

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as award number 13/140/05. The contractual start date was in October 2015. The draft report began editorial review in September 2021 and was accepted for publication in October 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this manuscript.
Permissions
Copyright statement
Copyright © 2024 Coyle et al. This work was produced by Coyle et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Coyle et al.
Chapter 1 Introduction
This chapter contains some text reproduced from the study protocol ‘Early switch from intravenous to oral antibiotic therapy in patients with cancer who have low risk neutropenic sepsis (the EASI-SWITCH trial): study protocol for a randomised controlled trial’ published in Trials (2020). https://doi.org/10.1186/s13063-020-04241-1. 1
Neutropenic sepsis
Neutropenic sepsis (NS) is a long-recognised and common complication of systemic anticancer treatment (SACT). 2 The term broadly refers to a significant inflammatory response to a presumed bacterial infection, in a person with or without fever and a low blood neutrophil count. 3 A low neutrophil count occurs commonly following SACT with a trajectory that varies depending on type and timing of SACT, typically reaching a nadir around 7 days post SACT then recovering over 2–3 weeks. 4 There is significant variation in the definition of neutropenia and sepsis, with a lack of comparative data to guide threshold-setting for neutrophil count or fever in patients with potential NS. 3,5–15 Based on the available data, the National Institute for Health and Care Excellence (NICE) Guideline Development Group (GDG) concluded using a threshold of 0.5 × 109/l for neutrophil count and ≥ 38°C for temperature for diagnosis of NS reflected an acceptable trade-off between over- and under-treatment of what could be a potentially fatal infection. 3 However, in the NHS, NS care pathways commonly use a temperature threshold of ≥ 38°C and an absolute neutrophil count threshold of either ≤ 0.5 × 109/l or < 1.0 × 109/l and falling/expected to fall.
Robust predictors of NS risk are lacking in patients with cancer receiving SACT. It seems that NS is more common soon after treatment is initiated (within the first two cycles)16 and following administration of anthracycline or taxane-containing regimens in treatment of early-stage breast cancer. Other factors associated with risk of developing NS include age, performance status and a diagnosis of blood cancer rather than solid tumour. 17,18
Despite widespread adoption of prophylactic colony-stimulating factors (CSF) and fluoroquinolone antibiotics for patients at high risk of septic complications, NS remains potentially life-threatening, with an in-hospital mortality rate of approximately 9.5%19 and, in the setting of severe sepsis or septic shock, as high as 50%. 20 NS deaths recorded by the Office of National Statistics more than doubled in England and Wales between 2001 and 2010 to approximately two deaths per day (716 deaths in 2010),3 with significant increases in chemotherapy use likely contributing. 21
Significant patient morbidity can also occur through hospitalisation, with a strong desire not to be hospitalised during treatment cited as a common barrier to patients promptly seeking help for symptoms. 22 An episode of NS can also result in dose delays and reductions to patients’ planned SACT, potentially compromising treatment efficacy in certain tumour types and settings. 3,23–25 There are associated financial implications on healthcare systems managing NS episodes, with hospital, antibiotic, diagnostic and additional therapeutic costs involved resulting in an estimated average cost per inpatient admission in the NHS ranging from approximately £257226 to £3163. 27
Empirical management of NS
Neutropenic sepsis continues to be viewed as a time-critical medical emergency with widespread agreement that early recognition and prompt administration of broad-spectrum empirical antibiotics are essential to successful treatment. 3,21,28,29 However, there is much less consensus on optimal patient management thereafter, including when to switch from intravenous (i.v.) to oral antibiotics, and duration of antibiotic treatment and hospital admission. Widely variable practice has been noted among cancer centres in the UK. 30 A review of 51 English and Welsh centres, prior to the introduction of a national UK NS clinical guideline, highlighted the heterogeneity that existed in almost all aspects of NS management, commenting that there was ‘surprisingly little agreement’ and ‘dramatic variations’ in clinical practice. These findings were consistent with previously published audits of both adult and paediatric haemato-oncology practice. 6,31,32
The NICE GDG recommended use of empirical beta-lactam monotherapy (piperacillin/tazobactam) as immediate treatment in patients with suspected NS in the absence of local microbiological indications to use an alternative agent or combination therapy (such as addition of an aminoglycoside) based on local resistance patterns. 3 Evidence supporting a specific duration of treatment was found to be lacking by the Group but the principle of switch to oral antibiotics following risk assessment after 48 hours of i.v. antibiotic therapy could be considered. While current European guidelines suggest that following initial assessment, including prompt institution of empirical broad-spectrum antibiotics, patients identified as low risk may be suitable for inpatient oral antibiotic treatment, the authors noted clinician preference was often to continue i.v. treatment for at least 48 hours and then consider a change to oral antibiotics if fever resolves. 28
Risk stratification
A spectrum of NS severity exists, encompassing a heterogeneous group of patients with variable risk of septic complications such as organ failure, need for critical care support and death. 33
At the low-risk end of the spectrum, there are patients who do not demonstrate clear clinical or microbiological evidence of proven infection, have uncomplicated hospital admissions and are at low risk of developing septic complications. These patients potentially receive overtreatment, with the associated distress of hospitalisation and additional burden to the healthcare system. 34
Risk stratification tools have therefore been developed in an attempt to identify patients predicted to be at low risk of an adverse outcome. The Multinational Association of Supportive Care in Cancer (MASCC) score (Table 1) is the most widely validated risk score for SACT-induced NS. 3,28,29,33
| Characteristic | Weight |
|---|---|
| Burden of febrile neutropenia: no or mild symptomsa | 5 |
| Burden of febrile neutropenia: moderate symptomsa | 3 |
| No hypotension (systolic BP > 90 mmHg) | 5 |
| No chronic obstructive pulmonary disease | 4 |
| Solid tumour or no previous fungal infection | 4 |
| No dehydration requiring parenteral fluids | 3 |
| Outpatient status | 3 |
| Age < 60 years | 2 |
In the original validation set, a risk index score of 21 or greater out of 26 identified low-risk patients with a positive predictive value of 91%, a sensitivity of 71% and a specificity of 68%. A low rate of adverse outcomes (6% serious medical complication, 1% mortality) was observed in patients with a risk index score ≥ 21 compared with 49% in those with a risk score of < 21. 35
Oral antibiotic therapy as treatment
A UK single-centre prospective randomised controlled trial (RCT) investigated the effectiveness of oral antibiotics (ciprofloxacin and co-amoxiclav) with early hospital discharge for patients with low-risk NS in comparison to standard i.v. antibiotics (tazobacam-piperacillin and gentamicin) with hospital admission. One hundred and twenty-six NS episodes from 102 patients were evaluated for the ‘success and safety’ primary end point, which comprised: lysis of fever, resolution of symptoms and signs, absence of modification of antibiotic regimen, absence of recurrence within 7 days and occurrence of serious complications or deaths. Treatment success was 90% in the i.v. arm and 84.8% in the oral arm. Re-admission was required in five episodes (7.6%) and deemed unrelated to the episode of NS in one. It was recognised that these results were obtained within a single specialist centre but that the findings supported undertaking a multicentre RCT evaluating oral antibiotics and early discharge. 36
A Cochrane review37 of oral versus i.v. antibiotics for NS, evaluating 22 trials comprising 3142 neutropenic episodes in 2372 patients, concluded that it is not likely that significant differences exist in treatment failure or mortality rates between oral antibiotic and i.v. antibiotic strategies. There was a trend towards more adverse events (AEs) in patients receiving oral antibiotics, typically gastrointestinal events, which did not necessitate treatment discontinuation. The majority of studies did not utilise any formal risk stratification tools but excluded high-risk patients with acute leukaemia, haemodynamic instability, evidence of organ failure or localising signs of infection.
The Cochrane review therefore broadly supported the early use of oral antibiotics in low-risk NS, but it was noted most trials were small in sample size, often single-centre and with methodological concerns and so a robust recommendation for upfront or early oral antibiotic therapy could not be made. It was suggested that ‘the combination of a quinolone and a second drug active against Gram-positive bacteria (for example ampicillin-clavulanate) seems prudent’. 37 This group also recommended that this therapeutic approach should be formally evaluated in patients with low-risk NS. The NICE GDG also considered oral antibiotic therapies but were unable to make a specific recommendation given variation in local microbiological resistance patterns and variation in use of prophylactic antibiotics. 3
Outpatient management of low-risk NS
The NICE GDG reviewed the evidence for inpatient versus outpatient management of NS and concluded outpatient management can be considered for selected low-risk patients, taking into account their individual clinical and social circumstances. 3 Although the metaregression undertaken by the GDG suggested early discharge (before 24 hours) may be associated with increased likelihood of re-admission or therapy change, the quality of evidence supporting outpatient management was low to moderate. The available data were limited by a lack of reporting of key outcomes such as critical care admission or clinically documented infection and a very low event rate for adverse outcomes including death. 3 Similarly, there is negligible literature relating to impact on quality of life for different models of care, including immediate use of oral antibiotics and non-admission to hospital, with a single study suggesting role function improved more for inpatients than home care patients but that emotional function declined with hospital admission. 38 It was therefore proposed that if a short period of hospital admission was found to be safe and effective for selected patients with NS, it could provide considerable improvements in quality of life and health resource usage.
Rationale for the trial
NICE therefore recommended that a randomised trial should be undertaken to evaluate the effectiveness of switching from i.v. to oral antibiotics within the first 24 hours of treatment in patients receiving i.v. antibiotics for NS. The early switch to oral antibiotic therapy in patients with low-risk NS (EASI-SWITCH) trial was developed in response to this recommendation and a commissioned call from the UK National Institute for Health and Care Research (NIHR) Health Technology Assessment (HTA) programme to address this evidence gap. It aimed to establish the clinical and cost effectiveness of an early switch to oral antibiotics 12–24 hours after i.v. antibiotic treatment commences in low-risk cancer patients with NS.
Acute oncology service development
While prospective RCTs evaluating upfront or early oral antibiotics remain lacking from publication of the NICE guidance, there have been significant service developments in response to acute care pressures and increasing demands due to increases in cancer incidence and available treatments with the aim of developing novel models of care for meeting cancer patients’ complex needs. 39 Ambulatory care has been introduced in acute medical and elderly care NHS settings with growing interest in developing care pathways in cancer services in recent years. This has included management of low-risk NS although it has only been implemented by a limited number of UK cancer centres. 40,41 Some have reported results from longitudinal patient series. Marshall et al. 42 reported a series of 100 patients from a large UK tertiary cancer centre over a 2-year period with NS who were assessed and given a first dose of i.v. antibiotics then managed on an ambulatory low-risk NS pathway. Patients were stratified using MASCC score and National Early Warning Score (NEWS) score and following observation for at least 4 hours were discharged for outpatient follow-up (repeat clinical assessment and routine bloods) within 48 hours. 42 Six of the 100 patients (8.8%) required re-admission within 7 days, typically with positive blood cultures, but none required critical care support. Brunner et al. 43 reported a low-risk NS ambulatory care pathway case series from another UK centre. One hundred and twenty-three patients presented with NS over a 2-year period, 41% of whom were deemed low risk based on MASCC score. Of these, 24 were managed on the ambulatory care pathway with a first dose of i.v. antibiotic and discharge with oral antibiotics and proactive telephone follow-up. A further 24 patients were admitted but had early discharge. Again, no serious complications occurred and the re-admission rate was 10%. However, despite the investment in establishing the ambulatory care pathway, approximately 80% of patients with NS were still admitted. Similarly, other international centres have reported real-world data where only a minority of patients are managed on ambulatory pathways or considered for same-day discharge. 44 For example, in a large US emergency department (ED) only 5% of NS patients were discharged home, with most low-risk patients admitted for inpatient antibiotics. 45 A subsequent large-scale review of approximately 350,000 US ED visits with NS confirmed this finding, with 94% of visits resulting in hospitalisation. 46 Cost analysis data from real-world data sets are also lacking.
Chapter 2 Methods
This chapter contains some text reproduced from the study protocol ‘Early switch from intravenous to oral antibiotic therapy in patients with cancer who have low risk neutropenic sepsis (the EASI-SWITCH trial): study protocol for a randomised controlled trial’ published in Trials (2020). https://doi.org/10.1186/s13063-020-04241-1. 1
Trial design
EASI-SWITCH was a UK prospective Phase 3, randomised, open-label, non-inferiority trial to evaluate whether early switch to oral antibiotics is non-inferior to standard care in adult patients with cancer with NS at low risk of complications.
The main aim was to determine the clinical effectiveness of early switch to oral antibiotics 12 to 24 hours after commencement of empirical i.v. antibiotics compared to standard care, which comprises continuation of i.v. treatment for at least 48 hours, based on treatment failure rate. Treatment failure was defined by a composite measure incorporating a number of important clinical outcomes assessed at day 14 of follow-up.
The trial included an embedded pilot study across four UK sites in order to test the recruitment and adherence assumptions which had informed the trial design.
Trial objectives
Primary objective
To determine whether early switch to oral antibiotic therapy is non-inferior to standard care therapy in terms of treatment failure measured at day 14.
Treatment failure was defined as a composite measure incorporating the following important treatment outcomes:
-
persistence, recurrence or new onset of fever (temperature ≥ 38°C) after 72 hours of starting i.v. antibiotic treatment
-
physician-directed escalation from protocol antibiotic treatment
-
re-admission to hospital (related to infection or antibiotic treatment)
-
critical care admission
-
death.
Secondary objectives
To assess the effect of early switch to oral antibiotics on:
-
short-term change in health-related quality of life (HRQoL), using EuroQoL-5 Dimensions, five-level version (EQ-5D-5L) as the measurement tool, at baseline and 14 days
-
cost-effectiveness, based on the cost per treatment failure avoided at 14 days and a cost–utility analysis (CUA) estimating the cost per QALY at 14 days
-
time to resolution of fever from initial i.v. antibiotic administration
-
AEs related to antibiotics
-
hospital discharge and total length of hospital stay
-
re-admission to hospital within 28 days
-
death within 28 days
-
adjustment to the subsequent scheduled cycle of chemotherapy within 28 days
-
patient preferences for antibiotic treatment assessed at day 14.
Research hypotheses
-
Early oral switch (within 12–24 hours after commencing i.v. antibiotic therapy) in cancer patients with low-risk NS is non-inferior to standard care (continuation of i.v. antibiotic therapy for at least 48 hours).
-
The incremental cost effectiveness of early oral switch is significant compared to standard care.
-
AEs are comparable between the two study arms.
-
Patients’ preference will be for early oral switch.
Study conduct
Ethics, regulatory and research and development approvals
The trial was approved by the Medicines Healthcare and Regulatory Agency (MHRA) on 31 July 2015. It was approved by the Northern Island (NI) Research Ethics Committee (REC) on 6 October 2015. At each participating site, local research and development (R&D) approval was obtained prior to patient enrolment to the trial. The trial was conducted in accordance with the principles of good clinical practice (GCP), the requirements and standards set out by the WU Directive 2001/20/EC and the applicable regulatory requirements in the UK, the Medicines for Human Use (Clinical Trials) Regulations 2001 and subsequent amendments and the Research Governance Framework.
Sponsorship
EASI-SWITCH was sponsored by the Belfast Health and Social Care Trust (BHSCT).
Trial management
Clinical trial management was undertaken by the Northern Ireland Clinical Trials Unit (NICTU). Additional trial oversight committees were convened by the trial including a Trial Management Group (TMG), Trial Steering Committee (TSC) and Data Monitoring and Ethics Committee (DMEC).
The TMG comprised the co-chief investigators, other clinical investigators, the trial manager/co-ordinator, the trial statistician, the trial health economist, the sponsor pharmacovigilance representative and the patient and public representative. The TMG met monthly to review site set-up, screening and recruitment, trial conduct, AEs and any other issues relating to trial conduct. A TMG charter detailed the terms of reference of the TMG including roles/responsibilities.
The TSC provided oversight for the progress of the trial on behalf of the sponsor and funder. The TSC was appointed by the NIHR and comprised an independent chair (a microbiologist), an independent oncologist, an independent statistician, at least one patient/public representative and TMG members. The remit of the TSC was progression of the trial including recruitment and adherence, the well-being, safety and rights of trial participants and ensuring trial conduct was appropriate. A TSC charter described the terms of reference of the TSC including membership and roles/responsibilities.
The DMEC provided independent review of the trial. Its role was to safeguard the rights and safety of participants, to review trial data related to recruitment, protocol compliance, safety and efficacy and to recommend to the TSC whether the trial should continue or not based on ethical or safety reasons. DMEC appointments were approved by NIHR and included an independent chair (an oncologist), an independent clinician, an independent statistician and a patient/public representative. DMEC reports were provided by the trial statistician to include recruitment, AE and outcome data along with any other information requested by the Committee. These reports were confidential and not shared with the trial investigators. A DMEC charter described the terms of reference of the DMEC including memberships and roles/responsibilities.
Trial set-up
In total 19 sites in hospitals and cancer centres across the four UK nations were opened to patient recruitment. A list of these sites can be found in the Acknowledgements. Potential sites were asked to complete an eligibility questionnaire that assessed clinical trial experience and local capability and capacity for the study. Local antimicrobial guidelines and treatment care pathways for NS were also requested from sites at this stage to identify and address any potential issues with protocol compliance. Prior to sites opening to recruitment a face-to-face site initiation visit was undertaken by trial team members to provide training on trial procedures to local research team members. Additional training, where needed, was provided by teleconference. The trial team maintained regular communication with sites by e-mail and teleconferencing to provide any ongoing training needed, answer any queries arising at site and support sites in identifying and resolving barriers to recruitment.
Patient information and consent
Potentially eligible patients were those who had commenced treatment with i.v. antibiotics for NS. Patients were identified at each study site daily through local acute admission/handover processes dependent on the unscheduled care admission pathways at site. Patients meeting these criteria were discussed with their treating physician on that day prior to enrolment to confirm their agreement to patient participation. This also provided an opportunity to confirm that their treating physician would be willing to follow the treatment strategy outlined in either arm of the trial. Patients were approached by a member of the research team and a patient information sheet was provided. Patients were given time to review the patient information sheet although this time period was < 24 hours given the acute care setting and timing of the intervention.
As enrolment was occurring at ward level and patients had already been initiated on treatment, patients being approached were clinically stable and viewed as competent to give informed consent in this setting. Patients who were unable to give informed consent, for any reason, were not recruited. Patients who indicated they were unwilling or unable to make a decision within the 24-hour time period were not recruited. Regulatory approvals were obtained for patient-facing materials additional to the patient information sheet to be used at site to make patients aware of the trial. These included a summary information sheet about the trial that could be included in the standard SACT patient education materials about NS and a poster to be displayed in SACT clinics and treatment units. All of these materials were prepared in collaboration with the trial patient representatives.
Informed consent for participation was sought from patients by appropriately trained research nurses (RNs) and medically trained investigators at site supported by the site principal investigator (PI) and local infrastructure. If patients required any further clarification about the risks and benefits of participation, this was provided by other research team members or an independent senior physician (one nominated in advance at each trial site). The PI (or designee) taking informed consent was required to have completed GCP training, be suitably qualified and experienced and be delegated this duty by the PI on the delegation log.
Screening and randomisation procedures
Electronic trial screening and recruitment logs, submitted by sites to the clinical trial unit (CTU) on a monthly basis, aimed to capture all patients who received a patient information sheet and whether they proceeded to consent and randomisation. Research teams were asked to provide a reason for non-participation if patients were not recruited.
After informed consent was obtained and eligibility was confirmed, participants were allocated to intervention or standard care groups using an automated randomisation system (sealed envelopes). Blocked randomisation with randomly permuted block sizes was used and a 1 : 1 allocation ratio. There were no factors for stratification. Access to the randomisation sequence was restricted and not accessible to site staff who enrolled patients or assigned interventions. Only the allocation of the intervention was blinded. As this was a pragmatic trial, it was felt that blinding clinical teams, researchers and trial participants to the intervention would limit the ability of the trial to measure the impact of the intervention on routine care pathways. Additional support from this approach came from our patient and public involvement (PPI) representatives, who viewed that participants would be highly likely to reveal their treatment allocation during discussion with healthcare providers and outcome assessors, making it unlikely these groups could remain blinded.
Trial treatment
Patients eligible to participate were aged over 16 years, receiving SACT for a cancer diagnosis and were receiving standard-dose i.v. piperacillin/tazobactam or meropenem as initial antibiotic treatment for suspected NS for < 24 hours. Patients were only permitted to be enrolled in the trial on one occasion in line with consensus guidelines. 47 All protocolised antibiotics were considered to be investigational medicinal products (IMPs) for the purpose of the trial: co-amoxiclav 500 mg/125 mg film coated tablets; ciprofloxacin 250 mg, 500 mg, 750 mg film coated tablets; meropenem 1 g powder for solution for injection or infusion and tazocin 4 g/0.5 g powder for solution for infusion.
Standard care arm
Participants in the standard care group were allocated to continue current treatment with i.v. antibiotics for a minimum of 48 hours. This was selected based on the NICE guidance recommendations. 3 Subsequent antibiotic management was at the discretion of the treating physician, who could switch to oral antibiotics or stop antibiotics at any point thereafter, reflecting the variation encountered in routine clinical practice. 30
Intervention arm
Participants randomised to the intervention group switched from i.v. antibiotic treatment within 12–24 hours after starting treatment, to co-amoxiclav 625 mg three times daily and ciprofloxacin 750 mg twice daily, to complete at least 5 days antibiotic treatment in total. The combination of a quinolone and a second drug active against Gram-positive bacteria (e.g. co-amoxiclav) was based on the conclusions of the Cochrane review. 37
Other treatments
Any other treatments or investigations that patients required were carried out in accordance with standard care. It was recognised that escalation from protocol-specified antibiotic treatment might be required in the event of clinical deterioration, progression of the presumed infection, a microbiological indication based on microbiological culture results or an adverse reaction (AR) to the prescribed antibiotics. A change from protocol-specified antibiotics, including additional antibiotic treatment other than the study drugs, or persistent/recurrent fever (> 38°C) after 72 hours was within the definition of treatment failure, with such participants reaching the trial’s primary end point.
Patients were discharged home from hospital once their treating physician was content to do so, with a patient diary to record any further temperatures and oral antibiotic compliance. Due to the pragmatic nature of the trial, specific discharge criteria were not protocolised, but it was assumed the patient’s overall clinical condition and psychosocial circumstances would be considered by the treating clinician in line with their normal clinical practice.
Patient population
Patients commenced on i.v. antibiotics within 24 hours after starting treatment for low-risk NS were recruited from sites across England, Scotland, Wales and NI, comprising both large cancer centres and cancer units, to ensure that the sample is broadly representative of patients developing NS in the UK.
Inclusion criteria
-
Age over 16 years.
-
Receiving SACT for a diagnosis of cancer.
-
Started on empirical i.v. piperacillin/tazobactam or meropenem, for suspected NS, for < 24 hours.
-
Absolute neutrophil count ≤ 1.0 × 109/l with either a temperature of at least 38°C or other signs or symptoms consistent with clinically significant sepsis, for example hypothermia. Self-measurement at home or earlier hospital assessment of temperature is acceptable provided this is documented in medical notes and is within 24 hours prior to i.v. antibiotic administration.
-
Expected duration of neutropenia < 7 days.
-
Low risk of complications using a validated risk score (MASCC score ≥ 21).
-
Able to maintain adequate oral intake and take oral medication.
-
Adequate hepatic [aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) < 5 × upper limit of normal (ULN)] and renal function (serum creatinine < 3 × ULN) within the 24 hours prior to randomisation.
-
Physician in charge of care willing to follow either the intervention or standard care protocol per randomisation, at enrolment, including not treating with CSF. Prophylactic CSF is not an exclusion criterion.
Exclusion criteria
-
Underlying diagnosis of acute leukaemia or haematopoietic stem cell transplant.
-
Hypotension (systolic pressure < 90 mmHg on > 1 measurement) within the 24 hours prior to randomisation.
-
Prior allergy, serious AR or contraindication to any study drug.
-
Enrolled in this trial with prior episode of NS.
-
Previously documented as being colonised with an organism resistant to a study drug regimen, for example MRSA.
-
Localising signs of severe infection (pneumonia, soft-tissue infection, central-venous access device infection, presence of purulent collection).
-
Patients unable to provide informed consent.
-
Pregnant women, women who have not yet reached the menopause (no menses for ≥ 12 months without an alternative medical cause) who test positive for pregnancy, are unwilling to take a pregnancy test prior to trial entry or are unwilling to undertake adequate precautions to prevent pregnancy for the duration of the trial.
-
Breastfeeding women.
Co-enrolment
Patients who were enrolled in other Phase I IMP studies and other antimicrobial IMP studies were excluded. Patients enrolled in other Phase II–IV IMP or observational studies were eligible for enrolment in this study at the PI’s discretion and when the burden on participants was not considered to be onerous.
Withdrawal of consent
Participants were able to withdraw consent to participate in the trial at any time. If the participant withdrew consent during protocolised treatment, no further treatment within the trial was given and the clinician responsible for their care determined the safest and most appropriate continued management plan. A withdrawal of consent form identified which parts of the trial the patient wished to withdraw from: protocol-specified antibiotic therapy; future data collection (all data collected or data collected at day 14 and/or day 28 follow-up). Participants could be withdrawn from the study at the discretion of the investigator if any safety concerns arose.
Data management
Trial database
The EASI-SWITCH trial database is an electronic clinical trial database (MACRO) held by the NICTU. Trial data were entered on to a web-based case report form (CRF) with imposed rules for data entry with valid responses and linkage of dates and trial identification numbers by trained and delegated site personnel. Data were processed in accordance with the trial Data Management Plan and CTU Standard Operating Procedures. Data queries were ‘raised’ electronically via MACRO where clarification was needed for data entries or to complete missing data and staff at site ‘responded’ electronically to queries and amended database entries where applicable. A final review for missing or inconsistent data was carried out by the trial statistician with subsequent opportunity for query resolution / data completion prior to the database lock for end of trial analyses. All essential documentation and trial records were stored securely with access restricted to authorised staff only.
Data quality
Data management within the CTU was governed by Standard Operating Procedures to ensure standardisation and compliance with International Conference of Harmonisation Good Clinical Practice (ICH-GCP) guidance and regulatory requirements. NICTU provided site staff training in data collection and CRF completion. On-site monitoring visits during the trial checked accuracy of CRF entries against source documents in addition to protocol and trial procedure adherence. Discrepancy reports were generated after data entry to identify inconsistent or out-of-range data and protocol deviations based on data validation checks programmed into the clinical trial database.
Data collection
Data were collected by delegated research team members. Each participant was allocated a unique Participant Study Number at randomisation, alongside their initials for identification for the duration of the trial. Data were collected from the time of trial entry until day 28 (± 1 day) in accordance with the schedule of assessments shown in Table 2. Baseline data collection occurred in the hospital setting. Primary and secondary outcome data were collected via a review of patient medical notes (including laboratory results), submission of participant questionnaires, patient diary, GP records and telephone calls with patients. Participants discharged before day 14 were asked to complete a diary noting administration of oral antibiotics, any new medications and a temperature diary (if required) until day 14. Questionnaires were administered face-to-face or via telephone (if discharged or no scheduled outpatient visit) at day 14 (± 1 day).
| Time point | t-24 hours | Day 0 | Study visits and procedures | ||||
|---|---|---|---|---|---|---|---|
| Pre-consent (standard care) | Pre-randomisation | Randomisation | Day 1–2 | Day 3–5 | Day 6–14 | Day 28 | |
| Pre-consent eligibility screening | |||||||
| Eligibility screening as appropriate (per standard care) for example full blood count, blood culturea | ✗ | ||||||
| Informed consent | |||||||
| Informed consent obtained | ✗ | ||||||
| Pre-randomisation eligibility and assessments | |||||||
| Eligibility screening as appropriate (non-standard care) for example pregnancy test, MASCC score, max temp ≤ 24 hours prior to randomisation. | ✗ | ||||||
| EQ-5D-5L | ✗ | ||||||
| Randomisation | |||||||
| Standard care antibiotic administrationb | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Intervention (early switch) antibiotic administrationb | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Send GP letter | ✗ | ||||||
| Baseline assessments to be recorded on CRF after eligibility is confirmed | |||||||
| Demographics | ✗ | ||||||
| Vital signs (HR, RR and BP) | ✗ | ||||||
| Medical historyc | ✗ | ||||||
| Symptoms indicative of mild localised infection | ✗ | ||||||
| Cancer assessmentd | ✗ | ||||||
| SACT administered prior to presentatione | ✗ | ||||||
| Relevant microbiological results | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Hospital admission details | ✗ | ||||||
| Concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Daily data collection | |||||||
| Antibiotic regimenf | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Highest daily temperatureg | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Protocol compliance | |||||||
| Adherence to protocol specified intervention | ✗ | ✗ | |||||
| Patient follow-up | |||||||
| EQ-5D-5L | ✗ | ||||||
| Patient follow-up questionnaire | ✗ | ||||||
| Follow-up contact | ✗ | ✗ | |||||
| Survival status | ✗ | ✗ | |||||
| New medications | ✗ | ||||||
| Changes to next planned SACT | ✗ | ||||||
| Hospital discharge/re-admission/critical care admission details | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Recording and reporting of AEs | ✗ | ✗ | ✗ | ✗ | ✗ | ||
Adverse event reporting
Directly observed or patient-reported AEs that were not related to underlying medical conditions were recorded by the site PI or designee. AEs clearly related to SACT administration (such as peripheral neuropathy) were not required to be recorded; however, if an AE could be due to SACT, NS or antibiotic therapy (such as fever or gastrointestinal symptoms), then it was required to be recorded. Initially the AE reporting period for the trial was from enrolment until 28 days after randomisation. This was amended subsequently to 14 days from enrolment until 14 days after randomisation in recognition that antibiotic AEs generally occurred within this time frame and that patients were typically receiving a further course of SACT within the 14–28 day window, resulting in AEs that were more likely to be SACT-related or a new episode of NS rather than related to the episode of NS that had resulted in trial entry or antibiotic therapy.
The PI (or designee) was required to make an assessment of expectedness of any AE deemed possibly, probably or definitely related to any of the trial IMPs based on the relevant Summary of Product Characteristics (SPCs).
AEs related to IMP exposure were deemed ARs. ARs were classified as expected (consistent with IMP side effects listed in the SPC) or unexpected (not consistent with the SPC).
Serious adverse event
In the trial, a serious adverse event (SAE) was defined as any AE that:
-
resulted in death
-
was life-threatening
-
required hospitalisation or prolongation of existing hospitalisation
-
resulted in persistent or significant disability or incapacity
-
consisted of a congenital anomaly or birth defect
-
was any other important medical event(s) that carried a real, not hypothetical, risk of one of the outcomes above.
All deaths that occurred within 28 days of randomisation were recorded and reported as a SAE regardless of the nature of the event (even if due to progressive cancer). PIs were required to report SAEs to NICTU using the trial-specific SAE form within 24 hours after becoming aware of the event.
Treatment failure due to persistence, recurrence or new occurrence of fever after 72 hours of antibiotic commencement and/or clinician-directed escalation of protocolised antibiotic therapy during the first 14 days was captured as part of the primary outcome measure. This was only reported as an AE when categorised as a SAE.
Suspected unexpected serious adverse reaction
Adverse reactions that were unexpected (not consistent with the relevant SPC) and met the criteria for seriousness were deemed suspected unexpected serious adverse reactions (SUSARs) and required expedited safety reporting. NICTU reporting procedures report SUSARs to relevant competent authorities within 7–15 calendar days in accordance with UK regulations.
Serious breaches
A serious breach was defined as an occurrence which represented a deviation from the trial protocol that was likely to result in significant effect on the safety of a trial participant or the scientific value of the trial.
The PI (or designee) was responsible for direct reporting of serious breaches to the trial sponsor with onward reporting to the relevant competent authorities in accordance with UK regulations.
Protocol amendments
All amendments to the trial protocol, patient information sheet, informed consent form and other key documents were submitted to the relevant regulatory authorities for approval prior to implementation. Local research and development approval was also obtained at each site. Version 2 of the trial protocol was the protocol approved for use as trial commencement and subsequent substantial protocol amendments are summarised below.
July 2016
Version 3 of the protocol was submitted to the regulatory authorities. This incorporated changes to the eligibility criteria in order to align better with the NICE guidance and routine practice in the NHS setting. The NICE guidance stated ‘Diagnose neutropenic sepsis in patients having anticancer treatment whose neutrophil count is 0.5 × 109 per litre or lower and who have either: a temperature higher than 38°C or other signs or symptoms consistent with clinically significant sepsis’. On initial design, the trial had only incorporated the objective definitions (i.e. temperature and neutrophil count) because of possible difficulty in defining ‘signs or symptoms consistent with clinically significant sepsis’ for a trial population. This protocol amendment permitted recruitment of patients with either:
-
a neutrophil count of ≤ 0.5 × 109 per litre who have either a temperature of at least 38°C or other signs or symptoms consistent with clinically significant sepsis (to align fully with the NICE guidance); or
-
a neutrophil count of < 1.0 × 109 per litre, and falling or expected to fall, who have a temperature of at least 38°C (to reflect usual NHS practice). Given the pragmatic nature of the trial this amendment was felt to allow a more realistic evaluation of the intervention in routine care.
August 2016
Versions 4 and 5 of the protocol incorporated an extension to the pilot phase of the study including the addition of up to four new sites, the resultant change in the overall study duration and clarification of eligibility criteria and outcome measure definitions. The key change within these amendments was the extension of the embedded pilot study to 12 months to assess the impact of the change in eligibility criteria on recruitment, which had been lower than anticipated after the trial commenced.
April 2017
Protocol version 7 was submitted to the regulatory authorities (protocol version 6 was a non-substantial amendment). The purpose of this amendment was to clarify use of non-protocolised antibiotics and reporting of AEs.
June 2018
Protocol version 8 was submitted to the regulatory authorities. This incorporated revisions to the study design following review of the pilot study and discussions with the research team, trial sites, trial oversight committees. While the extended pilot study did meet the pre-specified recruitment target for study continuation, the trial team and oversight committees concluded that it would not be possible to recruit the originally planned sample size of 628 patients within a meaningful time frame to ensure the results remained relevant to clinical practice. Both the trial team and oversight committees continued to view the research question as important and relevant to current practice; opinion sought from the oncology clinical community and PPI representatives nationally also supported this view. The DMEC confirmed in November 2017 that there were no concerns regarding treatment adherence, separation between treatment groups or the observed treatment failure rate. Therefore, the assumptions that had underpinned the original trial design remained valid but, in retrospect, the choice of non-inferiority margin and statistical analysis may have been too stringent for a pragmatic trial where the risk of treatment failure was unlikely to result in serious risk to patients (in particular critical care admission or death). Following extensive stakeholder discussion and consideration of alternative assumptions for sample size recalculation, the sample size was recalculated using a 15% non-inferiority margin as this was felt to maintain an acceptable trade-off between the possibility and consequences of treatment failure for this low-risk patient population. This amendment included a sample size recalculation that comprised both a widening of the non inferiority margin from 10% to 15% and a change in the one-sided confidence interval (CI) from 97.5 % to 95%. This was recommended by the TSC independent statistician and following discussion with the wider TSC felt to be reasonable given the original 97.5% CI had been based on regulatory agency recommendations for licensing studies of new treatments which require a greater degree of certainty than was felt warranted for a pragmatic trial testing antibiotics already licensed or within routine clinical use for the same indication. This amendment also included an increase in the total number of study sites and an extension to the project duration.
Embedded pilot study
The trial contained an embedded pilot study involving four UK sites to test the recruitment and adherence assumptions underpinning the study design. Progression to the full study was based on the following criteria:
-
Recruitment rate:
-
progression without major modification if at least 75% of target reached
-
progression with addition of further trial sites if between 50% and 75% of target reached
-
progression unlikely if < 50% of target reached – discussion with trial oversight committees and funder required.
-
-
Adherence to protocol-specified intervention:
-
progression without major modification if at least 75% adherence in both trial arms
-
if adherence was between 50% and 75% of target, progression would be supported by a detailed analysis of the process and decision-points that led to non-adherence and a recognised strategy to address this identified
-
progression unlikely if < 50% adherence in either arm.
-
-
Separation:
-
separation in terms of the timing of antibiotic switch of at least 24 hours between the trial arms to enable progression was required.
-
The four-site pilot study was expected to run for 6 months between February and July 2016. Recruitment was < 50% target threshold for progression and the study was halted to review progress and proposals to address under-recruitment. This review identified the stringent eligibility criteria as a barrier to recruitment and not reflective of the routine management of patients with suspected NS. A protocol amendment (July 2016, described above) was submitted to address this. Recruitment was resumed with a 3-month extension to the pilot study (December 2016 to February 2017) with an improvement in recruitment rates (11 patients of an anticipated 13.5 recruited). On the recommendation of the HTA Programme Director, a larger pilot phase extension followed from April until November 2017. Progression with addition of new sites continued from this point until the end of the study.
Statistical analysis plan
Non-inferiority design and non-inferiority margin
A non-inferiority design was felt to be appropriate as it was not anticipated that early switch to oral antibiotics would offer superior treatment efficacy to standard care (i.v. antibiotics for at least 48 hours based on NICE guidance). It would also enable the evaluation of an intervention of broadly comparable efficacy to standard care based on available literature but with potential for reduced cost and improved quality of life.
It was estimated that the treatment failure rate in the control arm would be 15% based on data from three studies with patient populations most comparable to the proposed EASI-SWITCH population. Selecting the non-inferiority margin, the maximum clinically acceptable extent of non-inferiority, was challenging due to the limited evidence available to help guide this selection. A 10% non-inferiority margin was originally chosen to reflect the recommendations of a published expert consensus in NS antibiotic trials but this did not take into consideration risk stratification. 47 Input from patient representatives was also considered, but it is important to note that feedback initially came predominantly from our PPI co-applicant rather than a wider range of patient representatives, who felt if an extra 10% failed an early switch, in addition to the expected 15% treatment failures that occur with standard care, the advantage of 75% of early switch patients having successful treatment outweighed this.
Sample size
The original target sample size for the trial was 628 patients. This was based on the assumed 15% treatment failure rate in the standard care arm and a 10% non-inferiority margin, at 90% power (one-sided 97.5% CI), which would require 269 patients per arm. A dropout rate of up to 5% was also accounted for based on previously reported NS trial data and a crossover rate of up to 10% from the control to the intervention arm, giving 314 patients per arm (628 in total).
Review of study design post-pilot phase and revision of sample size
As outlined above, a protocol amendment related to review of the study design following the embedded pilot phase was submitted in June 2018. This included a review of the assumptions that had underpinned the original trial design, including the rationale for the choice of non-inferiority margin and statistical analyses. As previously described, extensive discussion took place with stakeholders including the research team, trial oversight committees, funder, sponsor and clinicians and PPI representatives nationally, in relation to the study design. The consequent revisions to the study design had implications for the sample size, which was recalculated using the same treatment failure rate in the standard care group (15%), power (90%), dropout rate (5%) and crossover rate from control to intervention (10%) while widening the non-inferiority margin from 10% to 15% and using a one-sided 95% CI. This gave a revised sample size of 230 patients.
Statistical methods
The primary analysis was conducted on both the per-protocol (PP) population and the intention-to-treat (ITT) population. Given the potential risk of bias arising from either of these analyses alone within a non-inferiority trial, it was pre-specified that non-inferiority of the intervention would only be proven if demonstrated in both the PP and ITT groups.
Analyses were one-sided and at a significance level of 0.05. The difference in treatment failure rate (one-sided 95% confidence limit) was compared to the non-inferiority margin of 15%. As this was a non-inferiority trial, the null hypothesis was that the degree of inferiority of the intervention to the control was greater than the non-inferiority margin of 15%. The alternative hypothesis was therefore that the intervention was inferior to the control by less than the non-inferiority margin of 15%. Therefore, non-inferiority would be established by showing that the upper limit of a 90% CI for Intervention – Control is < 15%.
A secondary comparison of the primary and other binary outcomes between the two groups was investigated using logistic regression, adjusting for covariates (such as extent of neutropenia). Comparison of continuous outcomes between the two groups was investigated using independent t-tests or Mann–Whitney. Statistical diagnostic methods were used to check for violations of the assumptions, and transformations were performed where required.
Baseline characteristics, follow-up measurements and safety data were described using appropriate descriptive summary measures depending on the scale of measurement and distribution.
Subgroup analyses
Exploratory subgroup analyses were pre-specified using 99% CI. Logistic regression was used with interaction terms (treatment group by subgroup) for the following subgroups:
-
tumour type (solid tumour vs. lymphoma)
-
neutrophil count at randomisation (≤ 0.5 × 109/l vs. > 0.5 × 109/l < 1.0 × 109/l)
-
maximum temperature on the day of presentation (< 38°C vs. ≥ 38°C).
An additional post hoc subgroup analysis was requested for those patients who had a positive blood culture versus those who did not at baseline.
Survey design and delivery
A series of surveys were undertaken to obtain stakeholder feedback on important questions relating to the trial. All electronic surveys were conducted using the SurveyMonkey tool (www.surveymonkey.co.uk). Ethical approval was not required for these projects as they sought only to define clinicians’ current standard of care and were categorised as service evaluation rather than research according to UK Health Research Authority definitions. All were designed pragmatically for data collection and to encourage responses. They were piloted informally by clinical and PPI colleagues at the lead site prior to wider dissemination to improve clarity and understanding but did not undergo formal reliability or validity testing. Participation was voluntary, with potential responders assured that no site or personal information would be publicly shared. Onward e-mail circulation to appropriate colleagues was encouraged to maximise responses, accepting this makes estimation of response rates challenging. At least one e-mail reminder was sent also to encourage responses, with all surveys open for completion for at least 8 weeks in total.
The analyses of the survey responses are presented descriptively, with percentages rounded to the nearest 1%. If the survey contained questions where respondents had the opportunity to leave comments, these are presented thematically, with representative illustrative quotes.
Site interviews
To understand factors impacting trial recruitment at sites, a series of purposeful semistructured interviews with key personnel at each site, including PIs and lead RNs, were conducted by the trial Clinical Research Fellow over a 2-month period (April to May 2018). Qualitative research has previously been demonstrated as one of the most helpful tools in identifying and overcoming barriers to recruitment. 48,49 The aim of the interviews was to explore clinician’s experience of recruiting and delivering the trial at their site and identify barriers to recruitment.
An initial e-mail invitation describing the nature and purpose of the interviews was sent to all PIs and lead RNs at all 12 open sites in March 2018. Participation was voluntary, and participants were reassured that no individual or site-specific information would be identified in summary reports. Interviews could be conducted either face to face or via telephone depending on participant preference. On receipt of a positive response, an interview was arranged and all were conducted in April and May 2018. Verbal consent was confirmed at the commencement of each interview. A short interview guide was prepared based on broad themes that had already emerged from previous discussions about the trial’s progress and difficulty recruiting. Participants were first asked in an open-ended question to simply comment on their experience recruiting to the trial and then highlight any challenges encountered. If not already discussed, they were then prompted to provide feedback on the trial’s screening and recruitment processes, including eligibility criteria. They were also prompted to comment on any issues encountered with local support for the study, research capacity, participant follow-up and data collection. Finally, interviewees were asked to highlight any strategies they felt had facilitated recruitment at their individual site. Interviews ranged from 15 to 50 minutes in length.
With the participant’s permission field notes were taken during the interview to capture key responses. A summary of the main findings was then verbally confirmed by the participant at the end of each interview to ensure content validation. Data saturation (i.e. no new information raised in later interviews) was reached during the interviews.
Thematic analysis of the qualitative data was then undertaken50 to generate a summary list of reported barriers to trial recruitment. The field notes for each interview were first scrutinised individually and organised into themes relating to barriers to recruitment. This systematic process was repeated for all interviews and then combined to produce a final group analysis summary list. Post analysis of the data, the co-chief investigators reviewed the data for completeness, reporting coherence between the data and reported themes to ensure robustness of the interpretation of the data.
Patient and public involvement
The trial has benefitted from having an experienced PPI member in the team, Mrs Margaret Grayson, from study conception to completion. The trial was developed in response to a commissioned call, meaning the main research question was pre-defined; however, input from the Northern Ireland Cancer Research Consumer Forum (NICRCF) via Mrs Grayson was that this was an important research question of value to patients where potential overtreatment and prolonged hospital admission may negatively impact upon quality of life. From this position, Mrs Grayson contributed to the trial design with particular input in the following areas: (1) helping to define the composite outcome measure and define secondary outcome measures important to patients and the health service; (2) defining the non-inferiority margin to incorporate patients’ views on acceptable trade-offs for treatment de-escalation; and (3) providing the patient viewpoint on the most appropriate method to obtain day 14 outcome data balancing the need for data quality with burden on patients. Additional support was given by the readers panel of the NICRCF at this stage through development and review of the trial protocol, the patient information sheet, the informed consent form and other patient-facing materials, for example brief summary flyers and posters for sites to use in SACT treatment units to make patients aware of the trial.
As the trial progressed, there was ongoing input from Mrs Grayson through both her membership of the TSC and participation in the TMG. Her input was critical when the overall study design was reviewed during the course of the trial, co-ordinating PPI opinion on the proposed changes and contributing to the final amended design. Through her linkages with the NICRCF and nationally (with the Independent Cancer Patient Voice group) she sought opinion on the ongoing importance of the research question and whether there was support for continuation of the trial with changes to the study design. As part of this process, she co-developed with the study team a PPI survey outlining proposed changes to the study design with particular focus on whether additional uncertainty around treatment effectiveness arising from a change in the non-inferiority margin and sample size would be acceptable to patients. She then collated responses, reporting to the study team the support of the majority of respondents and providing written and verbal communication with the funder in decision-making about these proposed changes.
The trial was also supported by additional independent PPI representation on the TSC and DMEC. Input from these representatives in review of trial data and progress was critical at decision-making points relating to trial progression and potential amendments to study procedures. Dissemination of results is an ongoing area of activity for all of the PPI team trial team members.
Chapter 3 Pilot study results and review of study design
This chapter contains some text reproduced from the study protocol ‘Early switch from intravenous to oral antibiotic therapy in patients with cancer who have low risk neutropenic sepsis (the EASI-SWITCH trial): study protocol for a randomised controlled trial’ published in Trials (2020). https://doi.org/10.1186/s13063-020-04241-1. 1
Pilot study aim
The aim of the internal pilot study was to carefully evaluate the recruitment and adherence assumptions underpinning the main study design. The main parameters of interest were:
-
recruitment rates
-
adherence to the protocol-specified treatment
-
separation in terms of timing of the antibiotic switch between the two arms.
These criteria were set to guide trial progress and inform the procedures to be utilised in the delivery of the main trial.
Recruitment rates
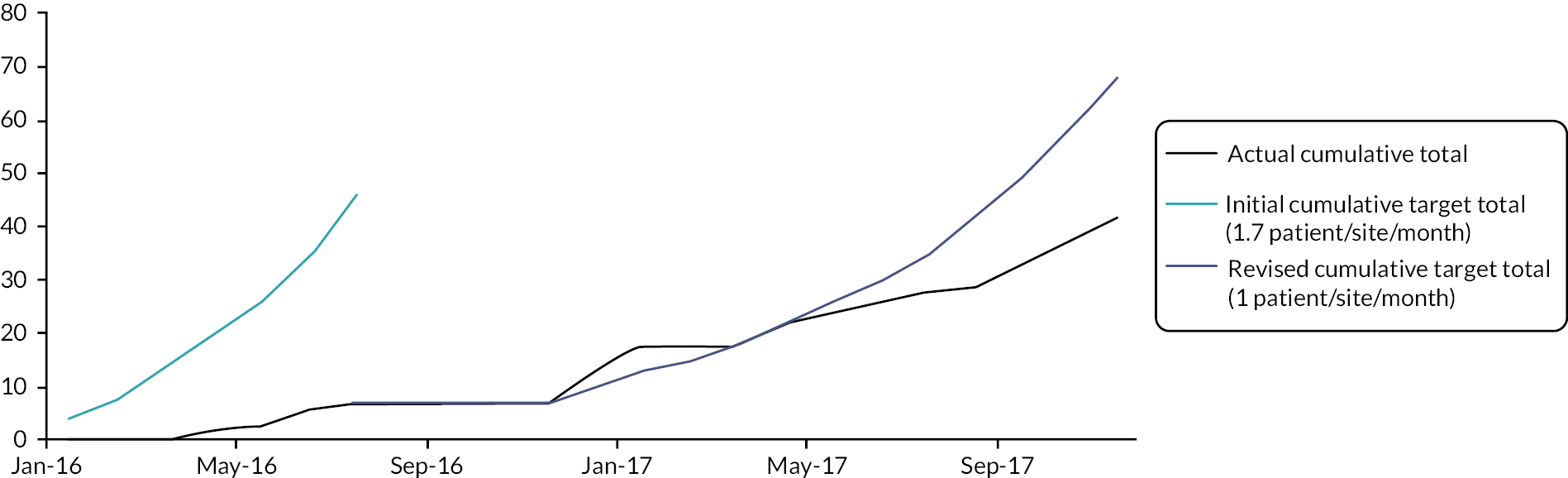
A target recruitment rate of 1.7 patients per site per month was set based on historical published data30 and service evaluation data from two of the pilot sites. Progression of the trial without modification beyond the embedded pilot study was contingent upon at least 75% of this recruitment target being met. It was otherwise pre-specified that progression would continue with the addition of further trial sites if 50–75% of this target was met but that progression at a recruitment rate lower than 50% of this target rate would require review of the trial and discussion with oversight committees, funder and sponsor for trial progression. The embedded pilot study was intended to last 6 months with 4 participating sites but was extended to 17 months and 10 sites in total, as summarised in Table 3.
| Dates | Sites | Recruitment target | Expected recruitment | Actual recruitment | Cumulative trial recruitment | |
|---|---|---|---|---|---|---|
| Initial pilot phase | February to July 2016 (6 months) | 4 pilot sites | 1.7 patients/month/site | 22 | 7 | 7 |
| Extended pilot phase (I) | December 2016 to February 2017 (3 months) | 4 pilot sites | 1.7 patients/month/site | 13.5 | 11 | 18 |
| Extended pilot phase (II) | April to November 2017 (8 months) | 10 4 pilot sites 6 additional sites |
1 patient/month/site | 49 | 24 | 42 |
Initial pilot phase (February to July 2016)
The internal four site pilot study was expected to run for 6 months. It commenced on the 17 February 2016, when Site 1 opened and completed on the 21 July 2016. Progress is summarised in Table 4.
| Site | Date open | Number screened | Duration of recruitment (months) | Expected recruitment | Actual recruitment | Recruitment rate (patients/site/month) |
|---|---|---|---|---|---|---|
| 1 | 17 February 2016 | 32 | 5 | 7 | 1 | 0.2 |
| 2 | 14 March 2016 | 26 | 4 | 6 | 2 | 0.5 |
| 3 | 4 May 2016 | 5 | 3 | 4 | 1 | 0.3 |
| 4 | 31 March 2016 | 6 | 4 | 5 | 3 | 0.75 |
| Total | 22 | 7 | ||||
Recruitment of 31 patients was originally projected if all four sites had been open to recruitment for the full 6 months. This was based on a recruitment rate of 1.7 patients per month per site, allowing for a 50% reduction in recruitment at each site during the first 3 months as sites became established. However, delays encountered at site level resulted in only one of the four sites being open for the full 6 month period. Taking this into account recruitment of 22 patients was anticipated, but only 7 were recruited. In view of this, the study was halted to enable review of screening and recruitment activity and recruitment target.
The original accrual target was based on historically published NS surveys and audit data from two sites, suggesting approximately 20 patients were admitted per month with NS in 2011 and 2013. This was consistent with the NICE guidance,3 which suggested active specialist units admitted at least 20 patients with NS per month. Allowing for exclusion of high-risk patients (approximately 30%51) and trial ineligibility it was assumed that 10 patients per month at each site would be eligible and recruitment of two of these appeared an achievable target.
On review of screening and recruitment logs the number of NS admissions was lower than expected. This was consistent with updated audit data from sites and may reflect changes in standard care relating to growing use of targeted therapies and immunotherapies in place of cytotoxic chemotherapy and continued improvements in the care of patients with suspected sepsis, including NS. However, even within this it was clear that the majority of patients screened for study entry were considered ineligible, as highlighted by activity at Sites 1 and 2 in the previous table where 1 of 32 patients and 2 of 26 patients were screened and recruited, respectively. The reasons for exclusion are summarised in Table 5; as expected around 30% of patients were high risk based on MASCC score, but for the remainder it seemed that the eligibility criteria did not reflect the patient population receiving treatment for NS in routine care pathways.
| Principal reason for exclusion | Site 1 (n) | Site 2 (n) |
|---|---|---|
| High risk (MASCC score < 21) | 9 | 7 |
| Pyrexial but absolute neutrophil count (ANC) between 0.5 and 1.0 × 109/l | 4 | 5 |
| ANC between 0.5 and 1.0 × 109/l Apyrexial but other signs and symptoms |
9 | 6 |
| Penicillin allergy | 6 | 3 |
| Recent prophylactic ciprofloxacin/other oral antibiotics | 2 | 3 |
| Total number of patients excluded | 30 | 24 |
From discussion with the clinical and research teams at study sites, specific issues with the trial eligibility criteria mainly related to the stringent requirements for fever, neutropenia, use of other antimicrobials and organ function, summarised in Table 6.
| Eligibility criteria | Impact on recruitment |
|---|---|
| 1. Neutrophil criteria |
|
| 2. Fever criteria |
|
| 3. Prophylactic antibiotics and recent antibiotic use |
|
| 4. Organ function |
|
| 5. Hypotension |
|
Review of eligibility criteria
It was therefore concerning from screening data that the trial population was not going to fully reflect the patient population being managed as low-risk NS, with a significant proportion of patients commenced on NS care pathways who did not meet the study eligibility criteria, negatively affecting recruitment. Moreover, as this was intended to be a pragmatic trial within the setting of standard care therapies (all agents used within the trial were already licensed for treatment of NS or used within routine care for this indication), it was also felt important that the trial was not restrictive around organ dysfunction beyond the parameters of standard care.
On initial trial design, the fever and neutrophil thresholds had been aligned with the objective elements of the NICE definitions of NS with the rationale that it might be difficult to define the non-objective elements of the NICE definition in which a diagnosis of NS may be appropriate in patients without documented fever but with other ‘signs or symptoms consistent with clinically significant sepsis’. It was clear this excluded a significant number of patients commencing treatment for NS.
Adjustments to the eligibility criteria were therefore proposed focused on ensuring they were less stringent and more pragmatic, as summarised in Table 7. These adjustments would widen the pool of eligible patients and ensure the trial population more accurately reflected patients being commenced on NS pathways in standard clinical practice, providing a more realistic evaluation of the intervention in routine care.
| Original eligibility criteria (Version 2.0, 30 July 2015) | Adjusted eligibility criteria (Version 3.0, 14 July 2016) |
|---|---|
| Absolute neutrophil count < 0.5 × 109/l | Absolute neutrophil count ≤ 1.0 × 109/l with either
|
| Fever > 38°C | |
| Self-measurement at home or hospital assessment acceptable provided this is documented in the medical notes and within 24 hours of i.v. antibiotic administration | |
| Received i.v. piperacillin/tazobactam or meropenem for < 24 hours | Systemic antibiotic administered prior to randomisation is not a reason for exclusion Patients who have been started on additional antimicrobial drugs (e.g. gentamicin or teicoplanin) are eligible provided the physician in charge of their care is willing to stop this additional antimicrobial at the time of enrolment |
| Adequate hepatic (AST and/or ALT < 2.5 × ULN) function within 24 hours prior to randomisation | Adequate hepatic (AST and/or ALT < 5 × ULN) function within 24 hours prior to randomisation |
| Physician in charge of care willing to follow either the intervention or standard care protocol per randomisation at enrolment, including not treating with CSF | Highlighted that prophylactic use of CSF is not an exclusion criterion |
| Hypotension (systolic pressure < 90 mmHg) within 24 hours of randomisation | Hypotension (systolic pressure < 90 mmHg on greater than one measurement) within 24 hours of randomisation |
| Treatment with fluoroquinolone or penicillin antibiotics in preceding 14 days | No longer an exclusion criterion |
With the adjusted eligibility criteria, a further 15 patients at Site 1 and 14 patients at Site 2 would have been potentially suitable during this 6 month pilot phase. It was therefore expected the revised eligibility criteria would increase the number of eligible patients to 1–2 patients per site per month. A 6-month extension to the pilot phase was proposed to assess the impact of the adjusted eligibility criteria on recruitment.
Extended pilot phase I (December 2016 to February 2017)
A 3-month, four-site extension to the pilot study was delivered from 1 December 2016 to 28 February 2017, using the adjusted eligibility criteria, summarised in Table 8.
| Site | Date open | Number screened | Duration of recruitment (months) | Target recruitment | Total recruitment | Recruitment rate (patients/site/month) |
|---|---|---|---|---|---|---|
| 1 | 1 December 2016 | 10 | 3 | 5 | 8 | 2.7 |
| 2 | 3 January 2017 | 0 | 2 | 3.5 | 0 | 0 |
| 3 | 1 December 2016 | 3 | 3 | 5 | 3 | 1.0 |
| 4 | 1 February 2017 | 1 | 1 | 0 | 0 | 0 |
| Total | 13.5 | 11 | 0.9 | |||
The expected recruitment rate was 1.7 patients per site per month, with again a 50% reduction permitted for the first 3 months of site opening. With recruitment on hold from 21 July 2016 until 1 December 2016 there was significant loss of momentum at sites, which was difficult to recover and combined with local site capacity issues resulted in a very short window of potential recruitment at two sites. Consequently, both of these sites failed to recruit during this extension phase, despite having been active in screening and recruitment during the first phase of the pilot study. The other two sites achieved an average monthly recruitment between the two sites of 1.8 patients per month, similar to the predicted recruitment rate, with review of screening data suggesting a positive impact of the adjusted eligibility criteria on recruitment. It was, however, again noted that the number of NS admissions remained lower than historical data, with an average of six admissions per month.
Extended pilot phase II (April 2017 to November 2017)
Due to the longer than anticipated pilot phase, under-recruitment was now inevitable if the project timeline was not modified, even if the number of recruiting sites for the main trial was expanded significantly from 12 to 20. To maximise the potential for successful completion of the trial, with the research question as fully addressed as possible, the preferred option was to increase the number of recruiting sites to 20 and extend the project timeline by approximately 1 year. To mitigate risk, it was agreed first to further extend the pilot phase until November 2017, aiming to open an additional seven sites during this period. This would allow an assessment of the potential recruitment at a broader selection of sites. A revised recruitment target was set of one patient per month per site to account for the lower frequency of neutropenic admissions.
A further 8 month period of recruitment to the pilot phase of the trial occurred between April and November 2017. Six additional sites were opened during this phase and a summary of recruitment by site is shown in Table 9. Even with the revised recruitment target of one patient per site per month, only 50% of the expected recruitment was met, with 24 patients recruited compared with the predicted 49 (average monthly recruitment rate across all sites was 0.4 patients).
| Site | Start date | Target recruitment | Total recruitment | Recruitment rate (patients/site/month) |
|---|---|---|---|---|
| 1 | 17 February 2016 | 8 | 11 | 1.4 |
| 2 | 14 March 2016 | 8 | 1 | 0.1 |
| 3 | 4 May 2016 | 8 | 2 | 0.3 |
| 4 | 31 March 2016 | 8 | 6 | 0.8 |
| 5 | 22 June 2017 | 4 | 1 | 0.1 |
| 6 | 17 July 2017 | 4 | 0 | 0 |
| 7 | 26 June 2017 | 5 | 3 | 0.4 |
| 9 | 18 September 2017 | 2 | 0 | 0 |
| 10 | 28 September 2017 | 2 | 0 | 0 |
| 11 | 13 November 2017 | 0 | 0 | 0 |
| Total | 49 | 24 | 0.4 | |
Summary of pilot phase recruitment
By 30 November 2017, there had been 17 months of active recruitment to the study, with as planned 10 sites open to recruitment and an eleventh due to open shortly. Forty-two patients had been recruited compared with a revised target of 68 patients as summarised in Figure 1.
FIGURE 1.
Summary of recruitment activity at the end of the pilot phase (February 2016 to November 2017): total recruitment of 42 patients.
Adherence to the protocol-specified treatment
Adherence to the protocol-specified intervention was assessed by whether patients switched to co-amoxiclav and ciprofloxacin, 12–24 hours after starting i.v. meropenem or piperacillin tazobactam i.v. therapy for at least 5 days total treatment. Adherence to the standard care arm required receipt of at least 48 hours of i.v. piperacillin/tazobactam or meropenem. The pre-specified threshold for adherence of 75% in both arms was achieved with adherence 86% and 83% in the intervention and control arms, respectively.
Separation between trial arms
The final criteria for trial progression was evidence of adequate separation in terms of the timing of antibiotic switch of at least 24 hours between the trial arms. The time on i.v. antibiotics was calculated for each patient and then the mean calculated for each trial arm and the difference between the two arms assessed. The mean duration of i.v. antibiotics was 19 hours in the intervention arm and 48 hours in the control arm; hence, mean separation in terms of timing of antibiotic switch was acceptable at 29 hours.
Summary of embedded pilot study results
The internal pilot phase of the EASI-SWITCH trial demonstrated that while recruitment was challenging there were no other concerns relating to treatment failure, protocol adherence or safety concerns. Based on recruitment between April and November 2017, it was likely that on average each site would only be able to recruit one patient every 2 months. The pilot phase had demonstrated that recruitment to the original proposed sample size of 628 patients for the main trial was not going to be achievable. Recruitment had originally been scheduled to complete at the end of December 2018. With 10 sites open and recruiting on average one patient every 2 months, based on current recruitment activity total recruitment was estimated to complete at approximately 100 patients. This would result in a significantly underpowered study, with results unlikely to have any significant impact and the evidence gap for an early oral switch identified by NICE remaining unanswered. It was clear important assumptions underpinning the trial design would have to be urgently reviewed to decide about whether to progress beyond the pilot phase to main trial delivery.
Determining continued importance of the research question
In view of the recruitment difficulties and the longer than anticipated pilot phase, an updated understanding of how low-risk NS patients were currently being managed across the UK was critical to assess the continued importance of the research question and assess whether there remained equipoise between the trial arms.
Between January and June 2018, NS management policies were reviewed and a survey of practice was undertaken nationally. Local NS policies from throughout the UK were sought via an e-mail request distributed to acute oncology nurse specialists within United Kingdom Oncology Nursing Society (UKONS) and EASI-SWITCH team members. Policies were reviewed for compliance with NICE recommendations, with particular focus on those advocated as key priorities for implementation in the guideline and their approach to low-risk management.
It is likely that a wide range of factors, including awareness of guidance, personal treatment preferences and clinical experience, will influence individual compliance with local policies when delivering NS care. A complementary electronic survey aimed to more fully reflect clinicians’ daily clinical practice, attitudes and preferences when managing NS. The survey link was disseminated via the UKONS, Royal College of Pathologists’ electronic newsletter and the clinicians involved in the EASI-SWITCH trial with onward dissemination to colleagues encouraged. In the survey, respondents were encouraged to reflect on their individual routine practice, rather than what their local institution’s NS policy or national guidelines might recommend.
A review of standard practice, completed for NICE in 2012, suggested approximately a third of 51 English and Welsh centres surveyed included the option of empirical oral antibiotics for lower-risk adult patients, with the intention of immediate discharge or earlier discharge compared with i.v. inpatient antibiotics. 3 It was unknown if this remained the status quo in the years following the publication of NICE guidance, with no national data available regarding centres’ standard approach to low-risk NS patients.
An updated understanding of management of low-risk NS patients was essential to assess whether there remained equipoise between the trial arms. If clinical practice had already largely shifted towards the intervention arm of EASI-SWITCH, there may be less impetus to continue to dedicate patient time and resources towards the trial. An updated audit of paediatric NS policies in 2015 had shown a lack of adherence, however, to NICE guidance, with continued variability in diagnostic criteria and management. 52 The impact of the NICE recommendations on adult NS management was unknown and it was unclear whether the publication reduced variation in practice for adult cancer patients.
A total of 53 adult NS policies were returned and reviewed: six cancer network policies and 47 acute trust policies, with representation from all four UK nations. As the request for policies was often forwarded to colleagues within a healthcare trust, it was not possible to determine an overall response rate. Most policies (94%) provided advice for both oncology and haematology patients. All policies, which were dated, had been updated post 2012, with no policy obviously issued before the NICE guidance was published and 57% specifically referenced that guideline.
A total of 235 responses were received from the electronic survey of individual practice. After excluding those containing only demographic data or minimal responses, there were 217 evaluable surveys. Again, there was representation from all four nations, with 69% of respondents based in England. The majority (72%) worked in oncology (predominantly as oncology consultants, acute oncology specialist nurses and advanced nurse practitioners), 23% worked in haematology and the remaining 5% were consultants mainly based in EDs or acute or general medicine. Forty-three per cent worked in university hospitals or regional cancer centres, 52% worked in acute hospitals with SACT day case units on site, and the remaining 5% worked in smaller acute hospitals which did not deliver SACT.
Of major interest was understanding current practice regarding routine risk stratification and any specific low-risk protocols or practices. The policies varied in their adoption of the NICE recommendations focused on assessing patients’ risk of septic complications and in decision-making about when, or if, to switch to oral antibiotics and about hospital discharge.
When practice was reviewed in 2012 by the NICE GDG, approximately a third of adult policies included the option of empirical oral antibiotics for lower-risk patients, with the intention of immediate discharge or earlier discharge compared with i.v. inpatient antibiotics (NICE30). In contrast, approximately half of current polices (53%) encourage identification of low-risk patients, with potential discharge on oral antibiotics within 48 hours after hospital admission. Almost half the policies (45%) suggest stratification based on MASCC score. Risk stratification is often only performed by senior oncology/haematology doctors or acute oncology specialists, if available.
Only 40% of clinicians reported routinely risk-stratifying patients within 24 hours of presentation. The MASCC score was most commonly used (by 70%), with the remaining 30% of clinicians using a local institutional risk-scoring system, a Modified Early Warning Score (EWS; Subbe et al. 53) or the Clinical Index of Stable Febrile Neutropenia (CISNE; Carmona-Bayonas et al. 54).
A wide range of approaches to early oral antibiotics and outpatient management are described in policies and by clinicians. Only 9% of policies clearly describe the option of starting empirical oral antibiotics for low-risk patients from the outset and most of these admit patients for 24–48 hours before discharge. Similarly, only a small number of survey respondents (5%) suggested they would start upfront oral antibiotics rather than i.v. antibiotics initially for low-risk patients. A further three haematology consultants suggested this would be considered for patients with difficult i.v. access or who were in the terminal phase of illness. Most policies do not define a period of observation following the start of oral antibiotics, although 13% of policies specify at least a 24-hour admission to hospital. The most common ‘low-risk’ oral antibiotic regimen is co-amoxiclav (625 mg three times daily) and ciprofloxacin, (either 500 mg or 750 mg twice daily; 16 of 20 policies specifying a regimen) for 5 to 10 days in duration.
A number of the policies referred to the extensive additional clinical criteria that the American Society of Clinical Oncology (ASCO) suggest should exclude patients from outpatient management, including ensuring patients do not live alone, they have access to a telephone and transport and can return to hospital promptly. 7,29
NICE commented in 2012 that the criteria for considering either stopping or switching from i.v. to oral antibiotics greatly varied. This work showed continuing variation in ongoing inpatient management of uncomplicated NS. Sixty-eight per cent of policies provided advice about when it might be suitable to switch from i.v. to oral antibiotics, in addition to guidance about early oral antibiotics. The majority (57%) recommend considering a switch from i.v. to oral antibiotics at 48 hours. The European Society for Medical Oncology (ESMO) guideline offers further advice about a range of factors to consider at 48 hours for switching to oral antibiotics and duration of treatment, which have been incorporated into many policies. 28 This includes whether the patient is still febrile and the neutrophil count is > 0.5 × 109/l.
One hundred and sixty-three survey respondents provided some indication as to when they would routinely switch low-risk uncomplicated patients to oral antibiotics and discharge home. These questions were not answered by clinicians who treat with oral antibiotics from the outset. Thirty-seven per cent reported switching to oral antibiotics typically within 24 hours of admission, 44% within or at 48 hours and 20% after 48 hours of admission. Various standard times to discharge after switching patients to oral antibiotics were also reported, ranging from immediate discharge in 27% to after at least 24 hours of observation in 53%.
It was evident that clinicians generally do not recalculate MASCC scores after admission. They confirmed, however, that the most important criteria, apart from initial risk scores, for determining when to switch to oral antibiotics are duration of apyrexia (70%) and if symptoms are improving (62%). Over half the respondents consider the patient’s neutrophil count (56%) and availability of microbiology results (52%) as important. Fewer consider the duration of i.v. antibiotics the patient received (31%) and expected duration of neutropenia (12%).
Survey respondents highlighted common reasons preventing early discharge of low-risk patients before 48 hours of hospitalisation (see Table 10), most commonly a clinically well patient experiencing ongoing fever.
| Reason preventing early discharge | % (n) |
|---|---|
| Patients are clinically well but have ongoing fever | 47 (103) |
| Patients have other clinical issues keeping them in hospital, for example, other anticancer treatment toxicities | 45 (97) |
| Patients do not feel clinically improved | 33 (71) |
| Clinicians are not keen for discharge | 33 (71) |
| The centre does not have an agreed protocol for early discharge of low-risk patients | 30 (66) |
| The centre does not have an agreed pathway for follow-up of low-risk patients discharged early | 26 (56) |
| Patients’ initial microbiology results are not available, for example, blood cultures | 24 (52) |
| Patients or carers are not keen for discharge | 16 (35) |
It would therefore seem that despite the introduction of NICE guidance risk stratification is not routine practice for many NHS centres or clinicians. A small number of specialist teams are prescribing upfront oral antibiotics for low-risk patients, but this practice appears limited. There has been some increase in the number of policies which include the option of discharging low-risk patients on oral antibiotics before 48 hours but approximately half of policies still advise initial i.v. antibiotics and subsequent oral switch after 48 hours or later. Even more clinicians, approximately two-thirds, in their routine practice choose to switch from i.v. to oral antibiotics at 48 hours or later. It is therefore evident that for at least half of centres and clinicians surveyed routine practice is in accordance with the standard care arm in the EASI-SWITCH trial. There has not yet been widespread adoption of risk stratification tools or routine early oral antibiotics in low-risk patients. A number of factors appear to have slowed progress, including the limited evidence base highlighted in the NICE guidance and in some cases dissatisfaction with current risk stratification tools, which has resulted in centre, clinician and patient hesitance about early oral switch approaches.
The majority of policies encourage initial investigations and empirical antibiotic regimens that accurately reflect NICE recommendations and therefore the initial management approach assumed in the EASI-SWITCH trial. Eighty-five per cent of polices promote initial beta-lactam monotherapy, compared to only 36% in NICE’s 2012 review. There has been a general reduction in the routine use of empirical aminoglycosides and glycopeptides for patients with central lines: 13% of current policies include gentamicin as part of an empirical dual antibiotic regimen, compared with 63% in 2012. Four of the current policies suggest initial doses of gentamicin but that these only be continued if there are signs of severe sepsis or for haematology patients. Local organism antibiotic resistance patterns are likely to be a contributing factor for those policies where gentamicin is routinely administered to all patients and is permitted under NICE guidance. Only 4% of current policies suggest empiric use of glycopeptides if a central line is present but a line infection not suspected, compared to 15% in 2012. Clinical practice therefore supported the standard care i.v. antibiotics being utilised in the EASI-SWITCH trial.
Finally, comparing diagnostic thresholds to the NICE guidance, only 19% of policies and 24% of survey respondents define neutropenia as a neutrophil count of ≤ 0.5 × 109/l as suggested by NICE, with most employing a broader definition. While there has been a reduction in policies simply defining neutropenia as < 1.0 × 109/l (51% compared with approximately 70% of adult policies in 2012) this is offset by those that include patients whose neutrophil count is < 1 × 109/l and expected to fall. More policies now encourage starting empirical treatment in patients who have had a single temperature ≥ 38°C (78% compared with 50% in 2012), in keeping with NICE criteria. Similarly, a patient presenting with a single temperature of 38°C or above would meet the temperature criteria used by 96% of clinicians and just over a third (36%) of these clinicians would start empirical treatment if lower temperature thresholds were recorded (e.g. single/sustained temperature of ≥ 37.5°C/≥ 37.7°C). The importance of considering NS in patients who are generally unwell, irrespective of their temperature, was stressed in the majority of policies, including prompts for signs and symptoms such as rigors, altered mental state and haemodynamic instability. Overall the review of current policy and practice supported the proposed adjustments to the eligibility criteria to the trial.
Review of assumptions underpinning study design
The review of current NS practice confirmed the continued importance of the research question nationally and the potential impact the trial could have on routine NS management in the UK. A careful review of the assumptions underpinning the original trial design and sample size calculation was therefore undertaken to determine whether the delivery of a smaller study could still have a meaningful clinical impact.
On initial design, the trial was powered at 90% and with a one-sided 97.5% CI, to detect non-inferiority of the early antibiotic intervention within a margin of 10%, assuming a 15% treatment failure rate in the standard care arm. This would require 269 patients per group. Allowing for a 5% dropout rate and up to 10% crossover from control to intervention, 314 participants would be required for each group, resulting in an overall target sample size of 628 patients.
The assumed 15% treatment failure rate for patients receiving standard care was derived from the three studies thought to best reflect the proposed control arm in relation to the populations included and duration of i.v. antibiotic treatment administered. 36,55,56
The pilot results raised no concerns regarding treatment adherence, separation between the treatment arms or the observed treatment failure rates in each arm. It was therefore presumed that the assumptions relating to those sample size parameters remain valid but, in retrospect, the choice of non-inferiority margin and statistical analysis may have been too stringent for a pragmatic trial where treatments were being used within their licensed indications or established clinical practice and where the risk of treatment failure was unlikely to result in serious risk to patients (in particular critical care admission or death).
Review of the non-inferiority margin
Selecting the non-inferiority margin was challenging due to the limited evidence available to help guide this selection. The rationale for the originally selected 10% margin was based on:
-
A historical consensus guideline advocating the use of a 10% margin in trials of antibiotic treatment among patients with NS. 47
-
Advice from the trial’s PPI co-applicant and representatives that this was an acceptable trade-off, for the potential overall gain in quality of life expected within this margin of uncertainty for clinical effectiveness.
It is important to note the consensus recommendation for a 10% margin relates to the overall NS population, with no consideration for stratification by risk of septic complications. This approach may be over-simplistic given the significant differences in clinical outcomes in low- and high-risk patients. The EASI-SWITCH target population are patients at low risk of septic complications, selected through their MASCC score and additional study eligibility criteria. Treatment failure in this population, as highlighted in two Cochrane reviews, therefore typically results in persistence or recurrence of fever, leading to prolongation of admission or re-admission for further i.v. antibiotic treatment. There has been no association noted between mortality and oral antibiotic treatment in low-risk patients. 37,57 This contrasts sharply with patients classified at high risk of complications at presentation, where treatment failure is likely to have more significant consequences and potentially result in organ failure, critical care admission or even death.
It was therefore felt appropriate to explore the acceptability of a larger non-inferiority margin considering the low-risk patient population, as this could maintain an acceptable trade-off between the possibility and consequences of treatment failure versus the potential quality of life and economic benefits associated with early switch. Therefore, the opinions of PPI and clinician stakeholders was sought on whether a review of design including widening of the non-inferiority margin would remain acceptable for assessment of the primary outcome.
1. Patient and public opinion
Input was sought from a range of PPI representatives. A summary of the study design and progress, including the rationale and implications for reviewing the sample size and specifically the non-inferiority margin, was prepared and distributed via e-mail to the membership of two PPI forums (see Appendix 1): (1) the Northern Ireland Cancer Research Consumer Forum, which has a range of patient and carer members from across Northern Ireland who work in partnership with cancer researchers; and (2) the Independent Cancer Patient Voice (ICPV), a national independent patient advocate group.
Respondents were requested to reply via e-mail as to whether they supported the proposed change in non-inferiority margin or not, with additional comments also welcome. Consent was obtained from all respondents for their comments to be made publicly available.
Twenty-one survey replies from patient and public representatives were received, with the majority of these supportive of increasing the non-inferiority margin from 10% to 15% (n = 19). Additional comments from seven of these respondents along with comments from one uncertain respondent and one who was not supportive are detailed in Appendix 1. Respondents’ comments demonstrated a good understanding of the key issues. They highlighted that earlier discharge if feasible is an important quality-of-life issue for many patients. The concerns raised about patients having adequate support at home and being able to be re-admitted quickly again if required, as well as the small risk of an adverse clinical outcome, are extremely appropriate.
2. UK clinicians’ opinion
Opinion was also sought from clinicians nationally regarding the proposed widening of the non-inferiority margin. A short description of the trial, key study design considerations and feedback to date on the proposed revisions from PPI representatives was summarised for clinicians. Again, respondents were asked a simple yes/no question, with additional comments welcome (see Appendix 2). Trial co-investigators and site PIs disseminated the survey via e-mail throughout their clinical networks. Attendees at a Scottish Clinical Trials showcase meeting (January 2018) were also surveyed using a paper copy of the questionnaire.
Almost all clinicians who provided feedback were supportive of widening the non-inferiority margin from 10% to 15% and continuing the study. In total, 50 responses were received from 39 consultants, 9 clinical fellows/speciality trainees and 2 RNs, with representation from the four UK nations. All but one of the respondents were supportive of the revision. Anonymised clinician comments in support of widening the non-inferiority are detailed in Appendix 2, and typically highlighted the continued validity of the research question and the potential impact upon practice. Notably, clinicians within centres that consider early/upfront oral antibiotics were supportive of continuation of a smaller achievable study; although this would involve altering the current standard of care in these centres, this would be an acceptable approach for the proposed time period given the potential to generate results that could make a compelling argument for further acute oncology service development. The cost-effectiveness analysis to be undertaken within the study was also perceived to be important, with the potential to show a reduction in length of stay cited in support of study continuation.
Additionally, the EASI-SWITCH trial is part of the National Cancer Research Institute’s Colorectal Cancer Clinical Studies Group (CSG) portfolio; hence, the proposed change to study design was presented at the Adjuvant and Advanced Disease CSG Subgroup meeting in January 2018. Membership of this group includes UK-wide representation from oncology (nine consultant medical oncologists) and other disciplines, including a statistician and clinical trials unit director. This group was also unanimously supportive of the proposed revision to the non-inferiority margin.
3. Trial Oversight Committees’ opinion
The EASI-SWITCH DMEC and TSC (met 22 November 2017 and 1 December 2017, respectively) both supported this approach and recommended continuing the study with a revised non-inferiority margin. The TSC were influenced in particular by the continued importance of the research question to the NHS and the strong support from PPI respondents when making their recommendation.
Review of statistical analysis plan
The TSC independent statistician also recommended review of the selected CI. The original choice of a one-sided 97.5% CI was based on recommendations by regulatory agencies for trials of novel therapies that have not been licensed for a specific clinical setting. This recommendation is focused on the provision of evidence for decisions about the licensing of drugs. This requires a greater degree of certainty than decisions about a strategic progression within the current use and licensed indication of an established treatment, which is the case with EASI-SWITCH. International recommendations for NS clinical trials also emphasise that the use of 95% CIs is acceptable. 47 There is obviously no expectation that the intervention will have superior efficacy to standard care, which is why a one-sided CI has been selected from the outset. Therefore, a one-sided 95% CI was felt appropriate for the EASI-SWITCH primary analysis and was unlikely to be detrimental to the impact of the trial’s findings.
The revised sample size calculation with a 15% non-inferiority margin and one-sided 95% CI requires 230 patients with retention of 90% power, a 5% dropout rate and a 10% crossover rate from control to intervention.
Progression to main trial
In addition to this stakeholder support, the study sponsor and HTA programme director were also supportive of continuation with the proposed revisions in study design. Based on the recruitment during the pilot phase, the revised target recruitment rate was 0.5 patients per site per month. Accrual of the revised sample size of 230 based on this target would be achieved by increasing the number of trial sites (to a total of 20) and the duration of recruitment to allow for the halt during the pilot phase and its subsequent prolongation.
Chapter 4 Main trial progression
Screening and recruitment post pilot phase
The internal pilot phase of EASI-SWITCH highlighted lower than anticipated recruitment rates, even with the revision of eligibility criteria and associated positive impact on recruitment. Further revision to study design was deemed acceptable to the trial stakeholders with the aim of delivering a clinically meaningful study. On review of updated audit data from two study sites, it was clear there was a sustained downward trend in NS admissions, consequently a revised recruitment target of 0.5 patients per site per month was set with an accompanying increase in number of recruiting sites to 20.
Post pilot phase, the EASI-SWITCH trial continued recruitment for another 24 months. In total, 19 out of a planned 20 sites opened to recruitment, which were a combination of cancer centres and units. This included 14 sites in England, 3 in Northern Ireland, 1 in Scotland and 1 in Wales (see Appendix 3, Table 37). In April and May 2019, 4 sites requested closure due to inactivity, leaving 15 active sites at the time of trial closure.
Identification of interested and appropriate sites was challenging. A range of methods were employed to promote the trial including the NIHR clinical research networks, relevant professional groups and national meetings. NIHR regulations around TSC and DMEC independence precluded participation of some interested sites because of associations with TSC and DMEC members. Table 11 summarises reasons for site non-participation.
| Reason for site non-participation | N = |
|---|---|
| Research team resource capacity | 3 |
| Clinical team resource capacity | 5 |
| Standard care more aligned with the intervention arm of EASI-SWITCH | 3 |
| Standard care in conflict with CSF use | 4 |
In total, 827 patients were screened for trial entry. The main reasons for exclusion (in order of frequency) were patients not fulfilling the diagnostic criteria of NS, a diagnosis of acute leukaemia, deemed at high risk of complications using the MASCC score (score < 21), history of penicillin (or other IMP) allergy, the physician in charge was not willing to support entry into either trial arm or not treat with CSFs, signs of severe localising infection present or the patient had already received > 24 hours of i.v. antibiotics. In total, 129 participants were recruited out of the target sample size of 230. In November 2019, the TSC recommended trial closure due to under-recruitment. A summary of recruitment activity across the trial phases is provided in Table 12 and Figure 2.
| Phase | Pilot phase | Main trial | ||
|---|---|---|---|---|
| Dates | February to July 16 | December 16 to February 17 | April to November 17 | December 17 to November 19 |
| Months | 1–6 | 7–9 | 10–18 | 19–45 |
| Total number open sites | 4 | 4 | 10 | 10–19 (15 sites active November 19) |
| Cumulative number of patients screened | 70 | 97 | 154 | 813 |
| Cumulative recruitment | 7 | 18 | 42 | 129 |
FIGURE 2.
Cumulative actual recruitment vs. predicted recruitment for the EASI-SWITCH trial (April 2017 to November 2019 inclusive). Total recruitment of 129 patients (November 2019). With the adjusted recruitment rate of 0.5 patients per site per month from January 2018 predicted recruitment was 216 patients by November 2019, based on the number of open sites.

Identification of barriers to recruitment
In view of the lower than anticipated target population, there was a need to understand the barriers to recruitment experienced by sites as the trial progressed and identify potential solutions in order to maximise the likelihood of successful trial delivery. This was particularly important in recognition of the narrow window of opportunity for patients to be recruited, whereby screening and recruitment both had to occur within 24 hours of commencing i.v. antibiotics for NS.
To understand factors impacting recruitment at sites, two approaches were taken as the trial progressed post pilot phase. These were (1) semistructured interviews with key site personnel and (2) an investigator survey of barriers to recruitment.
Semistructured interview feedback from key site personnel
A series of semistructured interviews with site PIs and lead RNs were conducted (April 2018) to explore clinician’s experience of recruiting and delivering the trial at their site (see Appendix 4).
All clinicians who were invited to take part via e-mail agreed to participate with representation from all 12 sites open to recruitment, including 12 PIs and 11 RNs (one open site had no RN in post due to illness). A summary list of reported barriers to trial recruitment is presented in Table 13.
| Trial design | Investigator | Patient | |
|---|---|---|---|
| Site | Clinician | ||
|
|
|
|
The most common barrier consistently noted by all sites unsurprisingly was the lower than anticipated number of patients being admitted with NS and therefore reduced pool of potentially eligible patients. It was noted at several sites that some patients required repeated hospitalisation with NS and other SACT-related toxicities during treatment, further reducing the number of potentially eligible patients.
Logistical issues at the site were more frequently identified as barriers compared to individual clinician barriers. It was noted that this trial, as an acute oncology trial across multiple tumour types, differed from the majority of oncology/haematology trials in their research portfolio, where patients were identified and managed as outpatients by tumour site-specific research teams, meaning the daily requirement for screening and patient identification could be challenging. Recruitment was also more challenging if the RNs were solely responsible for screening with minimal engagement or support from front-line clinical staff. The unpredictable nature of NS admissions and the requirement to be available at short notice made it difficult to plan workload.
Clinical and research team members’ interest in the study and the perceived importance of the trial and the research question it is trying to address were cited as an important barrier. While enthusiasm was often noted to be high when the trial opened, long periods of actively screening but not recruiting impacted morale and general willingness to continue to actively screen, refer and recruit patients. Maintaining prioritisation and interest in the study was reported as challenging with short-term clinical staff, for example, rotating junior medical staff or changes in inpatient nursing staff. Research teams therefore needed to regularly promote the trial and train new staff. Specialist registrars or clinical fellows, if available at sites, were noted to be well placed to help with consent, confirm eligibility and institute the allocated management plan but needed responsive site training and delegation to be available given typically short-term rotations.
The experience and confidence of the team member first introducing the study to patients was also noted to be important. RNs in particular noted recruitment was easiest when experienced and enthusiastic senior clinicians, for example senior acute oncology nurse specialists, consultants or clinical fellows, promptly reviewed patients post admission and considered their suitability for the trial, introducing trial participation as a treatment option from the outset for patients. Several RNs highlighted their limited experience reviewing inpatients, including patients with NS.
Three of the 12 sites (predominantly PIs) also importantly noted a preference by some clinicians for the intervention arm compared with standard care. All of the these sites prior to trial opening had utilised MASCC scoring and had a specific low-risk policy in place. They had, however, been supportive of the trial being undertaken and agreeable to recruit patients. Undoubtedly, some of the low-risk NS admissions have been treated with an approach similar to the intervention arm, rather than being offered the EASI-SWITCH trial. One site described how ‘bed pressures’ had resulted in some patients being managed with what they perceived to be an intermediate approach between the intervention and standard care trial arms. Conversely, some RNs indicated that certain clinicians were less supportive of certain groups of patients participating, whom they viewed as ‘higher risk’. This included certain tumour types, for example haematology and sarcoma patients or patients presenting with severe neutropenia (ANC < 0.1 × 109/l), despite there being no clear evidence to support this approach.
While barriers related to trial design were infrequently cited, these related to the inability to recruit penicillin-allergic patients and the sponsor requirement for confirmation of eligibility and prescription of protocolised therapy by a medical professional. Otherwise, it appeared that the previous revisions to the eligibility criteria had facilitated recruitment and the trial population was now reflective of the low-risk NS population seen in routine practice. While patient follow-up was viewed favourably, it was noted that the 28-day follow-up could be difficult to achieve if patients had received a further cycle of SACT within this period, particularly if toxicities related to this were experienced and/or re-admission with a new episode of NS or other SACT toxicity occurred, as this tended to impact patients’ recall of the NS episode related to the preceding cycle of treatment. Reporting of AEs beyond the day 14 primary outcome measure was felt to be not clinically relevant to the episode of NS in view of this and, along with capturing concomitant medications which could be multiple in this patient population, was a potential burden.
Site staff felt that the trial was well received by the majority of patients but, as anticipated, the short time to consider participation and consult with family or friends was the most common barrier for patients. Otherwise, patients’ previous experiences with SACT toxicity (and unscheduled care admissions arising from this) and symptom burden were also relevant – previous experiences, other toxicities, lack of improvement in symptoms from initial admission and difficulty coping with existing burden of information and uncertainty from an individual patient’s cancer care plan even prior to considering trial entry were all considered barriers to recruitment.
Overall, the interviews provided a clearer, in-depth understanding of the perspectives of key site personnel trying to recruit patients to the EASI-SWITCH trial. With equal representation from all sites, selection and non-respondent bias was avoided. It was evident a range of barriers to recruitment were present at several levels at trial design, site, individual clinician and patient levels, some of which could potentially be addressed.
Investigator survey of barriers to recruitment
As accrual to the trial continued to be challenging a further assessment of the key barriers to recruitment was undertaken in March 2019, through an electronic anonymous survey of personnel involved in trial recruitment. It aimed to assess whether the barriers identified from the interviews (April 2018) remained the most important and relevant to those currently trying to deliver the study, almost 1 year later. An online survey of recruitment experience was deemed the best strategy in order to obtain feedback from a wider range of personnel involved in the trial’s delivery and potentially identify additional factors through use of anonymised responses.
Forty complete responses were obtained with representation from all active sites except one (i.e. 15 of 16 open sites). Forty-eight per cent were RNs, 18% were site PIs and 33% were subinvestigators; 90% had been involved in the trial since it opened and 83% had been actively involved in recruiting participants.
Table 14 details the factors identified as barriers to recruitment by 30% or more of respondents. They are listed in order of their weighted score, providing an indication as to which barriers were felt most significant. The percentage of respondents who considered it a barrier at all (from somewhat to highly significant) is also provided.
| Ranked in order of weighted score | Identified barrier | Per cent of responses identifying this barrier (n) |
|---|---|---|
| 1 | Number of NS admissions to sites | 75 (30) |
| 2 | Potential participants are unscheduled admissions | 68 (27) |
| 3 | Eligibility criteria for patients to participate | 73 (29) |
| 4 | Having to consent and randomise within 24 hours of i.v. antibiotics commencing | 63 (25) |
| 5 | The acceptability of the trial to patients | 58 (23) |
| 6 | Identification of suitable patients | 58 (23) |
| 7 | Other clinicians prefer standard care | 50 (20) |
| 8 | Expected recruitment rate for your site | 43 (17) |
| 9 | Patients prefer standard care | 43 (17) |
| 10 | Other clinicians prefer the intervention | 38 (15) |
| 11 | Other patients prefer the intervention | 38 (15) |
| 12 | The acceptability of the trial to other clinicians | 45 (18) |
| 13 | Engagement and enthusiasm of other potential recruiters at your site | 43 (17) |
| 14 | Locally available resources and infrastructure to support the trial | 40 (16) |
| 15 | The clinician being surveyed prefer the intervention | 30 (12) |
| 16 | Administration burden and workload involved in recruiting a patient | 30 (12) |
The most commonly reported barriers of reduced NS admissions, the unscheduled patient population and short window for recruitment, were the same as those that had been identified in the semistructured interviews:
I think that we have a motivated team who are keen to recruit but we have very few numbers actually coming into our oncology assessment unit and ED with neutropenic sepsis (far smaller numbers than I had realised).
Difficulty in recognising potential participants in the time frame.
A small number have been missed due to lack of availability of research nurses within the time frame.
The trial’s eligibility criteria were also highly ranked as a barrier, more prominently than had emerged in the interviews. Apart from inability to recruit penicillin-allergic patients, this probably refers to CSFs being prohibited once on study. There is expert national and international consensus that this is not an indicated intervention in low-risk patients but nevertheless remains common practice. While survey respondents continued to express their enthusiasm and commitment to the trial it is interesting that the lack of engagement and enthusiasm of other colleagues was felt to be more significant at this stage of the trial than previously had been reported through the interviews. Table 15 summarises the results reported for all of the listed barriers categorised according to the ORCA key recruitment domains.
In general, the electronic survey further supported the multiple barriers that had emerged from the semistructured interviews, confirming that they were ongoing issues, which continued to impact recruitment. No new barriers were identified that had not previously been present. The variation in equipoise between the trial arms experienced by different clinicians and patients was, however, more prominently described in the survey than in the interviews. While the interviews had alluded to preferences for the intervention or standard care arms, it was clearer from the survey that there could in fact be significant preferences from investigators, clinical colleagues and patients for one arm over the other, impacting recruitment. Thirty per cent of survey respondents were inclined to prefer the intervention (early antibiotic switch), whereas clinical colleagues could prefer either standard care (cited by 49% respondents) or the intervention arm (cited by 38% respondents), hindering recruitment. Similarly, some patients’ strong preference for standard care (cited by 43% respondents) and others for the early oral intervention (cited by 38% respondents) had the potential to prevent trial recruitment:
The only barrier has been eligible patients declining entry to the study as their preference was inpatient antibiotic treatment.
Sometimes patients have turned down the study because they did not want a 50:50 chance of having to stay in on i.v. for 48 hrs if low risk.
The electronic survey was a quicker and easier strategy to obtain feedback about recruitment from a wider range of responders with different roles across different sites with variable recruitment performances. The ORCA recruitment domains provided a framework to guide the survey’s questions and ensure key barriers were not missed, although not all listed factors were overly applicable, and some benefitted from tailoring to the individual EASI-SWITCH setting. The survey importantly avoided interviewer bias and allowed respondents to express their views freely and anonymously. As a result of this a greater understanding of the variation in preference for one trial arm across the study was obtained. Despite a good response rate, potential limitations of the survey were obviously respondent non-response, selective responder bias and misinterpretation of questions.
Additional strategies to enhance recruitment
As the trial progressed and recruitment failed to meet expectations, a pragmatic shift in recruitment strategies occurred, with additional approaches added to try and address the modifiable barriers identified. As previously discussed, revision of eligibility criteria was made during and after the pilot phase to ensure the study was pragmatic and reflected the patient population routinely treated for NS. Further minor adjustments were made based on investigator interview feedback in May 2018, including: (1) recruitment of patients given additional systemic antibiotics at the time of presentation (such as a single dose of gentamicin) provided it was clinically appropriate to discontinue these at the time of enrolment, to include patients presenting to non-specialist units; (2) addition of suspension oral antibiotics to protocolised antibiotic therapy for patients unable to swallow tablets; (3) clarification of use of short-acting CSF to include patients who had a course of short-acting prophylactic CSFs prescribed as part of their SACT regimen but continuing to exclude those commenced on CSF as treatment for the presenting episode of NS.
In response to feedback about clinical workload, AE reporting was simplified, reducing the reporting period from 28 days to 14 days, with reporting of AEs beyond the 14-day primary outcome follow-up felt unlikely to add anything additional, in terms of understanding AEs related to the early switch antibiotic intervention. Shortening the reporting period was felt to benefit research teams, as it was not uncommon for patients to be re-admitted to hospital again within the current 28-day reporting period, once they had received a further cycle of chemotherapy. These hospital admissions were unrelated to the patient’s EASI-SWITCH episode and treatment but required research teams to report them as a serious AE within 24 hours of becoming aware. By shortening the reporting period to just the primary follow-up period of interest, it was hoped it would allow teams to prioritise time for recruiting new patients by reducing the intensity of patient follow-up required.
In the electronic survey distributed in March 2019, investigators were also asked to share any strategies they had found useful at their site in facilitating recruitment, so they could be shared with other sites. All comments related to optimising awareness of the trial, with clinical staff first assessing and managing new admissions and trying to create a suitable pool of staff who could provide consistent screening of patients and perform research-related tasks.
Main trial progress
While revisions to the eligibility criteria and trial design appeared to have a positive impact on recruitment initially, it is apparent from the recruitment graph in Figure 2 that, by the end of 2018, recruitment was consistently falling behind target and appeared to be plateauing thereafter, suggesting achieving even a smaller study in a timely manner would be challenging. Consequently, the DMEC recommended study closure on grounds of under-recruitment in November 2019. Despite clinician and researcher support for the study, persistent challenges to recruitment in relation to the numbers of potential patients and the challenges of identifying and enrolling patients within a narrow time window following admission remained. It is also possible that as time progressed, the variation in equipoise between trial arms demonstrated in the investigators’ survey (March 2019) in comparison to the site team interviews (April 2018) became more prominent and was potentially accompanied by changes in clinicians’ treatment approaches.
Chapter 5 Clinical effectiveness of early oral switch
This chapter contains some text reproduced from ‘Early switch to oral antibiotic therapy in patients with low risk neutropenic sepsis (EASI-SWITCH): a randomized non-inferiority trial’ published in Clinical Microbiology and Infection (2023). https://doi:10.1016/j.cmi.2023.07.021. 58
Recruitment
One hundred and twenty-nine patients were enrolled in EASI-SWITCH when trial closure was recommended by the DMEC in November 2019. In total, 827 patients were screened for study entry; the most common reasons for screen failure are described in the previous chapter and listed in Appendix 5. Recruitment numbers by site are summarised in Table 16 with a list of sites provided in Appendix 3, Table 37.
| Site | Total number of patients recruited (%) |
|---|---|
| s01 | 50 (38.8) |
| s02 | 4 (3.1) |
| s03 | 15 (11.6) |
| s04 | 13 (10.1) |
| s05 | 2 (1.6) |
| s06 | 5 (3.9) |
| s07 | 5 (3.9) |
| s09 | 9 (7.0) |
| s10 | 1 (0.8) |
| s11 | 7 (5.4) |
| s12 | 7 (5.4) |
| s13 | 3 (2.3) |
| s15 | 1 (0.8) |
| s16 | 4 (3.1) |
| s17 | 2 (1.6) |
| s19 | 1 (0.8) |
Consolidated Standards of Reporting Trials flow diagram
In total, 129 patients were randomised from 19 sites. Sixty-five patients were randomised to the early switch intervention and 64 to the standard care control arm (see Figure 3, CONSORT diagram). Of these, 125 patients were included in the primary ITT analysis (comprising 61 patients allocated to intervention and the 64 patients allocated to control). Four patients were excluded (all allocated to intervention) for the following reasons: one patient was randomised in error, one withdrew from continued participation and two patients were lost to follow-up. Twelve patients included in the primary ITT analysis were excluded from the PP analysis; consequently, the primary PP population comprised 53 patients in the intervention group and 60 in the standard care group. The reasons for exclusion of the eight patients in the intervention group were: premature discontinuation of antibiotic treatment (n = 4), interruption to antibiotic treatment involving at least two consecutive missed doses (n = 2) and insufficient initial i.v. antibiotic treatment (n = 1). All four excluded patients in the standard care group had received < 48 hours of i.v. antibiotic treatment.
FIGURE 3.
CONSORT diagram.

Baseline characteristics
Table 17 describes the patient, tumour and treatment characteristics in the intervention and control groups. As anticipated, only a minority of participants had a haematological malignancy (6.2%) in comparison with solid tumours (93.8%), of which the most common cancer type was breast cancer (54.6%). The majority of patients were receiving treatment in the neoadjuvant or adjuvant setting (60.4%) rather than palliative setting (27.9%). In general, baseline characteristics were well balanced across the intervention and control groups in relation to cancer type, anticancer treatment and symptoms and signs of infection (including fever, absolute neutrophil count and MASCC score). Use of prophylactic Granulocyte colony stimulating factor (GCSF) support was noted to be higher in the standard care arm (34.4%) in comparison to the intervention arm (15.6%).
| Baseline characteristics | Treatment group | ||
|---|---|---|---|
| Standard care (n = 64) | Intervention (early switch) (n = 65) | Total (n = 129) | |
| Age (years) | 56.2 (15.1) | 56.6 (13.4) | 56.4 (14.2) |
| Gender: | |||
| Male | 21 (32.8%) | 20 (30.8%) | 41 (31.8%) |
| Female | 43 (67.2%) | 45 (69.2%) | 88 (68.2%) |
| Malignancy: | |||
| Solid tumour | 59 (92.2%) | 62 (95.4%) | 121 (93.8%) |
| Haematological | 5 (7.8%) | 3 (4.6%) | 8 (6.2%) |
| Solid tumour type: | n = 59 | n = 62 | n = 121 |
| Breast | 30 (50.9%) | 36 (58.1%) | 66 (54.6%) |
| Lung | 4 (6.8%) | 3 (4.8%) | 7 (5.8%) |
| Gastrointestinal/hepatobiliary | 4 (6.8%) | 5 (8.1%) | 9 (7.4%) |
| Germ cell | 3 (5.1%) | 0 (0.0%) | 3 (2.5%) |
| Genitourinary | 5 (8.5%) | 5 (8.1%) | 10 (8.3%) |
| Head and neck | 2 (3.4%) | 0 (0.0%) | 2 (1.7%) |
| Gynae | 4 (6.8%) | 3 (4.8%) | 7 (5.8%) |
| Sarcoma | 4 (6.8%) | 4 (6.5%) | 8 (6.6%) |
| Other | 3 (5.1%) | 6 (9.7%) | 9 (7.4%) |
| Haematological malignancy type: | n = 5 | n = 3 | n = 8 |
| Hodgkin lymphoma | 4 (80.0%) | 2 (66.6%) | 6 (75%) |
| Other | 1 (20.0%) | 1 (33.3%) | 2 (25.0%) |
| Cancer treatment intention | |||
| Radical | 9 (14.1%) | 3 (4.6%) | 12 (9.3%) |
| Palliative | 16 (25.0%) | 20 (30.8%) | 36 (27.9%) |
| Adjuvant/neoadjuvant | 38 (59.4%) | 30 (61.5%) | 78 (60.4%) |
| Other | 1 (1.6%) | 2 (3.1%) | 3 (2.3%) |
| Cancer treatment line | n = 64 | n = 128 | |
| 1st line | 54 (84.4%) | 55 (85.9%) | 109 (85.2%) |
| 2nd line | 7 (10.9%) | 7 (10.9%) | 14 (10.9%) |
| 3rd line and beyond | 3 (4.7%) | 2 (3.1%) | 5 (3.9%) |
| Maximum temperature | 38.2 (0.6) | 38.2 (0.8) | 38.2 (0.7) |
| Absolute neutrophil count | 0.3 (0.3) | 0.3 (0.3) | 0.3 (0.3) |
| Symptoms of mild local infection | |||
| Cough | 15 (23.4%) | 21 (32.3%) | 36 (27.9%) |
| Sore mouth/throat | 29 (45.3%) | 22 (33.8%) | 55 (39.5%) |
| Dysuria | 3 (4.7%) | 5 (7.7%) | 8 (6.2%) |
| Nausea/vomiting | 11 (17.2%) | 10 (15.4%) | 21 (16.3%) |
| Diarrhoea | 10 (15.6%) | 9 (13.9%) | 19 (14.7%) |
| Other symptoms | 32 (50.0%) | 29 (44.6%) | 61 (47.3%) |
| MASCC score | 24.0 (1.8) | 24.3 (1.8) | 24.2 (1.8) |
| Prophylactic GCSF administered | |||
| Yes | 22 (34.4%) | 10 (15.6%) | 32 (25.0%) |
| No | 42 (65.6%) | 54 (84.4%) | 96 (75.0%) |
| C-reactive protein (mg/l) | 48.6 (49.2) | 50.3 (52.7) | 49.5 (50.8) |
| Blood cultures: | |||
| Positive | 20 (10.9%) | 16 (7.6%) | 36 (9.1%) |
| Negative | 163 (89.1%) | 196 (92.5%) | 359 (90.9%) |
Treatment after trial entry
All patients (n = 64) randomised to the standard care arm commenced allocated study treatment; however, four patients did not complete allocated treatment, receiving < 48 hours of i.v. antibiotic therapy. Two patients randomised to the early switch intervention (n = 65) did not proceed to allocated study treatment due to ineligibility (n = 1) and withdrawal of consent (n = 1). Therefore, 63 patients in the intervention arm commenced allocated treatment; however, protocolised treatment was not completed in eight patients for a number of reasons. These included premature switch to oral antibiotics (after < 12 hours of i.v. antibiotic therapy; n = 1); late switch to oral antibiotics (after 24 hours of i.v. therapy; n = 2); completion of < 5 days antibiotic treatment in total (n = 1) and a combination of late oral switch (after 12 hours) and short duration (< 5 days) of treatment in total (n = 2).
The mean duration of i.v. antibiotic therapy (and SD) was 54.8 hours (24.2) and 22.6 hours (26.9) in the standard care and early switch intervention groups, respectively.
No patients in the standard care arm were lost to follow-up for the day 14 primary outcome measure. Three patients is the intervention arm were lost to follow-up at day 14; however, it was still possible to determine the primary outcome for one of these patients, who had a treatment failure before the day 14 time point.
In total, of 65 patients allocated to intervention, 61 were included in the primary outcome ITT analysis and 53 in the PP analysis. Of the 64 patients allocated to standard care, all were included in the ITT analysis and 60 included in the PP analysis.
Protocol deviations
In total, 93 protocol deviations were reported. The most common reason for these was assessments taking place outside of schedule (45.2%). These typically related to the timing of study questionnaires at baseline, day 14 and day 28 of follow-up. This might be anticipated, particularly for the follow-up questionnaires, given this patient population are likely to have multiple hospital attendances related to continued SACT or management of complications arising from treatment or their underlying disease. Protocol deviations by number and type across treatment groups are listed in Table 18.
| Category | Treatment group | Total | |
|---|---|---|---|
| Standard care | Intervention (early switch) | ||
| Eligibility (%) | 1 (2.3) | 3 (6.1) | 4 (4.3) |
| IMP issues (%) | 3 (6.8) | 8 (16.3) | 11 (11.8) |
| Assessment outside schedule (%) | 19 (43.2) | 23 (46.9) | 42 (45.2) |
| Randomisation (%) | 1 (2.3) | 0 (0) | 1 (1.1) |
| Opportunistic research blood sampling (%) | 5 (11.4) | 2 (4.1) | 7 (7.5) |
| Other (%) | 15 (34.1) | 13 (26.5) | 28 (30.1) |
| Total | 44 | 49 | 93 |
Treatment outcomes
Primary outcome
Both ITT and PP analyses were conducted for the primary outcome measure of treatment failure. It was pre-specified that equal weighting would be given to both analyses such that a definitive conclusion of non-inferiority required both analyses to concur. A one-sided 95% confidence limit placed about the difference in treatment failure rate was compared to the non-inferiority margin of 15%. Conclusion of non-inferiority required that the upper bound of the one-sided 95% CI placed about the difference in treatment failure rates (intervention minus control) should not exceed 15% (see Figure 4). The 95% one-sided confidence limit was derived from the upper bound of a two-sided 90% CI. The Pearson chi-square test was used to test significance of observed differences between trial arms. Table 19 presents the number (%) and the differences in proportions (95% CI) in relation to the primary outcome in the ITT and PP analyses.
FIGURE 4.
Forest plot of differences (90% CI) by analyses performed.
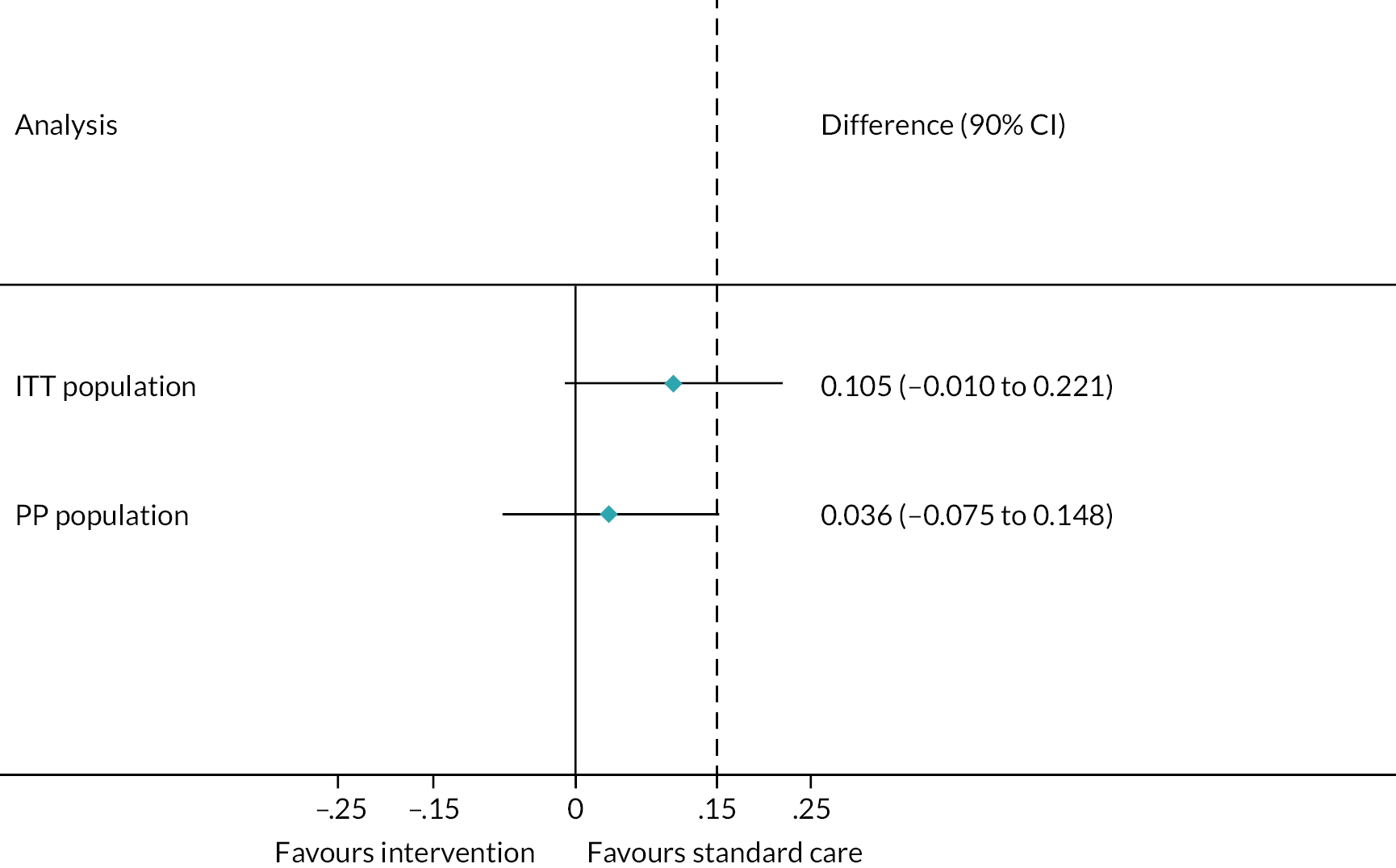
| Standard care (n = 64) | Intervention (n = 61) | Risk difference (90% CI) | p-value | |
|---|---|---|---|---|
| Treatment failure ITT (N = 125) | ||||
| Yes | 9 (14.1%) | 15 (24.6%) | 0.11 (−0.01 to 0.22) | 0.14 |
| No | 55 (85.9%) | 46 (75.4%) | ||
| Treatment failure PP (N = 113) | ||||
| Yes | 8 (13.3%) | 9 (17.0%) | 0.04 (−0.07 to 0.148) | 0.59 |
| No | 52 (86.7%) | 44 (83.0%) | ||
In the ITT analysis, 9 of the 64 patients (14.1%) in the standard care arm met the primary end point of treatment failure, compared with 15 of 61 (24.6%) in the early oral switch arm. Based on the pre-specified 15% non-inferiority margin, the risk difference of 10.5% (one-sided 95% CI −1% to 22%; p = 0.14) was such that the intervention was not found to be non-inferior to standard care in the ITT population.
The result of the PP analysis was not consistent with the ITT analysis. In the PP population, 8 of the 60 patients (13.3%) in the standard care arm met the primary end point of treatment failure, compared with 9 of 53 (17%) in the early oral switch arm. The risk difference of 3.7% (one-sided 95% CI −7% to 14.8%; p = 0.59) was such that the intervention was found to be non-inferior to standard care in the PP population.
Non-inferiority for the early switch intervention could not be concluded in the absence of concurrence of both the ITT and PP analyses and, even if concurrence had been demonstrated, findings would remain limited due to relative under-powering of the analyses. The main constituents of the composite primary outcome measure that accounted for patients reaching the treatment failure end point by day 14 were persistence/recurrence of fever and/or physician-directed escalation from the protocolised antibiotic regimen. None of the treatment failure events recorded in either arm were attributable to the need for critical care support or death before day 14. Tables 20 and 21 present the constituents of the primary outcome measure in the ITT and PP populations, respectively. The number of patients for whom each component of the composite outcome measure was available is expressed, by trial arm, for that measure and the p-value given is from significance testing of observed differences between trial arms using the Pearson chi-square test.
| Standard care (n = 64) | Intervention (n = 61) | Difference (95% CI) | p-value | |
|---|---|---|---|---|
| Treatment failure ITT | ||||
| Yes | 9 (14.1%) | 15 (24.6%) | 0.105 (−0.010 to 0.221) | 0.14 |
| No | 55 (85.9%) | 46 (75.4%) | ||
| Persistence/recurrence of fever (T ≥ 38°C) after 72 hours of i.v. antibiotic initiation | n = 62 | n = 60 | ||
| Yes | 4 (6.5%) | 10 (16.7%) | 0.102 (−0.010 to 0.215) | 0.08 |
| No | 58 (93.6%) | 50 (83.3%) | ||
| Physician-directed escalation from protocol-specified antibiotic treatment | ||||
| Yes | 6 (9.4%) | 10 (16.4%) | 0.070 (−0.047 to 0.187) | 0.24 |
| No | 58 (90.6%) | 51 (83.6%) | ||
| Day 14: CCU admission | n = 62 | |||
| Yes | 0 (0.0%) | 0 (0.0%) | ||
| No | 62 (100%) | 61 (100%) | ||
| Day 14: Re-admission to hospital | n = 62 | |||
| Yes | 2 (3.2%) | 2 (3.3%) | 0.001 (−0.062 to 0.063) | 0.99 |
| No | 60 (96.8%) | 59 (96.7%) | ||
| Day 14: Patient survival status | ||||
| Alive | 64 (100%) | 61 (100%) | ||
| Deceased | 0 (0.0%) | 0 (0.0%) | ||
| Standard care (n = 60) | Intervention (n = 53 | Difference (95% CI) | p-value | |
|---|---|---|---|---|
| Treatment failure | ||||
| Yes | 8 (13.3%) | 9 (17.0%) | 0.036 (−0.075 to 0.148) | 0.59 |
| No | 52 (86.7%) | 44 (83.0%) | ||
| Persistence/recurrence of fever (T ≥ 38°C) after 72 hours of i.v. antibiotic initiation | ||||
| Yes | 4 (6.8%) | 6 (11.5%) | 0.048 (−0.060 to 0.156) | 0.38 |
| No | 55 (93.2%) | 46 (88.5%) | ||
| Physician-directed escalation from protocol-specified antibiotic treatment | ||||
| Yes | 5 (8.4%) | 5 (9.4%) | 0.011 (−0.094 to 0.116) | 0.84 |
| No | 55 (91.7%) | 48 (90.6%) | ||
| Day 14: CCU admission | n = 58 | |||
| Yes | 0 (0.0%) | 0 (0.0%) | ||
| No | 58 (100%) | 53 (100%) | ||
| Day 14: Re-admission to hospital | n = 58 | |||
| Yes | 2 (3.5%) | 2 (3.8%) | 0.003 (−0.066 to 0.073) | 0.93 |
| No | 56 (96.6%) | 51 (96.2%) | ||
| Day 14: Patient survival status | ||||
| Alive | 60 (100%) | 53 (100%) | ||
| Deceased | 0 (0.0%) | 0 (0.0%) | ||
A post hoc exploratory analysis using generalised estimating equations (GEE) to account for possible clustering of observations within participating centres produced a risk difference (RD; 95% CI) of 0.106 (−0.032 to 0.244); p = 0.13, indicating no significant difference in risk of treatment failure when examining study site as a random effect.
Secondary outcomes
Among patients for whom secondary outcome data were available, there was a reduction in median duration of inpatient admission. Time to fever resolution, re-admission to hospital to day 28, survival to day 28, or changes to the originally intended SACT regimen appeared similar between the two arms (see Table 22).
| Standard Care (n = 64) | Intervention (n = 65) | p-value | |
|---|---|---|---|
| Day 14: time to fever resolution from initial i.v. antibiotic administration (hours)a | n = 36 | n = 37 | 0.52 |
| 25.6 (8.5, 46.0) | 18.5 (9.5, 39.6) | ||
| Day 28: Re-admission to hospital (related to infection or antibiotic treatment) | n = 64 | n = 61 | |
| Yes | 6 (9.4%) | 11 (18.0%) | 0.16 |
| No | 58 (90.6%) | 50 (82.0%) | |
| Day 28: Change in subsequent planned SACT | n = 61 | n = 59 | |
| Yes | 22 (36.1%) | 22 (37.3%) | 0.89 |
| No | 39 (63.9%) | 37 (62.7%) | |
| Day 28: Patient survival statusb | n = 62 | n = 61 | |
| Alive | 61 (98.4%) | 58 (95.1%) | 0.30 |
| Deceased | 1 (1.6%) | 3 (4.9%) | |
| Duration of inpatient admission (days)a | n = 58 | n = 52 | 0.002 |
| 3 (2,4) | 2 (2,3) |
Subgroup analyses
Subgroup analyses were pre-planned in the ITT population in the following subgroups: (1) tumour type (solid tumour vs. haematological malignancy); (2) neutrophil count at randomisation (≤ 0.5 × 109/l vs. > 0.5 × 109/l) and (3) maximum temperature on the day of presentation (< 38°C vs. > 38°C). A post hoc subgroup analysis based on blood culture results (positive or negative) was also performed. The first pre-specified analysis was not undertaken due to the small number of patients with haematological malignancy enrolled in the study. Risk differences and 99% CI from the treatment × subgroup interaction models are presented in Table 23 for the remaining subgroups for the primary outcome of TFR. The p-values presented are from a global test for interaction and indicate no significant interactions.
| Treatment group | ||||
|---|---|---|---|---|
| Treatment failure rate n (%) | Standard care | Intervention | RD (99% CI) | Interaction p-value |
| Neutrophil count at randomisation | ||||
| ≤ 0.5 × 109/l (n = 92) | 7 (14.6%) | 11 (25.0%) | 0.10 (−0.11 to 0.32) | 0.51 |
| > 0.5 × 109/l (n = 33) | 2 (12.5%) | 4 (23.5%) | 0.11 (−0.23 to 0.45) | |
| Maximum temperature on the day of presentation | ||||
| < 38°C (n = 41) | 3 (13.0%) | 4 (22.2%) | 0.09 (−0.22 to 0.40) | 0.50 |
| ≥ 38°C (n = 84) | 6 (14.6%) | 11 (25.6%) | 0.11 (−0.11 to 0.33) | |
| Positive blood culture | ||||
| No (n = 99) | 7 (13.7%) | 11 (22.9%) | 0.09 (−0.11 to 0.29) | 0.39 |
| Yes (n = 21) | 2 (20.0%) | 4 (36.4%) | 0.16 (−0.33 to 0.66) | |
Safety
Adverse events
In total, 106 AEs were observed from randomisation to the day 14 primary outcome measure end point. Of these, 46 occurred in the intervention arm. As might be anticipated, gastrointestinal disorders were the most commonly reported AEs, occurring in 33 patients (25.6%), of whom 12 patients were in the intervention arm.
There were 29 serious AEs, 12 of which occurred in the intervention arm. Of these, one was fatal but deemed unrelated to protocolised treatment. Eleven SAEs (8.5%) were due to blood and lymphatic system disorders, which may have reflected complications arising in patients who received a further cycle of SACT within the study reporting period.
Overall the AE profiles were similar between intervention and control groups. Table 24 summarises the total number of AEs, ARs, SAEs, serious adverse reactions (SARs) and SUSARs according to the number of events/patients by treatment group.
| Number of events | Number of patients | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Total | Standard care | Intervention | Total (n = 129) (%) | Standard care (n = 64) (%) | Intervention (n = 65) (%) | RR (95% CI) | p-value | ||
| AE/AR, SAE/SAR and SUSAR | Total number of AEs | 106 | 60 | 46 | 56 (43.4) | 30 (46.9) | 26 (40.0) | 0.85 (0.57 to 1.27) | 0.48 |
| Total number of ARs | 40 | 24 | 16 | 23 (17.8) | 14 (21.9) | 9 (13.9) | 0.63 (0.30 to 1.36) | 0.26 | |
| Total number of SAEs | 29 | 12 | 17 | 28 (21.7) | 12 (18.8) | 16 (24.6) | 1.31 (0.68 to 2.55) | 0.52 | |
| Total number of SARs | 1 | 1 | 0 | 1 (0.8) | 1 (1.6) | 0 (0.0) | |||
| Total number of events related to study drug and unexpected (SUSAR) | 0 | 0 | 0 | ||||||
| AEs | Blood and lymphatic system disorders | 12 | 6 | 6 | 12 (9.3) | 6 (9.4) | 6 (9.2) | 0.98 (0.34 to 2.89) | 1.00 |
| Gastrointestinal disorders | 48 | 31 | 17 | 33 (25.6) | 21 (32.8) | 12 (18.5) | 0.56 (0.30 to 1.05) | 0.07 | |
| General disorders and administration site conditions | 15 | 7 | 8 | 14 (10.9) | 6 (9.4) | 8 (12.3) | 1.31 (0.48 to 3.57) | 0.78 | |
| Infections and infestations | 6 | 3 | 3 | 6 (4.7) | 3 (4.7) | 3 (4.6) | 0.98 (0.21 to 4.70) | 1.00 | |
| Investigations | 8 | 5 | 3 | 4 (3.1) | 2 (3.1) | 2 (3.1) | 0.98 (0.14 to 6.78) | 1.00 | |
| Musculoskeletal and connective tissue disorders | 1 | 0 | 1 | 1 (0.8) | 0 (0.0) | 1 (1.5) | |||
| Nervous system disorders | 5 | 2 | 3 | 5 (3.9) | 2 (3.1) | 3 (4.6) | 1.48 (0.26 to 8.55) | 1.00 | |
| Psychiatric disorders | 1 | 0 | 1 | 1 (0.8) | 0 (0.0) | 1 (1.5) | |||
| Respiratory, thoracic and mediastinal disorders | 4 | 2 | 2 | 3 (2.3) | 1 (1.6) | 2 (3.1) | 1.97 (0.18 to 21.18) | 1.00 | |
| Skin and subcutaneous tissue disorders | 3 | 2 | 1 | 3 (2.3) | 2 (3.1) | 1 (1.5) | 0.49 (0.05 to 5.30) | 0.62 | |
| Vascular disorders | 3 | 2 | 1 | 3 (2.3) | 2 (3.1) | 1 (1.5) | 0.49 (0.05 to 5.30) | 0.62 | |
| SAEs | Blood and lymphatic system disorders | 11 | 6 | 5 | 11 (8.5) | 6 (9.4) | 5 (7.7) | 0.82 (0.26 to 2.55) | 0.76 |
| Gastrointestinal disorders | 2 | 1 | 1 | 2 (1.6) | 1 (1.6) | 1 (1.5) | 0.98 (0.06 to 15.4) | 1.00 | |
| General disorders and administration site conditions | 9 | 3 | 6 | 9 (7.0) | 3 (4.7) | 6 (9.2) | 1.97 (0.51 to 7.54) | 0.49 | |
| Infections and infestations | 3 | 2 | 1 | 3 (2.3) | 2 (3.1) | 1 (1.5) | 0.49 (0.05 to 5.30) | 0.62 | |
| Investigations | 1 | 0 | 1 | 1 (0.8) | 0 (0.0) | 1 (1.5) | |||
| Nervous system disorders | 1 | 0 | 1 | 1 (0.8) | 0 (0.0) | 1 (1.5) | |||
| Psychiatric disorders | 1 | 0 | 1 | 1 (0.8) | 0 (0.0) | 1 (1.5) | |||
| Respiratory, thoracic and mediastinal disorders | 1 | 0 | 1 | 1 (0.8) | 0 (0.0) | 1 (1.5) | |||
Chapter 6 Health economic and patient preference analyses
This chapter contains some text reproduced from ‘Early switch to oral antibiotic therapy in patients with low risk neutropenic sepsis (EASI-SWITCH): a randomized non-inferiority trial’ published in Clinical Microbiology and Infection (2023). https://doi:10.1016/j.cmi.2023.07.021. 58
Background
A within-trial economic evaluation was performed to assess the cost-effectiveness of early switch to oral antibiotics compared with usual care in the treatment of NS in patients with cancer. This included a cost-effectiveness analysis (CEA) consistent with the primary outcome measure to estimate the cost per treatment failure avoided at day 14 and a CUA to estimate the cost per QALY at day 14. The primary measure used in these analyses was the QALY, estimated from the EQ-5D-5L questionnaire administered at baseline and at day 14 of follow-up (either in person or using the validated telephone version). In recognition that the EQ-5D-5L only measures the potential effect on health of an early switch from i.v. to oral antibiotics and does not reflect patients’ preferences for non-health effects of the intervention, such as early discharge from hospital, a Patient Follow-up Questionnaire was used to collect additional information on this.
Methods
As the study treatments are for infection, not cancer, a short-term time horizon reduced the chance of costs and effects being contaminated by the impact of underlying disease or cancer therapy. A 14-day time horizon was adopted and the analysis was performed from a hospital perspective. Discounting of costs and outcomes was not necessary due to the time horizon being < 1 year. Patients’ use of hospital resources was collected over the study period on the case-report form using data from the day 14 interview and review of medical records, including treatments and medication received during the primary admission and any associated re-admissions. Costs were calculated by attaching appropriate unit costs from publicly available sources59–61 (e.g. Department of Health National Schedule of Reference Costs) and are listed in Appendix 6, Table 38. The final year of data collection was taken as the cost year (2018/2019).
For the CUA patients’ HRQoLs were measured at baseline and at day 14 of follow-up using the EQ-5D-5L (either in person or using the validated telephone version) and the 3L mapping function62 was used to convert responses into a single utility value, as currently recommended by NICE. 63 The area-under-the-curve method was used to estimate patient-specific QALYs accrued over the study duration. For the cost-effectiveness analysis, treatment failures were categorised as described in the previous chapter.
Since the EQ-5D-5L only measures the potential effect on health of an early switch from i.v. to oral antibiotics and does not reflect patients’ preferences for non-health effects of the intervention, such as early discharge from hospital, a Patient Follow-up Questionnaire was used to collect additional information on this.
Following database lock, individual patient data were evaluated to measure costs and QALYs related to early oral switch and standard care. Descriptive statistics were used to summarise the hospital resource use, associated costs and outcomes as means with 95% CIs for each group. Significance was judged where the CIs of differential means excluded zero or p < 0.05. The study was powered for the primary outcome of treatment failure but not for costs, QALYs or cost effectiveness.
The mean differences in costs, treatment failures and QALYs between groups were estimated and incremental cost-effectiveness ratios (ICERs) were calculated to estimate the cost per QALY gained (CUA) and the cost per treatment failure avoided (CEA) and the non-parametric bootstrapping was used to resample with replacement the cost and outcome data from the original sample to generate 1000 replicate ICERs. These were then plotted on the cost-effectiveness plane to display their joint distribution. Cost-effectiveness acceptability curves (CEAC) were constructed from the data by calculating the proportion of the ICER replicates which would be considered cost-effective at various thresholds of willingness-to-pay (WTP) for an additional QALY and to avoid a treatment failure. In general NICE consider interventions with an ICER of < £20,000/QALY to be cost-effective;64 however, no such threshold exists for treatment failures avoided.
The net monetary benefit (NMB) was also used to aid interpretation. The NMB is a summary statistic representing the value of an intervention in monetary terms when a WTP threshold for a unit of benefit is known. A positive NMB indicates the intervention is cost-effective. A range of threshold values were used for each analysis. All analysis was carried out using Stata 15/IC (StatCorp) for Windows®.
Sensitivity analyses
The following sensitivity analyses were performed:
-
In both the CUA and CEA costs and outcomes were adjusted for baseline age, gender and EQ-5D-5L score using multiple regression.
-
Where missing data levels were > 5% multiple imputation was used to predict missing values. As the levels of missing data in the CUA for QALYs were 13% (16% intervention, 9% control), QALY data points were filled using multiple imputation by chained equations and predictive mean matching to generate 20 imputed data sets. Treatment group, baseline EQ-5D-5L score, age and gender were entered into the model as predictors of missing data. Multiple imputation was not required for the CEA as missing data for treatment failures were under 2%.
-
PP analyses were carried out for both the CUA and CEA, including only patients who received treatment as detailed in the protocol and excluding those who did not.
-
An unplanned sensitivity analysis was conducted to exclude a patient in the CEA who had a prolonged ICU admission.
Non-health outcomes
A follow-up questionnaire was designed to determine what participants thought about the antibiotic treatment they received and also to find about their hypothetical preferences for future antibiotic treatment should they develop NS again (see Appendix 7). The questionnaire contained 14 questions, including questions to measure agreement towards specific statements (agree/disagree/uncertain), closed questions about treatment choice and home care, and one open-ended question to gather rationale for preferred treatment method. The number (%) of patients who agree or disagree with each statement in the questionnaire is presented and analysed.
Results
One hundred and twenty-six patients were eligible for the economic analysis; 62 in the intervention group and 64 in the standard care group. The results are presented in two parts: the CUA based on using the QALY as the outcome using complete data; and the cost effectiveness using treatment failures avoided, also only using complete data. Results using all available data are included in Appendix 8, Tables 39–41.
Cost–utility analysis
To maintain the correlation structure of the data the primary CUA analysis only included those participants with complete cost and QALY data, therefore if EQ-5D-5L data were not collected at baseline and day 14, or cost data were missing, they were excluded. These data were available for 110 patients (52 in the intervention group and 58 in the standard care group), with a higher proportion in the intervention arm having missing data (16% vs. 9%). Use of hospital services within 14 days of randomisation, including the primary admission and any subsequent re-admission(s), is presented in Table 25. The mean length of stay in participants’ primary admission was 0.52 days longer for standard care, as might have been anticipated from an early oral switch intervention. The mean length of stay during re-admission was less than half for standard care patients compared to those in the intervention arm. None of the differences between groups were statistically significant.
| Service | Intervention (n = 52) | Standard care (n = 58) | Mean difference (95% CI) | ||
|---|---|---|---|---|---|
| n (%) | Mean (95% CI) | n (%) | Mean (95% CI) | ||
| Primary admission | |||||
| Ward days | 52 (100.00) | 2.62 (1.99 to 3.24) | 58 (100.00) | 3.14 (2.71 to 3.57) | −0.52 (−1.26 to 0.22) |
| Emergency-department attendancea | 2 (3.85) | 0.04 (−0.02 to 0.09) | 2 (3.45) | 0.03 (−0.01 to 0.08) | 0.00 (−0.07 to 0.08) |
| Re-admission | |||||
| Ward days | 4 (7.69) | 0.46 (−0.13 to 1.06) | 2 (3.45) | 0.19 (−0.08 to 0.46) | 0.27 (−0.35 to 0.89) |
| Emergency-department visits | 4 (7.69) | 0.08 (0.00 to 0.15) | 2 (3.45) | 0.03 (−0.01 to 0.08) | 0.04 (−0.04 to 0.13) |
| Outpatient visits | 2 (3.85) | 0.04 (−0.02 to 0.09) | 2 (3.45) | 0.03 (−0.01 to 0.08) | 0.00 (−0.07 to 0.08) |
The corresponding costs of these services, along with drug costs, are presented in Table 26. The additional time spent as an inpatient led to a mean difference in cost of £250. The mean cost of study drugs was over £30 higher in the standard care arm, due to the lower unit cost of ciprofloxacin and co-amoxiclav, and the difference was statistically significant. The mean total cost was £1726 in the standard care arm compared to £1565 for intervention, a mean difference of £161, which was not statistically significant.
| Intervention (n = 52) | Standard care (n = 58) | Mean difference (95% CI) | |
|---|---|---|---|
| Mean cost £ (95% CI) | Mean cost £ (95% CI) | ||
| Primary admission | |||
| Ward days | 1250.81 (951.16 to 1550.46) | 1500.72 (1294.76 to 1706.67) | −249.91 (−603.17 to 103.35) |
| Emergency-department attendance | 7.54 (−3.06 to 18.13) | 6.76 (−2.73 to 16.24) | 0.78 (−13.24 to 14.80) |
| Re-admission | |||
| Ward days | 220.73 (−63.81 to 505.27) | 90.70 (−37.14 to 218.55) | 130.03 (−167.57 to 427.63) |
| Emergency-department visits | 15.08 (0.39 to 29.76) | 6.76 (−2.73 to 16.24) | 8.32 (−8.61 to 25.25) |
| Outpatient visits | 5.19 (−2.11 to 12.49) | 4.66 (−1.88 to 11.19) | 0.54 (−9.12 to 10.19) |
| Medication | |||
| Study drug | 52.18 (44.00 to 60.35) | 83.19 (68.68 to 97.70) | −31.01 (−48.00 to −14.02) |
| Concomitant medication | 13.34 (2.33 to 24.35) | 33.40 (2.02 to 64.77) | −20.06 (−54.44 to 14.33) |
| Total | 1564.86 (1171.52 to 1958.21) | 1726.18 (1453.16 to 1999.19) | −161.31 (−626.74 to 304.11) |
Mean HRQoL scores are reported in Table 27. There were no significant differences in baseline or follow-up scores in EQ-5D-5L or visual analogue scale. Both measures showed increases in both groups by similar amounts, with the base figure being higher in the intervention arm for each method. The incremental QALY gain was small and not statistically significant: 0.002 QALYs is equivalent to 0.73 days in full health per year, but given that the follow-up period was 14 days, any difference would be small.
| Variable | Intervention (n = 52) | Standard care (n = 58) | Difference (95% CI) |
|---|---|---|---|
| Mean (95% CI) | Mean (95% CI) | ||
| EQ-5D-5L utilities | |||
| Baseline | 0.78 (0.71 to 0.84) | 0.74 (0.68 to 0.80) | 0.03 (−0.05 to 0.12) |
| 14 days | 0.82 (0.77 to 0.87) | 0.77 (0.72 to 0.82) | 0.05 (−0.03 to 0.12) |
| EQ-5D VAS | |||
| Baseline | 65.94 (59.99 to 71.90) | 59.31 (53.97 to 64.65) | 6.63 (−1.25 to 14.52) |
| 14 days | 76.12 (71.13 to 81.10) | 69.98 (65.37 to 74.60) | 6.13 (−0.58 to 12.84) |
| QALYs | 1 (0.029 to 0.032) | 0.029 (0.027 to 0.031) | 0.002 (−0.001 to 0.004) |
Although the differences are small, in cases such as this where the intervention is cost-saving and leads to more positive outcomes the intervention is said to be the dominant strategy (see Table 28). The ICER is not calculated as it would be a negative and would not convey any meaning. 65 Figure 5 demonstrates the result of the bootstrapped ICERs and the majority of points fall in the south-east quadrant, indicating that in the majority of cases the intervention is associated with cost savings and QALY gains, albeit small and not statistically significant. The CEAC in Figure 6 shows at a WTP threshold of £20,000/QALY there is a 78% probability the intervention is cost-effective compared to standard care. The NMB was positive for all WTP thresholds (see Table 29), indicating the intervention was cost-effective compared to standard care, while the CEAC constructed (see Figure 7) from this method showed a marginal difference in cost-effectiveness probability (79%).
| Mean incremental total health service costs (£) (95% CI) | Mean incremental QALY gain (95% CI) | Incremental cost-effectiveness ratioa | |
|---|---|---|---|
| Primary analysis (nI = 52, nSC = 58) |
−161.31 (−630.59 to 307.96) | 0.002 (−0.001 to 0.004) | Dominant strategy |
| Sensitivity analysis – controlling baseline characteristics (nI = 52, nSC = 58) |
−125.45 (−612.33 to 361.44) | 0.001 (−0.000 to 0.002) | Dominant strategy |
| Sensitivity analysis – multiple imputation of QALY values (nI = 62, nSC = 64) |
18.42 (−520.09 to 556.93) | 0.001 (−0.001 to 0.004) | £11,437.45 |
| PP analysis (nI = 45, nC = 54) |
−319.70 (−795.55 to 156.16) | 0.002 (−0.001 to 0.004) | Dominant strategy |
FIGURE 5.
Cost-effectiveness plane for the primary cost–utility analysis showing 1000 bootstrapped replications of mean incremental costs and QALY gain and the willingness-to-pay threshold of £20,000/QALY.
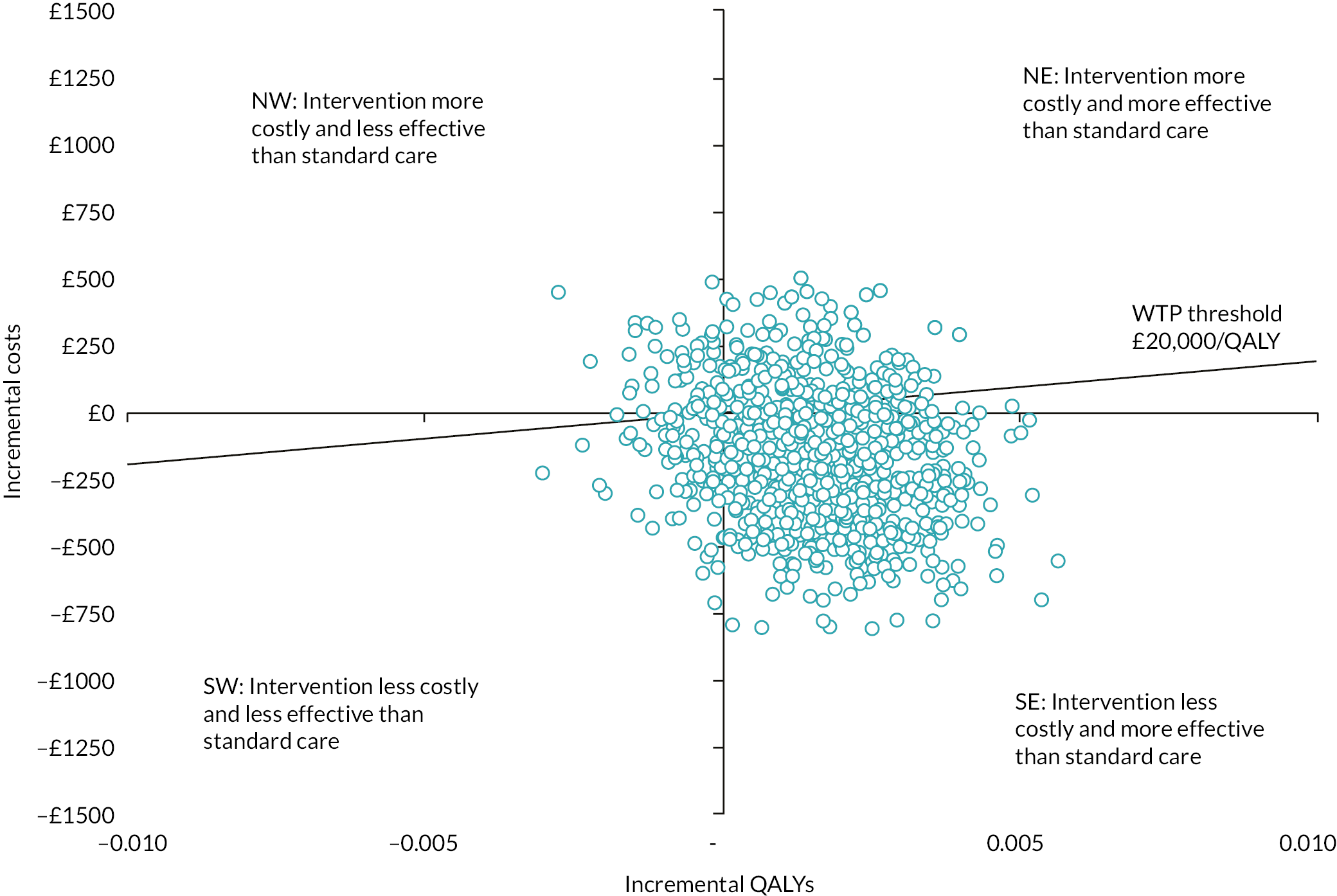
FIGURE 6.
Cost-effectiveness acceptability curves showing the probability of intervention being cost-effective compared to standard care for the primary and sensitivity analyses using QALYs as the outcome.

| Willingness to pay per additional QALY | Incremental net benefit (mean, 95% CI) |
|---|---|
| £0 | 161.31 (−304.11 to 626.74) |
| £1000 | 162.83 (−303.16 to 628.81) |
| £5000 | 168.87 (−299.51 to 637.25) |
| £10,000 | 176.43 (−295.24 to 648.11) |
| £20,000 | 191.55 (−287.71 to 670.82) |
| £50,000 | 236.91 (−272.52 to 746.34) |
FIGURE 7.
Cost-effectiveness acceptability curve showing the probability of intervention being cost-effective compared to standard care using the net monetary benefit method.

Sensitivity analyses (cost–utility analysis)
The results of the sensitivity analyses in the CUA are presented in Table 29. Adjusting for baseline characteristics (age, gender, baseline EQ-5D-5L score) reduced the size of the difference in costs, but the intervention arm still exhibited a lower mean cost and positive outcomes, and so remained dominant.
Using multiple imputation to estimate QALY scores for those not completing the EQ-5D-5L at day 14 resulted in the intervention arm’s mean cost being more than £18 higher than standard care and a slight increase in QALYs. This generated an ICER of £11,437/QALY, and a 54% probability of being cost-effective at a ceiling ratio of £20,000/QALY, much lower than the primary analysis, which can be partly attributed to the reversal in total costs. Those receiving the intervention who were missing these follow-up data were much heavier resource users than their counterparts and due to incomplete data were not included in the primary analysis.
The PP analysis showed the intervention to once again be the dominant option. The difference in QALYs remained small but the intervention was shown to be almost £320 less costly than standard care, with the revised CEAC estimating a 93% probability of the intervention being cost-effective (see Figure 6).
Cost-effectiveness analysis
Complete data were available for 124 patients (98%) for this analysis (100% standard care, 97% intervention), resulting in a slightly different population to the CUA. Table 30 shows the use of hospital resources. For those re-admitted to ward, the mean length of stay was more than double for intervention patients, 0.4 compared to 0.17 days, in part due to two of these patients both remaining hospitalised for 11 days. Notably in the intervention arm, one patient was transferred to critical care 5 days after starting antibiotics and was still there at the 14 day follow-up. This patient was excluded from the CUA as they did not complete the EQ-5D-5L at day 14.
| Service | Intervention (n = 60) | Standard care (n = 64) | Mean difference (95% CI) | ||
|---|---|---|---|---|---|
| n (%) | Mean (95% CI) | n (%) | Mean (95% CI) | ||
| Primary admission | |||||
| Ward days | 60 (100.00) | 2.78 (2.18 to 3.38) | 64 (100.00) | 3.17 (2.73 to 3.61) | −0.39 (−1.12 to 0.34) |
| Critical care days | 1 (1.67) | 0.15 (−0.15 to 0.45) | 0 (0) | – | 0.15 (−0.14 to 0.44) |
| Emergency department | 2 (3.33) | 0.03 (−0.01 to 0.08) | 2 (3.13) | 0.03 (−0.01 to 0.08) | 0.00 (−0.06 to 0.07) |
| Re-admission | |||||
| Ward days | 4 (6.67) | 0.40 (−0.11 to 0.91) | 2 (3.13) | 0.17 (−0.07 to 0.41) | 0.23 (−0.32 to 0.78) |
| Emergency-department visits | 4 (6.67) | 0.07 (0.00 to 0.13) | 2 (3.13) | 0.03 (−0.01 to 0.08) | 0.04 (−0.04 to 0.11) |
| Outpatient visits | 2 (3.13) | 0.03 (−0.01 to 0.08) | 2 (3.13) | 0.03 (−0.01 to 0.08) | 0.00 (−0.06 to 0.07) |
Costs of hospital resources and drugs are illustrated in Table 31. As was the case in the CUA, the difference in cost of study drug is statistically significant. Total costs were on average £22 higher in the intervention arm using these data – a difference that was not statistically significant.
| Intervention (n = 60) | Standard care (n = 64) | Mean difference (95% CI) | |
|---|---|---|---|
| Mean cost £ (95% CI) | Mean cost £ (95% CI) | ||
| Primary admission | |||
| Ward days | 1331.13 (1044.22 to 1618.03) | 1516.95 (1307.80 to 1726.10) | −185.82 (−534.10 to 162.46) |
| Critical care | 139.98 (−140.12 to 420.07) | 0 | 139.98 (−128.25 to 408.21) |
| Emergency department | 6.53 (−2.63 to 15.70) | 6.13 (−2.46 to 14.71) | 0.41 (−12.01 to 12.83) |
| Re-admission | |||
| Ward days | 191.30 (−54.95 to 437.55) | 82.20 (−33.52 to 197.91) | 109.10 (−154.78 to 372.98) |
| Emergency-department visits | 13.07 (0.33 to 25.80) | 6.13 (−2.46 to 14.71) | 6.94 (−8.09 to 21.97) |
| Outpatient visits | 4.50 (−1.81 to 10.81) | 4.22 (−1.70 to 10.13) | 0.28 (−8.27 to 8.84) |
| Medication | |||
| Study drug | 52.28 (44.83 to 59.73) | 80.81 (67.45 to 94.16) | −28.52 (−43.94 to −13.11) |
| Concomitant medication | 20.29 (3.54 to 37.05) | 40.26 (7.84 to 72.68) | −19.97 (−56.83 to 16.89) |
| Total | 1759.08 (1283.51 to 2234.65) | 1736.68 (1466.92 to 2006.44) | 22.40 (−510.16 to 554.96) |
In contrast to the CUA, these data have shown the cost to be marginally greater in the intervention arm, and the outcome to be negative due to higher treatment failure rate (see Table 32), although no differences were statistically significant. When this occurs the intervention is dominated by standard care. Figure 8 shows a large percentage of the bootstrapped ICERs in the north-west quadrant with higher costs and negative outcomes. As before, negative ICERs have not been calculated.
| Analysis | Mean incremental total health service costs (£) (95% CI) | Incremental treatment failures avoided (95% CI) | Incremental cost-effectiveness ratioa |
|---|---|---|---|
| Treatment failure avoided – primary analysis (nI = 60, nSC = 64) |
22.40 (−508.92 to 553.71) | −0.09 (−0.23 to 0.05) | Dominated by standard care |
| Sensitivity analysis – controlling baseline characteristics (nI = 60, nSC = 64) |
122.87 (−461.66 to 707.41) | −0.10 (−0.25 to 0.04) | Dominated by standard care |
| Sensitivity analysis – excluding patient who had prolonged ICU admission (nI = 59, nSC = 64) |
−131.57 (−579.67 to 316.53) | −0.08 (−0.22 to 0.06) | £1650.22 |
| PP analysis (nI = 52, nSC = 60) |
−149.44 (−694.28 to 395.39) | −0.02 (−0.15 to 0.11) | £7285.40 |
FIGURE 8.
Cost-effectiveness plane for the primary cost-effectiveness analysis showing 1000 bootstrapped replications of mean incremental costs and treatment failures avoided.
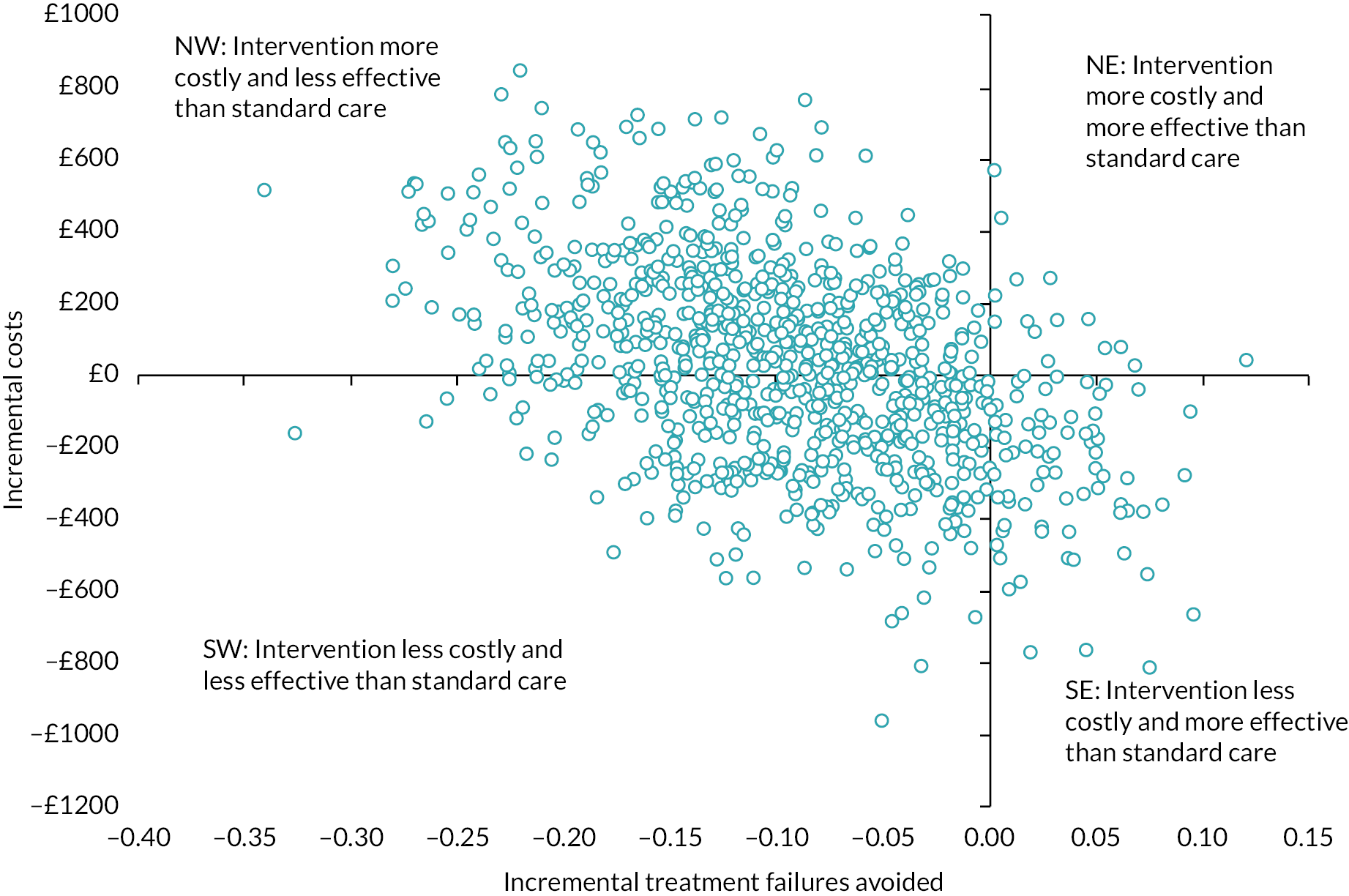
There is no generally accepted WTP threshold for treatment failure avoided but the CEAC (see Figure 9) shows the probability of being cost-effective at varying amounts: at £1000 it was 36%, decreasing to 13% at £10,000. The CEAC does not cut the y-axis as some of the joint density includes cost savings (SE and SW quadrants of Figure 8) and asymptotes to zero because the density does not involve health gains. 66
FIGURE 9.
Cost-effectiveness acceptability curves showing the probability of intervention being cost-effective compared to standard care for the primary and sensitivity analyses, using treatment failure rate as the outcome.
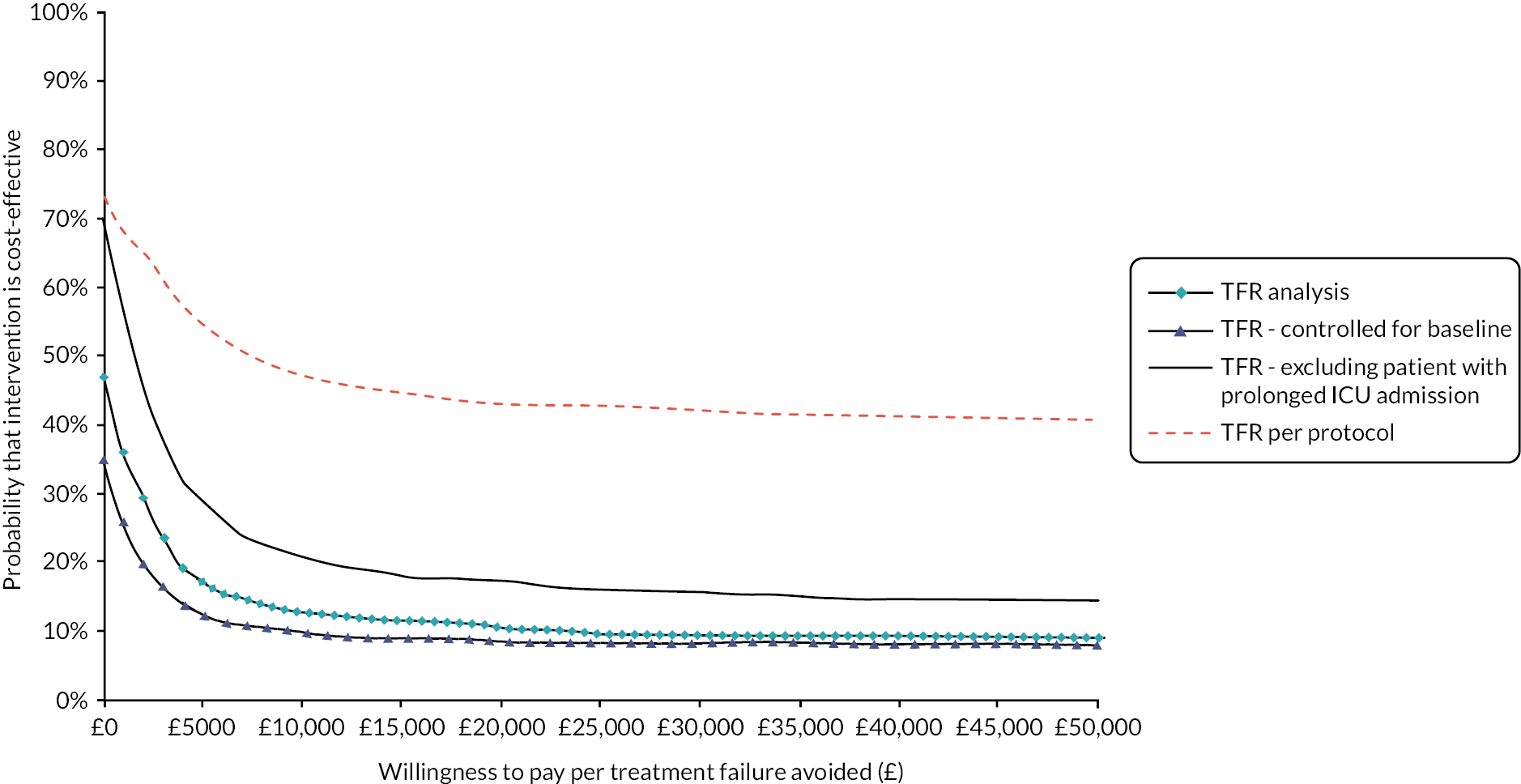
When using NMB at varying thresholds of WTP, the result was positive (meaning the intervention is cost-effective) for values up to £240/treatment failure avoided, and negative from £250 and above (see Table 33). The CEAC using NMB data (see Figure 10) showed a 39% probability of the intervention being cost-effective over standard care at a WTP threshold of £1000/treatment failure avoided.
| Willingness to pay for treatment failure avoided | Incremental net benefit (mean, 95% CI) |
|---|---|
| £0 | 22.40 (−510.16 to 554.96) |
| £200 | 3.86 (−515.83 to 523.55) |
| £250 | −0.78 (−517.43 to 515.88) |
| £1000 | −70.31 (−551.19 to 410.57) |
| £10,000 | −904.68 (−2122.83 to 313.46) |
| £20,000 | −1831.77 (−4384.27 to 720.73) |
FIGURE 10.
Cost-effectiveness acceptability curve showing the probability of intervention being cost-effective compared to standard care using the net monetary benefit method.
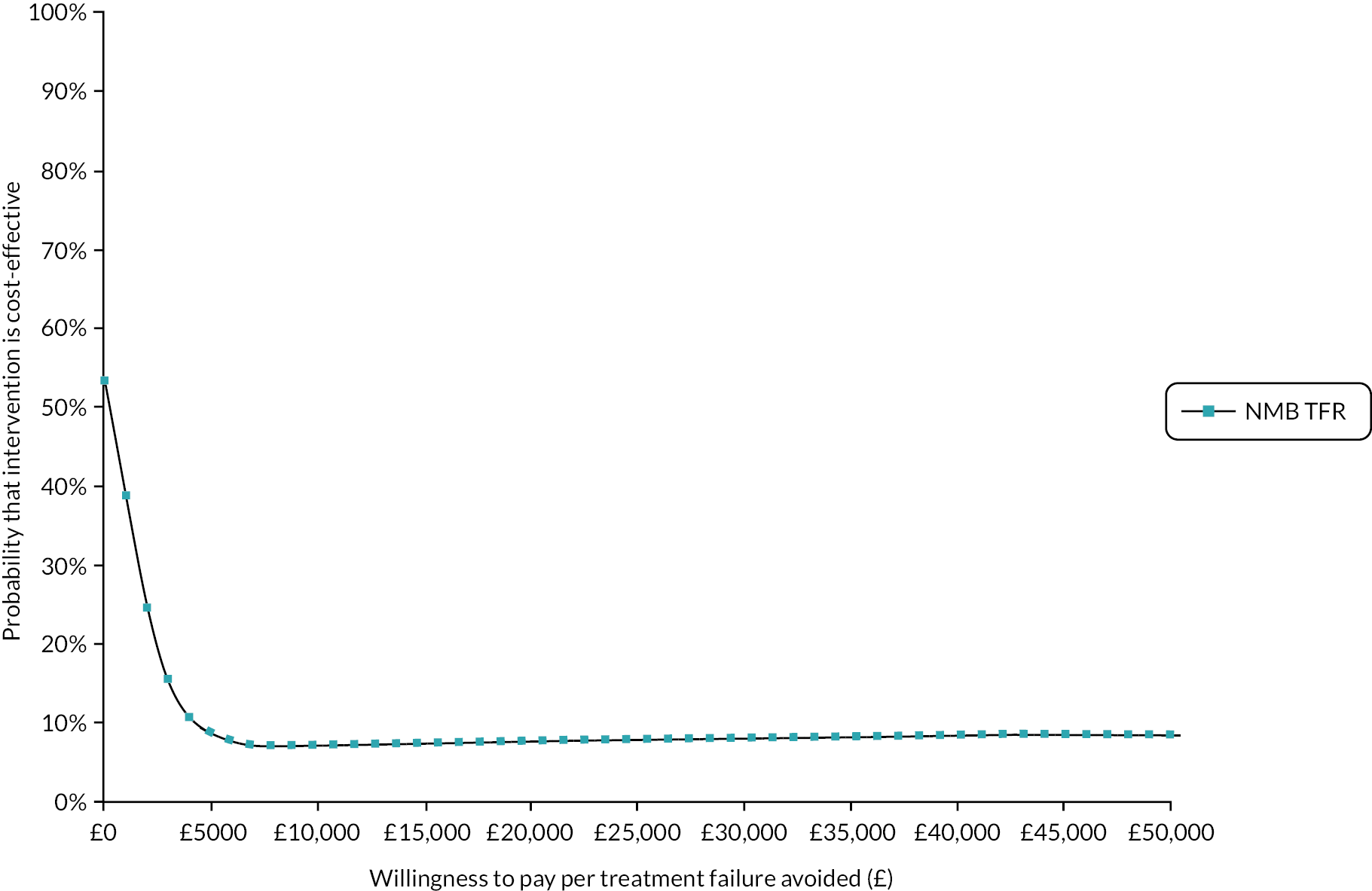
Sensitivity analyses (cost-effectiveness analysis)
For all sensitivity analyses, there continued to be no statistically significant differences in costs or outcomes. Baseline characteristics (age, gender, baseline EQ-5D-5L score) were adjusted for using multiple regression. The overall decision was unchanged: the incremental costs were five times higher than in the primary analysis, and outcomes were slightly more negative, and so the intervention was dominated by standard care (as previously presented in Table 32). When the analysis was performed excluding the patient who had a prolonged ICU admission the results showed although there were more treatment failures in the intervention arm, it was also now on average £132 less expensive per patient compared to standard care, giving an ICER of £1650.52 per treatment failure avoided. This turnaround in mean costs shows how heavily the primary analysis was influenced by this one high resource user. The adjusted CEAC exhibits increased probability of the intervention being cost-effective at all thresholds; at a threshold of £1000/treatment failure avoided it increases to 57%.
The PP analysis also showed the intervention arm to be less expensive by almost £150 per patient and although there were still on average more treatment failures in this arm (0.02), it was less than in the primary analysis. The probability of the intervention being cost-effective increased to 68% at a threshold of £1000/treatment failure avoided.
Patient preferences
A total of 114 patients (60 in the standard care group and 54 in the intervention group) provided responses, with slightly fewer for probability-based questions. The overwhelming majority of patients (95%) in both arms were satisfied with the care received and level of hospital support, and were happy with their method of treatment, believing it to be effective (see Table 34). Approximately, 70% of patients in each arm disagreed that they were concerned their treatment would not work beforehand and a total of 16 patients were concerned about how treatment for NS would affect family and friends (intervention 15%; standard care 13%). Of these 16, 88% would be willing to accept the risk of re-admission (15%) for early discharge, and 81% said they would still accept the risk even if this doubled to 30%, indicating a strong preference to be discharged as soon as possible.
| Statement | Intervention (n = 54), n (%) | Standard care (n = 60), n (%) |
|---|---|---|
| Satisfied with care received | ||
| Agree | 52 (96.30) | 59 (98.33) |
| Disagree | 0 (0) | 0 (0) |
| Uncertain | 2 (3.70) | 1 (1.67) |
| Satisfied with level of hospital support | ||
| Agree | 53 (98.15) | 58 (96.67) |
| Disagree | 1 (1.85) | 1 (1.67) |
| Uncertain | 0 (0) | 1 (1.67) |
| Believe treatment received was effective | ||
| Agree | 52 (96.30) | 57 (95.00) |
| Disagree | 0 (0) | 0 (0) |
| Uncertain | 2 (3.70) | 3 (5.00) |
| Happy with the place treatment received | ||
| Agree | 53 (98.15) | 58 (96.67) |
| Disagree | 0 (0) | 1 (1.67) |
| Uncertain | 1 (1.85) | 1 (1.67) |
| Concerned beforehand that treatment would not work | ||
| Agree | 6 (11.11) | 10 (16.67) |
| Disagree | 38 (70.37) | 41 (68.33) |
| Uncertain | 10 (18.52) | 9 (15.00) |
| Concerned beforehand how treatment would affect family/friends | ||
| Agree | 8 (14.81) | 8 (13.33) |
| Disagree | 42 (77.78) | 43 (71.67) |
| Uncertain | 4 (7.41) | 9 (15.00) |
A greater proportion of those in the intervention arm thought it was important to be discharged 1–2 days earlier, 59% in comparison to 35% in the control arm (see Table 35). This shows a tendency for patients to choose the response in keeping with the treatment they actually received. Patients in both arms agreed it was important not to be re-admitted to hospital although with slightly more in the intervention group agreeing this (85% vs. 72%). The majority of patients in both groups agreed they would accept some risk of re-admission in order to be discharged early (72% in the intervention group and 65% in the control group). When asked which treatment they would choose, 71% of patients in the intervention arm said they would choose the early switch to oral antibiotics, compared with 46% of standard care patients, again suggesting allegiance to the treatment received.
| Statement | n | Intervention, n (%) | n | Standard care, n (%) |
|---|---|---|---|---|
| Important to be discharged 1–2 days early | 54 | 60 | ||
| Agree | 32 (59.26) | 21 (35.00) | ||
| Disagree | 9 (16.67) | 25 (41.67) | ||
| Uncertain | 13 (24.07) | 14 (23.33) | ||
| Important to not be re-admitted to hospital for further treatment | 54 | 60 | ||
| Agree | 46 (85.19) | 43 (71.67) | ||
| Disagree | 7 (12.96) | 16 (26.67) | ||
| Uncertain | 1 (1.85) | 1 (1.67) | ||
| Willing to accept small risk of re-admission if it meant discharge 1–2 days early | 54 | 60 | ||
| Agree | 39 (72.22) | 39 (65.00) | ||
| Disagree | 7 (12.96) | 11 (18.33) | ||
| Uncertain | 8 (14.81) | 10 (16.67) | ||
| Preferred method of treatment for NS: | 52 | 57 | ||
| i.v. in hospital for at least 48 hours then switch to oral | 15 (28.85) | 31 (54.39) | ||
| i.v. in hospital for 12–24 hours then switch to oral | 37 (71.15) | 26 (45.61) | ||
| If the risk of re-admission increased from 15% to 30%, would you choose to be discharged 1–2 days sooner? | 53 | 56 | ||
| Yes | 39 (73.58) | 36 (64.29) | ||
| No | 14 (26.42) | 20 (35.71) |
When presented with the hypothetical scenario of risk of re-admission doubling from 15% to 30%, 74% and 64% of intervention and standard care patients respectively would still opt for early discharge.
There was some evidence of completion errors regarding these hypothetical risk-based questions observed in a small number of responses, showing some difficulty in understanding and interpreting these scenarios.
In both groups, the bulk of respondents did not receive home care on a daily basis or have dependents, as shown in Table 36. Of the 24 participants who stated they receive daily care at home, there was a strong inclination to get home: 22 (92%) would accept the risk to be discharged early; 19/22 (86%) would accept the higher risk of re-admission in order to be discharged early. There were seven patients who said they received daily care at home as well as having to care for children or dependents.
| Intervention (n = 54) | Standard care (n = 60) | |
|---|---|---|
| Do you receive care in your home on a daily basis? | ||
| Yes | 13 (24.07) | 11 (18.33) |
| No | 41 (75.93) | 49 (81.67) |
| Do you have children or dependents you care for? | ||
| Yes | 16 (29.63) | 15 (25.00) |
| No | 38 (70.37) | 45 (75.00) |
Conclusion
The primary CUA showed that although the early switch to oral antibiotics led to small gains in HRQoL and small cost savings the differences were not statistically significant. The probability of the intervention being cost-effective was highest for all WTP thresholds in the PP sensitivity analysis. Although two subsequent sensitivity analyses estimated the intervention as slightly more costly than standard care, the ICERs were favourable (well under £20,000/QALY). There was a higher number of treatment failures in the intervention group within the CEA, meaning the intervention was dominated by standard care. However, subsequent analysis removing a high resource outlier showed a small and not statistically significant cost saving for those receiving the intervention. This one patient, who had a prolonged ICU episode for an uncommon condition that was not related to treatment allocation in this trial, appeared to bias the costs allocated to the intervention arm. Given that this patient was not representative of patients with low-risk NS, it was felt appropriate to consider the effect of removing this patient’s data.
The results of the CUA and CEA are not consistent, which may reflect the difference in individual patient data used in the analyses as a result of missing data. While the CEA showed a greater number of treatment failures in the intervention arm, there was a small but not statistically significant gain in QALYs for this arm. This can be seen by the large number of bootstrapped ICERs in the NE and SE quadrants of Figure 5, suggesting the intervention did indeed impact on patient HRQoL, albeit in a small way. Interestingly the incremental QALYs went in the opposite direction to incremental treatment failures. Although the trial was unable to conclude that the intervention is non-inferior to standard care, the CUA showed both treatments had a similar impact on HRQoL. Therefore, although treatment failures as the primary outcome are clinically important and relate to the intended purpose of treatment for NS, these are not synonymous with HRQoL, as reflected in patient EQ-5D-5L responses, presenting a disparity between what is valued by clinicians and patients.
The aim of the patient preference questionnaire was to elicit preferences for non-health outcomes, and the responses showed the majority of patients were content with the treatment they received, regardless of the group they were randomised to. This suggests both treatment options for NS are acceptable to patients, and could support clinicians in helping patients make choices. Patients who were worried about how their treatment would affect family and those receiving care at home were found to be more prepared to accept higher levels of risk of re-admission in order to have an earlier discharge. These data were particularly revealing since they indicate that patients have a much higher tolerance for the possibility of treatment failure in order to enable early discharge for their primary admission.
A major strength of these analyses was the choice of perspective. By taking a hospital perspective, it meant there was the ability to collect daily data and details of any re-admissions using hospital records. The short follow-up duration also meant less of a burden for research staff and resulted in complete cost data for all patients. There was complete QALY data for 87% of patients and over 90% of patients provided responses to the patient preference questionnaire.
The health economics analysis had some limitations. Firstly, due to the challenges of collecting outcome measures from patient questionnaires versus routine data retrieval from hospital records, there was a higher number of patients with complete data for analysis in the CEA compared to the CUA (n = 124 and n = 110, respectively). The impact on staff time reduction for administration of oral versus i.v. antibiotics was not assessed, as given the time difference of only 24 hours, differences would be small. However, the additional time spent in hospital and associated costs per hospital bed already includes staff costs. The preferences questionnaire was administered mostly over the phone at day 14 (unless the patient had not been discharged or had a visit), and given the complexity of some of the questions, processing them could understandably lead to completion errors. To minimise error and increase acceptability, there is a need to aid patients’ understanding of risk. This may have wider implications in relation to the consent process in trials and practice, and considering whether or not risk has been satisfactorily explained to patients or those consenting on their behalf. Furthermore, it would be preferable if patients were asked for treatment preference before allocation as the results showed an allegiance to the assigned method and may have been biased by patients’ experience of treatment.
Chapter 7 Discussion
Aim of the study
The NICE guidance in 2012 provided the first UK consensus guideline for treatment of NS while also highlighting a number of outstanding questions regarding optimal management. NICE were unable to recommend switching from empirical i.v. to oral antibiotics at < 48 hours even in patients at low risk of infective complications because of uncertainty about whether this achieved comparable outcomes to standard practice of longer-duration i.v. antibiotics. To bridge this evidence gap, the EASI-SWITCH trial, commissioned by the UK NIHR, aimed to provide a definitive randomised trial evaluating the effectiveness of early oral switch in low-risk NS patients.
Main findings and interpretation of results
The within-trial review of NS management enabled an evaluation of current management of NS in the UK following publication of the NICE guidance, showing many local policies had been updated to accurately reflect many of the recommendations. Clinicians also reported standard clinical practice that appears to be compliant with many of the key aspects of the guidance but there appear to be no standardised sepsis diagnostic criteria in widespread use and instead there is a range of approaches to incorporating infectious/sepsis criteria into NS diagnostic and care pathways. There has been increased use of routine risk assessment and consideration of early outpatient oral antibiotics for low-risk patients but there appears to be a wide range of approaches to risk stratification, duration of treatment and discharge from hospital. Consequently, despite challenges with recruitment, there was support for continuation of EASI-SWITCH from clinicians across the UK due to its potential impact on current UK practice. For clinicians and centres that already considered early oral antibiotics and discharge it was felt the trial had the potential to validate their practice evolution and make a compelling argument for further acute oncology service investment and modernisation.
Despite this support, recruitment remained challenging, resulting in early termination of the trial. Multisite trial activity in the area of low-risk NS has been challenging to deliver in the UK. Prior to the EASI-SWITCH trial, the ORANGE trial (ORal Antibiotics for Neutropenic sepsis Giving Early hospital discharge), which opened in August 2007, aimed to evaluate immediate upfront oral antibiotic therapy in low-risk NS patients,67 but closed in 2009 due to poor recruitment (27 patients registered and 12 randomised). The trial team reported that recruitment of investigator sites and patients was extremely difficult, with sites unable to participate due to local admission and care pathways and conflicting clinical management and local antibiotic protocols. 68 While these barriers were overcome to some degree within EASI-SWITCH, similar themes emerged from the surveys and interviews undertaken during the trial and it is likely that a combination of these challenges and a shift in equipoise as the trial progressed towards upfront or early oral therapy for patients with NS resulted in the plateau in accrual seen prior to closure.
The consequence of the smaller than anticipated sample size is that non-inferiority for early oral switch cannot be proven. The conflicting results of the ITT and PP analyses for the primary outcome may reflect this but also highlight the potential for analysis of different patient data sets to deliver conflicting results. Because of this potential, we determined a priori that a firm conclusion on the non-inferiority of the intervention would be reached only if both the ITT and PP analyses were in agreement. While analysis of either population alone may lead to bias, there is particular potential for increased risk of bias in ITT analyses due to non-adherence to treatment or deviations from the protocol resulting in a higher likelihood of concluding non-inferiority incorrectly. 69,70 Within our data set, of the 11 ITT patients who were excluded from the PP population, 8 had been allocated to the intervention arm; of these, 4 had their antibiotic treatment stopped prematurely, 2 had substantial interruption to treatment (at least two consecutive missed doses), and 1 had less than the minimum of 12 hours initial i.v. treatment. The three excluded patients in the standard care arm had received < 48 hours of i.v. antibiotics. As the ITT population included these patients, who did not receive the complete protocolised intervention, an ITT analysis risks underestimating the efficacy of the intervention; by comparison a PP analysis may overcome this issue.
The trial was pragmatic in design; decisions such as non-blinding of patients and staff were guided by the opportunity to assess the effect of early switch on timing of hospital discharge as well as advice from our patient representatives that blinding was unlikely to be maintained in the course of patient care. Open-label treatment has the potential to introduce performance bias; in this instance it may be that physicians have a lower threshold for antibiotic escalation from oral therapy, resulting in treatment failure and risk of bias towards concluding inferiority of the intervention. Similarly, non-blinding of outcome assessment risks introduction of detection bias; however, the components of the composite outcome measure were largely objective measurements of treatment failure rather than subjective components or qualitative measures.
While non-inferiority of early switch could not be proven within this trial, it may be that an early switch approach results in a shorter initial admission for treatment of NS although with the trade-off that the risk of re-admission for treatment failure is more likely than with standard care therapy. Reassuringly, and accepting the smaller than anticipated sample size, the risk of treatment failure and requirement for antibiotic treatment escalation or re-admission with early switch was not associated with serious consequences in terms of critical care admission or death. AEs were generally as anticipated in this patient population and were reported at similar rates between the early oral switch and standard care groups. Overall, therefore, it would seem that early switch could be a safe management option for selected patients with low-risk NS.
Although the results did not permit a definitive conclusion on clinical efficacy of early oral switch in relation to treatment failure, there were small, albeit not significant, gains in HRQoL and patients found it an acceptable treatment approach. While the primary outcome measure of treatment failure is clearly an important efficacy outcome for clinicians and patients, the observed increased rate of treatment failure in the early switch arm was not reflected in the HRQoL results, highlighting the differences in outcome measures valued by clinicians and patients.
The results suggest that early oral switch may be a cost-effective approach within existing NHS NS care pathways. The primary CUA showed the early switch to oral antibiotics led to small, non-significant cost savings and improvements in HRQoL compared to standard care. The probability of the intervention being cost-effective was highest for all WTP thresholds in the PP sensitivity analysis. Although two subsequent sensitivity analyses estimated the intervention as slightly more costly than standard care, the ICERs were favourable (well under £20,000/QALY). There was a higher number of treatment failures in the intervention group within the CEA, meaning the intervention was dominated by standard care. However, subsequent analysis removing a high resource outlier showed a small, non-significant cost saving for those receiving the intervention. A major strength of these analyses was the choice of perspective. By taking a hospital perspective, it meant there was the ability to collect daily data and details of any re-admissions using hospital records. The short follow-up duration also meant less of a burden for research staff and resulted in complete cost data for all patients. However, due to the challenges of collecting outcome measures from patient questionnaires versus routine data retrieval from hospital records, there was a higher number of patients with complete data for analysis in the CEA compared to the CUA (n = 124 and n = 110, respectively), with complete QALY data for 87% of patients.
In addition to global HRQoL, a patient preference questionnaire was used to better understand the non-health outcome measures that are important to patients and their willingness to accept different treatment approaches in NS management. Over 90% of patients provided responses to the patient preference questionnaire, and the responses showed the majority of patients were content with the treatment they received, regardless of the group they were randomised to. Patients who were worried about how their treatment would affect family and those receiving care at home were found to be more prepared to accept higher levels of risk of re-admission in order to have an earlier discharge. This approach did have some limitations. The preferences questionnaire was administered mostly over the phone at day 14 (unless the patient had not been discharged or had a visit), and given the complexity of some of the questions, processing them could understandably lead to completion errors. To minimise error and increase acceptability, there is a need to aid patients’ understanding of risk. This may have wider implications in relation to the consent process in trials and practice, and considering whether or not risk has been satisfactorily explained to patients or those consenting on their behalf. Furthermore, it would preferable if patients were asked for treatment preference before allocation as the results showed an allegiance to the assigned method and may have been biased by patients’ experience of treatment. However, overall the data are revealing, indicating that patients have a much higher acceptance of the possibility of treatment failure in order to enable early discharge for their primary admission than might have been anticipated by clinicians.
Conclusion
A definitive conclusion about the clinical effectiveness of early switch to oral antibiotics in low-risk NS was unable to be reached from the study findings due to the limited sample size.
While non-inferiority of early oral switch could not be definitively concluded, it may be that early switch results in a shorter initial duration of hospitalisation for treatment of low-risk NS. However, it is uncertain whether there is a consequent increased likelihood of re-admission to hospital compared to standard care treatment.
Treatment failure or requirement for antibiotic escalation or re-admission was not associated with serious outcomes such as critical care admission or death. There was no unexpected or increased risk of AEs observed with early switch compared to standard care.
There were small, non-significant gains in HRQoL for early oral switch and patients found it acceptable. Patients, particularly those with caregiver roles, were more prepared to accept the higher levels of risk of re-admission in order to have an earlier discharge.
Given the findings, early oral switch may be an acceptable strategy for some patients who can adhere to such a regimen and would prefer reduced duration of hospitalisation while accepting a potentially increased risk of treatment failure, resulting in re-admission to hospital.
Implications for future research
EASI-SWITCH is the first UK multicentre RCT comparing early oral switch to the NICE recommendation of 48 hours i.v. antibiotic therapy in low-risk NS. Delivery of the trial has highlighted the difficulties with recruitment of patients to trials in the unscheduled care setting. Evidence supporting specific interventions to improve recruitment in this setting is lacking71 and further work evaluating strategies such as verbal provision of trial information and obtaining verbal consent is warranted across supportive care trials in cancer patients, not just in the setting of NS.
A number of specialist cancer centres in the NHS have already established acute oncology services and/or ambulatory care pathways to deliver early oral switch or upfront oral antibiotics. 39–41 This results in tension between delivering prospective trials evaluating management approaches for NS and accepting that current ambulatory care pathways or antimicrobial strategies utilising upfront or early switch to oral antibiotics are typically applied to highly selected patient populations. This shift in practice and our experience in delivering this trial, accompanied by the declining NS rates, suggest further randomised trials in this patient population will remain challenging to recruit to. However, it is clear there remains scope to optimise and unify management strategies for low-risk NS, in particular how best to identify patients suitable for a de-escalation or ambulatory care approach from initial presentation. While the MASCC score remains the most widely used within specialist cancer centres, others have reported that stratification tools such as MASCC score or CISNE are too cumbersome to be applied in real-life ED practice. 46 Other scores that encompass ‘clinical judgement’ are similarly felt less applicable in ED or acute medical settings than validated decision rules or algorithms. Further research should explore tools for patient stratification for low-risk de-escalation or ambulatory pathways, including use of biomarkers and/or point-of-care rapid viral, bacterial and fungal testing as an adjunct to clinical decision-making tools. This could include application to shorter-duration antimicrobial therapy in line with antimicrobial stewardship studies in other settings.
Evaluation of any future stratification tools, treatment strategies or ambulatory care pathways should include a formal economic analysis in addition to assessment of clinical efficacy. This is particularly important where novel algorithms or ambulatory care pathways are established but restricted to specialist oncology services rather than general acute care or ED settings.
The EASI-SWITCH trial has highlighted the importance of patients’ perspectives in setting research questions that encompass patients’ willingness to accept risk of treatment failure as a trade-off for less intensive treatment and shorter duration of admission. The results of the patient preference questionnaire could be used as a precursor for designing a future discrete-choice experiment to assess patient preferences for treatment for NS. This method is used in eliciting strength of preferences in health care by presenting hypothetical choices varying treatment options and levels of key attributes (e.g. life expectancy, side effects), and in doing so respondents make trade-offs. 72 They have been effectively used in other cancer populations73 and it has been long thought by experts that NICE should recommend their use in incorporating patient preferences,74 but while their US counterparts the FDA have endorsed their use, NICE have yet to do so.
Acknowledgements
The authors would like to thank all of the patients who took part in the EASI-SWITCH trial. They also wish to thank all of the research and clinical team members at each of the trial sites for screening, recruitment, management and follow-up of patients within the trial. Thanks are also extended to the staff of NICTU who contributed to trial management and the Belfast HSC Trust Research Office for undertaking trial sponsorship. Particular thanks are extended to Mrs Margaret Grayson, patient and public representative, who has been an integral member of the study team from conception through to delivery and dissemination. Mrs Grayson’s input was particularly invaluable in collating wider PPI opinion on the study design and continued importance of the research question as the study progressed and the design revised. Additional thanks are extended to Dr Caroline Forde, Clinical Research Fellow, who contributed significantly to study set-up and day-to-day trial management and to Mrs Margaret McFarland for pharmacovigilance support.
The authors are also very grateful to the independent TSC and DMEC members for their continued advice and guidance throughout the trial:
TSC:
Professor Kathy Bamford (chairperson, Imperial College Healthcare Trust, London, UK)
Capt. Robert Gray (independent PPI member),
Dr Ed Goodall (independent PPI member)
Dr Richard Jackson (independent statistician, University of Liverpool, Liverpool, UK)
Professor Christian Ottensmeier (University of Southampton, Southampton, UK)
DMEC:
Dr Mark Saunders (chairperson, The Christie NHS Foundation Trust, Manchester, UK)
Dr Ashley Jones (independent statistician, University of Liverpool, Liverpool, UK)
Professor Paul Dark (Salford Royal NHS Foundation Trust, Salford, UK)
The authors would also like to acknowledge the site investigators and their teams from 19 sites across the UK who enrolled patients in the trial: Belfast Health and Social Care Trust Professor Richard Wilson, Dr Audrey Fenton University Hospitals of Leicester Professor Anne Thomas The Newcastle Upon Tyne Hospitals NHS Foundation Trust Professor Ruth Plummer Velindre NHS Trust Professor Richard Adams, Dr Hilary Williams NHS Greater Glasgow and Clyde (Beatson) Dr Dawn Storey Royal Marsden NHS Foundation Trust Dr Ian Chau South Eastern Health and Social Care Trust Dr Lois Mulholland Salisbury NHS Foundation Trust Dr Effie Grand Hampshire NHS Foundation Trust Dr Luke Nolan Western Health and Social Care Trust Dr Sonali Dasgupta The Mid Yorkshire Hospitals NHS Trust Dr Kostas Konstantinos Heart of England NHS Foundation Trust Dr Joyce Thompson Derby Teaching Hospitals NHS Foundation Trust Dr Mike Smith-Howell Worcester Acute Hospitals NHS Trust Dr Bart Kurec Poole Hospital NHS Foundation Trust Dr Amelie Harle The Dudley Group NHS Foundation Trust Dr Jeffrey Neilson North Middlesex University Hospital NHS Trust Dr Fharat Raja Whittington Health NHS Trust Dr Pauline Leonard Singleton Hospital Swansea Dr Ceri Powell
Contributions of authors
Vicky Coyle (https://orcid.org/0000-0001-9566-4130) (Senior Lecturer in Medical Oncology), trial co-chief investigator, contributed to the concept and design of the trial, protocol development, recruitment, trial management and interpretation of results.
Caroline Forde (https://orcid.org/0000-0001-5021-9973) (Clinical Fellow in Medical Oncology) was the trial clinical fellow and contributed to trial design, protocol development and day-to-day trial management and undertook the review of NS practice as the study progressed.
Richard Adams (https://orcid.org/0000-0003-3915-7243) (Professor of Clinical Oncology and Cancer Clinical Trials) contributed to the trial design, protocol development, patient recruitment and interpretation of results.
Ashley Agus (https://orcid.org/0000-0001-9839-6282) (Senior Health Economist), trial health economist, contributed to trial design and development and led the concept and design of the health economics evaluation including data analysis, interpretation of results and writing of Chapter 6.
Rosemary Barnes (https://orcid.org/0000-0002-0574-8670) (Professor of Medical Microbiology) contributed to the trial design, protocol development and interpretation of results.
Ian Chau (https://orcid.org/0000-0003-0286-8703) (Consultant Medical Oncologist) contributed to the trial design, protocol development, patient recruitment and interpretation of results.
Mike Clarke (https://orcid.org/0000-0002-2926-7257) (Professor of Research Methodology and Director MRC Methodology Hub), trial methodologist, contributed to the trial design, protocol development and interpretation of results.
Annmarie Doran (https://orcid.org/0000-0002-9802-008X) (Trial Coordinator) contributed to protocol development and study set-up and was responsible for trial co-ordination and day-to-day management.
Margaret Grayson (https://orcid.org/0000-0002-1861-8541) (Chair Northern Ireland Cancer Research Consumer Forum), trial PPI co-investigator, contributed to the trial design, trial development and management, review of patient-facing materials, lay communication and co-ordinated PPI opinion on revision of the trial design.
Danny McAuley (https://orcid.org/0000-0002-3283-1947) (Professor of Intensive Care Medicine) contributed to the trial design, protocol development and interpretation of results.
Cliona McDowell (https://orcid.org/0000-0002-7644-7197) (Senior Statistician), trial statistician, contributed to development of the protocol and statistical analysis plan and confidential data analysis for the DMEC and led the analysis of primary and secondary outcome data.
Glenn Phair (https://orcid.org/0000-0001-8137-0959) (Health Economist), trial health economist, contributed to the health economic data analysis, interpretation of results and writing of Chapter 6.
Ruth Plummer (https://orcid.org/0000-0003-0107-1444) (Professor of Experimental Cancer Medicine) contributed to the trial design, protocol development, patient recruitment and interpretation of results.
Dawn Storey (https://orcid.org/0000-0002-6546-3183) (Consultant Medical Oncologist) contributed to the trial design, protocol development, patient recruitment and interpretation of results.
Anne Thomas (https://orcid.org/0000-0003-1998-3134) (Professor of Cancer Therapeutics) contributed to the trial design, protocol development, patient recruitment and interpretation of results.
Richard Wilson (https://orcid.org/0000-0001-8018-7730) (Professor of Gastrointestinal Oncology) contributed to the trial design, protocol development, patient recruitment and interpretation of results.
Ronan McMullan (https://orcid.org/0000-0001-6760-6072) (Professor of Medical Microbiology), co-chief investigator, contributed to the concept and design of the trial, protocol development, trial management and interpretation of results.
Data-sharing statement
Requests for access to de-identified patient data can be made by researchers to the chief investigators, and data will be shared subject to any constraints. Requests for access should be accompanied by a proposal describing the aims and scope of the research, details of the data requested and data analysis plan. Proposals will be considered by the Trial Management Group, co-investigators and sponsor who will make a decision regarding data access. A data-sharing agreement will be signed between the researchers, principal investigators and sponsor.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it is important that there are safeguards to make sure that they are stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives. You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This manuscript presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- Forde C, McMullan R, Clarke M, Wilson RH, Plummer R, Grayson M, et al. Early switch from intravenous to oral antibiotic therapy in patients with cancer who have low-risk neutropenic sepsis (the EASI-SWITCH trial): study protocol for a randomized controlled trial. Trials 2020;21. https://doi.org/10.1186/s13063-020-04241-1.
- Schimpff S, Satterlee W, Young VM, Serpick A. Empiric therapy with carbenicillin and gentamicin for febrile patients with cancer and granulocytopenia. N Engl J Med 1971;284:1061-5. https://doi.org./10.1056/NEJM197105132841904.
- NICE . Clinical Guideline, Neutropenic Sepsis: Prevention and Management of Neutropenic Sepsis in Cancer Patients 2012. www.nice.org.uk/guidance/cg151/resources/neutropenic-sepsis-prevention-and-management-in-people-with-cancer-pdf-35109626262469 (accessed 20 July 2020).
- Holmes FA, Jones SE, O’Shaughnessy J, Vukelja S, George T, Savin M, et al. Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol 2002;13:903-9. https://doi.org/10.1093/annonc/mdf130.
- de Naurois J, Novitzky-Basso I, Gill MJ, Marti Marti F, Cullen MH, Roila F. Management of febrile neutropenia: ESMO Clinical Practice Guidelines. Ann Oncol 2010;21:v252-6. https://doi.org/10.1093/annonc/mdq196.
- Clarke RT, Warnick J, Stretton K, Littlewood TJ. Improving the immediate management of neutropenic sepsis in the UK: lessons from a national audit. Br J Haematol 2011;153:773-9. https://doi.org/10.1111/j.1365-2141.2011.08693.x.
- Flowers CR, Seidenfeld J, Bow EJ, Karten C, Gleason C, Hawley DK, et al. Antimicrobial prophylaxis and outpatient management of fever and neutropenia in adults treated for malignancy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2013;31:794-810. https://doi.org/10.1200/JCO.2012.45.8661.
- Ammann RA, Hirt A, Lüthy AR, Aebi C. Identification of children presenting with fever in chemotherapy-induced neutropenia at low risk for severe bacterial infection. Med Pediatr Oncol 2003;41:436-43. https://doi.org/10.1002/mpo.10320.
- Apostolopoulou E, Raftopoulos V, Terzis K, Elefsiniotis I. Infection Probability Score, APACHE II and KARNOFSKY scoring systems as predictors of bloodstream infection onset in hematology-oncology patients. BMC Infect Dis 2010;10. https://doi.org/10.1186/1471-2334-10-135.
- Ha YE, Song J-H, Kang WK, Peck KR, Chung DR, Kang C-I, et al. Clinical factors predicting bacteremia in low-risk febrile neutropenia after anti-cancer chemotherapy. Support Care Cancer 2011;19:1761-7. https://doi.org/10.1007/s00520-010-1017-4.
- Hakim H, Flynn PM, Srivastava DK, Knapp KM, Li C, Okuma J, et al. Risk prediction in pediatric cancer patients with fever and neutropenia. Paediatr Infect Dis 2010;29:53-9. https://doi.org/10.1097/INF.0b013e3181c3f6f0.
- Klaassen RJ, Goodman TR, Pham B, Doyle JJ. ‘Low-risk’ prediction rule for pediatric oncology patients presenting with fever and neutropenia. J Clin Oncol 2000;18:1012-9. https://doi.org/10.1200/JCO.2000.18.5.1012.
- Santolaya ME, Alvarez AM, Becker A, Cofré J, Enríquez N, O’Ryan M, et al. Prospective, multicentre evaluation of risk factors associated with invasive bacterial infection in children with cancer, neutropenia and fever. J Clin Oncol 2001;19:3415-21. https://doi.org/10.1200/JCO.2001.19.14.3415.
- Tezcan G, Kupesiz A, Ozturk F, Ogunc D, Gultekin M, Yesilipek A, et al. Episodes of fever and neutropenia in children with cancer in a tertiary medical care center in Turkey. Pediatr Hematol Oncol 2006;23:217-29. https://doi.org/10.1080/08880010500506719.
- West DC, Marcin JP, Mawis R, He J, Nagle A, Dimand R. Children with cancer, fever and treatment-induced neutropenia: risk factors associated with illness requiring the administration of critical care therapies. Pediatr Emerg Care 2004;20:79-84. https://doi.org/10.1097/01.pec.0000113875.10140.40.
- Lyman GH, Lyman CH, Agboola O. Risk models for predicting chemotherapy-induced neutropenia. Oncologist 2005;10:427-37. https://doi.org/10.1634/theoncologist.10-6-427.
- Martin M, Pienkowski T, Mackey J, Pawlicki M, Guastalla JP, Weaver C, et al. Adjuvant docetaxel for node-positive breast cancer. N Engl J Med 2005;22:2302-13. https://doi.org/10.1056/NEJMoa043681.
- Head J, Archer C, Harper-Wynne C, Sinha R, Ring A, Banner R, et al. Rates of Neutropaenic Sepsis With the Use of Adjuvant FEC100-Docetaxel (FEC100-T) Chemotherapy in High-Risk Node-Positive Patients With Early Breast Cancer; A UK Perspective 2008. www.ncri.org.uk/ncriconference/2008abstracts/abstracts/B64.htm (accessed 9 September 2021).
- Kuderer NM, Dale DC, Crawford J, Cosler LE, Lyman GH. Mortality, morbidity, and cost associated with febrile neutropenia in cancer patients. Cancer 2006;106:2258-66. https://doi.org/10.1002/cncr.21847.
- Legrand M, Max A, Peigne V, Mariotte E, Canet E, Debrumetz A, et al. Survival in neutropenic patients with severe sepsis or septic shock. Crit Care Med 2012;40:43-9. https://doi.org/10.1097/CCM.0b013e31822b50c2.
- NCAG (National Chemotherapy Advisory Group) . Chemotherapy Services in England: Ensuring Quality and Safety 2009. https://webarchive.nationalarchives.gov.uk/20130104173757/http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/DH_104500 (accessed 1 August 2020).
- Clarke R, Bird S, Kakuchi I, Littlewood TJ, van Hamel Parsons V. The signs, symptoms and help-seeking experiences of neutropenic sepsis patients before they reach hospital: a qualitative study. Support Care Cancer 2015;23:2687-94. https://doi.org/10.1007/s00520-015-2631-y.
- Chirivella I, Bermejo B, Insa A, Pérez-Fidalgo A, Magro A, Rosello S, et al. Optimal delivery of anthracycline-based chemotherapy in the adjuvant setting improves outcome of breast cancer patients. Breast Cancer Res Treat 2009;114:479-84. https://doi.org/10.1007/s10549-008-0018-1.
- Herbst C, Naumann F, Kruse E-B, Monsef I, Bohlius J, Schulz H, et al. Prophylactic antibiotics or G-CSF for the prevention of infections and improvement of survival in cancer patients undergoing chemotherapy. Cochrane Database Syst Rev 2009. https://doi.org/10.1002/14651858.CD007107.pub2.
- Lalami Y, Klastersky J. Impact of chemotherapy-induced neutropenia (CIN) and febrile neutropenia (FN) on cancer treatment outcomes: An overview about well-established and recently emerging clinical data. Crit Rev Oncol Hematol 2017;120:163-79. https://doi.org/10.1016/j.critrevonc.2017.11.005.
- Morgan A, Sutton A, Wailoo A. The Risk and Costs of Febrile Neutropenia in Patients With Non-Small Cell Lung Cancer Treated With Docetaxel 2007. http://nicedsu.org.uk/wp-content/uploads/2016/03/Erlotinib-DSU-final-report1.pdf (accessed 1 August 2020).
- Talbot T, Dangoor A, Shah R, Naik J, Talbot D, Lester JF, et al. The burden of neutropenic sepsis in patients with advanced non-small cell lung cancer treated with single-agent docetaxel: A retrospective study. Lung Cancer 2017;113:115-20. https://doi.org/10.1016/j.lungcan.2017.09.014.
- Klastersky J, de Naurois J, Rolston K, Rapoport B, Maschmeyer G, Aapro M, et al. Management of febrile neutropaenia: ESMO clinical practice guidelines. Ann Oncol 2016;27:v111-8. https://doi.org/10.1093/annonc/mdw325.
- Taplitz R, Kennedy EB, Bow EJ, Crews J, Gleason C, Hawley DK, et al. Outpatient management of fever and neutropenia in adults treated for malignancy: American Society of Clinical Oncology and Infectious Diseases Society of America Clinical Practice Guideline Update. J Clin Oncol 2018;36:1443-53. https://doi.org/10.1200/JCO.2017.77.6211.
- Simmons T. Prevention and Management of Neutropenic Sepsis in Cancer Patients: Full Needs Assessment Report 2012. www.nice.org.uk/guidance/cg151/evidence/needs-assessment-pdf-188303583 (accessed 1 August 2020).
- Bate J, Gibson F, Selwood K, Skinner R, Phillips B, Chisholm JC. A reaudit of current febrile neutropenia practice in UK paediatric oncology centres prior to implementation of NICE guidance. Arch Dis Child 2013;98:315-6. https://doi.org/10.1136/archdischild-2013-303810.
- Phillips B, Selwood K, Lane SM, Skinner R, Gibson F, Chisholm JC. United Kingdom Children's Cancer Study Group . Variation in policies for the management of febrile neutropenia in United Kingdom Children’s Cancer Study Group centres. Arch Dis Child 2007;92:495-8. https://doi.org/10.1136/adc.2006.102699.
- Klastersky J, Paesmans M. The Multinational Association for Supportive Care in Cancer (MASCC) risk index score: 10 years of use for identifying low-risk febrile neutropenic cancer patients. Support Care Cancer 2013;21:1487-95. https://doi.org/10.1007/s00520-013-1758-y.
- Rubenstein EB, Rolston K, Benjamin RS, Loewy J, Escalante C, Manzullo E, et al. Outpatient treatment of febrile episodes in low-risk neutropenic patients with cancer. Cancer 1993;71:3640-6. https://doi.org/10.1002/1097-0142(19930601)71:11<3640::aid-cncr2820711128>3.0.co;2-h.
- Klastersky J, Paesmans M, Rubenstein EB, Boyer M, Elting L, Feld R, et al. The Multinational Association for Supportive Care in Cancer risk index: a multinational scoring system for identifying low-risk febrile neutropenic cancer patients. J Clin Oncol 2000;18:3038-51. https://doi.org/10.1200/JCO.2000.18.16.3038.
- Innes H, Smith DB, O’Reilly SM, Clark PI, Kelly V, Marshall E. Oral antibiotics with early hospital discharge compared with in-patient intravenous antibiotics for low-risk febrile neutropenia in patients with cancer: a prospective randomised controlled single centre study. Brit J Cancer 2003;89:43-9. https://doi.org/10.1038/sj.bjc.6600993.
- Vidal L, Ben Dor I, Paul M, Eliakim-Raz N, Pokroy E, Soares-Weiser K, et al. Oral versus intravenous antibiotic treatment for febrile neutropenia in cancer patients. Cochrane Database of Systematic Reviews 2013. https://doi.org/10.1002/14651858.CD003992.pub3.
- Talcott J, Yeap BY, Clark JA, Siegel RD, Loggers ET, Lu C, et al. Safety of early discharge for low-risk patients with febrile neutropenia: a multicenter randomized controlled trial. J Clin Oncol 2011;29:3977-83. https://doi.org/10.1200/JCO.2011.35.0884.
- Cooksley T, Rice T. Emergency oncology: development, current position and future direction in the USA and UK. Support Care Cancer 2017;25:3-7. https://doi.org/10.1007/s00520-016-3470-1.
- Cooksley T, Holland M, Klastersky J. Ambulatory outpatient management of patients with low risk febrile neutropenia. Acute Medicine 2015;14:178-81.
- Cooksley T, Marshall W, Ahn S, Lasserson DS, Marshall E, Rice TW, et al. Ambulatory emergency oncology: a key tenet of future emergency oncology care. Int J Clin Pract 2020;74. https://doi.org/10.1111/ijcp.13436.
- Marshall W, Campbell G, Knight T, Al-Sayed T, Cooksley T. Emergency ambulatory management of low-risk febrile neutropenia: Multinational Association for Supportive Care in Cancer fits real-world experience from a UK cancer center. J Emerg Med 2020;58:444-8. https://doi.org/10.1016/j.jemermed.2019.09.032.
- Brunner C, Botten J, Wennike N, Ford L, Michaels K, Stephens A, et al. Early supported discharge for patients with febrile neutropenia – experience at a large district hospital in the UK. Acute Medicine 2019;18:14-9.
- Dzienis M, Shahidzadeh Mahani A. Report on outpatient management of patients with neutropenic fever in a tertiary hospital. Intern Med J 2017;47:122-3. https://doi.org/10.1111/imj.13313.
- Baugh CW, Wang TJ, Caterino JM, Baker ON, Brooks GA, Reust AC, et al. Emergency department management of patients with febrile neutropenia: guideline concordant or overly aggressive?. Acad Emerg Med 2017;24:83-91. https://doi.org/10.1111/acem.13079.
- Baugh CW, Faridi MK, Mueller EL, Camargo CA, Pallin DJ. Near-universal hospitalization of US emergency department patients with cancer and febrile neutropenia. PLoS One 2019;14. https://doi.org/10.1371/journal.pone.0216835.
- Feld R, Paesmans M, Freifeld AG, Klastersky J, Pizzo PA, Rolston KVI, et al. Methodology for clinical trials involving patients with cancer who have febrile neutropenia: updated guidelines of the Immunocompromised Host Society/Multinational Association for Supportive Care in Cancer, with Emphasis on Outpatient Studies. Clin Infect Dis 2002;35:1463-8. https://doi.org/10.1086/344650.
- Donovan J, Mills N, Smith M, Brindle L, Jacoby A, Peters T, et al. Quality improvement report: improving design and conduct of randomised trials by embedding them in qualitative research: ProtecT (prostate testing for cancer and treatment) study. Commentary: presenting unbiased information to patients can be difficult. BMJ 2002;325:766-70. https://doi.org/10.1136/bmj.325.7367.766.
- Fletcher B, Gheorghe A, Moore D, Wilson S, Damery S. Improving the recruitment activity of clinicians in randomised controlled trials: a systematic review. BMJ 2012;2.
- Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol 2006;3:77-101. https://doi.org/10.1191/1478088706qp063oa.
- Okera M, Chan S, Dernede U, Larkin J, Popat S, Gilbert D, et al. A prospective study of chemotherapy-induced febrile neutropenia in the South West London Cancer Network. Interpretation of study results in light of NCAG/NCEPOD findings. Br J Cancer 2011;104:407-12. https://doi.org/10.1038/sj.bjc.6606059.
- Herd F, Bate J, Chisholm J, Johnson E, Phillips B. Variation in practice remains in the UK management of paediatric febrile neutropenia. Arch Dis Child 2016;101:410-1. https://doi.org/10.1136/archdischild-2015-310294.
- Subbe C, Kruger M, Rutherford P, Gemmel L. Validation of a modified Early Warning Score in medical admissions. QJM 2001;94:521-6. https://doi.org/10.1093/qjmed/94.10.521.
- Carmona-Bayonas A, Jiménez-Fonseca P, Virizuela Echaburu J, Antonio M, Font C, Biosca M, et al. Prediction of serious complications in patients with seemingly stable febrile neutropenia: Validation of the clinical index of stable febrile neutropenia in a prospective cohort of patients from the FINITE study. J Clin Oncol 2015;33:465-71. https://doi.org/10.1200/JCO.2014.57.2347.
- Paganini H, Sarkis CM, De Martino MG, Zubizarreta PA, Casimir L, Fernandez C, et al. Oral administration of cefixime to lower risk febrile neutropenic children with cancer. Cancer 2000;88:2848-52. https://doi.org/10.1002/1097-0142(20000615)88:12<2848::aid-cncr27>3.0.co;2-2.
- Shenep J, Flynn PM, Baker DK, Hetherington SV, Hudson MM, Hughes WT, et al. Oral Cefixime is similar to continued intravenous antibiotics in the empirical treatment of febrile neutropenic children with cancer. Clin Infect Dis 2001;32:36-43. https://doi.org/10.1086/317552.
- Rivas-Ruiz R, Villasis-Keever M, Miranda-Novales G, Castelán-Martínez OD, Rivas-Contreras S. Outpatient treatment for people with cancer who develop a low-risk febrile neutropaenic event. Cochrane Database Syst Rev 2019;3. https://doi.org/10.1002/14651858.CD009031.pub2.
- Coyle V, Forde C, McAuley DF, Wilson RH, Clarke M, Plummer R, et al. EASI-SWITCH Investigators . Early switch to oral antibiotic therapy in patients with low risk neutropenic sepsis (Easi-Switch): A randomized non-inferiority trial. Clinical Microbiol Infect 2023;28. https://doi:10.1016/j.cmi.2023.07.021.
- Department of Health, Reference Costs 2018–19, Department of Health, 2018 n.d. www.england.nhs.uk/wp-content/uploads/2020/08/1_-_NCC_Report_FINAL_002.pdf (accessed 24 March 2020).
- Curtis L, Burns A. Unit Costs of Health and Social Care 2018, Personal Social Services Research Unit. Canterbury: University of Kent; 2018.
- Joint Formulary Committee . British National Formulary (online) n.d. www.medicinescomplete.com (accessed 25 March 2020).
- van Hout B, Janssen MF, Feng Y-S, Kohlmann T, Busschbach J, Golicki D, et al. Interim scoring for the EQ-5D-5L: mapping the EQ-5D-5L to EQ-5D-3L value sets. Value Health 2012;15:708-15. https://doi.org/10.1016/j.jval.2012.02.008.
- National Institute for Health and Care Excellence . Position Statement on Use of the EQ-5D-5L Value Set for England [online] 2019. www.nice.org.uk/about/what-we-do/our-programmes/nice-guidance/technology-appraisal-guidance/eq-5d-5l (accessed 24 March 2020).
- National Institute for Health and Care Excellence . Guide to the Methods of Technology Appraisal 2013 2013.
- Glick HA, Doshi JA, Sonnad SS, Polsky D. Economic Evaluation in Clinical Trials. Oxford: Oxford University Press; 2007.
- Fenwick E, O’Brien BJ, Briggs A. Cost-effectiveness acceptability curves – facts, fallacies and frequently asked questions. Health Econ 2004;13:405-15. https://doi.org/10.1002/hec.903.
- Marshall E, . 2009. https://abstracts.ncri.org.uk (accessed 1 August 2020).
- Marshall E. 2014. www.theacp.org.uk/userfiles/file/acute_oncology_annual_update/neutropenic-sepsis-marshall.pdf (accessed 1 August 2020).
- European Medicines Agency . ICH Topic E 9 Statistical Principles for Clinical Trials 1998. www.ema.europa.eu/en/documents/scientific-guideline/ich-e-9-statistical-principles-clinical-trials-step-5_en.pdf (accessed 25 March 2020).
- U.S. Department of Health and Human Services Food and Drug Administration . Non-Inferiority Clinical Trials to Establish Effectiveness Guidance for Industry 2016. www.fda.gov/media/78504/download (accessed 25 March 2020).
- Rowlands C, Rooshenas L, Fairhurst K, Rees J, Gamble C, Blazeby JM. Detailed systematic analysis of recruitment strategies in randomised controlled trials in patients with an unscheduled admission to hospital. BMJ Open 2018;8. https://doi.org/10.1136/bmjopen-2017-018581.
- Ryan M. Discrete choice experiments in health care. BMJ 2004;328:360-1. https://doi.org/10.1136/bmj.328.7436.360.
- Sculpher M, Bryan S, Fry P, de Winter P, Payne H, Emberton M. Patients’ preferences for the management of non-metastatic prostate cancer: discrete choice experiment. BMJ (Clinical Research ed.) 2004;328. https://doi.org/10.1136/bmj.37972.497234.44.
- Johnson FR, Zhou M. Patient preferences in regulatory benefit-risk assessments: a US perspective. Value Health 2016;19:741-5. https://doi.org/10.1016/j.jval.2016.04.008.
Appendix 1 PPI survey on revision of the EASI-SWITCH trial non-inferiority margin
Summary of trial and context provided to PPI representatives
Neutropenic sepsis (NS), or infection that develops in the setting of a low white blood cell count, is a complication of chemotherapy treatment in patients with cancer. The ability to identify patients at low risk of complication from infection offers the opportunity to avoid unnecessary treatment and hospital stay which disadvantages both patients and the NHS.
We are carrying out a trial of patients at low risk of complications, to evaluate whether changing from intravenous (i.v.) to oral antibiotics on the first day of treatment is clinically and cost-effective in comparison with standard longer-duration i.v. antibiotics. The trial will study two patient groups: one group will switch from i.v. to oral antibiotics 24 hours after i.v. treatment commences; in the other group, patients will receive standard care i.v. antibiotic for at least 48 hours with subsequent antibiotic continuation or switch to oral therapy at physician discretion based on their usual practice. We will then compare the groups to see how effective the two different treatment strategies are.
The outcome that we will use to assess how well each treatment strategy works is ‘failure of treatment’ and we will measure this on day 14 of follow-up. This is defined by the occurrence of one of several undesirable outcomes that are all of importance to patients. These include: recurrence of fever; re-admission to hospital; antibiotic side-effects; quality of life and death. However, the most common way that patients’ treatment fails is that their fever comes back and they may need to be admitted to hospital again; by comparison, death is uncommon in patients with NS who are at low risk of complications (the group we are studying).
Question for PPI consideration: how many patients to study?
We have designed this trial based on the findings of other small studies. These indicate that it is reasonable to assume that early switch to oral treatment would have similar effectiveness to continuing on i.v. treatment for longer. We are thinking about the number of patients we might need to include in a large trial to confirm these findings. For example, with only one patient in each group, even if both had successful treatment, we would not be confident that this would be reproduced if all patients received this treatment across the NHS. By comparison, if we studied 10,000 patients in each group and found both treatments to be the same, we would be highly certain that this would be reproduced in NHS practice – but the trial would be impossible to do and cost tens of millions of pounds.
We are not trying to show that early oral switch is more effective than usual i.v. antibiotic treatment but are focusing on whether it is not much less effective treatment. This type of trial design is a ‘non-inferiority’ design in which there are statistical rules and tests to show how certain we are that the new treatment being tested is not much less effective than the usual treatment. Statisticians express how certain they are about study results using a 95% confidence range either side of the result. This means that researchers have very high certainty (95% certainty) that the true effectiveness of treatment lies within that range.
We are asking you to help us agree how wide this confidence range should be – or in other words what level of trade-off between the benefits of shorter duration of treatment and the risk of treatment failure is acceptable for patients. We also need to balance having enough patients to obtain meaningful results and not having so many patients that the study is not achievable.
We propose setting the limit of the 95% confidence range for this at 15%. This means that, if we show that both early oral antibiotic switch and longer courses of i.v. treatment effectively treat 85% of patients, we would have very high certainty (95% certainty) that the early oral switch approach had a ‘true’ effectiveness of between 70% and 100%. In other words, the study might conclude that the two treatments are similar but acknowledge that early oral switch is at least 70% effective while standard i.v. treatment is 85% effective.
In other words, for every 100 patients treated with an early oral switch strategy, all would benefit from early discharge home. However, while 15 would be expected to need re-admission (whether they had early switch or not), we would be acknowledging that early oral switch could lead to an extra 15 patients needing re-admission (in comparison with those who receive prolonged i.v. treatment in the first instance). Therefore, the trade-off we are asking you to consider is whether the possibility that an extra 15 patients may need re-admission is out-weighed by the advantage of the 100 patients receiving oral switch and being discharged early.
The main question is whether this possible difference in effectiveness between the two treatment options is acceptable. We had initially planned the study to assess whether the difference in effectiveness was no more than 10%; however, we have found that recruiting the number of patients needed to do this makes the study infeasible. Therefore, we are considering making a change to assess whether the difference in treatment effectiveness is no more than 15%, since that requires fewer patients and makes the study achievable.
We need to decide whether it is better to continue the study using the 15% confidence range or abandon the study altogether.
The main question is whether in your opinion it would still be useful to do the study if the confidence range is increased from 10% to 15%. In other words, do you think patients would be similarly convinced that switching to oral treatment earlier (to allow them to go home sooner) is a good idea if the additional risk of needing re-admission may be up to 15%, rather than 10%?
Given that the advantage of early oral switch is likely to be early discharge from hospital and the main consequence of the treatment being ineffective is re-admission we would like you to consider whether this trade-off would be to patients’ advantage overall.
2. PPI responses to sample size/NI margin question
Input from a range of PPI representatives was sought on this proposed change via our PPI co-applicant, Mrs Margaret Grayson, who distributed a summary of the study design and study progress including the rationale for reviewing the sample size and specifically the NI margin. The information was distributed to the Northern Ireland Cancer Consumer Forum and the Independent Cancer Patient Voice (a national independent patient advocate group which has the support of the professional members of the NCRI breast cancer Clinical Study Group).
Respondents were posed the question that if the NI margin was increased to 15% whether this level of trade-off between the benefit of less intensive treatment and the risk of treatment failure would be acceptable to patients; comments on the change were also welcomed.
There were 21 replies received and the majority of these supported this change (n = 19) with additional comments received from seven of these respondents included below. One respondent did not support this change and one was uncertain (comments from both included below).
PPI comments:
-
We all know that there is a finite amount of funding and that the professionals are trying to do the best for all within these limits. The fact that those who would require re-admission are being closely monitored is very positive so having considered the points made in your paper I feel that 15% re-admissions would be an acceptable level.
-
Very interesting study and I think well worth doing. I believe that for most patients going home early is key. Recovery at home is almost always preferably to being in hospital and I would definitely accept the very small extra risk providing good home back-up is in place.
-
I would recommend this study even if the risk of re-admission may be up to 15%. Having personally seen this condition with my daughter when she had become neutropenic when she was being treated for breast cancer. I believe she would have much preferred the oral method of treatment and getting home sooner. As in her case she had to spend 2/3 days in hospital. She did not like this. But I must add as only one person’s experience she may or may not have had to go back into hospital perhaps to have i.v. treatment. Personally as her mother and carer I can see that it would be a very good study.
-
This seems like a hard decision as it boils down to either doing the study or not doing the study. I wonder why recruitment was such a problem for the original study. I suppose we just have to accept that the study in its original form is not possible.
-
It would certainly be advantageous for patients to be discharged early as no one wants to spend any more time than necessary as an inpatient. My husband hated it and was always agitating to get home! However, doubling the percentage of patients that then need re-admitted, seems, on the face of it, rather a high number. Do these patients then need more protracted treatment, will their sepsis be more difficult to treat, are they at risk of becoming more seriously ill? In essence, how much additional danger are patients facing if such a change in treatment is implemented? Does it mean that we would be content for 30% of patients to need re-admission? Sorry, I seem to be asking questions rather than giving an opinion – but this is what is going around in my mind as I read the description. Maybe it’s only after a study that such questions can be answered. For that reason alone, I would tend to be supportive of the trial going ahead, though with reservations. I would expect that, should the results begin to show a greater risk to patients, that the study would be stopped. Sorry if these thoughts are not particularly helpful.
-
I think it would be useful to proceed with the trial if the confidence range is increased from 10% to 15%. Switching to oral treatment earlier will allow the patient to go home earlier and this in itself could aid recovery (psychological benefit) and reduce the risk of infection (less exposure to a wider population at home than in hospital). I would assume patients on oral treatment will be closely monitored at home with an identifiable easy/quick pathway for re-admission and i.v. antibiotics if necessary, as opposed to being processed through A&E. The patient’s home situation would be conducive to enabling close monitoring for indicators of infection occurring (maybe the presence of a carer?) and that there are no concerns regarding the home situation itself or quality of care at home. My feeling is that this study would be worthwhile.
-
Margaret, I have read this and think that, on balance, it IS worth while making the change and trying for the lower level but I honestly feel that there is really a decision for the researcher and the ethics committee rather than a PPI decision.
-
As you know I’m a fan of EASI-SWITCH, and believe that even with the increase in confidence range it is worth continuing the Trial.
-
Very difficult. Clearly good to reduce the amount of i.v. chemotherapy, but what would spook me is the, albeit remote, possibility of death (certainly an ‘undesirable outcome’), for the sake of cutting the stay in hospital by a day. But I can see that for someone with family responsibilities, the reduction in the time in hospital might outweigh that risk. I don’t think that the switch to 85% confidence, rather than 90%, would make any difference – but is it not possible to continue with the trial and as long as the risks are clearly explained, give patients the choice? If the take-up is low, then the trial would have to be abandoned.
-
I am not convinced that the gains to be had here are worth the trial. I would also want to see a health economic argument as re-admission to hospital may be more costly than reducing the initial stay by a day. But then this may be one of the trial outcomes. People who have sepsis are often very poorly for a long time and don’t always fully recover. I would want the treatment that’s going to give me the best chance of recovery.
Appendix 2 UK clinician survey
Opinion was sought from clinicians nationally regarding the proposed extension of the non-inferiority margin. The study co-investigators and site PIs were asked to disseminate the survey by e-mail throughout their clinical network and beyond as widely as possible. A paper survey of attendees at the Scottish Clinical Trials Showcase meeting was also undertaken. The supporting information provided by both e-mail and hard copy is included at the end of this appendix. Finally, as the study is part of the portfolio of the National Cancer Research Institute (NCRI) Colorectal Cancer Clinical Studies Group (CSG), support was also sought from the relevant subgroup of the CSG.
Overall, almost all clinicians surveyed were supportive of extending the non-inferiority margin and continuing the study. In total, responses were received from 38 consultants, 9 clinical fellows/specialty trainees and 2 RNs. All but one of the respondents were supportive of the proposed revision. The proposal was also discussed at the Adjuvant and Advanced Disease CSG Subgroup meeting on 16 January 2018. Membership of this group includes UK-wide representation from oncology and other disciplines (including a statistician/CTU director). This group was also supportive of this revision to study design and a letter of support from the Chair is also included.
An anonymised summary of the comments and respondents are included below:
-
Very happy with this approach – sensible ethical and appropriate.
-
An important question to try and answer, especially in current climate of bed shortage. No clinical risk. Support continuing with 15% NI margin.
-
I agree that the pragmatic solution is to expand the NI margin to 15%. A successful switch to oral therapy for 70% of low-risk patients would still be a valuable outcome.
-
This remains a critically important trial, the need for which was highlighted in the 2012 NICE neutropaenic sepsis guidelines. A potential failure rate of 30% in low-risk neutropenic fever is acceptable, given the safeguards built into the protocol.
-
As you know from the outset I did have concerns about this study recruiting as years ago as an SpR in Glasgow we failed to recruit well to the Liverpool-led ORANGE study (asking the same questions) – is early switch and discharge safe in low-risk neutropenic patients using the MASCC score. That study closed. I think the main two issues are: (1) we already make this switch in patients we perceive to be low risk (prob not using MASCC score though) to reduce inpatient stay. (2) There are data from the US studies showing this approach is safe. It is also part of ASCO guidelines 2013 and the infectious diseases society of US guidance 2011 that MASCC score low-risk patients should be managed as outpatients or discharged on orals after a brief inpatient stay. So I think its hard to randomise a patient to more i.v.s when you want to do early switch and discharge based on guidance. However what is a good thing about EASI-SWITCH is it will give us UK data and I think we should try to complete the study and if switching to smaller non-inferiority margin makes no.s needed less then I would support this and continue to look for patients.
-
Most appropriate option especially given the suggestions within the survey findings, this remains a relevant question and would still offer the potential to change practice dependent upon the final results.
-
As long as the trial is set up to show that (a) as long as sensible protocols are followed, death rates/morbidity is no higher in comparable groups and (b) bed days are saved overall I would support this change. The big picture shouldn’t be whether some end up needing a switch to i.v. antibiotics, but on whether bed days are saved/health economics is favourable.
-
I am worried about raising the bar of non-inferiority to 15% as the data interpretation shall become less meaningful and may lose its clinical impact. The other issue is high anxiety levels of clinicians in the past have led to failure of similar trials in low-risk neutropenic patients. If the consensus is accepting the 15% as standard I am happy to with it.
Appendix 3 Trial sites and recruitment
| Site | Site/trust name | Date opened | Number recruited (%) |
|---|---|---|---|
| 01 | Belfast Health and Social Care Trust | 17 February 2016 | 50 (38.8) |
| 02 | University Hospitals of Leicester | 14 March 2016 | 4 (3.1) |
| 03 | The Newcastle Upon Tyne Hospitals NHS Foundation Trust | 4 May 2016 | 15 (11.6) |
| 04 | Velindre NHS Trust | 31 March 2016 | 13 (10.1) |
| 05 | NHS Greater Glasgow and Clyde (Beatson) | 22 June 2017 | 2 (1.6) |
| 06 | Royal Marsden NHS Foundation Trust | 17 July 2017 | 5 (3.9) |
| 07 | South Eastern Health and Social Care Trust | 26 June 2017 | 5 (3.9) |
| 08 | Not allocated | n/a | n/a |
| 09 | Salisbury NHS Foundation Trust | 18 September 2017 | 9 (7.0) |
| 10 | Hampshire NHS Foundation Trust | 28 September 2017 | 1 (0.8) |
| 11 | Western Health and Social Care Trust | 13 November 2017 | 7 (5.4) |
| 12 | The Mid Yorkshire Hospitals NHS Trust | 18 January 2017 | 7 (5.4) |
| 13 | Heart of England NHS Foundation Trust | 5 December 2017 | 3 (2.3) |
| 14 | Derby Teaching Hospitals NHS Foundation Trust | 1 May 2018 | |
| 15 | Worcester Acute Hospitals NHS Trust | 25 September 2018 | 1 (0.8) |
| 16 | Poole Hospital NHS Foundation Trust | 1 November 2018 | 4 (3.1) |
| 17 | The Dudley Group NHS Foundation Trust | 1 November 2018 | |
| 18 | North Middlesex University Hospital NHS Trust | 18 July 2019 | 2 (1.6) |
| 19 | Whittington Health NHS Trust | 1 July 2019 | 1 (0.8) |
| 20 | Singleton Hospital Swansea | 15 July 2019 |
Appendix 4 Semistructured interview guide for EASI-SWITCH investigators regarding barriers to recruitment
Opening questions:
-
How have you found recruitment to the EASI-SWITCH trial?
-
Have you encountered any challenges in recruiting patients to the EASI-SWITCH trial?
Prompts:
-
Have there been any difficulties at your site with:
-
– screening and identifying suitable patients
-
– consenting and randomising patients successfully
-
– any aspects of the protocol or trial-related materials
-
– data management or follow-up
-
– local resources or infrastructure
-
– research or clinical team members
-
– communication with CTU?
-
Appendix 5 Reasons for study screening failure
Eight hundred and twenty-seven patients were screened for study entry in total, of whom 698 were excluded. The most common reason for exclusion was failure to meet the eligibility criteria (n = 581) and specific reasons for this are detailed below. Of the remaining patients, six were unable to provide informed consent and the remainder failed screening for other reasons (n = 111).
Screen failures based on specific eligibility criteria (n = 581):
-
patient 16 years or under (n = 1)
-
patient not receiving SACT for diagnosis of cancer (n = 4)
-
patient does not have a fever, that is, temperature > 38°C within 24 hours prior to randomisation (n = 38)
-
patient does not have either a temperature of at least 38°C or other sign or symptoms consistent with clinically significant sepsis, for example, hypothermia (n = 70)
-
patient does not have neutropenia, that is, absolute neutrophil count not ≤ 0.5 × 109/l within 24 hours prior to randomisation (during initial 6 month pilot; n = 15)
-
patient does not have an absolute neutrophil count ≤ 1.0 × 109/l, and falling or expected to fall, with a temperature of at least 38°C (following pause to recruitment; n = 301)
-
expected duration of neutropenia ≥ 7 days (n = 3)
-
patient has hypotension < 90 mmHg within 24 hours prior to randomisation (n = 11)
-
patient has hypotension (systolic pressure < 90 mmHg or reduction of > 40 mmHg from known baseline on > 1 measurement) within the 24 hours prior to randomisation (n = 5)
-
patient has received i.v. piperacillin/tazobactam or meropenem for more than 24 hours (n = 16)
-
patient was at risk of complications using MASCC score (i.e. MASCC score < 21; n = 28)
-
patient is not able to maintain adequate oral intake and take oral medication (n = 7)
-
patient does not have adequate hepatic (AST and/or ALT ≥ 5 × ULN) and renal function (serum creatinine ≥ 3 × ULN) within 24 hours prior to randomisation (n = 7)
-
patient’s physician in charge of care is not willing to follow either the intervention or standard care protocol per randomisation, at enrolment, including not treating with CSF. Prophylactic use of CSF is not an exclusion criterion if prescribed routinely as an integral component of a specific SACT regimen (n = 25)
-
patient has an underlying diagnosis of acute leukaemia (n = 38)
-
patient has an underlying diagnosis of haematopoietic stem cell transplant (n = 3)
-
patient has been enrolled in this trial with a prior episode of NS (n = 2)
-
patient has prior allergy, serious AR or contraindication to any study drug (n = 22)
-
patient is a pregnant woman (n = 1)
-
patient has received treatment with fluoroquinolone or penicillin in preceding 14 days. *Question no longer asked in non-randomisation (n = 7)
-
patient has localising signs of severe infection (pneumonia, soft-tissue infection, central-venous access device infection, presence of purulent collection; n = 15). 72,73
Appendix 6 Unit costs of hospital services and study drugs
Unit costs from the NHS Schedule of Reference Costs 2018–19,56 PSSRU Unit Costs of Health and Social Care57 and British National Formulary (BNF)58 were combined with the individual-level resource use to estimate a total hospital cost for each trial participant.
| Resource item | Unit cost (£) | Source | Details |
|---|---|---|---|
| Ward bed-day | 478.25 | Department of Health NHS Costs 2018/19 | Weighted average length of stay and cost of non-elective long stays (WJ06A–WJ06J) |
| Critical care bed-day | 933.19 | Department of Health NHS Costs 2018/19 | Adult Critical Care (XC06Z) |
| Outpatient attendance | 135 | PSSRU Unit Costs 2019 | |
| Emergency department | 196 | PSSRU Unit Costs 2019 | See, treat and refer |
| Piperacillin/tazobactam (2 g/250 mg) | 7.65 | BNF Drug Tariff March 2020 | Powder for solution for infusion vials (pack of one) |
| Meropenem (1 g) | 17.78 | BNF Drug Tariff March 2020 | Powder for solution for infusion vials. Cost per unit based on a pack of 10 (£177.80/10 = 17.78) |
| Ciprofloxacin (750 mg) | 0.80 | BNF Drug Tariff March 2020 (£8.00/10) | Cost per 750 mg tablet based on a pack of 10 (£8.00/10 = £0.80) |
| Co-amoxiclav (500 mg/125 mg) | 0.12 | BNF Drug Tariff March 2020 (£2.50/21) | Cost per 500 mg/125 mg tablet based on a pack of 21 (£2.50/21 = £0.12) |
Appendix 7 Patient preference questionnaire
Patient Follow-up Questionnaire

Interviewer
You were recently admitted to hospital with NS – an infection you developed during your anticancer treatment. You kindly agreed to take part in the EASI-SWITCH trial. We would now like to ask you some questions to find out what you think about the method of antibiotic treatment you received and also find out your hypothetical preferences for future antibiotic treatment should you develop NS again. The information you give us will be confidential and will only be used for the EASI-SWITCH trial. Your answers will not influence the health care you are receiving now or any health care you might receive in the future.
I am now going to read out some statements to you about the most recent treatment for NS you received. For each one, I would like you say whether you AGREE or DISAGREE with the statement, or if you are UNCERTAIN about it. Please think about each one carefully.
Please read out each statement and ask the respondent ‘Do you Agree or Disagree with this statement or are you UNCERTAIN?’
| Agree | Disagree | Uncertain | |
|---|---|---|---|
| 1. I am satisfied with the medical care I received for NS | ❑ | ❑ | ❑ |
| 2. I am satisfied with the level of support I received from the hospital | ❑ | ❑ | ❑ |
| 3. I believe that the antibiotic treatment I received was effective | ❑ | ❑ | ❑ |
| 4. I am happy with the place I received my antibiotic treatment | ❑ | ❑ | ❑ |
| 5. Before I was treated, I was concerned that the antibiotic treatment would not work | ❑ | ❑ | ❑ |
| 6. Before I was treated, I was concerned about how the antibiotic treatment I received would affect my family/friends | ❑ | ❑ | ❑ |
When you agreed to take part in the EASI-SWITCH trial you were told about the possible advantages and disadvantages of each method of antibiotic treatment. Just to remind you of them – when patients are switched early from i.v. to oral antibiotics, they may be discharged from hospital 1 to 2 days earlier than those who continue on i.v. antibiotics. However, there is also a small risk that their infection may not clear up as quickly as those who continue on i.v. antibiotics. As a result, they may need to be re-admitted to hospital for further i.v. antibiotics.
Please keep this information in mind as I read out some statements about any future treatment for NS you may need. Please remember, your responses will not influence the treatment you may actually receive. Like before, I would like you to say whether you AGREE or DISAGREE with the statement, or if you are UNCERTAIN about it. Please think about each one carefully.
Please read out each statement and ask the respondent ‘Do you Agree or Disagree with this statement or are you UNCERTAIN?’
| Agree | Disagree | Uncertain | |
|---|---|---|---|
| 7. It would be important to me to be discharged 1–2 days early | ❑ | ❑ | ❑ |
| 8. It would be important to me that I was not re-admitted to hospital for further treatment | ❑ | ❑ | ❑ |
| 9. I would accept the small risk of re-admission if it meant I was discharged 1–2 days early | ❑ | ❑ | ❑ |
10. If patients were to be offered a choice about which way they could be treated for NS in future, which antibiotic treatment would you choose?
-
❑ Intravenously in hospital for at least 48 hours and then switching to oral antibiotics if necessary
-
❑ Intravenously in hospital for 12–24 hours and then switching to oral antibiotics
11. Can you tell us why you would choose this method?
-
————————————————
-
————————————————
-
————————————————
There is a small risk of hospital re-admission for patients who have i.v. antibiotics (1–2 patients for every 10 treated). The risk of hospital re-admission for patients who are switched early may be the same as this or slightly higher but we will not know what this risk level really is until the EASI-SWITCH trial is finished.
12. If the risk of re-admission was three in every 10 patients switched early (30%) instead of 1–2 patients (15%) would you choose to be discharged home on oral antibiotics 1–2 days sooner?
-
❑ Yes
-
❑ No
Finally, the last two questions will help us to understand the care situation in your home.
13. Do you receive care in your own home on a daily basis?
-
❑ Yes
-
❑ No
14. Do you have children or dependents that you care for?
-
❑ Yes
-
❑ No
Thank you very much for taking the time to answer these questions for us today.
Please write down in the box below any feedback which will help us improve this questionnaire. E.g. did the respondent find any question difficult to understand? Did they question why they were being asked the information?
——————————————
——————————————
——————————————
Appendix 8 Health economics analyses using all available data
| Variable | Intervention | Standard care | Difference (95% CI) | ||
|---|---|---|---|---|---|
| n | Mean (95% CI) | n | Mean (95% CI) | ||
| EQ-5D-5L utilities | |||||
| Baseline | 59 | 0.77 (0.72 to 0.83) | 62 | 0.74 (0.68 to 0.79) | 0.04 (−0.04 to 0.11) |
| 14 days | 54 | 0.81 (0.76 to 0.86) | 59 | 0.77 (0.72 to 0.83) | 0.04 (−0.04 to 0.11) |
| EQ-VAS | |||||
| Baseline | 59 | 64.14 (58.59 to 69.68) | 62 | 58.95 (53.82 to 64.08) | 5.18 (−2.28 to 12.65) |
| 14 days | 56 | 74.70 (69.71 to 79.69) | 59 | 70.32 (65.74 to 74.91) | 4.37 (−2.32 to 11.07) |
| QALYs | 52 | 0.03 (0.03 to 0.03) | 58 | 0.03 (0.03 to 0.03) | < 0.00 (−0.00 to 0.00) |
| Service | Intervention (n = 62) | Standard care (n = 64) | Mean difference (95% CI) | ||
|---|---|---|---|---|---|
| n (%) | Mean (95% CI) | n (%) | Mean (95% CI) | ||
| Primary admission | |||||
| Ward days | 62 (100.00) | 2.73 (2.14 to 3.31) | 64 (100.00) | 3.17 (2.73 to 3.61) | −0.45 (−1.17 to 0.27) |
| Critical care days | 1 (1.61) | 0.15 (−0.15 to 0.44) | 0 (0) | n/a | 0.15 (−0.14 to 0.43) |
| Emergency department | 2 (3.23) | 0.03 (−0.01 to 0.08) | 2 (3.13) | 0.03 (−0.01 to 0.08) | 0.00 (−0.06 to 0.06) |
| Re-admission | |||||
| Ward days | 4 (6.45) | 0.39 (−0.11 to 0.89) | 2 (3.13) | 0.17 (−0.07 to 0.41) | 0.22 (−0.33 to 0.76) |
| Emergency department visits | 4 (6.45) | 0.06 (0.00 to 0.13) | 2 (3.13) | 0.03 (−0.01 to 0.08) | 0.03 (−0.04 to 0.11) |
| Outpatient visits | 2 (3.23) | 0.03 (−0.01 to 0.08) | 2 (3.13) | 0.03 (−0.01 to 0.08) | 0.00 (−0.06 to 0.06) |
| Intervention (n = 62) | Standard care (n = 64) | Mean difference (95% CI) | |
|---|---|---|---|
| Mean cost £ (95% CI) | Mean cost £ (95% CI) | ||
| Primary admission | |||
| Ward days | 1303.62 (1023.56 to 1583.67) | 1516.95 (1307.80 to 1726.10) | −213.33 (−557.91 to 131.24) |
| Critical care days | 135.46 (−135.41 to 406.34) | 0 | −135.46 (−128.40 to 399.33) |
| Emergency department | 6.32 (−2.54 to 15.19) | 6.13 (−2.46 to 14.71) | 0.20 (−12.02 to 12.41) |
| Re-admission | |||
| Ward days | 185.13 (−53.11 to 423.37) | 82.20 (−33.52 to 197.91) | 102.93 (−156.72 to 362.58) |
| Emergency department visits | 12.65 (0.32 to 24.97) | 6.13 (−2.46 to 14.71) | 6.52 (−8.27 to 21.31) |
| Outpatient visits | 4.35 (−1.75 to 10.46) | 4.22 (−1.70 to 10.13) | 0.14 (−8.28 to 8.55) |
| Medication | |||
| Study drug | 51.09 (43.66 to 58.52) | 80.81 (67.45 to 94.16) | −29.72 (−44.98 to −14.45) |
| Concomitant medication | 19.64 (3.41 to 35.87) | 40.26 (7.84 to 72.68) | −20.62 (−56.88 to 15.64) |
| Total | 1718.26 (1254.92 to 2181.60) | 1736.68 (1466.92 to 2006.44) | −18.42 (−545.13 to 508.28) |
List of abbreviations
- (G)CSF
- (granulocyte) colony-stimulating factor
- AE
- adverse event
- ALT
- alanine aminotransferase
- AR
- adverse reaction
- ASCO
- American Society of Clinical Oncology
- AST
- aspartate aminotransferase
- BHSCT
- Belfast Health and Social Care Trust
- CEA
- cost-effectiveness analysis
- CEAC
- cost-effectiveness acceptability curves
- CI
- confidence interval
- CISNE
- clinical index of stable febrile neutropenia
- CRF
- case report form
- CUA
- cost–utility analysis
- DMEC
- data monitoring and ethics committee
- EQ-5D-5L
- EuroQoL-5 Dimensions, five-level version
- ESMO
- European Society for Medical Oncology
- GCP
- good clinical practice
- GDG
- guideline development group
- GEE
- generalised estimating equations
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- IMP
- investigational medicinal product
- ITT
- intention-to-treat
- i.v.
- intravenous
- MASCC
- Multinational Association for Supportive Care in Cancer
- NICE
- National Institute for Health and Care Excellence
- NICTU
- Northern Ireland Clinical Trials Unit
- NIHR
- National Institute for Health and Care Research
- NMB
- net monetary benefit
- NS
- neutropenic sepsis
- PI
- principal investigator
- PP
- per-protocol
- PPI
- patient and public involvement
- QALY
- quality-adjusted life-year
- R&D
- research and development
- RD
- risk difference
- RN
- research nurse
- SACT
- systemic anticancer therapy
- SAE
- serious adverse event
- SAR
- serious adverse reaction
- SUSAR
- suspected unexpected serious adverse reaction
- SPC
- summary of product characteristics
- TMG
- trial management group
- TSC
- trial steering committee
- UKONS
- United Kingdom Oncology Nursing Society
- ULN
- upper limit of normal
- WTP
- willingness-to-pay