

Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the Evidence Synthesis Programme on behalf of NICE as project number NIHR135138. The contractual start date was in August 2021. The draft report began editorial review in February 2022 and was accepted for publication in February 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2024 Edwards et al. This work was produced by Edwards et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Edwards et al.
Chapter 1 Background
Description of health problem
Brief statement describing the health problem
Atopic dermatitis (AD), often referred to as atopic eczema, is a chronic relapsing inflammatory skin condition. 1 One of the most common skin disorders in children, AD typically manifests before the age of 5 years, but can develop at any age. AD is characterised by dry, inflamed skin accompanied by intense itchiness (pruritus). Oozing, weeping lesions can occur in more severe forms of the condition. Scratching constantly due to pruritus disturbs sleep patterns and is considered an important factor in the transition from acute to chronic AD. Bleeding and splitting of the skin, and increased prevalence of skin ‘superinfection’ (infection in addition to AD), are also hallmark features of AD in most people with xerosis (dry skin). 1 Due to repeated episodes of skin infections, extensive antibiotic prescriptions are common among AD patients.
Aetiology, pathology and prognosis
Although a commonly occurring skin disorder in children, around 75% of those with onset of AD in childhood will experience spontaneous remission of symptoms before reaching adolescence. Some whose symptoms do not resolve in childhood continue to suffer from AD symptoms at varying degrees of severity, into adulthood. Some will experience constant symptoms of AD, whereas others will follow a chronic relapsing course of disease. 2
Atopic dermatitis is a multifaceted condition, the underlying cause of which has yet to be firmly established. Genetics, environmental factors, abnormal inflammatory responses to allergens and disrupted function of the natural skin barrier all have roles in the development and extent of AD. 2,3 The risk of developing AD is higher for those with a family member who also has this condition or another atopic disease, particularly for children whose parent or parents have AD: where both parents have AD, about 80% of children will develop AD compared with 60% of children with one parent affected. 4 In addition to other hereditary risks of developing AD, presence of mutations in genes encoding structural components of the skin barrier and cells involved in the innate immune response is known to predispose an individual to the development of AD. 5
The stratum corneum, which is the outermost layer of the skin, is formed of skin cells (corneocyte) held together by lipids and acts as a barrier to maximise retention of moisture by the body and to prevent entry of external toxins through the skin. 6 The protein filaggrin, which connects structural proteins in the outermost skin cells, is essential for maintaining the integrity of the skin barrier. 6 In AD, loss-of-function mutations in the gene encoding filaggrin are considered to be the key genetic indicator for predisposition for development of AD, being linked with early-onset, severe disease. 7,8 Specific proteases, protease inhibitors and cytokines are also involved in maintaining the structural integrity of the skin, and mutations in the genes encoding some of these components also lead to structural abnormalities of and dysfunction of the skin barrier. 5
Defects in the skin barrier enable allergens to penetrate the skin. On passing through the skin, allergens interact with local immune cells, which trigger the release of AD-related pro-inflammatory cytokines. 9 There are two modes of immune response, the innate and the adaptive response. Innate immunity is the immunity that is present from birth. By contrast, adaptive immunity is acquired after exposure to an allergen, more specifically a person develops ‘antigen-specific memory’ to an allergen, which is a key feature of adaptive immunity. Keratinocytes are the most abundant cells of the skin outer layer and contribute to the innate immune defence by producing antimicrobial peptides, as well as mediators (chemokines), in response to presence of allergens or pathogens (e.g. virus, fungi, bacteria). The antimicrobial peptides and chemokines then direct effector white blood cells (T lymphocytes or T cells) into the skin. Dendritic cells (in the skin called Langerhans cells or dermal dendritic cells) link components of the innate and adaptive immune response. On encountering allergens or pathogens, dendritic cells trigger activation of effector immune cells. The type of allergen or pathogen encountered elicits production of specific signalling molecules, with the resulting signalling cascade subsequently activating cellular mechanisms to eliminate the allergen or pathogen. Abnormal innate and adaptive responses to the presence of allergens/pathogens are thought to have a role in the complex immune network that exacerbates defects in skin barrier dysfunction and also facilitates the inflammatory responses characteristic of AD. 9 Consensus has not been reached on whether epidermal dysfunction precedes immune dysregulation or vice versa. 9
Children with AD are at risk of having concomitant asthma, food allergy and hay fever (rhinitis allergica), all of which are triggered by allergens and are also associated with an abnormal immune response. 9 A child with moderate-to-severe AD may be at a 50% risk of developing asthma and 75% risk of developing hay fever. 3 Allergens that are associated with triggering a flare of AD include house dust mites, pollen, pet hair/dander, moulds and some foods. Allergen and non-allergen triggers, such as cigarette smoke, exposure to cold or hot temperatures and sweating, can exacerbate the symptoms of AD and trigger flares.
Epidemiology
Incidence and/or prevalence
As many as 1 in 5 children and 1 in 10 adults in the UK are estimated to have AD,3,10 with about 18% of cases of childhood AD categorised as moderate and 2% as severe. 3 Of adults with AD, it has been reported that 5% of cases are severe. 11 Of the people who need treatment for AD, 7% are estimated to have moderate-to-severe disease, and 27% of those receiving treatment will require systemic therapy to elicit sufficient symptom relief. 12,13
Impact of health problem
Significance for patients in terms of ill-health (burden of disease)
Although the impact of AD varies with age and disease severity, common across ages is that AD affects various aspects of day-to-day living, including emotional and mental well-being, and family and social interactions. 14 As a disease predominantly affecting children, AD can consequently have a substantial impact on parents and other family members due to potential changes in lifestyle management, such as diet and family routine, to help manage symptoms. The treatment regimen required to sooth symptoms can be time intensive, and children are likely to require assistance from older siblings or adults to apply the topical treatments at the intervals needed for optimal effectiveness. In addition to the physical symptoms of AD, many children and adults experience sleeplessness, anxiety, depression and other mental health problems related to their AD. 10,14 Adults with AD frequently report decreased work productivity. 14
People with AD may also face a financial burden arising from extra costs associated with purchasing cleaning and laundry detergents, and bathing products tailored to sensitive skin. 3 Other costs could be incurred from travelling to appointments for assessment or treatment, and for emollients and moisturisers potentially not provided by the NHS. A study focusing on adults with AD that encompassed nine European countries, including the UK, reported that AD was associated with an annual cost to the patient of about £800 (GBP). 15
Significance for the NHS
In 2006, around 24% of people in England and Wales visited their general practitioner (GP) with a skin disorder, which is equivalent to 12.9 million people. 16 Of those presenting to primary care with a skin disorder, 0.8 million (6.1%) are referred for specialist advice, with most (92%) attending appointments with dermatologists within the NHS. 16 After diagnosis and establishing a treatment plan, the majority of care occurs at home, with topical treatments forming the mainstay of care, and many of which are purchased by the patient. Thus, it has been reported that, despite skin disorders being common, the cost of skin disease to the NHS is modest. In 2005–6, the direct cost to the NHS in England and Wales for managing skin disease was reported to be around £1820 million. 16 With the introduction of biological therapies for AD, which is one of the more common skin disorders, it is likely that the cost to the NHS for managing skin disease will rise in coming years.
Measurement of disease
Diagnosis of AD is based on the clinician’s assessment together with patient history. 17 No laboratory test is available to diagnose AD. National Institute for Health and Care Excellence (NICE) guidance indicates that AD is likely if the following criteria are fulfilled, but alternative diagnoses may need to be excluded for different age groups:17
-
An itchy skin condition (or parental report of scratching) plus three or more of the following:
-
visible flexural eczema involving the skin creases, such as the bends of the elbows or behind the knees (or visible eczema on the cheeks and/or extensor areas in children aged 18 months or younger)
-
personal history of flexural eczema (or eczema on the cheeks and/or extensor areas in children aged 18 months or younger)
-
personal history of dry skin in the last 12 months
-
personal history of asthma or allergic rhinitis (or history of atopic disease in a first-degree relative of a child aged under 4 years)
-
onset of signs and symptoms before the age of 2 years (this criterion should not be used in children younger than 4 years of age).
-
In clinical practice, assessment of the degree of severity of AD is based on clinical judgement of the appearance, location and extent of lesions, patient-reported symptoms and quality-of-life (QoL) outcomes. 18 Various clinical scales and patient-reported outcomes are available to assess whether a prescribed treatment is improving symptoms (Table 1). The scales vary considerably in the characteristics of AD evaluated to categorise severity of disease, which makes cross-comparison of the resulting categorisations applied in studies challenging. The Harmonising Outcomes for Eczema (HOME) initiative recommends the Eczema Area and Severity Index (EASI) to assess severity of clinical signs of AD. 19 A visual analogue scale (VAS) of itch and sleep loss due to AD are two parameters that are important components of some composite scores, and are often used on their own to assess therapy efficacy. To account for patient preference and experience, the patient-reported Dermatology Life Quality Index (DLQI) is also captured in clinical practice, with an improvement in score of at least 4 points recommended to be clinically meaningful:20 the DLQI is not specific to AD, but it is tailored to evaluate QoL in skin diseases. In a clinical trial setting, additional tools used to assess severity of AD are the Investigator Global Assessment (IGA) and Scoring Atopic Dermatitis (SCORAD) index. Classification of disease as moderate or severe according to the various scales are:
-
EASI: moderate AD, score of 6.0–22.9, severe AD score of 23.0–7221
-
IGA: moderate AD, score of 3, severe AD score of 418
-
SCORAD: moderate AD, score of 25–50, severe AD, score of > 50. 18
| Scale | Description |
|---|---|
| Disease severity | |
| EASI | The body is divided into four regions:
|
| The extent of AD in each region is assessed and a score assigned based on the percentage of the region affected, scoring from 0 (no active eczema) to 6 (90–100% of the region is involved). | |
Severity of disease is assessed on a 4-point scale, from none (0) to severe (3), where each region is evaluated for the intensity of:
|
|
| The severity score is multiplied by the area score and a designated ‘multiplier’ for the individual regions. The final EASI score is the total of the separate scores for the four regions, with a maximum EASI score of 72. | |
Severity strata for EASI reported by Chopra et al.:21
|
|
| Response to treatment is the percentage reduction from baseline score. | |
| SCORAD | Determines extent and severity of AD and includes a patient-reported assessment of itch and sleeplessness. The SCORAD score for an individual is calculated using the equation: A/5 + 7B/2 + C. |
Measures the extent of AD. The affected sites are shaded on a drawing of the body, with each part of the body assigned a different proportion:
|
|
| The score for A is the sum of the individual parts of the body, with a maximum score of 100%. | |
B assesses the intensity of disease. A representative area of AD is selected and, in that area, the intensity of the specific signs is assessed on a 4-point scale (0 = none through to 3 = severe). Signs evaluated:
|
|
| The score for B is the total of all intensity scores, with a maximum score of 18. C captures the symptoms of itch and sleep loss. The patient scores each symptom on a VAS from 0 (no symptom) to 10 (worst imaginable). The scores for each symptom are added together. The maximum SCORAD score is 103. |
|
Severity is defined as:
|
|
| IGA | Assessment based on the overall appearance of lesions at a given point in time. Five-point score categorised as clear (0), almost clear (1), mild (2), moderate (3) and severe (4). Moderate is categorised as, ‘Clearly perceptible erythema (dull red), clearly perceptible induration/papulation, and/or clearly perceptible lichenification. Oozing and crusting may be present’. Severe is defined as, ‘Marked erythema (deep or bright red), marked induration/papulation, and/or marked lichenification. Disease is widespread in extent. Oozing or crusting may be present’. |
| Quality of life | |
| DLQI | Most commonly used QoL tool in dermatology. A self-administered, dermatology-specific questionnaire comprising 10 items that focus on 6 dimensions: symptoms; daily activities; leisure; work; personal relationships; and treatment. Designed to gauge the patients’ perception of the impact of their skin disease on QoL over the previous week. Each question is scored on a 4-point scale from not at all (0) to very much (3). Maximum score of 30. The CDLQI is the children’s version of the DLQI and is completed by the child with the help of a parent or guardian. The CDLQI has the same format as the DLQI but the bands for categorisation of the level of impact of AD on QoL differ between the two tools. |
| POEM | A self-administered disease-specific questionnaire, focusing on the illness as experienced by the patient. Involves seven questions about the frequency of eczema symptoms over the last week from no days (0), 1–2 days (1), 3–4 days (2), 5–6 days (3), to every day (4). Symptoms evaluated are itch; sleep loss; bleeding; oozing/weeping; cracking of skin; flaking of skin; and skin feels dry/rough to the touch. POEM score is the total of scores reported for each question, with a maximum score of 28. Scores of 8–16, 17–24 and 25–28 represent moderate, severe and very severe AD, respectively. |
| Worst Pruritus NRS | WP-NRS is a single-item patient-reported outcome questionnaire designed to determine itch severity in the past 24 hours. Peak pruritis (worst itch) is evaluated using a rating scale from no itch (0) to worst imaginable itch (10). A change of 2–4 points in WP-NRS has been suggested as a clinically relevant, within-person response to treatment.22 |
Current service provision
Management of disease
Atopic dermatitis is currently uncurable, and the goal of treatment is to improve symptoms and achieve long-term disease control. Those with mild AD, who form the majority of cases, are predominantly managed in primary care. 23 Guidance for GPs outlines a step-by-step approach to disease management for a person presenting with AD, starting with preparation of an individualised management plan. 23 Initial treatment focuses on topical therapy with emollients and moisturisers, which, as noted earlier, are the mainstay of therapy but their effectiveness is reliant on the patient applying the emollient as per the recommendations, which can be challenging. For those with mild AD, to achieve relief from dry skin, regular use of emollients is usually effective in controlling symptoms. Additionally, advice is given on identifying and controlling specific triggers of flare (a worsening of symptoms), for example overuse of irritants including shampoo and detergents. Children (< 12 years) and adolescents (aged 12–18 years) typically receive the same treatments as adults.
For someone presenting to primary care with a flare, initial treatment is typically a topical corticosteroid (TCS) to suppress inflammation, if this is an acceptable treatment option to the patient. TCSs can be prescribed in different strengths, depending on the severity of the disease and the areas of skin affected:24
-
very mild (hydrocortisone)
-
moderate (e.g. betamethasone valerate and clobetasone butyrate)
-
strong (e.g. higher dose of betamethasone valerate and betamethasone dipropionate)
-
very strong (e.g. clobetasol propionate and diflucortolone valerate).
The topical immunomodulators tacrolimus and pimecrolimus, both of which are calcineurin inhibitors, are additional treatment options in primary care for those patients whose preference is not to use a TCS (Figure 1). 23 Topical immunomodulators are also an option for AD affecting areas for which TCSs are not recommended, for example the eyelids and periorbital skin, and for when there are signs of skin atrophy.
FIGURE 1.
Overview of the treatment steps in AD.

Those with moderate-to-severe AD that only partially responds to treatment, and those presenting with severe disease, are referred to secondary care for more specialised therapy, where phototherapy [predominantly ultraviolet B (UVB)] is frequently the first treatment option (see Figure 1). If phototherapy is unsuccessful, subsequent treatment typically consists of systemic therapies such as ciclosporin A (CsA), methotrexate, dupilumab and, more recently, baricitinib.
Systemic immunosuppressants with marketing authorisation for use in AD are:
-
oral corticosteroids
-
CsA
-
dupilumab
-
baricitinib.
Additional systemic therapies used to manage AD and that are used outside of their marketing authorisations are:
-
azathioprine
-
mycophenolate mofetil
-
methotrexate.
The order of systemic treatment is determined on a case-by-case basis, with treatment choice influenced by clinician and patient preference, and patient comorbidities. Non-response to systemic therapy could potentially indicate a more severe form of AD, which could influence prognosis and response to subsequent treatment. The immunosuppressant CsA has been among the first choice of systemic treatment, but some clinicians now favour methotrexate in the first-line setting. When CsA is given, it is administered for a relatively short term, with an advised maximum duration of treatment of 4 months. 25 However, if a patient is responding and does not show signs of adverse effects, treatment with CsA could be continued for up to a year. Both CsA and azathioprine increase the risk of developing non-melanoma skin cancer and some other neoplasias, and there has been a decline in their use in clinical practice for the management of AD. Should a patient have inadequate response to first-line systemic immunosuppressant, the biological therapy dupilumab and the Janus kinase (JAK) inhibitor baricitinib are recommended by NICE as second-line treatment options. 12,13 Subsequent treatment of therapy-resistant cases is also influenced by location of treating centre, with some sites being able to offer an inpatient service during which a patient would receive intense topical treatment. Where such services are not available, the patient may be treated with another systemic therapy, including CsA, or with best supportive care (BSC), the definition of which varies from practice to practice.
Current service cost
Typically, TCSs and emollients are low-cost treatments, varying between £2.58 and £12.42, with topical calcineurin inhibitors (TCIs) costing £45.56 (see Concomitant medication costs for further details). Systemic treatments incur higher costs, with the list price of CsA up to £41.59 depending on capsule size, baricitinib priced at £805.56 per pack and dupilumab costing £1264.89 per two prefilled pens or syringes. It should be noted that patient access scheme (PAS) discounts are in place for baricitinib and dupilumab.
Variation in services and/or uncertainty about best practice
The diversity in the symptoms experienced and the course of AD can make the condition challenging to diagnose and to treat. A diagnosis of atopic, rather than non-atopic (sometimes also referred to as intrinsic AD), dermatitis is often based on the clinical history of the patient. However, to differentiate AD from intrinsic AD, some centres may test for sensitisation to allergens, specifically immunoglobulin E (IgE). Intrinsic AD is characterised by failure to detect IgE in serum. Other non-atopic types of AD, which do not run in families, can be caused by direct contact with an irritant or contact allergen, which is a delayed type of hypersensitivity and is not mediated by IgE antibodies. Consideration of AD versus other types of AD (contact AD, irritant AD) and potentially intrinsic AD is important because some systemic therapies (e.g. dupilumab and tralokinumab) act through inhibition of signalling molecules and other targets involved in the atopic pathway, and, therefore, might be less clinically effective in other forms of dermatitis.
Although guidelines are available on the management of aspects of AD, with a focus on primary care,23,26 the Evidence Assessment Group’s (EAG’s) clinical experts highlighted that clarity is lacking on clinical practice in some areas, for example the frequency of use of and withdrawal schedule for TCS, and the use of TCS in combination with emollients and systemic therapies. Recommended use of TCSs is typically as an interval treatment and as a once-daily (OD) application, but advice to patients on how to use TCS varies considerably, depending on the treating dermatologist. Additionally, guidance on the use of TCIs is lacking, and, thus, there is disparity across centres in administration of TCIs.
Uncertainty around the relative clinical effectiveness of systemic therapies considered to be traditional systemic treatments (i.e. CsA, azathioprine, methotrexate and mycophenolate mofetil) in adolescents and adults has led to variation in clinical practice in their use in the management of AD. CsA is often effective in controlling AD symptoms but, because of the known adverse effects, recommendations on maximum duration of treatment vary, with some centres limiting use to 6 months compared with a maximum of 1 year in others. Clinical practice in the use of systemic corticosteroids and biological systemic therapies (i.e. baricitinib and dupilumab) also varies across centres, with areas of uncertainty including length of treatment with systemic corticosteroids, when to switch to a biological systemic therapy, choice of biological therapy (due to a lack of head-to-head data), the level of monitoring required for dupilumab and identifying which patients are benefitting from treatment (responder vs. non-responder).
Access to phototherapy varies across England. Use of the device that administers phototherapy requires specialised training and treatment is typically supervised by a consultant dermatologist. Shortage of trained staff in some centres limits the number of patients to whom phototherapy can be offered. Severe cases of AD may require intensive topical therapy, and centres with a dedicated dermatology ward can offer this service as a routine admission, but this is rare. Most centres do not have a dermatology ward and are only able to admit patients with severe AD on an emergency basis.
Relevant national guidelines, including National Service Frameworks
Although there is guidance for the treatment of AD in primary care, few guidelines address the management of AD in secondary care settings. 23,26 The NICE pathway for management of AD outlines that, on inadequate response to first-line systemic immunosuppressant therapy, baricitinib and dupilumab are available treatment options. 17 However, recommendations on the course of treatment on lack of response to second-line systemic therapy are not, at the time of writing, available.
Description of technology under assessment
Summary of intervention
Systemic treatment options available within the NHS for the management of AD in line with their marketing authorisations are CsA in the first-line setting, and baricitinib and dupilumab as subsequent therapies, both of which are predominantly given in combination with TCS. The three interventions for which an evaluation of the clinical and cost effectiveness in the treatment of AD form the basis of this report are abrocitinib, tralokinumab and upadacitinib, all of which have been evaluated in clinical trials as a monotherapy and in combination with TCS. An overview of the characteristics of the interventions is provided below.
Available treatment options
Ciclosporin A
Ciclosporin A is a calcineurin antagonist that prevents the nuclear translocation of the nuclear factor of activated T cells (NFAT), which inhibits the production of cytokines involved in the regulation of T-cell activation: activation of T cells is thought to have a key role in the mechanism underlying development of AD. In dermatology, CsA only has marketing authorisation for the treatment of psoriasis and AD in those ≥ 16 years old. 25 CsA is taken orally, typically twice daily, and various formulations and doses are available to clinicians in England. There is no recommended starting dose or maintenance dosing schedule for CsA, which are at the discretion of the treating clinician. An induction dose of 2.5–3 mg/kg daily of CsA is typical, increasing to 5 mg/kg daily if necessary. Alternatively, people may start on a higher dose of 5 mg/kg of CsA, decreasing to 3 mg/kg. Due to known adverse effects on kidney function and blood pressure, CsA is usually prescribed for a period of 2–4 months. Monitoring kidney function and blood pressure at fortnightly intervals in the initial stages of treatment is recommended, with a reduction in frequency of testing to every 2–3 months reported to be adequate on stabilisation of the dose of CsA.
Baricitinib
Baricitinib (Olumiant®, Eli Lilly and Company, Indianapolis, IN, USA) is a OD, oral treatment for moderate-to-severe AD that acts selectively and reversibly to inhibit JAK family of protein tyrosine kinases, specifically JAK1 and JAK2. JAKs are enzymes that mediate the transduction of intracellular signals also involved in the process of inflammatory responses. Baricitinib is recommended by NICE as an option for treating moderate-to-severe AD in adults if the disease has not responded to at least one systemic immunosuppressant, such as CsA, methotrexate, azathioprine and mycophenolate mofetil, or these are not suitable. 13 The recommended dose of baricitinib for AD is 4 mg OD. Down-titration to 2 mg is appropriate for some patients, such as those aged 75 years or older, and may be appropriate for patients with a history of chronic or recurrent infections. In clinical practice in England, baricitinib is most likely to be given in combination with TCS. NICE recommends assessing response from 8 weeks and stopping treatment with baricitinib if there has not been an adequate response at 16 weeks, defined as a reduction of at least:13
-
fifty per cent in the EASI from when treatment started; and
-
four points in the DLQI from when treatment started.
Dupilumab
Dupilumab [Dupixent®, Sanofi (Paris, France) and Regeneron Pharmaceuticals, Inc. (Tarrytown, NY, USA)] is a fully human monoclonal antibody. Dupilumab binds to the shared α chain subunit of the receptors for the cytokines interleukin (IL)-4 and IL-13, thereby inhibiting signalling of the two cytokines, both of which are thought to be important drivers of atopic diseases, such as AD. Dupilumab is recommended by NICE as an option for treating moderate-to-severe AD in adults if the disease has not responded to at least one other systemic therapy, such as CsA, methotrexate, azathioprine and mycophenolate mofetil, or these are contraindicated or not tolerated. 12 Dupilumab is given via subcutaneous (SC) injection into the thigh or abdomen. Treatment with dupilumab should be initiated by healthcare professionals experienced in the diagnosis and treatment of AD but can be self-administered in the longer term. An initial loading dose of 600 mg dupilumab (two 300 mg injections) is given, followed by 300 mg once every 2 weeks (Q2W). As with other systemic treatments, in England, dupilumab is most likely to be given in combination with TCS. NICE recommends stopping treatment with dupilumab if there has not been an adequate response at 16 weeks, defined as a reduction of at least:
-
fifty per cent in the EASI from when treatment started; and
-
four points in the DLQI from when treatment started.
Interventions to be assessed
Abrocitinib
Abrocitinib (CIBINQO®, Pfizer, New York, NY, USA) is a OD, oral treatment for moderate-to-severe AD for those aged 12 years and older, with a recommended daily dose of 100 or 200 mg. The company advises a starting dose of 200 mg OD for most patients, with a dose of 100 mg OD recommended for those aged ≥ 65 years. Abrocitinib is a selective JAK1 inhibitor. Abrocitinib has been studied in clinical trials as a monotherapy or in combination with TCS and compared with placebo or dupilumab in people with moderate-to-severe AD that is not adequately controlled with topical therapies or for whom topical treatments are not appropriate, or who are candidates for systemic therapy. 27–32 Based on the report submitted by the company as part of the multiple technology appraisal (MTA) process, the populations of interest to the project are adolescents and adults who are aged over 12 years with moderate-to-severe AD and who have received one prior systemic therapy.
Contraindications included in the draft summary of product characteristics (SmPC) for abrocitinib are:
-
hypersensitivity to the active substance or to any of the excipients;
-
active serious systemic infections, including tuberculosis;
-
severe hepatic impairment;
-
pregnancy and lactation.
Tralokinumab
Tralokinumab (Adtralza®, Leo Pharma UK, Hurley, UK) is a fully human IgG4 monoclonal antibody that binds to circulating IL-13, which is thought to be one of the key cytokines involved in triggering the signs and symptoms of AD. 33 Administered subcutaneously, tralokinumab has been evaluated in studies:
-
as a monotherapy compared with placebo in adolescents34 and adults,35 with moderate-to-severe AD;
-
in combination with topical therapies compared with placebo in adults with moderate-to-severe AD;36,37
-
in combination with topical therapies compared with placebo in adults with severe AD that is not adequately controlled with CsA or for whom CsA is contraindicated. 38
In the studies evaluating tralokinumab, tralokinumab was given initially at a loading dose of 600 mg followed by tralokinumab 300 mg Q2W for a period of 16 weeks, the induction phase. After the induction phase, in some studies, those achieving a response, as defined in the study, could either remain on the Q2W regimen or move to tralokinumab 300 mg every 4 weeks (Q4W). The population and setting relevant to the MTA presented here is adults who have had an inadequate response, cannot tolerate or are contraindicated to their first systemic treatment.
Upadacitinib
Upadacitinib (Rinvoq®, AbbVie, Lake Bluff, IL, USA) is a OD oral treatment for AD in those aged 12 years and older. The recommended daily dose of upadacitinib is 15 mg for adolescents and 15 or 30 mg for adults. Targeting JAKs, upadacitinib is a selective and reversible, second-generation JAK inhibitor. Upadacitinib preferentially inhibits signalling by JAK1 or JAK1/3 with functional selectivity over cytokine receptors that signal via pairs of JAK2. Upadacitinib has been assessed in clinical trials:
-
as a monotherapy compared with placebo in people aged 12 years and over with moderate-to-severe chronic AD;39
-
as a monotherapy compared with dupilumab in adults with moderate-to-severe AD;40
-
in combination with TCS compared with placebo in people aged 12 years and over with moderate-to-severe chronic AD. 41
Upadacitinib is proposed as an option for adolescents and adults with moderate-to-severe AD:
-
as first-line systemic therapy for those having inadequate response to topical treatments;
or
-
as a subsequent systemic therapy on failure to respond to first-line systemic treatment, or for those who cannot tolerate or are contraindicated to other systemic therapies.
Identification of important subgroups
The final scope issued by the NICE for the project specifies the population to be those with moderate-to-severe AD, with no specification of previous treatment. 42 However, a subgroup of interest is specified as people for whom systemic therapies have been inadequately effective, not tolerated or contraindicated. Thus, for the purposes of this MTA, the clinical and cost effectiveness of abrocitinib, tralokinumab and upadacitinib are evaluated in the relevant setting and populations proposed by the companies, as outlined in the section Interventions to be assessed. Clinical effectiveness of abrocitinib, tralokinumab and upadacitinib when given as a monotherapy and when administered with concomitant TCS is evaluated.
As AD is a common disease of childhood, the subgroup of adolescents (aged 12–18 years) is of particular relevance and evidence is presented separately for this group. Skin colour is also of interest as research suggests that certain ethnic groups are at greater risk of developing AD. 43 However, evidence of clinical effectiveness based on ethnicity was reported in only one identified study evaluating dupilumab44 and so will not be covered by this project.
Current usage in the NHS
Recent resource impact reports for baricitinib45 and dupilumab46 estimated that there are between 7500 and 7650 people in England with moderate-to-severe AD with a history of systemic therapy failure that are eligible for treatment. Of those eligible for second-line systemic treatment, annual uptake of baricitinib and dupilumab is expected to be around 25%45 and 60%,46 respectively.
As AD is incurable, patients are likely to be on some type of treatment for life. Furthermore, systemic treatments may be sequenced according to clinician and patient preference to maximise likely response to treatment to remaining options available to a patient. Thus, the EAG’s clinical experts agree that there is no typical patient treatment journey and high variation in prescribing practices exist.
Anticipated costs associated with intervention
The interventions under assessment as part of this MTA are abrocitinib, tralokinumab and upadacitinib. Each of the interventions has a proposed PAS discount in place. The list prices for abrocitinib, upadacitinib and tralokinumab are presented in Table 2. Each of the interventions can be given as a monotherapy or in combination with TCS (mometasone 0.1% ointment), with a cost of TCS per 100 g of £2.58.
| Intervention | Pack size | Pack cost |
|---|---|---|
| Upadacitinib, Rinvoq 15 mg modified-release tablets (AbbVie Ltd) | 28 | £805.56 |
| Upadacitinib, Rinvoq 30 mg modified-release tablets (AbbVie Ltd) | 28 | £1611.12 |
| Abrocitinib, CIBINQO 100 and 200 mg tablets (Pfizer) | 28 | £893.76 |
| Tralokinumab, Adtralza 150 mg pre-filled syringes (Leo Pharma UK) | 4 | £1070.00 |
Chapter 2 Definition of the decision problem
Decision problem
The final scope issued by the NICE outlined the parameters of interest for the MTA that is presented in this report. 42 As detailed in the final scope issued by NICE, the three treatments that are the focus of the project – abrocitinib, tralokinumab and upadacitinib – are systemic therapies that are potential additions to the treatments for AD currently available to the NHS. Scoping searches were carried out to gain an insight into the evidence base available based on the EAG’s inclusion criteria (Table 3). The initial searches identified a systematic literature review (SLR) (search date August 2019) that evaluated systemic treatments for moderate-to-severe AD and presented results from a network meta-analysis (NMA), which the EAG used as a source of randomised controlled trials (RCTs) published up to and including August 2019. 47 The systematic review included all interventions listed in the final scope issued by NICE. 42 Additionally, the companies seeking approval for abrocitinib, tralokinumab and upadacitinib submitted evidence as requested by the EAG from RCTs yet to be published in a peer-reviewed journal.
| Factor | Inclusion criteria |
|---|---|
| Study designa | RCTs |
| Population | People with moderate-to-severe AD |
| Interventions | The interventions below are considered as monotherapy or in combination with TCS:
|
| Comparators | Specified interventions versus each other or BSC |
| Outcomes |
|
Abrocitinib and upadacitinib both have marketing authorisations for the treatment of moderate-to-severe AD in adults and adolescents aged 12 years and over and who are candidates for systemic therapy, whereas the marketing authorisation for tralokinumab restricts its use to adults with moderate-to-severe AD and eligible for systemic therapy. The EAG considers the populations of relevance to be adolescents aged 12–18 years and adults aged 18 years and older, and, where possible, data are presented separately for the two groups. In the MTA, as requested by NICE, the clinical and cost effectiveness of abrocitinib, tralokinumab and upadacitinib have been evaluated for the position in the treatment pathway for moderate-to-severe AD proposed by the companies in their submissions to the single technology appraisal (STA) process (Figure 2), which are restricted populations compared with the individual marketing authorisations. The proposed populations are:
FIGURE 2.
Proposed positioning of abrocitinib, tralokinumab and upadacitinib in the treatment pathway.

-
Abrocitinib:
-
second-line systemic therapy for adolescents
-
first-line systemic therapy for adults
-
second-line systemic therapy for adults.
-
-
Tralokinumab:
-
second-line systemic therapy for adults.
-
-
Upadacitinib:
-
adolescents
-
first-line systemic therapy for adults
-
second-line systemic therapy for adults.
-
Candidates for systemic therapy can be those who are not responding to topical interventions and those who have already received systemic treatment. For the purposes of the MTA, first-line systemic therapy denotes those who are eligible for systemic treatment on inadequate response to topical treatments and who have not received prior systemic therapy, and second-line systemic therapy captures those who achieve inadequate response to, cannot tolerate or are contraindicated to their first systemic therapy (often CsA, azathioprine or methotrexate), which, for the MTA, is limited to CsA (based on studies identified during scoping and expert clinical opinion). After discussion with clinical experts advising the EAG, to reflect clinical practice in England, the EAG deviated from the final scope issued by NICE in terms of the comparators evaluated. Given the positioning of abrocitinib, tralokinumab and upadacitinib as either first-line or second-line systemic therapies in adolescents or adults, depending on their proposed positioning, the EAG’s advisors considered phototherapy and oral corticosteroids not to be relevant comparators, which is reflected in the EAG’s eligibility criteria for the systematic review of the literature (see Table 3). 48 In adults, the EAG considers the comparators of interest for abrocitinib, tralokinumab and upadacitinib to be:
-
First-line systemic treatment:
-
CsA.
-
-
Second line after prior systemic therapy/immunosuppressant:
-
dupilumab with or without concomitant TCS;
-
baricitinib with or without concomitant TCS.
-
Clinical effectiveness of abrocitinib, tralokinumab and upadacitinib is evaluated when given as a monotherapy and when administered with concomitant TCS. The EAG’s experts advised that, in clinical practice, systemic therapies are likely to be predominantly given concomitantly with TCS. Estimates of clinical effectiveness are reported for abrocitinib, tralokinumab and upadacitinib (as monotherapy or in combination with TCS) compared with treatments currently available in clinical practice in England. Where interventions are evaluated as a monotherapy, the intervention is compared with relevant monotherapies and not in combination with TCS, and vice versa.
For the purposes of the MTA, the EAG has focused on outcomes of clinical effectiveness that inform the economic evaluation, rather than address all the outcomes specified in the final scope issued by NICE. 42 In line with preferences expressed by the NICE Committee when evaluating the Single Technology Appraisals for dupilumab and baracitinib,12,13 a composite outcome of reduction in EASI score of 50% and improvement in DLQI of at least four points (EASI 50 + ΔDLQI ≥ 4) is the primary clinical outcome for the MTA. Clinical experts fed back that the patient-reported DLQI component of EASI 50 + ΔDLQI ≥ 4 renders the composite outcome open to recall bias. Consequently, although EASI 50 + ΔDLQI ≥ 4 informs assessment of treatment response, improvement in EASI by 75% is also considered, and is therefore captured as a secondary outcome. Additionally, the DLQI is not specific to AD but is tailored to evaluate QoL in skin diseases. An extensively validated generic QoL instrument is the EuroQol-5 dimensions (EQ-5D), which, as a generic tool, facilitates comparisons of QoL across patient groups and health conditions. EQ-5D is the tool preferred by NICE to inform the reference case in economic evaluations,49 and, thus, change from baseline in EQ-5D is evaluated.
Clinical experts informed the EAG that the outcomes listed in the final scope issued by NICE of disease-free period, maintenance of remission, time to relapse and prevention of relapse are not terms that are commonly used in clinical practice in AD and are not defined for AD. 42 End points that could inform the duration of treatment response include:
-
number of days free from TCS during treatment;
-
proportion of people maintaining for a set period of time the level of response (as defined in the study) initially achieved.
During the scoping stage, the EAG noted that many studies were designed such that people responding to their initial allocated treatment entered a long-term follow-up phase that may or may not have included a control group, and frequently involved rerandomisation. Thus, comparative results for treatment versus comparator are not consistently available for the pre-specified outcome of the proportion of people maintaining, for a set period of time, the level of response (as defined in the study) initially achieved. As data are not available for most of the included studies, and comparative effectiveness across interventions of interest cannot be assessed, the EAG decided not to report the limited details available for the outcome of maintenance of response. Data were captured at the end-of-treatment time point as reported in individual studies or as provided by the companies, together with longer term or maintenance of treatment effect. Where data are available, the clinical outcomes evaluated are:
-
proportion of people achieving EASI 50 + ΔDLQI ≥ 4;
-
proportion of people achieving EASI 75;
-
change in EQ-5D score from baseline;
-
proportion of people who discontinue treatment (including those who discontinue treatment after a response at a set time point as defined in the study);
-
proportion of people requiring use of rescue therapy during treatment;
-
number of days free from TCS during treatment;
-
serious adverse effects of treatment.
Overall aims and objectives of the assessment
The research objectives of the MTA are to appraise the clinical and cost effectiveness of abrocitinib, tralokinumab and upadacitinib within their marketing authorisations as alternative therapies for treating moderate-to-severe AD.
Chapter 3 Assessment of clinical effectiveness
Method for reviewing effectiveness
A review of the evidence on the clinical effectiveness of abrocitinib, tralokinumab and upadacitinib in the treatment of moderate-to-severe AD was undertaken systematically following the general principles recommended in the preferred reporting items for systematic reviews and meta-analyses (PRISMA) statement. 50 Flow diagrams illustrating the flow of information through the systematic review process are presented in the section Quantity of research available, according to the PRISMA reporting guidelines. 50
Identification of studies
During scoping, the EAG identified a systematic review reporting a NMA of systemic treatments for moderate-to-severe AD that searched records up to August 2019. 47 The EAG considers the review to have been carried out systematically and following accepted systematic review methodology. The systematic review identified completed and ongoing studies evaluating all interventions and comparators of interest to the MTA outlined here.
The identified review evaluated systemic immunosuppressive and immunomodulatory therapies used in the management of AD, and therefore implemented broad search terms relating to interventions. 47 For the purposes of the current MTA, the EAG designed the search strategies to incorporate terms specific to the interventions of interest. Search strategies were designed to include Medical Subject Headings (MeSH) and free-text terms for the condition and all interventions.
As the identified review retrieved studies on all interventions of interest to the MTA, the EAG’s searches were restricted to records published from 1 August 2019. 47 Multiple electronic databases were searched, including MEDLINE (searched via OVID), EMBASE (searched via EMBASE) and CENTRAL. Search terms were tailored to the database searched and the platform used to carry out the search. Search filters developed and validated by the Scottish Intercollegiate Guidelines Network (SIGN) were used to identify RCTs in MEDLINE, and the strategy developed by Glanville et al. 51 was used to retrieve records in EMBASE. Full details of the terms used in the search are presented in the supplementary material. Electronic database searches were carried out on 8 July 2021 and an update search run on 29 November 2021. No language restrictions were applied to the search strategy.
The EAG evaluated the studies identified in the systematic review against the inclusion criteria for this MTA, presented in Table 3. Bibliographies of retrieved studies (RCTs and other systematic reviews) identified as relevant were manually reviewed for potentially eligible studies. Ongoing clinical trials were identified by searching the clinical trial registries ClinicalTrials.gov and the EU Clinical Trials Register. In addition, clinical experts advising the EAG were contacted with a request for details of additional published and unpublished studies of which they had knowledge. Furthermore, submissions provided by companies were assessed for unpublished data and the companies were contacted with a request for relevant data not available within the submissions.
Based on the scoping search and the RCTs reported in the identified systematic review, the EAG considered that data on the clinical outcomes of interest to the MTA would likely not be available from RCTs for CsA. An additional search was therefore conducted, concurrent with the RCT search, for observational and non-comparative studies of CsA in moderate-to-severe AD. Search filters developed and validated by SIGN were used for the observational search.
Inclusion and exclusion criteria
Eligibility criteria for the review of clinical effectiveness were as specified in the decision problem and summarised in Table 3. Two reviewers independently screened all titles and abstracts retrieved from the database and trial registry searches. Full-paper manuscripts of titles/abstracts that were deemed relevant were obtained and the relevance of each study assessed. Evidence submissions provided by the company for each of the interventions (abrocitinib, tralokinumab and upadacitinib) and committee papers for the comparators [dupilumab (TA534)12 and baricitinib (TA681)13] were screened for unpublished data. Discrepancies were resolved by consensus, with involvement of a third reviewer when necessary.
Data abstraction strategy
Full papers were ordered for all included references. Data were extracted independently by two reviewers using a standardised data extraction form. Information extracted included details of the study’s design and methodology, baseline characteristics of participants and data on outcomes of interest, both clinical effectiveness outcomes and adverse events (AEs). Where there was incomplete information, the companies of the interventions of interest were contacted for additional details. Discrepancies were resolved by discussion, with involvement of a third reviewer when necessary. Data extraction forms for the included studies are provided in Report Supplementary Material 1.
Critical appraisal strategy
The quality of the clinical effectiveness studies was assessed by one reviewer, and independently checked for agreement by a second reviewer. Any disagreements were resolved by consensus and, if necessary, a third reviewer was consulted. The quality of RCTs was assessed according to the Cochrane Risk of Bias Tool, version 2, for randomised studies. 52 Details of quality assessment for each included study are presented in structured tables (see Report Supplementary Material 1) and an overall assessment of study quality is provided as a narrative summary (see Quality of research available). The possible effects of study quality on the clinical effectiveness data and review findings are discussed where relevant.
Methods of data synthesis
Details of results on clinical effectiveness for each included study are presented in structured tables in the supplementary material.
The data were analysed by the pre-specified subgroups based on age and line of therapy, in line with the populations in the economic model:
-
adults with moderate-to-severe AD and inadequate response to topical treatments receiving first-line systemic treatment;
-
adults with moderate-to-severe AD receiving second-line systemic treatment after inadequate response to CsA, or where CsA was not tolerated or was contraindicated;
-
adolescents, irrespective of prior therapy.
The effectiveness of the interventions in subgroups based on skin colour was also captured and reported, where available, but no analysis of relative effectiveness versus the comparators of interest were conducted due to paucity of data. Refer to Report Supplementary Material 1 for further details.
The SLR did not identify any studies investigating the clinical effectiveness of each of the interventions (abrocitinib, tralokinumab, and upadacitinib) with the comparators of interest (dupilumab, baricitinib, and CsA) in the populations considered in the economic model (listed above). Therefore, NMAs were conducted for each population, with results presented for comparisons with dupilumab, baricitinib and CsA but not comparing the interventions with each other. The methods used for the NMA followed the guidance described in the NICE Decision Support Unit’s (DSU’s) technical support documents (TSDs) for Evidence Synthesis. 53,54 NMAs were performed using a Bayesian Markov Chain Monte Carlo (MCMC) simulation using OpenBUGS. 55 NMAs were conducted using three chains with results based on 50,000 iterations after a ‘burn in’ of 50,000 iterations. Convergence was assessed by visual inspection of Brooks–Gelman–Rubin (BGR) diagnostic plots, which assesses convergence by comparing within- and between-chain variability. The BGR diagnostic should gradually shrink to one as convergence is approached.
Fixed-effect (FE) and random-effect (RE) models were carried out for all analyses. All networks were expected to be populated with the results from a small number of studies in a ‘star-shape’ with few or no ‘loops’. In simple networks with a paucity of trials, it is likely there will be insufficient data to accurately estimate between-study heterogeneity. The EAG attempted to minimise some differences in the patient populations across the trials by focusing on the pre-specified subgroups based on age and line of therapy. However, other differences across studies at the trial level were expected to potentially introduce heterogeneity into the network, such as strength of TCS used and washout period prior to enrolment. In a Bayesian RE NMA, there is a risk that the prior selected for the between-study heterogeneity will dictate the heterogeneity in the posterior distribution when the number of studies per comparison is low, as in the NMAs in this MTA.
To inform the prior estimate of the between-study heterogeneity, external evidence on the likely extent of this heterogeneity was incorporated. A predictive distribution for the degree of between-study heterogeneity was chosen from Turner et al. based on type of intervention comparison and outcome. 56 Turner et al. presents prior distributions for between-trial heterogeneity based on pair-wise meta-analyses but the priors in the paper can also be applied to NMA provided comparisons are within one category. The most relevant category of intervention comparison was deemed to be ‘pharmacological versus placebo/control’, as the majority of the studies in the networks were placebo controlled. As the outcomes for the NMAs were EASI 50 + DLQI ≥ 4 and EASI 75, the most relevant outcome type was deemed to be the subjective outcome of ‘signs/symptoms reflecting continuation/end of condition’. The predictive distribution for the between-study heterogeneity for this combination of intervention comparison and outcome was a mean and variance on the natural log scale of −2.06 and 1.51,2 respectively. Vague or uninformed prior distributions were used for the relative treatment effects and trial-specific baselines.
Given the potential differences between the trials, the small number of trials expected in the networks and the likely small number of patients from the trials (given the subpopulations required), the EAG has a preference for a RE model with an informed prior over a FE model. Model fit of the FE and RE analyses were compared based on deviance information criterion (DIC). However, a difference in DIC of 5 or less was not considered significant and judged to indicate a similar model fit, that is, a FE model would have to have a DIC > 5 lower than the RE DIC to be considered a better statistical fit. Model fit was also assessed by comparing the residual deviance, which is a measure of how similar (or not) the model would predict the data used for the analysis, with the number of unconstrained data points.
As all outcome data analysed were dichotomous, treatment effects are presented as odds ratios (ORs) with 95% credible interval (CrI). The median ORs are presented in the clinical section, as these are easier to interpret and more applicable to individual patients, whereas the mean estimates are more informative on a population level and therefore inform the economic model (as log ORs).
Inconsistency in the NMAs was assessed where loops were present allowing a comparison of the direct and indirect effect estimates. The presence of inconsistency was assessed using the Bucher method for single loops of evidence as described in DSU TSD4. 57
Based on TA534 and TA681, the EAG was aware there could be different censoring rules, around patients who receive rescue medication during treatment, in the trials used for the analysis of clinical effectiveness. For the purposes of the research presented here, the EAG defines the population for the primary analysis to include those who receive rescue medication during treatment because, based on advice from clinical experts, the use of rescue medication more closely reflects what occurs in clinical practice in England, that is, the primary NMAs are based on using all observed data, regardless of rescue medication use to determine response, where possible. A sensitivity analysis was planned, where feasible, where patients requiring rescue medication were considered non-responders and censored following initiation of rescue therapy.
For dichotomous outcomes where the number of patients experiencing an outcome (n), for example the number of responders, was not reported but the proportion with a response (%) and total number of patients (N) were available, n was imputed by multiplying % and N and rounding to the nearest integer. Missing data were analysed as a treatment failure for all outcomes, that is, for EASI 50 + DLQI ≥ 4 and EASI 75, people lost to follow-up were considered not to have achieved response.
The low number of studies included in each comparison within the network precluded the evaluation of publication bias and/or small study effects. The potential limitations of the NMAs, together with the associated influence on the generated estimates of effect, are discussed in the strengths and limitations of the report (see Strengths and limitations).
Results
Quantity and quality of research available
Quantity of research available
As noted in the section Identification of studies, the EAG identified a systematic review reporting a NMA of systemic treatments for moderate-to-severe AD that searched records up to August 2019. 47 The EAG considers the review to have been carried out systematically and to follow accepted systematic review methodology. The systematic review appraised completed and ongoing studies for all systemic therapies used in the management of AD, and captured studies on all interventions and comparators of interest to the MTA reported here. The EAG re-evaluated the studies included by the review against the inclusion criteria presented in Table 3.
Searches of electronic databases to update the search of the identified review (search date 1 August 2019 to 8 July 2021) retrieved 1365 records (post deduplication) that were of possible relevance to the review (see Report Supplementary Material 1). First-pass appraisal of the 1365 unique records led to exclusion of 1244 records. Full publications for 121 references from the EAGs’ literature review were ordered, of which publications for two records (both conference abstracts) could not be obtained. 58,59 Manual searching of the identified systematic review forming the basis of the update search identified 18 supplementary records for full-text appraisal. 60–77 The EAG’s SLR retrieved records for an additional seven systematic reviews that evaluated one or more systemic interventions of interest to the MTA:78–84 one of the identified reviews is a ‘living’ systematic review and as such is continually updated. 79 Cross-referencing of bibliographies of the seven systematic reviews identified one additional record85 to those retrieved by the EAG and the original review, giving a total of 138 full text publications screened for inclusion in the review. Additionally, two sets of committee papers outlining recommendations from NICE for the use of dupilumab12 and baricitinib13 in the management of AD were also identified by searching the NICE website. The EAG’s literature search captured all studies presented in the committee papers for baricitinib and dupilumab. As noted in the section Definition of the decision problem, the EAG had access to submissions to the STA process from the individual companies producing abrocitinib, tralokinumab and upadacitinib, with documents available including the original submission and responses to requests for data from the EAG. The EAG’s literature search identified all relevant studies reported in the company submissions.
Of the 138 full articles evaluated, 38 publications describing 23 studies were relevant to the review (including 4 errata; Table 4): citation details for conference abstracts identified during the EAG’s literature review and related to full publications are provided for completeness. Six additional studies for which full-text publications were not available at the time of writing were identified from searches of trial registries (ClinicalTrials.gov and EU Clinical Trials Register). The six studies each evaluated one of abrocitinib, tralokinumab or upadacitinib, and relevant results were provided by the companies during clarification (see Table 4). Summaries of the studies included in the review are presented by key characteristics of studies (see Table 4). A list of publications screened but subsequently excluded (with reasons for exclusion) from the review is available in Report Supplementary Material 1.
| Study | Population | Intervention(s) | Comparator | Duration of treatment and follow-up | Additional information and related references |
|---|---|---|---|---|---|
| Interventions yet to be recommended by NICE for AD | |||||
| Abrocitinib (oral) | |||||
| Monotherapy | |||||
| Phase IIb27 | Adults with moderate-to-severe AD (EASI score ≥ 12, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical or systemic therapies. | Abrocitinib 200 mg (N = 55) Abrocitinib 100 mg (N = 56) Abrocitinib 30 mg (N = 51) Abrocitinib 10 mg (N = 49) |
Placebo (N = 56) | 12-week treatment phase | Related conference abstract60 Erratum to full publication101 |
| JADE MONO-128 | Adolescents and adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical or systemic therapies. | Abrocitinib 200 mg (N = 154) Abrocitinib 100 mg (N = 156) |
Placebo (N = 77) | 12-week treatment phase | Related conference abstracts102,103 JADE MONO-1 and JADE MONO-2 are independent multicentre RCTs that were run in parallel. Use of rescue medication was not permitted. |
| JADE MONO-229 | Abrocitinib 200 mg (N = 155) Abrocitinib 100 mg (N = 158) |
Placebo (N = 78) | |||
| Combination with TCS | |||||
| JADE TEEN30,31 | Adolescents with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical or systemic therapies. | Abrocitinib 200 mg (N = 94) Abrocitinib 100 mg (N = 95) |
Placebo (N = 96) | 12-week treatment phase | Topical therapies allowed during the trial included low- or medium-potency TCS, TCIs and topical phosphodiesterase 4-inhibitors. People were allowed to use more than one topical therapy. At the time of writing, results from JADE TEEN are not available in a published peer-reviewed journal. Reported details have been extracted from conference abstracts and from information provided by the company during the MTA process. Use of rescue medication was not permitted. |
| JADE COMPARE32 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical or systemic therapies. | Abrocitinib 200 mg (N = 226) Abrocitinib 100 mg (N = 238) |
Dupilumab 300 mg Q2W (N = 242) Placebo (N = 131) Dupilumab: 600 mg loading dose |
20-week treatment phase, with subsequent long-term extension | Related conference abstract.104 Topical therapies allowed during the trial included low- or medium-potency TCS, TCIs and topical phosphodiesterase 4-inhibitors. People were allowed to use more than one topical therapy. Use of rescue medication was not permitted. |
| JADE DARE90 | Adults with moderate-to-severe AD who required systemic therapy or had inadequate response to topical therapies. | Abrocitinib 200 mg (N = 362) | Dupilumab 300 mg Q2W (N = 365) | 26-week treatment phase | Topical therapies allowed during the trial included low- or medium-potency TCS, TCIs, and topical phosphodiesterase 4-inhibitors. Use of rescue medication was not permitted. |
| Tralokinumab (SC injection) | |||||
| Monotherapy | |||||
| ECZTRA 135 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical or systemic therapies. | Tralokinumab 300 mg (N = 603) Tralokinumab: 600 mg loading dose |
Placebo (N = 199) | 16-week treatment phase. Those achieving a clinical response at week 16 (defined as IGA of 0 or 1 or at least 75% reduction EASI score from baseline) moved onto maintenance treatment that continued until week 52. Patients in the tralokinumab arm who achieved EASI 75 or IGA 0/1 were rerandomised to receive tralokinumab 300 mg either Q2W or Q4W, or placebo. Patients in the placebo arm who achieved EASI 75 or IGA 0/1 continued to receive placebo. The remaining patients received open-label tralokinumab Q2W and had the option of adding TCS. |
ECZTRA 1 and ECZTRA 2 are independent multicentre RCTs that were run in parallel. |
| ECZTRA 235 | Tralokinumab 300 mg (N = 593) Tralokinumab: 600 mg loading dose |
Placebo (N = 201) | |||
| ECZTRA 592 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical therapies. | Tralokinumab 300 mg (N = 107) Tralokinumab: 600 mg loading dose |
Placebo (N = 108) | 16-week treatment phase followed by 14-week off-treatment follow-up period for the assessment of safety. Dependent on eligibility, people could transfer to an open-label, long-term trial at week 16 or later. | The objective of the study was to assess whether tralokinumab can affect the body’s immune response to vaccines. At the time of writing, results from ECZTRA 5 are not available in a published peer-reviewed journal. Reported details have been extracted from the study entry on ClinicalTrials.gov and from information provided by the company during the MTA process. |
| Combination with TCS | |||||
| Phase IIb91 | Adults with moderate-to-severe AD (EASI score ≥ 12, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months. | Tralokinumab 300 mg (N = 52) Tralokinumab 150 mg (N = 51) Tralokinumab 45 mg (N = 50) |
Placebo (N = 51) | 12-week treatment phase. | Related conference abstract.105 Unclear from full publication whether those enrolled in the tralokinumab group were given a loading dose of tralokinumab. |
| ECZTRA 336 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical therapies. | Tralokinumab 300 mg (N = 252) Tralokinumab: 600 mg loading dose |
Placebo (N = 126) | 16-week treatment phase. After 16 weeks, people could continue in an extension phase in which, depending on response, people could receive one of tralokinumab 300 mg Q2W, tralokinumab 300 mg Q4W or placebo. |
Related conference abstract.106 Concomitant TCS was mometasone furoate 0.1%. |
| ECZTRA 738 | Adults with moderate-to-severe AD (EASI score ≥ 20, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical therapies and either no previous CsA exposure and not currently a candidate for CsA treatment or previous exposure to CsA and had an inadequate response. | Tralokinumab 300 mg (N = 140) Tralokinumab: 600 mg loading dose |
Placebo (N = 137) | 26-week treatment phase. | Concomitant TCS was mometasone furoate 0.1%. At the time of writing, results from ECZTRA 7 are not available in a published peer-reviewed journal. Reported details have been extracted from the study entry on ClinicalTrials.gov and from information provided by the company during the MTA process. |
| Upadacitinib (oral) | |||||
| Monotherapy | |||||
| Phase IIb93 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months. | Upadacitinib 30 mg (N = 42) Upadacitinib 15 mg (N = 42) Upadacitinib 7.5 mg (N = 42) |
Placebo (N = 41) | 16-week treatment phase followed by 72-week double-blind, randomised withdrawal period. | Conference abstract reporting longer-term follow-up results.107 |
| HEADS UP40 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) and with a history of inadequate response to topical therapies or for whom topical therapies were medically inadvisable. | Upadacitinib 30 mg (N = 325) | Dupilumab 300 mg Q2W (N = 325) | 24-week treatment period followed by 12-week follow-up. | At the time of writing, results from HEADS UP are not available in a published peer-reviewed journal. Reported details have been extracted from the information provided by the company during the MTA process and the study entry on ClinicalTrials.gov. |
| MEASURE UP139 | Adolescents and adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) and with a history of inadequate response to topical therapies or for whom topical therapies were medically inadvisable. | Upadacitinib 30 mg (N = 285) Upadacitinib 15 mg (N = 281) |
Placebo (N = 281) | 16-week treatment phase followed by blinded extension period for up to 120 weeks of treatment. At week 16, people in the placebo group were randomised to upadacitinib 30 or 15 mg for the blinded extension period. |
Erratum to full publication.108 MEASURE UP1 and MEASURE UP2 are independent multicentre RCTs that were run in parallel. |
| MEASURE UP239 | Upadacitinib 30 mg (N = 282) Upadacitinib 15 mg (N = 276) |
Placebo (N = 278) | |||
| Combination with TCS | |||||
| AD UP41 | Adolescents and adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 36 months. | Upadacitinib 30 mg (N = 297) Upadacitinib 15 mg (N = 300) |
Placebo (N = 304) | 16-week treatment phase followed by blinded extension period for up to 120 weeks of treatment. | Related conference abstract.109 Erratum to full publication.110 Initial concomitant TCS was of medium potency (clinician choice), moving to low potency for 7 days once lesions became ‘clear’ or ‘almost clear’ or after 3 weeks, whichever occurred sooner. |
| RISING UP94 | Adolescents and adults with moderate-to-severe AD and with a history of inadequate response to topical therapies or for whom topical therapies were medically inadvisable. | Upadacitinib 30 mg (N = ?) Upadacitinib 15 mg (N = ?) |
Placebo (N = ?) | 16-week treatment phase followed by a long-term extension study. | Study carried out in Japan and enrolled 272 people. At the time of writing, results from RISING UP are not available in a published peer-reviewed journal. Reported details have been extracted from the study entry on ClinicalTrials.gov. |
| Interventions recommended as treatment options by NICE for the management of moderate-to-severe AD | |||||
| Baricitinib (oral) 13 | |||||
| Monotherapy | |||||
| BREEZE-AD195 | Adults with moderate-to-severe AD (EASI score ≥ 16, vIGA-AD score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months prior to screening, and documented history of inadequate response to topical or systemic therapies within 6 months before screening. | Baricitinib 4 mg (N = 125) Baricitinib 2 mg (N = 123) Baricitinib 1 mg (N = 127) |
Placebo (N = 249) | 16-week treatment phase and follow-up at 4 weeks after treatment. | Related conference abstract.111 Long-term extension study enrolling those with partial or full response from BREEZE-AD1 and BREEZE-AD2 (BREEZE-AD3).98 BREEZE-AD1 and BREEZE-AD2 are independent multicentre RCTs that were run in parallel. |
| BREEZE-AD295 | Baricitinib 4 mg (N = 123) Baricitinib 2 mg (N = 123) Baricitinib 1 mg (N = 125) |
Placebo (N = 244) | |||
| Combination with TCS | |||||
| Phase II77 | Adults with moderate-to-severe AD (EASI score ≥ 12, and ≥ 10% BSA involvement) for at least 24 months prior to screening, and documented history of inadequate response to topical or systemic therapies. | Baricitinib 4 mg (N = 38) Baricitinib 2 mg (N = 37) |
Placebo (N = 49) | 16-week treatment phase. | Concomitant TCS was triamcinolone 0.1%. |
| BREEZE-AD496 | Adults with moderate-to-severe AD (EASI score ≥ 16, vIGA-AD score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months and a history of inadequate response to topical therapy and a history of intolerance to, contraindication to, or inadequate response to CsA. | Baricitinib 4 mg (N = 92) Baricitinib 2 mg (N = 185) Baricitinib 1 mg (N = 93) |
Placebo (N = 93) | 5-week wash-out. 52-week treatment period (followed by a 52-week double-blind long-term extension which included a down-titration substudy for responders and rerandomisation for non-responders). 4-week post-treatment follow-up. |
At the time of writing, results from BREEZE AD4 are not available in a published peer-reviewed journal. Reported details have been extracted from the study entry on ClinicalTrials.gov and data available in TA681.13 Background TCS therapy with moderate-potency and/or low-potency TCS. |
| BREEZE-AD797 | Adults with moderate-to-severe AD (EASI score ≥ 16, vIGA-AD score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months prior to screening, and documented history of inadequate response to topical or systemic therapies within 6 months before screening. | Baricitinib 4 mg (N = 111) Baricitinib 2 mg (N = 109) |
Placebo (N = 109) | 16-week treatment phase and follow-up at 4 weeks after treatment. | Patients were allowed to use concomitant TCS that were of moderate or low potency. |
| Dupilumab (SC injection)12 | |||||
| Monotherapy | |||||
| Phase IIb75,85 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 36 months prior to screening, and documented history of inadequate response to topical or systemic therapies within 6 months before screening. | Dupilumab 300 mg Q4W (N = 65) Dupilumab 300 mg Q2W (N = 64) Dupilumab 300 mg QW (N = 63) Dupilumab 200 mg Q2W (N = 61) Dupilumab 100 mg Q4W (N = 65) Dupilumab: 600 mg loading dose. |
Placebo (N = 61) | 16-week treatment phase. | N/A |
| LIBERTY AD-ADOL99 | Adolescents with moderate-to-severe AD for at least 12 months prior to screening, and inadequately controlled by topical treatment or for whom topical treatment was medically inadvisable. | Dupilumab 300 mg Q4W (N = 84) Dupilumab 200 or 300 mg Q2W (N = 82) |
Placebo (N = 85) | 16-week treatment phase. | Related conference abstracts.31,112,113 In the dupilumab Q2W group, dose was weight based, with those weighing < 60 kg receiving 200 mg Q2W after a loading dose of 400 mg. Those weighing ≥ 60 kg received 300 mg Q2W after a loading dose of 600 mg. |
| LIBERTY AD SOLO-174 | Adults with moderate-to-severe AD (IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 36 months prior to screening, and documented history of inadequate response to topical or systemic therapies within 6 months before screening. | Dupilumab 300 mg Q2W (N = 224) Dupilumab 300 mg QW (N = 223) Dupilumab: 600 mg loading dose |
Placebo (N = 224) | 16-week treatment phase. People achieving an IGA score of 0 or 1 or EASI 75 at 16 weeks were rerandomised to dupilumab 300 mg at various intervals (QW, Q2W, Q4W or Q8W) or to placebo (SOLO-CONTINUE). |
Pooled analysis.114 Long-term extension SOLO-CONTINUE.115 Assessment of efficacy of dupilumab in different racial subgroups.44 SOLO-1 and SOLO-2 are independent multicentre RCTs that were run in parallel. |
| LIBERTY AD SOLO-274 | Dupilumab 300 mg Q2W (N = 233) Dupilumab 300 mg QW (N = 239) Dupilumab: 600 mg loading dose. |
Placebo (N = 236) | |||
| Combination with TCS | |||||
| LIBERTY AD CAFE100 | Adults with moderate-to-severe AD (EASI score ≥ 20, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 12 months, documented history of inadequate response to topical therapies and either no previous CsA exposure and not currently a candidate for CsA treatment or previous exposure to CsA and an inadequate response. | Dupilumab 300 mg Q2W (N = 107) Dupilumab 300 mg QW (N = 110) Dupilumab: 600 mg loading dose. |
Placebo (N = 108) | 16-week treatment phase. | Initial concomitant TCS was of medium potency applied OD to active lesions. Low-potency TCS could be applied to areas of thin skin. |
| LIBERTY AD CHRONOS70 | Adults with moderate-to-severe AD (EASI score ≥ 16, IGA score of ≥ 3 and ≥ 10% BSA involvement) for at least 36 months prior to screening, and documented history of inadequate response to topical or systemic therapies within 6 months before screening. | Dupilumab 300 mg Q2W (N = 106) Dupilumab 300 mg QW (N = 319) Dupilumab: 600 mg loading dose |
Placebo (N = 315) | 52-week treatment phase and 12 weeks of follow-up. Efficacy at week 16 was the study’s primary objective. |
Topical therapies allowed during the trial included low- or medium-potency TCS and TCI. People were allowed to use more than one topical therapy. Initial concomitant TCS was of medium potency, moving to low potency for 7 days once lesions became ‘clear’ or ‘almost clear’. |
The update search of the literature carried out on 29 November 2021 identified an additional 377 unique records. Given that only 5 months had passed since the EAG’s original search, the EAG limited its inclusion criteria to full publications of RCTs not previously found. Of the 377 titles and abstracts appraised, only one record identified a novel RCT. 86 The study compares dupilumab 300 mg Q2W versus placebo with clinical efficacy assessed at 16 weeks in Chinese patients. Patients were required to be aged ≥ 18 years old and have moderate-to-severe AD for ≥ 3 years, which could not be adequately controlled with topical medications or for which topical treatment was inadvisable. Additional criteria for eligibility were EASI score ≥ 16, IGA score ≥ 3 and ≥ 10% body surface area (BSA) affected by AD. Based on the full-text publication, it is unclear whether people who had previously received systemic therapy were included, and, thus, data are not available for those having inadequate response to systemic therapy prior, or who cannot tolerate or are contraindicated to systemic therapy. Additionally, the study enrols only adults, and focuses on monotherapy. As data are not available by prior line of therapy, the study would not inform the EAG’s analyses and has not been described in full.
Seven publications61–67 describing four studies of CsA versus placebo were identified by the systematic review forming the basis of the EAG’s literature review. 47 The published studies met the inclusion criteria of the EAG’s SLR for RCTs. However, because none of the studies of CsA reported data on the clinical outcomes of interest to the review and the studies were carried out 15 years ago or longer, the studies are not discussed further, and study characteristics are not reported. As no RCT for CsA was available to inform a NMA, the EAG carried out a SLR of observational studies evaluating CsA in the treatment of AD (described in greater detail in the section Systematic review of existing cost-effectiveness evidence).
The company submission for upadacitinib reported results from a search to identify observational studies on CsA that could inform a NMA. The company’s review identified one study describing an indirect comparison of CsA with dupilumab plus TCS to generate an estimate of comparative clinical effectiveness for EASI 75. 87 The EAG considered the company’s literature review to be robust and chose to carry out an update search to identify additional potentially relevant studies.
Searches of electronic databases to update the company’s search of the identified review (search date 1 January 2019 to 8 July 2021) retrieved 746 records (post deduplication) that were of possible relevance to the review (see Report Supplementary Material 1). First-pass appraisal of the 746 unique records led to exclusion of 743 records. Full publications for three references from the EAG’s literature review were reviewed,87–89 one of which was the study described in the company submission for upadacitinib (Ariens et al.). 87 One publication was identified as a Letter to the Editor detailing the drug survival of dupilumab and was excluded. 89 The second publication described a retrospective observational study with a primary outcome of drug survival of dupilumab and CsA when used to treat moderate-to-severe AD in adults (N = 251), with EASI 75 captured as a secondary outcome (Dal Bello et al.). 88 Both full-text publications report data on proportion of people achieving EASI 75 with dupilumab and with CsA and so both meet the EAG’s inclusion criteria. 87,88 However, in Ariens et al.,87 the authors applied regression models to adjust data for people receiving CsA to those of people receiving dupilumab 300 mg Q2W with TCS from LIBERTY AD CHRONOS. 70 The authors repeated the analysis in reverse, that is, adjusting results from those treated with dupilumab from CHRONOS to those given CsA in the registry. No type of adjustment was reported in the second publication identified by the EAG. 88 Ariens et al. 87 reported the adjusted proportion of people achieving EASI 75 after 12–16 weeks of treatment with CsA to be 52%, compared with 75% as observed in the dupilumab group from CHRONOS. By contrast, Dal Bello et al. 88 reported achievement of EASI 75 at 16 weeks by 44.3% (66/149) of those in the dupilumab group compared with 24.5% (25/102) of people treated with CsA. The EAG acknowledges that there is a considerable difference in estimates for dupilumab between the two studies. As data reported by Ariens at el. 87 for dupilumab are derived from the RCT, CHRONOS, rather than a registry, and CHRONOS is included in the network to generate estimates of first-line treatment, the EAG considered the adjusted estimate for CsA reported by Ariens et al. 87 to be more the appropriate choice to inform a NMA of RCTs.
Interventions to be assessed
As noted above, the EAG identified 23 studies (reported in 38 publications) of relevance to the MTA. Below, the EAG provides a summary of the key characteristics of the studies included in its review. The EAG considers it important to note that the identified studies predominantly include mixed populations of people with moderate-to-severe AD, with some studies comprising both adolescents and adults, as well as a combination of people receiving systemic therapy as a first-line or second-line regimen. Thus, data informing the NMAs for the populations and outcomes of interest to the MTA (see Assessment of clinical effectiveness) are predominantly derived from post hoc subgroups, which introduces bias and uncertainty around the robustness of the results of any analyses. Further discussion on the limitations in using post hoc subgroup data is available in the section Potential sources of heterogeneity and limitations of the available evidence base.
For abrocitinib, tralokinumab and upadacitinib, where data were not available in the submissions to the STA process, data were supplied by the companies during a clarification stage. For dupilumab and baricitinib, the relevant data were primarily extracted from the committee papers accompanying the NICE recommendations for their use in the NHS. 12,13 The overview of the included studies, together with the accompanying quality assessment, pertains to the design and conduct of the RCT from which data are derived, and not the post hoc subgroups and subsequent analyses.
Six studies evaluating abrocitinib at the recommended dose (200 or 100 mg orally, OD) in the treatment of adolescents and adults with moderate-to-severe AD were included in the MTA (see Table 4). 27–32 Of the six studies, five are Phase III RCTs,28–32,90 and one is a Phase IIb dose-ranging study. 27 Five studies included a group receiving placebo. 27–32
Three studies assessed clinical effectiveness of abrocitinib as a monotherapy, with one study focusing on an adult population (Phase IIb27), with the two remaining studies enrolling adolescents as well as adults (JADE MONO-128 and JADE MONO-229). Of the three studies evaluating abrocitinib in combination with TCS, one study recruited adolescents only (JADE TEEN30,31), and the other two enrolled adults only (JADE COMPARE,32 JADE DARE90) and included an active comparator group of dupilumab 300 mg Q2W with TCS. Topical therapies allowed in JADE TEEN JADE COMPARE and JADE DARE included low- or medium-potency TCS, TCIs and topical phosphodiesterase 4-inhibitors. People were allowed to use more than one topical therapy. Use of rescue therapy was not permitted in any of the included Phase III RCTs evaluating abrocitinib. 28–32,90 Across the included studies, primary efficacy end points were assessed at 12 weeks. The treatment phase lasted 12 weeks in all studies except for JADE COMPARE in which the treatment phase lasted 20 weeks and JADE DARE in which the treatment phase was 26 weeks.
Six studies were identified that included tralokinumab at the recommended dose (loading dose of 600 mg followed by 300 mg subcutaneously Q2W) either as a monotherapy or in combination with TCS and compared with placebo (see Table 4):35,36,38,91,92 results from the ECZTRA 1 and ECZTRA 2 RCTs were reported in the same publication. 35 The EAG identified one ongoing RCT, ECZTRA 8, that is located in Japan and compares tralokinumab in combination with TCS versus placebo. 37 Results for ECZTRA 8 are not yet available. The EAG notes that clinical practice on use of high-potency TCS differs between Japan and England, with use of high-potency TCS more common in Japan. Use of different potency of TCS across studies could introduce clinical heterogeneity into any analyses. The EAG considers inclusion of results from ECZTRA 8 could increase heterogeneity and uncertainty into a NMA.
All included studies enrolled adults only, which aligns with the population for which the company sought a recommendation from NICE in their submission to the STA process. Results for three of the studies identified have yet to be published in peer-reviewed journals, including the key ECZTRA 7 study,38 with the public record of the studies taken from the clinical trial registry ClinicalTrials.gov. 92 Additional details were available in the company submission to the STA process and were supplemented with information provided by the company on request.
ECZTRA 1 and ECZTRA 2 (reported in the same publication) are independent multicentre RCTs that were run in parallel and evaluated tralokinumab as a monotherapy. 35 In addition to ECZTRA 1 and ECZTRA 2, a Phase IIb study91 and ECZTRA 592 also evaluated tralokinumab as a monotherapy, with the remaining two studies assessing tralokinumab in combination with TCS. 36–38 The objective of ECZTRA 5 was to assess whether tralokinumab affects the body’s immune response to vaccines: clinical outcomes of interest, such as EASI 75, were captured as secondary outcomes. In response to the EAG’s request for data for ECZTRA 5 for the subgroup of adults receiving tralokinumab monotherapy after inadequate response to, inability to tolerate, or contraindicated to CsA, the company declined to provide the data, commenting that:
The number of ECZTRA 7-like patients in the ECZTRA 5 study was limited. The study was designed to assess whether treatment with tralokinumab can affect the body’s immune response to vaccines. Given limited time, we have presented only the baseline characteristics of all patients for reference.
Similarly, data for the relevant subgroup from the Phase IIb study are not available, with the company replying, ‘The Phase IIb dose ranging study was not powered to include analyses of an ECZTRA-7-like subgroup and efficacy was only assessed up to week 12’. Given the likely small number of events and patients forming the relevant subgroups for the Phase IIb study and ECZTRA 5, the EAG considers that the omission of the data is unlikely to have had a substantial impact on the results generated from the NMAs involving tralokinumab.
Of the two studies evaluating tralokinumab in combination with TCS, ECZTRA 3 and ECZTRA 7, ECZTRA 7 is a key study of relevance to the MTA, assessing treatment of patients with severe AD who had not had adequate control with, or had intolerance or contraindications to, CsA. 38 In ECZTRA 7, efficacy and safety were assessed over a 26-week treatment phase, whereas all other studies assessed primary clinical outcomes at 16 weeks after the start of treatment.
Six studies were included that assessed upadacitinib (30 or 15 mg orally, OD) in adolescents and adults (see Table 4):39–41,93,94 results from the MEASURE UP1 and MEASURE UP2 RCTs were reported in the same publication. 39 Of the six studies, five were Phase III RCTs,39–41,94 with one study being a Phase IIb design. 93 MEASURE UP1, MEASURE UP2 and the Phase IIb study all evaluated upadacitinib as a monotherapy versus placebo, each assessing clinical effectiveness at 16 weeks of treatment. Additionally, HEADS UP40 compared upadacitinib versus dupilumab as a monotherapy but with a treatment period of 24 weeks.
Atopic dermatitis UP is the only study for which data are available on the clinical effectiveness of upadacitinib in combination with TCS, and reports outcomes after 16 weeks of treatment. 41 RISING UP is a RCT carried out in Japan for which data are not yet available. 94 In their response to the EAG’s request for data from RISING UP, the company commented that, ‘Results from the AD UP, MEASURE UP1, MEASURE UP2 and HEADS UP were prioritised for this response’. Given the likely low number of events and of patients in the relevant subgroup, together with the fact that clinical practice in Japan on use of TCS differs from that in England, the EAG considers that omission of the results from RISING UP is unlikely to have a substantial impact on the results of the NMA involving upadacitinib in combination with TCS.
Available treatment options
Five studies (four publications) evaluating the efficacy and safety of baricitinib (4 or 2 mg orally, OD) for the treatment of moderate-to-severe AD in adults were identified by the EAG’s literature review (see Table 4). 77,95–97 BREEZE-AD1 and BREEZE-AD2 are two Phase III RCTs conducted in parallel that evaluated baricitinib monotherapy versus placebo and captured treatment response as 16 weeks, the results of which are available in the same publication. 95 A long-term extension study enrolling those with partial or full response from BREEZE-AD1 and BREEZE-AD2 was also identified (BREEZE-AD3). 98 Two additional Phase III studies, BREEZE-AD496 and BREEZE-AD7,97 together with a Phase II study,77 compared baricitinib in combination with TCS versus placebo. Of note, BREEZE-AD4 enrolled adults with moderate-to-severe AD and a history of intolerance to, contraindication to or inadequate response to CsA.
Five RCTs (four publications70,74,99,100) and one Phase IIb75,85 study compared dupilumab (300 mg Q2W given subcutaneously) with placebo in people with moderate-to-severe AD (see Table 4). Together with the Phase IIb study, three RCTs evaluated dupilumab as a monotherapy, with one enrolling adolescents only (AD ADOL99), and two focusing on adults (SOLO 174 and SOLO 274). SOLO 1 and SOLO 2 were of similar design and results were available in the same publication. 74 The remaining two RCTs – CAFÉ100 and CHRONOS70 – evaluated efficacy and safety of dupilumab in combination with TCS in an adult population. To be eligible for entry into CAFÉ, patients were required to have a history of intolerance, inadequate response or contraindication to CsA. All studies involved patients with moderate-to-severe AD whose disease was not adequately controlled with topical medications or for whom topical treatment was medically inadvisable and reported primary results after 16 weeks of treatment.
As noted in the section Quantity of research available, four studies evaluating clinical effectiveness of CsA versus placebo were identified,61–67 but none of the studies reported data on the clinical outcomes of interest to the MTA. Of the studies retrieved from a systematic search of the observational literature, the EAG selected Ariens et al. 87 to inform a NMA of first-line treatments for moderate-to-severe AD. Ariens et al. 87 describe an indirect comparison of CsA versus dupilumab plus TCS to generate an estimate of comparative clinical effectiveness for EASI 50 and EASI 75. For effectiveness of CsA, the authors sourced patient-level data from a registry of those treated with CsA at the University Medical Center (UMC), Utrecht, the Netherlands. Concomitant use of TCS was allowed as needed for patients treated with CsA. Effectiveness of dupilumab (300 mg Q2W) plus TCS was derived from the results of the CHRONOS RCT. 70 The EAG notes that the mean baseline EASI score in the group treated with CsA was considerably lower than that in the dupilumab group, with mean EASI scores of 19.3 [standard deviation (SD) 8.4] and 33.6 (SD 13.3) for the CsA and dupilumab groups, respectively. The lower EASI score observed in the CsA group reflects feedback from the EAG’s expert advisors that people seen in clinical practice are more likely to present with moderate, rather than severe, AD, whereas those enrolled in clinical trials predominantly have more severe AD.
As those receiving CsA were treated in clinical practice rather than as part of a clinical trial, there were no fixed clinic visits, and so the analysis of EASI 50 and EASI 75 was based on time periods rather than a strict number of weeks of treatment. 87 Data on EASI 50 and EASI 75 were collated for people treated between weeks 12 and 16, and between weeks 24 and 30. To facilitate the comparison with CsA, EASI 50 and EASI 75 scores from CHRONOS were those recorded at weeks 16 and 28 in the scheduled follow-up assessments. To derive the estimate of effect for achieving EASI 75, the authors used regression models to adjust data for people receiving CsA to those of people allocated to dupilumab 300 mg Q2W plus TCS in CHRONOS. 70 The authors also adjusted results from those treated with dupilumab from CHRONOS to those given CsA. Given that data from CHRONOS are derived from a RCT, the EAG used the results from the adjustment of people receiving CsA to those allocated to dupilumab in CHRONOS to inform the NMA for first-line treatment. The dependent variable was EASI 50 or EASI 75 (achieved or not achieved), and the focal regressor was a treatment indicator for CsA versus dupilumab use. Missing data were imputed by means of the last observation carried forward (LOCF) method for both populations. Additional regressors in the model were sex, baseline EASI and baseline thymus and activation-regulated chemokine (TARC) level. Adjusted weighting was carried out according to the baseline data.
Potential sources of heterogeneity and limitations of the available evidence base
Inclusion criteria across included studies were predominantly comparable, with studies enrolling either adults or adolescents and adults with moderate-to-severe AD. Differences were noted in the level of baseline severity of AD required at baseline in terms of EASI score. Most studies specified an EASI score of ≥ 16 for eligibility, with three studies requiring a score of ≥ 12 and two of ≥ 20 (see Table 4). Most studies also required baseline IGA score of ≥ 3, and ≥ 10% BSA involvement (see Table 4). Duration of AD at enrolment ranged from at least 12 months to a minimum of 36 months. The EAG notes that the mean baseline EASI score was around 29 in most studies (baseline characteristics available in Report Supplementary Material 1), which, based on the EASI score categories (available in Table 1), denotes severe AD. Clinical experts advising the EAG commented that the patients enrolled in the clinical studies have more severe AD than would typically be seen in clinical practice, with most patients presenting with disease that would be categorised as moderate severity. The primary outcome for clinical effectiveness for the MTA is the composite outcome of EASI 50 + DLQI ≥ 4. The EAG notes that some studies enrolled participants with a baseline DLQI score of 3, and, as such, these patients would not be able to contribute to the composite outcome. The EAG notes that, based on number of people included in analyses for EASI 50 + DLQI ≥ 4, most people enrolled in the studies had a baseline score of DLQI 4 and so the EAG considers the impact on the robustness of the outcome to be minimal.
Despite some differences in inclusion criteria relating to disease severity, mean baseline EASI score and the proportion of people with an IGA score of 3 or 4 were comparable across the included studies. Most studies required a documented history of inadequate response to topical or systemic therapies. Two studies (ECZTRA 738 and CAFÉ100) specified that people either had not been exposed to CsA and were not a candidate for CsA treatment or had previous exposure to CsA and had an inadequate response.
Although baseline characteristics for the full-trial populations are comparable, as noted earlier, most studies, the exceptions being ECZTRA 738 and CAFÉ,100 included a blended population and clinical data to inform the NMAs are derived from post hoc subgroups. The use of post hoc subgroups increases the comparability and applicability of the analyses, but also introduces bias and uncertainty to the results generated by the NMAs. The limitations and potential sources and types of heterogeneity for the populations of interest to the MTA are discussed in greater detail in the discussion of the interpretation of the results from the relevant NMA.
As expected, variation was noted across studies in the use of a washout period for TCS before randomisation to treatment, with some studies not including a washout period and, for those that did, the time allocated for washout varied (details available in Report Supplementary Material 1). In studies evaluating treatment in combination with TCS, differences were noted in the type and potency of concomitant TCS (low or medium). In studies allowing use of rescue therapy, rescue treatment was given at the discretion of the investigator and typically comprised use of TCS or higher-potency TCS in studies evaluating combination treatment or systemic immunosuppressant. Notably, in BREEZE AD7, high- or ultra-high-potency TCSs were permitted as rescue therapy. The use of rescue therapy was prohibited in trials evaluating abrocitinib (JADE COMPARE, JADE DARE, JADE MONO-1 and MONO-2, and JADE TEEN).
In most studies, patients receiving topical rescue treatment continued treatment with the study drug. By contrast, those receiving systemic rescue therapy discontinued the study drug, either permanently or until a pre-determined period of time after the last dose of systemic rescue treatment. The use of and the level of potency of topical rescue therapy may impact on the treatment and placebo response achieved in individual studies. Disparity in type of rescue therapy used across studies could potentially lead to clinical heterogeneity in observed placebo response, which may introduce bias and uncertainty into the NMA. The EAG evaluated the potential impact of placebo response on estimates of effect generated from the NMAs (see Assessment of clinical effectiveness).
In addition to most of the data informing the NMAs being derived from post hoc subgroups, the EAG notes that much of the data for baricitinib were unavailable at the time of writing. Results for studies evaluating baricitinib are not yet published in peer-reviewed journals and data submitted to the STA process were redacted from the committee papers accompanying the recommendation by NICE. Lack of data for baricitinib on clinical outcomes of interest to the MTA precluded inclusion of baricitinib in most of the relevant NMAs. However, data on clinical effectiveness of baricitinib in combination with TCS were available for EASI 75, which allowed inclusion of baricitinib in a NMA. No randomised evidence was identified to inform the efficacy of CsA, which is the relevant comparator in the first-line setting. However, an observational study carrying out an indirect comparison of CsA with or without TCS versus dupilumab plus TCS (based on CHRONOS) was identified. 87 Based on the reported methods and the populations analysed, the EAG considers the observational study to represent the best available evidence to facilitate comparison of upadacitinib and dupilumab in the first-line setting.
Quality of research available
Most of the studies included in the assessment of clinical effectiveness are considered to be well-conducted and well-designed Phase III RCTs, and, as such, are at an overall low risk of bias (summary of quality assessment in Report Supplementary Material 1). All studies are described as randomised, aside from the single observational study that provides evidence on CsA. 87 For most studies, the method of randomisation and allocation concealment for the included RCTs was deemed to be adequate. However, details on the methods used to generate the randomisation sequence were not available for five studies and so risk of allocation bias was judged to be unclear for these trials (see Report Supplementary Material 1).
Limited details were available in the full publications on the methods implemented to initially conceal allocation and to subsequently maintain masking of treatment from clinicians and participants. However, additional information was available from other sources, including clinical trial registries, the company submissions to the STA process and committee papers for recommended treatment options (dupilumab and baricitinib). The tools used to assess severity of the signs and symptoms of AD, and therefore level of improvement, are subjective in nature and so most recorded outcomes are at increased risk of bias. Most of the included studies employed a double-blind process to mitigate against introducing bias into outcome assessment. However, for most studies described as double blind, it was unclear whether the outcome assessor was the treating clinician and, if not, whether the outcome assessor was masked to treatment. Follow-up at the end of treatment (12–16 weeks) was high across most studies, with several studies categorised as a low risk of attrition bias for the outcomes evaluated. No study was assessed as high risk of selective reporting, with results routinely reported for all pre-specified outcomes.
The quality of the observational study87 used to facilitate comparison of upadacitinib and dupilumab in the first-line setting was assessed using the case–control component of the Newcastle–Ottawa Scale (NOS) for assessing the quality of non-randomised studies. 116 Based on the NOS, the observational study scored a point for most aspects of selection, comparability and exposure. The EAG acknowledges that the NOS is not an ideal tool to assess quality as Ariens et al. 87 does not include a control group, with both groups receiving an active treatment. Data on patients receiving CsA were acquired from secure records and, with the exception of EASI 75, the groups had comparable baseline characteristics. As noted in the section Ciclosporin A, the lower baseline EASI score of those receiving CsA reflects the patient population likely to present in clinical practice with moderate-to-severe AD, whereas people enrolled in clinical trials typically have more severe AD. Overall, the EAG considers the data presented by Ariens et al. 87 to represent the most robust evidence available to inform the NMA.
As highlighted in the section Quantity of research available, despite the low overall risk of bias of the studies, much of the data informing the analyses of comparative clinical effectiveness are derived from post hoc subgroups, which introduces uncertainty, the level of which cannot be quantified, into the analyses.
Assessment of clinical effectiveness
NMAs were conducted for the primary outcome EASI 50 + DLQI ≥ 4 and for EASI 75. Results for QoL (captured as change from baseline in EQ-5D), use of rescue medication and the number of days free from TCS are described narratively in Report Supplementary Material 1.
The primary NMAs were based on using all observed data, regardless of rescue medication use to determine response. Sensitivity analyses were conducted where patients requiring rescue medication were considered non-responder. Data based on this censoring rule were available for baricitinib, dupilumab, tralokinumab and upadacitinib, but not for CsA, and for abrocitinib, rescue therapy was not allowed.
For abrocitinib, the primary analysis was based on the subgroup of second-line patients who have failed on CsA, in line with the populations defined for this MTA. This subgroup, which is referred to as the ‘restricted’ population, was very small in all abrocitinib trials. Sensitivity analyses were therefore conducted using the ‘generalisable’ population for abrocitinib. The generalisable population included patients who were previously treated with at least one systemic treatment for AD. This subgroup was slightly larger than the restricted population. The generalisable population may be more reflective of the population who are likely to receive abrocitinib in UK clinical practice, but this population is less comparable to the populations in the comparator trials than the restricted population.
The companies for abrocitinib, tralokinumab and upadacitinib explored the impact of differences in baseline risk (placebo response) on the results of the NMAs in their evidence submission. However, the results of the baseline-risk adjustment analyses did not inform the any company’s base case for any of the interventions. The EAG therefore explored, as a sensitivity analysis, differences in placebo response, where possible, and where variation in placebo response was observed. The methods detailed in NICE DSU TSD3 were followed, in which heterogeneity in baseline risk is accounted for by centring the placebo response at the mean placebo response. 117 However, the EAG is aware that NICE DSU TSD5 recommends a different approach. 118
Given the small number of trials in the networks, the small number of patients from the trials, and the potential difference between the trials, the EAG had a preference for a RE model with informed prior over a FE model for the NMAs. Comparisons of the DIC between the RE and FE models show a difference of < 5 for all analyses, confirming the EAG’s preference for RE model, as the model fit is ‘similar’ between the two approaches. Therefore, a RE model with informed prior for the between-trial heterogeneity was chosen for the primary analysis, for all NMAs in all populations.
EASI 75 network meta-analysis: first-line systemic treatments – adult population
Upadacitinib and abrocitinib are both proposed as a first-line systemic therapy for adults having inadequate response to topical treatments.
The most relevant comparator in the first-line setting is CsA; however, no relevant RCTs of CsA were identified that could be linked in a NMA to the upadacitinib or abrocitinib trials. In the broader search for evidence to inform a comparison with CsA, Ariens et al. 87 was identified. As described in the section Quantity and quality of research available, Ariens et al. 87 provides the results of a regression analysis of patient-level data for patients treated with dupilumab in the placebo-controlled RCT CHRONOS and patients treated with CsA in daily practice at the Department of Dermatology and Allergology, UMC Utrecht, the Netherlands. Data for EASI 50 + ΔDLQI ≥ 4 were not available from Ariens et al. 87 and so EASI 75 became the primary outcome for this population.
Concomitant use of TCS was permitted as needed for all patients treated with CsA. CsA effectiveness presented in Ariens et al. 87 was therefore adjusted to data for dupilumab in combination with TCS from CHRONOS. Consequently, the EAG considered the appropriate comparator to be upadacitinib in combination with TCS from the AD UP trial, and abrocitinib in combination with TCS from the trials JADE COMPARE and JADE DARE. A NMA assessing these interventions versus CsA without concomitant TCS was not possible.
Time of assessment in CHRONOS, AD-UP, JADE COMPARE and JADE DARE was 16 weeks, whereas EASI scores were available for the range between weeks 12 and 16 for CsA.
The primary analysis focused on the post hoc subgroup of patients in the upadacitinib and abrocitinib trials for whom the trial intervention was their first-line systemic therapy. Of the CsA-treated patients in Ariens et al.,87 70% had no history of previous treatment with oral immunosuppressive drugs, though outcome data for this specific subgroup were not available and this cohort was therefore compared with the full population of the CHRONOS trial treated with dupilumab. Of the dupilumab-treated patients in CHRONOS, 41% had previously received systemic immunosuppressants to treat AD. A sensitivity analysis was conducted using the full trial population for AD UP, 52–58% of which had previously received systemic therapy. The difference in prior systemic therapy introduces clinical heterogeneity into the analysis, which is likely to favour upadacitinib because those with prior treatment are more severe at baseline.
The dupilumab data from CHRONOS reported in the Ariens et al. 87 analysis and in the committee papers for TA534 differ slightly; an additional two patients had a response (EASI 75) according to Ariens et al. 87 compared with TA534. The difference may be due to different handling of missing data with a LOCF analysis performed in cases of missing follow-up EASI values in Ariens et al.,87 whereas the dupilumab data from CHRONOS in TA534 either included all observed data or censored patients who received rescue medication. In the primary analysis, the data from Ariens et al. 87 (CsA vs. dupilumab) and from CHRONOS (dupilumab vs. placebo) were therefore analysed as two separate studies. However, to assess the possible impact of including the same dupilumab arm twice (once in CHRONOS and once in Ariens et al. 87), a sensitivity analysis was conducted, where the CsA arm from Ariens et al. 87 was considered an additional arm of the CHRONOS study. A different sensitivity analysis was also conducted where patients requiring rescue medication were considered non-responders. These data were available for AD UP and CHRONOS but not for the observational CsA data.
The network of trials contributing to the NMA in the first-line adult population are presented in Figure 3.
FIGURE 3.
Network plot first-line adult population, combination therapy, EASI 75. Abro, abrocitinib; Dup, dupilumab; Upa, upadacitinib.

For the NMA in the first-line adult population, the RE and FE models for the primary analysis and the sensitivity analyses were similar in terms of goodness of model fit (similar DIC), but for the primary analysis the residual deviance for the REs model was closer to the number of unconstrained data points than the FE model (see Report Supplementary Material 1).
The results of the NMA showed that abrocitinib, upadacitinib, dupilumab and CsA were all more effective than placebo, that is, leading to more responders (patients reaching EASI 75). The difference versus placebo was statistically significant for abrocitinib (both doses), upadacitinib (both doses) and dupilumab, but not for CsA, irrespective of the use of fixed or REs model. Results from the NMA were in agreement with findings from standard pair-wise analyses, in which all interventions analysed were found to be statistically significantly more effective than placebo.
Both doses of upadacitinib were shown to be more effective than CsA, with a larger OR for upadacitinib 30 mg than for upadacitinib 15 mg. The point estimates were similar for the fixed and REs models and the results were statistically significant except for the lower upadacitinib dose using the REs model. Both doses of abrocitinib were also shown to be more effective than CsA, with a dose-dependent response. However, only when using the FE model for the comparison of abrocitinib 200 mg were the results statistically significant.
Analysing the CsA data from Ariens et al. 87 and dupilumab data from CHRONOS (as reported in TA534)12 as one multiarm trial resulted in similar results to the primary analysis but with narrower CrIs. Similarly, using the full-trial population in AD UP rather than focusing on the first-line population (patients who had not received CsA) had limited impact on the results compared with the primary analysis. The sensitivity analysis using data where patients who received rescue therapy in AD UP and CHRONOS were censored also gave similar results to the primary analysis.
The placebo response in AD UP and CHRONOS were similar and thus no baseline risk adjustment sensitivity analysis was conducted for the first-line adult population.
No comparator data for CsA used without concomitant TCS were identified and therefore a NMA comparing upadacitinib and CsA used as monotherapies was not possible.
EASI 50 + ΔDLQI ≥ 4 network meta-analysis: monotherapies as second-line treatment – adult population
NMAs of interventions used as monotherapies in the second-line setting (for patients who have failed on CsA) could be carried out for both EASI 50 + ΔDLQI ≥ 4 and EASI 75, and, although results are reported for both, the primary outcome for this appraisal is EASI 50 + ΔDLQI ≥ 4. See Report Supplementary Material 1 for EASI 75 results used in scenario analyses.
Although relevant trials for baricitinib used as a monotherapy were identified, the results of these were redacted and baricitinib could, therefore, not be included in the monotherapy networks assessing EASI 50 + ΔDLQI ≥ 4 or EASI 75. For upadacitinib, the dose-finding trial reported by Guttmann-Yassky et al. in 2020 could have informed the NMA for EASI 75, as it provides the relevant outcome data at 16 weeks for upadacitinib 15 mg, 30 mg and for placebo. However, the company did not provide the relevant subgroup data for this trial at the clarification stage, and it was therefore excluded from the analysis. Data for dupilumab were informed by the post hoc subgroup data from SOLO 1 and SOLO 2 presented in TA534. For both end points in the monotherapy NMA, results for SOLO 1 and SOLO 2 were reported pooled across both studies and so has been considered as a single study, referred to as ‘SOLO CAFÉ-like’.
The primary analysis for the monotherapy NMAs was based on using all observed data, regardless of rescue medication use to determine response, with a sensitivity analysis conducted where patients requiring rescue medication were considered non-responders. Patients were not allowed rescue therapy in the abrocitinib trials.
The relevant subgroup in the abrocitinib studies, the restricted population, which was used for the primary analysis, was small. A sensitivity analysis using the generalisable population, which includes people who have had any prior systemic immunotherapy (not limited to prior CsA), was therefore also conducted. Due to variation across studies in placebo response, for both EASI 50 + ΔDLQI ≥ 4 and EASI 75, sensitivity analyses were also conducted to assess the impact of adjusting for these differences.
The network of trials contributing to the NMA of monotherapies on EASI 50 + ΔDLQI ≥ 4 in the second-line adult population is presented in Figure 4.
FIGURE 4.
Network plot second-line adult population, monotherapy, EASI 50 + ΔDLQI ≥ 4. Abro, abrocitinib; Dup, dupilumab; Tralo, tralokinumab; Upa, upadacitinib.
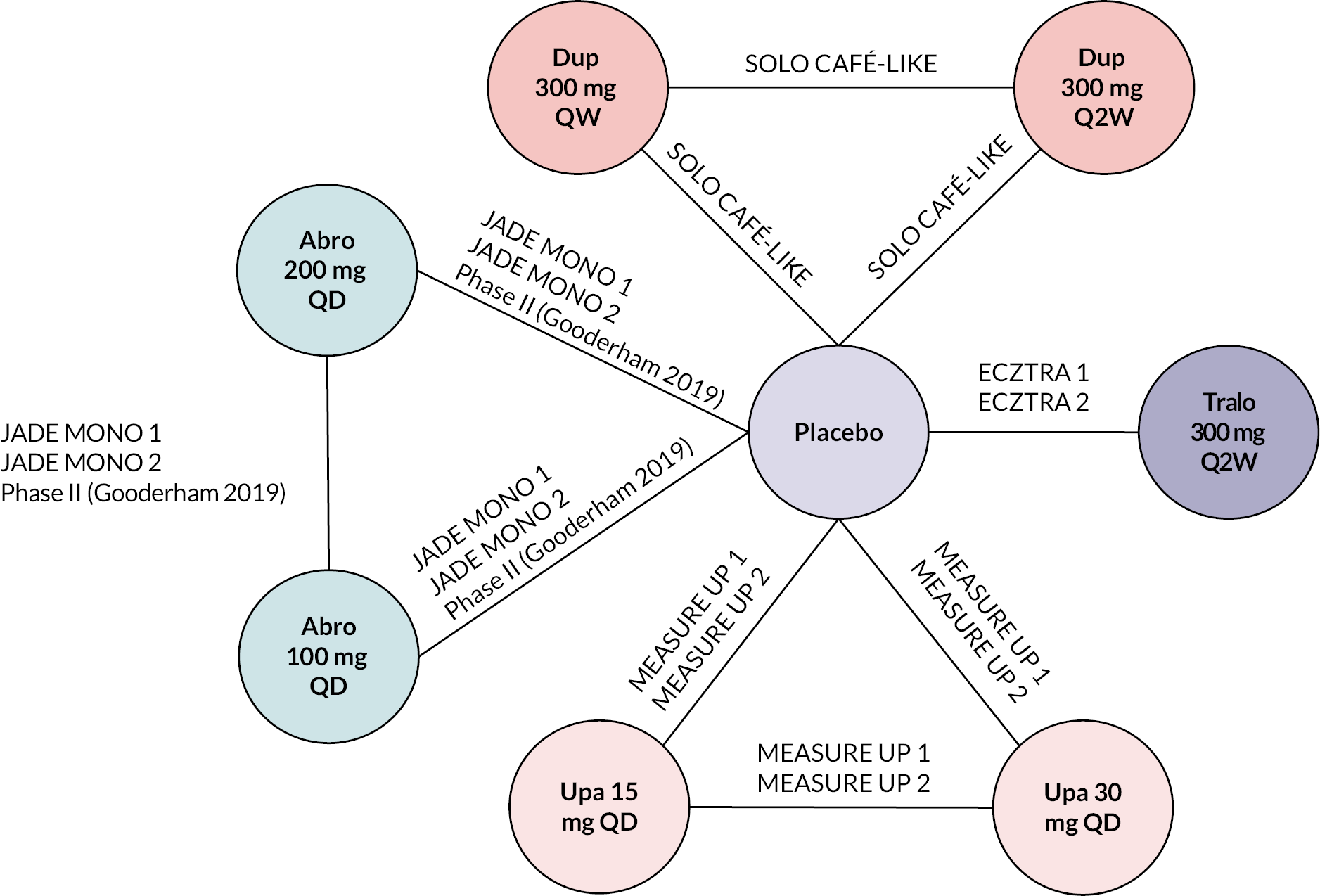
For the NMAs of EASI 50 + ΔDLQI ≥ 4, the RE and FE models for the primary and all sensitivity analyses were similar in terms of goodness of model fit (similar DIC) (see Report Supplementary Material 1). However, for both RE and FE models, the residual deviance showed a relatively poor prediction of the data used in each analysis, especially for the sensitivity analysis using the generalisable abrocitinib population.
The primary analysis, which focused on response irrespective of rescue therapy use, showed that treatment with any of the interventions assessed (abrocitinib, dupilumab, tralokinumab or upadacitinib) led to a statistically significant improvement in EASI 50 + ΔDLQI ≥ 4 compared with placebo. Results from the NMA were in agreement with findings from standard pair-wise analyses, in which all interventions analysed were found to be statistically significantly more effective than placebo. However, for abrocitinib 200 mg, the NMA resulted in a substantially higher OR compared with the underlying trial data.
A dose-dependent numerical benefit was observed for upadacitinib 15 mg and upadacitinib 30 mg over dupilumab; however, the differences did not reach statistical significance. Similarly, abrocitinib showed a dose-dependent effect versus dupilumab. Neither analysis showed a statistically significant difference versus dupilumab. Tralokinumab therapy resulted in a lower response than dupilumab, although, as for the other interventions, the difference was not statistically significant.
Censoring patients receiving rescue therapy in the dupilumab, tralokinumab and upadacitinib trials led to a smaller benefit of abrocitinib 200 mg, upadacitinib 15 and 30 mg compared with dupilumab, whereas the benefit of dupilumab over tralokinumab therapy increased and it also became more beneficial than abrocitinib 100 mg, though none of the relative differences between the interventions and dupilumab were statistically significant.
The sensitivity analysis based on the generalisable population for abrocitinib show relatively similar results to the primary analysis, although the ORs for both doses of abrocitinib were more favourable and the 95% CrIs were narrower for the generalisable population as the sample sizes were larger.
There was variation in placebo response across the included trials, from no responders to a third of patients on placebo being responders at 12 or 16 weeks: treatment effectiveness was captured at 12 weeks in studies evaluating abrocitinib and at 16 weeks in all other studies. However, the largest variation in placebo response rates was in the abrocitinib trials, which had very low numbers of included patients (six patients in each of the placebo arms). The sensitivity analysis adjusting for heterogeneity in placebo response gave a lower DIC than the primary, unadjusted analysis, indicating a better model fit. However, the total residual deviance, for this analysis, was lower than the number of unconstrained data points, indicating that the model may be ‘overfitting’ the data, that is, the model predicts the underlying trial data extremely well (and hence a lower DIC) but is likely to be less generalisable to the population of interest than the unadjusted analysis using observed data. The EAG is concerned that the results are unreliable for use in the cost-effectiveness analyses and, in keeping with the companies’ approach, the EAG used the observed data to inform the primary cost-effectiveness analysis.
EASI 50 + ΔDLQI ≥ 4 network meta-analysis: second-line systemic treatments in combination with topical corticosteroid – adult population
For the NMAs of interventions used in combination with TCS in the second-line setting (patients who have failed on CsA), post hoc subgroups were used for all studies apart from the dupilumab trial CAFÉ and the tralokinumab study ECZTRA 7. However, the data for dupilumab were informed by the pooled results of CAFÉ and the relevant post hoc subgroup data from CHRONOS presented in TA534. The pooled data have been considered as a single study. Two baricitinib trials were relevant for inclusion in these analyses, BREEZE-AD4 and BREEZE AD7. The majority of the results from these trials were redacted but baricitinib could be included in the analysis of EASI 75 based on data from BREEZE-AD4.
Network meta-analyses were possible to perform for both EASI 50 + ΔDLQI ≥ 4 and EASI 75, and although results are reported for both, the key outcome for this appraisal is EASI 50 + ΔDLQI ≥ 4. See Report Supplementary Material 1 for EASI 75 results used in scenario analyses.
The primary analysis for the combination therapy NMAs is based on using all observed data, regardless of rescue medication use to determine response, with a sensitivity analysis conducted where patients requiring rescue medication were considered non-responders. Sensitivity analyses were also conducted using the generalisable, rather than restricted, population for abrocitinib. A baseline risk-adjusted sensitivity analysis was conducted but the models did not converge despite attempts to increase convergence by thinning the sampling and increasing the number of model iterations.
The network of trials contributing to the NMA of combination therapies on EASI 50 + ΔDLQI ≥ 4 in the second-line adult population is presented in Figure 5.
FIGURE 5.
Network plot second-line adult population, combination therapy, EASI 50 + ΔDLQI ≥ 4. Abro, abrocitinib; Dup, dupilumab; Tralo, tralokinumab; Upa, upadacitinib.

For the NMAs of EASI 50 + ΔDLQI ≥ 4, the RE and FE models for the primary and all sensitivity analyses were similar in terms of goodness of model fit (similar DIC) and residual deviance (see Report Supplementary Material 1).
The FEs primary analysis showed that treatment with abrocitinib, dupilumab, tralokinumab or upadacitinib led to a statistically significant improvement in EASI 50 + ΔDLQI ≥ 4 compared with placebo, in agreement with findings from pair-wise meta-analyses. Using a REs model, the 95% CrIs were wider and, although treatment with any of the interventions was favoured over placebo, the results for tralokinumab did not reach statistical significance.
When compared with dupilumab, there were no comparisons that were statistically significant using the REs model. With the FEs model, the results were statistically significant for upadacitinib 30 mg, in favour of upadacitinib, and for tralokinumab, in favour of dupilumab. The OR of upadacitinib 1 mg, abrocitinib 100 and 200 mg were closer to 1, favouring dupilumab for both of the lower doses and favouring abrocitinib for the higher dose.
Censoring patients receiving rescue therapy in the dupilumab, tralokinumab and upadacitinib trials had a limited impact on the comparison of abrocitinib 100 or 200 mg versus dupilumab. However, the benefit of upadacitinib 30 mg over dupilumab was substantially smaller than for the primary analysis, the benefit of dupilumab over tralokinumab was statistically significant, and although not statistically significant, dupilumab therapy was favoured over upadacitinib 15 mg.
The sensitivity analysis based on the generalisable population for abrocitinib showed similar results to the primary analysis for upadacitinib 15 mg, 30 mg and tralokinumab; however, the benefit of dupilumab therapy over tralokinumab therapy reached statistical significance. For abrocitinib 100 and 200 mg, the direction of effect compared with dupilumab was the same as when using the restricted population (primary analysis), but the treatment effect favouring abrocitinib 200 mg over dupilumab was larger and the treatment effect favouring dupilumab over abrocitinib 100 mg was also more pronounced.
Placebo response varied between 25% and 61% in the studies contributing to the composite outcome. However, the models for the baseline risk-adjusted sensitivity analysis did not converge despite attempts to increase convergence by thinning the sampling and increasing the number of model iterations. Therefore, no results are presented for this sensitivity analysis.
EASI 75 network meta-analysis: adolescents
The companies for abrocitinib and upadacitinib are seeking a recommendation by NICE for the use of their drugs for the treatment of AD in adolescents. Of the treatment options for AD available in the NHS, baricitinib does not hold a marketing authorisation for use in adolescents and CsA is only for people aged 16 years and over and is thereby not available for a large proportion of the adolescent population. Thus, neither baricitinib nor CsA is a relevant comparator for treatment of adolescents. Additionally, although dupilumab has only been assessed and recommended by NICE for an adult population, its marketing authorisation encompasses adolescents, and dupilumab is funded for use in adolescents under the NHS England Medicines for Children Policy as part of specialised commissioning. 119 An adolescent is eligible for treatment with dupilumab if they are seen within a specialised treatment centre and they meet the criteria set out within TA534, for use of dupilumab in adults. 119 Dupilumab is therefore the key comparator for the adolescent population.
Trial evidence was available for the use of upadacitinib and abrocitinib both as monotherapies (JADE MONO 1 and 2, MEASURE UP 1 and 2) and in combination with TCS (JADE TEEN and AD UP) in the adolescent population. However, comparator data for dupilumab were only identified as a monotherapy (AD ADOL) but no trial was identified assessing dupilumab in combination with TCS in an adolescent population. A NMA assessing abrocitinib and upadacitinib versus dupilumab, all in combination with TCS, was therefore not possible, but the results of the combination therapy trials for abrocitinib and upadacitinib are presented alongside the monotherapy NMA results below for completeness.
Data on the composite outcome EASI 50 + DLQI ≥ 4 were not presented in the dupilumab trial (AD ADOL) and therefore only EASI 75 could be assessed for this population.
The abrocitinib trials did not allow use of rescue medications, whereas the dupilumab and upadacitinib trials included in the NMA for the adolescent population did. However, the dupilumab trial only reported results where patients were censored if/when they received rescue therapy. So, the primary analysis for the NMA in the adolescent population is based on upadacitinib data where patients were included in the analysis even if they required rescue medication, dupilumab data where patients were censored when they received rescue medication and abrocitinib data where patients did not receive rescue medication. A sensitivity analysis was conducted using upadacitinib data where patients were censored when receiving rescue medication, similar to the dupilumab study.
Due to variation in the placebo response between the trials included in the analysis, a sensitivity analysis adjusting for heterogeneity in placebo response was also conducted.
The NMA results are focused on the doses of the interventions recommended for adolescents, which are abrocitinib 100 and 200 mg and upadacitinib 15 mg. The network of trials contributing to the NMA in the adolescent population are presented in Figure 6.
FIGURE 6.
Network plot adolescent population, monotherapy, EASI 75. Abro, abrocitinib; Dup, dupilumab; Upa, upadacitinib.

For the NMA in the adolescent population, the FE and RE models of the primary and sensitivity analyses were similar in terms of goodness of model fit (similar DIC, see Report Supplementary Material 1), but the residual deviance for the RE models were closer to the number of unconstrained data points in all analyses, which reinforces the EAG’s preference for the RE model.
The primary analysis shows that treatment with abrocitinib (either dose), dupilumab or upadacitinib 15 mg was associated with a statistically significant improvement in EASI 75 compared with placebo. Results from the NMA were in agreement with findings from standard pair-wise analyses for each of the trial, in which all the interventions analysed were found to be statistically significantly more effective than placebo. However, for abrocitinib 200 and 100 mg, the NMA resulted in substantially higher ORs compared with the underlying trial data.
When compared with dupilumab, treatments with abrocitinib 200 mg, abrocitinib 100 mg and upadacitinib 15 mg were all more effective than dupilumab, although these comparisons were not statistically significant.
Censoring patients receiving rescue therapy in the dupilumab and upadacitinib trials led to relatively similar ORs compared with the primary analysis and no statistically significant difference compared with dupilumab.
Placebo response varied from 0% to 22% in the studies contributing to the analysis for the adolescent population. The DIC for the sensitivity analysis adjusting for heterogeneity in placebo response was markedly lower than the DIC for the primary analysis, indicating a better fitting model. However, similar to the sensitivity analysis conducted for the second-line adult monotherapy analysis, residual deviance for this NMA was lower than the number of data points used in the analysis, potentially indicating ‘overfitting’ to the underlying data used in the model. As such, due to concerns around generalisability of the results of the sensitivity analysis, the NMAs based on the observed data were preferred to inform the cost-effectiveness analyses.
No data were identified on dupilumab used with concomitant TCS in an adolescent population. Therefore, a NMA comparing upadacitinib and abrocitinib with dupilumab, all in combination with TCS, was not possible. However, data on the efficacy of upadacitinib and abrocitinib in combination with TCS and compared with placebo in adolescents were available. The results showed that both upadacitinib and abrocitinib with TCS are statistically significantly more effective than placebo with TCS, in terms of EASI 75. However, the relative benefit over placebo was substantially smaller when the interventions were used in combination with TCS than as monotherapies.
Safety
The safety of the interventions and comparators during treatment up to 16 weeks are reported for the full-trial populations and not separated by line of therapy. The EAG has focused on serious adverse events (SAEs) and on specific AEs (irrespective of severity) in line with those included in TA534 and TA681; injection-site reaction (for dupilumab and tralokinumab), conjunctivitis and allergic conjunctivitis, upper respiratory tract infections, acne and oral herpes.
These are not necessarily the most common AEs for each of the treatments, but each has been found to be associated with at least one of the treatments and were considered the most important to include by the EAG’s clinical experts.
In terms of both SAEs and the specific AEs of any severity, the numbers were generally small, indicating that the short-term use of these interventions, as monotherapy and in combination with TCS, was well tolerated. The results are therefore not discussed separately for monotherapy and combination therapy, or for adults and adolescents. AE data from individual studies are provided in Report Supplementary Material 1.
Abrocitinib
The short-term data (16 weeks) showed a dose-related increase in acne events with abrocitinib compared with placebo.
Tralokinumab
The overall frequency of SAEs during randomised treatment (16 weeks) was low, with slightly lower incidence with tralokinumab compared with placebo. Among the most frequent AEs in the initial treatment period were urinary tract infection (URTI), conjunctivitis (allergic and infectious) and injection-site reactions. The results of the trials indicate that tralokinumab therapy is associated with higher rates of conjunctivitis and of injection-site reactions than patients who received placebo, and potentially with a lower rate of oral herpes than placebo. There was no consistent trend across studies towards URTI being more common among patients treated with tralokinumab (±TCS) than among those receiving placebo.
Upadacitinib
The overall frequency of SAEs during randomised treatment (16 weeks) was low and relatively even between upadacitinib and placebo. However, the company for upadacitinib reports that there is a reasonable possibility that some AEs are related to upadacitinib treatment.
Dupilumab
There appeared to be increased rates of conjunctivitis and allergic conjunctivitis with dupilumab compared with placebo. Dupilumab therapy was also associated with higher rates of injection-site reactions compared with placebo. The rates of SAEs were slightly lower with dupilumab than placebo across the dupilumab trials.
Baricitinib
In the company submission, it is reported that the rates of SAEs were lower with baricitinib 4 mg than with placebo across most BREEZE-AD trials. Some unredacted data are only available for BREEZE-AD 4 and BREEZE-AD 7, both of which had low numbers of SAEs, and for BREEZE-AD 4 the number was lower with baricitinib than placebo.
Discussion
Summary of key results
The comparative clinical effectiveness of abrocitinib, tralokinumab and upadacitinib at their recommended dose or doses, both as monotherapy and in combination with TCS, versus treatment options available in the NHS for moderate-to-severe AD, was evaluated in the positions in the treatment pathway proposed by the individual companies.
Due to a lack of data for some interventions for EASI 50 + ΔDLQI ≥ 4, which is the primary outcome of interest to the MTA, the EAG also evaluated clinical effectiveness in achieving EASI 75. Estimates of comparative clinical effectiveness for abrocitinib, tralokinumab and upadacitinib versus treatment options available in the NHS were derived from NMAs as direct evidence for these comparisons was limited: some studies included dupilumab as an active comparator, but only as monotherapy.
Experts advising the EAG commented that, in clinical practice for the management of AD, all systemic therapies are likely to be used in combination with TCS rather than as monotherapies. However, monotherapy will still be relevant for a proportion of patients who cannot tolerate or do not want to use TCSs. Although comparisons of relevant treatments in combination with TCS are of most importance to this MTA, comparisons of monotherapies were explored for all populations, where possible. The EAG’s primary analyses are based on using all observed data, regardless of rescue medication as receipt of rescue therapy more closely reflects clinical practice in England.
Experts advising the EAG commented that, in clinical practice for the management of AD in adults, abrocitinib, tralokinumab and upadacitinib are likely to be used as alternatives to dupilumab and baricitinib, which are NICE-recommended treatment options after inadequate response, inability to tolerate or contraindication to first-line systemic therapy.
For the NMAs, there were considerable amounts of uncertainty, and the vast majority of results were not statistically significant. However, there were consistent trends across the outcomes (EASI 50 + ΔDLQI ≥ 4 and EASI 75), interventions (combination therapy or monotherapy) and populations (adults in the first- or second-line setting and adolescents) which are summarised below.
Abrocitinib
The NMA results indicate that treatment of adults in the second-line setting with abrocitinib 200 mg leads to a better response, assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75, than dupilumab treatment. The benefit in favour of abrocitinib 200 mg was larger when both treatments were used as monotherapies and less pronounced when used in combination with TCS. The effect was also greater when response was assessed as EASI 75 compared with when assessed as EASI 50 + ΔDLQI ≥ 4. The effectiveness of abrocitinib was dose dependent. As for abrocitinib 200 mg, the benefit of abrocitinib 100 mg compared with dupilumab was greatest for adults in the second-line setting using both treatments as monotherapies and when assessing response as EASI 75. The effectiveness of abrocitinib 100 mg compared with dupilumab favoured dupilumab when the two treatments were used in combination with TCS. The effectiveness of abrocitinib 100 mg was similar to dupilumab when used as monotherapies and response assessed as EASI 50 + ΔDLQI ≥ 4. Abrocitinib 200 and 100 mg with concomitant TCS were both more effective than baricitinib 4 mg with TCS in terms of EASI 75 for adults in the second-line setting and in the adolescent population both doses of abrocitinib were more effective than dupilumab, also in terms of EASI 75.
Rescue therapy was not permitted in the abrocitinib trials, unlike the trials for the other interventions included in the MTA. However, the use of systemic rescue therapy was low in the studies for the interventions where it was allowed. The lack of rescue therapy may lead to lower absolute response rates (in all trial arms) in the abrocitinib trials compared with clinical practice. However, it is unclear how the lack of rescue therapy may affect the relative treatment effect and therefore what impact the difference in rescue therapy has had on the results of the primary analysis. Sensitivity analysis censoring patient who needed rescue therapy in the trials informing the other interventions in the networks (dupilumab, tralokinumab and upadacitinib) had limited impact on the effectiveness of abrocitinib (either dose) on EASI 75 in the adolescent population and on EASI 50 + ΔDLQI ≥ 4 or EASI 75 in the adult population when all treatments given in combination with TCS in the second-line setting. However, for adults given the treatments as monotherapies in the second-line setting, the effectiveness of abrocitinib versus dupilumab (assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75) decreased substantially when patients receiving rescue therapy were censored.
The restricted population, which was used for the primary analysis of adults in the second-line setting, is in line with the populations used for dupilumab and baricitinib; however, it constituted a very small proportion of the abrocitinib trial populations and hence there is substantial uncertainty around the assessment of abrocitinib. The sensitivity analysis based on expanding the population receiving abrocitinib from those receiving only CsA as the first systemic therapy (restricted population) to include those who had received any type of systemic therapy at first line (generalisable population) was also uncertain and the 95% CrIs were wide, partly because the sample size of the generalisable population was still small. The sensitivity analysis gave similar results to the primary analysis for the composite outcome and EASI 75 for abrocitinib used in combination with TCS and for abrocitinib monotherapy when response was assessed as EASI 50 + ΔDLQI ≥ 4. However, for EASI 75, the benefit of abrocitinib monotherapy compared with dupilumab monotherapy was substantially reduced, favouring dupilumab over abrocitinib 100 mg, but still favouring abrocitinib 200 mg over dupilumab.
Tralokinumab
The NMA results indicate that treatment of adults in the second-line setting with tralokinumab leads to a better response treatment, assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75, than placebo but an inferior response to dupilumab treatment and baricitinib 4 mg treatment (only available for EASI 75). The benefit in favour of dupilumab over tralokinumab was larger when both treatments were used in combination with TCS and less pronounced when used as monotherapies. Sensitivity analysis censoring patient who required rescue therapy in the dupilumab, tralokinumab and upadacitinib trials informing the networks had limited impact on the effectiveness of tralokinumab compared with dupilumab or baricitinib on EASI 50 + ΔDLQI ≥ 4 or EASI 75 in the adult population when all treatments given in combination with TCS or as monotherapies in the second-line setting.
Upadacitinib
The NMA results indicate that treatment of adults in the second-line setting with upadacitinib 30 mg is more effective than dupilumab treatment when assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75. The benefit in favour of upadacitinib 30 mg over dupilumab was relatively similar for EASI 50 + ΔDLQI ≥ 4 or EASI 75 when both treatments were used as monotherapies. For upadacitinib 30 mg treatment in combination with TCS, the benefit over dupilumab was substantially larger when response was assessed as EASI 50 + ΔDLQI ≥ 4, and smaller when assessed as EASI 75, compared with the results for upadacitinib 30 mg as a monotherapy.
The effectiveness of upadacitinib was dose dependent with upadacitinib 15 mg consistently being less effective than upadacitinib 30 mg. Compared with dupilumab, upadacitinib 15 mg was more effective than dupilumab when both were given as monotherapies, but broadly similar effectiveness or even favouring dupilumab when given in combination with TCS. Upadacitinib 30 and 15 mg were both more effective than baricitinib 4 mg in terms of EASI 75 for adults in the second-line setting and more effective than CsA for adults in the first-line setting. In the adolescent population, upadacitinib 15 mg was also more, although a variation was observed in the placebo response across several of the networks, sensitivity analysis effective than dupilumab, also in terms of EASI 75.
Sensitivity analysis censoring patients who required rescue therapy in the dupilumab, tralokinumab and upadacitinib trials informing the networks had limited impact on the effectiveness of upadacitinib 30 and 15 mg compared with dupilumab, irrespective of outcome (EASI 50 + ΔDLQI ≥ 4 or EASI 75), population (first- or second-line adults or adolescent population) and if used as monotherapy or in combination with TCS. However, for the NMA of treatments in combination with TCS, the results for EASI 50 + ΔDLQI ≥ 4 differed substantially between the primary analysis and the sensitivity analysis censoring patients who received rescue therapy.
Placebo response
For all but the network for the first-line adult population, there was relatively large variability in placebo response for the treatments in each network. This indicates that there may be imbalances in prognostic factors, treatment effect modifiers or differences in the conduct between the trials. If the difference is due to imbalance in treatment effect modifiers, this can have an impact on the relative efficacy of the treatments. All companies explored the impact of differences in baseline risk (placebo response) on the results of their NMAs. However, the results of the baseline-risk adjustment analyses did not inform the base case for any of the interventions. The EAG also attempted to adjust for heterogeneity in placebo response rates; however, this was not possible for all networks and outcomes. Models assessed either did not converge on the posterior distribution or were considered by the EAG to overfit the underlying data. The EAG is concerned that the results produced are unreliable for use in the cost-effectiveness analyses and, in keeping with the companies’ approach, the EAG considers the analyses based on the observed data to be the most appropriate to inform the cost-effectiveness analyses.
Other outcomes
Quality of life measured using EQ-5D showed that treatment with abrocitinib, tralokinumab and upadacitinib for 12–16 weeks leads to an improvement in general QoL compared with placebo irrespective of dose, concomitant TCS or place in the treatment pathway (for adolescents, as a first-line or second-line systemic treatment for adults). The size of the benefit varied and, although based on direct evidence, there was substantial uncertainty around the results.
As mentioned previously, rescue medication was not allowed in the abrocitinib trials, but for tralokinumab and upadacitinib, use of rescue medication was lower than for patients given placebo. Use of rescue medication was also lower when tralokinumab and upadacitinib were used in combination with TCS compared to when used as monotherapies.
The trials assessing abrocitinib, tralokinumab and upadacitinib with concomitant TCS generally showed a higher number of days free from TCS with active therapy compared with placebo. The exception was for the restricted population for abrocitinib where the patient numbers were very low.
The incidence of both SAEs and specific AEs (injection-site reaction, conjunctivitis, upper respiratory tract infections, acne and oral herpes) of any severity was generally low, indicating that short-term use of these interventions, as monotherapy and in combination with TCS, was well tolerated.
For further details on other outcomes, refer to Report Supplementary Material 1.
Generalisability
Clinical experts advising the EAG commented that, based on baseline EASI scores, the patients enrolled in the RCTs identified as relevant to the MTA have more severe AD than would typically be seen in clinical practice, with most patients presenting with disease in clinical practice categorised as moderate severity. No analysis was possible to explore potential differences in effectiveness of abrocitinib, tralokinumab and upadacitinib based on disease severity. As such, the efficacy of these interventions seen in patients with more severe AD in the clinical trials may be different to the effect in patients with more moderate AD in clinical practice.
Evidence informing the NMAs is predominantly derived from post hoc subgroups for the populations relevant to the MTA. As discussed in the section Strengths and limitations, there are limitations associated with using post hoc subgroups that have an impact on the robustness of the results from the NMA.
For adults, the post hoc subgroups informing the EAG’s preferred analyses for second-line systemic treatment are clinically homogeneous in terms of people having inadequate response to, not being able to tolerate or being contraindicated to CsA, and in line with the population underpinning the recommendation for baricitinib and dupilumab. Both dupilumab and baricitinib are recommended by NICE for patients whose disease has not responded to at least one other systemic therapy, such as ciclosporin, methotrexate, azathioprine and mycophenolate mofetil, or these are not suitable. While the populations analysed in the MTA represent a subgroup of the population likely to be treated in clinical practice, they are ‘equally’ representative of the populations used for decision-making with dupilumab and baricitinib.
For the analysis of first-line systemic treatment, data for upadacitinib informing the NMA were supplied by the company in response to a request from the EAG for data on the subgroup of adults who were naïve to systemic therapy in the relevant studies. Thus, the EAG considers the population to be generalisable to the patient population who would likely be eligible for first-line treatment. However, the population informing the comparator, CsA, was not limited to those who were naïve to systemic therapy. It is unclear how this difference in the populations may affect the results of the analysis and the generalisability of the results to the systemic naïve patients in clinical practice.
For adolescents, the EAG considers the populations enrolled in the various studies to be generalisable to the adolescents in England who would likely be eligible for treatment with abrocitinib or upadacitinib. However, the EAG notes that comparisons with dupilumab were only possible for abrocitinib and upadacitinib monotherapy, whereas both treatments are likely to be used in combination with TCS by the majority of adolescents. Based on the results of the adult populations, it is possible that both upadacitinib and abrocitinib will be more efficient when used in combination with TCS than when used as monotherapies also for adolescents.
Strengths and limitations
A strength of the MTA is that a SLR has been carried out to identify the relevant evidence. The systematic review was carried out in line with established methods and principles. The RCTs identified by the EAG as relevant to the MTA were considered to be well-designed and well-conducted, and, generally, at a low risk of bias. However, only two RCTs provided direct evidence for a single population of interest to the MTA, with most RCTs predominantly including mixed populations of people with moderate-to-severe AD. Some studies comprised both adolescents and adults, as well as a combination of people receiving systemic therapy as a first-line or second-line regimen. Thus, data informing the NMAs for the populations and outcomes of interest to the MTA are primarily derived from post hoc subgroups, which introduces bias and uncertainty around the results generated by the NMAs, and is a considerable limitation that impacts on the robustness and confidence in the estimates of effect for clinical effectiveness. Use of post hoc subgroups reduces the sample size for analysis and also breaks the randomisation component of a RCT. While breaking randomisation can cause an imbalance in observed baseline characteristics, these were not apparent to the EAG based on the information supplied by the companies (see Report Supplementary Material 1). However, there remains the potential imbalances in the unobserved baseline characteristics.
However, the strength of the analyses is that the populations informing the comparison in the second-line setting are clinically homogeneous in terms of people having inadequate response to, not being able to tolerate or being contraindicated to CsA.
Methodological heterogeneity between the trials in the networks is likely to have contributed to the uncertainty in the results. Sources of methodological heterogeneity included variation across studies in the use of a washout period for TCS before randomisation to treatment, the type and potency of concomitant TCS used in studies evaluating treatment in combination with TCS, and the type and potency of rescue medication used. These differences, especially the disparity in use of and potency of rescue therapy used across studies, may lead to clinical heterogeneity in observed placebo response, which, consequently, may introduce bias and uncertainty into the NMA. Although variation was observed in the placebo response across several of the networks, sensitivity analysis adjusting for differences in placebo response was either not possible (due to lack of convergence) or the data were overfitted. Therefore, the unadjusted analyses were used to inform the economic model. The disparity in data available for the primary analysis and sensitivity analysis based on censoring of patients who needed rescue therapy is also likely to have introduced some bias in the results. However, despite the lack of consistency in the available data for some of the analyses, the difference between the primary analysis and the rescue therapy sensitivity analysis was small for most comparisons.
The EAG considers it important to note that the sample sizes informing the NMAs equate to a small proportion of the overall trial populations from which the subgroups are created, particularly for abrocitinib. The effect of small sample size on the results of the NMA is apparent in the wide 95% CrIs, which indicate considerable uncertainty around the true estimate of comparative effectiveness.
In addition to most of the data informing the NMAs being derived from post hoc subgroups, the EAG notes that much of the data for baricitinib were unavailable at the time of writing. Results for studies evaluating baricitinib are not yet published in peer-reviewed journals and data submitted to the STA process were redacted from the committee papers accompanying the recommendation by NICE. Lack of data for baricitinib on clinical outcomes of interest to the MTA precluded inclusion of baricitinib in most of the relevant NMAs. Additionally, no randomised evidence was identified to inform the efficacy of CsA, which is the relevant comparator in the first-line setting. Thus, results for the comparison with upadacitinib in the first-line setting are derived from observational data, which is associated with the bias inherent in observational studies and the results should be interpreted with caution.
Another limitation of the MTA are the data gaps; for the adolescent population and the adult population in the first-line setting, data to inform only EASI 75 could be assessed as data on EASI 50 + ΔDLQI ≥ 4 were not available. In addition, for the adolescent population, relevant comparisons could only be made for abrocitinib and upadacitinib as monotherapies, although the interventions are likely to be primarily used with concomitant TCS in clinical practice.
The analyses in this MTA focused on the efficacy and safety of patients after 12–16 weeks of treatment. Sixteen weeks is the time point when response, in terms of EASI 50 + ΔDLQI ≥ 4, is evaluated for dupilumab and baricitinib to determine if treatment should be continued or stopped. However, the EAG notes that longer-term follow-up data for efficacy and safety of these treatments are important and that they are lacking for most of them. It is therefore unclear if the level of response seen in the data will be maintained after 16 weeks.
Chapter 4 Assessment of cost effectiveness
Systematic review of existing cost-effectiveness evidence
Methods
A SLR was undertaken in July 2021 to identify published economic evaluations of biological treatments of moderate-to-severely active AD. A separate search was conducted to identify studies reporting health-related quality-of-life (HRQoL) data in patients with moderate-to-severely active AD.
Multiple electronic databases were searched including MEDLINE, EMBASE, the International Network of Agencies for Health Technology Assessment (INAHTA) and the Cost-Effectiveness Analysis (CEA) Registry. Further to the database searches, health technology appraisal (HTA) websites including Canadian Agency for Drugs and Technologies in Health (CADTH), NICE and Scottish Medicines Consortium (SMC) were searched to identify relevant appraisals. In addition, experts in the field were contacted with a request for details of relevant published and unpublished studies and reference lists of key identified studies were also reviewed for any potentially relevant studies.
The Centre for Reviews and Dissemination (CRD) databases were not searched as the CRD stopped adding records to the HTA database in March of 2018 and the Database of Abstracts of Reviews of Effects (DARE) and NHS Economic Evaluations Database (NHS EED) in March of 2015. The EAG considers it unlikely that relevant studies will be missed from the CRD databases as the INAHTAHTA has taken over the responsibility for the production of the HTA database. During protocol development, clinical experts also advised that they are unaware of any economic evaluations or HRQoL studies published prior to March of 2015 that will be of relevance to this review.
The search strategy for economic evaluations combined terms capturing the interventions or comparators of interest, the target condition (AD) and health economic terms (adapted from the CADTH search filter for economic evaluations). The search strategy for HRQoL data was not restricted by treatment, and combined terms capturing the target population with HRQoL terms (adapted from Arber et al. 120).
Limits were applied to the search strategies to remove animal studies. Additionally, a start date of 2014 was applied to the search strategies as clinical experts advised the EAG that clinical practice started to change following the publication of the first dupilumab RCT in 2014, with the most marked changes in UK clinical practice taking place after approval of dupilumab by NICE in 2018. As such, a start date of 2014 is considered inclusive. No language (to assess volume of foreign language studies available), setting or country restrictions were applied to the search strategies. Full details of the search strategies are presented in the supplementary material.
The titles and abstracts of papers identified through the searches were independently assessed for inclusion using pre-defined eligibility criteria. The inclusion and exclusion criteria for each review are outlined in Report Supplementary Material 1 for economic evaluations and for studies reporting HRQoL data. For the economic evaluation search, systematic reviews identified as potentially relevant were manually reviewed for potentially eligible studies, and then only primary sources were included. Additionally, for both searches, the EAG reviewed the companies’ submissions (including results of their SLRs) for additional references.
Results – economic evaluations
The electronic database searches identified 712 potential publications. Upon removal of duplicates, 674 publications were screened against the eligibility criteria. Of these, 14 publications were included and further assessed against the same eligibility criteria with 7 additional studies separately identified from HTA websites, reference lists and clinical experts. Overall, 10 publications were included, 4 were UK full-text publications (2 NICE technology appraisals12,13 and 2 SMC appraisals121,122), 5 were non-UK full-text publications123–127 and 1 was a non-UK abstract. 128 The PRISMA flow diagram (see Report Supplementary Material 1) details the inclusion and exclusions of studies at each stage of the review.
Of the interventions of interest to this review, all 10 publications included standard care, 7 included dupilumab only (monotherapy or with TCS),12,121,123–126,128 2 included baricitinib only (monotherapy or with TCS)13,122 and 1 included abrocitinib, baricitinib, dupilumab, tralokinumab and upadacitinib. 127 However, the definition of standard care was not consistent across the studies.
The type of economic evaluation in each of the 10 included publications was a cost-utility analysis, where the incremental cost-effectiveness ratio (ICER) was expressed as the cost per quality-adjusted life-year (QALY) gained. The most common type of model structure was a short-term decision tree, which modelled induction of treatment, followed by a long-term Markov model (MM), which modelled the maintenance phase of treatment. However, in five of the publications, a MM structure was solely used with either model cycle length adjusted to 4 months in the first year or the use of tunnel states to account for short-term treatment induction phase. 13,122,125–127 The time horizon used in nine of the publications was lifetime,12,13,121–126,128 with one publication implementing a 5-year time horizon. 127 Model cycle lengths ranged from 4 months to 1 year, although one study implemented a 4-week cycle length. 13 The time horizon for models with a short-term decision tree component, was typically 52 weeks, but one study limited the decision tree component to 16 weeks. 124
Response to treatment was assessed at week 16 in 9 of the publications (insufficient detail on response provided in abstract by Fanelli et al., 2020128). Treatment response was primarily based on percentage change in baseline EASI scores. However, across included publications, the threshold for change in baseline EASI scores varied, with nine studies opting for a minimum of 50% or more improvement in EASI score compared with baseline (EASI 50) or 75% or more improvement in EASI score compared with baseline (EASI 75). In addition to treatment response of EASI 50, three of the included publications defined treatment response to also include an improvement in the DLQI of at least four, resulting in a composite outcome of EASI 50 plus DLQI > 4 at week 16.
Modelling of long-term treatment response post week 16 varied in the included publications. Long-term response was estimated from responders at week 16, either as a conditional response probability or as a conditional discontinuation (preferred NICE TA68113). After year 2, all studies used treatment discontinuation in combination with various treatment-waning assumptions applied to estimate the proportion of patients per cycle who transition to BSC.
A summary of the 10 extracted publications is provided in Table 5 and detailed data extractions can be found in Report Supplementary Material 1.
| Study | Population | Model type | Intervention/comparator | Outcomes |
|---|---|---|---|---|
| CADTH 2020123 | Patients aged 12 years or older with moderate-to-severe AD for whom topical prescription therapies failed to achieve effective disease control or were not advisable. | Short-term 1-year decision tree followed by a long-term maintenance MM. The MM included annual cycles with half-cycle correction. | Intervention: dupilumab plus SOC. Comparator: SOC, assumed to be topical therapy (type of topical treatments not listed in study). |
Treatment response at week 16 based on EASI 50, with scenario using EASI 75. Treatment response at week 52 based on conditional response for responders at week 16. Treatment discontinuation applied annually for dupilumab patients in long-term model. |
| Kuznik et al. 2017124 | Adult patients with moderate-to-severe AD | Short-term (16-week) decision tree followed by a lifetime horizon MM. A 4-month cycle length was used for the MM. | Intervention: dupilumab plus emollients. Comparator: SOC, assumed to be emollients as required. |
Treatment response at week 16 based on EASI 75. Treatment response at week 52 based on conditional discontinuation (previously responding patients discontinued by 52 weeks). |
| Fanelli et al. 2020128 (abstract) | Adolescents (aged 12–17 years) with uncontrolled moderate-to-severe AD | Short-term 1-year decision tree followed by a lifetime horizon MM. | Intervention: dupilumab. Comparator: current supportive care. |
Not reported. |
| Zimmermann, et al. 2018125 | Adults with moderate-to-severe AD inadequately controlled with topical therapy, or for whom topical therapies were medically inadvisable. | Lifetime MM with 4-month cycles. | Intervention: dupilumab. Comparator: usual care (emollients). |
Treatment response in the model was defined as an initial response to treatment with a reduction in the EASI score of at least 50%, ≥ 75% or ≥ 90%, stratified by severity. Treatment discontinuation applied annually for dupilumab and usual care patients in long-term model. |
| NICE TA534, 201812 | Adult patients with moderate-to-severe AD who are contraindicated to, intolerant of, had an inadequate response to or for whom it is otherwise medically inadvisable to receive treatment with a systemic immunosuppressant. | Short-term 1-year decision tree followed by a long-term MM. | Intervention: dupilumab. Comparator: BSC, which includes emollients, low- to mid-potency TCSs and rescue therapy which may include higher potency TCSs, oral corticosteroids, TCIs, phototherapy or psychological support. |
Treatment response at week 16 based on composite outcome for EASI 50 + DLQI ≥ 4. Treatment response at week 52 based on conditional response to treatment at week 16. Treatment discontinuation applied annually for dupilumab patients in long-term model. Assumptions around loss of response also included for both arms of the model. |
| NICE TA681, 202113 | Adult patients with moderate-to-severe AD who have previously failed one or more systemic therapies. | A four-state, lifetime MM. The model cycle length was 4 weeks. | Intervention: baricitinib in combination with TCSs. Comparators: dupilumab and BSC, which includes emollients, low- to mid-potency TCSs, phototherapy, psychological support and rescue therapy. |
Treatment response at week 16 based on composite outcome for EASI 50 + DLQI ≥ 4. Treatment response at week 52 based on all-cause discontinuations applied to responders at week 16. Treatment discontinuation applied annually for dupilumab and BSC patients in long-term model. |
| Healthcare Improvement Scotland SMC2011 (2018) and SMC2232 (2019)121,129 | Patients who have had an inadequate response to existing systemic immunosuppressants such as ciclosporin, or in whom such treatment is considered unsuitable. | Short-term 1-year decision tree followed by a long-term (lifetime) MM with annual cycles. | Intervention: dupilumab. Comparator: BSC (not defined). |
Treatment response at week 16 based on composite outcome for EASI 50 + DLQI ≥ 4. Treatment discontinuation applied annually for dupilumab patients in long-term model. |
| Healthcare Improvement Scotland SMC2337, 2021122 | Adult patients with moderate-to-severe AD who are candidates for systemic therapy who have failed at least one current systemic immunosuppressant due to intolerance, contraindication or inadequate disease control. | Lifetime four-state MM. | Intervention: baricitinib. Comparators: dupilumab, BSC (not defined). |
Treatment response at week 16 based on EASI 75. Treatment response at week 52 based on conditional response for responders at week 16. After year 1, all-cause discontinuation rate at week 52 was used to calculate a constant rate of discontinuation. |
| Institute for Clinical and Economic Review, 2017126 | Adults with moderate-to-severe AD inadequately controlled with topical therapy, or for whom topical therapies were medically inadvisable. | Lifetime MM with 4-month cycles. | Intervention: dupilumab. Comparator: usual care (emollients). |
Treatment response in the model was defined as an initial response to treatment with a reduction in the EASI score of at least 50%, ≥ 75% or ≥ 90%, stratified by severity. Treatment discontinuation applied annually for dupilumab and usual care patients in long-term model. |
| Institute for Clinical and Economic Review, 2021127 | Patients with moderate-to-severe AD. | 5-year MM with 4-month cycles. | Interventions:
|
Treatment response in the model was defined as an initial response to treatment with a reduction in the EASI score of at least 50%, ≥ 75% or ≥ 90%, stratified by severity. Treatment specific per-cycle treatment discontinuation rates (all cause) for the first year after initial treatment and then for all subsequent years over the model time horizon where data was available was used in the model. |
Results – health-related quality of life
The electronic database searches identified 1741 potential publications. Upon removal of duplicates, 1632 publications were screened against the eligibility criteria. Of these, 79 publications were included and further assessed against the same eligibility criteria with 4 additional studies separately identified from HTA websites. Following this, 20 publications were included. The PRISMA flow diagram (see Report Supplementary Material 1) details the inclusion and exclusions of studies at each stage of the review.
Of the 20 included publications, 4 were HTA submissions,12,13,121,122 12 were full-text publications85,125,130–139 and 4 were abstracts. 140–143 The four HTA submissions include two NICE technology appraisals and two SMC technology appraisals (each including one appraisal for dupilumab and one appraisal for baricitinib). The 20 publications represent 16 unique studies (3 studies were reported in abstract and full-text form130,135,136 and 1 study was reported in 2 publications85,138). Although the HTA submissions to NICE and SMC for the same drug are based on the same study data, they are considered separately in this review as different methods and processes were used to analyse and assess the EQ-5D data provided by the company (thus, different results are reported).
Of the 20 included publications, 15 reported EQ-5D data12,13,85,121,122,125,130–134,137,138,141,142 and 1 reported EQ-5D data and time trade-off (TTO) data. 139 The remaining four publications reported short-form 6-dimension (SF-6D) data or TTO data. 135,136,140,143 Of the 16 publications which reported EQ-5D data, 9 collected EQ-5D-5L data13,122,130–132,134,139,141,142 and 4 collected EQ-5D-3L data. 12,85,137,138 The remaining three publications did not clearly specify which levels of the EQ-5D were used. 121,125,133 Of the nine publications which collected EQ-5D-5L data, two mapped EQ-5D-5L responses to EQ-5D-3L13,122 responses using the van Hout 2021 algorithm. 144
The majority of publications reported heath-state utility values (HSUVs) according to severity (based on the PO-SCORAD score, SCORAD score or self-reported severity) or response (based on improvements in the EASI score or EASI + DLQI score). A summary of the 20 included publications (16 unique studies) is provided in Table 6 and detailed data extractions can be found in the supplementary material.
| Study | Author, Year | Country | Measure | Valuation | HSUVs according to |
|---|---|---|---|---|---|
| 1 | Andersen, 2020130 | Europe (France, Germany, the UK) and the USA | EQ-5D-5L | Unclear (respective weights) | Severity and country |
| Nyberg, 2018 (abstract)142 | Europe (France, Germany, the UK) and the USA | EQ-5D-5L | Unclear | Severity and country | |
| 2 | Hsieh, 2021131 | Taiwan | EQ-5D-5L | UK weights | Severity |
| 3 | Kwatra, 2021132 | USA | EQ-5D-5L | Unclear | Comorbidity |
| 4 | Misery, 2018133 | France | EQ-5D | Unclear | Severity |
| 5 | Girolomoni, 2021134 | EU5 (France, Germany, Italy, Spain and the UK) | EQ-5D-5L | Unclear | Comorbidity |
| 6 | Retzler, 2019135 | UK | TTO | TTO | Regimen intensity |
| Retzler, 2018 (abstract)143 | Unclear | TTO | TTO | Regimen intensity | |
| 7 | Silverberg, 2019136 | USA | SF-6D | US weights | Severity |
| Silverberg, 2019 (abstract)140 | USA | SF-6D | Unclear | Severity | |
| 8 | Simpson, 2017137 | Multiple study locations | EQ-5D-3L | Unclear | Response and treatment |
| 9 | Simpson, 201685 | Multiple study locations | EQ-5D-3L | UK weights | Treatment |
| Simpson, 2016138 | Multiple study locations | EQ-5D-3L | UK weights | Overall only | |
| 10 | Song, 2019139 | Korea | EQ-5D-5L and TTO | Korean weights | Response |
| 11 | Vietri, 2017 (abstract)141 | France, Germany, the UK | EQ-5D-5L | Unclear | Severity |
| 12 | Zimmerman, 2018125 | USA | EQ-5D | Unclear | Severity and response |
| 13 | SMC2011, 2018129 | Multiple study locations | EQ-5D | Unclear | Response and treatment |
| 14 | SMC2237, 2021122 | Multiple study locations | EQ-5D-5L mapped to EQ-5D-3L | UK weights | Response |
| 15 | NICE TA534, 201812 | Multiple study locations | EQ-5D-3L | UK weights | Response and treatment |
| 16 | NICE TA681, 202113 | Multiple study locations | EQ-5D-5L mapped to EQ-5D-3L | UK weights | Response |
Independent economic assessment
Methods
Populations
As described in the section Decision problem, the population relevant to the MTA are those with moderate-to-severe AD, irrespective of previous treatment and of age, with a subgroup of interest being those for whom systemic therapies have been inadequately effective, not tolerated or contraindicated.
As per guidance from NICE, the EAG has considered each of the companies proposed positions in the treatment pathway as part of the assessment for the CEA. Thus, clinical data in the NMA were analysed by pre-specified subgroups based on the age and line of therapy and TCS use (see Methods of data synthesis). Consequently, the modelled populations are as follows:
-
Adults who are eligible for systemic treatment (CsA) on inadequate response to topical treatments (referred to hereafter as the adult first-line systemic treatment population).
-
Adults who achieve inadequate response to, cannot tolerate or are contraindicated to CsA – monotherapy and combination therapy (referred to hereafter as the adult second-line systemic treatment population).
-
Adolescents, irrespective of prior therapy.
Trials assessing AD treatments in the adolescent population included a mix of patients at all lines of systemic treatment (see Method for reviewing effectiveness). Due to small numbers in the trials that include adolescents, a robust subgroup analyses by line of treatment for this population is not possible; therefore, adolescents are considered in the model irrespective of prior line of therapy. Furthermore, due to limitations in the clinical evidence, combination therapy data are unavailable for the adolescent population and monotherapy data are unavailable for the adult first-line systemic treatment population (see Assessment of clinical effectiveness). Further details on available treatment effectiveness data, interventions and proposed positions in the treatment pathway for the CEA are given in the sections Treatment effectiveness and Interventions and comparators, respectively.
Baseline characteristics
In each of the companies’ economic models, baseline characteristics were taken from the key trials for the drugs, which is appropriate for a STA model. However, for a MTA model, a consistent approach using common assumptions and baseline characteristics is required. Due to the heterogeneity of baseline characteristics of those enrolled across the key trials of interest for abrocitinib, tralokinumab and upadacitinib, the EAG selected trials that most closely reflect the populations of interest to the MTA model based on feedback from the EAG clinical experts. The EAG clinical experts considered that the upadacitinib trials were appropriate to inform the baseline characteristics in the model. Table 7 presents the upadacitinib trials used to inform the baseline characteristics used for each population in the economic model.
| Population | Trial |
|---|---|
| First-line systemic treatment – adults, combination therapy | AD UP |
| Second-line systemic treatment – adults, monotherapy | Measures UP 1 and 2 |
| Second-line systemic treatment – adults, combination therapy | AD UP |
| Adolescents, monotherapy | Measures UP 1 and 2 |
Interventions and comparators
The interventions of interest as part of this MTA are abrocitinib, tralokinumab and upadacitinib. As per the final protocol,48 the EAG has considered each of the companies proposed position in the treatment pathway as part of the CEA. As such, the interventions are compared with the recommended treatment in the proposed position of the treatment pathway, outlined in Table 8. The recommended first-line systemic treatment for adults is CsA but does not hold a marketing authorisation for use in adolescents. However, dupilumab is funded for use in adolescents under the NHS England Medicines for Children Policy and is included as the comparator for this population. Recommended second-line systemic treatments for adults include dupilumab and baricitinib.
| Population | Intervention | Comparator |
|---|---|---|
| Adults | ||
| First-line systemic therapy for those having inadequate response to topical treatments | Upadacitinib + TCS Abrocitinib + TCS |
CsA + TCS |
| Second-line systemic therapy for those who achieve inadequate response to, cannot tolerate or are contraindicated to CsA | Abrocitinib (±TCS) | Dupilumab (±TCS) Baricitinib + TCS |
| Tralokinumab (±TCS) | ||
| Upadacitinib (±TCS) | ||
| Adolescents | ||
| Adolescents, irrespective of prior therapy | Abrocitinib | Dupilumab |
| Upadacitinib | ||
Each of the treatments is considered as a monotherapy and in combination with TCS in the cost-effectiveness analyses. However, as described in the section Method for reviewing effectiveness, results are only available from the NMA for combination therapy for the adult first-line systemic treatment population, while results for monotherapy are available for the adolescent population and for baricitinib for the adult second-line systemic treatment population (see Table 8).
Table 9 presents an overview of the treatment regimens and the EAG approach to inclusion of the different treatment regimens in the model.
| Treatment | Dose | Administration and frequency | SmPC guidance | EAG approach |
|---|---|---|---|---|
| Abrocitinib | 200 mg | Oral tablet, OD. | Recommended starting dose for most adults. | As both doses are recommended in the draft SmPC, each are evaluated separately in the CEA over a lifetime horizon. |
| 100 mg | Oral tablet, OD. | Recommended starting dose for patients aged 65 years and over and other patients who may benefit from a lower starting dose. | ||
| Baricitinib | 4 mg | Oral tablet, OD. | Recommended starting dose for patients. | In TA681,13 the committee considered that 4 mg was the licensed dose relevant for most patients and was used as the basis for the recommendation. For consistency, the EAG only considers the 4 mg dose. |
| 2 mg | Oral tablet, OD. | Appropriate for patients aged 75 years and over and those with a history of chronic or recurrent infections. A dose of 2 mg OD should be considered for patients who have achieved sustained control of disease activity with 4 mg OD. | ||
| CsA | 5 mg/kg for the first 6 weeks, followed by 3 mg/kg thereafter. | Oral tablet, OD for a maximum of 1 year. | Recommended dose range is 2.5–5 mg/kg/day given in two divided oral doses. However, due to the variability of this condition, treatment must be individualised. | The EAG’s clinical experts advised there is no clinical consensus on the appropriate CsA regimen, as this is individualised as per SmPC guidance. As such, the accepted dose in TA534 is included in the EAG base case. Alternative dosing explored in scenario analyses. |
| Dupilumab | 300 mg with an initial loading dose of 600 mg. | SC injection, once every 2 weeks. | Recommended dose for adults and adolescents weighing > 60 kg. | Included as per SmPC recommendation. |
| Tralokinumab | 300 mg with an initial loading dose of 600 mg. | SC injection, once every 2 weeks. | Recommended dose and frequency for all patients. At prescriber’s discretion, frequency of dose can be reduced to once every 4 weeks for patients who achieve clear or almost clear skin after 16 weeks of treatment. | The base case assumed that all patients stay on Q2W dosing. A scenario analysis was run where a proportion of responders (assumed to be the proportion achieving EASI 75 or more) move to once every 4 weeks dosing. |
| Upadacitinib | 15 mg | Oral tablet, OD. | The recommended dose of upadacitinib is 15 or 30 mg OD based on individual patient presentation. | For the EAG base case, both doses evaluated for adults and only the 15 mg dose evaluated for adolescents. |
| 30 mg | Oral tablet, OD. | Recommended dose of upadacitinib is 15 mg for adolescents weighing at least 30 kg and patients ≥ 65 years of age, the recommended dose is 15 mg OD.
|
Model structure
For the CEA, a hybrid economic model was developed comprising a short-term (1 year) decision tree component, to capture the treatment induction phase and treatment response assessments, followed by a long-term (lifetime), three-state MM. The comparator in the analysis for first-line systemic treatment is CsA and for second-line systemic treatment, comparators are dupilumab and baricitinib. All treatments in the model are evaluated as both monotherapies and in combination with TCS. The development of the model structure was informed by published models identified in the SLR, supplied company submissions and the approaches accepted for TA53412 and TA681. 13 Additionally, the EAG has developed two short-term decision tree models to reflect the differences in first- and second-line systemic treatments. Each component of the model is discussed in turn below.
Short-term decision tree – first-line systemic treatment (adults)
Abrocitinib and upadacitinib are the only treatments that have a proposed position in the first-line systemic treatment pathway and as such will be compared against CsA, which is only recommended for a maximum of 1 year, after which all patients discontinue to BSC.
Figure 7 presents the model schematic for first-line systemic treatment for adults. All patients enter the short-term model, starting either abrocitinib/upadacitinib or CsA (all in combination with TCS) and remain on treatment for 16 weeks (treatment induction phase). At week 16, treatment response is assessed, defined as achieving EASI 50 + DLQI ≥ 4 and responders remain on treatment until week 52. Non-responders at week 16 discontinue treatment and receive BSC.
FIGURE 7.
First-line systemic treatment short-term decision tree model structure – adults.

Between week 16 and 52, responders may lose response to treatment or discontinue treatment for other reasons and will enter the long-term MM in the BSC health state. Abrocitinib/upadacitinib responders who sustain their response between week 16 and 52 and are still on treatment enter the long-term MM in the maintenance health state. For patients on CsA, the maximum recommended treatment duration is 12 months; thus, they discontinue to BSC in the long-term MM. Sustained response for abrocitinib/upadacitinib at week 52 is based on conditional discontinuation data, defined as the proportion of patients discontinuing treatment at week 52 from those who achieve response at week 16.
In the short-term model, the BSC health state is composed of responders and non-responders and these proportions are informed by week 16 data response data (see Method for reviewing effectiveness and Treatment effectiveness). This approach was accepted in TA681 as an appropriate way to capture the waxing and waning nature of response to BSC treatment.
Short-term decision tree – second-line systemic treatment (adults)/adolescents (all lines of treatment)
Figure 8 presents the model schematic for second-line systemic treatment (adults) and adolescents (refer to the section Populations for further details about line of therapy for adolescents). Adult patients enter the short-term model starting treatment on one of the five second-line systemic treatments (abrocitinib, baricitinib, dupilumab, tralokinumab or upadacitinib). Adolescent patients enter the model on abrocitinib, dupilumab or upadacitinib. Patients remain on treatment for 16 weeks (treatment induction phase), after which point treatment response is assessed, defined as achieving EASI 50 + (C)DLQI ≥ 4. Responders at week 16 remain on treatment until week 52. Non-responders at week 16 discontinue treatment and receive BSC.
FIGURE 8.
Second-line systemic treatment short-term decision tree model structure (adults)/adolescents (all lines of treatment).

Between weeks 16 and 52, responders may lose response to treatment or discontinue treatment for other reasons and will enter the long-term MM in the BSC health state. Responders who sustain their response between weeks 16 and 52 and are still on treatment enter the long-term MM in the maintenance health state. Sustained response at week 52 is based on conditional discontinuation data, defined as the proportion of patients discontinuing treatment at week 52 from those who achieve response at week 16.
In the short-term model, the BSC health state is composed of responders and non-responders and these proportions are informed by week 16 data response data (see Method for reviewing effectiveness and Treatment effectiveness). As mentioned previously, this approach was accepted in TA681 as an appropriate way to capture the waxing and waning nature of response to BSC treatment.
Long-term Markov model
At the end of each of the short-term decision trees (start of year 2), patients enter a long-term three-health state MM. Health states in the model include maintenance, BSC and death (Figure 9).
FIGURE 9.
Long-term MM schematic.

Patients who have maintained a response at week 52 and still on treatment enter the MM in the maintenance health state and remain there until loss of response (via treatment waning) or if they discontinue treatment for any reason (all cause discontinuation) whereupon they transition to the BSC health state.
Patients who have discontinued treatment to BSC in the short-term decision tree enter the MM in the BSC health state and remain there until death. As with the BSC health state in the short-term decision tree model, the MM BSC health state is composed of responders and non-responders and these proportions are informed by week 16 data response data (see Method for reviewing effectiveness and Treatment effectiveness), in line with approach accepted in TA681. 13
At any time in the model, patients can transition to the death state. As treatment for AD is not expected to affect mortality, transitions to the death state are informed by general population mortality rates.
In the long-term model, an annual cycle length has been implemented and half-cycle correction applied.
Time horizon, perspective and discounting
The time horizon of the model is lifetime (up to a maximum age of 100 years). The perspective of the analysis is the NHS in England. Costs and QALYs have been discounted at 3.5%, as per the NICE reference case. Scenarios were conducted limiting the time horizon to 5 years after the mean age and 75 years of age for the adult analyses and 18 years of age for the adolescent analyses.
Treatment effectiveness
The primary treatment outcome assessed in the MTA model is treatment response at week 16, defined using a composite outcome of EASI 50 + DLQI ≥ 4. In TA53412 and TA681,13 the composite outcome was preferred by the committee as it was deemed to be sensitive to changes in treatment outcomes and more clinically relevant than EASI 75. As a result, the composite outcome was carried forward in each of the company models submitted as part of the MTA.
Log ORs from the NMA (see Method for reviewing effectiveness) were used to estimate week 16 treatment response probabilities used in the model. However, as noted in the section Quantity and quality of research available, there were several data limitations in the NMA that meant outcome data (composite or EASI 75) were unavailable for some of the populations. Table 10 presents an overview of the NMA outcome data available for each population.
| Population | Monotherapy | Combination therapy | ||
|---|---|---|---|---|
| EASI 50 + DLQI ≥ 4 | EASI 75 | EASI 50 + DLQI ≥ 4 | EASI 75 | |
| Adults – first-line systemic treatment | ✗ | ✗ | ✗ | ✓ |
| Adults – Second-line systemic treatmenta | ✓ | ✓ | ✓ | ✓ |
| Adolescents | ✗ | ✓ | ✗ | ✗ |
The composite outcome from the NMA was obtained for the adult second-line systemic subgroup for both monotherapy and combination therapy analyses and EASI 75 was explored in scenario analyses. However, the EAG was unable to obtain composite outcome data for baricitinib as these data are redacted in TA681 (and the company declined to provide them to the EAG). Furthermore, EASI 75 data for baricitinib were only available as combination therapy for the adult second-line systemic population. Therefore, any comparisons with baricitinib in the adult second-line systemic treatment subgroup are presented as scenario analyses.
The composite outcome could not be obtained for treatments relevant to the adolescent and adult first-line systemic treatment subgroups for both monotherapy and combination therapy analyses. However, the EASI 75 outcome was available for the adolescent subgroup for the monotherapy analyses and adult first-line systemic treatment subgroup for the combination analyses, and as such this outcome is used for the base case in these populations.
The committee for TA534 and TA681 considered that in clinical practice, dupilumab and baricitinib will likely be used as combination therapies rather than monotherapies and this view is reflected by the EAG’s clinical experts when considering the three new treatments under consideration. Thus, for the adult first-line systemic treatment subgroup, the combination analyses are likely to be more relevant than the monotherapy analyses. Furthermore, upadacitinib is the only treatment where the company has proposed its use in the adult first-line systemic treatment subgroup. For the adolescent population, the EAG’s clinical experts explained that they would be treated in the same way as adults; thus, combination therapy is more relevant than monotherapy.
As the combination therapy analyses are more relevant for clinical practice, the EAG considered that missing monotherapy data are not critical for decision-making for the adult first-line systemic treatment subgroup. However, for the adolescent population, monotherapy analyses may potentially underestimate the effectiveness of the treatments when used in combination with TCS in clinical practice and may reflect a conservative view of cost effectiveness. The EAG has included treatment response outcomes one-way sensitivity analyses to capture the uncertainty around the estimates, but the upper bound estimate for the adolescent population may be useful to help understand what the cost effectiveness of combination therapy might be for this population.
Implementation of network meta-analysis outputs
To calculate the probability of response at week 16 for each of the treatments, a baseline level of treatment response for patients who would have otherwise been on BSC was needed. In TA534, TA681 and the company models, placebo response from the key trials of the drug under consideration was used. However, as placebo response varies in each of the trials, the EAG consulted with its clinical experts to select a trial which had baseline characteristics that were representative of the population who would be treated in the NHS.
The trials informing the baseline characteristics used in the model are described in the section Baseline characteristics. For the adolescent and the second-line adult monotherapy analyses, the EAG’s clinical experts considered that the upadacitinib Measure UP 1 and 2 trials were appropriate to use for the baseline characteristics of the model and placebo response. As the populations in Measure UP 1 and 2 were considered comparable, for each subgroup, the EAG pooled placebo trial data. For the adult first- and second-line combination analyses, the upadacitinib AD-UP trial was considered appropriate to use for the baseline characteristics and placebo response by the EAG’s experts. It should be noted that the baseline treatment response is also used for the weighted average of responders and non-responders in the BSC health state to estimate costs and QALYs.
The baseline week 16 treatment response was converted into log-odds to be applied to the log-ORs from the NMA (representing treatment vs. placebo) to estimate baseline-adjusted log-odds for each treatment. The baseline-adjusted log-odds for each treatment were then exponentiated and transformed to calculate the probability of patients responding to treatment at week 16.
Described in the section Quantity of research available, the log-ORs for each treatment are based on the all-observed data from the trials, that is, patients were not censored from the analysis upon receipt of rescue treatment. Furthermore, for the abrocitinib analyses in the adult second-line systemic treatment analyses, the EAG used data from the relevant JADE trials for patients who previously failed or were intolerant to ciclosporin (referred to by the company as the restricted population in the abrocitinib company submission). The EAG considers that the abrocitinib-restricted population more closely reflects the definition of the adult second-line systemic treatment population, as described in the section Assessment of clinical effectiveness, but the patient numbers are small. The NMA sensitivity analysis based on the generalisable population for abrocitinib (defined by the company as patients who were previously treated with at least one systemic treatment for AD) show relatively similar results to the primary analysis (see Assessment of clinical effectiveness). Nonetheless, the EAG performed a scenario analysis exploring log-ORs based on data from the relevant JADE trials for the generalisable population.
Additionally, the EAG ran a sensitivity analysis in the NMA where patients were censored for rescue therapy (see Assessment of clinical effectiveness), which had a substantial impact on the treatment effect for dupilumab in the adult second-line systemic treatment monotherapy analyses and for upadacitinib 15 mg in the adolescent monotherapy analyses. Consequently, the EAG explored a scenario implementing the log-ORs based on the NMA for rescue therapy censoring for the adult second-line systemic treatment monotherapy analyses.
For the EAG base case, the results from the NMA using the observed data from studies were used, consistent with TA534, TA681 and the companies’ submissions. However, the EAG ran a sensitivity analysis in the NMA exploring an adjustment for potential baseline heterogeneity in placebo response. Described in the section Assessment of clinical effectiveness, the EAG’s sensitivity analysis failed to converge for the first- and second-line adult analyses for combination treatment but did converge for the adolescent and adult second-line monotherapy analyses. Thus, the EAG explored a scenario implementing the log-ORs based on the placebo response adjustment NMA for the adult second-line systemic treatment and adolescent monotherapy analyses.
Week-52 treatment response outcomes
At week 52, a proportion of treatment responders may not continue on to long-term maintenance treatment. In TA534, the committee preferred week 52 treatment response outcomes to be modelled using conditional response data, defined as the proportion of week 16 responders who were still responding to treatment at week 52. However, in TA681, the committee considered that loss of response is not the only reason for treatment discontinuation in week 16 responders and that sustained response at 52 weeks should be based on all cause stopping rate for people whose condition responded to treatment at week 16 but withdrew from treatment at week 52 (conditional discontinuation).
Of the company submissions supplied to the EAG, the abrocitinib model was the only one to use conditional discontinuation data to estimate the proportion of responders still on treatment at week 52. As noted earlier, the upadacitinib model was developed prior to the publication of TA681 and therefore the company’s base case used conditional response data to model week 52 outcomes, as per TA534. However, the tralokinumab model also based week-52 outcomes on conditional response data, which was used to inform a NMA for response to treatment at week 52, although the recommendations for TA681 had recently been published prior to the company’s submission to NICE.
The EAG has used conditional discontinuation data, consistent with TA681, to estimate the probability of week-16 responders transitioning to long-term maintenance treatment at week 52. The EAG requested conditional discontinuation data from the companies for abrocitinib, tralokinumab and upadacitinib, as well as extracting relevant data from TA534. It should be noted that discontinuation data in TA681 were redacted, and the company declined to provide them to the EAG.
Based on the data supplied by the companies, it precluded carrying out a NMA for week 52 conditional discontinuation for the treatments of interest. In the studies evaluating abrocitinib and upadacitinib, on completion of the 16-week treatment phase, those receiving placebo or active comparator and achieving response in the parent trial were rerandomised to a dose of the investigational treatment evaluated, that is, either abrocitinib or upadacitinib. Thus, there are no data available in the longer term for placebo, which means that there is no common comparator across the studies to provide a connected network suitable for analysis. Additionally, the data supplied by the company on conditional discontinuation of abrocitinib were for the whole trial population, which is predominantly formed of those receiving first-line systemic treatment. In addition, data for baricitinib are redacted in TA681 and so could not be included.
In lieu of NMA results for conditional discontinuation at week 52, the EAG implemented the available conditional discontinuation data in the model, but assumptions were made where data gaps existed. Table 11 presents an overview of available conditional discontinuation data at week 52 for dupilumab.
| Treatment | Conditional discontinuation at week 52 | Source/assumptions |
|---|---|---|
| Base case | ||
| Monotherapy – adults, EASI 50 + DLQI ≥ 4 | 3.7% | Assumed to be the same as dupilumab combination therapy |
| Combination therapy – adults, EASI 50 + DLQI ≥ 4 | 3.7% | TA534. Estimate accepted by the committee. Data based on annual discontinuation in CHRONOS, defined as non-completers in the 52-week treatment period among responders at week 16. |
| Monotherapy – adolescents (EASI 75) | 5.1% | TA534. Data based on annual discontinuation in CHRONOS, defined as non-completers in the 52-week treatment period among EASI 75 responders at week 16 (n/N = 4/78). |
| EASI 75 scenario (adults) | ||
| Monotherapy – adults, EASI 75 | 5.1% | Assumed to be the same as dupilumab combination therapy |
| Combination therapy – adults, EASI 75 | 5.1% | TA534. Data based on annual discontinuation in CHRONOS, defined as non-completers in the 52-week treatment period among EASI 75 responders at week 16 (n/N = 4/78). |
The company for tralokinumab only provided conditional discontinuation data for monotherapy administered once Q2W and once Q4W based on EASI 75 at week 52. As such, the EAG assumed conditional discontinuation data for tralokinumab monotherapy were the same as the combination therapy and composite outcome due to lack of data. It should be noted that the company for tralokinumab provided annual treatment discontinuation data based on the composite outcome and EASI 75 for tralokinumab from the ECZTEND study for the ECZTRA-7-like population. However, in ECZTEND, TCS use was optional and Q4W dosing was not an option. Furthermore, ECZTEND also included patients from ECZTRA 4, 5 and 6. To maintain a consistent approach with the estimation of conditional discontinuation across treatments, the EAG only considered the ECZTRA-7-like data from ECZTEND in a scenario analysis.
For dupilumab and tralokinumab, conditional discontinuation was assumed to be the same for monotherapy and combination therapy as the type of monoclonal antibody appears to be more important than the addition of TCS when considering sustained treatment response. For abrocitinib and baricitinib, the EAG assumed the same conditional discontinuation as upadacitinib as they are all JAK inhibitors. However, in the adolescent population, as only upadacitinib 15 mg is applicable, the conditional discontinuation rate has been assumed for abrocitinib 200 mg. The company for abrocitinib did provide conditional discontinuation data from JADE EXTEND for the adolescent population, but as an overall percentage rather than supplying patient numbers as requested by the EAG. The EAG is therefore unclear how many adolescent patients from JADE MONO-1/2 entered EXTEND and therefore inform the company’s estimates of conditional discontinuation.
The drug class approach maps available high- and low-dose data for upadacitinib to high- and low-dose JAK inhibitors. The EAG acknowledges this is a simplification and potentially results in conservative cost-effectiveness results for comparisons with baricitinib 4 mg (high dose), which is least efficacious of the JAK inhibitors. However, comparisons with baricitinib do not form part of the EAG base case and are only considered in scenario analyses.
Long-term treatment discontinuation
No long-term treatment discontinuation data are available for any of the treatments considered in the model. In TA534, the annual treatment discontinuation rate for dupilumab (3.7%) was based on the observed probability of week-16 responders discontinuing treatment by week 52, which was accepted by the committee. In TA681, the Evidence Review Group (ERG) preferred the use of all-cause discontinuation data observed between weeks 16 and 52 to model a single discontinuation rate across after week 16 for the remainder of the time horizon for baricitinib (data are redacted). However, it should be noted that in both TA534 and TA681, the annual discontinuation data based on conditional discontinuations are for the period between week 16 and week 52 and therefore represents 36-week data and not 52-week data.
The company for tralokinumab followed the same approach to long-term treatment discontinuation accepted in TA534 and TA681 in their economic model. Specifically, 56-week conditional treatment discontinuation data for tralokinumab from the full-trial population in ECZTEND (which includes patients from ECZTRA 1, 2 and 3 as well as from ECZTRA 4, 5 and 6) were used in the model. The annual rate of discontinuation from tralokinumab due to AEs or lack of efficacy in ECZTEND was 2.3% among patients who achieved EASI 50 and ΔDLQI ≥ 4 at week 16 in their parent study.
In the upadacitinib model, the company used 52-week treatment discontinuation data (based on response status at week 16) from AD UP to model annual discontinuations. The annual rate of treatment discontinuation for upadacitinib 15 and 30 mg was based on responders to EASI 75 and non-responders in the intention-to-treat population in AD UP. It should be noted that upadacitinib conditional discontinuation data from AD UP in the EAG base case are also based on the 52-week data cut but specifically for responders to the composite outcome and EASI 75 for scenario analyses. In the abrocitinib model, the company used the conditional discontinuation data at week 52, converted to an annual probability to inform the long-term treatment discontinuation rate.
For consistency, the EAG has adopted the approach accepted in TA534 and TA681 for long-term treatment discontinuation for the base case. Specifically, it is assumed for the economic model that the long-term treatment discontinuation rate is equal to the conditional discontinuation rate for each treatment. The EAG recognises that using conditional discontinuation data does not represent an annual rate, but rather a 36-week rate. However, there is a lack of long-term data on treatment discontinuation to suggest whether rates are likely to increase for patients with a long-term response to treatment. Furthermore, as discussed in the section Treatment waning, the EAG has assumed that treatment waning also results in treatment discontinuation. Nonetheless, the EAG explored a scenario where conditional discontinuation data are converted to an annual rate for completeness. Additionally, annual treatment discontinuation data from ECZTEND for the ECZTRA-7-like population was only considered in scenario analyses due to limitations with the data, as discussed in the section Week-52 treatment response outcomes.
Treatment waning
Over time, patients may lose response to treatment, whether on active treatment with biologics or BSC. In TA534, assumptions around treatment effect waning were included in the company’s economic model and accepted by the committee. The TA534 final appraisal document (FAD) states:
In the dupilumab maintenance state, the company assumed that 2% of the benefit would be lost in year 2, 5% in year 3, 7% in year 4, and 8% in year 5 and beyond. It used these estimates to adjust down the proportion of people who continued to have dupilumab (that is, those who lost the benefit of dupilumab moved to the best supportive care state and then accrued the utility associated with that state).
In the company submissions for abrocitinib, tralokinumab and upadacitinib and in TA681, treatment-waning assumptions were influenced by the approach in TA534 due to a lack of data in the key trials for the drugs. However, the assumptions and implementation of active treatment waning in TA534 have been interpreted in various ways by the companies for baricitinib (TA681), abrocitinib, tralokinumab and upadacitinib.
Active treatment-waning proportions in the abrocitinib and upadacitinib models were taken from TA534. In the abrocitinib model, by year 5 and beyond, 8% of patients on active treatment would lose response. However, between years 2 and 5 in the upadacitinib model, up to 8% of patients experienced treatment waning and from year 6 up to year 10, a further 1% per year were assumed to lose response, with no further waning beyond year 10. For the tralokinumab base case, it was assumed that between 2% and 3% of patients would lose treatment response annually up to year 4, with 1% losing treatment response annually from year 5 onwards.
The implementation of active treatment waning in the tralokinumab and upadacitinib models was similar to TA534, with patients discontinuing to BSC upon treatment waning. However, in the upadacitinib model, when patients on active treatment move to the BSC health state upon treatment waning, they first incur utility of BSC non-responders and then gradually return to the baseline utility following BSC non-responders’ treatment-waning rates (see Report Supplementary Material 1). In TA681 and the abrocitinib model, patients on active treatment were assumed to incur a utility loss. In TA681, patients returned to baseline utility upon treatment waning and in the abrocitinib model, non-responder utilities were applied to patients who lost response.
For the MTA model, the EAG has adopted the active treatment-waning approach accepted in TA534. Specifically, the EAG has assumed that in years 2, 3, 4 and 5 onwards, 2%, 5%, 7% and 8% of patients, respectively, will lose response to active treatment and discontinue to BSC. As soon as patients no longer achieve EASI 50 + DLQI ≥ 4, they are considered non-responders. The EAG acknowledges that there may be overlap between the proportion of patients losing response to treatment and long-term all-cause treatment discontinuation, as lack of efficacy is included as a reason to stop treatment. However, the size of the overlap between treatment waning and all-cause discontinuation is unknown and as such the EAG approach can be considered conservative. The EAG included long-term all-cause treatment discontinuation and treatment-waning proportions in one-way sensitivity analysis (OWSA) to determine if these parameters are key drivers of cost effectiveness (see Results). Furthermore, the EAG included a scenario analysis where no active treatment-effect waning was assumed.
Unlike TA534, TA681 and the company economic models, BSC is not a comparator in the EAG model but a single health state that patients transition to due to lack or loss of response to treatment or treatment discontinuation for other reasons. As described in the section Model structure, BSC is modelled as a weighted average of responders and non-responders to BSC to reflect the waxing and waning nature of AD and thus captures treatment effectiveness fluctuations. As such, the EAG has assumed no additional treatment waning for the BSC health state, which is in line with the ERG’s preferred approach in TA681 (see Report Supplementary Material 1). The committee for TA681 considered that the ERG’s approach represented different patients moving in and out of disease control over time, but treatment waning would be between the ERG’s approach (no waning, BSC modelled as a single health state of 50% responders and 50% non-responders) and the company’s approach based on TA534 (up to 97% of BSC patients lose response). However, the committee for TA681 did not give further direction to handle treatment waning in the BSC health state. To explore the uncertainty around BSC waning, baseline placebo response has been included in the OWSA (which informs the BSC health state), as well as scenarios exploring shorter time horizons.
Further information on the BSC treatment-waning assumptions adopted in TA534 and TA681, and assumptions used in the company models can be found in Report Supplementary Material 1.
Mortality
Treatments for moderate-to-severe AD are not expected to affect mortality. Thus, the EAG has used Office of National Statistics (ONS) National Life Tables for England and Wales to estimate age-adjusted all-cause mortality in the economic model.
Adverse events
Adverse events included in the EAG’s economic model are in line with those included in TA534 and TA681 and were considered the most important to include by the EAG’s clinical experts. In TA534, the most frequent and serious AEs reported in the dupilumab trials were included and these were injection-site reaction, allergic conjunctivitis, infectious conjunctivitis and oral herpes. TA681 also included the most frequent and serious AEs reported in the baricitinib trials, as well as those from TA534, with the only addition to the included AEs being upper respiratory tract infection. The company for tralokinumab only included the TA534 AEs in their economic model. In the abrocitinib and upadacitinib models, AEs with an incidence of > 5% in the intervention trials, dupilumab trials, TA534 and TA681 were included but no detail was provided for the severity of the AEs. Furthermore, the company for upadacitinib excluded oral herpes from included AEs as clinical advice suggested that patients with oral herpes would self-medicate with over-the-counter medication. However, in TA534, the cost associated with oral herpes was for a GP visit and the EAG considers it should be included in the CEA. Refer to Report Supplementary Material 1 for a comparison of AEs included in TA534, TA681 and the company models.
The EAG’s approach to AEs is generally in line with TA534, TA681 and the company models and includes serious AEs with an incidence of > 5% in any treatment arm. The EAG reviewed and extracted data on AEs from publications included in the clinical SLR, company submissions and appendices, company clarification responses and committee papers from TA534 and TA681. The available clinical study reports (CSRs) for abrocitinib, tralokinumab and upadacitinib studies were also searched for the AEs of interest where there were missing data. As the EAG included specific AEs that may not be applicable or captured for all treatments in the model, a NMA on individual AEs was not deemed to be appropriate.
The AEs included in the EAG’s economic model are injection-site reaction, allergic conjunctivitis, infectious conjunctivitis, oral herpes, upper respiratory tract infection and acne. In the upadacitinib model, skin infections were included, and in the abrocitinib model, folliculitis, headache, nausea, pharyngitis and nasopharyngitis were included, but these were excluded from the EAG model as the severity of these events could not be determined. These AEs are easily treated, and a cost to the NHS is rarely incurred. The EAG considered that the definition of infectious conjunctivitis in the companies’ models is based on the Medical Dictionary for Regulatory Activities (MedDRA) term ‘conjunctivitis’ (system organ class of infections and infestations), which is likely to reflect infectious conjunctivitis. To maintain consistency across the analysis, the EAG extracted data for conjunctivitis to inform the AE rate for infectious conjunctivitis.
Data on AEs for CsA were unavailable. In TA534, the committee accepted assuming zero AEs for CsA in the short term as treatment is only given for 1 year before patients move to BSC and this assumption has been carried forward in the EAG economic model.
Adverse events for BSC were based on placebo data from AD-UP. As mentioned in the section Baseline characteristics, baseline characteristics from the upadacitinib trials were deemed to be representative of the population who would be treated in clinical practice in England according to the EAG’s clinical experts. The EAG has assumed that the BSC AE rate for monotherapy is the same as combination therapy because in clinical practice BSC includes TCS.
Table 12 presents the 16-week AE rates included in the EAG economic model. Weekly and annual rates of AEs were calculated from the 16-week data for the short- and long-term model. The rates of AEs were used to estimate the costs to treat an AE only as it was assumed that the HRQoL data collected in the trials and used in the model would capture the acute impact of AEs. The EAG’s approach is in line with TA534 and TA681, as well as the approach adopted in the company models for abrocitinib, tralokinumab and upadacitinib. Refer to the section Costs of managing adverse events for AE costs used in the economic model.
| Treatment | Injection-site reaction (%) | Allergic conjunctivitis (%) | Infectious conjunctivitis (%) | Oral herpes (%) | Upper respiratory tract infection (%) | Acne (%) | Source/assumptions |
|---|---|---|---|---|---|---|---|
| Monotherapy – adults | |||||||
| Dupilumab | 10.97 | 3.01 | 4.30 | 3.66 | 2.80 | 0.00 | Pooled data from SOLO 1 and SOLO 2 |
| Upadacitinib 15 mg | 0.00 | 0.21 | 0.62 | 2.49 | 6.85 | 5.44 | Pooled data from Measure UP 1 and Measure UP 2 |
| Upadacitinib 30 mg | 0.00 | 0.41 | 1.02 | 4.49 | 9.18 | 16.46 | Pooled data from Measure UP 1 and Measure UP 2 |
| Combination therapy – adults | |||||||
| Baricitinib | 0.00 | 0.00 | 0.00 | 3.60 | 2.70 | 3.60 | BREEZE AD 7 (Reich 2020)97 |
| BSC | 0.00 | 0.33 | 1.65 | 1.65 | 6.93 | 1.98 | Placebo data from AD UP |
| CsA | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | Assumption accepted in TA53412 |
| Dupilumab | 5.53 | 10.60 | 5.53 | 2.76 | 3.69 | 0.00 | Pooled data from CHRONOS and CAFE |
| Upadacitinib 15 mg | 0.00 | 0.00 | 1.15 | 3.83 | 7.66 | 9.58 | AD UP |
| Upadacitinib 30 mg | 0.00 | 0.77 | 0.77 | 8.85 | 7.31 | 13.85 | AD UP |
| Monotherapy – adolescents | |||||||
| BSC | 0.00 | 0.00 | 0.00 | 0.00 | 2.56 | 0.00 | AD UP |
| Dupilumab | 8.54 | 3.66 | 4.88 | 0.00 | 12.20 | 0.00 | AD ADOL (Simpson et al.99) |
| Upadacitinib 15 mg | 0.00 | 1.33 | 0.00 | 0.00 | 14.67 | 13.33 | Pooled data from Measure UP 1 and Measure UP 2 |
Flares
During treatment for moderate-to-severe AD, patients may experience acute exacerbations of symptoms, called flares. The rate of flare can vary depending on the treatment received by a patient but treatments for flare are similar.
Flare rate was not an end point in the key studies described in the section Quantity and quality of research available. In TA534 and TA681, the receipt of rescue medication was accepted as a proxy for flare. Furthermore, the companies for tralokinumab and upadacitinib used receipt of rescue medications from their key trials to inform the rate of flare used in their economic models. However, the company for abrocitinib had data on protocol-defined flares from REGIMEN and this was used to inform their economic model.
The EAG requested 16-week data on the receipt of rescue medication (used as a proxy for flare, in line with TA534 and TA681) or rate of flare. Each company supplied the requested data (Table 13) and the data have been used in the EAG’s economic model. The flare rate for dupilumab was extracted from TA534 and data for baricitinib were obtained from Reich et al. 97 Similar to AEs, the flare rate for BSC was based on placebo data from AD-UP. However, unlike the data for AEs, flare data are split by first- and second-line systemic treatment. As mentioned in the section Baseline characteristics, baseline characteristics from the upadacitinib trials were deemed to be representative of the population who would be treated in clinical practice in England according to the EAG’s clinical experts. The EAG has assumed the BSC flare rate for monotherapy is the same as combination therapy because in clinical practice BSC includes TCS. Furthermore, due to a paucity of data, the EAG assumed flare rates for CsA were the same as upadacitinib.
| Treatment | Rate of flare | Source/assumptions | ||
|---|---|---|---|---|
| 16-week (%) | Weekly (%) | Annual (%) | ||
| Monotherapy – adults | ||||
| Abrocitinib 100 mg | N/A | 1.06 | 42.60 | Receipt for rescue medication was not permitted in JADE MONO 1/2. 40-week data from REGIMEN used in long-term model, taken from the company submission. Annual data used to calculate weekly rate. |
| Abrocitinib 200 mg | N/A | 0.40 | 18.90 | Receipt for rescue medication was not permitted in JADE MONO 1/2. 40-week data from REGIMEN used in long-term model, taken from the company submission. Annual data used to calculate weekly rate. |
| Dupilumab | 17.94 | 1.23 | 47.53 | Pooled 16-week data from SOLO1 (n/N = 47/224) and SOLO2 (n/N = 35/233), reported in TA534 (pooled n/N = 82/457). |
| Combination therapy – adults | ||||
| Abrocitinib 100 mg | N/A | 1.06 | 42.60 | Receipt for rescue medication was not permitted in JADE COMPARE. Forty-week data from REGIMEN used in long-term model, taken from the company submission. Annual data used to calculate weekly rate. |
| Abrocitinib 200 mg | N/A | 0.40 | 18.90 | Receipt for rescue medication was not permitted in JADE COMPARE. Forty-week data from REGIMEN used in long-term model, taken from the company submission. Annual data used to calculate weekly rate. |
| Baricitinib | 5.41 | 0.35 | 16.58 | 16-week data from BREEZE-AD7 (n/N = 6/111)97 |
| Dupilumab | N/A | 0.34 | 16.04 | 52-week data from CHRONOS, reported in TA534 (n/N = 17/106). |
| Monotherapy – adolescents | ||||
| Abrocitinib 100 mg | N/A | 1.06 | 42.60 | Receipt for rescue medication was not permitted in JADE MONO 1/2. Forty-week data from REGIMEN used in long-term model, taken from the company submission. Annual data used to calculate weekly rate. |
| Abrocitinib 200 mg | N/A | 0.40 | 18.90 | Receipt for rescue medication was not permitted in JADE MONO 1/2. Forty-week data from REGIMEN used in long-term model, taken from the company submission. Annual data used to calculate weekly rate. |
| Dupilumab | 20.73 | 1.44 | 53.12 | 16-week data from AD ADOL (n/N = 17/82)99 |
For abrocitinib, receipt of rescue medication in JADE COMPARE, MONO 1 and MONO 2 was prohibited. However, protocol-defined flare data at 40 weeks from REGIMEN were available for abrocitinib (reported in the company submission) and the data were used to inform the annual rate of flare for both adults (second-line systemic treatment) and adolescents for monotherapy and combination therapy, in line with the company’s preferred approach. Additionally, the annual rate of flare for abrocitinib was used to calculate a weekly rate of flare to be used in the short-term part of the economic model.
The rate of flare was applied in the short-term model by converting the available treatment-specific data into weekly rates. For the long-term model, annual rates of flare were estimated from the 16-week data, except for dupilumab and abrocitinib where annual flare data were already available. Flare rates were used to estimate the costs to treat a flare only as it was assumed that the HRQoL data collected in the trials and used in the model would capture the acute impact of flares. The EAG’s approach is in line with TA534 and TA681, as well as the approach adopted in the company models for abrocitinib, tralokinumab and upadacitinib. Refer to the section Costs of managing flares for flare costs used in the economic model.
Health-related quality of life
For each of the drugs considered in the MTA, their key trials collected EQ-5D-5L data, which in the companies’ submissions were mapped to the EQ-5D-3L using the van Hout cross-walk algorithm,144 as per the NICE reference case. 145 Each of the companies developed regression models to estimate utility values to be used in their models. They also estimated baseline, responder and non-responder values, according to subgroup and measure of response. The companies for abrocitinib and tralokinumab estimated utility values by type of therapy (combination therapy or monotherapy). Disutility values associated with AEs and flares were not included in the companies’ models as per TA534 and TA681.
Treatment-specific utility values were adopted by the companies for abrocitinib and tralokinumab, as this approach was accepted in TA534 and TA681. Furthermore, in TA534, it was accepted that non-responders on dupilumab accrued the average utility of a dupilumab non-responder and BSC non-responder (0.82) at week 16 after starting treatment, and after week 52 accrued the utility value of BSC non-responders (0.77). The TA534 approach was adopted by the companies for abrocitinib and tralokinumab. However, in the abrocitinib model, utility values were used to capture the benefit of early response to systemic treatment prior to the week-16 assessment point. In the upadacitinib model, utility values were not treatment specific; thus, no weighting of systemic treatment non-responder and BSC non-responder values was implemented. However, the utility values were used to capture the benefit of early response to upadacitinib treatment prior to the week-16 assessment point.
Due to the variation in the approach to utilities across the companies’ models, TA681 and TA534, a consistent and conservative approach to utilities for all treatments is adopted in the MTA model. It is worth noting that unlike in TA534, TA681 and the companies’ models, BSC is not a comparator in the EAG’s model. As mentioned previously, BSC is modelled as an average of responders and non-responders to BSC treatment to capture the waxing and waning nature of response. The implication of BSC as a comparator in the previous models is that it was reasonable to assume that being on systemic treatment but not achieving response was still likely to result in an improvement in HRQoL over and above BSC in the short term, hence the assumption being accepted in TA534.
However, as the three new drugs are only being compared against currently available systemic treatments in the MTA model and BSC is only a health state and not an independent comparator, the EAG took a conservative approach to use treatment-specific baseline utilities for weeks 0–16, treatment-specific responder utility values for those who achieve and maintain response to treatment and the weighted average utility of responders and non-responders to BSC for non-responders to treatment and those who discontinue treatment. As such, the benefit of systemic treatment remains only for those who respond to treatment. Given the remit of the MTA and the approach to the BSC, the EAG considers this a necessary deviation from the approach accepted in TA534 and can be considered conservative.
The EAG investigated the utility regressions and resulting utility values from each of the company submissions to assess the suitability of the data for the MTA model but found that that definition of the populations used to estimate utilities in each of the companies’ models was not aligned with definitions used in the EAG analysis (see Methods of data synthesis and Populations). Furthermore, monotherapy- and combination therapy-specific utility values were not available in the upadacitinib company submission. Thus, from each of the companies, the EAG requested HSUVs based on the relevant subgroups from the key trials, by response category (composite and EASI 75), dose (where applicable) and type of therapy (monotherapy and combination therapy). Additionally, the EAG requested data on the number of observations at each assessment point to gauge the size of the data sets informing the utility regressions and aid choice of which utilities should inform the drug class estimates.
All three companies provided the requested utility data to the EAG in time for the development of the economic model. The utility data provided by the company for abrocitinib was subject to several issues which made the suitability for use in the MTA model limited. Utility data for abrocitinib in the adolescent population using the EASI 75 measure of response were not provided, which is relevant for the adolescent analyses. The company did provide utility data for adult second-line systemic population for both the composite and EASI 75 outcomes and monotherapy and combination therapy. The abrocitinib utility analyses use data from the full-trial populations of the relevant JADE trials and apply the baseline characteristics of relevant populations (generalisable and restricted) to generate utility values. However, as mentioned in the section Quantity of research available, a fundamental issue with the JADE trial programme is that most of the trial populations are not relevant to the decision problem (patients are predominantly naïve to systemic treatment). Thus, the patient numbers informing the post hoc subgroups that are relevant to this appraisal are small and may potentially result in unreliable estimates of utility.
The companies for tralokinumab and upadacitinib provided complete data for the populations that are relevant to proposed position in the pathway for their drugs. The utilities provided warranted further examination by the EAG considering other available data.
As part of the HRQoL SLR (see Results – Health-related quality-of-life), the EAG extracted utility data from TA534 and TA681 for dupilumab and baricitinib. The baricitinib values were available for the composite outcome and combination therapy for the adult second-line systemic treatment, but in the MTA analyses, outcome data for baricitinib are only available for EASI 75. Additionally, the committee for TA681 concluded that, given the flaws with the company’s utility values, the utility values from TA534 were preferable. From TA534, utility values for dupilumab were available for monotherapy and combination therapy for the adult second-line systemic treatment population, but no data were available for adolescents.
As such, to account for limitations associated with missing data, uncertainty due to small numbers and relevance of the populations for utility values, the EAG decided to adopt a drug class approach for utility values in the model. The drug class approach was considered to be appropriate as the drugs considered in the economic model fall into two classes: JAK inhibitors (abrocitinib, baricitinib and upadacitinib); and monoclonal antibodies (dupilumab and tralokinumab). The EAG considers that HRQoL is unlikely to be affected by different treatments within a drug class but there could be differences across different drug classes due to the mode of action and administration of treatment. Furthermore, as JAK inhibitors are associated with both high and low doses, type of dose given may be indicative of severity of AD. As such, company’s utility values for tralokinumab were used to inform the base case for the monoclonal antibody drug class and upadacitinib utility values were used for the JAK inhibitors. Furthermore, the EAG employed a simplification for JAK inhibitors, using available high and low dose and mapping values to high- and low-dose treatments. Notably, the analyses that include baricitinib are only considered in a scenario and not the EAG base case due to lack of the composite outcome for baricitinib.
With regard to CsA, utility values were assumed to be the same as upadacitinib 15 or 30 mg (depending on the comparison). In the adolescent population, as there are no monoclonal antibody utility values, the EAG assumed the tralokinumab monotherapy adult second-line systemic treatment utility values for dupilumab monotherapy.
The weighted utility values for BSC responders and non-responders were based on the upadacitinib placebo utility values for the relevant population as baseline characteristics and BSC response status reflect the upadacitinib trials. Refer to Report Supplementary Material 1 for an overview of the companies’ regression models used to estimate the utility values used in the EAG base-case analysis. The EAG also explored a scenario where utility values for TA534 (presented in Report Supplementary Material 1) were used for all the populations.
Disutilities associated with AEs have not been included in the EAG’s economic model in line with TA534, TA681 and the companies’ models. It has been assumed that due to the frequency of capturing EQ-5D data in the upadacitinib and tralokinumab trials, the impact of AEs on HRQoL will be captured in the data.
Utility values in the model are adjusted for age based on UK population norms using the multiplicative method detailed in Ara and Brazier 2010. 146
Resource use and costs
The following cost categories are included in the model:
-
drug acquisition costs (see Drug acquisition costs)
-
drug administration costs (see Drug acquisition costs)
-
concomitant medication costs (see Concomitant medication costs)
-
healthcare resource use costs (monitoring costs) [see Healthcare resource use costs (monitoring costs)]
-
costs of managing flares (see Costs of managing flares)
-
costs of managing AEs (see Costs of managing adverse events).
The economic analysis is conducted from an NHS and personal social services perspective and therefore only includes costs that would be incurred by the NHS and personal social services. Costs are reported in pound sterling for a 2019–20 cost year. Drug costs have been sourced from the British National Formulary (BNF) and electronic drug marketing tool (eMIT), while service costs have been sourced from the National Schedule of NHS Costs and Personal Social Services Research Unit (PSSRU).
Drug acquisition costs
The drug acquisition costs included in the model are given in Table 14. The BNF was used to inform the cost of CsA, dupilumab, baricitinib, tralokinumab and upadacitinib 15 mg. Company submissions were used to inform the cost of upadacitinib 30 mg and abrocitinib 100 and 200 mg in the absence of publicly available costs. Confidential PAS are in place for all drugs, except CsA.
| Treatment | Pack size | List price | |
|---|---|---|---|
| Pack cost | Cost per unit | ||
| Oral | |||
| CsA, 100 mg capsules | 30 | £41.59 | £1.39 |
| CsA, 50 mg capsules | 30 | £21.80 | £0.73 |
| CsA, 25 mg capsules | 30 | £11.14 | £0.37 |
| CsA, 10 mg capsules | 60 | £12.75 | £0.21 |
| Baricitinib, Olumiant 2 and 4 mg tablets (Eli Lilly and Company Ltd) | 28 | £805.56 | £28.77 |
| Upadacitinib, Rinvoq 15 mg modified-release tablets (AbbVie Ltd) | 28 | £805.56 | £28.77 |
| Upadacitinib, Rinvoq 30 mg modified-release tablets (AbbVie Ltd) | 28 | £1611.12 | £57.54 |
| Abrocitinib, CIBINQO 100 and 200 mg tablets (Pfizer) | 28 | £893.76 | £31.92 |
| SC injection | |||
| Dupilumab, Dupixent 300 mg/2 ml solution for injection pre-filled pens or syringes (Sanofi) | 2 | £1264.89 | £632.45 |
| Tralokinumab, Adtralza 150 mg pre-filled syringes (Leo Pharma UK) | 4 | £1070.00 | £267.50 |
For weight-based dosing of CsA, baseline weight reported in adults in the AD-UP study of 77.2 kg is used in the model. As described in the section Interventions and comparators, the recommended dose range for CsA is 2.5–5 mg/kg/day, and treatment is individualised. 147 According to the EAG’s clinical experts, there is no clinical consensus on a typical CsA dose for patients with moderate-to-severe AD and depends largely on the treating clinician. In TA534, the dose accepted by the committee was 5 mg/kg for 6 weeks followed by 3 mg/kg for 46 weeks (maximum treatment duration of 1 year) and this has been used for the EAG base case. After 1 year of treatment with CsA, patients discontinue to BSC for the remainder of the model time horizon. The following alternative CsA doses are explored in sensitivity analysis:
-
clinical expert opinion – 3 mg/kg for 16 weeks followed by 5 mg/kg for 36 weeks
-
Ariens et al. 201987 – 5 mg/kg for 3 weeks followed by 2 mg/kg for 48 weeks.
The treatments under consideration can be given as monotherapies or in combination with TCS. The costs associated with TCS can be found in the section Concomitant medication costs.
The recommended treatment regimen as per the SmPC for tralokinumab is 300 mg Q2W and this is used for the base case. A scenario for tralokinumab was explored where a percentage of patients on tralokinumab switched to Q4W treatment regimen, presented in the section Scenario analyses. The tralokinumab treatment switching scenario was explored as the SmPC for tralokinumab states that at the prescriber’s discretion, frequency of dose can be reduced to Q4W for patients who achieve clear or almost clear skin after 16 weeks of treatment. Data on the number of patients entering maintenance phase by dose in ECZTRA 3 (ECZTRA 7-like subgroup) for combination therapy and pooled data on the ECZTRA-7-like population from ECZTRA 1 and ECZTRTA 2 for monotherapy were extracted from the tralokinumab company submission to calculate the proportion of patients who switched to the Q4W regimen.
Drug administration costs
Based on the resource use assumptions from previous technology appraisals (TA534 and TA681) and verified with the EAG’s clinical experts, it is assumed that patients treated with SC formulations (dupilumab and tralokinumab) receive training on how to self-administer treatment. It is assumed that each patient only receives one self-injection training session, requiring 30 minutes of patient contact with a hospital-based Band 6 nurse at a cost of £62.50 (PSSRU 2020,148 note: each hour spent with a client requires 2.5 paid hours). This cost is incurred when the SC treatment is prescribed (i.e. the first model cycle).
Leo Pharma has indicated that training on how to self-administer tralokinumab will be provided to the NHS free of charge. As such, no administration costs are incurred by tralokinumab-treated patients in the base-case analysis.
Orally administered drugs (CsA, baricitinib, upadacitinib and abrocitinib) are assumed to incur no administration costs in the model.
Concomitant medication costs
Based on the resource use assumptions accepted in TA681 and verified with the EAG’s clinical experts, it is assumed that patients receive concomitant medications, consisting of:
-
emollient products
-
mid-potency background TCS (mometasone 0.1% ointment)
-
TCI (protopic 0.1% ointment).
When TA534 was published, bathing products were frequently used in clinical practice to manage the symptoms of moderate-to-severe AD. Following RCT evidence149 suggesting bathing products offer no benefits, there has been a significant reduction in the use of bathing products and most NHS trusts no longer prescribe them. Furthermore, the committee for TA68113 preferred to exclude the costs of bathing products from the CEA for baricitinib. As such, the cost of bathing products is excluded in the economic model.
Based on the resource use assumptions accepted from previous technology appraisals (TA534 and TA681) and verified with the EAG’s clinical experts, it is assumed that:
-
the weekly cost of emollients is derived by averaging the weekly cost of the most commonly prescribed emollients;
-
responders to systemic treatment have a 50% reduction of resource use for concomitant emollients and TCS compared to non-responders;
-
responders do not require TCI.
Based on feedback from the EAG’s clinical experts, it is assumed that there is no reduction in use of emollients and TCS for patients who discontinue systemic maintenance treatment and go on to BSC.
The concomitant medication costs included in the model are summarised in Table 15. Further details on the sources used to inform concomitant medication costs can be found in Report Supplementary Material 1. As mentioned in the section Model structure, costs for the BSC health stated are weighted by the proportion of responders and non-responders to BSC at the week-16 assessment point. The weighted concomitant costs applied to BSC in the base-case analysis are given in Table 16.
| Medication | Cost | Source | Responders to systemic treatment | Non-responders | Responders to BSC | |||
|---|---|---|---|---|---|---|---|---|
| Amount per weeka | Cost per week | Amount per weeka | Cost per week | Amount per weeka | Cost per week | |||
| TCI | ||||||||
| Protopic 0.1% ointment (cost per 60 g, g per week) | £45.56 | BNF150 | 0.00 | £0.00 | 1.75 | £1.33 | 0.00 | £0.00 |
| TCS | ||||||||
| Mometasone 0.1% ointment (cost per 100 g, g per week) | £2.58 | eMIT151 | 56.70 | £1.46 | 112.04 | £2.89 | 112.04 | £2.89 |
| Emollient (cost per pack, packs per week) | ||||||||
| Aveeno cream | £6.47 | BNF150 | 0.50 | £3.24 | 1.00 | £6.47 | 1.00 | £6.47 |
| Cetraben ointment | £5.39 | 0.50 | £2.70 | 1.00 | £5.39 | 1.00 | £5.39 | |
| Dermol cream | £6.63 | 0.50 | £3.32 | 1.00 | £6.63 | 1.00 | £6.63 | |
| Diprobase ointment | £5.99 | 0.50 | £3.00 | 1.00 | £5.99 | 1.00 | £5.99 | |
| Epaderm ointment | £12.42 | 0.25 | £3.11 | 0.50 | £6.21 | 0.50 | £6.21 | |
| Hydromol ointment | £8.31 | 0.25 | £2.08 | 0.50 | £4.16 | 0.50 | £4.16 | |
| White soft paraffin 50%/Liquid paraffin 50% ointment | £4.32 | 0.50 | £2.16 | 1.00 | £4.32 | 1.00 | £4.32 | |
| Oilatum cream | £5.28 | 0.25 | £1.32 | 0.50 | £2.64 | 0.50 | £2.64 | |
| Total cost per week | £4.08 | £9.45 | £8.12 | |||||
| Total cost per year | £212.66 | £492.83 | £423.49 | |||||
| Response status | Annual cost | Weekly cost |
|---|---|---|
| First-line systemic treatment – adults, monotherapy | ||
| Responder | £423.49 | £8.12 |
| Non-responder | £492.83 | £9.45 |
| Weighted costa | £469.94 | £9.01 |
| Second-line systemic treatment – adults, monotherapy | ||
| Responder | £423.49 | £8.12 |
| Non-responder | £492.83 | £9.45 |
| Weighted costa | £478.96 | £9.18 |
| Second-line systemic treatment – adults, combination | ||
| Responder | £423.49 | £8.12 |
| Non-responder | £492.83 | £9.45 |
| Weighted costa | £464.05 | £8.89 |
| Adolescents, monotherapy | ||
| Responder | £423.49 | £8.12 |
| Non-responder | £492.83 | £9.45 |
| Weighted costa | £482.80 | £9.25 |
Healthcare resource use costs (monitoring costs)
In the model, healthcare resource use depends on:
-
the stage of treatment (induction vs. maintenance);
-
the treatment response (responder vs. non-responder); and
-
the treatment received (BSC and CsA are associated with more visits and test than biologics).
Based on feedback from the EAG’s clinical experts, adolescents typically follow the same treatment pathway as adults and therefore healthcare resource use is assumed to be the same for adults and adolescents.
Healthcare resource use in the economic model is based on the ERG estimates for TA534 and the company estimates for TA681, which were accepted by their relevant appraisal committees and have been verified by the EAG’s clinical experts. The types of visits and tests considered in the economic model include:
-
outpatient visits to a dermatologist
-
outpatient visits to a dermatology nurse
-
visits to a GP
-
visits to accident and emergency (A&E)
-
hospital admissions
-
hospital day case visits
-
full blood counts (FBCs) (an additional test for patients on CsA, JAKis or BSC)
-
phototherapy (an additional service for patients who are non-responders to BSC)
-
psychological support (an additional service for patients who are non-responders to BSC).
When any systemic treatment is initiated, patients are assumed to visit their dermatologist twice during the induction period. These visits are in addition to the ongoing monitoring a dermatologist will provide. The ongoing healthcare resource use data applied in the economic model, according to response status, are given in Table 17. Further details on the sources used to inform monitoring costs can be found in Report Supplementary Material 1.
| Visit/test | Unit cost | Number per annum | Cost per annum | Number per week | Cost per week | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Responder (MAB) | Responder (BSC/ JAKi) | Non-responder (BSC) | Responder (MAB) | Responder (BSC/ JAKi) | Non-responder (BSC) | Responder (MAB) | Responder (BSC/ JAKi) | Non-responder (BSC) | Responder (MAB) | Responder (BSC/ JAKi) | Non-responder (BSC) | ||
| Dermatologist outpatient consultation | £124.83 | 4.320 | 4.320 | 6.000 | £539.27 | £539.27 | £748.98 | 0.083 | 0.083 | 0.115 | £10.34 | £10.34 | £14.35 |
| Dermatologist nurse visit | £31.25 | 0.350 | 0.350 | 0.460 | £10.94 | £10.94 | £14.38 | 0.007 | 0.007 | 0.009 | £0.21 | £0.21 | £0.28 |
| GP consultation | £39.00 | 6.150 | 6.150 | 12.810 | £239.85 | £239.85 | £499.59 | 0.118 | 0.118 | 0.246 | £4.60 | £4.60 | £9.57 |
| A&E visit | £170.98 | 0.021 | 0.021 | 0.082 | £3.59 | £3.59 | £14.02 | 0.000 | 0.000 | 0.002 | £0.07 | £0.07 | £0.27 |
| Hospitalisation | £1611.14 | 0.017 | 0.017 | 0.130 | £27.39 | £27.39 | £209.45 | 0.000 | 0.000 | 0.002 | £0.52 | £0.52 | £4.01 |
| Day case | £439.00 | 0 | 0.000 | 0.200 | £0 | £0 | £87.80 | 0 | 0.000 | 0.004 | £0 | £0.00 | £1.68 |
| FBC | £2.58 | 0 | 4.000 | 4.000 | £0 | £10.32 | £10.32 | 0 | 0.077 | 0.077 | £0 | £0.20 | £0.20 |
| Phototherapy | £107.24 | 0 | 0.000 | 0.060 | £0 | £0 | £6.43 | 0 | 0.000 | 0.001 | £0 | £0.00 | £0.12 |
| Psychological support | £324.88 | 0 | 0.000 | 0.070 | £0 | £0 | £22.74 | 0 | 0.000 | 0.001 | £0 | £0.00 | £0.44 |
| Total cost | £821.03 | £831.35 | £1613.71 | £15.74 | £15.93 | £30.93 | |||||||
Healthcare resource use is stratified by induction (weekly frequency in year 1) and maintenance (annual frequency in year 2+) to ensure that the right frequency of visits or tests is captured in the appropriate period in the short- and long-term models. It is assumed that resource use in the induction phase of the short-term model is based on non-responders until the initial treatment assessment point (Table 18). This is a deviation from the approach in TA681, where responder resource use estimates were applied in the treatment induction phase (weeks 0–16). However, the EAG considered that assuming non-responder resource use until treatment response is assessed at week 16 is a conservative assumption for all treatment arms.
| Visit/test | Unit cost | Number per annum | Number per week | Cost per week |
|---|---|---|---|---|
| Dermatologist outpatient consultation | £124.83 | 6.000 | 0.115 | £14.35 |
| Dermatologist nurse visit | £31.25 | 0.460 | 0.009 | £0.28 |
| GP consultation | £39.00 | 12.810 | 0.246 | £9.57 |
| A&E visit | £170.98 | 0.082 | 0.002 | £0.27 |
| Hospitalisation | £1611.14 | 0.130 | 0.002 | £4.01 |
| Day case | £439.00 | 0.200 | 0.004 | £1.68 |
| Total cost | £30.17 | |||
It should be noted that for patients on JAK inhibitors, additional FBC monitoring is required. In TA681, the committee accepted that four FBCs per annum would be required for patients on baricitinib. Furthermore, in the company submissions for abrocitinib and upadacitinib, four FBCs per annum were assumed for the base-case analyses. As such, the EAG has assumed that patients on treatment with a JAK inhibitor incur the costs of four FBCs per annum (including the induction period).
It is assumed that non-responders to systemic treatment incur the healthcare resource use costs associated with BSC when they transition to the BSC health state. As mentioned in the section Model structure, costs in the BSC health state are weighted by the proportion of responders and non-responders to BSC at the week-16 assessment point. The weighted monitoring costs applied to BSC in the base-case analysis are given in Table 19.
| Population | Weighted annual cost | Weighted weekly cost |
|---|---|---|
| First-line systemic treatment – adults, monotherapy | £1355.42 | £25.98 |
| Second-line systemic treatment – adults, monotherapy | £1457.24 | £27.93 |
| Second-line systemic treatment – adults, combination therapy | £1288.96 | £24.70 |
| Adolescents, monotherapy | £1500.47 | £28.76 |
Ciclosporin A requires additional monitoring for potentially severe side effects, including nephrotoxicity. 152 Thus, regular monitoring of blood pressure, renal function, FBC and liver function is recommended. 150 The company for TA534 considered CsA as a comparator in scenario analysis. To reflect the increased burden of CsA monitoring in this scenario analysis, 15 FBCs were costed in the first year of treatment, as per the BNF requirement that, in psoriasis and AD, serum creatinine should be monitored Q2W for first 3 months and then every month. This results in around eight FBCs for weeks 1–16 (months 1–4), followed by around seven FBCs for weeks 17–52 (months 5–12). It is likely that some of these tests will be combined with routine dermatology appointments and GP visits. As such, six additional nurse visits are costed in the induction period (eight FBCs in the induction phase minus the two dermatologist visits in the induction phase). The additional monitoring costs associated with CsA are summarised in Table 20.
| Visit/test | Unit cost | Weeks 1–16 | Weeks 17–52 | ||
|---|---|---|---|---|---|
| Number | Cost | Number | Cost | ||
| Dermatologist nurse visit | £31.25 | 6 | £187.50 | 0 | £0.00 |
| FBC | £2.58 | 8 | £20.64 | 7 | £18.06 |
| Total cost | £208.14 | £18.06 | |||
Costs of managing flares
The treatments used and associated costs to manage a flare are given in Table 21. These treatments are generally in line with those accepted in TA534 and TA681 and are in line with the companies’ economic models. Data on flare treatment distributions were obtained from TA534 for dupilumab and Reich et al. 97 for baricitinib. The companies for tralokinumab and upadacitinib supplied data on flare treatment distributions upon request from the EAG. No data on flare treatment distributions were available for abrocitinib as receipt of rescue medication was prohibited in the JADE COMPARE, MONO 1 and MONO 2 trials. As such, the company for abrocitinib assumed the flare treatment distribution from TA534. However, the EAG has assumed the flare treatment distribution data for adult second-line systemic treatment and adolescents for abrocitinib 100 and 200 mg (monotherapy and combination therapy) are the same as upadacitinib 15 and 30 mg (monotherapy and combination therapy), as both treatments are the same drug class (JAK inhibitors). Furthermore, adolescent flare treatment distribution data for upadacitinib 15 mg were also assumed for dupilumab, as data from AD ADOL99 were unavailable.
| Medication | Cost per pack | Packs per flare | Cost per flare | |
|---|---|---|---|---|
| TCS potent | ||||
| Betamethasone valerate cream | £2.71 | 1 | £2.71 | £16.83 |
| Cutivate 0.005% ointment | £4.24 | 3.33 | £14.12 | |
| TCS very potent | ||||
| Eumovate 0.05% ointment | £5.44 | 1 | £5.44 | £13.34 |
| Dermovate 0.05% cream | £7.90 | 1 | £7.90 | |
| Systemic steroid | ||||
| Prednisolone 5 mg | £0.40 | 1 | £0.40 | £0.40 |
| TCI | ||||
| Protopic 0.1% ointment | £45.56 | 0.4 | £18.22 | £18.22 |
The flare treatment distribution for BSC was based on placebo data from AD-UP, split by first and second line. As mentioned in the section Baseline characteristics, baseline characteristics from the upadacitinib trials were deemed to be representative of the population who would be treated in clinical practice in England according to the EAG’s clinical experts.
The costs associated with flare treatments are multiplied by the distributions of flare treatments (Table 22) to estimate a treatment-specific flare cost. The treatment-specific flare costs are then multiplied by the treatment-specific rate of flare (see Table 13 in the section Flares) to estimate weekly and annual treatment-specific flare costs for the short- and long-term parts of the economic model. In the short-term part of the model, it is assumed that non-responders to systemic treatment incur the flare costs associated with BSC.
| Treatment | TCS potent | TCS very potent | Systemic steroida | TCIb | Cost of flare treatment | Source |
|---|---|---|---|---|---|---|
| Monotherapy – adults | ||||||
| Dupilumab | 0.0% | 0.0% | 15.9% | 0.0% | £0.06 | TA534 |
| Combination therapy – adults | ||||||
| Baricitinib | 0.0% | 66.7% | 33.3% | 0.0% | £9.03 | BREEZE-AD7 (Reich 202097). |
| CSA | 0.0% | 80.0% | 20.0% | 0.0% | £10.75 | Assumed to be the same as upadacitinib 30 mg. |
| Dupilumab | 42.0% | 23.0% | 29.0% | 0.0% | £10.25 | TA534. For TCIs, the rate was reported as 0% in TA534; however, EAG’s experts considered TCI use would be the same as BSC. |
In scenario analysis, the EAG explored using flare treatment distributions from TA534 to estimate a single cost of flare treatment for patients on systemic treatment and BSC.
Costs of managing adverse events
The unit cost to manage each AE in the model is given in Table 23. These unit costs and sources are generally in line with those applied in TA534 and TA681.
| Adverse event | Unit cost | Source148,150,153 |
|---|---|---|
| Injection-site reaction | £124.83 | National Schedule of NHS Costs – Year 2019–20 – NHS trusts and NHS foundation trusts. Service code 330, dermatology, consultant-led, weighted average WF01A-WF01D, WF02A-WF02D. |
| Allergic conjunctivitis | £39.00 | Unit Costs of Health and Social Care 2020. GP per surgery consultation lasting 9.22 minutes. Including direct care staff costs and qualifications. |
| Infectious conjunctivitis | £53.33 | Unit Costs of Health and Social Care 2020. GP per surgery consultation lasting 9.22 minutes. Including direct care staff costs and qualifications. £39.00 (80% weight from TA681). National Schedule of NHS Costs – Year 2019–20 – NHS trusts and NHS foundation trusts. Service code 130, ophthalmology, consultant-led, weighted average WF01A-WF01D, WF02A-WF02D. £110.66 (20% weight from TA681). |
| Oral herpes | £39.00 | Unit Costs of Health and Social Care 2020. GP per surgery consultation lasting 9.22 minutes. Including direct-care staff costs and qualifications (£39.00). 1-week aciclovir 5% cream (£6.77, BNF). |
| Upper respiratory tract infection | £39.00 | Unit Costs of Health and Social Care 2020. GP per surgery consultation lasting 9.22 minutes. Including direct-care staff costs and qualifications. |
| Acne | £248.43 | Unit Costs of Health and Social Care 2020. GP per surgery consultation lasting 9.22 minutes. Including direct-care staff costs and qualifications (£39.00). National Schedule of NHS Costs – Year 2019–20 – NHS trusts and NHS foundation trusts. Service code 330, dermatology, consultant-led, weighted average WF01A-WF01D, WF02A-WF02D (£124.83). 3 months Epiduo (£19.53 per month, BNF) and oral lymecycline (£8.67 per month, BNF). |
In the short-term model, it is assumed that non-responders to systemic treatment incur the AE costs associated with BSC and that CsA-treated patients incur no AE costs as CsA treatment is limited to 1 year and patients are likely to discontinue CsA should any AEs develop.
The unit costs associated with each AE (see Table 23) are multiplied by the weekly (short-term model) and annual (long-term model) proportion of patients experiencing each AE calculated based on 16-week data reported in Table 12 in the section Adverse events to estimate weekly and annual treatment-specific AE costs (Table 24).
| Treatment | Weekly cost | Annual cost |
|---|---|---|
| Monotherapy – adults | ||
| Dupilumab | £1.57 | £70.54 |
| Upadacitinib 15 mg | £1.25 | £60.01 |
| Upadacitinib 30 mg | £3.57 | £144.55 |
| Combination therapy – adults | ||
| Baricitinib | £0.81 | £39.64 |
| BSC | £0.64 | £31.75 |
| CsA | - | - |
| Dupilumab | £1.22 | £57.54 |
| Upadacitinib 15 mg | £2.12 | £95.40 |
| Upadacitinib 30 mg | £3.09 | £130.57 |
| Monotherapy – adolescents | ||
| BSC | £0.06 | £3.17 |
| Dupilumab | £1.49 | £67.84 |
| Upadacitinib 15 mg | £2.95 | £123.34 |
List of assumptions
The EAG base-case assumptions are summarised in Table 25.
| EAG base-case assumptions | Justification |
|---|---|
| Baseline characteristics and placebo response from upadacitinib trials. | EAG’s clinical experts considered that the upadacitinib trials were appropriate to inform the baseline characteristics and response in the EAG economic model. |
| BSC modelled as a single health state, weighted by responders and non-responders. | Preferred approach in TA681. |
| First-line CsA patients discontinue to BSC. | Simplification as patients likely to get dupilumab after discontinuing treatment. |
| The primary treatment outcome assessed in the model is response to treatment at week 16, defined using a composite outcome of EASI 50 + DLQI ≥ 4. | In TA534 and TA681, the composite outcome was preferred by the committee as it was deemed to be sensitive to changes in treatment outcomes and more clinically relevant than EASI 75. |
| Conditional discontinuation data for tralokinumab monotherapy assumed to be the same as the combination therapy and composite outcome. | Lack of composite outcome and combination therapy conditional discontinuation data from ECZTRA 7 and ECZTRA 7-like population for tralokinumab. |
| Conditional discontinuation for abrocitinib and baricitinib assumed to be same as upadacitinib. | Lack of data on conditional discontinuation for abrocitinib and baricitinib, as such assumed a JAK inhibitor class effect. |
| Long-term treatment discontinuation rate is equal to the conditional discontinuation rates. | In line with the approach accepted in TA534 and TA681. Same in the abrocitinib and upadacitinib company models. |
| Active treatment-waning results in discontinuation to BSC | Approach accepted by the committee in TA534. |
| No treatment-waning assumptions applied to the BSC health state. | As BSC is modelled as a single health state, weighted by responders and non-responders, it captures the waxing and waning nature of a patient’s response to BSC treatment for moderated to severe AD. |
| No AEs assumed for CsA. | Data on AEs for CsA were unavailable. In TA534, the committee accepted assuming zero AEs for CsA in the short term as treatment is only given for 1 year before patients move to BSC. |
| AEs for BSC assumed based on placebo safety data from the upadacitinib trials. | To maintain alignment with the source for baseline characteristics and placebo response in the model. |
| BSC flare rate for monotherapy is the same as combination therapy. | In clinical practice, BSC includes TCS. |
| Flare treatment distribution data for adult second-line systemic treatment and adolescents for abrocitinib 100 and 200 mg (monotherapy and combination therapy) are the same as upadacitinib 15 and 30 mg (monotherapy and combination therapy). | In the JADE trial programme for abrocitinib, rescue treatment was not permitted as such there are no data to inform the flare rate. Upadacitinib data were assumed for abrocitinib as both treatments are the same drug class (JAK inhibitors). |
| Adolescent flare treatment distribution data for upadacitinib 15 mg were assumed for dupilumab. | Data from AD ADOL99 for dupilumab were unavailable. |
| Utilities based on drug class implemented for the base case. | To account for limitations associated with missing data, uncertainty due to small numbers and relevance of the populations for utility values. |
| Disutilities associated with AEs have not included in the base case. | In line with TA534, TA681 and the companies’ models. It has been assumed that due to the frequency of capturing EQ-5D data in the upadacitinib and tralokinumab trials, the impact of AEs on HRQoL will be captured in the data. |
| Utility values for CsA were assumed to be the same as upadacitinib 15 or 30 mg (depending on the comparison). | Lack of utility data for CsA. |
| CsA dose based on TA534. | The recommended dose range for CsA is 2.5–5 mg/kg/day and treatment is individualised.147 According to the EAG’s clinical experts, there is no clinical consensus on a typical CsA dose for patients with moderate-to-severe AD and depends largely on the treating clinician. In TA534, the dose accepted by the committee was 5 mg/kg for 6 weeks followed by 3 mg/kg for 46 weeks (maximum treatment duration of 1 year) |
| No administration costs for tralokinumab. | Company has indicated that training on how to self-administer tralokinumab will be provided to the NHS free of charge. |
| Resource use in the induction phase of the short-term model is based on non-responders until the initial treatment assessment point. | In TA681, responder resource use estimates were applied in the treatment induction phase (weeks 0–16). However, the EAG considered that a conservative assumption for all treatment arms is to assume non-responder resource use until treatment response is assessed at week 16. |
| Responders on maintenance treatment who discontinue to BSC have no reduction in resource use of emollients and TCS. | According to the EAG’s clinical experts, emollients and TCS are key components of BSC and no reduction in use should be assumed if a patient loses response to systemic treatment. |
| Costs of bathing products excluded from the model. | RCT evidence suggests that bathing products offer no benefits, and most NHS trusts no longer prescribe them.149 Furthermore, the committee for TA68113 preferred to exclude the costs of bathing products |
| Monitoring costs are the same for adults and adolescents. | Based on feedback from the EAG’s clinical experts, adolescents typically follow the same treatment pathway as adults and therefore healthcare resource use is assumed to be the same for adults and adolescents. |
| Non-responders to systemic treatment incur AE costs associated with BSC. | Once systemic treatment is stopped and BSC is initiated, AE profile will reflect treatments given in BSC. |
Results
Deterministic results
List price ICERs are presented in Table 26 for the adult first-line systemic treatment, Table 27 for the adult second-line systemic treatment and in Table 28 for the adolescent populations. As abrocitinib and upadacitinib have different doses, the EAG has ordered these first in the presentation of results. The EAG notes that incremental QALYs were relatively small and incremental costs were relatively large for each treatment in each population resulting in the sensitive ICERs.
| ICER (£/QALY) | Abro 100 mg + TCS | Abro 200 mg + TCS | Upadacitinib 15 mg + TCS | Upadacitinib 30 mg + TCS |
|---|---|---|---|---|
| vs. CsA + TCS | £91,156 | £79,392 | £79,969 | £146,465 |
| ICER (£/QALY) | Abro 100 mg | Abro 200 mg | Upa 15 mg | Upa 30 mg | Tralo |
|---|---|---|---|---|---|
| Monotherapy – EASI 50 + DLQI ≥ 4 (list price) | |||||
| vs. dupilumab | Dominant | Dominant | Dominant | £66,324 | £406,187a |
| Combination therapy – EASI 50 + DLQI ≥ 4 (list price) | |||||
| vs. dupilumab | £169,480a | Dominant | £181,649a | £130,198 | £220,333a |
| ICER (£/QALY) | Abrocitinib 100 mg | Abrocitinib 200 mg | Upadacitinib 15 mg |
|---|---|---|---|
| vs. dupilumab | Dominant | Dominant | Dominant |
Probabilistic results
The EAG conducted a probabilistic sensitivity analysis (PSA) to assess the impact of the combined uncertainty from all parameters in the model. This was performed by sampling from distributions of the uncertain parameters 1000 times, to generate the equivalent number of sampled ICERs. Conditional discontinuation data, utility values, AEs and flare rates were varied using a beta distribution. Costs were varied using a gamma distribution. Variation for the week-16 treatment response was based on 1000 CODA samples from the NMA (see Methods of data synthesis for NMA methods).
List price probabilistic ICERs are presented in Table 29 for the adult first-line systemic treatment, Table 30 for the adult second-line systemic treatment and in Table 31 for the adolescent populations. For the cost-effectiveness acceptability curves (CEACs), refer to the Report Supplementary Material 1 document. It should be noted that for each population and intervention, PSAs were run separately due to the structure of the model and therefore the sampling from parameter distributions for the comparator provides slightly different mean estimates for each pairwise comparison. However, total costs and QALYs for the comparator are similar for the PSA results. Additionally, the EAG notes that incremental QALYs were relatively small and incremental costs were relatively large for each treatment in each population resulting in the sensitive ICERs.
| Treatment | vs. CsA + TCS | |
|---|---|---|
| PSA ICER (£/QALY) | Deterministic ICER (£/QALY) | |
| Abrocitinib 100 mg + TCS | £94,520 | £91,156 |
| Abrocitinib 200 mg + TCS | £79,270 | £79,392 |
| Upadacitinib 15 mg + TCS | £80,342 | £79,969 |
| Upadacitinib 30 mg + TCS | £152,134 | £146,465 |
| Treatment | vs. dupilumab | |
|---|---|---|
| PSA ICER (£/QALY) | Deterministic ICER (£/QALY) | |
| Monotherapy – EASI 50 + DLQI ≥ 4 | ||
| Abrocitinib 100 mg | Dominant | Dominant |
| Abrocitinib 200 mg | Dominant | Dominant |
| Upadacitinib 15 mg | Dominant | Dominant |
| Upadacitinib 30 mg | £66,539 | £66,324 |
| Tralokinumab | £337,165a | £406,187a |
| Combination therapy – EASI 50 + DLQI ≥ 4 | ||
| Abrocitinib 100 mg | £182,263a | £169,480a |
| Abrocitinib 200 mg | Dominant | Dominant |
| Upadacitinib 15 mg | £185,877a | £181,649a |
| Upadacitinib 30 mg | £107,339 | £130,198 |
| Tralokinumab | £220,640a | £220,333a |
| Treatment | vs. dupilumab | |
|---|---|---|
| PSA ICER (£/QALY) | Deterministic ICER (£/QALY) | |
| Abrocitinib 100 mg | Dominant | Dominant |
| Abrocitinib 200 mg | Dominant | Dominant |
| Upadacitinib 15 mg | Dominant | Dominant |
One-way sensitivity analysis
One-way sensitivity analysis was conducted by varying key model parameters between the upper and lower values of the expected value used in the deterministic base case. The key model parameters include:
-
week-16 response
-
conditional discontinuation (used to inform the week-52 response and annual discontinuation)
-
treatment waning
-
utility values
-
monitoring/healthcare resource use (frequency)
-
AEs (frequency)
-
flares (frequency)
-
age.
The response at week 16 was varied for each treatment using the 95% CrI estimated by the NMA. The response to BSC at week 16 was also varied, as this parameter is used to weight costs and utilities when patients discontinue active treatment and transition to BSC.
Conditional discontinuation rates, AE rates and flare rates were varied individually for each treatment by their 95% confidence intervals (CIs). No estimates of precision were available for treatment waning, utility or monitoring/healthcare resource use, and therefore the SE was assumed to equal ±20% of the mean value. Utility values were varied individually for each treatment, while treatment waning and monitoring/healthcare resource use parameters were varied simultaneously for the intervention and comparator. Age data (including variation) for the post hoc subgroups were presented by treatment arm rather than an overall mean. As such, the EAG calculated a mean age for each subgroup (presented in the section Baseline characteristics) and this was varied by ± 20% of the mean value. Drug acquisition costs and service costs were not varied as these are assumed to be fixed values. Alternative discount rates and time horizons were explored in scenario analysis (see Scenario analyses).
The below paragraph summarises the results of the OWSA for the top two or three parameters for the adult first-line systemic treatment population, adult second-line systemic treatment population and adolescents. For tornado diagrams of the top 10 parameters, see Report Supplementary Material 1.
The key drivers in the adult first-line systemic treatment population are the week-16 response for BSC and the week-52 response for upadacitinib 15 and 30 mg. As noted in the section Week-52 treatment response outcomes, the week-52 response represents all-cause discontinuation for people whose condition responded to treatment at week 16 but withdrew from treatment at week 52. Additionally, the upadacitinib week-52 response is used to inform the abrocitinib week-52 response. The key drivers in the adult second-line systemic monotherapy and combination therapy populations also include the week-16 response and the week-52 response. In addition to response at weeks 16 and 52, the key driver for the adolescent population also included the BSC non-responder HSUV, and lower non-responder BSC utility values favour dupilumab. As noted in the section Health-related quality-of-life, the overall BSC utility value is derived by weighting the BSC non-responder and responder values by the proportion of responders to BSC.
Scenario analyses
Adults’ first-line systemic treatment population
In all scenarios (presented in Table 32), the resultant ICERs are above £30,000 per QALY gained and therefore conclusions are unlikely to change. The scenarios which led to the largest increases in the ICER include using TA534 inputs (combined scenario and utility values).
| Scenario | ICER vs. CsA + TCS (£/QALY) | |||
|---|---|---|---|---|
| Abrocitinib 100 mg + TCS | Abrocitinib 200 mg + TCS | Upadacitinib 15 mg + TCS | Upadacitinib 30 mg + TCS | |
| Base case | £91,156 | £79,392 | £79,969 | £146,465 |
| No active treatment waning | £86,848 | £77,284 | £76,668 | £143,199 |
| CsA dose – Ariens 2019 | £91,780 | £79,691 | £80,521 | £146,725 |
| CsA dose – clinical expert opinion | £90,669 | £79,159 | £79,538 | £146,262 |
| Discount rate – costs and benefits 1.5% | £89,560 | £78,469 | £78,735 | £144,956 |
| Time horizon – limit to 5 years from mean age | £96,580 | £84,281 | £84,119 | £154,432 |
| Time horizon – limit to age 75 years | £91,156 | £79,387 | £79,969 | £146,453 |
| TA534 utility values | £136,119 | £124,082 | £119,414 | £228,910 |
| Annual discontinuation rates adjusted for 36-week rates | £94,892 | £80,215 | £82,845 | £147,809 |
Adults’ second-line systemic treatment population – monotherapy
Results of the scenario analyses are presented in Table 33. Except for tralokinumab, the ICERs in for the scenario exploring the alternative EASI 75 response definition are more in favour of dupilumab than the base-case analysis.
| Scenario | ICER vs. dupilumab (£/QALY) | ||||
|---|---|---|---|---|---|
| Abrocitinib 100 mg | Abrocitinib 200 mg | Upadacitinib 15 mg | Upadacitinib 30 mg | Tralokinumab | |
| Base case | Dominant | Dominant | Dominant | £66,324 | £406,187 a |
| EASI 75 response definition | Dominant | £7354 | Dominant | £72,901 | £470,051a |
| NMA – abrocitinib generalisable population | Dominant | Dominant | Dominant | £66,221 | £404,342a |
| Alternative tralokinumab annual discontinuation data – CS (2.3%) | NA | NA | NA | NA | £831,456a |
| Alternative tralokinumab discontinuation data – MTA CQ (6.2%) | NA | NA | NA | NA | £392,675a |
| Proportion of patients transition from tralokinumab Q2W to Q4W at week 16 | NA | NA | NA | NA | £407,709a |
| No active treatment waning | Dominant | Dominant | Dominant | £64,991 | £377,337a |
| Discount rate – costs and benefits 1.5% | Dominant | Dominant | Dominant | £66,023 | £398,381a |
| Time horizon – limit to 5 years from mean age | Dominant | Dominant | Dominant | £67,718 | 461,296 |
| Time horizon – limit to age 75 | Dominant | Dominant | Dominant | £66,330 | £406,270 |
| TA534 scenario | Dominant | Dominant | Dominant | £304,614 | £299,795a |
| TA534 utility values | Dominant | Dominant | Dominant | £321,223 | £236,033a |
| Annual discontinuation rates adjusted for 36-week rates | Dominant | Dominant | Dominant | £65,541 | £393,673a |
Other scenarios with large impacts on the ICER include reducing the time horizon to 75 years of age, using TA534 inputs (combined scenario and utility values) and censoring patients who receive rescue therapy. The alternative NMAs were generally more in favour of the interventions than the base-case analysis. Enabling a proportion of patients from tralokinumab Q2W to tralokinumab Q4W also had a large impact in favour of tralokinumab.
Adults’ second-line systemic treatment population – combination therapy
Data were not available for baricitinib using the EASI 50 + DLQI ≥ 4 response definition, and so it was not included in the base-case analysis and results using EASI 75 are presented as a scenario. Furthermore, the EAG used a drug class approach for data gaps (such as conditional discontinuation), where high-dose JAK inhibitor data based on upadacitinib 30 mg was used for baricitinib 4 mg, though the latter is the least efficacious of the JAK inhibitors. Thus, results of this scenario should be considered as illustrative and potentially conservative. Results for baricitinib using the EASI 75 response definition are provided in Table 34.
| ICER | Incremental value (£/QALY) | ||||
|---|---|---|---|---|---|
| Abro 100 mg + TCS | Abro 200 mg + TCS | Upa 15 mg + TCS | Upa 30 mg + TCS | Tralo + TCS | |
| vs. baricitinib | Dominateda | £81,431 | Dominateda | £187,893 | £551,116 |
The results of the scenario analyses are presented in Table 35. Except for dupilumab, all treatments produce more QALYs using the EASI 75 response definition than the EASI 50 + DLQI ≥ 4 response definition. Furthermore, the ICERs in this scenario are generally more in favour of dupilumab than the base-case analysis.
| Scenario | ICER vs. dupilumab (£/QALY) | ||||
|---|---|---|---|---|---|
| Abrocitinib 100 mg | Abrocitinib 200 mg | Upadacitinib 15 mg | Upadacitinib 30 mg | Tralokinumab | |
| Base case | £169,480 a | Dominant | £181,649 a | £130,198 | £220,333 a |
| EASI 75 response definition | £152,130a | Dominant | £165,090a | £126,037 | £204,059a |
| NMA – abrocitinib generalisable population | £169,202a | Dominant | £181,386a | £128,483 | £218,113a |
| NMA – censored for rescue therapy | £169,534a | Dominant | £182,497a | £124,066 | £216,629a |
| Alternative tralokinumab annual discontinuation data – CS (2.3%) | NA | NA | NA | NA | £273,706a |
| Alternative tralokinumab discontinuation data – MTA CQ (6.2%) | NA | NA | NA | NA | £216,844a |
| Proportion of patients transition from tralokinumab Q2W to Q4W at week 16 | NA | NA | NA | NA | £247,445a |
| No active treatment waning | £176,524a | Dominant | £184,502a | £114,747 | £215,430a |
| Discount rate – costs and benefits 1.5% | £170,953a | Dominant | £182,149a | £127,509 | £218,728a |
| Time horizon – limit to 5 years from mean age | £161,739a | Dominant | £180,847a | £140,307 | £231,109 |
| Time horizon – limit to age 75 | £169,417a | Dominant | £181,606a | £130,286 | £220,333a |
| TA534 scenario | £19,253,139a | Dominant | £7,687,765a | £510,130 | £225,885a |
| TA534 utility values | £244,803a | £1,188,371 | £258,627a | £2,115,553 | £205,627a |
| Annual discontinuation rates adjusted for 36-week rates | £168,112a | Dominant | £178,807a | £128,276 | £216,839a |
Other scenarios with large impacts on the ICER include reducing the time horizon to 75 years of age and using TA534 inputs (combined scenario and utility values). Removing treatment waning also had a large impact on abrocitinib.
Adolescents
The scenarios with the largest impact on the ICER include reducing the time horizon to 18 years of age and using TA534 inputs (combined scenario and utility values). In all other scenarios, the interventions continue to dominate dupilumab (Table 36).
| Results per patient | ICER vs. dupilumab (£/QALY) | ||
|---|---|---|---|
| Abrocitinib 100 mg | Abrocitinib 200 mg | Upadacitinib 15 mg | |
| Base case | Dominant | Dominant | Dominant |
| NMA – censored for rescue therapy | Dominant | Dominant | Dominant |
| No active treatment waning | Dominant | Dominant | Dominant |
| Discount rate – costs and benefits 1.5% | Dominant | Dominant | Dominant |
| Time horizon – limit to age 75 | Dominant | Dominant | Dominant |
| Time horizon – limit to age 18 | Dominant | Dominant | Dominant |
| TA534 scenario | £61,356 | £106,593 | £87,617 |
| TA534 utility values | £219,440a | £267,817a | £248,853a |
| Annual discontinuation rates adjusted for 36-week rates | Dominant | Dominant | Dominant |
Model validation
A senior health economist was responsible for the specification and development of the MTA model. A principal health economist was responsible for validating model assumptions and performing a detailed quality assurance of the MTA model. A third health economist, not involved in the MTA project, performed an independent review of the MTA model, including face validity checks and black and white box testing of the model.
The EAG’s clinical experts were involved with validating key assumptions in the model to ensure clinical validity of model inputs and outputs as well as peer review of the report.
Discussion
Summary of key results
The purpose of this MTA was to assess the cost effectiveness of abrocitinib, tralokinumab and upadacitinib individually as monotherapy and in combination with TCS for treatment of moderate-to-severe AD. In the MTA, as requested by NICE, the cost effectiveness of abrocitinib, tralokinumab and upadacitinib has been evaluated for the proposed position in the treatment pathway for moderate-to-severe AD as presented by the companies in their submissions to the STA process. All results shown in this report are based on list prices for all drugs.
The companies for abrocitinib and upadacitinib have a proposed position for the entire indication, that is for adolescents irrespective of prior treatment and adults as both first- and second-line systemic treatment. Upadacitinib is available in two doses, 15 and 30 mg. Upadacitinib 15 mg is approved for use in adolescents and both doses are approved for adults. In the adolescent population, upadacitinib 15 mg dominates dupilumab. In the adult first-line systemic treatment population, compared with CsA with TCS, upadacitinib 15 and 30 mg in combination with TCS are associated with probabilistic ICERS of £80,342 and £152,134. In the adult second-line systemic treatment population, upadacitinib 15 mg as monotherapy dominates dupilumab and in combination with TCS is less costly and less effective than dupilumab (south-west quadrant ICER of £185,877). Upadacitinib 30 mg as monotherapy and in combination with TCS is more expensive and more effective than dupilumab, with probabilistic ICERs of £66,539 and £107,339, respectively, for the adult second-line systemic treatment population.
Abrocitinib is available in two doses, 100 and 200 mg, with both approved for use in adolescents and adults. In the adolescent population, abrocitinib 100 and 200 mg dominate dupilumab. In the adult first-line systemic treatment population, compared with CsA with TCS, abrocitinib 100 and 200 mg in combination with TCS are associated with probabilistic ICERS of £94,520 and £79,270. In the adult second-line systemic treatment population, abrocitinib 100 mg in combination with TCS is less costly and less effective than dupilumab in combination with TCS (south-west quadrant probabilistic ICER of £182,263). However, as a monotherapy for both doses and as combination therapy with the 200 mg dose, abrocitinib dominates dupilumab in the adult second-line systemic treatment population.
The proposed position in the treatment pathway by the company for tralokinumab was for the adult second-line systemic population. Compared with dupilumab as a monotherapy and combination therapy, tralokinumab was less expensive and less effective, resulting in south-west quadrant probabilistic ICERs of £337,165 for monotherapy and £220,640 for combination therapy.
For all treatments in all populations, probabilistic ICERs were consistent with deterministic ICERs. Variation in the probabilistic ICERs compared to the deterministic ICERs were seen for tralokinumab monotherapy, abrocitinib 100 mg, upadacitinib 15 and 30 mg in the adult second-line systemic combination treatment population. The EAG notes that in all analyses, incremental QALYs were relatively small for each treatment resulting in sensitive ICERs. Furthermore, the sensitivity in the ICERs was seen in the OWSA and scenarios (discussed below) with changes in magnitude but rarely direction of results.
The EAG cautions the interpretation of the cost-effectiveness results presented in the MTA report as they are based on list prices for abrocitinib, baricitinib, dupilumab, tralokinumab and upadacitinib as all have confidential PAS discounts in place.
In the OWSA for all populations, the key drivers of cost effectiveness were week-16 response probabilities and conditional discontinuation probabilities (used to inform the week-52 response and annual discontinuation), which are as expected as these are the key effectiveness estimates in the model. In particular, the NMA for week-16 response was associated with substantial uncertainty, in particular, for abrocitinib due to small numbers informing the network.
The EAG conducted a range of scenarios to test the impact on the ICER of alternative assumptions and data inputs for key parameters. In the adult second-line systemic treatment population, baricitinib is also a comparator, but it could not be included in the EAG base-case analyses as response data using the composite outcome were not made available to the EAG. However, EASI 75 response data were available for baricitinib combination therapy. Thus, the EAG conducted a scenario comparing the three new drugs against baricitinib as combination therapy using the EASI 75 response outcome. When compared with baricitinib in combination with TCS, upadacitinib 15 mg in combination with TCS is less costly and less effective (south-west quadrant deterministic ICER of £19,830) and in the 30 mg dose is more costly and more effective (deterministic ICER of £321,819). Both tralokinumab and abrocitinib 100 mg are dominated by baricitinib. However, abrocitinib 200 mg is more costly and more effective than baricitinib (deterministic ICER of £331,473). It should be noted that while abrocitinib and upadacitinib (both doses) are more effective than baricitinib, long-term discontinuations are more favourable for baricitinib compared with the low doses of abrocitinib and upadacitinib and are similar for the high doses. Furthermore, baricitinib is less expensive than all three new drugs. However, as mentioned previously, the results are based on list prices for interventions and results based on inclusion of confidential PAS discounts are more appropriate for decision-making.
The EAG explored the use of the EASI 75 response outcome for comparisons against dupilumab in the adult second-line systemic treatment population. For both the monotherapy and combination therapy analyses, ICERs were consistent with the base case, although total QALYs were higher for all treatments (except dupilumab combination therapy).
Focus on the composite outcome for the EAG base-case analyses was informed by the recommendations of TA534 and TA681, where the committee considered that EASI 50 and an improvement in the DLQI of at least four are sensitive to changes in treatment outcomes and more clinically relevant than an EASI 75. The EAG’s clinical experts fed back that EASI 50 + ΔDLQI ≥ 4 does inform their assessment of response to treatment, but they went on to caution that the subjective nature of the DLQI, as a patient-assessed tool that is open to recall bias, is also borne in mind and, consequently, their preference to assess clinical effectiveness is changed in EASI by 75%. Furthermore, the EAG’s clinical experts considered that EASI 75 is much harder to achieve compared to the composite outcome.
The majority of the QALYs generated for each treatment are derived from occupation in the BSC health state and over a lifetime most patients end up on BSC. Patients on BSC will experience periods of response and relapse and data on the disease course is limited. As such, the EAG explored a scenario reducing the time horizon of the model to 5 years for the adult analyses, which had a substantial impact on the results in the second-line population analyses. For both monotherapy and combination therapy for all interventions, ICERs become substantially more cost-effective. In particular for abrocitinib 100 mg (both monotherapy and combination therapy) and abrocitinib 200 mg monotherapy, ICERs were within the range to be considered cost-effective using a £30,000 per QALY threshold. Both tralokinumab and upadacitinib 15 mg dominated dupilumab for both monotherapy and combination therapy analyses. However, the ICERs for upadacitinib 30 mg (monotherapy and combination therapy) and abrocitinib 200 mg monotherapy were still above the £30,000 threshold, but substantially lower than the base case. The ERG notes that for the 5-year time horizon scenario, incremental QALYs increased for all treatments compared with dupilumab, suggesting that longer time horizon gives less-effective treatments time to ‘catch up’ in terms of overall QALYs accrued.
For the adolescent analyses, reducing the time horizon to 18 years of age resulted in the ICERs for all treatments remaining dominant.
Across all populations, using parameter utility values, conditional discontinuation and flare treatment estimates from TA534 had a substantial impact on the ICERs, in particular, using utility values from TA534 was a key driver of cost effectiveness. However, the TA534 scenario did not change the direction of the results, except for the abrocitinib 100 mg comparison in the adult second-line systemic treatment population (changed from south-west quadrant ICER to dominated by dupilumab) and all treatments in the adolescent population (changed from dominant compared with dupilumab to south-west quadrant ICERs). However, the TA534 scenario should be considered as illustrative as data from the key trials for each of the three new drugs are available and can be considered as more appropriate for the base case compared to values in TA534 that just reflect dupilumab only.
Most of the other scenarios across all populations resulted in a change in the magnitude of the ICER, but not in the direction of the results. The scenario using the NMA where censoring for rescue therapy was included resulted in a change in the ICER for upadacitinib 15 mg from dominant to south-west quadrant in the adult second-line systemic treatment population. However, the EAG considers that in clinical practice, patients would receive rescue therapy while on treatment and this was also noted in the FAD for baricitinib. As such, the scenario using the NMA where patients were censored for rescue therapy can be considered only illustrative. Furthermore, rescue therapy was not allowed in abrocitinib trials and very few patients received rescue therapy in the upadacitinib and tralokinumab studies.
Generalisability of results
The perspective of the cost-effectiveness analysis reflects the NHS in England and thus results are generalisable to the patients in England with moderate-to-severe AD. To ensure consistency with current clinical practice, the EAG has used relevant NICE guidance (TA534 and TA680) to inform key assumptions and parameters within the MTA model. Furthermore, the EAG consulted with its clinical experts to determine the trials which are most representative of the patient population in England to inform the baseline characteristics and effectiveness used in the model.
Strengths and limitations of analysis
The primary strength of the EAG’s analysis of the three new drugs compared with current practice for each of the subpopulations is that the results have been produced using a consistent approach to the CEA. Specifically, common assumptions have been used across all comparators, such that results facilitate a consistent basis for decision-making. The EAG has utilised available trial data for each of the interventions to ensure results are as robust as possible and has defined the populations under consideration to reflect the NICE final scope and the treatment pathway which closely reflect patients who would be seen in clinical practice. Furthermore, the EAG has specified a single NMA for each population for all relevant treatments to produce consistent effectiveness estimates to use in the MTA model.
The EAG’s approach to the CEA can be considered conservative, in particular, the implementation of the drug class approach to fill gaps in the data. The strength of the drug class approach can be seen when considering utility values as it makes the inherent assumption that HRQoL is unlikely to be affected by different treatments within a drug class but there could be differences across different drug classes due to the mode of action and administration of treatment.
Despite the strengths of the EAG’s approach, there were several limitations with the analyses that required assumptions to be made where possible and where not possible, omissions within the analysis. The most significant limitation with the analysis is that the composite outcome of EASI 50 + DLQI ≥ 4 could not be obtained for the adolescent and adult first-line systemic treatment populations due to a paucity of data for dupilumab informing the adolescent NMA and CsA data informing the adult first-line systemic treatment NMA. Furthermore, even though combination therapy data were available for abrocitinib and upadacitinib, only monotherapy could be assessed for the adolescent population as combination data for dupilumab were unavailable to inform the NMA. Conversely, only combination therapy could be assessed for the adult first-line systemic treatment population as monotherapy data were unavailable.
The EAG considered the feasibility of estimating an ‘adjustment factor’ to estimate combination therapy outcomes based on monotherapy outcomes (and vice versa) to fill the data gaps. Treatment response data presented in the section Treatment effectiveness suggest that combination therapy is more effective than monotherapy for adults in the second-line systemic treatment subgroup. However, there is not a consistent trend in terms of inflation of benefit when comparing combination therapy and monotherapy across the treatments. Additionally, the populations considered in the MTA are heterogeneous and an adjustment factor based on one population for one treatment may not reflect the true outcomes for population where data are unavailable. Therefore, the EAG found that there was not a robust method to estimate the missing data.
As the combination therapy analyses are more relevant for clinical practice, the EAG considered missing monotherapy data are unlikely to be critical for decision-making for the adult first-line systemic treatment subgroup. However, for the adolescent population, monotherapy analyses may potentially underestimate the effectiveness of the treatments when used in combination with TCS in clinical practice. Therefore, adolescent monotherapy analyses may reflect a conservative view of clinical effectiveness.
Another significant limitation with the analysis is that comparisons with baricitinib could only be included in scenario analysis. In TA681, baricitinib was assessed in the adult second-line systemic population using the composite outcome but data were redacted, and the company did not provide these data for inclusion in the MTA NMA analyses. As such, only EASI 75 data for baricitinib combination therapy could be included in the NMA for the adult second-line systemic treatment population. The downstream implication of a lack of base-case result for the three new drugs compared with baricitinib is that incremental analysis of dupilumab and baricitinib against each of the three new drugs could not be presented and any recommendations for the adult second-line systemic population based on the primary composite outcome are limited to comparisons with dupilumab.
Analyses exploring increasing or decreasing dose for abrocitinib and upadacitinib were not possible as efficacy data based on titrating dose are unavailable. However, the SmPC guidance for both abrocitinib and upadacitinib does take into consideration circumstances where moving to the lower or higher dose of each drug may be beneficial and this is likely to happen in clinical practice.
Another consideration for clinical practice that could not be explored in the current analyses was treatment sequencing. Currently, there is a lack of clinical data on the effectiveness of sequences of AD treatments, especially changing drug class (e.g. starting on a JAK inhibitor and then moving to a monoclonal antibody). As such, more clinical data are needed to understand how patients respond to subsequent lines of systemic treatment and the resulting cost effectiveness of treatment sequences.
Chapter 5 Assessment of factors relevant to the NHS and other parties
The EAG considers that all factors relevant to the NHS and other parties are captured within the clinical and cost-effectiveness analyses. However, the EAG acknowledges that the outcome of the MTA may result in potentially more treatment options being made available to patients with moderate-to-severe AD and thus the cost effectiveness of treatment sequencing becomes a relevant consideration.
As stated in the final protocol for the MTA, there is a lack of clinical data on the effectiveness of sequences of AD treatments. Furthermore, in agreement with the NICE, the remit of the initial phase of the MTA prior to the first Appraisal Committee Meeting (ACM) is to compare each of the treatments against current treatment options to obtain a view on the cost effectiveness of the new drugs. As such, the EAG considers that further analysis of the cost effectiveness of treatment sequences, based on assumptions in lieu of clinical data, can be provided for discussion at the second ACM if considered appropriate by the committee.
Chapter 6 Discussion
Statement of principal findings
The clinical and cost effectiveness of abrocitinib, tralokinumab and upadacitinib versus treatment options available in the NHS for moderate-to-severe AD was evaluated in the positions in the treatment pathway proposed by the individual companies as follows:
-
Abrocitinib:
-
second-line systemic therapy for adolescents
-
second-line systemic therapy for adults.
-
-
Tralokinumab:
-
second-line systemic therapy for adults.
-
-
Upadacitinib:
-
adolescents
-
first-line systemic therapy for adults
-
second-line systemic therapy for adults.
-
The EAG focused on outcomes of clinical effectiveness that inform the economic evaluation of this MTA. In line with preferences expressed by the NICE Committee for dupilumab and baricitinib in TA534 and TA681,12,13 a composite outcome of reduction in EASI score of 50% and improvement in DLQI of at least four points (EASI 50 + ΔDLQI ≥ 4) is the primary clinical outcome for the MTA. Clinical experts fed back that the patient-reported DLQI component of EASI 50 + ΔDLQI ≥ 4 renders the composite outcome open to recall bias. In addition, data on EASI 50 + ΔDLQI ≥ 4 were not available for all comparisons in all populations and, consequently, improvement in EASI by 75% was also evaluated.
The EAG’s experts advised that, in clinical practice, systemic therapies are likely to be predominantly given concomitantly with TCS. However, treatment with systemic treatments as a monotherapy are relevant for a proportion of patients who cannot tolerate or do not want to use TCSs. Therefore, clinical effectiveness of abrocitinib, tralokinumab and upadacitinib was evaluated when given as a monotherapy and when administered with concomitant TCS.
The primary clinical effectiveness analysis included patients who received rescue medication during treatment because, based on advice from clinical experts, use of rescue medication more closely reflects what occurs in clinical practice in England, that is, the primary NMAs were based on using all observed data, regardless of rescue medication use to determine response, where possible.
Experts advising the EAG commented that, in clinical practice for the management of AD in adults, abrocitinib, tralokinumab and upadacitinib are likely to be used as alternatives to dupilumab and baricitinib, which have been assessed as treatment options after inadequate response, inability to tolerate or contraindication to first-line CsA. For abrocitinib, tralokinumab and upadacitinib, the primary analyses of adults in the second-line setting were based on the subgroup of patients who aligned with the populations informing the efficacy of dupilumab, baricitinib. For abrocitinib, in particular, this subgroup had a very small sample size.
There were considerable amounts of uncertainty and most results for EASI 50 + ΔDLQI ≥ 4 and EASI 75 were not statistically significant. However, there were consistent trends across the outcomes (EASI 50 + ΔDLQI ≥ 4 and EASI 75), interventions (combination therapy or monotherapy) and populations (adults in the first- or second-line setting, and adolescents).
Treatment with abrocitinib 200 mg leads to a better response, assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75, than dupilumab treatment, whereas there was less of a difference in effectiveness between dupilumab and the lower dose of abrocitinib (100 mg) with some comparisons showing a benefit in favour of dupilumab and others in favouring abrocitinib 100 mg. Both doses of abrocitinib were more effective than baricitinib 4 mg (EASI 75 for adults in the second-line setting) and in the adolescent population both doses of abrocitinib were more effective than dupilumab (EASI 75). The sensitivity analyses based on expanding the population receiving abrocitinib from those that had failed/not tolerated CsA as the first systemic therapy (restricted population) to include those who were previously treated with at least one systemic treatment for AD (generalisable population) gave similar results to the primary analysis for the composite outcome and EASI 75 for abrocitinib used in combination with TCS and for abrocitinib monotherapy when response was assessed as EASI 50 + ΔDLQI ≥ 4. However, for EASI 75, the benefit of abrocitinib monotherapy compared with dupilumab monotherapy was substantially reduced, favouring dupilumab over abrocitinib 100 mg, but still favouring abrocitinib 200 mg over dupilumab.
Although significantly better than placebo, tralokinumab treatment was numerically, but not statistically significantly, less effective than treatment with either dupilumab or baricitinib 4 mg (response assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75).
Like abrocitinib, treatment with upadacitinib 30 mg led to a better response, assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75, than dupilumab treatment, whereas there was less of a difference in effectiveness between dupilumab and the lower dose of upadacitinib (15 mg) with some comparisons showing a benefit in favour of dupilumab and others favouring upadacitinib 15 mg. Both doses of upadacitinib were more effective than baricitinib 4 mg (EASI 75 for adults in the second-line setting). In the adolescent population, upadacitinib 15 mg was more effective than dupilumab (EASI 75).
Rescue therapy was not permitted in the abrocitinib trials, though sensitivity analysis censoring patients who needed rescue therapy in the trials informing the dupilumab, tralokinumab, and upadacitinib had limited impact for most comparisons. However, for adults given the treatments as monotherapies in the second-line setting, the efficacy of abrocitinib versus dupilumab (assessed as either EASI 50 + ΔDLQI ≥ 4 or EASI 75) decreased substantially when patients receiving rescue therapy were censored, and for the NMA of treatments in combination with TCS, the results for EASI 50 + ΔDLQI ≥ 4 differed substantially for upadacitinib between the primary analysis and the sensitivity analysis censoring patients who received rescue therapy.
For all but the network for the first-line adult population, there was a variability in placebo response for the treatments in each network. This indicates that there may be imbalances in treatment effect modifiers between the trials, which can have an impact on the relative efficacy of the treatments. The EAG attempted to adjust for the difference in placebo response rates; however, this was not possible for all networks and outcomes. The models for EASI 50 + ΔDLQI ≥ 4 and EASI 75 for the second-line systemic treatments in combination with TCS did not converge and therefore no results could be presented for these, which means that substantial uncertainly remains which could not be adjusted for. The baseline-adjusted models for the NMAs of the treatments used as monotherapies in the second-line setting and in the adolescent population did converge and had a better statistical fit than for the unadjusted data. However, the data for these seemed to be overfitted. As the clinical results were produced to inform the health economic model, but results adjusted for baseline differences could not be produced for all outcomes and populations feeding into the base case, the EAG has a strong preference for the unadjusted analyses.
The NICE typically considers interventions a cost-effective use of the NHS resources if the ICER sits within a £20,000–30,000 threshold. The decision rule is reversed if an intervention is less costly and less effective, such that if the ICER is > £20,000–30,000 threshold, it can be considered a cost-effective use of NHS resources.
The following could be considered cost-effective uses of NHS resources: both doses of abrocitinib and upadacitinib 15 mg in the adolescent population; upadacitinib 15 mg and tralokinumab in the adult second-line monotherapy population; and abrocitinib 100 mg, upadacitinib 15 mg and tralokinumab in the adult second-line combination therapy population. For the adult first-line systemic treatment population, upadacitinib may not be considered a cost-effective use of NHS resources.
The EAG cautions the interpretation of the cost-effectiveness results presented in the MTA report as they are based on list prices for abrocitinib, baricitinib, dupilumab, tralokinumab and upadacitinib, but all have confidential PAS discounts in place.
The key drivers of cost effectiveness for all populations were week-16 response probabilities and conditional discontinuation probabilities (used to inform the week-52 response and annual discontinuation), which are as expected as these are the key effectiveness estimates in the model. In particular, the NMA for week-16 response was associated with substantial uncertainty, in particular, for abrocitinib due to small numbers informing the network.
The majority of the QALYs generated for each treatment are derived from occupation in the BSC health state and over a lifetime most patients end up on BSC. Patients on BSC will experience periods of response and relapse and data on the disease course is limited. Reducing the time horizon of the model to 5 years for the adult analyses had a substantial impact on the results in the second-line systemic treatment population.
For both monotherapy and combination therapy for all interventions, ICERs become substantially more cost-effective. In particular, for abrocitinib 100 mg (both monotherapy and combination therapy) and abrocitinib 200 mg monotherapy, ICERs were within the range to be considered cost-effective using a £30,000 threshold. Both tralokinumab and upadacitinib 15 mg dominated dupilumab for both monotherapy and combination therapy analyses. However, the ICERs for upadacitinib 30 mg (monotherapy and combination therapy) and abrocitinib 200 mg monotherapy were still above the £30,000 threshold, but substantially lower than the base case. The ERG notes that for the 5-year time horizon scenario, incremental QALYs increased for all treatments compared with dupilumab, suggesting that longer time horizon gives less-effective treatments time to ‘catch up’ in terms of overall QALYs accrued.
For the adolescent analyses, reducing the time horizon to 18 years of age resulted in ICERs for all treatments moving from dominant to north-east quadrant but still within the range considered to be cost-effective uses of NHS resources.
In the adult second-line systemic treatment population, baricitinib is also a comparator, but it could not be included in the EAG base-case analyses as response data using the composite outcome were not made available to the EAG. However, EASI 75 response data were available for baricitinib combination therapy. Thus, the EAG conducted an illustrative scenario comparing the three new drugs against baricitinib as combination therapy using the EASI 75 response outcome. Under this scenario, none of the three interventions could be considered cost-effective uses of NHS resources.
The EAG explored the use of the EASI 75 response outcome for comparisons against dupilumab in the adult second-line systemic treatment population. For both the monotherapy and combination therapy analyses, ICERs were consistent with the base case, although total QALYs were higher for all treatments (except dupilumab combination therapy).
Across all populations, using parameter utility values, conditional discontinuation and flare treatment estimates from TA534 had a substantial impact on the ICERs, in particular, using utility values from TA534 was a key driver of cost effectiveness. However, the TA534 scenario did not change the direction of the results, except for the abrocitinib 100 mg comparison in the adult second-line systemic treatment population (changed from south-west quadrant ICER to dominated by dupilumab) and all treatments in the adolescent population (changed from dominant compared with dupilumab to south-west quadrant ICERs). However, the TA534 scenario should be considered as illustrative as data from the key trials for each of the three new drugs are available and can be considered as more appropriate for the base case compared to values in TA534 that just reflect dupilumab only.
Most of the other scenarios across all populations resulted in a change in the magnitude of the ICER, but not in the direction of the results. The scenario using the NMA where censoring for rescue therapy was included resulted in a change in the ICER for upadacitinib 15 mg from dominant to south-west quadrant in the adult second-line systemic treatment population. However, the EAG considers that in clinical practice, patients would receive rescue therapy while on treatment and this was also noted in the FAD for baricitinib. As such, the scenario using the NMA where patients were censored for rescue therapy can be considered only illustrative. Furthermore, rescue therapy was not allowed in abrocitinib trials and very few patients received rescue therapy in the upadacitinib and tralokinumab studies.
Strengths and limitations of the assessment
The primary strength of the EAG’s analysis of the three new drugs compared with current practice for each of the subpopulations is that the results have been produced using a consistent approach to the clinical and CEA. Specifically, common assumptions have been used across all comparators, such that results facilitate a consistent basis for decision-making. Furthermore, the EAG has utilised available trial data for each of the interventions in the model to ensure results are as robust as possible.
A strength of the EAG’s clinical analysis is that the trial populations informing the comparisons in the first- and second-line settings were consistently defined across all interventions and in line with the clinical data informing TA534 and TA681. The second-line population was defined as patients who achieved inadequate response to, could not tolerate or were contraindicated to CsA. However, in some studies, contraindication to CsA was not captured, and for abrocitinib, the definition of the population eligible for treatment at second line meant a very small sample size could be included in the primary analysis as the majority of patients in the trials had not received prior systemic therapy or failed/not tolerated systemic therapies other than CsA. That is, the majority of patients in the abrocitinib trials were eligible for first-line systemic therapy according to the EAG’s definition; the population eligible for first-line systemic therapy was patients who were eligible for systemic treatment on inadequate response to topical treatments and who had not received prior systemic therapy.
Although the consistent definitions of the first- and second-line populations is a strength of the analysis, the evidence informing the NMAs for these populations was predominantly derived from post hoc subgroups. The use of post hoc subgroups reduces the sample size for analysis and also breaks the randomisation component of a RCT. A consequence of breaking randomisation is the potential creation of imbalances in both observed and unobserved baseline characteristics. The use of post hoc subgroups introduces bias and uncertainty around the results generated by the NMAs and is a considerable limitation that impacts on the robustness and confidence in the estimates of effect for clinical effectiveness, which is propagated to the cost-effectiveness analysis.
The EAG’s approach to the CEA can be considered conservative, in particular, the implementation of the drug class approach to fill gaps in the data. The strength of the drug class approach can be seen when considering utility values as it makes the inherent assumption that HRQoL is unlikely to be affected by different treatments within a drug class, but there could be differences across different drug classes due to the mode of action and administration of treatment.
Despite the strengths of the EAG’s approach, there were several limitations with the analyses that required assumptions to be made where possible and where not possible, omissions within the analysis. The most significant limitation with the analysis is that the composite outcome of EASI 50 + DLQI ≥ 4 could not be obtained for the adolescent and adult first-line systemic treatment populations due to a paucity of data for dupilumab informing the adolescent NMA and CsA data informing the adult first-line systemic treatment NMA. Furthermore, even though combination therapy data were available for abrocitinib and upadacitinib, only monotherapy could be assessed for the adolescent population as combination data for dupilumab were unavailable to inform the NMA. Conversely, only combination therapy could be assessed for the adult first-line systemic treatment population as monotherapy data were unavailable. As the combination therapy analyses are more relevant for clinical practice, the EAG considered missing monotherapy data are unlikely to be critical for decision-making for the adult first-line systemic treatment subgroup. However, for the adolescent population, monotherapy analyses may potentially underestimate the relative effectiveness of the treatments when used in combination with TCS in clinical practice. Therefore, adolescent monotherapy analyses may reflect a conservative view of clinical effectiveness.
A significant limitation with the analysis is that comparisons with baricitinib could only be included in scenario analysis. In TA681, baricitinib was assessed in the adult second-line systemic population using the composite outcome, but data were redacted. As such, only EASI 75 data for baricitinib combination therapy could be included in the NMA for the adult second-line systemic treatment population. The downstream implication of a lack of base-case result for the three new drugs compared with baricitinib is that incremental analysis of dupilumab and baricitinib against each of the three new drugs could not be presented and any recommendations for the adult second-line systemic population based on the primary composite outcome are limited to comparisons with dupilumab.
Another limitation of the analysis for upadacitinib is the lack of RCT data for CsA in the first-line setting. Thus, results for the comparison of upadacitinib and CsA in the first-line setting are derived from observational data, which is associated with the bias inherent in observational studies and the clinical and cost-effectiveness results for upadacitinib versus CsA should be interpreted with caution.
Analyses exploring increasing or decreasing dose for abrocitinib and upadacitinib were not possible as efficacy data based on titrating dose are unavailable. However, the SmPC guidance for both abrocitinib and upadacitinib does take into consideration circumstances where moving to the lower or higher dose of each drug may be beneficial and this is likely to happen in clinical practice.
A consideration for clinical practice that could not be explored in the current analyses was treatment sequencing. Currently, there is a lack of clinical data on the effectiveness of sequences of AD treatments, especially changing drug class (e.g. starting on a JAK inhibitor and then moving to a monoclonal antibody). As such, more clinical data are needed to understand how patients respond to subsequent lines of systemic treatment and the resulting cost effectiveness of treatment sequences.
Uncertainties
The generalisability of the clinical data informing the analysis is a key area of uncertainty. Clinical experts advising the EAG commented that, based on baseline EASI scores, the patients enrolled in the RCTs identified as relevant to the MTA have more severe AD than would typically be seen in clinical practice, with most patients presenting with disease in clinical practice categorised as moderate severity. No analysis was possible to explore potential differences in efficacy of abrocitinib, tralokinumab and upadacitinib based on disease severity. As such, the efficacy of these interventions seen in patients with more severe AD in the clinical trials may be different to the effect in patients with more moderate AD in clinical practice.
A key uncertainty in the CEA is around the long-term effectiveness of treatments for maintaining response, which is a key driver of cost effectiveness in the MTA model. All of the key trials for each of the new drugs only report short-term data on treatment response and discontinuation. As such, the EAG made assumptions about long-term response in the MTA model based on available short-term data conditional treatment discontinuation data, which is subject to a substantial amount of uncertainty but has been appropriately explored in sensitivity and scenario analyses.
Related to the uncertainty about long-term effectiveness of treatments is the question of how clinical similar treatments for moderate-to-severe AD are to each other. For the NMAs, there were considerable amounts of uncertainty, and the vast majority of results were not statistically significant. There are several reasons for the large uncertainty (wide 95% CrIs) in the clinical results, including the use of post hoc subgroups and small sample sizes, especially for abrocitinib. The EAG is aware that non-significant results from NMAs have been used to substantiate an assumption of clinical equivalence. However, the EAG does not consider this appropriate in this MTA given the magnitude of uncertainty in the results. Additionally, direct evidence from the trials of abrocitinib and upadacitinib demonstrate a statistically significant difference between the lower and higher doses; as such, it is not clinically plausible to consider that they would all have the same effectiveness as a comparator. Thus, the EAG could not reliably assume that treatments were clinically equivalent.
Patient and public involvement
As part of the commissioning process, patient and public involvement (PPI) was conducted by NICE for the technology appraisal process, and so additional PPI was not required for this research.
Other relevant factors
The EAG acknowledges that the outcome of the MTA may result in potentially more treatment options being made available to patients with moderate-to-severe AD, but there is a lack of clinical data on the effectiveness of sequences of AD treatments. Nonetheless, the cost effectiveness of treatment sequencing may become a relevant consideration in the future.
Chapter 7 Conclusions
Implications for service provision
As a result of this MTA, more treatments for moderate-to-severe AD may be made available to patients. However, currently, there is a lack of clinical data on the effectiveness of sequences of AD treatments, especially changing drug class (e.g. starting on a JAK inhibitor and then moving to a monoclonal antibody). As such, more clinical data are needed to understand how patients respond to subsequent lines of systemic treatment and the resulting cost effectiveness of treatment sequences.
Suggested research priorities
Although abrocitinib has a marketing authorisation for the treatment of moderate-to-severe AD in adults and adolescents aged 12 years and over and who are candidates for systemic therapy, the company has positioned abrocitinib as a treatment option for adolescents and for adults in the second-line setting, where second-line systemic therapy captures those who achieve inadequate response to, cannot tolerate or are contraindicated to their first systemic therapy, which, for the MTA, was limited to CsA. However, the majority of patients in the abrocitinib trials were systemic therapy naïve and not relevant for the company’s proposed positioning of abrocitinib. More clinical data, in the form of confirmatory RCT evidence in the second-line setting, are therefore needed.
The remit of this MTA was to assess abrocitinib, tralokinumab and upadacitinib against current treatments in the companies’ proposed positions in the treatment pathway. However, where there are multiple new treatments in a proposed position (for instance, second-line systemic treatment for adults), comparing the new drugs against one another in addition to current practice in an incremental CEA would be beneficial to provide a robust view on which treatments are the most cost-effective. Furthermore, the incremental CEA could feed into further analyses around cost-effective treatment pathways. As noted in the section Implications for service provision, the impact of this MTA is that more treatments for moderate-to-severe AD may be made available to patients. However, currently, there is a lack of clinical data on the effectiveness of sequences of AD treatments; thus, more research in this area is required to perform robust analyses of the cost effectiveness of treatment sequences.
The UK-Irish Atopic Eczema Systemic Therapy Register (A-STAR) is a currently ongoing observational study seeking to understand the safety, effectiveness and health economic implications of systemic immunomodulators in people with AD. Enrolment onto A-STAR for all patients starting these biological treatments will provide useful real-world evidence to inform future clinical and cost-effectiveness studies of systemic treatments for AD.
Acknowledgements
The Evidence Assessment Group would like to thank Victoria Wakefield (Principal Clinical Evidence Analyst, BMJ-TAG), Ben Mayer (Senior Clinical Evidence Analyst, BMJ-TAG) and Conor Hickey (Associate Health Economist, BMJ-TAG) for their assistance with title and abstract appraisal and data extraction and quality assurance of the model.
Contribution of authors
Steven J Edwards (https://orcid.org/0000-0002-9049-3421) was the project lead: supervised the production of the final report; critical appraisal of the clinical evidence; and critical appraisal of the economic evidence.
Charlotta Karner (https://orcid.org/0000-0003-3855-4853) was involved in study selection, data extraction, critical appraisal of the clinical evidence; carried out network meta-analyses; and contributed to writing the clinical sections of the report.
Tracey Jhita (https://orcid.org/0000-0002-8041-0346) was involved in study selection; data extraction; critical appraisal of the economic evidence; development of the conceptual model; quality assurance of the economic model; and writing the economic sections of the report.
Samantha Barton (https://orcid.org/0000-0001-7051-5112) devised and carried out the literature searches on clinical effectiveness; contributed to study selection, data extraction, critical appraisal of the clinical evidence; and contributed to writing the clinical sections of the report.
Gemma Marceniuk (https://orcid.org/0000-0001-9365-0384) devised and carried out the economic literature searches; study selection; data extraction; critical appraisal of the economic evidence. Development of the economic model and carried out the economic analysis and contributed to writing the economic sections of the report.
Zenas Z N Yiu (https://orcid.org/0000-0002-1831-074X) supplied expert clinical advice throughout the project – including reading and commenting on the final report.
Miriam Wittmann (https://orcid.org/0000-0003-2328-4926) supplied expert clinical advice throughout the project – including reading and commenting on the final report.
All authors read and commented on draft versions of the ERG report. Requests for access to data should be addressed to the corresponding author. Ethics approval was not required as this is secondary research conducted as part of the NICE technology assessment programme.
Data-sharing statement
Further information and requests for access to the data used in this report can be obtained from the corresponding author.
Ethics statement
No ethical approval was needed as all included data were from secondary published sources.
Disclaimers
This report presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- Kapur S, Watson W, Carr S. Atopic dermatitis. Allergy Asthma Clin Immunol 2018;14.
- Thomsen SF. Atopic dermatitis: natural history, diagnosis, and treatment. ISRN Allergy 2014;2014.
- Cork MJ, Danby SG, Ogg GS. Atopic dermatitis epidemiology and unmet need in the United Kingdom. J Dermatolog Treat 2020;31:801-9.
- Patient . Atopic Dermatitis and Eczema 2021. https://patient.info/doctor/atopic-dermatitis-and-eczema#nav-0 (accessed 24 September 2021).
- Al-Shobaili HA, Ahmed AA, Alnomair N, Alobead ZA, Rasheed Z. Molecular genetic of atopic dermatitis: an update. Int J Health Sci (Qassim) 2016;10:96-120.
- Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci 2009;122:1285-94.
- Baurecht H, Irvine AD, Novak N, Illig T, Buhler B, Ring J, et al. Toward a major risk factor for atopic eczema: meta-analysis of filaggrin polymorphism data. J Allergy Clin Immunol 2007;120:1406-12.
- Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006;38:441-6.
- Figueras-Nart I, Palomares-Gracia O. Atopic Dermatitis: From Physiopathology to the Clinics 2019. www.intechopen.com/chapters/69553 (accessed 30 September 2021).
- British Association of Dermatologists . Atopic Eczema 2020. www.bad.org.uk/shared/get-file.ashx?id=69&itemtype=document (accessed 31 March 2021).
- Silverwood RJ, Forbes HJ, Abuabara K, Ascott A, Schmidt M, Schmidt SAJ, et al. Severe and predominantly active atopic eczema in adulthood and long term risk of cardiovascular disease: population based cohort study. BMJ 2018;361.
- National Institute for Health and Care Excellence (NICE) . Dupilumab for Treating Moderate to Severe Atopic Dermatitis: Technology Appraisal Guidance [TA534] 2018. www.nice.org.uk/guidance/TA534/chapter/1-Recommendations (accessed 18 October 2021).
- National Institute for Health and Care Excellence (NICE) . Baricitinib for Treating Moderate to Severe Atopic Dermatitis 2021. www.nice.org.uk/guidance/TA681 (accessed 18 October 2021).
- Koszoru K, Borza J, Gulacsi L, Sardy M. Quality of life in patients with atopic dermatitis. Cutis 2019;104:174-7.
- Zink AGS, Arents B, Fink-Wagner A, Seitz IA, Mensing U, Wettemann N, et al. Out-of-pocket costs for individuals with atopic eczema: a cross-sectional study in nine European Countries. Acta Derm Venereol 2019;99:263-7.
- Schofield J, Grindlay D, Williams H. Skin Conditions in the UK: A Health Care Needs Assessment 2009. www.nottingham.ac.uk/research/groups/cebd/documents/hcnaskinconditionsuk2009.pdf (accessed 30 September 2021).
- National Institute for Health and Care Excellence (NICE) . Eczema 2021. https://pathways.nice.org.uk/pathways/eczema (accessed 29 October 2021).
- Gooderham MJ, Bissonnette R, Grewal P, Lansang P, Papp KA, Hong CH. Approach to the assessment and management of adult patients with atopic dermatitis: a consensus document. Section II: tools for assessing the severity of atopic dermatitis. J Cutan Med Surg 2018;22:10S-6S.
- Harmonising Outcomes for Eczema . Core Outcomes for Trials 2021. www.homeforeczema.org/research/index.aspx (accessed 30 June 2021).
- Basra MK, Salek MS, Camilleri L, Sturkey R, Finlay AY. Determining the minimal clinically important difference and responsiveness of the Dermatology Life Quality Index (DLQI): further data. Dermatology 2015;230:27-33.
- Chopra R, Vakharia PP, Sacotte R, Patel N, Immaneni S, White T, et al. Severity strata for Eczema Area and Severity Index (EASI), modified EASI, scoring atopic dermatitis (SCORAD), objective SCORAD, atopic dermatitis severity index and body surface area in adolescents and adults with atopic dermatitis. Br J Dermatol 2017;177:1316-21.
- Yosipovitch G, Reaney M, Mastey V, Eckert L, Abbe A, Nelson L, et al. Peak Pruritus Numerical Rating Scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol 2019;181:761-9.
- Primary Care Dermatology Society . Eczema – Atopic Aczema 2019. www.pcds.org.uk/clinical-guidance/atopic-eczema (accessed 1 June 2021).
- NHS . Atopic Eczema 2021. www.nhs.uk/conditions/atopic-eczema/treatment/ (accessed 31 March 2021).
- Berth-Jones J, Exton LS, Ladoyanni E, Mohd Mustapa MF, Tebbs VM, Yesudian PD, et al. British Association of Dermatologists guidelines for the safe and effective prescribing of oral ciclosporin in dermatology 2018. Br J Dermatol 2019;180:1312-38.
- Scottish Intercollegiate Guidelines Network . Management of Atopic Eczema in Primary Care 2011. www.sign.ac.uk/media/2029/sign125.pdf (accessed 29 October 2021).
- Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Efficacy and safety of oral Janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol 2019;155:1371-9.
- Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet 2020;396:255-66.
- Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol 2020;156:863-73.
- Eichenfield L, Flohr C, Sidbury R, Szalai Z, Galus R, Yao Z, et al. Efficacy and safety of abrocitinib in adolescent patients with moderate-to-severe atopic dermatitis (AD): results from the phase 3 JADE TEEN study. J Allergy Clin Immunol 2021;147.
- Eichenfield LF, Silverberg JI, Gadkari A, Guillemin I, Chen Z, Bansal A, et al. 15013 Dupilumab improves signs, symptoms, and quality of life in adolescents with moderate to severe atopic dermatitis. J Am Acad Dermatol 2020;83.
- Bieber T, Simpson EL, Silverberg JI, Thaçi D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med 2021;384:1101-12.
- Bieber T. Interleukin-13: targeting an underestimated cytokine in atopic dermatitis. Allergy 2020;75:54-62.
- ClinicalTrials.gov . Tralokinumab Monotherapy for Adolescent Subjects With Moderate to Severe Atopic Dermatitis – ECZTRA 6 (ECZema TRAlokinumab Trial No. 6) 2021. https://clinicaltrials.gov/ct2/show/NCT03526861 (accessed 2 June 2021).
- Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol 2021;184:437-49.
- Silverberg JI, Toth D, Bieber T, Alexis AF, Elewski BE, Pink AE, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol 2021;184:450-63.
- ClinicalTrials.gov . Tralokinumab in Combination With Topical Corticosteroids in Japanese Subjects With Moderate-to-Severe Atopic Dermatitis (ECZTRA 8) 2021. https://clinicaltrials.gov/ct2/show/NCT04587453 (accessed 21 October 2021).
- ClinicalTrials.gov . Tralokinumab in Combination With Topical Corticosteroids in Subjects With Severe Atopic Dermatitis – ECZTRA 7 (ECZTRA 7) 2021. https://clinicaltrials.gov/ct2/show/NCT03761537?term=tralokinumab&draw=2&rank=5 (accessed 2 June 2021).
- Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet 2021;397:2151-68.
- ClinicalTrials.gov . A Study to Compare Safety and Efficacy of Upadacitinib to Dupilumab in Adult Participants With Moderate to Severe Atopic Dermatitis (Heads Up) 2021. https://clinicaltrials.gov/ct2/show/NCT03738397 (accessed 2 June 2021).
- Reich K, Teixeira HD, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021;397:2169-81.
- National Institute for Health and Care Excellence (NICE) . Abrocitinib, Tralokinumab and Upadacitinib for Treating Moderate to Severe Atopic Dermatitis. Final Scope 2021. www.nice.org.uk/guidance/gid-ta10856/documents/final-scope (accessed 2 November 2021).
- Ben-Gashir MA, Hay RJ. Reliance on erythema scores may mask severe atopic dermatitis in black children compared with their white counterparts. Br J Dermatol 2002;147:920-5.
- Alexis AF, Rendon M, Silverberg JI, Pariser DM, Lockshin B, Griffiths CEM, et al. Efficacy of dupilumab in different racial subgroups of adults with moderate-to-severe atopic dermatitis in three randomized, placebo-controlled phase 3 trials. J Drugs Dermatol 2019;18:804-13.
- National Institute for Health and Care Excellence (NICE) . Resource Impact Report: Baricitinib for Treating Moderate to Severe Atopic Dermatitis (TA681) 2021. www.nice.org.uk/guidance/ta681/resources/resource-impact-report-pdf-9021362653 (accessed 11 October 2021).
- National Institute for Health and Care Excellence (NICE) . Resource Impact Report: Dupilumab for Treating Moderate to Severe Atopic Dermatitis (TA534) 2018. www.nice.org.uk/guidance/ta534 (accessed 11 October 2021).
- Sawangjit R, Dilokthornsakul P, Lloyd-Lavery A, Lai NM, Dellavalle R, Chaiyakunapruk N. Systemic treatments for eczema: a network meta-analysis. Cochrane Database Syst Rev 2020;9.
- BMJ-TAG . Abrocitinib, Tralokinumab and Upadacitinib for Treating Moderate to Severe Atopic Dermatitis. Protocol for Multiple Technology Appraisal 2021. www.nice.org.uk/guidance/gid-ta10856/documents/final-protocol (accessed 2 November 2021).
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal 2013 2013. www.nice.org.uk/process/pmg9/resources/guide-to-the-methods-of-technology-appraisal-2013-pdf-2007975843781 (accessed 17 December 2021).
- Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 2009;339.
- Glanville J, Foxlee R, Wisniewski S, Noel-Storr A, Edwards M, Dooley G. Translating the Cochrane EMBASE RCT filter from the Ovid interface to Embase.com: a case study. Health Info Libr J 2019;36:264-77.
- Sterne JAC, Savovic J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366.
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 1: Introduction to Evidence Synthesis for Decision Making 2012. https://www.sheffield.ac.uk/nice-dsu/tsds/evidence-synthesis (accessed 3 June 2021).
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials 2011. https://www.sheffield.ac.uk/nice-dsu/tsds/evidence-synthesis (accessed 3 June 2021).
- OpenBUGS v3.2.3 rev1012 2014.
- Turner RM, Jackson D, Wei Y, Thompson SG, Higgins JP. Predictive distributions for between-study heterogeneity and simple methods for their application in Bayesian meta-analysis. Stat Med 2015;34:984-98.
- Dias S WN, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: Inconsistency in Networks of Evidence Based on Randomised Controlled Trials 2011. https://www.sheffield.ac.uk/nice-dsu/tsds/evidence-synthesis.
- Reich K, Szepietowski JC, Smith CH, Irvine AD, Ogg G, Tsianakas A, et al. Safety of dupilumab in moderate-to-severe atopic dermatitis patients inadequately controlled with, intolerant to, or inadvisable for cyclosporine-A: laboratory findings from the liberty-ad-café trial. J Dermatol Nurses Assoc 2020;12.
- Zheng YI, Li C. Efficacy and safety of dupilumab for the treatment of moderate-to-severe atopic dermatitis in adults. J Dermatol Nurses Assoc 2020;12.
- Gooderham M, Forman S, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. The Janus kinase 1 (JAK1) inhibitor PF-04965842 reduces signs and symptoms of moderate to severe atopic dermatitis (AD). J Investig Dermatol 2018;138.
- Sowden JM, Berth-Jones J, Ross JS, Motley RJ, Marks R, Finlay AY, et al. Double-blind, controlled, crossover study of cyclosporin in adults with severe refractory atopic dermatitis. Lancet 1991;338:137-40.
- Allen BR. Royal Society of Medicine International Congress and Symposium Series Vol 192 Athens. Greece: Cyclosporin A and the skin: proceedings, satellite symposium to the 2nd congress of the European Academy of Dermatology & Venereology; 1991.
- Berth Jones J. Cyclosporin in severe adult atopic dermatitis – a multi-centre, double-blind, placebo-controlled crossover study. Br J Dermatol 1991;125.
- Munro CS, Levell NJ, Shuster S, Friedmann PS. Maintenance treatment with cyclosporin in atopic eczema. Br J Dermatol 1994;130:376-80.
- Salek MS, Finlay AY, Luscombe DK, Allen BR, Berth-Jones J, Camp RD, et al. Cyclosporin greatly improves the quality of life of adults with severe atopic dermatitis. A randomized, double-blind, placebo-controlled trial. Br J Dermatol 1993;129:422-30.
- van Joost T, Heule F, Korstanje M, van den Broek MJ, Stenveld HJ, van Vloten WA. Cyclosporin in atopic dermatitis: a multicentre placebo-controlled study. Br J Dermatol 1994;130:634-40.
- Wahlgren CF, Scheynius A, Hagermark O. Antipruritic effect of oral cyclosporin A in atopic dermatitis. Acta Derm Venereol 1990;70:323-9.
- Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med 2014;371:130-9.
- Bieber T, Thaci D, Graham N, Pirozzi G, Teper A, Ren H, et al. Dupilumab monotherapy in adults with moderate-to-severe atopic dermatitis: a 12-week, randomized, double-blind, placebo-controlled study. J Allergy Clin Immunol 2014;133.
- Blauvelt A, de Bruin-Weller M, Gooderham M, Cather JC, Weisman J, Pariser D, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet 2017;389:2287-303.
- Blauvelt A, Simpson EL, Tyring SK, Purcell LA, Shumel B, Petro CD, et al. Dupilumab does not affect correlates of vaccine-induced immunity: a randomized, placebo-controlled trial in adults with moderate-to-severe atopic dermatitis. J Am Acad Dermatol 2019;80:158-167.e1.
- Guttman-Yassky E, Bissonnette R, Ungar B, Suarez-Farinas M, Ardeleanu M, Esaki H, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol 2019;143:155-72.
- Hamilton J, Ren H, Weinstein SP, Pirozzi G, Graham N, Radin A. Dupilumab improved all domains of Eczema Area and Severity Index (EASI) and 5-D pruritus scale in adults with atopic dermatitis in a phase 2 study. J Investig Dermatol 2014;134.
- Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. SOLO 1 and SOLO 2 investigators . Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med 2016;375:2335-48.
- Thaci D, Simpson EL, Beck LA, Bieber T, Blauvelt A, Papp K, et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: a randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet 2016;387:40-52.
- Tsianakas A, Luger TA, Radin A. Dupilumab treatment improves quality of life in adult patients with moderate-to-severe atopic dermatitis: results from a randomized, placebo-controlled clinical trial. Br J Dermatol 2018;178:406-14.
- Guttman-Yassky E, Silverberg JI, Nemoto O, Forman SB, Wilke A, Prescilla R, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol 2019;80:913-921.e9.
- Arora CJ, Khattak FA, Yousafzai MT, Ibitoye BM, Shumack S. The effectiveness of Janus kinase inhibitors in treating atopic dermatitis: a systematic review and meta-analysis. Dermatol Ther 2020;33.
- Drucker AM, Ellis AG, Bohdanowicz M, Mashayekhi S, Yiu ZZN, Rochwerg B, et al. Systemic immunomodulatory treatments for patients with atopic dermatitis: a systematic review and network meta-analysis. JAMA Dermatol 2020;156:659-67.
- Kerschbaumer A, Smolen JS, Nash P, Doerner T, Dougados M, Fleischmann R, et al. Points to consider for the treatment of immune-mediated inflammatory diseases with Janus kinase inhibitors: a systematic literature research. RMD Open 2020;6.
- Lam M, Zhu JW, Maqbool T, Adam G, Tadrous M, Rochon P, et al. Inclusion of older adults in randomized clinical trials for systemic medications for atopic dermatitis: a systematic review. JAMA Dermatol 2020;156:1240-5.
- Seger EW, Wechter T, Strowd L, Feldman SR. Relative efficacy of systemic treatments for atopic dermatitis. J Am Acad Dermatol 2019;80:411-416.e4.
- Siegels D, Heratizadeh A, Abraham S, Binnmyr J, Brockow K, Irvine AD, et al. European Academy of Allergy, Clinical Immunology Atopic Dermatitis Guideline Group . Systemic treatments in the management of atopic dermatitis: a systematic review and meta-analysis. Allergy 2021;76:1053-76.
- Silverberg JI, Thyssen JP, Fahrbach K, Mickle K, Cappelleri JC, Romero W, et al. Comparative efficacy and safety of systemic therapies used in moderate-to-severe atopic dermatitis: a systematic literature review and network meta-analysis. J Eur Acad Dermatol Venereol 2021;35:1797-810.
- Simpson EL, Gadkari A, Worm M, Soong W, Blauvelt A, Eckert L, et al. Dupilumab therapy provides clinically meaningful improvement in patient-reported outcomes (PROs): a phase IIb, randomized, placebo-controlled, clinical trial in adult patients with moderate to severe atopic dermatitis (AD). J Am Acad Dermatol 2016;75:506-15.
- Zhao Y, Wu L, Lu Q, Gao X, Zhu X, Yao X, et al. The efficacy and safety of dupilumab in Chinese patients with moderate-to-severe atopic dermatitis: a randomized, double-blind, placebo-controlled study. Br J Dermatol 2022;186:633-41.
- Ariens LFM, Gadkari A, Van Os-Medendorp H, Ayyagari R, Terasawa E, Kuznik A, et al. Dupilumab versus cyclosporine for the treatment of moderate-to-severe atopic dermatitis in adults: indirect comparison using the Eczema Area and Severity Index. Acta Derm Venereol 2019;99:851-7.
- Dal Bello G, Maurelli M, Schena D, Girolomoni G, Gisondi P. Drug survival of dupilumab compared to cyclosporin in moderate-to-severe atopic dermatitis patients. Dermatol Ther 2020;33.
- Spekhorst LS, Ariëns LFM, van der Schaft J, Bakker DS, Kamsteeg M, Oosting AJ, et al. Two-year drug survival of dupilumab in a large cohort of difficult-to-treat adult atopic dermatitis patients compared to cyclosporine A and methotrexate: results from the BioDay registry. Allergy 2020;75:2376-9.
- ClinicalTrials.gov . Study of Abrocitinib Compared With Dupilumab in Adults With Moderate to Severe Atopic Dermatitis on Background Topical Therapy 2020. www.cochranelibrary.com/central/doi/10.1002/central/CN-02089738/full (accessed 1 August 2020).
- Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J Allergy Clin Immunol 2019;143:135-41.
- ClinicalTrials.gov . Vaccine Responses in Tralokinumab-Treated Atopic Dermatitis – ECZTRA 5 (ECZema TRAlokinumab Trial No. 5) (ECZTRA 5) 2021. https://clinicaltrials.gov/ct2/show/NCT03562377?term=tralokinumab&cond=Atopic±Dermatitis&draw=2&rank=7 (accessed 2 June 2021).
- Guttman-Yassky E, Thaçi D, Pangan AL, Hong HC, Papp KA, Reich K, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol 2020;145:877-84.
- ClinicalTrials.gov . A Study to Evaluate Safety of Upadacitinib in Combination With Topical Corticosteroids in Adolescent and Adult Participants With Moderate to Severe Atopic Dermatitis (Rising Up) 2020. https://clinicaltrials.gov/ct2/show/NCT03661138 (accessed 22 April 2021).
- Simpson EL, Lacour JP, Spelman L, Galimberti R, Eichenfield LF, Bissonnette R, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol 2020;183:242-55.
- ClinicalTrials.gov . A Long-Term Study of Baricitinib (LY3009104) With Topical Corticosteroids in Adults With Moderate to Severe Atopic Dermatitis That Are Not Controlled With Cyclosporine or for Those Who Cannot Take Oral Cyclosporine Because It Is Not Medically Advisable (BREEZE-AD4) 2018. https://clinicaltrials.gov/ct2/show/study/NCT03428100 (accessed 18 October 2021).
- Reich K, Kabashima K, Peris K, Silverberg JI, Eichenfield LF, Bieber T, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol 2020;156:1333-43.
- Silverberg JI, Simpson EL, Wollenberg A, Bissonnette R, Kabashima K, Delozier AM, et al. Long-term efficacy of baricitinib in adults with moderate to severe atopic dermatitis who were treatment responders or partial responders: an extension study of 2 randomized clinical trials. JAMA Dermatol 2021;157:691-9.
- Simpson EL, Paller AS, Siegfried EC, Boguniewicz M, Sher L, Gooderham MJ, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: a phase 3 randomized clinical trial. JAMA Dermatol 2020;156:44-56.
- de Bruin-Weller M, Thaçi D, Smith CH, Reich K, Cork MJ, Radin A, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical trial (LIBERTY AD CAFÉ). Br J Dermatol 2018;178:e366-e366.
- Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Erratum: efficacy and safety of oral Janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial (JAMA Dermatology (2019) 155: 12 (1371–1379) DOI: 10.1001/jamadermatol.2019.2855). JAMA Dermatol 2020;156. 10.1001/jamadermatol.2019.2855.
- Simpson EL, Sinclair R, Forman S, Wollenberg A, Asch R, Cork M, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: results from the phase III, JADE MONO-1 study. Br J Dermatol 2020;183:e105-6.
- Silverberg J, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: results from the phase III JADE MONO-2 study. Br J Dermatol 2020;183.
- Thac-i D, Bieber T, Simpson EL, Silverberg JI, Paul C, Sinclair R, et al. A phase III study to investigate the efficacy and safety of abrocitinib and dupilumab in comparison with placebo in adults with moderate-to-severe atopic dermatitis. Br J Dermatol 2021;184:e72-3.
- Guttman‐Yassky E, Silverberg J, Roepke M, Wollenberg A. Tralokinumab, an anti-interleukin-13 monoclonal antibody, reduces Staphylococcus aureus colonization of the skin and systemic levels of inflammatory biomarkers in atopic dermatitis patients. J Am Acad Dermatol 2019;81.
- Alexis AF, Zirwas M, Pinter A, Adam DN, Chiricozzi A, Pink AE, et al. Progressive and sustained improvements in the extent and severity of atopic dermatitis with tralokinumab in combination with topical corticosteroids as needed in moderate-to-severe atopic dermatitis. Br J Dermatol 2021;184:e82-3.
- Guttman‐Yassky E, Beck LA, Anderson JK, Hu X, Gu Y, Teixeira HD, et al. Upadacitinib treatment withdrawal and retreatment in patients with moderate-to-severe atopic dermatitis from a phase 2b, randomized, controlled trial. J Am Acad Dermatol 2019;81.
- Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet 2021;397:2151-68.
- Reich K, Teixeira HD, De Bruin-Weller M, Bieber T, Soong W, Kabashima K, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis: results from the pivotal phase III, randomized, double-blind, placebo-controlled AD Up study. Br J Dermatol 2021;184:e59-60.
- Reich K, Teixeira HD, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2021;397:2169-81.
- Wollenberg A, Manuel Carrascosa J, Lacour J, Reich K, Eichenfield L, Rubel D, et al. Summary of efficacy, impact on work productivity and activity, and safety of baricitinib in moderate-to-severe atopic dermatitis from two monotherapy phase III trials. Br J Dermatol 2020;183.
- Paller A, Blauvelt A, Pariser D, Worrell R, Soong W, Hong C, et al. Dupilumab for adolescents with moderate-to-severe atopic dermatitis: results from a phase 3, randomized, double-blinded trial. Australas J Dermatol 2019;60.
- Paller AS, Blauvelt A, Pariser DM, Soong W, Hong HC‐H, Zhang R, et al. Dupilumab for adolescents with moderate-to-severe atopic dermatitis: results from a phase 3, randomized, double-blinded trial. J Am Acad Dermatol 2019;81.
- Thaçi D, Simpson EL, Deleuran M, Kataoka Y, Chen Z, Gadkari A, et al. Efficacy and safety of dupilumab monotherapy in adults with moderate-to-severe atopic dermatitis: a pooled analysis of two phase 3 randomized trials (LIBERTY AD SOLO 1 and LIBERTY AD SOLO 2). J Dermatol Sci 2019;94:266-75.
- Worm M, Simpson EL, Thaci D, Bissonnette R, Lacour J-P, Beissert S, et al. Efficacy and safety of multiple dupilumab dose regimens after initial successful treatment in patients with atopic dermatitis: a randomized clinical trial. JAMA Dermatol 2020;156:131-43.
- Wells GA, Shea B, O’Connell D, Peterson J, Welch V, Losos M, et al. The Newcastle–Ottawa Scale (NOS) for Assessing the Quality of Nonrandomised Studies in Meta-Analyses 2021. www.ohri.ca/programs/clinical_epidemiology/oxford.asp (accessed 1 December 2021).
- Dias S, Sutton AJ, Welton NJ, Ades AE. Evidence synthesis for decision making 3: heterogeneity – subgroups, meta-regression, bias, and bias-adjustment. Med Decis Making 2013;33:618-40.
- Dias S, Welton NJ, Sutton AJ, Ades AE. Evidence synthesis for decision making 5: the baseline natural history model. Med Decis Making 2013;33:657-70.
- England N. Early Access to Medicines Scheme – Dupilumab in the Treatment of Adolescent Patients ≥ 12 to ≪ 18 Years of Age With Severe Atopic Dermatitis Who Have Responded Inadequately to at Least One Systemic Therapy or Where the Available Systemic Therapies Are Not Recommended or Are Not Tolerated 2019. https://www.ekhuftformulary.nhs.uk/docs/SSC%202082%20letter%20Early%20Access%20to%20Medicines%20Scheme%20%20Dupilumab%20in%20the%20treatment%20of%20adolescent%20patient.pdf.
- Arber M, Garcia S, Veale T, Edwards M, Shaw A, Glanville JM. Performance of Ovid MEDLINE search filters to identify health state utility studies. Int J Technol Assess Health Care 2017;33:472-80.
- Health Improvement Scotland (HIS) . SMC2232 Dupilumab 200 Mg and 300 Mg Solution for Injection in Pre-Filled Syringe (Dupixent®) 2019. www.scottishmedicines.org.uk/media/4976/dupilumab-dupixent-abbreviated-final-december-2019-for-website.pdf (accessed 1 August 2021).
- Health Improvement Scotland (HIS) . SMC2337 Baricitinib 2 Mg and 4 Mg Film-Coated Tablets (Olumiant®) 2018. www.scottishmedicines.org.uk/media/6030/baricitinib-olumiant-final-may-2021-for-website.pdf (accessed 1 August 2021).
- Anonymous . CADTH Drug Reimbursement Review Dupilumab (Dupixent) 2018. https://www.cadth.ca/dupilumab.
- Kuznik A, Bego-Le-Bagousse G, Eckert L, Gadkari A, Simpson E, Graham CN, et al. Economic evaluation of dupilumab for the treatment of moderate-to-severe atopic dermatitis in adults. Dermatol Ther (Heidelb) 2017;7:493-505.
- Zimmermann M, Rind D, Chapman R, Kumar V, Kahn S, Carlson J. Economic evaluation of dupilumab for moderate-to-severe atopic dermatitis: a cost–utility analysis. J Drugs Dermatol 2018;17:750-6.
- Institute for Clinical and Economic Review (ICER) . Dupilumab and Crisaborole for Atopic Dermatitis: Effectiveness and Value 2017. https://icer.org/wp-content/uploads/2020/10/MWCEPAC_ATOPIC_EVIDENCE_REPORT_051217.pdf (accessed 1 August 2021).
- Institute for Clinical and Economic Review (ICER) . JAK Inhibitors and Monoclonal Antibodies for the Treatment of Atopic Dermatitis: Effectiveness and Value 2021. https://icer.org/wp-content/uploads/2020/12/ICER_Atopic-Dermatitis_Draft-Evidence-Report_051421.pdf (accessed 4 November 2021).
- Fanelli F, Pedone MP, Serra A, Bitonti R, Furneri G. PBI11 Cost-effectiveness analysis of dupilumab for the treatment of atopic dermatitis in adolescent patients in Italy. Value Health 2020;23.
- Health Improvement Scotland (HIS) . SMC2011 Dupilumab 300 mg Solution for Injection in Pre-Filled Syringe (Dupixent®) 2018. www.scottishmedicines.org.uk/media/3751/dupilumab-dupixent-final-august-2018-for-website.pdf (accessed 1 August 2021).
- Andersen L, Nyeland ME, Nyberg F. Higher self-reported severity of atopic dermatitis in adults is associated with poorer self-reported health-related quality of life in France, Germany, the U.K. and the U.S.A. Br J Dermatol 2020;182:1176-83.
- Hsieh B-J, Shen D, Hsu C-J, Chan TC, Cho Y-T, Tang C-H, et al. The impact of atopic dermatitis on health-related quality of life in Taiwan. J Formos Med Assoc 2021;21:269-77.
- Kwatra SG, Gruben D, Fung S, DiBonaventura M. Psychosocial comorbidities and health status among adults with moderate-to-severe atopic dermatitis: a 2017 US National Health and Wellness Survey Analysis. Adv Ther 2021;38:1627-37.
- Misery L, Seneschal J, Reguiai Z, Merhand S, Héas S, Huet F, et al. Patient burden is associated with alterations in quality of life in adult patients with atopic dermatitis: results from the ECLA study. Acta Derm Venereol 2018;98:713-4.
- Girolomoni G, Luger T, Nosbaum A, Gruben D, Romero W, Llamado LJ, et al. The economic and psychosocial comorbidity burden among adults with moderate-to-severe atopic dermatitis in Europe: analysis of a cross-sectional survey. Dermatol Ther (Heidelb) 2021;11:117-30.
- Retzler J, Smith A, Reaney M, Rout R, Hudson R. Process utilities for topical treatment in atopic dermatitis. Qual Life Res 2019;28:2373-81.
- Silverberg JI, Gelfand JM, Margolis DJ, Boguniewicz M, Fonacier L, Grayson MH, et al. Health utility scores of atopic dermatitis in US adults. J Allergy Clin Immunol Pract 2019;7:1246-52.e1.
- Simpson EL. Dupilumab improves general health-related quality-of-life in patients with moderate-to-severe atopic dermatitis: pooled results from two randomized, controlled phase 3 clinical trials. Dermatol Ther (Heidelb) 2017;7:243-8.
- Simpson EL, Bieber T, Eckert L, Wu R, Ardeleanu M, Graham NMH, et al. Patient burden of moderate to severe atopic dermatitis (AD): insights from a phase 2b clinical trial of dupilumab in adults. J Am Acad Dermatol 2016;74:491-8.
- Song HJ, Park H, Park S-Y, Lee E-K, Ha S-Y, Park S-Y, et al. Estimation of health utilities based on the response to treatment in atopic dermatitis: a population-based study. Clin Ther 2019;41:700-13.
- Silverberg J, Gelfand J, Margolis D, Boguniewicz M, Fonacier L, Grayson M, et al. Societal burden of atopic dermatitis in the US population. J Am Acad Dermatol 2019;81.
- Vietri J, Nyberg F, Varol N. The impact of severity of atopic dermatitis on patient-reported outcomes of adults with moderate-to-severe atopic dermatitis. J Am Acad Dermatol 2017;76.
- Nyberg F, Hansen JB, Andersen L. Greater severity of atopic dermatitis (AD) is associated with poorer patient reported outcomes among adults with moderate-to-severe AD. Pharmacoepidemiol Drug Saf 2018;27.
- Retzler J, Smith AB, Reaney M, Rout R, Hudson R. Process utilities for topical treatment in atopic dermatitis. Value Health 2018;21.
- van Hout B, Janssen MF, Feng Y-S, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15.
- National Institute for Health and Care Excellence (NICE) . Guide to the Methods of Technology Appraisal 2013 2013. https://www.nice.org.uk/process/pmg9/chapter/foreword.
- Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health 2010;13:509-18.
- Electronic Medicines Compendium (emc) . Neoral Soft Gelatin Capsules. Summary of Product Characteristics 2021. www.medicines.org.uk/emc/product/1034/smpc#gref (accessed 4 November 2021).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2020, Personal Social Services Research Unit, University of Kent, Canterbury 2020. www.pssru.ac.uk/project-pages/unit-costs/unit-costs-2020/ (accessed 31 March 2021).
- Santer M, Rumsby K, Ridd MJ, Francis NA, Stuart B, Chorozoglou M, et al. Adding emollient bath additives to standard eczema management for children with eczema: the BATHE RCT. Health Technol Assess 2018;22:1-116.
- Joint Formulary Committee . British National Formulary 2021. https://bnf.nice.org.uk/ (accessed 1 August 2021).
- Department of Health and Social Care . Drugs and Pharmaceutical Electronic Market Information Tool (eMIT) 2021. www.gov.uk/government/publications/drugs-and-pharmaceutical-electronic-market-information-emit (accessed 31 March 2021).
- Wollenberg A, Barbarot S, Bieber T, Christen-Zaech S, Deleuran M, Fink-Wagner A, et al. European Dermatology Forum (EDF), the European Academy of Dermatology and Venereology (EADV), the European Academy of Allergy and Clinical Immunology (EAACI), the European Task Force on Atopic Dermatitis (ETFAD), European Federation of Allergy and Airways Diseases Patients’ Associations (EFA), the European Society for Dermatology and Psychiatry (ESDaP), the European Society of Pediatric Dermatology (ESPD), Global Allergy and Asthma European Network (GA2LEN) and the European Union of Medical Specialists (UEMS) . Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: part II. J Eur Acad Dermatol Venereol 2018;32:850-78.
- NHS Improvement . National Schedule of NHS Costs – Year 2019–20 – NHS Trusts and NHS Foundation Trusts 2020. www.england.nhs.uk/national-cost-collection/ (accessed 1 August 2021).
List of abbreviations
- ACM
- Appraisal Committee Meeting
- AD
- atopic dermatitis
- AE
- adverse event
- A&E
- accident and emergency
- BGR
- Brooks–Gelman–Rubin
- BNF
- British National Formulary
- BSA
- body surface area
- BSC
- best supportive care
- CADTH
- Canadian Agency for Drugs and Technologies in Health
- CEA
- cost-effectiveness analysis
- CRD
- Centre for Reviews and Dissemination
- CrI
- credible interval
- CsA
- ciclosporin A
- DIC
- deviance information criterion
- DLQI
- Dermatology Life Quality Index
- DSU
- Decision Support Unit
- EAG
- Evidence Assessment Group
- EASI
- Eczema Area and Severity Index
- eMIT
- electronic drug marketing tool
- EQ-5D
- EuroQol-5 dimensions
- ERG
- Evidence Review Group
- FAD
- final appraisal document
- FBC
- full blood count
- FE
- fixed effect
- GP
- general practitioner
- HRQoL
- health-related quality of life
- HSUV
- heath-state utility value
- HTA
- health technology appraisal
- ICER
- incremental cost-effectiveness ratio
- IGA
- Investigator Global Assessment
- IgE
- immunoglobulin E
- IL
- interleukin
- INAHTA
- International Network of Agencies for Health Technology Assessment
- JAK
- Janus kinase
- LOCF
- last observation carried forward
- MM
- Markov model
- MTA
- multiple technology appraisal
- NICE
- National Institute for Health and Care Excellence
- NMA
- network meta-analysis
- NOS
- Newcastle–Ottawa Scale
- OD
- once daily
- OR
- odds ratio
- OWSA
- one-way sensitivity analysis
- PAS
- patient access scheme
- PPI
- patient and public involvement
- PRISMA
- preferred reporting items for systematic reviews and meta-analyses
- PSA
- probabilistic sensitivity analysis
- PSSRU
- Personal Social Services Research Unit
- Q2W
- every 2 weeks
- Q4W
- every 4 weeks
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- QW
- every week
- RCT
- randomised controlled trial
- RE
- random effects
- SAE
- serious adverse event
- SC
- subcutaneous
- SCORAD
- Scoring Atopic Dermatitis
- SD
- standard deviation
- SF-6D
- short-form 6-dimension
- SIGN
- Scottish Intercollegiate Guidelines Network
- SLR
- systematic literature review
- SMC
- Scottish Medicines Consortium
- SmPC
- summary of product characteristics
- STA
- single technology appraisal
- TCI
- topical calcineurin inhibitor
- TCS
- topical corticosteroid
- TSD
- technical support document
- TTO
- time trade-off
- UMC
- University Medical Center
- URTI
- urinary tract infection
- VAS
- visual analogue scale
- WTP
- willingness to pay
Glossary
- Dominant
- Treatment is less expensive and more effective than the comparator.
- Dominated
- Treatment is more expensive and less effective than the comparator.
- North-east quadrant
- The incremental cost-effectiveness ratio lies in the north-east quadrant of the cost-effectiveness plane, which means it is more expensive and more effective than the comparator.
- South-west quadrant
- The incremental cost-effectiveness ratio lies in the south-west quadrant of the cost-effectiveness plane, which means it is less expensive and less effective than the comparator.
Note about the research
This monograph is based on the Multiple Technology Assessment Report produced for National Institute for Health and Care Excellence (NICE). The full report contained a considerable number of data that were deemed confidential and were used by the Advisory Committee at NICE in their deliberations. The full version of the report with the confidential information removed is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/LEXB9006).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.