.1038/eye.2015.164.
- Sun JK, Lin MM, Lammer J, Prager S, Sarangi R, Silva PS, et al. Disorganization of the retinal inner layers as a predictor of visual acuity in eyes with center-involved diabetic macular edema. JAMA Ophthalmol 2014;132:1309-16. https://doi.org/10.1001/jamaophthalmol.2014.2350.
- Sun JK, Radwan SH, Soliman AZ, Lammer J, Lin MM, Prager SG, et al. Neural retinal disorganization as a robust marker of visual acuity in current and resolved diabetic macular edema. Diabetes 2015;64:2560-70. https://doi.org/10.2337/db14-0782.
- Chan EW, Eldeeb M, Sun V, Thomas D, Omar A, Kapusta MA, et al. Disorganization of retinal inner layers and ellipsoid zone disruption predict visual outcomes in central retinal vein occlusion. Ophthalmol Retina 2019;3:83-92. https://doi.org/10.1016/j.oret.2018.07.008.
- The Central Vein Occlusion Study Group . Natural history and clinical management of central retinal vein occlusion. The Central Vein Occlusion Study Group. Arch Ophthalmol 1997;115:486-91. https://doi.org/10.1001/archopht.1997.01100150488006.
- Hayreh S. Central retinal vein occlusion. Ophthalmol Clin North Am 1998;11:559-90. https://doi.org/10.1016/S0896-1549(05)70079-1.
- Tsui I, Kaines A, Havunjian MA, Hubschman S, Heilweil G, Prasad PS, et al. Ischemic index and neovascularization in central retinal vein occlusion. Retina 2011;31:105-10. https://doi.org/10.1097/IAE.0b013e3181e36c6d.
- Nicholson L, Vazquez-Alfageme C, Clemo M, Luo Y, Hykin PG, Bainbridge JW, et al. Quantifying retinal area in ultra-widefield imaging using a 3-dimensional printed eye model. Ophthalmol Retina 2018;2:65-71. https://doi.org/10.1016/j.oret.2017.03.011.
- McIntosh RL, Rogers SL, Lim L, Cheung N, Wang JJ, Mitchell P, et al. Natural history of central retinal vein occlusion: an evidence-based systematic review. Ophthalmology 2010;117:1113-23.e15. https://doi.org/10.1016/j.ophtha.2010.01.060.
- Arditi A, Cagenello R. On the statistical reliability of letter-chart visual acuity measurements. Invest Ophthalmol Vis Sci 1993;34:120-9.
- Beck RW, Maguire MG, Bressler NM, Glassman AR, Lindblad AS, Ferris FL. Visual acuity as an outcome measure in clinical trials of retinal diseases. Ophthalmology 2007;114:1804-9. https://doi.org/10.1016/j.ophtha.2007.06.047.
- Scott IU, VanVeldhuisen PC, Ip MS, Blodi BA, Oden NL, Awh CC, et al. Effect of bevacizumab vs. aflibercept on visual acuity among patients with macular edema due to central retinal vein occlusion: the SCORE2 randomized clinical trial. JAMA 2017;317:2072-87. https://doi.org/10.1001/jama.2017.4568.
- Figueroa MS, Contreras I. Potential anti-vascular endothelial growth factor therapies for central retinal vein occlusion. Drugs 2012;72:2063-71. https://doi.org/10.2165/11640820-000000000-00000.
- Ogura Y, Roider J, Korobelnik JF, Holz FG, Simader C, Schmidt-Erfurth U, et al. Intravitreal aflibercept for macular edema secondary to central retinal vein occlusion: 18-month results of the phase 3 GALILEO study. Am J Ophthalmol 2014;158:1032-8. https://doi.org/10.1016/j.ajo.2014.07.027.
- Pielen A, Clark WL, Boyer DS, Ogura Y, Holz FG, Korobelnik JF, et al. Integrated results from the COPERNICUS and GALILEO studies. Clin Ophthalmol 2017;11:1533-40. https://doi.org/10.2147/OPTH.S140665.
- Heier JS, Clark WL, Boyer DS, Brown DM, Vitti R, Berliner AJ, et al. Intravitreal aflibercept injection for macular edema due to central retinal vein occlusion: two-year results from the COPERNICUS study. Ophthalmology 2014;121:1414-20.e1. https://doi.org/10.1016/j.ophtha.2014.01.027.
- Braithwaite T, Nanji AA, Lindsley K, Greenberg PB. Anti-vascular endothelial growth factor for macular oedema secondary to central retinal vein occlusion. Cochrane Database Syst Rev 2014;5. https://doi.org/10.1002/14651858.CD007325.pub3.
- Brown DM, Campochiaro PA, Singh RP, Li Z, Gray S, Saroj N, et al. Ranibizumab for macular edema following central retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology 2010;117:1124-33.e1. https://doi.org/10.1016/j.ophtha.2010.02.022.
- Campochiaro PA, Sophie R, Pearlman J, Brown DM, Boyer DS, Heier JS, et al. Long-term outcomes in patients with retinal vein occlusion treated with ranibizumab: the RETAIN study. Ophthalmology 2014;121:209-19. https://doi.org/10.1016/j.ophtha.2013.08.038.
- Chatziralli I, Theodossiadis G, Moschos MM, Mitropoulos P, Theodossiadis P. Ranibizumab versus aflibercept for macular edema due to central retinal vein occlusion: 18-month results in real-life data. Graefes Arch Clin Exp Ophthalmol 2017;255:1093-100. https://doi.org/10.1007/s00417-017-3613-1.
- Ford JA, Clar C, Lois N, Barton S, Thomas S, Court R, et al. Treatments for macular oedema following central retinal vein occlusion: systematic review. BMJ Open 2014;4. https://doi.org/10.1136/bmjopen-2013-004120.
- Pielen A, Feltgen N, Isserstedt C, Callizo J, Junker B, Schmucker C. Efficacy and safety of intravitreal therapy in macular edema due to branch and central retinal vein occlusion: a systematic review. PLOS ONE 2013;8. https://doi.org/10.1371/journal.pone.0078538.
- Heier JS, Campochiaro PA, Yau L, Li Z, Saroj N, Rubio RG, et al. Ranibizumab for macular edema due to retinal vein occlusions: long-term follow-up in the HORIZON trial. Ophthalmology 2012;119:802-9. https://doi.org/10.1016/j.ophtha.2011.12.005.
- Spaide RF, Chang LK, Klancnik JM, Yannuzzi LA, Sorenson J, Slakter JS, et al. Prospective study of intravitreal ranibizumab as a treatment for decreased visual acuity secondary to central retinal vein occlusion. Am J Ophthalmol 2009;147:298-306. https://doi.org/10.1016/j.ajo.2008.08.016.
- Scott IU, VanVeldhuisen PC, Ip MS, Blodi BA, Oden NL, King J, et al. Baseline factors associated with 6-month visual acuity and retinal thickness outcomes in patients with macular edema secondary to central retinal vein occlusion or hemiretinal vein occlusion: SCORE2 study report 4. JAMA Ophthalmol 2017;135:639-49. https://doi.org/10.1001/jamaophthalmol.2017.1141.
- Bressler NM, Chang TS, Fine JT, Dolan CM, Ward J. Anti-VEGF Antibody for the Treatment of Predominantly Classic Choroidal Neovascularization in Age-Related Macular Degeneration (ANCHOR) Research Group. Improved vision-related function after ranibizumab vs. photodynamic therapy: a randomized clinical trial. Arch Ophthalmol 2009;127:13-21. https://doi.org/10.1001/archophthalmol.2008.562.
- Chang TS, Bressler NM, Fine JT, Dolan CM, Ward J, Klesert TR. MARINA Study Group . Improved vision-related function after ranibizumab treatment of neovascular age-related macular degeneration: results of a randomized clinical trial. Arch Ophthalmol 2007;125:1460-9. https://doi.org/10.1001/archopht.125.11.1460.
- Schmidt-Erfurth U, Kaiser PK, Korobelnik JF, Brown DM, Chong V, Nguyen QD, et al. Intravitreal aflibercept injection for neovascular age-related macular degeneration: ninety-six-week results of the VIEW studies. Ophthalmology 2014;121:193-201. https://doi.org/10.1016/j.ophtha.2013.08.011.
- Poku E, Rathbone J, Everson-Hock E, Essat M, Wong R, Pandor A, et al. Bevacizumab in Eye Conditions: Issues Related to Quality, Use, Efficacy and Safety n.d. http://nicedsu.org.uk/wp-content/uploads/2016/03/Bevacizumab-report-NICE-published-version-11.04.13.pdf (accessed 27 May 2013).
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Wordsworth S, et al. IVAN Study Investigators . Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology 2012;119:1399-411. https://doi.org/10.1016/j.ophtha.2012.04.015.
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. CATT Research Group . Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011;364:1897-908. https://doi.org/10.1056/NEJMoa1102673.
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al. IVAN study investigators . Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet 2013;382:1258-67. https://doi.org/10.1016/S0140-6736(13)61501-9.
- Curtis LH, Hammill BG, Schulman KA, Cousins SW. Risks of mortality, myocardial infarction, bleeding, and stroke associated with therapies for age-related macular degeneration. Arch Ophthalmol 2010;128:1273-9. https://doi.org/10.1001/archophthalmol.2010.223.
- Carneiro AM, Barthelmes D, Falcão MS, Mendonça LS, Fonseca SL, Gonçalves RM, et al. Arterial thromboembolic events in patients with exudative age-related macular degeneration treated with intravitreal bevacizumab or ranibizumab. Ophthalmologica 2011;225:211-21. https://doi.org/10.1159/000323943.
- National Institute for Health and Care Excellence . Age-Related Macular Degeneration 2018. www.nice.org.uk/guidance/ng82 (accessed 7 April 2020).
- Thulliez M, Angoulvant D, Pisella PJ, Bejan-Angoulvant T. Overview of systematic reviews and meta-analyses on systemic adverse events associated with intravitreal anti-vascular endothelial growth factor medication use. JAMA Ophthalmol 2018;136:557-66. https://doi.org/10.1001/jamaophthalmol.2018.0002.
- Solomon SD, Lindsley K, Vedula SS, Krzystolik MG, Hawkins BS. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst Rev 2019;3. https://doi.org/10.1002/14651858.CD005139.pub4.
- Royal Courts of Justice . Judicial Review (2018) High Court Approved Judgement, Case No: CO 5288 2017 n.d. www.judiciary.uk/wp-content/uploads/2018/09/bayer-and-novartis-v-nhs-darlington-ccg-judgment.pdf (accessed 12 May 2020).
- Bressler NM, Beaulieu WT, Maguire MG, Glassman AR, Blinder KJ, Bressler SB, et al. Early response to anti-vascular endothelial growth factor and two-year outcomes among eyes with diabetic macular edema in protocol T. Am J Ophthalmol 2018;195:93-100. https://doi.org/10.1016/j.ajo.2018.07.030.
- Larsen M, Waldstein SM, Boscia F, Gerding H, Monés J, Tadayoni R, et al. Individualized ranibizumab regimen driven by stabilization criteria for central retinal vein occlusion: twelve-month results of the CRYSTAL study. Ophthalmology 2016;123:1101-11. https://doi.org/10.1016/j.ophtha.2016.01.011.
- Larsen M, Waldstein SM, Priglinger S, Hykin P, Barnes E, Gekkieva M, et al. Sustained benefits from ranibizumab for central retinal vein occlusion with macular edema: 24-month results of the CRYSTAL study. Ophthalmol Retina 2018;2:134-42. https://doi.org/10.1016/j.oret.2017.05.016.
- Casselholm de Salles M, Amrén U, Kvanta A, Epstein DL. Injection frequency of aflibercept versus ranibizumab in a treat-and-extend regimen for central retinal vein occlusion: a randomized clinical trial. Retina 2019;39:1370-6. https://doi.org/10.1097/IAE.0000000000002171.
- Scott IU, VanVeldhuisen PC, Ip MS, Blodi BA, Oden NL, Altaweel M, et al. SCORE2 Investigator Group . Comparison of monthly vs. treat-and-extend regimens for individuals with macular edema who respond well to anti-vascular endothelial growth factor medications: secondary outcomes from the SCORE2 randomized clinical trial. JAMA Ophthalmol 2018;136:337-45. https://doi.org/10.1001/jamaophthalmol.2017.6843.
- Lotfy A, Solaiman KAM, Abdelrahman A, Samir A. Efficacy and frequency of intravitreal aflibercept versus bevacizumab for macular edema secondary to central retinal vein occlusion. Retina 2018;38:1795-800. https://doi.org/10.1097/IAE.0000000000001782.
- Hoerauf H, Feltgen N, Weiss C, Paulus EM, Schmitz-Valckenberg S, Pielen A, et al. Clinical efficacy and safety of ranibizumab versus dexamethasone for central retinal vein occlusion (COMRADE C): a European label study. Am J Ophthalmol 2016;169:258-67. https://doi.org/10.1016/j.ajo.2016.04.020.
- Low A, Faridi A, Bhavsar KV, Cockerham GC, Freeman M, Fu R, et al. Comparative effectiveness and harms of intravitreal antivascular endothelial growth factor agents for three retinal conditions: a systematic review and meta-analysis. Br J Ophthalmol 2019;103:442-51. https://doi.org/10.1136/bjophthalmol-2018-312691.
- Sangroongruangsri S, Ratanapakorn T, Wu O, Anothaisintawee T, Chaikledkaew U. Comparative efficacy of bevacizumab, ranibizumab, and aflibercept for treatment of macular edema secondary to retinal vein occlusion: a systematic review and network meta-analysis. Expert Rev Clin Pharmacol 2018;11:903-16. https://doi.org/10.1080/17512433.2018.1507735.
- Sivaprasad S, Hykin P, Prevost AT, Vasconcelos J, Riddell A, Ramu J, et al. Intravitreal aflibercept compared with panretinal photocoagulation for proliferative diabetic retinopathy: the CLARITY non-inferiority RCT. Efficacy Mech Eval 2018;5. https://doi.org/10.3310/eme05050.
- Great Britain . Data Protection Act 1998 1998.
- Rosser DA, Cousens SN, Murdoch IE, Fitzke FW, Laidlaw DA. How sensitive to clinical change are ETDRS logMAR visual acuity measurements?. Invest Ophthalmol Vis Sci 2003;44:3278-81. https://doi.org/10.1167/iovs.02-1100.
- Ferris FL, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol 1982;94:91-6. https://doi.org/10.1016/0002-9394(82)90197-0.
- Babiuch AS, Han M, Conti FF, Wai K, Silva FQ, Singh RP. Association of disorganization of retinal inner layers with visual acuity response to anti-vascular endothelial growth factor therapy for macular edema secondary to retinal vein occlusion. JAMA Ophthalmol 2019;137:38-46. https://doi.org/10.1001/jamaophthalmol.2018.4484.
- Das R, Spence G, Hogg RE, Stevenson M, Chakravarthy U. Disorganization of inner retina and outer retinal morphology in diabetic macular edema. JAMA Ophthalmol 2018;136:202-8. https://doi.org/10.1001/jamaophthalmol.2017.6256.
- Bolz M, Schmidt-Erfurth U, Deak G, Mylonas G, Kriechbaum K, Scholda C. Diabetic Retinopathy Research Group Vienna . Optical coherence tomographic hyperreflective foci: a morphologic sign of lipid extravasation in diabetic macular edema. Ophthalmology 2009;116:914-20. https://doi.org/10.1016/j.ophtha.2008.12.039.
- Framme C, Schweizer P, Imesch M, Wolf S, Wolf-Schnurrbusch U. Behavior of SD-OCT-detected hyperreflective foci in the retina of anti-VEGF-treated patients with diabetic macular edema. Invest Ophthalmol Vis Sci 2012;53:5814-18. https://doi.org/10.1167/iovs.12-9950.
- Nicholson L, Vazquez-Alfageme C, Ramu J, Triantafyllopoulou I, Patrao NV, Muwas M, et al. Validation of concentric rings method as a topographic measure of retinal nonperfusion in ultra-widefield fluorescein angiography. Am J Ophthalmol 2015;160:1217-25. https://doi.org/10.1016/j.ajo.2015.09.003.
- Nicholson L, Vazquez-Alfageme C, Patrao NV, Triantafyllopolou I, Bainbridge JW, Hykin PG, et al. Retinal nonperfusion in the posterior pole is associated with increased risk of neovascularization in central retinal vein occlusion. Am J Ophthalmol 2017;182:118-25. https://doi.org/10.1016/j.ajo.2017.07.015.
- Medicines and Healthcare products Regulatory Agency, Department of Health and Social Care . Good Manufacturing Practice and Good Distribution Practice n.d. www.gov.uk/guidance/good-manufacturing-practice-and-good-distribution-practice (accessed June 2020).
- ICH harmonized tripartite guideline: guideline for good clinical practice. J Postgrad Med 2001;47:45-50.
- Royal College of Ophthalmologists . Ophthalmic Service Guide. Intravitreal Injection Therapy n.d. www.rcophth.ac.uk/wp-content/uploads/2018/02/Intravitreal-Injection-Therapy-August-2018-2.pdf (accessed June 2020).
- Martin DF, Maguire MG, Fine SL, Ying GS, Jaffe GJ, Grunwald JE, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology 2012;119:1388-98. https://doi.org/10.1016/j.ophtha.2012.03.053.
- White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ 2011;342. https://doi.org/10.1136/bmj.d40.
- International conference on harmonisation; guidance on statistical principles for clinical trials; availability – FDA . Notice. Fed Regist 1998;63:49583-98.
- Lesaffre E. Superiority, equivalence, and non-inferiority trials. Bull NYU Hosp Jt Dis 2008;66:150-4.
- Garrett AD. Therapeutic equivalence: fallacies and falsification. Stat Med 2003;22:741-62. https://doi.org/10.1002/sim.1360.
- White IR, Carpenter J, Horton NJ. Including all individuals is not enough: lessons for intention-to-treat analysis. Clin Trials 2012;9:396-407. https://doi.org/10.1177/1740774512450098.
- White IR, Thompson SG. Adjusting for partially missing baseline measurements in randomized trials. Stat Med 2005;24:993-1007. https://doi.org/10.1002/sim.1981.
- Nicholson L, Vazquez-Alfageme C, Sen P, Patrao N, Peto T, Yang Y, et al. The clinical relevance of ultra-widefield angiography findings in patients with central retinal vein occlusion and macular oedema receiving anti-VEGF therapy [published online ahead of print May 25 2021]. Eye (Lond) 2021. https://doi.org/10.1038/s41433-021-01553-7.
- National Institute for Health and Care Excellence . Guide to the Methods of Technology Appraisal 2013 n.d. www.nice.org.uk/process/pmg9/ (accessed 18 June 2019).
- National Institute for Health and Care Excellence . Developing NICE Guidelines: The Manual 2014. www.nice.org.uk/process/pmg20/ (accessed 18 June 2019).
- Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. Cost Eff Resour Alloc 2013;11. https://doi.org/10.1186/1478-7547-11-6.
- Ramsey SD, Willke RJ, Glick H, Reed SD, Augustovski F, Jonsson B, et al. Cost-effectiveness analysis alongside clinical trials II – An ISPOR Good Research Practices Task Force report. Value Health 2015;18:161-72. https://doi.org/10.1016/j.jval.2015.02.001.
- Hykin P, Sivaprasad S, Prevost AT, Vasconcelos JC, Murphy C, Kelly J, et al. Protocol 14PRT 06545: A Multicentre Phase 3 Double-Masked Randomised Controlled Non-Inferiority Trial Comparing the Clinical and Cost Effectiveness of Intravitreal Therapy With Ranibizumab (Lucentis) Vs. Aflibercept (Eylea) Vs. Bevacizumab (Avastin) for Macular Oedema Due to Central Retinal Vein Occlusion (LEAVO Trial) n.d. www.thelancet.com/protocol-reviews/14PRT-06545 (accessed 25 May 2020).
- Alshreef A, Brazier J, Poku E, Hykin P, Sivaprasad S. LEAVO Health Economic and Decision Modelling Analysis Plan (HEDMAP) 2019. https://figshare.shef.ac.uk/articles/LEAVO_Health_Economic_and_Decision_Modelling_Analysis_Plan_HEDMAP_/7988303/1 (accessed 13 May 2019).
- Moher D, Liberati A, Tetzlaff J, Altman DG. PRISMA Group . Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 2009;339. https://doi.org/10.1136/bmj.b2535.
- Kaltenthaler E, Tappenden P, Paisley S, Squires H. NICE DSU Technical Support Document 13: Identifying and Reviewing Evidence to Inform the Conceptualisation and Population of Cost-Effectiveness Models 2011. http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD-13-model-parameters.pdf (accessed 19 July 2019).
- Coyle D, Lee K, Donaldson C, Mugford M, Vale L. Evidence-based Health Economics: From Effectiveness to Efficiency in Systematic Review. London: BMJ Publishing Group; 2002.
- Critical Appraisal Skills Programme . CASP (Systematic Review) Checklist 2018 n.d. https://casp-uk.net/wp-content/uploads/2018/01/CASP-Systematic-Review-Checklist_2018.pdf (accessed 20 June 2019).
- Novartis International AG . LUMINOUS™: Study to Observe the Effectiveness and Safety of Ranibizumab through Individualized Patient Treatment and Associated Outcomes 2017. www.pei.de/SharedDocs/Downloads/DE/awb/nis-0101-0200/0170-abschlussb.pdf?__blob=publicationFile&v=3 (accessed 24 June 2019).
- Pennington R, Alshreef A, Flight A, Metry A, Poku E, Hykin PG, et al. Cost-effectiveness of ranibizumab vs. aflibercept vs. bevacizumab for the treatment of macular oedema due to central retinal vein occlusion: the LEAVO study [published online ahead of print April 26 2021]. PharmacoEconomics 2021. https://doi.org/10.1007/s40273-021-01026-5.
- Brown DM, Campochiaro PA, Bhisitkul RB, Ho AC, Gray S, Saroj N, et al. Sustained benefits from ranibizumab for macular edema following branch retinal vein occlusion: 12-month outcomes of a phase III study. Ophthalmology 2011;118:1594-602. https://doi.org/10.1016/j.ophtha.2011.02.022.
- Claxton L, Hodgson R, Taylor M, Malcolm B, Pulikottil Jacob R. Simulation modelling in ophthalmology: application to cost effectiveness of ranibizumab and aflibercept for the treatment of wet age-related macular degeneration in the United Kingdom. PharmacoEconomics 2017;35:237-48. https://doi.org/10.1007/s40273-016-0459-z.
- Klein BE, Klein R, Moss SE. Change in visual acuity associated with cataract surgery. The Beaver Dam Eye Study. Ophthalmology 1996;103:1727-31. https://doi.org/10.1016/S0161-6420(96)30434-X.
- Hayreh SS, Podhajsky PA, Zimmerman MB. Natural history of visual outcome in central retinal vein occlusion. Ophthalmology 2011;118. https://doi.org/10.1016/j.ophtha.2010.04.019.
- Fleurence RL, Hollenbeak CS. Rates and probabilities in economic modelling: transformation, translation and appropriate application. PharmacoEconomics 2007;25:3-6. https://doi.org/10.2165/00019053-200725010-00002.
- Bertelsen M, Linneberg A, Christoffersen N, Vorum H, Gade E, Larsen M. Mortality in patients with central retinal vein occlusion. Ophthalmology 2014;121:637-42. https://doi.org/10.1016/j.ophtha.2013.07.025.
- Office for National Statistics . National Life Tables: UK 2018. www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/datasets/nationallifetablesunitedkingdomreferencetables (accessed 18 June 2019).
- Briggs AH, Weinstein MC, Fenwick EA, Karnon J, Sculpher MJ, Paltiel AD. ISPOR-SMDM Modeling Good Research Practices Task Force . Model parameter estimation and uncertainty: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force – 6. Value Health 2012;15:835-42. https://doi.org/10.1016/j.jval.2012.04.014.
- Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD. National Eye Institute Visual Function Questionnaire Field Test Investigators . Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol 2001;119:1050-8. https://doi.org/10.1001/archopht.119.7.1050.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Longworth L, Yang Y, Young T, Mulhern B, Hernández Alava M, Mukuria C, et al. Use of generic and condition-specific measures of health-related quality of life in NICE decision-making: a systematic review, statistical modelling and survey. Health Technol Assess 2014;18. https://doi.org/10.3310/hta18090.
- Tosh J, Brazier J, Evans P, Longworth L. A review of generic preference-based measures of health-related quality of life in visual disorders. Value Health 2012;15:118-27. https://doi.org/10.1016/j.jval.2011.08.002.
- Brazier J, Czoski-Murray C, Roberts J, Brown M, Symonds T, Kelleher C. Estimation of a preference-based index from a condition-specific measure: the King’s Health Questionnaire. Med Decis Making 2008;28:113-26. https://doi.org/10.1177/0272989X07301820.
- Rentz AM, Kowalski JW, Walt JG, Hays RD, Brazier JE, Yu R, et al. Development of a preference-based index from the National Eye Institute Visual Function Questionnaire-25. JAMA Ophthalmol 2014;132:310-18. https://doi.org/10.1001/jamaophthalmol.2013.7639.
- Kowalski JW, Rentz AM, Walt JG, Lloyd A, Lee J, Young TA, et al. Rasch analysis in the development of a simplified version of the National Eye Institute Visual-Function Questionnaire-25 for utility estimation. Qual Life Res 2012;21:323-34. https://doi.org/10.1007/s11136-011-9938-z.
- van Hout B, Janssen MF, Feng YS, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15. https://doi.org/10.1016/j.jval.2012.02.008.
- National Institute for Health and Care Excellence . Position Statement on Use of the EQ-5D-5L Valuation Set 2017. www.nice.org.uk/Media/Default/About/what-we-do/NICE-guidance/NICE-technology-appraisal- guidance/eq5d5l_nice_position_statement.pdf (accessed 16 February 2019).
- Wailoo AJ, Hernandez-Alava M, Manca A, Mejia A, Ray J, Crawford B, et al. Mapping to estimate health-state utility from non-preference-based outcome measures: an ISPOR Good Practices for Outcomes Research Task Force Report. Value Health 2017;20:18-27. https://doi.org/10.1016/j.jval.2016.11.006.
- Pennington BM, Hernández-Alava M, Hykin P, Sivaprasad S, Flight L, Alshreef A, et al. Mapping from visual acuity to EQ-5D, EQ-5D with vision bolt-on and VFQ-UI in patients with macular edema in the LEAVO trial. Value Health 2020;23:928-35. https://doi.org/10.1016/j.jval.2020.03.008.
- University of Sheffield . Utility Calculator for Visual Acuity n.d. https://figshare.shef.ac.uk/articles/Utility_calculator_for_visual_acuity/9873731 (accessed 18 June 2019).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2018. Canterbury: Personal Social Services Research Unit, University of Kent; 2018.
- Joint Formulary Committee . British National Formulary n.d. www.medicinescomplete.com (accessed 13 May 2019).
- Department of Health and Social Care . Reference Costs 2017 18 2018.
- Thorn JC, Brookes ST, Ridyard C, Riley R, Hughes DA, Wordsworth S, et al. Core items for a standardized resource use measure: expert Delphi consensus survey. Value Health 2018;21:640-9. https://doi.org/10.1016/j.jval.2017.06.011.
- Royal National Institue of Blind People . The Criteria for Certification 2019. www.rnib.org.uk/eye-health/registering-your-sight-loss/criteria-certification (accessed 31 May 2019).
- Colquitt JL, Jones J, Tan SC, Takeda A, Clegg AJ, Price A. Ranibizumab and pegaptanib for the treatment of age-related macular degeneration: a systematic review and economic evaluation. Health Technol Assess 2008;12. https://doi.org/10.3310/hta12160.
- Meads C, Hyde C. What is the cost of blindness?. Br J Ophthalmol 2003;87. https://doi.org/10.1136/bjo.87.10.1201.
- National Institute for Health and Care Excellence . Adalimumab and Dexamethasone for Treating Non-Infectious Uveitis 2017. www.nice.org.uk/guidance/ta460 (accessed 18 June 2019).
- Hatswell AJ, Bullement A, Briggs A, Paulden M, Stevenson MD. Probabilistic sensitivity analysis in cost-effectiveness models: determining model convergence in cohort models. PharmacoEconomics 2018;36:1421-6. https://doi.org/10.1007/s40273-018-0697-3.
- Drummond M, Brandt A, Luce B, Rovira J. Standardizing methodologies for economic evaluation in health care. Practice, problems, and potential. Int J Technol Assess Health Care 1993;9:26-3. https://doi.org/10.1017/s0266462300003007.
- Faria R, Gomes M, Epstein D, White IR. A guide to handling missing data in cost-effectiveness analysis conducted within randomised controlled trials. PharmacoEconomics 2014;32:1157-70. https://doi.org/10.1007/s40273-014-0193-3.
- Willan AR, Briggs AH, Hoch JS. Regression methods for covariate adjustment and subgroup analysis for non-censored cost-effectiveness data. Health Econ 2004;13:461-75. https://doi.org/10.1002/hec.843.
- Alshreef A, Wailoo AJ, Brown SR, Tiernan JP, Watson AJM, Biggs K, et al. Cost-effectiveness of haemorrhoidal artery ligation versus rubber band ligation for the treatment of grade II-III haemorrhoids: analysis using evidence from the HubBLe trial. Pharmacoecon Open 2017;1:175-84. https://doi.org/10.1007/s41669-017-0023-6.
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96. https://doi.org/10.1002/hec.944.
- Department of Health and Social Care . NHS Reference Costs 2016 17 2017.
- Margrain TH. Minimising the impact of visual impairment. BMJ 1999;318. https://doi.org/10.1136/bmj.318.7197.1504.
- Galaria II, Casten RJ, Rovner BW. Development of a shorter version of the geriatric depression scale for visually impaired older patients. Int Psychogeriatr 2000;12:435-43. https://doi.org/10.1017/s1041610200006554.
- Owen CG, Fletcher AE, Donoghue M, Rudnicka AR. How big is the burden of visual loss caused by age related macular degeneration in the United Kingdom?. Br J Ophthalmol 2003;87. https://doi.org/10.1136/bjo.87.3.312.
- Deramo VA, Cox TA, Syed AB, Lee PP, Fekrat S. Vision-related quality of life in people with central retinal vein occlusion using the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol 2003;121:1297-302. https://doi.org/10.1001/archopht.121.9.1297.
- Ford JA, Shyangdan D, Uthman OA, Lois N, Waugh N. Drug treatment of macular oedema secondary to central retinal vein occlusion: a network meta-analysis. BMJ Open 2014;4. https://doi.org/10.1136/bmjopen-2014-005292.
- Giuffre G, Randazzo-Papa G, Palumbo C. Central retinal vein occlusion in young people. Doc Ophthalmol 1992;80:127-32. https://doi.org/10.1007/BF00161238.
- May DR, Klein ML, Peyman GA, Raichand M. Xenon arc panretinal photocoagulation for central retinal vein occlusion: a randomised prospective study. Br J Ophthalmol 1979;63:725-34. https://doi.org/10.1136/bjo.63.11.725.
- Sigford DK, Reddy S, Mollineaux C, Schaal S. Global reported endophthalmitis risk following intravitreal injections of anti-VEGF: a literature review and analysis. Clin Ophthalmol 2015;9:773-81. https://doi.org/10.2147/OPTH.S77067.
- Xu Y, Tan CS. Safety and complications of intravitreal injections performed in an Asian population in Singapore. Int Ophthalmol 2017;37:325-32. https://doi.org/10.1007/s10792-016-0241-4.
- Bradshaw SE, Gala S, Nanavaty M, Shah A, Mwamburi M, Kefalas P. Systematic literature review of treatments for management of complications of ischemic central retinal vein occlusion. BMC Ophthalmol 2016;16. https://doi.org/10.1186/s12886-016-0282-5.
- Braithwaite T, Nanji AA, Greenberg PB. Anti-vascular endothelial growth factor for macular edema secondary to central retinal vein occlusion. Cochrane Database Syst Rev 2010;10. https://doi.org/10.1002/14651858.CD007325.pub2.
- Brand C. Luminous: results from the 2014 interim analysis to provide further real-world evidence for clinical ranibizumab use. Ophthalmologica 2014;232:4-5.
- Chatziralli I, Theodossiadis G, Chatzirallis A, Parikakis E, Mitropoulos P, Theodossiadis P. Ranibizumab for retinal vein occlusion: predictive factors and long-term outcomes in real-life data. Retina 2018;38:559-68. https://doi.org/10.1097/IAE.0000000000001579.
- Cornel S, Adriana ID, Mihaela TC, Speranta S, Algerino S, Mehdi B, et al. Anti-vascular endothelial growth factor indications in ocular disease. Rom J Ophthalmol 2015;59:235-42.
- Daradbounis D, Nanos P, Patsiamanidi M. Long term results of ranibizumab treatment in patients with macular oedema due to retinal venous occlusive disease. Ophthalmologica 2014;232.
- DeCroos FC, Todorich B, Alshareef R, Khuthaila M, Fekrat S, Ho AC, et al. Neovascular events in eyes with central retinal vein occlusion undergoing serial bevacizumab or ranibizumab intravitreal injections: a retrospective review. J Ophthalmic Vis Res 2014;9:461-8.
- Deonandan R, Jones S. Anti-Vascular Endothelial Growth Factor Drugs for the Treatment of Retinal Conditions: A Review of the Safety. Ottawa, ON: Canadian Agency for Drugs and Technologies in Health; 2017.
- Edwards SJ, Barton S, Trevor N, Lois N, Nherera L, Hamilton V. PSS2 Comparisons of the clinical effectiveness of treatments for macular oedema (MO) caused by retinal vein occlusion (RVO). Value Health 2012;15. https://doi.org/10.1016/j.jval.2012.08.2049.
- Freund KB, Korobelnik JF, Devenyi R, Framme C, Galic J, Herbert E, et al. Treat-and-extend regimens with anti-vegf agents in retinal diseases: a literature review and consensus recommendations. Retina 2015;35:1489-506. https://doi.org/10.1097/IAE.0000000000000627.
- Gallego-Pinazo R, Dolz-Marco R, Marín-Lambíes C, Díaz-Llopis M. Safety and efficacy of ranibizumab in macular edema following retinal vein occlusion. Ophthalmol Eye Dis 2012;4:15-21. https://doi.org/10.4137/OED.S7264.
- Gerding H, Monés J, Tadayoni R, Boscia F, Pearce I, Priglinger S. Ranibizumab in retinal vein occlusion: treatment recommendations by an expert panel. Br J Ophthalmol 2015;99:297-304. https://doi.org/10.1136/bjophthalmol-2014-305041.
- Glanville J, Patterson J, McCool R, Ferreira A, Gairy K, Pearce I. Efficacy and safety of widely used treatments for macular oedema secondary to retinal vein occlusion: a systematic review. BMC Ophthalmol 2014;14. https://doi.org/10.1186/1471-2415-14-7.
- Hernando M, López M, Sánchez M, Manco F, Alonso J, Manzanas L. 4CPS-187 Effect of ranibizumab in vision-related quality of life in patients diagnosed with retinal pathology. Eur J Hosp Pharm 2018;25. https://doi.org/10.1136/ejhpharm-2018-eahpconf.277.
- Jager RD, Aiello LP, Patel SC, Cunningham ET. Risks of intravitreous injection: a comprehensive review. Retina 2004;24:676-98. https://doi.org/10.1097/00006982-200410000-00002.
- Jiang Y, Mieler WF. Update on the use of anti-VEGF intravitreal therapies for retinal vein occlusions. Asia Pac J Ophthalmol 2017;6:546-53. https://doi.org/10.22608/APO.2017459.
- Jumper JM, Dugel PU, Chen S, Blinder KJ, Walt JG. Anti-VEGF treatment of macular edema associated with retinal vein occlusion: patterns of use and effectiveness in clinical practice (ECHO study report 2). Clin Ophthalmol 2018;12:621-9. https://doi.org/10.2147/OPTH.S163859.
- Kinge B, Stordahl PB, Forsaa V, Fossen K, Haugstad M, Helgesen OH, et al. Efficacy of ranibizumab in patients with macular edema secondary to central retinal vein occlusion: results from the sham-controlled ROCC study. Am J Ophthalmol 2010;150:310-14. https://doi.org/10.1016/j.ajo.2010.03.028.
- Konidaris V, Al-Hubeshy Z, Tsaousis KT, Gorgoli K, Banerjee S, Empeslidis T. Outcomes of switching treatment to aflibercept in patients with macular oedema secondary to central retinal vein occlusion refractory to ranibizumab. Int Ophthalmol 2018;38:207-13. https://doi.org/10.1007/s10792-017-0512-8.
- Kornhauser T, Schwartz R, Goldstein M, Neudorfer M, Loewenstein A, Barak A. Bevacizumab treatment of macular edema in CRVO and BRVO: long-term follow-up. (BERVOLT study: Bevacizumab for RVO long-term follow-up). Graefes Arch Clin Exp Ophthalmol 2016;254:835-44. https://doi.org/10.1007/s00417-015-3130-z.
- Kumar P, Banarji A, Patyal S, Gurunadh VS, Ahluwalia TS, Oli A, et al. A clinical study to evaluate the efficacy of intravitreal anti-VEGF therapy in treating macular edema due to retinal venous occlusions. Med J Armed Forces India 2013;69:260-7. https://doi.org/10.1016/j.mjafi.2013.02.001.
- Liu M, Eichenbaum D, Taylor S, Wang P, Quezada-Ruiz C. Branch and central retinal vein occlusion: clinical pearls from trials of ranibizumab. Invest Ophthalmol Vis Sci 2017;58:4624-4.
- Mohamed Q, McIntosh RL, Saw SM, Wong TY. Interventions for central retinal vein occlusion: an evidence-based systematic review. Ophthalmology 2007;114:507-19. https://doi.org/10.1016/j.ophtha.2006.11.011.
- ClinicalTrials.gov . A Study Evaluating Dosing Regimens for Treatment With Intravitreal Ranibizumab Injections in Subjects With Macular Edema Following Retinal Vein Occlusion 2011. https://clinicaltrials.gov/ct2/show/NCT01277302 (accessed 18 June 2019).
- Nghiem-Buffet S, Baillif S, Regnier S, Skelly A, Yu N, Sodi A. Treatment patterns of ranibizumab intravitreal injection and dexamethasone intravitreal implant for retinal vein occlusion in the USA. Eye 2017;31:551-9. https://doi.org/10.1038/eye.2016.269.
- Nicolò M, Bonetto M, Rosa R, Musetti D, Musolino M, Traverso CE, et al. Real-life management of patients with retinal vein occlusion using I-Macula web platform. J Ophthalmol 2017;2017. https://doi.org/10.1155/2017/5601786.
- Nuzzi R, Tridico F. Local and systemic complications after intravitreal administration of anti-vascular endothelial growth factor agents in the treatment of different ocular diseases: a five-year retrospective study. Semin Ophthalmol 2015;30:129-35. https://doi.org/10.3109/08820538.2013.835833.
- Pacella E, Pacella F, La Torre G, Impallara D, Malarska K, Brillante C, et al. Testing the effectiveness of intravitreal ranibizumab during 12 months of follow-up in venous occlusion treatment. Clin Ter 2012;163:e413-22.
- Patel A, Nguyen C, Lu S. Central retinal vein occlusion: a review of current evidence-based treatment options. Middle East Afr J Ophthalmol 2016;23:44-8. https://doi.org/10.4103/0974-9233.173132.
- Penedones A, Mendes D, Alves C, Batel Marques F. Safety monitoring of ophthalmic biologics: a systematic review of pre- and postmarketing safety data. J Ocul Pharmacol Ther 2014;30:729-51. https://doi.org/10.1089/jop.2013.0206.
- Poku E, Rathbone J, Wong R, Everson-Hock E, Essat M, Pandor A, et al. The safety of intravitreal bevacizumab monotherapy in adult ophthalmic conditions: systematic review. BMJ Open 2014;4. https://doi.org/10.1136/bmjopen-2014-005244.
- Qian T, Zhao M, Xu X. Comparison between anti-VEGF therapy and corticosteroid or laser therapy for macular oedema secondary to retinal vein occlusion: a meta-analysis. J Clin Pharm Ther 2017;42:519-29. https://doi.org/10.1111/jcpt.12551.
- Rayess N, Rahimy E, Storey P, Shah CP, Wolfe JD, Chen E, et al. Postinjection endophthalmitis rates and characteristics following intravitreal bevacizumab, ranibizumab, and aflibercept. Am J Ophthalmol 2016;165:88-93. https://doi.org/10.1016/j.ajo.2016.02.028.
- Regnard MA, Glacet-Bernard A, Mascali R, Souied EH. Anti-VEGF treatment of macular edema using a treat-and-extend regimen in retinal vein occlusion in clinical practice. Invest Ophthalmol Vis Sci 2016;57:4158-8.
- Risard SM, Pieramici DJ, Rabena MD, Basefsky JC, Avery RL, Castellarin AA, et al. Intravitreal ranibizumab for macular edema secondary to central retinal vein occlusion. Retina 2011;31:1060-7. https://doi.org/10.1097/IAE.0b013e3181fbce76.
- Scott IU, Figueroa MJ, Oden NL, Ip MS, Blodi BA, VanVeldhuisen PC. SCORE2 Investigator Group . SCORE2 report 5: vision-related function in patients with macular edema secondary to central retinal or hemiretinal vein occlusion. Am J Ophthalmol 2017;184:147-56. https://doi.org/10.1016/j.ajo.2017.10.008.
- Sharma S. Canadian Luminous Investigators. Baseline characteristics of Canadian patients with neovascular age-related macular degeneration (nAMD), diabetic macular edema (DME) and retinal vein occlusion (RVO) enrolled in the LUMINOUS study. Invest Ophthalmol Vis Sci 2015;56:5356-6.
- Sophie R, Hafiz G, Scott AW, Zimmer-Galler I, Nguyen QD, Ying H, et al. Long-term outcomes in ranibizumab-treated patients with retinal vein occlusion; the role of progression of retinal nonperfusion. Am J Ophthalmol 2013;156:693-705. https://doi.org/10.1016/j.ajo.2013.05.039.
- Tabandeh H, Boscia F, Sborgia A, Ciracì L, Dayani P, Mariotti C, et al. Endophthalmitis associated with intravitreal injections: office-based setting and operating room setting. Retina 2014;34:18-23. https://doi.org/10.1097/IAE.0000000000000008.
- Taylor M, Serbetci E, Ferreira A, Gairy K, Lewis L, Blouin J, et al. A United Kingdom-based economic evaluation of ranibizumab for patients with retinal vein occlusion (RVO). J Med Econ 2014;17:423-34. https://doi.org/10.3111/13696998.2014.909435.
- Thulliez M, Angoulvant D, Le Lez ML, Jonville-Bera AP, Pisella PJ, Gueyffier F, et al. Cardiovascular events and bleeding risk associated with intravitreal antivascular endothelial growth factor monoclonal antibodies: systematic review and meta-analysis. JAMA Ophthalmol 2014;132:1317-26. https://doi.org/10.1001/jamaophthalmol.2014.2333.
- Vorum H, Olesen TK, Zinck J, Størling Hedegaard M. Real world evidence of use of anti-VEGF therapy in Denmark. Curr Med Res Opin 2016;32:1943-50. https://doi.org/10.1080/03007995.2016.1221803.
- Wang JK. A review of randomized trials of approved pharmaceutical agents for macular edema secondary to retinal vein occlusion. Asia Pac J Ophthalmol 2016;5:159-64. https://doi.org/10.1097/APO.0000000000000168.
- Wecker T, Ehlken C, Bühler A, Lange C, Agostini H, Böhringer D, et al. Five-year visual acuity outcomes and injection patterns in patients with pro-re-nata treatments for AMD, DME, RVO and myopic CNV. Br J Ophthalmol 2017;101:353-9. https://doi.org/10.1136/bjophthalmol-2016-308668.
- Yeh S, Kim SJ, Ho AC, Schoenberger SD, Bakri SJ, Ehlers JP, et al. Therapies for macular edema associated with central retinal vein occlusion: a report by the American Academy of Ophthalmology. Ophthalmology 2015;122:769-78. https://doi.org/10.1016/j.ophtha.2014.10.013.
- Yuan A, Ahmad BU, Xu D, Singh RP, Kaiser PK, Martin DF, et al. Comparison of intravitreal ranibizumab and bevacizumab for the treatment of macular edema secondary to retinal vein occlusion. Int J Ophthalmol 2014;7:86-91. https://doi.org/10.3980/j.issn.2222-3959.2014.01.15.
- Ziemssen F, Feltgen N, Holz FG, Guthoff R, Ringwald A, Bertelmann T, et al. Demographics of patients receiving Intravitreal anti-VEGF treatment in real-world practice: healthcare research data versus randomized controlled trials. BMC Ophthalmol 2017;17. https://doi.org/10.1186/s12886-017-0401-y.
- Turner J, O’Cathain A, Knowles E, Nicholl J, Tosh J, Sampson F. Evaluation of NHS 111 Pilot Sites. Final Report to the Department of Health 2012.
Appendix 1 The LEAVO study group
LEAVO study group
The LEAVO study group thanks all the patients who participated in the study, and all of the site investigators and research teams.
| Site | Principal investigator |
|---|---|
| Moorfields Eye Hospital NHS Foundation Trust, London | Sobha Sivaprasad |
| King’s College Hospital, London | Haralabos Eleftheriadis |
| New Cross Hospital, Wolverhampton & Midland Counties Eye Infirmary, Wolverhampton | Yit Yang |
| Royal Liverpool and Broadgreen University Hospitals NHS Trust, Liverpool | Michael Briggs |
| University Hospital Southampton NHS Foundation Trust, Southampton | Andrew Lotery |
| Royal Victoria Hospital, Belfast, and Queen’s University Belfast | Michael Williams |
| Department of Ophthalmology, Royal Blackburn Hospital, Blackburn | Salwa Abugreen |
| Bradford Ophthalmology Research Network, Bradford Teaching Hospitals NHS Foundation Trust, Bradford | Faruque Ghanchi |
| Sussex Eye Hospital, Brighton | Edward Hughes |
| Bristol Eye Hospital, Bristol | Adam Ross |
| Department of Ophthalmology, West Suffolk NHS Foundation Trust, Suffolk | Nitin Gupta |
| Ophthalmology Department, Torbay Hospital, Devon | Stephen Turner Yinka Osoba |
| Essex County Hospital, Colchester | Jignesh Patel |
| Macular Unit, Hospital of St. Cross, Rugby | Sergio Pagliarini |
| Birmingham and Midlands Eye Centre, Birmingham | Peck-Lin Lip |
| Kent and Canterbury Hospital, Canterbury | Nishal Patel Afsar Jafree |
| Ophthalmology Department, Frimley Park Hospital NHS Foundation Trust, Surrey | Geeta Menon |
| Whipps Cross University Hospital, Barts Health NHS Trust, London | Sudeshna Patra |
| James Paget University Hospital, Norfolk | Ben Burton |
| Department of Ophthalmology, Royal Surrey County Hospital, Guildford, Surrey | Simon Taylor |
| Harrogate and District NHS Foundation Trust, Harrogate, North Yorkshire | Sarah Mackenzie |
| York Teaching Hospital NHS Foundation Trust, York | Richard Gale |
| Darlington Memorial Hospital, County Durham and Darlington NHS Foundation Trust | Komala Vadivelu |
| St James’s University Hospital, Leeds | Martin McKibbin |
| Ophthalmology Department, Hillingdon Hospitals NHS Foundation Trust, London | Sheena George |
| Maidstone and Tunbridge Wells NHS Trust, Kent | Goncalo Almeida |
| Central Manchester Hospital, Manchester University NHS Foundation Trust, Manchester | Yvonne D’Souza |
| Royal Victoria Infirmary, Newcastle upon Tyne | James Talks |
| Luton and Dunstable NHS University Hospital, Hertfordshire | Venki Sundaram |
| University Hospital of Wales, Cardiff | Sanjiv Banerjee |
| Sunderland Eye Infirmary, Sunderland | Maged Habib |
| Royal Glamorgan Hospital, North Glamorgan NHS Trust | Raghu Ram |
| Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield | Christopher Brand |
| Addenbrooke’s Hospital, Cambridge | Doug Newman |
| Department of Ophthalmology, Gartnavel General Hospital, Glasgow | David Gilmour |
| Ophthalmology Department, Bolton NHS Foundation Trust, Bolton | Simon Kelly |
| Calderdale Royal Hospital, Halifax | Rehna Khan |
| University Hospitals of Leicester NHS Trust, Leicester | Theo Empeslidis |
| Department of Ophthalmology, Norfolk and Norwich Hospital, Norwich | Colin Jones |
| Cheltenham General Hospital, Gloucestershire | Emily Fletcher |
| Department of Ophthalmology, Hull and East Yorkshire Hospitals NHS Trust, Hull | Louise Downey |
| Western Eye Hospital, London | Saad Younis |
| James Cook University Hospital, South Tees NHS Foundation Trust, South Tees | Philip Severn |
| Princess Alexandra Hospital, Harlow, Essex | Priya Prakash |
Appendix 2 The LEAVO study committees
We would like to thank the following for their valuable contribution to this study:
Trial Steering Committee members – Susan Downes (chairperson, Oxford Eye Hospital, UK), Irene Stratton (Gloucestershire Hospitals NHS Foundation Trust, UK), Hiten Dodhia (Lambeth and Southwark Councils, Public Health, London, UK), Greg Fell (Sheffield Council, Public Health, Sheffield, UK), Riaz Asaria (Royal Free London NHS Foundation Trust, London, UK), Jonathan Byrne (King’s College NHS Foundation Trust, London, UK), Vanessa Burgess (NHS Lambeth Clinical Commissioning Group, London, UK), Alison Powling (Community Diabetes, Barts Health NHS Trust, London, UK) and Mrs Melba Ryde (lay representative).
Data Monitoring Committee members – Sarah Walker (chairperson, Oxford University, Oxford, UK), Consuela Moorman (Stoke Mandeville Hospital, Buckinghamshire Healthcare NHS Trust) and Baljean Dhillon (Centre for Clinical Brain Sciences, University of Edinburgh).
Appendix 3 Additional data: tables and figures
| Type of variable | Outcome | Method |
|---|---|---|
| Continuous | BCVA at 52 weeks | LME model |
| Mean OCT CST at 52 and 100 weeks | LME model | |
| Macular volume at 52 and 100 weeks | LME model | |
| VFQ-25 composite score, distance and near subscales at 52 and 100 weeks | LME model | |
| Number of injections by 100 weeks | Difference in means with 95% CI | |
| Change in retinal non-perfusion at week 100 as assessed by disc areas of non-perfusion (in approximately 27 sites) | Difference in medians with 95% CI | |
| Categorical | Participants with a ≥ 15 and ≥ 10 ETDRS letter improvement, < 15 ETDRS letter loss and ≥ 30 ETDRS letter loss (severe visual loss) at 52 and 100 weeks | Differences in proportions with 95% CI |
| Participants scoring ≥ 73 ETDRS letters, ≤ 58 ETDRS letters and ≤ 19 letters at 52 and 100 weeks | Differences in proportions with 95% CI consistent with a chi-squared test | |
| Participants with OCT CST of < 320 µm at 52 and 100 weeks | Differences in proportions with 95% CI consistent with a chi-squared test | |
| Persistent non-responder participants at 52 and 100 weeks | Differences in proportions with 95% CI | |
| Participants developing ocular neovascularisation by 52 and 100 weeks | Differences in proportions with 95% CI | |
| Participants with OCT anatomical features (e.g. diffuse intraretinal oedema, subretinal fluid, vitreomacular interface abnormality, EZ disruption, DRIL) at 52 and 100 weeks | Differences in proportions with 95% CI | |
| Participants with change in area of retinal non-perfusion | Differences in proportions with 95% CI | |
| Prevalence of local and systemic side effects | Differences in proportions with 95% CI |
| Assessment | Screening | Baseline | Week | Withdrawal visit | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 8 | 12 | 16 | 20 | 24 | 28–48 | 52 | 56–72 | 76 | 80–96 | 100 | ||||
| Variable treatment visits | 4–8 weeks | 4–8 weeks | 4–8 weeks | 13–97 weeks | |||||||||||
| Informed consent | ✗ | ||||||||||||||
| Inclusion/exclusion criteria (✗1 if on different daya) | ✗ | ✗1 | |||||||||||||
| Randomisation | ✗ | ||||||||||||||
| Urine pregnancy test for women of child-bearing age | ✗ | ||||||||||||||
| Patient demographics and medical and ophthalmic history | ✗ | ||||||||||||||
| AEs | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Concomitant medication review | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Blood pressure | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| BCVA (ETDRS) in both eyes (✗2 if with refraction) | ✗2 | ✗ | ✗ | ✗ | ✗2 | ✗ | ✗ | ✗2 | ✗ | ✗2 | ✗ | ✗2 | ✗ | ✗2 | ✗2 |
| Standard ophthalmic examination of both eyes | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| OCT on both eyes | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Wide-angle or seven-field colour fundus photography | ✗ | ✗ | ✗ | ✗ | |||||||||||
| Wide-angle or seven-field FFA | ✗ | ✗ | ✗ | ||||||||||||
| VFQ-25, EQ-5D and EQ-5D-V | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||
| Resource use questionnaire | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||||
| Treatment allocation guess form | ✗ | ✗ | |||||||||||||
| Administer IMP (✗3 if pro re nata treatment) | ✗ | ✗ | ✗ | ✗ | ✗3 | ✗3 | ✗3 | ✗3 | ✗3 | ✗3 | ✗3 | ✗3 | |||
| Amendment number | Purpose | Sponsor classification | MHRA date approved | REC date approved | HRA date approved | Changes to documents |
|---|---|---|---|---|---|---|
| SA#1 |
|
Substantial | 24 July 2014 | 4 September 2014 | N/A |
|
| SA#2 |
|
Substantial | 27 February 2015 | 10 November 2014 | N/A |
|
| SA#3 | New principal investigator at existing site, removal of site, addition of new site | Substantial | 16 March 2015 | 17 February 2015 | N/A |
|
| SA#4 | Adding sites: Calderdale Royal Hospital, Leicester Royal Infirmary, Norfolk and Norwich Hospital, Cheltenham General Hospital | Substantial | N/A | 2 June 2015 | N/A | None |
| SA#5 | Adding sites: Hull Royal Infirmary, Gartnavel General Hospital, Western Eye Hospital, James Cook University Hospital, Princess Alexandra Hospital, Aberdeen Royal Infirmary; new principal investigator at existing site: Cheltenham General Hospital | Substantial | N/A | 4 August 2015 | N/A | None |
| SA#6 | Changes to protocol, patient information sheet, ICF | Substantial | 14 March 2016 | 11 February 2016 | 16 May 2016 |
|
| SA#7 | New principal investigator at existing site: Darlington Memorial Hospital | Substantial | N/A | 11 August 2016 | N/A | None |
| SA#8 | New principal investigator at existing site: Canterbury Hospital | Substantial | N/A | 19 June 2017 | 20 June 2017 | None |
| SA#9 | Change of SPC regarding reference safety information | Substantial | 2 August 2017 | 25 July 2017 | 22 August 2017 | None |
| SA#10 | New principal investigator at existing site: Darlington Memorial Hospital | Substantial | N/A | 16 July 2018 | 16 July 2018 | None |
| SA#11 | New principal investigator at existing site: Torbay Hospital | Substantial | N/A | 4 September 2018 | 4 September 2018 | None |
| Time point | Trial arm (n) | Total (n) | ||
|---|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | ||
| Baseline | 3 | 0 | 0 | 3 |
| 4 weeks | 1 | 0 | 2 | 3 |
| 8 weeks | 2 | 1 | 0 | 3 |
| 12 weeks | 0 | 2 | 0 | 2 |
| 16 weeks | 0 | 1 | 0 | 1 |
| 20 weeks | 0 | 0 | 2 | 2 |
| 24 weeks | 1 | 1 | 0 | 2 |
| 28 weeks | 3 | 0 | 1 | 4 |
| 32 weeks | 1 | 2 | 0 | 3 |
| 36 weeks | 0 | 0 | 0 | 0 |
| 40 weeks | 2 | 1 | 2 | 5 |
| 44 weeks | 0 | 0 | 0 | 0 |
| 48 weeks | 0 | 0 | 1 | 1 |
| 52 weeks | 0 | 2 | 2 | 4 |
| 56 weeks | 2 | 1 | 0 | 3 |
| 60 weeks | 0 | 0 | 0 | 0 |
| 64 weeks | 0 | 2 | 2 | 4 |
| 68 weeks | 0 | 3 | 1 | 4 |
| 72 weeks | 1 | 3 | 0 | 4 |
| 76 weeks | 1 | 1 | 1 | 3 |
| 80 weeks | 0 | 1 | 0 | 1 |
| 84 weeks | 0 | 0 | 0 | 0 |
| 88 weeks | 1 | 0 | 0 | 1 |
| 92 weeks | 1 | 0 | 1 | 2 |
| 96 weeks | 1 | 0 | 0 | 1 |
| Total | 20 | 21 | 15 | 56 |
| Date withdrawn | Date randomised | Weeks in trial (n) | Reason for withdrawal | Trial arm |
|---|---|---|---|---|
| 30 June 2015 | 9 April 2015 | 12 | Health deterioration | Ranibizumab |
| 14 September 2015 | 24 June 2015 | 12 | Participant no longer wish to take part | Aflibercept |
| 6 November 2015 | 10 September 2015 | 8 | Unable to locate/contact participant | Bevacizumab |
| 6 November 2015 | 25 September 2015 | 6 | Participant no longer wish to take part | Ranibizumab |
| 8 December 2015 | 8 December 2015 | 0 | Other | Ranibizumab |
| 8 January 2016 | 31 March 2015 | 40 | Other | Bevacizumab |
| 12 April 2016 | 19 May 2015 | 47 | Participant no longer wishes to take part | Bevacizumab |
| 26 May 2016 | 1 September 2015 | 38 | Participant no longer wishes to take part | Aflibercept |
| 1 June 2016 | 23 December 2015 | 23 | Participant no longer wishes to take part | Bevacizumab |
| 7 June 2016 | 13 October 2015 | 34 | AE | Ranibizumab |
| 21 June 2016 | 23 September 2015 | 39 | Patient moving away from area | Aflibercept |
| 22 July 2016 | 16 June 2015 | 57 | Participant no longer wishes to take part | Ranibizumab |
| 29 July 2016 | 18 April 2016 | 15 | Other | Ranibizumab |
| 19 August 2016 | 9 June 2016 | 10 | Death of participant | Ranibizumab |
| 30 August 2016 | 29 January 2016 | 31 | Unable to locate/contact participant | Aflibercept |
| 26 September 2016 | 3 November 2015 | 47 | Health deterioration | Ranibizumab |
| 12 October 2016 | 17 June 2015 | 69 | Patient moving away from area | Bevacizumab |
| 17 October 2016 | 28 August 2015 | 59 | Participant no longer wishes to take part | Ranibizumab |
| 19 October 2016 | 11 December 2015 | 45 | Participant no longer wishes to take part | Ranibizumab |
| 29 October 2016 | 18 February 2016 | 36 | Death of participant | Aflibercept |
| 31 October 2016 | 14 April 2016 | 29 | Death of participant | Bevacizumab |
| 8 November 2016 | 25 April 2016 | 28 | Participant no longer wishes to take part | Aflibercept |
| 9 November 2016 | 8 April 2015 | 83 | Unable to locate/contact participant | Aflibercept |
| 26 November 2016 | 26 January 2016 | 44 | Participant no longer wishes to take part | Bevacizumab |
| 18 December 2016 | 13 June 2016 | 27 | Death of participant | Aflibercept |
| 3 January 2017 | 26 August 2016 | 19 | Death of participant | Aflibercept |
| 3 January 2017 | 26 August 2015 | 71 | Health deterioration | Aflibercept |
| 12 January 2017 | 17 September 2015 | 69 | Unable to locate/contact participant | Aflibercept |
| 1 February 2017 | 13 April 2016 | 42 | Participant no longer wishes to take part | Ranibizumab |
| 9 February 2017 | 6 November 2015 | 66 | Participant no longer wishes to take part | Bevacizumab |
| 20 February 2017 | 23 October 2015 | 69 | Other | Aflibercept |
| 2 March 2017 | 28 April 2016 | 44 | Participant no longer wishes to take part | Ranibizumab |
| 9 March 2017 | 22 October 2015 | 72 | Death of participant | Ranibizumab |
| 10 March 2017 | 23 October 2015 | 72 | Participant no longer wishes to take part | Bevacizumab |
| 21 March 2017 | 27 October 2015 | 73 | AE | Aflibercept |
| 15 May 2017 | 3 March 2016 | 63 | Death of participant | Ranibizumab |
| 25 May 2017 | 31 December 2015 | 73 | Participant no longer wishes to take part | Aflibercept |
| 19 June 2017 | 16 October 2015 | 87 | Death of participant | Aflibercept |
| 1 August 2017 | 12 October 2015 | 94 | Death of participant | Bevacizumab |
| 5 September 2017 | 22 March 2016 | 76 | Health deterioration | Bevacizumab |
| 14 September 2017 | 25 February 2016 | 81 | AE | Bevacizumab |
| 10 November 2017 | 14 November 2016 | 52 | Participant no longer wishes to take part | Aflibercept |
| 13 November 2017 | 2 June 2016 | 76 | Unable to locate/contact participant | Ranibizumab |
| 17 November 2017 | 21 October 2015 | 108 | Unable to locate/contact participant | Ranibizumab |
| 27 November 2017 | 14 June 2016 | 76 | Death of participant | Aflibercept |
| 4 December 2017 | 28 October 2016 | 57 | Death of participant | Bevacizumab |
| 17 January 2018 | 28 October 2016 | 64 | Death of participant | Bevacizumab |
| 1 March 2018 | 17 June 2016 | 89 | Participant no longer wishes to take part | Ranibizumab |
| 29 March 2018 | 23 June 2016 | 92 | Participant no longer wishes to take part | Aflibercept |
| 5 May 2018 | 18 October 2016 | 81 | Death of participant | Aflibercept |
| 4 June 2018 | 10 October 2016 | 86 | Patient moving away from area | Aflibercept |
| 13 August 2018 | 24 November 2016 | 90 | AE | Bevacizumab |
| 13 September 2018 | 11 October 2016 | 100 | Participant no longer wishes to take part | Ranibizumab |
| 5 October 2018 | 29 November 2016 | 96 | Health deterioration | Aflibercept |
| 13 November 2018 | 30 November 2016 | 102 | AE | Ranibizumab |
| 27 November 2018 | 4 November 2016 | 108 | Unable to locate/contact participant | Ranibizumab |
| Time point | Treatment A | Treatment B | Adjusted difference between groupsa (95% CI) |
|---|---|---|---|
| Aflibercept vs. ranibizumab | |||
| Aflibercept | Ranibizumab | ||
| Mean (SE) | |||
| At screening | 12.3 (0.2) | 13.0 (0.2) | |
| At 52 weeks | 9.1 (0.2); n = 140 | 9.2 (0.2); n = 138 | |
| At 100 weeks | 8.6 (0.1); n = 133 | 8.9 (0.1); n = 135 | |
| Adjusted difference | |||
| At 52 weeks | –0.1 (–0.6 to 0.4) | ||
| At 100 weeks | –0.2 (–0.6 to 0.3) | ||
| Bevacizumab vs. ranibizumab | |||
| Bevacizumab | Ranibizumab | ||
| Mean (SE) | |||
| At screening | 12.8 (0.2) | 13.0 (0.2) | |
| At 52 weeks | 9.4 (0.2); n = 135 | 9.2 (0.2); n = 138 | |
| At 100 weeks | 9.1 (0.2); n = 135 | 8.9 (0.1); n = 135 | |
| Adjusted difference | |||
| At 52 weeks | 0.2 (–0.3 to 0.7) | ||
| At 100 weeks | 0.3 (–0.2 to 0.7) | ||
| Parameter | Distribution | Mean (SE) | Source of mean | Source for SE |
|---|---|---|---|---|
| Intervention and related costs | ||||
| Ranibizumab injection | N/A | £551.00 | BNF117 | N/A |
| Aflibercept injection | N/A | £816.00 | BNF117 | N/A |
| Bevacizumab injection | N/A | £28.00 | Judicial review53 | N/A |
| CST cost | Gamma | £108.21 |
|
|
| First visit cost | Gamma | £140.04 |
|
|
| Follow-up visit cost | Gamma | £105.19 |
|
|
| Costs associated with resource use | ||||
| A&E visit cost | Gamma | £160.23 (£9.34) |
|
|
| Visit cost of ocular A&E | Gamma | £118.02 (£2.67) |
|
|
| Visit cost of eye consultant | Gamma | £95.13 (£1.85) |
|
|
| Call cost to ophthalmologist | Gamma | £28.20 (£4) |
|
|
| Visit cost of optometrist/optician | Gamma | £76.50 (£10.50) |
|
|
| Visit cost for low-vision appointment | N/A | £153.00 | Estimated to be double the visit cost of optometrist/optician | |
| Visit cost of GP | Gamma | £37.40 (£3.74) | Curtis and Burns116 | 10% assumption around the mean |
| Visit cost of practice nurse | Gamma | £17.79 (£1.78) | ||
| Call cost to GP | Gamma | £28.00 (£2.80) | ||
| Resource use parameters (every 3 months) | ||||
| A&E visit: WSE | Multinormal | –0.001 | Analysis of LEAVO data | |
| A&E visit: constant | 0.103 | |||
| Eye A&E visit: WSE | Multinormal | –0.002 | ||
| Eye A&E visit: constant | 0.183 | |||
| GP visit: WSE | Multinormal | –0.004 | ||
| GP visit: constant | 0.441 | |||
| GP call: WSE | Multinormal | –0.001 | ||
| GP call: constant | 0.082 | |||
| Eye consultant visit: WSE | Multinormal | –0.004 | ||
| Eye consultant visit: constant | 1.163 | |||
| Low-vision appointment: WSE | Multinormal | –0.002 | ||
| Low-vision appointment: constant | 0.137 | |||
| Nurse appointment: WSE | Multinormal | –0.001 | ||
| Nurse appointment: constant | 0.083 | |||
| Optometrist appointment: WSE | Multinormal | 0.000 | ||
| Optometrist: constant | 0.054 | |||
| Ophthalmologist call: mean | Normal | 0.013 (0.007) | ||
| Helpline call: mean | Normal | 0.025 (0.009) | ||
| Blindness costs | ||||
| Percentage requiring community care | Beta | 6% (0.6%) | Colquitt et al.121 | 10% assumption around mean |
| Percentage requiring hip replacement | Beta | 5% (0.5%) | Colquitt et al.121 | 10% assumption around mean |
| Percentage requiring low-vision aids | Beta | 33% (0.05%) | Colquitt et al.121 | Margrain131 |
| Percentage requiring low-vision rehabilitation | Beta | 11% (1.1%) | Colquitt et al.121 | 10% assumption around mean |
| Percentage requiring residential care | Beta | 30% (3%) | Colquitt et al.121 | 10% assumption around mean |
| Percentage requiring treatment for depression | Beta | 39% (5.8%) | Colquitt et al.121 | Galaria et al.132 |
| Percentage requiring blindness registration | Beta | 95% (0.05%) | Colquitt et al.121 | Owen et al.133 |
| Cost of community care (annual) | Gamma | £10,060.95 (£1006.10) | Curtis and Burns116 | 10% assumption around mean |
| Cost of hip replacement (annual) | Gamma | £4170.00 (£417.00) |
|
10% assumption around mean |
| Cost of low-vision aids (one-off) | Gamma | £194.41 (£19.44) | Meads,122 Curtis116 | 10% assumption around mean |
| Cost of low-vision rehabilitation (one-off) | Gamma | £153 | Estimated to be double the visit cost of optometrist/optician | |
| Cost of residential care (annual) | Gamma | £6000.80 (£600.08) | Curtis and Burns116 | 10% assumption around mean |
| Cost of treatment for depression (annual) | Gamma | £2430.58 (£243.06) | NICE123 | 10% assumption around mean |
| Cost of blindness registration (one-off) | Gamma | £60.50 (£6.05) | Curtis and Burns116 | 10% assumption around mean |
| AEs | ||||
| Cost of AE | Gamma | £317.96 (£2.58) | Department of Health and Social Care118 | Weighted variance from NHS reference costs118 |
| Weibull distribution: shape parameter | Multinormal | 0.745 | Analysis of LEAVO data | |
| Weibull distribution: scale parameter – constant | –2.271 | |||
| Weibull distribution: scale parameter – aflibercept | –0.271 | |||
| Weibull distribution: scale parameter – bevacizumab | –0.049 | |||
| Withdrawal | ||||
| Weibull distribution: shape parameter | Multinormal | 0.326 | Analysis of LEAVO data | |
| Weibull distribution: scale parameter – constant | –2.966 | |||
| Weibull distribution: scale parameter – aflibercept | 0.126 | |||
| Weibull distribution: scale parameter – bevacizumab | –0.227 | |||
| Mortality: hazard ratios for CRVO | ||||
| Female: aged 0–49 years | Log-normal | 0.83 (2.89) | Bertelsen et al.101 | Calculated from CIs |
| Female: aged 50–59 years | Log-normal | 1.49 (1.86) | Bertelsen et al.101 | Calculated from CIs |
| Female: aged 60–69 years | Log-normal | 1.94 (1.27) | Bertelsen et al.101 | Calculated from CIs |
| Female: aged 70–79 years | Log-normal | 0.94 (1.25) | Bertelsen et al.101 | Calculated from CIs |
| Female: aged ≥ 80 years | Log-normal | 1.04 (1.23) | Bertelsen et al.101 | Calculated from CIs |
| Male: aged 0–49 years | Log-normal | 1.49 (1.88) | Bertelsen et al.101 | Calculated from CIs |
| Male: aged 50–59 years | Log-normal | 1.71 (1.54) | Bertelsen et al.101 | Calculated from CIs |
| Male: aged 60–69 years | Log-normal | 1.17 (1.3) | Bertelsen et al.101 | Calculated from CIs |
| Male: aged 70–79 years | Log-normal | 1.24 (1.14) | Bertelsen et al.101 | Calculated from CIs |
| Male: aged ≥ 80 years | Log-normal | 1.26 (1.22) | Bertelsen et al.101 | Calculated from CIs |
| BCVA and CST modelling | ||||
| BCVA: baseline age/10 on intercept | Normal | –0.19728 (0.049) | Analysis of LEAVO data | |
| BCVA: baseline BCVA/10 on intercept | Normal | 0.56235 (0.041) | ||
| BCVA: aflibercept on intercept | Normal | 0.18927 (0.155) | ||
| BCVA: bevacizumab on intercept | Normal | 0.03001 (0.154) | ||
| BCVA: baseline age/10 on slope | Normal | –0.25323 (0.06) | ||
| BCVA: baseline BCVA/10 on slope | Normal | –0.15787 (0.047) | ||
| BCVA: aflibercept on slope | Normal | –0.04577 (0.186) | ||
| BCVA: bevacizumab on slope | Normal | –0.06674 (0.18) | ||
| BCVA: days since injection at 12 weeks | Normal | –0.00083 (0.005) | ||
| BCVA: days since injection at 24 weeks | Normal | –0.00536 (0.001) | ||
| BCVA: days since injection at 52 weeks | Normal | 0.00069 (0.001) | ||
| BCVA: days since injection at ≥ 76 weeks | Normal | –0.00026 (0.0001) | ||
| BCVA: number of injection at 12 weeks | Normal | 0.10891 (0.072) | ||
| BCVA: number of injection at 24 weeks | Normal | 0.06345 (0.035) | ||
| BCVA: number of injection at 52 weeks | Normal | –0.00871 (0.021) | ||
| BCVA: number of injection at ≥ 76 weeks | Normal | –0.01121 (0.019) | ||
| BCVA: intercept | Multinormal | 4.811 | ||
| BCVA: slope | Multinormal | 2.878 | ||
| CST: baseline age/10 on intercept | Normal | –0.1953 (0.048) | ||
| CST: baseline CST/10 on intercept | Normal | 0.13111 (0.029) | ||
| CST: aflibercept on intercept | Normal | –0.46501 (0.151) | ||
| CST: bevacizumab on intercept | Normal | 0.22923 (0.149) | ||
| CST: baseline age/10 on slope | Normal | 0.29301 (0.067) | ||
| CST: baseline CST/10 on slope | Normal | –0.04915 (0.039) | ||
| CST: aflibercept on slope | Normal | 0.36749 (0.205) | ||
| CST: bevacizumab on slope | Normal | –0.02506 (0.197) | ||
| CST: days since injection at 12 weeks | Normal | 0.00231 (0.007) | ||
| CST: days since injection at 24 weeks | Normal | 0.02045 (0.003) | ||
| CST: days since injection at 52 weeks | Normal | 0.00239 (0.001) | ||
| CST: days since injection at ≥ 76 weeks | Normal | 0.00144 (0.001) | ||
| CST: number of injection at 12 weeks | Normal | –0.00612 (0.103) | ||
| CST: number of injection at 24 weeks | Normal | –0.0594 (0.056) | ||
| CST: number of injection at 52 weeks | Normal | 0.06798 (0.027) | ||
| CST: number of injection at ≥ 76 weeks | Normal | 0.06327 (0.022) | ||
| CST: intercept | Multinormal | 3.76348 | ||
| CST: slope | Multinormal | –2.75221 | ||
| Annual BCVA change | ||||
| Age 55–64 years: mean | Normal | 0.0200 (0.002) | Klein et al.98 | 10% assumption around mean |
| Age 55–64 years: SD | Normal | 0.0400 (0.004) | Klein et al.98 | 10% assumption around mean |
| Age 65–74 years: mean | Normal | –0.2600 (0.026) | Klein et al.98 | 10% assumption around mean |
| Age 65–74 years: SD | Normal | 0.0400 (0.004) | Klein et al.98 | 10% assumption around mean |
| Age 65–74 years: mean | Normal | –0.7600 (0.076) | Klein et al.98 | 10% assumption around mean |
| Age 65–74 years: SD | Normal | 0.0602 (0.060) | Klein et al.98 | 10% assumption around mean |
| Utility parameters: VFQ-UI | ||||
| Component 1: BSE/10 | Multinormal | –0.00025 | Analysis of LEAVO data | |
| Component 1: WSE/10 | –0.00033 | |||
| Component 1: age/10 | 0.00922 | |||
| Component 1: male | 0.00110 | |||
| Component 1: constant | 0.88490 | |||
| Component 2: BSE/10 | 0.02353 | |||
| Component 2: WSE/10 | 0.01637 | |||
| Component 2: age/10 | 0.03448 | |||
| Component 2: male | 0.00751 | |||
| Component 2: constant | 0.18926 | |||
| Component 3: BSE/10 | 0.00372 | |||
| Component 3: WSE/10 | –0.00187 | |||
| Component 3: age/10 | 0.00638 | |||
| Component 3: male | –0.00413 | |||
| Component 3: constant | 0.83403 | |||
| Probability of component 1 membership: BSE/10 | 0.25197 | |||
| Probability of component 1 membership: WSE/10 | 0.23102 | |||
| Probability of component 1 membership: constant | –2.31366 | |||
| Probability of component 2 membership: BSE/10 | –0.41024 | |||
| Probability of component 2 membership: WSE/10 | –0.04126 | |||
| Probability of component 2 membership: constant | 4.00996 | |||
| Component 1: log-sigma | –4.78402 | |||
| Component 2: log-sigma | –2.24672 | |||
| Component 3: log-sigma | –3.49052 | |||
| Utility parameters: EQ-5D | ||||
| Component 1: BSE/10 | Multinormal | 0.01626 | Analysis of LEAVO data | |
| Component 1: WSE/10 | 0.01022 | |||
| Component 1: age/10 | –0.02851 | |||
| Component 1: male | 0.02663 | |||
| Component 1: constant | 0.86003 | |||
| Component 2: BSE/10 | 0.01693 | |||
| Component 2: WSE/10 | –0.02069 | |||
| Component 2: age/10 | 0.04236 | |||
| Component 2: male | 0.20485 | |||
| Component 2: constant | 0.01774 | |||
| Probability of component 1 membership: BSE/10 | 0.39593 | |||
| Probability of component 1 membership: WSE/10 | 0.24805 | |||
| Probability of component 1 membership: constant | –2.76469 | |||
| Component 1: log-sigma | –1.99075 | |||
| Component 2: log-sigma | –1.32132 | |||
| Utility parameters: EQ-5D-V | ||||
| Component 1: BSE/10 | Multinormal | 0.00378 | Analysis of LEAVO | |
| Component 1: WSE/10 | –0.00730 | |||
| Component 1: age/10 | 0.04348 | |||
| Component 1: male | 0.20676 | |||
| Component 1: constant | 0.03574 | |||
| Component 2: BSE/10 | 0.02012 | |||
| Component 2: WSE/10 | 0.01255 | |||
| Component 2: age/10 | –0.01937 | |||
| Component 2: male | 0.01592 | |||
| Component 2: constant | 0.73587 | |||
| Probability of component 1 membership: BSE/10 | –0.53561 | |||
| Probability of component 1 membership: WSE/10 | –0.20177 | |||
| Probability of component 1 membership: constant | 3.77924 | |||
| Component 1: log-sigma | –1.25309 | |||
| Component 2: log-sigma | –1.93060 | |||
FIGURE 21.
Within-trial analysis: confidence ellipses VFQ; VFQ-UI measure. (a) Aflibercept vs. bevacizumab; (b) aflibercept vs. ranibizumab; and (c) bevacizumab vs. ranibizumab.
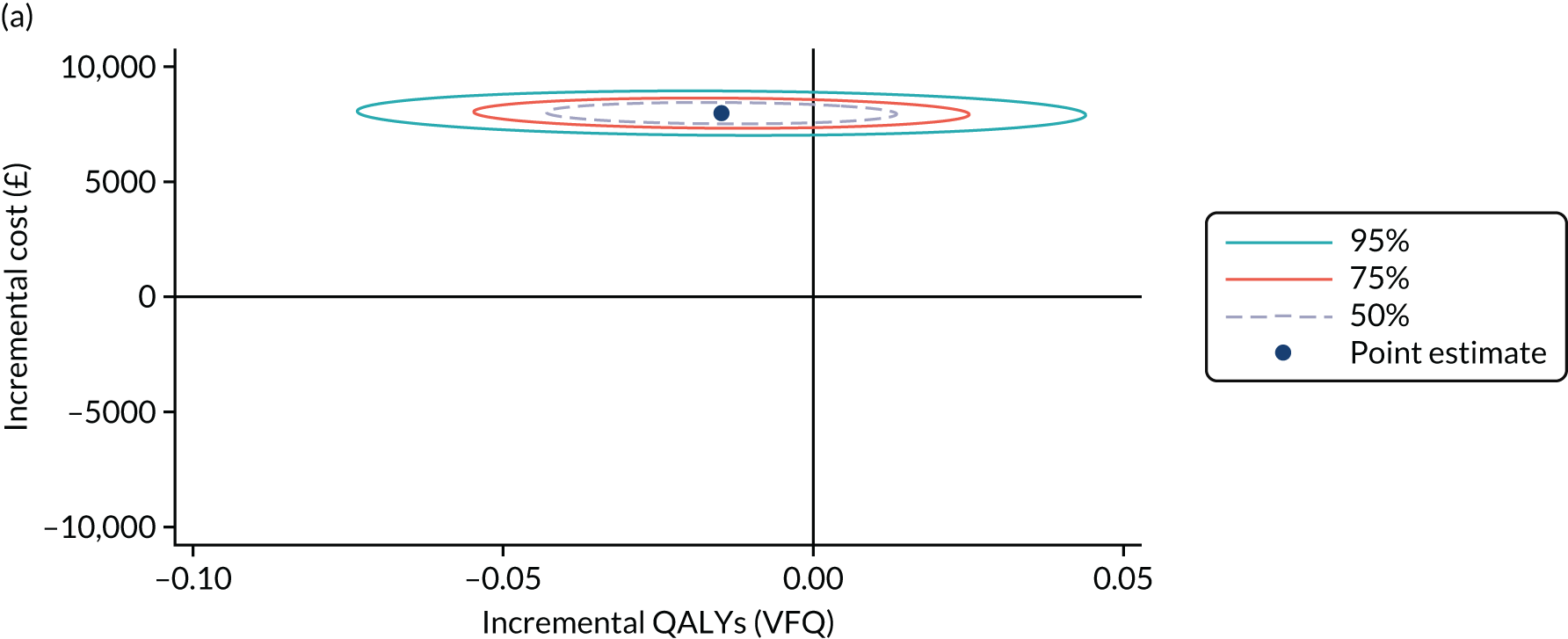

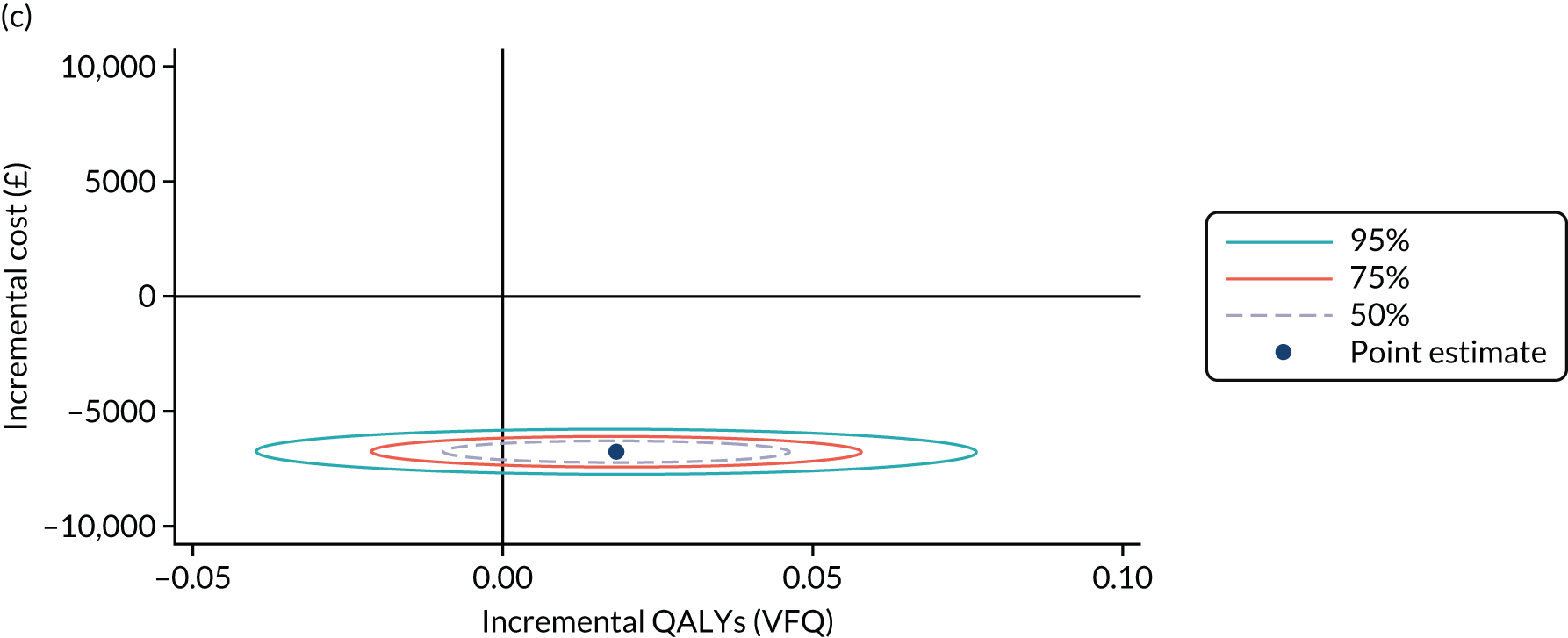
Appendix 4 Procedure for assessing the primary outcome
Refracted visual acuity was performed by a certified optometrist who had signed and dated the site delegation log before study participation and was masked to patient treatment allocation. All procedures were performed in a certified visual acuity lane. The visual acuity examiners received the participants into the visual acuity lanes with a visual acuity worksheet form, trial number and detail of trial eye and non-trial eye to be refracted, but with no previous subject records or worksheet forms. BCVA was measured following refraction at screening and at 12, 24, 52, 76 and 100 weeks (and unscheduled visits if they were to be considered as milestone visits, including a withdrawal visit) in all participants in both eyes. At all other visits, visual acuity was recorded by masked personnel using the refraction results from the previous refraction visit.
Equipment and room set-up
The ETDRS chart R was used for refraction. The lightbox was illuminated with two cool daylight 20-W fluorescent tubes. New tubes were kept on for 96 hours before use. Room lights were turned off, and the chart lights were turned on. Any windows were covered. The illumination of the room was such that, with the room set up for testing, but with the chart light switched off, not more than 161.4 lux fell on the centre of the chart. The height of the chart needed to be such that the top of the third row of letters was 124.5 cm (± 5 cm) from the floor. Full aperture trial lenses were used with a trial frame.
Refraction
The right eye was refracted first, with the participant seated 4 m from the chart. The fellow eye was occluded with a pad and tape. At the baseline visit, the initial acuity was measured with the participant’s own spectacles or unaided if the participant did not have distance spectacles. The spectacles were analysed with a focimeter. Retinoscopy was performed to provide a starting point for subjective refraction. At follow-up visits, the previous refraction was used as the starting point. If the initial acuity was ≥ 6/60 (four letters read correctly), refraction was performed at 4 m. If the acuity was < 6/60, refraction was performed at 1 m. Subjective refraction was performed using the format below. Plus/minus was offered in intervals appropriate to the level of acuity.
The sphere was checked as follows. Plus was added if it improved or made no difference to the visual acuity. This was continued until the offered plus blurred the visual acuity. Minus was added only if the subject read at least one more letter and the plus was rechecked. The cylinder axis was rechecked using a round letter on a row one or two lines above the lowest row the participant could read. The cylinder power was rechecked using a round letter on the lowest row the subject could read. The sphere was refined as before, offering plus, minus, then plus. The refraction recorded was the 4-m result. If the participant was tested at 1 m, 0.75 dioptre sphere (DS) was taken from the result to adjust for the 4-m distance. The procedure was repeated for the left eye.
Protocol for measuring ETDRS acuities
Best corrected visual acuity was measured using ETDRS chart 1 for the right eye and chart 2 for the left eye. Participants were not shown the charts until the test began. Each eye was tested at 4 m initially, even if the refraction had been performed at 1 m. The right eye was tested first, followed by the left.
The participant was seated 4 m from the chart. The distance was marked with clear and permanent floor markings. The left eye was occluded with a pad and tape and the lens correction from the subjective refraction was placed in the trial frame. What was required of the participant was then explained: there were 5 letters on each row, the letters were to be read slowly, there were no numbers on the chart, even if they were unsure of a letter they should guess, they could not go back and change their mind once they had attempted the next letter, and they could move their head or eye to give the best possible visual acuity as long as they did not lean forward.
The participant began by reading the top row of the chart and continued by reading every letter on each smaller line. The examiner recorded the results, circling each letter read correctly, putting a cross through each letter read incorrectly and leaving unmarked any letter for which no attempt was made. Participants were permitted to change their mind about a letter provided that the subsequent letter had not already been read. If a participant gave a choice of two letters, the examiner asked them to select one response only. The examiner did not read any letters out loud during the test, nor did they tell the participant if a letter had been identified correctly. If the participant lost their place, the examiner pointed to the next line to be read, but then moved away from the chart. The participant was asked and encouraged to move on to the next line as long as they correctly identified at least one letter on the previous line. The test was stopped when the participant could no longer guess, provided that mistakes had been made on previous guesses. Ideally, the aim was for four letters to be missed in a row.
If a participant could not read ≥ 20 letters at 4 m, the test was repeated at 1 m. In this case, only the first six rows needed to be attempted, and 0.75 DS was added to the prescription in the trial frame to correct for the shorter test distance. A rigid measuring device was used to ensure that the distance was correct, and care was taken to ensure that the participant did not move forward during testing. The visual acuity score was the number of letters read correctly at 4 m, plus the number of letters read correctly at 1 m. If a participant did not need to be tested at 1 m, that is they could read ≥ 20 letters at 4 m, then the score was the number of letters read correctly at 4 m, plus 30. The participant was given the credit for the 30 letters at 1 m, even though they did not have to read them. The approximate Snellen equivalent was also recorded (in metres). This was taken as the lowest row with one or no errors. If a participant could not read any letters on the ETDRS chart at 1 m, then their ability to detect hand movements or light perception was measured.
Testing for hand movement vision
The examiner held their hand steady approximately 0.5 m in front of the participant with all of their fingers outstretched. A light was shone directly on the hand from behind the participant. The fellow eye was completely occluded with a pad and tape. The examiner moved their hand from side to side or up and down at a constant speed of one back and forth presentation per second. The participant was asked ‘In which direction am I moving my hand?’. This was to be repeated five times. Four out of five correct responses indicated hand movement vision. If this was not achieved, light perception was to be tested for.
Testing for light perception/no light perception
Light perception should be measured with an indirect ophthalmoscope in a darkened room. The indirect ophthalmoscope is focused at 1 m, with the rheostat on maximum voltage. The beam was directed in and out of the eye at least four times, and the participant was asked to respond when they see the light. Light perception was recorded if the examiner was convinced that the participant saw the light. If not, the acuity was ‘no light perception’.
Appendix 5 Optical coherence tomography and fundus fluorescein angiography image grading
Specific grading of individual morphological optical coherence tomography features
Normal macula cross-sectional architecture with a Spectralis OCT device is shown in Figure 22, and key abnormal macula morphological features are shown in Figure 23.
FIGURE 22.
Normal macula architecture with a Spectralis OCT device.

FIGURE 23.
Abnormal macula morphological features on a Spectralis OCT device.

-
Vitreomacular interface abnormality:
-
Epiretinal membrane was defined as present if one or more of the following conditions were met – a macular pseudohole, a difference in optical reflectivity between membrane and retina, or a visible membrane tuft or edge.
-
Vitreomacular traction was present if a highly reflective band was observed on the surface of the retina at specific sites and elevated off the surface elsewhere, whether continuous or not with the posterior vitreous surface.
-
-
Disorganisation of the retinal inner layers was defined as an area of the inner retina where the boundary between the ganglion cell layer, inner plexiform layer complex, inner nuclear layer and outer plexiform layer could not be separately identified in the central five line scans. The total amount of DRIL in each line scan was added and the average extent per line scan was calculated. If the total exceeded 50%, DRIL was graded positive. Lesser amounts and no DRIL were graded absent, and if shadowing prevented assessment, it was deemed ungradable. The averaged horizontal extent of DRIL per line scan was recorded.
-
Macular oedema was classified as:
-
Diffuse retinal thickening, defined as sponge-like retinal swelling with reduced intraretinal reflectivity and the absence of hyporeflective spaces.
-
Cystoid macula oedema, defined as intraretinal cystoid spaces of low reflectivity with highly reflective septa separating cystoid-like cavities. Intraretinal cysts were further defined based on the greatest horizontal diameter of the largest cyst (small cysts, < 250 mm; medium cysts, ≥ 250 mm to < 500 mm; and large cysts, ≥ 500 mm).
-
The mixed pattern was graded present if diffuse retinal thickening and cystoid macula oedema were present together.
-
-
Hyper-reflective foci: intraretinal abnormally bright dots distributed throughout all retinal layers, without a characteristic intraretinal location and optimally visualised under ‘black-on-white’ options. Any number of HRF was graded as ‘present’; if none was visible, the grading was ‘absent’. 69,70
-
External limiting membrane: the faint narrow line superior to the EZ was graded as intact if visible throughout the entire foveal line scan, not intact if disrupted or completely absent under high-contrast settings, and ungradable if there was shadowing of the oedematous retina.
-
Ellipsoid zone: the EZ is synonymous with the third hyper-reflective band and is a distinct band just above the high-reflectance layer of the retinal pigment epithelium–choriocapillaris complex and COST line (see below), best detected in greyscale mode, and was graded as intact if visible throughout the foveal centre line scan, not intact if disrupted or completely absent based on continuity under high-contrast settings, or ungradable if there was shadowing of the oedematous retina.
-
Cone outer segment tips: the COST line was defined as the hyper-reflective band between the retinal pigment epithelium and EZ bands and was graded as intact if visible throughout the entire foveal line scan, not intact if disrupted or absent in part or all of the central line scan and ungradable if image quality precluded grading.
-
Subretinal detachment: this was characterised as present by a shallow elevation of the retina, with an optically clear space between the retina and the retinal pigment epithelium.
Fundus fluorescein angiography grading
Standard fundus fluorescein angiography grading
For the standard 13-sector ETDRS retinal grading grid, see Figure 24. The size and extent of the macula and zones are given in the figure, and the contained table summarises a two-step change in capillary non-perfusion.
FIGURE 24.
The 13-sector ETDRS retinal grid for grading retinal non-perfusion. CNP, capillary non-perfusion.

Novel concentric ring template for calculating retinal non-perfusion
For the novel concentric ring retinal template for calculating non-perfusion, see Figure 25. This was modified to a concentric ring template suited to the central Optos ultra-widefield image. The superior and inferior segments of rings 3 and 4, which are usually ungradable, were removed to ensure consistent measurements. Each cell of the grid was individually graded by determining whether or not the area of retina within the sector was perfused. A glassy, homogeneous appearance to the retina with pruning or absence of retinal capillaries was used to confirm a diagnosis of non-perfusion and each cell was graded as either ‘ischaemic’ (i.e. > 50% of total area non-perfused) or ‘perfused’ (i.e. < 50% of total area non-perfused).
FIGURE 25.
Novel concentric ring retinal template. Reproduced with permission from Nicholson et al. 83 This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/. The figure includes minor additions and formatting changes to the original figure.
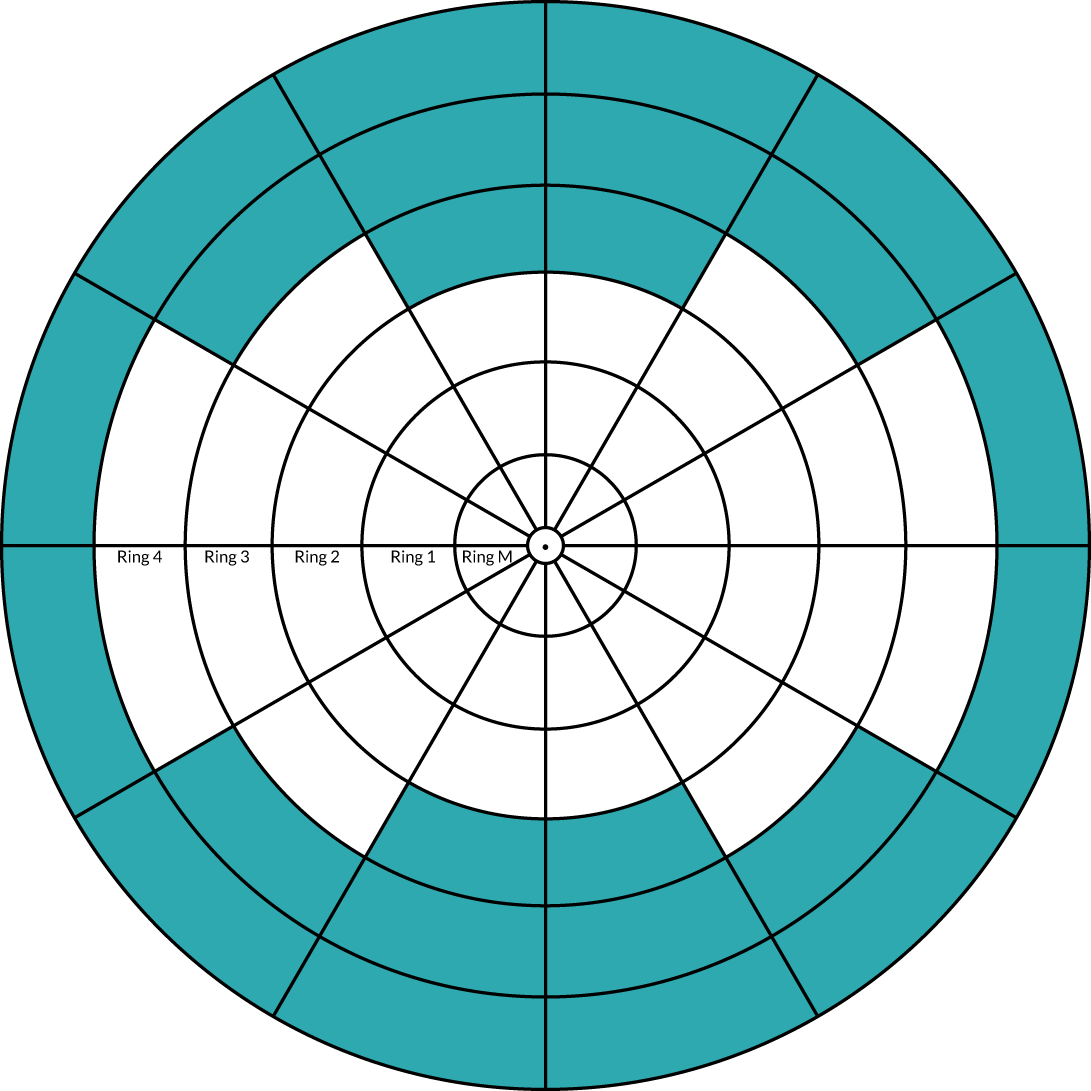
Appendix 6 Health economics: additional information
Systematic literature review to support the long-term health economic model
Background
Description of the health problem
Central retinal vein occlusion is a vascular condition of the eye associated with significant visual loss and impaired quality of life. 36,134 RVO is the second most common cause of visual loss. 1 The obstruction causes a reduction in blood supply to the eye and results in a condition referred to as retinal ischaemia. In severe cases, blood supply may be entirely cut off, leading to non-perfusion of the retinal capillaries. Reduced perfusion of the retinal vessels triggers an increased production of VEGF and other mediators, which leads to the formation of new vessels (neovascularisation) and increased permeability and leakage in parts of the retina (MO). 37 MO is the most important cause of visual loss in patients with CRVO. 37 It is the most notable complication of CRVO, followed by retinal ischaemia. 16 Other complications of CRVO include vitreous haemorrhage, NVG and tractional retinal detachment.
Epidemiology
Prevalence rates of CRVO range from 0.1% to 0.5%, with a 15-year incidence rate of 0.1% to 0.2%. 1 A systematic review3 reporting a pooled analysis from population-based studies (15 studies, 68,751 participants) conducted in the USA, Asia, Australia and Europe estimated that there are approximately 2.5 million (CI 1.9 million to 3.1 million) people living with CRVO. The review also demonstrated that the prevalence and incidence of CRVO increases with age. 1
Subtypes of central retinal vein occlusion
Both CRVO and BRVO are generally classified into non-ischaemic or ischaemic type. 16 This classification is based on the area of capillary non-perfusion and is essential in the prognosis of CRVO. Although ischaemic CRVO has been defined in the Central Retinal Vein Occlusion Study20 as ‘fluorescein angiographic evidence of 410 disc areas of capillary nonperfusion on seven-field fundus fluorescein angiography’, there is currently no agreed consensus on its definition. Better functional prognosis has been reported for patients with the non-ischaemic subtype than for those with ischaemic eyes. 16 Patients with ischaemic CRVO tend to have poorer vision (visual acuity of < 6/60) following treatment, whereas those with non-ischaemic CRVO may experience resolution of the condition without complications. 16
Anti-vascular endothelial growth factor agents treatments
The introduction of therapies that target and block the activities of VEGF (referred to hereafter as anti-VEGFs) has significantly transformed the management options for patients with CRVO. Available intravitreal treatments include Lucentis (ranibizumab), Eylea (aflibercept) and Avastin (bevacizumab), the last of which is used currently used as an off-label intravitreal injection. The clinical effectiveness and safety of intravitreal anti-VEGFs in patients with ocular conditions have been studied extensively. 32,36,53,135 However, significant differences in study characteristics and methodological designs exist; furthermore, there is a lack of head-to-head comparisons and long-term data. LEAVO is a pivotal trial that shows promise in addressing some of the previously mentioned concerns.
Review methods
Aim
The aim of this review was to systematically identify evidence to inform inputs and assumptions for the long-term (> 2 years) economic model of LEAVO. Data requirements for patients with MO secondary to CRVO treated with intravitreal injections of ranibizumab (0.5 mg/0.05 ml), aflibercept (2.0 mg/0.05 ml) or bevacizumab (1.25 mg/0.05 ml) included:
-
relative clinical effectiveness and safety (including withdrawals and mortality)
-
HRQoL estimates
-
resource use and costs related to treatment, clinic visits, staffing and equipment
-
presence of ischaemic CRVO at baseline
-
prior treatment for CRVO at baseline
-
trial eye OCT CST
-
trial eye BCVA
-
non-trial eye OCT CST
-
non-trial eye BCVA
-
new-onset MO
-
injection frequency.
Identification of studies
The review was undertaken in line with current recommendations. 90,91
Electronic database searches
Studies were identified through electronic database searches and supplementary searches. The following databases were searched from the date of inception up to 28 June 2018:
-
MEDLINE and MEDLINE In-Process & Other Non-Indexed Citations
-
Cochrane Central Register of Controlled Trials
-
Cochrane Database of Systematic Reviews
-
Database of Abstracts of Reviews of Effects
-
HTA database
-
NHS EED
-
EMBASE
-
CINAHL
-
Web of Science Core Collection (Science Citation Index, Social Sciences Citation Index, Conference Proceedings Citation Index – Science and Conference Proceedings Citation Index – Social Science & Humanities).
Free-text terms and subject headings relating to MO, RVO, ranibizumab, aflibercept and bevacizumab, plus relevant synonyms for each concept, were used to develop a search strategy. The search strategy was then cross-checked against a Cochrane review of anti-VEGF for MO secondary to CRVO32 to ensure that all relevant terms were included. Methodological search filters were applied in selected databases to identify systematic reviews, randomised trials, observational studies and economic studies (including quality-of-life studies). No search filters were applied to the CINAHL search as a limited number of references (n = 73) was retrieved. Furthermore, the search in Web of Science was refined by document type (review, article, proceedings paper, meeting abstract) because there are no available search filters. No additional limits were applied to the searches; details of the search strategy in MEDLINE are presented (see Search strategy).
Supplementary searches
Reference lists of key studies identified as potentially relevant were checked for additional references. Grey literature and authors of potentially eligible studies were also consulted. Literature-searching was iterative to ensure that sufficient data had been retrieved to populate the model. All records obtained were uploaded to an electronic bibliographic database, EndNote X8 [Clarivate Analytics (formerly Thomson Reuters), Philadelphia, PA, USA].
Study selection
Study selection was informed by inclusion criteria (Table 35); the process was completed using a two-stage method. One researcher screened the titles and abstracts of retrieved records. Potentially relevant full-text articles were then obtained for detailed examination. A second reviewer checked selection decisions at each stage. Disagreements were resolved through consensus. In the case of multiple publications of the same study, the most recent version with up-to-date information was considered for inclusion.
| Characteristic | Eligibility criteria |
|---|---|
| Population | Patients with MO due to CRVO, aged ≥ 18 years |
| Interventions |
|
| Comparisons | Comparisons between any of the interventions listed above |
| Comparisons between listed interventions and no active treatment, best supportive care, placebo, sham | |
| Combination of any of the above comparisons | |
| Outcomes |
|
| Time horizon | > 2 years |
| Study design | Systematic reviews, randomised trials and observational studies reporting economic evaluations and quality of life |
Eligible data sources were studies with comparative or non-comparative study designs (systematic reviews, RCTs and non-RCTs) reporting long-term (i.e. ≥ 2 years) outcomes of clinical effectiveness, safety and quality of life in patients with MO secondary to CRVO who were treated with ranibizumab (Lucentis) (0.5 mg/0.05 ml), aflibercept (Eylea) (2.0 mg/0.05 ml) or bevacizumab (Avastin) (1.25 mg/0.05 ml). Patients were included if treatment was administered as a monotherapy compared with a control (e.g. another active treatment or sham injection). Prospective uncontrolled before-and-after studies were also reviewed for inclusion. In addition, studies reporting the natural history of CRVO were sought for inclusion.
The hierarchy of evidence recommendation relating to evidence of data sources for economic models by Coyle and Lee92 (see Recommendations for data sources) informed the selection of full-text articles. Narrative reviews, case reports, non-human studies, editorials, expert opinions and non-English-language papers were excluded. Papers with insufficient and unclear information to ascertain the study aim(s), participant characteristics, CRVO diagnosis, treatment schedules and relevant outcome measurements were also excluded from the review.
Data extraction, quality assessment and data synthesis
Data relating to study characteristics (first author’s name, publication date, study type, setting, follow-up duration), population characteristics (sample size, recruitment or identification, age, sex and comorbidities), interventions administered and reported outcomes of interest were abstracted into summary tables by one reviewer. Reported outcomes according to study eye (primary treated eye), non-study eye (secondary treated eye), BSE and WSE or both eyes were obtained, when these were reported. Data extraction of outcomes of interest was double-checked by a second researcher. Discrepancies were resolved by discussion.
After applying the rating of hierarchies of evidence of data sources for economic models in study selection (see Table 55),92 the most appropriate quality checklist endorsed the CASP93 was used to assess the methodological quality of included studies. Tabular and narrative syntheses were presented because the available data could not be meta-analysed.
Results
Quantity and quality of available research
A total of 1338 unique records were retrieved through literature searches. Of these, three articles24,34,94 provided potentially relevant evidence for inclusion in this review. Figure 26 shows an outline of the selection of included studies. Table 36 presents the available relevant data sources and their methodological ratings. A summary of excluded full-text papers with reasons for exclusion is provided in Excluded studies with reasons.
FIGURE 26.
Systematic literature review: flow diagram of study selection.

| Characteristic | Campochiaro et al.34 | Novartis International AG94 | McIntosh et al.24 |
|---|---|---|---|
| Sample size (CRVO) | 32 patients | 1048 patients | 3271 eyes |
| Intervention(s) | IVR | IVR | Not reported |
| Treatment schedule | TER | Not reported | Not reported |
| Study design | Non-RCT (open-label extension of CRUISE) | Non-RCT (observational, non-interventional, multicentre, open-label, single-arm study) | Systematic review of various study types |
| Funding | Genentech, Inc. | Novartis International AG | Allergan plc |
| Duration of follow-up (years) | 4 | 5 | 3 |
| Baseline characteristics of patients (mean) |
|
|
Not applicable |
| Primary study outcomes |
|
|
|
| Secondary outcomes |
|
|
Not reported |
| Evidence rating (Coyle and Lee92) | 4a | 4;a 2 to 3;b 1c | 3b |
| Methodological quality (CASP) | Unclear quality | Unclear quality | Good quality |
Relative clinical effectiveness and safety
Included studies: long-term efficacy and safety
No evidence relating to the long-term (i.e. > 2 years’ follow-up) head-to-head comparison of intravitreal injections of ranibizumab (0.5 mg/0.05 ml), aflibercept (2.0 mg/0.05 ml) and bevacizumab (1.25 mg/0.05 ml) in patients with MO secondary to CRVO was found. Two studies34,94 contributed data to the review of clinical effectiveness of intravitreal ranibizumab injection:
-
the RETAIN study,34 a prospective, open-label, single-arm multicentre extension study34
-
the LUMINOUS study,94 a post-authorisation multicentre safety study for all licensed indications of ranibizumab.
Both studies included patients with ocular conditions other than CRVO. Hereafter, patients with CRVO in the RETAIN34 and LUMINOUS94 studies are referred to as ‘patients’, unless otherwise specified.
The RETAIN study: study and patient characteristics (central retinal vein occlusion subset)
The RETAIN study enrolled a subset of patients with BRVO (n = 34) and CRVO (n = 32), originating from two pivotal multicentre Phase III RCTs9,33,96 and a subsequent follow-up (HORIZON) trial. 38 Patients who completed CRUISE (392 participants, 95 locations)9,33 or the BRAVO study (397 participants, 93 locations)96 could immediately enter the RVO cohort of HORIZON. 38 HORIZON included 304 patients (intravitreal ranibizumab 0.5 mg, n = 99; intravitreal ranibizumab 0.3 mg, n = 107; and sham, n = 98) who had completed 12 months of follow-up in CRUISE. 9,33 From months 12 to 24, patients were reviewed every 3 months, or more frequently if needed, and were eligible to receive 0.5 mg of intravitreal ranibizumab on an ‘as-needed’ (pro re nata) basis. 38 The duration of follow-up for patients in HORIZON varied (mean duration 14 ± 4.7 months, range 1–24 months) because the study was terminated early following FDA approval for ranibizumab for treating RVO. 38
The mean time from HORIZON study exit to RETAIN study entry was 92.7 days (range 68–150 days). 34 Patients with CRVO who entered the RETAIN study were representative of those enrolled in CRUISE. 34 In the RETAIN study, the mean age, visual acuity and central point thickness at baseline of enrolled patients with CRVO were 66.9 years, 50.0 ETDRS letters and 639.8 µm, respectively. Primary outcomes of the RETAIN study were mean improvement in BCVA and proportion of patients with resolved MO (defined as no intraretinal or subretinal fluid in the macula for at least 6 months after the last injection). Other reported outcomes were as follows:
-
percentage of patients gaining or losing ≥ 15 letters from baseline
-
percentages of patients with Snellen-equivalent BCVA of ≥ 20/40 or ≤ 20/200
-
mean change from baseline in CFT, as measured by the Stratus OCT device
-
percentage of patients with CFT of ≤ 250 µm at each study visit
-
incidence and severity of ocular and systemic AEs.
Follow-up of patients was completed monthly for 12 months and then every 3 months for the duration of the study. At each study visit, patients were eligible to receive therapies based on treatment criteria protocol (Table 37).
| Criteria | Treatment |
|---|---|
| 1. Presence of intraretinal fluid involving the fovea | IVR (0.5 mg) |
| 2. Intraretinal fluid persisting after two consecutive IVR and BCVA < 20/30 | IVR (0.5mg) + scatter panretinal photocoagulation |
Scatter photocoagulation and grid laser therapy were administered as adjunctive treatments in patients who needed repeated ranibizumab injections at consecutive visits. The mean follow-up period of the RETAIN study was 49.7 months (with a maximum follow-up of 60 months). 34 Of 32 enrolled patients, 27 (84%) completed 2 years of follow-up. Reasons for loss to follow-up or withdrawal were death (n = 2), resolution of MO (n = 2) and persistence of MO (n = 1). 34
Patients who entered the RETAIN study had previously completed two pivotal multicentre Phase III RCTs9,33,96 and a subsequent follow-up trial (HORIZON). 38 Patients who had received laser photocoagulation for MO 4 months before entering the study were excluded from CRUISE. 33 Further details of prior treatments in patients were not available in the extension study. 34 The mean number of prior injections with ranibizumab per eye was not reported.
There was limited information relating to previous medical and ocular conditions in enrolled patients.
LUMINOUS: study and patient characteristics (central retinal vein occlusion subset)
The LUMINOUS study was a 5-year international multicentre post-authorisation study (43 countries, 494 centres) that evaluated the long-term effectiveness and safety of ranibizumab for all its indications in the real-world setting. The study population included patients with neovascular (wet) AMD, visual impairment due to MO secondary to CRVO or BRVO, DMO and choroidal neovascularisation secondary to pathological myopia. Of the entire study population in LUMINOUS (n = 30,153), patients with CRVO made up 3.5% (n = 1048). The mean age of patients was 69.7 years. Women made up 41.5% of the subgroup with CRVO. Baseline BCVA and CFT were 44.7 ETDRS letters and 551.5 µm, respectively.
The primary outcomes were mean change in BCVA, mean change in central retinal thickness (CRT) and the frequency and severity of ocular and systemic AEs among patients during the study period. Secondary outcomes of interest included the VFQ-25 scores (change from baseline), total number of injections, number of visits and re-treatments, time interval between injections, and reasons for re-treatment or treatment termination.
Safety and effectiveness outcomes in the LUMINOUS study were presented according to various analysis sets and patient subgroups. Table 38 shows a summary of relevant classifications.
| Analysis set | Definition |
|---|---|
| Enrolled set | All consenting patients with at least one baseline assessment |
| Safety set | Patients in the enrolled set who had received at least one dose of ranibizumab prior to study entry or during the study, and had at least one safety assessment after the first ranibizumab dose |
| Primary treated eye set |
|
| Secondary treated eye set |
|
| Fellow treated eye set | The fellow eye referred to a non-ranibizumab-treated eye, including information prior to study entry visit/treatment history |
| Treatment-naive eyes | Eyes that had not been pre treated with any intravitreal treatmentsa |
| Treatment non-naive (ranibizumab) eye | Eyes previously treated with at least one injection of ranibizumab, regardless of other treatments |
| Treatment non-naive (other ocular treatments) eyes | Eyes previously treated with at least one ocular treatment other than ranibizumab |
At baseline, 55.1% of patients with CRVO (primary treated eye set) had received ranibizumab. The mean number of prior injections with ranibizumab per eye was 4.5 (SD 4.29 injections) over a mean treatment duration of 38.7 weeks (SD 45.8 weeks). Additional previous intravitreal treatments other than ranibizumab were other anti-VEGFs (in 16.5% of patients, mean of 7.2 treatments per eye) and steroids (in 11.5% of patients, mean of 1.8 treatments per eye). Approximately 22.3% of all patients in the CRVO subgroup had received laser therapy, the most common type being panretinal photocoagulation. 94
A medical history of systemic comorbidity was reported for 4–61.3% of patients with CRVO. Previous ocular conditions were reported for 6.0–16.5% of participants.
Up to 70.8% of patients [62.7% treatment-naive eyes, 75.2% of treatment non-naive (ranibizumab) eyes and 71.5% of treatment non-naive (other ocular treatments) eyes] received non-ocular medicines or non-drug treatments during the study. Ocular concomitant medicines and non-drug treatments in the primary treated eye were administered in 37.1% of patients [31.5% of treatment-naive eyes, 40.6% of treatment non-naive (ranibizumab) eyes and 36.1% of treatment non-naive (other ocular treatments) eyes]. 94
Table 39 presents the baseline characteristics of patients in the RETAIN study and LUMINOUS.
| Study characteristic | RETAIN study30,34 | LUMINOUS94 | |
|---|---|---|---|
| All patients | Reported according to pre treatment in primary treated eye | ||
| Sample size (n patients) | 32 | 1048a,b |
|
| Age (years), mean (SD) | 66.9 (SD not reported) | 69.7 (12.32)b |
|
| Female (%) | NR | 41.51b |
|
| Duration of disease at baseline, mean (SD) | mean 4.6 months | 391.5 (626.83) days;a n = 1048 |
|
| BCVA (ETDRS letters), mean (SD) | 50 | 44.7 (23.88)b |
|
| SD-OCT (µm), mean SD | 639.8 | 463.5 (212.53);a n = 656 |
|
| Ocular history | NR | RVO (16.5%), glaucoma (10.4%), cataract operation (9.1%) and cataract (6.0%) | – |
| Medical history | NR | Cardiovascular risk factors,c 4–61.3% | |
| Percentage of patients with ischaemic CRVO | NR | NR | NR |
| Percentage of patients with MO | NR | NR | NR |
| VFQ-25 composite score, mean (SD) | NR | 73.0 (20.62) | |
Efficacy outcomes
Outcomes of interest included relevant functional and anatomical outcomes reported in the RETAIN34 study and in LUMINOUS. 94
Visual outcomes (the RETAIN study)34
The mean baseline visual acuity in the RETAIN study for the study population was 50.0 letters. 94 The mean improvement in BCVA was 14.0 ETDRS letters from CRUISE baseline (32 patients), resulting in a final visual acuity score of 64 letters (20/50) in patients with available data at year 4 of the RETAIN study. However, this improvement was not statistically significant when compared with the improvement of 13.1 letters (p = 0.3) from the end of CRUISE (i.e. the HORIZON baseline) until the end of the RETAIN study. 34 It is unclear whether this outcome is based on a per-protocol analysis because the authors also reported that patients who completed the RETAIN study had a mean BVCA of 61.3 letters (20/63), an increase of 12.6 letters from the CRUISE baseline. Overall, 43.8% of patients had a final visual acuity of 20/40 after 51.4 months of follow-up, with 53.1% of patients experiencing a 15-letter gain in visual acuity at the end of the study. Patients with resolved MO had better visual outcomes than those with unresolved MO [BCVA at the year 4 visit 73.2 letters (20/32) vs. 56.1 letters (20/80), respectively; p = 0.1]. A final visual acuity of ≥ 20/40 was reported in 64.3% of patients with resolved MO, in contrast to 27.8% of patients with unresolved MO (p = 0.04), translating into a visual acuity gain of 25.2 letters versus 4.3 letters (p = 0.002) in the respective subgroups. Six patients experienced reduced vision ranging from a loss of 3 to 33 ETDRS letters during the study. For reported visual outcomes in the RETAIN study, see Tables 40 and 41.
| Visual outcome | Baseline | Month | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 6 | 12 | 18 | 24 | 30 | 36 | 42 | 48 | ||
| Mean BCVA, score from CRUISE study entry (ETDRS) | |||||||||
| All patients | 50.0 (n = 32) | 63.1 (n = 32) | 64.4 (n = 32) | 62.8 (n = 32) | 62.7 (n = 32) | 62.9 (n = 31) | 64.2 (n = 29) | 60.8 (n = 29) | 64.0 (20/50) (n = 28) |
| Patients with resolved MO (n = 13) | 49.2 | 69.2 | 70.0 | 69.6 | 69.7 | 68.8 | 71.8 | 69.7 | 73.2 (20/32) |
| Patients with unresolved MO (n = 15) | 50.4 | 55.4 | 60.3 | 54.9 | 54.1 | 55.1 | 56.3 | 52.3 | 56.1 (20//80) |
| Visual outcome | Value |
|---|---|
| Mean BCVA, end of the RETAIN study, from CRUISE study entry (ETDRS), all patients | 61.3 (20/63) |
| Mean BCVA, score, end of the RETAIN study, from CRUISE study entry (ETDRS)a | 64.0 (20/50) |
| % of patients with BVCA of ≥ 20/40 (n = 32), mean follow-up 51.4 months | 43.80 |
| % of patients gaining ≥ 15 letters from baseline (n = 32), mean follow-up 51.4 months | 53.10 |
| Improvement in BCVA, from CRUISE baseline (ETDRS)b | 12.60 |
| Improvement in BCVA, from CRUISE baseline (ETDRS)a | 14.00 |
| Improvement in BCVA, at the end of CRUISE (ETDRS)b | 13.60 (p = 0.5) |
| Improvement in BCVA, at the end of CRUISE (ETDRS)a | 13.10 (p = 0.3) |
Visual outcomes (LUMINOUS)
The mean BCVA for all patients with CRVO (1048 patients) at baseline was 44.7 letters. 94 Baseline visual acuity for patients in the treatment-naive, treatment non-naive (ranibizumab) and treatment non-naive (other ocular treatments) subgroups were 40.6 letters, 49.5 letters and 35.9 letters, respectively. Less than 5% (30/1048) of patients provided relevant data for visual acuity outcomes beyond 2 years of follow-up. Overall, between two and eight patients across different subgroups gained > 10 or 15 letters in visual acuity, with only one patient achieving a gain of > 10 letters or a final BCVA of ≥ 73 letters in the treatment non-naive (ranibizumab) subgroup after long-term follow-up (up to 48 months). 94 Table 42 presents the mean change in visual acuity from baseline for patients with CRVO in LUMINOUS. 94
| Visual acuity, change in ETDRS letters | Month | |||
|---|---|---|---|---|
| 12 | 24 | 36 | 48 | |
| CRVO, primary treated eye set | ||||
| Gain of ≥ 10 letters (%) | 34.9 | 38.1 | NR | NR |
| Gain of ≥ 15 letters (%) | 28.8 | 28.1 | NR | NR |
| Treatment naive | ||||
| Primary treated eyes with baseline and post-baseline values (n) | 152.0 | 48.0 | 9.0 | 0.0 |
| Gain of ≥ 5 letters, n (%) | 96 (63.2) | 28 (58.3) | 8 (88.9) | 0.0 |
| Gain of ≥ 10 letters, n (%) | 77 (50.7) | 20 (41.7) | 7 (77.8) | 0.0 |
| Gain of ≥ 15 letters, n (%) | 66 (43.4) | 13 (27.1) | 5 (55.6) | Not assessed |
| Visual acuity of ≥ 73 letters, n (%) | 38 (25.0) | 10 (20.8) | 3 (33.3) | 0.0 |
| Treatment non-naive (ranibizumab) | ||||
| Primary treated eyes with baseline and post-baseline values (n) | 297.0 | 71.0 | 14.0 | 1.0 |
| Gain of ≥ 5 letters, n (%) | 113 (38.0) | 30 (42.3) | 7 (50.0) | 1 (100) |
| Gain of ≥ 10 letters, n (%) | 77 (25.9) | 22 (31.0) | 6 (42.9) | 1 (100) |
| Gain of ≥ 15 letters, n (%) | 59 (19.9) | 19 (26.8) | 4 (28.6) | Not assessed |
| Visual acuity of ≥ 73 letters, n (%) | 59 (19.9) | 16 (22.5) | 4 (28.6) | 1 (100) |
| Treatment non-naive (other ocular treatments) | ||||
| Primary treated eye with baseline and post-baseline values (n) | 90.0 | 41.0 | 4.0 | 0.0 |
| Gain of ≥ 5 letters, n (%) | 48 (53.3) | 20 (48.8) | 2 (50.0) | 0.0 |
| Gain of ≥ 10 letters, n (%) | 34 (37.8) | 19 (46.3) | 2 (50.0) | 0.0 |
| Gain of ≥ 15 letters, n (%) | 30 (33.3) | 13 (31.7) | 2 (50.0) | Not assessed |
| Visual acuity of ≥ 73 letters, n (%) | 9 (10.0) | 3 (7.3) | 0.0 | 0.0 |
Central foveal thickness and macular oedema outcomes (the RETAIN study)
The assessment of MO in the RETAIN study was based on the mean change in CFT from baseline, measured by a Stratus OCT device, at each study visit; the proportion of patients with CFT of ≤ 250 µm at each study visit; and the proportion of patients with resolved MO. Resolution of MO was defined as the absence of intraretinal or subretinal fluid in the macula for ≥ 6 months after the last injection. At study entry into CRUISE, enrolled patients had a mean baseline CFT of 639.8 µm, which reduced to 253.6 µm at month 24 (n = 32 patients). Of the 32 patients enrolled in the RETAIN study, 14 experienced MO resolution (43.8%). Mean CFT reported at year 4 in the RETAIN study was 171.3 µm, 263.4 µm and 220.6 µm for patients with resolved MO, patients with unresolved MO and all patients, respectively. A statistically significant difference in CFT change was noted between patients with resolved MO and those with unresolved MO (p = 0.01) (Table 43a and b). Patients with resolved MO had worse CFT at baseline than those with unresolved MO [mean 616.6 µm (SD 238.4 µm) vs. mean 497.9 µm (SD 218.8 µm); p = 0.04, respectively]. Two patients with resolved MO left the study prematurely. Of those with resolved MO (n = 14), eight patients (57%), two patients (14%), three patients (21%) and one patient (7%) received their last injection in years 1, 2, 3 and 4 of the follow-up period, respectively. 34
| Patient category | Baseline | Month | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 6 | 12 | 18 | 24 | 30 | 36 | 42 | 48 | ||
| Mean CFT (µm), from CRUISE study entry | |||||||||
| All patients | 639.8 (n = 32) | 268.5 (n = 32) | 201.4 (n = 32) | 272.8 (n = 32) | 253.6 (n = 32) | 255.5 (n = 31) | 217.4 (n = 29) | 266.4 (n = 29) | 220.6 (n = 28) |
| Patients with resolved MO (n = 13) | 706.2 | 164.8 | 151.3 | 189.7 | 166.5 | 168.4 | 170.7 | 165.4 | 171.3 |
| Patients with unresolved MO (n = 15) | 601.7 | 392.4 | 261.3 | 362.3 | 355.8 | 344.8 | 263.5 | 346.5 | 263.4 |
| Patients with | Mean CFT (µm) | p-value |
|---|---|---|
| Resolved MO | 171.30 | 0.01 |
| Unresolved MO | 263.40 |
Central retinal thickness and macular oedema outcomes (LUMINOUS)
In the primary treated eye set, the mean CRT was 551.5 µm (SD 219.95 µm) for patients with data at baseline (n = 224). 94 At 36 months, CRT (seven patients) was 290.3 µm (SD 129.2 µm). Although there was a trend of CRT reducing over time, the greatest decrease was observed at month 36 in both the treatment-naive and treatment non-naive (other ocular treatments) subgroups. A similar rate of resolution was not observed in patients who had received previous injections of ranibizumab (Table 44).
| Primary treated eye | Baseline | Month | |||
|---|---|---|---|---|---|
| 12 | 24 | 36 | 48 | ||
| Treatment naive (n) | 224 | 101 | 32 | 7 | NR |
| CRT (µm) (SD) | |||||
| Value at visit | 551.5 (219.95) | 399.6 (218.10) | 372.9 (151.35) | 290.3 (129.20) | NR |
| Mean change | N/A | –176.4 (219.83) | –186.0 (225.46) | –257.1 (179.91) | NR |
| Treatment non-naive (ranibizumab) (n) | 341 | 192 | 45 | 11 | NR |
| CRT (µm) (SD) | |||||
| Value at visit | 393.3 (184.69) | 347.1 (159.35) | 304.9 (117.71) | 411.2 (172.10) | NR |
| Mean change | N/A | –49.0 (205.18) | –97.7 (210.46) | 6.1 (249.31) | NR |
| Treatment non-naive (other ocular treatments) (n) | 91 | 51 | 19 | 2 | NR |
| CRT (µm) (SD) | |||||
| Value at visit | 510.2 (199.62) | 382.8 (147.71) | 321.5 (119.54) | 375.0 (77.78) | NR |
| Mean change | N/A | –157.4 (207.74) | –231.1 (163.44) | –277.0 (135.76) | NR |
Figure 27 shows trends in CFT change in patients included in the RETAIN34 study and in LUMINOUS. 94
FIGURE 27.
Systematic literature review: trends in CFT change over time in patients with MO due to CRVO. a, Study groups: 1, LUMINOUS, treatment naive (CRT); 2, LUMINOUS, treatment non-naive (ranibizumab) (CRT); 3, LUMINOUS, treatment non-naive (other ocular treatments) (CRT); 4, RETAIN study, all patients (CFT); 5, RETAIN study, resolved MO (CFT); 6, RETAIN study, unresolved MO (CFT).
Number of injections (the RETAIN study)34
The mean number of injections of ranibizumab (0.5 mg) administered in the RETAIN study was 19.2 over 54 months of follow-up (28 patients). The mean number of injections administered in years 2, 3 and 4 of the study was 4.5, 3.6 and 3.3, respectively. Fewer injections were administered to patients with resolved MO than to those with unresolved MO (Table 45a–c). At the end of the RETAIN study, there was a statistically significant difference in the total mean number of injections received by patients with unresolved MO, compared with those with resolved MO (28.5 vs. 8.7 injections; p < 0.01). 34
| Mean number of injections | Baseline to month 5 | Months | ||||||
|---|---|---|---|---|---|---|---|---|
| 6–11 | 12–17 | 18–23 | 24–29 | 30–35 | 36–41 | 42–47 | ||
| All patients | 4.15 (n = 32) | 4.0 (n = 32) | 2.4 (n = 32) | 2.1 (n = 32) | 1.8 (n = 31) | 1.8 (n = 29) | 1.6 (n = 29) | 1.7 (n = 28) |
| Patients with resolved MO (n = 13) | 3.70 | 2.30 | 0.90 | 0.50 | 0.40 | 0.60 | 0.20 | 0.00 |
| Patients with unresolved MO (n = 15) | 4.30 | 5.30 | 3.80 | 3.30 | 3.00 | 2.80 | 2.80 | 3.10 |
| Year | Mean number of injections |
|---|---|
| 1 | 4.5 |
| 2 | 3.6 |
| 3 | 3.3 |
| Group | Mean number of injections | p-value |
|---|---|---|
| Mean follow-up 54.0 months | 19.20 | |
| Unresolved MO | 28.50 | 0.001 |
| Resolved MO | 8.70 |
Number of injections (LUMINOUS)94
A total of 6224 ranibizumab injections were administered to patients with CRVO, of which 239 injections were administered in the secondary treated eye set. Although the majority of patients received treatment in only one eye, an estimated 3% were treated in both eyes. Treatment was administered over a mean duration of 323.5 days (primary treated eye set: 1048 patients). The mean treatment duration according to pre-treatment status in primary treated eye was 290.3 days, 337.8 days and 345.5 days for eyes that were treatment naive, treatment non-naive (ranibizumab) and treatment non-naive (other ocular treatments), respectively. The mean duration between consecutive injections (primary treated eye set) was 10.6 weeks. The shortest treatment interval was reported for treatment-naive patients (Table 46a and b). By month 48, the mean number of injections per patient was 5.9. Differences in pre-treatment status did not influence the number of injections received between subgroups.
| Patient category | Up to month | ||||
|---|---|---|---|---|---|
| 3 | 12 | 24 | 36 | 48 | |
| CRVO (all) n = 1048 | 2.0 (0.99) | 4.2 (2.78) | 5.6 (4.55) | 5.9 (5.13) | 5.9 (5.25) |
| Treatment naive, n = 327 | 2.4 (0.87) | 4.3 (2.53) | 5.5 (4.18) | 5.8 (4.82) | 5.8 (5.01) |
| Treatment non-naive (ranibizumab), n = 577 | 1.6 (0.94) | 4.1 (2.83) | 5.5 (4.53) | 5.8 (4.95) | 5.8 (5.05) |
| Treatment non-naive (other ocular treatments), n = 144 | 2.3 (0.97) | 4.5 (3.09) | 6.2 (5.37) | 6.6 (6.37) | 6.7 (6.40) |
| Patient category | Mean number of injections |
|---|---|
| Mean number of injections, per patient, secondary treated eye | |
| CRVO (all), n = 1048 | 5.6 |
| Mean (SD) duration between consecutive injections (weeks) | |
| CRVO (all), n = 1048 | 10.57 (9.16) |
| Treatment naive, n = 327 | 9.28 (6.83) |
| Treatment non-naive (ranibizumab), n = 577 | 11.12 (9.63) |
| Treatment non-naive (other ocular treatments), n = 144 | 11.61 (11.68) |
Reasons provided for administering treatment in the primary treated eye set during the treatment period (baseline to month 60) were abnormal OCT findings (82.3%), abnormal FFA findings (8.2%), ‘no further confirmation of disease activity beyond unstable visual acuity’ (15.2%) and ‘none of the above’ (31.6%). 94 Common reasons for treatment termination in the primary treated eye set were treatment-switching to another anti-VEGF other than ranibizumab, and the decision of the treating physician or patient. Data for the secondary treated eye set were limited or not available from the report. 94
Safety outcomes
Adverse event data were provided in two studies: the RETAIN34 study (n = 32 patients) and LUMINOUS94 (n = 1048 patients). Overall, the reported safety outcomes were identical in both studies. Ocular and non-ocular AEs were reported and were similar in both studies. Based on the updated health economic and decision-modelling analysis plan, only ocular AEs are reported here.
Safety outcomes (the RETAIN study)34
A small number of severe AEs were observed in patients with BRVO and CRVO in the RETAIN study. There was insufficient information to identify whether AEs occurred during the CRUISE or HORIZON studies or afterwards.
Ocular adverse events
Four severe ocular AEs were reported, the most serious event being a superior hemiretinal vein occlusion in one patient. The remaining events were vitreous traction (two patients) and severe reaction to a local antiseptic (one patient). However, it is unclear whether or not these events occurred in patients with CRVO only. On the other hand, six patients with CRVO in the RETAIN study experienced visual loss, ranging from 3 to 33 ETDRS letters (Table 47). Reasons for visual loss included persisting or recurrent MO, the presence of an epiretinal membrane and poor visual improvement during the CRUISE and HORIZON studies. 34
| Ocular AE | Sham arm, n (%) | Ranibizumab (0.5 mg) arm, n (%) | |||
|---|---|---|---|---|---|
| Day 0 to month 6 (n = 129) | Sham/0.5 mg, months 6–12 (n = 110) | Sham/0.5 mg, months 12–24 (n = 96) | Day 0 to month 12 (n = 129) | Months 12–24 (n = 99) | |
| Endophthalmitis | 0 | 0 | 0 | 0 | 0 |
| Rhegmatogenous retinal detachment | 0 | 0 | NR | 0 | NR |
| Retinal tear | 0 | 2 (1.8)a | NR | 2 (1.6) | NR |
| Any intraocular inflammation event (iridocyclitis, iritis, vitritis) | 5 (3.9) | 2 (1.8) | NR | 2 (1.6) | NR |
| Vitreous haemorrhage | 9 (7.0)a | 2 (1.8)a | 1 (1.0) | 7 (5.4) | 0 |
| Lens damageb | 0 | 0 | 0 | 0 | 0 |
| Cataractc | 0 | 2 (1.8)a | 0 | 9 (7.0) | 0 |
| IOP increased | NR | NR | 0 | NR | 0 |
| MO | NR | NR | 1 (1.0) | NR | 2 (2.0) |
| Visual acuity reduced | NR | NR | 3 (3.1) | NR | 1 (1.0) |
Safety outcomes (LUMINOUS)94
The mean duration of observed period for safety outcomes in the patients with CRVO was 530.0 days, with a cumulative duration of 1521.7 person-years for the primary treated eye set. Available data on systemic and ocular AEs in LUMINOUS are presented in Tables 47 and 48.
| Reported AEs | All patients (N = 1048) | Treatment naive (N = 327) | Treatment non-naive (ranibizumab) (N = 577) | Treatment non-naive (other ocular treatments) (N = 144) |
|---|---|---|---|---|
| Ocular AEs | ||||
| Ocular AEs (all) (incidence rate per 100-person years) | 164 | 53 | 87 | 24 |
| Ocular AEs, % (n) | ||||
| All | 10.4 (109) | 11.32 (37) | 9.71 (56) | 11.11 (16) |
| Ocular SAEs | 0.95 (10) | 1.22 (4) | 0.69 (4) | 1.39 (2) |
| Ocular severe AEs | 1.05 (11) | 1.22 (4) | 1.21 (7) | 0 |
| Infectious endophthalmitis | NR | NR | NR | NR |
| Retinal detachment | NR | NR | NR | NR |
| Retinal (pigment epithelium) tear | NR | NR | NR | NR |
| Anterior chamber reactiona | NR | NR | NR | NR |
| Conjunctival haemorrhage | 0.57 (6) | 0.92 (3) | 0.52 (3) | 0 |
| Vitreous haemorrhage | 0.38 (4) | 0.31 (1) | 0.52 (3) | 0 (0) |
| Cataract | 1.91 (20) | 1.22 (4) | 1.91 (11) | 3.47 (5) |
| Glaucoma | 0.95 (10) | 1.53 (5) | 0.87 (5) | 0 |
| Ocular hypertension (raised IOP of > 21 mmHg) | 0.57 (6) | 1.22 (4) | 0.17 (1) | 0.69 (1) |
| Increased IOP | 0.86 (9) | 0.61 (2) | 1.04 (6) | 0.69 (1) |
| Visual loss | 0.57 (6) | 0.61 (2) | 0.52 (3) | 0.69 (1) |
| Retinal ischaemia | 0.19 (2) | 0.31 (1) | 0. (0) | 0.69 (1) |
| Retinal neovascularisation | 0.19 (2) | 0 (0) | 0.17 (1) | 0.69 (1) |
| MO | 0.57 (6) | 0.31 (1) | 0.87 (5) | 0 |
Ocular adverse events LUMINOUS94
Ocular AEs were reported in 10.4% (109/1048) of patients with CRVO in the primary treated eye set [treatment naive, 11.32% (37/327); treatment non-naive (ranibizumab), 9.71% (56/577); and treatment non-naive (other ocular treatments), 11.11% (16/144)]. Among these, 0.95% of AEs were considered to be severe. Cataracts were the most common AE in the primary treated eye set, affecting 20 eyes (1.91%). The incidence of severe ocular AEs and serious ocular AEs in the primary treated eye was low (1.05% and 0.95%, respectively). Twenty-three patients (2.2%) experienced ocular AEs in the primary treated eye suspected to be related to ranibizumab and/or ocular injection. Data relating to ocular AEs in the secondary treated eye and fellow treated eye sets were not available.
| Reported AEs | % (n) | |||
|---|---|---|---|---|
| All patients (N = 1048) | Treatment naive (N = 327) | Treatment non-naive (ranibizumab) (N = 577) | Treatment non-naive (other ocular treatments) (N = 144) | |
| Ocular severe AEs, possibly treatment-related | 1.05 (11) | 0.38 (4) | 0.67 (7) | NR |
| Ocular AEs, possibly treatment-related (ranibizumab or other ocular treatment) | 2.20 (23) | 3.36 (11) | 1.56 (9) | 2.08 (3) |
| Ocular AEs, possibly treatment-related (ranibizumab only) | 0.67 (7) | 0.92 (3) | 0.52 (3) | 0.69 (1) |
| Conjunctival haemorrhage | 0.48 (5) | 0.92 (3) | 0.35 (2) | 0 |
| Cataract | 0.095 (1) | 0 | 0.17 (1) | 0 |
| Ocular hypertension | 0.57 (6) | 1.22 (4) | 0.17 (1) | 0.69 (1) |
| Increased IOP | 0.10 (1) | 0 | 017 (1) | 0 |
| Visual field defect | 0.095 (1) | 0.31 (1) | 0 | 0 |
Discontinuation of treatment (LUMINOUS)
Discontinuation of treatment with ranibizumab in the LUMINOUS study was reported as withdrawals (Table 50) and discontinuation of treatment as a result of AEs (Table 51). 94 It is unclear whether or not the discontinuation rates at specified time points included the proportion of patients who discontinued treatment because of AEs. Up to 87.5% (7/8) of patients stopped treatment prematurely by year 4. The most common reason was loss to follow-up (42.9%). Ranibizumab discontinuation because of AEs was rare. Discontinuation was more commonly related to systemic AEs (1.24%) than to ocular AEs (0.38%). 94
| Discontinuation of treatment | Year | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Patients (n) | 1047 | 481 | 119 | 8 |
| Withdrawals, n (%) | 241 (23.0) | 208 (43.2) | 71 (59.7) | 7 (87.5) |
| Common reasons for withdrawala |
|
|
|
|
| AEs | % (n) | |||
|---|---|---|---|---|
| All patients (N = 1048) | Treatment naive (N = 327) | Treatment non-naive (ranibizumab) (N = 577) | Treatment non-naive(other ocular treatments) (N = 144) | |
| Any AE (systemic and ocular) | 1.62 (17) | 2.14 (7) | 1.56 (9) | 0.69 (1) |
| Systemic AEsa | 1.24 (13) | 1.84 (6) | 1.04 (6) | 0.69 (1) |
| Ocular AEs | 0.38 (4) | 0.31 (1) | 0.52 (3) | 0 |
| Details of ocular AEs | ||||
| Retinal haemorrhage | 0.10 (1) | 0 | 0.17 (1) | 0 |
| Vitreous haemorrhage | 0.10 (1) | 0 | 0.17 (1) | 0 |
| Tachyphylaxis | 0.10 (1) | 0 | 0.17 (1) | 0 |
| Retinal injury | 0.10 (1) | 0.31 (1) | 0 | 0 |
Health-related quality-of-life outcomes: LUMINOUS
Data relating to the HRQoL of patients with CRVO treated with intravitreal ranibizumab were obtained from the LUMINOUS94 study (Table 52). HRQoL was assessed at baseline and then at yearly intervals in the safety set, using the VFQ-25 non-preference-based scoring system. 104 Slight improvements in VFQ-25 composite scores were reported for patients in the treatment-naive and treatment non-naive subgroups from baseline to the 12-month follow-up time point. From month 24 to month 36, small to moderate decreases in HRQoL scores were observed in a decreasing number of patients with baseline and post-baseline data (number of patients, 1 to 31 out of 214 patients included in the safety set). The mean VFQ-25 composite score at month 36 was higher than the baseline score only for patients in the treatment non-naive (ranibizumab) subgroup [mean 85.7 points (SD 12.20 points) vs. 79.9 points (SD 17.90 points), representing an improvement of 3.6 points (SD 10.70 points)].
| HRQoL scores | Baseline | Month | ||
|---|---|---|---|---|
| 12 | 24 | 36 | ||
| Treatment naive, primary treated eyes with baseline and post-baseline value (n) | 214 | 70 | 19 | 5 |
| VFQ-25 composite score, mean (SD) | 73.0 (20.62) | 74.4 (22.89) | 71.6 (19.66) | 67.6 (20.89) |
| Mean (SD) change from baseline in VFQ-25 score | 1.5 (10.47) | –2.3 (11.56) | –7.4 (23.15) | |
| Treatment non-naive (ranibizumab), primary treated eyes with baseline and post-baseline value (n) | 306 | 86 | 31 | 12 |
| VFQ-25 composite score, mean (SD) | 79.9 (7.90) | 82.9 (17.85) | 82.6 (17.83) | 85.7 (12.20) |
| Mean (SD) change from baseline in VFQ-25 score | –0.8 (11.65) | –0.0 (11.89) | 3.6 (10.70) | |
| Treatment non-naive (other ocular treatments), primary treated eye with baseline and post-baseline value (n) | 104 | 42 | 21 | 1 |
| VFQ-25 composite score, mean (SD) | 71.4 (20.38) | 72.1 (24.49) | 62.4 (21.17) | 35.3 |
| Mean (SD) change from baseline in VFQ-25 score | 1.0 (9.38) | –8.3 (15.47) | –49.3 | |
Resource use: patients’ visits and concurrent treatment
Evidence relating to resource use and costs was reported in the RETAIN34 and LUMINOUS94 studies. Data for the number of injections received by patients in both studies were presented earlier (see Appendix 6, Table 45a and b, for the RETAIN study34 and Appendix 6, Table 46a and b, for LUMINOUS94).
Number of visits (LUMINOUS)
The authors of LUMINOUS94 indicated that patients with CRVO (i.e. the safety set) had a mean of 11.6 visits by month 48 [treatment naive, 11.0 visits; treatment non-naive (ranibizumab), 11.4 visits; and treatment non-naive (other ocular treatments), 13.7 visits]. The comparability of number of visits at months 36 and 48 was noted (Table 53). 94
| Patients | Mean (SD) number of visits up to month | |||
|---|---|---|---|---|
| 12 | 24 | 36 | 48 | |
| All patients (N = 1048) | 7.4 (3.51) | 10.8 (6.08) | 11.5 (6.91) | 11.6 (7.06) |
| Treatment naive (N = 327) | 7.2 (3.46) | 10.2 (5.72) | 10.9 (6.44) | 11.0 (6.63) |
| Treatment non-naive (ranibizumab) (N = 577) | 7.5 (3.50) | 10.7 (6.05) | 11.4 (6.76) | 11.4 (6.91) |
| Treatment non-naive (other ocular treatments) (N = 144) | 7.9 (3.62) | 12.3 (6.75) | 13.6 (8.13) | 13.7 (8.18) |
Concomitant treatments
Two patients with CRVO received scatter photocoagulation in the RETAIN study. 34 However, further details were missing. On the other hand, data from LUMINOUS94 showed that 37.1% of the CRVO primary treated eye set [31.5% of the treatment-naive eyes, 40.6% of the treatment non-naive (ranibizumab) eyes and 36.1% of the treatment non-naive (other ocular treatments) eyes] received ocular concomitant medications and significant non-drug therapies (not specified). Concurrent systemic medications and significant non-drug therapies (not specified) reported in the CRVO safety set were administered more frequently than ocular treatments [70.8% of all patients: 62.7% of treatment-naive, 75.2% of treatment non-naive (ranibizumab) and 71.5% of treatment non-naive (other ocular treatments) subgroups]. 94
Natural history (McIntosh et al.24)
Evidence relating to the natural history of CRVO was obtained from a systematic review by McIntosh et al. 24 The review conducted literature searches up to November 2008 and included English-language articles (53 studies, 57 citations). Eligible studies were limited to observational studies of the natural history of RVO and all clinical trials evaluating interventions for CRVO. A total of 31 studies (3271 eyes) were assessed as of adequate quality and included studies evaluating patients with different CRVO subtypes [585 ischaemic eyes (20%), 1495 ischaemic subtype – unspecified (50%), 730 non-ischaemic eyes (25%) and 149 hemi-CRVO (5%)]. 24
Natural history outcomes
For reported outcomes related to the natural history of CRVO as outlined below, see Table 54:
-
baseline visual acuity
-
MO development
-
MO resolution
-
development of neovascularisation
-
development of NVG
-
development of vitreous haemorrhage
-
conversion from non-ischaemic to ischaemic RVO
-
fellow eye involvement.
| Natural history outcome | Findings | Evidence | Importance of clinical outcomea |
|---|---|---|---|
| VA | Initial VA generally poor (20/40) and generally decreases over time. I-CRVO has lower mean initial VA (< 20/200) and lower subsequent VA over timeb |
II II |
A A |
| Developmentc and resolution of ME | MO resolves in approximately 30% of non-ischaemic CRVO eyes | II | A |
| MO resolution in up to 73% of ischaemic CRVO by 15 months post-occlusion | II | B | |
| Development of NV | NV develops in up to 33% of NI-CRVO eyes 12 to 15 months post-occlusion | II | A |
| NV develops in up to 20% of ischaemic CRVO eyes by 8 to 9 months post-occlusion | III | A | |
| Development of NVG | NVG development occurs in 23%-60% of ischaemic CRVO by 12 to 15 months post-occlusion | III | A |
| Development of VH | Development of VH occurs in 10% of CRVO by 9 months post-occlusion | III | B |
| Conversion from NI-CRVO to I-CRVO | Conversion to i-CRVO occurs in up to 27% of niCRVO eyes within 10 weeks to 13 months post-occlusion | II | A |
| Fellow eye involvement | Bilateral RVO is present in 0.4% to 43% of CRVOs at presentation | II | C |
| 1.4% of patients with CRVO develop a CRVO in the fellow eye within 3 years | III | B | |
| 5% of patients with CRVO develop a BRVO in the fellow eye within 30 months | III | C | |
| 5% of patients with CRVO develop any RVO in the fellow eye within 1 year | III | B |
Visual acuity observed in patients at the onset of CRVO is initially low, ranging from 20/40 to 20/200. Conversion from non-ischaemic CRVO to ischaemic CRVO occurs in up to 27% of patients. In general, patients with ischaemic CRVO present with worse vision than those with the non-ischaemic subtype. 24 Although most patients with CRVO at the time of presentation have MO, up to 73% experience resolution of this complication within 15 months of CRVO onset (i.e. post occlusion). CRVO in both eyes has been reported in 0.4% to 43% of patients. Within 1–3 years, up to 5% of patients with unilateral CRVO may develop a RVO in the fellow eye. 24
Discussion
The review was conducted to identify evidence to inform inputs and assumptions for the long-term (> 2 years) economic model of LEAVO, a non-inferiority clinical trial comparing the clinical effectiveness and cost-effectiveness of intravitreal injections of ranibizumab (0.5 mg/0.05 ml), aflibercept (2.0 mg/0.05 ml) and bevacizumab (1.25 mg/0.05 ml) in patients with MO secondary to CRVO. No relevant long-term comparative evidence was identified by the systematic review. Although long-term data were identified for patients treated with ranibizumab, there was a lack of long-term evidence for patients with MO secondary to CRVO who received aflibercept and bevacizumab. Three studies provided evidence that was considered potentially useful to inform the long-term model. One systematic review provided natural history data24 and two non-randomised studies reported outcomes beyond 24 months for patients treated with ranibizumab. 34,94 Major concerns with these sources of evidence include the dearth of recent natural history evidence,24 small sample sizes beyond 2 years of treatment,24,34,94 and heterogeneity in clinical study design and reporting. 34,94 The RETAIN study34 was a 4-year extension trial of a Phase III trial (CRUISE),9,33 which followed up less than one-tenth of patients [8.2% (32/392)] enrolled in the original study. Furthermore, only 3% (30/1048) of patients evaluated in the 5-year observational, non-interventional, multicentre, open-label, single-arm study (LUMINOUS)94 provided relevant visual acuity data beyond 2 years of follow-up.
In general, patients with CRVO have reduced visual acuity (44.7 letters94 to 50 letters,34 or 20/40 to 20/40024) and signs of MO24 (mean SD-OCT CST, 463.5 µm94 to 639.8 µm34) at the time of presentation. Although there is some evidence that treatment with intraocular ranibizumab improves visual acuity beyond 2 years (a 15-letter gain in visual acuity for 53.1% of patients; mean follow-up of 51.4 months), it is likely that greater improvement may be experienced in the earlier phase of treatment. 34 Visual acuity improved by 14.0 ETDRS letters from the CRUISE baseline (32 patients), resulting in a final visual acuity score of 64 letters (20/50) in patients with available data at year 4. 34 However, this improvement was not statistically significant when compared with the improvement of 13.1 letters (p = 0.3) from the end of the CRUISE study until the end of the RETAIN study. 34 Differences in treatment regimens such as fixed-dose regimen of injections every 6 months and pro re nata dosing may explain this observation. Available data suggest that earlier treatment is likely to result in greater functional improvement than delayed therapy. 35 This could explain the lack of statistically significant difference in BCVA in the CRUISE and RETAIN studies. The presence of MO was a predictor of visual acuity outcome. Improvements in visual acuity tend to be greater in patients with resolved MO than in those with unresolved MO [year 4 visit, 73.2 letters (20/32) vs. 56.1 letters (20/80); p = 0.1]. 34 A statistically significantly higher proportion of treated patients achieved better final visual acuity of ≥ 20/40 (64.3% and 27.8% for resolved MO and unresolved MO respectively; p = 0.04) and greater visual acuity gain (25.2 and 4.3 ETDRS letters for resolved MO and unresolved MO, respectively; p = 0.002). 34
Up to 43.8% of patients in the RETAIN34 study had resolved MO at year 4. A statistically significant difference in CFT change was noted between patients with resolved MO and those with unresolved MO (p = 0.01). 34 Shorter-term real-world data comparing patients with CRVO receiving ranibizumab with patients with CRVO receiving aflibercept reported complete resolution in 50% and 42.9% of patients in the ranibizumab and aflibercept treatment groups, respectively. 35 Although clinical trials tend to provide better outcomes than real-world data,35 fewer patients in the RETAIN34 study experienced resolution of MO than patients in the shorter, real-world study. This may be explained by the differences in CFT at baseline: (RETAIN), 639.8 µm;34 real-world study,35 573.8 µm (ranibizumab group) versus 599.1 µm (aflibercept group).
After 3 years of treatment, patients receiving ranibizumab tended to experience improved quality of life [VFQ-25 composite score, change from baseline 3.6 (SD 10.70)]. 94 Reported mortality was generally low across all three included studies. 24,34,94 The rate of systemic or ocular AEs was approximately 10%. Previous studies,24,32,34,36,51,94,135,138,139 albeit with short-term data addressing the safety of intraocular anti-VEGF use in the treatment of MO due to CRVO, have not demonstrated major systemic and ocular AEs between anti-VEGF treatments. Although existing literature for long-term data suffers from inadequate sample sizes, inconsistent definitions and other methodological weaknesses, the findings of this review were in line with those of earlier work.
The mean number of injections of ranibizumab (0.5 mg) administered in the RETAIN study was 19.2 over 54 months of follow-up (n = 28 patients). 34 At the end of the RETAIN study, there was a statistically significant difference in the total mean number of injections received by patients with unresolved MO, and the total number received by those with resolved MO (28.5 vs. 8.7 injections, respectively; p < 0.01). 34 By contrast, by month 48, the mean number of injections per patient in LUMINOUS was 5.9. Differences in pre-treatment status did not influence the number of injections received between subgroups. This was similar to the mean number of injections reported in real-life data elsewhere evaluating 62 (62 eyes) treatment-naive patients with CRVO treated with intravitreal ranibizumab (0.5 mg) (6.8 injections) and aflibercept (0.2 mg) (6.1 injections). 35 Common reasons for treatment termination, such as treatment-switching to an anti-VEGF other than ranibizumab, could explain the observed difference in the number of injections in the RETAIN study34 and LUMINOUS. 94 A recently published network meta-analysis found no difference between ranibizumab, aflibercept, bevacizumab and triamcinolone in improving vision. 135 The authors noted that treating physicians may tend to prefer aflibercept over other anti-VEGFs because it requires fewer injections. 94
Visual acuity outcome is largely dependent on initial acuity. 94 In addition, visual acuity at baseline is a strong predictor of visual acuity at 3 years for eyes with good vision and eyes with poor vision, but a poor predictor for intermediate acuities. 20 Visual acuity was low in patients at the onset of CRVO, ranging from 20/40 to 20/200. 94 Conversion from non-ischaemic CRVO to ischaemic CRVO occurs in up to 27% of patients. In general, patients with ischaemic CRVO present with worse vision than patients with the non-ischaemic subtype. 94 Further evidence shows that patients with ischaemic CRVO tend to have poorer vision (visual acuity of < 6/60) following treatment, whereas those with non-ischaemic CRVO may experience resolution of the condition without complications. 16 Patients with ischaemic CRVO were not eligible for enrolment in CRUISE,9 but there was uncertainty about the conversion rate of non-ischaemic eyes to ischaemic eyes in the extension study, RETAIN, and whether or not efficacy outcomes may have been influenced by a number of confounders (e.g. concomitant treatments and comorbidities) in the long term.
A majority of patients with CRVO have MO at the time of presentation; however, up to 73% experience resolution within 15 months of CRVO onset (i.e. post occlusion). 24 CRVO in both eyes has been reported in 0.4–43% of patients. Within 1–3 years, up to 5% of patients with unilateral CRVO may develop a RVO in the fellow eye. 24 Included studies34,94 did not provide sufficient data to assess fellow eye involvement.
It is important to note the strengths of this systematic review, which was conducted in line with standard recommendations and informed by a multidisciplinary team comprising an information specialist, a systematic reviewer, health economists and cost-effectiveness modellers. On the other hand, a few limitations are noted here. Data relating to potential model inputs were checked by a second researcher; one researcher selected studies and performed data extraction and synthesis. The last search was carried out in June 2018. For these reasons, it is possible that key studies may have been missed.
Conclusion
Overall, the approach to identify evidence for the long-term LEAVO model was robust. There was limited evidence to inform the long-term clinical effectiveness and cost-effectiveness of anti-VEGFs used in the management of MO secondary to CRVO. A proficient understanding of comparative and long-term efficacy and safety of anti-VEGFs is needed.
Search strategy
Database searched: MEDLINE/MEDLINE In Process & Other Non-Indexed Citations.
Date range searched: from inception.
Date of search: 18 June 2018.
-
exp Macular Edema/
-
exp Macula Lutea/
-
exp EDEMA/
-
(macula* adj3 oedema).tw.
-
(macula* adj3 edema).tw.
-
6. (CME or CMO).tw.
-
or/1-6
-
exp Retinal Vein Occlusion/
-
exp Retinal Vein/
-
((vein* or venous or occlu* or obstruct* or clos* or stricture* or steno* or block* or embolism*) adj3 (central or retina*)).tw.
-
(CRVO or CVO or RVO).tw.
-
or/8-11
-
7 and 12
-
exp Angiogenesis Inhibitors/
-
exp Angiogenesis Inducing Agent
-
exp Endothelial Growth Factors/
-
exp Vascular Endothelial Growth Factors/
-
exp RANIBIZUMAB/
-
(ranibizumab or lucentis or rhuFab*).tw.
-
ZL1R02VT79.rn.
-
(aflibercept or eylea).tw.
-
15C2VL427D.rn.
-
exp BEVACIZUMAB/
-
(bevacizumab or avastin).tw.
-
2S9ZZM9Q9 V.rn.
-
(anti adj2 VEGF*).tw.
-
(endothelial adj2 growth adj2 factor*).tw.
-
or/14–28
-
13 and 28.
Recommendations for data sources
Excluded studies, with reasons
| First author and year | Title | Reason(s) for exclusion/notes |
|---|---|---|
| Bradshaw 2016140 | Systematic literature review of treatments for management of complications of ischemic central retinal vein occlusion |
|
| Braithwaite 2010141 | Anti-vascular endothelial growth factor for macular edema secondary to central retinal vein occlusion | Follow-up of < 2 years |
| Braithwaite 201432 | Anti-vascular endothelial growth factor for macular oedema secondary to central retinal vein occlusion | Follow-up of < 2 years |
| Brand 2014142 | Luminous: results from the 2014 interim analysis to provide further real-world evidence for clinical ranibizumab use |
|
| Brown 201033 | Ranibizumab for macular edema following central retinal vein occlusion: six-month primary end point results of a Phase III study |
|
| Central Vein Occlusion Study Group 199720 | Natural history and clinical management of central retinal vein occlusion | Natural history, data available in included review reported by McIntosh et al.24 |
| Chatziralli 2018143 | Ranibizumab for retinal vein occlusion: predictive factors and long-term outcomes in real-life data | Natural history; full text not available |
| Cornel 2015144 | Anti-vascular endothelial growth factor indications in ocular disease | Follow-up of < 2 years |
| Daradbounis 2014145 | Long-term results of ranibizumab treatment in patients with macular oedema due to retinal venous occlusive disease |
|
| Deramo 2003134 | Vision-related quality of life in people with central retinal vein occlusion using the 25-item National Eye Institute Visual Function Questionnaire |
|
| Casselholm de Salles 201957 | Injection frequency of aflibercept versus ranibizumab in a treat-and-extend regimen for central retinal vein occlusion: a randomized clinical trial |
|
| DeCroos 2014146 | Neovascular events in eyes with central retinal vein occlusion undergoing serial bevacizumab or ranibizumab intravitreal injections: a retrospective review |
|
| Deonandan 2017147 | Anti-vascular endothelial growth factor drugs for the treatment of retinal conditions: a review of the safety | Follow-up of < 2 years |
| Edwards 2012148 | Comparisons of the clinical effectiveness of treatments for macular oedema (MO) caused by retinal vein occlusion (RVO) | Follow-up of < 2 years |
| Figueroa 201228 | Potential anti-vascular endothelial growth factor therapies for central retinal vein occlusion |
|
| Ford 201436 | Treatments for macular oedema following central retinal vein occlusion: systematic review | Follow-up of < 2 years |
| Ford 2014135 | Drug treatment of macular oedema secondary to central retinal vein occlusion: a network meta-analysis | Follow-up of < 2 years |
| Freund 2015149 | Treat-and-extend regimens with anti-VEGF agents in retinal diseases: a literature review and consensus recommendations |
|
| Gallego-Pinazo 2012150 | Safety and efficacy of ranibizumab in macular edema following retinal vein occlusion | No usable data |
| Gerding 2015151 | Ranibizumab in retinal vein occlusion: treatment recommendations by an expert panel | No usable data |
| Glanville 2014152 | Efficacy and safety of widely used treatments for macular oedema secondary to retinal vein occlusion: a systematic review | No usable data |
| Heier 201238 | Ranibizumab for macular edema due to retinal vein occlusions: long-term follow-up in the HORIZON trial |
|
| Hernando 2018153 | Vision-related quality of life in patients diagnosed with retinal pathology |
|
| Jager 2004154 | Risks of intravitreous injection: a comprehensive review | No usable data |
| Jiang 2017155 | Update on the use of anti-VEGF intravitreal therapies for retinal vein occlusions |
|
| Jumper 2018156 | Anti-VEGF treatment of macular edema associated with retinal vein occlusion: patterns of use and effectiveness in clinical practice (ECHO study report 2) |
|
| Kinge 2010157 | Efficacy of ranibizumab in patients with macular edema secondary to central retinal vein occlusion: results from the sham-controlled ROCC study |
|
| Konidaris 2018158 | Outcomes of switching treatment to aflibercept in patients with macular oedema secondary to central retinal vein occlusion refractory to ranibizumab |
|
| Kornhauser 2016159 | Bevacizumab treatment of macular edema in CRVO and BRVO: long-term follow-up. (BERVOLT study: Bevacizumab for RVO long-term follow-up) |
|
| Kumar 2013160 | A clinical study to evaluate the efficacy of intravitreal anti-VEGF therapy in treating macular oedema due to retinal venous occlusions |
|
| Larsen 201655 | Individualized ranibizumab regimen driven by stabilization criteria for central retinal vein occlusion: twelve-month results of the CRYSTAL study | No usable data |
| Liu 2017161 | Branch and central retinal vein occlusion: clinical pearls from trials of ranibizumab |
|
| Mohamed 2007162 | Interventions for central retinal vein occlusion: an evidence-based systematic review | No usable data |
| NCT01277302 2011163 | A study evaluating dosing regimens for treatment with intravitreal ranibizumab injections in subjects with macular edema following retinal vein occlusion |
|
| Nghiem-Buffet 2017164 | Treatment patterns of ranibizumab intravitreal injection and dexamethasone intravitreal implant for retinal vein occlusion in the USA |
|
| Nicolò 2017165 | Real-life management of patients with retinal vein occlusion using I-Macula Web platform | Follow-up of < 2 years |
| Nuzzi 2015166 | Local and systemic complications after intravitreal administration of anti-vascular endothelial growth factor agents in the treatment of different ocular diseases: a five-year retrospective study |
|
| Pacella 2012167 | Testing the effectiveness of intravitreal ranibizumab during 12 months of follow-up in venous occlusion treatment | Follow-up of < 2 years |
| Patel 2016168 | Central retinal vein occlusion: a review of current evidence-based treatment options |
|
| Penedones 2014169 | Safety monitoring of ophthalmic biologics: a systematic review of pre- and post-marketing safety data | No usable data |
| Pielen 201337 | Efficacy and safety of intravitreal therapy in macular edema due to branch and central retinal vein occlusion: a systematic review | Follow-up of < 2 years |
| Poku 2014170 | The safety of intravitreal bevacizumab monotherapy in adult ophthalmic conditions: systematic review | No usable data |
| Qian 2017171 | Comparison between anti-VEGF therapy and corticosteroid or laser therapy for macular oedema secondary to retinal vein occlusion: a meta-analysis | Follow-up of < 2 years |
| Rayess 2016172 | Post injection endophthalmitis rates and characteristics following intravitreal bevacizumab, ranibizumab, and aflibercept | No usable data |
| Regnard 2016173 | Anti-VEGF treatment of macular edema using a treat-and-extend regimen in retinal vein occlusion in clinical practice |
|
| Risard 2011174 | Intravitreal ranibizumab for macular edema secondary to central retinal vein occlusion | Follow-up of < 2 years; no baseline data |
| Scott 2017175 | SCORE2 report 5: vision-related function in patients with macular edema secondary to central retinal or hemiretinal vein occlusion | No usable data |
| Sharma 2015176 | Baseline characteristics of Canadian patients with neovascular age-related macular degeneration (nvAMD), diabetic macular oedema (DMO) and retinal vein occlusion (RVO) enrolled in the LUMINOUS study | No usable data |
| Sigford 2015138 | Global reported endophthalmitis risk following intravitreal injections of anti-VEGF: a literature review and analysis | No usable data |
| Sophie 2013177 | Long-term outcomes in ranibizumab-treated patients with retinal vein occlusion; the role of progression of retinal nonperfusion |
|
| Spaide 200939 | Prospective study of intravitreal ranibizumab as a treatment for decreased visual acuity secondary to central retinal vein occlusion | No usable data |
| Tabandeh 2014178 | Endophthalmitis associated with intravitreal injections: office-based setting and operating room setting |
|
| Taylor 2014179 | A United Kingdom-based economic evaluation of ranibizumab for patients with retinal vein occlusion (RVO) |
|
| Thulliez 2014180 | Cardiovascular events and bleeding risk associated with intravitreal anti-vascular endothelial growth factor monoclonal antibodies: systematic review and meta-analysis | No usable data |
| Thulliez 201851 | Overview of systematic reviews and meta-analyses on systemic adverse events associated with intravitreal anti-vascular endothelial growth factor medication use | No usable data |
| Vorum 2016181 | Real world evidence of use of anti-VEGF therapy in Denmark |
|
| Wang 2016182 | A review of randomized trials of approved pharmaceutical agents for macular edema secondary to retinal vein occlusion | No usable data |
| Wecker 2017183 | Five-year visual acuity outcomes and injection patterns in patients with pro-re-nata treatments for AMD, DME, RVO and myopic CNV | No usable data |
| Xu 2017139 | Safety and complications of intravitreal injections performed in an Asian population in Singapore | No usable data |
| Yeh 2015184 | Therapies for macular edema associated with central retinal vein occlusion: a report by the American Academy of Ophthalmology |
|
| Yuan 2014185 | Comparison of intravitreal ranibizumab and bevacizumab for the treatment of macular edema secondary to retinal vein occlusion | No usable data |
| Ziemssen 2017186 | Demographics of patients receiving intravitreal anti-VEGF treatment in real-world practice: healthcare research data versus randomized controlled trials | No usable data |
Model-based analysis methods: additional data
FIGURE 28.
Model-based analysis: stabilisation graphs. (a) QALYs; and (b) costs. Reproduced with permission from Pennington et al. 95 This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. The figure includes minor additions and formatting changes to the original figure.

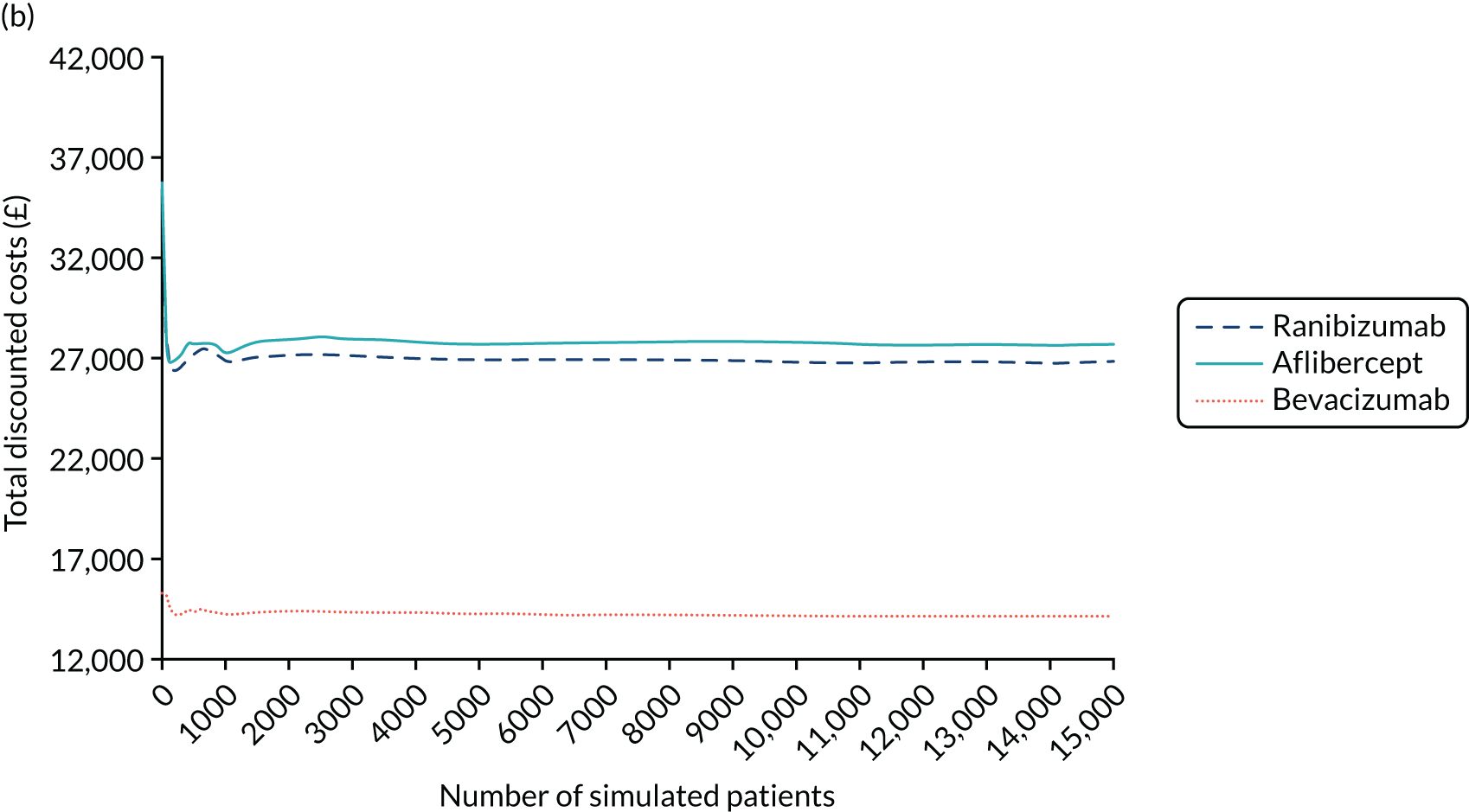
Within-trial analysis methods: additional data
Table 57 presents the unit cost of each resource in the within-trial analysis.
| Description | Unit cost (£) | Source | HRG code | Notes |
|---|---|---|---|---|
| Intervention costs | ||||
| Ranibizumab | 551.00 | BNF117 | – | 1.65 mg/0.165 ml solution for injection in pre-filled syringes (Novartis, supplied from routine NHS hospital stock) |
| Aflibercept | 816.00 | BNF117 | – | 2 mg/50 µl solution for injection vials (Bayer Pharmaceuticals AG, supplied from routine NHS hospital stock) |
| Bevacizumab | 28.00 | Judicial review53 | – | Cost per pre-filled syringe 1.25 mg/0.05 ml (Roche, supplied by the Royal Liverpool and Broadgreen Pharmacy Aseptic Unit) |
| Baseline appointment | 140.04 | Department of Health and Social Care118 | WF02B | Consultant led, ophthalmology |
| Follow-up appointments | 105.19 | Department of Health and Social Care118 | WF02A | Consultant led, ophthalmology |
| OCT | 108.21 | Department of Health and Social Care118 | BZ87A | Outpatient procedure |
| Colour fundus photography | 116.23 | Department of Health and Social Care118 | BZ89A |
|
| FFA | 108.21 | Department of Health and Social Care118 | BZ87A | Outpatient procedure |
| Procedures and hospital admissions | ||||
| Hospital admission | 337.36 | Department of Health and Social Care118 | Index: NEL_XS | Non-elective inpatients excess bed-days |
| Mental health admission | 420.62 | Department of Health and Social Care118 |
MHCC MHCCIA MHCC02 |
|
| Nose bleed | 1257.40 | Department of Health and Social Care118 |
CA12Z CA13A |
Weighted average of elective inpatient and non-elective long stay |
| Outpatient procedures | ||||
| Panretinal photocoagulation | 120.66 | Department of Health and Social Care118 | BZ86B | |
| Intravitreal steroid injection (non-study eye) | 108.21 | Department of Health and Social Care118 | BZ87A | |
| Intravitreal anti-VEGF injection (non-study eye) | 213.04 | Department of Health and Social Care118 | BZ87A WF02A | |
| Cyclodiode laser treatment | 143.26 | Department of Health and Social Care118 | BZ95Z | |
| Argon laser | 120.66 | Department of Health and Social Care118 | BZ86B | |
| Entropion repair | 137.52 | Department of Health and Social Care118 | BZ45B | |
| Epilation | 125.53 | Department of Health and Social Care118 | BZ46A | |
| Incision and curettage | 125.53 | Department of Health and Social Care118 | BZ46A | |
| Left needling of bleb with 5-fluorouracil | 143.26 | Department of Health and Social Care118 | BZ95Z | |
| Lester Jones tube insertion | 125.53 | Department of Health and Social Care118 | BZ46A | |
| Paracentesis | 143.26 | Department of Health and Social Care118 | BZ95Z | |
| Medial canthoplasty | 137.52 | Department of Health and Social Care118 | BZ45B | |
| Foreign-body removal | 117.21 | Department of Health and Social Care118 | BZ65Z | |
| Elective procedures | ||||
| Vitrectomy | 2319.38 | Department of Health and Social Care118 | BZ80A–BZ84B | |
| Cataract surgery | 1636.57 | Department of Health and Social Care118 | BZ32A–BZ34C | |
| Trabeculectomy | 2215.03 | Department of Health and Social Care118 |
|
|
| Tube shunt surgery | 2341.09 | Department of Health and Social Care118 |
|
|
| Blepharoplasty | 2768.81 | Department of Health and Social Care118 | BZ41B | |
| Peripheral iridotomy | 860.28 | Department of Health and Social Care118 | BZ04 A,B | Inflated costs using HCHS index |
| Yag capsulotomy | 860.28 | Department of Health and Social Care118 | BZ04 A,B | Inflated costs using HCHS index |
| Low-vision aids | 194.41 | Department of Health and Social Care118 | – |
|
| Blind registration | 60.50 | Meads and Hyde122 | – |
|
| Continuous care and support | ||||
| Care home cost | 1154.00 | Curtis and Burns116 | – |
|
| Home help (social services) | 27.64 | Curtis and Burns116 | – | Average taken over weekday, weekend and day, night |
| Day centre | 58.00 | Curtis and Burns116 | – | Local authority own–provision day care for older people (aged ≥ 65 years) |
| Health-care contacts | ||||
| Visit | ||||
| Ophthalmology consultant | 95.13 | Department of Health and Social Care118 | WF01A | |
| GP | 37.40 | Curtis and Burns116 | – |
|
| Practice nurse | 17.79 | Curtis and Burns116 | – |
|
| A&E/emergency department | 160.32 | Department of Health and Social Care118 | VB01Z– VB09Z, VB11Z | Weighted average of all codes of those admitted and those non-admitted |
| Consultant at glaucoma clinic | 118.02 | Department of Health and Social Care118 | WF01B | Consultant-led ophthalmology |
| Eye casualty | ||||
| Rapid access and optician | ||||
| Ophthalmology outpatient doctor | 86.55 | Department of Health and Social Care118 | WF01B | Non-consultant-led ophthalmology |
| Optician or optometrist | 76.50 | Department of Health and Social Care118 | WF01B | Non-consultant-led optometry |
| Blood sample | 2.83 | Department of Health and Social Care118 | DAPS08 | |
| Eye clinic | 73.95 | Department of Health and Social Care118 | WF01A | Non-consultant-led ophthalmology |
| Mental health department | 159.51 | Department of Health and Social Care118 | MHSTOTHA | |
| Diabetic community eye screening/retinopathy clinic | 56.79 | Department of Health and Social Care118 | WF02B | Non-consultant-led optometry |
| Neurologist for double vision | 284.66 | Department of Health and Social Care118 | WF01B |
|
| High-street optician | 25.00 | www.boots.com/opticians-service/eyetest (accessed 7 September 2020) | – | |
| Eye hospital for a low-vision appointment | 153.00 | Department of Health and Social Care118 | WF01A |
|
| Call | ||||
| Helpline | 28.66 | Curtis and Burns116 | Band 6 specialist nurse (hospital based), 15.5-minute consultation | |
| A&E eye department | ||||
| Staff nurse in eye clinic | ||||
| Ophthalmologist | 28.20 | Department of Health and Social Care118 | WF01D | Non-consultant-led ophthalmology |
| Optometrist | 21.61 | Department of Health and Social Care118 | WF01C | Non-consultant-led optometry |
| Optician | ||||
| Orthoptist | 37.60 | Department of Health and Social Care118 | WF01D | Non-consultant-led orthoptist |
| Macular service | 23.25 | Curtis and Burns116 | – | Band 5 nurse (hospital based), 15.5-minute consultation |
| Clinical nurse | ||||
| Eye clinic staff | ||||
| NHS 24 | 13.59 | Turner et al.187 | ||
| GP | 28.00 | Curtis and Burns116 |
|
|
| Practice nurse | 6.90 | Curtis and Burns116 |
|
|
| Home visit | ||||
| GP | 56.16 | Curtis and Burns116 | ||
| Practice nurse | 32.89 | Curtis and Burns116 | – | |
| Optometrist | 76.50 | Department of Health and Social Care118 | ||
| Concomitant medications | ||||
| Acetazolamide 250 mg tablets | 13.87 | BNF117 | – | |
| Acyclovir 200 mg tablets | 0.97 | BNF117 | – | |
| Acyclovir 400 mg tablets | 2.35 | BNF117 | – | |
| Acular 0.5% eye drops | 3.00 | BNF117 | – | |
| Alphagan 0.2% eye drops | 2.55 | BNF117 | – | |
| Amikacin 100 mg/2 ml solution for injection vials | 10.33 | BNF117 | – | |
| Artelac Night-time 0.2% eye gel | 2.80 | BNF117 | – | |
| Aspirin 75 mg gastroresistant tablets | 0.61 | BNF117 | – | |
| Atropine 1% eye drops | 131.88 | BNF117 | – | |
| Azarga 10 mg/ml/5 mg/ml eye | 11.05 | BNF117 | – | |
| Azopt 10 mg/ml eye drops | 2.00 | BNF117 | – | |
| Betnesol-N ear/eye/nose drops | 3.39 | BNF117 | – | |
| Bimatoprost 300 µg/ml eye drops | 10.30 | BNF117 | – | |
| Brinzolamide 10 mg/ml eye drops | 2.00 | BNF117 | – | |
| Brochlor 1% eye ointment | 1.96 | BNF117 | – | |
| Celluvisc 0.5% eye drops 0.4 ml unit dose | 4.80 | BNF117 | – | |
| Celluvisc 1% eye drops 0.4 ml unit dose | 3.00 | BNF117 | – | |
| Chloramphenicol 0.5% eye drops | 1.44 | BNF117 | – | |
| Clinitas 0.4% eye drops 0.5 ml unit dose | 5.70 | BNF117 | – | |
| Clinitas Carbomer 0.2% eye gel | 2.80 | BNF117 | – | |
| Clopidogrel 75 mg tablets | 1.31 | BNF117 | – | |
| Co-codamol 30 mg/500 mg caplets | 3.23 | BNF117 | – | |
| Codeine 15 mg tablets | 0.77 | BNF117 | – | |
| Codeine 30 mg tablets | 0.87 | BNF117 | – | |
| Cosopt 20 mg/ml/5 mg/ml eye drops | 1.85 | BNF117 | – | |
| Diamox Sodium Parenteral 500 mg powder for solution for injection vials | 17.71 | BNF117 | – | |
| Diamox SR 250 mg capsules | 16.66 | BNF117 | – | |
| Diclofenac 0.074% mouthwash sugar-free | 12.95 | BNF117 | – | |
| Dorzolamide 20 mg/ml eye drops | 2.04 | BNF117 | – | |
| Dropodex 0.1% eye drops 0.4 ml unit | 10.48 | BNF117 | – | |
| DuoTrav 40 µg/ml/5 mg/ml eye | 13.95 | BNF117 | – | |
| Evolve Carmellose 0.5% eye drops preservative free | 4.99 | BNF117 | – | |
| Exocin 0.3% eye drops | 2.17 | BNF117 | – | |
| Ganfort 0.3 mg/ml/5 mg/ml eye drops | 14.16 | BNF117 | – | |
| Hyabak 0.15% eye drops preservative free | 7.99 | BNF117 | – | |
| Hydromoor 0.3% eye drops 0.4 ml unit dose preservative free | 5.75 | BNF117 | – | |
| Hylo-Forte 0.2% eye drops preservative free | 9.50 | BNF117 | – | |
| Hylo-Tear 0.1% eye drops preservative free | 8.50 | BNF117 | – | |
| Hypromellose 0.3% eye drops | 1.21 | BNF117 | – | |
| Ibuprofen 400 mg tablets | 0.84 | BNF117 | – | |
| Ilube 5% eye drops | 16.90 | BNF117 | – | |
| Iopidine 5 mg/ml eye drops | 10.88 | BNF117 | – | |
| Lacri-lube eye ointment | 3.01 | BNF117 | – | |
| Latanoprost 50 µg/ml/Timolol 5 mg/ml eye drops | 6.37 | BNF117 | – | |
| Latanoprost 50 µg/ml eye drops | 5.89 | BNF117 | – | |
| Levofloxacin 5 mg/ml eye drops | 6.95 | BNF117 | – | |
| Liquifilm Tears 1.4% eye drops | 1.93 | BNF117 | – | |
| Liquivisc 0.25% eye gel | 4.50 | BNF117 | – | |
| Lumigan 100 µg/ml eye drops | 11.71 | BNF117 | – | |
| Macushield | 27.18 | Amazon (Amazon.com, Inc., Bellevue, WA, USA) | – | |
| Maxidex 0.1% eye drops | 1.42 | BNF117 | – | |
| Maxitrol eye drops | 1.68 | BNF117 | – | |
| Maxitrol eye ointment | 1.44 | BNF117 | – | |
| Minims artificial tears 0.44% eye drops 0.5 ml unit | 9.33 | BNF117 | – | |
| Minims fluorescein sodium 1% eye drops 0.5 ml unit dose | 9.25 | BNF117 | – | |
| Minims oxybuprocaine hydrochloride 0.4% eye drops 0.5 ml unit dose | 10.56 | BNF117 | – | |
| Minims phenylephrine hydrochloride 2.5% eye drops 0.5 ml unit dose | 11.87 | BNF117 | – | |
| Minims proxymetacaine 0.5% eye drops 0.5 ml unit dose | 12.12 | BNF117 | – | |
| Minims saline 0.9% eye drops 0.5 ml unit dose | 7.43 | BNF117 | – | |
| Minims tropicamide 0.5% eye drops 0.5 ml unit dose | 11.18 | BNF117 | – | |
| Mitomycin 2 mg powder for solution for injection | 55.89 | BNF117 | – | |
| Monopost 50 µg/ml eye drops 0.2 ml unit dose | 8.49 | BNF117 | – | |
| Moxivig 0.5% eye drops | 9.80 | BNF117 | – | |
| Mydrilate 0.5% solution | 8.08 | BNF117 | – | |
| Opatanol 1 mg/ml eye drops | 4.68 | BNF117 | – | |
| Optive 0.5% eye drops | 7.49 | BNF117 | – | |
| Ozurdex 700 µg intravitreal implant in applicator | 870.00 | BNF117 | – | |
| Paracetamol 1 g tablets | 2.49 | BNF117 | – | |
| Pevanti 5 mg tablets (Advanz Pharma) Prednisolone 5 mg tablets | 0.67 | BNF117 | – | |
| Pilocarpine hydrochloride 2% eye drops | 22.12 | BNF117 | – | |
| Pred Forte 1% eye drops | 1.82 | BNF117 | – | |
| Predsol 0.5% ear/eye drops | 2.00 | BNF117 | – | |
| Simbrinza 10 mg/ml/2 mg/ml eye drops | 9.23 | BNF117 | – | |
| Sodium cromoglicate 2% eye drops | 9.25 | BNF117 | – | |
| Tears Naturale eye drops | 1.89 | BNF117 | – | |
| Timolol 0.5% eye drops | 0.88 | BNF117 | – | |
| Tiopex 1 mg/g eye gel 0.4 g unit dose | 7.49 | BNF117 | – | |
| Tobradex 3 mg/ml/1 mg/ml eye drops | 5.37 | BNF117 | – | |
| Travatan 40 µg/ml eye drops | 3.26 | BNF117 | – | |
| Trusopt 20 mg/ml eye drops 0.2 ml unit dose preservative free | 2.04 | BNF117 | – | |
| Vancocin 500 mg powder for solution for infusion vials | 5.49 | BNF117 | – | |
| Virgan 0.15% eye gel | 19.99 | BNF117 | – | |
| Viscotears 2 mg/g liquid gel | 2.80 | BNF117 | – | |
| Vitaros 3 mg/g cream | 40.00 | BNF117 | – | |
| Xailin 0.2% eye gel | 2.80 | BNF117 | – | |
| Xailin Fresh 0.5% eye drops 0.4 ml unit dose | 4.80 | BNF117 | – | |
| Xailin HA 0.2% eye drops | 7.19 | BNF117 | – | |
| Xalacom eye drops | 6.37 | BNF117 | – | |
| Yellox 900 µg/ml eye drops | 8.50 | BNF117 | – | |
| Zovirax 3% ophthalmic ointment | 9.34 | BNF117 | – | |
Model-based analysis results: additional data
| Analysis | Total (95% CI) | Incremental (95% CI) | ICER (£) (95% CI) | ||
|---|---|---|---|---|---|
| Costs (£) | QALYs | Costs (£) | QALYs | ||
| Base-case analysis | |||||
| Bevacizumab | 14,302 | 10.3642 | |||
| Ranibizumab | 27,015 | 10.3564 | 12,712 | –0.0078 | Dominated |
| Aflibercept | 27,894 | 10.3607 | 13,592 | –0.0035 | Dominated |
| Scenario analysis: EQ-5D for utilities | |||||
| Bevacizumab | 14,302 | 8.8834 | |||
| Ranibizumab | 27,015 | 8.8753 | 12,712 | –0.0081 | Dominated |
| Aflibercept | 27,894 | 8.8906 | 13,592 | 0.0072 | 1,891,888 |
| Scenario analysis: EQ-5D-V for utilities | |||||
| Bevacizumab | 14,302 | 8.5432 | |||
| Ranibizumab | 27,015 | 8.5327 | 12,712 | –0.0105 | Dominated |
| Aflibercept | 27,894 | 8.5464 | 13,592 | 0.0032 | 4,209,328 |
| Scenario analysis: 100-week time horizon | |||||
| Bevacizumab | 6278 | 1.6749 | |||
| Ranibizumab | 15,881 | 1.6748 | 9603 | –0.0001 | Dominated |
| Aflibercept | 16,711 | 1.6806 | 10,432 | 0.0057 | 1,820,265 |
| Scenario analysis: 5-year time horizon | |||||
| Bevacizumab | 6278 | 1.6749 | |||
| Ranibizumab | 15,881 | 1.6748 | 9603 | –0.0001 | Dominated |
| Aflibercept | 16,711 | 1.6806 | 10,432 | 0.0057 | 1,820,265 |
| Scenario analysis: 10-year time horizon | |||||
| Bevacizumab | 11,725 | 6.5389 | |||
| Ranibizumab | 23,900 | 6.5400 | 12,175 | 0.0011 | 10,710,733 |
| Aflibercept | 24,600 | 6.5491 | 700 | 0.0091 | 77,086 |
| Scenario analysis: bevacizumab list price from the BNF117 (£243) | |||||
| Bevacizumab | 20,947 | 10.3642 | |||
| Ranibizumab | 27,015 | 10.3564 | 6068 | –0.0078 | Dominated |
| Aflibercept | 27,894 | 10.3607 | 6948 | –0.0035 | Dominated |
| Outcome | Intervention, mean (SD) | Comparator, mean (SD) | Difference, mean (95% CI) |
|---|---|---|---|
| Ranibizumab vs. bevacizumab | |||
| Cost (£) | 30,226 (9582) | 18,353 (6520) | 11,873 (11,458 to 12,288) |
| QALY | 9.635 (1.395) | 9.678 (1.219) | –0.044 (–0.074 to –0.013) |
| ICER (£) | Dominated (INMB: –14,316 to –12,067) | ||
| Aflibercept vs. ranibizumab | |||
| Cost (£) | 35,026 (11,820) | 30,226 (9582) | 4,800 (4445 to 5154) |
| QALY | 9.569 (1.599) | 9.635 (1.395) | –0.065 (–0.097 to –0.033) |
| ICER (£) | Dominated (INMB: –7917 to –5603) | ||
| Aflibercept vs. bevacizumab | |||
| Cost (£) | 35,026 (11,820) | 18,353 (6520) | 16,673 (16,036 to 17,310) |
| QALY | 9.569 (1.599) | 9.678 (1.219) | –0.109 (–0.161 to –0.057) |
| ICER (£) | Dominated (INMB: –21,864 to –18,040) | ||
| Outcome | Intervention, mean (SD) | Comparator, mean (SD) | Difference, mean (95% CI) |
|---|---|---|---|
| Ranibizumab vs. bevacizumab | |||
| Cost (£) | 30,226 (9582) | 18,353 (6520) | 11,873 (11,458 to 12,288) |
| QALY | 8.795 (0.468) | 8.782 (0.476) | 0.013 (0.008 to 0.018) |
| ICER (£) | 908,532 (659,881 to 1,476,254) | ||
| Aflibercept vs. ranibizumab | |||
| Cost (£) | 35,026 (11,820) | 30,226 (9582) | 4800 (4445 to 5154) |
| QALY | 8.832 (0.478) | 8.795 (0.468) | 0.037 (0.032 to 0.043) |
| ICER (£) | 128,513 (110,116 to 152,663) | ||
| Aflibercept vs. bevacizumab | |||
| Cost (£) | 35,026 (11,820) | 18,353 (6520) | 16,673 (16,036 to 17,310) |
| QALY | 8.832 (0.478) | 8.782 (0.476) | 0.050 (0.044 to 0.057) |
| ICER (£) | 330,697 (292,449 to 381,601) | ||
| Outcome | Intervention, mean (SD) | Comparator, mean (SD) | Difference, mean (95% CI) |
|---|---|---|---|
| Ranibizumab vs. bevacizumab | |||
| Cost (£) | 30,226 (9582) | 18,353 (6520) | 11,873 (11,458 to 12,288) |
| QALY | 8.351 (1.960) | 8.346 (0.731) | 0.005 (–0.007 to 0.017) |
| ICER (£) | 2,491,676 (INMB: –12,327 to –11,155) | ||
| Aflibercept vs. ranibizumab | |||
| Cost (£) | 35,026 (11,820) | 30,226 (9582) | 4800 (4445 to 5154) |
| QALY | 8.639 (0.913) | 8.351 (1.960) | 0.018 (0.000 to 0.045) |
| ICER (£) | 268,963 (INMB: –4930 to –3602) | ||
| Aflibercept vs. bevacizumab | |||
| Cost (£) | 35,026 (11,820) | 18,353 (6520) | 16,673 (16,036 to 17,310) |
| QALY | 8.639 (0.913) | 8.346 (0.731) | 0.023 (–0.001 to 0.047) |
| ICER (£) | 737,383 (INMB: –17,033 to –14,981) | ||
| Outcome | Intervention, mean (SD) | Comparator, mean (SD) | Difference, mean (95% CI) |
|---|---|---|---|
| Ranibizumab vs. bevacizumab | |||
| Cost (£) | 15,254 (3324) | 6349 (638) | 8905 (8650 to 9161) |
| QALY | 1.641 (0.115) | 1.641 (0.115) | 0.000 (0.000 to 0.001) |
| ICER (£) | 34,067,841 (217,070 to 10,420,696) | ||
| Aflibercept vs. ranibizumab | |||
| Cost (£) | 18,844 (4629) | 15,254 (3324) | 3590 (3400 to 3780) |
| QALY | 1.646 (0.112) | 1.641 (0.115) | 0.005 (0.004 to 0.005) |
| ICER (£) | 793,348 (688,418 to 926,352) | ||
| Aflibercept vs. bevacizumab | |||
| Cost (£) | 18,844 (4629) | 6349 (638) | 12,495 (12,119 to 12,871) |
| QALY | 1.646 (0.112) | 1.641 (0.115) | 0.005 (0.004 to 0.006) |
| ICER (£) | 2,610,554 (2,199,924 to 3,200,947) | ||
| Outcome | Intervention, mean (SD) | Comparator, mean (SD) | Difference, mean (95% CI) |
|---|---|---|---|
| Ranibizumab vs. bevacizumab | |||
| Cost (£) | 30,226 (9582) | 23,530 (7372) | 6696 (6400 to 6992) |
| QALY | 9.635 (1.395) | 9.678 (1.219) | –0.044 (–0.074 to –0.013) |
| ICER (£) | –153,559 (INMB: –9084 to –6937) | ||
| Aflibercept vs. ranibizumab | |||
| Cost (£) | 35,026 (11,820) | 30,226 (9582) | 4800 (4445 to 5154) |
| QALY | 9.569 (1.599) | 9.635 (1.395) | –0.065 (–0.097 to –0.033) |
| ICER (£) | Dominated (INMB: –7917 to –5603) | ||
| Aflibercept vs. bevacizumab | |||
| Cost (£) | 35,026 (11,820) | 23,530 (7372) | 11,496 (10,961 to 12,030) |
| QALY | 9.569 (1.599) | 9.678 (1.219) | –0.109 (–0.161 to –0.057) |
| ICER (£) | –105,573 (INMB: –16,636 to –12,905) | ||
Within-trial analysis results: additional data
| Parameter | n (%) | Difference in percentage missing | |||||
|---|---|---|---|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | Total | Aflibercept vs. ranibizumab | Bevacizumab vs. ranibizumab | Aflibercept vs. ranibizumab | |
| Baseline utility (EQ-5D without vision) | 4 (0.03) | 6 (0.04) | 4 (0.03) | 14 (0.03) | 0.013 | 0.000 | 0.013 |
| Baseline utility (EQ-5D-V) | 12 (0.08) | 19 (0.12) | 18 (0.12) | 49 (0.11) | 0.045 | 0.039 | 0.006 |
| Baseline utility (VFQ-UI) | 6 (0.04) | 6 (0.04) | 4 (0.03) | 14 (0.03) | 0.000 | –0.013 | 0.013 |
| QALYs (EQ-5D without vision) | 42 (0.27) | 46 (0.30) | 55 (0.36) | 143 (0.31) | 0.026 | 0.084 | –0.058 |
| QALYs (EQ-5D-V) | 67 (0.44) | 75 (0.49) | 74 (0.48) | 216 (0.47) | 0.052 | 0.045 | 0.006 |
| QALYs (VFQ-UI) | 32 (0.21) | 45 (0.29) | 52 (0.34) | 129 (0.28) | 0.084 | 0.130 | –0.045 |
| Total cost | 89 (0.58) | 83 (0.54) | 87 (0.56) | 259 (0.56) | –0.039 | –0.013 | –0.026 |
FIGURE 29.
Within-trial analysis: pattern of missing cost data. Green shading indicates missing data for one or more patients. Y, year.
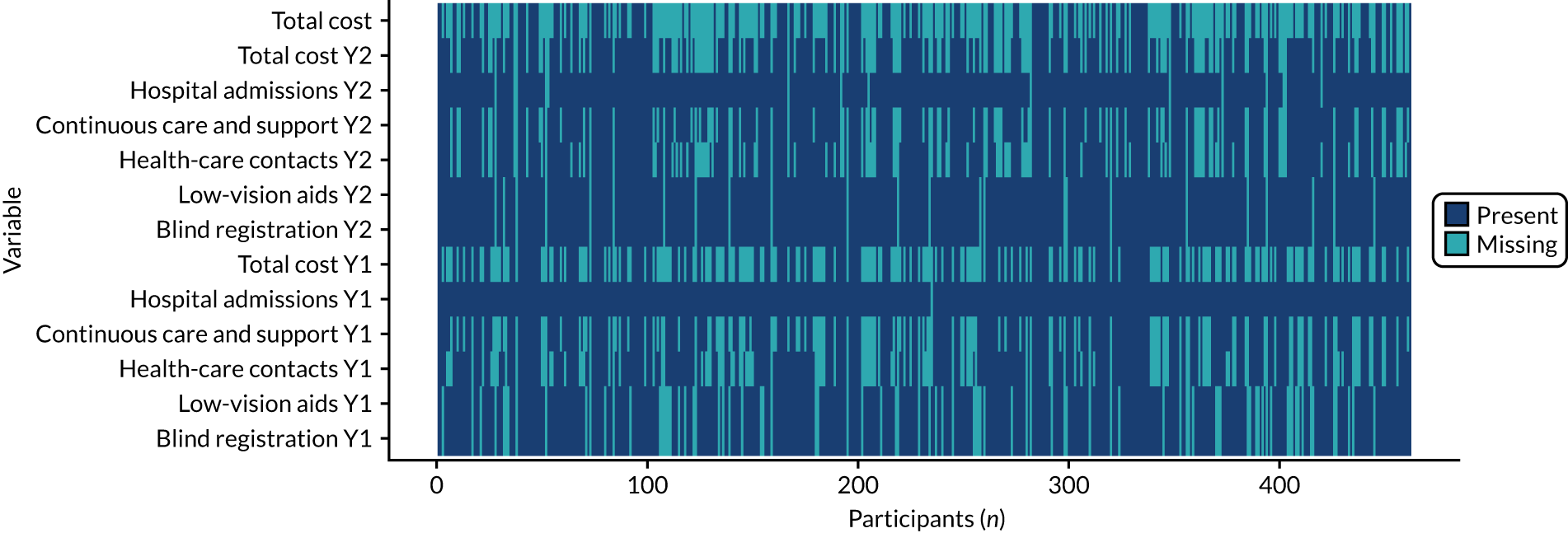
FIGURE 30.
Within-trial analysis: pattern of missing VFQ-UI data. Green shading indicates missing data for one or more patients.
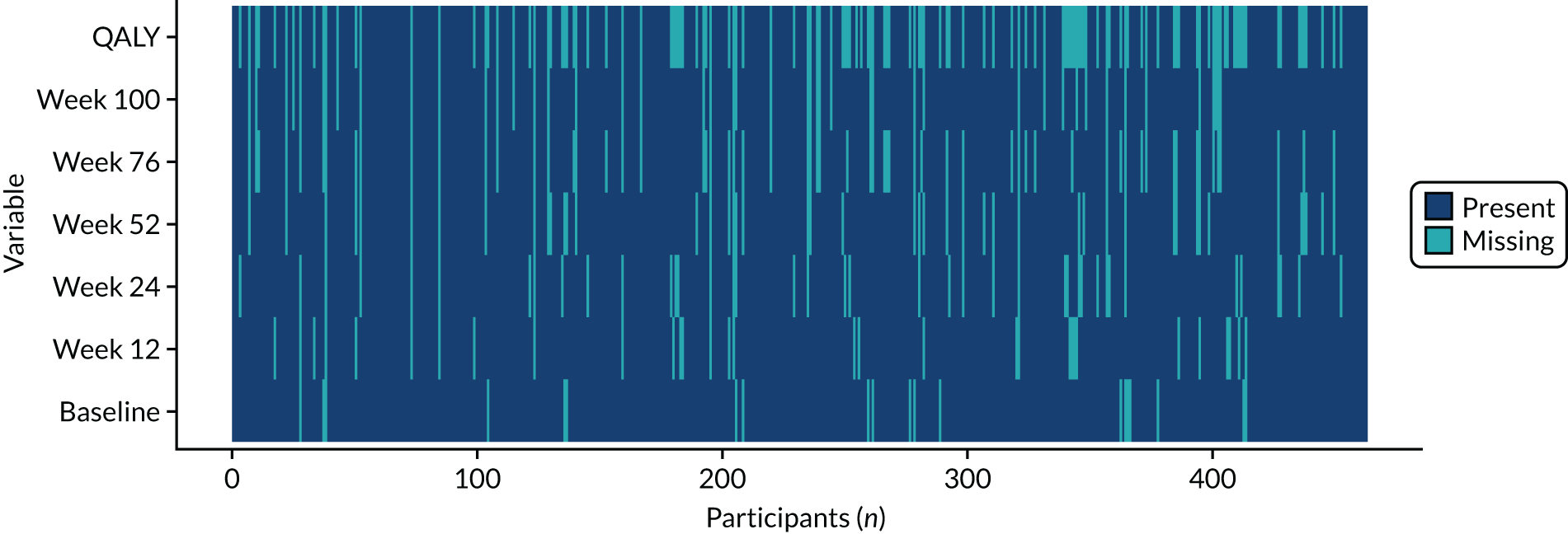
FIGURE 31.
Within-trial analysis: pattern of missing EQ-5D (without the vision bolt-on) data. Green shading indicates missing data for one or more patients.
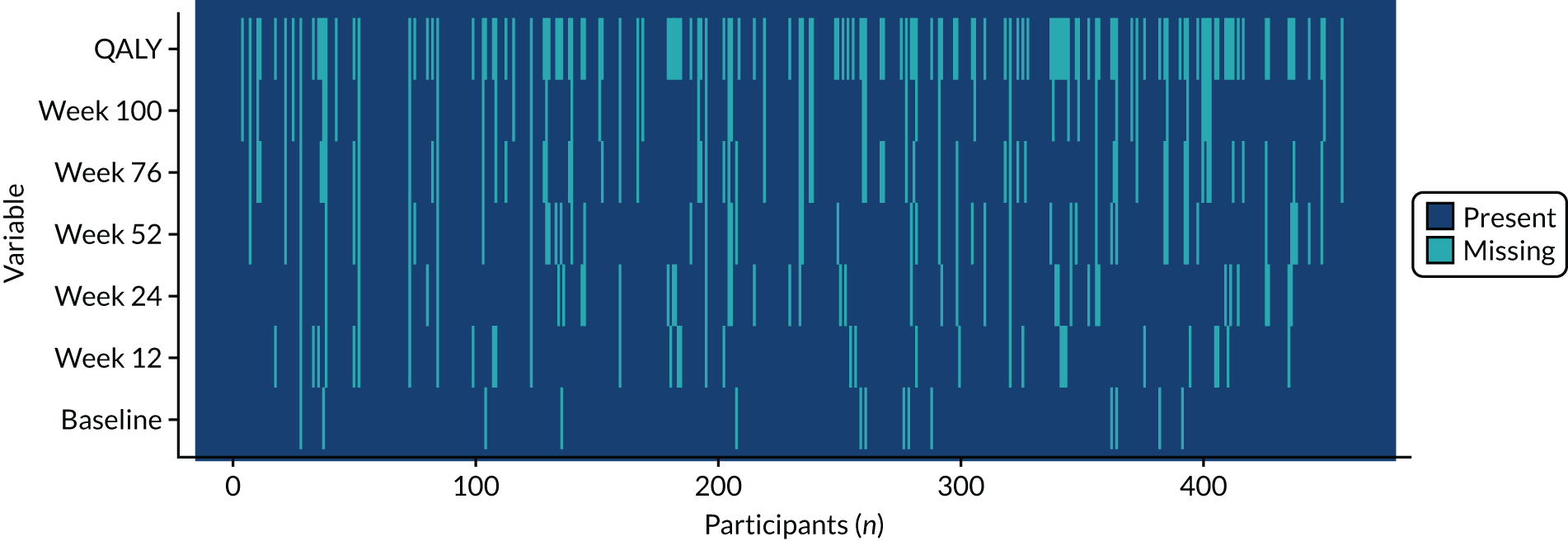
FIGURE 32.
Within-trial analysis: pattern of missing EQ-5D-V data. Green shading indicates missing data for one or more patients.
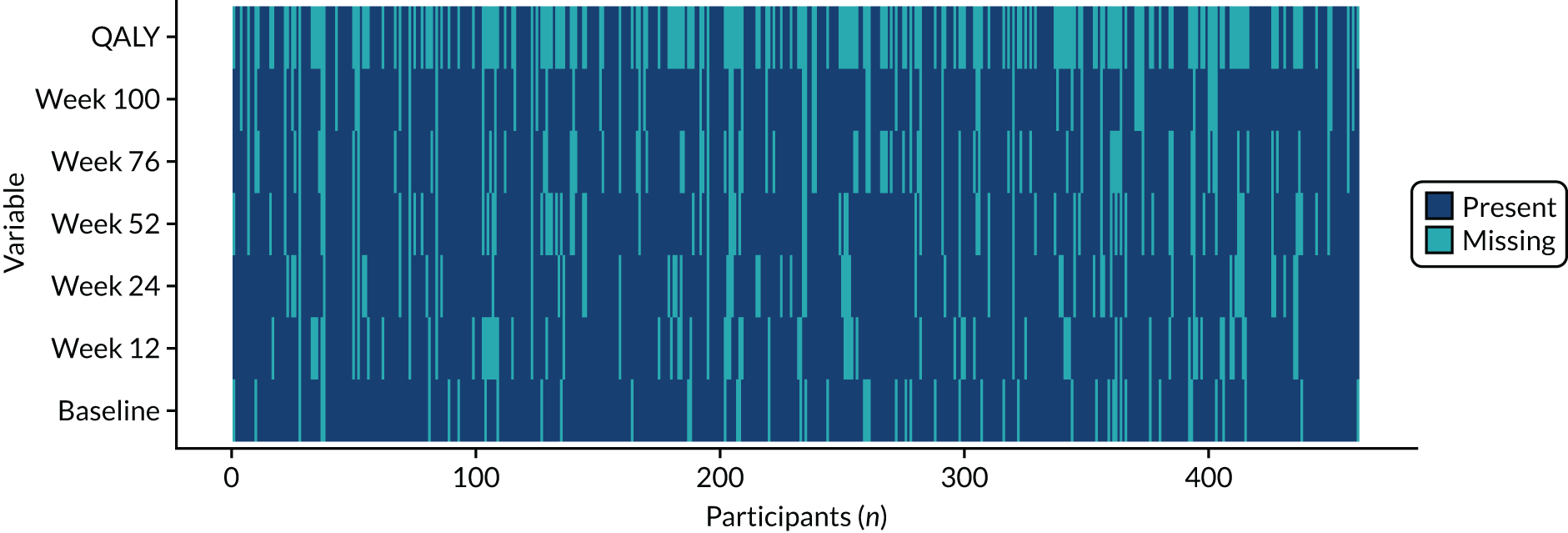
FIGURE 33.
Within-trial analysis: histograms of complete-case cost data by treatment arm. (a) Ranibizumab (n = 65); (b) aflibercept (n = 71); and (c) bevacizumab (n = 67).



| HRQoL questionnaire | Mean (SD); n patients | ||
|---|---|---|---|
| Ranibizumab | Aflibercept | Bevacizumab | |
| VFQ-UI | |||
| Baseline (complete case) | 0.849 (0.1322); 148 | 0.868 (0.1295); 147 | 0.869 (0.1156); 148 |
| QALY (imputed) | 1.627 (0.2471); 154 | 1.651 (0.2374); 154 | 1.666 (0.2426); 154 |
| EQ-5D (without vision bolt-on) | |||
| Baseline (complete case) | 0.790 (0.2118); 150 | 0.813 (0.2204); 148 | 0.801 (0.2055); 150 |
| QALY (imputed) | 1.472 (0.3666); 154 | 1.516 (0.3856); 154 | 1.500 (0.3757); 154 |
| EQ-5D-V | |||
| Baseline (complete case) | 0.767 (0.2065); 142 | 0.783 (0.2029); 135 | 0.739 (0.2410); 136 |
| QALY (imputed) | 1.513 (0.3744); 154 | 1.560 (0.3801); 154 | 1.535 (0.3759); 154 |
FIGURE 34.
Within-trial analysis: mean utility scores using the EQ-5D (without vision bolt-on) over 100 weeks. Table shows number of observations at each time point. Reproduced with permission from Pennington et al. 95 This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. The figure includes minor additions and formatting changes to the original figure.
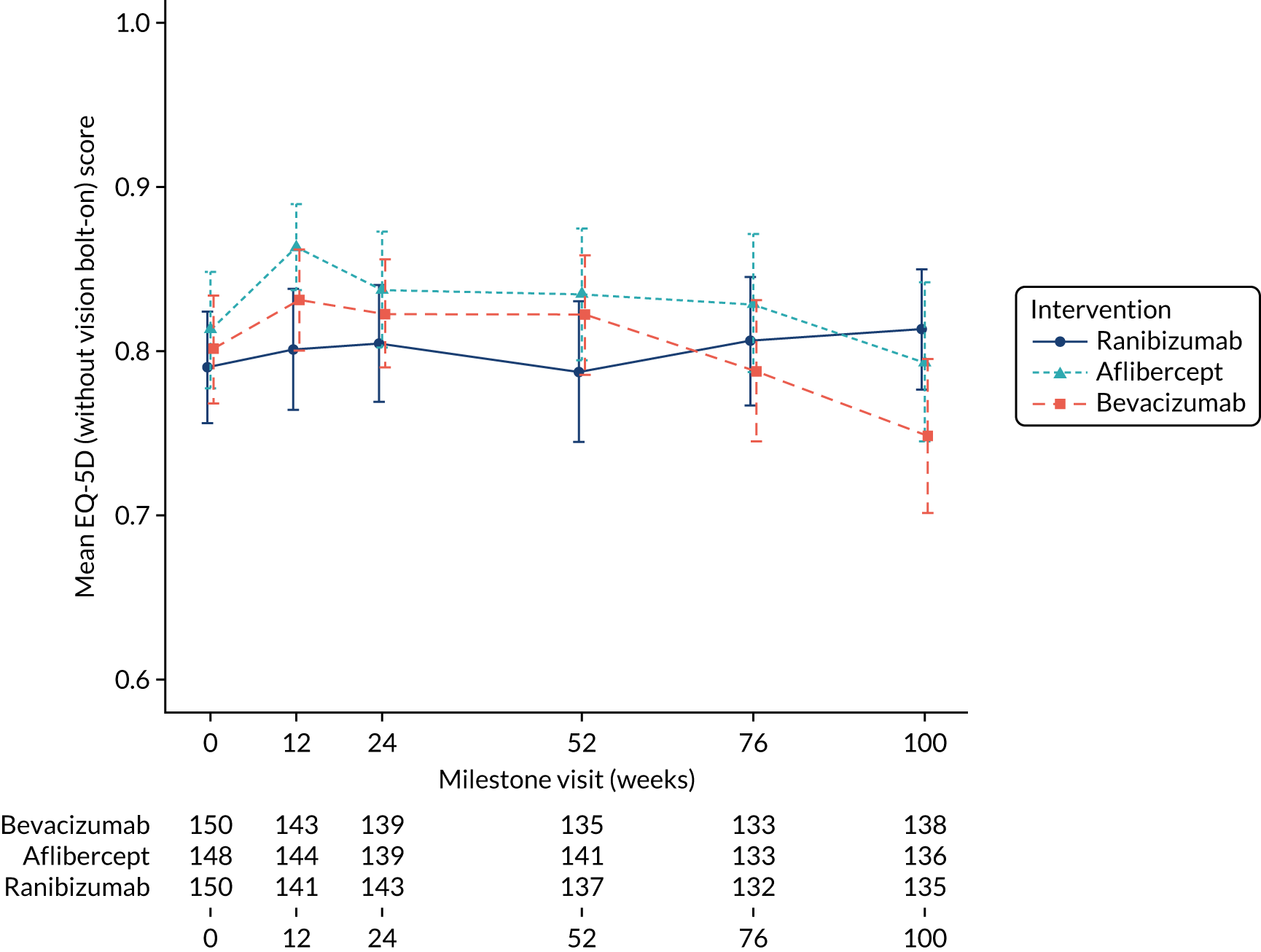
FIGURE 35.
Within-trial analysis: mean utility scores using the EQ-5D-V over 100 weeks. Table shows number of observations at each time point. Reproduced with permission from Pennington et al. 95 This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/. The figure includes minor additions and formatting changes to the original figure.
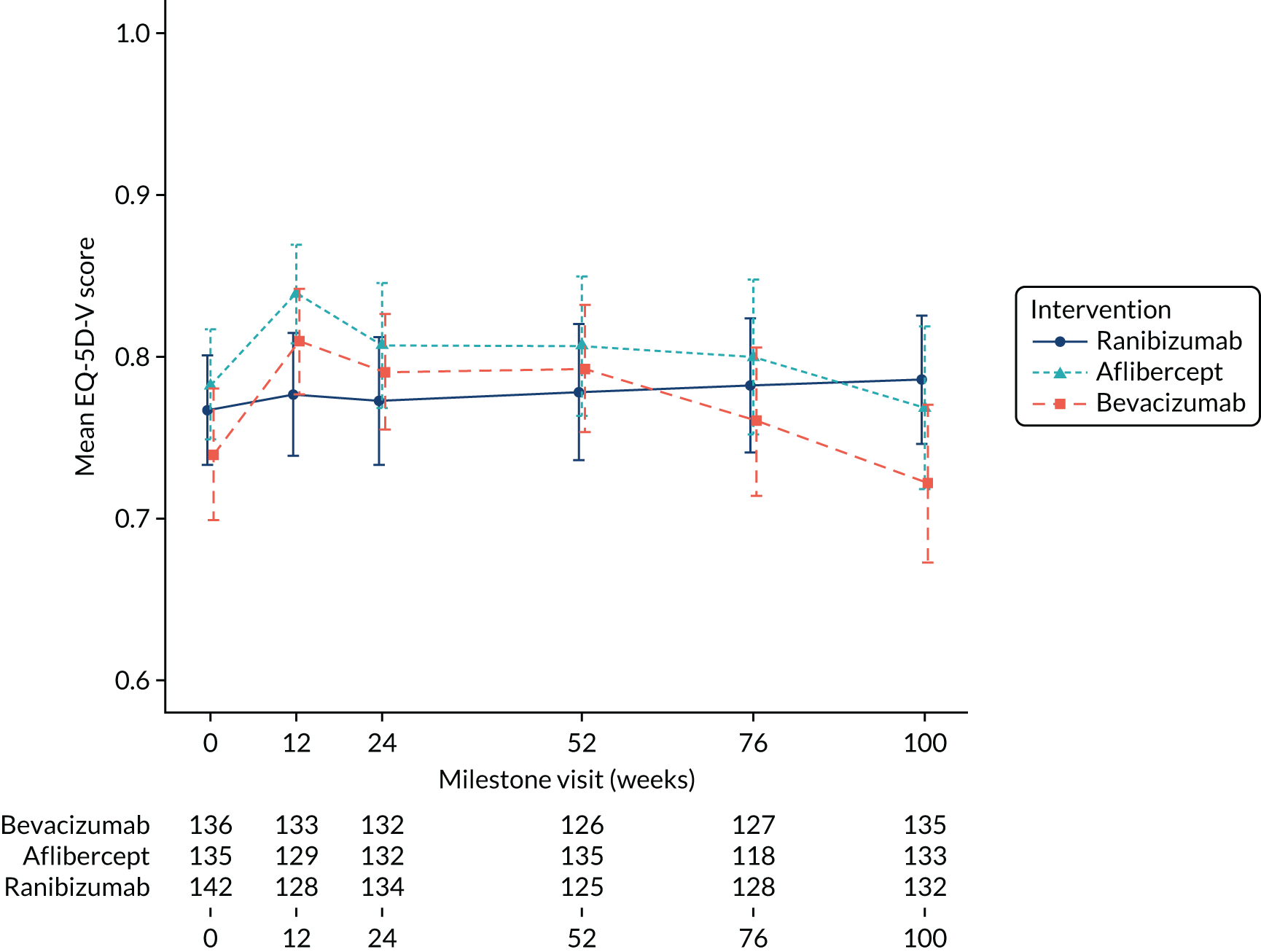
FIGURE 36.
Within-trial analysis: NMB and 95% CIs. (a) Aflibercept vs. ranibizumab; (b) aflibercept vs. bevacizumab; and (c) bevacizumab vs. ranibizumab. Cor, covariance between total costs and QALYs; dC, mean difference in costs; dQ, mean difference in QALYs; sd(dC), standard error of mean differential costs; sd(dQ), standard error of mean differential QALYs.
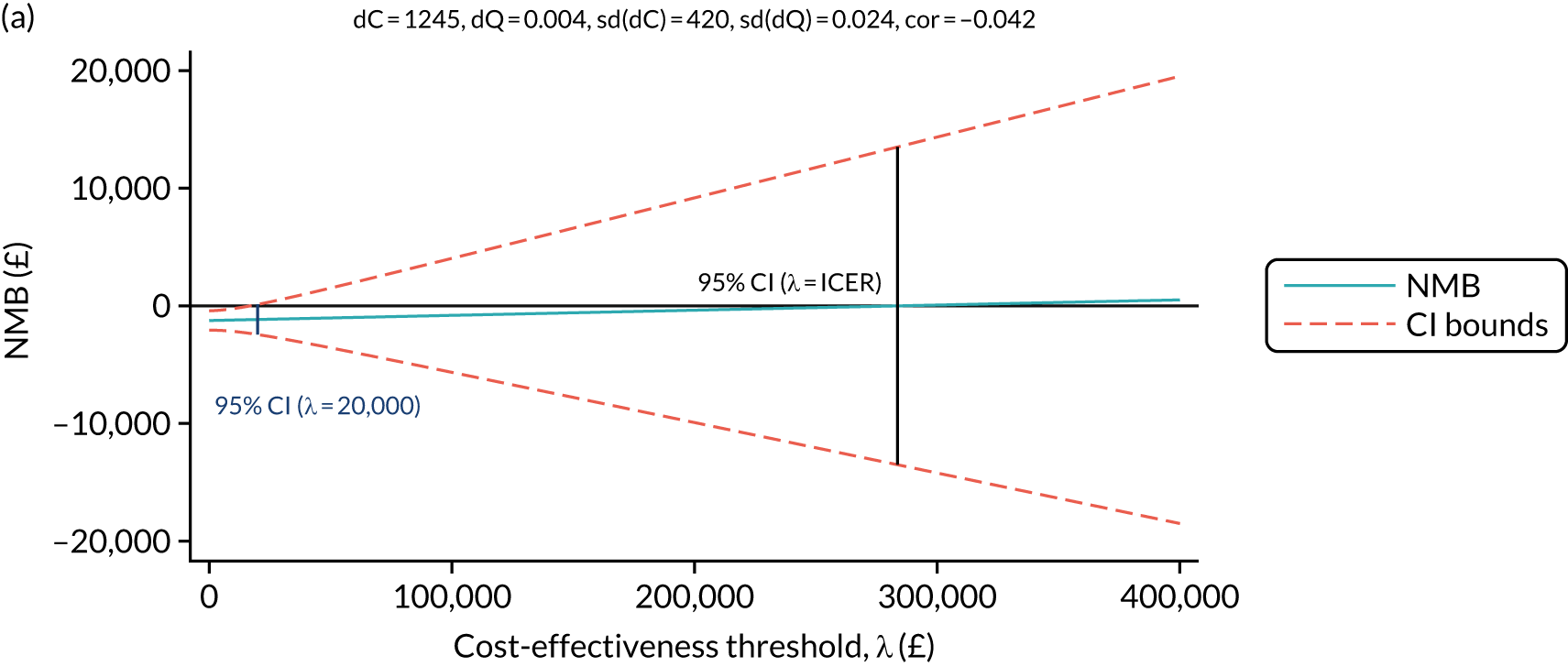
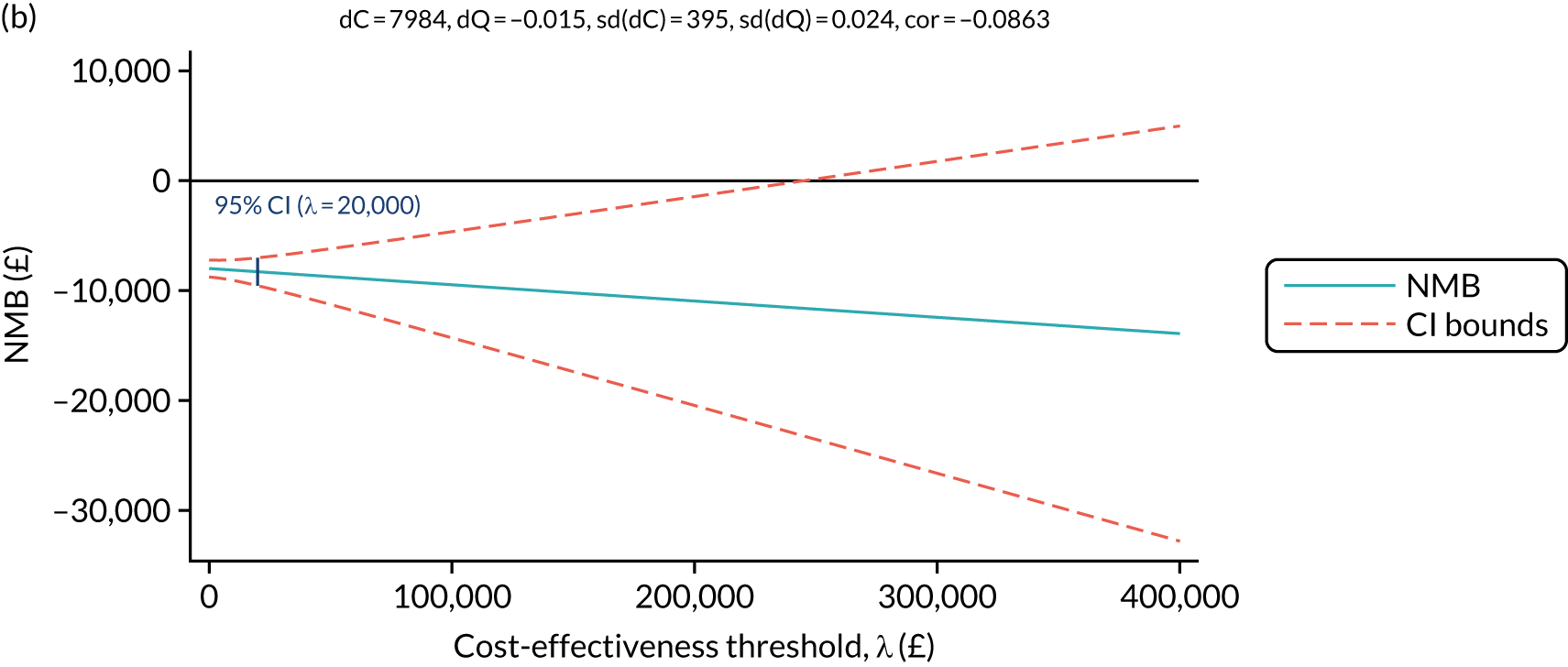

| Mean total (SD) | Incremental (95% CI)a | ICER (£)/dominance | |||
|---|---|---|---|---|---|
| Cost (£) | QALY | Cost (£) | QALY | ||
| VFQ-UI | |||||
| Bevacizumab | 6292 (3371) | 1.666 (0.2426) | – | – | – |
| Ranibizumab | 13,014 (3605) | 1.627 (0.2471) | 6734 (5970 to 7498) | –0.019 (–0.065 to 0.0284) | Dominated |
| Aflibercept | 14,328 (3773) | 1.651 (0.2374) | 7984 (7209 to 8759) | –0.015 (–0.0618 to 0.0322) | Dominated |
| EQ-5D (without vision bolt-on) | |||||
| Bevacizumab | 6273 (3384) | 1.535 (0.3759) | – | – | – |
| Ranibizumab | 13,068 (3636) | 1.513 (0.3744) | 6769 (5987 to 7550) | –0.0102 (–0.0710 to 0.0504) | Dominated |
| Aflibercept | 14,271 (3857) | 1.560 (0.3801) | 8035 (7246 to 8824) | 0.008 (–0.0529 to 0.0683) | 104,1476 |
| EQ-5D-V | |||||
| Bevacizumab | 6268 (3368) | 1.500 (0.3757) | – | – | – |
| Ranibizumab | 13,000 (3661) | 1.472 (0.3666) | 6748 (5948 to 7547) | –0.035 (–0.1172 to 0.0480) | Dominated |
| Aflibercept | 14,273 (3720) | 1.516 (0.3856) | 8012 (7232 to 8793) | 0.0032 (–0.0837 to 0.0902) | 2,483,943 |
| Outcome | Intervention, mean (SD); n | Comparator, mean (SD); n | Difference, mean (95% CI)a | Probability of being cost-effective at £20,000 (£30,000) per QALY |
|---|---|---|---|---|
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 14,328 (3883); 154 | 13,013 (3673); 154 | 1245 (406 to 2085) | – |
| QALY | 1.651 (0.2426); 154 | 1.627 (0.2471); 154 | 0.004 (–0.0430 to 0.0518) | – |
| ICER (£) | 284,255 | 0.04 (0.10) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 8933 (3474); 154 | 13,013 (3673); 154 | –4103 (–4949 to –3257) | – |
| QALY | 1.666 (0.2374); 154 | 1.627 (0.2471); 154 | 0.018 (–0.0282 to 0.0648) | – |
| ICER (£) | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 14,328 (3883); 154 | 8933 (3474); 154 | 5342 (4552 to 6133) | – |
| QALY | 1.651 (0.2426); 154 | 1.666 (0.2374); 154 | –0.015 (–0.0618 to 0.0322) | – |
| ICER (£) | Aflibercept is dominated | 0.00 (0.00) | ||
| Outcome | Intervention, mean (SD); n | Comparator, mean (SD); n | Difference, mean (95% CI)a | Probability of being cost-effective at £20,000 (£30,000) per QALY |
|---|---|---|---|---|
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 14,013 (3507); 66 | 12,608 (2342); 65 | 1405 (204 to 2606) | – |
| QALY | 1.691 (0.1931); 66 | 1.656 (0.1605); 65 | 0.011 (–0.0413 to 0.0629) | – |
| ICER (£) | 130,020 | 0.07 (0.14) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 6459 (3045); 62 | 12,608.2 (2342.29); 65 | –6149 (–7369 to –4929) | – |
| QALY | 1.651 (0.1507); 62 | 1.656 (0.1605); 65 | –0.007 (–0.0596 to 0.0458) | – |
| ICER (£) | 890,736 | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 14,013 (3507); 66 | 6459 (3045); 62 | 7554 (6338 to 8769) | – |
| QALY | 1.691 (0.1931); 66 | 1.651 (0.1507); 62 | 0.018 (–0.0345 to 0.0704) | – |
| ICER (£) | 426,551 | 0.00 (0.00) | ||
| Outcome | Intervention, mean (SD); n | Comparator, mean (SD); n | Difference, mean (95% CI)a | Probability of being cost-effective at £20,000 (£30,000) per QALY |
|---|---|---|---|---|
| Aflibercept vs. ranibizumab | ||||
| Cost (£) | 9214 (2235); 154 | 8164 (2163); 154 | 1002 (516 to 1487) | – |
| QALY | 0.8798 (0.1208); 154 | 0.865 (0.1230); 154 | 0.004 (–0.0178 to 0.0256) | – |
| ICER (£) | 256,547 | 0.00 (0.02) | ||
| Bevacizumab vs. ranibizumab | ||||
| Cost (£) | 3621 (2017); 154 | 8164 (2163); 154 | –4546 (–4999 to –4093) | – |
| QALY | 0.8842 (0.1171); 154 | 0.865 (0.1230); 154 | 0.007 (–0.0143 to 0.0290) | – |
| ICER (£) | Bevacizumab is dominant | 1.00 (1.00) | ||
| Aflibercept vs. bevacizumab | ||||
| Cost (£) | 9214 (2235); 154 | 3621 (2017); 154 | 5560 (5082 to 6039) | – |
| QALY | 0.8798 (0.1208); 154 | 0.8842 (0.1171); 154 | –0.004 (–0.0256 to 0.0168) | – |
| ICER (£) | Aflibercept is dominated | 0.00 (0.00) | ||
List of abbreviations
- A&E
- accident and emergency
- AE
- adverse event
- AIC
- Akaike information criterion
- ALDVMM
- adjusted limited dependent variable mixture model
- AMD
- age-related macular degeneration
- APTC
- Antiplatelet Trialists’ Collaboration
- BCVA
- best corrected visual acuity
- BIC
- Bayesian information criterion
- BRAVO
- RanibizumaB for the treatment of macular edema following BRAnch Retinal Vein Occlusion
- BRVO
- branch retinal vein occlusion
- BSE
- better-seeing eye
- CASP
- Critical Appraisal Skills Programme
- CATT
- Comparison of Age-related macular degeneration Treatments Trials
- CEAC
- cost-effectiveness acceptability curve
- CFP
- colour fundus photography
- CFT
- central foveal thickness
- CI
- confidence interval
- CINAHL
- Cumulative Index to Nursing and Allied Health Literature
- CLRN
- Comprehensive Local Research Network
- COST
- cone outer segment tip
- CRF
- case report form
- CRT
- central retinal thickness
- CRUISE
- Ranibizumab for the Treatment of Macular Edema after Central Retinal Vein OcclUsIon Study: Evaluation of Efficacy and Safety
- CRVO
- central retinal vein occlusion
- CST
- central subfield thickness
- CUA
- cost–utility analysis
- CVI
- Certificate of Vision Impairment
- CVOS
- Central Vein Occlusion Study
- DD
- disc diameter
- DMEC
- Data Monitoring and Ethics Committee
- DMO
- diabetic macula oedema
- DRIL
- disorganisation of the retinal inner layers
- DS
- dioptre sphere
- eCRF
- electronic case report form
- ELM
- external limiting membrane
- EMA
- European Medicines Agency
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-3L
- EuroQol-5 Dimensions, three-level version
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- EQ-5D-V
- EuroQol-5 Dimensions with vision bolt-on
- ETDRS
- Early Treatment Diabetic Retinopathy Study
- EZ
- ellipsoid zone
- FDA
- Food and Drug Administration
- FFA
- fundus fluorescein angiography
- GP
- general practitioner
- HCHS
- Hospital and Community Health Service
- HORIZON
- An Open-Label, Multicentre Extension Study to Evaluate the Safety and Tolerability of Ranibizumab in Subjects with Choroidal Neovascularization Secondary to Age-Related Macular Degeneration or Macular Oedema Secondary to Retinal Vein Occlusion Who Have Completed a Genentech-Sponsored Ranibizumab
- HRF
- hyper-reflective foci
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ICH
- International Conference on Harmonisation
- IMP
- investigational medicinal product
- INMB
- incremental net monetary benefit
- IQR
- interquartile range
- ITT
- intention to treat
- IVAN
- Inhibit VEGF in Age-related choroidal Neovascularisation
- KCTU
- King’s Clinical Trials Unit
- LEAVO
- a multicentre, Phase III, double-masked, randomised controlled non-inferiority trial comparing the clinical effectiveness and cost-effectiveness of intravitreal therapy with ranibizumab (Lucentis) versus aflibercept (Eylea) versus bevacizumab (Avastin) for macular oedema due to central retinal Vein Occlusion
- LME
- linear mixed effects
- M
- macular ring
- MAE
- mean absolute error
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MO
- macular oedema
- NetwORC
- Network of Ophthalmic Reading Centres
- NHS EED
- NHS Economic Evaluation Database
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NMB
- net monetary benefit
- nvAMD
- neovascular age-related macular degeneration
- NVD
- neovascularisation disc
- NVE
- neovascularisation elsewhere
- NVG
- neovascular glaucoma
- NVI
- neovascularisation of the iris
- OCT
- optical coherence tomography
- PAS
- Patient Access Scheme
- PIN
- patient identification number
- QALY
- quality-adjusted life-year
- QMUL
- Queen Mary University of London
- R
- ring
- RAPD
- relative afferent pupillary defect
- RCT
- randomised controlled trial
- RETAIN
- extended follow-up of patients with macular edema due to bRanch rETinal vein occlusion or centrAl retinal veIn occlusioN previously treated with intravitreal ranibizumab
- RMSE
- root-mean-square error
- RVO
- retinal vein occlusion
- SA
- substantial amendment
- SAE
- serious adverse event
- SCORE2
- Study of Comparative Treatments for Retinal Vein Occlusion 2
- SD
- standard deviation
- SD-OCT
- spectral-domain optical coherence tomography
- SE
- standard error
- SUR
- seemingly unrelated regression
- SUSAR
- suspected unexpected serious adverse reaction
- TA
- technology appraisal
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- VEGF
- vascular endothelial growth factor
- VFQ-25
- Visual Function Questionnaire-25 items
- VFQ-UI
- Visual Function Questionnaire-Utility Index
- WSE
- worse-seeing eye