

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as award number 09/91/21. The contractual start date was in June 2011. The draft manuscript began editorial review in December 2021 and was accepted for publication in March 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this article.
Permissions
Copyright statement
Copyright © 2024 Collinson et al. This work was produced by Collinson et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Collinson et al.
Chapter 1 Introduction
Scientific background
Despite significant advances in the treatment of advanced renal cell carcinoma (RCC) and improvements in survival, it remains a difficult disease to treat. Despite frequent initial responses to a variety of available systemic therapies, most patients subsequently progress and die from their disease. It is therefore paramount to develop more efficacious treatment, but also optimise treatment strategies aiming to maximise quality and quantity of life, while minimising toxicity. There has been increasing interest in drug-free interval strategy (DFIS), or planned treatment breaks, aiming to do this.
Renal cell carcinoma
Renal cell carcinoma constitutes 4% of adult malignancies and 90% of kidney cancers. Over the last 10 years, the incidence has increased by a third. Despite a number of new treatments now available, it is still the 13th commonest cause of cancer-related death, responsible for around 3% of all cancer deaths in the UK. The annual incidence of RCC in the UK is approximately 13,100 cases, with around 4600 deaths. 1
Approximately 56% of cases present with localised disease (Stage I/II) at diagnosis, and 44% with more advanced disease (Stage III/IV). 1 Additionally, between 30% and 50% of patients with apparent localised and locally advanced disease at the time of diagnosis will subsequently develop metastatic disease. The 5-year survival for metastatic RCC (mRCC) is only around 10%, although this figure is increasing, particularly in the subset of patients now treated with immunotherapy. 2
Standard treatment options at STAR trial conception (c.2009)
The options for systemic treatment of mRCC have changed significantly over recent years. At the time of the initial design of the STAR trial, National Institute for Health and Care Excellence (NICE) approval for any tyrosine kinase inhibitor (TKI) was still pending and the UK standard of care remained interferon-α (IFNα), with an 11–15% objective response rate (RR) in appropriately selected individuals and an improvement in overall survival (OS) of around 4 months compared to best supportive care. 3,4
The strategy of targeting angiogenic pathways produced positive results in advanced RCC. TKIs, for example, sunitinib and sorafenib and monoclonal antibodies (e.g. bevacizumab with IFNα), have all demonstrated improvements in terms of progression-free survival (PFS) and also OS. 5,6
Tyrosine kinase inhibitors
Sunitinib
Sunitinib selectively targets multiple protein receptor tyrosine kinases including vascular endothelial growth factor (VEGF) receptor and platelet-derived growth factor receptor. TKIs are thought to ‘starve’ tumours of blood and nutrients needed for growth, which leads to death of the cancer cells. These drugs also potentially have a direct effect on the tumour cells. Sunitinib was an early success story of targeted therapies as cancer treatments. 7
The landmark randomised controlled blinded first-line trial of 750 patients [Eastern Cooperative Oncology Group (ECOG) performance status (PS) 0 or 1] with mRCC directly compared sunitinib and IFNα, with PFS as the primary end point. 5 The trial was unblinded after a second interim analysis demonstrated significant benefit in patients treated with sunitinib. This subsequently led to crossover of a number of patients from IFNα to sunitinib. Updated results were published in 2009 demonstrating in the intention-to-treat (ITT) population, a median PFS of 11 months with sunitinib and 5 months with IFNα (p < 0.001). 6
Adverse events (AEs), in the sunitinib arm, were as expected from other studies, including hypertension (12%), fatigue (11%), diarrhoea (9%) and hand–foot syndrome (9%). OS was 26.4 months with sunitinib and 21.8 months with IFNα [hazard ratio (HR) 0.821, 95% confidence interval (CI), 0.673 to 1.001; p-value 0.051], although this likely underestimated the true OS benefit due to the significant crossover that occurred in the study population. Sunitinib was also associated with improved RRs over IFNα with 3% versus 1% complete response (CR), 44% versus 11% partial response (PR) and 40% versus 54% stable disease (SD) as the best responses seen.
Early in 2009, sunitinib was approved in the UK by NICE for use in the first-line treatment of advanced and/or mRCC in patients with a good PS (ECOG 0 or 1) until evidence of disease progression or unacceptable toxicity. 8 This was after reappraisal under the ‘end-of-life’ criteria with the assessment of the value of the health gain to meet conventional cost-effectiveness criteria. This changed the standard of care in RCC to first-line sunitinib in appropriately fit patients.
The recommended cycle of sunitinib is 50 mg orally once daily (OD) on days 1–28, followed by a 14-day period treatment-free. Standard practice dictates that these cycles are repeated without interruption (with regular radiological assessment) until disease progression or unacceptable toxicity [the approach in the conventional continuation strategy (CCS) arm of the STAR trial]. Sunitinib is however associated with a significant side-effect burden. The landmark first-line trial reported that 8% of patients discontinued sunitinib due to AEs, 32% of patients required a dose reduction and 38% a dose interruption. 6 In the sunitinib open access programme, 8% of patients discontinued the drug due to toxicity, with a further 33% requiring a dose reduction to 37.5 mg, with a further 13% requiring a subsequent dose reduction to 25 mg. 9 The longer-term impacts of sunitinib-associated toxicities are recognised to be increasingly important as patients are living longer; individualised treatment strategies are necessary to optimise benefit and cost effectiveness while minimising toxicity. 10
Pazopanib
Pazopanib is another TKI which works in a similar way to sunitinib but is given daily (without interruption) at 800 mg OD continuous dosing. It was approved by NICE as an alternative first-line treatment option for patients with advanced RCC in early 2011. This was based on a phase III study comparing pazopanib to placebo,11 conditional on pricing and further data from the pharmaceutical company GlaxoSmithKline, including the results from the COMPARZ trial. 12
At the time of commencing the STAR trial, the introduction of pazopanib was already anticipated for the Phase III part of the trial. There was no evidence as to the relative clinical effectiveness of the two TKI drugs, but the COMPARZ trial was ongoing which was directly comparing the two drugs 1 : 1 in fit [Karnofsy Performance Status (KPS) > 70] patients with locally advanced/mRCC. The trial was designed as a non-inferiority (NI) trial of PFS, at a HR margin of 1.25 (upper 95% CI); however, the EMA-defined primary end point was NI in PFS with a HR margin of 1.22 (upper 95% CI). Over 1000 patients were randomised, 557 to the pazopanib arm and 553 to the sunitinib arm.
The initial data from this study were presented at European Society for Medical Oncology in October 2012. 13 Analysis of the primary end point (with independent review) demonstrated a PFS HR of 1.047 (0.898–1.220); hence achievement of the primary end point demonstrating NI of pazopanib to sunitinib. There was no significant difference in OS between the arms with HR 0.908 (0.762–1.082), p-value 0.275. Clinical benefit rate (CR + PR + SD) was similar between the arms, 79% for pazopanib and 69% for sunitinib, although there was a slight increase in the rate of objective responses (CR + PR) seen with pazopanib (31% vs. 25%, p-value 0.032).
Although the two drugs had many toxicities in common, there was significantly more fatigue (63% vs. 55%), hand–foot syndrome (50% vs. 29%), taste changes (36% vs. 26%) and thrombocytopenia (34% vs. 10%) with sunitinib, but more hair changes (30% vs. 10%) and increases in alanine transaminase (ALT) (31% vs. 18%) with pazopanib. The median duration of treatment was similar in both arms (8.0 months pazopanib and 7.6 months sunitinib). Importantly, the number of dose reductions (44% pazopanib and 51% sunitinib) and discontinuations due to AEs (24% pazopanib and 19% sunitinib) were substantial in both arms. These data confirmed the relevance of the STAR trial in investigating planned treatment breaks with both sunitinib and pazopanib (S/P) due to the potential benefits to patients in terms of reduced toxicity and improved quality of life (QoL), in addition to cost benefits to theNHS.
In the COMPARZ study, the QoL data presented (FACIT-fatigue) demonstrated reduced QoL in the sunitinib arm compared to pazopanib. However, the time points used for comparison were day 28 of each 42-day cycle, the time when the difference between QoL on the drugs will be maximised as sunitinib toxicity peaks around this time due to the 4 weeks on/2 weeks off schedule, compared to the toxicity seen with pazopanib which is more uniform within each cycle due to continuous dosing. 14 The differences seen in COMPARZ between S/P appear less marked than those from the previously reported patient preference PISCES study, possibly as within PISCES each treatment was only taken for 10 weeks and this study was based on significantly fewer patients. 15
Inclusion of sunitinib and pazopanib in the STAR trial
STAR had originally mandated that sunitinib was the only drug permitted for use in the phase II part, with pre-planned reconsideration prior to opening the phase III part, based on available data at that time. Early consideration of the data available in October 2012 (midway through recruitment into the phase II part of the trial) and discussions with investigators found that a number of sites wanted to be able to offer pazopanib as an alternative to sunitinib. Following discussion with the funder [National Institute for Health and Care Research Health Technology Assessment (NIHR HTA)], the Trial Steering Committee (TSC) and the Data Monitoring and Ethics Committee (DMEC), from protocol version 4.0 dated 15 February 2013 the Trial Management Group (TMG) introduced the option of using either sunitinib or pazopanib into the phase II part of the study. The TKI used was also included as an additional stratification factor.
The STAR trial was always designed to be a pragmatic trial testing the strategy of introducing planned treatment breaks, aligned with standard practice at that time which required careful amendment to include both the TKIs which were approved and widely used. More recently, in 2018, additional TKIs, tivozanib16 and cabozantinib,17 were approved by NICE, but as recruitment was complete at that time, these approvals did not impact on the trial.
Intermittent treatment strategies in systemic cancer treatment
There is increased interest in DFIS in oncology with evidence that these approaches are associated with reduced toxicity and increased QoL, without significantly compromising previously demonstrated survival benefits. This approach is most studied in colorectal cancer (CRC), where there is a considerable evidence base that treatment breaks can be introduced (utility of a DFIS) without a clinically significant survival deficit, but with evidence of a QoL advantage.
In an early trial, 354 patients with metastatic CRC were treated with 5-fluorouracil (5FU) and folinic acid (FA) (de Gramont schedule) or continuous infusion 5FU or raltitrexed. Those who had stable or responding disease at 12 weeks were then randomised to continue therapy until progression or to stop, with the option to restart the same chemotherapy on progression. There was no evidence of a difference in OS between the two groups (intermittent or continuous chemotherapy), with both groups having a 1-year survival rate of approximately 45%. 18 There was evidence of a QoL advantage for those patients having intermittent chemotherapy over continuous therapy.
In the UK, this concept was further investigated with the COIN trial. 19 This was a large, randomised trial of 1639 patients receiving oxaliplatin plus fluoropyrimidine-based chemotherapy. Patients were randomly assigned (1 : 1) to continuous chemotherapy until progression (arm A) or intermittent chemotherapy (arm C). In arm C, chemotherapy was stopped at 12 weeks (until evidence of clinical or radiological disease progression) for patients who were responding, or who had SD. While the results did not demonstrate NI of intermittent compared with continuous chemotherapy in terms of OS, it was concluded that intermittent treatment could still be considered in informed patients based on reduced toxicity and improved QoL. Treating patients with CRC with pre-planned chemotherapy breaks remains standard practice in most UK centres. 19
Other trials have also demonstrated equivalence between intermittent and continuous therapy. OPTIMOX1 compared 6 cycles of FOLFOX7 (3 weekly bolus oxaliplatin, FA and 5FU) followed by continuous 5FU/FA alone (for a maximum of 24 weeks) before re-introduction of FOLFOX7 to FOLFOX4 (2 weekly bolus oxaliplatin, 5FU and FA) until progression in 623 patients with metastatic CRC. The indication to restart oxaliplatin in the intermittent arm was evidence of progression compared to the baseline scan, not progression compared to best response. Duration of disease control was similar between both arms (10.6 and 9.0 months, respectively), as were PFS and OS. 20 Of note, almost 60% of patients on the intermittent arm did not have oxaliplatin reintroduced (protocol violations), but those who did tended to have a better prognosis. 21
Leading on from this, the OPTIMOX2 trial compared FOLFOX7 for 12 weeks and then continued 5FU/FA until progression, at which point oxaliplatin was re-introduced, to FOLFOX7 for 12 weeks and then a complete break from chemotherapy until progression. The trial recruited 202 patients but was prematurely closed with bevacizumab becoming available as a first-line treatment option. The median duration of disease control was 13.1 months in the maintenance arm and 9.2 months in the intermittent arm, with respective OS of 23.8 months and 19.5 months. 22 There were however significant design issues within this trial and the results are not clear cut. The primary end point of duration of disease control has been criticised as treatment was not mandated to be restarted until the disease reached baseline size, hence introducing variation in the time of restarting treatment. The statistical plan was also not adapted to account for the reduced sample size from 600 to 216. The extensive criticism of this trial has meant that definitive conclusions cannot be drawn and a DFIS is still widely practised,23 backed two meta-analyses of the relevant trials. 24,25
During the COVID-19 pandemic in the UK, the advice regarding treatment breaks to minimise visits to hospital and treatment-associated risks was supported by the rapidly published NICE guidelines and widened outwith chemotherapy alone to include targeted therapies, for example epidermal growth factor inhibitors in combination with chemotherapy in advanced CRC. 26
Intermittent treatment strategy with tyrosine kinase inhibitors
Similar data for the use of planned treatment breaks with TKIs are sparse. There is one randomised Phase III trial in gastrointestinal stromal tumours (GISTs) treated with imatinib. It reported that although most patients treated with a DFIS after a year of treatment progressed, of the 26 patients who progressed, 24 responded again on re-exposure to imatinib and there was no significant detriment in terms of OS. 27 Imatinib is however associated with a minimal toxicity profile; hence there was less incentive to adopt an intermittent scheduling approach in this setting.
There are a number of additional studies supporting our rationale for the DFIS with sunitinib or pazopanib in advanced RCC. In one study, 23 patients with advanced RCC who had initially responded to sunitinib and then progressed were treated with other second-line therapies (median duration 6.7 months). These patients were then rechallenged with sunitinib with a further median PFS of 7.2 months. 28 This suggests that initial resistance to sunitinib therapy can be reversible and adds support for the rationale for this study. Importantly, no additional or increased toxicities were observed upon rechallenge. An observational French study also found evidence of further response with rechallenge sunitinib treatment in this population and also concluded that initial progression may not be associated with irreversible resistance. 29
Another recent small retrospective analysis studied the effects of stopping sunitinib therapy in 11 patients who had a CR after sunitinib alone (n = 5) or after sunitinib followed by a residual metastasectomy (n = 6). At median follow-up of 8.5 months, five patients had recurrent disease, but in all cases re-introduction of sunitinib was effective, providing additional support to the reuse of sunitinib after an initial response. 28 A published case series also demonstrated a re-introduction of sunitinib sensitivity after changing from the standard dosing schedule (50 mg daily day 1–28 every 42 days) to a lower continuous daily dose (37.5 mg). 30
Finally, data from one other randomised phase II study present further support to the hypothesis that a DFIS could be used for sunitinib or pazopanib. In this study, 202 patients with mRCC were treated with sorafenib (an alternative TKI). After 12 weeks of treatment, 73 patients had a PR and 65 patients had SD. The patients with SD were then randomly assigned to sorafenib (n = 32) or placebo (n = 33). At 24 weeks, 50% of patients continuing sorafenib were progression-free compared with 18% of placebo-treated patients (p-value 0.0077) and median PFS from randomisation was significantly longer in sorafenib-treated patients. When sorafenib was re-administered in 28 placebo-treated patients whose disease had progressed, further progression was delayed for a median of 24 weeks. The researchers concluded that the re-stabilisation of progressive disease (PD) in patients whose disease had progressed on placebo and were switched to sorafenib resulted in comparable median summative PFS as for patients who had no gap in sorafenib treatment. This suggests that patients were not disadvantaged from a brief period of placebo treatment, providing further ethical support for this design. 31
Subsequent to the STAR trial being designed another phase II non-randomised study has also reported. Patients with advanced clear cell RCC with > 10% reduction in tumour burden after 4 cycles of sunitinib treatment underwent a planned treatment break until there was a subsequent increase in tumour burden of > 10%. This was repeated until Response Evaluation Criteria of Solid Tumours (RECIST) defined PD while receiving sunitinib treatment. Of the 20 patients who had at least one treatment break, there was a median PFS of 22.4 months and OS of 34.8 months, again supporting the feasibility and efficacy of treatment breaks. 32
Change in treatments available for advanced renal cell carcinoma after initial design
Summary of advances in new agents to treat renal cell carcinoma
Since the start of the STAR trial in 2012, there have been a number of treatment advances in systemic therapy for metastatic renal cancer. These include the routine use of additional TKIs such as axitinib and tivozanib and agents which are combined TKI and c-MET inhibitors (small molecules which inhibit activity of c-MET tyrosine kinase, the receptor for hepatocyte growth factor, HGF/SFs) such as cabozantinib, new combinations such as everolimus and lenvatinib (a newer TKI), all given orally. 33,34
Renal cancer is an immunosensitive disease. Over recent years, there has been great success in a number of cancers from immunotherapeutic agents, for example in melanoma, lung cancer and bladder cancer. Treatment with PD-1 and PD-L1 inhibitors and CTLA4 (cytotoxic T-lymphocyte-associated protein 4) inhibitors has now also become the standard of care in renal cancer. 35 There is however still a role for first-line TKI treatment in a number of patients.
Progressive disease-1 and PD-L1 inhibitors are a group of checkpoint inhibitor anticancer drugs that block the activity of PD-1 and PD-L1 immune checkpoint proteins present on the surface of cells. CTLA-4 is a protein receptor that functions as an immune checkpoint and downregulates immune responses. CTLA-4 is constitutively expressed in regulatory T cells but only upregulated in conventional T cells after activation. This phenomenon is particularly notable in cancers. Initially, agents such as nivolumab were given as monotherapy in second-line metastatic renal cancer treatment. However, more recently, ipilimumab (CTLA-4 inhibitor) and nivolumab (PD-1 inhibitor) in combination has been given as standard of care in the first-line metastatic setting in renal cancer (approved by NICE in 2019). 36
Over the last 2–3 years, immunotherapy agents combined with TKI therapy have also become a standard of care in the first-line setting in mRCC. 35 Such immunotherapies are given intravenously and include agents such as nivolumab, ipilimumab, pembrolizumab and avelumab.
Currently, for some patients, TKIs such as pazopanib and sunitinib remain first-line therapy, either because patients prefer to start with oral therapies or because they are unsuitable for combination immunotherapy, or TKI with immunotherapy, due to contraindications such as pre-existing autoimmune disease or comorbidities making them not suitable for combination therapy. In addition, those patients who are treated with immunotherapy in the first-line setting are likely to receive TKI therapy as second, third or fourth line, depending on their fitness for further therapy. Notably, with immunotherapy alone or in combination with TKIs, questions regarding treatment breaks similar to those addressed in the STAR trial remain very relevant, as these combinations are expensive, have significant toxicity and are currently continued until disease progression.
Although in 2021 first-line treatment of advanced renal cancer remains non-curative, the hope is that with further treatment advances, survival will increase.
Adjuvant therapy in renal cell carcinoma
Patients may initially present with metastatic renal cancer at the time of diagnosis or following potentially curative nephrectomy for localised renal cancer. In the latter group which is particularly common in intermediate or high-risk Leibovitch groups, individuals may develop metastases. In high-risk groups, the risk is up to 50% over a 5-year period. Until recently, most adjuvant trials have not convincingly demonstrated significant advantages of giving TKI therapy in the adjuvant setting, so the standard of care has been regular follow-up and imaging. However, with the increasing success of immunotherapy in treating metastatic renal cancer, encouraging signals are starting to emerge using immunotherapy in the adjuvant setting in intermediate- or high-risk renal cancer where there is a substantial risk of developing metastatic disease in the next 5 years following nephrectomy (as defined by Leibovitch score at nephrectomy). 37–39 In this situation for those patients who progress on adjuvant immunotherapy, TKIs may also remain the standard of care in the first-line metastatic setting.
Prognostic risk score
At the start of the STAR trial, the Motzer score remained the main prognostic index in RCC and was therefore used for stratification in control and treatment arms for this study. The Motzer score is used as a tool to determine survival and is based on data from a study reported in 1999 in which 670 patients were treated at Memorial Sloan Kettering Institute in the USA. The Motzer score looks at five key indices, scoring one for each ‘yes’ category for the following: PS (Karnofsky score) < 80%; time from diagnosis to systemic therapy < 12 months; haemoglobin less than the lower limit of the normal range; corrected calcium levels > 10 mg/dL and lactate dehydrogenase (LDH) > 1.5 × upper limit of normal (ULN). This divides the patient population with metastatic renal cancer into three prognostic groups: ‘favourable’ who have a Motzer score of 0, ‘intermediate’ who have a score of 1 or 2 and ‘poor’ with a score ≥ 3, with calculated survival reducing from favourable to poor groups. This score was however developed in patients treated with cytokines and interferon and not with more modern treatments such as TKIs and immunotherapy.
More recently, the International Metastatic RCC Database Consortium (IMDC) Risk Score for RCC has been introduced and predicts OS in patients treated with systemic therapy. It has become increasingly used as it has a number of advantages in predicting survival in prospective studies over the Motzer score using more modern treatments. This IMDC score was developed by Daniel Heng from Tom Baker Cancer Centre, Calgary, Canada, who developed a new set of variables that included neutrophil count. 40 Upon validation in a kidney cancer trial, Heng’s model came closest to predicting death after a 2-year period compared with a number of other models. IMDC is now one of the standard models currently used for mRCC.
The factors included in the IMDC model are shown in Appendix 1 with the scores that correlate with each factor. The total score is then categorised into favourable (0 risk factors), intermediate (1–2 risk factors) and poor (> 2 risk factors).
Risk categories in advanced renal cancer according to the IMDC score
More recently, a further adaption has also been made to the original Motzer score, which has been extended to include not just the original five factors but also primary radiotherapy and ≥ 2 metastatic sites (the Mekhail Extension41); however, it remains broadly similar.
For the STAR trial, in terms of the statistical prospective analysis, the IMDC score was also calculated (as well as the original Motzer score) for individual patients and included as a subgroup analysis for the primary analyses. This was felt important as, with development of immunotherapy and combined TKI therapy, determination of risk score in individual patients has become more important to guide treatment choice.
Further details on tyrosine kinase inhibitors that have become available during the duration of the STAR study
Historically sorafenib has been available for many years but, on efficacy grounds, S/P have remained the favoured first-line option. The newer TKIs include the following.
Axitinib
Axitinib is an oral multitargeted TKI with antitumour activity. It selectively inhibits VEGF receptors 1, 2 and 3, platelet-derived growth factor receptor, and c-kit, which may inhibit angiogenesis in tumours. Axitinib alone is not currently recommended or NICE-approved for the first-line setting in advanced RCC. However, more recently, it has been considered in combination with immunotherapy first line.
A large trial (AXIS) assessed axitinib for the second-line treatment of patients with advanced RCC. AXIS was a Phase III, international, multicentre, randomised, open-label, active-controlled trial comparing axitinib with sorafenib for treating advanced or mRCC after failure of first-line systemic therapy. 42 This study resulted in axitinib being recommended as an option for treating adults with advanced RCC, after failure of treatment with a first-line TKI or a cytokine, and resulted in it becoming part of standard second-line treatment in RCC. It is given orally, initially starting at a dose of 5 mg twice daily, but can be increased or titrated to blood pressure (BP) readings in the patient to 7 or 10 mg twice daily. Similarly, it can be decreased in those with toxicity to 3 mg or a minimum dose of 2 mg twice daily as needed.
Tivozanib
The main clinical evidence for routine use of tivozanib came from TIVO-1, an open-label randomised controlled trial (RCT) that primarily investigated whether tivozanib (n = 260) prolongs time to disease progression compared with sorafenib (n = 257). 43 Most of these patients were recruited from Eastern Europe rather than the UK. Upon disease progression, patients in the sorafenib group could switch (cross over) to treatment with tivozanib. As a result of this study, tivozanib was approved by NICE for its use as a first-line treatment for adult patients with advanced RCC. 17 It is given for 3 weeks on and 1 week off for a 4-week cycle. The starting dose of 1340 mg with (as in other TKIs) treatment continues until disease progression or unacceptable toxicity. Only one dose reduction is allowed. The cost of treating RCC with tivozanib is likely to be lower than the cost of treating with sunitinib or pazopanib (mainly because of drug costs), but tivozanib is also likely to be less effective on the basis of the trial data. However NICE felt that the estimated cost savings are high enough to compensate for the estimated lower effectiveness (using the tivozanib open access scheme) and it was approved by NICE in 2018. This approval is relevant for future NICE considerations of TKI intermittent treatment strategies in this setting. The toxicity pattern is slightly different to pazopanib and sunitinib, but it has not proven to be better and survival data also appeared to be inferior to S/P. 16
Cabozantinib
Cabozantinib is a combined MET and AXL inhibitor, in addition to inhibiting VEGF receptors. It is given orally with a standard approved dose of 60 mg daily and allows two dose reductions for toxicity to 45 mg and then 20 mg daily. Initial approval from NICE was given in August 2017 as an option for treating advanced RCC in adults, after VEGF-targeted therapy, as a subsequent line of therapy. During this time, axitinib and nivolumab were also available post first line in the advanced RCC setting. Therefore, NICE allowed cabozanitinib to be used after one or two lines of prior therapy in the advanced setting.
This NICE approval44 was largely based on data from the METEOR study comparing cabozantinib with everolimus after failing initial TKI therapy, which showed PFS superiority in the cabozantinib arm compared with everolimus (median 7.4 and 3.9 months, respectively; HR 0.51, 95% CI 0.41 to 0.62; p < 0.0001). OS improved with cabozantinib compared with everolimus (median 21.4 and 16.5 months, respectively; HR 0.66, 95% CI 0.53 to 0.83; p-value 0.00026). This led to the widespread use of cabozantinib in either the second- or third-line setting in advanced RCC.
Subsequently, the CARBOSUN Phase 2 study compared sunitinib with cabozantinib in the first-line advanced RCC setting. Initial results showed improved investigator-assessed PFS, with a median PFS of 8.2 months for cabozantinib compared with 5.6 months for sunitinib (p = 0.012). Updated results from the CABOSUN trial showed cabozantinib significantly prolonged PFS per the independent review committee compared with sunitinib as first-line therapy for advanced RCC of poor or intermediate risk. 45
These data made a major contribution to the NICE approval of cabozantinib in the first-line setting in 2017. However, in practice, many clinicians have preferred to continue to use cabozantinib in the second- or third-line setting rather than first line to increase the number of treatment options available to patients and also as CABOSUN was a Phase II, not a Phase III, trial.
Lenvatinib in combination with everolimus
Lenvatinib targets VEGFR1, 2 and 3, PDGFRα, fibroblast growth factor receptor (FGFR) and the KIT and RET tyrosine kinases and was initially developed for use in differentiated thyroid carcinoma refractory to standard therapy. 46 Evidence for its efficacy in treatment of RCC came from a randomised Phase II trial of 153 patients with metastatic or unresectable, locally advanced, ccRCC who had received prior antiangiogenic therapy. 47
Three arms with 1 : 1 : 1 randomisation were compared: patients received lenvatinib alone (24 mg/day) or everolimus alone (10 mg/day) or lenvatinib (18 mg/day) plus everolimus (5 mg/day) in 28-day cycles until progression or unacceptable toxicity. The lenvatinib/everolimus combination resulted in significant improvement in median PFS (14.6 months) compared with everolimus alone (5.5 months), but not compared with lenvatinib alone (7.4 months). Lenvatinib alone significantly improved PFS versus everolimus alone. However, toxicity events Grade > 2 occurred in more patients in the lenvatinib alone arm (79%) and in the combined arm (71%) than in the everolimus alone arm (50%).
This combination was approved by NICE as a second-line treatment option in 2018. The evidence from a single clinical trial suggests that, on average, people live around 10.1 months longer if they have lenvatinib plus everolimus rather than everolimus alone. 47 In the trial, lenvatinib plus everolimus caused side effects, leading many patients to interrupt or even stop treatment. This is despite the patients enrolled in the trial being relatively fit (i.e. they had an ECOG PS score of 0 or 1), so it is a treatment option for the fittest patients only.
Immunotherapy in advanced renal cell carcinoma
Immune suppression within RCC occurs via several pathways and immune checkpoint inhibitors as highlighted earlier have become standard of care in mRCC. 35 There is also emerging positive data in the adjuvant setting, for example, with pembrolizumab. 37–39 These drugs are very expensive and have significant immunotoxicities which can occur. Currently, as standard of care, they are also given until progression which can be for several years in some patients.
Initially, nivolumab was approved by NICE for second-line therapy following the Checkmate 025 trial48 comparing nivolumab and everolimus, which showed an improvement of median OS in the nivolumab arm (25 vs. 19.6 months; HR 0.73, 98.5% CI 0.57 to 0.93; p = 0.0018).
There followed a number of innovative combinations of immunotherapy agents or immunotherapy agents plus TKIs. 35,49 The combination of nivolumab with ipilimumab was approved by NICE as a first-line treatment for advanced RCC in patients in intermediate- or poor-risk groups following the results of the Checkmate 214 trial36 which showed that this combination was superior to sunitinib with 30-month OS 60% versus 47% (p < 0001); objective response rate (ORR) 42% versus 27% and complete response rate (CR) 11% versus 1% (p < 0.001). However, such combination therapy is only possible for fitter patients who are willing to have intravenous (IV) therapy and do not have significant autoimmune diseases or other contraindications to treatment.
Over the last few years, the combination of immunotherapy with a TKI in the first-line setting has also been explored with, for example, axitinib + pembrolizumab versus sunitinib in the KEYNOTE 426 study,50 which showed 12-month OS 90% versus 78% (p < 0.0001), ORR 59.3% versus 35.7%, CR 5.8% versus 1.9%. The JAVELIN RENAL 101 study51 (avelumab + axitinib vs. sunitinib) and the IMMOTION 151 study52 (atezolizumab + bevacizumab vs. sunitinib) also demonstrated ORR and CR benefits from the combination but failed to show significant OS benefits versus sunitinib.
Very recently, the ongoing Phase III CHECKMATE 9ER study53 (nivolumab + cabozantinib vs. sunitinib) reported that the probability of OS at 12 months was 85.7% (95% CI 81.3 to 89.1) with nivolumab plus cabozantinib and 75.6% (95% CI 70.5 to 80.0) with sunitinib (HR for death, 0.60, 98.89% CI 0.40 to 0.89; p-value 0.001). Studies with tivozanib + nivolumab are ongoing.
Although it is great news that there are now more treatment options and combinations emerging in RCC, it should be remembered that not all patients are fit enough for all therapies. In the future, patients who have undergone adjuvant immunotherapy may then go back to first-line TKI in the first-line advanced setting. Similarly, patients treated with immunotherapy in the first-line metastatic setting will currently receive second-line TKI therapy. Thus, TKI therapy is likely to remain a cornerstone of therapy in RCC for some considerable time and the STAR trial results will have relevance in both the first line and subsequent lines of TKI therapy. Similarly, the questions around treatment break will be relevant to studies of immunotherapy and immunotherapy/TKI combinations in the future.
Summary of rationale for the STAR trial
The STAR trial is a pragmatic randomised trial of a sunitinib or pazopanib CCS compared to a sunitinib or pazopanib DFIS.
In the UK, NICE approval for the use of sunitinib or pazopanib in the first-line treatment for patients with locally advanced and/or mRCC was a major step forward in the management of this disease. Over recent years, and continuing now with newer therapies, there has been increased interest in intermittent treatment strategies with the potential to reduce toxicity and improve QoL and cost effectiveness, without compromising treatment efficacy significantly. Additional benefits of a DFIS are hypothesised to include delaying or reducing the development of drug resistance.
Sunitinib and pazopanib are both associated with significant side-effect burden. The initial first-line trial for sunitinib reported that 8% of patients discontinued treatment due to AEs. 6 In the reported sunitinib open access programme, 8% of patients discontinued the drug due to serious adverse events (SAEs) and a further 33% had at least one dose reduction (13% had two dose reductions). 9 The previously mentioned Phase III study of pazopanib compared to placebo reported a discontinuation rate of 14% due to AEs and dose reductions due to AEs in 24% of patients. The COMPARZ study suggested that pazopanib was associated with similar substantial patterns of dose reductions and discontinuations as sunitinib. 12 A treatment strategy incorporating a DFI, assuming no survival disadvantage, would potentially give patients periods of time when symptoms attributable to sunitinib or pazopanib would be alleviated and would therefore have the potential to improve overall QoL and also cost-effectiveness.
In 2009 (at the time of development of the STAR trial and NICE approval of sunitinib in this setting), the average cost per cycle of sunitinib was £3700 per 6-week cycle, equating to an average cost of £47,000 per patient and a total annual cost of around £75 million for 1600 patients to the NHS. Estimates from our simulation show a likely reduction of approximately 21% in the duration of sunitinib treatment with a DFIS. This would correspond to a saving of approximately £9870 per patient, which when extrapolated to annual NHS costs in England produces a simulated annual saving of approximately £16 million.
Even now, 12 years after the initial design of the STAR trial was proposed, there is no clearly defined, evidence-based, optimal treatment strategy for any targeted therapy. Research in this field is crucial for both patients and the NHS. Evidence for the cost effectiveness of S/P remains poor, and standard decision criteria did not support their implementation in the NHS. Introduction of their first-line use in this setting likely displaced more health than it produced at a population level.
The STAR trial was designed to address the need to gather robust evidence on the costs, QoL and clinical outcomes of S/P both in the dosing schedule used in routine clinical practice (CCS) and in the DFIS. If successful, the design and implementation may be applicable to other drugs across a wide range of diseases.
Chapter 2 Trial design and methods
Trial design
The STAR trial was a seamless Phase II/III randomised controlled, UK multicentre, two-arm trial in advanced RCC. The trial was designed to investigate a TKI DFIS compared to a CCS in advanced (inoperable loco-regional or metastatic) RCC.
The trial was initially designed to determine whether a sunitinib DFIS was non-inferior in terms of 2-year OS and quality-adjusted life-years (QALYs) compared to CCS in patients with advanced RCC. The NI margins of 7.5% (OS) and 10% (QALYs) were decided on following collaboration between trial and recruiting clinicians along with patient representatives. The lesser used QALYs were guided by collaboration between experts in Health Economics and Statistics who were experienced in analysing patient QoL data.
The approval of pazopanib, an alternative TKI, midway through trial recruitment led to the trial being amended to investigate a DFIS using either drug. 6
Due to the novelty of the DFIS approach, the trial had a seamless Phase II/III design with two stages incorporated into the Phase II component (Stages A and B) and the Phase III component including all stages (Stages A, B and C).
The Phase II component was conducted in 16 UK renal cancer trial sites. The objective of Stage A was to establish the feasibility of performing the trial, in terms of average monthly recruitment. This was to ensure that sufficient participants were recruited for the trial to enable its completion in a timely manner. The objective of Stage B was to assess preliminary efficacy data by comparing time to strategy failure (TSF) in both arms and test for NI between the approaches to assess comparability (see Outcomes).
The primary objective for Stage C was to assess OS and QALYs averaged over trial recruitment and follow-up. Participants from all three stages contributed to the final Phase III analysis.
The secondary objectives were to evaluate how utilisation of a DFIS compared to utilisation of a CCS impacts on:
-
Summative progression-free interval (SPFI)5
-
TSF5
-
Time to treatment failure (TTF)5
-
Toxicity [common terminology criteria for adverse events (CTCAE) v.4.0]
-
QoL [FSKI-15, Functional Assessment of Cancer Therapy-G (FACT-G), EuroQol-5 Dimensions, three-level version (EQ-5D-3LTM) and EQ-Visual Analog Scale (VAS)TM]
-
Cost effectiveness
-
PFS.
The study also included three ancillary substudies:
-
The Patient Preference and Understanding Study was an embedded qualitative substudy designed to help understand participants’ experiences of taking part in the STAR trial and the impact of their treatment decisions on their physical and psychological health and well-being. Details of this study were provided in a separate protocol (REC reference: 11/YH/0261).
-
The dynamic contrast-enhancedmagnetic resonance imaging (DCE-MRI) substudy was undertaken at St James University Hospital, Leeds, and was optional for participants approached at that site. The substudy was designed to investigate the utility of tumour vascularity measured by DCE-MRI post randomisation, at around 10 weeks, and at 4 weeks after initiation of sunitinib or pazopanib to predict PD.
-
The computerised tomography (CT) substudy was open to participants at all sites where the appropriate imaging was performed routinely and was optional for participants approached at those sites. The sub-study was designed to define the interoperator variability (reliability) and hence the robustness of contrast-enhanced computed tomography (CECT) as a potential biomarker in this setting by performing a test–retest comparison (dual reporting) and to prospectively evaluate the utility of CECT modified Choi criteria (mChoi) assessed to predict for PD.
The trial also included the collection of archival diagnostic pathology tissue samples (from nephrectomy or from a diagnostic biopsy) from consenting randomised patients. The samples were collected by the NRS Lothian Bioresource in Edinburgh for future research from constructed STAR tissue micro-arrays (TMAs). More information is provided in Appendix 2.
Ethical and regulatory approval and research governance
Ethical approval for the study was given by the Liverpool Central Research Ethics Committee in June 2011 (reference number 11/NW/0246). Medicines and Healthcare products Regulatory Agency (MHRA) approval was given in May 2011.
The trial was registered with the International Standard Randomised Controlled Trial Register (ISRCTN) under the reference number 06473203 and with the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) under the reference number 2011-001098-16. Summaries of the most significant changes made to the original protocol are given below.
Protocol v4.0 amendment (approved April 2013)
STAR had originally mandated that sunitinib was the only drug permitted for use in the Phase II trial with a pre-planned reconsideration of pazopanib (another TKI drug under consideration by NICE and the subject of a comparative study against sunitinib, COMPARZ) prior to opening the Phase III part of the STAR trial, based on the available data at that time. However, the COMPARZ trial reported data early (October 2012) which showed pazopanib to be non-inferior to sunitinib in terms of its primary end-point PFS and with no significant difference in OS. NICE approved pazopanib for first-line treatment in 2011. Based on these data, it appeared likely that some clinicians would wish to offer pazopanib in standard practice, as an alternative to sunitinib, which would potentially reduce the number of participants taking sunitinib and therefore be eligible to participate in the STAR trial. Therefore, the decision was made [following discussion with the funder (NIHR HTA), TSC, DMEC, key investigators and patient representatives] that the protocol be amended (v4.0) to include the option of using pazopanib, with the type of TKI as a stratification factor.
Protocol v7.0 amendment (approved September 2014)
Following a review of the interim analysis results in July 2014 (end of Phase II), the trial oversight committees concluded that both the Stage A and B end points had been met, including no evidence that a DFIS was inferior to a CCS arm in terms of TSF (Stage B). Therefore, the DMEC advised continuation of the trial to Phase III (with both sunitinib and pazopanib) and the TSC approved trial continuation.
It was also decided in consultation with the clinical advisors to remove the requirement for participants to meet the maximal radiological response threshold prior to commencing a treatment break in the DFIS arm. Experience from the Phase II aspect of the study demonstrated that it would not be feasible for local sites to reliably determine maximal radiological response. In addition, only around 5% of participants had not reached the maximal radiological response threshold at 6 months. This change was approved by the TSC and DMEC.
Protocol v10.0 amendment (approved July 2017)
In 2016, the DMEC and TSC carried out a re-evaluation of some of the key assumptions made within the original sample size calculations. Although not a pre-planned review of the sample size estimates, this was deemed appropriate due to the fact that the trial had accumulated a significant amount of follow-up data since first opening to recruitment and thus had more accurate information for some of the required sample size estimation values, specifically the 2-year survival estimate in the CCS arm, the (extended) period of recruitment and the overall dropout rate. As a result of this, the sample size was reduced from 1000 to 920 participants, where 720 events were required for the analysis to have 80% power.
Protocol v11.0 amendment (approved July 2019)
Recruitment closed on 12 September 2017 following the recruitment of 920 participants. The trial was planned to continue follow-up until the target of 720 OS events had been observed.
A review of the number of events (death from any cause) for the co-primary end point of OS showed that the number of events was lower than expected and it was considered highly unlikely that the target of 720 events would be reached by the planned end of follow-up in September 2019. An analysis based on data estimated to be collected until September 2019 was considered to have between 72% and 73% power (as opposed to the target of 80% power) for OS, meaning that the trial would have insufficient evidence to be able to demonstrate NI.
On discussion with the Chief Investigator and Co-Chief Investigator, their opinion was that the main reason for the lower event rate was due to the increasing availability of second- and third-line treatment options (nivolumab and cabozantinib), which therefore improved OS and reduced the number of events seen within a specified time. To enable the final analysis to be carried out with the appropriate 80% power, the follow-up period was extended up to an additional 15 months (based on event rate modelling by Renfro et al. 54) in order to observe the required 720 events.
Protocol v12.0 (approved January 2021)
Ongoing monitoring of the event rate showed that, due to multifactorial reasons, as discussed previously, the event rate continued to decrease and a repeat of the modelling, carried out in July 2020, shifted the predicted time frame to observe 720 survival events.
As the study was in the tails of any applied distribution, it was not possible to accurately predict when 720 events would be observed; however, it was clear that it would not be until significantly beyond the planned end of follow-up. As such, it was not feasible to extend the trial for a further fixed duration and the TMG, DMEC and TSC agreed that follow-up would be completed on 31 December 2020 as planned, regardless of whether 720 survival events had been observed.
The end of trial definition was amended to be defined as the date of the collection of the last tissue sample or 31 December 2021, whichever came sooner. This change allowed tissue block collection, which had been interrupted by the COVID pandemic, to proceed beyond the end of follow-up until no later than 31 December 2021 (with the possibility that it could end before this if the TMG considered that it was not feasible to collect any further tissue blocks).
The trial schema is shown in Appendix 3.
Participants
A total of 920 participants were recruited for the trial from 24 February 2012 to 4 September 2017.
The trial recruited patients with locally advanced or metastatic clear cell RCC who had received no prior systemic therapy for locally advanced/metastatic disease.
Eligibility waivers to inclusion and exclusion criteria were not permitted.
Inclusion criteria
Patients were permitted to participate if they met all of the following criteria:
-
Male or female aged ≥ 18 years.
-
Histological confirmation of a component of clear cell RCC.
-
Inoperable loco-regional or metastatic disease.
-
No prior systemic therapy for advanced disease (inoperable loco-regional and/or metastatic disease). Previous treatment in the placebo arm of the SORCE study was permitted.
-
ECOG PS 0–1 assessed prior to randomisation and within 16 days of starting treatment with either sunitinib or pazopanib. 55
-
Uni-dimensionally measurable disease as defined by RECIST criteria.
-
Full blood count56 was performed prior to randomisation and within 16 days of starting treatment with either sunitinib or pazopanib.
-
Haemoglobin (Hb) ≥ 9 g/dL57 (blood transfusions were acceptable).
-
Absolute neutrophil count (ANC) ≥ 1 × 109/L.
-
Platelets ≥ 80 × 109/L.
-
-
Renal biochemistry58 was performed prior to randomisation and within 16 days of starting treatment with either sunitinib or pazopanib. Measured or calculated glomerular filtration rate (GFR) ≥ 30 ml/minute was permitted. (Cockcroft and Gault or Wright formula were used according to local practice.)
-
Hepatobiliary function59 was performed prior to randomisation and within 16 days of starting treatment with either sunitinib or pazopanib.
-
Aspartate transaminase (AST) or ALT ≤ 2.5 × ULN.
-
Bilirubin (BR) ≤ 1.5 × ULN, or in patients with Gilbert syndrome BR ≤ 3 × ULN and direct BR ≤ 35%.
-
-
Provided written informed consent prior to any trial-specific procedures.
-
Able and willing to comply with the terms of the protocol including:
-
commencement of sunitinib or pazopanib within 5 (actual not working) days of randomisation;
-
temporarily stopping sunitinib or pazopanib if randomised to the DFIS arm;
-
capable of oral self-medication;
-
commencement of sunitinib or pazopanib within 42 days of the baseline CT scan;
-
capable of reporting toxicity and completing QoL and medical resource utilisation (MRU)/Health Economics questionnaires.
-
-
If female and of child-bearing potential, must:
-
have a negative pregnancy test within 72 hours prior to randomisation, and should not be breastfeeding;
-
agree to use adequate, medically approved, contraceptive precautions (oral or barrier contraceptive) under the supervision of a general practitioner (GP) or Family Planning Clinic during and for 30 days after the last dose of sunitinib or pazopanib.
-
-
If male with a partner of child-bearing potential, must agree to use adequate, medically approved, contraceptive precautions (oral or barrier contraceptive) under the supervision of a GP or Family Planning Clinic during and for 30 days after the last dose of sunitinib or pazopanib.
-
Requirement to start first-line therapy with either sunitinib or pazopanib and decision already made as to which TKI to be used according to local standard practice.
Allowed situations included:
-
primary renal cancer in situ or previous nephrectomy;
-
previous brain metastases treated with complete surgical resection. Stereotactic brain radiation therapy (SBRT) or Gamma Knife with no subsequent evidence of progression (patients treated only with whole brain radiotherapy are not eligible);
-
previous radiotherapy and/or previous/ongoing bisphosphonates or bone antiresorptive drugs for the treatment of symptomatic bony metastasis. Care should be taken to follow dental guidelines for the antibone resorptive drug.
Exclusion criteria
Patients were excluded from participation if they met any of the following criteria:
-
Pulmonary or mediastinal disease causing obstruction or clinically significant bleeding/haemoptysis.
-
Patients with an estimated life expectancy of < 6 months.
-
Known contraindications to the particular TKI to be used (i.e. sunitinib or pazopanib).
-
Any previous treatment with sunitinib, pazopanib or other TKI (including in the adjuvant setting).
-
Untreated brain metastases60: patients were eligible if previous brain metastases were treated with complete surgical resection, SBRT or Gamma Knife with no subsequent evidence of progression. Patients were not eligible if brain metastases were treated only with whole-brain radiotherapy.
-
Any concurrent or previous other invasive cancer that could confuse diagnosis or end points. Allowed situations included (but not limited to): non-melanomatous skin cancer or superficial bladder cancer.
-
Hypersensitivity to the particular TKI to be used (i.e. sunitinib or pazopanib).
-
Any concomitant medication or substances forming part of local ongoing care known to significantly affect, or have the potential to significantly affect, the activity or pharmacokinetics of the particular TKI to be used (i.e. sunitinib or pazopanib).
-
Poorly controlled hypertension despite maximal medical therapy. 7 It was recommended that subjects should have a systolic BP of either < 150 mmHg, and/or a diastolic BP of < 90 mmHg. Antihypertensive drugs could be used to achieve these values.
-
Any other serious medical or psychiatric condition which in the opinion of the investigator could affect participation in the STAR trial, including gastrointestinal abnormalities limiting the effectiveness of orally administrated drugs, uncontrolled infections, current or recent history of clinically significant cardiovascular disease, significant haemorrhage or gastrointestinal perforation or fistula which, in the opinion of the local investigator, would render the patient unsuitable for standard sunitinib or pazopanib therapy.
Recruitment procedure
Patients were approached during routine oncology appointments and were provided with verbal and written details about the trial. The verbal explanation of the trial and the version of the participant information sheet (PIS) and consent form (CF) appropriate for the TKI recommended for use (sunitinib or pazopanib) were provided by the patient’s clinical team.
An optional DCE-MRI substudy was undertaken at St James University Hospital, Leeds, and participants approached at this site were also provided with an additional PIS and CF regarding the DCE-MRI substudy.
Following information provision, patients were given a minimum of 24 hours to consider trial participation. Assenting patients were then formally assessed for eligibility and invited to provide written informed consent.
Informed consent
Informed, written consent was obtained prior to randomisation into the study, subject to the patient meeting the eligibility criteria. A record of the consent process detailing the date of consent and all those present was kept in the participant’s notes. The original signed and dated CFs were held securely as part of the trial site file; copies were filed in the hospital notes (as per local practice) and copies were returned to the Clinical Trials Research Unit (CTRU).
Registration for DCE-MRI substudy participants
Patients participating in the DCE-MRI substudy were required to undergo a baseline DCE-MRI scan prior to the commencement of sunitinib or pazopanib treatment on the STAR trial. Given the narrow window specified between randomisation and commencement of sunitinib or pazopanib, baseline DCE-MRI substudy scans were scheduled prior to randomisation. Patients agreeing to participate in this substudy were therefore registered with the CTRU prior to their baseline DCE-MRI scan in order to confirm their eligibility for the main trial, consent and participation in the substudy.
Registration was carried out by the CTRU automated 24-hour telephone randomisation service. Participants were allocated a unique trial identification number at registration which was used at randomisation and throughout their study participation.
Randomisation
Randomisation took place as soon as possible after consent and confirmation of eligibility and was no more than 5 days prior to the start date of sunitinib or pazopanib treatment. The decision regarding the TKI (sunitinib or pazopanib) to be used was at the discretion of the treating clinician and was made prior to randomisation.
Randomisation was carried out by the CTRU automated 24-hour telephone randomisation service. Participants were randomised in a 1 : 1 ratio between the CCS and DFIS treatment arms.
A computer-generated minimisation programme that incorporated a random element was used to ensure that treatment groups are well balanced by:
-
Motzer/MSKCC (Memorial Sloan-Kettering Cancer Centre) prognostic group. 61
-
Favourable risk (0 factors).
-
Intermediate risk (1–2 factors).
-
Poor risk (≥ 3 factors).
-
-
Trial site.
-
Gender.
-
Age:
-
< 60 years.
-
≥ 60 years.
-
-
Disease status at the time of randomisation:
-
Metastatic.
-
Locally advanced.
-
Previous nephrectomy:
-
Yes.
-
No.
-
-
TKI:
-
Sunitinib.
-
Pazopanib.
-
Participants who had not been registered for the study were allocated a unique trial identification number at randomisation.
Participants, medical staff and clinical trial staff were informed of the allocated treatment arm. The treatment allocation was not blinded as accurate radiological evaluations were fundamental to the Stage B end point, and for this reason all radiological evaluations in the Phase II component of the study (Stages A and B) were performed centrally. Central reporting of radiological evaluations was not carried out in Phase III of the study.
Interventions
Participants received either pazopanib or sunitinib for at least 24 weeks (4 cycles) of treatment. The protocol defined treatment with S/P was as follows:
-
One cycle of sunitinib treatment: 50 mg (starting dose) on days 1–28, repeated every 42 days.
-
One cycle of pazopanib treatment: 800 mg (starting dose) on days 1–42, repeated every 42 days.
Participants were not permitted to change from sunitinib to pazopanib or vice versa after randomisation. If this was required, the participant was considered to have discontinued trial treatment.
After 4 cycles of treatment, participants took up their randomised treatment allocation:
-
Control arm: CCS
Participants continued on sunitinib or pazopanib with regular radiological assessments every 12 weeks until they met protocol-defined PD according to RECIST criteria, experienced unacceptable cumulative toxicity, decided to stop treatment or withdraw from the study.
-
Research arm: DFIS
Participants stopped treatment and continued 6-weekly active surveillance (clinical assessment) and 12-weekly radiological assessment. At the point of protocol-defined PD according to RECIST criteria, participants recommenced treatment with sunitinib or pazopanib for a minimum of 4 cycles. Assuming ongoing disease control, participants were permitted to take further treatment breaks following the same schedule. This DFIS (planned treatment-break strategy) was continued until either PD occurred during S/P treatment or the participant experienced cumulative toxicity or the participant decided to stop treatment or withdraw from the study.
Dose modifications were permitted for both sunitinib and pazopanib and were made according to local practice, with reductions occurring in 12.5 mg stages for sunitinib and in 200 mg stages for pazopanib. A maximum of two dose reductions were allowed in the trial. Participants requiring dose reduction to less than 25 mg/day sunitinib or to < 400 mg/day pazopanib (i.e. more than two dose reductions) were required to permanently stop trial treatment. However, this was not the case for non-haematological toxicities of greater than or equal to grade 3 (haemorrhage/bleeding/coagulopathy, venous thrombosis, fatigue, hand–foot syndrome), where a dose reduction of one level was recommended for all subsequent cycles.
Dose re-escalation following a dose reduction was permitted if considered appropriate by the treating investigator.
All participants continued with their allocated sunitinib or pazopanib treatment strategy as per protocol (PP) (with dose reductions as required):
-
until disease progression (RECIST) occurs while taking sunitinib or pazopanib; or until
-
unacceptable toxicity;
-
participant chooses to stop protocol treatment;
-
end of study follow-up.
In the very rare circumstance where there was substantial ongoing response during the treatment break in the DFIS arm, the latest best response scan [minimal sum of the longest diameters (SLD)] was used to define progression rather than the usual new baseline scan (the scan performed immediately prior to the commencing a treatment break), if this was clinically appropriate.
All participants permanently stopping protocol-defined treatment or prescribed alternative treatment continued to attend follow-up assessments as per the STAR protocol, unless consent was withdrawn. Participants were recorded as having reached the strategy failure end point but were continued to be followed up for QoL and survival.
After disease progression on sunitinib or pazopanib as PP (i.e. not on a treatment break), participants permanently stopped protocol-defined treatment but patients were permitted to begin further systemic therapy or supportive care, as considered appropriate.
Trial assessments
Baseline assessments
Participants were required to have cross-sectional imaging (chest, abdomen and pelvis were strongly recommended) within 42 days before the start of protocol treatment. A contrast CT scan (chest abdomen pelvis) was the preferred modality of cross-sectional imaging. If this was not possible (e.g. in the case of contrast allergy or renal insufficiency), then a non-contrast CT (chest abdomen pelvis) scan was performed, assuming the disease was evaluable by this method. If the disease was not evaluable using a non-contrast CT scan, a MRI scan of the abdomen and pelvis and a non-contrast CT scan of the chest were performed. All subsequent follow-up scans were required to be in the same modality (CT or MRI) and performed using the same technique.
Following informed consent and prior to randomisation (within 16 days prior to commencing trial treatment) patients were assessed with regard to medical history, physical examination (including height, weight, ECOG PS, vital signs, heart rate and BP), full blood count, biochemistry [urea and electrolytes (UE) including urea, creatinine, sodium and potassium], liver function tests (LFTs) including alkaline phosphatase (ALP), ALT/AST, total BR and albumin, LDH, thyroid function tests (TFTs), bone profile (calcium) for calculation of Motzer score and pregnancy test (if woman of child-bearing potential) within 72 hours prior to randomisation.
In addition, if a bone scan was carried out as part of standard local practice, this was performed in accordance with routine time frames but was not mandated by the protocol.
The baseline QoL (booklet A) (FACT-G and FSKI-15, EQ-5D-3L/EQ-VAS and MRU/Health Economics questionnaires) was completed prior to randomisation, as close as possible to commencement of treatment.
Treatment assessments
Irrespective of the allocated treatment arm (DFIS or CCS), participants were assessed clinically for symptoms and toxicity 6-weekly at the start of each treatment cycle.
The following assessments were conducted within 5 days prior to each treatment cycle and/or clinical review (while on planned treatment break for DFIS arm participants): clinical assessment including weight, ECOG PS and vital signs (HR and BP), AE reporting/toxicity assessment (CTCAE v.4.0), full blood count, UE, LFT and TFT (q 12 weeks).
More frequent monitoring of liver function was required for participants receiving pazopanib at timings recommended in the pazopanib Summary of Product Characteristics62 at weeks 3, 5, 7 and 9, then at months 3 and 4, with additional tests as clinically indicated. Periodic testing should then continue after month 4.
Imaging
Computerised tomography scan imaging was carried out prior to commencement of cycle 3 and every 12 weeks thereafter. During Phase II of the trial, central reporting of scans was performed to ensure consistency. During Phase III, scans were reported locally according to RECIST.
The timing of the radiological assessments was the same in both the CCS and DFIS arms. However, if the scan scheduling fell out of sync with the cycles of treatment (e.g. a delay due to toxicity or other medical reasons), the scan could be delayed by up to 4 weeks to allow the scan to coincide with the usual treatment cycles.
In the event of clinical evidence of disease progression at a time other than that when radiological reassessment is due, radiological assessments were performed to confirm progression, unless there was a compelling reason that this was not possible.
All scans performed during the treatment break were compared to the scan immediately before the treatment break started as per the standard RECIST reporting guidelines.
Follow-up
After permanent discontinuation of protocol treatment, participants were followed up until the end of the trial follow-up period. SAEs, serious adverse reactions (SARs) and suspected unexpected serious adverse reactions (SUSARs) collected for 30 days following the end of the trial follow-up period were included in the final analysis. If it was not possible for the participant to attend clinic at this time point, events were collected by telephone call if considered to be appropriate by the treating clinician.
Participants were seen in clinic 6 months after permanently discontinuing protocol treatment and annually thereafter until the end of follow-up on 31 December 2020. Details of the participant’s status and any subsequent treatment received for renal cancer were collected at the follow-up visits along with the EQ-5D-3L/EQ-VAS QoL questionnaire.
All randomised participants were followed up for survival unless consent was withdrawn for further data collection.
Quality of life
Information from all questionnaires (FACT-G, FSKI-15 and EQ-5D-3L/EQ-VAS) was collected at clinic visits at baseline (before the participant was informed of their randomisation allocation) and at day 1 of cycles 2, 3 and 4 during which time participants on both arms received sunitinib or pazopanib as clinically appropriate.
After cycle 4 (24 weeks post randomisation), the EQ-5D-3L/EQ-VAS questionnaires were collected every 2 weeks and were completed by participants at home. This intensive QoL collection continued until 48 weeks post randomisation and covered the 24-week period after participants had taken up their randomised treatment allocation (DFIS or CCS). After this point, questionnaires were once again collected in clinic on a 6-weekly basis.
The FSKI-15 and FACT-G questionnaires were collected every 6 weeks at clinical assessment visits for the duration of trial treatment.
The 2-weekly questionnaire completion was acknowledged to be a significant burden for participants; however, it was considered key to informing the QALY co-primary end point as differences between the treatment strategies were likely to be the greatest immediately following participants taking up their randomised treatment allocation.
In addition, 2-weekly QoL was considered relevant for participants receiving sunitinib as it was given over 28 days followed by a 14-day off-treatment period. In comparison, pazopanib was administered for the full 42 days of the 6-weekly treatment cycles.
In order to capture any differences in QALYs between the arms after treatment strategy failure, EQ-5D-3L/EQ-VAS information was collected for all participants (where possible) until the end of follow-up.
Due to the importance of QoL data in this trial, measures were taken to ensure maximum compliance of questionnaire completion. Participants consented to receive e-mail or text message reminders from the research team at CTRU (this was optional) over the 24-week period where participants were required to complete 2-weekly QoL questionnaires at home. Where a QoL questionnaire was missed at a hospital clinic visit, the local research team posted the questionnaire to the participant’s home (after checking the participant’s status to establish it was appropriate to do so).
Outcomes
Phase II
Stage A
The feasibility of performing the Phase III trial was assessed using the average recruitment rate. This was measured on a per-month and per-site basis to adjust for the increase in participating sites when moving on to Phase III.
The assessment of this outcome was to be over the 10th–21st months of recruitment, with the first 9 months of recruitment discounted to allow for site set-up. However, this assessment was delayed to allow for the implementation of protocol v4.0 and allow pazopanib into the trial. The final assessment was conducted from 6 weeks after the first site implemented the new protocol until the recruitment target of 210 was met or the 12-month period was reached.
Stage B
The efficacy of the strategies, during Phase II, was assessed using TSF. TSF was defined as the time from randomisation until:
-
death;
-
disease progression while on sunitinib or pazopanib;
-
disease progression with no disease response or stabilisation from subsequent sunitinib or pazopanib treatment;
-
participant required the use of a new systemic anticancer RCC treatment;
-
clinical deterioration, assumed to be due to renal cell (RC) progression, excluding any comorbidities, that is sufficient to warrant cessation of sunitinib or pazopanib treatment or precludes restarting treatment, if on the DFIS arm, without it being clinically appropriate to arrange a radiological confirmation of progression.
Phase III
Primary
The two co-primary end points for Phase III were OS and QALYs.
Overall survival was defined as the time from randomisation to death by any cause. Any participants lost to follow-up or still alive at the time of analysis were censored at the time at which they were last known to be alive.
Quality-adjusted life-years is a measure that considers both survival and QoL. In STAR, the QALYs for each participant were determined by the area under the curve (AUC) of the utility scores from the EQ-5D-3L questionnaire, assuming a linear change in utility in the time between questionnaires. These questionnaires were collected at baseline every 2 weeks for 24 weeks and every 6 weeks thereafter. The questionnaires were also collected until the end of follow-up, with one 6 months after the end of treatment and annually thereafter. Any participant who died prior to analysis was treated as if they had scored zero in their subsequent utility scores as per the EQ-5D-3L scoring manual. 63
Secondary
The trial also included several secondary outcomes.
Note that throughout this section, disease progression refers to both radiological and clinical progression. If disease progression is defined radiologically (RECIST), then the date of progression was taken as the date of the scan which concluded PD. However, in the rare circumstance that disease progression was determined clinically, due to global deterioration in clinical status attributable to disease progression in the view of the investigator, then the date of progression will be defined as the date of stopping treatment due to clinical suspicion of disease progression.
Time to strategy failure
Time to strategy failure is defined as the time from randomisation until the first occurrence of one of the following events:64
-
death;
-
disease progression while on treatment;
-
disease progression assuming no further disease response or stabilisation occurs in the DFIS arm;
-
participant required the use of a new systematic anticancer agent for RCC (end point measured at the first of either time of disease progression or time of initiation of new agent).
If an individual never started treatment following randomisation, they were classed as having an event at time zero.
Individuals who stopped trial treatment and did not experience one of the events above during follow-up were censored at the date they were last assessed during follow-up or, if applicable, the date they withdrew from trial follow-up.
Any individual still on trial at the time of analysis was censored at their last scan which confirmed that they were still responding to the treatment strategy (alive and progression-free). In the event that a DFIS participant’s last scan resulted in the decision to ‘restart trial treatment’ following progression while on a break and there were no further scans which confirmed further response, their end point was censored at the scan date.
In the event that an individual came off trial treatment due to toxicity and was not followed up for 6 months following the end of their study treatment, they were censored at their date of last dose.
A flow diagram of TSF can be found in Report Supplementary Material 1, Figure 1.
Time to treatment failure
Time to treatment failure was defined as the time from randomisation until permanent protocol-based treatment discontinuation for any reason64 (including toxicity, withdrawal, death or progression on trial provided there is no further response in the DFIS arm). If an individual stopped trial treatment due to withdrawal, their event was taken at the latest date from their date last dose of sunitinib or pazopanib and date of withdrawal of trial treatment. An individual was censored at their last on-study assessment date if they were still on trial treatment at the time of the analysis. If an individual never started treatment following randomisation they were classed as having an event at time zero.
The following rules were also applied to DFIS participants:
-
If an individual was on a treatment break at the time of the final analysis, then the end point was censored at the scan date which confirmed that they should continue on their current break.
-
If an individual had been told to restart trial treatment according to the scan form but the treatment data are missing, then they were censored at the scan date which resulted in the decision to restart treatment.
A flow diagram of TTF can be found in Report Supplementary Material 1, Figure 2.
Progression-free survival
Progression-free survival was defined as the time from randomisation to first disease progression (irrespective of future disease stabilisation in the DFIS arm) or death from any cause. PFS for participants who came off trial treatment without experiencing a progression was measured up until their first record of disease progression off treatment, as recorded on the follow-up forms.
Participants who had not progressed or died at the time of analysis were censored at the last date they were known to be alive and progression-free.
A flow diagram of PFS can be found in Report Supplementary Material 1, Figure 3.
Summative progression-free interval
Summative progression-free interval was defined as the sum of the intervals during which the participant was defined to be progression-free, allowing for participants in the DFIS to respond to the trial treatment following a progression on a treatment break.
For the CCS arm, this was defined as the time from randomisation to the first documented evidence of disease progression. For the DFIS arm, the first interval was defined the same way as for the CCS arm. Subsequent intervals were defined as the time from the date of the CT scan that provided evidence of disease control (SD, PR or CR) to disease progression. SPFI was then calculated as the sum of these intervals.
If a participant permanently came off trial treatment for reasons other than progression (e.g. toxicity/withdrawal), their (current) progression-free interval was measured up until their first record of disease progression off treatment or the start of another systemic anticancer therapy as recorded on the follow-up forms. If a DFIS participant did not respond to the treatment following progression on a treatment break, their (current) interval was measured up to the progression date which required them to restart treatment.
Participants who had not progressed at the time of analysis were censored at the last date they were known to be alive and progression-free. Participants in the DFIS arm who had progressed on a treatment break but had yet to be assessed for response were censored at the date of their progression which required them to go back to treatment.
Note that if a participant died during an interval, then their current interval stopped at their date of death and their SPFI was censored.
A flow diagram of SPFI can be found in Report Supplementary Material 1, Figure 4 for the CCS arm and Report Supplementary Material 1, Figure 5 for the DFIS arm.
Toxicity
Toxicity was measured through the collection of AEs, SAEs, SARs and SUSARs.
Adverse events were collected regularly on the 6-weekly on-study case report forms (CRFs). These specifically looked for any occurrences of pre-specified AEs of interest, for example, hypotension and fatigue. The AEs were graded according to the CTCAE V4 seriousness criteria, which led to the identification of SAEs, SARs and SUSARs. AEs were reported from the start of treatment until 30 days post permanent end of treatment.
Serious adverse events, SARs and SUSARs had their own expedited reporting process and were reported from the start of the treatment until either 30 days post permanent end of treatment (SAEs) or the end of follow-up (i.e. the end of the trial) (SARs and SUSARs).
Quality of life
Quality of life was measured with the EQ-5D-3L, EQ-VAS, FACT-G and FSKI-15 questionnaires. All questionnaires were completed at the baselines and on weeks 6, 12 and 18. From week 24, the EQ-5D-3L and EQ-VAS were completed every 2 weeks and FACT-G and FSKI-15 completed every 6 weeks, for 24 weeks. For the rest of the treatment, all questionnaires were collected every 6 weeks. EQ-5D-3L and EQ-VAS were collected during follow-up with one completion at 6 months from cessation of protocol treatments and then annually thereafter. Questionnaires were scored according to their scoring criteria to give overall scores [EQ-5D-3L Utility index, FACT-G Overall Score, Functional Assessment of Cancer Therapy – Kidney Symptom Index (FKSI-15) Overall Score] and the corresponding subscales (FKSI-DRS, FACT-G Physical well-being, FACT-G Social/Family well-being, FACT-G emotional well-being, FACT-G functional well-being). 63,65–69
Cost-effectiveness was evaluated using the QALY outcome, healthcare resource use and treatment costs and is fully defined in Chapter 4.
Sample size
Phase II
Stage A
A formal power calculation was not conducted for the Stage A interim analysis. However, it was pre-specified that the number of ‘full’ and ‘half’ sites open to recruitment during the entire 12-month period would be determined by the TMG and that the expected rate of recruitment was one participant per month per full site over the 12-month period.
Stage B
The Stage B analysis required 210 participants. This value was based off simulations. The simulations considered the Stage B primary endpoint TSF. Four key assumptions were applied within the calculations. Firstly, it was assumed that a difference of ≤ 15% in TSF at 15 months between the two arms was an acceptable NI margin (equivalent to a HR of 0.540). Secondly, it was assumed, using the literature that at 15 months the probability of TSF in the CCS arm was 0.8. 5 Thirdly it was assumed that TSF would meet the proportional hazards (PH) assumption. Finally, it was assumed that 47.5% of participants would take up their randomisation allocation at week 24. The simulations required 97 participants to take up their randomisation at week 24 for theinterim analysis to have 80% power to detect NI, with a one-sided 2.5% significance level. Therefore, using the assumption that 47.5% of participants would take up their allocation a minimum of 210 participants were required to be randomised so that at least 97 remained at week 24.
Evaluating this end point required the pooling of both the sunitinib and pazopanib data, shortly after pazopanib had been added to the trial. As such, it was pre-specified in protocol version 4 that a minimum of 80 participants should be receiving pazopanib on the trial. This would give an approximate 3 : 2 split of sunitinib to pazopanib participants, which was deemed to be sufficient ratio in order to have a sufficient amount of data on the pazopanib participants (assuming approximately 45–50% of participants reach/take up their randomisation allocation at 6 months) and to have confidence in the combined results from the Stage B analysis. Should the number of participants receiving pazopanib on the trial be < 80 by the end of Stages A and B, recruitment was to continue until 80 participants were receiving pazopanib.
Phase III
Original sample size
The sample size was originally estimated to be 1000 participants, allowing for a loss to follow-up of 10%. This was determined using the OS outcome, with the aim of 80% power at a 2.5% significance level for NI. A difference of < 7.5%, between DFIS and CCS, was assumed to be the NI margin and the probability of surviving o 2 years on CCS was assumed to be 54%. This suggested that the number of events should be at least 665, requiring 4.5 years of recruitment and 2 years of follow-up.
For the QALY outcome, a difference of < 10% was deemed suitable to show NI between the two strategies. From simulations, the mean QALY for CCS participants was estimated at 1.3436 years. A HR of 0.9 was assumed in favour of CCS over DFIS, which was used in simulations alongside the sample size of 1000, with 4.5 years to recruitment and 2 years of follow-up to give a power estimate of 85.22%. There were also additional assumptions used in the simulations. The median PFS in the CCS was assumed to be 11 months during treatment, and 7.2 months during follow-up. It was also assumed that 31.9% of participants would die at disease progression. The mean (SD) of the QoL utilities was assumed to be 0.570 (0.210) and 0.680 (0.190) while on treatment and off treatment, respectively. This choice was informed by the results of the Japanese trial. Finally, the duration of the second and any subsequent treatment intervals in the DFIS arm was assumed to be 6 months.
Final sample size
In February 2017, at the recommendation of the DMEC and TSC, the assumed dropout rate was reduced from 10% to 5% in light of the observed dropout rate of 2%. This gave a new sample size of 920 participants. This was again determined using the OS outcome, with the aim of 80% power at a 2.5% significance level for NI. Additionally, the recruitment period was adjusted to allow for pazopanib’s inclusion and the survival rate of participants on CCS was lowered from 54% to 48.5% using a model-based approach. With the NI margin of < 7.5%, this suggested that the number of events should be at least 720, requiring 5.83 years of recruitment and 2 years of follow-up.
The new sample size and times, along with the original NI margin and assumed HR, gave a CCS QALY estimate of 1.4156 and a power of 77.63%.
A second update to the estimates of the power of the QALY outcome adjusted for the 15-month extension to the trial. Simulations gave a new estimate of the CCS QALY of 1.56 years. Along with a NI margin of < 10%, an assumed 0.9 HR in favour of CCS, a sample size of 920, 5.83 years of recruitment and 3.25 years of follow-up this gave a power of 69.94%. The additional assumptions remained the same as the original estimate.
Statistical methods
Before any statistical analyses were undertaken, a full statistical analysis plan (SAP) was written by the Leeds CTRU STAR trial statistician and agreed upon by the: supervising statistician; Chief Investigator; CTRU principal Investigator and Senior Trial Co-ordinator.
All analysis described here was conducted using SAS 9.4.
Analysis populations
The analysis required the following populations: ITT, PP, safety, EQ5D, FKSI and FACT-G. All randomised participants were considered for inclusion in each population. Participants were excluded from the ITT population if they did not have RCC. Participants were excluded from the PP population if they were deemed to be major protocol violators by the TMG or they reached 6 months post randomisation but do not take up their randomisation allocation. Note that participants in Phase II were required to reach maximum radiological response (MRR) prior to taking up their first treatment break. Any participants in the Phase II part of the trial who did not go on a treatment break because they did not achieve MRR will not be excluded from this population. Participants were excluded from the safety population if they did not receive any of their protocol treatment (sunitinib or pazopanib). Participants were excluded from the QoL populations (EQ5D, FKSI, FACT-G) if their baseline questionnaire could not be scored. With the exception of the safety population, all analysis was conducted according to what participants were randomised to receive. For the safety population if a participant in the DFIS arm declined or did not take up a treatment break in error at 6 months post randomisation and it is not rectified by their next scan they were summarised and included in the CCS arm.
Interim analyses
Stage A
The average monthly recruitment rate was estimated across those recruited within the formal monitoring period, 1 June 2013 to 31 May 2014. The recruitment rate was calculated per month per whole trial site open, to account more sites to be added in Phase III. The number of whole sites was weighted by the populations of each site's catchment areas and adjusted for sites that were not open for the entire duration of the monitoring period. The estimate was compared to the desired recruitment, rate of one patient per trial site per month, using a 95% CI, calculated based upon the number of open sites. To demonstrate the feasibility of recruitment it was pre-specified that the observed rate should be greater than the lower bound of the CI.
Stage B
For Stage B of the interim analysis, the number and proportion of TSF events were summarised overall and by randomisation allocation along with the reasons for the events. TSF survival curves with median survival and corresponding 95% CIs were plotted using the Kaplan–Meier method. The log-rank test, and adjusted log-rank test, was used to compare differences between the treatment strategy arms (DFIS vs. CCS). However, the analysis of primacy was a Cox regression analysis accounting for the minimisation factors of the trial, except for randomising centre. If the PH assumption was not met it was pre-specified that other forms of analysis would be considered.
Non-inferiority between the two treatment arms (DFIS vs. CCS) was to be concluded if the lower bound of the two-sided 95% CI around the HR for the treatment covariate was ≥ 0.54. This corresponded to a ≤ 15% difference in TSF between the two strategies at 15 months. Figure 1 summarises the interpretation of NI conclusions for TSF.
FIGURE 1.
Non-inferiority of TSF.
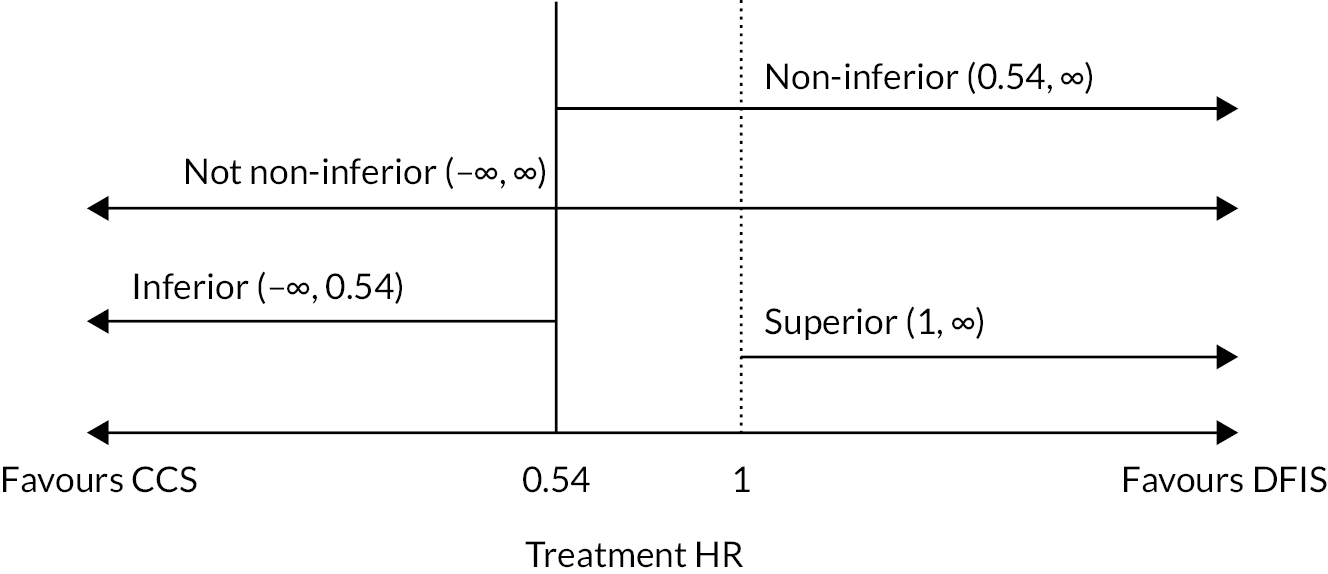
As there was no current evidence of a similar efficacy between the two TKIs, a 60% CI was calculated around the HR for the TSF point estimate of the sunitinib participants in the ITT population. It was pre-specified that if the HR for the TSF point estimate for the pazopanib participants lay within this CI, and there are no obvious indications of their differences after evaluating all clinical information, then the treatments would be concluded to be similar enough for the data to be pooled to evaluate the Stage B end point.
A pre-planned summary of the utility data also took place at the end of Phase II. Mean and standard errors for the EQ-5D-3L utility estimate were derived for the on and off treatment periods and examined to determine whether the estimates used in the simulations for QALYs were suitable.
Descriptive analysis
Key baseline characteristics [minimisation factors of the trial (excluding centre), age, gender, ethnicity, PS, haemoglobin, neutrophils, platelets, calcium, LDH bone involvement and time since diagnosis] were compared between randomisation arms in all analysis populations. In addition, the treatment participants received on trial, along with the results of their on-study assessments, and the treatment they received in follow-up were summarised between randomisation arms in the ITT population.
Co-primary end-point analysis
As both primary outcomes were to assess NI, these analyses were conducted in both the PP and ITT populations. If DFIS could show NI in both OS and QALY in both populations, then the analysis would conclude that DFIS was non-inferior to CCS.
Overall survival
The number and proportion of participants who died at the time of analysis were summarised along with the causes both by randomisation allocation and by TKI received. OS curves with median survival and corresponding 95% CIs were plotted using the Kaplan–Meier method, and OS estimates (i.e. the proportion of participants alive) at each year following randomisation were presented for each treatment arm, along with their corresponding 95% CIs.
Cox regression analysis was used to formally compare OS between the treatment arms, accounting for the minimisation factors, except for randomising centre. The PH assumption was assessed using the supremum test where if violated it was pre-specified that additional analysis methods would be implemented.
Non-inferiority between the two treatment arms (DFIS vs. CCS) was to be concluded if there was a ≤ 7.5% difference in OS between the two strategies in both the ITT and PP populations. Using the assumption that survival at 2 years was 48.5% in the CCS arm (see Phase III), to conclude NI survival in the DFIS arm was required to be at least 41%, leading to a HR of 0.812. Therefore, NI was to be concluded at the 2.5% significance level if the lower bound of the two-sided 95% CI around the HR for the treatment covariate was ≥ 0.812.
Figure 2 summarises the interpretation of NI conclusions for OS.
FIGURE 2.
Non-inferiority of OS.
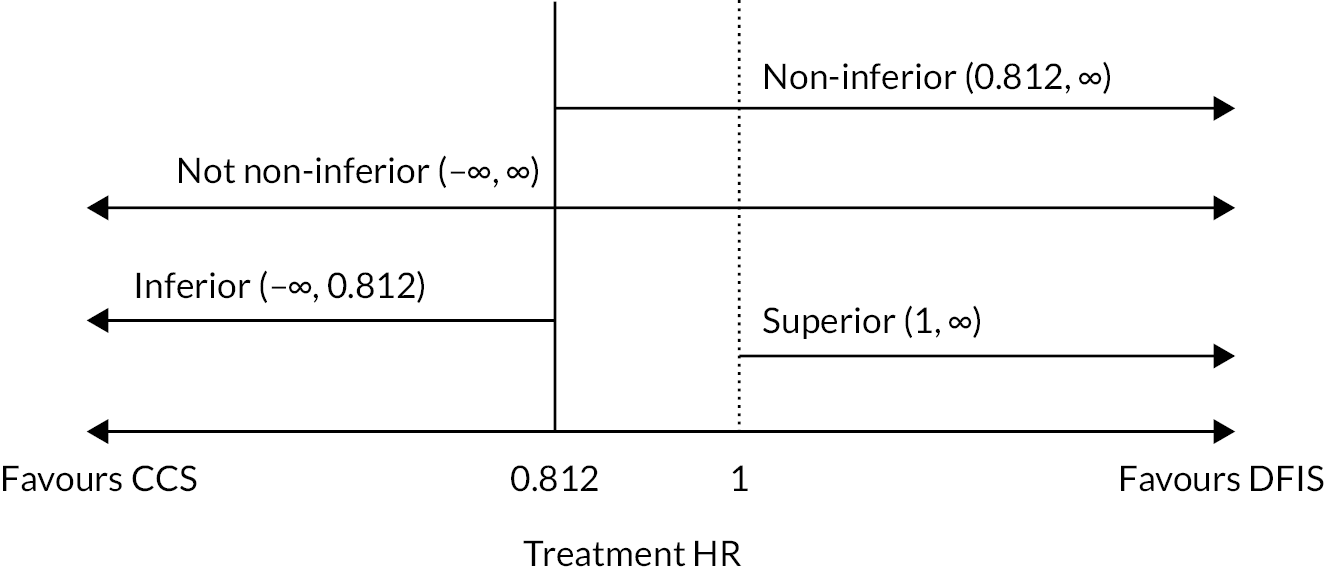
Quality-adjusted life-years
Summary statistics [mean, 95% CI, median, interquartile range (IQR)] for the QALYs were calculated using imputed data. A finite mixture model (FMM) with two components was applied to the QALYs adjusted for the minimisation factors of the trial, excluding trial site, and the results marginalised to give a point estimate for randomisation allocation in to assess the NI conclusion. The appropriateness of the FMM was assessed by investigating whether the knowledge of a participants ‘component’ improved the fit of a multivariable linear regression model. 70
Non-inferiority between the two arms was to be concluded if the regression coefficient, derived from the primary analysis results, for treatment allocation corresponded to a ≤ 10% difference in mean QALYs between the two strategies in both ITT and PP populations. Using the assumption that mean QALYs in the CCS arm was 0.156 (see Phase III), this equated to the lower bound of the two-sided 95% CI around the treatment covariate being ≥ −0.156.
Figure 3 summarises the NI conclusions for the QALYs end point.
FIGURE 3.
Non-inferiority of QALYs.

Secondary end-point analysis
For time to event secondary end points (TSF, TTF, PFS and SPFI), analysis was conducted on the ITT population. The number and proportion of events at the time of analysis were summarised both by randomisation allocation and by TKI received. Time-to-event curves with median survival and corresponding 95% CIs were plotted using the Kaplan–Meier method, and event estimates (i.e. the proportion of participants event-free) at each year following randomisation were presented for each treatment arm, along with their corresponding 95% CIs.
Cox regression analysis was used to formally compare the end points between the treatment arms, accounting for the minimisation factors, except for randomising centre. The PH assumption was assessed using the supremum test, where, if violated, it was pre-specified that additional analysis methods would be implemented.
The safety analysis summarised the observed AEs, SAEs, SARs and SUSARs for the safety population. Summaries included overall statistics as well as by treatment strategy (DFIS or CCS) and TKI treatment (sunitinib or pazopanib). The confirmed cases of osteonecrosis of the jaw (ONJ) and any notifications of trial participant pregnancies or their partners were also reported.
Each QoL measure (total FKSI, FKSI Disease-Related Subscale, FACT-G total and subscales, EQ-5D-3L index and EQ-VAS) was summarised using the median and IQR within the relevant QoL population at each possible time point. Additional analysis was conducted for all of the measures except the EQ-5D-3L index and the EQ-5D-3L VAS. For the additional analysis, the measure was considered in a multilevel repeated measures model. Time, treatment strategy, baseline QoL and an interaction for treatment strategy by time were included as fixed effects with participant and participant by time included as random effects. The model was also adjusted for the minimisation factors of the trial, excluding the centre. Note that time, measured in 6-weekly intervals, was treated as continuous.
Ancillary analysis
For OS the following ancillary analysis was conducted:
-
A piecewise hazards model was applied to account for participants in both arms being treated the same for the first 6 months of the trial, provided the piecewise hazards model is not used within the analysis of primacy.
-
The analysis described in the section Co-primary end-point analysis was repeated where the Motzer stratification factor was combined into two groups rather than three.
For QALYs the following ancillary analysis was conducted:
-
The analysis described in the section Co-primary end-point analysis was repeated where QALYs were measures from week 24, the point where participants were due to take up their randomisation allocation.
-
The analysis described in the section Co-primary end-point analysis was repeated where QALYs were measured up to:
-
12 months post randomisation.
-
24 months post randomisation.
-
36 months post randomisation.
-
-
A multivariable linear regression model was applied on the imputed data.
-
The analysis described in the section Co-primary end-point analysis was repeated using only complete case data.
-
The analysis described in the section Co-primary end-point analysis was repeated on data where observations thought to be missing not at random (MNAR), defined to be those with the reason for missing ‘Inappropriate to give to participant/participant too unwell’, were imputed to be the worst health state (−0.59).
For both PFS and TTF, a piecewise hazards model was considered with both two (splitting at week 24) and three intervals (splitting at week 24 and approximately week 42, the end of the average length of a treatment break).
In addition, for TTF, an ancillary analysis, the same as described in the section Secondary end-point analysis, was conducted which derived TTF, excluding the time spent on a treatment break in the DFIS arm. For the CCS arm, this was the same as TTF as defined above. For this DFIS arm, this was calculated as the TTF defined above, minus the sum of the treatment breaks. The duration of a treatment break was defined as the time between the expected end date of the last cycle prior to the break (cycle start date + 42 days) and the start of the next treatment cycle following the treatment break. For this sensitivity analysis, if a participant was still on a treatment break at the time of analysis or had been told to restart trial treatment following a break but the treatment information was outstanding, then their end point was censored at the scan date which told them to start their current treatment break.
For the FKSI-15 and FACT-G questionnaires, the analysis described in the section Secondary end-point analysis was repeated where QoL was measured up to:
-
12 months post randomisation.
-
24 months post randomisation.
-
36 months post randomisation.
Subgroup analysis
Exploratory subgroup analysis was conducted for both co-primary end points within the PP population using the correct stratification factors. The subgroups investigated were:
-
Body mass index (BMI) as recorded at baseline (underweight or normal < 25, overweight or obese ≥ 25).
-
Comorbidities as recorded at baseline (0, 1, 2 or more).
-
Age at randomisation (> 70 or ≤ 70 and > 75 or ≤ 75).
-
Bone involvement as recorded at baseline (Yes/No).
-
Liver metastases as recorded at baseline (Yes/No).
-
IMDC score (Favourable, Intermediate, Poor).
-
The minimisation factors of the trial.
For OS the subgroup analysis considered the number of participants who died within each subgroup category as well as the 2-year OS estimate and 95% CI using the Kaplan–Meier method. The analysis was then extended to consider a Cox regression model similar to that described within the section Co-primary end-point analysis with the addition of a term for the subgroup being investigated (except for when the minimisation factor is the subgroup under consideration) and an appropriate interaction term. Heterogeneity was determined by a likelihood ratio test on the inclusion of the interaction term.
For QALYs the mean and 95% CI of QALYs, calculated over the course of the trial, for participants in each subgroup category will be calculated by randomisation using the imputed data. The difference in means QALYs between the randomisation arms was also calculated.
Missing data
Missing data were considered for the EQ-5D-3L utility index due to its involvement in the derivation of QALYs. However, due to the extensive collection of questionnaire data, missing data were only considered to be an issue during follow-up when the questionnaire collection is more infrequent. However, if a baseline questionnaire was missing, it was imputed using mean imputation. 71
The pattern of missingness was investigated along with the key baseline demographics of those with and without questionnaires at each follow-up time point. The distribution of the utility index was plotted during follow-up and imputed using predictive mean matching in a multiple imputation by chained equations framework. 72–74 The imputation model included:
-
Randomisation allocation.
-
The observed/imputed EQ-5D-3L utility score at the previous follow-up time points.
-
The value of the utility score at the participant’s last on-study review (F05/a) at which the EQ-5D-3L questionnaire was completed.
-
The minimisation factors of the trial (excluding randomising centre).
The number of imputed data sets was selected to be the maximum percentage of missing questionnaires across the time points which were being imputed, where imputation was conducted in time order and followed the following rules:
-
If a participant had withdrawn from QoL completion or was lost to follow-up, then questionnaires were imputed up to the time they were last known to be alive.
-
If a participant was not lost to follow-up, missing questionnaires were imputed for the entire follow up period or until death.
-
Only participants still alive at the relevant time point were included in each imputation model.
Trace plots were used to investigate how well the imputation converged.
Chapter 3 Trial results
This section details the final analysis results; the interim analysis concluded that the trial should proceed to Phase III. A summary of the interim analysis results which lead to the conclusion to continue to Phase III is included in Appendix 4, Tables 36–41 and Appendix 4, Figures 24–25.
Participant flow
Clinical pathway
The trial recruited participants between 13 January 2012 and 12 September 2017. Follow-up continued until 31 December 2020. The Consolidated Standards of Reporting Trials (CONSORT) diagram showing how participants flowed through the clinical aspect of the study is shown in Figure 4. Overall, 2197 potential participants were screened for trial entry with 920 (461 CCS, 459 DFIS) participants being randomised.
FIGURE 4.
Consolidated Standards of Reporting Trials diagram for the STAR Trial. a, Indicates the number of participants who received each TKI rather than the number who were randomised under each TKI. b, Indicates that the reasons given are none mutually exclusive.

During the course of the trial, 878 (95.4%, n = 920) [CCS: 453 (98.3%, n = 461), DFIS: 425 (92.6%, n = 459)] participants discontinued their trial treatment prior to the end of trial follow-up (31 December 2020). This equates to 42 participants (4.6%%, n = 920) [CCS: 8 (2.7%, n = 461), DFIS: 34 (7.4%, n = 459)] still receiving trial treatment at the end of the trial. Of these, 432 participants discontinued prior to week 24. This was the case for a similar proportion of participants in both arms [CCS: 221 (47.9%, n = 461): DFIS 211 (46.0%, n = 459)]. For the majority of participants, radiological disease progression played a role in them discontinuing trial treatment (see Report Supplementary Material 1, Table 1). This was the case for a similar proportion of participants in both arms prior to week 24 (CCS: 35.2%, DFIS: 37.1%). However, unsurprisingly, post week 24 the CCS arm had a higher proportion of participants stopping treatment due to radiological progression (CCS: 60.7%, DFIS: 40.7%), whereas the DFIS arm had a higher proportion of cessation due to death (CCS: 0.7%, DFIS: 8%) and clinical lead withdrawal (CCS: 3.3%, DFIS: 9.1%). The other reasons for discontinuing trial strategy are listed in Report Supplementary Material 1, Table 2. Of those who continued past week 24, 52.9% (n = 488) were randomised under the Motzer/MSKCC prognostic group of intermediate or poor risk.
Quality of life
Figure 5 displays how participants moved through the QoL study, the number of expected questionnaires and the number returned at a questionnaire booklet level. Recall that Booklet A was due at baseline, Booklet B at weeks 6, 12 and 18 from randomisation, Booklet C at weeks 24, 30, 36 and 42, Booklet D at 2-weekly intervals between weeks 24 and 46, Booklet E at 6-weekly intervals from week 48 while the participant was still on treatment and Booklet F at 6 months following the end of trial treatment and annually thereafter. Time point-specific information can be found in Report Supplementary Material 1, Table 3. The reasons for missing questionnaires by booklet are shown in Table 1. The other reasons for missing questionnaires include completed on an incorrect date, destroyed by site, misplaced by site English-language barriers.
FIGURE 5.
Overview of how participants moved through the QoL aspect of the STAR trial.
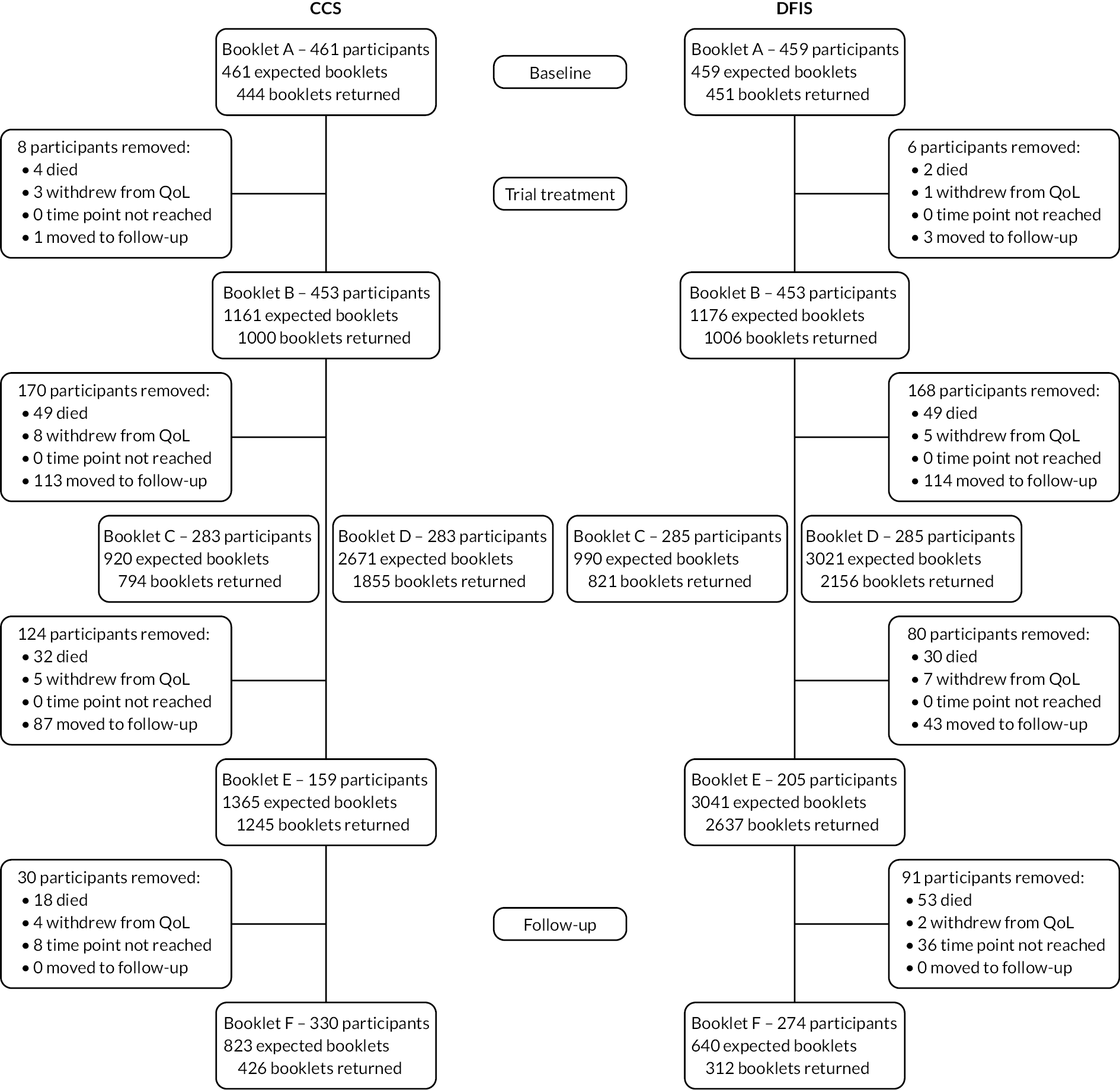
| Booklet A | Booklet B | Booklet C | Booklet D | Booklet E | Booklet F | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCS | DFIS | Total | CCS | DFIS | Total | CCS | DFIS | Total | CCS | DFIS | Total | CCS | DFIS | Total | CCS | DFIS | Total | |
| Inappropriate to give to participant/participant too unwell | 0 (0.00) | 0 (0.00) | 0 (0.00) | 11 (6.83) | 23 (13.53) | 34 (10.27) | 11 (8.73) | 5 (2.96) | 16 (5.42) | 23 (2.82) | 1 (0.12) | 24 (1.43) | 5 (4.17) | 11 (2.72) | 16 (3.05) | 37 (9.32) | 36 (10.98) | 73 (10.07) |
| Missed by site in error | 4 (23.53) | 2 (25.00) | 6 (24.00) | 116 (72.05) | 103 (60.59) | 219 (66.16) | 61 (48.41) | 92 (54.44) | 153 (51.86) | 165 (20.22) | 134 (15.49) | 299 (17.79) | 44 (36.67) | 122 (30.20) | 166 (31.68) | 75 (18.89) | 49 (14.94) | 124 (17.10) |
| Participant refused to or did not complete | 3 (17.65) | 1 (12.50) | 4 (16.00) | 17 (10.56) | 8 (4.71) | 25 (7.55) | 13 (10.32) | 20 (11.83) | 33 (11.19) | 45 (5.51) | 47 (5.43) | 92 (5.47) | 14 (11.67) | 61 (15.10) | 75 (14.31) | 15 (3.78) | 11 (3.35) | 26 (3.59) |
| Participant died before follow-up visit | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 3 (0.91) | 3 (0.41) |
| Not known if booklet given | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 12 (3.02) | 8 (2.44) | 20 (2.76) |
| Participant not seen in clinic (before posting of questionnaires) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 16 (4.03) | 4 (1.22) | 20 (2.76) |
| Participant not seen in clinic | 0 (0.00) | 0 (0.00) | 0 (0.00) | 1 (0.62) | 6 (3.53) | 7 (2.11) | 0 (0.00) | 7 (4.14) | 7 (2.37) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 16 (3.96) | 16 (3.05) | 2 (0.50) | 0 (0.00) | 2 (0.28) |
| Reason unknown | 2 (11.76) | 1 (12.50) | 3 (12.00) | 3 (1.86) | 5 (2.94) | 8 (2.42) | 9 (7.14) | 10 (5.92) | 19 (6.44) | 49 (6.00) | 69 (7.98) | 118 (7.02) | 3 (2.50) | 12 (2.97) | 15 (2.86) | 30 (7.56) | 12 (3.66) | 42 (5.79) |
| Missed due to COVID-19 | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 16 (3.96) | 16 (3.05) | 4 (1.01) | 4 (1.22) | 8 (1.10) |
| Completed and posted but never received at CTRU | 1 (5.88) | 0 (0.00) | 1 (4.00) | 0 (0.00) | 3 (1.76) | 3 (0.91) | 1 (0.79) | 0 (0.00) | 1 (0.34) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 4 (1.01) | 3 (0.91) | 7 (0.97) |
| Other | 1 (5.88) | 0 (0.00) | 1 (4.00) | 1 (0.62) | 1 (0.59) | 2 (0.60) | 2 (1.59) | 0 (0.00) | 2 (0.68) | 8 (0.98) | 0 (0.00) | 8 (0.48) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| Missing | 6 (35.29) | 4 (50.00) | 10 (40.00) | 12 (7.45) | 21 (12.35) | 33 (9.97) | 29 (23.02) | 35 (20.71) | 64 (21.69) | 526 (64.46) | 614 (70.98) | 1140 (67.82) | 54 (45.00) | 166 (41.09) | 220 (41.98) | 202 (50.88) | 198 (60.37) | 400 (55.17) |
| Total | 17 | 8 | 25 | 161 | 170 | 331 | 126 | 169 | 295 | 816 | 865 | 1681 | 120 | 404 | 524 | 397 | 328 | 725 |
Withdrawal
In total, 63 participants withdrew from some aspect of the trial across 64 occurrences (see Report Supplementary Material 1, Table 4). Report Supplementary Material 1, Figure 6 shows how participants could withdraw consent from different aspects of the trial. The majority of instances withdrew from QoL (71.9%), a higher proportion in the CCS arm (84.4%) than the DFIS arm (59.4%). Of the 28 (43.8%) who withdrew from trial follow-up, only 7 of them allowed data to be collected at standard visits. This resulted in 21 (2.82%, n = 920) participants being formally lost to follow-up. An additional one participant in the DFIS arm was lost to follow-up due to the participant moving away but this was not formally recorded. However, participants’ long-term follow-ups were included in all analyses. If recorded, the reasons for withdrawal are given in Report Supplementary Material 1, Table 5.
Protocol violations
In total, 76 protocol violations were observed from 71 participants. The majority of protocol violations (65.8%) were due to a breach in eligibility criteria (see Report Supplementary Material 1, Table 6). The eligibility criteria breached are shown in Report Supplementary Material 1, Table 7.
Baseline data
The key demographic and disease-related characteristics for each of the populations by randomisation allocation are presented in Table 2 for the ITT and PP populations. This information is presented for the remaining analysis populations in Report Supplementary Material 1, Tables 8–11. The same information is presented by randomised under TKI for the ITT and PP populations only in Report Supplementary Material 1, Tables 12 and 13.
| ITT population | PP population | |||||
|---|---|---|---|---|---|---|
| CCS (n = 461) |
DFIS (n = 458) |
Total (n = 919) |
CCS (n = 453) |
DFIS (n = 418) |
Total (n = 871) |
|
| Ethnic origin | ||||||
| White | 445 (96.5%) | 440 (96.1%) | 885 (96.3%) | 438 (96.7%) | 402 (96.2%) | 840 (96.4%) |
| Mixed – white and black Caribbean | 1 (0.2%) | 0 (0.0%) | 1 (0.1%) | 1 (0.2%) | 0 (0.0%) | 1 (0.1%) |
| Other mixed background | 2 (0.4%) | 0 (0.0%) | 2 (0.2%) | 2 (0.4%) | 0 (0.0%) | 2 (0.2%) |
| Asian – Indian | 3 (0.7%) | 2 (0.4%) | 5 (0.5%) | 3 (0.7%) | 2 (0.5%) | 5 (0.6%) |
| Asian – Pakistani | 2 (0.4%) | 2 (0.4%) | 4 (0.4%) | 2 (0.4%) | 2 (0.5%) | 4 (0.5%) |
| Other Asian background | 0 (0.0%) | 1 (0.2%) | 1 (0.1%) | 0 (0.0%) | 1 (0.2%) | 1 (0.1%) |
| Black – Caribbean | 2 (0.4%) | 1 (0.2%) | 3 (0.3%) | 2 (0.4%) | 1 (0.2%) | 3 (0.3%) |
| Black – African | 1 (0.2%) | 1 (0.2%) | 2 (0.2%) | 0 (0.0%) | 1 (0.2%) | 1 (0.1%) |
| Other black background | 1 (0.2%) | 0 (0.0%) | 1 (0.1%) | 1 (0.2%) | 0 (0.0%) | 1 (0.1%) |
| Other ethnic group | 2 (0.4%) | 2 (0.4%) | 4 (0.4%) | 2 (0.4%) | 2 (0.5%) | 4 (0.5%) |
| Not stated | 2 (0.4%) | 9 (2.0%) | 11 (1.2%) | 2 (0.4%) | 7 (1.7%) | 9 (1.0%) |
| Age (years) | ||||||
| Median (range) | 65.00 (38.00–87.00) |
67.00 (22.00–90.00) |
66.00 (22.00–90.00) |
65.00 (38.00–87.00) |
67.00 (22.00–88.00) |
66.00 (22.00–88.00) |
| IQR | 59.00–72.00 | 59.00–72.00 | 59.00–72.00 | 59.00–72.00 | 59.00–72.00 | 59.00–72.00 |
| Missing | 0 | 0 | 0 | 0 | 0 | 0 |
| F04 stratification factor: sex | ||||||
| Male | 336 (72.9%) | 332 (72.5%) | 668 (72.7%) | 330 (72.8%) | 304 (72.7%) | 634 (72.8%) |
| Female | 125 (27.1%) | 126 (27.5%) | 251 (27.3%) | 123 (27.2%) | 114 (27.3%) | 237 (27.2%) |
| ECOG PS | ||||||
| 0 | 246 (53.4%) | 258 (56.3%) | 504 (54.8%) | 244 (53.9%) | 237 (56.7%) | 481 (55.2%) |
| 1 | 215 (46.6%) | 196 (42.8%) | 411 (44.7%) | 209 (46.1%) | 177 (42.3%) | 386 (44.3%) |
| Missing | 0 (0.0%) | 4 (0.9%) | 4 (0.4%) | 0 (0.0%) | 4 (1.0%) | 4 (0.5%) |
| Disease present in bones | ||||||
| Yes | 108 (23.4%) | 94 (20.5%) | 202 (22.0%) | 107 (23.6%) | 83 (19.9%) | 190 (21.8%) |
| No | 352 (76.4%) | 364 (79.5%) | 716 (77.9%) | 345 (76.2%) | 335 (80.1%) | 680 (78.1%) |
| Missing | 1 (0.2%) | 0 (0.0%) | 1 (0.1%) | 1 (0.2%) | 0 (0.0%) | 1 (0.1%) |
| Time since initial diagnosis (years) | ||||||
| Mean (SD) | 2.67 (4.27) | 2.75 (4.45) | 2.71 (4.36) | 2.64 (4.29) | 2.84 (4.57) | 2.74 (4.42) |
| Missing | 1 | 1 | 2 | 1 | 1 | 2 |
| Haemoglobin (g/dL) | ||||||
| Mean (SD) | 13.35 (4.25) | 13.00 (1.92) | 13.18 (3.31) | 13.37 (4.28) | 13.03 (1.93) | 13.21 (3.37) |
| Missing | 0 | 0 | 0 | 0 | 0 | 0 |
| ANC (×109/L) | ||||||
| Mean (SD) | 5.44 (2.37) | 5.41 (2.20) | 5.42 (2.29) | 5.44 (2.38) | 5.42 (2.25) | 5.43 (2.32) |
| Missing | 0 | 0 | 0 | 0 | 0 | 0 |
| Platelets (×10 9 /L) | ||||||
| Mean (SD) | 292.97 (107.03) | 298.62 (117.02) | 295.78 (112.09) | 292.81 (107.09) | 298.53 (118.37) | 295.55 (112.61) |
| Missing | 0 | 0 | 0 | 0 | 0 | 0 |
| Corrected serum calcium (mmol/L) | ||||||
| Mean (SD) | 2.41 (0.17) | 2.39 (0.14) | 2.40 (0.16) | 2.41 (0.16) | 2.39 (0.14) | 2.40 (0.15) |
| Missing | 54 | 55 | 109 | 53 | 52 | 105 |
| Lactate dehyrogenase (IU/L) | ||||||
| Mean (SD) | 326.54 (204.69) | 320.69 (199.34) | 323.63 (201.96) | 326.96 (206.02) | 313.64 (187.28) | 320.57 (197.25) |
| Missing | 5 | 6 | 11 | 5 | 5 | 10 |
| Randomised under stratification factor: Motzer/MSKCC prognostic group | ||||||
| Favourable risk (0 factors) | 202 (43.8%) | 203 (44.3%) | 405 (44.1%) | 199 (43.9%) | 185 (44.3%) | 384 (44.1%) |
| Intermediate risk (1–2 factors) | 224 (48.6%) | 223 (48.7%) | 447 (48.6%) | 219 (48.3%) | 202 (48.3%) | 421 (48.3%) |
| Poor risk (≥ 3 factors) | 35 (7.6%) | 32 (7.0%) | 67 (7.3%) | 35 (7.7%) | 31 (7.4%) | 66 (7.6%) |
| Randomised under stratification factor: age group | ||||||
| < 60 | 122 (26.5%) | 122 (26.6%) | 244 (26.6%) | 119 (26.3%) | 110 (26.3%) | 229 (26.3%) |
| ≥ 60 | 339 (73.5%) | 336 (73.4%) | 675 (73.4%) | 334 (73.7%) | 308 (73.7%) | 642 (73.7%) |
| Randomised under stratification factor: disease status | ||||||
| Metastatic | 451 (97.8%) | 448 (97.8%) | 899 (97.8%) | 443 (97.8%) | 408 (97.6%) | 851 (97.7%) |
| Locally advanced | 10 (2.2%) | 10 (2.2%) | 20 (2.2%) | 10 (2.2%) | 10 (2.4%) | 20 (2.3%) |
| Randomised under stratification factor: previous nephrectomy | ||||||
| Yes | 347 (75.3%) | 345 (75.3%) | 692 (75.3%) | 339 (74.8%) | 316 (75.6%) | 655 (75.2%) |
| No | 114 (24.7%) | 113 (24.7%) | 227 (24.7%) | 114 (25.2%) | 102 (24.4%) | 216 (24.8%) |
| Randomised under stratification factor: TKI received | ||||||
| Sunitinib | 195 (42.3%) | 193 (42.1%) | 388 (42.2%) | 191 (42.2%) | 177 (42.3%) | 368 (42.3%) |
| Pazopanib | 266 (57.7%) | 265 (57.9%) | 531 (57.8%) | 262 (57.8%) | 241 (57.7%) | 503 (57.7%) |
| Randomised under stratification factor: sex | ||||||
| Male | 336 (72.9%) | 332 (72.5%) | 668 (72.7%) | 330 (72.8%) | 304 (72.7%) | 634 (72.8%) |
| Female | 125 (27.1%) | 126 (27.5%) | 251 (27.3%) | 123 (27.2%) | 114 (27.3%) | 237 (27.2%) |
Treatment received
Treatment cycles
The median (IQR) number of treatment cycles was 4 (2, 10) overall and similar between the two arms [CCS: 5(2, 10), DFIS: 4 (2, 9)]. Prior to week 24, the median (IQR) number of treatment cycles was identical between the two arms 4 (2, 4). Post week 24, the median (IQR) was again similar between the two arms with the medians being identical [CCS: 6 (2, 13), DFIS: 6 (3, 12)]. In terms of short cycles, cycle delays and dose reductions, the percentage of short cycles was similar across the randomisation arms both overall (CCS: 25.7%, n cycles = 3602, DFIS: 26.3%, n cycles = 3285) and pre and post cycle 4 (pre CCS: 32.3%, n cycles = 1439, DFIS: 31.1%, n cycles = 1449, post CCS: 21.3%, n cycles = 2163, DFIS: 22.5%, n cycles = 1836), as were the percentage of delays (overall — CCS: 13.8%, n cycles = 3602, DFIS: 13.4%, n cycles = 3285, pre CCS: 14.1%, n cycles = 1439, DFIS: 13.0%, n cycles = 1449, post CCS: 13.5%, n cycles = 2163, DFIS: 13.8%, n cycles = 1836), and the percentage of dose reductions (overall — CCS: 7.4%, n cycles = 3602, DFIS: 8.3%, n cycles = 3285, pre CCS: 12.6%, n cycles = 1439, DFIS: 13.6%, n cycles = 1449, post CCS: 3.9%, n cycles = 2163, DFIS: 4%, n cycles = 1836). However, the median (95% CI) time to first delay and time to first dose reduction was longer in the DFIS arm compared to the CCS arm [delay — CCS: 6 months (4, 7), DFIS: 10 months (7, 11), reduction – CCS: 6 months (6, 8), DFIS: 10 months (5, 14)] accounting for the fact that when on a treatment break participants cannot have treatment delayed or reduced.
Treatment breaks
In total, 248 participants (56.2%, n = 459) in the DFIS arm continued on trial after 6 months. Of these, 210 (84.7%) started their first treatment break according to the protocol at week 24. A similar proportion of participants on each TKI [sunitinib: 89 (84.0%, n = 106), pazopanib: 121 (85.2%, n = 142)]. Thirty-eight participants took their first treatment break later than protocol advised, where 15 of these (39.5%) did not take up a treatment break despite continuing at week 24. These participants continued from week 24 onwards in error due to the clinician’s decision and then either withdrew from trial treatment or experienced radiological progression on a later scan. Of the 38, 12 (31.6%) took up their first break at 36 weeks, 6 (15.8%) at 48 weeks, 2 (5.3%) at 60 weeks and 3 (7.9%) at other time points. The three other time points at which the treatment break was taken up were week 168, week 30 and week 42. For the majority, the reasons for continuing past week 24 were due to clinical decisions or during Phase II of the trial that maximal radiological response had not been reached.
The median number of treatment breaks was one in both TKI groups. The maximum number of breaks taken by a participant was nine (see Table 3). The median (IQR) length of a treatment break was 87 (84–119) days and similar between the two TKIs [sunitinib: 85.5 (84–112), pazopanib 87.5 (84–137)].
| Sunitinib | Pazopanib | Total | |
|---|---|---|---|
| According to the scan and treatment data, how many breaks did the participant start? | |||
| 0 | 7 (6.6%) | 8 (5.6%) | 15 (6.0%) |
| 1 | 51 (48.1%) | 76 (53.5%) | 127 (51.2%) |
| 2 | 17 (16.0%) | 21 (14.8%) | 38 (15.3%) |
| 3 | 15 (14.2%) | 13 (9.2%) | 28 (11.3%) |
| 4 | 11 (10.4%) | 8 (5.6%) | 19 (7.7%) |
| 5 | 3 (2.8%) | 11 (7.7%) | 14 (5.6%) |
| 6 | 0 (0.0%) | 1 (0.7%) | 1 (0.4%) |
| 7 | 1 (0.9%) | 3 (2.1%) | 4 (1.6%) |
| 8 | 0 (0.0%) | 1 (0.7%) | 1 (0.4%) |
| 9 | 1 (0.9%) | 0 (0.0%) | 1 (0.4%) |
| Total | 106 (100%) | 142 (100%) | 248 (100%) |
Anticancer treatment post trial
Overall, 61.6% of participants received any systemic anticancer therapy treatment during follow-up: a slightly higher proportion in the CCS arm (68.5%, n = 461) compared to the DFIS arm (54.6%, n = 458), in addition to a higher proportion of those on sunitinib (65.5%, n = 385) compared to pazopanib (58.8%, n = 534). The remaining types of treatment recorded post trial (radiotherapy, surgery, palliative care) were similar across the two randomisation arms (see Table 4) and TKI received (see Report Supplementary Material 1, Table 14). The medication names for the anticancer therapy received, distinct per participant, are given by randomisation allocation in Report Supplementary Material 1, Tables 15 and 16. These were categorised into first-line TKI, TKI, immunotherapy (alone or in combination), Mammalian target of rapamycin (alone or in combination), other cancer treatment or investigational product by a TMG clinician. Note that anticancer therapies considered not to aim to improve survival were not included in this re-categorisation. The number of distinct types of therapy a participant received is shown in Report Supplementary Material 1, Table 17. Note that zero in Report Supplementary Material 1, Table 17 refers to only STAR trial treatment received. The number of participants who received each type of therapy at least once is shown in Table 5. Note that immunotherapy was received by a similar proportion of participants in both arms.
| CCS (n = 461) |
DFIS (n = 458) |
Total (n = 919) |
|
|---|---|---|---|
| Is the participant recorded as having any systemic anticancer treatment during follow-up? | |||
| Yes | 316 (68.5%) | 250 (54.6%) | 566 (61.6%) |
| No | 92 (20.0%) | 111 (24.2%) | 203 (22.1%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| Is the participant recorded as having any radiotherapy treatment during follow-up? | |||
| Yes | 126 (27.3%) | 113 (24.7%) | 239 (26.0%) |
| No | 282 (61.2%) | 248 (54.1%) | 530 (57.7%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| Is the participant recorded as having any anticancer surgery during follow-up? | |||
| Yes | 32 (6.9%) | 35 (7.6%) | 67 (7.3%) |
| No | 376 (81.6%) | 326 (71.2%) | 702 (76.4%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| Is the participant recorded as having palliative care during follow-up? | |||
| Yes | 174 (37.7%) | 151 (33.0%) | 325 (35.4%) |
| No | 234 (50.8%) | 210 (45.9%) | 444 (48.3%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| CCS (n = 461) |
DFIS (n = 458) |
Total (n = 919) |
|
|---|---|---|---|
| Only STAR treatment recorded | |||
| Yes | 146 (31.7%) | 209 (45.6%) | 355 (38.6%) |
| No | 315 (68.3%) | 249 (54.4%) | 564 (61.4%) |
| First-line TKI received during follow-up | |||
| Yes | 142 (30.8%) | 103 (22.5%) | 245 (26.7%) |
| No | 266 (57.7%) | 258 (56.3%) | 524 (57.0%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| TKI received during follow-up | |||
| Yes | 179 (38.8%) | 139 (30.3%) | 318 (34.6%) |
| No | 229 (49.7%) | 222 (48.5%) | 451 (49.1%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| Immunotherapy received during follow-up (alone or in combination) | |||
| Yes | 117 (25.4%) | 108 (23.6%) | 225 (24.5%) |
| No | 291 (63.1%) | 253 (55.2%) | 544 (59.2%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| mTor received during follow-up (alone or in combination) | |||
| Yes | 44 (9.5%) | 35 (7.6%) | 79 (8.6%) |
| No | 364 (79.0%) | 326 (71.2%) | 690 (75.1%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
| Other cancer treatment or investigational product received in follow-up | |||
| Yes | 5 (1.1%) | 3 (0.7%) | 8 (0.9%) |
| No | 403 (87.4%) | 358 (78.2%) | 761 (82.8%) |
| N/A | 53 (11.5%) | 97 (21.2%) | 150 (16.3%) |
Impact of COVID-19
Recruitment into the trial had ceased when the COVID-19 pandemic began, and the trial was well into its follow-up period with 72.1% of the overall follow-up time completed (12 September 2017–30 January 2020).
In total, 41 participants (DFIS: 32, CCS: 9) were affected across 115 visits (see Table 6). The majority of visits (62, 53.9%) confirmed that the physical and/or blood assessments required by the trial protocol were not completed. The other reasons included one participant having a treatment break from 4 cycles, additional bisphosphonates due to COVID-19, a participant declining to restart treatment due to COVID-19 and toxicity assessments not done due to a participant’s COVID-19 diagnosis. Table 6 suggests that the randomisation arms were disproportionately affected by the pandemic. However, it is important to note that as per section Time to treatment failure participants in the CCS arm were on trial treatment for a shorter time than participants in the DFIS arm and therefore will have had fewer opportunities to be affected by the pandemic.
| CCS (n = 9) |
DFIS (n = 32) |
Total (n = 41) |
|
|---|---|---|---|
| Physical and/or blood assessments not done | 8 (34.8%) | 54 (58.7%) | 62 (53.9%) |
| On study assessment missed | 0 (0.0%) | 2 (2.2%) | 2 (1.7%) |
| Confirmed telephone assessment | 9 (39.1%) | 18 (19.6%) | 27 (23.5%) |
| Scan missed | 4 (17.4%) | 9 (9.8%) | 13 (11.3%) |
| Scan delayed | 2 (8.7%) | 2 (2.2%) | 4 (3.5%) |
| Other | 0 (0.0%) | 7 (7.6%) | 7 (6.1%) |
| Total | 23 (100%) | 92 (100%) | 115 (100%) |
In terms of the co-primary and secondary end points, few questionnaires were reported to be missing due to COVID-19 (see Table 1). In addition, COVID-19 was not reported to be the cause of any deaths (see Report Supplementary Material 1, Table 18). Therefore, it is unlikely that the co-primary end points have been affected by the pandemic. However, secondary end points which include progression as an event may have been, due to the delays in scan assessments, albeit for a small proportion of participants, which may have resulted in missed or delayed reporting of progressions.
Numbers analysed
Intention to treat
Of the 920 randomised, 919 were included in the ITT population (CCS: 461, DFIS: 458). One participant in the DFIS arm was excluded as they did not have RCC.
Per-protocol population
Of the 920 randomised, 871 (94.7%) were included in the PP population (CCS: 453, DFIS: 418). A higher proportion of participants randomised to the CCS arm (98.3%, n = 461) compared to those randomised to the DFIS arm (91.1%, n = 459). The reasons for exclusion from the PP population are shown in Table 7.
| CCS (n = 8) |
DFIS (n = 41) |
Total (n = 49) |
|
|---|---|---|---|
| Inclusion criteria breached | 1 (12.50) | 2 (4.88) | 3 (6.12) |
| Never took up a treatment break | 0 (0.00) | 15 (36.59) | 15 (30.61) |
| No treatment received | 1 (12.50) | 3 (7.32) | 4 (8.16) |
| Overdose or underdose of treatment | 5 (62.50) | 5 (12.20) | 10 (20.41) |
| Withdrew due to randomised strategy | 1 (12.50) | 2 (4.88) | 3 (6.12) |
| Continued at week 24 in error | 0 (0.00) | 14 (34.15) | 14 (28.57) |
Safety population
Nine hundred and sixteen (99.6%) of all randomised were included in the safety population (CCS: 485, DFIS: 431). Four participants, three in the DFIS arm and one in the CCS arm, were excluded due to never commencing their TKI treatment. In addition, 24 (5.2%, n = 459) participants randomised to the DFIS arm are included in the CCS safety arm.
Quality-of-life populations
Eight hundred and sixty-nine (94.5%) randomised participants were included in the EQ-5D-3L population (CCS: 438, DFIS: 431). Fifty-one participants were excluded due to having baseline questionnaires which could not be scored. Similarly, 856 (93.0%) randomised participants were included in the FACT-G population (CCS: 425, DFIS: 431). Finally, 882 (95.9%) randomised participants were included in the FKSI population (CCS: 436, DFIS: 446).
Outcomes and estimation
Overall survival
Per-protocol population
Number and causes of death
In total, 648 (74.4%) of participants in the PP population (n = 871) died prior to the end of follow-up on 31 December 2020. Note that this is less than the 720 required for 80% power in the OS comparison. Of the 648, 330 were from the CCS arm, accounting for 72.8% of those in the PP population randomised to CCS (n = 453). The remaining 318 were from the DFIS arm, accounting for 76.1% of those in the PP population randomised to DFIS (n = 418). In terms of TKI, 280 survival events were randomised under sunitinib (76.1%, n = 368) and 368 (73.2%, n = 503) under pazopanib. Renal cancer was related to the cause of 607 (93.7%) deaths [CCS: 314 (95.2%, n = 330), DFIS: 293 (92.1%, n = 318); sunitinib: 264 (94.3%, n = 280), pazopanib: 343 (93.2%, n = 368)]. In total, 526 (81.2%) deaths [CCS: 274 (83%, n = 330), DFIS: 252 (79.2%, n = 318); sunitinib: 234 (83.6%, n = 280), pazopanib: 292 (79.3%, n = 368)] were caused by renal cancer alone. Table 8 shows the non-mutually exclusive reasons by randomisation allocation. The other reasons are listed in Report Supplementary Material 1, Table 18. The same information is presented by randomised under TKI in Report Supplementary Material 1, Tables 19 and 20.
| CCS (n = 330) |
DFIS (n = 318) |
Total (n = 648) |
|
|---|---|---|---|
| Renal cancer | 274 (83.0%) | 252 (79.2%) | 526 (81.2%) |
| Renal cancer, other | 35 (10.6%) | 26 (8.2%) | 61 (9.4%) |
| Unknown | 7 (2.1%) | 9 (2.8%) | 16 (2.5%) |
| Other | 6 (1.8%) | 9 (2.8%) | 15 (2.3%) |
| Renal cancer, cardiovascular related | 1 (0.3%) | 8 (2.5%) | 9 (1.4%) |
| Renal cancer, cardiovascular related, other | 2 (0.6%) | 3 (0.9%) | 5 (0.8%) |
| Cardiovascular related | 1 (0.3%) | 2 (0.6%) | 3 (0.5%) |
| Trial toxicity | 1 (0.3%) | 2 (0.6%) | 3 (0.5%) |
| Renal cancer, trial toxicity | 0 (0.0%) | 2 (0.6%) | 2 (0.3%) |
| Renal cancer, trial toxicity, cardiovascular related, other | 1 (0.3%) | 1 (0.3%) | 2 (0.3%) |
| Trial toxicity, cardiovascular related | 0 (0.0%) | 2 (0.6%) | 2 (0.3%) |
| Trial toxicity, other | 1 (0.3%) | 1 (0.3%) | 2 (0.3%) |
| Renal cancer, trial toxicity, other | 0 (0.0%) | 1 (0.3%) | 1 (0.2%) |
| Renal cancer, unknown | 1 (0.3%) | 0 (0.0%) | 1 (0.2%) |
Time to event analysis
For the PP population, median (IQR) follow-up for OS is 58 (46 to 72) months. Median survival (95% CI) in the CCS arm was 28 (24 to 32) and 27 (23 to 31) in the DFIS arm. At 24 months post randomisation, there was a similar proportion (95% CI) of participants alive in both arms [CCS: 55.2% (50.5% to 59.7%), DFIS: 53.1% (48.2% to 57.8%)]. Figure 6 shows the Kaplan–Meir curve for OS by randomisation allocation.
FIGURE 6.
Kaplan–Meier figure for OS by randomisation allocation in the PP population.
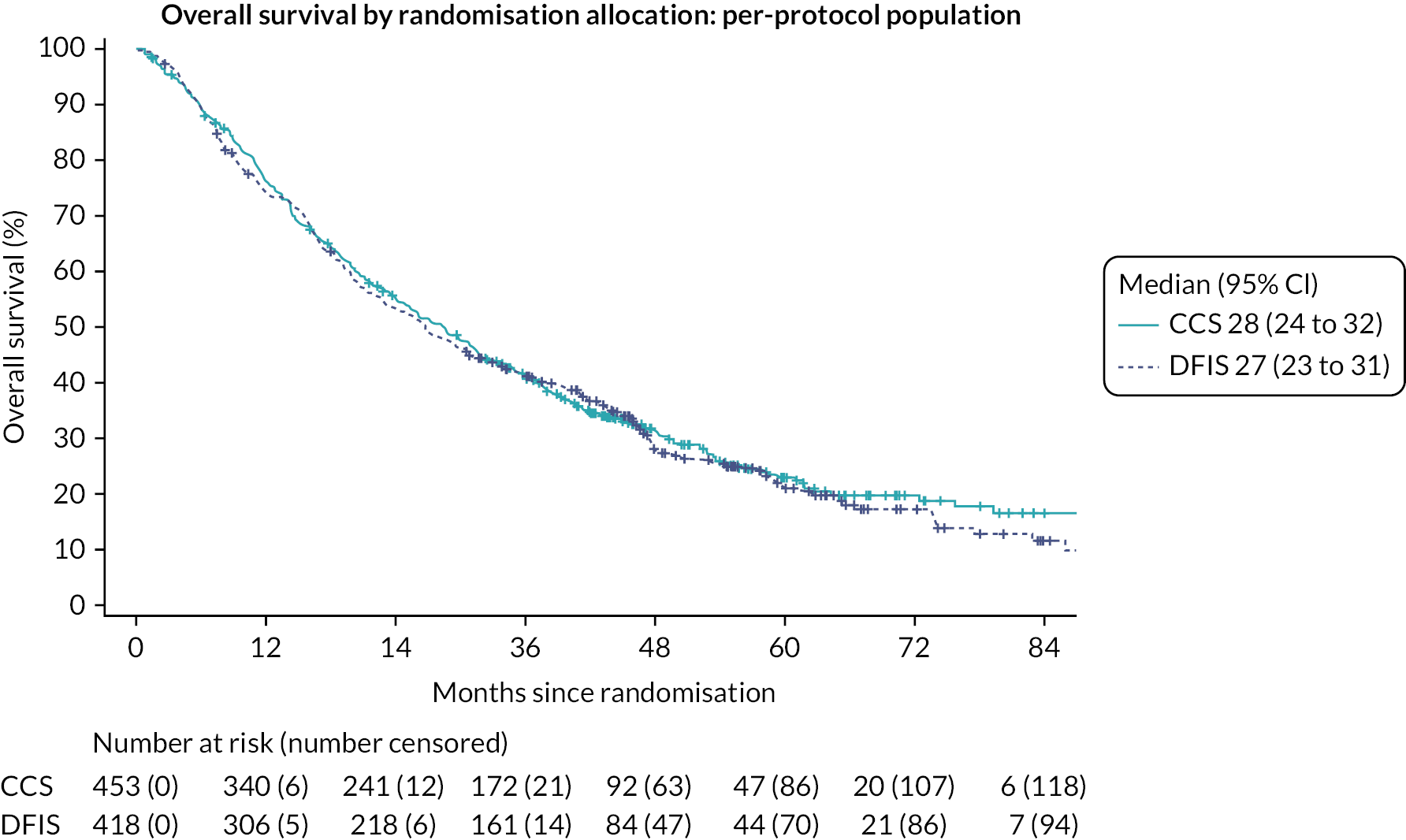
Table 9 shows the Cox PH model results for OS. The HR for OS suggests that the CCS arm has a risk of death 0.94 times the risk of death in the DFIS arm. The CI suggests that at most the CCS arm has a risk of death of 0.80 times the risk of death in the DFIS arm. This compares to the 7.5% NI margin where the assumption of a 48.5% survival rate in the CCS arm resulted in a NI boundary of 0.812. At the 2.5% significance level, we do not reject the null hypothesis that DFIS is not non-inferior to CCS in terms of OS. There is insufficient evidence to conclude NI. The supremum test for PH showed that at the 1% significance level only Motzer violated PH, and this is investigated through sensitivity analysis in the section Overall Survival – Motzer.
| DF | Estimate | Standard error | HR estimate | 95% CI for HR | Test statistic | p-value | |
|---|---|---|---|---|---|---|---|
| Randomisation treatment | 1 | 0.69 | 0.406 | ||||
| CCS vs. DFIS | −0.07 | 0.08 | 0.94 | (0.80 to 1.09) | |||
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2 | 31.21 | < 0.001 | ||||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | 0.33 | 0.09 | 1.39 | (1.15 to 1.67) | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | 0.99 | 0.18 | 2.68 | (1.89 to 3.81) | |||
| Randomised under stratification factor: sex | 1 | 0.12 | 0.734 | ||||
| Female vs. male | 0.03 | 0.09 | 1.03 | (0.87 to 1.23) | |||
| Randomised under stratification factor: age group | 1 | 0.02 | 0.881 | ||||
| ≥ 60 vs. < 60 | 0.01 | 0.09 | 1.01 | (0.85 to 1.21) | |||
| Randomised under stratification factor: disease status | 1 | 0.17 | 0.677 | ||||
| Metastatic vs. locally advanced | −0.11 | 0.27 | 0.89 | (0.52 to 1.52) | . | . | |
| Randomised under stratification factor: previous nephrectomy | 1 | 10.57 | 0.001 | ||||
| Yes vs. no | −0.35 | 0.11 | 0.70 | (0.57 to 0.87) | |||
| Randomised under stratification factor: TKI received | 1 | 0.29 | 0.592 | ||||
| Pazopanib vs. sunitinib | 0.04 | 0.08 | 1.04 | (0.89 to 1.22) |
Intention-to-treat population
Number and causes of death
In total, 678 (73.8%) of participants in the ITT population (n = 919) died prior to the end of follow-up on 31 December 2020. Note that this is less than the 720 required for 80% power in the OS comparison. Of the 678, 335 were from the CCS arm, accounting for 72.7% of those in the ITT population randomised to CCS (n = 461). The remaining 343 were from the DFIS arm, accounting for 74.9% of those in the ITT population randomised to DFIS (n = 458). In terms of TKI, 293 survival events were randomised under sunitinib (75.5%, n = 388) and 385 (72.5%, n = 531) under pazopanib. Renal cancer was related to the cause of 634 (93.5%) deaths [CCS: 317 (94.6%, n = 335), DFIS: 317 (92.4%, n = 343); sunitinib: 275 (93.9%, n = 293), pazopanib: 359 (93.2%, n = 385)]. In total, 550 (81.1%) deaths [CCS: 276 (82.4%, n = 335), DFIS: 274 (79.9%, n = 343); sunitinib: 245 (83.6%, n = 293), pazopanib: 305 (79.2%, n = 385)] were caused by renal cancer alone. Table 10 shows the mutually exclusive reasons by randomisation allocation. Line listings for the other causes of death can be found in Report Supplementary Material 1, Table 21. The mutually exclusive causes of death by randomised under TKI are shown in Report Supplementary Material 1, Table 22 along with the line listings for other reasons (see Report Supplementary Material 1, Table 23).
| CCS (n = 335) |
DFIS (n = 343) |
Total (n = 678) |
|
|---|---|---|---|
| Renal cancer | 276 (82.4%) | 274 (79.9%) | 550 (81.1%) |
| Renal cancer, other | 36 (10.7%) | 28 (8.2%) | 64 (9.4%) |
| Other | 8 (2.4%) | 9 (2.6%) | 17 (2.5%) |
| Unknown | 7 (2.1%) | 9 (2.6%) | 16 (2.4%) |
| Renal cancer, cardiovascular related | 1 (0.3%) | 8 (2.3%) | 9 (1.3%) |
| Renal cancer, cardiovascular related, other | 2 (0.6%) | 3 (0.9%) | 5 (0.7%) |
| Cardiovascular related | 1 (0.3%) | 3 (0.9%) | 4 (0.6%) |
| Trial toxicity | 1 (0.3%) | 2 (0.6%) | 3 (0.4%) |
| Renal cancer, trial toxicity | 0 (0.0%) | 2 (0.6%) | 2 (0.3%) |
| Renal cancer, trial toxicity, cardiovascular related, other | 1 (0.3%) | 1 (0.3%) | 2 (0.3%) |
| Trial toxicity, cardiovascular related | 0 (0.0%) | 2 (0.6%) | 2 (0.3%) |
| Trial toxicity, other | 1 (0.3%) | 1 (0.3%) | 2 (0.3%) |
| Renal cancer, trial toxicity, other | 0 (0.0%) | 1 (0.3%) | 1 (0.1%) |
| Renal cancer, unknown | 1 (0.3%) | 0 (0.0%) | 1 (0.1%) |
Time to event analysis
For the ITT population, median (IQR) follow-up for OS is 58 (46 to 73) months. Median survival (95% CI) in the CCS arm was 28 (24 to 32) and 27 (23 to 33) in the DFIS arm. At 24 months post randomisation, there was a similar proportion (96% CI) of participants alive [CCS: 55.5% (50.3% to 59.5%), DFIS: 54.2% (49.5% to 58.7%)].
Figure 7 shows the Kaplan–Meier curve for OS by randomisation allocation. Table 11 shows Cox PH model results for OS. The HR for OS suggests that the CCS arm has a risk of death 0.97 times the risk of death in the DFIS arm. The CI suggests that at most the CCS arm has a risk of death of 0.83 times the risk of death in the DFIS arm. This is compared to the 7.5% NI margin where the assumption of a 48.5% survival rate in the CCS arm resulted in a NI boundary of 0.812. At the 2.5% significance level we reject the null hypothesis and conclude that DFIS is non-inferior to CCS in terms of OS in the ITT population. Note that while we conclude NI in the ITT population, this is a different conclusion to that in the analysis conducted in the PP population (see Per-protocol population) and therefore we cannot conclude NI for OS. Similar to the PP analysis, the supremum test for PH showed that at the 1% significance level, only Motzer violated the PH assumption.
FIGURE 7.
Kaplan–Meier figure for OS by randomisation allocation in the ITT population.

| DF | Estimate | Standard error | HR estimate | 95% CI for HR | Test statistic | p-value | |
|---|---|---|---|---|---|---|---|
| Randomisation treatment | 1 | 0.20 | 0.652 | ||||
| CCS vs. DFIS | −0.03 | 0.08 | 0.97 | (0.83 to 1.12) | |||
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2 | 35.99 | < 0.001 | ||||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | 0.35 | 0.09 | 1.42 | (1.18 to 1.69) | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | 1.04 | 0.18 | 2.83 | (2.00 to 3.99) | |||
| Randomised under stratification factor: sex | 1 | 0.12 | 0.733 | ||||
| Female vs. male | 0.03 | 0.09 | 1.03 | (0.87 to 1.22) | . | . | |
| Randomised under stratification factor: age group | 1 | 0.08 | 0.778 | ||||
| ≥ 60 vs. < 60 | 0.02 | 0.09 | 1.03 | (0.86 to 1.22) | |||
| Randomised under stratification factor: disease status | 1 | 0.22 | 0.636 | ||||
| Metastatic vs. locally advanced | −0.13 | 0.27 | 0.88 | (0.52 to 1.50) | |||
| Randomised under stratification factor: previous nephrectomy | 1 | 9.23 | 0.002 | ||||
| Yes vs. No | −0.32 | 0.11 | 0.72 | (0.59 to 0.89) | |||
| Randomised under stratification factor: TKI received | 1 | 0.14 | 0.712 | ||||
| Pazopanib vs. sunitinib | 0.03 | 0.08 | 1.03 | (0.88 to 1.20) |
Quality-adjusted life-years
Imputation
In total, 51 participants (23 CCS, 28 DFIS) did not complete or did not sufficiently complete a baseline EQ5D questionnaire. These were imputed to be 0.78 using mean imputation. Across the follow-up time points, the maximum percentage of missing questionnaires was 70% at 78 months of follow-up. However, only 10 questionnaires were expected to be returned at this point. Across follow-up, the missing data pattern was observed to be a mixture of monotone and non-monotone. The key baseline characteristics split by missing status for the baseline questionnaire and all follow-up time points were considered. The populations of those who completed and did not complete a questionnaire at each time point are similar in terms of the key baseline characteristics (see Report Supplementary Material 1, Tables 24–31). However, after 42 months of follow-up, no one with a Motzer Category of ‘poor risk’ at baseline was considered and no one with a disease status of ‘locally advanced’ completed a questionnaire. The distribution of the EQ-5D-3L utility score was observed to be not normally distributed (see Report Supplementary Material 1, Figures 7–20). Therefore, predictive mean matching was considered to be appropriate. Due to the small sample size at the later follow-up time points, the EQ-5D-3L utility index was imputed up to month 48 only using 52 imputed data sets to reflect the 52.38% missing data at this time point. The trace plots showing convergence of the imputation method are shown in Report Supplementary Material 1, Figures 21–24.
Per-protocol population
Summary statistics
The summary statistics for QALYs, derived over trial and follow-up in the PP population, were combined across the 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 1.73 (1.60 to 1.86) and 1.80 (1.65 to 1.95) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 1.28 to 1.39 in the CCS arm and 1.38 to 1.48 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM for the PP population are shown in Table 12. On average, DFIS increases QALYs by 0.04 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0.14 points. Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and therefore the 10% margin is 0.156. At the 2.5% significance level as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs in the PP population.
| Estimate | Standard error | 95% CI | |
|---|---|---|---|
| Intercept | 1.34 | 0.24 | (0.86 to 1.81) |
| Randomisation allocation | |||
| DFIS vs. CCS | 0.04 | 0.09 | (−0.14 to 0.21) |
| Randomised under stratification factor: Motzer/MSKCC prognostic group | |||
| Intermediate risk (1–2 factors) vs. poor risk (≥ 3 factors) | 0.57 | 0.22 | (0.13 to 1.00) |
| Favourable risk (0 factors) vs. poor risk (≥ 3 factors) | 0.74 | 0.24 | (0.26 to 1.22) |
| Randomised under stratification factor: sex | |||
| Female vs. male | −0.15 | 0.10 | (−0.34 to 0.04) |
| Randomised under stratification factor: age group | |||
| < 60 vs. ≥ 60 | −0.01 | 0.10 | (−0.21 to 0.18) |
| Randomised under stratification factor: disease status | |||
| Locally advanced vs. metastatic | −0.10 | 0.34 | (−0.75 to 0.56) |
| Randomised under stratification factor: previous nephrectomy | |||
| No vs. yes | −0.25 | 0.13 | (−0.51 to 0.01) |
| Randomised under stratification factor: TKI received | |||
| Pazopanib vs. sunitinib | −0.17 | 0.09 | (−0.35 to 0.00) |
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 25–27. In Report Supplementary Material 1, Figure 26 we can see that the residuals are violating the normal distribution assumption in the tails. This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. Report Supplementary Material 1, Figure 27 shows increased variation in the residuals as the predicted value increases. These are improved compared to the linear model fitted in the section Quality-adjusted life-years – multivariate linear regression analysis.
Intention-to-treat population
Summary statistics
The summary statistics for QALYs, derived over trial and follow-up in the ITT population, were combined across the 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 1.73 (1.59 to 1.86) and 1.83 (1.69 to 1.98) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 1.28 to 1.38 in the CCS arm and 1.38 to 1.49 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM for the ITT population are shown in Table 13. On average, the DFIS increases QALYs by 0.06 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0.11 points. Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and therefore the 10% margin is 0.156. At the 2.5% significance level as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs in the ITT population. As the analysis conducted in the ITT population and the PP population (see Per-protocol population) concluded NI we conclude NI overall for the QALY end point.
| Estimate | Standard error | 95% CI | |
|---|---|---|---|
| Intercept | 1.30 | 0.24 | (0.83 to 1.76) |
| Randomisation allocation | |||
| DFIS vs. CCS | 0.06 | 0.09 | (−0.11 to 0.23) |
| Randomised under stratification factor: Motzer/MSKCC prognostic group | |||
| Intermediate risk (1–2 factors) vs. poor risk (≥ 3 factors) | 0.58 | 0.22 | (0.16 to 1.01) |
| Favourable risk (0 factors) vs. poor risk (≥ 3 factors) | 0.78 | 0.24 | (0.31 to 1.24) |
| Randomised under stratification factor: sex | |||
| Female vs. male | −0.13 | 0.10 | (−0.32 to 0.05) |
| Randomised under stratification factor: age group | |||
| < 60 vs. ≥ 60 | 0.02 | 0.10 | (−0.18 to 0.21) |
| Randomised under stratification factor: disease status | |||
| Locally advanced vs. metastatic | −0.12 | 0.34 | (−0.78 to 0.54) |
| Randomised under stratification factor: previous nephrectomy | |||
| No vs. yes | −0.25 | 0.13 | (−0.50 to 0.01) |
| Randomised under stratification factor: TKI received | |||
| Pazopanib vs. sunitinib | −0.16 | 0.09 | (−0.34 to 0.01) |
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 28–30. Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 29). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. In addition, there was increased variation in the residuals as the predicted value increased (see Report Supplementary Material 1, Figure 30). These are similar to the PP analysis conducted above.
Time to strategy failure
Time to strategy failure is defined in Report Supplementary Material 1, Figure 1. In total, 850 (92.5%) participants in the ITT population (n = 919) died, progressed or required another anticancer systemic treatment prior to the end of follow-up on 31 December 2020. Of these, 438 were from the CCS arm, accounting for 95.0% of those in the ITT population randomised to CCS (n = 461). The remaining 412 were from the DFIS arm, accounting for 90.0% of those in the ITT population randomised to DFIS (n = 458). In terms of TKI, 356 TSF events were randomised under sunitinib (91.8%, n = 388) and 494 (93.0%, n = 531) under pazopanib.
Median TSF (95% CI) in the CCS arm was 8 months (8 to 9) and 11 months (9 to 13) in the DFIS arm. At 24 months, there was a higher proportion (95% CI) of participants event-free in the DFIS arm compared to the CCS arm [CCS: 15.6% (12.4% to 19.1%), DFIS: 26.3% (22.3% to 30.4%)]. Figure 8 shows the Kaplan–Meier curve for TSF by randomisation allocation.
FIGURE 8.
Kaplan–Meier figure for TSF by randomisation allocation in the ITT population.
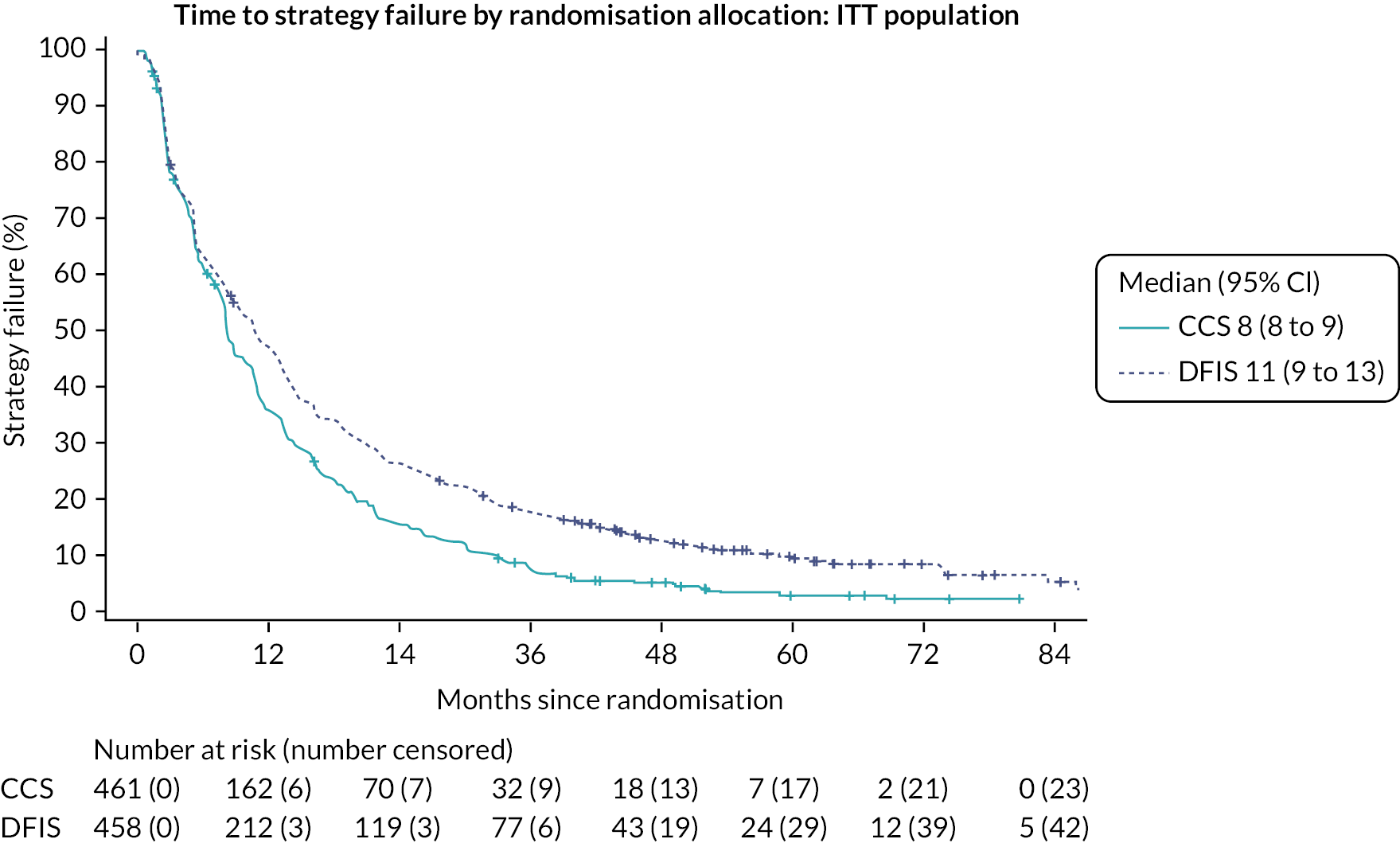
Table 14 shows the Cox PH model results for TSF. The HR for randomisation allocation suggests that the DFIS arm has a risk of strategy failure 0.75 times the risk of strategy failure in the CCS arm. This comparison is statistically significant at the 1% significance level (p < 0.001). The supremum test for PH showed that no variables included in the regression model violated the PH assumption at the 1% significance level.
| DF | Estimate | Standard error | HR estimate | 95% CI for HR | Test statistic | p-value | |
|---|---|---|---|---|---|---|---|
| Randomisation treatment | 1 | 17.19 | < 0.001 | ||||
| DFIS vs. CCS | −0.29 | 0.07 | 0.75 | (0.65 to 0.86) | |||
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2 | 22.98 | < 0.001 | ||||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | 0.22 | 0.08 | 1.25 | (1.07 to 1.46) | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | 0.77 | 0.16 | 2.15 | (1.57 to 2.95) | |||
| Randomised under stratification factor: sex | 1 | 0.48 | 0.487 | ||||
| Female vs. male | 0.05 | 0.08 | 1.05 | (0.91 to 1.23) | |||
| Randomised under stratification factor: age group | 1 | 0.00 | 0.947 | ||||
| ≥ 60 vs. < 60 | 0.01 | 0.08 | 1.01 | (0.86 to 1.17] | |||
| Randomised under stratification factor: disease status | 1 | 0.44 | 0.506 | ||||
| Metastatic vs. locally advanced | −0.16 | 0.23 | 0.86 | (0.54 to 1.35) | |||
| Randomised under stratification factor: previous nephrectomy | 1 | 0.69 | 0.405 | ||||
| Yes vs. no | −0.08 | 0.10 | 0.92 | (0.76 to 1.12) | |||
| Randomised under stratification factor: TKI received | 1 | 1.37 | 0.241 | ||||
| Pazopanib vs. sunitinib | 0.08 | 0.07 | 1.09 | (0.95 to 1.24) |
Time to treatment failure
Time to treatment failure is defined in Report Supplementary Material 1, Figure 2. In total, 877 (95.4%) participants in the ITT population (n = 919) stopped their trial strategy prior to the end of follow-up on 31 December 2020. Of these, 453 were from the CCS arm, accounting for 98.3% of those in the ITT population randomised to CCS (n = 461). The remaining 424 were from the DFIS arm, accounting for 92.6% of those in the ITT population randomised to DFIS (n = 458). In terms of TKI, 371 TTF events were randomised under sunitinib (95.6%, n = 388) and 506 (95.3%, n = 531) under pazopanib.
Median TTF (95% CI) in the CCS arm was 7 months (5 to 8) and 8 (6 to 9) months in the DFIS arm. At 24 months, there was a higher proportion (95% CI) of event-free participants in the DFIS arm compared to the CCS arm [CCS: 12.1% (9.4% to 15.3%), DFIS: 22.5% (18.8% to 26.4%)]. Figure 9 shows the Kaplan–Meier curve for TTF by randomisation allocation. Table 15 shows the Cox PH model results for TSF. The HR for randomisation allocation suggests that the DFIS arm has a risk of treatment failure 0.75 times the risk of treatment failure in the CCS arm. This comparison is statistically significant at the 1% significance level (p < 0.001). The supremum test for PH showed that both randomisation allocation and randomised under TKI violated the PH assumption at the 1% significance level. Therefore, piecewise hazards models were applied both with two intervals (see Report Supplementary Material 1, Table 32), separating at week 24, and three intervals (see Report Supplementary Material 1, Table 33) separating at week 24 and approximately week 42, the end of the average length of a treatment break. In both cases, the randomisation allocation comparison remains statistically significant at the 1% significance level [HR (95% CI); two intervals: 0.71 (0.62 to 0.82), three intervals: 0.72 (0.63 to 0.82)]. However, the goodness-of-fit statistic for both piecewise models was significant at the 1% model suggesting that the piecewise model is not a good fit to the data. As this did not change the conclusions to the Cox regression model, no further investigations were conducted.
FIGURE 9.
Kaplan–Meier figure for TTF by randomisation allocation in the ITT population.

| DF | Estimate | Standard error | HR estimate | 95% CI for HR | Test statistic | p-value | |
|---|---|---|---|---|---|---|---|
| Randomisation treatment | 1 | 17.72 | < 0.001 | ||||
| DFIS vs. CCS | −0.29 | 0.07 | 0.75 | (0.65 to 0.86) | |||
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2 | 15.14 | < 0.001 | ||||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | 0.21 | 0.08 | 1.24 | (1.06 to 1.44) | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | 0.59 | 0.16 | 1.80 | (1.32 to 2.46) | |||
| Randomised under stratification factor: sex | 1 | 5.05 | 0.025 | ||||
| Female vs. male | 0.17 | 0.08 | 1.19 | (1.02 to 1.38) | |||
| Randomised under stratification factor: age group | 1 | 0.82 | 0.366 | ||||
| ≥ 60 vs. < 60 | 0.07 | 0.08 | 1.07 | (0.92 to 1.25) | |||
| Randomised under stratification factor: disease status | 1 | 1.09 | 0.296 | ||||
| Metastatic vs. locally advanced | −0.24 | 0.23 | 0.78 | (0.50 to 1.24) | |||
| Randomised under stratification factor: previous nephrectomy | 1 | 0.19 | 0.659 | ||||
| Yes vs. no | −0.04 | 0.10 | 0.96 | (0.79 to 1.16) | |||
| Randomised under stratification factor: TKI received | 1 | 0.57 | 0.449 | ||||
| Pazopanib vs. sunitinib | 0.05 | 0.07 | 1.05 | (0.92 to 1.21) |
Progression-free survival
Note that this section refers to the first progression recorded on the trial, as defined in Report Supplementary Material 1, Figure 3, irrespective of whether it resulted in cessation or recommencement of trial treatment.
In total, 868 (94.5%) participants in the ITT population (n = 919) progressed or died prior to the end of follow-up on 31 December 2020. Of these, 431 were from the CCS arm, accounting for 93.5% of those in the ITT population randomised to CCS (n = 461). The remaining 437 were from the DFIS arm, accounting for 95.4% of those in the ITT population randomised to DFIS (n = 458). In terms of TKI, 368 PFS events were randomised under sunitinib (94.8%, n = 388) and 500 (94.2%, n = 531) under pazopanib.
Median PFS (95% CI) in the CCS arm was 11 months (9 to 11) and 8 months (8 to 8) in the DFIS arm. At 24 months, there was a higher proportion (95% CI) of participants in the CCS arm event-free compared to the DFIS arm [CCS: 21.4% (17.8% to 25.3%), DFIS: 10.2% (7.6% to 13.2%)]. Figure 10 shows the Kaplan–Meier curve for PFS by randomisation allocation. Table 16 shows the Cox PH model results for PFS. The HR for randomisation allocation suggests that the DFIS arm has a risk of progression or death 1.37 times the risk of progression or death in the CCS arm. This comparison is statistically significant at the 1% significance level (p < 0.001). The supremum test for PH showed that both randomisation allocation and randomised under Motzer score violated the PH assumption at the 1% significance level. Therefore, piecewise hazards models were applied both with two intervals (see Report Supplementary Material 1, Table 34), separating at week 24, and three intervals (see Report Supplementary Material 1, Table 35) separating at week 24 and approximately week 42, the end of the average length of a treatment break. In both cases, the randomisation allocation comparison remains statistically significant at the 1% significance level [HR (95% CI); two intervals: 1.40 (1.22 to 1.60), three intervals: 1.33 (1.16 to 1.52)]. However, the goodness-of-fit statistic for both piecewise models was significant at the 1% level suggesting that the piecewise models are not a good fit to the data. As this did not change the conclusions to the Cox regression model, no further investigations were conducted.
FIGURE 10.
Kaplan–Meier figure for PFS by randomisation allocation in the ITT population.

| DF | Estimate | Standard error | HR estimate | 95% CI for HR | Test statistic | p-value | |
|---|---|---|---|---|---|---|---|
| Randomisation treatment | 1 | 20.57 | < 0.001 | ||||
| DFIS vs. CCS | 0.31 | 0.07 | 1.37 | (1.19 to 1.57) | . | . | |
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2 | 14.30 | < 0.001 | ||||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | 0.18 | 0.08 | 1.20 | (1.02 to 1.40) | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | 0.61 | 0.16 | 1.85 | (1.34 to 2.55) | |||
| Randomised under stratification factor: sex | 1 | 2.20 | 0.138 | ||||
| Female vs. male | −0.11 | 0.08 | 0.89 | (0.77 to 1.04) | |||
| Randomised under stratification factor: age group | 1 | 0.49 | 0.486 | ||||
| >= 60 vs. < 60 | . | −0.05 | 0.08 | 0.95 | (0.81 to 1.10) | ||
| Randomised under stratification factor: disease status | 1 | 5.18 | 0.023 | ||||
| Metastatic vs. locally advanced | −0.52 | 0.23 | 0.60 | (0.38 to 0.93) | |||
| Randomised under stratification factor: previous nephrectomy | 1 | 3.54 | 0.060 | ||||
| Yes vs. no | −0.19 | 0.10 | 0.83 | (0.68 to 1.01) | |||
| Randomised under stratification factor: TKI received | 1 | 0.29 | 0.593 | ||||
| Pazopanib vs. sunitinib | −0.04 | 0.07 | 0.96 | (0.84 to 1.10) |
Summative progression-free interval
Summative progression-free interval is defined in Report Supplementary Material 1, Figures 4 and 5. In total, 783 (85.2%) participants in the ITT population (n = 919) ended their last SPFI with an event prior to the end of follow-up on 31 December 2020. Of these, 416 were from the CCS arm, accounting for 90.2% of those in the ITT population randomised to CCS (n = 461). The remaining 367 were from the DFIS arm, accounting for 80.1% of those in the ITT population randomised to DFIS (n = 458). In terms of TKI, 341 SPFI events were randomised under sunitinib (87.9%, n = 388) and 442 (83.2%, n = 531) under pazopanib.
Median SPFI (95% CI) in the CCS arm was 8 months (8 to 10) and 10 months (9 to 11) in the DFIS arm. At 24 months post randomisation, there was a higher proportion of participants (95% CI) event-free in the DFIS arm compared to the CCS arm [CCS: 14.9% (11.7% to 18.5%), DFIS: 24.6% (20.5% to 28.9%)]. Figure 11 shows the Kaplan–Meier curve for SPFI by randomisation allocation. Table 17 shows the Cox PH model results for SPFI. The HR for randomisation allocation suggests that the DFIS arm has a risk of SPFI ending in an event 0.77 times the risk of SPFI ending in an event in the CCS arm. This comparison is statistically significant at the 1% significance level (p < 0.001). The supremum test for PH showed that no variables included in the regression model violated the PH assumption at the 1% significance level.
FIGURE 11.
Kaplan–Meier Figure for SPFI by randomisation allocation in the ITT population.
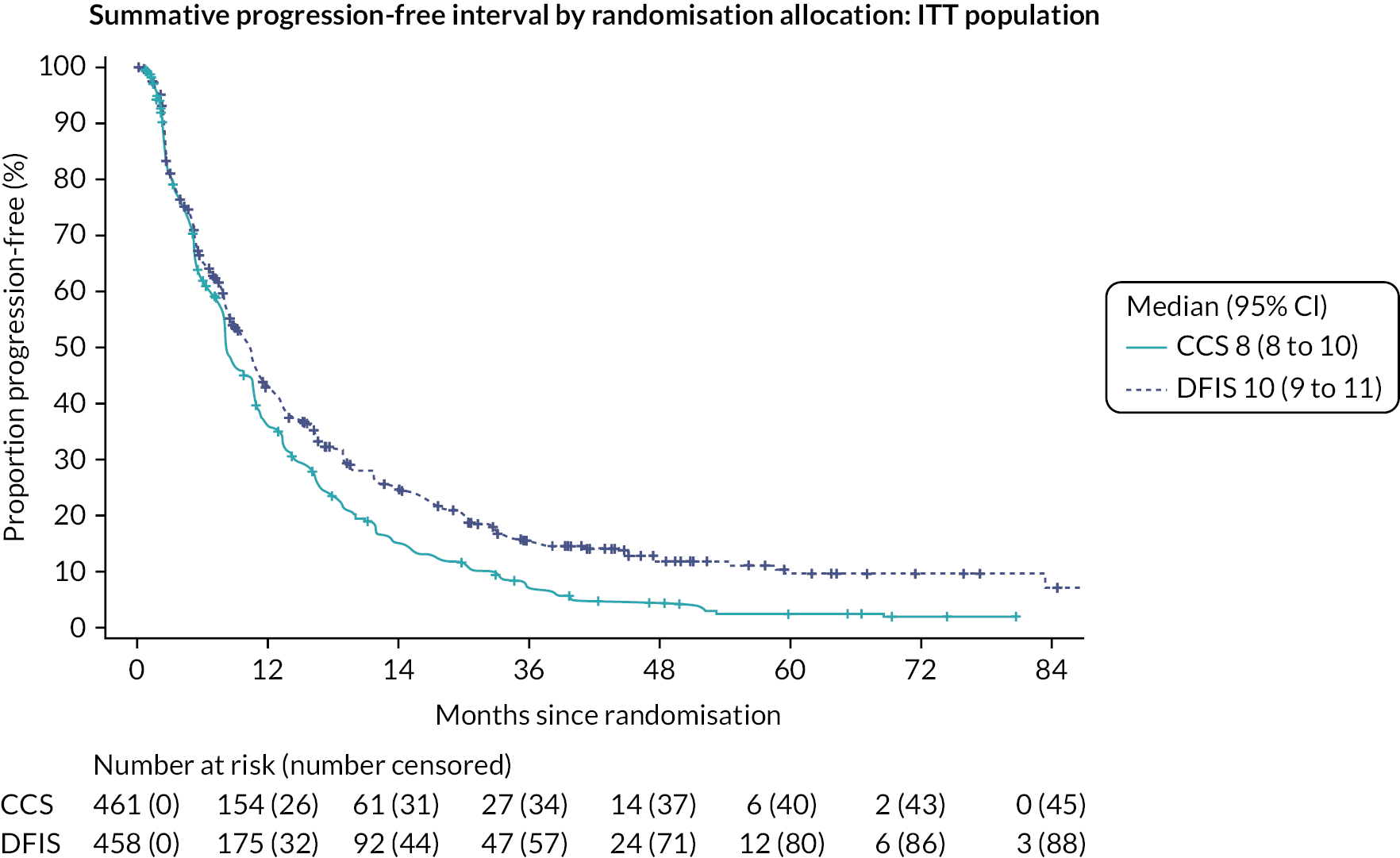
| DF | Estimate | Standard error | HR estimate | 95% CI for HR | Test statistic | p-value | |
|---|---|---|---|---|---|---|---|
| Randomisation treatment | 1 | 12.70 | < 0.001 | ||||
| DFIS vs. CCS | −0.26 | 0.07 | 0.77 | (0.67 to 0.89) | |||
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2 | 13.42 | 0.001 | ||||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | 0.18 | 0.08 | 1.19 | (1.02 to 1.40) | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | 0.63 | 0.18 | 1.88 | (1.33 to 2.65) | |||
| Randomised under stratification factor: sex | 1 | 0.03 | 0.861 | ||||
| Female vs. male | 0.01 | 0.08 | 1.01 | (0.87 to 1.19) | |||
| Randomised under stratification factor: age group | 1 | 0.06 | 0.802 | ||||
| ≥ 60 vs. < 60 | −0.02 | 0.08 | 0.98 | (0.84 to 1.15) | |||
| Randomised under stratification factor: disease status | 1 | 2.06 | 0.151 | ||||
| Metastatic vs. locally advanced | −0.35 | 0.24 | 0.71 | (0.44 to 1.13) | |||
| Randomised under stratification factor: previous nephrectomy | 1 | 0.06 | 0.801 | ||||
| Yes vs. no | −0.03 | 0.10 | 0.97 | (0.80 to 1.19) | |||
| Randomised under stratification factor: TKI received | 1 | 0.14 | 0.713 | ||||
| Pazopanib vs. sunitinib | 0.03 | 0.07 | 1.03 | (0.89 to 1.18) |
Quality of life
FKSI-15
The median scores with IQR of the FKSI-15 score for all QoL time points for participants in the FKSI population are shown in Figures 12 and 13. Recall that a higher FKSI-15 score indicates fewer symptoms or less severe symptoms, where a score of 60 indicates that the participant is asymptomatic. The size of the populations at each time point that the FKSI-15 questionnaire was collected (every 6 weeks) is shown below, split into counts of returned questionnaires and missing questionnaires. The total number of missing questionnaires also includes questionnaires where the relevant subscale could not be calculated. Figure 12 shows little change or difference in QoL over the first 42 weeks of treatment. It is worth noting that this is expected for the first 24 weeks as the two strategies are identical up to this point. Figure 13 shows that this little difference continues until around week 200 after which the median QoL for the CCS arm is slightly lower. However, there are considerably fewer participants on the CCS arm compared to the DFIS arm at this time point, so this trend may be due to chance or particular characteristics of those eight participants. The remaining participants in the DFIS arm appear to have a median score above the average for the rest of the time points though only seven or so participants are included in these calculations. The population decreasing faster in the CCS arm than in the DFIS arm is consistent with DFIS improving the TTF. Report Supplementary Material 1, Figures 31 and 32 show the same information for the FSKI-15 disease-related subscale, where a score of 36 indicates that the participant is asymptomatic, where again there is little difference observed between the two strategies prior to week 200.
FIGURE 12.
FKSI-15 overall score summary statistics, median and IQR, by randomisation allocation – (A) Booklets A, B and C.

FIGURE 13.
FKSI-15 overall score summary statistics, median and IQR, by randomisation allocation – (B) Booklet E.

Table 18 shows the results of the modelling process for the FKSI-15 score. Note that this was modelled up to week 312 where both strategies had participants on treatment. Overall, a slight but reducing over time improvement is suggested in favour of the DFIS arm. Similar results were observed for the FKSI-15 DRS subscale (see Report Supplementary Material 1, Table 36).
| Estimate | Standard error | Degrees of freedom | Test statistic | p-value | |
|---|---|---|---|---|---|
| Number of observations used: 7424 | |||||
| Intercept | 16.25 | 2.19 | |||
| QoL time point | 0.06 | 0.03 | 1, 646 | 0.04 | 0.845 |
| FKSI-15 total score at baseline | 0.59 | 0.03 | 1, 5994 | 465.09 | < 0.001 |
| Randomisation treatment | 1, 5994 | 6.51 | 0.011 | ||
| DFIS vs. CCS | 1.21 | 0.47 | |||
| QoL Time point for DFIS vs. CCS | −0.10 | 0.04 | 1, 5994 | 7.36 | 0.007 |
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2, 5994 | 0.93 | 0.395 | ||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | −0.51 | 0.54 | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | −1.44 | 1.12 | |||
| Randomised under stratification factor: sex | 1, 5994 | 0.57 | 0.449 | ||
| Female vs. male | −0.40 | 0.53 | |||
| Randomised under stratification factor: age group | 1, 5994 | 0.10 | 0.754 | ||
| ≥ 60 vs. < 60 | 0.16 | 0.52 | |||
| Randomised under stratification factor: disease status | 1, 5994 | 1.41 | 0.236 | ||
| Metastatic vs. locally advanced | 1.96 | 1.65 | |||
| Randomised under stratification factor: previous nephrectomy | 1, 5994 | 2.91 | 0.088 | ||
| Yes vs. no | −1.11 | 0.65 | |||
| Randomised under stratification factor: TKI received | 1, 5994 | 10.74 | 0.001 | ||
| Pazopanib vs. sunitinib | −1.53 | 0.47 | |||
Functional Assessment of Cancer Therapy-G
The median scores with IQR of the overall FACT-G score for all QoL time points, for participants in the FACT-G population, are shown in Figures 14 and 15. Recall that a higher score indicates better QoL. The size of the populations at each time point that the FACT-G questionnaire was collected (every 6 weeks) is shown below, split into counts of returned questionnaires and missing questionnaires. The total number of missing questionnaires also includes questionnaires where the relevant score could not be calculated. There is little observed difference in terms of median and IQR for the two treatment strategies, which is partially expected as both strategies are identical until week 24. This continues in Figure 15 until around week 264 when the difference in medians increases and few participants remain in either strategy. The population decreasing faster in the CCS arm than in the DFIS arm is consistent with DFIS improving the TTF. This was consistent across the various FACT-G subscales: Social/Family Well-being (see Report Supplementary Material 1, Figures 33 and 34), Physical Well-Being (see Report Supplementary Material 1, Figures 35 and 36), Emotional Well-being (see Report Supplementary Material 1, Figures 37 and 38) and Functional Well-being (see Report Supplementary Material 1, Figures 39 and 40).
FIGURE 14.
FACT-G overall score summary statistics, median and IQR, by randomisation allocation – (A) Booklets A, B and C.
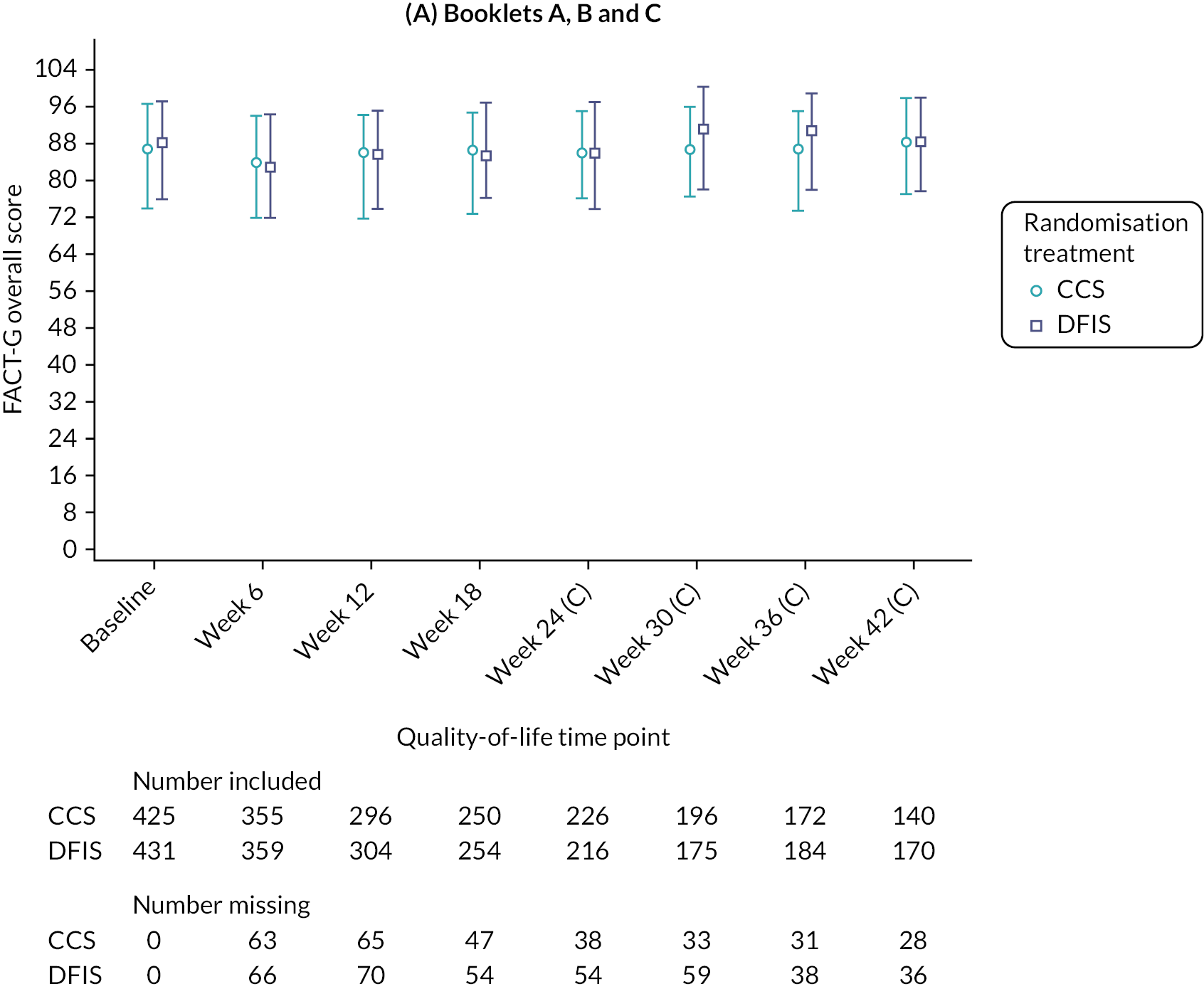
FIGURE 15.
FACT-G overall score summary statistics, median and IQR, by randomisation allocation – (B) Booklets E.

Table 19 shows the results of the baseline-adjusted mixed modelling for the FACT-G overall subscale. Overall, small and non-significant differences were found between the randomisation treatment arms. Similar results were found for all subscales (see Report Supplementary Material 1, Tables 37–40).
| Estimate | Standard error | Degrees of freedom | Test statistic | p-value | |
|---|---|---|---|---|---|
| Number of observations used: 7027 | |||||
| Intercept | 23.52 | 3.62 | |||
| QoL time point | 0.09 | 0.05 | 1, 621 | 0.48 | 0.490 |
| FACT-G total score at baseline | 0.68 | 0.03 | 1, 5650 | 725.75 | < 0.001 |
| Randomisation treatment | 1, 5650 | 2.37 | 0.124 | ||
| DFIS vs. CCS | 1.17 | 0.76 | |||
| QoL time point for DFIS vs. CCS | −0.22 | 0.06 | 1, 5650 | 11.84 | < 0.001 |
| Randomised under stratification factor: Motzer/MSKCC prognostic group | 2, 5650 | 1.60 | 0.202 | ||
| Intermediate risk (1–2 factors) vs. favourable risk (0 factors) | −1.38 | 0.87 | |||
| Poor risk (≥ 3 factors) vs. favourable risk (0 factors) | −2.53 | 1.78 | |||
| Randomised under stratification factor: sex | 1, 5650 | 0.49 | 0.483 | ||
| Female vs. male | −0.60 | 0.85 | |||
| Randomised under stratification factor: age group | 1, 5650 | 0.84 | 0.360 | ||
| ≥ 60 vs. < 60 | 0.78 | 0.85 | |||
| Randomised under stratification factor: disease status | 1, 5650 | 1.94 | 0.163 | ||
| Metastatic vs. locally advanced | 3.78 | 2.71 | |||
| Randomised under stratification factor: previous nephrectomy | 1, 5650 | 3.05 | 0.081 | ||
| Yes vs. no | −1.84 | 1.05 | |||
| Randomised under stratification factor: TKI received | 1, 5650 | 4.32 | 0.038 | ||
| Pazopanib vs. sunitinib | −1.57 | 0.76 | |||
EQ-5D-3L
The median and IQR for the EQ-5D-3L utility index at each of the time points for participants in the EQ-5D-3L population are shown in Report Supplementary Material 1, Figures 41–43. Recall that a higher score indicates an improved QoL. The number of participants included in each calculation is shown below, split into counts of returned questionnaires and missing questionnaires. The total number of missing questionnaires also includes questionnaires where relevant subscale could not be calculated. We can see that there is little difference between the two arms across the majority of all time points, where the IQR for each arm overlaps. Differences between the time points become noticeable in Report Supplementary Material 1, Figure 42 when there are only a few participants remaining on trial treatment in the CCS arm, where no participants in the CCS arm remained on trial treatment after week 316. This reflects the time on trial results observed in the section Time to treatment failure.
EQ-VAS
The median and IQR for the EQ-5D-3L VAS at each of the time points for participants in the EQ-5D-3L population are shown in Report Supplementary Material 1, Figures 43 and 44. The number of participants included in each calculation is shown below, split into counts of returned questionnaires and missing questionnaires. The total number of missing questionnaires also includes questionnaires where relevant subscale could not be calculated. We can see that there is little difference between the two arms across the majority of all time points, where the IQR for each arm overlaps. Differences between the time points become noticeable in Report Supplementary Material 1, Figure 44 when there are only a few participants remaining on trial treatment in the CCS arm, where no participants in the CCS arm remained on trial treatment after week 316. This reflects the time on trial results observed in the section Time to treatment failure.
Ancillary analysis
Overall survival – piecewise model
A piecewise hazards model for OS was fitted in the PP population with two intervals, split at week 24 (see Report Supplementary Material 1, Table 41). The HR for randomisation allocation suggests that the CCS arm has a risk of death 0.94 times the risk of death in the DFIS arm. The CI suggests that at most the CCS arm has a risk of death of 0.80 times the risk of death in the DFIS arm. Comparing this to the 7.5% NI margin where the assumption of a 48.5% survival rate in the CCS arm resulted in a NI boundary of 0.812. At the 2.5% significance level, we do not reject the null hypothesis that DFIS is not non-inferior to CCS in terms of OS. There is insufficient evidence to conclude NI. This is the same conclusion as was observed in the analysis of primacy in the section Overall survival. The p-value for the goodness-of-fit statistic was 0.515, suggesting that the model is a good fit to the data.
Overall survival – Motzer
A Cox regression model, where the stratification factor Motzer score was re-categorised into two groups rather than three, was fitted in both the PP population (see Report Supplementary Material 1, Table 42) and the ITT population (see Report Supplementary Material 1, Table 43).
In the PP population, the HR for randomisation allocation suggests that the CCS arm has a risk of death 0.95 times the risk of death in the DFIS arm. The CI suggests that at most the CCS arm has a risk of death of 0.81 times the risk of death in the DFIS arm. Comparing this to the 7.5% NI margin where the assumption of a 48.5% survival rate in the CCS arm resulted in a NI boundary of 0.812. At the 2.5% significance level, we reject the null hypothesis and conclude that DFIS is non-inferior to CCS in terms of OS in the PP population. Note that this is a different conclusion to that in the analysis of primacy in the section Overall survival. The supremum test for PH showed that at the 1% significance level, Motzer continues to violate the PH assumption when considered as a two-factor categorical variable rather than three.
In the ITT population, the HR for randomisation allocation suggests that the CCS arm has a risk of death 0.98 times the risk of death in the DFIS arm. The CI suggests that at most the CCS arm has a risk of death of 0.84 times the risk of death in the DFIS arm. Comparing this to the 7.5% NI margin where the assumption of a 48.5% survival rate in the CCS arm resulted in a NI boundary of 0.812. At the 2.5% significance level, we reject the null hypothesis and conclude that DFIS is non-inferior to CCS in terms of OS in the ITT population. Note that this is a different conclusion to that in the analysis of primacy in the section Overall survival. Similar to the analysis of primacy, the supremum test for PH showed that at the 1% significance level Motzer continues to violate the PH assumption when considered as a two-factor categorical variable rather than three.
Overall survival – subgroup analysis
The results from the pre-specified subgroup analysis conducted within the PP population are shown in Figure 16. Note that the correct stratification factors were used in this analysis. In Figure 16, if the horizontal lines cross the solid vertical line, then there is no statistically significant difference between the two arms at the 5% significance level. Alternatively, if the horizontal line crosses the dotted vertical line, then there is insufficient evidence to conclude NI between the two arms at the 2.5% significance level. From this, we can see that the comparison between randomisation arms is borderline statistically significant in favour of CCS for those on pazopanib or those with two or more comorbidities. In addition, DFIS is concluded to be non-inferior to CCS in those who are overweight or obese and are in the intermediate-risk Motzer prognostic group. However, only the Motzer prognostic group at baseline has a significant interaction effect with randomisation allocation (p-value = 0.026).
FIGURE 16.
Subgroup analysis results for OS.
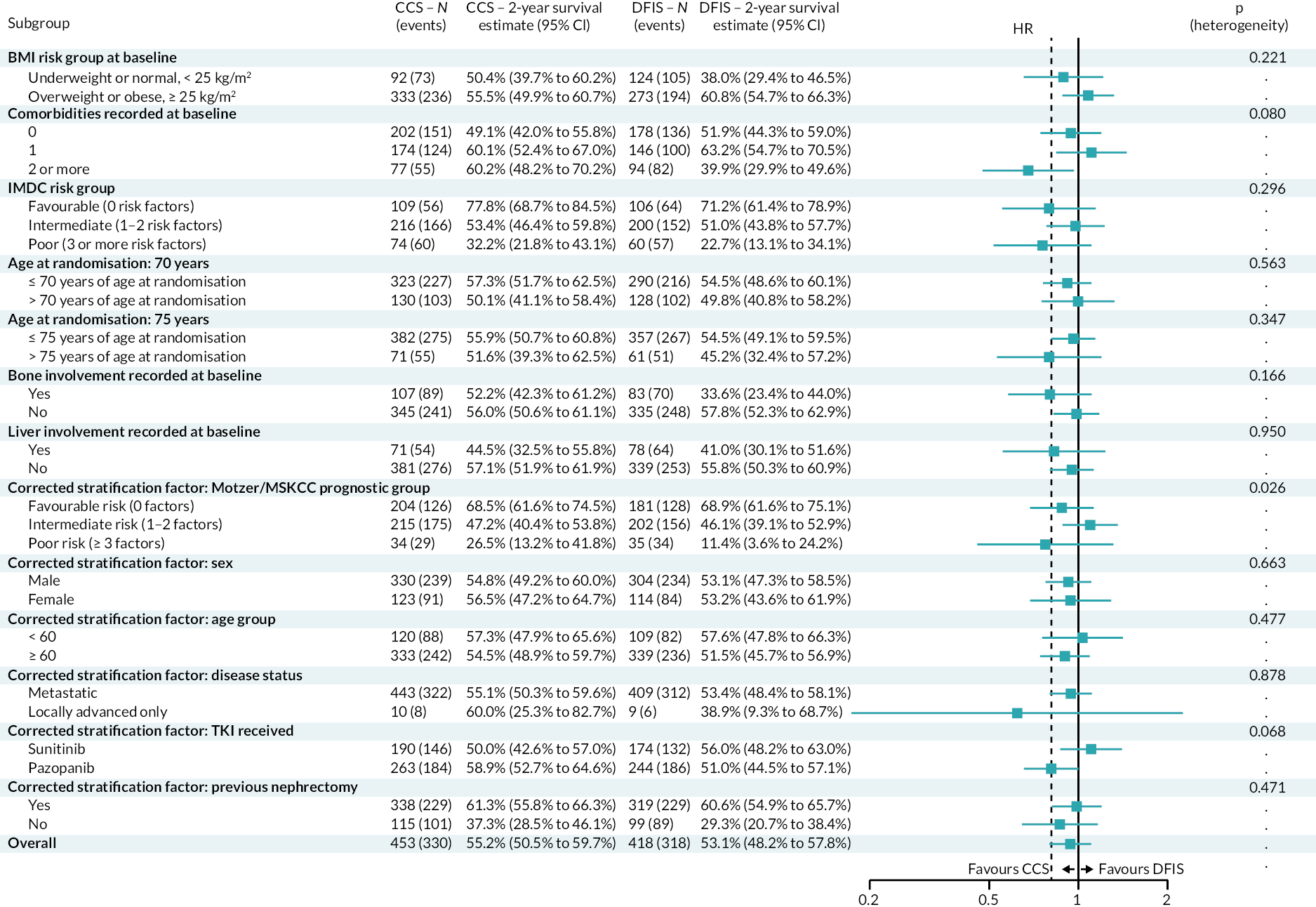
Quality-adjusted life-years – derived from week 24
Summary statistics
The summary statistics for QALYs, derived from week 24, over trial and follow-up in the PP population, were combined across the 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 1.83 (1.64 to 2.01) and 2.17 (1.95 to 2.39) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 1.48 to 1.61 in the CCS arm and 1.85 to 2.02 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM showed that on average the DFIS increases QALYs by 0.27 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0 points (see Report Supplementary Material 1, Table 44). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and therefore the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs in the PP population when measured from week 24.
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 47–49. Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 48). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. In addition, there was increased variation in the residuals as the predicted values increased (see Report Supplementary Material 1, Figure 49). However, this is much less marked than the diagnostics observed in the analysis of primacy for the QALY end point in the section Intention-to-treat population.
Quality-adjusted life-years – 12 months follow-up
Summary statistics
The summary statistics for QALYs, derived up to 12 months post randomisation in the PP population, were combined across the 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 0.47 (0.45 to 0.50) and 0.50 (0.47 to 0.53) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 0.49 to 0.49 in the CCS arm and 0.52 to 0.52 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM showed that on average DFIS increases QALYs by 0.02 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0.02 points (see Report Supplementary Material 1, Table 45). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and therefore the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs, measured up to 12 months post randomisation, in the PP population.
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figure 50–52. Overall, the residuals violated the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 51). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. Note that Report Supplementary Material 1, Figure 52 shows groups of residuals due to the way the data was transformed to perform the model diagnostics.
Quality-adjusted life-years – 24 months follow-up
Summary statistics
The summary statistics for QALYs derived up to 24 months post randomisation in the PP population were combined across the 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 0.83 (0.78 to 0.88) and 0.89 (0.83 to 0.95) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 0.77 to 0.77 in the CCS arm and 0.81 to 0.83 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM showed that on average DFIS improves QALYs by 0.02 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0.04 points (see Report Supplementary Material 1, Table 46). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and therefore the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs, derived up to 24 months post randomisation, in the PP population.
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 53–55. Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 54). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. Note that Report Supplementary Material 1, Figure 55 shows groups of residuals due to the way the data was transformed to perform the model diagnostics.
Quality-adjusted life-years – 36 months follow-up
Summary statistics
The summary statistics for QALYs, derived up to 36 months post randomisation in the PP population, were combined across the 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 1.09 (1.01 to 1.16) and 1.20 (1.11 to 1.29) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 0.87 to 0.88 in the CCS arm and 1.01 to 1.01 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM showed that on average DFIS improves QALYs by 0.03 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0.06 points (see Report Supplementary Material 1, Table 47). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and, therefore, the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs calculated up to 36 months follow-up in the PP population.
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 56–59. Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 57). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. Note that Report Supplementary Material 1, Figure 58 shows groups of residuals due to the way the data was transformed to perform the model diagnostics.
Quality-adjusted life-years – multivariate linear regression analysis
Analysis
The results of a multivariate linear regression model within the PP population combined across the 52 imputed data sets showed that DFIS improves QALYs by 0.07 points compared to DFIS, where at most DFIS reduces QALYs compared to CCS by 0.13 points (see Report Supplementary Material 1, Table 48). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and, therefore, the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs in the PP population – the same conclusion as the analysis of primacy in the section Intention-to-treat population.
Model diagnostics
The residual plots for the multivariate linear regression model diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 59–61. Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 60). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. In addition, there was increased variation in the residuals as the predicted value increased (see Report Supplementary Material 1, Figure 61). On comparison with the FMM diagnostics in the section Intention-to-treat population, we can see that while the FMM does not remove all limitations observed in the multivariate linear regression model, it does improve upon them.
Quality-adjusted life-years – complete case analysis
Summary statistics
In the PP population using complete case data, the median QALY (IQR) in the CCS arm was 1.02 (0.44–2.39) and 1.20 (0.40–2.54) in the DFIS arm. Note that 12 people were excluded from this analysis (7 CCS, 5 DFIS) due to insufficient data. The distribution of QALYs over trial and follow-up was observed to be non-normal.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM showed that on average DFIS improves QALYs by 0.02 points compared to CCS, where at most DFIS reduces QALYs compared to CCS by 0.15 points (see Report Supplementary Material 1, Table 49). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and therefore the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs derived using complete case data in the PP population.
Model diagnostics
The residual plots for the FMM diagnostics for complete case data are shown in Report Supplementary Material 1, Figures 62–64. Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 63). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level. In addition, there was increased variation in the residuals as the predicted value increased (see Report Supplementary Material 1, Figure 64).
Imputation – worst case
Summary statistics
The summary statistics for QALYs, derived over trial and follow-up in the PP population, were combined across 52 imputed data sets using Rubin’s rules. The combined mean QALY (95% CI) in the CCS arm was 1.67 (1.54 to 1.81) and 1.73 (1.58 to 1.89) in the DFIS arm. Across the 52 imputed data sets, the median ranged from 1.18 to 1.27 in the CCS arm and 1.23 to 1.32 in the DFIS arm. The distribution of the QALYs was observed to be non-normal and similar across the imputed data sets.
Analysis
The results of the marginal model derived from the combined results of the two-component FMM showed that on average DFIS increases QALYs by 0.04 points compared to DFIS, where at most DFIS reduces QALYs compared to CCS by 0.13 points (see Report Supplementary Material 1, Table 50). Comparing this to the 10% NI margin where the average QALYs in the CCS arm was assumed to be 1.56 and, therefore, the 10% margin is 0.156. At the 2.5% significance level, as the lower bound of the CI is above −0.156, we conclude that the DFIS arm is non-inferior to the CCS arm in terms of QALYs in the PP population under MNAR scenario one. As this does not change the conclusion of the analysis of primacy in the section Intention-to-treat population, no further MNAR scenarios were considered.
Model diagnostics
The residual plots for the FMM diagnostics across the imputed data sets are shown in Report Supplementary Material 1, Figures 65–67). Overall, the residuals were seen to violate the normal distribution assumption in the tails (see Report Supplementary Material 1, Figure 66). This was confirmed through the Shapiro–Wilk test for normality p-value being significant at the 1% level for all imputed data sets. In addition, there was increased variation in the residuals as the predicted value increased (see Report Supplementary Material 1, Figure 67).
Quality-adjusted life-years subgroup analysis
The results from the pre-specified subgroup analysis conducted within the PP population are shown in Figure 17. Note that the correct stratification factors were used. In Figure 17, if the horizontal lines cross the solid vertical line, then there is no statistically significant difference between the means of the two arms at the 5% significance level. Alternatively, if the horizontal line crosses the dotted vertical line, then there is insufficient evidence to conclude NI between the two arms at the 2.5% significance level. From this, a number of subgroups suggest NI between the two arms, including those on sunitinib; however, it is important to remember that this analysis is not stratified so it does not account for other factors which may influence outcome.
FIGURE 17.
Quality-adjusted life-year subgroup analysis results.
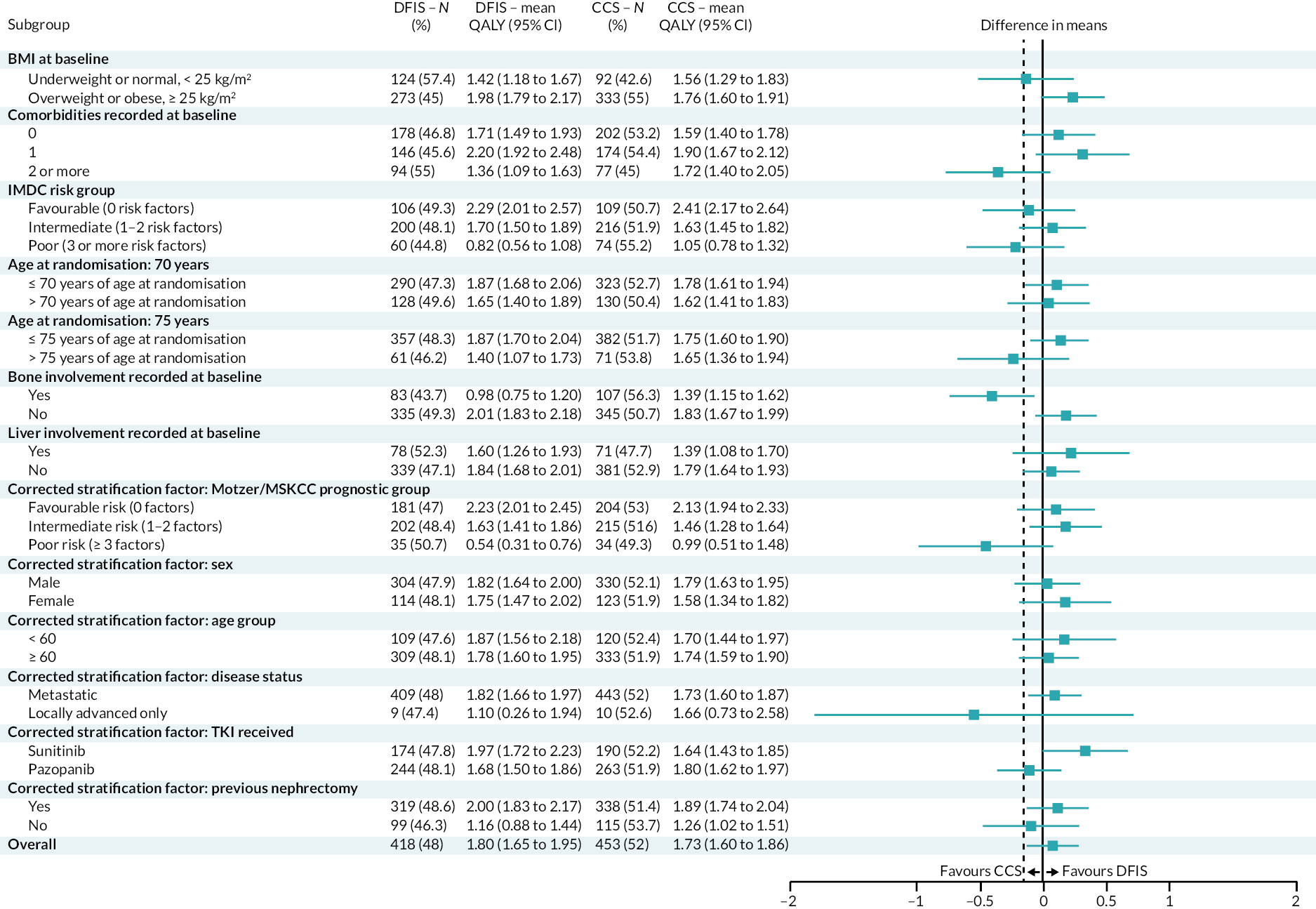
Time to treatment failure – treatment breaks
When a formal treatment break is considered to be time off treatment, the median TTF (95% CI) in the CCS arm was 7 months (5 to 8) and 7 months (6 to 8) in the DFIS arm (see Report Supplementary Material 1, Figure 68). On application of a Cox PH model, the HR for randomisation allocation suggests that the DFIS arm has a risk of treatment failure 0.97 times the risk of treatment failure in the CCS arm (see Report Supplementary Material 1, Table 51). This comparison is not statistically significant. The supremum test for PH showed that randomised under TKI violated the PH assumption at the 1% significance level. As this was a secondary end-point ancillary analysis, no additional analysis was conducted.
FKSI-15 – 12, 24 and 36 months post randomisation
The analysis conducted in the section FKSI-15 was repeated using only QoL data collected 12, 24 and 36 months post randomisation. Note that this analysis was not pre-specified and measured up to week 54, week 102 and week 156 respectively to fit with the questionnaire schedule. Interestingly, the significant results observed in the analysis of primacy in the section FKSI-15 for both randomisation allocation and the interaction term between randomisation allocation and time point are not observed until 36 months post randomisation (overall: Report Supplementary Material 1, Table 52, DRS subscale: Report Supplementary Material 1, Table 53). Neither effect was significant at 12 months post randomisation (overall: Report Supplementary Material 1, Table 56, DRS subscale: Report Supplementary Material 1, Table 57) and only randomisation allocation was significant in the FKSI-DRS subscale at 24 months post randomisation (overall: Report Supplementary Material 1, Table 54, DRS subscale: Report Supplementary Material 1, Table 55).
FACT-G – 12, 24 and 36 months post randomisation
The analysis conducted in FACT-G was repeated using only QoL data collected 12, 24 and 36 months post randomisation (Report Supplementary Material 1, Tables 58–72). Note that this analysis was not pre-specified and measured up to week 54, week 102 and week 156 respectively to fit with the questionnaire schedule. For the overall score, only the 36 months (see Report Supplementary Material 1, Table 68) post randomisation had a similar significant interaction as that in the analysis of primacy. For the Physical Well-Being subscale, none of the analyses showed the same significant effects in terms of both randomisation allocation and the interaction term. However, the interaction term was significant at both 12 months (see Report Supplementary Material 1, Table 59) and 36 months (see Report Supplementary Material 1, Table 69). For the Social/Family Well-being subscale, the results remained consistent with the analysis of primacy. For the Emotional Well-being subscale, only 12 months post randomisation (see Report Supplementary Material 1, Table 61) differed from the analysis of primacy where the interaction term between randomisation and QoL time point was no longer statistically significant. Finally, the significant interaction effect observed in the analysis of primacy for the Functional Well-Being subscale was not observed for any of the additional analyses.
Harms
Adverse events
There were 913 participants in the safety population who experienced AEs across the trial with only 3 not experiencing any. Out of 485 participants, 484 (99.8%) on the CCS experienced an AE, while 429 (99.5%) out of 431 participants on the DFIS experienced an AE. All 384 participants on sunitinib experienced an AE and 529 (99.4%) of the participants on pazopanib experienced an AE. During cycles 1–4 of the treatment, both strategies were identical. During this time, the number of participants who experienced an AE is identical to the overall summary. Only 488 participants in the safety population continued on to cycle 5 or further. Of these, 484 (99.2%) experienced an AE, 262 (98.9%) on the CCS and 222 (99.6%) on the DFIS out of a total of 265 and 223, respectively. Two hundred and twenty-two participants on sunitinib continued past cycle 5 and 220 (99.1%) experienced an AE at this point. Two hundred and sixty-four (99.2%) out of 266 participants on pazopanib experienced an AE from cycle 5 onwards.
Overall, 671 (73.3%) out of 916 participants in the safety population experienced an AE of grade 3 or above, with 343 (70.7%) out of 485 on the CCS and 378 (76.1%) out of 431 on the DFIS. Two hundred and eighty-two (73.4%) of participants on sunitinib and 389 (73.1%) of participants on pazopanib experienced AEs of CTCAE grade 3 or higher. From cycles 1 to 4, both treatment strategies were identical. During this time, 539 (58.8%) out of 916 participants experienced an AE of CTCAE of grade 3 or higher. 278 (57.3%) out of 485 participants on the CCS and 261 (60.6%) on the DFIS experienced an AE of CTCAE of grade 3 or above. Two hundred and twenty-one (57.6%) and 318 (59.8%) of those on sunitinib and pazopanib, respectively, experienced an AE of grade 3 or higher during this time. Of the 488 participants who continued trial treatment past cycle 4, 275 (56.4%) experienced an AE of grade 3 or above. These were experienced by 134 (50.6%) participants on the CCS and 141 (63.2%) participants on the DFIS. One hundred and eighteen (53.2%) and 157 (59.0%) participants on sunitinib and pazopanib, respectively, experienced AEs of grade 3 or above from cycle 5 onwards.
Of the AEs which were pre-specified to be of interest, noticeably most participants experienced fatigue (88.3%) and hypertension (69.2%) (see Report Supplementary Material 1, Table 73). In the trial, there was little difference in the reporting between randomisation allocations. However, pazopanib had a noticeably higher proportion of participants experiencing hypertension (pazopanib: 73.1%, sunitinib: 63.8%) and hepatotoxicity (pazopanib: 49.2%, sunitinib: 22.4%). Alternatively, sunitinib had a noticeably higher proportion of participants experiencing neutropenia (pazopanib: 11.1%, sunitinib: 31.3%), thrombocytopenia (pazopanib: 16.4%, sunitinib: 29.4%) and mucositis/stomatitis (pazopanib: 44.4%, sunitinib: 66.9%). Considering the maximum grade of each CTCAE (see Report Supplementary Material 1, Table 74), it seems that the differences between TKI received occur in the higher CTCAE grades.
Overall, 451 (93%, n = 485) participants on the CCS and 414 (96.1%, n = 431) on the DFIS reported other AEs. Overall, 865 (94.4%, n = 916) of participants experienced an other AE. Three hundred and seventy-one (96.6%, n = 384) of participants on sunitinib and 494 (92.9%, n = 532) of participants on pazopanib experienced an other AE. In total 13,007 other AEs were reported with 6643 occurrences from participants on the CCS and 6364 occurrences from participants on the DFIS. The majority of other AEs were of grade 1, the lowest grade, 4881 (73.5%) from the CCS and 4567 (71.8%) from the DFIS (see Report Supplementary Material 1, Table 75). Six thousand one hundred and thirteen and 6894 other AEs came from participants on sunitinib and pazopanib, respectively. There was little observed difference between the grades of the AEs from the two TKIs. Other AEs of grade 3 or above were re-categorised to the CTCAE term (see Report Supplementary Material 1, Table 76). The most commonly reported other AE of grade 3 or above was back pain (4.5%), followed by lung infection (4.5%) and abdominal pain (4.0%). Other AEs grade 3 or above which could not be coded to a CTCAE term are listed in Report Supplementary Material 1, Table 77.
Serious adverse events
Expectedness
Overall, 744 SAEs were reported on the trial, 343 (46.1%) from participants on the CCS and 401 (53.9%) on the DFIS. Considering by TKI received, there were 316 (42.5%) SAEs reported from participants on sunitinib and 428 (57.5%) from those on pazopanib. Of the 744 SAEs, 226 (30.3%) were suspected to be related to trial treatment and categorised as SARs (CCS: 31.2%, n = 343, DFIS: 29.7%, n = 401). In terms of expectedness, there were 213 (28.6%, n = 744) SAEs, suspected to be related to treatment that was expected, with 102 (29.7%, n = 343) and 111 (27.7%, n = 401) on CCS and DFIS, respectively. There were also 13 (1.7%, n = 744) SAEs, suspected to be related to treatment that was unexpected (SUSARs). Eight (2.0%, n = 401) of these were from DFIS participants and five (1.5%, n = 343) were from CCS participants. For participants on sunitinib, 108 (34.2%, n = 316) were suspected and expected, while 4 (1.3%, n = 316) were suspected to be related to treatment but not expected. For the participants on pazopanib, there were 9 (2.1%, n = 428) SAEs that were suspected but also unexpected and 105 (24.5%, n = 428) that were suspected and also expected.
Incidences of serious adverse events
In the safety population, 455 (49.7%, n = 916) participants experienced at least one SAE. This includes 222 (45.8 %, n = 485) of those on the CCS and 233 (54.1%, n = 431) of those on the DFIS. Two hundred and four (53.1%, n = 384) and 251 (47.2%, n = 532) participants on sunitinib and pazopanib, respectively, experienced a SAE. During cycles 1–4, when both treatment strategies were identical, 312 (34.1%, n = 916) of participants experienced a SAE with 159 (32.8%, n = 485) and 153 (35.5%, n = 431) on the CCS and DFIS, respectively. One hundred and ninety-five (40%) of the 488 participants who continued treatment past cycle 4 experienced at least one SAE. Of these, there were 87 (32.8%, s = 265) on the CCS in comparison to 108 (48.4%, n = 223) on the DFIS. When we consider events which were suspected to be related to treatment (SARs), there were 192 (21.0%, n = 916) participants in the safety population who experienced a SAR, 91 (18.8%, n = 485) in participants on the CCS and 101 (23.4%, n = 431) on the DFIS. There were 94 (24.5%, n = 384) and 98 (18.4%, 532) participants who experienced a SAR on sunitinib and pazopanib, respectively. During treatment cycles 1–4, when both strategies are identical, there were 64 (13.2%, n = 485) SARs on the CCS, 83 (19.3%, n = 431) on the DFIS, 74 (19.3%, n = 384) on sunitinib and 73 (13.7%, n = 532) on pazopanib. Within the 488 participants who carried on treatment past cycle 5, 52 (10.7%) experienced at least one SAR. There were 31 (11.7%, n = 265) participants on CCS, 21 (9.4%, n = 223) on DFIS, 23 (10.4%, n = 222) on sunitinib and 29 (10.2%, n = 226) on pazopanib who have experienced a SAR.
During cycles 1–4, there were 420 SAEs in total, with 207 (49.3%) and 213 (50.7%) on CCS and DFIS, respectively. There were 324 SAEs reported during treatment after cycle 5, with 136 (42.0%) in CCS in comparison to 188 (58.0%) in DFIS. In terms of both raw numbers and percentages, DFIS has more SAEs than CCS. This may be due to differences in the length of time spent on the study, giving participants on DFIS more time to experience and report SAEs. Therefore, cycle 5 onwards could represent a much longer period of time than cycles 1–4 which can explain why there are almost as many SAEs post cycle 4 as there were pre cycle 4 despite there being fewer participants, in this time period. Alternatively, when we consider the subset of SAEs and SARs which were reported for the duration of the trial during cycles 1–4 when both strategies were identical, there were 167 SARs with 71 (42.5%) on the CCS and 96 (57.5%) on the DFIS. There were 59 SARs that occurred post cycle 4: 36 (61.0%) from participants on the CCS and 23 (39.0%) on the DFIS. We can see that for related safety events post cycle 4 more were reported from participants in the CCS arm.
The mean (SD) number of SAEs reported for the participants who reported at least one SAE is 1.64 (1.08), with a mean of 1.55 (1.00) for participants on the CCS and 1.72 (1.15) for DFIS participants. The median number of SAEs, 1 (1, 10), is identical across both arms. The mean number of SAEs for participants on sunitinib who had reported at least one SAE was 1.55 (0.86) in comparison to 1.71 (1.24) for those on pazopanib. Alternatively, the mean number of SAEs reported for all participants in the safety population is 0.81 (1.12), for participants on the CCS the mean was 0.71 (1.02) and 0.93 (1.21) for DFIS participants. The median number of SAEs reported for the total population and for the participants on the CCS was 0 (0, 10). For the DFIS participants, this was 1 (0, 10). The median number of SAEs from participants on sunitinib, 1 (0, 5), was higher than that for pazopanib, 0 (0, 10), though their ranges are overlapping. Considering this only for related safety events (SARs), the mean (SD) numbers of SARs reported across participants who reported at least one SAR is 1.18 (0.45) across both arms, with 1.18 (0.38) for CCS participants and 1.18 (0.50) for DFIS participants. The median (range) number of SARs reported was 1 (1, 4) both overall and from DFIS participants. The median for CCS was 1 (1, 2). The mean and median number of SARs reported for participants on sunitinib who experienced at least one SAR were 1.19 (0.47) and 1 (1, 4). For participants on pazopanib, this was 1.16 (0.42) and 1 (1, 3). Alternatively, the mean number of SARs for all participants in the safety population was 0.25 (0.52), with 0.22 (0.49) for CCS participants and 0.28 (0.55) for DFIS participants. The median (range) number of SARs was 0 (0, 4) in both the total population and those on the DFIS. For those on CCS, it was 0 (0, 2). The mean and median number of SARs reported for participants on sunitinib were 0.29 (0.56) and 0 (0, 4), respectively. For participants on pazopanib, this was 0.21 (0.49) and 0 (0, 3).
CTCAE and MedDRA grading, outcome and seriousness of serious adverse events
The most common grade of SAEs was 3 with 404 (54.3%) of all SAEs, 190 (55.4%) of CCS SAEs, 214 (53.4%) of DFIS SAEs, 173 (54.7%) of sunitinib SAEs and 231 (54.0%) of pazopanib SAEs (see Report Supplementary Material 1, Table 78). CTCAE grade 3 remained the most common grade when only related events (SARs) were considered (see Report Supplementary Material 1, Table 79). The most common MedDRA categories for SAEs were gastrointestinal disorders; infections and infestations; and respiratory, thoracic and mediastinal disorders with 155 (15.5%), 112 (15.1%) and 89 (12.0%) SAEs, respectively (see Report Supplementary Material 1, Table 80). For SARs, these were gastrointestinal disorders; infections and infestations and vascular disorders with 70 (31.0%), 23 (10.2%) and 20 (8.8%), respectively (see Report Supplementary Material 1, Table 81).
The majority of SAEs, 80.1% (n = 744), were recovered from or recovered from with sequelae (see Report Supplementary Material 1, Table 82). A total of 35 (4.7%, n = 744) SAEs resulted in the participant’s death, 14 (4.1%, n = 343) of these were from participants on the CCS and 21 (5.2%, n = 401) from those on the DFIS. Ninety-nine (13.3%) of all SAEs were ongoing at the time of death. Thirty-nine (11.4%, n = 343) of CCS SAEs and 60 (15.0, 401%) of DFIS SAEs were ongoing at the time of death. For participants on sunitinib and pazopanib, there were 16 (5.1%, n = 316) and 19 (4.4%, n = 428) that led to death, respectively. Sixty-eight (15.9%, n = 428) of SAEs were ongoing at the time of death for participants on pazopanib, in comparison to 31 (9.8%, 316) for participants on sunitinib. Similarly, for SARs, 87.6% (n = 226) were recovered from or recovered from with sequelae (see Report Supplementary Material 1, Table 83). There were 12 (5.3%, n = 226) SARs which resulted in death with 3 (2.8%, n = 107) from those on CCS and 9 (7.6%, n = 119) on DFIS. Ten (4.4%, n = 226) of SARs were ongoing at the time of death, 6 (5.6%, n = 107) of which were on the CCS and 4 (3.4%, 119) of which were on DFIS. The number of SARs with an outcome of death from participants on sunitinib and pazopanib were 3 (2.7%, n = 112) and 9 (7.9%, n = 114), respectively. There were 4 (3.6%, n = 112) SARs ongoing at the time of death from participants on sunitinib and 6 (5.3%, n = 114) from those on pazopanib.
The majority of SAEs reported requiring or prolonging hospitalisation (89.7%, n = 796), with a similar proportion between randomisation arms (CCS: 89.9%, n = 367, DFIS: 89.5%, n = 429). Seventeen (2.1%) of all serious criteria were reported as life threatening, with 7 (1.9%) on CCS and 10 (2.3%) on DFIS. Two (0.5%) of all serious criteria reported on the DFIS were reported as jeopardising the participant or requiring an intervention to prevent hospitalisation, death or incapacity (see Report Supplementary Material 1, Table 84). This information was similar when only events which were reported to be related to TKI (SARs) were considered (see Report Supplementary Material 1, Table 85). Note that a single event can fit multiple seriousness criteria; therefore, the total serious criteria are increased compared to the number of events.
Line listings of the SUSARs can be found in Report Supplementary Material 1, Tables 86–88.
Osteonecrosis of the jaw
There have been four cases of ONJ reported during trial treatment; all four cases were in participants on sunitinib with two participants on the CCS and two on the DFIS. The full details of each case are given in Report Supplementary Material 1, Table 89.
Pregnancies
One pregnancy was reported on the trial. This was the partner of a trial participant who was receiving pazopanib and was on the DFIS arm. The pregnancy was 40 weeks and resulted in a healthy birth.
Chapter 4 Health economic evaluation
Introduction
An economic evaluation was undertaken to estimate the cost effectiveness of DFIS compared to CCS. The evaluation adhered to the (NICE75) reference case where possible. The primary analysis adopted a health and social care provider perspective and a supplementary analysis incorporated patient and carer costs and productivity losses.
Objectives
The evaluation was a cost-effectiveness analysis of the DFIS compared to the CCS in the management of patients with locally advanced and/or metastatic clear RCC receiving treatment with sunitinib or pazopanib.
The economic evaluation involved two sets of analyses:
-
Within-trial analyses using the observed data (2-year horizon).
-
Model-based analyses, extrapolating the trial results over a longer time period (lifetime horizon).
-
Value of Information analysis.
Methods
Within-trial cost-effectiveness analysis
Measurement of outcomes
The primary outcome for the within-trial analysis was cost per QALY gained for DFIS compared to CCS at 2 years post randomisation. The 2-year time point reflects the minimum follow-up length for trial participants beyond which resource use capture was limited. We chose this as our primary trial end point for the following reasons: after 2 years, greater levels of imputation are required; after treatment strategy failure, resource use was not collected and EQ-5D-3L data were collected at different time points for patients, making analyses and imputation more challenging. In consideration of these factors, the decision model was the preferred mechanism to capture longer-term outcomes.
The utility capture, valuation and QALY estimation procedures are described in the statistical analysis section. The health economists and statisticians calculated the QALYs over the whole trial independently, the estimated values were found to be the same, to two decimal places.
Measurement of resource use
NHS and social care perspective
Self-reported data on primary (e.g. GP and nurse contacts) and secondary (e.g. hospital outpatient visits and inpatient stays) healthcare use and medicines use were collected through MRU CRF at each on-study review appointment. These data were collected at baseline and then at 6-weekly intervals until treatment strategy failure.
Unit costs for resources were taken from the British National Formulary (BNF),76 Personal Social Services Research Unit (PSSRU) unit costs of Health and Social Care,77 the Department of Health’s National Schedule of Reference Costs78 and Marie Curie Cancer Care. 79 Unit costs for hospice care were also cross-checked with Public Health England’s end-of-life care economic tool80 and those for S/P were cross-checked with another previous study. 81 Appendix 5, Figure 26 includes data on the number of participants who completed selected resource use questions at each data collection point (weeks 6–40). Resource unit costs are provided in Appendix 5, Table 42 and trial medication costs in Appendix 5, Table 43. Summary data on self-reported resource use collected in the resource use questionnaires are provided in Appendix 5, Table 45.
Sunitinib and pazopanib were costed based on the cycles patients received, accounting for dose reductions. Full pack prices were costed even where cycles were not completed. Unit costs for concomitant medications are listed in an online document (n > 400), with a very large proportion of these being low-cost items. In a supplementary analysis, we costed the use of subsequent anticancer therapies; these costs are included in Appendix 5, Table 44.
Other resource uses included in the analyses were the 6-weekly on-study review appointments.
Societal perspective
Prior to the treatment strategy failure, data were also collected on out-of-pocket expenses, carer time and productivity loss (time off work). These costs were combined along with costs to the healthcare system to enable a wider cost perspective. Productivity loss was costed using the human capital approach. 82 Median daily earnings (£117.20)83 were multiplied by time off work but adjusted downwards by an elasticity factor of 0.8. 84
All costs are presented in Great British pounds (price year 2020–1) with values inflated using the NHS cost inflation index (and the hospital and community health services index for any pre-2015–6 prices) if required. All costs incurred during the second year of the trial period were discounted at 3.5%.
Missing data
The nature and pattern of the missingness of QALY and cost data were assessed. 71 Missing baseline EQ-5D-3L values were imputed using mean imputation. While the resource use measurement schedule (every 6 weeks) and form recall period (6 weeks) meant that full capture of data was possible, there was a substantial degree of missing self-reported data in both resource use items and forms, with only a very small proportion of patients providing all the necessary responses. Given this, the pragmatic approach of extrapolating backwards for missing 6-week periods was taken and a range of analyses were presented using this approach (extrapolating observed responses 12, 18 and 24 weeks backwards). The latter (24 weeks retrospective extrapolation) was adopted as the ‘complete case’ base case (n = 352). In line with the statistical analysis, patients with only one EQ-5D-3L completion were excluded from all analyses (n = 16).
Under an assumption of missing at random (MAR), multiple imputation using chained equations was implemented in Stata. This produced multiple estimates of QALYs and costs which were combined according to Rubin’s rules. 73 A range of variables were tested for inclusion in the imputation model. Included in the final imputation model were those variables used in the statistical analysis: randomisation allocation, gender, age (< 60, ≥ 60), TKI, Motzer/MSKCC group, disease status at time of randomisation and previous nephrectomy. In line with the statistical analysis, all imputation was done using the ITT population.
Cost-effectiveness analyses
The primary trial evaluation at 2 years adopted an ITT approach, with supplementary analyses exploring (PP; defined in Statistical method) analyses. This is in contrast to the main primary statistical analysis (PP with ITT as sensitivity analysis) as that was predicated on testing NI and PP is hence a more conservative approach in that circumstance.
Incremental costs and QALYs for the DFIS strategy compared to CCS were estimated using a seemingly unrelated regression (SUR)85 approach accounting for the correlation between costs and outcomes. The final model was adjusted for the stratification factors used by the trial statisticians.
Uncertainty
Nonparametric bootstrapping was used to determine the level of sampling uncertainty around the incremental cost-effectiveness ratio (ICER) by generating 10,000 estimates of incremental costs and benefits. These estimates were plotted on the cost-effectiveness plane. The cost-effectiveness acceptability curve (CEAC)86 (see Appendix 5, Figure 27) was derived by plotting the probability that bootstrapped estimates of incremental net benefit were positive. Net monetary benefit (NMB) was estimated as follows:
where λ is the willingness to pay per QALY gain. The strategy with the highest NMB (incremental NMB > 0) should be recommended if the decision-maker’s objective is to protect population health. These analyses were conducted using two different values of λ: £12,93687 and £20,000.
Sensitivity analyses
The primary within-trial analysis (multiple imputations) used backwards extrapolation of resource use over 24 weeks, multiple imputations for other missing data and estimated ICERs at 2 years’ follow-up from the healthcare provider perspective (n = 904).
Within-trial supplementary analyses reported in Table 20 were:
| Primary analysis | Supplementary analyses | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ITT population using full imputed data set (n = 904)a | ITT population using full imputed data set with societal perspective (n = 904) | PP population using full imputed data set (n = 869)a | Cases with complete QALY data (n = 856)a | Cases with complete QALYs and with up to 24 weeks extrapolation for costs (n = 352)a | ||||||
| CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | |
| Sample size | 453 | 451 | 453 | 451 | 446 | 413 | 422 | 434 | 146 | 206 |
| Mean values at 2 years | ||||||||||
| QALYs | 0.958 | 1.008 | 0.958 | 1.008 | 0.957 | 0.996 | 0.983 | 1.025 | 0.942 | 1.108 |
| Total costs (£) | 25,589.70 | 22,354.70 | 27,153.29 | 23,846.73 | 25,571.50 | 21,696.99 | n/a | n/a | 32,818.14 | 26,410.55 |
| Treatment costs (£) | 19,623.94 | 16,331.65 | 19,623.94 | 16,331.65 | 19,587.54 | 15,741.94 | 19,736.26 | 16,586.94 | 29,000.28 | 21,661.34 |
| Inpatient care costs (£) (Q1) | 2193.64 | 1789.34 | 2193.64 | 1789.35 | 2207.08 | 1809.31 | n/a | n/a | 1816.16 | 2208.33 |
| Outpatient care costs (£) (Q2) | 572.24 | 680.16 | 572.24 | 680.17 | 574.86 | 688.39 | n/a | n/a | 532.74 | 687.37 |
| Radiology unit costs (£) (Q2A) | 560.15 | 720.13 | 560.15 | 720.13 | 561.37 | 698.32 | n/a | n/a | 859.33 | 1100.92 |
| Primary and community care costs (£) (Q3) | 510.15 | 557.46 | 510.16 | 557.47 | 513.69 | 537.82 | n/a | n/a | 483.35 | 610.68 |
| Other medication costs (£) | 106.60 | 121.60 | 106.60 | 121.60 | 107.16 | 119.98 | 114.44 | 126.37 | 126.26 | 141.87 |
| On-study review costs (£) | 2022.95 | 2154.32 | 2022.96 | 2154.32 | 2019.76 | 2101.21 | ||||
| Societal costs (£) | Excluded | 1563.59 | 1492.02 | Excluded | Excluded | Excluded | ||||
| ICER and net benefit | ||||||||||
| Incremental QALY (95% CI) | 0.049 (−0.031 to 0.132) | 0.049 (−0.031 to 0.132) | 0.039 (−0.0453 to 0.122) | 0.042 | 0.165 (0.0194 to 0.3114) | |||||
| Incremental costs (95% CI) | −3235.00 (−5517.32 to −952.69) | −3306.56 (−5713.44 to −899.61) | −3874.50 (−6223.58 to −1525.41) | n/a | −6407.59 (−9882.08 to −2933.108) | |||||
| ICER (unadjusted) | −64,940.77 | −66,377.36 | −99,861.29 | n/a | −38,735.12 | |||||
| ICER (adjusted in SUR) | −62,922.54 | −63,832.11 | −98,888.39 | n/a | −41,857.21 | |||||
| Net benefit using a £12,936 threshold (95% CI) | £3879.40 (1833.91 to 5924.90) | £3924.07 (1736.49 to 6111.64) | £4376.41 (2257.93 to 6494.88) | n/a | £8907.99 (6453.69 to 11362.29) | |||||
| Probability of DFIS being cost-effective using a £12,936 threshold | 99.9% | 99.9% | 99.9% | n/a | 99.9% | |||||
| Net benefit using a £20,000 threshold (95% CI) | £4231.29 (2095.93 to 6366.66) | £4302.86 (2028.69 to 6577.01) | £4650.48 (2433.25 to 6867.70) | n/a | £9716.01 (7039.83 to 12,392.20) | |||||
| Probability of DFIS being cost-effective using a £20,000 threshold | 99.9% | 99.9% | 99.9% | n/a | 99.9% | |||||
-
Using backwards extrapolation of resource use over 24 weeks (complete case) and estimating ICERs at 2 years’ follow-up from the healthcare provider perspective but without further imputation of costs and EQ-5D-3L data (n = 352).
-
Adding a wider cost perspective (societal perspective) to the above analyses (n = 904).
-
PP analysis.
In order to test the robustness of the results, additional sensitivity analyses reported in Appendix 5, Table 46 and Figure 28 included:
-
Using alternative periods of backwards extrapolation of resource use (6 and 12 weeks).
-
Assessing plausible MNAR scenarios;71 these are based on the judgement that patients with missing data might be more likely to have experienced worse health outcomes and higher healthcare costs. In each scenario, costs and QALYs that were collected in the study (i.e. they are observed in the primary analysis) remain unchanged, but those who were imputed in the primary analysis are adjusted as follows:
-
imputed costs were increased in year 1 by 10–50% and subsequent years by 10% in the DFIS arm;
-
imputed costs were adjusted in the same way but in both arms;
-
imputed QALYs were reduced by 10–50% in year 1 and 10% in subsequent years in the DFIS arm;
-
imputed QALYs were adjusted in the same way in both arms.
-
-
Alternative approaches to costing inpatient care (as detailed in Appendix 5, Table 42)
-
Inclusion of costs of subsequent lines of anticancer medications.
Decision economic model analysis
A decision-analytic model (DAM) was developed to estimate the outcomes occurring beyond the 2-year follow-up and outwith the trial period (to a lifetime horizon). These were combined with the observed costs and benefits accumulated up to 2 years.
Model structure and parameters
The DAM was a semi-Markov model with three health states: progression-free, progressed disease and dead. This is the most common structure for oncology models88 and was adopted in the NICE appraisals of both sunitinib8 and pazopanib89 (although the latter was stated to be a partitioned survival model). Our model structure did not attempt to capture on/off treatment periods via dedicated health states; principally since, at the point of extrapolation, very few patients remained on the randomised treatment strategy. Furthermore, an individual-level simulation model would likely be required to precisely model all the transitions between on- and off-treatment states efficiently. The additional modelling complexity was not deemed worthwhile since the primary analysis modelled costs and outcomes from year 3 onwards by which time a vast majority of these transitions had already occurred (and were therefore captured in the trial data). However, the impact of treatment intervals is represented in the current model as we allow the progression-free health state to comprise proportions of patients on and off treatment.
The health states and possible transitions are illustrated in the influence diagram (Appendix 5, Figure 29). Individuals move from progression-free to progressed disease and could die in both these states. Progression was defined as treatment strategy failure (i.e. in both arms, clinical progression while on active treatment). The progression-free state was associated with higher health-related quality of life (HRQoL; EQ-5D-3L), lower costs and lower mortality risk than the progressed disease state. Being on treatment was associated with higher costs and lower HRQoL. The proportion of treatment and duration of treatment was estimated from the trial data and was considered fixed. However, separate probabilistic parameters were estimated for the mean number of treatment cycles per year across arms. The cycle length was 1 month and, in the primary analysis, the model ran from the end of year 2 to the expected lifetime of the cohort (determined to be 100 years with the mean cohort starting age of 67).
The model parameters (see Appendix 5, Table 47) were derived solely from the trial data. These included health state utility values (generated using ordinary least squares regression) and costs (generated using generalised linear model regression); and proportions on treatment, mean treatment cycles per year and probability of a grade 3 or 4 AE. Three sets of health state transitions were required: Progression-Free to Progressed-Disease [PD_PF]; Progression-Free to Dead [D_PF]; and Progressed-Disease to Dead [D_PD]. Parametric survival curves were applied to the trial data to estimate cycle-specific transition probabilities. The choice of distribution was based on fit statistics (see Appendix 5, Table 48), visual fit (see Appendix 5, Figure 30) and plausibility of the predictions (see Appendix 5, Table 49), as recommended90 by the NICE Decision Support Unit. Lognormal was used for each curve in the base case with log–logistic curves used for sensitivity analyses.
Overall survival and post-progression survival (PPS) were estimated separately, including the randomisation arm as a covariate. PD_PF were directly estimated using the time to progression survival analysis. D_PF was taken to be OS minus PPS.
For all non-fixed parameters, appropriate distributions were specified to enable a probabilistic sensitivity analysis (PSA). 91
Analysis and sensitivity analysis
Expected costs, outcomes, ICERs and net benefits were estimated over a lifetime following discounting and a half-cycle correction. Several deterministic sensitivity analyses were conducted to test the impact of parameters and modelling choices on model outputs. A PSA was also conducted using Monte Carlo simulations from parameter distributions to estimate the total parameter uncertainty in the model. Where necessary, the PSA took account of parameter correlations using a Cholesky decomposition. 91 Results from the PSA are presented in terms of the cost-effectiveness plane and CEAC.
We also used the PSA outputs to estimate the value of additional research using the Value of Information framework. 92 We recorded model outputs and associated simulated parameter values and used the non-parametric approach to estimate the expected value of perfect information (EVPI)93 and the expected value of partial perfect information (EVPPI). The relevant UK population for the decision problem was estimated to be 6550 based on an annual incidence of 13,100 and assuming 50% of these will go on to develop metastatic disease. 1
Model validation
We validated the decision model internally by conducting extreme value tests. The external validity of the model was assessed by comparing model predictions (especially those relating to expected survival for this population) to those reported by other sources such as observational studies with long follow-ups.
A payer willingness to pay per incremental QALY of £20,000 was adopted in both trial and model analyses. All analyses discounted future costs and benefits at 3.5% p.a. post 12 months randomisation. All analyses were conducted in Microsoft Excel© and Stata® 14.2.
Results
Within-trial analysis
The results from the primary analysis (ITT) and sensitivity analyses (listed in Numbers analysed including PP, and complete case analyses) are included in Table 20 and Appendix 5, Table 46. In all analyses, DFIS is found to be both cost saving and providing additional QALYs over CCS (i.e. it is a dominant strategy). The differences in total costs appear to be largely driven by the additional medicine costs in the CCS arm. There are high levels of missing data on self-reported resource use (ranging from 19% of observations for items listed under primary and community care to 45% for items listed under outpatient and inpatient care), with slightly more missing data in the CCS than in the DFIS arm (Appendix 5, Table 45). However, the general pattern across these analyses is that the greater the level of imputation or extrapolation, the smaller the differences in costs and QALYs become. This indicates that the imputation and extrapolation methods used in our primary analysis have not led us to overestimate the QALY gains or cost savings associated with DFIS.
The DFIS appears to be the optimal strategy, regardless of the sample used in the analysis and remains robust when analysis assumptions are relaxed. Figure 18 shows the cloud of ICERs on the cost-effectiveness plane; the simulated ICERs fall predominantly in the south-east quadrant meaning that DFIS is highly likely to reduce costs and increase HRQoL compared to CCS. The bootstrap analysis found that DFIS would have a 99% chance of being cost-effective. Given these results, the CEAC was not considered informative and is not included.
FIGURE 18.
Cost-effectiveness plane for the primary analysis.
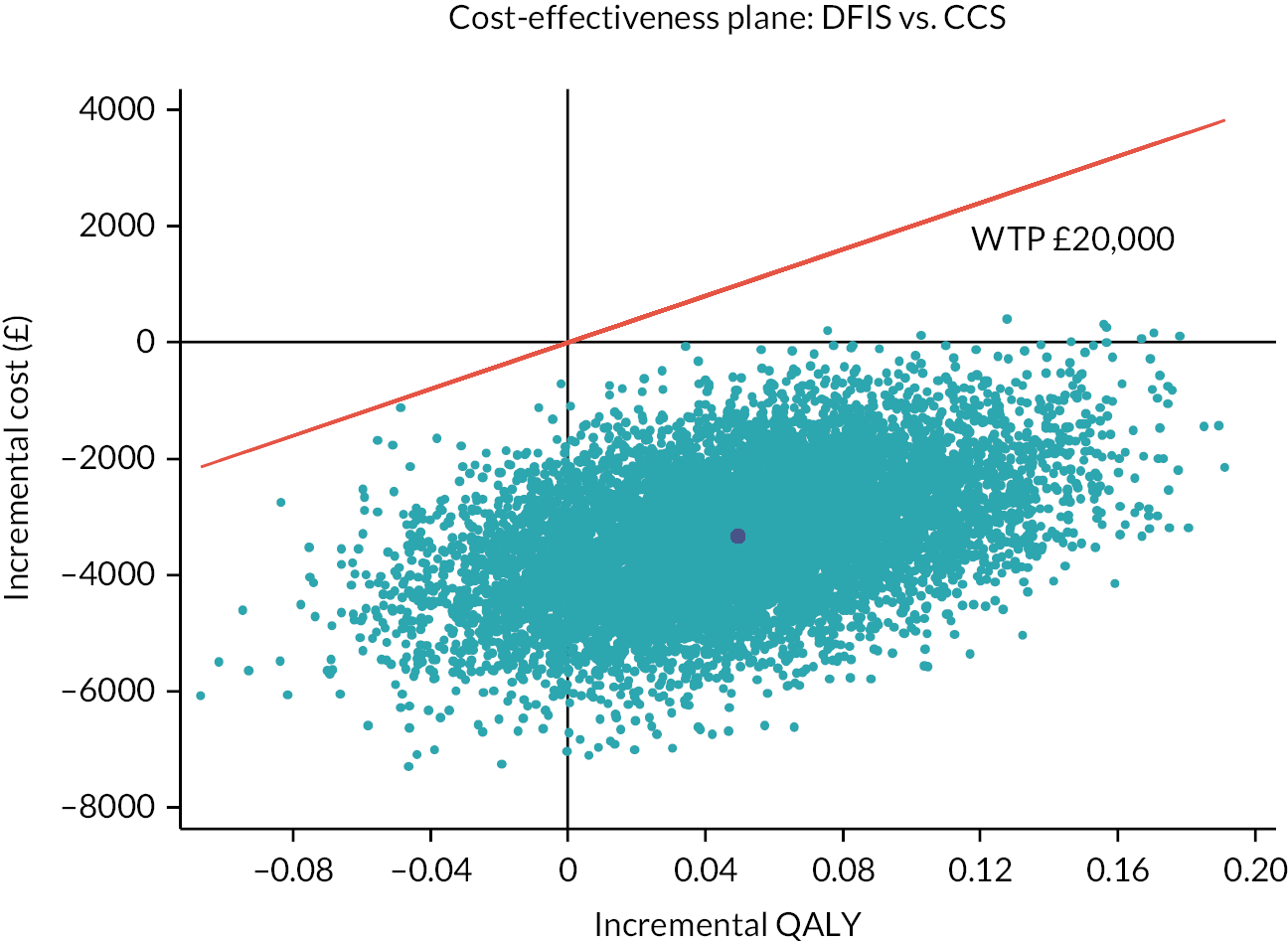
The MNAR sensitivity analyses indicated the sensitivity of the results to the MAR assumption except where changes were applied to both arms (see Appendix 5, Figure 28).
Decision model analysis
The cost-effectiveness results yielded by the lifetime decision model are included in Table 21. In all analyses, DFIS is found to be both cost saving and yielding QALY gains over CCS. This finding is robust to changes in the modelling period, discount rates and survival curve estimation. The PSA indicates that, at a willingness-to-pay threshold of £20,000 per QALY gained, DFIS has a 95% chance of being cost-effective. DFIS has a 96% chance of being cost saving and a 68% chance of yielding QALY gains. The cost-effectiveness plane, CEAC and net benefit distributions generated by the lifetime analysis are included in Appendix 5, Figures 31–33.
| Analysis | Costs | Incremental cost | QALYs | Incremental QALY | ICER | INMB | ||
|---|---|---|---|---|---|---|---|---|
| CCS | DFIS | CCS | DFIS | |||||
| Supplementary and sensitivity analyses | Costs | Incremental Cost | QALYs | Incremental QALY | ICER | INMB | ||
| CCS | DFIS | CCS | DFIS | |||||
| Base-case deterministic | £32,623 | £29,636 | −£2987 | 2.55 | 2.63 | 0.08 | DFIS dominates | £4510 |
| Base-case probabilistic | £32,039 | £29,620 | −£2420 | 2.49 | 2.57 | 0.08 | DFIS dominates | £4018 |
| Modelling whole period | £24,176 | £22,992 | −£1184 | 2.78 | 2.82 | 0.04 | DFIS dominates | £1928 |
| Including subsequent medicines | £87,854 | £75,955 | −£11,898 | 2.55 | 2.63 | 0.08 | DFIS dominates | £13,421 |
| Log-logistic curves for survival | £32,673 | £29,682 | −£2991 | 2.54 | 2.61 | 0.07 | DFIS dominates | £4309 |
| Cost/QALY discount rate = 1% | £34,031 | £31,040 | −£2991 | 2.88 | 2.96 | 0.08 | DFIS dominates | £4630 |
The EVPI (£135 per patient; £884,250 per population in year 1) is low and reflects the degree of certainty regarding which strategy is optimal. The partial EVPI bar chart indicates the absence of decision uncertainty across parameters. Only the mean (distribution scale) parameter for the OS curve is determined to have a positive EVPPI (Appendix 5, Figure 34). However, these values are low (£21.66 for the CCS OS mean) and over the relevant population (£141,773). In tandem with the overall EVPI results, given the likely patient population, the cost of decision uncertainty does not appear to warrant additional research.
Discussion
The Health Economic Evaluation of the trial and combined trial and modelled estimates of cost-effectiveness indicate that DFIS is the optimal strategy. Indeed, in all base-case and most sensitivity analyses, DFIS is shown to be cost saving and provides QALYs gains over CCS (i.e. dominant). Note that the discussions here reflect the HE analysis and not the main trial QoL analysis which considers different analysis methods.
In the modelled period of the analysis, QALY gains are increased for DFIS, likely as patients spend longer in the progression-free state, enjoying better QoL. In contrast, cost savings are reduced in the modelled period with the likely explanation being that patients in DFIS spend longer on the trial medicines. Including costs from subsequent treatments in the analysis had the effect of increasing the relative value for money of DFIS – presumably, as patients receiving intervals were less likely to progress on to expensive additional lines of therapy (such as checkpoint inhibitors). The value of information analysis indicated a low value of further research given the lack of remaining decision uncertainty.
The economic evaluation was slightly impaired by data gaps generated by unplanned missing data and by design as we did not collect healthcare resource use during follow-up. The use of routine care databases in future should provide a solution in such scenarios. Tests of the impact of the MAR assumption not holding indicate the results are relatively robust to the adopted approach. Only systematic overestimates of QALYs (by > 10%) and underestimates of costs (by > 20%) in the DFIS arm imputations would affect the results of the economic evaluation.
Future research is required on developing methodological approaches to adjusting outcomes following subsequent treatments in trials. It is possible that the full QoL impact of treatment intervals has not been captured here, in part due to the assessment schedule, missing data and the breadth and recall of the HRQoL measure we used. Thus, future research seeking to estimate the value of treatment breaks and dose reductions should adopt a more nuanced approach to QoL capture. This approach to capturing benefits should also explore patients’ preferences for such strategies, including their willingness to trade off the associated risks and benefits.
Conclusion
Drug-free interval strategy in this setting, compared to CCS, is very likely to be cost-effective and cost saving. This strategy is unlikely to lead to a reduction in survival or QoL from a health economic perspective.
Chapter 5 Qualitative assessment
Introduction
During trial design, there was concern that patients would not accept an extended treatment break because they would be stopping an effective treatment. Furthermore, there is little evidence about the psycho-social impact of stopping a potentially life-extending treatment (albeit for a limited time of up to 12 weeks) in patients with renal cancer. This uncertainty about whether stopping a successful treatment could be viewed as distressing by patients required further exploration. The QUART study94 provided evidence that an ‘integral in practice study’ usually a qualitative exploration of key issues tailored to the context of each trial could provide valuable insights to interpret the process of a study and its results.
Therefore, STAR included a qualitative assessment incorporating two studies designed as part to explore:
-
whether patients would agree to take part (study 1);
-
the experience and impact of the DFIS for modified sunitinib/pazopanib as part of the STAR trial (study 2).
Aim
The embedded qualitative studies were designed to:
-
understand whether patients would accept the offer of the STAR trial;
-
explore the experiences and impact of the novel treatment strategy for patients with RCC on physical and psychological health and well-being.
Method
Design
A qualitative inductive study design using in-depth interviews with a pre-designed topic guide was employed.
Patient and public involvement
Patients with RCC were consulted about adapting information including information sheets and topic guides so they were patient friendly. A patient recruitment video was developed for research nurses to use and to demonstrate the patient perspective.
Topic guides
The topic guides (see Appendix 6) contained questions to explore why patients had declined to take part in the study, their reasons for agreeing to join the trial and their experience of the treatment and extended breaks.
Participants
We aimed to approach 24 patients from three clinical research sites (Leeds, Manchester, Cambridge) for each study. This was increased to four sites to increase recruitment numbers (Hull). For study 2, participants would be selected at variable points in the trial, for example, some prior to their planned treatment break, some during their first or subsequent treatment breaks and all were recruited on a consecutive basis.
Inclusion criteria
For study 2, participants who had been randomised to the modified sunitinib/pazopanib arm of the STAR trial in the DFIS were invited to interview. They had to be well enough to take part in an interview either face to face or over the telephone, able to provide written, informed consent and be willing to talk about their experiences in the trial.
Exclusion criteria
Patients were excluded if they were unable to provide written informed consent, were unwell or declined to take part.
Recruitment method
Patients were approached by their clinical team and given an information sheet and a demographic questionnaire. Verbal consent to pass personal details to researchers was obtained. Patients had a week to decide to take part or decline by completing a reply slip, sending an e-mail or by telephone. The interview was scheduled and signed consent was obtained. This recruitment strategy was preferred because it minimises response bias and potentially increases the methodological rigour of the research. 95
Data management and analysis
Interviews were transcribed verbatim and managed using Nvivo. 96 Data were stored according to standard procedures at the University of Leeds. An analysis plan and coding frame were developed a priori.
A thematic analysis was used,97,98_ENREF_98. Interview data were analysed after each interview to aid additional prompts at the interview. Transcripts were read and coded for themes and subthemes. The analysis was refined by using a constant comparison and contrastive approach and identifying negative cases to examine similarities and differences between participants. 98,99 The analytical process, the themes and subthemes and the interpretations resulting from it were refined and agreed in discussions with a second researcher (JH).
Results
Study 1: acceptability of STAR trial
Study 1 employed qualitative interviews and a self-completion questionnaire to obtain views of those who declined to take part in the trial. Only one participant was referred to be interviewed for study 1. They declined trial participation due to perceived health reasons, not the treatment break. Recruitment to the STAR trial itself was very successful, so the study of decliners – intended to facilitate recruitment – was discontinued in Phase 3.
Study 2
Participant sample
Seventeen participants were invited to interview, six chose to opt out and received no further contact. Of the patients who declined to take part, most felt too unwell to do the interview, one did not want to talk about treatment or illness in an interview and one experienced a bereavement. During the interviews that were conducted, 2/11 became upset and distressed when focusing on side effects and personal loss of work and social lives due to illness. The research nurses also experienced problems identifying patients for the substudy due to competing studies, patients being too unwell or with complex needs and staff absence.
Eleven participants, median age of 69 years (54–79 years), took part. The original sample of 24 patients was not achieved, but after 9 patients, we were not collecting any new themes, so we thought we had good information saturation at that point. We recruited a further two patients to check this.
Table 22 describes characteristics of participants interviewed and Table 23 the sites and number of extended treatment breaks. Patients were interviewed after their treatment breaks and some had more than one. The accounts of patients were positive regardless of the number of breaks (see Study limitations).
| No | Age | Sex | Education | Occupation | Religion | Marital status | Ethnic Group |
|---|---|---|---|---|---|---|---|
| 001 | 56 | M | University | Professional | None | Married | White British |
| 002 | 70 | F | University | Retired | None | Married | White British |
| 003 | 61 | M | Secondary | Skilled manual | Christian | Married | White British |
| 004 | 58 | F | Secondary | Professional | Christian | Married | White British |
| 005 | 54 | M | Secondary | Skilled manual | None | Divorced | White British |
| 006 | 76 | M | Secondary | Retired | Christian | Married | White British |
| 007 | 79 | M | University | Retired previously professional | Christian | Married | White British |
| 008 | 69 | M | College/diploma | Retired | Christian | Married | White British |
| 009 | 67 | F | Secondary | Retired | Catholic | Widowed | White British |
| 010 | 70 | M | Postgraduate | Professional | No religious beliefs | Married | White British |
| 011 | 70 | M | College/diploma | Retired, previously professional | Christian | Married | White British |
| No | Site | Treatment break |
|---|---|---|
| 001 | 2 | Had one break |
| 002 | 3 | Prior to first break |
| 003 | 3 | Had one break |
| 004 | 1 | On third treatment break |
| 005 | 1 | Had two treatment breaks |
| 006 | 1 | Had one break |
| 007 | 1 | Had one break |
| 008 | 4 | Had one break |
| 009 | 4 | Had two breaks |
| 010 | 1 | Had one break |
| 011 | 1 | Had one break |
More men (8) than women (3) were interviewed which reflected the percentage of males to females in the sample that consented to take part in the trial. Although the qualitative assessment was aimed to purposively select participants based on ethnic and cultural background, those who agreed to take part were all white and British. Participants across all four sites had different educational and professional backgrounds.
Interviews
Ten interviews were conducted face to face and one over the telephone. Two out of 11 participants were upset during the interview with recording stopped and only resumed if the participant wished to continue. Follow-up discussions with nurses were made in both cases.
Analysis and themes identified
The initial coding related to the questions on the topic guide. Further refinement of themes identified three themes:
-
Rationale and decision to take part in the STAR trial.
-
QoL while on treatment.
-
QoL during the extended treatment breaks.
Rationale and decision to take part in the STAR trial
Participants were clear about the options being offered, the comparison between treatment strategies and that the allocation of the treatment arm was done by a computer.
Concerns about taking part in the trial
The main concerns in terms of taking part were whether it was better to continue treatment if the treatment was working:
well I was mildly concerned as to whether being on the trial with a break was a bad thing because you were having a break … it was there at the back of my mind and to an extent it still is, as to whether the prolonged treatment would be better.
PU1020 (n = 1 of 3)
However, the participants were aware that chemotherapy treatment could take a toll:
Yes. And I thought that would be beneficial, you know, to have that respite if it was, if the effects were considerably adverse, it would seem sensible to have a break.
PU 0110 (n = 2 of 3)
Some sought reassurance that there would be regular monitoring as part of the trial:
I mean, the information assured that we would get the same check-ups and the same treatment as we would have got if we’d continued on the drug, and if there were any concerns that I could … go back onto the drug or, you know, have further tests and I was confident that I wouldn’t suffer for being off the drug for a lengthy period.
PU1002 (n = 1 of 4)
Deciding to take part in the STAR trial
Participants’ reasons for taking part reflected those recorded in the literature. They wanted to help others and access the best clinical care.
Two reasons. One because I thought it might help other people in the future because you’re a little bit of a guinea pig to a certain extent and two, I actually thought I would receive more attention, you know, examinations and tests by being on the study …
PU2001 (n = 1 of 8)
Those in this study described having great trust in the health professionals providing their care and involved in the trial:
I trust the people that are treating me , and if they reckon I need to be treated more, you know, that’ll happen. And I know that if I have any symptoms that are worrying. I can pick the phone up and speak to somebody and it’ll be dealt with.
PU 0102 (n = 1 of 11)
Quality of life on treatment with sunitinib
All participants in this study described extensive side effects while being on treatment. This included fatigue, sickness and diarrhoea, sore mouth and feet, changes to appetite and hair changing colour amongst other things. All reported three or more side effects (see Table 24).
| Pt no. | Sore feet and hands | Sore mouth | Diarrhoea | Loss of appetite/changes to taste | Sickness | Changes to hair | Fatigue | Anxiety | Hypertension | Indigestion |
|---|---|---|---|---|---|---|---|---|---|---|
| 001 | x | x | x | x | x | |||||
| 002 | x | x | x | x | ||||||
| 003 | x | x | x | x | x | |||||
| 004 | x | x | x | x | ||||||
| 005 | x | x | x | x | ||||||
| 006 | x | x | x | x | ||||||
| 007 | x | x | x | |||||||
| 008 | x | x | x | x | ||||||
| 009 | x | x | x | x | x | x | ||||
| 010 | x | x | x | x | x | x | ||||
| 011 | x | x | x | x |
I also feel, on occasions, a bit lethargic so yeah, I just get tired and a bit weary. No, but it’s just unpleasant to eat and then you think, well why bother!
PU1411 (n = 1 of 18)
the sort of side effects you get is I get sore mouth, loss of taste, the taste buds go, sore mouth, very sore mouth, sore feet, I’ve had to, in the past I’ve had to use a walking stick because it’s like walking on glass, I get sores round me backside and round me scrotum, and sores on me hands in the joints of the fingers, see the little red bit there?
PU1331 (n = 2 of 18)
The participants were aware that they would have to put up with side effects as part of treatment. With the help of over-the-counter medications, they could cope with most of these. Some had their dose reduced to reduce the impact of the side effects. The longer they were on treatment, the greater the side effects and some said it took longer for the symptoms to resolve.
As part of standard treatment, participants could be offered a short break from treatment to help alleviate the side effects (depending on clinical status). Other studies on S/P describe participants having to stop treatment due to the severity of the side effects. 6 The short break was welcomed but some participants had only just recovered from the side effects and this was not thought to be long enough to improve QoL:
I think I would have got fed up of being on the treatment you know, being constantly on it and not being monitored as much, I don’t know …. I didn’t want to be on it all the time, that was me only you know, sort of grudge, I didn’t want to be in it constantly, I think I would have got worn down.
PU2413 (n = 1 of 5)
I think with hindsight, I’m not sure I would be able to, would really want to tolerate being on the treatment all the time because at the end of the four, weeks it was a bit grim I thought, so in terms of quality of life I think, I’m glad I’m on this side of the trial.
PU5111 (n = 2 of 5)
Quality of life during extended breaks
Chemotherapy treatment with extended break – benefits and harms
The main problem with the extended treatment break was regrowth or disease progression and the extent of this and what it meant in terms of treatment or care:
One of the side effects after I’d stopped, I think it was after I’d stopped taking the tablets, or thereabouts anyway, was that I started to pass blood in my urine, which is the effect of the tumour in my bladder.
PU1122 (n = 1 of 6)
I was a bit disappointed with the results after being on the three months off because it had spread to my liver which I didn’t have cancer on before, and the other ones had increased, one had doubled in size, and I thought, ‘oh heavens’, we thought, I expected after the break stuff to start moving you know, the ones I have get bigger again, the ones that had been decreased, but even [Dr X], I don’t think he was expecting a spread to somewhere else.
PU2841 (n = 1 of 9)
Both participants were in touch with clinical teams and wanted to continue with extended breaks.
The degree to which the disease had progressed was concerning and some monitored the percentage change in their tumours each time:
Well it was best part of nine months, … the tumours … in my chest had shrunk by about 10%, yeah, and after the three months …. Of course, they’d grown again by 10% but I thought to myself [sighs] and …, then after the first four, … Immediately they’d gone back down 9, 10% so I’m thinking, phew, at this moment in time it’s in control, you know.
PU1121 (n = 1 of 6)
So when I’ve got to my 24th week I’d lost nearly 50% of the mass so eh, that’s good so they decided there and then because of the trial and whatnot, it would be good to stop, fine, not a problem, so I had me 12 weeks off than I had me scan and then I’d put nearly 50% back on so I started the treatment again and it’s the same, I’d lose 50%, put 50% back on.
PU4513 (n = 2 of 6)
These changes were associated with the next scan which was seen as a source of anxiety and worry for participants:
On occasions I’ve found the scans not too much fun and I think it’s, I think it’s a contrast thing, that it doesn’t have any dramatic effect but I just feel unwell so that’s a bit of a downside.
PU1112 (n = 3 of 6)
… it was just, it was just a bit of a body blow but you pick yourself up and you get on with it.
PU1512 (n = 4 of 6)
Despite these issues, all those interviewed had a positive view of the extended treatment breaks, although this was balanced with concerns about tumour growth during the treatment breaks. Despite the risk that the tumour could increase in size during the breaks, they were all keen for some respite from treatment with significant side effects and impact on their daily lives. In addition, it is important to note that any anxiety and concerns around scans may reflect the views of participants in both arms as the scan schedule was the same and reflects standard practice.
there are no disadvantages, I think they’re all advantages of having the bigger break because it enables your body to get used to being back to normal, doing normal things, even in your mind doing normal things and getting yourself and your body ready and your mind in the right set for the next onslaught of the next pills that come along.
PU2331 (n = 1 of 12)
well, I mean like I say, when I get to that, that 24th week and they say ‘Right, you’re on an extended break’ it’s ker-ching, it’s good times, it fills you, it gives you some hope.
PU 4512 (n = 1 of 12)
Discussion
Recruitment to the trial and the substudy
Recruitment and retention figures in the STAR trial were high. The accounts of participants showed that anticipated benefits of QoL became a reality. The recruitment figures speak for themselves: many eligible patients were content with the treatment alternatives represented by STAR. The qualitative work, which must be interpreted in that context, affords insight into some more specific reasons for acceptability. Participants were asked directly why they had taken part, but responses to other questions also threw light on the appeal of participation from their point of view.
The risk of harm continued to be acknowledged and was seldom far from respondents’ minds, but they did not regret their decision to take part in the trial, even if they had personally experienced the return of unpleasant symptoms or clinical progression. They understood that allocation to the DFIS arm of the STAR trial would entail an early and fairly predictable improvement in daily life. They were less clear about how long treatment might be effective for them, as it was tailored to their clinical circumstances. The question of whether the break might prove later to have been enjoyed at the expense of an unpredictable reduction in treatment effectiveness was not at the forefront of their minds. They were happy that two treatment strategies were being assessed and that the health professionals caring for them had their best interests in mind.
The initial context for STAR was favourable: participants trusted their doctors not to suggest anything that was against their interests. As in other studies, ‘wanting to give something back’ and altruism featured in accounts,100 as did having few other options. In STAR itself, if allocated to the DFIS arm, participants knew they would be monitored closely and reinstated on drug treatment if the need arose. They had high levels of resilience (evidenced by strategies for getting on with usual activities) and good support networks.
Also relevant was the nature of the treatment under evaluation: unlike non-crossover intervention trials (common), in the experimental DFIS arm of STAR, at the point of disease progression off treatment, participants were planned to re-start previous treatment and could then continue without treatment breaks as per participant choice. Of note, a number of participants chose to have multiple planned treatment breaks (see Treatment breaks).
Quality of life in treatment breaks
The accounts suggest that the DFIS was not just acceptable, it was an attractive option to many and a definite preference for some. They welcomed the opportunity to feel more able to resume valued activities in their lives, even if only temporarily. It follows that STAR recruitment may have been enhanced because a treatment option preferred by many patients was not available outside of the trial. Preference trials such as ProtecT, comparing very different treatment modalities, in this case, active monitoring, surgery and radiotherapy, would not benefit from this kind of effect as all arms were available outside of the trial and patients would choose their preferred arm rather than being randomised to one of the three options. 101
Study limitations
A highly selected group of participants in the DFIS arm of the STAR trial were interviewed. These participants had maintained resilience, hope and personal social networks of support. Participants on the continuous arm, those who dropped out or participants for whom treatment was no longer working were not interviewed. It is possible that an alternative research design that incorporated the perspectives of those on the continuous arm would enable a more in-depth comparison of long to short breaks. This may have illustrated additional psychosocial issues. A further design issue is sampling patients with different numbers of treatment breaks. Ideally, you would stratify the sample to have equal numbers in each category, so this limited any comparison of those experiences.
We were not able to reach the recruitment target, although we believe we reached sufficient information saturation about the issues being evaluated. The main pressure on recruitment was the limited number of sites for the qualitative substudy and staff changes at the site. A high number of participants who agreed to be contacted declined an interview (6/17). It is possible that the qualitative assessment did not include the less resilient or those living alone or who were deemed not well enough to take part due to the protective actions of the nurses caring for them. The additional burden of taking part in an interview should not be underestimated. Those trial participants who took part in the interviews were required to focus on the adversity of diagnosis and treatment and it was made clear that interview participation was entirely voluntary.
Implications for care
If such a strategy was implemented in practice, some thought should be given to how patients should be supported during the extended break to cope with and alleviate worries. The overall trial results will help with this, as no substantial detrimental effect on OS was shown. It will be important to support patients to maintain resilience and receive adequate information about disease progression and tumour regrowth along with professional and peer support. As part of this, patients on a treatment break require close monitoring and access to rapid clinical assessment if they become unwell.
Implications for trials
For understandable reasons, much of the literature on the added value of qualitative work to the conduct of trials has focused on recruitment,102 and initially, that was also the focus in STAR. When recruitment proved not to be a problem, attention switched to understanding the experience of receiving the novel treatment and how that might relate to the acceptability of DFIS as a distinct departure from usual care.
By collecting data on the experience of being in the intervention arm, STAR extends the methodological literature on the role of qualitative research in trials. The data throw light on the acceptability, even attractiveness, of the intervention and the reasons for that acceptability, which in turn help explain the high recruitment rate. A distinctive feature of STAR is that eligible patients could relate to both arms of the trial: the DFIS form of ‘active monitoring’ to such experienced patients held considerable appeal and contributed to their willingness to take part in the trial.
More generally, the experience of STAR is a reminder that even when a patient’s treatment options entail materially different trade-offs between process and outcome, some people will wish to weigh up of pros and cons – including those relating to uncertainty – before a treatment decision is made. The usual care approach of maximising effectiveness within the limits of tolerability will undoubtedly be the preferred option for some, but it is unlikely to be the preferred option for all.
Quality note for this chapter
All themes and subthemes that were used for this chapter had to have at least three narratives from different participants to be agreed as a theme, most themes had many more than that. The results reported here are an edited version with a single quote to illustrate each.
Chapter 6 Magnetic resonance imaging substudy
Introduction
Imaging substudies are increasingly being embedded within RCTs for cancer treatment, as this provides a unique opportunity to perform translational studies within it, with the aim to identify key imaging factors that may provide information regarding early prediction of treatment response for responders and non-responders to sunitinib or pazopanib. In this translational substudy, DCE-MRI was used to see if it was possible to predict patients who will respond to sunitinib or pazopanib and those who will not, earlier than with the current 12-weekly CT scanning approach. If this enables accurate prediction of responses, this would allow non-responding patients to stop treatment earlier, hence limiting exposure to unnecessary toxicity and earlier access to second- and third-line treatments. The standard imaging assessment used to assess response to cancer treatments is usually based on CT. However, MRI as a functional imaging tool offers the advantages of no ionising radiation exposure with better contrast resolution. DCE-MRI can be used to assess tumour perfusion.
The MRI imaging substudy nested within STAR explored whether early DCE-MRI parameters could be used as a biomarker to predict PD at 24 weeks after initiation of TKI therapy in patients with advanced RCC treated with sunitinib or pazopanib. The DCE-MRI technique assessed the change in perfusion of the target lesions at baseline (prior to TKI treatment), 4 weeks and 10 weeks after treatment of advanced RCC with sunitinib or pazopanib. DCE-MRI-based parameters assessed included perfused tumour volume, the transfer constant Ktrans, extracellular volume (ECV) and extracellular mean transit time (MTT).
Methods
Design
Participant recruitment and intervention
All participants in the substudy were identified from participants of the STAR trial recruited from a single tertiary cancer centre. Participation was optional and consent for the substudy was sought at trial registration.
Inclusion criteria
-
Measurable disease within the abdomen or pelvis.
-
For patients with bony metastases, only those with a measurable soft tissue component were included.
-
Consent to substudy participation.
Exclusion criteria
-
Non-measurable disease within the abdomen or pelvis.
-
Unwilling to take part.
All participants were required to have both baseline CECT and DCE-MRI scans before the commencement of TKI treatment.
The treatment response at 6 months was assessed by CECT using RECIST version 1.1 criteria.
Imaging assessment with dynamic contrast-enhanced magnetic resonance imaging
All DCE-MRI examinations were performed on a Siemens (Erlangen, Germany) 1.5 T system. The DCE-MRI assessed up to five target lesions (the largest five) within the abdomen and pelvis identified at baseline (before TKI initiation), 4 weeks (post initiation of TKI therapy) (± 4 days) and 10 weeks (± 4 days) after STAR trial randomisation.
A detailed description of the dedicated imaging technique and parameters was published. 103
Post dynamic contrast-enhanced magnetic resonance imaging acquisition image analysis
The DCE-MRI imaging data were anonymised and post-processed using the software Platform for Research in Medical Imaging Version 0.4 (PMI 0.4). The post-DCE-MRI acquisition image analysis to assess the parameters such as perfused tumour volume, Ktrans, ECV and MTT were described in detail103 by two experienced observers.
Statistical analysis
Two-tailed paired t-tests were used to analyse the change in the DCE-MRI parameters between the three time points (baseline and 4 weeks, 4 weeks and 10 weeks and baseline and 10 weeks). The differences in DCE-MRI parameters between the participants with PD at 24 weeks and those with no progression were evaluated using an independent samples t-test for normally distributed data and the Mann–Whitney U test was used for non-normally distributed data determined by using a Kolmogorov–Smirnov normality test. For participants with more than one lesion identified on the DCE-MRI, only the largest lesion was selected to analyse the changes to tumour perfusion characteristics over the three time points in relation to the primary end point as these were least affected by partial volume effect. Receiver operating characteristic (ROC) curve analysis and AUC values were calculated for parameters that were associated with PD at 24 weeks. The statistical significance level was set at p < 0.05. All statistical tests were performed using SPSS Statistics software (Version 21.0; IBM Corp., Armonk, New York, USA). Interobserver agreement was assessed using the intraclass correlation coefficient (ICC) with ICC values scored as excellent (> 0.81), good (0.61–0.80), moderate (0.41–0.60), fair (0.21–0.40) and poor agreement (< 0.2).
Results
A total of 14 participants were included in the translational MRI substudy of the STAR trial, after the exclusion of five participants due to claustrophobia (n = 2) and non-measurable diffuse disease on MRI (n = 3) (see Figure 19). All participants included in this DCE-MRI substudy were on TKI treatment prior to taking up their randomised allocation.
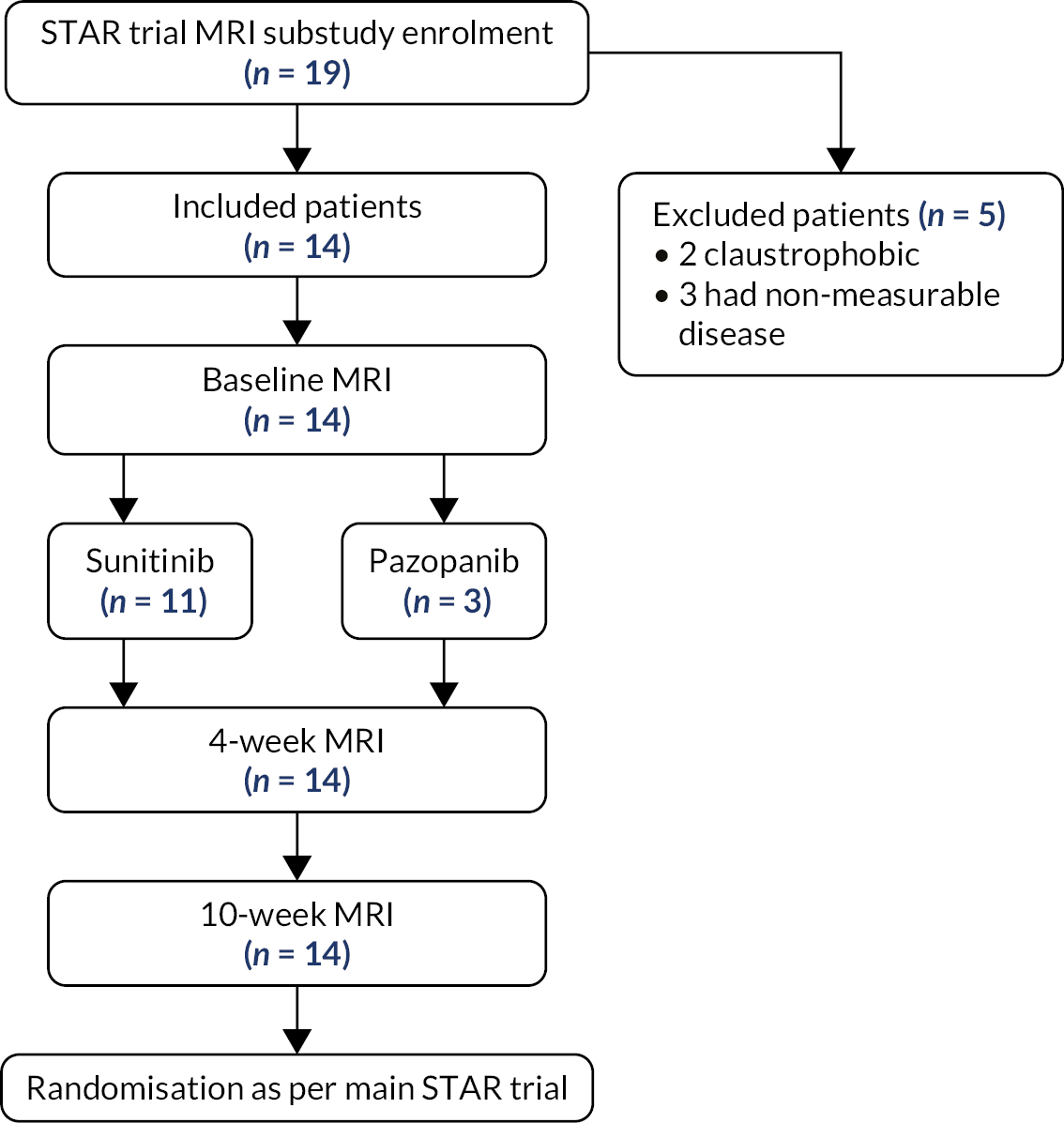
There were 12 male and 2 female participants with a median age 64 years (range 52–77). Their median Karnofsky performance was 90% (range 80–100) and their baseline treatment information is shown in Table 25. Within this cohort, 10 had SD, 1 had a PR and 3 participants had PD at 24 weeks.
| Participant | Prior nephrectomy | Sites of disease/index lesions | TKI therapy | PD at 24 weeks |
|---|---|---|---|---|
| 1 | Yes | Nodal | Sunitinib | No |
| 2 | Yes | Spleen/stomach | Sunitinib | No |
| 3 | Yes | Nodal | Sunitinib | No |
| 4 | Yes | Liver (2) | Sunitinib | Yes |
| 5 | Yes | Nodal | Sunitinib | Yes |
| 6 | No | Kidney | Sunitinib | No |
| 7 | No | Kidney | Pazopanib | No |
| 8 | No | Kidney | Sunitinib | No |
| 9 | No | Kidney | Pazopanib | No |
| 10 | Yes | Nephrectomy bed/nodal | Sunitinib | No |
| 11 | Yes | Nodal (2) | Sunitinib | No |
| 12 | Yes | Kidney (2)/liver/pancreas (2) | Pazopanib | No |
| 13 | No | Kidney/pancreas | Sunitinib | Yes |
| 14 | No | Kidney | Sunitinib | No |
There were 23 separate measurable tumours and the target lesion sites were: kidney (n = 8), nodal (n = 6), liver (n = 3), pancreas (n = 3), stomach (n = 1), spleen (n = 1) and renal bed (n = 1). The time-intensity curves for each tumour volume segmented were produced103 to which a single compartment model was fitted to provide estimates of the perfusion parameters. The largest lesion per participant was selected for further analysis of the perfusion parameters. The perfused tumour volume (cm3), Ktrans (minute−1), ECV (ml/100 ml) and ECV MTT (s) estimates per participant for each tumour at every study time point with percentage changes were published in detail. 103
The median perfused baseline tumour volume was 77.5 cm3 (range 2.5–880). Across the time points from baseline to 4 and 10 weeks, the median perfused tumour volumes were variable with a reduction of perfused volume; the median percentage change was −48% from baseline to 4 weeks, (range −92 to +8.6%) (p-value < 0.001) and −32.8% from baseline to 10 weeks (range −93 to 83%) (p-value 0.01).
There was a statistically significant reduction of mean Ktrans (minute−1) (± SD) from baseline (0.96 ± 0.63) to 4 weeks (0.37 ± 0.24) (p-value 0.006) and from baseline to 10 weeks (0.46 ± 0.51) (p-value 0.033) (see Figure 20). In addition, there was a statistically significant difference in the mean absolute change in Ktrans between 4 and 10 weeks in the 24 weeks disease progression group when compared to the non-disease progression group at 24 weeks, + 0.44 minute−1 and −0.004 minute−1, respectively (p-value 0.038).
FIGURE 20.
Mean Ktrans (minute−1) at baseline, 4 weeks and 10 weeks of the substudy population.

The DCE-MRI parameters that were found to be associated with early disease progression at 24 weeks: the percentage change in the perfused tumour volume from baseline to 4 weeks (p-value 0.016), the change in Ktrans from 4 to 10 weeks (p-value 0.038) and the percentage ECV change from 4 to 10 weeks (p-value 0.009).
Interobserver agreement
The interobserver agreement was excellent; perfused tumour volume [ICC (95% CI): 0.928; (0.869 to 0.959)]; Ktrans [ICC (95% CI): 0.949; (0.918 to 0.969)] and ECV [ICC (95% CI): 0.910; (0.800 to 0.961)].
Discussion
Renal cancer biology is characterised by angiogenesis and increased vascularity as a result of increased expression of VEGF leading to endothelial proliferation and neo-vessel formation. Therefore, RCC is an optimal target for measuring tumour perfusion, and it is particularly relevant in the context of evaluating the efficacy of anti-angiogenic TKIs which inhibit VEGF receptor signalling, and reduction in the microvascular density. The changes in microvascular density have been shown to correlate with treatment response and resistance to anti-angiogenic therapy.
Dynamic contrast-enhanced magnetic resonance imaging-derived quantitative parameter, Ktrans, may serve as a surrogate for tumour blood flow and provide non-invasive imaging assessment of microvascular function. The important findings of this translational DCE-MRI substudy were the absolute and relative changes in DCE-MRI-derived quantitative parameters (perfused tumour volume, Ktrans and ECV) at the 4 and 10 weeks post TKI initiation and were correlated with early PD at 24 weeks.
This is the first clinical study to use longitudinal serial assessments to detect changes in quantitative DCE-MRI biomarkers following sunitinib or pazopanib treatment in mRCC. The decrease in Ktrans measurement at 4 and 10 weeks when compared to baseline after TKI therapy is similar to previous studies. The perfused tumour volume reduction at 4 weeks could be due to early changes in microvasculature caused by TKI therapy, which can occur within 3 days post initiation of treatment. Previous studies have shown a reduction in Ktrans with tumour response post TKI therapy. 104,105 These findings suggest that biomarkers of angiogenesis inhibition could be an important independent predictor of outcome.
In the DCE-MRI substudy cohort of participants, the increase in Ktrans between 4 and 10 weeks was correlated with disease progression at 24 weeks, despite all these participants still having SD by RECIST criteria based on CT assessment at 10 weeks. This finding has again supported the potential capability of Ktrans to be an early imaging biomarker of treatment response before a change in tumour size is observed. For those three participants with early disease progression, the rising Ktrans between 4 and 10 weeks may indicate early signs of TKI resistance and/or disease relapse.
The strength of this translational study is its inclusion within a large-scale Phase III clinical trial with high-quality and robust data management. Limitations include the small sample size of the substudy, the use of target lesions from a variety of organ sites and a number of the target lesions being close to other well-perfused structures, for example, spleen, liver or abdominal arteries leading to some difficulty in accurate segmentation. The STAR trial included two TKI therapies, S/P; however, based on previous DCE-MRI studies, this was not expected or seen to alter the trends in the reduction of Ktrans due to being the same class of drug with equivalent efficacy. 13
Conclusions
This feasibility study has shown DCE-MRI-derived biomarkers of tumour perfusion (perfused tumour volume, Ktrans and ECV) as potential surrogate biomarkers to predict early disease progression following TKI therapy in advanced RCC. The study has also demonstrated these DCE-MRI assessments to be reproducible. Further larger prospective clinical studies are required to test its wider application in the context of routine clinical practice.
Chapter 7 Computerised tomography substudy
Introduction
Imaging-based evaluation of response with CT is the mainstay for therapy assessment in mRCC. 106,107 Initial landmark trials reported over 10 years ago led to TKI becoming the standard first-line systemic treatment for patients with advanced RCC due to improvements in PFS and OS. 5,12,108,109 More recently, further trials have established the utility of alternative agents including immunotherapies. 48,110
Anti-angiogenic multitarget receptor TKIs such as sunitinib, pazopanib and cabozantinib block VEGF-1,2,3 (VEGF receptors) as well as other receptors including platelet-derived growth factor receptor (PDGFR), c-kit, c-MET (hepatocyte growth factor receptor) and rearranged during transfection (RET). Although these therapies may induce tumour devascularisation and necrosis,111 there may be a delay in the reduction of absolute tumour size, as opposed to traditional cytotoxic agents, potentially undermining the ability of standard size-based response criteria such as RECIST112 in evaluating early benefit at the earliest. Categorisation of PR relies on a decrease of ≥ 30% in the sum of the long-axis diameters of up to five target lesions (measurable solid tumours ≥ 10 mm). 112 Yet, this degree of size change may not be seen in patients deriving clinical benefit.
There is an unmet need for robust alternative surrogate imaging markers to characterise and potentially predict response/non-response earlier in treatment and allow for re-evaluation of therapeutic strategy, as needed. Given the often profound devascularisation seen, alternative response criteria which take into account both a reduction in tumour enhancement as well as size have been recognised as a clinical gap. The first was proposed by Choi et al. in the setting of metastatic GISTs treated with imatinib. 113 Here, they found that a decrease in tumour size ≥ 10% or CT attenuation (in Hounsfield unit) ≥ 15% had a sensitivity of 97% for tumour response when correlated with 18F-fluorodeoxyglucose positron emission tomography/CT (18F-FDG PET/CT) as the reference standard, compared to a sensitivity of 52% using RECIST. 113 Since then, Choi and other criteria (see Appendix 7) have been investigated by several groups in the setting of mRCC, and have been found to be putative indicators of improved outcome. 114–118 Yet, there is limited information with respect to observer variation,119 as well as the impact of the phase of CT acquisition, given the higher conspicuity of mRCC in the arterial versus portal venous phase. 120 Further prospective evaluation of proposed response criteria is required in a multicentre setting.
Recent research has also shown a potential role for additional radiomic analysis as a response marker in the context of advanced malignancy including RCC. 121,122 Quantitative analysis of the relationship of pixel spatial and grey-level distribution within an image may provide surrogate markers for intra-tumoural heterogeneity. 123,124 Alteration of these quantitative measures during treatment may provide objective information on changes in tumour heterogeneity that might not be reflected by unidimensional size or mean or median Hounsfield unit changes. Initial studies have found Gaussian-filtered first-order features such as increasing uniformity or decreasing entropy during TKI therapy, with potential relationships to TTP and OS respectively. 121,122
Our hypothesis was that mChoi criteria combining both size and enhancement change may provide a better categorisation of response/non-response to therapy than RECIST or Choi criteria and may predict early disease progression at 24 weeks. We also hypothesised that additional assessment of pixel heterogeneity may augment standard response assessment by providing spatial information of response/non-response.
In this prospective substudy of the STAR trial, we aimed to assess the performance of mChoi criteria at 24 weeks post initiation of treatment to predict ongoing response/progression at 12 weeks versus RECIST and Choi criteria. The secondary aims were to assess the impact of the phase of CT acquisition (arterial or portal venous phase enhancement) on response categorisation by Choi and mChoi criteria and to explore the ability of radiomic analysis including first-order histogram and fractal analysis to demonstrate the heterogeneity of response.
Methods
Participants and treatment
Participants who were eligible and who consented to the STAR trial125 provided additional (optional) consent to participate in this CT substudy. Participants with suitable CT scans were included in the analysis defined as RECIST measurable disease at one or more sites on baseline pre-treatment imaging and IV contrast-enhanced CT imaging. Participants were not included in the analysis if there was non-measurable disease at baseline; non-contrast CT at the required time points; or incomplete imaging data sets.
Computerised tomography imaging
Computerised tomography imaging was performed pre-treatment and 12 weekly thereafter for the main trial. CT scanning parameters required for inclusion in the substudy population are summarised in Appendix 8.
Image analysis
For this substudy, contrast-enhanced CT scans from baseline, 12 weeks and 24 weeks of therapy were reanalysed centrally. Image analysis was carried out by an experienced radiologist (7 years of body imaging) on a workstation using commercial software (Syngo 2012c, Siemens Healthcare, Erlangen, Germany). Target lesions were selected, as per RECIST 1.1. guidelines (≥ 10 mm long axis in size or ≥ 15 mm short axis if nodal, up to two lesions per organ, up to five per patient), and their locations recorded.
Target lesions were assessed in the arterial or portal phase and for a subset of lesions where both arterial and portal venous phase imaging was acquired through the lesion; assessment was performed in both phases. Assessment of tumour size (maximum target lesion longest dimension or nodal short-axis dimension) and attenuation (in Hounsfield units, HU) were undertaken using a semi-automated process. Whole lesion measurements were obtained by drawing a freehand region of interest around the lesion perimeter which automatically propagated to subsequent slices and could be manually corrected for contouring accuracy.
A region-of-interest (ROI) was also placed within the descending aorta at the level of the diaphragmatic hiatus and the aortic attenuation was recorded for the purpose of signal normalisation. The process was repeated for the same lesions in follow-up studies.
Radiomic analysis was performed using in-house software based on MATLAB (Matlab 2013, The Mathworks, Inc., Natick, MA, USA) that has been validated as part of the International Biomarker Standardisation Initiative. 126 Again, freehand ROIs were drawn around each target lesion and a range of radiomic features including locoregional second- and high-order features were extracted automatically by the software.
Statistical analysis
End points and sample size estimation
The primary end point of this study was the correlation of 12-week response categorisation with RECIST 1.1 defined disease progression at 24 weeks. The secondary end points were difference in response categorisation with the phase of CT contrast enhancement and association between radiomic features and response/non-response.
Utility of response criteria for predicting disease progression at 24 weeks
All analyses considered the association between the 12-week response categorisation and outcome at 24 weeks. The outcome at 24 weeks was considered using two different approaches. The first used the original categorisation of the outcome and the second approach considered a reduced number of outcome categories, either PD or not.
The first stage of the analysis was to examine the separate association between each factor and the outcome. For both approaches, these analyses were performed using the chi-squared test.
Subsequently, factors associated with the outcome in the first stage of the analysis were considered jointly upon the outcome in a multivariable analysis. For the first approach, with the outcome on the original scale of measurement, the analysis was performed using ordinal logistic regression to allow for the ordered nature of the outcome categories. For the second approach, whether PD or not, this stage of the analysis was performed using binary logistic regression.
In all analyses, when there were a small number of responses in some categories, these categories were combined with a similar category in order to boost the numbers in each category and thus the power of the analysis. Analysis was undertaken for arterial phase imaging and for the subset of patients with lesions included in both arterial and portal venous phase imaging.
Exploratory baseline radiomic prediction of response categorisation at 24 weeks
Baseline lesional radiomic variables were considered in this analysis. Radiomic features with nil variance were excluded. Associations between variables and 24-week outcomes were assessed using two-sided tests of Spearman correlation. The asymptotic t approximation to the test statistic was employed. Significance was adjusted for multiple comparisons using false-discovery rate control, and 95% CIs were estimated for Spearman’s rho via bootstrapping with replacement. Significant associations were defined according to the cut-off at α ≤ 0.05.
All statistical analyses were undertaken in R and Rstudio.
Results
Participants
In total, 182 participants were enrolled from 27 sites. Fifty-three participants were excluded, the majority for missing data or corrupt imaging data, leaving 129 participants [94 male, 35 female, mean ± SD age 64 ± 9 years (range: 40–85 years)] for final analysis. The participant flowchart is shown in Figure 21.
FIGURE 21.
Participant flowchart.
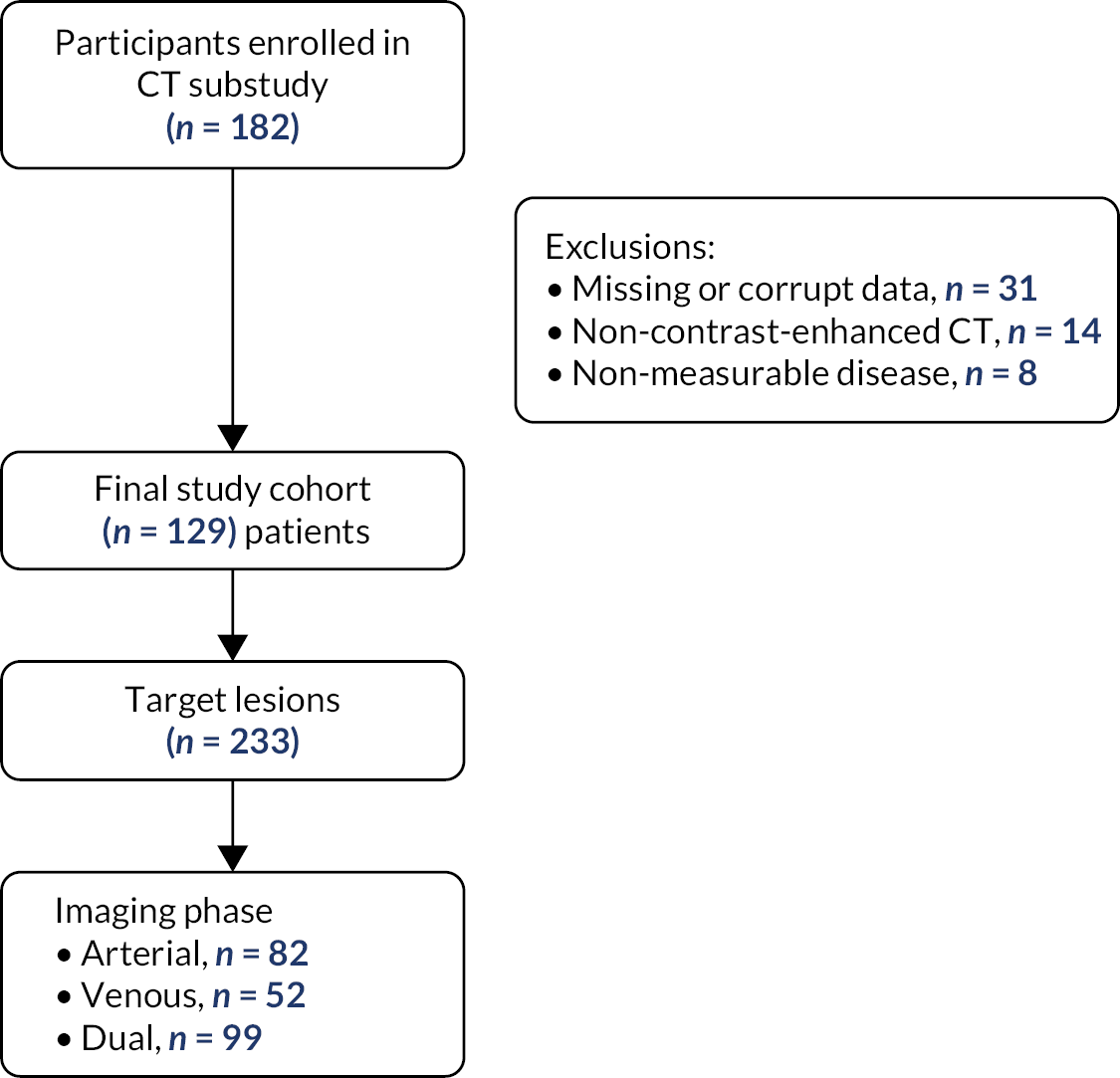
Participant characteristics are summarised in Table 26. The majority of participants (71%, 92/129) had undergone prior nephrectomy. Of these, 56% (72/129) received sunitinib therapy, the remainder pazopanib. In total, 233 target lesions were evaluated; the majority were nodal in location (29%, 69/233). Of these target lesions, 42% (99/233) were imaged in both phases, 35% (82/233) were imaged in the arterial phase only and 22% (52/233) in the portal phase only.
| Summary characteristics | n (%) |
|---|---|
| Total patient number | 129 |
| Sex | |
| Male | 94 (72) |
| Female | 35 (27) |
| Mean ± SD age (range)(years) | 64 ± 9.2 (40–85) |
| Karnofsky scale | |
| > 80% | 127 (98.5) |
| < 80% | 2 (1.5) |
| Previous nephrectomy | 92 (71) |
| Metastatic disease sites | |
| Lung | 29 (12) |
| Liver | 19 (8) |
| Bone | 17 (7) |
| Node | 69 (29) |
| Other | 46 (19) |
| Treatment received | |
| Sunitinib | 72 (56) |
| Pazopanib | 57 (44) |
Imaging response categorisation and outcome prediction
Arterial phase CT imaging
Response categorisation at 12 weeks using RECIST, Choi and mChoi criteria are summarised in Table 27.
| Variable | Category | Number | Percentage |
|---|---|---|---|
| RECIST at 12 weeks | PD | 0 | 0 |
| SD | 70 | 72 | |
| PR | 27 | 28 | |
| CR | 0 | 0 | |
| CHOI at 12 weeks | PD | 1 | 1 |
| SD | 9 | 9 | |
| PR | 87 | 90 | |
| CR | 0 | 0 | |
| mChoi at 12 weeks | PD | 1 | 1 |
| SD | 42 | 43 | |
| PR | 54 | 56 | |
| CR | 0 | 0 | |
| RECIST at 24 weeks | PD | 13 | 13 |
| SD | 67 | 69 | |
| PR | 17 | 18 | |
| CR | 0 | 0 |
An example of a participant with a RECIST SD lesion, but which would be classified as showing a PR with both Choi and mChoi criteria, is shown in Figure 22.
FIGURE 22.
Axial CT image (left), corresponding surface histogram plot (middle) and 3D-volume (right) at baseline (top row) and 12 weeks following initiation of TKI therapy (bottom row) showing no significant size change (RECIST SD) but a reduction in attenuation as well as increasing uniformity (yellow vs. red signal on the histogram plot; peak reduction).

Univariable associations with outcomes at 24 weeks are summarised in Table 28.
| Variable | Category | Outcome at 24 weeks | p-value | ||
|---|---|---|---|---|---|
| PD N (%) |
SD N (%) |
PR N (%) |
|||
| RECIST | SD | 12 (17) | 50 (71) | 8 (11) | 0.02 |
| PR | 1 (4) | 17 (63) | 3 (33) | ||
| CHOI | Progressive/SD | 3 (30) | 7 (70) | 0 (0) | 0.12 |
| PR | 10 (11) | 60 (70) | 17 (20) | ||
| mChoi | Progressive/SD | 7 (16) | 33 (77) | 3 (7) | 0.05 |
| PR | 6 (11) | 34 (63) | 14 (26) | ||
The results suggested there was a significant association between the RECIST criterion and outcome at 24 weeks. Those with a PR for this criterion were more likely to have a PR at 24 weeks, compared to those with SD on the criterion. There was also evidence of an association for the mChoi criteria with 24-week outcome, but this result was only of borderline statistical significance. There was no significant association between CHOI criteria and 24-week outcome.
The second stage in the analysis considered the joint association between the RECIST and mChoi criteria upon the outcome in a multivariable analysis. The results are summarised in Table 29. The figures are the odds ratios from the regression analyses together with their corresponding CIs. The odds ratios represent the odds of being in the next highest outcome category (e.g. PR rather than SD, SD rather than progression disease) in each category, relative to the odds in a baseline category.
| Variable | Category | Odds ratio (95% CI) | p-value |
|---|---|---|---|
| RECIST | SD | 1 | 0.05 |
| PR | 3.25 (1.02 to 10.4) | ||
| mChoi | Progressive/SD | 1 | 0.43 |
| PR | 1.53 (0.53 to 4.37) |
After adjusting for RECIST, there was no evidence that the mChoi measure was associated with outcome, that is mChoi criteria did not provide any additional information in predicting outcome on top of RECIST.
Analyses were also repeated with outcomes either as PD or otherwise. Initially, the separate association between each measure and this categorised outcome was examined. The results are summarised in Table 30.
| Variable | Category | Outcome at 24 weeks | p-value | |
|---|---|---|---|---|
| No PD N (%) |
PD N (%) |
|||
| RECIST | SD | 58 (83) | 12 (17) | 0.08 |
| PR | 26 (96) | 1 (4) | ||
| CHOI | Progressive/SD | 7 (70) | 3 (30) | 0.10 |
| PR | 77 (89) | 10 (11) | ||
| mChoi | Progressive/SD | 36 (84) | 7 (16) | 0.46 |
| PR | 48 (89) | 6 (11) | ||
As no factors were found to be associated with PD, no multivariable analysis was performed.
Arterial plus portal venous phase CT imaging
Response categorisation at 12 weeks using RECIST, Choi and mChoi criteria are summarised in Table 31.
| Variable | Category | Number | Percentage |
|---|---|---|---|
| RECIST at 12 weeks | PD | 0 | 0 |
| SD | 41 | 73 | |
| PR | 15 | 27 | |
| CR | 0 | 0 | |
| CHOI at 12 weeks | PD | 3 | 5 |
| (arterial) | SD | 6 | 11 |
| PR | 47 | 84 | |
| CR | 0 | 0 | |
| CHOI at 12 weeks | PD | 3 | 5 |
| (venous) | SD | 8 | 14 |
| PR | 45 | 80 | |
| CR | 0 | 0 | |
| mChoi at 12 weeks | PD | 3 | 5 |
| (arterial) | SD | 20 | 36 |
| PR | 33 | 59 | |
| CR | 0 | 0 | |
| mChoi at 12 weeks | PD | 3 | 5 |
| (venous) | SD | 19 | 34 |
| PR | 34 | 61 | |
| CR | 0 | 0 | |
| Outcome at 24 weeks | PD | 6 | 11% |
| SD | 36 | 64% | |
| PR | 14 | 25% | |
| CR | 0 | 0% |
Univariable associations with outcome at 24 weeks are summarised in Table 32.
| Variable | Category | Outcome at 24 weeks | p-value | ||
|---|---|---|---|---|---|
| PD N (%) |
SD N (%) |
PR N (%) |
|||
| RECIST | SD | 6 (15) | 30 (73) | 5 (12) | 0.001 |
| PR | 0 (0) | 6 (40) | 9 (60) | ||
| CHOI | Progressive/SD | 2 (22) | 7 (78) | 0 (0) | 0.12 |
| (arterial) | PR | 4 (9) | 29 (62) | 14 (30) | |
| CHOI | Progressive/SD | 2 (18) | 9 (82) | 0 (0) | 0.09 |
| (venous) | PR | 4 (9) | 27 (60) | 14 (31) | |
| mChoi | Progressive/SD | 3 (13) | 20 (87) | 0 (0) | 0.001 |
| (arterial) | PR | 3 (9) | 16 (48) | 14 (42) | |
| mChoi | Progressive/SD | 2 (9) | 19 (86) | 1 (5) | 0.01 |
| (venous) | PR | 4 (12) | 17 (50) | 13 (38) | |
The data suggested that the RECIST criterion and both the arterial and venous mChoi measures were significantly associated with the outcome. However, the results did not reach statistical significance for either the arterial or venous CHOI variables. For all three significant variables, a PR on the criteria was associated with a higher chance of a PR at 24 weeks.
The factors significant in the univariable analyses were considered together in multivariable analyses. The results of two different analyses are shown in Table 33. The first analysis shows the results when all three variables were included in the analysis. The second analysis omits one of the factors not found to be significant in the first analysis, in order to simplify the analysis.
| Analysis | Variable | Category | Odds ratio (95% CI) | p-value |
|---|---|---|---|---|
| 1 | RECIST | SD | 1 | 0.01 |
| PR | 7.69 (1.54 to 38.3) | |||
| mChoi | Progressive/SD | 1 | 0.08 | |
| (arterial) | PR | 5.42 (0.82 to 35.9) | ||
| mChoi | Progressive/SD | 1 | 0.35 | |
| (venous) | PR | 0.44 (0.08 to 2.49) | ||
| 2 | RECIST | SD | 1 | 0.02 |
| PR | 6.40 (1.37 to 29.8) | |||
| mChoi | Progressive/SD | 1 | 0.14 | |
| (arterial) | PR | 3.20 (0.68 to 14.9) |
As there was no evidence that the venous mChoi variable added any additional information to the other two measures in Analysis 1, this variable was omitted from Analysis 2, which suggested only the RECIST criterion was significantly associated with the outcome, and that the arterial mChoi was not additionally significant after adjusting for RECIST.
The outcome was also considered as either PD or not. The univariable results are summarised in Table 34.
| Variable | Category | Outcome at 24 weeks | p-value | |
|---|---|---|---|---|
| No PD N (%) |
PD N (%) |
|||
| RECIST | SD | 35 (85) | 6 (15) | 0.12 |
| PR | 15 (100) | 0 (0) | ||
| CHOI | Progressive/SD | 7 (78) | 2 (22) | 0.22 |
| (arterial) | PR | 43 (91) | 4 (9) | |
| CHOI | Progressive/SD | 9 (82) | 2 (18) | 0.37 |
| (venous) | PR | 41 (91) | 4 (9) | |
| mChoi | Progressive/SD | 20 (87) | 3 (13) | 0.64 |
| (arterial) | PR | 30 (91) | 3 (9) | |
| mChoi | Progressive/SD | 20 (91) | 2 (9) | 0.75 |
| (venous) | PR | 30 (88) | 4 (12) | |
As none of the measures were associated with PD, no multivariable analysis was performed.
Prediction of 24-week response categorisation using baseline radiomic variables
Exploratory analysis was undertaken for 116 target lesions from 76 participants; 81 baseline radiomic variables were considered. Nine baseline radiomic features were found to be significantly associated with patient response (see Table 35). Associated features were all derived from grey-level dependence matrix (GLDM) and the grey-level co-occurrence matrix (GLCM).
| Radiomic feature | Spearman ρ (95% CI) |
p-value (FDR-adjusted) |
|---|---|---|
| GLCM_Contrast | 0.30 (0.12 to 0.45) | 0.03 |
| GLCM_Difference_Entropy | 0.27 (0.09 to 0.42) | 0.03 |
| GLCM_Difference_Variance | 0.30 (0.11 to 0.45) | 0.03 |
| GLCM_Dissimilarity | 0.27 (0.10 to 0.42) | 0.03 |
| GLCM_Inverse_Difference_Moment_Normalised | −0.28 [−0.43 to −0.10] | 0.03 |
| GLDM_Mean | 0.27 [0.11 to 0.43] | 0.03 |
| GLDM_Entropy | 0.27 [0.08 to 0.43] | 0.03 |
| GLDM_Variance | 0.28 [0.11 to 0.44] | 0.03 |
| GLDM_Contrast | 0.30 [0.12 to 0.45] | 0.03 |
Discussion
There is an ongoing need to improve imaging response assessment following targeted therapy in advanced RCC, especially to predict response/non-response earlier in treatment and allow for re-evaluation of therapeutic strategy as appropriate. To date, a number of strategies have been proposed. These have included (1) redefining the threshold of size change for response which may differ between first- and second-line therapy;127–129 (2) introduction of alternative response criteria incorporating attenuation change as well as lower thresholds for size change for mRCC, for example, Choi,114,130 mChoi115,116 and MASS;118 and (3) exploration of novel biomarkers including vascular tumour burden density;131 radiomic analysis;121,122 or quantitative analysis from dynamic contrast-enhanced imaging. 132 Most studies to date have been post hoc analyses of completed clinical trials, retrospective analyses or single-centre exploratory/pilot studies, with data from prospective multicentre studies lacking.
In our study, response categorisation changed compared to RECIST with Choi and mChoi criteria. One participant was defined as having PD at an earlier time point by Choi and mChoi criteria at 12 weeks, based on an increase in size ≥ 10% and/or attenuation ≥ 15%, compared to none with RECIST. While 72% and 28% of participants were categorised as having SD or PR by RECIST, with Choi this was reversed at 9% and 90%, respectively, and for mChoi this was 43% and 56%, respectively, for lesions imaged in the arterial phase. No participant had a CR by all three criteria. These findings are in line with previous publications. 114–116,130 When response categorisation was compared for a subset of lesions imaged in both the arterial and portal venous phase, the proportion of participants with PD, SD or PR did not differ substantially, suggesting the acquisition phase does not have a significant impact.
By 24 weeks, 10% of participants had PD as defined by RECIST criteria, while 87% of participants had SD (69%) or a PR (18%). Unlike the Choi criteria which was not associated with outcome at 24 weeks, there was a borderline association of mChoi criteria for the categorisation of ongoing benefit (non-progression) at 24 weeks for lesions assessed in the arterial phase of imaging, but this was not an independent predictor at multivariate analysis, that is mChoi criteria did not provide additional information in predicting response on top of RECIST. In terms of predicting PD at 24 weeks, none of the criteria (RECIST, Choi, mChoi) were associated with progression, though the small number of progressors (13/129) is a limitation. Further analysis of target lesions imaged in both the arterial and portal phases again suggested that both RECIST and mChoi criteria (arterial and portal venous) categorisation of PR was associated with a higher chance of a PR at 24 weeks, but with multivariate analysis indicating no additional significance for mChoi criteria following adjustment for RECIST categorisation. Again, none of the criteria were associated with early PD at 24 weeks. These data complement published literature which has focused on TTP115 or the association with PFS133 or OS130 rather than prediction of response/non-response at this earlier time point of 24 weeks post initiation of treatment.
With respect to heterogeneity of tumour enhancement, initial assessment of baseline parameters suggested an association of some GLCM and GLDM features with 24-week outcome. Nevertheless, this type of analysis remains exploratory due to limited number of participants with disease progression in this cohort.
Conclusion
In summary, in a prospective multicentre study, we have confirmed that assessment of enhancement as well as size change alters the categorisation of response. Use of mChoi criteria may allow for earlier detection of PD, and more representative separation of participants with PR versus SD. While published literature has suggested an association of mChoi criteria with TTP, PFS and OS in terms of prediction of early progression within 24 weeks of treatment initiation, no association was shown in this cohort. Assessment of tumour heterogeneity may complement standard response assessment but current data remain limited and further work is still required to further the field.
Chapter 8 Overall discussion
Summary of findings
The STAR trial represents over 12 years of work from conception to presentation and publication and is the largest academic UK trial to date in advanced RCC, recruiting 920 patients over 70 months. The novel co-primary hypothesis tested by the overall trial was to determine whether a DFIS was non-inferior to a CCS in terms of both OS and QALYs, that is if a DFIS did not reduce OS or QALYs compared to a CCS by a margin which was pre-specified as ≤ 7.5% for OS and ≤ 10% for QALYs.
In terms of the co-primary end points QALY NI was demonstrated in both ITT and PP populations and for OS, NI was demonstrated for ITT population, but not for the PP population, using the pre-chosen inferiority margin. Conventionally, rigorous application of NI criteria requires that the condition is met for both the ITT and the PP populations since, although ITT analysis may be satisfactory for superiority trials, including dropouts in the analysis for NI trials may bias the results towards equivalence. The PP analysis, which includes all patients who satisfactorily complied with the assigned treatment, is more likely to identify any strategy differences. 134 Therefore, we cannot formally conclude NI for both OS and QALYs. Informally, there does not appear to be any clinically meaningful difference in OS between the two arms. For example, the median OS values for patients in the DFIS and CCS arms were 27 and 28 months, respectively, for both the ITT and PP populations.
Additionally, over 40% of patients in the DFIS arm who continued post week 24 had at least two treatment breaks, with 27% receiving greater than or equal to three breaks. Given that taking more than one break was voluntary, this demonstrates that treatment breaks were considered to be acceptable (and desirable) to both patients and health professionals. The evidence may also imply that there may be a subset of patients who may be more appropriate to receive this approach, and further work to define this population is ongoing.
Overall, participants in the DFIS arm received a similar amount of treatment to those in the CCS arm, but over a longer period of time, the number of treatment-related safety events (SARs) were higher in the CCS arm when participants were on trial strategy. In addition, the economic evaluation indicated that the DFIS was highly likely to be cost-effective compared to the CCS.
Parallel to the delivery of the STAR trial, the landscape of RCC treatment has changed. For many patients, standard first-line therapy has now changed to include immunotherapy (IO), either alone or in combination with a TKI. However, despite this, single-agent TKI remains appropriate first-line therapy for a significant proportion of patients and second-line therapy for many more. These data support and facilitate an informed discussion with these patients regarding a DFIS strategy and planned treatment breaks.
Strengths and limitations
Trial design and analysis
The STAR trial utilised a novel study design, in particular relating to its powering on both, OS and QALYs and the inclusion of a number of end points specific for intermittent treatment strategies.
For a study spanning almost 12 years from concept to completion with a large number of participants in a large number of sites, there was inevitably a need for the study to be able to respond to changing external circumstances, such as new drug approvals. A major strength of the STAR trial was the ability to maintain, against this background, rigorous conduct and reporting according to CONSORT recommendations, with analyses conducted according to a predefined SAP agreed with the TMG and reviewed by the DMEC.
The STAR trial was designed to be pragmatic, aiming to recruit a real-world population of patients receiving TKI therapy as first-line treatment for advanced RCC. The inclusion criteria were therefore kept as broad as possible. The intervention of a DFIS changed only the timing of treatment cycles and hence was not predicted to cause any additional toxicity to participants or any significant logistical issues in delivery at the site. We, therefore, enabled sunitinib (and later sunitinib or pazopanib) to be used in line with local practice to ensure that the majority of patients suitable for treatment with first-line TKI would also be eligible to participate in STAR. This was done to ensure the generalisability of the trial results and the baseline characteristics of the participants confirm the real-world population represented.
The trial was robustly designed to answer the overall Phase III aims. It was however recognised during conception that there were significant challenges to being able to do this, for example, in terms of recruitment of the sample size required to demonstrate NI, the duration of recruitment required, ensuring that the approach of planned treatment breaks was deemed acceptable to clinicians and health professionals and the limited data available to inform the SAP. For these reasons, the three-stage trial was proposed with a Phase II to Phase III seamless design: Phase II including Stage A to address feasibility in terms of recruitment rate, Stage B to address efficacy in terms of TSF in addition to Phase II secondary end points to confirm the accuracy of the assumptions made to inform the Phase III sample size. This was a key strength of the study design as it permitted the Phase II data to provide assurance that the study should continue on to a Phase III study.
Careful consideration was made in selecting all the trial end points and a strength of the study is in the selection of those appropriate to an intermittent treatment strategy. Standard end points such as PFS are not appropriate in intermittent strategy trials as earlier progression is expected with the inclusion of a treatment break, the question relates to whether disease control can be regained on retreatment. Time to strategy failure (also referred to as time to failure of strategy) was proposed as an appropriate end point for intermittent strategies. 64,135 For this reason, TSF was selected to be the primary outcome of Stage B.
The large sample size, and the fact that renal cancer is termed a ‘rare’ cancer, required a relatively long duration of recruitment from a high number of sites. This prolonged recruitment meant that there were changes in the treatment options available for RCC during the time it was open. Although STAR was planned to be a pragmatic study, it was essential to ensure that any agreed amendments would not compromise the trial integrity and/or interpretation of the results. When considering the results now in line with current practice, it is also important to account for the situation and the time of trial design and delivery.
This ability to respond to the changing treatment landscape was another strength of the STAR trial. For example, during initial trial development, the potentially practice-changing first-line trial of sunitinib compared to IFNα5 had been published, and sunitinib in this setting was under consideration by NICE and was expected by the clinical community, but was yet to be formally approved (this occurred March 2009). There were accumulating data that alternative TKIs such as pazopanib may provide similar benefits in the first-line setting potentially while causing less toxicity, but data confirming this were awaited. It was appreciated by the TMG that in order for the trial to be feasible, the CCS arm needed to align with standard UK practice. Horizon scanning anticipated pazopanib approval (occurred February 2011) and consideration of inclusion of pazopanib alongside sunitinib was planned pre-opening the Phase III part of the trial. During recruitment into Phase II, it became apparent that not allowing clinicians to utilise pazopanib as an alternative TKI would limit recruitment after the publication of the COMPARZ trial (October 2012)13 which demonstrated comparability with sunitinib. Waiting to include pazopanib only in Phase III would have therefore jeopardised demonstrating the feasibility of continuing to Phase III and caused premature closure of the trial. After careful consideration of the pros and cons, and with the full support of key investigators, patient representatives, TSC, DMEC and the NIHR HTA, the eligibility criteria were updated to allow this in April 2013. The TKI used was added in as an additional stratification factor.
STAR was one of the first Phase III trials to have a patient-reported outcome measure feed into the co-primary end point (QALY). Overall, the return rate of QoL questionnaires was excellent [13,147 out of 16,726 (78.6%) questionnaire booklets were returned during the trial]. However, the nature of the end point and the trial meant that careful consideration was required during the analysis. Missing data in patient-reported outcome measures typically cannot be chased and therefore plans to address missingness were included in the SAP as summarised in the section Missing data. The results of the primary analysis which imputed missing data during the follow-up period reached the same conclusion as the complete case analysis and alternative imputation methods. Therefore, it is likely that missing questionnaire data were due to chance (MAR) rather than for underlying reasons related to the QoL of patients (MNAR). A way to determine between data that are MAR and MNAR is to collect the reasons for missing questionnaires. A limitation of this study is that this information was not available for the majority of questionnaires. However, given the concordance in the results for the QALY analysis where both the PP and ITT analyses concluded NI, this is less of a concern for this study. We would recommend that any future studies considering patient- reported outcome data, either as a primary or secondary end point, collect the reasons for missing questionnaires from the outset of the trial to aid their analysis.
The decision to impute questionnaires during the follow-up period was supported by the frequent collection of QoL questionnaires during treatment which resulted in an accurate measure of QALYs even when questionnaires were missing. However, this will have increased the burden on the research staff at the site as all questionnaires were completed at a site on paper. Since trial conception, more up-to-date methods of collection have been adopted where participants can complete questionnaires at home on phones, computers or tablets. We would recommend that future trials adopt these more modern methods of QoL collection.
A limitation of the secondary QoL analysis is that multiple models were fitted onto the same data set. However, no adjustments for multiplicity were made. This was deemed appropriate as they were unpowered secondary end points and on consideration of the results, while statistically significant effects were observed, the effect sizes were not clinically meaningful.
As highlighted previously, when the STAR trial was designed there was a very real concern that planned treatment breaks may not be acceptable to patients and/or clinicians. In fact, this was not the case, supported by the qualitative outcomes with few patients withdrawing from the trial stating a wish for continuous treatment as the reason. However, during trial design, these concerns led to the STAR trial mandating only one planned treatment break in the DFIS arm, with subsequent breaks taken at the discretion of the patient and clinician. The median number of planned treatment breaks was one, but a broad range was observed up to nine. A consideration in similar trials in the future would be to test the ongoing strategy to include multiple planned treatment breaks as PP.
Radiological reporting
Another strength of the trial design was that it also permitted a number of changes to be implemented around the time of transition to the Phase III part of the trial. A key change was the removal of the requirement for central radiological reporting and the transition to local radiological reporting. Central radiological reporting was required in the Phase II part to reassure a number of clinicians who, at the time of trial conception, were concerned that participants on the DFIS arm who at 24 weeks had a disease that was continuing to respond (i.e. still shrinking) should not have their TKI stopped until the response stabilised. For participants to be eligible to take up their treatment break in Phase II, they were required to have achieved MRR; this required comparison to the previous scan rather than baseline (the latter is standard for RECIST reporting).
In practice, the implementation of central radiological reporting was very challenging. A key issue was the short turnaround time available between imaging and the clinic appointment for informing patients of the results. At the time, there was no way that images could be sent electronically, so each scan had to be anonymised and downloaded onto a CD at the site and then sent centrally to Leeds. A considerable amount of work was involved in the anonymisation of data and transfer of images and reports between sites; associated with this were logistical difficulties with the collection and delivery of images and timely central reporting, so it was not always possible to have the central report at the time of clinic appointment and meant that clinicians at the site were sometimes required to make treatment decisions without this.
The central reporting had not been planned to continue into Phase III and it was apparent that it would not be feasible to make radiologists at the site report scans in a new way (by comparing to the previous scan). Reassuringly, when the data from the central reporting were reviewed at the end of Phase II, fewer than 5% of participants had not had MRR at 24 weeks. This provided reassurance to clinicians that simplifying the pathway was clinically justifiable and that participants in the DFIS who had not progressed by 24 weeks could take up their planned treatment break at this point (unless a strong clinical rationale to continue). This proposal was reviewed by the TMG and DMEC and ratified by the TSC.
PPI input and patient perceptions
The STAR trial included significant PPI input throughout, from initial focus groups to throughout the study with PPI membership on both the TMG and TSC. As mentioned previously, there was concern regarding whether patients would be willing to stop a treatment known to be working. Consideration was therefore given to the timing of approach to participate and randomisation as well as to how best to present the trial to patients. Based on clinical considerations from other relevant studies and patient discussion, the decision was made to perform randomisation at baseline prior to patients receiving any TKI, with patients taking up their randomisation at 24 weeks assuming no progression. A DVD was also developed for patients assisted with the presentation of the trial. The benefits of this approach were demonstrated by achieving the recruitment rate required in Phase II to enable progression to Phase III and the high take-up rate of allocated treatment arm.
The decision to not attempt to blind the trial to patients, medical staff or clinical trial staff was deemed to be justified in view of the additional cost and logistical issues which outweighed the benefits.
Amendments to sample size and event rate
In the trial design, the required sample size was 1000 participants (see Phase III). This assumed that 2-year survival in the CCS arm was 54% and that a maximum of 10% of participants would be lost to follow-up. As a result of the recruitment rate for the trial slowing down in 2016, these assumptions were re-assessed. It was found that the 2-year survival rate in the CCS arm was 48.5% and only 2% of patients had been lost to follow-up. Therefore, following the DMEC and TSC agreement, the sample size was updated to reflect these estimates requiring 920 participants and 720 events.
As discussed previously, during the lifespan of the trial, the landscape of treatment for advanced renal cancer changed significantly. From a situation at the start with only one TKI approved (sunitinib), there are now multiple treatment options spanning 3+ lines of therapy, and with these changes, there has been a significant improvement in outcomes for patients. This is evidenced through the 2-year survival estimates in the CCS arm of 55.5% in the ITT population and 55.2% in the PP population, a higher proportion than the assumed 48.5%. For the trial, this meant that fewer events (deaths) occurred that were predicted during follow-up, which had a consequent impact on the overall power of the study. Follow-up was extended to include more events, but this was not sufficient, and a very significant further extension would have been required to attain the number of events required to reach 80% power. Therefore, after careful consideration, this was not felt to be justifiable in terms of resources for a minimal increase in potential power. This was a limitation as it meant that the overall power of the trial was slightly reduced. Clearly, this slight reduction in power may have been a reason why the study only fell short of demonstrating NI in OS for the PP population. In addition, the change in the event rate could have motivated a change in the NI margin. However, as it was pre-specified to conclude NI based on the relative difference of 0.812, rather than the absolute difference of 7.5% and relative differences are less affected by changes in event rate,136 0.812 was kept as the NI margin. Note that because a similar proportion of patients in both arms received immunotherapy treatment post trial (see Anticancer treatment post trial), subsequent therapy was not adjusted for in any ancillary analysis.
Generalisability
STAR was planned from initial development to be a pragmatic study, representative of the population as a whole rather than a fitter subset and due to the intervention being relatively simple (planned treatment breaks), also to be deliverable across any centre/unit treating patients with TKI.
Overall, the trial succeeded with this. The large number of sites that STAR was open at, and the high number of patients recruited, demonstrates the fact that the trial eligibility criteria were not overly restrictive and enabled recruitment of the great majority of patients who would be treated with TKI therapy in the clinic, the caveat being that there are a different population of patients treated with single-agent TKIs now compared to when the trial was open and recruiting.
The population from STAR is representative of the previous trial populations for TKI trials in terms of sex (males: 72.7% STAR, 71.5% Motzer et al. 5 and 73% COMPARZ12). There were slightly more participants of the MSKCC-favourable prognostic group in the STAR trial (favourable; intermediate; poor; unknown: 44.3%; 48.4%; 7.3%; NA STAR, 36%; 57.5%; 6.5%; NA Motzer et al. 5 and 27%; 58.5%; 10.5%; 3.5% COMPARZ12) and a slightly higher proportion of ECOG PS one participants in the STAR trial (45% STAR, 38.5% Motzer et al. 5 and 26% in COMPARZ12). The population appears to be representative of the UK population at the time. The median number of treatment cycles received was 4 overall and similar between the two arms (24 weeks in total). This is also comparable to the original publications (e.g. Motzer et al. 5 months, slightly lower than in the COMPARZ study 7.8 months).
It should be emphasised that approximately 40% of patients, randomised at baseline, did not proceed to take up their randomised arm after 24 weeks, largely due to progression during that period. However, a large number of patients did take up their allocated arms which would be anticipated to ensure balance between the arms. From an ethical perspective, the decision to randomise at baseline rather than at 24 weeks was taken due to concern that if randomisation was performed just prior to the time that treatment could be stopped (DFIS arm), then potentially more patients with toxicity (i.e. keen to have a treatment break) may choose to participate and fewer with no toxicity.
Regarding survival in the STAR trial median OS was 27.5 months (28 months CCS and 27 months DFIS), which is again comparable to that in the original Motzer publication 26.4 months Motzer et al. 6 and the COMPARZ study 28.4 months/29.3 months for pazopanib and sunitnib. 12 This OS is significantly lower than that in more contemporary trials of first-line treatments (e.g. Checkmate 214137). However, as discussed elsewhere, this relates to the improvements in survival attributable to IO treatments, the availability of second- and third-line treatments and only a proportion of fitter patients being suitable for treatment with IO drugs.
While it is clear that the STAR conclusions regarding the benefits of treatment breaks remain widely applicable to patients with RCC receiving TKIs, the results strongly justify consideration of treatment breaks in more recent RCC treatments, for example IO alone or TKI/IO combinations. This also applies to the use of treatment breaks in other cancers and in systemic anticancer therapies more generally.
Interpretation
Outcomes of main study
The STAR trial was one of the largest non-commercial studies in advanced RCC ever carried out in the UK. It has provided extensive evidence demonstrating patient acceptability of treatment breaks. As outlined above, the appetite of patients for a treatment break was surprisingly high such that only 12 patients in the DFIS arm (2.6%, n = 459) withdrew from the study so they could have continuous treatment. In addition, although only one treatment break was mandated, more than 43% of patients opted to take two or more treatment-breaks and the qualitative substudy (see Qualitative assessment) clearly showed patient acceptability for the treatment break strategy, although the knowledge that they were being closely monitored during a break remained crucial. In a trusted and supportive clinical context, patients welcomed the opportunity that a break afforded to resume valued activities in their lives, even if only temporarily.
The trial also demonstrated the successful use of the seamless Phase II/Phase III design, with all patients contributing to the final Phase III outcome data as well as using Phase II data to demonstrate adequate recruitment rate and early indication of efficacy via TSF.
The formal co-primary end point required that both the OS end point and the QALY end point meet the pre-set boundary conditions set out in the grant application in both PP and ITT populations in order to be able to conclude NI. For the OS end point, although the ITT population met this condition, the PP population marginally failed to do so (Cox HRs 0.83 and 0.80, respectively, compared to a requirement of ≥ 0.81 in order to show NI, based on the pre-stated ≤ 7.5% difference in OS, Figure 23). For the QALY end point, this condition was met for both the PP and ITT populations, based on the pre-stated ≤ 10% difference in QALY. Because the PP population did not meet the OS requirement, this means that formally we cannot conclude that the NI condition was met. This difference may be explained through the lack of power for the comparison due to the reduced number of events observed in the PP population potentially explained by the changing treatment landscape throughout the trial and the nature of the PP population.
FIGURE 23.
Comparison of lower CI bounds to NI margin – OS.

During the STAR trial, other systemic therapies were introduced into the RCC treatment landscape, and OS improved for STAR participants by the introduction of immunotherapy and new therapies such as nivolumab and cabozantinib in the second- and third-line setting. While this improvement in OS is very welcome, it has resulted in a lower event rate (death) for STAR patients. With the agreement of the DMEC and TSC, trial follow-up ceased before the required number of survival events (720) were observed for 80% power. In addition, due to the nature of the strategy, 49 (5.3%) of patients were excluded from the PP population, more in the DFIS arm compared to the CCS arm (see Figure 4) resulting in less power for the PP comparison to the ITT comparison (720 events were required, 678 events were observed in the ITT population and 648 events were observed in the PP population).
Nevertheless, it may be seen from Figure 7 that for the ITT population, the Kaplan–Meier OS plots for the two arms are almost superimposable, except for the small numbers of patients with OS longer than about 6 years and there is no clinically meaningful difference in the median survivals (28 months in the CCS arm, compared with 27 months in the DFIS arm). Similarly, for the PP population, again the Kaplan–Meier OS plots for the two arms are almost superimposable (see Figure 6), except for the small numbers of patients with OS longer than about 6 years and there is no clinically meaningful difference in median survivals (28 months in the CCS arm, compared with 27 months in the DFIS arm). Notably also, the respective median OS values are the same in the ITT and PP populations, yet the ITT population met the pre-defined NI margin, but the PP population marginally did not.
The NI margin of ≤ 7.5% was chosen following discussions with the UK and US communities and NCRI Renal Clinical Studies Group (CSG). Earlier designs had proposed a difference of ≤ 10% to be clinically acceptable. However, this was not accepted due to the data available on sunitinib over IFNα (54% compared to 46% at 2 years). During the discussions around the determination of the NI margin, it was agreed that a limited reduction in OS would be clinically acceptable if benefits in terms of reduced drug toxicity and improvements in QoL and health economic parameters were observed. Given the results observed, while NI cannot be concluded, the DFIS is considered to be clinically acceptable based on these parameters.
We also carried out sensitivity analysis for OS using a piecewise model. This analysis accounted for the fact that patients in both arms were treated identically for the first 24 weeks after randomisation. These analyses (see Overall survival – piecewise model) showed similar results to the main primary analysis where NI could not be concluded in the PP population by a small margin (95% CI Lower Bound 0.80 < 0.812).
The cost-effectiveness analysis showed that, at 2 years, the DFIS arm was associated with substantial cost savings (£6408 and £3235 per patient in complete case and imputation analyses, respectively). These savings are driven by overall reduced treatment costs. We also estimated QALY gains for DFIS versus CCS over the same period (0.049 and 0.165 for imputation and complete case analyses, respectively). These findings were mirrored by the decision modelling analyses over a lifetime horizon. Both analyses concluded that DFIS was highly likely to be the most cost-effective strategy. Sensitivity analyses indicate this conclusion is robust to changes in various assumptions and analytical approaches. Cost-effectiveness estimates were increased in favour of DFIS when the costs of subsequent therapies were included in the analyses.
Although not a primary end point for the overall Phase III study, the ‘Time to Strategy Failure’ (defined as progression on sunitinib/pazopanib treatment, need for a change in systemic treatment or death) proved to be valuable as an interim Phase II end point and also a secondary Phase III end point. In essence, this was a measure of the time for which the trial strategy (CCS or DFIS) was working. Figure 8 shows a plot for the ITT population, demonstrating a clear and significant increase in TSF for the DFIS arm (HR 0.75, 95% CI 0.65 to 0.86); p-value < 0.001 compared with the CCS arm (median 11 months vs. 8 months), although this does not ultimately translate into an OS advantage.
It is clear that PFS cannot be used in the conventional way in a trial with treatment breaks as the conventional PFS does not take into account the successful rechallenge with sunitinib/pazopanib following progression on a treatment break. Figure 10 illustrates this point where it is clear that, for the first 6 months, the PFS curves are superimposable for the CCS and DFIS arms, but the DFIS arm shows shorter PFS following the treatment breaks. A better way of comparing the two arms is to use summative PFS (see Figure 11), where it is clear that the proportion of participants progression-free is significantly higher in the DFIS arm than in the CCS arm.
Comparison of toxicity data is also complex because raw AE and SAE data do not take into account that participants in the DFIS arm were recording them for longer due to their greater time on trial treatment. However, on consideration of SAEs (see Serious adverse events) deemed to be related to TKI treatment (SARs) which are recorded in the same period for both arms, a smaller proportion of participants in the DFIS arm experienced an event than in the CCS arm and participants in the DFIS arm accounted for fewer of the overall events compared to the CCS arm, illustrating the potential safety benefits of the DFIS arm.
Substudies
The substudies add valuable detail and context to the overall STAR trial which have been fully discussed above. The key messages were:
-
A key conclusion from the qualitative substudy is that the data throw additional light on the acceptability and attractiveness of the intervention and the reasons for that acceptability, which in turn help explain the high recruitment rate. A distinctive feature of STAR is that eligible patients felt able to relate to both arms of the trial: the DFIS form of ‘active monitoring’ to such patients held considerable appeal and contributed to their willingness to take part in the trial.
-
The quantitative DCE-MRI feasibility clinical study associated with the STAR trial was the first to use longitudinal serial assessments to detect changes in biomarkers following sunitinib or pazopanib treatment in mRCC. It showed that DCE-MRI-derived biomarkers of tumour perfusion were potential surrogate biomarkers to predict early disease progression following TKI therapy in advanced RCC. The study also demonstrated that these DCE-MRI assessments were reproducible and that further larger prospective clinical studies are justified to test its wider application in the context of routine clinical practice.
-
The CT substudy took advantage of the STAR trial to conduct a prospective multicentre study to assess the possibility of predicting response/non-response earlier in treatment, thus allowing for re-evaluation of therapeutic strategy as appropriate. The study confirmed that assessment of enhancement as well as size change alters the categorisation of response. Use of mChoi criteria may allow for earlier detection of PD, and more representative separation of participants with PR versus SD. Notably, while published literature has suggested an association of mChoi criteria with TTP, PFS and OS in terms of prediction of early progression within 24 weeks of treatment initiation, no association was shown in the STAR cohort. Assessment of changes in tumour heterogeneity may complement standard response assessment but current data remain limited and further work is still required to advance this field.
Potential benefits and harms
Because of the nature of the STAR research question, much of the above discussion already addresses the potential benefits and harms of a treatment-break strategy. The qualitative substudy showed perceived patient benefits in the opportunity to take treatment breaks which was also reflected in the low number of patients (12 in total) opting to withdraw at the randomisation point in order to have continuous treatment. In terms of OS, STAR has shown that there is no clinically meaningful difference between the DFIS and CCS arms when considering median OS (27 and 28 months, respectively). Further, ongoing analysis is looking at whether we can predict which patients will most benefit from treatment breaks and multiple treatment breaks. We plan to further analyse the STAR data in this regard. The health economic evaluations of cost-effectiveness indicate that DFIS is the optimal strategy and that, in all base cases and most sensitivity analyses, DFIS was shown to be cost saving and providing QALYs gains over CCS. Overall, the potential benefits of DFIS appear to outweigh any potential harms.
The STAR study, the largest of its kind which includes an OS end point, is expected to make a substantial contribution to the literature on the concept of treatment breaks, especially since it combines extensive assessment of patient preferences with quantitative outcome data. In RCC, while the STAR study was ongoing, a single-centre Phase II study by Rini and co-workers32 also provided evidence for the benefits of treatment breaks.
Other ongoing trials which have taken the STAR design into account include the REFINE study138 which is looking at treatment breaks in a range of cancer types including RCC patients on immunotherapy and studies such as DANTE139 which is exploring treatment breaks in immunotherapy treatment of melanoma.
Recommendations for future research
The broad and wide-ranging outcomes from this trial and related substudies lead to a number of future research questions worthy of consideration.
Relating to the main trial
The main trial demonstrated that treatment breaks are acceptable to patients and clinicians, by virtue of > 40% of patients who continued past week 24 having greater than or equal to two treatment breaks and 27% greater than or equal to three treatment breaks. This suggests that there may be a subset of patients who are more appropriate for and will benefit most from the utilisation of a DFIS. There is work ongoing currently, aiming to define this population. The trial protocol mandated only one treatment break; however, it would be interesting when designing similar trials in the future to test the DFIS fully by including multiple treatment breaks rather than only the initial one. Another population of interest are the subset of patients who experienced exceptionally prolonged durations of disease control during treatment breaks, and it will be useful to further define these participants. Further research is warranted to consider if there are certain biological factors which result in some patients benefiting from a treatment break more than others.
As described at length previously, patient-reported outcomes were key outcomes in this trial. We would recommend that any future studies considering patient-reported outcome data, either as a primary or secondary end point, collect the reasons for missing questionnaires from the outset of the trial to aid their analysis. We would also suggest that in future trials, consideration is given to more contemporaneous methods of data collection enabling participants to complete questionnaires at home on phones, computers or tablets. These more convenient collection methods would hopefully reduce missing data. Thought should also be given to prompts to remind patients to complete questionnaires.
Since the conception of the STAR trial, treatments for advanced RCC have changed, such that it is a minority of patients who are treated with single-agent TKI treatment. TKI remains an important treatment in advanced RCC, and this may increase further first line with the recent approval of adjuvant IO in a subset of patients. Further research should now be considered into the potential benefits of treatment breaks in more contemporary RCC treatment, for example IO monotherapy or TKI/IO combination therapy.
In addition, more broad learning around intermittent treatment strategy trials should be taken from this trial and used to extend this research area to other types of cancer and their treatments more generally. With the number of available systemic treatments in cancer increasing with associated high costs in terms of finance and side effects, research to define appropriate treatment duration is increasingly important. Such trials are frequently large, costly and can be challenging to deliver; hence, ensuring that they are designed in the most appropriate way is essential. The STAR trial has demonstrated patient and health professional interest and support for exploring these alternative approaches.
Relating to health economics
The use in the STAR trial of a QALY-based primary end point remains novel. Its accuracy was reliant in part on PROMSs and hence was potentially impacted due to the assessment schedule, missing data and the breadth and recall of the HRQoL measure that we used. It is possible that the full QoL impact of treatment intervals has not been captured here due to these reasons; thus future research seeking to estimate the value of treatment breaks and dose reductions should adopt a more nuanced approach to QoL capture. This approach of capturing benefits should also explore patients’ preferences for such strategies, including their willingness to trade off the associated risks and benefits. Another area of future research identified while performing the STAR analyses was the need to develop methodological approaches to adjusting outcomes following subsequent treatments in trials.
Relating to qualitative work
The STAR qualitative work is related primarily to recruitment. However, if a DFIS approach is implemented in practice, thought should be given to how patients should be supported during the extended break to cope with and alleviate worries. The overall trial results will help with this, but additional qualitative work with patients who are approaching or on a treatment break would be useful to aid understanding of how patients are feeling during this time and could inform how patients could best be supported.
Relating to computerised tomography substudy
The STAR CT substudy confirmed that assessment of enhancement, as well as well-recognised size change, alters the categorisation of response. Use of mChoi criteria may allow for earlier detection of PD, and more representative separation of participants with PR versus SD. Assessment of changes in tumour heterogeneity may complement standard response assessment but current data remain limited. Further work is required to explore how to incorporate these findings alongside standard size assessments for response and also to explore the clinical relevance of these findings, in terms of the impact of earlier changes in treatment.
Relating to magnetic resonance imaging substudy
This small feasibility substudy has shown DCE-MRI-derived biomarkers of tumour perfusion (perfused tumour volume, Ktrans and ECV) as potential surrogate biomarkers to predict early disease progression following treatment with TKI therapy in advanced RCC, and in addition, demonstrated reproducibility of these DCE-MRI biomarkers. Going forward, larger prospective clinical studies are required to confirm these biomarkers as predictive markers of early progression, and importantly lead on from this and test their wider application and clinical relevance.
Chapter 9 Conclusion
The STAR trial provides clear evidence that a treatment-break strategy for patients with RCC as part of their TKI therapy is feasible, has both patient and NHS economic benefits, and does not meaningfully reduce life expectancy. However, NI between the two arms cannot be concluded from the trial due to the reasons discussed above. From the trial results, following 6 months of continuous TKI therapy, at least one treatment break should be considered, with additional treatment breaks thought to be reasonable provided that the patient has not progressed while on treatment. In addition, where there is a patient or healthcare need to disrupt treatment (e.g. during the COVID pandemic), STAR provides reassurance that this is not likely to have a detrimental effect on patient outcomes.
Further research should now be considered into the potential benefits of treatment breaks in more recent RCC treatment, for example, IO monotherapy or TKI/IO combination therapy. In addition, further research is also warranted to consider if there are certain biological factors which result in some patients benefiting from a treatment break more than others. In addition, learning around intermittent treatment strategy, trials should be taken from this trial and used to extend this research area to other types of cancer and their treatments more generally. With the number of available systemic treatments in cancer increasing with associated high costs, in terms of finance and side effects, research to define appropriate treatment duration is increasingly important. Such trials are frequently large, costly and can be challenging to deliver, hence ensuring that they are designed in the most appropriate way is essential.
Additional information
Contributions of authors
Fiona Collinson (https://orcid.org/0000-0001-6964-6406) was co-Chief Investigator.
Kara-Louise Royle (https://orcid.org/0000-0003-0225-1199) conducted statistical analysis.
Jayne Swain (https://orcid.org/0000-0003-2729-3029) conducted trial management.
Christy Ralph (https://orcid.org/0000-0001-5581-2987) conducted trial design and management.
Anthony Maraveyas (https://orcid.org/0000-0003-4176-5176) conducted trial design.
Tim Eisen (https://orcid.org/0000-0001-9663-4873) conducted trial design.
Paul Nathan (https://orcid.org/0000-0002-2327-3250) conducted trial design.
Robert Jones (https://orcid.org/0000-0002-2904-6980) conducted trial design.
David Meads (https://orcid.org/0000-0003-1369-2483) was the lead of the health economics analysis.
Tze Min Wah (https://orcid.org/0000-0001-5670-5454) was the lead of the MRI substudy, conducted trial design and management and was lead of the central CT reporting.
Adam Martin (https://orcid.org/0000-0002-2559-6483) conducted health economics analysis.
Janine Bestall (https://orcid.org/0000-0001-6765-6379) conducted patient preference and understanding substudy data collection and analysis.
Christian Kelly-Morland (https://orcid.org/0000-0002-1197-0382) conducted CT substudy data analysis.
Christopher Linsley (https://orcid.org/0000-0002-3940-1739) conducted data management.
Jamie Oughton (https://orcid.org/0000-0002-2047-804X) conducted trial management.
Kevin Chan (https://orcid.org/0000-0002-4865-2388) was a trial physician.
Elisavet Theodoulou (https://orcid.org/0000-0002-1094-3630) was a trial physician.
Gustavo Arias-Pinilla (https://orcid.org/0000-0002-0137-8377) was a trial physician.
Amy Kwan (https://orcid.org/0000-0003-1046-854X) was a trial physician.
Luis Daverede (https://orcid.org/0000-0002-5018-6275) was a trial physician.
Cat Handforth (https://orcid.org/0000-0001-5171-4917) was a trial physician.
Sebastian Trainor (https://orcid.org/0000-0003-3840-4142) was a trial physician.
Abdulazeez Salawu (https://orcid.org/0000-0002-4420-0958) was a trial physician.
Christopher McCabe (https://orcid.org/0000-0001-5728-4129) conducted health economics design.
Vicky Goh (https://orcid.org/0000-0002-2321-8091) was lead of the CT substudy.
David Buckley (https://orcid.org/0000-0001-6659-8365) conducted the MRI substudy.
Jenny Hewison (https://orcid.org/0000-0003-3026-3250) was lead of the patient preference and understanding substudy.
Walter Gregory (https://orcid.org/0000-0003-2641-8416) conducted statistical design.
Peter Selby (https://orcid.org/0000-0002-3782-069X) conducted trial design and management.
Julia Brown (https://orcid.org/0000-0002-2719-7064) conducted trial design and management, input into QoL and statistical design and analysis.
Janet Brown (https://orcid.org/0000-0003-4960-3032) was Chief Investigator.
Acknowledgements
We would like to thank the NCRI Bladder and Renal Oncology Group for their invaluable support and advice.
Participants
We thank our 920 participants for their willingness to take part in research in order to inform future generations. Through ongoing clinical trials, we continue to strive for better outcomes for patients with resected RCC.
Trial Steering Committee
Professor Barry Hancock (chairperson), Dr Bernard Escudier, Dr Wedi Qian, Jackie Low (PPI).
Data Monitoring and Ethics Committee
Mr James Paul (chairperson), Dr Richard Jackson, Dr Peter Hall, Dr Uschi Hoffmann.
Trial Management Group
Professor Janet Brown (chairperson), Dr Fiona Collinson, Professor Julia Brown, Dr Christy Ralph, Professor Peter Selby, Professor Jenny Hewison, Dr David Meads, Dr Janine Bestall, Dr Adam Martin, Dr Tze Min Wah, Dr Pat Hanlon, Kara-Louise Royle, Jayne Swain, Chris Linsley.
Previous members: Dr Cat Handforth, Dr Kevin Chan, Dr Abdulazeez Salawu, Dr Sebastian Trainor, Professor Walter Gregory, Dr Luis Daverede, Dr Sandy Tubeuf, Silviya Nikolova, Dr Helen Howard, Lucy McParland, Emma Best, Emma Batman, Laura Allen, Vicky Hiley, Katie Neville, Heather Cook, Jamie Oughton, Cait Kielty-Adey, Alex Smith.
We would like to acknowledge the significant contribution of Pat Hanlon, our TMG patient and public involvement representative, who sadly passed away in January 2020. Pat had a huge commitment to kidney cancer research. He played an important role on the TMG and we will miss him very much.
We would also like to acknowledge the contribution of Kate Hayward who provided invaluable guidance during trial design.
MRI substudy group
Dr Tze Min Wah, Professor David Buckley, Dr Jim Zhong.
The MRI substudy was funded by the Leeds Hospitals Charity at St James’s Hospital, Leeds.
CT substudy group
Professor Vicky Goh, Dr Christian Kelly-Morland.
Investigator sites
We would like to thank all the trial teams members involved in the study at the participating sites, and it would not have been possible to carry out this work without their support.
The following sites and Principal Investigators were involved in the recruiting and treating trial participants:
Nottingham University Hospital (Nottingham University Hospitals Trust); Professor Poulam Patel.
Weston Park Hospital (Sheffield Teaching Hospitals NHS Foundation Trust); Dr Omar Din.
The Clatterbridge Cancer Centre NHS Foundation Trust; Dr Judith Carser (former Principal Investigator), Dr Richard Griffiths.
The Royal Marsden NHS Foundation Trust; Professor James Larkin.
St James’ University Hospitals NHS Foundation Trust; Professor Janet Brown (former Principal Investigator), Dr Christy Ralph.
Castle Hill Hospital (Hull University Teaching Hospitals NHS Trust); Professor Anthony Maraveyas.
The Christie NHS Foundation Trust; Professor Fiona Thistlewaite (former Principal Investigator), Dr Tom Waddell.
Mount Vernon Hospital (East and North Hertfordshire NHS Foundation Trust); Dr Paul Nathan.
Addenbrooke’s Hospital (Cambridge University Hospitals NHS Foundation Trust); Professor Tim Eisen.
The Beatson West of Scotland Cancer Centre (NHS Greater Glasgow and Clyde); Professor Rob Jones.
Southampton General Hospital (University Hospital Southampton NHS Foundation Trust); Dr Matthew Wheater.
Northern Centre for Cancer Care (The Newcastle upon Tyne Hospitals NHS Foundation Trust); Dr Rhona McMenemin (former Principal Investigator), Dr Ashraf Azzabi.
St Bartholomew’s Hospital (Barts Health NHS Trust); Professor Thomas Powles.
Scarborough General Hospital (York and Scarborough Teaching Hospitals NHS Foundation Trust); Dr Mohan Hingorani (former Principal Investigator), Dr Khaliq Rehman (former Principal Investigator), Dr Mohammad Khan.
Belfast City Hospital (Belfast Health and Social Care Trust); Dr James McAleer (former Principal Investigator), Dr Alison Clayton.
Yeovil District Hospital NHS Foundation Trust; Dr Geoffrey Sparrow (former Principal Investigator), Dr Urmila Barthakur (former Principal Investigator), Dr Emma Gray (former Principal Investigator), Dr Erica Beaumont (former Principal Investigator).
Royal Cornwall Hospitals NHS Trust; Dr Alastair Thomson.
Royal United Hospital (Royal United Hospitals Bath NHS Foundation Trust); Professor Mark Beresford.
Queen Alexandra Hospital (Portsmouth Hospitals University NHS Trust); Dr Joanna Gale.
Royal Bournemouth Hospital (University Hospitals Dorset NHS Foundation Trust); Dr Thomas Geldart.
Essex County Hospital (East Suffolk and North Essex NHS Foundation Trust); Dr Dakshinamoorthy Muthukumar.
Royal Free London NHS Foundation Trust; Professor Thomas Powles (former Principal Investigator), Dr Ekaterini Boleti.
Royal Shrewsbury Hospital (Shrewsbury and Telford Hospitals NHS Trust); Dr Narayanan Srihari.
Royal Devon and Exeter NHS Foundation Trust; Dr Denise Sheehan (former Principal Investigator), Dr Rajaguru Srinivasan.
Great Western Hospital (Great Western Hospitals NHS Foundation Trust); Dr Omar Khan.
Birmingham Heartlands Hospital (University Hospitals Birmingham NHS Foundation Trust); Dr Anjali Zarkar.
Norfolk and Norwich University Hospitals NHS Foundation Trust; Dr Gaurav Kapur.
Kent and Canterbury Hospital (East Kent Hospitals University NHS Foundation Trust); Dr Carys Thomas.
Maidstone Hospital (Maidstone and Tunbridge Wells NHS Trust); Dr Sharon Beesley (former Principal Investigator), Dr Kathryn Lees.
Medway Maritime Hospital (Medway NHS Foundation Trust); Dr Henry Taylor (former Principal Investigator), Dr Christos Mikropoulos (former Principal Investigator), Professor Stergios Boussios.
Cheltenham General Hospital (Gloucestershire Hospitals NHS Foundation Trust); Dr David Farrugia (former Principal Investigator), Dr Marios Decatris.
Cumberland Infirmary (North Cumbria Integrated Care NHS Foundation Trust); Dr Anil Kumar.
Blackpool Victoria Hospital (Blackpool Teaching Hospitals NHS Foundation Trust); Dr Falalu Danwata.
King’s Mill Hospital (Sherwood Forest Hospitals NHS Foundation Trust); Dr Santhanam Sundar.
Royal Preston Hospital (Lancashire Teaching Hospitals NHS Foundation Trust); Dr Natalie Charnley.
Musgrove Park Hospital (Somerset NHS Foundation Trust); Dr Mohini Varughese (former Principal Investigator), Dr Emma Gray.
Broomfield Hospital (Mid and South Essex NHS Foundation Trust); Dr Gopalakrishnan Srinivasan (former Principal Investigator), Dr Abdel Hamid.
Charing Cross Hospital (Imperial College Healthcare NHS Trust); Dr Naveed Sarwar.
University Hospital Coventry (University Hospitals Coventry and Warwickshire NHS Trust); Dr Andrew Stockdale (former Principal Investigator), Dr Jane Worlding (former Principal Investigator), Professor Stergios Boussios.
Eastbourne District General Hospital (East Sussex Healthcare NHS Trust); Dr Caroline Manetta.
Conquest Hospital (East Sussex Healthcare NHS Trust); Dr Caroline Manetta.
Ninewells Hospital (NHS Tayside); Dr Angela Scott (former Principal Investigator), Dr Mark Baxter.
Western General Hospital (NHS Lothian); Professor Duncan McLaren (former Principal Investigator), Dr Aravindhan Sundaramurthy.
Dorset County Hospital (Dorset County Hospital NHS Foundation Trust); Dr Richard Osborne (former Principal Investigator), Dr Renata Dega (former Principal Investigator), Dr Melanie Harvey.
Darent Valley Hospital (Dartford and Gravesham NHS Trust); Dr Roy Vergis (former Principal Investigator), Professor Seshadri Sriprasad (former Principal Investigator), Dr Patryk Brulinski (former Principal Investigator), Dr Amanda Clarke.
Leicester Royal Infirmary (University Hospitals of Leicester NHS Trust); Dr Guy Faust.
Glan Clwyd Hospital (Betsi Cadwaladr University Health Board); Professor Nicholas Stuart (former Principal Investigator), Dr Carey MacDonald-Smith.
Ysbyty Gwynedd (Betsi Cadwaladr University Health Board); Professor Nicholas Stuart (former Principal Investigator), Dr Carey MacDonald-Smith (former Principal Investigator), Dr Anna Mullard (former Principal Investigator), Dr Pasquale Innominato.
James Cook University Hospital (South Tees Hospitals NHS Foundation Trust); Dr Janine Graham.
Velindre Cancer Centre (Velindre University NHS Trust); Dr Jason Lester (former Principal Investigator), Dr Nachi Palaniappan.
Derriford Hospital (University Hospitals Plymouth NHS Trust); Dr Martin Highley.
Royal Derby Hospital (University Hospitals of Derby and Burton NHS Foundation Trust); Dr Prabir Chakraborti (former Principal Investigator), Dr Prantik Das.
Royal Surrey County Hospital (Royal Surrey NHS Foundation Trust); Dr Agnieszka Michael.
Torbay Hospital (Torbay and South Devon NHS Foundation Trust); Dr Anna Lydon.
Guy’s Hospital (Guy’s and St Thomas’ NHS Foundation Trust); Dr Sarah Rudman.
Patient data statement
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it is important that there are safeguards to make sure that they are stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review. For access, data requestors will need to sign a data access agreement.
Ethics statement
Ethical approval for the study was given by Liverpool Central Research Ethics Committee in June 2011 (reference number 11/NW/0246).
Preparation of the participant DVD
Dame Leslie Fallowfield and Professor Valerie Jenkins the team at Sussex Health Outcomes Research and Education in Cancer (SHORE-C) at the University of Sussex.
Information governance statement
University of Leeds is committed to handling all personal information in line with the UK Data Protection Act (2018) and the General Data Protection Regulation (EU GDPR) 2016/679. Under the Data Protection legislation, University of Leeds is the Data Controller, and you can find out more about how we handle personal data, including how to exercise individual rights and the contact details for our Data Protection Officer here: https://ctru.leeds.ac.uk/privacy-cookies/how-we-use-personal-data/.
Disclosure of interests of authors
Full disclosure of interests: Completed ICMJE forms for all authors, including all related interests, are available in the toolkit on the NIHR Journals Library report publication page at https://doi.org/10.3310/JWTR4127.
Primary conflicts of interest: Christy Ralph declared travel support from BMS and Eisai. Anthony Maraveyas declared payment or honoraria for lectures from Eisai. Tim Eisen declared research grants from AstraZeneca, Bayer and Pfizer; employment by Roche and formerly by AstraZeneca; a leadership role at Macmillan Cancer Support and Kidney Cancer UK; AstraZeneca and Roche stock and other interests in AstraZeneca, Roche, University of Cambridge and Cambridge University Hospitals NHS Trust. Paul Nathan declared consulting fees from Novartis, BMS, Merck and Pfizer; payment or honoraria for lectures from Novartis; support for attending meetings from Pfizer and participation on a Data Safety Monitoring Board or Advisory Board for COMBI-I, YKST and ACHILLES and a leadership role at Melanoma Focus and CTRT. Robert Jones declared research grants from Roche and Exelixis; consulting fees from Roche, Pfizer, MSD, Merck Serono, Ipsen, EUSA, Novartis; payment or honoraria for lectures from Roche, Pfizer, MSD, Merck Serono, Ipsen, EUSA; support for attending meetings from MSD and Ipsen and participation on a Data Safety Monitoring Board or Advisory Board for Roche. Tze Min Wah declared support from Angiodynamics, Boston Scientific; research grants from Boston Scientific, Angiodynamics, British Society of Interventional Radiology, HistoSonics, Johnson & Johnson; payment or honoraria for lectures from Angiodynamics and a leadership role at Interventional Oncology UK, British Society of Interventional Radiology, CIRSE Membership Sub-Committee. Elisavet Theodoulou declared a fellowship from Yorkshire Cancer Research UK. Sebastian Trainor declared a grant from WCRF; travel support from Lily Oncology and Novartis. David Meads was a member of the HTA EESC methods group (2014–17); HTA EESC panel (2013–17). He is a member of the NIHR PGfAR funding panel (2016–present) and of the NICE technology appraisal committee (2016–present). David Buckley received grants from EPSRC and British Heart Foundation. Christopher McCabe declared research grants from AstraZeneca, Boehringer, Inglemheim, GlaxoSmithKline, Roche, Takeda; consulting fees from AstraZeneca, Merck, Novartis and a leadership role at Board Observer International Network Health Technology Assessment Agencies (INAHTA). Vicky Goh declared research grants from Siemens Healthcare; payment or honoraria for lectures from Siemens Healthcare, European School of Radiology and support for attending meetings from Siemens Healthcare. Jenny Hewison declared research awards from the NIHR HTA Programme, NIHR Programme Grants for Applied Research, NIHR Health Services and Development Research Programme. Jenny Hewison reports membership of the following: NIHR Technology Assessment Programme Clinical Trials Board (Deputy Chair until 2012), NIHR CTU Standing Advisory Committee (2012–17); NIHR Programme Grants for Applied Research (Chair of Subpanel 2008–07), NIHR Global Health Research Programme Selection Panel (2017–18) and the TARS Contract Retender committee (2014). Walter Gregory declared research grants from Janssen and Abbvie, consulting fees from Abbvie and payment or honoraria for lectures from Abbvie. Peter Selby declared support from NIHR; participation on a Data Safety Monitoring Board or Advisory Board for Scientific Advisory Board at Lincoln International Institute for Rural Health, University of Lincoln, Chair of Scientific Advisory Board for NIHR Research for Patient Benefit Grant, Chair of Scientific Advisory Board for Pinpoint Scientific, Electronic Frailty Index in frail patients undergoing chemotherapy, Faculty member of European School of Oncology and Advisor to the European Cancer Organisation, Emeritus Professor at the University of Leeds. Julia Brown declared research grants from Yorkshire Cancer Research, NIHR HTA, NIHR EME, Cancer Research UK, Bowel Cancer UK, F Hoffman La Roche Ltd; payment or honoraria for lectures from NIHR HTA General Funding Committee Chair and NIHR Senior Investigator and participation on a Data Safety Monitoring Board or Advisory Board for Chair NIHR Global Surgery Unit Steering Committee, Chair NIHR HTA ByBand Trial Steering Committee, NIHR HTA General Funding Committee, NIHR HTA Remit and Competitiveness Group, NIHR Post-funding Committee Teleconference, NIHR HTA Funding Committee Policy Group, NIHR HTA Clinical Evaluation and Trials Committee, NIHR HTA Programme Oversight Committee, NIHR HTA Fast-Track Committee and a leadership role in CTUs Funding by NIHR Committee and COVID-19 Reviewing. Janet Brown reports grants from NIHR (RP-PG-1016-20007) and Bayer; Personal fees from Novartis, Ipsen, Amgen, MSD, BMS, and Bayer; Travel support from Ipsen; Trustee of the Bone Research Trust; Assistance with Abstract from Ipsen.
Disclaimers
This article presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- UK CR . Kidney Cancer Statistics n.d. www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/kidney-cancer#heading-Zero (accessed 23 March 2023).
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with Nivolumab. JAMA Oncol 2019;5:1411-20.
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet 1999;353:14-7.
- Pyrhonen S, Salminen E, Ruutu M, Lehtonen T, Nurmi M, Tammela T, et al. Prospective randomized trial of interferon alfa-2a plus vinblastine versus vinblastine alone in patients with advanced renal cell cancer. J Clin Oncol 1999;17:2859-67.
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 2007;356:115-24.
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:3584-90.
- Pytel D, Sliwinski T, Poplawski T, Ferriola D, Majsterek I. Tyrosine kinase blockers: new hope for successful cancer therapy. Anticancer Agents Med Chem 2009;9:66-7.
- National Institute for Health and Care Excellence (NICE) . Sunitinib for the First-Line Treatment of Advanced and Or Metastatic Renal Cell Carcinoma 2009. www.nice.org.uk/guidance/TA169 (accessed 23 March 2023).
- Gore ME, Szczylik C, Porta C, Bracarda S, Bjarnason GA, Oudard S, et al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: an expanded-access trial. Lancet Oncol 2009;10:757-63.
- Shepard DR, Garcia JA. Toxicity associated with the long-term use of targeted therapies in patients with advanced renal cell carcinoma. Expert Rev Anticancer Ther 2009;9:795-80.
- Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 2010;28:1061-8.
- Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013;369:722-31.
- Motzer RHT, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, et al. Randomized, Open-label, Phase III Trial of Pazopanib Versus Sunitinib in First-line Treatment of Patients with Metastatic Renal Cell Carcinoma (MRCC): Results of the COMPARZ Trial. Vienna: European Society for Medical Oncology; 2012.
- European Society for Medical Oncology. Vienna: Elsevier Inc.; 2012.
- Escudier B, Porta C, Bono P, Powles T, Eisen T, Sternberg CN, et al. Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J Clin Oncol 2014;32:1412-8.
- National Institute for Health and Care Excellence (NICE) . Tivozanib for Treating Advanced Renal Cell Carcinoma 2018. www.nice.org.uk/Guidance/TA512 (accessed 23 March 2023).
- National Institute for Health and Care Excellence (NICE) . Cabozantinib for Untreated Advanced Renal Cell Carcinoma 2018. www.nice.org.uk/Guidance/TA542 (accessed 23 March 2023).
- Maughan TS, James RD, Kerr DJ, Ledermann JA, Seymour MT, Topham C, et al. Medical Research Council Colorectal Cancer Group . Comparison of intermittent and continuous palliative chemotherapy for advanced colorectal cancer: a multicentre randomised trial. Lancet 2003;361:457-64.
- Adams RA, Meade AM, Seymour MT, Wilson RH, Madi A, Fisher D, et al. MRC COIN Trial Investigators . Intermittent versus continuous oxaliplatin and fluoropyrimidine combination chemotherapy for first-line treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet Oncol 2011;12:642-53.
- Tournigand C, Cervantes A, Figer A, Lledo G, Flesch M, Buyse M, et al. OPTIMOX1: a randomized study of FOLFOX4 or FOLFOX7 with oxaliplatin in a stop-and-Go fashion in advanced colorectal cancer – a GERCOR study. J Clin Oncol 2006;24:394-400.
- de Gramont A, Buyse M, Abrahantes JC, Burzykowski T, Quinaux E, Cervantes A, et al. Reintroduction of oxaliplatin is associated with improved survival in advanced colorectal cancer. J Clin Oncol 2007;25:3224-9.
- Chibaudel B, Maindrault-Goebel F, Lledo G, Mineur L, Andre T, Bennamoun M, et al. Can chemotherapy be discontinued in unresectable metastatic colorectal cancer? The GERCOR OPTIMOX2 Study. J Clin Oncol 2009;27:5727-33.
- Maughan T, Adams R, Wilson R, Seymour M, Meade A, Kaplan R. Chemotherapy-free intervals for patients with metastatic colorectal cancer remain an option. J Clin Oncol 2010;28:e275-6.
- Pereira AA, Rego JF, Munhoz RR, Hoff PM, Sasse AD, Riechelmann RP. The impact of complete chemotherapy stop on the overall survival of patients with advanced colorectal cancer in first-line setting: a meta-analysis of randomized trials. Acta Oncol 2015;54:1737-46.
- Berry SR, Cosby R, Asmis T, Chan K, Hammad N, Krzyzanowska MK. Cancer Care Ontario's Gastrointestinal Disease Site Group . Continuous versus intermittent chemotherapy strategies in metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol 2015;26:477-85.
- National Institute for Health and Care Excellence (NICE) . COVID-19 Rapid Guideline: Delivery of Systemic Anticancer Treatments 2020. www.nice.org.uk/guidance/ng161/resources/covid19-rapid-guideline-delivery-of-systemic-anticancer-treatments-pdf-66141895710661 (accessed 23 March 2023).
- Blay JY, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 2007;25:1107-13.
- Zama IN, Hutson TE, Elson P, Cleary JM, Choueiri TK, Heng DY, et al. Sunitinib rechallenge in metastatic renal cell carcinoma patients. Cancer 2010;116:5400-6.
- Oudard S, Geoffrois L, Guillot A, Chevreau C, Deville JL, Falkowski S, et al. Clinical activity of sunitinib rechallenge in metastatic renal cell carcinoma-Results of the REchallenge with SUnitinib in MEtastatic RCC (RESUME) Study. Eur J Cancer 2016;62:28-35.
- Kahl C, Hilgendorf I, Freund M, Casper J. Continuous therapy with sunitinib in patients with metastatic renal cell carcinoma. Onkologie 2008;31.
- Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24:2505-12.
- Ornstein MC, Wood LS, Elson P, Allman KD, Beach J, Martin A, et al. A Phase II Study of intermittent sunitinib in previously untreated patients with metastatic renal cell carcinoma. J Clin Oncol 2017;35:1764-9.
- Fogli S, Porta C, Del Re M, Crucitta S, Gianfilippo G, Danesi R, et al. Optimizing treatment of renal cell carcinoma with VEGFR-TKIs: a comparison of clinical pharmacology and drug-drug interactions of anti-angiogenic drugs. Cancer Treat Rev 2020;84.
- Atkins MB, Tannir NM. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat Rev 2018;70:127-37.
- Deleuze A, Saout J, Dugay F, Peyronnet B, Mathieu R, Verhoest G, et al. Immunotherapy in renal cell carcinoma: the future is now. Int J Mol Sci 2020;21.
- Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. CheckMate 214 Investigators . Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N Engl J Med 2018;378:1277-90.
- Taguchi S, Buti S, Fukuhara H, Otsuka M, Bersanelli M, Morikawa T, et al. Benefit of adjuvant immunotherapy in renal cell carcinoma: a myth or a reality?. PLOS ONE 2017;12.
- Gul A, Rini BI. Adjuvant therapy in renal cell carcinoma. Cancer 2019;125:2935-44.
- Martini A, Fallara G, Pellegrino F, Cirulli GO, Larcher A, Necchi A, et al. Neoadjuvant and adjuvant immunotherapy in renal cell carcinoma. World J Urol 2021;39:1369-76.
- MD+CALC . IMDC (International Metastatic RCC Database Consortium) Risk Score for RCC 2021. www.mdcalc.com/imdc-international-metastatic-rcc-database-consortium-risk-score-rcc (accessed 23 March 2023).
- Mekhail TM, Abou-Jawde RM, Boumerhi G, Malhi S, Wood L, Elson P, et al. Validation and extension of the Memorial Sloan-Kettering prognostic factors model for survival in patients with previously untreated metastatic renal cell carcinoma. J Clin Oncol 2005;23:832-41.
- Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378:1931-9.
- Mehta A, Sonpavde G, Escudier B. Tivozanib for the treatment of renal cell carcinoma: results and implications of the TIVO-1 trial. Future Oncol 2014;10:1819-26.
- National Institute for Health and Care Excellence (NICE) . Cabozantinib for Previously Treated Advanced Renal Cell Carcinoma 2017. www.nice.org.uk/guidance/ta463 (accessed 26 September 2022).
- Choueiri TK, Hessel C, Halabi S, Sanford B, Michaelson MD, Hahn O, et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): progression-free survival by independent review and overall survival update. Eur J Cancer 2018;94:115-25.
- Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 2015;372:621-30.
- Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol 2015;16:1473-82.
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. CheckMate 025 Investigators . Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13.
- Rassy E, Flippot R, Albiges L. Tyrosine kinase inhibitors and immunotherapy combinations in renal cell carcinoma. Ther Adv Med Oncol 2020;12.
- Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. KEYNOTE-426 Investigators . Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1116-27.
- Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1103-15.
- Flippot R, Escudier B, Albiges L. Immune checkpoint inhibitors: toward new paradigms in renal cell carcinoma. Drugs 2018;78:1443-57.
- Bedke J, Albiges L, Capitanio U, Giles RH, Hora M, Lam TB, et al. The 2021 Updated European association of urology guidelines on renal cell carcinoma: immune checkpoint inhibitor-based combination therapies for treatment-naive metastatic clear-cell renal cell carcinoma are standard of care. Eur Urol 2021;80:393-7.
- Renfro LA, Grothey AM, Paul J, Floriani I, Bonnetain F, Niedzwiecki D, et al. Projecting event-based analysis dates in clinical trials: an illustration based on the international duration evaluation of adjuvant chemotherapy (IDEA) collaboration. Projecting analysis dates for the IDEA collaboration. Forum Clin Oncol 2014;5:1-7.
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. TARGET Study Group . Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125-34.
- Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol 2009;27:3312-8.
- Escudier B, Szczylik C, Hutson TE, Demkow T, Staehler M, Rolland F, et al. Randomized phase II trial of first-line treatment with sorafenib versus interferon alfa-2a in patients with metastatic renal cell carcinoma. J Clin Oncol 2009;27:1280-9.
- Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Ou SS, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol 2008;26:5422-8.
- Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. AVOREN Trial investigators . Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet 2007;370:2103-11.
- Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427-34.
- Motzer RJ, Mazumdar M, Bacik J, Berg W, Amsterdam A, Ferrara J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530-40.
- Ltd NPU . Votrient 200 mg Film Coated Tablets – Summary of Product Characteristics 2019. www.medicines.org.uk/emc/product/7861/smpc (accessed 14 November 2019).
- EuroQol Research Foundation . EQ-5D-3L User Guide 2018. https://euroqol.org/publications/user-guides/ (accessed 23 March 2023).
- Allegra C, Blanke C, Buyse M, Goldberg R, Grothey A, Meropol NJ, et al. End points in advanced colon cancer clinical trials: a review and proposal. J Clin Oncol 2007;25:3572-5.
- Dolan P, Gudex C, Kind P, Williams A. A Social Tariff for EuroQol: Results from a UK General Population Survey. Discussion Paper Number 138. New York: Centre for Health Economics, University of York; 1995.
- Group F. FKSI-DRS Scoring Downloads 2021. www.facit.org/measures-scoring-downloads/fksi-drs-scoring-downloads (accessed 23 March 2023).
- Group F. FKSI-15 Scoring Downloads 2021. www.facit.org/measures-scoring-downloads/fksi-15-scoring-downloads (accessed 23 March 2023).
- Group F. Scoring of the FACIT Measures 2021. www.facit.org/scoring (accessed 23 March 2023).
- Group F. FACT-G Scoring Downloads 2021. www.facit.org/measures-scoring-downloads/fact-g-scoring-downloads (accessed 23 March 2023).
- Introducing the FMM procedure for finite mixture models. SAS Global Forum 2012;2012. https://support.sas.com/resources/papers/proceedings12/328-2012.pdf (accessed 23 March 2023).
- Faria R, Gomes M, Epstein D, White IR. A guide to handling missing data in cost-effectiveness analysis conducted within randomised controlled trials. PharmacoEconomics 2014;32:1157-70.
- Simons CL, Rivero-Arias O, Yu LM, Simon J. Multiple imputation to deal with missing EQ-5D-3L data: should we impute individual domains or the actual index?. Qual Life Res 2015;24:805-15.
- White IR, Royston P, Wood AM. Multiple imputation using chained equations: issues and guidance for practice. Stat Med 2011;30:377-99.
- Morris TP, White IR, Royston P. Tuning multiple imputation by predictive mean matching and local residual draws. BMC Med Res Methodol 2014;14.
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal 2013 2013. www.nice.org.uk/process/pmg9/chapter/foreword (accessed 4 April 2013).
- National Institute for Health and Care Excellence (NICE) . British National Formulary (BNF) 2020. https://bnf.nice.org.uk/ (accessed 2 September 2021).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2016. Canterbury, UK: Personal Social Services Research Unit, The University of Kent; 2016.
- Health Do . NHS Reference Costs 2015–2016 2017. www.gov.uk/government/publications/nhs-reference-costs-2015-to-2016 (accessed 15 December 2016).
- Care MCC. Understanding the Cost of End of Life Care in Different Settings 2012. www.mariecurie.org.uk/globalassets/media/documents/commissioning-our-services/publications/understanding-cost-end-life-care-different-settingspdf (accessed 23 March 2023).
- England PH. End of Life Care Economic Tool 2017. www.gov.uk/government/publications/end-of-life-care-economic-tool (accessed 23 March 2023).
- Amdahl J, Diaz J, Sharma A, Park J, Chandiwana D, Delea TE. Cost-effectiveness of pazopanib versus sunitinib for metastatic renal cell carcinoma in the United Kingdom. PLOS ONE 2017;12.
- Zhang W, Bansback N, Anis AH. Measuring and valuing productivity loss due to poor health: a critical review. Soc Sci Med 2011;72:185-92.
- Statistics OfN . Employee Earnings in the UK: 2020 2020. https://www.ons.gov.uk/employmentandlabourmarket/peopleinwork/earningsandworkinghours/bulletins/annualsurveyofhoursandearnings/2020 (accessed 23 March 2023).
- Koopmanschap MA, Rutten FF, van Ineveld BM, van Roijen L. The friction cost method for measuring indirect costs of disease. J Health Econ 1995;14:171-89.
- Willan AR, Lin DY, Manca A. Regression methods for cost-effectiveness analysis with censored data. Stat Med 2005;24:131-45.
- Barton GR, Briggs AH, Fenwick EA. Optimal cost-effectiveness decisions: the role of the cost-effectiveness acceptability curve (CEAC), the cost-effectiveness acceptability frontier (CEAF), and the expected value of perfection information (EVPI). Value Health 2008;11:886-97.
- Claxton K, Martin S, Soares M, Rice N, Spackman E, Hinde S, et al. Methods for the estimation of the National Institute for Health and Care Excellence cost-effectiveness threshold. Health Technol Assess 2015;19:1-504.
- Bullement A, Cranmer HL, Shields GE. A review of recent decision-analytic models used to evaluate the economic value of cancer treatments. Appl Health Econ Health Policy 2019;17:771-80.
- National Institute for Health and Care Excellence (NICE) . Pazopanib (Votrient) for the First-Line Treatment of Patients With Advanced Renal Cell Carcinoma (RCC) 2010. www.nice.org.uk/guidance/ta215/documents/renal-cell-carcinoma-first-line-metastatic-pazopanib-manufacturer-submission-submission2.
- Latimer NR. Survival analysis for economic evaluations alongside clinical trials – extrapolation with patient-level data: inconsistencies, limitations, and a practical guide. Med Decis Making 2013;33:743-54.
- Edlin R, McCabe C, Hulme P, Wright J. Cost Effectiveness Modelling for Health Technology Assessment: A Practical Course. New York: Springer; 2015.
- Claxton KP, Sculpher MJ. Using value of information analysis to prioritise health research: some lessons from recent UK experience. PharmacoEconomics 2006;24:1055-68.
- Strong M, Oakley JE, Brennan A. Estimating multiparameter partial expected value of perfect information from a probabilistic sensitivity analysis sample: a nonparametric regression approach. Med Decis Making 2014;34:311-26.
- O’Cathain A, Thomas KJ, Drabble SJ, Rudolph A, Goode J, Hewison J. Maximising the value of combining qualitative research and randomised controlled trials in health research: the QUAlitative Research in Trials (QUART) study – a mixed methods study. Health Technol Assess 2014;18:1v-197vi.
- Hewison J, Haines A. Overcoming barriers to recruitment in health research. BMJ 2006;333:300-2.
- QSR International Pty Ltd. NVivo 2015 version 11 computer programme 2015.
- Boyatzis RE. Transforming Qualitative Information: Thematic Analysis and Code Development. London: SAGE Publications Ltd; 1998.
- Hoffe H YJ, Marks David F, Yardley Lucy. Research Methods for Clinical and Health Psychology. London: SAGE Publications Ltd; 2004.
- Boyatzis RE. Transforming Qualitative Information. London: SAGE Publications Ltd; 1988.
- McCann SK, Campbell MK, Entwistle VA. Reasons for participating in randomised controlled trials: conditional altruism and considerations for self. Trials 2010;11.
- Lane JA, Donovan JL, Davis M, Walsh E, Dedman D, Down L, et al. ProtecT study group . Active monitoring, radical prostatectomy, or radiotherapy for localised prostate cancer: study design and diagnostic and baseline results of the ProtecT randomised phase 3 trial. Lancet Oncol 2014;15:1109-18.
- Rooshenas L, Paramasivan S, Jepson M, Donovan JL. Intensive triangulation of qualitative research and quantitative data to improve recruitment to randomized trials: The QuinteT approach. Qual Health Res 2019;29:672-9.
- Zhong J, Palkhi E, Buckley DL, Collinson FJ, Ralph C, Jagdev S, et al. Feasibility study on using dynamic contrast enhanced MRI to assess the effect of tyrosine kinase inhibitor therapy within the star trial of metastatic renal cell cancer. Diagnostics (Basel) 2021;11.
- Hahn OM, Yang C, Medved M, Karczmar G, Kistner E, Karrison T, et al. Dynamic contrast-enhanced magnetic resonance imaging pharmacodynamic biomarker study of sorafenib in metastatic renal carcinoma. J Clin Oncol 2008;26:4572-8.
- Sweis RF, Medved M, Towey S, Karczmar GS, Oto A, Szmulewitz RZ, et al. Dynamic contrast-enhanced magnetic resonance imaging as a pharmacodynamic biomarker for pazopanib in metastatic renal carcinoma. Clin Genitourin Cancer 2017;15:207-12.
- Bex A, Fournier L, Lassau N, Mulders P, Nathan P, Oyen WJ, et al. Assessing the response to targeted therapies in renal cell carcinoma: technical insights and practical considerations. Eur Urol 2014;65:766-77.
- Rossi SH, Prezzi D, Kelly-Morland C, Goh V. Imaging for the diagnosis and response assessment of renal tumours. World J Urol 2018;36:1927-42.
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. RECORD-1 Study Group . Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372:449-56.
- Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. METEOR Investigators . Cabozantinib versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1814-23.
- McDermott DF, Drake CG, Sznol M, Choueiri TK, Powderly JD, Smith DC, et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving Nivolumab. J Clin Oncol 2015;33:2013-20.
- Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24:16-24.
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47.
- Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 2007;25:1753-9.
- van der Veldt AA, Meijerink MR, van den Eertwegh AJ, Haanen JB, Boven E. Choi response criteria for early prediction of clinical outcome in patients with metastatic renal cell cancer treated with sunitinib. Br J Cancer 2010;102:803-9.
- Nathan PD, Vinayan A, Stott D, Juttla J, Goh V. CT response assessment combining reduction in both size and arterial phase density correlates with time to progression in metastatic renal cancer patients treated with targeted therapies. Cancer Biol Ther 2010;9:15-9.
- Thian Y, Gutzeit A, Koh DM, Fisher R, Lote H, Larkin J, et al. Revised Choi imaging criteria correlate with clinical outcomes in patients with metastatic renal cell carcinoma treated with sunitinib. Radiology 2014;273:452-61.
- Smith AD, Lieber ML, Shah SN. Assessing tumor response and detecting recurrence in metastatic renal cell carcinoma on targeted therapy: importance of size and attenuation on contrast-enhanced CT. AJR Am J Roentgenol 2010;194:157-65.
- Smith AD, Shah SN, Rini BI, Lieber ML, Remer EM. Morphology, Attenuation, Size, and Structure (MASS) criteria: assessing response and predicting clinical outcome in metastatic renal cell carcinoma on antiangiogenic targeted therapy. AJR Am J Roentgenol 2010;194:1470-8.
- Krajewski KM, Nishino M, Franchetti Y, Ramaiya NH, Van den Abbeele AD, Choueiri TK. Intraobserver and interobserver variability in computed tomography size and attenuation measurements in patients with renal cell carcinoma receiving antiangiogenic therapy: implications for alternative response criteria. Cancer 2014;120:711-21.
- Jain Y, Liew S, Taylor MB, Bonington SC. Is dual-phase abdominal CT necessary for the optimal detection of metastases from renal cell carcinoma?. Clin Radiol 2011;66:1055-9.
- Goh V, Ganeshan B, Nathan P, Juttla JK, Vinayan A, Miles KA. Assessment of response to tyrosine kinase inhibitors in metastatic renal cell cancer: CT texture as a predictive biomarker. Radiology 2011;261:165-71.
- Haider MA, Vosough A, Khalvati F, Kiss A, Ganeshan B, Bjarnason GA. CT texture analysis: a potential tool for prediction of survival in patients with metastatic clear cell carcinoma treated with sunitinib. Cancer Imaging 2017;17.
- Gillies RJ, Kinahan PE, Hricak H. Radiomics: images are more than pictures, they are data. Radiology 2016;278:563-77.
- Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Carvalho S, et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun 2014;5.
- Collinson FJ, Gregory WM, McCabe C, Howard H, Lowe C, Potrata D, et al. The STAR trial protocol: a randomised multi-stage phase II/III study of Sunitinib comparing temporary cessation with allowing continuation, at the time of maximal radiological response, in the first-line treatment of locally advanced/metastatic renal cancer. BMC Cancer 2012;12.
- Zwanenburg A, Vallieres M, Abdalah MA, Aerts H, Andrearczyk V, Apte A, et al. The image biomarker standardization initiative: standardized quantitative radiomics for high-throughput image-based phenotyping. Radiology 2020;295:328-38.
- Thiam R, Fournier LS, Trinquart L, Medioni J, Chatellier G, Balvay D, et al. Optimizing the size variation threshold for the CT evaluation of response in metastatic renal cell carcinoma treated with sunitinib. Ann Oncol 2010;21:936-41.
- Oudard S, Thiam R, Fournier LS, Medioni J, Lamuraglia M, Scotte F, et al. Optimisation of the tumour response threshold in patients treated with everolimus for metastatic renal cell carcinoma: analysis of response and progression-free survival in the RECORD-1 study. Eur J Cancer 2012;48:1512-8.
- Krajewski KM, Franchetti Y, Nishino M, Fay AP, Ramaiya N, Van den Abbeele AD, et al. 10% Tumor diameter shrinkage on the first follow-up computed tomography predicts clinical outcome in patients with advanced renal cell carcinoma treated with angiogenesis inhibitors: a follow-up validation study. Oncologist 2014;19:507-14.
- Krajewski KM, Guo M, Van den Abbeele AD, Yap J, Ramaiya N, Jagannathan J, et al. Comparison of four early posttherapy imaging changes (EPTIC; RECIST 1.0, tumor shrinkage, computed tomography tumor density, Choi criteria) in assessing outcome to vascular endothelial growth factor-targeted therapy in patients with advanced renal cell carcinoma. Eur Urol 2011;59:856-62.
- Smith AD, Zhang X, Bryan J, Souza F, Roda M, Sirous R, et al. Vascular tumor burden as a new quantitative CT biomarker for predicting metastatic RCC response to antiangiogenic therapy. Radiology 2016;281:484-98.
- Hudson JM, Bailey C, Atri M, Stanisz G, Milot L, Williams R, et al. The prognostic and predictive value of vascular response parameters measured by dynamic contrast-enhanced-CT, -MRI and -US in patients with metastatic renal cell carcinoma receiving sunitinib. Eur Radiol 2018;28:2281-90.
- Matoori S, Thian Y, Koh DM, Sohaib A, Larkin J, Pickering L, et al. Contrast-enhanced CT density predicts response to sunitinib therapy in metastatic renal cell carcinoma patients. Transl Oncol 2017;10:679-85.
- Hahn S. Understanding noninferiority trials. Korean J Pediatr 2012;55:403-7.
- Chibaudel B, Bonnetain F, Shi Q, Buyse M, Tournigand C, Sargent DJ, et al. Alternative end points to evaluate a therapeutic strategy in advanced colorectal cancer: evaluation of progression-free survival, duration of disease control, and time to failure of strategy – an Aide et Recherche en Cancerologie Digestive Group Study. J Clin Oncol 2011;29:4199-204.
- Head SJ, Kaul S, Bogers AJJC, Kappetein AP. Non-inferiority study design: lessons to be learned from cardiovascular trials. Eur Heart J 2012;33:1318-24.
- Motzer RJTN, McDermott DF, Burotto M, Choueiri TK, Hammers HJ, Plimack ER, et al. Conditional Survival and 5-Year Follow-up in CheckMate 214: First-line nivolumab + ipilimumab (N+I) versus sunitinib (S) in Advanced Renal Cell Carcinoma (aRCC). European Society for Medical Oncology; 16 Sep 2021. Paris: European Society for Medical Oncology; 2021.
- MRC Clinical Trials Unit . REFINE: REduced Frequency ImmuNE Checkpoint Inhibition in Cancers: University College London 2021. www.ctu.mrc.ac.uk/studies/all-studies/r/refine/ (accessed 28 June 2021).
- Coen O, Corrie P, Marshall H, Plummer R, Ottensmeier C, Hook J, et al. The DANTE trial protocol: a randomised phase III trial to evaluate the Duration of ANti-PD-1 monoclonal antibody Treatment in patients with metastatic mElanoma. BMC Cancer 2021;21.
Appendix 1 Factors included in the IMDC Risk Score40

Appendix 2 Tissue substudy
The STAR trial planned to collect renal cancer tissue samples from patients receiving TKIs for validation of tissue biomarkers, particularly to develop markers which will predict response to the new generation of treatments which are being developed and toxicity.
Diagnostic pathology samples are routinely taken from all patients with suspected renal cancer, either at the time of nephrectomy or from a diagnostic biopsy and therefore these samples already existed for patients entering the STAR trial. Patients were given the option to consent to this tissue collection when approached regarding trial participation. Out of the 920 patients recruited, 901 patients consented to the collection of their archived tissue.
Tissue samples are collected as formalin-fixed paraffin-embedded tissue blocks by the Lothian NRS Bioresource biobank in Edinburgh. The sample collection process is co-ordinated by the CTRU in Leeds. All tissue collection from participating sites will be completed by 31 December 2021.
The tissue samples will be used to prepare TMAs for use in future research studies. This processing and all subsequent research are performed out with the STAR trial. The STAR Translational Committee will manage the resource, with a peer review process to prioritise science. Histopathology and appropriate anonymised clinical outcome data will be electronically available, and TMA data will be made available online using TMANavigator.org.
Appendix 3 STAR trial schema
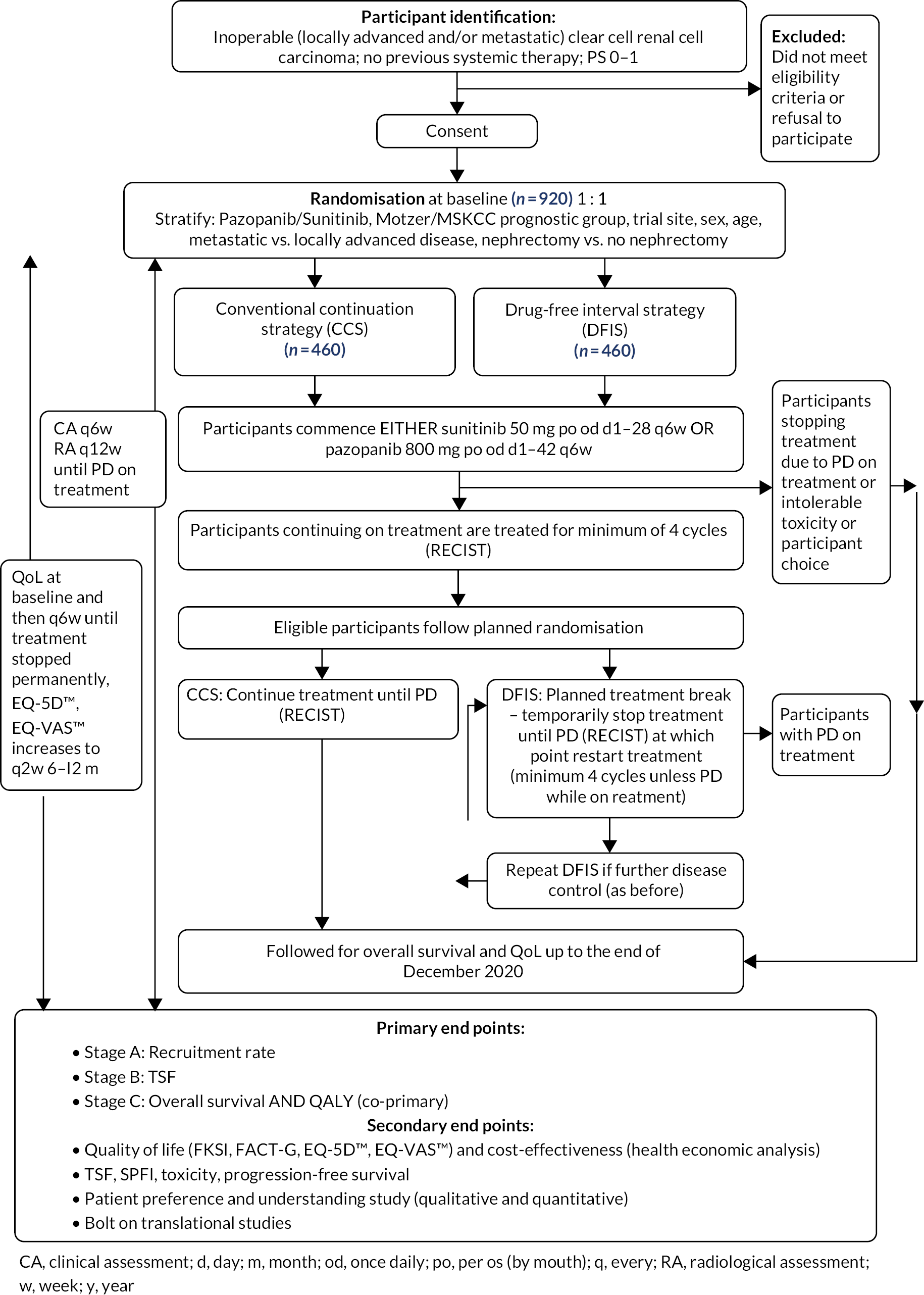
Appendix 4 Summary of interim analysis
Stage A
On average 12.7 sites were open to recruitment during the 12-month formal monitoring period:
-
12.5 sites for 3 months between June and August (inclusive).
-
13.5 sites for 2 months between September and October (inclusive).
-
12.5 sites for 7 months between November and May (inclusive).
The expected total and monthly recruitment rate assuming a recruitment rate of one patient per month is:
-
Total: 12.7 sites × 1 patient × 12 months = 152 patients.
-
Monthly = 152 patients/12 months = 12.7 patients per month.
The 95% CI around this monthly recruitment rate is 0.846–1.155.
Thus, 129 (152 × 0.846) was the minimum total number of patients required to demonstrate the feasibility of recruitment, that is 10.7 patients per month (12.7 × 0.846).
In total, 136 patients were recruited in total within the formal monitoring period, 11.3 patients per month. Therefore, the Stage A end point was reached.
Stage B
Intention-to-treat analysis
All 219 participants were included in the ITT analysis population, 110 CCS and 109 DFIS participants. Of these, 139 participants were receiving treatment with sunitinib (63.5%) and 80 with pazopanib (36.5%). In the ITT population, 77 participants (35.2%) had failed the treatment strategy. A higher proportion of participants in the CCS arm (39.1%) compared to the DFIS arm (31.2%) (see Table 36).
| CCS N (%) |
DFIS N (%) |
Total N (%) |
|
|---|---|---|---|
| Has the participant failed the strategy? | |||
| Yes | 43 (39.1%) | 34 (31.2%) | 77 (35.2%) |
| No | 67 (60.9%) | 75 (68.8%) | 142 (64.8%) |
| Total | 110 (100%) | 109 (100%) | 219 (100%) |
The majority of participants failed the treatment strategy due to disease progression (77.9%), a slightly higher proportion of those participants who failed in the DFIS arm (79.4%) compared to the CCS arm (76.7%) (see Table 37). Three participants failed due to receiving a new systemic anticancer agent, details of which are presented Table 38.
| CCS N (%) |
DFIS N (%) |
Total N (%) |
|
|---|---|---|---|
| Reason for strategy failure | |||
| Death | 8 (18.6%) | 6 (17.6%) | 14 (18.2%) |
| Disease progression | 33 (76.7%) | 27 (79.4%) | 60 (77.9%) |
| Use of a new systemic anticancer agent for RCC | 2 (4.7%) | 1 (2.9%) | 3 (3.9%) |
| Total | 43 (100%) | 34 (100%) | 77 (100%) |
| Obs | Randomisation allocation | Medication name | Was this in relation to another trial? | Trial name | Follow-up time point |
|---|---|---|---|---|---|
| 1 | CCS | Axitinib | N/A | 6 months | |
| 1 | Axitinib 5 mg | N/A | Yearly | ||
| 1 | Axitinib 7 mg | N/A | Yearly | ||
| 2 | CCS | Everolimus | N/A | 6 months | |
| 3 | DFIS | Tasquinimod | Yes | Tasquinimod/ABR215050 | 6 months |
| 3 | Tasquinimod | Yes | Tasquinimod Abrzisoso | Yearly |
Figure 24 presents the Kaplan–Meier curves for the time from randomisation to strategy failure in weeks stratified by randomisation allocation.
FIGURE 24.
Kaplan–Meier curves for the time from randomisation to strategy failure in weeks (ITT population) – interim analysis.
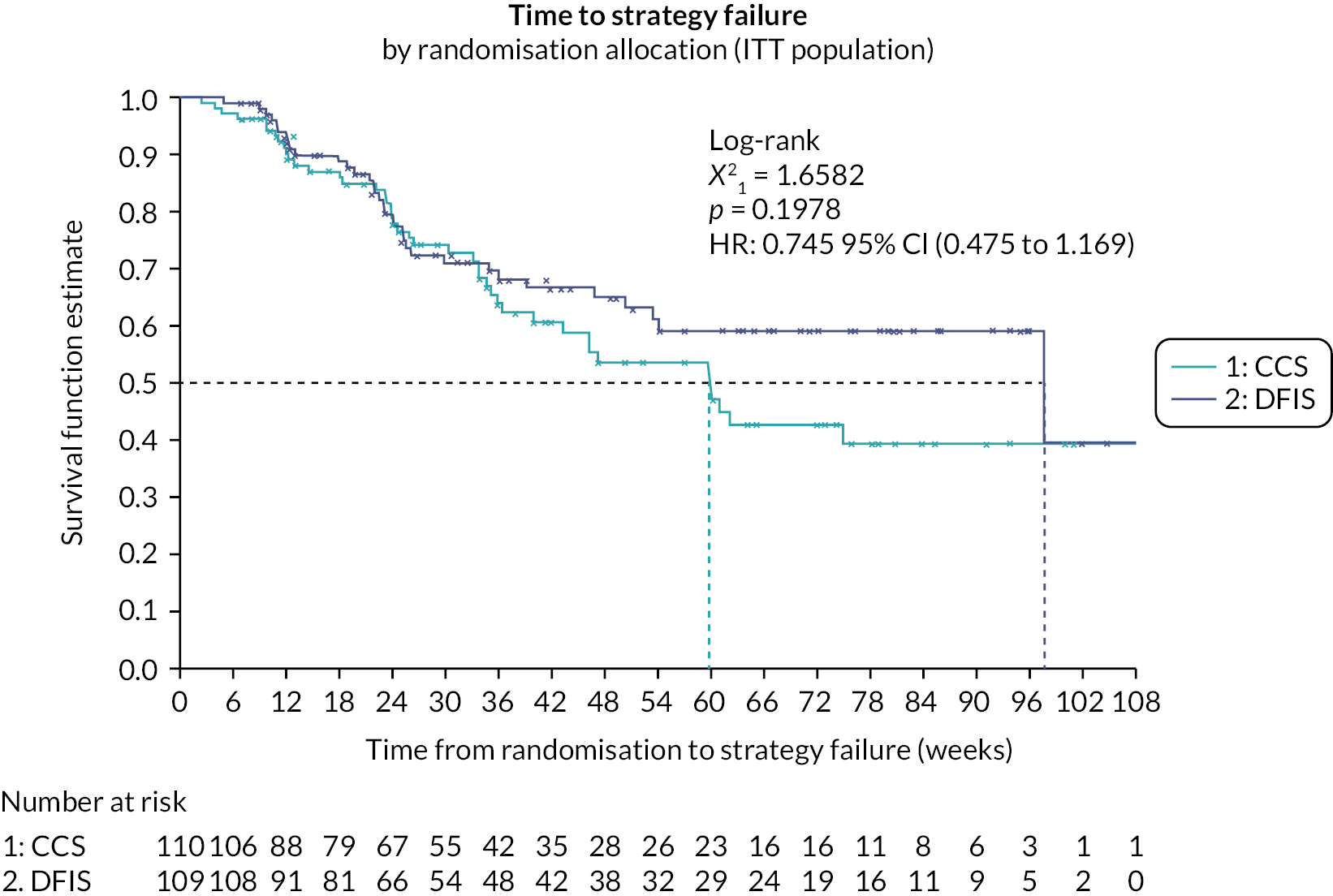
Participants who had not failed their treatment strategy were censored at the time of the analysis. Two participants (one in each strategy arm) withdrew from trial treatment and follow-up and as such were censored at the point of withdrawal. One participant who did not receive any treatment was censored at time zero.
The median TSF was greater in the DFIS arm compared with the CCS arm. The log-rank test was used to compare the differences between the strategy arms (CCS vs. DFIS). The difference between the two strategy arms was non-significant in the ITT population with a p-value 0.1978 at the 5% significance level.
A multivariate Cox’s PH analysis was performed to compare the difference between the two strategy arms after adjusting for the minimisation factors (excluding trial site and disease status as only one participant had locally advanced disease).
The PH assumption was checked for all covariates included in the model. The Kolmogorov-type supremum test for the PH assumption indicated that PH assumption was not violated for any of the covariates and such the Cox PH model was adequate.
For randomisation allocation a HR of 1.37 (0.87, 2.17) was observed. Implying that participants in the DFIS arm are less likely to fail their treatment strategy than participants in the CCS arm, although this difference is non-significant at the 5% level as the 95% CI around the HR contains 1. As the lower bound of this interval is above 0.54, a DFIS is non-inferior to a CCS in terms of TSF in the ITT population.
Per-protocol analysis
The PP population included 84 participants, 44 CCS and 40 DFIS participants. A total of 135 participants were excluded from the PP analysis. In the PP population, 21 participants (25.0%) failed their treatment strategy (see Table 39). A higher proportion of participants in the CCS arm (34.1%) compared to the DFIS arm (15.5%).
| CCS N (%) |
DFIS N (%) |
Total N (%) |
|
|---|---|---|---|
| Has the participant failed the strategy? | |||
| Yes | 15 (34.1%) | 6 (15.0%) | 21 (25.0%) |
| No | 29 (65.9%) | 34 (85.0%) | 63 (75.0%) |
| Total | 44 (100%) | 40 (100%) | 84 (100%) |
The majority of participants failed the treatment strategy due to disease progression (85.7%), a slightly higher proportion of participants who failed in the CCS arm (86.7%) compared to the DFIS arm (83.3%) (see Table 40). One participant failed due to receiving a new systemic anticancer agent (Axitinib) post STAR trial treatment.
| CCS N (%) |
DFIS N (%) |
Total N (%) |
|
|---|---|---|---|
| Reason for strategy failure | |||
| Death | 1 (6.7%) | 1 (16.7%) | 2 (9.5%) |
| Disease progression | 13 (86.7%) | 5 (83.3%) | 18 (85.7%) |
| Use of a new systemic anticancer agent for RCC | 1 (6.7%) | 0 (0.0%) | 1 (4.8%) |
| Total | 15 (100%) | 6 (100%) | 21 (100%) |
Figure 25 presents the Kaplan–Meier curves for the time from randomisation to strategy failure in weeks stratified by randomisation allocation. Participants who had not failed their treatment strategy were censored at the time of the analysis. Median TSF could not be derived as there were insufficient events. The log-rank test was used to compare the differences between the strategy arms (CCS vs. DFIS). The difference between the two strategy arms was significant in the PP population with a p-value 0.0327 at the 5% significance level in favour of the DFIS arm.
FIGURE 25.
Kaplan–Meier curves for the time from randomisation to strategy failure in weeks (PP population) – interim analysis.
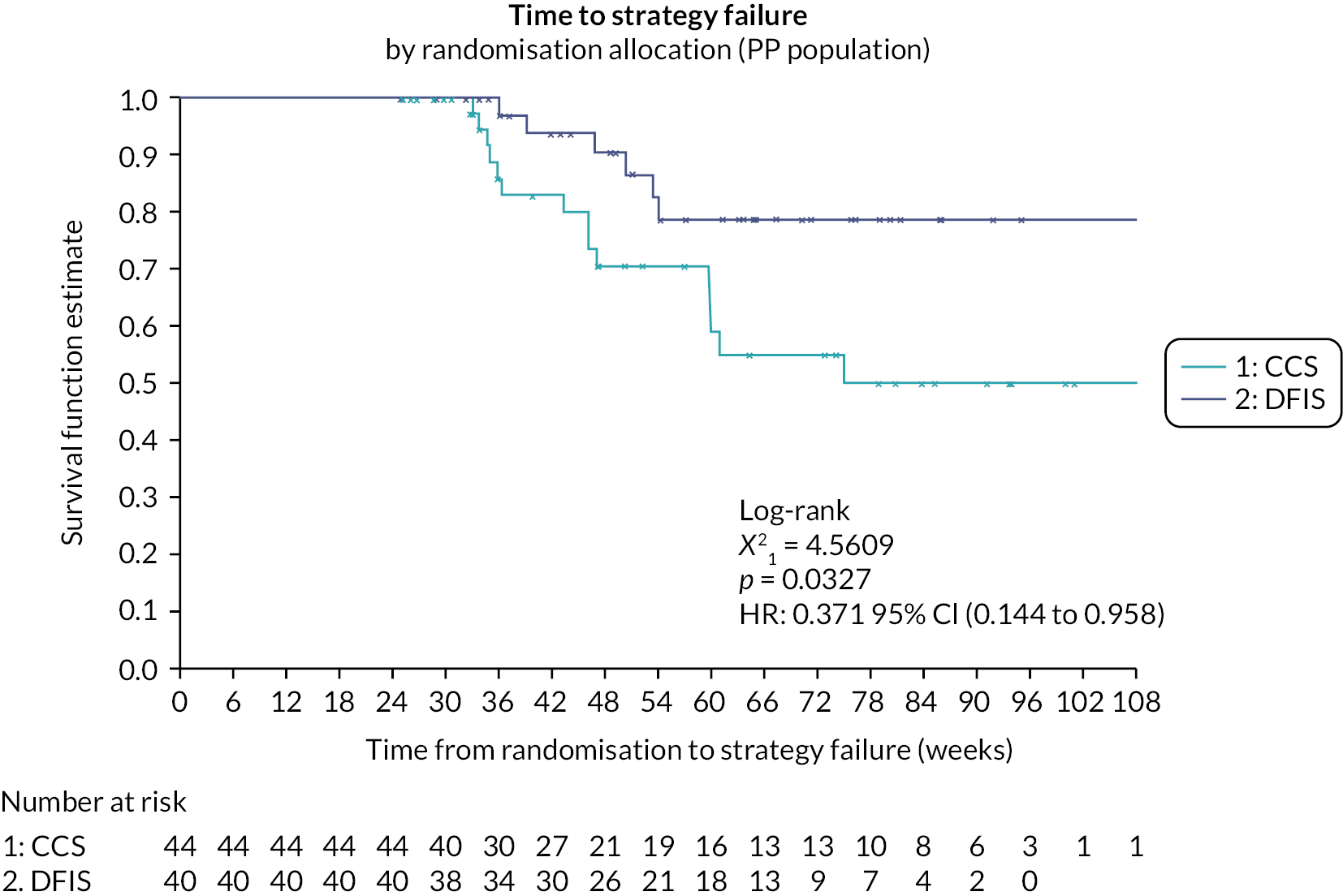
A multivariate Cox’s PH analysis was performed to compare the difference between the two strategy arms after adjusting for the minimisation factors (excluding trial site and disease status as only one participant had locally advanced disease). The PH assumption was checked for all covariates included in the model. The Kolmogorov-type supremum test for the PH assumption indicated that PH assumption was not violated for any of the covariates and such the Cox PH model was adequate.
For randomisation allocation a HR of 4.54 (1.39, 16.67) was observed. Implying that participants in the DFIS arm are less likely to fail their treatment strategy than participants in the CCS arm and this difference is significant at the 5% level with p-value 0.0130. As the lower bound of this interval is more than 0.54, a DFIS is non-inferior to a CCS in terms of TSF in the PP population.
As NI was shown in both the ITT and PP populations the Stage B end point was concluded to be met.
Pooling of sunitinib and pazopanib data
The HRs and their 60% CIs are shown in Table 41. As the HR for pazopanib lies within the CI for sunitinib, it was deemed appropriate to pool the data together.
| TKI | N | Parameter | HR (60% CI) |
|---|---|---|---|
| Sunitinib | 136 | Randomisation allocation: DFIS vs. CCS | 0.768 (0.618 to 0.955) |
| Pazopanib | 80 | Randomisation allocation: DFIS vs. CCS | 0.709 (0.463 to 1.085) |
Utility data
For participants on mean (SD) sunitinib, the EQ5D utility score was 0.73 (0.29) for participants on treatment and 0.81 (0.22) for participants off treatment. Similarly for participants on pazopanib, the on treatment mean (SD) was 0.68 (0.14) and 0.76 (0.17) for off treatment. Comparing these to the original estimates in the simulations 0.57 (0.21) for periods on-treatment and 0.68 (0.19) for periods off-treatment. It was deemed acceptable for the original estimates in the sample size to hold and QALYs to remain a co-primary end point of the trial.
Appendix 5 Health economics supplementary tables
Figure 28 shows the impact on the probability that DFIS is cost-effective if data were MNAR for QALYs or for costs. The probability that DFIS is cost-effective is stable at values close to 1 if the imputed costs and QALY are changed in both trial arms. However, changes in imputed costs and QALYs have an impact on the probability of cost effectiveness if the change is implemented only in patients with missing data randomised to the DFIS group, but probability remains above 50% so long as the reduction in QALYs in the first year does not exceed 20% and the increase in costs in the first year does not exceed 35%.
| Resource item | Unit cost (2020–1 prices) | Source/notes |
|---|---|---|
| Inpatient care | ||
| Oncology (hospital) | ||
| First day (primary analysis) |
£379.58 | National Schedule of NHS Costs Adjusted from 2019 to 2020 prices Inpatient specialist palliative care (SD01A) (cross-checked with Public Health England’s end-of-life care model) |
| First day (sensitivity analysis) |
£698.12 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Malignant, Hepatobiliary or Pancreatic Disorders, without Interventions, with CC Score 3–5 (GC12H) |
| Subsequent days (primary analysis) |
£392.07 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Weighted average of all elective inpatient excess bed-days |
| Subsequent days (sensitivity analysis) |
£313.29 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Malignant, Hepatobiliary or Pancreatic Disorders, without Interventions, with CC Score 3-5 (GC12H) |
| Oncology (hospice) | ||
| First day and subsequent days | £484.18 | Marie Curie Cancer Care Adjusted from 2012 to 2013 prices (cross-checked with Public Health England’s end-of-life care model) |
| General medicine | ||
| First day | £830.91 | National Schedule of NHS Costs Adjusted from 2019 to 2020 prices Weighted average of all day cases |
| Subsequent days | £392.07 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Weighted average of all elective inpatients excess bed-days |
| General surgery | ||
| First day | £830.91 | National Schedule of NHS Costs Adjusted from 2019 to 2020 prices Weighted average of all day cases |
| Subsequent days | £392.07 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Weighted average of all elective inpatients excess bed-days |
| HDU (high dependence unit) | ||
| First day and subsequent days | £1120.64 | National Schedule of NHS Costs Adjusted from 2019 to 2020 prices Adult Critical Care (0 organs supported) XC07Z |
| ICU (intensive care unit) | ||
| First day and subsequent days | £1842.05 | National Schedule of NHS Costs Adjusted from 2019 to 2020 prices Adult Critical Care (2 organs supported) XC05Z |
| Other | ||
| First day | £379.58 | National Schedule of NHS Costs Adjusted from 2019 to 2020 prices Inpatient specialist palliative care (SD01A) |
| Subsequent days | £392.07 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Weighted average of all elective inpatients excess bed-days |
| Outpatient care | ||
| Oncology | £165.95 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Medical oncology (outpatient attendance) Service code: 370 |
| Any medical speciality apart from oncology | £183.45 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices General medicine (outpatient attendance) Service code: 800 |
| Psychology | £158.90 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Clinical Psychology (outpatient attendance) Service code: 656 |
| Physiotherapy | £53.07 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Physiotherapy (outpatient attendance) Service code: 650 |
| Outpatient visit to a hospice | £484.18 | Marie Curie Cancer Care Adjusted from 2012 to 2013 prices (cross-checked with Public Health England’s end-of-life care model) |
| Other | £165.95 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Outpatient attendance (average) |
| Assessments | ||
| CT scan | £102.94 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices CT scan of one area, without contrast (RD20A) |
| MRI scan | £157.56 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Magnetic resonance imaging scan of one area, without contrast (RD01A) |
| X-ray | £75.24 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Dexa scan (RD50Z) |
| Ultrasound | £57.39 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Ultrasound scan with duration of less than 20 minutes, without contrast (RD40Z) |
| Bone scan | £264.66 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Nuclear bone scan of two or three phases (RN15A) |
| Other | £94.31 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Weighted average of all diagnostic imaging |
| Primary and Community Care | ||
| GP | ||
| Face to face in surgery | £47.70 | PSSRU Unit Costs of Health and Social Care Adjusted from 2015 to 2016 prices Per GP/patient contact lasting 11.7 minutes |
| Face to face in clinic | £70.46 | PSSRU Unit Costs of Health and Social Care Adjusted from 2015 to 2016 prices Per GP/patient contact lasting 17.2 minutes |
| Face to face at home | £48.78 | PSSRU Unit Costs of Health and Social Care Adjusted from 2015 to 2016 prices Per GP/patient contact lasting 11.7 minutes |
| E-mail or telephone contact | £29.27 | PSSRU Unit Costs of Health and Social Care Adjusted from 2015 to 2016 prices Per GP/telephone consultation lasting 7.1 minutes |
| Practice nurse | £6.71 | PSSRU Unit Costs of Health and Social Care Adjusted from 2015 to 2016 prices Band 5 nurse (£39.03 per hour) Assumed 17.2-minute appointment |
| District nurse | £41.17 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices District nurse, adult, face to face |
| Macmillan/palliative care nurse | £99.54 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Specialist nursing, palliative/respite care, adult, face to face |
| Physiotherapist | £53.05 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Physiotherapist, adult, one to one |
| Occupational therapist | £85.14 | National Schedule of NHS Costs Adjusted from 2015 to 2016 prices Occupational therapist, adult, one to one |
| Social worker | £10.63 | PSSRU Unit Costs of Health and Social Care Adjusted from 2015 to 2016 prices Social worker (£57 per hour) Assumed 17.2-minute appointment |
| Drug/item | Pack | Price | Source/notes |
|---|---|---|---|
| Pazopanib | |||
| Pazopanib 200 mg |
30 tablets | £560.50 | BNF list prices (NHS indicative price – hospital only) (November 2020) Not listed on Emit |
| Pazopanib 400 mg |
30 tablets | £1121.00 | |
| Cost per 6-week cycle per mg | £4.20 | This assumes that pazopanib is taken daily during each 6-week cycle. It also assumes that three 30-tablet packs can be used without wastage across two 6-week cycles (~90 days). This figure is multiplied by the mg dosage reported in the trial data. It equates to a cost of £3363.00 per 6-week cycle for a (typical) 800 mg daily dose. |
|
| Sunitinib | |||
| Sunitinib 12.5 mg |
28 capsules | £784.70 | BNF list prices (NHS indicative price – hospital only) (November 2020) Not listed on Emit |
| Sunitinib 25 mg |
28 capsules | £1569.40 | |
| Sunitinib 50 mg |
28 capsules | £3138.80 | |
| Cost per 6-week cycle per mg | £62.78 | This assumes that, in each 6-week cycle, sunitinib is taken daily for 4 weeks followed by a 2-week break. This figure is multiplied by the mg dosage reported in the trial data. It equates to a cost of £3318.80 per 6-week cycle for a (typical) 50 mg daily dose. |
|
| Dispensing cost | |||
| Per prescription | £16.33 | £14.74 in 2014 prices reported in: https://doi.org/10.1371/journal.pone.0175920 | |
FIGURE 26.
Number of participants with complete resource use data at each data collection point (weeks 6–40). Note: In this figure, participants are defined as having complete resource use data if they have responded to the relevant question on the resource use questionnaire or if they have died (where resource use is zero). Missing resource use data occur when participants do not complete the relevant question on the resource use questionnaire.
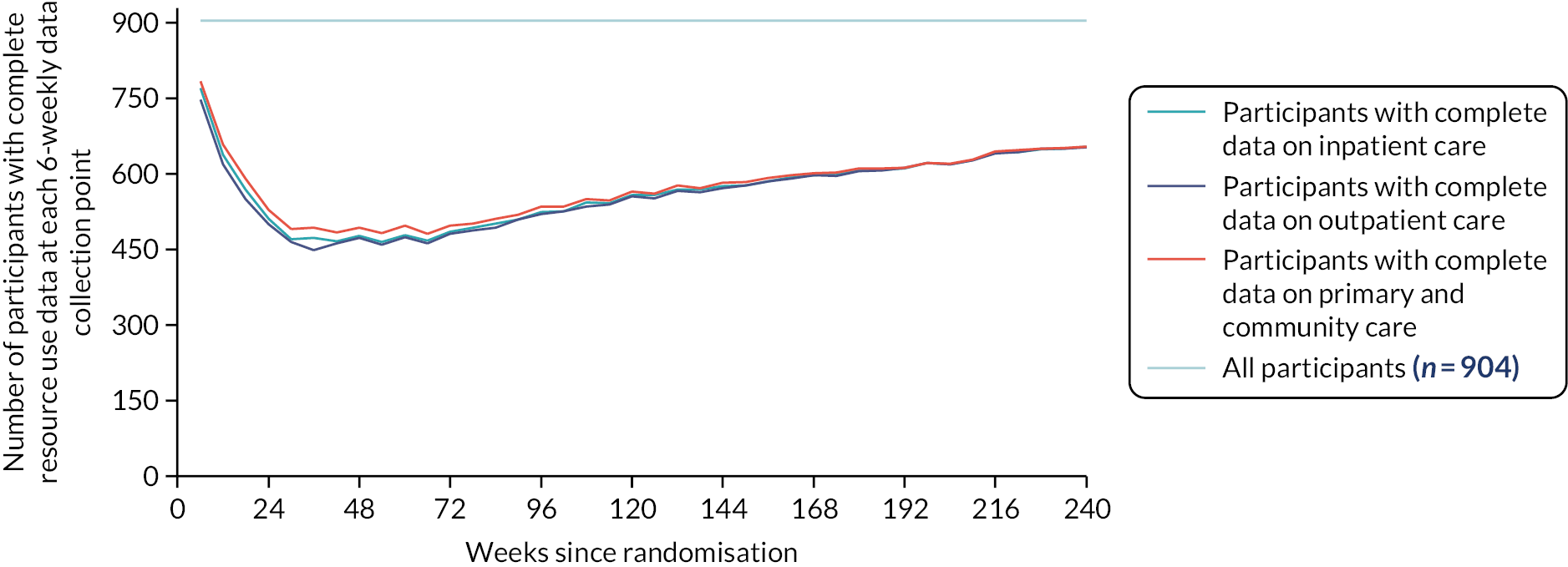
| Q1 Inpatient care | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total (n = 904; N = 16,272) | CCS (n = 453; N = 8154) | DFIS (n = 451; N = 8118) | ||||||||||
| Yes | No | Due to death (if no) | Missing | Yes | No | Due to death (if no) | Missing | Yes | No | Due to death (if no) | Missing | |
| Q2 Outpatient care | ||||||||||||
| Total (n = 904; N = 16,272) | CCS (n = 453; N = 8154) | DFIS (n = 451; N = 8118) | ||||||||||
| Yes | No | Due to death (if no) | Missing | Yes | No | Due to death (if no) | Missing | Yes | No | Due to death (if no) | Missing | |
| Q3 Primary and community care | ||||||||||||
| Total (n = 904; N = 16,272) | CCS (n = 453; N = 8154) | DFIS (n = 451; N = 8118) | ||||||||||
| Yes | No | Due to death (if no) | Missing | Yes | No | Due to death (if no) | Missing | Yes | No | Due to death (if no) | Missing | |
| Total | 257 (1.58%) | 8887 (54.61%) | 4009 (24.64%) | 7128 (43.81%) | 124 (1.52%) | 4198 (51.48%) | 2019 (24.76%) | 3832 (47.00%) | 133 (1.64%) | 4689 (57.76%) | 1990 (24.51%) | 3296 (40.60%) |
| Oncology (hospital) | 103 (0.63%) | 9036 (55.53%) | 7133 (43.84%) | 52 (0.64%) | 4268 (52.34%) | 3834 (47.02%) | 51 (0.63%) | 4768 (58.73%) | 3299 (40.64%) | |||
| Oncology (hospice) | 6 (0.04%) | 9130 (56.11%) | 7136 (43.85%) | 3 (0.04%) | 4316 (52.93%) | 3835 (47.03%) | 3 (0.04%) | 4814 (59.30%) | 3301 (40.66%) | |||
| General medicine | 61 (0.38%) | 9075 (55.77%) | 7136 (43.85%) | 25 (0.31%) | 4294 (52.66%) | 3835 (47.03%) | 35 (0.43%) | 4781 (58.89%) | 3301 (40.66%) | |||
| General surgery | 47 (0.29%) | 9088 (55.85%) | 7137 (43.86%) | 23 (0.28%) | 4296 (52.69%) | 3835 (47.03%) | 24 (0.30%) | 4792 (59.03%) | 3302 (40.68%) | |||
| HDU | 8 (0.05%) | 9128 (56.10%) | 7136 (43.85%) | 5 (0.06%) | 4315 (52.92%) | 3834 (47.02%) | 3 (0.04%) | 4813 (59.29%) | 3302 (40.68%) | |||
| ICU | 5 (0.03%) | 9131 (56.11%) | 7136 (43.85%) | 2 (0.02%) | 4317 (52.94%) | 3835 (47.03%) | 3 (0.04%) | 4814 (59.30%) | 3301 (40.66%) | |||
| Other | 59 (0.36%) | 9077 (55.78%) | 7136 (43.85%) | 27 (0.33%) | 4293 (52.65%) | 3834 (47.02%) | 32 (0.39%) | 4784 (58.93%) | 3302 (40.68%) | |||
| Total | 1116 (6.86%) | 7894 (48.51%) | 4009 (24.64%) | 7262 (44.63%) | 470 (5.76%) | 3784 (46.41%) | 2019 (24.76%) | 3900 (47.83%) | 646 (7.96%) | 4110 (50.62%) | 1990 (24.51%) | 3362 (41.41%) |
| Oncology | 651 (4.00%) | 8322 (51.14%) | 7299 (44.86%) | 262 (3.21%) | 3976 (48.76%) | 3916 (48.03%) | 389 (4.79%) | 4346 (53.54%) | 3383 (41.67%) | |||
| Other speciality | 147 (0.90%) | 8809 (54.14%) | 7316 (44.96%) | 55 (0.67%) | 4174 (51.19%) | 3925 (48.14%) | 92 (1.13%) | 4635 (57.10%) | 3391 (41.77%) | |||
| Psychology | 13 (0.08%) | 8937 (54.92%) | 7322 (45.00%) | 4 (0.05%) | 4220 (51.75%) | 3930 (48.20%) | 9 (0.11%) | 4717 (58.11%) | 3392 (41.78%) | |||
| Physio-therapy | 18 (0.11%) | 8932 (54.89%) | 7322 (45.00%) | 7 (0.09%) | 4220 (51.75%) | 3927 (48.16%) | 11 (0.14%) | 4712 (58.04%) | 3395 (41.82%) | |||
| Outpatient hospice visit | 29 (0.18%) | 8923 (54.84%) | 7320 (44.99%) | 5 (0.06%) | 4220 (51.75%) | 3929 (48.18%) | 24 (0.30%) | 4703 (57.93%) | 3391 (41.77%) | |||
| Other | 324 (1.98%) | 8646 (53.13%) | 7302 (44.87%) | 148 (1.82%) | 4086 (50.11%) | 3920 (48.07%) | 176 (2.17%) | 4560 (56.17%) | 3382 (41.66%) | |||
| Total | 2621 (16.11%) | 6778 (41.65%) | 4009 (24.64%) | 6873 (42.24%) | 1149 (14.09%) | 3297 (40.43%) | 2019 (24.76%) | 3708 (45.47%) | 1472 (18.13%) | 3481 (42.88%) | 1990 (24.51%) | 3165 (38.99%) |
| Yes | Mean number (if yes) | No | Missing | Yes | Mean number (if yes) | No | Missing | Yes | Mean number (if yes) | No | Missing | |
| GP | 1570 (9.65%) | 1.45 | 7828 (48.11%) | 6874 (42.24%) | 697 (8.55%) | 1.43 | 3748 (23.03%) | 3709 (22.79%) | 873 (10.75%) | 1.46 | 4080 (25.07%) | 3165 (19.45%) |
| Practice nurse | 1177 (7.23%) | 1.85 | 8222 (50.53%) | 6873 (42.24%) | 518 (6.35%) | 1.83 | 3928 (24.14%) | 3708 (22.79%) | 659 (8.12%) | 1.87 | 4294 (26.39%) | 3165 (19.45%) |
| District nurse | 258 (1.59%) | 3.13 | 9140 (56.17%) | 6874 (42.24%) | 130 (1.59%) | 3.59 | 4315 (26.52%) | 3709 (22.79%) | 128 (1.58%) | 2.67 | 4825 (29.65%) | 3165 (19.45%) |
| Macmillan/PC nurse | 172 (1.06%) | 1.8 | 9226 (56.70%) | 6874 (42.24%) | 84 (1.03%) | 1.55 | 4361 (26.80%) | 3709 (22.79%) | 88 (1.08%) | 2.03 | 4865 (29.90%) | 3165 (19.45%) |
| Physiotherapist | 95 (0.58%) | 2 | 9303 (57.17%) | 6874 (42.24%) | 36 (0.44%) | 1.78 | 4409 (27.10%) | 3709 (22.79%) | 59 (0.73%) | 2.14 | 4894 (30.08%) | 3165 (19.45%) |
| Occupational therapist | 28 (0.17%) | 1.54 | 9370 (57.58%) | 6874 (42.24%) | 9 (0.11%) | 1.33 | 4436 (27.26%) | 3709 (22.79%) | 19 (0.23%) | 1.63 | 4934 (30.32%) | 3165 (19.45%) |
| Social worker | 8 (0.05%) | 1.25 | 9390 (57.71%) | 6874 (42.24%) | 3 (0.04%) | 1.33 | 4442 (27.30%) | 3709 (22.79%) | 5 (0.06%) | 1.2 | 4948 (30.41%) | 3165 (19.45%) |
| Complete cases with 6-week extrapolation (n = 162) | Complete cases with 12-week extrapolation (n = 233) | Alternative inpatient care costs (n = 904) | Inclusion of subsequent treatment costs (n = 904) | |||||
|---|---|---|---|---|---|---|---|---|
| CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | |
| Sample size | 56 | 106 | 56 | 106 | 453 | 451 | 453 | 451 |
| Mean values at 2 years | ||||||||
| QALYs | 1.259 | 1.471 | 1.259 | 1.471 | 0.958 | 1.008 | 0.958 | 1.008 |
| Total costs (£) | 44,572.94 | 29,970.19 | 38,251.25 | 28,234.53 | 27,231.59 | 24,251.06 | 41,907.48 | 36,681.00 |
| Treatment costs (£) | 40,815.3 | 25,804.42 | 34,818.79 | 24,011.97 | 19,623.94 | 16,331.65 | 19,623.94 | 16,331.65 |
| Inpatient care costs (£) (Q1) | 1529.74 | 1473.26 | 1497.31 | 1769.58 | 2136.72 | 1867.11 | 2193.64 | 1789.35 |
| Outpatient care costs (£) (Q2) | 587.46 | 703.86 | 517.42 | 627.35 | 573.02 | 688.66 | 572.24 | 680.16 |
| Radiology unit costs (£) (Q2A) | 975.21 | 1344.32 | 835.09 | 1164.08 | 560.15 | 720.13 | 560.15 | 720.13 |
| Primary and community care costs (£) (Q3) | 548.26 | 546.47 | 462.14 | 544.77 | 512.10 | 562.27 | 512.10 | 562.27 |
| Other medication costs (£) | 116.94 | 97.84 | 120.47 | 116.75 | 106.60 | 121.60 | 106.60 | 121.60 |
| On-study review costs (£) | Excluded | Excluded | 3719.02 | 3959.62 | 2022.96 | 2154.32 | ||
| Non-STAR anticancer medications (£) | n/a | n/a | n/a | n/a | n/a | n/a | 16,315.85 | 14,321.52 |
| Incremental QALY | 0.212 | 0.191 | 0.049 (−0.031 to 0.132) | 0.049 (−0.031 to 0.132) | ||||
| Incremental costs | −14,602.75 | −10,016.72 | −2980.53 (−5352.35 to −608.71) | −5226.48 (−8675.55 to −1777.42) | ||||
| ICER (unadjusted) | −68,756.99 | −52,415.34 | −59,832.43 | −10,4918.59 | ||||
| ICER (adjusted in SUR) | −73,721.59 | −53,290.68 | −57,886.99 | −99,962.251 | ||||
FIGURE 27.
Cost-effectiveness acceptability curve for the primary analysis.
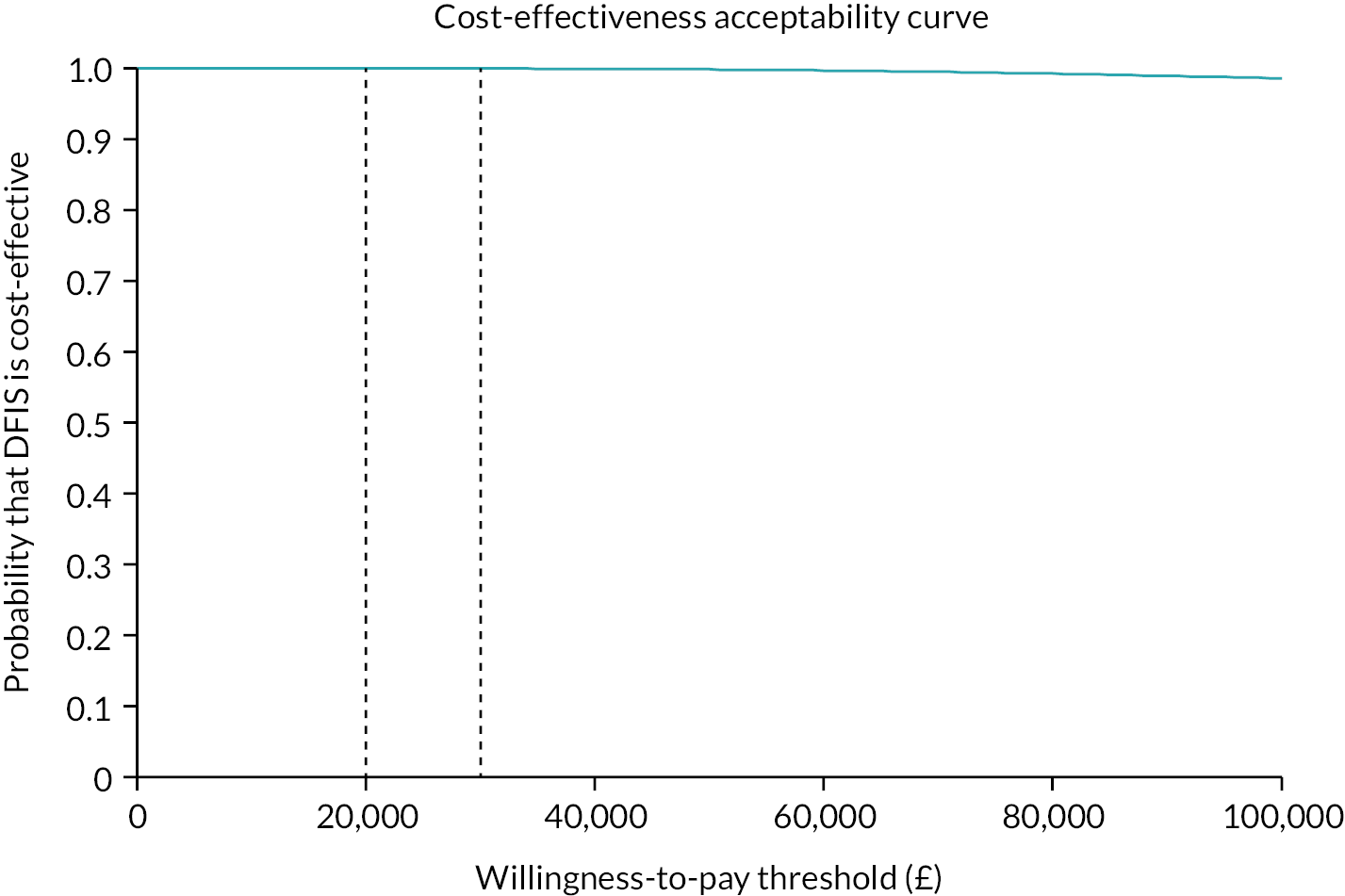
FIGURE 28.
Sensitivity analyses: MNAR scenarios.

FIGURE 29.
Decision-analytic model.
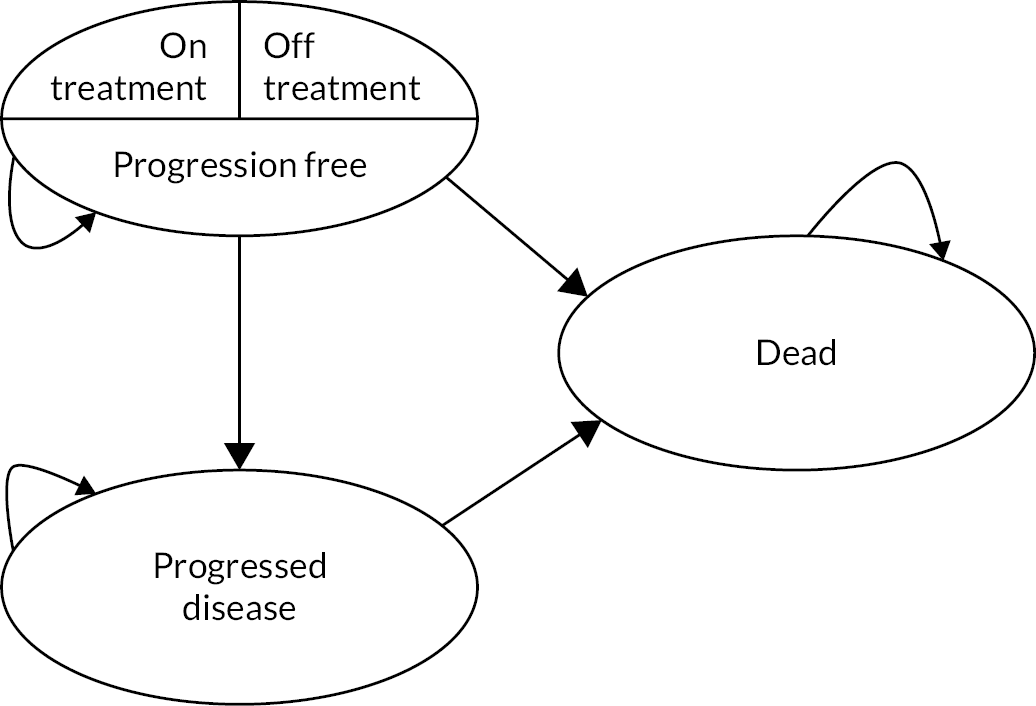
FIGURE 30.
Kaplan–Meier and parametric survival curve comparisons. (continued)

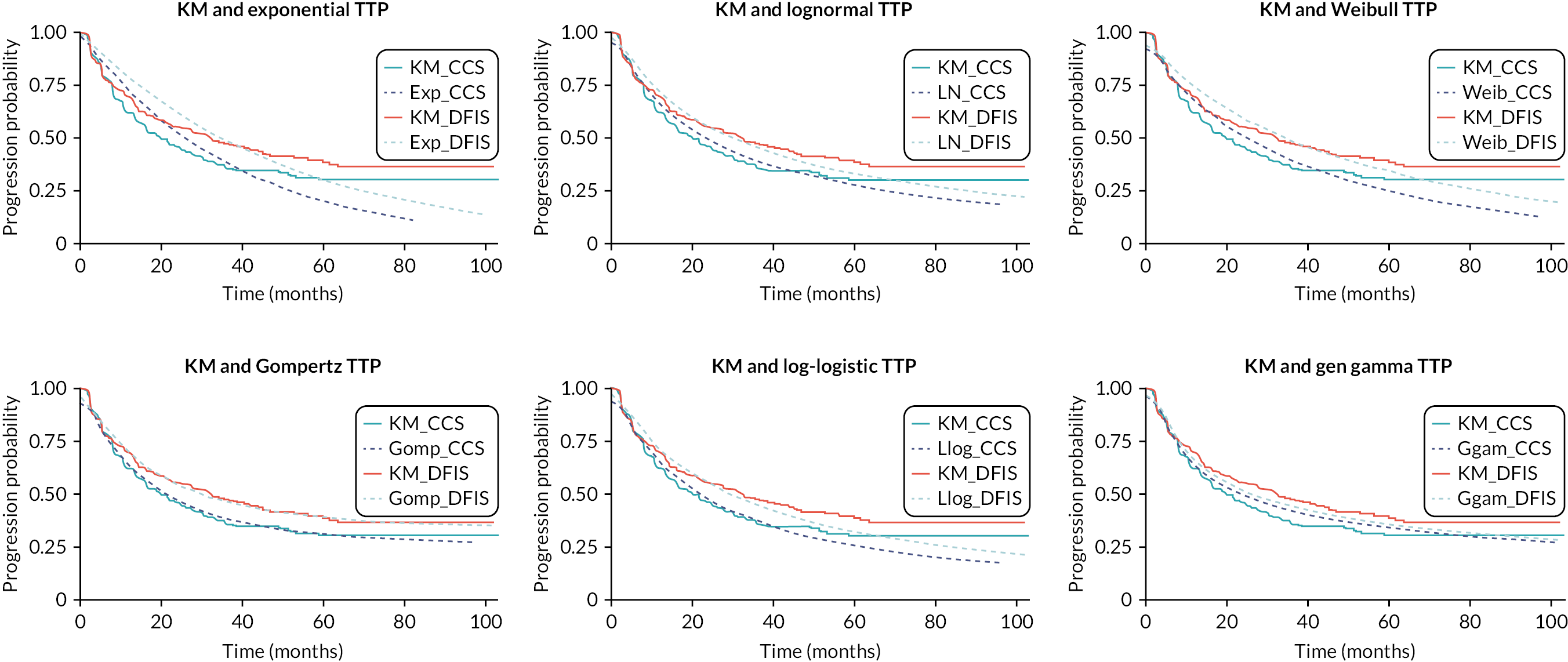

| Parameter | Arm | Value | PSA distribution | Parameter 1 | Parameter 2 |
|---|---|---|---|---|---|
| Lambda | Both | £20,000.00 | N/A | ||
| Cohort age | Both | N/A | |||
| Discount rate QALYs | Both | £0.04 | N/A | ||
| Discount rate costs | Both | £0.04 | N/A | ||
| % on treatment (Sun/Paz) | |||||
| Year 1 | CCS | 0.981 | N/A | ||
| Year 2 | CCS | 0.304 | N/A | ||
| Year 3 | CCS | 0.126 | N/A | ||
| Year 4 | CCS | 0.043 | N/A | ||
| Year 5 | CCS | 0.022 | N/A | ||
| Year 6 | CCS | 0.011 | N/A | ||
| Year 7 | CCS | 0.002 | N/A | ||
| Year 8 | CCS | 0.000 | N/A | ||
| Year 1 | DFIS | 0.989 | N/A | ||
| Year 2 | DFIS | 0.362 | N/A | ||
| Year 3 | DFIS | 0.192 | N/A | ||
| Year 4 | DFIS | 0.111 | N/A | ||
| Year 5 | DFIS | 0.041 | N/A | ||
| Year 6 | DFIS | 0.022 | N/A | ||
| Year 7 | DFIS | 0.004 | N/A | ||
| Year 8 | DFIS | 0.002 | N/A | ||
| Prob G3/4 AE | Both | 0.152 | Beta | α | β |
| 671 | 4403 | ||||
| Survival estimation | |||||
| PFS – Meanlog | CCS | 3.144 | Lognormal | ||
| PFS – SDlog | CCS | 1.594 | Lognormal | ||
| PPS – Meanlog | CCS | 2.523 | Lognormal | ||
| PPS – SDlog | CCS | 1.438 | Lognormal | ||
| OS – Meanlog | CCS | 3.312 | Lognormal | ||
| OS – SDlog | CCS | 1.217 | Lognormal | ||
| PFS – Meanlog | DFIS | 3.396 | Lognormal | ||
| PFS – SDlog | DFIS | 1.594 | Lognormal | ||
| PPS – Meanlog | DFIS | 2.091 | Lognormal | ||
| PPS – SDlog | DFIS | 1.438 | Lognormal | ||
| OS – Meanlog | DFIS | 3.309 | Lognormal | ||
| OS – SDlog | DFIS | 1.217 | Lognormal | ||
| Costs-resource use (per month) | μ | σ | |||
| Progression free | Both | £223.15 | Lognormal | 5.81 | 0.04 |
| Progressed disease | Both | £256.11 | Lognormal | 3.58 | 0.80 |
| Death | Both | 0 | Fixed | ||
| On treatment (Sun/Paz per cycle) | Both | £3250.90 | Fixed | ||
| μ | σ | ||||
| Cycles per year | CCS | 4.435 | Lognormal | 1.36 | 0.50 |
| DFIS | 3.603 | Lognormal | 1.18 | 0.44 | |
| Cost of grade 3–4 AE | Both | £73.99 | Lognormal | 4.65 | 0.33 |
| Utility | α | β | |||
| Constant (progression free, off treat) | Both | 0.806 | Beta | 693.83 | 167.16 |
| Progressed disease (decrement) | Both | −0.123 | Gamma | 3288.75 | 0.00 |
| On treatment (decrement) | Both | −0.035 | Gamma | 7703.23 | 0.00 |
| Model | Obs | ll (null) | ll (model) | df | AIC | BIC |
|---|---|---|---|---|---|---|
| OS | ||||||
| exp | 920 | −1325.386 | −1325.354 | 2 | 2654.707 | 2664.356 |
| lognormal | 920 | −1308.216 | −1308.216 | 3 | 2622.431 | 2636.905 |
| weib | 920 | −1323.97 | −1323.939 | 3 | 2653.877 | 2668.351 |
| gomp | 920 | −1324.04 | −1324.006 | 3 | 2654.012 | 2668.485 |
| llog | 920 | −1305.847 | −1305.845 | 3 | 2617.69 | 2632.163 |
| ggamma | 920 | −1306.049 | −1306.044 | 4 | 2620.089 | 2639.386 |
| Time to progression | ||||||
| exp | 920 | −1288.18 | −1282.879 | 2 | 2569.758 | 2579.407 |
| lognormal | 920 | −1215.968 | −1213.625 | 3 | 2433.25 | 2447.723 |
| weib | 920 | −1265.605 | −1261.515 | 3 | 2529.029 | 2543.502 |
| gomp | 920 | −1221.383 | −1218.413 | 3 | 2442.826 | 2457.299 |
| llog | 920 | −1232.876 | −1229.896 | 3 | 2465.793 | 2480.266 |
| ggamma | 920 | −1188.255 | −1187.705 | 4 | 2383.411 | 2402.708 |
| Post-progression survival | ||||||
| exp | 492 | −833.7808 | −825.6305 | 2 | 1655.261 | 1663.658 |
| lognormal | 492 | −804.4259 | −799.2094 | 3 | 1604.419 | 1617.014 |
| weib | 492 | −821.1549 | −814.5189 | 3 | 1635.038 | 1647.633 |
| gomp | 492 | −808.8813 | −803.1003 | 3 | 1612.201 | 1624.796 |
| llog | 492 | −806.4837 | −800.7954 | 3 | 1607.591 | 1620.186 |
| ggamma | 492 | −804.3266 | −798.8804 | 4 | 1605.761 | 1622.555 |
| Year | Kaplan–Meier | Exponential | Lognormal | Weibull | Gompertz | Log-Logistic | Gen Gamma | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | CCS | DFIS | |
| OS | ||||||||||||||
| 2 | 0.548 | 0.539 | 0.577 | 0.571 | 0.544 | 0.543 | 0.587 | 0.581 | 0.565 | 0.558 | 0.551 | 0.549 | 0.555 | 0.552 |
| 5 | 0.252 | 0.235 | 0.253 | 0.246 | 0.260 | 0.259 | 0.246 | 0.240 | 0.259 | 0.252 | 0.249 | 0.248 | 0.253 | 0.251 |
| 10 | N/A | N/A | 0.064 | 0.061 | 0.113 | 0.112 | 0.054 | 0.051 | 0.086 | 0.082 | 0.110 | 0.109 | 0.096 | 0.094 |
| 20 | N/A | N/A | 0.004 | 0.004 | 0.037 | 0.037 | 0.002 | 0.002 | 0.016 | 0.015 | 0.044 | 0.043 | 0.024 | 0.023 |
| Time to progression | ||||||||||||||
| 2 | 0.459 | 0.555 | 0.527 | 0.620 | 0.491 | 0.554 | 0.511 | 0.596 | 0.470 | 0.545 | 0.477 | 0.554 | 0.495 | 0.517 |
| 5 | 0.303 | 0.384 | 0.202 | 0.303 | 0.275 | 0.331 | 0.249 | 0.342 | 0.309 | 0.389 | 0.260 | 0.323 | 0.340 | 0.357 |
| 10 | N/A | N/A | 0.041 | 0.092 | 0.151 | 0.191 | 0.090 | 0.156 | 0.266 | 0.345 | 0.145 | 0.188 | 0.252 | 0.265 |
| 20 | N/A | N/A | 0.002 | 0.008 | 0.071 | 0.096 | 0.015 | 0.040 | 0.260 | 0.339 | 0.076 | 0.101 | 0.186 | 0.195 |
| Post-progression survival | ||||||||||||||
| 2 | 0.350 | 0.22 | 0.359 | 0.216 | 0.324 | 0.225 | 0.354 | 0.223 | 0.325 | 0.206 | 0.318 | 0.214 | 0.326 | 0.221 |
| 5 | 0.150 | 0.08 | 0.077 | 0.022 | 0.137 | 0.082 | 0.107 | 0.040 | 0.151 | 0.070 | 0.135 | 0.083 | 0.132 | 0.075 |
| 10 | N/A | N/A | 0.006 | 0.000 | 0.058 | 0.030 | 0.019 | 0.003 | 0.105 | 0.042 | 0.064 | 0.038 | 0.051 | 0.025 |
| 20 | N/A | N/A | 0.000 | 0.000 | 0.020 | 0.009 | 0.001 | 0.000 | 0.096 | 0.037 | 0.029 | 0.017 | 0.015 | 0.006 |
FIGURE 31.
Cost-effectiveness plane (lifetime horizon).
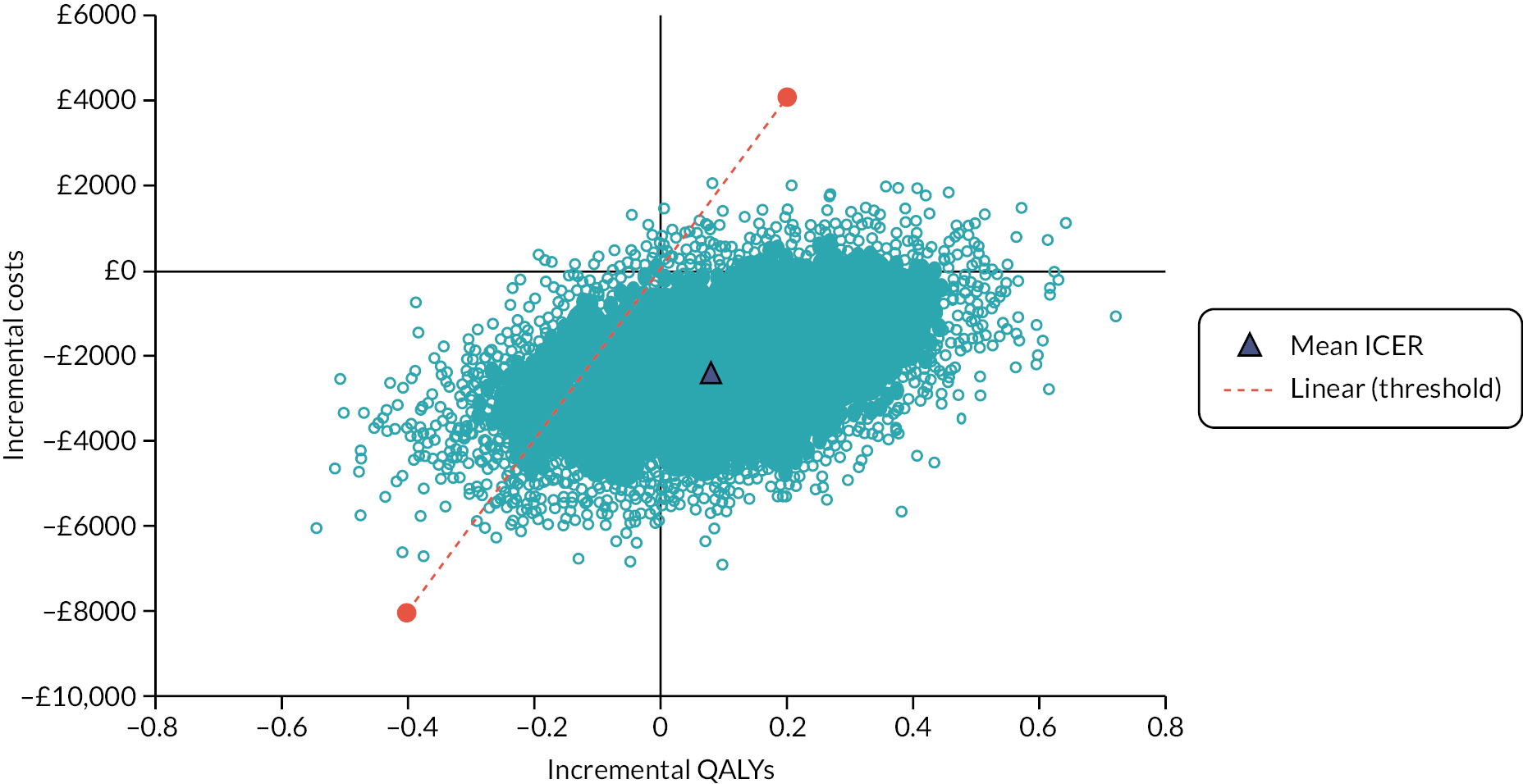
FIGURE 32.
Cost-effectiveness acceptability curve (lifetime horizon).
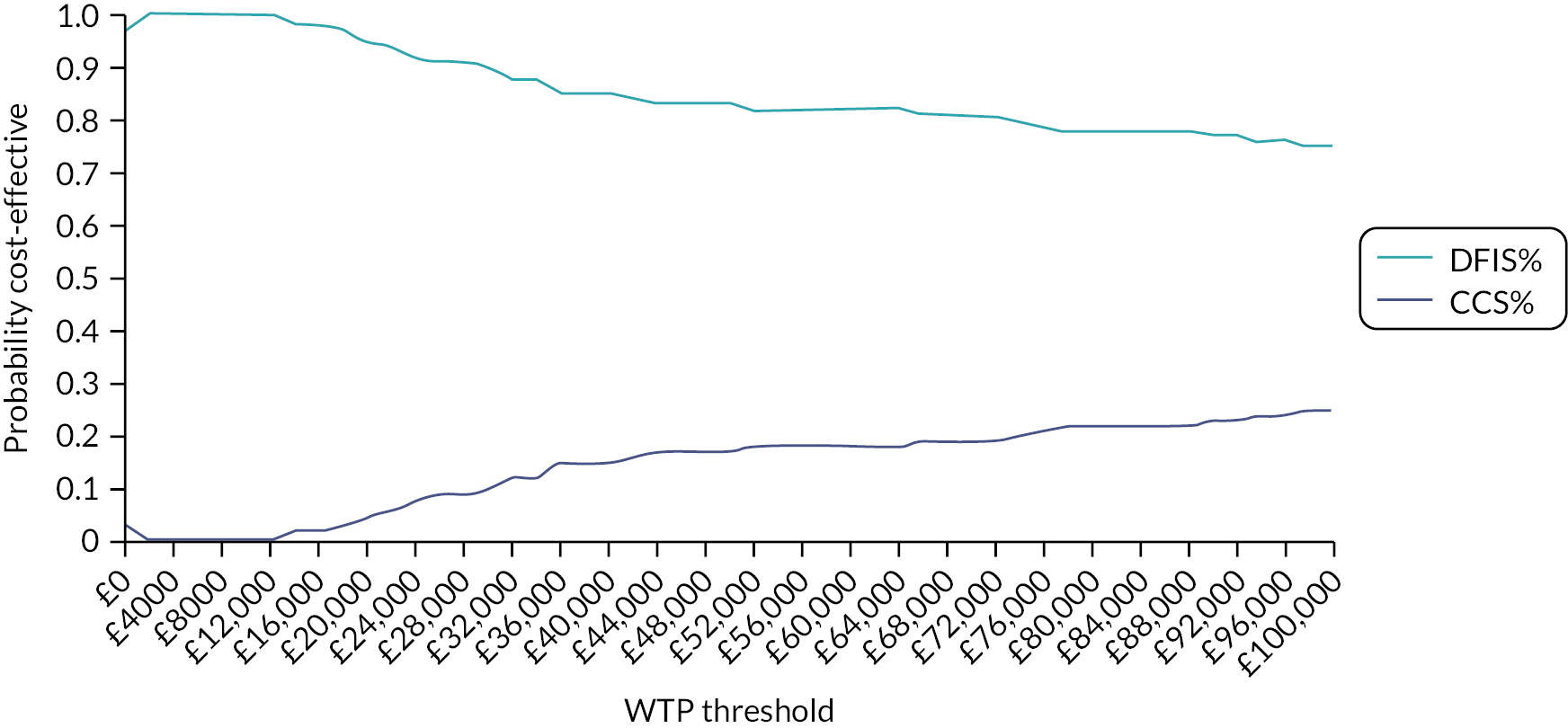
FIGURE 33.
Net benefit distributions.
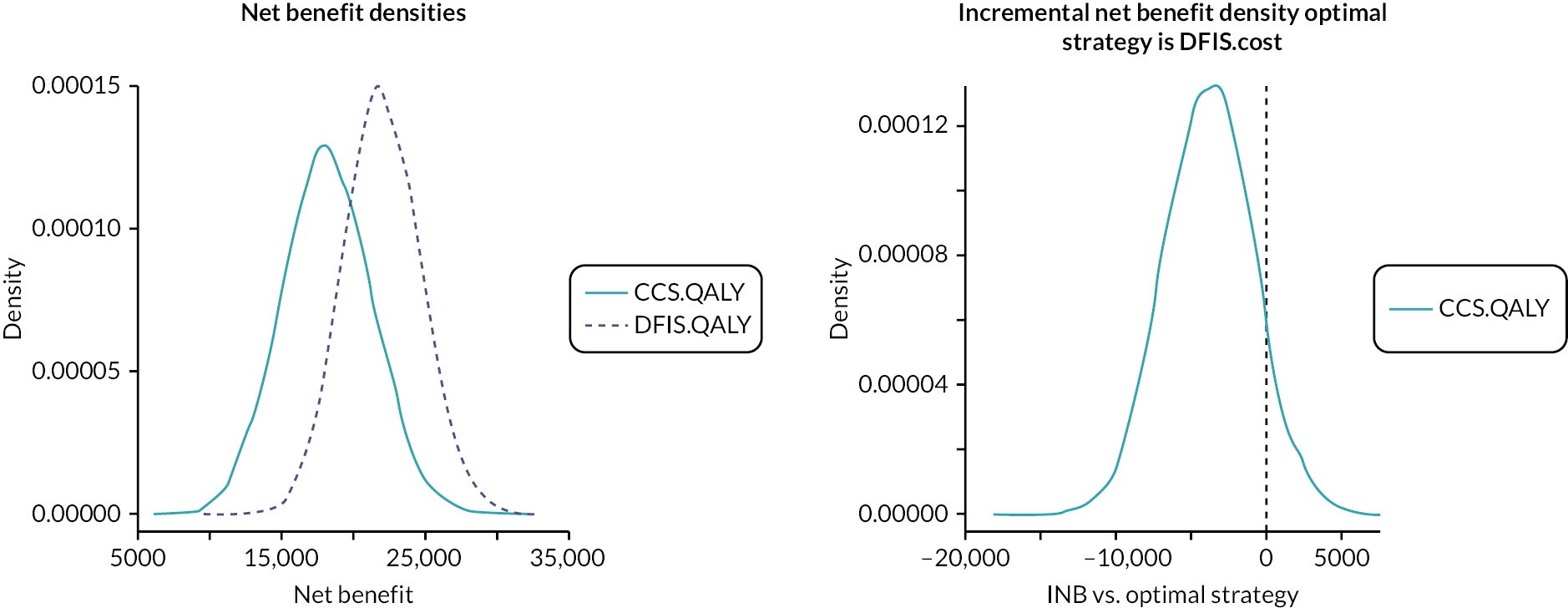
FIGURE 34.
Expected value of partial perfect information.

Appendix 6 Qualitative topic guides
Interview topic guide for study: patients’ preference in the STAR study
The interview will start with a brief introduction to the project and the aims. The participants will be again reminded that they are not obliged to take part at all. They will be told that there are no right and wrong answers to the questions. If they do not hear or understand a particular question, they are invited to ask for clarification. They will also be informed that they can choose not to answer a particular question, without needing to give a reason. Finally, they will be informed that we are happy to explain why we are asking a particular question, but we will need to do this at the end of the interview in order not to influence their answers.
-
Please explain in your own words what the STAR study is about/Identifying actual understandings and misunderstandings about the trial
-
Why did you decide not to participate in the study? (Possible prompts: did not want to risk being assigned to the modified arm/did not want to risk having to stop the Sutent, feared it would take too much time, received too much information, it was not clear what the trial was about etc.)
-
Who explained the trial to you? (what was good, what was bad, what was incomplete, did the patient trust a person who explained the trial etc.)
-
Have you read the PIS (watched DVD)? What did you like about PIS (DVD)? What did you dislike (was incomplete, incomprehensible etc.)? How could it be improved?
-
Have you talked to anybody about taking part in the study? (GP, friends, family etc) If yes, what advice have you been given about taking part? Have you looked anywhere else to find out about this or similar studies (e.g. Internet)/identifying information sources?
-
What do you think are advantages and disadvantages of this study?/Identify pros and cons for trial participation. Explore how the participants weight pros and cons. How much more did you think you would need to do if you decided to participate in the trial – time, effort, money/study burden?
-
Was there anything in particular that put you off taking part?
Interview topic guide for study: ‘Patients’ understanding of modified Sutent arm in the STAR study’
The interview will start with a brief introduction to the project and the aims. The participants will be again reminded that they are not obliged to participate at all. They will be told that there are no right and wrong answers to the questions. If they do not hear a particular question, or if they do not understand a particular question, they are invited to ask for clarification. They will also be informed that they can choose not to answer a particular question, without needing to give a reason. Finally, we will inform participants that we are happy to explain why we are asking a particular question, but we will need to do this at the end of the interview, in order not to influence their answers. The information on stage of the trial (e.g. treatment break – which one?) is also collected.
-
What were your reasons for deciding to take part in the STAR study initially? How were you told about the trial? (Where did you receive information – clinical team, PIS, DVD)?/deciding about study participation, perception of information tools
-
Did you think about what was likely to happen to you while you were taking part in the study?
-
Did you think about any extra time that may be taken up by taking part in the study?/expected study burden
-
Can you explain the differences between how you take your Sutent in the STAR study, compared to how you would have taken it had you not taken part in the STAR study? While you were considering taking Sutent as part of the study, did you think about how you were going to feel when it came to having a longer planned treatment break that is temporarily stopping Sutent? If so, what did you think about this?/understanding of the study, expectations
-
How did you feel (are feeling) when your treatment actually stopped? What were you thinking (are thinking)? Was there a difference between first break and other breaks?
-
How were your overall experiences of taking part in the STAR study? What have been the easiest and most difficult thing relating to taking the Sutent (or anything related to the taking the Sutent)? How do you think participating in this study arm (modified STAR trial arm) differs in both positive and negative aspects from participating in the standard arm?/perceived differences between standard and modified study arm
-
Refer to the problem or issue that a participant has mentioned/especially in relation to treatment break/and ask: What do you do when X happens? What would help you in that situation?/coping strategies
-
Could anything else have been done while you have been taking part in this study to help you and your family (by Drs, nurses social services etc)?/support needs
-
Is there anything that you now wish you had known about taking part in the STAR study before you agreed, that you weren’t told? Have you got any suggestions for anything that your medical team could do better to help you while you are taking Sutent in this way with planned treatment breaks or in general while you are taking part in the study?/patients’ recommendations, supportive care.
Appendix 7 Response criteria for assessing therapy in metastatic renal cell carcinoma
| RECIST | Choi | mChoi | MASSa | |
|---|---|---|---|---|
| PD | Increase in sum of longest target lesion diameters ≥ 20% Development of new lesions Unequivocal non-target lesion progression |
Increase in lesion size ≥ 10% Development of new lesions New or enlarging intratumoural nodule |
Increase in lesion size ≥ 10% Development of new lesions New or enlarging intratumoural nodule |
Increase in lesion size ≥ 20% Development of new lesions Absence of central necrosis or marked decrease attenuation |
| PR | ≥ 30% decrease in sum of longest target lesion diameters | Decrease in target lesion CT attenuation in the portal venous phase ≥ 15% or sum of longest target lesion diameters ≥ 10% | Decrease in target lesion CT attenuation ≥ 15% in the arterial phase and sum of longest target lesion diameters ≥ 10% | Decrease in target lesion CT attenuation ≥ 40HU in the portal venous phase or sum of longest target lesion diameters ≥ 20% |
| CR | Disappearance of all target lesions and resolution of lymphadenopathy (< 10 mm) | Disappearance of all target lesions | Disappearance of all target lesions | Disappearance of all target lesions |
| SD | None of the above | None of the above | None of the above | None of the above |
Appendix 8 Computerised tomography scanning parameters
| Technique | Volumetric helical acquisition following iodinated IV contrast administration |
|---|---|
| Phase of enhancement | Combination of arterial (25–35 seconds post contrast injection) and portal venous (65–75 seconds post contrast injection) phase imaging of the chest, abdomen and pelvis |
| kVp/mAs | 90–120/auto-modulated mA |
| Slice thickness (mm) | 1–5 |
| Field of view (mm) | 350 |
| Matrix | 512 × 512 |
List of abbreviations
- AE
- adverse event
- ALT
- alanine transaminase
- ANC
- absolute neutrophil count
- AST
- aspartate transaminase
- AUC
- area under the curve
- BNF
- British National Formulary
- BP
- blood pressure
- CCS
- conventional continuation strategy
- CEAC
- cost-effectiveness acceptability curve
- CECT
- contrast-enhanced computed tomography
- CF
- consent form
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CR
- complete response
- CRC
- colorectal cancer
- CRF
- case report form
- CT
- computerised tomography
- CTCAE
- common terminology criteria for adverse events
- CTLA-4
- cytotoxic T-lymphocyte-associated protein 4
- CTRU
- Clinical Trials Research Unit
- DAM
- decision-analytic model
- DCE-MRI
- dynamic contrast-enhanced magnetic resonance imaging
- DFIS
- drug-free interval strategy
- DMEC
- data monitoring and ethics committee
- ECOG
- Eastern Cooperative Oncology Group
- ECV
- extracellular volume
- EVPI
- expected value of perfect information
- FA
- folinic acid
- FACT-G
- Functional Assessment of Cancer Therapy-G
- FKSI
- Functional Assessment of Cancer Therapy – Kidney Symptom Index
- FMM
- finite mixture model
- FU
- fluorouracil
- GIST
- gastrointestinal stromal tumours
- GLCM
- grey-level co-occurrence matrix
- GLDM
- grey-level dependence matrix
- GP
- general practitioner
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- ICC
- intraclass correlation coefficient
- ICER
- incremental cost-effectiveness ratio
- IFNα
- interferon-α
- IMDC
- International Metastatic RCC Database Consortium
- IQR
- interquartile range
- ITT
- intention-to-treat
- IV
- intravenous
- LDH
- lactate dehydrogenase
- LFT
- liver function test
- MAR
- missing at random
- mChoi
- modified Choi
- MNAR
- missing not at random
- mRCC
- metastatic renal cell carcinoma
- MRI
- magnetic resonance imaging
- MRR
- maximum radiological response
- MRU
- medical resource utilisation
- MSKCC
- Memorial Sloan-Kettering Cancer Centre
- MTT
- mean transit time
- NI
- non-inferiority
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health and Care Research
- NMB
- net monetary benefit
- ONJ
- osteonecrosis of the jaw
- OS
- overall survival
- PD
- progressive disease
- PFS
- progression-free survival
- PH
- proportional hazards
- PIS
- participant information sheet
- PP
- per-protocol
- PR
- partial response
- PS
- performance status
- PSA
- probabilistic sensitivity analysis
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RC
- renal cell
- RCC
- renal cell carcinoma
- RCT
- randomised controlled trial
- RECIST
- Response Evaluation Criteria of Solid Tumours
- RET
- rearranged during transfection
- ROI
- region-of-interest
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SAR
- serious adverse reaction
- SBRT
- stereotactic brain radiation therapy
- SD
- stable disease
- SLD
- sum of the longest diameters
- S/P
- sunitinib and pazopanib
- SPFI
- summative progression-free interval
- SUR
- seemingly unrelated regression
- SUSAR
- suspected unexpected serious adverse reaction
- TFT
- thyroid function test
- TKI
- tyrosine kinase inhibitor
- TMA
- tissue micro-array
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- TSF
- time to strategy failure
- TTF
- time to treatment failure
- ULN
- upper limit of normal
- VAS
- Visual Analogue Scale
- VEGF
- vascular endothelial growth factor
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/JWTR4127).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.