

Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the Evidence Synthesis Programme on behalf of NICE as award number NIHR135477. The protocol was agreed in May 2022. The draft manuscript began editorial review in December 2022 and was accepted for publication in June 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this article.
Permissions
Copyright statement
Copyright © 2024 Llewellyn et al. This work was produced by Llewellyn et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Llewellyn et al.
Chapter 1 Background and definition of the decision problem
Description of health problem
Prostate cancer (PCa) is the most commonly diagnosed cancer in men in the UK; it accounts for more than a quarter (27%) of all male cancer diagnoses in 2016–8. 1 It is the second most common cause of cancer death in males in the UK, accounting for 14% of all cancer deaths. The estimated lifetime risk of a PCa diagnosis is one in eight for males born in the UK. 2,3 Over 57,000 new cases were diagnosed in 2018, with an estimated 10-year survival rate of 77.6%. Since the early 1990s, estimates of PCa incidence rates have increased by nearly half (48%) in males in the UK (2016–8) and are projected to rise by 12% between 2014 and 2035, resulting in 233 cases per 100,000 males by 2035. 3
Early-stage diagnosis is associated with improved survival outcomes compared with patients diagnosed at the latest stage of the disease. PCa primarily affects people aged 50 years or more, and the risk of developing PCa increases with age. 3 In England and Wales, 87% of people diagnosed with PCa are aged 60 years or older,4 and on average each year around a third of new cases (34%) were in males aged 75 and older. 2 People from an African family background and individuals with a family history of PCa are at higher risk of PCa. 5,6
Prostate cancer might be suspected if any of the following symptoms cannot be attributed to other health conditions: lower urinary tract symptoms, such as frequency, urgency, hesitancy, terminal dribbling and/or overactive bladder; erectile dysfunction; haematuria; lower back or bone pain; lethargy and weight loss.
The descriptor ‘clinically significant’ (CS) is widely used to differentiate PCa that may lead to morbidity or death from types of PCa that do not. This distinction is important as insignificant PCa that does not cause harm is common. 7 Autopsy studies, in men who died of causes other than PCa, indicate that there is a significant prevalence of non-CS prostate in the general male population, which increases with age. 7 PCa screening may therefore lead to overdiagnosis, by identifying cancers that are not destined to cause morbidity or mortality. Men with these cancers are at risk of being harmed by early detection and unnecessary treatment,8,9 such as radical prostatectomy or radiotherapy with no additional mortality benefit compared to an active surveillance approach, which includes regular monitoring of prostate-specific antigen (PSA) levels and digital rectal examination (DRE). On the other hand, individuals with undetected cancer or with lesions incorrectly classed as benign may miss out on relevant treatment. Clinical guidelines have focused efforts to address the risk of overtreatment and undertreatment of PCa, notably with recent updates to diagnosis pathways and refinements to risk stratification of cancer lesions. 10–12
Care pathways for the diagnosis and management of prostate cancer
Referral to suspected cancer pathway
There is no screening programme in the UK for PCa, although PSA testing is available for asymptomatic individuals above 50 years of age requesting this test. 13 For people presenting to primary care with certain clinical signs and symptoms that may indicate PCa, National Institute for Health and Care Excellence (NICE)’s guideline for suspected cancer recognition and referral advises to consider a PSA test and DRE to assess for PCa in men with: any lower urinary tract symptoms (such as nocturia, urinary frequency, hesitancy, urgency or retention) or erectile dysfunction or visible haematuria. 14 The guideline recommends men should be referred using a suspected cancer pathway (for an appointment within 2 weeks) for PCa if their PSA levels are above the age-specific reference range or if their prostate feels malignant (hard, or lumpy) on DRE. The NHS Faster Diagnosis Standard requires that patients are diagnosed or have cancer ruled out within 28 days of being referred urgently by their general practitioner (GP) for suspected cancer,15 and NICE requires that GPs should have direct access to appropriate imaging tests. 16
Figure 1 summarises the EAG’s interpretation of the pathway for the diagnosis and care of individuals with suspected PCa according to NICE guidance (NG) 131 and the NHS timed PCa pathway, which was validated by clinical advisers to the EAG. 12,17
Figure 1.
Diagnostic and care pathway for individuals with suspected prostate cancer. a, per mL of prostate volume. MDT, multidisciplinary team.

Magnetic resonance imaging for suspected cancer
National Institute for Health and Care Excellence’s guideline for diagnosis and management of PCa advises that, in patients with suspected clinically localised PCa, multiparametric magnetic resonance imaging (mpMRI) should be offered as the first-line investigation, but not to those patients who would not be able to have radical treatment. 12 This guidance superseded prior guidance which recommended transrectal ultrasound (TRUS)-guided systematic biopsy as first-line test. Introduced in the 2019 review of the guidelines, the recommendation to offer first-line mpMRI followed the results of PROMIS and PRECISION studies which found a greater negative predictive value (NPV) with mpMRI as first-line diagnostic test compared with the traditional standard-of-care use of TRUS-guided systematic biopsy. 18,19
The results of the MRI can be reported using a 5-point Likert scale as recommended in NICE Guideline 131 (NG131), which estimates the risk that an area seen on the MRI scan may be a cancer or not. The prostate imaging – reporting and data system (PI-RADS) is an alternative to the Likert scale assessment of MRI results. 20–22 Here, each lesion is assigned a score from 1 to 5, with higher scores, usually PI-RADS 4 and 5, indicating a higher likelihood of CS cancer.
Multiparametric magnetic resonance imaging and compliance with National Institute for Health and Care Excellence guidance
Uptake of MRI, prior to biopsy in England and Wales, has significantly increased in recent years, from 37% in 2017 to 87% in 2019. Data from 10 of 14 trusts in Scotland also indicate that uptake of a pre-biopsy bi-parametric MRI (bpMRI) or mpMRI as first-line diagnostic ranged from 75% to 100% across centres, although most trusts have not yet met the new NHS Scotland target of 95%. 23,24 TRUS biopsy is still offered as first-line investigation for some patients, although the practice is becoming increasingly rare. 4 Clinical advice to the EAG noted that in some hospitals, patient presenting with an overtly malignant feeling prostate gland (T4) and high PSA may proceed directly to TRUS and biopsy before having MRI to speed up diagnosis. Reasons for deviating from the recent NICE guidance include challenges in meeting waiting targets and the limited availability of mpMRI slots. The COVID-19 pandemic has also disrupted the implementation of the guidance. 23,24
Clinical advisers to the EAG highlighted that bpMRI is sometimes used in current practice where mpMRI is not available. Although the 2019 National Prostate Cancer Audit (NPCA) indicated that 98% of NHS organisations were able to offer mpMRI on site, challenges in meeting the 28-day diagnostic waiting target have been reported. 25 However, there is no evidence that the accuracy of mpMRI and bpMRI differ in treatment-naive patients. 26
Although uptake of mpMRI as first-line diagnostic test has increased in recent years, it is unclear to what extent this is implemented in the NHS, and whether and to what extent other alternative pathways may be followed.
Biopsy
The decision to collect biopsy samples is informed by the MRI, as well as specific risk factors (such as PSA density, family history and ethnicity) and individual clinician preference. One or more prostate biopsies may be performed to rule out or confirm the presence of PCa. Different methods exist for sampling the prostate tissue. The site(s) for biopsy can be targeted for people who have a suspicious lesion identified by the MRI scan. Tissue samples or cores are only collected from the areas identified in the MRI scan as suspicious. The biopsies can also be systematic, where multiple samples are taken in a systematic fashion from different regions of the prostate according to a predefined scheme rather than guided by the MRI results. A systematic only biopsy approach may be taken for instance where clinical suspicion is high but not reflected in the MRI (typically with a LikApplicability of Urostation to KOELISTrinity is unknown.ert/PI-RADS score of 2 or less), although there is regional variation in this practice.
Prostate biopsies may be performed via the transrectal route or the transperineal route. Both routes use a TRUS probe inserted into the anus to generate a live image of the prostate. With TRUS prostate biopsy, a biopsy needle is inserted through the rectal wall via the anus. TRUS biopsies are usually performed under local anaesthesia, although it can also be carried out under general anaesthesia (e.g. if the patient is unlikely to tolerate the procedure otherwise). In a transperineal biopsy (TP), the biopsy needle is inserted through the perineum. Historically, TPs were always conducted under general anaesthesia. However, recent developments in TP techniques have made the procedure more tolerable, and it is now routinely performed under local anaesthesia. 27 NICE draft guidance has recently recommended local anaesthetic transperineal (LATP) prostate biopsy, using the freehand needle positioning devices PrecisionPoint, EZU-PA3U device, Trinity Perine Grid, and UA1232 puncture attachment, as options for diagnosing PCa. 28,29 Furthermore, patients may receive a spinal block prior to the biopsy being taken, although practice will vary between centres. Spinal anaesthesia may be conducted in an outpatient office30 or operating theatre. 31
When a prostate biopsy is performed, tissue cores from the prostate are obtained for histological examination. The number of cores sampled primarily depends on the biopsy technique, but may also vary based on whether the patient has a previous negative biopsy. In a systematic biopsy, the number of cores sampled can range from 6 to 12 or 14. When more samples are obtained, a greater volume of the prostate gland is sampled, potentially increasing the detection rate. Obtaining any further cores is associated with a limited increase in diagnostic yield,32 but an increased risk in the incidence of complications, such as bleeding (haematuria, haematospermia, haemoejaculate, haematochezia or rectal bleeding), infections [e.g. urinary tract infection (UTI)], pain, urinary retention and erectile dysfunction. 33 In MRI-guided biopsies, fewer cores can be obtained, as sampling can be targeted at the areas where there is a high suspicion of cancer. The NICE guidelines do not specify the number of cores that should be obtained from each suspicious area; European guidelines state that multiple (three to five) biopsy cores per lesions should be taken to reduce the chance of missing or under sampling lesions,34 whereas guidance from the American Urological Association and the Society of Abdominal Radiology’s Prostate Cancer Disease-Focused Panel35 notes that at least two target cores per region of interest should be obtained. Clinical advisers to the EAG indicated that a minimum of two cores per targeted lesion were typically taken in NHS practice, and that for most patients, only one lesion (typically the largest) was targeted.
National Institute for Health and Care Excellence NG131 recommend that a targeted, MRI-influenced prostate biopsy should be offered to people whose Likert score is 3 or more. 10 Currently, MRI-influenced prostate biopsy may use one of three different approaches:
-
cognitive fusion (CF or visual estimation), in which the operator interprets the MRI imaging before the biopsy and manually targets the area of interest using TRUS as a guide; additional samples are also taken in a systematic way according to a pre-defined protocol
-
software fusion (SF), which automatically overlays the MRI image onto the real-time TRUS therefore allowing for real-time visualisation of the area of interest where targeted samples are taken additional samples are also taken in a systematic way according to a pre-defined protocol
-
in-bore biopsy, or ‘in-gantry’ biopsy, a technique that involves performing the prostate biopsy in the MRI scanner, where the diagnostic MRI is fused with real-time MRI using the MR images taken immediately after each needle placement to guide the biopsy.
Cognitive fusion is the current standard of care (SOC). Clinical advisers to the EAG noted that different versions of SF are currently used in a number of NHS centres. In-bore biopsies, and MRI-fusion software that integrates AI-driven diagnosis of PCa, are not used in standard clinical practice.
Software fusion and CF prostate biopsy can be performed with or without the addition of systematic biopsy. The European Association of Urology (EAU) guidelines on PCa recommends combining targeted and systematic biopsy in people with a PI-RADS score of 3 or more who have not had a prior biopsy. 34 In UK clinical practice, after targeting sites of interest for biopsy in eligible people, additional biopsy cores may be taken from the area around the target lesion and a systematic biopsy is performed in addition to the targeted biopsy. Although not strictly recommended by NICE, their guideline on the diagnostic and management of PCa (NG131) notes that most often, MRI-influenced biopsies will be performed in combination with systematic biopsies. 10 However, there is variation in practice dependent on local protocols in terms of whether off-target cores are sampled or not, the number of samples taken and the sampling pattern for the systemic component of combined biopsies. For people whose Likert score is 1 or 2, omitting a prostate biopsy should be considered but only after discussing the risks and benefits with the person and reaching a shared decision. If a person opts to have a biopsy, systematic prostate biopsy (whereby multiple samples are taken in a systematic fashion from different regions of the prostate according to a predefined scheme) is offered. NHS England guidance17 states that people with a Likert or PI-RADS score of 1 or 2 and people with a Likert or PI-RADS score of 3, who also have a PSA density < 0.15 ng (or 0.12 ng in some centres) of PSA per mL of serum per mL of prostate volume may be discharged, taking account of risk factors and patient preferences.
For those patients whose MRI-influenced biopsy is negative, results will be reviewed by a urological cancer multi disciplinary team (MDT), typically including a urologist and a radiologist, and the possibility of significant disease discussed with the patient. However, clinical advice to the EAG noted that in practice, not all hospitals are able to perform a MDT review of all negative MRI-influenced biopsies, in which case results may be sent for individual clinician review. A decision to offer a repeat biopsy is based on individual risk factors, including whether the biopsy showed high-grade prostatic intra-epithelial neoplasia, atypical small acinar proliferation or whether the DRE result was abnormal. 12,17,34 Clinical advice to the EAG noted that factors determining eligibility for, and timing of, repeat biopsy may vary across centres and will depend on individual risk factors, although patients with a negative biopsy and PI-RADS/Likert scores of 4 or 5, larger suspicious lesions on MRI and fitter patients are more likely to undergo repeat biopsy within 12 months. If a repeat biopsy is not offered, patients could instead undergo active surveillance with PSA testing or may be discharged depending on the MRI and histology findings. 17 Patients whose repeat biopsy result is positive may be offered active surveillance or radical treatment, depending on individual patient characteristics and preferences (see Software fusion prostate biopsy). Patients with a negative repeat biopsy may be discharged, or have their PSA levels monitored if cancer is still suspected. Antibiotics, combined with PSA monitoring, may be administered to rule out prostatitis, which may show as false positive on MRI. In some rare cases, further tests such as an additional repeat biopsy, template biopsy, or a positron emission tomography (PET) scan may be conducted to definitely rule out cancer.
Following the biopsy, a pathologist will look at the biopsy samples and assign a Gleason score (GS). The GS is a grading system which estimates the aggressiveness of the PCa, based on the pattern of the cancer cells and the extent of cell differentiation. Gleason grade 1 cells look like normal prostate tissue, and Gleason grade 5 cells have mutated to such an extent that they do not resemble typical prostate cells. A primary grade is given to describe the cells that make up the largest area of the tumour and a secondary grade is given to describe the cells of the next largest area. For example, a GS written as 3 + 4 = 7 indicates that most of the tumour is grade 3 and the next largest section of the tumour is grade 4. The two most common patterns of cells (e.g. Gleason grades 3 and 4) are added together to determine a GS. GSs can range from 2 to 10, with a score of 6 being the lowest grade cancer. To help with outcome prediction and patient communication, GSs ≤ 6, 3 + 4, 4 + 3, 8 and 9–10, respectively, can be reported as five risk groups defined by the International Society of Urological Pathology (ISUP), that is, ISUP grades 1–5, respectively. 36
Although the exact definition of CSPCa varies across studies, it commonly refers to organ-confined cancer above a specific GS (or grade) and maximum cancer core length, indicating PCa that may cause excess morbidity or death. 34 European guidelines state that lesions with a GS between 2 and 6 can be considered clinically insignificant. Recent studies have commonly defined CSPCa as above a GS of 7 (3 + 4), some have used a narrower definition, including above 7 (4 + 3). 19,37–39 Some publications provide more than one definition within a single study, reflecting the lack of consensus and difficulty in defining clinical significance. 40,41
People diagnosed with PCa are assigned a Cambridge Prognostic Group (CPG) risk category. The CPG score is assigned based on the person’s PSA levels, the GS of the lesion(s) (based on histological analysis of the biopsy) and the clinical stage of the disease. 10 The EAU guidance states that further tests, such as abdominopelvic imaging and bone scans, may be required to determine clinical stage of the disease when there is suspicion that the cancer has spread to the lymph nodes or the bone marrow. 34 The CPG risk category and definition is described in Table 1.
| CPG | Risk category | Definition |
|---|---|---|
| CPG 1 | Low risk | GS 6 (GG 1) AND PSA < 10 ng/ml AND stages T1–T2 |
| CPG 2 | Favourable intermediate risk | GS 3 + 4 = 7 (GG 2) OR PSA 10–20 ng/ml AND stages T1–T2 |
| CPG 3 | Unfavourable intermediate risk | GS 3 + 4 = 7 (GG 2) AND PSA 10–20 ng/ml AND stages T1–T2 OR GS 4 + 3 = 7 (GG 3) AND stages T1–2 |
| CPG 4 | High risk | One of: GS 8 (GG 4) OR PSA > 20 ng/ml OR Stage T3 |
| CPG 5 | Very high risk | Any combination of: GS 8 (GG 4), PSA > 20ng/ml or Stage T3. OR GS 9–10 (GG 5) OR Stage T4 |
These risk categories, along with the outcome of discussion with patients regarding the benefits and harms of the treatment options, determine which treatment option is chosen. This ranges from active surveillance, for patients with CPG 1 or 2, to radical prostatectomy or radical radiotherapy for people with localised cancer and CPG ≥ 2. Patients with locally advanced PCa and CPG 4 or 5 may also be offered docetaxel (DTX) chemotherapy. The recommendation to use the CPG five-tier risk prediction model was included in the NICE NG131 2021 update10 and superseded a three-tier risk classification model including low-, intermediate- and high-risk/locally advanced groups, which did not differentiate between favourable intermediate risk (CPG 2) and unfavourable intermediate risk (CPG 3). Another important difference between the two classifications is that CPG 1 includes more men than the low-risk group in the previously recommended risk classification; some men who previously would have been in the intermediate-risk group are now classified as CPG 1. This change in risk prediction model aims to reduce under- and over-treatment in people who are at either end of the tiers, following evidence from the NICE’s surveillance programme that indicated that active surveillance may not be appropriate in patients with unfavourable intermediate PCa, and that patients with favourable intermediate risk and lower risk may be over-treated. 10,12,42,43
Software fusion prostate biopsy
Using a digital overlay, SF biopsies allow operators to view a real-time ultrasound image alongside the patient’s MRI. This requires a period of preparation, to obtain and annotate the MRI images prior to biopsy. 44 MRI images are first downloaded onto a dedicated processing software before they are annotated by contouring the edge of the prostate and the regions of interest. Clinical advice to the EAG suggests that, for an experienced practitioner, this contouring can take around 5–7 minutes. The annotated MRI scans are then uploaded onto a fusion software platform and are fused with the real-time ultrasound image. Updates to the fusion software are possible, and, depending on the fusion device, are covered by a service contract or can be purchased with a one-off payment.
Use of SF prostate biopsy systems may potentially improve detection rate of CSPCa compared with CF, while reducing the number of samples taken, potentially reducing pain and risk of sepsis associated with the procedure. It could improve the accuracy of assignment of prognostic scores such as Gleason, which influences subsequent treatment and associated patient outcomes. The technology could reduce the number of repeat biopsies for those patients with a negative index biopsy, avoiding unnecessary travel and anxiety for the person. Some fusion technologies also allow operators to keep records of previous biopsy sites to allow the urologist to return to those areas with greater precision for follow-up or additional testing.
However, the accuracy of a prostate biopsy may be impacted by a number of factors. Movement during the procedure (which could stem from patient pain),45 operator experience,46 difference in bladder size or prostate deformation may impact the accuracy of the biopsy, as the MRI image may not accurately reflect the prostate shape at the point of biopsy. Mechanisms using ‘elastic’ prostate registration, where the MRI image alters to fit the ultrasound image, have been designed to account for prostate deformation and allow for more accurate targeting of the lesions of interest. 47 Errors during the fusion of images, specifically incorrect image registration or discordance between the MRI and ultrasound image planes, especially around the base of the prostate, can lead to biopsy failure. 48
The mechanism by which SF techniques may lead to improved accuracy relates notably to a better targeting of suspicious prostate lesions, including in locations that are more challenging to diagnose, such as anterior and posterior lesions. 49,50 However, evidence for the accuracy of SF biopsy systems compared with CF methods is limited. Watts et al. 51 and Sathianathen et al. 52 found no statistically significant difference between SF and CF in PCa detection, while Bass et al. 53 found no evidence that SF was superior to CF at detecting CSPCas. An older review found that SF biopsies detect more CS cancers, using fewer biopsy cores. 54 Between-study heterogeneity ranged from moderate51 to high,53 although review methods and selection criteria varied.
Prostate cancer management: active surveillance, watchful waiting and radical treatment options
Active surveillance is a monitoring strategy for people with localised PCa for whom radical treatments (such as radical prostatectomy or radical radiotherapy) are suitable; it allows avoiding or deferring these treatments when disease progression is likely to be slow, while maintaining the possibility to initiate timely curative treatment. Current NICE guidance suggests a schedule of active surveillance involving regular monitoring of PSA levels and kinetics, and annual DREs. Reassessment with mpMRI and/or re-biopsy can be triggered if concerns about clinical or PSA changes emerge at any time during active surveillance; a positive result (GS 3 + 4 or above) on re-biopsy would then result in offering radical treatment.
For people with CPG 1, active surveillance is offered (radical treatments can be considered if active surveillance is not suitable or acceptable to the person). For people with CPG 2, a choice between radical radiotherapy with androgen deprivation (anti-hormone therapy), radical prostatectomy or active surveillance is given. For people with CPG 3, localised PCa, radical prostatectomy or radical radiotherapy with androgen deprivation is offered, and active surveillance can be considered for people who choose not to have immediate radical treatment. This recommendation is informed by a randomised trial that found that PCa-specific mortality is low (approximately 1%) at 10 years follow-up and does not differ significantly between active surveillance, prostatectomy or radical radiotherapy in individuals with localised PCa, although surgery and radiotherapy resulted in lower incidences of disease progression and metastatic disease compared with active monitoring. Radical prostatectomy may also be associated with worse urinary and erectile dysfunction outcomes compared with active surveillance and radical radiotherapy at up to 6 years follow-up. 55 People with CPG 4 and 5, localised or locally advanced PCa, should be offered a combination of radical radiotherapy and androgen deprivation. Evidence from an individual patient data (IPD) meta-analysis shows that the addition of androgen deprivation therapy (ADT) to radiotherapy significantly improves metastasis-free survival. 56 Brachytherapy (a form of radiotherapy where radiation is directly targeted on the tumour by inserting radioactive pellets into the prostate) in combination with external beam radiotherapy should also be considered for people with CPG 2, 3, 4 and 5 localised or locally advanced PCa. 57 Randomised controlled trial (RCT) evidence shows a reduction in biochemical failure (such as local recurrence or distant metastases) associated with the use of low-dose-rate brachytherapy plus external beam radiotherapy at 6.5 years follow-up for people with high-risk (CPG 4 and 5) localised PCa. 58
Radical prostatectomy or radical radiotherapy is offered to people with CPG 4 and 5 localised and locally advanced PCa, when it is likely that the person’s cancer can be controlled in the long term. DTX chemotherapy may also be considered for these patients. This recommendation follows RCT evidence indicating that clinical progression-free survival (PFS) was prolonged in individuals with hormone-sensitive high-risk PCa receiving DTX compared to standard care alone. 59–61
Finally, some patients with metastatic disease, where the cancer has spread outside the prostate may still undergo targeted biopsy to aid decision-making for localised treatment where the patient may receive some symptomatic benefit.
People with localised PCa, who do not wish to undergo potentially curative treatment with radical prostatectomy or radical radiotherapy (or for whom this is not suitable), can be managed with watchful waiting. This is a monitoring strategy that aims to achieve disease control rather than cure. It is less formal and intensive than active surveillance and involves fewer tests (e.g. typically an annual PSA level measurements not leading to a MRI or biopsy10) and is more likely to be offered to older, frailer populations. With watchful waiting, treatment is generally only considered in response to symptoms. Since MRI as first-line test is only recommended for patients fit for radical treatment, only a small subset of patients who received a MRI for suspected prostate lesions, such as those with worsening health since initial investigation and a PCa diagnosis, are expected to undergo watchful waiting in practice. Some patients who are not fit enough or eligible for curative treatment may also be offered a MRI because their lack of eligibility for radical treatment is not identified prior to undergoing imaging.
Description of technologies under assessment
This assessment will evaluate SF technologies matching the following criteria:
-
intended for use in people with suspected PCa
-
available in the UK
-
holds a CE-mark
-
compatible with MRI scanners of 1.5 tesla field strength or above.
This includes; ARTEMIS (InnoMedicus ARTEMIS), BioJet (Healthcare Supply Solutions Ltd), BiopSee (Medcom), bkFusion (BK Medical UK Ltd and MIM Software Inc), Fusion Bx 2.0 (Focal Healthcare), FusionVu (Exact Imaging), iSR’obotTM Mona Lisa (Biobot iSR’obot), KOELIS Trinity (KOELIS and Kebomed) and UroNav Fusion Biopsy System (Phillips). Table 23, Appendix 1, presents a brief summary of the characteristics of these nine technologies.
Software fusion devices can have a variety of different features, which means they vary in the way in which they operate.
-
Positioning of the ultrasound probe: An ultrasound probe can be cradled and held stationary using a device called a stepper which is attached to a workstation (also known as a stabilised approach). It can be supported by a semi-robotic arm, which allows for the ultrasound probe to be manoeuvred, while maintaining a stable pressure on the prostate. The semi-robotic arm can be used as a stepper for stabilised biopsies or can allow complete freedom of movement for use during a freehand biopsy. Finally, the ultrasound probe can be held by hand (using a freehand technique).
-
Core sampling technique: Different techniques can be used to take the cores, especially in the case of transperineal biopsies. First, a grid or template can be used, which is attached to a stepper and placed in front of the perineum. The grid is marked with a number of holes, which correspond to a letter and a number to allow for multiple cores to be taken in a systematic way. Alternatively, a coaxial needle can be used. In this technique, a larger introductory needle is used to puncture the perineum before the biopsy needles is passed through. This biopsy needle can be angled to take multiple biopsies without creating multiple puncture wounds to the perineum. The coaxial needle is used with the freehand technique, where it is attached to the ultrasound probe, or in a double freehand technique, where the needle is held by hand.
-
Image registration: During SF, the mpMRI images are fused with the ultrasound images during the biopsy procedure. The mpMRI image can be fixed (known as rigid registration) and will not move when the prostate is deformed, either by patient movement or by the insertion of a needle; or elastic, which means the mpMRI image adjusts to match the ultrasound image to account for prostate deformation.
A description of the principal features of the technologies is given in Appendix 1.
Other interventions
‘In-bore’ biopsy, or ‘in-gantry’ biopsy, is a technique that involves performing the prostate biopsy in the MRI scanner, using the MR images taken immediately after each needle placement to guide the biopsy. The use of in bore MRI and artificial intelligence (AI)-driven software are beyond the scope of this assessment.
Place of the intervention in the diagnostic and care pathway
Software fusion targeted biopsy, for people with suspected PCa, takes place at the same two points in the diagnostic pathway as targeted CF biopsy, the current SOC.
Patients having a first targeted biopsy
Software fusion biopsy (with or without systematic biopsy) would be offered as an alternative to targeted CF biopsy to people with a Likert/PI-RADS score of 3 or more following a MRI, after having been referred to secondary care with suspected PCa (with PSA levels above the age-specific reference range or those whose prostate is suspicious of malignancy based on rectal examination). Clinical advisers to the EAG indicated that biopsy-naive patients represented the large majority (more than 90%) of patients with suspected PCa undergoing targeted biopsy.
Patients having a repeat targeted biopsy
Patients offered a repeat biopsy, following a prior negative biopsy, could also be offered a SF biopsy as an alternative to targeted CF. As discussed in Care pathways for the diagnosis and management of prostate cancer, NG131 recommends that an MDT decides on whether to offer a repeat biopsy based on individual risk factors, although not all centres may be able to perform a MDT review of all negative MRI-influence biopsies, and eligibility and timing of repeat biopsy may vary in practice. In clinical practice, repeat biopsies are likely to be offered to patients whose mpMRI results were not consistent with the biopsy (i.e. mpMRI of 4–5 and no PCa detected on biopsy). NG131 does not recommend repeat MRI for patients requiring a repeat biopsy; instead a repeat targeted biopsy can be conducted based on the initial MRI report. EAG clinical advisers suggested this subgroup would make up <10% of patients with suspected PCa.
Potential pathway positions out of scope for the current assessment
Although SF may also be used to monitor patients and inform treatment for individuals with a PCa diagnosis in active surveillance, this population is beyond the scope of this assessment.
Relevant comparator
The comparator for this assessment is targeted transperineal or transrectal prostate biopsy using CF with or without systematic biopsy, under local or general anaesthesia, in which the operator interprets the MRI imaging before the biopsy and manually targets the area of interest using TRUS as a guide. Clinical advisers to the EAG highlighted that the expertise of the person performing the biopsy may affect the accuracy and procedure time of CF.
Chapter 2 Aims and objectives
The aim of the study was to assess the clinical and cost-effectiveness of SF biopsy systems in people with suspected localised and locally advanced PCa, by addressing the following protocol-specified objectives:
Clinical effectiveness
-
To perform a systematic review of the diagnostic accuracy and clinical efficacy of nine SF systems compared with CF targeted biopsy and with each other, in people with suspected PCa who have had a MRI scan that indicates a lesion.
-
To compare the diagnostic accuracy of different SF biopsy systems with each other and with CF targeted biopsy in people with suspected PCa who have had a MRI scan that indicates a lesion using meta-analytical methods and to combine the diagnostic accuracy of different SF systems where appropriate.
-
To perform a narrative systematic review of the clinical efficacy, safety and practical implementation of SF targeted biopsy. This includes assessment of intermediate outcomes, mortality and morbidity, patient-centred outcomes, adverse events (AEs), and acceptability to clinicians and patients.
Cost-effectiveness
-
To conduct a systematic review and critical appraisal of relevant cost-effectiveness evidence of the use of SF biopsy systems compared to CF for targeted biopsy in people with suspected PCa who have had a MRI scan indicating a lesion.
-
To develop and validate a decision-analytic model to estimate the cost-effectiveness of SF targeted biopsy systems in people with suspected PCa who have had an MRI scan indicating a lesion compared to targeted biopsy using CF. This will require linking intermediate outcomes, such as the diagnostic accuracy of SF biopsy systems to subsequent management decisions and to final health outcomes including morbidity and mortality associated with alternative treatment options. The analysis will take the perspective of the NHS and Personal Social Services (PSS), consistent with the current manual for health technology evaluations by the NICE. Final health outcomes will be evaluated in terms of quality-adjusted life-years (QALYs).
-
To populate the model using the most appropriate available evidence. This evidence is likely to be identified from published literature, routine data sources and potentially using data elicited from relevant clinical experts and companies.
-
To estimate the incremental cost-effectiveness of the SF biopsy systems compared to the current SOC for the population of interest (CF biopsy), based on an assessment of long-term NHS and PSS costs and quality-adjusted survival. The time horizon of the model will be sufficient to capture both the short-term and longer-term outcomes.
-
To characterise the parameter uncertainty in the data used to populate the model and present the resulting uncertainty in the results to decision-makers. To this purpose, we will perform comprehensive (probabilistic and deterministic) sensitivity analyses varying parameter inputs, and structural assumptions of the model, as appropriate.
-
Where possible and applicable, to assess the impact of potential sources of heterogeneity on cost-effectiveness, including subgroup analyses (e.g. patients with previous negative biopsy results within 12 months) and consideration of other factors that may affect diagnostic accuracy.
Chapter 3 Assessment of diagnostic accuracy and clinical effectiveness
This section presents the methods and results of the systematic review of diagnostic accuracy and clinical effectiveness. Systematic review methods (study selection, data extraction, quality assessment) details the systematic review methods, and Data synthesis methods presents the data synthesis methods. Quantity and quality of evidence summarises the quantity and quality of evidence included in the systematic review, Diagnostic accuracy results presents the diagnostic accuracy results of the systematic review and meta-analysis; results for all other outcomes included in the systematic review are presented in Clinical effectiveness results. Diagnostic accuracy and clinical effectiveness: summary and conclusions summarises the key findings from the systematic review, and Additional evidence to inform model structure and parameterisation presents a summary of additional evidence identified to inform the economic model.
Systematic review methods (study selection, data extraction, quality assessment)
Searches
The aim of the literature search was to systematically identify published and unpublished studies of prostate biopsies utilising either SF or CF.
An information specialist (MH) developed a search strategy in Ovid MEDLINE using textword searches of the title and abstracts of database records along with relevant subject heading searches. The search strategy consisted of: (1) terms for PCa AND, (2) terms for MRI AND, (3) terms relating to fusion techniques AND, (4) terms for prostate biopsy. The terms used to describe fusion techniques were found to vary in the literature with some articles lacking any terms for fusion techniques in the title, abstract or subject headings of the database record. Therefore, related terms such as targeted biopsy, focal biopsy or MRI-guided biopsy were added to the strategy along with some proximity searching to capture phrases in the title and abstracts of records around the use of MRI prior to a prostate biopsy. Named SF software and hardware were also included in the strategy (e.g. Fusion Bx, BioJet, KOELIS Trinity, bkFusion).
A date limit was applied (from 2008 onwards), due to the relatively recent nature of the technologies under assessment, and as informed by results of scoping searches and previous systematic reviews. 51,53,62,63 No language or study design restrictions were applied to the searches. The MEDLINE strategy was agreed with the review team and checked by a second information specialist using aspects of the PRESS checklist. 64 The final MEDLINE strategy was adapted for use in all resources searched.
The following databases were searched in May 2022: MEDLINE ALL (Ovid), Cochrane Controlled Register of Trials (Wiley), Cochrane Database of Systematic Reviews (Wiley), Cumulative Index to Nursing and Allied Health (Ebsco), Database of Abstracts of Reviews of Effects (CRD databases), EconLit (Ovid), EMBASE (Ovid), Health Technology Assessment (HTA) database (CRD databases), Health Management Information Consortium (Ovid), International Health Technology Assessment (INAHTA) database, Latin American and Caribbean Health Sciences Literature (LILACS) database, NHS Economic Evaluation Database (CRD databases), and Science Citation Index (Web of Science).
Further ongoing and unpublished studies were identified through searches of: ClinicalTrials.gov, Conference Proceedings Citation Index: Science (Web of Science), European Union Clinical Trials Register, Open Access Theses and Dissertations, Proquest Dissertations and Theses A&I, PROSPERO, and World Health Organization (WHO) International Clinical Trials Registry Platform portal.
A search for relevant guidelines was carried out via the following websites: NICE, ECRI Guidelines Trust, Guidelines International Network (GIN) international guideline library and the Trip database. Full search strategies for all resources can be found in Appendix 2.
Additionally, company websites were searched to identify relevant publications and other materials relating to the technology, and companies registered with NICE at the time of the protocol submission were contacted for further details about their respective technologies. Reference lists of included studies and relevant systematic reviews were scanned to identify any further potentially relevant studies.
An update search was carried out on 2 August 2022 to capture any recently published studies. The update search was undertaken on the following four databases: MEDLINE ALL (Ovid), Cochrane Controlled Register of Trials (Wiley), Embase (Ovid) and the Science Citation Index (Web of Science). Search results were downloaded from each database and added to the EndNote library of original search results for deduplication.
Selection criteria
All titles and abstracts were screened independently by two reviewers (AL and LB). Full-text papers of any titles and abstracts deemed to be relevant were obtained where possible, and the relevance of each study assessed independently by two reviewers according to the criteria below. Disagreements were resolved by consensus, or where necessary, by consulting a third reviewer. Conference abstracts were considered to be eligible if they provide sufficient information for inclusion, and attempts were made to contact authors for further data. The eligibility criteria that were used to identify relevant studies are listed below.
Population
People with suspected PCa who have had a MRI scan that indicates a significant lesion (Likert or PI-RADS score of 3 or more). This included people who were biopsy naive and those who are referred for a repeat biopsy following a previous negative prostate biopsy. No time limit since the first negative biopsy was set for inclusion of studies including patients with repeat biopsies, although applicability with respect to the scope was considered as part of the quality assessment.
Studies primarily focused on people who do not have a lesion visible on their magnetic resonance image, people on an active surveillance care pathway, and people with relapsing PCa were excluded. Patients who could not have a MRI scan were also excluded. Studies including a small subset of individuals with a Likert or PI-RADS score of 2 or less were included if they provided data primarily for the eligible population; their applicability was assessed during quality assessment.
Interventions
Studies evaluating SF alone or in combination with CF or systematic biopsy, under local or general anaesthesia were eligible. No exclusions were made based on the biopsy route. The included SF technologies are described in Appendix 1. Where applicable, earlier versions of these technologies were also included, and their applicability was accounted for during quality assessment.
Comparators
Eligible comparators were targeted transperineal or transrectal prostate biopsy using CF with or without systematic biopsy, under local or general anaesthesia. Although systematic biopsies and ‘in-bore’ biopsies are outside the scope of this review, studies that evaluate these methods were included if they provide separate data to compare targeted biopsies using SF against CF. Studies evaluating several SF technologies against one another were also eligible for inclusion.
Reference standard
Total cancer cases in diagnostic accuracy studies are commonly identified using a combination of SF, CF and systematic biopsies as ‘reference standard’. 51,53
In those studies, diagnostic accuracy estimates of SF and CF are therefore inherently dependent on the accuracy of mpMRI, TRUS and fusion approaches, as well as the accuracy of the biopsy method, which may vary by type and route. Reference standards that use histopathology from biopsy samples, rather than radical prostatectomy, may also miss positive cases. Reference standards that include results from samples identified by SF and/or CF are at risk of incorporation bias (when results of an index test are used to establish the final diagnosis). Reference standards that use histopathology from radical prostatectomy are usually only reported for those who have been classified as high risk and have had radical prostatectomy. In addition, histopathology, although commonly used as the gold standard test for cancer detection and grading, may also misclassify a small proportion (approximately 2%) of negative PCa cases as positive. 65
Template-guided biopsy, including transperineal template-guided mapping biopsy (TTMB), also called template-guided saturation biopsy (TSB), is seen as a more optimal reference standard, compared with standard 12-core systematic biopsy. TTMB is a transperineal TRUS-guided biopsy of the prostate using a 5-mm brachytherapy grid, with at least one biopsy from each hole. TSB includes 20 or more transperineal or transrectal TRUS-guided biopsies of the prostate performed to comprehensively sample the whole prostate, according to a predefined core distribution pattern. Template-guided biopsies using a uniform grid and taken at 5 mm intervals can technically only miss tumours that are smaller than the distance between the adjacent cores. 66 Although template-guided biopsy is imperfect, notably due to the fact that test accuracy depends on the intensity of cores taken and core trajectory,66 it is superior to standard systematic biopsy as a reference standard as it aims to comprehensively sample all zones of the prostate. However, template-guided biopsies are invasive and may not be used in diagnostic accuracy studies, therefore combinations of reference standards with lower diagnostic accuracy (e.g. CF with SF and systematic biopsies with fewer than 20 cores) were also eligible for inclusion.
A positive biopsy was defined as histopathological confirmation of one of the target conditions within the biopsy cores.
Outcomes
The following intermediate outcomes were eligible:
-
measures of diagnostic accuracy (including sensitivity, specificity, test positive/negative rates)
-
cancer detection rates (number of patients with detected cancer by SF or CF divided by the total number of patients with confirmed cancer)
-
CS cancer detection rates (all definitions)
-
clinically insignificant cancer detection rates (all definitions)
-
cancer detection rates by prognostic score (such as CPG 1 to 5 or other similar classification that can be mapped into the CPG classification) and/or GS
-
biopsy positivity rate (ratio of positive biopsies out of total number of biopsy samples)
-
biopsy sample suitability/quality
-
number of biopsy samples taken
-
procedure completion rates
-
software failure rate
-
time to diagnosis
-
length of hospital stay (emergency department and inpatient stay)
-
time taken for MR image preparation
-
time taken for biopsy procedure
-
number of repeat biopsies within 12 months
-
subsequent PCa management (such as no treatment, active surveillance, radical prostatectomy, radical radiotherapy and hormone therapy).
The following clinical outcomes were eligible:
-
rates of biopsy-related complications and AEs, including infection, sepsis and haematuria, urinary retention, erectile dysfunction, and bowel function
-
hospitalisation events after biopsy
-
survival
-
PFS
-
AEs from treatment.
Patient- and carer-reported outcomes were eligible, including:
-
health-related quality of life (HRQoL)
-
other self-reported outcomes including tolerability, embarrassment and loss of dignity.
The following implementation end points were eligible:
-
operator preferences
-
barriers and facilitators to implementation.
The following cost outcomes were eligible:
-
costs of MRI fusion software and any proprietary hardware (including the workstation, ultrasound systems, probe holders, replacement parts, consumables such as guides, and maintenance)
-
cost of staff time (including MR image interpretation time and biopsy procedure time) and of any associated training
-
medical costs arising from the biopsy such as anaesthetic, sedation, hospital admissions and stays
-
costs related to using intervention (including any time analysing and storing data)
-
costs of histopathology biopsy samples analysis
-
cost of treatment of cancer (including costs of any AEs)
-
costs relating to follow-up
-
costs of subsequent biopsies
-
costs arising from watchful waiting
-
costs arising from active surveillance.
Study designs
Prospective studies comparing SF against CF biopsy that report the results of both SF and CF biopsy separately were considered. Studies including within-patient comparisons (where SF and CF biopsy are compared within the same patient) and between-patient comparisons (where participants receive either SF or CF biopsy) were included.
Where no prospective evidence could be found to inform the diagnostic accuracy of an eligible SF technology, retrospective studies that met all other selection criteria were included.
No restriction by healthcare setting was made.
Indirect evidence
Where the interventions of interests did not form a connected network to allow comparison of each intervention against every other, prospective, within-patient comparisons or RCTs between SF and systematic biopsy, and between CF and systematic biopsy, were also eligible to inform indirect comparisons, provided they reported numbers or rates of patients with no cancer (NC), all PCa and CS cancers for either SF or CF against systematic biopsy or template biopsy, and the combination of software or CF with systematic biopsy or template biopsy.
Data extraction
Information on study details (including study design, sample size), patient characteristics (e.g. age, PSA, PI-RADS/Likert score and version, reason for referral, whether first biopsy, repeat biopsy and lesion location), intervention characteristics (including SF technology type and version, MRI technology and magnet strength, biopsy route (transrectal or transperineal) whether the procedure used fixed/free hand; local/general anaesthetic and was based on biparametric or mpMRI, the use and number of targeted and systematic core biopsy samples, operator experience), outcomes data and definitions of outcomes were extracted by at least one reviewer (AL or LB) using a standardised data extraction form and independently checked by a second reviewer (AL or LB). Discrepancies were resolved by discussion, with involvement of a third reviewer (SD) where necessary.
Where required and appropriate, attempts were made to contact companies for additional information, including unpublished data, missing data, relevant subgroup data and more granular outcome data (e.g. matrices reporting a breakdown of detection rates by cancer prognostic score). Data from relevant studies, with multiple publications, were extracted and reported as a single study. The most recent or most complete publication were used in situations where the possibility of overlapping populations could not be excluded. Where not reported (NR), rates of clinically insignificant cancers were imputed by subtracting the number of CS cancers from the total number of cancers detected (as per Bass, et al.). 53
Critical appraisal
The quality of the diagnostic accuracy studies was assessed using the tools Quality Assessment tool of Diagnostic Accuracy Studies (QUADAS)-2 and QUADAS-C tools. 67,68 The QUADAS-2 tool evaluates both risk of bias (associated with the population selection, index test, reference standard and patient flow) and study applicability (population selection, index test and reference standard) of individual studies to the review question. The QUADAS-C tool is designed to assess risk of bias in test comparisons undertaken in studies that evaluate two or more index tests. QUADAS-C is an extension of QUADAS-2 and includes all domains covered by QUADAS-2. Each QUADAS-C domain is informed by each QUADAS-2 judgement for each test and additional signalling questions that are specific for comparisons to produce a risk of bias judgement for the comparison. The quality assessment focused on the risk of bias and applicability of cancer detection outcomes only. Since the review focused on the relative accuracy of two index tests, QUADAS-2 risk of bias assessments were not presented. All studies were quality assessed and checked by a second reviewer. Disagreements were resolved through discussion. Decisions with rationale for judgements were presented in tables.
Data synthesis methods
Meta-analysis
The meta-analyses aimed to compare four types of prostate biopsy approaches: CF, SF, CF with concomitant systematic biopsies, and SF with systematic biopsies. When relative effects comparing more than one intervention are of interest, a network meta-analysis (NMA) should be conducted to allow comparison of all interventions to each other. 69 NMA is an extension of pairwise (two-treatment) meta-analysis to allow comparisons across more than two treatments by producing relative effects for every pair of treatments in a connected network. Direct evidence from studies comparing two interventions directly is pooled with indirect evidence from studies that have a common comparator thus allowing consistent estimates of relative effects to be produced that account for all relevant evidence and are typically more precise. Common- (fixed-) or random-effects models can be used. 70
Since many studies compared one or more of the four biopsy types of interest to systematic biopsy alone, this biopsy type was also included in the network of interventions in order to allow more comparisons to be made and to increase precision in the estimated relative effects. 69
Network meta-analyses were conducted using a Bayesian framework estimated through Markov chain Monte Carlo methods. In an attempt to minimise bias, only prospective studies reporting within-patient comparisons, or RCTs reporting comparative results for two or more of the interventions of interest (SF, CF, systematic biopsy or a combination of software/CF with systematic biopsy), were included in the synthesis.
Model convergence was assessed by running two independent chains with different starting values looking at history plot and through inspection of Gelman–Rubin diagnostic plots. Due to data sparseness (few studies per comparison and not all studies reporting all outcomes) only fixed-effects models were fit to the data. Model fit was assessed by comparing the mean total residual deviance to the number of independent data points contributing to the analysis. 71
Network plots were drawn in R72 using the netmeta package. 73
Multinomial synthesis model
To adequately distinguish between the different biopsy methods and SF devices, it is necessary not only to describe how they differ in classifying patients as having PCa or not, but also how they differ in classifying patients as having PCa at different Gleason grades, as that determines further treatment strategies. To inform post-biopsy patient management in the economic model, data are modelled by ISUP grade, where reported.
In order to best describe the differences between biopsy methods for each diagnostic category, a multinomial logistic regression model was fitted where the odds of being categorised in each of the different categories in Table 2 compared to the reference category (no PCa) are allowed to vary by biopsy type. This model is conceptually equivalent to four binomial logistic regressions comparing category r > 1 with category 1 (no PCa), for each different biopsy type compared to the reference, cognitive biopsy.
| Categories | Gleason | ISUP grade |
|---|---|---|
| 1 | – | NCa |
| 2 | 3 + 3 | 1 |
| 3 | 3 + 4 | 2 |
| 4 | 4 + 3 | 3 |
| 5 | 8 – 10 | 4–5 |
The multinomial logistic regression model accounts for the ordered nature of the categories, which is important since a higher or lower detection of higher-grade cancers may have an impact on the cost-effectiveness of each device. However, the model does not take into account that some of the included studies reported results from different biopsies techniques performed on the same patients. 74 The study arms are treated as independent. This is a limitation of this model, which may inflate the uncertainty in the estimates. Models and code that can incorporate non-independent data (measured on the same patients) with ordered categories are not readily available.
Studies that only report the number of individuals in collapsed categories, for example the number of individuals with NC, non-CS cancer (Gleason 3 + 3) and CS cancer (Gleason > 3 + 3) provide information only on the odds ratio of being classified in the first two categories (NC, non-CS cancer). The model has been adapted to allow these studies to be included. However, they provide only limited information to the network compared to studies that report a finer breakdown of GSs.
Models were fitted in WinBUGS 1.4.3. 75 CF prostate biopsy was chosen as the reference intervention, and ‘no cancer’ as the reference category. Full details of the model and WinBUGS code are given in Appendix 3.
The relative effects produced by the model are the odds ratios for being classified in category r, instead of category 1 (‘no cancer’), using intervention X (SF, systematic biopsy or a combination of software/CF with systematic biopsy), compared to cognitive fusion biopsy. Interpretation of these relative effects is complex since it relates to both a reference treatment and reference category. To aid interpretation, absolute probabilities of being classified in each category, using each intervention, are also reported. Details of how these are calculated are given in Appendix 3.
Analyses are presented assuming all SF devices share a common effect, that is they all have the same odds ratio compared to CF biopsy (Model 1a) and assuming individual device effects (Model 1b).
Cancer detection network meta-analysis models
The odds ratios of cancer detection, for different biopsy methods compared to each other, were also pooled. The number of cancers detected were modelled using the NMA model for binomial data with a logit link described in NICE technical support document 2,71 fitted in R72 using the package gemtc. 76
Model convergence was assessed through inspection of Gelman–Rubin diagnostic plots. Both fixed-effect and random-effect models were fitted to the data. Non-informative prior distributions were used for all effect parameters and a Uniform (0,5) prior distribution was selected for the between-study standard deviation (SD) in random-effects models. 71 Model fit was assessed through mean total residual deviance and inspection of residual deviance contribution for each study arm. Heterogeneity was assessed by inspecting the size of the between-study SD and its 95% credible interval (CrI), and by comparing the Deviance Information Criteria (DIC) for fixed-effect and random-effects models. Where DIC differed by < 3 points the simplest model (fixed effect) was chosen. Consistency between direct and indirect evidence was assessed by fitting an unrelated mean effects model and where that suggested potential inconsistency, further investigation of the location of inconsistency was carried out by fitting node-split models. 77
Any cancer detection network meta-analysis
The odds ratios of detecting any PCa (both CS and non-CS, i.e. Gleason ≥ 3 + 3) for different biopsy methods compared to each other were pooled. Analyses are presented assuming all SF devices share a common effect (Model 2a) and for individual device effects (Model 2b).
Clinically significant cancer detection NMA model
The odds ratios of detecting CSPCa (Gleason > 3 + 3), as opposed to NC or Gleason 3 + 3, for different biopsy methods compared to each other were also pooled, for studies that reported it. Analyses are presented assuming all SF devices share a common effect (Model 3a) and for individual device effects (Model 3b).
Narrative synthesis
Results of studies that were not eligible for inclusion in the NMAs, and results of all studies reporting protocol-specified outcomes other than diagnostic accuracy, were synthesised narratively following published guidelines. 78
Outcomes were presented following the order listed in the protocol, then by comparison. Effect estimates, including metrics, measures of variance, statistical significance (at conventional threshold of p = 0.05), and direction of effect were presented narratively and/or in tables at patient-level, unless only data per lesion could be extracted. Studies were grouped based on direction of effect and statistical significance. Where NR, outcomes including detection rates, test positive rates and biopsy positivity rates were imputed. No formal statistical methods were used to assess heterogeneity. Results were narratively compared with the meta-analyses, and limitations of the evidence (e.g. inconsistency, risk of bias) informed findings summaries and conclusions.
Quantity and quality of evidence
Figure 2 presents an overview of the study selection process in a Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram. The literature searches identified a total of 6289 unique records. After title and abstract screening, 247 references were retrieved and a total of 23 unique studies were included in the systematic review. 31,79–101 Fourteen studies were included in the quantitative synthesis,31,79,80,82,84,86–88,92–94,96,97,99 while nine studies were included in the narrative synthesis only. 81,83,85,89–91,95,98,100,101
FIGURE 2.
Study selection process (PRISMA flow diagram).

Evidence was included for all SF technologies specified in the scope and protocol (all versions) except for Fusion Bx (Focal Healthcare) and ExactVu (Exact Imaging). Report Supplementary Material 1 presents a summary of the evidence for Fusion Bx and ExactVu that was considered for inclusion and ultimately excluded, and a list of studies excluded from the systematic review, grouped by reason for exclusion.
Description of studies included in the systematic review of diagnostic accuracy and clinical effectiveness
Table 25, Appendix 4, presents the characteristics of the 23 studies included in the systematic review. The majority of studies were conducted in Europe,31,80,81,83,86,89,92–95,98,101 and five studies were conducted in the USA. 87,88,90,96,97
Twelve studies compared SF against CF; of those, three used a within-patient comparison design (where participants underwent biopsy with both SF and CF within the same session),88,93,97 and nine compared separate cohorts who received either SF or CF biopsy (between-patient design). 31,82,85,89,90,95,98,100,101 Three studies compared two or more SF software against one another. 79,86,99 Five studies compared SF against systematic biopsy,80,87,92,94,96 and three studies compared CF with systematic biopsy. 79,86,99
Three RCTs were included; of those, two compared SF against CF,31,82 and one compared three SF devices. 83 All other studies were non-randomised trials or observational; of those, four studies used a retrospective design. 85,90,100,101
The following SF technologies were evaluated: ARTEMIS (five studies),82,84,88,96,97 BioJet (four studies),81,83,84,89 BiopSee (two studies),31,95 BK (two studies, referred to as Predictive Fusion Software in one study85 and MIM fusion software in another),100 iSR’obot Mona Lisa (one study)101 and UroNav (one study). 90 One study evaluated KOELIS Trinity,83 and six studies evaluated, KOELIS Urostation, an earlier version of the software which used a third-party ultrasound. 80,81,92–94,98
Table 26, Appendix 4, maps the evidence by SF technology, biopsy route, anaesthesia method and registration method, and highlights a number of limitations in reporting and gaps in the evidence. Of the 20 studies that evaluated a SF technology, 7 studies used SF for a TP,31,84,85,89,95,100,101 and there was no evidence for ARTEMIS, KOELIS and UroNav used in the context of a TP. BiopSee was only evaluated under general anaesthesia,31,95 and 10 studies did not report their method of anaesthesia. 80,81,89,91–94,96,98,100 Image registration methods (rigid vs. elastic) were NR or could not be inferred in five studies. 84,88,89,95,96
Table 27, Appendix 4, summarises the characteristics of the patients in the included studies. Across all included studies, a total of 3733 patients who received SF and 2154 individuals who underwent CF were analysed and informed estimates of PCa detection. Where reported, the median age ranged from 62 to 73.1 years, median PSA levels ranged from 4.2 ng/mL to 10.7 ng/mL, and all patients had a PI-RADS or Likert score of 3 or more. Seven studies only included biopsy-naive patients,79,81,82,85,88,95,98 four studies only included patients who received a repeat biopsy following one or more prior negative biopsies31,86,94,99 and eight studies included a mix of patients with no prior biopsy and individuals undergoing a repeat biopsy following a prior negative biopsy. 80,83,84,87,89–93,101 Three studies included a subset of patients under active surveillance and reported separate results biopsy naive and/or repeat biopsy with prior negative result. 96,97,100 Where reported, all operators were experienced in biopsy procedures, although levels of expertise varied across the studies. Table 28, Appendix 4, summarises information from the studies on operator experience.
Quality of included studies
Results of the quality and applicability assessment are reported in Figure 3, and further details on the rationale for decisions are reported in Appendix 5. All studies were at high risk of bias for at least one of the following domains: patient selection, index test, reference standard and flow and timing. Eight studies were at high risk of patient selection bias; all were non-randomised comparisons. 81,83,89,90,95,98,100,101 Three studies were at unclear risk of selection bias;31,84,85 including the two RCTs,31,84 and all other studies were at low risk of selection bias. Eight studies had a high risk of bias related to the comparison of index tests,31,81,84,88,89,93,97,101 and all other 15 studies were at low risk of bias for this domain.
FIGURE 3.
Risk of bias and applicability assessment summary of studies included in the systematic review.
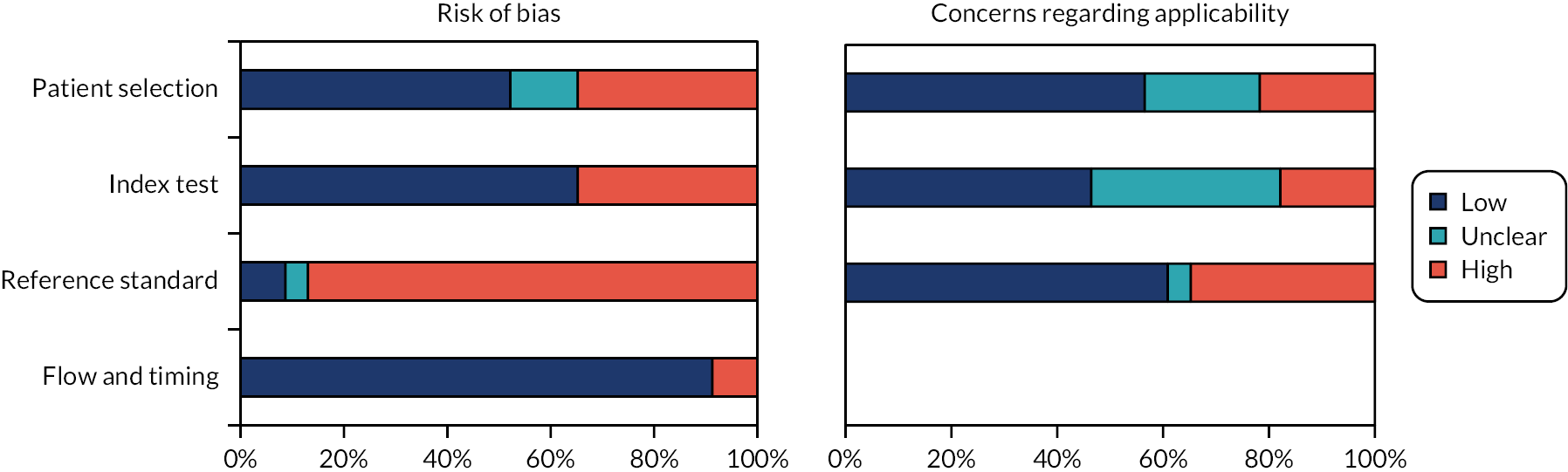
Twenty studies were at high risk of bias associated with the reference standard. 31,79–87,89,90,92,94–96,98–101 For between-patient comparisons, this was primarily due to the fact that total cancer positive cases in each study arm or cohort were derived from different biopsy methods; in within-patient comparisons, as all biopsy methods were performed within the same examination, it was not feasible for studies to truly blind operators from tracks of preceding biopsy methods (true blinding would several biopsy sessions per patient, which would be unethical). Participants in all within-patient comparison studies received SF, CF and/or systematic biopsy within the same examination; the order in which the different biopsy methods were implemented varied where reported, therefore the overall direction of bias due to the lack of operator blinding could not be determined.
Of the 15 studies that compared SF with CF or with another SF device,31,81–85,88–90,93,95,97,98,100,101 7 did not use systematic biopsy or include systematic biopsy results as part of a reference standard test. 31,81,83–85,97,101 Of the studies that included systematic biopsy as part of a reference standard test, only one reported blinding the systematic biopsy operator to the MRI report. 88 This is an important design limitation, since knowledge of the MRI report may have influenced the placing of systematic biopsy cores. Clinical advisers to the EAG confirmed that lack of blinding to MRI reports may have improved the accuracy of systematic biopsies relative to targeted biopsies. Therefore, for most of the evidence for systematic biopsy included in this review, there is a risk that the detection of PCa from systematic biopsy may have been overestimated compared with true random, standard systematic biopsy. This said, the lack of blinding to MRI report when using systematic biopsy concomitant with targeted biopsy is reflective of current practice. Blinding of the histopathologists, who analysed the biopsy samples, was generally NR, and none of studies used TTMB. Two studies were at high risk of bias due to missing outcomes data (flow and timing domain),93,95 and all other studies were at low risk of bias for this domain.
Three studies raised no concerns about their applicability to the review question. 79,82,88 Five studies included a population that was deemed not applicable (NA) to the review question (high concern),31,86,90,94,99 and five included a significant proportion (approximately half) of patients undergoing repeat biopsy following a prior negative biopsy. 87,89,92,93,101 Although patients with a prior negative biopsy were eligible in this systematic review, clinical advisers to the EAG noted that they made up only a minority (approximately under 10%) of the total population undergoing targeted biopsy who are not under active surveillance. All other studies included mostly biopsy-naive patients and had a population that was considered broadly representative. Five studies used an intervention that was not considered applicable to the review question,31,84,89,95,101 primarily due to the use of general anaesthesia in all procedures. Clinical advisers to the EAG noted that general anaesthesia is normally only used in a minority of patients, although it may facilitate biopsy targeting due to the lack of patient movement. The applicability of SF was uncertain in 10 studies. 80,81,90,92–94,96,98–100 In four cases, this was due to insufficient reporting about biopsy routes and anaesthesia methods,90,96,99,100 and in six studies, a KOELIS device with no integrated ultrasound was evaluated, and the applicability of their results to KOELIS Trinity was uncertain. 80,81,92–94,98 Following request for further information from the EAG, the company did not clarify or provide evidence that the diagnostic accuracy of older versions of KOELIS was equivalent to KOELIS Trinity. Eight studies raised concerns about the applicability of the reference standard test. 31,81,83–85,95,97,101
Diagnostic accuracy results
This section presents the evidence included in the meta-analyses and structure of the networks of evidence (see Studies included in the meta-analysis and network structure), the results of the NMAs (see Meta-analysis results), and results of studies not included in the meta-analyses (see Narrative synthesis results).
Studies included in the meta-analysis and network structure
Model 1a: multinomial synthesis model (base case)
Thirteen studies, identified by the systematic review, with data suitable for inclusion in the NMA are presented in Table 24, Appendix 3 and form the network in Figure 4. Rabah et al. 84 is excluded as it compared two SF devices, assumed to have identical effects, and therefore does not contribute to the analysis. The multinomial synthesis model was used to synthesise comparative information on the probabilities of being classified at the various ISUP grades of PCa (see Meta-analysis results). Resulting estimates are then used in the base-case economic model.
FIGURE 4.
Network of biopsy types compared, under the assumption of a common effect for different SF devices. Lines represent comparisons made in studies, numbers on the lines show how many studies included that comparison and shaded areas represent multiarm studies. SB, systematic biopsy.

Due to data sparseness, we assumed that there is no difference in relative effects of the various SF biopsy devices compared to cognitive biopsy and only fixed-effect models could be fitted. This assumption is supported by the limited direct evidence comparing different fusion devices and clinical advice to the EAG. However, the different costs of each device will still be taken into account in the economic model. This assumption will be relaxed in an additional analysis (Model 1b: Multinomial synthesis model, individual device effects).
Although the network in Figure 4 is fully connected (there is a path connecting every intervention to every other), not all studies reported the breakdown of cancers detected by ISUP grade (see Appendix 3, Table 24). This resulted in a de facto disconnect in the network for comparisons of CF + SB and SF + SB for ISUP grades > 2. Relative effects comparing disconnected components of the network cannot be estimated and are reported separately.
Calculating absolute probabilities
As noted in Meta-analysis results, odds ratios estimated from this model are hard to interpret. We will therefore also present results on the absolute probability scale to aid interpretation. To calculate the absolute probabilities of being classified in each category using each intervention, we need to assume a set of underlying baseline probabilities of being classified in each category on one of the included interventions. For ease of interpretation, in this section these underlying baseline probabilities will be assumed to be fixed, that is, to have no uncertainty. All other probabilities are then obtained by applying the estimated odds ratios to these probabilities, as described in Appendix 2. These baseline probabilities should be as representative as possible of the population of interest. A targeted review was carried out to determine a good source of evidence on these probabilities (see Review of additional prevalence, test results and diagnostic accuracy evidence and Appendix 8 Distribution of test results obtained with cognitive fusion or software fusion biopsy).
The two studies with the largest sample size that were identified and deemed most representative of NHS practice were considered as a source of evidence for the baseline probabilities: Filson et al. 96 and PAIREDCAP (2019). 88 Two subgroups of patients are of interest: biopsy-naive patients and those undergoing a repeat biopsy after a negative result. Filson (2016)96 reported probabilities for these two subgroups separately and for two interventions of interest, SF using ARTEMIS and combined SF (ARTEMIS) with systematic biopsy, allowing the same source of baseline probabilities to be used for both disconnected components of the network.
However, Filson et al. 96 does not report separate data for ISUP grades 3 and 4–5, as required by the model. We approximated the probabilities of patients being in grade 3 and 4–5 by splitting the combined patients according to the proportions in each category reported in PAIREDCAP (2019)88 (approximately 60/40).
In a sensitivity analysis for the subgroup of biopsy-naive patients, the distribution of test results from PAIREDCAP (2019)88 (which only include biopsy-naive patients) was used to inform the baseline probabilities in the first part of the network. In the absence of other suitable sources of evidence, data on biopsy-naive patients from Filson et al. 96 will continue to inform the baseline probabilities in the combined biopsy (software/CF plus systematic biopsy) network.
Absolute probabilities were therefore reported for:
-
subgroup of biopsy-naive patients (based on Filson et al. 96 biopsy-naive data)
-
subgroup of previous negative biopsy patients (based on Filson et al. 96 previous negative biopsy data)
-
a sensitivity analysis using alternative baseline probabilities for the biopsy-naive subgroup (based on biopsy-naive data from PAIREDCAP (2019)88 and Filson et al. 96).
Results will be reported separately for comparisons of CF, SF and systematic biopsy, and comparisons of combined cognitive/SF with systematic biopsy.
Model 1b: multinomial synthesis model, individual device effects
Fourteen studies identified by the systematic review with data suitable for inclusion in the NMA are presented in Table 24, Appendix 3 and form the network in Figure 5. The multinomial synthesis model was used to synthesise comparative information on the probabilities of being classified at the various ISUP grades of PCa (see Meta-analysis results).
FIGURE 5.
Network of biopsy types and devices compared. Lines represent comparisons made in studies, numbers on the lines show how many studies included that comparison and shaded areas represent multiarm studies. SB, systematic biopsy.

Although the network in Figure 5 is fully connected (there is a path connecting every intervention to every other), not all studies reported the breakdown of cancers detected by ISUP grades. This resulted in a de facto disconnect in the network for comparisons of some devices for higher ISUP grades (see Table 24, Appendix 3). Relative effects comparing disconnected components of the network cannot be estimated and are reported separately, where possible.
Calculating absolute probabilities
Absolute probabilities will be reported for:
-
subgroup of biopsy-naive patients (based on Filson et al. 96 biopsy-naive data);
-
subgroup of previous negative biopsy patients (based on Filson et al. 96 previous negative biopsy data).
As many network components are disconnected for high ISUP grades in this analysis, absolute probabilities are only reported where they can be reliably obtained, which limits the interpretation of results.
Model 2a: cancer detection
Data from the studies identified by the systematic review (see Figure 4) were pooled in a NMA to compare the proportion of PCas (CS and non-CS, i.e. Gleason ≥ 3 + 3) detected by the different biopsy strategies. Data were obtained by adding the relevant ISUP grades in Table 24, Appendix 3, and are presented in Table 30, Appendix 6.
In Model 2a we assumed that there is no difference in relative effects of the various SF biopsy devices compared to cognitive biopsy. This assumption relaxed in Model 2b where the individual device effects are estimated. Both fixed- and random-effects models were considered.
Model 2b: cancer detection, individual device effects
Data from the studies, identified by the systematic review (see Figure 5 and Table 30, Appendix 6), were pooled in a NMA to compare the proportion of PCas (CS and non-CS) detected by the different biopsy strategies. Both fixed- and random-effects models were considered.
Model 3a: clinically significant cancer detection
Data from the studies identified by the systematic review were pooled in aNMA to compare the proportion of CSPCas (Gleason > 3 + 3) detected by the different biopsy strategies. Only 10 studies reported the number of CS cancers detected, obtained by adding the relevant ISUP grades in Table 24 (see Appendix 3), and are presented in Table 31, Appendix 6. In addition, Rabah et al. 84 is excluded as it compared two SF devices, assumed to have identical effects in Model 3a, and therefore does not contribute to this analysis. Nine studies were included in the network (see Figure 13, Appendix 6). Both fixed- and random-effects models were considered.
Model 3b: clinically significant cancer detection, individual device effects
Data from 10 studies reporting the number of CSPCas detected by the different biopsy strategies (see Table 31 and Figure 14, Appendix 6) were pooled in a NMA. Both fixed- and random-effects models were considered.
Meta-analysis results
Model 1a: multinomial synthesis model (base-case)
Models were sampled for 100,000 iterations from 2 independent chains (50,000 iterations on each chain) after checking that convergence was achieved after a burn-in of 50,000 iterations.
Results from fitting Model 1a to the data in Table 24, Appendix 3 (network in Figure 4) are presented in Table 3. One study (Gomez-Ortiz et al. 99) had a higher than expected contribution to the mean residual deviance (15 compared to its expected contribution of 6) but overall the model fitted the data well with a posterior mean of the residual deviance of 77.4, which is close to the 75 data points included.
| ISUP grade | Compared to CF biopsy | Compared to cognitive fusion + systematic biopsy | ||||
|---|---|---|---|---|---|---|
| SB | SF | SF + SB | ||||
| No cancer | Reference | |||||
| 1 | 1.57 | (1.09 to 2.26) | 1.98 | (1.28 to 3.06) | 1.20 | (0.72 to 1.99) |
| 2 | 2.24 | (1.45 to 3.47) | 1.34 | (0.80 to 2.25) | 2.57 | (0.95 to 7.97) |
| 3 | 1.40 | (0.82 to 2.38) | 1.25 | (0.66 to 2.33) | 0.66 | (0.12 to 2.92) |
| 4–5 | 1.54 | (0.83 to 2.84) | 1.58 | (0.90 to 2.77) | 4.33 | (0.45 to 158.38) |
Compared to CF biopsy, there is evidence of higher odds of being categorised in ISUP grade 1 instead of NC when using SF (OR 1.98 95% CrI 1.28 to 3.06, Table 3). There is no evidence of more patients being categorised as ISUP 2, 3 or 4–5 instead of NC for SF biopsy compared to CF biopsy (see Table 3). More patients are categorised as having non-CS cancer (ISUP grade 1) (OR 1.57 95% CrI 1.09 to 2.26) and as having a CS cancer with ISUP grade 2 (OR 2.24 95% CrI 1.45 to 3.47), instead of having NC when using systematic biopsy compared to CF biopsy. There is no clear evidence of more patients being categorised as ISUP 3 or 4–5 instead of NC for systematic biopsy compared to CF biopsy (see Table 3). However, we note the large uncertainty in all results, particularly for higher ISUP grades, due to limited data broken down by higher ISUP grades. As discussed in Quality of included studies, most of the evidence for systematic biopsy was not blinded to MRI reports. This may have inflated the accuracy of systematic biopsy compared with SF and CF.
Compared to CF plus systematic biopsy, there is no clear evidence of more patients being categorised as having cancer (ISUP grades 1 to 4–5) instead of NC for SF plus systematic biopsy. However, we note the large uncertainty in all results, particularly for the highest category. This is due to few studies reporting data broken down by higher ISUP grades and the small number of patients categorised as ISUP 4–5 using any of the two biopsy types (see Table 24, Appendix 3).
Absolute probabilities of being classified as having NC or at different ISUP grades for the two subgroups of interest: biopsy-naive patients and patients with a previous negative biopsy based on data from Filson et al. 96 are presented for ease of interpretation. A sensitivity analysis for the biopsy-naive subgroup is presented in Table 32, Appendix 6.
Absolute probabilities: biopsy-naive patients
Using baseline probabilities for SF biopsy and SF plus systematic biopsy from the biopsy-naive subgroup in Filson et al.,96 and applying the odds ratios in Table 3 the probabilities of being classified as having NC or at different ISUP grades are given in Table 4.
| ISUP | ARTEMIS probabilities from Filson et al.96 biopsy-naive data | ARTEMIS + SB probabilities from Filson et al.96 biopsy-naive data | ||||||
|---|---|---|---|---|---|---|---|---|
| Cognitive | Systematic | Softwarea | Cognitive + SB | Software + SBa | ||||
| No cancer | 0.55 | (0.48 to 0.62) | 0.42 | (0.37 to 0.47) | 0.47 | 0.41 | (0.21 to 0.56) | 0.36 |
| 1 | 0.17 | (0.13 to 0.22) | 0.21 | (0.17 to 0.25) | 0.16 | 0.21 | (0.10 to 0.33) | 0.22 |
| 2 | 0.12 | (0.08 to 0.16) | 0.20 | (0.16 to 0.24) | 0.20 | 0.10 | (0.03 to 0.23) | 0.22 |
| 3 | 0.09 | (0.06 to 0.14) | 0.10 | (0.06 to 0.15) | 0.11 | 0.21 | (0.06 to 0.59) | 0.12 |
| 4–5 | 0.06 | (0.03 to 0.10) | 0.07 | (0.04 to 0.12) | 0.06 | 0.02 | (0.00 to 0.18) | 0.08 |
For biopsy-naive patients, Model 1a suggests that compared to SF biopsy, patients undergoing CF biopsy may have (see Table 4):
-
a higher probability of being classified as not having cancer (55% vs. 47%)
-
similar probability of being classified as having non-CS cancer (ISUP 1, 17% vs. 16%)
-
lower probability of being classified at higher ISUPs, particularly ISUP 2.
Probabilities for systematic biopsy are similar to those for SF biopsy for NC and all ISUP grades. However, results are uncertain.
Results were similar when systematic biopsy was added to software and CF, although there may be a higher probability of patients being classified at ISUP grade 2 with software plus systematic biopsy compared to cognitive plus systematic biopsy, and versus lower probability for ISUP grade 3 (see Table 4), although results are imprecise due to the small number of observed events. The proportion of patients classified at ISUP grades 4–5 are similar. However, these results are very uncertain.
Absolute probabilities: previous negative biopsy patients
Using baseline probabilities for SF biopsy and SF plus systematic biopsy from the subgroup of patients with a previous negative biopsy in Filson et al.,96 and applying the odds ratios in Table 3 the probabilities of being classified as having NC or at different ISUP grades are given in Table 5.
| ISUP | ARTEMIS probabilities from Filson et al.96 previous negative biopsy data | ARTEMIS + SB probabilities from Filson et al.96 previous negative biopsy data | ||||||
|---|---|---|---|---|---|---|---|---|
| Cognitive | Systematic | Softwarea | Cognitive + SB | Software + SBa | ||||
| NC | 0.75 | (0.69 to 0.80) | 0.64 | (0.59 to 0.69) | 0.69 | 0.63 | (0.38 to 0.76) | 0.58 |
| 1 | 0.08 | (0.06 to 0.11) | 0.11 | (0.09 to 0.14) | 0.09 | 0.13 | (0.07 to 0.21) | 0.15 |
| 2 | 0.06 | (0.04 to 0.08) | 0.11 | (0.09 to 0.13) | 0.10 | 0.05 | (0.02 to 0.12) | 0.12 |
| 3 | 0.06 | (0.04 to 0.10) | 0.08 | (0.05 to 0.12) | 0.08 | 0.14 | (0.04 to 0.47) | 0.09 |
| 4–5 | 0.04 | (0.02 to 0.07) | 0.05 | (0.03 to 0.09) | 0.05 | 0.01 | (0.00 to 0.13) | 0.06 |
For patients with a previous negative biopsy, given a 69% probability of being classified as not having cancer with SF biopsy,96 the probability of being classified as not having cancer is higher for patients undergoing cognitive biopsy (75% 95% CrI 69% to 80%) but lower for patients undergoing systematic biopsy (64% 95% CrI 59% to 69%). As there is high probability that patients with a prior negative biopsy will again be classified as having NC with SF, CF or systematic biopsy, the probabilities of being classified at different ISUP grades are small and similar across these biopsy strategies (see Table 5).
Similar results were obtained when adding a systematic biopsy to software and CF.
Model 1b: multinomial synthesis model, individual device effects
Models were sampled for 100,000 iterations from 2 independent chains (50,000 iterations on each chain) after checking that convergence was achieved after a burn-in of 50,000 iterations.
Results from fitting Model 1b to the data in Table 24, Appendix 3 (network in Figure 5) are presented in Table 33, Appendix 6. One study (Gomez-Ortiz et al. 99) had a higher than expected contribution to the mean residual deviance (16 compared to its expected contribution of six). Other studies had deviances in the range expected, although the posterior mean of the residual deviance was 89.2, which is higher than the number points included (79). Often a model fit can be poor when data are sparse as many parameters cannot be reliably estimated. However, more complex models, such as random-effects models, cannot be considered due to data sparseness. We would advise caution when interpreting the results from this model.
No odds ratios can be estimated for SF biopsy using UroNav or UroNav plus systematic biopsy since the only study comparing this device does not report details of classifications broken down by category (see Table 24, Appendix 3).
Compared to CF biopsy, there is only evidence of higher odds of being categorised in ISUP grade 1 instead of NC when using systematic biopsy (OR 1.54 95% CrI 1.06 to 2.24, Table 33, Appendix 6). There is some evidence that more patients are categorised as ISUP grade 2 instead of having NC when using systematic biopsy, ARTEMIS or Urostation, compared to CF biopsy. There is no clear evidence of more patients being categorised as ISUP 3 or 4–5 instead of NC for systematic biopsy or ARTEMIS compared to CF biopsy. No relative effects are estimable for the other devices and there is large uncertainty in all results.
Compared to CF plus systematic biopsy, there is no clear evidence of more patients being categorised as having cancer (ISUP grades 1 to 4–5) instead of NC for ARTEMIS or Urostation plus systematic biopsy. However, we note the large uncertainty in all results which led to some relative effects not being estimable (see Table 33, Appendix 6).
Absolute probabilities of being classified as having NC or at different ISUP grades for the two subgroups of interest can only be reported where the odds ratios are estimable for all ISUP grades. Therefore, these are only presented for CF, systematic biopsy and SF using ARTEMIS (assumed underlying baseline probabilities), and when adding systematic biopsy, for biopsy-naive patients and patients with a previous negative biopsy based on data from Filson et al. 96 (see Tables 34 and 35, Appendix 6).
Model 2a: cancer detection
Fixed- and random-effects models were fitted. Based on the model fit statistics (see Table 36, Appendix 6) both the fixed- and random-effects models fitted the data well and differences in DIC were small. Therefore, the fixed-effect model was selected. The fixed-effect unrelated mean effects model suggested no evidence of inconsistency between direct and indirect evidence based on both the model fit statistics and deviance plots (see Table 36 and Figure 15, Appendix 6).
Results from fitting Model 2a to the data in Table 30, Appendix 6 (network in Figure 4) are presented in Figure 6 and all pairwise comparisons are reported in Table 37, Appendix 6.
FIGURE 6.
Odds ratio of detection (median and 95% CrI) of cancer. OR, odds ratio; SB, systematic biopsy.
Model 2a suggests SF biopsy may classify more patients as having cancer (any ISUP), than CF biopsy (OR 1.30 95% CrI 1.06 to 1.61; Figure 6). However, note that this cannot be directly compared to the results from ISUP 1 for Model 1a, since Model 2a is comparing detection of any cancer, that is, all ISUP 1 to 5 combined, and not only the detection of non-CS cancer. The increase in the ORs for detection of any cancer is driven by the increase in the probability of categorisation at ISUP>1, which in this case is driven by increases at ISUP 2 [see Model 1a: multinomial synthesis model (base-case)]. Results for the random-effects model are presented as a sensitivity analysis in Table 37 and Figure 16, Appendix 6. As discussed in Quality of included studies, the accuracy of systematic biopsy may have been inflated due to study design limitations.
Model 2b: cancer detection, individual device effects
Fixed- and random-effects models were fitted. Based on the model fit statistics (see Table 36, Appendix 6) both the fixed- and random-effects models fitted the data well and differences in DIC were small. Therefore, the fixed-effects model was selected. The fixed-effects unrelated mean effects model suggested no evidence of inconsistency between direct and indirect evidence based on both the model fit statistics and deviance plots (see Table 36 and Figure 15, Appendix 6).
Results from fitting Model 2b to the data in Table 30, Appendix 6 (network in Figure 4) are presented in Figure 7 and all pairwise comparisons are reported in Table 38, Appendix 6.
FIGURE 7.
Odds ratio of detection (median and 95% CrI) of cancer, individual device effects. OR, odds ratio; SB, systematic biopsy.

Compared to CF biopsy, there is evidence that SF biopsy with BioJet, Urostation and ARTEMIS, and Urostation, UroNav or cognitive biopsy combined with systematic biopsy may detect more cancers. Results for the random-effects model are presented as a sensitivity analysis in Table 38 and Figure 16, Appendix 6.
Model 3a: clinically significant cancer detection
Fixed- and random-effects models were fitted. Based on the model fit statistics (see Table 36, Appendix 6) the random-effects model had a better fit to the data and the difference in DIC was > 3. Therefore, the random-effects model was selected. The random-effects unrelated mean effects model suggested no evidence of inconsistency between direct and indirect evidence based on both the model fit statistics and deviance plots (see Table 36 and Figure 15, Appendix 6).
Results from fitting Model 3a to the data in Table 31, Appendix 6 (network in Figure 13) are presented in Figure 6 and all pairwise comparisons are reported in Table 37, Appendix 6. The posterior median of the between-study heterogeneity SD was 0.313 (95% CrI 0.132 to 0.634), which is moderate on the log odds ratio scale. The full posterior distribution of the between-study SD is presented in Figure 16, Appendix 6.
While Model 2a suggested SF biopsy may classify more patients as having cancer (ISUP 1 to 5) than CF biopsy (see Figure 5), Model 3a also suggests SF biopsy may classify more patients as having CS cancer (ISUP 2, 3, 4–5), as opposed to NC or ISUP 1, than CF biopsy, but with a wider confidence interval (CI) that includes the null effect (OR 1.35 95% CrI 0.86, 2.10; Figure 6). By using odds and collapsing ISUP grades, the statistical model has higher power to detect statistically significant differences against NC than against non-CS cancer (which pools NC with ISUP 1). These results are consistent with the findings from Models 1a and 2a. There is some evidence that adding systematic biopsy to cognitive or SF increases CS cancer detection.
Model 3b: clinically significant cancer detection, individual device effects
Fixed- and random-effects models were fitted. Based on the model fit statistics (see Table 36, Appendix 6) the random-effects model had a better fit to the data and the difference in DIC was > 3. Therefore, the random-effects model was selected. The random-effects unrelated mean effects model suggested no evidence of inconsistency between direct and indirect evidence based on both the model fit statistics and deviance plots (see Table 36 and Figure 15, Appendix 6).
Results from fitting Model 3b to the data in Table 31, Appendix 6 (network in Figure 13) are presented in Figure 7 and all pairwise comparisons are reported in Table 39, Appendix 6. The posterior median of the between-study heterogeneity SD was 0.304 (95% CrI 0.048 to 0.769), which is similar to the posterior heterogeneity from Model 3a. This suggests there is moderate heterogeneity (log odds ratio scale) and that splitting the device effects did not explain the between-study variability. The full posterior distribution of the between-study SD is presented in Figure 16, Appendix 6.
Compared to CF biopsy, there is no evidence that SF with ARTEMIS, BiopSee, Urostation, or systematic biopsy detect more CS cancers. However, there is evidence that SF with BioJet or adding systematic biopsy to cognitive or SF with ARTEMIS or Urostation may increase CS cancer detection.
Narrative synthesis results
Nine studies reported data on PCa detection but were not included in a meta-analysis, due to reasons specified in Meta-analysis. 81,83,85,89–91,95,98,100 None of these studies had a within-patient comparison, and none used a randomised comparison between SF and CF or between two or more eligible SF technologies. Therefore, these studies were considered at higher risk of confounding compared with studies included in the NMA. This section presents a narrative summary of their results.
All nine studies reported a comparison between separate cohorts. Five used a prospective design,81,83,89,95,98 and four were retrospective. 85,90,91,100 Only one study used propensity score matching to adjust for differences in participant characteristics,81 and one study performed a comparison between software and CF using conditional logistic regression. 98 All other studies reported naive, unadjusted comparisons.
Six studies compared SF alone with CF alone85,89–91,95,98 and two studies reported a comparison between SF with concomitant systematic biopsy against CF with systematic biopsy. 89,100 Two studies compared different SF technologies against one another; one compared two technologies (BioJet with Urostation),81 and another compared three (BioJet, KOELIS Trinity and UroNav). 83 The following SF technologies were evaluated: BioJet (three studies),81,83,89 BiopSee (one study),95 bkFusion (two studies)85,100 and iSR’obot Mono Lisa (one study). 91 Three studies included a SF technology manufactured by KOELIS, including Trinity (one study),83 Urostation (two studies)81,98
The diagnostic accuracy results of studies not included in the meta-analayses are summarised by comparisons in Narrative synthesis results. 1 (SF vs. CF), 4.4.3.2 (SF vs. SB) and Software fusion versus software fusion (SF vs. SF) with further details presented in Tables 40–44, Appendix 7. Subgroups presents a narrative synthesis of diagnostic accuracy results by lesion location, patient type (biopsy naive and experienced), impact of operator experience and PI-RADS scores of all studies included in the systematic review.
Software fusion versus cognitive fusion
Prostate cancer
Five studies compared SF with CF and reported PCa rates. 85,90,91,95,98 All three studies that reported a definition of PCa used the same threshold (GS of 6). Their results are presented in Table 40, Appendix 7.
Three studies reported higher test-positive rates of PCa for subjects receiving SF compared with CF; two of those reported that the difference was statistically significant,91,98 and one did not report measures of statistical significance. 95 One study found no statistically significant difference between CF and SF,85 and one study reported higher test-positive rates for CF but no measures of statistical significance. 90
Overall, these five studies broadly agree with the findings of the NMA which showed SF was associated with more PCa detection than CF. However, the evidence from these five studies is inconsistent and also at high risk of confounding, notably due to the lack of paired or randomised comparison.
Clinically significant prostate cancer
Five studies compared SF with CF and reported data on CSPCa test-positive rates. 85,90,91,95,98 All studies defined CS cancer as GS of 7 (3 + 4) or higher. Their results are presented in Table 41, Appendix 7.
Two studies reported no statistically significant difference in test-positive rates of CSPCa between SF and CF,85,91 whereas one study reported a statistically significant difference in test positive rates favouring SF. 98 One study reported a higher rate of CSPCa for CF compared with SF, although it did not report whether this difference was statistically significant. 91 One study reported similar rates of CS cancers between SF and CF,90 and comparable rates of missed, upstaged and equivalent CS biopsy results identified by each targeted biopsy method against concurrent 14-core, systematic biopsy (SF, p = 0.172).
Although outcomes between these studies are inconsistent and are at high risk of bias overall, they do not show evidence of a significant difference in rates of CSPCa detection between SF and CF. This evidence is broadly reflective of the meta-analysis findings.
Software fusion and systematic biopsy versus cognitive fusion and systematic biopsy
Two studies that were excluded from the meta-analyses compared PCa testpositive rates between SF with concomitant systematic biopsy, against CF with systematic biopsy. 89,100 Results are summarised in Table 42, Appendix 7. There was no statistically significant difference in rates of overall PCa and CS cancer detection between the two methods.
Software fusion versus software fusion
Two studies that were not included in the meta-analyses compared biopsy test-positive rates between SF technologies. 81,83 One study compared BioJet with KOELIS Urostation, and one study evaluated three devices: BioJet, KOELIS Trinity and UroNav. Results are summarised in Table 43, Appendix 7. Both studies found no statistically significant difference in test-positive rates of PCa and CSPCa between SF devices. Overall, this evidence is consistent with the findings of the meta-analyses.
Subgroups
Three subgroups were prespecified in the NICE scope and review protocol: patients with anterior lesions, patients with posterior lesions, and individuals who have had a previous negative prostate biopsy and are referred for a repeat biopsy within 12 months. The review protocol also specified that the following potential factors affecting diagnostic accuracy would be investigated in subgroup analyses: biopsy-naive patients, and operator experience. Test-positive rates by PI-RADS groups (3, 4 and 5) were also summarised, although this subgroup was no pre-specified.
Network meta-analyses for biopsy-naive or prior negative-biopsy subgroups were not conducted due to the limited number of studies included. Absolute probabilities of being classified as having NC or being at different ISUP grades are presented for biopsy-naive or patients with a previous negative biopsy in the meta-analysis results Model 1a: multinomial synthesis model (base case).
Due to the limited evidence, results for the other subgroups (lesion location, operator experience and PI-RADS) are summarised narratively only.
Lesion location
One study31 reported test-positive estimates by lesion location (anterior, posterior), and found no significant differences in test-positive rates of PCa and CSPCa between SF (BiopSee) and CF for posterior and anterior located lesions. The results are summarised in Table 45, Appendix 7. Test-positive rates were also stratified by other locations (peripheral and transition zones, NR here) and showed no statistically significant differences between the two methods.
Repeat biopsy and biopsy-naive patients
Test-positive rates for patients receiving a repeat biopsy following a prior negative biopsy and for biopsy-naive patients are presented in Appendix 7, Tables 46 and 47 respectively. Overall, there was no evidence that SF had higher test-positive rates compared with CF in either subgroup. While it is expected that these characteristics may influence the number of positive cancers detected (due to a different underlying prevalence of cancer in the different populations), there is no evidence that they may affect the relative diagnostic accuracy across biopsy types.
Impact of operator experience
One study evaluated how operator experience impacts the cancer biopsy positivity rates. 89 The results are reported in Table 48, Appendix 7. Stabile et al. 89 evaluated the learning curve for the probability of detecting CSPCa from three urologists, who each used a different biopsy approach on separate patient cohorts: transrectal cognitive biopsy (operator 1), transrectal SF biopsy (operator 2), and transperineal SF biopsy (operator 3). Each urologist had performed at least 200 prostate biopsies but were naive to targeted biopsy techniques. The total number of targeted biopsies performed by operator 1, 2 and 3 were 87, 70 and 87 respectively. Operator experience was defined as the progressive number of targeted biopsies performed by each operator. Stabile et al. 89 found that there was a sharp increase in the CSPCa biopsy positivity rates in the first 60 procedures, where it plateaued, regardless of the biopsy approach. Operator experience was a significant predictor of the CSPCa biopsy positivity rate in targeted cores, which was more pronounced for the operator who conducted transrectal SF biopsy compared with the other two biopsy approaches.
Prostate imaging – reporting and data system
Six studies reported test positive rates of PCa stratified by PI-RADS score (3, 4 or 5). All four studies that reported any PCa rates for SF and CF found no statistically significant differences by PI-RADS score between the two methods. 31,85,88,95 Similarly, the two studies that compared CS rates between software and CF subgroups found no difference across PI-RADS subgroups. 31,95 One study81 found that test positive rates of any PCa cancer and CSPCa were comparable between KOELIS Urostation and BioJet after stratifying for PI-RADS score except for PI-RADS Score 4, where the rate of any PCa was higher in the Urostation group compared with BioJet (80% vs. 58.1%, respectively for EF and RF groups, p = 0.025), and one study84 found that rates of CSPCa were higher for PI-RADS 4 patients undergoing TP with BioJet compared with transrectal biopsy with ARTEMIS (43.4% vs. 33.3%), but similar for PI-RADS 3 and 5 subgroups. These results are all based on small (n < 100) subgroups and may not be reliable.
Clinical effectiveness results
Biopsy positivity rates
Four studies reported biopsy positivity rates outcomes;31,84,88,98 their results are presented in Table 49, Appendix 7. Three studies compared SF with CF and one compared different SF biopsies. None of the studies reported what threshold was used to define biopsy positivity rates. Biopsy positivity rates varied widely, from 21.1% to 75% for SF, and from 33.3% to 67% for CF.
Overall, there is no evidence that biopsy positivity rates differ significantly between SF and CF. Evidence comparing biopsy positivity rates between SF devices is inconclusive, as it limited to one study at high risk of confounding.
Software fusion versus cognitive fusion
Of the three studies that compared SF with CF, two studies did not find any significant difference in biopsy positivity rates between the two methods;31,88 one study found a statistically significant difference in biopsy positivity rates that favoured SF,98 although its results may be confounded due to the lack of matching or adjustment between the two study arms.
Comparisons between software fusion technologies
One study84 found that the biopsy positivity rate of BioJet was significantly higher than that of ARTEMIS (43.5% vs. 21.1% respectively, p = 0.0002). However, this finding is at high risk of confounding, due to differences in biopsy route (transrectal for ARTEMIS, and transperineal for BioJet) and anaesthesia (local for ARTEMIS, and general for BioJet) between the two study arms.
Time taken for biopsy procedure
Two studies compared the time required to complete biopsies between different SF devices. The results of these studies are presented in Table 6. Procedure completion duration varied widely, from an average of 13 minutes to 41 minutes; this variation is likely due in part to differences in biopsy and anaesthesia methods.
| Study | Design | Pop. | Biopsy method | Sample size | N cores per ROIa | N ROI targeted | Effect estimates | p-value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Type | Route | Anaesthesia | ||||||||
| Rabah (2021)84 | RCT, between patient | BN, RB | SF: ARTEMIS, SF: BioJet | ARTEMIS: TR BioJet: TP |
ARTEMIS: LA BioJet: GA |
ARTEMIS: 165 BioJet: 142 |
2–4 cores | All ROI | Mean (SD) BioJet: 41.2 minutes (±0.7) ARTEMIS: 13 minutes (±2.3) |
p < 0.001 |
| Sokolakis (2021)83 | Prospective cohort, between patient | BN, RB | SF: BioJet SF: KOELIS SF: UroNav |
TR (all) | LA (all) | BioJet: 20 KOELIS: 20 UroNav: 20 |
2–3 cores | All ROI | Median (IQR) BioJet: 16 minutes (15–18) KOELIS: 28 minutes (26–29) UroNav: 17 minutes (15–20) |
p < 0.001 |
Overall, there is evidence suggesting that duration of biopsy procedures performed transrectally under local anaesthesia, using BioJet or UroNav (rigid registration) is significantly shorter than with KOELIS Trinity (elastic registration). However, this finding is based on a single, small study and is not conclusive.
Both the studies found statistically significant differences in procedure time between SF devices. Sokolakis et al.,83 found biopsies conducted transrectally under local anaesthesia were significantly faster using BioJet and UroNav devices (both with rigid registration), compared with the KOELIS Trinity device (elastic registration). In Rabah et al. 84 the time taken to conduct the biopsy procedure was significantly shorter using the ARTEMIS device, compared to the BioJet device, although this comparison is at high risk of confounding due to differences in biopsy route and anaesthesia method: biopsies conducted with ARTEMIS were performed transrectally under local anaesthesia, whereas biopsies with BioJet were done transperineally under general anaesthesia.
Sokolakis et al.,83 also compared the time taken to conduct the biopsy procedure by operator experience. Four urologists [two trainees who had completed around 40 TRUS-guided biopsies (junior urologists) and two senior urologists who had completed more than 250 TRUS-guided biopsies, but none had any experience of SF] conducted five biopsies with each system. Overall, operative time for the rigid registration fusion devices was shorter for the senior urologists compared to the junior urologists, but there were minimal differences in operating time for the elastic registration fusion device.
Complications and adverse events
Five studies evaluated the AEs and complications arising from the prostate biopsy procedure. 31,83–85,90 Of those, three studies compared complication rates and AEs of SF and CF, and two compared different SF devices.
Overall, there is no evidence of a significant difference in safety outcomes between biopsies conducted with SF and CF, although the evidence is limited by poor reporting and at high risk of confounding due to differences in biopsy routes and anaesthesia methods.
Software fusion versus cognitive fusion
Table 50, Appendix 7, presents the results of the three studies that compared safety events between SF and CF. Of those, two found no difference in safety outcomes (severity NR) between the two fusion methods,85,90 and one found higher rates of grade 1–2 AEs for patients undergoing CF transrectal biopsy under local anaesthesia compared with SF transperineal biopsy under spinal/general anaesthesia. As discussed in Quality of included studies, the comparison in this study is at high risk of confounding due to the different biopsy routes and anaesthesia methods.
Comparisons between software fusion technologies
Table 51, Appendix 7, summarises the results of the two studies that compared safety outcomes between SF technologies. 83,84 Both studies found similar rates of AEs. Rabah et al. 84 found no difference between the rates of urinary retention or haematuria (p = 0.56, p = 0.6, respectively) between two SF biopsy devices (ARTEMIS and BioJet), although these results are at high risk of confounding due to differences in biopsy route. Sokolakis et al. 83 found no severe peri- or post-operative AEs, but mild AEs were reported in most participants, although this was not evaluated statistically.
Operator preferences between software fusion technologies
One study83 evaluated the usability of SF biopsy which found evidence suggesting that rigid systems (BioJet and UroNav) are easier to use, compared to the elastic registration system (KOELIS) for transrectal biopsies under local anaesthesia, although this finding is based on a single small study at high risk of bias and is therefore not conclusive.
Sokolakis et al. 83 compared the impact of operator experience on the usability of three SF devices, using a system usability scale: a 100-point scale measuring the learnability and user-friendliness of a given technology, with higher values indicating a device or technology is easier to use. 102 Senior urologists also found that the SF devices had better usability compared to the junior urologists. Sokolakis et al. 83 also compared the usability of the three SF devices and found that the rigid systems (BioJet and UroNav) were significantly easier to use compared to the elastic registration system (KOELIS). Further results are presented in Table 52, Appendix 7.
Other outcomes
No evidence was found for the following outcomes specified in the protocol: biopsy sample suitability/quality, number of repeat biopsies performed, procedure completion rates, software failure rate, time to diagnosis, length of hospital stay, re-biopsy rate, hospitalisation, overall survival (OS), PFS, patient- and carer-reported outcomes (including tolerability and HRQoL), barriers and facilitators to implementations, or cost outcomes.
Diagnostic accuracy and clinical effectiveness: summary and conclusions
The evidence identified by the systematic review included a total of 3733 patients who received SF and 2154 individuals with CF from 23 studies. Evidence was included for all devices specified in the protocol, except for Fusion Bx 2.0 and FusionVu. Overall, the evidence for all devices was at high risk of bias. Up to 14 studies were included in NMAs. Analyses compared the relative diagnostic accuracy of SF, CF, CF with concomitant systematic biopsies, SF with systematic biopsies, and systematic biopsies alone.
Our main NMAs looked at how CF compares to SF in classifying patients across the range of ISUP grades. Results must be cautiously interpreted due to the high risk of bias, but suggest that patients undergoing software biopsy may show: (1) a lower probability of being classified as not having cancer, (2) similar probability of being classified as having non-CS cancer (ISUP grade 1) and (3) higher probability of being classified at higher ISUP grades, particularly ISUP 2. Similar results were obtained when comparing between same biopsy methods where both were combined with systematic biopsy.
Additional meta-analyses of cancer detection rates suggest that, compared with CF biopsy, SF may identify more PCa (any grade) (OR 1.30; 95% CrI 1.06 to 1.61). Adding systematic biopsy to cognitive or SF may increase the detection of all PCa and of CS cancer, and from this evidence there is no suggestion that SF with concomitant systematic biopsy is superior to CF with systematic biopsy.
Meta-analyses by individual device showed that compared with CF biopsy, BioJet and Urostation are associated with a higher detection of PCa overall, and that BioJet is associated with more CS cancer, although only one study of BioJet was included. The evidence for all other software devices was insufficient to evaluate their accuracy compared with CF reliably, or to assess whether some SF technologies are more accurate than others.
There was large uncertainty in all estimates due to the limited evidence, particularly for higher ISUP grades and by individual device. Results from studies, excluded from the meta-analyses, broadly reflected these findings. Compared with CF, there was no evidence that the accuracy of SF may differ by lesion location, or between biopsy naive and prior negative biopsy patients, or according to operator experience.
The applicability of the evidence for KOELIS Trinity is uncertain, as it was almost entirely informed by evaluations of a previous version (KOELIS Urostation) without integrated ultrasound. The applicability of the evidence for BiopSee is also limited due to the lack of evaluations under local anaesthesia. There is no evidence comparing the accuracy of Fusion Bx 2.0 and FusionVu with CF, and no evidence for these devices was eligible for inclusion in the indirect comparisons.
Evidence for all other protocol specified outcomes was limited and inconclusive. Overall, there is no evidence that biopsy positivity rates differ significantly between SF and CF, or between SF devices. There was some evidence that systems with rigid registration (BioJet or UroNav) are easier and faster to use than elastic registration (KOELIS Trinity), although this is informed by a single, small study and is not conclusive. Overall, there is no evidence of a significant difference in safety outcomes between biopsies conducted with SF and CF or between SF devices, although the evidence is limited by poor reporting and at high risk of confounding.
No relevant evidence was found for the following outcomes: biopsy sample suitability/quality, number of repeat biopsies performed, procedure completion rates, software failure rate, time to diagnosis, length of hospital stay, time taken for MR image preparation, subsequent PCa management, re-biopsy rate, hospitalisation, OS, PFS, patient- and carer-reported outcomes (including tolerability and HRQoL), barriers and facilitators to implementations.
Additional evidence to inform model structure and parameterisation
Additional evidence was required to inform a number of economic parameters, including (1) PCa prevalence; (2) distribution of test results for cognitive and SF broken down by Gleason grade; (3) test accuracy of cognitive and SF and (4) long-term evidence on outcomes from management strategies for patients with PCa. In addition to the systematic review of diagnostic accuracy and clinical effectiveness, targeted reviews were conducted to identify the most relevant evidence to inform these parameters.
Review of additional prevalence, test results and diagnostic accuracy evidence
Studies included in the systematic review of diagnostic accuracy and clinical effectiveness were reviewed to identify suitable evidence to inform the following economic model parameters: (1) PCa prevalence, estimated from a ‘gold-standard’ test (template mapping or saturation biopsy with at least 20 cores) and with sufficient granularity (by ISUP grade); (2) distribution of test results for cognitive or SF MRI in PI-RADS 3 + by ISUP grade and (3) accuracy of cognitive or SF MRI in PI-RADS 3 + patients against a ‘gold-standard’ test, that is comparative studies against template mapping or saturation biopsy for which a composite end point could be derived from the results of both tests.
Due to the lack of evidence from ‘gold-standard’ tests identified in the systematic review, additional targeted, pragmatic searches were conducted. References from a recent Cochrane systematic review, which included studies on the diagnostic accuracy of MRI‐targeted biopsy against template‐guided biopsy, were checked for further evidence. 66 As the searches in the Drost et al. 66 review were limited to July 2018, pragmatic searches of PubMed and Google Scholar were conducted to identify more recent studies. This search included the following search terms: [(template mapping) OR (saturation) AND (biopsy) AND (prostate)] AND (fusion biopsy).
Studies were prioritised according to the applicability of their population to the NHS. Ten studies were considered potentially eligible to inform at least one of the model parameters of interest. Their characteristics are summarised in Table 53, Appendix 8. Further details on the prioritisation and limitations of studies informing each of the three model parameters are available in Appendix 8.
Review of long-term evidence
To inform economic model parameters on morbidity and mortality outcomes for PCa patients, a targeted, pragmatic review was conducted. Searches included reference checking of evidence reviews informing NICE guidance on the management of PCa (NG131),10 references included in the PROMIS economic analysis, targeted searches for relevant Cochrane reviews in Cochrane Database of Systematic Reviews (CDSR) and citation searches to identify the most up-to-date follow-up data. Studies evaluating long-terms survival and disease progression outcomes in PCa patients according to prognosis status, either under active surveillance or receiving radical treatment recommended by NICE12 and described in Prostate cancer management: active surveillance, watchful waiting and radical treatment options, were included. Priority was given to larger RCTs with at least 2 years of follow-up, individual patient data (IPD) meta-analyses and large UK cohort studies. Fourteen studies, including 12 RCTs,55,59–61,103–110 1 IPD meta-analysis111 and 1 cohort study112 were identified and are listed in Table 60, Appendix 8. Table 61, Appendix 8, provides a brief summary of key trials considered most reflective of current NHS practice. The process for prioritising the final set of studies included in the model is described in Clinical effectiveness results.
Three RCTs evaluated the effect of radical prostatectomy in relation to an observation-based strategy in clinically localised PCa: SPCG4, PIVOT and ProtecT. 55,109,110 The comparators differed across trials between observation (PIVOT), watchful waiting (SPCG4) and active monitoring (PROTecT). Both SPCG4 and PROTecT included patients with localised, non-metastatic cancer, and PIVOT included low-to high- risk PCa patients. PROTecT was conducted in the UK, PIVOT in the USA and SPCG4 in Sweden, Finland and Iceland. Follow-up duration ranged from 10 years (PROTecT) to 29 years (SPCG4). PROTecT was the most recent study (1999 to 2009, compared with 1994–2002 for PIVOT and 1989–9 for SPCG4). None of the studies used mpMRI to diagnose patients.
Only SPCG4 found a significant effect for prostatectomy on OS, with the more contemporary studies not identifying an effect on all-cause mortality. PROTecT, which compared radical prostatectomy, radiotherapy and a passive management strategy (active monitoring) found that despite surgery and radiotherapy being associated with lower incidences of disease progression and metastases than active monitoring, at a median of 10 years, PCa–specific mortality was low irrespective of the treatment assigned, with no significant difference among treatments.
Of the trials identified that focused on treatments for intermediate- to high-risk disease, Systemic Therapy in Advancing or Metastatic Prostate Cancer: Evaluation of Drug Efficacy (STAMPEDE) was the largest, most recent and only study conducted in the UK. 59 STAMPEDE evaluated treatments for high-risk or metastatic or recurring cancer. 113 A large UK-based RCT of 2962 men, conducted between 2005 and 2013 with a median follow-up of 6.5 years, evaluated three drug treatment combinations for high-risk or metastatic cancer including zoledronic acid and DTX, as used in addition to SOC. While zoledronic acid showed no evidence of survival improvement, DTX led to improved survival and an increase in AEs.
Other trials with high-risk and/or metastatic disease included HYPO-RT-PC, GETUG-12 and TAX-3501. 60,61,107 HYPO-RT-PC compared hypofractionated radiotherapy with conventional radiotherapy in 1180 intermediate- to high-risk cancer patients and found that hypofractionated radiotherapy was non-inferior in terms of failure-free survival. GETUG-12 and TAX-3501. GETUG-12 evaluated the effectiveness of adding DTX, zoledronic acid/estramustine, or both to first-line long-term hormone therapy in patients with high-risk PCa (Gleason 8–10) and TAX-3501 evaluated the addition of DTX to leuprolide against leuprolide alone in metastatic patients following radical prostatectomy. Both were smaller (GETUG-12 included 413 participants, and TAX-3501 had 228 participants). At median follow-up of 12 years, GETUG-12 found that DTX chemotherapy reduces the risk of clinical relapse or mortality in high-risk PCa. TAX-3501 was terminated at 3.4 years and was underpowered to detect differences in PFS between study arms.
Additionally, evidence was sought on UK studies reporting outcomes by the five-stage CPG risk stratification system that currently supports treatment decisions in the UK NHS. 12 Only one large cohort study was identified. 112 The study included diagnostic data from 10,139 men with non-metastatic PCa from the Public Health England National Cancer Registration Service and had a median follow-up of 6.9 years, and found that a five-stratum risk stratification system outperformed the previous three-stratum risk stratification system used in the UK in predicting the risk of PCa death at diagnosis in men with primary non-metastatic PCa.
Overall, there is relevant evidence on the effectiveness of radical versus ‘conservative’ treatment options in delaying progression to metastatic disease, despite the limited observed impacts on mortality. The most contemporary and relevant evidence is from PROTecT, a recent, UK-based study. 114 Although there is UK-based evidence favouring the prognostic ability of a 5-level score for PCa mortality, there is no evidence on treatment effectiveness stratified by CPG scores.
Chapter 4 Assessment of evidence on the cost-effectiveness of software fusion biopsy
Overview
In the next sections, we provide an overview of published cost-effectiveness studies on the use of SF biopsy systems in comparison with CF for targeted prostate biopsy (see Methodology of the cost-effectiveness of software fusion biopsy for suspected prostate cancer and Results of the review of the cost-effectiveness of MRI fusion biopsy for suspected prostate cancer), to determine generalisability of the evidence to inform this assessment’s decision problem. In addition, this chapter presents a targeted review of diagnostic cost-effectiveness studies (see Methodology of the additional targeted reviews to support model conceptualisation and Results of the additional targeted reviews to support model conceptualisation), which model prostate biopsy procedures to identify aPCa (same point in the diagnostic pathway as the interventions in this assessment). This targeted review is done with the aim to support the conceptualisation and parameterisation of a de novo decision-analytic model.
Methodology of the cost-effectiveness of software fusion biopsy for suspected prostate cancer
The methodology of the systematic review of published cost-effectiveness studies comparing SF biopsy systems with CF for targeted prostate biopsy in men with suspected PCa is described below. The review aimed to assess the generalisability of existing evidence to the decision problem defined by the NICE DAR scope, and provide a brief overview of the model structure, parameterisation and results. Titles identified for inclusion in this review, are subsequently included in the review to inform the conceptualisation and development of the de novo model alongside other studies.
Literature searches
The results of the systematic literature searches carried out to inform the clinical effectiveness of technologies described in Systematic review methods (study selection, data extraction, quality assessment) were used to identify relevant cost-effectiveness studies of SF systems compared to CF for targeted biopsy in men with suspected PCa.
Study selection
Full economic evaluations that consider both costs and consequences (including cost-effectiveness, cost–utility and cost–benefit analyses) were considered for inclusion. A broad range of economic evidence on the use of MRI fusion systems was considered eligible, including economic evaluations conducted alongside trials, studies using modelling approaches and analyses of administrative databased. The inclusion criteria also defined the:
-
population as men with an elevated PSA level and/or abnormal DRE who had suspicious lesion(s) detected by (bi- or multiparametric) MRI
-
interventions as targeted transperineal or transrectal prostate biopsy using MRI fusion software with or without systematic biopsy, under local or general anaesthesia intervention
-
comparators as targeted transperineal or transrectal prostate biopsy using CF with or without systematic biopsy, under local or general anaesthesia.
Studies reporting only resource use, costs or HRQoL were excluded from the review, but considered to support the parametrisation of the de novo model.
The information submitted by the companies in response to NICE and the EAG’s requests for information was also reviewed to identify economic studies that complied with the inclusion criteria described above.
Studies identified by the search strategies (see Appendix 1) were screened and selected through a two-stage process: (1) titles and abstracts identified by the bibliographic search were screened for possible inclusion, and (2) full texts of potentially relevant records were obtained and screened for inclusion. The process was performed independently by two researchers (HP and AD) with any disagreement resolved by consensus.
Quality appraisal
Cost-effectiveness evidence selected for inclusion was quality assessed using a checklist tool developed for the assessment of model-based economic evaluations of diagnostic tests. 115
Synthesis of evidence
The characteristics and key findings of the included economic evidence were narratively summarised and tabulated for comparison. The extracted information included:
-
the perspective of analysis;
-
the comparators and its positioning in the diagnostic pathway, study population and setting, main analytic approaches (e.g. analysis of individual patient data/decision-analytic model), primary outcomes of the economic analysis;
-
details of adjustment for HRQoL, resource usage (direct and indirect costs);
-
estimates of incremental cost-effectiveness and how uncertainty was quantified (e.g. deterministic/probabilistic sensitivity analysis).
The relevance of existing economic evidence to the current decision problem in the NICE DAR scope was assessed based on:
-
consistency with the decision problem being considered in this assessment, including relevance to the UK
-
relevance of outputs for decision-making (i.e. to estimate long-term NHS costs and QALYs based on morbidity and mortality associated with PCa tailoring according to patient prognosis and preferences)
-
the model flexibility which allows the consideration of different subgroups (e.g. patients with previous negative biopsy results) and potential effect modifiers of diagnostic accuracy (e.g. operator experience).
Methodology of the additional targeted reviews to support model conceptualisation
Given an expected dearth of evidence on the cost-effectiveness of biopsies using SF biopsy systems compared to biopsies using CF in the UK context, we performed additional targeted reviews of cost-effectiveness evidence of diagnostic strategies at the point of biopsy to support the model conceptualisation. These aimed to (1) identify value components of the biopsy approaches, (2) characterise alternative mechanisms of evidence linkage from disease prevalence, diagnostic accuracy, choice of treatment to final outcomes and (3) identify any UK relevant sources of evidence.
Literature searches
We screened cost-effectiveness modelling studies identified by the main search described in Systematic review methods (study selection, data extraction, quality assessment) to identify evaluations of diagnostic strategies in the same diagnostic pathway position proposed for SF biopsy systems (i.e. at the point of biopsy), but which do not fulfil the full inclusion criteria for the population, interventions and comparators defined for the main cost-effectiveness review (see Methodology of the cost-effectiveness of software fusion biopsy for suspected prostate cancer). We also considered for inclusion cost-effectiveness modelling studies identified in the cost-effectiveness reviews conducted for a previous assessment of the cost-effectiveness of TP for diagnosing PCa recently developed to inform NICE guidance. 116 Studies included in the review of cost-effectiveness studies in scope with this assessment (see Methodology of the cost-effectiveness of software fusion biopsy for suspected prostate cancer) were also included in the targeted review.
Study selection
We included studies considered potentially informative for the model conceptualisation and for the identification of relevant input sources of evidence with a particular emphasis on those used in UK-based or UK generalisable models. The relevance of these studies to inform the model conceptualisation under the current decision problem was assessed as described in Results of the review of the cost-effectiveness of MRI fusion biopsy for suspected prostate cancer.
Quality appraisal
Given the pragmatic nature of this review and its aims, identified studies did not undergo a formal quality appraisal.
Synthesis of evidence
The studies identified as potentially relevant were summarised in tabular form. A subset of the studies identified was selected for detailed extraction, if they were model-based cost-effectiveness studies which complied with at least the following criteria:
-
UK-relevant evaluations of alternative prostate biopsy approaches
-
UK policy-relevant assessments of diagnostic tests for PCa or
-
evaluations comparing alternative MRI-influenced biopsy approaches.
The value of diagnostic technologies is to a large extent dependent on how downstream clinical management choices based on diagnostic information impact on final outcomes. Therefore, most of these value components rely on indirect mechanisms of value accrual to determine trade-offs in final outcomes, health system costs or both, the balance of which determines the net value of the technologies.
For the subset of studies considered most relevant for the conceptualisation, we synthesised narratively the following types of evidence:
-
key components of value, that is, ways in which the diagnostic technologies may lead to impacts on individuals’ health and/or system cost compared to their alternatives (i.e. the comparators)
-
characterisation of the modelling/evidence-linkage approaches used to quantify the key indirect components of value, identifying underlying structural assumptions
-
value drivers, that is, factors expected to have a considerable impact on cost-effectiveness
-
main areas of uncertainty and evidence scarcity, as well as approaches taken to deal with these issues
-
sources of heterogeneity, and approaches taken to handle heterogeneity
-
data sources relevant to the UK decision making-context.
The focus of the narrative synthesis was placed on the characterisation of value accrual mechanisms that may be relevant to the current assessment of SF biopsy systems, rather than exhaustive characterisation of all value components.
Methodology of the review of economic evidence provided by the companies
We reviewed the economic evidence submitted by the companies in response to requests for information (RFIs) by NICE and the EAG. We listed this economic evidence grouped into three categories:
-
full economic evaluations that consider both costs and consequences (including cost-effectiveness, cost–utility and cost–benefit analyses)
-
resource use and cost data
-
other.
Full economic evaluations were considered for inclusion in one of the two other economic reviews (see Methodology of the cost-effectiveness of software fusion biopsy for suspected prostate cancer or Methodology of the additional targeted reviews to support model conceptualisation) as appropriate given their study characteristics.
Resource use and cost data were considered for the parametrisation of the de novo model.
Results of the review of the cost-effectiveness of magnetic resonance imaging fusion biopsy for suspected prostate cancer
Search and studies identified
Records from the searches described in Systematic review methods (study selection, data extraction, quality assessment) were examined to identify potentially relevant economic records. Figure 17 in Appendix 9 shows the PRISMA flow diagram for this review which details results at each stage of the review. A total of 27 studies were identified as being potentially relevant to the assessment of cost-effectiveness of SF biopsy versus CF biopsy. After screening the titles and/or abstracts, 26 studies were excluded. One full-text publication was retrieved and assessed for inclusion, Pahwa et al. 117 This study met the full set of inclusion criteria and was included in this review of SF biopsy for suspected PCa.
We note that the economic evidence submitted by the companies in response to information requests (RFIs) by NICE and the EAG largely consisted of resource use and cost data (mostly acquisition, maintenance, and training costs) on the SF they commercialise. This evidence was considered for the parameterisation of the model and is discussed in Biopsy procedure adverse events costs.
In addition to this, KOELIS and Kebomed also submitted economic evidence consisting of:
-
a cost-analysis in a Japanese setting
-
two business case analysis
-
a slide set describing what is referred to as a cost–benefit analysis comparing MRI-influenced biopsy using KOELIS Trinity with TRUS-guided biopsy in the US healthcare setting.
This evidence is not considered further in this report, as the economic analyses did not comply with the inclusion criteria of this review. For example, the cost–benefit analysis presented in the slide set did not appear to include HRQoL outcomes (only cost and diagnostic outcomes). Furthermore, the evidence provided lacked sufficient detail to be informative for the model parameterisation (e.g. the methodology, sources of evidence and assumptions were not clearly described in the business case analyses) and it was not peer-reviewed.
Review of Pahwa et al.
The Pahwa et al. 117 study is summarised in Table 7. The quality assessment of this study is reported in Table 62 (see Appendix 9).
| Study country, perspective | Population | Population characteristics | Diagnostic strategies | Analytical approach, time horizon | Outcomes |
|---|---|---|---|---|---|
| USA, not stated | Biopsy-naive men with indication for biopsy due to elevated PSA levels or CS DRE findings | Mean age 65 years PCa prevalence 50% Probability of CSPCa (given PCa) 50% |
1. Systematic TRUS biopsy for all. 2–4. Non-contrast mpMRI for all followed by MRI-influenced biopsy (2. CF, 3. MRI fusion or 4. in-bore) for those with clinically suspect lesions on mpMRI. Those without mpMRI detected suspicious lesions do not receive biopsy. 5–7. Non-contrast mpMRI followed by MRI-influenced biopsy (5. CF, 7. MRI fusion or 7. in-bore) for those with clinically suspect lesions on mpMRI. Those without MRI detected lesions receive systematic TRUS biopsy |
Cohort decision tree model Lifetime horizon |
Costs QALYs NHB ICER |
Pahwa et al. 117 evaluated the cost-effectiveness of mpMRI followed by MRI-influenced biopsy using alternative MRI-influenced methods (SF, CF and in-bore MRI biopsy) compared to systematic TRUS biopsy in individuals with suspected PCa in the US healthcare system. The study’s perspective is not explicitly stated, but the costs included suggest a societal perspective.
The study population consisted of biopsy-naive men with elevated PSA levels and/or CS DRE findings. In the base-case analysis, the cohort had a mean age of 65 years, and a PCa prevalence of 50%; this prevalence estimate was varied in subgroup analyses by age groups. Cancer prevalence by age was sourced from a study which reviewed US cancer statistics and autopsy data; it is unclear if this estimate is reflective of a biopsy-naive population. The probability that PCa is CS cancer [defined as tumour volume > 0.5 cm3, a GS higher than 6, or with a Gleason pattern of 4 or 5 (if GS ≤ 6) or not confined to the prostate] was assumed to be 50%, based on a previous cost-effectiveness study.
The study compared three diagnostic strategy types with the following test sequences: (1) systematic biopsy for all individuals, (2) mpMRI for all individuals followed by MRI-influenced biopsy for those with clinically suspicious lesions detected on mpMRI (positive mpMRI) and no further testing for those with negative MRI findings and (3) mpMRI for all individuals followed by MRI-influenced biopsy for those with a positive mpMRI result and TRUS systematic biopsy for those with a negative mpMRI result. Each strategy type with a MRI-influenced component (2 and 3) was evaluated separately for each alternative MRI-influenced method (SF, CF and in-bore MRI biopsy). Individuals who did not undergo biopsy or had a negative result did not receive treatment. Those who undergo biopsy and have a positive result are classified according to cancer significance and receive treatment consisting of a mix of active surveillance, watchful waiting, ADT and radical treatments. mpMRI was described as non-contrast and biopsy as TRUS; no further details on the specifications of the test were provided.
The decision model consisted of a cohort decision tree structure which characterised diagnostic pathways, treatment allocation and assigned lifetime payoffs by classification and treatment allocated. It started by classifying individuals according to their true disease status including clinical significance [no PCa, clinically non-significant (CNS) or significant PCa]. Individuals were subsequently classified according to the diagnostic accuracy of test sequences in each strategy according to diagnosis results and their true underlying disease status (including disease significance).
The metrics of diagnostic accuracy for the different biopsy approaches included the sensitivity to detect (1) cancer (for systematic biopsy only), (2) CS cancer (only for targeted biopsies), (3) clinically insignificant cancer (only for targeted biopsies) and (4) a probability of correctly identifying the tumour aggressiveness. In addition, all biopsy approaches were assumed to be 100% specific to detect PCa. The diagnostic accuracy of SF biopsy was NR as specific to any particular software fusion technology. The evidence used to inform the sensitivity of SF to detect clinically insignificant cancer was pooled from various MRI-fusion systems, while the sensitivity to detect CNS cancer was informed by evidence on ARTEMISTM ProFuse.
The costs considered in the model included the costs of MRI, biopsies (systematic, CF, SF or in-gantry), histopathological evaluation, workdays lost, biopsy complications and lifetime treatment (cost payoffs). The cost of SF (mean US$ 731 including physician fees) applied in the model was not technology specific.
The model does not consider the impact on HRQoL of biopsy complications.
Treatments considered in the model included radical prostatectomy, external beam radiation therapy, brachytherapy, ADT, active surveillance and watchful waiting. Treatment distributions conditional on diagnosed clinical significance were sourced from a US registry and supplemented by assumptions.
The QALY pay-offs at each terminal node are conditional on the cancer presence (and its clinical significance), treatment status (treated, untreated), and type of treatment (independent of the clinical significance of cancer). The lifetime QALY pay-offs for treated patients are mostly derived from a previous cost-effectiveness study118 which used a state transition Markov model to compare expectant management (active surveillance or watchful waiting) with initial treatments (brachytherapy, intensity-modulated radiation, radical prostatectomy) on men with low-risk, clinically localised PCa. The studies pooled to inform the treatment effectiveness in the external model are not clearly described. The Markov model captures disease progression and recurrence, short- and long-term AEs from treatment choice on lifetime quality-adjusted life expectancies (QALEs). The Pahwa et al. 117 model does not capture the probability of developing new cancer during the lifetime for men with NC.
The cost pay-offs are conditional on the diagnostic status (diagnosed, undiagnosed/later diagnosed), treatments received (for diagnosed patients) and the clinical significance of cancer (for undiagnosed or later diagnosed patients). The lifetime costs are also derived from Hayes et al. model. 118 As the risk of developing new cancer is not considered, no lifetime cost is assigned to men with NC.
Cost-effectiveness results are expressed as fully incremental cost-effectiveness ratios (ICERs) and net health benefits (NHBs) at US $50,000 per additional QALY. Sensitivity analysis included probabilistic sensitivity analysis, one-way sensitivity analyses and scenario analysis. The scenario analysis considers the cost-effectiveness of each strategy at three alternative Gleason cut-off scores for CS cancer (3 + 4, 4 + 3, ≥8). The authors also present subgroup analysis by three age subgroups (41–50 years; 51–60 years; and 61–70 years), with prevalence and life expectancy varying across subgroups.
Pahwa et al. cost-effectiveness results
The cost-effectiveness base-case results are summarised in Table 8. Strategy 4, consisting of mpMRI followed by in-bore biopsy for those who test positive on imaging and no further biopsy for those with a negative imaging result, had the highest NHB at US $50,000 per additional QALY.
| Total costs (US$) | Total QALYs | ICER (US$ per QALY) | NHB (QALYs)a (95% CI) | |
|---|---|---|---|---|
| Strategy 2: mpMRI, CF biopsy, no systematic biopsy if negative | 17,630 | 9.250 | – | 8.997 (7.34 to 10.21) |
| Strategy 4: mpMRI, in-bore biopsy, no systematic biopsy if negative | 17,870 | 9.308 | $4147 | 8.950 (7.54 to 10.21) |
| Strategy 3: mpMRI, SF biopsy, no systematic biopsy if negative | 18,608 | 9.198 | Dominated | 8.826 (7.33 to 10.19) |
| Strategy 5: mpMRI, cognitive biopsy, systematic biopsy if negative | 18,802 | 9.269 | Dominated | 8.893 (7.45 to 10.18) |
| Strategy 7: mpMRI, in-bore biopsy, systematic biopsy if negative | 19,042 | 9.326 | $65,111 | 8.946 (7.60 to 10.17) |
| Strategy 6: mpMRI, SF biopsy, systematic biopsy if negative | 19,780 | 9.217 | Dominated | 8.822 (7.43 to 10.16) |
| Strategy 1: Systematic biopsy | 19,133 | 9.082 | Dominated | 8.699 (7.08 to 10.15) |
Strategies with CF components (2 and 5) have higher NHB than the corresponding strategies with SF biopsy (3 and 6) than those of MRI-influenced fusion biopsy in both the base-case analysis and for scenario analysis where the definition of CS disease is varied. SF biopsy generally results in lower total QALYs and higher total costs compared to cognitive biopsy.
The authors claimed that the one-way sensitivity analysis results suggest that the cost-effectiveness drivers are cancer prevalence, the proportion of CS cancer and the sensitivity of MRI. However, we note that results are not presented and that the ranges within which the model parameters were varied do not seem to follow any other rationale other than assuming great parameter uncertainty and testing extreme input values. Scenario and subgroup analysis results were consistent with those of the base-case analysis.
Generalisability and relevance of the Pahwa et al. study to the decision problem in the current assessment
The Pahwa et al. 117 study has several features that limit its generalisability and relevance to the decision problem in the current assessment.
Firstly, the study’s perspective does not correspond to the NICE reference case, as it seems to take a US societal perspective rather than that of NHS and PSS. This difference in perspective implies that the opportunity costs considered in Pahwa et al. 117 are unlikely to be comparable to those relevant to this assessment. It also means that the range of included costs in Pahwa et al. 117 are not directly generalisable to this assessment.
Another area where there is a lack of alignment between this assessment and Pahwa et al. 117 is the study population considered and how this links to the position of the tests in the diagnostic pathway. Since the study predates the routine use of MRI to screen individuals with suspected PCa for biopsy, the study population is not limited to individuals with a MRI Likert or PI-RADS score ≥ 3. The study population is also limited to those individuals without a prior biopsy. Population characteristics such as prevalence, a cost-effectiveness driver in Pahwa et al.,117 are, therefore, likely to differ between this study’s population and the population defined by the scope of this assessment, thus limiting the generalisability of the study findings to this assessment.
The diagnostic pathway in the study also differs from the one currently recommended in UK clinical practice, as it does not allow for repeat biopsies.
The way in which diagnostic accuracy was modelled in Pahwa et al. 117 is another limitation, as the tests classified individuals according to PCa presence and its clinical significance. Clinical recommendations for management of PCa in the UK are made based on prognostic risk (characterised via a five-tier risk score), rather than clinical significance of disease alone. Therefore, the diagnostic classification in the study is insufficiently granular to allow linking classification to clinical management choices in the UK context.
Another issue in Pahwa et al. 117 is that it did not model a specific SF technology. The way in which the direct costs and diagnostic accuracy of SF were modelled implies that these estimates are equivalent across different technologies. This assumption is not justified, but the equivalence of the direct costs of alternative technologies is debatable, even if diagnostic accuracy can be assumed equivalent, given the similar functioning of these software systems. The study also does not model or discuss potential diagnostic accuracy and/or cost modifying factors, such as the method of estimation (rigid vs. elastic), the biopsy sampling method (targeted alone vs. combined), the biopsy approach (transperineal vs. transrectal, local anaesthesia vs. general anaesthesia), etc. These factors have been identified in the scope of this assessment as features of interest and may impact on the cost-effectiveness results.
Finally, the evidence linkage between clinical management and final outcomes in the Pahwa et al. 117 model lacks flexibility to allow adaptation to other jurisdictions, since these outcomes are modelled as pay-offs estimated from an external US-Markov model. It is unclear whether the distribution of treatments used to weigh the costs and QALYs pay-offs in the study is likely to match what is observed in a UK setting. However, even if the treatment distribution was reflective of UK clinical practice, the external Markov model also quantifies lifetime outcomes specific to the US setting. Therefore, it is not possible to easily implement alternative UK relevant treatment choices and reflect the impact of these on long-term cost and HRQoL outcomes.
Therefore, the EAG concludes that the Pahwa et al. 117 study cannot directly inform, or be adapted to inform, the decision problem in the current assessment.
Results of the additional targeted reviews to support model conceptualisation
The results of the searches are given in detail in Appendix 1. In total, 15 cost-effectiveness models116,119–133 were considered potentially relevant to inform the de novo model conceptualisation for inclusion. These studies are summarised in Table 63, Appendix 9.
Of the 15 cost-effectiveness models identified at the first stage of the review, 9 were selected for a more in-depth review, as these were identified as the most appropriate to support the conceptualisation of the de novo model given the relevance of:
-
the comparisons and position in the diagnostic pathway – studies which compared biopsies conducted with MRI-influence methods (i.e. targeted and/or combined biopsies) for PCa diagnosis;119,120,124,129,130
Studies included in the model conceptualisation review
A summary description of the subset of identified studies116,119–121,123–126,129,130 included in the model conceptualisation review is provided in this section, followed by a critical review (see Critical review). A summary table of these studies is presented in Appendix 9 (Table 64) alongside further details on the studies.
Scope of the study
The population in the majority of studies comprises individuals with suspected PCa, who enter a secondary care diagnostic pathway,116,119,121,123,125,126,129,130 while other studies consider patients being screened for PCa. 120,124
A variety of biopsy approaches were compared in the studies; these differ by route of access (transrectal vs. transperineal), type of anaesthesia used (general vs. local), sample collection method (targeted vs. systematic vs. mapping or saturation biopsy) and MRI-influenced methods (SF, CF and in-bore MRI). Two models are of particular interest for UK policy. Souto-Ribeiro et al. 116 reports a previous DAR by the Southampton EAG. This study established two main comparisons between biopsy approaches: (1) local anaesthetic transperineal (LATP) biopsy (with any type of biopsy device) versus local anaesthesia transrectal ultrasound (LATRUS) biopsy and general anaesthesia transperineal (GATP) biopsy and (2) LATP with specific freehand devices versus LATRUS and versus transperineal transrectal biopsy conducted with a grid and stepping device conducted under local or general anaesthetic. The NICE CG131 model123 evaluated alternative follow-up strategies of individuals with suspected PCa and placed little emphasis on alternative biopsy approaches.
Some studies modelled the possibility of repeat biopsies. 116,119,121,125,126 These studies varied in how they specified: who would receive a repeat biopsy, what proportion of those eligible would receive one (or more) repeat biopsies, the type of biopsy received, and the number of subsequent biopsies allowed (if more than one).
Classification
In most studies, the diagnostic accuracy of the biopsy procedure classifies individuals as not having PCa or having non-CS or CSPCa. 116,119,121,123–126,129,130 The exception was the study by Hao et al., in which classification is done by ISUP grade. 120 Both types of classification are usually defined by histopathological features of the biopsied lesions (graded according to GSs).
The specificity of biopsy, to detect PCa, is assumed perfect across most models, therefore individuals without PCa cannot be misclassified as having the disease. However, some studies considered the possibility of individuals with CNS PCa misclassified as CS. 124,129,130
Choice of clinical management
Decisions on patient management at diagnosis could be determined by the biopsy diagnostic outcomes alone125,126,129,130 or with other factors also influencing treatment allocation. 116,119–121,123,124
In three models125,126,129,130 patient management was attributed according to individuals’ classification in terms of disease presence and clinical significance of disease. This classification was established based on the diagnostic accuracy of the biopsy approaches. Some models tracked the individuals’ underlying cancer prognostic risk and used this information, jointly with the diagnostic outcomes, to allocate treatment. For example, the Southampton DAR model116 allocated treatments based on disease presence, clinical significance of disease and underlying cancer risk distribution.
For patients diagnosed with PCa, the primary treatment allocation was conditional on:
-
diagnosed clinical significance of disease, true cancer risk category and disease spread116,123
-
GS, PSA level and age124
-
type of biopsy (targeted or systematic), cancer risk category and age. 119
A range of evidence sources were used to inform the distribution of treatments for diagnosed PCa. Amongst these, the following are relevant in the UK context:
-
the Southampton DAR model116 based treatment distribution by risk category on UK clinical guidance and observed treatment allocation from national audit data134
-
the NICE NG131 model123 used observed primary treatment distributions by risk category from UK registry data112
-
the PROMIS trial125,126 assumed that treatment choice was guided by diagnosed disease clinical significance alone.
Individuals diagnosed as not having PCa were discharged to follow-up,121,123,125,126 or returned to the screening schedule. 120,124 One study116 conditioned the individuals’ subsequent management after a no PCa diagnosis on whether they had been misclassified [true negative (TN) results led to discharge and false negative (FN) results (patients with PCa of any risk category) to routine PSA monitoring]. This assumption was not justified, and it is not clear how in clinical practice the two groups of individuals (TN and FN) would be distinguished so that distinct treatment decisions could be made for each group.
Outcomes
The evidence linkage approaches applied in the identified studies to connect patient classification and subsequent treatment choices with longer-term outcomes differed in whether PCa progression was explicitly modelled as an intermediate outcome or not.
Only two studies did not model disease progression. 129,130 Pahwa et al. 129 conditioned lifetime QALYs and cost payoffs on diagnostic status (i.e. whether cancer had been diagnosed or remained undiagnosed), underlying true disease status (no PCa, CNS or CSPCa) and type of treatment received. Venderink et al. 130 used a long-term Markov model that only allowed for transitions from alive to death states, with survival conditional on type of treatment received and the underlying true disease clinical significance, with the diagnostic status (diagnosed vs. undiagnosed cancer) determining whether individuals received treatment. 130
All other models considered disease progression from localised to metastatic disease, although health states and possible state transitions varied across models. 116,119,121,123–126 Some studies modelled progression from localised to metastatic disease, and conditioned disease progression on underlying risk category and being correctly diagnosed/treatment received. 119,121,125,126 Other studies modelled sequential disease progression across disease risk categories (from low- to intermediate-risk and from the latter to high-risk disease) for localised disease followed by progression from the high-risk localised to metastatic disease. In these models, the probabilities of transitioning to later disease stages were conditioned on the underlying true disease status (including risk category) and being diagnosed as having CS or non-significant disease. 116,123 The screening studies modelled progression differently in the preclinical stage and in the clinical states. 120,124
All the disease progression models shared the assumption that PCa mortality only applied to patients with metastatic disease. Treatment for patients identified as having cancer reduced disease progression to metastatic cancer compared to untreated patients, and thus reduced the probability of dying from PCa for these patients. The transition probabilities for treated and untreated patients in the Markov disease progression were estimated by calibration or partially observable Markov model decision processes (as progression is an unobservable process). The data sources and calibration methods, used to estimate these transition probabilities, differed across models and are reviewed below for the two most relevant UK models. Details on the remaining models are in Appendix 9.
The PROMIS model125,126 calibrated the probability of progressing from localised to metastatic disease by risk category and treatment received, combining risk-stratified survival data and proportion of patients with metastases from the PCa Intervention versus Observation Trial (PIVOT),109 with the mortality in the metastatic subgroup of the STAMPEDE trial. 113 The PIVOT observation arm was used to inform the transition probabilities for individuals with PCa who did not receive active treatment (due to correct classification on misclassification depending on the risk category). The PIVOT radical prostatectomy arm was used to inform the transition probabilities for those treated with active treatment (true positives with intermediate and high-risk cancer). The ‘treatment’ effects of being diagnosed on disease progression were thus informed by randomised comparative efficacy evidence.
The model used in the previous DAR116 and in the NICE NG131 model123 disaggregated disease progression by cancer risk categories and used calibration to estimate transition probabilities. The calibration method estimated transition probabilities first for the transition from high-risk to metastatic disease, then from intermediate- to high-risk disease, and finally from low-risk to intermediate-risk disease can be derived. The calibration was done separately for the undetected and detected cancers using different data sources. Transition probabilities for the undetected cancers used cumulative metastases risk rates by cancer risk category from the watchful waiting arm in the Scandinavian Prostate Cancer Group Study Number 4 (SPCG4) trial135 jointly with and Swedish life-table data (from 1999 to reflect background mortality in the trial). For the diagnosed cancers, the data sources for calibration included: cancer-specific survival by risk category sourced from a UK registry study,112 all-cause survival for people with metastatic PCa from the STAMPEDE trial,59 and UK life-table (from 2010 to 2022 to reflect background trial mortality in STAMPEDE). Thus, this calibration approach relies on an indirect naive comparison to derive the ‘treatment’ effects of being diagnosed on disease progression, which may introduce bias on the probabilities of disease progression used in the model.
In general, disease progression models, survival outcomes for individuals with PCa were conditional on having metastatic disease and age. Two models116,123 further conditioned mortality on whether metastatic disease was diagnosed (and therefore, received treatment for metastatic cancer) or not. Metastatic mortality data sources of relevance to the UK context include different publications of the STAMPEDE study, a UK-based trial which compared the survival outcomes of men with newly diagnosed metastatic, high-risk or node-positive cancer treated with alternative cancer treatments. The PROMIS and related models estimated the probability of metastatic death using early (median follow-up of 20 months) survival data of men with newly diagnosed metastatic PCa from the control arm (who received SOC consisting of androgen depleting therapy) of the STAMPEDE trial. The NICE NG131 and related models used a later survival data cut (median follow-up 43 months) from the DTX and control arms of the STAMPEDE trial that includes individuals with metastatic and non-metastatic disease. 59
Health-related quality-of-life outcomes of patients with PCa were most frequently conditioned on having metastatic disease,116,119–121,123–126 age116,119–121,123–126 and treatment received and time since treatment initiation,120,124,130 although other factors having been considered in select models (see Appendix 9). The UK-relevant utility sources for patients with PCa in the long-term outcome models include Torvinen et al. 136 – for the disutility of metastatic disease, Ara and Brazier, 2010137 – for the disutility of ageing, Mowatt et al. 133 – for the disutility of treatment-related AEs (combined with rates of AEs) from Donovan et al. 138.
Most models considered the cost of treatment for patients with diagnosed localised or locally advanced PCa (radical treatment or active surveillance)116,119–121,123–126,129,130 and management of treatment AEs. 116,121,123,125,126 Patients with undiagnosed PCa would incur the costs of routine follow-up116,119,121,123,125,126,129 or of delayed radical treatment. 129 The studies also considered the costs of metastatic disease treatment with or without staging and follow-up tests. 116,119,121,123–126 Two models assumed diagnosed metastatic disease would be treated differently if diagnosed (DTX would be added to androgen depleting therapy) compared to undiagnosed metastatic disease and that treatment with DTX would vary with age. 116,123 Some models included an end-of-life cost for patients who died from PCa,116,119,120,123,124 with one study conditioning the end-of-life costs on age at death. 124
The costs of individuals who did not have PCa were not clearly reported for most models, but, where reported, consisted of the costs of routine follow-up. 116,119,123,124
In UK-relevant models, treatment and follow-up resource use was informed mainly by UK [clinical and technology appraisal (TA)] guidance, as well as other published data (e.g. a randomised control trial informed AE rates of treatment138) and supplemented with assumptions. End-of-life costs were uprated to the relevant price year based on Round et al. 139 Unit costs were sourced mainly from national published sources.
Critical review
Value components
The value components of the biopsy tests, in the studies included in the conceptualisation review, are summarised in Table 65 (see Appendix 9), which distinguishes between value components that require evidence linkage and those that are direct impacts of the tests. Direct value components of biopsy included the costs of the procedure, and its AEs (with associated complication costs and negative health impacts). The indirect value components identified here are linked to diagnostic accuracy.
All studies in the conceptualisation review modelled two common value components requiring evidence linkage to be quantified; these are an improvement of outcomes resulting from an increased and/or earlier detection of PCa and of CSPCa. To capture the value of increased/earlier detection of CSPCa, the majority of models determined a single clinical management strategy for each biopsy classification option. Classification (under an assumed clinical management strategy), together with true disease status (either true cancer risk category, e.g. NICE NG131 model123), or cancer grade, for example, Hao et al. 120 was then linked to the outcomes. Clinical management strategies either consisted of a single treatment option125,126 or a particular mix of treatments. 123
Only three studies explicitly modelled the impact on outcomes resulting from improved detection of CNS PCa. 124,129,130 Although the evidence linkage requirements for modelling this value component are similar to those described above for the increased and/or earlier detection of CSPCa, these are the only models in which the parameterisation of biopsy diagnostic accuracy allowed for CNS PCa to be misclassified as CS. Individuals who have been misclassified thus incur the costs and harms of unnecessary radical treatment but have limited ability to benefit in the long-term from treatment, compared to those who have CS disease.
Another value component relates to the costs and/or harms incurred for individuals who undergo a repeat biopsy conditional on the result of the index (or subsequent to index) biopsy. Although these costs and harms are a direct impact of the biopsy, this is classified as an indirect value component because the decision to repeat the biopsy is conditional on the classification of the index biopsy in the testing strategy and, therefore, requires evidence via linkage. Differences in diagnostic accuracy between biopsy approaches partially determine the proportion of individuals classified as eligible for a repeat biopsy, that is the proportion of those who will incur the costs and harms of an additional biopsy. In addition to the linkage via classification, modelling this value component also requires a decision rule to define who is eligible for a repeat biopsy (e.g. all or a proportion of the individuals classified as not having CS cancer at the previous biopsy in the test sequence). One study further assumed (in scenario analysis only) that with one type of biopsy a smaller proportion of individuals initially classified by the previous biopsy in the test sequence as eligible for a repeat biopsy would receive repeat biopsies compared to the alternative biopsy approach. 116
The biopsy value components with direct impact on outcomes modelled in the studies included the costs of the biopsy procedure, and the costs of managing AEs of biopsy, as well as the detrimental health impacts of AEs.
Evidence linkage
The evidence linkage used to model the indirect value components relied in most studies on a common model structure whereby a decision tree approach to track individuals’ diagnostic outcomes (and, in some models, biopsy AEs) was linked to a Markov model to capture long-term outcomes.
In most models, diagnostic classification categorised individuals (correctly or not) as having (1) no PCa, (2) CNS or (3) CSPCa. The definition of clinical significance differed across models but was generally defined in terms of a GS threshold or a three-tier cancer risk categorisation (defined in terms of GS, PSA levels and cancer stage). This stratification reflects differences in diagnostic accuracy and prognostic for individuals in the different risk categories. In general, the low-risk disease category was assumed to correspond to true non-CSPCa, while the intermediate- and high-risk cancer categories corresponded to CS disease.
Treatment allocation for each diagnostic classification group was usually determined. This could be a single treatment option for each group (such as in PROMIS125,126 where all of those identified with CS cancer received radical treatment). Or it could be a pre-defined mix of treatments, where the distribution of treatments differs by group (e.g. with a higher proportion of radical treatments for those at higher cancer risk). 123 In either case, the linkage does not aim to disentangle the outcome for the diagnosed/treated by treatment received.
In most studies, the impact of being correctly or incorrectly classified by the biopsy was modelled as an effect on disease progression to metastatic cancer, and PCa death only affected individuals who were in metastatic disease health states.
Chapter 5 Independent economic assessment: York model
Diagnostic strategies
The model evaluated two strategies for two alternative comparisons: (1) targeted SF biopsy versus targeted cognitive biopsy and (2) combined (targeted and systematic) SF biopsy versus combined cognitive biopsy. The four strategies could not be incrementally compared due to the mechanism of evidence generation for the diagnostic accuracy, which relied on separate evidence networks.
The test sequence and clinical management for each strategy:
-
all patients receive the index biopsy:
-
If biopsy result suggests no PCa or ISUP grade 1, a proportion of patients undergo repeat biopsy. Patients who do not undergo repeat biopsy are managed in accordance with their diagnosed ISUP grade/CPG or discharged to routine monitoring.
-
If biopsy result suggests ISUP grade 2 or greater, the individual receives treatment according to CPG.
-
-
for the patients who receive repeat biopsy:
-
Individuals are clinically managed according to the highest ISUP grade/CPG score between the two biopsy results or discharged to routine monitoring if the biopsy suggests no PCa.
-
Model development
Conceptualisation
The value components identified in the review supporting conceptualisation (see Results of the additional targeted reviews to support model conceptualisation) were:
-
direct value components of biopsy, including the costs of the procedure and its AEs (with associated complication costs and negative health impacts) and
-
indirect value components, including the increased or earlier detection of any PCa, of CSPCa, or of non-CS cancer, and the reduction of repeat biopsies.
From the review, supporting the conceptualisation (see Results of the additional targeted reviews to support model conceptualisation), we have identified several key aspects to consider in the conceptualisation of the de novo model, which we describe below and pertain to the diagnostic accuracy, the concept of under- and overdiagnosis, the modelling of disease progression and issues with outcome evidence sources.
The histopathological biopsy results are expressed in terms of GS (see Description of health problem) and sometimes including lesion core length or cores positivity. In order to estimate the diagnostic accuracy measures applied in the models, the results of the biopsy are typically collapsed into one no PCa and two PCa categories (CNS and CS). The collapse of diagnostic information into these categories implies an information loss, as the granularity of biopsy results is not preserved in the classification according to biopsy accuracy (GS ranges from 2 to 10). It also implies a judgement on the definition of CS disease at a specific Gleason threshold, with some models using a Gleason threshold of 3 + 3 and others 3 + 4.
Furthermore, making clinical management choices between active surveillance and a range of radical surgical treatments and/or radiotherapy requires information provided by the biopsy diagnostic accuracy, but also information with prognostic value like PSA levels and disease stage at diagnosis. In clinical practice, patient preference is also another factor influencing the choice of management strategy. Due to this, several models made assumptions on how to map from the two PCa classification into three-tier risk cancer prognostic risk classifications. Current UK clinical guidance,10 for the management of newly diagnosed localised or locally advanced PCa, recommends an even more granular prognostic risk classification, the CPG system, which uses the same type of information as the previous risk classification but classifies patients into five categories. The most recent update of the NICE CG131 defines four alternative clinical management strategies for individuals diagnosed in the different groups (same treatment strategy for CPG 4 and 5), whereas previous guidance defined three management strategies (one for each risk category).
The concepts of under-/overtreatment are not clearly defined in the literature. In general terms, overtreatment seems to arise when patients with PCa of favourable prognostic receive radical treatment (e.g. radical prostatectomy or radiotherapy) instead of active surveillance. In contrast, undertreatment would arise when patients with worse disease prognosis receive active surveillance, rather than radical treatment. So under-/overtreatment can occur if the clinical management approach taken is not commensurate with the true disease prognostic risk, which may be due to:
-
disease not been correctly classified in terms of its underlying prognostic risk; and/or
-
the prognostic risk categorisation not being accurately predictive; and/or
-
treatment decision rules not being followed due to clinical variation and/or patient preference.
The move from the three-tier to the five-group classification aims to improve the identification of patients who have slow progressing disease and should be managed with active surveillance. For these patients, the harms (and costs) of radical treatment are likely to offset its long-term benefits.
The misclassification of individuals in the lower-risk categories/groups as having a higher prognostic risk (overdiagnosis) may result in net health losses if it leads to unnecessarily radical treatment (overtreatment). Therefore, reducing overtreatment is an important value component of biopsy. The few previous studies which modelled this value component did so by capturing misclassification of CNS as significant cancer and linking this to the outcomes of more radically treated patients. This is an imperfect link, as it lacks the flexibility to identify individuals with CS who are at the lower end of the prognostic risk spectrum (i.e. CPG 2 or favourable intermediate risk), and, thus, quantify the net benefit of providing active surveillance to this group.
Most studies modelled the reduction of underdiagnosis, that is, the value of increased or earlier detection of PCa in individuals whose disease will progress at a faster rate if not managed with radical treatment. This value component was modelled by capturing misclassification of CS as non-significant cancer (or NC) and linking this to the outcomes of patients undiagnosed for CS cancer. Since this classification does not allow the identification of individuals with favourable intermediate risk, it may overestimate the net benefit of treating with more radical treatment individuals with true CSPCa.
While most studies modelled longer-term outcomes as a function of PCa disease progression, we identified two alternative structural choices to model the unobservable disease progression: (1) directly between localised (or locally advanced disease) to metastatic disease and (2) sequentially progression across three health states defined by category of true underlying prognostic risk. These two approaches also differ in terms of evidence requirements for parameterisation, with the second approach requiring more data and/or more structural assumptions to be imposed in the model. We also identified alternative methods to estimate unobservable transitions probabilities, namely calibration and partially observed Markov process models.
We have also identified issues with outcomes evidence. Some models used naive/unadjusted comparisons, that is, used different data sources to describe outcomes for different groups. This may result on bias. Additionally, all models used data sources to describe outcomes according to true disease that use an imperfect reference standard (typically PSA results).
These key aspects grounded the de novo model conceptualisation, an overview of which is provided below.
Risk stratification: In terms of risk stratification, and given that the current UK clinical guidance10 recommends a five-category prognostic risk classification, the CPG system, there is the need to consider this more granular classification system in the modelling. Despite this being a five-tier classification system, only four alternative clinical management strategies are recommended in the NICE Guideline (same treatment strategy for CPG 4 and 5), therefore CPG 4–5 can be reasonably collapsed in analysis. However, broader evidence does not typically use the CPG system, for example, we found no diagnostic studies reporting results using CPG, and therefore ISUP grade was used in the diagnostic component to reflect CPG tiers.
Determining diagnostic accuracy: The review work (see Systematic review methods (study selection, data extraction, quality assessment)) focused on identifying and synthesising studies (RCTs and within-patient comparisons) comparing CF and SF targeted prostate biopsy methods. The multinomial model used in the synthesis of this evidence (see Multinomial synthesis model) compares the alternative biopsy methods in how they classify individuals across the following categories: 1 (no PCa), 2 (ISUP grade 1), 3 (ISUP grade 2), 4 (ISUP grade 3) and 5 (ISUP grade 4 or 5 pooled together). This allows a more complete consideration of evidence across ISUP grades, extending from previous approaches that focus on either cancer detection rates (typically defined as NC vs. ISUP grade ≥ 1) or detection rates of CS cancer (typically defined as NC or ISUP grade 1 vs. ISUP grade 2 or above). 116,117,119–121,123–126,130
The synthesis model considers the distribution of individuals by ISUP grades and relates this distribution across technologies using a set of odds ratios, the quantities pooled across studies. Note that such a model does not identify concordance between methods in biopsy test results (further explanation in Appendix 10). The application of the synthesised odds ratios to an externally derived distribution of probabilities of test results for one of the tests (say SF) retrieves the expected distribution of probabilities for the other test (CF). This calculation of absolute probabilities is described in Appendix 10
The evidence synthesis model does not consider the accuracy of either method in relation to a reference standard (by virtue of the evidence available for inclusion), that is, it does not consider the extent of misclassification with either any of the modelled methods. This has important implications for economic modelling as, in the absence of a robust and representative outcomes RCT, evidence linkage is required, facilitated by knowing the extent of misclassification of the different tests in relation to true disease status, to allow determine its consequences to health and economic outcomes.
To consider accuracy evidence, a structural approach is required that extends the synthesis model to integrate such evidence. The approach developed here is described in Diagnostic pathway.
Diagnostic pathway and repeat biopsy: The need and the accuracy of repeat biopsies is a potential value component for SF methods, in relation to CF. This may arise indirectly from improved diagnostic accuracy of the method used for the first biopsy, that is, a more accurate identification from a first biopsy can lead to a decreased pool of individuals eligible for re-biopsy. We did not identify comparative evidence suggesting differences in the rates of repeat biopsy between cognitive and SF. However, the clinical advisers to the EAG suggested that a potential value component for SF, is that by consulting the stored cartograms produced by MRI systems, the MDT could better target re-biopsy. There is, however, a lack of evidence to parameterise impact beyond what can be captured via diagnostic accuracy. We will explore the potential value of such a case in scenario analyses.
Treatment of PCa: There is UK-relevant evidence on the distribution of treatments for patients identified at different CPG groups. Our model will therefore be reflective of the different mixes of treatments used at different CPG levels (see Treatment of prostate cancer).
Modelling of long-term outcomes: To reflect the value of increased/earlier detection, the long-term outcomes component of the model will need to condition on true disease status and the diagnosed disease category (given the PCa management strategy determined by the diagnosed disease category). None of the existing long-term models have been developed using the five-category prognostic risk classification based on CPG system, recommended in the current UK clinical guidelines. 10 Therefore, a de novo inference model will be developed for this assessment. For its structure, and given that this assessment focuses on the diagnostic pathway, considering PCa disease progression over time and incidence is not as relevant as for the NG131 model, which aimed to model monitoring strategies. Therefore, the increased complexity of the structure used in the NG131 model123 (and in the Southampton DAR116) may not be justified for the purpose of modelling biopsy within the diagnostic pathway. Additionally, evidence to support such a complex structure is sparse (if existing at all), and therefore its parameterisation would rely on a number of assumptions that cannot be verified. However, the added complexity of such a structure would allow for the time profile of treatment costs on those that leave the diagnostic pathway under a monitoring strategy to be better captured.
In terms of evidence to quantify the impact of alternative treatments on outcomes, comparative effectiveness evidence will be preferred to avoid bias. The most contemporary evidence available will be used to inform the inference submodel.
Further details on the inference model and on how this will be incorporated in the cost-effectiveness decision model are provided in Modelling of long-term outcomes.
Model structure and parameterisation
Modelling of first biopsy results
Determining diagnostic accuracy
As identified above (see Conceptualisation), the fact that the evidence synthesis conducted as part of this assessment does not consider the accuracy of the different biopsy methods in relation to a reference standard has important implications for economic modelling. In the absence of a robust and representative outcomes RCT, economic modelling relies on evidence linkage facilitated by knowing the extent of misclassification of the different tests in relation to true disease status and determining its consequences to health and economic outcomes.
The extent of misclassification can, however, be made explicit by the accuracy matrix, the elements of which reflect the probabilities of obtaining a particular test result with one method conditional on a particular level of (true) disease status. Together with prevalence estimates, this matrix determines the distribution of test results, shown at the top of Figure 8.
FIGURE 8.
Illustration of the relationship between prevalence, and the accuracy and distribution of test results across five categories, for two hypothetical tests.
Note that, due to the nature of biopsy and histological examination of the biopsy specimen, it is reasonable to assume that false-positive results are not possible, that is, if cancer is histologically identified, then it is present. This implies that biopsy methods cannot identify a higher category than true disease status, and therefore zero probability is attributed to such cases in the above accuracy matrices.
Where multiple methods are of interest, the problem becomes more complex for two reasons. First, the prevalence (i.e. the true distribution across categories) is independent of test results and therefore a common prevalence estimate needs to ground all distributions of test results, and be consistent with these. Second, explicit accounts of accuracy need to respect both the prevalence estimates and the marginal distribution of test results derived from the synthesis. Therefore, a structural approach is required for determining accuracy from the marginal distributions obtained through application of the synthesis model.
Summary of approaches used in previous cost-effectiveness models
From the conceptualisation reviews (see Results of the additional targeted reviews to support model conceptualisation), two cost-effectiveness reviews have focused on a similar context where no accuracy evidence was synthesised.
A previous DAR,116 from now on referred as the Southampton DAR, conducted a meta-analysis on cancer detection rates [using relative risks (RRs)] including studies comparing the biopsy methods of interest to the decision problem (e.g. LATP vs. LATRUS), and did not include evidence comparing either method to a reference standard. In this work, the authors sourced the baseline distribution for LATRUS and its accuracy matrix, from an external diagnostic accuracy study (the PROMIS study125,126). The authors then applied the synthesised RRs of cancer detection for LATP biopsy (derived for marginal distributions) directly to both (1) the conditional probability of LATRUS identifying CS cancer conditional on true disease status, and to the (2) conditional probability of identifying CNS cancer (assumption imposed in the base case). The conditional probability of NC given true disease status was then adjusted to be one minus the remaining. The way the RRs were applied in the model is not consistent with the way in which they were derived, in that the RR derived from the synthesis model refers to the relative increase in detection rate with one method in relation to another; the RRs were therefore derived on marginal probabilities and not on conditional probabilities. Their application to conditional probabilities in such a way implies that the increase in accuracy of detecting cancer with a particular test is independent of whether the cancer was CS or non-CS, and that the increase in accuracy of detecting non-CS cancer given the patient has non-CS cancer is equal to the increase in accuracy of detecting CS cancer, given the patient has CS cancer.
An alternative study, Wilson et al. 121 also investigating LATP in relation to LATRUS, assumed no difference in the expected accuracy of the biopsy methods in the comparison of interest. Therefore, the authors sourced prevalence and accuracy estimates for LATRUS from the PROMIS study125,126 and used it to represent the expected results for both biopsy methods. In reflecting uncertainty, the authors sampled from the accuracy matrix directly, taking two independent samples to represent the two different biopsy methods, and therefore generate differences in the accuracy matrix between the methods, due to randomness only.
None of the existing approaches has direct applicability in the current assessment, where a disaggregation by ISUP grade is required.
Summary of methods
The approach used in the current assessment was designed to:
-
be grounded on the results of the evidence synthesis model
-
return a true distribution across ISUP grade categories (prevalence) that is internally valid, that is not lower than the estimated ISUP Grade detection rates of the different biopsy methods
-
be grounded on available evidence on the likely accuracy of targeted MRI fusion conditional on ISUP grade
-
define accuracy matrices for the remaining biopsy methods of interest that are consistent with both prevalence and the distributions of biopsy results from the evidence synthesis.
To achieve this, an extension to the synthesis model was developed, drawing on the broader evidence in Review of additional prevalence, test results and diagnostic accuracy evidence. To allow for an internally consistent approach, we grounded our methodology on the distribution of test results obtained with MRI-influenced methods and their accuracy. Given that disease prevalence is fully determined by these two results, the prevalence evidence identified in Review of additional prevalence, test results and diagnostic accuracy evidence will not be explicitly incorporated in our analyses but will instead be used qualitatively to put our results into context.
The methodology is summarised below. A more comprehensive description of the methods used is presented in Appendix 10.
Distribution of test results
The distributions of test results across the disease categories for the relevant biopsy methods within each disconnected component of the network in Model 1a were computed by applying network-specific baseline distributions to the results of the NMA. Building from the analyses in the evidence synthesis section, the baseline distributions were sampled from a multinomial likelihood with an uninformative Dirichlet prior distribution for its hyperparameters, to allow for uncertainty in describing the data from the empirical studies.
Accuracy matrix for software fusion
Evidence on the accuracy matrix for SF, sourced from the literature, was used to characterise the elements of the accuracy matrix probabilistically in the model. A multinomial likelihood was used to describe the distribution of test results conditional on each particular level of true disease status (each line in the matrix in Figure 8) with Dirichlet uninformative prior distributions.
Prevalence
The derivation of prevalence followed two steps, the first consisted of the analytical derivation of an initial prevalence estimate from the marginal distribution and accuracy matrix for SF. The second step entailed applying a constraint to ensure that the prevalence is always higher than the detection rates (by ISUP grade) observed across all tests.
Accuracy matrix for remaining biopsy methods
The diagonals of the accuracy matrices for the remaining biopsy methods were determined by the prevalence and the test-specific distribution of results. To define the remaining non-zero and free elements of the matrix, uninformative beta distributions were used, constrained so that their multiplication by the prevalence retrieves the test results estimated within the evidence synthesis.
The extension to the synthesis model, developed to determine accuracy, was implemented alongside the synthesis model in a Bayesian framework estimated through Markov chain Monte Carlo methods using WinBUGS 1.4.3. 140 Due to the sparseness of evidence in other networks, this was applied to Model 1a [see Model 1a: Multinomial synthesis model (base case)] which includes SF, CF and systematic biopsy in a first connected network, and the combination of software and CF with systematic biopsy in a second connected network. As in the evidence synthesis, model convergence was assessed where possible by running two independent chains with different starting values looking at a history plot and through inspection of Gelman–Rubin diagnostic plots. Model fit was assessed by comparing the mean total residual deviance to the number of independent data points contributing to the analysis. 71
Given that the approach proposed here is heavily data driven, sensitivity analyses focused on varying the data sources for the baseline distributions and accuracy matrix.
Results
The extension to the synthesis model reflects the data sources described in Model 1a: Multinomial synthesis model (base-case) for the baseline distribution of test results for SF, the reference method. The extension model also required data to characterise the accuracy matrix for the reference biopsy method, two sources for these data were available (see Section Review of additional prevalence, test results and diagnostic accuracy evidence and Appendix 8, Distribution of test results obtained with cognitive fusion or software fusion biopsy) and were used here. According to the data sources used, the following analyses were conducted:
-
Main analysis, for the subgroup of biopsy-naive individuals: baseline distribution of test results for SF sourced from biopsy-naive data from Filson et al. 96 relative accuracy data from the multinomial evidence synthesis model (see Multinomial synthesis model) which was incorporated in this extension, and accuracy data from Mortezavi et al. 141 Mortezavi et al. 141 was chosen for the main analysis over Zhou et al. 142 as it more closely reflects the lower accuracy observed in UK-specific evidence sources.
-
Subgroup analysis for previous negative-biopsy individuals: all sources were equal to those used in the main analysis except the baseline distribution of test results for SF which was sourced from previous negative-biopsy data from Filson et al. 96
-
Sensitivity analysis to data source on baseline distribution: all sources were equal to those used in the main analysis except the baseline probabilities, which were based on biopsy-naive data from PAIREDCAP (2019),88 for network 1.
-
Sensitivity analysis to data source on accuracy matrix: all sources were equal to those used in the main analysis except accuracy data which was sourced from Zhou et al. 142
Note that given the two networks are disconnected, results are reported separately for comparisons of CF and SF – network 1, and for comparisons of combined cognitive/SF with systematic biopsy – network 2. Note that while network 1 includes systematic biopsy, results for this biopsy method are NR here.
Main analyses (biopsy naive)
Table 9 shows the results of the structured approach applied to the main analysis for the subgroup of biopsy-naive patients. Results are internally consistent, and consistent with the sources of evidence these drew upon. They mirror the high level of uncertainty in the evidence base.
| Network 1 | (Distribution of test results) | (Distribution of test results) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.516 (0.416 to 0.615) | 0.186 (0.131 to 0.249) | 0.136 (0.068 to 0.211) | 0.098 (0.052 to 0.157) | 0.064 (0.031 to 0.114) | 0.457 (0.403 to 0.513) | 0.173 (0.137 to 0.214) | 0.196 (0.157 to 0.233) | 0.108 (0.079 to 0.144) | 0.066 (0.043 to 0.095) | ||
| CF | SF | ||||||||||
| (Accuracy matrix) | (Accuracy matrix) | ||||||||||
| (Prevalence) | ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 |
| Network 1 | (Distribution of test results) | (Distribution of test results) | |||||||||
| 0.460 (0.335 to 0.583) | 0.250 (0.152 to 0.356) | 0.127 (0.034 to 0.261) | 0.131 (0.046 to 0.231) | 0.033 (0.001 to 0.107) | 0.348 (0.273 to 0.418) | 0.223 (0.179 to 0.273) | 0.232 (0.168 to 0.311) | 0.115 (0.081 to 0.152) | 0.082 (0.054 to 0.114) | ||
| Combined CF and systematic biopsy | Combined SF and systematic biopsy | ||||||||||
| (Accuracy matrix) | (Accuracy matrix) | ||||||||||
| (Prevalence) | ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 |
| 0.121 (0.007 to 0.238) | NC | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 0.318 (0.212 to 0.452) | 1 | 0.829 (0.529 to 0.994) | 0.171 (0.006 to 0.471) | 0 | 0 | 0 | 0.671 (0.538 to 0.796) | 0.329 (0.204 to 0.462) | 0 | 0 | 0 |
| 0.262 (0.193 to 0.341) | 2 | 0.300 (0.016 to 0.64) | 0.362 (0.083 to 0.674) | 0.338 (0.111 to 0.55) | 0 | 0 | 0.251 (0.167 to 0.347) | 0.204 (0.128 to 0.288) | 0.544 (0.443 to 0.64) | 0 | 0 |
| 0.183 (0.119 to 0.265) | 3 | 0.189 (0.006 to 0.526) | 0.140 (0.005 to 0.422) | 0.192 (0.008 to 0.537) | 0.479 (0.213 to 0.804) | 0 | 0.224 (0.121 to 0.343) | 0.059 (0.012 to 0.138) | 0.207 (0.112 to 0.322) | 0.510 (0.387 to 0.65) | 0 |
| 0.116 (0.077 to 0.174) | 4 or 5 | 0.125 (0.004 to 0.389) | 0.111 (0.004 to 0.357) | 0.111 (0.004 to 0.362) | 0.101 (0.002 to 0.332) | 0.552 (0.299 to 0.882) | 0.111 (0.046 to 0.199) | 0.047 (0.011 to 0.112) | 0.130 (0.063 to 0.217) | 0.140 (0.068 to 0.226) | 0.573 (0.467 to 0.687) |
| 0.121 (0.007 to 0.238) | NC | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 0.318 (0.212 to 0.452) | 1 | 0.709 (0.289 to 0.987) | 0.291 (0.013 to 0.711) | 0 | 0 | 0 | 0.528 (0.206 to 0.824) | 0.472 (0.176 to 0.794) | 0 | 0 | 0 |
| 0.262 (0.193 to 0.341) | 2 | 0.249 (0.01 to 0.689) | 0.437 (0.07 to 0.836) | 0.314 (0.028 to 0.78) | 0 | 0 | 0.078 (0.001 to 0.273) | 0.152 (0.011 to 0.384) | 0.770 (0.523 to 0.975) | 0 | 0 |
| 0.183 (0.119 to 0.265) | 3 | 0.126 (0.002 to 0.488) | 0.124 (0.003 to 0.449) | 0.134 (0.002 to 0.482) | 0.616 (0.148 to 0.981) | 0 | 0.132 (0.005 to 0.441) | 0.135 (0.005 to 0.411) | 0.130 (0.004 to 0.403) | 0.603 (0.338 to 0.92) | 0 |
| 0.116 (0.077 to 0.174) | 4 or 5 | 0.195 (0.004 to 0.618) | 0.187 (0.005 to 0.603) | 0.173 (0.004 to 0.561) | 0.163 (0.004 to 0.543) | 0.281 (0.006 to 0.865) | 0.069 (0.001 to 0.282) | 0.070 (0.001 to 0.265) | 0.066 (0.001 to 0.27) | 0.071 (0.001 to 0.266) | 0.724 (0.402 to 0.98) |
The prevalence estimates inferred by the extended synthesis model are in line with those available in the literature (see Review of additional prevalence, test results and diagnostic accuracy evidence and Appendix Distribution of test results obtained with cognitive fusion or software fusion biopsy), perhaps closer to the lowest available estimate of cancer prevalence (i.e. low probability of NC). This is, however, expected, as the inferred prevalence in the extended model is bounded by a composite of all five tests, and is sampled from a distribution that allows for even higher cancer prevalences than those identified by the composite of all five tests.
In terms of distribution of test results, the results obtained here (presented at the top of each accuracy matrix in Table 9) are consistent with those in the synthesis section (see Tables 68–71, Appendix 10 for detailed comparisons). In summary, within network 1 (which includes cognitive and SF), the results suggest that SF may retrieve a higher detection of cancer at ISUP grade 2 and above when compared to CF, with the detection at ISUP grade 2 being highest. These results are not statistically significant, in that CrIs overlap significantly. Table 10 presents the information on distribution of test results converted onto detection rates at thresholds of categories. This information highlights that: SF presents a similar level of detection at ISUP grades 4–5, slightly increased detection of ISUP grade 3 or above of 1.3%, increased detection of ISUP grade 2 or above of 7.1% and increased detection at ISUP grade 1 or above of 5.9%.
| ISUP grade | Estimated detection rates with the different biopsy methods | |||
|---|---|---|---|---|
| Network 1 | Network 2 | |||
| CF | SF | Combined CF and systematic biopsy | Combined SF and systematic biopsy | |
| 4 or 5 | 0.064 (0.031 to 0.114) | 0.066 (0.043 to 0.095) | 0.033 (0.001 to 0.107) | 0.082 (0.054 to 0.114) |
| 3 to 5 | 0.162 (0.102 to 0.237) | 0.175 (0.135 to 0.217) | 0.164 (0.064 to 0.27) | 0.197 (0.152 to 0.243) |
| 2 to 5 | 0.299 (0.209 to 0.396) | 0.370 (0.322 to 0.424) | 0.290 (0.173 to 0.428) | 0.429 (0.358 to 0.502) |
| 1 to 5 | 0.484 (0.385 to 0.584) | 0.543 (0.487 to 0.597) | 0.540 (0.417 to 0.665) | 0.652 (0.582 to 0.727) |
In terms of accuracy, the results for network 1 suggest that SF is more accurate at detecting the correct category (the diagonal of the accuracy matrix is always higher for SF), with higher differences at ISUP grades 1 and 2.
The accuracy matrix results show that despite cognitive presenting a higher likelihood of an ISUP grade 1 result, there is an increased accuracy of SF at ISUP grade 1. This is due to, with CF, individuals at higher ISUP categories being misclassified as grade 1. The accuracy matrix shows increased accuracy at ISUP grade 2 for SF, but retains a significant proportion inaccuratelyclassified as ‘NC’ [with a probability of 0.25 95% CrI (0.17 to 0.34)] which is higher than the proportion innacurately classified as ISUP grade 1 [with a probability of 0.20 95% CrI (0.13 to 0.29)]. A similar effect is observed in ISUP grade 3, where the likelihood of being classified as ‘NC’ is higher for SF than for CF – probabilities 0.224 95% CrI (0.121 to 0.343) versus 0.189 95% CrI (0.006 to 0.526). This is a result of the increased detection at ISUP grade 2 not being matched by a similar level of detection at ISUP grade 1.
By multiplying the prevalence by the respective element of the accuracy matrix, the joint probability matrix is obtained (see Table 67, Appendix 10). This matrix identifies, for a cohort with the mix of ISUP grades as per the prevalence estimates, the probability of both events, that is the probability of a particular ‘true’ ISUP grade and a particular test result. This matrix identifies that, at all grades, the probability of an accurate result is 0.524 95% CrI (0.411, 0.628) for SF – 0.12 at NC, 0.10 at ISUP grade 1, 0.14 at ISUP grade 2, 0.09 at ISUP grade 3 and 0.07 at ISUP grade 4 or 5. The probability of an accurate result is 0.413 95% CrI (0.256, 0.583) for CF – 0.12 at NC, 0.05 at ISUP grade 1, 0.09 at ISUP grade 2, 0.09 at ISUP grade 3 and 0.06 at ISUP grade 4 or 5. The highest difference between software and cognitive is observed at ISUP grades 1 and 2 (approximately 5% increase in each with software). Notably, in terms of misclassification, the overall proportion of ISUP grade 3 identified as ‘no cancer’ is higher with SF 4.2% than with CF (3.5%). This implies that the key trade-offs for SF are the benefits achieved by the general increase in detection, but particularly for ISUP grades 1 and 2, at the expense of a slightly higher proportion of grade 3s that will not be detected as cancerous.
Network 2 (including software and CF combined with systematic biopsy) shows higher identification in the distribution of test results (due to the baseline used) to but qualitative results are similar to those in network 1 to noting that there is substantial uncertainty in these results. Detection rates at thresholds of categories show that cancer detection is expected to be higher with combined software to at all levels to but particularly at ISUP grade 2 or above where detection is 13.9% higher than with combined CF and at ISUP grade 1 or above where detection is 9.2% higher that with combined CF.
In terms of accuracy to at all grades to the probability of an accurate result is 0.655 95% CrI (0.471 to 0.816) for combined SF and 0.438 95% CrI (0.218 to 0.665) for combined CF. For both combined strategies to the likelihood of a ‘no cancer’ result for ISUP grades 2 and 3 is still relatively high to but this is now comparable to the likelihood of an ISUP grade 1 result.
Subgroup analysis (previous negative biopsy)
We conducted a subgroup analysis to where the baseline distribution of test results for SF was sourced from Filson et al. 96 but using the group of individuals recruited into this study that had previous negative-biopsy results. However, the diagnostic accuracy evidence synthesis and the accuracy matrix are still sourced as per the main analysis to grounded on evidence over biopsy-naive and repeat biopsy patients. Summary results of distribution of test results for the subgroup analysis are presented in Table 72 (Appendix 10) alongside their interpretation. Prevalence probabilities and results of the accuracy matrices are also presented in Tables 73 and 74 to Appendix 10.
Sensitivity analysis
Sensitivity analyses change the main sources of evidence of the main analyses (on biopsy-naive patients): a first sensitivity analysis uses an alternative baseline distribution of test results for SF [from PAIREDCAP (2019)],88 and a second analysis uses an alternative source for accuracy matrix evidence [from Zhou et al.]. 142
In both these analyses, results for the accuracy matrices could only be presented for the first network because of increased uncertainty.
The summary results in Tables 75 and 76, Appendix 10, for the first sensitivity analysis, indicate that results are sensitive to the distribution of test results. The PAIREDCAP study distribution showed a higher proportion of ‘no cancer’ identified with SF (31% vs. 46% in the main analysis grounded on Filson, Table 9), identical in ISUP grade 1, and higher proportions across all remaining ISUP categories (26%, 16% and 10%, respectively for ISUP grades 2, 3 and 4 or 5, vs. 20%, 11% and 7% in the main analysis grounded on Filson, Table 9). The distribution of test results for ISUP grade 4 or 5 are similar between software and CF, but are significantly increased for software at ISUP grade 2, slightly increased at ISUP grade 3 and slightly reduced for ISUP grade 1.
The summary results in Tables 77 and 78, Appendix 10, for the second sensitivity analysis indicate that results on the distribution of test results are only slightly sensitive to the source of evidence on the accuracy matrix in Filson (see Table 9). The main difference distribution of test results for ISUP grades 4 or 5 are slightly higher for SF in this analysis in relation to the main analysis in Table 9. The estimates of the accuracy matrices (in Appendix 10) show increased accuracy (in classifying individuals in the right category) for both technologies in relation to the main analysis in Table 9, which reflect the data from Zhou et al. 142 However, differences between the technologies in the accuracy matrices are encountered in individuals with true ISUP grade 4 or 5 where the misclassified have an equal chance across being identified across all other categories in cognitive but are slightly less likely to be identified as NC or ISUP grade 1 with SF. For those in ISUP grade 2, sensitivity analysis indicates a low likelihood of the misclassified being identified as grade 1 with SF (and therefore being more likely to be classified as ‘no cancer’), which was not observed in the main analysis.
Diagnostic pathway
The diagnostic pathway is structured as a decision tree that captures AEs, repeat biopsies and classifies individuals according to the result of the biopsy (or biopsies), and the true disease status (see Diagnostic pathway), defined as ISUP grade for those with PCa (ISUP grades 1, 2, 3, 4–5). Figure 9 shows a simplified schematic of the decision tree illustrating biopsy-related mortality, sequence of biopsies, and cost and HRQoL pay-offs which apply for each strategy. The diagram does not show the biopsy-related non-fatal events, as these do not modify the probability of moving forward in the diagnostic pathway. The probabilities of AEs are applied as weights to adjust the branch costs and HRQoL pay-offs. The diagram also does not show how the classification is established conditional on the true disease state and test accuracy at each biopsy, or how the classification conditions the probability of repeat biopsy; this is illustrated in Appendix 11, Table 79.
FIGURE 9.
Decision-tree schematics. •, probability node; , terminal node; #, complement probability (1-probability); Bx, biopsy; c_, cost; du_, disutility; p1, probability of surviving the first biopsy; p2, probability of repeat biopsy (is conditional on first biopsy result); p3, probability of surviving second biopsy.
All individuals who undergo the first biopsy are at risk of biopsy-related non-fatal and fatal AEs. The mortality risk corresponds to the complement of probability (p1). For those who survive the first biopsy, the probability of receiving a repeat biopsy (p2) is conditional on the result of the first biopsy. Individuals who test positive at first biopsy (biopsy result ISUP grade ≥ 2) and survived the first biopsy receive no further testing (p2 = 0). Those who test negative (no PCa or ISUP grade 1) and survived the first biopsy have a probability of undergoing repeat biopsy (p2), with the remaining individuals receiving no further testing. The individuals who receive a repeat biopsy are again exposed to biopsy mortality risk (p1-p3), and to a probability of having non-fatal biopsy AEs. Time is not modelled within the decision tree, so events are assumed to occur instantaneously (or in rapid succession prior to long-term model entry); this is in line with the other cohort models examined in Results of the additional targeted reviews to support model conceptualisation.
The decision-tree models repeat biopsies for a proportion of individuals who have a negative-first biopsy result. In the base case, this proportion is not conditional on whether the strategy includes a cognitive or SF component. The base-case analysis assumes that the proportion of repeat biopsy is only conditional on the result of the first biopsy (15.45% and 5%, if the result of the first biopsy indicated a lesion with ISUP grade 1 and no PCa, respectively) as per a previous DAR. 116
Similar to a previous DAR,116 we assume the same rates of biopsy complications per biopsy approach for the first and repeat biopsies. However, because we assume a different distribution between transperineal and transrectal biopsy, for the first and repeat biopsies in the diagnostic pathway, the repeat biopsy complication rates reflect a higher proportion of TP (10% GATP and 60% LATP) compared to first biopsy (65% LATP) (see Biopsy procedure costs).
In the base-case scenario, the diagnostic performance of the repeat biopsy is assumed the same as of the first biopsy. The model allows exploring a degradation in the diagnostic performance of repeat when compared to first biopsy; the impact of applying this alternative assumption is assessed through scenario analysis.
We note the (first and repeat) biopsy results are assigned in the decision tree immediately before the biopsy mortality risk is applied, meaning the proportion of individuals in each category is adjusted by the proportion who survived the biopsy procedure (assuming the same mortality risk applies to all individuals regardless of true disease category and biopsy result). Similarly, we assumed that the biopsy AEs apply to all individuals who undergo a biopsy procedure.
The costs and QALY pay-offs in the decision tree capture the short-term impacts of first and repeat biopsy. First biopsy cost pay-offs apply to all branches and include the cost of the biopsy procedure and of associated AEs. Similarly, the QALY pay-offs of the first biopsy also apply to all decision-tree branches. These QALY pay-offs aim to quantify the QALY loss associated with biopsy procedural complications. The repeat biopsy-related costs (including the same cost categories as for the first biopsy) and repeat biopsy complications QALY loss apply only to the decision-tree branches which include a repeat biopsy.
The costs of the biopsy procedure vary by strategy to reflect the differences in cost between CF and SF with each of the MRI fusion systems modelled (see Biopsy procedure costs for the estimation of biopsy procedure costs). The biopsy procedure and AEs costs are both specific to the biopsy approach (LATP, GATP or LATRUS); these costs are estimated as a weighted average of the costs by biopsy approach (where the weights correspond to the proportion of LATP, GATP and LATRUS for each biopsy in the strategy). The QALY loss from biopsy-related complications also varies by biopsy approach to reflect the different biopsy complication rates by biopsy route of access (transperineal or transrectal) and, therefore, is also estimated as a weighted average by biopsy approach.
Clinical management conditional on biopsy final classification
There are 15 possible final classifications at the end of the diagnostic pathway, which are as follows:
-
For individuals correctly classified:
-
Diagnosed as having no PCa and without PCa;
-
Diagnosed as ISUP grade 1 and with ISUP grade 1;
-
Diagnosed as ISUP grade 2 and with ISUP grade 2;
-
Diagnosed as ISUP grade 3 and with ISUP grade 3;
-
Diagnosed as ISUP grades 4–5 and with ISUP grades 4–5;
-
-
For individuals misclassified:
-
Diagnosed as having no PCa and with:
-
ISUP grade 1;
-
ISUP grade 2;
-
ISUP grade 3;
-
ISUP grades 4–5;
-
-
Diagnosed as ISUP grade 1 and with:
-
ISUP grade 2;
-
ISUP grade 3;
-
ISUP grades 4–5;
-
-
Diagnosed as ISUP grade 2 and with:
-
ISUP grade 3;
-
ISUP grades 4–5;
-
-
Diagnosed as ISUP grade 3 and with:
-
ISUP grades 4–5.
-
-
The clinical management for each of these possible classifications is dependent on the diagnosed category. As detailed in Care pathways for the diagnosis and management of prostate cancer, current clinical guidance10 recommends that individuals, diagnosed as having localised or locally advanced disease (henceforth referred to as localised disease for simplicity), are involved in decisions about the management of their disease, with the range of management options offered varying as a function of their prognostic risk. Thus, patients with lower CPG scores (better prognosis) are offered more conservative management (active surveillance) with option to undergo radical treatment, while those with higher CPG scores are offered radical treatment as the preferred management option.
The diagnostic performance evidence only allows classifying patients according to their histopathological information (i.e. ISUP grade), which is only part of the prognostic information used to determine the CPG scores. Therefore, we made a simplifying assumption that ISUP grade can be used as a proxy for the individuals’ CPG score (e.g. CPG1 = ISUP grade 1), to allow establishing the evidence linkage between classification and clinical management and subsequently from this to treatment outcomes. Henceforth, we refer to the classification in the model in terms of CPG score, assuming interchangeability between ISUP grades and CPG scores. The treatment options for localised disease include active surveillance or radical treatment. Radical treatment includes radiotherapy [consisting of the model of brachytherapy or external beam radiotherapy for costing purposes (see Prostate cancer treatment costs – metastatic disease)], and radical prostatectomy.
For individuals identified as having PCa, the model assigns varying proportions of active surveillance and radical treatment, according to diagnosed CPG score (see Treatment of localised prostate cancer). All patients in the localised disease health states receive monitoring, with the set of monitoring tests and schedule varying according to whether they are receiving active surveillance or radical treatment. Individuals without a PCa diagnosis also receive monitoring, but its regime is less intensive compared to individuals diagnosed with PCa and is time limited (maximum of 10 years).
Prostate cancer treatment is associated with AEs, such as sexual, urinary and bowel dysfunction, with rates of AEs varying by treatment (see Localised treatment adverse events). AEs from PCa management are associated with disutility and costs of managing these events, which are quantified within the long-term model.
Modelling of long-term outcomes
Overview of the decision-analytic model
The long-term outcomes of the model cohort conditional on latent true disease status, the diagnosed disease category and PCa management assigned are quantified in a state transition Markov model. The model has yearly cycles (with a half-cycle correction applied) and a lifetime time horizon (40 years).
The core structure of the model is illustrated in Figure 10. Individuals who survived the biopsy procedure(s) in the diagnostic pathway can enter the model through the no PCa state if they are disease free or the localised (and locally advanced) disease state if they have PCa. Patients with PCa at model entry can remain in the localised disease health state or transition to the metastatic disease state at each yearly model cycle. The individuals who died due to the diagnostic procedure enter the ‘other cause’ death state, one of the two absorbent states in the model (highlighted in grey in Figure 10). Transitions to the other-cause death state are possible from the ‘no PCa’, localised and metastatic disease health states, with the same probability as the general population (see Other-cause mortality). The only other possible transition for the ‘no PCa’ state is to remain in the same state (i.e. the model does not consider that individuals can develop PCa, so disease progression is not modelled for those who do not have the disease at model entry). The metastatic health state is modelled as three tunnel health states (not illustrated in this diagram, Prostate cancer treatment adverse event costs – localised disease, Figure 12), where individuals can only stay in the two first tunnels states for a maximum of 1 year. Patients who transition to the metastatic health state can only remain in that health state or die. PCa mortality only applies to patients in the metastatic disease states.
FIGURE 10.
Long-term outcomes Markov model structure.

There are 15 possible localised disease health states (illustrated in the box below the model schematics), each reflecting the final classification (here expressed as CPG scores) attributed by the diagnostic pathway and the different treatments assigned conditional on the diagnosed category in the final classification.
Over the next subsections we provide details on the parameterisation of long-term transition probabilities.
Inference sub-model (disease progression by Cambridge Prognostic Group and treatment intensity)
The decision-analytic PCa model requires consideration of the impact of treatment decisions according to diagnostic accuracy. Treatment decisions are currently grounded on the identification of CPG groups, and therefore the outcomes component of the model aims to reflect: (1) differences in outcomes across the CPG risk groups that underlie treatment decisions in clinical practice and (2) the impact of different treatment intensities on each of these risk groups. Our conceptualisation review has not identified any previous cost-effectiveness model where treatment outcomes for five-level CPG groups have been considered (see Results of the additional targeted reviews to support model conceptualisation). Therefore, an estimation strategy was developed in this assessment grounded on the targeted review of evidence on the long-term outcomes of PCa (see Review of long-term evidence).
The brief overview of the wider literature highlights that, while there is evidence on the effectiveness of radical versus ‘conservative’ treatment options in delaying progression to metastatic disease, there are limited mortality benefits observed within the follow-up of clinical trials in this area. Also, we did not find evidence on treatment effectiveness stratified by CPG scores, despite the prognostic ability of the five-level score for PCa-specific death having been demonstrated in a large UK-based observational study. 112
The aim of the inference model is therefore to pull existing evidence together to predict differences in progression to metastatic disease by five-level CPG score and by treatment. Given this has not been directly observed, a calibration model was developed to infer these. The calibration model uses the structure of the decision-analytic model in Figure 10, but without considering the ‘no PCa’ health state, which has, thus, been faded out in the diagram.
The model structure is underpinned by the following assumptions. All individuals are assumed to begin with localised disease. They can continue to have localised disease, progress to metastatic disease or die from causes other than PCa. The speed of progression to metastatic disease is expected to depend on CPG group and is given by λi, where the index i reflects the CPG group. Other-cause mortality is age-specific and is determined by δage. Those with metastatic disease may (1) continue to live with metastatic disease, (2) die from PCa or (3) die from other causes. Following the NICE NG131 model,123 it was assumed that death from PCa could only occur after metastatic disease. The model was parameterised for each CPG score of interest to this assessment (CPG 1, 2, 3, and 4 and 5 combined).
The inference procedure is undertaken in two parts.
Part 1: identifying rates of progression to metastatic disease by CPG, λi
This part uses calibration. For any calibration process, two sets of parameters are of interest. The first concerns model parameters, some of which are unobserved and the target of inference, and others are observed and therefore evidence directly informs these. The second set concerns calibration targets, which are functions of the model parameters that have been observed and are used to identify the unobserved parameters under the model structure and other observed inputs. Table 11 lists the calibration parameters and targets and presents the results of the calibration model. A more detailed description of these parameters and their evidence sources is presented in the subsequent subsections.
| Description | Source | Parameter value | Results | |
|---|---|---|---|---|
| PART 1 | Calibration targets | |||
| 10-year PC death by CPG group at diagnosis | Gnanapragasam et al.112 pooled results for testing and training sets | 10-year PC survival (SE) (a, b parameters of a beta distribution): G1: 0.968 (0.007) (586, 19) G2: 0.938 (0.010) (577, 38) G3: 0.871 (0.016) (356, 53) G4/5: 0.763 (0.052) (50, 16) |
– | |
| Calibration model parameters | ||||
| Unobserved rate of progression from localised to metastatic disease, by CPG: λ1, λ2, λ3, λ4 | Unobserved (calibration parameters) | NA | Rate (SE): G1: 0.0101 (0.00236) G2: 0.0229 (0.00403) G3: 0.0645 (0.01058) G4/5: 0.1788 (0.08641) |
|
| Observed rate of PC mortality from metastatic disease, γ | STAMPEDE143 | Yearly rate of PC mortality in ADT arm of 0.162 (SE 0.0073), calculated from 5-year PC mortality | – | |
| Observed rate of death from other causes, age-specific | ONS life tables (2000–2)144 | Assumed mean age for each CPG group G1: 66.2 G2: 68.14 G3: 71.13 G4/5: 72.18 |
– | |
| PART 2 | Proportions under radical treatment vs. conservative management, by CPG | Gnanapragasam112 | G1: 0.53 G2: 0.70 G3: 0.81 G4/5: 0.95 |
– |
| Rate ratio for the development of metastasis of radical vs. conservative treatment | ProtecT55 | Rate ratio = 0.43 95% CI (0.26 to 0.72), log rate ratio mean = –0.834, SE = 0.2545 |
– | |
| Rate of progression from localised to metastatic disease, by CPG and by treatment | Unobserved | NA | Conservative management λ1(0) = 0.0143 (0.00357) λ2(0) = 0.0380 (0.00832) λ3(0)= 0.1197 (0.02812) λ4(0) = 0.3950 (0.22287) Radical treatment: λ1(1) = 0.0063 (0.00184) λ2(1) = 0.0165 (0.00357) λ3(1)= 0.0516 (0.00964) λ4(1) = 0.1674 (0.08066) |
|
Calibration targets
Our calibration target is 10-year PCa-specific mortality according to CPG group at diagnosis of localised disease, as reported in Gnanapragasam et al. 112 Our analysis combines groups 4 and 5, as the recommended treatment is the same for both groups. 10 We used a single data point for each CPG group of interest, at 10-year follow-up, in the calibration model. We used WebPlotDigitizer145 to extract point estimates and upper and lower CIs for PCa survival at 10 years (3652 days) from both training and validation sets in Gnanapragasam et al. 112 (from figures 1a and 2a). Standard errors (SEs) were calculated by considering the average distance between the point estimate and the upper and lower confidence limits (where both were available). The figures were then combined across data sets to derive a single estimate for each CPG, by weighting according to the inverse of their precision (analogously to a fixed-effect meta-analysis). Values for the combined CPG 4 and 5 group were derived by pooling the distributions, that is, assuming that the variance of the combined group is the weighted sum of the variances in each group. To describe the survival probabilities probabilistically, the parameters of beta distributions were specified using the method of moments. The estimates of 10-year PCa survival and the parameters of the beta distributions used to describe this in the calibration model are presented in Table 11. When simulating from the beta distribution to run the calibration, we preserved the ordering ensuring survival is highest in group 1 then group 2, group 3 and groups 4–5.
Calibration model parameters
The rates of progression from localised to metastatic disease by CPG score (λi, where i represent the CPG score groups of interest) were the unobserved parameters we sought to achieve inference on.
The remaining model parameters were observed. PCa specific mortality was assumed to only be possible after progression to metastatic disease, and therefore to inform this model parameter we used outcomes reported from STAMPEDE, a UK study. 143 Data from STAMPEDE’s control arm were used, as long-term hormonal treatment was the SOC at the time the study informing the calibration target was conducted. The individual patient data were reconstructed from the published Kaplan-Meier curve using the Guyot algorithm146 and a Weibull distribution was fitted using the flexsurv package in R. 147 PCa survival at 5 years, predicted by the fitted Weibull function, was 40.6% (95% CI from 43.9 to 47.0%), which was converted onto a rate assuming constant hazard. This resulted in a mean hazard of 0.162 and a 95% CI from 0.1777 to 0.1492. Assuming a symmetrical distribution, this implies a SE of 0.0073.
Office for National Statistics (ONS) life tables for men144 were used to parameterise the transitions to death from other causes (both from localised disease and from metastatic disease). Life tables were used for the years 2000–2 to approximate the mortality at the time of the Gnanapragasam study (2000–10). The average age at the start of that study differs by risk group according to data reported in the NICE model. 123 Using linear interpolation, we extended the three risk groups reported in the NICE report to the four risk groups we are considering. The ages assumed were: 66.2 years for Group 1; 68.14 years for Group 2; 71.13 years for Group 3 and 72.18 years for Groups 4–5. Due to the large sample size underlying the life tables, we did not consider this parameter uncertain.
Analysis methods
Using the parameters and targets described above, we ran the calibration analysis in the software package R, according to the algorithm below:
-
Sample a value from the uncertainty distribution for the target (PCa mortality at 10 years, for each risk group) and the known model inputs (metastatic mortality rate).
-
For each risk group, identify the value of the rate of transition from localised to metastatic disease (λi) that is consistent with the PCa mortality at 10 years sampled in step 1. Record the result.
-
Repeat steps 1 and 2, 10,000 times.
The optim function in R was used for the second step in this algorithm. 36 To find the rate consistent with the target, we defined a discrete time Markov model with the structure in Figure 11, and determined that its predicted 10-year survival should be compared against the target value. The loss function used was the squared distance from the proposed value to the target value. The Brent method was used with lower and upper bounds of 0 and 10, respectively. 148
FIGURE 11.
Calibration model structure and parameters.

Results
The results from the calibration procedure regarding the unobserved rate of progression from localised to metastatic disease by CPG are shown in Table 11. Comparisons of calibration parameter estimates with those from recent UK cost-effectiveness models are presented in Tables 80 and 81, Appendix 11.
Part 2: identifying the effect of treatment on the rates of metastasis
The estimated rate of progression to metastatic disease from the calibration exercise above reflect outcomes with current practice, which comprises a mix of radical and conservative treatment. In part 2, we back-calculate how these rates differ for the proportions treated with radical and conservative treatment observed in Gnanapragasam et al. 112 using an external estimate of effect for radical treatment.
Gnanapragasam et al. 112 reported the treatment mix by risk group observed in UK clinical practice during the years 2000–10. The treatment categories considered were: conservative management, brachytherapy, primary ADT, radical prostatectomy and radical radiotherapy. We further grouped treatments into two categories: conservative management and all other options, which we considered ‘radical treatment’ (see Appendix 11 for further details and comparison to NICE guidance). The split by risk group is shown in Table 11.
The rates of progression to metastatic disease inferred in the calibration step (part 1) reflect the treatment allocations in Gnanapragasam et al. 112 (see Table 11). To consider the rates of progression with and without radical treatment, we disentangle the effect of treatment by considering that the pooled estimate of the rate of progression is a weighted average of the rates under radical treatment and conservative management (weighted by the proportions treated). The rates under radical treatment are assumed to be the rates under conservative treatment multiplied by a rate ratio sourced from external evidence. For such, we use the treatment effect from ProTecT,55 the most recent UK study identified in the targeted review of the literature (see Review of long-term evidence). The rate ratio data for radical treatment pooled the PROTecT results for radical prostatectomy and radiotherapy, retrieving an estimate of 0.43 (95% CI from 0.26 to 0.72). Note that this estimate is similar to the US-based PIVOT study which estimated a HR for developing bone metastasis for radical prostatectomy of 0.40 (0.22–0.70).
Results
The results from the second part of the inference model, regarding the rate of progression from localised to metastatic disease by CPG and by treatment, are shown in Table 11.
Parameterisation of the prostate cancer health states transition probabilities
The transition probabilities from each of the 15 localised disease health states to metastatic disease were informed by calibration as described above [see Inference sub-model (disease progression by Cambridge Prognostic Group and treatment intensity)]. The calibration estimated the transition rates by true disease status and treatment assigned (active surveillance or radical treatment). Transition probabilities were subsequently estimated by weighting the annual transition rates according to the treatments assigned based on diagnosed category (see Treatment of localised prostate cancer), and then converted to annual transition probabilities assuming constant hazards over time (i.e. an exponential time to event distribution).
For patients in the metastatic disease health state transitions to PCa death were informed by PCa-specific death from the UK-STAMPEDE trial. 143 As described previously, a Weibull distribution was fitted using the flexsurv package in R data to the reconstructed individual-level PCa mortality data for the SOC (ADT) arm (metastatic patient subgroup) in Clarke et al. 143 The choice of parametric distribution was in line with a recent NICE TA evaluating enzalutamide in combination with ADT for hormone-sensitive metastatic cancer and based on a visual fit assessment conducted by the EAG (a full assessment of survival curve fit was considered out of scope for this assessment, so a targeted approach was taken). This baseline probability was parametrised in the executable model based on the flexsurv estimated Weibull coefficients (with a multivariate normal distribution fitted using the corresponding Cholesky decomposition for the PSA) and then adjusted by the effectiveness of contemporaneous combination treatments (i.e. in addition to ADT) weighted HR according to the current treatment distribution (see Treatment of localised prostate cancer). The weighted hazard ratios (HRs), for three combinations used in current clinical practice for first-line metastatic PCa compared to ADT alone, were applied to the baseline probability of PCa death to derive the metastatic to PCa death transition probability. The combination treatments considered in the model included DTX (HR vs. ADT 0.78, 95% CI: 0.66 to 0.93),143 enzalutamide (HR vs. ADT 0.66, 95% CI: 0.53 to 0.81)149 and apalutamide (HR vs. ADT 0.65, 95% CI: 0.53 to 0.79). 150 Lognormal distributions were fitted to each HR in the probabilistic model setup.
Other-cause mortality
Age-dependent other-cause mortality rates for men from Office for National Statistics (ONS) lifetables (2018–20 collection period)144 was used to estimate other-cause death probabilities in the long-term model. Parameter uncertainty was not considered for these inputs, due to the large sample size of the source data set.
Treatment of prostate cancer
In Prostate cancer management: active surveillance, watchful waiting and radical treatment options, we stated that the clinical management (choice component) of individuals with a localised and locally advanced PCa diagnosis in the model was conditional on (1) diagnosed CPG score for the treatment component and (2) on the type of PCa treatment (active surveillance or radical treatment) received for the routine monitoring component. For those in the metastatic health state, treatment includes androgen deprivation alone or in combination with other treatments. Here we present further details on the treatment distribution inputs in the model for (1) localised disease and (2) metastatic disease.
Treatment of localised prostate cancer
National Institute for Health and Care Excellence NG131 makes separate treatment recommendations by CPG score and conditional on patient preference and/or suitability for radical treatment (see Prostate cancer management: active surveillance, watchful waiting and radical treatment options) for individuals diagnosed with localised and locally advanced PCa. In order to reflect treatment allocation based on the diagnosed CPG score and the patient-level factors, we have sourced treatment allocation from Parry et al. 151 a study on the differences in localised and locally advanced treatment according to CPG in clinical practice in England. Our approach parameterising the treatment distribution contrasts to the approach taken in a previous DAR. 116 First, in the York model the distribution of active surveillance and radical treatments is conditional on the diagnosed disease status, whereas the Southampton DAR model conditioned this distribution on the ‘true’ disease category. Second, this previous model sourced the treatment distribution mostly from Gnanapragasam et al. 112 with further assumptions imposed on this distribution based on NPCA data. Both, Parry et al. 151 and Gnanapragasam et al. 112 reported treatment distribution by CPG for cohorts of newly diagnosed with non-metastatic cancer. However, we preferred to source the treatment distribution from Parry et al. 151 to Gnanapragasam et al. 112 because the data collection period is more recent (2014–7 vs. 2000–10) and had a higher sample size (61,999 vs. 10,139). Furthermore, Parry et al. 151 collected evidence from England, whereas Gnanapragasam et al. 112 was limited to data collected within the East of England Cancer Network area. We therefore considered the evidence in Parry et al. 151 study to be more contemporaneous and likely to be more reflective of current clinical practice than Gnanapragasam et al. 112
Table 12 contrasts the PCa management options distribution in the current and previous DAR. We note that the estimates applied in the York model, for individuals diagnosed with CPG 2 and 3, suggest less use of radiotherapy and more use of radical prostatectomy compared to what was applied to individuals with intermediate risk disease in the Southampton DAR model. 116 There are also differences between studies in the proportion of individuals receiving active surveillance and watchful waiting.
| Treatment choice based on | Southampton DAR model116 | York model | |||||
|---|---|---|---|---|---|---|---|
| ‘True’ disease status | Diagnosed disease status | ||||||
| Low-riska (%) | Intermediate-riska (%) | High-riska (%) | CPG1 (%) | CPG2 (%) | CPG3 (%) | CPG4–5 (%) | |
| Active surveillance | 95 | 12.7 | 0 | 88.7 | 51.6 | 33.7 | 24.1 |
| Radical prostatectomy | 2 | 21.9 | 17.6 | 6.6 | 27.2 | 26.3 | 22.8 |
| Radiotherapy | 3 | 52.8 | 52.4 | 4.7 | 21.3 | 40.0 | 53.1 |
| External radiotherapy | 2.3 | 48.7 | 52.5 | 3.6 | 19.0 | 38.2 | 52.3 |
| Brachytherapy | 0.7 | 4.1 | 0.9 | 1.1 | 2.3 | 1.8 | 0.8 |
| Watchful waiting | 0 | 12.7 | 29 | 0 | 0 | 0 | 0 |
In the York model, we assumed that individuals would not be treated with watchful waiting, because this is a monitoring strategy for individuals for whom potentially curative treatment is not suitable (or do not wish to undergo this type of treatments). mpMRI to inform prostate biopsy decisions is currently only recommended for people who can undergo radical treatment,10 so the exclusion of this treatment option was considered clinically plausible. We, therefore, assumed that individuals who were not treated with radical treatment underwent active surveillance.
Parry et al. 151 did not report the proportion of individuals who were treated with brachytherapy, a radiotherapy that is more costly than external therapy. We assumed that the proportion of individuals treated with radiotherapy who underwent brachytherapy by CPG was the same as in Gnanapragasam et al. 112 with the remaining patients receiving external therapy. Furthermore, we assumed that all patients treated with radiotherapy also received ADT (length of treatment conditional on diagnosed CPG score), as per the Southampton DAR. 116 We note that current clinical guidance recommends 6 months of ADT before, during or after radiotherapy for individuals with CPG 2 to 5, and for treatment to continue for up to 3 years for people with CPG 4 and 5.
There is also an important structural difference in the choice component of the York model compared to the Southampton DAR. 116 As stated in Inference sub-model (disease progression byCambridge Prognostic Groupand treatment intensity) and Parameterisation of the prostate cancer health states transition probabilities, the York model has flexibility to reflect different treatment distributions between conservative (active surveillance) and radical treatment on disease progression, as the calibration model estimates disease progression rates by type of treatment and the derived transition probabilities for each localised disease health state are adjusted as a function of the treatment distribution per diagnosed CPG. In contrast, the calibrated disease progression probabilities in the Southampton DAR model116 reflect the treatments received by the individuals in the outcome data used to derive them [i.e. Gnanapragasam et al. 112 for the diagnosed states and Bill-Axelson et al. 135 (observation arm)] and changes in the parameterisation of the treatment distribution only change how the cost and disutility of localised disease management are weighed in the model.
We fitted a Dirichlet probability distribution to the disaggregated observed count data by treatment type in Parry et al. 151 in the probabilistic parameterisation of the model.
Treatment of metastatic prostate cancer
Metastatic disease is treated initially with ADT alone or in combination, while disease is hormone-sensitive. Once disease progresses to hormone resistance, ADT is stopped and individuals will receive subsequent treatments.
Initial metastatic disease treatment (hormone-sensitive metastatic cancer) was assumed to consist of a mix of ADT alone and in combination with DTX, enzalutamide and apalutamide, similarly to a previous DAR. 116 We updated the distribution of metastatic treatments in the Southampton DAR to reflect the 74% reduction in the use of DTX between 2019 and 2020 suggested by the NPCA 2021 report. 4 Therefore, in the York model we assumed that 9% of individuals with hormone-sensitive metastatic cancer would be treated with DTX in combination with ADT (in contrast with the 36% in the Southampton DAR). We assumed that the difference in the proportion of treated with DTX between the two models (27%) would receive enzalutamide instead, since the NPCA 2021 report4 also suggested a considerable increase on the use of this alternative treatment. We have sourced the proportion of treatment with ADT alone and in combination with apalutamide directly from the Southampton DAR. 116 The metastatic treatment distribution applied in the two models is reported in Table 82, Appendix 11.
As mentioned in Parameterisation of the prostate cancer health states transition probabilities, the distribution of hormone-sensitive metastatic cancer treatments was reflected in the transition probability from metastatic to PCa death, by weighing the treatment of effect of combination therapy according to the relative distribution of treatments. This was also in contrast with the Southampton DAR model,116 which did not link metastatic treatment distribution to the metastatic treatment effectiveness.
Subsequent metastatic treatment (for hormone-resistant metastatic cancer) was also considered in the York model, for the proportion of individuals who survived the first 2 years in the metastatic health state (see Treatment of metastatic prostate cancer). The treatments considered included monotherapy with abiraterone, DTX and enzalutamide, and best supportive care, and the treatment distribution was conditional on the type of treatment received at first line (i.e. for hormone-sensitive metastatic cancer). We sourced the hormone-resistant metastatic treatment distribution from the Southampton DAR model116 (see Table 82, Appendix 11). While the hormone-sensitive metastatic treatment distribution is linked to treatment costs, treatment effectiveness and AE costs, the hormone-resistant metastatic treatment distribution in both models is applied only to estimate the costs of metastatic treatment. While this structural decision was not justified in the Southampton DAR,116 we considered that extending the model to establish these additional links would be of limited value to this assessment. Therefore, the York model does not consider the effectiveness and safety of hormone-resistant metastatic treatment.
Given that the distribution of metastatic cancer treatments was informed by assumptions, these parameters were not set up probabilistically (i.e. probability distributions were not fitted to these parameters).
Adverse events
Biopsy procedure-related adverse events
The biopsy procedure is associated with AEs such as urinary retention, infections, sepsis, haematuria and death. The cost and HRQoL impacts of these AEs vary according to their severity and the level of healthcare resource use required to treat them.
The review in Systematic review methods (study selection, data extraction, quality assessment) could not establish differences in the type and the rates of AEs (i.e. the safety profile) between software and CF, as well as between different SF systems. This was because either comparative safety evidence was not presented, was confounded by the biopsy route of access or the observational nature of the studies limited the ability to attribute differences to the intervention. Furthermore, there is a clear biological mechanism (e.g. clear difference in the number of cores for each MRI-influenced method or a marked increase in procedural time that might increase the likelihood of AEs from anaesthesia) that suggests the safety profile of cognitive and SF is different.
The Southampton DAR116 modelled differences in safety profile between biopsy procedure by route of access and type of anaesthesia. In their revised base case, the biopsy complications considered for LATP/GATP and LATRUS were mild AEs (more frequent with transperineal biopsies), AEs leading to non-elective hospital admission within 28 days of the procedure and peri-procedural death (also within 28 days of the procedure). Transperineal biopsies had a higher rate of mild AEs and slightly lower rates of non-elective admission and peri-procedural death. 116,152 Table 83 in Appendix 11 summarises the AE rates and sources used to parameterise the current report base-case analysis (which correspond to the revised base-case estimates in the Southampton DAR). 116
We note that the AE rates estimated for the Southampton DAR116,152 did not distinguish between biopsies in terms of sample collection method, so it is unclear whether these estimates are reflective of the safety profile of systematic, targeted or combined biopsies. In the base case, we assume that the biopsy safety parameterisation of the Southampton DAR is applicable to targeted biopsies and that there are no differences in biopsy complications between these and combined biopsies; this assumption is relaxed in sensitivity analysis.
Parameter uncertainty in the AE rates was modelled by fitting beta distributions to these parameters.
Localised treatment adverse events
Individuals diagnosed as having PCa will receive treatment for localised disease (active surveillance or radical treatment) in the long-term model according to their diagnosed CPG category, while those diagnosed as not having the disease are assumed to receive monitoring (see Treatment of localised prostate cancer). Both radical and conservative (active surveillance) treatment are assumed to have associated AEs.
In line with the Southampton DAR and the NICE NG131 model,116,123 our base-case analysis includes the following categories of AEs for radical and conservative treatment: (1) erectile dysfunction, (2) urinary incontinence and (3) bowel dysfunction. The rates of AEs for radical prostatectomy, radiotherapy and active surveillance were sourced from Table 64, in the Southampton DAR,116 which was informed by a single trial comparing all three treatments (PROTecT trial138). While all patients receiving radiotherapy are assumed to also received ADT (see Treatment of localised prostate cancer), the Southampton DAR assumed no AEs from hormone therapy;116 we also applied this assumption in the York model.
Parameter uncertainty in the AE rates was modelled by fitting beta distributions to these parameters.
Metastatic disease treatment adverse events
Similarly, to the Southampton DAR model,116 we only modelled AEs of treatment for hormone-sensitive metastatic disease. AE rates per type of AE were sourced from Table 64, in the Southampton DAR,116 which obtained the rates from three pivotal trials59,150,153 comparing ADT alone to each of the three combination therapies modelled.
Parameter uncertainty in the AE rates was modelled by fitting beta distributions to these parameters.
Health-related quality of life
Health-related quality of life outcomes, estimated from an NHS and PSS perspective, in the model are expressed as QALYs and discounted at 3.5% annual rate.
Biopsy procedure disutility
The model considers the disutility of biopsy-related AEs. In line with the Southampton DAR116 a disutility weight was attributed to each type of AE (mild, leading to non-elective hospital admissions and death) and then adjusted for duration of the event to generate a QALY loss per type of AE. The biopsy procedure QALY loss in the model is then adjusted to reflect the different safety profile between transperineal and transrectal biopsy. The disutility weights and AE duration per type of AE are reported in Table 84 in Appendix 11, and were sourced from the Southampton DAR. 116 We did not consider parameter uncertainty in the disutility weights or AEs duration inputs, given lack of information on their variance.
Health state utilities and treatment disutilities
Health state utilities and treatment disutilities were applied as per the Southampton DAR,116 but adapted for the delayed radical treatment at 2 years in the model for misdiagnosed cases.
Resource use and costs
The resource use and costs, considered in the diagnostic pathway, include those associated with the biopsy procedure and its adverse eves. The long-term model quantifies the costs of monitoring individuals following the diagnostic procedures in the diagnostic model, the costs of PCa treatment and end of life. Costs in the model are expressed as 2020–1 Great British pounds, estimated from a NHS and PSS perspective, and discounted at a 3.5% annual discount rate.
The resource use and cost in the long-term model (costs associated with monitoring, PCa treatment, treatment AEs and end of life) was largely informed by the Southampton DAR,116 as were the unit costs sources (updated or inflated to 2020–1 price year as appropriate). Therefore, descriptions of these categories of cost and resource use are brief and refer back to the Southampton DAR model. 116 Emphasis is put into describing elements where our assumptions and/or parameter sources differ from those of the Southampton DAR model. 116
Parameter uncertainty in resource use and costs inputs was not considered for the large majority of the inputs due to lack of information on their variance and the reliance on assumptions to define parameter quantities.
Biopsy procedure costs
This section reports the costs associated with the biopsy procedure, which include the following components:
-
Cost of the SF system – costs of the fusion software and, in some cases, a workstation (or cart). This cost only applies to the diagnostic strategies which include a SF component.
-
Cost of the ultrasound – cost of the ultrasound probe/transducer, and any required software. This cost applies to diagnostic strategies with either software or cognitive function components, but some SF systems are not compatible with third-party ultrasounds.
-
Cost of SF system installation – cost of connecting the SF system to the NHS trust IT system. This cost only applies to the diagnostic strategies which include a SF component.
-
Cost of SF system maintenance – costs of service contracts to maintain the technology and keep software up to date. This cost only applies to the diagnostic strategies which include a SF component.
-
Costs of SF system training – staff time costs required to train NHS professionals to perform biopsies. The use of SF methods requires additional training compared to CF, but the cost of training also varies across biopsy approaches (by route of access).
-
Cost of staff time to perform the biopsy procedure – cost of urologists, nurses and anaesthetist (for procedures requiring general anaesthesia). This cost varies across biopsy approaches (by route of access and type of anaesthesia), but there is also a difference in procedural time between SF and CF.
-
Cost of the biopsy setting – costs of the setting in which the biopsy procedure takes place (outpatient room, theatre session); it varies by route of access, type of anaesthesia, and MRI-influenced method.
-
Costs of other biopsy devices and consumables – cost of (a) devices and equipment (e.g. freehand needle positioning devices, lithotomy beds and biopsy guns) and (b) needles and other materials requiring replacement (immediate or after a certain number of uses). These costs are often specific to the biopsy approach [transrectal or transperineal (stabilised, freehand or double freehand)], and may differ across MRI-influenced methods and across SF systems, due to compatibility issues.
-
Cost of histopathology analysis and report – costs of processing the biopsy sample and communicating the results to the patient in a consultation. This cost applies to all strategies but may differ for strategies using different sampling methods (combined vs. targeted-only biopsy), as these may result in different number of cores being sampled.
The evidence considered to estimate these components of costs (and their calculation) is detailed in Tables 85–95, Appendix 11. Table 13 summarises the aggregated cost per biopsy for each technology and by biopsy approach, with further breakdown of costs in Tables 96–98 in Appendix 11.
| Technology | Biopsy approach | |||
|---|---|---|---|---|
| LATRUS (£) | GATP (£) | LATP (£) | ||
| Technology specific | bkFusion | 147.48 | 380.67 | 231.68 |
| FusionVu | 169.47 | 402.67 | 253.68 | |
| KOELIS Trinity | 150.37 | 384.86 | 235.87 | |
| BiopSee | 89.51 | 323.52 | 179.18 | |
| Fusion Bx 2.0 | 158.10 | 391.72 | 242.73 | |
| CF | 48.44 | 260.00 | 133.07 | |
| Non-technology specific | 209.95 | 634.15 | 239.25 | |
| Total cost per biopsy | bkFusion | 356.53 | 914.82 | 470.93 |
| FusionVu | 378.53 | 936.82 | 492.93 | |
| KOELIS Trinity | 359.43 | 919.01 | 475.12 | |
| BiopSee | 298.56 | 857.67 | 418.43 | |
| Fusion Bx 2.0 | 367.15 | 925.87 | 481.98 | |
| CF | 257.49 | 794.15 | 372.32 |
As stated in Modelling of long-term outcomes, we assumed for the first biopsy in the diagnostic pathway, 65% of biopsies were conducted with LATP and the remainder with LATRUS. For the repeat biopsy, we assume that 60% are LATP, 30% are LATRUS and 10% are GATP (to reflect those individuals where there was concern that first biopsy may not have been accurate due to patient moving excessively during the procedure). We weighted the costs per biopsy approach by the corresponding proportions for first and repeat biopsy to estimate their costs in the model; these costs are reported in Table 14.
| Technology | 1st biopsy cost (£) | Repeat biopsy cost (£) |
|---|---|---|
| bkFusion | 430.89 | 481.00 |
| FusionVu | 452.89 | 503.00 |
| KOELIS Trinity | 434.62 | 484.80 |
| BiopSee | 376.47 | 426.39 |
| Fusion Bx 2.0 | 441.79 | 491.92 |
| Average cost SF | 427.33 | 477.42 |
| CF | 332.13 | 380.05 |
There are a number of uncertainties in the biopsy procedure costs. These pertain to:
-
the set of essential components that are integral part of each technology and the lifespan for all components
-
the potential commercial discounts that may be offered by the companies, what is included in the commercial arrangements and how do these apply to each technology
-
what additional costs may stem from compatibility issues with existing equipment and accessories in use in the NHS
-
the additional time required to perform SF
-
how training for the use of SF is delivered (to whom and for how long), and if the training requirements differ substantially between SF technologies.
Given these uncertainties and that it was not possible to calculate diagnostic performance evidence by individual SF devices at the granularity of classification (ISUP G1, ISUP G2, ISUP G3, and ISUP G4 or 5) required by the economic model, it was considered the biopsy procedure costs for each individual technology was potentially misleading to decision-makers. Thus, we apply the average biopsy cost across all SF technologies in this assessment for which cost data were submitted by the companies. Given this and the numerous uncertainties in the cost estimation of each SF technology, it was not considered appropriate to compare each SF technology against each other and CF in the model. Instead, in the base-case analysis, we apply the average cost per biopsy across all SF. Individual SF technology costs are presented alongside the base-case analysis results to illustrate how their individual costs would impact on the estimates of cost-effectiveness.
Biopsy procedure adverse events costs
The biopsy procedure-related AE costs, considered in the diagnostic pathway model, were estimated by multiplying the AE rate by the unit cost for each type of AE. The unit costs for each type of AE were derived from the Southampton DAR sources {updated for the 2020–1 price year by either using the corresponding versions of national tariffs [e.g. Personal Social Services Research Unit (PSSRU) and NHS reference costs] or inflating costs using the NHS Cost Inflation Index (NHSCII),154 as appropriate} and using the same assumptions (e.g. on resource use required to treat a mild AEs);116 further details are presented on in Table 99 in Appendix 11.
Monitoring costs
Routine monitoring costs at model entry apply to all patients who enter the long-term model. In the model, the set of monitoring tests and schedule varying according to whether the individuals have been diagnosed:
-
localised and locally advanced PCa, and if so, monitoring also varies with:
-
the diagnosed CPG category (CPG 1, CPG 2–3 or CPG 4–5)
-
treatment assigned (active surveillance or radical treatment)
-
and time in the model (first, second or subsequent years).
-
-
or not, and if so, monitoring only varies with the underlying true disease status (no PCa or CPG1–5).
Table 100 in Appendix 11 summarises the resource use and cost per year of the monitoring tests considered in the model for patients in the diagnosed as localised (and locally advanced) disease health states. These costs are applied from model entry (cycle 0) and while individuals remain in the localised disease health states.
We assumed that individuals without a PCa diagnosis would also undergo routine monitoring, regardless of whether they had PCa. In contrast, the Southampton DAR116 only attributed a cost of monitoring to those with localised PCa who had not been identified as having PCa. We changed this assumption in the York model, because in principle these two groups of individuals would be indistinguishable, as true disease status would be unknown to clinicians. We assume that in both groups individuals receive the same monitoring schedule when they are discharged to primary care: an annual PSA test (velocity test at a threshold of 75 ng/ml/year) for up to 10 years, performed at a 10-minute nurse-led appointment, and followed by a CF biopsy (costed at £477.75; assumes 35% LATRUS and 65% LATP) if the PSA test results is positive. As per the Southampton DAR,116 the probability of testing positive in the PSA test for those with PCa was assumed to be 0.69, which corresponds to the sensitivity of the corresponding PSA velocity test used in NICE NG131 model. We further assumed that the probability of testing positive in the PSA test for those without PCa corresponded to one minus the specificity of the same test (1–0.56 = 0.44). The testing schedule is similar to what was modelled in the Southampton DAR116 for those with PCa who were diagnosed as not having the disease, but the first PSA test is assumed in the York model to occur within 1 year in the long-term model (rather than 6 months). The annual cost per year of monitoring applied in the York model was £342.50 and £223.06 for those with and without PCa, respectively. These costs are applied from model entry (cycle 0) and for up to 10 years in the entry health states.
After 2 years in the model, individuals in the local disease health states are assumed to be correctly identified at their true disease status, and move to the monitoring regime that matches their true disease status.
Individuals who enter the metastatic health state incur a one-off monitoring cost of £577.83, corresponding to the same resource use as in the Southampton DAR116 (i.e. one CT and bone scan).
Prostate cancer treatment costs – localised disease
Individuals identified as having localised PCa are assumed to receive treatment at long-term model entrance according to their diagnosed CPG (see distribution of treatments by diagnosed CPG in Treatment of localised prostate cancer). Individuals who receive active surveillance is assumed to not incur any treatment costs (only monitoring costs as detailed in Prostate cancer treatment costs – localised disease), so costs of treatment are only incurred by those who undergo radical treatment.
Radical treatment resource use and costs vary according to the type of radical treatment (radical prostatectomy, external radiotherapy or brachytherapy). The cost of each type of radical treatment procedure applied in the York model is reported in Table 101, Appendix 11, alongside details on resource use and unit costs. We note that the cost of brachytherapy has increased considerably in relation to the one used in the Southampton model (£9156.96 vs. £3106.02); these differences are driven by an increase in the unit cost of delivering brachytherapy in an outpatient setting (as well as increased activity for the corresponding currency code) in 2020–1 compared to 2019–20. The costs of the radical treatment procedures were applied as one-off costs at long-term model entry (cycle 0). For those who were misdiagnosed and treated with conservative treatment, it is assumed that they receive radical treatments according to their true disease status after 2 years in the model.
In addition to the medical procedures, we also included the cost of ADT for those patients who treated with radiotherapy, according to NICE guidance. 10 ADT in the localised disease setting was assumed to consist of the same treatments as in the Southampton model, that is, bicalutamide 50 mg for 21 days followed by luteinising hormone-releasing hormone (LHRH) agonists [either leuprorelin 11.25 mg (every 3 months), triptorelin 11.25 mg (every 3 months) or goserelin 3.6 mg (every 28 days)]. Similarly, to the Southampton model, the duration of LHRH treatments was varied according to category of prognostic risk; we assumed LHRH treatment duration would be 3 and 6 months for those diagnosed in CPG 1 and CPG 2–3 categories. For those diagnosed in the CPG 4–5 category, we updated the duration of treatment for 3 years, in line with the current NICE guidance for that prognostic risk group (see Care pathways for the diagnosis and management of prostate cancer). 10 The costs of ADT included drug acquisition and administration costs and were costed as per the Southampton DAR (updated to the current price year).
Prostate cancer treatment costs – metastatic disease
Metastatic disease treatment was assumed to consist of hormone-sensitive disease treatment for the first 2 years in the metastatic health states, followed by hormone-resistant disease treatment. Costs of metastatic treatment are summarised in Table 102, Appendix 11; these include drug acquisition and administration costs.
In line with the Southampton DAR, hormone-sensitive metastatic treatment was modelled as a blended treatment consisting of ADT alone (but not identical to the regimes described for the localised disease setting, as course of bicalutamide 50 mg is longer), or in combination with either DTX, apalutamide or enzalutamide. We updated the distribution of treatments for the hormone-sensitive metastatic treatment, as described in Treatment of metastatic prostate cancer. We note that yearly costs of metastatic treatment have increased considerably in the York model compared to the Southampton model (e.g. metastatic hormone-sensitive first year cost increased to £15,603.87 from £8388.63), due to the increased proportion of individuals treated with ADT combined with enzalutamide, due to the high cost of enzalutamide. Furthermore, although we apply the same DTX treatment regimen [i.e. six cycles (delivered every 3 weeks) at a dose of 75 mg/m2; body surface area 1.91] as in the Southampton model, in the York model the 2-year DTX treatment costs are assumed to be distributed evenly between two model cycles (constant annual cost).
The Southampton DAR states that ADT alone or in combination was taken until disease progression, which was assumed to occur after 2 years. We also make their stated assumption, but we implemented it in a different way. In the Southampton model, a cost for first and second year is estimated for metastatic treatment (both treatment for hormone-sensitive and hormone-resistant disease) and applied to individuals in the metastatic disease state at first and second year (modelled in a way akin to tunnel states), respectively. Thus, in the Southampton model the cost of hormone-sensitive and hormone-resistant treatment is applied to the same set of individuals. In the York model, we explicitly model a set of three tunnel health states representing the first, second and subsequent years of metastatic disease (Figure 12). We applied the costs of hormone-sensitive metastatic treatment to individuals in the first and second year of metastatic tunnel health states, and the costs of hormone-resistant metastatic treatment costs are applied as a one-off cost to individuals who enter the ‘metastatic subsequent years’ health state.
FIGURE 12.
Diagram of metastatic tunnel health states and death states.
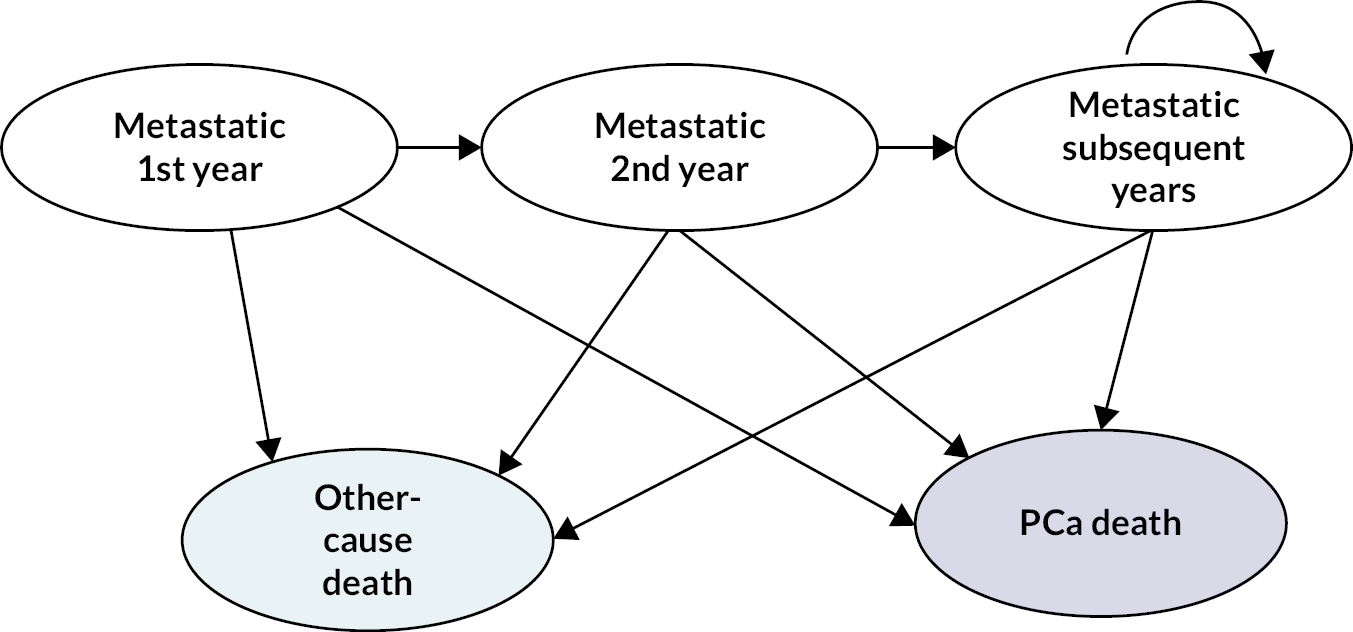
Another difference between models is that in the York model metastatic treatment (and monitoring) is assumed to apply to all patients with metastatic disease, as we do not distinguish between diagnosed and undiagnosed metastatic disease (the latter does not appear to incur treatment costs in the Southampton model). Thus, we implicitly assume that all individuals with metastatic disease have been diagnosed.
All metastatic treatments costs are applied as an average of the costs of the different types of treatments weighted by their treatment distribution (see treatment distribution in Table 12, Treatment of localised prostate cancer).
Prostate cancer treatment adverse event costs – localised disease
The model considers the costs of managing the AEs from active surveillance, radical prostatectomy and radiotherapy for localised PCa. These costs were estimated by multiplying the AE rates (see Localised treatment adverse events) by the unit cost of the corresponding AE (see Table 103, Appendix 11). The costs are applied in the model as a one-off at localised disease health states to the proportion of patients who receives each treatment (see treatment distribution in Table 12, Treatment of localised prostate cancer).
Prostate cancer treatment adverse event costs – metastatic disease
The costs of managing metastatic treatment-related AEs was applied in the model. Similarly, to the Southampton model only AEs of treatment for hormone-sensitive metastatic disease were included. The AE costs for androgen therapy alone and in three alternative combinations (with DTX, apalutamide or enzalutamide) were estimated by applying the unit cost per type of AE (see Table 103, Appendix 11) by the corresponding rate (see Metastatic disease treatment adverse events). The resulting costs per treatment were then applied as a one-off cost at entrance to the ‘metastatic 1st year’ health state. The one-off cost was estimated by weighing each treatment cost by the metastatic treatment distribution (see treatment distribution in Table 82 in Appendix 11).
End-of-life costs
End-of-life costs are applied to all individuals who die in the model of other-cause or PCa death, but not to those who have died of peri-procedural biopsy complications (see Biopsy procedure related adverse events). This one-off cost is applied to individuals who enter the death states at each cycle in the model, and it was sourced from Round et al. 139 and inflated to 2020–1 price year. 154
Analytic methods
Overview
The diagnostic and long-term model is evaluated deterministically and probabliistically for the base-case analysis (1000 Monte Carlo simulations) to incorporate the joint parameter uncertainty across all of the model inputs according to the probability distributions assigned to each. The parameters set up probabilistically in the model are identified in Table 104 in Appendix 11.
Following conventional decision rules for cost-effectiveness, the mean costs and QALYs for the two strategies (cognitive or software fusion) for two set of comparisons (targeted biopsy alone or combined with systematic biopsy) are presented and cost-effectiveness compared by estimating the ICERs, as appropriate. A NHB approach is also applied, for which the unambiguous decision rule. Net benefits can be expressed on the effect scale (NHB), which is calculated at the two cost-effectiveness thresholds at the lower and upper bound of the range used by NICE to guide decision-making (i.e. £20,000 and £30,000 per additional QALY). The formula to estimate NHBs is presented below:
Heterogeneity is partly explored in a subgroup analysis detailed in Subgroup analysis. Uncertainty regarding the appropriate source of data, and other assumptions are explored by scenario analysis and threshold analysis, as detailed in Threshold analysis on costs of software fusion and Scenario analyses.
Base-case analysis
The base-case analysis considers two alternative set of comparisons. The first comparison is established between targeted SF and targeted CF, while the second is established between combined SF and combined CF. Therefore, we consider a dual base-case analysis with results presented separately for (1) targeted biopsy alone and (2) combined (targeted + systematic) biopsy.
The dual base case is defined by the following data sources and assumptions:
-
the main analysis extension to the evidence synthesis for the subgroup of biopsy-naive individuals, which uses:
-
the baseline distribution of test results for SF sourced from biopsy-naive data from Filson et al.;96
-
relative accuracy data from the multinomial evidence synthesis model (Model 1a) which was incorporated into the extension to the evidence synthesis – network 1 was used to inform the targeted biopsy comparison, while network 2 informed the combined biopsy comparison;
-
accuracy data from Mortezavi et al. 107 extension to the evidence synthesis;
-
-
the only differences between combined and targeted biopsy stem from the data used in the extension to the evidence synthesis [i.e. they are assumed to have the same profile of AEs and biopsy procedure costs (note that both set of comparisons consider SF and CF biopsy)];
-
the cost of first and repeat biopsy with SF is modelled as an average of these costs for each technology (headline cost-effectiveness results for the individual technologies are presented for the base case analysis) and
-
the cost of first biopsy assumed procedures are conducted as a mix of LATP and LATRUS; similarly, repeat biopsy is a mix of LATP, GATP and LATRUS. These proportions were assumed the same for SF and cognitive biopsy.
-
Structural assumptions:
-
only individuals classified in the CPG 1 or ‘no cancer’ categories are eligible for repeat biopsy, and of those a fixed proportion received repeat biopsy in the model (15.45% and 5% for those classified CPGI and ‘no cancer’, respectively);
-
while the model considers different progression rates by true CPG (as modified by radical treatment effect in accordance with the diagnosed CPG), progression across CPG scores is not modelled – only progression between each local disease status and the metastatic health state are possible;
-
after 2 years in the misclassified localised disease health status, all individuals who remain in the corresponding states and have not yet received radical treatment will receive radical treatment according to their true disease status, incurring the costs and disutility of radical treatment then and receiving monitoring commensurate with their true disease status from that point onwards.
-
Threshold analysis on costs of software fusion
We highlighted in Resource use and costs uncertainties and areas of evidence scarcity, relating to the costs of the biopsy procedure, particularly for the SF technologies. We reiterate that given these uncertainties and that it was not possible to calculate diagnostic performance evidence by individual SF devices with the necessary classification granularity required by the economic model, it was considered the biopsy procedure costs for each individual technology were potentially misleading to decision-makers.
Thus, we apply the average biopsy cost across all SF technologies in this assessment for which cost data were submitted by the companies. We also perform a threshold analysis in which we estimate what is the cost per biopsy procedure with SF at which it is no longer likely that the new technologies will be cost-effective at the conventional range of opportunity costs considered by NICE. This threshold analysis applies the same assumptions and data sources of the base-case analysis, but assumes that:
-
all biopsies are LATP procedures
-
excludes the cost of the third-party ultrasounds from the biopsy cost calculations (to disentangle the cost of cognitive and SF).
These assumptions are necessary in order to run a threshold analysis varying a single parameter (i.e. cost of SF biopsy).
Subgroup analysis
As mentioned in Results, the extension of the evidence synthesis included a subgroup analysis for previous negative-biopsy individuals. We performed a subgroup analysis for the same group of patients, which mirrors the subgroup analysis in Results. In brief, this subgroup analysis used the same evidence sources to inform the extension to the synthesis, except the baseline distribution of test results for SF. This was sourced from previous negative biopsy data from Filson et al. 96
Scenario analyses
The scenario analyses are summarised in Table 15. In brief, the aim of the scenario analysis is:
| Scenario number and label | Element of uncertainty | Base case | Scenario variation |
|---|---|---|---|
| 1. PAIREDCAP (2019) baseline | Extension of the evidence synthesis model | Data sources for the extension to evidence synthesis: •baseline distribution of test results for SF from biopsy-naive data in Filson et al. •Relative accuracy data from the multinomial evidence synthesis model (Model 1a, network 1 + 2 – targeted and combined biopsy). •Accuracy data from Mortezavi et al. |
Data sources for the extension to evidence synthesis: •baseline distribution of test results for SF from biopsy-naive data in biopsy-naive data from PAIREDCAP (2019) for network 1. •Relative accuracy from the multinomial evidence synthesis model (Model 1a, network 1 only – targeted biopsy). •Accuracy data as for base-case analysis. |
| 2. Zhou (2018) diagnostic | Data sources for the extension to evidence synthesis: •Baseline distribution of test results for SF as for base-case analysis. •Relative accuracy from the multinomial evidence synthesis model (Model 1a, network 1 only – targeted biopsy). •Acccuracy data from Zhou et al.142(2018) |
||
| 3. Degradation of repeat biopsy accuracy | Diagnostic performance of MRI-influenced repeat biopsy | Repeat biopsy is as accurate as first biopsy for both cognitive and SF. | •Probability of correctly classifying individuals as having cancer at each CPG category is reduced by 80% at repeat biopsies (changes in diagnostic accuracy are distributed equally across all other possible CPG classifications for each true disease CPG). |
| 4. SF as quality assurance | Diagnostic performance of MRI-influenced biopsy and selection for repeat biopsy | Diagnostic performance of MRI-influenced biopsy is informed by the extension of the evidence synthesis model 1a (network 1 and 2) and only a proportion of those classified at first biopsy as having NC or CPG1 receive repeat biopsy. | •No difference in overall diagnostic performance of CF vs. SF •Individuals eligible for repeat biopsy are those: ○Who have been misclassified as CPG1or NC at first biopsy with SF ○Who have been classified (correctly or not) as CPG1or NC at first biopsy with SF |
| 5. Radical treatment for all identified CPG ≥ 2 and conservative treatment for CPG 1 | Distribution of treatment for localised disease | The distribution of radical treatment for localised disease is sourced from Parry et al.151 | All individuals diagnosed CPG ≥ 2 are treated at long-term model entrance with radical treatment (maintaining the distribution between radical prostatectomy and radiotherapy as per the base case) and those diagnosed with CPG1 receive conservative treatment (and do not switch for radical treatment). |
| 6.1 Throughput (150/year) | Annual biopsy throughput | 300 biopsies per year | •150 biopsies per year: 50% lower than base case |
| 6.2 Throughput (450/year) | 300 biopsies per year | •450 biopsies per year: 50% higher than base case |
-
Scenario analyses 1 and 2: to mirror the sensitivity analysis performed around the sources of data informing the sensitivity analyses of the evidence synthesis extension (see Results), and explore their impact on the cost-effectiveness estimates.
-
Scenario analysis 3: to explore the impact of lowering the diagnostic accuracy of repeat biopsy, as considered in the PROMIS, NICE NG131 and Southampton DAR models.
-
Scenario 4: to model the use of SF biopsy as quality assurance, as this was suggested by clinical advisers to the EAG as a potential value component of software. The clinical advisers commented that they would be more confident that a negative biopsy result with SF biopsy following a positive MRI result would not require a confirmatory biopsy compared to CF, and that this confidence did not arise from any perceived gains in diagnostic accuracy of SF versus CF biopsy. Thus, we set the diagnostic accuracy of SF to be equal to the base-case accuracy for CF (implying that the sole value of SF is to inform the selection of cases for repeat biopsy), and we changed the eligibility criteria for repeat biopsy with SF as described in Table 15.
-
Scenario 5: aims to approximate the assumptions on localised disease treatment conditional on final classification to those of the PROMIS model.
-
Scenarios 6.1 and 6.2: aim to explore the impact of using SF in NHS trusts with lower (6.1) and higher patient throughput (6.2) than that assumed to correspond in the base-case analysis to the national average throughput.
Results
Base-case analysis
The deterministic and probabilistic cost-effectiveness results for the base-case analysis are presented in Tables 16 and 105, Appendix 12, respectively. Base case results for each individual technology are also presented in Tables 114 and 115, Appendix 12. The data sources used to derive prevalence and true disease status (see Table 110, Appendix 12) in this analysis refer to a biopsy-naive population. For both the targeted biopsy (informed by network 1 of Model 1a) and the combined biopsy (informed by network 2 of Model 1a) comparisons, the SF strategy seems to on average be costlier and to yield greater QALYs than the CF strategy, resulting in a deterministic ICER of £5623 and £1826 per additional QALY, respectively. These ICERs are below the lower bound of the cost-effectiveness threshold range recommended by NICE, suggesting that it may be cost-effective compared to CFs in both the targeted and the combined comparisons. However, these results should be interpreted cautiously given the uncertainties in the relative diagnostic accuracy evidence which informs the model.
The probabilistic analysis suggests a higher probability of cost-effectiveness for SF versus CF at the range of cost-effectiveness thresholds recommended by NICE (0.64 and 0.68 at £20,000 and £30,000 per additional QALY for targeted SF biopsy). The probabilistic and deterministic cost-effectiveness results for each set of comparisons are similar. Henceforth and for subsequent analysis, we focus on the deterministic results, as these are easier to compare across base case, threshold, subgroup and scenario analyses.
For the targeted biopsy (network 1), the SF strategy results in average higher costs (£543 vs. £443) and slightly lower QALY loss due to biopsy AEs (–0.00175 vs. 0.00176) compared to CF in the diagnostic model. The higher costs are driven by the cost of performing biopsy with SF, which on average costs £92 and £97 more than with CF, for first and repeat biopsy, respectively. The SF strategy appears to lead to fewer repeat biopsies due to its higher correct detection rate at categories CPG 2 to CPG 4–5 compared to CF; this has a small impact on incremental costs and QALY loss. This small impact on costs and benefits is due to the reduction in repeat biopsy with SF compared to CF being small (0.055 vs. 0.050) and the only differences in rates of biopsy AEs between MRI-influenced methods stemming from differences in the proportion of repeat biopsy for each strategy.
The targeted SF strategy appears to increase correct classification (see Table 106, Appendix 12) across all CPGs compared to targeted CF at the end of the diagnostic pathway (final classification), particularly for CPG 2 (correctly classified 15% vs. 10%, out of a true disease prevalence for this category of 26%) and to a lesser extent for category CPG 1 (correctly classified 0.108 vs. 0.057, out of a true disease prevalence for this category of 32%). This is consistent with the results of the extension to the evidence synthesis (see Results) and suggests that even with repeat biopsy for the cases classified as CPG 1 or no PCa at first disease, the remaining true disease CPG 2 cases misclassified are likely to be largely classified as having no PCa. For those with true disease CPG 3 (prevalence for this category of 18.3%), the increase in correct detection with SF versus CF is modest (from 9% to 9.5%). The likelihood of being CPG 3 and being misclassified by the CF strategy as NC, CPG 1 or CPG 2 is 33%, 23% and 36%, respectively, whereas with SF these proportions are 40%, 10% and 39%, respectively (results not shown; extracted directly from model). Disaggregated results for the diagnostic model are presented in Table 107, Appendix 12.
In the long-term model, the targeted SF strategy appears to be accompanied by small life-year and QALY gain (0.02 life years and 0.01 QALYs) compared to CF in the long-term model. Some of the higher incremental diagnostic costs of the SF biopsy strategy versus CF appear to be offset by the lower costs accrued for this strategy compared to CF in the long-term model (£28,885 vs. £27,919, costs disaggregated by CPG are shown in Table 108, Appendix 12). The higher health outcomes with the SF technology compared to CF are likely to stem from a slight increase in time spent in the localised disease health state (as suggested by the higher life years and baseline QALYs accrued in the model and lower metastatic disease QALY loss; see Table 109, Appendix 12), which is partially offset by the higher upfront QALY loss from immediate localised radical treatment with the SF strategy versus CF. This is due to more patients being correctly identified in the diagnostic model with the SF strategy. The increased correct classification with SF also results in higher upfront costs from radical treatment and its AEs, but lower costs of managing metastatic disease and of monitoring.
For combined biopsy (network 2), the incremental costs and QALY loss of the SF strategy versus CF in the diagnostic model are fairly similar to those observed for the targeted biopsy. However, there seems to be greater cost savings and health outcomes benefits in the long-term model for SF compared to CF in the combined biopsy analysis, which result in cost-effectiveness results more favourable to the SF strategy. Disaggregated results for combined biopsy are presented in Tables 111–113, Appendix 12.
The level of correct classification across all grades in the combined biopsy diagnostic pathway is increased for the SF strategy compared to CF (45.5% vs. 68%, more so than for targeted biopsy). The results suggest that when compared to combined CF strategy, SF retrieves a higher proportion of CPG 4–5 (8.5% vs. 3.4%), CPG 2 (20.7% vs. 8.9%), CPG 1 (15.4% vs. 9.6%). Overall, this suggests that 16.8% more individuals are correctly identified with combined SF versus CF at CPG 2 or above, the threshold above which radical treatment is a treatment option according to current clinical guidance.
The correct higher detection at CPG results warranting radical treatment results in higher costs of upfront radical treatment for combined SF compared to CF, but also the health benefits in the long-term model (due to slower disease progression). It also reduces the costs of metastatic treatment for the SF strategy versus CF. The impact on total costs and QALYs in the long-term model is still limited, as the increased correct detection concentrates on those who have a true CPG 2 and are less likely to benefit from radical treatment than those at CPG 3–4 (where increases in correct classification for combined SF vs. CF are less marked). Nevertheless, the cost savings (–£49) and small incremental increase on QALYs (0.03 QALYs) for combined SF compared to CF result in an ICER favourable to combined SF (see Table 16).
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs | Total LYsa | Total QALYsa | Total costsa | Total LYsa | Total QALYsa | Total costsa | ICERb | NHB at £20,000b |
NHB at £30,000b |
|
| Targeted CF | –0.00176 | £445 | 11.45 | 8.29 | £27,919 | 11.45 | 8.29 | £28,364 | 6.87 | 7.34 | |
| Targeted SF | –0.00175 | £543 | 11.46 | 8.30 | £27,885 | 11.46 | 8.30 | £28,428 | 6.88 | 7.35 | |
| Targeted | Inc QALY loss | Inc costs | Inc LYs a | Inc QALYs a | Inc costs a | Inc LYs a | Inc QALYs a | Inc costs a | INHB at £20,000b |
INHB at £30,000b | |
| SF vs. CF | 0.00001 | £98 | 0.02 | 0.01 | –£34 | 0.02 | 0.01 | £63 | £5623 | 0.01 | 0.01 |
| Strategy | QALY loss | Total costs | Total LYs a | Total QALYs a | Total costs a | Total LYs a | Total QALYs a | Total costs a | ICERb | NHB at, £20,000b | NHB at £30,000b |
| Combined CF | –0.00177 | £448 | 11.44 | 8.28 | £27,889 | 11.44 | 8.28 | £28,337 | 6.86 | 7.33 | |
| Combined SF | –0.00176 | £544 | 11.49 | 8.31 | £27,840 | 11.49 | 8.30 | £28,384 | 6.89 | 7.36 | |
| Combined | Inc QALY loss | Inc costs | Inc Lys a | Inc QALYs a | Inc costs a | Inc Lys a | Inc QALYs a | Inc costs a | INHB at £20,000b | INHB at £30,000b | |
| SF vs. CF | 0.00002 | £95 | 0.05 | 0.03 | –£49 | 0.05 | 0.03 | £47 | £1,826 | 0.02 | 0.02 |
Further exploration of the base-case analysis
This subsection reports further results from the base-case model on the comparison of targeted strategies, that aim to identify the cost-effectiveness drivers and aid decision-making when trying to integrate the uncertainties over the clinical evidence with the overall cost-effectiveness.
This is important because of the complexity of the classification of disease, treatment allocation rules, combined with the impacts of the different treatments, makes it difficult to establish how the misclassification of suspected PCa lesions across different categories drives the value of SF compared to CF. To clarify this issue we present the final diagnostic accuracy for the strategies as implied by the test sequences modelled, the disaggregated cost-effectiveness results (corresponding to aggregated results for targeted biopsy in Table 16) by true CPG, and the trade-offs between different degrees of misclassification and correct classification. The results presented here are deterministic.
Diagnostic accuracy of the test sequences in the decision model
Table 17 illustrates the distribution of test results and conditional accuracy probabilities at final classification for cognitive and SF biopsies (includes first biopsy and repeat biopsy for a proportion of individuals). The difference in diagnostic accuracy between SF and CF by each classification is shown in brackets in Table 17 (increased and detection are highlighted in green and red, respectively).
| Prev (%) | CPG | Distribution test results | Distribution of test results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 50.5% | 18.3% | 14.4% | 10.2% | 6.6% | 44.6% | 17.2% | 20.3% | 11.1% | 6.7% | ||
| CF biopsy | SF biopsy | ||||||||||
| Accuracy matrix | Accuracy matrix | ||||||||||
| NC (%) | 1 (%) | 2 (%) | 3 (%) | 4 or 5 (%) | NC (%) | 1 (%) | 2 (%) | 3 (%) | 4 or 5 (%) | ||
| 12 | NC | 100 | 100 (0) |
||||||||
| 32 | 1 | 82 | 18 | 66 (–16) |
34 (+16) |
||||||
| 26 | 2 | 29 | 35 | 36 | 24 (–5) |
19 (–16) |
57 (+21) |
||||
| 18 | 3 | 18 | 13 | 20 | 49 | 22 (+3) |
5 (–7) |
21 (1) |
52 (+3) |
||
| 12 | 4 or 5 | 12 | 10 | 11 | 10 | 56 | 11 (–1) |
4 (–6) |
13 (+2) |
14 (+4) |
58 (+2) |
The diagnostic accuracy at final classification is consistent with the results for the first biopsy for both strategies (targeted CF and SF), suggesting that SF increases the correct classification across all CPGs (cells along the diagonal line) compared to CF, particularly for CPG 2 (21% more) and CPG 1 (16% more). For those with true CPG 2, the greatest reduction in misclassification is observed at diagnosed CPG 1 (16% less).
Estimation of disaggregated base-case results
Table 18 presents the base-case analysis incremental NHB (INHB) at £20,000 per additional QALY of SF versus CF as a total estimate [0.01 QALY (0.00810 with further decimal cases), as in Table 16] and disaggregated by true disease category (where the prevalence for each true disease category is set to 100%). The total INHB corresponds to the sum of the INHB by true disease category weighted by its corresponding prevalence.
| CPG | Prevalence (weights) (%) | INHB by CPG | INHB by CPG × prevalencea |
|---|---|---|---|
| NC | 12.1 | –0.00500 | –0.00061 |
| 1 | 31.8 | –0.01631 | –0.00519 |
| 2 | 26.2 | 0.02890 | 0.00757 |
| 3 | 18.3 | 0.01907 | 0.00349 |
| 4 or 5 | 11.6 | 0.02435 | 0.00283 |
| Total INHBa | 0.00810 | ||
The results suggest:
-
The disaggregated INHB estimates are negative for the ‘no cancer’ and CPG 1 categories, which suggests that the increased correct detection of CPG 1 with SF does not result in net health gains in relation to CF.
-
The disaggregated INHB estimates suggest higher net health gains for SF compared to CF for CPG ≥ 2. Once prevalence is considered (column 3), the largest effective contribution to the total INHB arises from CPG 2.
To aid interpretation of these results, in Table 19 further disaggregates the base-case results by model component, and within the long-term model component by health state. The grey shading highlights estimates unweighted by prevalence (totals correspond to prevalence weighted values).
| CPG | Diagnostic model | Long-term model | ||||
|---|---|---|---|---|---|---|
| INHB | INHB | Inc | Inc | INHB | INHB | |
| Total | Total | QALYs | Costs | Localised | Metastatic + EoL | |
| NC | –0.0050 | – | – | – | – | – |
| 1 | –0.0056 | –0.0107 | –0.0017 | £180 | –0.0075 | –0.0032 |
| 2 | –0.0043 | 0.0332 | 0.0198 | –£268 | 0.0636 | –0.0304 |
| 3 | –0.0046 | 0.0237 | 0.0178 | –£117 | 0.0327 | –0.0090 |
| 4 or 5 | –0.0046 | 0.0289 | 0.0290 | £2 | 0.0392 | –0.0103 |
| Totala | –0.0049 | 0.0130 | 0.0113 | –£34 | 0.0248 | –0.0118 |
In the diagnostic model, the INHB of SF is negative across all CPGs. Since the INHB of SF in the model overall (diagnostic + long term) is positive, this suggests that the costs and harms of SF in the diagnostic model are only offset by long-term costs HRQoL outcomes that result from the subsequent clinical management of individual conditional on final biopsy classification. The different diagnostic INHB for each category reflect only differences in the proportion of repeat biopsies across true disease categories.
The long-term model INHB estimates follow the same pattern across true disease categories as observed for the full model results (see Table 18). For true disease categories CPG 2 and above, the INHB for SF versus CF is positive; the greater contribution to the INHB stems from the CPG 2 category. Compared to CF, QALY gains occur for CPG 2 and above with SF, and these are accompanied by cost savings for CPG 2 and 3.
The INHB for CPG 1 is negative in the long-term model due to higher costs and lower QALYs compared to CF; this is due to the increased correct detection of CPG 1 leading to more individuals receiving immediate (conservative or radical) treatment of localised disease with associated costs and AEs (if they had been misclassified as NC they would have received only monitoring in the first 2 years in the model) for SF compared to CF, which are not offset by the benefits of early treatment. The annual probability of progression from localised to metastatic disease is similar for CPG 1 misclassified compared to correctly classified CPG 1 (0.14 vs. 0.13), so the benefits from increased correct detection at this category are limited.
The localised and metastatic INHB estimates also suggest that the increased correct detection in category CPG 2 with SF is contributing more to the total long-term model INHB. We note that while the metastatic INHB is negative across all cancer categories, this does not mean that there are higher net health losses with SF compared to CF in the metastatic health states because the INHBs are not estimated by individual in the model. Since individuals spend less time in the metastatic health states with SF compared to CF due to overall slower progression to metastatic disease with SF (e.g. for CPG 2, 3.55 and 3.70 undiscounted life-years are accrued in the metastatic health states for SF and CF, respectively), SF accrues overall fewer QALYs than CF in the metastatic health states. Despite the lower costs accrued with SF in the metastatic states, the metastatic INHB is always negative.
Disaggregated estimates of cost-effectiveness by final classification category
We estimated the NHB that could be achieved in the long-term model if all individuals were identified in a particular final classification category (see Table 20) to understand how shifts in classification may impact on cost-effectiveness estimates.
| CPG | NC | 1 | 2 | 3 | 4 or 5 |
|---|---|---|---|---|---|
| No cancer | 8.966 (9.435, –0.469) | ||||
| 1 | 7.996 (8.121, –0.125) | 7.930 (8.074, –0.145) | |||
| 2 | 7.072 (6.756, 0.316) | 6.855 (6.578, 0.277) | 7.066 (6.927, 0.139) | ||
| 3 | 5.215 (4.077, 1.139) | 5.117 (4.027, 1.090) | 5.468 (4.571, 0.897) | 5.707 (4.925, 0.782) | |
| 4 or 5 | 3.476 (1.740, 1.737) | 3.408 (1.689, 1.719) | 3.642 (2.007, 1.635) | 3.816 (2.245, 1.571) | 3.912 (2.385, 1.527) |
Results suggest that there will be more (long-term) NHB loss in misclassifying CPG ≥ 2 as CPG 1 than as ‘no cancer’. The highest increase in NHB for CPG ≥ 2 can be achieved with technologies that shift misclassification from CPG 1 to the correct classification. When shifting between adjacent categories, the highest NHB gain can be generated when someone with CPG 3 misclassified as CPG 1 with one technology is identified as CPG 3 with the alternative (+0.351 QALYs). The lowest NHB gain between adjacent categories is generated for those with true CPG 4–5, when they ‘move’ from a CPG 3 to a correct diagnosis (+0.096 QALY).
The incremental value of one technology will depend on how it changes the distribution across classification categories for each true disease category compared to the alternative technology, and on the prevalence per true disease category. For SF compared to CF, the INHB will be positive for the classification categories where it increases detection and negative for those where detection is decreased (see Table 17 for differences between diagnostic accuracy matrices).
This can be illustrated with an example for true disease category CPG 2. For SF versus CF:
-
the reduction in detection of CPG 2 as ‘no cancer’, the category with highest NHB for CPG 2 (7.072 QALYs) is small (–5%), so the INHB is –0.339 QALYs
-
the reduction in detection of CPG 2 as CPG 1 (NHB = 6.885 QALYs) is –16% resulting in an INHB of –1.086 QALYs
-
the increased correct detection of CPG 2 (+21%, NHB = 7.066 QALYs) is sufficient to offset the negative INHBs from the alternative classifications (as ‘no cancer’ and CPG 1), and the total INHB across this disease category is 0.033 QALYs
-
since the prevalence of CPG 2 is 26%, the relative contribution from changes in detection rates for this category is 0.009.
The NHB gains for correctly identifying lesions at CPG3 and CPG 4–5 compared to missing them (i.e. identifying them as CPG 0) is positive (> 0.4 QALYs for both CPG groups). This suggests that it is worth radically treating individuals with CPG ≥ 3 PCa early, as radical treatment reduces disease progression proportionally more for these individuals compared to those with lower CPG. This delay to disease progression translates into longer time spent in the localised PCa state with higher HRQoL and lower mortality compared to the metastatic disease state, and without incurring the costs of metastatic treatment. These benefits off set the costs and harms of early radical treatment.
Given the uncertainties and limitations of the clinical evidence informing the diagnostic accuracy NMA and its extension, the information in Tables 18–20 can be used by decision-makers to consider how their judgements on what are the plausible differences in the prevalence and relative diagnostic accuracy between SF and CF can be translated into cost-effectiveness impacts.
Threshold analysis on costs of software fusion
Given the uncertainties in the costing of SF we conducted a threshold analysis to identify the SF biopsy cost at which there would be a shift in the decision to accept SF as a good use of NHS resources. Since the base-analysis suggests that the SF strategy might be cost-effective compared to CF, the point of decision shift is identified as the cost per SF (holding the cost of CF constant) at which the incremental of NHB of the SF biopsy compared to CF becomes negative (i.e. SF is not likely to be cost-effective). The threshold analysis is conducted under the assumption that all biopsies are LATP and excluding the cost of the ultrasound components from the cost of CF. Under these assumptions the cost per biopsy is £448.50 and £331.00 per SF and cognitive fusio biopsy, respectively.
The threshold analysis results (see Figure 18, Appendix 12) suggest that the decision inversion point is located at a cost per targeted SF biopsy of £586 and £695 at £20,000 and £30,000 per additional QALY, respectively. For combined SF biopsy (see Figure 19, Appendix 12), the inversion point cost per biopsy was estimated as of £874 and £1116 at £20,000 and £30,000 per additional QALY, respectively.
Subgroup analysis
The deterministic cost-effectiveness results of the subgroup analysis for previous negative biopsy individuals are presented in Table 21 with full breakdown presented in Table 116–123, Appendix 12. We note that this analysis only differs from the base-case analysis in the source for the baseline distribution of test results for SF (sourced from previous negative biopsy data from Filson et al. 96 rather than the biopsy naive in the base-case analysis). The estimated prevalence of PCa disease in this subgroup is lower than in the base-case analysis (57% vs. 88%), while the diagnostic accuracy matrices for both targeted and combined biopsies in the subgroup analysis (see Appendix 10) are similar to those estimated for biopsy-naive individuals (as expected).
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs | Total LYsa | Total QALYsa | Total costsa | Total LYsa | Total QALYsa | Total costsa | ICERb | NHB at £20,000b |
NHB at £30,000b |
|
| Targeted CF | –0.00176 | £444 | 11.75 | 8.68 | £22,014 | 11.75 | 8.68 | £22,457 | 7.56 | 7.93 | |
| Targeted SF | –0.00175 | £542 | 11.76 | 8.69 | £21,994 | 11.76 | 8.69 | £22,536 | 7.56 | 7.94 | |
| Targeted | Inc QALY loss | Inc costs | Inc LYs a | Inc QALYs a | Inc costs a | Inc LYs a | Inc QALYs a | Inc costs a | INHB at £20,000b |
INHB at £30,000b | |
| SF vs. CF | 0.00000 | £99 | 0.01 | 0.01 | -£20 | 0.01 | 0.01 | £79 | £9285 | 0.00 | 0.01 |
| Strategy | QALY loss | Total costs | Total LYs a | Total QALYs a | Total costs a | Total LYs a | Total QALYs a | Total costs a | ICERb | NHB at £20,000b |
NHB at £30,000b |
| Combined CF | –0.00177 | £446 | 11.75 | 8.68 | £22,001 | 11.75 | 8.68 | £22,447 | 7.55 | 7.93 | |
| Combined SF | –0.00176 | £545 | 11.77 | 8.69 | £22,000 | 11.77 | 8.69 | £22,545 | 7.57 | 7.94 | |
| Combined | Inc QALY loss | Inc costs | Inc LYs a | Inc QALYs a | Inc costs a | Inc LYs a | Inc QALYs a | Inc costs a | INHB at £20,000b |
INHB at £30,000b | |
| SF vs. CF | 0.00001 | £98 | 0.03 | 0.02 | -£1 | 0.03 | 0.02 | £98 | £5946 | 0.01 | 0.01 |
In the subgroup analysis, there is an increased likelihood of correctly classifying individuals with PCa across all CPGs for software versus CF in both the targeted and combined biopsy analysis. However, the lower prevalence means that there are fewer individuals in the model who are more likely to benefit from radical treatment (e.g. prevalence at CPG 4–5 for the prior biopsy subgroup is 8.5% compared to 11.6% in the biopsy naive). Consistently with this, the prior biopsy subgroup cost savings and QALY gains in the long-term model for SF versus CF strategies appear to be smaller than for the base case (particularly so for combined biopsy strategies), resulting in increased ICERs compared to the biopsy naive.
Scenario analysis
The summary results of the scenario analysis are presented in Table 22, with full breakdown presented in Table 124–139, Appendix 12.
| Scenario | Inc LYsa | Inc QALYsa | Inc costsa (£) | ICER per QALY (£) |
|---|---|---|---|---|
| Targeted biopsy | ||||
| Base case | 0.02 | 0.01 | 63 | 5623 |
| 1. PAIREDCAP (2019) baseline | 0.02 | 0.01 | 39 | 4428 |
| 2. Zhou et al.142 diagnostic | 0.03 | 0.03 | 83 | 3105 |
| 3. Degradation of repeat biopsy accuracy | 0.02 | 0.01 | 63 | 5477 |
| 4. SF as quality assurance | 0.000100 | 0.000099 | 87 | 875,042 |
| 5. Radical treatment for all identified CPG ≥ 2 | 0.04 | 0.03 | –117 | Dominates |
| 6.1 Throughput (150/year) | 0.02 | 0.01 | 129 | 11,425 |
| 6.2 Throughput (450/year) | 0.02 | 0.01 | 42 | 3689 |
| Combined biopsy | ||||
| Base case | 0.04 | 0.02 | 49 | 2199 |
| 1. PAIREDCAP (2019) baseline | – | – | – | – |
| 2. Zhou et al.142 diagnostic | – | – | – | – |
| 3. Degradation of repeat biopsy accuracy | 0.05 | 0.03 | 46 | 1801 |
| 4. SF as quality assurance | 0.000141 | 0.000139 | 81 | 582,123 |
| 5. Radical treatment for all identified CPG ≥ 2 and conservative treatment for CPG 1 | 0.08 | 0.05 | –300 | Dominates |
| 6.1 Throughput (150/year) | 0.05 | 0.03 | 110 | 4275 |
| 6.2 Throughput (450/year) | 0.05 | 0.03 | 26 | 1009 |
The cost-effectiveness results for both set of comparisons (targeted and combined biopsy) appear to be robust to variations of the elements of uncertainty in all scenario analyses, with the exception of scenario 5. We discuss below the scenarios in which data sources of the evidence synthesis extension were modified and scenario 5 given its high impact on the estimates of cost-effectiveness. The remaining scenarios are not discussed further.
In scenario 1, the prevalence of PCa is higher (at all CPGs except CPG1) than for the corresponding base-case analysis (targeted comparison), which means that there are proportionally more individuals who can potentially benefit from early treatment. The diagnostic accuracy of the targeted SF is also higher than that of CF strategy, but more so to correctly identify those with CPG2. Overall, this translates into increased cost savings in the long-term model for the targeted SF versus CF compared to the base case (–£58 vs. –£34), which lead to a lower ICER.
In scenario 2, the prevalence of PCa is lower (at all CPGs except CPG2) than for the corresponding base case analysis (targeted comparison), but the diagnostic accuracy is higher for SF compared to CF for all categories of CPG, which overall reduces the ICER for the targeted SF strategy compared to CF to £3689 per additional QALY.
Scenario 5 shows that if there is no difference in diagnostic accuracy between SF and CF, even if some repeat biopsies can be avoided with SF due to it being less prone to operator inexperience, the ICERs for SF compared to CF (targeted and combined biopsy analysis) are far above the upper bound of the cost-effectiveness threshold range recommended by NICE. This is because the small incremental benefits from fewer repeat biopsies are insufficient to offset the higher costs of SF biopsy compared to CF.
Chapter 6 Discussion
Statement of principal findings
The systematic review of clinical evidence included a total of 3733 patients who received SF and 2154 individuals with CF from 23 studies. Evidence was included for all devices specified in the protocol, except for Fusion Bx 2.0 and FusionVu. Fourteen studies were included in the network meta-analyses.
Overall, the evidence for all devices was at high risk of bias and therefore the quantitative synthesis results must be interpreted with caution. Results from our main analysis (looking across ISUP grades) suggest that patients undergoing software biopsy may have: (1) a lower probability of being classified as not having cancer, (2) similar probability of being classified as having non-CS cancer (ISUP grade 1) and (3) higher probability of being classified at higher ISUP grades, particularly ISUP 2. Similar results were obtained when comparing between same biopsy methods where both were combined with systematic biopsy.
Additional meta-analyses of cancer detection rates suggest that, compared with CF biopsy, SF may identify more PCa (any grade) (OR 1.30; 95% CrI 1.06, 1.61). Adding systematic biopsy to cognitive or SF may increase the detection of all PCa and of CS cancer, and from this evidence there is no suggestion that SF with concomitant systematic biopsy is superior to CF with systematic biopsy.
Meta-analyses by individual device showed that compared with CF biopsy, BioJet and Urostation may be associated with a higher detection of PCa overall, and BioJet may be associated with a higher rate of CS cancers, although only one study of BioJet was included in the meta-analyses. Evidence for all other software devices was insufficient to reliably compare their accuracy with CF, or to determine whether some SF technologies are more accurate than others. Evidence for bkFusion, iSR’obot Mona Lisa and KOELIS Trinity was included in the systematic review but not in the meta-analyses. Compared with CF, there was no evidence that the accuracy of SF may differ by lesion location, or between biopsy-naive and prior negative-biopsy patients, or according to operator experience, although the number and quality of the studies informing the potential effect modifiers were limited.
Overall, there is no evidence that biopsy positivity rates and safety outcomes differ significantly between SF and CF, or between SF devices. There was some evidence that systems with rigid registration (BioJet or UroNav) are easier and faster to use than elastic registration (KOELIS Trinity), although this is informed by a single, small study and is not conclusive.
The base-case cost-effectiveness analysis suggests for the targeted biopsy and the combined biopsy comparisons, that SF strategy is on average costlier and yields greater QALYs than the CF strategy, resulting in a probabilistic ICER of £6197 and £2199 per additional QALY for each comparison, respectively. These ICERs are below the lower bound of the cost-effectiveness threshold range recommended by NICE, suggesting that SF may be cost-effective compared to CFs in both the targeted and the combined comparisons. However, these results should be interpreted cautiously, given the uncertainties in the relative diagnostic accuracy evidence which informs the model. The probabilistic analysis suggests a higher probability of cost-effectiveness for SF versus CF at the range of cost-effectiveness thresholds recommended by NICE (0.64 and 0.68 at £20,000 and £30,000 per additional QALY for targeted SF biopsy).
The key findings on the drivers of economic value of SF compared to CF are:
-
The costs (and harms) of SF biopsy in the diagnostic model component can only be offset in the long-term model component, which will only arise from differences in diagnostic accuracy between software and CF.
-
The value gains for SF appear to stem from increased detection at CPG ≥ 2 and, once we adjust for prevalence by CPG category, the greatest contribution to the cost-effectiveness of SF compared to cognitive results from increased correct detection at CPG 2.
-
Increased detection at CPG 1 due to reduced detection of ‘no cancer’ results in value losses at all cancer grades [i.e. there are net losses from shifting classification from ‘no cancer’ to CNS cancer (CPG 1)].
-
The magnitude of value realised for SF versus CF from the balance between different degrees of misclassification and correct classification with the two technologies also depends on the prevalence at each cancer grade.
Given the uncertainties in the costing of SF, we conducted a threshold analysis to identify the SF biopsy cost at which there would be a shift in the decision to accept SF as a good use of NHS resources. This suggested that at the cost of each the five individual technologies for which there was cost data, the recommendation decision would not change.
The base-case cost-effectiveness results were not sensitive to variations to alternative data sources and assumptions, except when no difference in diagnostic accuracy is assumed between SF and CF. Under this assumption, the ICERs for SF compared to CF (targeted and combined biopsy analysis) far exceed the upper bound of the cost-effectiveness threshold range recommended by NICE.
Strengths and limitations of the assessment
This is the first systematic review to formally compare the relative accuracy of SF and CF, with and without systematic biopsy, as well as different SF devices, using both direct and indirect evidence in a formal NMA. In order to best estimate differences between biopsy methods for each PCa grade, a multinomial logistic regression model was fitted, where the odds of being categorised in each of the different ISUP grades were allowed to vary by biopsy type.
Our findings are consistent with those of recent systematic reviews that found no significant difference between SF and CF at detecting non-CSPCas,51–53 although unlike recent evidence,51,53 our NMA found that SF increased detection of CS cancer compared with CF. This result might be explained by differences in review and synthesis methods.
Our review has a number of limitations. Despite attempts to reduce bias by excluding unpaired, non-randomised studies, the evidence included in the meta-analysis remains at high risk of bias. Although within-patient comparisons remove much of the risk of confounding from imbalances in participant characteristics, true blinding from tracks of preceding biopsy methods within the same examination is not feasible (or would require two separate biopsy sessions per patient, which would be unethical). So far, no high-quality RCTs have been published.
There was variation across the studies in patient characteristics. In particular, a number of studies included patients with prior negative-biopsy and biopsy-naive patients, who form the large majority of patients eligible for targeted biopsy, were under-represented. Some variation and gaps in reporting were observed in MRI acquisition methods, criteria for referral to biopsy, biopsy routes and anaesthesia methods. Definitions of PCa and CS cancer varied across the studies. There was insufficient evidence to explore the impact of a number of potential effect modifiers, including lesion location, operator experience, biopsy routes and anaesthesia methods.
The results of the synthesis models require careful interpretation, as they refer to comparisons between different cancer grades. The interpretation of the multinomial models on the absolute probability scale results is more intuitive and directly relevant to clinical practice. Overall, results are concordant across analyses and concordant with the data. Only the multinomial results are used in the economic model, as the value of diagnostic information provided by each test is dependent on the subsequent clinical decisions based on test results, and clinical management is conditional on cancer grades (jointly with other prognostic information).
Most estimates from the NMAs were imprecise, particularly in the multinomial synthesis and at higher ISUP grades where data were most sparse. The NMA relied on a number of assumptions. CF was assumed to be equivalent across studies. The risk and extent to which the accuracy of CF may vary by centre and operator experience are uncertain due to lack of evidence. It was also assumed that data from within-patient studies were independent. A model that accounted for the full structure of the data was not available, although it could have added precision to the estimates.
There were few studies per comparison and not all studies reported outcomes by all cancer grades. Therefore, only fixed-effect models were fit to the data. Data were sparse for most SF devices, and few studies included more than one SF technology, making it difficult to draw conclusions for relative accuracy of individual devices.
While our review identified several relevant studies, many could not be included in the synthesis due to lack of reporting of key data. For example, studies comparing software and CF to systematic biopsy reported data on both targeted technologies jointly, and few studies reported a sufficient breakdown of biopsy results by ISUP grades (or equivalent breakdown) to inform the evidence synthesis required for the economic model. In addition, where studies included a mixed population of patients, a lack of reporting of biopsy results for the relevant population led to their exclusion from the meta-analysis. We were therefore limited in the models we could consider due to data sparseness, and results are uncertain.
Studies not included in the meta-analyses mostly reported test-positive rates (positive cases as percentage of all patients). As this measure is dependent on disease prevalence rather than diagnostic accuracy, results from these studies may be influenced by differences in PCa rates between cohorts and may not be reliable.
The above-mentioned limitations in the evidence are not captured in the quantitative evidence synthesis, which is used to inform the economic analysis.
The cost-effectiveness analysis relies on the evidence informing it. Beyond the evidence sourced from the synthesis, this includes evidence on the long-term outcomes of treating PCa and the cost data on each SF technology. This evidence is limited.
Uncertainties
No evidence was found for most of this assessment’s prespecified outcomes: biopsy sample suitability/quality, number of repeat biopsies performed, procedure completion rates, software failure rate, time to diagnosis, length of hospital stay, time taken for MR image preparation, subsequent PCa management, re-biopsy rate, hospitalisation, OS, PFS, patient- and carer-reported outcomes (including tolerability and HRQoL), barriers and facilitators to implementations.
There was large uncertainty in all estimates due to the limited evidence. Meta-analyses showed moderate heterogeneity that could not be explained by differences in individual SF devices. The evidence for all SF devices was at high risk of bias, and the diagnostic accuracy of systematic biopsy relative to SF and cognitive may have been overestimated in the meta-analyses. The applicability of the evidence for KOELIS Trinity and BiopSee is uncertain. There is no evidence comparing the accuracy of Fusion Bx 2.0 and FusionVu with CF, and no evidence for these devices were eligible for inclusion in the indirect comparisons.
None of the studies included in the systematic review of diagnostic accuracy used template mapping biopsy as a reference standard, and many studies did not use standard 12-core systematic biopsy in addition to targeted biopsy methods. This means that the absolute true rate of PCa lesions was underestimated and is uncertain. However, the lack of a gold-standard test is likely to have affected comparisons between all devices similarly, and therefore is unlikely to have biased relative estimates of PCa detection.
The evidence supporting recent consensuses for classifying the clinical significance of PCa is not without limitations, despite recent improvements in imaging. 34,112,151 Trial evidence indicates that survival outcomes for some patients with more severe grades of localised PCa (CPG 3 and above) are favourable, and that the detriment associated with active monitoring may be small. 55,114 This raises further uncertainty regarding the added clinical (and economic) value of SF. Where reported, the number of targeted cores performed with software and CF were broadly comparable between the studies. However, not all studies reported data on number of targeted cores to fully assess the risk of confounding from a possible difference in number of targeted cores between software and CF. Evidence for all other protocol-specified outcomes was limited and inconclusive.
The economic value of SF seems to be driven by (1) comparative diagnostic accuracy derived where evidence is particularly sparse (cancer grades above 2), and (2) by prevalence, which is also affected by evidence sparsity.
Structural assumptions were applied to allow estimating the economic value of SF given the evidence gaps in the diagnostic accuracy of SF and in the longer-term outcomes of PCa. The structural uncertainty of these assumptions was explored in scenario analysis, but could not be jointly captured with parameter uncertainty. This means that the probabilistic results presented in this report are likely to underestimate the overall uncertainty.
Other relevant factors
Participants of studies included in the systematic review of diagnostic accuracy and clinical effectiveness had elevated PSA and/or abnormal DRE results and were referred to targeted biopsy following a PI-RADS or Likert score of three or more on MRI. This is reflective of NICE guidance, which recommends that men should be referred for mpMRI if their PSA levels are above the age-specific reference range or if their prostate feels malignant on DRE. However, other organisations have recommended that PSA levels should be used as part of a risk prediction tool, potentially leading to better targeting of patients referred to mpMRI. It is unclear how a change in referral criteria may affect the applicability of this assessment’s findings. 13
Equality, diversity and inclusion
This study was carried out by a multidisciplinary team, consisting of an information specialist, a statistician, systematic reviewers and health economists. Junior members of staff contributed according to their skills and had the opportunity to undertake tasks that furthered their training. Clinical experts that provided advice included a nurse and senior consultants.
This report was prepared to support NICE guidance to help reduce health inequalities, improve access to health care and encourage health improvement (www.nice.org.uk/about/who-we-are/policies-and-procedures/nice-equality-scheme).
Patient and public involvement
This report was prepared to support NICE guidance. Lay people, and organisations representing their interests, have opportunities to contribute to developing NICE guidance, advice and quality standards, and support their implementation (www.nice.org.uk/about/nice-communities/nice-and-the-public/public-involvement/public-involvement-programme/patient-public-involvement-policy).
Chapter 7 Conclusions
Compared to CF biopsy, patients undergoing SF biopsy may have a lower probability of being classified as not having cancer, similar probability of being classified as having non-CS cancer, and a higher probability of being classified at higher ISUPs, particularly ISUP 2. Both SF and CF biopsy can miss CS cancer lesions, and the addition of a standard-systematic biopsy increases the detection of all PCa and CS cancer for both fusion methods. There is insufficient evidence to conclude on the relative accuracy and clinical effectiveness of different software devices.
Cost-effectiveness estimates comparing software to CF were generally favourable to SF, except where the technologies were assumed to have the same diagnostic accuracy. The drivers of economic value of SF, comparative diagnostic accuracy and prevalence, are affected by unquantified uncertainty. Judgements on the economic value of SF require integration of the uncertainties over the clinical evidence with the overall cost-effectiveness.
Suggested research priorities
High-quality, sufficiently powered RCT evidence, comparing SF and CF with or without systematic biopsy in trained operators, is needed to address the limitations of the diagnostic accuracy evidence identified in this study. Ideally, a trial should be sufficiently powered to detect long-term oncological outcomes (PFS, PCa mortality, overall mortality), although we acknowledge that such a study may not be feasible. IP7-PACIFIC (NCT05574647),155 a large UK-based randomised trial, will aim to determine whether SF biopsy is superior to CF at detecting CSPCa in patients with suspicious MRI in patients randomised to either mpMRI or bpMRI. It is hoped that this trial will provide more precise diagnostic accuracy estimates, although it is not clear which specific SF devices will be used, and the protocol indicates that estimates of diagnostic accuracy will not be informed by a gold standard test.
Full reporting of ISUP grades for each randomised arm is recommended, and for within-patient comparison studies, full reporting of cross-tabulation tables, where the classification of patients’ cancer by ISUP grade for each biopsy type is described and the relative accuracy of the interventions can be derived. In mixed population studies, reporting by key patient characteristics, such as PI-RADS score, whether biopsy naive or experienced, and route of referral for MRI (e.g. following clinical concerns, routine surveillance, screening etc.) are required to inform decision-making. Availability of more granular data, from already published studies, would enable future syntheses to make use of a larger body of evidence. Qualitative evidence on the acceptability of SF to patients, notably where biopsy procedure time might be significantly increased, is needed.
Additional information
Contributions of authors
Alexis Llewellyn (https://orcid.org/0000-0003-4569-5136) (Systematic Reviewer) contributed to the protocol, performed the systematic review, wrote the background and most of the sections on clinical effectiveness.
Thai Han Phung (https://orcid.org/0000-0001-6193-4673) (Health Economist) assisted with the economic evidence reviews and with the development of the economic model, its analyses and validation, and contributed to the writing of the cost-effectiveness sections.
Marta O Soares (https://orcid.org/0000-0003-1579-8513) (Health Economist) developed the methods to extend the evidence synthesis for use in the economic model and conducted the required analyses over the extended synthesis model. She provided oversight and contributed to the development of the inference sub model to inform the economic model. She wrote the cost-effectiveness sections of the protocol and main report, and provided leadership support to the economic sections.
Lucy Shepherd (https://orcid.org/0000-0001-6803-5566) (Systematic Reviewer) contributed to the protocol, performed the systematic review and wrote parts of the clinical sections.
David Glynn (https://orcid.org/0000-0002-0989-1984) (Health Economist) reviewed the prostate cancer long-term outcome evidence and developed the inference sub model to inform the economic model.
Melissa Harden (https://orcid.org/0000-0003-2338-6869) (Information Specialist) designed and ran all searches for the study, managed the library of references, and wrote the search sections of the report.
Ruth Walker (https://orcid.org/0000-0003-2765-7363) (Systematic Reviewer) contributed to the protocol and background materials.
Ana Duarte (https://orcid.org/0000-0002-0528-4773) (Health Economist) developed the economic model, conducted analyses over this model and validated it. Ana had overall responsibility for the cost-effectiveness sections of the report. She wrote the cost-effectiveness sections of the protocol and of the report.
Sofia Dias (https://orcid.org/0000-0002-2172-0221) (Statistician) contributed to the protocol, adapted the network meta-analysis model and performed the meta-analyses, wrote up the meta-analysis section results, and oversaw the conduct and writing of the clinical effectiveness sections and the report as a whole. Sofia has overall responsibility for the clinical effectiveness sections of the report.
Acknowledgements
We would like to thank specialist DAC members Dr Oliver Hulson, Consultant Radiologist at Leeds Teaching Hospital Trust, Ms Lillian White, Clinical Nurse Specialist at NHS Ayshire and Arran, and Mr Hide Yamamoto, Consultant Urological Surgeon at Maidstone and Tunbridge Wells Trust for providing helpful advice during this study. We also thank Ms Lillian White for commenting on an early draft of the report. We would also like to thank Prof Hashim Ahmed for providing helpful advice during this study; Mr Connor Evans, Research Trainee at CRD, for helping to edit the final version of this report; and Prof Stephen Palmer for advice on the conceptualisation of the economic model.
We thank Dr Bhash Naidoo and the NICE Centre for Guidelines who developed the NICE NG131 health economics model and for sharing it. The review of the electronic version of the model contributed to inform the York-model conceptualisation. Similarly, we would like to thank Dr Joanne Lord and the Southampton TAR group who developed the Southampton model and for sharing it. The electronic version of the Southampton model was used to support both model conceptualisation and parameterisation of the York model.
Data-sharing statement
All data are provided in appendices to this report. Additional requests for access to data should be addressed to the corresponding author.
Ethics statement
Due to the nature of this study (systematic review of aggregate data and economic analysis), no ethical approval was required.
Information governance statement
This study did not handle any personal information.
Disclosure of interests
Full disclosure of interests: Completed ICMJE forms for all authors, including all related interests, are available in the toolkit on the NIHR Journals Library report publication page at https://doi.org/10.3310/PLFG4210.
Primary conflict of interest: Marta O Soares is a member of the NIHR Health Technology Assessment (HTA) General Committee.
Disclaimers
This article presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- Cancer Research UK . Ten Most Common Cancers in Males 2021. www.cancerresearchuk.org/health-professional/cancer-statistics/incidence/common-cancers-compared#heading-One (accessed 20 June 2022).
- Public Health England . Cancer Registration Statistics, England: Final Release 2018. www.gov.uk/government/statistics/cancer-registration-statistics-england-2018-final-release/cancer-registration-statistics-england-final-release-2018 (accessed 2 April 2022).
- Cancer Research UK . Prostate Cancer Statistics: Prostate Cancer Incidence 2017. www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/prostate-cancer#heading-Zero (accessed 23 March 2022).
- National Prostate Cancer Audit (NPCA) Team . Eighth Year Annual Report of the NPCA Prospective Audit in England and Wales for Men Diagnosed from 1 April 2019 to 31 March 2020 and the Impact of COVID-19 in England During 2020 2022.
- Jones AL, Chinegwundoh F. Update on prostate cancer in black men within the UK. Ecancermedicalscience 2014;8.
- Kiciński M, Vangronsveld J, Nawrot TS. An epidemiological reappraisal of the familial aggregation of prostate cancer: a meta-analysis. PLOS ONE 2011;6.
- Guo Z, He J, Pan J, Huang L, Cao J, Bai Z, et al. Prevalence and risk factors for incidental prostate cancer in patients after transurethral resection of the prostate with negative results on prostate biopsy: a retrospective study. Investig Clin Urol 2022;63:201-6.
- Moynihan R, Doust J, Henry D. Preventing overdiagnosis: how to stop harming the healthy. BMJ 2012;344.
- McCaffery K, Nickel B, Pickles K, Moynihan R, Kramer B, Barratt A, et al. Resisting recommended treatment for prostate cancer: a qualitative analysis of the lived experience of possible overdiagnosis. BMJ Open 2019;9.
- National Institute for Health and Care Excellence . Prostate Cancer: Diagnosis and Management. [I] Evidence Reviews for Risk Stratification of Localised Prostate Cancer 2021.
- National Institute for Health and Care Excellence . Diagnostics Assessment Programme. Diagnostics Consultation Document: Transperineal Biopsy for Diagnosing Prostate Cancer 2022.
- National Institute of Health and Care Excellence . Prostate Cancer: Diagnosis and Management 2019.
- Prostate Cancer UK . Consensus Statements on PSA Testing in Asymptomatic Men in the UK: Information for Health Professionals – Prostate Cancer UK 2016. https://prostatecanceruk.org/PSAconsensusHP (accessed 24 October 2022).
- National Institute for Health and Care Excellence . Suspected Cancer: Recognition and Referral 2015.
- NHS England . Faster Diagnosis Standard n.d. www.england.nhs.uk/cancer/faster-diagnosis/#:~:text=The%20Faster%20Diagnosis%20Standard%20(FDS,cancer%20have%20a%20timely%20diagnosis (accessed 24 October 2022).
- National Institute for Health and Care Excellence . Quality Statement 1: Direct Access to Diagnostic Tests – Suspected Cancer: Quality Standard [QS124] 2016. www.nice.org.uk/guidance/qs124/chapter/Quality-statement-1-Direct-access-to-diagnostic-tests (accessed 24 October 2022).
- NHS England . Implementing a Timed Prostate Cancer Diagnostic Pathway: A Handbook for Local Health and Care Systems 2018.
- Ahmed HU, Bosaily AES, Brown LC, Kaplan RS, Colaco-Moraes Y, Ward K, et al. The PROMIS study: a paired-cohort, blinded confirmatory study evaluating the accuracy of multi-parametric MRI and TRUS biopsy in men with an elevated PSA. J Clin Oncol 2016;34.
- Kasivisvanathan V, Rannikko AS, Borghi M, Panebianco V, Mynderse LA, Vaarala MH, et al. PRECISION Study Group Collaborators . MRI-targeted or standard biopsy for prostate-cancer diagnosis. N Engl J Med 2018;378:1767-77.
- Barentsz JO, Richenberg J, Clements R, Choyke P, Verma S, Villeirs G, et al. European Society of Urogenital Radiology . ESUR prostate MR guidelines 2012. Eur Radiol 2012;22:746-57.
- Turkbey B, Choyke PL. Annual Review of Medicine. Palo Alto: Annual Reviews; 2019.
- Weinreb JC, Barentsz JO, Choyke PL, Cornud F, Haider MA, Macura KJ, et al. PI-RADS prostate imaging: reporting and data system: 2015, version 2. Eur Urol 2016;69:16-40.
- West of Scotland Cancer Network . Audit Report: Prostate Cancer Quality Performance Indicators – Clinical Audit Data: 01 July 2019 to 30 June 2020 2021.
- North Cancer Alliance . Quality Performance Indicators Audit Report: Prostate Cancer 2022.
- Barrett T, Slough R, Sushentsev N, Shaida N, Koo BC, Caglic I, et al. Three-year experience of a dedicated prostate mpMRI pre-biopsy programme and effect on timed cancer diagnostic pathways. Clin Radiol 2019;74:894.e1-9.
- Alabousi M, Salameh JP, Gusenbauer K, Samoilov L, Jafri A, Yu H, et al. Biparametric vs. multiparametric prostate magnetic resonance imaging for the detection of prostate cancer in treatment-naïve patients: a diagnostic test accuracy systematic review and meta-analysis. BJU Int 2019;124:209-20.
- Lopez JF, Campbell A, Omer A, Stroman L, Bondad J, Austin T, et al. Local anaesthetic transperineal (LATP) prostate biopsy using a probe-mounted transperineal access system: a multicentre prospective outcome analysis. BJU Int 2021;128:311-8.
- National Institute for Health and Care Excellence . NICE Recommends New Diagnostic Devices for Men With Suspected Prostate Cancer in Draft Guidance 2022. www.nice.org.uk/news/article/nice-recommends-new-diagnostic-devices-for-men-with-suspected-prostate-cancer-in-draft-guidance (accessed 9 May 2022).
- National Institute for Health and Care Excellence . Transperineal Biopsy for Diagnosing Prostate Cancer: In Development [GID-DG10043] n.d. www.nice.org.uk/guidance/indevelopment/gid-dg10043 (accessed 23 May 2022).
- Kucur M, Goktas S, Kaynar M, Apiliogullari S, Kilic O, Akand M, et al. Selective low-dose spinal anesthesia for transrectal prostate biopsy: a prospective and randomized study. J Endourol 2015;29:1412-7.
- Wegelin O, Exterkate L, van der Leest M, Kummer JA, Vreuls W, de Bruin PC, et al. The FUTURE trial: a multicenter randomised controlled trial on target biopsy techniques based on magnetic resonance imaging in the diagnosis of prostate cancer in patients with prior negative biopsies. Eur Urol 2019;75:582-90.
- Bjurlin MA, Taneja SS. Standards for prostate biopsy. Curr Opin Urol 2014;24:155-61.
- Das CJ, Razik A, Sharma S, Verma S. Prostate biopsy: when and how to perform. Clin Radiol 2019;74:853-64.
- European Association of Urology . EAU Guidelines on Prostate Cancer 2022.
- Fulgham PF, Rukstalis DB, Rubenstein JN, Taneja SS, Carroll PR, Pinto PA, et al. Standard Operating Procedure for Multiparametric Magnetic Resonance Imaging in the Diagnosis, Staging and Management of Prostate Cancer. American Urological Association; 2019.
- Prostate Cancer Foundation . About Prostate Cancer 2022. www.pcf.org/about-prostate-cancer/ (accessed 24 October 2022).
- Rouviere O, Puech P, Renard-Penna R, Claudon M, Roy C, Mege-Lechevallier F, et al. MRI-FIRST Investigators . Use of prostate systematic and targeted biopsy on the basis of multiparametric MRI in biopsy-naïve patients (MRI-FIRST): a prospective, multicentre, paired diagnostic study. Lancet Oncol 2019;20:100-9.
- van der Leest M, Cornel E, Israel B, Hendriks R, Padhani AR, Hoogenboom M, et al. Head-to-head comparison of transrectal ultrasound-guided prostate biopsy vs. multiparametric prostate resonance imaging with subsequent magnetic resonance-guided biopsy in biopsy-naïve men with elevated prostate-specific antigen: a large prospective multicenter clinical study. Eur Urol 2019;75:570-8.
- Emmett L, Buteau J, Papa N, Moon D, Thompson J, Roberts MJ, et al. The additive diagnostic value of prostate-specific membrane antigen positron emission tomography computed tomography to multiparametric magnetic resonance imaging triage in the diagnosis of prostate cancer (PRIMARY): a prospective multicentre study. Eur Urol 2021;80:682-9.
- Ahmed HU, El-Shater Bosaily A, Brown LC, Gabe R, Kaplan R, Parmar MK, et al. PROMIS Study Group . Diagnostic accuracy of multi-parametric MRI and TRUS biopsy in prostate cancer (PROMIS): a paired validating confirmatory study. Lancet 2017;389:815-22.
- Thompson J, Savdie R, Ponsky L, Brenner P, Shnier R, Moses D, et al. Magnetic resonance imaging detects significant prostate cancer and could be used to reduce unnecessary biopsies: initial results from a prospective trial of MRI in men planned for diagnostic biopsy. Urology 2013;82:S26-7.
- Parry MG, Boyle JM, Nossiter J, Morris M, Sujenthiran A, Berry B, et al. Determinants of variation in radical local treatment for men with high-risk localised or locally advanced prostate cancer in England [Published online ahead of print September 7 2021]. Prostate Cancer Prostatic Dis 2021;26:257-63. https://doi.org/10.1038/s41391-021-00439-9.
- Morris M. Exploring the Use of a New Risk Grouping to Assess ‘Over-Treatment’ for the National Prostate Cancer Audit. National Prostate Cancer Audit, Royal College of Surgeons of England; 2022.
- Bjurlin MA, Rosenkrantz AB, Taneja SS. MRI-fusion biopsy: the contemporary experience. Transl Androl Uro 2017;6:483-9.
- Yan P, Wang XY, Huang W, Zhang Y. Local anesthesia for pain control during transrectal ultrasound-guided prostate biopsy: a systematic review and meta-analysis. J Pain Res 2016;9:787-96.
- Lee D, Chung BH, Lee KS. Effect of training and individual operator’s expertise on prostate cancer detection through prostate biopsy: implications for the current quantitative training evaluation system. Investig Clin Urol 2021;62:658-65.
- Costa DN, Pedrosa I, Donato F, Roehrborn CG, Rofsky NM. MR imaging-transrectal US fusion for targeted prostate biopsies: implications for diagnosis and clinical management. Radiographics 2015;35:696-708.
- Cash H, Gunzel K, Maxeiner A, Stephan C, Fischer T, Durmus T, et al. Prostate cancer detection on transrectal ultrasonography-guided random biopsy despite negative real-time magnetic resonance imaging/ultrasonography fusion-guided targeted biopsy: reasons for targeted biopsy failure. BJU Int 2016;118:35-43.
- Al-Ahmadie HA, Tickoo SK, Olgac S, Gopalan A, Scardino PT, Reuter VE, et al. Anterior-predominant prostatic tumors: zone of origin and pathologic outcomes at radical prostatectomy. Am J Surg Pathol 2008;32:229-35.
- Bott SRJ, Young MPA, Kellett MJ, Parkinson MC. Contributors to the UCL Hospitals’ Trust Radical Prostatectomy Database . Anterior prostate cancer: is it more difficult to diagnose?. BJU Int 2002;89:886-9.
- Watts KL, Frechette L, Muller B, Ilinksy D, Kovac E, Sankin A, et al. Systematic review and meta-analysis comparing cognitive vs. image-guided fusion prostate biopsy for the detection of prostate cancer. Urol Oncol 2020;38:734.e19-25.
- Sathianathen NJ, Butaney M, Bongiorno C, Konety BR, Bolton DM, Lawrentschuk N. Accuracy of the magnetic resonance imaging pathway in the detection of prostate cancer: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis 2019;22:39-48.
- Bass EJ, Pantovic A, Connor MJ, Loeb S, Rastinehad AR, Winkler M, et al. Diagnostic accuracy of magnetic resonance imaging targeted biopsy techniques compared to transrectal ultrasound guided biopsy of the prostate: a systematic review and meta-analysis. Prostate Cancer Prostatic Dis 2021;25:174-9.
- Valerio M, Donaldson I, Emberton M, Ehdaie B, Hadaschik BA, Marks LS, et al. Detection of clinically significant prostate cancer using magnetic resonance imaging-ultrasound fusion targeted biopsy: a systematic review. Eur Urol 2015;68:8-19.
- Hamdy FC, Donovan JL, Lane JA, Mason M, Metcalfe C, Holding P, et al. PROTecT Study Group . 10-year outcomes after monitoring, surgery, or radiotherapy for localized prostate cancer. N Engl J Med 2016;375:1415-24.
- Kishan AU, Sun Y, Hartman H, Pisansky TM, Bolla M, Neven A, et al. MARCAP Consortium Group . Androgen deprivation therapy use and duration with definitive radiotherapy for localised prostate cancer: an individual patient data meta-analysis. Lancet Oncol 2022;23:304-16.
- National Institute for Health and Care Excellence . Low Dose Rate Brachytherapy for Localised Prostate Cancer (IPG 132) 2005.
- Morris WJ, Tyldesley S, Rodda S, Halperin R, Pai H, McKenzie M, et al. Androgen suppression combined with elective nodal and dose escalated radiation therapy (the ASCENDE-RT trial): an analysis of survival endpoints for a randomized trial comparing a low-dose-rate brachytherapy boost to a dose-escalated external beam boost for high- and intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys 2017;98:275-85.
- James ND, Sydes MR, Clarke NW, Mason MD, Dearnaley DP, Spears MR, et al. STAMPEDE Investigators . Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016;387:1163-77.
- Fizazi K, Faivre L, Lesaunier F, Delva R, Gravis G, Rolland F, et al. Androgen deprivation therapy plus docetaxel and estramustine vs. androgen deprivation therapy alone for high-risk localised prostate cancer (GETUG 12): a phase 3 randomised controlled trial. Lancet Oncol 2015;16:787-94.
- Schweizer MT, Huang P, Kattan MW, Kibel AS, de Wit R, Sternberg CN, et al. Adjuvant leuprolide with or without docetaxel in patients with high-risk prostate cancer after radical prostatectomy (TAX-3501): important lessons for future trials. Cancer 2013;119:3610-8.
- Wegelin O, van Melick HHE, Hooft L, Bosch J, Reitsma HB, Barentsz JO, et al. Comparing three different techniques for magnetic resonance imaging-targeted prostate biopsies: a systematic review of in-bore vs. magnetic resonance imaging-transrectal ultrasound fusion vs. cognitive registration. Is there a preferred technique?. Eur Urol 2017;71:517-31.
- Drost F, Osses D, Nieboer D, Steyerberg E, Bangma C, Roobol M, et al. Prostate MRI, with or without MRI‐targeted biopsy, and systematic biopsy for detecting prostate cancer. Cochr Datab Syst Rev 2019;4.
- McGowan J, Sampson M, Salzwedel DM, Cogo E, Foerster V, Lefebvre C. PRESS peer review of electronic search strategies: 2015 guideline statement. J Clin Epidemiol 2016;75:40-6.
- Beltran L, Ahmad AS, Sandu H, Kudahetti S, Soosay G, Møller H, et al. Transatlantic Prostate Group . Histopathologic false-positive diagnoses of prostate cancer in the age of immunohistochemistry. Am J Surg Pathol 2019;43:361-8.
- Drost FJ, Osses D, Nieboer D, Bangma C, Steyerberg E, Roobol M, et al. Prostate MRI, with or without targeted biopsy, and standard biopsy for detecting prostate cancer: a Cochrane systematic review and meta-analysis. J Urol 2019;18:e728-9.
- Whiting PF, Rutjes AW, Westwood ME, Mallett S, Deeks JJ, Reitsma JB, et al. QUADAS-2 Group . QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med 2011;155:529-36.
- Yang B, Mallett S, Takwoingi Y, Davenport CF, Hyde CJ, Whiting PF, et al. QUADAS-C Group . QUADAS-C: a tool for assessing risk of bias in comparative diagnostic accuracy studies. Ann Intern Med 2021;174:1592-9.
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 1: Introduction to Evidence Synthesis for Decision Making. Sheffield: Decision Support Unit, ScHARR, University of Sheffield; 2011.
- Dias S, Caldwell DM. Network meta-analysis explained. Arch Dis Child Fetal Neonatal Ed 2019;104:F8-F12.
- Dias S, Welton NJ, Sutton AJ, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-analysis of Randomised Controlled Trials. Sheffield: Decision Support Unit, ScHARR, University of Sheffield; 2011.
- R Core Team . R: A Language and Environment for Statistical Computing [Software]. 2022. www.R-project.org/ (accessed 25 October 2022).
- Rücker G, Schwarzer G, Krahn U, König J. Netmeta: Network Meta-Analysis Using Frequentist Methods. Version 2.5-0 Ed. [Software] 2022. http://CRAN.R-project.org/package=netmeta (accessed 25 October 2022).
- Gelman A, Carlin JB, Stern HS, Dunson DB, Vehtari A, Rubin DB. Bayesian Data Analysis. Boca Raton, FL: CRC Press; 2013.
- Lunn D, Jackson C, Best N, Thomas A, Spiegelhalter D. The BUGS Book. Boca Raton, FL: CRC Press; 2013.
- van Valkenhoef G, Kuiper J. gemtc: Network Meta-analysis Using Bayesian Methods. R Package. 1.0-1 ed. [Software]. CRAN; 2021.
- Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: Inconsistency in Networks of Evidence Based on Randomised Controlled Trials. Sheffield: Decision Support Unit, ScHARR, University of Sheffield; 2011.
- Campbell M, McKenzie JE, Sowden A, Katikireddi SV, Brennan SE, Ellis S, et al. Synthesis without meta-analysis (SWiM) in systematic reviews: reporting guideline. BMJ 2020;368.
- Thangarasu M, Jayaprakash SP, Selvaraj N, Bafna S, Paul R, Mahesh C, et al. A prospective study on the efficacy of cognitive targeted transrectal ultrasound prostate biopsy in diagnosing clinically significant prostate cancer. Res Rep Urol 2021;13:207-13.
- Alberts AR, Schoots IG, Bokhorst LP, Drost FH, van Leenders GJ, Krestin GP, et al. Characteristics of prostate cancer found at fifth screening in the European randomized study of screening for prostate cancer Rotterdam: can we selectively detect high-grade prostate cancer with upfront multivariable risk stratification and magnetic resonance imaging?. Eur Urol 2018;73:343-50.
- Ferriero M, Tuderti G, Muto GL, Fiori C, Bove AM, Mastroianni R, et al. Diagnostic performance of fusion (US/MRI guided) prostate biopsy: propensity score matched comparison of elastic vs. rigid fusion system. World J Urol 2022;40:991-6.
- Izadpanahi MH, Elahian A, Gholipour F, Khorrami MH, Zargham M, Mohammadi Sichani M, et al. Diagnostic yield of fusion magnetic resonance-guided prostate biopsy vs. cognitive-guided biopsy in biopsy-naïve patients: a head-to-head randomized controlled trial. Prostate Cancer Prostatic Dis 2021;24:1103-9.
- Sokolakis I, Pyrgidis N, Koneval L, Krebs M, Thurner A, Kubler H, et al. Usability and diagnostic accuracy of different MRI/ultrasound-guided fusion biopsy systems for the detection of clinically significant and insignificant prostate cancer: a prospective cohort study. World J Urol 2021;39:4101-8.
- Rabah D, Al-Taweel W, Khan F, Arafa M, Mehmood S, Mokhtar A, et al. Transperineal vs. transrectal multi-parametric magnetic resonance imaging fusion targeted prostate biopsy. Saudi Med J 2021;42:649-54.
- Liang L, Cheng Y, Qi F, Zhang L, Cao D, Cheng G, et al. A comparative study of prostate cancer detection rate between transperineal COG-TB and transperineal FUS-TB in patients with PSA <=20 ng/mL. J Endourol 2020;34:1008-14.
- Kulis T, Zekulic T, Alduk AM, Lusic M, Bulimbasic S, Ferencak V, et al. Targeted prostate biopsy using a cognitive fusion of multiparametric magnetic resonance imaging and transrectal ultrasound in patients with previously negative systematic biopsies and non-suspicious digital rectal exam. Croat Med J 2020;61:49-54.
- Wajswol E, Winoker JS, Anastos H, Falagario U, Okhawere K, Martini A, et al. A cohort of transperineal electromagnetically tracked magnetic resonance imaging/ultrasonography fusion-guided biopsy: assessing the impact of inter-reader variability on cancer detection. BJU Int 2020;125:531-40.
- Elkhoury FF, Felker ER, Kwan L, Sisk AE, Delfin M, Natarajan S, et al. Comparison of targeted vs systematic prostate biopsy in men who are biopsy naïve: the prospective assessment of image registration in the diagnosis of prostate cancer (PAIREDCAP) study. JAMA Surg 2019;154:811-8.
- Stabile A, Dell’Oglio P, Gandaglia G, Fossati N, Brembilla G, Cristel G, et al. Not all multiparametric magnetic resonance imaging-targeted biopsies are equal: the impact of the type of approach and operator expertise on the detection of clinically significant prostate cancer. Eur Urol Oncol 2018;1:120-8.
- Monda SM, Vetter JM, Andriole GL, Fowler KJ, Shetty AS, Weese JR, et al. Cognitive vs. software fusion for MRI-targeted biopsy: experience before and after implementation of fusion. Urology 2018;119:115-20.
- Kaufmann S, Russo GI, Thaiss W, Notohamiprodjo M, Bamberg F, Bedke J, et al. Cognitive vs. software-assisted registration: development of a new nomogram predicting prostate cancer at MRI-targeted biopsies. Clin Genitourin Cancer 2018;16:e953-60.
- Fourcade A, Payrard C, Tissot V, Perrouin-Verbe MA, Demany N, Serey-Effeil S, et al. The combination of targeted and systematic prostate biopsies is the best protocol for the detection of clinically significant prostate cancer. Scand J Urol 2018;52:174-9.
- Cornud F, Roumiguie M, de Longchamps NB, Ploussard G, Bruguiere E, Portalez D, et al. Precision matters in MR imaging-targeted prostate biopsies: evidence from a prospective study of cognitive and elastic fusion registration transrectal biopsies. Radiology 2018;287:534-42.
- Albisinni S, Aoun F, Noel A, El Rassy E, Lemort M, Paesmans M, et al. Are concurrent systematic cores needed at the time of targeted biopsy in patients with prior negative prostate biopsies?. Prog Urol 2018;28:18-24.
- Hansen NL, Barrett T, Kesch C, Pepdjonovic L, Bonekamp D, O’Sullivan R, et al. Multicentre evaluation of magnetic resonance imaging supported transperineal prostate biopsy in biopsy-naïve men with suspicion of prostate cancer. BJU Int 2018;122:40-9.
- Filson CP, Natarajan S, Margolis DJ, Huang J, Lieu P, Dorey FJ, et al. Prostate cancer detection with magnetic resonance-ultrasound fusion biopsy: the role of systematic and targeted biopsies. Cancer 2016;122:884-92.
- Wysock JS, Rosenkrantz AB, Huang WC, Stifelman MD, Lepor H, Deng FM, et al. A prospective, blinded comparison of magnetic resonance (MR) imaging-ultrasound fusion and visual estimation in the performance of MR-targeted prostate biopsy: the PROFUS trial. Eur Urol 2014;66:343-51.
- Delongchamps NB, Peyromaure M, Schull A, Beuvon F, Bouazza N, Flam T, et al. Prebiopsy magnetic resonance imaging and prostate cancer detection: comparison of random and targeted biopsies. J Urol 2013;189:493-9.
- Gomez-Ortiz D, Garza-Gangemi AM, Oropeza-Aguilar M, Rangel-Suarez S, Espinosa-Cruz V, Villegas-Hernandez AC, et al. Routine systematic prostate biopsies not replaced by magnetic resonance imaging-targeted biopsy [published online ahead of print June 7 2022]. Rev Invest Clin 2022;74:212-8. https://doi.org/10.24875/RIC.22000084.
- Lockhart K, Martin J, White M, Raman A, Grant A, Chong P. Fusion vs. cognitive MRI-guided prostate biopsies in diagnosing clinically significant prostate cancer [published online ahead of print May 13 2022]. J Clin Urol n.d. https://doi.org/10.1177/20514158221085081.
- Kaufmann S, Russo GI, Bamberg F, Lowe L, Morgia G, Nikolaou K, et al. Prostate cancer detection in patients with prior negative biopsy undergoing cognitive-, robotic- or in-bore MRI target biopsy. World J Urol 2018;36:761-8.
- Jordan PWTBMIL, Weerdmeester B, Brooke J. Usability Evaluation in Industry. London: CRC Press; 1996.
- Rodda S, Tyldesley S, Morris WJ, Keyes M, Halperin R, Pai H, et al. ASCENDE-RT: an analysis of treatment-related morbidity for a randomized trial comparing a low-dose-rate brachytherapy boost with a dose-escalated external beam boost for high- and intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys 2017;98:286-95.
- Incrocci L, Wortel RC, Alemayehu WG, Aluwini S, Schimmel E, Krol S, et al. Hypofractionated vs. conventionally fractionated radiotherapy for patients with localised prostate cancer (HYPRO): final efficacy results from a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol 2016;17:1061-9.
- Catton CN, Lukka H, Gu CS, Martin JM, Supiot S, Chung PWM, et al. Randomized trial of a hypofractionated radiation regimen for the treatment of localized prostate cancer. J Clin Oncol 2017;35:1884-90.
- Dearnaley D, Syndikus I, Mossop H, Khoo V, Birtle A, Bloomfield D, et al. CHHiP Investigators . Conventional vs. hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: 5-year outcomes of the randomised, non-inferiority, phase 3 CHHiP trial. Lancet Oncol 2016;17:1047-60.
- Widmark A, Gunnlaugsson A, Beckman L, Thellenberg-Karlsson C, Hoyer M, Lagerlund M, et al. Ultra-hypofractionated vs. conventionally fractionated radiotherapy for prostate cancer: 5-year outcomes of the HYPO-RT-PC randomised, non-inferiority, phase 3 trial. Lancet 2019;394:385-95.
- Marzi S, Saracino B, Petrongari MG, Arcangeli S, Gomellini S, Arcangeli G, et al. Modeling of alpha/beta for late rectal toxicity from a randomized phase II study: conventional vs. hypofractionated scheme for localized prostate cancer. J Exp Clin Cancer Res 2009;28.
- Wilt TJ, Brawer MK, Jones KM, Barry MJ, Aronson WJ, Fox S, et al. Prostate Cancer Intervention vs. Observation Trial (PIVOT) Study Group . Radical prostatectomy vs. observation for localized prostate cancer. N Engl J Med 2012;367:203-13.
- Bill-Axelson A, Holmberg L, Ruutu M, Garmo H, Stark JR, Busch C, et al. SPCG-4 Investigators . Radical prostatectomy vs. watchful waiting in early prostate cancer. N Engl J Med 2011;364:1708-17.
- Hegde JV, Demanes DJ, Veruttipong D, Raince J, Park SJ, Raman SS, et al. Pretreatment 3T multiparametric MRI staging predicts for biochemical failure in high-risk prostate cancer treated with combination high-dose-rate brachytherapy and external beam radiotherapy. Brachytherapy 2017;16:1106-12.
- Gnanapragasam VJ, Lophatananon A, Wright KA, Muir KR, Gavin A, Greenberg DC. Improving clinical risk stratification at diagnosis in primary prostate cancer: a prognostic modelling study. PLOS Med 2016;13.
- James ND, Spears MR, Clarke NW, Dearnaley DP, De Bono JS, Gale J, et al. Survival with newly diagnosed metastatic prostate cancer in the ‘docetaxel era’: data from 917 patients in the control arm of the STAMPEDE trial (MRC PR08, CRUK/06/019). Eur Urol 2015;67:1028-38.
- Bryant RJ, Oxley J, Young GJ, Lane JA, Metcalfe C, Davis M, et al. PROTecT Study Group . The PROTecT trial: analysis of the patient cohort, baseline risk stratification and disease progression. BJU Int 2020;125:506-14.
- Yang Y, Abel L, Buchanan J, Fanshawe T, Shinkins B. Use of decision modelling in economic evaluations of diagnostic tests: an appraisal and review of Health Technology Assessments in the UK. PharmacoEcon Open 2019;3:281-91.
- Souto-Ribeiro I, Woods L, Maund E, Scott DA, Lord J, Picot J, et al. Transperineal Biopsy in People with Suspected Prostate Cancer: A Systematic Review and Economic Evaluation. London: NICE; 2022.
- Pahwa S, Schiltz NK, Ponsky LE, Lu Z, Griswold MA, Gulani V. Cost-effectiveness of MR imaging-guided strategies for detection of prostate cancer in biopsy-naïve men. Radiology 2017;285:157-66.
- Hayes JH, Ollendorf DA, Pearson SD, Barry MJ, Kantoff PW, Lee PA, et al. Observation vs. initial treatment for men with localized, low-risk prostate cancer: a cost-effectiveness analysis. Ann Intern Med 2013;158:853-60.
- Cheng LJ, Soon SS, Tan TW, Tan CH, Lim TSK, Tay KJ, et al. Cost-effectiveness of MRI targeted biopsy strategies for diagnosing prostate cancer in Singapore. BMC Health Serv Res 2021;21.
- Hao S, Karlsson A, Heintz E, Elfstrom KM, Nordstrom T, Clements M. Cost-effectiveness of magnetic resonance imaging in prostate cancer screening: a microsimulation study. Value Health 2021;24:1763-72.
- Wilson ECF, Wreford A, Tamer P, Leonard K, Brechka H, Gnanapragasam VJ. Economic evaluation of transperineal vs. transrectal devices for local anaesthetic prostate biopsies. PharmacoEcon 2021;5:737-53.
- Getaneh AM, Heijnsdijk EA, de Koning HJ. Cost-effectiveness of multiparametric magnetic resonance imaging and MRI-guided biopsy in a population-based prostate cancer screening setting using a micro-simulation model. Cancer Medicine 2021;10:4046-53.
- National Institute for Health and Care Excellence . NG131 Health Economic Model Report: Prostate Cancer Update 2019.
- Barnett CL, Davenport MS, Montgomery JS, Wei JT, Montie JE, Denton BT. Cost-effectiveness of magnetic resonance imaging and targeted fusion biopsy for early detection of prostate cancer. BJU Int 2018;122:50-8.
- Faria R, Soares MO, Spackman E, Ahmed HU, Brown LC, Kaplan R, et al. Optimising the diagnosis of prostate cancer in the era of multiparametric magnetic resonance imaging: a cost-effectiveness analysis based on the prostate MR imaging study (PROMIS). Eur Urol 2018;73:23-30.
- Brown LC, Ahmed HU, Faria R, El-Shater Bosaily A, Gabe R, Kaplan RS, et al. Multiparametric MRI to improve detection of prostate cancer compared with transrectal ultrasound-guided prostate biopsy alone: the PROMIS study. Health Technol Assess 2018;22:1-176.
- Patel S, Rongen JJ, Futterer JJ, Boltyenkov A, Rovers MM. The role of multiparametric magnetic resonance imaging in active surveillance for men with low-risk prostate cancer: a cost-effectiveness modeling study. Eur Urol Oncol 2018;1:476-83.
- Sathianathen NJ, Kuntz KM, Alarid-Escudero F, Lawrentschuk NL, Bolton DM, Murphy DG, et al. Incorporating biomarkers into the primary prostate biopsy setting: a cost-effectiveness analysis. J Urol 2018;200:1215-20.
- Pahwa S, Schiltz NK, Ponsky LE, Lu Z, Griswold MA, Gulani V. MR imaging-guided strategies for detection of prostate cancer in biopsy-naïve men: response. Radiology 2017;285:157-66.
- Venderink W, Govers TM, de Rooij M, Futterer JJ, Sedelaar JPM. Cost-effectiveness comparison of imaging-guided prostate biopsy techniques: systematic transrectal ultrasound, direct in-bore MRI, and image fusion. AJR: Am J Roentgenol 2017;208:1058-63.
- Cerantola Y, Dragomir A, Tanguay S, Bladou F, Aprikian A, Kassouf W. Cost-effectiveness of multiparametric magnetic resonance imaging and targeted biopsy in diagnosing prostate cancer. Urol Oncol 2016;34:119.e1-9.
- de Rooij M, Crienen S, Witjes JA, Barentsz JO, Rovers MM, Grutters JP. Cost-effectiveness of magnetic resonance (MR) imaging and MR-guided targeted biopsy vs. systematic transrectal ultrasound-guided biopsy in diagnosing prostate cancer: a modelling study from a health care perspective. Eur Urol 2014;66:430-6.
- Mowatt G, Scotland G, Boachie C, Cruickshank M, Ford JA, Fraser C, et al. The diagnostic accuracy and cost-effectiveness of magnetic resonance spectroscopy and enhanced magnetic resonance imaging techniques in aiding the localisation of prostate abnormalities for biopsy: a systematic review and economic evaluation. Health Technol Assess 2013;17:1-281.
- Royal College of Surgeons of England . NPCA Annual Report 2020 2021.
- Bill-Axelson A, Holmberg L, Garmo H, Rider JR, Taari K, Busch C, et al. Radical prostatectomy or watchful waiting in early prostate cancer. N Engl J Med 2014;370:932-42.
- Torvinen S, Färkkilä N, Sintonen H, Saarto T, Roine RP, Taari K. Health-related quality of life in prostate cancer. Acta Oncol 2013;52:1094-101.
- Ara R, Brazier JE. Populating an economic model with health state utility values: moving toward better practice. Value Health 2010;13:509-18.
- Donovan JL, Hamdy FC, Lane JA, Mason M, Metcalfe C, Walsh E, et al. PROTecT Study Group . Patient-reported outcomes after monitoring, surgery, or radiotherapy for prostate cancer. N Engl J Med 2016;375:1425-37.
- Round J, Jones L, Morris S. Estimating the cost of caring for people with cancer at the end of life: a modelling study. Palliat Med 2015;29:899-907.
- Lunn DJ, Thomas A, Best N, Spiegelhalter D. WinBUGS: a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000;10:325-37.
- Mortezavi A, Marzendorfer O, Donati OF, Rizzi G, Rupp NJ, Wettstein MS, et al. Diagnostic accuracy of multiparametric magnetic resonance imaging and fusion guided targeted biopsy evaluated by transperineal template saturation prostate biopsy for the detection and characterization of prostate cancer. J Urol 2018;200:309-18.
- Zhou Y, Zhou Z, Li Q, Xu Y, Sun H, Xiao Y, et al. Diagnostic accuracy of magnetic resonance-guided prostate biopsy and template-guided transperineal saturation biopsy. Medicine 2018;97.
- Clarke NW, Ali A, Ingleby FC, Hoyle A, Amos CL, Attard G, et al. Addition of docetaxel to hormonal therapy in low- and high-burden metastatic hormone sensitive prostate cancer: long-term survival results from the STAMPEDE trial. Ann Oncol 2019;30:1992-2003.
- Office for National Statistics . National Life Tables: UK 2021. www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/datasets/nationallifetablesunitedkingdomreferencetables (accessed 29 September 2022).
- Ankit Rohatgi . WebPlotDigitizer 2022. https://automeris.io/WebPlotDigitizer/index.html (accessed 27 October 2022).
- Guyot P, Ades AE, Ouwens MJNM, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol 2012;12.
- Jackson C. flexsurv: a platform for parametric survival modeling in R. J Stat Softw 2016;70:1-33.
- Brent R. Algorithms for Minimization Without Derivatives 2022.
- Wallis CJ. ESMO 2021: Final Overall Survival Analysis from ARCHES: A Phase 3, Randomized, Double-blind, Placebo-Controlled Study of Enzalutamide + ADT in Men with mHSPC. UroToday.com; 2021.
- Chi KN, Chowdhury S, Bjartell A, Chung BH, Pereira de Santana Gomes AJ, Given R, et al. Apalutamide in patients with metastatic castration-sensitive prostate cancer: final survival analysis of the randomized, double-blind, phase III TITAN study. J Clin Oncol 2021;39:2294-303.
- Parry MG, Cowling TE, Sujenthiran A, Nossiter J, Berry B, Cathcart P, et al. Risk stratification for prostate cancer management: value of the Cambridge Prognostic Group classification for assessing treatment allocation. BMC Med 2020;18.
- Souto-Ribeiro I, Woods L, Maund E, Scott DA, Lord J, Picot J, et al. Transperineal Biopsy in People with Suspected Prostate Cancer: A Systematic Review and Economic Evaluation. Addendum 1. London: NICE; 2022.
- Armstrong AJ, Iguchi T, Azad AA, Szmulewitz RZ, Holzbeierlein J, Villers A, et al. LBA25 final overall survival (OS) analysis from ARCHES: a phase III, randomized, double-blind, placebo (PBO)-controlled study of enzalutamide (ENZA) + androgen deprivation therapy (ADT) in men with metastatic hormone-sensitive prostate cancer (mHSPC). Ann Oncol 2021;32:S1300-1.
- Jones K, Burns A. Unit Costs of Health and Social Care 2021. Canterbury: Personal Social Services Research Unit, University of Kent; 2021.
- Imperial College London . Imperial Prostate 7: Prostate Assessment Using Comparative Interventions – Fast MRI and Image-Fusion for Cancer (IP7-PACIFIC). ClinicalTrials.Gov 2022. https://clinicaltrials.gov/ct2/show/NCT05574647 (accessed 30 October 2022).
- Kruschke JK. Doing Bayesian Data Analysis: A Tutorial with R and BUGS. London: Academic Press; 2010.
- Briggs AH, Ades AE, Price MJ. Probabilistic sensitivity analysis for decision trees with multiple branches: use of the Dirichlet distribution in a Bayesian framework. Med Decis Making 2003;23:341-50.
- Cash HH, Shore SL, Pavlovich N, Bulang CP, Schostak S, Planken M. E Prostate cancer detection by novice micro-ultrasound users enrolled in a training program. Soc Int Urol J 2022;3:62-8.
- Exterkate L, Wegelin O, Barentsz JO, van der Leest MG, Kummer JA, Vreuls W, et al. Is there still a need for repeated systematic biopsies in patients with previous negative biopsies in the era of magnetic resonance imaging-targeted biopsies of the prostate?. Eur Urol Oncol 2020;3:216-23.
- Kalavagunta C, Zhou X, Schmechel SC, Metzger GJ. Registration of in vivo prostate MRI and pseudo-whole mount histology using Local Affine Transformations guided by Internal Structures (LATIS). J Magn Reson Imaging 2015;41:1104-14.
- Wegelin O, Exterkate L, van der Leest M, Kelder JC, Ruud Bosch JLH, Barentsz JO, et al. Complications and adverse events of three magnetic resonance imaging-based target biopsy techniques in the diagnosis of prostate cancer among men with prior negative biopsies: results from the FUTURE trial, a multicentre randomised controlled trial. Eur Urol Oncol 2019;2:617-24.
- Mannaerts CK, Engelbrecht MRW, Postema AW, van Kollenburg RAA, Hoeks CMA, Savci-Heijink CD, et al. Detection of clinically significant prostate cancer in biopsy-naïve men: direct comparison of systematic biopsy, multiparametric MRI- and contrast-ultrasound-dispersion imaging-targeted biopsy. BJU Int 2020;126:481-93.
- Simmons LAM, Kanthabalan A, Arya M, Briggs T, Barratt D, Charman SC, et al. Accuracy of transperineal targeted prostate biopsies, visual estimation and image fusion in men needing repeat biopsy in the PICTURE trial. J Urol 2018;200:1227-34.
- Hansen N, Patruno G, Wadhwa K, Gaziev G, Miano R, Barrett T, et al. Magnetic resonance and ultrasound image fusion supported transperineal prostate biopsy using the Ginsburg protocol: technique, learning points, and biopsy results. Eur Urol 2016;70:332-40.
- Kesch C, Radtke JP, Popeneciu IV, Gasch C, Dieffenbacher SC, Klein T, et al. TOP: Prospective evaluation of a volume based, computer assisted method for transperineal optimized prostate biopsy. Urol Int 2017;99:149-55.
- Hansen NL, Caglic I, Berman LH, Kastner C, Doble A, Barrett T. Multiparametric prostate magnetic resonance imaging and cognitively targeted transperineal biopsy in patients with previous abdominoperineal resection and suspicion of prostate cancer. Urology 2016;96:8-14.
- Oh J, Tyldesley S, Pai HH, McKenzie MR, Halperin RM, Duncan GG, et al. An updated analysis of survival endpoints for ASCENDE-RT, a randomized trial comparing a low-dose-rate brachytherapy boost to a dose-escalated external beam boost for high- and intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys 2020;108.
- Hedley M. Multi-parametric magnetic resonance imaging before prostate biopsy. Cancer Nurs Pract 2017;16:21-5.
- de Vries KC, Wortel RC, Oomen-de Hoop E, Heemsbergen WD, Pos FJ, Incrocci L. Hyprofractionated vs. conventionally fractionated radiation therapy for patients with intermediate- or high-risk, localized, prostate cancer: 7-year outcomes from the randomized, multicenter, open-label, phase 3 HYPRO trial. Int J Radiat Oncol Biol Phys 2020;106:108-15.
- David PD, Clare G, Isabel S, Vincent K, Alison Jane B, Ananya C, et al. Eight-year outcomes of a phase III randomized trial of conventional vs. hypofractionated high-dose intensity modulated radiotherapy for prostate cancer (CRUK/06/016): update from the CHHiP Trial. J Clin Oncol 2020;38.
- Fransson P, Nilsson P, Gunnlaugsson A, Beckman L, Tavelin B, Norman D, et al. Ultra-hypofractionated vs. conventionally fractionated radiotherapy for prostate cancer (HYPO-RT-PC): patient-reported quality-of-life outcomes of a randomised, controlled, non-inferiority, phase 3 trial. Lancet Oncol 2021;22:235-45.
- Wilt TJ, Vo TN, Langsetmo L, Dahm P, Wheeler T, Aronson WJ, et al. Radical prostatectomy or observation for clinically localized prostate cancer: extended follow-up of the prostate cancer intervention vs. observation trial (PIVOT). Eur Urol 2020;77:713-24.
- Bill-Axelson A, Holmberg L, Garmo H, Taari K, Busch C, Nordling S, et al. Radical prostatectomy or watchful waiting in prostate cancer: 29-year follow-up. N Engl J Med 2018;379:2319-29.
- Fizazi K, Carmel A, Joly F, Delva R, Gravis G, Rolland F, et al. Updated results of GETUG-12, a phase III trial of docetaxel-based chemotherapy in high-risk localized prostate cancer, with a 12-year follow-up. Ann Oncol 2018;29.
- Rosario DJ, Lane JA, Metcalfe C, Donovan JL, Doble A, Goodwin L, et al. Short term outcomes of prostate biopsy in men tested for cancer by prostate specific antigen: prospective evaluation within Protect study. BMJ 2012;344. https://doi.org/10.1136/bmj.d7894.
- Pepe P, Aragona F. Morbidity after transperineal prostate biopsy in 3000 patients undergoing 12 vs 18 vs more than 24 needle cores. Urology 2013;81:1142-6.
- Tamhankar AS, El-Taji O, Vasdev N, Foley C, Popert R, Adshead J. P9-7 The clinical and financial implications of a decade of prostate biopsies in the NHS: interrogation of the Hospital Episode Statistics (HES) Data 2008-2019 [BAUS abstracts]. J Clin Urol 2020;13.
- NHS England . 2018/19/National/Cost/Collection/Data/Publication 2021. www.england.nhs.uk/publication/2018-19-national-cost-collection-data-publication/ (accessed 27 September 2022).
- NHS England . 2019/20/National/Cost/Collection/Data/Publication 2021. www.england.nhs.uk/publication/2019-20-national-cost-collection-data-publication/ (accessed 27 September 2022).
- NHS England . 2020/2021/National/Cost/Collection/Data 2022. www.england.nhs.uk/costing-in-the-nhs/national-cost-collection/#ncc1819 (accessed 27 September 2022).
- NPCA Organisational Audit 2021 . Table of NHS Provider-Level Results: National Prostate Cancer Audit 2022. www.npca.org.uk/reports/npca-organisational-audit-2021/ (accessed 29 September 2022).
- National Institute for Health and Care Excellence . NICE Impact Prostate Cancer 2020.
- Exchange Rates UK . US Dollar (USD) to British Pound (GBP) Exchange Rate History: Wednesday 9 03 2022 to Saturday 3 09 2022 2022. www.exchangerates.org.uk/USD-GBP-exchange-rate-history.html (accessed 5 September 2022).
- Drugs and Pharmaceutical Electronic Market Information Tool (eMIT) 2021. www.gov.uk/government/publications/drugs-and-pharmaceutical-electronic-market-information-emit (accessed 29 September 2022).
- British National Formulary (BNF) . BMJ Group and the Royal Pharmaceutical Society of Great Britain 2022. https://bnf.nice.org.uk/ (accessed 27 September 2022).
Appendix 1 Software fusion technologies’ principal features
| Software system | Manufacturer | Hardware system | Fixation for biopsies | Elastic or rigid estimation | Was there a submission for the DAR? |
|---|---|---|---|---|---|
| ARTEMIS | InnoMedicus ARTEMIS | ARTEMIS | Stabilised, freehand unknown, semi-robotic arm | Both | No |
| BioJet | Healthcare Supply Solutions Ltd | Third-party ultrasounds | Stabilised, freehand (without tracking movement) | Both | No |
| BiopSee | Medcom | MedSta or third-party ultrasounds | Stabilised, freehand | Both | Yes |
| bkFusion | BK Medical UK Ltd and MIM Software Inc. | BK3000 or BK5000 | Stabilised, freehand | Rigid | Yes |
| Fusion Bx 2.0 | Focal Healthcare | Third-party ultrasounds | Stabilised, freehand, robotic arm | Both | Yes |
| FusionVu | Exact Imaging | ExactVu | Stabilised, freehand | Rigid | Yes |
| iSR’obot™ MonaLisa | Biobot iSR’obot | iSR’obot™ Mona Lisa | Stabilised, freehand unknown, robotic arm | Elastic | No |
| KOELIS Trinity | KOELIS and Kebomed | TRINITY ultrasound system | Stabilised, freehand | Elastic | Yes |
| UroNav Fusion Biopsy System | Phillips | Third- party ultrasounds | Stabilised, freehand | Unknown | No |
ARTEMIS (InnoMedicus ARTEMIS)
The ARTEMIS fusion biopsy system comprises a semi-robotic mechanical arm and a mobile workstation. The system includes the ProFuse radiology software for preparation of MRI data for fusion and reporting findings on the ARTEMIS biopsy system. The system allows both elastic and rigid estimation to account for prostate deformation, and supports both transrectal and transperineal biopsies. The mechanical arm is used to track the prostate in real time and guide the biopsy needle.
At the time of writing the EAG report, the company had not registered with NICE, and therefore did not provide information on this technology’s compatibility with a picture archiving and communication system (PACS), image measurement capabilities and ability to produce archivable cartograms.
BioJet (Healthcare Supply Solutions Ltd)
The BioJet MR Fusion system comprises MRI fusion software, a mobile workstation and is compatible with third-party ultrasounds. The system uses elastic estimations and is compatible with both transrectal and transperineal biopsies and supports both stabilised and freehand biopsy approaches.
The software enables image measurements and generates reports displaying the location of sampled areas. BioJet can be connected to a local PACS.
BiopSee (Medcom)
The BiopSee consists of the BiopSee software and the MedSta cart (workstation) and is compatible with third-party ultrasounds. The system supports both elastic and rigid estimation to account for prostate deformation, and allows both transrectal and transperineal biopsies. The system can be used for stabilised and freehand biopsy approaches. A stabilising arm is available for transperineal stabilised biopsies. Patient movement is tracked through the stepper during stabilised biopsies, or through a magnetic tracker, which is attached to the probe during freehand biopsies. The system can automatically adjust for patient movement, or the user can manually adjust the contours when a patient moves.
The BiopSee records all positions of the needle and shows the coverage of the prostate. Image measurements such as prostate and lesion volumes are also possible. The data are stored locally and can be connected to a PACS for import and export of images.
bkFusion (BK Medical UK Ltd and MIM Software Inc.)
BK Medical UK Ltd offers three versions of bkFusion software: one for transrectal, one for freehand transperineal and one for stabilised transperineal biopsies. The software can be integrated into either the bk3000 or bk5000 ultrasounds. The bkFusion system uses rigid estimation to account for prostate deformation. Predictive Fusion software re-orientates the MRI image before the biopsy. The transrectal and freehand transperineal fusion systems comprise a magnetic field generator and sensor to track the probe position.
Image measurements such as prostate volume are possible. A detailed report of the biopsy can be saved locally, or transferred to a PACS.
Fusion Bx 2.0 (Focal Healthcare)
The Fusion Bx 2.0 is a biopsy device that includes a counter balanced, semi-robotic arm that is mounted to a mobile cart. The Fusion Bx 2.0 comprises Fusion MR software which is compatible with third-party ultrasounds. The system uses both elastic and rigid estimation to account for prostate deformation, and supports both transrectal and transperineal biopsies. Patient movements are tracked with sensors inside the semi-robotic arm.
The software allows image measurements such as prostate volume and distances can be calculated. Data on the biopsied samples and the regions of interest are recorded on a 3D image of the prostate. The system can connect to PACS using a wired Ethernet or Wi-Fi connection.
FusionVu (Exact Imaging)
The ExactVu device includes a micro-ultrasound (high-resolution ultrasound at > 20MHz) and a FusionVu feature that enables SF biopsy. A stabiliser arm or stepper is available for stabilised biopsies, and freehand biopsies are also possible. The system uses rigid estimation followed by real-time visualisation of the lesions using micro-ultrasound, and supports both transperineal and transrectal biopsies. The system tracks and adjusts for patient movement using data from a movement sensor together with the live ultrasound images.
The software provides image measurements such as prostate volume and lesion size. Information on the orientation of all images and video frames are recorded so that the same position can be found if a repeat biopsy is performed. The system is PACS compatible, but a separate software (Weasis DICOM viewer) is available in the case that a PACS is not available.
iSR’obot Mona Lisa (Biobot iSR’obot)
The iSR’obot Mona Lisa is a robotic transperineal prostate biopsy system with MRI-ultrasound fusion capability. The system uses UroFusion software to highlight regions of interest on MR images and fuses the MRI model with the ultrasound model. The robotic needle guide allows automated positioning and depth control of the biopsy needle to the targeted biopsy core. The system uses elastic estimation to account for prostate deformation.
Reports are generated with 3D-images and co-ordinates are recorded of each biopsy sample. At the time of writing the EAG report, the company had not registered with NICE, and therefore did not provide information on the tracking of patient movement, whether freehand biopsies can be done, PACS compatibility and image measurement capabilities of this system.
KOELIS Trinity (KOELIS and Kebomed)
The KOELIS Trinity is a mobile ultrasound system with mapping fusion software, which comprises PROMAP 3D-Prostate Suite software and the TRINITY ultrasound system (workstation, RECFIRE ultrasound probes, guides specific to transperineal or transrectal biopsies and a Steady Pro probe holder). The system uses elastic estimation to account for prostate deformation, and supports both transrectal and transperineal biopsies. It enables both stabilised and freehand probe biopsies. The Organ-Based Tracking Fusion software identifies and compensates for patient movements and prostate deformations to record each core location.
The PROMAP software produces a 3D map of the prostate recording the position of MRI lesion targets and location of biopsy samples. The KOELIS Trinity provides image measurements such as prostate volume, exact measurements of the regions of interest and other quantitative measurements of the image. Data can be transferred to a PACS.
UroNav Fusion Biopsy System (Phillips)
The UroNav Fusion Biopsy System includes an electromagnetic tracking system, a mobile workstation and DynaCAD Prostate fusion software. The system is compatible with third-party ultrasounds. It supports both transperineal and transrectal biopsies, with stabilised or freehand approaches. UroNav uses both rigid and elastic registration methods to create and maintain 3D registration of MR/US images and compensate for patient movement. The system can be used with the UroNav mobile stepper system and the two navigation sensors to track patient movement.
The UroNav Fusion Biopsy system provides the core location data, images and videos. At the time of writing the EAG report, the company had not registered with NICE, and therefore did not submit any information on image estimation methods for prostate deformation, patient movement tracking feasibility for freehand biopsies, PACS compatibility and image measurement capabilities of this system.
Appendix 2 Literature search strategies
Database search strategies
MEDLINE ALL
(includes: Epub Ahead of Print, In-process and Other Non-Indexed Citations, Ovid MEDLINE Daily and Ovid MEDLINE)
Via Ovid http://ovidsp.ovid.com/
Date range: 1946 to 13 May, 2022
Date searched: 16 May 2022
Records retrieved: 3129
MEDLINE ALL was searched again on 2 August 2022. 3218 studies were retrieved.
-
exp Prostatic Neoplasms/ (142378)
-
Prostatic Intraepithelial Neoplasia/ (1399)
-
((prostate$ or prostatic or intraprostatic) adj4 (cancer$ or neoplas$ or tumour$ or tumor$ or malignan$ or metasta$ or carcinoma$ or adenocarcinoma$ or lesion$ or nodul$ or sarcoma$ or lymphoma$)).ti,ab. (165600)
-
(PCa or sPCa or csPCa or PrCa).ti,ab. (52571)
-
(((atypical adj3 proliferation) or ASAP) and prostat$).mp. (292).
-
or/1-5 (224791)
-
Magnetic Resonance Imaging/ (453356)
-
Multiparametric Magnetic Resonance Imaging/ (961)
-
(magnetic resonance or MRI or MR imag$ or MR scan$).ti,ab. (560471)
-
(mpMRI or mp-MRI or mpMR imag$ or mpMR scan$ or mp-MR imag$ or mp-MR scan$ or bpMRI or bp-MRI or bpMR imag$ or bpMR scan$ or bp-MR imag$ or bp-MR scan$).ti,ab. (2060)
-
or/7-10 (721668)
-
Image Interpretation, Computer-Assisted/ (47627)
-
(fusion$ or fuse$ or fusing$).ti,ab. (299284)
-
cognitive$.ti,ab. (424900)
-
(visual$ adj3 (estimat$ or direct$ or align$ or guid$ or influenc$)).ti,ab. (28436)
-
registration$.ti,ab. (161125)
-
(elastic or rigid or nonrigid).ti,ab. (138219)
-
Software/ (120348)
-
(software or hardware).ti,ab. (224399)
-
or/12-19 (1355053)
-
Prostate/ (39209)
-
(prostate$ or prostatic).ti,ab. (234214)
-
21 or 22 (238231)
-
Biopsy/ (185156)
-
Image-Guided Biopsy/ (5020)
-
Endoscopic Ultrasound-Guided Fine Needle Aspiration/ (3254)
-
Biopsy, Fine-Needle/ (14970)
-
Biopsy, Large-Core Needle/ (2307)
-
Biopsy, Needle/ (49647)
-
(biopsy or biopsie$ or rebiopsy or rebiopsie$).ti,ab. (427177)
-
or/24-30 (548867)
-
23 and 31 (26179)
-
6 and 11 and 20 and 32 (1621)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (biopsy or biopsie$)).ti,ab. (860)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 prebiops$)).ti,ab. (160)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (ultrasound$ or ultrasonic$ or ultrasonograph$ or TRUS or transperineal$ or transrectal$)).ti,ab. (773)
-
or/34-36 (1626)
-
6 and 37 (662)
-
(target$ adj4 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).ti,ab. (3800)
-
(focal adj2 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).ti,ab. (636)
-
39 or 40 (4405)
-
6 and 41 (1842)
-
(target$ adj4 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).ti,ab. (4003)
-
(focal adj2 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).ti,ab. (546)
-
43 or 44 (4534)
-
6 and 32 and 45 (1125)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj3 (guid$ or influenc$ or direct$ or align$)).ti,ab. (11942)
-
6 and 32 and 47 (951)
-
((MRI stratified or magnetic resonance imaging stratified) adj3 pathway$)).ti,ab. (3)
-
33 or 38 or 42 or 46 or 48 or 49 (3265)
-
(MRGB or MR-GB or MRIGB or MRI-GB).ti,ab. (75)
-
(MRIFB or MRI-FB).ti,ab. (3)
-
(MRFTB or MRF-TB).ti,ab. (9)
-
(MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx).ti,ab. (96)
-
FBx.ti,ab. (94)
-
(FUSTB or FUS-TB or TB-FUS).ti,ab. (9)
-
Fusion TB.ti,ab. (21)
-
(MRI-TRUS or MRI-TRUSB or MRI-TPB).ti,ab. (189)
-
(COG-TB or TB-COG or CBx).ti,ab. (530)
-
TRUS-TB.ti,ab. (3)
-
(‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’).ti,ab. (306)
-
or/51-61 (1105)
-
6 and 62 (437)
-
50 or 63 (3292)
-
(fusion$ adj3 (software or hardware or computer$ or device$ or system$ or technolog$ or machine$ or platform$)).ti,ab. (5680)
-
6 and 65 (294)
-
64 or 66 (3331)
-
KOELIS.ti,ab. (23)
-
Fusion Bx.ti,ab. (1)
-
BioJet.ti,ab. (28)
-
(Trinity or PROMAP).ti,ab. (1329)
-
Fusion MR.ti,ab. (8)
-
(bkFusion or bk Fusion or BK3000 or BK 3000 or BK5000 or BK 5000 or Predictive Fusion).ti,ab. (7)
-
or/70-73 (1371)
-
6 and 74 (20)
-
68 or 69 or 75 (38)
-
BiopSee .ti,ab. (6)
-
UroNav.ti,ab. (17)
-
(‘iSR’obot’ or iSRobot or iSR obot or UroFusion or UroBiopsy).ti,ab. (2)
-
(FusionVu$ or ExactVu$).ti,ab. (12)
-
DynaCAD.ti,ab. (9)
-
(ARTEMIS or ProFuse).ti,ab. (4760)
-
Mona Lisa.ti,ab. (106)
-
or/81-83 (4874)
-
6 and 84 (54)
-
or/77-80 (34)
-
85 or 86 (81)
-
67 or 76 or 87 (3362)
-
exp animals/not humans.sh. (5007245)
-
88 not 89 (3357)
-
limit 90 to yr=‘2008 -Current’ (3129)
Key
-
/ = subject heading (MeSH heading)
-
sh = subject heading (MeSH heading)
-
exp = exploded subject heading (MeSH heading)
-
$ = truncation
-
ti,ab = terms in title or abstract fields
-
mp = multi-purpose field search – terms in title, original title, abstract, name of substance word, or subject heading word
-
adj3 = terms within three words of each other (any order)
Cochrane Controlled Register of Trials (CENTRAL)
Via Wiley http://onlinelibrary.wiley.com/
Issue: Issue 4 of 12, April 2022
Date searched: 16 May 2022
Records retrieved: 425
CENTRAL was searched again on 2 August 2022. 434 studies were retrieved.
-
MeSH descriptor: [Prostatic Neoplasms] explode all trees 6115
-
MeSH descriptor: [Prostatic Intraepithelial Neoplasia] this term only 47
-
((prostate* or prostatic or intraprostatic) near/4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*)):ti,ab,kw 15719
-
(PCa or sPCa or csPCa or PrCa):ti,ab,kw 5554
-
((atypical near/3 proliferation) or ASAP) and prostat*)):ti,ab,kw 21
-
#1 or #2 or #3 or #4 or #5 20099
-
MeSH descriptor: [Magnetic Resonance Imaging] this term only 7831
-
MeSH descriptor: [Multiparametric Magnetic Resonance Imaging] this term only 11
-
(‘magnetic resonance’ or MRI or (MR next imag*) or (MR next scan*)):ti,ab,kw 41256
-
(mpMRI or mp-MRI or (mpMR next imag*) or (mpMR next scan*) or (mp-MR next imag*) or mp-MR scan* or bpMRI or bp-MRI or (bpMR next imag*) or (bpMR next scan*) or bp-MR imag* or bp-MR scan*):ti,ab,kw 260
-
#7 or #8 or #9 or #10 41264
-
MeSH descriptor: [Image Interpretation, Computer-Assisted] this term only 875
-
(fusion* or fuse* or fusing*):ti,ab,kw 8635
-
cognitive*:ti,ab,kw 80126
-
(visual* near/3 (estimat* or direct* or align* or guid* or influenc*)):ti,ab,kw 2089
-
registration*:ti,ab,kw 66768
-
(elastic or rigid or nonrigid):ti,ab,kw 6102
-
MeSH descriptor: [Software] this term only 1008
-
(software or hardware):ti,ab,kw 26282
-
#12 or #13 or #14 or #15 or #16 or #17 or #18 or #19 180581
-
MeSH descriptor: [Prostate] this term only 975
-
(prostate* or prostatic):ti,ab,kw 23298
-
#21 or #22 23298
-
MeSH descriptor: [Biopsy] this term only 3365
-
MeSH descriptor: [Image-Guided Biopsy] this term only 119
-
MeSH descriptor: [Endoscopic Ultrasound-Guided Fine Needle Aspiration] this term only 156
-
MeSH descriptor: [Biopsy, Needle] explode all trees 1270
-
(biopsy or biopsie* or rebiopsy or rebiopsie*):ti,ab,kw 32970
-
#24 or #25 or #26 or #27 or #28 33007
-
#23 and #29 2832
-
#6 and #11 and #20 and #30 211
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) near/6 (prior or previous* or preced* or before* or earlier or first or initial*) near/6 (biopsy or biopsie*)):ti,ab,kw 95
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) near/6 prebiops*):ti,ab,kw 34
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) near/6 (prior or previous* or preced* or before* or earlier or first or initial*) near/6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*)):ti,ab,kw 84
-
(target* near/4 (biopsy or biopsie* or rebiopsy or rebiopsie*)):ti,ab,kw 573
-
(focal near/2 (biopsy or biopsie* or rebiopsy or rebiopsie*)):ti,ab,kw 22
-
#32 or #33 or #34 or #35 or #36 715
-
#6 and #37 324
-
(target* near/4 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI)):ti,ab,kw 453
-
(focal near/2 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI)):ti,ab,kw 38
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) near/3 (guid* or influenc* or direct* or align*)):ti,ab,kw 923
-
#39 or #40 or #41 1299
-
#6 and #30 and #42 279
-
((‘MRI stratified’ or ‘magnetic resonance imaging stratified’) near/3 pathway*):ti,ab,kw 0
-
#31 or #38 or #43 or #44 430
-
(MRGB or MR-GB or MRIGB or MRI-GB or MRIFB or MRI-FB or MRFTB or MRF-TB or MRFTB or MRF-TB or MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx or FBx or FUSTB or FUS-TB or TB-FUS or ‘Fusion TB’ or MRI-TRUS or MRI-TRUSB or MRI-TPB or COG-TB or TB-COG or CBx or TRUS-TB or ‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’):ti,ab,kw 136
-
#6 and #46 82
-
#45 or #47 431
-
(fusion* near/3 (software or hardware or computer* or device* or system* or technolog* or machine* or platform*)):ti,ab,kw 267
-
#6 and #49 57
-
#48 or #50 434
-
(KOELIS or ‘Fusion Bx’ or BioJet):ti,ab,kw 18
-
(Trinity or PROMAP or ‘Fusion MR’ or bkFusion or ‘bk Fusion’ or BK3000 or ‘BK 3000’ or BK5000 or ‘BK 5000’ or ‘Predictive Fusion’):ti,ab,kw 161
-
#6 and #53 3
-
#52 or #54 19
-
(BiopSee or UroNav or ‘iSR’obot’ or iSRobot or ‘iSR obot’ or UroFusion or UroBiopsy or FusionVu* or ExactVu*):ti,ab,kw 19
-
(DynaCAD or ARTEMIS or ProFuse or ‘Mona Lisa’):ti,ab,kw 283
-
#6 and #57 9
-
#56 or #58 27
-
#51 or #55 or #59 with Publication Year from 2008 to 2022, in Trials 425
-
#51 or #55 or #59 in Cochrane Reviews, Cochrane Protocols 1
Key
-
MeSH descriptor = subject heading (MeSH heading)
-
* = truncation
-
ti,ab,kw = terms in title, abstract or keyword fields
-
near/3 = terms within three words of each other (any order)
-
next = terms are next to each other
Cochrane Database of Systematic Reviews (CDSR)
Via Wiley http://onlinelibrary.wiley.com/
Issue: Issue 5 of 12, May 2022
Date searched: 16 May 2022
Records retrieved: 1
See above under CENTRAL for search strategy.
Cumulative Index to Nursing and Allied Health (CINAHL Plus)
Via Ebsco http://onlinelibrary.wiley.com/
Date range: Inception to 20220516
Date searched: 16 May 2022
Records retrieved: 916
-
(MH ‘Prostatic Neoplasms+’) 34,206
-
TI ((prostate* or prostatic or intraprostatic) N4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*)) OR AB ((prostate* or prostatic or intraprostatic) N4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*)) 35,654
-
TI ((PCa or sPCa or csPCa or PrCa)) OR AB ((PCa or sPCa or csPCa or PrCa)) 7320
-
TI (((atypical N3 proliferation) or ASAP) and prostat*) OR AB (((atypical N3 proliferation) or ASAP) and prostat*) 29
-
S1 OR S2 OR S3 OR S4 48,489
-
(MH ‘Magnetic Resonance Imaging’) 136,332
-
TI ((‘magnetic resonance’ or MRI or (MR N1 imag*) or (MR N1 scan*))) OR AB ((‘magnetic resonance’ or MRI or (MR N1 imag*) or (MR N1 scan*))) 123,908
-
TI (((mpMRI or mp-MRI or (mpMR N1 imag*) or (mpMR N1 scan*) or (mp-MR N1 imag*) or (mp-MR N1 scan*) or bpMRI or bp-MRI or (bpMR N1 imag*) or (bpMR N1 scan*) or (bp-MR N1 imag*) or (bp-MR N1 scan*))) OR AB (((mpMRI or mp-MRI or (mpMR N1 imag*) or (mpMR N1 scan*) or (mp-MR N1 imag*) or (mp-MR N1 scan*) or bpMRI or bp-MRI or (bpMR N1 imag*) or (bpMR N1 scan*) or (bp-MR N1 imag*) or (bp-MR N1 scan*))) 631
-
S6 OR S7 OR S8 181,020
-
(MH ‘Image Interpretation, Computer Assisted’) 9454
-
TI (fusion* or fuse* or fusing*) OR AB (fusion* or fuse* or fusing*) 26,160
-
TI cognitive* OR AB cognitive* 154,740
-
TI (visual* N3 (estimat* or direct* or align* or guid* or influenc*)) OR AB (visual* N3 (estimat* or direct* or align* or guid* or influenc*)) 4578
-
TI registration* OR AB registration* 64,987
-
TI (elastic or rigid or nonrigid) OR AB (elastic or rigid or nonrigid) 12,473
-
(MH ‘Software’) 31,273
-
TI (software or hardware) OR AB (software or hardware) 59,300
-
S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S16 OR S17 341,260
-
(MH ‘Prostate’) 3816
-
TI (prostate* or prostatic) OR AB (prostate* or prostatic) 45,719
-
S19 OR S20 46,101
-
(MH ‘Biopsy’) 35,975
-
(MH ‘Biopsy, Needle’) 11,989
-
TI (biopsy or biopsie* or rebiopsy or rebiopsie*) OR AB (biopsy or biopsie* or rebiopsy or rebiopsie*) 59,743
-
S22 OR S23 OR S24 84,744
-
S21 AND S25 4603
-
S5 AND S9 AND S18 AND S26 463
-
TI ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N6 (prior or previous* or preced* or before* or earlier or first or initial*) N6 (biopsy or biopsie*)) OR AB ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N6 (prior or previous* or preced* or before* or earlier or first or initial*) N6 (biopsy or biopsie*)) 254
-
TI ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N6 prebiops*) OR AB ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N6 prebiops*) 45
-
TI ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N6 (prior or previous* or preced* or before* or earlier or first or initial*) N6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*)) OR AB ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N6 (prior or previous* or preced* or before* or earlier or first or initial*) N6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*)) 289
-
TI (target* N4 (biopsy or biopsie* or rebiopsy or rebiopsie*)) OR AB (target* N4 (biopsy or biopsie* or rebiopsy or rebiopsie*)) 961
-
TI (focal N2 (biopsy or biopsie* or rebiopsy or rebiopsie*)) OR AB (focal N2 (biopsy or biopsie* or rebiopsy or rebiopsie*)) 136
-
S28 OR S29 OR S30 OR S31 OR S32 1512
-
S5 AND S33 591
-
TI (target* N4 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)) OR AB (target* N4 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)) 880
-
TI (focal N2 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)) OR AB (focal N2 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)) 257
-
TI ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N3 (guid* or influenc* or direct* or align*)) OR AB ((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) N3 (guid* or influenc* or direct* or align*)) 3420
-
S35 OR S36 OR S37 4296
-
S5 AND S26 AND S38 533
-
TI ((‘MRI stratified’ or ‘magnetic resonance imaging stratified’) N3 pathway*) OR AB ((‘MRI stratified’ or ‘magnetic resonance imaging stratified’) N3 pathway*) 2
-
S27 OR S34 OR S39 OR S40 909
-
TI (MRGB or MR-GB or MRIGB or MRI-GB or MRIFB or MRI-FB or MRFTB or MRF-TB or MRFTB or MRF-TB or MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx or FBx or FUSTB or FUS-TB or TB-FUS or ‘Fusion TB’ or MRI-TRUS or MRI-TRUSB or MRI-TPB or COG-TB or TB-COG or CBx or TRUS-TB or ‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’) OR AB (MRGB or MR-GB or MRIGB or MRI-GB or MRIFB or MRI-FB or MRFTB or MRF-TB or MRFTB or MRF-TB or MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx or FBx or FUSTB or FUS-TB or TB-FUS or ‘Fusion TB’ or MRI-TRUS or MRI-TRUSB or MRI-TPB or COG-TB or TB-COG or CBx or TRUS-TB or ‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’) 185
-
S5 AND S42 126
-
S41 OR S43 915
-
TI (fusion* N3 (software or hardware or computer* or device* or system* or technolog* or machine* or platform*)) OR AB (fusion* N3 (software or hardware or computer* or device* or system* or technolog* or machine* or platform*)) 832
-
S5 AND S45 86
-
S44 OR S46 922
-
TI (KOELIS or ‘Fusion Bx’ or BioJet) OR AB (KOELIS or ‘Fusion Bx’ or BioJet) 17
-
TI (Trinity or PROMAP or ‘Fusion MR’ or bkFusion or ‘bk Fusion’ or BK3000 or ‘BK 3000’ or BK5000 or ‘BK 5000’ or ‘Predictive Fusion’) OR AB (Trinity or PROMAP or ‘Fusion MR’ or bkFusion or ‘bk Fusion’ or BK3000 or ‘BK 3000’ or BK5000 or ‘BK 5000’ or ‘Predictive Fusion’) 482
-
S5 AND S49 2
-
S48 OR S50 18
-
TI (BiopSee or UroNav or ‘iSR’obot’ or iSRobot or ‘iSR obot’ or UroFusion or UroBiopsy or FusionVu* or ExactVu*) OR AB (BiopSee or UroNav or ‘iSR’obot’ or iSRobot or ‘iSR obot’ or UroFusion or UroBiopsy or FusionVu* or ExactVu*) 11
-
TI (DynaCAD or ARTEMIS or ProFuse or ‘Mona Lisa’) AND AB (DynaCAD or ARTEMIS or ProFuse or ‘Mona Lisa’) 32
-
S5 AND S53 0
-
S52 OR S54 11
-
S47 OR S51 OR S55 925
-
S47 OR S51 OR S55 Limiters – Published Date: 20080101-20221231 916
Key
-
MH = CINAHL subject heading
-
+ = exploded CINAHL subject heading
-
* = truncation
-
TI = terms in the title
-
AB = terms in the abstract
-
N3 = terms within three words of each other (any order)
Database of Abstracts of Reviews of Effects (DARE)
Via http://onlinelibrary.wiley.com/
Date range: Inception – 31 March 2015
Date searched: 16 May 2022
Records retrieved: 7
-
MeSH DESCRIPTOR Prostatic Neoplasms EXPLODE ALL TREES 709
-
MeSH DESCRIPTOR Prostatic Intraepithelial Neoplasia 2
-
((prostate* or prostatic or intraprostatic) NEAR4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*)) 891
-
(PCa or sPCa or csPCa or PrCa) 44
-
((atypical NEAR3 proliferation) or ASAP) AND (prostat*) 1
-
#1 OR #2 OR #3 OR #4 OR #5 935
-
MeSH DESCRIPTOR Magnetic Resonance Imaging 693
-
MeSH DESCRIPTOR Multiparametric Magnetic Resonance Imaging 0
-
(‘magnetic resonance’ or MRI or MR imag* or MR scan*) 1337
-
(mpMRI or mp-MRI or mpMR imag* or mpMR scan* or mp-MR imag* or mp-MR scan* or bpMRI or bp-MRI or bpMR imag* or bpMR scan* or bp-MR imag* or bp-MR scan*) 2
-
#7 OR #8 OR #9 OR #10 1337
-
MeSH DESCRIPTOR Image Interpretation, Computer-Assisted 27
-
(fusion* or fuse* or fusing* or cognitive or registration* or elastic or rigid or nonrigid) 3376
-
(visual* NEAR3 (estimat* or direct* or align* or guid* or influenc*)) 23
-
MeSH DESCRIPTOR Software 76
-
(software or hardware) 812
-
#12 OR #13 OR #14 OR #15 OR #16 4163
-
MeSH DESCRIPTOR Prostate 82
-
(prostate* or prostatic) 1283
-
#18 OR #19 1283
-
MeSH DESCRIPTOR Biopsy 248
-
MeSH DESCRIPTOR Image-Guided Biopsy 11
-
MeSH DESCRIPTOR Endoscopic Ultrasound-Guided Fine Needle Aspiration 19
-
MeSH DESCRIPTOR Biopsy, Fine-Needle 83
-
MeSH DESCRIPTOR Biopsy, Large-Core Needle 8
-
MeSH DESCRIPTOR Biopsy, Needle 164
-
(biopsy or biopsie* or rebiopsy or rebiopsie*) 1457
-
#21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27 1473
-
#20 AND #28 137
-
#6 AND #11 AND #17 AND #29 4
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR6 (biopsy or biopsie*)) 39
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR6 prebiops*) 0
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*)) 121
-
(target* NEAR4 (biopsy or biopsie* or rebiopsy or rebiopsie*)) 10
-
(focal NEAR2 (biopsy or biopsie* or rebiopsy or rebiopsie*)) 0
-
#31 OR #32 OR #33 OR #34 OR #35 155
-
#6 AND #36 10
-
(target* NEAR4 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI)) 3
-
(focal* NEAR2 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI)) 1
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR3 (guid* or influenc* or direct* or align*)) 65
-
#38 OR #39 OR #40 67
-
#6 AND #29 AND #41 5
-
((‘MRI stratified’ or ‘magnetic resonance imaging stratified’) NEAR3 pathway*) 0
-
(MRGB or MR-GB or MRIGB or MRI-GB or MRIFB or MRI-FB or MRFTB or MRF-TB or MRFTB or MRF-TB or MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx or FBx or FUSTB or FUS-TB or TB-FUS or ‘Fusion TB’ or MRI-TRUS or MRI-TRUSB or MRI-TPB or COG-TB or TB-COG or CBx or TRUS-TB or ‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’) 1
-
(fusion* NEAR3 (software or hardware or computer* or device* or system* or technolog* or machine* or platform*)) 34
-
#44 OR #45 35
-
#6 AND #46 2
-
#30 OR #37 OR #42 OR #43 OR #47 11
-
(Trinity or PROMAP or ‘Fusion MR’ or bkFusion or ‘bk Fusion’ or BK3000 or ‘BK 3000’ or BK5000 or ‘BK 5000’ or ‘Predictive Fusion’) 2
-
(DynaCAD or ARTEMIS or ProFuse or ‘Mona Lisa’) 8
-
#49 OR #50 10
-
#6 AND #51 0
-
(KOELIS or ‘Fusion Bx’ or BioJet or BiopSee or UroNav or ‘iSR’obot’ or iSRobot or ‘iSR obot’ or UroFusion or UroBiopsy or FusionVu* or ExactVu*) 0
-
#48 OR #52 OR #53 11
Key
-
MeSH DESCRIPTOR = subject heading (MeSH heading)
-
* = truncation
-
NEAR3 = terms within three words of each other (order specified)
EconLit
Via Ovid http://ovidsp.ovid.com/
Date range: 1886 to 5 May, 2022
Date searched: 16 May 2022
Records retrieved: 0
-
((prostate$ or prostatic or intraprostatic) adj4 (cancer$ or neoplas$ or tumour$ or tumor$ or malignan$ or metasta$ or carcinoma$ or adenocarcinoma$ or lesion$ or nodul$ or sarcoma$ or lymphoma$)).mp. (114)
-
(PCa or sPCa or csPCa or PrCa).mp. (541)
-
(((atypical adj3 proliferation) or ASAP) and prostat$).mp. (0)
-
or/1-3 (651)
-
(magnetic resonance or MRI or MR imag$ or MR scan$).mp. (188)
-
(mpMRI or mp-MRI or mpMR imag$ or mpMR scan$ or mp-MR imag$ or mp-MR scan$ or bpMRI or bp-MRI or bpMR imag$ or bpMR scan$ or bp-MR imag$ or bp-MR scan$).mp. (0)
-
5 or 6 (188)
-
(fusion$ or fuse$ or fusing$).mp. (643)
-
cognitive$.mp. (17030)
-
(visual$ adj3 (estimat$ or direct$ or align$ or guid$ or influenc$)).mp. (75)
-
registration$.mp. (1925)
-
(elastic or rigid or nonrigid).mp. (4352)
-
(software or hardware).mp. (15832)
-
or/8-13 (39541)
-
(prostate$ or prostatic).mp. (141)
-
(biopsy or biopsie$ or rebiopsy or rebiopsie$).mp. (17)
-
15 and 16 (4)
-
4 and 7 and 14 and 17 (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (biopsy or biopsie$)).mp. (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 prebiops$).mp. (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (ultrasound$ or ultrasonic$ or ultrasonograph$ or TRUS or transperineal$ or transrectal$)).mp. (0)
-
(target$ adj4 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).mp. (0)
-
(focal adj2 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).mp. (0)
-
(target$ adj4 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).mp. (2)
-
4 and 24 (0)
-
(focal adj2 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).mp. (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj3 (guid$ or influenc$ or direct$ or align$)).mp. (9)
-
4 and 27 (0)
-
((MRI stratified or magnetic resonance imaging stratified) adj3 pathway$).mp. (0)
-
(MRGB or MR-GB or MRIGB or MRI-GB).mp. (0)
-
(MRIFB or MRI-FB).mp. (0)
-
(MRFTB or MRF-TB).mp. (0)
-
(MRFTB or MRF-TB).mp. (0)
-
(MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx).mp. (0)
-
FBx.mp. (0)
-
(FUSTB or FUS-TB or TB-FUS).mp. (0)
-
Fusion TB.mp. (0)
-
(MRI-TRUS or MRI-TRUSB or MRI-TPB).mp. (0)
-
(COG-TB or TB-COG or CBx).mp. (1)
-
4 and 39 (0)
-
TRUS-TB.mp. (0)
-
(‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’).mp. (0)
-
(fusion$ adj3 (software or hardware or computer$ or device$ or system$ or technolog$ or machine$ or platform$)).mp. (26)
-
4 and 43 (0)
-
(KOELIS or Fusion Bx).mp. (0)
-
(BioJet or Trinity or PROMAP or Fusion MR or bkFusion or bk Fusion or BK3000 or BK 3000 or BK5000 or BK 5000 or Predictive Fusion).mp. (356)
-
4 and 46 (0)
-
(BiopSee or UroNav or ‘iSR’obot’ or iSRobot or iSR obot or UroFusion or UroBiopsy or FusionVu$ or ExactVu$).mp. (0)
-
(DynaCAD or ARTEMIS or ProFuse or Mona Lisa).mp. (24)
-
4 and 49 (0)
-
18 or 19 or 20 or 21 or 22 or 23 or 25 or 26 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 40 or 41 or 42 or 44 or 45 or 47 or 48 or 50 (0)
Key
-
$ = truncation
-
mp = multi-purpose field search – terms in title, original title, abstract, name of substance word, or subject heading word
-
adj3 = terms within three words of each other (any order)
EMBASE
Via Ovid http://onlinelibrary.wiley.com/
Date range: 1974 to 13 May 2022
Date searched: 16 May 2022
Records retrieved: 6221
Embase was searched again on 2 August 2022. After conference abstracts were removed, 3318 studies were retrieved.
-
exp prostate tumor/ (271321)
-
((prostate$ or prostatic or intraprostatic) adj4 (cancer$ or neoplas$ or tumour$ or tumor$ or malignan$ or metasta$ or carcinoma$ or adenocarcinoma$ or lesion$ or nodul$ or sarcoma$ or lymphoma$)).ti,ab. (244110)
-
(PCa or sPCa or csPCa or PrCa).ti,ab. (77312)
-
(((atypical adj3 proliferation) or ASAP) and prostat$).mp. (644)
-
or/1-4 (351675)
-
nuclear magnetic resonance imaging/ (903701)
-
multiparametric magnetic resonance imaging/ (6477)
-
(magnetic resonance or MRI or MR imag$ or MR scan$).ti,ab. (819806)
-
(mpMRI or mp-MRI or mpMR imag$ or mpMR scan$ or mp-MR imag$ or mp-MR scan$ or bpMRI or bp-MRI or bpMR imag$ or bpMR scan$ or bp-MR imag$ or bp-MR scan$).ti,ab. (4373)
-
or/6-9 (1163227)
-
computer assisted diagnosis/ (41296)
-
(fusion$ or fuse$ or fusing$).ti,ab. (361890)
-
cognitive$.ti,ab. (585952)
-
(visual$ adj3 (estimat$ or direct$ or align$ or guid$ or influenc$)).ti,ab. (35918)
-
registration$.ti,ab. (163670)
-
(elastic or rigid or nonrigid).ti,ab. (152621)
-
software/ or imaging software/ or nuclear magnetic resonance scanner software/ or ultrasound imaging system software/ (139562)
-
(software or hardware).ti,ab. (363110)
-
or/11-18 (1695591)
-
exp prostate/ (54557)
-
(prostate$ or prostatic).ti,ab. (336120)
-
20 or 21 (339264)
-
biopsy/ (174400)
-
image guided biopsy/ (6935)
-
endoscopic ultrasound guided fine needle biopsy/ (5968)
-
exp needle biopsy/ (79356)
-
biopsy technique/ (7739)
-
tumor biopsy/ (43525)
-
(biopsy or biopsie$ or rebiopsy or rebiopsie$).ti,ab. (685526)
-
or/23-29 (782015)
-
22 and 30 (43352)
-
prostate biopsy/ or exp transperineal biopsy/ or exp transrectal biopsy/ (24654)
-
31 or 32 (48987)
-
5 and 10 and 19 and 33 (3137)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (biopsy or biopsie$)).ti,ab. (1707)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 prebiops$).ti,ab. (248)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (ultrasound$ or ultrasonic$ or ultrasonograph$ or TRUS or transperineal$ or transrectal$)).ti,ab. (1370)
-
or/35-37 (2954)
-
5 and 38 (1359)
-
(target$ adj4 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).ti,ab. (7633)
-
(focal adj2 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).ti,ab. (1195)
-
40 or 41 (8750)
-
5 and 40 (3525)
-
(target$ adj4 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).ti,ab. (6907)
-
(focal adj2 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).ti,ab. (959)
-
44 or 45 (7838)
-
5 and 31 and 46 (2297)
-
mri guided biopsy/ (246)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj3 (guid$ or influenc$ or direct$ or align$)).ti,ab. (18601)
-
48 or 49 (18743)
-
5 and 31 and 50 (1937)
-
((MRI stratified or magnetic resonance imaging stratified) adj3 pathway$).ti,ab. (3)
-
magnetic resonance imaging ultrasound fusion biopsy/ (128)
-
image guided noninferiority targeted biopsy/ (1)
-
cognitive biopsy/ (4)
-
software based targeted biopsy/ (1)
-
visually directed targeted biopsy/ (1)
-
ultrasound fusion targeted biopsy/ (3)
-
or/52-58 (140)
-
34 or 39 or 43 or 47 or 51 or 59 (6166)
-
(MRGB or MR-GB or MRIGB or MRI-GB).ti,ab. (132)
-
(MRIFB or MRI-FB).ti,ab. (8)
-
(MRFTB or MRF-TB).ti,ab. (36)
-
(MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx).ti,ab. (168)
-
FBx.ti,ab. (226)
-
(FUSTB or FUS-TB or TB-FUS).ti,ab. (11)
-
Fusion TB.ti,ab. (29)
-
(MRI-TRUS or MRI-TRUSB or MRI-TPB).ti,ab. (485)
-
(COG-TB or TB-COG or CBx).ti,ab. (829)
-
TRUS-TB.ti,ab. (8)
-
(‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’).ti,ab. (777)
-
or/61-71 (2124)
-
5 and 72 (1009)
-
60 or 73 (6215)
-
(fusion$ adj3 (software or hardware or computer$ or device$ or system$ or technolog$ or machine$ or platform$)).ti,ab. (7446)
-
5 and 75 (707)
-
magnetic resonance imaging-ultrasound fusion-guided prostate biopsy device/ (245)
-
74 or 76 or 77 (6346)
-
KOELIS.ti,ab,dv. (180)
-
Fusion Bx.ti,ab,dv. (16)
-
BioJet.ti,ab,dv. (105)
-
(Trinity or PROMAP).ti,ab,dv. (2121)
-
Fusion MR.ti,ab,dv. (13)
-
(bkFusion or bk Fusion or BK3000 or BK 3000 or BK5000 or BK 5000 or Predictive Fusion).ti,ab,dv. (60)
-
or/81-84 (2295)
-
5 and 85 (148)
-
79 or 80 or 86 (307)
-
BiopSee .ti,ab,dv. (52)
-
UroNav.ti,ab,dv. (163)
-
(‘iSR’obot’ or iSRobot or iSR obot or UroFusion or UroBiopsy).ti,ab,dv. (31)
-
(FusionVu$ or ExactVu$).ti,ab,dv. (84)
-
DynaCAD.ti,ab,dv. (73)
-
(ARTEMIS or ProFuse).ti,ab,dv. (6586)
-
Mona Lisa.ti,ab,dv. (162)
-
or/92-94 (6817)
-
5 and 95 (247)
-
88 or 89 or 90 or 91 or 96 (506)
-
78 or 87 or 97 (6483)
-
(animal/ or animal experiment/ or animal model/ or animal tissue/ or nonhuman/) not exp human/ (6457016)
-
98 not 99 (6455)
-
limit 100 to yr=‘2008 -Current’ (6221)
Key
-
/ = subject heading (Emtree heading)
-
exp = exploded subject heading (Emtree heading)
-
$ = truncation
-
ti,ab = terms in title or abstract fields
-
mp = multi-purpose field search – terms in title, original title, abstract, name of substance word, or subject heading word
-
dv = terms in the device trade name field
-
adj3 = terms within three words of each other (any order)
Health Management and Information Consortium (HMIC)
Via Ovid http://onlinelibrary.wiley.com/
Date range: 1979 to March 2022
Date searched: 16 May 2022
Records retrieved: 0
-
((prostate$ or prostatic or intraprostatic) adj4 (cancer$ or neoplas$ or tumour$ or tumor$ or malignan$ or metasta$ or carcinoma$ or adenocarcinoma$ or lesion$ or nodul$ or sarcoma$ or lymphoma$)).mp. (736)
-
(PCa or sPCa or csPCa or PrCa).mp. (74)
-
(((atypical adj3 proliferation) or ASAP) and prostat$).mp. (0)
-
or/1-3 (792)
-
(magnetic resonance or MRI or MR imag$ or MR scan$).mp. (483)
-
(mpMRI or mp-MRI or mpMR imag$ or mpMR scan$ or mp-MR imag$ or mp-MR scan$ or bpMRI or bp-MRI or bpMR imag$ or bpMR scan$ or bp-MR imag$ or bp-MR scan$).mp. (0)
-
5 or 6 (483)
-
(fusion$ or fuse$ or fusing$).mp. (94)
-
cognitive$.mp. (2602)
-
(visual$ adj3 (estimat$ or direct$ or align$ or guid$ or influenc$)).mp. (23)
-
registration$.mp. (4038)
-
(elastic or rigid or nonrigid).mp. (258)
-
(software or hardware).mp. (1828)
-
or/8-13 (8757)
-
(prostate$ or prostatic).mp. (914)
-
(biopsy or biopsie$ or rebiopsy or rebiopsie$).mp. (303)
-
15 and 16 (36)
-
4 and 7 and 14 and 17 (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (biopsy or biopsie$)).mp. (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 prebiops$).mp. (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous$ or preced$ or before$ or earlier or first or initial$) adj6 (ultrasound$ or ultrasonic$ or ultrasonograph$ or TRUS or transperineal$ or transrectal$)).mp. (0)
-
(target$ adj4 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).mp. (0)
-
(focal adj2 (biopsy or biopsie$ or rebiopsy or rebiopsie$)).mp. (0)
-
(target$ adj4 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).mp. (1)
-
4 and 24 (0)
-
(focal adj2 (MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI)).mp. (0)
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj3 (guid$ or influenc$ or direct$ or align$)).mp. (22)
-
4 and 27 (0)
-
((MRI stratified or magnetic resonance imaging stratified) adj3 pathway$).mp. (0)
-
(MRGB or MR-GB or MRIGB or MRI-GB).mp. (0)
-
(MRIFB or MRI-FB).mp. (0)
-
(MRFTB or MRF-TB).mp. (0)
-
(MRFTB or MRF-TB).mp. (0)
-
(MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx).mp. (0)
-
FBx.mp. (0)
-
(FUSTB or FUS-TB or TB-FUS).mp. (0)
-
Fusion TB.mp. (0)
-
(MRI-TRUS or MRI-TRUSB or MRI-TPB).mp. (0)
-
(COG-TB or TB-COG or CBx).mp. (0)
-
TRUS-TB.mp. (0)
-
(‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’).mp. (0)
-
(fusion$ adj3 (software or hardware or computer$ or device$ or system$ or technolog$ or machine$ or platform$)).mp. (1)
-
4 and 42 (0)
-
(KOELIS or Fusion Bx or BioJet).mp. (0)
-
(Trinity or PROMAP or Fusion MR or bkFusion or bk Fusion or BK3000 or BK 3000 or BK5000 or BK 5000 or Predictive Fusion).mp. (12)
-
4 and 45 (0)
-
(BiopSee or UroNav or ‘iSR’obot’ or iSRobot or iSR obot or UroFusion or UroBiopsy or FusionVu$ or ExactVu$).mp. (0)
-
(DynaCAD or ARTEMIS or ProFuse or Mona Lisa).mp. (12)
-
4 and 48 (0)
-
18 or 19 or 20 or 21 or 22 or 23 or 25 or 26 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 43 or 44 or 46 or 47 or 49 (0)
Key
-
$ = truncation
-
mp = multi-purpose field search – terms in title, original title, abstract, name of substance word, or subject heading word
-
adj3 = terms within three words of each other (any order)
Health Technology Assessment (HTA) database
Via www.crd.york.ac.uk/CRDWeb/
Date range: Inception – 31 March 2018
Date searched: 16 May 2022
Records retrieved: 2
See under DARE for search strategy used.
International Health Technology Assessment (INAHTA) database
Via http://onlinelibrary.wiley.com/
Date searched: 16 May 2022
Records retrieved: 38
-
((((biopsy OR biopsie* OR rebiopsy OR rebiopsie*)[Title] OR (biopsy OR biopsie* OR rebiopsy OR rebiopsie*)[abs] OR (biopsy OR biopsie* OR rebiopsy OR rebiopsie*)[Keywords]) OR (‘Biopsy, Needle’[mh]) OR (‘Biopsy, Large-Core Needle’[mh]) OR (‘Biopsy, Fine-Needle’[mh]) OR (‘Endoscopic Ultrasound-Guided Fine Needle Aspiration’[mh]) OR (‘Image-Guided Biopsy’[mh]) OR (‘Biopsy’[mh])) AND (((prostate* OR prostatic)[Title] OR (prostate* OR prostatic)[abs] OR (prostate* OR prostatic)[Keywords]) OR (‘Prostate’[mh]))) AND (((software OR hardware)[Title] OR (software OR hardware)[abs] OR (software OR hardware)[Keywords]) OR (‘Software’[mh]) OR ((visual* AND (estimat* OR direct* OR align* OR guid* OR influenc*))[Title] OR (visual* AND (estimat* OR direct* OR align* OR guid* OR influenc*))[abs] OR (visual* AND (estimat* OR direct* OR align* OR guid* OR influenc*))[Keywords]) OR ((fusion* OR fuse* OR fusing* OR cognitive OR registration* OR elastic OR rigid OR nonrigid)[Title] OR (fusion* OR fuse* OR fusing* OR cognitive OR registration* OR elastic OR rigid OR nonrigid)[abs] OR (fusion* OR fuse* OR fusing* OR cognitive OR registration* OR elastic OR rigid OR nonrigid)[Keywords]) OR (‘1mage 1nterpretation, Computer-Assisted’[mh])) AND (((mpMRI OR mp-MRI OR mpMR imag* OR mpMR scan* OR mp-MR imag* OR mp-MR scan* OR bpMRI OR bp-MRI OR bpMR imag* OR bpMR scan* OR bp-MR imag* OR bp-MR scan*)[Title] OR (mpMRI OR mp-MRI OR mpMR imag* OR mpMR scan* OR mp-MR imag* OR mp-MR scan* OR bpMRI OR bp-MRI OR bpMR imag* OR bpMR scan* OR bp-MR imag* OR bp-MR scan*)[abs] OR (mpMRI OR mp-MRI OR mpMR imag* OR mpMR scan* OR mp-MR imag* OR mp-MR scan* OR bpMRI OR bp-MRI OR bpMR imag* OR bpMR scan* OR bp-MR imag* OR bp-MR scan*)[Keywords]) OR ((‘magnetic resonance’ OR MRI OR MR imag* OR MR scan*)[Title] OR (‘magnetic resonance’ OR MRI OR MR imag* OR MR scan*)[abs] OR (‘magnetic resonance’ OR MRI OR MR imag* OR MR scan*)[Keywords]) OR (‘Multiparametric Magnetic Resonance Imaging’[mh]) OR (‘Magnetic Resonance Imaging’[mh])) AND (((ASAP AND prostat*)[Title] OR (ASAP AND prostat*)[abs] OR (ASAP AND prostat*)[Keywords]) OR ((atypical AND proliferation AND prostat*)[Title] OR (atypical AND proliferation AND prostat*)[abs] OR (atypical AND proliferation AND prostat*)[Keywords]) OR ((PCa OR sPCa OR csPCa OR PrCa)[Title] OR (PCa OR sPCa OR csPCa OR PrCa)[abs] OR (PCa OR sPCa OR csPCa OR PrCa)[Keywords]) OR (((cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Title] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[abs] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Keywords]) AND (((prostate* OR prostatic OR intraprostatic))[Title] OR ((prostate* OR prostatic OR intraprostatic))[abs] OR ((prostate* OR prostatic OR intraprostatic))[Keywords])) OR (‘Prostatic Intraepithelial Neoplasia’[mh]) OR (‘Prostatic Neoplasms’[mhe])) 4 hits
-
((((target* OR focal) AND (biopsy OR biopsie* OR rebiopsy OR rebiopsie*))[Title] OR ((target* OR focal) AND (biopsy OR biopsie* OR rebiopsy OR rebiopsie*))[abs] OR ((target* OR focal) AND (biopsy OR biopsie* OR rebiopsy OR rebiopsie*))[Keywords]) OR (((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (ultrasound* OR ultrasonic* OR ultrasonograph* OR TRUS OR transperineal* OR transrectal*))[Title] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (ultrasound* OR ultrasonic* OR ultrasonograph* OR TRUS OR transperineal* OR transrectal*))[abs] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (ultrasound* OR ultrasonic* OR ultrasonograph* OR TRUS OR transperineal* OR transrectal*))[Keywords]) OR ((((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND prebiops*)[Title] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND prebiops*)[abs] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND prebiops*)[Keywords])) OR (((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND biops*)[Title] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND biops*)[abs] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND biops*)[Keywords])) AND (((ASAP AND prostat*)[Title] OR (ASAP AND prostat*)[abs] OR (ASAP AND prostat*)[Keywords]) OR ((atypical AND proliferation AND prostat*)[Title] OR (atypical AND proliferation AND prostat*)[abs] OR (atypical AND proliferation AND prostat*)[Keywords]) OR ((PCa OR sPCa OR csPCa OR PrCa)[Title] OR (PCa OR sPCa OR csPCa OR PrCa)[abs] OR (PCa OR sPCa OR csPCa OR PrCa)[Keywords]) OR (((cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Title] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[abs] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Keywords]) AND (((prostate* OR prostatic OR intraprostatic))[Title] OR ((prostate* OR prostatic OR intraprostatic))[abs] OR ((prostate* OR prostatic OR intraprostatic))[Keywords])) OR (‘Prostatic Intraepithelial Neoplasia’[mh]) OR (‘Prostatic Neoplasms’[mhe])) 9 hits
-
((((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (guid* OR influenc* OR direct* OR align*))[Title] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (guid* OR influenc* OR direct* OR align*))[abs] OR ((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (guid* OR influenc* OR direct* OR align*))[Keywords]) OR (((target* OR focal) AND (MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI))[Title] OR ((target* OR focal) AND (MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI))[abs] OR ((target* OR focal) AND (MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI))[Keywords])) AND ((((biopsy OR biopsie* OR rebiopsy OR rebiopsie*)[Title] OR (biopsy OR biopsie* OR rebiopsy OR rebiopsie*)[abs] OR (biopsy OR biopsie* OR rebiopsy OR rebiopsie*)[Keywords]) OR (‘Biopsy, Needle’[mh]) OR (‘Biopsy, Large-Core Needle’[mh]) OR (‘Biopsy, Fine-Needle’[mh]) OR (‘Endoscopic Ultrasound-Guided Fine Needle Aspiration’[mh]) OR (‘Image-Guided Biopsy’[mh]) OR (‘Biopsy’[mh])) AND (((prostate* OR prostatic)[Title] OR (prostate* OR prostatic)[abs] OR (prostate* OR prostatic)[Keywords]) OR (‘Prostate’[mh]))) AND (((ASAP AND prostat*)[Title] OR (ASAP AND prostat*)[abs] OR (ASAP AND prostat*)[Keywords]) OR ((atypical AND proliferation AND prostat*)[Title] OR (atypical AND proliferation AND prostat*)[abs] OR (atypical AND proliferation AND prostat*)[Keywords]) OR ((PCa OR sPCa OR csPCa OR PrCa)[Title] OR (PCa OR sPCa OR csPCa OR PrCa)[abs] OR (PCa OR sPCa OR csPCa OR PrCa)[Keywords]) OR (((cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Title] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[abs] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Keywords]) AND (((prostate* OR prostatic OR intraprostatic))[Title] OR ((prostate* OR prostatic OR intraprostatic))[abs] OR ((prostate* OR prostatic OR intraprostatic))[Keywords])) OR (‘Prostatic Intraepithelial Neoplasia’[mh]) OR (‘Prostatic Neoplasms’[mhe])) 5 hits
-
((MRGB OR MR-GB OR MRIGB OR MRI-GB OR MRIFB OR MRI-FB OR MRFTB OR MRF-TB OR MRFTB OR MRF-TB OR MRTB OR MR-TB OR MRITB OR MRI-TB OR MRTBx OR MR-TBx OR MRITBx OR MRI-TBx OR FBx OR FUSTB OR FUS-TB OR TB-FUS OR ‘Fusion TB’ OR MRI-TRUS OR MRI-TRUSB OR MRI-TPB OR COG-TB OR TB-COG OR CBx OR TRUS-TB OR ‘MRI/TRUS’ OR ‘mpMRI/TRUS’ OR ‘MR/US’ OR ‘MRI/TRUS-TB’)[Title] OR (MRGB OR MR-GB OR MRIGB OR MRI-GB OR MRIFB OR MRI-FB OR MRFTB OR MRF-TB OR MRFTB OR MRF-TB OR MRTB OR MR-TB OR MRITB OR MRI-TB OR MRTBx OR MR-TBx OR MRITBx OR MRI-TBx OR FBx OR FUSTB OR FUS-TB OR TB-FUS OR ‘Fusion TB’ OR MRI-TRUS OR MRI-TRUSB OR MRI-TPB OR COG-TB OR TB-COG OR CBx OR TRUS-TB OR ‘MRI/TRUS’ OR ‘mpMRI/TRUS’ OR ‘MR/US’ OR ‘MRI/TRUS-TB’)[abs] OR (MRGB OR MR-GB OR MRIGB OR MRI-GB OR MRIFB OR MRI-FB OR MRFTB OR MRF-TB OR MRFTB OR MRF-TB OR MRTB OR MR-TB OR MRITB OR MRI-TB OR MRTBx OR MR-TBx OR MRITBx OR MRI-TBx OR FBx OR FUSTB OR FUS-TB OR TB-FUS OR ‘Fusion TB’ OR MRI-TRUS OR MRI-TRUSB OR MRI-TPB OR COG-TB OR TB-COG OR CBx OR TRUS-TB OR ‘MRI/TRUS’ OR ‘mpMRI/TRUS’ OR ‘MR/US’ OR ‘MRI/TRUS-TB’)[Keywords]) AND (((ASAP AND prostat*)[Title] OR (ASAP AND prostat*)[abs] OR (ASAP AND prostat*)[Keywords]) OR ((atypical AND proliferation AND prostat*)[Title] OR (atypical AND proliferation AND prostat*)[abs] OR (atypical AND proliferation AND prostat*)[Keywords]) OR ((PCa OR sPCa OR csPCa OR PrCa)[Title] OR (PCa OR sPCa OR csPCa OR PrCa)[abs] OR (PCa OR sPCa OR csPCa OR PrCa)[Keywords]) OR (((cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Title] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[abs] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Keywords]) AND (((prostate* OR prostatic OR intraprostatic))[Title] OR ((prostate* OR prostatic OR intraprostatic))[abs] OR ((prostate* OR prostatic OR intraprostatic))[Keywords])) OR (‘Prostatic Intraepithelial Neoplasia’[mh]) OR (‘Prostatic Neoplasms’[mhe])) 14 hits
-
((Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’)[Title] OR (Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’)[abs] OR (Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’)[Keywords]) AND (((ASAP AND prostat*)[Title] OR (ASAP AND prostat*)[abs] OR (ASAP AND prostat*)[Keywords]) OR ((atypical AND proliferation AND prostat*)[Title] OR (atypical AND proliferation AND prostat*)[abs] OR (atypical AND proliferation AND prostat*)[Keywords]) OR ((PCa OR sPCa OR csPCa OR PrCa)[Title] OR (PCa OR sPCa OR csPCa OR PrCa)[abs] OR (PCa OR sPCa OR csPCa OR PrCa)[Keywords]) OR (((cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Title] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[abs] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Keywords]) AND (((prostate* OR prostatic OR intraprostatic))[Title] OR ((prostate* OR prostatic OR intraprostatic))[abs] OR ((prostate* OR prostatic OR intraprostatic))[Keywords])) OR (‘Prostatic Intraepithelial Neoplasia’[mh]) OR (‘Prostatic Neoplasms’[mhe]))
-
((fusion* AND (software OR hardware OR computer* OR device* OR system* OR technolog* OR machine* OR platform*))[Title] OR (fusion* AND (software OR hardware OR computer* OR device* OR system* OR technolog* OR machine* OR platform*))[abs] OR (fusion* AND (software OR hardware OR computer* OR device* OR system* OR technolog* OR machine* OR platform*))[Keywords]) AND (((ASAP AND prostat*)[Title] OR (ASAP AND prostat*)[abs] OR (ASAP AND prostat*)[Keywords]) OR ((atypical AND proliferation AND prostat*)[Title] OR (atypical AND proliferation AND prostat*)[abs] OR (atypical AND proliferation AND prostat*)[Keywords]) OR ((PCa OR sPCa OR csPCa OR PrCa)[Title] OR (PCa OR sPCa OR csPCa OR PrCa)[abs] OR (PCa OR sPCa OR csPCa OR PrCa)[Keywords]) OR (((cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Title] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[abs] OR (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)[Keywords]) AND (((prostate* OR prostatic OR intraprostatic))[Title] OR ((prostate* OR prostatic OR intraprostatic))[abs] OR ((prostate* OR prostatic OR intraprostatic))[Keywords])) OR (‘Prostatic Intraepithelial Neoplasia’[mh]) OR (‘Prostatic Neoplasms’[mhe])) 2 hits
key
-
[abs] = abstract
-
[mh] = subject heading (MeSH heading)
-
[mhe] = exploded subject heading (MeSH heading)
-
* = truncation
Latin American and Caribbean Health Sciences Literature (LILACS)
Via https://pesquisa.bvsalud.org/portal/advanced/?lang=en
Date searched: 16 May 2022
Records retrieved: 98
-
1. Search of title, abstract, subject heading fields: (prostat$) AND (cancer$ OR neoplas$ OR tumour$ OR tumor$ OR malignan$ OR metasta$ OR carcinoma$ OR adenocarcinoma$ OR lesion$ OR nodul$ OR sarcoma$ OR lymphoma$) AND (‘magnetic resonance’ OR MRI OR MR imag$ OR MR scan$ OR mpMRI OR mp-MRI OR mpMR imag$ OR mpMR scan$ OR mp-MR imag$ OR mp-MR scan$ OR bpMRI OR bp-MRI OR bpMR imag$ OR bpMR scan$ OR bp-MR imag$ OR bp-MR scan$) AND (fusion$ OR fuse$ OR fusing$ OR cognitive$ OR visual$ OR registration$ OR elastic OR rigid OR nonrigid OR software OR hardware OR target$ OR focal OR guid$ OR influenc$ OR direct$ OR align$) AND (biopsy OR biopsie$ OR rebiopsy OR rebiopsie$ OR prebiopsy$)
-
Limit: 2008–2022
-
35 hits
-
2. Search of title, abstract, subject heading fields: (prostat$) AND (cancer$ OR neoplas$ OR tumour$ OR tumor$ OR malignan$ OR metasta$ OR carcinoma$ OR adenocarcinoma$ OR lesion$ OR nodul$ OR sarcoma$ OR lymphoma$) AND (MRI OR MR OR magnetic resonance OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) AND (ultrasound$ OR ultrasonic$ OR ultrasonograph$ OR TRUS OR transperineal$ OR transrectal$)
-
Limit: 200–2022
-
53 hits
-
3. Search of title, abstract, subject heading fields: (prostat$) AND (cancer$ OR neoplas$ OR tumour$ OR tumor$ OR malignan$ OR metasta$ OR carcinoma$ OR adenocarcinoma$ OR lesion$ OR nodul$ OR sarcoma$ OR lymphoma$) AND (MRGB OR MR-GB OR MRIGB OR MRI-GB OR MRIFB OR MRI-FB OR MRFTB OR MRF-TB OR MRFTB OR MRF-TB OR MRTB OR MR-TB OR MRITB OR MRI-TB OR MRTBx OR MR-TBx OR MRITBx OR MRI-TBx OR FBx OR FUSTB OR FUS-TB OR TB-FUS OR ‘Fusion TB’ OR MRI-TRUS OR MRI-TRUSB OR MRI-TPB OR COG-TB OR TB-COG OR CBx OR TRUS-TB OR ‘MRI/TRUS’ OR ‘mpMRI/TRUS’ OR ‘MR/US’ OR ‘MRI/TRUS-TB’)
-
Limit: 2008–2022
-
9 hits
-
4. Search of title, abstract, subject heading fields: (KOELIS OR ‘Fusion Bx’ OR BioJet OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu$ OR ExactVu$)
-
Limit: 2008–2022
-
0 hits
-
5. Search of title, abstract, subject heading fields: (Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’) AND (prostat$)
-
Limit: 2008–2022
-
1 hit
NHS Economic Evaluations Database (NHS EED)
Via www.crd.york.ac.uk/CRDWeb/
Date range: Inception – 31 March 2015
Date searched: 16 May 2022
Records retrieved: 2
See under DARE for search strategy used.
Science Citation Index
Va Web of Science, Clarivate Analytics https://clarivate.com/
Date range: 1900 – present
Date searched: 16 May 2022
Records retrieved: 3616
The Science Citation Index and the Conference Proceedings Citation Index-Science were both searched using the strategy below. Numbers of records retrieved are therefore the total number from searching both databases.
The Science Citation Index only was searched again on 2 August 2022. An amount of 3561 studies were retrieved.
-
48 #45 OR #41 OR #37 3616
-
47 #45 or #41 or #37 3857
-
46 #45 OR #41 OR #37 3857
-
45 #42 OR #44 69
-
44 #43 AND #4 42
-
43 TS = (DynaCAD or ARTEMIS or ProFuse or ‘Mona Lisa’) 5737
-
42 TS = (BiopSee or UroNav or ‘iSR’obot’ or iSRobot or ‘iSR obot’ or UroFusion or UroBiopsy or FusionVu* or ExactVu*) 34
-
41 #40 OR #38 41
-
40 #39 AND #4 19
-
39 TS = (BioJet or Trinity or PROMAP or ‘Fusion MR’ or bkFusion or ‘bk Fusion’ or BK3000 or ‘BK 3000’ or BK5000 or ‘BK 5000’ or ‘Predictive Fusion’) 2748
-
38 TS = (KOELIS or ‘Fusion Bx’) 25
-
37 #36 OR #34 OR #32 3825
-
36 #35 AND #4 471
-
35 TS = (fusion* NEAR/3 (software or hardware or computer* or device* or system* or technolog* or machine* or platform*)) 24,330
-
34 #33 AND #4 451
-
33 TS = (MRGB or MR-GB or MRIGB or MRI-GB or MRIFB or MRI-FB or MRFTB or MRF-TB or MRFTB or MRF-TB or MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx or FBx or FUSTB or FUS-TB or TB-FUS or ‘Fusion TB’ or MRI-TRUS or MRI-TRUSB or MRI-TPB or COG-TB or TB-COG or CBx or TRUS-TB or ‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’) 1351
-
32 #31 OR #30 OR #25 OR #18 3620
-
31 TS=((‘MRI stratified’ or ‘magnetic resonance imaging stratified’) NEAR/3 pathway*) 3
-
30 #29 AND #17 AND #4 2,016
-
29 #26 OR #27 OR #28 22800
-
28 TS=((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR/3 (guid* or influenc* or direct* or align*)) 17,122
-
27 TS = (focal NEAR/2 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI)) 1243
-
26 TS = (target* NEAR/4 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI)) 5682
-
25 #24 AND #4 2567
-
24 #23 OR #22 OR #21 OR #20 OR #19 6484
-
23 TS = (focal NEAR/2 (biopsy or biopsie* or rebiopsy or rebiopsie*)) 666
-
22 TS = (target* NEAR/4 (biopsy or biopsie* or rebiopsy or rebiopsie*)) 4437
-
21 TS=((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR/6 (prior or previous* or preced* or before* or earlier or first or initial*) NEAR/6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*)) 858
-
20 TS=((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR/6 prebiops*) 179
-
19 TS=((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) NEAR/6 (prior or previous* or preced* or before* or earlier or first or initial*) NEAR/6 (biopsy or biopsie*)) 963
-
18 #17 AND #14 AND #7 AND #4 1832
-
17 #15 AND #16 28,427
-
16 TS = (biopsy or biopsie* or rebiopsy or rebiopsie*) 379,853
-
15 TS = (prostate* or prostatic) 336,855
-
14 #13 OR #12 OR #11 OR #10 OR #9 OR #8 2,737,360
-
13 TS = (software or hardware) 906,626
-
12 TS = (elastic or rigid or nonrigid) 624,588
-
11 TS = (registration*) 134,030
-
10 TS = (visual* NEAR/3 (estimat* or direct* or align* or guid* or influenc*)) 43,631
-
9 TS = cognitive* 514,118
-
8 TS = (fusion* or fuse* or fusing*) 589,649
-
7 #5 OR #6 757,071
-
6 TS = (mpMRI or mp-MRI or ‘mpMR imag*’ or ‘mpMR scan*’ or ‘mp-MR imag*’ or ‘mp-MR scan*’ or bpMRI or bp-MRI or ‘bpMR imag*’ or ‘bpMR scan*’ or ‘bp-MR imag*’ or ‘bp-MR scan*’) 2175
-
5 TS=(‘magnetic resonance’ or MRI or ‘MR imag*’ or ‘MR scan*’) 756,868
-
4 #1 OR #2 OR #3 332,891
-
3 TS=(((atypical NEAR/3 proliferation) or ASAP) and prostat*) 317
-
2 TS = (PCa or sPCa or csPCa or PrCa) 101,467
-
1 TS=((prostate* or prostatic or intraprostatic) NEAR/4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*)) 246,739
Key
-
TS = topic tag; searches in title, abstract, author keywords and keywords plus fields
-
* = truncation
-
NEAR/3 = terms within three words of each other (any order)
On-going, unpublished or grey literature search strategies
ClinicalTrials.gov
https://clinicaltrials.gov/ct2/
Date searched: 23 May 2022
Records retrieved: 572
Targeted search screen
-
87 Studies found for: (prostate OR prostatic OR intraprostatic) AND (neoplasm OR cancer OR tumour OR tumor OR lesion OR nodule OR adenocarcinoma) [condition] | (biopsy OR biopsied OR rebiopsy OR rebiopsied) AND (MRI OR MR OR ‘magnetic resonance’ OR biparametric OR multiparametric OR bpMRI OR mpMRI OR bp-MRI OR mp-MRI)[title]
-
238 Studies found for: (prostate OR prostatic OR intraprostatic) AND (neoplasm OR cancer OR tumour OR tumor OR lesion OR nodule OR adenocarcinoma) [condition] | (biopsy OR biopsied OR rebiopsy OR rebiopsied) AND (MRI OR MR OR ‘magnetic resonance’ OR biparametric OR multiparametric OR bpMRI OR mpMRI OR bp-MRI OR mp-MRI) [intervention]
-
53 Studies found for: (prostate OR prostatic OR intraprostatic) AND (neoplasm OR cancer OR tumour OR tumor OR lesion OR nodule OR adenocarcinoma) [condition] | (biopsy OR biopsied OR rebiopsy OR rebiopsied) AND (targeted) [title]
-
129 Studies found for: (prostate OR prostatic OR intraprostatic) AND (neoplasm OR cancer OR tumour OR tumor OR lesion OR nodule OR adenocarcinoma) [condition] | (biopsy OR biopsied OR rebiopsy OR rebiopsied) AND (targeted) [intervention]
Main search screen
-
5. 21 Studies found for: KOELIS OR ‘Fusion Bx’ OR BioJet OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu OR ExactVu [other terms]
-
6. 44 Studies found for: Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’ [other terms] | (prostate OR prostatic OR intraprostatic) [condition]
Conference proceedings citation index – Science (CPCI-Science)
Via Web of Science, Clarivate Analytics https://clarivate.com/
Date range: 1990–present (CPCI-Science)
Date searched: 16 May 2022
See above under Science Citation Index for search strategy used. The number of records retrieved from CPCI-Science is not available as both Science Citation Index and CPCI-Science were searched together retrieving 3616 records in total from both databases.
EU Clinical Trials Register
via https://www.clinicaltrialsregister.eu/ctr-search/search
Search date: 15 June 2022
Records retrieved: 86
-
68 result(s) found for: (prostate OR prostatic OR intraprostatic) AND (biopsy OR rebiopsy OR re-biopsy) AND (neoplasm OR cancer OR tumour OR tumor OR lesion OR nodule OR adenocarcinoma) AND (MRI OR MR OR ‘magnetic resonance’ OR biparametric OR multiparametric OR bpMRI OR mpMRI OR bp-MRI OR mp-MRI) date range: 2015-01-01 to 2022-06-15
-
18 result(s) found for: (prostate OR prostatic OR intraprostatic) AND (biopsy OR rebiopsy OR re-biopsy) AND (neoplasm OR cancer OR tumour OR tumor OR lesion OR nodule OR adenocarcinoma) AND targeted date range: 2015-01-01 to 2022-06-15
-
0 result(s) found for: (KOELIS OR ‘Fusion Bx’ OR BioJet OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu OR ExactVu) date range: 2015-01-01 to 2022-06-15
-
0 result(s) found for: (Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’) AND (prostate OR prostatic OR intraprostatic) date range: 2015-01-01 to 2022-06-15
Open Access Theses and Dissertations (OATD)
Date searched: 16 May 2022
Records retrieved: 74
3 search queries used:
Query 1
(Prostat* AND biops*) AND (fusion* OR cognitive* OR software) AND (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*) AND (‘magnetic resonance’ OR MRI OR biparametric OR multiparametric)
50 hits
Query 2
(cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*) AND (prostat*) AND (BioJet OR Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’)
23 hits
Query 3
KOELIS OR ‘Fusion Bx’ OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu* OR ExactVu*
1 hit
Key
-
* = truncation
ProQuest Dissertations and Theses A&I
Date searched: 16 May 2022
Records retrieved: 207
-
((TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) OR TI,AB,SU,IF(((atypical NEAR/3 proliferation) OR ASAP) AND prostat*)) AND (TI,AB,SU,IF(‘magnetic resonance’ OR MRI OR MR imag* OR MR scan*) OR TI,AB,SU,IF(mpMRI OR mp-MRI OR mpMR imag* OR mpMR scan* OR mp-MR imag* OR mp-MR scan* OR bpMRI OR bp-MRI OR bpMR imag* OR bpMR scan* OR bp-MR imag* OR bp-MR scan*)) AND (TI,AB,SU,IF(prostate* OR prostatic) AND TI,AB,SU,IF(biopsy OR biopsie* OR rebiopsy OR rebiopsie*)) AND (TI,AB,SU,IF(fusion* OR fuse* OR fusing* OR cognitive* OR registration* OR elastic OR rigid OR nonrigid OR software OR hardware) OR TI,AB,SU,IF(visual* NEAR/3 (estimat* OR direct* OR align* OR guid* OR influenc*))) limit: 2008-01-01 to 2022-05-16 33 Hits
-
(TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) AND TI,AB,SU,IF((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) NEAR/6 (biopsy OR biopsie* OR prebiops* OR ultrasound* OR ultrasonic* OR ultrasonograph* OR TRUS OR transperineal* OR transrectal*)) limit: 2008-01-01 to 2022-05-16 67 hits
-
(TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) AND (TI,AB,SU,IF((target* OR focal) NEAR/4 (biopsy OR biopsie* OR rebiopsy OR rebiopsie*)) OR TI,AB,SU,IF((target* OR focal) NEAR/4 (MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI))) limit: 2008-01-01 to 2022-05-16 53 hits
-
((TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) OR TI,AB,SU,IF(((atypical NEAR/3 proliferation) OR ASAP) AND prostat*)) AND (TI,AB,SU,IF(prostate* OR prostatic) AND TI,AB,SU,IF(biopsy OR biopsie* OR rebiopsy OR rebiopsie*)) AND TI,AB,SU,IF((MRI OR MR OR ‘magnetic resonance’ OR mpMRI OR mp-MRI OR bpMRI OR bp-MRI) NEAR/3 (guid* OR influenc* OR direct* OR align*)) limit: 2008-01-01 to 2022-05-16 20 hits
-
((TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) OR TI,AB,SU,IF(((atypical NEAR/3 proliferation) OR ASAP) AND prostat*)) AND TI,AB,SU,IF(MRGB OR MR-GB OR MRIGB OR MRI-GB OR MRIFB OR MRI-FB OR MRFTB OR MRF-TB OR MRFTB OR MRF-TB OR MRTB OR MR-TB OR MRITB OR MRI-TB OR MRTBx OR MR-TBx OR MRITBx OR MRI-TBx OR FBx OR FUSTB OR FUS-TB OR TB-FUS OR ‘Fusion TB’ OR MRI-TRUS OR MRI-TRUSB OR MRI-TPB OR COG-TB OR TB-COG OR CBx OR TRUS-TB OR ‘MRI/TRUS’ OR ‘mpMRI/TRUS’ OR ‘MR/US’ OR ‘MRI/TRUS-TB’) limit: 2008-01-01 to 2022-05-16 6 hits
-
((TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) OR TI,AB,SU,IF(((atypical NEAR/3 proliferation) OR ASAP) AND prostat*)) AND TI,AB,SU,IF(fusion* NEAR/3 (software OR hardware OR computer* OR device* OR system* OR technolog* OR machine* OR platform*)) limit: 2008-01-01 to 2022-05-16 26 hits
-
TI,AB,SU,IF(KOELIS OR ‘Fusion Bx’ OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu* OR ExactVu*) 0 hits
-
((TI,AB,SU,IF((prostate* OR prostatic OR intraprostatic) NEAR/4 (cancer* OR neoplas* OR tumour* OR tumor* OR malignan* OR metasta* OR carcinoma* OR adenocarcinoma* OR lesion* OR nodul* OR sarcoma* OR lymphoma*)) OR TI,AB,SU,IF(PCa OR sPCa OR csPCa OR PrCa)) OR TI,AB,SU,IF(((atypical NEAR/3 proliferation) OR ASAP) AND prostat*)) AND TI,AB,SU,IF(BioJet OR Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’) limit: 2008-01-01 to 2022-05-16 2 hits
Key
-
TI,AB,SU,IF = search of title, abstract, subject heading and keyword fields
-
* = truncation
-
NEAR/3 = terms within three words of each other (any order)
PROSPERO
Via https://www.crd.york.ac.uk/prospero/
Date searched: 23 May 2022
Records retrieved: 78
-
MeSH DESCRIPTOR Prostatic Intraepithelial Neoplasia 0
-
MeSH DESCRIPTOR Prostatic Neoplasms EXPLODE ALL TREES 406
-
(prostate* or prostatic or intraprostatic) adj4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*) 1351
-
((prostate* or prostatic or intraprostatic) adj4 (cancer* or neoplas* or tumour* or tumor* or malignan* or metasta* or carcinoma* or adenocarcinoma* or lesion* or nodul* or sarcoma* or lymphoma*)):TI 740
-
((prostate* or prostatic or intraprostatic)):TI 1080
-
PCa or sPCa or csPCa or PrCa 335
-
#1 OR #2 OR #4 OR #6 951
-
#1 OR #2 OR #3 OR #6 1509
-
#5 OR #2 OR #1 1092
-
MeSH DESCRIPTOR Magnetic Resonance Imaging 458
-
MeSH DESCRIPTOR Multiparametric Magnetic Resonance Imaging 6
-
‘magnetic resonance’ or MRI or MR imag* or MR scan* 5234
-
(‘magnetic resonance’ or MRI or MR imag* or MR scan*):TI 773
-
((mpMRI or mp-MRI or mpMR imag* or mpMR scan* or mp-MR imag* or mp-MR scan* or bpMRI or bp-MRI or bpMR imag* or bpMR scan* or bp-MR imag* or bp-MR scan*)):TI 8
-
(mpMRI or mp-MRI or mpMR imag* or mpMR scan* or mp-MR imag* or mp-MR scan* or bpMRI or bp-MRI or bpMR imag* or bpMR scan* or bp-MR imag* or bp-MR scan*) 59
-
#10 OR #11 OR #12 OR #15 5259
-
#10 OR #11 OR #13 OR #14 887
-
MeSH DESCRIPTOR Image Interpretation, Computer-Assisted 4
-
MeSH DESCRIPTOR Software 31
-
fusion* or fuse* or fusing* or cognitive or registration* or elastic or rigid or nonrigid 17958
-
visual* adj3 (estimat* or direct* or align* or guid* or influenc*) 274
-
software or hardware 48745
-
#18 OR #19 OR #20 OR #21 OR #22 60890
-
MeSH DESCRIPTOR Prostate 102
-
prostate* or prostatic 1862
-
(prostate* or prostatic):TI 1080
-
#24 OR #25 1881
-
#24 OR #26 1102
-
(MeSH DESCRIPTOR Biopsy):TI 0
-
(MeSH DESCRIPTOR Image-Guided Biopsy EXPLODE ALL TREES):TI 0
-
(MeSH DESCRIPTOR Biopsy, Needle EXPLODE ALL TREES):TI 0
-
MeSH DESCRIPTOR Biopsy, Needle EXPLODE ALL TREES 50
-
MeSH DESCRIPTOR Biopsy, Needle EXPLODE ALL TREES 50
-
MeSH DESCRIPTOR Biopsy 103
-
MeSH DESCRIPTOR Image-Guided Biopsy EXPLODE ALL TREES 27
-
biopsy or biopsie* or rebiopsy or rebiopsie* 2655
-
(biopsy or biopsie* or rebiopsy or rebiopsie*):TI 251
-
#32 OR #34 OR #35 OR #36 2678
-
#32 OR #34 OR #35 OR #37 295
-
#8 AND #16 AND #23 AND #27 AND #38 54
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous* or preced* or before* or earlier or first or initial*) adj6 (biopsy or biopsie*)):TI 1
-
((MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 prebiops*):TI 0
-
((MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous* or preced* or before* or earlier or first or initial*) adj6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*)):TI 0
-
(target* adj4 (biopsy or biopsie* or rebiopsy or rebiopsie*)):TI 15
-
(focal* adj2 (biopsy or biopsie* or rebiopsy or rebiopsie*)):TI 1
-
(MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous* or preced* or before* or earlier or first or initial*) adj6 (biopsy or biopsie*) 9
-
(MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 prebiops* 0
-
(MRI or MR or magnetic resonance or mpMRI or mp-MRI or bpMRI or bp-MRI) adj6 (prior or previous* or preced* or before* or earlier or first or initial*) adj6 (ultrasound* or ultrasonic* or ultrasonograph* or TRUS or transperineal* or transrectal*) 0
-
target* adj4 (biopsy or biopsie* or rebiopsy or rebiopsie*) 48
-
focal* adj2 (biopsy or biopsie* or rebiopsy or rebiopsie*) 1
-
#46 OR #47 OR #48 OR #49 OR #50 55
-
#8 AND #51 44
-
#52 OR #40 66
-
target* adj4 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) 22
-
focal* adj2 (MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) 2
-
(MRI or MR or ‘magnetic resonance’ or mpMRI or mp-MRI or bpMRI or bp-MRI) adj3 (guid* or influenc* or direct* or align*) 139
-
#54 OR #55 OR #56 154
-
#8 AND #27 AND #38 AND #57 38
-
#53 OR #58 76
-
MRGB or MR-GB or MRIGB or MRI-GB or MRIFB or MRI-FB or MRFTB or MRF-TB or MRFTB or MRF-TB or MRTB or MR-TB or MRITB or MRI-TB or MRTBx or MR-TBx or MRITBx or MRI-TBx or FBx or FUSTB or FUS-TB or TB-FUS or ‘Fusion TB’ or MRI-TRUS or MRI-TRUSB or MRI-TPB or COG-TB or TB-COG or CBx or TRUS-TB or ‘MRI/TRUS’ or ‘mpMRI/TRUS’ or ‘MR/US’ or ‘MRI/TRUS-TB’ 17
-
fusion* adj3 (software or hardware or computer* or device* or system* or technolog* or machine* or platform*) 75
-
#60 OR #61 91
-
#8 AND #62 16
-
#63 OR #59 76
-
KOELIS OR ‘Fusion Bx’ OR BioJet OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu* OR ExactVu* 7
-
Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’ 489
-
#66 AND #8 4
-
#64 OR #65 OR #67 78
Key
MeSH DESCRIPTOR = subject heading (MeSH heading)
* = truncation
adj3 = terms within 3 words of each other (order specified)
WHO International Clinical Trials Registry Platform (ICTRP)
https://trialsearch.who.int/AdvSearch.aspx
Date searched: 23 May 2022
Records retrieved: 378
Advanced search screen. Recruitment status set to ALL
-
1. Title field: (biops* OR rebiops* OR re-biops*) AND (MRI OR MR OR ‘magnetic resonance’ OR biparametric OR multiparametric OR bpMRI OR mpMRI OR bp-MRI OR mp-MRI)
-
Condition field: (prostate* OR prostatic OR intraprostatic) AND (neoplas* OR cancer* OR tumour* OR tumor* OR lesion* OR nodul* OR adenocarcinoma*)
-
117 hits
-
2. Intervention field: (biops* OR rebiops* OR re-biops*) AND (MRI OR MR OR ‘magnetic resonance’ OR biparametric OR multiparametric OR bpMRI OR mpMRI OR bp-MRI OR mp-MRI)
-
Condition field: (prostate* OR prostatic OR intraprostatic) AND (neoplas* OR cancer* OR tumour* OR tumor* OR lesion* OR nodul* OR adenocarcinoma*)
-
106 hits
-
3. Title field: (biops* OR rebiops* OR re-biops*) AND target*
-
Condition field: (prostate* OR prostatic OR intraprostatic) AND (neoplas* OR cancer* OR tumour* OR tumor* OR lesion* OR nodul* OR adenocarcinoma*)
-
68 hits
-
4. Intervention field: (biops* OR rebiops* OR re-biops*) AND target*
-
Condition field: (prostate* OR prostatic OR intraprostatic) AND (neoplas* OR cancer* OR tumour* OR tumor* OR lesion* OR nodul* OR adenocarcinoma*)
-
64 hits
-
5. Title field: (Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’)
-
Condition field: (prostate* OR prostatic OR intraprostatic) AND (neoplas* OR cancer* OR tumour* OR tumor* OR lesion* OR nodul* OR adenocarcinoma*)
-
4 hits
-
6. Intervention field: (Trinity OR PROMAP OR ‘Fusion MR’ OR bkFusion OR ‘bk Fusion’ OR BK3000 OR ‘BK 3000’ OR BK5000 OR ‘BK 5000’ OR ‘Predictive Fusion’ OR DynaCAD OR ARTEMIS OR ProFuse OR ‘Mona Lisa’)
-
Condition field: (prostate* OR prostatic OR intraprostatic) AND (neoplas* OR cancer* OR tumour* OR tumor* OR lesion* OR nodul* OR adenocarcinoma*)
-
3 hits
Basic search screen
-
7. KOELIS OR ‘Fusion Bx’ OR BioJet OR BiopSee OR UroNav OR ‘iSR’obot’ OR iSRobot OR ‘iSR obot’ OR UroFusion OR UroBiopsy OR FusionVu* OR ExactVu*
-
16 hits
Key
-
* = truncation
Guideline website searches
Simple searches were carried out on the guideline websites listed below and any results were browsed for relevance. Relevant guidelines identified were checked against the endNote library of results and added to the library if they had not already been found through previous searches.
ECRI guidelines trust
Date searched: 23 May 2022
-
prostate or prostatic – 39 results browsed – 9 relevant
GIN international guideline library
https://guidelines.ebmportal.com/
Date searched: 23 May 2022
-
prostate cancer – 36 results browsed – 8 relevant
National Institute of Health and Care Excellence (NICE)
Date searched: 23 May 2022
-
Browsed 43 items on the prostate cancer guidance page https://www.nice.org.uk/guidance/conditions-and-diseases/cancer/prostate-cancer
- 4 relevant
Trip database
Date searched: 23 May 2022
Two further guidelines found through searching the Trip database.
-
Prostate cancer AND MRI OR ‘magnetic resonance’ OR biparametric OR multiparametric – 5 guideline results – browsed for relevance – 4 relevant – all in EndNote library already.
-
Prostate cancer AND fusion OR cognitive OR software – 0 guideline results
-
Prostate cancer AND imag* – 6 guideline results – browsed for relevance – 3 relevant – all in EndNote library already.
-
Prostate cancer AND diagnos* – 10 guideline results – browsed for relevance – 8 relevant – 6 in EndNote library already.
Appendix 3 Multinomial network meta-analysis model
A multinomial logistic regression model was used where the odds of being categorised in each of the different categories in Table 2 compared to the reference category (no PCa) are allowed to vary by biopsy type. 74,75,156,157 This model is conceptually equivalent to R-1 binomial logistic regressions comparing category r > 1 with category 1 (no PCa), for each different biopsy type compared to the reference, cognitive biopsy.
Define
-
i – study index
-
k – study arm index
-
r – category index
-
R – number of categories
Data from the N studies are modelled with a multinomial likelihood with probability vector qikr
-
yik,1:R – vector of observed events in arm k of study i
-
Mik – number of patients in arm k of study i
Category probabilities for arm k of study i are defined as
Log-odds ratio for category r relative to category 1, for arm k in study i:
with air representing the baseline log-odds for being classified in category r, instead of category 1, in study i and δikr=dti1tik,r=d1tik,r−d1ti1,r representing the additional effect for being classified in category r, instead of category 1, using the intervention in arm k, compared to the intervention in arm 1.
We set
Note that
Hence
We model ϕikr, the odds ratio for category r relative to category 1, for arm k in study i as
Calculating absolute probabilities
To calculate the absolute probabilities of being classified in category r using intervention k, Tkr we note:
Using equation (2), and defining Ar as the log-odds of being classified in category r using the reference intervention, we have
and using equation (1) we have
External data inform Tk1 which are used to calculate Ar and calculate all the other probabilities
Using equations (3) and (4), we have
| Study | Biopsy type | Number of patients | Category 1, No cancer | Category 2, ISUP grade 1 | Category 3, ISUP grade 2 | Category 4, ISUP grade 3 | Category 5, ISUP grades 4–5 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | |
| PAIREDCAP (2019)88 | CF | SB | ARTEMIS | 248 | 248 | 248 | 94 | 52 | 71 | 38 | 46 | 43 | 52 | 87 | 70 | 39 | 37 | 40 | 25 | 26 | 24 |
| Izadpanahi (2021)82 | CF + SB | ARTEMIS + SB | NA | 100 | 99 | NA | 69 | 55 | NA | 19 | 25 | NA | 6 | 13 | NA | 5 | 3 | NA | 1 | 3 | NA |
| Wajswol (2020)87 | SB | UroNav | UroNav + SB | 169 | 169 | 169 | 53 | 49 | 36 | 116 | 120 | 133 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Thangarasu (2021)79 | CF | SB | CF + SB | 75 | 75 | 75 | 41 | 35 | 32 | 34 | 40 | 43 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Kulis (2020)86 | CF | SB | CF + SB | 63 | 63 | 63 | 30 | 33 | 25 | 33 | 30 | 38 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Cornud (2018)93 | CF | Urostation | NA | 88 | 88 | NA | 57 | 48 | NA | 31 | 40 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| FUTURE (2019)31 | CF | BiopSee | NA | 78 | 79 | NA | 44 | 40 | NA | 8 | 12 | NA | 26 | 27 | NA | NA | NA | NA | NA | NA | NA |
| PROFUS (2014)97 | CF | ARTEMIS | NA | 125 | 125 | NA | 85 | 80 | NA | 16 | 16 | NA | 24 | 29 | NA | NA | NA | NA | NA | NA | NA |
| Albisinni (2018)94 | SB | Urostation | Urostation + SB | 74 | 74 | 74 | 41 | 39 | 32 | 12 | 10 | 13 | 21 | 25 | 29 | NA | NA | NA | NA | NA | NA |
| Fourcade (2018)92 | SB | Urostation | Urostation + SB | 191 | 191 | 191 | 103 | 106 | 85 | 36 | 25 | 34 | 52 | 60 | 72 | NA | NA | NA | NA | NA | NA |
| Gomez-Ortiz (2022)99 | CF | SB | CF + SB | 111 | 111 | 111 | 69 | 81 | 65 | 19 | 9 | 20 | 23 | 21 | 26 | NA | NA | NA | NA | NA | NA |
| Rabah (2021)84 | ARTEMIS | BioJet | NA | 165 | 142 | NA | 117 | 78 | NA | 27 | 18 | NA | 21 | 46 | NA | NA | NA | NA | NA | NA | NA |
| Alberts (2018)80 | SB | Urostation | Urostation + SB | 48 | 48 | 48 | 23 | 20 | 16 | 11 | 11 | 13 | 10 | 13 | 13 | 4 | 4 | 6 | NA | NA | NA |
| Filson (2016)96 | SB | ARTEMIS | ARTEMIS + SB | 538 | 538 | 538 | 294 | 310 | 252 | 114 | 68 | 100 | 74 | 81 | 92 | 56 | 79 | 94 | NA | NA | NA |



Appendix 4 Characteristics of studies included in the systematic review of clinical evidence
| Study | Country | Design | N | Population investigated | MRI type | MRI magnet strength (T) | SF technology | Comparison | Biopsy route | N of cores per lesion (targeted biopsy)a | Number of ROI targeted | N of cores (SB) | Anaesthesia | Definition of CSPCa | Definition of PCa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SF vs. cognitive fusion: prospective | |||||||||||||||
| Cornud (2018)93 | France | Prospective, within-patient | 88 | BN, RB | mpMRI | 1.5 | Urostation Touch (KOELIS) | SF vs. CFb | TR | 2 | NR | NR | NR | NA | Gleason 3 + 3 |
| Delongchamps (2013)98 | France | Consecutive series, between patient | SF: 82 CF: 54 |
BN | mpMRI | 1.5 | Urostation Touch (KOELIS)c | SF vs. CF vs. SB | TR | ≥2 | NR | 10–12 | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| FUTURE (2019)31,166 | The Netherlands | RCT, between patient | SF: 79, CF: 78 | RB | mpMRI | 3 | BiopSee (MedCom) | SF vs. CF | SF: TP, CF: TR | Median (IQR). SF: 4 (3–5), CF: 3 (3–4) | All | NA | General/spinal | Gleason ≥ 3 + 4 | NR |
| Hansen (2018)95 | UK, Germany, Australia | Prospective, between patient | SF: 395 CF: 176 |
BN | mpMRI | 1.5 or 3 | BiopSee (Medcom) | SF vs. CF vs. SB | TP | At least 2 | All ROI | 18–24d | General | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Izadpanahi (2021)82 | Iran | RCT, between patient | SF: 99 CF: 100 | BN | mpMRI | 3 | ARTEMIS (InnoMedicus ARTEMIS) | SF vs. CF, ± SB | TR | SF: 1–2 CF: 1-2 |
2 | 4 | Local | Gleason ≥ 3 + 4, or 3 + 3 with ≥ 4 mm core length | GS 3 + 3 with < 4 mm core length |
| PAIREDCAP (2019)88 | USA | Prospective, within-patient | 248 | BN | mpMRI | 3 | ARTEMIS (InnoMedicus ARTEMIS) | SF vs. CF vs. SB | TR | SF: 3 CF: 3 |
1 | 12 | Local | Gleason ≥ 3 + 4 | Gleason ≥ 3 + 3 |
| PROFUS (2014)97 | USA | Prospective, within patient | 101 (BN, RB) | BN, RB, AS | mpMRI | 3 | ARTEMIS (InnoMedicus ARTEMIS) | SF vs. CF | TR | SF: 2 CF: 2 |
2 | 12d | Local | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Stabile (2018)89 | Italy | Prospective, between patient | SF: 157 CF: 87 |
BN, RB | mpMRI | 1.5 | BioJet | SF + SB vs. CF + SB | SF: TP/TR CF: TR |
Median (range) SF: 3 (2–3); CF: 2 (2–5) |
All ROI | 12 | NR | Gleason ≥ 3 + 4 | NR |
| SF vs. CF: retrospective | |||||||||||||||
| Kaufmann (2018)91,101 | Germany, Italy | Retrospective, between patient | SF: 191 CF: 87 |
BN, RB | mpMRI | 3 | iSR’obot Mona Lisa (Biobot Surgical) | SF vs. CF | SF: TP CF: TR |
4 | 1 | 14d | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Liang (2020)85 | China | Retrospective, between patient | SF: 92 CF: 71 |
BN | bpMRI | 3 | Predictive Fusion Software (BK) | SF vs. CF | TP | 4 | All ROI | NA | Local | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Lockhart (2022)100 | Australia | Retrospective, between patient | SF: 131 CF: 224 |
BN | mpMRI | 3 | MIM Fusion Software (with BK 3000 US) | SF + SB vs. CF + SB | TP | NRe | NR | NR | NR | Gleason ≥ 3 + 4 | NR |
| Monda (2018)90 | USA | Retrospective; before and after study | SF: 348 CF: 162 |
BN, RB (+ve/–ve) | mpMRI | 3 | UroNav (Invivo Corporation) | SF vs. CF vs. SB | TR | NR | NR | 12 | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Software fusion vs. software fusion | |||||||||||||||
| Ferriero (2022)81 | Italy | Prospective cohort, between patient | Urostation: 103 BioJet: 232 |
BN | mpMRI | NR | Urostation (KOELIS) BioJet (Healthcare Supply Solutions Ltd) |
SF vs. SF | Urostation: TR; BioJet: NR | Median (IQR) Unmatched Urostation: 4 (4–6) BioJet: 6 (5–6) Matched Urostation: 4 (4–6) BioJet: 6 (4–6) |
NR | NA | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Rabah (2021)84 | Saudi Arabia | RCT, between patient | Artermis: 165 BioJet: 142 |
BN, RBf | mpMRI | NR | ARTEMIS (InnoMedicus ARTEMIS) BioJet (Healthcare Supply Solutions Ltd) |
SF vs. SF vs. SB | ARTEMIS: TR BioJet: TP |
2–4 | All ROI | 12 | ARTEMIS: Local BioJet: General |
NR | NR |
| Sokolakis (2021)83 | Germany | Prospective, between patient | BioJet: 20 Urnoav: 20 KOELIS Trinity: 20 |
BN, RB | mpMRI | 3 | BioJet (Healthcare Supply), UroNav (Phillips), KOELIS Trinity | SF vs. SF | TR | 2–3 | All ROI | 12d | Local | NR | Gleason 3 + 3 |
| SF vs. systematic biopsy vs. SF and systematic biopsy | |||||||||||||||
| Alberts (2018)80 | The Netherlands | Prospective, within patient | 48g | BN, RB | mpMRI | NR | Urostation (KOELIS) | SF vs. SB vs. SF + SB | TR | 2 | All ROIs | 12 | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Albisinni (2018)94 | Belgium | Prospective, within-patient | 74 | RB | mpMRI | 3 | Urostation (KOELIS) | SF vs. SB vs. SF + SB | TR | 2–4 | 1 | 12–14 | NR | Gleason ≥ 3 + 4 and/or cancer core length ≥ 6 mm (UCL) | NR |
| Filson (2016)96 | USA | Prospective, within-patient | 538 (PI-RADS ≥ 3, excl AS) | BN, RB, AS (NR) | mpMRI | 3 | ARTEMIS (InnoMedicus ARTEMIS) | SF vs. SB vs. SF + SB | NR | 1 | NR | 12 | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Fourcade (2018)92 | France | Prospective, within-patient | 191 | BN, RB | mpMRI | 3 | Urostation (KOELIS) | SF vs. SB vs. SF + SB | TR | 2–4 | All ROI | 12 | NR | NR | NR |
| Wajswol (2020)87 | USA | Prospective, within-patient | 169 (PI-RADS ≥ 3) | BN, RB | mpMRI | 3 | UroNav (Phillips) | SF vs. SB vs. SF + SB | TP | 4–6 | All ROI | 12 | Local | Gleason ≥ 3 + 4 | NR |
| CF vs. systematic biopsy vs. CF and systematic b632iopsy | |||||||||||||||
| Gomez-Ortiz (2022)99 | Mexico | Prospective, within-patient | 111 | RB | NR | 1.5 | N/A | CB vs. SB vs. CB + SB | TR | 2–4 | All ROI | 12 | NR | Gleason ≥ 3 + 4 | Gleason 3 + 3 |
| Kulis (2020)86 | Croatia | Prospective, within-patient | 63 | RB | mpMRI | 3 | N/A | CB vs. SB vs. CB + SB | NR | 6 | Up to 2 | 12 | Localh | Gleason ≥ 3 + 4 | GS ≤ 6 |
| Thangarasu (2021)79 | India | Prospective, within-patient | 75 | BN | mpMRI | 3 | N/A | CB vs. SB vs. CB + SB | TR | 2 | All ROI | 12 | Localh | Gleason ≥ 3 + 4 | NR |
| Device | Author | N | Biopsy route | Anaesthesia | Image registration | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TR | TP | NR | Local | General | NR | Rigid | Elastic | NR | |||
| ARTEMIS (InnoMedicus) | Filson, 2016 | 538 | X | X | X | ||||||
| Izadpanahi, 2021 | 99 | X | X | X | |||||||
| PAIRED CAP, 2019 | 248 | X | X | X | |||||||
| PROFUS, 2014 | 101 | X | X | X | |||||||
| Rabah, 2021 | 165 | X | X | X | |||||||
| TOTAL | 1151 | 4 | 0 | 1 | 4 | 0 | 1 | 0 | 2 | 3 | |
| BiopSee (Medcom) | FUTURE, 2019 | 79 | X | X | X | ||||||
| Hansen, 2018 | 395 | X | X | X | |||||||
| TOTAL | 474 | 0 | 2 | 0 | 0 | 2 | 0 | 1 | 0 | 1 | |
| BK | Liang, 2020 | 92 | X | X | X | ||||||
| Lockhart, 2022 | 131 | X | X | X | |||||||
| TOTAL | 223 | 0 | 2 | 0 | 1 | 0 | 1 | 2 | 0 | 0 | |
| KOELIS (earlier versions) | Alberts, 2018 | 48 | X | X | X | ||||||
| Albisinni, 2018 | 74 | X | X | X | |||||||
| Cornud, 2018 | 88 | X | X | X | |||||||
| Delongchamps, 2013 | 82 | X | X | X | |||||||
| Ferriero, 2022 | 103 | X | X | X | |||||||
| Fourcade, 2018 | 191 | X | X | X | |||||||
| TOTAL | 586 | 5 | 0 | 1 | 0 | 0 | 6 | 0 | 6 | 0 | |
| BioJet | Ferriero, 2022 | 232 | X | X | X | ||||||
| Rabah, 2021 | 142 | X | X | X | |||||||
| Sokolakis 2021 | 20 | X | X | X | |||||||
| Stabile, 2018 | 157 | X | X | X | X | ||||||
| TOTAL | 551 | 2 | 2 | 1 | 1 | 1 | 2 | 2 | 0 | 2 | |
| iSR’obot Mona Lisa | Kaufmann, 2018 | 191 | X | X | X | ||||||
| TOTAL | 191 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | |
| UroNav | Monda, 2018 | 348 | X | X | X | ||||||
| Sokolakis, 2021 | 20 | X | X | X | |||||||
| Wajswol, 2020 | 169 | X | X | X | X | ||||||
| TOTAL | 537 | 2 | 1 | 0 | 2 | 0 | 1 | 2 | 2 | 0 | |
| KOELIS Trinity | Sokolakis, 2021 | 20 | X | X | X | ||||||
| TOTAL | 20 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | |
| Study | N | Population investigated | Recruitment criteria | Age (years) | PSA (ng/ml) | PI-RADS version | PI-RADS score | Lesion location | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 5 | ||||||||
| SF vs. CF | ||||||||||
| Cornud (2018)93 | 88 | BN, RB | PI-RADS ≥ 3a | Med (IQR) 63 (60–69) |
Med (IQR) 8.2 (6.0–10.9) |
NR | NR | NR | ||
| Delongchamps (2013)167b | mpMRI + ve SF: 82 CF: 54 |
BN | PSA ≥ 4 ng/ml, and/or suspicious DRE | Mean (SD)c SF: 64.5 (7.9) CF: 62.7 (7.4) |
Mean (SD)c SF: 8.3 (4.1) CF: 9 (3.9) |
NR | NR | NR | ||
| FUTURE (2019)31 | SF: 79 CF: 78 |
RB | Repeat SB (< 4 years), PSA ≥ 4 (ng/ml) and/or suspicious DRE | Mean (SD) SF: 64.6 (6.9) CF: 66.5 (6.3) |
Mean (SD) SF: 11.6 (9.0) CF: 11.0 (7.1) |
v2 | SF: 23 CF: 21 |
SF: 34 CF: 32 |
SF: 22 CF: 25 |
SF: 35 Post, 37 Ant CF: 46 Post, 25 Ant |
| Hansen (2018)95 | PI-RADS ≥ 3 SF: 395 CF: 176 |
BN | PSA ≤ 30 ng/mL, ≤79 years | Median (IQR)+ Centre 1 (SF): 64 (57–69) Centre 2 (SF): 65 (60–70) Centre 3 (CF): 65 (60–70) |
Median (IQR)+ Centre 1 (SF): 6.6 (4.6–9.0) Centre 2 (SF): 6.9 (5.2–9.1) Centre 3 (CF): 5.9 (4.6–8.0) |
v1-2 | Centre 1 (SF): 34, Centre 2 (SF): 91, Centre 3 (CF): 28 |
Centre 1 (SF): 99, Centre 2 (SF): 171, Centre 3 (CF): 148 | NR | |
| Izadpanahi (2019)82 | SF: 99; CF: 100 |
BN | PSA 2-10 ng/dL, PI-RADS ≥ 3 | Mean (SD) SF: 61.9 (7.4) CF: 61.9 (7.4) |
Mean (SD) SF: 6.1 (1.3) CF: 5.9 (1.3) |
v2 | NR | NR | ||
| PAIREDCAP (2019)31 | 248 | BN | Elevated PSA (serum PSA < 25 ng/mL) or abnormal DRE | Mean (SD) 65.5 (7.7) |
Med (IQR) 6.2 (4.6–8.20) |
v2 | 56 | 91 | 101 | Ant: 93 |
| PROFUS (2014)97 | 125 (101 BN, RB) | BN, RB, AS | NR | NR (range) 65 (56.3–71.0) |
NR (range) | v2 | NR | Post: 140 Ant: 32 |
||
| Stabile (2018)89 | SF: 157 CF: 87 |
BN, RB | NR | Median (IQR) SF: 67 (61–73) CF: 62 (58–70) |
Median (IQR) SF: 7.3 (5.2–10.5) CF: 6 (4–9) |
NR | SF: 59, CF: 35 |
SF: 98 CF: 52 |
NR | |
| SF vs. CF – Retrospective | ||||||||||
| Kaufman (2018)91,101 | SF: 191 CF: 87 |
BN, RB | Rising and/or persistently elevated PSA | Median (IQR): 69.0 (63.0–74.0) |
Median (IQR): 8.0 (5.87–12.0) |
v2 | NR | NR | ||
| Liang (2020)85 | SF: 92 CF: 71 |
BN | PSA level of ≤ 20 ng/mL | Mean (SD) SF: 69.17 (9.18) CF: 67.59 (8.45) |
Median (IQR) SF: 8.03 (0.66–19.78) CF: 7.66 (0.67–18.81) |
v2 | NR | NR | ||
| Lockhart (2022)100 | Total: 355 (SF: 131, CF: 224); BN only: 283 (SF: 97; CF: 186) | BN, AS | NR | Mean (range) SF: 65 (41–80) CF: 66.6 (44–85) |
Mean SF: 5.8 CF: 7.64 |
NR | NR | NR | ||
| Monda (2018)90 | SF: 348 CF: 162 |
BN, RB (+ve/–ve) | NR | Mean (SD) SF: 65.0 (7.2) CF: 63.9 (7.8) |
Mean (SD) 7.8 (7.8) 7.9 (7.8) |
v2 | NR | NR | ||
| SF vs. SF | ||||||||||
| Ferriero (2022)81 | Unmatched Urostation: 103 BioJet: 232 Matched: Urostation: 83 BioJet: 83 |
BN | PI-RADS ≥ 3 | Median (IQR) Unmatched Urostation: 67 (59, 72) BioJet: 60 (65, 75) Matched Urostation: 69 (60, 72) BioJet: 65 (61, 71) |
Median (IQR) Unmatched Urostation: 7 (4.9, 10.3) BioJet: 6.5 (5, 5.95) Matched Urostation: 7 (4.9, 10.3) BioJet: 6.6 (5, 10) |
NR | Unmatched: Urostation: 21 BioJet: 52. Matched: Urostation: 50 BioJet: 19. |
Unmatched: Urostation: 55 BioJet: 108 Matched: Urostation: 26 BioJet: 43 |
Unmatched: Urostation:27 BioJet: 51 Matched: Urostation: 15 BioJet: 21 |
NR |
| Rabah (2021)84 | Artermis: 165 BioJet: 142 |
BN, RB | PI-RADS ≥ 3, and PSA ≥ 3.5 ng/ml or abnormal DRE | Mean (SD) ARTEMIS: 65.1 (7.8) BioJet: 65 (8.5) |
Mean (SD) ARTEMIS: 14.2 (5) BioJet: 13.7 (25.9) |
v2 | ARTEMIS: 35 BioJet: 30 |
ARTEMIS: 19 BioJet: 25 |
ARTEMIS: 16 BioJet: 20 |
NR |
| Sokolakis (2021)83 | BioJet: 20 Urnoav: 20 KOELIS Trinity: 20 |
BN, RB | PI-RADS ≥ 3 | Median (IQR) BioJet: 66 (61, 67) UroNav: 64 (61, 74) Trinity: 64 (62, 67) |
Median (IQR) BioJet: 8 (6,9) UroNav: 6 (5,8) Trinity: 7 (5,8) |
v2 | BioJet: 4 UroNav: 6 Trinity: 6 |
BioJet: 12 UroNav: 7 Trinity: 9 |
BioJet: 4 UroNav: 7 Trinity: 5 |
NR |
| SF vs. systematic biopsy vs. SF and systematic biopsy | ||||||||||
| Alberts (2018)80 | 48 (who received TB and SB) | BN, RB | PI-RADS ≥ 3, and PSA ≥ 3.5 ng/ml | Median (IQR)c 73.1 (72.4–73.8) |
Median (IQR)c 4.2 (3.4–5.8) |
NR | NR | NR | ||
| Albisinni (2018)91 | 74 | RB | NR | Median (IQR) 65 (62–69) |
Median (IQR) 9.27 (6.84–13.4) |
v2 | NR | NR | ||
| Filson (2016)96 | 538 (PI-RADS ≥ 3, excl AS) | BN, RB, AS (NR) | Elevated PSA or abnormal DRE | Median (IQR) BN: 64.4 (58.5–69.4) RB: 65.7 (59.3–70.2) |
Median (IQR) BN: 5.8 (4.4–8.1) RB: 7.6 (5.0–11.5) |
v2 | BN: 129 RB:148 |
BN: 109 RB: 87 |
BN: 35 RB: 30 |
Anterior: BN: 148 RB: 100 |
| Fourcade (2018)92 | 191 | BN, RB | PSA > 4ng/mL and abnormal DRE | Median (range) 66 (47–80) |
Mean (range) 9 (0.7–48) |
v2 | NR | |||
| Wajswol (2020)87 | 169 (PI-RADS ≥ 3) | BN, RB | PI-RADS ≥ 2 (visible lesion), PSA > 2.5ng/mL | Median (range) 67.5 (44–89) |
Median (range) 8.25 (1.4–103.8) |
v2 | 26 | 76 | 67 | NR |
| CF vs. systematic biopsy vs. CF and systematic biopsy | ||||||||||
| Gomez-Ortiz (2022)99 | 111 | RB | PI-RADS ≥ 3 | Mean (SD) 66.27 (6.85) |
Median (IQR) 9.9 (1.21–26) |
2 | NR | NR | ||
| Kulis (2020)86 | 63 | RB | PI-RADS ≥ 3, PSA > 4 ng/mL | Median (range) 67 (57–84) |
Median (range) 10.70 (4.86–64.00) |
v2 | 12 | 35 | 16 | Central: 42 Peripheral: 9 Apical: 9 Anterior: 3 |
| Thangarasu (2021)79 | 75 | BN | PI-RADS ≥ 3, serum PSA > 4 and ≤ 20 ng/mL, suspected ≤ T2 stage on rectal examination | Mean (SD) 66.31 (7.9) |
Median (NR) 10.6 (4.5–20) |
v2 | 42 | 23 | 10 | NR |
Appendix 5 Quality assessment
| Study | Operator experience |
|---|---|
| Cornud (2018)93 | > 10 years in MRI and elastic SF |
| Delongchamps (2013)98 | ‘Experienced uroradiologist’ |
| FUTURE (2019)31,166 | ‘Performed by five urologists and expert-trained PhD candidates having at least 6 mo of experience, including 3 mo of experience under expert supervision’ |
| Hansen (2018)95 | SF (Centre 1): several years’ experience of TP biopsy. SF (Centre 2): Supervised Residents. CF: 1/5 urologists |
| Izadpanahi (2021)82 | ‘Experience of performing at least 2000 targeted prostate biopsies’ |
| PAIREDCAP (2019)88 | ‘Experienced’ |
| PROFUS (2014)97 | NR |
| Stabile (2018)89 | Urologists had performed at least 200 prostate biopsies but were naive for TB techniques. |
| Kaufman (2018)91,101 | NR |
| Liang (2020)85 | Experienced urologist with more than 1 year experience |
| Lockhart (2022)100 | Experienced radiologist |
| Monda (2018)90 | NR |
| Rabah (2021)84 | NR |
| Ferriero (2022)81 | 9 years experience |
| Sokolakis (2021)83 | 4 operators with no prior experience on mpMRI/TRUS fusion PB, 2 trainees who accomplished 40 TRUS-guided biopsies; and two senior urologists who had done over 250 TRUS-guided biopsies |
| Alberts (2018)80 | NR |
| Albisinni (2018)94 | Single operator who performs > 100 TBs each year with 20 + years experience |
| Filson (2016)96 | NR |
| Fourcade (2018)92 | NR |
| Wajswol (2020)87 | NR |
| Gomez-Ortiz (2022)99 | NR |
| Kulis (2020)86 | NR |
| Thangarasu (2021)79 | NR |
| Study | Tests | Reference std or tests to estimate total positive rates | Risk of bias (QUADAS-C) |
Applicability concerns (QUADAS-2) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| P | I | R | FT | Comments | P | I | R | Comments | |||
| Alberts (2018)80 | SF (KOELIS Urostation) | SF + SB | ✓ | ✓ | ✗ | ✓ | SF performed after SB within the same examination, by the same operator; no blinding. | ✓ | ? | ✓ | Equivalence of Urostation (out of scope) with KOELIS Trinity (in scope) is uncertain. Anaesthesia method NR. |
| SB | |||||||||||
| Albisinni (2018)94 | SF (KOELIS Urostation) | SF + SB | ✓ | ✓ | ✗ | ✓ | SF performed after SB within the same examination, by the same operator; no blinding. | ✗ | ? | ✓ | All patients had one prior negative TRUS. Equivalence of Urostation (out of scope) with KOELIS Trinity (in scope) is uncertain. Anaesthesia method NR. |
| SB | |||||||||||
| Cornud (2018)93 | SB | SF + CF ± SB | ✓ | ✗ | ✓ | ✗ | Although SF and CF were conducted by a separate operator, both were conducted within the same session and tracks from the first method (CF) may have been visible to the SF operator. 12 out of 100 patients were not considered for analysis because of missing data (n = 6) or difficulties in extracting the information from the Digital Imaging and Communications in Medicine archives of the biopsy procedure (n = 6). |
? | ✓ | ? | 47% referred following a prior negative SB. Urostation (TR) is not within scope. Equivalence with KOELIS Trinity (in scope) is uncertain. Reference standard informed by both index tests + SB in unknown number of patients. |
| CF | ? | ||||||||||
| Delongchamps (2013)98 | SF (Urostation Touch, KOELIS) | SF + SB | ✗ | ✓ | ✗ | ✓ | Consecutive series, unpaired, no matching. Targeted biopsies performed after SB within the same examination, by the same operator; no blinding. Different reference standards were used in relative comparisons (CF + SB vs. SF + SB). |
✓ | ? | ✓ | Applicability of KOELIS Urostation to KOELIS Trinity is uncertain. Anaesthesia method NR. |
| CF | CF + SB | ✓ | |||||||||
| Elkhoury (2019)88 (PAIREDCAP) | SF (ARTEMIS) | SF + CF + SB | ✓ | ✗ | ✓ | ✓ | SB, followed by CF, then SF by same operator in the same session. SB operator blinded to MRI report, but no blinding of SF operator to CF tracks. | ✓ | ✓ | ✓ | |
| CF | |||||||||||
| Ferriero (2022)81 | SF (Urostation, KOELIS) | SF (Urostation, KOELIS) | ✗ | ✗ | ✗ | ✓ | Significant differences in characteristics of two study cohorts (including age, positive DRE and n of target cores), although attempts were made to adjust with propensity score matching (PSM). After adjustment, significant differences remained in median n of target cores [4 (IQR 4–6) for Urostation, vs 6 (4–6) for BioJet]. N following PSM reduced from 103 to 83 (Urostation) and 211 to 83 (BioJet). Unclear if anaesthesia and biopsy routes differed between the two index tests. Different reference standard between study arms, only informed by one of two index tests. |
✓ | ? | ✗ | Applicability of Urostation to KOELIS Trinity is unknown. Anaesthesia type unclear. Biopsy positivity rates were not informed by SB, but only by SF biopsies. |
| SF (BioJet) | SF (BioJet) | ||||||||||
| Filson (2016)96 | SF (ARTEMIS) | SF + SB | ✓ | ✓ | ✗ | ✓ | SB performed after SF within the same examination, by the same operator; no blinding. | ✓ | ? | ✓ | Biopsy route and anaesthesia method NR. |
| SB | |||||||||||
| Fourcade (2018)92 | SF (KOELIS Urostation) | SF + SB | ✓ | ✓ | ✗ | ✓ | No blinding; biopsy method order NR. | ? | ? | ✓ | Half of the patients had a prior negative biopsy. Biopsy route and anaesthesia method NR. Applicability of Urostation to KOELIS Trinity is unknown. |
| SB | |||||||||||
| FUTURE31 | SF (BiopSee) | SF | ? | ✗ | ✗ | ✓ | RCT, no reporting of allocation concealment; higher proportion of posterior lesions in cog (59%) vs. SF arm (44%). Different routes and anaesthesia methods between arms (TP and GA for SF, s. TR and LA for CF) No SB; test positivity informed by index test, which by design differed between the two arms. |
✗ | ✗ | ✗ | Only includes individuals with prior negative SB. SF conducted under GA. Positivity rate was only informed by targeted biopsy (index test). |
| CF | CF | ✓ | |||||||||
| Gomez-Ortiz (2022)99 | CF | CF + SB | ✓ | ✓ | ✗ | ✓ | SB performed after CF within the same examination, by the same operator; no blinding. | ✗ | ? | ✓ | All patients had prior negative biopsy. Anaesthesia method NR. |
| SB | |||||||||||
| Hansen (2018)95 | SF (BiopSee) | SF + SB | ✗ | ✓ | ✗ | ✗ | Allocation to SF or CF according to study centre. Participant allocation not randomised, no matching. Different reference standards used between centres (CF + SB in 1 centre, SF + SB in 2 centres). Significant number of participants in centre III were excluded from the analysis due to process errors. |
✓ | ✗ | ✗ | All index test and reference standard biopsies performed under GA. |
| CF | CF + SB | ||||||||||
| Izadpanahi (2021)82 | SF (ARTEMIS) + SB | SF + SB | ✓ | ✓ | ✗ | ✓ | Different reference standard test between arms. | ✓ | ✓ | ✓ | |
| CF + SB | CF + SB | ||||||||||
| Kaufmann (2018)101 | SF (iSR’obot Mona Lisa) | SF | ✗ | ✗ | ✗ | ✓ | Assignment to SF (TP, GA) or CF (TR, LA) based on patient preference, and statistically significant differences between arms in PSA density, median lesion size and cancer positive rate, though nearest-neighbour matching was performed. SF conducted transperineally under GA, CF transrectally under LA. Different reference standards used between study arms (SF + SB or CF + SB). |
? | ✗ | ✗ | Large proportion of prior negative biopsy patients (40%). Positive DRE excluded. SF conducted under GA. Cancer rate was only informed by targeted biopsy (index test). |
| CF | CF | ✓ | |||||||||
| Kulis (2020)86 | CF | CF + SB | ✓ | ✓ | ✗ | ✓ | SB performed after CF within the same examination, by the same operator; no blinding. | ✗ | ✓ | ✓ | All patients had prior negative TRUS. |
| SB | |||||||||||
| Liang (2020)85 | SF (BK) | SF | ? | ✓ | ✗ | ✓ | No random allocation; criteria for assignment to SF and CF NR; no significant differences in characteristics between SF and CF arms. No systematic biopsy; cancer rates only informed by targeted biopsy, which by design differed between the study arms (either SF or CF). |
✓ | ✓ | ✗ | Positivity rate was only informed by targeted biopsy (index test). |
| CF | CF | ||||||||||
| Lockhart (2022)100 | SF (BK/MIM) | SF + SB | ✗ | ✓ | ✗ | ✓ | Retrospective, criteria for assignment to FS and CF NR; significant differences in characteristics between the two study arms, including mean PSA, AS, median ISUP, mean n of cores per case, CSPCa rates. No blinding; biopsy method order NR. Different reference standard used between arms (SF + SB, vs. CF + SB). |
✓ | ? | ✓ | Biopsy route and anaesthesia method NR. |
| CF | CF + SB | ||||||||||
| Monda (2018)90 | SF (UroNav) | SF + SB | ✗ | ✓ | ✗ | ✓ | Assignment to SF and CF determined by time of introduction of SF to practice. Significant difference in percentage of biopsy naive (SF: 36%; CF: 27%). Targeted and SB performed in same session, order NR, no blinding reported. Different reference standards between study arms due to design (SF + SB, or CF + SB). |
✗ | ? | ✓ | Only 36% of SF and 27% of CF were biopsy naive; 18% and 21% were on AS respectively. Biopsy route and anaesthesia method NR. |
| CF | CF + SB | ||||||||||
| PROFUS97 | SF (ARTEMIS) | SF + CF | ✓ | ✗ | ? | ✓ | Although CF was blinded to the SF targets and conducted by a separate operator, the risk that biopsy tracks from SF biopsy may have influenced the placement of CF cores cannot be excluded. Results for SF + SB and CF + SB, or comparisons between each targeted method with SF + CF + SB NR. |
✓ | ✓ | ✗ | Results for SF + SB and CF + SB, or comparisons between each targeted method with SF + CF + SB NR. |
| CF | |||||||||||
| Rabah (2021)84 | SF (BioJet) | SF (BioJet) | ? | ✗ | ✗ | ✓ | Insufficient details on random allocation method and allocation concealment; unclear why a larger number of patients was randomised to TRUSBx (n = 165) than TPBx (n = 142); no baseline imbalances reported, although no data on lesions location reported. GA was peformed for the TPBx arm only; N of biopsies taken was higher in TRUSBx arm (n = 403) compared with TPBx (n = 338). Positive rates only informed by one index test in each arm. Each arm had a different software fusion method, route and anaesthesia type. |
✓ | ✗ | ✗ | All BioJet biopsies performed under GA. Positive rates only informed by one index test in each arm. SB (12 core) were conducted for all patients but not included as part of ref std. |
| SF (ARTEMIS) | SF (ARTEMIS) | ✓ | |||||||||
| Sokolakis (2021)83 | SF (BioJet) | SF (BioJet) | ✗ | ✓ | ✗ | ✓ | No randomisation, consecutive series. Small sample size in each arm; no statistically significant differences in reported characteristics, though difference in % with previous biopsy (0 in Trinity arm, vs. 40% in UroNav and 22% in BioJet arm. Different test for positive rate estimates for each cohort; SB was not incorporated to the results. |
✓ | ✓ | ✗ | Positivity rate was only informed by targeted biopsy (index test). |
| SF (UroNav) | SF (UroNav) | ||||||||||
| SF (KOELIS Trinity) | SF (KOELIS Trinity) | ||||||||||
| Stabile (2018)89 | SF (BioJet) | SF + SB | ✗ | ✗ | ✗ | ✓ | Unpaired, unmatched design; choice of TB method (including route) at operator’s discretion; statistically significant difference in age, PSA, median n of targets per lesion, and previous biopsy between SF and cog fusion cohorts (p < 0.05). Median target cores per MRI was higher in the SF cohort [3, IQR (2–3)] than the cognitive biopsy cohort [2 (2–5)] (p < 0.001), which may favour the fusion biopsy group. Different reference standards between arms (SB + cog vs. SB + SF) and no blinding of SB operator. |
? | ✗ | ✓ | 46% prior negative biopsy. All three urologists were naive to targeted biopsy techniques. Evidence of significant learning curve provided for all targeted biopsy approaches. Anaesthesia method NR. |
| CF | CF + SB | ||||||||||
| Thangarasu (2021)79 | CF | CF + SB | ✓ | ✓ | ✗ | ✓ | SB performed after CF within the same examination, by the same operator; no blinding. | ✓ | ✓ | ✓ | |
| SB | |||||||||||
| Wasjwol 202087 | SF (UroNav) | SF + SB | ✓ | ✓ | ✗ | ✓ | SB performed after SF within the same examination, by the same operator; no blinding. | ? | ✓ | ✓ | 49% had prior negative biopsy. |
| SB | |||||||||||
Appendix 6 Additional network meta-analysis data and results
Data for additional analyses
| Study | Intervention | Number of patients | Number of cancers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | |
| PAIREDCAP (2019)88 | CF | SB | ARTEMIS | 248 | 248 | 248 | 154 | 196 | 177 |
| Izadpanahi (2021)82 | CF + SB | ARTEMIS + SB | NA | 100 | 99 | NA | 31 | 44 | NA |
| Wajswol (2020)87 | SB | UroNav | UroNav + SB | 169 | 169 | 169 | 116 | 120 | 133 |
| Thangarasu (2021)79 | CF | SB | CF + SB | 75 | 75 | 75 | 34 | 40 | 43 |
| Kulis (2020)86 | CF | SB | CF + SB | 63 | 63 | 63 | 33 | 30 | 38 |
| Cornud (2018)93 | CF | Urostation | NA | 88 | 88 | NA | 31 | 40 | NA |
| FUTURE (2019)31 | CF | BiopSee | NA | 78 | 79 | NA | 34 | 39 | NA |
| PROFUS (2014)97 | CF | ARTEMIS | NA | 125 | 125 | NA | 40 | 45 | NA |
| Albisinni (2018)94 | SB | Urostation | Urostation + SB | 74 | 74 | 74 | 33 | 35 | 42 |
| Fourcade (2018)92 | SB | Urostation | Urostation + SB | 191 | 191 | 191 | 88 | 85 | 106 |
| Gomez-Ortiz (2022)99 | CF | SB | CF + SB | 111 | 111 | 111 | 42 | 30 | 46 |
| aRabah (2021)84 | ARTEMIS | BioJet | NA | 165 | 142 | NA | 48 | 64 | NA |
| Alberts (2018)80 | SB | Urostation | Urostation + SB | 48 | 48 | 48 | 25 | 28 | 32 |
| Filson (2016)96 | SB | ARTEMIS | ARTEMIS + SB | 538 | 538 | 538 | 244 | 228 | 286 |
| Study | Intervention | Number of patients | Number of CS cancers | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | Arm 1 | Arm 2 | Arm 3 | |
| PAIREDCAP (2019)88 | CF | SB | ARTEMIS | 248 | 248 | 248 | 116 | 150 | 134 |
| Izadpanahi (2021)82 | CF + SB | ARTEMIS + SB | NA | 100 | 99 | NA | 12 | 19 | NA |
| FUTURE (2019)31 | CF | BiopSee | NA | 78 | 79 | NA | 26 | 27 | NA |
| PROFUS (2014)97 | CF | ARTEMIS | NA | 125 | 125 | NA | 24 | 29 | NA |
| Albisinni (2018)94 | SB | Urostation | Urostation + SB | 74 | 74 | 74 | 21 | 25 | 42 |
| Fourcade (2018)92 | SB | Urostation | Urostation + SB | 191 | 191 | 191 | 52 | 60 | 106 |
| Gomez-Ortiz (2022)99 | CF | SB | CF + SB | 111 | 111 | 111 | 23 | 21 | 46 |
| aRabah (2021)84 | ARTEMIS | BioJet | NA | 165 | 142 | NA | 21 | 46 | NA |
| Alberts (2018)80 | SB | Urostation | Urostation + SB | 48 | 48 | 48 | 14 | 17 | 19 |
| Filson (2016)96 | SB | ARTEMIS | ARTEMIS + SB | 538 | 538 | 538 | 130 | 160 | 186 |
FIGURE 13.
Network of biopsy types and devices compared for CSPCa detection, under the assumption of a common effect for different SF devices. Lines represent comparisons made in studies, numbers on the lines show how many studies included that comparison and shaded areas represent multi arm studies. SB, systematic biopsy.

FIGURE 14.
Network of biopsy types and devices compared for CSPCa detection. Lines represent comparisons made in studies, numbers on the lines show how many studies included that comparison and shaded areas represent multi arm studies. SB, systematic biopsy.

Results from additional analyses: tables
| ISUP | ARTEMIS probabilities from Filson et al.96 biopsy-naive data | ARTEMIS + SB probabilities from Filson et al.96 biopsy-naive data | ||||
|---|---|---|---|---|---|---|
| Cognitive | Systematic | Softwarea | Cognitive + SB | Software + SBa | ||
| NC | 0.36 (0.29 to 0.44) | 0.25 (0.21 to 0.29) | 0.29 | 0.41 (0.21 to 0.56) | 0.36 | |
| 1 | 0.20 (0.15 to 0.25) | 0.21 (0.17 to 0.26) | 0.17 | 0.21 (0.10 to 0.33) | 0.22 | |
| 2 | 0.18 (0.13 to 0.25) | 0.28 (0.23 to 0.33) | 0.28 | 0.10 (0.03 to 0.23) | 0.22 | |
| 3 | 0.15 (0.10 to 0.23) | 0.15 (0.10 to 0.22) | 0.16 | 0.21 (0.06 to 0.59) | 0.12 | |
| 4-5 | 0.10 (0.06 to 0.17) | 0.10 (0.06 to 0.17) | 0.10 | 0.02 (0.00 to 0.18) | 0.08 | |
| ISUP | Compared to CF biopsy | Compared to CF biopsy plus systematic biopsy | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SB | ARTEMIS | BioJet | BiopSee | Urostation | ARTEMIS + SB | Urostation + SB | ||||||||
| NC | REFERENCE | REFERENCE | ||||||||||||
| 1 | 1.54 | (1.06 to 2.24) | 1.04 | (0.72 to 1.52) | 1.04 | (0.49 to 2.24) | 1.65 | (0.61 to 4.73) | 1.21 | (0.67 to 2.15) | 1.17 | (0.71 to 1.94) | 1.28 | (0.65 to 2.53) |
| 2 | 2.28 | (1.50 to 3.47) | 1.92 | (1.26 to 2.95) | NE | NE | NE | NE | 3.03 | (1.04 to 8.94) | 2.57 | (0.94 to 8.13) | 3.31 | (0.78 to 15.77) |
| 3 | 1.41 | (0.82 to 2.41) | 1.31 | (0.78 to 2.22) | NE | NE | NE | NE | NE | NE | 0.65 | (0.12 to 2.90) | NE | NE |
| 4-5 | 1.54 | (0.83 to 2.86) | 1.22 | (0.65 to 2.29) | NE | NE | NE | NE | NE | NE | 4.41 | (0.46 to 150.05) | NE | NE |
| ISUP | ARTEMIS probabilities from Filson et al.96 biopsy-naive data | ARTEMIS + SB probabilities from Filson et al.96 biopsy-naive data | ||||||
|---|---|---|---|---|---|---|---|---|
| Cognitive | SB | ARTEMISa | Cognitive + SB | ARTEMIS + SBa | ||||
| NC | 0.54 | (0.46 to 0.61) | 0.41 | (0.35 to 0.47) | 0.47 | 0.41 | (0.21 to 0.56) | 0.36 |
| 1 | 0.18 | (0.13 to 0.24) | 0.21 | (0.17 to 0.26) | 0.16 | 0.21 | (0.10 to 0.33) | 0.22 |
| 2 | 0.12 | (0.08 to 0.16) | 0.20 | (0.16 to 0.25) | 0.20 | 0.10 | (0.03 to 0.23) | 0.22 |
| 3 | 0.09 | (0.06 to 0.14) | 0.10 | (0.06 to 0.15) | 0.11 | 0.21 | (0.06 to 0.59) | 0.12 |
| 4-5 | 0.06 | (0.03 to 0.10) | 0.07 | (0.04 to 0.12) | 0.06 | 0.02 | (0.00 to 0.17) | 0.08 |
| ISUP | ARTEMIS probabilities from Filson et al.96 previous negative-biopsy data | ARTEMIS + SB probabilities from Filson et al.96 previous negative-biopsy data | ||||||
|---|---|---|---|---|---|---|---|---|
| Cognitive | SB | ARTEMISa | Cognitive + SB | ARTEMIS + SBa | ||||
| NC | 0.74 | (0.68 to 0.80) | 0.63 | (0.57 to 0.69) | 0.69 | 0.63 | (0.38 to 0.76) | 0.58 |
| 1 | 0.09 | (0.07 to 0.12) | 0.12 | (0.09 to 0.15) | 0.09 | 0.14 | (0.07 to 0.21) | 0.15 |
| 2 | 0.06 | (0.04 to 0.08) | 0.11 | (0.09 to 0.14) | 0.10 | 0.05 | (0.02 to 0.12) | 0.12 |
| 3 | 0.07 | (0.04 to 0.10) | 0.08 | (0.05 to 0.12) | 0.08 | 0.14 | (0.04 to 0.47) | 0.09 |
| 4-5 | 0.04 | (0.02 to 0.07) | 0.05 | (0.03 to 0.09) | 0.05 | 0.01 | (0.00 to 0.12) | 0.06 |
| Model | Any cancer | CS cancer | ||||||
|---|---|---|---|---|---|---|---|---|
| Model 2a | Model 2b | Model 3a | Model 3b | |||||
| ResDeva | DIC | ResDevb | DIC | ResDevc | DIC | ResDevd | DIC | |
| NMA random effects | 33.56 | 55.35 | 36.60 | 66.13 | 23.11 | 42.76 | 26.51 | 49.33 |
| NMA fixed effect | 37.54 | 54.57 | 43.21 | 67.26 | 40.29 | 53.32 | 34.47 | 52.65 |
| UME randomeffects | NA | NA | NA | NA | 23.30 | 43.53 | 25.83 | 48.49 |
| UME fixedeffect | 37.95 | 56.96 | 44.35 | 71.48 | NA | NA | NA | NA3 |
| Device Y compared to X | Any cancer (model 2a) | CS cancer (model 3a) | |||||
|---|---|---|---|---|---|---|---|
| Fixed-effect NMA | Random-effects NMA | Random-effects NMA | |||||
| X | Y | Median | (95% CrI) | Median | (95% CrI) | Median | (95% CrI) |
| Cognitive | Systematic | 1.37 | (1.11 to 1.68) | 1.32 | (0.99 to 1.70) | 1.18 | (0.72 to 1.89) |
| Cognitive | Software | 1.30 | (1.06 to 1.61) | 1.29 | (1.00 to 1.67) | 1.35 | (0.86 to 2.10) |
| Cognitive | Cognitive + SB | 1.56 | (1.16 to 2.12) | 1.54 | (1.08 to 2.16) | 2.47 | (1.20 to 4.98) |
| Cognitive | Software + SB | 2.05 | (1.60 to 2.61) | 2.03 | (1.49 to 2.75) | 2.71 | (1.56 to 4.71) |
| Systematic | Software | 0.95 | (0.82 to 1.11) | 0.98 | (0.81 to 1.24) | 1.15 | (0.80 to 1.66) |
| Systematic | Cognitive + SB | 1.15 | (0.86 to 1.53) | 1.17 | (0.84 to 1.66) | 2.09 | (1.07 to 4.10) |
| Systematic | Software + SB | 1.50 | (1.27 to 1.77) | 1.54 | (1.24 to 1.99) | 2.29 | (1.56 to 3.52) |
| Software | Cognitive + SB | 1.20 | (0.88 to 1.63) | 1.19 | (0.82 to 1.70) | 1.82 | (0.90 to 3.64) |
| Software | Software + SB | 1.57 | (1.32 to 1.86) | 1.57 | (1.25 to 1.98) | 2.00 | (1.34 to 3.07) |
| Cognitive + SB | Software + SB | 1.31 | (0.96 to 1.78) | 1.32 | (0.92 to 1.91) | 1.10 | (0.56 to 2.22) |
| Device Y compared to X | Fixed-effect NMA | Random-effects NMA | |||
|---|---|---|---|---|---|
| X | Y | Median | (95% CrI) | Median | (95% CrI) |
| Cognitive | SB | 1.39 | (1.11 to 1.73) | 1.31 | (0.92 to 1.78) |
| Cognitive | ARTEMIS | 1.24 | (0.98 to 1.58) | 1.20 | (0.81 to 1.75) |
| Cognitive | BioJet | 2.49 | (1.47 to 4.27) | 2.43 | (1.08 to 5.24) |
| Cognitive | BiopSee | 1.26 | (0.67 to 2.38) | 1.26 | (0.56 to 2.81) |
| Cognitive | Urostation | 1.45 | (1.05 to 2.01) | 1.41 | (0.88 to 2.22) |
| Cognitive | UroNav | 1.55 | (0.93 to 2.62) | 1.47 | (0.67 to 3.06) |
| Cognitive | Cognitive + SB | 1.56 | (1.15 to 2.13) | 1.53 | (1.01 to 2.30) |
| Cognitive | ARTEMIS + SB | 2.00 | (1.51 to 2.65) | 2.01 | (1.23 to 3.33) |
| Cognitive | Urostation + SB | 2.18 | (1.51 to 3.13) | 2.10 | (1.23 to 3.49) |
| Cognitive | UroNav + SB | 2.35 | (1.37 to 4.07) | 2.24 | (1.00 to 4.77) |
| SB | ARTEMIS | 0.90 | (0.74 to 1.09) | 0.92 | (0.64 to 1.36) |
| SB | BioJet | 1.80 | (1.08 to 3.00) | 1.85 | (0.85 to 4.06) |
| SB | BiopSee | 0.91 | (0.47 to 1.78) | 0.96 | (0.41 to 2.35) |
| SB | Urostation | 1.04 | (0.79 to 1.38) | 1.08 | (0.73 to 1.63) |
| SB | UroNav | 1.12 | (0.70 to 1.79) | 1.12 | (0.57 to 2.23) |
| SB | Cognitive + SB | 1.13 | (0.84 to 1.51) | 1.17 | (0.80 to 1.77) |
| SB | ARTEMIS + SB | 1.44 | (1.16 to 1.79) | 1.53 | (1.01 to 2.52) |
| SB | Urostation + SB | 1.57 | (1.15 to 2.13) | 1.60 | (1.04 to 2.50) |
| SB | UroNav + SB | 1.69 | (1.04 to 2.80) | 1.71 | (0.85 to 3.46) |
| ARTEMIS | BioJet | 2.01 | (1.25 to 3.22) | 2.01 | (1.01 to 3.98) |
| ARTEMIS | BiopSee | 1.02 | (0.52 to 2.00) | 1.05 | (0.43 to 2.58) |
| ARTEMIS | Urostation | 1.17 | (0.84 to 1.62) | 1.17 | (0.69 to 1.97) |
| ARTEMIS | UroNav | 1.25 | (0.76 to 2.08) | 1.22 | (0.55 to 2.63) |
| ARTEMIS | Cognitive + SB | 1.26 | (0.91 to 1.74) | 1.27 | (0.79 to 2.07) |
| ARTEMIS | ARTEMIS + SB | 1.61 | (1.29 to 2.01) | 1.66 | (1.06 to 2.76) |
| ARTEMIS | Urostation + SB | 1.75 | (1.22 to 2.50) | 1.75 | (0.99 to 3.04) |
| ARTEMIS | UroNav + SB | 1.89 | (1.12 to 3.24) | 1.86 | (0.83 to 4.07) |
| BioJet | BiopSee | 0.51 | (0.22 to 1.16) | 0.52 | (0.17 to 1.62) |
| BioJet | Urostation | 0.58 | (0.32 to 1.03) | 0.58 | (0.25 to 1.39) |
| BioJet | UroNav | 0.62 | (0.31 to 1.25) | 0.61 | (0.22 to 1.71) |
| BioJet | Cognitive + SB | 0.63 | (0.35 to 1.11) | 0.63 | (0.28 to 1.48) |
| BioJet | ARTEMIS + SB | 0.80 | (0.47 to 1.35) | 0.83 | (0.37 to 1.97) |
| BioJet | Urostation + SB | 0.87 | (0.48 to 1.58) | 0.87 | (0.36 to 2.12) |
| BioJet | UroNav + SB | 0.94 | (0.46 to 1.93) | 0.93 | (0.32 to 2.62) |
| BiopSee | Urostation | 1.15 | (0.56 to 2.32) | 1.11 | (0.44 to 2.81) |
| BiopSee | UroNav | 1.23 | (0.54 to 2.80) | 1.16 | (0.38 to 3.41) |
| BiopSee | Cognitive + SB | 1.24 | (0.61 to 2.49) | 1.21 | (0.49 to 3.01) |
| BiopSee | ARTEMIS + SB | 1.58 | (0.79 to 3.13) | 1.59 | (0.63 to 4.14) |
| BiopSee | Urostation + SB | 1.73 | (0.83 to 3.56) | 1.66 | (0.63 to 4.26) |
| BiopSee | UroNav + SB | 1.86 | (0.80 to 4.30) | 1.78 | (0.57 to 5.26) |
| Urostation | UroNav | 1.07 | (0.62 to 1.86) | 1.04 | (0.47 to 2.28) |
| Urostation | Cognitive + SB | 1.08 | (0.73 to 1.60) | 1.09 | (0.64 to 1.86) |
| Urostation | ARTEMIS + SB | 1.38 | (0.97 to 1.95) | 1.42 | (0.81 to 2.62) |
| Urostation | Urostation + SB | 1.50 | (1.10 to 2.05) | 1.49 | (0.96 to 2.29) |
| Urostation | UroNav + SB | 1.62 | (0.92 to 2.88) | 1.59 | (0.70 to 3.51) |
| UroNav | Cognitive + SB | 1.01 | (0.58 to 1.74) | 1.04 | (0.48 to 2.34) |
| UroNav | ARTEMIS + SB | 1.29 | (0.76 to 2.14) | 1.36 | (0.62 to 3.20) |
| UroNav | Urostation + SB | 1.40 | (0.80 to 2.45) | 1.42 | (0.64 to 3.25) |
| UroNav | UroNav + SB | 1.51 | (0.92 to 2.51) | 1.52 | (0.75 to 3.07) |
| Cognitive + SB | ARTEMIS + SB | 1.28 | (0.92 to 1.76) | 1.31 | (0.81 to 2.20) |
| Cognitive + SB | Urostation + SB | 1.39 | (0.92 to 2.11) | 1.37 | (0.76 to 2.42) |
| Cognitive + SB | UroNav + SB | 1.50 | (0.85 to 2.69) | 1.46 | (0.64 to 3.23) |
| ARTEMIS + SB | Urostation + SB | 1.09 | (0.75 to 1.58) | 1.05 | (0.55 to 1.88) |
| ARTEMIS + SB | UroNav + SB | 1.18 | (0.69 to 2.03) | 1.12 | (0.47 to 2.50) |
| Urostation + SB | UroNav + SB | 1.08 | (0.61 to 1.94) | 1.07 | (0.46 to 2.43) |
| Device Y compared to X | Random-effects NMA | ||
|---|---|---|---|
| X | Y | Median | (95% CrI) |
| Cognitive | SB | 1.30 | (0.71 to 2.24) |
| Cognitive | ARTEMIS | 1.44 | (0.80 to 2.47) |
| Cognitive | BioJet | 4.79 | (1.56 to 14.56) |
| Cognitive | BiopSee | 1.04 | (0.38 to 2.85) |
| Cognitive | Urostation | 1.65 | (0.72 to 3.64) |
| Cognitive | Cognitive + SB | 2.41 | (1.10 to 5.18) |
| Cognitive | ARTEMIS + SB | 2.32 | (1.13 to 5.28) |
| Cognitive | Urostation + SB | 3.71 | (1.55 to 7.91) |
| SB | ARTEMIS | 1.10 | (0.65 to 1.90) |
| SB | BioJet | 3.69 | (1.23 to 11.40) |
| SB | BiopSee | 0.80 | (0.26 to 2.61) |
| SB | Urostation | 1.27 | (0.72 to 2.27) |
| SB | Cognitive + SB | 1.86 | (0.89 to 3.91) |
| SB | ARTEMIS + SB | 1.78 | (0.98 to 3.78) |
| SB | Urostation + SB | 2.85 | (1.56 to 4.94) |
| ARTEMIS | BioJet | 3.34 | (1.28 to 8.88) |
| ARTEMIS | BiopSee | 0.72 | (0.23 to 2.34) |
| ARTEMIS | Urostation | 1.15 | (0.52 to 2.53) |
| ARTEMIS | Cognitive + SB | 1.68 | (0.75 to 3.75) |
| ARTEMIS | ARTEMIS + SB | 1.62 | (0.86 to 3.50) |
| ARTEMIS | Urostation + SB | 2.59 | (1.12 to 5.45) |
| BioJet | BiopSee | 0.22 | (0.05 to 0.99) |
| BioJet | Urostation | 0.34 | (0.10 to 1.18) |
| BioJet | Cognitive + SB | 0.51 | (0.14 to 1.77) |
| BioJet | ARTEMIS + SB | 0.48 | (0.15 to 1.70) |
| BioJet | Urostation + SB | 0.77 | (0.21 to 2.59) |
| BiopSee | Urostation | 1.59 | (0.43 to 5.71) |
| BiopSee | Cognitive + SB | 2.32 | (0.64 to 8.11) |
| BiopSee | ARTEMIS + SB | 2.24 | (0.66 to 8.19) |
| BiopSee | Urostation + SB | 3.56 | (0.92 to 12.33) |
| Urostation | Cognitive + SB | 1.47 | (0.57 to 3.75) |
| Urostation | ARTEMIS + SB | 1.41 | (0.62 to 3.66) |
| Urostation | Urostation + SB | 2.25 | (1.23 to 3.86) |
| Cognitive + SB | ARTEMIS + SB | 0.96 | (0.47 to 2.23) |
| Cognitive + SB | Urostation + SB | 1.53 | (0.58 to 3.80) |
| ARTEMIS + SB | Urostation + SB | 1.60 | (0.59 to 3.53) |
Results from additional analyses: Figures
FIGURE 15.
Plots of residual deviance contributions for the NMA (consistency) and unrelated mean effects model. UME, unrelated mean effects.

FIGURE 16.
Posterior densities of the between-study SD for random-effects models.

Appendix 7 Additional results from studies included in the systematic review
| Population | SF technology | Routea | Anaesthesiaa | Sample size | PCa definition | Effect estimates | Statistical significance | ||
|---|---|---|---|---|---|---|---|---|---|
| Delongchamps (2013)98 | BN | Urostation Touch (KOELIS)b | TR | NR | SF: 82 CF: 54 |
NR | NRc | SF vs. SB: p = 0.006d CF vs. SB: p = 0.22 |
|
| Hansen (2018)95 | BN | BiopSee | TP | GA | SF: 395 CF: 176 |
NR | SF: 53% CF: 38% |
NR | |
| Kaufmann (2018)91 | BN, RB | BioJet | SF: TP CF: TR |
NR | SF: 191 CF: 87 |
GS: 6 | SF: 58.1% CF: 43.7% |
p = 0.02 | |
| Liang (2020)85 | BN | bkFusione | TP | LA | SF: 92 CF: 71 |
GS: 6 | SF: 51.08% CF: 60.56% |
p = 0.228 | |
| Monda (2018)90 | BN, RB | UroNav | TR | NR | SF: 348 CF: 162 |
GS: 6 | SF: 14.4% CF: 22.8% |
NR | |
| Population | SF technology | Routea | Anaesthesia | Sample size | CSPCa definition | Effect estimates | p-value | |
|---|---|---|---|---|---|---|---|---|
| Delongchamps (2013)98 | BN | Urostation Touch (KOELIS)a | TR | NR | SF: 82 CF: 54 |
GS ≥ 3 + 4 | SF: 44% CF: 45% |
SF vs. SB: p = 0.01b CF vs. SB: p = 0.6 |
| Hansen (2018)95 | BN | BiopSee | TP | GA | SF: 395 CF: 176 |
GS ≥ 3 + 4 | SF: 56% CF: 70% |
NR |
| Kaufmann (2018)91 | BN, RB | BioJet | SF: TP CF: TR |
NR | SF: 191 CF: 87 |
GS ≥ 3 + 4 | SF: 80.4% CF: 84.6% |
p = 0.55 |
| Liang (2020)85 | BN | bkFusionc | TP | LA | SF: 92 CF: 71 |
GS ≥ 3 + 4 | SF: 35.87% CF: 39.43% |
p = 0.641 |
| Monda (2018)90 | BN, RB | UroNav | TR | NR | SF: 162 CF: 348 |
GS ≥ 3 + 4 | SF: 27.9%, CF: 27.2% |
NR |
| Population | SF technology | Route | Anaesthesia | Sample size | Outcome | Effect estimates | Statistical significance | |
|---|---|---|---|---|---|---|---|---|
| Lockhart (2022)100 | BN | bkFusiona | TP | NR | SF + SB: 97 CF + SB: 186 |
GS ≥ 3 + 4 | SF + SB: 53% CF + SB: 66.7% |
NR |
| Stabile (2018)89 | BN, RB | BioJet | SF: TP or TR CF: TR |
NR | SF: 157 CF: 87 |
PCa (not defined) | SF + SB: 68.2% CF + SB: 58.6% | p = 0.2 |
| CSPCa (GS ≥ 3 + 4) | SF + SB: 58% CF + SB: 44.8% | p = 0.07 |
| Population | SF technology | Routea | Anaesthesiaa | Sample size | Outcome | Effect estimates | Statistical significance | |
|---|---|---|---|---|---|---|---|---|
| Ferriero (2022)81 | BN, RB | BioJet Urostation | Urostation: TR BioJet: NR |
NR | SF: 103 (83)b SF: 211 (83)b |
PCa (NR) Per target |
SF(Urostation): 69.8% SF (BioJet): 56.6% |
p = 0.077 |
| CSPCa (NR) Per target |
SF(Urostation): 50.6% SF (BioJet): 50.6% |
p = 1 | ||||||
| Sokolakis (2021)83 | BN, RB | BioJet KOELIS Trinity UroNav |
TR | LA | BioJet: 20 Trinity: 20 UroNav: 20 |
PCa | BioJet: 65% Trinity: 70% UroNav: 65% |
p > 0.99 |
| CSPCa (GS ≥ 3 + 4) | BioJet: 50% Trinity: 55% UroNav: 50% |
p > 0.99 |
| Study | Design | Pop. | Tests | N | Outcome (metric) | Summary | Test-positive estimates | Direction of effect/p-value |
|---|---|---|---|---|---|---|---|---|
| Delongchamps (2013)98 | Consecutive series, between-patient | BN | SF (KOELIS –Urostation Touch) vs. SBa | 82 | CSPCa: Gleason ≥ 3 + 4 (N) | SF alone detected 35 of the 44 cancers detected by SB as well as 27 undetected by SB, of which 8 had a GS of > 6. All 9 cancers missed by SF but detected by SB had a GS of 6, of which 7 involved < 5 mm of the biopsy core. | SF: 44% CF: 45% |
Favours SF vs. SB p = 0.01 |
| CB vs. SB | 54 | PCa (OR) Definition NR |
CF alone detected 37 of the 55 cancers detected by SB as well as 3 undetected by RB, of which 2 had a GS of > 6. Of the 18 cancers missed by SF but detected by RB, 16 had a GS of 6 and 15 involved < 5 mm of the biopsy core. Conditional logistic regression analysis showed that CF was not significantly better at detecting PCa compared to systematic biopsy (OR NR) |
NR | No significant difference (p = 0.66) | |||
| Hansen (2018)95 | Prospective, between-patient | BN | SF vs. CF; SB vs. SF + CF + SB | SF: 395 CF: 176 |
PCa (PI-RADS 3) | Favours combination biopsy over targeted biopsy alone. No significant difference between combination biopsy and systematic biopsy | SB: 53% CF + SF: 38% SB + SF + CF: 56% |
p < 0.001 (TB vs. SB + TB) P = 0.063 (SB vs. SB + TB) |
| PCa (PI-RADS 4–5) | Favours combination biopsy over systematic biopsy or targeted biopsy alone | SB: 80% CF + SF: 73% SB + SF + CF: 88% |
p < 0.001 (both) | |||||
| CSPCa: Gleason ≥ 3 + 4 (PI-RADS 3) | Favours combination biopsy over targeted biopsy alone. No significant difference between combination biopsy and systematic biopsy | SB: 37% SF + CF: 21% SB + SF + CF: 30% |
p < 0.001 (TB vs. SB + TB) p = 0.125 (SB vs. SB + TB) |
|||||
| CSPCa: Gleason ≥ 3 + 4 (PI-RADS 4–5) | Favours combination biopsy over systematic biopsy or targeted biopsy alone | SB: 61% CF + SF: 59% SB + SF + CF: 71% |
p < 0.001 (both) | |||||
| Ferriero (2022)81 | Prospective cohort, between patients | BN + RB | SF (Urostation) vs. SF (BioJet) | Urostation: 103 BioJet: 211 |
PCa per target (%) Definition NR |
No significant differences between the two SF types | SF (Urostation): 69.8%, SF (BioJet): 56.6% | Not significant p = 0.077 |
| CSPCa per (%) Definition NR |
No significant differences between the two SF types | SF (Urostation): 50.6%, SF (BioJet): 50.6% | Not significant p = 1.0 |
|||||
| Sokolakis (2021)83 | Prospective cohort, between patients | BN + RB | SF (BioJet) vs. SF (Trinity) vs. SF (UroNav) | BioJet: 20 Trinity: 20 UroNav: 20 |
ISUP 1 (N) | No significant difference between the three software types | BioJet: 2, Trinity: 3, UroNav: 3 | No significant difference. p > 0.99 |
| ISUP 2 (N) | BioJet: 4, Trinity: 4, UroNav: 4 | |||||||
| ISUP 3 (N) | BioJet: 4, Trinity: 3, UroNav: 3 | |||||||
| ISUP 4 (N) | BioJet: 1, Trinity: 2, UroNav: 2 | |||||||
| ISUP 5 (N) | BioJet: 1, Trinity: 2, UroNav: 1 | |||||||
| Liang (2020)85 | Retrospective cohort, between patients | BN | SF (Predictive Fusion Software) vs. CF | SF: 92 CF: 71 |
ISUP 1 (%) | Similar detection rates (within 5%) | SF = 17%, CF = 21% | Significance NR |
| ISUP 2 (%) | SF = 14%, CF = 13% | |||||||
| ISUP 3 (%) | SF = 9%, CF = 11% | |||||||
| ISUP 4 (%) | SF = 8%, CF = 13% | |||||||
| ISUP 5 (%) | SF = 3%, CF = 3% | |||||||
| Lockhart (2022)100 | Retrospective cohort, between patients | BN, AS | SF (MIM Fusion Software) vs. CF | SF: 131 CF: 223 |
ISUP 2 | Multinomial logistic regression analysis was performed to explore potential factors affecting CSPCa detection rates. Fusion or cognitive biopsy made no difference to CSPCa detection rates | NR | p = 0.729 |
| Monda (2018)90 | Retrospective cohort, between patients | BN + RB | SF (UroNav) vs. CF vs. SB (concurrent) | SF/SB: 162 CF/SB: 348 |
Gleason 6 (%) | Higher rate of PCa detection with cognitive targeted biopsy | SF: 14.4%, CF: 22.8% | Significance NR |
| Gleason 7 (%) | Similar rates of detection | SF: 20.1%, CF: 18.5% | Significance NR | |||||
| Gleason 8 (%) | Similar rates of detection | SF: 3.4%, CF: 3.1% | Significance NR | |||||
| Gleason 9–10 (%) | Similar rates of detection | SF: 4.3%, CF: 5.6% | Significance NR | |||||
| Missed targeted biopsy (%) TB < 7 and SB > 7 |
Similar rates (within 5%) | SF: 5.5%, CF: 9.9% | Not significant p = 0.172 |
|||||
| Equivalent (%) TB and SB ≥ 7 or TB and SB < 7 |
SF: 85.1%, CF: 82.1% | Not significant p = 0.172 |
||||||
| Upstage (%) TB ≥ 7 and SB < 7 |
SF: 9.5%, CF: 8.0% | Not significant p = 0.172 |
| Study | SF technology | Route | Anaesthesia | Lesion location | N of lesions | Outcome (definition) | Test-positive rates | Statistical significance |
|---|---|---|---|---|---|---|---|---|
| FUTURE (2019)31 | BiopSee | SF: TP CF: TR |
SF: GA CF: LA |
Anterior | SF: 37 CF: 25 |
PCa (NR) | SF: 62.2% CF: 60.0% |
p > 0.9 |
| CSPCa (GS: ≥3 + 4) | SF: 48.6% CF: 44.0% |
p = 0.6 | ||||||
| Posterior | SF: 35 CF: 46 |
PCa (NR) | SF: 40.0% CF: 26.1% |
p = 0.12 | ||||
| CSPCa (GS: ≥3 + 4) | SF: 20.0% CF: 26.1% |
p = 0.7 |
| Study | Population | SF technology | Routea | Anaesthesiaa | Number of patients | Outcome (definition) | Biopsy positive rates | Statistical significance |
|---|---|---|---|---|---|---|---|---|
| FUTURE (2019)31 | Prior negative SB within median 8 months (IQR 4–23) | BiopSee | SF: TP CF: TR |
SF: GA CF: LA |
SF: 79 CF: 78 |
PCa (NR) | SF: 49.4% CF: 43.6% |
p = 0.4 |
| CsPCa (GS: ≥3 + 4) | SF: 34.2% CF: 33.3% |
p > 0.9 | ||||||
| PROFUS (2014)97 | Prior negative biopsy (no further details) | ARTEMIS | TR | LA | SF and CFb: 34 | PCa (NR) | SF: 29.4% CF: 23.5% |
NR |
| CsPCa (GS: ≥3 + 4) | SF: 20.6% CF: 14.7% |
NR |
| Study | SF technology | Routea | Anaesthesiaa | Number of patients | Outcome (definition) | Test positive rate | Statistical significance |
|---|---|---|---|---|---|---|---|
| Delongchamps (2013)98 | Urostation Touch (KOELIS)b | TR | NR | SF: 82 CF: 54 |
PCa (NR) | NRc | SF vs. SB: p = 0.006 CF vs. SB: p = 0.22 |
| CSPCa (NR) | NRc | SF vs. SB: p = 0.001 CF vs. SB: p = 0.6 |
|||||
| Ferriero (2022)81 | Urostation; BioJet | Urostation: TR; BioJet: NR | NR | Urostation: 103 BioJet: 232 (1:1 PS matched cohort, n = 83) |
PCa (GS 6) | Urostation: 69.8% BioJet: 56.6% |
p = 0.077 |
| CSPCa (GS ≥ 7) | Urostation: 50.6% BioJet: 50.6% |
p = 1 | |||||
| Hansen (2018)95 | BiopSee | TP | GA | SF: 395 CF: 176 |
PCa (NR) | SF: 53% CF: 38% |
NR |
| CSPCa () | SF: 56% CF: 70% |
NR | |||||
| Izadpanahi (2021)82 | ARTEMIS | TR | LA | SF: 99 CF: 100 |
PCa (GS 6 and < 4-mm core length) | SF: 44.4% CF: 31.0% |
p = 0.035 |
| CSPCa (GS ≥ 7 or GS 6 and ≥ 4mm core length) | SF: 33.3% CF: 19.0% |
p = 0.016 | |||||
| Liang (2020)31,85 | bkFusiond | TP | LA | SF: 92 CF: 71 |
PCa (GS 6) | SF: 51.08% CF: 60.56% |
p = 0.228 |
| CSPCa () | SF: 35.87% CF: 39.43% |
p = 0.641 | |||||
| Lockhart (2022)100 | bkFusion^ | TP | NR | SF + SB: 97 CF + SB: 186 |
CSPCa (GS ≥ 7) | SF + SB: 53% CF + SB: 66.7% |
NR |
| PAIREDCAP (2019)88 | ARTEMIS | TR | LA | 248 | PCa (GS 6) | SF: 17.3% CF: 15.3% |
NR |
| CSPCa (GS ≥ 7) | SF: 54.0% CF: 46.8% |
NR | |||||
| PROFUS (2014)97 | ARTEMIS | TR | LA | 67 | PCa (GS 6) | SF: 35.8% CF: 34.3% |
NR |
| CSPCa (GS ≥ 7) | SF: 28.4% CF: 26.9% |
NR |
| Study | Pop. | SF technology | Route | Anaesthesia | Sample size | Number of targeted biopsies | N cores per ROI | Effect estimates | p-value |
|---|---|---|---|---|---|---|---|---|---|
| Stabile (2018)89 | BN, RB | BioJet | SF: TP or TR CF: TR |
NR | SF: 157 CF: 87 |
TR SF: 70 TP SF: 87 TR CF: 87 |
Med (range) SF: 3 (2–3); CF: 2 (2–5) |
Learning curve CSPCa detection by operator experience: OR 1.03, 1.06, and 1.01 for operators 1, 2, and 3, respectively CSPCa biopsy positivity rate at first procedure to 60th procedure: Operator 1 (TR CF): 30–57% Operator 2 (TR SF): 15–78% Operator 3 (TP-SF): 70–83% |
p < 0.04 |
| Study | Design | Pop. | Biopsy method | Sample size (participants) | Total n of cores | N cores | N ROI targeteda | Biopsy positivity definition | Effect estimates | p-value | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | Route | Anaesthesia | ||||||||||
| SF vs. CF | ||||||||||||
| Delongchamps (2013)98 | Consecutive series, between-patient | BN | SF (Urostation Touch)b CF | SF and CF: TR | NR | mpMRI + ve KOELIS: 82 CF: 54 |
NR | Med (range) SF: 3 (2–5) CF: 4 (3–10) |
NR | NR | Median % (IQR) SF: 75% (33–100) CF: 67% (20–86) |
p = 0.003 |
| FUTURE (2019)31 | RCT, between patient | RB | SF (BiopSee); CF | SF: TP, CF: TR | NR | 157 (SF: 79, CF: 78) |
SF: 358 CF: 275 |
Med (IQR) SF: 4 (3–5) CF: 3 (3–4) |
All ROI | NR | Mean % (SD) SF: 31.3% (37.8) CF: 33.3% (42.1) |
NR |
| PAIREDCAP (2019)88 | Prospective cohort, within patient | BN | SF (ARTEMIS); CF; SB | NR | NR | 248 | SF: 741 CF: 744 |
3 cores | Index ROI | NR | SF: 38.1% CF: 33.3% SB: 15.7%c |
SF vs. CF: NSc |
| Software fusion vs. software fusion | ||||||||||||
| Rabah (2021)84 | RCT, between patient | BN, RB | SF (ARTEMIS), SF (BioJet) | ARTEMIS: TR BioJet: TP |
ARTEMIS: LA BioJet: GA |
307 | ARTERMIS: 403 BioJet: 338 |
2–4 cores | All ROI | NR | BioJet: 43.5% ARTEMIS: 21.1% | p = 0.0002 |
| Study | Design | Pop. | Biopsy method | Sample size | N cores per ROIa | Total N of cores | Effect estimates | p-value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type | Route | Anaesthesia | ||||||||||
| FUTURE (2019)31,168 | RCT, between patient | RB | SF (BiopSee, TP) vs. CF (TR) | SF: TP, CF: TR | SF: GA/spinal anaesthesia CF: LA |
SF: 79 CF: 78 |
Median (IQR) SF: 4 (3–5) CF: 3 (3–4) |
SF: 358 CF: 275 |
SF vs. CF: OR 2.27 (95% CI 1.04 to 5.00) (Grade 1–2) Grade 1 AEs (SF: 65.8%; CF: 74.4%); Grade 2 AEs (SF: 5.1%; CF: 10.3%) Grade 1–2 AEs |
p < 0.05 | ||
| SF (%) | CF (%) | |||||||||||
| Haematuria | 50.6 | 74.4 | ||||||||||
| Haematospermia | 35.4 | 50.0 | ||||||||||
| Rectal bleeding | 2.5 | 5.1 | ||||||||||
| UTI | 1.3 | 6.4 | ||||||||||
| Fever | 2.5 | 5.1 | ||||||||||
| Urinary retention | 3.8 | 5.1 | ||||||||||
| Haematoma | 3.8 | – | ||||||||||
| Lower back pain | 1.3 | – | ||||||||||
| Atrial fibrillation | 1.3 | – | ||||||||||
| Liang (2020)85 | Retrospective, between patients | BN | SF (BK Predictive Software) vs. CF (Both TP) | TP | LA | SF: 92 CF: 71 |
4 | NR | SF: 2 AEs (1 post-biopsy fever, 1 bacteraemia). CF: 2 AEs (2 post-biopsy fever). AE grade NR. No patients developed severe bleeding, dysuria, vasovagal reactions, or other complications that required to be addressed. |
NR | ||
| Monda (2018)90 | Retrospective; before and after study | BN, RB (+ve/–ve) | SF: UroNav vs. CF. (Both TR) |
NR | NR | SF: 348 CF: 162 |
NR | NR | % patients with complications: CF: 8.6%; SF: 7.2% AE grade NR. |
p = 0.564 | ||
| Study | Design | Pop. | Biopsy method | Sample size | N cores per ROIa | Total N of cores | Effect estimates | p-value | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Type | Route | Anaesthesia | ||||||||
| Rabah (2021)84 | RCT, between patient | BN, RB | SF: ARTEMIS (TR) and BioJet (TP) | ARTEMIS: TR BioJet: TP |
ARTEMIS: Local BioJet: General |
ARTEMIS:165 BioJet: 142 |
2–4 | Artemis: 403 BioJet: 338 |
Haematuria: 2 ARTEMIS 1 BioJet Urinary retention 7 ARTEMIS 8 BioJet Rectal bleeding 6 ARTEMIS AE grade NR |
p = 0.6 p = 0.56 |
| Sokolakis (2021)83 | Prospective cohort, between patient | BN, RB | SF: BioJet vs. KOELIS vs. UroNav (All TR) |
TR | Local | BioJet: 20 KOELIS: 20 UroNav: 20 |
2–3 | NR | No severe peri- or post operative AEs. Transient AEs common (haematuria, haematospermia and haematochezia) |
NR |
| Study | Design | Pop. | Biopsy methods | Sample size | N cores per ROIa | Total N of cores | Effect estimates | ||
|---|---|---|---|---|---|---|---|---|---|
| Type | Route | Anaes-thesia | |||||||
| Sokolakis (2021)83 | Prospective, between patient | BN, RB | SF: BioJet, KOELIS, UroNav | TR (all) | LA (all) | BioJet: 20 KOELIS: 20 UroNav: 20 |
2–3 | NR | System Usability Scale [Median (IQR)] Total BioJet: 65 (63.8, 68.1); KOELIS: 38.8 (37.5,45); UroNav: 72.5 (63.8, 80.6) Junior Urologists BioJet: 65 (65, 65); KOELIS: 38.8 (38.1, 39.4); UroNav: 62.5 (61.2, 63.8) Senior urologists BioJet: 68.8 (64.4, 73.1); KOELIS: 48.8 (43.1, 54.4); UroNav: 81.2 (80.6, 81.9) p-values NR |
Appendix 8 Studies informing model parametrisation and structure
| Study | Study design | Sample size | Population | Biopsy test 1 | Biopsy test 2 |
|---|---|---|---|---|---|
| Mannaerts (2020)169 | Prospective, Within patient |
142 | Naive | SF (ARTEMIS) | SB |
| PAIREDCAP (2019)88 | Prospective, Within patient |
248 | Naive | SF (ARTEMIS) | CF |
| Izadpanahi (2021)82 | RCT, Between patient |
199 | Naive | SF (ARTEMIS) + SB | CF + SB |
| Filson (2016)96 | Prospective, Within-patient |
538a,b (273 naive) | Naive, repeat or active surveillancec | SF (ARTEMIS) | SB |
| Alberts (2018)80 | Prospective, Within-patient |
48 | Naive, repeatc | SF (UroStation) | SB |
| Mortezavi (2018)141 | Retrospective | 291b | Naive, repeat or active surveillance | SF (BiopSee) | TTMB |
| Zhou (2018)142 | Prospective, Between patient |
153 | NR | SF (Hitachi) or CF | TTMB |
| Simmons (2018)170 | Prospective, Within patient |
200 | Repeat | SF (SmartTarget) or CF | TTMB |
| Hansen (2016)171 | Retrospective | 289a,b | Naive, repeat or active surveillancec | SF (BiopSee) | TTMB |
| Kesch (2017)172 | Prospective, Within-patient |
172 | Naive, repeat or active surveillance | MRI targeted (both software and cognitive) | TTMB |
Prevalence
We were unable to identify any population-level evidence on the prevalence of PCa by ISUP grade. From the 10 studies identified in our targeted review, 5 studies compared MRI-targeted biopsy compared to a template‐guided biopsy (template mapping or saturation biopsy (see Table 54, Appendix 8). 141,142,170–172 The template-guided biopsy does not present perfect accuracy, as the test’s accuracy depends on the intensity of cores taken and core (see Reference standard). Therefore, to approximate prevalence, and given the assumption of negligible false-positive results to biopsy, we used a ‘composite’ reference standard combining the template-guided biopsy with the other biopsy method investigated in each study. The results from the five studies included are shown in Table 54, Appendix 8.
The results show considerable variation between studies with, for example, the prevalence of NC varying between 7.5% and 34% across studies and the prevalence of ISUP grade 4 or 5 cancer from 4% to 20%. The reasons for this heterogeneity are unclear, and may arise from the significant clinical diversity across studies, including in participants (settings of care), diagnostic tests (and in the protocols for their implementation) and outcomes, and/or from the methodological diversity across studies, including variability in study design and risk of bias. The results from Hansen et al. 171 suggest that the position of patients in the pathway may be a significant source of heterogeneity.
| ISUP | Hansen et al.171 Proportion (N) |
Zhou (2018)142 Proportion (N) |
Simmons et al.170 Proportion (N) |
Mortezavi et al.141 Proportion (N) |
Kesch et al.172 Proportion (N) |
||
|---|---|---|---|---|---|---|---|
| BN | RB | AS | |||||
| 0 | 0.306 (26) | 0.461 (94) | 0.101 (9) | 0.340 (52) | 0.075 (15) | 0.237 (69) | 0.276 (35) |
| 1 | 0.235 (20) | 0.181 (37) | 0.393 (35) | 0.163 (25) | 0.210 (42) | 0.12 (35) | 0.173 (22) |
| 2 | 0.212 (18) | 0.191 (39) | 0.270 (24) | 0.190 (29) | 0.675 (135) | 0.285 (83) | 0.378 (48) |
| 3 | 0.129 (11) | 0.083 (17) | 0.157 (14) | 0.131 (20) | 0.155 (45) | 0.079 (10) | |
| 4 or 5 | 0.118 (10) | 0.083 (17) | 0.079 (7) | 0.176 (27) | 0.04 (8) | 0.203 (59) | 0.094 (12) |
| N | 85 | 204 | 89 | 153 | 200 | 291 | 127 |
Distribution of test results obtained with cognitive fusion or software fusion biopsy
The 10 studies identified in the targeted review were potentially relevant to inform the distribution of test results obtained with CF or SF biopsy. Four studies were initially excluded because their population was not considered representative of the NHS. 80,141,170,172 Mortezavi et al. 107 and Kesch et al. 111 included patients under active surveillance. Simmons et al. 109 only included patients with a repeat biopsy, and patients in Alberts et al. 80 were selected from a population-wide screening programme.
The distribution of test results obtained from a targeted biopsy for the remaining five studies are presented in Table 55, Appendix 8. There is considerable heterogeneity in the proportion of patients identified in each Gleason grade group (GG) across the studies.
Therefore, to ensure that the distribution of Gleason grades is representative to the NHS population, the remaining six studies’ eligibility criteria were compared to determine which was most representative to NHS practice. According to the NICE guideline NG131 and the PCa diagnostic pathway, patients are referred if their prostate-specific antigen levels are above the age-specific reference range (which, for men aged 50–69 is a PSA level of >3.0 ng/ml) or if their prostate feels malignant (hard, or lumpy) on DRE. 10,17 Furthermore, this DAR is focused on patients with mpMRI visible lesions (PI-RADS 3+), who are biopsy naive, or are undergoing a repeat biopsy (after a negative result). Table 56, Appendix 8 summarises the study eligibility criteria and participant characteristics, and the decisions for inclusion/exclusion.
Two studies82,142 were not deemed to be appropriate for use in this analysis. Izadpanahi et al. 82 limited their population to patients with a PSA > 10 ng/mL; and the population in Zhou et al. 142 had a considerably higher baseline PSA compared to the other studies. In addition, the settings of these studies (Iran and China) may not be reflective of NHS practice.
| ISUP | Mannaerts169 | PAIREDCAP88 | Izadpanahi82 | Zhou142 | Hansen (RB)171 | Filson (BN)96 | Filson (RB)96 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SF (ARTEMIS) | CF | SF (ARTEMIS) | CF + SB | SF + SB | Mixed CF/SF |
SF (BiopSee) | SF (ARTEMIS) |
SF + S | SF | SF + SB | |
| 0 | 0.140 (7) | 0.379 (94) | 0.286 (71) | 0.690 (69) | 0.556 (55) | 0.503 (77) | 0.642 (131) | 0.469 (128) | 0.355 (97) | 0.687 (182) | 0.585 (155) |
| 1 | 0.040 (2) | 0.153 (38) | 0.173 (43) | 0.190 (19) | 0.253 (25) | 0.105 (16) | 0.103 (21) | 0.165 (45) | 0.220 (60) | 0.087 (23) | 0.151 (40) |
| 2 | 0.380 (19) | 0.21 (52) | 0.282 (70) | 0.060 (6) | 0.131 (13) | 0.118 (18) | 0.167 (34) | 0.198 (54) | 0.223 (61) | 0.102 (27) | 0.117 (31) |
| 3 | 0.280 (14) | 0.157 (39) | 0.161 (40) | 0.050 (5) | 0.03 (3) | 0.111 (17) | 0.088 (18) | 0.168 (46) | 0.201 (55) | 0.125 (33) | 0.147 (39) |
| 4/5 | 0.160 (8) | 0.101 (25) | 0.097 (24) | 0.010 (1) | 0.03 (3) | 0.163 (25) | 0.118 (10) | ||||
| N | 50 | 248 | 248 | 100 | 99 | 153 | 204 | 273 | 273 | 265 | 265 |
| Study | Country | PSA 3level | DRE exam | Naive/repeat | Considerations for inclusion or exclusion | |
|---|---|---|---|---|---|---|
| Eligibility criteria | Included patients | |||||
| Mannaerts (2020)169 | The Netherlands | ≥ 3.0 – 20 ng/mL | Median (IQR) 6.2 (4.7–8.0) |
Suspicious DRE | 100% BN | Similar referral pathway: appropriate PSA cut-off and suspicious DRE |
| PAIREDCAP (2019)88 | USA | < 25 ng/mL | Median (IQR) 6.2 (4.6–8.2) |
Suspicious DRE | 100% BN | Similar referral pathway: appropriate PSA cut-off and suspicious DRE |
| Filson (2016)96 | USA | ‘Elevated PSA’ | Median (IQR) Naive: 5.8 (4.4–8.1) Repeat: 7.6 (5.0–11.5) |
Suspicious DRE | 33% BN 32% RB 35% AS |
Unclear PSA cut-off, but similar PSA of included patients. Less granularity in Gleason grades (only data on Grade 3+). |
| Hansen (2016)171 | UK | ‘Elevated PSA’ | Median (IQR) Naive: 6.2 (4.8–8.6) Repeat: 7.8 (4.8-8.6) |
Suspicious DRE | 20% BN 55% repeat 25% AS |
Unclear PSA cut-off, but similar PSA of included patients. Greater proportion of patients with repeat biopsy, and number of naive patients is small. |
| Zhou (2018)142 | China | > 4 ng/mL | Median (IQR) 9.5 (6.5–15.5) |
Suspicious DRE | 100% BN | Similar referral pathway: appropriate PSA cut-off and suspicious DRE. Concerns regarding high baseline PSA levels in the included patients. Differences in healthcare systems between UK and China. |
| Izadpanahi (2019)82 | Iran | > 2–10 ng/dL | Mean (SD) 6.1 ng/dL (1.3) |
Suspicious DRE | 100% BN | Concerns regarding reporting of PSA-levels (report ng/dL). Limiting PSA levels to < 10 ng/dL was not deemed representative of UK practice. Differences in healthcare systems between UK and Iran. |
The remaining four studies88,96,169,173 were deemed to be most similar to NHS practice, based on population eligibility criteria. All studies applied focused on patients with an elevated PSA and included patients who were referred for suspicious DRE. We considered that only biopsy-naive patients should be included in the analysis, as the vast majority (~90%) of patients in NHS practice will be receiving a first biopsy. Therefore, in the studies where separable data were available, we only included the biopsy-naive patients, as the proportion of patients with repeat biopsy was often high.
Accuracy of cognitive fusion or software fusion biopsy
In order to determine the accuracy of CF or SF biopsy, studies which compared MRI-targeted biopsy (SF and/or CF) against template or saturation biopsy were identified. To determine true disease status as closely as possible, patients were reclassified according to a composite reference standard from both tests. Out of four studies,141,142,170,171 two provided test accuracy data with the required granularity by ISUP grade. 141,142 The characteristics of these two studies are summarised in Table 53, Appendix 8. Zhou et al. 142 compared SF biopsies [including both SF biopsy (29% of patients) and CF biopsy (71% of patients)] with template-guided transperineal prostate saturation biopsy, although the study did not provide accuracy data for SF biopsy and CF biopsy separately. Mortezavi et al. 141 on the other hand, does provide data on accuracy specifically for SF biopsy compared to transperineal template saturation prostate biopsy. Mortezavi et al. 141 includes patients who are on active surveillance, who are likely to have a different GS distribution compared with biopsy-naive and prior negative-biopsy patients. However, as accuracy evidence is conditional on true disease status, any such differences in the patient population included are not likely to have a significant impact on conditional accuracy estimates.
Table 57, Appendix 8, provides the computed conditional (accuracy) probabilities of patients being identified at a particular grade with MRI-fusion given a particular true disease status given by the composite TMB and MRI fusion results.
The results show significant heterogeneity, with Zhou identifying a higher accuracy at ISUP grade 3 and above. To aid interpretation of these results, we next describe the two further studies which are UK-based and therefore have higher representativeness than both Zhou (2018) or Mortezavi (2018).
Two UK-based studies – Simmons et al. 170 and Hansen et al. 171 – did not report results with the necessary disaggregation of Gleason grade. Simmons et al. 170 reports a within-patient comparison (secondary analysis of PICTURE trial), of TMP biopsy and targeted biopsy (mixture of cognitive and software fusion) but only reported Gleason Grade 1, 2–3 and 4–5 (reported in Table 58, Appendix 8). The full accuracy matrix by ISUP grade could not be retrieved for Hansen et al.,171 but sensitivity at ISUP grade thresholds of 1 or above, 2 or above and 3 or above could be calculated, against a composite reference standard. The table below compares these sensitivity values, with the results from the studies for which fuller reporting of the accuracy matrices was available (see Table 59, Appendix 8). As Table 59 shows, there is also some variation in the sensitivity results between the UK-based studies. The results from Mortezavi (2018),141 are more similar to Hansen et al. 171 at Gleason grade 3 or above, whereas the results of Zhou et al. 142 are more similar to Simmons (2018)170 at the lower GGs. It is therefore unclear what are the relevant source(s) for the between-study heterogeneity observed between Zhou et al. 142 and Mortezavi et al.,141 and the representativeness of both these studies to the UK context is uncertain.
| Composite reference standard | Zhou (2018) 142 | Mortezavi et al.141 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (Software and CF) | ||||||||||
| NC | ISUP1 | ISUP 2 | ISUP 3 | ISUP 4 or 5 | NC | ISUP 1 | ISUP 2 | ISUP 3 | ISUP 4 or 5 | |
| NC | 52/52 (1) | 0 | 0 | 0 | 0 | 69/69 (1) | 0 | 0 | 0 | 0 |
| ISUP 1 | 11/25 (0.44) | 14/25 (0.56) | 0 | 0 | 0 | 24/35 (0.69) | 11/35 (0.31) | 0 | 0 | 0 |
| ISUP 2 | 13/29 (0.45) | 1/29 (0.03) | 15/29 (0.52) | 0 | 0 | 21/83 (0.25) | 17/83 (0.20) | 45/83 (0.54) | 0 | 0 |
| ISUP 3 | 1/20 (0.05) | 1/20 (0.05) | 2/20 (0.10) | 16/20 (0.80) | 0 | 10/45 (0.22) | 2/45 (0.04) | 9/45 (0.20) | 24/45 (0.53) | 0 |
| ISUP 4 or 5 | 0/27 (0) | 0/27 (0) | 1/27 (0.04) | 1/27 (0.04) | 25/27 (0.93) | 6/59 (0.1) | 2/59 (0.03) | 7/59 (0.12) | 8/59 (0.14) | 36/59 (0.61) |
| Composite reference standard | MRI fusion biopsy (mix of software and cognitive) | |||
|---|---|---|---|---|
| NC | ISUP grade 1 | ISUP grade 2 or 3 | ISUP grade 4 or 5 | |
| NC | 15/15 (1) | 0 | 0 | 0 |
| ISUP grade 1 | 25/42 (0.60) | 17/42 (0.40) | 0 | 0 |
| ISUP grade 2 or 3 | 15/135 (0.11) | 21/135 (0.16) | 99/135 (0.73) | 0 |
| ISUP grade 4 or 5 | 1/8 (0.13) | 0/8 (0.00) | 2/8 (0.25) | 5/8 (0.63) |
| Sensitivity against composite reference standard | ||||
|---|---|---|---|---|
| Hansen et al.171 | Simmons et al.170 | Mortezavi et al.141 | Zhou et al.142 | |
| ISUP grade ≥ 1 | 0.670 (73/109) | 0.778 (144/185) | 0.725 (161/222) | 0.752 (76/101) |
| ISUP grade ≥ 2 | 0.712 (52/73) | 0.741 (106/143) | 0.690 (129/187) | 0.789 (60/76) |
| ISUP grade ≥ 3 | 0.529 (18/34) | NA | 0.654 (68/104) | 0.894 (42/47) |
| Study | Design | Population | Treatment | Comparator | Outcome |
|---|---|---|---|---|---|
| Radical radiotherapy | |||||
| ACENDE-RT103,174,175 | RCT n = 398 |
Intermediate–high risk. CPG 4–5 | Low-dose-rate brachytherapy + esxternal beam radiotherapy | Dose-escalated external beam radiation therapy | Local recurrence, distant metastases, OS (KM). F-u up to 10years |
| HYPRO104,176 | RCT, n = 820 | Intermediate–high risk | Hypofractionated radiotherapy | Conventional radiotherapy | OS, 7-year relapse free survival, AE F-u up to 10years |
| PROFIT105 | RCT, n = 1206 | Intermediate | Hypofractionated radiotherapy | Conventional radiotherapy | OS, biochemical failure, AE, f-u up to 5years HRQOL-48 weeks |
| CCHiP106,177 | RCT, n = 3216 | Intermediate–high risk | Hypofractionated radiotherapy | Conventional radiotherapy | OS, relapse-free survival, AE F-u up to 8 years |
| HYPO-RT-PC107,178 | RCT n = 1180 |
Intermediate–high risk | Ultra-hypofractionation | Conventional fractionated radiotherapy | Failure free survival and PCa-specific survival (5year) QoL (6years) |
| Marzi (2009)108 | RCT, n = 162 | Intermediate–high-risk Gleason 7–10 | Hypofractionated radiotherapy | Conventional radiotherapy | OS. f-u 30 months |
| Radiotherapy + ADT vs. radiotherapy alone | |||||
| Kishan, (2022)111 | IPD M-A | Intermediate–high risk | Radiotherapy + ADT (incl. as prolongation therapy) | Radiotherapy alone | Metastasis-free survival (KM) OS (KM). 11.4 years f-u. Biochemical recurrence, distant metastasis. |
| Prostatectomy vs. observation | |||||
| PIVOT109,179 | RCT | Low, intermediate and high | Radical prostatectomy | Watchful waiting | OS, PCa death, distant metastases, AEs f-u 22.1years |
| SPCG4110,180 | RCT | Localised, non-metastatic | Radical prostatectomy | Watchful waiting | Overall mortality, PCa death, distant metastases, AEs, QoL F-u: 29 years |
| Radical prostatectomy vs. radical radiotherapy vs. observation | |||||
| PROTecT55,114 | RCT n = 1643 |
Localised, non-metastatic | Radical prostatectomy, radical radiotherapy | Active monitoring | PFS, patient-centred outcomes F-u: median 10years |
| DTX and hormone-sensitive therapy | |||||
| STAMPEDE59,143 | RCT, n = 1776 | High-risk PCa (Gleason 8–10) and metastatic | ADT plus DTX and estramustine | ADT alone | OS, PFS. F-u: 6.5years |
| GETUG 1260,181 | RCT, n = 413 | High-risk PCa (Gleason 8–10) | Addition of DTX, zoledronic acid/estramustine, or both to first-line long-term hormone therapy | Long-term hormone therapy | OS, PFS F-u: 12 years |
| TAX-350161 | RCT n = 228 |
Metastatic, post-radical prostatectomy | DTX and leuprolide | Leuprolide alone | OS, PFS, AEs, f-u 3.4 years |
| Study | Patient group, enrolment period | Location | Design, interventions if RCT | Mortality and disease progression-related outcomes, maximum FU | Conclusions |
|---|---|---|---|---|---|
| Bill-Axelson (2011) (SPCG4}110 | Localised disease (1989–9) | Sweden, Finland, Iceland | RCT, watchful waiting vs. radical prostatectomy | Reported at 15 years: – all-cause mortality – PCa death – distant metastases – local progression |
Radical prostatectomy was associated with a reduction in the rate of death from PCa. |
| Wilt (2012) (PIVOT) 109 | Localised disease (1994–2002) | USA | RCT, observation vs. radical prostatectomy | Reported at 10 years: – all-cause mortality – PCa death – bone metastases |
Prostatectomy did not significantly reduce all-cause or PCa mortality. |
| James (2015) (STAMPEDE)113 | Metastatic disease (2005 and 2014) | UK and Switzerland | RCT, SOC arm (androgen deprivation therapy) |
Reported at 5 years: – failure-free survival – all-cause mortality |
Survival remains disappointing in men presenting with M1 disease who are started on only long-term androgen deprivation therapy. |
| James (2016) (STAMPEDE)59 | High-risk and metastatic disease (2005 and 2013) | As above | RCT, SOC as above vs. SOC + zoledronic acid vs. SOC + DTX, vs. SOC + zoledronic acid and DTX |
Reported at 7 years: – OS – failure-free survival |
Zoledronic acid showed no evidence of survival improvement DTX showed evidence of improved survival accompanied by an increase in AEs. |
| Clarke (2019) (STAMPEDE)143 | Metastatic disease (2005 and 2013) | As above | RCT, SOC arm (androgen deprivation therapy) |
Reported at 6.5 years: – metastatic burden – OS – failure-free survival |
The survival benefit for upfront DTX is maintained at longer follow-up. No evidence that the benefit differs by metastatic burden. |
| Hamdy (2016) (ProtecT) 55 | Localised disease (1999–2009) | UK | RCT, active monitoring vs. radical prostatectomy, vs. radiotherapy | Reported at 10 years: – adherence – PCa death – all-cause mortality – metastases – disease progression |
No significant difference among – active monitoring; – radical prostatectomy; and – radiotherapy. |
| Bryant (2020) (ProtecT)114 | As above | As above | As above | Reported at 10 years: – disease progression |
There are differences in risk categorisation between men who progressed during PROTecT and those that did not. Different grade, low/intermediate/high risk. |
| Widmark (2019) (HYPO-RT-PC)107 | Intermediate- to high-risk aPCa (2005–15) | Sweden and Denmark | –Ultra-hypofractionated vs. conventionally fractionated radiotherapy | Reported at 5 years: – failure-free survival – disease-free survival – PCa survival – OS |
Ultra-hypofractionated radiotherapy is non-inferior to conventionally fractionated radiotherapy. |
| Gnanapragasam (2016)112 | Localised or locally advanced disease (2000–10) |
UK | Observational, exploring the prognostic ability of five levels of CPG scores | Reported at 13.7 years: – PCa death Reported at 9.6 years: – all-cause mortality |
The five-stratum CPG system outperforms the standard three-stratum risk system in predicting the risk of PCa death. |
Appendix 9 Review of cost-effectiveness evidence
FIGURE 17.
PRISMA flow diagram for cost-effectiveness of SF systems review.
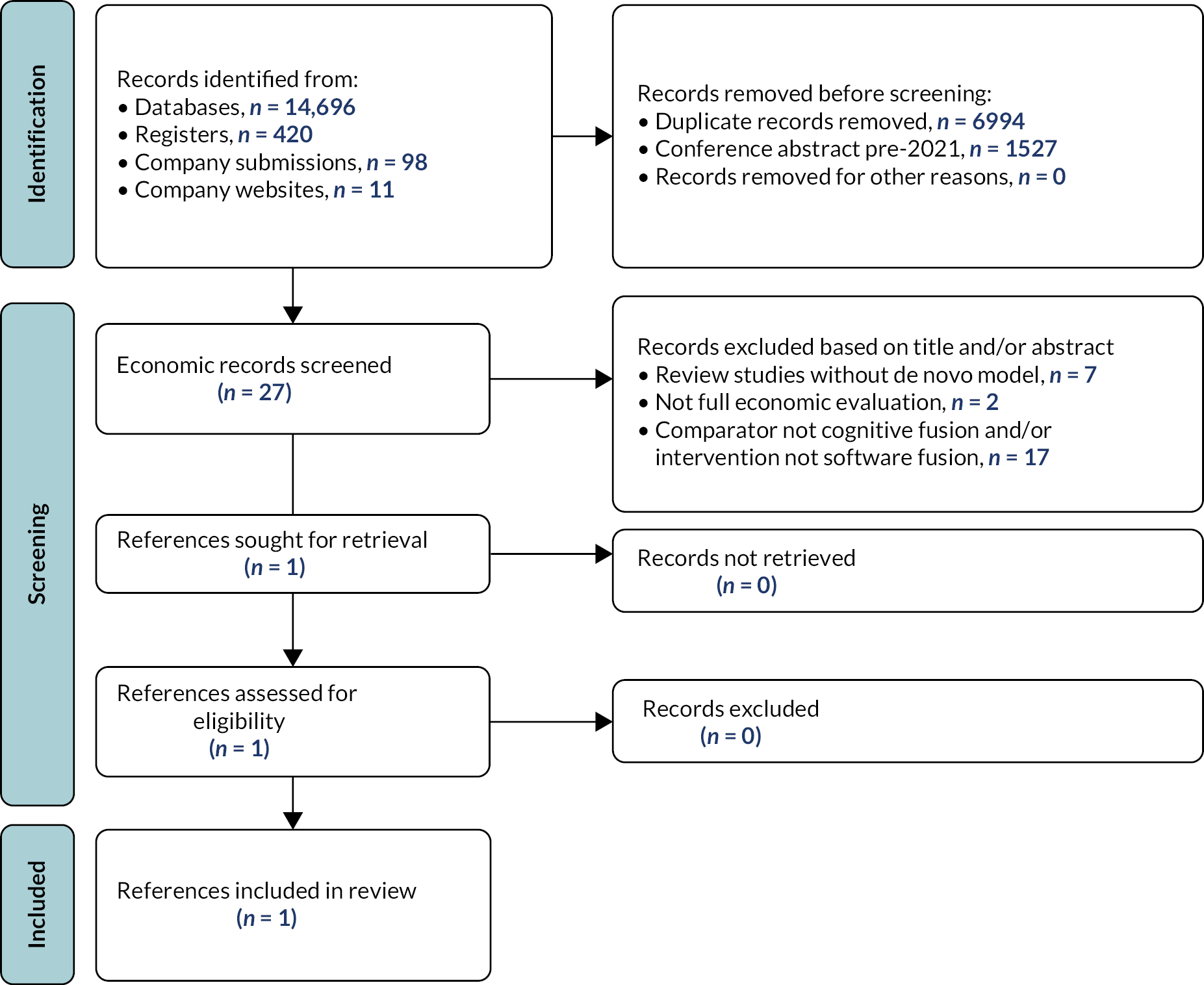
Critical appraisal of cost-effectiveness studies of MRI Fusion systems Pahwa et al. (2017)
| Response (Y, N or NA) | Comments | |
|---|---|---|
| 1. Decision problem and scope specified | ||
| Is there a clear statement of the decision problem? | Y | |
| Is the perspective of the model stated clearly? | N | |
| Has the target population been identified? | Y | |
| Are the model inputs consistent with the stated perspective? | NA | Perspective not stated clearly. |
| Are the primary outcomes of the model consistent with the perspective, scope and overall objective of the model? | NA | Perspective not stated clearly. |
| 2. Identification and description of the comparators | ||
| Have all the feasible and practical options been identified? | Unclear | Authors do not state whether there are other feasible and relevant alternatives. |
| Have the comparators being evaluated been clearly described? | Y | |
| If comparators have been excluded from the evaluation, have these exclusions been justified? | NA | |
| 3. Appropriate data identification | ||
| Are the data identification methods transparent, systematic and appropriate given the objectives of the model? | N | The data identification methods are not described. |
| 4. Sufficient detail for data incorporation | ||
| Have all data incorporated into the model been described and referenced in sufficient detail? | Y | |
| Where choices have been made between data sources, are these justified appropriately? | N | |
| Are transition probabilities calculated appropriately? | NA | Not a state transition model. |
| Has discounting been conducted? | Y | |
| 5. Quality and incorporation of test accuracy data | ||
| Has the quality of the test accuracy data been assessed? | N | |
| Have diagnostic accuracy data been derived from high quality data sources (hierarchy of evidence)? | NA | Sources of data to inform data accuracy are not described in sufficient detail to establish quality of data. |
| Are tests in sequence treated dependently, where appropriate? | N | Dependencies between tests in a sequence not modelled (implicit assumption of independence between tests in each sequence). |
| 6. Quality and incorporation of treatment data | ||
| Has the quality of the treatment effect data been assessed? | N | Linkage to long-term outcomes is done via lifetime pay-offs applied to diagnostic decision tree – relative treatment effects are not applied in the model. |
| Have relative treatment effects been derived from high-quality data sources (hierarchy of evidence)? | NA | |
| 7. Source and incorporation of cost data | ||
| Has the source of cost data been presented clearly? | Y | |
| Have costs been inflated to a specific year, where appropriate? | Y | |
| 8. Source and incorporation of utility data | ||
| Is the source for the utility weights referenced and justified? | N | Assumption that 1 LY corresponds to 1 QALY in healthy individuals (no PCa) is not supported by empirical data. |
| Are the utilities incorporated into the model appropriately? | Unclear | Most QALYs are estimated directly from an external Markov model. |
| 9. Model structure | ||
| Have the reasons behind the type of decision-analytic model chosen been fully described and justified? | N | |
| Has a systematic review of existing economic evaluations been carried out? | N | |
| Is the structure of the model consistent with a coherent theory of the health condition under evaluation? | NA | The structure of the model is not sufficiently described or depicted to assess whether it is consistent with the health condition. |
| Are the structural assumptions underpinning the model transparent and justified? | Partly | Not all assumptions are justified, and some assumptions are not explicit (e.g. independence between results of tests in a sequence). |
| Have the methods used to extrapolate short-term results to final outcomes been documented and justified? | NA | Linkage to long-term outcomes is done via lifetime pay offs applied to diagnostic decision tree. |
| Has the time horizon been stated and justified? | Y | |
| Has cycle length of Markov models been justified? | NA | Not a Markov model. |
| 10. Uncertainty | ||
| Has parameter uncertainty been addressed via sensitivity analysis? | Y | One-way sensitivity analysis. |
| Has probabilistic sensitivity analysis been carried out? If not, has this omission been justified? | Y | |
| If data are incorporated as point estimates, are the ranges used for sensitivity analysis stated clearly and justified? | Partly | The ranges used are not clearly justified for most parameters. |
| If data have been incorporated as distributions, has the choice of distribution for each parameter been described and justified? | N | Probability distributions for each parameter are not described. |
| Have structural uncertainties been addressed via sensitivity analysis? | N | |
| Have alternative assumptions related to final outcomes been explored through sensitivity analysis? | N | |
| Has value of information analysis been done? | N | |
| 11. Validity | ||
| Has the face validity been reviewed by someone external to the model developers? | N | Not described |
| Has the mathematical logic of the model been assessed? (e.g. using null and extreme values) | N | Not described |
| Have the model and its results been compared to the findings of other models and studies, and any disagreements or inconsistencies been explained (cross-validity)? | Y |
Results of the additional targeted reviews to support model conceptualisation
The searches described in Systematic review methods (study selection, data extraction, quality assessment) identified 27 titles of which 16 did not meet the inclusion criteria based on title and/or abstract. Full-text publications were obtained for the remaining 10 records. 119,120,122,124–126,129–132 In addition, economic studies were identified from a systematic review in a previous DAR by the Southampton EAG. 116 We identified five additional publications from the previous DAR economic evidence review. 121,123,127,128,133
In total, 16 titles comprising 15 cost-effectiveness models116,119–133 were considered potentially relevant to inform the de novo model conceptualisation for inclusion. We note that the Wilson et al. 121 model is structurally similar (and shares many common evidence sources) to the cost-effectiveness model developed in the context of the PROMIS trial125,126 (henceforth referred to as the PROMIS model), although it does not model the full range of strategies in PROMIS. Similarly, the Southampton DAR model116 is an extension of the model developed in the context of the 2019 update of the NICE CG131123 (henceforth referred to as the NICE CG131 model). These studies are summarised in Table 63.
The majority of identified studies aimed to evaluate the cost-effectiveness of strategies for initial PCa diagnosis involving biopsy approaches. 116,119,121,123,125,126,129–132 The study populations in some of these diagnostic studies included only biopsy-naive individuals121,125,126,129–132 while others included biopsy-naive individuals and those with a previous negative biopsy. 116,123 The population in Mowatt et al. 133 included only individuals who had had a previous negative biopsy. Three studies evaluated alternative prostate diagnostic strategies in the context of PSA based screening. 120,122,124 One study examined alternative protocols of active surveillance for those diagnosed with low-risk PCa. 127 One study examined the use of mpMRI and MRI-influenced biopsy as an alternative in the evaluation of PCa biomarkers. 128
In the majority of the identified studies a cohort simulation modelling approach using a combined decision tree and Markov model structure was applied. 116,119,121,123,125–130,132,133 In these models, the decision tree component modelled the diagnostic/screening pathway to classify individuals according to their true disease and diagnostic outcomes, while the Markov model component linked the diagnostic outcomes (and subsequent clinical management decisions) to the long-term effects on outcomes. Other cohort models relied solely on a decision tree structure129 or a Markov model structure131 to evaluate the cost-effectiveness of the alternative strategies. One cohort model was described as a partially observable Markov model,124 and distinguishes between unobservable pretreatment [or preclinical (i.e. prior to the presentation of any disease signs or symptoms)] and observable (or clinical) states. Two studies used a continuous-time microsimulation (i.e. patient level) model calibrated to registry data. These models had two main model components to reflect 1) PCa natural history (from preclinical to clinical cancer) and 2) its diagnostic and treatment pathways. 120,122
| Study: First author (year), country Study aim |
Diagnostic strategies | Definition of CSPCa | Biopsy diagnostic accuracy outcomes | Model structure and modelling approach | Evidence linkage to long-term outcomes |
|---|---|---|---|---|---|
| Souto-Ribeiro (2022),116 UK To assess the CE of LATP vs. LATRUS and GATP in men with suspected PCa for whom prostate biopsy is indicated |
First biopsy (+): treated (–): % discharged/monitored and % Second biopsy; →second biopsy (–): discharged/monitored; second biopsy (+): treated Biopsy options: First biopsy – LATP (w/wo specific freehand devices)/GATP/LATRUS; second biopsy – LATRUS |
Histopathological definition: GS ≥ 4 and/or a cancer core length ≥ 4 mm Clinical definition: - CNSPCa: GS ≤ 6, PSA ≤ 10 ng/ml and T1–T2a stage (=LR) - CSPCa: Gleason = 7, PSA 10–20 ng/ml and T2b stage (=IR); or GS ≥ 8, PSA > 20 ng/ml and ≥T2c stage (=HR) |
Probabilities of TRUS detecting CNS and CSPCa (stratified by LR, IR, HR) RRs for PCa detection rates for LATP and GATP vs. LATRUS are applied to baseline probabilities (with LATRUS) Specificity of detecting PCa |
Decision tree: classifies patients according to diagnostic accuracy, true disease status and underlying risk category. Tree also captures biopsy complications + Markov model capturing treatment allocation conditional on classification and longer-term outcomes Health states: No PCa; unDx LR; unDx IR; unDx HR; unDx metastatic; Dx LR; Dx IR; Dx HR; Dx metastatic; PCa death; other-cause death. |
Via Markov model capturing sequential disease progression from lower to higher risk category (LR→IR→HR) of localised disease and from HR to metastatic disease. PCa mortality only applies to metastatic disease. |
| Wilson (2021),121 UK To assess the CE of LATP vs. LATRUS for men at risk of PCa who are referred to secondary care investigations |
mpMRI (No/CNSPCa): discharged/monitored; mpMRI (CSPCa); →first biopsy (CSPCa): treated; first biopsy (No/CNS PCa) →second biopsy (No/CNS PCa): discharged/monitored; second biopsy (CSPCa): treated Biopsy alternatives: LATP or LATRUS |
Histopathological definition: NR Clinical definition: Text suggests LR is equivalent to CNS PCa, and IR/HR to CSPCa, but the risk categories are not defined. |
Probabilities of detecting No PCa, LR, IR or HR conditional on true disease status and previous test results (mpMRI/biopsy) Specificity of detecting PCa |
Decision tree: classifies patients according to diagnostic accuracy, true disease status and underlying risk category. Tree also captures biopsy complications and treatment allocation. + Markov model capturing longer-term outcomes Health states: no PCa (?); progression-free, metastatic disease, death |
Via Markov model capturing disease progression from localised disease to metastatic disease. PCa mortality only applies to metastatic disease. |
| Cheng (2021),119 Singapore To assess the CE of diagnostic strategies involving combined biopsy in sequences (with/wo SBx/TPMB) for men with suspected PCa based on elevated PSA and/or abnormal DRE |
Test sequence in each strategy: 1. Combined biopsy 2. Combined biopsy→(–): SBx 3. Combined biopsy→(–): TPMB 4. Combined biopsy→(–): SBx→(–): TPMB 5. SBx→(–): Combined biopsy 6. SBx→(–): Combined biopsy→(–): TPMB Where combined biopsy means: mpMRI → PI-RADS 1,2: % no biopsy and % SBx; PI-RADS 3+: Combined biopsy; individuals with biopsy(+) receive treatment |
Histopathological definition: NR; Clinical definition - CNSPCa: GS < 7, PSA < 10 ng/ml; and T1–T2a stage (=LR) - CSPCa: GS = 7, or PSA 10–20ng/ml; or T2b stage (=IR); or GS > 7, PSA > 20ng/ml; or ≥ T2c stage (=HR) |
For SBx, TBx and combined biopsy: Probabilities of detecting LR, IR, HR conditional on true disease status and prior test results For TPMB: Specificity of detecting PCa, sensitivity to detect LR, IR, HR |
Decision tree: classifies patients according to diagnostic accuracy, true disease status and underlying risk category + Markov model capturing treatment allocation conditional on classification and longer-term outcomes Health states: No PCa, unDx localised PCa, metastatic PCa, correctly Dx localised LR (3 separate treatment health states: WW, AS, RTx ± ADT) localised IR Dx LR (3 separate treatment health states: WW, AS, RTx ± ADT), correctly Dx localised LR (2 separate treatment health states: WW, RTx ± ADT), PCa death, all-cause death. |
Via Markov model capturing: - Primary treatment allocation and subsequent treatment changes - disease progression from localised to metastatic disease PCa mortality only applies to metastatic disease. |
| Hao (2021), Sweden To assess the CE of diagnostic strategies involving TBx, SBx or combined biopsy for men undergoing (or eligible for) quadrennial PSA screening |
1. No PSA screening (assumes average 2. SBx for symptomatic identification) Screening strategies If PSA ≥ 3 ng/mL: 2. SBx 3. mpMRI → PI-RADS < 3: rescreening; PI-RADS ≥ 3: TBx 4. mpMRI → PI-RADS < 3: rescreening; PI-RADS ≥ 3: Combined biopsy 5. mpMRI → PI-RADS < 3: SBx; PI-RADS ≥ 3: Combined biopsy Where individuals with biopsy (+) receive treatment, and those with biopsy (–) return to screening |
NA | FN rates conditional on the true disease status (ISUP GG1 or GG ≥ 2) Specificity of detecting PCa |
Continuous time microsimulation PCa natural history model Health states: – No PCa .Preclinical states: ISUP GG1, T1–T2; ISUP GG1, T3–T4; ISUP GG1 metastatic; ISUP GG2–3, T1–T2; ISUP GG2–3, T3–T4; ISUP GG2–3 metastatic; ISUP GG4–5,T1–T2; ISUP GG4–5, T3–T4; ISUP GG4–5, metastatic – Clinical states: ISUP GG1, ISUP GG1,T1–T2; ISUP GG1, T3–T4; ISUP GG1 metastatic; ISUP GG2–3,T1–T2; ISUP GG2–3, T3–T4; ISUP GG2–3 metastatic; ISUP GG4–5, T1–T2; ISUP GG4-5, T3–T4; ISUP GG4–5, metastatic. – Diagnosis and treatment submodel for clinical states: diagnosis; localised T1, T2, T3, T4, ISUP GG1 or GG2 + treatment (AS, RP and /or RT, post treatment follow-up), metastatic (treatment, palliative care, terminal illness) – other-cause death; PCa death |
Via microsimulation model capturing – disease onset and progression from preclinical to clinical PCa. – preclinical states reflect disease onset by ISUP GG and progression by T stage to metastatic PCa (from T1–T2→T3–T4→metastatic PCa) – disease progression in clinical states seems to be from localised to metastatic – Primary treatment allocation and subsequent treatment changes PCa mortality only applies to metastatic disease in clinical states. |
| Getaneh (2021),122 The Netherlands To assess the CE of adding mpMRI as triage test between PSA and biopsy for population-based triennial screening |
Screening strategies: 1. (not described) PSA screening protocol involving TRUS 2. PSA → PSA < 3 ng/mL: no further assessment; PSA ≥ 3 ng/mL: mpMRI → PI-RADS < 3: no biopsy; PI-RADS ≥ 3: TBx Individuals with biopsy (+) receive treatment, and those with biopsy (–) return to screening (not explicit) |
NR | TBx: – Sensitivity to detect LG and HGa PCa – Misclassification rate (HG classified as LG) TRUS: – Biopsy sensitivity (not specified whether it applies to PCa or PCa significance) – Misclassification rate (HG classified as LG) |
Microsimulation screening analysis; life history model w/wo screening Health states: – No PCa – Preclinical states: T1, GS < 7; T1, GS = 7; T1, GS > 7; T2, GS < 7; T2, GS = 7; T2, GS > 7; T3, GS < 7, T3, GS = 7; T3, GS > 7; each state can be local-regional or distant metastatic (18 health states in total) – Clinical states: T1, GS < 7; T1, GS = 7; T1, GS > 7; T2, GS < 7; T2, GS = 7; T2, GS > 7; T3, GS < 7, T3, GS = 7; T3, GS > 7; each state can be local-regional or distant metastatic; death (18 health states in total plus death) |
Via microsimulation model capturing disease onset and progression from preclinical to clinical PCa by screening or clinical diagnosis preclinical states reflects disease onset at T1–GS < 7 or T1–GS > 7; then progression by T stage (T1→T2→T3→T4) and GS (GS < 7→GS = 7→GS > 7); any state can progress from local-regional state to distant state – Disease progression in clinical state is not modelled – Primary treatment allocation based on age, T stage, GS PCa mortality only applies at clinical states. |
| NICE (2019),123 UK To assess the CE of follow-up protocols for people who have a raised PSA, MRI(–) and/or (–) biopsy |
Alternative follow-up protocols, defined according to: - Type of screening test and the related threshold (e.g. PSA derivatives); - Frequency of the screening test; - Type of biopsy if the previous test positive (e.g. TRUS or TPMB); - Stopping rule – defines the duration of follow-up for each strategy. |
Histopathological definition: GS ≥ 3 + 4 or cancer core length ≥ 4mm Clinical definition: - CNSPCa: Gleason scor < 7 or cancer core length < 4 mm or PSA ≤ 10 ng/mL (=LR) - CSPCa: GS = 7 or cancer core length ≥ 4 mm; PSA 10–20 ng/mL (=IR); or GS ≥ 8 or cancer core length ≥ 4 mm; PSA > 20 ng/mL (=HR) |
Sensitivity to detect CNS and CSPCa for SBx, and: adjusted by relative sensitivity of TBx vs. SBx, if TBx) adjusted by relative sensitivity of first vs. subsequent biopsy if second biopsy |
Decision tree: classifies patients according to diagnostic accuracy, true disease status and underlying risk category + Markov model capturing treatment allocation conditional on classification and longer-term outcomes Health states: No PCa; unDx LR; unDx IR; unDx HR; unDx metastatic; Dx LR; Dx IR; Dx HR; Dx metastatic; PCa death; other-cause death. |
Via Markov model capturing disease onset and sequential disease progression from lower to higher risk category (LR→IR→HR) of localised disease and from HR to metastatic disease. PCa mortality only applies to metastatic disease. |
| Faria (2018)125/Brown (2018),126 UK To assess the CE of combinations of mpMRI, TRUS, TPMB for the diagnosis of PCa in men referred to secondary care investigations |
383 strategies with alternative combinations of mpMRI, TRUS, and TPMB, which differ in terms of: - whether or not, and when (to guide TRUS or to inform repeat biopsy) to use mpMRI; - the type of biopsy (TRUS-guided or TPM); - whether repeat biopsy is allowed and who receives it conditional on previous test results; - definition of suspicious lesion on mpMRI (4 alternative cut-offs) - definitions of CSPCa (2 alternatives) |
Histopathological definition: 1. dominant Gleason pattern ≥ 4 and/or any Gleason pattern 5 and/or cancer core length ≥ 6mm; or 2. any Gleason pattern ≥ 4 and/or cancer core length ≥ 4 mm Clinical definition: - CNS PCa: PSA ≥ 10 ng/ml and GS ≥ 6 (=LR) - CSPCa: PSA 10-15 ng/ml and GS (=IR); or GS ≥ 8 (=HR) |
Probability of detecting PCa, CNS or CSPCa conditional on true risk category of LR, IR, HR Specificity of detecting PCa |
Decision tree: classifies patients according to diagnostic accuracy, true disease status and underlying risk category + Markov model capturing treatment allocation and longer-term outcomes Health states: no PCa(?), localised PCa, metastatic disease, death |
Via Markov model capturing disease progression from localised disease to metastatic disease. PCa mortality only applies to metastatic disease. |
| Barnett (2018),124 US To assess the CE of diagnostic strategies involving MRI and TBx (alone or combined) for men undergoing biennial PSA screening |
1. No PSA screening Screening strategies If PSA > 4 ng/mL: 2. SBx 3. MRI → PI-RADS < 3: SBx; PI-RADS 3+: TBx 4. MRI → PI-RADS < 3: no biopsy; PI-RADS 3+: TBx 5. mpMRI → PI-RADS < 3: SBx; PI-RADS 3+: Combined biopsy 6. mpMRI → PI-RADS < 3: no biopsy; PI-RADS 3+: Combined biopsy TBx performed with MRI fusion Individuals with biopsy (+) receive treatment, and those with biopsy (–) return to screening (not explicit) |
Histopathological definition: - high-volume tumour and GS 3 + 4 or GS ≥ 4 + 3 (high grade disease) Clinical definition: any GS ≥ 7 |
SBx: – Sensitivity of detecting PCa – Probability of incorrect grading for (+) biopsy TBx and combined biopsy: – sensitivity and specificity for high-grade cancer |
Partially observable Markov model capturing screening/diagnostic outcomes (via implicit decision treeb embedded in the model), treatment allocation and longer-term outcomes. Health states: – no PCa; other-cause death; – pretreatment PCa states (unobservable): organ confined GS < 7, organ confined GS = 7, organ confined GS > 7, EPLN – detected PCa: PCa treatment (AS or RP), no recurrence following treatment (NRFT), possible recurrence following treatment, metastatic PCa, PCa death. |
Via partially observable Markov model capturing: – Onset of PCa – Primary treatment allocation – Disease progression from localised to metastatic disease PCa mortality only applies to metastatic disease in detected states. |
| Patel (2018),127 Netherlands To assess the CE of AS strategies for men with LR |
1. 3-yearly SBx biopsy → biopsy (–): AS; biopsy (+): treated 2. 3-yearly mpMRI → mpMRI (–): AS; mpMRI (+): TBx → biopsy (–): AS; biopsy (+): treated 3. 3-yearly mpMRI → mpMRI (–): AS; mpMRI (+): treated mpMRI (+)/(–) defined in relation to presence of HR. All biopsies are performed via TRUS |
Histopathological definition: GS ≥ 7 Clinical definition: NR, but text suggests that LR (PSA < 10 ng/ml, GS < 6, and stage T2a) is equivalent to CNS PCa and HR (GS ≥ 7) to CSPCa |
Sensitivity and specificity of detecting HR | Markov model capturing diagnostic outcomes (via implicit decision tree embedded in the Markov model) and longer-term outcomes Health states: LR, HR, survival after treatment LR, survival after treatment HR, death (due to PCa or other causes) |
Via Markov model capturing disease progression from LR to HR. PCa mortality only applies to individuals with HR. |
| Sathianathen (2018),128 US To assess the CE of biomarkers in determining the need for biopsy in men with elevated PSA |
1. SBx 2-5. biomarker → (< cut-off): followed-up (not explicit); (≥ cut-off): SBx 6. mpMRI → mpMRI(–): followed-up; mpMRI(+): TBx Biomarkers: phi, 4Kscore®, SelectMDx™ and the EPI [ExoDx™ Prostate (Intelli-Score)] Where individuals with biopsy (+) receive treatment, and those with biopsy (–) are followed up. SBx is performed via TRUS. mpMRI (+)/(–) is not defined |
NR | Sensitivity of detecting LG and HG PCa | Decision tree: classifies patients according to diagnostic accuracy, true disease status + Markov model for Dx PCa (not described) + State transition model (not described) for unDx PCa capturing risk of clinical diagnoses due to symptoms and risk of metastasis by clinical diagnosis Health states: NR |
NR |
| Pahwa et al.,129 US To assess the CE of SBx and TBx (with alternative MRI-influence method (MRI fusion, CF or in-bore) for biopsy-naive men with elevated PSA and /or CS DRE |
1. SBx 2–4. mpMRI→(no suspicious lesions): discharged; (suspicious lesions): TBx 5–7. mpMRI→(no suspicious lesions): SBx; (suspicious lesions): TBx Where individuals with biopsy (+) receive treatment, and those with biopsy (–) are discharged. All biopsies are performed via TRUS |
Histopathological definition(?): CNS PCa: GS < 6 and tumour volume < 0.5 cm3 Clinical definition: NR, but text suggests that CNS PCa is equivalent to LR and CSPCa to HR |
Sensitivity for detecting PCa, CNS and CSPCa Specificity for PCa Probability of correctly classifying tumour aggressiveness |
Decision tree classifies patients according to diagnostic accuracy, true disease status and allocates primary treatment | Via lifetime health and cost payoffs conditional on diagnostic status (diagnosed/missed), primary treatment, and age Pay-offs are informed by outcomes of an external Markov model (supplemented with assumptions for patient management options not examined in the external model) |
| Venderink (2017),130 Netherlands To assess the CE of SBx and TBx (with alternative MRI-influence method used (MRI-fusion or in-bore) for biopsy-naive men with elevated PSA and /or abnormal DRE |
1. SBx 2–3. mpMRI → (no suspicious lesions): discharged; (suspicious lesions): TBx (2. MRI fusion and 3. In-bore) Where individuals with biopsy (+) receive treatment, and those with biopsy (–) are discharged. All biopsies are performed via TRUS |
Histopathological definition(?): GS ≥ 3 + 4 high-volume (IR/HR). | Sensitivity to detect CNS and CSPCa Specificity for PCa Probability of false CNS and CS |
Decision tree: classifies patients according to diagnostic accuracy, true disease status (CS and CNS PCa) and treatment allocation + Markov model capturing longer-term outcomes Health states: No PCa, status after RP, status after RT, status after AS, death |
Via Markov model capturing long-term outcomes |
| Cerantola (2016),131 Canada To assess the CE of using MRI and TBx for biopsy-naive men with elevated PSA and abnormal DRE |
1. SBx 2. mpMRI → (PI-RADS < 3): followed-up; (PI-RADS ≥ 3): TBx Where individuals with biopsy (+) receive treatment, and those with biopsy (–) are followed-up. SBx is performed via TRUS |
NR CSPCa is not defined but manuscript suggests that it is equivalent to IR/HR |
TBx: - Rate of biopsy (+) - Rate of CS among biopsy (+) SBx: - Rate of biopsy (+) - Rate of FN - Rate of CS among biopsy (+) |
Markov model capturing diagnostic (via implicit decision tree embedded in the Markov model) and longer-term outcomes Health states: two set of health states 1. mpMRI, TBx, 2. SBx, SBx(+), (1) or (2) plus follow-up, LR PCa, IR/HR PCa, AS, curative treatment, biochemical recurrence, CRPC, PCa death, other-cause death |
Via Markov model capturing: 1. biopsy alternatives: TBx or SB (+) 2. biopsy outcomes: No PCa (captured in follow-up), LR PCa, HR PCa; 3. Primary treatment allocation; 4. disease progression from localised disease (LR, IR/HR to relapse) to metastasis (CRPC) PCa mortality only applies to metastatic disease. |
| de Rooij (2014),132 The Netherlands To assess the CE of using MRI and TBx for biopsy-naive men with elevated PSA |
1. SBx 2. mpMRI → (no suspicious lesions): followed-up; (suspicious lesions): TBx re individuals with biopsy (+) receive treatment, and those with biopsy (–) are followed-up. SBx is performed via TRUS. |
Histopathological definition: CNS PCa: GS ≥ 3 + 4 or large tumour with GS 3 + 3 | Sensitivity and specificity for detecting PCa Probability of correctly classifying tumour aggressiveness |
Decision tree: classifies patients according to diagnostic accuracy, true disease status (CS and CNS PCa) and treatment allocation + Markov model capturing longer-term outcomes Health state: alive, dead |
Via Markov model capturing long-term outcomes |
| Mowatt (2013),133 UK To assess the CE of using alternative MRS/MRI sequences to target TRUS biopsy, compared with SBx in individuals with suspected PCa and a previous (–) biopsy |
1. SBx (extended (14–16) core TRUS) 2. MRI/MRS → MRI/MRS (–): followed-up; MRI/MRS (+): TBx 3. MRI/MRS → MRI/MRS (–): SBx; MRI/MRS (+): TBx Individuals with biopsy (+) receive treatment, and those with biopsy (–) follow-up (with a repeat saturation at 12 months if FN) |
NA | Sensitivity and specificity of detecting PCa | Markov model capturing diagnostic (via implicit decision tree embedded in the Markov model) and longer-term outcomes Health states: No PCa or undetectable PCa, Dx localised T1–2 PCa (LR), Dx localised PCa (IR), Dx localised PCa (HR), Dx locally advanced T3 PCa (or extraprostatic cancer), unDx localised T1–2 PCa (LR), unDx localised PCa (IR), unDx localised PCa (HR), unDx locally advanced T3 PCa, Dx metastatic PCa, PCa death, other-cause death |
Via Markov model capturing PCa onset, and disease progression from 1) localised to metastatic PCa, and 2) from locally advanced to metastatic PCa. PCa mortality only applies to individuals with metastatic cancer. |
The biopsy diagnostic outcomes applied across studies allow classification of patients according to the presence of PCa alone (i.e. no PCa/PCa),133 or based on disease presence and its clinical significance (i.e. clinically nonsignificant/significant PCa). 127,129,130 One study classified patients according to ISUP grades into three categories: no PCa (ISUP grade 0), PCa of ISUP grade 1, or 2 and higher. 120 It is worth noting, that biopsy results only provide histopathological information, usually expressed in terms GS and/or pattern (or as ISUP grade) and maximum core length. However, ascertaining the disease clinical significance for the purposes of guiding patient management requires knowledge of further prognostic information (e.g. T stage and PSA levels), as more radical treatment is only indicated for cancer with worse prognosis (i.e. those likely to progress at a quicker rate from localised to metastatic disease). The definition of clinical significance applied in the models to classify individuals according to biopsy results is based on the histopathological definition of clinical significance only. The full clinical definition of disease significance which is applied in the models to select patient management is conditional on biopsy results and other prognostic information. Establishing a link between histopathological and clinical definitions of disease significance (usually requiring judgements on how to map across definitions and/or risk stratification) is thus a feature of most models. However, not all studies make a clear distinction between the two types of definitions of clinical significance. 121,127,129,130,132 In some studies, the definition of clinical significance is not provided. 122,128,131
Some studies116,119,121,123,125,126 further classify PCa according to three-tier cancer risk classifications, which are generally similar (generally low-risk, intermediate-risk, high-risk PCa) despite some minor differences across classification in how each category is defined. While the exact definition of the risk categories varies across studies, individuals with PCa are in general assigned to a risk category on the basis of their PSA levels, histopathological presentation and disease (T) stage.
In the majority of identified studies, the link between diagnostic outcomes and subsequent treatment choice was established via a Markov or partially observable Markov model component. 116,119,121,123–133 The structure of most of these models allows capturing disease progression to metastatic disease116,119,121,123–127,131 or high-risk disease. 127 The model by Barnett et al. (2018) allowed for progression in patients with undetected PCa (preclinical states) across health states defined by GS and whether disease localised, and from any of these states to metastatic disease. For patients with detected PCa who underwent radical prostatectomy the progression to metastatic disease was done via a cancer recurrence health state. 124 In all of these disease progression models, PCa mortality only applies to individuals with metastatic disease116,119,121,123–127,131,133 or high-risk disease. 127 Two Markov models did not consider disease progression, with long-term outcomes directly conditioned on true disease status, diagnostic status (diagnosed or undiagnosed cancer) and primary treatment received. 130,132
Hao et al. 120 and Getaneh et al. 122 modelled disease progression (and onset) within a calibrated microsimulation model. In Hao et al. 120 disease progression occurred sequentially from disease stage T1–T2 to T3–T4 and from T3 to T4 to metastatic disease in preclinical states and from localised to metastatic disease in clinical states. PCa mortality only applied to individuals with metastatic disease in clinical states. In Getaneh et al. 122 disease onset was assumed to imply a T1 tumour stage; disease progression would occur sequentially from the T1 stage to T2, and from this to T3. At each tumour stage, individuals also progressed across GSs (lower than 7 → equal to 7 → > 7). Individuals in each preclinical state could progress from local-regional to distant metastasis, but PCa mortality only applied to individuals in clinical states.
In one study, long-term outcomes were quantified by the decision tree alone, which assigned lifetime QALY and cost pay-offs to each terminal node, conditional on true disease status, diagnostic status (diagnosed or missed) and allocated treatment. 129
Of the 16 studies identified at the first stage of the review, 9 were selected for a more in-depth review, as these were identified as the most appropriate to support the conceptualisation of the de novo model given the relevance of:
-
the comparisons and position in the diagnostic pathway – studies which compared biopsies conducted with MRI-influence methods (i.e. targeted and/or combined biopsies) for PCa diagnosis119,120,124,129,130
Although Mowatt et al. 133 were considered to have UK-policy relevance, it was not considered for the second stage of this review, given that diagnostic accuracy in this study only allowed classifying individuals according to PCa presence. Therefore, the evidence linkage in this study is unlikely to be suitable for the current decision problem, as the choice of PCa management needs to be linked as a minimum to some level of prognostic information (e.g. clinical significance of disease).
Studies included in the model conceptualisation review
Table 64 summarises the subset of identified studies included in the model conceptualisation review. A detailed description is provided next.
Population
The population in the majority of studies comprises individuals with suspected PCa who enter a secondary care diagnostic pathway,116,119,121,123,125,126,129,130 while other studies consider patients being screened for PCa. 120,124
Some of the studies on patients with suspected PCa consider a single homogeneous population in terms of disease (and CS disease) prevalence,129,130 others model different baseline populations defined by their diagnostic story (MRI results, number of previous biopsies)116,123 and underlying cancer risk category. 116,123,125,126 One study further considers subgroups defined by age brackets, with increased disease prevalence for older individuals (but the same CS prevalence for all subgroups). 129
Hao et al. 120 considered a population eligible for PSA-based PCa screening. The manuscript mentions that individual heterogeneity is considered in the natural history model (informed by Swedish registry data) but does not clearly state which individual characteristics are modelled beyond PSA levels.
Biopsy approaches
A variety of biopsy approaches were compared in the studies; these differ by route of access (transrectal vs. transperineal), type of anaesthesia used (general vs. local anaesthesia), sample collection method (targeted vs. systematic vs. mapping or saturation biopsy) and MRI-influenced methods (SF, CF, and in-bore MRI).
In the studies, which compared alternative MRI-influenced methods with each other, one compared MRI followed by targeted biopsy approaches for those who tested positive on imaging with (1) all three129 or (2) just two methods (in-bore and SF)130 versus systematic biopsy (without prior MRI) for all patients. None of these studies specified the SF system modelled.
The study by Cheng et al. 119 evaluated sequences of prostate biopsies with alternative combinations of (1) systematic, (2) template mapping and (3) combined targeted and systematic biopsy. The MRI-influenced method used for the combined biopsies was not specified. Another study considered a wide number of diagnostic strategies for patients with suspected PCa, which included systematic, targeted and template mapping biopsies. 125,126 No MRI-influenced method was specified for the targeted biopsy approaches in either study.
Two other studies compared diagnostic strategies with a MRI-influenced component (targeted alone or combined with systematic biopsy) versus systematic biopsy, but in the context of PSA-based screening. 120,124 One study120 did not specify whether MRI-influenced biopsies were performed with SF, CF or in-bore methods. In the other study124 MRI-influenced biopsies were conducted with SF, but the technology used was not specified.
| Study: First author (year), country Type of model |
Population | Biopsy approaches modelled | Classification (via biopsy diagnostic accuracy) | Choice component | Evidence linkage to longer-term outcomes | |
|---|---|---|---|---|---|---|
| PCa | No PCa | |||||
| Souto-Ribeiro (2022),116 UK Diagnostic |
Main population: Biopsy-naive individuals with mpMRI Likert3 + for suspected localised PCa. Other populations: biopsy-naive mpMRI Likert 1,2; previous negative biopsy and mpMRI Likert 3+; previous negative biopsy and mpMRI Likert 1,2 |
LATP vs. LATRUS vs. GATP biopsy Repeat biopsy: with LATRUS for a proportion of those diagnosed as No PCa or CNS PCa (max: 1) |
No PCa CNS PCa CSPCa |
– No PCa: discharge if true negative (TN); PSA monitoring if FN – CNS PCa: either AS or radical treatment – CSPCaa: – intermediate risk: offered radical treatment, with option of AS; %WW (if no curative intent) – high risk: % Radical treatment; %WW (if no curative intent) – Metastatic PCa: ADT ± Chemo |
Intermediate outcome: disease progression to metastatic disease – varies by underlying true risk category and being diagnosed as having CS or CNS PCa Survival: metastatic disease, diagnostic status of metastatic disease, age HRQoL: metastatic disease, age, AEs from treatment Costs: disease spread, age, diagnostic status, treatment received, EoL |
Surv: Age HRQoL: NR Costs: Monitoring |
| Wilson (2021),121 UK Diagnostic |
Individuals with suspected PCa presenting for mpMRI | LATP vs. LATRUS biopsy Repeat biopsy: all diagnosed no PCa at previous biopsy (max: 1) |
No PCa CNS PCa CSPCa |
– No PCa: discharged back to primary care – CNS PCa: AS – CSPCaa: intermediate or high risk: AS or radical prostatectomy |
Intermediate outcome: disease progression to metastatic disease – varies by underlying true risk category and treatment received Surv: metastatic disease, age HRQoL: metastatic disease, age Costs: treatment received |
Surv: Age HRQoL: Age Costs: NR |
| Cheng (2021),119 Singapore Diagnostic |
Biopsy-naive individuals with elevated PSA level and/or abnormal DRE findings | Combined vs. systematic (12-core) vs. saturation (20-core) biopsy Repeat biopsy: all diagnosed no PCa at previous biopsy (# of repeat biopsies is strategy dependent, max: 2) |
No PCa CNS PCa CSPCa |
– No PCa: monitoring – CNS PCa: AS, WW or radical treatment – CSPCaa: intermediate or high risk: WW or radical treatment. WW only offered if no curative intent |
Intermediate outcome: disease progression to metastatic disease – varies by underlying true risk category and diagnostic status Surv: metastatic disease, age HRQoL: metastatic disease, castration-resistant disease, age, treatment, underlying true risk category Costs: metastatic disease, castration-resistant disease; treatment received, EoL |
Surv: Age HRQoL: Age Costs: Monitoring |
| Hao (2021),120 Sweden Screening + diagnostic |
Men eligible (55–69 years old) for quadrennial PSA screening of PCa | Targeted biopsy vs. systematic biopsy vs. combined biopsy Repeat biopsy: not modelled as part of the diagnostic component |
ISUP GG0 ISUP GG1 ISUP GG ≥ 2 |
–ISUP GG0: return to screening –ISUP GG1 and GG2+: AS or radical prostatectomy and/or radiation therapy Metastatic PCa: metastatic drug treatment Treatment allocation also seems to consider disease stage at diagnosis (T1–T2, T3–T4). |
Intermediate outcome: disease progression to metastatic disease – varies by underlying ISUP GG and T stage and diagnostic status Surv: Metastatic disease, other factors NR HRQoL: metastatic disease, age, treatment and time since treatment initiation received, being diagnosed, EoL Costs: treatment received, EoL |
Surv: NR HRQoL: NR Costs: NR |
| NICE (2019),123 UK Diagnostic |
Individuals with raised PSA, negative MRI and/or a previous negative prostate biopsy | TPMB vs. TRUS Repeat biopsy: no consecutive biopsies allowed |
No PCa CNS PCa CSPCa |
– No PCa: monitoring (tests and testing schedule differ across strategies) – CNS or CSPCaa: mix of AS, brachytherapy, hormone therapy, radical prostatectomy, external radiotherapy with the distribution of treatments varying by underlying category of risk (low, intermediate or high risk). – Metastatic PCa: ADT ± Chemo |
Intermediate outcome: disease progression to metastatic disease – varies by underlying true risk category and being diagnosed as having CS or CNS PCa Surv: Metastatic disease, diagnostic status of metastatic disease, age HRQoL: metastatic disease, age, AEs from treatment Costs: disease spread, age, diagnostic status, treatment received, EoL |
Surv: Age HRQoL: Age Costs: NR |
| Faria (2018)125 and Brown (2018),126 UK Diagnostic |
Biopsy-naive individuals with suspected localised PCa | TRUS vs. TPMB Repeat biopsy: who receives it (No PCa or CNS PCa) varied by strategy (max 1) |
No PCa CNS PCa CSPCa |
– No PCa: follow-up primary care – CNS PCa: AS – CSPCa: intermediate or high-risk radical prostatectomy |
Intermediate outcome: disease progression to metastatic disease – varies by underlying true risk category and treatment received Surv: metastatic disease, age HRQoL: metastatic disease, age Costs: treatment received |
Surv: Age HRQoL: Age Costs: NR |
| Barnett (2018),124 US Screening + diagnostic |
Men eligible (55–69 years old) for annual PSA based screening of PCa | TRUS systematic vs. TRUS MRI fusion vs. TRUS combined biopsy Repeat biopsy: not modelled in the diagnostic component |
No PCa CNS PCa CSPCa |
– No PCa: routine screening – CNS PCa: if GS ≤ 6 – % AS, % radical prostatectomy; – CSPCaa: if GS ≥ 7 – radical prostatectomy; if PSA > 20 ng/mL or a Gleason score ≥ 8 – bone scan and a CT scan for staging – PCa (CNS and CS) and age > 80 years: WW |
Intermediate outcome: disease progression to metastatic disease– varies by treatment received and indirectly by location of disease (organ confined vs. extraprostatic or with lymph node) Survb: metastatic disease, age, HRQoL: metastatic disease; being diagnosed; treatment received and time since treatment initiation time post radical prostatectomy, EoL Costs: disease spread, treatment received, EoL by age |
Surv: Age HRQoL: Age Costs: Monitoring |
| Pahwa et al.,129 US Diagnostic |
Biopsy-naive patients with elevated PSA level/abnormal DRE findings. Subgroups: 41–50, 51–60, 61–70 years old |
Systematic TRUS, targeted CF, targeted MRI-fusion, targeted MRI in-bore. Repeat biopsy: not modelled |
No PCa CNS PCa CSPCa |
– No PCa: NR – CNS or CSPCa: mix of AS, WW, radiation therapy, brachytherapy, prostatectomy, ADT; treatment distribution varies by diagnosed clinical significance with a higher proportion of more aggressive treatment assumed for CSPCa |
Surv: Diagnostic status, age, treatment type, underlying true disease status (including clinical significance) HRQoL: being diagnosed, age, treatment received and underlying true disease status (including clinical significance) Costs: diagnostic status, treatment received and underlying true disease status (including clinical significance) |
Surv: Age HRQoL: NR Costs: NR |
| Venderink (2017),130 The Netherlands Diagnostic |
Biopsy-naive patients with elevated PSA level/abnormal DRE findings | Systematic TRUS, targeted TRUS MRI-fusion, targeted in-bore MRI biopsy Repeat biopsy: not modelled |
No Pca CNS PCa CSPCa |
– No PCa: NR – CNS or CSPCa: mix of AS, WW, radiation therapy, brachytherapy, prostatectomy, ADT; the distribution of treatments varies by diagnosed clinical significance with a higher proportion of more aggressive treatment assumed for CSPCa |
Surv: diagnostic status, treatment received, and underlying true disease (including clinical significance) HRQoL: being diagnosed, treatment received and time since treatment initiation Costs: treatment received |
Surv: Age HRQoL: NR Costs: NR |
The type of anaesthesia under which biopsies are performed is only specified for the studies which focus their comparison on transperineal vs. transrectal biopsy approaches. 116,121 One assumes local anaesthesia for all biopsied patients regardless of biopsy route of access,121 while the other considers local anaesthesia for those biopsied via the transrectal route and either general or local anaesthesia for TP. 116
Souto-Ribeiro et al. 116 a previous DAR by the Southampton EAG, established two main comparisons between biopsy approaches: (1) LATP biopsy (with any type of biopsy device) versus local anaesthesia transrectal (LATRUS) biopsy and GATP biopsy and (2) LATP with specific freehand devices versus LATRUS and versus transperineal transrectal biopsy conducted with a grid and stepping device conducted under local or general anaesthetic.
The NICE CG131 model123 evaluated alternative follow-up strategies of individuals with suspected PCa and placed little emphasis on alternative biopsy approaches. The main analysis presented results only for strategies which used transrectal biopsy, although strategies with transperineal mapping biopsy were considered in extended analyses only.
Another feature of the biopsy approaches modelled is whether repeat biopsies were allowed, the number of subsequent biopsies modelled and who would receive these. In the studies which considered the possibility of repeat biopsies, this has been modelled in the following ways:
-
All patients with a no PCa diagnosis at previous biopsy were assumed to receive repeat biopsy with a maximum of one repeat biopsy allowed in the model (assumption not justified). It is not clear whether the repeat biopsy would follow the same biopsy approach as the index biopsy for all strategies, as only one strategy is fully illustrated. 121
-
All patients with a no PCa diagnosis at previous biopsy were assumed to receive a repeat biopsy, in the subset of strategies allowing repeat biopsy. 119 Strategies were defined in terms of the number of repeat biopsies allowed (up to a maximum of 2) and on the sample collection method (combined, systematic or saturation) conditional on the method of the previous biopsy in the testing sequence. Repeat biopsies were assumed to always follow a sample collection method different from the one in previous biopsies in the testing sequence.
-
A proportion of patients with a no PCa or CNS PCa diagnosis receive one repeat biopsy with LATRUS (regardless of biopsy approach for the index biopsy). 116 The proportion of patients receiving a repeat biopsy was informed by the literature (single-centre observational study comparing TRUS, LATP and GATP biopsy) for the biopsy-naive populations, and by assumptions for those with previous biopsies (a lower proportion of repeat biopsy was assumed for the latter population). While the proportion of repeat biopsies was assumed to be the same across biopsy approaches in the base-case analysis for LATP, GATP, LATRUS, this assumption was relaxed in scenario analysis where LATRUS was assumed to result in more repeat biopsies than the TP approaches (LATP and GATP).
-
Repeat biopsy was allowed across most strategies but depending on the strategy the biopsy would be performed in those diagnosed at index biopsy with (1) NC, (2) CNS cancer or both NC and CNS cancer. The type of biopsy approach (template mapping, systematic or targeted) would also vary across strategy, but no strategy allowed more than one repeat biopsy. 125,126
Some studies did not model the possibility of repeat biopsy. 129,130 In other studies, the possibility of repeat biopsy was not modelled within the diagnostic component of the strategies, but repeat biopsies for individuals who returned to screening and were identified again for biopsy via screening. 120,124 The NICE CG131 model also did not consider consecutive biopsies in the diagnostic strategies. 123 All individuals with a ‘no cancer’ biopsy result returned to follow-up, but individuals could receive more than one biopsy if they tested positive again to the screening tests in their follow-up protocol.
Classification
In most studies, the diagnostic accuracy of the biopsy procedure classifies individuals as not having PCa or having non-CS or CSPCa. 116,119,121,123–126,129,130 The exception was the study by Hao et al. in which classification is done by ISUP grade. 120 Both types of classification are usually defined by histopathological features of the biopsied lesions (graded according to GSs).
The specificity of biopsy to detect PCa is assumed perfect across most models, so individuals without PCa cannot be misclassified as having the disease. However, studies differ in terms of other types of misclassification allowed for patients tested with biopsy procedures. Misclassification types allowed in the studies via both the structure and the parameterisation of the diagnostic accuracy for the biopsy approach include:
Choice of clinical management
Decisions on patient management at diagnosis could be determined by the biopsy diagnostic outcomes alone125,126,129,130 or with other factors also influencing treatment allocation. 116,119–121,123,124
In three models125,126,129,130 patient management was attributed according to individuals’ classification in terms of disease presence and clinical significance of disease. This classification was established based on the diagnostic accuracy of the biopsy approaches.
Some models tracked the individuals’ underlying cancer prognostic risk and used this information jointly with the diagnostic outcomes to allocate treatment. For example, the Southampton DAR model116 allocated treatments based on disease presence, clinical significance of disease and underlying cancer risk distribution. In order to classify patients according to these factors, the model stratified individuals with PCa into three cancer risk categories (low, intermediate, and high risk) according to the lesion’s GS, disease stage and PSA levels in separate diagnostic sub-decision trees for individuals in each risk category (plus a sub-decision tree for individuals without PCa). Low-risk disease was assumed to correspond to CNS disease (as determined by the diagnostic accuracy – that is based on GS alone), and intermediate- and high-risk disease to CS disease.
Disease spread at diagnosis (localised vs. metastatic) was also considered a factor for treatment allocation in some studies,116,120,123 which assumed that a proportion of individuals in the baseline population would have metastatic disease and, if disease was detected, received treatment with chemotherapy and/or androgen depleting therapy.
One study considered age and PSA levels alongside GS to determine PCa treatment allocation. 124 Patients older than 80 years old diagnosed with PCa of any clinical significance were treated with watchful waiting. Patients diagnosed with CS cancer and PSA levels higher than 20 ng/mL or GS >8 would undergo tests for staging purposes. It is not clear how treatment was then allocated conditional on the results of staging.
In the model by Cheng et al. 119 treatment allocation was determined by diagnosed disease clinical significance, age (with palliative care for those 75 years old or older) and cancer risk category. Although the text suggests that the distribution of treatments varies by diagnosed risk category, it is unclear how this is done since the biopsy only classifies patients according to clinical significance.
In summary, for patients diagnosed with PCa, the primary treatment allocation was conditional on:
-
diagnosed clinical significance of disease, true cancer risk category and disease spread;116,123
-
GS, PSA level and age;124
-
type of biopsy (targeted or systematic), cancer risk category and age. 119
In one study, the mechanism of treatment allocation for patients with diagnosed with cancer was not clear, but it may have been conditioned by ISUP grade (established by the biopsy diagnosis accuracy), disease T stage and spread. The manuscript suggests that the treatment pathways were informed by Swedish registry data, but does not describe how this was done. 120
A range of evidence sources were used to inform the distribution of treatments for diagnosed PCa. Amongst these the following are relevant in the UK context:
-
the Southampton DAR model116 based treatment distribution by risk category on UK clinical guidance and observed treatment allocation from national audit data;134
-
the NICE NG131 model123 used observed primary treatment distributions by risk category from UK registry data;112
-
the PROMIS trial125,126 assumed that treatment choice was guided by diagnosed disease clinical significance alone.
Individuals diagnosed as not having PCa were discharged to follow-up,121,123,125,126 or returned to the screening schedule. 120,124 One study,116 conditioned the individuals’ subsequent management after a no PCa diagnosis on whether they had been misclassified (TN results led to discharge and FN results [patients with PCa of any risk category] to routine PSA monitoring). This assumption was not justified and it is not clear how in clinical practice the two groups of individuals (TN and FN) would be distinguished so that distinct treatment decisions could be made for each group.
Outcomes
The evidence linkage approaches applied in the identified studies to connect patient classification and subsequent treatment choices with longer-term outcomes differed in whether PCa progression was explicitly modelled as an intermediate outcome or not.
Only two studies did not model disease progression. 129,130 Pahwa et al. 129 conditioned lifetime QALYs and cost pay-offs on diagnostic status (i.e. whether cancer had been diagnosed or remained undiagnosed), underlying true disease status (no PCa, CNS or CSPCa) and type of treatment received. The model applied a life-expectancy multiplier, to adjust payoffs according to alternative starting ages (scenario analysis). The lifetime pay-offs were mainly derived from an external Markov model118 comparing alternative treatments for patients with low-risk localised PCa. The long-term Markov model in Venderink et al. 130 only allowed for transitions from alive to death states. Individuals with PCa health states were defined in terms of the primary treatment received (status after 1) active surveillance, (2) radical prostatectomy or (3) radiotherapy) or no treatment (for those who had been misclassified as not having cancer). In these patients, survival was conditional on type of treatment received and the underlying true disease clinical significance, with the diagnostic status (diagnosed vs. undiagnosed cancer) determining whether individuals received treatment. 130 In both these models, treatment had a direct impact on survival. 129,130
All other models considered disease progression from localised to metastatic disease, although health states and possible state transitions varied across models. 116,119,121,123–126 Some studies modelled progression from localised to metastatic disease, and conditioned disease progression on underlying risk category and being correctly diagnosed/treatment received. 119,121,125,126 Other studies modelled sequential disease progression across disease risk categories (from low to intermediate-risk and from the latter to high-risk disease) for localised disease followed by progression from the high-risk localised to metastatic disease. In these models, the probabilities of transitioning to later disease stages were conditioned on the underlying true disease status (including risk category) and being diagnosed as having CS or non-significant disease. 116,123 The screening studies modelled progression differently in the preclinical and clinical states. 120 In the microsimulation model,120 individuals with PCa could transition between preclinical states defined in terms of ISUP grade, tumour stage and metastasis; within each ISUP grade individuals progressed sequentially from stage T1–T2 to T3–T4 and from T3 to T4 to metastatic disease. In the clinical states (for those whose PCa was detected) disease progression occurred from localised to metastatic disease. In the partially observed Markov model,124 disease progression in the preclinical states could occur (1) sequentially between three localised disease health states defined according to GS (<7, =7, >7) (2) from any of the localised disease states to extra-prostatic or lymph node-positive cancer, or (3) from any of the preclinical states to observable (clinical) metastatic cancer. The rate of progression to metastatic cancer was the same for all pre-clinical states. In the clinical states, patients treated with radical prostatectomy could transition to one of the two post-treatment states: no recurrence following treatment (NRFT) or possible recurrence following treatment (PRFT) health states. Progression to metastatic cancer was only possible for individuals in the PRFT state, with those in the NRFT state assumed cured. The probability of transitioning from the PCa treatment health state to the post-treatment states was conditional on disease location (organ confined vs. extra prostatic or lymph node-positive cancer) and treatment received. Patients who were treated with active surveillance could progress to metastatic disease at the same rate as those who were untreated, unless they transitioned to surgical treatment. The model appears to track progression over time across GSs and disease location for those under active surveillance, in a manner similar to what happened in the pre-clinical states.
All the disease progression models shared the assumption that PCa mortality only applied to patients with metastatic disease. Treatment for patients identified as having cancer reduced disease progression to metastatic cancer compared to untreated patients, and thus reduced the probability of dying from PCa for these patients. The transition probabilities for treated and untreated patients in the Markov disease progression were estimated by calibration or partially observable Markov model decision processes (as progression is an unobservable process). The data sources and calibration methods used to estimate these transition probabilities differed across models, and are reviewed below.
The PROMIS model125,126 calibrated the probability of progressing from localised to metastatic disease by risk category and treatment received, combining risk-stratified survival data and proportion of patients with metastases from the PCa Intervention versus Observation Trial (PIVOT),109 with the mortality in the metastatic subgroup of the STAMPEDE trial. 113 The PIVOT observation arm was used to inform the transition probabilities for individuals with PCa who did not receive active treatment (due to correct classification on misclassification depending on the risk category). The PIVOT radical prostatectomy arm was used to inform the transition probabilities for those treated with active treatment (true positives with intermediate- and high-risk cancer). The ‘treatment’ effects of being diagnosed on disease progression were thus informed by randomised comparative efficacy evidence.
The models which disaggregated disease progression by cancer risk categories, also used calibration to estimate transition probabilities. 116,123 The calibration method estimated transition probabilities first for the transition from high-risk to metastatic disease, then from intermediate- to high-risk disease, and finally from low-risk to intermediate-risk disease can be derived. The calibration was done separately for the undetected and detected cancers using different data sources. Transition probabilities for the undetected cancers used cumulative metastases risk rates by cancer risk category from the watchful waiting arm in the Scandinavian Prostate Cancer Group Study Number 4 (SPCG4) trial135 jointly with and Swedish life-table data (from 1999 to reflect background mortality in the trial). For the diagnosed cancers, the data sources for calibration included: cancer-specific survival by risk category sourced from a UK registry study,112 all-cause survival for people with metastatic PCa from the STAMPEDE trial,59 and UK life-table (from 2010 to 2012 to reflect background trial mortality in STAMPEDE). Thus, this calibration approach relies on an indirect naive comparison to derive the ‘treatment’ effects of being diagnosed on disease progression, which may introduce bias on the probabilities of disease progression used in the model.
The screening model by Hao et al. 120 does not describe the calibration method used to parameterise disease progression transitions, mentioning only that the model is calibrated to UK registry data. The other screening model124 also used calibration to estimate the transition probability from localised and extra-prostatic or lymph node-positive cancer (preclinical states). The authors varied the metastasis rate in 10-year periods and calibrated the values so that the resulting age-dependent risk of PCa-specific death under routine screening matched the values estimated from historical US cancer registry data. For the clinical states, the authors state that the probability of transitioning from recurrent to metastatic disease was informed by another US cancer registry data and using the methodology of an external partially observed Markov model. It is not clear how the methodology described for the external model was applied in the model developed by Barnett (2018). It is also not clear why the transition probabilities for the preclinical and clinical states were estimated by two different methods (i.e. calibration and partially observed Markov decision process).
One model119 does not describe how transition probabilities were estimated and does not fully report the data sources used to inform these parameters.
In general disease progression models, survival outcomes for individuals with PCa were conditional on having metastatic disease and age. Two models116,123 further conditioned mortality on whether metastatic disease was diagnosed (and therefore, received treatment for metastatic cancer) or not. Metastatic mortality data sources of relevance to the UK context include different publications of the STAMPEDE study, a UK-based trial which compared the survival outcomes of men with newly diagnosed metastatic, high-risk or node-positive cancer treated with alternative cancer treatments. The PROMIS and related models estimated the probability of metastatic death using early (median follow-up of 20 months) survival data of men with newly diagnosed metastatic PCa from the control arm (who received SOC consisting of androgen depleting therapy) of the STAMPEDE trial. The NICE NG131 and related models used a later survival data cut (median follow-up 43 months) from the DTX and control arms of the STAMPEDE trial that includes individuals with metastatic and non-metastatic disease. 59
HRQoL outcomes of patients with PCa were conditional on:
-
having metastatic disease116,119–121,123–126 – negative impact on HRQoL
-
having castration-resistant metastatic disease119 – negative impact on HRQoL
-
being diagnosed with PCa120,129,130 – negative impact on HRQoL
-
receiving radical treatment119 – positive impact on HRQoL
-
underlying true disease status (including clinical significance)129 – negative impact on HRQoL of having PCa, which is worsened by presence of CS disease
-
AEs with radical treatment by true risk category116,123 – negative impact on HRQoL
-
treatment received and time since treatment initiation120,124,130 – initial negative impact on HRQoL with improvement in post-treatment period
The UK-relevant utility sources for patients with PCa in the long-term outcome models include:
-
Torvinen et al. (2013)136 – for the disutility of metastatic disease
-
Ara and Brazier et al. (2010)137 – for the disutility of ageing
-
Mowatt et al. (2013)133 – for the disutility of treatment-related AEs [combined with rates of AEs from Donovan et al.(2016)]. 138
The long-term HRQoL outcomes of patients without PCa were dependent on age in most models,119,121,123–126 with Ara and Brazier (2010)137 the most frequently used source to inform age-adjusted utilities.
Most models considered the cost of treatment for patients with diagnosed localised or locally advanced PCa (radical treatment or active surveillance)116,119–121,123–126,129,130 and management of treating AEs. 116,121,123,125,126 Patients with undiagnosed PCa would incur the costs of routine follow-up116,119,121,123,125,126,129 or of delayed radical treatment. 129 The studies also considered the costs of metastatic disease treatment with or without staging and follow-up tests. 116,119,121,123–126 Two models assumed diagnosed metastatic disease would be treated differently if diagnosed (DTX would be added to androgen depleting therapy) compared to undiagnosed metastatic disease and that treatment with DTX would vary with age. 116,123 Some models included an end-of-life cost for patients who died from PCa,116,119,120,123,124 with one study conditioning the end-of-life costs on age at death. 124
The costs of individuals who did not have PCa were not clearly reported for most models, but, where reported, consisted of the costs of routine follow-up. 116,119,123,124
In UK-relevant models, treatment and follow-up resource use was informed mainly by UK (clinical and TA) guidance, as well as other published data (e.g. a randomised control trial informed AE rates of treatment138) and supplemented with assumptions. End-of-life costs were uprated to the relevant price year based on Round et al. 139 Unit costs were sourced mainly from national published sources.
Value components
| Value components requiring evidence linkage | Studies [first author (year)] | Mechanism |
|---|---|---|
| Improved outcomes due to increased/earlier detection of cancer, that is fewer PCa classified as no PCa | Souto-Ribeiro (2022);116 Wilson (2021);121 Cheng (2021);119 Hao (2021);120 NICE (2019);123 Faria (2018)125/Brown (2018);126 Barnett (2018);124 Pahwa et al.;117 Venderink (2017)130 | via diagnostic accuracy identifying true cancer status and treatment outcomes |
| Reduction of undertreatment: improved outcomes due to increased/earlier detection of CSPCa, that is fewer CSPCa treated as CNS PCa | Souto-Ribeiro (2022);116 Wilson (2021);121 Cheng (2021);119 Hao (2021);120 NICE (2019);123 Faria (2018);125 Brown (2018);126 Barnett (2018);124 Pahwa et al.;117 Venderink (2017)130 | via diagnostic accuracy and assumptions on treatment distribution and impact of treatment on outcomes, which is conditioned on true clinical significance of PCa, true cancer risk category or cancer grade |
| Reduction in overtreatment: improved outcomes due to improved detection of CNS PCa, that is fewer CNS PCa treated as CSPCa | Barnett (2018);124 Pahwa et al.;117 Venderink (2017)130 | via diagnostic accuracy and assumptions on treatment distribution and impact of treatment on outcomes, which is conditioned on true clinical significance of PCa |
| Change the number of repeat biopsies with impacts on biopsy costs and AEs | Souto-Ribeiro (2022);116 Wilson (2021);121 Cheng (2021);119 Faria (2018);125 Brown (2018)126 | via diagnostic accuracy and decision rule on which individuals are eligible for a repeat biopsy |
| Value components with direct impacts | ||
| Biopsy procedure costs | Souto-Ribeiro (2022);116 Wilson (2021);121 Cheng (2021);119 Hao (2021);120 NICE (2019);123 Faria (2018);125 Brown (2018);126 Barnett, (2018);124 Pahwa et al.;117 Venderink (2017)130 | – |
| Harms and/or costs of biopsy AEs | Souto-Ribeiro (2022);116 Wilson (2021);121 Cheng (2021);119 NICE (2019);123 Faria (2018);125 Brown (2018);126 Barnett (2018);124 Venderink (2017)130 | – |
Appendix 10 Extension of the evidence synthesis to determine diagnostic accuracy
Methods
Description of methods
Methods were developed to provide an internally consistent framework for evidence on the distribution of test results across a number of technologies (from the evidence synthesis), and data on the extent of misclassification of the technologies in relation to (true) disease status.
This framework relies on expressing the natural probability relationships between the different quantities of interest. The extent of misclassification is made explicit by the accuracy matrix. The accuracy matrix was expressed using conditional probabilities, with its elements being the probability of obtaining a particular test result with one method conditional on a particular level of (true) disease status, that is, the probability of a test (A) retrieving a particular result × in patients with a particular disease (D) level y – P[A = x|D = y] – or, using simplified notation, p(A) x|y·. The set of conditional probabilities that fully define accuracy are shown in the matrix in Table 66. Together with prevalence estimates, P[D = y], or py in the simplified notation shown at the left side of Table 66 this matrix determines the distribution of test results, P[A = x], shown at the top of Table 66 using the simplified notation of p(A) x.
Note that, due to the nature of biopsy and histological examination of the biopsy specimen, it is reasonable to assume that false-positive results are not possible, that is, if cancer is histologically identified, then it is present. This implies that biopsy methods cannot identify a higher ISUP Grade than true disease status, and therefore zero probability is attributed to such cases in the above accuracy matrix.
Where two methods are of interest, the problem becomes more complex. Table 66 formalises the problem by depicting the quantities of interest for two alternative biopsy methods, including the prevalence (i.e. the true distribution across ISUP grades), which is independent of test results, the two conditional accuracy matrices, and the (marginal) distributions of test results, which are themselves a function of prevalence and accuracy. The key relationships that introduce complexity are:
-
prevalence is independent of test results and therefore a common prevalence estimate needs to ground all distributions of test results, and be consistent with these
-
explicit accounts of accuracy need to respect both the prevalence estimates and the marginal distribution estimates derived from the synthesis.

|
Where the distribution of test results has been related across tests without consideration for their accuracy against a reference standard, a structured approach is therefore required for characterising accuracy to ensure that probability relationships are maintained.
Note that such a model does not identify concordance between methods in biopsy test results. To consider concordance, the synthesis model would have had to be grounded on the underlying joint or conditional probabilities of classification across tests that, that is, the likelihood of identifying individuals in a particular category using one method and in another category using a different method (joint probabilities) or the likelihood of individuals identified in a particular category by one method being classified in another by a different method (conditional probabilities). Joint/conditional probabilities determine the potential concordance between tests, which cannot be ascertained by the marginal distributions alone, that is, the same marginal distributions can be retrieved under very different levels of concordance between tests.
The approach developed for the current assessment was designed to:
-
be grounded on the results of the evidence synthesis model
-
return a true distribution across ISUP grade categories (prevalence) that is internally valid, that is, that is not lower than the estimated ISUP grade detection rates of the different biopsy methods
-
be grounded on available evidence on the likely accuracy of MRI fusion conditional on ISUP grade
-
define accuracy matrices for the remaining biopsy methods of interest that are consistent with both prevalence and the distributions of biopsy results from the evidence synthesis.
To achieve this, an extension to the synthesis model was developed in WinBUGS,140 drawing on the broader evidence in Multinomial synthesis model. To allow for an internally consistent approach, we grounded our methodology on evidence of the distribution of test results obtained with targeted-MRI methods, and of their accuracy. Given that disease prevalence is fully determined by these two results, the prevalence evidence identified in Review of additional prevalence, test results and diagnostic accuracy evidence and Distribution of test results obtained with cognitive fusion or software fusion biopsy will not be explicitly incorporated in our analyses but will instead be used qualitatively to put our results into context.
Describing distribution of test results
The distributions of test results across the disease categories for the relevant biopsy methods within each disconnected network of Model 1a were computed by applying network-specific baseline distributions to the results of the NMA. Building from the analyses in the evidence synthesis section, the baseline distributions were assumed uncertain by using a multinomial likelihood to describe the data from the empirical studies and an uninformative Dirichlet prior for its hyperparameters. The Dirichlet prior was implemented via a series of conditional beta distributions to facilitate the later use of constraints.
Note that the scope of this assessment is to compare targeted-biopsy methods; therefore, results on systematic biopsy, used in isolation, will not be shown here (by not including the broader literature on the accuracy of systematic biopsy, the results are also not relevant to support decision-making).
Describing the accuracy matrix for software fusion
Evidence on the accuracy of SF in identifying disease status according to the categories of interest was used to characterise this probabilistically in the model. A multinomial likelihood was used to describe the distribution of test results conditional on each particular level of true disease status (each line in the matrix in Table 66). The hyperparameters of the multinomial were attributed an uninformative Dirichlet distribution, implemented via a series of conditional beta distributions to facilitate the later use of constraints.
Deriving the prevalence distribution
The derivation of prevalence followed two steps.
Analytical derivation of prevalence from the marginal distribution and accuracy matrix for cognitive fusion
The prevalence and the accuracy matrix for a particular technology fully define the marginal distribution of test results for that technology. If represented in matrix form, the prevalence vector, p, multiplied by the accuracy matrix, M, retrieves the test result marginal distribution, p(i), that is p · M = p(i). We have used this relationship to derive the distribution of prevalence, that is p = p(i)/M. Because of the reverse calculation, a constraint was implemented to ensure prevalence results across categories would sum to 1.
Derivation and application of constraints for the prevalence distribution
Given the absence of false-positive results (i.e. that biopsy cannot retrieve results of ISUP grade higher than the true value), the true distribution of disease across ISUP grades is constrained by the marginal distributions of test results obtained across tests. This is because the prevalence of higher-grade tumours is expected to be at least equal to the maximum proportion in those groups identified across all tests. This means that:
-
the true prevalence of ISUP grade 4 or 5 (j = 5) is equal or higher than the maximum proportion of ISUP grade 4 or 5 identified across all tests – p5 ≥ maxi(p(i)5)
-
the true prevalence of histology ISUP grade 3 and above (j = 4 or j = 5) is equal or higher than the maximum proportion of ISUP grade 3 and above identified across all tests – p5 + p4 ≥ maxi(p(i)5 + p(i)4)
-
the true prevalence of histology ISUP grade 2 and above (j = 3, j = 4 or j = 5) is equal or higher than the maximum proportion of ISUP grade 2 and above identified across all tests – p5 + p4 + p3 ≥ maxi(p(i)5 + p(i)4 + p(i)3)
-
the true prevalence of histology ISUP grade 1 and above (j = 2, j = 3, j = 4 or j = 5) is equal or higher than the maximum proportion of ISUP grade 1 and above identified across all tests – p5 + p4 + p3 + p2 ≥ maxi(p(i)5 + p(i)4 + p(i)3 + p(i)2).
The true prevalence distribution should meet these conditions. The boundaries for each of the inequalities defined (i.e. the values at equality) can be determined recursively (with calculations starting at the highest grade). These conditions were implemented in WinBUGS using inequality constrains (see code below).
Derivation of accuracy matrix for other technologies
The accuracy matrix for the remaining technologies is determined by the prevalence estimates and by their marginal distributions. The diagonal cells in each of the accuracy matrices were therefore defined as a function of prevalence, probability of test result and other relevant elements in the accuracy matrices, by using the structural relationships between these parameters. For example, for category 4, the diagonal of the accuracy matrix for biopsy method k was defined as:
which subtracts those from category 5 that were incorrectly identified as 4’s from the total with category 4 test results.
The remaining free elements of each line in the matrix were sampled from an uninformative Dirichlet distribution (defined as a set of conditional beta distributions). Given that the diagonal cells relating prevalence with distribution of test results used the non-diagonal elements of the matrix, information is already conveyed on these parameters, and therefore final inference on these will not be fully uninformative. All accuracy parameters were constrained to be between 0 and 1, as the inverse matrix calculation, on its own, does not ensure that.
Implementation
The extension to the evidence synthesis model was developed in WinBUGS and was appended to the synthesis model code to draw on the inferences from the synthesised log odds ratios. The constraints implemented within the code extension need the log odds ratios in the synthesis model to be influenced by these. This will ensure that the inferences on the log odds ratio from the extended model are plausible with the data incorporated (accuracy matrices and baseline distribution of test results) and with the structural relationships between the quantities of interest. To evaluate the influence over the unconstrained evidence synthesis inferences, we will compare the probabilities of test results derived from the synthesis model used in isolation [see Model 1a: Multinomial synthesis model (base case)] with those derived from the extended synthesis and accuracy model.
Additionally, non-diagonal elements of the accuracy matrices inferred by the model were simulated from a stochastic distribution, with information on them conveyed indirectly via the diagonal elements. For this reason, retrieving test results from inferences over the prevalence and accuracy matrix approximates, but does not equal, the distribution of test results retrieved by the synthesis model. Results were therefore also compared to determine the magnitude of differences.









Additional results
| Network 1 | (Distribution of test results) | (Distribution of test results) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.516 (0.416 to 0.615) | 0.186 (0.131 to 0.249) | 0.136 (0.068 to 0.211) | 0.098 (0.052 to 0.157 | 0.064 (0.031 to 0.114) | 0.457 (0.403 to 0.513) | 0.173 (0.137 to 0.214) | 0.196 (0.157 to 0.233) | 0.108 (0.079 to 0.144) | 0.066 [0.043 to 0.095] | ||
| (Prevalence) | CF | SF | |||||||||
| (Joint probability matrix) | (Joint probability matrix) | ||||||||||
| ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 | |
| Network 2 | (Distribution of test results) | (Distribution of test results) | |||||||||
| 0.460 (0.335 to 0.583) | 0.250 (0.152 to 0.356) | 0.127 (0.034 to 0.261) | 0.131 (0.046 to 0.231) | 0.033 (0.001 to 0.107) | 0.348 (0.273 to 0.418) | 0.223 (0.179 to 0.273) | 0.232 (0.168 to 0.311) | 0.115 (0.081 to 0.152) | 0.082 (0.054 to 0.114) | ||
| (Prevalence) | Combined CF and systematic biopsy | Combined SF and systematic biopsy | |||||||||
| (Joint probability matrix) | (Joint probability matrix) | ||||||||||
| ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 | |
| 0.121 (0.007 to 0.238) | NC | 0.121 (0.007 to 0.238) | 0 | 0 | 0 | 0 | 0.121 (0.007 to 0.238) | 0 | 0 | 0 | 0 |
| 0.318 (0.212 to 0.452) | 1 | 0.265 (0.136 to 0.413) | 0.053 (0.002 to 0.141) | 0 | 0 | 0 | 0.215 (0.122 to 0.335) | 0.103 (0.063 to 0.149) | 0 | 0 | 0 |
| 0.262 (0.193 to 0.341) | 2 | 0.08 (0.004 to 0.184) | 0.094 (0.022 to 0.178) | 0.088 (0.026 to 0.145) | 0 | 0 | 0.066 (0.038 to 0.104) | 0.054 (0.03 to 0.084) | 0.142 (0.102 to 0.185) | 0 | 0 |
| 0.183 (0.119 to 0.265) | 3 | 0.035 (0.001 to 0.108) | 0.026 (0.001 to 0.081) | 0.035 (0.002 to 0.105) | 0.086 (0.032 to 0.146) | 0 | 0.042 (0.016 to 0.079) | 0.011 (0.002 to 0.029) | 0.038 (0.017 to 0.071) | 0.092 (0.062 to 0.129) | 0 |
| 0.116 (0.077 to 0.174) | 4 or 5 | 0.015 (0.000 to 0.048) | 0.013 (0.000 to 0.044) | 0.013 (0.000 to 0.043) | 0.012 (0.000 to 0.04) | 0.064 (0.031 to 0.114) | 0.013 (0.005 to 0.026) | 0.006 (0.001 to 0.014) | 0.015 (0.006 to 0.031) | 0.016 (0.007 to 0.032) | 0.066 (0.043 to 0.095) |
| 0.121 (0.007 to 0.238) | NC | 0.121 (0.007 to 0.238) | 0 | 0 | 0 | 0 | 0.121 (0.007 to 0.238) | 0 | 0 | 0 | 0 |
| 0.318 (0.212 to 0.452) | 1 | 0.227 (0.08 to 0.382) | 0.091 (0.004 to 0.212) | 0 | 0 | 0 | 0.171 (0.051 to 0.306) | 0.147 (0.054 to 0.219) | 0 | 0 | 0 |
| 0.262 (0.193 to 0.341) | 2 | 0.066 (0.002 to 0.185) | 0.114 (0.017 to 0.229) | 0.082 (0.007 to 0.208) | 0 | 0 | 0.021 (0.000 to 0.080) | 0.041 (0.003 to 0.122) | 0.199 (0.141 to 0.251) | 0 | 0 |
| 0.183 (0.119 to 0.265) | 3 | 0.023 (0.000 to 0.093) | 0.022 (0 to 0.09) | 0.025 (0.000 to 0.097) | 0.112 (0.023 to 0.212) | 0 | 0.025 (0.001 to 0.098) | 0.026 (0.001 to 0.092) | 0.025 (0.001 to 0.089) | 0.106 (0.063 to 0.146) | 0 |
| 0.116 (0.077 to 0.174) | 4 or 5 | 0.023 (0.000 to 0.078) | 0.022 (0.001 to 0.078) | 0.02 (0.000 to 0.069) | 0.019 (0.000 to 0.067) | 0.033 (0.001 to 0.107) | 0.009 (0.000 to 0.042) | 0.009 (0.000 to 0.037) | 0.008 (0.000 to 0.037) | 0.009 (0.000 to 0.038) | 0.082 (0.054 to 0.114) |
Influence of the use of constraints on the network meta-analysis estimates
Comparison of inferences on distribution of test results with the synthesis code used in isolation and the synthesis code including the extension.
| Model | Synthesis model | Extended synthesis and accuracy model | |||
|---|---|---|---|---|---|
| Assumptions over baseline probability | Deterministic | Deterministic | Probabilistic | Probabilistic | |
| Calculation of distribution of test results | Directly from synthesis | Directly from synthesis | Directly from synthesis | Back calculated from prevalence and accuracy matrix | |
| Biopsy method | Category | ||||
| Network 1 | |||||
| CF | NC | 0.552 (0.475 to 0.624) | 0.545 (0.477 to 0.612) | 0.531 (0.446 to 0.616) | 0.513 (0.414 to 0.610) |
| 1 | 0.174 (0.132 to 0.223) | 0.177 (0.135 to 0.225) | 0.185 (0.129 to 0.249) | 0.185 (0.132 to 0.245) | |
| 2 | 0.118 (0.081 to 0.164) | 0.121 (0.085 to 0.165) | 0.120 (0.078 to 0.173) | 0.139 (0.070 to 0.217) | |
| 3 | 0.094 (0.058 to 0.143) | 0.095 (0.059 to 0.142) | 0.098 (0.055 to 0.160) | 0.097 (0.056 to 0.159) | |
| 4 or 5 | 0.062 (0.034 to 0.104) | 0.062 (0.035 to 0.099) | 0.066 (0.032 to 0.117) | 0.065 (0.032 to 0.115) | |
| SF | NC | 0.469 | 0.469 | 0.457 (0.403 to 0.509) | 0.457 (0.403 to 0.513) |
| 1 | 0.165 | 0.165 | 0.172 (0.137 to 0.212) | 0.173 (0.137 to 0.214) | |
| 2 | 0.198 | 0.198 | 0.195 (0.159 to 0.236) | 0.196 (0.157 to 0.233) | |
| 3 | 0.105 | 0.105 | 0.109 (0.080 to 0.146) | 0.108 (0.079 to 0.144) | |
| 4 or 5 | 0.063 | 0.063 | 0.067 (0.045 to 0.095) | 0.066 (0.043 to 0.095) | |
| Network 2 | |||||
| Combined CF and systematic biopsy | NC | 0.402 (0.210 to 0.559) | 0.451 (0.345 to 0.562) | 0.455 (0.343 to 0.570) | 0.460 (0.335 to 0.583) |
| 1 | 0.211 (0.102 to 0.326) | 0.246 (0.16 to 0.349) | 0.249 (0.156 to 0.362) | 0.250 (0.152 to 0.356) | |
| 2 | 0.109 (0.031 to 0.238) | 0.136 (0.054 to 0.256) | 0.134 (0.052 to 0.255) | 0.127 (0.034 to 0.261) | |
| 3 | 0.241 (0.058 to 0.586) | 0.135 (0.048 to 0.245) | 0.130 (0.045 to 0.230) | 0.131 (0.046 to 0.231) | |
| 4 or 5 | 0.037 (0.001 to 0.172) | 0.033 (0.001 to 0.107) | 0.032 (0.001 to 0.107) | 0.033 (0.001 to 0.107) | |
| Combined SF and systematic biopsy | NC | 0.355 | 0.355 | 0.359 (0.305 to 0.413) | 0.346 (0.274 to 0.408) |
| 1 | 0.220 | 0.220 | 0.223 (0.178 to 0.273) | 0.222 (0.177 to 0.273) | |
| 2 | 0.223 | 0.223 | 0.221 (0.177 to 0.270) | 0.234 (0.170 to 0.313) | |
| 3 | 0.118 | 0.118 | 0.115 (0.082 to 0.154) | 0.116 (0.084 to 0.153) | |
| 4 or 5 | 0.083 | 0.083 | 0.082 (0.055 to 0.114) | 0.082 (0.054 to 0.114) | |
Results from this comparison show that for network 1 the structural extension model does not significantly influence synthesis estimates. For network 2, estimates of category 4 for the non-reference treatment (combined CF and systematic biopsy) are reduced in the extended model, which suggests a conflict between the structural extension (including data sources added) and the uncertainty derived from the multinomial log odds model implemented in the synthesis. For this category, there is only one study providing a direct comparison of combined software versus combined CF with very few patients classified in categories 4 or 5,82 providing very sparse information. This study reports a proportion of 5% of test results in category 4 with combined cognitive, versus 3% in combined SF. Therefore, uncertainty is very wide for this category and the constrained model restricts the distribution of this category the most.
| Model | Synthesis model | Extended synthesis and accuracy model | |||
|---|---|---|---|---|---|
| Assumptions over baseline probability | Deterministic | Deterministic | Probabilistic | Probabilistic | |
| Calculation of distribution of test results | Directly from synthesis | Directly from synthesis | Directly from synthesis | Back calculated from prevalence and accuracy matrix | |
| Biopsy method | ISUP grade | ||||
| Network 1 | |||||
| CF | NC | 0.750 (0.688 to 0.803) | 0.744 (0.686 to 0.798) | 0.719 (0.643 to 0.788) | 0.703 (0.618 to 0.776) |
| 1 | 0.085 (0.062 to 0.114) | 0.088 (0.065 to 0.12) | 0.105 (0.069 to 0.153) | 0.105 (0.071 to 0.155) | |
| 2 | 0.057 (0.038 to 0.082) | 0.058 (0.04 to 0.079) | 0.061 (0.039 to 0.091) | 0.077 (0.035 to 0.138) | |
| 3 | 0.065 (0.039 to 0.101) | 0.066 (0.038 to 0.101) | 0.068 (0.036 to 0.116) | 0.068 (0.033 to 0.120) | |
| 4 or 5 | 0.043 (0.023 to 0.074) | 0.044 (0.024 to 0.07) | 0.047 (0.021 to 0.086) | 0.046 (0.021 to 0.085) | |
| SF | NC | 0.687 | 0.687 | 0.661 (0.611 to 0.710) | 0.661 (0.611 to 0.709) |
| 1 | 0.087 | 0.087 | 0.103 (0.075 to 0.137) | 0.103 (0.076 to 0.135) | |
| 2 | 0.102 | 0.102 | 0.107 (0.082 to 0.136) | 0.107 (0.082 to 0.136) | |
| 3 | 0.078 | 0.078 | 0.080 (0.055 to 0.110) | 0.080 (0.055 to 0.111) | |
| 4 or 5 | 0.047 | 0.047 | 0.049 (0.031 to 0.073) | 0.049 (0.031 to 0.073) | |
| Network 2 | |||||
| Combined CF and systematic biopsy | NC | 0.615 (0.382 to 0.76) | 0.658 (0.557 to 0.75) | 0.664 (0.56 to 0.761) | 0.659 (0.561 to 0.752) |
| 1 | 0.135 (0.073 to 0.208) | 0.152 (0.098 to 0.221) | 0.155 (0.093 to 0.240) | 0.157 (0.096 to 0.241) | |
| 2 | 0.053 (0.015 to 0.120) | 0.067 (0.025 to 0.135) | 0.065 (0.023 to 0.136) | 0.067 (0.015 to 0.152) | |
| 3 | 0.171 (0.036 to 0.468) | 0.095 (0.03 to 0.181) | 0.090 (0.028 to 0.163) | 0.091 (0.027 to 0.165) | |
| 4 or 5 | 0.027 (0.000 to 0.125) | 0.029 (0.001 to 0.099) | 0.027 (0.001 to 0.087) | 0.027 (0.001 to 0.081) | |
| Combined SF and systematic biopsy | NC | 0.585 | 0.585 | 0.591 (0.536 to 0.647) | 0.583 (0.513 to 0.649) |
| 1 | 0.151 | 0.151 | 0.154 (0.114 to 0.201) | 0.155 (0.114 to 0.198) | |
| 2 | 0.117 | 0.117 | 0.113 (0.080 to 0.147) | 0.120 (0.074 to 0.181) | |
| 3 | 0.086 | 0.086 | 0.084 (0.056 to 0.118) | 0.083 (0.057 to 0.117) | |
| 4 or 5 | 0.061 | 0.061 | 0.058 (0.036 to 0.084) | 0.058 (0.037 to 0.084) | |
Sensitivity analysis
| Model | Synthesis model | Extended synthesis and accuracy model | |||
|---|---|---|---|---|---|
| Assumptions over baseline probability | Deterministic | Deterministic | Probabilistic | Probabilistic | |
| Calculation of distribution of test results | Directly from synthesis | Directly from synthesis | Directly from synthesis | Back calculated from prevalence and accuracy matrix | |
| ISUP | |||||
| Network 1 | |||||
| CF | NC | 0.363 (0.294 to 0.435) | 0.364 (0.293 to 0.44) | 0.392 (0.308 to 0.482) | 0.368 (0.248 to 0.473) |
| 1 | 0.197 (0.148 to 0.255) | 0.199 (0.152 to 0.253) | 0.192 (0.133 to 0.263) | 0.191 (0.140 to 0.256) | |
| 2 | 0.182 (0.130 to 0.245) | 0.184 (0.130 to 0.25) | 0.169 (0.111 to 0.242) | 0.196 (0.101 to 0.306) | |
| 3 | 0.156 (0.100 to 0.226) | 0.152 (0.095 to 0.219) | 0.147 (0.084 to 0.232) | 0.145 (0.079 to 0.228) | |
| 4 or 5 | 0.102 (0.057 to 0.167) | 0.102 (0.059 to 0.154) | 0.100 (0.051 to 0.176) | 0.101 (0.052 to 0.176) | |
| SF | NC | 0.286 | 0.286 | 0.313 (0.271 to 0.360) | 0.314 (0.271 to 0.362) |
| 1 | 0.173 | 0.173 | 0.170 (0.135 to 0.209) | 0.169 (0.137 to 0.207) | |
| 2 | 0.282 | 0.282 | 0.262 (0.218 to 0.310) | 0.263 (0.218 to 0.308) | |
| 3 | 0.161 | 0.161 | 0.158 (0.117 to 0.203) | 0.157 (0.117 to 0.204) | |
| 4 or 5 | 0.097 | 0.097 | 0.097 (0.064 to 0.137) | 0.098 (0.064 to 0.140) | |
| Model | Synthesis model | Extended synthesis and accuracy model | ||||
|---|---|---|---|---|---|---|
| Assumptions over baseline probability | Deterministic | Deterministic | Probabilistic | Probabilistic | ||
| Calculation of distribution of test results | Directly from synthesis | Directly from synthesis | Directly from synthesis | Back calculated from prevalence and accuracy matrix | ||
| ISUP | ||||||
| Network 1 | ||||||
| CF | NC | 0.552 (0.475 to 0.624) | 0.555 (0.488 to 0.626) | 0.531 (0.445 to 0.621) | 0.525 (0.433 to 0.620) | |
| 1 | 0.174 (0.132 to 0.223) | 0.179 (0.132 to 0.232) | 0.191 (0.132 to 0.264) | 0.190 (0.131 to 0.256) | ||
| 2 | 0.118 (0.081 to 0.164) | 0.117 (0.082 to 0.161) | 0.112 (0.070 to 0.169) | 0.122 (0.062 to 0.201) | ||
| 3 | 0.094 (0.058 to 0.143) | 0.093 (0.059 to 0.137) | 0.099 (0.055 to 0.160) | 0.098 (0.053 to 0.158) | ||
| 4 or 5 | 0.062 (0.034 to 0.104) | 0.056 (0.032 to 0.083) | 0.066 (0.034 to 0.109) | 0.065 (0.033 to 0.106) | ||
| SF | NC | 0.469 | 0.469 | 0.449 (0.396 to 0.503) | 0.450 (0.400 to 0.509) | |
| 1 | 0.165 | 0.165 | 0.176 (0.139 to 0.220) | 0.175 (0.140 to 0.217) | ||
| 2 | 0.198 | 0.198 | 0.189 (0.149 to 0.234) | 0.189 (0.147 to 0.236) | ||
| 3 | 0.105 | 0.105 | 0.112 (0.082 to 0.149) | 0.112 (0.082 to 0.146) | ||
| 4 or 5 | 0.063 | 0.063 | 0.075 (0.053 to 0.105) | 0.075 (0.053 to 0.103) | ||
Detailed results for subgroup analysis on previous negative-biopsy individuals
In this analysis, the baseline distribution of test results for SF was sourced from Filson et al.,96 but using the group of individuals recruited into this study that had previous negative-biopsy results. However, the diagnostic accuracy evidence synthesis and the accuracy matrix are still sourced as per the main analysis, grounded on evidence over biopsy-naive patients. Table 72 presents summary results of distribution of test results and prevalence probabilities and results of the accuracy matrices are presented in Appendix 10.
The summary results in Table 72 illustrate that, for individuals with a previous negative biopsy, a significantly increased proportion of ‘no cancer’ results are expected in relation to biopsy-naive individuals. This impacts the (implicit) prevalence estimates: for those with previous negative biopsy, the probability of NC is 43% (95% CrI 26% to 53%), while for biopsy naive it is 12% (95% CrI 0.7% to 24%). In comparing software with CF biopsy strategies, across both networks, we observe similar probabilities of ISUP grade 1, 3 and 4 or 5 results, and a slightly higher probability of ISUP grade 2 results for software strategies. This differs from the results of the synthesis model for ISUP grade 3 only, where the probability under combined CF was slightly higher than for combined fusion software (see Table 4). The accuracy matrix estimates (reported in Appendix 10) are similar to those estimated for biopsy-naive individuals (main analysis, Table 9).
| Prevalence | ISUP | Distribution of test results | |||
|---|---|---|---|---|---|
| Network 1 | Network 2 | ||||
| CF | SF | Combined CF and systematic biopsy | Combined SF and systematic biopsy | ||
| Subgroup analysis (previous negative biopsy) | |||||
| 0.428 (0.259 to 0.529) | NC | 0.703 (0.618 to 0.776) | 0.661 (0.611 to 0.709) | 0.659 (0.561 to 0.752) | 0.583 (0.513 to 0.649) |
| 0.224 (0.138 to 0.39) | 1 | 0.105 (0.071 to 0.155) | 0.107 (0.082 to 0.136) | 0.157 (0.096 to 0.241) | 0.155 (0.114 to 0.198) |
| 0.132 (0.091 to 0.188) | 2 | 0.077 (0.035 to 0.138) | 0.107 (0.082 to 0.136) | 0.067 (0.015 to 0.152) | 0.120 (0.074 to 0.181) |
| 0.131 (0.079 to 0.199) | 3 | 0.068 (0.033 to 0.120) | 0.080 (0.055 to 0.111) | 0.091 (0.027 to 0.165) | 0.083 (0.057 to 0.117) |
| 0.085 (0.053 to 0.127) | 4 or 5 | 0.046 (0.021 to 0.085) | 0.049 (0.031 to 0.073) | 0.027 (0.001 to 0.081) | 0.058 (0.037 to 0.084) |
| Sensitivity analysis to baseline distribution for biopsy naive (PAIREDCAP’s baseline, Mortezavi’s accuracy) | |||||
| 0.031 (0.001 to 0.092) | NC | 0.368 (0.248 to 0.473) | 0.314 (0.271 to 0.362) | NA | NA |
| 0.226 (0.163 to 0.319) | 1 | 0.191 (0.140 to 0.256) | 0.169 (0.137 to 0.207) | NA | NA |
| 0.322 (0.222 to 0.42) | 2 | 0.196 (0.101 to 0.306) | 0.263 (0.218 to 0.308) | NA | NA |
| 0.252 (0.154 to 0.37) | 3 | 0.145 (0.079 to 0.228) | 0.098 (0.064 to 0.140) | NA | NA |
| 0.169 (0.104 to 0.254) | 4 or 5 | 0.101 (0.052 to 0.176) | 0.098 (0.064 to 0.140) | NA | NA |
| Sensitivity analysis to accuracy matrix for biopsy naive (Filson’s baseline, Zhou’s accuracy) | |||||
| 0.170 (0.023 to 0.280) | NC | 0.525 (0.433 to 0.620) | 0.450 (0.400 to 0.509) | NA | NA |
| 0.279 (0.196 to 0.400) | 1 | 0.190 (0.131 to 0.256) | 0.175 (0.140 to 0.217) | NA | NA |
| 0.300 (0.211 to 0.436) | 2 | 0.122 (0.062 to 0.201) | 0.189 (0.147 to 0.236) | NA | NA |
| 0.155 (0.109 to 0.223) | 3 | 0.098 (0.053 to 0.158) | 0.112 (0.082 to 0.146) | NA | NA |
| 0.095 (0.067 to 0.136) | 4 or 5 | 0.450 (0.400 to 0.509) | 0.075 (0.053 to 0.103) | NA | NA |
| Network 1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.703 (0.618 to 0.776) | 0.105 (0.071 to 0.155) | 0.077 (0.035 to 0.138) | 0.068 (0.033 to 0.12) | 0.046 (0.021 to 0.085) | 0.661 (0.611 to 0.709) | 0.103 (0.076 to 0.135) | 0.107 (0.082 to 0.136) | 0.080 (0.055 to 0.111) | 0.049 (0.031 to 0.073) | ||
| CF | SF | ||||||||||
| ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 | |
| Network 2 | |||||||||||
| 0.659 (0.561 to 0.752) | 0.157 (0.096 to 0.241) | 0.067 (0.015 to 0.152) | 0.091 (0.027 to 0.165) | 0.027 (0.001 to 0.081) | 0.583 (0.513 to 0.649) | 0.155 (0.114 to 0.198) | 0.120 (0.074 to 0.181) | 0.083 (0.057 to 0.117) | 0.058 (0.037 to 0.084) | ||
| Combined CF and systematic biopsy | Combined SF and systematic biopsy | ||||||||||
| ISUP | NC | 1 | 2 | 3 | 4 or 5 | 1 | 2 | 3 | 4 | 5 | |
| 0.428 (0.259 to 0.529) | NC | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 0.224 (0.138 to 0.39) | 1 | 0.857 (0.575 to 0.995) | 0.143 (0.005 to 0.425) | 0 | 0 | 0 | 0.698 (0.557 to 0.826) | 0.302 (0.174 to 0.443) | 0 | 0 | 0 |
| 0.132 (0.091 to 0.188) | 2 | 0.324 (0.026 to 0.722) | 0.374 (0.066 to 0.718) | 0.302 (0.049 to 0.557) | 0 | 0 | 0.264 (0.183 to 0.35) | 0.201 (0.13 to 0.288) | 0.536 (0.436 to 0.626) | 0 | 0 |
| 0.131 (0.079 to 0.199) | 3 | 0.195 (0.005 to 0.571) | 0.130 (0.003 to 0.428) | 0.208 (0.007 to 0.579) | 0.466 (0.181 to 0.793) | 0 | 0.225 (0.129 to 0.344) | 0.054 (0.011 to 0.125) | 0.195 (0.103 to 0.301) | 0.526 (0.404 to 0.648) | 0 |
| 0.085 (0.053 to 0.127) | 4 or 5 | 0.136 (0.002 to 0.439) | 0.109 (0.002 to 0.324) | 0.115 (0.003 to 0.363) | 0.093 (0.004 to 0.315) | 0.547 (0.283 to 0.902) | 0.110 (0.044 to 0.193) | 0.046 (0.01 to 0.114) | 0.122 (0.055 to 0.216) | 0.141 (0.071 to 0.226) | 0.581 (0.462 to 0.694) |
| 0.428 (0.259 to 0.529) | NC | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 0.224 (0.138 to 0.39) | 1 | 0.693 (0.251 to 0.984) | 0.307 (0.016 to 0.749) | 0 | 0 | 0 | 0.497 (0.153 to 0.845) | 0.503 (0.155 to 0.847) | 0 | 0 | 0 |
| 0.132 (0.091 to 0.188) | 2 | 0.272 (0.009 to 0.709) | 0.448 (0.058 to 0.864) | 0.281 (0.017 to 0.791) | 0 | 0 | 0.087 (0.002 to 0.315) | 0.176 (0.012 to 0.46) | 0.736 (0.408 to 0.971) | 0 | 0 |
| 0.131 (0.079 to 0.199) | 3 | 0.136 (0.001 to 0.515) | 0.121 (0.002 to 0.474) | 0.140 (0.001 to 0.515) | 0.603 (0.126 to 0.982) | 0 | 0.138 (0.003 to 0.439) | 0.132 (0.002 to 0.44) | 0.131 (0.003 to 0.418) | 0.600 (0.298 to 0.938) | 0 |
| 0.085 (0.053 to 0.127) | 4 or 5 | 0.207 (0.003 to 0.629) | 0.196 (0.004 to 0.577) | 0.147 (0.004 to 0.546) | 0.135 (0.003 to 0.434) | 0.315 (0.014 to 0.899) | 0.071 (0.001 to 0.251) | 0.074 (0.001 to 0.284) | 0.073 (0.001 to 0.266) | 0.083 (0.001 to 0.311) | 0.699 (0.367 to 0.966) |
| Network 1 | (Distribution of test results) | (Distribution of test results) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.703 (0.618 to 0.776) | 0.105 (0.071 to 0.155) | 0.077 (0.035 to 0.138) | 0.068 (0.033 to 0.12) | 0.046 (0.021 to 0.085) | 0.661 (0.611 to 0.709) | 0.103 (0.076 to 0.135) | 0.107 (0.082 to 0.136) | 0.080 (0.055 to 0.111) | 0.049 (0.031 to 0.073) | ||
| CF | SF | ||||||||||
| (Joint probability matrix) | (Joint probability matrix) | ||||||||||
| (Prevalence) | ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 |
| Network 2 | (Distribution of test results) | (Distribution of test results) | |||||||||
| 0.659 (0.561 to 0.752) | 0.157 (0.096 to 0.241) | 0.067 (0.015 to 0.152) | 0.091 (0.027 to 0.165) | 0.027 (0.001 to 0.081) | 0.583 (0.513 to 0.649) | 0.155 (0.114 to 0.198) | 0.120 (0.074 to 0.181) | 0.083 (0.057 to 0.117) | 0.058 (0.037 to 0.084) | ||
| Combined CF and systematic biopsy | Combined SF and systematic biopsy | ||||||||||
| (Joint probability matrix) | (Joint probability matrix) | ||||||||||
| (Prevalence) | ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 |
| 0.428 (0.259 to 0.529) | NC | 0.428 (0.259 to 0.529) | 0 | 0 | 0 | 0 | 0.428 (0.259 to 0.529) | 0 | 0 | 0 | 0 |
| 0.224 (0.138 to 0.39) | 1 | 0.194 (0.092 to 0.363) | 0.03 (0.001 to 0.085) | 0 | 0 | 0 | 0.159 (0.082 to 0.313) | 0.065 (0.037 to 0.098) | 0 | 0 | 0 |
| 0.132 (0.091 to 0.188) | 2 | 0.044 (0.003 to 0.103) | 0.049 (0.009 to 0.095) | 0.04 (0.006 to 0.076) | 0 | 0 | 0.035 (0.02 to 0.057) | 0.026 (0.014 to 0.043) | 0.071 (0.047 to 0.104) | 0 | 0 |
| 0.131 (0.079 to 0.199) | 3 | 0.026 (0.001 to 0.087) | 0.017 (0 to 0.056) | 0.028 (0.001 to 0.085) | 0.06 (0.02 to 0.113) | 0 | 0.03 (0.013 to 0.061) | 0.007 (0.001 to 0.019) | 0.026 (0.011 to 0.047) | 0.068 (0.041 to 0.101) | 0 |
| 0.085 (0.053 to 0.127) | 4 or 5 | 0.012 (0 to 0.041) | 0.009 (0 to 0.029) | 0.01 (0 to 0.031) | 0.008 (0 to 0.03) | 0.046 (0.021 to 0.085) | 0.009 (0.003 to 0.018) | 0.004 (0.001 to 0.011) | 0.01 (0.004 to 0.021) | 0.012 (0.005 to 0.023) | 0.049 (0.031 to 0.073) |
| 0.121 (0.007 to 0.238) | NC | 0.428 (0.259 to 0.529) | 0 | 0 | 0 | 0 | 0.428 (0.259 to 0.529) | 0 | 0 | 0 | 0 |
| 0.318 (0.212 to 0.452) | 1 | 0.159 (0.041 to 0.344) | 0.065 (0.004 to 0.145) | 0 | 0 | 0 | 0.118 (0.025 to 0.3) | 0.106 (0.039 to 0.16) | 0 | 0 | 0 |
| 0.262 (0.193 to 0.341) | 2 | 0.037 (0.001 to 0.101) | 0.059 (0.007 to 0.122) | 0.036 (0.002 to 0.11) | 0 | 0 | 0.012 (0 to 0.048) | 0.024 (0.001 to 0.072) | 0.096 (0.054 to 0.136) | 0 | 0 |
| 0.183 (0.119 to 0.265) | 3 | 0.018 (0 to 0.072) | 0.016 (0 to 0.063) | 0.018 (0 to 0.071) | 0.079 (0.015 to 0.154) | 0 | 0.019 (0 to 0.074) | 0.018 (0 to 0.064) | 0.018 (0 to 0.068) | 0.075 (0.04 to 0.115) | 0 |
| 0.116 (0.077 to 0.174) | 4 or 5 | 0.018 (0 to 0.057) | 0.017 (0 to 0.056) | 0.012 (0 to 0.045) | 0.012 (0 to 0.044) | 0.027 (0.001 to 0.081) | 0.007 (0 to 0.027) | 0.007 (0 to 0.029) | 0.007 (0 to 0.027) | 0.008 (0 to 0.031) | 0.058 (0.037 to 0.084) |
Detailed results of sensitivity analyses
| Network 1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ISUP | 0.368 (0.248 to 0.473) | 0.191 (0.14 to 0.256) | 0.196 (0.101 to 0.306) | 0.145 (0.079 to 0.228) | 0.101 (0.052 to 0.176) | 0.314 (0.271 to 0.362) | 0.169 (0.137 to 0.207) | 0.263 (0.218 to 0.308) | 0.157 (0.117 to 0.204) | 0.098 (0.064 to 0.14) | |
| CF | SF | ||||||||||
| NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 | ||
| 0.031 (0.001 to 0.092) | NC | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 0.226 (0.163 to 0.319) | 1 | 0.754 (0.355 to 0.990) | 0.246 (0.010 to 0.645) | 0 | 0 | 0 | 0.634 (0.506 to 0.773) | 0.366 (0.227 to 0.494) | 0 | 0 | 0 |
| 0.322 (0.222 to 0.42) | 2 | 0.300 (0.025 to 0.642) | 0.288 (0.071 to 0.532) | 0.412 (0.133 to 0.687) | 0 | 0 | 0.229 (0.155 to 0.305) | 0.197 (0.123 to 0.276) | 0.575 (0.484 to 0.67) | 0 | 0 |
| 0.252 (0.154 to 0.37) | 3 | 0.175 (0.007 to 0.520) | 0.121 (0.004 to 0.402) | 0.183 (0.006 to 0.492) | 0.521 (0.27 to 0.862) | 0 | 0.191 (0.105 to 0.294) | 0.058 (0.013 to 0.121) | 0.222 (0.124 to 0.331) | 0.530 (0.412 to 0.659) | 0 |
| 0.169 (0.104 to 0.254) | 4 or 5 | 0.121 (0.004 to 0.392) | 0.086 (0.001 to 0.285) | 0.105 (0.003 to 0.342) | 0.088 (0.002 to 0.31) | 0.599 (0.324 to 0.920) | 0.102 (0.041 to 0.184) | 0.044 (0.009 to 0.107) | 0.127 (0.06 to 0.216) | 0.144 (0.069 to 0.236) | 0.583 (0.449 to 0.700) |
| Network 1 | (Distribution of test results) | (Distribution of test results) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.368 (0.248 to 0.473) | 0.191 (0.14 to 0.256) | 0.196 (0.101 to 0.306) | 0.145 (0.079 to 0.228) | 0.101 (0.052 to 0.176) | 0.314 (0.271 to 0.362) | 0.169 (0.137 to 0.207) | 0.263 (0.218 to 0.308) | 0.157 (0.117 to 0.204) | 0.098 (0.064 to 0.14) | ||
| CF | SF | ||||||||||
| (Prevalence) | ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 |
| 0.031 (0.001 to 0.092) | NC | 0.031 (0.001 to 0.092) | 0 | 0 | 0 | 0 | 0.031 (0.001 to 0.092) | 0 | 0 | 0 | 0 |
| 0.226 (0.163 to 0.319) | 1 | 0.171 (0.07 to 0.271) | 0.055 (0.002 to 0.151) | 0 | 0 | 0 | 0.143 (0.098 to 0.202) | 0.083 (0.043 to 0.129) | 0 | 0 | 0 |
| 0.322 (0.222 to 0.42) | 2 | 0.099 (0.006 to 0.235) | 0.092 (0.021 to 0.173) | 0.131 (0.042 to 0.21) | 0 | 0 | 0.074 (0.042 to 0.114) | 0.063 (0.036 to 0.097) | 0.185 (0.125 to 0.246) | 0 | 0 |
| 0.252 (0.154 to 0.37) | 3 | 0.045 (0.001 to 0.144) | 0.03 (0.001 to 0.095) | 0.047 (0.001 to 0.142) | 0.13 (0.055 to 0.219) | 0 | 0.048 (0.021 to 0.088) | 0.015 (0.003 to 0.035) | 0.057 (0.024 to 0.1) | 0.132 (0.081 to 0.181) | 0 |
| 0.169 (0.104 to 0.254) | 4 or 5 | 0.021 (0.001 to 0.079) | 0.015 (0 to 0.054) | 0.018 (0 to 0.064) | 0.015 (0 to 0.056) | 0.101 (0.052 to 0.176) | 0.018 (0.006 to 0.038) | 0.008 (0.001 to 0.02) | 0.022 (0.008 to 0.048) | 0.025 (0.009 to 0.049) | 0.098 (0.064 to 0.14) |
| Network 1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.525 (0.433 to 0.62) | 0.190 (0.131 to 0.256) | 0.122 (0.062 to 0.201) | 0.098 (0.053 to 0.158) | 0.065 (0.033 to 0.106) | 0.450 (0.400 to 0.509) | 0.175 (0.140 to 0.217) | 0.189 (0.147 to 0.236) | 0.112 (0.082 to 0.146) | 0.075 (0.053 to 0.103) | ||
| CF | SF | ||||||||||
| ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 | |
| 0.170 (0.023 to 0.280) | NC | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| 0.279 (0.196 to 0.400) | 1 | 0.787 (0.435 to 0.992) | 0.213 (0.008 to 0.565) | 0 | 0 | 0 | 0.472 (0.321 to 0.631) | 0.528 (0.369 to 0.679) | 0 | 0 | 0 |
| 0.300 (0.211 to 0.436) | 2 | 0.327 (0.03 to 0.693) | 0.362 (0.065 to 0.646) | 0.312 (0.127 to 0.518) | 0 | 0 | 0.415 (0.268 to 0.569) | 0.048 (0.006 to 0.129) | 0.537 (0.392 to 0.691) | 0 | 0 |
| 0.155 (0.109 to 0.223) | 3 | 0.152 (0.003 to 0.448) | 0.113 (0.003 to 0.367) | 0.147 (0.003 to 0.477) | 0.588 (0.292 to 0.919) | 0 | 0.094 (0.013 to 0.257) | 0.077 (0.013 to 0.203) | 0.144 (0.035 to 0.31) | 0.685 (0.512 to 0.846) | 0 |
| 0.095 (0.067 to 0.136) | 4 or 5 | 0.083 (0.001 to 0.326) | 0.074 (0.001 to 0.278) | 0.080 (0.002 to 0.284) | 0.080 (0.001 to 0.296) | 0.683 (0.391 to 0.975) | 0.037 (0.001 to 0.128) | 0.034 (0.001 to 0.113) | 0.075 (0.011 to 0.189) | 0.068 (0.01 to 0.175) | 0.787 (0.65 to 0.915) |
| Network 1 | (Distribution of test results) | (Distribution of test results) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.525 (0.433 to 0.62) | 0.190 (0.131 to 0.256) | 0.122 (0.062 to 0.201) | 0.098 (0.053 to 0.158) | 0.065 (0.033 to 0.106) | 0.450 (0.400 to 0.509) | 0.175 (0.140 to 0.217) | 0.189 (0.147 to 0.236) | 0.112 (0.082 to 0.146) | 0.075 (0.053 to 0.103) | ||
| CF | SF | ||||||||||
| (Prevalence) | ISUP | NC | 1 | 2 | 3 | 4 or 5 | NC | 1 | 2 | 3 | 4 or 5 |
| 0.170 (0.023 to 0.280) | NC | 0.170 (0.023 to 0.28) | 0 | 0 | 0 | 0 | 0.170 (0.023 to 0.28) | 0 | 0 | 0 | 0 |
| 0.279 (0.196 to 0.400) | 1 | 0.221 (0.101 to 0.355) | 0.059 (0.002 to 0.158) | 0 | 0 | 0 | 0.134 (0.068 to 0.24) | 0.145 (0.103 to 0.192) | 0 | 0 | 0 |
| 0.300 (0.211 to 0.436) | 2 | 0.102 (0.009 to 0.261) | 0.106 (0.019 to 0.194) | 0.092 (0.039 to 0.154) | 0 | 0 | 0.127 (0.064 to 0.235) | 0.015 (0.001 to 0.042) | 0.158 (0.111 to 0.205) | 0 | 0 |
| 0.155 (0.109 to 0.223) | 3 | 0.024 (0 to 0.077) | 0.018 (0 to 0.058) | 0.023 (0 to 0.077) | 0.09 (0.042 to 0.153) | 0 | 0.015 (0.002 to 0.042) | 0.012 (0.002 to 0.033) | 0.023 (0.004 to 0.057) | 0.105 (0.074 to 0.142) | 0 |
| 0.095 (0.067 to 0.136) | 4 or 5 | 0.008 (0 to 0.035) | 0.007 (0 to 0.029) | 0.008 (0 to 0.029) | 0.008 (0 to 0.028) | 0.065 (0.033 to 0.106) | 0.004 (0 to 0.013) | 0.003 (0 to 0.012) | 0.007 (0.001 to 0.021) | 0.007 (0.001 to 0.018) | 0.075 (0.053 to 0.103) |
Appendix 11 Model parameterisation
Classification in the diagnostic pathway
Table 79 illustrates the set of possible results (classification) of first, repeat biopsy (for the proportion of individuals who undergo a repeat biopsy), and final classification, according to the joint probabilities of being classified in ISUP grade j with test k conditional on being in true latent category i. The final classification is assumed to correspond to the highest result of the two biopsies, since we are assuming that misclassification at a higher category is not possible. Misclassification at the terminal nodes (final classification) of the model is highlighted in italics in Table 79.
| True disease state | First biopsy classification | Repeat biopsy | Repeat biopsy classification | Final classification |
|---|---|---|---|---|
| No PCa | No PCa | 95%a No | – | No PCa |
| 5%a Yes | No PCa | No PCa | ||
| No PCa | 95%a No | – | No PCa | |
| ISUP grade 1 | 5%a Yes | No PCa | No PCa | |
| (GS 3 + 3) | ISUP grade 1 | 85%a No | – | ISUP grade 1 |
| 15%a Yes | No PCa | ISUP grade 1 | ||
| ISUP grade 1 | ISUP grade 1 | |||
| ISUP grade 2 | No PCa | 95%aNo | – | No PCa |
| (GS 3 + 4) | 5%a Yes | No PCa | No PCa | |
| ISUP grade 1 | 85%a No | – | ISUP grade 1 | |
| 15%a Yes | No PCa | ISUP grade 1 | ||
| ISUP grade 1 | ISUP grade 1 | |||
| ISUP grade 2 | ISUP grade 2 | |||
| ISUP grade 2 | No | – | ISUP grade 2 | |
| ISUP grade 3 | ||||
| (GS 4 + 3) | No PCa | 95%aNo | – | No PCa |
| 5%a Yes | No PCa | No PCa | ||
| ISUP grade 1 | 85%a No | – | ISUP grade1 | |
| 15%a Yes | No PCa | ISUP grade1 | ||
| ISUP grade 1 | ISUP grade1 | |||
| ISUP grade 2 | ISUP grade2 | |||
| ISUP grade 3 | ISUP grade 3 | |||
| ISUP grade 2 | No | – | ISUP grade 2 | |
| ISUP grade 3 | No | – | ISUP grade 3 | |
| ISUP grade 4–5 | No PCa | 95%aNo | – | No PCa |
| (GS ≥ 8) | 5%a Yes | No PCa | No PCa | |
| ISUP grade 1 | 85%a No | – | ISUP grade 1 | |
| 15%a Yes | No PCa | ISUP grade 1 | ||
| ISUP grade 1 | ISUP grade 1 | |||
| ISUP grade 2 | ISUP grade 2 | |||
| ISUP grade 3 | ISUP grade 3 | |||
| ISUP grade 4–5 | ISUP grade 4–5 | |||
| ISUP grade 2 | No | – | ISUP grade 2 | |
| ISUP grade 3 | No | – | ISUP grade 3 | |
| ISUP grade 4–5 | No | – | ISUP grade 4–5 |
Comparison of calibration parameter estimates with those from recent UK cost-effectiveness models
The review in Results of the additional targeted reviews to support model conceptualisation identified that cost-effectiveness models typically consider the increased, or earlier, identification of PCa cases to affect health outcomes by modifying the likelihood of progression to metastatic disease (via earlier, or more appropriate, cancer treatment). Two approaches are used in the long-term outcome component of these models. The first way is to condition speed of progression on true risk group at the time of (or close to) model start. Models that use this approach typically focus on the diagnostic pathway (leading on to treatment decisions). Implicitly, future changes in disease status or in further treatment are implicitly considered in the evidence informing the likelihood of progression over time. One such model is the PROMIS long-term model. 125,126
The second approach implemented is to model explicitly progression across risk groups over time spent in model. Such explicit modelling of progression allows more granularity in the evaluation of monitoring, observation or watchful waiting type strategies, which in turn will determine future treatment decisions. Of the UK cost-effectiveness models, the long-term inference model developed to inform the NICE NG131 model123 (also used in the Southampton DAR116) is of such a kind.
We compared predictions of PCa specific mortality at 2, 5 and 10 years by risk group and treatment from our inference model with those of other UK-relevant models: the NICE NG131 model123 (used in the Southampton DAR116) and the PROMIS model. 125,126
In summary, the outcomes component of the PROMIS model125,126 calibrated the probability of progressing to metastatic disease by risk category and treatment received. Calibration targets were survival data and proportion of patients with metastases by treatment arm from PIVOT109 (risk stratified) considering mortality in the metastatic subgroup from the STAMPEDE trial. 113
The NICE NG131 model also used calibration to derive transitions between risk groups and to metastatic disease (over time), under the assumption that patients would have to be high risk before developing metastatic disease. 123 The calibration targets (risk stratified) were, for undiagnosed cancers, metastases risk from the watchful waiting arm in SPCG4135 and, for diagnosed cancers, cancer-specific survival from Gnanapragasam et al. 112 For both groups, mortality in the metastatic subgroup from the STAMPEDE trial59 was considered.
To compare the different models, results were conditioned on risk group and treatment. To condition on risk group, across all models, prevalence was set to 100% for each of the risk groups in turn. To condition on treatment, diagnostic accuracy was either set at 100% (to secure all patients are diagnosed and treated) or at 0% (to reflect the values if all patients are undiagnosed and untreated). Where relevant, diagnosis due to symptom presentation was not allowed. Where relevant, treatment allocation was set to 100% conservative management or, alternatively, to 100% radical treatment. To derive PCa specific mortality in the PROMIS model,125,126 we only considered mortality in individuals with metastatic disease, and subtracted general mortality. The results of these analyses are presented in Table 80, Appendix 11.
| True disease status | Final classification/treatment | PCa mortality at … | ||
|---|---|---|---|---|
| 2 years (%) | 5 years (%) | 10 years (%) | ||
| NICE NG131 model123 | ||||
| LR | No PCa | < 0.1 | 0.7 | 7.1 |
| LR | < 0.1 | 0.1 | 1.6 | |
| IR | No PCa/LR | 0.3 | 3.9 | 19.3 |
| IR | < 0.1 | 0.7 | 5.2 | |
| HR | No PCa/LR | 1.6 | 9.5 | 28.9 |
| IR/HR | 0.6 | 4.0 | 14.7 | |
| PROMIS125,126 | ||||
| LR | WW | 0.1 | 0.9 | 3.0 |
| IR | WW | 0.3 | 2.1 | 6.4 |
| RP | 0.1 | 0.8 | 2.6 | |
| HR | WW | 0.3 | 2.6 | 7.8 |
| RP | 0.1 | 0.9 | 2.9 | |
| De novo inference model by treatment | ||||
| CPG1 | Conservative | 0.2 | 1.5 | 4.4 |
| Radical | 0.1 | 0.6 | 2.0 | |
| CPG2 | Conservative | 0.5 | 3.5 | 9.8 |
| Radical | 0.2 | 1.6 | 4.6 | |
| CPG3 | Conservative | 1.3 | 8.6 | 20.7 |
| Radical | 0.6 | 4.1 | 10.8 | |
| CPG4 or 5 | Conservative | 3.5 | 18.9 | 36.2 |
| Radical | 1.7 | 10.7 | 24.6 | |
| De novo inference model with weighted treatment estimates | ||||
| CPG1 | Weighted | 0.1 | 0.4 | 3.1 |
| CPG2 | Weighted | 0.3 | 2.2 | 6.2 |
| CPG3 | Weighted | 0.7 | 5.0 | 13.0 |
| CPG4 or 5 | Weighted | 1.8 | 11.2 | 25.6 |
This table highlights that there are marked differences between the predictions, which are primarily due to the sources of long-term outcome evidence these inference models relied upon, which differed. Table 81 depicts PCa mortality at 2, 5 and 10 years observed within the studies that served as calibration targets for the different models.
| Study | Population + treatment | PCa mortality at … | |||
|---|---|---|---|---|---|
| 2 years (%) | 5 years (%) | 10 years (%) | |||
| PIVOT109 | LR | Obs | < 0.1 | 1.8 | 2.2 |
| RP | < 0.1 | 1.0 | 1.5 | ||
| IR | Obs | < 0.1 | 1.7 | 8.3 | |
| RP | 1.2 | 2.4 | 5.3 | ||
| HR | Obs | 0.5 | 3.1 | 17.2 | |
| RP | 2.6 | 5.1 | 5.1 | ||
| SPCG4135 | LR | WW | < 0.1 | < 0.1 | 4.5 |
| RP | < 0.1 | 0.9 | 3.3 | ||
| IR | WW | < 0.1 | 3.8 | 17.8 | |
| RP | < 0.1 | 1.9 | 7.8 | ||
| HR | WW | 1.9 | 7.1 | 22.6 | |
| RP | < 0.1 | 3.9 | 16.9 | ||
| Gnanapragasam (2016),112 3-tier risk groupa | LR | As per clinical practice | < 0.1 | 0.1 | 3.1 |
| IR | 0.1 | 2.0 | 8.6 | ||
| HR | 2.1 | 10.0 | 23.4 | ||
| Gnanapragasam (2016),112 5-tier risk groupa | CPG1 | As per clinical practice | < 0.1 | 1.0 | 4.2 |
| CPG2 | < 0.1 | 1.7 | 7.0 | ||
| CPG3 | < 0.1 | 3.5 | 13.2 | ||
| CPG4 | 0.1 | 5.3 | 17.7 | ||
| CPG5 | 5.7 | 19.4 | 38.1 | ||
| CPG4 and 5b | 2.1 | 10.2 | 24.8 | ||
Treatments distribution
| Metastatic hormone-sensitive treatment | Southampton DAR (%) | Current DAR (%) |
|---|---|---|
| Metastatic hormone-sensitive treatment | Southampton DAR (%) | Current DAR (%) |
| Metastatic hormone-relapsed treatment | Previously treated with | |
| ADT alone (%) | Enzalutamide + ADT (%) | |
| ADT alone | 50 | 50 |
| DTX + ADT | 36 | 9 |
| Enzalutamide + ADT | 7 | 34 |
| Apalutamide + ADT | 7 | 7 |
| Abiraterone | 35 | 0 |
| DTX | 10 | 60 |
| Enzalutamide | 35 | 0 |
| BSC | 20 | 40 |
Adverse events
| AE | AE rates | |||
|---|---|---|---|---|
| LATRUS (%) | Source | LATP (%) | Source | |
| Mild AE | 1.31 | Rosario et al., 2012182 | 9.13 | Pepe and Aragona et al., 2013183 – emergency visits all patients |
| Non-elective admissiona | 3.74 | Tamhankar et al., 2020 | 3.54 | Tamhankar et al., 2020184 |
| Deatha | 0.07 | 0.05 | ||
Health-related quality of life
Resource use and costs
Biopsy procedure costs
This section details the costs associated with the biopsy procedure, which include the following components:
-
Cost of the SF system – costs of the fusion software and, in some cases, a workstation (or cart). This cost only applies to the diagnostic strategies which include a SF component.
-
Cost of the ultrasound – cost of the ultrasound probe/transducer, and any required software. This cost applies to diagnostic strategies with either software or cognitive function components, but some SF systems are not compatible with third-party ultrasounds.
-
Cost of SF system installation – cost of connecting the SF system to the NHS trust IT system. This cost only applies to the diagnostic strategies which include a SF component.
-
Cost of SF system maintenance – costs of service contracts to maintain the technology and keep software up to date. This cost only applies to the diagnostic strategies which include a SF component.
-
Costs of SF system training – staff time costs required to train NHS professionals to perform biopsies. The use of SF methods requires additional training compared to CF, but the cost of training also varies across biopsy approaches (by route of access).
-
Cost of staff time to perform the biopsy procedure – cost of urologists, nurses and anaesthetist (for procedures requiring general anaesthesia). This cost varies across biopsy approaches (by route of access and type of anaesthesia), but there is also a difference in procedural time between SF and CF.
-
Cost of the biopsy setting – costs of the setting in which the biopsy procedure takes place (outpatient room, theatre session); it varies by route of access, type of anaesthesia, and MRI-influenced method.
-
Costs of other biopsy devices and consumables – cost of (1) devices and equipment (e.g. freehand needle positioning devices, lithotomy beds and biopsy guns), and (2) needles and other materials requiring replacement (immediate or after a certain number of uses). These costs are often specific to the biopsy approach [transrectal or transperineal (stabilised, freehand or double freehand)], and may differ across MRI-influenced methods and across SF systems, due to compatibility issues.
-
Cost of histopathology analysis and report – costs of processing the biopsy sample and communicating the results to the patient in a consultation. This cost applies to all strategies but may differ for strategies using different sampling methods (combined vs. targeted-only biopsy), as these may result in different number of cores being sampled. These costs are reported in the Appendix 11, as they are not SF specific.
In the subsequent sections we start by discussing patient throughput and then provide more detail in each component of cost described above, with emphasis in those costs that vary by MRI-influenced method and/or across SF systems. Further information is provided in Appendix 11. All costs presented are exclusive of value-added tax, unless otherwise stated.
Patient throughput
The annual patient throughput represents the average annual number of targeted biopsies (alone or in combination with systematic biopsy) per NHS trust. The annual patient throughput is determinant to calculate the cost of biopsy. The EAG did not identify a source directly reporting this estimate. The evidence considered and the calculations used to inform our base case assumption of throughput are described below.
We considered the estimates of throughput applied in the Southampton DAR,116 which assumed 18 weekly and 1000 annual biopsies (not distinguishing throughput between systematic and targeted biopsies). Clinical advisers to the EAG considered that the annual estimate is likely to overestimate the average total number of biopsies per NHS trust and may be more reflective of a very high throughput centre.
We also examined prostate biopsy activity numbers across all HRGs in the main schedule of NHS reference costs across three financial years (2018–9, 2019–20, 2020–1)185–187 for the prostate biopsy currency codes across all HRG data [LB76Z (transrectal ultrasound guided biopsy of prostate) and LB77Z (transperineal template biopsy of prostate)] and contrasted these figures against those reported for the latest available NPCA annual report,4 as illustrated in Table 85, Appendix 11. We did not consider earlier versions of the NPCA annual reports due to changes in the reporting style and high level of missing data, which hinder establishing meaningful comparisons across time. We note as a limitation of the NHS reference data that the TP currency code suggests these were transperineal template biopsies, so it is unclear how other types of transperineal biopsies were captured in the data set.
Although the NPCA reports data for both England and Wales, the total number of biopsies reported is lower than that reported for a similar period in the main schedule NHS reference costs; this is due to missing data issues. To estimate the average number of biopsies per NHS England trust and/or Welsh University Health board, we assumed the number of institutions from which the NCPA collected data in 2019–20 (127 NHS trusts and 5 University Health Boards). 188 Although clinical guidance has recommended performing a mpMRI before any biopsy is offered at least since 2019, NICE has identified data suggesting that in 2019189 only 87% of biopsies were preceded by a mpMRI in England and Wales. Thus, we used the 87% estimate (varied in scenario analysis to 100%, Multiparametric magnetic resonance imaging and compliance with National Institute for Health and Care Excellence guidance, to explore the impact of complete compliance with clinical guidance) to adjust the average annual number of biopsies by NHS trust. Finally, we estimated the average annual number of targeted biopsies by assuming that 72.6% of biopsies preceded by a mpMRI had a Likert or PI-RADS score of at least 3, as this is the threshold at which targeted biopsy is recommended. The 72.6% was obtained by pooling the proportion of patients in two relevant RCTs [71.8% in PROMIS (UK) and 72.6% PRECISION (11 countries)]19,126 who had a mpMRI result of at least 3 (Likert or PI-RADS).
| Data collection period | Data source | |||||
|---|---|---|---|---|---|---|
| NHS reference costs; all HRG data | NPCA annual report4 | |||||
| 2018–9 financial year184 | 2019–20 financial year185 | 2020–1 financial year186 | April 2019–March 2020 | |||
| Country | England | England | Wales | England and Wales | ||
| Biopsy route | ||||||
| TP biopsy | 39,211 | 30,451 | 11,492 | 20,623 | 969 | 21,592 |
| TR biopsy | 2,1424 | 21,674 | 22,332 | 13,756 | 300 | 13,756 |
| Total biopsies per year | 60,635 | 52,125 | 33,824 | 34,379 | 1269 | 35,348 |
| Estimated annual number of biopsies preceded by a MRI/NHS trusta | 52,752 | 45,349 | 29,427 | 29,910 | 1104 | 30,753 |
| Estimated annual number of biopsies/NHS trusta | 415 | 357 | 232 | 236 | 221 | 235 |
| Estimated annual number of targeted biopsies/NHS trusta | 300 | 258 | 168 | 170 | 160 | 170 |
The evidence considered suggests the average annual number of targeted biopsies (alone or in combination with systematic biopsy) per NHS trust in England is in a range within 168 and 300. However, the two latest data cuts of NHS-reference costs185–187 are likely to be affected to some extent the impact of COVID-related constraints on NHS service provision. Therefore, we consider that the expected patient throughput is likely to be closer to the upper bound of the estimated range and consider an annual throughput of 300 targeted biopsies in the base-case analysis.
Cost of the software fusion system and ultrasound components
The MRI-fusion systems under comparison differ in terms of their compatibility with third-party ultrasound devices (and are, therefore, sold without an ultrasound component), with the ultrasound component being an integral part of the SF system for some technologies (e.g. KOELIS Trinity). Therefore, the capital costs of the SF systems and ultrasound components are reported jointly in this section.
Only five companies provided information on the costs of the technologies under comparison; these were BK Medical UK Ltd (with MIM Software Inc. for bkFusion), Exact Imaging (for FusionVu), Focal Healthcare (for Fusion Bx 2.0), KOELIS (with Kebomed for KOELIS Trinity), and MedCom (BiopSee). No information was provided for the costs of ARTEMIS, iSR’obot Mona Lisa, and UroNav Fusion Biopsy System. The capital costs of the SF systems and ultrasound components for bkFusion, FusionVu, Fusion Bx 2.0, KOELIS Trinity and BiopSee are summarised in Table 86, alongside the lifespan of the equipment.
For three SF systems (bkFusion, FusionVu, and KOELIS Trinity) the SF component is integral to the ultrasound component (or the micro-ultrasound component for FusionVu). In the other two systems (Fusion Bx 2.0 and BiopSee) the fusion software is installed on a standalone workstation (or cart), which is integral to the SF system, but does not comprise an ultrasound system. Fusion Bx 2.0 and BiopSee require third-party ultrasounds and transducers to perform prostate biopsies, for which the costs are NR in Table 86 (as sold by third party). Both Fusion Bx 2.0 and BiopSee include a cart; the cart is an integral part of each technology.
For SF systems that are compatible with third-party ultrasounds (i.e. BiopSee and Fusion Bx 2.0), we assume the same cost for the ultrasound components as for CF. In the base-case, this cost was derived from the cost of the three standalone ultrasound machines in the Southampton DAR116 (FUJIFILM transducer and Ultrasound System [inflated to 2020–21 price year according to the NHSCII;154BK ultrasound system and urology software with transducer; Trinity® 3D Prostate Suite plus KOELIS Sidefire Ultrasound probe). We averaged across the costs of these three technologies (with costs updated based on the information provided by bkMedical and KOELIS and Kebomed in the context of the current DAR for the BK ultrasound and Trinity ultrasound components) to estimate an average annual capital cost for ultrasound of £10,846 and £10,974 for transrectal and transperineal biopsies, respectively.
| Type of SF system | Technology | SF costs | Ultrasound costs | Lifespan (years) |
|---|---|---|---|---|
| Fully integrated system | bkFusion | Cart and software: 52,250a | bk3000 ultrasound: £37,500 Prostate procedural application: £1800 DICOM standard with encryption: £1700 Leakage test kit: £332 Transducer: £15,000 Sensor clamp for the transducer: £200 |
8 (4 for transducer and sensor clamp) |
| FusionVu | £124,958b | 5 | ||
| KOELIS Trinity | £23,620 | Ultrasound: £45,000 Transrectal software: £39,948c Transperineal software: £41,754c |
5 | |
| Compatible with third party= ultrasounds | BiopSee | Transrectal: - Software: £15,000 - Cart: £12,000 Transperineal: - Software: £20,000 - Cart for stabilised biopsy: £8000 - Cart for freehand biopsy: £20,000 |
NA | 10 |
| Fusion Bx 2.0 | Software: £24,244d Cart: £96,974d |
NA | 10 | |
For bkFusion, lifespan estimates provided by the company for the transducer (3–5 years) and leakage test kit (8 years) are said to be end-user dependent. We assumed that the transducer lifespan corresponded to the midpoint of the range provided by the company (i.e. 4 years). The lifespan of the sensor clamp for the transducer was not provided by the company despite the EAG request to provide this information for all components, so we assumed it was the same as for the transducer.
Commercial discounts may be available for bkFusion. The company stated that We have a 5 years fixed service contract (excluding any civco and mim products) called Priority Care – at point of sale if the service contract is purchased we provide a 10% discount to the priority care quote. If priority care is not purchase within the systems first 2 years of life you can not access this contract again. Alternative contracts are available. However, the company did not detail what was included in the Priority Care quote and how much it costs. It is also not clear if the discount applies to maintenance or equipment costs, as we do not know what is covered by the Priority Care quote. Therefore, the information provided by the company is insufficient to implement the discount in the model, and this is not considered by the EAG.
FusionVu uses micro-ultrasound technology, and therefore, does not require ultrasound components. The cost presented for this technology in Table 86 reflects the cost of all equipment and software components. We note that the company stated that they are willing to offer a discount to the UK NHS but that they could not finalise it within the timelines of this DAR.
KOELIS and Kebomed also stated that they can offer discount for multi-unit purchases of KOELIS Trinity, but these depend on the number of units purchased, method of purchase and specification of the units (response to EAG’s RFI, question 11). The company did not provide further details on the discounts available, and therefore this is not modelled.
Commercial discounts may also be available for BiopSee according to MedCom, who states that these discounts are usually handled by distributors. As no further information on the applicability and size of the discounts was provided, we could not model discounted costs for BiopSee.
The costs of some SF systems and/or ultrasound components were specific to the biopsy approach in terms of route of access (transrectal or transperineal) and/or the fixation method [stabilised, (single) freehand and double-freehand], so for KOELIS Trinity and BiopSee costs will vary conditional on the diagnostic strategy they are being used in. The costs provided by the company for bkFusion were reported solely for a transperineal procedure, despite the EAG request to provide costs by biopsy approach. Therefore, it was assumed that the costs of bkFusion are the same across biopsy approaches.
The software costs assumed for Fusion Bx 2.0 (£24,244) assume the purchase of a perpetual licence. The company also provided the cost of an annual licence costing a third of the perpetual licence. Given the lifespan of Fusion Bx 2.0 exceeds 3 years (point beyond which the annual cost of a perpetual license becomes lower than the annual license), we did not consider annual licences as an option. The company stated that a discount on the software and hardware components of Fusion Bx 2.0 of up to 30% could be offered to the NHS, depending on the number of systems purchased. We did not implement this discount on our base-case analysis, as the company did not specify the level of discount applied conditional on number of units purchased.
Some SF systems had optional probe holders and software components, which were not considered in the costs of the ultrasound components, as these are not essential components of the technology. We note that the cost of Fusion Bx 2.0 includes one probe holder as an integral part of the system, and therefore, this cost was not excluded.
The costs of SF systems and ultrasound components were annuitised at a 3.5% discount. Annuitised costs and costs per biopsy are reported in Table 87.
Cost of installation of software fusion systems
One company (Medcom) reported the time required to install the software fusion technology to the NHS trust IT system as ranging between 30 and 60 minutes. We assumed that this results in a one-off staff time cost, which is applicable to all SF systems. The cost of installation was estimated assuming it would take 45 minutes (midpoint of the time range provided by Medcom) of an IT worker time [costed at £35.67 per hour (average working hour of band 4 hospital-based scientific and professional staff)154]. The cost was distributed over the annuitised (3.5% annual rate) average lifespan of the five SF systems for which the companies had submitted costing information. The resulting annual cost and cost per biopsy were estimated to be £3.97 and £0.01, respectively.
Cost of software fusion system maintenance
The costs of maintaining the SF systems mostly consist of the costs of service contracts. These contracts also include maintenance of the ultrasound components when the ultrasound components are integral to the SF system. The maintenance contracts are summarised in Table 88 alongside the annual cost estimate applied in the model.
| Technology | Biopsy approach | Annuatized cost | Cost per biopsy | ||||
|---|---|---|---|---|---|---|---|
| TR | TP stabilised | TP freehand | TR | TP stabilised | TP freehand | ||
| Type of system | |||||||
| bkFusion | Fully integrateda | £17,152 | £57.17 | ||||
| FusionVu | Fully integrateda | £26,740 | £89.13 | ||||
| KOELIS Trinity | Fully integrateda | £23,233 | £23,619 | £23,619 | £77.44 | £78.73 | £78.73 |
| BiopSee | SF alone | £13,982 | £14,227 | £15,621 | £46.61 | £47.42 | £52.07 |
| Fusion Bx 2.0 | SF alone | £24,928 | £25,057 | £83.09 | £83.52 | ||
| bkFusion | CF | £10,846 | £10,974 | £36.15 | £36.58 | ||
| Technology | Maintenance contract duration and cost | Costs of maintenance applied in the model | |
|---|---|---|---|
| Annual cost | Cost per biopsy | ||
| bkFusion | 5 years: £66,975.00 | £13,395.00 | £44.65 |
| FusionVu | NR | £12,206.12 | £37.20 |
| KOELIS Trinity | Essential – 1 year: £5,500.00 | £11,017.24 | £29.76 |
| Comfort – 1 year: £7 465.52 | |||
| Serenity – 1 year: £11,017.24 | |||
| BiopSee | NR | – | – |
| Fusion Bx 2.0 | 1 year: £9697.44a | £9697.44 | £32.32 |
Three companies provided information on the duration and cost of the maintenance contracts. Most contracts had an annual duration; only bkFusion had a 5-year maintenance contract. Given the lifespan of bkFusion is > 5 years, we distribute this cost equally over time and apply it as a cost of £13,395 per annum in the model. We note that there are discounts available for alternative maintenance contracts for bkFusion, which have been described above.
KOELIS Trinity has three levels of maintenance contract, which differ in terms of annual costs; the levels are: Essential, Comfort and Serenity. According to the company Serenity is the level most often purchased (50%) followed by Essential (34%), and Comfort (16%). In the model, we assume the annual cost of the contract to be a weighted average of the three contract levels by the corresponding ‘market share’, resulting in an annual cost and a cost per biopsy of £8926.90 and £29.76, respectively.
We note that the maintenance service contract cost for Fusion Bx 2.0 is an approximate estimate provided by the company, who stated that this would typically cost US $12,000 or less and that they plan to enlist a UK distributor to perform this service. Alternatively, the maintenance could be conducted by hospital staff who are responsible for performing preventative maintenance of other medical devices, and who would need to undergo annual on-site maintenance training (1–2 hours). The company also stated in the responses submitted to NICE that if the maintenance contract is longer than 1 year, the cost would be discounted accordingly. However, this does not provide information on the level of discount over time, so this potential discount cannot be implemented without more detail. In the model, we consider only the approximate cost of an annual maintenance service delivered by the company or their distributors.
No maintenance costs were provided for FusionVu and BiopSee. The company who commercialises FusionVu stated that their technology is serviced through a local distributor in the UK under annual or more contracts, but could not yet provide a cost estimate for the contract. Therefore, we have assumed that the FusionVu maintenance contract costs is an average of the two SF systems with fully integrated SF system and ultrasound components (bkFusion and KOELIS Trinity). Medcom stated that BiopSee does not require any maintenance, as damaged parts can be repaired on demand and reported cost range for repairing accessory equipment (e.g. £200–600 to replace a mouse or an accidentally damaged cable, and £100–3000 to replace a damaged stepper or stabiliser). However, it is unknown how often damage to different components is likely to occur, and so estimate a maintenance cost on a per damaged part basis. We could have assumed a maintenance cost similar to that of Fusion Bx 2.0 (the other SF system that does not have an integral ultrasound component); however, we note that the maintenance cost for Fusion Bx 2.0 is an approximate estimate provided by the company and assumes that there is a service contract (not available for BiopSee). In our base-case analysis, we assume that there is no cost attached to maintaining BiopSee.
The cost of software updates is included within the maintenance contract for most technologies. One of the exceptions is bkFusion, which only includes software malfunction fixes. No software update costs were provided for bkFusion, but we note that the lifespan of the hardware and software components for this technology are generally the same. The cost of software updates for BiopSee SF software is 50% of the software cost and new versions are usually released annually, according to the company. The company also stated they do not plan to withdraw the current version being used in the UK NHS. Therefore, we assume that no additional costs for software updates need to be considered for any of the technologies.
Cost of software fusion training
The technology specific cost of SF systems training consists of the cost of staff time to attend the training sessions, as companies do not charge for training provision. Each company provides a core training programme composed of different elements. The information provided by the companies on the NHS staff who should undergo core training and the time required per training component is summarised in Table 89.
| Technology | NHS staff | Training components | Duration |
|---|---|---|---|
| bkFusion | Urologists, radiologists, radiation oncologists, sonographers and assisting staff | Not described | 1 or 2 days |
| FusionVu | Urologists, radiologists, nurses and sonographers | eLearning | 2 hours |
| On-site training | 1 hour | ||
| Live expert support | 10–15 cases | ||
| KOELIS Trinity | End user, consultant, radiologist, CNS | Pre-installation training | 3 hours |
| OPD staff, theatre staff, ODP | Installation training | 1 hour | |
| End user, consultant, radiologist, CNS | Theatre List | 4 or 5 cases | |
| BiopSee | Urologists/radiologists | Not described | 3 hours |
| Nurses | 1 hour | ||
| Fusion Bx 2.0 | Urologists, nurses and/or sonographers | Video training | 1 hour |
| Hands-on training with phantom prostate | 0.5–0.75 hour | ||
| Support to clinical cases | 10–20 casers over 2–3 days |
To estimate an annual cost of training for the use of SF per trust for each technology, we assumed that core training would be delivered to two urologists, two nurses, one radiologist and one sonographer, and training would remain up to date for 5 years (the shorter lifespan across fusion software). We used the training duration provided by the companies to estimate staff time requirements, and assumed the same time for all categories of staff unless the company stated different times by category of staff. Where the companies provided training duration as a range we assumed the staff time requirement would correspond to the midpoint of that range. We did not include any staff time for theatre list or support to clinical cases, as we assumed that this would not result in additional time requirements in relation to the procedure time. The information used to estimate the costs of SF systems training is presented in Table 90 alongside the annuitised annual cost (at 3.5% per annum) of training for each technology. Unit costs were sourced from the PSSRU (2021) unit costs report. 154
We also considered the cost of training to perform biopsy procedures more generally; these were assumed to vary by biopsy access route (transperineal vs. transrectal) in line with the Southampton DAR. We assumed the same level resource use per biopsy approach as was assumed in the Southampton DAR and updated the unit costs to reflect our analysis price year. 154
FusionVu has a free-of-charge optional training programme, the Mastery programme. The company did not provide clear information on the staff time requirements to undergo this optional training, or clarify to whom it would be delivered. The company stated that the effectiveness of the Mastery programme was studied in Cash et al. 158 but it was not possible to ascertain based on the information provided if the Mastery programme described by the company corresponded to the training programme assessed in this publication. Given this and the optional nature of this training component, we have not included this cost in our cost-effectiveness analysis.
| NHS staff time | Annuitised annual cost | Cost per biopsy | Unit cost | |||||
|---|---|---|---|---|---|---|---|---|
| Urologist | Nurse | Radiologist | Sonographer | Staff | Cost per working hour | |||
| Technology specific | ||||||||
| bkFusion | 2 × 11.25 hoursa | 2 × 11.25 hoursa | 1 × 11.25 hoursa | 1 × 11.25 hoursa | £1029.97 | £3.43 | Urologist and radiologist | £87.50, average of hospital-based registrar and medical consultant PSSRU (2021)154 |
| FusionVu | 2 × 3 hours | 2 × 3 hours | 1 × 3 hours | 1 × 3 hours | £274.66 | £0.92 | ||
| KOELIS Trinity | 2 × 3 hours | 2 × 3 hours | 1 × 3 hours | 1 × 4 hoursb | £285.86 | £0.95 | Nurse | £46.00, average of hospital-based nurse specialist/team leader (band 6) and nurse advanced/team manager (band 7) PSSRU (2021)154 |
| BiopSee | 2 × 3 hours | 2 × 1 hour | 1 × 3 hours | 1 × 1 hour | £203.90 | £0.68 | Sonographer | £52.33, average hospital-based scientific and professional staff (band 6) PSSRU (2021)154 |
| Fusion Bx 2.0 | 2 × 1.625 hours | 2 × 1.625 hours | 1 × 1 hourc | 1 × 1.625 hours | £137.07 | £0.46 | ||
| Biopsy approach specific | ||||||||
| Transrectal | 5 × 1 hour | – | – | – | £437.50d | £1.46 | Urologist | £123, medical consultant PSSRU (2021)154 |
| Transperineal | 5 × 8 hours | – | – | – | £3500.00d | £11.67 | ||
Cost of staff time to perform the biopsy procedure
The staff costs associated with the biopsy procedure are likely to vary depending on the biopsy route of access and the type of anaesthesia. We based our estimates of staff time requirements to conduct the biopsy on the Southampton DAR. 116
In the Southampton DAR, each local anaesthesia biopsy was assumed to require one urologist and two nurses, with general anaesthesia biopsy further requiring one anaesthetist. The time required for LATP biopsy was sourced from the published literature for two devices (0.41 and 0.33 hour for CamPROBE and PrecisionPoint™, respectively), with an average of the two assumed for the devices for which there was no published evidence. For GATP biopsy, the procedure time was assumed to be 1 hour. To estimate the procedure time of local anaesthesia transrectal (LATRUS) biopsy, the EAG applied a ratio of procedure time between the transrectal and transperineal approach (0.84) derived from the literature to the time estimates by transperineal device.
In this study, we have assumed the procedure time of LATP conducted with PrecisionPoint, as the diagnostics consultation document (DCD) for the Southampton DAR29 suggests that CamPROBE will not be recommended for use in the NHS UK. We applied the same LATRUS/LATP time ratio as in the Southampton DAR to the LATP time estimate to calculate the LATRUS procedure time and assumed GATP would take 1 hour. We also assumed that 50% of procedures would be undertaken by urologists and 50% by sonographers. The remaining staff requirements were assumed the same as in the Southampton DAR (i.e. two nurses plus one anaesthetist if GATP). We applied the same unit cost as those used to cost training costs (see Table 90) and assumed the same unit cost for anaesthetist time as for the urologist time. The procedure time costs by route of access and type of anaesthesia are summarised in Table 91.
Procedure time may further increase when this is performed with fusion software compared to CF. This additional time is due to the need (1) to contour the prostate in the MRI and ultrasound images, and (2) to connect the MRI-fusion system. The companies provided different estimates of how much time would be added to the biopsy procedure when using their technologies (see Table 92), but these were not supported by published evidence. As discussed in Clinical effectiveness results, the procedure time estimates in the diagnostic literature do not allow disentangling the additional procedure time due to software compared to CF from procedure time differences associated with the biopsy approaches. The clinical advisers to the EAG commented that the additional procedure time for SF (vs. CF) should be approximately 10 minutes in a high- throughput centre, 5 minutes of which would correspond to additional time to import and obtain the appropriate MRI sequences (radiologist time) and 5 minutes during the actual biopsy (urologist/sonographer and nurse time) to connect the SF systems and contouring the ultrasound. They also noted that these time estimates could be longer when the use of these interventions is first rolled out, due to lack of experience.
| Biopsy approach | Procedure time (hours) |
NHS staff | Cost per biopsy (£) | |||
|---|---|---|---|---|---|---|
| Urologist | Nurse | Sonographer | Anaesthetist | |||
| LATP | 0.33 | 0.5 | 2 | 0.5 | – | 60.36 |
| GATP | 1 | 1 | 270.42 | |||
| LATRUS | 0.28 | – | 50.70 | |||
| Fusion system | MRI contouring | Connect fusion system to ultrasound | Contouring ultrasound |
|---|---|---|---|
| bkFusion | 3–5 minutes | NR | -a |
| FusionVu | 1 minute | NR | 10 seconds |
| KOELIS Trinity | 5 minutes | NR | 5 minutes |
| BiopSee | 1–2 minutes | NR | < 1 minute |
| Fusion Bx 2.0 | 8–10 minutes | 30 seconds | 5–10 minutes |
We calculated the additional staff time costs, required to conduct SF, based on the time estimates provided by the clinical advisers to the EAG. We assumed the same staff requirements per type of biopsy approach, as for the core biopsy procedure time, and further accounted for the additional time requirements for one radiologist. We applied the same unit cost as those used to cost training costs (see Table 90) and assumed the same unit cost for anaesthetist time as for the urologist time. The additional procedure staff time costs for SF compared to cognitive by route of access and type of anaesthesia are summarised in Table 93.
Cost of the biopsy setting
The Southampton DAR116 examining the cost-effectiveness of LATP, GATP and LATRUS considered a cost for the setting on which the biopsy took place, with LATRUS and LATP being conducted in an outpatient room, and GATP in a theatre session. These costs were sourced from an unpublished study submitted by the sponsor of one of the technologies under assessment in the Southampton DAR116 and suggested a unit cost of £43 and £129 per hour for the outpatient room and theatre session, respectively. These unit costs were inflated to 2020–1 price year using the NHSCII,154 applied to the duration of the procedures for each biopsy approach, to estimate the cost of the setting.
The micro-costing study is not described in sufficient detail to understand what is included in the costs of biopsy setting. This DAR’s EAG has decided to include the cost of setting for consistency with the Southampton DAR,116 but notes the opacity of the cost estimates as a potential limitation.
The cost of biopsy setting, applied in the model for strategies using CF, was calculated by multiplying the time of the procedure by biopsy approach (see Table 91) by the unit costs by setting (inflated to 2020–1 price year)154 in the Southampton DAR. 116 For strategies using SF, we assumed that the procedure would take 10 additional minutes (in line with the assumptions to estimate the additional staff time to conduct SF and assuming that the MRI is also done in an outpatient setting). Costs associated with biopsy setting by biopsy approach and MRI-influenced method, as well as the model inputs to estimate these, are summarised in Table 94.
Costs of other biopsy devices and consumables
The biopsy procedure requires other devices and consumables which may vary by biopsy approaches (GATP, LATP, LATRUS). While these devices and materials are not required to conduct biopsy procedures with either software or CF, some technologies have compatibility issues which mean that costs of technology-specific materials may have to be considered where appropriate to fully account for differences in costs between the different SF systems. For example, FusionVu is only compatible with needle guides commercialised by Exact Imaging, meaning that in principle, you cannot use other needle guides that would be suitable for CF or other SF systems without compatibility issues. In our base-case analysis, we apply a simplifying assumption that the costs of the biopsy devices do not vary by MRI-influenced methods.
| approach | NHS staff time (minutes) | Additional cost per SF biopsy | ||||
|---|---|---|---|---|---|---|
| Radiologist | Urologist | Nurse | Sonographer | Anaesthetist | ||
| LATP | 1 × 5 | 0.5 × 5 | 2 × 5 | 0.5 × 5 | – | £22.53 |
| GATP | 5 × 1 | £29.83 | ||||
| LATRUS | – | £22.53 | ||||
| Approach | Procedure time (hours) | Unit cost (per hour) (£) | Cost per biopsy | ||
|---|---|---|---|---|---|
| CF | SF | Cognitive fusion (£) | SF (£) | ||
| LATP | 0.33 | 0.50 | 44.32 | 14.63 | 22.01 |
| GATP | 1.00 | 1.17 | 132.97 | 132.97 | 155.14 |
| LATRUS | 0.28 | 0.44 | 44.32 | 12.29 | 19.67 |
Transperineal biopsies can be conducted with a (1) grid and stepper unit; (2) freehand device (the Southampton DAR116 assessed five of these devices) or (3) coaxial needle (one such device assessed in the Southampton DAR). Grid and stepper units are used for stabilised biopsies with the stepping unit usually fixed to a stabiliser (mounted onto a table or supported by a floor stand). The stepper is a reusable device used to hold the ultrasound probe, while a (single use or reusable) grid is used to guide the needle insertion. Grid and stepper units can be used to perform transperineal biopsies under or local general anaesthesia. Recent LATP techniques are performed using an access needle guide (or equivalent) to pierce the perineum and through which the biopsy needle passes to sample the prostate. These techniques can be performed using (1) freehand devices attached to the ultrasound probe or (2) a co-axial needle not attached to the probe (also known as double freehand technique).
We based the costs of a grid and stepper unit for stabilised GATP and LATP biopsy on the estimates used in the Southampton DAR for this cost element with adjustments to reflect our throughput estimates. We assumed a cost of reprocessing reusable materials of £5.15 (cost of cleaning and sterilising), sourced from the Southampton DAR and inflated to 2020–1 price year using the NHSCII. 154
The information considered by the EAG for costing the freehand biopsy devices is summarised in Table 95.
For costing transperineal biopsies with (single) freehand techniques with CF, we have considered the costs of the five freehand devices evaluated in the Southampton DAR116 [PrecisionPoint (BXTAccelyon), EZU-PA3U (Hitachi), UA1232 (BK Medical), Trinity® Perine (KOELIS and Kebomed), and SureFire (Delta Surgical); inflated to 2020–1 price year using the NHSCII]. 154 We have updated the costs of Trinity Perine device based on the cost of the reusable Perine Grid 18G provided by KOELIS and Kebomed in the context of the current DAR (£779.31; 100 uses). We note that KOELIS and Kebomed also commercialise single use Trinity Perine grids (costed as £62.04 and £86.20 for a Mini grid and a Full grid, respectively – not modelled); this are not included in the model but would yield higher costs per biopsy than the single use devices. We included a £5.15 cost of reprocessing for the reusable devices [EZU-PA3U (Hitachi), UA1232 (BK Medical), Trinity® Perine (KOELIS and Kebomed)]. In the base case, the cost of freehand devices is an average of the costs for the five devices and applies equally to cognitive and SF.
| Device | Manufacturer | Compatible with | Cost of device | Number of uses | Reprocessing | Co-axial needle | Source |
|---|---|---|---|---|---|---|---|
| PrecisionPoint | BXTAccelyon | KOELIS Trinity, BiopSee, Fusion Bx 2.0 | £206.16 | 1 | – | – | Southampton DAR;116 Inflated to 2020–1 price year154 |
| £250.00 | NR | NR | NR | KOELIS and Kebomed response to NICE and/or EAG RFI | |||
| £350.00 | NR | NR | NR | Focal Healthcare response to NICE and/or EAG RFI | |||
| £150–250 | NR | NR | NR | Medcom response to NICE and/or EAG RFI | |||
| FusionVu guide | Exact Imaging | FusionVu | £1333 | 24 | – | – | Exact Imaging response to EAG RFI |
| EZU-PA3 | Hitachi | ? | £1971.66a | 100b | £5.15 | £22.06 | Southampton DAR;116 Inflated to 2020–1 price year154 |
| UA1232 | Bk Medical | bkFusionc | £1443.12 | 100b | £5.15 | – | Southampton DAR;116 Inflated to 2020–1 price year154 |
| Trinity Perine | KOELIS and Kebomed | KOELIS Trinity | £777.64 | 100 | £5.15 | – | Southampton DAR;116 Inflated to 2020–1 price year154 |
| Perine Grid 18G | £779.31 | 100 | NR | used with or without a guide needle | KOELIS and Kebomed response to NICE and/or EAG RFI | ||
| Full Grid 18G | £1303.44 | 100 | NR | ||||
| Perine Mini Grid | £86.20 | 1 | |||||
| Perine Full Grid | £62.04 | 1 | |||||
| SureFire | Delta Surgical | Fusion Bx 2.0 | £123.70 | 1 | – | Southampton DAR;116 Inflated to 2020/2021 price year154 | |
| £125.00 | NR | NR | NR | Focal Healthcare response to NICE and/or EAG RFI | |||
| Unnamed reusable device | NR | BiopSee | £700.00 | NR | NR | NR | Medcom response to NICE and/or EAG RFI |
The costs of transperineal devices applied in the model for the base-case analysis were, thus, £90.44 and £81.86 for stabilised and freehand biopsy, respectively.
We did not consider the costs of LATP with double-freehand technique, as the provisional DCD for the previous DAR does not recommend the use of double-freehand devices to conduct LATP. We also did not consider any device costs to conduct LATRUS in line with the Southampton DAR. 116
We included the annuitised cost of a lithotomy bed (£10,308, 10-year lifespan) in the calculations of the cost per biopsy of TP; this cost was sourced from the Southampton DAR,152 and inflated to 2020–1 price year using the NHSCII. 154
The costs of general consumables by biopsy approach were also sourced from the previous DAR,116,152 where they are detailed (see Table 113 of the Southampton DAR). We applied a cost per biopsy of £80.7, £65.55, and £79.10 for LATP, GATP and LATRUS, respectively.
Cost of histopathology analysis and report
The Southampton DAR152 assumed that the cost of histopathology analysis was dependent on the number of cores sampled and each biopsy involved sampling 12 cores.
There was limited comparative evidence to inform any differences in the number of cores sampled between cognitive and software fusion identified in the clinical review, as most diagnostic accuracy studies performed a fixed pre-specified number of cores per biopsy. One RCT31 reported the median number of cores per subject undergoing a targeted biopsy; 4 [interquartile range (IQR): 3–5, n = 79] and 3 (IQR: 3–3; n = 78) for software and CF, respectively. This suggests that fewer cores than 12 would require analysis per targeted biopsy, and that differences between MRI-influenced methods are small. However, the study had a small sample size and this was not a primary outcome, so it is unlikely that the study was powered to identify any differences in this particular outcome between MRI-influenced methods.
The unit cost of histopathology analysis, of the cores sampled through biopsy, was sourced in the Southampton DAR116,152 initially from a histopathology pricing document by the University of Surrey, and then corrected to a HRG cost (£36.58; currency code DAPS02: Directly Accessed Pathology Services – Histopathology and histology). 186 The resulting cost for the analysis of a 12-core biopsy was £438.96 in the Southampton revised base-case analysis, which assumed the unit costs applied to each core tested. This level of resource used applied is more in line with some systematic biopsies (see Biopsy).
In the York model, we assumed that the NHS reference cost applied in the Southampton model, also applied to a single targeted biopsy (with fewer than 12 cores sampled per biopsy). We sourced the same HRG currency cost (£16.29) from the latest version of the NHS reference costs187 and applied it to each targeted biopsy. We also did not identify comparative evidence on the number of cores sampled for targeted and combined biopsies, so no differences were assumed. We note that if we have underestimated the histopathology analysis cost of biopsy (targeted or combined) that this would only be likely to impact the cost-effectiveness estimates if there were considerable differences in the rates of subsequent biopsy between the intervention and comparator.
We also considered the cost of reporting to the patient the biopsy result. In line with the previous DAR,152 this was assumed to require a 30-minute appointment with a urologist (medical consultant, £123 per hour),154 resulting in a cost per biopsy of £61.50.
Costs per software and cognitive fusion biopsy
| LATRUS | bkFusion (£) | FusionVu (£) | KOELIS Trinity (£) | BiopSee (£) | Fusion Bx 2.0 (£) | CF (£) |
|---|---|---|---|---|---|---|
| Technology specific | ||||||
| MRI fusion and US | 57.17 | 89.13 | 77.44 | 46.61 | 83.09 | 36.15 |
| Installation | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| Maintenance | 44.65 | 37.20 | 29.76 | 32.32 | ||
| Training | 3.43 | 0.92 | 0.95 | 0.68 | 0.46 | |
| Procedure time | 22.53 | 22.53 | 22.53 | 22.53 | 22.53 | |
| Biopsy setting | 19.67 | 19.67 | 19.67 | 19.67 | 19.67 | 12.29 |
| TP biopsy devices | ||||||
| Total | 147.48 | 169.47 | 150.37 | 89.51 | 158.10 | 48.44 |
| Not technology specific | ||||||
| Training | 1.46 | |||||
| Procedure time | 50.70 | |||||
| General consumables | 79.10 | |||||
| Lithomy bed | ||||||
| Histology | 77.79 | |||||
| Total | 209.05 | |||||
| Total per biopsy | 356.53 | 378.53 | 359.43 | 298.56 | 367.15 | 257.49 |
| LATP | bkFusion (£) | FusionVu (£) | KOELIS Trinity (£) | BiopSee(£) | Fusion Bx 2.0 (£) | CF (£) |
|---|---|---|---|---|---|---|
| Technology specific | ||||||
| MRI fusion and US | 57.17 | 89.13 | 78.73 | 52.07 | 83.52 | 36.58 |
| Installation | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | |
| Maintenance | 44.65 | 37.20 | 29.76 | 32.32 | ||
| Training | 3.43 | 0.92 | 0.95 | 0.68 | 0.46 | |
| Procedure time | 22.53 | 22.53 | 22.53 | 22.53 | 22.53 | |
| Biopsy setting | 22.01 | 22.01 | 22.01 | 22.01 | 22.01 | 14.63 |
| TP biopsy devices | 81.86 | 81.86 | 81.86 | 81.86 | 81.86 | 81.86 |
| Total | 231.68 | 253.68 | 235.87 | 179.18 | 242.73 | 133.07 |
| Not technology specific | ||||||
| Training | 11.67 | |||||
| Procedure time | 60.36 | |||||
| General consumables | 85.44 | |||||
| Lithomy bed | 3.99 | |||||
| Histology | 77.79 | |||||
| Total | 239.25 | |||||
| Total per biopsy | 470.93 | 492.93 | 475.12 | 418.43 | 481.98 | 372.32 |
| GATP | bkFusion (£) | FusionVu (£) | KOELIS Trinity (£) | BiopSee (£) | Fusion Bx 2.0 (£) | CF (£) |
|---|---|---|---|---|---|---|
| Technology specific | ||||||
| MRI fusion and US | 57.17 | 89.13 | 78.73 | 47.42 | 83.52 | 36.58 |
| Installation | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| Maintenance | 44.65 | 37.20 | 29.76 | 32.32 | ||
| Training | 3.43 | 0.92 | 0.95 | 0.68 | 0.46 | |
| Procedure time | 29.83 | 29.83 | 29.83 | 29.83 | 29.83 | |
| Biopsy setting | 155.14 | 155.14 | 155.14 | 155.14 | 155.14 | 132.97 |
| TP biopsy devices | 90.44 | 90.44 | 90.44 | 90.44 | 90.44 | 90.44 |
| Total | 380.67 | 402.67 | 384.86 | 323.52 | 391.72 | 260.00 |
| Not technology specific | ||||||
| Training | 11.67 | |||||
| Procedure time | 270.42 | |||||
| General consumables | 170.29 | |||||
| Lithomy bed | 3.99 | |||||
| Histology | 77.79 | |||||
| Total | 534.15 | |||||
| Total per biopsy | 914.82 | 936.82 | 919.01 | 857.67 | 925.87 | 794.15 |
Biopsy adverse event costs
| Biopsy AEs | Cost (£) | Resource use and unit costs |
|---|---|---|
| Mild AE | 49.78 | Resource use for outpatient urinary infection (Wilson, (2021),121 including: •GP visit: £39.23 – PSSRU (2021)154 GP – unit costs; per patient contact lasting 9.22 minutes •Urinalysis: £10.18 – NHS reference costs 2020–1187 – Direct Access Pathology Services: currency code DAPS07, Microbiology •7-day trimethoprim: £0.37 – eMIT (2021)191 – trimethoprim 200 mg × 14 tablets |
| Non-elective admissiona | Transrectal: 2580.24 Transperineal: 1952.98 |
Tamhankar (2020),184 inflated to 2020–1 price year154 |
| Deatha | 9560.56 | NHS reference costs 2020–1187 – Non-Elective: currency code WJ06A, Sepsis with multiple interventions, CC Score 9 + (weighted average of short stay and long stay patients) |
Prostate cancer management costs
| Treatment assigned | Active surveillance | Radical treatment | Resource use and unit costs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time | First year | Subsequent years | First year | Second year | Subsequent years | |||||
| Diagnosed CPG | CPG 1 | CPG2–3 | CPG4–5 | CPG1–5 | CPG 1 | CPG2–3 | CPG4–5 | CPG1–5 | CPG1–5 | |
| Resource use | ||||||||||
| PSA test | 4 | 2 | 2 | 2 | 1 | £1.85 – NHS reference costs 2020–21187 – currency code DAPS04, Clinical Biochemistry, Direct Access Pathology Services | ||||
| Nurse-led outpatient appointment | 4 | 2 | 2 | 2 | 1 | £11.00 – assumed as cost per 10 minutes, adjusted from cost per hour of band 7 community-based nurse – PSSRU (2021)154 | ||||
| DRE | 1 | 1 | 0 | 0 | 0 | £78.46 – assumed as cost per approximately 20 minutes of GP appointment – PSSRU (2021)154 adjusted from GP – unit costs; per patient contact lasting 9.22 minutes | ||||
| mpMRI | 1 | 0 | 0 | 0 | 0 | £294.70 – NHS reference costs 2020–1187 – currency code RD03Z, Diagnostic Imaging, MRI Scan of One Area, with Pre- and Post-Contrast | ||||
| CT scan | 0 | 0.5 | 0.7 | 0 | 0 | 0.5 | 0.7 | 0 | 0 | £150.62 – NHS reference costs 2020–1187 – currency code RD21A, Diagnostic Imaging, Computerised Tomography Scan of One Area, with Post-Contrast Only, 19 years and over |
| Bone scan | 0 | 0.5 | 0.7 | 0 | 0 | 0.5 | 0.7 | 0 | 0 | £427.21 – NHS reference costs 2020–21187 – currency code RN15A, Nuclear Medicine, Nuclear Bone Scan of Two or Three Phases, 19 years and over |
| Cost per year | £424.56 | £713.48 | £829.05 | £104.16 | £25.70 | £314.62 | £430.18 | £25.70 | £12.85 | |
| Radical treatment | Cost of procedure and follow-up (£) | Resource use and unit costs |
|---|---|---|
| Radical prostatectomy | 11,625.37 | Robotic surgery: £11,245.08 – NHS reference costs 2020–1187 – Elective inpatient, currency code LB69Z: Major Robotic, Prostate or Bladder Neck Procedures (Male) |
| First surgery appointment: £87.14 – NHS reference costs 2020–1187 – Outpatient procedure, currency code WF01B: Non-Admitted Face-to-Face Attendance, First (General surgery) | ||
| Two follow-up appointments: 2 x £146.58 – NHS reference costs 2020–1187 – Outpatient procedure, currency code WF01A: Non-Admitted Face-to-Face Attendance, Follow-up (General surgery) | ||
| External radiotherapy | 5341.81 | Preparation: £1721.79 – NHS reference costs 2020–1187 Preparation of for Intensity Modulated Radiation Therapy, weighted average of currency codes DC40Z and DC41Z (Total HRGs) |
| Fraction delivery – 20 x £181.00 – NHS reference costs 2020–1187 – Deliver a Fraction of Treatment on a Superficial or Orthovoltage Machine, currency code SC12Z (Total HRGs) | ||
| Brachytherapy | 9156.96 | Preparation: £1550.22– NHS reference costs 2020–1187 – Preparation for Interstitial Brachytherapy, weighted average of currency code SC55Z over day case, inpatient, outpatient and other setting |
| Fraction delivery: £7606.74 – NHS reference costs 2020–1187 – Deliver a Fraction of Intraluminal Brachytherapy, weighted average of currency code SC30Z over day case, inpatient outpatient, and other setting |
| Treatment | Cost (£) | Treatments included | Source of unit cost |
|---|---|---|---|
| Metastatic hormone sensitive – year 1 | 15,603.87 | - ADT: £973.76 for LHRHa (leuprorelin 11.25 mg, every 3 months; triptorelin 11.25 mg; or goserelin 3.6 mg, every 28 days) + £1.00 one-off bicalutamide 50 mg for 28 days (in year 1 only) .- ADT + DTX: £973.76 for LHRHa (as above) + £1404.00 (6 cycles of DTXb at a dose of 75 mg/m2; a cycle every 3 weeks – divided equally over 2 years) + £1.00 one-off bicalutamide 50 mg for 28 days (in year 1 only) .- ADT + apalutamide: £973.76 for LHRH (as above) + £35,677.10 (apalutamide 240 mg daily) + £1.00 one-off bicalutamide 50 mg for 28 days (in year 1 only) - ADT + enzalutamide: £973.76 for LHRHa (as above) + £35,672.79 (enzalutamide 160 mg daily) + £1.00 one-off bicalutamide 50 mg for 28 days (in year 1 only) |
BNF 2022,192 eMIT 2022,191 PSSRU 2021,154 NHS reference costs 2020–1187 |
| Metastatic hormone sensitive – year 2 | 15,602.88 | ||
| Metastatic hormone resistant – year 1 | 14,907.45 | - Abiraterone: £23,784.73 (1000 mg daily, 8 months) - DTXb: £4509.64 (9.5 cycles of DTX at a dose of 75 mg/m2; a cycle every 3 weeks) - Enzalutamide: £41,618.26 (160 mg daily, 14 months) |
BNF 2022,192 eMIT 2022,191 PSSRU 2021,154 NHS reference costs 2020–1187 |
Treatment adverse event costs
| Treatment for | AE | Unit cost (£) | Source |
|---|---|---|---|
| Localised PCa | Erectile dysfunction | 328.58 | NHS reference costs 2020–1187 – treatment of Erectile Dysfunction weighted average of the currency code LB43Z General Surgery, Genitourinary Medicine, Plastic Surgery, Urology |
| Urinary incontinence | 317.54 | NICE NG131123 – managed by containment pads. Inflated to 2020–1 price year154 | |
| Bowel dysfunction | 1941.19 | NICE NG131123 – mean weighted cost including costs associated with sigmoidoscopy, laser therapy, enemas and blood transfusion. Inflated to 2020–1 price year154 | |
| Hormone-sensitive metastatic PCa | Blood disorder | 2428.70 | NHS reference costs 2020–1187 – weighted average of currency codes SA03G–SA03H, SA08G–SA08J, SA12G–SA12K non-elective long stay and non-elective short stay |
| Cardiac disorder | 2042.04 | NHS reference costs 2020–1187 – weighted average of currency codes EB10A–EB10E non-elective long stay and non-elective short stay | |
| Endocrine disorder | 328.58 | Assume the same as erectile dysfunction (as in Southampton DAR)116 | |
| Gastrointestinal disorder | 2019.47 | NHS reference costs 2020–1187 – weighted average of currency codes FD10A–FD10M non-elective long stay and non-elective short stay | |
| General disorder | 39.90 | - GP visit per patient contact lasting 9.22 minutes: £39.23 – GP – unit costs; PSSRU 2021154 - 3-day Trimethoprim: £0.67 – eMIT 2021191 – trimethoprim 200 mg × 6 tablets |
|
| Musculoskeletal disorder | 26.58 | NHS reference costs 2020–1187 – weighted average of currency codes HD26D–HD26G non-elective long stay and non-elective short stay | |
| Nervous system disorder | 1933.29 | NHS reference costs 2020–1187 – weighted average of currency codes AA26C–AA26H non-elective long stay and non-elective short stay | |
| Neutropenia | 9842.93 | NHS reference costs 2020–1187 – weighted average of currency codes PM45A–PM45D non-elective long stay and non-elective short stay | |
| Renal disorder | 49.78 | Assume the same as urinary infection (as in Southampton DAR)116 | |
| Respiratory disorder | 971.68 | NHS reference costs 2020–1187 – weighted average of currency codes DZ19H–DZ19N non-elective long stay and non-elective short stay | |
| Skin disorder | 2191.91 | NHS reference costs 2020–1187 – weighted average of currency codes JD07A–JD07K non-elective long stay and non-elective short stay |
Base-case parameterisation
| Parameter | Value | Probabilistic setup | Source | |
|---|---|---|---|---|
| Population characteristics | ||||
| Age | 66 years | NA | Southampton DAR116 | |
| Prevalence and distribution across ISUP grade | ||||
| No PCa | 0.12 | Calculated from each 1000 iterations of network 1 and 2 | See Modelling of first biopsy results | |
| ISUP grade 1 | 0.32 | |||
| ISUP grade 2 | 0.26 | |||
| ISUP grade 3 | 0.18 | |||
| ISUP grade 4–5 | 0.12 | |||
| Diagnostic performance | ||||
| First biopsy and repeat biopsy with CF | ||||
| Probability of (diagnosis) | (true disease) | Targeted | Combined | ||
| ISUP grade 4–5 | ISUP grade 4–5 | 0.552 | 0.573 | Calculated from each 1000 iterations of network 1 for targeted and network 2 for combined | See Modelling of first biopsy results |
| ISUP grade 3 | ISUP grade 4–5 | 0.101 | 0.140 | ||
| ISUP grade 2 | ISUP grade 4–5 | 0.111 | 0.130 | ||
| ISUP grade 1 | ISUP grade 4–5 | 0.111 | 0.047 | ||
| No PCa | ISUP grade 4–5 | 0.125 | 0.111 | ||
| ISUP grade 3 | ISUP grade 3 | 0.479 | 0.510 | ||
| ISUP grade 2 | ISUP grade 3 | 0.192 | 0.207 | ||
| ISUP grade 1 | ISUP grade 3 | 0.140 | 0.059 | ||
| No PCa | ISUP grade 3 | 0.189 | 0.224 | ||
| ISUP grade 2 | ISUP grade 2 | 0.338 | 0.544 | ||
| ISUP grade 1 | ISUP grade 2 | 0.362 | 0.204 | ||
| No PCa | ISUP grade 2 | 0.300 | 0.251 | ||
| ISUP grade 1 | ISUP grade 1 | 0.171 | 0.329 | ||
| No PCa | ISUP grade 1 | 0.829 | 0.671 | ||
| No PCa | No PCa | 1.000 | 1.000 | ||
| First biopsy and repeat biopsy with SF | ||||
| Probability of (diagnosis) | (true disease) | Targeted | Combined | ||
| ISUP grade 4–5 | ISUP grade 4–5 | 0.281 | 0.724 | Calculated from each 1000 iterations from network 1 for targeted and network 2 for combined | See Modelling of first biopsy results |
| ISUP grade 3 | ISUP grade 4–5 | 0.163 | 0.071 | ||
| ISUP grade 2 | ISUP grade 4–5 | 0.173 | 0.066 | ||
| ISUP grade 1 | ISUP grade 4–5 | 0.187 | 0.070 | ||
| No PCa | ISUP grade 4–5 | 0.195 | 0.069 | ||
| ISUP grade 3 | ISUP grade 3 | 0.616 | 0.603 | ||
| ISUP grade 2 | ISUP grade 3 | 0.134 | 0.130 | ||
| ISUP grade 1 | ISUP grade 3 | 0.124 | 0.135 | ||
| No PCa | ISUP grade 3 | 0.126 | 0.132 | ||
| ISUP grade 2 | ISUP grade 2 | 0.314 | 0.770 | ||
| ISUP grade 1 | ISUP grade 2 | 0.437 | 0.152 | ||
| No PCa | ISUP grade 2 | 0.249 | 0.078 | ||
| ISUP grade 1 | ISUP grade 1 | 0.291 | 0.472 | ||
| No PCa | ISUP grade 1 | 0.709 | 0.528 | ||
| No PCa | No PCa | 1.000 | 1.000 | ||
| Probability of repeat biopsy | ||||
| if diagnosed as No PCa | 5% | NA | Southampton DAR assumption116 | |
| if diagnosed as ISUP grade 1 | 15.45% | Beta distribution: α = 95; β = 520 | Southampton DAR116 | |
| Biopsy AEs rates | ||||
| Mild AEs with TR biopsy | 1.31% | Beta distribution: α = 15; β = 1132 | Southampton DAR116,152 | |
| Mild AEs with TP biopsy | 9.13% | Beta distribution: α = 274; β = 2726 | ||
| Leading to NEL with TR biopsy | 3.74% | Beta distribution: α = 2845; β = 73,261 | ||
| Leading to NEL with TR biopsy | 3.54% | Beta distribution: α = 1314; β = 35,763 | ||
| TR mortality | 0.07% | Beta distribution: α = 53; β = 76,053 | ||
| TP mortality | 0.05% | Beta distribution: α = 19; β = 37,058 | ||
| Distribution by biopsy approach at first biopsy | ||||
| LATRUS | 35% | NA | Assumption informed by NHS reference data 2018–9185 | |
| LATP | 65% | NA | ||
| Distribution by biopsy approach at repeat biopsy | ||||
| LATRUS | 30% | NA | Assumption informed by NHS reference data 2018–9185 and clinical advice | |
| LATP | 60% | NA | ||
| GATP | 10% | NA | ||
| Long-term model transitions | ||||
| Progression Localised/Locally advanced to Metastatic | ||||
| Lambda CPG 1 with observation | 0.0143 | Sampled from 1000 simulations of the calibration model joint output for the 4 CPG categories and treatment received | Calibrated (see Modelling of long-term outcomes) | |
| Lambda CPG 2 with observation | 0.0379 | |||
| Lambda CPG 3 with observation | 0.1197 | |||
| Lambda CPG 4-5 with observation | 0.3997 | |||
| Lambda CPG 1 with radical treatment | 0.0063 | |||
| Lambda CPG 2 with radical treatment | 0.0164 | |||
| Lambda CPG 3 with radical treatment | 0.0514 | |||
| Lambda CPG 4-5 with radical treatment | 0.1683 | |||
| Metastatic to PCa death | ||||
| Weibull | γ = 1.26; λ = 0.11 | Multivariate lognormal | The PCa death curve for the control arm in Clarke (2019)143 was digitised by using WebPlotDigitizer;145 a pseudo-IPD was reconstructed by using Guyot algorithm,146 Weibull distribution was then fitted to the pseudo-IPD to obtain γ, λ and variance–covariance matrix using flexsurv package in R147 | |
| Mortality HR for DTX + ADT vs. ADT alone | 0.78 | Log-normal, 95% CI (0.66 to 0.93) | James (2016)59 | |
| Mortality HR for Enzalutamide + ADT vs. ADT alone | 0.66 | Log-normal, 95% CI (0.53 to 0.81) | ARCHES study149 | |
| Mortality HR for Apalutamide + ADT vs. ADT alone | 0.65 | Log-normal, 95% CI (0.53 to 0.79) | TITAN study150 | |
| Other-cause mortality | Age dependent | NA | ONS lifetables 2018–20144 | |
| Treatment distributions | ||||
| Localised disease disease | ||||
| Radical treatment and diagnosed (ISUP grade 4–5) | 75.9% | Dirichlet distribution | Calculated as sum of proportions of radical prostatectomy and radiotherapy; Parry (2020)151 |
|
| Radical treatment and diagnosed (ISUP grade 3) | 66.3% | |||
| Radical treatment and diagnosed (ISUP grade 2) | 48.4% | |||
| Radical treatment and diagnosed (ISUP grade 1) | 11.3% | |||
| Radical treatment and diagnosed (No PCa) | 0% | NA | Assumption | |
| Metastatic cancer | ||||
| ADT | 50.0% | NA | Assumption informed by Southampton DAR116 and NPCA report 2021188 | |
| ADT + DTX | 9.4% | NA | ||
| ADT + apalutamide | 6.6% | NA | ||
| ADT + enzalutamide | 34.0% | NA | ||
| Treatment AE rates | ||||
| Radical prostatectomy | ||||
| Sexual dysfunction | 85.39% | Beta distribution: α = 304; β = 52 | Southampton DAR116 | |
| Bowel dysfunction | 2.47% | Beta distribution: α = 9; β = 355 | ||
| Urinary dysfunction | 26.24% | Beta distribution: α = 95; β = 267 | ||
| Radiotherapy | ||||
| Sexual dysfunction | 62.39% | Beta distribution: α = 219; β = 132 | Southampton DAR116 | |
| Bowel dysfunction | 5.85% | Beta distribution: α = 21; β = 338 | ||
| Urinary dysfunction | 3.63% | Beta distribution: α = 21; β = 345 | ||
| Active surveillance | ||||
| Erectile dysfunction | 50.88% | Beta distribution: α = 173; β = 167 | Southampton DAR116 | |
| Bowel dysfunction | 1.68% | Beta distribution: α = 6; β = 352 | ||
| Urinary incontinence | 4.20% | Beta distribution: α = 15; β = 342 | ||
| Metastatic treatment | ||||
| ADT | ||||
| Blood disorder | 0.00% | Southampton DAR116 | ||
| Cardiac disorder | 2.96% | Beta distribution: α = 35; β = 1149 | ||
| Endocrine disorder | 12.25% | Beta distribution: α = 145; β = 1039 | ||
| Gastrointestinal disorder | 3.04% | Beta distribution: α = 36; β = 1148 | ||
| General disorder | 3.89% | Beta distribution: α = 46; β = 1138 | ||
| Musculoskeletal disorder | 5.83% | Beta distribution: α = 69; β = 1115 | ||
| Nervous system disorder | 1.69% | Beta distribution: α = 20; β = 1164 | ||
| Neutropenia | 1.77% | Beta distribution: α = 21; β = 1163 | ||
| Renal disorder | 6.00% | Beta distribution: α = 71; β = 1113 | ||
| Respiratory disorder | 2.28% | Beta distribution: α = 27; β = 1157 | ||
| Skin disorder | 0.00% | |||
| ADT + DTX | ||||
| Blood disorder | 0.00% | Southampton DAR116 | ||
| Cardiac disorder | 2.91% | Beta distribution: α = 16; β = 534 | ||
| Endocrine disorder | 10.36% | Beta distribution: α = 57; β = 493 | ||
| Gastrointestinal disorder | 8.18% | Beta distribution: α = 45; β = 505 | ||
| General disorder | 6.18% | Beta distribution: α = 34; β = 516 | ||
| Musculoskeletal disorder | 5.82% | Beta distribution: α = 32; β = 518 | ||
| Nervous system disorder | 3.45% | Beta distribution: α = 19; β = 531 | Southampton DAR116 | |
| Neutropenia | 27.27% | Beta distribution: α = 150; β = 400 | ||
| Renal disorder | 4.18% | Beta distribution: α = 23; β = 527 | ||
| Respiratory disorder | 5.27% | Beta distribution: α = 29; β = 521 | ||
| Skin disorder | 0.00% | |||
| ADT + apalutamide | ||||
| Blood disorder | 2.10% | Beta distribution: α = 11; β = 513 | Southampton DAR116 | |
| Cardiac disorder | 8.40% | Beta distribution: α = 44; β = 480 | ||
| Endocrine disorder | 0.00% | |||
| Gastrointestinal disorder | 1.15% | Beta distribution: α = 6; β = 518 | ||
| General disorder | 3.44% | Beta distribution: α = 18; β = 506 | ||
| Musculoskeletal disorder | 6.49% | Beta distribution: α = 34; β = 490 | ||
| Nervous system disorder | 0.19% | Beta distribution: α = 1; β = 523 | ||
| Neutropenia | 0.00% | |||
| Renal disorder | 0.76% | Beta distribution: α = 4; β = 520 | ||
| Respiratory disorder | 0.00% | |||
| Skin disorder | 6.49% | Beta distribution: α = 34; β = 490 | ||
| ADT + enzalutamide | ||||
| Blood disorder | 0.00% | Southampton DAR116 | ||
| Cardiac disorder | 4.90% | Beta distribution: α = 28; β = 544 | ||
| Endocrine disorder | 0.35% | Beta distribution: α = 2; β = 570 | ||
| Gastrointestinal disorder | 0.52% | Beta distribution: α = 3; β = 569 | ||
| General disorder | 2.80% | Beta distribution: α = 16; β = 556 | ||
| Musculoskeletal disorder | 4.37% | Beta distribution: α = 25; β = 547 | ||
| Nervous system disorder | 2.10% | Beta distribution: α = 12; β = 560 | ||
| Neutropenia | 0.35% | Beta distribution: α = 2; β = 570 | ||
| Renal disorder | 0.00% | |||
| Respiratory disorder | 0.00% | |||
| Skin disorder | 0.35% | Beta distribution: α = 2; β = 570 | ||
| HRQoL | ||||
| Disutility of biopsy AEs | ||||
| Mild AEs | –0.289 | NA | Southampton DAR;116 assumed duration 3 days | |
| Leading to NEL | –0.490 | NA | Southampton DAR;116 assumed duration 30 days | |
| Death | –0.490 | NA | Southampton DAR;116 assumed duration 30 days | |
| Baseline health state utility | Age and sex dependent | NA | Ara and Brazier (2010)137 | |
| Localised treatment disutility | ||||
| Sexual dysfunction | –0.0230 | No-mild symptoms: Beta distribution: α = 578; β = 93 Moderate–severe symptoms: Beta distribution: α = 452; β = 87 |
Calculated as the difference between no-mild symptoms and moderate-severe symptoms (as per Southampton DAR)116 | |
| Urinary dysfunction | –0.0950 | No-mild symptoms: Beta distribution: α = 1013; β = 154 Moderate–severe symptoms: Beta distribution: α = 131; β = 39 |
||
| Bowel dysfunction | –0.2090 | No-mild symptoms: Beta distribution: α = 1097; β = 176 Moderate–severe symptoms: Beta distribution: α = 62; β = 33 |
||
| Metastatic disutility | –0.137 | Localised 1: Beta distribution: α = 102; β = 11 Localised 2: Beta distribution: α = 404; β = 50 Localised 3: Beta distribution: α = 841; β = 126 Metastatic: Beta distribution: α = 165; β = 58 |
Calculated as the difference between metastatic and the average across localised 1, 2, 3 (as per Southampton DAR)116 | |
| Resource use and costs | ||||
| Annual patient throughput | 300 | NA | Assumed based on NHS reference costs 2018–9185 | |
| Cost per first CF biopsy (targeted or combined) | £332.13 | NA | Calculated | |
| Cost per first SF (targeted or combined) | £427.33 | NA | Calculated | |
| Cost per repeat CF biopsy (targeted or combined) | £380.05 | NA | Calculated | |
| Cost per repeat SF (targeted or combined) | £477.42 | NA | Calculated | |
| Cost of localised treatment | ||||
| Cost of radical prostatectomy | £11,625.37 | NA | Calculated | |
| Cost of radiotherapy for those who diagnosed as CPG1 | £6283.42 | NA | Calculated | |
| Cost of radiotherapy for those who diagnosed as CPG2 | £5754.11 | NA | Calculated | |
| Cost of radiotherapy for those who diagnosed as CPG3 | £5510.29 | NA | Calculated | |
| Cost of radiotherapy for those who diagnosed as CPG4–5 | £5402.04 | NA | Calculated | |
| Cost of ADT | £973.76 | NA | Calculated (see Table 102) | |
| Cost of bicalutamide | £1.49 | NA | 21 days course of bicalutamide – BNF 2022192 bicalutamide 50mg × 28 tablets | |
| Cost of metastatic treatment | ||||
| Cost of first year hormone-sensitive treatment | £15,603.87 | NA | Calculated (see Table 102) | |
| Cost of second year hormone-sensitive treatment | £15,602.88 | NA | ||
| Cost of metastatic treatment in subsequent years (one-off) | £14,907.45 | NA | ||
| Cost of monitoring/active surveillance | ||||
| Cost of AS for those who diagnosed as CPG1 in first year | £424.56 | NA | Calculated | |
| Cost of AS for those who diagnosed as CPG2–3 in first year | £713.48 | NA | Calculated | |
| Cost of AS for those who diagnosed as CPG4–5 in first year | £829.05 | NA | Calculated | |
| Cost of AS for those who diagnosed as any CPG in subsequent years | £104.16 | NA | Calculated | |
| Cost of monitoring those who diagnosed as CPG1 receiving RT, in first year | £25.70 | NA | Calculated | |
| Cost of monitoring those who diagnosed as CPG2–3 receiving RT, in first year | £314.62 | NA | Calculated | |
| Cost of monitoring those who diagnosed as CPG4–5 receiving RT, in first year | £430.18 | NA | Calculated | |
| Cost of monitoring those who diagnosed as any CPG receiving RT, in second year | £ 25.70 | NA | Calculated | |
| Cost of monitoring those who diagnosed as any CPG receiving RT, in 2 + year | £12.85 | NA | Calculated | |
| Cost of monitoring those who have No PCa diagnosed as No PCa | £158.99 | NA | Calculated | |
| Cost of monitoring those who have No PCa diagnosed as ISUP grade 1 | £242.02 | NA | Calculated | |
| Cost of monitoring metastatic patients (one off) | £577.83 | NA | Calculated | |
| Cost of managing AEs | ||||
| Cost of managing AEs of biopsy procedure | ||||
| Cost per mild AE | £49.78 | NA | Calculated | |
| Cost per NEL event with LATRUS | £2580.24 | NA | Calculated | |
| Cost per NEL event with LATP/GATP | £1952.98 | NA | Calculated | |
| Cost per biopsy death | £9560.56 | NA | Calculated | |
| Cost of managing AEs of | ||||
| Active surveillance | £213.06 | NA | Calculated | |
| Radical prostatectomy | £411.91 | NA | Calculated | |
| Radiotherapy | £330.09 | NA | Calculated | |
| Metastatic treatment | ||||
| ADT | £397.49 | See probabilistic setup for AE rates of ADT | Calculated | |
| ADT + DTX | £3067.24 | See probabilistic setup for AE rates of ADT + DTX | Calculated | |
| ADT + enzalutamide | £196.62 | See probabilistic setup for AE rates of ADT + enzalutamide | Calculated | |
| ADT + apalutamide | £394.96 | See probabilistic setup for AE rates of ADT + apalutamide | Calculated | |
| End of life costs | £16,546.08 | NA | Round (2015);139 inflated to 2020–1 price year154 | |
Appendix 12 Additional cost-effectiveness results
Base-case analysis
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa | Total QALYsa | Total costsa (£) | Total LYsa | Total QALYsa | Total costsa (£) | ICERb (£) | NHB at £20,000b |
NHB at £30,000b |
Probability CE at £20,000b | Probability CE at £30,000b | |
| Targeted CF | –0.00176 | 445 | 11.46 | 8.30 | 27,734 | 11.46 | 8.30 | 28,179 | 6.89 | 7.36 | 0.36 | 0.32 | |
| Targeted SF | –0.00175 | 543 | 11.48 | 8.31 | 27,702 | 11.48 | 8.31 | 28,245 | 6.90 | 7.37 | 0.64 | 0.68 | |
| Targeted | Inc QALY loss | Inc costs | Inc LYs a | Inc QALYs a | Inc costs a | Inc LYs a | Inc QALYs a | Inc costs a |
INHB at
£20,000 b |
INHB at £30,000b | |||
| SF vs. CF | 0.00001 | 98 | 0.02 | 0.01 | –32 | 0.02 | 0.01 | £65 | 6197 | 0.01 | 0.01 | ||
| Strategy | QALY loss | Total costs | Total LYs a | Total QALYs a | Inc costs a | Inc LYs a | Total QALYs a | Total costs a | ICER b |
NHB
at
£20,000 b |
NHB
at
£30,000 b |
Probability CE at £20,000 b | Probability CE at £30,000 b |
| Combined CF | –0.00177 | 448 | 11.46 | 8.30 | 27,716 | 11.46 | 8.30 | 28,164 | 6.89 | 7.36 | 0.27 | 0.25 | |
| Combined SF | –0.00176 | 544 | 11.50 | 8.33 | 27,669 | 11.50 | 8.32 | 28,213 | 6.91 | 7.38 | 0.73 | 0.75 | |
| Combined | Inc QALY loss | Inc costs | Inc LYs a | Total QALYs a | Total costs a | Inc LYs a | Inc QALYs a | Inc costs a |
INHB at
£20,000 b |
INHB at £30,000 b | |||
| SF vs. CF | 0.00002 | 96 | 0.04 | 0.02 | –47 | 0.04 | 0.02 | 49 | 2199 | 0.02 | 0.02 | ||
| Strategy | Prevalence | Proportion correctly classifieda | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CPG 4-5 | CPG G3 | CPG 2 | CPG 1 | No PCa | CPG 4-5 | CPG G3 | CPG 2 | CPG 1 | No PCa | All categories | |
| CF | 0.116 | 0.183 | 0.262 | 0.318 | 0.121 | 0.066 | 0.090 | 0.095 | 0.057 | 0.121 | 0.428 |
| SF | 0.067 | 0.095 | 0.149 | 0.108 | 0.121 | 0.540 | |||||
| Strategy | Proportion repeat biopsy | Proportion AEs | Cost | AEs QALY loss | |||||
|---|---|---|---|---|---|---|---|---|---|
| All | Unnecessarya | Death | Mild | Repeat biopsy | First biopsy (£) | Repeat biopsy (£) | AEs (£) | ||
| CF | 0.055 | 0.038 | 0.001 | 0.068 | 0.038 | 332 | 21 | 92 | –0.00176 |
| SF | 0.050 | 0.035 | 0.001 | 0.067 | 0.038 | 427 | 24 | 92 | –0.00175 |
| Strategy | Local disease – radical treatment | Local disease AEs | Metastatic disease | Monitoring (£) | EoL (£) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immediate | Delayed | Immediate | Delayed | ||||||||||||||
| All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | Treatment (£) | AEs (£) | ||||||
| CF | 1844 | 103 | 280 | 568 | 252 | 688 | 12 | 84 | 276 | 146 | 17,241 | 456 | 948 | 16,510 | |||
| SF | 2158 | 76 | 260 | 395 | 202 | 850 | 10 | 78 | 192 | 117 | 17,008 | 449 | 1047 | 16,510 | |||
| Strategy | LYs | Baseline QALYs | QALY loss | ||
|---|---|---|---|---|---|
| Immediate radical treatment | Delayed radical treatment | Metastatic disease | |||
| CF | 16.22 | 10.99 | –0.09 | –0.13 | –0.52 |
| SF | 16.25 | 11.01 | –0.13 | –0.10 | –0.51 |
| Strategy | Prevalence | Proportion correctly classifieda | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | All categories | |
| CF | 0.116 | 0.183 | 0.262 | 0.318 | 0.121 | 0.034 | 0.115 | 0.089 | 0.096 | 0.121 | 0.455 |
| SF | 0.085 | 0.113 | 0.207 | 0.154 | 0.121 | 0.680 | |||||
| Strategy | Proportion repeat biopsy | Proportion AEs | Cost | AEs QALY loss (£) | |||||
|---|---|---|---|---|---|---|---|---|---|
| All | Unnecessarya | Death | Mild | Repeat biopsy | First biopsy (£) | Repeat biopsy (£) | AEs (£) | ||
| CF | 0.062 | 0.043 | 0.001 | 0.068 | 0.038 | 332 | 23 | 93 | –0.00177 |
| SF | 0.051 | 0.036 | 0.001 | 0.067 | 0.038 | 427 | 25 | 92 | –0.00176 |
| Strategy | Local disease – radical treatment | Local disease AEs | Metastatic disease | Monitoring (£) | EoL (£) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immediate | Delayed | Immediate | Delayed | |||||||||||
| All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | Treatment (£) | AEs (£) | |||
| CF | 1835 | 169 | 205 | 574 | 214 | 723 | 20 | 62 | 280 | 124 | 17,172 | 454 | 1016 | 16,510 |
| SF | 2547 | 57 | 216 | 181 | 158 | 1048 | 7 | 65 | 88 | 92 | 16,705 | 441 | 1167 | 16,510 |
| Strategy | LYs | Baseline QALYs | QALY loss | ||
|---|---|---|---|---|---|
| Immediate radical treatment | Delayed radical treatment | Metastatic disease | |||
| CF | 16.21 | 10.98 | –0.11 | –0.12 | –0.52 |
| SF | 16.29 | 11.03 | –0.18 | –0.07 | –0.50 |
Results of base-case by software fusion technology
In Table 114, Appendix 12 we show the deterministic base-case analysis results of targeted SF by individual technology in pairwise comparison versus targeted cognitive. Corresponding results for the combined comparison are presented in Appendix 12.
| Strategy | Diagnostic model | Overall results | |||||
|---|---|---|---|---|---|---|---|
| Inc costs (£) | Total LYsa | Total QALYsa | Total costsa (£) | ICER vs. CFb (£) | NHB at £20,000b |
NHB at £30,000b |
|
| Targeted CF | – | 11.45 | 8.29 | 28,364 | 6.87 | 7.34 | |
| Targeted software fusion | 98 | 11.46 | 8.30 | 28,428 | 5623 | 6.88 | 7.35 |
| Targeted bkFusion | 101 | 28,431 | 5954 | 6.88 | 7.35 | ||
| Targeted FusionVu | 125 | 28,454 | 8001 | 6.88 | 7.35 | ||
| Targeted KOELIS Trinity | 105 | 28,435 | 6302 | 6.88 | 7.35 | ||
| Targeted Fusion Bx 2.0 | 113 | 28,443 | 6968 | 6.88 | 7.35 | ||
| Targeted BiopSee | 44 | 28,374 | 890 | 6.88 | 7.35 | ||
| Strategy | Diagnostic model | Overall results | |||||
|---|---|---|---|---|---|---|---|
| Inc costs (£) | Total Lysa | Total QALYsa (£) | Total costsa (£) | ICER vs. CFb (£) | NHB at £20,000b | NHB at £30,000b | |
| Targeted CF | – | 11.75 | 8.68 | 22,457 | – | 7.56 | 7.93 |
| Combined SF | 99 | 11.76 | 8.69 | 22,536 | 9285 | 7.56 | 7.94 |
| Combined bkFusion | 103 | 22,540 | 9725 | 7.56 | 7.94 | ||
| Combined FusionVu | 126 | 22,563 | 12,443 | 7.56 | 7.94 | ||
| Combined KOELIS Trinity | 106 | 22,544 | 10,187 | 7.56 | 7.94 | ||
| Combined Fusion Bx 2.0 | 114 | 22,551 | 11,072 | 7.56 | 7.94 | ||
| Combined BiopSee | 45 | 22,483 | 2998 | 7.57 | 7.94 | ||
The pairwise ICERs of the targeted SF strategies versus CF range between £28,374 and £28,454 per additional QALY for BiopSee and FusionVu, respectively. Results for the combined biopsy comparison show the same pattern. The only incremental difference between individual SF technologies strategies are the incremental costs in the diagnostic model.
FIGURE 18.
Targeted SF cost threshold analysis.

FIGURE 19.
Combined SF cost threshold analysis.
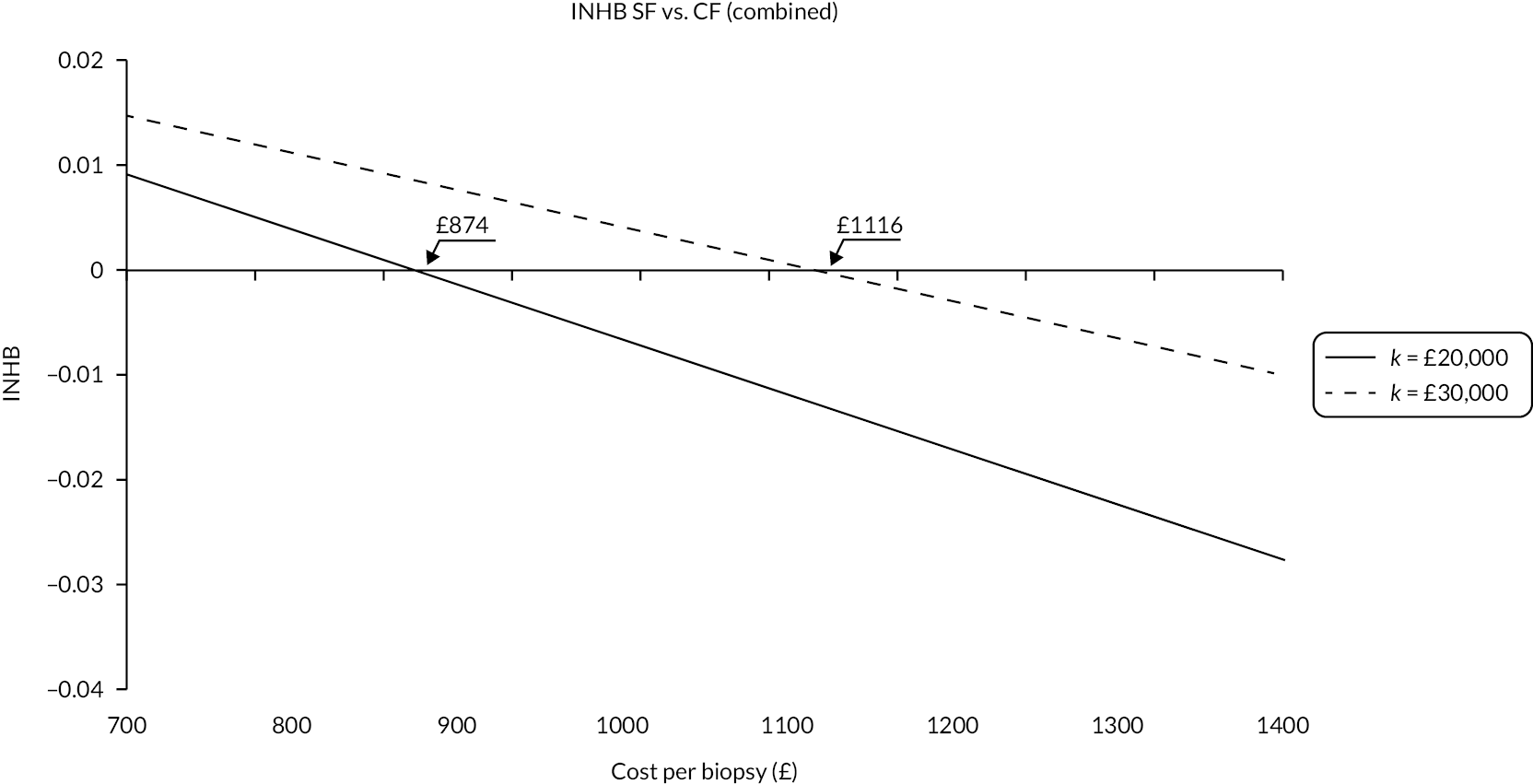
Subgroup analyses
| Strategy | Prevalence | Proportion correctly classifieda | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | All categories | |
| CF | 0.085 | 0.131 | 0.132 | 0.224 | 0.428 | 0.048 | 0.063 | 0.043 | 0.033 | 0.428 | 0.614 |
| SF | 0.050 | 0.070 | 0.074 | 0.070 | 0.428 | 0.692 | |||||
| Strategy | Proportion repeat biopsy | Proportion AEs | Cost | AEs QALY loss | |||||
|---|---|---|---|---|---|---|---|---|---|
| All | Unnecessarya | Death | Mild | NEL | First biopsy (£) | Repeat biopsy (£) | AEs (£) | ||
| CF | 0.052 | 0.042 | 0.001 | 0.067 | 0.038 | 332 | 20 | 92 | –0.00176 |
| SF | 0.049 | 0.040 | 0.001 | 0.067 | 0.038 | 427 | 23 | 92 | –0.00175 |
| Strategy | Local disease – radical treatment | Local disease AEs | Metastatic disease | Monitoring (£) | EoL (£) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immediate | Delayed | Immediate | Delayed | |||||||||||
| All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | Treatment (£) | AEs (£) | |||
| CF | 1185 | 78 | 202 | 303 | 183 | 416 | 9 | 61 | 148 | 107 | 11,439 | 302 | 1109 | 16,509 |
| SF | 1394 | 55 | 183 | 204 | 148 | 522 | 7 | 55 | 99 | 86 | 11,287 | 298 | 1177 | 16,509 |
| Strategy | LYs | Baseline QALYs | QALY loss | ||
|---|---|---|---|---|---|
| Immediate radical treatment | Delayed radical treatment | Metastatic disease | |||
| CF | 16.72 | 11.27 | –0.05 | –0.09 | –0.35 |
| SF | 16.74 | 11.28 | –0.08 | –0.06 | –0.34 |
| Strategy | Prevalence | Proportion correctly classifieda | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | All categories | |
| CF | 0.085 | 0.131 | 0.132 | 0.224 | 0.428 | 0.028 | 0.081 | 0.040 | 0.071 | 0.428 | 0.648 |
| SF | 0.060 | 0.081 | 0.100 | 0.115 | 0.428 | 0.784 | |||||
| Strategy | Proportion repeat biopsy | Proportion AEs | Cost | AEs QALY loss | |||||
|---|---|---|---|---|---|---|---|---|---|
| All | Unnecessarya | Death | Mild | NEL | First biopsy (£) | Repeat biopsy (£) | AEs (£) | ||
| CF | 0.057 | 0.046 | 0.001 | 0.068 | 0.038 | 332 | 22 | 92 | –0.00177 |
| SF | 0.053 | 0.043 | 0.001 | 0.068 | 0.038 | 427 | 25 | 92 | –0.00176 |
| Strategy | Local disease – radical treatment | Local disease AEs | Metastatic disease | Monitoring (£) | EoL (£) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immediate | Delayed | Immediate | Delayed | |||||||||||
| All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | Treatment (£) | AEs (£) | |||
| CF | 1187 | 124 | 152 | 306 | 147 | 448 | 14 | 46 | 149 | 85 | 11,385 | 301 | 1172 | 16,508 |
| SF | 1612 | 45 | 157 | 105 | 104 | 633 | 6 | 47 | 51 | 61 | 11,127 | 294 | 1272 | 16,508 |
| Strategy | Life years (LYs) | Baseline QALYs | QALY loss | ||
|---|---|---|---|---|---|
| Immediate radical treatment | Delayed radical treatment | Metastatic disease | |||
| CF | 16.71 | 11.27 | –0.07 | –0.08 | –0.34 |
| SF | 16.76 | 11.29 | –0.11 | –0.05 | –0.34 |
Scenario analyses
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00175 | 442 | 11.17 | 7.99 | 32,490 | 11.17 | 7.99 | 32,932 | 6.34 | 6.89 | |
| SF | –0.00174 | 538 | 11.19 | 8.00 | 32,432 | 11.19 | 7.99 | 32,970 | 6.35 | 6.90 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00001 | 96 | 0.02 | 0.01 | –58 | 0.02 | 0.01 | 39 | 4428 | 0.01 | 0.01 |
| Strategy | Prevalence | Proportion correctly classifieda | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | CPG 4–5 | CPG G3 | CPG 2 | CPG 1 | No PCa | All categories | |
| CF | 0.169 | 0.252 | 0.322 | 0.226 | 0.031 | 0.103 | 0.135 | 0.140 | 0.058 | 0.031 | 0.467 |
| SF | 0.100 | 0.136 | 0.193 | 0.086 | 0.031 | 0.544 | |||||
| Strategy | Proportion repeat biopsy | Proportion AEs | Cost | AEs QALY loss | |||||
|---|---|---|---|---|---|---|---|---|---|
| All | Unnecessarya | Death | Mild | NEL | First biopsy (£) | Repeat biopsy (£) | AEs (£) | ||
| CF | 0.048 | 0.028 | 0.001 | 0.067 | 0.038 | 332 | 18 | 92 | –0.00175 |
| SF | 0.042 | 0.025 | 0.001 | 0.067 | 0.038 | 427 | 20 | 91 | –0.00174 |
| Strategy | Local disease – radical treatment | Local disease AEs | Metastatic disease | Monitoring (£) | EoL (£) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immediate | Delayed | Immediate | Delayed | |||||||||||
| All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | All CPG (£) | CPG 4–5 (£) | CPG 3 (£) | CPG 2 (£) | CPG 1 (£) | Treatment (£) | AEs (£) | |||
| CF | 2638 | 134 | 350 | 626 | 162 | 970 | 16 | 105 | 304 | 94 | 21,381 | 565 | 934 | 16,512 |
| SF | 2937 | 107 | 325 | 449 | 136 | 1120 | 14 | 98 | 218 | 79 | 21,154 | 559 | 998 | 16,512 |
| Strategy | Life years (LYs) | Baseline QALYs | QALY loss | ||
|---|---|---|---|---|---|
| Immediate radical treatment | Delayed radical treatment | Metastatic disease | |||
| CF | 15.78 | 10.73 | –0.13 | –0.10 | –0.65 |
| SF | 15.80 | 10.75 | –0.16 | –0.09 | –0.64 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00176 | 446 | 11.55 | 8.41 | 26,652 | 11.55 | 8.40 | 27,098 | 7.05 | 7.50 | |
| SF | –0.00175 | 543 | 11.58 | 8.43 | 26,638 | 11.58 | 8.43 | 27,180 | 7.07 | 7.52 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00001 | 97 | 0.03 | 0.03 | –14 | 0.03 | 0.03 | 83 | 3105 | 0.02 | 0.02 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00176 | 445 | 11.44 | 8.29 | 27,922 | 11.44 | 8.29 | 28,367 | 6.87 | 7.34 | |
| SF | –0.00175 | 543 | 11.46 | 8.30 | 27,887 | 11.46 | 8.30 | 28,429 | 6.88 | 7.35 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00001 | 98 | 0.02 | 0.01 | –35 | 0.02 | 0.01 | 63 | 5477 | 0.01 | 0.01 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00177 | 448 | 11.44 | 8.28 | 27,892 | 11.44 | 8.28 | 28,340 | 6.86 | 7.33 | |
| SF | –0.00176 | 544 | 11.49 | 8.31 | 27,843 | 11.49 | 8.30 | 28,386 | 6.88 | 7.36 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00002 | 95 | 0.05 | 0.03 | –49 | 0.05 | 0.03 | 46 | 1801 | 0.02 | 0.02 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00176 | 445 | 11.45 | 8.29 | 27,864 | 11.45 | 8.29 | 28,310 | 6.87 | 7.34 | |
| SF | –0.00174 | 537 | 11.45 | 8.29 | 27,859 | 11.45 | 8.29 | 28,396 | 6.87 | 7.34 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00002 | 92 | 0.00 | 0.00 | –6 | 0.00 | 0.00 | 87 | 874,744 | 0.00 | 0.00 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | -0.00177 | 448 | 11.44 | 8.28 | 27,833 | 11.44 | 8.28 | 28,282 | 6.86 | 7.34 | |
| SF | -0.00174 | 538 | 11.44 | 8.28 | 27,824 | 11.44 | 8.28 | 28,363 | 6.86 | 7.33 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00003 | 90 | 0.00 | 0.00 | –9 | 0.00 | 0.00 | 81 | 581,847 | 0.00 | 0.00 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00176 | 445 | 11.55 | 8.37 | 28,816 | 11.55 | 8.37 | 29,261 | 6.90 | 7.39 | |
| SF | –0.00175 | 543 | 11.59 | 8.40 | 28,601 | 11.59 | 8.40 | 29,144 | 6.94 | 7.43 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000b | INHB at £30,000b | ||
| SF vs. CF | 0.00001 | 98 | 0.04 | 0.03 | –215 | 0.04 | 0.03 | –117 | Dominates | 0.04 | 0.03 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00177 | 448 | 11.55 | 8.36 | 28,786 | 11.55 | 8.35 | 29,234 | 6.89 | 7.38 | |
| SF | –0.00176 | 544 | 11.63 | 8.41 | 28,390 | 11.63 | 8.41 | 28,934 | 6.96 | 7.44 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00002 | 95 | 0.08 | 0.05 | –396 | 0.08 | 0.05 | –300 | Dominates | 0.07 | 0.06 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00176 | 495 | 11.45 | 8.29 | 27,956 | 11.45 | 8.29 | 28,451 | 6.87 | 7.34 | |
| SF | –0.00175 | 661 | 11.46 | 8.30 | 27,919 | 11.46 | 8.30 | 28,580 | 6.87 | 7.35 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000b | INHB at £30,000b | ||
| SF vs. CF | 0.00001 | 166 | 0.02 | 0.01 | –37 | 0.02 | 0.01 | 129 | 11,425 | 0.00 | 0.01 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00177 | 498 | 11.44 | 8.28 | 27,924 | 11.44 | 8.28 | 28,422 | 6.86 | 7.33 | |
| SF | –0.00176 | 662 | 11.49 | 8.31 | 27,870 | 11.49 | 8.30 | 28,532 | 6.88 | 7.35 | |
| Inc QALY loss | Inc costs (£) | Inc LYsa (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00002 | 164 | 0.05 | 0.03 | –54 | 0.05 | 0.03 | 110 | 4,275 | 0.02 | 0.02 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00177 | 432 | 11.44 | 8.28 | 27,878 | 11.44 | 8.28 | 28,309 | 6.86 | 7.34 | |
| SF | –0.00176 | 504 | 11.49 | 8.31 | 27,831 | 11.49 | 8.30 | 28,335 | 6.89 | 7.36 | |
| Inc QALY loss | Inc costs (£) | Inc LYs a (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000b | INHB at £30,000b | ||
| SF vs. CF | 0.00002 | 73 | 0.05 | 0.03 | –47 | 0.05 | 0.03 | 26 | 1009 | 0.02 | 0.02 |
| Strategy | Diagnostic model | Long-term model | Overall results | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| QALY loss | Total costs (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | Total LYsa (£) | Total QALYsa (£) | Total costsa (£) | ICERb (£) | NHB at £20,000b | NHB at £30,000b | |
| CF | –0.00176 | 428 | 11.45 | 8.29 | 27,907 | 11.45 | 8.29 | 28,335 | 6.87 | 7.34 | |
| SF | –0.00175 | 503 | 11.46 | 8.30 | 27,873 | 11.46 | 8.30 | 28,377 | 6.88 | 7.35 | |
| Inc QALY loss | Inc costs (£) | Inc LYs a (£) | Inc QALYs a | Inc costsa (£) | Inc LYs a | Inc QALYs a | Inc costsa (£) | INHB at £20,000 b | INHB at £30,000 b | ||
| SF vs. CF | 0.00001 | 75 | 0.02 | 0.01 | –33 | 0.02 | 0.01 | 42 | 3689 | 0.01 | 0.01 |
Glossary
- Active surveillance
- Monitoring of a person following a diagnosis of prostate cancer, with a view to switching to radical treatment if the cancer progresses. Aims to prevent the risk of overtreatment by avoiding immediate radical intervention. Active surveillance typically includes regular monitoring of prostate-specific antigen (PSA) levels and digital rectal examination.
- Cognitive fusion biopsy
- When the operator views both sets of MRI and ultrasound images and mentally translates the MRI target lesions onto the real-time ultrasound images during the biopsy procedure, to guide the placement of biopsy needles. Also referred to as visual estimation or visual registration.
- Double freehand
- A transperineal biopsy technique whereby the ultrasound probe is held in the hand, rather than being supported by a stepping device. Unlike the freehand technique, the introducer needle is not attached to the ultrasound probe and is held in the other hand.
- Elastic registration
- During software fusion with elastic registration, the MRI image is altered to match the ultrasound image, to adjust for potential deformation to the prostate during the biopsy. Also referred to as non-rigid registration.
- Freehand
- A biopsy in which the ultrasound probe is held in the hand, rather than being supported by a stepping device. This allows the probe to be moved in all directions. A needle attached to the ultrasound probe is then used to puncture the perineum before the biopsy needle is passed through. The biopsy needle can be pivoted to take the samples, reducing the number of puncture sites on the perineum.
- Gleason system
- A system used to grade prostate cancer cells to estimate how quickly they are likely to grow (Gleason grade). Grade Group 1 is the least aggressive, indicating that the cancer is likely to grow very slowly, if at all. Grade Group 5 is the most aggressive, indicating the cells look very abnormal and the cancer is likely to grow quickly. Since prostate tumours are often made up of cancerous cells that have different grades, two grades are assigned for each patient. A primary grade is given to describe the cells that make up the largest area of the tumour and a secondary grade is given to describe the cells of the next largest area. For example, a Gleason score written as 3 + 4 = 7 indicates that most of the tumour is grade 3 and the next largest section of the tumour is grade 4. To help with outcome prediction and patient communication, Gleason scores ≤ 6, 3 + 4, 4 + 3, 8 and 9–10, respectively, can be reported as five risk groups defined by the International Society of Urological Pathology (ISUP), that is, ISUP grades 1–5.
- Grid and stepping device
- A stepping device used in prostate biopsy to cradle the ultrasound probe. On this device, a grid can be attached. A grid (or template) is used in transperineal biopsies. The grid, which is placed in front of the perineum, includes a number of holes in which the biopsy needle can be inserted. Each hole is correlated to numbers and letters which allow for precise sampling of prostate. Also referred to as a template (the grid) and a stepper (stepping device).
- In-bore biopsy
- Technique that involves performing the prostate biopsy in the MRI scanner, where the needle is inserted within the MRI machine, and placement is guided by the MRI images in real time. Also referred to as in-gantry biopsy.
- ISUP Gleason grades
- Grouping of Gleason scores into risk groups defined by the International Society of Urological Pathology (ISUP) to help with outcome prediction and patient communication.
- Likert score
- A Likert score is reported using a 5-point Likert scale. The Likert scale, when used in the diagnosis of prostate cancer, accounts for clinical factors and lesion size on the MRI. A score of 1 indicates prostate cancer is very unlikely and a score of 5 indicates prostate cancer is very likely. Likert scores are used to help decide whether or not to have a prostate biopsy at the current time. The Likert score differs from the PI-RADS score in that it accounts for clinical factors and does not require the MRI to be conducted in a particular sequence.
- PI-RADS score
- prostate imaging – reporting and data system (PI-RADS) score is a system whereby each lesion, identified by MRI, is assigned a score from 1 to 5 to indicate the likelihood of clinically significant cancer (where 1 is very unlikely and 5 is very likely). PI-RADS v2 is the current validated version. It differs from the Likert score in that it does not account for clinical factors and it requires the MRI to be conducted in a particular order.
- Rigid registration
- During software fusion with rigid registration, the MRI image is fixed, and is not adjusted to match the ultrasound image when potential deformation to the prostate may occur during the biopsy.
- Route of access
- A route employed to reach the prostate with a biopsy needle. Can be either via the rectum (transrectal) or the perineum (transperineal). Also referred to as biopsy route.
- Semi-robotic arm
- Used in prostate biopsies, the semi-robotic arm is attached to the ultrasound probe. It allows the operator to manoeuvre the probe into the position of interest while ensuring a consistent level of pressure on the prostate to reduce prostate deformation.
- Software fusion biopsy
- Software fusion is software-based technology used to fuse pre-biopsy MRI image and real-time ultrasound images to create a detailed 3D image. Software fusion biopsy refers to biopsies where software fusion is used to guide and record the placement of biopsy needles. Also referred to as MRI fusion.
- Systematic biopsy
- Biopsy method where samples are taken in a systematic fashion from different regions of the prostate according to a predefined scheme. The number of cores sampled can range from 6 to 14, and is most commonly 12. Also referred to as random biopsy or 12-core biopsy.
- Targeted biopsy
- Biopsy where the site (or sites) for sampling is (or are) targeted based on the location of one or more potentially cancerous lesions identified by a MRI scan. Includes software fusion biopsy, cognitive fusion biopsy, and in-bore biopsy. Also referred to as MRI-targeted.
- Template biopsy
- Biopsy method where samples are taken in a systematic fashion from different regions of the prostate using a grid template. The minimum number of cores is typically 20. Also referred to as template prostate mapping.
- Transrectal ultrasound (TRUS) biopsy
- Where a biopsy needle is inserted through the rectal wall via the anus, and positioning is informed by ultrasound imaging.
- Watchful waiting
- Monitoring of a person, diagnosed with prostate cancer, where any potential treatment offered aims to control rather than cure the prostate cancer (palliative rather than curative intent).
List of abbreviations
- ADT
- androgen deprivation therapy
- AE
- adverse event
- bpMRI
- bi-parametric magnetic resonance imaging
- CF
- cognitive fusion
- CNS
- clinically non-significant
- CPG
- Cambridge Prognostic Group
- CrI
- credible interval
- CS
- clinically significant
- CSPCa
- clinically significant prostate cancer
- DCD
- diagnostics consultation document
- DRE
- digital rectal examination
- DTX
- docetaxel
- EAG
- External Assessment Group
- EAU
- European Association of Urology
- FN
- false negative
- GATP
- general anaesthesia transperineal
- GG
- grade group
- GIN
- Guidelines International Network
- GP
- general practitioner
- GS
- Gleason score
- HR
- hazard ratio
- HRG
- healthcare resource group
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- INHB
- incremental net health benefit
- IQR
- interquartile range
- ISUP
- International Society of Urological Pathology
- LATP
- local anaesthetic transperineal
- LATRUS
- local anaesthesia transrectal ultrasound
- mpMRI
- multiparametric magnetic resonance imaging
- MRI
- magnetic resonance imaging
- NA
- not applicable
- NC
- no cancer
- NG131
- NICE Guideline 131
- NHB
- net health benefit
- NHSCII
- NHS Cost Inflation Index
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health and Care Research
- NMA
- network meta-analysis
- NPCA
- National Prostate Cancer Audit
- NPV
- negative predictive value
- NR
- not reported
- NRFT
- no recurrence following treatment
- OS
- overall survival
- PACS
- picture archiving and communication system
- PCa
- prostate cancer
- PFS
- progression-free survival
- PI-RADS
- prostate imaging – reporting and data system
- PRFT
- possible recurrence following treatment
- PSA
- prostate-specific antigen
- PSS
- Personal Social Service
- QALE
- quality-adjusted life expectancy
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RFI
- response to information request
- RR
- Relative risk/risk ratio
- SF
- software fusion
- SOC
- standard of care
- TA
- technology appraisal
- TN
- true negative
- TP
- transperineal biopsy
- TRUS
- transrectal ultrasound
- TSB
- template-guided saturation biopsy
- TTMB
- template-guided mapping biopsy
- UTI
- urinary tract infection
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/PLFG4210).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.