

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as award number 15/57/02. The contractual start date was in March 2017. The draft manuscript began editorial review in March 2023 and was accepted for publication in February 2024. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this article.
Permissions
Copyright statement
Copyright © 2024 Curry et al. This work was produced by Curry et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Curry et al.
Chapter 1 Introduction
Trauma and major haemorrhage
Trauma is one of the biggest contributors to the global burden of disease, with over 5 million deaths each year, and is responsible for as many lost functional life-years as cardiovascular disease and cancer. 1 It is also one of the few conditions that is on the rise globally, as population growth, socioeconomic inequalities and climate change lead to increased road-, occupation-, violence- and conflict-related injuries. Approximately half of all trauma deaths are due to bleeding. 2 Bleeding can cause death rapidly from exsanguination and subsequently from the effects of insufficient blood supply to the heart, brain and other organs. 3
Trauma-induced coagulopathy
Bleeding is complicated by a complex clotting disorder induced by injury and resuscitation, termed trauma-induced coagulopathy (TIC). 4 TIC reduces the ability of blood to form stable clots, and this leads to increased bleeding and impairs surgical attempts to stop the haemorrhage. Subsequent rebound changes can lead to increased clotting, a tendency to thrombose and further organ dysfunction.
Early intervention to stop bleeding and correct TIC has led to dramatic reductions in mortality over the last decade. 3 Since the discovery of TIC, approaches to resuscitation have changed completely. Most hospitals now have a major haemorrhage protocol (MHP) that is activated for patients who are suspected of having active bleeding. 5 This brings additional specialist resources and expertise to the patient. A new ‘haemostatic resuscitation’ paradigm focuses on rapid bleeding control and the targeted correction of coagulopathy. 6 This focuses on controlling bleeding and protecting the body’s ability to form a clot. Clear fluids such as saline are no longer used, as they dilute the body’s clotting factors. Instead, plasma is given alongside red blood cell (RBC) transfusions to replace lost blood volume. 7 Other agents are also used to prevent or treat different aspects of TIC. Tranexamic acid is given early to reduce clot breakdown. 8 Platelet concentrates are given, typically later in the bleeding course, to restore platelet numbers and function. 9 In addition, most protocols include some later replacement of fibrinogen, either with cryoprecipitate transfusions or with fibrinogen concentrates. 10,11
Fibrinogen in trauma-induced coagulopathy
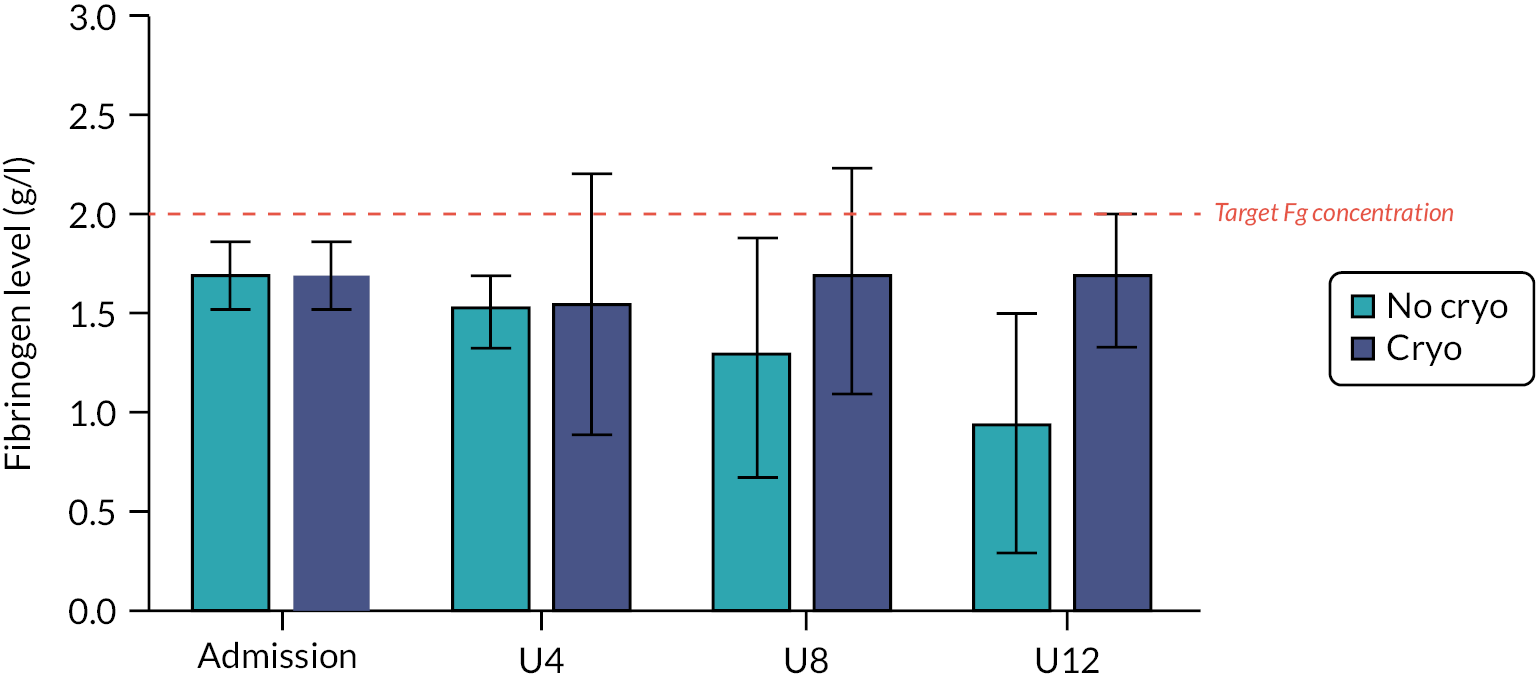
Fibrinogen is an essential pro-coagulant factor that is needed for stable clot formation and effective haemostasis. 11 Fibrinogen is rapidly lost in bleeding trauma patients. In TIC, it is consumed during clot formation and directly broken down through fibrinogenolysis. 12 Alterations in fibrinogen metabolism, including differential effects on synthesis and breakdown, also contribute to decreased fibrinogen availability. 13 Low fibrinogen levels on hospital admission have been found to be independently associated with in-hospital, 24-hour and 28-day mortality. 14 Studies have shown that patients with fibrinogen levels below 1 g/l have a greater than threefold increase in the odds of dying compared with those with normal fibrinogen concentrations of 1.5–3.5 g/l. European and other guidance recommend maintaining fibrinogen levels above 2 g/l during haemorrhage. 15,16 Therefore, the effective replacement of fibrinogen is important for managing TIC and preventing further bleeding and an increase in morbidity and mortality (Figure 1).
FIGURE 1.
Effects of cryoprecipitate on fibrinogen levels following traumatic haemorrhage. Fg, fibrinogen.
Fibrinogen replacement
When treating bleeding disorders caused by low fibrinogen levels, there are two main options: cryoprecipitate and fibrinogen concentrate. 17 Cryoprecipitate is derived from fresh-frozen plasma (FFP) and contains several proteins, including fibrinogen, factor VIII, von Willebrand factor, factor XIII, fibronectin and alpha-2 antiplasmin, which may have additional effects on trauma haemorrhage. 18 Cryoprecipitate is typically pooled from five or six single plasma donations and undergoes two or three freeze–thaw cycles before being transfused. Variability in clotting factor levels in blood donors means that the fibrinogen concentration in cryoprecipitate can vary. Fibrinogen replacement is also available as a concentrate. Fibrinogen concentrate is a purified and standardised product that is often used in Europe. Fibrinogen concentrate is made by extracting fibrinogen from pooled plasma and then purifying and concentrating it. A systematic review comparing the efficacy of cryoprecipitate and fibrinogen concentrate found no difference in fibrinogen increment, transfusion requirement, thromboembolic events or bleeding across the four studies included. 19 In vitro and ex vivo studies have shown that these two products lead to similar, but not identical, improvements in clot structure and quality. 20 Currently, cryoprecipitate is the more frequently used replacement in the UK and the USA, while fibrinogen concentrate is favoured more in Europe.
Empiric therapy versus guided therapy
One key concern with fibrinogen therapy in traumatic haemorrhage is that the time interval between identifying low fibrinogen levels and administering the replacement may be too long for the replacement to exert an effect. In our national study of existing MHPs, on average, patients received their first cryoprecipitate 3 hours after admission to hospital. 10 We know that a large proportion of bleeding trauma patients will have died by this point. Others may have developed such a profound coagulopathy that treatment is likely to be ineffective, and failure to stop or reduce haemorrhage by that point may lead to irreversible cell and organ damage.
Most hospitals in England and around the world will guide fibrinogen therapy using laboratory-based assays of fibrinogen activity. However, these assays are slow to be processed, and the results are usually not available for at least 1 hour after sampling. Point-of-care assessments of fibrinogen levels are not widely available, and so some protocols suggest administering fibrinogen products empirically. These products are usually given later during bleeding, for example after 8 or 10 units of RBC transfusions have been given. 3 The question remains whether the early administration of fibrinogen to all at-risk patients will be of benefit in reducing death rates and other outcomes after traumatic haemorrhage.
Studies of small clinical trials of fibrinogen supplementation have suggested a benefit to replacing fibrinogen, although they have not been powered for a primary mortality end point. We studied the potential for cryoprecipitate in the CRYOSTAT-1 feasibility trial (ISRCTN55509212),21 which provided pilot data for CRYOSTAT-2. This trial demonstrated that it was feasible to deliver cryoprecipitate within 90 minutes of admission and suggested that early cryoprecipitate therapy maintained blood fibrinogen levels during resuscitation, which may lead to reduced mortality. Other pilot studies have used fibrinogen concentrate. FiiRST (NCT02203968),22 RETIC (NCT01545635)23 and E-FIT1 (ISRCTN67540073)24 set coagulation results as the main end points. These trials have demonstrated that early infusion of supplemental fibrinogen leads to an increase in plasma fibrinogen concentration. The European prehospital trial FIinTIC (NCT01475344) found that physicians were able to administer fibrinogen concentrate in the field, resulting in an improved coagulation profile of the patient on arrival at the hospital. 25
Overall study objective
Based on the results of CRYOSTAT-1 and these other studies, we postulated that patients thought to have active haemorrhage would benefit from the early empiric administration of cryoprecipitate as soon as possible after admission to hospital. Early administration would treat those who presented with low fibrinogen levels and provide enough reserve fibrinogen such that levels would not fall to critical levels during haemorrhage. We therefore hypothesised that early empiric cryoprecipitate administration would improve outcomes following traumatic haemorrhage and reduce medium- and long-term mortality.
Chapter 2 Methods
Trial design
CRYOSTAT-2 was a randomised, parallel-group, unblinded, multicentre, international trial evaluating the effects of early high-dose cryoprecipitate in adult patients with major trauma haemorrhage requiring MHP activation. The trial is registered at ISRCTN, with reference number ISRCTN14998314 (https://doi.org/10.1186/ISRCTN14998314),26 Sections of this chapter are based on the trial protocol published in Transfusion Medicine,27 and the original study protocol document,28 available from the National Institute for Health and Care Research (NIHR) Funding and Awards site.
Approvals
The Medical Research and Ethics Committee and Health Research Authority reviewed the protocol and supporting documents for the CRYOSTAT-2 trial and provided a favourable ethics opinion on 26 May 2017 (Research Ethics Committee reference 17/SC/0164).
Six substantial amendments and eight non-substantial amendments were approved during the project, as detailed in Table 1.
| Amendment number | Date of approval | Comments |
|---|---|---|
| Substantial 02 | 10 September 2018 | Temporary halt at a participating site following a serious breach of the protocol |
| Substantial 03 | 28 November 2018 | Restart at a participating site following a serious breach of the protocol |
| Substantial 04 | 31 January 2020 | Application to the CAG seeking Section 251 support to enable linkage of participant identifiable data from a subset of patients from whom it was not possible to collect signed, informed consent. This was to provide information to NHS Digital and TARN for patient matching |
| Substantial 05 | 3 April 2020 | Public-facing study poster |
| Substantial 06 | 14 June 2021 | Extension to the study approved by funder and sponsor. Approval for sites in Northern Ireland |
| Substantial 07 | 5 May 2022 | Extending the number of participants for whom we used Section 251 support for to collect data where appropriate and in line with conditions of CAG support. The NIHR and sponsor agreed that it would be possible to recruit more patients in the UK to make up for the shortfall in expected US patients due to COVID-19. Updates to the statistical section of the protocol including the per-protocol analysis population and updates to administrative sections |
| Non-substantial 01 | 31 August 2017 | Additions and changes to principal investigators at participating sites |
| Non-substantial 04 | 2 September 2019 | Approval for sites in Wales. Additions and changes to principal investigators at participating sites |
| Non-substantial 05 | 6 April 2020 | Non-notifiable amendment: temporary pause in recruitment due to COVID-19, approved by the sponsor |
| Non-substantial 06 | 1 May 2020 | Non-notifiable amendment: reopening to recruitment due to COVID-19, approved by the sponsor |
| Non-substantial 07 | 29 May 2020 | Change of principal investigator at participating site |
| Non-substantial 08 | 29 June 2020 | Change in recruitment end date |
| Non-substantial 09 | 19 April 2021 | Change in recruitment end date definition from a date to until the target number of participants is reached |
| Non-substantial 10 | 7 October 2021 | Increase in sample size due to a larger number of participants exiting the study than expected |
The Confidentiality Advisory Group (CAG) reviewed the application and supporting documents for Section 251 support to process confidential patient information without consent and provided approval on 28 January 2020 (CAG reference 19/CAG/0161).
Participants (inclusion and exclusion)
Patients were eligible for this trial if:
-
The patient was judged to be an adult, was aged ≥ 16 years in the UK (or according to local guidance) and had sustained severe traumatic injury.
-
The patient was deemed by the attending clinician to have active haemorrhage.
And required:
-
Activation of the local MHP for the management of severe blood loss.
And had started or had received:
-
At least one unit of any blood component.
Patients were not eligible for this trial if they fulfilled one or more of the following criteria:
-
They had been transferred from another hospital.
-
The trauma team leader deemed the injuries incompatible with life.
-
More than 3 hours had elapsed from the time of injury.
Consent
Patients with major trauma haemorrhage have a life-threatening injury. On arrival at the emergency department, patients are usually unconscious, and those who are not are usually in pain, in distress and/or under the influence of medication. Patients were enrolled into the study using an ‘emergency waiver of consent’, whereby the treating senior clinician (the trauma team leader) assessed the patient for eligibility and made the decision whether or not to enter them into the trial. This is an established method of enrolling patients without capacity into emergency medicine trials.
UK law allows patients who need emergency treatment and lack capacity to consent to have a ‘consultee’, who can recommend whether the participant would like to continue in or would object to being in the trial. This consultee can be the treating clinician who is not part of the research team (a professional consultee) or a relative/friend (a personal consultee). This study recommended that a professional consultee be sought in the first instance to allow the participant to move from the emergency waiver to consultee declaration and remain in the study until such time that a personal consultee or the participant could be approached about the study. The aim of the consent process in this trial was always to obtain informed signed consent from participants. The parents/legal guardians of participants who were found to be below 16 years of age after the point of entry were approached as personal consultees. See Appendix 1, Figure 11, for an overview of the consent procedure used in the UK.
Participants enrolled at international sites were subject to the regional or national ethical code of practice for conducting research in incapacitated adults.
There were occasions where a participant died soon after their arrival at hospital, and there had been no opportunity for contact between the research team and a personal consultee. In these situations, it was accepted that approaching the consultee to ask their opinion was likely to be distressing and without benefit. Families of bereaved participants were not contacted by the research team after the participant’s death to obtain personal consultee advice. In these cases, advice from a professional consultee was sought so that data collection could be completed at site. Recruiting hospitals were informed that participation in the trial should be disclosed if a coroners’ court hearing was convened. This process was agreed after receiving advice from the Research Ethics Committee.
The COVID-19 pandemic began during this trial. This made the consent process more complex because of COVID-19-related restrictions in hospitals, particularly when participants were on a COVID-19 ward where no visitors or staff visits were allowed. It was agreed that initial verbal consent could be taken from COVID-19-positive individuals or those on restricted wards in hospital if this was documented appropriately in the medical notes. When it was safe to do so, the participant was approached to sign an informed consent form. If the participant had been discharged, they were contacted at home by telephone or by post. Consent was obtained using an electronic form or via a physical form returned to the hospital. This process was also followed when seeking consultee declarations during this time. The PANDO application30 was used at some participating sites to electronically transfer patient-signed informed consent forms from COVID-19-restricted wards to the research team.
It became apparent shortly after the trial began that, for a subset of participants, research staff were at times not able to start or complete the consent process. The reasons for this included:
-
patients unexpectedly being discharged (overnight or weekends)
-
patients unexpectedly self-discharging (including absconding from authorities)
-
participants being rapidly discharged to police custody or returned to the custody of His Majesty’s Prison Service
-
participants being rapidly transferred to mental health trusts under section
-
participants having been repatriated to a non-participating hospital or residential nursing care home or rehabilitation centre
-
participants providing false details or refusing to give any details to hospital staff
-
participants being homeless and having no listed general practitioner
-
participants in hospital being abusive, aggressive or violent, and whom the research team had been advised not to approach
-
participants who would have been extremely distressed if contacted due to the nature of their trauma (e.g. if they had been in an accident in which a loved one had died)
-
participants who never regained capacity due to the nature of their injuries (brain injury or coma)
-
participants having considerable language difficulties where it was not possible to provide an NHS translator or to ask a family member to translate information about the trial.
To enable follow-up data to be collected for these participants, we applied to the HRA for Section 251 support via the CAG, and this was granted.
The participant or their consultee was free to withdraw their consent or change their opinion about participating in the trial at any time. Participants were withdrawn with or without permission for follow-up data collection. If participants withdrew and did not provide permission for continued data collection, data collected up to the point of withdrawal were retained. Although the participant was not required to give a reason for withdrawing consent, a reasonable effort was be made to establish this reason while fully respecting the participant’s rights.
Randomisation
The allocation sequence was produced by the trial statistician using SAS® statistical software (SAS Institute Inc, Cary, NC, USA). The allocation sequence had a varying block size that was not disclosed and was stratified by centre. Participants were allocated in a 1 : 1 ratio to the intervention (early cryoprecipitate + standard MHP) or standard care (standard MHP) arm. Allocation cards were prepared and placed in sequentially numbered opaque envelopes, 100% QA checked and sealed with tamperproof tape. Each envelope contained a randomisation number and the allocated treatment and were opened at sites in sequential order. Envelopes were released by the Clinical Trials Unit (CTU) in small batches sufficient to support projected recruitment and replace used envelopes. Envelopes were securely stored in a locked cupboard at each site, access to which was controlled by the research team. In February 2020, an issue was identified with the creation of the allocation sequence, such that there was a tendency for one arm to appear first more often in each block in the early part of the randomisation list, leading to concerns about a small imbalance by the end of the trial. A revised list was created and used when issuing new randomisation envelopes from that point on.
Randomisation took take place in the emergency department or the transfusion laboratory/blood bank, as agreed with the participating hospital. The recruiting staff completed an enrolment log each time an envelope was taken for use, and signed and dated the envelope to confirm that the next available and lowest numbered envelope of the batch had been taken, was unopened and bore no evidence of tampering. The participant’s initials, date of birth and hospital number were also written on the envelope prior to opening (if the initials and date of birth were not known, the unique identifiers used at the participating hospital for unknown patients were used). The correct and sequential use of envelopes was strictly audited by the site research team and CTU staff at site monitoring visits.
If the randomised intervention differed from that which the participant received, a reason for this was requested. These processes allowed for clear and regular auditing of the randomisation process through comparison with the randomisation list held by the statistician. At the end of recruitment, sites returned the randomisation envelopes to the CTU, with participant details obscured.
This was an unblinded trial. As the study control arm was standard care (MHP), it was not possible to blind the intervention of early cryoprecipitate as there was no comparative transfusion of a placebo. Members of the trauma team in emergency department were therefore aware of the treatment arm to which the participant had been randomised. However, the risk to trial integrity was minimised, given that the primary outcome of 28-day mortality was a hard end point.
Trial intervention
The intervention was 3 pools of cryoprecipitate (equivalent to 15 single units of cryoprecipitate or 6 g of fibrinogen supplementation), infused as rapidly as possible, within 90 minutes of arrival at hospital. The intervention was given in addition to the standard MHP.
The comparator was standard MHP only. The standard treatment of major traumatic haemorrhage involves administering RBCs, FFP and platelets. Each participating site followed its local standardised MHP that aligned with the current accepted best practice for transfusion therapy, that is damage control resuscitation, the use of tranexamic acid and the empiric (automatic and unguided) delivery of RBCs, FFP and platelets. The MHP comprises a balanced resuscitation with blood products and was standardised as much as possible across participating sites; however, some degree of variation was inevitable and pragmatic, reflecting actual clinical practice. The transfusion products given vary to some extent from the target ratio according to blood product availability and the participant’s ongoing clinical condition.
Early cryoprecipitate (3 pools = 6 g of fibrinogen) was stored in its frozen state and defrosted in accordance with the local standard operating procedures for each blood bank/transfusion laboratory. In the UK, these procedures will be in accordance with the national guidelines. 31 The cryoprecipitate was made available to the emergency department as quickly as possible. The study team provided trial-specific, yellow-labelled bags for the intervention to differentiate trial cryoprecipitate from any cryoprecipitate given as standard of care. As an alternative, and depending on local approval for use, pre-thawed cryoprecipitate was also permitted during the trial. The early cryoprecipitate was administered as rapidly as possible via an intravenous line and in accordance with local practice. The cryoprecipitate was not mixed with platelets prior to infusion.
There were no restrictions on treatments that could be given during this trial, as long as the treatments were part of standard care and were not administered as part of another trial [unless authorised by the Trial Management Group (TMG) or delegated co-investigator].
Sites
This study was conducted in participating major trauma centres in the UK and the USA. The list of study sites is in Appendix 2, Table 17.
Data collection
Screening data
Site research teams collected data in a screening log to allow an assessment of the proportion of eligible patients recruited and the reasons why eligible patients were not recruited to CRYOSTAT-2. This log recorded data for all patients considered eligible for enrolment in the trial and included age, sex, inclusion/exclusion criteria, and other reasons for non-enrolment.
Outcome data
Once patients were randomised into CRYOSTAT-2, data were collected on paper case report forms (CRFs) by the local research team, and these were posted to the CTU at day 28, discharge or death, for data entry into the MACRO database. Background data (participant characteristics) were collected, including participant age, participant sex, mechanism of injury and injury type. Clinical data were also collected, which included vital signs on arrival at the emergency department (systolic blood pressure, heart rate and Glasgow Coma Scale), blood components given (pre-hospital, in the emergency department and 24 hours from arrival in the emergency department) and cryoprecipitate administration.
At day 28, discharge or death, the Injury Severity Score (ISS), EuroQol-5 Dimensions, five-level version (EQ-5D-5L) and Glasgow Outcome Scale (GOS), hospital stay, consent status, and discharge and mortality status of participants were collected on the CRF. Patient-reported outcome measures are routinely collected from eligible patients at all major trauma centres across the UK during the first hospital admission and at 6 months from injury as part of an ongoing (Trauma Audit and Research Network (TARN) project.
Follow-up data
Data on participants’ quality of life at 6 months post admission were collected through linkage with TARN for participants with informed consent in place. Data on participants’ survival status up to 1 year post admission were collected through linkage with NHS Digital for participants with patient informed consent in place or those covered by Section 251 approval.
Monitoring
CRYOSTAT-2 was assessed as moderate risk; therefore, visits were conducted twice per year at each site. Investigators and their institutions provided direct access to source data for monitoring, auditing and regulatory inspections. We planned to monitor 20% of UK participants through on-site source data verification, but from March 2020, owing to restrictions during the COVID-19 pandemic, as most sites had been visited multiple times monitoring was conducted remotely, with site research teams using a checklist to reconfirm key data. We monitored 30% of UK participants on site. Key data were monitored centrally on a bi-monthly basis as part of the TMG report. This included recruitment, withdrawals, serious adverse events, data queries and completion and consent.
Outcome measures
Primary outcome measures
The primary outcome measure was all-cause mortality at 28 days.
Secondary outcome measures
Secondary outcome measures were all-cause mortality at 6 hours, 24 hours, 6 months and 12 months from admission; death from bleeding at 6 and 24 hours; transfusion requirements for RBC, platelets, FFP and cryoprecipitate at 24 hours from admission; destination of participant at discharge; quality-of-life measurements (EQ-5D-5L and GOS) at discharge/day 28 and 6 months after injury; and hospital resource use up to discharge or day 28 (including ventilator-days, hours spent in critical care and inpatient stays).
Adverse events
Data on symptomatic thrombotic events (venous thromboembolism and arterial thrombotic events), death and serious transfusion-related adverse reactions were collected from randomisation up to day 28 or discharge. All serious adverse events were reviewed by the chief investigators or delegated clinical members of the TMG.
Changes to the protocol
Table 2 summarises the changes made to the protocol throughout the lifespan of the trial.
| Version | Author | Date | Reason for revision |
|---|---|---|---|
| 2.0 | TMG | 3 July 2019 | Clarification of consent processes for England and Wales. Clarification of continued follow-up data in accordance with General Data Protection Regulation and CAG approval |
| 3.0 | TMG | 25 March 2021 | Change in planned study duration. Addition of Northern Ireland specific study processes. Addition of verbal and electronic consent of personal consultees and participants where obtaining a signed copy of the personal consultee declaration form or participant informed consent form would put staff and/or participants at risk of contracting COVID-19. Addition of the use of the PANDO application to transfer consultee declaration form and participant informed consent forms |
| 3.1 | TMG | 5 October 2021 | Sample size changed to 1600; changes to the statistics section to reflect this |
| 4.0 | TMG | 15 February 2022 | To reflect that ISSs will be collected from TARN to ensure consistency with nationally reported scores. Additional data collection of baseline quality-of-life data from TARN where the research team have not been able to collect it in hospital |
Sample size
The CRYOSTAT-2 study was designed to detect an absolute mortality difference of 7% from a baseline mortality rate of 26%, that is a reduction to 19%. The baseline mortality was based on the feasibility study CRYOSTAT-1, which reported a 28% mortality rate; the national epidemiological study of trauma transfusion practice, which reported a 39% mortality rate among patients receiving ≥ 10 units of RBC in 24 hours (classified as having ‘massive haemorrhage’); and the PROPPR study of bleeding trauma patients conducted at level 1 trauma centres in North America, which had a baseline mortality rate of 26%.
CRYOSTAT-2 used 90% power to detect a reduction in 28-day all-cause mortality of 7% using a 5% level of significance and a two-tailed test. An initial blinded analysis after the first 300 participants had been recruited and followed up to 28 days was used to reassess sample size requirements and recruitment rates, and at the request of the Data Monitoring Committee (DMC) this was repeated after 750 participants had been recruited.
A group sequential design was used to allow for the DMC reviewing the primary outcome for evidence of harm or benefit (but not futility) after 500 and 1000 participants had been followed up for 28 days. The design used O’Brien–Fleming stopping guidelines29 determined at the study design stage to ensure an overall Type I error of 5% at the end of the trial. O’Brien–Fleming guidelines were used because of the low chance of terminating the trial at early interim analysis, and because of the minimal change to the alpha used at final analysis. The stopping guidelines were used to help guide the DMC’s decision-making alongside other safety data available to the committee. Allowing for the interim analyses in this way, the required sample size to meet specified power requirements was 1530 participants in total. This was initially increased by 2.5% to allow for dropout, but as dropout was higher than anticipated this was later increased to 4.4%, resulting in a total of 1600 participants.
Statistical methods and analysis plan
The Statistical Analysis Plans (SAPs) for 28-day outcomes and for longer-term outcomes are available in Report Supplementary Material 1 and 2. All analyses were on an ‘intention-to-treat’ (ITT) basis and included all randomised participants (including those randomised in error) for whom values of a response variable were obtained, analysed according to randomised group. All analyses were two-sided and the significance level was 5%.
The primary outcome, all-cause mortality at 28 days from arrival at hospital, was determined as the proportion of participants in each treatment arm who died within 28 days. The odds ratio (OR) for death within 28 days [with 95% confidence interval (CI) and p-value for the treatment arm term] was presented, and adjusted for centre using a marginal model, and this was the primary analysis of the outcome. This was supplemented by logistic regression analysis so that account could be taken of any prespecified factors that might have differed between the treatment arms and had a statistically significant association with 28-day mortality. Mortality rates according to timing of cryoprecipitate administration were also analysed, using categories of ≤ 45 minutes, 46–60 minutes, 61–90 minutes and > 90 minutes from arrival at hospital. Mortality rates among those who did or not receive cryoprecipitate in the cryoprecipitate arm were also analysed.
As no placebo was used in the study, a per-protocol analysis that excluded patients who did not receive trial treatment would differentially exclude those in the intervention arm. Per-protocol analysis therefore focused on the cohort of patients who could have benefitted from the intervention and excluded only protocol deviations unrelated to the details of cryoprecipitate administration, randomisations in error, those who died within 90 minutes of admission and those who did not receive any blood products after arrival at hospital (an indication that they had already stopped bleeding). A per-protocol analysis was conducted for all mortality end points up to 28 days (6 hours, 24 hours and 28 days), transfusion requirements, hospital stay and thrombotic events.
Multiple imputation based on full conditional specification was used to impute values of potential risk adjustment factors. The set of variables used in the multiple imputation model was specified in the SAP and included the primary outcome variable. Primary and secondary outcome measures were not imputed, and these were treated as missing data. The methods used for the analysis of secondary outcomes in the form of proportions were similar to those described for the primary outcome. Survival times and rates were estimated using the Kaplan–Meier method and compared using Cox proportional hazards regression. Transfusion requirements were summarised as the median and interquartile range (IQR) of the number of units administered from injury to 24 hours post arrival at hospital, and the mean products transfused per participant per hour over the first 24 hours compared between the arms. Hospital stay, critical care stay and ventilator-days were estimated using a competing risks analysis,32,33 with discharge/extubation as the event and death as the competing risk.
The number of symptomatic thrombotic events up to day 28 were presented overall and by treatment arm. In particular, the number of venous thromboembolisms (pulmonary embolism, deep-vein thrombosis) and arterial thrombotic events (myocardial infarction, stroke) were calculated.
Subgroup analyses
The primary outcome analysis was repeated to assess the heterogeneity of treatment effects for the following subgroups:
-
UK participants versus non-UK participants
-
head Abbreviated Injury Score (AIS) < 4 versus ≥ 4
-
participant sex
-
participant age < 70 versus ≥ 70
-
injury type, blunt versus penetrating.
The secondary outcome analysis of 6- and 24-hour mortality was repeated for subgroup analysis (b), head AIS < 4 versus ≥ 4.
Economic evaluation methods
An economic analysis model was developed to analyse the cost-effectiveness of early cryoprecipitate plus standard of care versus standard of care alone from an NHS and Personal Social Services perspective. Full details of the methods and results are provided in Chapter 4.
Patient and public involvement
The principles set out by the INVOLVE advisory group (active until 2020) were used to guide our approach to patient and public involvement and experience (PPIE) engagement. The research team also built on their pre-existing patient and public involvement (PPI) links that had been set up prior to the start of the feasibility study, CRYOSTAT-1, in 2012. The initial focus of CRYOSTAT-2 PPIE was to engage with patient and public stakeholders to inform the design of the study. Advertising documents (posters/e-mails/website adverts) were sent out both to patients who had previously been injured and to all the lay members of one participating site's trust mailing list to ask for interested volunteers to join a meeting to discuss the study. In total, 2 face-to-face meetings were held (2014, 2016) and 1 survey was distributed to 50 members of the public, asking for views about 2 important trial design questions: what they felt would be the most relevant outcome to test in the proposed CRYOSTAT-2 study (e.g. survival, functional status, or other) and what their views were about consenting participants to a study for which most eligible patients lacked capacity.
Following the second meeting, which specifically focused on consent, in 2016 a dedicated PPI group Patient/Public Advisors for Injury Research (PAIR) was formed to support CRYOSTAT-2 trial development. This group provided ongoing support to the CRYOSTAT-2 trial in the following ways: (1) most importantly, they helped the study team shape the trial design with regard to the primary end point and most acceptable method of gaining consent; (2) the group nominated a member to sit on the Trial Steering Committee (TSC) and this member actively contributed to each TSC meeting throughout the study (a second, more experienced PPI representative was also invited to sit on the TSC); (3) the PAIR group collectively co-wrote the plain language summary for the application to the NIHR Health Technology Assessment (HTA) programme as well as the CRYOSTAT-2 trial protocol and will continue to help with the dissemination of information; (4) the group helped to draft and subsequently edited the patient-facing trial documents such as the patient/patient representative information leaflets and consent/assent forms; and (5) the TSC members helped to develop the NIHR HTA funding application as an integral part of the research team. We will ask the PAIR group for assistance in disseminating the results of this study more widely through social media and patient forums.
Chapter 3 Results
Recruitment and baseline results
The first patient was enrolled on 23 August 2017 and the last patient was enrolled on 2 November 2021. Recruitment was stopped temporarily between 6 April 2020 and 1 May 2020 due to the COVID-19 pandemic. Trial-specific data collection continued until 1 December 2021 in the UK and until 5 July 2022 in the USA. Central data linkage for mortality continued until 27 May 2022 in the UK.
The original planned sample size was 1568, later amended to 1600. A planned sample size re-estimation was conducted after 300 participants had been randomised and repeated after 750 participants had been randomised following a DMC request. In both cases, the committee recommended that the study continue with the original sample size. The final sample size was subsequently increased to 1600 after the TMG reviewed lost to follow-up rates. Two pre-planned interim analyses for harm or benefit after the recruitment of 500 and 1000 participants were conducted using data on 578 and 1027 participants, respectively. In both cases, the test statistic did not cross the O’Brien–Fleming early stopping boundary, and the DMC recommended that the trial continue to the full sample size. Due to delayed randomisation notifications, recruitment closed after the recruitment of 1604 participants. In total, 1555 participants were recruited across 25 UK centres, and 49 participants were recruited in one US centre (see Appendix 2, Table 17).
The Consolidated Standards of Reporting Trials diagram is shown in Figure 2. A total of 9036 patients were assessed for eligibility, of whom 2756 (31%) were found to be eligible for the study; 1604 (58%) of these 2756 patients were randomised. The most common reason for not randomising eligible patients was that the research team was unavailable for patients presenting at sites out of hours.
FIGURE 2.
Summary of trial entry, randomisation and treatment. a, Standard major haemorrhage therapy: empiric high ratio RBC, FFP and platelet transfusions.
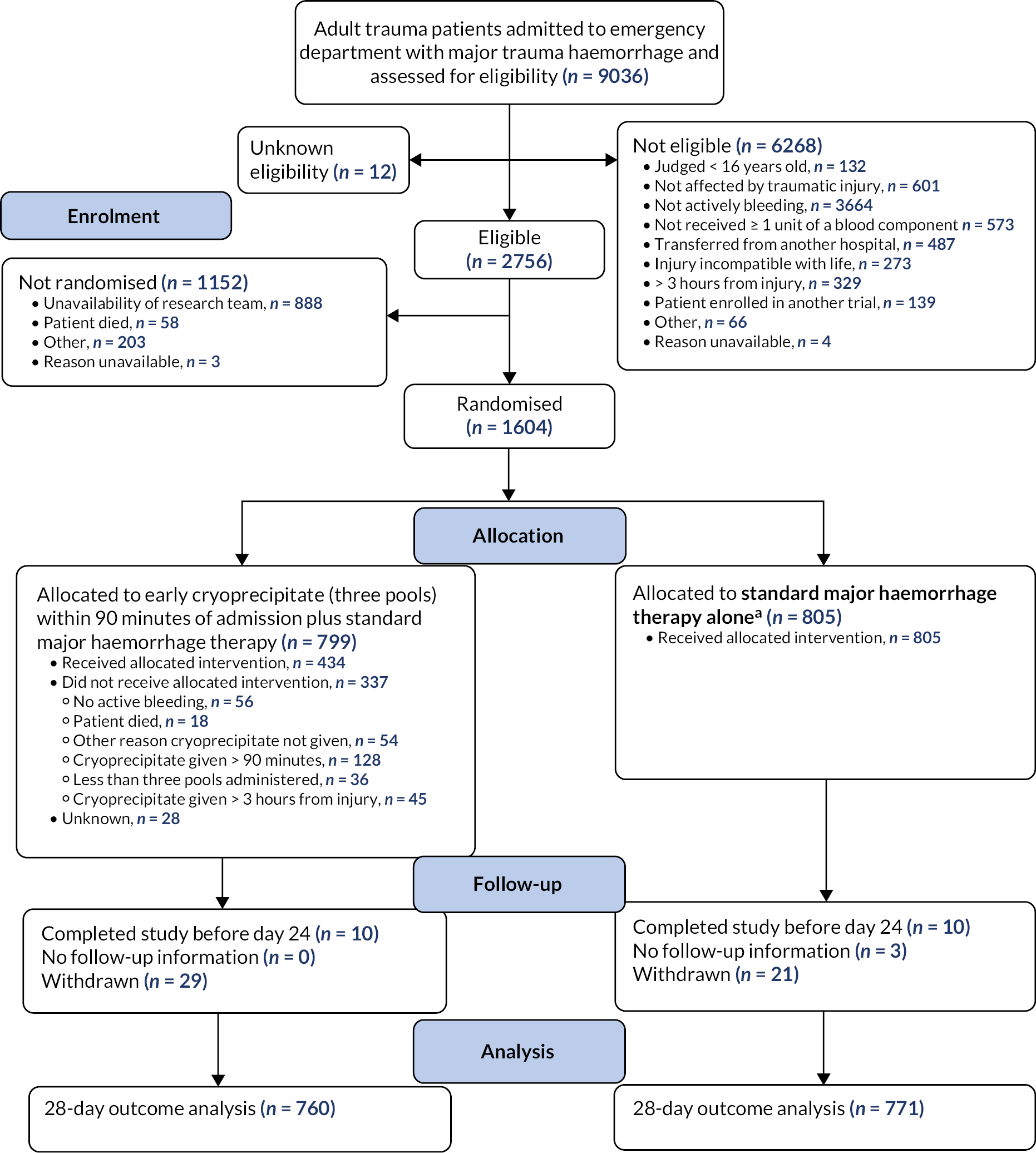
The 1604 patients were randomly assigned to receive either early cryoprecipitate in addition to the standard MHP (n = 799, 49.8%) or the standard MHP alone (n = 805, 50.2%).
Sixty-nine participants were randomised in error (intervention arm, n = 29; standard care arm, n = 40), having found to be ineligible for the study after randomisation, most commonly because they had not received at least one unit of any blood component prior to randomisation. All patients randomised in error were included in the ITT analysis if they had primary outcome data.
A total of 88 patients (intervention arm, n = 50; standard care arm, n = 38) withdrew consent after randomisation, but, of these, primary outcome data were missing for only 50 (intervention arm, n = 29; standard care arm, n = 21). For the remaining 38 patients, primary outcome data were available due to either the timing of withdrawal or the participants’ agreement to continued data collection after withdrawal.
Primary outcome data were missing for a further 23 participants (intervention arm, n = 10; standard care arm, n = 13) for whom no follow-up data were available at all, or available only beyond day 24 (as the 28-day follow-up form had a reporting window of ± 4 days patients reported alive at day 24 were assumed alive at day 28 as specified in the SAP).
Baseline characteristics were similar in both arms (Table 3). Seventy-nine per cent of participants were male and the median age was 39 years (IQR 26–55 years). Sixty-four per cent of patients sustained a blunt injury, 26% had a head AIS ≥ 4 and the median ISS was 29 (IQR 18–43). Forty-three per cent were administered a blood component and 79% were administered tranexamic acid prior to hospital arrival.
| Characteristic | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) |
|---|---|---|---|
| Male | 633/796 (80) | 618/785 (79) | 1251/1581 (79) |
| Age (years) | 40 (26–55) | 38 (25–55) | 39 (26–55) |
| Time from injury to admission to emergency department (minutes) | 77 (55–100) | 75 (55–99) | 76 (55–100) |
| Time from admission to randomisation (minutes) | 14 (7–27) | 15 (7–28) | 15 (7–27) |
| Injuries and physiology at admission to emergency department | |||
| Blunt injury | 519/796 (65) | 495/785 (63) | 1014/1581 (64) |
| ISS | 29 (18–43) | 29 (17–43) | 29 (18–43) |
| Head AIS ≥ 4 | 191/664 (29) | 157/665 (24) | 348/1329 (26) |
| Systolic blood pressure (mmHg) | 103 (83–126) | 102 (84–124) | 103 (83–125) |
| Systolic blood pressure < 90 mmHg | 250/738 (34) | 230/724 (32) | 480/1462 (33) |
| Heart rate (per minute) | 108 (88–127) | 108 (88–126) | 108 (88–127) |
| In cardiac arrest | 17/735 (2) | 12/717 (2) | 29/1452 (2) |
| Glasgow Coma Scale score | 13 (3–15) | 14 (3–15) | 14 (3–15) |
| Pre hospital | |||
| Administered any blood components | 348/795 (44) | 323/783 (41) | 671/1578 (43) |
| RBC (units) | 0 (0–2) | 0 (0–2) | 0 (0–2) |
| FFP (units) | 0 (0–1) | 0 (0–1) | 0 (0–1) |
| Crystalloids (ml) | 0 (0–250) | 0 (0–250) | 0 (0–250) |
| Colloids (ml) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
| TXA administered | 639/796 (80) | 615/783 (79) | 1254/1579 (79) |
As expected in the absence of a placebo, protocol deviations were more common in the intervention arm. One patient was transfused the wrong component in error and a further 337 of the 799 patients (42%) in the intervention arm did not receive the full intervention per protocol. Of those 337 patients, 128 did not receive any early cryoprecipitate (16% of those randomised to early cryoprecipitate) because the patient had died, no active bleeding was identified at the time, or it was reported that haemostasis or correction of coagulopathy had been achieved. Other reasons for protocol deviations were administering cryoprecipitate more than 90 minutes after admission (e.g. due to delayed activation of the MHP), administering fewer than three pools of cryoprecipitate or administering cryoprecipitate more than 3 hours after injury.
Four patients in the standard care arm had protocol deviations. Two were randomised when recruitment at the site was paused due to COVID-19-related pressures, one was randomised when human error resulted in production of the randomisation envelope that showed the incorrect treatment, and one patient was randomised twice [in the emergency department and intensive care unit (ICU), randomised to the same arm, only one set of data was collected and analysed].
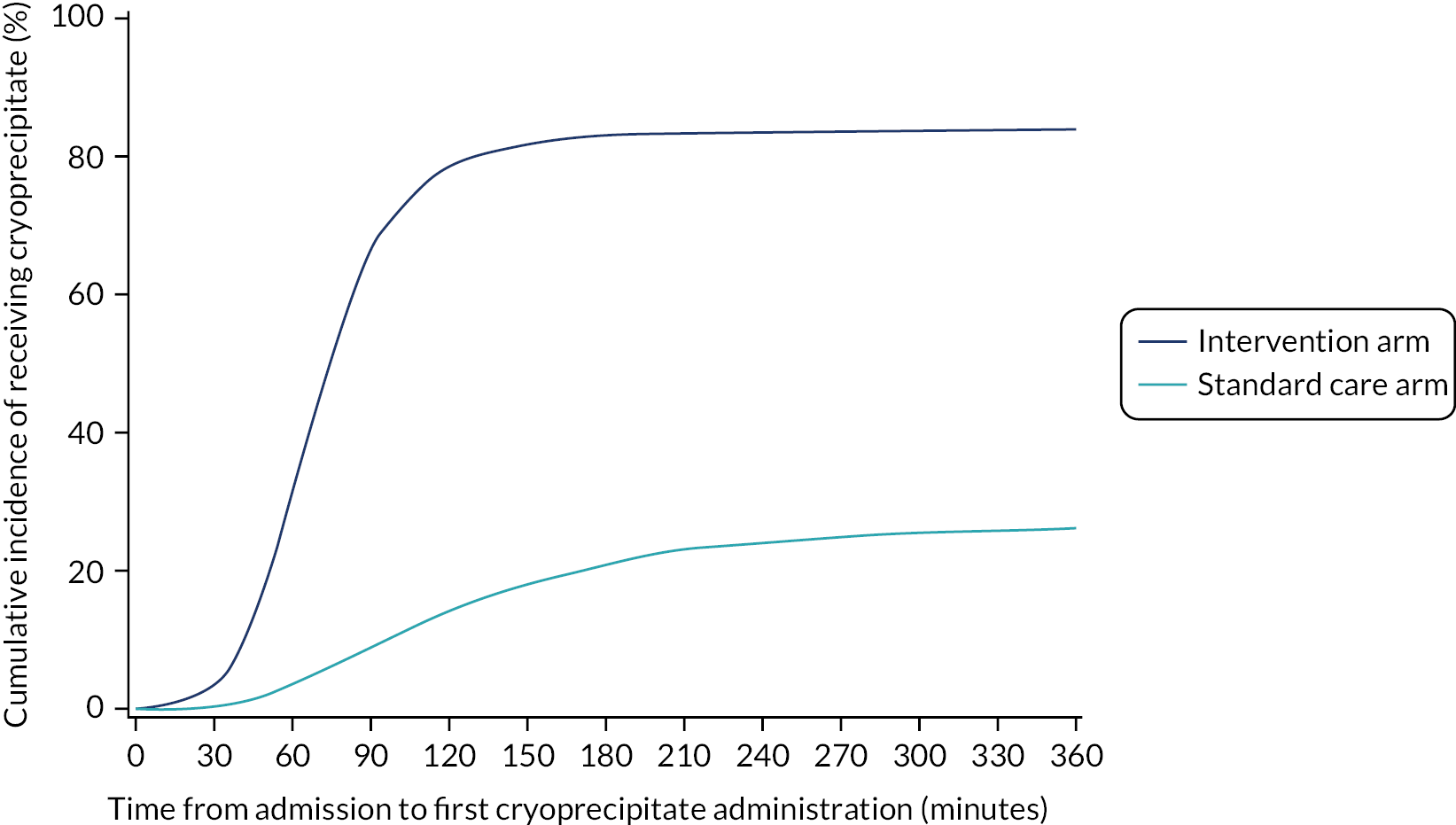
Eighty-five per cent of those in the intervention arm and 32% of those in the standard care arm received cryoprecipitate within the first 24 hours of arrival at hospital. Sixty-eight per cent of participants in the intervention arm received it within 90 minutes of arrival at hospital, compared with 9% in the standard care arm (p < 0.0001; Figure 3). Median (IQR) time to first cryoprecipitate (among those who received it) was 68 (53–85) minutes in the intervention arm and 120 (79–184) minutes in the standard care arm.
FIGURE 3.
Cumulative incidence curve of time from admission to first cryoprecipitate administration (including all patients) by treatment arm.
Primary outcome
We obtained primary outcome data for 760 patients in the intervention arm (95%) and 771 participants (96%) in the standard care arm. Patients discharged from hospital prior to 28 days were assumed alive at 28 days, together with anyone reported alive at day 24 or later.
Table 4 presents the ITT analysis of the primary outcome. In the intervention arm, 25.3% died within 28 days of admission compared with 26.1% in the standard care arm. The OR was 0.96 (95% CI 0.75 to 1.23) for 28-day mortality for early cryoprecipitate versus standard MHP, with a p-value of 0.7406 for the difference between the arms. The relative risk was 0.97 (95% CI 0.81 to 1.17).
| Outcome | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value |
|---|---|---|---|---|
| Participants who died on or before day 28 from admission, n/N (%) | 201/771 (26.1) | 192/760 (25.3) | 393/1531 (25.7) | |
| Relative riska (95% CI) | 0.97 (0.81 to 1.17) | |||
| ORb (95% CI) | 0.96 (0.75 to 1.23) | 0.7406 | ||
| OR also adjusted for participant factorsc (95% CI) | 1.15 (0.93 to 1.42) | |||
| Participants for whom 28-day vital status was not available from any source, n/N (%) | 34/805 (4.2) | 39/799 (4.9) | 73/1604 (4.6) | |
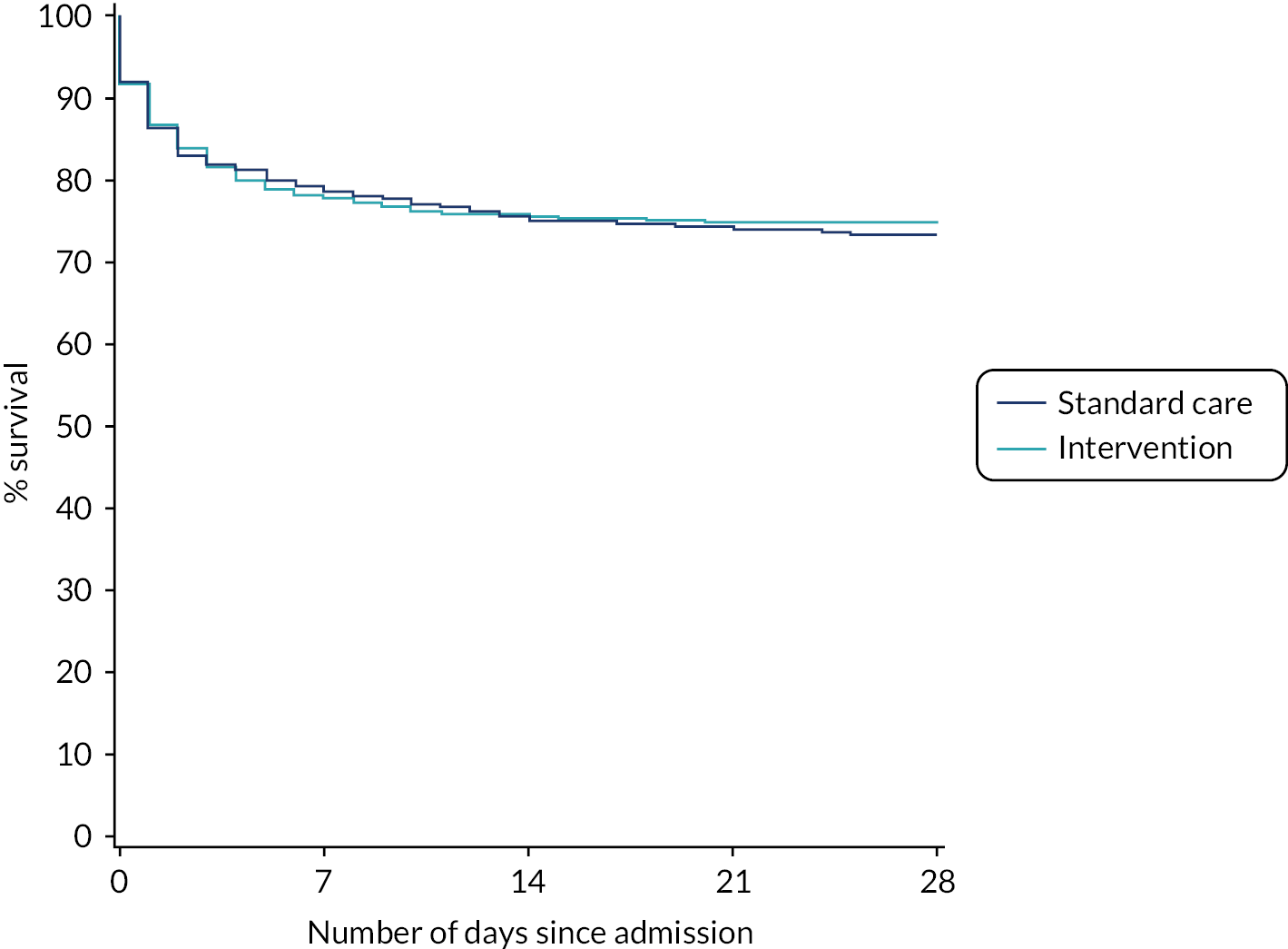
After risk adjustment for statistically significant patient factors (see Appendix 2, Table 20, Figures 11 and 12, for the details of this model), the OR for mortality was 1.15 (95% CI 0.93 to 1.42). Figure 4 presents an unadjusted Kaplan–Meier plot of survival to 28 days by treatment arm.
FIGURE 4.
Kaplan–Meier survival plot up to 28 days from admission by treatment arm.
Similar results were obtained in a per-protocol analysis (those excluded from the per-protocol analysis are summarised in Appendix 2, Table 21). In the per-protocol analysis, 23.1% of those in the intervention arm and 22.5% in the standard care arm died within 28 days of admission (OR 1.03, 95% CI 0.77 to 1.37; p = 0.8272). The relative risk was 1.02 (95% CI 0.82 to 1.27). After adjustment for significant participant factors (as per ITT analysis), the OR was 1.24 (95% CI 1.00 to 1.55).
Similar results were obtained from other sensitivity analyses, including an ITT analysis unadjusted for centre (OR 0.96, 95% CI 0.76 to 1.21) and a sensitivity analysis that assumed that 2% of those discharged died before day 28 (OR 0.95, 95% CI 0.75 to 1.21). A forest plot is presented in Appendix 2, Figure 13.
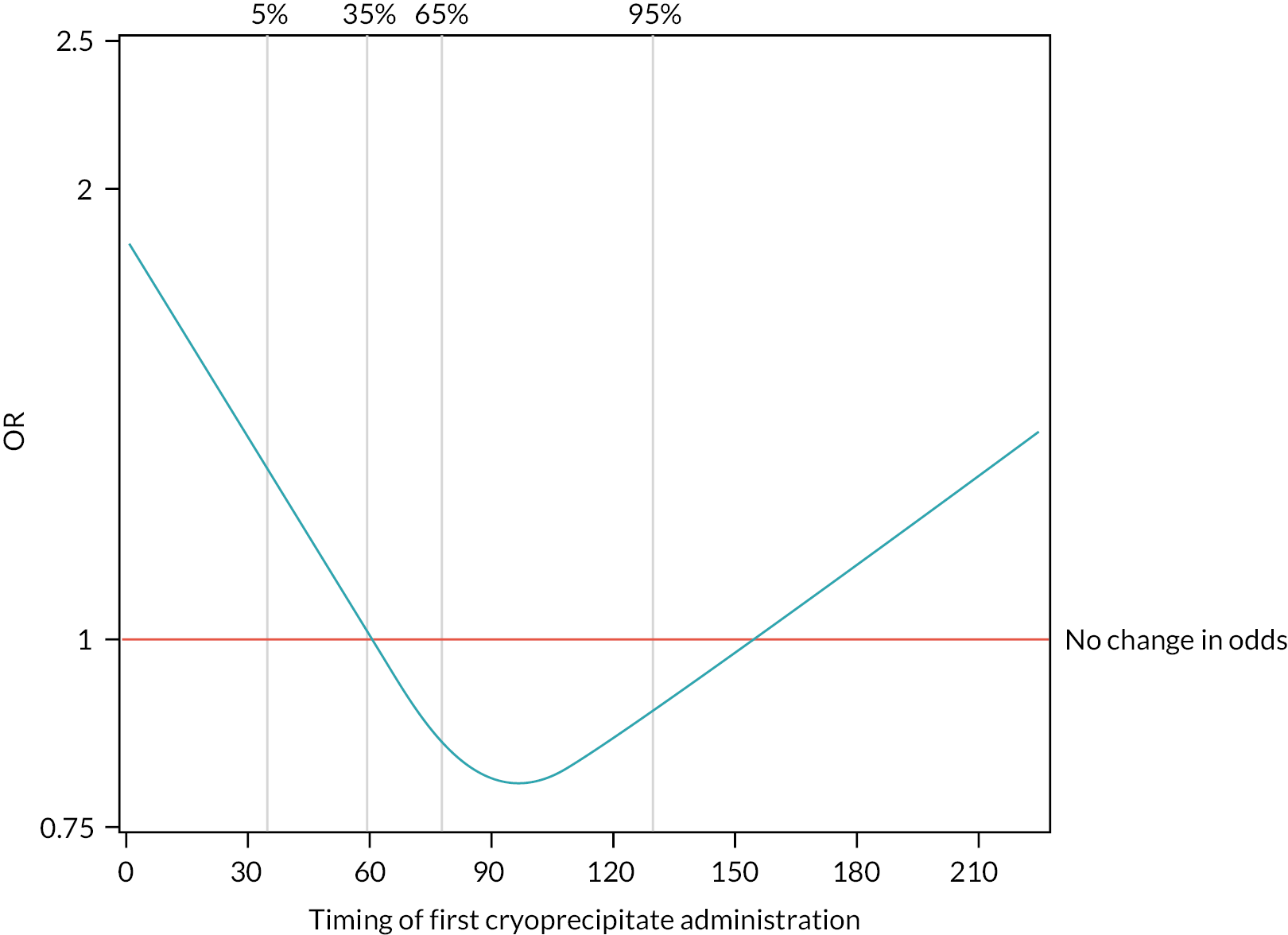
The 28-day mortality rate ranged between 16.5% and 34.4% according to when early cryoprecipitate was given (see Table 5 and Figure 5). Those given early cryoprecipitate 61–90 minutes after admission had significantly lower 28-day mortality than all those in the standard arm (p = 0.0093). Differences between the standard care arm and the other categories of cryoprecipitate timing were non-significant. Figure 6 presents a plot showing the effect of the timing of first cryoprecipitate administration on the odds of 28-day mortality after adjustment for patient factors described in Appendix 2, Table 20. The timing term was fitted as a restricted cubic spline as there was evidence of non-linearity. The plot demonstrates the U-shaped relationship also seen in Table 5.
FIGURE 5.
Kaplan–Meier survival plot up to 28 days from admission by treatment arm and time of first cryoprecipitate dose.
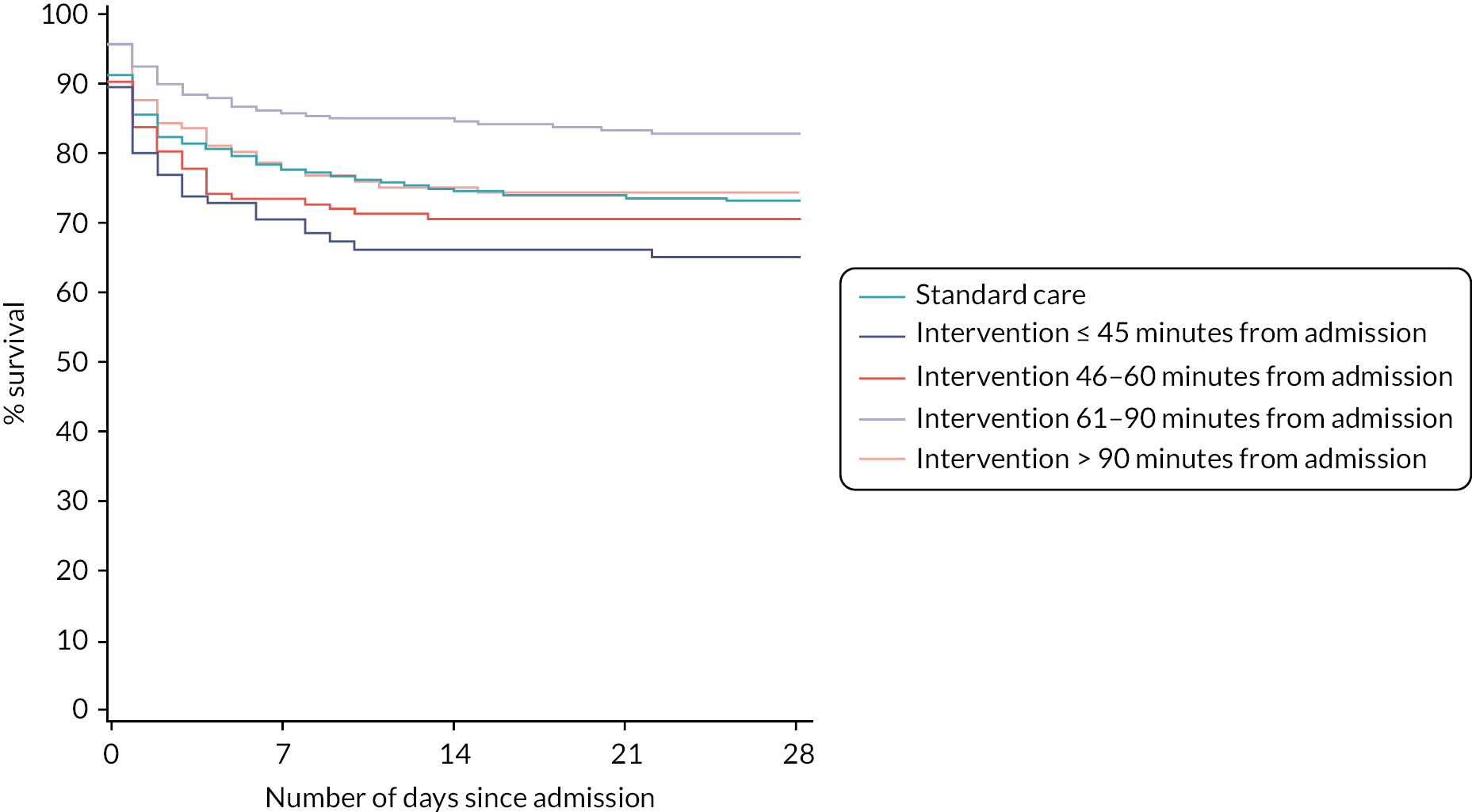
FIGURE 6.
Effect of timing of first cryoprecipitate administration on 28-day mortality, relative to a baseline participant with first cryoprecipitate administration at 60 minutes, after adjustment for Glasgow Coma Scale score, ISS, participant age and systolic blood pressure (vertical lines indicate the distribution of the data and where the knots were applied for the restricted cubic spline).
| Cryoprecipitate administered at | Mortality rate, n/N (%) | Relative risk (95% CI)a | OR (95% CI)a | p-valueb |
|---|---|---|---|---|
| Standard care arm | 201/771 (26.1) | 1.00 | 1.00 | |
| Intervention arm ≤ 45 minutes from admission | 33/96 (34.4) | 1.29 (0.94 to 1.77) | 1.45 (0.91 to 2.31) | 0.1195 |
| Intervention arm 46–60 minutes from admission | 42/144 (29.2) | 1.11 (0.84 to 1.48) | 1.16 (0.78 to 1.73) | 0.4550 |
| Intervention arm 61–90 minutes from admission | 44/267 (16.5) | 0.65 (0.46 to 0.91) | 0.57 (0.38 to 0.87) | 0.0093 |
| Intervention arm > 90 minutes from admission | 31/123 (25.2) | 1.00 (0.71 to 1.41) | 1.00 (0.62 to 1.60) | 0.9870 |
In a post hoc analysis, the baseline characteristics of participants who were given cryoprecipitate at different times were examined (see Appendix 2, Table 18), and those given cryoprecipitate early were found to be more severely injured and shocked on admission. As there was no placebo, there is no equivalent ‘timing’ in the standard arm, and so a comparison can only be made against the whole standard MHP group, rather than with a similarly severely injured subgroup.
Appendix 2, Table 22 presents the 28-day mortality rate for those in the standard care arm compared with those in the intervention arm who did or did not receive early cryoprecipitate. The 28-day mortality rate was 31.7% among those in the intervention arm who did not receive early cryoprecipitate and 24.0% among those who did. There were no statistically significant differences compared with the standard care arm. However, Appendix 2, Figure 14, demonstrates that in the group who did not receive early cryoprecipitate there were many very early deaths, indicating that the higher mortality rate may be because participants died before cryoprecipitate could be administered. This was confirmed when the analysis was repeated on the per-protocol cohort, from which early deaths were excluded (23.3% mortality among those in the intervention arm who did not receive early cryoprecipitate and 23.1% among those who did).
Table 6 presents the cause of death for the 393 participants who died within 28 days. The primary cause of death in both arms was traumatic brain injury (37%), followed by uncontrolled bleeding (22%).
| Cause of death | Standard care arm, n/N (% of those who died) (N = 805) | Intervention arm, n/N (% of those who died) (N = 799) | Overall, n/N (% of those who died) (N = 1604) |
|---|---|---|---|
| Total deaths within 28 days, n/N (% of those randomised) | 201/771 (26) | 192/760 (25) | 393/1531 (26) |
| Multiorgan failure | 26/201 (13) | 21/192 (11) | 47/393 (12) |
| Multiple injury | 38/201 (19) | 31/192 (16) | 69/393 (18) |
| Myocardial infarction | 0/201 (0) | 3/192 (2) | 3/393 (1) |
| Pulmonary embolism | 1/201 (0) | 0/192 (0) | 1/393 (0) |
| Sepsis | 4/201 (2) | 0/192 (0) | 4/393 (1) |
| Stroke | 0/201 (0) | 5/192 (3) | 5/393 (1) |
| Traumatic brain injury | 78/201 (39) | 67/192 (35) | 145/393 (37) |
| Uncontrolled bleeding | 41/201 (20) | 46/192 (24) | 87/393 (22) |
| Other | 13/201 (6) | 19/192 (10) | 32/393 (8) |
| All | 201/201 (100) | 192/192 (100) | 393/393 (100) |
Subgroup analysis
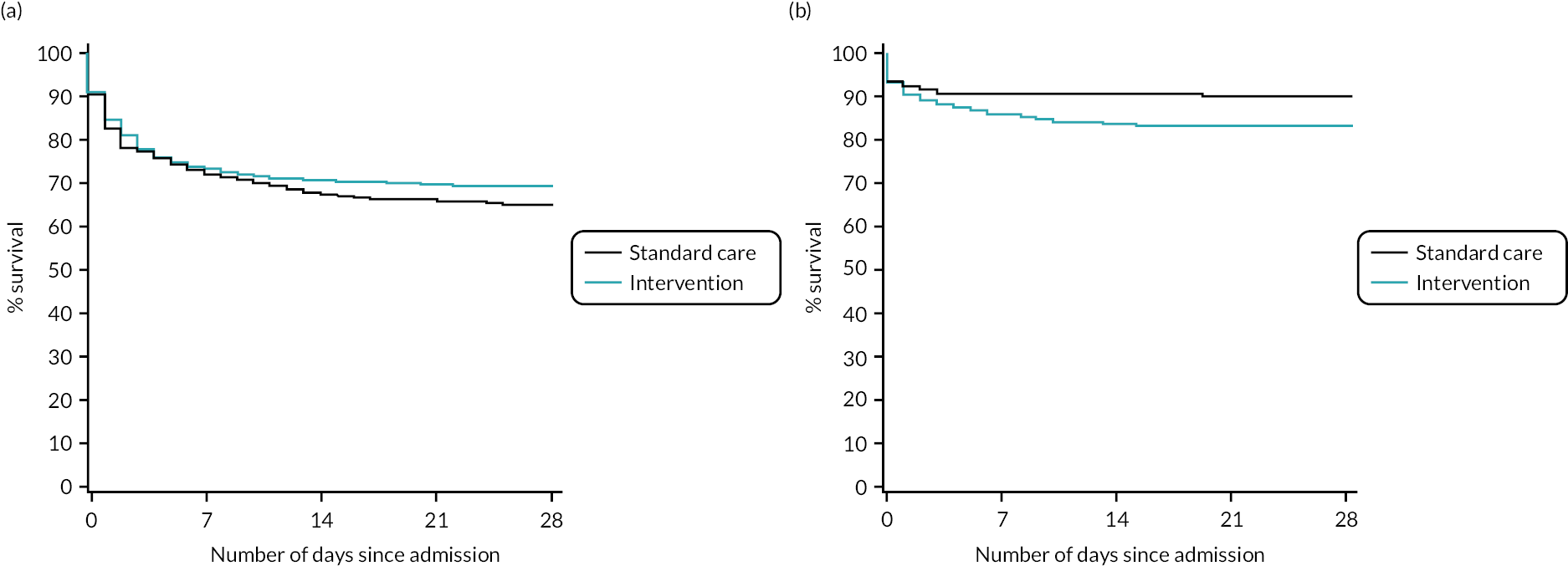
There was no evidence of a differential effect of early cryoprecipitate on 28-day mortality for four of the five prespecified subgroup analyses: UK versus non-UK participants, head AIS < 4 versus ≥ 4, participant sex, and participant age < 70 versus ≥ 70 years (see Appendix 2, Tables 23–26 and Figures 15–18). However, there was evidence of a differential effect according to whether the participant’s injury was blunt or penetrating (see Table 7 and Figure 7). For penetrating injuries, early cryoprecipitate increased the odds of death compared with those who received only the standard MHP (OR 1.74, 95% CI 1.20 to 2.51). For blunt injuries, early cryoprecipitate decreased the odds of death, but this was not statistically significant (OR 0.82, 95% CI 0.62 to 1.09). There were no apparent differences in baseline characteristics between the two subgroups (see Appendix 2, Table 19). Head AIS < 4 versus ≥ 4 also did not have a differential effect on 6- or 24-hour mortality. Subgroup analysis results are also presented in the forest plot in Appendix 2, Figure 13.
FIGURE 7.
Kaplan–Meier survival plots up to 28 days from admission by treatment arm for (a) blunt and (b) penetrating injuries.
| Outcome | Blunt | Penetrating | ||
|---|---|---|---|---|
| Standard care arm (N = 519) | Intervention arm (N = 495) | Standard care arm (N = 277) | Intervention arm (N = 290) | |
| Participants who died on or before day 28 from admission, n/N (%) | 174/500 (34.8) | 147/483 (30.4) | 27/271 (10.0) | 45/277 (16.2) |
| Relative riska (95% CI) | 0.88 (0.72 to 1.06) | 1.62 (1.17 to 2.23) | ||
| ORa (95% CI) | 0.82 (0.62 to 1.09) | 1.74 (1.20 to 2.51) | ||
| p-value for subgroupa | 0.1634 | 0.0058 | ||
| p-value for interaction termb | 0.0040 | |||
| Participants for whom 28-day vital status was not available from any source, n/N (%) | 19/519 (3.7) | 12/495 (2.4) | 6/277 (2.2) | 13/290 (4.5) |
Secondary outcomes
Tables and figures relating to secondary outcomes are presented in Appendix 2 or below. All results relate to the ITT analysis. Per-protocol analysis of certain secondary outcomes was planned in the protocol, and for these outcomes the results were very consistent with those of the ITT analysis.
Mortality data
There were no statistically significant differences between arms in all-cause mortality and deaths from bleeding at 6 and 24 hours (see Appendix 2, Table 27). The median (IQR) time to death from bleeding among those who bled was 191 (81–445) and 86 (40–205) minutes in the intervention and standard care arms, respectively.
Figure 8 presents an unadjusted Kaplan–Meier plot of survival to 12 months by treatment arm. The estimated mortality rate at 6 months (using the Kaplan–Meier method) was 26.1% for the intervention arm and 27.3% for the standard care arm (see Appendix 2, Table 31). This gave a hazard ratio of 0.96 (95% CI 0.79 to 1.17) for early cryoprecipitate compared with standard MHP and a p-value of 0.6748 for the difference between the arms. After risk adjustment for statistically significant patient factors (Glasgow Coma Scale score, ISS, age, systolic blood pressure and sex), the hazard ratio was 1.08 (95% CI 0.89 to 1.32).
FIGURE 8.
Kaplan–Meier survival plot up to 12 months from admission by treatment arm: ITT.
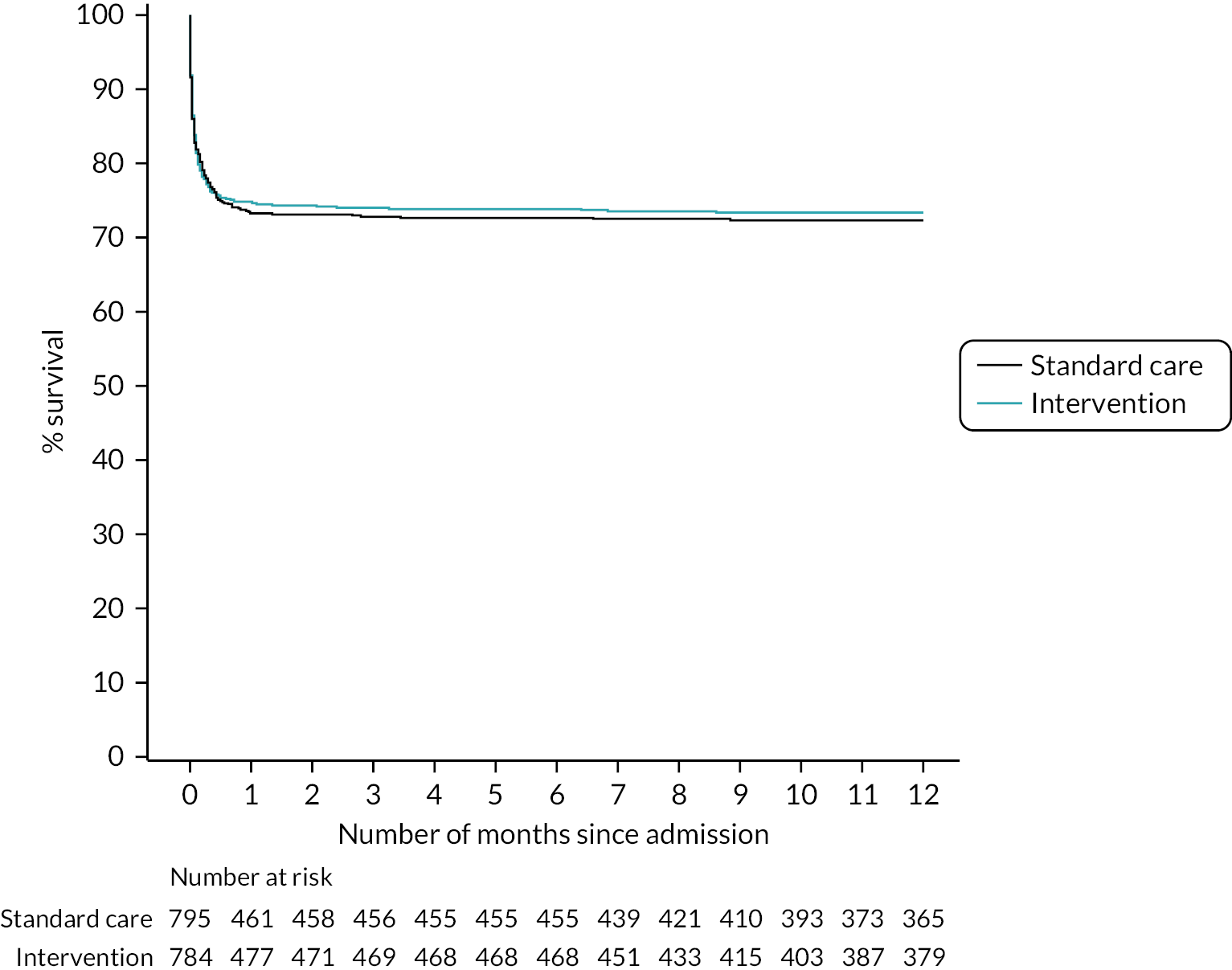
The estimated mortality rate at 12 months was 26.6% for the intervention arm and 27.7% for the standard care arm (see Appendix 2, Table 31). The primary analysis gave the same hazard ratio as at 6 months, with a p-value of 0.7120 for the difference between arms. After risk adjustment for the same statistically significant patient factors, the hazard ratio was 1.09 (95% CI 0.90 to 1.32).
Transfusion requirements
Participants in the intervention arm received more cryoprecipitate per hour from injury to 24 hours (p < 0.0001), but the use of other blood components was similar in the two arms (see Appendix 2, Table 28 and Figures 19 and 20).
Quality of life
Data completeness for the EQ-5D-5L was poor at discharge (59%) and at 6 months (11%). The median index value at discharge was 0.50 in the standard care arm and 0.51 in the intervention arm, and the median self-evaluated health score at discharge was 60 in the standard care arm and 50 in the intervention arm; the difference in medians was not statistically significant for either measure. There was also no statistically significant difference in GOS at discharge between the two arms. At 6 months, the median index value was 0.66 in the standard care arm and 0.76 in the intervention arm and the median self-evaluated health score was 65 in the standard care arm and 75 in the intervention arm (see Appendix 2, Table 29).
Hospital resource use and destination of participant at time of discharge from hospital
There was no statistically significant difference between arms in median ventilator-days, critical care stay or hospital stay (see Appendix 2, Table 30). Forty-nine per cent of participants in each arm were discharged before the end of the study, and the discharge destinations of participants in the two arms were very similar.
Adverse events
The numbers of thromboembolic events and arterial thromboembolic events were similar in the two treatment arms (Table 8). There were three serious transfusion-related adverse events, which all occurred in the intervention arm. Two were anaphylaxis (one reported as unlikely to be related to the intervention and the other reported to be possibly related) and one serious adverse event was reported as potential transfusion-associated circulatory overload.
| Outcome | Standard care arm (N = 805) | Intervention arm (N = 799) | Overall (n = 1604) | p-value |
|---|---|---|---|---|
| Thrombotic events, n | 85 | 85 | 170 | |
| Venous thromboembolism | 59 | 58 | 117 | |
| Pulmonary embolus | 36 | 38 | 74 | |
| Deep-vein thrombosis | 23 | 20 | 43 | |
| Participants affected, n/N (%) | 57/805 (7.1) | 55/799 (6.9) | 112/1604 (7.0) | |
| Arterial thrombotic events | 26 | 27 | 53 | |
| Myocardial infarction | 4 | 4 | 8 | |
| Stroke | 11 | 11 | 22 | |
| Other occlusion of any other artery | 11 | 12 | 23 | |
| Participants affected, n/N (%) | 26/805 (3.2) | 26/799 (3.3) | 52/1604 (3.2) | |
| Cumulative incidence of thrombotic events at day 28, % (95% CI)a | 12.9 (10.2 to 15.8) | 12.7 (10.1 to 15.6) | 12.8 (10.9 to 14.8) | 0.8852 |
| Serious transfusion-related adverse events, n/N (%)b | 0/805 (0) | 3/799 (0.4) | 3/1604 (0.2) | 0.1234 |
Chapter 4 Economic evaluation
Aim
The aim of this study was to evaluate the cost-effectiveness of early high-dose cryoprecipitate versus standard-of-care in adult patients with major trauma haemorrhage requiring MHP activation.
Methods
Overview of economic evaluation
We undertook a cost–utility analysis to estimate the within-trial cost-effectiveness of early high-dose cryoprecipitate versus standard-of-care in adult patients with major trauma haemorrhage requiring MHP activation at 28 days following randomisation. The analysis was performed using individual patient-level data from the CRYOSTAT-2 trial. The sample size for the economic analysis was 1581 patients. The outcome measure was the quality-adjusted life-year (QALY), which combines length of life and quality of life, based on the National Institute for Health and Care Excellence (NICE) recommendations. 34 Cost-effectiveness was expressed as the incremental cost per QALY gained based on the differences in costs and QALYs between the intervention and standard care arms. The trial was conducted in the UK (25 sites) and the USA (1 site); data from all participants in the study were included, from the UK and the USA. The perspective for the economic analysis was the UK NHS and Personal Social Services. Costs were calculated in 2022–3 Great British pounds and inflated where necessary using the Consumer Price Index. 35 The time horizon was 28 days. Extrapolation beyond the end of the trial was not undertaken because the within-trial analysis found no evidence of significant differences in costs or benefits between the intervention and standard care arms; 28 days was long enough to reflect all important differences in costs or outcomes between treatments with early high-dose cryoprecipitate and standard of care. Given the time horizon, discounting was not applied to costs or outcomes.
Resource use and costs
For every patient we calculated costs up to 28 days after randomisation. We included the following items of healthcare resource use that a priori were identified as potentially differing between the two treatment arms:
-
length of hospital stay
-
number of days in the ICU
-
number of days in the high-dependency unit (HDU)
-
number of days on an inpatient ward
-
-
use of blood products
-
use of RBCs
-
use of FFP
-
use of cryoprecipitate
-
use of platelets
-
use of colloids
-
use of crystalloids.
-
Resource use data were collected at 28 days post randomisation using trial CRFs, completed by the trial nurses and patients. The CRF used to collect these data, which are included in Report Supplementary Material 3. 36–39
To calculate length of stay in each hospital unit (ICU, HDU, ward), we used the date of admission to that unit and the following date of admission to another unit, or death or discharge. If the patient was not admitted to another unit, or had died or been discharged by the 28th day after randomisation, we assumed that they remained in the latest reported unit until the 28th day. In cases where the patient was known to have been discharged by the 28th day but the discharge date was missing or the duration of stay in a unit was unknown, we assumed that the length of stay in that unit was the mean length of stay observed across all patients. Length of stay after randomisation for both the initial hospitalisation and any re-admissions were included provided that these occurred in the 28 days after randomisation. All length-of-stay calculations were up to 28 days; therefore, any hospital stays beyond this time were not included, even if the patient had not been discharged by 28 days.
Unit costs were obtained from published sources and inflated where appropriate35–38 (Table 9). For length of hospital stay, we multiplied the daily cost by the number of days spent in each type of unit/ward. For blood products, we multiplied the number of units of each product received by the cost per unit.
| Cost component | Unit | Unit cost (£, 2022–3) | Source of unit cost data |
|---|---|---|---|
| Blood products | |||
| RBC | Per unit of product | 153.30 | NHS Blood and Transplant37 |
| FFP | Per unit of product | 42.59 | NHS Blood and Transplant37 |
| Cryoprecipitate | Per pooled bag | 221.85 | NHS Blood and Transplant37 |
| Platelets | Per unit of product | 240.90 | NHS Blood and Transplant37 |
| Colloids | Per 500 ml of product | 3.44 | Campbell et al.38 |
| Crystalloids | Per 500 ml of product | 0.92 | Campbell et al.38 |
| Hospital stay | |||
| ICU | Per day | 1838 | Walsh et al.,33 NICE,34 ONS,35 Agus et al.,36 Massou and Morris (personal communication) |
| HDU | Per day | 1189 | Walsh et al.,33 NICE,34 ONS,35 Agus et al.,36 Massou and Morris (personal communication) |
| Ward | Per day | 557 | Walsh et al.,33 NICE,34 ONS,35 Agus et al.,36 Massou and Morris (personal communication) |
Utilities and quality-adjusted life-years
Generic health status was described at 28 days post randomisation using the EQ-5D-5L descriptive system. 40 The EQ-5D-5L contains five dimensions: mobility; self-care; usual activities; pain and discomfort; and anxiety and depression. For each dimension of the EQ-5D-5L, there are five levels (no problems, slight problems, moderate problems, severe problems and unable to/extreme problems). 33 As patients recruited to the trial were initially critically ill following major trauma, completion of the EQ-5D-5L at randomisation was not possible. Therefore, baseline utility was assumed to be zero for all patients. This assumption was made in previous studies in which baseline utility measurement was not possible. 41 EQ-5D-5L health states were converted into utility values using a formula that attaches weights to each level in each dimension based on valuations by UK general population samples;42 this formula was also used for the small proportion of US trial participants. Utility values of one represent full health, values of zero are equivalent to death and negative values represent states worse than death. Patients who died during the 28-day follow-up period were assigned a utility value of zero at 28 days. A utility profile was constructed for every patient, assuming a straight-line relation between their utility values at randomisation (assumed to be zero) and 28 days (measured). QALYs for every patient from baseline to 28 days were calculated as the area under the utility profile. The implications of our assumptions were therefore as follows. For those patients who survived up to 28 days, QALYs up to 28 days were calculated using the EQ-5D-5L scores measured at 28 days, assuming an EQ-5D-5L score of zero at randomisation, and a linear interpolation between randomisation and 28 days. For decedents between randomisation and 28 days, we assumed zero QALYs. We assigned all the EQ-5D-5D data that were collected to the 28-day measurement point, irrespective of the precise time when this was actually measured.
Statistical methods
We addressed missing values of the volume of blood products received and EQ-5D-5L scores at 28 days using multiple imputation for each treatment arm separately. 43,44 Patient age, sex, injury type (blunt/penetrating), early receipt of tranexamic acid, ISS and Glasgow Coma Scale were used in the multiple imputation models as additional explanatory variables. We used an iterative Markov chain Monte Carlo procedure based on multivariate normal regression, generating 20 imputed data sets.
We investigated the incremental costs and incremental effectiveness of the intervention using regression analysis based on the participant-level costs and outcomes data. We regressed costs, separately for the costs of hospital stay and cost of blood products, and all cost components combined, against an indicator variable for the intervention arm using generalised linear model regression (gamma family and log link) to account for the skewness of the cost data. 45 We regressed QALYs (with and without imputation) against an indicator variable for the intervention arm using ordinary least squares regression. In these regression models, we did not adjust for covariates, and we report the coefficient and marginal effect of the intervention arm (for the analysis of QALYs gained the marginal effect is the coefficient).
The incremental cost per QALY gained was calculated as the incremental cost divided by the QALYs gained; this was computed using the results from the regression analyses as the marginal effect of the coefficient on the indicator variable for the intervention arm in the cost regression model divided by the coefficient on the indicator variable for the intervention arm in the QALY regression model.
We used the ‘multiple imputation nested within bootstrapping’ approach suggested by Brand et al. 44 We calculated the mean value for each observation across the 20 imputed data sets and ran the regression analyses described below on this data set. We then reran this analysis 1000 times, using non-parametric bootstrapping, resampling the mean values with replacement.
We repeated the above process investigating cost-effectiveness for subgroups and participants differentiated by sex, age (< 70 years vs. ≥ 70 years) and injury type (blunt vs. penetrating).
We created a scatterplot to display the 1000 bootstrapped replications using the incremental QALYs with imputation. A cost-effectiveness acceptability curve showing the probability that early cryoprecipitate would be cost-effective compared with standard care at a range of values for the maximum willingness to pay for a QALY was generated based on the proportion of these 1000 bootstrap replications with positive incremental net monetary benefits (calculated as the mean incremental QALYs per patient with early cryoprecipitate multiplied by the maximum willingness to pay for a QALY minus the mean incremental cost per patient with early cryoprecipitate).
Results
Resource use and costs
In our sample, the overall mean length of stay in the ICU across all patients was 6.5 days [standard deviation (SD) 8.5 days] and the median was 3 days (IQR 0–10 days) (Table 10). The overall mean length of stay in the HDU was 1.8 days (SD 3.5 days) and the median was 0 days (IQR 0–3 days). The mean length of stay on the ward overall was 5.8 days (SD 7.0 days) and the median was 3 days (IQR 0–10 days). The mean and median total lengths of stay overall across all patients were 14.1 days (SD 11.0 days) and 12 days (IQR 3–28 days), respectively. All length of hospital stay values were similar in the intervention and standard care arms.
| Obs | Mean | SD | Median | IQR | Minimum | Maximum | |
|---|---|---|---|---|---|---|---|
| Hospital stay | |||||||
| ICU (days) | |||||||
| Overall | 1581 | 6.5 | 8.5 | 3 | 0–10 | 0 | 28 |
| Intervention arm | 785 | 6.4 | 8.5 | 3 | 0–9 | 0 | 28 |
| Standard care arm | 796 | 6.6 | 8.6 | 3 | 0–10 | 0 | 28 |
| HDU (days) | |||||||
| Overall | 1581 | 1.8 | 3.5 | 0 | 0–3 | 0 | 28 |
| Intervention arm | 785 | 1.9 | 3.7 | 0 | 0–3 | 0 | 28 |
| Standard care arm | 796 | 1.7 | 3.4 | 0 | 0–3 | 0 | 24 |
| Ward (days) | |||||||
| Overall | 1581 | 5.8 | 7.0 | 3 | 0–10 | 0 | 28 |
| Intervention arm | 785 | 5.7 | 7.0 | 3 | 0–10 | 0 | 28 |
| Standard care arm | 796 | 5.8 | 7.1 | 3 | 0–10 | 0 | 28 |
| Total (days) | |||||||
| Overall | 1581 | 14.1 | 11.0 | 12 | 3–28 | 0 | 28 |
| Intervention arm | 785 | 14.1 | 10.9 | 12 | 4–28 | 0 | 28 |
| Standard care arm | 796 | 14.2 | 11.1 | 13 | 3–28 | 0 | 28 |
| Blood products | |||||||
| RBC (units) | |||||||
| Overall | 1577 | 6.5 | 7.7 | 4 | 2–8 | 0 | 92 |
| Intervention arm | 784 | 6.6 | 7.7 | 4 | 2–8 | 0 | 52 |
| Standard care arm | 793 | 6.4 | 7.7 | 4 | 2–7 | 0 | 92 |
| FFP (units) | |||||||
| All patients | 1577 | 5.4 | 6.9 | 4 | 2–7 | 0 | 78 |
| Intervention arm | 784 | 5.5 | 7.0 | 3 | 2–7 | 0 | 52 |
| Standard care arm | 793 | 5.3 | 6.8 | 4 | 2–7 | 0 | 78 |
| Cryoprecipitate (pooled bags) | |||||||
| Overall | 1577 | 2.0 | 2.5 | 3 | 0–3 | 0 | 22 |
| Intervention arm | 784 | 3.1 | 2.4 | 3 | 3–3 | 0 | 22 |
| Standard care arm | 793 | 1.0 | 2.0 | 0 | 0–2 | 0 | 22 |
| Platelets (units) | |||||||
| Overall | 1577 | 0.8 | 1.5 | 0 | 0–1 | 0 | 19 |
| Intervention arm | 784 | 0.8 | 2.0 | 0 | 0–1 | 0 | 16 |
| Standard care arm | 793 | 0.8 | 1.6 | 0 | 0–1 | 0 | 19 |
| Crystalloids (ml) | |||||||
| Overall | 1561 | 2108 | 2171 | 1506 | 100–3100 | 0 | 14,905 |
| Intervention arm | 775 | 2167 | 2087 | 1778 | 500–3142 | 0 | 14,905 |
| Standard care arm | 786 | 2021 | 2235 | 1473 | 0–3000 | 0 | 14,250 |
| Colloids (ml) | |||||||
| Overall | 1561 | 99 | 521 | 0 | 0–0 | 0 | 11,500 |
| Intervention arm | 775 | 95 | 541 | 0 | 0–0 | 0 | 11,500 |
| Standard care arm | 786 | 103 | 500 | 0 | 0–0 | 0 | 8000 |
In terms of the volume of blood products received, the mean and median number of RBC units overall across all patients were 6.5 (SD 7.7) and 4 (IQR 2–8), respectively. The mean and median number of FFP units overall were 5.4 (SD 6.9) and 4 (IQR 2–7), respectively. For cryoprecipitate these values were 2.0 (SD 2.5) and 3 (IQR 0–3), respectively. For platelets they were 0.8 (SD 1.5) and 0 (IQR 0–1), respectively. For crystalloids the mean and median volumes in millilitres overall were 2108 (SD 2171) and 1506 (IQR 100–3100), respectively. For colloids they were 99 (SD 521) and 0 (IQR 0–0), respectively. All blood product values were similar for the intervention and standard care arms, with the exception of cryoprecipitate, where the mean and median values were 3.1 (SD 2.4) and 3 (IQR 3–3) in the intervention arm and 1.0 (SD 2.0) and 0 (IQR 0–2) in the standard care arm.
The mean total cost per patient (including the costs of length of hospital stay and blood products) was £19,477 (SD £16,207) in the intervention arm and £19,236 (SD £16,681) in the standard care arm (Table 11). In both arms, approximately 90% of the total cost was accounted for by the cost of hospital stay, and 10% by the cost of blood products. ICU costs accounted for most of the cost of hospital stay, accounting for approximately 70% of the cost of hospital stay and 62% of the total cost.
| Obs | Mean | SD | Minimum | Maximum | |
|---|---|---|---|---|---|
| Hospital stay | |||||
| ICU (£) | |||||
| Overall | 1581 | 12,029 | 15,789 | 0 | 51,479 |
| Intervention arm | 785 | 11,799 | 15,700 | 0 | 51,479 |
| Standard care arm | 796 | 12,255 | 15,883 | 0 | 51,479 |
| HDU (£) | |||||
| Overall | 1581 | 2192 | 4256 | 0 | 33,318 |
| Intervention arm | 785 | 2276 | 4417 | 0 | 33,318 |
| Standard care arm | 796 | 2109 | 4091 | 0 | 33,318 |
| Ward (£) | |||||
| Overall | 1581 | 3241 | 3959 | 0 | 15,622 |
| Intervention arm | 785 | 3230 | 3937 | 0 | 15,622 |
| Standard care arm | 796 | 3251 | 3983 | 0 | 15,622 |
| Total (£) | |||||
| Overall | 1581 | 17,462 | 16,241 | 0 | 51,479 |
| Intervention arm | 785 | 17,306 | 16,062 | 0 | 51,479 |
| Standard care arm | 796 | 17,616 | 16,425 | 0 | 51,479 |
| Blood products | |||||
| RBC (£) | |||||
| Overall | 1577 | 999 | 1178 | 0 | 14,103 |
| Intervention arm | 784 | 1017 | 1176 | 0 | 7971 |
| Standard care arm | 793 | 982 | 1181 | 0 | 14,103 |
| FFP (£) | |||||
| Overall | 1577 | 232 | 293 | 0 | 3322 |
| Intervention arm | 784 | 235 | 298 | 0 | 2214 |
| Standard care arm | 793 | 229 | 287 | 0 | 3322 |
| Cryoprecipitate (£) | |||||
| Overall | 1577 | 454 | 543 | 0 | 48,807 |
| Intervention arm | 784 | 690 | 526 | 0 | 4880 |
| Standard care arm | 793 | 221 | 452 | 0 | 4880 |
| Platelets (£) | |||||
| Overall | 1577 | 198 | 365 | 0 | 4577 |
| Intervention arm | 784 | 200 | 368 | 0 | 3854 |
| Standard care arm | 793 | 196 | 362 | 0 | 4577 |
| Crystalloids (£) | |||||
| Overall | 1561 | 4 | 4 | 0 | 27 |
| Intervention arm | 775 | 4 | 4 | 0 | 27 |
| Standard care arm | 786 | 4 | 4 | 0 | 26 |
| Colloids (£) | |||||
| Overall | 1561 | 1 | 3 | 0 | 79 |
| Intervention arm | 775 | 1 | 3 | 0 | 79 |
| Standard care arm | 786 | 1 | 3 | 0 | 55 |
| Total (£) | |||||
| Overall | 1578 | 1888 | 2127 | 0 | 21,937 |
| Intervention arm | 784 | 2148 | 2140 | 0 | 16,259 |
| Standard care arm | 794 | 1632 | 2083 | 0 | 21,937 |
| Total costs (£) | |||||
| Hospital stay plus blood products | |||||
| Overall | 1578 | 19,356 | 16,443 | 0 | 64,658 |
| Intervention arm | 784 | 19,477 | 16,207 | 0 | 64,658 |
| Standard care arm | 794 | 19,236 | 16,681 | 0 | 61,333 |
Utilities and quality-adjusted life-years
Mean EQ-5D-5L utility scores at 28 days overall across all patients were 0.294 (SD 0.339) (Table 12). Mean QALYs overall were 0.011 (SD 0.013) with no imputation and 0.012 (SD 0.013) with imputation. Both mean EQ-5D-5L utility scores at 28 days and mean QALYs were similar in the intervention and standard care arms.
| Obs | Mean | SD | Minimum | Maximum | |
|---|---|---|---|---|---|
| EQ-5D-5L utility scores at 28 days | |||||
| Overall | 1061 | 0.294 | 0.339 | −0.285 | 1.000 |
| Intervention arm | 516 | 0.293 | 0.337 | −0.285 | 1.000 |
| Standard care arm | 545 | 0.294 | 0.341 | −0.285 | 1.000 |
| QALYs | |||||
| QALYs (no imputation) | |||||
| Overall | 1061 | 0.011 | 0.013 | −0.010 | 0.038 |
| Intervention arm | 516 | 0.011 | 0.012 | −0.010 | 0.038 |
| Standard care arm | 545 | 0.011 | 0.013 | −0.010 | 0.038 |
| QALYs (with imputation) | |||||
| Overall | 1581 | 0.012 | 0.013 | −0.011 | 0.038 |
| Intervention arm | 785 | 0.012 | 0.013 | −0.011 | 0.038 |
| Standard care arm | 796 | 0.013 | 0.013 | −0.010 | 0.038 |
Incremental cost–utility analysis
The results of the regression analyses for costs across the whole sample indicate that there were significant differences in the cost of blood products between the intervention and standard care arms (costs were £519 higher in the intervention arm; p < 0.0001; Table 13). The differences between the costs associated with the length of hospital stay and the total costs were not statistically significant in either of the two arms (both p > 0.7). The results of the regression analyses for QALYs (without and with imputation) indicate that the between-arm differences in QALYs were small (both QALYs gained < 0.0002) and not statistically significant (both p > 0.7).
| Coefficient | 95% CI | p-value | Marginal effect | |
|---|---|---|---|---|
| Total cost of hospital stay | ||||
| Intervention arm | −0.017 | −0.109 to 0.073 | 0.705 | −310 |
| Total cost for blood products | ||||
| Intervention arm | 0.276 | 0.163 to 0.389 | < 0.0001 | 519 |
| Total cost | ||||
| Intervention arm | 0.011 | −0.074 to 0.096 | 0.803 | 210 |
| QALYs (without imputation) | ||||
| Intervention arm | 0.00004 | −0.0016 to 0.0015 | 0.848 | |
| QALYs (with imputation) | ||||
| Intervention arm | 0.0005 | −0.002 to 0.008 | 0.455 | |
The incremental cost per QALY gained for intervention versus standard care is £1,121,509 based on the QALYs gained calculated without imputation and £1,348,181 based on the QALYs gained calculated with imputation (numbers may not be directly calculated from the values in Table 13 due to rounding). The high, unattractive incremental cost-effectiveness ratios associated with early cryoprecipitate compared with standard MHP are due to the higher cost of blood products that is not offset by lower costs elsewhere, and the negligible QALY gains/losses.
In terms of the subgroup analyses (Tables 14–16), the regression analyses were largely qualitatively the same as for the whole sample: the cost of blood products was significantly higher for the intervention arm; for all other cost variables and the QALY variables there were no significant differences between early cryoprecipitate and standard MHP, and the QALYs gained were negligible in magnitude, making the interpretation of the incremental cost per QALY gained difficult.
| Sex | Coefficient | 95% CI | p-value | Marginal effect | |
|---|---|---|---|---|---|
| Female (n = 330) | Total cost of hospital stay | ||||
| Intervention arm | −0.050 | −0.237 to 0.137 | 0.600 | −864 | |
| Total cost for blood products | |||||
| Intervention arm | 0.201 | −0.031 to 0.432 | 0.089 | 365 | |
| Total cost | |||||
| Intervention arm | −0.026 | −0.204 to 0.152 | 0.773 | −500 | |
| QALYs (without imputation) a | |||||
| Intervention arm | 0.0003 | −0.0026 to 0.0031 | 0.862 | ||
| QALYs (with imputation) | |||||
| Intervention arm | 0.001 | −0.001 to 0.004 | 0.361 | ||
| Male (n = 1251) | Total cost of hospital stay | ||||
| Intervention arm | −0.009 | −0.116 to 0.098 | 0.866 | −161 | |
| Total cost for blood products | |||||
| Intervention arm | 0.296 | 0.166 to 0.455 | < 0.01 | 560 | |
| Total cost | |||||
| Intervention arm | 0.206 | −0.070 to 0.111 | 0.656 | 399 | |
| QALYs (without imputation) b | |||||
| Intervention arm | −0.0001 | −0.0019 to 0.0017 | 0.912 | ||
| QALYs (with imputation) | |||||
| Intervention arm | −0.001 | −0.002 to 0.001 | 0.238 | ||
| Ιnjury type | Coefficient | 95% CI | p-value | Marginal effect | |
|---|---|---|---|---|---|
| Blunt (n = 1014) | Total cost of hospital stay | ||||
| Intervention arm | 0.015 | 0.089 to 0.119 | 0.779 | 308 | |
| Total cost for blood products | |||||
| Intervention arm | 0.178 | 0.034 to 0.322 | 0.015 | 325 | |
| Total cost | |||||
| Intervention arm | 0.028 | −0.067 to 0.123 | 0.560 | 633 | |
| QALYs (without imputation) a | |||||
| Intervention arm | 0.006 | −0.001 to 0.002 | 0.498 | ||
| QALYs (with imputation) | |||||
| Intervention arm | 0.003 | −0.001 to 0.002 | 0.676 | ||
| Penetrating (n = 567) | Total cost of hospital stay | ||||
| Intervention arm | −0.075 | −0.116 to 0.098 | 0.866 | −874 | |
| Total cost for blood products | |||||
| Intervention arm | 0.437 | 0.254 to 0.620 | < 0.01 | 857 | |
| Total cost | |||||
| Intervention arm | −0.001 | −0.168 to 0.166 | 0.988 | −17 | |
| QALYs (without imputation) b | |||||
| Intervention arm | −0.003 | −0.006 to 0.000 | 0.082 | ||
| QALYs (with imputation) | |||||
| Intervention arm | −0.003 | −0.005 to −0.001 | 0.023 | ||
| Age group | Coefficient | 95% CI | p-value | Marginal effect | |
|---|---|---|---|---|---|
| < 70 years (n = 1414) | Total cost of hospital stay | ||||
| Intervention arm | −0.037 | −0.133 to 0.058 | 0.441 | −668 | |
| Total cost for blood products | |||||
| Intervention arm | 0.283 | 0.160 to 0.406 | < 0.01 | 543 | |
| Total cost | |||||
| Intervention arm | −0.006 | −0.092 to 0.079 | 0.866 | −124 | |
| QALYs (without imputation) a | |||||
| Intervention arm | 0.0003 | −0.002 to 0.001 | 0.752 | ||
| QALYs (with imputation) | |||||
| Intervention arm | −0.001 | −0.002 to 0.001 | 0.247 | ||
| ≥ 70 years (n = 167) | Total cost of hospital stay | ||||
| Intervention arm | 0.143 | −0.188 to 0.473 | 0.397 | 2007 | |
| Total cost for blood products | |||||
| Intervention arm | 0.156 | −0.128 to 0.441 | 0.281 | 236 | |
| Total cost | |||||
| Intervention arm | 0.144 | −0.164 to 0.451 | 0.359 | 2242 | |
| QALYs (without imputation) b | |||||
| Intervention arm | −0.0003 | −0.003 to 0.002 | 0.848 | ||
| QALYs (with imputation) | |||||
| Intervention arm | 0.0004 | −0.003 to 0.003 | 0.819 | ||
The scatterplot showing the bootstrap replications is shown in Figure 9. The cost-effectiveness acceptability curve is shown in Figure 10. This shows that the probability that early cryoprecipitate is cost-effective versus standard MHP is just under 0.4 across a range of plausible values of the cost-effectiveness threshold.
FIGURE 9.
Scatterplot showing 1000 bootstrap replications of the incremental costs and QALYs associated with early cryoprecipitate vs. standard MHP.
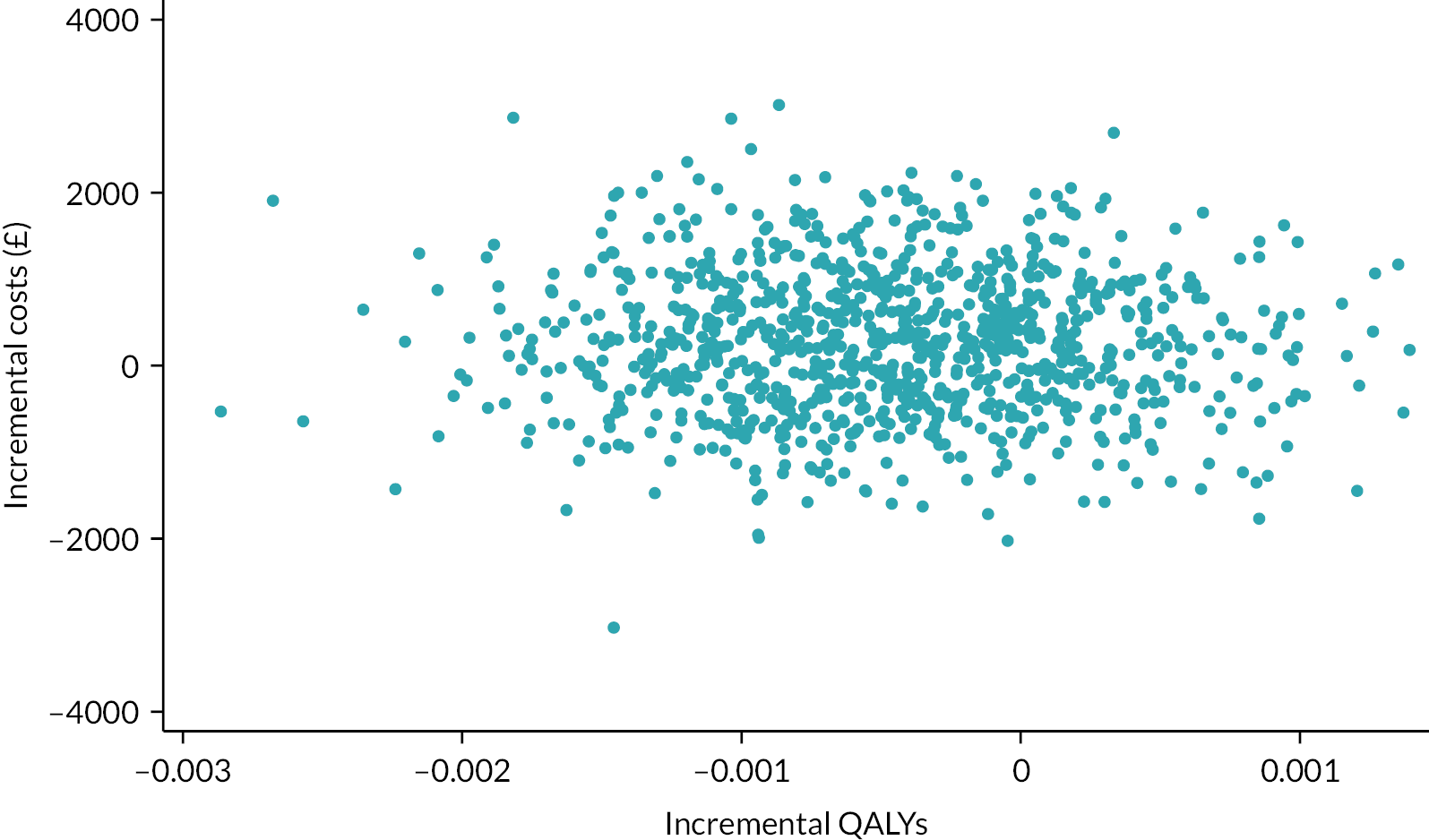
FIGURE 10.
Cost-effectiveness acceptability curve showing the probability that early cryoprecipitate is cost-effective vs. standard MHP for a range of cost-effectiveness thresholds.
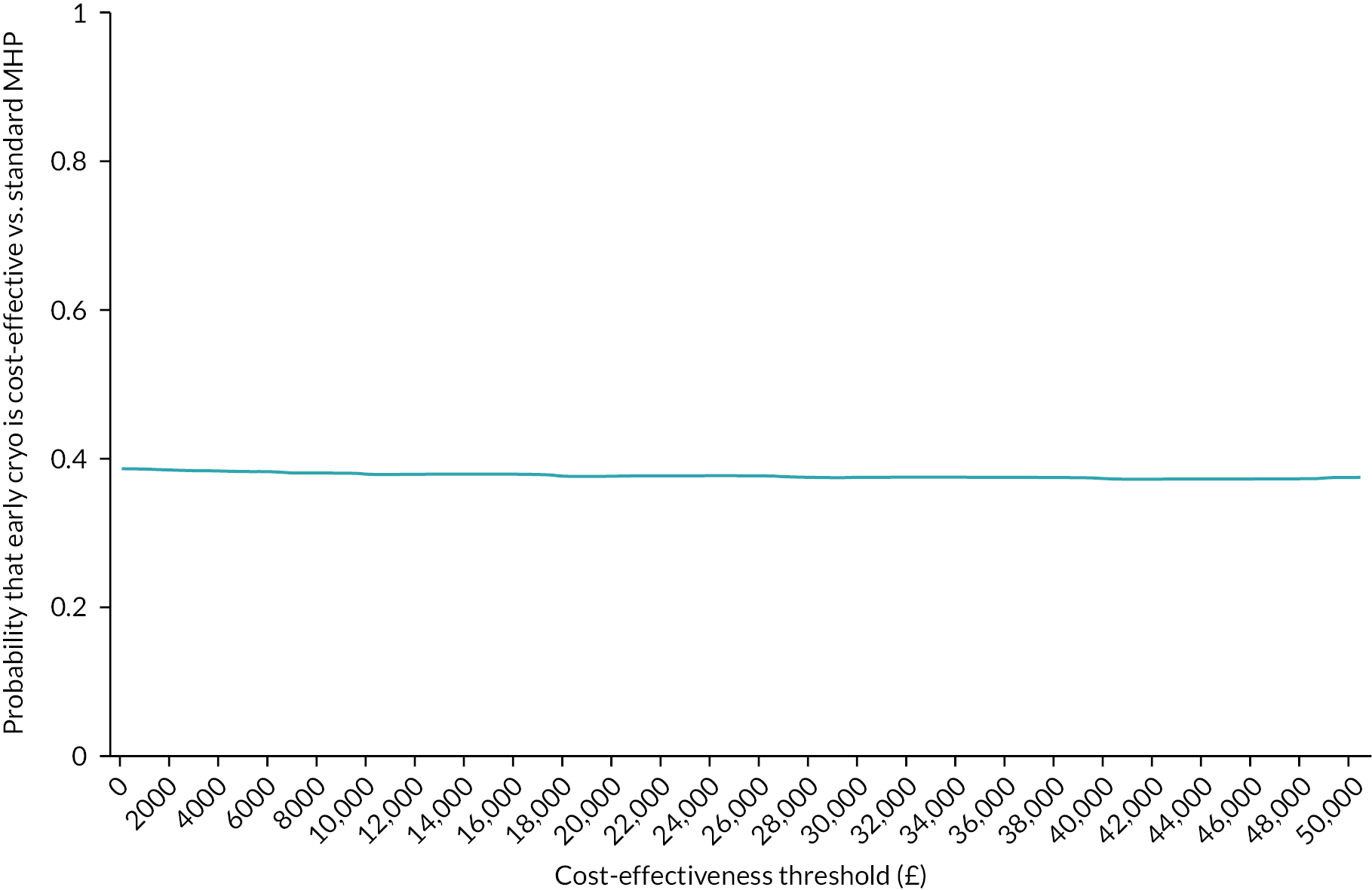
Summary
Our economic analysis to evaluate the cost-effectiveness of using early high-dose cryoprecipitate versus standard of care in adult patients with major trauma haemorrhage requiring MHP activation showed that there are no differences in terms of costs and outcomes. The findings mean that there is no reason to prefer early high-dose cryoprecipitate to standard of care based on cost or value-for-money grounds.
Chapter 5 Discussion
Overall interpretation and generalisability
The main result from this study is that the use of early high-dose cryoprecipitate given to all patients pre-emptively as an additional treatment for severe injury-related bleeding did not confer an improvement in overall survival when compared with standard treatment alone. The population enrolled in the study was representative of a severely injured cohort of patients and was in line with our prior data and trial design estimates. Our findings do not support empiric fibrinogen supplementation for all patients suspected of bleeding. Fibrinogen is known to be critical to the clotting process, and further research is required to understand whether selected critically injured patients with low fibrinogen levels can be identified who might benefit from fibrinogen supplementation. The findings question the approach of transfusion support that is unselected and aimed at all trauma patients irrespective of their baseline characteristics.
To the best of our knowledge, CRYOSTAT-2 is, to date, the largest interventional study, and also the first transatlantic study, of critically bleeding trauma patients. Research in the field of trauma is challenging, and it necessitates the co-ordination of clinicians, and, in this study, blood transfusion staff, in a 24/7, time-critical setting. The trial successfully enrolled at all MTCs in the UK and delivered this high-quality study during a period of disruption due to the COVID-19 pandemic. The UK major trauma networks represent an important and substantial resource of future trauma trials.
Implications for research
Fibrinogen replacement in the presence of low fibrinogen levels is known to be important. 14,17 CRYOSTAT-2 administered cryoprecipitate to an unselected group of patients. It remains possible that fibrinogen therapy will be effective in subgroups of patients. Eligibility for CRYOSTAT-2 was made on the presence of clinical features, for example evidence of significant injury and active bleeding, including haemodynamic parameters and clinical acumen. Blood fibrinogen levels were not required for entry into CRYOSTAT-2. Previous smaller studies (CRYOSTAT-1,21 E-FIT,24 FiiRST22) reported that fibrinogen levels at admission are typically low (e.g. median fibrinogen of 1.6–1.9 g/l) in this patient group. There might be some support for targeting fibrinogen supplementation from the results of the recent iTACTIC trial, which compared MHP therapy guided by either standard clotting tests or point-of-care viscoelastic haemostasis assays in a similar population of patients as in CRYOSTAT-2. 46 Only around one-quarter of those entered into the iTACTIC study were coagulopathic. In this study, viscoelastic haemostasis assay-treated participants received more fibrinogen therapy than those in the standard clotting test group, and there was a suggestion that the coagulopathic participants had better outcomes with viscoelastic haemostasis assay-guided treatment. Importantly, at admission the groups had median (IQR) fibrinogen levels of 2.0 g/l (1.4–2.4 g/l) (standard clotting test arm) and 1.9 g/l (1.5–2.4 g/l) (viscoelastic haemostasis assay arm), with coagulopathic participants accounting for 32% and 25%, of each cohort, respectively. 46 Further research is required to explore the effectiveness and safety of fibrinogen therapy at different levels of hypofibrinogenaemia.
FEISTY-2 (Fibrinogen Early In Severe Trauma StudY-2), which is recruiting in Australia (NCT05449834),47 will evaluate the differential effect using a primary end point of ‘days alive out of hospital at day 90 after injury’ of cryoprecipitate or fibrinogen concentrate. Eligible trauma patients will be selected for study entry according to similar clinical criteria to CRYOSTAT-2 but also on the basis of having a low fibrinogen level. A point-of-care fibrinogen testing device that could be used at scene would also be an avenue to explore for rapid, guided treatment of severely injured, bleeding patients. 48
There may also be a differential effect of fibrinogen replacement according to mechanism of injury. In one of the prespecified subgroup analyses of our trial, there was a signal of potential harm seen in patients with penetrating injury who were allocated to receive early high-dose cryoprecipitate. Participants with penetrating injury alone had higher mortality with early cryoprecipitate (cryoprecipitate vs. standard care 16.2% vs. 10.0%; OR 1.74, 95% CI 1.20 to 2.51), whereas for those affected by blunt injury alone, there was a suggestion that 28-day mortality was lower (cryoprecipitate vs. standard care 30.4 % vs. 34.8%; OR 0.82, 95% CI 0.62 to 1.09). This finding persisted after risk adjustment and is an area for future research. It is possible also that these groups may represent different severities of TIC and therefore different degrees of hypofibrinogenaemia.
The selection of primary outcome, whether all-cause mortality or death due to bleeding, has been extensively discussed in the literature of trials of tranexamic acid. 49 We noted that deaths due to bleeding varied between arms, perhaps suggesting that the clots that formed after administration of cryoprecipitate may be more resistant to clot breakdown. Further adequately powered trials would be needed to address the outcome of deaths due to bleeding, alongside mechanistic work, at the level of the developing clot, to learn more about the differential importance of clot formation balanced against clot breakdown. 20,50
Other considerations for further research
The optimal dose and efficacy of cryoprecipitate and fibrinogen administration in trauma patients remain unclear given our results. It is also recognised that many patients receive plasma transfusions that contain less concentrated amounts of fibrinogen. In addition to exploring the correction of coagulopathy, future research should explore the prevention of development of coagulopathy, including new therapeutic approaches to the inflammatory and metabolic derangements that result in TIC and increased bleeding. 51 A more exploratory route for future research includes the potential for transfusing manipulated blood components, for example sources of fibrinogen that are less resistant to breakdown in vivo.
This study found a 59% and then 11% capture rate of patient-reported outcomes at discharge and 6 months, respectively. Future research could be directed towards further PPIE work exploring how to optimise patient-reported outcome data.
Research in the context of other clinical settings of hypofibrinogenaemia
The role of fibrinogen replacement or supplementation has been tested in other settings of major bleeding. In major obstetric haemorrhage, low fibrinogen blood levels at the start of major bleeding are a predictor of large volume blood loss and poor outcomes. 52–54 However, and similar to our finding in this study, administration of fibrinogen replacement in a non-guided manner (e.g. without a blood test result) does not improve outcomes. 55 Importantly, there are only a few distinct causes of postpartum haemorrhage (in particular placental abruption or intrauterine death) that are strongly associated with low fibrinogen levels early in the course of postpartum haemorrhage. 56 Randomised controlled trial data on postpartum haemorrhage have also shown that if a woman experiencing postpartum haemorrhage has a blood fibrinogen level of > 2 g/l, there is no benefit (defined as a reduction in transfusion need) in giving fibrinogen replacement. 57
Implications for decision-makers
This study supports the ongoing use of current MHP practices, whereby concentrated fibrinogen therapies such as cryoprecipitate are given often in the second ‘MHP pack’, or reactively in response to repeated monitoring of fibrinogen levels.
There are two main ways to administer concentrated doses of fibrinogen: cryoprecipitate and fibrinogen concentrate. Clinical evaluation of these two products, aiming to determine whether one product is superior to the other, has led – so far at least – to the conclusion that there is no difference in terms of efficacy, transfusion outcome or safety concerns. 19,58 It therefore seems reasonable to surmise that the results of CRYOSTAT-2 would have been similar had fibrinogen concentrate been used as the replacement source.
Limitations
CRYOSTAT-2 was designed to be pragmatic, to facilitate participant recruitment in a challenging and time-critical clinical environment and to ensure that, in rapidly changing clinical situations, the clinicians responsible for each participant were able to treat their patient as safely as possible. The pragmatic design allowed site teams to enrol patients successfully and consistently into the trial despite the demands of the environment. However, the lack of additional interventions such as blood sampling, and particularly not knowing the baseline fibrinogen level prior to study intervention, means that we are limited in our ability to explore the reasons for a lack of difference between treatment arms, and effects of cryoprecipitate in patients with and without TIC, and the impact on restoration of fibrinogen levels. Additionally, there was no possible placebo for this trial, and this has limited our ability to compare efficacy when patient populations varied – most notably across the different time periods of intervention.
Only 434 (54%) of the 799 participants in the intervention arm started the cryoprecipitate within 90 minutes and received all three pools. This reflects the difficulty of administering an intervention that takes time to be defrosted and transported to the patient, as well as the changing clinical situation such that many patients did not have all three pools due to their bleeding stopping.
Blood fibrinogen levels were not collected as part of this study, following analysis of the CRYOSTAT-1 trial as well as data from a large European observational study (ACIT2: Activation of Coagulation and Inflammation in Trauma-2; ISRCTN12962642). Owing to the size of CRYOSTAT-2, and the aim for every major trauma centre to be able to actively recruit eligible patients without the need for research nurse support 24/7, a balance between pragmatism and study procedures was sought. A choice therefore was made not to take and store coagulation blood samples before patient recruitment. However, central trial data collection of baseline fibrinogen blood sample results, where they were taken for clinical reasons, would have added to the ability to interrogate the admission fibrinogen level and associations with outcomes.
Each recruiting site followed its local MHP, and this may have included the use of viscoelastic testing to guide transfusion therapy. No data were collected about which sites used, or did not use, viscoelastic testing for MHP guidance in patients. We cannot exclude the possibility that the MHP in some recruited patients was additionally guided by viscoelastic testing results and therefore those patients might have received cryoprecipitate earlier than patients in centres where viscoelastic testing was not available.
These caveats do not detract from CRYOSTAT-2’s primary intention, to evaluate the real-world potential for early empiric cryoprecipitate supplementation in the context of MHP activation, which was not impacted by these limitations. Despite our wide geographic coverage of the UK, we were able to open only one site in the USA due to the lengthy regulatory approvals process required. While the results from the US site are in line with those from the UK, a broader representation would have been more desirable to demonstrate wider applicability.
A not uncommon challenge in trauma-related research is the high percentage of ‘loss-to follow-up’ data for the longer-term outcomes. Our low rate of questionnaire returns reflected this and we were not able to fully analyse the QALY using a 6- or 12-month time frame. Moving forward, future studies will need to think about more innovative ways to collect long-term data on participants.
Conclusions
The results from the CRYOSTAT-2 study demonstrate that in a very complex and heterogenous population of patients, unselected early administration of high-dose cryoprecipitate will not have a survival benefit in addition to MHP. Cryoprecipitate and other forms of fibrinogen supplementation should probably be reserved for those patients with a known or suspected deficiency, although further research is needed to explore this.
Equality, diversity and inclusion
Participant representation
CRYOSTAT-2 was a trial that was conducted at every MTC in England, as well as additional sites in Northern Ireland and the USA. By including every possible site across England, the study team hoped to be as inclusive and as representative of the study population as possible.
Severe injury is more frequently encountered by men, and our trial numbers reflect this, with approximately 80% of the participants being male. There were no exclusion criteria that meant that men were more readily recruited than women (e.g. no exclusions were stipulated for pregnant women or for women of childbearing age).
Data on ethnicity were not collected as part of the study participant characteristics, except in the single US site, and therefore it is not possible to be certain that the study was fully representative of the local populations at each site.
Reflections on the research team and wider involvement
The core research team was made up of near-equal numbers of women and men and spanned several clinical and ethnic backgrounds. More widely, at the major trauma centre sites the study team facilitated the inclusion of both clinical and laboratory teams, which brought professionals together at each hospital who had previously not been commonly in contact. At the TMG meetings, there was support for more junior members of the team, with career development opportunities provided. Investigator meetings were held that focused on the research experiences of the sites involved and gave the clinical and laboratory teams a platform to enable the sharing of good trial practices. Members of our PPI group sat on the TSC.
Patient and public involvement
The CRYOSTAT-2 trial design was significantly shaped by PPIE. There were two questions that were of particular importance to the trial design and were directly influenced by the views of our PPI group and a survey of the wider public. These were the primary end point (e.g. survival, functional status, or other) and the most acceptable process for obtaining consent into an emergency study in which the majority of eligible patients lacked capacity. In 2014, the PPI group (which included mostly previous patients who had been affected by significant injury requiring blood transfusion) met initially in Oxford to discuss these two important trial design questions. Following this meeting, a survey was conducted to evaluate the views of a larger number of trauma patients and members of the public. The consensus from these two PPIE events was that survival was the most important outcome, and this view was shared across previously injured patients and lay members of the public (n = 50). However, there was a lack of agreement about the most acceptable consenting process, for example whether a legal representative or a waiver of consent should be used. In the light of these results, in April 2016 a follow-on face-to-face PPI meeting was conducted in London, which focused on consent. The CRYOSTAT-2 team gave a presentation (‘Consenting: how best to do it in emergency studies’) and a morning of interactive discussion was held. The outcome of this meeting was twofold: (1) the consent process for the study was defined and subsequently used in CRYOSTAT-2 and (2) the specific PPI group for CRYOSTAT-2 was formed: PAIR.
The face-to-face meetings, during which free and open discussion was possible, were the most informative for the clinical research team. The clinical team worked hard to include previously injured patients in the PPI meetings – a group of patients who can be hard to engage – and it is possible that an earlier observational trauma study that the clinical research team had been involved with helped to encourage this engagement. The views of both previously injured patients and members of the public – who provided interesting insights into how gaining consent for patients lacking capacity might be best approached – complemented each other and enabled a balanced view to be sought for our specific trial questions. These engagements worked well. Furthermore, a snapshot of a larger number of members of the public using a simple survey also worked well for less in-depth questions.
Two PPI members sat on our TSC. One was an experienced PPI advocate, and the other was a less experienced member. Our TSC chairperson was always mindful of the need to discuss trial matters in an accessible manner for all members of the committee. Training in trial methodology/oversight for our PPI TSC members was not easily accessible, and this is an area where more support could be offered to individuals.
Further reflecting on the PPIE involvement in CRYOSTAT-2, another area where we may be able to improve is the engagement of a PPIE specialist who perhaps could be employed within the trial and act as an advisor to the clinical research team to direct ongoing PPIE involvement, including training the PPI group members, hosting PPIE events and disseminating trial results.
Dissemination
CRYOSTAT-2 results will be disseminated in several ways. The primary publication has been published in JAMA, and dissemination has included webinars to publicise results to coincide with the publication of the primary results paper. The results will be discussed with all the trial teams from the major trauma centres in UK (12 March 2024). The results will also be presented and discussed at scientific trauma/haematology/transfusion conferences, which will inform clinicians and scientists further about this study.
The trial website was a well-used resource during the trial itself and is a platform from which the published papers available by request to corresponding author. Additional websites led by the chief investigators for CRYOSTAT-2 as well as the NIHR social media platforms and alternative forms of social media, including Twitter/X, and/or the Science Media Centre, were used as routes for the dissemination of medical data to the wider public. The results will also be discussed at the PAIR and Oxford Blood Group PPI meetings to explore ways to disseminate and contextualise our findings for the wider public and policy-makers.
Additional information
Contributions of authors
Nicola Curry (https://orcid.org/0000-0002-3849-0688) (Consultant Haematologist, Co-Investigator) was involved in the design, conduct, analysis and reporting phases of the study.
Ross Davenport (https://orcid.org/0000-0002-8593-6582) (Consultant Trauma and Vascular Surgeon, Co-Investigator) was involved in the design, conduct, analysis and reporting phases of the study.
Helen Thomas (https://orcid.org/0000-0002-7017-7739) (Head of Clinical Trial Statistics) was involved in the design, conduct, analysis and reporting phases of the study.
Erin Fox (https://orcid.org/0000-0002-8843-5054) (Study Management in US) was involved in the conduct phase of the study.
Joanne Lucas (https://orcid.org/0000-0002-3390-9654) (Clinical Trial Manager) was involved in the conduct and reporting phases of the study.
Amy Evans (https://orcid.org/0000-0002-6664-9342) (Clinical Trial Manager) was involved in the conduct and reporting phases of the study.
Efthalia Massou (https://orcid.org/0000-0003-0488-482X) (Research Associate in Statistics) was involved in the analysis and reporting phases of the study.
Rupa Sharma (https://orcid.org/0009-0000-5414-0859) (Clinical Data Manager) was involved in the conduct phase of the study.
Shaminie Shanmugaranjan (https://orcid.org/0009-0009-2091-4014) (Statistician) was involved in the analysis and reporting phases of the study.
Claire Rourke (https://orcid.org/0000-0002-7631-9275) (Clinical Operations Manager) was involved in the design, conduct and reporting phase of the study.
Alice Newton (https://orcid.org/0009-0009-1437-7953) (Statistician) was involved in the conduct and analysis phases of the study.
Alison Deary (https://orcid.org/0000-0001-8351-4186) (Head of Clinical Operations) was involved in the design and conduct phases of the study.
Nikki Dallas (https://orcid.org/0009-0009-9604-027X) (Data Management Support Officer) was involved in the conduct phase of the study.
Chloe Fitzpatrick-Creamer (https://orcid.org/0009-0000-8064-6108) (Clinical Trial Administrator) was involved in the conduct phase of the study.
Jeanette M Podbielski (https://orcid.org/0000-0003-4053-8553) (Study Management in US) was involved in the conduct phase of the study.
Charles E Wade (https://orcid.org/0000-0003-0055-5885) (Trauma Surgeon) was involved in the conduct phase of the study.
Antoinette Edwards (https://orcid.org/0000-0003-1427-0725) (Executive Director, TARN) was involved in the design and conduct of the study.
Jonathan Benger (https://orcid.org/0000-0001-6131-0916) (Professor of Emergency Care, Co-investigator) was involved in the design, conduct and reporting phases of the study.
Stephen Morris (https://orcid.org/0000-0002-5828-3563) (Professor of Health Services Research) was involved in the design, conduct, analysis and reporting phases of the study.
Bryan A Cotton (https://orcid.org/0000-0003-4184-6742) (Trauma Surgeon) was involved in the conduct phase of the study.
James Piercy (https://orcid.org/0000-0003-4891-7259) (expert lay representative) was involved in the design, conduct and reporting phases of the study.
Laura Green (https://orcid.org/0000-0003-4063-9768) (Consultant Haematologist) Co-Investigator, was involved in the design, conduct and reporting phases of the study.
Karim Brohi (https://orcid.org/0000-0003-0643-8866) (Professor of Trauma Sciences, Chief Investigator) was involved in the design, conduct, analysis and reporting phases of the study.
Simon Stanworth (https://orcid.org/0000-0002-7414-4950) (Consultant Haematologist, Co-Chief Investigator) was involved in the design, conduct, analysis and reporting phases of the study.
Acknowledgements
We would like to acknowledge and thank the significant contribution made by our oversight committees in their oversight and governance of the trial.
Trial Steering Committee
Professor Beverley Hunt OBE (Chairperson), Professor Jason L Sperry (Clinical Specialist), Dr Alastair Nimmo (Clinical Specialist), James Piercy (Patient Representative) and Kathryn Orchard (Patient Representative).
Data Monitoring Committee
Professor Timothy Coats (Chairperson), Professor Gavin Murphy (Clinical Specialist) and Professor Paul White (Independent Statistician).
We would like to acknowledge the contribution of Claire Foley (NHS Blood and Transplant Clinical Trials Unit) in the design and conduct phases of the study.
Participating sites
We would like to thank the principal investigators and transfusion leads at the participating hospitals for supporting the trial. We also wish to thank all the research teams at sites, without whom the trial would not have been possible.
Cambridge University Hospital: Dr Nathan Howes, Dr Gioacchino Cracolici, Katherine Philpott and Claire Newsam.
St Mary’s Hospital: Mr Dan Firth, Mr Christopher Aylwin and Dr Feteha Chowdhury.
St George’s Hospital: Dr Phil Moss, Professor Heather Jarman and Steve Wilshire.
Royal London Hospital: Dr Ross Davenport and Dr Laura Green.
King’s College London: Dr Fleur Cantle, Dr Jeff Keep and Kenneth Amenyah.
Leeds General Infirmary: Dr Jonathan Thornley, Dr Michael Harrison, Dr Alice Downes, Dr Marina Karakantza and Gulraiz Hussain.
James Cook University Hospital: Dr Richard Procter and Chris Elliott.
Royal Victoria Infirmary: Dr Jonathan Shelton and Gemma Smithson.
Aintree University Hospital: Dr Abdo Sattout, Rebecca Wright and Jan Gorry.
Royal Preston Hospital: Dr Kirsty Challen and Alan Noyon.
Salford Royal Hospital: Dr Dan Horner and Lydia Baxter.
Manchester Royal Infirmary: Professor Simon Carley, Dr Kate Pendry and Thomas Trimble.
John Radcliffe Hospital: Dr Melanie Darwent and Julie Staves.
Southampton General Hospital: Dr Suzanne Kellett, Dr Bentley Waller and Kerry Dowling.
Royal Sussex County Hospital: Dr Robert Kong and Julie Cole.
Southmead Hospital: Dr Jason Kendall, Professor Edd Carlton and Tim Wreford-Bush.
Derriford Hospital: Dr Tony Kehoe, Professor Jason Smith, Martin Binney and Caroline Lowe.
Queen Elizabeth Hospital: Dr Ansar Mahmood and Dr Suzanne Morton.
University Hospital of Coventry and Warwickshire: Dr Caroline Leech, Dr Maria Mushkbar and Julie Northcote.
University Hospital of North Staffordshire: Dr Richard Hall, Rebecca Sivers and Jane Graham.
Queen’s Medical Centre: Dr Adam Brooks and Dr Cherry Chang.
Hull Royal Infirmary: Dr William Townend, Robert Elshaw and Mandy Bryan.
Northern General Hospital: Dr Justin Squires and Brian Taylor.
University Hospital of Wales: Dr Raza Alikhan.
Royal Victoria Hospital, Belfast: Dr Adeel Akhtar and Stephen Kane.
University of Texas Healthcare Center at Houston: Dr Bryan A Cotton, Dr Erin E Fox, Janette Podbielski and Charles Wade.
We would like to recognise the team at the University of Texas Healthcare Center at Houston team (Erin Fox, Bryan Cotton, Charles Wade and Janette Podbielski) for preparing and co-ordinating the submission to the US Food and Drug Administration to allow the trial to open in the USA.
We would especially like to thank the patients and their families for participating in this trial.
Patient data statement
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that they are stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Ethics statement
The South Central – Oxford C Research Ethics Committee reviewed the protocol and supporting documents for the CRYOSTAT-2 trial and provided a favourable ethics opinion on 26 May 2017 (Research Ethics Committee reference 17/SC/0164).
Information governance statement
Queen Mary University of London and NHS Blood and Transplant are committed to handling all personal information in line with the UK Data Protection Act (2018) and the General Data Protection Regulation (EU GDPR) 2016/679.
Under the Data Protection legislation, Queen Mary University of London and NHS Blood and Transplant are joint Data Controllers; NHS Blood and Transplant is the Data Processor and process personal data in accordance with the Data Controllers’ instructions. You can find out more about how they handle personal data, including how to exercise your individual rights and the contact details for the Data Protection Officer, here: www.nhsbt.nhs.uk/privacy/; www.qmul.ac.uk/privacy/.
Disclosure of interests
Full disclosure of interests: Completed ICMJE forms for all authors, including all related interests, are available in the toolkit on the NIHR Journals Library report publication page at https://doi.org/10.3310/JYTR6938.
Primary conflicts of interest: Karim Brohi and Simon Stanworth report grants from the National Institute for Health and Care Research (NIHR) Health Technology Assessment programme during the conduct of the study. Karim Brohi also reports grants from Barts Charity (London, UK). Simon Stanworth is employed by NHS Blood and Transplant (NHSBT), which manufactures cryoprecipitate. Ross Davenport reports membership of a Data Safety Monitoring Committee and grants from HemoSonics (Durham, NC, USA), Werfen (Barcelona, Spain), Barts Charity and NHSBT. Stephen Morris reports former membership of the NIHR Health and Social Care Delivery Research (HS&DR) Commissioning Board (June 2014–March 2016), the NIHR HS&DR Evidence Synthesis Sub-Board (May 2016–October 2016), the NIHR Public Health Research Research Funding Board (June 2011–June 2017), the NIHR HS&DR Funding Committee (June 2014–June 2019) and NIHR HS&DR Sub-Committee Unmet Need (November 2019). Stephen Morris and Efthalia Massou received funding for the present manuscript. Nicola Curry reports participation in an advisory board (Octapharma) and being part of the International Society on Thrombosis and Haemonstatis Scientific Subcommittee for Perioperative Care. Charles E Wade reported receiving grants from Barts Charity during the conduct of the study and holding stock in Decisio Health (Houston, TX, USA), receiving grants from Athersys (Cleveland OH, USA) and Grifols (Barcelona, Spain), and receiving personal fees from Cellphire (Rockville, MD, USA) outside the submitted work. Bryan A Cotton reported receiving grants from Barts Charity during the conduct of the study and personal fees from Cerus (Concord, CA, USA) outside the submitted work. Erin Fox reports receiving grants or contracts from Queen Mary University of London, the US Department of Defense, the US National Institutes of Health and Grifols. Jeanette M Podbielski received funding from Queen Mary University of London for the current manuscript and had contracts with the US Department of Health and Human Services, the US National Institutes of Health and Grifols. They also report consulting fees from Arsenal Medical (Waltham MA, USA), Avania Clinical (Boston, MA, USA), JSL Innovations LLC (Houston, TX, USA) and CCJ Medical (Davidson, NC, USA). Laura Green received an honorarium from the University of Oslo and received support from the Haematology Society of Australia and New Zealand for attendance at its annual meeting (2022). James Piercy received payments from Cambridge Biological Research Centre and NIHR Brain Injury MedTech Co-operative and reports being a trustee of the UK Brain Injury Forum.
Publications
Marsden M, Benger J, Brohi K, Curry N, Foley C, Green L, et al.; CRYOSTAT-2 investigators. Coagulopathy, cryoprecipitate and CRYOSTAT-2: realising the potential of a nationwide trauma system for a national clinical trial. Br J Anaesth 2019;122:164–9. https://doi.org/10.1016/j.bja.2018.10.055
Curry N, Davenport R, Lucas J, Deary A, Benger J, Edwards A, et al. The CRYOSTAT2 trial: the rationale and study protocol for a multi-Centre, randomised, controlled trial evaluating the effects of early high-dose cryoprecipitate in adult patients with major trauma haemorrhage requiring major haemorrhage protocol activation. Transfus Med 2023;33:123–31. https://doi.org/10.1111/tme.12932
Davenport R, Curry N, Fox EE, Thomas H, Lucas J, Evans A, et al. Early and empirical high-dose cryoprecipitate for hemorrhage after traumatic injury: the CRYOSTAT-2 randomized clinical trial. JAMA 2023;330:1882–91. https://doi.org/10.1001/jama.2023.21019
Disclaimers
This article presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- GBD 2019 Diseases and Injuries Collaborators . Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020;396:1204-22. https://doi.org/10.1016/S0140-6736(20)30925-9.
- Kalkwarf KJ, Drake SA, Yang Y, Thetford C, Myers L, Brock M, et al. Bleeding to death in a big city: an analysis of all trauma deaths from hemorrhage in a metropolitan area during 1 year. J Trauma Acute Care Surg 2020;89:716-22. https://doi.org/10.1097/TA.0000000000002833.
- Cole E, Weaver A, Gall L, West A, Nevin D, Tallach R, et al. A decade of damage control resuscitation: new transfusion practice, new survivors, new directions. Ann Surg 2021;273:1215-20. https://doi.org/10.1097/SLA.0000000000003657.
- Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers 2021;7. https://doi.org/10.1038/s41572-021-00264-3.
- Khan S, Allard S, Weaver A, Barber C, Davenport R, Brohi K. A major haemorrhage protocol improves the delivery of blood component therapy and reduces waste in trauma massive transfusion. Injury 2013;44:587-92. https://doi.org/10.1016/j.injury.2012.09.029.
- Duchesne JC, Islam TM, Stuke L, Timmer JR, Barbeau JM, Marr AB, et al. Hemostatic resuscitation during surgery improves survival in patients with traumatic-induced coagulopathy. J Trauma 2009;67:33-7; discussion 37. https://doi.org/10.1097/TA.0b013e31819adb8e.
- Johansson PI, Oliveri RS, Ostrowski SR. Hemostatic resuscitation with plasma and platelets in trauma. J Emerg Trauma Shock 2012;5:120-5. https://doi.org/10.4103/0974-2700.96479.
- Roberts I, Shakur H, Afolabi A, Brohi K, Coats T, Dewan Y, et al. CRASH-2 Collaborators . The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011;377:1096-101, 1101.e1. https://doi.org/10.1016/S0140-6736(11)60278-X.
- Hallet J, Lauzier F, Mailloux O, Trottier V, Archambault P, Zarychanski R, et al. The use of higher platelet: RBC transfusion ratio in the acute phase of trauma resuscitation: a systematic review. Crit Care Med 2013;41:2800-11. https://doi.org/10.1097/CCM.0b013e31829a6ecb.
- Stanworth SJ, Davenport R, Curry N, Seeney F, Eaglestone S, Edwards A, et al. Mortality from trauma haemorrhage and opportunities for improvement in transfusion practice. Br J Surg 2016;103:357-65. https://doi.org/10.1002/bjs.10052.
- Winearls J, Reade MC, McQuilten Z, Curry N. Fibrinogen in traumatic haemorrhage. Curr Opin Anaesthesiol 2021;34:514-20. https://doi.org/10.1097/ACO.0000000000001027.
- Chapin JC, Hajjar KA. Fibrinolysis and the control of blood coagulation. Blood Rev 2015;29:17-24. https://doi.org/10.1016/j.blre.2014.09.003.
- Martini WZ. Coagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availability. J Trauma 2009;67:202-8; discussion 208. https://doi.org/10.1097/TA.0b013e3181a602a7.
- Rourke C, Curry N, Khan S, Taylor R, Raza I, Davenport R, et al. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J Thromb Haemost 2012;10:1342-51. https://doi.org/10.1111/j.1538-7836.2012.04752.x.
- Rossaint R, Afshari A, Bouillon B, Cerny V, Cimpoesu D, Curry N, et al. The European guideline on management of major bleeding and coagulopathy following trauma: sixth edition. Crit Care 2023;27. https://doi.org/10.1186/s13054-023-04327-7.
- Stanworth SJ, Dowling K, Curry N, Doughty H, Hunt BJ, Fraser L, et al. Transfusion Task Force of the British Society for Haematology . Haematological management of major haemorrhage: a British Society for Haematology Guideline. Br J Haematol 2022;198:654-67. https://doi.org/10.1111/bjh.18275.
- Grottke O, Mallaiah S, Karkouti K, Saner F, Haas T. Fibrinogen supplementation and its indications. Semin Thromb Hemost 2020;46:38-49. https://doi.org/10.1055/s-0039-1696946.
- Nascimento B, Levy JH, Tien H, Da Luz LT. Cryoprecipitate transfusion in bleeding patients. Can J Emerg Med 2020;22:S4-11. https://doi.org/10.1017/cem.2019.409.
- Jensen NH, Stensballe J, Afshari A. Comparing efficacy and safety of fibrinogen concentrate to cryoprecipitate in bleeding patients: a systematic review. Acta Anaesthesiol Scand 2016;60:1033-42. https://doi.org/10.1111/aas.12734.
- Morrow GB, Carlier MSA, Dasgupta S, Craigen FB, Mutch NJ, Curry N. Fibrinogen replacement therapy for traumatic coagulopathy: does the fibrinogen source matter?. Int J Mol Sci 2021;22. https://doi.org/10.3390/ijms22042185.
- Curry N, Rourke C, Davenport R, Beer S, Pankhurst L, Deary A, et al. Early cryoprecipitate for major haemorrhage in trauma: a randomised controlled feasibility trial. Br J Anaesth 2015;115:76-83. https://doi.org/10.1093/bja/aev134.
- Nascimento B, Callum J, Tien H, Peng H, Rizoli S, Karanicolas P, et al. Fibrinogen in the initial resuscitation of severe trauma (FiiRST): a randomized feasibility trial. Br J Anaesth 2016;117:775-82. https://doi.org/10.1093/bja/aew343.
- Innerhofer N, Treichl B, Rugg C, Fries D, Mittermayr M, Hell T, et al. On Behalf of the Retic Study Group . First-line administration of fibrinogen concentrate in the bleeding trauma patient: searching for effective dosages and optimal post-treatment levels limiting massive transfusion-further results of the RETIC study. J Clin Med 2021;10. https://doi.org/10.3390/jcm10173930.
- Curry N, Foley C, Wong H, Mora A, Curnow E, Zarankaite A, et al. Early fibrinogen concentrate therapy for major haemorrhage in trauma (E-FIT 1): results from a UK multi-centre, randomised, double blind, placebo-controlled pilot trial. Crit Care 2018;22. https://doi.org/10.1186/s13054-018-2086-x.
- Ziegler B, Bachler M, Haberfellner H, Niederwanger C, Innerhofer P, Hell T, et al. FIinTIC study group . Efficacy of prehospital administration of fibrinogen concentrate in trauma patients bleeding or presumed to bleed (FIinTIC): a multicentre, double-blind, placebo-controlled, randomised pilot study. Eur J Anaesthesiol 2021;38:348-57. https://doi.org/10.1097/EJA.0000000000001366.
- Early Cryoprecipitate in Major Trauma Haemorrhage: CRYOSTAT-2 n.d. https://doi.org/10.1186/ISRCTN14998314 (accessed 6 March 2023).
- Curry N, Davenport R, Lucas J, Deary A, Benger J, Edwards A, et al. The CRYOSTAT2 trial: the rationale and study protocol for a multi-centre, randomised, controlled trial evaluating the effects of early high-dose cryoprecipitate in adult patients with major trauma haemorrhage requiring major haemorrhage protocol activation. Transfus Med 2023;33:123-31. https://doi.org/10.1111/tme.12932.
- CRYOSTAT-2: A Multi-Centre, Randomised, Controlled Trial Evaluating the Effects of Early High-Dose Cryoprecipitate in Adult Patients With Major Trauma Haemorrhage Requiring Major Haemorrhage Protocol (MHP) Activation (Protocol) n.d. https://fundingawards.nihr.ac.uk/award/15/57/02 (accessed 17 June 2024).
- O’Brien PC, Fleming TR. A multiple testing procedure for clinical trials. Biometrics 1979;35:549-56. https://doi.org/10.2307/2530245.
- PANDO Network . Framework Agreement for Clinical Communication Tools for NHS Organisations n.d. https://hellopando.com/nhs (accessed 5 March 2023).
- Joint United Kingdom (UK) Blood Transfusion and Tissue Transplantation Services Professional Advisory Committee . Guidelines for the Blood Transfusion and Tissue Transplantation Services in the UK n.d. www.transfusionguidelines.org/red-book (accessed 5 March 2023).
- Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc 1999;94:496-509. https://doi.org/10.2307/2670170.
- Walsh TS, Stanworth S, Boyd J, Hope D, Hemmatapour S, Burrows H, et al. The Age of BLood Evaluation (ABLE) randomised controlled trial: description of the UK-funded arm of the international trial, the UK cost-utility analysis and secondary analyses exploring factors associated with health-related quality of life and health-care costs during the 12-month follow-up. Health Technol Assess 2017;21:1-118. https://doi.org/10.1186/s13054-018-2086-x.
- Guide to the Methods of Technology Appraisal. London: NICE; 2013.
- Office for National Statistics . Inflation and Price Indices 2023. www.ons.gov.uk/economy/inflationandpriceindices (accessed 4 March 2023).
- Agus A, Hulme C, Verghis RM, McDowell C, Jackson C, O’Kane CM, et al. Simvastatin for patients with acute respiratory distress syndrome: long-term outcomes and cost-effectiveness from a randomised controlled trial. Crit Care 2017;21. https://doi.org/10.1186/s13054-017-1695-0.
- NHS Blood and Transplant . Price List 2022 23 n.d. https://nhsbtdbe.blob.core.windows.net/umbraco-assets-corp/26343/price_list_bc_nhs_cost-per-item_2022-23_nocic.pdf (accessed 4 March 2023).
- Campbell HE, Stokes EA, Bargo DN, Curry N, Lecky FE, Edwards A, et al. Quantifying the healthcare costs of treating severely bleeding major trauma patients: a national study for England. Crit Care 2015;19. https://doi.org/10.1186/s13054-015-0987-5.
- CRYOSTAT-2 . CRF Datasheet, Used to Infer Health Economic Results n.d.
- EuroQol Group . EQ-5D-5L Quality of Life Questionnaire n.d. https://euroqol.org/eq-5d-instruments/eq-5d-5l-about/ (accessed 4 March 2023).
- Improve Trial Investigators . Endovascular strategy or open repair for ruptured abdominal aortic aneurysm: one-year outcomes from the IMPROVE randomized trial. Eur Heart J 2015;36:2061-9. https://doi.org/10.1093/eurheartj/ehv125.
- van Hout B, Janssen MF, Feng YS, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15. https://doi.org/10.1016/j.jval.2012.02.008.
- van Buuren S, Boshuizen HC, Knook DL. Multiple imputation of missing blood pressure covariates in survival analysis. Stat Med 1999;18:681-94. https://doi.org/10.1002/(SICI)1097-0258(19990330)18:6<681::AID-SIM71>3.0.CO;2-R.
- Brand J, van Buuren S, le Cessie S, van den Hout W. Combining multiple imputation and bootstrap in the analysis of cost-effectiveness trial data. Stat Med 2019;38:210-20. https://doi.org/10.1002/sim.7956.
- Barber J, Thompson S. Multiple regression of cost data: use of generalised linear models. J Health Serv Res Policy 2004;9:197-204. https://doi.org/10.1258/1355819042250249.
- Baksaas-Aasen K, Gall LS, Stensballe J, Juffermans NP, Curry N, Maegele M, et al. Viscoelastic haemostatic assay augmented protocols for major trauma haemorrhage (ITACTIC): a randomized, controlled trial. Intensive Care Med 2021;47:49-5. https://doi.org/10.1007/s00134-020-06266-1.
- NCT05449834 . FEISTY II (Fibrinogen Early In Sever Trauma StudY-2) n.d. www.feisty.org.au/ (accessed 28 February 2023).
- Hikida Y, Sumikura H, Okada H, Fujino T, Tanaka M, Sakai Y, et al. The value of the portable fibrinogen measuring device – a case report of severe postpartum hemorrhage with obstetric disseminated intravascular coagulation. JA Clin Rep 2021;7. https://doi.org/10.1186/s40981-021-00426-y.
- Brenner A, Belli A, Chaudhri R, Coats T, Frimley L, Jamaluddin SF, et al. CRASH-3 trial collaborators . CRASH-3 trial collaborators. Understanding the neuroprotective effect of tranexamic acid: an exploratory analysis of the CRASH-3 randomised trial. Crit Care 2020;24. https://doi.org/10.1186/s13054-020-03243-4.
- Morrow GB, Feller T, McQuilten Z, Wake E, Ariëns RAS, Winearls J, et al. Cryoprecipitate transfusion in trauma patients attenuates hyperfibrinolysis and restores normal clot structure and stability: results from a laboratory sub-study of the FEISTY trial. Crit Care 2022;26. https://doi.org/10.1186/s13054-022-04167-x.
- Crombie N, Doughty HA, Bishop JRB, Desai A, Dixon EF, Hancox JM, et al. RePHILL collaborative group . Resuscitation with blood products in patients with trauma-related haemorrhagic shock receiving prehospital care (RePHILL): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Haematol 2022;9:e250-61. https://doi.org/10.1016/S2352-3026(22)00040-0.
- Collins PW, Lilley G, Bruynseels D, Laurent DB, Cannings-John R, Precious E, et al. Fibrin-based clot formation as an early and rapid biomarker for progression of postpartum hemorrhage: a prospective study. Blood 2014;124:1727-36. https://doi.org/10.1182/blood-2014-04-567891.
- Cortet M, Deneux-Tharaux C, Dupont C, Colin C, Rudigoz RC, Bouvier-Colle MH, et al. Association between fibrinogen level and severity of postpartum haemorrhage: secondary analysis of a prospective trial. Br J Anaesth 2012;108:984-9. https://doi.org/10.1093/bja/aes096.
- De Lloyd L, Collins PW, Kaye A, Collis RE. Early fibrinogen as a predictor of red cell requirements during postpartum haemorrhage. Int J Obstet Anesth 2012;21. www.obstetanesthesia.com/article/S0959-289X(12)00045-3/pdf (accessed 28 February 2023).
- Wikkelsø AJ, Edwards HM, Afshari A, Stensballe J, Langhoff-Roos J, Albrechtsen C, et al. FIB-PPH trial group . Pre-emptive treatment with fibrinogen concentrate for postpartum haemorrhage: randomized controlled trial. Br J Anaesth 2015;114:623-33. https://doi.org/10.1093/bja/aeu444.
- Green L, Knight M, Seeney FM, Hopkinson C, Collins PW, Collis RE, et al. The epidemiology and outcomes of women with postpartum haemorrhage requiring massive transfusion with eight or more units of red cells: a national cross-sectional study. BJOG 2016;123:2164-70. https://doi.org/10.1111/1471-0528.13831.
- Collins PW, Cannings-John R, Bruynseels D, Mallaiah S, Dick J, Elton C, et al. Viscoelastometric-guided early fibrinogen concentrate replacement during postpartum haemorrhage: OBS2, a double-blind randomized controlled trial. Br J Anaesth 2017;119:411-21. https://doi.org/10.1001/jama.2019.17312.
- Callum J, Farkouh ME, Scales DC, Heddle NM, Crowther M, Rao V, et al. FIBRES Research Group . Effect of fibrinogen concentrate vs cryoprecipitate on blood component transfusion after cardiac surgery: the FIBRES Randomized Clinical Trial. JAMA 2019;322:1966-76.
Appendix 1 Consent procedure overview
FIGURE 11.
Consent procedure overview.
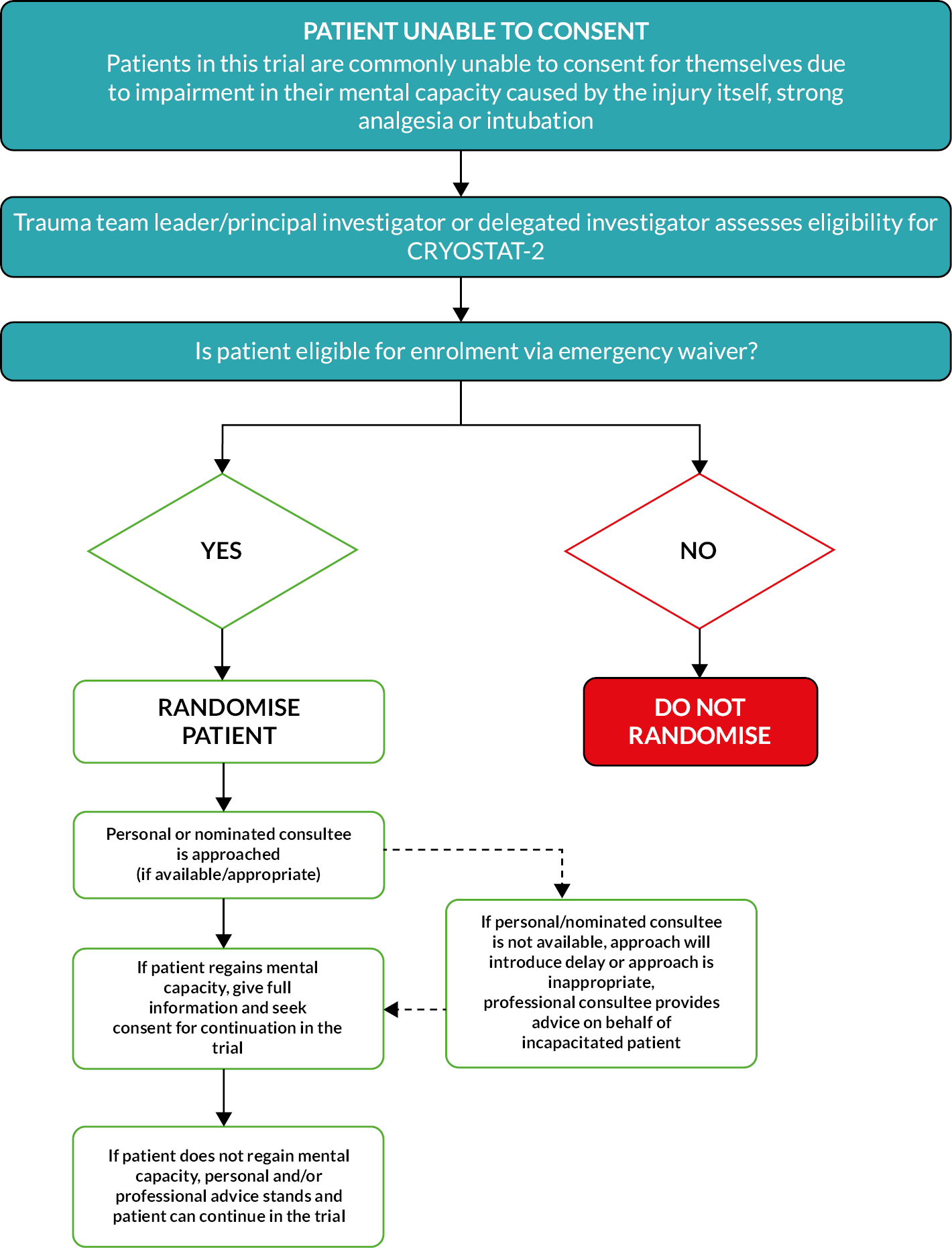
Appendix 2 Additional tables and figures
| Sites | Total number randomised |
|---|---|
| Royal London Hospital | 213 |
| John Radcliffe Hospital | 29 |
| University Hospital Southampton | 92 |
| St George’s Hospital | 141 |
| St Mary’s Hospital, London | 202 |
| King’s College Hospital, London | 49 |
| Derriford Hospital, Plymouth | 28 |
| Addenbrooke’s Hospital, Cambridge | 39 |
| Southmead Hospital, Bristol | 49 |
| James Cook University Hospital, Middlesbrough | 57 |
| Leeds General Infirmary, Leeds | 71 |
| Queen’s Medical Centre, Nottingham | 62 |
| Royal Victoria Infirmary, Newcastle upon Tyne | 39 |
| Hull Royal Infirmary, Hull | 16 |
| Northern General Hospital, Sheffield | 46 |
| Queen Elizabeth Hospital, Birmingham | 126 |
| Royal Preston Hospital, Preston | 19 |
| Royal Sussex County Hospital, Brighton | 5 |
| University Hospital, Coventry | 56 |
| Royal Stoke University Hospital | 18 |
| Manchester Royal Infirmary | 52 |
| Salford Royal Hospital | 42 |
| Aintree University Hospital, Liverpool | 100 |
| University Hospital of Wales, Cardiff | 2 |
| Royal Victoria Hospital, Belfast | 2 |
| Houston, TX | 49 |
| Overall | 1604 |
Baseline characteristics for different subgroups
| Standard care arm (n = 805) | Intervention arm | Overall (n = 1604) | |||||
|---|---|---|---|---|---|---|---|
| EC < 45 (n = 101) | EC 46–60 (n = 147) | EC 61–90 (n = 273) | EC ≥ 90 (n = 128) | EC unknown (n = 150) | |||
| Subjects | |||||||
| Male | 633/796 (80) | 81/101 (80) | 115/147 (78) | 218/273 (80) | 97/128 (76) | 107/136 (79) | 1251/1581 (79) |
| Age (years) | 40 (26–55) | 41 (28–52) | 34 (24–53) | 39 (26–55) | 37 (25–55) | 41 (27–58) | 39 (26–55) |
| Time from injury to admission to emergency department (minutes) | 77 (55–100) | 89 (69–107) | 76 (56–100) | 72 (52–96) | 70 (53–91) | 83 (60–104) | 76 (55–100) |
| Injuries and physiology at admission to emergency department | |||||||
| Blunt injury | 519/796 (65) | 60/101 (59) | 87/147 (59) | 174/273 (64) | 82/128 (64) | 92/136 (68) | 1014/1581 (64) |
| ISS | 29 (18–43) | 33 (17–43) | 29 (17–45) | 29 (17–43) | 29 (18–43) | 27 (16–42) | 29 (18–43) |
| Head AIS ≥ 4 | 191/664 (29) | 20/82 (24) | 32/132 (24) | 47/235 (20) | 24/111 (22) | 34/105 (32) | 348/1329 (26) |
| Systolic blood pressure (mmHg) | 103 (83–126) | 98 (78–121) | 104 (80–126) | 99 (84–122) | 104 (84–126) | 107 (92–130) | 103 (83–125) |
| Heart rate (per minute) | 108 (88–127) | 110 (91–130) | 110 (92–128) | 109 (86–128) | 108 (85–125) | 103 (85–121) | 108 (88–127) |
| In cardiac arrest | 17/735 (2) | 3/88 (3) | 2/132 (2) | 3/252 (1) | 2/122 (2) | 2/123 (2) | 29/1452 (2) |
| Glasgow Coma Scale score | 13 (3–15) | 3 (3–14) | 12 (3–14) | 14 (3–15) | 15 (7–15) | 12 (3–15) | 14 (3–15) |
| Pre hospital | |||||||
| RBC (units) | 0 (0–2) | 1 (0–2) | 0 (0–2) | 0 (0–1) | 0 (0–1) | 0 (0–1) | 0 (0–2) |
| FFP (units) | 0 (0–1) | 0 (0–2) | 0 (0–1) | 0 (0–1) | 0 (0–0) | 0 (0–1) | 0 (0–1) |
| Crystalloids (ml) | 0 (0–250) | 0 (0–300) | 0 (0–350) | 0 (0–250) | 0 (0–250) | 0 (0–300) | 0 (0–250) |
| Colloids (ml) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
| TXA administered | 639/796 (80) | 86/100 (86) | 118/147 (80) | 209/272 (77) | 92/128 (72) | 110/136 (81) | 1254/1579 (79) |
| Blunt | Penetrating | Overall (n = 1604) | |||
|---|---|---|---|---|---|
| Standard care arm (n = 519) | Intervention arm (n = 495) | Standard care arm (n = 277) | Intervention arm (n = 290) | ||
| Subjects | |||||
| Male | 384/519 (74) | 355/495 (72) | 249/277 (90) | 263/290 (91) | 1251/1581 (79) |
| Age (years) | 46 (30–60) | 46 (30–60) | 30 (22–43) | 30 (22–42) | 39 (26–55) |
| Time from injury to admission to emergency department (minutes) | 88 (67–108) | 84 (66–106) | 57 (44–78) | 59 (41–83) | 76 (55–100) |
| Injuries and physiology at admission to emergency department | |||||
| Blunt injury | 519/519 (100) | 495/495 (100) | 0/277 (0) | 0/290 (0) | 1014/1581 (64) |
| ISS | 38 (27–50) | 36 (26–48) | 18 (11–26) | 17 (10–26) | 29 (18–43) |
| Head AIS ≥ 4 | 176/453 (39) | 143/437 (33) | 15/211 (7) | 14/228 (6) | 348/1329 (26) |
| Systolic blood pressure (mmHg) | 104 (82–128) | 100 (84–125) | 102 (84–126) | 104 (84–124) | 103 (83–125) |
| Heart rate (per minute) | 107 (88–126) | 108 (89–126) | 109 (87–129) | 107 (87–128) | 108 (88–127) |
| In cardiac arrest | 9/479 (2) | 7/453 (2) | 8/256 (3) | 5/264 (2) | 29/1452 (2) |
| Glasgow Coma Scale score | 6 (3–15) | 12 (3–15) | 15 (11–15) | 14 (8–15) | 14 (3–15) |
| Pre hospital | |||||
| RBC (units) | 0 (0–2) | 0 (0–2) | 0 (0–1) | 0 (0–1) | 0 (0–2) |
| FFP (units) | 0 (0–1) | 0 (0–1) | 0 (0–1) | 0 (0–1) | 0 (0–1) |
| Crystalloids (ml) | 0 (0–250) | 0 (0–300) | 0 (0–100) | 0 (0–100) | 0 (0–250) |
| Colloids (ml) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) | 0 (0–0) |
| TXA administered | 439/519 (85) | 406/495 (82) | 200/277 (72) | 209/288 (73) | 1254/1579 (79) |
Sensitivity analyses
| Risk factor | ORa (95% CI) | p-value |
|---|---|---|
| Glasgow Coma Scale score (per one-unit increase) | 0.80 (0.77 to 0.84) | < 0.0001 |
| ISS score (per one-unit increase) | 1.03 (1.02 to 1.04) | < 0.0001 |
| Participant age (spline term) | See Figure 9 | < 0.0001 |
| Systolic blood pressure (spline term) | See Figure 10 | < 0.0001 |
| Intervention armb | 1.15 (0.93 to 1.42) | 0.1984 |
FIGURE 12.
Risk-adjusted OR by participant age, relative to a baseline participant at age 40 years.
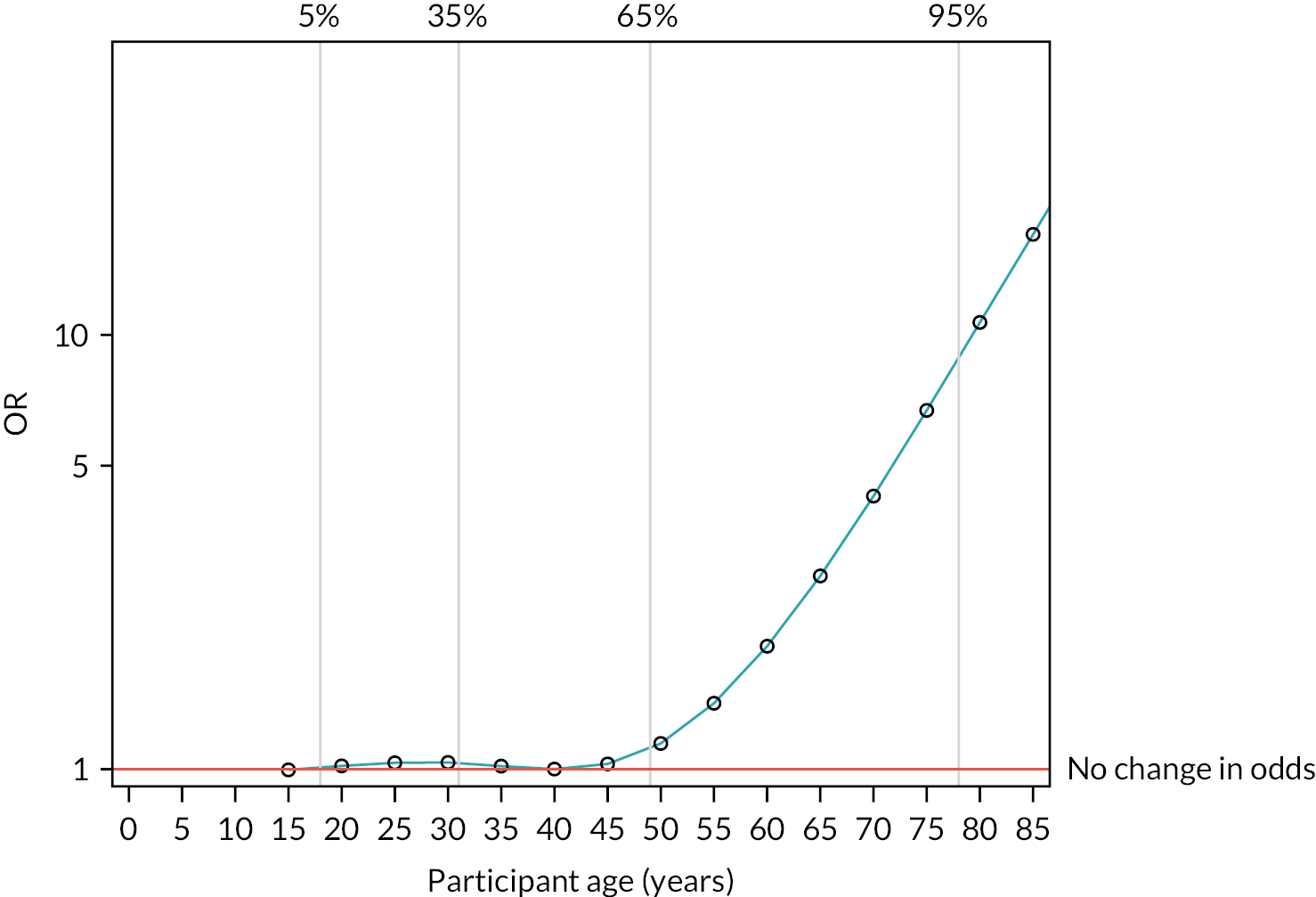
FIGURE 13.
Risk-adjusted OR by systolic blood pressure, relative to a baseline participant with systolic blood pressure of 100 mmHg.
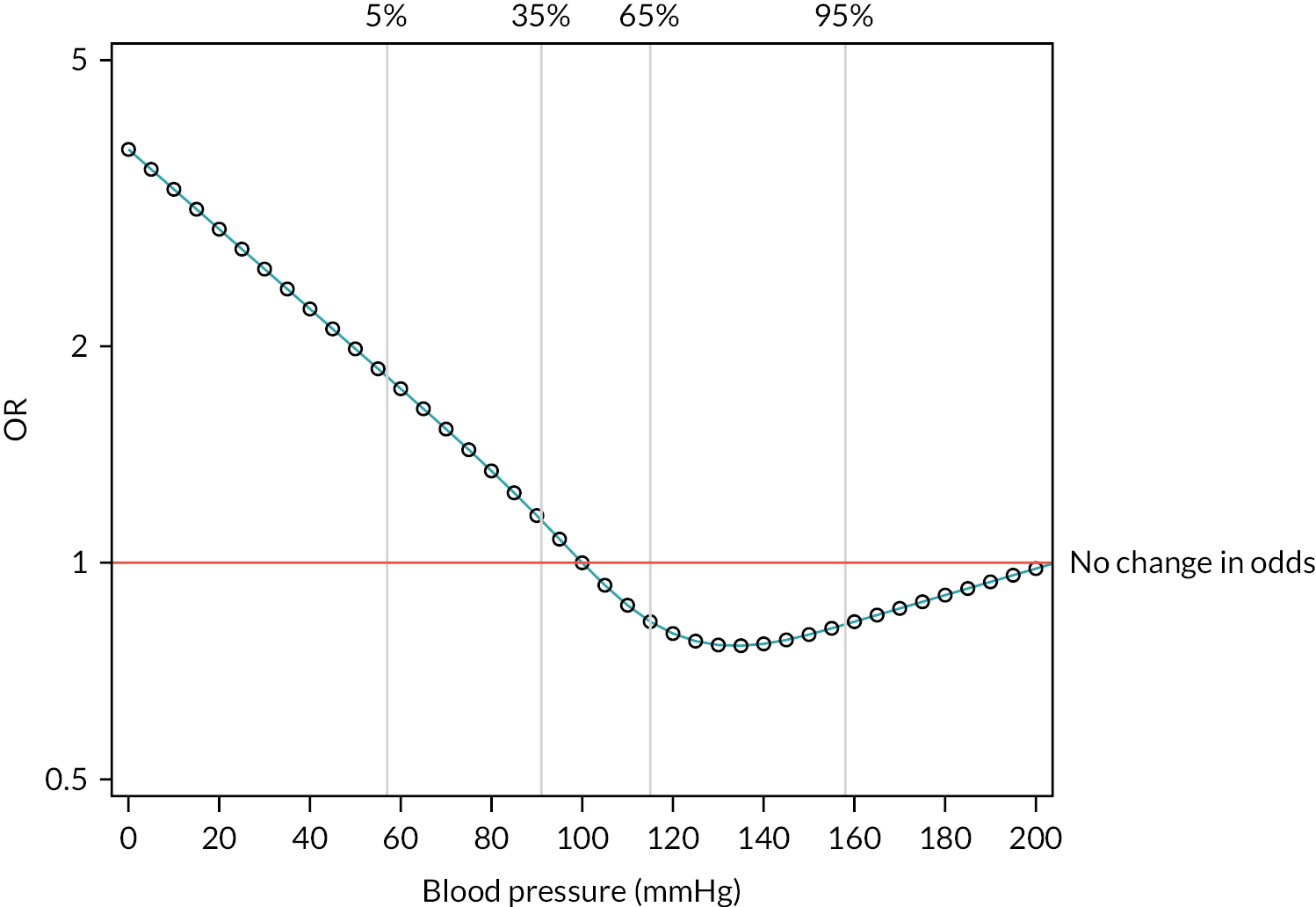
| Reason for exclusion | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) |
|---|---|---|---|
| Randomised in error | 40/805 (5.0) | 29/799 (3.6) | 69/1604 (4.3) |
| Clinically significant protocol deviation | 3/805 (0.4) | 1/799 (0.1) | 4/1604 (0.2) |
| Died within 90 minutes of arrival | 46/805 (5.7) | 37/799 (4.6) | 83/1604 (5.2) |
| No blood products after arrival | 32/805 (4.0) | 33/799 (4.1) | 65/1604 (4.1) |
| Included in per-protocol analysis | 707/805 (87.8) | 727/799 (91.0) | 1434/1604 (89.4) |
FIGURE 14.
Forest plot of ORs and CIs for main ITT and per-protocol analyses of the primary outcome, subgroup analyses and sensitivity analyses.
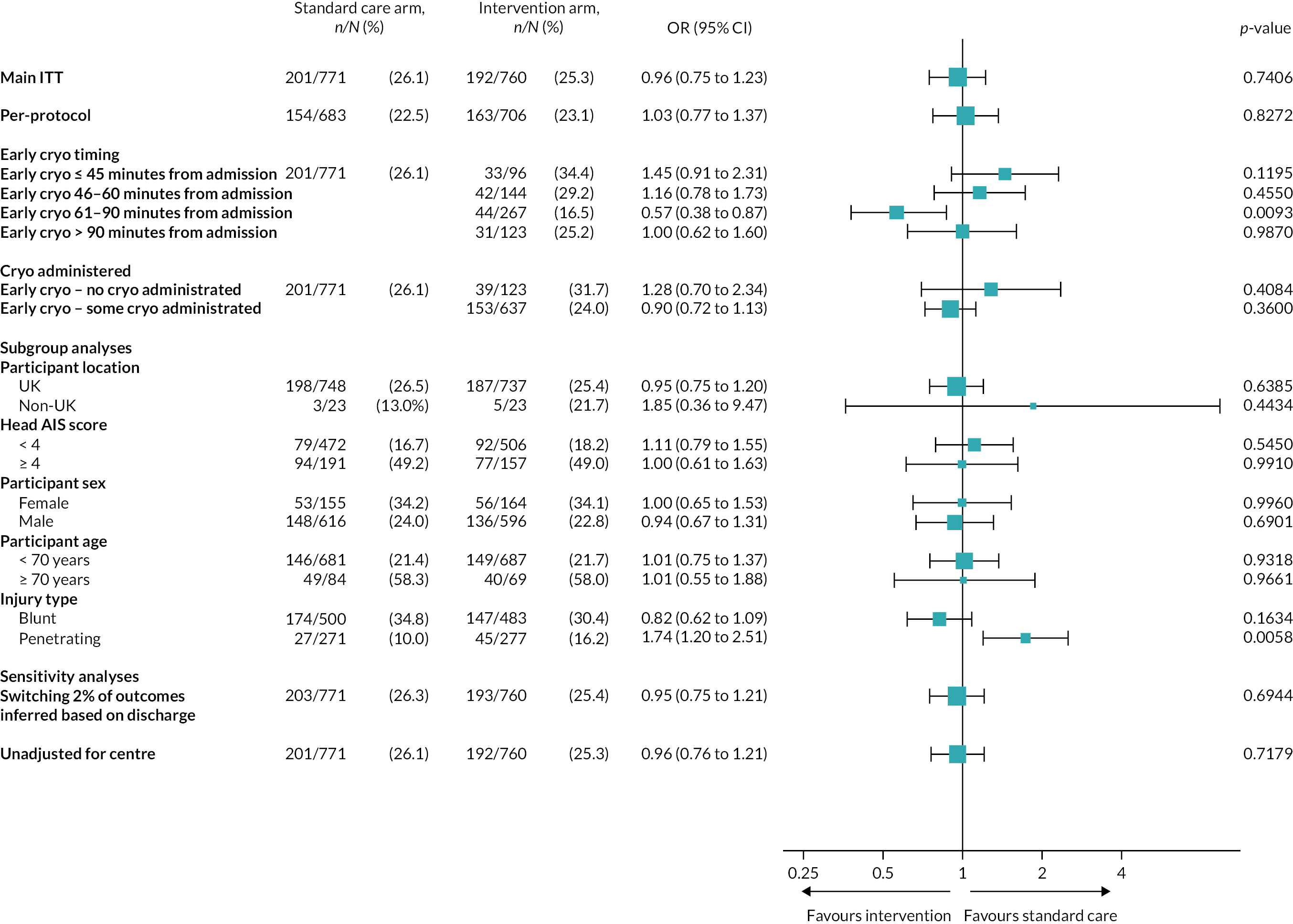
| Cryoprecipitate group | Mortality rate, n/N (%) | Relative risk (95% CI)a | OR (95% CI)a | p-valueb |
|---|---|---|---|---|
| Standard care arm | 201/771 (26.1) | 1.00 | 1.00 | |
| Intervention arm: no cryoprecipitate received | 39/123 (31.7) | 1.19 (0.79 to 1.81) | 1.28 (0.70 to 2.34) | 0.4084 |
| Intervention arm: some cryoprecipitate received | 153/637 (24.0) | 0.93 (0.79 to 1.09) | 0.90 (0.72 to 1.13) | 0.3600 |
FIGURE 15.
Kaplan–Meier survival plot up to 28 days from admission by treatment arm and whether or not any cryoprecipitate given.
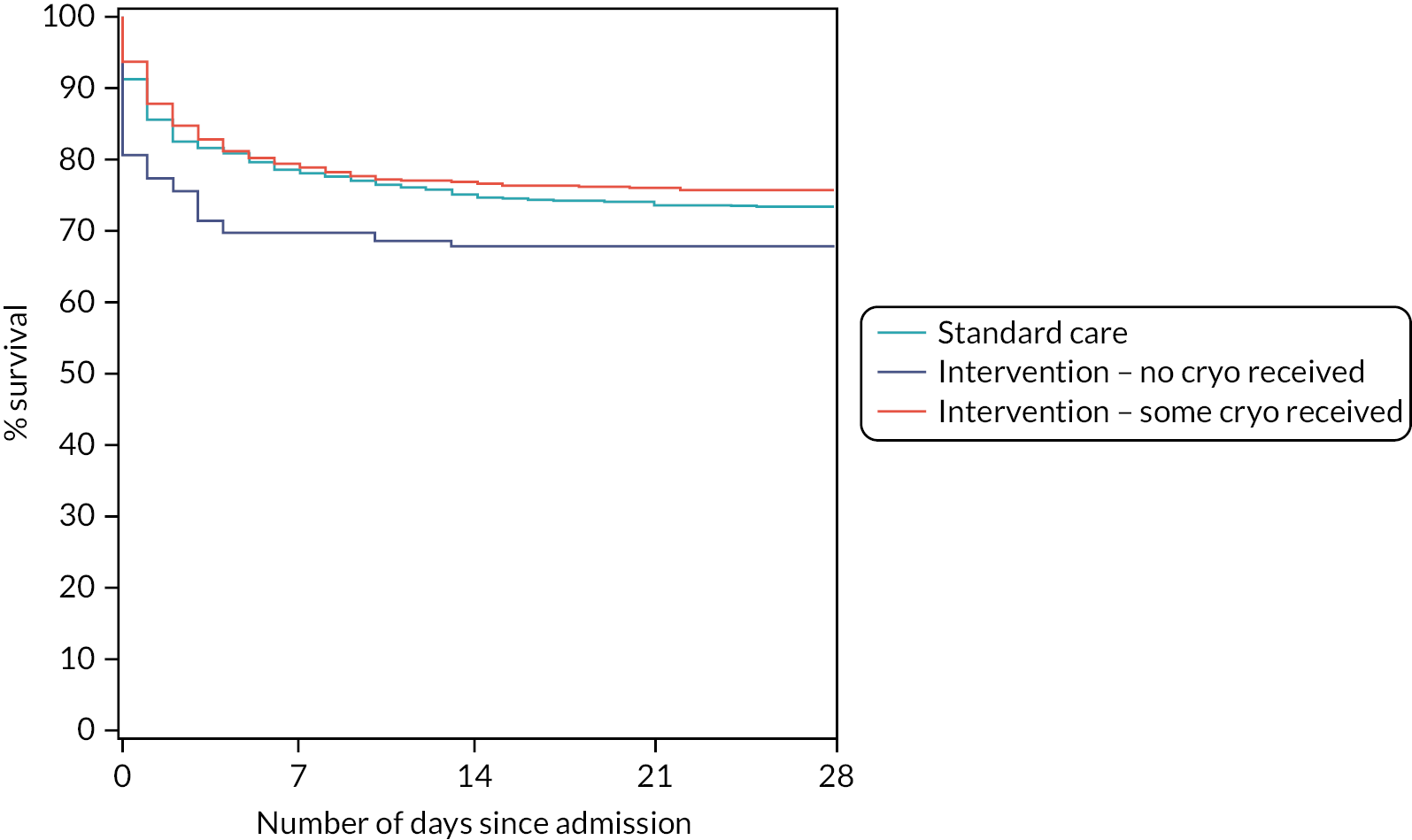
Subgroup analyses
| Outcome | UK participants | USA participants | ||
|---|---|---|---|---|
| Standard care arm (n = 780) | Intervention arm (n = 775) | Standard care arm (n = 25) | Intervention arm (n = 24) | |
| Participants who died on or before day 28 from admission, n/N (%) | 198/748 (26.5) | 187/737 (25.4) | 3/23 (13.0) | 5/23 (21.7) |
| Relative riska (95% CI) | 0.96 (0.81 to 1.14) | 1.67 (0.42 to 6.55) | ||
| ORa (95% CI) | 0.95 (0.75 to 1.20) | 1.85 (0.36 to 9.47) | ||
| p-value for subgroup | 0.6385 | 0.4434 | ||
| p-value for interaction termb | 0.4092 | |||
| Participants for whom 28-day vital status was not available from any source, n/N (%) | 32/780 (4.1) | 38/775 (4.9) | 2/25 (8.0) | 1/24 (4.2) |
FIGURE 16.
Kaplan–Meier survival plots up to 28 days from admission by treatment arm for (a) UK and (b) USA.
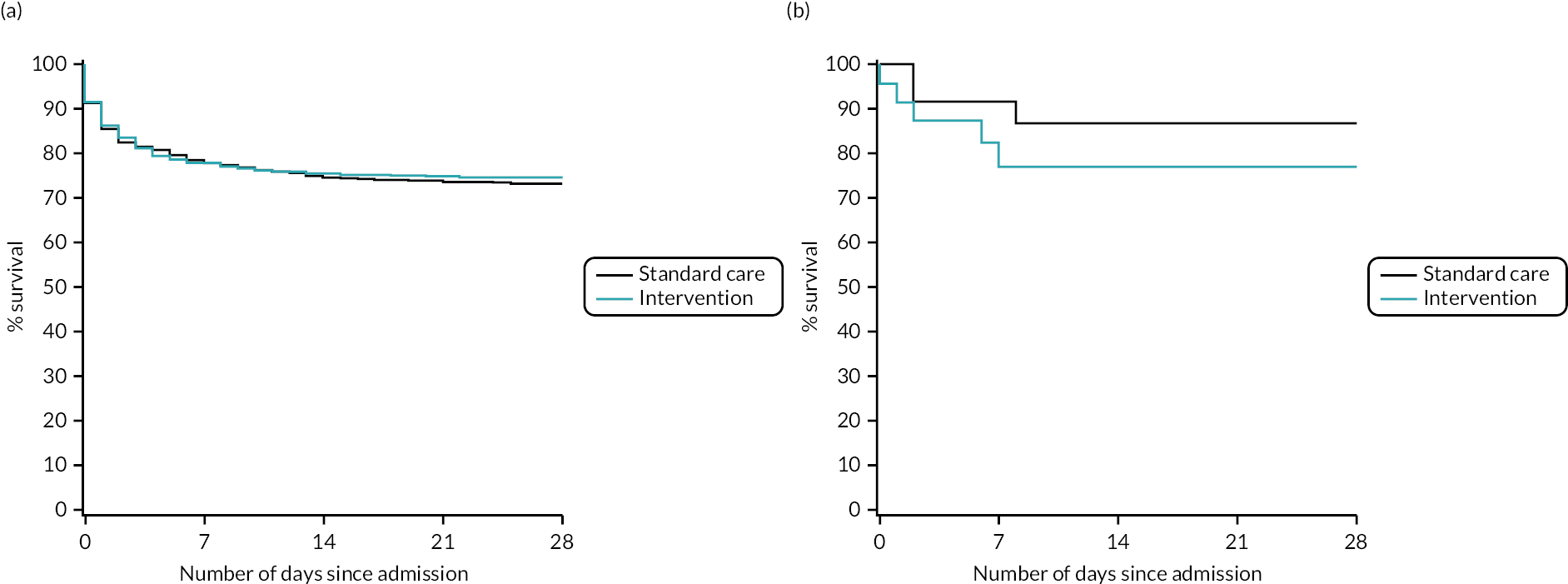
| Outcome | Head AIS < 4 | Head AIS ≥ 4 | ||
|---|---|---|---|---|
| Standard care arm (n = 473) | Intervention arm (n = 508) | Standard care arm (n = 191) | Intervention arm (n = 157) | |
| Participants who died on or before day 28 from admission, n/N (%) | 79/472 (16.7) | 92/506 (18.2) | 94/191 (49.2) | 77/157 (49.0) |
| Relative riska (95% CI) | 1.09 (0.82 to 1.44) | 1.00 (0.78 to 1.28) | ||
| ORa (95% CI) | 1.11 (0.79 to 1.55) | 1.00 (0.61 to 1.63) | ||
| p-value for subgroup | 0.5450 | 0.9910 | ||
| p-value for interaction termb | 0.7408 | |||
| Participants for whom 28-day vital status was not available from any source, n/N (%) | 1/473 (0.2) | 2/508 (0.4) | 0/191 (0.0) | 0/157 (0.0) |
FIGURE 17.
Kaplan–Meier survival plots up to 28 days from admission by treatment arm for (a) head AIS < 4 and (b) head AIS ≥ 4.
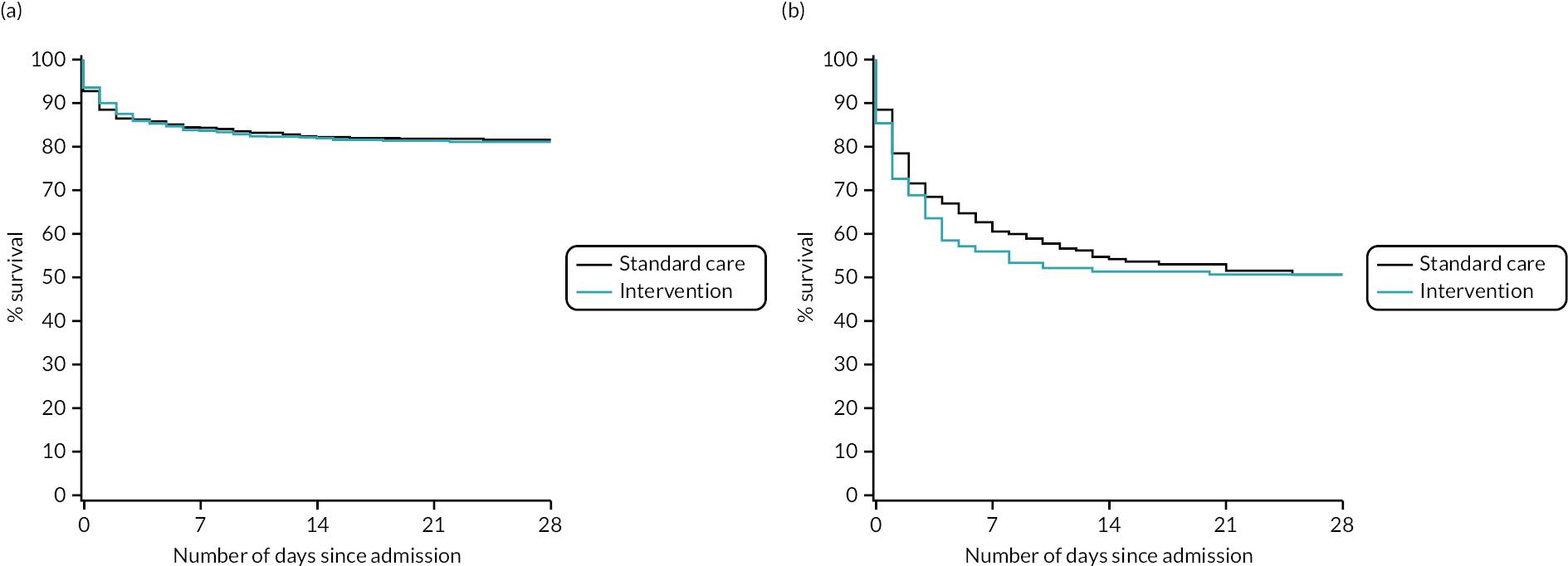
| Outcome | Male | Female | ||
|---|---|---|---|---|
| Standard care arm (n = 633) | Intervention arm (n = 618) | Standard care arm (n = 163) | Intervention arm (n = 167) | |
| Participants who died on or before day 28 from admission, n/N (%) | 148/616 (24.0) | 136/596 (22.8) | 53/155 (34.2) | 56/164 (34.1) |
| Relative riska (95% CI) | 0.95 (0.74 to 1.23) | 1.00 (0.76 to 1.32) | ||
| ORa (95% CI) | 0.94 (0.67 to 1.31) | 1.00 (0.65 to 1.53) | ||
| p-value for subgroup | 0.6901 | 0.9960 | ||
| p-value for interaction termb | 0.8204 | |||
| Participants for whom 28-day vital status was not available from any source, n/N (%) | 17/633 (2.7) | 22/618 (3.6) | 8/163 (4.9) | 3/167 (1.8) |
FIGURE 18.
Kaplan–Meier survival plots up to 28 days from admission by treatment arm for (a) male and (b) female patients.
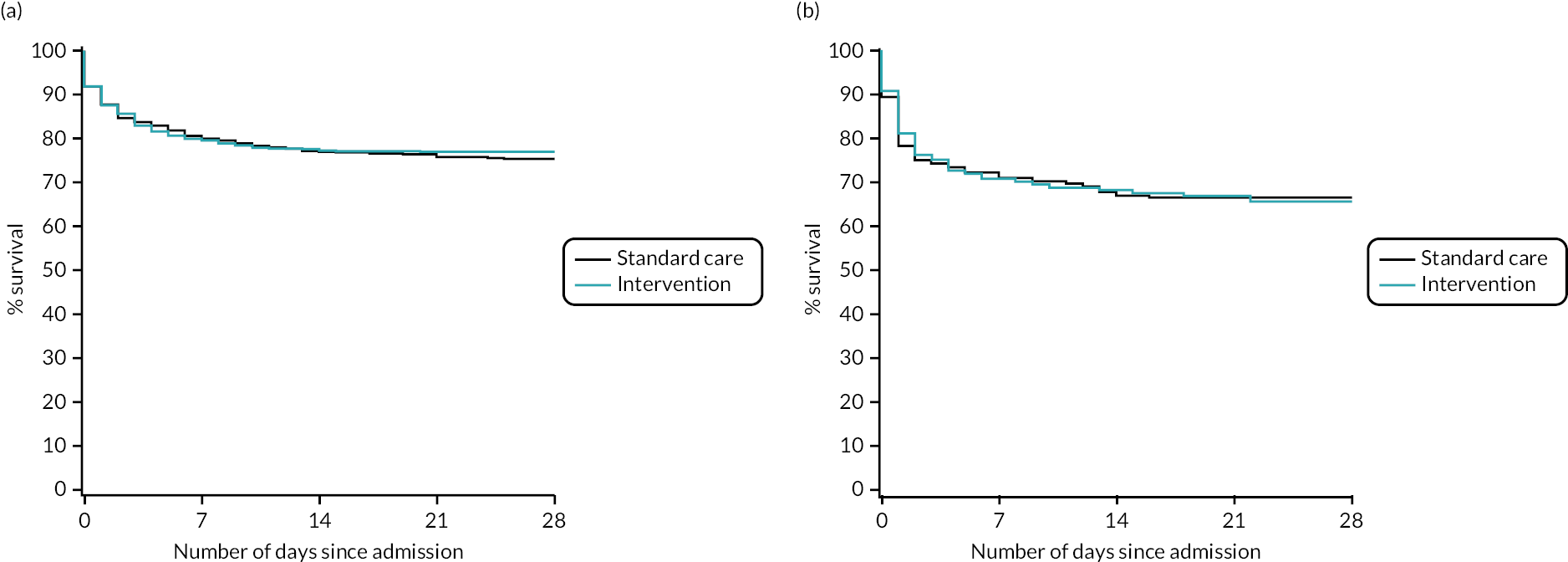
| Outcome | < 70 years | ≥ 70 years | ||
|---|---|---|---|---|
| Standard care arm (n = 704) | Intervention arm (n = 710) | Standard care arm (n = 86) | Intervention arm (n = 71) | |
| Participants who died on or before day 28 from admission, n/N (%) | 146/681 (21.4) | 149/687 (21.7) | 49/84 (58.3) | 40/69 (58.0) |
| Relative riska (95% CI) | 1.01 (0.80 to 1.28) | 1.01 (0.78 to 1.30) | ||
| ORa (95% CI) | 1.01 (0.75 to 1.37) | 1.01 (0.55 to 1.88) | ||
| p-value for subgroup | 0.9318 | 0.9661 | ||
| p-value for interaction termb | 0.9999 | |||
| Participants for whom 28-day vital status was not available from any source, n/N (%) | 23/704 (3.3) | 23/710 (3.2) | 2/86 (2.3) | 2/71 (2.8) |
FIGURE 19.
Kaplan–Meier survival plots up to 28 days from admission by treatment arm for (a) age < 70 and (b) age ≥ 70 years.
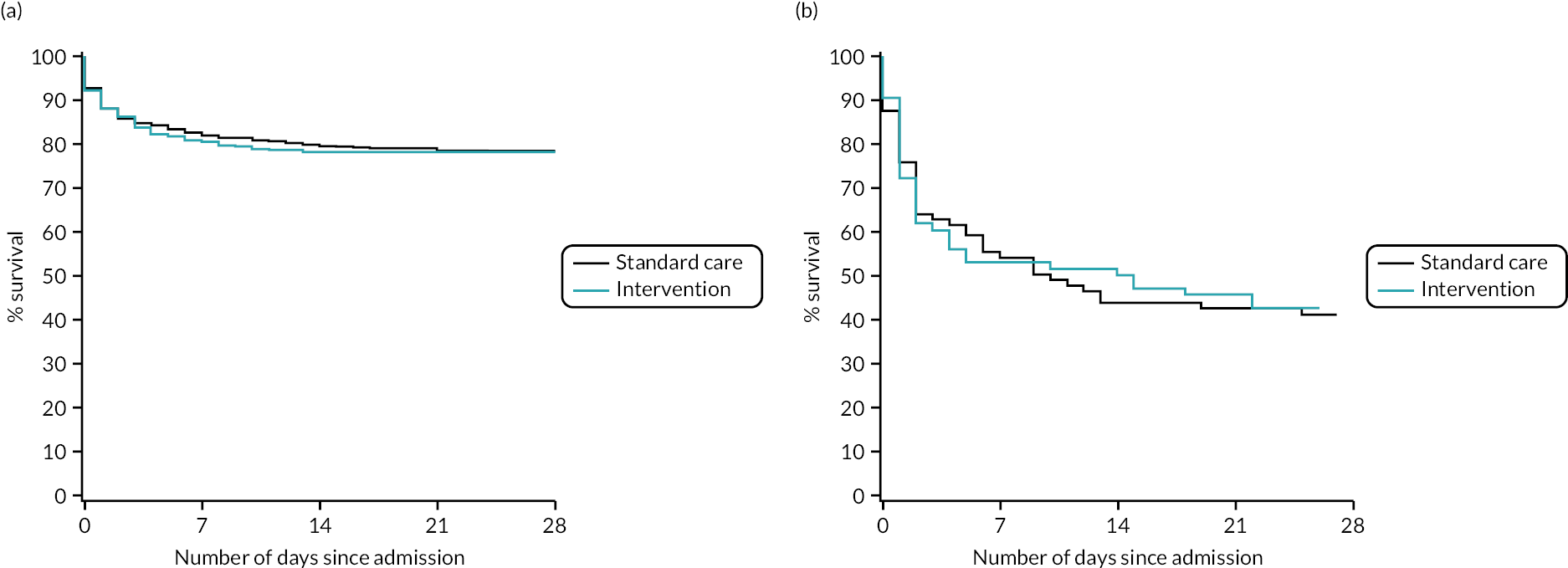
Secondary outcomes
| Outcome | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value |
|---|---|---|---|---|
| Mortality at 6 hours from admission, n/N (%) | 68/795 (8.6) | 56/784 (7.1) | 124/1579 (7.9) | |
| OR (95% CI) for mortality at 6 hours from admissiona | 0.82 (0.58 to 1.17) | 0.2595 | ||
| Mortality at 24 hours from admission, n/N (%) | 97/794 (12.2) | 88/783 (11.2) | 185/1577 (11.7) | |
| OR (95% CI) for mortality at 24 hours from admissiona | 0.91 (0.63 to 1.31) | 0.6064 | ||
| Death from bleeding at 6 hours from admission, n/N (%) | 35/795 (4.4) | 32/784 (4.1) | 67/1579 (4.2) | |
| OR (95% CI) for death from bleeding at 6 hours from admissiona | 0.93 (0.54 to 1.58) | 0.7675 | ||
| Death from bleeding at 24 hours from admission, n/N (%) | 39/794 (4.9) | 43/783 (5.5) | 82/1577 (5.2) | |
| OR (95% CI) for death from bleeding at 24 hours from admissiona | 1.13 (0.62 to 2.05) | 0.6776 | ||
| Median (IQR) time to death from bleeding among those who bled, in minutesb | 86 (40–205) | 191 (81–445) | 132 (59–353) | 0.0212 |
| Outcome | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value |
|---|---|---|---|---|
| Median (IQR) products transfused per participant from injury to 24 hours | ||||
| RBC (units) | 5 (3–8) | 5 (3–9) | 5 (3–9) | |
| Platelets (units) | 0 (0–1) | 0 (0–1) | 0 (0–1) | |
| FFP (units) | 4 (2–8) | 4 (2–8) | 4 (2–8) | |
| Cryoprecipitate (units) | 0 (0–2) | 3 (3–3) | 2 (0–3) | |
| Total blood products (units) | 10 (5–18) | 12 (7–21) | 11 (6–20) | |
| Crystalloids (ml) | 1600 (250–3200) | 2000 (700–3500) | 1958 (500–3384) | |
| Colloids (ml) | 0 (0–0) | 0 (0–0) | 0 (0–0) | |
| Mean products transfused per participant per hour over the first 24 hours | ||||
| RBC (units) | 0.65 | 0.63 | 0.64 | 0.7588 |
| Platelets (units) | 0.05 | 0.05 | 0.05 | 0.8475 |
| FFP (units) | 0.47 | 0.48 | 0.48 | 0.9963 |
| Cryoprecipitate (units) | 0.06 | 0.17 | 0.11 | < 0.0001 |
| Total blood products (units) | 1.23 | 1.33 | 1.28 | 0.4364 |
| Crystalloids (ml)2 | 101.94 | 123.71 | 112.73 | 0.0802 |
| Colloids (ml)2 | 4.39 | 7.60 | 5.98 | 0.8326 |
FIGURE 20.
Box-and-whisker plots to summarise transfusions administered from injury up to 24 hours from admission, by treatment arm. (a) RBC; (b) platelets; (c) FFP; (d) cryoprecipitate; (e) total blood products; and (f) crystalloids. These box-and-whisker plots show the median (line inside box), IQR (boundary of box), mean (diamond), minimum and maximum (whiskers) and outliers (values 1.5 × IQR above Q3 or below Q1). Extreme outliers for all products, defined as the top 1% of data, were excluded from these plots. Note the differing y-axes for each product.
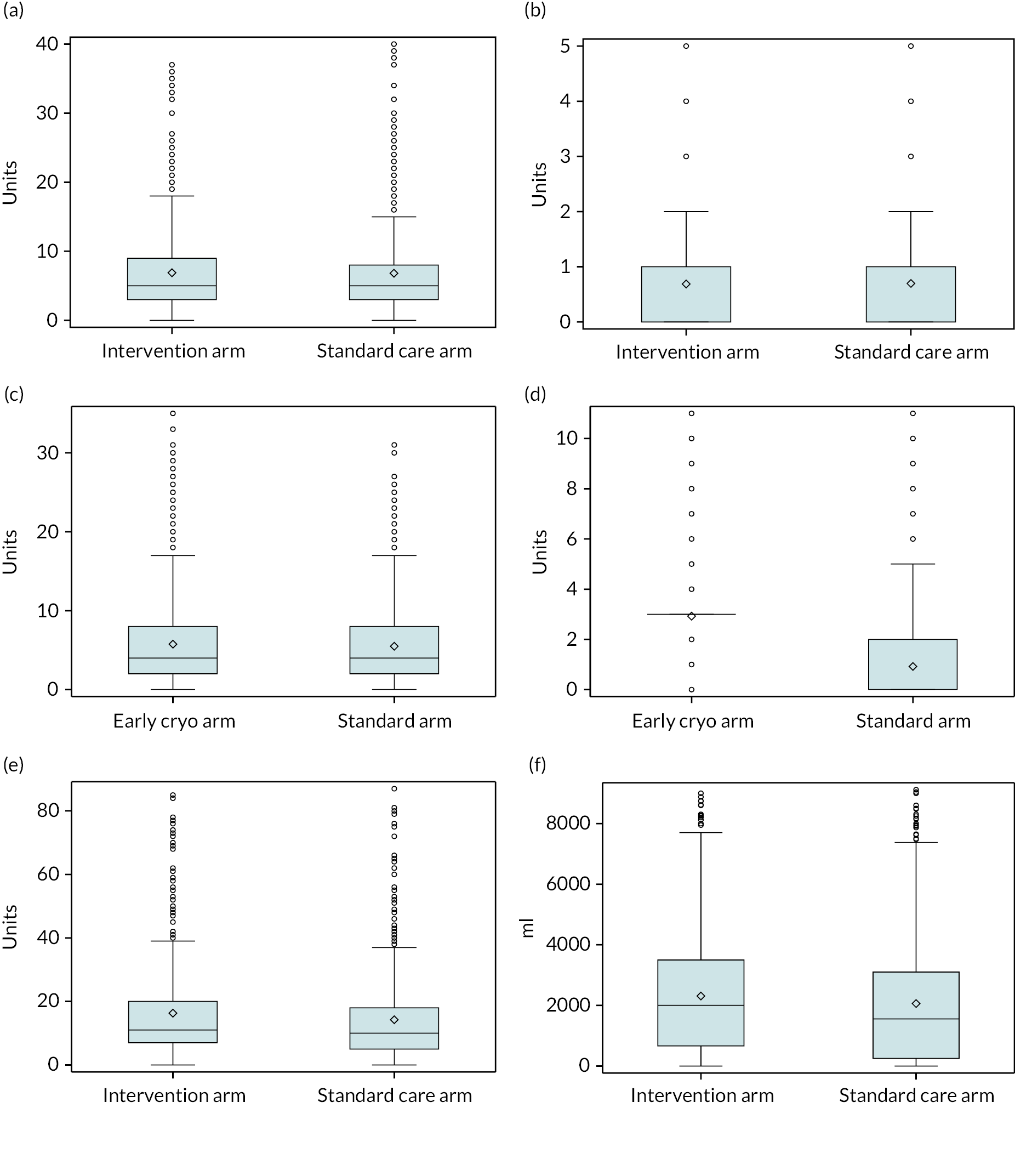
| Outcome | At discharge or day 28 where alive | At 6-month follow-up | ||||||
|---|---|---|---|---|---|---|---|---|
| Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value | |
| Participants who completed EQ-5D-5L index value questions, n/N (%) | 347/571 (61) | 324/569 (57) | 671/1140 (59) | 47/455 (10) | 52/468 (11) | 99/923 (11) | ||
| Participants who completed EQ-5D-5L ‘health today’ question, n/N (%) | 322/571 (56) | 306/569 (54) | 628/1140 (55) | 46/455 (10) | 51/468 (11) | 97/923 (11) | ||
| Median (IQR) index valuea | 0.50 (0.20–0.73) | 0.51 (0.26–0.72) | 0.50 (0.22–0.73) | 0.8024 | 0.66 (0.36–0.78) | 0.76 (0.63–0.93) | 0.70 (0.41–0.86) | N/A |
| Cohen’s d for index value (95% CI) | 0.01 (−0.14 to 0.17) | 0.47 (0.07 to 0.87) | ||||||
| Median (IQR) self-evaluated health scorea | 60 (40–70) | 50 (30–70) | 53 (35–70) | 0.1015 | 65 (50–75) | 75 (51–85) | 70 (50–80) | N/A |
| Cohen’s d for self-evaluated health score (95% CI) | −0.13 (−0.29 to 0.03) | 0.31 (−0.09 to 0.70) | ||||||
| Participants who completed GOS questionnaire at discharge n/N (%) | 712/805 (88) | 705/799 (88) | 1417/1604 (88) | |||||
| GOS at discharge, n/N (%)b | ||||||||
| Low disability | 221/712 (31) | 226/705 (32) | 447/1417 (32) | 0.5540 | ||||
| Moderate disability | 129/712 (18) | 129/705 (18) | 258/1417 (18) | |||||
| Severe disability | 153/712 (21) | 155/705 (22) | 308/1417 (22) | |||||
| Persistent vegetative state | 27/712 (4) | 21/705 (3) | 48/1417 (3) | |||||
| Death | 182/712 (26) | 174/705 (25) | 356/1417 (25) | |||||
| Outcome | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value |
|---|---|---|---|---|
| Median (IQR) ventilator-daysa,b | 1 (0–7) | 1 (0–6) | 1 (0–6) | 0.8996 |
| Median (IQR) hospital staya,b (days) | 11 (3–27) | 11 (3–27) | 11 (3–27) | 0.8810 |
| Median (IQR) first critical care staya,b (days) | 4 (1–13) | 4 (1–12) | 4 (1–12) | 0.8538 |
| Participants discharged from hospital, n/N (%) | 374/771 (49) | 375/761 (49) | 749/1532 (49) | |
| Destination, n/N (% of those discharged) | ||||
| Home | 278/374 (74) | 280/375 (75) | 558/749 (74) | |
| Nursing home/rehabilitation facility | 8/374 (2) | 9/375 (2) | 17/749 (2) | |
| Another hospital | 72/374 (19) | 63/375 (17) | 135/749 (18) | |
| Other | 16/374 (4) | 23/375 (6) | 39/749 (5) |
| Outcome | Standard care arm (n = 805) | Intervention arm (n = 799) | Overall (n = 1604) | p-value |
|---|---|---|---|---|
| Estimated mortality rate at 6 months from admission,a % (95% CI) | 27.3 (24.3 to 30.7) | 26.1 (23.2 to 29.4) | 26.7 (24.6 to 29.1) | |
| Hazard ratiob (95% CI) | 0.96 (0.79 to 1.17) | 0.6748 | ||
| Hazard ratio also adjusted for participant factorsc (95% CI) | 1.08 (0.89 to 1.32) | |||
| Estimated mortality rate at 12 months from admission,a % (95% CI) | 27.7 (24.6 to 31.1) | 26.6 (23.6 to 30.0) | 27.2 (25.0 to 29.5) | |
| Hazard ratiob (95% CI) | 0.96 (0.79 to 1.17) | 0.7120 | ||
| Hazard ratio also adjusted for participant factorsc (95% CI) | 1.09 (0.90 to 1.32) | |||
| Participants with missing survival data at all time points, n/N (%) | 10/805 (1) | 15/799 (2) | 25/1604 (2) | |
List of abbreviations
- AIS
- Abbreviated Injury Score
- CAG
- Confidentiality Advisory Group
- CRF
- case report form
- CTU
- Clinical Trials Unit
- DMC
- Data Monitoring Committee
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- FFP
- fresh-frozen plasma
- GOS
- Glasgow Outcome Scale
- HTA
- Health Technology Assessment
- HDU
- high-dependency unit
- ICU
- intensive care unit
- ISS
- Injury Severity Score
- ITT
- intention to treat
- MHP
- major haemorrhage protocol
- NIHR
- National Institute for Health and Care Research
- PAIR
- Patient/Public Advisors for Injury Research
- PPI
- patient and public involvement
- PPIE
- patient and public involvement and experience
- QALY
- quality-adjusted life-year
- RBC
- red blood cell
- SAP
- statistical analysis plan
- TARN
- Trauma Audit and Research Network
- TIC
- trauma-induced coagulopathy
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/JYTR6938).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.