
Notes
Article history
The research reported in this issue of the journal was funded by PGfAR as project number RP-PG-0707-10101. The contractual start date was in January 2009. The final report began editorial review in February 2016 and was accepted for publication in June 2017. As the funder, the PGfAR programme agreed the research questions and study designs in advance with the investigators. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The PGfAR editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Peter J Selby has an international patent (application no. PCT/GB2014/050768; biomarker; University of Leeds) issued. Rosamonde E Banks reports grants from various non-commercial funding sources during the conduct of the study; in addition, she has a international patent (application no. PCT/GB2014/050768; biomarker; University of Leeds) issued. Jenny Hewison is a panel member of the National Institute for Health Research (NIHR) Clinical Trials Unit Standing Advisory Committee. Grant D Stewart has a patent (1408091.5) pending to the Medical Research Council (MRC) Human Genetics Unit (HGU). James Neuberger is a consultant to Astellas Pharma, Inc., and has received speaker support funding from Astellas and Novartis UK. Claire Hulme has received grants from the NIHR during the conduct of the study and is a panel member of the Health Technology Assessment (HTA) Commissioning Board. Andrew Lewington has received personal fees from Fresenius Medical Care, Baxter Healthcare Ltd, AM-Pharma, BioPorto Diagnostics A/S and GE Healthcare, outside the submitted work; in addition, he has a patent (ACY-1 biomarker) pending. Naveen S Vasudev has received personal fees from Bristol-Myers Squibb and GlaxoSmithKline plc, outside the submitted work. William Rosenberg was an inventor of the enhanced liver fibrosis (ELF) test when he was an employee of the University of Southampton. His rights were transferred to Siemens Healthcare Diagnostics Ltd by the University of Southampton. He does not receive any payment in relation to sales of the test by the manufacturer Siemens Helathcare Diagnostics. He has received grant support and speaker fees from Siemens Healthcare Diagnostics and is a director of iQur Ltd (Southampton, UK), a company that provides ELF testing. In the context of this NIHR-funded study, all ELF testing provided by iQur Ltd was performed on a not-for-profit, cost-recovery basis. William Rosenberg is married to Julie Parkes, who is a co-investgator on the programme. Julie Parkes reports receiving support from the Speakers’ Bureau for Siemens Healthcare Diagnostics, outside the submitted work, and is married to William Rosenberg (co-applicant). Jonathan J Deeks is a panel member of the HTA Commissioning Board.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Selby et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introducing new biomarkers for renal and hepatic diseases into health-care systems
Background
Protein biomarkers in body fluids are now being regularly identified using new techniques and are associated with the presence and activity of diseases and treatment benefits/toxicities. They are accessible, measurable in real time and inexpensive to test for. However, their potential benefit to the NHS is not being realised because of the absence of a defined pathway linking biomarker research to health services research. This programme aimed to establish a process for the stringent evaluation of promising biomarkers, encompassing methodological developments, clinical evaluation and a randomised controlled trial (RCT) to enable assessment of their impact on clinical outcomes, the process of care, resource use and service configurations.
Our specific objectives were to:
-
evaluate and develop methodology for the optimal use of biomarkers for disease monitoring and to optimise benefits, for patients and the NHS
-
establish a sample and clinical data bank together with a robust system for the evaluation of promising markers to facilitate their rapid assessment prior to large-scale trialling in the NHS
-
conduct a RCT of an established panel of biomarkers of potential value in chronic liver disease (CLD) to diagnose cirrhosis at an early stage when interventions may reduce dangerous complications and to determine patient and NHS benefits.
Biomarkers have major potential benefits for patients and the NHS, particularly in contributing to ‘personalised’ and/or ‘stratified’ medicine and improved safety. They may supplement or replace invasive procedures or imaging tests for:
-
accurate and early diagnosis
-
measurement of the activity and extent of disease
-
indication of prognosis
-
selection and prediction of optimal treatments
-
monitoring for treatment response/toxicity or disease progression.
In addition, biomarker information may inform patient counselling on lifestyle issues (e.g. alcohol avoidance or diet) and motivate patients towards healthy alternatives. Overall, better biomarkers should lead to improvements in outcomes and more efficient, cost-effective and evidence-based use of NHS resources.
With ongoing technological developments, particularly in proteomics, the rate of identification of potential new biomarkers may be expected to increase. Various stages in the ‘biomarker pipeline’ have been defined, but the translational work needed to progress through these – involving technology transfer, methodological considerations and aspirations of different stakeholders – presents challenges. The lack of a clear evaluative infrastructure means that the route from stringent evaluation and/or validation to clinical implementation and then to evaluation of impact on outcomes and health care is not yet established, representing a major threat to achieving full patient benefit. 1–9 As an indication, the number of new protein markers approved by the US Food and Drug Administration (FDA) has gradually declined, with only 10 approved in the period from 1994 to 2002, only two of which were approved in 1998–2002. 10 Encouragingly, the need for national strategies for the rapid evaluation and introduction of new biomarker tests is now better appreciated, for example by the National Institutes of Health (NIH) in the USA11 and by the Royal College of Pathologists in the UK. 12
A framework is, therefore, required for the pipeline to justify and guide the introduction of biomarkers, including specification and establishment of the infrastructure to acquire such evidence, appropriate assessment of test results, identification of whether or not RCTs are required and the means of deciding when and how biomarker development and introduction should be accelerated.
Selected diseases
Chronic liver disease and renal diseases provided ideal subjects with which to work up such a framework. Liver and renal diseases generate huge and increasing burdens on patients and the NHS. The care of patients would be transformed with improved outcomes, more appropriate use of complex and expensive therapies and avoidance of expensive and invasive investigations if biomarkers of real health-care value could be found.
Chronic liver disease
Chronic liver disease is the fifth most common cause of death in the UK and the second most common cause of death in men aged 35–54 years. It is usually associated with alcoholic liver disease, fatty liver disease or hepatitis C infection, any of which may lead to fibrosis, cirrhosis and hepatocellular carcinoma (HCC). 13–15 Life-threatening complications include variceal bleeding, recurrent ascites and hepatic encephalopathy. Once cirrhosis has developed, HCC arises in 1–6% of patients per annum. 16,17 Social issues such as an inability to work constitute a huge health-care and financial burden. Evidence shows that earlier cirrhosis detection results in better survival and reduced morbidity rates.
Acute renal transplant
Currently, in the UK there are 23,000 patients with functioning transplants (see Chapter 10). Annually, almost 3080 renal transplants are performed. Transplantation represents the best therapy for improving survival and quality of life and is the most cost-effective, saving the NHS > £490M per year compared with dialysis. Acute rejection (AR; 25% of patients) and delayed graft function (DGF; 40% of patients) significantly reduce short- and long-term graft survival. Early diagnosis of AR/DGF is critical for optimal treatment. The biomarker serum creatinine is slow to respond and is insensitive. Currently, renal biopsy is required for a definitive diagnosis, which is invasive and may not be available immediately. 18–24 Biomarkers allowing the earlier diagnosis of DGF and AR and discrimination of subgroups, a strategic priority of the American Society of Nephrology,25 would allow earlier and more appropriate therapeutic intervention.
Renal cell carcinoma
Accounting for approximately 3% of adult malignancies, the incidence of renal cell carcinoma (RCC) is increasing, with approximately 330,000 new cases each year worldwide and 10,000 new cases each year in the UK and > 140,000 deaths worldwide (see Chapter 10). 26,27 Locally advanced or metastatic disease affects > 50% of patients, for which treatments are limited. New drugs have improved response rates and relapse-free survival (RFS) but they are expensive and markers for the diagnosis, prognosis and selection of expensive therapy are desperately needed. 28–31
Protein biomarkers
Protein biomarkers in body fluids have substantial potential to improve the quality of health care. There are active pipelines identifying them in both the commercial and the non-commercial sectors, but robust methodological approaches and well-organised rapid clinical and health service evaluation is still limited.
In a clinical setting, the value of a protein biomarker depends on test performance and its relation to health improvements. Methods of reporting, and hence judging, test performance are well developed for biomarkers and other kinds of measures when these are used in a prognostic role and in a diagnostic role. 32,33 The methodology for evaluating biomarker test performance when used in an individual patient monitoring role is, however, poorly developed.
After the initial discovery and preliminary evaluation of a protein biomarker, robust evaluation of its clinical characteristics [sensitivity, specificity, receiver operating characteristic (ROC), etc.] is often performed slowly and in limited sample numbers. We set out to establish a robust system using samples and clinical data in adequate numbers to rapidly evaluate markers that may be useful in the NHS and select those that justify formal evaluation.
Chronic liver disease can frequently progress to cirrhosis and to life-threatening complications. Early diagnosis of cirrhosis with appropriate management can reduce the incidence of these complications, and a panel of protein biomarkers that directly evaluate fibrosis has excellent clinical characteristics [ADVIA Centaur® Enhanced Liver Fibrosis (ELF) test; Siemens Healthcare Diagnostics Ltd, Camberley, UK]. We proposed and have conducted a RCT to establish whether or not its use can substantially improve patient outcomes and health-care provision. Previous work on the ELF test shows that it has a proven clinical association with cirrhosis and its complications. This provides justification for its evaluation to see whether or not it can be used to monitor patients and allow the diagnosis of cirrhosis at a time when interventions will reduce the morbidity and mortality associated with these complications. 34–37
Monitoring studies
Considerable research has been carried out into the use of biomarkers for prognosis (including prediction of response to treatment). In contrast, there is a relatively small amount of literature on the use of biomarkers for monitoring. 38,39 Methodological work has not yet been conducted on the design and interpretation of studies with repeated measurements of biomarkers, and we used value of information analysis in this context.
Monitoring may be undertaken for various purposes. In chronic diseases, it may assess whether or not interventions are keeping the disease and symptoms under control; assess the rate of progression of disease (e.g. the ELF test in our RCT); detect recurrence of disease; or evaluate the efficacy of treatments. Monitoring may also allow adverse effects to be avoided. A strategy is required for the frequency of testing and rules for clinical actions (including retesting). Although each test result can be judged on its own, there is the potential also to learn from the change since the previous test or the rate of change over time or to devise and calibrate a model of change over time. Rules may be devised using suitable cut-off points, changes between values or values of fitted model parameters or confidence intervals (CIs) thereof. In addition, it will be important to understand natural variability because of measurement error, prognostic factor-related trends (e.g. in age) and the risks of incorrect decisions.
Few biomarkers have been studied in this context. Examples in cancer include cancer antigen 125 (CA-125) for monitoring patients with ovarian cancer for recurrence and prostate-specific antigen (PSA) for monitoring men at risk of prostate cancer and for monitoring prostate cancer for recurrence. 40,41 Serum creatinine measurements are used to monitor patients with chronic kidney disease (CKD) and transplants. An important question for monitoring by repeated measurement is whether prognosis relates to the actual marker level or the change over time or more complex models. 42
Two different questions need to be considered, relating first to the interpretation of specific marker values and sequences of such values, and second to the implementation of such information, for example to determine how frequently measurements should be taken. There is a need for large, high-quality data sets with repeated measurements to inform the development of decision rules for monitoring.
Choice of the Enhanced Liver Fibrosis test for the ELUCIDATE randomised controlled trial
Within our range of liver and renal diseases, we identified the ELF test as having ideal characteristics for our RCT because:
-
the ELF test has convincing clinical evaluation data showing an association with cirrhosis (e.g. ROC of > 0.8) but the clinical and health service benefits from monitoring CLD for cirrhosis are untested
-
the burden of CLD and cirrhosis on patients and the NHS is huge and increasing
-
early diagnosis of cirrhosis allows effective surveillance and the use of interventions to improve clinical outcomes and care
-
a simple blood test could radically improve and provide cost-effective care for CLD patients
-
this model will provide an excellent prototype for health service biomarker research and vital data sets for our methodologists.
In the vast majority of cases, liver fibrosis is asymptomatic and cirrhosis develops insidiously with non-specific symptoms, so that opportunities for disease modification or cure are missed. Standard biochemical tests of liver function are not specific or sensitive. Liver biopsy is hazardous and inaccurate and subject to sampling error and variation in interpretation. 43–45 Imaging has a major role in the detection and assessment of liver fibrosis. However, all imaging modalities, including ultrasound, elastography and cross-sectional imaging with X-rays or magnetic resonance, require access to expensive technology and skilled operators. 46,47
With cirrhotic diseases of the liver, we are in the relatively fortunate position of having a number of treatments (such as beta-blocker therapy or surgery for low-volume HCC) that are known to be effective at reducing complications, if the cirrhotic condition is detected early enough. The ELF test seeks to identify a ‘pool’ of patients with a slowly progressing disorder who can be treated prophylactically and in whom the incidence of severe complications can be reduced.
Evidence shows that the early detection of varices and treatment with prophylactic use of beta-blockers to reduce portal hypertension or band ligation reduces morbidity and increases survival, and respected guidelines recommend surveillance because of its benefits and health economic justification. 48–52 Similarly, the early detection of ascites and treatment has been shown to reduce the morbidity associated with bacterial peritonitis from 17% to 2%. 53 The case for surveillance and early detection of HCC is more contentious, with some RCTs showing evidence of benefit and others showing none. International guidelines now advocate surveillance. 54–56 Retrospective analyses have identified criteria, essentially the presence of small tumours, that are associated with better outcomes of HCC resection and liver transplantation, but many patients are diagnosed after the growth of their tumours has ruled them out for curative resection or transplantation. 57,58
Studies and systematic reviews have demonstrated that single direct markers are less accurate than panels of markers for the detection of liver fibrosis. 59,60 One such panel of direct markers is the ELF test, the only CE (Conformité Européene)-marked [European Union (EU) regulatory approval] test for liver fibrosis measuring constituents of liver matrix [hyaluronic acid (HA) and procollagen III amino-terminal peptide (PIIINP)] and a molecule critical to the regulation of matrix remodelling [tissue inhibitor of metalloproteinase 1 (TIMP-1)] using sensitive automated enzyme-linked immunosorbent assays (ELISAs) designed and manufactured specifically for this purpose. 61 The three individual biomarkers were selected as being optimal from among 20 candidates. The results of the individual assays are combined in an algorithm that was derived and validated in > 1000 cases of liver fibrosis to generate a score that correlates with the severity of liver fibrosis on liver biopsy. ELF values have been shown to be highly predictive of clinical outcomes, including variceal bleeding, ascites, HCC and mortality. Subsequent validation studies in hepatitis C, fatty liver disease, human immunodeficiency virus (HIV)–hepatitis C virus (HCV) co-infection and primary biliary cirrhosis have confirmed the performance of the test. 34–37 Although performance is best in the detection of advanced fibrosis and cirrhosis, the test can also detect mild and moderate degrees of fibrosis accurately, with area under the curve (AUC) ROCs of 0.83 for Ishak fibrosis stages of 0–3 compared with 4–6 and 0.86 for Ishak fibrosis stages of 0–4 compared with 5–6. The ELF test is excellent at detecting advanced fibrosis/cirrhosis in a range of CLDs and is, thus, well suited for use in screening populations at risk for cirrhosis. The ELF test has been developed by Siemens Healthcare Diagnostics Ltd (formerly Bayer Healthcare) in conjunction with the University of Southampton and iQur Ltd (Southampton, UK).
The overall shape of this programme of work is provided in Figure 1.
FIGURE 1.
The programme of work. NEQAS, National External Quality Assessment Service.

Chapter 2 Introduction to the methodology workstream (workstream 1)
In this chapter we introduce the methodology workstream on monitoring tests, described in Chapters 3–9.
Monitoring is the repeated application of a test, or set of tests, over time to assist in the management of a disease or condition. It is a fundamental element of patient care, comprising much of the clinical workload. 62 Often thought of in terms of treatment titration and maintenance, in which the aim is to keep a marker within predefined limits until treatment can be discontinued or an alternative treatment is required, monitoring is also used to manage individuals with a disease or condition that is likely to progress or recur at some time in the future, allowing timely decisions to be made regarding patient management. Patients are usually asymptomatic or mildly symptomatic but not yet receiving treatment, or they may experience symptoms of a disease that puts them at risk of developing other conditions. Monitoring often involves a general clinical assessment and physical examination of patients but is likely to include the application of specific tests, from tools assessing functional or psychological status, to blood or urine tests, physiological measurements such as blood pressure, imaging tests or more invasive assessments such as colonoscopy or biopsy. Although subsequent chapters of this report focus on protein biomarkers in particular, the methodological considerations that underlie the development and evaluation of monitoring strategies are relatively universal and can be applied regardless of type of test.
Historically, methodological research around test evaluation has lagged behind methodological research around interventions. Over the last 10–15 years, however, considerable research effort has focused on identifying optimal methods for establishing diagnostic test accuracy and, more recently, on the evaluation of diagnostic tests and strategies in terms of their impact on patient management and outcomes. 63,64 Monitoring tests, particularly in terms of treatment titration, are now beginning to receive attention in the literature. 38,65 Our particular interest is in monitoring patients for disease progression or recurrence.
The development and evaluation of tests for monitoring purposes bears many resemblances to the development and evaluation of diagnostic tests but with a few key differences. 62,66,67 First, unlike tests for diagnosis, tests for monitoring often do not aim to detect present disease but rather some marker of preclinical or early-stage disease that precedes the development of clinical disease progression or recurrence. Vitally, this latent stage of disease must be of a reasonable duration to make the repeated application of a monitoring test worthwhile – too short and recurrence or progression may be missed regardless of the monitoring schedule adopted, too long and frequent monitoring may not be of clinical benefit. Second, although diagnostic tests are often applied once, perhaps with a repeat application to confirm diagnosis, by the very nature of monitoring, monitoring tests are applied repeatedly over an indefinite period of time and according to some predetermined schedule. With patients’ true disease status often not established until clinical disease progression occurs, a cross-sectional evaluation of the diagnostic accuracy of a test can be impossible to establish and more longitudinal measures to capture how well a test predicts clinical outcome have been suggested. 65 The same principle of detecting true disease (recurrence and progression) while limiting false-positive results applies, with the further consideration that the test should be able to differentiate long-term change in disease status from short-term measurement variability. The further in advance of the clinical event of interest the marker is measured, the less predictive it may be and the greater the potential influence from measurement variability on false-positive and false-negative results. More complex decision rules to determine the point at which some clinical action should be taken may also be relevant for monitoring. Although each test result can be judged on its own, as in a diagnostic context, serial values over time may provide valuable information. Rules may be devised using individual thresholds, the change in measurement since the previous test, the rate of change in measurement values, values of fitted model parameters or CIs thereof. Finally, whereas a diagnostic test may be applied to assist in the ruling in or out of a number of differential diagnoses and any number of therapeutic approaches may be indicated, a positive monitoring test often only increases the probability of a particular future clinical event and, furthermore, a series of further investigations may be initiated before a particular treatment approach is applied.
Many of these considerations are particularly true of protein biomarkers, for which changes in biomarker levels may occur before any clinical symptoms or signs become apparent. Measuring biomarkers in blood or urine is relatively simple and safe for patients, making them an attractive alternative or complement to more complex, invasive or expensive tests. However, initially promising results at the biomarker identification stage do not necessarily translate into clinical benefit in practice.
Our aim was to identify and describe general methodological considerations for the development and evaluation of testing strategies to monitor for disease progression or recurrence, reviewing current best practice and exploring methodological innovations.
Chapters 3–9 all address aspects of the evaluation of monitoring biomarkers, but they do so from different starting points, depending on the amount and nature of the relevant literature being used as the starting point for the work. Each chapter, therefore, begins with a brief introduction, presenting the background elements most relevant to explaining the work to be reported. This approach has created a degree of repetition, but does enable the chapters to be read separately without the need for extensive cross-referencing to material presented elsewhere in the report. The chapters do also show some stylistic differences, reflecting in part the nature of the work reported, but also the discipline background of their lead authors.
Chapter 3 reviews monitoring strategies recommended in available clinical guidelines, with specific reference to the use of PSA for the detection of recurrent prostate cancer. PSA was chosen because of the extensive literature surrounding it and, in the event, there was so much material to consider, and so many generalisable considerations were emerging, we decided to focus entirely on PSA rather than pursue our original plan to add a number of ‘mini’ case studies from other clinical areas. The particular focus was on the degree of consistency between guidelines, the explicit consideration of factors important for specifying a monitoring strategy and the use of supporting evidence to justify any recommendations.
Ultimately, monitoring strategies are employed to allow timely decisions to be made regarding patient management, thereby improving patient outcomes, for example through earlier initiation of treatment to prevent or delay some clinical outcome. The RCT design is considered to be the gold standard approach to the evaluation of patient benefit from therapeutic interventions; however, testing strategies are complex interventions with many components, with their evaluation presenting considerable challenges. Chapter 4 reports a methodological review of RCTs of monitoring strategies to consider how successfully the design has been used to identify patient benefit from monitoring.
The methodological research is reviewed in Chapter 5. Although it is generally acknowledged that methodological work around monitoring tests has been lacking, there are areas of research that could be used or adapted for the development and evaluation of monitoring strategies for monitoring for disease progression or recurrence.
Chapter 6 focuses on the wider impact of monitoring on patients. Ferrante di Ruffano et al. 68 have produced a framework to assist those designing and evaluating trials of diagnostic tests to understand the ways in which changes to testing strategies can affect patient outcomes. We have adapted this framework to tests for monitoring, in light of our review of randomised trials. In this chapter we consider the potential for benefit and harm from monitoring in broad terms, before considering the ways in which patient outcomes can be mediated by particular aspects of the monitoring care pathway, noting the similarities and differences between diagnostic and monitoring tests.
Chapter 7 considers how simulation modelling can be used to identify optimal monitoring strategies, prior to or alongside a randomised trial. Simulation offers a powerful tool to design and evaluate monitoring rules. However, such models are data intensive, requiring many pieces of information to allow their construction. For many tests and diseases, limitations in the available data may affect the reliability of the final model. We explored how information obtained during an ongoing study [the ELF to Uncover Cirrhosis as an Indication for Diagnosis and Action for Treatable Event (ELUCIDATE) trial] could be incorporated into a simulation model of the ELF biomarker panel for monitoring patients with known liver fibrosis. The aim was to optimise monitoring rules to allow earlier detection of liver cirrhosis and to consider whether or not any resulting adaptations to the design of the ongoing study that were suggested by the model could be implemented without compromising the validity or clinical value of the trial.
Chapter 8 takes a health economic approach, modelling a method of optimising a monitoring test to meet a cost-effectiveness target and exploring the feasibility of using value of information analysis to inform biomarker research and development (R&D).
The final chapter in workstream 1, Chapter 9, brings together our findings and reports on a consultation with patient and public representatives, considering what we know from current practice in monitoring for disease progression and recurrence, what we have learned in terms of understanding the monitoring process and how this should inform the future development and evaluation of monitoring strategies.
Chapter 3 How is evidence being used to make recommendations about monitoring?: the example of prostate-specific antigen
The work described in this chapter has been published in Dinnes et al. 69
Introduction
Monitoring involves the scheduled, repeated use of a test or tests in an individual over time to make decisions about the management of a disease or condition. It is a central activity in the management of patients, making up a considerable part of the clinical workload and associated cost. 62 In contrast, the volume of published literature on the evaluation and use of tests for monitoring purposes is relatively small.
Mant66 and others62,67 have provided a framework for developing and evaluating a monitoring strategy. The framework has four main steps: (1) deciding whether or not to monitor, (2) choosing a test, (3) specifying and assessing the monitoring strategy to be used and (4) an implementation phase. Underlying this is the key concept that the ‘signal’ from the test, reflecting the status of the underlying condition, should be greater than the surrounding ‘noise’, or measurement variability, that may affect test interpretation. 66,67 If the ‘noise’ around a test measurement is too high in relation to the signal, one’s certainty in a given test result will be considerably reduced.
The repeated measurement of PSA in men who have undergone primary treatment of prostate cancer is an apparently successful example of a rule-based monitoring strategy. The behaviour of PSA following radical treatment varies, but, in general, recurrence of disease is associated with the presence of PSA (following radical prostatectomy) or some rise in [following radical radiotherapy (RTX)] PSA level. 70 When a predefined level of PSA is reached, biochemical failure is said to have occurred. The usefulness of PSA as a monitoring test is based on the assumption that biochemical failure predates clinical failure within some clinically meaningful time frame. The decision to initiate treatment for recurrence, however, will depend on multiple factors (M-Factors) rather than on a single PSA value alone. 71
We undertook a review of clinical guidelines on monitoring with PSA testing for the detection of recurrent prostate cancer to determine the extent to which they take into account key factors that should inform rule-based strategies for monitoring. Our particular focus was on the degree of consistency between guidelines, the explicit consideration of factors important for specifying a monitoring strategy and the use of supporting evidence to justify any recommendations.
Methods
Inclusion criteria
Guidelines that considered the use of PSA measurement as a test for monitoring patients treated with either radical prostatectomy or RTX for localised prostate cancer were eligible. Guidelines that considered only screening or treatment were excluded. Guideline recommendations regarding PSA measurement following other potentially curative treatments or as part of active surveillance were not considered.
Literature searches
MEDLINE was searched from 1999 to July 2009 using the medical subject heading terms (‘Prostatic Neoplasms’ OR ‘Prostate-Specific Antigen’) AND ‘Practice Guideline’, limited to English-language publications. The National Library of Guidelines, the Trip database and The Cochrane Library were also accessed and reference lists of retrieved papers were checked. Titles and abstracts of retrieved records were assessed for inclusion by two authors independently (JD and JJD), with discrepancies resolved by consensus.
Data extraction
Recommendations or statements relating to the use of PSA testing following treatment with curative intent were extracted and references to any supporting evidence were noted. Guideline methods were assessed using the Appraisal of Guidelines for Research and Evaluation (AGREE) framework, which contains 23 key items organised into six domains. 72 We applied only the seven items included in the ‘rigour of development’ domain (Table 1). We replaced the fourth item in this domain with one relevant to using tests for monitoring as opposed to consideration of benefits and harms of interventions.
| Criteria | Details | Example |
|---|---|---|
|
1. Systematic search methods used |
Details of the strategy used to search for evidence should be provided including search terms used, sources consulted and dates of the literature covered | Sources may include electronic databases [e.g. MEDLINE, EMBASE, Cumulative Index to Nursing and Allied Health Literature (CINAHL)], databases of systematic reviews [e.g. The Cochrane Library, Database of Abstracts of Reviews of Effects (DARE)], handsearching journals, conference proceedings and other guidelines (e.g. the US National Guideline Clearinghouse, the German Guidelines Clearinghouse). Further point for judgement on the completeness of the search |
|
2. Selection criteria clearly described |
Criteria for including/excluding evidence identified by the search should be provided. These criteria should be explicitly described and reasons for including and excluding evidence should be clearly stated | For example, guideline authors may decide to include only evidence from randomised clinical trials and to exclude articles not written in English. Further point for judgement on the application of the criteria |
|
3. Formulation of recommendations clearly described |
There should be a description of the methods used to formulate the recommendations and how final decisions were arrived at. Areas of disagreement and methods of resolving them should be specified | Methods include, for example, a voting system, formal consensus techniques (e.g. Delphi, Glaser techniques) |
|
4. Considers relevant issues for monitoring in recommendationsa |
The guideline should consider factors relevant to the monitoring test | Variability in measurements/need for repeat testing, rationale presented for interval frequency and PSA threshold and acknowledgement of the uncertainties in the natural history of PSA following radical treatment |
|
5. Explicit link with supporting evidence |
There should be an explicit link between the recommendations and the evidence on which they are based. Each recommendation should be linked with a list of references on which it is based | An explicit link between the recommendations and the evidence on which they are based should be included in the guideline. The guideline user should be able to identify the components of the body of evidence relevant to each recommendation |
|
6. Pre-publication external review |
A guideline should be reviewed externally before it is published. A description of the methodology used to conduct the external review should be presented, which may include a list of the reviewers and their affiliations | Reviewers should not have been involved in the development group and should include some experts in the clinical area and some methodological experts. Patients’ representatives may also be included |
|
7. Update procedure described |
Guidelines need to reflect current research. There should be a clear statement about the procedure for updating guidelines | For example a timescale has been given or a standing panel receives regularly updated literature searches and makes changes as required |
A maximum score of 4 points was attached to each of the seven items, giving a maximum score of 28 points. A generous approach to scoring items was used. For example, if a systematic search was reported to have been carried out but was not reported in detail, the guideline would score 3 out of a possible 4 points. If a discussion of evidence was provided that appeared to relate to a recommended monitoring schedule, an explicit link with evidence was judged to have been provided, without closer examination of the actual evidence cited. We did not make a judgement about the acceptability of any rationale presented for test frequency or threshold, but indicated whether a rationale was presented or not.
Synthesis
A narrative synthesis was undertaken.
Results
Guidelines (n = 7) or best practice statements (n = 2) from nine organisations were identified. 75–83 Four were North American in origin,77,79,82,83 four were from Europe75,78,80,81 and one was from Australia. 76 Eight of the guidelines75–79,81–83 scored poorly on the framework criteria, with scores between 9 and 16 out of a possible 28 points (Figure 2). The National Institute for Health and Care Excellence (NICE) guideline80 scored considerably higher, with 22 points. The highest scoring item overall was the use of systematic searches, which was reported in most guidelines, even if it was often not described in any detail. Methods for recommendation formulation were described in only three guidelines. 78–80 Only one guideline80 fully considered relevant issues for monitoring tests; this was the only guideline to consistently provide clear links between its recommendations and the underlying evidence base. It also reported its methods in more detail than most of the other guidelines in the sample.
FIGURE 2.
Rigour of guideline development. Adapted with permission from Dinnes et al. 69 Aus CN, Australian Cancer Network; AUA, American Urological Association; DUA, Dutch Urological Association; EAU, European Association of Urology; NCCN, National Comprehensive Cancer Network; NCI PDQ, National Cancer Institute – Physician Data Query; UK PCWG, UK Prostate Cancer Working Group.

Table 2 shows the lack of consistency in guideline recommendations regarding the frequency of follow-up assessments and thresholds; there does not appear to be any clear pattern in recommendations over time.
| Recommendation | Guideline | Number of guidelines | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| UK PCWG 199975 | Aus CN 200276 | AUA 200777 | aDUA 200778 | NCI PDQ 200879 | aNICE 200880 | AUA 200983 | aEAU 200981 | aNCCN 200982 | ||
| 1. Frequency of follow-up visits after radical treatment | 6/9 | |||||||||
| Quarterly for 1 or 2 years, then 6 monthly or annually | ✓,b ASTROc | – | – | ✓✓,d 3 primary studies (level 3) | – | – | – | ✓✓,d none cited (grade B) | – | 3 |
| Every 6 months (for 2 or 5 years) then annual | – | – | ✓,e none cited | – | – | ✓✓,b no direct evidence (consensus) | – | – | ✓✓,e 1 primary study (level 2a) | 3 |
| 2.1 Threshold for ‘intervention’ following prostatectomy | 9/9 | |||||||||
| Any detectable PSA | ✓, none cited | – | – | – | ✓, 3 primary studies | – | – | – | ✓✓, none cited (level 2a) | 3 |
| PSA > 0.2 ng/ml | – | – | – | ✓✓, 1 primary study, 1 review (level 4) | – | ✓, 3 primary studies, 2 reviews | ✓✓, 1 primary studies, 1 review | ✓✓, 4 primary studies, 2 reviews (grade B) | – | 4 |
| No definite threshold recommended | – | ✓, 1 primary study | ✓, none cited | – | – | – | – | – | – | 2 |
| 2.2 Threshold for ‘intervention’ following radiotherapy | 9/9 | |||||||||
| Three consecutive increases in PSA (ASTROc) | ✓, ASTROc | – | – | ✓✓, ASTROc (level 4) | – | – | – | – | – | 2 |
| PSA nadir + 2 ng/ml (Phoenixc) | – | – | – | – | – | – | ✓✓, 2 primary studies, Phoenixc | ✓✓, 2 primary studies, ASTRO,c Phoenixc (grade B) | ✓✓, Phoenixc (level 2a) | 3 |
| PSA nadir + 4 ng/ml | – | – | – | – | – | ✓, 3 primary studies, 1 review, Phoenixc | – | – | – | 1 |
| No specific recommendation | – | ✓, 2 primary studies, 1 review | ✓, none cited | – | ✓, 2 primary studies, ASTRO,c Phoenixc | – | – | – | – | 3 |
| 3. Sources of PSA variability acknowledged and/or remedial action recommendedd | 8/9 | |||||||||
| Technical variability possible | ✓ | ✓, 1 primary study | – | ✓, 1 review | ✓, 1 primary study | ✓✓ (consensus) | ✓, 3 primary studies | ✓, 4 primary studies | – | 7 |
| Biological variability possible | – | ✓ | ✓ | ✓, 3 primary studies | – | – | ✓, 18 primary studies | ✓, 8 primary studies | – | 5 |
| Remedial action recommended | – | – | – | ✓✓, repeat at 1–2 months | Same assay | Same assay | Same assay, 3–6 weeks after biopsy | – | 4 | |
| 4. Acknowledgement of uncertainties in the natural history of PSA and prostate cancer following primary treatment | 3/9 | |||||||||
| – | – | ✓✓, 1 review | – | ✓✓, 4 primary studies | ✓✓, 1 primary study, 1 review | – | – | – | ||
Eight75–81,83 of the nine guidelines acknowledged that PSA levels may be affected by technical or biological variability but in most cases this was presented in the introductory sections of the guidelines, with only one guideline78 tempering its recommendations with reference to the fact that a single PSA measurement may be unreliable (recommending retesting within 2 months). Three guidelines79,80,83 acknowledged the potential impact from technical variation, recommending that the same assay be used at each measurement. Four guidelines75,78,80,82 made some attempt to justify the interval between tests and three75,79,80 discussed relevant issues affecting the choice of threshold. A further three76,77,79 stated that it was not possible to provide a recommendation on the most appropriate biochemical failure definition. Only three77,79,80 of the nine guidelines commented on the difficulty of using PSA as a monitoring tool because of the uncertainties in its behaviour following radical treatment, with two79,80 clearly recognising that not all men with biochemical failure go on to experience clinical failure, such that evidence of the former alone may not be sufficient to alter treatment.
Many recommendations on frequency or threshold were made with no or few supporting citations (see Table 1). Only one guideline78 cited a primary study in support of its recommended monitoring intervals and only four78,80–82 of the nine indicated the level of evidence supporting their recommendations. The levels of evidence suggested ranged from ‘Guideline Development Group consensus only’ to ‘well-conducted clinical studies’ (see Table 1), suggesting that different guideline groups had varying views on the quality of the evidence available.
Despite the general lack of citations in individual guideline documents, a wide range of papers were cited across the guidelines. In total, 49 papers were cited71,84–131 (Tables 3–5); 31% (15/49) were reviews or consensus statements and the remainder were primary studies, almost exclusively retrospective in nature. Of the primary studies, we judged half to have studied the natural history of PSA following treatment and one-quarter to have evaluated the effect of different biochemical failure definitions on clinical outcomes. Only two primary studies examining measurement variability were cited.
| Study | Study design/aim (extracted from abstract) | Focus of study | Used to support guideline statements on | ||||
|---|---|---|---|---|---|---|---|
| Frequency | Threshold (RP) | Threshold (RT) | Variability | Natural history | |||
| ASTRO 199784 | Consensus statement providing guidelines for PSA monitoring following RT | ASTRO consensus statement | UK PCWG | UK PCWG, DUA, NCI PDQ | |||
| Cox 199991 | Report of ASTRO consensus panel to develop evidence-based guidelines for (1) prostate rebiopsy after radiation and (2) RT with rising PSA levels after radical prostatectomy in the management of patients with localised prostatic cancer | ASTRO consensus statement | NCCN | ||||
| Roach 200685 | Reports second consensus conference to revise the ASTRO definition of BF | ASTRO consensus statement | NICE, AUA 2009, EAU, NCCN, NCI PDQ | ||||
| Carroll 200192 | Best practice statement (AUA) | Best practice statement | NICE | ||||
| Aus 200693 | Review of high-intensity focused ultrasound and cryosurgery as the primary treatment option in patients with prostate cancer | Review | EAU | ||||
| Bott 200494 | Review of management of recurrence following RP | Review | EAU | ||||
| Catton 200395 | Review/comment paper examining follow-up strategies | Review | NICE | ||||
| Cookson 200771 | AUA review of the variability in published definitions of biochemical recurrence; recommends a standard definition in patients treated with RP | Review | NICE, AUA 2009 | AUA 2007 | |||
| Edelman 199796 | Review of available data on follow-up strategies | Review | NICE | ||||
| Lee 200597 | Review of PSA kinetics in addition to clinical factors in the selection of patients for salvage local therapy | Review | NCCN | ||||
| Nelson 200398 | Review of RP for prostate cancer | Review | DUA | DUA | |||
| Polascik 199999 | Review of PSA | Review | EAU | ||||
| Selley 1997100 | HTA review of prostate cancer management | Review | Aus CN | ||||
| Vicini 200590 | Review of PSA for monitoring patients after radical treatment | Review | NICE | NICE | |||
| Yao 2003101 | Review/comment paper examining follow-up strategies | Review | NICE | ||||
| Albertsen 2004102 | Retrospective(?) study of 1136 men undergoing surgery or RT to document patterns of PSA recurrence and quantify the extent to which increasing PSA levels predict death | Natural history of patients post treatment | EAU | ||||
| Amling 2001103 | Retrospective(?) analysis of 2782 men who had undergone RP to attempt to determine the best PSA cut-off point for defining BF | Testing definitions of BF | EAU | ||||
| Booker 2004104 | Study of telephone follow-up led by a specialist nurse for patients undergoing RT | Follow-up acceptability | NICE | ||||
| Buyyounouski 2005105 | Retrospective(?) review of 688 men who had undergone RT to compare three definitions of BF in terms of sensitivity, specificity, etc. for detecting clinical progression | Testing definitions of BF | DUA | ||||
| Cathala 2003106 | Feasibility study of 140 patients undergoing RP to determine the acceptability of an internet follow-up service | Follow-up acceptability | NICE | ||||
| Cheung 2005107 | Retrospective(?) analysis of 101 men who received salvage RT for BF after RP to compare outcomes for patients who received RT alone and for those who received combined RT and hormonal therapy | Prognosis following salvage treatment | NCCN | ||||
| Crook 1997108 | Prospective study of 207 patients to correlate the failure pattern after RT with pretreatment PSA and post-RT nadir PSA levels | Natural history of patients post treatment | UK PCWG | ||||
| D’Amico 2004109 | Retrospective review of 8669 men who had undergone radical treatment to determine whether or not a short post-treatment PSA doubling time is a suitable surrogate end point for prostate cancer-specific mortality | Natural history of patients without treatment | NICE | ||||
| Eastham 2003110 | Retrospective analysis of an unscreened population of 972 men over 4 years to determine whether or not year-to-year fluctuations in PSA levels are due to natural variation, rendering a single PSA test result unreliable | Measurement variability | EAU | ||||
| Frazier 1993111 | Analysis of 226 patients who underwent radical perineal prostatectomy to identify whether or not a raised serum PSA level infers failure of the procedure | Natural history of patients post treatment | NCI PDQ | NCI PDQ | |||
| Horwitz 200587 | Determined the sensitivity and specificity of several BF definitions using pooled data on 4839 patients treated with external-beam RT alone | Testing definitions of BF | AUA 2007, AUA 2009, NICE | ||||
| Klotz 2005112 | Reports PSA doubling time in a series of 299 patients undergoing active surveillance for prostate cancer | Natural history of patients post treatment | DUA | ||||
| Kuban 200686 | Primary study of 2693 patients treated with a radioisotopic implant as solitary treatment for T1–T2 prostatic adenocarcinoma. Multiple PSA failure definitions were tested for their ability to predict clinical failure | Testing definitions of BF | AUA 2007, AUA 2009, NICE | NCI PDQ | |||
| Leibman 1995113 | Retrospective review of 628 patients who underwent RP to determine whether or not prostate cancer recurrence can occur without an increase in serum PSA levels | Natural history of patients post treatment | EAU | ||||
| Nielsen 2008114 | Retrospective review of data from 2570 men who had undergone RP to examine the effect of applying the 2005 ASTRO definition of BF (for RT patients) to surgical series | Testing definitions of BF | AUA 2009 | ||||
| Niwakawa 2002115 | Study of 221 patients treated with RP to determine the optimal frequency and method of follow-up to minimise medical costs | Follow-up – optimal frequency | DUA | ||||
| Oefelein 1995116 | Retrospective review of data from 394 men who underwent RP to characterise the incidence of recurrent carcinoma despite undetectable serum PSA levels | Natural history of patients post treatment | EAU | ||||
| Patel 2005117 | Retrospective review of 48 patients who had undergone salvage RT for biochemical relapse after RP to determine whether or not PSA velocity is a suitable selection criterion for salvage radiotherapy | Prognosis following salvage treatment | NCCN | ||||
| Pickles 2006118 | Analysis of a ‘prospective’ database of 2030 patients who underwent external-beam RT or brachytherapy to determine the false-call rate for PSA relapse according to nine different PSA relapse definitions after a PSA bounce has occurred | Testing definitions of BF | NICE | ||||
| Pound 199989 | Retrospective review of a large surgical series (n = 1997) to examine the natural history of progression to distant metastases in men with a raised PSA level following surgery | Natural history of patients post treatment | DUA, NCCN | NCI PDQ, DUA, NICE, EAU | NCI PDQ, NICE | ||
| Ragde 1997119 | Study of 126 patients with localised prostate cancer to determine the efficacy of treatment with iodine-125 radionuclides (two definitions of PSA failure used) | Testing definitions of BF | Aus CN | ||||
| Ray 2006120 | Retrospective(?) review of 4839 patients treated definitively with RT to determine the significance of PSA nadir and time to PSA nadir in predicting biochemical or clinical DSF | Natural history of patients post treatment | EAU | ||||
| Ritter 1992121 | Study of the prognostic value of PSA in pretreatment evaluation and post-treatment follow-up in 63 patients undergoing RT for localised prostate cancer | Natural history of patients post treatment | Aus CN | ||||
| Rose 1996122 | To identify patients’ symptoms following completion of RT for common cancers by a nurse-managed telephone interview (n = 111) | Follow-up acceptability | NICE | ||||
| Sandler 2000123 | Retrospective database study of 1844 patients who had undergone RT and had a minimum of two post-RT PSA measurements separated by at least 1 week to determine the significance of BF (i.e. in terms of survival) | Natural history of patients post treatment | NCI PDQ | NCI PDQ | |||
| Sartor 1997124 | Primary study of 400 patients treated with RT to determine whether or not the rate of PSA rise could differentiate future local vs. metastatic failure | Natural history of patients post treatment | UK PCWG | ||||
| Stamey 1989125 | Study of pre- and post-treatment serum PSA levels in 102 men who underwent RP to determine the usefulness of PSA as a preoperative marker | Natural history of patients post treatment | NCI PDQ, EAU | ||||
| Stephan 2006126 | Assessed five frequently used commercial assay combinations in sera from 314 patients with prostate cancer and 282 men with no evidence of prostate cancer to identify the interchangeability of the PSA values | Measurement variability | EAU | ||||
| Stephenson 2004127 | Retrospective review of 501 patients who underwent salvage RT following RP to identify those variables indicative of a durable response | Prognosis following salvage treatment | NCCN | ||||
| Stephenson 200688 | Tested 10 definitions of BF on 3125 patients who underwent RP, to identify the one that best explains metastatic progression | Testing definitions of BF | AUA 2009, EAU, NICE | ||||
| Trapasso 1994128 | Primary study of 601 patients undergoing radical retropubic prostatectomy followed by serial PSA measurement. Evaluated rate of detectable PSA (> 0.4 ng/ml) as an indicator of cancer progression | Natural history of patients post treatment | EAU | EAU | |||
| Trock 2008129 | Retrospective analysis of a cohort of 635 men undergoing RP and who experienced biochemical and/or local recurrence to determine the effect of salvage RT and to identify subgroups for whom salvage treatment is most beneficial | Prognosis following salvage treatment | NCCN | ||||
| Ward 2004130 | Retrospective cohort study of 211 men with detectable PSA levels following RP to determine whether or not PSA doubling time predicts outcomes following salvage RT | Natural history of patients post treatment | NCCN | ||||
| Zagars 1997131 | Analysis of 841 men with serial PSA measurements who underwent external-beam RT without androgen ablation to determine the kinetics of serum PSA after RT and to evaluate whether or not such kinetics provide prognostic information | Natural history of patients post treatment | UK PCWG | ||||
| Type of study | Number of studies per group | Used to support guideline recommendations on | ||||
|---|---|---|---|---|---|---|
| Test frequency | Threshold (RP) | Threshold (RT) | Variability | Uncertainty in natural history | ||
| ASTRO consensus statements | 3 | ✗ | ✗ | |||
| Best practice statement | 1 | ✗ | ||||
| Reviews | 11 | ✗ | ✗ | ✗ | ✗ | ✗ |
| Primary studies | 34 | ✗ | ✗ | ✗ | ✗ | |
| Follow-up (acceptability of) | 3 | ✗ | ||||
| Follow-up (optimal frequency) | 1 | ✗ | ||||
| Natural history of PSA post treatment | 15 | ✗ | ✗ | ✗ | ✗ | |
| Natural history of PSA without treatment | 1 | ✗ | ||||
| Salvage RTX outcomes | 4 | ✗ | ||||
| Testing BF definitions | 8 | ✗ | ✗ | ✗ | ||
| Measurement variability | 2 | ✗ | ||||
| Number of guidelines citing evidence | 4 | 6 | 7 | 2 | 3 | |
| Study | Study design/aim (extracted from abstract) | Used to support statements on | Number of times cited |
|---|---|---|---|
| Roach 200685 | Reports second consensus conference to revise the ASTRO definition of BF | Threshold (RTX) | 5 |
| Pound 199989 | Retrospective review of a large surgical series (n = 1997) to examine the natural history of progression to distant metastases in men with a raised PSA level following surgery | Frequency, threshold (RP), natural history | 5 |
| Kuban 200686 | Primary study of patients treated with a radioisotopic implant as solitary treatment for localised prostate cancer (n = 2693). Multiple PSA failure definitions were tested for their ability to predict clinical failure | Threshold (RTX), natural history | 4 |
| ASTRO 199784 | Consensus statement providing guidelines for PSA testing following radiation therapy | Frequency, threshold (RTX) | 3 |
| Cookson 200771 | AUA review of the variability in published definitions of biochemical recurrence; recommends a standard definition in patients treated with RP | Threshold (RP), natural history | 3 |
| Horwitz 200587 | Determined the sensitivity and specificity of several BF definitions using pooled data on 4839 patients treated with external-beam radiotherapy alone | Threshold (RTX) | 3 |
| Stephenson 200688 | Tested 10 definitions of BF on 3125 patients who underwent RP, to identify the one that best explains metastatic progression | Threshold (RP) | 3 |
Most studies were cited by only one or two of the guidelines, but a handful of studies were cited three or more times (see Table 5). Two consensus statements84,85 were among the most frequently cited studies, as was a review of biochemical failure definitions. 71 The four primary studies had among the largest sample sizes of all of the cited primary studies: three86–88 evaluated the use of different biochemical failure definitions and one89 studied the natural history of disease progression in men with raised PSA.
The findings are summarised in Table 6.
| Study | Brief description | Rating on the evaluation instrument | Comment | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Systematic search methods used | Selection criteria clearly described | Formulation of recommendations clearly described | Considers relevant issues for monitoring in recommendations | Explicit link with supporting evidence | Pre-publication external review | Update procedure described | Total | |||
| AUA 200777 | Localised prostate cancer management guideline | 2 | 3 | 1 | 1 | 1 | 2 | 3 | 13 | One database used, search poorly reported; inclusion criteria described but grounds for later exclusion of papers not clear; limited description of recommendation formulation; no basis for interval between measurements; evidence-based recommendations for threshold not possible; natural history uncertainty acknowledged in recommendations, variability acknowledged but not in recommendations; no link between recommendations and evidence; external review carried out but not described; update recommended and to include only RCT evidence |
| AUA 200983 | PSA best practice statement | 1 | 1 | 2 | 1 | 2 | 2 | 1 | 10 | No systematic search; inclusion criteria not described; some description of recommendation formulation; no interval between measurements recommended; consensus definition of threshold used; natural history uncertainty not acknowledged; variability acknowledged but not in recommendations; some supporting evidence cited; peer review carried out but not described in detail; no mention of update |
| Aus CN 200276 | Localised prostate cancer management evidence-based recommendations | 4 | 1 | 2 | 1 | 2 | 2 | 2 | 14 | Comprehensive and systematic search described; inclusion criteria not described; no interval between measurements recommended; states no widely accepted biochemical range applicable; natural history uncertainty not acknowledged; variability acknowledged but not in recommendations; some supporting evidence cited; internal review carried out but not described in detail; update recommended but procedure not described |
| DUA 200778 | Prostate cancer management guideline | 3 | 2 | 3 | 2 | 2 | 2 | 2 | 16 | Systematic search carried out but not fully described; some description of inclusion criteria; interval between measurements based on evidence; consensus threshold used for post RT, no justification for threshold post RP; natural history uncertainty not acknowledged; variability acknowledged but not in recommendations; some link to supporting evidence; external review partly described; update recommended but procedure not described |
| EAU 200981 | Prostate cancer management guideline | 2 | 1 | 1 | 1 | 2 | 2 | 1 | 10 | Systematic search carried out but only partly described; no description of inclusion criteria; formulation of recommendations not described; no basis for interval between measurements; consensus thresholds used; natural history uncertainty not acknowledged; variability acknowledged but not in recommendations; some link to supporting evidence; external review conducted but not described; no mention of update |
| NCI PDQ 200879 | Prostate cancer treatment evidence-based summary for health professionals | 1 | 1 | 3 | 3 | 2 | 1 | 2 | 13 | No search described; no description of inclusion criteria; formulation of recommendations not described; no interval between measurements recommended; evidence-based recommendations for threshold post RT not possible, basis for post-RP threshold given; natural history uncertainty acknowledged in recommendations; the importance of variability was acknowledged, but no specific recommendations were made about the handling of variability and the associated uncertainty; some links to supporting evidence; external review not described; limited description of update procedures |
| NCCN 200982 | Prostate cancer management guideline | 1 | 1 | 1 | 2 | 2 | 1 | 1 | 9 | No search described; no description of inclusion criteria; no description of formulation of recommendations; some justification given for interval between measurements; consensus threshold used for post RT, no justification for post-RP threshold; natural history uncertainty and variability not acknowledged; some links to supporting evidence; external review not described; no mention of update |
| NICE 200880 | Prostate cancer diagnosis and treatment guideline | 4 | 3 | 3 | 4 | 3 | 2 | 3 | 22 | Systematic search carried out and fully described; inclusion criteria developed for each question but not reported; recommendation formulation described but methods used to deal with disagreement not reported; attempted to find evidence to justify interval between measurements; relevant discussion regarding choice of thresholds; natural history uncertainty and variability acknowledged in recommendations; clear link to supporting evidence; external review not described; an update of the guideline was recommended and some details of the procedure required are described |
| Royal College of Radiologists’ Clinical Oncology Information Network, British Association of Urological Surgeons 199975 | Prostate cancer management guideline | 3 | 1 | 2 | 2 | 2 | 1 | 1 | 12 | Systematic search carried out and fully described; inclusion criteria developed for each question but not reported; recommendation formulation described but methods used to deal with disagreement not reported; interval between measurements justified; relevant discussion regarding choice of threshold post RT only; natural history uncertainty not acknowledged, variability acknowledged but not in recommendations; clear link to supporting evidence; external review not described; update recommended but and procedure described |
Discussion
We found considerable inconsistency in the recommendations in guidelines for the use of PSA as a monitoring test, even when they were published within a few years of each other. Factors considered to be important when specifying a monitoring strategy were given limited attention and were not well supported with reference to primary literature.
Recommendations on when to test and what action to take following a given test result were very much considered in isolation from each other. ‘When to test’ appeared to be almost exclusively determined by standard follow-up schedules rather than having any scientific basis. Although most guidelines acknowledged the potential presence of measurement variability, they did not attempt to account for its potential effect on test interpretation. A systematic review of biological variation in PSA levels has found a mean biological variability of 20%. 132 Using reference change value (RCV) methodology it concluded that, to be 95% sure that a change in total PSA level is not the result of random variation, the change needs to be around 50% of the previous measurement. 132 This review was not cited by any of the eight guidelines subsequently published.
Recommendations on when to take action were based on consensus statements or retrospective case series, with little attention paid to variations in the definition of the threshold, the definition of clinical failure and the frequency and length of follow-up between studies, all of which can affect the accuracy of any given cut-off point. Sensitivity and specificity are also known to be affected by differences in patient case mix between studies. 133 This was not acknowledged by any of the identified guidelines. However, a 2005 review of PSA measurement for monitoring prostate cancer90 found it impossible to recommend any single definition of biochemical failure following either surgery or radiotherapy for the reasons listed above. This review was cited by only one of the nine guidelines,80 possibly because it was not a full systematic review. Given the lack of description of inclusion criteria used in the guidelines, it is difficult to reconcile why an individual study or review was included or not.
Reviews of guidelines in other areas have shown similar findings regarding the presentation of evidence for recommended monitoring schedules. 134,135 Reviews of treatment and diagnostic guidelines have identified a similar inconsistency in recommendations between guidelines and variation in evidence cited, with some referring to a substantial body of evidence and others presenting very little evidence. 136–140
A number of factors are likely to contribute to these findings. In the first instance, although this area is beginning to receive more attention, there is a lack of high-quality evidence, and indeed lack of clear methodological guidance, on what to consider when establishing monitoring strategies. 38 It is, therefore, perhaps not surprising that relevant evidence has not been used to inform guidelines.
Second, the various pieces of information needed to inform a monitoring strategy are not usually available from a single study. Ideally, one or more monitoring strategies should be evaluated in a RCT or some form of prospective comparative study. When there is high-quality evidence, greater consensus between guideline recommendations and stronger guideline recommendations have been found. 136 Randomised trials of monitoring, however, have their own challenges and are consequently relatively rare. Instead, evidence has to be gathered from various sources. Although the diversity of evidence needed to inform coherent monitoring strategies makes the identification of relevant pieces of evidence a challenge for guideline developers and likely adds to the inconsistency in recommendations between guidelines, guideline developers have a responsibility to highlight recommendations for which there is a lack of evidence or the evidence is inconsistent.
Efforts to improve the evidence base for monitoring are ongoing. For example, a Bayesian hierarchical change-point model has been used to simulate multiple post-radiotherapy PSA series from primary data; the sensitivity and specificity of different definitions of biochemical failure were then compared, allowing characteristics that might affect accuracy to be controlled for. 141 More pertinently, statistical models using estimates of mean change and variability in a measurement over time to suggest optimal monitoring intervals are being developed. A review142 of four case studies143–146 found that for each topic the results suggested overfrequent monitoring. There is clear potential for the extension of this work to monitoring in other settings.
Finally, general failings in the guideline development process are likely to contribute significantly to the variations between published guidelines. In a review of hypertension guidelines, Campbell et al. 138 found a lack of methodological rigour in the guideline development process. In our sample, NICE80 and the Australian Cancer Network76 were the only organisations to cite a published handbook on guideline development,147 which may explain their higher ratings on the evaluation instrument; those clearly based on expert consensus tended to score considerably lower. 83 Others suggest that the greater the involvement of clinical experts in the development process of the guideline, the less the recommendations reflect the research evidence. 137 It is likely that, in the absence of clear methodologies for assessing monitoring strategies, greater involvement of methodologists on guideline panels would be beneficial.
Strengths and limitations
Our research has some limitations. Our literature search was limited to one major medical database, supplemented with searches of more specialist resources, and records were limited to English-language studies only. We believe, however, that we have identified key guidelines that provide a good representation of the methodologies in use by well-known agencies. Although other guidelines may be available, they are unlikely to have used alternative methods or to have reported on evidence that the included guidelines have missed.
Second, our use of the original AGREE instrument may be criticised given that it was published in 2003; however, at the time that the framework was chosen, the update to the original instrument148 and other potentially useful frameworks were not yet available. 146,149,150 Nevertheless, our approach to assessing the development and content of the guidelines was systematic and provides a reasonable means of comparison between guidelines.
We were not able to comment fully on the state of the underlying literature cited in support of the monitoring schedules as we did not retrieve copies of all of the cited primary studies. Furthermore, our ‘generous’ approach to associating citations to recommendations may have inadvertently led to citations being incorrectly associated with recommendations. This may have led to some bias in favour of the guidelines. However, this could be avoided only by carrying out a full review of all of the evidence cited or by direct contact with the guideline authors to determine which aspect of the recommendations were supported by the citations given, both of which were outside the scope of this review.
Finally, our use of only one case study may limit the generalisability of our results to other topic areas. However, we have no reason to believe that the picture would be any better or any worse for other fields and, indeed, Moschetti et al. 135 found similar results for monitoring in cardiovascular disease.
Our systematic approach to assessing the development and content of the guidelines provides a valuable insight into how strategies for monitoring are developed and reported and we were able to present a general picture of the types of evidence that have been cited. The true picture may be even worse given our attempt to attribute citations to recommendations whenever possible.
Conclusions
Our findings highlight the lack of a scientific or systematic approach to the development of monitoring schedules for the use of PSA testing, as reported in clinical guidelines. This is the result of both inadequacies in the evidence base and inappropriate use of the available evidence, resulting in considerable inconsistencies between guidelines.
Guideline developers should be encouraged to adopt systematic approaches to guideline development, such as those developed in Australia149 and the USA,150 and should take care to explicitly consider each element of a recommended monitoring schedule (interval, threshold and action to be taken on crossing that threshold) and the standard of its evidence base.
Chapter 4 Has the randomised controlled trial design been successfully used to evaluate strategies for monitoring disease progression or recurrence? An assessment of experience to date
Introduction
Clinical consultations between patient and clinician usually involve the use of tests, often starting with a general clinical assessment and physical examination but also including the application of specific tests, from tools assessing functional or psychological status, to blood or urine tests, physiological measurements of, for example, blood pressure, imaging tests or more invasive assessments such as a colonoscopy or biopsy. Testing can inform a diagnosis or can be used for monitoring whereby a test, or set of tests, is applied repeatedly over time to assist in the management of a disease or condition. Our particular interest is in monitoring individuals with (or at risk of) a disease or condition that is likely to progress or recur at some time in the future. This is distinct from monitoring in a treatment titration context, in which the aim is to keep a marker within predefined limits until treatment can be discontinued or an alternative treatment is required. Although monitoring for disease progression or recurrence can serve many purposes, including providing reassurance to patients or clinicians, it is usually undertaken to allow timely decisions to be made regarding patient management. Management decisions include the initiation of treatment to prevent some clinical outcome from occurring (e.g. variceal bleeding subsequent to cirrhosis of the liver or infertility as a result of Turner syndrome), delay a clinical event [e.g. progression to acquired immune deficiency syndrome (AIDS) in HIV infection] or otherwise improve outcome (e.g. through earlier treatment of cancer recurrence); additionally, the goal may be to avoid or delay treatment in those who may not need it (e.g. surveillance of mild hip dysplasia in infants).
Whatever the goal, monitoring is a central activity for patient and disease management and, just as for therapeutic interventions and for tests used in a diagnostic context, it is important to identify its impact on patient outcomes: ‘the primary purpose of using . . . tests should . . . be to prevent premature death and suffering and restore functional health’. 151 Given the advantages of the RCT design for the evaluation of therapeutic interventions, it is tempting to assume that the same approach must be the gold standard for the evaluation of all monitoring strategies.
Trials of monitoring regimes present considerable challenges, however. As for diagnostic tests, trials of monitoring evaluate a particular strategy, with tests applied at specific intervals, defined thresholds for changing patient management and prescription of effective interventions, all of which should be specified in advance and ideally supported by previous research. The complexity of such strategies, and in particular the serial nature of testing, and consequent potential for ‘interactions between tests, repeated tests, test results and the decisions based on these results’ may necessitate unfeasibly large sample sizes to detect an effect on important patient outcomes. 152 Furthermore, even with careful planning it may be difficult to capture in a RCT the wider patient impact of testing, whether it is used for diagnostic, screening or monitoring purposes. Ferrante di Ruffano et al. 68 have outlined a range of effects from testing in a diagnostic context, including emotional, cognitive and behavioural effects, which also have applications in monitoring.
We conducted a methodological review of RCTs of monitoring to gain some insight into how successfully the design has been used to identify patient benefit from monitoring.
Methods
Literature search
Our target sample size was 60 RCTs. The Cochrane Central Register of Controlled Trials (CENTRAL) was searched to retrieve relevant records (last updated 21 July 2011; details available from authors). The search was supplemented by screening all RCTs funded by the National Institute for Health Research (NIHR) Health Technology Assessment programme and those published in the Trials journal (to December 2011). After assimilation of studies meeting the inclusion criteria, a search of the US NIH ClinicalTrials.gov database was carried out (using the keywords monitor#, surveill# or early or immediate treatment) and the results were purposively sampled to include trials conducted in topic areas that would complement those already identified. The sampling was not carried out on the basis of trial quality. Attempts were made to identify publications related to these trials using Google Scholar and by contacting trial principal investigators to request copies of their protocols or trial reports.
Inclusion criteria
Trials were eligible for inclusion if they considered monitoring of a disease or condition that is likely to progress or recur at some time in the future. RCTs in which the main purpose of monitoring was treatment titration or improvement in adherence to a treatment regimen or those evaluating methods of delivering monitoring were excluded as were trials of tests used for population-based screening or for diagnosis. Trials had to compare at least one formal monitoring strategy with no formal monitoring, an alternative monitoring strategy or an immediate treatment option. All clinical topics, test types and outcomes were eligible.
Trials reported only as protocols were included, but those available only in abstract form and non-English-language papers were excluded. Multiple reports of a single trial were assimilated through cross-referencing.
The search was conducted by and search results were screened by one reviewer (JaD).
Data extraction and analysis
A data extraction form was designed and piloted. Data were extracted on items including the study population and topic area, monitoring strategies or interventions in the experimental and control arms (including details of the testing frequency, threshold and intervention), the citation of evidence to support these features of the monitoring strategies, study design and validity criteria (see Table 1). Details of the primary outcomes used were also extracted, with outcomes classified as patient, process or composite outcomes; if not clearly reported the outcome used in the study’s power calculation was extracted or, failing that, the outcome most closely related to the study aim was extracted. When final analyses were reported, the result for the primary outcome was extracted and, when possible, this was compared graphically with that predicted in the sample size calculation. For 30 trials, data were extracted independently by two authors (JaD and AS or JP); for the remaining 28 trials, data were extracted by one author (JaD) and were checked by a second (AS or JP). Any disagreements were resolved by consensus.
Studies were considered according to topic area, types of tests and monitoring strategies, the study aim and change in patient care that was evaluated, study validity and primary outcomes and results.
Results
The CENTRAL search retrieved 4697 potentially eligible records (Figure 3), of which 119 titles were selected for further evaluation, along with nine trials identified from the Health Technology Assessment database and the Trials journal. Following full-text review, 49 trials published in 58 publications were selected for inclusion. Twenty trials identified from the ClinicalTrials.gov database were selected and the trial principal investigators were contacted. Documents related to 12 of these were successfully retrieved, of which seven met the inclusion criteria. Reference list screening identified a further two eligible trials. Of the 58 included trials, five were reported in two publications153,154 and 19 previous or related publications were also identified (for a total of 74 papers153–225). Figure 4 shows that there was a general upwards trend in the number of trials published per year.
FIGURE 3.
Flow diagram of the trial selection process. HTA, Health Technology Assessment.
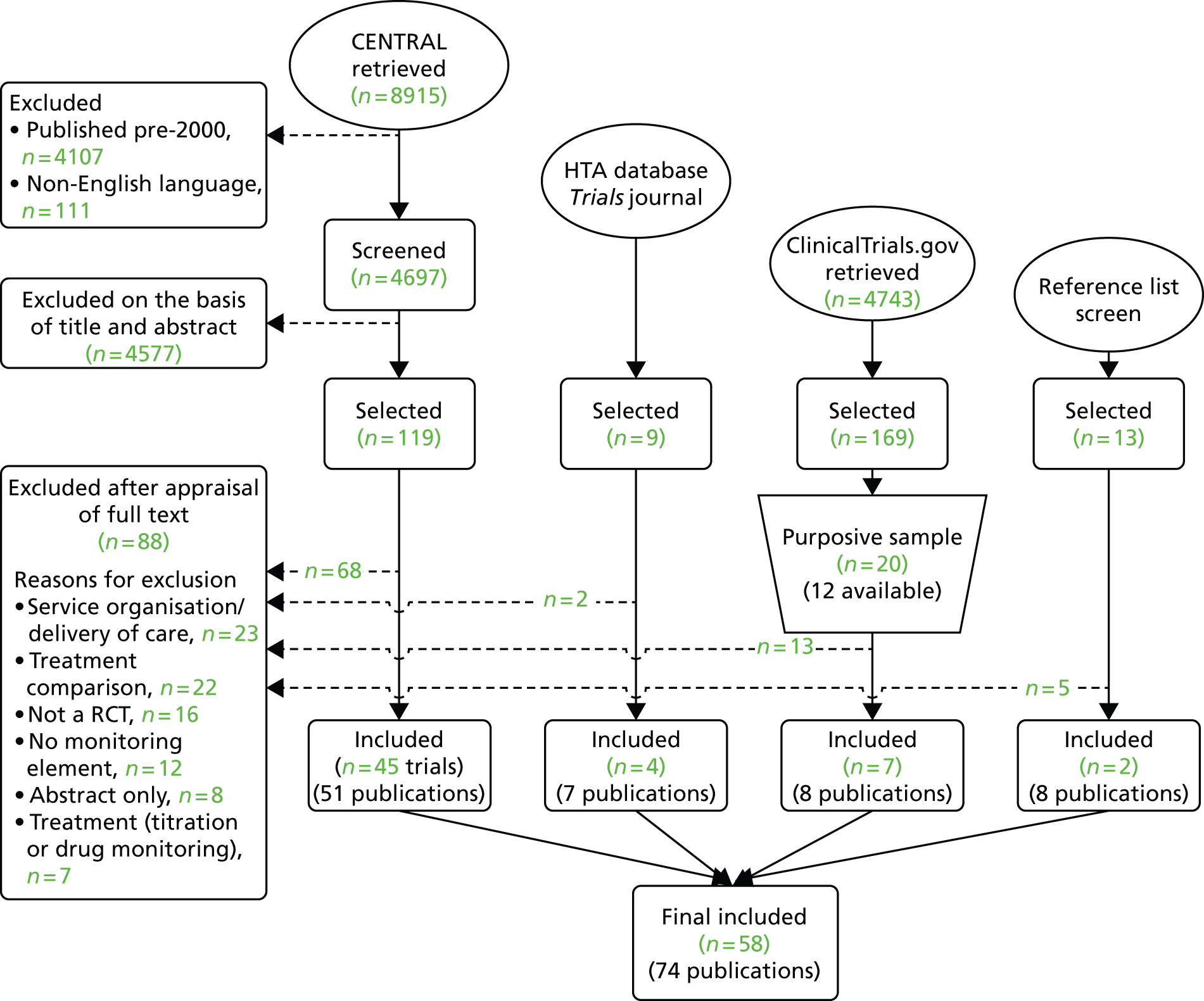
FIGURE 4.
Number of trials published over time. The seven trials identified from ClinicalTrials.gov were published in 2007 (n = 1), 2009 (n = 1), 2010 (n = 3) and 2011 (n = 2).

General description of the included trials
The trials were primarily conducted in the fields of cancer (29%), cardiovascular disease (16%) and renal disease (16%) (Table 7). A further 9% of trials were conducted in patients with aneurysm – either abdominal aortic or cranial – and 9% of trials were conducted in transplant recipients, including stem cell and bone marrow transplant recipients. Most were parallel in design, except for six multiarm trials; the total number of experimental arms was 68.
| Characteristic | Number of trials | Percentage of trials | Notes |
|---|---|---|---|
| Patient group | |||
| Cancer | 17 | 29 | |
| Cardiovascular disease | 9 | 16 | All with implanted devices |
| Renal disease | 9 | 16 | All haemodialysis recipients |
| Aneurysm | 5 | 9 | 4 abdominal aortic aneurysm, 1 intracranial aneurysm |
| Transplant recipients | 5 | 9 | 3 stem cell/bone marrow, 2 solid organ |
| Other (fewer than three trials per group) | 13 | 22 | |
| Number of study arms | |||
| 2 | 52 | 90 | |
| 3 | 2 | 3 | |
| 4 | 4 | 7 | |
| Publication type | |||
| Full trial report | 34 | 59 | |
| Interim analysis | 11 | 19 | 9/11 stopped early |
| Protocol | 13 | 22 | 3/13 stopped early |
| Trial early-stopping reasons (n = 12) | |||
| Recruitment difficulties | 3 | 25 | |
| Technology issues | 3 | 25 | |
| Interim analyses showed (n = 12) | |||
| Early superiority | 1 | 8 | |
| No evidence of benefit | 1 | 8 | |
| Low event rate in control group | 4 | 33 | |
| Sample size calculations | |||
| Reported | 45 | 78 | |
| Full trial reports (n = 34) | 25 | 74 | |
| Interim analyses (n = 10) | 8 | 80 | |
| Protocol only (n = 14) | 12 | 86 | |
| Study funding | |||
| Industry sponsored | 14 | 24 | |
| Non-industry sponsored | 33 | 57 | |
| Not reported | 11 | 19 | |
| Total number of participants randomised, median (IQR; range)a | 272 (120–599; 64–4439) | ||
| Length of follow-up (months), median (IQR; range)a | 21 (12–60; 1–240) | ||
Of the 58 trials, 34 were available as full trial reports, 10 as interim analyses and 14 as trial protocols. Twelve trials were stopped early (21%); interim analyses were available for nine of these trials and a protocol only was available for the remaining three trials. The most common reason for a trial stopping was a lower than expected event rate in the control group (33%; 4/12). Three-quarters of the trials reported sample size calculations (78%; 45/58) and one-quarter were industry sponsored (24%; 14/58). The median sample size was 272 [interquartile range (IQR) 120–599] and the median follow-up was 21 months (IQR 12–60).
Description of monitoring strategies
A total of 139 tests were applied in the control arms of 55 RCTs, excluding those with no formal surveillance or those in which all patients were treated (Table 8). After clinical examination, imaging tests were the most commonly used tests (39%). The 55 ‘new’ tests applied in the experimental arms included biochemical (35%), imaging (34%) and physiological (16%) tests and implanted devices (9%).
| Details | Trial arm | |||
|---|---|---|---|---|
| Control (N = 139 testsa) | Experimental (N = 55 testsb) | |||
| n | % | n | % | |
| Type of test | ||||
| Biochemical | 15 | 11 | 19 | 35 |
| Clinical | 53 | 38 | 0 | 0 |
| Cytological | 0 | 0 | 2 | 4 |
| Histological | 1 | 1 | 1 | 2 |
| Imaging | 46 | 33 | 16 | 29 |
| Imaging (invasive) | 8 | 6 | 3 | 5 |
| Implanted device | 4 | 3 | 5 | 9 |
| Physiological | 12 | 9 | 9 | 16 |
| Type of threshold used (excluding clinical assessment) | ||||
| Not reported | 41 | 48 | 18 | 33 |
| Simple threshold | 30 | 35 | 21 | 38 |
| Change from previous measurement | 1 | 1 | 4 | 7 |
| Algorithm | 6 | 7 | 6 | 11 |
| More than one threshold | 8 | 9 | 6 | 11 |
| Trial arm | ||||
| Control (N = 86 testsc) | Experimental (N = 55 tests) | |||
| n | % | n | % | |
| Repeat measure taken to confirm abnormal result (excluding clinical assessments) | ||||
| All abnormal | 8 | 9 | 4 | 7 |
| Indeterminate only | 7 | 8 | 4 | 7 |
| Not repeated | 0 | 0 | 1 | 2 |
| Not reported | 71 | 83 | 46 | 84 |
| Change in patient management following positive monitoring test (N = 58 trials) | ||||
| Confirmatory testing (non-invasive) | 10d | 17 | 10 | 17 |
| Confirmatory testing (invasive) | 8 | 14 | 14 | 24 |
| Treatment | 26 | 45 | 26e | 45 |
| Treatment or further investigation | 2 | 3 | 1 | 2 |
| More intensive surveillance + treatment option | 1 | 2 | 2 | 3 |
| Not described | 11 | 19 | 5 | 9 |
Figure 5 shows that, although the frequency of application of the tests was well reported, the method of application of the tests was provided for only two-thirds of the experimental arms and fewer than half of the control arms. The test thresholds, describing when a change is patient management is indicated, were reported for 79% and 51% of the experimental and control arms, respectively. A simple threshold approach (i.e. in which the patient crosses a predefined threshold on a single test measurement) was used to judge an abnormal test result in 35% of control tests and 38% of experimental tests (see Table 8). Few trials reported using test measurements over time to define an abnormal result, although the percentages were higher for the experimental tests: the change from the previous measurement was reported for 1% of control tests and 7% of experimental tests and a more complex algorithm taking account of more than one test result was reported for 7% and 11% of the control and experimental tests, respectively. There was also limited reporting of repeated testing to confirm abnormal or indeterminate test results [reported for < 20% of tests in both arms (see Table 8)].
FIGURE 5.
Adequate description of monitoring strategy elements.
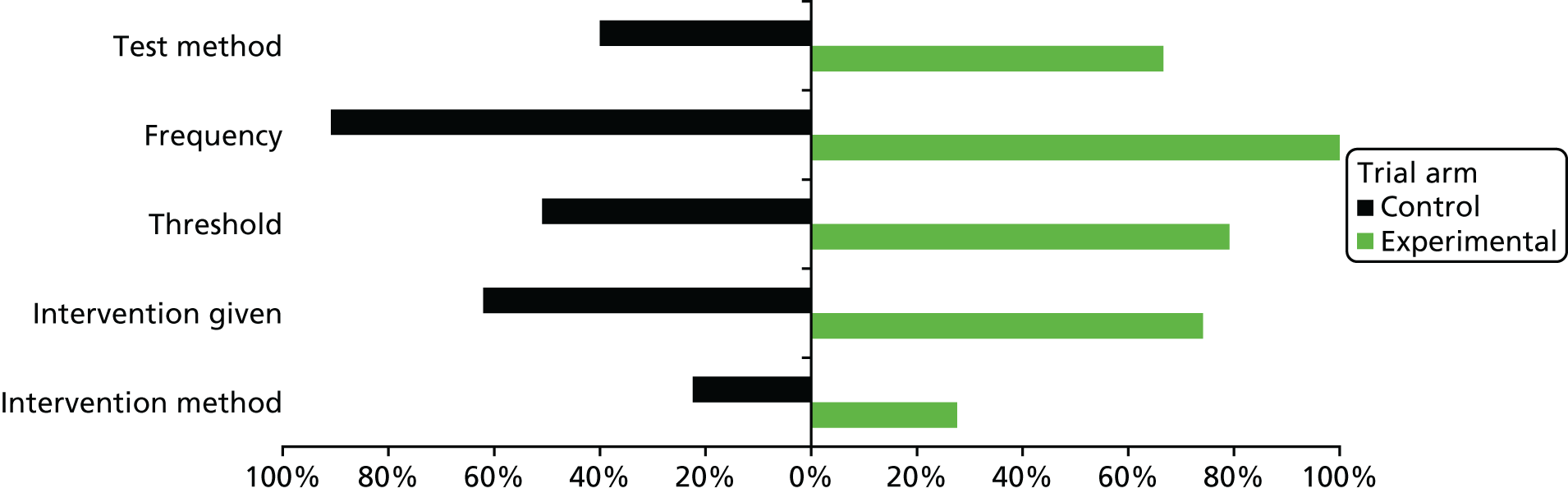
The recommended change in patient management following a positive monitoring test was the same in the experimental and control groups for 69% of trials. The change in management prescribed was usually treatment (45% of both the control and the experimental arms). Some form of confirmatory testing was indicated in 31% and 41% of the control and experimental arms, respectively (see Table 8), with many studies also recommending the treatment to be used following a positive confirmatory test. Details of the actual intervention given and method of application of the intervention were provided for 74% and 28% of the experimental arms, respectively, compared with only 62% and 22% of the control arms, respectively (see Figure 5).
More evidence was cited to support the various elements of the monitoring strategies for the experimental arms than for the control arms (Figure 6); however, many of the previous studies were cited in the introduction or discussion sections of the papers rather than being cited to explicitly support particular test frequencies, test thresholds or interventions reported in the methods sections.
FIGURE 6.
Citation of evidence to support features of the monitoring strategies. NA, not applicable; Rx, treated.
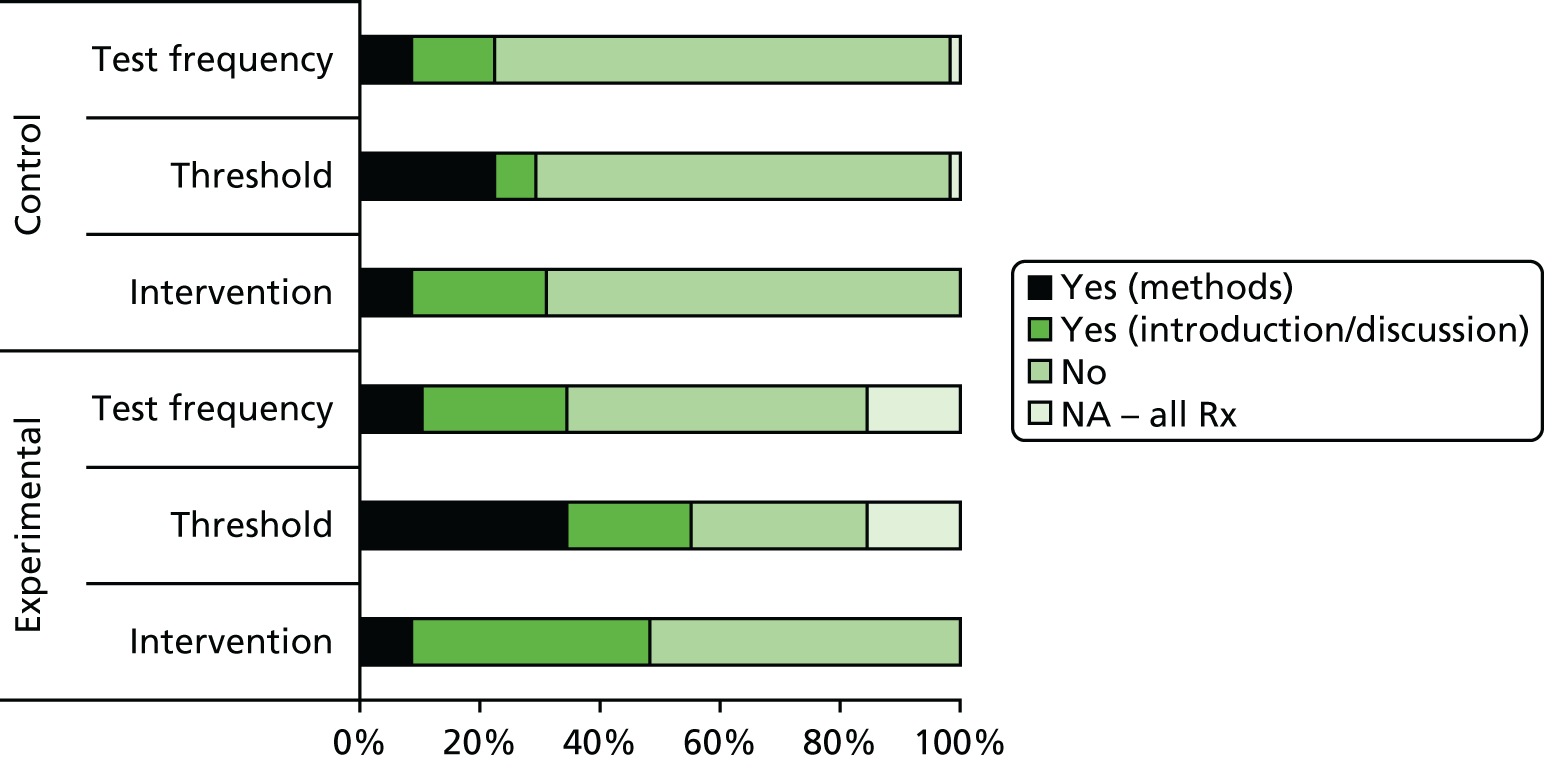
What was the aim of the monitoring evaluation and what change in patient care was implemented?
In 78% (45/58) of the trials, usual care in the control arm was based on some form of clinical assessment (Table 9), often with a focus on one main test (12/58) or more commonly a battery of other tests (21/58). Across the 68 experimental arms, the most common change in monitoring was the addition of a new test to an existing monitoring strategy (n = 29) or as triage to a more invasive test (n = 3). In 12 experimental arms, there was no change in the tests used, but test frequency was increased (n = 5) or decreased (n = 7). In 53% of the experimental arms (36/68), the frequency of outpatient visits undertaken by patients was the same as for the control arm.
| Control arms (n = 58a) | Total, n | % of total (n = 8) | Experimental arms (n = 68a) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Change in testing strategy | Effect on frequency of office visits | |||||||||||
| Addition of test | As triage to existing test | Replacement test | Same test(s) (change in test frequency) | Fewer tests (no monitoring) | Immediate treatment | More | Same | Less | Not reported | |||
| No formal monitoring | 2 | 3 | 4 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 |
| Monitoring focused on single test | 11 | 19 | 2 | 2 | 5 | 5 | 0 | 0 | 3 | 6 | 5 | 0 |
| Clinical assessment | 12 | 21 | 10 | 0 | 0 | 0 | 0 | 2 | 1 | 8 | 1 | 2 |
| Clinical assessment plus one main test | 12 | 21 | 4 | 0 | 0 | 4 | 0 | 5 | 0 | 4 | 4 | 5 |
| Clinical assessment plus multiple testsb | 21 | 36 | 9 | 1 | 5 | 4 | 1 | 5 | 0 | 18 | 2 | 5 |
| Total | 58 | 29 | 3 | 10c | 13d | 1 | 12e | 8 | 36 | 12 | 12 | |
| % of experimental arms (n = 68) | 43 | 4 | 15 | 19 | 1 | 18 | 12 | 53 | 18 | 3 | ||
For most trials, the change in patient management was intended to improve patient outcomes (75%; 51/68), either through earlier initiation of treatment or better selection of patients requiring treatment (Figure 7). This was generally achieved through the addition of a new test to an existing monitoring strategy (55%; 28/51), although, in 12 (24%) studies, triallists evaluated earlier treatment by enrolling patients who had not yet reached the standard (implicit or explicit) threshold for treatment and randomising them to either an immediate treatment option or continued surveillance. In four studies,155–157,226 there was an explicit underlying evaluation of a monitoring strategy as patients in both groups were monitored with a specific test following recruitment and randomised only when their test result crossed a predefined threshold for intervention.
FIGURE 7.
Aim of monitoring evaluation (N = 68 experimental arms). Rx, treated.

For the remainder of the studies, the goal was to maintain the same patient outcomes (25%; 17/68) but to reduce the amount of testing undertaken, either by reducing the number of tests (n = 9) or reducing the amount of invasive testing carried out (n = 7), or introduce surveillance to avoid treatment (n = 1) (see Figure 7). This was achieved by replacing an existing test (n = 4), reducing the frequency of testing (n = 8), adding a new triage test (n = 3) to select patients for a more invasive test or reducing the number of tests performed at each visit (n = 1). The remaining study was a non-inferiority trial that aimed to demonstrate that additional testing did not improve the survival of patients with colorectal cancer. 211
In terms of study validity, sequence generation and allocation concealment were judged to be adequate in 48% and 45% of the studies, respectively (Figure 8). Blinding of study participants, study personnel and outcome assessment was rarely implemented; in the majority of the studies, blinding was not described and a judgement had to be made as to the likelihood of blinding being present or not. Uniform and unbiased outcome assessment (in which the primary outcome was assessed in the same way in both arms and was not determined by the monitoring test under evaluation) was carried out in 83% of studies. In eight trials, however, the presence of the primary outcome was clearly measured by the monitoring test, so that the outcome was defined differently between arms.
FIGURE 8.
Trial validity.

Primary outcomes and results
Approximately half of the trials chose to evaluate patient-related primary outcomes (Table 10), of which one-third (n = 9/31) aimed to assess the impact of monitoring on mortality and over half aimed to detect either new (n = 9/31) or recurrent (n = 7/31) disease. Process outcomes evaluated were primarily related to the numbers of patients treated between arms (n = 8/13) or the time to treatment (n = 4/13).
| Topic | n | % |
|---|---|---|
| Type of primary outcome measures | ||
| Patient | 31 | 53 |
| Process | 13 | 22 |
| Composite | 10 | 17 |
| Unclear/not described | 4 | 7 |
| Patient outcomes used | ||
| Clinical | 4 | 13 |
| Function | 1 | 3 |
| New disease rate | 9 | 29 |
| Recurrent disease rate | 7 | 23 |
| Mortality | 9 | 29 |
| Psychological morbidity | 1 | 3 |
| Process outcomes used | ||
| Diagnostic yield | 1 | 8 |
| Therapeutic yield | 8 | 62 |
| Timing of care | 4 | 31 |
| Was the effect on the primary outcome statistically significant? (n = 34a) | ||
| Yes | 12 | 35 |
| No | 20 | 59 |
| Not reported | 2 | 6 |
Around one-third of the full trial reports (12/34) reported statistically significant effects on the primary outcome (Table 11). The 16 trials reporting power calculations and reporting results as risk differences generally found smaller effects on the primary outcome than were predicted (Figure 9).
| Study features assessed for full trial reports | Results | Chi-squared test | |
|---|---|---|---|
| Non-significant (N = 22) | Statistically significant (N = 12) | ||
| Mean sample size, n | 405 | 778 | |
| Median sample size (range) | 274 (64–1340) | 225 (79–4439) | |
| Industry funded, n/N (%) | 4/22 (18) | 4/12 (33) | p = 0.912 |
| Median follow-up (months) | 35 | 12 | |
| Adequate sequence generation, n/N (%) | 18/22 (82) | 2/12 (17) | p < 0.001 |
| Adequate allocation concealment, n/N (%) | 12/22 (55) | 4/12 (33) | p = 0.24 |
| Outcome assessment blinded, n/N (%) | 3/22 (14) | 3/12 (25) | p = 0.41 |
| No uniform outcome assessment, n/N (%) | 3/22 (14) | 2/12 (17) | p = 0.81 |
| Primary outcome – patient outcome, n/N (%) | 16/22 (73) | 5/12 (42) | p = 0.07 |
| Primary outcome – process outcome, n/N (%) | 4/22 (17) | 5/12 (41) | p = 0.14 |
| Evidence cited (methods) for experimental arm, n/N (%) | 11/22 (50) | 4/12 (33) | p = 0.35 |
| Evidence cited (methods) for control arm, n/N (%) | 6/22 (27) | 3/12 (25) | p = 0.89 |
| Evidence cited (methods) in either arm, n/N (%) | 11/22 (50) | 5/12 (42) | p = 0.64 |
| Any evidence cited for experimental arm, n/N (%) | 16/22 (73) | 9/12 (75) | p = 0.89 |
| Any evidence cited for control arm, n/N (%) | 11/22 (50) | 4/12 (33) | p = 0.35 |
| Any evidence cited in either arm, n/N (%) | 17/22 (77) | 10/12 (83) | p = 0.67 |
FIGURE 9.
Comparison of observed vs. predicted effects.

Discussion
This is the first review that we know of to have examined the use of the randomised trial design to evaluate monitoring tests. We found that triallists have made valiant attempts to evaluate a wide range of monitoring strategies in various medical fields and clinical settings and the number of studies published per year appears to be increasing over time. Many of the strategies examined were relatively complex, involving the addition or replacement of a test within an existing battery of tests or changing the frequency of one or more tests, presenting considerable challenges for their evaluation. Only a small proportion of trials reported statistically significant results for the primary outcome; various possible reasons for this and the impact of other key features of the trials are worth exploring. From data presented in the Hopewell et al. 227 publication bias paper, between 55% and 75% of published trials reviewed in five studies demonstrated statistically significant effects.
In the first instance, a striking lack of scientific basis for the monitoring strategies that were evaluated was presented. Just as reviews of monitoring strategies specified in clinical guidelines have shown,69,134,135 any existing evidence base for the strategies was poorly cited. Potential issues with the tests, thresholds or interventions, which could have been avoided with a thorough evaluation of each component of the new monitoring strategies in the context in which they were to be applied, were identified only in retrospect. 228
Key to this is the consideration of relevant evidence related to the test(s) to be used (in terms of both accuracy and the ability to predate the appearance of clinically relevant disease), the interval between test applications (which should be influenced by the expected natural history of disease and by the degree of measurement variability associated with the test in the given setting) and the change in patient management that is to be implemented (particularly in terms of establishing the effectiveness of any intervention in the trial population). Providing evidence (or, at the very minimum, a rationale) for each of these components is key to the interpretation of trials of monitoring; as others have pointed out, ‘a mediocre test could improve outcomes when it is coupled with effective management; similarly a quality test could fail to improve outcomes in the absence of effective management’. 151
It appears, however, that insufficient attention was paid to establishing test properties and intervention effectiveness in the populations of interest before trials were undertaken. For example, although one might expect both the rate of disease progression and the degree of measurement variability associated with a given test to be taken into account when setting test frequency, test intervals were apparently determined by convenience or by fear of missing a key clinical event. 38 There was little acknowledgement of the potential for false-positive results, limited use of repeated testing to confirm abnormal or indeterminate test results and examples of ‘personal tailoring’ of decisions based on risk from previous tests, for example by changing the frequency of testing or altering thresholds for intervention on the basis of previous test results, were rare. A small number of studies did acknowledge problems with technical aspects of the tests evaluated, or with the test thresholds evaluated,159–162 with one reporting a change to the threshold in the trial protocol (to minimise the number of biopsies undertaken in the experimental arm). 162 Others found that, although the new monitoring strategies identified potential disease earlier in the disease pathway, as was intended, the disease was detected too early to warrant intervention, the intervention itself may have been ineffective or the lack of observed benefit from monitoring may have resulted from the effectiveness of treatment in the control group. 163–165 It is difficult to assess how widespread such issues might be without a more in-depth examination of each topic area; however, it is clear that, before a trial is undertaken, the specifics of the monitoring strategies to be evaluated should be established using appropriate methods and in a similar population to that eligible for the trial.
The second characteristic of the trials was the lack of detailed description of the protocols for monitoring, particularly for the control groups. Overall, the methods of application of the tests and interventions, and of how test results should be used to inform downstream management, were particularly poorly reported. Given the multiple components of a monitoring strategy and the multitude of ways in which these might interact to affect outcomes, a clearly defined protocol for testing and subsequent management is essential, not only so that the ‘intervention’ can be replicated in the future, but also so that the mechanisms by which outcomes are affected can be better understood and the generalisability of the results beyond the trial can be judged. 151,228 The reasons behind the lack of description are not clear. It may be partly down to poor reporting but could also be related to a lack of acknowledgement of testing strategies as complex interventions or an unwillingness to standardise a complex intervention. 229
Even for those elements of the intended monitoring strategies that were described, we found some evidence of lack of compliance, for example trials reporting the use of the experimental test or the use of extensive additional testing in the control arm; failure to administer treatment despite positive test results; and the prescribing of treatment despite negative test results. 160,163,166–168 This apparent lack of ‘buy in’ by clinicians is difficult to interpret. One explanation might be that a monitoring test is considered more of a guide to potential changes in management rather than as a definitive indication for a particular intervention. The often lengthy time frame of monitoring may also increase the likelihood of trial fatigue, making the intended intervention difficult to implement over long periods. Delaney et al. 228 advocate that a variety of methods might be used to ensure that complex interventions are delivered reliably over a period of time. Alternative designs might also be considered, particularly when monitoring relies primarily on a single test. For example, monitoring of all enrolled patients with randomisation to immediate treatment or a continued surveillance option on crossing a particular threshold leaves less to chance in terms of downstream management. 155–157,169
In terms of study validity, trials of tests should be subject to the same validity standards of randomisation and allocation concealment as the wider RCT literature; however, in many instances, the same standards of blinding of patients, and particularly clinicians, will be difficult to achieve (Box 1). In theory, patient or clinician blinding might be more easily implemented for simpler tests such as blood tests or when tests are applied by a non-treating clinician such as a radiographer, but in many contexts blinding could be more difficult and even inappropriate given that it is not just the test per se that is being evaluated but its interaction with other components of the overall monitoring process. Although we found two examples in which stringent attempts at blinding patients mitigated the potential benefit from monitoring (both using implanted devices),170,171 the full impact of not using blinding in a monitoring trial is as yet unclear. Nevertheless, its impact may be mitigated through the use of objective outcome measures and blinded and uniform outcome assessment. Blinded outcome assessment should be feasible for most trials, but its use was reported for less than one-fifth of our sample and in over half of these trials the blinding related to outcome adjudicators rather than those collecting the outcome data. Furthermore, we observed a small number of trials with a ‘fatal flaw’ in terms of the outcome assessment, whereby the presence of the primary outcome, for example recurrence of cancer, was defined by a different test in each arm, thereby introducing an additional source of bias. It is fundamentally important that the primary outcome is uniformly defined in the same way between groups, ideally at the same point in time.
The adequacy of random sequence generation and allocation concealment was judged according to The Cochrane Collaboration’s risk of bias assessment tool:230
-
Sequence generation:
-
Adequate – random number table, computer random number generator, coin tossing, shuffling of cards/envelopes, throwing dice, drawing lots, minimisation.
-
-
Allocation concealment:
-
Adequate – central allocation (including telephone, web-based and pharmacy-controlled randomisation), sequentially numbered, opaque sealed envelopes.
-
The presence of blinding was judged using the risk of bias assessment tool supplemented with the instructions for estimating unclearly reported blinding status outlined by Akl et al. :231
-
Blinding [assessed for four groups: (1) patients, (2) treating clinicians (i.e. those making subsequent management decisions), (3) non-treating clinicians (e.g. those undertaking the monitoring test) and (4) outcome assessment (both primary and secondary, e.g. outcome adjudicators)]:
-
Explicit statement that a group was blinded – definitely yes.
-
Explicit statement that a group was not blinded – definitely no.
-
Explicit statement that investigators were blinded – definitely yes for clinicians and outcome assessors.
-
Explicit description of the trial as ‘open’ or ‘unblinded’ – definitely no.
-
If no explicit statement about blinding status – probably no.
-
Described as single or double blinded – use best judgement to assign ‘probably yes’ to one or more groups as appropriate.
-
The method of assessment/definition of the primary outcome was appraised for both groups:
-
Uniform outcome assessment:
-
Present if the primary outcome was defined/measured in the same way in all groups or if attempts were made to ensure that the primary outcome definition captured relevant events in both groups.
-
Absent if differences in measurement of the primary outcome were likely to have led to bias between the groups (i.e. if the monitoring test result was used to define the primary outcome, e.g. when the presence of recurrence was defined by an existing test in one arm and by the new test in the experimental arm).
-
Unclear – if insufficient information was available to judge.
-
A final characteristic of these monitoring trials was an apparent lack of power to detect significant effects. This is not a phenomenon limited to trials of tests; previous authors have found sample size calculations to be based on inaccurate assumptions for the control group, with others suggesting that up to 10–20% of trials might be inappropriately discontinued because of a perceived insufficient rate of recruitment. 232–234 However, statistically significant effects are easiest to achieve when two groups are allocated to different treatments; in trials of testing strategies all ‘test-positive’ patients usually undergo the same treatment, reducing the potential to demonstrate clear differences in outcome between groups. We have not yet conducted an in-depth look at the sample size calculations of trials in our sample and cannot yet comment on the assumptions made around the predicted benefit from monitoring; however, we did find evidence of lower than expected control group event rates both in trials that were stopped early and other trials, with some forced to revise their sample size calculations or change their primary outcome from overall survival (OS) to a surrogate outcome of number of recurrences treated surgically with curative intent. 156,170–173
Our research has some limitations. We are unlikely to have retrieved all of the available eligible trials. This is in part because of our focus on one main database, although this was supplemented with searches of more specialist resources, and in part because of a lack of standard terminology for trials of monitoring, which made searching for trials a challenge. However, an exhaustive search is not as important for a methodological review as for a systematic review of effectiveness. We did not aim to systematically identify all of the available trials but to retrieve a sample of trials that provide a good representation of those available. Although other trials may be available, it seems likely that our review has flagged up many of the key issues.
Our review has identified a range of problems with available randomised trials of monitoring, raising real questions regarding the feasibility and appropriateness of this approach for evaluating monitoring. Trial investigators have perhaps underestimated the complexity of the interventions that they were trying to evaluate and the multitude of ways in which the effect of an intended change in a monitoring strategy might be mediated by other factors. The recommendations in Box 2 provide guidance for future researchers evaluating monitoring strategies.
Triallists should:
-
provide a scientific basis, or at a minimum a carefully considered rationale, for the monitoring strategy to be evaluated, including the –
-
test interval
-
test threshold
-
intervention(s) to be used following a positive test result
-
-
ensure that the test(s) operate as expected and that the interventions are effective in the intended patient population
-
provide clear guidance to clinicians taking part in the trial regarding how they are expected to respond to a positive, indeterminate or negative monitoring test result
-
make stringent attempts to avoid known biases, for example using –
-
proper randomisation
-
adequate concealment of allocation
-
blinded outcome assessment
-
-
avoid additional bias from non-uniform outcome assessment between study arms [i.e. the (primary) outcome must not be determined by the monitoring test under evaluation]
-
ensure that the trial follow-up is sufficiently long to allow important events to occur
-
be realistic with estimates used in sample size calculations
-
follow the Consolidated Standards of Reporting Trials (CONSORT) guidelines for trial reporting235 and clearly report the care provided in both the experimental and the control arms of trials.
Chapter 5 A review of the monitoring-related methodology literature
Introduction
Monitoring strategies used to direct the care of patients with potential recurrent or progressive disease are rarely evidence based. 69 Monitoring strategies specify the frequency of observations, the duration of monitoring, the decision rule and the threshold for a positive test result, with a positive result prompting a change in patient management. There is a need for monitoring strategies to be developed based on evidence of how a disease will progress and the performance of the monitoring test to be used. Too often test frequencies are based on routine care schedules with decision rules and thresholds chosen in an ad hoc manner.
Methods
Methodological information related to monitoring was first sought from the first edition of the book Evidence-Based Medical Monitoring,38 edited by Paul Glasziou, Les Irwig and Jeffrey Aronson. Key textwords related to monitoring methodology were identified and purposive searches of MEDLINE were undertaken from 2000 to 2010 (searches conducted on 26 March 2010). Reference tracking and citation tracking using Science Citation Index were used to identify additional relevant literature.
A variety of searches were performed to identify relevant literature using various combinations of the following text words:
-
monitor*
-
measure* or biomarker* or marker*
-
serial or repeat* or periodic or longitudinal or trajectory*
-
recurrence or progression
-
rule* or threshold* or trigger
-
statistical process control or control chart* or reference change value or critical difference
-
screen* with frequenc* or intensit* or interval*
When necessary, the results were filtered to select:
-
statistical or epidemiological journal titles (Biostatistics, Biometrics, Statistics in Medicine, Methods of Information in Medicine, Lifetime Data Analysis, Journal of Clinical Epidemiology, American Journal of Epidemiology and Annals of Epidemiology)
-
RCTs (sensitive search).
Additional searches were undertaken to identify literature related to statistical process control, RCVs and the methodology of health screening.
The papers selected for review are summarised in Table 12 and in the following sections.
| Study | Design | Analysis | Citationsa (n) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Test frequency | Test thresholds | Decision rules | Review of methods | Other | General data structure | Linear mixed-effects modelling/SNR | Joint modelling | Cost-effectiveness | Review of methods | Other | ||
| Monitoring | ||||||||||||
| Ahdieh-Grant 2003236 | ✓ | ✓ | 32 | |||||||||
| Bell 2008237 | ✓ | 19 | ||||||||||
| Bell 2009238 | ✓ | 36 | ||||||||||
| Bell 2009239 | ✓ | ✓ | 13 | |||||||||
| Bell 2011240 | ✓ | 8 | ||||||||||
| Bellera 2008241 | ✓ | 12 | ||||||||||
| Bellera 2008242 | ✓ | ✓ | ✓ | 2 | ||||||||
| Bellera 2009141 | ✓ | ✓ | ✓ | 5 | ||||||||
| Buclin 2011146 | ✓ | ✓ | ✓ | ✓ | 6 | |||||||
| Cole 2004243 | ✓ | 28 | ||||||||||
| DeLong 1985244 | ✓ | 20 | ||||||||||
| Glasziou 2007245 | ✓ | 195 | ||||||||||
| Glasziou 2008143 | ✓ | 55 | ||||||||||
| Inoue 2004246 | ✓ | 23 | ||||||||||
| Keenan 2009144 | ✓ | 40 | ||||||||||
| Li 2012247 | ✓ | ✓ | 1 | |||||||||
| Oke 2012248 | ✓ | ✓ | 2 | |||||||||
| Powers 2011249 | ✓ | 50 | ||||||||||
| Proust-Lima 2009250 | ✓ | 25 | ||||||||||
| Proust-Lima 2014251 | ✓ | ✓ | 3 | |||||||||
| Slate 2000252 | ✓ | 44 | ||||||||||
| Sölétormos 2000253 | ✓ | ✓ | 7 | |||||||||
| Stevens 2010142 | ✓ | ✓ | ✓ | 8 | ||||||||
| Subtil 2010254 | ✓ | 3 | ||||||||||
| Takahashi 2010255 | ✓ | ✓ | 16 | |||||||||
| Takahashi 2012256 | ✓ | ✓ | ✓ | 2 | ||||||||
| Taylor 2005257 | ✓ | ✓ | 31 | |||||||||
| Thiébaut 2003258 | ✓ | ✓ | 18 | |||||||||
| Thompson 1990259 | ✓ | 26 | ||||||||||
| When To Start Consortium 2009260 | ✓ | ✓ | ✓ | 389 | ||||||||
| Wolbers 2010261 | ✓ | ✓ | 22 | |||||||||
| Biomarker design, analysis and development | ||||||||||||
| Baker 2000262 | ✓ | 58 | ||||||||||
| Baker 2006263 | ✓ | ✓ | 41 | |||||||||
| Baker 2009264 | ✓ | ✓ | 15 | |||||||||
| Lumbreras 2008265 | ✓c | 5 | ||||||||||
| Parker 2010266 | ✓d | 8 | ||||||||||
| Pepe 2001267 | ✓ | ✓ | 670 | |||||||||
| Pepe 2008268 | ✓ | ✓ | 206 | |||||||||
| Ransohoff 20071 | ✓ | ✓ | 83 | |||||||||
| Ransohoff 2010269 | ✓ | ✓ | 73 | |||||||||
| Sturgeon 2009270 | ✓b | 28 | ||||||||||
| Sturgeon 2010271 | ✓ | ✓ | 16 | |||||||||
| Screening | ||||||||||||
| Day 1984272 | ✓ | 119 | ||||||||||
| Etzioni 1997273 | ✓ | 9 | ||||||||||
| Frame 1998274 | ✓ | 27 | ||||||||||
| Lee 1998275 | ✓ | 31 | ||||||||||
| Lee 2004276 | ✓ | 13 | ||||||||||
| McIntosh 2002277 | ✓ | 40 | ||||||||||
| McIntosh 2003278 | ✓ | 36 | ||||||||||
| Walter 1983279 | ✓ | 91 | ||||||||||
| Zelen 1993280 | ✓ | 49 | ||||||||||
| Time-dependent ROC curves | ||||||||||||
| Cai 2006281 | ✓ | 26 | ||||||||||
| Etzioni 1999282 | ✓ | 42 | ||||||||||
| Parker 2003283 | ✓ | 14 | ||||||||||
| Pepe 2008284 | ✓ | ✓ | 24 | |||||||||
| Slate 2000252 | ✓ | 44 | ||||||||||
| Subtil 2009285 | ✓ | 3 | ||||||||||
| Zheng 2004286 | ✓ | 21 | ||||||||||
| Variability | ||||||||||||
| Biosca 2006287 | ✓ | ✓ | 3 | |||||||||
| Clerico 2004288 | ✓ | 239 | ||||||||||
| Fraser 1990289 | ✓ | 57 | ||||||||||
| Fraser 2001290 | ✓ | – | ||||||||||
| Klee 2010291 | ✓ | 39 | ||||||||||
| Macaskill 2008292 | ✓ | ✓ | ✓ | 0 | ||||||||
| Omar 2008293 | ✓ | ✓ | 10 | |||||||||
| Petersen 2005294 | ✓ | – | ||||||||||
| Petersen 2012295 | ✓ | 5 | ||||||||||
| Smellie 2008296 | ✓ | ✓ | 15 | |||||||||
| Sölétormos 2000297 | ✓ | ✓ | 3 | |||||||||
| Statistical process control | ||||||||||||
| Gavit 2009298 | ✓ | 3 | ||||||||||
| Macaskill 2008292 | ✓ | ✓ | ✓ | 0 | ||||||||
| Tennant 2007299 | ✓e | 18 | ||||||||||
| Thor 2007300 | ✓ | ✓e | 90 | |||||||||
| Decision-analytic models | ||||||||||||
| Baker 1998301 | ✓ | ✓ | 8 | |||||||||
| Karnon 2007302 | ✓ | ✓ | ✓ | ✓ | 20 | |||||||
| Parmigiani 1997303 | ✓ | ✓ | 10 | |||||||||
| Sutton 2008304 | ✓ | ✓ | ✓ | 26 | ||||||||
| Real options approaches | ||||||||||||
| Driffield 2007305 | ✓ | ✓ | 17 | |||||||||
| Lasserre 2006306 | ✓ | ✓ | 6 | |||||||||
| Meyer 2012307 | ✓ | ✓ | 1 | |||||||||
| Palmer 2000308 | ✓ | ✓ | 66 | |||||||||
| Shechter 2010309 | ✓ | ✓ | 0 | |||||||||
| Whynes 1995310 | ✓ | ✓ | 2 | |||||||||
Results: development and evaluation of monitoring strategies
Limited methodological literature was identified that provides guidance on the design of studies to evaluate monitoring tests. Work has focused more on analytical techniques to assist with the design of monitoring strategies, primarily through analysis of existing data in order to make recommendations on monitoring frequency or decision rules, or simulation work, with both approaches being specific to the disease area researched.
Designing studies to evaluate monitoring strategies
A small number of papers from the biomarker development field cover study design issues of relevance to the monitoring field; however, these largely take the form of commentary papers highlighting study design issues often not considered in biomarker development, rather than reporting empirical work.
Pepe et al. 267 discuss the five stages of biomarker development for cancer: preclinical exploratory studies, clinical assay development for clinical disease, retrospective longitudinal repository studies, prospective screening studies and cancer control studies. The initial stage is the primary search for promising biomarkers; this stage is preclinical and generally involves comparison of diseased and non-diseased tissue for many potential biomarkers. At this initial stage, biomarkers are assessed on their ability to produce stable results and to discriminate between disease and non-disease. The next stage sees the development of a clinical assay that can be carried out non-invasively, with the discriminative ability of the assay being assessed. The third stage of the process involves comparing the ability of the biomarker to differentiate between samples taken from patients with disease prior to the diagnosis of disease and samples taken from patients who are free of disease, with the aim of this stage being to evaluate whether or not the biomarker can detect preto clinical disease and understand what biomarker values should be used to classify a result as positive. The fourth stage uses the biomarker to prospectively screen patients, with those having a positive result also receiving definitive diagnostic tests; this stage of the process allows the stage of disease that the biomarker is able to detect to be identified, as well as the likely positive and false-positive yield of the test. The final stage of the process is to test the benefit of screening to the population in terms of reducing mortality. In this last stage participants should be representative of the population to be screened and a RCT approach can be taken to understand the difference between groups offered screening with the biomarker and those not undertaking screening.
Biomarker development studies have been criticised on a number of counts:1 for producing novel findings that are often unreliable and not replicable and being open to bias, both before and after the laboratory receives the study samples, primarily because of a lack of standardisation. A number of papers have discussed issues of bias in both randomised and non-randomised study designs and, to improve the development and evaluation of biomarkers, have provided recommendations on both study design and analysis. 1,263,264 Baker et al. ,263 Baker,264 Ransohoff1 and Ransohoff and Gourlay269 provide further discussion of other issues related to biomarker development studies.
Lumbreras et al. 265 present a tool for evaluating the quality of test accuracy studies of new biomarker (or ‘-omics’) technologies and Parker et al. 266 report an evaluation of the QUADOMICS tool [an adaptation of the Quality Assessment of Diagnostic Accuracy Assessment Studies (QUADAS) tool311].
In terms of specific study designs, in an overview paper of study design and methods for evaluating biomarkers for the early detection of cancer, Baker et al. 263 cover four different types of prospective study design:
-
a cohort-type design in which asymptomatic patients are tested with a biomarker and followed up to clinical diagnosis may be an option when there is limited available evidence of benefit of the biomarker and no ethical grounds to make decisions based on the test result
-
a longitudinal accuracy study in which all asymptomatic patients are tested with a biomarker and all patients receive a biopsy or definitive testing; however, some concerns have been raised around this design because of overdiagnosis
-
a randomised trial design in which sensitivity is estimated using the number of positive tests and the number who develop disease within the time frame
-
a design in which only those asymptomatic patients with a positive biomarker result receive definitive testing, with discussion of the issue of patients with a negative result not receiving definitive testing.
Pepe et al. 268 further discuss a study design entitled the PRoBE design (prospective-specimen-collection, retrospective-blinded-evaluation). In this paper they suggest the prospective nested case–control design as a means of ensuring that biomarker development is rigorous and robust. In the first instance, specimens should be prospectively collected from a randomly selected cohort reflective of the population in which the proposed biomarker would be used, and then stored, prior to development of the outcome of interest. Cases and controls (i.e. those who do/do not experience the clinical event of interest) are then randomly selected and their specimens are retrieved from storage and tested for the biomarker of interest, blinded to case/control status. The authors also suggest that the performance that would be required of a new biomarker be established in a clinical context. The design has clear potential for application in a monitoring context, with the possibility of obtaining repeat biomarker measurements on a relatively frequent schedule, so that the best threshold and test interval can be determined from the data.
A further design proposed for the evaluation of a biomarker as a screening test, which is, therefore, potentially applicable to the monitoring of disease, is the paired design, in which different screening strategies are introduced at different centres and screening performance is assessed by comparing the number of interval cases (symptomatic cases detected in the interval after screening) observed. 264
Analytical approaches to developing monitoring strategies
The literature in the area of monitoring focuses on modelling approaches (linear mixed modelling, joint modelling and non-linear modelling). The review of the literature also identified some simulation studies and some work on the evaluation of monitoring strategies.
Linear mixed-effects models and estimation of signal-to-noise ratio
Glasziou et al. 245 question the need for RCTs of monitoring under certain circumstances, pointing to the need to understand the background variation and evaluate the signal-to-noise ratio when assessing treatment effects. They suggest that large estimated treatment effects would be required to demonstrate an effect.
General description of models
Stevens et al. 142 have reviewed statistical models used for the control phase of monitoring and explain how models can be fitted to observed monitoring data, providing details of maximum likelihood methods, moment-based methods and literature-based methods, with parameter estimates obtained from reviewing the literature. They introduce a generic model for monitoring data, defining Yit as the observed monitoring values, including assay noise and variability, and Uit as the ‘true’ underlying and unobserved values:
where αi is the true value at time 0, βi,t is the change in the true value over time and ωi,t is random error.
Signal and noise
Modelling methods are used with repeated test data in the hope of distinguishing ‘signal’ from ‘noise’. The ‘noise’ is normal fluctuation in test results for patients (caused by the measurement variability of the test) and the ‘signal’ is a change in test results signifying a true change in disease state.
Thompson and Pocock259 present their findings following the analysis of repeated serum cholesterol measurements in 14,600 men and women. Their work focused on the impact of within-individual variability on screening and monitoring. Using the cholesterol data, a single observed measurement did not reflect the true underlying value. They showed how the probability of a measure being classed as ‘high’ varied with the true underlying value and whether the classification was based on a single measure or the mean value of multiple measures. The use of multiple measures was shown to improve classification. The authors identified regression to the mean when analysing multiple measures and variability in the measures for untreated individuals over time, leading them to doubt whether or not repeated measures would be able to identify the benefit of treatment. The authors state that the use of repeated measures could be ‘very discouraging’ for some patients.
Buclin et al. 146 defined two decision rules that could be used to guide the treatment of patients with HIV infection with antiretroviral therapy based on cluster of differentiation 4 (CD4) cell measurements using a review of longitudinal analyses of CD4 cell trajectories. The first decision rule is a ‘snap-shot rule’ – dependent on a single CD4 measure – and the other is a ‘track-shot rule’ – in which multiple CD4 measurements are required. The devised rules are then tested using clinical data, with the view of minimising false findings, and recommendations are made regarding the frequency of testing.
Bell et al. 237 developed a framework to identify when monitoring of initial response to treatment would be beneficial using data from RCTs. The findings showed that monitoring of initial response to treatment would be useful only when there is variation in the treatment effect between patients and not all treated patients achieved results at the level targeted.
Other examples of the use of mixed modelling and signal-to-noise ratio estimation to understand when it is appropriate to monitor response to treatment, thresholds or monitoring frequency include for the monitoring of cholesterol, bone mineral density, blood pressure, lipids and diabetes mellitus. 143,144,237–240,248,249,255,256
Joint modelling of longitudinal and outcome data
Joint latent class models
When fitting a joint latent class model subjects are split into a finite number of latent subgroups. The trajectory of biomarker measurements and the risk of an event are specific to each latent class, meaning that the joint latent class model allows for the dependency of biomarker values and the risk of the event. Biomarker measurements and time to event are conditionally dependent, given the latent class. More specifically, a multinomial logistic regression model is used to assign subjects to subgroups. A linear mixed model is then used to model repeated biomarker measurements given the assigned latent class of the subject and a survival model is used to model the time to event, again given the latent class of the subject. The model is fitted using maximum likelihood estimation. 251
Examples of the use of joint latent class models can be seen in work by Proust-Lima and Taylor250 and Li and Gatsonis,247 both applied to monitoring with PSA for prostate cancer recurrence.
Proust-Lima and Taylor250 discuss the derivation of a posterior probability of recurrence from a joint latent class model to identify a ‘dynamic prognostic tool of recurrence’. The posterior probability obtained from the joint latent class model gives the probability of an event occurring between time s and time s + t (with the subject being event free at time s). Estimating the probability of an event after a certain time requires fitting survival models to subjects at each time being estimated with only covariates available at time s. As biomarker data are often discrete, imputation techniques are used to allow predictions of an event to be obtained at multiple time points. Proust-Lima and Taylor250 also discuss the validation of predictive tools and the lack of consensus in this area.
Li and Gatsonis247 use a joint latent class model to develop a strategy that modifies monitoring intervals. They use a two-stage approach when fitting the joint latent class model in which the model used to identify latent classes is fitted separately. The Bayesian information criterion is used to select the number of classes. The two-stage approach has the advantage of being less computationally intensive and the expectation–maximisation (EM) algorithm can be used to estimate parameters at each point of monitoring. The uncertainty of latent class assignment is evaluated using multiple imputation, assuming that latent class is missing completely at random. For prospective studies the two-stage procedure is repeated as new information is collected (measures, events and study end). Li and Gatsonis247 demonstrate the method using simulated PSA measurements for 150 patients with prostate cancer with testing to identify recurrence. Predictions from the model inform a utility function, which is used to identify the appropriate monitoring intervals for each patient. The expected value of the utility function used is Ũ (t) = aP(event at time t), where a is a negative value if the event occurs and zero otherwise. The optimal monitoring interval can be identified for individuals or groups of patients; the authors advocate optimising by latent class as these intervals can then be adapted for new patients.
Bayesian hierarchical change-point models
Bayesian hierarchical change-point models model the trajectory of test results prior to the onset of disease, the onset of disease and the trajectory of test results after the onset of disease simultaneously. These models also allow for the within-individual correlation, as individuals have multiple test measurements, between-subject variation in trajectories and the random change-point. Bayesian hierarchical change-point models use a piecewise or segmented linear model in which the parameters of the model are the trajectory of test results prior to the change point, the test result value at the time of the change point, the time of the change point and the trajectory of test results after the change point; each of the parameters is a random effect within the model. Non-informative prior distributions are used for the parameters in the model, with the parameters describing the distributions of the parameters used in the model being drawn from non-informative prior distributions. 241,252
Slate and Turnbull252 discuss and demonstrate the use of Bayesian hierarchical change-point models using PSA data from the Nutritional Prevention of Cancer Trial. They state that the advantages of using Bayesian hierarchical change-point models are the ‘borrowing of strength’ when estimating parameters specific to individuals whilst also accounting for the correlation of measures and, by obtaining posterior distributions using Gibbs sampling, the model can give the probability that an individual has reached the change point.
Bellera et al. 241 also demonstrate the use of Bayesian hierarchical change-point modelling using PSA data. They state that the additional advantages of this type of modelling are the ability of the model to provide precise estimates compared with simpler models, that the parameters used by the model are all of clinical importance, that estimates of test measurement variability can be estimated as a function of the test result value and that the model is flexible and can be easily adapted. Bellera et al. 241 do, however, comment that the model can be influenced by the timing of and the number of test measurements for individuals, with the potential for this to cause bias, as participants with more test results will provide more information for the model and participants with more test results may be different from those with fewer test results. Subsequent work by Bellera et al. 241 uses an empirical simulation approach with Bayesian hierarchical change-point modelling to evaluate and compare different rules used in detecting the recurrence of prostate cancer based on PSA measurements. 141 Bayesian hierarchical change-point models were used to identify whether or not the rules used in practice were able to adequately classify patients with real progression of PSA measurements and those with stable PSA values.
Inoue et al. 246 combined longitudinal PSA measurements from three different studies using a non-linear Bayesian hierarchical model. At the individual level a non-linear model is used to model PSA over time and the hierarchical model component then accounts for the variability between studies.
Non-linear mixed models
Non-linear mixed models allow more flexibility in modelling as linearity of the parameters is not necessary, which may be appropriate for modelling longitudinal test data in some conditions. Multiple measures for each individual can also be accounted for by non-linear mixed models with the incorporation of random effects.
Examples of the use of non-linear mixed models can be seen in work by Subtil and Rabilloud254 and Taylor et al. 257
Alternative modelling approaches
Alternative modelling approaches are used by Thiébaut et al. 258 and Wolbers et al. 261 in the area of CD4 cell monitoring to understand when to initiate treatment for patients with HIV infection.
Machine learning methods
We have also considered machine learning methods. These essentially arise from a set of pattern recognition processes or algorithms. They are primarily concerned with non-continuous data, such as radiographic images or electronic health records, and for machine learning methods to provide high-quality predictions these pattern recognition processes would usually be applied to large data sets. 312,313 The biomarker data are continuous and a limited number of data are available. The statistical analysis and modelling approach also enables testing of different trial design strategies, as discussed extensively in Chapter 7. In conclusion, we believe that machine learning technologies may have a future role in shaping and supporting monitoring strategies. However, it is too soon to say in what form this role may be expected to develop. Machine learning methods are still in the developmental stage and may become useful in the field of biomarkers in the future, as the field develops. Issues remain about such methods not always providing estimates of uncertainty and being hard to interpret. The logic and mathematics to underpin these processes are still being developed and are not trivial. The task of modelling these data sets is very complex and requires many assumptions, which need verifying, as for statistical approaches. 312,314 Use of machine learning methods is currently limited by the lack of availability of simple off-the-shelf application packages.
Monitoring simulation studies
With knowledge of the progression of disease and the variability of the test used to monitor the disease, data can be simulated allowing the evaluation and comparison of decision rules and testing frequency. Simulation approaches have been used by Sölétormos et al. 253 and Bellera et al. 242
Evaluation of monitoring strategies
How the performance of a monitoring strategy is measured will be different from, and more complex than, the measurement of the performance of testing at a single time point, because of repeated testing and the potential for patients to change disease state. DeLong et al. 244 discuss the sensitivity and specificity of monitoring tests and Li and Gatsonis247 provide guidance on the evaluation of monitoring strategies.
Pooled analysis of prospective cohort studies
In some disease areas data sets from multiple cohort studies exist and it is possible to combine the data and analyse the pooled group of patients. When the data in multiple cohort studies are combined, the data set may be useful to allow analyses that compare groups of patients, correct for case mix and allow investigators to infer the findings, which might otherwise require a RCT. The cohort approach has been used to evaluate the appropriate CD4 cell level at which to begin antiretroviral treatment for patients with HIV infection by the When To Start Consortium260 (using a method introduced by Cole et al. 243) and Ahdieh-Grant et al. 236
Results
Screening literature
The aim of screening is to benefit patients by detecting disease prior to the onset of symptoms, as is the case with monitoring. The detectable preclinical stage of disease is the time when screening may detect asymptomatic disease; this is also known as the sojourn time. The delay time is the period of the sojourn time when the screening has not detected disease and the lead time is the period of the sojourn time after screening has detected disease. The greater the lead time, the greater the potential benefit of screening.
Walter and Day279 discuss the biases that need to be considered when analysing screening data. First, the population participating in screening may vary from the population not participating in screening, having a higher or lower risk of having the disease that the screening process aims to detect. This is likely to be less of an issue for monitoring populations, although it is conceivable that there will be differences between those who do participate in monitoring and those who either drop out (perceiving themselves to be at low risk of the event in question) or who demand some form of treatment (perceiving themselves to be at high risk of the event in question). Other biases that may affect monitoring studies include length-biased sampling and lead time bias. Length-bias sampling occurs as patients with more aggressive disease will be in the preclinical phase of disease when screening will detect disease (sojourn time) for a shorter length of time than those with less aggressive disease. Screening is most likely to detect cases with a longer sojourn time, hence cases of less aggressive disease, which will likely have a better prognosis. Lead time bias is when survival times for screened cases appear to be greater than survival times for cases identified by different means when there is actually no difference in survival; the only difference is that cases identified by screening are detected earlier.
There is a body of work in the area of screening that focuses on estimating the duration of the preclinical stage of disease,272,273,279 which enabled further work into the optimal frequency of screening. 274–276,280 Others have considered how to set the optimal decision rule for a screening strategy using a new test when the length of the sojourn period is not known. 277,278
Biomarker development process
Methods for analysis and study design used in the biomarker development process
The methods for the analysis and design of studies of biomarker development are discussed by Baker et al. ,263 Pepe et al. 268 and Sturgeon et al. 271 Baker262 discusses a method for evaluating multiple biomarkers for selection for further study. Baker et al. ,263 Baker,264 Ransohoff1 and Ransohoff and Gourlay269 discuss the issues around biomarker development studies.
Sturgeon et al. 270 provide information on the biomarkers that have been developed to identify cancer and the extent of their use in practice.
Time-dependent receiver operating characteristic curves
When a test provides a binary result (positive or negative) the performance of the test is usually assessed by calculating sensitivity and specificity. When the result of a test is a continuous value the performance of the test is evaluated for various cut-off points by calculating the sensitivity and specificity of the test for each possible result value and plotting sensitivity against 1 – specificity, a ROC plot. The ROC plot and the AUC produced can then be used to assess the performance of the test and identify optimal thresholds for use of the test in practice. When allowing for time in ROC analysis, time-dependent ROC methods are used.
Sensitivity and specificity
Pepe et al. 284 undertook a review of time-dependent ROC curves. The definition of the sensitivity of a test is dependent on the time when the test is performed. As it is assumed that diseased cases will present with positive test values early in the testing process, it is thought that sensitivity will decrease with time. Pepe et al. 284 also discussed cumulative sensitivity, which would provide the sensitivity of a test for an interval of time, and how this can be derived. The false-positive fraction, or 1 – specificity, is problematic to define as the disease status of individuals can change over time, making it difficult to classify individuals as diseased or non-diseased, especially in situations in which all individuals will have an event at some point. One approach is to choose a time point specific to the context being assessed, with individuals treated as non-diseased if they are free of the event at the specified time (the static false-positive fraction). Another approach is to allow the false-positive fraction to vary with the time since the test was performed (the dynamic false-positive fraction). When using the dynamic false-positive fraction test performance may be misleading as a positive result will be falsely positive shortly before an individual develops disease. For tests with continuous results, time-dependent ROC curves compare individuals with and without disease at each time point. If using the dynamic false-positive fraction, ROC curves are difficult to interpret because of the non-diseased group changing over time.
Cai et al. 281 present equivalent time-dependent definitions of sensitivity and 1 – specificity, but with the emphasis on the time that an event occurs, defining sensitivity and 1 – specificity as functions of time relative to the time of disease or time of an event. The authors state that most research in this area assumes that the test and assessment of disease status are carried out simultaneously, raising the issue of the predictive accuracy of a test being dependent on the time that it is carried out in comparison to the onset of disease, assuming an increase in accuracy if the test is carried out closer to the time of an event. Cai et al. 281 also fit semiparametric models using longitudinal test data to separately estimate sensitivity and 1 – specificity. Zheng and Heagerty286 discuss sensitivity and specificity for time-dependent ROC analysis as functions of both the time of testing and the time of an event. They also discuss the difference between estimating incident and estimating prevalent ROC curves, restricting their work to incident ROC curves.
Subtil et al. 285 discuss how estimation of incident sensitivity requires a test to be performed a given number of days prior to the onset of disease and offers a way of taking into account the variation in time between individuals receiving a test and individuals developing disease. They introduce a Bayesian method to allow for the interval-censored measurements. The results from using this method compared with the method without adjustment suggest that the ‘crude’ method underestimates sensitivity.
Parker and DeLong283 provide a method to convert estimates of sensitivity and specificity for monitoring tests for ROC curve analysis. The estimates of sensitivity and specificity used are those introduced by DeLong et al. ,244 which are derived using partial likelihood estimation under the assumption that diseased participants can have at most one test result when in the diseased state.
Modelling to produce time-dependent receiving operating characteristic curves
Slate and Turnbull252 review methods used to analyse repeated test data when the test is used to screen or monitor for the onset of disease in a population. These methods are used to estimate the ROC curve for each test and the resulting ROC curves are compared. The review discusses the use of time-dependent Cox proportional hazards modelling, joint modelling of longitudinal test data and time of diagnosis, Weibull methods to model two time events, random-effects models and integrated Onstein–Uhlnbeck stochastic processes, multistate models and Markov models and change-point models.
Zheng and Heagerty286 discuss a semiparametric regression approach used to estimate ROC curves and an approach based on asymptotic distribution theory, which will allow covariates to change the distributional shape of test results.
Etzioni et al. 282 introduce and demonstrate two methods for modelling the effect of lead time on the ROC curve. The first approach requires modelling of longitudinal test data; then, using parameter estimates from the model the ROC curve can be estimated at varying time points. The second approach directly models the ROC curve as a function of covariates, including the time of the test relative to the time of diagnosis. Etzioni et al. 282 discuss how the methods can be adapted to compare two tests. The first method requires separate fitting of models using data for the two tests followed by comparison of the derived ROC curves, whereas the approach of modelling the ROC curve directly more easily allows for a comparison of tests and the difference between tests to be assessed. Other advantages of the direct modelling approach are fewer distributional assumptions with the method using the ranking of data points, robustness and flexibility and ease of implementation.
Differentiating measurement change from measurement variability
Variability: reference change values and coefficients of variation
The variability of repeated test measures for an individual can be broken down into three components: pre-analytical variability, analytical variability and individual variability. Analytical variability is the variation in results caused by the laboratory test and individual variation is the within-patient variability. Pre-analytical variability can be minimised by ensuring that the collection and storage of the samples is carried out in a standard way and so is not regularly considered when calculating the total variability of successive measurements. Analytical and individual variability are combined using:
where SD2A represents analytical variation, SD2I represents individual variation and SD2T represents the total variation of repeated measures for an individual. The coefficient of variation (CV) is calculated by dividing the standard deviation (SD) by the mean and is commonly used in place of SDs as it allows for a RCV to be calculated to reflect percentage changes rather than absolute changes. Normality is assumed and the RCV is given by:
where CVA is the analytical variation, expressed as a CV, CVI is the within-individual variability, expressed as a CV, and Z refers to the z-statistic (value from the standard normal distribution). Given the values of analytical and within-individual variation, a difference between two results greater than the RCV suggests a real change in condition. 292 Sölétormos et al. 297 used this rule in a computer model for monitoring progression to metastatic breast cancer with cancer antigen 15-3 (CA-15-3), carcinoembryonic antigen (CEA) and tissue polypeptide antigen.
The use of RCVs is discussed further by Smellie,296 Petersen,294 Fraser,290 Fraser et al. 289 Klee,291 Petersen et al. 295 and Omar et al. 293 Biosca et al. 287 report a study of biological variability to identify the appropriate RCV to use in their specific clinical situation and Clerico and Emdin288 highlight differences in analytical sensitivity across studies carried out in differing populations.
Statistical process control and statistical rules for the interpretation of sequential tests
Statistical process control methods (first developed by Shewhart) are often used in manufacturing and can be used for medical applications when a process can be measured directly or using a biomarker. 292 Statistical process control procedures measure variability across time, with variability divided into common-cause and special-cause variability (or assignable-cause variability). Special-cause variability is akin to signal and signifies true change in the disease state of an individual. Common-cause variability, as noise, reflects random variability in measures. 292
X-bar charts are used to display measurements over time for an individual. If a process is stable, measurements are expected to fluctuate around the mean and the SD of observed measures is expected to be constant over time. Estimates of the mean (µ) and SD (σ) can be taken from stable processes, with an unbiased estimate of the SD obtained using a moving range (the difference between consecutive measures) and dividing the mean of the moving range estimates by a constant (d2 = 1.128). Estimates of the mean and SD of a stable process can then be used to identify control limits. The control limits can be identified using many criteria and should be modified depending on the situation; it may be that target values are safety driven. Moving range charts and exponentially weighted moving average charts (moving averages are calculated with greater weight given to the most recent observations) are also used in similar ways. The variability of a process can be quantified using the capability index, the difference between the upper and lower limit divided by 6σ. The off-target ratio, ST=(µ−T)σ, where T is the target value, measures how far the process is from the specified target value in terms of SDs. Process control charts use the assumption of independent normally distributed outcomes and generally require at least 20–25 observations. 292
Tennant et al. 299 reviewed studies in which patients are monitored using statistical process control methods and compared the use of statistical control methods with currently used rules and guidelines. Clinical areas found to use process control methods are peak flow measurements for patients with asthma, blood pressure measurements for patients with hypertension and serum creatinine measurements for patients after undergoing a kidney transplant. Thor et al. 300 also reviewed studies using statistical process control methods to monitor patients and highlight the disadvantages of using these methods. They discuss how, in some studies, methods had been employed for which there was a clear lack of understanding. The authors also comment on issues around autocorrelated measures, the collection of data and the application of the methods.
Gavit et al. 298 discuss a slightly different approach to process control using change-point analysis. Change-point analysis uses cumulative sum charts of the difference between the mean value and the recorded value. Change points are then analysed as bootstrapping methods are used to generate a CI for the change point. The change-point method can also be used to identify differences in variability. An advantage of the change-point method is the ability to analyse non-normal data because of the lack of distributional assumptions. Gavit et al. 298 also claim that the change-point method is able to identify subtle changes that would not be picked up by control charts.
Health economic approaches
Decision-analytic models
Decision-analytic modelling evaluates the costs, outcomes and cost-effectiveness of interventions. In the case of repeated testing appropriate techniques need to be used for this evaluation.
Karnon et al. 302 review models for measuring the cost-effectiveness of screening regimes, featuring the Baker301 and Parmigiani303 approaches. Sutton et al. 304 introduce comprehensive decision modelling.
Real options approaches
Palmer and Smith308 introduce real options approaches, inspired by methods used in financial markets, which aim to include the uncertainty around the use of a new technology along with health economic evaluation. The approach uses the potential to delay introducing a new technology (akin to a change in management) and the irreversibility of using a new technology. Analyses factor in deferring using a technology and the better evidence that may be available after deferral using expected value of perfect information methods.
Real options approaches are further discussed and expanded on by Driffield and Smith,305 Meyer and Rees307 and Shechter et al. ,309 whereas Whynes310 and Lasserre et al. 306 discuss a similar method.
Summary and conclusions
This review has revealed that there is limited methodological literature on the design of monitoring strategies. Work has focused primarily on the analysis of data, allowing subsequent recommendations to be made for monitoring frequency or decision rules, or simulation work, with both approaches being specific to the disease area being researched. Methods have been developed in the area of screening, with the focus being on identifying the optimal frequency of screening, which could be used for designing monitoring strategies. Some work has been carried out on the design of biomarker development studies, which could potentially be adapted to allow for the evaluation of a monitoring strategy using previously collected specimens. It appears that thresholds are often developed by analysis of the variability of the test being used, identified by the literature describing signal-to-noise ratios, biomarker development studies, statistical process control and RCVs.
The study by Buclin et al. 146 shows an approach in which decision rules were devised by a review of the literature and, then, using an obtained data set and analysis of signal and noise the rules were refined to minimise false results. Following this, recommendations on the decision rule and frequency of monitoring could be made. Takahashi et al. 255,256 and Oke et al. 248 also used signal and noise methods when analysing data and subsequently recommendations could be made for future monitoring strategies.
A number of applications of the signal and noise approach were identified, largely in the area of treatment titration. 143,144,146,237–240,245,248,249,255,256,259 The limitations of this approach for monitoring disease progression or recurrence are that rules and thresholds are devised purely by analysing the variability of test measures and the minimisation of false findings rather than the detection of disease at the earliest point possible and the impact on patients.
The simulation approach proposed by Li and Gatsonis247 uses a joint latent class model that combines predictions from the model with a utility function to identify optimal monitoring frequencies. The results of a simulation study reported by Li and Gatsonis247 appear promising; however, the approach has not been widely adopted, perhaps because of the complex nature of the model. Other simulation approaches may also have potential under certain circumstances, particularly if measurement error and a link between biomarker values and true disease state can be included.
The biases that are well documented in the screening literature are also applicable to the area of monitoring. Length time bias and lead time bias should be considered when analysing monitoring data and when designing monitoring studies. There is also the issue of post-screening noise, which is again important to take into consideration when evaluating a monitoring strategy; the time point at which monitored and non-monitored patients are compared should be selected to minimise the issue of incidence after the final testing point and should also consider the number of likely events. Harm to patients is vitally important in screening and monitoring as this harm may occur at several time points and this must be thought of when designing strategies.
A further consideration in the analysis of monitoring data concerns the number of test measurements and the timing of test measurements: people with more results will contribute more data to the model but they may be very different from those with fewer results. 241 Measurement error and particularly biological variability should also be considered. Studies have shown that RCVs from biological variability studies of healthy participants are not necessarily reflective of the true RCV for a diseased population. As methods to derive test thresholds used in monitoring rely heavily on the variability of test results it is important that estimates from biological variability studies are accurate. In addition, the quality of studies undertaken when developing new biomarkers is not always rigorous; however, there is literature concerning the design of these studies268 and the evaluation of quality of these studies. 265
Chapter 6 How can monitoring impact on patient outcomes?
Much of the test evaluation literature centres on establishing key test properties such as test accuracy. However, the ultimate use of any test in clinical practice should be based on the knowledge that testing does more good than harm to patients. Comparison of patient outcomes resulting from different interventions is ideally assessed using a RCT design and the same design can be applied to the evaluations of tests. RCTs are less commonly used for assessing medical tests but are increasing in number both for diagnostic68 and for monitoring tests (see Chapter 4), such that a thorough understanding of the ways in which testing can affect patient outcomes is important.
Patient monitoring is undertaken for many purposes, most obviously within the context of ongoing treatment as the main tool for treatment titration and maintenance, with the goal being to maintain test results within certain limits of a given marker until such a time as treatment can be discontinued or an alternative treatment is needed. Our particular interest is in monitoring people who have a known disease or condition that is likely to progress or recur at some point in the future but that does not yet require treatment. Patients are usually asymptomatic (e.g. following primary treatment for the first occurrence of a disease), but may be mildly symptomatic but not yet receiving treatment or may experience symptoms of a disease that puts them at risk of developing other conditions. The primary goal is usually earlier treatment or the avoidance or delay of treatment, with the crux of monitoring being to detect the need for a change in patient management in a timely manner.
Particular challenges to evaluating the impact of monitoring tests on patient outcomes are, first, that the effect on outcomes will be relatively small, thus requiring large samples of patients to demonstrate statistically significant effects, and, second, that changing patient outcomes is reliant on patients and clinicians following potentially complex protocols both for testing and for treatment.
Over the last 10–15 years a number of framework papers related to the development and evaluation of tests for screening, diagnosis, prognosis and treatment monitoring purposes have been published, many of which have been comprehensively reviewed by previous authors. 315,316 We have selected three frameworks of particular relevance to the consideration of patient outcomes in monitoring. The first, by Adriaensen et al. ,317 presents a stepwise evaluation process for new screening strategies, which includes a consideration of the trade-off between the harms and the benefits from a new test. The second, by Ferrante di Ruffano et al. ,68 aims to assist those evaluating diagnostic tests to understand the ways in which changes to testing strategies can affect patient outcomes. The third, by Lord et al. ,318 considers the circumstances in which randomised evidence of patient impact from a new diagnostic test may be needed. With these in mind, our aim was to consider the potential impact of monitoring on patient outcomes, illustrated by our review of randomised trials of monitoring strategies.
Methods
A monitoring care pathway was outlined to identify in simple terms the points at which monitoring might affect outcomes (Figure 10). The three identified frameworks68,317,318 were reviewed in terms of their relevance to this monitoring context and 58 trials from a review of RCTs in which monitoring was carried out in at least one arm of the trial (see Chapter 4) were used for illustration purposes. The trials were grouped into three main categories in terms of the change in patient care under evaluation and the intended impact on patient outcome:
-
A new monitoring strategy compared with an existing monitoring strategy, such that the current monitoring test might be replaced by a new and more accurate test, a new test might be added to the strategy or the currently used test might be applied at a different intensity or with an alternative threshold for intervention. Depending on the associated change in patient care, the new monitoring strategy may be intended to detect patients at an earlier stage of disease, to more accurately detect those in need of treatment or to reduce the invasiveness or frequency of testing.
-
A monitoring strategy compared with immediate treatment of all patients at risk of an adverse outcome, in which monitoring may be used to avoid or delay treatment in those who do not need it.
-
A monitoring strategy compared with no monitoring, in which patients are usually treated on the basis of clinical presentation only and the likely aim of monitoring is the detection and treatment of disease at an earlier stage.
FIGURE 10.
Monitoring care pathway: (a) pathway; and (b) detail of ongoing monitoring process. Adapted from Ferrante di Ruffano et al. 69 with permission. +ve, positive; –ve, negative.
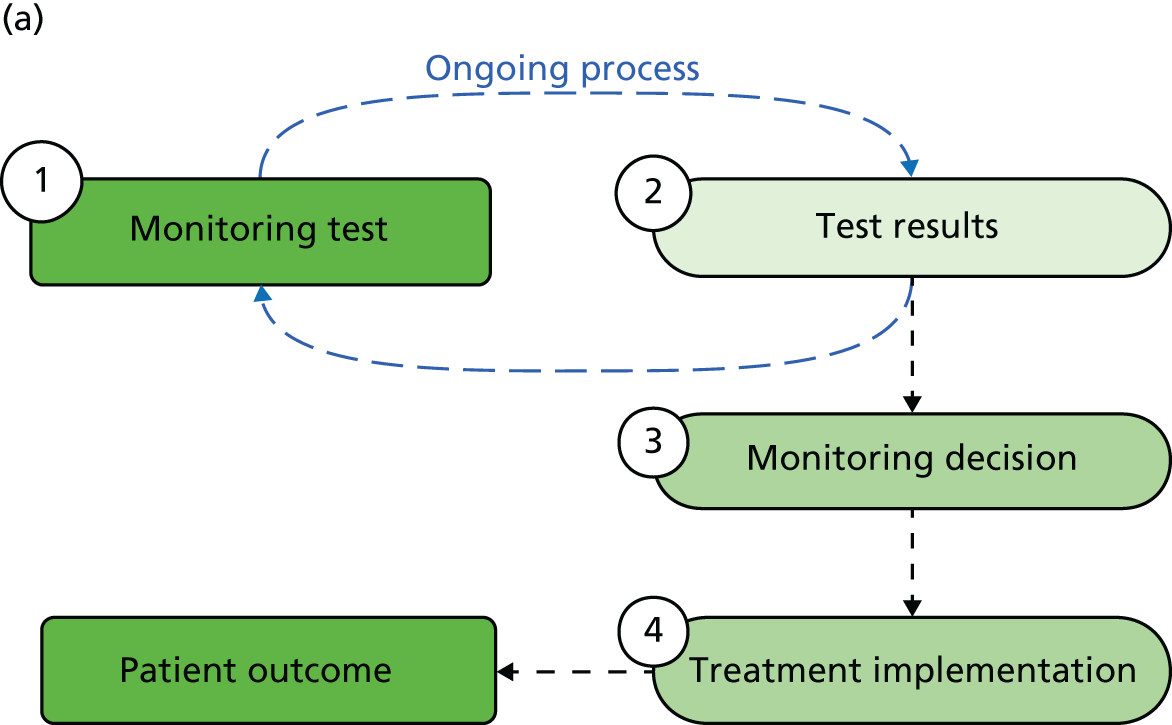

In the following sections, we first consider the similarities and differences between monitoring, screening and diagnosis, before broadly outlining the potential for benefit and harm from monitoring and considering the ways in which patient outcomes can be mediated by particular aspects of the monitoring care pathway according to the aim of monitoring and the change in strategy under evaluation.
Monitoring compared with screening or diagnosis
The monitoring care pathway outlined in Figure 10 bears close resemblance to that for diagnosis and for screening. 68,317 In a monitoring context, (1) a test is administered according to a predetermined schedule to detect a target condition or some precursor or marker of that condition, (2) the test result is considered (often in relation to previous measurements and with the potential for repeat testing to confirm abnormal or indeterminate results), (3) the test result is considered alongside other evidence (usually including the results of further investigations) to decide whether or not therapeutic intervention is needed and (4) the necessary intervention is implemented.
Where the pathway diverges from that for diagnosis is with the added dimension of repeated testing over time and a merging of the ‘diagnostic’ and ‘management’ decisions outlined in the diagnostic care pathway of Ferrante di Ruffano et al. 68 The serial nature of testing for monitoring purposes can affect patient outcomes in a number of ways, most obviously by increasing the physical and psychological burden of testing on patients but also potentially impacting on other outcomes, for example through patient and physician compliance with testing protocols. Furthermore, although a diagnostic test informs both a ‘diagnostic decision’ (often when more than one differential diagnosis may be available) and a ‘management decision’ (assisting in the choice of a range of therapeutic options), a monitoring test is often relatively less definitive, providing more of a guide to the need for changes in patient management. A positive monitoring test result frequently triggers further investigation to determine whether or not and when a particular treatment should be implemented, rather than informing the choice of one of a range of therapeutic options. In this respect, monitoring is more akin to screening, in which a test is applied repeatedly over time to detect and treat a particular clinical condition, rather than to differentiate between diagnoses, with the caveat that monitoring populations have a higher risk of the clinical event of interest occurring and that, although the ultimate goal of a new screening programme is usually a reduction in disease-specific mortality, monitoring can be implemented for a range of reasons.
Potential benefits and harms from monitoring
Figure 11 uses the concept of a 2 × 2 contingency table to illustrate that assigning potential benefits and harms from a monitoring test is not as straightforward as might be imagined. For simplicity, the following is set mainly in the context of a new monitoring strategy to allow earlier detection and treatment.
FIGURE 11.
Summary of the potential harms and benefits from a new monitoring strategy. Harms and benefits are similar to those identified from a screening context by Adriaensen et al. 317 FN, false neagative; FP, false positive; TN, true negative; TP, true positive.
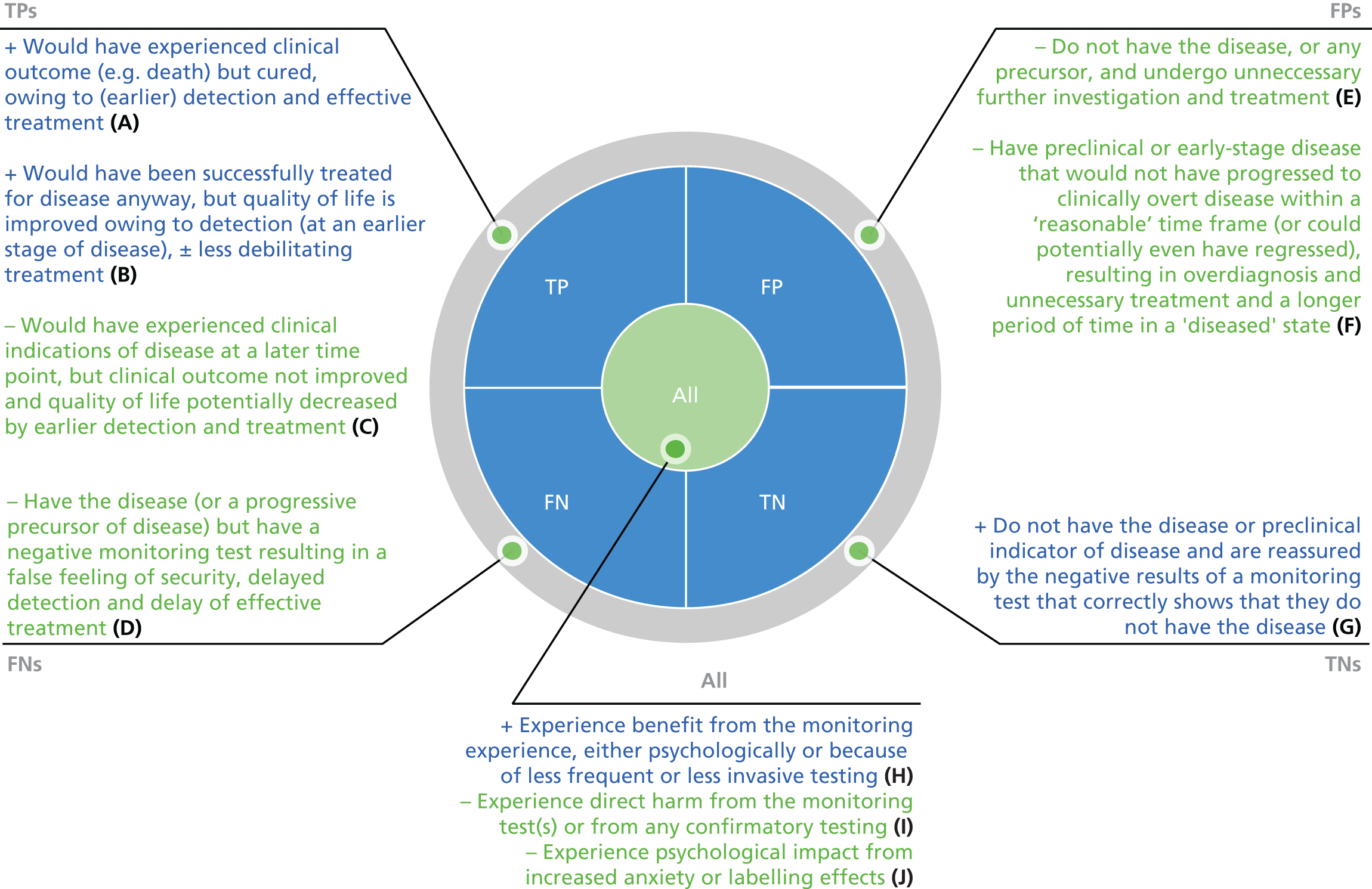
In general terms, for those patients with a ‘true’ result, benefits accrue both to those who would otherwise have experienced a poor clinical outcome but for the new test (A) and to those who would have been detected clinically and successfully treated but the new test allows this to happen at an earlier point in the disease process (B). Benefit also occurs for those with no disease and for whom a negative monitoring test result has a reassurance value, increasing a patient’s sense of control over the disease (G). Positive benefits might also be experienced by all patients, for example with the use of a less invasive test or less frequent testing (H).
Patients who have a ‘false’ monitoring test result will experience harm from the new testing strategy in a similar manner to that experienced after false-negative or false-positive diagnostic test results. False-negative test results can lead to a false feeling of security, delayed detection of disease and potentially a delay in effective treatment until the disease becomes clinically apparent (D). False-positive test results can lead to unnecessary further investigation and/or unnecessary treatment (E). For monitoring tests that aim to detect preclinical or very-early-stage disease, ‘early’ false-positive results (i.e. in patients whose disease would not have progressed to clinically overt disease within a clinically meaningful time frame) will lead to a longer period of time in a diseased state and potentially in overdiagnosis and unnecessary treatment (F). Similarly, ‘early’ positive monitoring tests in those with a true-positive test result (C) can cause harm for those patients who go on to experience a poor clinical outcome and who undergo a longer period of treatment (with associated side effects and a longer period of time in a ‘diseased’ state).
More direct harms can also be incurred by the testing experience, relating to repeated applications of the monitoring test, to any confirmatory testing and to any intervention that is implemented (I and J). These can be physical or psychological in nature and may result from either positive or negative test results, with the serial nature of testing in a monitoring context necessarily multiplying the potential impact. In particular, the ongoing monitoring process (even with repeated negative results) may raise general levels of anxiety and distress and can also have a ‘labelling’ effect that can have a negative influence on patients’ perceptions of themselves and their disease. 319,320
When is randomised evidence needed?
The RCT is the gold standard approach to assessing impact on patient outcomes but, given the challenges to implementing this design for the assessment of test impact, its use requires careful consideration.
Lord et al. 318 determined the need for randomised evidence for a new diagnostic test based, first, on whether or not the cases detected by the new test represent a similar spectrum of disease to those detected by the old test and, second, whether or not treatment has been shown to be, or can be assumed to be, as effective in the new group of patients, regardless of disease spectrum. Incorporating the time dimension of monitoring into this framework, for a new monitoring strategy one must determine, first, whether or not the cases detected by the new strategy represent a similar spectrum of disease, both in terms of the biological characteristic that is measured by the test and in terms of the time point in the disease process at which disease recurrence or progression is identified, and, second, whether or not treatment has been shown to be, or can be assumed to be, as effective in the new group of patients, regardless of disease spectrum and the timing of detection in relation to the stage of disease.
Notwithstanding the simple appeal of this approach, testing strategies are necessarily complex interventions, with various components and possible interactions that can combine to affect patient outcomes; even a ‘perfect’ test and highly effective treatment will not necessarily improve patient outcomes. Ferrante di Ruffano et al. 68 developed a framework to consider how testing can affect health outcomes. This has been adapted for the monitoring context in Table 13, considering factors such as timing, test properties, treatment effectiveness, potential to change practice and the patient experience. 68 Many of these can apply equally to patients undergoing monitoring; however, some require more or less emphasis or need to be adapted to the monitoring context.
| Care pathway component | Definition | ||
|---|---|---|---|
| No. | Attribute | ||
| Test delivery | |||
| 1 | Test feasibility | Completion of the test process, where reasons for non-completion might include:
|
|
| 2 | Test procedure | Patients’ interaction with the test procedure, potentially causing physical or psychological harms or benefits | |
| 3 | Test frequency | Patients’ response to serial testing, potentially multiplying the impact of any physical harms and incurring additional psychological impact | |
| Test result | |||
| 4 | Interpretability | After successful completion of the test process, the likelihood of high frequencies of indeterminate or unreadable test results (distinct from the measurement variability associated with an individual test result, which will affect the ability of the test to detect true changes in disease status) | |
| 5 | Clinical validity | The ability of a test to predict the presence of, or development of, clinical or overt disease | |
| 6 | Timing of test result | The ability of the test to predict overt disease within a clinically meaningful time frame | |
| 7 | Detection of long-term change | The ability of a test to differentiate true changes in patients’ disease status from short-term variations | |
| Management decision | |||
| 8 | Added clinical value | The degree to which the test contributes to a change in management:
|
|
| 9 | Time frame of management decision | The time frame within which patients undergo a change in management | |
| 10 | Clinical confidence | The degree of confidence that clinicians have in the validity or applicability of a test result | |
| Treatment implementation | |||
| 11 | Timing of treatment | The time frame within which patients receive treatment | |
| 12 | Efficacy | The ability of the intervention to improve patient outcomes at the particular stage of disease detected | |
| 13 | Adherence | The extent to which patients participate in the management plan, as advised by their physician, in order to attain the therapeutic goal | |
We considered the extent to which these factors might affect a monitoring evaluation according to the aim of the new strategy and the way in which it fits with standard care, to demonstrate how this could inform the need for randomised evidence (Table 14).
| Goal of strategy and comparison | Change in testing | Key effects on patient outcomes | Example | Is a RCT necessary? |
|---|---|---|---|---|
| Earlier detection and treatment | ||||
| New monitoring strategy vs. existing monitoring strategy | Add or replace test |
Timing Test properties – limit early false-positive results Treatment effectiveness – at earlier stage of disease? Added clinical value – what does test or change in threshold/frequency add to clinical decision making? Patient experience – test more/less invasive? |
Addition of PET scans at 3 and 15 months to standard surveillance for the detection of recurrent colon or rectal cancer (includes CT scans at 3 and 15 months)175 |
Longitudinal studies could identify additional cases detected by the new test if the management decision made after standard surveillance is compared with the management decision made following the new test or if it is ethical to blind the results of additional (interim) investigations and act only on those carried out according to the original follow-up schedule RCTs are needed to determine the effect on patient outcomes of earlier treatment if randomised evidence does not already exist |
| Change an existing test threshold | aSurveillance of patients with HIV infection and treatment at a higher CD4 threshold157 | |||
| More frequent testing | Increased frequency of Doppler US for the detection of HCC in patients with compensated cirrhosis of the liver163 | |||
| Monitoring vs. no monitoring | New testing strategy |
Timing Test properties – limit false-positive results Treatment effectiveness – at earlier stage of disease? Added clinical value Patient experience – potential adverse effects from invasive test; psychological impact |
Introduction of new endoscopic surveillance compared with endoscopy on demand to allow the earlier detection of oesophageal carcinoma in patients with Barrett’s oesophagus176 | Akin to screening context; randomised evidence is needed to determine the benefit of formalised monitoring |
| Reduce the invasiveness of testing | ||||
| New monitoring strategy vs. existing monitoring strategy | Replacement test |
Timing Test properties – should be similar to existing strategy Treatment effectiveness Added clinical value – effect on patient confidence? Patient experience – less invasive, safer test |
Less invasive biopsy approach for patients at risk of colorectal cancer177 | No. Unlikely to be a change in disease spectrum. The accuracy/predictive ability of the new biopsy approach is key; the value of the new test may be inferred from an assessment of safety and/or cost |
| Triage test |
Timing – potential increase in time to treatment Test properties – low false-negative rate needed Treatment effectiveness – outcomes in those not treated key Added clinical value – patient/clinician confidence in triage test Patient experience – less invasive, safer test |
Gene expression profiling as triage to endomyocardial biopsy to detect cardiac transplant rejection162 | Yes. Only a subset of patients will be identified by the new test. Treatment will be as effective in the subset of patients but clinical outcomes will be unknown in those not selected by the new test | |
| Reduce the volume of testing | ||||
| New (or no) monitoring strategy vs. existing monitoring strategy | Fewer tests or less frequent testing |
Timing – potential increase in time to treatment Test properties Treatment effectiveness – possible treatment at a later stage Added clinical value – patient/clinician confidence in less testing Patient experience – possible psychological impact |
Reduction in the number of CT scans from five over 36 months to two in 12 months for the detection of recurrence of non-seminoma testicular cancer180 |
Yes. Beyond 3 months’ follow-up the spectrum will change to the later stage of disease, especially for those whose disease has not recurred by 12 months. Treatment options/effectiveness are likely to be similar for those detected in the first 12 months A longitudinal study could indicate the number of positive tests that would be missed by reducing the test frequency but could not compare clinical outcomes between strategies |
| Reduce overtreatment | ||||
| New monitoring strategy vs. existing monitoring strategy | Add or replace test |
Timing Test properties – similarly predictive with fewer false-positive results Treatment effectiveness – does test detect same marker? Added clinical value – clinician confidence key Patient experience – impact depends on nature of new test |
DNA-based test vs. antigenemia test to allow pre-emptive instead of prophylactic treatment of HCMV infection following solid organ transplantation181 | No. The two tests reflect different aspects of virus replication but there is apparently no indication of a differential treatment response in this example. A RCT is needed only if treatment on the basis of the new test is expected to result in a different treatment response |
| Delay/avoid treatment by introducing a new surveillance strategy | ||||
| New monitoring strategy vs. treatment | New testing strategy |
Timing – possible unnecessary delay in treatment Test properties – should minimise false-negative results Treatment effectiveness – possible treatment at later stage of disease Added clinical value – patient/clinician confidence key Patient experience – increase in testing but reduction in treatment |
Sonographic surveillance vs. immediate treatment of mild hip dysplasia in newborns158 | Yes. The spectrum changes to more severe disease as some cases resolve without treatment. A RCT may be needed to establish whether or not later/no treatment leads to poorer outcomes |
Earlier detection and treatment
Evaluations of monitoring strategies that aim to detect and treat disease at an earlier stage or time point will generally take the form of a new monitoring regime compared with an existing monitoring regime or the initiation of monitoring when no previous monitoring was undertaken. In this setting, the goal of earlier detection almost necessarily implies that those patients detected and treated by a new monitoring strategy will have a different spectrum from those previously treated. Although patient outcomes may be affected by all of the identified mechanisms, it is the clinical validity of the test(s) used, their ability to detect long-term change within a clinically meaningful time frame and the effectiveness of the treatment at the particular stage of disease that are of overarching importance. Longitudinal studies can establish test properties and identify the spectrum of patients detected by the test(s). If no randomised evidence for treatment in this group of patients exists or if it is not clear whether or not the existing evidence will apply in the new group of patients, a new RCT may be needed, as for example in a trial evaluating a lower CD4 threshold for the initiation of antiviral treatment in HIV infection. 157 Alternatively, the evidence may be such that no trial is indicated. A trial of ultrasound to detect small HCCs in patients with cirrhosis of the liver found that many of the lesions detected were too small to warrant treatment, with some even regressing rather than progressing. 163 This high rate of ‘early false-positive’ results could potentially have been identified in a longitudinal study without the need for a RCT.
Even when clinically valid, timely monitoring tests and effective treatments are available, the potential for a monitoring strategy to have a positive impact on patients will be influenced both by the degree to which it can substantively add clinical value over and above that of usual clinical practice and by the degree of clinical and patient confidence in the new strategy. When an individual monitoring test provides a clear guide to future management, as in the CD4 example above, the added contribution of the change in strategy might be relatively easy to discern. However, when a new monitoring test is one component of a bigger surveillance programme, for example the addition of biochemical tests and/or imaging tests to an existing surveillance programme, its added value may be more difficult to ascertain. In some circumstances, the accuracy of the confirmatory test could also mitigate any impact of the monitoring test, especially if the new test is able to detect very-early-stage disease that is not detectable by the confirmatory test. 182–184,321
Clinician and patient confidence in and compliance with a prescribed monitoring strategy can be vital to the success of a monitoring evaluation. For example, if clinicians have a high degree of faith in a new test, its ‘off-protocol’ use in the control arm of a trial may dilute the observed effect. This was observed in a trial of ultrasound for the detection of HCC, which also attempted to evaluate the added value of serial alpha-fetoprotein (AFP) measurements; high rates of serum AFP assay use in the two groups not randomised to AFP (60.5% and 54.8%, respectively) precluded reliable interpretation of the data and led to a final analysis restricted to ultrasound randomisation only. 163
The effect of the patient experience of monitoring on outcome will depend on the nature of the test(s) involved and of the care that would otherwise have been received, especially if no monitoring was previously carried out. In the TOMBOLA (Trial Of Management of Borderline and Other Low-grade Abnormal smears) trial, for example, non-attendance was higher in the cytological surveillance arm: 10.6% did not attend the first cytological surveillance appointment whereas 6.8% did not attend for immediate colposcopy. 178 Of the 10% who did not attend the first cytological surveillance appointment, 2% did not attend at all and 8% attended > 6 months after the test was due. Some tests (or subsequent confirmatory testing) will carry a risk of immediate or long-term physical harm, for example the introduction of routine endoscopic surveillance with biopsies in patients with Barrett’s oesophagus to allow the earlier detection of oesophageal carcinoma or the initiation of 6-monthly computed tomography (CT) in patients at risk of colorectal cancer recurrence, with the potential to affect patient compliance. 176 Strategies that afford patients more control over a disease, however, might increase adherence to treatment regimens, as in a trial of daily foot skin temperature monitoring by patients with diabetes mellitus in which those who were compliant with the monitoring strategy for at least 50% of the time were significantly less likely to develop a foot ulcer. 174
Patients will also experience a sometimes complex psychological impact from the monitoring experience. A qualitative study of patients’ attitudes to and understanding of CA-125 testing for the monitoring of ovarian cancer recurrence found that both negative and positive test results can reassure patients if the result appears to legitimise patients’ own subjective experience of the disease, with a positive result confirming their worst suspicion or a negative result providing reassurance that they are as ‘well’ as they feel. 322 Positive, or rising, test results can mediate a patient’s experience of any clinical symptoms, with the knowledge that he or she is probably experiencing a relapse potentially making symptoms more unbearable or causing him or her to reinterpret earlier ‘symptoms’ that had previously been discounted. 322 Similar findings in other monitoring situations have also been observed. 320
Reduce the invasiveness of testing
Patients’ exposure to invasive tests can be reduced in two ways: (1) by replacing an existing test with a less invasive one or (2) by introducing a triage test to select the most appropriate patients for invasive testing. For the former option, assuming that the new test aims to detect disease at a similar stage or time point, it will be important to establish its properties in relation to the existing test and to ensure that it identifies a similar spectrum of patients. If so, treatment can be expected to be similarly effective and the impact of the test on patient outcomes will result from the less invasive nature of the test, as for example with the use of a less invasive biopsy approach in patients at risk of colorectal cancer. 177
If a new triage test is introduced to prevent harm from the testing process, such as gene expression profiling as triage for endomyocardial biopsy when monitoring for acute cardiac transplant rejection162 or the introduction of cytological surveillance to reduce the number of colposcopies undertaken in women with mild dyskaryosis,178 the need for a randomised trial will be greater. Only a subgroup of patients will be selected for further investigation and treatment, such that treatment effectiveness could vary and outcomes in those no longer selected for treatment must also be assessed. In this setting, the properties of the new test (in particular the rate of false-negative results), the timing of detection and treatment effectiveness are likely to be of overarching importance. Patient and clinician confidence in the new triage test will also be needed to ensure that any potential benefits are realised in practice. An element of ‘trust’ that the new, less invasive test will accurately identify those in need of the more invasive test is required on both parts. Patient preference is not always as intuitive as it might appear, for example gene testing was favoured over endomyocardial biopsy in the example above,162 whereas patients at risk of bladder cancer recurrence preferred an immediate result from a more invasive test (cystoscopy) to the result from a less invasive (urine) test a week later. 179
Reduce the volume of testing
Sometimes new monitoring strategies aim to reduce the number or frequency of tests without adversely having an impact on patient outcome, as is often the goal when monitoring for cancer recurrence. 180,184 One example is the proposal to reduce the number of CT scans from five over a 3-year period to two in the first year of follow-up following primary treatment of non-seminoma testicular cancer. 180 Under the new regime, there could be a change in spectrum to later-stage disease in those detected at the 12-month follow-up point and detection of those patients whose disease recurs beyond 12 months would rely on clinical relapse or detection by biochemical markers or chest radiography. In this situation, the number of cases that would be missed and the stage of disease at detection could be identified using a longitudinal study design but the clinical outcomes for the different strategies could not be directly compared without a RCT. If, however, sufficient evidence exists for treatment at the various stages of disease, this could be linked to data from a longitudinal study using a decision-analytic-type model. 318
A trial of less frequent fetal surveillance of small-for-gestational-age fetuses demonstrates the sometimes complex responses of clinicians and patients to monitoring. Over half of the experimental group in this trial attended for ultrasound more frequently than scheduled and underwent additional tests of fetal well-being, suggesting that clinicians were not always comfortable with the planned reduced frequency of fetal surveillance and making it difficult to assess whether or not the apparent safety of less frequent monitoring may have been in part because of this additional surveillance. 158 At the same time, 17% of women in the twice-weekly surveillance group attended less frequently than requested, suggesting a patient perception of over-frequent monitoring. 185
Reduce overtreatment
In some monitoring contexts, a new test can be introduced to replace another simply to better select the right patients, for example a new deoxyribonucleic acid (DNA)-based test to detect human cytomegalovirus infection following stem cell transplantation compared with the existing antigenemia test. 167 This is more akin to a diagnostic test context in which the goal is to use the most sensitive and/or specific test, as the time dimension of monitoring is less relevant than the properties of the test concerned. Although the new test may detect a different biochemical marker, if there is no indication of a differential treatment response according to the marker used, and randomised evidence exists for the effectiveness of treatment in the patients identified, then a study to establish the properties of the tests concerned may be sufficient evidence for the introduction of the new test.
To fully affect patient outcomes, however, clinicians must have confidence to act on the results of the new test. Only half of patients who tested positive on the DNA-based test in the example above actually underwent treatment, whereas, in a trial of a new galactomannan assay in patients at risk of invasive aspergillosis following stem cell transplantation, two-thirds of those treated in the experimental arm had a negative monitoring test, perhaps because clinicians had previously relied on clinical assessment as the basis for treatment decisions. 167,168
Delay or avoid treatment when it is not required
The final scenario is one in which monitoring is introduced as an alternative to immediate treatment. In this circumstance, patient outcome might be affected by later treatment in the surveillance arm, further delays to necessary treatment because of false-negative test results, the effectiveness of treatment at a later stage of disease and the need for clinician and patient confidence in the monitoring regime. When immediate treatment is the standard care option, evidence for treatment at a later stage of disease may not be available, so the onus is not only on demonstrating that the monitoring test used is clinically valid and able to detect the point at which treatment is needed, but also on evidencing treatment effectiveness in the surveillance group. In a small trial in infants with mild hip dysplasia, sonographic surveillance allowed abduction treatment to be delayed or avoided with no significant difference in radiological outcomes at 1 year compared with immediate abduction treatment; < 50% of those in the surveillance arm underwent treatment during the course of the trial. 158
Conclusion
The impact of a monitoring strategy is driven not only by the properties and timing of testing and the effectiveness of treatment but also by patients’ responses to the type and frequency of testing and clinicians trust in, and willingness to comply with, the monitoring protocol. Reitsma et al. 323 advocate that what is more important for clinical decision-making than the level or change in a given marker is the confidence with which that marker can be used to inform patient management. A move towards a test validation paradigm is advocated, by which a number of methods (including establishing test properties) are used to determine whether or not the results of a test are meaningful in practice. In some circumstances, randomised evidence will be needed to fully assess the impact of a test, but this level of evidence will not be needed in every circumstance.
For example, the feasibility of testing and interpretability of test results can be estimated in the development phase of a test, as long as the technical properties of the test are established in clinically relevant populations rather than in laboratory-based studies alone. 316 Patients’ interaction with the testing experience and their likely adherence to monitoring or to subsequent management can be assessed in qualitative studies. Feasibility or pilot studies can help identify what a new test adds to current clinical practice, particularly in terms of clinicians’ interaction with and likely adherence to a new monitoring strategy, and can help identify potential barriers to implementation, as has been recommended for trials of complex interventions. 228 Estimates of key aspects of test performance can be obtained from non-randomised, preferably longitudinal studies comparing tests against a (delayed) reference standard or clinical outcome. 65 The efficacy of treatment is the only mechanism that requires evaluation in a RCT per se; however, the combined effect of the individual mechanisms that come into play may be fully assessable only in a RCT.
Any decision to undertake a RCT should be informed at a minimum by good evidence of the natural history of the disease, the establishment of test properties (in terms of clinical validity and estimation of long-term change in disease status) and evidence (or lack) of treatment efficacy in those patients who are identified by the new monitoring strategy.
Chapter 7 Simulating monitoring data and evaluating monitoring strategies
Introduction
Tests are used in health care to monitor, and subsequently manage, a variety of chronic conditions. The focus of this research is the monitoring of progressive or recurrent conditions, in which the aim of monitoring is to identify early signs of recurrence or progression, prompting a change in management, typically the initiation of treatment or further testing.
Monitoring strategies are complex interventions combining a test, a schedule, a decision rule and further diagnostic or therapeutic action. Monitoring strategies stipulate the frequency of testing and the ‘monitoring rule’ used to identify when a change in patient management is necessary. A monitoring rule indicates the value or values that would trigger a change in management. Monitoring rules can be simple, such as when a single value above a threshold will prompt a change in management (a ‘snapshot rule’), or more complex, such as when a patient requires a series of test results observed at different time points that fit certain criteria (such as a relative increase from previous measures) to initiate a change in management (a ‘track-shot rule’). 146
Although patient monitoring is a fundamental function of health care, resulting in considerable costs to health-care providers, the area of monitoring is under-researched and there is an increased need for monitoring strategies to be systematically developed with knowledge of the likely progression of disease and the performance of the monitoring test to be used. 62,324 Dinnes et al. ,69 in Chapter 3, reviewed the evidence base for PSA monitoring to identify the recurrence of prostate cancer. The review identified the lack of a systematic approach to developing a monitoring strategy, with monitoring intervals based on standard follow-up schedules and limited evidence of consensus over the thresholds used to initiate treatment.
Stevens et al. 142 discussed various statistical models of the transition between the maintenance and the re-established control phases of monitoring (the process of detecting when a disease is out of control, leading to a change in management, e.g. treatment or more intensive monitoring) and identified a general statistical model for the evolution of monitoring data over time, outlining possible sources of variation. This general statistical model proposes the form of monitoring data based on the observed values of sequential monitoring tests, the values of measurement error and other sources of variability and the true disease state, which can be modelled based on epidemiological evidence but never observed.
This general model, along with existing data and evidence gathered from the literature, can be used to simulate monitoring data and allow for the evaluation of strategies for a given target condition. The potential effect of monitoring strategies can then be evaluated and ranked, prior to full-scale investigation. 325
The example presented here investigates the use of the ELF biomarker in monitoring patients with known liver fibrosis, alongside the ELUCIDATE trial,326 a prospective multicentre randomised trial. The ELUCIDATE trial is evaluating the ELF test for the early detection of progression from liver fibrosis to liver cirrhosis compared with routine care, with the aim of enabling earlier treatment and providing potentially improved patient outcomes. It is described fully in Chapters 16–24.
Aims and objectives
The aim of this study was to identify the optimal monitoring strategy, from candidate monitoring strategies, for patients known to have liver fibrosis receiving repeated testing using the ELF biomarker.
Candidate strategies were selected and evaluated to:
-
compare the alternative frequencies of monitoring (6-month or 12-month intervals)
-
evaluate the benefit of using targeted retesting compared with no retesting
-
compare decision rules [positive results based on crossing a threshold determined by a single value (snapshot simple threshold rule) and positive results dependent on track-shot rules based on absolute or relative increases from the first test value, absolute or relative increases from the last test value and prediction from a linear regression model].
Methods
First, we describe the model used to generate the underlying and unobserved disease progression, incorporating estimates of disease progression and the variability of these estimates, for a cohort of simulated individuals. Then, the process of obtaining observed test result values using the true disease progression values and estimates of test performance is described. Finally, the methods used to evaluate and compare selected monitoring strategies, using both the observed test values and the true disease status, are given. An explanation of the notation used in the model is provided in Table 15.
| Description | Notation |
|---|---|
| Individual number | ii |
| Number of initially simulated individuals | nn |
| Number of simulated individuals eligible for randomisation | NN |
| Fibrosis stage | ss |
| Time within fibrosis stage | xjsxjs |
| Time across fibrosis stages | tt |
| Monitoring time points | TT |
| Fibrosis progression | pipi |
| Mean fibrosis progression | µpµp |
| SD of fibrosis progression | σpσp |
| Starting fibrosis stage | SiSi |
| Probability of starting in each fibrosis stage | ρsρs |
| ELF value at each stage of fibrosis | EsEs |
| Mean ELF value at each fibrosis stage | µsµs |
| SD of ELF value at each fibrosis stage | σsσs |
| Observed mean ELF value at each fibrosis stage | µYsµYs |
| Observed SD of ELF value at each fibrosis stage | σYsσYs |
| ELF value progression between fibrosis stages | EisEis |
| Values from the standard normal distribution | zizi |
| Gradient of ELF progression | βisβis |
| Time point when patients progress in fibrosis stage | πisπis |
| True ELF value by time in fibrosis stage | EijsEijs |
| True ELF value across fibrosis stages | EitEit |
| True ELF value over the period of the trial | UitUit |
| Time at registration | τriτri |
| Time at randomisation | τtiτti |
| Total observation error | ωitωit |
| SD of total observation error | σωσω |
| Observed ELF value | YitYit |
| Observed ELF value at monitoring points | YiTYiT |
| Entry ELF criteria | Yr*Yr* |
| Frequency of observations | θθ |
| Range for targeted retesting | ΔΔ |
| Time period before entering compensation cirrhosis when a patient is considered diseased | δδ |
| Simple decision rule threshold | Y*Y* |
| Simple decision rule threshold at the point of randomisation | Y0*Y0* |
| Absolute increase from start value decision rule threshold | YD*YD* |
| Absolute increase from last value decision rule threshold | YE*YE* |
| Relative increase from start value decision rule threshold | YF*YF* |
| Relative increase from last value decision rule threshold | YG*YG* |
| Linear regression decision rule threshold | YH*YH* |
| Maximum time in cirrhosis before trial entry | c1c1 |
| Maximum time in fibrosis before trial entry | c2c2 |
| Time between registration and randomisation | c3c3 |
| Trial duration | c4c4 |
| Time between test and retest measures | εε |
Simulation of true disease progression
The model simulated true disease progression, generating a random slope and random intercept in terms of fibrosis stage for each patient. The model then converted the fibrosis stages to ELF values.
Fibrosis progression: random slope
The rate at which patients progress through the fibrosis stages was assumed to be constant throughout the stages of fibrosis and normally distributed:
where i = 1, . . ,n and n is the number of simulated individuals, µp is the mean fibrosis progression and σp is the SD of fibrosis progression. Fibrosis progression was restricted to only positive values, by fixing pi at 0.01 if pi ≤ 0, meaning that only increases in fibrosis stage were simulated, with no patients having decreasing fibrosis stages; however, as the increase is just 0.01 fibrosis units per year, this effectively means that these patients are in a stable fibrosis state.
Fibrosis stage at entry: random intercept
Patients recruited to a trial would be in varying stages of disease at entry. Using data on the likely distribution of fibrosis stage for a population with known liver fibrosis, a multinomial distribution was used to simulate a starting stage for each individual:
where ρs is the probability of starting in each stage, s is the fibrosis stage and s = 0, . . ,4.
Enhanced Liver Fibrosis score link to fibrosis stage
For each stage of fibrosis, the distribution of true ELF values within fibrosis stage was assumed to follow a normal distribution:
where s is the fibrosis stage and s = 0, . . ,4, µS is the mean value of ELF at each fibrosis stage and σs is the SD of the mean ELF value at each fibrosis stage.
Enhanced Liver Fibrosis progression between fibrosis stages
The model used fibrosis stage on a continuous scale rather than on a discrete scale. To generate ELF values for each patient at all stages of fibrosis, ELF progression between consecutive integer fibrosis stages was assumed to be linear. It was assumed that patients would have ELF values at the same point of the normal distribution for each fibrosis stage (patients would remain a given number of SDs from the mean). To randomly select the point of the normal distribution that patients would follow, a value from the standard normal distribution was generated for each patient, zi∼N (0,1). The ELF value for each participant at each stage of fibrosis was Eis = µs + (ziσs) (Figure 12a).
FIGURE 12.
(a) Fibrosis units linked to ELF value; (b) ELF value progression through time; and (c) starting stage adjusted ELF value progression through time.



Enhanced Liver Fibrosis progression: random slope
The ELF values at the beginning of each fibrosis stage for each individual (Eis) and the rate at which each simulated participant progresses through fibrosis (pi) were combined to calculate the increase in ELF per year. The gradient of ELF progression was βis = (Ei,s + 1 − Ei,s)pi, for s = 0, . . ,3. The gradient of ELF progression after stage 4 was assumed to be the same as the gradient between stages 3 and 4 (βi3 = βi4). The time point signalling when each individual would progress to the next fibrosis stage was πis=spi. βis is the random slope in terms of ELF progression.
The underlying and true ELF progression for each stage and all time points from the onset of fibrosis was then calculated as:
where xjs is time within stage s and:
For stage 4, s = 4 and xjs = 0, . . , ∞. The true ELF values for each individual across time could also be expressed as:
where t is time across all stages (t = 0, . . , ∞). This allowed the simulation of lifetime progression data for a cohort of patients (see Figure 12b). ELF values were truncated at 0 if a negative value was simulated.
Enhanced Liver Fibrosis value at entry: random intercept
The time at registration for each participant (τri) was a randomly selected time point from the time period when the individual was in his or her generated fibrosis stage at study entry (Si), that is, a random value from the interval [πi,si,πi,si+1), where Si is the starting stage for each individual and Si = 0, . . ,3. If the participant was in stage 4 of fibrosis at entry, Si = 4 and τri was generated by identifying a random value from the interval [πi,4,πi,4 + c1], where c1 is the longest given amount of time that a patient can be in stage 4 of liver fibrosis before entering the trial. τri also has a maximum value of c2, where c2 is the longest amount of time that a patient can have fibrosis on registration to the trial. If, for a simulated individual, τri>πi,4+c1 or τri>c2, the data for that individual were not used in the analysis. This was done to prevent patients being included when they would be confirmed as having cirrhosis or were at a point of fibrosis that they would not have reached in their lifetime because of their simulated progression rate. The c1 time used means that participants registering in the trial who are in stage 4 of liver fibrosis have been in stage 4 for a maximum of c1 years. The c2 time used means that participants have liver fibrosis for a maximum of c2 years before being registered in the trial.
In the ELUCIDATE trial, a registration ELF test was given to each patient to assess eligibility. The first ELF test included in the trial data was taken at the point of randomisation. If the start of the trial occurred c3 time units after registration, then the time at randomisation was τti=τri+c3. The random intercept in terms of ELF is αi = Eit for t=τti.
Random slope and random intercept model in terms of the Enhanced Liver Fibrosis test
The underlying disease progression for the simulated individuals over the time of the trial was Uit = Eit for τti≤t<τti+c4, where c4 is the duration of the trial and i denotes simulated patients with an eligible registration ELF value (i = 1, . . ,N, where N is the number of simulated patients available for randomisation) (see Figure 12c).
Simulation of observed values
The true underlying ELF measurements were converted to observed ELF measures by the addition of error.
Error
The error at each observation point (ωit) was formed of within-individual variation and analytical variation. Error was assumed to be normally distributed with a mean of zero:
The observed ELF measurement at any given time was:
Values were adjusted to equal 0 if a negative observed ELF value was simulated.
Entry criteria
To fulfil trial entry criteria the observed ELF measurement at registration had to be greater than the preset value of Y*r; thus, the equation:
had to be satisfied for each simulated participant to be included in the trial data. An example of simulated observed ELF measures can be seen in Figure 13.
FIGURE 13.
Observed ELF values.

Data sources
Data sources were used to estimate the fibrosis progression rate, fibrosis stage at trial entry, measurement error and ELF value link to fibrosis stage. Additional information regarding the data sources used is provided in Table 16.
| Estimate required | Data | Estimates used in the model |
|---|---|---|
| Fibrosis progression rate | Estimate of median fibrosis progression (Scheuer fibrosis units per year): 0.133 (95% CI 0.125 to 0.143)327 |
Estimate calculated from Poynard et al. :327 pi ∼ N (0.13, 0.172) Estimate after adjustment (to be used in sensitivity analyses) – estimate of fibrosis progression was increased to reflect expert opinion: pi ∼ N (0.27, 0.172) |
| ELF stage at entry to trial | Cross-sectional data set61 – estimated proportion of patients in each stage: stage 0, 0.25; stage 1, 0.35; stage 2, 0.13; stage 3, 0.15; stage 4, 0.12 | The cross-sectional data set was used: ρ0 = 0.25, ρ1 = 0.35, ρ2 = 0.13, ρ3 = 0.15, ρ4 = 0.12 |
| Measurement error |
Longitudinal data set – estimate of the SD of the measurement error of 0.81 Siemens – estimate of the SD of the total measurement error of 0.11 ELUCIDATE trial registration and randomisation data: estimate of the SD of the total measurement error of 0.47 |
Estimate obtained from the ELUCIDATE trial was used: ωit ∼ N (0, 0.472) |
| ELF value link to fibrosis stage | Cross-sectional data set61 – estimates of ELF mean (SD) value at each fibrosis stage: stage 0, 8.82 (0.87); stage 1, 9.18 (0.96); stage 3, 9.55 (1.00); stage 4, 11.32 (1.47) | After adjustment – measurement error is accounted for to give the true unobserved ELF values and modified to represent values for each stage: E0 ∼ N (8.63, 0.732), E1 ∼ N (9.00, 0.842), E2 ∼ N (9.36, 0.892), E3 ∼ N (9.91, 1.222), E4 ∼ N (10.80, 1.392) |
Fibrosis progression rate
An estimate of the median rate of fibrosis progression based on data for 1157 patients was obtained from Poynard et al. ;327 the estimate of the median was assumed to be equal to the mean and the 95% CI for the median was used to calculate the SD. When consulting clinical experts it was suggested that the estimate provided by Poynard et al. 327 was identified in a population that was not comparable with that of the ELUCIDATE trial (participants in the Poynard study were thought to have less severe disease). The estimate from Poynard et al. 327 was used primarily in the simulation model, with an adjusted estimate used for sensitivity analyses. Estimates of fibrosis progression are given as Scheuer fibrosis units per year, where Scheuer scores range from 0 to 4 and measure the severity of liver disease, with stage 0 showing no fibrosis and stage 4 showing liver cirrhosis. 328
Fibrosis stage at entry
As the purpose of the study was to simulate and evaluate trial data, not all participants would enter the trial at the same stage of fibrosis. A cross-sectional data set with ELF results and Scheuer fibrosis scores following liver biopsy for 921 patients was used to identify the distribution of fibrosis stages in the cohort. 61
Measurement error
To estimate the error associated with each observed ELF test value, three data sources were considered. First, a longitudinal data set with repeat ELF measurements (baseline and 3 months) for 220 patients was subjected to analysis of variance (ANOVA) to identify variability at the individual level. The manufacturer of the test also provided information on the performance of the ELF test. 329 Because of discrepancies between the estimates from the two sources, data were obtained directly from the ELUCIDATE trial. Registration and randomisation ELF values for 112 eligible participants were again subjected to ANOVA to identify the variability at the individual level. The estimate obtained from the ELUCIDATE trial data was used in the simulation model.
Enhanced Liver Fibrosis value link to fibrosis stage
The cross-sectional data set was used to provide an estimate of the observed ELF value for patients at each level of fibrosis, with a corresponding measure of variability. 61 To estimate the true and unobserved SD of the ELF value at each fibrosis stage, the measurement error that would have been included in these observed measures, inflating the variability, was accounted for. As σ2s and σ2ω are independent in the simulation:
where σYs is the observed SD at each stage of fibrosis. To estimate the true and unobserved mean ELF value at each stage of fibrosis, the observed estimates were assumed to give the mean value for the midpoint of the corresponding fibrosis stage and, thus, were altered to reflect an ELF value for the point when a patient initially enters each stage of fibrosis:
where µYs is the observed mean ELF value at each stage of fibrosis; when:
σ2s is the between-individual variability at each fibrosis stage.
Implementation of a monitoring strategy
The effect of implementing different monitoring strategies was predicted using simulated observed values of ELF. The specified monitoring strategy (decision rule, use of retesting and frequency of testing) changed the simulated observed values that would be measured and the how the value or values for each individual would be interpreted.
Monitoring data required
The observed values that would have been measured under each monitoring strategy were extracted; the exact values required were dependent on both the frequency and the duration of monitoring. The duration of monitoring was specified by the duration of the trial, c4. If the frequency of monitoring was every θ time units after randomisation, the observed values used to guide the management of participants would be Yit for t=τti+θT, where T=0,1,. . . ,c4θ and c4θ is the number of observation points additional to randomisation. The subscript used for time is now simplified to indicate monitoring time points; this will be T, where T=t−πtiθ.
To incorporate a targeted retest component an additional test would be carried out ε time units after the scheduled tests for patients with an observed value at that time point within a specified range (Δ) of the value used to trigger a positive test. When a patient required retesting, the mean of the original test and the retest result was calculated and this value was subjected to the decision rules to identify positive participants:
Patients with a value above the upper limit of the range were classed as positive on the initial test without further testing and patients below the limit of the retesting range were classed as negative using just the initial test.
Monitoring strategies
Monitoring strategies are defined by the decision rule for identifying a positive result and the data that the decision rule is applied to, which is dependent on the frequency of monitoring and the use of retesting.
Simple decision rule (strategy A)
The simplest decision rule was based on a single-value threshold (snapshot rule). The threshold value, Y*, was specified and any single observed value over this threshold indicated a positive result for that participant at that time point. A result was considered positive when YiT > Y*.
Retesting (strategy B)
Patients with an initial test value within Δ of the threshold value, Y*, were subjected to retesting. Patients required retesting when Y* − Δ > YiT < Y* + Δ and these patients were considered positive when YriT>Y*. When patients did not require retesting, YiT ≤ Y* − Δ or YiT ≥ Y* + Δ, patients were considered positive when YiT > Y*. The retesting component could be used with any of the alternative decision rules explained (strategies D–H).
Frequency of monitoring (strategy C)
The frequency of monitoring was every θ time units after the initial test at randomisation. By decreasing or increasing the value of θ the timing of the monitoring tests became more or less frequent, respectively. When varying the frequency of monitoring, the time points and, hence, the observations evaluated changed. When changing the frequency of monitoring, the value of θ determines the data evaluated, as:
and patients are considered positive when YiT > Y*. Varying the frequency of monitoring could be used in conjunction with any of the alternative decision rules explained (strategies D–H).
Alternative decision rules
Decision rules incorporating previous test results as well as the current result (track-shot rules) to identify positive patients were also considered. Absolute and relative increases from the randomisation ELF value or from the last recorded ELF value were investigated. A rule using predictions from a linear regression model fitted using all available observed data points was also considered.
Decisions rules based on absolute and relative increases and the linear regression method required at least two observations to declare a participant as positive. A simple threshold rule was used to identify participants at the point of randomisation using Yi0 > Y*0. Y*D, Y*E, Y*F, Y*G and Y*H are specified thresholds for the corresponding decision rule method.
A result was considered positive when the absolute difference between the test value and the first recorded value for the patient was greater than the threshold:
A result was considered positive when the absolute difference between the test value and the last observed test value for that patient was greater than the threshold:
A result was considered positive when the relative difference between the test value and the first recorded test value for the patient was greater than the threshold:
A result was considered positive when the relative difference between the test value and the last observed test value for that patient was greater than the threshold:
The linear regression decision rule involved the fitting of a linear regression model for each participant at each time point, using all available measures for that participant at the time point; the prediction from the model was then used to identify the patient as test positive or test negative. A result was considered positive when the prediction from the linear regression model was greater than Y*H.
Evaluation of a monitoring strategy
To evaluate each strategy, the decision made from implementing that monitoring strategy using the simulated observed values and the corresponding true underlying values was assessed. With knowledge of the true underlying disease state of each participant, the performance of a variety of monitoring strategies was evaluated.
Comparison of observed results with the underlying disease state
Participants had a positive or negative test result based on the simulated observed data and the decision rule employed. The test result was then found to be either true or false depending on the underlying disease state. The purpose of the ELF test was to identify when patients enter compensated cirrhosis, stage 4. From the simulation of underlying true ELF values, the time point when each individual enters stage 4 was πi,4. As it may be beneficial to identify patients prior to time πi,4, participants were classed as ‘diseased’ δ units of time prior to πi,4 and onwards. If at a testing point a participant was diseased (≥ πi,4 − δ), a positive result would be a true positive and a negative result would be a false negative. If at a testing point the patient was not diseased (< πi,4 − δ), a negative result would be a true negative and a positive result would be a false positive. As a positive result (truly or falsely) caused a change in management and cessation of monitoring, patients who achieved a positive result did not have a test result at subsequent monitoring times. Figure 14 illustrates how a strategy with a simple threshold decision rule can be evaluated.
FIGURE 14.
Results of implementing a monitoring strategy.
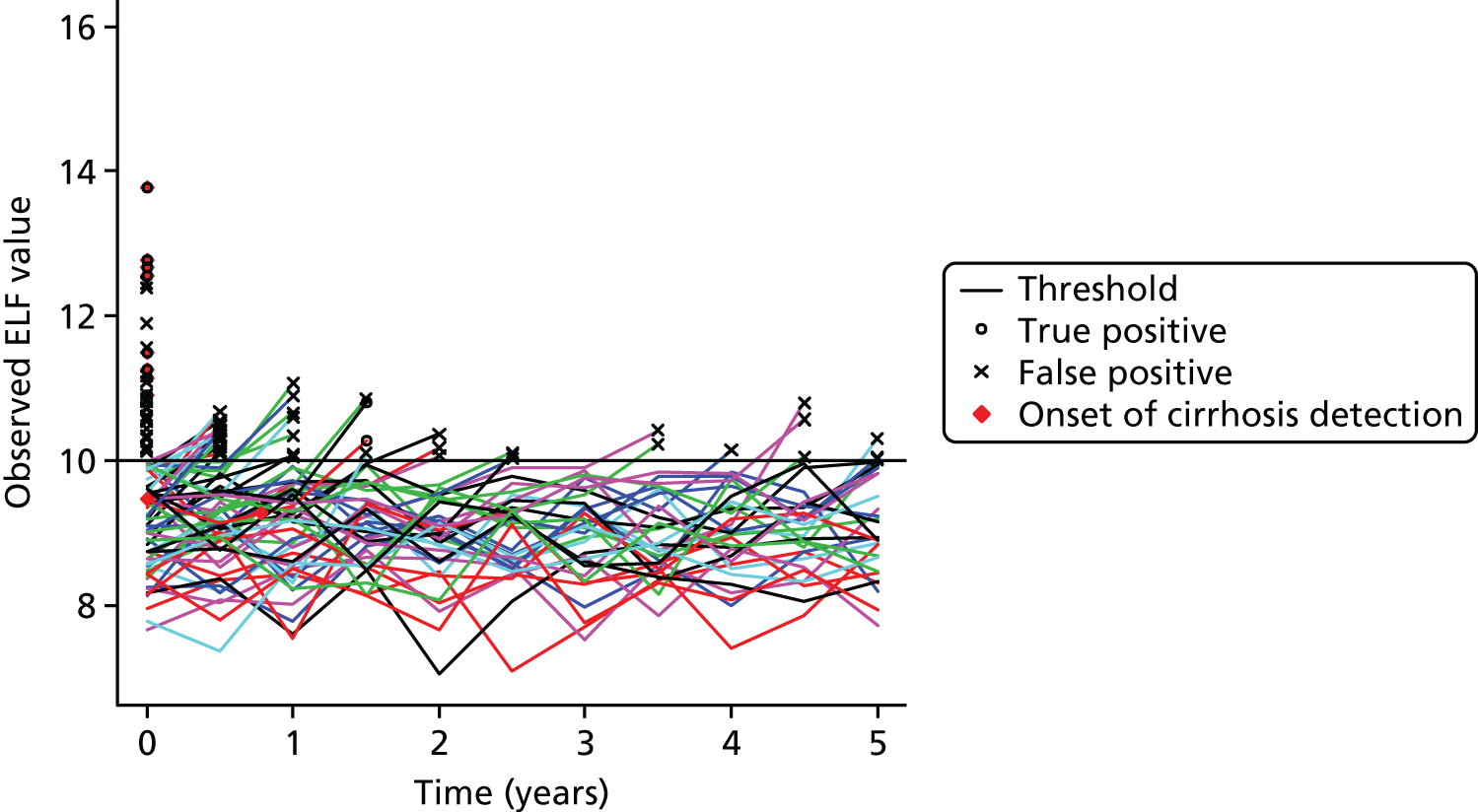
Measuring the performance of a monitoring strategy
The performance of a monitoring strategy was assessed at each monitoring point by calculating the number of patients at each monitoring test visit and specifically the number of true-positive, false-positive, true-negative and false-negative test results. The number of tests carried out across the duration of the strategy was used to represent resource use. The positive predictive value (PPV) was used to investigate how likely it was for an individual with a positive result to be diseased. To measure patient harm the delay from onset of disease to the point of diagnosis was calculated; this was the time between the onset of compensated cirrhosis (πi,4) and a patient having a positive test result.
When comparing strategies the number of tests per person for the duration of monitoring, the PPV (for all tests over the duration of monitoring) and the percentage of patients with a delayed diagnosis (delay from onset of disease to diagnosis of > 12 months) were used to measure performance. To allow for comparisons to be made between strategies in which only two of the three measures of performance ranged, thresholds used by monitoring strategies were varied to obtain a PPV of 25%. A PPV of 25% was chosen as this would be an acceptable PPV in practice.
Evaluation of strategies
The strategy evaluated first was the simple threshold strategy, with observations every 6 months and no retest component (reference strategy). Alternative strategies were evaluated in which individual components of the reference strategy were varied: the frequency of monitoring (θ), the decision rule and whether or not a retest value was used. The same simulated data were used when evaluating strategies A–H.
Sensitivity analyses were carried out to estimate the effect of inaccurate information regarding test performance and progression of liver disease. Estimates used in the simulation of data were altered (halved and doubled) and the reference strategy was evaluated with all aspects of the strategy kept constant (including the threshold value). Results for the reference strategy with the threshold varied to give a PPV of 25% using data with altered estimates were also obtained. Further sensitivity analyses were undertaken in which the fibrosis progression rate was adjusted based on expert opinion; these were analyses of strategies A–H as for the main analysis (with PPV held at 25%) and the reference strategy using varied estimates to generate monitoring data.
To assess the accuracy of the model, the mean randomisation ELF values and SDs were calculated and compared for the ELUCIDATE and simulated data sets. ANOVA was used to assess between-individual and within-individual variability in the ELF values recorded for patients in the trial and the simulated results. Multilevel models were fitted using the simulated observed values and the observed values from the ELUCIDATE trial (for participants with two or more ELF measures post registration) and the results from these models were compared. In the ELUCIDATE trial ELF measurements were not taken in the majority of cases after participants had an ELF value of ≥ 9.5. To allow for this, the ELUCIDATE and simulated data sets were modified so that each patient with an ELF value of ≥ 9.5 did not undergo any subsequent measurements.
Simulations were based on a cohort of 20,000 patients to give adequate precision. With 20,000 test results, if one of the performance measures gave an estimate of 15%, the corresponding 95% CI would range from 14.5% to 15.5%; for an estimate of 1.5% the 95% CI would range from 1.3% to 1.7%.
Results
The estimates from various data sources and details of the data used in the simulation model can be found in Table 16. In addition, the estimates regarding the trial used in the simulation are shown in Table 17. When evaluating strategies patients were considered truly positive if they received a positive result 3 months prior to entering compensated cirrhosis (δ = 0.25). When using targeted retesting, patients with initial values within ±1 of the threshold value have a retest (Δ = 1).
| Description | Estimate used in simulation modelling |
|---|---|
| Maximum time in cirrhosis before trial entry | To avoid simulating patients who are in advanced cirrhosis, the maximum amount of time that a patient has been cirrhotic was set to 2 years: c1 = 2 |
| Maximum time in fibrosis before entry to the trial | To avoid patients being simulated at a point of disease that they would not have reached given their fibrosis progression rate the maximum amount of time that a patient has had fibrosis for at the time of entering the trial was set at 20 years: c2 = 20 |
| Time between registration and randomisation | The time between the registration ELF test and the randomisation ELF test was estimated to be 3 months: c3 = 0.25 |
| Trial duration | The duration of the trial used in all simulations was 5 years: c4 = 5 |
| Time between test and retest measures | The time between a patient having a test and a patient having a retest (if the original test were in the target range) was estimated to be 1 week: ε = 0.02 |
The same simulated data were used when evaluating strategies A–H. For the simulated cohort of 20,000 patients, 5314 (26.6%) would develop cirrhosis if there were no intervention during the period of the trial.
Reference monitoring strategy (strategy A)
Table 18 shows the performance of the reference monitoring strategy at each testing time point. For the reference monitoring strategy (simple threshold, observations 6-monthly and no retest component), the threshold required to maintain the PPV at 25% was an ELF value of 10.715. The sensitivity and PPV calculated for the strategy were highest at the initial observation point and the percentage of tests with a positive result was also larger, because of cases being identified from a prevalent population at the initial testing point. The percentage of false-negative results generally increased as the strategy continued over time. Over the duration of the monitoring strategy 7.64 tests per person (152,724 tests in total) were performed and 6.10% of all patients experienced a delay to diagnosis.
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1162 (5.8) | 2236 (11.2) | 771 (3.9) | 15,831 (79.2) | 3398 (17.0) | 1933 (9.7) | 34.2 | 60.1 |
| 6 | 16,602 | 207 (1.2) | 920 (5.5) | 733 (4.4) | 14,742 (88.8) | 1127 (6.8) | 940 (5.7) | 18.4 | 22.0 |
| 12 | 15,475 | 147 (0.9) | 661 (4.3) | 740 (4.8) | 13,927 (90.0) | 808 (5.2) | 887 (5.7) | 18.2 | 16.6 |
| 18 | 14,667 | 102 (0.7) | 558 (3.8) | 783 (5.3) | 13,224 (90.2) | 660 (4.5) | 885 (6.0) | 15.5 | 11.5 |
| 24 | 14,007 | 113 (0.8) | 495 (3.5) | 786 (5.6) | 12,613 (90.0) | 608 (4.3) | 899 (6.4) | 18.6 | 12.6 |
| 30 | 13,399 | 104 (0.8) | 446 (3.3) | 813 (6.1) | 12,036 (89.8) | 550 (4.1) | 917 (6.8) | 18.9 | 11.3 |
| 36 | 12,849 | 127 (1.0) | 458 (3.6) | 805 (6.3) | 11,459 (89.2) | 585 (4.6) | 932 (7.3) | 21.7 | 13.6 |
| 42 | 12,264 | 121 (1.0) | 429 (3.5) | 828 (6.8) | 10,886 (88.8) | 550 (4.5) | 949 (7.7) | 22.0 | 12.8 |
| 48 | 11,714 | 129 (1.1) | 449 (3.8) | 817 (7.0) | 10,319 (88.1) | 578 (4.9) | 946 (8.1) | 22.3 | 13.6 |
| 54 | 11,136 | 135 (1.2) | 390 (3.5) | 806 (7.2) | 9805 (88.0) | 525 (4.7) | 941 (8.5) | 25.7 | 14.3 |
| 60 | 10,611 | 125 (1.2) | 369 (3.5) | 814 (7.7) | 9303 (87.7) | 494 (4.7) | 939 (8.8) | 25.3 | 13.3 |
| All | 152,724 | 2472 (1.6) | 7411 (4.9) | 8696 (5.7) | 134,145 (87.8) | 9883 (6.5) | 11,168 (7.3) | 25.0 | 22.1 |
Comparing strategies with changes to individual components with the reference strategy
Table 19 and Figure 15 show the performance of various monitoring strategies. The results of each strategy by observation point are provided in Tables 20–26.
| Strategy | Monitoring strategy components | PPV, % | Testsa | Delay to diagnosisb | Test performance | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Decision rule | Threshold value | Observation interval (months) | Retest | Initial threshold | N | N per personc | Median IQR | N | % of alld | % of stage 4e | TP per personf | FP per persong | Positive, n (%) | Sensitivity, % | ||
| A | Simple threshold | 10.715 | 6 | False | – | 25 | 152,724 | 7.64 | 11 (3, 11) | 1220 | 6.10 | 22.96 | 0.12 | 0.37 | 9883 (6.47) | 22.13 |
| B | Simple threshold | 10.58 | 6 | True | – | 25 | 218,974h | 10.95 | 12 (6, 15) | 1300 | 6.50 | 24.46 | 0.12 | 0.35 | 9406 (6.11) | 21.15 |
| C | Simple threshold | 10.55 | 12 | False | – | 25 | 86,787 | 4.34 | 6 (2, 6) | 1249 | 6.25 | 23.5 | 0.13 | 0.38 | 10,053 (11.58) | 35.34 |
| D | Absolute increase from initial value | 1.295 | 6 | False | 10.715 | 25 | 155,648 | 7.78 | 11 (4, 11) | 1536 | 7.68 | 28.9 | 0.13 | 0.40 | 10,598 (6.81) | 19.85 |
| E | Absolute increase from last value | 1.46 | 6 | False | 10.715 | 25 | 172,363 | 8.62 | 11 (7, 11) | 2085 | 10.42 | 39.24 | 0.08 | 0.24 | 6305 (3.66) | 8.45 |
| F | Relative increase from initial value | 1.144 | 6 | False | 10.715 | 25 | 156,460 | 7.82 | 11 (4, 11) | 1630 | 8.15 | 30.67 | 0.13 | 0.38 | 10,266 (6.56) | 18.12 |
| G | Relative increase from last value | 1.1795 | 6 | False | 10.715 | 25 | 176,385 | 8.82 | 11 (9, 11) | 2217 | 11.09 | 41.72 | 0.07 | 0.20 | 5338 (3.03) | 6.68 |
| H | Linear regression | 10.495 | 6 | False | 10.715 | 25 | 150,478 | 7.52 | 11 (2, 11) | 1126 | 5.63 | 21.19 | 0.12 | 0.35 | 9342 (6.21) | 21.58 |
FIGURE 15.
Results of various monitoring strategies.
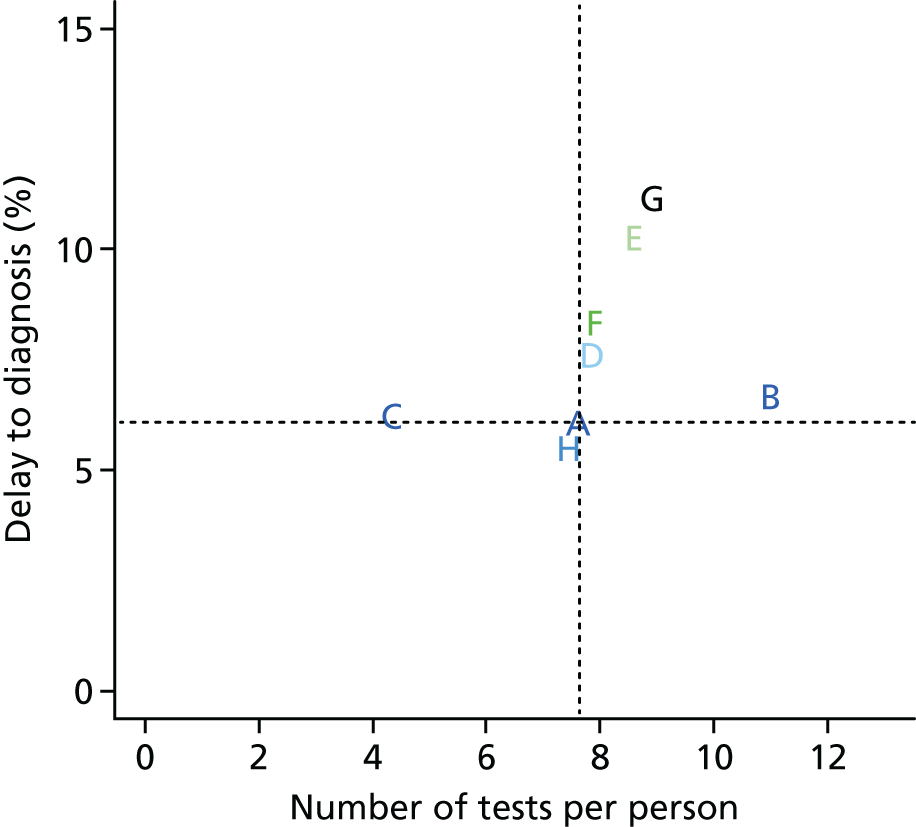
| Observation time (months) | Tests,a N/n | Results, n (%) | Diseased,b n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 28,357/20,000 | 1233 (6.2) | 2447 (12.2) | 700 (3.5) | 15,620 (78.1) | 3680 (18.4) | 1933 (9.7) | 33.5 | 63.8 |
| 6 | 22,924/16,320 | 144 (0.9) | 796 (4.9) | 711 (4.4) | 14,669 (89.9) | 940 (5.8) | 855 (5.2) | 15.3 | 16.8 |
| 12 | 21,549/15,380 | 125 (0.8) | 568 (3.7) | 734 (4.8) | 13,953 (90.7) | 693 (4.5) | 859 (5.6) | 18.0 | 14.6 |
| 18 | 20,617/14,687 | 97 (0.7) | 494 (3.4) | 775 (5.3) | 13,321 (90.7) | 591 (4.0) | 872 (5.9) | 16.4 | 11.1 |
| 24 | 19,913/14,096 | 117 (0.8) | 438 (3.1) | 776 (5.5) | 12,765 (90.6) | 555 (3.9) | 893 (6.3) | 21.1 | 13.1 |
| 30 | 19,167/13,541 | 103 (0.8) | 408 (3.0) | 803 (5.9) | 12,227 (90.3) | 511 (3.8) | 906 (6.7) | 20.2 | 11.4 |
| 36 | 18,555/13,030 | 105 (0.8) | 413 (3.2) | 816 (6.3) | 11,696 (89.8) | 518 (4.0) | 921 (7.1) | 20.3 | 11.4 |
| 42 | 17,927/12,512 | 105 (0.8) | 422 (3.4) | 849 (6.8) | 11,136 (89.0) | 527 (4.2) | 954 (7.6) | 19.9 | 11.0 |
| 48 | 17,272/11,985 | 108 (0.9) | 408 (3.4) | 858 (7.2) | 10,611 (88.5) | 516 (4.3) | 966 (8.1) | 20.9 | 11.2 |
| 54 | 16,674/11,469 | 126 (1.1) | 392 (3.4) | 855 (7.5) | 10,096 (88.0) | 518 (4.5) | 981 (8.6) | 24.3 | 12.8 |
| 60 | 16,019/10,951 | 88 (0.8) | 269 (2.5) | 887 (8.1) | 9707 (88.6) | 357 (3.3) | 975 (8.9) | 24.6 | 9.0 |
| All | 218,974/153,971 | 2351 (1.5) | 7055 (4.6) | 8764 (5.7) | 13,5801 (88.2) | 9406 (6.1) | 11,115 (7.2) | 25.0 | 21.2 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1247 (6.2) | 2735 (13.7) | 686 (3.4) | 15,332 (76.7) | 3982 (19.9) | 1933 (9.7) | 31.3 | 64.5 |
| 12 | 16,018 | 277 (1.7) | 1253 (7.8) | 739 (4.6) | 13,749 (85.8) | 1530 (9.6) | 1016 (6.3) | 18.1 | 27.3 |
| 24 | 14,488 | 243 (1.7) | 983 (6.8) | 775 (5.3) | 12,487 (86.2) | 1226 (8.5) | 1018 (7.0) | 19.8 | 23.9 |
| 36 | 13,262 | 247 (1.9) | 952 (7.2) | 782 (5.9) | 11,281 (85.1) | 1199 (9.0) | 1029 (7.8) | 20.6 | 24.0 |
| 48 | 12,063 | 246 (2.0) | 861 (7.1) | 806 (6.7) | 10,150 (84.1) | 1107 (9.2) | 1052 (8.7) | 22.2 | 23.4 |
| 60 | 10,956 | 252 (2.3) | 757 (6.9) | 809 (7.4) | 9138 (83.4) | 1009 (9.2) | 1061 (9.7) | 25.0 | 23.8 |
| All | 86,787 | 2512 (2.9) | 7541 (8.7) | 4597 (5.3) | 72,137 (83.1) | 10,053 (11.6) | 7109 (8.2) | 25.0 | 35.3 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1162 (5.8) | 2236 (11.2) | 771 (3.9) | 15,831 (79.2) | 3398 (17.0) | 1933 (9.7) | 34.2 | 60.1 |
| 6 | 16,602 | 50 (0.3) | 564 (3.4) | 890 (5.4) | 15,098 (90.9) | 614 (3.7) | 940 (5.7) | 8.1 | 5.3 |
| 12 | 15,988 | 66 (0.4) | 517 (3.2) | 1014 (6.3) | 14,391 (90.0) | 583 (3.6) | 1080 (6.8) | 11.3 | 6.1 |
| 18 | 15,405 | 95 (0.6) | 550 (3.6) | 1085 (7.0) | 13,675 (88.8) | 645 (4.2) | 1180 (7.7) | 14.7 | 8.1 |
| 24 | 14,760 | 116 (0.8) | 582 (3.9) | 1132 (7.7) | 12,930 (87.6) | 698 (4.7) | 1248 (8.5) | 16.6 | 9.3 |
| 30 | 14,062 | 169 (1.2) | 559 (4.0) | 1123 (8.0) | 12,211 (86.8) | 728 (5.2) | 1292 (9.2) | 23.2 | 13.1 |
| 36 | 13,334 | 187 (1.4) | 603 (4.5) | 1066 (8.0) | 11,478 (86.1) | 790 (5.9) | 1253 (9.4) | 23.7 | 14.9 |
| 42 | 12,544 | 174 (1.4) | 586 (4.7) | 1048 (8.4) | 10,736 (85.6) | 760 (6.1) | 1222 (9.7) | 22.9 | 14.2 |
| 48 | 11,784 | 212 (1.8) | 583 (4.9) | 955 (8.1) | 10,034 (85.1) | 795 (6.7) | 1167 (9.9) | 26.7 | 18.2 |
| 54 | 10,989 | 218 (2.0) | 591 (5.4) | 865 (7.9) | 9315 (84.8) | 809 (7.4) | 1083 (9.9) | 26.9 | 20.1 |
| 60 | 10,180 | 204 (2.0) | 574 (5.6) | 765 (7.5) | 8637 (84.8) | 778 (7.6) | 969 (9.5) | 26.2 | 21.1 |
| All | 155,648 | 2653 (1.7) | 7945 (5.1) | 10,714 (6.9) | 134,336 (86.3) | 10,598 (6.8) | 13,367 (8.6) | 25.0 | 19.8 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1162 (5.8) | 2236 (11.2) | 771 (3.9) | 15,831 (79.2) | 3398 (17.0) | 1933 (9.7) | 34.2 | 60.1 |
| 6 | 16,602 | 28 (0.2) | 342 (2.1) | 912 (5.5) | 15,320 (92.3) | 370 (2.2) | 940 (5.7) | 7.6 | 3.0 |
| 12 | 16,232 | 22 (0.1) | 281 (1.7) | 1086 (6.7) | 14,843 (91.4) | 303 (1.9) | 1108 (6.8) | 7.3 | 2.0 |
| 18 | 15,929 | 32 (0.2) | 256 (1.6) | 1230 (7.7) | 14,411 (90.5) | 288 (1.8) | 1262 (7.9) | 11.1 | 2.5 |
| 24 | 15,641 | 31 (0.2) | 256 (1.6) | 1378 (8.8) | 13,976 (89.4) | 287 (1.8) | 1409 (9.0) | 10.8 | 2.2 |
| 30 | 15,354 | 34 (0.2) | 240 (1.6) | 1538 (10.0) | 13,542 (88.2) | 274 (1.8) | 1572 (10.2) | 12.4 | 2.2 |
| 36 | 15,080 | 39 (0.3) | 248 (1.6) | 1682 (11.2) | 13,111 (86.9) | 287 (1.9) | 1721 (11.4) | 13.6 | 2.3 |
| 42 | 14,793 | 52 (0.4) | 228 (1.5) | 1860 (12.6) | 12,653 (85.5) | 280 (1.9) | 1912 (12.9) | 18.6 | 2.7 |
| 48 | 14,513 | 56 (0.4) | 223 (1.5) | 2014 (13.9) | 12,220 (84.2) | 279 (1.9) | 2070 (14.3) | 20.1 | 2.7 |
| 54 | 14,234 | 45 (0.3) | 204 (1.4) | 2212 (15.5) | 11,773 (82.7) | 249 (1.7) | 2257 (15.9) | 18.1 | 2.0 |
| 60 | 13,985 | 78 (0.6) | 212 (1.5) | 2423 (17.3) | 11,272 (80.6) | 290 (2.1) | 2501 (17.9) | 26.9 | 3.1 |
| All | 172,363 | 1579 (0.9) | 4726 (2.7) | 17,106 (9.9) | 148,952 (86.4) | 6305 (3.7) | 18,685 (10.8) | 25.0 | 8.5 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1162 (5.8) | 2236 (11.2) | 771 (3.9) | 15,831 (79.2) | 3398 (17.0) | 1933 (9.7) | 34.2 | 60.1 |
| 6 | 16,602 | 44 (0.3) | 641 (3.9) | 896 (5.4) | 15,021 (90.5) | 685 (4.1) | 940 (5.7) | 6.4 | 4.7 |
| 12 | 15,917 | 48 (0.3) | 539 (3.4) | 1041 (6.5) | 14,289 (89.8) | 587 (3.7) | 1089 (6.8) | 8.2 | 4.4 |
| 18 | 15,330 | 84 (0.5) | 525 (3.4) | 1125 (7.3) | 13,596 (88.7) | 609 (4.0) | 1209 (7.9) | 13.8 | 6.9 |
| 24 | 14,721 | 100 (0.7) | 558 (3.8) | 1193 (8.1) | 12,870 (87.4) | 658 (4.5) | 1293 (8.8) | 15.2 | 7.7 |
| 30 | 14,063 | 143 (1.0) | 524 (3.7) | 1217 (8.7) | 12,179 (86.6) | 667 (4.7) | 1360 (9.7) | 21.4 | 10.5 |
| 36 | 13,396 | 169 (1.3) | 519 (3.9) | 1184 (8.8) | 11,524 (86.0) | 688 (5.1) | 1353 (10.1) | 24.6 | 12.5 |
| 42 | 12,708 | 179 (1.4) | 528 (4.2) | 1164 (9.2) | 10,837 (85.3) | 707 (5.6) | 1343 (10.6) | 25.3 | 13.3 |
| 48 | 12,001 | 195 (1.6) | 568 (4.7) | 1099 (9.2) | 10,139 (84.5) | 763 (6.4) | 1294 (10.8) | 25.6 | 15.1 |
| 54 | 11,238 | 226 (2.0) | 528 (4.7) | 1009 (9.0) | 9475 (84.3) | 754 (6.7) | 1235 (11.0) | 30.0 | 18.3 |
| 60 | 10,484 | 220 (2.1) | 530 (5.1) | 918 (8.8) | 8816 (84.1) | 750 (7.2) | 1138 (10.9) | 29.3 | 19.3 |
| All | 156,460 | 2570 (1.6) | 7696 (4.9) | 11,617 (7.4) | 134,577 (86.0) | 10,266 (6.6) | 14187 (9.1) | 25.0 | 18.1 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1162 (5.8) | 2236 (11.2) | 771 (3.9) | 15,831 (79.2) | 3398 (17.0) | 1933 (9.7) | 34.2 | 60.1 |
| 6 | 16,602 | 16 (0.1) | 263 (1.6) | 924 (5.6) | 15,399 (92.8) | 279 (1.7) | 940 (5.7) | 5.7 | 1.7 |
| 12 | 16,323 | 9 (0.1) | 206 (1.3) | 1117 (6.8) | 14,991 (91.8) | 215 (1.3) | 1126 (6.9) | 4.2 | 0.8 |
| 18 | 16,108 | 18 (0.1) | 199 (1.2) | 1280 (7.9) | 14,611 (90.7) | 217 (1.3) | 1298 (8.1) | 8.3 | 1.4 |
| 24 | 15,891 | 13 (0.1) | 184 (1.2) | 1455 (9.2) | 14,239 (89.6) | 197 (1.2) | 1468 (9.2) | 6.6 | 0.9 |
| 30 | 15,694 | 16 (0.1) | 170 (1.1) | 1642 (10.5) | 13,866 (88.4) | 186 (1.2) | 1658 (10.6) | 8.6 | 1.0 |
| 36 | 15,508 | 13 (0.1) | 172 (1.1) | 1829 (11.8) | 13,494 (87.0) | 185 (1.2) | 1842 (11.9) | 7.0 | 0.7 |
| 42 | 15,323 | 22 (0.1) | 161 (1.1) | 2054 (13.4) | 13,086 (85.4) | 183 (1.2) | 2076 (13.5) | 12.0 | 1.1 |
| 48 | 15,140 | 25 (0.2) | 140 (0.9) | 2264 (15.0) | 12,711 (84.0) | 165 (1.1) | 2289 (15.1) | 15.2 | 1.1 |
| 54 | 14,975 | 20 (0.1) | 134 (0.9) | 2511 (16.8) | 12,310 (82.2) | 154 (1.0) | 2531 (16.9) | 13.0 | 0.8 |
| 60 | 14,821 | 21 (0.1) | 138 (0.9) | 2809 (19.0) | 11,853 (80.0) | 159 (1.1) | 2830 (19.1) | 13.2 | 0.7 |
| All | 176,385 | 1335 (0.8) | 4003 (2.3) | 18,656 (10.6) | 152,391 (86.4) | 5338 (3.0) | 19,991 (11.3) | 25.0 | 6.7 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1162 (5.8) | 2236 (11.2) | 771 (3.9) | 15,831 (79.2) | 3398 (17.0) | 1933 (9.7) | 34.2 | 60.1 |
| 6 | 16,602 | 280 (1.7) | 1488 (9.0) | 660 (4.0) | 14,174 (85.4) | 1768 (10.6) | 940 (5.7) | 15.8 | 29.8 |
| 12 | 14,834 | 146 (1.0) | 741 (5.0) | 650 (4.4) | 13,297 (89.6) | 887 (6.0) | 796 (5.4) | 16.5 | 18.3 |
| 18 | 13,947 | 92 (0.7) | 492 (3.5) | 685 (4.9) | 12,678 (90.9) | 584 (4.2) | 777 (5.6) | 15.8 | 11.8 |
| 24 | 13,363 | 87 (0.7) | 385 (2.9) | 706 (5.3) | 12,185 (91.2) | 472 (3.5) | 793 (5.9) | 18.4 | 11.0 |
| 30 | 12,891 | 93 (0.7) | 307 (2.4) | 742 (5.8) | 11,749 (91.1) | 400 (3.1) | 835 (6.5) | 23.2 | 11.1 |
| 36 | 12,491 | 85 (0.7) | 284 (2.3) | 772 (6.2) | 11,350 (90.9) | 369 (3.0) | 857 (6.9) | 23.0 | 9.9 |
| 42 | 12,122 | 68 (0.6) | 268 (2.2) | 838 (6.9) | 10,948 (90.3) | 336 (2.8) | 906 (7.5) | 20.2 | 7.5 |
| 48 | 11,786 | 100 (0.8) | 273 (2.3) | 859 (7.3) | 10,554 (89.5) | 373 (3.2) | 959 (8.1) | 26.8 | 10.4 |
| 54 | 11,413 | 107 (0.9) | 277 (2.4) | 880 (7.7) | 10,149 (88.9) | 384 (3.4) | 987 (8.6) | 27.9 | 10.8 |
| 60 | 11,029 | 111 (1.0) | 260 (2.4) | 906 (8.2) | 9752 (88.4) | 371 (3.4) | 1017 (9.2) | 29.9 | 10.9 |
| All | 150,478 | 2331 (1.5) | 7011 (4.7) | 8469 (5.6) | 132,667 (88.2) | 9342 (6.2) | 10,800 (7.2) | 25.0 | 21.6 |
Inferior strategies
The retest strategy (strategy B) and the strategies with decision rules based on absolute and relative increases from the first and last recorded value (strategies D–G) were inferior to the reference strategy, requiring more tests and causing more patients who had progressed to liver cirrhosis to experience a delay to diagnosis.
Compared with the reference strategy, the main effect of the retest strategy was to increase the number of tests performed (increase of 3.30 tests per person), with also a small increase in the percentage of patients with a delay to diagnosis (absolute increase of 0.40 percentage points), whereas the strategies with decision rules based on absolute and relative increases from the initial value showed only small increases in the number of tests required (increases of 0.14 and 0.18 tests per person, respectively) but larger increases in the percentage of patients with a delay to diagnosis (absolute increases of 1.58 and 2.05 percentage points, respectively). The absolute and relative increase from last recorded value decision rules both increased the number of tests required (by 0.98 and 1.18 per person, respectively) and increased the percentage of patients with a delay to diagnosis (to 10.42% and 11.09%, respectively).
‘Trade-off’ strategies
The reduced monitoring frequency strategy (strategy C) showed a ‘trade-off’ between delay to diagnosis and the number of tests required compared with the reference strategy. The number of tests required decreased by 3.30 tests per person and the percentage of patients with a delay to diagnosis increased by 0.15 percentage points (absolute increase) compared with the reference strategy.
Superior strategies
The reference strategy was found to be inferior to the linear regression strategy. The linear regression strategy used fewer tests (decrease of 0.12 tests per person) and resulted in a lower percentage of patients with a delay to diagnosis (absolute decrease of 0.47 percentage points) compared with the reference strategy.
Sensitivity analyses
Comparing results from the reference strategy when varying estimates of test performance and disease progression
Table 27 demonstrates the effect on the reference strategy of increasing (doubling) or decreasing (halving) various parameter estimates. The results at each monitoring time point using these alternative estimates are provided in Tables 28–39.
| Change in data simulation | Threshold value | PPV, % | Tests per person, na | Delayb (%) | Develop cirrhosis,c n (%) |
|---|---|---|---|---|---|
| None | 10.715 | 25.0 | 7.64 | 6.10 | 5314 (26.6) |
| Decreasedd measurement error | 10.715 | 29.6 (+4.6) | 8.37 (+0.73) | 7.40 (+1.30) | 5248 (26.2) [–66 (0.33)] |
| 10.450 | 25.0 | 7.70 (+0.06) | 5.73 (–0.37) | ||
| Increasede measurement error | 10.715 | 17.6 (–7.4) | 5.84 (–1.80) | 3.85 (–2.25) | 5421 (27.1) [+107 (0.54)] |
| 11.365 | 25.0 | 7.65 (+0.01) | 7.05 (+0.95) | ||
| Decreased between-individual variability | 10.715 | 33.6 (+8.6) | 8.55 (+0.91) | 3.98 (–2.12) | 5139 (25.7) [–175 (0.88)] |
| 10.463 | 25.0 | 7.84 (+0.20) | 2.28 (–3.82) | ||
| Increasede between-individual variability | 10.715 | 16.5 (–8.8) | 5.80 (–1.84) | 5.38 (–0.72) | 5272 (26.4) [–42 (0.21)] |
| 11.905 | 25.0 | 8.26 (+0.62) | 9.95 (+3.85) | ||
| Decreasedd fibrosis progression rate | 10.715 | 22.7 (–2.3) | 7.90 (+0.26) | 4.95 (–1.15) | 4440 (22.2) [–874 (4.37)] |
| 10.860 | 25.0 | 8.29 (+0.65) | 5.68 (–0.42) | ||
| Increasede fibrosis progression rate | 10.715 | 29.2 (+4.2) | 7.00 (–0.64) | 7.62 (+1.52) | 7689 (38.4) [+2375 (11.88)] |
| 10.460 | 25.0 | 6.21 (–1.43) | 5.63 (–0.47) |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1153 (5.8) | 1931 (9.7) | 750 (3.8) | 16,166 (80.8) | 3084 (15.4) | 1903 (9.5) | 37.4 | 60.6 |
| 6 | 16,916 | 113 (0.7) | 478 (2.8) | 810 (4.8) | 15,515 (91.7) | 591 (3.5) | 923 (5.5) | 19.1 | 12.2 |
| 12 | 16,325 | 92 (0.6) | 371 (2.3) | 881 (5.4) | 14,981 (91.8) | 463 (2.8) | 973 (6.0) | 19.9 | 9.5 |
| 18 | 15,862 | 109 (0.7) | 341 (2.1) | 934 (5.9) | 14,478 (91.3) | 450 (2.8) | 1043 (6.6) | 24.2 | 10.5 |
| 24 | 15,412 | 103 (0.7) | 331 (2.1) | 967 (6.3) | 14,011 (90.9) | 434 (2.8) | 1070 (6.9) | 23.7 | 9.6 |
| 30 | 14,978 | 93 (0.6) | 356 (2.4) | 1025 (6.8) | 13,504 (90.2) | 449 (3.0) | 1118 (7.5) | 20.7 | 8.3 |
| 36 | 14,529 | 129 (0.9) | 353 (2.4) | 1032 (7.1) | 13,015 (89.6) | 482 (3.3) | 1161 (8.0) | 26.8 | 11.1 |
| 42 | 14,047 | 128 (0.9) | 356 (2.5) | 1061 (7.6) | 12,502 (89.0) | 484 (3.4) | 1189 (8.5) | 26.4 | 10.8 |
| 48 | 13,563 | 112 (0.8) | 341 (2.5) | 1086 (8.0) | 12,024 (88.7) | 453 (3.3) | 1198 (8.8) | 24.7 | 9.3 |
| 54 | 13,110 | 137 (1.0) | 334 (2.5) | 1105 (8.4) | 11,534 (88.0) | 471 (3.6) | 1242 (9.5) | 29.1 | 11.0 |
| 60 | 12,639 | 164 (1.3) | 370 (2.9) | 1095 (8.7) | 11,010 (87.1) | 534 (4.2) | 1259 (10.0) | 30.7 | 13.0 |
| All | 167,381 | 2333 (1.4) | 5562 (3.3) | 10,746 (6.4) | 148,740 (88.9) | 7895 (4.7) | 13,079 (7.8) | 29.6 | 17.8 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1289 (6.4) | 2755 (13.8) | 614 (3.1) | 15,342 (76.7) | 4044 (20.2) | 1903 (9.5) | 31.9 | 67.7 |
| 6 | 15,956 | 114 (0.7) | 665 (4.2) | 645 (4.0) | 14,532 (91.1) | 779 (4.9) | 759 (4.8) | 14.6 | 15.0 |
| 12 | 15,177 | 93 (0.6) | 441 (2.9) | 686 (4.5) | 13,957 (92.0) | 534 (3.5) | 779 (5.1) | 17.4 | 11.9 |
| 18 | 14,643 | 97 (0.7) | 431 (2.9) | 726 (5.0) | 13,389 (91.4) | 528 (3.6) | 823 (5.6) | 18.4 | 11.8 |
| 24 | 14,115 | 83 (0.6) | 438 (3.1) | 761 (5.4) | 12,833 (90.9) | 521 (3.7) | 844 (6.0) | 15.9 | 9.8 |
| 30 | 13,594 | 111 (0.8) | 410 (3.0) | 772 (5.7) | 12,301 (90.5) | 521 (3.8) | 883 (6.5) | 21.3 | 12.6 |
| 36 | 13,073 | 100 (0.8) | 399 (3.1) | 785 (6.0) | 11,789 (90.2) | 499 (3.8) | 885 (6.8) | 20.0 | 11.3 |
| 42 | 12,574 | 108 (0.9) | 361 (2.9) | 815 (6.5) | 11,290 (89.8) | 469 (3.7) | 923 (7.3) | 23.0 | 11.7 |
| 48 | 12,105 | 108 (0.9) | 379 (3.1) | 818 (6.8) | 10,800 (89.2) | 487 (4.0) | 926 (7.6) | 22.2 | 11.7 |
| 54 | 11,618 | 133 (1.1) | 383 (3.3) | 817 (7.0) | 10,285 (88.5) | 516 (4.4) | 950 (8.2) | 25.8 | 14.0 |
| 60 | 11,102 | 118 (1.1) | 393 (3.5) | 815 (7.3) | 9776 (88.1) | 511 (4.6) | 933 (8.4) | 23.1 | 12.6 |
| All | 153,957 | 2354 (1.5) | 7055 (4.6) | 8254 (5.4) | 136,294 (88.5) | 9409 (6.1) | 10,608 (6.9) | 25.0 | 22.2 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1169 (5.8) | 3257 (16.3) | 812 (4.1) | 14,762 (73.8) | 4426 (22.1) | 1981 (9.9) | 26.4 | 59.0 |
| 6 | 15,574 | 369 (2.4) | 1897 (12.2) | 621 (4.0) | 12,687 (81.5) | 2266 (14.5) | 990 (6.4) | 16.3 | 37.3 |
| 12 | 13,308 | 213 (1.6) | 1360 (10.2) | 540 (4.1) | 11,195 (84.1) | 1573 (11.8) | 753 (5.7) | 13.5 | 28.3 |
| 18 | 11,735 | 141 (1.2) | 1130 (9.6) | 495 (4.2) | 9969 (85.0) | 1271 (10.8) | 636 (5.4) | 11.1 | 22.2 |
| 24 | 10,464 | 123 (1.2) | 881 (8.4) | 455 (4.3) | 9005 (86.1) | 1004 (9.6) | 578 (5.5) | 12.3 | 21.3 |
| 30 | 9460 | 99 (1.0) | 753 (8.0) | 430 (4.5) | 8178 (86.4) | 852 (9.0) | 529 (5.6) | 11.6 | 18.7 |
| 36 | 8608 | 93 (1.1) | 682 (7.9) | 408 (4.7) | 7425 (86.3) | 775 (9.0) | 501 (5.8) | 12.0 | 18.6 |
| 42 | 7833 | 88 (1.1) | 534 (6.8) | 404 (5.2) | 6807 (86.9) | 622 (7.9) | 492 (6.3) | 14.1 | 17.9 |
| 48 | 7211 | 86 (1.2) | 514 (7.1) | 391 (5.4) | 6220 (86.3) | 600 (8.3) | 477 (6.6) | 14.3 | 18.0 |
| 54 | 6611 | 78 (1.2) | 454 (6.9) | 388 (5.9) | 5691 (86.1) | 532 (8.0) | 466 (7.0) | 14.7 | 16.7 |
| 60 | 6079 | 77 (1.3) | 409 (6.7) | 375 (6.2) | 5218 (85.8) | 486 (8.0) | 452 (7.4) | 15.8 | 17.0 |
| All | 116,883 | 2536 (2.2) | 11,871 (10.2) | 5319 (4.6) | 97,157 (83.1) | 14,407 (12.3) | 7855 (6.7) | 17.6 | 32.3 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 848 (4.2) | 1562 (7.8) | 1133 (5.7) | 16,457 (82.3) | 2410 (12.0) | 1981 (9.9) | 35.2 | 42.8 |
| 6 | 17,590 | 368 (2.1) | 1081 (6.1) | 1001 (5.7) | 15,140 (86.1) | 1449 (8.2) | 1369 (7.8) | 25.4 | 26.9 |
| 12 | 16,141 | 274 (1.7) | 827 (5.1) | 942 (5.8) | 14,098 (87.3) | 1101 (6.8) | 1216 (7.5) | 24.9 | 22.5 |
| 18 | 15,040 | 189 (1.3) | 726 (4.8) | 913 (6.1) | 13,212 (87.8) | 915 (6.1) | 1102 (7.3) | 20.7 | 17.2 |
| 24 | 14,125 | 160 (1.1) | 583 (4.1) | 889 (6.3) | 12,493 (88.4) | 743 (5.3) | 1049 (7.4) | 21.5 | 15.3 |
| 30 | 13,382 | 139 (1.0) | 580 (4.3) | 881 (6.6) | 11,782 (88.0) | 719 (5.4) | 1020 (7.6) | 19.3 | 13.6 |
| 36 | 12,663 | 130 (1.0) | 596 (4.7) | 861 (6.8) | 11,076 (87.5) | 726 (5.7) | 991 (7.8) | 17.9 | 13.1 |
| 42 | 11,937 | 121 (1.0) | 473 (4.0) | 883 (7.4) | 10,460 (87.6) | 594 (5.0) | 1004 (8.4) | 20.4 | 12.1 |
| 48 | 11,343 | 129 (1.1) | 497 (4.4) | 877 (7.7) | 9840 (86.7) | 626 (5.5) | 1006 (8.9) | 20.6 | 12.8 |
| 54 | 10,717 | 119 (1.1) | 441 (4.1) | 883 (8.2) | 9274 (86.5) | 560 (5.2) | 1002 (9.3) | 21.2 | 11.9 |
| 60 | 10,157 | 125 (1.2) | 433 (4.3) | 882 (8.7) | 8717 (85.8) | 558 (5.5) | 1007 (9.9) | 22.4 | 12.4 |
| All | 153,095 | 2602 (1.7) | 7799 (5.1) | 10,145 (6.6) | 132,549 (86.6) | 10,401 (6.8) | 12,747 (8.3) | 25.0 | 20.4 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1171 (5.9) | 963 (4.8) | 639 (3.2) | 17,227 (86.1) | 2134 (10.7) | 1810 (9.0) | 54.9 | 64.7 |
| 6 | 17,866 | 290 (1.6) | 548 (3.1) | 514 (2.9) | 16,514 (92.4) | 838 (4.7) | 804 (4.5) | 34.6 | 36.1 |
| 12 | 17,028 | 185 (1.1) | 421 (2.5) | 500 (2.9) | 15,922 (93.5) | 606 (3.6) | 685 (4.0) | 30.5 | 27 |
| 18 | 16,422 | 181 (1.1) | 406 (2.5) | 464 (2.8) | 15,371 (93.6) | 587 (3.6) | 645 (3.9) | 30.8 | 28.1 |
| 24 | 15,835 | 128 (0.8) | 391 (2.5) | 447 (2.8) | 14,869 (93.9) | 519 (3.3) | 575 (3.6) | 24.7 | 22.3 |
| 30 | 15,316 | 117 (0.8) | 373 (2.4) | 449 (2.9) | 14,377 (93.9) | 490 (3.2) | 566 (3.7) | 23.9 | 20.7 |
| 36 | 14,826 | 127 (0.9) | 445 (3.0) | 434 (2.9) | 13,820 (93.2) | 572 (3.9) | 561 (3.8) | 22.2 | 22.6 |
| 42 | 14,254 | 117 (0.8) | 424 (3.0) | 436 (3.1) | 13,277 (93.1) | 541 (3.8) | 553 (3.9) | 21.6 | 21.2 |
| 48 | 13,713 | 127 (0.9) | 471 (3.4) | 431 (3.1) | 12,684 (92.5) | 598 (4.4) | 558 (4.1) | 21.2 | 22.8 |
| 54 | 13,115 | 136 (1.0) | 424 (3.2) | 435 (3.3) | 12,120 (92.4) | 560 (4.3) | 571 (4.4) | 24.3 | 23.8 |
| 60 | 12,555 | 124 (1.0) | 474 (3.8) | 442 (3.5) | 11,515 (91.7) | 598 (4.8) | 566 (4.5) | 20.7 | 21.9 |
| All | 170,930 | 2703 (1.6) | 5340 (3.1) | 5191 (3.0) | 157,696 (92.3) | 8043 (4.7) | 7894 (4.6) | 33.6 | 34.2 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1333 (6.7) | 1517 (7.6) | 477 (2.4) | 16,673 (83.4) | 2850 (14.2) | 1810 (9.0) | 46.8 | 73.6 |
| 6 | 17,150 | 259 (1.5) | 826 (4.8) | 333 (1.9) | 15,732 (91.7) | 1085 (6.3) | 592 (3.5) | 23.9 | 43.8 |
| 12 | 16,065 | 156 (1.0) | 655 (4.1) | 298 (1.9) | 14,956 (93.1) | 811 (5.0) | 454 (2.8) | 19.2 | 34.4 |
| 18 | 15,254 | 131 (0.9) | 558 (3.7) | 271 (1.8) | 14,294 (93.7) | 689 (4.5) | 402 (2.6) | 19.0 | 32.6 |
| 24 | 14,565 | 86 (0.6) | 562 (3.9) | 253 (1.7) | 13,664 (93.8) | 648 (4.4) | 339 (2.3) | 13.3 | 25.4 |
| 30 | 13,917 | 79 (0.6) | 552 (4.0) | 256 (1.8) | 13,030 (93.6) | 631 (4.5) | 335 (2.4) | 12.5 | 23.6 |
| 36 | 13,286 | 96 (0.7) | 576 (4.3) | 241 (1.8) | 12,373 (93.1) | 672 (5.1) | 337 (2.5) | 14.3 | 28.5 |
| 42 | 12,614 | 72 (0.6) | 529 (4.2) | 238 (1.9) | 11,775 (93.3) | 601 (4.8) | 310 (2.5) | 12.0 | 23.2 |
| 48 | 12,013 | 85 (0.7) | 575 (4.8) | 250 (2.1) | 11,103 (92.4) | 660 (5.5) | 335 (2.8) | 12.9 | 25.4 |
| 54 | 11,353 | 107 (0.9) | 565 (5.0) | 236 (2.1) | 10,445 (92.0) | 672 (5.9) | 343 (3.0) | 15.9 | 31.2 |
| 60 | 10,681 | 84 (0.8) | 535 (5.0) | 236 (2.2) | 9826 (92.0) | 619 (5.8) | 320 (3.0) | 13.6 | 26.2 |
| All | 156,898 | 2488 (1.6) | 7450 (4.7) | 3089 (2.0) | 143,871 (91.7) | 9938 (6.3) | 5577 (3.6) | 25.0 | 44.6 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1279 (6.4) | 5806 (29.0) | 656 (3.3) | 12,259 (61.3) | 7085 (35.4) | 1935 (9.7) | 18.1 | 66.1 |
| 6 | 12,915 | 104 (0.8) | 1193 (9.2) | 685 (5.3) | 10,933 (84.7) | 1297 (10.0) | 789 (6.1) | 8.0 | 13.2 |
| 12 | 11,618 | 78 (0.7) | 768 (6.6) | 707 (6.1) | 10,065 (86.6) | 846 (7.3) | 785 (6.8) | 9.2 | 9.9 |
| 18 | 10,772 | 81 (0.8) | 589 (5.5) | 734 (6.8) | 9368 (87.0) | 670 (6.2) | 815 (7.6) | 12.1 | 9.9 |
| 24 | 10,102 | 85 (0.8) | 468 (4.6) | 757 (7.5) | 8792 (87.0) | 553 (5.5) | 842 (8.3) | 15.4 | 10.1 |
| 30 | 9549 | 84 (0.9) | 392 (4.1) | 745 (7.8) | 8328 (87.2) | 476 (5.0) | 829 (8.7) | 17.6 | 10.1 |
| 36 | 9073 | 78 (0.9) | 368 (4.1) | 758 (8.4) | 7869 (86.7) | 446 (4.9) | 836 (9.2) | 17.5 | 9.3 |
| 42 | 8627 | 85 (1.0) | 356 (4.1) | 774 (9.0) | 7412 (85.9) | 441 (5.1) | 859 (10.0) | 19.3 | 9.9 |
| 48 | 8186 | 90 (1.1) | 302 (3.7) | 768 (9.4) | 7026 (85.8) | 392 (4.8) | 858 (10.5) | 23.0 | 10.5 |
| 54 | 7794 | 99 (1.3) | 285 (3.7) | 779 (10.0) | 6631 (85.1) | 384 (4.9) | 878 (11.3) | 25.8 | 11.3 |
| 60 | 7410 | 79 (1.1) | 305 (4.1) | 794 (10.7) | 6232 (84.1) | 384 (5.2) | 873 (11.8) | 20.6 | 9.0 |
| All | 116,046 | 2142 (1.8) | 10,832 (9.3) | 8157 (7.0) | 94,915 (81.8) | 12,974 (11.2) | 10,299 (8.9) | 16.5 | 20.8 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 890 (4.4) | 2368 (11.8) | 1045 (5.2) | 15,697 (78.5) | 3258 (16.3) | 1935 (9.7) | 27.3 | 46.0 |
| 6 | 16,742 | 117 (0.7) | 610 (3.6) | 1127 (6.7) | 14,888 (88.9) | 727 (4.3) | 1244 (7.4) | 16.1 | 9.4 |
| 12 | 16,015 | 98 (0.6) | 413 (2.6) | 1208 (7.5) | 14,296 (89.3) | 511 (3.2) | 1306 (8.2) | 19.2 | 7.5 |
| 18 | 15,504 | 102 (0.7) | 339 (2.2) | 1286 (8.3) | 13,777 (88.9) | 441 (2.8) | 1388 (9.0) | 23.1 | 7.3 |
| 24 | 15,063 | 81 (0.5) | 332 (2.2) | 1399 (9.3) | 13,251 (88.0) | 413 (2.7) | 1480 (9.8) | 19.6 | 5.5 |
| 30 | 14,650 | 78 (0.5) | 308 (2.1) | 1490 (10.2) | 12,774 (87.2) | 386 (2.6) | 1568 (10.7) | 20.2 | 5.0 |
| 36 | 14,264 | 114 (0.8) | 295 (2.1) | 1535 (10.8) | 12,320 (86.4) | 409 (2.9) | 1649 (11.6) | 27.9 | 6.9 |
| 42 | 13,855 | 103 (0.7) | 276 (2.0) | 1606 (11.6) | 11,870 (85.7) | 379 (2.7) | 1709 (12.3) | 27.2 | 6.0 |
| 48 | 13,476 | 122 (0.9) | 318 (2.4) | 1641 (12.2) | 11,395 (84.6) | 440 (3.3) | 1763 (13.1) | 27.7 | 6.9 |
| 54 | 13,036 | 103 (0.8) | 271 (2.1) | 1742 (13.4) | 10,920 (83.8) | 374 (2.9) | 1845 (14.2) | 27.5 | 5.6 |
| 60 | 12,662 | 130 (1.0) | 286 (2.3) | 1817 (14.4) | 10,429 (82.4) | 416 (3.3) | 1947 (15.4) | 31.2 | 6.7 |
| All | 165,267 | 1938 (1.2) | 5816 (3.5) | 15,896 (9.6) | 141,617 (85.7) | 7754 (4.7) | 17,834 (10.8) | 25.0 | 10.9 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1038 (5.2) | 2122 (10.6) | 698 (3.5) | 16,142 (80.7) | 3160 (15.8) | 1736 (8.7) | 32.8 | 59.8 |
| 6 | 16,840 | 172 (1.0) | 848 (5.0) | 631 (3.7) | 15,189 (90.2) | 1020 (6.1) | 803 (4.8) | 16.9 | 21.4 |
| 12 | 15,820 | 110 (0.7) | 603 (3.8) | 617 (3.9) | 14,490 (91.6) | 713 (4.5) | 727 (4.6) | 15.4 | 15.1 |
| 18 | 15,107 | 97 (0.6) | 580 (3.8) | 624 (4.1) | 13,806 (91.4) | 677 (4.5) | 721 (4.8) | 14.3 | 13.5 |
| 24 | 14,430 | 86 (0.6) | 448 (3.1) | 648 (4.5) | 13,248 (91.8) | 534 (3.7) | 734 (5.1) | 16.1 | 11.7 |
| 30 | 13,896 | 84 (0.6) | 402 (2.9) | 655 (4.7) | 12,755 (91.8) | 486 (3.5) | 739 (5.3) | 17.3 | 11.4 |
| 36 | 13,410 | 82 (0.6) | 459 (3.4) | 677 (5.0) | 12,192 (90.9) | 541 (4.0) | 759 (5.7) | 15.2 | 10.8 |
| 42 | 12,869 | 102 (0.8) | 413 (3.2) | 667 (5.2) | 11,687 (90.8) | 515 (4.0) | 769 (6.0) | 19.8 | 13.3 |
| 48 | 12,354 | 108 (0.9) | 416 (3.4) | 646 (5.2) | 11,184 (90.5) | 524 (4.2) | 754 (6.1) | 20.6 | 14.3 |
| 54 | 11,830 | 84 (0.7) | 371 (3.1) | 663 (5.6) | 10,712 (90.5) | 455 (3.8) | 747 (6.3) | 18.5 | 11.2 |
| 60 | 11,375 | 96 (0.8) | 351 (3.1) | 678 (6.0) | 10,250 (90.1) | 447 (3.9) | 774 (6.8) | 21.5 | 12.4 |
| All | 157,931 | 2059 (1.3) | 7013 (4.4) | 7204 (4.6) | 141,655 (89.7) | 9072 (5.7) | 9263 (5.9) | 22.7 | 22.2 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 960 (4.8) | 1707 (8.5) | 776 (3.9) | 16,557 (82.8) | 2667 (13.3) | 1736 (8.7) | 36.0 | 55.3 |
| 6 | 17,333 | 184 (1.1) | 730 (4.2) | 707 (4.1) | 15,712 (90.6) | 914 (5.3) | 891 (5.1) | 20.1 | 20.7 |
| 12 | 16,419 | 125 (0.8) | 516 (3.1) | 692 (4.2) | 15,086 (91.9) | 641 (3.9) | 817 (5.0) | 19.5 | 15.3 |
| 18 | 15,778 | 101 (0.6) | 466 (3.0) | 706 (4.5) | 14,505 (91.9) | 567 (3.6) | 807 (5.1) | 17.8 | 12.5 |
| 24 | 15,211 | 96 (0.6) | 395 (2.6) | 735 (4.8) | 13,985 (91.9) | 491 (3.2) | 831 (5.5) | 19.6 | 11.6 |
| 30 | 14,720 | 77 (0.5) | 393 (2.7) | 760 (5.2) | 13,490 (91.6) | 470 (3.2) | 837 (5.7) | 16.4 | 9.2 |
| 36 | 14,250 | 83 (0.6) | 427 (3.0) | 792 (5.6) | 12,948 (90.9) | 510 (3.6) | 875 (6.1) | 16.3 | 9.5 |
| 42 | 13,740 | 101 (0.7) | 374 (2.7) | 793 (5.8) | 12,472 (90.8) | 475 (3.5) | 894 (6.5) | 21.3 | 11.3 |
| 48 | 13,265 | 110 (0.8) | 381 (2.9) | 783 (5.9) | 11,991 (90.4) | 491 (3.7) | 893 (6.7) | 22.4 | 12.3 |
| 54 | 12,774 | 100 (0.8) | 379 (3.0) | 795 (6.2) | 11,500 (90.0) | 479 (3.7) | 895 (7.0) | 20.9 | 11.2 |
| 60 | 12,295 | 105 (0.9) | 342 (2.8) | 813 (6.6) | 11,035 (89.8) | 447 (3.6) | 918 (7.5) | 23.5 | 11.4 |
| All | 165,785 | 2042 (1.2) | 6110 (3.7) | 8352 (5.0) | 149,281 (90.0) | 8152 (4.9) | 10,394 (6.3) | 25.0 | 19.6 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1535 (7.7) | 2428 (12.1) | 947 (4.7) | 15,090 (75.4) | 3963 (19.8) | 2482 (12.4) | 38.7 | 61.8 |
| 6 | 16,037 | 269 (1.7) | 990 (6.2) | 909 (5.7) | 13,869 (86.5) | 1259 (7.9) | 1178 (7.3) | 21.4 | 22.8 |
| 12 | 14,778 | 200 (1.4) | 772 (5.2) | 930 (6.3) | 12,876 (87.1) | 972 (6.6) | 1130 (7.6) | 20.6 | 17.7 |
| 18 | 13,806 | 186 (1.3) | 649 (4.7) | 945 (6.8) | 12,026 (87.1) | 835 (6.0) | 1131 (8.2) | 22.3 | 16.4 |
| 24 | 12,971 | 163 (1.3) | 607 (4.7) | 938 (7.2) | 11,263 (86.8) | 770 (5.9) | 1101 (8.5) | 21.2 | 14.8 |
| 30 | 12,201 | 165 (1.4) | 557 (4.6) | 975 (8.0) | 10,504 (86.1) | 722 (5.9) | 1140 (9.3) | 22.9 | 14.5 |
| 36 | 11,479 | 196 (1.7) | 596 (5.2) | 969 (8.4) | 9718 (84.7) | 792 (6.9) | 1165 (10.1) | 24.7 | 16.8 |
| 42 | 10,687 | 171 (1.6) | 506 (4.7) | 974 (9.1) | 9036 (84.6) | 677 (6.3) | 1145 (10.7) | 25.3 | 14.9 |
| 48 | 10,010 | 204 (2.0) | 512 (5.1) | 979 (9.8) | 8315 (83.1) | 716 (7.2) | 1183 (11.8) | 28.5 | 17.2 |
| 54 | 9294 | 197 (2.1) | 440 (4.7) | 1009 (10.9) | 7648 (82.3) | 637 (6.9) | 1206 (13.0) | 30.9 | 16.3 |
| 60 | 8657 | 205 (2.4) | 413 (4.8) | 1009 (11.7) | 7030 (81.2) | 618 (7.1) | 1214 (14.0) | 33.2 | 16.9 |
| All | 139,920 | 3491 (2.5) | 8470 (6.1) | 10,584 (7.6) | 117,375 (83.9) | 11,961 (8.5) | 14,075 (10.1) | 29.2 | 24.8 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 271 (1.8) | 1308 (8.7) | 709 (4.7) | 12,676 (84.7) | 1579 (10.6) | 980 (6.5) | 17.2 | 27.7 |
| 12 | 13,385 | 183 (1.4) | 979 (7.3) | 699 (5.2) | 11,524 (86.1) | 1162 (8.7) | 882 (6.6) | 15.7 | 20.7 |
| 18 | 12,223 | 174 (1.4) | 743 (6.1) | 705 (5.8) | 10,601 (86.7) | 917 (7.5) | 879 (7.2) | 19.0 | 19.8 |
| 24 | 11,306 | 147 (1.3) | 691 (6.1) | 680 (6.0) | 9788 (86.6) | 838 (7.4) | 827 (7.3) | 17.5 | 17.8 |
| 30 | 10,468 | 139 (1.3) | 566 (5.4) | 693 (6.6) | 9070 (86.6) | 705 (6.7) | 832 (7.9) | 19.7 | 16.7 |
| 36 | 9763 | 139 (1.4) | 591 (6.1) | 699 (7.2) | 8334 (85.4) | 730 (7.5) | 838 (8.6) | 19.0 | 16.6 |
| 42 | 9033 | 146 (1.6) | 553 (6.1) | 684 (7.6) | 7650 (84.7) | 699 (7.7) | 830 (9.2) | 20.9 | 17.6 |
| 48 | 8334 | 159 (1.9) | 472 (5.7) | 689 (8.3) | 7014 (84.2) | 631 (7.6) | 848 (10.2) | 25.2 | 18.8 |
| 54 | 7703 | 162 (2.1) | 465 (6.0) | 706 (9.2) | 6370 (82.7) | 627 (8.1) | 868 (11.3) | 25.8 | 18.7 |
| 60 | 7076 | 163 (2.3) | 450 (6.4) | 712 (10.1) | 5751 (81.3) | 613 (8.7) | 875 (12.4) | 26.6 | 18.6 |
| All | 124,255 | 3387 (2.7) | 10150 (8.2) | 7754 (6.2) | 102,964 (82.9) | 13,537 (10.9) | 11,141 (9.0) | 25.0 | 30.4 |
Improved estimates of test performance (decreased measurement error and decreased between-individual variability) both improved the PPV (absolute increases of 4.6 and 8.7 percentage points, respectively) and increased the number of tests required (increases of 0.73 and 0.91 tests per person, respectively), with the decreased measurement error also increasing the percentage of patients with a delay to diagnosis (absolute increase of 1.30 percentage points). Both increased and decreased between-individual variability reduced the percentage of patients with a delay to diagnosis (absolute decrease of 0.72 and 2.12 percentage points, respectively). An increased rate of fibrosis progression led to both an increased PPV (absolute increase of 4.2 percentage points) and an increase in the percentage of patients with a delay to diagnosis (absolute increase of 1.52 percentage points) but decreased the number of tests required (decrease of 0.64 tests per person).
The largest difference in PPV was achieved by increasing the between-individual variability (absolute decrease of 8.8 percentage points); the largest difference in number of tests required was achieved by increasing the measurement error (decrease of 1.84 tests per person); and the largest difference in the percentage of patients with a delay to diagnosis was achieved by decreasing between-individual variability (absolute decrease of 2.12 percentage points).
Adjusted fibrosis progression rate
The results of evaluating strategies based on data with the adjusted estimate of fibrosis progression are provided in Tables 40–60 and Figure 16. Results for strategies compared with the reference strategy appeared similar to results when using the unadjusted estimate.
| Strategy | Monitoring strategy components | PPV, % | Testsa | Delay to diagnosisb | Test performance | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Decision rule | Threshold value | Observation interval (months) | Retest | Initial threshold value | N | N per personc | Median IQR | N | % of alld | % of stage 4e | TP per personf | FP per persong | Positive, n (%) | Sensitivity, % | ||
| A | Simple threshold | 10.46 | 6 | False | – | 25 | 124,255 | 6.21 | 6 (1, 11) | 1126 | 5.63 | 14.64 | 0.17 | 0.51 | 13,537 (10.89) | 30.40 |
| B | Simple threshold | 10.325 | 6 | True | – | 25 | 192,590h | 9.63 | 11 (2, 15) | 1194 | 5.97 | 15.53 | 0.16 | 0.49 | 13,048 (10.43) | 29.88 |
| C | Simple threshold | 10.265 | 12 | False | – | 25 | 72,001 | 3.60 | 4 (1, 6) | 1128 | 5.64 | 14.67 | 0.17 | 0.52 | 13,814 (19.19) | 47.02 |
| D | Absolute increase from initial value | 1.138 | 6 | False | 10.46 | 25 | 125,699 | 6.28 | 7 (1, 11) | 1415 | 7.07 | 18.40 | 0.18 | 0.54 | 14,509 (11.54) | 28.57 |
| E | Absolute increase from last value | 1.245 | 6 | False | 10.46 | 25 | 142,849 | 7.14 | 10 (1, 11) | 2220 | 11.10 | 28.87 | 0.13 | 0.40 | 10,587 (7.41) | 13.40 |
| F | Relative increase from initial value | 1.122 | 6 | False | 10.46 | 25 | 124,237 | 6.21 | 6 (1, 11) | 1450 | 7.25 | 18.86 | 0.18 | 0.55 | 14,565 (11.72) | 27.98 |
| G | Relative increase from last value | 1.154 | 6 | False | 10.46 | 25 | 151,266 | 7.56 | 11 (1, 11) | 2547 | 12.73 | 33.13 | 0.11 | 0.33 | 8673 (5.73) | 9.60 |
| H | Linear regression | 10.235 | 6 | False | 10.46 | 25 | 121,214 | 6.06 | 5 (1, 11) | 1039 | 5.20 | 13.51 | 0.16 | 0.48 | 12,930 (10.67) | 30.01 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 271 (1.8) | 1308 (8.7) | 709 (4.7) | 12,676 (84.7) | 1579 (10.6) | 980 (6.5) | 17.2 | 27.7 |
| 12 | 13,385 | 183 (1.4) | 979 (7.3) | 699 (5.2) | 11,524 (86.1) | 1162 (8.7) | 882 (6.6) | 15.7 | 20.7 |
| 18 | 12,223 | 174 (1.4) | 743 (6.1) | 705 (5.8) | 10,601 (86.7) | 917 (7.5) | 879 (7.2) | 19.0 | 19.8 |
| 24 | 11,306 | 147 (1.3) | 691 (6.1) | 680 (6.0) | 9788 (86.6) | 838 (7.4) | 827 (7.3) | 17.5 | 17.8 |
| 30 | 10,468 | 139 (1.3) | 566 (5.4) | 693 (6.6) | 9070 (86.6) | 705 (6.7) | 832 (7.9) | 19.7 | 16.7 |
| 36 | 9763 | 139 (1.4) | 591 (6.1) | 699 (7.2) | 8334 (85.4) | 730 (7.5) | 838 (8.6) | 19.0 | 16.6 |
| 42 | 9033 | 146 (1.6) | 553 (6.1) | 684 (7.6) | 7650 (84.7) | 699 (7.7) | 830 (9.2) | 20.9 | 17.6 |
| 48 | 8334 | 159 (1.9) | 472 (5.7) | 689 (8.3) | 7014 (84.2) | 631 (7.6) | 848 (10.2) | 25.2 | 18.8 |
| 54 | 7703 | 162 (2.1) | 465 (6.0) | 706 (9.2) | 6370 (82.7) | 627 (8.1) | 868 (11.3) | 25.8 | 18.7 |
| 60 | 7076 | 163 (2.3) | 450 (6.4) | 712 (10.1) | 5751 (81.3) | 613 (8.7) | 875 (12.4) | 26.6 | 18.6 |
| All | 124,255 | 3387 (2.7) | 10,150 (8.2) | 7754 (6.2) | 102,964 (82.9) | 13,537 (10.9) | 11,141 (9.0) | 25.0 | 30.4 |
| Observation time (months) | Tests,a N/n | Results, n (%) | Diseased,b n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 30,364/20,000 | 1805 (9.0) | 3694 (18.5) | 677 (3.4) | 13,824 (69.1) | 5499 (27.5) | 2482 (12.4) | 32.8 | 72.7 |
| 6 | 22,078/14,501 | 196 (1.4) | 1108 (7.6) | 656 (4.5) | 12,541 (86.5) | 1304 (9.0) | 852 (5.9) | 15.0 | 23.0 |
| 12 | 20,017/13,197 | 141 (1.1) | 827 (6.3) | 682 (5.2) | 11,547 (87.5) | 968 (7.3) | 823 (6.2) | 14.6 | 17.1 |
| 18 | 18,707/12,229 | 139 (1.1) | 691 (5.7) | 709 (5.8) | 10,690 (87.4) | 830 (6.8) | 848 (6.9) | 16.7 | 16.4 |
| 24 | 17,432/11,399 | 146 (1.3) | 585 (5.1) | 682 (6.0) | 9986 (87.6) | 731 (6.4) | 828 (7.3) | 20.0 | 17.6 |
| 30 | 16,494/10,668 | 132 (1.2) | 564 (5.3) | 706 (6.6) | 9266 (86.9) | 696 (6.5) | 838 (7.9) | 19.0 | 15.8 |
| 36 | 15,442/9972 | 153 (1.5) | 562 (5.6) | 698 (7.0) | 8559 (85.8) | 715 (7.2) | 851 (8.5) | 21.4 | 18.0 |
| 42 | 14,378/9257 | 145 (1.6) | 537 (5.8) | 686 (7.4) | 7889 (85.2) | 682 (7.4) | 831 (9.0) | 21.3 | 17.4 |
| 48 | 13,460/8575 | 161 (1.9) | 466 (5.4) | 683 (8.0) | 7265 (84.7) | 627 (7.3) | 844 (9.8) | 25.7 | 19.1 |
| 54 | 12,558/7948 | 141 (1.8) | 462 (5.8) | 703 (8.8) | 6642 (83.6) | 603 (7.6) | 844 (10.6) | 23.4 | 16.7 |
| 60 | 11,660/7345 | 100 (1.4) | 293 (4.0) | 766 (10.4) | 6186 (84.2) | 393 (5.4) | 866 (11.8) | 25.4 | 11.5 |
| All | 192,590/125,091 | 3259 (2.6) | 9789 (7.8) | 7648 (6.1) | 104,395 (83.5) | 13,048 (10.4) | 10,907 (8.7) | 25.0 | 29.9 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1818 (9.1) | 4227 (21.1) | 664 (3.3) | 13,291 (66.5) | 6045 (30.2) | 2482 (12.4) | 30.1 | 73.2 |
| 12 | 13,955 | 383 (2.7) | 1789 (12.8) | 650 (4.7) | 11,133 (79.8) | 2172 (15.6) | 1033 (7.4) | 17.6 | 37.1 |
| 24 | 11,783 | 314 (2.7) | 1327 (11.3) | 634 (5.4) | 9508 (80.7) | 1641 (13.9) | 948 (8.0) | 19.1 | 33.1 |
| 36 | 10,142 | 292 (2.9) | 1133 (11.2) | 654 (6.4) | 8063 (79.5) | 1425 (14.1) | 946 (9.3) | 20.5 | 30.9 |
| 48 | 8717 | 318 (3.6) | 995 (11.4) | 641 (7.4) | 6763 (77.6) | 1313 (15.1) | 959 (11.0) | 24.2 | 33.2 |
| 60 | 7404 | 331 (4.5) | 887 (12.0) | 651 (8.8) | 5535 (74.8) | 1218 (16.5) | 982 (13.3) | 27.2 | 33.7 |
| All | 72,001 | 3456 (4.8) | 10,358 (14.4) | 3894 (5.4) | 54,293 (75.4) | 13,814 (19.2) | 7350 (10.2) | 25.0 | 47.0 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 90 (0.6) | 867 (5.8) | 890 (5.9) | 13,117 (87.7) | 957 (6.4) | 980 (6.5) | 9.4 | 9.2 |
| 12 | 14,007 | 132 (0.9) | 816 (5.8) | 968 (6.9) | 12,091 (86.3) | 948 (6.8) | 1100 (7.9) | 13.9 | 12.0 |
| 18 | 13,059 | 172 (1.3) | 852 (6.5) | 1008 (7.7) | 11,027 (84.4) | 1024 (7.8) | 1180 (9.0) | 16.8 | 14.6 |
| 24 | 12,035 | 185 (1.5) | 754 (6.3) | 991 (8.2) | 10,105 (84.0) | 939 (7.8) | 1176 (9.8) | 19.7 | 15.7 |
| 30 | 11,096 | 222 (2.0) | 780 (7.0) | 966 (8.7) | 9128 (82.3) | 1002 (9.0) | 1188 (10.7) | 22.2 | 18.7 |
| 36 | 10,094 | 233 (2.3) | 785 (7.8) | 882 (8.7) | 8194 (81.2) | 1018 (10.1) | 1115 (11.0) | 22.9 | 20.9 |
| 42 | 9076 | 231 (2.5) | 794 (8.7) | 789 (8.7) | 7262 (80.0) | 1025 (11.3) | 1020 (11.2) | 22.5 | 22.6 |
| 48 | 8051 | 249 (3.1) | 732 (9.1) | 688 (8.5) | 6382 (79.3) | 981 (12.2) | 937 (11.6) | 25.4 | 26.6 |
| 54 | 7070 | 224 (3.2) | 599 (8.5) | 594 (8.4) | 5653 (80.0) | 823 (11.6) | 818 (11.6) | 27.2 | 27.4 |
| 60 | 6247 | 190 (3.0) | 566 (9.1) | 527 (8.4) | 4964 (79.5) | 756 (12.1) | 717 (11.5) | 25.1 | 26.5 |
| All | 125,699 | 3632 (2.9) | 10,877 (8.7) | 9081 (7.2) | 102,109 (81.2) | 14,509 (11.5) | 12,713 (10.1) | 25.0 | 28.6 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 63 (0.4) | 629 (4.2) | 917 (6.1) | 13,355 (89.2) | 692 (4.6) | 980 (6.5) | 9.1 | 6.4 |
| 12 | 14,272 | 61 (0.4) | 600 (4.2) | 1078 (7.6) | 12,533 (87.8) | 661 (4.6) | 1139 (8.0) | 9.2 | 5.4 |
| 18 | 13,611 | 69 (0.5) | 534 (3.9) | 1231 (9.0) | 11,777 (86.5) | 603 (4.4) | 1300 (9.6) | 11.4 | 5.3 |
| 24 | 13,008 | 76 (0.6) | 451 (3.5) | 1360 (10.5) | 11,121 (85.5) | 527 (4.1) | 1436 (11.0) | 14.4 | 5.3 |
| 30 | 12,481 | 89 (0.7) | 445 (3.6) | 1520 (12.2) | 10,427 (83.5) | 534 (4.3) | 1609 (12.9) | 16.7 | 5.5 |
| 36 | 11,947 | 83 (0.7) | 425 (3.6) | 1687 (14.1) | 9752 (81.6) | 508 (4.3) | 1770 (14.8) | 16.3 | 4.7 |
| 42 | 11,439 | 101 (0.9) | 431 (3.8) | 1852 (16.2) | 9055 (79.2) | 532 (4.7) | 1953 (17.1) | 19.0 | 5.2 |
| 48 | 10,907 | 135 (1.2) | 428 (3.9) | 2034 (18.6) | 8310 (76.2) | 563 (5.2) | 2169 (19.9) | 24.0 | 6.2 |
| 54 | 10,344 | 123 (1.2) | 345 (3.3) | 2256 (21.8) | 7620 (73.7) | 468 (4.5) | 2379 (23.0) | 26.3 | 5.2 |
| 60 | 9876 | 147 (1.5) | 316 (3.2) | 2423 (24.5) | 6990 (70.8) | 463 (4.7) | 2570 (26.0) | 31.7 | 5.7 |
| All | 142,849 | 2651 (1.9) | 7936 (5.6) | 17,136 (12.0) | 115,126 (80.6) | 10,587 (7.4) | 19,787 (13.9) | 25.0 | 13.4 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 87 (0.6) | 1020 (6.8) | 893 (6.0) | 12,964 (86.6) | 1107 (7.4) | 980 (6.5) | 7.9 | 8.9 |
| 12 | 13,857 | 133 (1.0) | 877 (6.3) | 975 (7.0) | 11,872 (85.7) | 1010 (7.3) | 1108 (8.0) | 13.2 | 12.0 |
| 18 | 12,847 | 159 (1.2) | 862 (6.7) | 1028 (8.0) | 10,798 (84.1) | 1021 (7.9) | 1187 (9.2) | 15.6 | 13.4 |
| 24 | 11,826 | 182 (1.5) | 784 (6.6) | 1019 (8.6) | 9841 (83.2) | 966 (8.2) | 1201 (10.2) | 18.8 | 15.2 |
| 30 | 10,860 | 227 (2.1) | 737 (6.8) | 994 (9.2) | 8902 (82.0) | 964 (8.9) | 1221 (11.2) | 23.5 | 18.6 |
| 36 | 9896 | 243 (2.5) | 759 (7.7) | 911 (9.2) | 7983 (80.7) | 1002 (10.1) | 1154 (11.7) | 24.3 | 21.1 |
| 42 | 8894 | 215 (2.4) | 731 (8.2) | 843 (9.5) | 7105 (79.9) | 946 (10.6) | 1058 (11.9) | 22.7 | 20.3 |
| 48 | 7948 | 255 (3.2) | 701 (8.8) | 737 (9.3) | 6255 (78.7) | 956 (12.0) | 992 (12.5) | 26.7 | 25.7 |
| 54 | 6992 | 241 (3.4) | 598 (8.6) | 637 (9.1) | 5516 (78.9) | 839 (12.0) | 878 (12.6) | 28.7 | 27.4 |
| 60 | 6153 | 196 (3.2) | 522 (8.5) | 558 (9.1) | 4877 (79.3) | 718 (11.7) | 754 (12.3) | 27.3 | 26.0 |
| All | 124,237 | 3642 (2.9) | 10,923 (8.8) | 9373 (7.5) | 100,299 (80.7) | 14,565 (11.7) | 13,015 (10.5) | 25.0 | 28.0 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 33 (0.2) | 455 (3.0) | 947 (6.3) | 13,529 (90.4) | 488 (3.3) | 980 (6.5) | 6.8 | 3.4 |
| 12 | 14,476 | 41 (0.3) | 429 (3.0) | 1135 (7.8) | 12,871 (88.9) | 470 (3.2) | 1176 (8.1) | 8.7 | 3.5 |
| 18 | 14,006 | 40 (0.3) | 379 (2.7) | 1321 (9.4) | 12,266 (87.6) | 419 (3.0) | 1361 (9.7) | 9.5 | 2.9 |
| 24 | 13,587 | 44 (0.3) | 327 (2.4) | 1508 (11.1) | 11,708 (86.2) | 371 (2.7) | 1552 (11.4) | 11.9 | 2.8 |
| 30 | 13,216 | 45 (0.3) | 311 (2.4) | 1741 (13.2) | 11,119 (84.1) | 356 (2.7) | 1786 (13.5) | 12.6 | 2.5 |
| 36 | 12,860 | 44 (0.3) | 290 (2.3) | 1978 (15.4) | 10,548 (82.0) | 334 (2.6) | 2022 (15.7) | 13.2 | 2.2 |
| 42 | 12,526 | 47 (0.4) | 282 (2.3) | 2254 (18.0) | 9943 (79.4) | 329 (2.6) | 2301 (18.4) | 14.3 | 2.0 |
| 48 | 12,197 | 63 (0.5) | 283 (2.3) | 2565 (21.0) | 9286 (76.1) | 346 (2.8) | 2628 (21.5) | 18.2 | 2.4 |
| 54 | 11,851 | 56 (0.5) | 212 (1.8) | 2923 (24.7) | 8660 (73.1) | 268 (2.3) | 2979 (25.1) | 20.9 | 1.9 |
| 60 | 11,583 | 52 (0.4) | 204 (1.8) | 3267 (28.2) | 8060 (69.6) | 256 (2.2) | 3319 (28.7) | 20.3 | 1.6 |
| All | 151,266 | 2169 (1.4) | 6504 (4.3) | 20,417 (13.5) | 122,176 (80.8) | 8673 (5.7) | 22,586 (14.9) | 25.0 | 9.6 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1704 (8.5) | 3332 (16.7) | 778 (3.9) | 14,186 (70.9) | 5036 (25.2) | 2482 (12.4) | 33.8 | 68.7 |
| 6 | 14,964 | 367 (2.5) | 2070 (13.8) | 613 (4.1) | 11,914 (79.6) | 2437 (16.3) | 980 (6.5) | 15.1 | 37.4 |
| 12 | 12,527 | 156 (1.2) | 1032 (8.2) | 607 (4.8) | 10,732 (85.7) | 1188 (9.5) | 763 (6.1) | 13.1 | 20.4 |
| 18 | 11,339 | 135 (1.2) | 703 (6.2) | 619 (5.5) | 9882 (87.2) | 838 (7.4) | 754 (6.6) | 16.1 | 17.9 |
| 24 | 10,501 | 126 (1.2) | 513 (4.9) | 597 (5.7) | 9265 (88.2) | 639 (6.1) | 723 (6.9) | 19.7 | 17.4 |
| 30 | 9862 | 99 (1.0) | 397 (4.0) | 649 (6.6) | 8717 (88.4) | 496 (5.0) | 748 (7.6) | 20.0 | 13.2 |
| 36 | 9366 | 97 (1.0) | 413 (4.4) | 684 (7.3) | 8172 (87.3) | 510 (5.4) | 781 (8.3) | 19.0 | 12.4 |
| 42 | 8856 | 125 (1.4) | 362 (4.1) | 702 (7.9) | 7667 (86.6) | 487 (5.5) | 827 (9.3) | 25.7 | 15.1 |
| 48 | 8369 | 130 (1.6) | 297 (3.5) | 737 (8.8) | 7205 (86.1) | 427 (5.1) | 867 (10.4) | 30.4 | 15.0 |
| 54 | 7942 | 151 (1.9) | 303 (3.8) | 764 (9.6) | 6724 (84.7) | 454 (5.7) | 915 (11.5) | 33.3 | 16.5 |
| 60 | 7488 | 147 (2.0) | 271 (3.6) | 801 (10.7) | 6269 (83.7) | 418 (5.6) | 948 (12.7) | 35.2 | 15.5 |
| All | 121,214 | 3237 (2.7) | 9693 (8.0) | 7551 (6.2) | 100,733 (83.1) | 12,930 (10.7) | 10,788 (8.9) | 25.0 | 30.0 |
| Change in data simulation | Threshold value | PPV, % | Number of tests per persona | Delay,b % | Develop cirrhosis,c n (%) |
|---|---|---|---|---|---|
| None | 10.460 | 25.0 | 6.21 | 5.63 | 7689 (38.45) |
| Decreasedd measurement error | 10.460 | 29.7 (+4.7) | 7.04 (+0.83) | 7.18 (+1.55) | 7664 (38.32) [–25 (0.13)] |
| 10.205 | 25.0 | 6.22 (+0.01) | 5.16 (–0.47) | ||
| Increasede measurement error | 10.460 | 19.9 (–5.1) | 4.64 (–1.57) | 3.86 (–1.77) | 7808 (39.04) [+119 (0.60)] |
| 10.97 | 25.0 | 6.01 (–0.21) | 6.60 (+0.97) | ||
| Decreased between-individual variability | 10.460 | 28.5 (+3.5) | 7.10 (+0.89) | 2.97 (–2.66) | 7531 (37.66) [–158 (0.79)] |
| 10.35 | 25.0 | 6.66 (+0.45) | 2.23 (–3.40) | ||
| Increasede between-individual variability | 10.460 | 19.7 (–5.3) | 4.78 (–1.43) | 5.63 (+0.00) | 7659 (37.85) [–120 (0.60)] |
| 11.32 | 25.0 | 6.68 (+0.47) | 10.08 (+4.45) | ||
| Decreasedd fibrosis progression rate | 10.460 | 20.7 (–4.3) | 6.85 (+0.64) | 4.72 (–0.91) | 5314 (26.57) [–2375 (11.88)] |
| 10.725 | 25.0 | 7.64 (+1.43) | 6.10 (+0.47) | ||
| Increasede fibrosis progression rate | 10.460 | 36.3 (+11.3) | 5.13 (–1.08) | 9.29 (+3.66) | 13967 (69.84) [+6278 (31.39)] |
| 9.765 | 25.0 | 3.14 (–3.07) | 2.81 (–2.82) |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1705 (8.5) | 2899 (14.5) | 752 (3.8) | 14,644 (73.2) | 4604 (23.0) | 2457 (12.3) | 37.0 | 69.4 |
| 6 | 15,396 | 152 (1.0) | 773 (5.0) | 802 (5.2) | 13,669 (88.8) | 925 (6.0) | 954 (6.2) | 16.4 | 15.9 |
| 12 | 14,471 | 157 (1.1) | 563 (3.9) | 845 (5.8) | 12,906 (89.2) | 720 (5.0) | 1002 (6.9) | 21.8 | 15.7 |
| 18 | 13,751 | 150 (1.1) | 543 (3.9) | 899 (6.5) | 12,159 (88.4) | 693 (5.0) | 1049 (7.6) | 21.6 | 14.3 |
| 24 | 13,058 | 161 (1.2) | 521 (4.0) | 895 (6.9) | 11,481 (87.9) | 682 (5.2) | 1056 (8.1) | 23.6 | 15.2 |
| 30 | 12,376 | 168 (1.4) | 490 (4.0) | 928 (7.5) | 10,790 (87.2) | 658 (5.3) | 1096 (8.9) | 25.5 | 15.3 |
| 36 | 11,718 | 165 (1.4) | 519 (4.4) | 941 (8.0) | 10,093 (86.1) | 684 (5.8) | 1106 (9.4) | 24.1 | 14.9 |
| 42 | 11,034 | 166 (1.5) | 502 (4.5) | 959 (8.7) | 9407 (85.3) | 668 (6.1) | 1125 (10.2) | 24.9 | 14.8 |
| 48 | 10,366 | 218 (2.1) | 490 (4.7) | 958 (9.2) | 8700 (83.9) | 708 (6.8) | 1176 (11.3) | 30.8 | 18.5 |
| 54 | 9658 | 191 (2.0) | 462 (4.8) | 978 (10.1) | 8027 (83.1) | 653 (6.8) | 1169 (12.1) | 29.2 | 16.3 |
| 60 | 9005 | 230 (2.6) | 427 (4.7) | 963 (10.7) | 7385 (82.0) | 657 (7.3) | 1193 (13.2) | 35.0 | 19.3 |
| All | 140,833 | 3463 (2.5) | 8189 (5.8) | 9920 (7.0) | 119,261 (84.7) | 11,652 (8.3) | 13,383 (9.5) | 29.7 | 25.9 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1869 (9.3) | 4113 (20.6) | 588 (2.9) | 13,430 (67.2) | 5982 (29.9) | 2457 (12.3) | 31.2 | 76.1 |
| 6 | 14,018 | 142 (1.0) | 926 (6.6) | 607 (4.3) | 12,343 (88.1) | 1068 (7.6) | 749 (5.3) | 13.3 | 19.0 |
| 12 | 12,950 | 135 (1.0) | 675 (5.2) | 632 (4.9) | 11,508 (88.9) | 810 (6.3) | 767 (5.9) | 16.7 | 17.6 |
| 18 | 12,140 | 133 (1.1) | 661 (5.4) | 657 (5.4) | 10,689 (88.0) | 794 (6.5) | 790 (6.5) | 16.8 | 16.8 |
| 24 | 11,346 | 119 (1.0) | 555 (4.9) | 655 (5.8) | 10,017 (88.3) | 674 (5.9) | 774 (6.8) | 17.7 | 15.4 |
| 30 | 10,672 | 139 (1.3) | 598 (5.6) | 664 (6.2) | 9271 (86.9) | 737 (6.9) | 803 (7.5) | 18.9 | 17.3 |
| 36 | 9935 | 138 (1.4) | 525 (5.3) | 660 (6.6) | 8612 (86.7) | 663 (6.7) | 798 (8.0) | 20.8 | 17.3 |
| 42 | 9272 | 121 (1.3) | 537 (5.8) | 673 (7.3) | 7941 (85.6) | 658 (7.1) | 794 (8.6) | 18.4 | 15.2 |
| 48 | 8614 | 170 (2.0) | 451 (5.2) | 647 (7.5) | 7346 (85.3) | 621 (7.2) | 817 (9.5) | 27.4 | 20.8 |
| 54 | 7993 | 165 (2.1) | 464 (5.8) | 653 (8.2) | 6711 (84.0) | 629 (7.9) | 818 (10.2) | 26.2 | 20.2 |
| 60 | 7364 | 190 (2.6) | 439 (6.0) | 624 (8.5) | 6111 (83.0) | 629 (8.5) | 814 (11.1) | 30.2 | 23.3 |
| All | 124,304 | 3321 (2.7) | 9944 (8.0) | 7060 (5.7) | 103,979 (83.6) | 13,265 (10.7) | 10,381 (8.4) | 25.0 | 32.0 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1790 (8.9) | 4412 (22.1) | 773 (3.9) | 13,025 (65.1) | 6202 (31.0) | 2563 (12.8) | 28.9 | 69.8 |
| 6 | 13,798 | 385 (2.8) | 2254 (16.3) | 608 (4.4) | 10,551 (76.5) | 2639 (19.1) | 993 (7.2) | 14.6 | 38.8 |
| 12 | 11,159 | 243 (2.2) | 1572 (14.1) | 532 (4.8) | 8812 (79.0) | 1815 (16.3) | 775 (6.9) | 13.4 | 31.4 |
| 18 | 9344 | 169 (1.8) | 1255 (13.4) | 484 (5.2) | 7436 (79.6) | 1424 (15.2) | 653 (7.0) | 11.9 | 25.9 |
| 24 | 7920 | 123 (1.6) | 892 (11.3) | 452 (5.7) | 6453 (81.5) | 1015 (12.8) | 575 (7.3) | 12.1 | 21.4 |
| 30 | 6905 | 130 (1.9) | 738 (10.7) | 432 (6.3) | 5605 (81.2) | 868 (12.6) | 562 (8.1) | 15.0 | 23.1 |
| 36 | 6037 | 105 (1.7) | 636 (10.5) | 406 (6.7) | 4890 (81.0) | 741 (12.3) | 511 (8.5) | 14.2 | 20.5 |
| 42 | 5296 | 95 (1.8) | 528 (10.0) | 380 (7.2) | 4293 (81.1) | 623 (11.8) | 475 (9.0) | 15.2 | 20.0 |
| 48 | 4673 | 99 (2.1) | 464 (9.9) | 372 (8.0) | 3738 (80.0) | 563 (12.0) | 471 (10.1) | 17.6 | 21.0 |
| 54 | 4110 | 89 (2.2) | 378 (9.2) | 374 (9.1) | 3269 (79.5) | 467 (11.4) | 463 (11.3) | 19.1 | 19.2 |
| 60 | 3643 | 101 (2.8) | 312 (8.6) | 363 (10.0) | 2867 (78.7) | 413 (11.3) | 464 (12.7) | 24.5 | 21.8 |
| All | 92,885 | 3329 (3.6) | 13,441 (14.5) | 5176 (5.6) | 70,939 (76.4) | 16,770 (18.1) | 8505 (9.2) | 19.9 | 39.1 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1468 (7.3) | 2728 (13.6) | 1095 (5.5) | 14,709 (73.5) | 4196 (21.0) | 2563 (12.8) | 35.0 | 57.3 |
| 6 | 15,804 | 464 (2.9) | 1604 (10.1) | 916 (5.8) | 12,820 (81.1) | 2068 (13.1) | 1380 (8.7) | 22.4 | 33.6 |
| 12 | 13,736 | 278 (2.0) | 1252 (9.1) | 871 (6.3) | 11,335 (82.5) | 1530 (11.1) | 1149 (8.4) | 18.2 | 24.2 |
| 18 | 12,206 | 222 (1.8) | 1015 (8.3) | 826 (6.8) | 10,143 (83.1) | 1237 (10.1) | 1048 (8.6) | 17.9 | 21.2 |
| 24 | 10,969 | 206 (1.9) | 811 (7.4) | 768 (7.0) | 9184 (83.7) | 1017 (9.3) | 974 (8.9) | 20.3 | 21.1 |
| 30 | 9952 | 177 (1.8) | 750 (7.5) | 760 (7.6) | 8265 (83.0) | 927 (9.3) | 937 (9.4) | 19.1 | 18.9 |
| 36 | 9025 | 168 (1.9) | 651 (7.2) | 735 (8.1) | 7471 (82.8) | 819 (9.1) | 903 (10.0) | 20.5 | 18.6 |
| 42 | 8206 | 145 (1.8) | 582 (7.1) | 723 (8.8) | 6756 (82.3) | 727 (8.9) | 868 (10.6) | 19.9 | 16.7 |
| 48 | 7479 | 173 (2.3) | 543 (7.3) | 709 (9.5) | 6054 (80.9) | 716 (9.6) | 882 (11.8) | 24.2 | 19.6 |
| 54 | 6763 | 159 (2.4) | 460 (6.8) | 721 (10.7) | 5423 (80.2) | 619 (9.2) | 880 (13.0) | 25.7 | 18.1 |
| 60 | 6144 | 139 (2.3) | 388 (6.3) | 731 (11.9) | 4886 (79.5) | 527 (8.6) | 870 (14.2) | 26.4 | 16.0 |
| All | 120,284 | 3599 (3.0) | 10,784 (9.0) | 8855 (7.4) | 97,046 (80.7) | 14,383 (12.0) | 12,454 (10.4) | 25.0 | 28.9 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1809 (9.0) | 1745 (8.7) | 546 (2.7) | 15,900 (79.5) | 3554 (17.8) | 2355 (11.8) | 50.9 | 76.8 |
| 6 | 16,446 | 349 (2.1) | 925 (5.6) | 397 (2.4) | 14,775 (89.8) | 1274 (7.7) | 746 (4.5) | 27.4 | 46.8 |
| 12 | 15,172 | 189 (1.2) | 771 (5.1) | 368 (2.4) | 13,844 (91.2) | 960 (6.3) | 557 (3.7) | 19.7 | 33.9 |
| 18 | 14,212 | 143 (1.0) | 731 (5.1) | 367 (2.6) | 12,971 (91.3) | 874 (6.1) | 510 (3.6) | 16.4 | 28.0 |
| 24 | 13,338 | 139 (1.0) | 668 (5.0) | 335 (2.5) | 12,196 (91.4) | 807 (6.1) | 474 (3.6) | 17.2 | 29.3 |
| 30 | 12,531 | 141 (1.1) | 660 (5.3) | 334 (2.7) | 11,396 (90.9) | 801 (6.4) | 475 (3.8) | 17.6 | 29.7 |
| 36 | 11,730 | 140 (1.2) | 695 (5.9) | 334 (2.8) | 10,561 (90.0) | 835 (7.1) | 474 (4.0) | 16.8 | 29.5 |
| 42 | 10,895 | 153 (1.4) | 665 (6.1) | 304 (2.8) | 9773 (89.7) | 818 (7.5) | 457 (4.2) | 18.7 | 33.5 |
| 48 | 10,077 | 132 (1.3) | 698 (6.9) | 332 (3.3) | 8915 (88.5) | 830 (8.2) | 464 (4.6) | 15.9 | 28.4 |
| 54 | 9247 | 153 (1.7) | 649 (7.0) | 326 (3.5) | 8119 (87.8) | 802 (8.7) | 479 (5.2) | 19.1 | 31.9 |
| 60 | 8445 | 143 (1.7) | 560 (6.6) | 341 (4.0) | 7401 (87.6) | 703 (8.3) | 484 (5.7) | 20.3 | 29.5 |
| All | 142,093 | 3491 (2.5) | 8767 (6.2) | 3984 (2.8) | 125,851 (88.6) | 12,258 (8.6) | 7475 (5.3) | 28.5 | 46.7 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1917 (9.6) | 2120 (10.6) | 438 (2.2) | 15,525 (77.6) | 4037 (20.2) | 2355 (11.8) | 47.5 | 81.4 |
| 6 | 15,963 | 297 (1.9) | 1119 (7.0) | 321 (2.0) | 14,226 (89.1) | 1416 (8.9) | 618 (3.9) | 21.0 | 48.1 |
| 12 | 14,547 | 161 (1.1) | 951 (6.5) | 290 (2.0) | 13,145 (90.4) | 1112 (7.6) | 451 (3.1) | 14.5 | 35.7 |
| 18 | 13,435 | 127 (0.9) | 849 (6.3) | 287 (2.1) | 12,172 (90.6) | 976 (7.3) | 414 (3.1) | 13.0 | 30.7 |
| 24 | 12,459 | 121 (1.0) | 757 (6.1) | 247 (2.0) | 11,334 (91.0) | 878 (7.0) | 368 (3.0) | 13.8 | 32.9 |
| 30 | 11,581 | 122 (1.1) | 722 (6.2) | 235 (2.0) | 10,502 (90.7) | 844 (7.3) | 357 (3.1) | 14.5 | 34.2 |
| 36 | 10,737 | 119 (1.1) | 757 (7.1) | 222 (2.1) | 9639 (89.8) | 876 (8.2) | 341 (3.2) | 13.6 | 34.9 |
| 42 | 9861 | 109 (1.1) | 721 (7.3) | 211 (2.1) | 8820 (89.4) | 830 (8.4) | 320 (3.2) | 13.1 | 34.1 |
| 48 | 9031 | 95 (1.1) | 696 (7.7) | 239 (2.6) | 8001 (88.6) | 791 (8.8) | 334 (3.7) | 12.0 | 28.4 |
| 54 | 8240 | 126 (1.5) | 672 (8.2) | 222 (2.7) | 7220 (87.6) | 798 (9.7) | 348 (4.2) | 15.8 | 36.2 |
| 60 | 7442 | 129 (1.7) | 605 (8.1) | 221 (3.0) | 6487 (87.2) | 734 (9.9) | 350 (4.7) | 17.6 | 36.9 |
| All | 133,296 | 3323 (2.5) | 9969 (7.5) | 2933 (2.2) | 117,071 (87.8) | 13,292 (10.0) | 6256 (4.7) | 25.0 | 53.1 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1779 (8.9) | 6916 (34.6) | 679 (3.4) | 10,626 (53.1) | 8695 (43.5) | 2458 (12.3) | 20.5 | 72.4 |
| 6 | 11,305 | 171 (1.5) | 1299 (11.5) | 683 (6.0) | 9152 (81.0) | 1470 (13.0) | 854 (7.6) | 11.6 | 20.0 |
| 12 | 9835 | 109 (1.1) | 808 (8.2) | 741 (7.5) | 8177 (83.1) | 917 (9.3) | 850 (8.6) | 11.9 | 12.8 |
| 18 | 8918 | 119 (1.3) | 631 (7.1) | 746 (8.4) | 7422 (83.2) | 750 (8.4) | 865 (9.7) | 15.9 | 13.8 |
| 24 | 8168 | 99 (1.2) | 544 (6.7) | 757 (9.3) | 6768 (82.9) | 643 (7.9) | 856 (10.5) | 15.4 | 11.6 |
| 30 | 7525 | 111 (1.5) | 446 (5.9) | 765 (10.2) | 6203 (82.4) | 557 (7.4) | 876 (11.6) | 19.9 | 12.7 |
| 36 | 6968 | 124 (1.8) | 390 (5.6) | 752 (10.8) | 5702 (81.8) | 514 (7.4) | 876 (12.6) | 24.1 | 14.2 |
| 42 | 6454 | 129 (2.0) | 412 (6.4) | 747 (11.6) | 5166 (80.0) | 541 (8.4) | 876 (13.6) | 23.8 | 14.7 |
| 48 | 5913 | 143 (2.4) | 346 (5.9) | 718 (12.1) | 4706 (79.6) | 489 (8.3) | 861 (14.6) | 29.2 | 16.6 |
| 54 | 5424 | 122 (2.2) | 305 (5.6) | 727 (13.4) | 4270 (78.7) | 427 (7.9) | 849 (15.7) | 28.6 | 14.4 |
| 60 | 4997 | 130 (2.6) | 286 (5.7) | 717 (14.3) | 3864 (77.3) | 416 (8.3) | 847 (17.0) | 31.2 | 15.3 |
| All | 95,507 | 3036 (3.2) | 12,383 (13.0) | 8032 (8.4) | 72,056 (75.4) | 15,419 (16.1) | 11,068 (11.6) | 19.7 | 27.4 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1406 (7.0) | 3876 (19.4) | 1052 (5.3) | 13,666 (68.3) | 5282 (26.4) | 2458 (12.3) | 26.6 | 57.2 |
| 6 | 14,718 | 190 (1.3) | 1002 (6.8) | 1109 (7.5) | 12,417 (84.4) | 1192 (8.1) | 1299 (8.8) | 15.9 | 14.6 |
| 12 | 13,526 | 141 (1.0) | 667 (4.9) | 1215 (9.0) | 11,503 (85.0) | 808 (6.0) | 1356 (10.0) | 17.5 | 10.4 |
| 18 | 12,718 | 125 (1.0) | 523 (4.1) | 1272 (10.0) | 10,798 (84.9) | 648 (5.1) | 1397 (11.0) | 19.3 | 8.9 |
| 24 | 12,070 | 117 (1.0) | 461 (3.8) | 1349 (11.2) | 10,143 (84.0) | 578 (4.8) | 1466 (12.1) | 20.2 | 8.0 |
| 30 | 11,492 | 132 (1.1) | 423 (3.7) | 1418 (12.3) | 9519 (82.8) | 555 (4.8) | 1550 (13.5) | 23.8 | 8.5 |
| 36 | 10,937 | 159 (1.5) | 441 (4.0) | 1437 (13.1) | 8900 (81.4) | 600 (5.5) | 1596 (14.6) | 26.5 | 10.0 |
| 42 | 10,337 | 130 (1.3) | 392 (3.8) | 1509 (14.6) | 8306 (80.4) | 522 (5.0) | 1639 (15.9) | 24.9 | 7.9 |
| 48 | 9815 | 161 (1.6) | 363 (3.7) | 1587 (16.2) | 7704 (78.5) | 524 (5.3) | 1748 (17.8) | 30.7 | 9.2 |
| 54 | 9291 | 196 (2.1) | 342 (3.7) | 1627 (17.5) | 7126 (76.7) | 538 (5.8) | 1823 (19.6) | 36.4 | 10.8 |
| 60 | 8753 | 183 (2.1) | 317 (3.6) | 1651 (18.9) | 6602 (75.4) | 500 (5.7) | 1834 (21.0) | 36.6 | 10.0 |
| All | 133,657 | 2940 (2.2) | 8807 (6.6) | 15,226 (11.4) | 106,684 (79.8) | 11,747 (8.8) | 18,166 (13.6) | 25.0 | 16.2 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 1290 (6.4) | 3094 (15.5) | 643 (3.2) | 14,973 (74.9) | 4384 (21.9) | 1933 (9.7) | 29.4 | 66.7 |
| 6 | 15,616 | 174 (1.1) | 1204 (7.7) | 606 (3.9) | 13,632 (87.3) | 1378 (8.8) | 780 (5.0) | 12.6 | 22.3 |
| 12 | 14,238 | 128 (0.9) | 833 (5.9) | 601 (4.2) | 12,676 (89.0) | 961 (6.7) | 729 (5.1) | 13.3 | 17.6 |
| 18 | 13,277 | 116 (0.9) | 693 (5.2) | 602 (4.5) | 11,866 (89.4) | 809 (6.1) | 718 (5.4) | 14.3 | 16.2 |
| 24 | 12,468 | 99 (0.8) | 608 (4.9) | 597 (4.8) | 11,164 (89.5) | 707 (5.7) | 696 (5.6) | 14.0 | 14.2 |
| 30 | 11,761 | 108 (0.9) | 532 (4.5) | 594 (5.1) | 10,527 (89.5) | 640 (5.4) | 702 (6.0) | 16.9 | 15.4 |
| 36 | 11,121 | 98 (0.9) | 540 (4.9) | 596 (5.4) | 9887 (88.9) | 638 (5.7) | 694 (6.2) | 15.4 | 14.1 |
| 42 | 10,483 | 110 (1.0) | 463 (4.4) | 593 (5.7) | 9317 (88.9) | 573 (5.5) | 703 (6.7) | 19.2 | 15.6 |
| 48 | 9910 | 106 (1.1) | 460 (4.6) | 572 (5.8) | 8772 (88.5) | 566 (5.7) | 678 (6.8) | 18.7 | 15.6 |
| 54 | 9344 | 90 (1.0) | 415 (4.4) | 583 (6.2) | 8256 (88.4) | 505 (5.4) | 673 (7.2) | 17.8 | 13.4 |
| 60 | 8839 | 90 (1.0) | 401 (4.5) | 587 (6.6) | 7761 (87.8) | 491 (5.6) | 677 (7.7) | 18.3 | 13.3 |
| All | 137,057 | 2409 (1.8) | 9243 (6.7) | 6574 (4.8) | 118,831 (86.7) | 11,652 (8.5) | 8983 (6.6) | 20.7 | 26.8 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 2473 (12.4) | 3320 (16.6) | 1123 (5.6) | 13,084 (65.4) | 5793 (29.0) | 3596 (18.0) | 42.7 | 68.8 |
| 6 | 14,207 | 447 (3.1) | 1394 (9.8) | 1097 (7.7) | 11,269 (79.3) | 1841 (13.0) | 1544 (10.9) | 24.3 | 29.0 |
| 12 | 12,366 | 344 (2.8) | 1097 (8.9) | 1096 (8.9) | 9829 (79.5) | 1441 (11.7) | 1440 (11.6) | 23.9 | 23.9 |
| 18 | 10,925 | 311 (2.8) | 933 (8.5) | 1088 (10.0) | 8593 (78.7) | 1244 (11.4) | 1399 (12.8) | 25.0 | 22.2 |
| 24 | 9681 | 335 (3.5) | 859 (8.9) | 1035 (10.7) | 7452 (77.0) | 1194 (12.3) | 1370 (14.2) | 28.1 | 24.5 |
| 30 | 8487 | 347 (4.1) | 766 (9.0) | 990 (11.7) | 6384 (75.2) | 1113 (13.1) | 1337 (15.8) | 31.2 | 26.0 |
| 36 | 7374 | 350 (4.7) | 746 (10.1) | 1019 (13.8) | 5259 (71.3) | 1096 (14.9) | 1369 (18.6) | 31.9 | 25.6 |
| 42 | 6278 | 386 (6.1) | 579 (9.2) | 1036 (16.5) | 4277 (68.1) | 965 (15.4) | 1422 (22.7) | 40.0 | 27.1 |
| 48 | 5313 | 395 (7.4) | 509 (9.6) | 1016 (19.1) | 3393 (63.9) | 904 (17.0) | 1411 (26.6) | 43.7 | 28.0 |
| 54 | 4409 | 421 (9.5) | 389 (8.8) | 949 (21.5) | 2650 (60.1) | 810 (18.4) | 1370 (31.1) | 52.0 | 30.7 |
| 60 | 3599 | 391 (10.9) | 277 (7.7) | 843 (23.4) | 2088 (58.0) | 668 (18.6) | 1234 (34.3) | 58.5 | 31.7 |
| All | 102,639 | 6200 (6.0) | 10,869 (10.6) | 11,292 (11.0) | 74,278 (72.4) | 17,069 (16.6) | 17,492 (17.0) | 36.3 | 35.4 |
| Observation time (months) | Tests, N | Results, n (%) | Diseased,a n (%) | PPV, % | Sensitivity, % | ||||
|---|---|---|---|---|---|---|---|---|---|
| TP | FP | FN | TN | Positive | |||||
| 0 | 20,000 | 3037 (15.2) | 6915 (34.6) | 559 (2.8) | 9489 (47.4) | 9952 (49.8) | 3596 (18.0) | 30.5 | 84.5 |
| 6 | 10,048 | 328 (3.3) | 2124 (21.1) | 454 (4.5) | 7142 (71.1) | 2452 (24.4) | 782 (7.8) | 13.4 | 41.9 |
| 12 | 7596 | 216 (2.8) | 1330 (17.5) | 403 (5.3) | 5647 (74.3) | 1546 (20.4) | 619 (8.1) | 14.0 | 34.9 |
| 18 | 6050 | 207 (3.4) | 990 (16.4) | 339 (5.6) | 4514 (74.6) | 1197 (19.8) | 546 (9.0) | 17.3 | 37.9 |
| 24 | 4853 | 153 (3.2) | 789 (16.3) | 295 (6.1) | 3616 (74.5) | 942 (19.4) | 448 (9.2) | 16.2 | 34.2 |
| 30 | 3911 | 139 (3.6) | 635 (16.2) | 277 (7.1) | 2860 (73.1) | 774 (19.8) | 416 (10.6) | 18.0 | 33.4 |
| 36 | 3137 | 135 (4.3) | 493 (15.7) | 284 (9.1) | 2225 (70.9) | 628 (20.0) | 419 (13.4) | 21.5 | 32.2 |
| 42 | 2509 | 158 (6.3) | 358 (14.3) | 262 (10.4) | 1731 (69.0) | 516 (20.6) | 420 (16.7) | 30.6 | 37.6 |
| 48 | 1993 | 151 (7.6) | 290 (14.6) | 247 (12.4) | 1305 (65.5) | 441 (22.1) | 398 (20.0) | 34.2 | 37.9 |
| 54 | 1552 | 126 (8.1) | 219 (14.1) | 234 (15.1) | 973 (62.7) | 345 (22.2) | 360 (23.2) | 36.5 | 35.0 |
| 60 | 1207 | 111 (9.2) | 155 (12.8) | 213 (17.6) | 728 (60.3) | 266 (22.0) | 324 (26.8) | 41.7 | 34.3 |
| All | 62,856 | 4761 (7.6) | 14,298 (22.7) | 3567 (5.7) | 40,230 (64.0) | 19,059 (30.3) | 8328 (13.2) | 25.0 | 57.2 |
FIGURE 16.
Results of strategies A–H using the adjusted fibrosis progression estimate data.

Comparison with ELUCIDATE trial data
The ELUCIDATE data contained 705 observations taken from 420 participants randomised to the ELF monitoring arm of the trial. After removing measurements following an ELF value of ≥ 9.5 for each individual (akin to the trial setting), the simulated data set contained 66,320 observations for 20,000 participants and the simulated data set with an adjusted fibrosis progression rate included 59,000 observations for 20,000 participants. Analysis of the ELF value at the point of randomisation for each of the data sets showed similar results: the mean (SD) value for the ELUCIDATE data was 9.57 (1.21), for the simulated data was 9.71 (1.15) and for the simulated data with an adjusted fibrosis progression rate was 9.83 (1.20), with the mean value being slightly lower for the ELUCIDATE data than for the two simulated data sets. The between-individual SD was higher for the ELUCIDATE data than for the simulated data sets (0.93 for the ELUCIDATE data vs. 0.76 for the simulated data and 0.82 for the simulated data with an adjusted fibrosis progression rate). The within-individual SD was similar for the ELUCIDATE data and both simulated data sets (0.53 for the ELUCIDATE data and 0.51 and 0.52 for the simulated and simulated with adjusted fibrosis progression data sets, respectively). The results of the analysis of the randomisation ELF value and ANOVA for ELF measurements at all recorded time points are provided in Table 61.
| Variable | Data | ||
|---|---|---|---|
| ELUCIDATE | Simulated | Simulated with adjusted fibrosis progression | |
| Randomisation point ELF value, mean (SD) | 9.57 (1.21) | 9.71 (1.15) | 9.83 (1.20) |
| ANOVA | |||
| Between-individual SD | 0.93 | 0.76 | 0.82 |
| Within-individual SD | 0.53 | 0.51 | 0.52 |
The ELUCIDATE data modelled consisted of 429 observations from 153 participants, with each participant having a minimum of two and a maximum of six ELF observations and an average number of observations per person of 2.8. The number of observation points used from the simulation model was, therefore, capped to give a similar mean number of observations per person to the value seen in the ELUCIDATE data. Allowing more observations per person would introduce bias as patients with more slowly progressing disease will have more ELF measurements prior to having a test result of ≥ 9.5. This bias relates to comments made by Bellera et al. 241 when analysing monitoring data. The model fitted to simulated data used 26,429 observation points for 9608 simulated participants and the model fitted to simulated data with an adjusted fibrosis progression rate used 23,972 observations for 8779 simulated participants. For the simulated data sets, the mean number of observations was 2.8 for the original data set with an unadjusted fibrosis progression rate and 2.7 for the data set with an adjusted fibrosis progression rate. The results of the modelling of the ELUCIDATE data, simulated data and simulated date with an adjusted fibrosis progression rate are provided in Table 62.
| Variable | Estimate | 95% CI | p-value |
|---|---|---|---|
| ELUCIDATE trial ELF value | |||
| Years | 0.31 | 0.22 to 0.39 | < 0.001 |
| Constant | 8.73 | 8.63 to 8.82 | < 0.001 |
| Between-individual SD | 0.43 | 0.36 to 0.51 | |
| Within-individual SD | 0.48 | 0.44 to 0.52 | |
| Simulated ELF value | |||
| Years | 0.24 | 0.23 to 0.26 | < 0.001 |
| Constant | 8.84 | 8.83 to 8.85 | < 0.001 |
| Between-individual SD | 0.42 | 0.41 to 0.43 | |
| Within-individual SD | 0.47 | 0.46 to 0.47 | |
| Simulated ELF value with adjusted fibrosis progression | |||
| Years | 0.28 | 0.27 to 0.30 | < 0.001 |
| Constant | 8.86 | 8.84 to 8.87 | < 0.001 |
| Between-individual SD | 0.42 | 0.41 to 0.43 | |
| Within-individual SD | 0.46 | 0.46 to 0.47 | |
Modelling of the ELUCIDATE data showed the increase in ELF value per year to be 0.31 (95% CI 0.22 to 0.39; p < 0.001). Modelling of the simulated data and the simulated data with an adjusted fibrosis progression rate showed the increase in ELF value per year to be comparable at 0.24 (95% CI 0.23 to 0.26; p < 0.001) and 0.28 (95% CI 0.27 to 0.30; p < 0.001), respectively.
Discussion
Reference strategy
At the initial testing point a monitoring strategy will be identifying cases from a prevalent population in which a large proportion of patients will have high ELF values. At subsequent time points those with a positive result will not be tested and the tested population will contain cirrhotic patients who either have been falsely negative at the previous testing point or have developed cirrhosis since the last testing point (incident cases), hence the difference in results between the initial monitoring time point and the other time points. The percentage of false-negative results generally increased with each time point as patients with a low ELF trajectory have reached compensated cirrhosis but as they have a low ELF value for their disease stage they are required to progress further to have a positive test result using the simple threshold decision rule. The increasing percentage of false-negative results as the testing points advanced suggests that the simple threshold should be reduced at later time points to account for the patients who have false-negative results using the original threshold.
Comparing strategies with changes to individual components with the reference strategy
Inferior strategies
It was anticipated that the strategy with retesting would result in an increase in the number of tests per person required compared with the reference strategy, but the percentage of patients with a delay to diagnosis also increased. For the patients in the target retest range their test result was determined by combining both the initial and the retest results and their corresponding measurement errors with the mean of the initial and retest result being used to classify a result as positive or negative. Because of the measurement error of both the initial and the retest results some patients would have been positive on their initial test (as with the reference strategy) but, using the mean of the initial test and retest, they have a negative result. The slight increase in time to diagnosis when using a retest strategy will also have a small effect on the percentage of participants with a delay to diagnosis.
The strategies using the absolute and relative increase from the last recorded value decision rules were notably worse than the strategies using the absolute and relative changes from the initial recorded value. When using a decision rule based on detecting a magnitude of change between one value and another, the two values used to calculate the change will both have measurement error. Comparisons with the initial value will consider increases in ELF across the entire monitoring period rather than increases since the previous monitoring point only. Differences from the initial value rather than the last value were better for detecting true change over measurement error (signal from noise).
The simple threshold strategy outperformed the strategies comparing current with previous values. This is in part because of the index of individuality, the ratio between within-individual and between-individual variation. If a test has a high index of individuality value, whereby an individual can have results spanning a wide range of the possible results for a group of people, comparison with constant thresholds will be more meaningful than for tests with low index of individuality values, whereby an individual will have test results spanning only part of the possible range of results and comparison with previous results will be more beneficial. 290
‘Trade-off’ strategies
The reduced test frequency strategy showed a large decrease in the number of tests per person used for a small increase in the percentage of people with a delay to diagnosis. It may be that for a substantial decrease in the number of tests required, and, therefore, the resource used, the slight potential for increased harm to patients (through later diagnosis) is acceptable.
Superior strategies
The linear regression strategy was the only strategy tested that showed a reduction in both the number of tests required and the percentage of patients with a delay to diagnosis. By fitting a regression model using all previous observations for an individual and obtaining a prediction from this, the linear regression method utilised all available data and some allowance was made for the fluctuation in results because of measurement error. The linear regression strategy, however, resulted in only small benefits compared with the reference strategy. This modest improvement in monitoring strategy performance may not merit the extra complexity involved when using the linear regression method.
Estimates of test performance and disease progression (sensitivity analyses)
The results obtained when varying estimates in the simulation model and evaluating the reference strategy highlight the importance of including accurate data. The increases and decreases in estimates of test performance (measurement error and between-individual variability) and fibrosis progression rate affected the three measures of performance in different ways.
Measurement error and between-individual variability
The measurement error of a test affects the number of false-positive test results, with larger measurement error resulting in more false-positive results and smaller measurement error resulting in fewer false-positive results. Between-individual variability will affect the underlying ELF values possible at each fibrosis stage. As long as the ELF value is related to fibrosis stage, if the between-individual variability is smaller it will be easier to correctly identify the fibrosis stage from the ELF value, resulting in fewer false-positive results and more true-positive results.
With fewer false-positive and more true-positive results, the PPV will increase and the number of tests required will increase as the reduction in number of false-positive results means that the number of patients correctly staying in the monitoring programme will increase. With reduced measurement error the observed values reflect more closely the underlying disease state of each patient; if the threshold does not adequately account for this, patients will need to progress for longer to have a test value over the threshold, indicating a positive result. When the between-individual variability is reduced, because of the increase in the number of true-positive results the percentage of patients with a delay to diagnosis will decrease.
Fibrosis progression rate
The fibrosis progression rate will affect the number of diseased patients. With an increased fibrosis progression rate more patients will have compensated cirrhosis, which will lead to an increase in the PPV. With an increased fibrosis progression rate patients have a positive result earlier in the strategy and the strategy will require fewer tests to be performed. If patients have an increased fibrosis progression rate more patients will have been in cirrhosis for > 12 months, meaning that more patients can be undetected for > 12 months.
Limitations
Data sources
The estimates from data sources used to inform the simulation model will have a large impact on the results of the simulation model. The suitability of the data was assessed by consultation with clinical colleagues and, when necessary, estimates were adjusted in sensitivity analyses. However, as the model was dependent on the information used, the quality and suitability of the data used will always be a limitation. Just one cross-sectional study provided information on both the link between ELF values and fibrosis stage and the distribution of fibrosis stages at entry to the trial. When looking to identify an estimate of measurement error several sources were identified, with the estimates from each found to be vastly different. The data linking ELF values to fibrosis stage defined fibrosis stage by biopsy. Even though biopsy is the reference standard for staging fibrosis, biopsy is known not to be accurate in some cases.
The ELUCIDATE trial data used to assess the simulation model were not completely appropriate as the data set contained repeated observations from only 153 participants, with many participants having only two observations; more observations per person would allow the model to better estimate the error terms and the changes over time. ELF measurements being taken only until the point of a measurement being classed as positive also hinders the ability of the data to estimate the true progression of ELF over time, as those with higher ELF values (and possibly more developed cirrhosis) cease to have their values recorded and so progression beyond this cannot be assessed. Patients with lower ELF measurements (< 9.5) continued to be monitored, meaning that they had more measurements taken and, therefore, contributed more data to the model; however, they were potentially very different from those who had fewer measurements taken, who were likely to be in a worse health state.
Assumptions
A major limitation of the simulation model is the vast number of assumptions required. Some of the estimates used to generate the monitoring data, such as fibrosis progression rate and measurement error, can be varied in magnitude and the results assessed to identify the impact of using data of insufficient quality or suitability in the model. However, many assumptions, made out of necessity, in the development of the model could not be varied and assessed so easily and the consequences explored. The model assumed that fibrosis progression is constant and requires patients to have positive fibrosis progression; the minimum fibrosis progression was set at 0.01 fibrosis units per year for the purposes of the simulation (which would indicate a stable disease state). The model assumed linear increases in ELF values between fibrosis stages, normally distributed ELF values within fibrosis stage and a constant fibrosis progression rate. The error associated with each observation was assumed to be normally distributed and a simple error term was used, with no distinction made between within-individual and analytical variation. The error used in the simulation may also be simplistic as the error term was randomly chosen from a distribution that not only is constant across individuals and time but also was not linked to the magnitude of the ELF value. As no alternative data or substantiated opinion were available to enable modelling of these factors in any other way, these assumptions were necessary for the development of the model. Longitudinal data sets with ELF values and biopsy results recorded in addition to data from a biological variability study of ELF would be required to test these assumptions.
Trial considerations
Several criteria were required to allow the simulation model to generate data for a trial (described in Table 17). Although these criteria were included to avoid anomalies and were based on clinical advice, there are no data to support them.
Further work
A greater variety of strategies could be evaluated with multiple components assessed simultaneously. More complex decision rules and frequencies could be explored, for example a simple threshold decision rule in which the threshold remains the same across patients but varies by time point within a monitoring strategy or changing the frequency of testing to be non-constant.
It may be possible for the simulation model to be adapted to account for usual care (and variation in usual care). If usual care could be modelled, it may be possible to compare the use of monitoring strategies (in addition to usual care) with usual care alone and with further simulation work estimate differences in patient outcomes between the approaches.
The model can be used to show lifetime progression for a time-matched cohort of patients with fibrosis (if the data are simulated with all patients starting at the onset of liver fibrosis). These data may be beneficial to the assessment of how a strategy would perform in practice rather than specifically in the trial setting, as they would provide information on how newly diagnosed patients would benefit from monitoring.
Conclusions
Simulation can be used to obtain monitoring data for candidate monitoring strategies and to enable an appropriate strategy to be selected for full-scale evaluation.
In the case of using ELF measurements to monitor liver fibrosis, only the linear regression monitoring strategy showed better performance than the simple threshold strategy and, given the additional complexity and small benefit of using the linear regression strategy, the simple threshold strategy may be most appropriate. Reducing the frequency of testing may be an alternative to the simple threshold strategy if the compromise between number of tests and delay to diagnosis is acceptable.
To generate monitoring data there has to be available evidence on the natural history of the disease and the performance of the monitoring test (measurement error and test accuracy); this evidence can be obtained from existing data sets, by reviewing the literature or from potentially expert opinion. If the data informing the simulation model are inaccurate, the results obtained from the evaluation of strategies will not reflect the truth. Inaccurate estimates will affect the results in a complex way. The results of the sensitivity analyses highlighted the importance, for this test and disease area, of having accurate estimates of test performance and progression.
When comparing data from the ELUCIDATE trial with the simulated data it was clear that, because of the design of the ELUCIDATE trial, the analysis would have to limit the bias of recorded ELF results. Comparison of the ELUCIDATE data and the simulated data provided similar results. Bias in monitoring data, particularly concerning the number of recorded results, should be considered when carrying out the analyses.
Chapter 8 Methodological considerations in the optimisation of monitoring biomarkers to meet value-based market access hurdles
Components of the work described in this chapter have been published in Longo et al. 330
Background
Biomarkers are a central component of the proposed revolution in health care. Sometimes called personal, precision or stratified medicine, the defining characteristic of personalised medicine is the use of molecular (including genetic) and imaging information on individual patients, that is, biomarkers, to guide decisions on their clinical management.
The potential clinical applications of personalised medicine include:
-
screening for the risk of developing disease
-
diagnosing the presence of disease
-
providing prognosis of an individual patient’s disease progression
-
identifying whether or not patients are likely to respond to particular treatments (pharmacogenomics)
-
identifying whether or not patients are at an elevated risk of adverse events from particular treatments (pharmacogenomics/toxicogenomics).
The biomedical knowledge that underlies personalised medicine also has significant potential in the discovery and translation of new therapies; however, consideration of these is outside the scope of this project.
Personalised medicine technologies are being developed at a time when developed health-care systems are under increasing pressure to increase efficiency, that is, to consider the value of new technologies in terms of what they produce in relation to how much they cost, and to review current clinical practices with a view to eliminate those activities that are of low or no value. The former process is implemented through health technology assessment processes, such as those undertaken by NICE in the UK, the Haute Autorité de Santé (HAS) in France, the Pharmaceutical Benefits Advisory Committee in Australia and the Canadian Agency for Drugs and Technologies in Health in Canada. In line with other new health technologies, personalised medicine innovations tend to come at a substantial financial cost. Commercially available tests such as Oncotype Dx® (Genomic Health, Inc., Redwood City, CA, USA) are magnitudes more expensive than conventional laboratory tests and co-dependent therapies, such as crizotinib (Xalkori, Pfizer Inc., New York, NY, USA), are even more expensive. These price tags mean that they are inevitably subject to formal health technology assessment prior to market access.
The review of current clinical practices with a view to eliminate those activities that are of low or no value is being driven internationally by the Choosing Wisely campaign. 331 The elimination of unnecessary tests is at the forefront of the campaign. As a result, there is an inevitable tension between the widespread adoption of additional tests that are required for personalised medicine and the societal and professional pressure to make less use of tests. In this context, biomarker tests will be adopted only if they can be demonstrated to be a high-value use of limited health-care resources. This relatively new pressure to assess the value of new tests before they are adopted aligns testing with the processes of health technology assessment that have been used for drugs and some devices for many years. The focus of this theme of the programme grant has been the development of methods for assessing the value of personalised medicine technologies, with a particular focus on monitoring tests, that is, tests that are applied repeatedly to the same patient over a period of time to inform sequential clinical care decisions.
The remainder of this chapter is structured as follows. In Cost-effectiveness in personalised medicine technologies we consider the difference between a conventional clinical utility for the individual patient approach to test optimisation and an approach based on the cost-effectiveness of the test from a population health perspective. A framework for characterising personalised medicine technologies describes a framework for characterising personalised medicine technologies. Extending the method of Phelps and Mushlin for monitoring tests describes an existing method for optimising diagnostic tests to meet cost-effectiveness targets and its extension for monitoring tests. Some observations on the estimation of the value of information for monitoring tests describes the formal extension of this framework for a monitoring test with ‘n’ administrations and provides an illustrative example in which there are six sequential administrations of the test. The conclusions section then sets out some conceptual issues related to the calculation of the value of information of additional research for a monitoring test.
Cost-effectiveness in personalised medicine technologies
As described in the previous section, biomarker tests are at the centre of the personalised medicine revolution. Although some tests are dichotomous, that is, they test the presence or absence of a particular biomedical characteristic, many tests measure a continuous biomedical parameter, for example blood glucose or forced expiratory volume. The interpretation of the test result converts the continuous variable into a categorical variable by defining a certain test result as the transition point between normal and abnormal. This transition point is referred to as the test cut-off point. Conventionally, the choice of cut-off point is selected on the basis of clinical utility. Clinical utility considers the risk–benefit ratio of a test from the perspective of the individual patient. Conditional on the treatment associated with a positive test result having no or only a low risk of an adverse event, maximising clinical utility leads to preferring a highly sensitive diagnostic test cut-off point over a highly specific diagnostic test cut-off point.
Figure 17 shows how shifting the cut-off point for a diagnostic test changes the proportions of patients who receive false-positive and false-negative test results. The upper half of the figures show the test score distribution for individuals who actually have the condition of interest. The lower half of the diagram shows the test score distribution for individuals who do not have the condition of interest. Between points A and B, for any given test result it is possible that an individual may have or be free of the condition of interest. The initial cut-off point is shown by the solid blue vertical line and the dark green portions show the proportions of the test results that are incorrect. Individuals who have the condition but who receive a test score below the cut-off point are defined as false negatives and individuals free of the condition who receive a test score above the cut-off point are defined as false positives.
FIGURE 17.
Relationship between cut-off point and test performance.
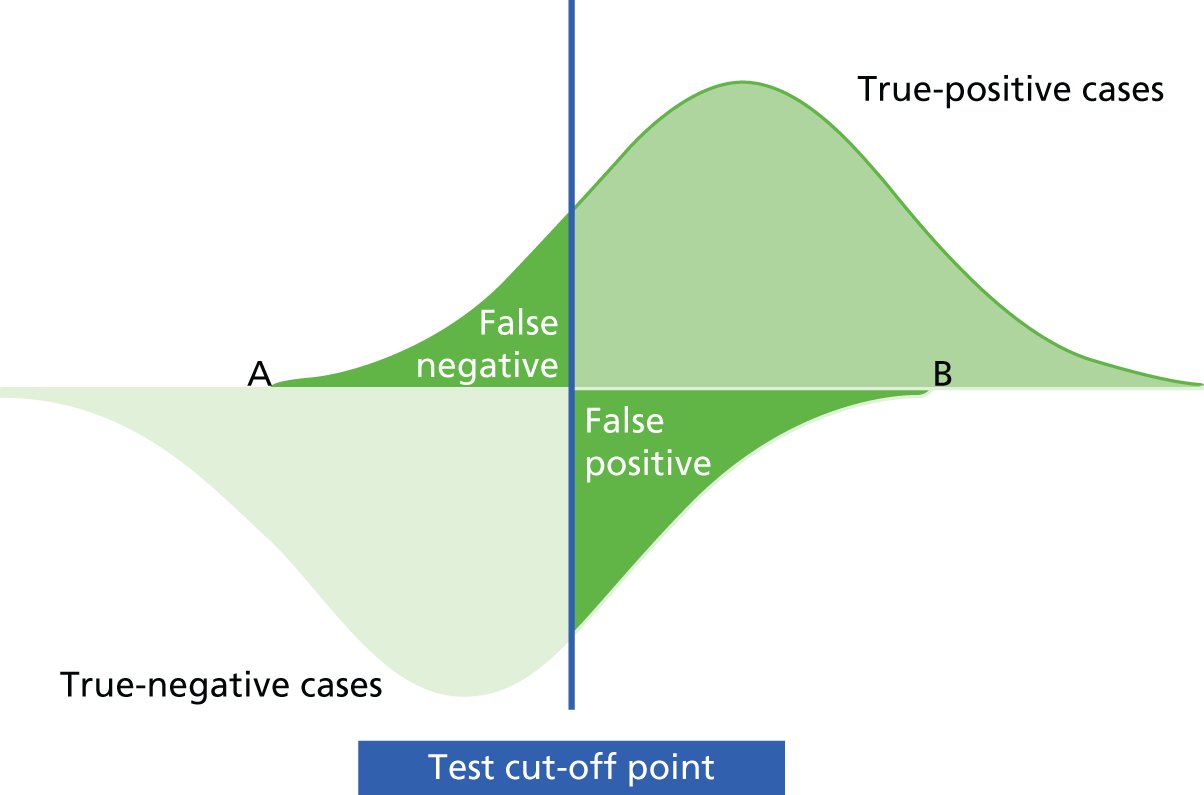
Figure 18 shows how the proportions of individuals who receive false-positive and false-negative test results change as the cut-off point is moved. As the test cut-off point moves to the left (lower), the proportion of false-negative results reduces but the proportion of false-positive results increases. By contrast, if the cut-off point moves to the right (higher), the proportion of individuals who receive a false-negative result increases whereas the proportion of individuals who receive a false-positive result decreases.
FIGURE 18.
Relationship between cut-off point and test performance. (a) Test cut-off point moves to the left (lower); and (b) test cut-off point moves to the right (higher).
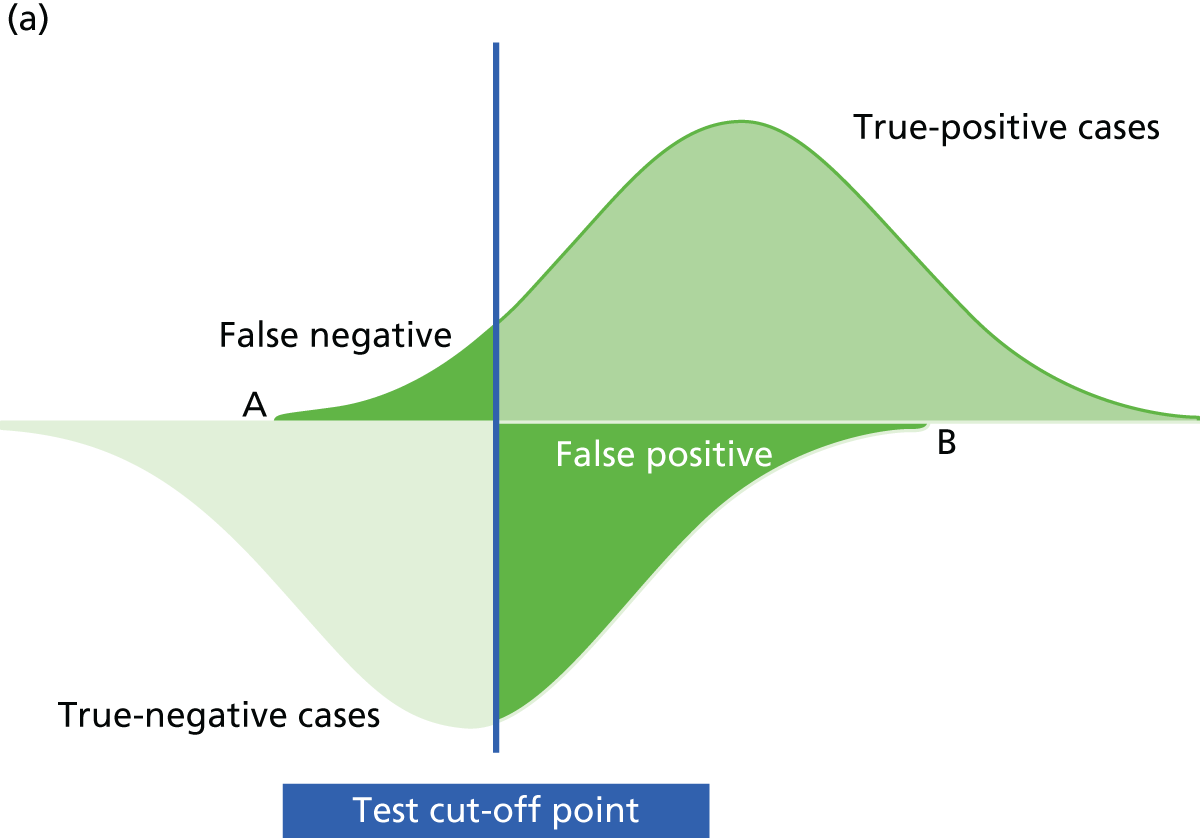

As described earlier, because clinical utility is evaluated from the individual patient perspective, the value attached to a false-positive results tends to be considerably less than the value attached to a false-negative result. A false-negative result deprives the individual of the opportunity to receive appropriate treatment in a timely manner. By contrast, a false-positive result exposes the individual to a treatment that they will not benefit from but that is not expected to do them harm. However, when consideration of the value of the test is expanded to include the resources consumed by the test and any treatments administered subsequent to the test result, as is the case for the health technology appraisal of tests, the perspective for establishing value moves from the individual patient to that of the population that the health-care system is responsible for.
From a population health perspective, in a health system that operates with a budget that is fixed, or quasi-fixed in real terms, the cost of a technology is the health forgone by others. We use the ‘bookshelf’ model developed by Culyer, McCabe and Edlin332–334 to illustrate how the cost-effectiveness threshold represents the health forgone as a result of the premium cost of new therapies.
The cost-effectiveness ‘bookshelf’ is a graphical representation of the health system in which each available health technology is represented as a unique ‘book’ on the bookshelf. A broad definition of ‘technology’ is adopted for this purpose, which includes any health-care intervention or service that consumes resources and provides value to the health system.
The width of each book represents each technology’s budget impact if funded (i.e. the incremental cost of providing the technology to all patients in the relevant indication), whereas the height of each book represents each technology’s incremental cost-effectiveness ratio (ICER). For the purpose of this study, the preferred unit of ‘effectiveness’ for a health technology was assumed to be the quality-adjusted life-year (QALY), such that the height of each book represents the incremental cost per additional QALY provided by the technology. The books are stacked next to each other along the bookshelf (the x-axis of Figure 19) and sorted so that the most desirable technologies (represented by the shortest books) are at the far left of the bookshelf and the least desirable technologies (the tallest books) are at the far right. With a fixed health budget not all technologies can be funded. In choosing which technologies to fund, the decision-maker maximises the value produced by the health system by funding the technology at the far left of the bookshelf first (technology A in in Figure 19). The decision-maker carries on funding each technology to its right in turn (B, C, D, etc.) until the budget is spent. The least desirable technology to be funded (G) is referred to as the ‘marginal’ technology.
FIGURE 19.
The cost-effectiveness bookshelf. Republished with permission of SAGE Publications, Inc., from Determinants of change in the cost-effectiveness threshold, Paulden et al. 333 vol. 37, iss. 2, 2018; permission conveyed through Copyright Clearance Center, Inc.
As the health system has a fixed budget, funding any new technology will inevitably displace one or more existing, funded technologies. For the purposes of this explanation, we assume that the health system’s objective is to improve population health with the available budget. Hence, a new technology will be funded only if it provides more QALYs than are forgone through the displacement of currently funded technologies. For a new technology to do this it must produce more QALYs per pound spent than the least valuable technology that is currently paid for. In our bookcase example, a new technology must have a cost per QALY (ICER) that is equal to or better than that of technology G, the marginal technology, if it is to produce more QALYs than it displaces. Hence, the health system budget provides an implicit value of health – the ICER of the marginal technology. This is the value that technologies, including personalised medicine, must target if they are to be attractive to decision-makers.
Now consider the relationship between the test cut-off point and cost-effectiveness. In Figure 17 we showed the four categories of test results: true positives and negatives and false positives and negatives. Table 63 provides illustrative costs and QALYs associated with each of the categories. The rows CP1 and CP2 provide the distribution of tested individuals between the four possible outcomes for the two different cut-off points, CP1 and CP2. In the sixth column we report the expected costs and QALYs for the test using CP1 and CP2. We can see that the expected cost of the test is £5000 using CP2 and £4400 using CP1. However, the expected QALYs from the test using CP2 are also higher – 0.74 compared with 0.64. From a population health perspective, is it worth moving from CP1 to CP2? We know that, for this to be the better-value choice, the incremental cost per QALY produced by this substitution must be lower than the cost per QALY of the marginal technology. In our example, the cost per QALY of the marginal technology is £50,000 (top right-hand cell in Table 63) and the incremental cost per QALY of CP2 compared with CP1 is calculated as follows:
| Variable | True positive | True negative | False positive | False negative | Expected value | Net monetary benefit (£) | Value of health (£) |
|---|---|---|---|---|---|---|---|
| QALYs | 0.9 | 1 | 0.7 | –0.5 | 50,000 | ||
| Costs (£) | 5000 | 500 | 8000 | 8000 | |||
| Distribution CP1 | 0.45 | 0.3 | 0.05 | 0.2 | 27,600 | ||
| Expected QALYs | 0.405 | 0.3 | 0.035 | –0.1 | 0.64 | ||
| Expected costs (£) | 2250 | 150 | 400 | 1600 | 4400 | ||
| Distribution CP2 | 0.5 | 0.2 | 0.2 | 0.1 | 32,000 | ||
| Expected QALYs | 0.45 | 0.2 | 0.14 | –0.05 | 0.74 | ||
| Expected costs (£) | 2500 | 100 | 1600 | 800 | 5000 |
As £6000 is considerably lower than £50,000, we would expect the implementation of the test using CP2 rather than CP1 to create more health than is displaced by the additional cost.
When using the cost-effectiveness decision criterion, the increase in population health is maximised by including new technologies up to the point at which the ICER for the new technology is exactly equal to the ICER of the marginal technology. Hence, in the cost-effectiveness framework it is possible to go further than merely identifying whether or not a specific test cut-off point is good value. The framework can be used to identify the most cost-effective cut-off point for a test. We describe how this is done in more detail in Extending the method of Phelps and Mushlin for monitoring tests.
A framework for characterising personalised medicine technologies
The umbrella of personalised medicine covers a wide range of technologies and combinations of technologies. Identifying the appropriate methods for the economic evaluation of a specific personalised medicine technology will be significantly helped by a systematic approach to characterising the components of the technology. In this section we describe a model for characterising personalised medicine technologies in terms of their constituent technologies, for the purposes of economic evaluation.
As described in the introduction, the foundation characteristic of personalised medicine is a test of the molecular, including genetic, characteristics of an individual or, or in the case of cancers, a disease. For screening and diagnostic tests, this may be the sole component of the technology. However, for prognostic tests and for test–treatment combinations, the technology will require combination of the molecular information and clinical expression (phenotypic) data. Prognostic technologies inherently link the molecular information to phenotypic information and, thus, are a combination of two testing technologies. The magnitude of an individual patient’s health benefit from treatment is dependent on the phenotypic expression of the disease. The relationship between the molecular characterisation of disease and phenotypic expression is uncertain in even the monogenetic disorders such as Gaucher’s disease, in which homozygous twins have been shown to have radically different times to symptomic presentation. In addition to the molecular and phenotypic test components, some personalised medicine technologies may have an additional pharmacogenomic test technology, to establish whether or not an individual will respond to the specific therapy, with HER2 (human epidermal growth factor receptor 2) testing for Herceptin® (trastuzumab; Roche Products Ltd, Welwyn Garden City, UK) therapy in breast cancer possibly being the most well-known example. The final potential component of a personalised medicine technology is the treatment itself. Some technologies, such as Kalydeco® [ivacaftor; Vertex Pharmaceuticals (UK) Ltd, London, UK], the gene-specific treatment for cystic fibrosis, are themselves personalised medicine technologies, whereas others are more conventional treatments. However, their economic evaluation will require consideration of at least the phenotypic expression test as well as possibly the molecular data.
Figure 20 provides a graphical representation of this framework.
FIGURE 20.
Characterising personalised medicine technologies. +ve, positive; –ve, negative; Dx, diagnosis.

The framework allows us to highlight the potential for correlations between the different components of a specific personalised medicine technology. For example, the effectiveness of treatment is likely to be systematically related to phenotypic expression. This relationship may be positive or negative depending on the nature of the treatment. For example, a treatment that stops further progression but does not resolve accumulated disability will have a less valuable effect the greater the phenotypic expression; in contrast, a treatment that resolves accumulated disability will have a more valuable effect in patients with greater disability.
Changes in the phenotypic expression of the population targeted for treatment will change the case mix of the patients to whom a pharmacogenomic test is administered, with potential implications for the test performance characteristics of the pharmacogenomic test and, by extension, the expected effectiveness of the therapy in ‘responders’ identified by that test. For cost-effectiveness analyses, understanding relationships that impact on the expected magnitude of benefit are clearly central.
In the next section we describe how to identify the optimum cut-off point for a diagnostic test and consider how this framework can be extended for a monitoring test.
Extending the method of Phelps and Mushlin for monitoring tests
Phelps and Mushlin335 use cost-effectiveness analysis to assess diagnostic technologies. In their work, they describe how to identify the optimal test cut-off point for a diagnostic test that meets a prespecified cost-effectiveness threshold. They consider a population in which each person has some probability of illness (f) and model the simple case in which a physician uses a dichotomous test to identify patients as being either sick or healthy depending on the test diagnosis. It is, however, recognised that the population in whom these diagnostic technologies are applied is heterogeneous and that the probability of each individual being sick may vary.
The benefits and harms often associated with the use of diagnostic technologies depend on the true state of health of the patients in whom these technologies are applied. However, it is unlikely for an individual’s true health state to be observed and, thus, the actual patient benefits and costs will depend on how well the diagnostic technologies identify these states of health. Ideally, tests will be optimised to identify individuals but, as resources in every health system are finite, the goal is that the use of diagnostic devices will optimise population health and this requires adding up the benefits and costs over the population eligible to use the test.
In developing the theoretical framework for assessing diagnostic devises, Phelps and Mushlin335 use the following notation:
-
f = probability that patient is sick
-
p = sensitivity
-
1 – q = specificity
-
UST(CST) = utility (cost) of sick person, treated
-
USN(CSN) = utility (cost) of sick person, not treated
-
UHT(CHT) = utility (cost) of healthy person, treated
-
UHN(CHN) = utility (cost) of healthy person, not treated.
Using the above information and a predetermined cost-effectiveness threshold (g), they determine the net benefit for a diagnostic technology and maximise this net benefit in an optimisation setting with respect to the test performance characteristics, specifically p and q. Phelps and Mushlin335 define an expression for the net benefit as:
The combination of p and q that maximises the net benefit can be obtained by varying the diagnostic test cut-off point used to establish the diagnosis (we can find combinations of p and q in which the net benefit remains the same by taking the differential of Equation 23 and allowing p and q to vary jointly, but holding the total change in net benefit to zero). For a given cost-effectiveness threshold, the optimal choice of p and q that maximises the expected net benefit is given by:
The slope βd corresponds to a cut-off point kd on the ROC curve for our diagnostic test technology of interest. Using the costs and outcomes of treatments of false positives and false negatives obtained from the literature, in addition to the ROC curve for our diagnostic test, a cost-effective test cut-off point can be identified for a given threshold.
As part of this NIHR programme grant we have extended the work of Phelps and Mushlin335 The framework developed by Phelps and Mushlin335 for a diagnostic test has been used to evaluate the same test, CA-125, as a monitoring test. Specifically, Phelps and Mushlin335 consider a two-period monitoring test in which clinicians readminister the same test to patients who are diagnosed as either positive or negative by the first test (for the reason that these patients display a clear predisposition for the condition of interest). Using the CA-125 test for monitoring for relapse in ovarian cancer, we show how the repeated use of the initial cut-off point can lead to a substantially increased false-negative rate compared with the monitoring cut-off point – over 4% higher in this example – with the associated harms for individual and population health.
In this monitoring scenario presented by Longo et al. ,330 each of the different subgroups of patients that are retested have a systematically different prevalence (probability of being sick) in the period in which they are tested compared with the initial population. 330 The prevalence in the subgroups in the second period depends on both the ability of the previous test to correctly classify patients as sick or healthy and the probability of developing the disease in the time between the two tests. We argue that, if the prevalences within the subgroups of the population that are being tested in the current period are unique and different, then, likewise, the optimal combination of test performance characteristics that maximise the net benefit among these subgroups will also be unique and differ from p and q of the initial test. As a result, a unique cost-effective test cut-off point can be identified for each subpopulation on a ROC curve that corresponds to their test performance characteristics. We illustrate this by first updating the prevalence in Equation 23 to obtain the unique slopes in each subpopulation and then identifying the unique cut-off points for each subpopulation, as shown below:
It is possible to build on this work to develop a framework that identifies the prevalence and the corresponding optimal cost-effective test cut-off points for subgroups in the monitoring period for a set of ‘n’ administrations of the test. For simplicity, and ease of computational burden, we use a discrete set of cut-off points between prespecified test score limits of 0 and 1. We obtain the optimal pathway for our n-period monitoring using a backward induction approach in a dynamic programming setting.
We consider a monitoring regime in which individuals with or at risk of a disease of interest, presumably a chronic disease, are followed for i periods. Those who are diagnosed as positive receive treatment whereas those diagnosed as negative do not receive treatment and we continue to monitor both the positive and negative subpopulations to the ith period. In each period, a physician decides on a test cut-off point [Tci,j, where i refers to the monitoring period and j refers to a specific subpopulation within a period (thus, in period n = 1, j = (1); in period i = 2, j = (1,2); in period i = 3, j = (1,2,3,4)] that will be used to test each subpopulation. Assuming that Tci,j is measured on a continuous scale and lies anywhere between 0 and 1, then individuals with test scores above Tci,j are diagnosed as positive and vice versa.
We considered a three-period monitoring regime. In this monitoring regime, a physician decides on Tc1,1 in period 1 and the initial population is tested with this cut-off point. Individuals whose test score falls above Tc1,1 are considered as our positive subpopulation and those with test scores below this are considered as the negative subpopulation. In period i = 2, the physician decides on two test cut off points (Tc2,1 and Tc2,2) that will be used to test the subgroups. The positive subgroup in period i = 2 is tested with a cut-off point Tc2,1 whereas the negative subgroup is tested with a cut-off point Tc2,2. Based on the test diagnosis of each individual in each of the two subpopulations, they are further stratified into four subgroups in period i = 3, as shown in Figure 21, and the physician has to decide on the test cut-off point for each of these four subgroups. Thus, for every ith period, a physician is faced with 2i – 1 subpopulations and decides the test cut-off point that must be applied to each. In Figure 21, every positive subpopulation includes all of the true positives and false positives whereas every negative subpopulation includes all of the true negatives and false negatives. The utilities and costs associated with individuals in each of these test outcomes is different [the utilities (costs) associated with true positives, false positives, true negatives and false negatives are UTP(CTP), UFP(CFP), UTN(CTN) and UFN(CFN), respectively].
FIGURE 21.
Decision tree for a three-period monitoring test.

The prevalence of a condition varies under sequential testing and this compels a difference in test cut-off points. 330,336 In the period i = 1, the prevalence in the initial population can in most cases be obtained from clinical experts, the registry or epidemiological literature. 337 Longo et al. 330 show that patients presenting for monitoring tests have a different prevalence compared with the general population presenting for the initial test. They estimate the prevalence for the positive and negative subgroups in period i = 2 to be r1 + (1 – r1) × ρ and (1 – s1) + s1 × ρ, respectively, where r1 and s1 represent the PPV and negative predictive value (NPV), respectively, and ρ is the probability that a patient is sick when presenting for a test in period i = 2, given that they were not sick when presenting for a test in period i = 1. Thus, the prevalence of a condition in the current period is a function of the test performance characteristics in the previous period and the rate of disease progression.
Developing on the framework we described in Longo et al. ,330 consider the case in which individuals are monitored for more than two periods. We define the following notations:
-
i = the monitoring period
-
j = set of subpopulations in period i
-
fi,j = prevalence for the jth subpopulation in the ith period.
Thus, from Figure 21, we can see that at period i = 2, j = (1, 2) and fi,j = (f2,1, f2,2) and at period i = 3, j = (1, 2, 3, 4) and fi,j = (f3,1, f3,2, f3,3, f3,4).
We also define k such that:
Given the above notation, the prevalence for a subpopulation j in period i can be generalised as:
If i = 1 then we have the case of a diagnostic test. For i ≥ 2, we have a monitoring scenario. It must also be noted that f1,1 is exogenous and can be obtained from the epidemiological literature.
For a two-period monitoring test, Longo et al. 330 show how to calculate the disease prevalence for each decision node of the second period using the PPV, the NPV and the disease progression rate from the first period. In our model we generalise on the results published by Longo et al. 330 and derive the formulas for calculating the respective prevalence at each decision node when the monitoring test is administered repeatedly in n periods. Then, we maximise the expected net benefits resulting from the repeated administration of the test by finding the optimal test cut-off scores in each period.
Our illustrative model uses the dynamic programming method to calculate the optimal test cut-off scores for a monitoring test with six periods. In this model the sensitivity and specificity are calculated at each decision node, which can be achieved by estimating the distributions of the test scores for the sick and healthy patients at the respective nodes. Next, we show an optimisation example for a monitoring test administered in six periods (n = 6). Let the model parameters be denoted as follows:
-
f = probability that patient is sick
-
UST(CST) = utility (cost) of sick person, treated
-
USN(CSN) = utility (cost) of sick person, not treated
-
UHT(CHT) = utility (cost) of healthy person, treated
-
UHN(CHN) = utility (cost) of healthy person, not treated
-
g = inverse of the cost-effectiveness threshold
-
ρ = rate of disease progression.
The parameters take the values reported in Table 64.
| Parameter | True positive (ST) | False negative (SN) | False positive (HT) | True negative (HN) |
|---|---|---|---|---|
| QALYs | 0.9 | –2 | 0.5 | 1 |
| Costs (US$) | 500 | 800 | 400 | 0 |
| g | 1/50,000 = 0.00002 | f 1 | 0.01 | |
| ρ | 0.01 |
Furthermore, let TS , 0 ≤ TS ≤ 1, denote the test scores of sick patients and TH , 0 ≤ TH ≤ 1, denote the test scores of healthy patients. We use beta distributions for TS and TH because the beta distribution is restricted to values in [0, 1] and can be used to model distributions that are symmetric or skewed. Let TS ≈β(2,5) and TH ≈ β(5,2) for all decision nodes in all periods. However, the model is developed such that the distributions for TS and TH can be defined distinctly for each decision node.
To solve the optimisation problem and find the optimal cut-off points for each decision node in each period, we have discretised TS and TH with the granularity 0.1. Thus, TS and TH ε {0,0.1,. . .,1]. Using the backward induction method of dynamic programming results in the optimal cut-off points depicted in Table 65.
| Period | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |||||
| Decision for first period | First period outcomes | Decisions for second period | Second period outcomes | Decisions for third period | Third period outcomes | Decisions for fourth period | Fourth period outcomes | Decisions for fifth period | Fifth period outcomes | Decisions for sixth period |
| 0.3 | + | 0.5 | + | 0.6 | + | 0.6 | + | 0.7 | + | 0.7 |
| – | 0.6 | |||||||||
| – | 0.6 | + | 0.6 | |||||||
| – | 0.5 | |||||||||
| – | 0.5 | + | 0.6 | + | 0.6 | |||||
| – | 0.5 | |||||||||
| – | 0.5 | + | 0.5 | |||||||
| – | 0.5 | |||||||||
| – | 0.4 | + | 0.5 | + | 0.6 | + | 0.6 | |||
| – | 0.5 | |||||||||
| – | 0.5 | + | 0.6 | |||||||
| – | 0.5 | |||||||||
| – | 0.4 | + | 0.5 | + | 0.6 | |||||
| – | 0.5 | |||||||||
| – | 0.4 | + | 0.5 | |||||||
| – | 0.4 | |||||||||
| – | 0.3 | + | 0.5 | + | 0.6 | + | 0.6 | + | 0.7 | |
| – | 0.6 | |||||||||
| – | 0.5 | + | 0.6 | |||||||
| – | 0.5 | |||||||||
| – | 0.5 | + | 0.5 | + | 0.6 | |||||
| – | 0.5 | |||||||||
| – | 0.4 | + | 0.5 | |||||||
| – | 0.4 | |||||||||
| – | 0.3 | + | 0.5 | + | 0.6 | + | 0.6 | |||
| – | 0.5 | |||||||||
| – | 0.5 | + | 0.5 | |||||||
| – | 0.4 | |||||||||
| – | 0 | + | 1 | + | 0.6 | |||||
| – | 0.5 | |||||||||
| – | 0.3 | + | 0.5 | |||||||
| – | 0.3 | |||||||||
Some observations on the estimation of the value of information for monitoring tests
The methods that we have described in the previous section develop Phelps and Mushlin’s335 work to take account of how the case mix of patients changes after each test administration. However, we assumed that the ROC curve is the same for each test administration. However, it is known that changing the case mix of the population to which a test is applied will change the sensitivity and specificity and, thus, the ROC curve for the test at second administration will be significantly different from the ROC curve for the test at the previous administration and for the individuals who tested positive compared with those who tested negative in the previous administration. Figures 22 and 23 illustrate how the data driving the sensitivity and the specificity for the second administration of a test would differ from the data driving the sensitivity and specificity for the first administration (strictly these figures are correct under the assumption that there is no disease progression).
FIGURE 22.
Data for ROC curve at second administration for test-positive individuals. Reprinted from Clinica Chimica Acta, vol. 427, Horvath AR, Lord SJ, StJohn A, Sandberg S, Cobbaert CM, Lorenz S, et al. From biomarkers to medical tests: the changing landscape of test evaluation, pp. 49–57, 2014,338 with permission from Elsevier.
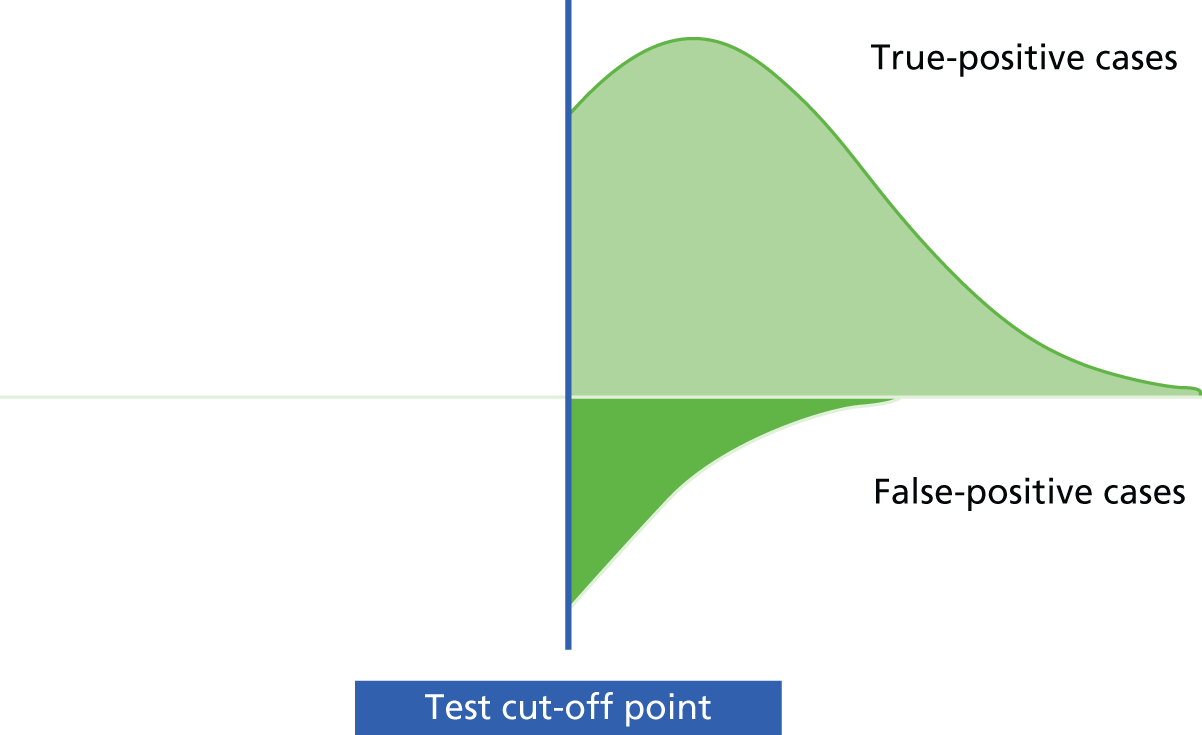
FIGURE 23.
Data for ROC curve at second administration for test-negative individuals.

Perfect information for a diagnostic test can be characterised as being able to illustrate the distribution of test scores for each true health state that the test is designed to measure. With this information for each test score it would be possible to define the probability that an individual who received a particular score was a true/false positive or true/false negative. Perfect information for a monitoring test requires the same information plus knowing with certainty how many individuals will have progressed from a true-negative state to a true-positive state in the time interval between the tests. This would allow the calculation of sensitivity and specificity, assuming that the test score distribution for the new true-positive individuals is the same as for the true-positive individuals at the time of the first administration of the test and, hence, allowing the construction of separate ROC curves for the two groups defined by the initial test.
It follows that there are three distinct sources of uncertainty in a monitoring test:
-
test score distribution for true-positive individuals
-
test score distribution for true-negative individuals
-
progression rate from true negative to true positive.
The choice of the value of health used in the analysis plays a more complex role in the estimation of the value of information for a monitoring technology than for an effectiveness parameter. This is because it is the choice of the value of health that determines the cut-off point for the initial and subsequent administrations of the test. In determining the initial cut-off point it determines the ROC curves for the subsequent test administrations. This points to a much more complex relationship between the value of health and the value of information than is observed for therapies.
Uncertainty about the true health state is unlikely to be evenly distributed across the range of test scores. Extreme test scores (either high or low) are systematically less likely to be false positives/false negatives than test scores in the mid-range. The implication of this is that the location of the initial cut-off point will have a direct effect on the contribution of the test score distributions for true positives and true negatives to the total value of information about the test performance. Furthermore, the choice of initial cut-off point will have an impact on the degree to which disease progression can lead to a change of status after the monitoring test and, hence, the value of additional research around this parameter.
Because of the pivotal role of the choice of the value of health in determining the initial cut-off point and, hence, the value of further research, value of information analyses will be health system specific to an even greater degree than is the case for research on the effectiveness and safety of therapeutic interventions. For monitoring test technologies that are developed for multiple markets, it may be more efficient to undertake substantial global pre-market studies that characterise all three parameters (test score distribution for true-positive and true-negative cases and the rate of progression) in some detail as this may be more efficient than multiple studies that target portions of the evidence base for each parameter.
Conclusions
The work described in this chapter and published in Longo et al. 330 provides a new insight into the use of biomarkers for monitoring tests when the principal evidence of benefit is the cost-effectiveness of the health-care services that will be managed in the context of the biomarker test results. In future work, rigorous study designs and careful attention to the choice of cut-off will be essential.
-
By applying Phelps and Mushlin’s335 framework to monitoring tests, we have shown that the test cut-off point that is cost-effective in monitoring may be quite different from the one that is cost-effective in the first administration of the test.
-
We show that the extent of this difference, and, hence, its importance to clinicians, patients and decision-makers, depends on the underlying prevalence of the condition, the test performance characteristics of the test at its first administration, the rate of disease progression and the interval between test administrations, as well as the target cost-effectiveness threshold.
-
Decision-makers are likely to be interested in the impact of changing the cost-effectiveness threshold on the optimal case definition cut-off value; hence, we present the cost-effectiveness threshold curve, which is able to identify the optimal case definition cut-off value corresponding to each cost-effectiveness threshold.
-
The framework developed by Phelps and Mushlin,335 and extensions such as the ones that we have described here, offer a formal decision-analytic approach to identify the cut-off points that optimise the contribution of these technologies to population health in a given health-care system.
Chapter 9 An overview and patient perspective on biomarker-guided research
Introduction
Does a marker-guided strategy lead to better outcomes for patients? Despite a wealth of literature on the development and validation of biomarkers, relatively few studies have directly addressed this question. The best evidence to answer this kind of question comes from a pragmatic RCT looking at comparative clinical effectiveness and cost-effectiveness. Few such trials have been conducted and there have been some major disappointments. In oncology, one of the most active areas for biomarker research, McShane et al. 32 concluded in 2005 that ‘the number of markers that have emerged as clinically useful is pitifully small’, an uncomfortable conclusion that has been echoed several times over the subsequent decade. 339–342 It seems, therefore, that, despite a very active biomarker discovery process, researchers cannot yet claim that the ‘biomarker pipeline’ is working successfully in patient benefit terms.
In this chapter we pull together the salient conclusions of the work in Chapters 2–8, to provide an overview from the perspective of both researchers and health-care professionals. However, in light of this, we were keen to explore the patient perspective on our overview of the ‘state of the art’ in this field. The main findings of the methodology workstream were distilled and presented to a group of patients and family members and their opinions sought. This chapter also describes that exercise.
The technical language used in methodological writing tends to make the key arguments inaccessible to a non-technical readership, so we decided to proceed in two main stages. In stage 1, the key points were identified in discussion between this chapter’s lead author (JH) and two health researchers (MT and JanB), chosen as they were experienced in patient and public involvement (PPI) but were not familiar with the methodological literature on biomarkers or test evaluation. This initial step entailed extracting key messages from nearly 100 pages of methodological writing. Jenny Hewison prepared a description of the main findings, which itself ran to several pages; Maureen Twiddy and Janine Bestall read the original chapters as well as JH’s summary and then, over three meetings, key concepts were agreed and provisional plans were drawn up for how the material might be presented to patient and public representatives. In stage 2, the points were first discussed with one of the programme’s highly experienced PPI representatives (JoanB) and amendments made, before the agreed material was presented (by MT and JanB) to a group of patients and family members from the Liver North patient support group.
Further details of the stage 2 work are provided towards the end of this chapter. The following section presents the results of the stage 1 work. For completeness, it incorporates a few points that chapter authors added after the PPI consultation exercise was completed; however, none of the additions was substantive enough to change the key messages.
The starting point of workstream 1 was that a marker is a type of test, so that, when we took a close look at the way that marker-guided strategies have been evaluated, we could draw on what is known about evaluating tests. This is a well-trodden path: most of the basic principles of test evaluation have been known for a long time. However, as the chapters in this section have shown, these basic principles are not consistently applied to the evaluation of a biomarker-guided patient management strategy. The impact of such oversight can be substantial, a reality that seems not to be widely appreciated. It may be that researchers are unclear about the mechanisms at work, and few patients or members of the public are likely to have ‘lifted the lid’ and questioned how the performance of a test may be judged. To ensure that the planned PPI consultation was meaningful, therefore, it was necessary to ensure, first, that the researchers had a good understanding of the workstream 1 outputs, in order for them to be able to explain in non-technical terms the background to the workstream 1 work, the results of the studies themselves and also some of the implications. The stage 1 document was the means to that end and is quite lengthy as a result. Methodologist readers may wish to skip the following section (and rejoin the chapter on p. 164), as it will present them with no surprises and indeed they may find much of the material self-evident.
Stage 1: understanding outputs from the methodology workstream and agreeing key areas for discussion with patient and public representatives
This section begins by rehearsing a few terminological basics to ensure that there is enough common ground for the discussion to follow.
The terminology being used is ACCE (analytical validity; clinical validity; clinical utility; and ethical, legal and social considerations), which divides the research pipeline into labelled components: analytical validity is followed by clinical validity, then clinical utility and, finally, ethical, legal and social considerations. 340,343,344
Analytical validity,’A’, is about the quality of the measure used for the biomarker and how best to handle the inevitability of measurement error. Analytical validity is in itself a complex construct, with three main components: pre-analytical variability (the effect on samples of different storage conditions, transportation times, etc.), analytical variability (differences between reagents, analysers, software, laboratory quality assurance procedures, etc.) and biological variability (within-patient differences, reflecting samples collected at different times of the day, times since the last meal, etc.). Assorted standardisation and calibration procedures can reduce the variability from these sources, but never eliminate it, so estimating residual measurement error is an important part of the ‘A’ phase of test development.
Clinical validity, here ‘C1’, is about the relationship between the measure and the clinical condition, summarised using a variety of parameters. Measurement variability in the marker will necessarily set a limit to the strength of any such relationship and, hence, to the achievable performance of a marker-based test.
-
The case-definition threshold (the cut-off point on a continuous measure) is used to divide the tested population into test positives and test negatives. Choosing a cut-off point always necessitates trading off detection rates and false alarms. The balancing act may be data driven, for example using ROC analysis to identify the cut-off point that minimises misclassifications, or there may be a policy-driven focus on one parameter, for example the test must detect at least 60% of cases.
-
The performance of the test at the chosen cut-off point can be reported using the familiar sensitivity (how well the test performs in relation to cases) and specificity (how well the test performs in relation to non-cases). ‘Test accuracy’ as conventionally calculated uses the same principles and essentially reports the proportion of the total sample (cases and non-cases) correctly classified.
-
If the sensitivity and specificity figures observed in one study are replicated in one or more very similar studies, clinical validity (C1) is considered to have been established, justifying the use of terms such as ‘a validated test’.
Clinical utility, the second ‘C’ in ACCE, refers to information on comparative clinical effectiveness and cost-effectiveness, information that is usually obtained in a pragmatic RCT.
The way that the ACC components of the pipeline work has major implications for the last component, ethical, legal and social considerations, so consideration of ‘E’ is deferred until later in this chapter.
Other authorities have labelled up the stages of biomarker development in different ways, but the basic notion of ‘a pipeline’ is widely accepted. Why then have so few clinically useful biomarkers emerged when researchers have followed this pathway?
Some of the answers to this question are familiar from the wider literature on evaluating tests. Evaluation of marker-guided care often requires a test-and-treat strategy, with an associated increase in sample size, etc. At its most basic level, ‘benefit’ from early identification and treatment depends not only on the effectiveness of treatment – the usual consideration when designing a RCT – but also on the number of people who could benefit if found, together with the ability of the test to find them. Standard treatment trials can take as read the number of people who could benefit if found and the ability of the test to find them, as these are incorporated into eligibility criteria and recruitment projections. ‘Test and treat’ trials on the other hand need to include in their planning estimates – or assumptions – the number of people who could benefit if found and the ability of the test to find them, including in their power calculations. If treatment as usual includes existing tests or clinical decision rules, then it follows that studies of comparative effectiveness will need to be larger still.
The working of the biomarker pipeline is returned to towards the end of the next section, following a brief rehearsal of the complications entailed when a test is used for monitoring purposes. Most of the above insights have come from methodological work on the evaluation of tests used for diagnostic purposes and guidelines for researchers in this area have been available for some time. Our present interest, however, lies elsewhere, namely in tests used for monitoring purposes, the evaluation of which has received, by comparison, little methodological attention.
Monitoring tests
Consistent with the wider test literature, the use of biomarkers for monitoring purposes has been very little studied, despite the widespread use of biomarkers in patient care. This section, therefore, begins by revisiting some of the basic considerations entailed in a test-and-treat clinical utility evaluation, as described in the previous section for a diagnostic test.
It is important in designing any clinical utility study to know the number of people who could benefit if found. In the diagnostic context, the relevant figure is the prevalence of the condition in the sample at the time of testing (‘time 1’). In the monitoring context, it follows that information is also needed on changes in prevalence between test occasions.
And what about the ability of the test to find the people who could benefit from treatment? In a diagnostic context, if the prevalence of the condition and the sensitivity of the test are both known, then the number of cases detected can be calculated. Real cases, correctly detected, are called ‘true positives’ in test parlance. But not all of the people with positive test results will be true positives. Some will be ‘false positives’ and their numbers can be calculated from the proportion of non-cases in the sample, together with the specificity of the test. If no confirmatory testing is carried out, false positives will not be identified as such and will not be returned to the monitoring sample. In these circumstances, all those testing positive, that is, false positives as well as true positives, will be managed in the same way, but it would be reasonable to assume that only the latter might be able to benefit. If the new management regime is associated with the potential for any kind of harm (further tests of an invasive nature, treatment side effects) then it must be remembered that false positives as well as true positives will be subject to these.
Any cases missed by the diagnostic test (the ‘false negatives’) will remain in the monitoring sample. Over time, these will be supplemented by new ‘cases’, arising as a result of disease progression. To estimate the number of cases in the sample on a subsequent monitoring occasion (‘time 2’), it is necessary to know three things: the false-negative rate at time 1, the progression rate of the disease and the elapsed time. Consequently, it cannot be assumed that prevalence at time 2 will be the same as that at time 1 – it could be higher or it could be lower. The performance of the monitoring test will reflect these new circumstances and the pattern will repeat itself over subsequent monitoring occasions.
The well-established route along the biomarker research pipeline does not readily generate all of the information indicated above or make use of it in the design of clinical utility studies, such as a trial to evaluate a monitoring strategy. Does this matter? If some figures are not available and plausible values have to be assumed, how much hangs on the assumed values being ‘about right’? And at what point in the pipeline should health economic considerations be addressed? Enhanced monitoring or the initiation of treatment for people unlikely to benefit will waste resources and can overburden health-care systems, so simply widening the definition of who is eligible for enhanced care is seldom the best approach, even if the enhanced regime is acceptable and relatively benign.
Workstream 1 aimed to address the above questions and help to bridge the gap between clinical validity and clinical utility studies. Our topic was the design of pragmatic trials aimed at evaluating the clinical utility of a marker used for monitoring purposes and our focus was the role of key test and patient parameters in the design of such studies. We approached the task in a number of different ways, and from a number of different academic perspectives, but some very similar messages emerged.
A case study
Chapter 3 asked if key test and patient parameters had informed published guidelines on the use of PSA for monitoring prostate cancer recurrence. Little evidence of such a systematic approach was found. When to test, for example, ‘appeared to be almost exclusively determined by standard follow-up schedules rather than being based on any scientific evidence’. Although the potential for variation in measurements was usually accepted by guideline developers, they showed little interest in the potential effect of such variation on the interpretation of test results. Relevant evidence was not always available, but, even when it was available, it was not always used. A systematic review of biological variation in levels of PSA found a mean variability of 20%. Based on this figure, it was calculated that, ‘to be 95% sure that a change in total PSA level is not the result of random variation, the change needs to be around 50% of the previous measurement’.
This review was not, however, cited by any of the seven guidelines subsequently published. Recommendations on when to take action were based on consensus statements or retrospective case series rather than on calculations of optimal values for cut-offs and monitoring frequency in the target population.
Trial evidence
Chapter 4 asked if the RCT design had been successfully used to evaluate strategies for monitoring disease progression or recurrence. Fifty-eight relevant trials were found, with the intervention usually taking the form of adding an assessment to an existing schedule of care. The reviewers observed that, ‘although one might expect both the rate of disease progression and the degree of measurement variability associated with a given test to be taken into account when setting test frequency, test intervals were apparently determined by convenience or by fear of missing a key clinical event’. The test strategy that delivers a high detection rate (i.e. few missed key events) will inevitably also deliver false alarms, but in the published trials ‘[t]here was little acknowledgement of the potential for false-positive results’. The reviewers also noted that many of the trials seemed to be underpowered. About one-fifth of the 58 trials were stopped early, often because of a lower than expected event rate in the control groups. For these and no doubt other reasons, ‘[o]nly a small proportion of trials reported statistically significant results for the primary outcome’.
The methodology literature
Chapter 5 found that the design and evaluation of strategies to monitor disease progression have received relatively little methodological attention. There is, however, relevant work on the statistical analysis of monitoring data and on the development and evaluation of screening strategies, with the starting point for most approaches being quite a detailed understanding of how test results fluctuate in the absence of disease progression or other ‘signals’ of interest.
How big does a change in marker scores need to be to justify clinical interest? In clinical validity studies, if patient groups known to differ in clinically important terms show a marker score difference of a particular magnitude, then consensus can emerge that that constitutes a ‘clinically important difference’. In analytical validity studies, marker scores alone may reveal patterns of interest. RCVs are a way of quantifying score fluctuations likely to occur in the absence of a real change in a patient’s underlying condition. Assuming that the statistics of the normal curve apply, then a difference between two results greater than the RCV is taken to indicate that a real change has occurred.
Some work has been carried out on using score variability to develop decision rules, but in the absence of a demonstrable link to patients’ clinical condition the contribution of ‘signal-to-noise’ approaches to choosing a monitoring strategy will remain limited. Overall, there is little evidence that methodological work has informed the functioning of the biomarker research pipeline.
Patient outcomes and monitoring
Chapter 6 explored two ways in which monitoring strategies may improve patient outcomes. If test results do not influence patient management, then a testing strategy is unlikely to improve patient outcomes. The purpose of introducing a new test needs first to be made explicit, however: is it to replace an existing test on the grounds of improved accuracy, or reduced invasiveness, for example? Or is earlier diagnosis the aim? Or identification of an ‘at-risk’ group who are to receive further investigation? The causal pathways through which benefit is achieved are not necessarily simple in any of these circumstances, however, so understanding how the relevant pathways operate and, if necessary, modifying the testing strategy in order to optimise them, may be necessary if maximum patient benefit is to be obtained. Three key approaches to structuring the problem in relation to tests for screening and for diagnosis were identified in the literature and applied to tests used for monitoring, drawing on 58 previously identified trials (see Chapter 9, Trial evidence) for illustrative purposes.
The focus of the first approach was the trade-off between benefits and harms under new testing regimes compared with standard care, when tests are introduced for different purposes. This approach highlighted the extent to which the impact of a monitoring strategy depends not only on the effectiveness of treatment, but also on the properties and timing of testing and, hence, the extent to which the size and the management of different subgroups of patients differ in practice between the new and the standard regimes. Prior to conducting a RCT, these figures should be identified from pilot or feasibility studies.
In the second approach, a new testing strategy was considered to be a complex intervention and its components were picked apart in those terms. Clinicians’ and patients’ trust in the monitoring protocol, and their willingness to comply with it, were identified as important considerations that should not simply be assumed, but should be assessed prior to a trial and, in some circumstances, should be the subject of research in their own right.
Lastly, the circumstances in which a RCT for the evaluation of patient benefit was both necessary and timely were scrutinised. Conclusions overlapped with those drawn in the two previous paragraphs: a RCT may indeed be required to evaluate the net impact of a new monitoring strategy, because the latter is likely to be exerting its effect via a network of individual mechanisms. However, that RCT will be most informative if it is based on good estimates of important population, test and treatment parameters.
The first modelling study
The aim of the first modelling study (see Chapter 7) was to see if modelling could help identify a ‘best bet’ monitoring strategy for potential evaluation in a subsequent trial. The approach was to look at different rules for defining a test-positive result and, on the assumption that true diagnostic status (‘caseness’) would later be known, compare the different rules in terms of important indicators such as delay to diagnosis. The statistical model that enabled informative comparisons to be made was found in sensitivity analyses to be heavily reliant on detailed information about disease progression and test performance. In Chapter 7, the latter was characterised in terms of two components: (1) measurement error and (2) between-individual variability. The nature and role of measurement error and – in the biomarker field – its relationship to analytical validity (‘A’) have been described earlier. Terminology is unfortunately not standardised in this area, so here it also needs to be noted that between-individual variability is one way of characterising clinical validity (‘C1’), as it refers to the variation in marker scores observed among individuals belonging – according to a reference standard – to the same diagnostic category (e.g. cirrhosis or a particular fibrosis stage). Such variation within a diagnostic category is likely to be associated with overlap in the score ranges observed in different categories, which in turn brings the potential for misclassification of individuals when that is based on marker score – the conventional measure of ‘test accuracy’ characterises the performance of a test essentially in terms of the overall amount of such misclassification.
Putting the disease progression and test performance parameters together, it can be seen that we are on familiar territory, because together they determine the number of ‘cases’ in the study sample at a point in time and also the biomarker test’s ability to identify them.
Regarding the test performance parameters, available information on ELF, generated through the traditional research route, proved to be insufficient for comparison purposes. On disease progression, longitudinal descriptive data were needed and even cross-sectional prevalence information would not have sufficed.
Using the simulated data to compare monitoring strategies for liver fibrosis, it was found that a simple threshold case rule performed very well. A more complex rule using linear regression to summarise changes in score over time did perform marginally better, but would have been potentially more difficult to apply in practice. It was emphasised that these comparisons did depend on the assumptions made. For example, tests that show only limited within-individual variation in their scores lend themselves to rules that incorporate changes over time, whereas comparison with a simple threshold is more meaningful for tests generating less stable scores.
The second modelling study
Because of its dependence on the ELUCIDATE trial, the details of this study are provided in the workstream 3 section of this report (see Chapter 21). However, most of the results were available in time for the workstream 1 patient consultation exercise, so key points are reported here. In this simulation study, the measure of success of the monitoring strategy was not time to diagnosis, but time to severe complications. Reference standard diagnostic information is not available in many monitoring contexts, so cannot be used to check the clinical validity of the test or be the determinant of patient management. This was the case in the ELUCIDATE trial and, furthermore, any ‘diagnostic’ information that became available would be defined differently in the two arms of the trial: clinically in the control arm and using the ELF test in the intervention arm. Consequently, test accuracy considerations were not part of this simulation. In addition, the clinical utility of any test-and-treat strategy is dependent not only on successful diagnosis but also on successful treatment, so simulating the success of the monitoring strategy as evaluated in the trial required the modelling of patient outcomes. The 2015 Health Technology Assessment report on non-invasive liver biomarkers noted the value of this approach, on the grounds that it would provide a hard end point without the need for liver biopsy. 345
The key modelled relationship – as in the previously published ELF data – was between ELF values and the rate of development of severe complications. Measurement error and the test performance of the ELF test would have contributed to that initial relationship, so were understood to take a similar role here and, hence, were not specifically addressed in the simulation, although the success of that approach does depend on the these parameters remaining similar in the two settings. In planning the trial, a cut-off point of 12.5 on the ELF test was initially adopted as the marker-defined ‘diagnostic’ threshold, but, as a clearer picture emerged of the ELF value distribution in the trial population, a decision was taken to adopt the lower value of 9.5 to define an ‘at-risk’ subgroup, eligible for enhanced care.
In this study, trial participants’ time to severe complications was modelled, using early information from the trial itself. In the ELF arm, the initial distribution of ELF values, together with the observed cumulative incidence of above-threshold scores), was used to model the relationship between a starting ELF value and the likelihood of passing the threshold of 9.5 after a given period of time. In the control arm, the relationship between starting ELF values and the observed cumulative incidence of clinically diagnosed cirrhosis was modelled, as well as the relationship between clinical diagnosis and concurrent ELF values. Previously published data on the relationship between ELF ranges and time to severe complications were then used to model the occurrence of the latter in the two arms of the trial. An assumed treatment effect was then applied to all ‘diagnosed’ patients and the two simulated arms were compared over different durations of follow up. This simulated trial enabled the achieved power of the actual trial to be calculated and provided an illustration of how the approach could be used for designing future trials.
Measurement error and test accuracy were both indirectly incorporated into this simulation model through the association reported in the literature between ELF values and the subsequent rate of development of cirrhosis. Subject to certain assumptions, therefore, it could be said that the traditional research route had provided enough of the evidence needed to plan a trial. It was, however, clear that it would have been preferable to have information on key change parameters available, including those relating to sample composition, prior to designing a trial, ideally in this case from a cohort study incorporating sequential ELF values. The conclusion about the need for better longitudinal data in order to model the likely numbers of ‘test positives’ more accurately is similar to one drawn in the first simulation study.
The third modelling study
In Chapter 8, a third modelling study had a different focus again. It was conducted from a health economic perspective and specifically addressed how to optimise the benefit to patients when using an imperfect test for monitoring. The starting point for such an exercise is that the anticipated effects on all four outcome groups of testing (true and false positives, true and false negatives) need to be included in the calculation of benefit. It follows that the number in each of the groups also needs to be known and that these numbers will depend not only on the numbers of actual cases but also on the definition of a test-positive result in terms of a threshold score.
In the context of diagnostic testing, the academic literature already contained a method for choosing a cut-off point in such a way that it would maximise overall patient benefit, taking into account the consequences for patients and budgets of incorrect as well as correct diagnoses. The third modelling study applied these methods to the different needs of monitoring tests and in so doing highlighted once again the importance of understanding how sample composition is likely to change over time.
Health economists can make reasonable estimates of the benefits and the harms, as well as the costs, consequent on each of the four possible outcomes of testing, namely that the patient has the tested-for condition and is correctly identified, the patient has the tested-for condition and it is missed, the patient is well and is correctly identified as such or the patient is mistakenly diagnosed as having the condition. The overall benefit to the patient population of different testing strategies is calculated by multiplying these ‘per-patient’ figures by the numbers of patients in the four outcome categories. These numbers will change according to the cut-off point used for the test, but also, and crucially, they depend on the proportion of patients in the tested sample who actually have the condition. It follows that the optimum cut-off point is sample dependent, because the lower the prevalence of the condition in the sample, the higher will be the proportion of ‘false alarms’ among the patients identified by the test as having the condition. How then might the composition of a monitored patient cohort change over time? And what would be the implications for the optimum cut-off point for the monitoring test?
First, the optimum ‘diagnostic’ threshold was calculated, using the best available information on test accuracy and the consequences for the four resulting categories: true and false positives and true and false negatives. Considering first the patients not identified as having relapsed (the ‘diagnostic test negatives’) at that time point, it could be anticipated that, at any subsequent surveillance point, this sample would contain a mixture of patients who had stayed well, new cases of cancer and cancer cases missed (‘false negatives’) at the earlier administration of the test. Estimating the numbers of new cases required information on the rate of disease progression as well as the time elapsed since the previous testing, whereas estimating the numbers of previously missed cases drew on knowledge of the accuracy of the diagnostic test, in particular the likelihood that a test-negative result would prove to be correct (a test parameter known as the NPV). Under plausible estimates of these values, the sample at the second testing occasion might have a higher or lower disease prevalence than the sample at the time of ‘diagnostic’ testing. A new optimal ‘monitoring’ threshold could then be calculated, reflecting the new prevalence value.
If the next step for the ‘diagnostic test-positive’ subgroup (i.e. patients identified as having relapsed according to the initial test) was to be continued monitoring, equivalent calculations could be performed, but would produce a different optimum cut-off point. It was noted that, in principle, the approach could be applied to monitoring across multiple occasions.
The potential of a value of information approach was explored towards the end Chapter 8. For a diagnostic test, ‘perfect information’ consists of the distribution of test scores in ‘cases’ and ‘non cases’. This information can be used to calculate the probability that a patient with a given test score has been correctly classified, noting that uncertainty about the patient’s true health state will be systematically greater for mid-range test scores than for extreme ones. Putting this argument in slightly different terms, misclassification rates are likely to be greater for mid-range test scores than for very low or very high test scores. As mid-range scores are also going to be more affected by the choice of cut-off point (choosing a lower cut-off point, for example, will increase the number of mid-range scorers testing positive), it follows that the distribution of uncertainty between the test positives and the test negatives will vary according to the cut-off point adopted.
In the monitoring context, true disease progression data are required, but additional testing complexities also need to be considered. First, the distribution of test scores in the new cases may differ from that observed in the cases present on the first testing occasion. Second, the likelihood that a previously test-negative individual will have become a test-positive individual will depend not only on their true change in health status, but also on how likely it is that their test score will have crossed the relevant cut-off point boundary, which in turn depends on where that boundary is set. Adoption of a low cut-off point on the first, ‘diagnostic’ occasion will lead to a reduced likelihood that such a stringently defined subgroup will have changed its real status the next time around, that is, it reduces the value of information in the monitoring of originally defined test negatives. Finally, as the modelling reported earlier in Chapter 8 showed, the optimum cut-off point will always reflect the chosen value of health, suggesting a very complex relationship between the value of information and the value of health in the monitoring context.
Implications for the design of clinical utility studies
The literature reviews and the case study identified similar issues. The ways in which factors such as disease progression rate, measurement error, choice of cut-off point and monitoring interval can affect clinical utility – and, hence, trials to evaluate clinical utility – were then clearly illustrated by the modelling studies.
All three of the modelling exercises concluded that information on changes in the tested sample over time would greatly improve the usefulness of the models. Disease progression is clearly a major element of such changes and the need to characterise it could perhaps be regarded as self-evident: if the aim of a monitoring intervention is to change the progress of a disease over time, then comparison data on the progress of the disease in the absence of the intervention are clearly a prerequisite for evaluation purposes. Disease progression data are not, however, provided by the conventional research pipeline, which may partly explain why their importance has been persistently overlooked.
A trial of a marker-guided monitoring strategy will entail specifying that strategy in terms of thresholds, monitoring intervals, etc. How, though, should those elements be chosen? And is it always necessary to know who were the true ‘cases’ and who were not? Modelling study 2, and the trial in workstream 3 of this programme, were based on identifying an ‘at-risk’ group rather than diagnosing ‘cases’ as such. Patients were defined as being at risk in terms of ELF value ranges previously observed to be associated with subsequent differences in patient outcomes. By applying an assumed treatment effect, a trial could be modelled and its size calculated, based in the usual way on the amount of benefit that it was judged important to detect. If the results of such an adequately powered trial suggested that it was clinically effective and cost-effective to monitor with the threshold and the interval used, then it would be tempting to conclude that the research pipeline was working. But how were the threshold and intervals chosen and were they the best ones? Maybe the threshold adopted to define an at-risk group was too low, tipping the balance too much towards detection of every possible case at the expense of increasing the numbers of false alarms? Maybe such frequent testing was unnecessary and longer intervals would have been perfectly adequate? And what if no evidence of clinical effectiveness or cost-effectiveness was found? Where might the explanation lie? And what would then be the way forward: more trials with different combinations of thresholds and intervals?
Modelling studies 1 and 3 suggest that a different approach might produce better value from the trial’s (clinical utility) budget. However, in addition to the disease progression information previously mentioned, this approach depends on eventual knowledge of true ‘caseness’ and so would require more and better test performance information (analytical validity and clinical validity) than the pipeline currently supplies. If available, the information could be used to estimate the effects of different monitoring strategies on patients much more precisely than is possible at present, enabling ‘best bets’ to be identified for subsequent evaluation in trials. Better estimates of measurement error, for example, would contribute to increased test accuracy, to reducing misclassifications and, hence, to increasing benefit (through fewer false negatives) without disproportionate cost or harm (from treating the false positives who do not benefit).
It may be that part of the problem in the functioning of the biomarkers research pipeline is the prominence given at the clinical validation stage to just two indicators of test performance: sensitivity (how well the test performs in relation to cases) and specificity (how well the test performs in relation to non-cases). Both metrics can be derived from the case–control studies that play a prominent role in test development and initial validation, and it is perhaps insufficiently appreciated that people using tests in practice – and also people evaluating test use in practice – are in a fundamentally different position. As in the ELUCIDATE trial, if it is the test result that is intended to be used by the people making patient management decisions, then test performance metrics will be needed that take that starting point into account. The test evaluation literature identifies two such metrics, capturing, first, how many test positives turn out to be cases (the PPV) and, second, how many test negatives turn out to be non-cases (the NPV). To illustrate how misleading reliance on sensitivity and specificity can be in this context – and remembering that even a very specific test will incorrectly identify a small proportion of non-cases as cases (i.e. it has a false-positive rate) – in a low-prevalence sample (i.e. one mainly consisting of non-cases), the basic arithmetic of applying a small percentage to a large number will result in many test-positive results being generated by non-cases and a consequent lowering of the proportion of the test positives coming from cases. In a higher-prevalence sample, arithmetic dictates that this proportion (the PPV) will be higher.
Applying the same logic to test negatives, in a low-prevalence sample, that is, one containing a small number of cases, a reasonably sensitive test will miss very few of these and, hence, add only a very small number of test-negative results to the large number generated by the non-cases. It follows that the proportion of test negatives who are indeed non-cases – the NPV – will decrease as prevalence increases, but also that this parameter will change very little across a range of low prevalence values. It also follows that, in a low-prevalence sample, a lot of the people receiving treatment will not be able to benefit because, even using a very accurate test with high specificity, although they tested positive, they were in fact false positives. Lowering the cut-off point used to define ‘caseness’ will increase the number of people testing positive, but without good prevalence data it is not possible to estimate what proportion of the test positives have the potential to respond to treatment.
As explained, both PPV and NPV change with the composition of the tested population, that is, they reflect prevalence, and that is why they cannot be calculated from case–control studies alone. With the correct input parameters, modelling of all of these effects would enable a much more realistic picture to be built up of the magnitude of benefit that could potentially accrue from a specific monitoring regime being applied to a specific patient population, although it might also raise challenging questions about the rationale for the monitoring intervals and the thresholds currently in use. From a research perspective, there would be clear advantages in applying such a model to the design of a clinical utility trial.
Therefore, should funders insist on better longitudinal data – on disease progression in the population that will be the subject of the trial and on how marker scores change over time? And what about the analytical validity of the marker as measured outside a research laboratory and test accuracy results, including PPV and NPV, from a relevant population? Should these be required before funding a monitoring trial? At the conclusion of the value of information section in Chapter 8, it was argued that the most efficient way forward for monitoring technologies might be the improved characterisation of test score distributions in cases and non-cases and of progression rates. The same could perhaps be said to those funding research on the evaluation of monitoring technologies: extra time and effort spent on providing better-quality information of this kind would almost certainly lead to a better-designed trial, and the overall research duration and budget might not be very different.
Stage 2: obtaining patient and public perspectives
Establishing a shared understanding
Following a number of meetings between the three researchers to agree understandings and discuss possible content, the key areas to be taken forward for discussion with PPI representatives were identified as:
-
assumptions about test development from the laboratory bench to the clinic
-
variations in test scores within and between individuals (signal to noise)
-
test accuracy (relationship to clinical condition)
-
detection of cases and non-cases (sensitivity and specificity)
-
prevalence of disease and using prevalence in interpreting tests
-
rate of disease progression and its impact on treatment strategies
-
development of treatment strategies to improve patient care
-
development of guidelines and communication about tests between professionals and patients.
Initial presentation to patient and public involvement representatives
An initial meeting with a PPI representative (one representative was unable to attend) was held in Leeds on 31 July 2015. The key concepts of test measurement, accuracy, performance and interpretation were highlighted in the context of the NIHR liver biomarkers programme. It was agreed that these issues were important and interesting for patients and the public to discuss. Preparation for a future workshop with a larger group of people was discussed.
LIVErNORTH workshop
Twelve people (nine women and three men) took part in the PPI workshop. A range of conditions was represented, including:
-
liver transplant (n = 3)
-
primary biliary cirrhosis (n = 3)
-
carers (n = 3)
-
non-alcoholic fatty liver disease (n = 1)
-
overdose with a potentially hepatoxic drug (n = 3)
-
primary sclerosing cholangitis (n = 3).
It became clear from questions asked and from the discussion following the presentation that patients were interested in methodological issues around biomarker evaluation. The group members understood the concepts and asked sophisticated questions about the biomarker test development process. The key discussion areas were as noted in Establishing a shared understanding.
Participants assumed that all parts of the biomarker pipeline from laboratory bench to clinic were equally researched and considered. Any new biomarker test that was evaluated in a trial or implemented into a service should be ‘fit for purpose’. Participants stated, ‘Don’t set up a test, then move on and think we’ve cracked it’. All aspects of test development and evaluation were thought to be equally important and should carry a similar weight in the research pipeline.
Participants stated that it should be made clear what the level of accuracy of a test is in practice and also the numbers of false positives and false negatives that the test produces. The signal-to-noise issue was thought to be important and participants indicated that in their opinion only tests that reached a certain level of performance should be taken forward for further evaluation.
The variation in test scores within and between patients was acknowledged, with several participants offering examples of how their test results had an impact on their care. At least one carer had noted fluctuations in test scores over time and he used these to monitor the patterns in the health of his partner, concluding that they meant very little on their own. He readily understood the notion of a false-positive test result and, when he thought about how particular test results could make a big difference to his partner’s care, he was alarmed to think that the likelihood of such an outcome might not be well understood by clinicians. Participants agreed that fluctuations in scores or changes in scores were poorly explained by doctors if at all. The fact that snapshot ‘how you are today’ test results were used to guide patient care and to instigate treatment changes was thought to be poor practice in the context of chronic conditions. All agreed that tests should be part of a wider clinical assessment. With the advent of new technology and the ability to monitor and log routine data it was thought that understanding patterns of change within and between a cohort of patients with liver disease over a series of different points in time was really very important. Participants could clearly extrapolate this example to other conditions as well.
Participants found it hard to believe that disease progression rates – of central importance to their experience as patients – were not routinely available and routinely used for research purposes. They were almost as taken aback by the lack of interest shown by researchers as well as clinicians in the practical implications of test inaccuracy and were ‘astonished’ that the routine monitoring of patients was not captured and made use of to support the assessment of new biomarker tests. Participants could pick up on the notions of false positives and false negatives without any difficulty and, because the implications for an individual (clinical but also psychological) of either sort of misclassification were very apparent, they expected high standards to prevail at every stage of the biomarker pipeline.
Participants stated that it was important that all of the information required to interpret a test result, such as variation in scores (signal to noise), accuracy, prevalence, performance in practice and evidence-based treatment strategies, should be communicated to patients. Any information that was put into guidelines should be developed with the involvement of patient and public representatives and someone with a plain English remit. Any guidelines developed should be undertaken at a national level and should be reviewed within a relevant time frame to account for regular updates. A lay summary should be available for patients if they would like to know more information.
Participants stated that the way in which test results are communicated to patients needs careful consideration and the involvement of patients and the public to make messages clear. The role of the doctor in promoting good communication about any monitoring test and about the ‘possibility’ of there being other interpretations and outcomes of test results should be part of their education programme. Participants wanted to know about how accurate a test was and the chance of false positives and negatives. How such results might have an impact on patients in an acute or emergency situation and a routine situation was considered. Supported decision-making was advocated for both situations. Further awareness of these issues involving patients and the public should be implemented, with careful thought being given to how information is presented and the types of examples that are used. Participants clearly stated that decision-making should be facilitated in partnership with health professionals.
Patient perspectives and the ‘E’ in ACCE
The ‘E’ in ACCE – the ethical, legal and social considerations attendant on biomarker testing – have not been addressed in this account so far. Emphasis in the literature to date has been on the potential implications of test results, for example genetic tests for late-onset conditions, rather than on the quality and efficiency of the research pipeline. But are there downsides for patients of the present, ‘let’s try it and see’ approach to biomarker development? The PPI work reported here suggests, first, that quality and efficiency matter to patients in relation to the research pipeline, as well as in direct care, and, second, that the widely recognised right of patients to make their own trade-off between length and quality of life when making treatment decisions may also need to be more consistently and transparently applied to decisions about tests. The health economic approach provides very useful tools of thought here in its explicit calculation of harms (physical and psychological) and benefits forgone, as well as the costs, of over- and under-investigation and treatment.
Patients quite rightly expect trials to be well designed, but it could be argued that, with regard to some of the aspects discussed here, they are not well served by the existing evidence pipeline. Research funding mechanisms are currently ‘tuned’ towards traditional A and C1 work in laboratories, and towards clinical trials, but descriptive longitudinal work is not currently attractive to funders, possibly because its contribution to bridging the evidence gap is insufficiently appreciated. Secondary use of data collected for other purposes may offer one practicable way forward.
Patient perspectives are returned to in Chapter 24.
The workstream 1 methodology studies were reported using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) 2009 checklist. 346
Chapter 10 Biomarker pipelines: ensuring clinical translation using renal cancer and kidney transplantation as exemplars
This chapter highlights the main aims of the clinical translation workstream, provides the background to this aspect of the programme and the clinical context and outlines the various elements of the work undertaken, which is further described in detail in the following chapters.
Main aims of the clinical translation workstream (workstream 2)
Within the context of the overall aim of the programme to develop strategies to enable the rapid evaluation and translation of promising protein biomarkers into the NHS for patient benefit with a main focus on renal and liver diseases, workstream 2 focuses on the clinical biochemistry aspects, with the main aim being to establish and maintain a multicentre sample and clinical data bank in CLD, renal cancer and renal transplantation (RT), together with the development of a robust system for the evaluation of promising emerging biomarkers and related assays, to facilitate rapid biomarker assessment prior to large-scale trialling in practice within the NHS.
Within this, specific objectives were to:
-
undertake systematic reviews of biomarker status in renal cancer particularly relating to prognosis and in RT relating to acute complications and long-term outcome with a focus on DGF
-
prioritise selected biomarkers for further evaluation at the multicentre level and develop and employ validation strategies for technical validation of relevant biomarker assays, including determination of significant pre-analytical issues
-
design and undertake studies examining the clinical utility of the selected biomarkers within the chosen disease areas and identify those biomarkers that justify evaluation in a clinical trial, leading to applications for funding
-
create a clinical sample and data resource to underpin studies within this programme but also to facilitate future rapid and cost-effective biomarker evaluation, with the infrastructure and strategies developed providing a blueprint for future similar studies in other disease areas.
Critical elements in the biomarker discovery and translation process
General concepts
With the massive investments in large-scale ‘-omics’ studies accompanying increased technological and bioinformatics capabilities, our understanding of the molecular changes and mechanisms underlying many diseases has increased dramatically. It is now understood that within a single disease phenotype or diagnosis there exists underlying molecular heterogeneity, giving rise to previously unrecognised disease subgroups. With this, there is a drive to use such information to tailor the treatment of each patient on an individual basis, recognising the heterogeneity within a disease, variously described as ‘personalised’, ‘precision’ or ‘stratified’ medicine. 347–351 Consequently, the development of ‘targeted therapies’ has revolutionised treatment in many diseases, particularly many cancers. 352 This vision is of course predicated on the use of biomarkers to optimise all aspects of the patient pathway, from early and accurate diagnosis, to determining the extent and activity of the disease and prognosis, predicting response to therapy allowing optimal treatment selection and monitoring for treatment response/toxicity or disease progression. Using biomarkers in this way has major potential benefits for patients and the NHS in terms of improving outcomes and providing more cost-effective care, for example supplementing or replacing invasive or expensive procedures or imaging tests, improving patient safety and quality of life and avoiding unnecessary or suboptimal treatment or toxicities. Interestingly, however, and using therapy-related biomarkers as an example, although it extends to other biomarkers types equally, there are currently only 26 FDA-approved ‘official’ companion diagnostics, 10 of which relate to HER2 testing for trastuzumab (Herceptin), although there are considerably more laboratory-developed tests (LDTs). 352,353 The need to refine existing methods used in economic evaluations of companion diagnostics to include additional characteristics of the test performance itself given the increasing complexities has also been highlighted. 354
Genomic analysis of tissue or cells is now routinely used not just in inherited diseases but increasingly also in many cancers to allow the selection of specific targeted therapies. However, many routinely measured biomarkers are proteins present in body fluids and as such remain the province of clinical biochemistry laboratories. Protein biomarkers have the advantages of being comparatively cheap and easy to measure, being relatively non-invasive and providing dynamic information. At the time of application for this programme in 2008, the depth of knowledge being generated, as described above, and the extent of its impact in terms of effectively revolutionising several aspects of patient care in some diseases areas were not apparent, although expectations were high. Likewise, with the plethora of potential novel protein biomarkers arising from the surge of discovery efforts in clinical proteomic studies, expectations surrounding the introduction of new biomarkers to clinical practice were high. However, even in 2008 this was accompanied by an increasing awareness that the translation of novel biomarkers into clinical practice was not being realised. A pivotal reason for this was the absence of a clearly defined pathway linking biomarker research to health services research, and this was compounded by the technical inadequacies of some of the studies negating any likely translation of the findings. The stages in the ‘biomarker pipeline’ have been variously defined, as illustrated in Figure 24.
FIGURE 24.
Linear depiction of the phases in the biomarker pathway. WS2, workstream 2.
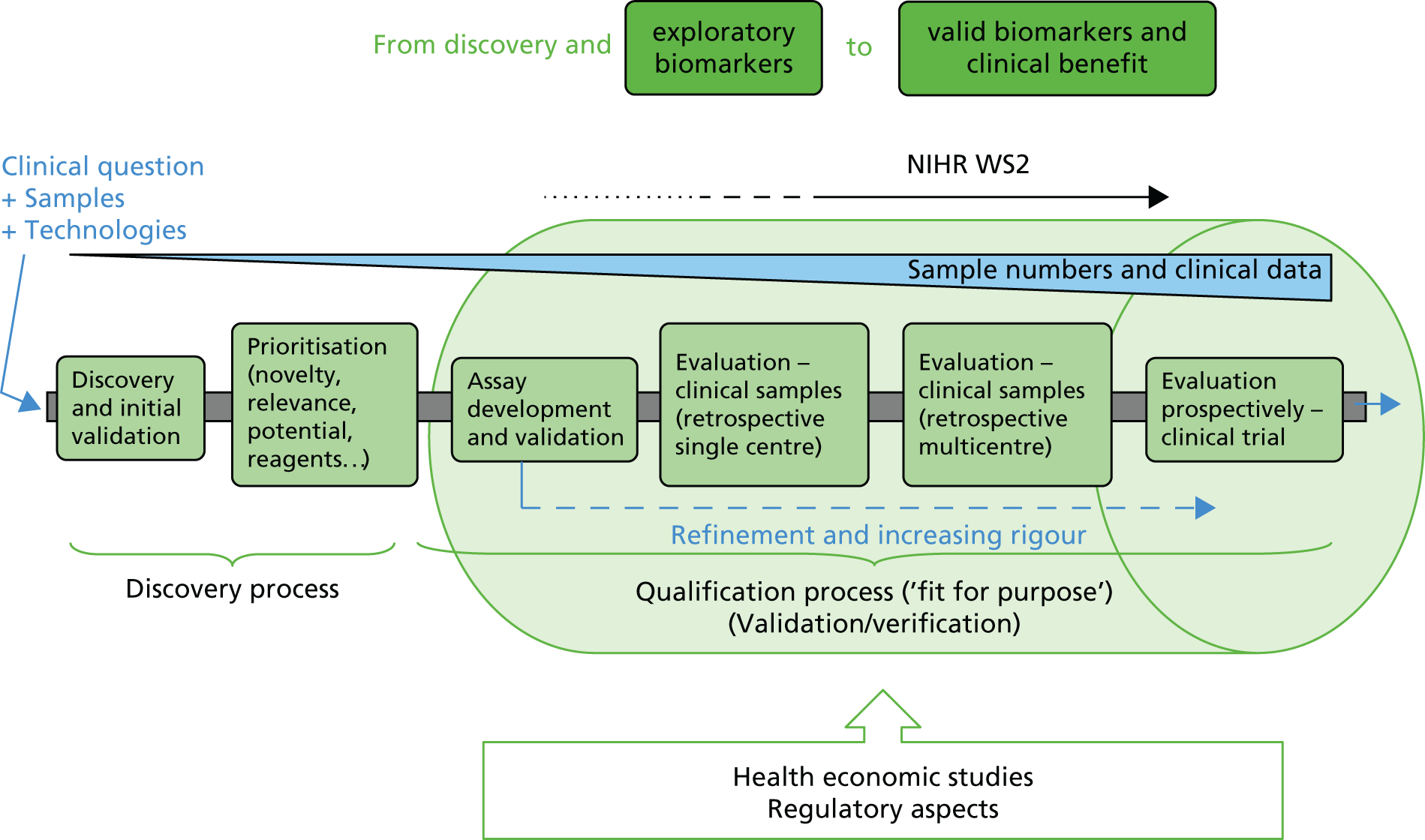
The dynamic nature of test evaluation in this pathway from biomarker to medical test is more apparent when summarised as a cyclical process (Figure 25), as recently described by the Test Evaluation Working Group of the European Federation of Clinical Chemistry Laboratory Medicine. 338
FIGURE 25.
Cyclical framework for the evaluation of in vitro medical tests. This framework illustrates that the key components of the test evaluation process are driven by the purpose and role of using a test in the clinical pathway. Reciprocally, the key test evaluation elements may influence or modify existing clinical pathways. The outer circle linking the various elements of the test evaluation cycle highlights the interplay between the various components and how, for example, analytical performance may impact clinical performance and, vice versa, how clinical performance or effectiveness of a test may call for improved analytical performance and sets new analytical goals for improving the clinical effectiveness and cost-effectiveness of the test–treatment pathway. Reprinted from Clin Chim Acta, vol. 427, Horvath AR, Lord SJ, St John A, Sandberg S, Cobbaert CM, Lorenz S, et al. , From biomarkers to medical tests: the changing landscape of test evaluation, pp. 49–57,338 copyright 2014, with permission from Elsevier.

Certainly, in 2008, the necessary evaluative framework involving multiple stakeholders, including academia, industry, health-care providers and regulatory authorities, and with wide-ranging considerations, including technology transfer, cost-effectiveness, methodological workflows and regulatory hurdles, was not clear or effective. Without a comprehensive evaluation framework, the route from stringent evaluation to clinical implementation (including evaluation of the impact on outcomes) and the realisation of the substantial potential of biomarkers to contribute to improving patient care and health service provision was recognised to be at risk, with various issues highlighted from multiple perspectives, including those of academia and industry. 1–9,342,355,356 Encouragingly, though, the need for national strategies to overcome this was also increasingly being appreciated at this time, for example by the NIH in the USA11 and by the Royal College of Pathologists in the UK. 12 This programme of research was essentially established to explore and address some of these issues, with health economic and trial methodology covered in workstream 1 and workstream 3, respectively, and with workstream 2 directed at exploring aspects of infrastructure related to sample banking and assay validation and focusing on renal cancer and RT as exemplars for biomarker testing.
So has the situation changed in the intervening period and decreased the value of this programme? The answer to this is a resounding no if the situation is reflected in the number of biomarker tests approved or cleared for marketing by the FDA. In a recent report generated by the consultancy company Amplion and using specific biomarker definitions, of necessity focused on FDA approval as this is the only agency to make comprehensive information publicly available, the number of 510(k) clearances of biomarker-based tests each year fell from just over 120 in 2004 to approximately one-third of this number in 2014. 353 Premarket approval was granted in zero to fewer than five cases each year in the same time period. Importantly, from 2003 to 2014, the average number of novel biomarker targets that went to market in FDA-reviewed tests was only three and the average time from discovery to clearance/approval was 15 years, although this was highly variable. 353 However, it should be recognised that this may be a large underestimate overall as there are a considerable number of LDTs available that are currently in use although not centrally registered. 352,353 This situation is likely to change in the near future with proposed compulsory registration with the FDA. Additionally, many tests gaining European approval through CE marking are not FDA approved, such as the ELF test, which is a central feature of workstream 3.
The perceived obstacles and pitfalls in taking a biomarker (or biomarker panel) along the pathway from laboratory to clinic essentially remain the same in more recent studies as those highlighted in earlier studies. 338,357–361 Focusing on the laboratory and clinical biochemistry perspectives, some key aspects are discussed further in the following section and form the subject of specific workstream 2 activities.
Specific laboratory-based/clinical biochemistry aspects
Although this programme covers the later parts of the biomarker pipeline, it is still appropriate to briefly consider some of the issues that are particularly challenging in biomarker discovery and those that contribute to the high level of biomarker attrition at the early stages. Particular technical challenges for novel biomarker discovery in clinical fluids such as serum or plasma include the vast dynamic range of protein concentrations, spanning a concentration of approximately 40–60 g/l for albumin to a concentration of < 1 ng/l for cytokines, a range exceeding the analytical capability of proteomic technologies and necessitating extensive fractionation/enrichment strategies, particularly with just 22 proteins constituting 99% of the entire plasma protein content. 10 For urine, low protein concentration and high salt content present challenges, although as approximately 70% of urinary proteins are thought to be kidney derived it is an attractive alternative source of enriched biomarkers, particularly for renal diseases. 362 However, normalisation of results is an issue; creatinine is most commonly used although this is not ideal as it is affected by many factors such as muscle mass and renal function. The results from many studies have not been confirmed, with a major contributory factor being the poor initial study design. This includes insufficient statistical power, inherent study bias because of a lack of attention to/awareness of potential confounding factors and the impact of pre-analytical factors on sample quality, although such issue are being addressed, with promising results emerging. 358–361,363–366 To improve study design and reporting, several guidelines have now been published in relation to diagnostic markers, such as the REporting recommendations for tumour MARKer prognostic studies (REMARK)367 and the STAndards for Reporting Diagnostic accuracy (STARD). 368
Other factors preventing further progression include the lack of the necessary tools or resources, such as the availability of suitable numbers of stringently collected clinical samples with the appropriate associated clinical data, including long-term follow-up data, which can take years to accumulate. Biobanking is not co-ordinated internationally, nor is there a complete central database holding such information about available resources, although there are very good examples of integrated activities within certain disease areas or countries, for example the Organisation of European Cancer Institutes (OECI) Pathobiology Working Group,369 with its maintenance of the OECI-TuBaFrost exchange platform, the String of Pearls Initiative in the Netherlands for CKD370 and specific large-scale more general and less disease-focused national initiatives in the USA, Japan, Iceland, South Korea and China, for example. 371 Similarly, examples of publications describing specific biobanks and including indicators of the quality of the samples are relatively few, with some notable exceptions, such as that from the Mayo Clinic in Arizona describing the RCC samples held within the Multidisciplinary Genitourinary Diseases Biospecimen Bank,372 and, hence, awareness of potential samples for validation purposes is limited. Encouragingly, given the importance of such resources, the impact of sample processing protocols, pre-analytical factors and quality on biomarker research is increasingly appreciated and the need for standardised approaches to processing, quality assessment and recording of critical variables through initiatives such as SPIDIA (standardisation and improvement of generic pre-analytical tools and procedures for in vitro diagnostics), BRISQ (Biospecimen Reporting for Improved Study Quality) and SPREC (Sample PREanalytical Code) are promoted. 363,373–375 Other aspects such as governance, ethical issues, patient involvement, ensuring long-term social, operational and financial sustainability and the importance of considering and collecting indicators of impact, all critical issues for funders as well as researchers, are also assuming a higher level of prominence. 371,376–380 Importantly, concerns over the issue of the underuse of samples have been raised in a survey of 456 biobanks in the USA and this adds weight to the importance of ensuring the visibility and accessibility of such resources to maximise their value and avoid the duplication of efforts. 381,382
Gradually, these issues these are being addressed. Another bottleneck is the lack of appropriately validated assays. Awareness of potential pitfalls in the level of validation of assays is increasing, including commercial assays, and the need for consolidation/harmonisation and the appropriate use of guidelines for validation is being highlighted. 383–393 Additionally, a disjoin in many cases between the ‘discovery’ researchers and laboratories and the test implementer laboratories, that is, the clinical biochemistry community with its wealth of experience in using assays and knowledge of the level of performance needed in a routinely available clinical test, has also contributed to the mismatch between expectations and delivery. Undoubtedly, the decline in academic activities of clinical biochemists in the UK, driven by the pressures to deliver an ever-increasing hospital workload and with little time to devote to biomarker-related research, has not helped the situation. 271,394 Thoughtful reviews by clinical biochemists and colleagues on biomarker progression also add the perspective of the final implementation in hospital laboratories, not just in terms of the need for evidence-based use but also in terms of the practical considerations important for routine adoption and the reimbursement considerations. 271,342,355
For the regulatory bodies there is a recognition of the need to be flexible and also to accelerate the approval processes, particularly with the increasing emergence of multiplex marker panels, but also in terms of retaining safety whilst not slowing drug development in the case of companion diagnostics. 353,395,396 Efforts by governments and funding bodies to increase the progress made in precision or stratified medicine generally and biomarkers specifically are evident from initiatives such as the development of the infrastructure required to help generate evidence on the clinical effectiveness and cost-effectiveness of a given in vitro test [e.g. MedTech and In vitro Diagnostics Co-operatives (MICs)397 and the Innovate UK Medicines Discovery Catapult398]. In the USA and spanning the whole of the biomarker pipeline, the recently established National Biomarker Development Alliance is a trans-sector initiative addressing the issues of a ‘dysfunctional and disjointed status of biomarker R&D [research and development]’ and aiming to develop widely accepted standards, best practices and guidelines through an ‘end-to-end systems approach’. 399,400
Establishing the pipeline in renal cancer and kidney transplantation
Renal cancer
Clinical context
Over one-third of a million cases of renal cancer are diagnosed annually worldwide, with > 143,000 deaths. 401 In the UK, it is the eighth most common cancer, with ≈ 10,000 new cases annually, and the incidence is increasing. For example, in the period between 1975–77 and 2009–11, rates have more than doubled, increasing by 132% overall and by 168% in women. 402 Occurring with a male-to-female ratio of 3 : 2, risk factors include smoking, obesity and hypertension. Most renal cancers (≈90%) are RCCs, cancers arising from the renal parenchyma, with the most common histological subtype (70–80%) being the conventional (clear-cell) RCC (ccRCC). Other main histological subtypes include papillary (10–15%), chromophobe (5–10%) and collecting duct (< 1%) tumours and benign oncocytomas (2%–5%), arising from various kidney cell types. Novel subtypes of RCC are continually being defined, largely on the basis of morphology, although genetic characterisation is providing further insights, with > 24 subtypes of renal cancer now included in the most recent Vancouver classification. 403–405 Subtypes have broadly differing clinical behaviour and underlying genetic changes although these are also heterogeneous and complex within a subtype. Major increases in our understanding of the underlying molecular genetic and epigenetic changes and the heterogeneity within the subtypes is leading to changes in the clinical management of patients, with the most marked impact currently being in the development of targeted therapies. 406,407
The majority of patients have few or no symptoms and a recent international prospective study involving 4288 patients with renal masses found that diagnosis was incidental in 67% of patients. 408 Approximately two-thirds of patients with RCC present with disease localised to the kidney and surgery or ablative therapies are the standard treatment. Although largely curative, 30–40% of patients will subsequently relapse. Renal cancer is inherently resistant to chemotherapy and radiotherapy and, for patients with metastatic disease or those at high risk and warranting adjuvant therapy, the increased biological knowledge has led to the rational design of therapies targeting specific pathways. In the majority (> 80%) of sporadic cases of the ccRCC subtype, the von Hippel–Lindau (VHL) tumour suppressor gene has been implicated. 409–412 A major role of the VHL protein relates to its role as a ubiquitin ligase, targeting proteins such as members of the transcription factor hypoxia-inducible factor (HIF) family for ubiquitination and subsequent proteasomal degradation. Loss of VHL protein function leads to accumulation of HIF with consequent transcriptional activation and upregulation of genes including that coding for vascular endothelial growth factor (VEGF). In the past 10 years, seven agents targeting VHL-related pathways either through kinase inhibition or antibody-based targeting, namely sorafenib (Nexavar®, Bayer, Leverteusen Germany), sunitinib (Sutent®, Pfizer New York, NY, USA), pazopanib (Votrient®, Novartis, Basel, Switzerland), axitinib (Inlyta®, Pfizer), everolimus (Afinitor®, Novartis), temsirolimus (Torisel®, Pfizer), bevacizumab (Avastin®, Genentech, San Francisco, CA, USA), cabozantinib (Cabometyx®, Exelixis Inc, San Francisco, CA, USA) and lenvatinib (Lenvima®, Eisai Co, Tokyo, Japan) (in combination with everolimus) have been approved for treatment of metastatic ccRCC. 406,413 Several other genes have also now been implicated along with epigenetic changes, which may lead to further insights into clinical behaviour and therapeutic exploitation. 404,414 Recent developments in immunotherapy with immune checkpoint inhibitors based on antibodies to cytotoxic T lymphocyte-associated protein 4, programmed cell death protein 1 and programmed death ligand 1 are also showing promise in RCC. 415 As studies start to define the underlying molecular changes in the non-clear-cell subtypes, it is likely that novel therapeutic targets may be found. 416–420 However, it is clear that no dominant driver gene equivalent to the VHL gene exists, although the mesenchymal–epithelial transition (MET) factor gene represents a promising possibility in papillary tumours. MET gene mutations are evident in only 12% of sporadic cases but copy number gain has been found in 81% and 46% of type I and type II papillary RCCs, respectively, and MET protein is overexpressed in up to 90% of cases. 419,421–423 A range of tyrosine kinase inhibitors with activity against MET, including crizotinib (Xalkori®, Pfizer), savolitinib (AstraZeneca, Cambridge, UK), capmatinib (Incyte Corporation, Wilmington, DE, USA) and volitinib (AstraZeneca), are currently undergoing clinical trial or clinical trials are planned.
Currently, no circulating biomarkers are routinely used in RCC although clear clinical needs have been identified and such developments are a priority research area. 424,425 Clearly, circulating biomarkers have the advantage of being accessible and of being measured relatively non-invasively and are able to provide information longitudinally, even prior to surgical or other treatment. Biomarkers could potentially have an impact on the patient pathway by enabling earlier diagnosis, determining prognosis for stratification of follow-up, detecting relapse and selecting patients for specific therapies by predicting response. A particular challenge, in terms of both diagnosis and prognosis, relates to the management of small renal masses (≤ 4 cm), up to 25% of which are benign, and the risks of surgery or ablative procedures, particularly in elderly patients with comorbidities, have to be balanced against the risk and time frame of progression of the tumour. 426,427
At the time of submission of the application for this programme grant there were few emerging diagnostic biomarkers or predictive biomarkers in RCC, although several prognostic biomarkers seemed to have apparent potential utility and, hence, the focus was on prognostic biomarkers. We have recently reviewed existing potential tissue and fluid biomarkers and there are now also some promising initial studies in the areas of diagnosis and prediction of response to therapy. 428 For example, urinary concentrations of the two proteins aquaporin-1 (AQP-1) and perilipin-2/adipophilin have been shown in several studies by the same group to be significantly elevated in patients with RCC compared with healthy, benign renal and surgical control subjects, declining post nephrectomy. 429–431 These findings have been extended to 720 patients undergoing CT scans for a variety of indications using a newly developed ELISA for AQP-1, in which a ROC AUC of 0.99 was achieved, although elevated concentrations were also seen in other malignancies. 432 Clearly, these and some other markers show promise, but lack of the necessary level of evidence in terms of numbers and sizes of studies, independent validation and availability of robust assays precludes a focus on exploring these further within this programme at present. However, it is anticipated that such biomarkers may be the subject of future studies utilising the sample banks assembled within this programme.
Determining prognosis in renal cell carcinoma
The ability to stratify patients according to risk is highly desirable. Detection of relapse in patients with localised disease is based on repeated imaging, in some cases with biopsy. This is expensive and has the associated risks of cumulative radiation exposure and the potential morbidity associated with the biopsy procedure. Although postoperative surveillance protocols have been developed, detection of relapse is often not optimal and the ability to rationally guide the surveillance regimens on an individual basis and also to identify high-risk patients for adjuvant therapy has obvious benefits. 433 A critical determinant of a patient’s prognosis is the stage of the cancer and currently this is determined using the TNM (tumour, node, metastasis) system based on the extent of the primary tumour (T), whether or not local nodes are affected (N) and whether or not metastatic disease is present (M). 434,435 Other recognised independent clinicopathological prognostic factors include tumour grade and the presence of necrosis and various prognostic models or algorithms have been developed incorporating these and other factors. 436 The problems with these include the subjective nature of some elements such as tumour grade and the grouping of patients into a limited number of risk groups, meaning that estimates of risk can be wide for individual patients. 437 This is exemplified in the widely used system for patients with localised disease developed at the Mayo Clinic [Stage, Size, Grade and Necrosis score (SSIGN)], which integrates pathological T stage, N stage, tumour size, nuclear grade and tumour necrosis. 438 This divides patients into low-, intermediate- and high-risk groups with estimated 5-year metastases-free survival rates of 97.1%, 73.8% and 31.2%, respectively. However, the challenge is being able to further stratify these patients, particularly those placed into the intermediate-risk category.
For patients with metastatic disease, the most widely used clinical prognostic model is that proposed by the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC), which similarly groups patients into risk categories depending on the number of poor prognostic features present (Karnofsky performance status of < 80%, < 1 year from diagnosis to treatment, anaemia, hypercalcaemia, neutrophilia and thrombocytosis). 439 For the favourable-, intermediate- and poor-risk groups, median OS of 43.2, 22.5 and 7.8 months, respectively, has been reported. 440
Preoperative nomograms that do not include histopathological features but that are based around parameters such as age, sex, symptoms and CT-determined tumour size, T stage and metastasis have also been developed, with the advantage of potentially being used to determine optimal treatment strategies both surgically and in terms of neoadjuvant therapies. 441 Tumour expression of selected proteins such as B7 homolog 1 (B7-H1) protein, survivin and Ki-67 has been shown to have independent prognostic significance for cancer-specific survival (CSS), either alone or within an algorithm ‘BioScore’,442 and to add prognostic value to established clinicopathological models, including the Mayo Clinic SSIGN score. 438 Other similar examples with prognostic utility include Ki-67, p53, endothelial vascular endothelial growth factor receptor-1 (VEGFR-1), epithelial VEGFR-1 and epithelial vascular endothelial growth factor D combined with performance status and T stage and, in metastatic RCC, carbonic anhydrase IX (CAIX), phosphatase and tensin homologue, vimentin and p53 combined with T stage and performance status. 443,444 Recent genomic and transcriptomic studies are adding to this, for example a 34-gene classifier, ClearCode34,445 assigns patients to good-risk (ccA) and poor-risk (ccB) groups and a 16-gene signature categorises patients into low-, intermediate- and high-risk groups. 446
In terms of circulating biomarkers, several routinely measured haematological and clinical biochemistry factors, including the neutrophil–lymphocyte ratio (NLR), thrombocytosis, haemoglobin and serum sodium, calcium and C-reactive protein (CRP) levels, have been found to be prognostic, as have several more directly tumour-related proteins, including VEGF and CAIX – attractive possibilities in terms of their ease of measurement, even preoperatively (reviewed in Chapter 12). Several of these have sufficient evidence to justify their exploration in a large-scale programme of this type and in particular to determine whether or not, as a multiplex panel or in combination with additional clinicopathological parameters or models, they could provide superior performance to that of existing models.
Kidney transplantation
Clinical context
Chronic kidney disease affects approximately 8–16% of the adult population, with this figure increasing dramatically, at least in part linked to global epidemics in diabetes mellitus, hypertension and obesity. 447–449 It is estimated that 2–4% of people with CKD will eventually develop end-stage kidney disease (ESKD) and require renal replacement therapy (RRT; dialysis or transplantation), with 2.6 million people receiving RRT in 2010 and a similar number being unable to access it and dying prematurely. 447 Kidney transplantation represents the gold standard treatment for patients with ESKD, providing improved quality of life and survival compared with commencing dialysis. 450–452 A functioning kidney transplant is able to re-establish many of the important functions that the kidneys perform beyond what dialysis can deliver. For example, the kidneys have important endocrine functions in terms of the regulation of blood pressure and producing erythropoietin (EPO) (stimulates red blood cell production) and activated vitamin D3 in addition to controlling electrolyte, acid/base and fluid balance. It has also been recognised that kidney transplantation is of economic benefit to society compared with treatment with dialysis. 453,454 In the first year the cost of kidney transplantation is equal to that of haemodialysis; however, in subsequent years the cost is halved. The average cost of maintaining a patient with ESKD on dialysis is £30,800 per year. The cost of a kidney transplant is £17,000 per patient per transplant, leading to a cost benefit in the second and subsequent years of £25,800 per annum. At the end of March 2009, > 23,000 people in the UK had a functioning kidney transplant, saving the NHS over £512M for that year in costs for dialysis that would have been needed if these patients did not have a functioning kidney transplant. 453 At a time of financial pressure in all health-care systems, this is an important aspect to consider.
There are a number of different sources of kidneys for transplantation. The current classification can be broadly divided into deceased donor and living donor kidney transplantation, depending on the source of the donor organ. 455,456 Deceased donation is further classified as donation after brain death (DBD) and donation after circulatory death (DCD). Kidneys donated after a circulatory death experience a longer period of warm ischaemia (reduced blood flow) prior to surgical retrieval than kidneys donated after brain death. The length of time between the surgical removal of the donated kidney and its implantation into the recipient is a major factor in determining whether there is immediate kidney function or delayed kidney function, referred to as DGF. Therefore, deceased donation is associated with a higher rate of DGF than living donation and DCD kidney transplantation has a higher rate of DGF than DBD kidney transplantation. Living donor kidney transplants are characterised as being genetically related between parents and children or between siblings (living related) or genetically non-related between husband and wife, partners and friends (living unrelated). More recently, there has been an increase in the number of altruistic donors who wish to donate a kidney to anyone in society who is on the kidney transplant waiting list.
The need for kidney transplantation is far greater than the availability of organs. In 2012 there were 2998 kidney transplants performed in the UK; however, there are > 6000 people on the UK kidney transplant waiting list. 457 The reality is that people will die unnecessarily whilst on the waiting list for a kidney transplant. Because of the ongoing shortage of kidney transplants the average waiting time for a deceased donor kidney transplant is 2–3 years. There have been a number of initiatives to increase the number of organ donors. These initiatives have included the recently introduced opt-out system in Wales, where the assumption is that people are willing to donate unless they opt out, and accepting kidneys from expanded-criteria donors (ECD). 456 ECDs are defined as those aged > 60 years or aged ≥ 50 years with two of the following: a terminal serum creatinine level of ≥ 1.5 mg/dl (132 µmol/l), a cerebral vascular accident as the cause of death or a donor history of hypertension. Patients receiving ECD kidney transplants are consequently at a higher risk of developing DGF. Pooled living kidney donation has been introduced to allow pairs of kidney donors and potential recipients who could not donate to one another because of immunological barriers to be matched with other pairs in the UK to whom they could donate. 455 Further advances in medicine have allowed kidney transplantation to occur across blood group and human leucocyte antigen (HLA) incompatibility. 458,459 The government has recognised the importance of kidney transplantation and has increased the number of nurses specialised in organ donation to help identify more potential donors.
Unfortunately, kidney transplants have a finite lifespan. A number of factors determine how long a transplanted kidney lasts. 455,456 These include donor age and the nature of the donation, for example living donation compared with deceased donation, immunological matching with respect to blood group and HLA compatibility, baseline function of the donated kidney, immediate graft function compared with DGF, the primary kidney disease and the health of the recipient. Overall, the average kidney transplant survival times are about 95% in the first year, 85–90% at 5 years and 75% by 10 years. 460 Living donor kidney transplants have better long-term outcomes than those from deceased donors. This is in part because of the controlled nature of the donor assessment and in part because of the reduced ischaemic time and subsequent DGF.
Following kidney transplantation a number of complications can occur. The early complications are those common to any surgical procedure, including postoperative chest or wound infection. 461 Early postoperative complications that are specific to kidney transplantation include renal vein thrombosis, DGF, AR and calcineurin inhibitor toxicity. All of these complications result in a failure of the transplanted kidney to function effectively. Renal vein thrombosis is detected on renal tract ultrasound scanning and is rarely reversible, with devastating consequences. A kidney transplant biopsy is required to distinguish between DGF, AR and calcineurin inhibitor toxicity. This procedure is not without risk and is resource intensive. Non-invasive diagnostic techniques are needed to allow rapid diagnosis and appropriate treatment.
Delayed graft function
Overall, DGF has a higher risk of mortality, transplant loss, need for dialysis, kidney biopsy and increased hospital stay. 462,463 Management strategies promoting renal function post transplant have major implications for patient benefit and resource savings. 464 DGF is secondary to the ischaemia–reperfusion injury (IRI) that follows retrieval of the kidney from the donor, with attendant loss of perfusion, and implantation into the recipient, with restoration of perfusion. During this, the kidney is stored on ice and transported to the designated kidney transplant unit. This period of time is referred to as the cold ischaemic time (CIT). Generally speaking, the longer the CIT the more likely the kidney will experience IRI, which is manifest clinically as DGF.
The multiple definitions of DGF have hindered the ability to characterise its incidence and outcomes. As many as 18 definitions have been used in the literature. 465 The most common definition proposed has been the receipt of dialysis within 7 days following transplantation. 462 However, this is still a crude definition and holds back the opportunity to stratify different degrees of IRI and outcomes. Within this programme, any analysis has used the following definitions and the creatinine reduction ratio [CRR = (day 0 serum creatinine – day 7 serum creatinine)/day 0 serum creatinine]: immediate graft function, in which there is a significant and sustained fall in serum creatinine within the first 48 hours (CRR ≥ 0.7), slow graft function, in which serum creatinine fails to fall significantly in the first 48 hours but the patient does not receive dialysis in the first week (CRR < 0.7) and DGF, in which the patient receives dialysis in the first week except for isolated hyperkalaemia.
Delayed graft function ranges from 5% to 50% following deceased donor transplantation and depends on many factors. 466,467 Not surprisingly, DGF is significantly lower in live donations, at 4–10%. 468 As previously described, the variations in the reported incidence will depend on the definition of DGF that has been used. 469 The United Network for Organ Sharing database470 recorded a rate of DGF in US patients of 21.3% in early 2011. A number of donor and recipient factors have been identified as contributing to the development of DGF. There has been an attempt to develop risk calculators for DGF. 471,472 These factors can be categorised as recipient-related factors, including sex, ethnicity, previous transplantation, presence of diabetes mellitus, HLA mismatch, peak panel reactive antibodies, previous blood transfusions, body mass index (BMI), duration of dialysis before transplantation, or donor-related factors, including donor age, CIT, warm ischaemic time (WIT), DBD compared with DCD, presence of hypertension, baseline serum creatinine level, cause of death and weight. 462,471–475
Delayed graft function is extremely important as an independent risk factor for both early and late kidney transplant loss and higher mortality. 22,476,477 DGF lasting for > 6 days strongly decreases long-term transplant survival. 24 It has been proposed that the kidney donor type may be more important than current DGF definitions in understanding the impact of DGF on longer-term outcomes. This is corroborated by the fact that the prediction of poor outcomes associated with DGF is largely independent of many of the definitions in use and provides further support for the stratified approach to defining DGF that is proposed. 469 It has been shown that the severity and duration of native acute kidney injury (AKI) secondary principally to IRI predicts the risk of CKD. 478 There are obvious parallels between DGF and AKI but with the caveats that there are other factors at play, such as immunological responses and immunosuppressant medications. 462
Delayed graft function is associated with other adverse outcomes such as AR. 23 AR is more likely to occur during an episode of DGF because of increased exposure of donor epitopes and has a significant impact on kidney transplant survival. 479 It is, therefore, important to identify the underlying cause of the DGF following kidney transplantation. It is usual to perform a kidney transplant biopsy to confirm the histological diagnosis of tubular injury from IRI and exclude the possibility of AR. The clinical management of DGF requires close attention to detail, with appropriate management of patients’ medication, acid/base, electrolyte and volume status. There can be a risk of volume overloading of patients, particularly if they are oliguric. The dose of specific immunosuppressants, for example calcineurin inhibitors, is usually lowered in the setting of IRI because of their vasoconstrictive effects, whereas in the case of AR the dose of immunosuppressants is increased.
In summary, the early identification of DGF and the specific underlying pathology has significant potential to improve immediate patient management, allowing fluid volume status optimisation and timely appropriate dialysis and the avoidance of unnecessary investigation and treatment. 480 With the increased use of donor kidneys from deceased donors and ECDs to meet the demand for transplants there is a concurrent need for new biomarkers to improve the assessment of the quality of donated kidneys prior to transplantation and enable more objective decisions to be made about viability. The opportunity to stratify patients and identify those with significant IRI may allow the individualisation of immunosuppressive regimens (e.g. the avoidance of or use of lowered doses of calcineurin inhibitors) at an earlier time point, which may in turn result in improved longer-term outcomes. Similarly, improved monitoring of kidney function or prediction pre or post transplant, earlier diagnosis of early postoperative complications such as DGF or AR or longer-term prognostic information would allow earlier intervention, avoidance of biopsies and tailoring of longer-term immunosuppression. Efforts to identify novel biomarkers of DGF to improve the use of serum creatinine are increasing and potential new biomarkers are reviewed in Chapter 12.
Overview of the work undertaken in the clinical translation workstream to develop and use these pipelines
An overview of the activities and key deliverables of workstream 2 is shown in Figure 26.
FIGURE 26.
An overview of workstream 2. Key activities and deliverables are shown together with the inter-relationships with workstream 1 and workstream 3. NEQAS, National External Quality Assessment Service.

Sample banks and clinical data
Essentially to overcome the inherent inertia in the later stages of the biomarker pipeline, where often readily available sample sizes are inadequate to provide sufficient statistical power, sample collection has been inconsistent, clinical data may be incomplete and follow-up time is not mature enough to produce enough events, a key element of workstream 2 was the assembly of fluid samples together with associated clinical data, including long-term follow-up data, from cohorts of patients with CLD, renal cancer or ESKD undergoing RT, the last two being major areas of interest in Leeds. This will provide a valuable underpinning resource for this programme and future collaborative biomarker studies aimed at evaluating whether or not specific putative novel biomarkers are likely to benefit patients and health services. This has been achieved through the RCT in workstream 3 for liver diseases using cross-sectional sample collection and through involving centres across the local research networks in longitudinal and cross-sectional sampling for RT and renal cancer (10 and 11 centres, respectively) and is described in detail in Chapter 11. In total, this has involved 1967 patients and 149 healthy volunteers and 5976 sample time points, with many of these sample time points also including multiple sample types being banked such as serum, plasma and urine and each sample being stored in multiple aliquots to maximise future use. Standard operating procedures (SOPs) were used for the collection and processing of samples and data were collected using specific case report forms (CRFs), managed by the Leeds Clinical Trials Research unit (CTRU), with samples shipped to Leeds and ultimately stored within a licensed research tissue bank (RTB) to maximise future use and benefits. The theoretical advantages to this and the level of stringency are, thus, similar to those of a clinical trial (Figure 27).
FIGURE 27.
Schematic of the integrated approach adopted within the programme to sample banking and clinical data collection. GCLP, Good Clinical Laboratory Practice; HTA, Health Technology Assessment.

These prospective samples provide the ability to evaluate biomarkers in the following contexts:
-
CLD – identification of patients at risk of the subsequent development of cirrhosis and major liver events such as HCC
-
renal transplantation – prediction/earlier diagnosis of patients with acute post-transplant complications such as DGF or AR and prognostic stratification for long-term outcomes
-
renal cancer – prognostic stratification and longitudinal monitoring with other possibilities including diagnosis and prediction of response to therapy
-
healthy control subjects – enabling determination of reference ranges and effects of factors including sex, age, ethnicity and diet.
Biomarker prioritisation, assay validation and evaluation of clinical utility
Focusing on the renal diseases, the identification of potential biomarkers to evaluate currently was based on our ongoing discovery activities relating to novel biomarkers, a systematic review of the relevant literature, which is the focus of Chapter 12, and approaches from other groups. A certain level of evidence must have existed already (e.g. significant independent association with prognosis), with selected biomarkers already having been the subject of initial validation studies and agreed by a subpanel of applicants to be of suitable scientific quality. When promising biomarkers exist but with a lower level of evidence or requiring further technical assay validation, further evidence has been sought in some cases using our own local Leeds multidisciplinary RTB. This phased approach ensures independent validation (or failure to validate) whilst conserving the NIHR programme-related samples for final multicentre validation studies and ultimately biomarkers emerging from this would be the subject of future clinical trials similar to that described in workstream 3. This strategy is illustrated in Figure 28.
FIGURE 28.
The phased approach to biomarker validation with evidence-based progression and utilisation of sample banks depending on the stage of the biomarker. HTA, Health Technology Assessment; LTHT, Leeds Teaching Hospitals NHS Trust; RfPB, NIHR Research for Patient Benefit programme.
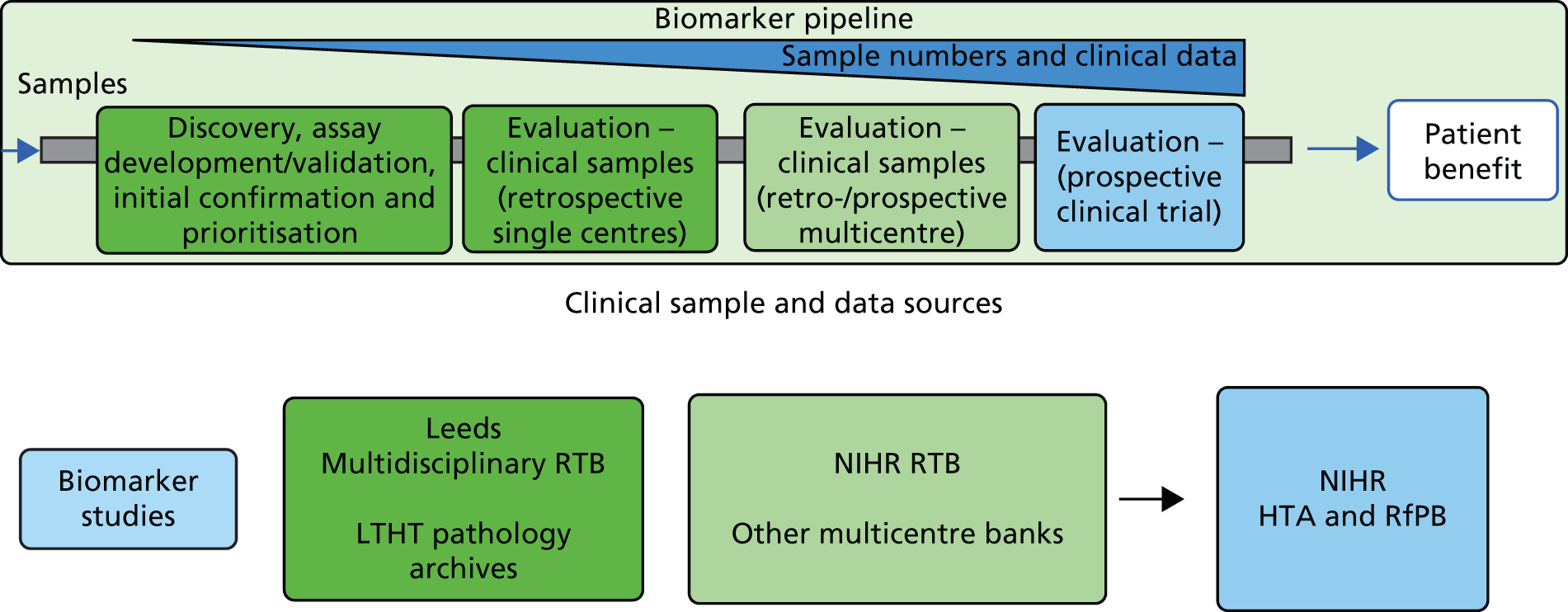
Within the time frame of this programme, assays for several potential biomarkers prioritised following systematic review have been systematically evaluated and validated, including in some cases analysis of specific pre-analytical aspects, as described in Chapter 13. Following this the prognostic use of serum and plasma VEGF, plasma osteopontin (OPN) and CAIX and serum CRP, alone or in combination, together with extensive clinicopathological variables, have been explored in RCC and this is reported in Chapter 14. In RT, suitable promising biomarkers have been similarly prioritised and, once the outcome data are mature enough in the next 2–3 years, appropriate studies will be taken forward.
Additional deliverables
The deliverables relating to the activities described above are summarised in Chapter 15, additionally highlighting others including industry partnerships, academic collaborations, generation of intellectual property, the additional utilisation of the RTB and plans for long-term sustainability of the resources generated.
Chapter 11 Establishment of multicentre prospective observational cohorts with sample banks for biomarker validation
This chapter describes the development of three multicentre prospective observational cohorts, with high-quality biospecimens, in RCC, RT and liver disease. These cohorts have been established to enable the rapid clinical validation of new biomarkers, as exemplified in Chapter 14. In addition to providing a summary of the final cohorts, this chapter details and discusses the issues pertaining to study design, management and governance, providing some generalisable learning for future researchers establishing similar resources.
Disease areas
Patients were recruited to provide samples and data for future biomarker research from the major diseases under study: RCC and RT, as described in the Chapter 10, and liver disease, through linkage to workstream 3 and the clinical trial. A cohort of healthy control subjects was also recruited to determine reference ranges and potential biological or technical confounding factors such as age, sex and length of storage. Over the duration of the programme the three studies recruited 2216 participants in total and 5976 samples. These included:
-
847 liver disease patients (847 serum samples)
-
514 patients on the transplant waiting list, including 312 who were subsequently transplanted (3806 samples, with each sample including multiple aliquots of serum, plasma and urine)
-
706 RCC patients (1132 samples, with each sample including multiple aliquots of serum, plasma, buffy coat and urine)
-
149 healthy volunteers (191 samples, with each sample including multiple aliquots of serum, plasma and urine).
Renal cell carcinoma
Objectives and end points
The primary objective for establishing this cohort was to provide prospectively collected high-quality clinical samples and data from multiple centres to validate the prognostic and longitudinal monitoring biomarkers of RCC. The end points for such studies would be to determine the association between these markers and outcome [disease-free survival (DFS), CSS and OS] and their ability to detect relapse, when measured longitudinally.
Eligibility criteria
Renal cell carcinoma inclusion criteria
-
Newly diagnosed suspected RCC (all stages).
-
All histological types of RCC.
-
No prior treatment for renal cancer.
-
Ability and willingness to provide written informed consent.
-
Ability and willingness to co-operate with study procedures, including blood and urine sampling.
-
Age ≥ 18 years.
Renal cell carcinoma exclusion criteria
-
Diagnosed familial RCC, for example VHL syndrome.
-
Renal cancer acquired following/during renal dialysis.
-
High risk of or known HIV/AIDS, hepatitis B virus (HBV) infection or HCV infection or similar infectious diseases.
Healthy volunteer inclusion criteria
-
Able to provide consent.
-
Willingness to co-operate with study procedures.
-
Age ≥ 18 years.
Health volunteer exclusion criteria
-
History of any cancer.
-
High risk of or known HIV/AIDS, HBV infection or HCV infection or similar infectious diseases.
-
History of diagnosed renal disease.
-
Current/recent (within the last 3 months) urinary tract infection (UTI).
Study design
A multicentre prospective observational cohort design for retrospective blinded biomarker validation was adopted. 268 Blood and urine samples were requested from eligible patients diagnosed with suspected RCC attending 11 participating centres (Table 66), according to the study site operating procedure (SSOP), as summarised below:
-
cross-sectional study (target of n = 500 prior to surgery or other treatment) –
-
a single baseline blood sample (12–18 ml for serum, plasma and buffy coat)
-
mid-stream urine sample
-
-
longitudinal monitoring study (target of n = 200 prior to nephrectomy) –
-
two separate baseline blood samples (for serum, plasma and buffy coat)
-
two separate baseline urine samples
-
additional blood and urine samples at 3–6, 12, 18 and 24 months post nephrectomy, ceasing at relapse if earlier, with a sample taken at that time.
-
| Site code | Hospital |
|---|---|
| N0000015 | Charing Cross Hospital – Imperial College Healthcare NHS Trust (London) |
| N0000039 | Nottingham City Hospital |
| N0000050 | St James’s University Hospital (Leeds) |
| N0000069 | Freeman Hospital (Newcastle) |
| N0000131 | Lister Hospital – East and North Hertfordshire NHS Trust |
| N0000132 | Northwick Park Hospital (London) |
| N0000153 | Churchill Hospital (Oxford) |
| N0000221 | Stepping Hill Hospital (Stockport) |
| N0000352 | University Hospital of Wales (Cardiff) |
| N0000361 | Western General Hospital (Edinburgh) |
| N0000537 | Gartnavel General Hospital (Glasgow) |
For each patient undergoing nephrectomy, a representative formalin-fixed paraffin-embedded (FFPE) block of tumour tissue was also collected. This was not included in the programme grant application and was funded from elsewhere but provided added value to the sample collection through enabling multiple centres to participate in tissue-based studies in the future, given that the clinical data were already being collected. In the Leeds Teaching Hospitals NHS Trust, frozen tissue was also stored as this was being undertaken routinely already as part of local research.
Cross-sectional blood and urine samples were also collected from healthy volunteers across the different centres (target of n = 200; relatives, hospital staff, etc.) to allow the determination of biomarker reference ranges and the effects of factors including sex, age, ethnicity and diet.
The study schema is outlined in Table 67.
| Activity | Time point | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Screening | Pre treatment (baseline) | Nephrectomy | 3–6 months | 12 months | 18 months | 24 months | 36 months | 48 months | 60 months | Relapse | |
| Consent | ✗ | ||||||||||
| Eligibility | ✗ | ||||||||||
| Cross-sectional cohort (n = 500) | |||||||||||
| Blood samplea | ✗ | ||||||||||
| Urine sample | ✗ | ||||||||||
| Longitudinal cohort (n = 200) | |||||||||||
| Blood samplea,b | ✗ (2) | ✗ | ✗ | ✗ | ✗ | ✗c | |||||
| Urine sampleb | ✗ (2) | ✗ | ✗ | ✗ | ✗ | ✗c | |||||
| Tissue sample | ✗ | ||||||||||
| Demographics | ✗ | ||||||||||
| Risk factors | ✗ | ||||||||||
| Symptoms | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Radiology data | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Pathology data | ✗d | ✗e | |||||||||
| Sites of disease at baseline and relapse | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Treatment details | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| ECOG PS | ✗ | ||||||||||
| Comorbidities | ✗ | ||||||||||
| Concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Haematology and biochemistry | ✗ | ✗b,f | ✗b | ✗b,f | ✗b | ||||||
| Survival assessment | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
Statistical considerations
The design and analysis/reporting of many prognostic marker studies has been criticised482–484 and we conformed to REMARK guidelines. 367 There are many imponderables involved in calculating the sample size for biomarker studies. Patient recruitment in terms of numbers has been powered based on a consideration of our experience of ongoing marker analysis, for example cathepsin D, for which a 15% difference was seen between groups at 2 years,485 and CRP, for which the difference was 40–50%. 486 Based on relapse rates ranging from 12.5% to 27.5% of the population diagnosed with ccRCC undergoing nephrectomy or ablation, we have modelled numbers providing from 80% power to 90% power to correctly separate prognostic groups. A marker with 15% separation between the groups would require 216–380 patients in total to provide a power of 90% assuming that event frequencies range from 12.5% to 27.5% at 2 years, whereas a marker with 40% separation would require a total of 72 patients to provide the same power. Given the projected figure of 400 ccRCC patients in total, these targets are easily met. Cox multivariate proportional hazards model analysis will be used to identify multiple marker combinations, with significance levels adjusted for the number of variables included. About four times as many patients are required to detect interaction affects for a pair of dichotomous variables as for an individual variable, so only combinations of markers with relatively large effects are likely to be identified definitively. Simulations and sensitivity analyses will be performed to validate and confirm significance levels for such analyses.
For the longitudinal monitoring, the methodology for evaluating biomarker test performance is more poorly developed. Experience suggests that, with up to five marker values throughout the follow-up period for 200 patients, we would have adequate numbers to show the predictive capacity of relevant markers in this setting and to develop mathematical models, as appropriate, to enhance our understanding of the disease process in these patients. 487
Renal transplant
Objectives and end points
The primary objective for establishing this cohort was to validate biomarkers for use in the monitoring and diagnosis of early/acute kidney transplant complications. The secondary objective was to validate biomarkers for use in predicting patient outcomes and loss of transplant function. The end points of such studies would be to determine the association between the concentration of these biomarkers and the diagnosis or prediction of disease/outcome.
Eligibility criteria
Inclusion criteria
-
All sites: active patients on the renal transplant waiting list.
-
Ability and willingness to provide written informed consent.
-
Ability and willingness to co-operate with study procedures, including blood and urine sampling.
-
Age ≥ 18 years.
Exclusion criteria
-
High risk of or known HIV/AIDS, HBV infection or HCV infection or similar infectious diseases.
-
Patients in the custody of Her Majesty’s Prison Service.
Study design
A multicentre prospective observational cohort design for retrospective blinded biomarker validation was adopted. 268 Blood and urine samples were requested from eligible patients attending participating centres who were on the waiting list for RT. The 10 centres participating in this study, shown in Table 68, were to recruit up to 850 renal transplant patients from the waiting list, aiming for a target of 300 deceased donor and 40 live transplantations within the recruitment period of the study. The study was focused on patients receiving deceased donor kidney transplants because of the increased risk of DGF and chronic transplant dysfunction longer term and, therefore, on providing sufficient samples and events for the assessment of biomarkers.
| Site code | Hospital |
|---|---|
| N0000046 | Royal Liverpool University Hospital |
| N0000050 | St James’s University Hospital (Leeds) |
| N0000065 | York Hospital |
| N0000069 | Freeman Hospital (Newcastle) |
| N0000078 | Hull Royal Infirmary |
| N0000110 | Queen Alexandra Hospital (Portsmouth) |
| N0000118 | Derriford Hospital (Plymouth) |
| N0000230 | Southmead Hospital (Bristol) |
| N0000232 | Northern General Hospital (Sheffield) |
| N0000299 | St Luke’s Hospital (Bradford) |
Blood and urine samples were to be obtained following consent, whilst on the waiting list. When possible, a second baseline sample was to be collected immediately pre transplant. Samples were then collected daily during the first week of the hospital stay (approximately five samples) and then at the following intervals post discharge: weekly for 1 month and then at 2, 3 and 6 months. The study schema is outlined in Table 69.
| Activity | Time point | ||||||
|---|---|---|---|---|---|---|---|
| Screening | Pre transplant | Daily during week 1 of hospital stay | At discharge from hospital | Weekly for next month | 2, 3 and 6 months post discharge | Annually for up to 5 years | |
| Consent | ✗ | ||||||
| Eligibility | ✗ | ||||||
| Research samplesa | |||||||
| Blood sampleb | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Urine sample | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Clinical data | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Follow-up data | ✗ | ✗ | ✗ | ||||
Statistical considerations
The frequency of sampling used in this study was designed to enable the clinical validation of biomarkers of potential use for distinguishing between DGF and AR, detecting DGF or rejection earlier and potentially predicting chronic kidney transplant dysfunction and patient outcomes. At the time of the application it was possible to provide only approximate estimates of sample sizes for this proposal. First, several different patterns could be examined in the data that may predict DGF, AR and later clinical or subclinical dysfunction, for instance a sudden rise in marker levels, marker levels elevated over time, a slow steady rise or a sharp rise followed by a sudden drop. Second, normal variability in putative markers is uncertain and will have to be determined for each individual biomarker. With 20 samples per patient, a slow steady rise, which is likely to be the most difficult pattern to distinguish, can be estimated fairly accurately. Preliminary simulations suggest that, in this case, assuming a linear increase in marker levels, with a ± 10% ‘measurement’ error (as a result of patient, pre-analytical and analytical factors), we could clearly detect a 10% increase over 6 months. Rates of increase can then be used as predictors of DGF, AR and later rejection/dysfunction. We have experience with alternative models, which can be accommodated with similar magnitudes of variability. Assuming that we have accurate parameters from such models, they can be used to predict DGF, AR or later clinical or subclinical rejection. Such predictions need to be highly discriminatory to make them clinically useful, for instance to be able to differentiate between patients with a 10% chance of DGF ± AR and patients with a 40% chance of DGF ± AR. This requires sample sizes of approximately 100 patients (with 90% power and a 5% false-positive rate), assuming that the model parameters are clearly estimated with a small standard error. We also have experience with other projects involving serial measurements, in which sample sizes of the order of 200 patients, and multiple measurements over time, were sufficient to enable us to distinguish clinically meaningful effects (see, for example, Migdal et al. 488).
Recognising the variation arising from multiple underlying aetiologies and that associated with different biomarkers (neither sources being readily quantifiable), we set out to recruit 340 patients over 6 months, with between 10 and 20 samples, to ensure that any such variation was accounted for.
Liver disease
Samples from patients with liver disease were obtained as part of the main ELUCIDATE trial (see Chapters 16–23). From the initial 878 randomised patients, 847 consented to take part in the translational research aspect of the trial and provided a single blood sample in a plain serum clot activator tube at baseline. The serum was then processed according to the ELUCIDATE SSOP, stored locally at –80°C and shipped on dry ice to the RTB in Leeds for storage. Patients were followed up and data collected as specified within the ELUCIDATE protocol. A detailed summary of this cohort is presented in Table 79.
Study management
Investigator authorisation
Patients were recruited once all appropriate authorisations for the centres were granted and the appropriate regulatory paperwork had been collected. This included but was not limited to:
-
investigator contact details, including contact details for research nurses
-
an up-to-date, signed and dated curriculum vitae for each individual on the signature and responsibilities log, including dates of Good Clinical Practice (GCP) training
-
written confirmation of local trust R&D approval for the study
-
a copy of the site-specific information (SSI) form, signed by the principal investigator
-
a signed principal investigator declaration
-
authorised signature logs.
Recruitment
Patients were recruited from UK centres, with slight overlap between our selected disease areas. For example, two of the 11 centres recruiting patients for the RCC study were also among the 10 centres recruiting patients for the RT study. Research centres were required to have completed a feasibility assessment, obtained local NHS management approvals and undertaken a site initiation meeting with the study/trial management team prior to the start of recruitment. The Leeds CTRU was responsible for the monitoring of patient recruitment and consent and also for the co-ordination and storage of clinical data and logging of sample collection information, whereas details of the actual samples received were maintained within our local laboratory information management system SENTRY.
Patient consent
Patients at each site were approached by clinicians or research nurses, trained in taking informed consent, and provided with study-specific patient information sheets regarding the study and consent forms. In all cases the information in the patient information sheet was explained fully to patients and they were given the opportunity to ask any questions. In the renal transplant study, some centres sent patients a letter along with the patient information sheet prior to them coming to the clinic, informing them of the research study and giving them time to consider their participation. The right of a patient to refuse consent without giving a reason was respected. Whenever possible, informed consent was obtained during a hospital visit before samples were obtained. However, because of the patterns of referral of many of these patients this was not possible in many instances. In such cases, a full explanation of the study was provided and patients were given as much time as they needed to make a decision. Healthy participants were also recruited at each centre by approaching visitors and hospital staff.
Broad consent was sought to store and use patient data and samples for this project and also for subsequent unspecified studies, given the intended storage within a RTB, if residual tissues and fluids remained, with examples of the types of subsequent studies provided in the patient information sheet. Patients were specifically asked to opt in or out, using a multilevel consenting procedure for more sensitive aspects of the research, including whole-genome sequencing studies; permission to contact their general practitioner regarding clinically relevant findings; permission to contact relatives regarding clinically relevant findings; and studies conducted by other groups and commercial partners.
No specific additional risks were involved for patients participating in this study as whenever possible blood samples were obtained at the time of routine venepuncture and no surgical procedures or treatment other than those planned as part of their standard treatment were received as part of the study.
Should a patient have required a translation of the study documentation, it was the responsibility of the individual investigator to translate the patient information leaflet and the consent form, using locally approved translators. The consent form then had to be appropriately signed and dated by the patient, the investigator and the translator.
Original copies of the consent form were retained in the investigator site file; a copy of the consent form was given to the patient, a second copy was filed in the patient’s health-care records (as per local practice) and a third copy was returned to the Leeds CTRU.
All patients were free to withdraw from the study at any time at their own request, without prejudice; they could also be withdrawn from the study at any time at the discretion of the investigator. Unused samples and data would, after the notice of withdrawal, be disposed of securely and respectfully. Part discontinuation or withdrawal involved using a subject’s samples already obtained up to the point of withdrawal. Full discontinuation or withdrawal involved having the subject’s samples destroyed. This required breaking the study code so that anonymised samples could be identified. In the event that analysis had already been performed on the sample(s), requests to destroy molecular data could not be honoured because the associated data could be required for audit purposes by a regulatory authority. Additionally, the sample may have been pooled with other samples such that it would not be possible to isolate a particular sample from the pool, and the pool could continue to be analysed until it was used up. A standard letter for withdrawing consent was included at recruitment alongside the patient information sheet.
Patient registration
Once a patient was confirmed as being eligible for the study and had given written informed consent, the investigator or designee completed the supplied registration CRF and contacted the Leeds CTRU’s 24-hour registration telephone line. The CTRU recorded basic patient details (date of birth, initials and confirmation of consent) and then allocated a unique trial number to the patient; this trial number was then recorded in the patient’s medical notes as well as on all study CRFs. Confirmation of the patient’s registration was then faxed/e-mailed back to the participating site and the original registration and eligibility form was sent to the CTRU by post.
Data collection and storage
Data collection was managed by the Leeds CTRU. Relevant clinical and demographic data were collected by research nurses and clinicians on standardised CRFs, appropriate to the participant group. These were then copied and the originals submitted to the CTRU and held securely in paper and electronic form. Participating sites were expected to maintain a file of essential study documentation and to keep copies of all completed forms for at least the duration of the study. All information collected during the course of the study was kept strictly confidential and the CTRU ensured compliance with all aspects of the Data Protection Act 1998. 481 On completion of the study, data will eventually be transferred to the Medical Research Council Informatics Centre in Leeds and will be held securely in compliance with all aspects of the Data Protection Act 1998 for a minimum of 10 years. Site files will be archived by the participating NHS trusts for 10 years and arrangements for confidential destruction will then be made.
Sample collection and storage
Blood and urine samples were processed promptly at each centre by either research nurses or staff of the clinical chemistry laboratories, according to SOPs (see Appendix 1); samples were frozen in multiple aliquots at –80°C until shipped. Details about the time of sampling and processing were recorded. Tissue samples (FFPE blocks and frozen tissue at Leeds Teaching Hospitals Trust only) were processed by staff in the pathology department at each centre as part of routine tissue processing procedures.
All samples (tissue and biological fluids) were shipped by courier at regular intervals and stored at a Human Tissue Authority (HTA)-licensed establishment (St James’s University Hospital/Chapel Allerton Hospital/University of Leeds – licence number 12279) in compliance with the licensing provisions of the Human Tissue Act 2004. 489 Each set of fluid samples was mirror banked between two secure monitored/logged –80°C freezers linked to a central alarm system to alert to temperature fluctuations. Frozen tissue (from Leeds only) was stored securely in the bank’s liquid nitrogen dewars. FFPE tissue blocks were stored in a secure filing system within the banking facility. In all cases samples were pseudonymised and assigned unique storage numbers. Their locations have been logged on a secure laboratory information management system (SENTRY) in Leeds.
Monitoring and quality assurance
All returned consent forms and CRFs were checked for compliance with the study protocol, inconsistent data and missing data and timings. Study staff were in regular contact with study centre personnel to check on progress and deal with any queries that they may have had. In the event of unclear data, the Leeds CTRU issued a manual query form, giving details of which information was missing or unclear. Responses to queries were made on the query forms, which were copied and returned. In addition, data cleaning and checking were undertaken by researchers prior to embarking on research studies.
A study steering committee met every 6 months during the recruitment phase and annually (meeting or telecom) during the follow-up phase and was responsible for the overall supervision of the study.
Study management results and discussion of issues
Consent
Within this programme a multilevel approach to consent was adopted, in which patients were given a choice about whether or not they wished to participate in certain more sensitive aspects of the research. This was considered to be the most appropriate method of respecting the wishes of patients whilst ensuring that their samples and data could be used in a wide a range of research projects.
Refusal to consent to the optional elements was low (< 10%), with the exception of 18% of patients in the RCC cohort who did not wish to consent to their relatives being contacted regarding any health findings if they themselves could not be contacted. This concern among patients reflects similar ongoing discussions in the ethical field concerning the implications of genetic testing for families and how to balance the obligation for health-care professionals to disclose someone’s disease status or not with an individual’s right to know a relative’s disease status. 490
The number of consenting issues was quite high, with issues in 79 out of 706 (11.19%) RCC forms and 41 out of 514 (7.98%) renal transplant forms. The vast majority of these issues concerned the optional consent elements not being appropriately deleted. The cost and time associated with chasing, correcting and administering the optional consent elements were considered by the trials unit to be more than for a typical study.
Taken together, these findings provide some evidence to support the case for a broad, as opposed to a multilevel, consent process, as the number of patients who would not have participated would have been low. These conclusions are supported by a recent workshop funded by the US NIH. 491 This reported that broad consent is acceptable and pragmatic as long as participants are provided with sufficient information to make a reasonably informed decision and that sufficient processes are in place to provide independent oversight and approval of future research. They argue that broad consent can protect the rights of donors whilst minimising the cost and administrative burden on researchers.
When multilevel consent is required careful attention should be paid to the design of the consent form. With hindsight, the delete options used in these forms were not clear enough, although they were what were mandated locally at that time. This was also very much an emerging area with regard to the use of RTBs and larger-scale genomic analysis becoming more widely used.
Sample processing
Technical pre-analytical factors, including specimen collection, processing, transport and storage, are a major source of measurement uncertainty in biomarker discovery, clinical trials and clinical laboratory medicine. 373,492,493 To minimise the effects of pre-analytical technical factors on samples, a multicentre localised approach was established, as opposed to a centralised processing strategy (e.g. UK Biobank). Localised strategies have several benefits over centralised strategies, including short processing and cryostorage times, leading to better preservation of biomarkers, and batch shipments, reducing the overall cost of transportation. However, localised strategies require appropriate facilities, equipment and staff at each centre, increasing the burden of training and quality control (QC).
To ensure consistency across all centres, standardised sample packs (Figures 29 and 30) were prepared centrally and shipped out to centres, all sites underwent an initiation and research staff were trained in sample processing and handling procedures. SSOPs were carefully prepared for all sample collections, taking into account our previous experience in Leeds in processing samples and developing protocols for biomarker discovery studies and published guidance when appropriate. 494–496 However, for some parameters, best practice guidelines were not deemed to be pragmatic for ‘real-world’ sample collection in the NHS and also some analytes are not optimally processed under such conditions. However, the most important aspect is consistency and recording conditions so that this can be accounted for in terms of suitability for specific analytes. Table 70 outlines some of the key pre-analytical parameters and highlights differences between examples of published standards and guidance and the SOPs. The International Agency for Research on Cancer (IARC) guidance496 has been developed specifically for biobanking whereas the Clinical and Laboratory Standards Institute (CLSI) procedures494,495 have been developed for use in the context of routine clinical chemistry analysis.
FIGURE 29.
Enhanced liver fibrosis sample and liver biobank sample kit. From top left clockwise: Safebox® (Royal Mail) for shipping the ELF sample; ELF test sample shipping form; 10 × 0.5-ml liver biobank tubes (Sarstedt Ltd, Leicester, UK); biobank blood tube label (CILS International, Worthing, UK); pastettes (Scientific Laboratory Supplies Ltd, Nottingham, UK); 7-ml Bijou (Scientific Laboratory Supplies Ltd); sample form; ELF test blood tube label (CILS International); and ELF test sample tube (Thermo Fisher Scientific, Loughborough, UK).

FIGURE 30.
Renal transplant and RCC sample kit. From top left clockwise: sample tube kit (FluidX Ltd, Nether Alderley, UK); pastettes (Scientific Laboratory Supplies Ltd, Nottingham, UK); sample form; sample tube caps (FluidX Ltd); 150-ml urine collection pot; 50-ml centrifuge tube; 20-ml barcoded universal tube; and 7-ml Bijou (× 2) (all Scientific Laboratory Supplies Ltd).

| Matrix | Pre-analytical factor | SOP | IARC496 | CLSI (H18-A4)494 | CLSI (GP16-A3)495 |
|---|---|---|---|---|---|
| Urine | Transport temperature | Room temperature | Ambient | Room temperature (if < 2 hours) | |
| Urine | Time to processing | < 2 hours | Minimum | < 2 hours | |
| Urine | Centrifugation speed | 2000 g | Not specified | Not specified | |
| Urine | Centrifugation time | 10 minutes | Not specified | Not specified | |
| Urine | Storage temperature | < –70°C | –80°C | Not specified | |
| Serum | Tube type | Plain clot activator | Without anticoagulant | Clot activator | |
| Serum | Tube volume | 8–10 ml | Not specified | Not specified | |
| Serum | Transport temperature | Room temperature | Room temperature | Room temperature | |
| Serum | Collection procedure | Invert 5× | Not specified | Invert 5–10× | |
| Serum | Clotting time | > 45 minutes | > 30 minutes | 5–30 minutes | |
| Serum | Venepuncture to storage | < 2 hours | < 1 hour | < 2 hours | |
| Serum | Centrifugation speed | 2000 g | 1500 g | Not specified | |
| Serum | Centrifugation time | 10 minutes | 10 minutes | Not specified | |
| Serum | Centrifugation temperature | Room temperature | Room temperature | 20–22°C | |
| Serum | Storage temperature | < –70°C | –80°C or liquid nitrogen | < –20°C | |
| Plasma | Tube type | EDTA | EDTA | Not specified | |
| Plasma | Transport temperature | Room temperature | Room temperature | 20–22°C | |
| Plasma | Collection procedure | Invert 5× | Not specified | Invert 5–10× | |
| Plasma | Venepuncture to processing | < 45 minutes | As soon as possible | Immediately | |
| Plasma | Venepuncture to storage | < 2 hours | < 1 hour | < 2 hours | |
| Plasma | Centrifugation speed | 2000 g | 815 g, then 2500 g | Not specified | |
| Plasma | Centrifugation time | 10 minutes | 10 minutes, then 10 minutes | Not specified | |
| Plasma | Centrifugation temperature | Room temperature | 4°C | 20–22°C | |
| Plasma | Storage temperature | < –70°C | –80°C or liquid nitrogen | < –20°C | |
| Buffy coat | Storage temperature | < –70°C | –70°C or liquid nitrogen | Not specified |
Compliance data were collected on key parameters, in particular sample processing times; a summary of these data is presented in Table 71, along with the percentages of samples meeting SSOP specifications. Compliance within the RCC cohort was high, with > 94% of samples being centrifuged within 2 hours and > 89% being frozen within 2 hours. Compliance within the renal transplant cohort was lower, especially for blood samples, with only 47% of samples being processed in < 2 hours. The primary reason for this became apparent during study set-up at several centres, where most inpatient routine blood samples are collected in the very early morning (at approximately 06:00), but where most of the local sample processing staff did not begin work until 08:00. Therefore, a pragmatic decision was made to process these blood samples in the shortest time frame possible. Compliance with SSOPs for urine samples was better as these were generally collected fresh by the research nurses.
| Cohort | Sample | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blood | Urine | |||||||||||
| Median time to centrifugation (hours:minutes) | Samples centrifuged within 45–120 minutes (%) | Missing data (%) | Median time to freezing (hours:minutes) | Samples frozen within 2 hours (%) | Missing data (%) | Median time to centrifugation (hours:minutes) | Samples centrifuged within 45–120 minutes (%) | Missing data (%) | Median time to freezing (hours:minutes) | Samples frozen within 2 hours (%) | Missing data (%) | |
| RCC | 01:11 | 98 | 2 | 01:20 | 96 | 16 | 01:03 | 94 | 14 | 01:20 | 89 | 23 |
| Renal transplant | 01:24 | 78 | 8 | 01:30 | 47 | 8 | 01:10 | 82 | 21 | 01:22 | 79 | 19 |
An inspection of some biomarker results highlighted that a single centre was using inappropriate blood tubes for the collection of serum samples and this is described in detail in Chapter 14. In this instance, the confusion stemmed from the tube manufacturer (Greiner Bio-One, Stonehouse, UK) making both a red top serum tube and a red top ethylenediaminetetraacetic acid (EDTA) plasma tube. In both the SOPs and the on-site training, careful attention was paid to ensure that sites selected the appropriate tube type for the collection of serum and plasma. At the time of CRF development it was thought to be overly burdensome to request sites to record the catalogue and lot numbers of each blood collection tube used; instead, researchers selected the manufacturer from a tick list. The lack of standardised colour coding of blood collection tubes has been previously highlighted as a patient safety issue and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) has called for harmonisation. 497 Our experiences support this view that the current heterogeneity in tube colours presents a significant pre-analytical risk for multicentre biomarker studies and trials and, until blood collection tubes are harmonised, researchers must remain vigilant. Unfortunately, it is difficult to standardise blood collection tubes by manufacturer within multicentre research studies, as the tubes must be compatible with the local venepuncture system. Patient preference and the terms of ethics approval usually dictate that research blood samples are collected within the same cycle of venepuncture, using the same venepuncture apparatus as for routine samples.
Clinical data
Compliance to the study protocol and schedule were generally high across both the RCC and the renal transplant cohorts, as demonstrated in Tables 72 and 73, respectively, by the > 90% and > 85% overall CRF compliance for the respective studies. However, in both cohorts, sampling fatigue and/or loss to follow-up was observed. This was more apparent in the RCC cohort than in the transplant cohort, with only 48% of RCC patients in the longitudinal cohort providing their final sample (at 2 years) compared with 74% of transplant patients (at 6 months). This is potentially because of both differences in the length of follow-up and whether or not follow-up occurs at the same hospital as recruitment and the higher frequency of hospital visits required by transplant patients. However, as follow-up is still ongoing, compliance may change over time.
| CRF | CRFs | ||
|---|---|---|---|
| Received (n) | Due (n) | Return rate (%) | |
| F01 Eligibility form | 705 | 706 | 99.9 |
| F02 Baseline assessment | 705 | 706 | 99.9 |
| F03 Surgery details | 697 | 706 | 98.7 |
| F04 Sample form (initial) | 699 | 706 | 99.0 |
| F04 Sample form (3–6 months) | 139 | 169 | 82.3 |
| F04 Sample form (12 months) | 107 | 148 | 72.3 |
| F04 Sample form (18 months) | 73 | 148 | 49.3 |
| F04 Sample form (24 months) | 70 | 147 | 47.6 |
| F06 Follow-up 1 year | 624 | 629 | 99.2 |
| F06 Follow-up 2 years | 359 | 468 | 76.7 |
| F06 Follow-up 3 years | 142 | 200 | 71.0 |
| F06 Follow-up 4 years | 15 | 47 | 31.9 |
| F06 Follow-up 5 years | 0 | 0 | NA |
| Overall compliance | 4335 | 4780 | 90.7 |
| CRF | CRFs | ||
|---|---|---|---|
| Received (n) | Due (n) | Return rate (%) | |
| F01 Baseline assessment | 419 | 433 | 96.8 |
| F01 RT eligibility and registration | 512 | 512 | 100.0 |
| F06 RT sample form (pre transplant) | 442 | 512 | 86.3 |
| F03 RT postoperative investigations | 291 | 297 | 98.0 |
| F06 RT sample form (hospital stay) | 227 | 298 | 76.2 |
| F04 RT duration of postoperative stay | 290 | 296 | 98.0 |
| F05 RT follow-up week 1 | 272 | 292 | 93.2 |
| F06 RT sample form week 1 | 252 | 292 | 86.3 |
| F05 RT follow-up week 2 | 272 | 289 | 94.1 |
| F06 RT sample form week 2 | 244 | 289 | 84.4 |
| F05 RT follow-up week 3 | 266 | 289 | 92.0 |
| F06 RT sample form week 3 | 244 | 289 | 84.4 |
| F05 RT follow-up week 4 | 268 | 288 | 93.1 |
| F06 RT sample form week 4 | 240 | 288 | 83.3 |
| F05 RT follow-up month 2 | 251 | 284 | 88.4 |
| F06 RT sample form month 2 | 223 | 284 | 78.5 |
| F05 RT follow-up month 3 | 232 | 280 | 82.9 |
| F06 RT sample form month 3 | 199 | 280 | 71.1 |
| F05 RT follow-up month 6 | 219 | 266 | 82.3 |
| F06 RT sample form month 6 | 197 | 266 | 74.1 |
| F05 RT follow-up year 1 | 159 | 203 | 78.3 |
| F05 RT follow-up year 2 | 71 | 97 | 73.2 |
| F05 RT follow-up year 3 | 13 | 15 | 86.7 |
| Overall compliance | 5803 | 6639 | 87.4 |
Recruitment to time and target
The recruitment rates to the RCC and renal transplant cohorts are shown in Figures 31 and 32, respectively. The accrual graphs clearly show that recruitment was slower than expected across both studies, with the RCC cohort aiming to have been completed by December 2012 and the renal transplant cohort by September 2013. As well as the slower than expected monthly recruitment rate, this was in part because of the length of time taken to set up individual centres (see Tables 77 and 78). However, 5 of 11 RCC centres and 8 of 10 renal transplant centres managed to achieve or exceed their target number of patients by the end of the study period, as shown in Figures 33 and 34, respectively. Over-recruitment was encouraged and two RCC and six transplant centres over-recruited by > 50% of their original target figure. However, we observed that there were limited incentives within the system to encourage over-recruitment and upwards revision of target recruitment figures. One centre in particular excelled at recruiting patients and hit its recruitment target within 6 months of opening, but declined to increase its target.
FIGURE 31.
Monthly and cumulative accrual of patients with suspected RCC. Figures are shown for both the longitudinal cohort and the cross-sectional cohort.
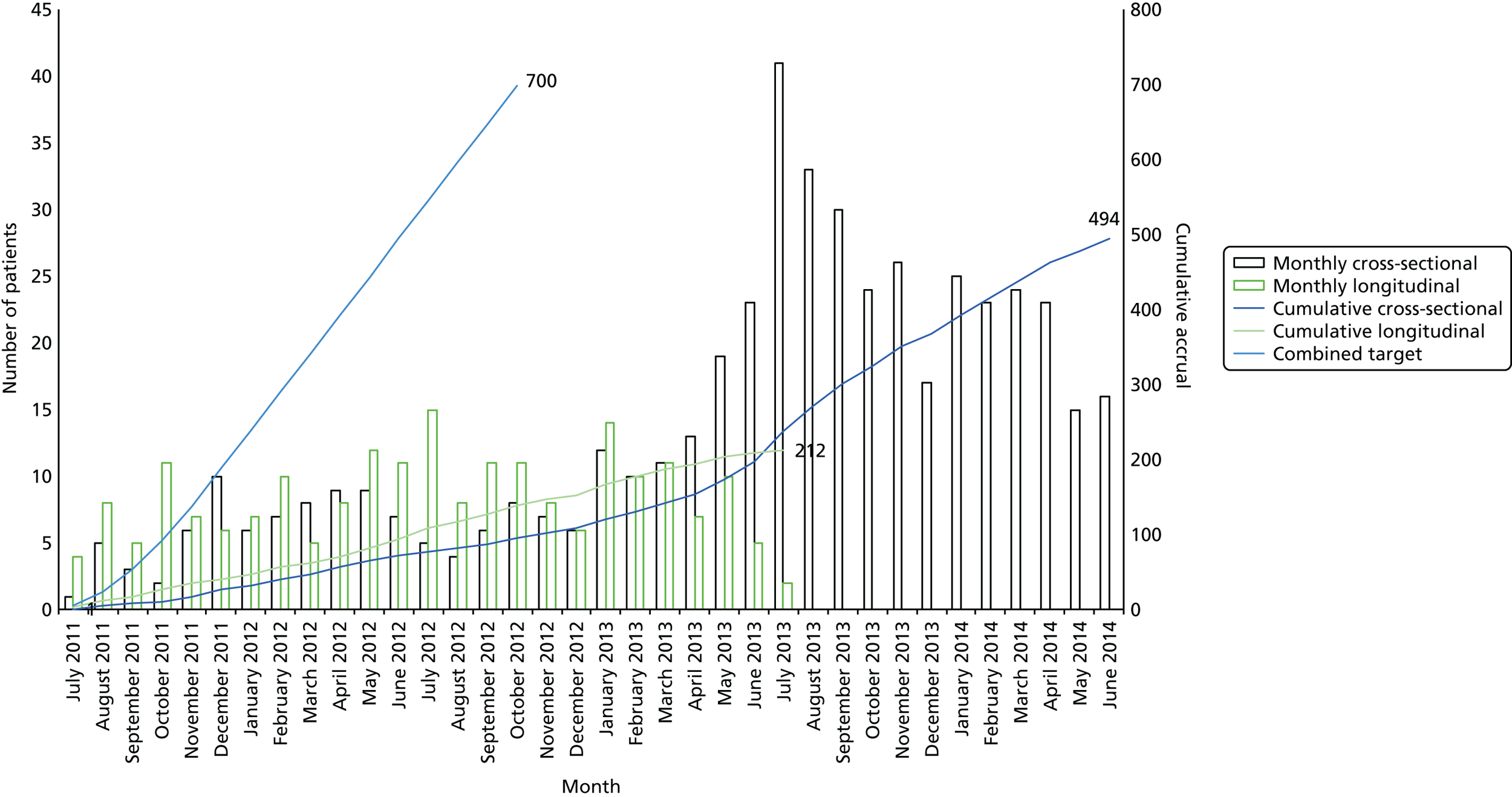
FIGURE 32.
Renal transplant: monthly and cumulative recruitment from the waiting list and transplantation.
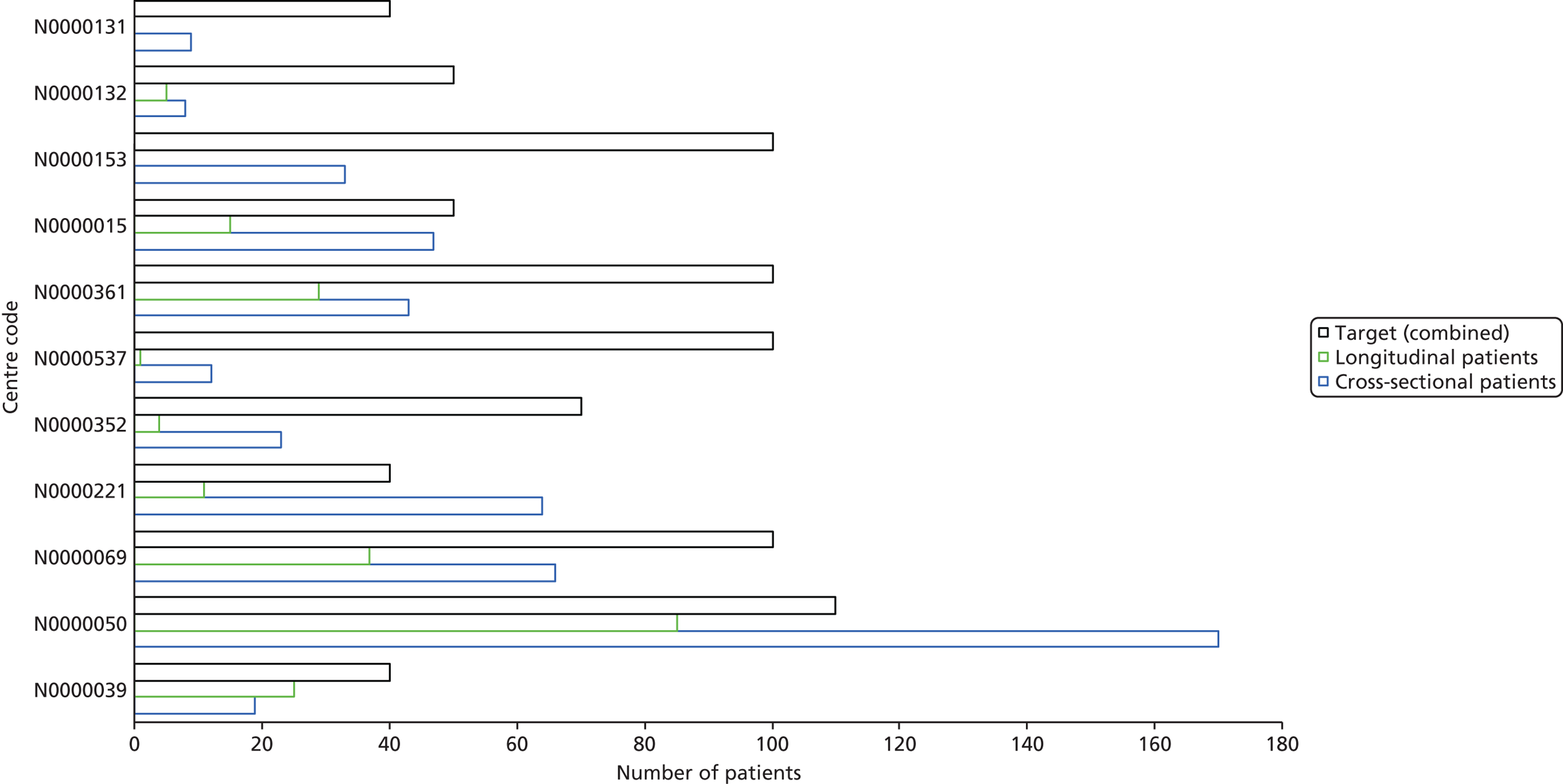
FIGURE 33.
Renal cell carcinoma targets and accrual by centre.
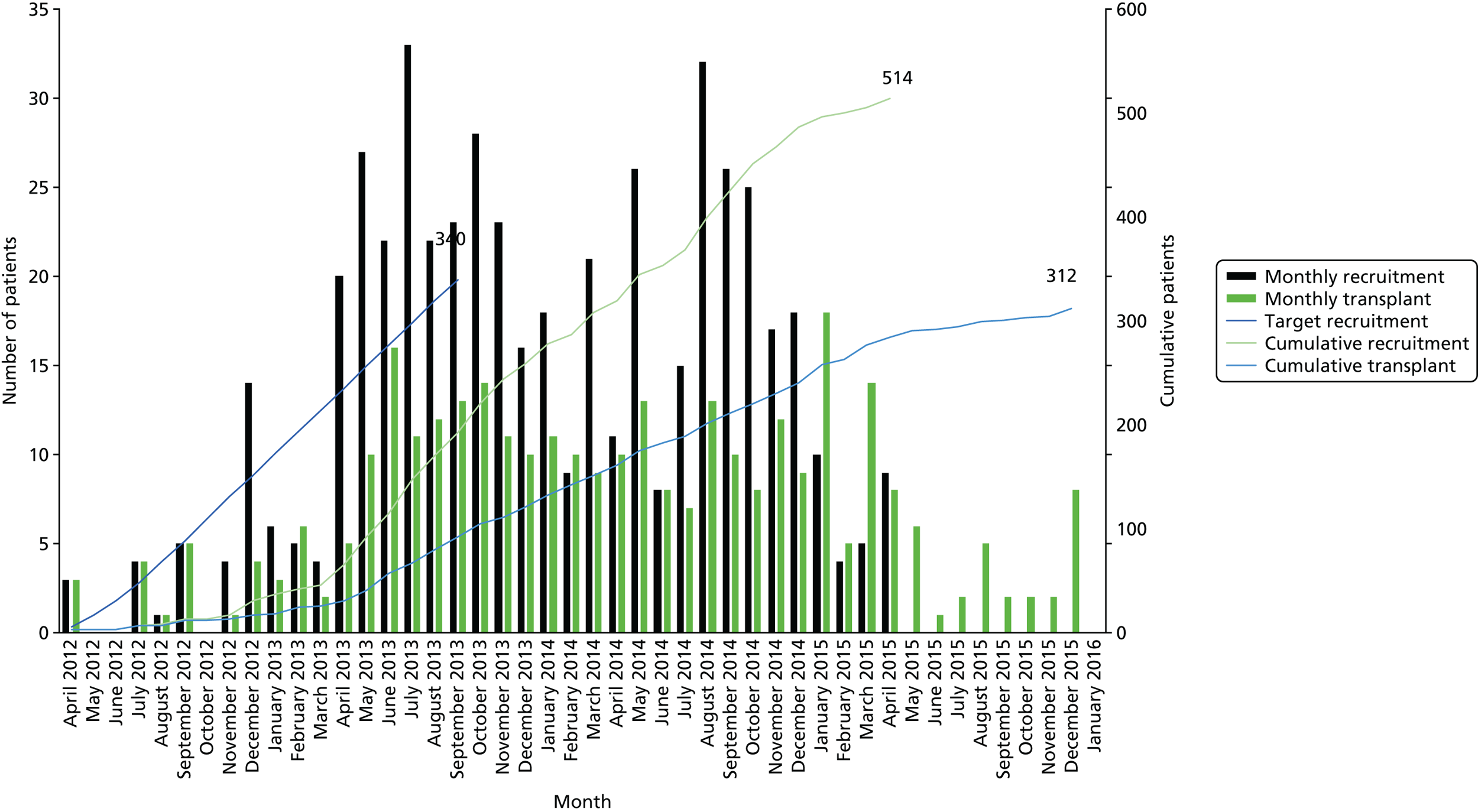
FIGURE 34.
Renal transplant targets and accrual by centre.

Study governance aspects
Ethical considerations
The studies were all performed in accordance with the recommendations for biomedical research involving human subjects adopted by the 18th World Medical Assembly, Helsinki, Finland, 1964, and subsequent amendments. 498 Permission was sought from each NHS organisation and national/local principles of research governance were adhered to. The possibility of samples being used in collaboration/partnerships with commercial companies was made explicitly clear in the patient information sheet and consent form. All procedures and processes were compliant with the Human Tissue Act 2004489 and the management of tissues and fluids stored centrally in Leeds was the responsibility of the RTB Management Committee.
Establishment of a research tissue bank
To establish the renal transplant and RCC cohorts, two separate approaches to gaining ethics approval were available at the time of set-up:
-
project-specific ethics approval
-
RTB ‘generic ethics approval’.
Traditionally, researchers would apply to a research ethics committee (REC) for project-specific ethics approval, which would enable them to use the material collected in the study for the duration and purpose of the study. However, if these materials had value beyond the duration of the initial ethics approval, researchers would need to do one of the following before the end of the study period:
-
apply for a renewal of project-specific approval
-
obtain a HTA licence and set up a RTB
-
transfer samples to a RTB.
Therefore, rather than applying for multiple project-specific approvals and setting up a RTB at the end, we opted to set up a RTB from the outset and to seek generic ethics approval for the collection and use of patient data and samples in the current and future projects.
Ethics approval was sought to establish the Leeds NIHR Biomarker Research Tissue Bank (reference number 10/H1306/6) and a Management Committee was appointed to oversee the collection, storage and release of material for research (chaired by Professor Banks and Professor Selby and including the local HTA-designated individual, quality assurance manager, RTB manager, nurse representative, patient representative, clinical representatives and others). This committee monitors and reviews the bank and related activities and considers applications for access and proposed use of the samples by other research groups once the samples have been used to meet the remit of this programme. Priority is given to collaborative applications and groups need to provide a clear and well-defined research plan according to the criteria laid out in the application process. Additionally, research groups have to complete a Material Transfer Agreement (MTA) and demonstrate provision of the correct storage of material, logging of material and records of storage as required. Groups who are granted permission to access samples from the bank are expected to meet the costs of having the samples shipped under appropriate conditions to their establishment. Samples will be provided only if appropriate consent has been given by the patient, that is, a sample would not be released for a project involving genetic analyses if the patient has not given consent for the sample to be used in such studies. The bank is promoted to researchers and patients through the Leeds Multidisciplinary Research Tissue Bank website. 499
The Human Tissue Act 2004489 requires that the storage of relevant material is licensed by the HTA. The relevant material is essentially cellular material for all:
. . . research in connection with disorders, or the functioning, of the human body.
Human Tissue Act 2004. 489
NHS RECs can give generic ethics approval for a RTB to collect, store and release tissues for use in research, providing the bank is on a HTA-licensed premises. This is advantageous in terms of maximising the use of samples for future research, ensuring consistent and high-level governance procedures and minimising administrative burden.
Local and national NHS approvals
The RCC and renal transplant studies both utilised the generic ethics approval of the RTB (reference number 10/H1306/6) from the start for recruiting patients and collecting samples. The liver disease serum samples were collected under the project-specific approval of the ELUCIDATE trial (reference number 10/H1313/2) but broad patient consent was sought for use of the samples in future biomarker research. The liver samples and data were then subsumed under the governance of the RTB following the completion of recruitment and sample collection.
Governance results and discussion of issues
Research RTBs are purported to have several advantages over project-specific approvals, notably the ability to collect, store and use samples for a wide range of applications without seeking specific ethics approval for individual projects and the fact that there is no requirement for local R&D approvals for tissue collection centres (Table 74).
| Areas | Project-specific approval | RTBs |
|---|---|---|
| Portfolio adoption | Yes | No |
| Human Tissue Act 2004 applies | No | Yes |
| Usage of samples | Within scope of original project-specific approval | Within scope of original RTB approval – but requires approval of RTB committee |
| Local NHS R&D approval | Required | Not required |
However, the reality was more complicated, presenting numerous challenges, as detailed in the following sections.
National Institute for Health Research Clinical Research Network Portfolio adoption
Adoption onto the NIHR Clinical Research Network (CRN) Portfolio is a fundamental enabler to run successful multicentre studies in the NHS. Portfolio adoption enables access to NHS service support, without which most recruiting centres would not have participated.
However, the guidelines for Portfolio eligibility500 specifically exclude the ‘banking of biological samples or data except where this activity is integral to a self-contained research project designed to test a clear hypothesis’. This meant that the RTB itself was deemed to be ineligible and, therefore, individual studies (RCC and RT) were submitted to the CRN Portfolio, using the ethics approval of the RTB.
NHS permissions
Although the individual projects were eventually adopted onto the Portfolio, the creation of projects meant that local NHS approval was required for every centre participating in the studies. One of the advantages of RTBs is that tissue collection centres do not need local approval. Furthermore, the unfamiliarity of local teams with RTBs and the complexity of the project approval process was a source of much confusion and delay at almost every NHS centre (see Tables 77 and 78).
Human Tissue Act 2004-relevant material
Tissue samples or buffy coats or any cellular material collected under a project-specific REC approval are not bound by the governance of the HTA for the duration of the project. However, such samples (‘relevant material’) collected within a RTB infrastructure are governed by the HTA. On the whole, HTA governance did not impact greatly on our procedures, as it is well aligned with best practice and GCP. However, one aspect mandated by the HTA, that is, storage incidental to transportation, was a significant cause for concern and incurred significant expense. The HTA defines storage as incidental to transportation if:
. . . tissue is held for a matter of hours or days (but never weeks) pending transfer to a licensed establishment.
Human Tissue Act 2004. 489
For example, as buffy coat samples (serum, plasma and urine are exempt providing acellular) were to be stored on site and shipped in batches to save money (£150 for one sample vs. £400 for 100 samples), the time spent on local sites would have exceeded the time that the HTA considers to be acceptable for storage incidental to transportation. For centres that had licensed premises this meant that all HTA-relevant materials needed to be transferred from the local clinic to the licensed premises within 7 days, incurring additional risk, time and cost. For centres that did not have licensed premises, this meant shipping all samples back to Leeds within 7 days of collection, significantly increasing the shipping costs and the amount of administration.
NHS service support costs
Prior to the introduction of the AcoRD (Attributing the costs of health & social care Research & Development) guidelines in 2012,501 there was ambiguity as to what would be classified as a research cost and what would be classified as a NHS service support cost. Also, the reimbursement costs for biomarker studies as opposed to clinical trials of investigational medicinal products (cTIMPs) were at a much lower level and did not vary depending on whether a single sample and little information was collected or multiple samples and multiple CRFs were collected. In 2009 when this programme grant was funded, many of the clinical research costs incurred by sites, including CRF completion, for example, were assumed to be covered by the NHS service support costs and were, therefore, not funded separately by the NIHR. For the sample and CRF-intensive renal transplant cohort, this meant that there was insufficient reimbursement to incentivise centres to participate. The CRN Renal Speciality Group advised that the research group should ask for fewer CRFs to be completed, making the study more attractive to potential centres. The impact of this amendment can be observed in the boost to recruitment following March 2013, shown in Figure 32. However, sampling fatigue was an ongoing issue (see Tables 72 and 73), suggesting that further incentives may help to improve compliance.
Local and national NHS approvals
The process of gaining ethics approval for the RCC and renal transplant studies, through the establishment of a RTB, took 5 months from REC submission (15 January 2010) to REC approval being granted (15 June 2010). This was in part because of the committee requesting further information and some revision of documentation on 8 February 2010 (response 3 March 2010) and again on 1 April 2010 (response 15 April 2010). All three studies gained adoption onto the National Institute for Health Research Clinical Research Network (NIHR CRN) Portfolio through the Coordinated System for gaining NHS Permission, ensuring access to local service support. The timescales for gaining national approval for each study are shown in Table 75. In April 2010 the NIHR introduced the CRN High Level Objectives,502 which aimed to increase the proportion of studies in the CRN Portfolio that deliver to their planned recruitment time and targets, reduce the time taken to achieve NHS permission and reduce the time taken to recruit the first participants. This includes a 40-day target for gaining NHS permission and a 30-day target for recruitment of the first patient following permission. These targets were not achieved for either the RCC cohort or the renal transplant cohort. In total, for the RCC and renal transplant cohorts it took 6 and 12 months, respectively, from CRN Portfolio submission to recruitment of the first patient.
| Cohort | Time period | |||
|---|---|---|---|---|
| CRN Portfolio approval (months) | NHS R&D approval (months) | Time from R&D approval to first patient (months) | Time from first to last patient recruited (months) | |
| RCC | 6 January 2011–24 February 2011 (1.6) | 2 February 2011–1 April 2011 (1.9) | 1 April 2011–14 July 2011 (3.4) | 14 July 2011–30 June 2014 (36) |
| Renal transplant | 19 April 2011–12 March 2012 (10.8) | 16 December 2011–5 March 2012 (2.6) | 5 March 2012–26 April 2012 (1.7) | 26 April 2012–30 April 15a (37) |
Adoption onto the CRN Portfolio was a challenge, with both the RTB status and also queries concerning peer review causing delays. Delays in study-wide national sign-off were mainly the result of these being some of the first RTB studies to come through the Comprehensive Local Research Network (CLRN), which was, at the time, unfamiliar with the process. The concept of having ethics approval for the Leeds NIHR Biomarker Research Tissue Bank but not for the specific studies caused confusion, as did having protocols for the specific studies that sat alongside the RTB protocol. At a local permission level, NHS trust R&D offices were also unfamiliar with the RTB ethics approval status, asking questions around the need for study-specific ethics approval. It is worth noting that, towards the end of the programme, some centres sent letters confirming that local R&D approval was not required because of the RTB status. RTB status also introduced another source of delay, as MTAs had to be signed off by the trust’s designated individual for research, as they are responsible under the Human Tissue Act 2004489 for any relevant materials. These individuals were often unknown to R&D departments or were not in post, were difficult to contact or simply refused to sign the agreements. Transfer of patient data at several centres was also a cause of delay, despite using the standard procedures of an accredited CTRU.
Participating NHS hospitals were identified initially by the Chief Investigators, but later through word of mouth and the invaluable support of CRN Cancer, Liver and Renal Specialty Groups. Tables 76 and 77 show the time taken by individual centres to submit their SSI form (research application), gain local R&D approval, open the centre following R&D approval and recruit the first patient for the RCC and renal transplant cohort, respectively. The main source of local delay for the RCC study was the R&D approval process, taking an average of 3.8 months (range 0.0–7.0 months), whereas the main source of local delay for the renal transplant study was the submission of the SSI form, taking an average of 4.2 months (range 0.6–7.8 months). This difference in timing of R&D approval may be a reflection of the introduction of the NIHR Performance in Initiating and Delivering Clinical Research exercise in the autumn of 2011. 503 This initiative set a 70-day benchmark for local NHS providers to recruit their first patient, following receipt of a valid research application. The two studies span this transition period, with the majority of RCC centres (73%) starting in 2011 and the majority of renal transplant centres (90%) starting in 2012/13. None of the RCC centres achieved the 70-day benchmark (mean 218 days, range 76–369 days), whereas 40% of renal transplant centres did (mean 115 days, range 26–287 days). By combining the two cohorts together a downwards trend in R&D approval times over time is observed (Figure 35). This supports the CRN’s own analysis, which reports that in 2014–15 83% of studies are now receiving NHS permission within 40 days at all sites. 504 It was queried during the study period whether or not this reduction in time to achieve local NHS permissions might in part be the result of ‘gaming’, where centres would withhold submitting their SSI form until the end of the process to improve their metrics. This hypothesis is supported to some extent with the increase in median SSI submission times between the cohorts (1.2 months for RCC vs 5.1 months for RT) and the trend towards increased SSI submission times as a percentage of local review and approval times (Figure 36). However, Figure 37 does suggest that there might be a trend towards shorter overall set-up times over time, suggesting that improvements in set-up time may be being achieved and that this initiative may be having the desired impact on pushing the pace of UK clinical research. It should be noted, however, that, although of interest, these observations are inconclusive and could be affected by a multitude of other factors, such as different centre make-up in the comparisons of the cohorts and differences in the complexity of study set-up for the cohorts. Further evaluation at a national level should be conducted, taking into account the time from transfer of SSI forms to submission, the data for which are logged within the Integrated Research Application System.
| Hospital | Time from (months) | Total time (months) | |||
|---|---|---|---|---|---|
| Transfer of SSI form to submission | Submission of SSI form to R&D approval | R&D approval to open for recruitment | Open for recruitment to recruitment of first patient | ||
| 1 | 4.3 | 0.0 | 1.3 | 2.3 | 6.8 |
| 2 | 3.6 | 1.2 | 2.3 | 5.0 | 12.1 |
| 3 | 5.8 | 4.9 | 1.8 | 0.7 | 13.2 |
| 4 | 0.6 | 3.3 | 2.0 | 1.7 | 7.6 |
| 5 | 0.0 | 5.8 | 0.9 | 5.4 | 12.2 |
| 6 | 7.0 | 3.9 | 1.6 | 1.8 | 14.3 |
| 7 | 0.7 | 2.5 | 1.3 | 0.2 | 4.8 |
| 8 | 1.7 | 7.0 | 3.4 | 0.5 | 12.5 |
| 9 | 1.7 | 7.0 | 3.4 | 0.5 | 12.5 |
| 9 | 0.0 | 2.3 | 2.3 | 0.5 | 5.1 |
| 10 | 0.3 | 3.5 | 0.3 | 0.1 | 4.1 |
| Median | 1.2 | 3.7 | 1.9 | 0.6 | 12.2 |
| Hospital | Time from (months) | Total time (months) | |||
|---|---|---|---|---|---|
| Transfer of SSI form to submission | Submission of SSI form to R&D approval | R&D approval to open for recruitment | Open for recruitment to recruitment of first patient | ||
| 1 | 0.6 | 0.7 | 3.4 | 0.3 | 4.9 |
| 2 | 7.8 | 2.4 | 0.0 | 1.7 | 11.9 |
| 3 | 2.8 | 0.1 | 1.1 | 0.6 | 4.5 |
| 4 | 0.6 | 3.7 | 2.0 | 3.7 | 10.0 |
| 5 | 0.7 | 5.4 | 1.4 | 0.7 | 8.2 |
| 6 | 5.8 | 0.8 | 2.6 | 0.6 | 9.9 |
| 7 | 5.3 | 0.9 | 0.4 | 0.4 | 6.9 |
| 8 | 7.0 | 0.1 | 0.5 | 1.0 | 8.6 |
| 9 | 6.7 | 1.4 | 0.7 | 0.4 | 9.2 |
| 10 | 4.9 | 0.0 | 0.2 | 0.6 | 5.7 |
| Median | 5.1 | 0.8 | 0.9 | 0.6 | 8.4 |
FIGURE 35.
Time taken for R&D approvals plotted over time.
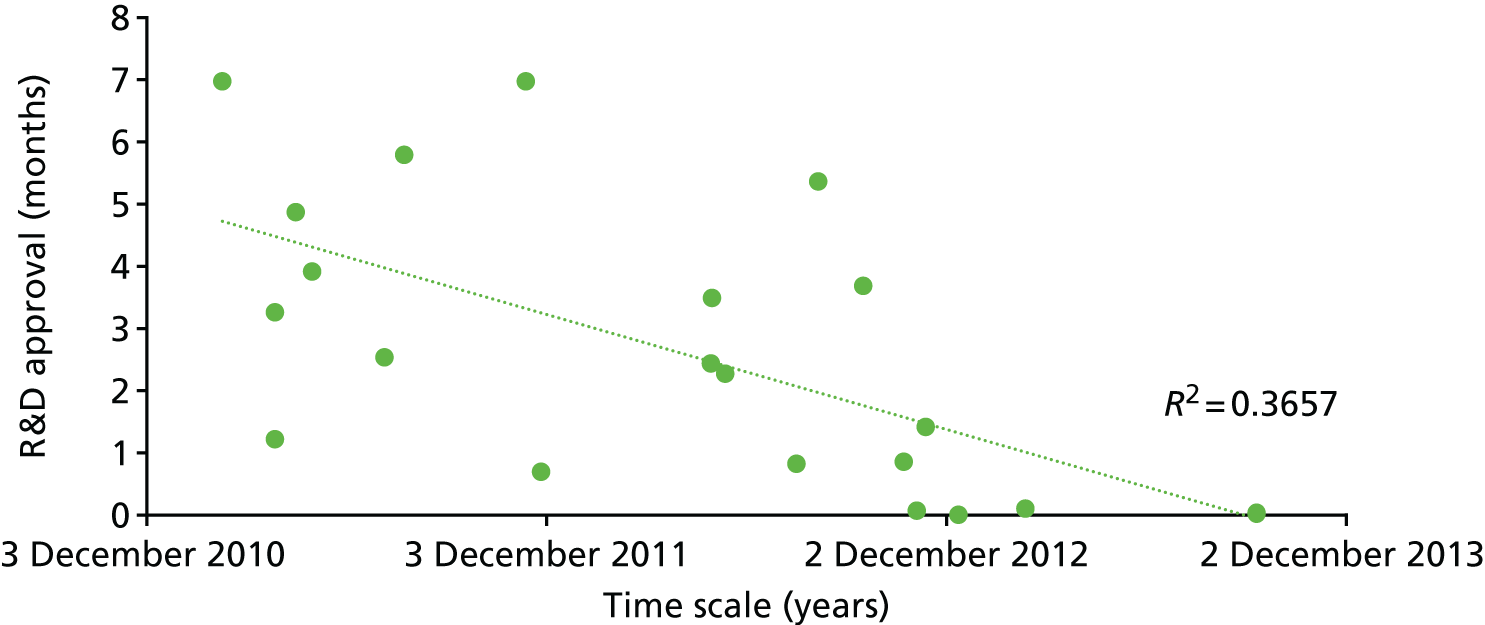
FIGURE 36.
Site-specific information form submission as a percentage of the total time for local review and approval (submission of SSI form + R&D approval).

FIGURE 37.
Combined timescales for overall study set-up plotted over time.
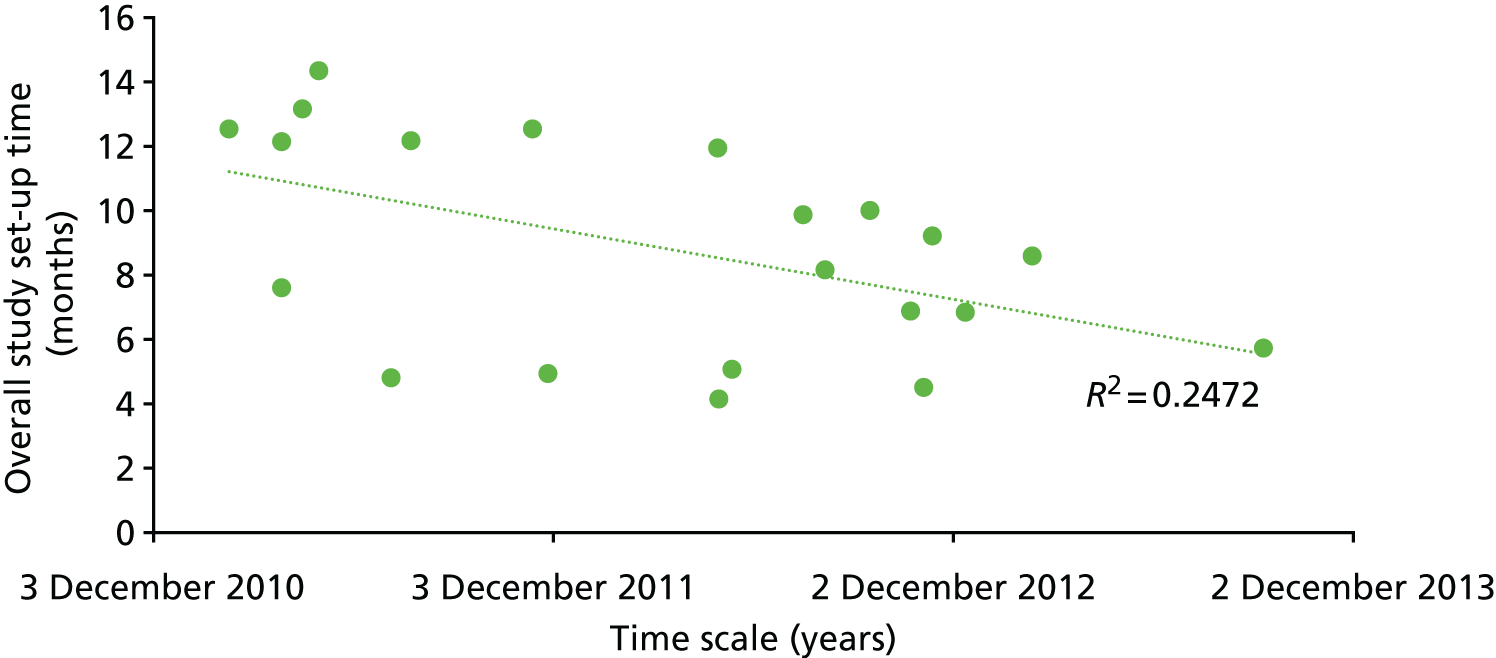
Summary of the final resource
Liver disease cohort summary statistics
The original recruitment target for the ELUCIDATE trial was 1000 participants. In total, 878 participants were eventually recruited, of whom 847 contributed a sample to the RTB. The summary statistics of the final liver biorepository cohort are shown in Table 78.
| Characteristic | Liver disease patients (n = 847) |
|---|---|
| Age (years), median (range) | 55 (23–75) |
| Sex, n (%) | |
| Male | 476 (56) |
| Female | 371 (44) |
| Weight (kg), median (range) | 81.3 (38–166) |
| Height (cm), median (range) | 169 (131–203) |
| Cause of fibrosis, n (%) | |
| Non-alcoholic fatty liver disease | 225 (27) |
| Viral liver disease | 338 (40) |
| Alcoholic liver disease | 59 (7) |
| Other/unknown | 225 (27) |
| ELF value, median (range) | |
| Registration | 9.36 (8.4–17.35) |
| Randomisation | 9.37 (7.13–17.84) |
Renal cell carcinoma and healthy control cohort summary statistics
The recruitment target for the RCC study was 700 patients with suspected RCC (500 cross-sectional cohort and 200 longitudinal cohort) and 200 healthy control subjects. In total, 706 RCC patients and 149 healthy control subjects were eventually recruited. The summary statistics of all patients suspected of having RCC are shown in Table 79 and for the ccRCC subgroup are shown in Table 80. The summary statistics of the healthy control subjects are shown in Table 81.
| Characteristic | All patients (n = 706) |
|---|---|
| Sex, n (%) | |
| Male | 447 (63) |
| Female | 259 (37) |
| Age (years), median (range) | 64 (29–92) |
| Body mass index (kg/m2), median (range) | 27.9 (15.6–74.4) |
| Symptoms, n (%) | |
| Local | 199 (28) |
| Systemic | 103 (15) |
| Both | 151 (21) |
| None | 253 (36) |
| Procedure, n (%) | |
| Radical nephrectomy | 465 (66) |
| Partial nephrectomy | 162 (23) |
| Radiofrequency ablation | 20 (3) |
| Cryoablation | 17 (2) |
| Biopsy only | 12 (2) |
| None | 29 (4) |
| Missing | 1 (< 1) |
| Histological type, n (%) | |
| Clear cell | 481 (68) |
| Papillary | 59 (8) |
| Chromophobe | 46 (7) |
| Oncocytoma | 27 (4) |
| Translocation carcinoma | 2 (< 1) |
| Unclassified | 12 (2) |
| Other | 39 (6) |
| Unknown | 39 (6) |
| Missing | 1 (< 1) |
| TNM stage, n (%) | |
| I | 336 (48) |
| II | 71 (10) |
| III | 143 (20) |
| IV | 56 (8) |
| Not applicablea | 93 (13) |
| Missing | 7 (1) |
| Interval between study blood sample and procedure (days), median (range) | 13 (0–737) |
| Characteristic | Clear-cell subtype patients (n = 481) |
|---|---|
| Sex, n (%) | |
| Male | 321 (67) |
| Female | 160 (33) |
| Age (years), median (range) | 64 (29–92) |
| Body mass index (kg/m2), median (range) | 28.3 (16–74.4) |
| Procedure, n (%) | |
| Radical nephrectomy | 345 (72) |
| Partial nephrectomy | 103 (21) |
| Radiofrequency ablation | 17 (4) |
| Cryoablation | 9 (2) |
| Biopsy only | 7 (1) |
| Tumour size (mm), median (range) | 55 (11–180) |
| Pathological T stage, n (%) | |
| 1 | 239 (50) |
| 2 | 50 (10) |
| 3 | 154 (32) |
| 4 | 5 (1) |
| Not applicablea | 33 (7) |
| Fuhrman grade, n (%) | |
| 1 | 13 (3) |
| 2 | 156 (32) |
| 3 | 231 (48) |
| 4 | 77 (16) |
| Missing | 4 (< 1) |
| Necrosis, n (%) | |
| Present | 146 (30) |
| Absent | 302 (63) |
| Missing | 33 (7) |
| Microvascular invasion, n (%) | |
| Present | 82 (17) |
| Absent | 365 (76) |
| Missing | 34 (7) |
| Sarcomatoid change, n (%) | |
| Present | 29 (6) |
| Absent | 418 (87) |
| Missing | 34 (7) |
| Leibovich risk group, n (%) | |
| Low | 155 (32) |
| Intermediate | 168 (35) |
| High | 80 (17) |
| Not applicableb | 78 (16) |
| TNM stage, n (%) | |
| I | 257 (53) |
| II | 45 (9) |
| III | 123 (26) |
| IV | 50 (10) |
| Missing | 6 (1) |
| Characteristic | Healthy control subjects (n = 149) |
|---|---|
| Sex, n (%) | |
| Male | 50 (33.6) |
| Female | 99 (66.4) |
| Age (years), median (range) | 45 (21–88) |
| Weight (kg), median (range) | 71 (41–153) |
| Height (cm), median (range) | 168 (148–203) |
| Body mass index (kg/m2), median (range) | 25 (16–52) |
| Illnesses, n (%) | |
| None | 113 (74.8) |
| Hypertension | 22 (14.6) |
| Diabetes mellitus | 6 (4.0) |
| Other | 10 (6.9) |
| Ethnicity, n (%) | |
| White | 121 (81.2) |
| Black Caribbean | 3 (2.0) |
| Asian Indian | 6 (4.0) |
| Black African | 7 (4.7) |
| Chinese | 1 (0.7) |
| Asian Pakistani | 1 (0.7) |
| Other Asian | 4 (2.7) |
| Asian Bangladeshi | 1 (0.7) |
| Mixed | 2 (1.3) |
| Other | 3 (2.0) |
| Menopausal status (females), n (%) | |
| Pre | 66 (66.7) |
| Post | 25 (25.3) |
| Peri | 8 (8.1) |
| Smoking status, n (%) | |
| Yes | 16 (10.7) |
| No | 105 (70.5) |
| Ex-smoker | 25 (16.8) |
| Passive smoker | 3 (2.0) |
| Alcohol consumption, n (%) | |
| Light | 136 (91.3) |
| Teetotal | 11 (7.4) |
| Unknown | 2 (1.3) |
| Diet, n (%) | |
| All | 136 (91.3) |
| Vegetarian | 9 (6.0) |
| Vegan | 1 (0.7) |
| Other | 3 (2.0) |
Renal transplant cohort summary statistics
The planed recruitment target for the renal transplant cohort was 340 transplanted patients, with a mix of around 300 deceased donor transplants and 40 live donor transplants. The total number of transplanted patients recruited was 312, of whom 214 received deceased donor transplants and 86 received live donor transplants. Within the cohort, acute complications were observed in at least 66 patients (AR, n = 10; DGF, n = 56). In terms of the causes of end-stage renal disease (ESRD), glomerulonephritis and inherited renal disease are over-represented in the cohort relative to their frequency in the renal disease population at large, which often happens in renal studies unless specifically focusing on other causes such as hypertension or diabetes mellitus. The frequency of DGF is slightly lower than expected, although several patients with missing data are known to have had dialysis in the first week after transplant and may be confirmed as having DGF later. This is also partly a reflection of the population recruited, with the live donor proportion being higher than in the transplant population overall. Efforts are ongoing to obtain the missing data values and the richness of this cohort will continue to improve over time with further data chasing and up to 5 years’ long-term follow-up planned. The summary statistics for the cohort are shown in Table 82.
| Characteristics | Renal transplant patients (n = 312) |
|---|---|
| Age at transplantation (years), median (range) (missing, n = 5) | 52 (19–80) |
| Transplants per centre, n (%) | |
| Liverpool | 21 (6.7) |
| Leeds | 150 (48.1) |
| York | 18 (5.8) |
| Newcastle | 29 (9.3) |
| Hull | 10 (3.2) |
| Portsmouth | 40 (12.8) |
| Plymouth | 2 (0.6) |
| Bristol | 17 (5.4) |
| Sheffield | 10 (3.2) |
| Bradford | 15 (4.8) |
| Sex, n (%) | |
| Male | 191 (61.2) |
| Female | 110 (35.3) |
| Missing | 11 (3.5) |
| Ethnicity, n (%) | |
| White | 271 (86.9) |
| Asian | 17 (5.4) |
| Black/African/Caribbean | 6 (1.9) |
| Other | 5 (1.6) |
| Missing | 13 (4.2) |
| Cause of ESRD, n (%) | |
| Pyelonephritis/reflux nephropathy | 28 (9.0) |
| Diabetes mellitus | 15 (4.8) |
| Glomerulonephritis | 85 (27.2) |
| Hypertension/vascular | 36 (11.5) |
| Obstructive | 3 (1.0) |
| Inherited | 70 (22.4) |
| Other/unknown | 61 (19.6) |
| Missing | 14 (4.5) |
| Transplant types, n (%) | |
| DBD | 126 (40.3) |
| DCD | 88 (28.2) |
| Live donor | 86 (27.6) |
| Missing | 12 (3.8) |
| Ischaemic time, median (range) (n = 293; 17 missing CIT and n = 278; 34 missing WIT) | |
| Cold (hours:minutes) | 12:17 (00:25–23:53) |
| Warm (hours:minutes) | 00:40 (00:03–02:23) |
| Acute complications, n (%) | |
| DGF | 56 (17.9) |
| Missing | 17 (5.4) |
| AR | 10 (3.2) |
| Missing | 14 (4.5) |
Learning and recommendations for future bioresources
Within this NIHR programme grant, three different patient cohorts have been established, consisting of 5976 sets of samples from 2116 patients across 50 NHS centres.
The parallel development of these three projects, in particular the diverse expertise of the teams and individual experiences gleaned from each, not only cross-fertilised each of the projects, but also provided some good learning points worthy of disseminating to future researchers wishing to embark on similar endeavours:
-
The successful delivery of these cohorts confirms that the UK has an outstanding national network of enthusiastic patients, clinical investigators and NHS centres willing to participate in research studies. It also verifies the widely shared view that the NIHR CRN’s systems for setting up national research studies work and, although clearly too slow initially, are improving with regard to both the reduction of complexity and increased pace. Although notable improvements have been made, the NIHR must continue to streamline, simplify and speed up the study approval and set-up process. Researchers are advised, when possible, to keep study elements as simple as possible, as unusual or complex factors may cause delays. At a local level, study/trial co-ordinators should push for SSI forms to be submitted to R&D offices quickly, to start the clock towards sign-off.
-
Another noteworthy highlight of the NIHR CRN is the unprecedented access that it provides to the smaller or less well-known research sites. The CRN specialty groups played a key role in identifying potential research centres, many of which went on to be outstanding recruiters to all three of our cohorts. Not only is this excellent for research, it also provides NHS patients outside the main centres of academic excellence with the opportunity to participate in and benefit from cutting-edge research. As many regional services, including transplant and cancer, operate around a hub and spoke model, researchers need to consider where patients will be identified, consented, treated and followed up. Multiple centres may be involved in a single patient’s journey and, therefore, good co-ordination of and communication with sites is essential. Losing patients at follow-up through discharge back to their initial referring hospital is certainly posing a challenge for the RCC cohort at present at some sites, for example.
-
The RTB ethics approval process may have several benefits over project-specific approvals for certain types of research, for example collecting surplus surgical tissue. However, for prospective multicentre observational cohorts, such as those within this programme, a project-specific approval process followed by transfer to a RTB may with hindsight prove to be easier and quicker to deliver. Project-specific approval would have negated the delays encountered during NIHR CRN Portfolio approval and the issues around the Human Tissue Act 2004, in particular the huge burden and cost associated with storage incidental to transportation. Within this initial phase of a multicentre research programme such as this, there appear to few benefits to using the RTB process, but certainly for long-term sustainability, governance and maximising the use of samples the RTB infrastructure will be invaluable.
-
At the start of this programme, the zeitgeist surrounding patient consent to biomarker research and biobanks was very much in favour of giving patients choice and the opportunity to opt in or out of different aspects. Although this is still a long-term goal, our experience and that of others suggests that the vast majority of patients do not take up most of these options and are happy to provide broad consent. 490 However, the administrative burden and error rate associated with managing such multilevel consent processes is high. Electronic patient management and consenting systems will probably reduce this burden and are under development. However, until such systems are the norm and deployable across multiple centres (a major challenge), researchers are advised only to consider a multilevel consent process when the use of broad consent will hinder recruitment. A consideration here, though, is to what extent this is possible as, in this case, this aspect was raised by the ethics committee at the time as being necessary. However, practice is evolving and experience of such aspects is greater and undoubtedly patients’ views will be important in this area. If multilevel consent must be used, it is important to consult with and thoroughly test the process with patients and health-care professionals.
-
Within the RCC cohort, 18% of patients did not consent to their relatives being contacted regarding any health findings, if they themselves could not be contacted. The precise reasons for this were not clear and warrant further analysis, but one might speculate that they did not wish their relatives to be informed of an identified genetic susceptibility to an illness. In the case of a serious and actionable genetic finding, a health-care professional may believe that he or she has a duty of care to the patient’s relative(s), which may justify breaching the confidentiality of the donor patient. 490 More research with patients is required in this area to refine patient information leaflets and consent forms, to ensure that patients adequately understand how their medical information may or may not be used with regard to their family’s care.
-
Pre-analytical factors are a major source of uncertainty in biomarker research, as verified in Chapter 13. We implemented standardised processes, local training, contingency planning and rigorous QC for these factors within the cohorts. However, future researchers may benefit from going further and monitoring/auditing sites for their sample-processing competency and compliance. Researchers should pay specific attention to blood collection tubes as the lack of standardisation of tube-top colours makes them especially vulnerable to errors. Investigators should also budget for sample processing errors within a percentage of the final sample number, for power calculations, etc., depending on the complexity of the study.
-
In comparison with single-centre sample banking, there are many additional costs associated with multicentre sample banking. Of particular note are the costs of the manufacturer and distribution of standardised sample kits, the provision of local freezer storage and temperature-controlled return shipping. The expense of providing high-quality sample banks and associated clinical data is little appreciated and often the financial support offered by funders is low compared with what is needed, a fact that goes against the drive to improve sample quality for research purposes.
-
Compliance with CRFs could have been improved within these cohorts. There is no doubt that the use of handwritten paper-based CRFs sent by post is complex and costly for all involved. The time delay for data entry into the database also made real-time monitoring and management of the study difficult. The use of electronic data capture and the increasing use of routinely collected data from electronic patient records, in particular pathology results, should be further developed and encouraged. Testing and early auditing of CRFs with research nurses is also advisable.
-
We observed a noticeable decline in the number of samples and follow-up forms being completed after the initial consent visit or discharge. Researchers could consider incentives or penalties to improve compliance with study protocols. Undoubtedly, this may in part reflect the lack of ‘policing’ in a biomarker study such as this compared with a cTIMP, in which regular inspections of site files and compliance with the regulatory requirements of the Medicines and Healthcare products Regulatory Agency is the norm. In addition, the consequences are quite different.
Concluding remarks
These cohorts form an invaluable resource that will underpin research studies validating biomarkers of renal cancer, renal transplant and liver disease for many years to come. A review of the learning outcomes above suggests that, although there is still much to do, many of the problems and challenges encountered within this programme have been or are already being addressed.
Chapter 12 Review and prioritisation of circulating biomarkers in renal cancer and renal transplantation
This chapter describes systematic reviews in two areas to enable serum and plasma protein biomarker prioritisation for subsequent studies, namely (1) biomarkers of prognosis in localised ccRCC and (2) biomarkers for the early detection and diagnosis of DGF following RT and long-term prognosis. The clinical context and rationale for these areas as being the initial focus have been presented in Chapter 10.
Renal cancer
Literature search strategy
During the early part of the programme, a review of the literature was undertaken to identify a small number of biomarkers with high potential. This was carried out to focus some of the initial technical studies and in some cases generate further supporting evidence to warrant eventual investigation using the multicentre bank of samples accrued during the programme (described in Chapter 13). Prior to the final selection of the biomarkers to evaluate in the first major study involving the assessment of prognosis in patients with localised ccRCC, a systematic review of the literature was carried out. The search for publications was carried out using PubMed. Search terms used were as broad as possible to maximise coverage (Figure 38), with subsequent manual filtering as indicated below to select the relevant references. Reasons for inclusion or exclusion at the various stages are shown in Figure 38. This strategy was selected following an iterative testing process, checking to see if selected known diverse references were detected, and for this reason the term ‘biomarker’ was not included as it was too restrictive in some cases. Exclusion during the search itself using the term ‘metastatic’ was not used either as it was recognised that many studies involved patients with both localised and metastatic disease and it was important to include these studies, with later manual filtering removing those studies solely involving patients with metastatic disease. The term ‘predictive’ was included as, although this should primarily refer to the response to treatment, it is still often used interchangeably with prognostic. Biomarkers with only single published studies were excluded as there was insufficient evidence to justify evaluation in a multicentre cohort at this stage, but some may be evaluated in the future using local samples and eventually the NIHR cohorts, as described in Chapter 10. To provide further background information or context for specific clinical or biological areas in which the biomarkers are discussed in this chapter, additional specific references were also searched for as needed but were not included in the final reference counts in Figure 38.
FIGURE 38.
The systematic literature review process adopted for circulating serum or plasma biomarkers of prognosis in ccRCC. The search for publications relating to circulating prognostic biomarkers in ccRCC was carried out on 23 June 2015 in PubMed. DNA, deoxyribonucleic acid; miRNA, microribonucleic acid.
Review of serum and plasma biomarkers of prognosis in clear-cell renal cell carcinoma
A number of analytes routinely available for measurement in hospital laboratories have demonstrated an association with outcome in patients presenting with localised RCC, although as yet they are not generally used clinically for that specific purpose. If sufficient evidence could be provided for any of these, either alone or as part of a panel, they have the advantages of being relatively easy and cheap to measure, with widely available and standardised assays in many cases. Although focusing on proteins, we have also included some other analytes in this category such as electrolytes. It is also important to bear in mind that, although not within the scope of this review, several routinely measured haematological variables have also shown promise. Given their routine measurement preoperatively, these variables, particularly the NLR, would certainly merit inclusion in any analyses. 505–511 In the following section, we review those protein biomarkers that are still currently measured only in the research laboratory environment.
Routinely measured analytes
Many such analytes, not unexpectedly, are associated with prognosis in cancers generally, particularly in the metastatic disease setting, and this is also true for metastatic RCC with regard to serum lactate dehydrogenase, calcium and haemoglobin, which, for example, are included within the Memorial Sloan Kettering Cancer Center nomogram. 512 However, even for localised disease, several such factors appear to have prognostic value, although they are not included as variables in many studies, presumably because of the perceived likely lack of specificity, and many studies are potentially confounded by the inclusion of all subtypes. For example, we first reported preoperative sodium as being independently prognostic for DFS and OS both in patients with localised RCC or when all stages were included (n = 212). This was true whether it was treated as a continuous variable or it was dichotomised to above and below the median value [139 mmol/l; hazard ratio (HR) 0.44, 95% CI 0.22 to 0.88; p = 0.014), with patients with values equal to or below the median value having significantly poorer survival. 513 No studies have yet explored this further although the association of hyponatraemia with worse survival has since been confirmed in patients with metastatic disease being treated with interleukin (IL)-2/interferon-based therapy or targeted therapies, but whether or not this is prognostic or predictive is not yet clear. 514–516 Interestingly, in a study investigating the outcomes of patient with tumour thrombus in either the renal vein or the inferior vena cava, sodium was not listed among the large number of preoperative laboratory variables analysed but high serum potassium was significantly associated with poor survival on multivariate analysis. 517
Hypercalcaemia has been reported to increase with increasing cancer stage, even in the absence of bone metastases;518 in a large study of 1707 patients with localised ccRCC, hypercalcaemia was found in 9% of patients and was significantly independently prognostic for CSS (risk ratio 1.64; p = 0.002), together with anaemia and ESR. 519 This was later confirmed in another large study, which also demonstrated the significant association of tissue parathyroid hormone-like hormone [PTHLH; also known as parathyroid hormone-related protein] messenger RNA levels with OS in ccRCC. PTHLH has been linked to the hypercalcaemia seen as part of the paraneoplastic syndrome in cancers such as RCC, for example. 520 An earlier study had shown a significant correlation between serum calcium and PTHLH, with calcium but not PTHLH being significantly associated with cancer stage, although the authors did highlight possible issues with long-term storage of samples and PTHLH measurement and the use of serum rather than plasma. 521 A further study demonstrated that a high serum PTHLH level was a significant adverse indicator in terms of survival at the univariate but not the multivariate level, although numbers were small (n = 51), with only seven patients having hypercalcaemia. 522
In a very early study, serum alkaline phosphatase (ALP), which exists in several isoforms, with major sources being liver and bone, was reported to be a better indicator of outcome than a bone scan (with routinely used assays not measuring specific forms of ALP). 523 In a study involving 365 patients with RCC, the incidence of paraneoplastic elevation of ALP was reported to be 21.1%, with no stage-related differences and a significant association with poorer survival. 524 A further similar-sized study examining a range of common laboratory variables found a significant association of ALP with progression-free survival (PFS) and, when results for the non-metastatic subgroup were examined, serum ALP was an independent prognostic factor for CSS. 525 However, the previously described study involving 1707 patients with localised ccRCC found an elevated ALP level in 8% of patients but no significant association with CSS. 519 Addressing very specific clinical groups within RCC populations, two studies evaluated outcome in patients with either local recurrence or extension into the inferior vena cava. 526,527 In the former, serum ALP at the time of recurrence was prognostic for poor outcome in terms of CSS but only at the univariate level, although numbers were small (n = 54); in the latter, preoperative ALP was prognostic in multivariate analysis in the whole group (n = 166) and in univariate analysis only if restricted to patients with non-metastatic disease. 526,527
Recently, gamma-glutamyltransferase has also been implicated as having independent prognostic significance in a large study involving 921 patients with RCC (all stages and subtypes),528 although, in a study involving 700 patients with non-metastatic disease only, gamma-glutamyltransferase was significant only in univariate analysis. 529
Obesity is a recognised risk factor for RCC and body mass index is associated with poor prognosis, with two recent studies independently showing significant associations between low preoperative serum total cholesterol levels and worse prognosis. 530,531 Both studies involved patients with metastatic and localised disease, with one including only the ccRCC subtype (n = 364)530 and the other including all subtypes (n = 867). 531 In both studies, serum total cholesterol level was a significant independent predictor of CSS and this was also the case in one study when only the subgroup of patients with localised disease was examined. 531 Interestingly, three studies examining nutritional status, employing varying indices but all of which included serum albumin level together with varying factors such as cholesterol level or lymphocyte count, found nutritional deficiency to be a significant independent prognostic factor for recurrence or for CSS and OS when restricted to patients with local or locoregional disease. 532–534
A small number of studies have examined ‘conventional’ tumour markers used in other cancer types. Serum beta-2-microglobulin was related to both stage and grade of RCC (n = 145) and was significantly inversely related to survival, although only at the univariate level. 535 Analysis of CEA, cancer antigen 50, cancer antigen 19-9, CA-125 and CA-15-3 found elevated serum levels for all except CEA in a cohort of 154 RCC patients, with correlations of the last two with stage and grade and independent prognostic value of CA-125. 536 In a more recent study, almost two decades later, CA-15-3, CA-125 and β2-microglobulin were all associated with stage and grade of RCC and CSS but only CA-15-3 was significant in multivariate analysis and also for PFS. 537 Whether or not the marked differences between the two studies regarding the relative merits of CA-125 and CA-15-3 reflect changes in assays, differences in the patient populations or differences in outcome (it is not clear in the earlier study if CSS or OS was examined) is not clear. Two studies by the same group have also examined the free beta-subunit of human chorionic gonadotropin (HCG) in 177 and 256 patients with RCC, respectively, with some patients possibly being included in both studies; the free beta-subunit of HCG was found to be an independent prognostic variable for CSS. 538,539 Evaluation of neuron-specific enolase has shown positive correlations with stage and grade of RCC in four papers,540–543 two of which were published by the same group,542,543 with a commercially available assay used in the later study. A significant independent prognostic association with survival was reported.
Abnormalities in coagulation occur widely in cancer patients. Initial findings of significantly higher plasma fibrinogen concentrations in RCC patients with metastatic disease544 have subsequently been confirmed and extended by two larger studies. 545. 546 In one study (n = 286), including all subtypes and stages of RCC, an independent association was found for fibrinogen with both DFS and OS. 545 In a larger study involving 994 patients with localised RCC of all subtypes, there was an association of plasma fibrinogen (measured prospectively the day before surgery) with tumour stage and grade, with independent prognostic significance for metastasis-free survival (MFS), CSS and OS, with HRs varying from 2.15 to 2.48 (p < 0.001 for all). 546 A further smaller study (n = 128) also found a significant association of plasma fibrinogen with CSS but additionally reported that D-dimer levels were negatively associated with OS. 547 Clearly, this represent an attractive possibility given the ready availability of standardised assays in clinical laboratories and these results could be relatively easily confirmed in further prospective studies on a multicentre basis.
C-reactive protein
C-reactive protein is an acute-phase protein that is elevated in many inflammatory diseases and often raised in patients with cancer. With the main source generally being the liver and expression regulated by cytokines such as IL-6 and tumour necrosis factor-alpha, it has been shown that IL-6 is produced by renal cancer cells and functions as an autocrine growth factor. 548 The level of circulating IL-6 is increased in patients with RCC, particularly in those patients with metastatic disease, and several studies have shown a significant correlation with serum CRP. 549–552 High serum IL-6 has been shown to be associated with poorer survival in univariate analyses but either this was not confirmed in multivariate analysis or further multivariate analysis was not conducted. 550,553 It is now known that CRP is also produced by renal tumour cells and, indeed, intra-tumoural CRP staining has been shown to be significantly associated with OS. 554,555 However, the extent to which this contributes to the circulating CRP pool is unclear as no significant normalisation of CRP or IL-6 was seen at approximately 3 months post surgery for RCC, although an earlier study had reported significant concentrations by 6 months post surgery. 552,556
Although one of the earliest studies 20 years ago analysing several acute-phase proteins including CRP found all to be significant at the univariate level but only orosomucoid (α1-acid glycoprotein) at the multivariate level, a large number of studies have now shown CRP to be a significant independent prognostic biomarker in RCC and a recent meta-analysis showed that this is the case across the urological cancers. 557 Many of the studies have investigated the value of preoperative serum CRP against the standard clinicopathological prognostic factors and demonstrated independent prognostic significance for DFS/RFS and/or CSS in patients with localised disease, although one study involving only patients with disease extending into the vena cava showed a significant association of CRP with CSS only at the univariate level. 553,558–563 The largest such study included 1161 patients (including 146 with M1 disease) across all subtypes and confirmed CRP as a significant independent prognostic factor for CSS and OS. 564 Interestingly, although preoperative serum CRP was demonstrated in one study to be a significant independent prognostic factor for RFS in patients with localised disease (n = 263), non-normalisation of CRP postoperatively rather than preoperative CRP was significantly associated with OS on multivariate analysis. The 5-year survival figures were 96.9% and 30% in patients whose CRP normalised or failed to normalise, respectively. 565
Several recent studies have also explored the complementarity or additive value of existing clinicopathological prognostic factors or scoring systems. In a study involving 83 patients with localised disease, CRP and also the UCLA Integrated Staging System (UISS) and SSIGN scores were all shown to be independent prognostic predictors of RFS. 566 This was confirmed in a similar subsequent study (n = 130) for SSIGN score and CRP but not the UISS, with only CRP and platelets being independent prognostic predictors of 1-year OS. 567 In a later study by this group, CRP was shown to remain significantly prognostic when lifestyle factors such as smoking and obesity were included in the model. 568 In a study of 313 patients (21% with metastatic disease), preoperative CRP treated as three categorical variables (≤ 4.0 mg/l, 4.1–23.0 mg/l and > 23.0 mg/l) was independently prognostic for CSS (p = 0.003). Importantly, CRP added to the UISS prognostic model improved its accuracy by 3.8% at 5 years (p < 0.001). 569 The TNM-C score, which is based on CRP and the TNM staging system alone, was developed based on 249 RCC patients with advanced and localised disease, with CRP dichotomised as < or ≥ 5 mg/l and combined with the TNM staging system to generate four risk groups, with CSS stratified from 99% to 18% 5-year survival across the groups. 570 External validation was achieved in a further 290 patients, with a C-index of 0.865,570 and subsequently in an additional 518 patients with ccRCC, with a C-index of 0.85. 571 More recently, in a cohort of patients with localised ccRCC (n = 403), preoperative CRP was independently significantly associated with DFS and increased the prognostic accuracy of the SSIGN score. 572 However, the ability to increase the prognostic accuracy in this way will depend very much on the initial performance of the scoring system and its components; the addition of CRP to a model including TNM stage, grade and Karnofsky index did not improve the model’s performance, which already had a high predictive value of 88.1%. 573
A study from our own group investigated whether or not CRP measurement prior to nephrectomy adds to a published model that is solely based on preoperative factors, in this case age, sex, symptoms, tumour size, clinical T stage and metastatic status. 441 Based on 286 patients (84% clear-cell subtype) and with CRP dichotomized as ≤ or > 15 mg/l, 5-year survival rates of 72% (95% CI 65% to 78%) and 33% (95% CI 23% to 44%), respectively, were shown, with CRP an independent prognostic factor for OS (p < 0.006) and CSS (p < 0.001) and adding significantly to the preoperative score. 511
Clearly, serum CRP has significant prognostic potential and has the major advantage of being easily measured in hospital laboratories using existing assays, although it is worth noting that, in the more recent studies, the availability of different generations of higher-sensitivity CRP assays will have allowed quantification over a wider range. Challenges moving forward will include the possible effects of comorbidities on CRP levels and the optimal cut-off points, with, for example, the studies above using a variety of cut-off points including 2.5, 3, 4, 5, 7.5, 10 and 15 mg/l. Alternatively, the possibility of treating CRP as a continuous variable should be explored. Only three studies have adopted this,564,567,572 with two of these studies concluding that treating CRP as a categorical variable was best. 567,572
Serum amyloid A
Significantly elevated concentrations of the acute-phase protein serum amyloid A (SAA) have been reported in patients with RCC, particularly in patients with metastatic disease, with patients with localised disease having values that are largely similar to those of healthy control patients. 574–577 Three studies have now shown that SAA has significant independent prognostic value for CSS, although these studies included only moderately sized cohorts (n = 72–119), but of all stages. 574,575,578 The two largest and most recent studies (including the one that we undertook involving only ccRCC patients) provided remarkably similar results (HR 2.46, 95% CI 1.17 to 5.15; p = 0.017578 vs. HR 2.51, 95% CI 1.09 to 5.78; p = 0.030575). However, when we also included CRP in our model, SAA was no longer independently significant. 578
Ferritin
Serum ferritin is another acute-phase protein whose level is reported to be increased in patients with RCC, with the level increasing with stage and significantly correlated with tumour volume and with some evidence supporting the tumour as being a possible source of some of the circulating ferritin. 579–581 In a further analysis of serum ferritin in 158 RCC patients of all stages, grouping on the basis of both preoperative and postoperative ferritin combined (n = 103) relative to normal healthy control values (i.e. high or normal), and stage, was significantly independently associated with survival. 581,582 Preoperative serum ferritin alone was significant only at the univariate level582 and this was further confirmed in a smaller study (n = 52),581 with renal vein ferritin being higher than peripheral vein ferritin and both being significantly associated with survival, but not in multivariate analysis. Interestingly, in some studies, patients were excluded if they were anaemic, had been transfused recently or had comorbidities that included liver disease because of possible effects on ferritin and this may preclude its usefulness.
Erythropoietin
An initial study with 57 patients with RCC found increased plasma EPO levels in 63% of cases but no correlation with stage or grade. 583 However, subsequent studies (n = 165 and n = 195) reported associations of serum EPO with grade and stage and survival, although this was not significant on multivariate analysis. 584,585 In a more recent study examining both serum EPO (n = 138) and tissue EPO receptor (n = 56) expression, the association of EPO with stage and grade was confirmed and also the association between higher EPO levels and lower survival, but examination of patients with localised disease only (n = 110) found no association of EPO with DFS. 583 However, grouping the 47 patients analysed by both serum EPO and tissue EPO receptor expression identified a group with high levels of both and with worse CSS, although the numbers included were small. 583
Vascular endothelial growth factor
With the role played by VEGF in angiogenesis and its regulation by VHL protein ,and early studies reporting elevated circulating concentrations of VEGF in patients with RCC, particularly those with metastatic disease, exploring its potential as a prognostic marker was logical. 586–589 However, mixed results were obtained in survival analyses, with analysis of serum VEGF preoperatively in all subtypes of RCC (n = 146)590 or just ccRCC (n = 45)591 finding no significant association with outcome, a slightly larger study (n = 161) finding a significant association but only at the univariate level592 and the most recent study, of a similar size and with a similar patient mix (n = 124), finding that, on multivariate analysis, VEGF was an independent marker associated with CSS and RFS. 593 There was some indication that concentrations differed between subtypes in two of these studies but this is unlikely to have accounted for the differences in findings here. 592,593 Analysis of serum VEGF in 83 patients with non-metastatic ccRCC found serum VEGF to be a significant independent predictor of recurrence (p = 0.013). 594 RFS was significantly lower in the cases who stained positively for VEGF or who had higher serum concentrations, although there was no significant association between VEGF staining and serum VEGF. Interestingly, serum VEGF has been shown to increase markedly in most patients (n = 66, including 48 with distant metastases) following nephrectomy and in this study preoperative VEGF or the pre- to postoperative changes in serum VEGF were not significantly related to outcome. 595
Studies have also analysed plasma VEGF concentrations and a significant correlation has been reported between plasma concentrations and cytoplasmic VEGF staining. However, although tissue VEGF concentration correlated with outcome, no such relationship was seen for circulating VEGF concentration. 596 Using carefully prepared citrated plasma samples to minimise release of VEGF from platelets, plasma VEGF concentration prior to surgery has been shown to be significantly associated with CSS (n = 74 patients, including 67 ccRCC patients and 22 stage IV patients), but this is lost on multivariate analysis. 597 Focusing on just the clear-cell subtype but including all stages (n = 102), plasma VEGF was associated with T stage and grade but not nodal or metastatic disease. However, both tissue and plasma VEGF concentrations were significantly associated with PFS and CSS. 598 A later study analysing plasma samples in ccRCC patients only (n = 68) reported higher VEGF concentrations in patients with nodal or metastatic spread but found no significant association with OS. 599
Clearly, although VEGF appears to have prognostic potential in several studies, there are inconsistencies in the findings, probably reflecting the heterogeneity of the studies in terms of RCC subtypes, whether or not studies were restricted to patients with localised disease only and, possibly, even more critically, whether serum or plasma was used and how samples were processed. It is now recognised that platelet release of VEGF is a main contributor to VEGF measured in serum, but equally this may also contribute to circulating VEGF concentrations measured in plasma, depending on the sample handling conditions. 600–603 Platelet number and serum VEGF concentrations have been reported to be highly significantly correlated in patients with advanced cancer and, hence, the prognostic value of serum VEGF may actually be related to the prognostic value of platelet number or thrombocytosis. 604,605 These and other pre-analytical aspects of VEGF measurement are considered further in Chapter 14.
Carbonic anhydrase IX
Interest in CAIX in RCC stemmed initially from the observation of the selective binding of a monoclonal antibody (G250) to RCC tissue but not normal proximal epithelium. 606 The antigen was later identified as CAIX, a hypoxia-inducible protein involved in the regulation of intra- and extracellular pH and upregulated as a consequence of VHL inactivation in RCC. 607 Studies examining the prognostic value of CAIX expression in tissue samples have largely found high expression to be related to better outcomes, although inconsistencies between studies have been found and it is still not clear whether or not CAIX has independent prognostic value, as we reviewed in 2016. 428 However, with the recognition that CAIX exists as a shed form in the blood and urine, several studies have explored soluble CAIX as a prognostic marker in RCC, with most using the same assay as the one that we have used in this programme, making interstudy comparison more feasible, as summarised by Závada et al. 608 In patients with ccRCC (n = 91; n = 79 with localised disease), mean serum CAIX concentrations were significantly higher in patients with metastatic disease (p = 0.004), with concentrations correlating with tumour grade, size and stage. 609 In univariate analysis, serum CAIX concentration was significantly associated with early relapse in patients with localised disease. 609 In a larger study involving 361 RCC patients with all subtypes, serum CAIX concentration was related to stage and grade but failed to reach significance as a prognostic marker for CSS when dichotomised around the median in the cohort of patients with ccRCC (n = 287) and was not a significant prognostic factor on multivariable analysis. 610 Our subsequent study analysed serum CAIX, CRP and plasma OPN prior to nephrectomy in 216 patients with ccRCC (24% with M1 disease) and found CAIX to be significantly associated with CSS, DFS and OS on univariate analysis but independently prognostic for OS only on multivariate analysis. 486 The combination of the three markers outperformed stage.
Matrix metalloproteinase-7
Matrix metalloproteinase-7 (MMP-7) (matrilysin) is a member of a family of zinc-containing enzymes involved in proteolytic degradation of many extracellular matrix components and, hence, involved in many of the pathological processes in cancer, particularly invasion and angiogenesis. Expression of MMP-7 in RCC tissue has been shown to be increased relative to normal kidney tissue and independently prognostic for CSS or OS. 611,612 Following a two-dimensional polyacrylamide gel electrophoresis study of a RCC cell line with screening of separated proteins using RCC patient sera to detect immunogenic reactivity, pro-MMP-7 was detected and subsequently reported as being elevated in serum from RCC patients. 613 Using an assay measuring pro-MMP-7, MMP-7 and TIMP-1 complexed forms (according to the manufacturer’s information), plasma concentrations were found to be significantly elevated in RCC patients (n = 97, including 45 patients with metastatic disease) compared with healthy control patients. This was particularly the case for patients with distant metastases and, on multivariate analysis, MMP-7 was found to be independently prognostic for CSS (HR 2.70, 95% CI 1.39 to 5.24; p = 0.003). 614 Pilot data from our group also support plasma MMP-7 as being associated with tumour size and stage (unpublished data, 2017).
Osteopontin
Osteopontin, also called secreted phosphoprotein 1, is a member of the small integrin-binding ligand N-linked glycoprotein family (SIBLING). One of the most abundant non-collagenous extracellular matrix proteins in bone, it is now known to have a widespread tissue distribution and plays a role in many processes including cell adhesion, remodelling, angiogenesis and inflammation. 379 Increased tissue expression has been reported for many cancer types, including renal cancer. 379,615 The first study to demonstrate an independent prognostic role for OPN in RCC involved 80 patients of all stages and subtypes. Plasma OPN was significantly elevated in patients with metastatic disease and was the only factor, together with the presence of metastases, out of several examined to retain independent prognostic significance for CSS in a multivariate model. 616 In a larger study involving 269 patients with renal cancer of all subtypes, analysis of OPN in plasma and serum samples (n = 75 and 116, respectively, with the ccRCC subtype) found higher concentrations in plasma (median 2.3-fold higher concentration than in serum) and independent prognostic significance for CSS in ccRCC, with higher concentrations linked to poorer survival, particularly for plasma, although almost 50% of patients included had metastatic disease. 617 The lower OPN concentrations in serum compared with plasma have been found in other studies and may be accounted for, at least in part, by the known cleavage by thrombin during clotting; this is discussed further in Chapter 13. 618 Surprisingly, neither stage nor grade were independently prognostic in this study. Subsequently, our study focusing on ccRCC only (n = 216) found preoperative plasma OPN to be significantly prognostic for OS, CSS and DFS but only at the univariate level and not the multivariate level. 486 This may have been because CRP was included in our model, which was strongly prognostic and correlated with OPN, and also because only 24% of participants had metastatic disease. Interestingly, a group of patients with low-stage RCC who had higher OPN concentrations were identified as being at high risk of death, mainly from non-cancer-related causes. 486 Importantly, and although not relevant to the initial study proposed here on localised RCC, OPN was one of the markers identified as being a strong prognostic marker for patients with metastatic RCC in the placebo arms of Phase 2 and 3 trials of pazopanib (Votrient; Novartis Pharmaceuticals UK Ltd, Camberley, UK), outperforming routine clinical indicators. 619
Immunosuppressive acidic protein
Serum immunosuppressive acidic protein (IAP) was first described in 1986 as being elevated in patients with high-stage RCC compared with low-stage disease,620 which was initially confirmed in a slightly larger study of 66 patients and 133 previously untreated RCC patients, in which IAP had an area under the ROC curve of 0.894 for metastatic disease. 621,622 IAP was also found to be associated with higher grade in 181 RCC patients, with higher IAP concentrations indicating a more than fourfold risk of higher grade. 623 In terms of survival, an initial study (n = 143) showed higher IAP concentrations to be associated with poorer survival at 3 years, although this was not analysed at the multivariate level;624 a subsequent smaller study involving 92 patients of mixed stages confirmed the association with survival, although this was not significant on multivariate analysis. 625 In the most recent study from 2006, IAP doubling time when measured longitudinally after nephrectomy in patients with localised disease who subsequently relapsed (n = 125) was independently prognostic for survival (p = 0.0026). 626 Using the cut-off point of a doubling time of > or < 200 days, 3-year survival was 58.9% and 12.5%, respectively. 626 A limitation of several of these studies, which has probably accounted for the restriction to Japanese studies and the long time period over which the studies span, is the availability of the assay, with all but the last study (which used a nephelometric assay) having used radial immunodiffusion to measure IAP. In addition, it is not clear for most studies which histological subtypes of RCC were included.
Tumour M2 pyruvate kinase and thymidine kinase
Serum concentrations of the glycolytic dimeric M2 isoenzyme form of tumour M2 pyruvate kinase (TuM2-PK) were initially reported simultaneously in two studies from 1999 to be increased in some patients with RCC compared with patents with benign diseases or healthy control patients, correlating with tumour stage and grade in both studies and with grade in one study. 627,628 These results have been extended, with more recent larger studies largely confirming these findings. 629,630 In patients with RCC of varying subtypes (n = 116), preoperative TuM2-PK concentration was also found to be a significant independent prognostic marker for disease recurrence (HR 7.3, 95% CI 1.1 to 47.8; p = 0.037), with a crude 5-year RFS of 55% for patients with elevated concentrations compared with 94% for patients with normal concentrations (p < 0.001). 629 This study also examined the prognostic potential of thymidine kinase 1, which had previously been reported to be associated with grade, stage and size in in a small study involving 27 patients,631 and found similar results as for TuM2PK in terms of independent prediction of disease recurrence, with a crude 5-year RFS of 21% and 90% (p = 0.002) for patients with elevated or normal concentrations of TK1, respectively. 629
Soluble interleukin-2 receptor
Serum concentrations of soluble interleukin-2 receptor (sIL-2R) have been shown to be significantly increased in RCC patients, with significant associations with clinical stage; higher sIL-2R concentrations were associated with poorer CSS, although this was not examined at a multivariate level and only 52 patients were included, with subtypes being unclear. 632 However, a more recent study involving only patients with ccRCC (n = 70) has confirmed these findings. 633 Significant correlations with stage were demonstrated; in particular, there were higher concentrations in patients with stage IV disease (p < 0.001) and the group with a higher sIL-2R concentration was associated with shorter CSS (p < 0.05). 633 Larger studies including multivariable analysis are now needed.
Basic fibroblast growth factor
Following early demonstrations of elevated serum basic fibroblast growth factor (bFGF) concentrations in approximately 50% of patients with RCC and a trend towards higher concentrations with increasing stage and grade,634,635 significant correlations with stage and grade were reported in a larger study involving 206 patients. 636 However, although higher bFGF concentrations were associated with poorer survival, they were not significantly associated with outcome on multivariate analysis. 636 In the most recent study performed in 2005 (n = 74), no significant association between serum bFGF and stage was apparent, although higher concentrations of bFGF were found in patients with metastatic disease. 540
Conclusions and prioritisation
Clearly, several circulating markers reflecting diverse aspects of RCC biology appear promising but need further systematic evaluation, including whether a multiplex panel would be most effective and the optimal combination of markers and whether or not these markers could add value to or outperform existing clinicopathological scoring systems. On the basis of the evidence presented in the previous sections, OPN, VEGF (serum and plasma), CAIX and CRP were prioritised to take forward in the initial prognostic study. Although fibrinogen appeared to be very promising, this marker was not feasible as it would need to be measured prospectively on freshly collected, rather than frozen, citrated plasma. MMP-7 also appeared to be promising but further work needs to be carried out to determine the effects of inhibitors such as TIMP-1 on the assays, the detectability of pro-MMP-7 and the relative suitability of plasma or serum for the assays. Lack of commercially available assays preclude investigating IAP and further studies are needed with larger numbers of patients to investigate TK1 and TuM2-PK. Several other markers do not seem to have been pursued for many years despite earlier promising findings and this may relate to a lack of assays currently. Several markers also appear to have promise although currently only at the single-study level, covering a wide range of tumour biology. These include insulin-like growth factor-1,637 soluble B7 family ligand (B7-H3),633 tumour-associated trypsin inhibitor638 and vitamin D. 639 In the future it is possible that these may be explored using the RTB assembled within this programme, once further supportive evidence is available.
Renal transplantation
We have restricted this review to circulating plasma or serum biomarkers. Although, intuitively, urine would appear to be the most obvious choice of biological fluid for the detection of biomarkers, in the early postoperative period following kidney transplantation several considerations reduce the clinical validity of urinary biomarkers. First, some patients, particularly those who receive a pre-emptive (prior to starting RRT) kidney transplant, maintain residual urine output from their native kidneys, which will confound the measured values of biomarkers. Second, patients who develop DGF and who do not have any residual urine output will have minimal urine output. Third, from an analytical and scientific perspective, there are a number of issues concerning the normalisation of urinary measurements of biomarkers.
Literature search strategy
Publications were searched using PubMed. Search terms used were as broad as possible to maximise coverage (Figure 39), with subsequent manual filtering as indicated to select the relevant references. Reasons for inclusion or exclusion at the various stages are as indicated. This strategy was selected following an iterative testing process, checking to see if selected known diverse references were detected, and for this reason the term ‘biomarker’ was not included as it was too restrictive in some cases. To provide further background information or context for specific clinical or biological areas in which the biomarkers were discussed, additional specific references were then searched for as needed.
FIGURE 39.
The systematic literature review process adopted for circulating biomarkers of DGF in RT. The search for publications relating to circulating biomarkers of DGF in RT to allow a focus on serum and plasma biomarkers was carried out on 11 October 2015 in PubMed. DNA, deoxyribonucleic acid; miRNA, microribonucleic acid.

Review of serum and plasma biomarkers of delayed graft function following renal transplantation: diagnosis and prognostic utility for long-term outcome
Although the focus of this programme was on protein biomarkers, given the relative paucity of biomarkers with a significant level of supporting evidence currently emerging in this area of RT, at the end of this section we also mention some promising non-protein studies.
Creatinine
Although not a protein biomarker, creatinine is of course widely available and routinely measured. Creatinine is a product of the metabolism of creatine, which is released from muscle. Creatine is non-enzymatically dehydrated to creatinine in the liver. Creatinine is freely filtered in the glomerulus and does not undergo significant metabolism or reabsorption in the kidney. 640 As such, it is the usual clinical standard for monitoring kidney function post transplantation, together with urinary output. Consequently, it is the benchmark against which other biomarkers are compared, which in itself is problematic as it is actually quite a poor biomarker in several ways. 641 For example, increases or decreases in its concentration lag behind true changes in kidney function, including by a number of days, and it is influenced by a wide variety of factors such as age, sex, muscle mass, level of nutrition (including protein intake) and liver function. 642 In addition, around 10% of creatinine is cleared by tubular secretion, which can be disrupted by particular medications, for example trimethoprim and cimetidine. Post transplantation, the use of serum creatinine suffers from all of these limitations. 643 Glomerular filtration rate (GFR) estimates based on serum creatinine, including the commonly used Modification of Diet in Renal Disease equations644 and the Cockcroft and Gault equation,645 are not useful unless a steady state is present, which of course is not the case post renal transplant. At lower GFRs, secretion of creatinine increases as a proportion of creatinine clearance, causing these equations to overestimate the GFR.
Cystatin C
Cystatin C is a 13.4-kDa cysteine protease inhibitor produced by all nucleated cells. It is an endogenous marker of GFR as it is freely filtered by the glomerulus and is not reabsorbed into the circulation or secreted. 646 In an early study of serum cystatin C, 41 consecutive deceased donor kidney transplant patients had concentrations measured before and 1, 3, 6 and 10 days post surgery. 647 The study demonstrated that in patients with DGF the serum cystatin C concentration did not fall. It is unclear from the paper how many patients developed DGF or what the shape of the ROC curve was. 647 In another study, serum cystatin C and other biomarkers [serum neutrophil gelatinase-associated lipocalin (NGAL) and IL-18] were measured for the first 3 days in 78 recipients of deceased donor kidney transplants, of whom 26 had DGF. 648 Serum cystatin C values were effective at distinguishing DGF from immediate graft function, with an AUC of 0.83 on day 1 postoperatively.
Fonseca et al. 649 investigated the potential of urinary NGAL to predict DGF and 1-year kidney transplant function in comparison with cystatin C. This prospective study measured serum cystatin C at days 0, 1, 2, 4 and 7 post transplant in 20 consecutive patients, of whom 18 developed DGF. Day 1 cystatin C concentration predicted DGF, with an AUC of 0.95. Cystatin C was further investigated as a comparative biomarker in a prospective study of malondialdehyde (MDA) as a predictor of DGF. 650 Plasma concentrations were measured preoperatively (day 0) and postoperatively (days 1, 2, 4 and 7) in 40 consecutive recipients of kidney transplants, of whom 18 developed DGF. Day 1 serum cystatin C concentrations accurately predicted DGF, with an AUC of 0.91. The same research group utilised a multiple biomarker approach to detect DGF, which included urinary NGAL, serum leptin, serum MDA and serum cystatin C. 651 This was a prospective cohort study of 40 consecutive patients, including deceased donor and living donor transplants, of whom 18 developed DGF. Serum cystatin C had an AUC of 0.914 at 8–12 hours post surgery. The most informative combination was a triple biomarker approach that included serum creatinine, MDA and serum cystatin C; this combination had an AUC of 0.96. A further analysis of the data demonstrated that there was a trend for cystatin C values on day 1 post transplant to correlate with kidney transplant function at 3 months when divided into upper, middle and lower tertiles. Serum cystatin C demonstrates very good utility as an early predictive biomarker of DGF following kidney transplantation.
Neutrophil gelatinase-associated lipocalin
In a study of 41 consecutive deceased donor kidney transplant patients, serum NGAL was measured before and 1, 3, 6 and 10 days post surgery. 647 The study demonstrated that, in patients with DGF, the serum NGAL concentration did not fall. It is unclear from the paper how many patients developed DGF or what the ROC curve was. However, the authors concluded that serum NGAL needed to be investigated further as a potential marker of DGF. 647 A prospective observational study measured plasma NGAL in 41 patients receiving a deceased donor (n = 39) or living donor (n = 2) kidney transplant. 652 DGF developed in 15 patients, all of whom had received a deceased donor kidney. The plasma NGAL ROC curve at 12 hours for predicting DGF demonstrated an AUC of 0.97. In another study, serum NGAL and other biomarkers (serum cystatin C and IL-18) were measured for the first 3 days in 78 recipients of deceased donor kidney transplants, of whom 26 had DGF. 648 Serum NGAL values were ineffective at predicting DGF. In contrast, in a further study, serum NGAL concentration at 24 hours post transplantation was shown to be an accurate predictor of DGF, which affected 6 of the 33 transplant patients, with an AUC of 0.82. 653 Of these 33 patients, 20 received a deceased donor kidney and 13 received a living donor kidney.
A retrospective study measured serum NGAL (and serum IL-18) in 59 recipients preoperatively and at days 1, 5 and 14 postoperatively. 654 The day 1 serum NGAL concentration had an AUC of 0.86 in the 14 patients who developed DGF. A further retrospective study analysed serum NGAL in 67 patients, of whom 27 received kidneys donated after a circulatory death. 655 The function in the DCD kidneys never recovered, which may account for the AUC of 0.99 for predicting DGF on the first day postoperatively. Hollmen et al. 656 measured serum NGAL in 176 consecutive deceased donor kidney transplant recipients utilising two different methods. Sixty-six patients developed DGF and serum NGAL was significantly higher in this group, with an AUC of 0.853 when measured on day 1. Serum NGAL also predicted DGF lasting > 14 days, with an AUC of 0.825. In a further study of 97 patients (17 living donor transplant recipients and 80 deceased donor transplant recipients), of whom 20 developed DGF, there was no correlation between donor plasma NGAL concentration (and urinary NGAL concentration) and post-transplant DGF. 657 In this study, plasma NGAL predicted DGF, with an AUC of 0.73 at 6 hours, 0.80 at 12 hours and 0.85 at 24 hours post transplant.
A different approach was taken in two studies that investigated the serum concentrations of NGAL in the kidney donors prior to surgery. Hollmen et al. 658 collected serum and urine samples from 99 consecutive deceased kidney donors prior to the operation and their 176 recipients. Serum NGAL concentrations failed to predict DGF. Muller et al. 659 performed a prospective, multicentre observational study that included 146 brain-dead donors, leading to 243 transplants, with 56 transplant recipients developing DGF. The concentrations of serum NGAL in the donors failed to predict DGF or normal transplant function in recipients.
There is now a significant body of evidence suggesting that serum NGAL (less so for plasma NGAL) has good potential for use as an early biomarker for predicting DGF when measured in the recipient but not in the donor.
Aminoacylase-1
Serum aminoacylase-1 (ACY-1) was identified as a potential outcome biomarker following mass spectrometry analysis of serum samples before and on day 2 post transplant from five patients with DGF and five with immediate transplant function. 660 Following development of an ELISA for ACY-1, analysis of the results from an initial validation cohort (n = 55 patients) showed a moderate predictive value for ACY-1 on day 1 or 2 post transplant, complementing cystatin C. A further validation cohort of 194 patients (54 patients with DGF) confirmed this association, with a day 1 AUC of 0.74 for ACY-1, 0.9 for cystatin C and 0.93 for the combination of ACY-1 and cystatin C. 660 Importantly, however, analysis of long-term follow-up data for 54 patients with DGF showed a highly significant association between day 1 or day 3 serum ACY-1 concentration and dialysis-free survival, mainly associated with kidney DBD and offering the potential for use in stratification of follow-up.
Other promising biomarkers
A number of additional biomarkers have been evaluated but either only at the single-study level or in small patient numbers. The most promising of these, all of which would require further validation before being considered for multicentre evaluation, are described briefly in the following sections.
Complement
Ischaemia–reperfusion injury in the kidney results in the activation of the complement cascade. A terminal panel of complement molecules (C3a, C5a and C5b-9/membrane attack complex) was analysed following kidney transplant reperfusion. 661 Seventy-five kidney transplant recipients were divided into early graft function, slow graft function and DGF groups. Blood samples were collected from the renal vein during reperfusion. Analysis revealed that C5b-9/membrane attack complex concentrations were two to three times higher in DGF patients than in patients with early and slow graft function (p < 0.005). In addition, C5b-9/membrane attack complex concentrations had a relatively high clinical sensitivity and specificity (70–87.5%) for the prediction of early and 1-year kidney transplant function. ROC curves were not calculated, which limits the interpretation of this study
C-terminal agrin fragment
The C-terminal agrin fragment (CAF) is a cleavage product of agrin, the major proteoglycan (PG) of the glomerular basement membrane. It has been proposed that elevated CAF values may be related to reduced glomerular filtration and clearance. Serum CAF and creatinine concentrations were measured in 96 healthy individuals and in 110 patients undergoing kidney transplantation, before and after transplantation. 662 Serum CAF concentrations at day 1 and day 3 were significantly associated with DGF (40 patients), with an AUC of 0.81. This small study demonstrates moderate clinical utility for CAF in the early prediction of DGF.
Fms-like tyrosine kinase
Ischaemia–reperfusion induces tubular epithelial and endothelial cell damage in the kidney transplant, which contribute to the development of DGF. Chapal et al. 663 prospectively assessed the kinetics of the soluble VEGF receptor, soluble Fms-like tyrosine kinase-1 (sFlt-1), in 136 consecutive kidney transplant patients. Patients with DGF had higher sFlt-1 concentrations at all time points during the first 7 days following kidney transplantation. Multivariate analysis demonstrated that a peak plasma sFlt-1 concentration of ≥ 250 pg/ml was associated with a 2.5-fold increase in the risk of DGF (p = 0.04). ROC curves were not calculated, which limits the interpretation of this study.
Immunoglobulin A antibodies to beta-2-glycoprotein 1
The prevalence of immunoglobulin A (IgA) anti-beta-2-glycoprotein 1 antibodies (IgA-aβ2GP1-ab) in patients on dialysis is elevated (> 30%) and these antibodies correlate with mortality and cardiovascular morbidity. Isolated IgA-aβ2GP1-ab are associated with thrombosis. A single-centre prospective study evaluated the effect of IgA-aβ2GP1-ab in 269 patients following kidney transplantation. 664 The presence of IgA-aβ2GP1-ab in pre-transplant serum was examined retrospectively. Eighty-nine patients were positive for IgA-aβ2GP1-ab. Multivariate analysis showed that the presence of IgA-aβ2GP1-ab was an independent risk factor for early graft loss (p = 0.04) and DGF (p = 0.04). ROC curves were not calculated, which limits the interpretation of this study.
Pregnancy-associated plasma protein A
Pregnancy-associated plasma protein A (PAPP-A) has been shown to be a marker of acute coronary syndrome and cardiovascular pathology. Blood samples were taken from 178 patients prior to receiving their first deceased donor kidney transplant. 665 Sixty-one patients subsequently developed DGF. Pre-transplant PAPP-A values were significantly elevated in the group of patients with DGF. Multivariate analysis showed that PAPP-A was an independent risk factor for DGF although ROC curves were not examined.
Interleukin-16
Alachkar et al. 666 analysed serum and urine samples from 61 patients 48 hours following kidney transplantation for a panel of 23 cytokines including IL-16, which has been implicated in IRI. Six patients developed DGF. The AUC was 0.74 for serum IL-16 in this small study and, therefore, does not provide any compelling evidence to support the use of IL-16 as an early predictor of DGF.
Interleukin-18
Interleukin-18 is a cytokine that mediates inflammation and ischaemic tissue injury in many organs including the proximal tubules in the kidney. 667 Hall et al. 648 compared alternative serum biomarkers with creatinine for predicting DGF. IL-18 and other biomarkers (serum cystatin C and serum NGAL) were measured prospectively for the first 3 days in 78 recipients of deceased donor kidney transplants, of whom 26 had DGF. Serum IL-18 measurements were unable to distinguish DGF from slow graft function or immediate graft function. A retrospective study measured serum IL-18 (and serum NGAL) in 59 recipients preoperatively and at days 1, 5 and 14 postoperatively. 654 Day 1 serum IL-18 had an AUC of 0.63 in the 14 patients who developed DGF and, therefore, had limited value. Serum IL-18 does not appear to be useful for predicting DGF.
Leptin
Leptin is removed from circulation primarily by the kidney and could be considered a surrogate marker for kidney function. A prospective study was performed to measure the concentrations of leptin in 40 consecutive patients at days 0, 1, 2, 4 and 7 following kidney transplantation. 668 Median leptin concentrations were significantly higher in patients developing DGF (n = 18) at all times points. The leptin reduction rate between pre transplant and 1 day postoperatively moderately predicted DGF, with an AUC of 0.73. The day 1 serum leptin concentration predicted DGF, with an AUC of 0.76. Separating the analysis by sex improved the performance of leptin in predicting DGF, with an AUC of 0.86 for male sex. A further prospective cohort study of 40 consecutive kidney transplant patients utilised a multiple biomarker approach including serum leptin. 651 Both deceased donors and living donors were included in the study, with 18 transplant recipients developing DGF. Serum leptin had an AUC of 0.76 at 8–12 hours post surgery. Serum leptin appears to be only moderately useful for predicting DGF.
Resistin
Brain death triggers a complex cascade of molecular and cellular events resulting in the release of inflammatory mediators. The level of resistin increases during several inflammatory diseases and after intracerebral bleeding or head trauma. It promotes endothelial activation and may initiate an inflammatory response. The potential role of plasma resistin values in the brain-dead kidney donors in predicting DGF in recipients was analysed in 63 kidney transplant patients. 669 Twenty-six recipients of kidneys from living donors were used as control subjects. Donor resistin values in the recipients of kidneys from brain-dead donors correlated with DGF, with an AUC of 0.765. Donor resistin values appear to be of only moderate clinical utility in predicting DGF.
Galbeta1,4GlcNAcalpha2,6-sialyltransferase
Galbeta1,4GlcNAcalpha2,6-sialyltransferase (ST6GalI) is an acute-phase reactant whose release from cells can be induced by proinflammatory cytokines. It has been hypothesised that patients with CKD may have circulating concentrations of ST6GalI, which might increase the risk of DGF. Serum concentrations of ST6GalI were measured in 70 patients immediately before receiving a kidney transplant. 670 The mean serum level of ST6GalI was significantly higher in the patients than in 19 control subjects. Twenty patients developed DGF and had significantly higher concentrations of ST6GalI pre transplant than 50 patients who had immediate graft function. In a multivariate analysis the ST6GalI level was found to be an independent risk factor for the development of DGF. ROC curves were not calculated, which limits the interpretation of this study.
Stem cell factor
Alachkar et al. 666 investigated whether or not a panel of serum and urinary cytokines could act as early biomarkers for predicting DGF and slow graft function. Serum and urine samples from 61 patients were collected 48 hours following kidney transplantation and analysed using a multiplex ELISA technique to measure concentrations of 23 cytokines. One of the cytokines included stem cell factor (SCF), which has been implicated in early inflammation and tissue fibrosis. Six patients developed DGF and eight developed slow graft function. The AUC was 0.88 for serum SCF; however, sampling was performed at 48 hours following kidney transplant, which would reduce its clinical utility as an early predictive biomarker of DGF.
Hydroxyeicosatetraenoic acids
Eicosanoids are the active metabolites of arachidonic acid and have been implicated in the pathogenesis of IRI in the kidney. 20-Hydroxyeicosatetraenoic acid (HETE) is one such active metabolite and is generated by cytochrome P450 enzymes. To assess the potential roles of eicosanoids the concentrations of lipoxygenase-derived 5-, 12- and 15-HETE concentrations were measured in 69 kidney transplant recipients. 671 The kidney transplant recipients were divided into early graft function, slow graft function and DGF groups. Blood was taken directly before and immediately following kidney transplant reperfusion. Application of newly proposed cut-off limits for 5-HETE, 12-HETE and 15-HETE resulted in 72.5–81.5% sensitivity and 50–54% specificity for slow graft function/DGF prediction. A mixed-model analysis revealed that recipients classified according to results of the 5-HETE and 15-HETE cut-off points were able to predict 1-year kidney transplant function. A further study measured 20-HETE concentrations during the first 5 minutes of kidney transplant reperfusion and analysed whether or not the concentrations were associated with post-transplant kidney function. 672 Sixty-nine kidney transplant recipients were divided, according to their outcome, into early graft function, slow graft function and DGF groups. The sensitivity, specificity, PPV and NPV of 20-HETE in discriminating early and slow graft function from DGF were 69%, 54%, 74% and 48%, respectively. Both of the studies included a relatively small group of patients and did not calculate the ROC curves, which limits the interpretation of these studies.
Malondialdehyde
Ischaemia–reperfusion injury results in cellular death mediated by a number of different pathways. Oxidative stress is one such pathway, which leads to the generation of reactive oxygen species. MDA is a marker of oxidative stress and has been investigated as a potential biomarker of DGF and transplant function at 1 year in a prospective study of 40 consecutive kidney transplant patients. 650 Plasma concentrations of MDA were measured preoperatively (day 0) and postoperatively (days 1, 2, 4 and 7). At all time points after transplantation, mean MDA concentrations were significantly higher in patients developing DGF (n = 18). Day 1 MDA concentrations accurately predicted DGF, with an AUC of 0.90; the performance of MDA was higher than that of serum creatinine (AUC of 0.73) and similar to that of cystatin C (AUC of 0.91). Multivariable analysis revealed that MDA concentrations on day 7 represented an independent predictor of 1-year graft function. Another prospective cohort study of 40 consecutive patients including deceased donors and living donors utilised a multiple biomarker approach to detect DGF, including urinary NGAL, serum leptin, serum MDA and serum cystatin C. 651 Serum MDA had an AUC of 0.90 at 8–12 hours post surgery. The most informative combination was a triple biomarker approach that included serum creatinine, MDA and serum cystatin C, with an AUC of 0.96. MDA used either alone or in combination with other biomarkers demonstrates good potential as an early biomarker of DGF.
Neutrophil–lymphocyte ratio
The NLR is an indicator of inflammatory status and has been used to assess outcome in critically ill surgical patients. A retrospective study was performed to investigate the effect of preoperative elevated NLR on the kidney transplant recipient with regard to the risk of DGF. 673 The preoperative white blood cell (WBC) count of 398 kidney transplant recipients was analysed. In total, 249 patients received kidneys from donors after brain death (DBD), 61 received kidneys from donors after circulatory death and 88 received kidneys from living donors. A NLR of > 3.5 was considered to be elevated. A total of 103 patients developed DGF, of whom 67 had a NLR of > 3.5. Multivariate analysis demonstrated that a NLR of > 3.5 had a HR of 10.673 (95% CI 6.151 to 18.518). ROC curves were not calculated, which limits the interpretation of this study.
Regulatory T-cells
Regulatory T-cells have been shown to be protective in models of AKI and their suppressive function is predictive of AKI following kidney transplantation. The role of regulatory T-cells as a biomarker of DGF has been explored in a prospective observational cohort study. 674 Fifty-three deceased donor kidney transplant recipients were divided into those who developed AKI (n = 37), including DGF and slow graft function, and those with immediate graft function (n = 16). Pre-transplantation peripheral blood CD4CD25FoxP3 regulatory T-cell frequency was quantified by flow cytometry. Regulatory T-cell suppressive function was measured by suppression of autologous effector T-cell proliferation by regulatory T-cells in co-culture. In univariate and multivariate analyses accounting for the effects of CIT and donor age, regulatory T-cell suppressive function accurately predicted AKI (DGF and slow graft function), with an AUC of 0.82. The same group also performed a prospective observational cohort study utilising flow cytometry to measure pre-transplant recipient circulating CD4+CD25+CD127lo/– and CD4+CD127lo/– tumour necrosis factor receptor 2 (TNFR2)+ regulatory T-cells in 76 deceased donor kidney transplant recipients, of whom 18 patients developed DGF. 675 The ROC curves demonstrated an AUC of 0.75 and 0.77, respectively, for the percentage and absolute number of CD4+CD127lo/–TNFR2+ regulatory T-cells in predicting DGF. Neither of these studies demonstrate good clinical utility of regulatory T-cells in predicting DGF. The first study had a good AUC but combined DGF with slow graft function.
Summary
The data presented in the systematic review demonstrate that a number of small studies have investigated a range of potential serum/plasma biomarkers to enable the early detection of DGF, but relatively few have looked at longer-term outcomes. The studies have been heterogeneous in terms of the populations studied and definitions of DGF, although most have applied the definition of DGF of receipt of haemodialysis in the first week following kidney transplantation. The most promising serum biomarkers for the early detection of DGF appear to be NGAL and serum cystatin C, with ACY-1 validated but only in a single centre to date. The use of cystatin C is becoming more widespread throughout the health-care system and with a growing familiarity it may well be utilised in the future as an earlier biomarker of DGF. In the case of NGAL, there remain issues of standardisation with respect to which cut-off values to recommend.
A number of emerging biomarkers have been studied in AKI outside kidney transplantation, with the most recent candidates being TIMP-2 and insulin-like growth factor-binding protein 7 (IGFBP7). It is only natural to assume that studies of these biomarkers will follow in the setting of kidney transplantation and predicting outcomes. The most recent study investigating urinary TIMP-2 and IGFBP7 was unfortunately rather unimpressive. 676 The most promising urinary biomarker to date is NGAL and a number of studies have demonstrated its potential utility. However, as discussed earlier, there are significant issues surrounding the use of urinary biomarkers post kidney transplant.
There is a distinct lack of studies in the literature that have investigated the clinical use of biomarkers in predicting longer-term outcomes. It has been proposed by many experts that panels of multiple biomarkers may be able to improve the predictive value but, again, such studies are lacking. The most obvious panel to utilise would include serum cystatin C and serum NGAL, which have both demonstrated very good utility in predicting DGF in the early phase following kidney transplantation; ACY-1 should also be explored further, particularly in view of its promising prognostic performance. There is now an excellent opportunity to validate existing biomarkers and investigate novel biomarkers in a cohort of kidney transplant patients with a well-described phenotype.
Chapter 13 Exploring technical aspects of biomarker assays: verification, validation and pre-analytical variables
Evaluation of the validity and performance of assays is fundamental to their introduction to clinical practice. This chapter describes some of the aspects and concepts of, and guidelines for, the technical evaluation of assay performance together with pre-analytical considerations, before describing the practices developed and results generated in a series of such studies undertaken within this programme, in preparation for the analysis of biomarkers in specific research studies.
Appraisal of assay performance
General concepts
Ensuring that assays are appropriately validated is critical in terms of ensuring that measurements of biomarkers are accurate and reproducible, both across time and between laboratories. It is important within this to ensure that assays are ‘fit for purpose’ and deliver the level of performance needed for the study phase or clinical situation, avoiding a dogmatic approach to guidelines that may not be completely relevant at some stages. 385,391,393 The level of validation may vary depending on whether the assay is for research use only or requires CE marking or FDA approval and is for use in a hospital laboratory or pharmaceutical laboratory, for example. However, many immunoassays are relatively easy to purchase and use and many studies are undertaken by researchers with the assumption that, because they are commercially available, they will be ‘fit for purpose’. It is increasingly recognised that this is not the case and examples affecting specific analytes include studies using an ELISA (USCN Life Science, Wuhan, China) for CUB and zona pellucida-like domains protein 1 (CUZD1), which actually measured CA-125,677 and an ELISA for soluble hemojuvelin from the same company that did not detect the specified target but some unknown protein. 678 A key issue that has been highlighted is the plethora of biotech companies that have sprung up, marketing a very wide range of immunoassays and antibodies, with several companies often using the same reagents, although this is not always clear, with very little apparent validation. 386 The extent of this problem was really made apparent with the testing of > 5000 commercially available antibodies using immunohistochemistry and Western blotting, in which almost 50% failed, although importantly not all had been certified by the manufacturers for these specific applications and only a generic protocol was used for all, with a limited range of sample types examined. 679 This type of problem is not restricted to antibodies, with other laboratory biological reagents also posing problems and with limited access to information to resolve such issues because of commercial sensitivities. 680 Although not having the same impact as for clinically used biomarkers, such issues when encountered in research laboratories are very costly in terms of inappropriate conclusions regarding clinical utility and utility and the money spent and samples and time used. Suggestions of how to minimise the impact of antibody-based problems have been made, covering the various stakeholders. 383,386 Clinical laboratories are not immune to such problems either, with pitfalls and a lack of consistent results identified for gastrin,681 growth hormone and insulin-like growth factor,682 cardiac troponin388 and even serum creatinine683 measurements, for example, depending on the assays used.
Guidelines
Numerous guidelines exist to provide a framework and set consistent standards for assay validation. The CLSI produces some of the most widely used and accepted standards and guidelines for clinical laboratory measurements, some of which are mandatory for certain regulatory bodies or accreditation. 684 These range from methods and performance standards for specific procedures to safety, laboratory and quality management system standards and include more than 25 Evaluation Protocols (EPs) for assay evaluation. These vary depending on the particular stage or aspect of the assay being examined. For example, EP05 is an extensive validation protocol covering the initial establishment de novo of the precision of an assay (i.e. agreement, although it is actually imprecision that is measured), for example when first developing or significantly changing an assay, whereas EP15 is a shorter verification protocol aimed at confirming stated imprecision results such as those provided by the manufacturer with a commercially available assay. 685 Both within-run (repeatability) and between-run assessment of precision needs to be determined, with the latter covering both within-laboratory precision and overall reproducibility, in which assessment involves the changed conditions across multiple laboratories. A list of the available CLSI EPs (as of December 2015) showing the breadth of coverage is shown in Table 83.
| Code | Title of CLSI EP |
|---|---|
| EP05-A3 | Evaluation of Precision of Quantitative Measurement Procedures; Approved Guideline – Third Edition 686 |
| EP06-A | Evaluation of the Linearity of Quantitative Measurement Procedures: a Statistical Approach; Approved Guideline 687 |
| EP07-A2 | Interference Testing in Clinical Chemistry; Approved Guideline – Second Edition 688 |
| EP09-A3 | Measurement Procedure Comparison and Bias Estimation Using Patient Samples; Approved Guideline – Third Edition 689 |
| EP10-A3-AMD | Preliminary Evaluation of Quantitative Clinical Laboratory Measurement Procedures; Approved Guideline – Third Edition 690 |
| EP12-A2 | User Protocol for Evaluation of Qualitative Test Performance; Approved Guideline – Second Edition 691 |
| EP14-A3 | Evaluation of Commutability of Processed Samples; Approved Guideline – Third Edition 692 |
| EP15-A3 | User Verification of Precision and Estimation of Bias; Approved Guideline – Third Edition 693 |
| EP17-A2 | Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline – Second Edition 694 |
| EP18-A2 | Risk Management Techniques to Identify and Control Laboratory Error Sources; Approved Guideline – Second Edition 695 |
| EP18-A2/EP23-A WS | Sources of Failure Template695,696 |
| EP19-Ed2 | A Framework for Using CLSI Documents to Evaluate Clinical Laboratory Measurement Procedures, Second Edition 697 |
| EP21-A | Estimation of Total Analytical Error for Clinical Laboratory Methods; Approved Guideline 698 |
| EP23-A™ | Laboratory Quality Control Based on Risk Management; Approved Guideline 699 |
| EP23-A WB | Laboratory Quality Control Based on Risk Management; Workbook 700 |
| EP23-A WS | A Sample Form for Laboratory Quality Control Based on Risk Management; Worksheet 701 |
| EP24-A2 | Assessment of the Diagnostic Accuracy of Laboratory Tests Using Receiver Operating Characteristic Curves; Approved Guideline – Second Edition 702 |
| EP25-A | Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline 703 |
| EP26-A | User Evaluation of Between-Reagent Lot Variation; Approved Guideline 704 |
| EP27-A | How to Construct and Interpret an Error Grid for Quantitative Diagnostic Assays; Approved Guideline 705 |
| EP28-A3c | Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory; Approved Guideline – Third Edition 706 |
| EP29-A | Expression of Measurement Uncertainty in Laboratory Medicine; Approved Guideline 707 |
| EP30-A | Characterization and Qualification of Commutable Reference Materials for Laboratory Medicine; Approved Guideline 708 |
| EP31-A-IR | Verification of Comparability of Patient Results Within One Health Care System; Approved Guideline (Interim Revision) 709 |
| EP32-R | Metrological Traceability and Its Implementation; a Report 710 |
| EP36-Ed1 | Harmonization of Symbology and Equations, First Edition 711 |
In addition, insights and educational resources are also provided through bodies such as the EFLM, exemplified by a recent paper concerning the assessment of quality of analytical methods and the various parameters to investigate. 712 Internationally recognised standards also exist, such as ISO 15189 for medical laboratories, providing particular requirements for quality and competence, and accreditation of laboratories will be based on meeting such standards and guidelines as stipulated.
Pre-analytical errors and variation
In addition to appropriate analytical validation of the assay for any specific biomarker, considering and minimising the impact of pre-analytical variables is arguably just as important to ensure clinically valid results. In the hospital clinical chemistry laboratory, where the total testing process can be broken down into pre-, intra- and post-analytical phases, it is generally considered that the majority of errors, possibly up to 75%, occur in the pre-analytical phase. 713–716 There are many anecdotal examples of errors, including falsely elevated urinary amylase concentrations as a result of contamination with salivary amylase from the nurses collecting the samples and haemolysed blood samples resulting from contamination with rainwater, but essentially all aspects of the pre-analytical phase can be affected, either by errors or by variability in the processes adopted. 717 Potential errors include requesting the incorrect test and failure to comply with any defined requirements, for example carrying out an assay at a particular time of day or after fasting, through to obtaining a specimen from the correct patient in an inappropriate tube, labelling a tube incorrectly or failing to process a sample according to agreed protocols, ready for analysis. Although historically the focus has often been on the analytical phase, this is now regarded as the ‘tip of the iceberg’ and more efforts in the future will be directed towards addressing quality aspects of the pre-analytical phase. 718–720 Within the EFLM, a Working Group for Preanalytical Phase has been established to promote these efforts; it published its findings on non-compliance with CLSI guidelines for phlebotomy in 2015. 718,721 Similarly, the Laboratory Errors and Patient Safety working group of the International Federation of Clinical Chemistry and Laboratory Medicine (IFCC) has included 35 quality indicators relating to the pre-analytical phase in its model of quality management for the total testing process. 720 In the UK, a Preanalytical Working Group within the Association for Clinical Biochemistry and Laboratory Medicine (ACB) has also begun to address the pre-analytical phase, with a recent UK-wide survey indicating wide variation in the recording of pre-analytical issues (e.g. recording of haemolysis, icterus and lipaemia in 80% of laboratories but sample mislabeling recorded in only 56.9% of laboratories). This provides evidence to support quality assurance schemes going forwards. A UK National External Quality Assessment Service (UK NEQAS) pilot scheme has been set up. 716 In the research community, the International Society for Biological and Environmental Repositories (ISBER) has a Biospecimen Science Working Group looking at aspects of defining and coding critical pre-analytical factors in biobanking (SPREC) and possible QC procedures and the BRISQ recommendations describe critical elements of the pre-analytical phase that should be included in publications to aid transparency. 375,722–724 The importance of the development of appropriate information and communication technology tools has also been raised. 725
Whenever possible in the hospital environment, automated procedures are used or are being introduced to reduce the chance of error. 713 To reduce variability, standards or guidelines have been produced by the CLSI covering all procedures, from venepuncture to patient and sample identification, tube specification and sample processing. 494,726–728 Using blood as an example, technical sources of such variability include factors such as type of blood collection tube and components used (even the presence of gel activators can markedly affect results), inadequate fill compromising sample-to-anticoagulant ratio, haemolysis, elapsed time between venepuncture and centrifugation, centrifugation speed and temperature, elapsed time between centrifugation and analysis and, for samples not analysed immediately, the duration and temperature of storage. 729 Storage duration and temperature are particularly important when banking samples for biomarker research studies. Recent reviews discuss this in greater detail, both from the perspective of biomarker discovery/validation studies in the research environment, where often the impact is poorly understood, with many proteins detected in proteomic studies having been little studied, and from the perspective of hospital laboratories, where it is often better understood for the specific panel of tests in routine use. 363,373,713,715,718,723,729,730 When the potential impacts cannot be controlled for, it is essential that they are at least appreciated and factored into study protocols and analysis to avoid potential confounding of the results. Such effects need to be considered in terms of understanding the pre-analytical uncertainty of measurements and any potential changes, for example the impact of venepuncture or tube type on measurement of a panel of 15 routinely measured serum analytes has been investigated;492 in addition, comparison of five tube types for serum preparation found clinically significant differences depending on tube type for several analytes, including creatinine, amylase and phosphate. 731 The consequences of pre-analytical errors or variation in research studies are generally not appreciated but include loss of time and wasted resources and further indirect effects through repetition and follow-on studies based on inaccurate results, all leading to massive financial consequences. More broadly, the irreproducibility in preclinical research in the life sciences generally has been estimated to cost > $28M per year in the USA alone. 732 In the clinical environment, failure to standardise pre-analytical conditions can have a critical impact on patient safety, for example in terms of a wrong or delayed diagnosis or inappropriate treatment, and of course there are also financial implications. 715 Within the development of a health economic model assessing the impact of pre-analytical errors, various clinical case study scenarios were used and the average cost of a pre-analytical error was remarkably similar in North America and Europe, at just over $200, accounting for up to 1.2% of hospital operating costs. 715
Biological variability must also be taken into account when considering the pre-analytical phase. Factors such as an age, sex, diet, time of day, comorbidities, effects of drugs, smoking status or alcohol consumption, body mass and, for females, menstrual cycle stage, pregnancy status and menopausal status can all potentially have an impact on biological markers. In addition, the acceptability criteria of an assay for use in a particular clinical context may vary depending on physiological or pathological variability. For example, a higher level of assay imprecision may be acceptable if a large effect size is being sought and given a sufficient sample size. Recently, and analogous to the STARD criteria,368 a checklist has been produced by the EFLM that specifies the key data that need to be reported to allow studies of biological variation to be interpreted and used effectively. 733 A databank of intra-subject and inter-subject variation for > 300 routinely measured analytes has been compiled from the literature and is available online,734,735 together with two guest essays describing how such data are important in clinical chemistry in defining quality specifications and desirable performance characteristics such as total error, imprecision and bias. 734,736–738 Although the concept of this database is recognised as being very valuable, recently issues have been highlighted that limit this value. These include the dubious quality of some of the studies from which the data were derived, the age of some of the studies, resulting in the use of methodology that is now outdated, the use of different units of measurement in different studies of the same analyte, the reliance in some cases on data from only a single study and the limitation of many studies to healthy individuals. 739 The importance of biological variability lies in its requirement for the calculation of the RCV, which enables the interpretation of the significance of a difference in successive results. It also provides information on whether or not reference intervals can be used to interpret an individual’s marker results, for example creatinine has a small within-subject variability and a large between-subject variability and so any changes should be interpreted in light of that individual’s reference interval. An important consideration to also bear in mind is how a clinician interprets assay results and to what extent the assay performance impacts on that. 740 There is often confusion about the terminology used and the ‘unfinished symphony’ of the evolution, meaning and relative merits of various reference values, such as decision limits, RCVs and reference intervals, has been discussed. 741
Development of verification protocols for commercially available immunoassays
Key technical validation (verification) elements and criteria
Key technical validation elements and criteria were developed within this programme based on a consideration of the various guidelines discussed in the previous section and the level of implementation of the assays, that is, in a Good Clinical Laboratory Practice-level research laboratory rather than in the diagnostic environment. Key elements to be considered and criteria of acceptability when validating existing immunoassays, often referred to as verification in that context compared with when establishing a new assay, were decided on, although the need for flexibility was also recognised. This is illustrated in the examples provided of studies undertaken within the programme, in which a range of problems were encountered, requiring some unique investigations that would not be needed generically. A similar flexible approach allowing adjustments or modifications as needed has been described for a range of pharmacodynamic assays, from their development within the National Cancer Institute to their deployment in multiple centres. 742 Similarly, the assumption is that samples have been handled under ideal standardised conditions and are, therefore, suitable for assessment of clinical validity utility, with relaxation of this being possible only once pre-analytical aspects have been investigated. If this is not the case, evaluation of some pre-analytical aspects may have to occur earlier in the process.
Essentially, the assay protocol specified by the manufacturer was followed, with any required adjustments recorded, for example alterations in timings or settings of any specific step. The aspects described below were investigated as standard, with some being dictated by the performance requirements of the assay, based on its intended clinical application and available information at the time, for example type of sample matrix intended for use and range of analyte concentrations.
Once the specific matrix had been decided on, a familiarisation and range-finding run was undertaken to determine if the initial assay protocol was satisfactory and to determine the selection of samples for use in the validation studies, such as those with high or low analyte concentrations. Appropriate samples from the assembled bank of samples (‘surplus diagnostic samples’) with a known high concentration of endogenous interferents were also needed, together with recombinant/purified proteins (analyte and proteins with known homology). The following aspects were then assessed for each matrix.
Standard assessments
-
Analytical range:
-
Limit of detection (LoD) – approximately 20 repetitions of zero standard over multiple plates (mean blank + 3 SDs).
-
Lower limit of quantification (LLoQ) – serial dilutions of low standard to the approximate LoD, analysed over multiple plates (n ≥ 3), to generate a precision profile; LLoQ is the lowest concentration from the profile, which can be measured with < 20% imprecision and inaccuracy.
-
Evaluation of the hook effect – the analyte is spiked into the sample at 100–1000 times the concentration of the highest standard and the reported value should be greater than that of the highest standard. The spiked sample is diluted back into the assay range and recovery is checked.
-
It is worth noting that a significant element of confusion is the variable use of terminology and methodology employed by users and manufacturers to determine the performance of assays, particularly at the lower concentrations. Values variably quoted include those for functional sensitivity, the limit of quantification (LoQ) or LLoQ, the LoD, the limit of blank (LoB), the minimal detectable concentration and sensitivity, making comparisons and interpretation difficult. In particular, LoD and LLoQ or LoQ are often used interchangeably and it is critical that a distinction is made between these values and that they are both assessed. This has been illustrated for LoB, LoQ and LoD. 743
-
-
-
Imprecision:
-
Intra-assay –
-
Inter-assay –
-
Assessed with a minimum of two QC samples (pooled or independent) with a high and a low analyte concentration using five or more independent determinations for each, over each of 5 days, that is, 25 determinations in total minimum. The intra-assay CV should be ≤ 10% and the inter-assay CV should be ≤ 15% (20% at LLoQ).
-
-
-
Accuracy:
-
Recovery – a spike of recombinant/purified analyte is added to three or more independent pools or base material of an appropriate matrix at three different concentrations; acceptable recovery is 80–120%.
-
Evaluation of suitable reference materials, if available (five or more determinations over three concentrations; < 20% imprecision and inaccuracy).
-
Comparison with a reference method if available.
-
-
Analytical specificity:
-
Cross-reactivity – identified proteins with homology to the analyte are spiked (recombinant/purified forms) into independent samples (n ≥ 2) at two concentrations spanning the pathophysiological cross-reactant range (if known).
-
Parallelism/dilution linearity (normal working dilution and three or more serial dilutions of a minimum of three samples). Assessed by back-calculating the diluted concentration of the four dilutions to the actual concentration, with an acceptability limit of ≤ 15%.
-
Common interferents – (e.g. rheumatoid factor, lipids, bilirubin, complement, haemolysate) –
-
The recombinant analyte is spiked into surplus diagnostic samples (n ≥ 3) with known moderate and high interferent concentrations and recovery is calculated, for example 150 and 300 µmol/l of total bilirubin or 10 and 25 mmol/l of triglycerides. Alternatively, stock interferents can be purchased and spiked into samples with known amounts of analytes. Final concentrations would be 50 or 150 µg/ml of bilirubin (conjugated and unconjugated, respectively) or 30 mg/ml of triglycerides. For testing the effects of haemolysis, samples containing known concentrations of analyte can be spiked with haemolysate to produce 5 mg/ml of haemoglobin for serum and plasma samples or 2.25 µg/ml (which equates to +++ on a urine dipstick as determined experimentally) for urine samples.
-
Dilutional linearity is also assessed for at least one spiked test sample.
-
If recovery is outside 80–120% or significantly different from that of previous samples, a dose–response series can be undertaken by spiking five or more concentrations of purified/recombinant/synthetic interferent into samples to further assess the effects (n ≥ 3).
-
-
-
Evaluation of curve-fitting model (five or more determinations over multiple runs):
-
Imprecision (< 10%; 20% at LLoQ).
-
Inaccuracy (< 10%; 20% at LLoQ) (> 80% of non-zero standards, including highest and lowest, must pass).
-
Additional assessments
These may be necessary depending on the phase of the study:
-
inter-laboratory imprecision (reproducibility)
-
reference ranges
-
analyte stability –
-
freeze–thaw
-
short-term bench stability
-
long-term storage stability (length and temperature)
-
-
pre-analytical variables –
-
biological, for example within-subject variability, stress, exercise, diet and alcohol, smoking status, drugs, pregnancy status, age, sex, comorbidities, race, sample timing
-
technical, for example phlebotomy technique, blood collection systems, blood collection tubes, sample preparation procedures, transportation conditions.
-
Specific biomarker technical (assay and pre-analytical) studies undertaken within the programme
The assays selected for validation/verification were for biomarkers that we had prioritised for potential analysis in either the RCC prognostic study undertaken within the time frame of this programme or in future planned studies in RT in the case of NGAL. Selection was based on published studies at that time and this has been reconfirmed through more recent studies, as reviewed in the previous chapter. Samples used for these initial studies were obtained with fully informed consent as part of the Leeds multidisciplinary RTB and had been collected and processed according to stringent in-house SOPs. For example, the Vacuette® system was used (Greiner Bio-One, Frickenhausen, Germany), with Z/serum clot-activator tubes (coated with micronised silica particles) and EDTA plasma tubes (K2EDTA). Samples were processed within 45–60 minutes of venepuncture, with centrifugation at 2000 g at 20°C for 10 minutes. Serum and plasma were aliquoted and stored at –80°C until used. The exceptions to this were samples obtained for interference studies with high concentrations of factors such as bilirubin and lipid, which were obtained from Leeds Teaching Hospitals Blood Sciences Laboratory as anonymised surplus diagnostic samples under project-specific ethics approval (reference number 10/H1313/12) and without patient consent. The specific technical studies that we undertook and the results, many of which have been published as indicated, are described in the following sections.
Osteopontin
Introduction
Osteopontin is a member of the SIBLING family, which includes bone sialoprotein. An extracellular matrix glycoprotein, OPN is produced by many cell types and is predominantly secreted, although intracellular and other forms of OPN with varying post-translational modifications have been reported. 744–746 With roles in cell adhesion through binding to integrins and CD44 splice variants, OPN can differentially affect adhesion and migration through cleavage by thrombin, with subsequent separation of the integrin- and CD44-binding domains. Together with effects on proliferation, apoptosis and differentiation, OPN has been implicated in many processes including tissue remodelling, inflammation and tumourigenesis and is associated with tumour aggressiveness in several cancer types. 744–746 This includes renal cancer in which tissue expression of OPN and plasma OPN are associated with several prognostic clinical variables and are prognostic themselves, although only plasma OPN has independent prognostic significance. 615,616 Given the promising nature of those findings we selected OPN to evaluate further prognostically and consequently we undertook an evaluation of an OPN ELISA prior to analysis of samples from a cohort of RCC patients within our Leeds multidisciplinary RTB. The results from that study confirming the prognostic utility of OPN have now been published486 and this study has been reviewed together with other relevant studies in the previous chapter and has led to OPN being one of the prioritised biomarkers for evaluation in this NIHR programme, as described in the following chapter.
Methodology
The OPN ELISA kit used was the Quantikine ELISA for human OPN from R&D Systems (Abingdon, UK). All samples were assayed in duplicate and according to the manufacturer’s protocol. Assessment of assay performance was based on our in-house protocol described earlier, including inter- and intra-assay imprecision, parallelism and recovery (using recombinant OPN from Abcam, Cambridge, UK) and interference. The main assessment was carried out using EDTA plasma samples as, during the clotting process, OPN is proteolytically cleaved by thrombin, which is widely thought to lead to the lower OPN concentrations found in serum. 618 EDTA plasma samples from patients with RCC were used for the main studies, with matched serum and EDTA plasma being used for the plasma–serum comparison that we undertook to confirm the plasma–serum differences previously reported. In addition, as one of the future local uses of this assay was for analysis of samples from patients in the Leeds melanoma cohort, whose samples were shipped by post, we included a small study examining stability over up to 4 days post venepuncture, as at that time there were no published data on the stability of OPN. For this purpose, blood samples were collected into EDTA tubes from five melanoma patients and four healthy volunteers with informed consent. For each individual, two 4-ml tubes of blood were collected, with one being processed immediately after venepuncture and one being processed after 4 days at room temperature. In each case, plasma was then removed, aliquoted and stored at −80°C until analysis.
Results
Serum compared with EDTA plasma
Results from a matched comparison of serum and EDTA plasma from six RCC patients clearly showed the marked differences between serum and plasma OPN concentrations (Table 84). Most serum values ranged between 40% and 65% of those obtained for plasma, with one exception for which a value of 93.1% was seen. These differences are presumed to be the result of the thrombin cleavage previously described and accordingly we used EDTA plasma for the assay evaluation studies.
| Sample | Patient | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Plasma OPN (ng/ml) | 45.11 | 71.79 | 36.73 | 286.65 | 269.8 | 99.7 |
| Serum OPN (ng/ml) | 21.45 | 31.30 | 18.75 | 137.03 | 251.2 | 63.5 |
Imprecision
Overall, intra-assay imprecision was 2.9%, with values of 3.1%, 2.8% and 2.8% for low, medium and high QC samples, respectively, with each being assayed five times in duplicate in a single assay run. Inter-assay imprecision was also < 10%.
Parallelism
Samples (n = 5 RCC samples) were titrated in parallel, with serial dilutions from 1 in 12.5 to 1 in 100, and parallelism CVs ranged from 6.8% to 12.9%, which is within our limits of acceptability (< 15%).
Recovery, interference and hook effect
As shown in Table 85, with the exception of one sample with a low spike for which the replicate CV was > 10% and for which the result was, therefore, not available, recovery was acceptable for all EDTA plasma samples (n = 6; n = 3 melanoma and n = 3 RCC samples), with low spikes of +217.5 ng/ml and high spikes of +389.5 ng/ml.
| Sample | Clinical group | % recovery | |
|---|---|---|---|
| Low spike | High spike | ||
| 1 | Melanoma | 102.3 | 93.1 |
| 2 | Melanoma | – | 96.0 |
| 3 | Melanoma | 104.9 | 109.6 |
| 4 | RCC | 86.5 | 87.1 |
| 5 | RCC | 82.6 | 92.9 |
| 6 | RCC | 120.0 | 116.7 |
No evidence of a hook effect was seen and bilirubin, haemolysis, triglycerides and rheumatoid factor showed no appreciable signs of interference in terms of measured OPN concentration or dilution linearity.
Lower limit of quantification
This was assessed as being < 78 pg/ml, equating to 1.95 ng/ml in a sample diluted 25-fold. Further evaluation of the LLoQ using lower OPN concentrations to determine an exact value was not carried out as all samples measured were well above this value.
Sample stability
As shown in Figure 40, there was no significant difference in OPN concentrations in EDTA plasma between blood samples (four healthy control subjects and five patients with melanoma) processed immediately and blood samples processed 4 days later (Wilcoxon matched-pairs signed-rank test, p = 0.07), with the majority of samples differing by < 3% between the two time points.
FIGURE 40.
The effects of immediate vs. delayed processing on plasma OPN concentrations (n = 9). In the delayed samples, the time elapsed between venepuncture and centrifugation of the EDTA blood samples was 4 days.

Discussion
The assay performed satisfactorily in all aspects evaluated, briefly referred to in subsequent published studies486,747 and confirming and extending the manufacturer’s documentation. The main issues to consider going forwards relate to the forms of OPN measured in the various assays. Although we confirmed in a small number of samples the marked serum–plasma differences in OPN concentration, which is commented on in several studies and in the manufacturer’s kit insert as being the result of thrombin cleavage of OPN, this is actually not clearly evidenced as definitely being the underlying mechanism when the various studies are reviewed in more detail, as here. At the time that we undertook this evaluation we clearly had not looked into this in as much detail as would have been ideal and had taken at face value papers stating that this was based on earlier solid evidence.
Certainly, OPN is cleaved by thrombin experimentally and analysis of the amino acid sequence shows such a cleavage site to be present. 618 For the R&D Systems kit used here there are no data on the epitopes recognised by the antibodies and so it is impossible to say for certain that this assay detects only the intact form of OPN, although the lower concentrations of OPN in serum may support this. Similar findings using this assay (or related R&D Systems reagents and protocol in the first study cited below) have been reported before by other groups, for example with samples from healthy controls657 or RCC patients557 with 3.8- to 4.8-fold higher or 2.34-fold higher median OPN concentrations, respectively, in EDTA plasma compared with serum. Using Western blotting to explore the differences in the serum and plasma forms recognised by one of the antibodies was not conclusive, although it appeared that the monoclonal coating antibody used recognised multiple forms of OPN, including the cleaved form. 748 However, this would not be quantified if the detection antibody was unable to recognise that fragment, which is not clear; in addition, the fragments may bind to the coating antibody and compete with the intact form and, hence, affect measurements indirectly. An alternative commercially available assay kit from Immuno-Biological Laboratories (Gunma, Japan) documents the antibody specificity, with the kit insert describing the coating antibody as recognising an epitope in the N-terminal region and the detection antibody binding to an epitope at the C-terminal side of the thrombin cleavage site. This kit should, therefore, not detect the cleaved form of OPN but it is very possible that the N-terminal fragment of cleaved OPN would bind to the coating antibody and competitively inhibit the binding of intact OPN, with unknown quantitative effects, as discussed above, certainly precluding the use of serum, as indicated by the manufacturer. For plasma this may not be an issue, assuming that there is no circulating endogenous thrombin-cleaved OPN. A definitive assay for intact OPN only that did not suffer from any competitive effects of the thrombin-cleaved form would have to incorporate a coating antibody that recognises an epitope that spans the region of the thrombin cleavage site and includes amino acids either side and which is accessible within the three-dimensional conformational structure of OPN under assay conditions. Antibodies to various domains in the OPN protein have been used in combination to design several ELISAs with different specificities for the different isoforms and truncated forms (although not as per the above suggestion) and to characterise OPN in cell supernatant and urine samples, demonstrating that, in the latter, few if any cleaved forms of OPN are present. 749 One or possibly two of these antibodies have been used in the Immuno-Biological Laboratories ELISA. 749
Interestingly, OPN has also been reported to bind complement factor H and detection of OPN in serum samples using an in-house competitive ELISA was possible only once such complexes had been disrupted through heating in a chaotropic buffer with a reducing agent. 750,751 Clearly, the effects of such complex formation on OPN concentrations measured will be dependent on the antibodies used in the assay, in the same way as for detection of intact compared with cleaved OPN. Whether or not this could also contribute to the differences between serum and plasma OPN concentrations is not clear but clearly spiking studies using complement factor H would be worth pursuing in future studies. In addition to the presence/absence of calcium-regulating thrombin activity affecting OPN cleavage, OPN also binds calcium and it is not inconceivable that the presence/absence of calcium in serum and plasma, respectively, could influence the detection of OPN, depending on calcium-dependent epitopes recognised by antibodies, as described later in this chapter for NGAL. If calcium is part of the underlying mechanism in this way, a possible explanation may lie in the fact that OPN undergoes calcium-dependent polymerisation mediated by transglutaminase 2, which may affect the accessibility of antibodies to binding sites and has been proposed to potentially contribute to differences in serum and EDTA plasma OPN concentrations. 748 Alternatively, direct sequestration by the clot during formation of serum may account for the findings. 748
In terms of stability, multiple freeze–thawing of plasma was not found in other studies to affect OPN concentrations measured using a new multiplex assay or the R&D Systems assay, at least in the latter case until the fifth freeze–thaw cycle. 752,753 However, a study using the Immuno-Biological Laboratories ELISA reported effects of freeze–thawing for both serum and to a lesser extent plasma, and even decreased OPN concentrations after storage at –80 °C for 1–4 weeks, although no details were provided as to the extent of these differences and whether or not these exceeded the variability of the assay. 754 Different storage conditions for plasma and serum prior to freezing have been reported to have little impact on OPN concentrations. 748 Whereas delaying centrifugation of whole blood prior to serum or plasma removal, for up to 1 hour at room temperature or 6 hours at 4°C, did not affect plasma OPN concentrations, similar to our results over a much longer time period, decreased OPN concentrations were detected in serum measured after 15 or 60 minutes at room temperature, which is presumably occurs during the clotting process. 754
It is difficult to understand whether serum or plasma measurements of OPN provide the optimal information clinically, particularly given the differences in fluid types used across studies and also with direct comparison of different assays yielding very different results, whether because of the forms of OPN present or measured or because of standardisation differences. 755 Both serum and plasma OPN have been shown to be prognostic in advanced non-small-cell lung cancer, for example, but in mesothelioma, with a matched sample comparison, plasma was superior to serum. 754,756–758 In a study of RCC published since our study,486 both serum and plasma samples were used for OPN measurement, depending on what was available for each patient, with independent prognostic value shown for both, although this included multiple RCC subtypes. 617 Interestingly, significantly higher OPN concentrations were found in men than in women, in both plasma and serum, although it is not clear whether or not this was corrected for clinical factors such as the stage and grade mix in the two groups, for example. This is under investigation in our RCC cohort, but we did not find any significant sex-specific differences in normal healthy control subjects and a study determining the reference range for OPN in 300 healthy individuals also found no significant effects of age or sex and a low biological within-subject variation of 8.2%. 753 This study also found similar precision results for the R&D Systems assay to those in our study.
Clearly, although OPN has shown value in many studies across various disease areas and findings have been shown to be reproducible in terms of clinical validity, further characterisation of the available immunoassays with regard to the possible effects proposed here would be of value and could be relatively easily achieved using spiking experiments, for example with complement factor H, cleaved OPN or serum with EDTA. This may address some of the variability across studies and provide a clear way forward as to the optimal way of determining OPN concentrations and importantly provide clarity as to which form(s) is the most relevant clinically in the different disease areas, allowing the potential of OPN as a biomarker to be more fully and robustly realised.
Carbonic anhydrase IX
Introduction
Carbonic anhydrase IX is a 46-kDa membrane protein that has been shown to exist in several forms as a result of alternative splicing, proteolytic cleavage, glycosylation and phosphorylation. 759–762 Playing an important role in regulating intracellular pH, allowing cell survival in hypoxic conditions, for example, increased expression in renal cancer was initially recognised through positive reactivity of a monoclonal antibody (clone G250), with the antigen later being identified as CAIX/MN protein. 606,607,763,764 It is now known that this upregulation is also present in other cancers and is mediated through hypoxia-inducible factor-1α (HIF-1α), which in RCC is increased as a downstream consequence of the VHL gene alterations, as described in Chapter 10. 765 The use of CAIX has been investigated both in diagnostic imaging and as a therapeutic target in antibody, vaccine or small molecule inhibitor-based strategies. 766,767 At the time that we commenced our studies relating to CAIX, several studies had shown tissue expression of CAIX to be prognostic, and soluble CAIX in serum or plasma from patients with RCC and also other cancers appeared to have both prognostic and predictive potential, although there was inconsistency across studies. 428,608–610,768–773 Given these findings and the biological relevance related to the VHL gene, we selected CAIX as a potential prognostic biomarker to explore further, initially in the Leeds multidisciplinary RTB cohort, which has now been published,486 and now with further studies using the NIHR cohort, as described in the previous chapter. We describe here the analytical verification of two commercially available immunoassays for CAIX and the issues encountered, which have important consequences for some published studies and highlight the importance of adequate validation in the first place and later verification. Our findings have been published in detail774 and are summarised here.
Methodology
A total of 17 sets of matched serum and EDTA plasma samples from patients with RCC of varying age, sex, stage and grade were used for the various parts of the verification study. The assays used were both commercially available sandwich ELISAs: the human MN/CAIX ELISA kit from Oncogene Science Diagnostics (Cambridge, MA, USA)/Siemens Healthcare Diagnostics and the Quantikine human CAIX/CA9 ELISA kit from R&D Systems (Minneapolis, MN, USA). The initial work was carried out using the Siemens assay only but, once it became apparent that there were some problems with this assay, the R&D Systems assay was also examined to help resolve the nature of the problems seen and also as a possible alternative to use in future studies. Samples were assayed in duplicate and the manufacturers’ protocols were followed in each case. The LoD of the Siemens and R&D Systems assays, as quoted by the manufacturers, are 2.5 and 2.28 pg/ml, respectively. Assay verification was based on our in-house protocol described earlier, including elements such as imprecision, parallelism, specificity and recovery. Following demonstration of specific issues with the Siemens assay, additional ad hoc investigations were undertaken, including direct comparisons of serum and plasma and the effects of metal ions, to determine the nature of the problems. 774
Results
Standardisation
Differences in standardisation were apparent, with analysis of recombinant CAIX (rCAIX) showing a ratio of Siemens-to-R&D assay values of 2.8. Reciprocal analysis of standards between the assays showed average ratios of 2.51 and 3.13, supporting this, with the slight differences presumably caused by the differing matrices of the standards between the two assays. However, analysis of 15 plasma samples showed Siemens-to-R&D ratios that varied from 1.95 to 17.3, with values for 15 matched serum samples varying from 0 to 7.1, indicating that, with clinical samples, additional factors were affecting the results.
Imprecision
Intra-and inter-assay imprecision for both assays was < 10% at concentrations of CAIX spanning the standard curve, although CVs of 14.5% and 18.6%, respectively, were found for the lowest concentration controls analysed using the Siemens assay. For any sample duplicate, the CV was generally < 5% of the mean and this was the same for both assays.
Parallelism
Issues were found with the Siemens assay, with one of five samples passing compared with five out of five samples using the R&D assay, as shown in Figure 41.
FIGURE 41.
Results from the assessment of parallelism of the CAIX assays. (a) Siemens assay; and (b) R&D Systems assay. Open symbols = serum and closed symbols = plasma. Back-calculated concentrations are plotted against serial doubling dilutions of the samples, with arrows indicating the normal working dilution used in each assay. Reproduced from Wind et al. 774 with permission.

Recovery and specificity
As shown in Table 86, recovery was acceptable for all EDTA plasma samples tested on both the Siemens assay and the R&D Systems assay. Serum was not tested in the Siemens assay because of emerging problems with this assay. For the R&D Systems assay, one of five serum samples showed poor recovery of both high and low spikes. Cross-reactivity with carbonic anhydrase II and carbonic anhydrase XII was minimal or non-existent for both assays.
| Assay | Sample | Sample | |||||
|---|---|---|---|---|---|---|---|
| EDTA plasma | Serum | ||||||
| Initial CAIX concentration (pg/ml) | % recovery | Initial CAIX concentration (pg/ml) | % recovery | ||||
| Low spike | High spike | Low spike | High spike | ||||
| Siemens | 1 | 516.5 | 102.2 | 88.4 | |||
| 2 | 830.9 | 93.3 | 81.0 | ||||
| 3 | 485.1 | 94.1 | 88.2 | ||||
| R&D Systems | 4 | 483.3 | 81.7 | 80.2 | 502.4 | 59.4 | 66.7 |
| 5 | 92.4 | 82.3 | 86.3 | 92.7 | 86.7 | 84.9 | |
| 6 | 26.9 | 81.7 | 83.0 | 27.4 | 81.5 | 91.2 | |
| 7 | 47.6 | 82.0 | 91.5 | 53.2 | 89.9 | 93.5 | |
| 8 | 84.0 | 97.8 | 90.7 | 88.0 | 94.0 | 86.6 | |
Measurement of carbonic anhydrase IX and effects of metal ions
No significant difference was seen between results obtained for matched EDTA plasma and serum samples in the R&D Systems assay (Figure 42), with a significant correlation (p < 0.001; R2 = 0.998) and a slope of 0.905. Using the R&D Systems assay, EDTA plasma concentrations ranged from 17.7 pg/ml to 482.9 pg/ml and serum concentrations ranged from 18.2 pg/ml to 436.6 pg/ml. However, with the Siemens assay, significantly higher CAIX concentrations were found in the EDTA plasma samples than in the serum samples (p < 0.001) and, although significantly correlated, (p < 0.001; R2 = 0.961), the slope of the line was only 0.538, with several outlying samples. EDTA plasma concentrations ranged from 34.5 pg/ml to 1476.4 pg/ml whereas in the matched serum samples concentrations ranged from < 2.5 pg/ml to 770.6 pg/ml.
FIGURE 42.
The relationship between concentrations of CAIX in EDTA plasma or serum. Results are shown for 15 matched sample pairs assessed using (a) the Siemens assay and (b) the R&D Systems assay. The dotted line shows the line of equivalence with a slope of 1. (c) shows the effects on the CAIX concentrations as measured using the Siemens assay of adding 20 mmol/l of CaCl2 to EDTA plasma or adding 1.8 mg/ml of EDTA to serum. (d) The reversibility of the effect is shown by the sequential addition of CaCl2 and EDTA to EDTA plasma or rCAIX. Reproduced from Wind et al. 774 with permission.

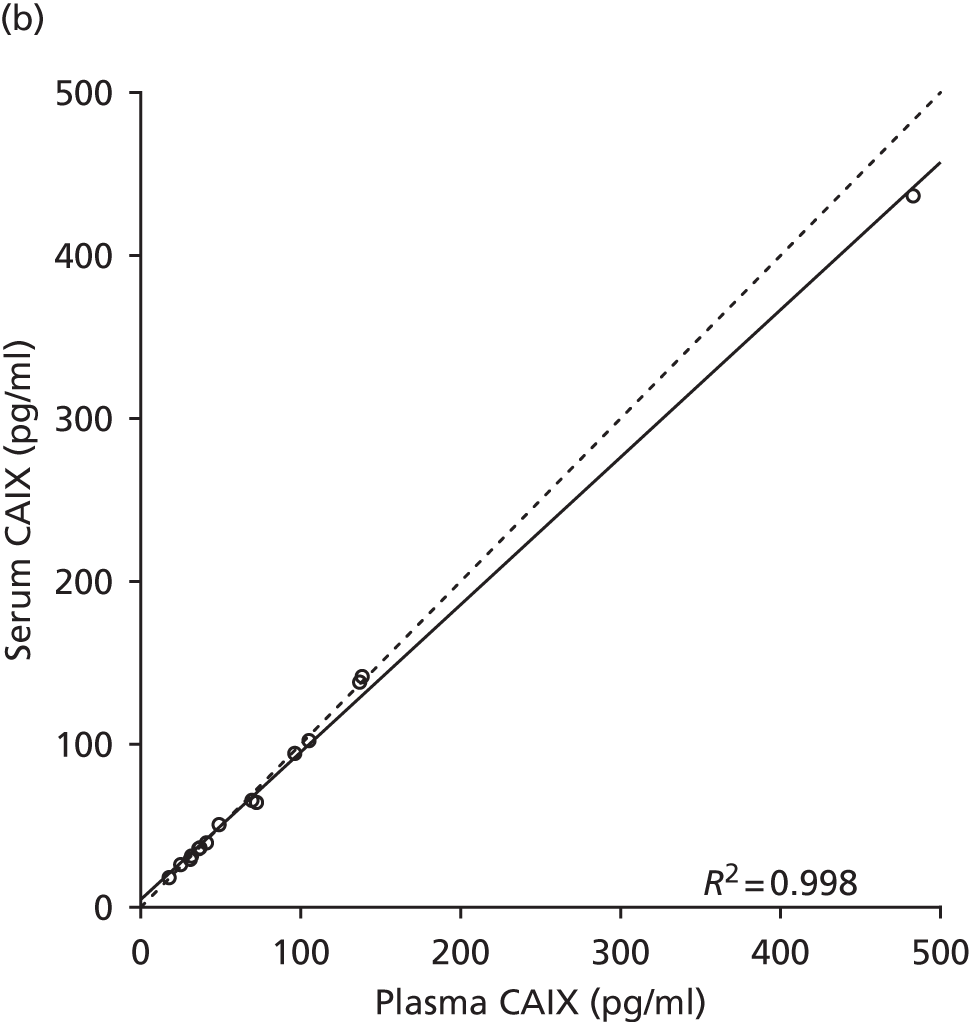


These differences between serum and plasma in the Siemens assay were originally assumed to be the result of clotting events causing a generic reduction in measurable CAIX in serum rather than this being assay specific as at that time we had not used the R&D Systems assays as a comparator. Addition of excess calcium to EDTA plasma to promote clot formation and effectively generating serum did reduce the CAIX concentrations measured, almost to those resulting from analysis of serum directly, as shown in Figure 42, supporting this possibility. However, this was reversible, with a marked elevation of CAIX detected when EDTA was added to the serum. Importantly, this was also seen with rCAIX. These results, together with the fact that the R&D Systems assay showed no difference between serum and plasma, clearly indicated that clotting per se was not responsible for the effects seen in the Siemens assay but that metal ions may be the cause. Magnesium and calcium were interchangeable in terms of causing this effect and no effects were seen with calcium or EDTA addition in the R&D Systems assay, indicating that this was an assay-specific effect.
Introducing the Siemens capture antibody into the R&D Systems assay did not change the R&D Systems pattern of results in terms of serum and plasma having similar measured CAIX concentrations. However, using the R&D Systems capture antibody in combination with the Siemens detection antibody resulted in marked differences in the values generated for serum and plasma (Figure 43), indicating that the Siemens detection antibody (M75 clone) is responsible for the effects seen. 775 The most likely explanation is that this antibody recognises a metal ion-dependent epitope.
FIGURE 43.
The effect of crossing over antibodies between assays to determine which antibody–antigen interaction accounts for the metal ion-dependent effects seen in the measurement of CAIX. Results are shown for four matched pairs of EDTA plasma and serum samples assayed using the R&D Systems assay, the Siemens assay or each of these but with the capture antibodies swapped between the assays. Absorbances were measured at 450 nm in both cases but background subtraction at 540 nm was carried out in the case of the R&D Systems assay. Reproduced from Wind et al. 774 with permission.
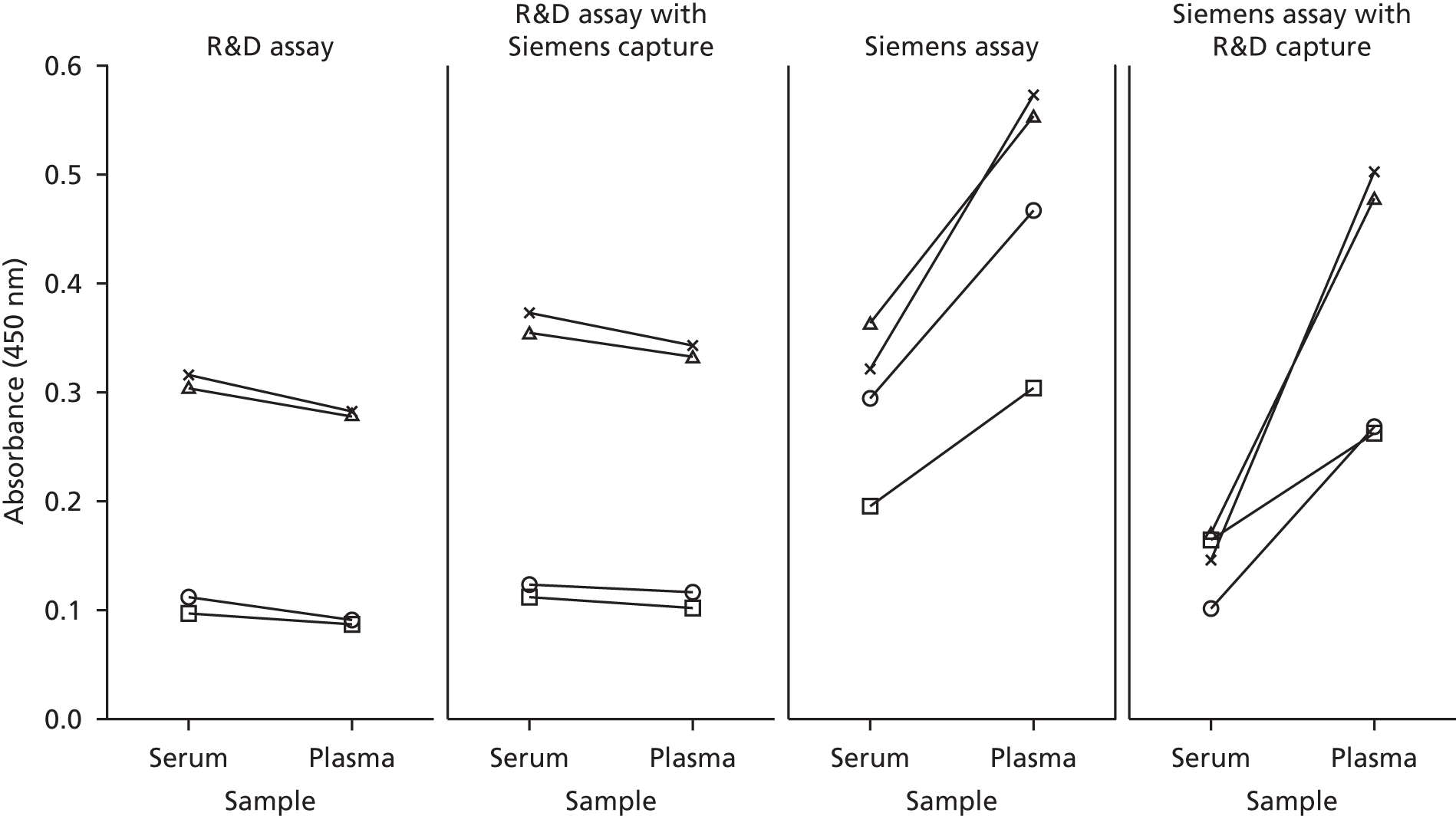
Discussion
No studies have been published that have undertaken verification of the available assays for soluble CAIX, despite there now being considerable interest in its potential clinical utility. Early studies, predominantly focused on RCC, have reported elevated serum or plasma concentrations, decreasing in some cases post surgery, and variable associations with tumour size, stage and prognosis. 608–610,768,770 There has also been interest in other cancers and, even since we undertook this validation study, particularly promising results have been obtained for CAIX within a diagnostic urinary biomarker panel for bladder cancer and further studies in RCC have been published, as reviewed in the previous chapter. 774,776–778 Clearly, our results show the need to undertake such studies so that results can be generated robustly and that the Siemens assay has not undergone adequate validation prior to becoming commercially available. In contrast, the R&D Systems assay performed well although confirmation of the manufacturer’s sensitivity data is needed as well as the potential effects of other pre-analytical factors.
The explanation of the metal ion effect that we have described on the binding of CAIX to antibodies within the Siemens assay is not completely clear. Cleavage of the extracellular region of CAIX generates at least two soluble forms (50 and 54 kDa). 608,762 This region contains the carbonic anhydrase (CA) catalytic domain, which has three metal-binding regions important for the catalytic activity, and the V10 capture antibody recognises a conformational epitope in this domain. 762,779,780 However, our evidence supports the phenomenon seen in our studies as arising through the M75 detection antibody, which recognises a linear epitope on the PG-like domain, also present in the extracellular region. 781 This could be explained by the finding that the catalytic activity of CAIX is also regulated by binding of multiple divalent cations to this negatively charged PG domain. It is conceivable, therefore, that the M75 antibody binds a metal ion-dependent epitope, with metal ions then either directly inhibiting binding of M75 to CAIX competitively or inhibiting binding by induction of a conformational change. 759 Although this markedly affects the serum results in particular, effects on plasma measurements when a chelating agent is present may also be affected to variable extents, depending on the variability in the final concentrations of EDTA, for example.
This issue was not apparent from the data given in the Siemens assay kit insert, but the kit insert did show a possible trend towards lower concentrations in serum, although slight, and only samples from healthy volunteers were used, with lower CAIX concentrations. Many published studies have used the R&D Systems assay but several studies in renal, ovarian, lung and bladder cancer have been undertaken with the Siemens assay or assays involving the M75 antibody and the results may, therefore, be questionable. 608,769–773 As the M75 antibody clone is also used in many immunohistochemical studies, it is possible that results obtained may depend on the buffers used and the presence/absence of metal ions. This effect of metal ions on epitope availability and analyte measurements is relatively rare, with possible examples, although not necessarily occurring through the same mechanism, including the calcium dependence of calretinin782 and S100A12783 measurements. Our results clearly demonstrate the need for careful independent verification of commercially available immunoassays, even from large diagnostics companies. As far as we are aware, the Siemens assay used in this study is still available, although from a different source, Nuclea Biotechnologies Inc. (Cambridge, MA, USA), as around the time of this study Wilex Inc. acquired the assets of Oncogene Science from Siemens Healthcare Diagnostics and in 2013 Nuclea Biotechnologies acquired Wilex. It is not readily apparent that the assay available currently has been modified in any way from the one that we used and it certainly uses the M75 antibody. Nuclea Biotechnologies has a major interest in CAIX and has recently announced that its CAIX automated immunohistochemisty (IHC) kit has been granted FDA class I in vitro diagnostic (IVD) status, which will be of potential utility in several cancer types, although particularly in determining the aggressiveness of head and neck squamous cell carcinomas. The question of whether it is the responsibility of the manufacturer or end user to ensure that assays are validated and verified more robustly was rasied in an editorial focusing on our study784 and it may be that the answer lies in more open partnerships between the manufacturers and researchers at early stages of biomarker research studies.
Neutrophil gelatinase-associated lipocalin
Introduction
One of the most promising emerging kidney-related biomarkers is NGAL, in particular in AKI, which is now recognised as a major health problem. 785,786 Originally isolated from neutrophils in 1993, NGAL was proposed as a novel urinary biomarker of ischaemic kidney injury produced predominantly by proximal tubule cells 10 years later, although subsequently an NGAL reporter mouse model has supported a distal tubule cellular origin. 787–789 Now known to be expressed by several tissues, the predominant form released by renal tubules is a 25-kDa monomer whereas the main neutrophil-derived form is a 45-kDa homodimer. 790 In addition, NGAL exists as a 125-kDa covalently complexed heterodimer with matrix metalloproteinase-9 (MMP-9) and other forms have been found. 787,791–793 Key functions are in protecting against MMP-9-mediated degradation, a role in bacteriostasis through mediating shuttling of iron through binding siderophores, and multiple effects on cell proliferation, differentiation and apoptosis. 790 Our interest in NGAL was stimulated by the rapidly growing level of interest surrounding NGAL in renal diseases, with urinary, plasma and serum concentrations of NGAL having been shown to be superior to creatinine diagnostically or prognostically in AKI in several studies when reviewed at that time and urinary NGAL outperforming kidney injury molecule-1, cystatin C, IL-18 and liver-type fatty acid-binding protein in a large prospective study in the emergency department setting. 794–797 Of particular interest was emerging evidence of an association with DGF following RT. 798,799
However, it was also apparent even then that, to provide definitive answers to its potential clinical utility, bigger prospective studies needed to be pursued and the impact of factors such as background levels of CKD on NGAL determined. In addition, there was a lot of uncertainty regarding the assays used and form of NGAL being measured, with only a small number of limited studies having been undertaken. 800–803 To contribute to the evidence, we undertook a study to independently examine five of the commercially available assays for NGAL, two CE-marked IVDs and three research use-only ELISAs, using urine samples, as most assays had been validated only for that matrix. This study has been published804 and the findings are described in brief in the following sections.
Methodology
Mid-stream urine samples from patients with AKI, RCC, renal stones, recurrent UTI or diabetic albuminuria or healthy control subjects (to ensure a variety of matrix backgrounds and NGAL concentrations; n = 78 in total) were banked for this study after processing according to local SOPs. The CE-marked assays evaluated were the NGAL Test™ from BioPorto Diagnostics A/S (Hellerup, Denmark), a particle-enhanced turbidimetric immunoassay that we used on the Siemens ADVIA® 1800 platform, and the ARCHITECT® Urine NGAL assay from Abbott Laboratories (Chicago, IL, USA), a two-step chemiluminescent microparticle assay that we used on the ARCHITECT i2000SR analyser. The research use-only assays evaluated were all of the sandwich ELISA format for human NGAL and were the NGAL ELISA (HK330) from Hycult Biotech (Uden, the Netherlands), the NGAL ELISA Kit 036 from BioPorto Diagnostics and the Quantikine Lipocalin-2/NGAL (DLCN20) immunoassay from R&D Systems. All assays were performed according to the manufacturers’ instructions. Assay verification was based on our in-house protocol described earlier, with assessment of imprecision, parallelism, recovery, selectivity, limit of quantitation, haemoglobin interference and high-dose hook effect, as described in detail in Kift et al. 804 Measurements were performed in singlicate on the ARCHITECT and ADVIA platforms and in duplicate for all ELISAs, and CVs of < 10% within replicates were considered acceptable.
Results
Our detailed findings are summarised below and in Table 87. As we found a much higher LLoQ with the BioPorto/ADVIA assay than expected from the manufacturer’s specification, we were unable to investigate recovery and effects of the NGAL/MMP-9 complex and haemoglobin with this assay as the endogenous NGAL concentrations were below the LLoQ determined in practice and so the baseline values could not be used.
| Test | Assay | ||||
|---|---|---|---|---|---|
| BioPorto/ADVIA | Abbott | R&D Systems | BioPorto ELISA | Hycult | |
| Assay standards (ng/ml), range | 150–5000 | 10–1500 | 0.156–10 | 0.01–1 | 0.4–100 |
| Dilution factor, range | NA | NA | 1/25–1/800 | 1/500–1/4000 | 1/20 |
| Imprecision % CV, range (median) | |||||
| Intra-assay (n = 5 for each of four QC samples) | 0.7–3.0 (2.0) | 0.8–10.8 (1.4) | 3.2–4.4 (3.6) | 0.6–5.1 (3.4) | 5.8–34.4 (8.8) |
| Inter-assay (n = 10 for each of four QC samples) | 1.9–7.9 (3.7) | 4.8–9.9 (7.6) | 3.2–10.1 (7.1) | 6.4–15.8 (12.6) | 26.1–33.3 (30.2) |
| % parallelism, range (median; n) | 1.9b (1.9; 1) | 2.2–5.8 (3.0; 3) | 1.9–7.9 (3.6; 3) | 3.2–49.8 (44.6; 3) | 17.8–30.2 (21.5; 3) |
| % recovery, range (median; n) | Not determinedb | 88.6–99.1 (95.6; 8) | 93.5–106.7 (98.9; 9) | 100.6–113.4 (104.1; 8) | 73.6–95.2 (88.1; 8) |
| Specificity | |||||
| + MMP-9 | Not determinedb | No effect | No effect | No effect | Inconclusive |
| + complex | Not determinedb | No effect | No effect | No effect | Inconclusive |
| LoQ (ng/ml) (including sample dilution factor) | 150 | 5 | 0.078 (2.0) | 0.01 (5.0) | 1 (20) |
| Haemoglobin interference | |||||
| 0.75, 1.125, 2.25 µg/ml (+/++/+++) | Not determinedb | No interference | No interference | No interference | Interference |
| 5.0 mg/ml | Not determinedb | Interference | Interference | Interference | Interference |
| Hook analysis | Hook effect | No effect | No effect | No effect | Inconclusive |
Imprecision
Intra-assay imprecision was generally acceptable across all platforms, with one exception of a CV of 34.4% with the low NGAL QC urine sample in the Hycult assay. Problems with the Hycult assay only were also seen for inter-assay imprecision, which was unacceptable, and this assay also showed poor agreement between replicates.
Parallelism
As shown in Figure 44, parallelism was demonstrated for the Abbott and R&D Systems assays but not for the Hycult assay and for two-thirds of the samples in the BioPorto ELISA. An issue with one sample was also seen on the BioPorto/ADVIA assay, although the issues relating to the LLoQ meant that this was inconclusive.
FIGURE 44.
Results from the assessment of parallelism for each of the five assays, comparing dilution-adjusted NGAL concentrations (log scale) against serial double dilutions of each of three samples represented by different colours: (a) BioPorto/ADVIA; (b) Abbott; (c) R&D; (d) BioPorto ELISA; and (e) Hycult. The initial dilution factor used is indicated next to each sample dilution. Fine dashed lines illustrate ±15% of the mean for each sample. ‘<LLoQ’ highlights sample dilutions that fall below the LLoQ determined by this study. Reproduced from Kift et al. 804 with permission.
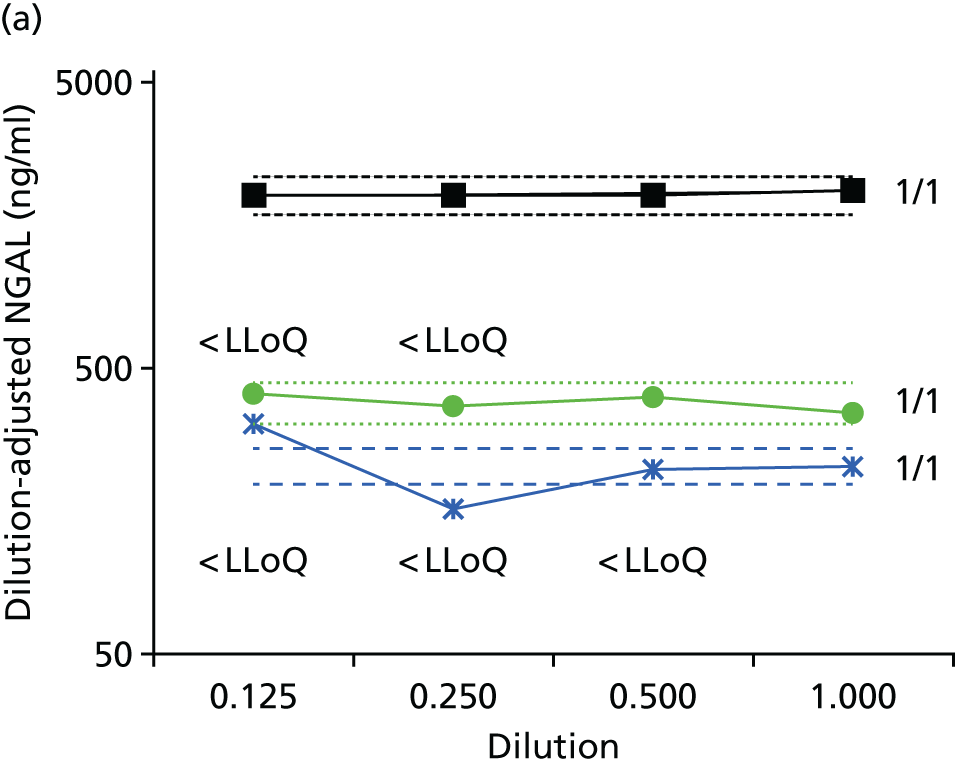

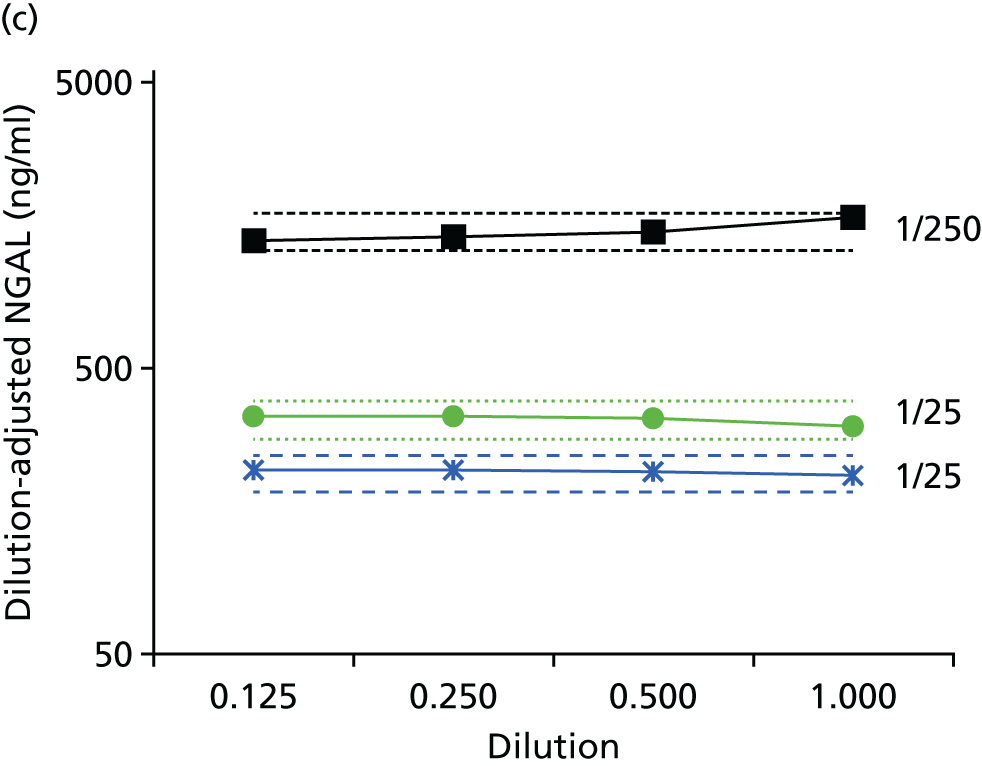
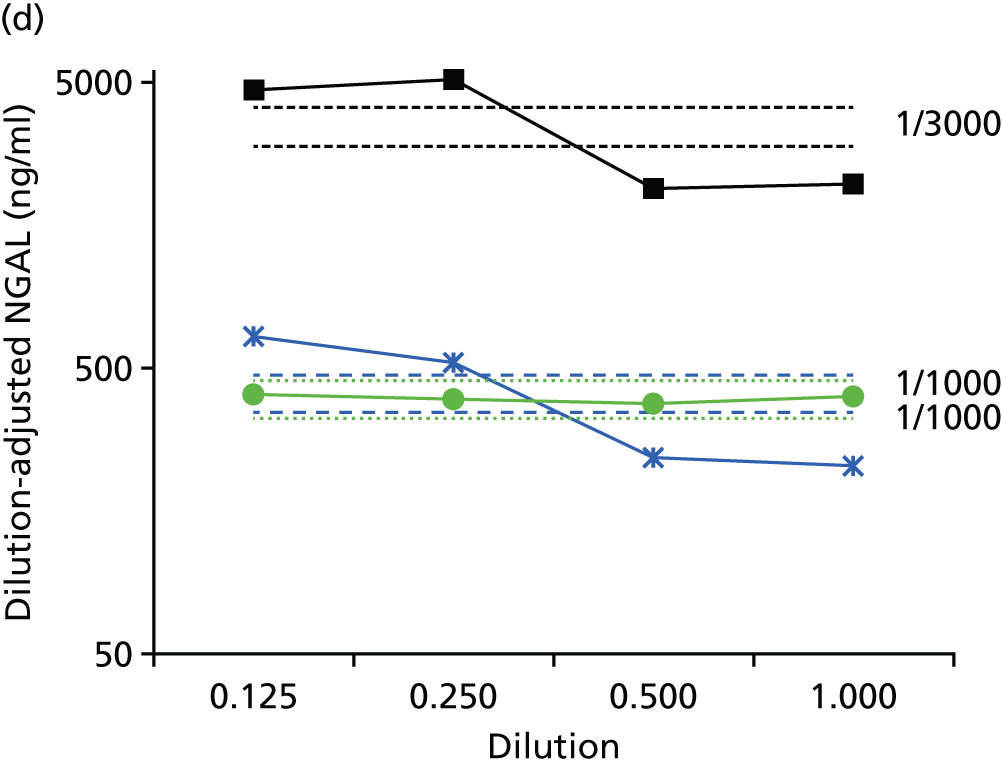
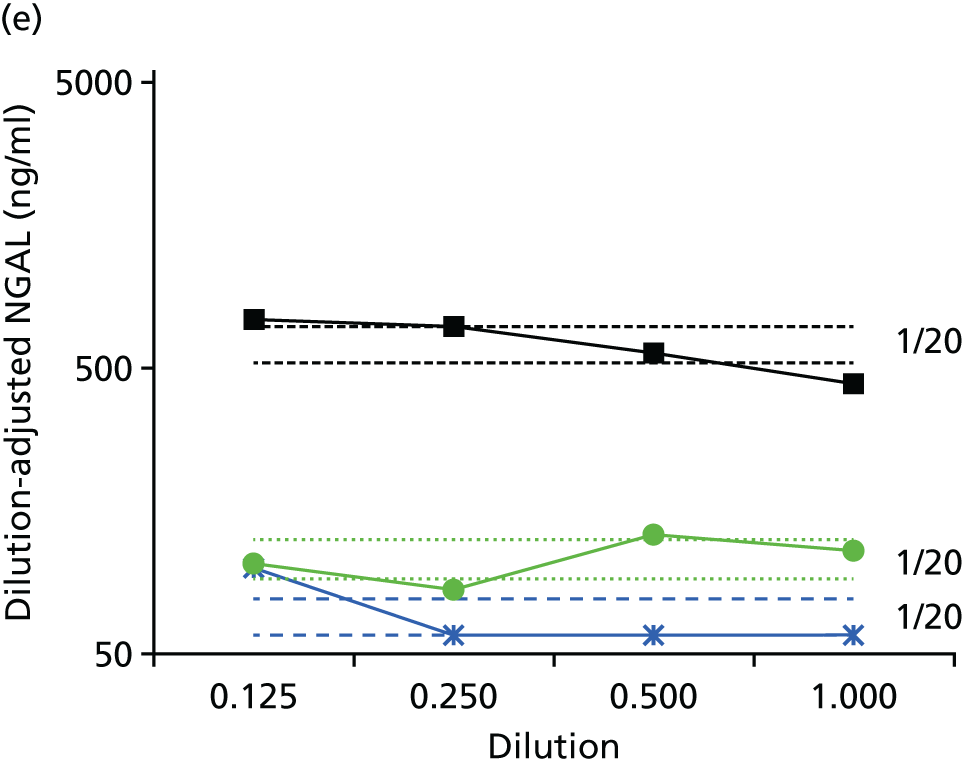
Lower limit of quantification
The LLoQ values for the Abbott assay and R&D Systems, BioPorto and Hycult ELISAs were 5, 2, 5 and 20 ng/ml, respectively, including any dilution factors. The LLoQ was 150 ng/ml for the BioPorto/ADVIA assay, which was much higher than expected given the manufacturer’s figure of 25 ng/ml for the lower end of the measuring range.
Recovery
A stock solution of recombinant NGAL (rNGAL; 1 mg/ml, with ≈80–90% monomeric form) gave quite different results across the assays, with near-quantitative results for the BioPorto ELISA but 31% recovery for the Hycult assay and 67–75% recovery for all other assays. This may reflect differences between the assays in specificity for the different NGAL forms or in standardisation. To overcome this, the recovery of spiked rNGAL material in urine samples was related to the assay-specific assigned concentrations for the rNGAL stock, essentially allowing the determination of relative recoveries and allowing assays to be compared. Acceptable recoveries were found with the Abbott assay and R&D Systems, BioPorto and Hycult ELISAs (with the exception of a recovery of 73.6% for one sample in the Hycult ELISA).
Selectivity
None of the assays detected the rNGAL/MMP-9 complex and neither rMMP-9 (recombinant matrix metalloproteinase 9) nor the complex affected the urinary NGAL results, except for the Hycult assay, where variable effects were seen.
Haemoglobin interference
The Hycult assay was affected by haemoglobin at all concentrations tested but all other assays were affected only at the highest concentration of haemoglobin of 5 mg/ml.
Hook effect
A hook effect was absent from the Abbott assay and the R&D Systems and BioPorto ELISAs. However, using the BioPorto/ADVIA assay, a typical high-dose hook effect was seen, although only at NGAL concentrations of > ≈70,000 ng/ml. With the Hycult assay an atypical effect was seen in which the assay appeared to plateau at NGAL concentrations of 20–25 ng/ml, although the range of the standard was up to 100 ng/ml (Figure 45).
FIGURE 45.
Results from the hook effect analysis. (a) Point-to-point line illustrates the high-dose hook effect seen in the BioPorto (in vitro diagnostic) assay, with the dotted line showing the upper limit of the assay range; and (b) Hycult assay data illustrating the plateauing/saturation effect, with the dotted line showing the upper limit of the assay range. Reproduced from Kift et al. 804 with permission.

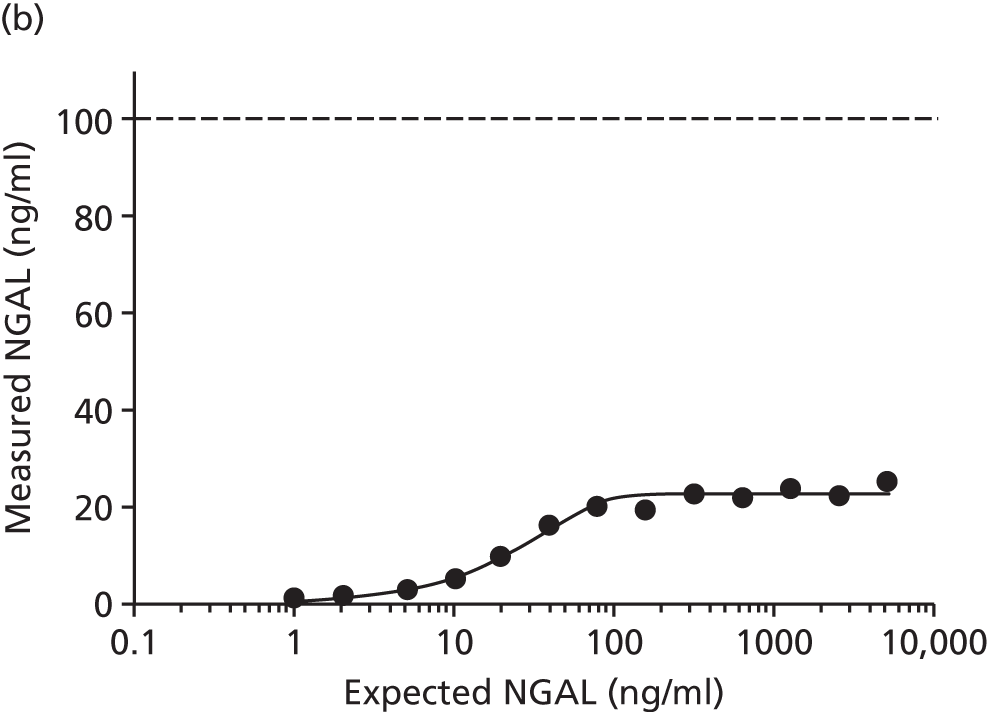
Inter-assay neutrophil gelatinase-associated lipocalin comparison
Using the BioPorto/ADVIA and Hycult assays, > 50% of the urine samples were below our determined LLoQ values and so could not be included in comparison results involving those assays. From the modified Bland–Altman plots (Figure 46), it can be seen that there was generally good agreement between the Abbott and R&D Systems assays, with only two samples falling outside 95% of overall bias and a mean bias of 14%. A Passing–Bablok analysis for comparison of these two assays only demonstrated that data lay on the line of equality. Good agreement between the BioPorto ELISA and the Abbott and the BioPorto/ADVIA assays was also seen, with a mean bias of 13% and 23%, respectively (the latter case included only 22 samples). The results from the two BioPorto assays tended to be higher than those obtained with the other assays. The Hycult assay showed a marked negative bias, with 29 of the 32 samples above the LLoQ showing NGAL concentrations of < 50% of those measured on the Abbott assay, with some values even being < 20%. With the Hycult assay, no urine samples demonstrated NGAL concentrations of > 20 ng/ml (400 ng/ml corrected for dilution), which mirrors the plateau effect of this assay shown in the hook effect studies.
FIGURE 46.
Modified Bland–Altman plots for various comparisons of the NGAL assays: (a) Abott vs. R&D Systems; (b) Abott vs. BioPorto/ADVIA; (c) Abott vs. BioPorto ELISA; (d) R&D Systems vs. BioPorto/ADVIA; (e) R&D Systems vs. BioPorto ELISA; (f) BioPorto/ADVIA vs. BioPorto ELISA; (g) Hycult vs. Abbott; (h) Hycult vs. R&D Systems; (i) Hycult vs. BioPorto/ADVIA; and (j) Hycult vs. BioPorto ELISA. Samples that fell below the LLoQ have been omitted from the relevant plots. In (g)–(j) the y-axis has been expanded to incorporate the differences seen. The solid green line indicates no bias, with the dotted lines indicating the mean bias and the limits of agreement (mean difference ±1.96 SDs of the differences). In each case the difference referred to on the y-axis is the second assay in the plot title subtracted from the first named assay. Reproduced from Kift et al. 804 with permission.
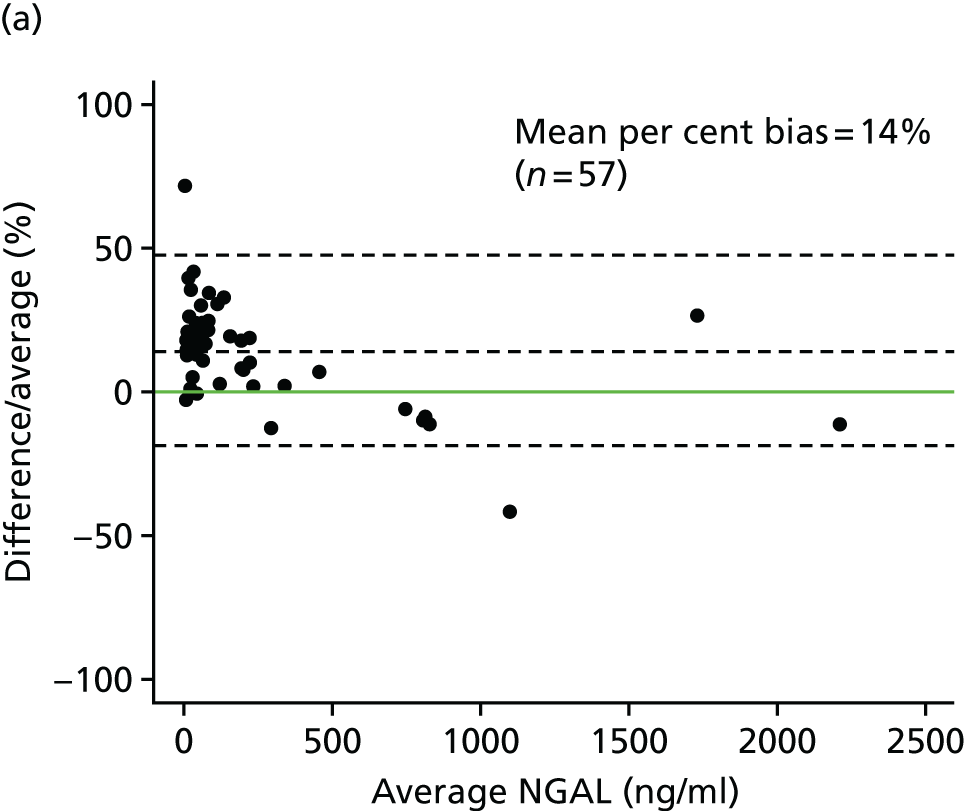
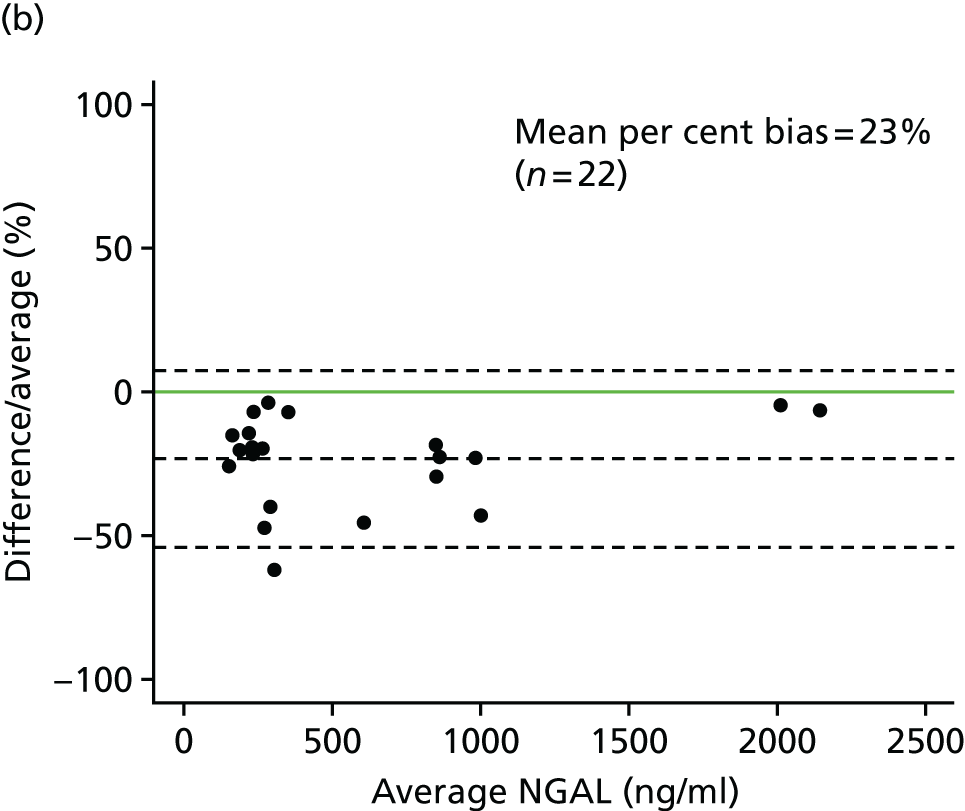



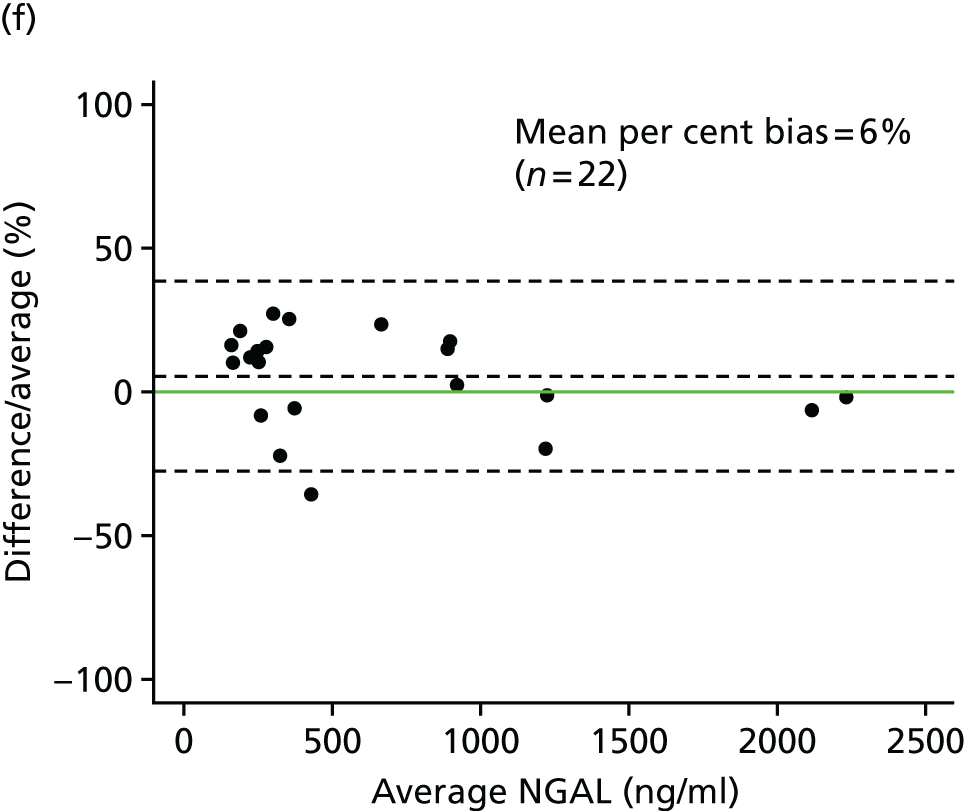


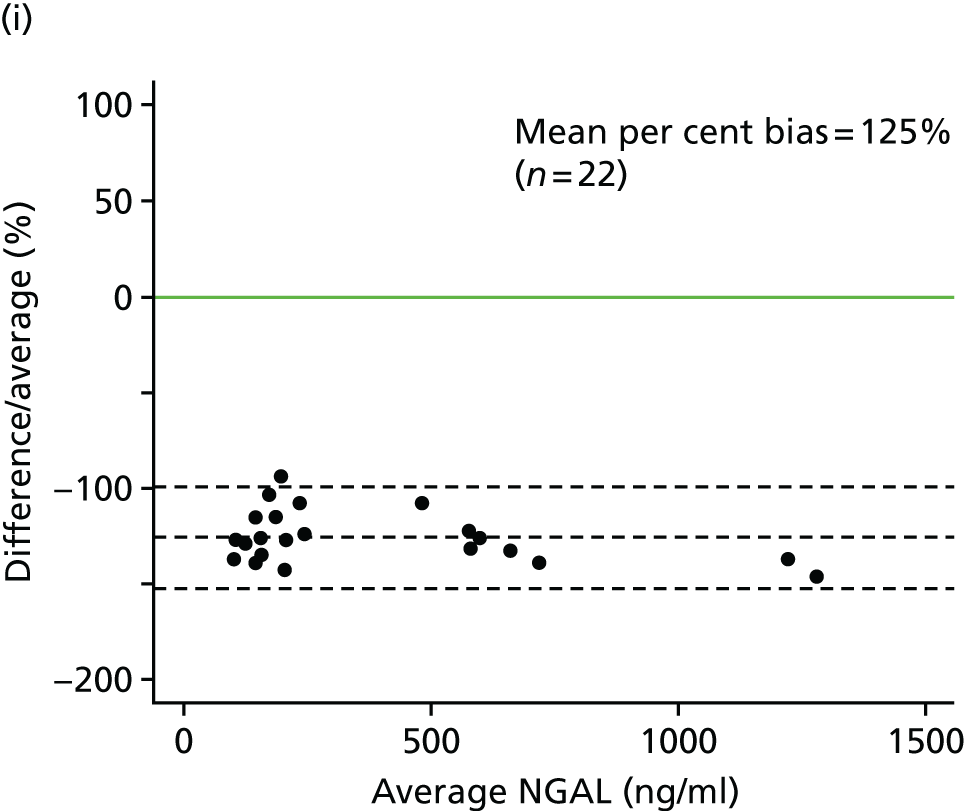

Discussion
Our independent verification findings support the Abbott and R&D Systems assays as having been validated adequately by the manufacturers and producing comparable results. A single published study evaluating the Abbott assay and sample stability had previously reported excellent reproducibility and precision, despite not examining other aspects such as recovery, linearity and selectivity, and found functional sensitivity (based on imprecision of < 20% alone in this case) to be < 2 ng/ml,800 which is similar to our determination of a LLoQ of < 5 ng/ml. Acceptable variability of the Abbott assay was also reported in a further small study. 803 Excellent performance characteristics had been previously reported for the BioPorto ELISA in an extensive verification study with both urine and plasma, although issues were reported for inter-batch variability and there was some evidence of non-parallelism. 802 The latter was not as pronounced as that seen in our study and this may reflect study-specific differences such as the urinary matrices (paediatric vs. adult), dilution factors or NGAL forms present. The fundamental reasons for the saturation of the Hycult assay, essentially rendering it unusable, are not clear but, again, this may reflect a combination of differences in standardisation and forms of NGAL recognised and potentially interfering.
Although broadly comparable to other assays, we found that the BioPorto assays were biased towards higher concentrations, although not as marked as the 65% bias previously reported in a comparison of the BioPorto assay on the Beckman Coulter platform with the Abbott assay. 801 Marked biases were also seen in a study comparing two Bioporto assays with the Abbott assay. 805 Whether such results arise because of differences in standardisation, assay design or NGAL forms measured (including possibly glycoforms) is not clear but we saw this with both recombinant and endogenous NGAL and, even with assays with good agreement, differences were apparent in some samples. Clearly, the forms of NGAL detected will depend on the antibodies used in the various assays but our results demonstrate that the NGAL/MMP-9 complex was not detected by and did not interfere with measurement of NGAL in the Abbott, BioPorto and R&D Systems assays. 791,793,806–808 The relative specificities of the assays for the monomeric and dimeric forms of NGAL were not determined, although a study reporting a significant association of monomeric forms with tubulointerstitial fibrosis in CKD found a significant correlation between urinary NGAL results obtained using the Abbott assay and the monomeric form detected by Western blotting. However, it appears that a systematic analysis of the absolute specificity for this form had not been undertaken. 792 Assays able to robustly differentiate between the NGAL forms would be useful in terms of determining potential contributory sources and providing further clinical insight and this is currently being addressed by several companies. For example, predominantly homodimeric NGAL is found in urine from patients with UTI compared with monomeric NGAL in patients with AKI and mainly elevations in homodimeric NGAL in urine in patients following cardiac surgery in the absence of AKI, suggesting a predominant activated neutrophil source. 793,806 Indeed, in a cohort of 5599 individuals from the general population recruited into the Copenhagen Heart Study, plasma NGAL measured using an in-house assay was significantly associated with several inflammatory indices, particularly neutrophil count and CRP, in addition to showing an inverse association with estimated glomerular filtration rate (eGFR), and was independently associated with outcome irrespective of eGFR. 809 Interestingly, similarly elevated plasma NGAL concentrations were found in anephric and anuric patients on dialysis compared with healthy individuals, providing support for the elevated NGAL levels in CKD being predominantly extra-renal, although the balance of increased production against decreased clearance in accounting for the elevated concentrations is not known. 810
The most surprising finding was the marked disparity between the manufacturer’s reported measuring range for the CE-marked BioPorto/ADVIA assay and our LLoQ findings, which essentially placed the LLoQ at the suggested optimal cut-off value for NGAL in diagnosing AKI, a situation that is far from ideal given the inherent greater variability in that area. 795 This may well be platform specific as previous studies had reported the performance of this assay to be acceptable on the Beckman Coulter AU 5822 platform, although LLoQ, hook effect and recovery were not investigated,801 and also on the Roche Cobas 6000 and Hitachi 917 platforms, although consistently higher results were obtained for EDTA plasma on the former. 811 Following our study, BioPorto issued a new version of the application note for the ADVIA platform, with a revised measuring range and LLoQ included among the changes.
The significance of the interference of very high concentrations of haemoglobin in all assays (although a previous study had not found this for the Abbot assay800) is not clear and the extent to which such high concentrations are found clinically in urine needs to be investigated. Certainly, haemolysis of whole blood is known to affect plasma NGAL measurements, although this could potentially reflect the presence of neutrophil-derived NGAL or interference by haemoglobin directly. 802 The importance of such technical and biological pre-analytical effects is beginning to be recognised for NGAL, with an association of leukocyturia with higher NGAL concentrations, much higher concentrations of NGAL being present in serum compared with matched EDTA plasma samples, most likely because of release of NGAL from neutrophils, which is of particular importance when historical studies are compared, and age- and sex-related effects on urinary NGAL being reported. 812–816 Stability during processing and storage does not seem to be an issue under the conditions examined. 817,818
The aspects of NGAL measurement highlighted in this chapter are crucial to an interpretation of studies examining the clinical validity and utility of NGAL measurements. However, the extent to which studies or reviews examining the clinical potential of NGAL785 consider such aspects is highly variable, with, for example, a review of the broader clinical applications of NGAL only briefly mentioning such aspects, an earlier meta-analysis of studies examining the potential of NGAL in AKI diagnosis including a consideration of the assays used786 and a recent review focusing on NGAL in predicting AKI providing a much more comprehensive overview, including a tabulated summary of the assays used in each study. 795 The last review, although supporting the promise of NGAL, also flagged up the issue that many of the studies had an inadequate study design and failed to follow guidelines such as the STARD criteria,368 issues to address if NGAL is to fulfil its potential. Indeed, although agreeing that NGAL is a promising biomarker, the idea that it is the ‘troponin of the kidney’ has recently been used as an example of one of the ‘false myths and legends’ in laboratory diagnostics,790 and further analytical and biological insights including understanding of the impact of comorbidities and specific assays for kidney-derived NGAL are needed to allow rational evaluation and optimal use. 796,819
Vascular endothelial growth factor: relative value of serum or plasma and quality control aspects
Introduction
Vascular endothelial growth factor-A (VEGF-A), often referred to as VEGF, although there are several family members, is a major angiogenic cytokine. Existing as several isoforms generated by alternative splicing, there has been considerable interest in VEGF, particularly in cancer, given its pivotal role in regulating angiogenesis. 820,821 With increased understanding of the underlying mechanisms involving hypoxia and HIF-1α in upregulating VEGF, numerous therapies have been developed targeting this pathway and are in use across many cancer types. 587 This has particularly been the case in ccRCC, given the widespread inactivation of the VHL gene and consequent stabilisation of HIFs and increased expression of VEGF and the ineffectiveness of conventional chemotherapy. 822 Accompanying this has been a raft of studies exploring the possible use of VEGF as a biomarker, in particular for either prognostic use or for predicting response to VEGF-related therapies. In renal cancer, several studies have reported predictive uses for VEGF or its receptors, although studies are small and heterogeneous and require further confirmation. 823 As reviewed in the previous chapter, VEGF has also shown promise in RCC prognostically, although this is complicated by studies using either serum or plasma, in which concentrations vary markedly. When we and others first reported such serum–plasma differences in VEGF, this was proposed to be the result of platelets containing and releasing VEGF, which was subsequently confirmed in several studies. 600,601,824 Based on the evidence reviewed in Chapter 12, VEGF was included as one of the prioritised biomarkers for the RCC prognostic study undertaken as part of this programme and we included both plasma and serum VEGF to determine which, if any, provides clinically useful information, particularly given the impact of processing, as reviewed extensively below. The results of the prognostic study are described in Chapter 14 but we describe here some of the technical aspects of the measurements.
Methodology
As described in the following chapter, VEGF concentrations were determined for the RCC patients in the prognostic cohort. Matched serum and EDTA plasma samples were analysed for each patient as available, with a total of 430 patients having both sample types with detectable VEGF in both. Samples were analysed using the Human VEGF Quantikine kit from R&D Systems, which is a sandwich ELISA specifically measuring VEGF-A. This assay is one of the most widely used commercially available assays for VEGF and we previously carried out validation studies of this assay, both when evaluating the importance of blood sample handling and also when describing the existence of a novel soluble VEGF receptor variant in amniotic fluid. 630,825 All aspects evaluated, including parallel dilution, within- and between-run precision and recovery, were acceptable and similar findings were reported in subsequent studies using this assay. 826,827 It was apparent that the assay measured the free form of VEGF and not VEGF complexed with the receptors and, although specific for VEGF-A, both VEGF121 and VEGF165 isoforms are detected. 825,827,828 The manufacturer’s protocol was followed, with all samples being analysed in duplicate and low, medium and high recombinant VEGF controls also being analysed. On inspection of the data, some results appeared anomalous in terms of almost no difference in values between serum and plasma. To investigate this further, additional data analysis was carried out and, following this, some of the samples were subjected to additional measurements of calcium and potassium, undertaken using routine assays in the Leeds Teaching Hospitals Clinical Chemistry Laboratories, to confirm the presence of potassium EDTA in the purported plasma aliquots and its absence in the serum aliquots. In addition, to assess stability during a freeze–thaw cycle, matched aliquots of serum and plasma from 20 patients with RCC were thawed at room temperature and after approximately 1 hour were refrozen. These 40 aliquots were then analysed together with 40 matched aliquots that had been stored frozen without any additional freeze–thaw step.
Results
As expected given platelet-derived VEGF being released during clotting, serum VEGF concentrations were higher than VEGF concentrations in matched plasma samples in most cases. The range for plasma VEGF was 5.2–1480.9 pg/ml (median 67.8 pg/ml), with corresponding values for serum VEGF being 9.9–4283.3 pg/ml (median 348.4 pg/ml), with one patient providing the sample with the unusually low values for both serum and plasma VEGF (i.e. 9.9 and 5.2 pg/ml, respectively), which were almost at the limits of the assay (manufacturer’s LoD 9 pg/ml and 10.9–12.7 pg/ml); these will be reanalysed and the functional sensitivity of the assay determined in our hands. 826 What was striking, however, was a number of samples in which there was very little difference between plasma and serum VEGF and a number of samples in which the plasma concentrations of VEGF were higher than the serum concentrations. This is depicted in Figure 47.
FIGURE 47.
Frequency distribution of plasma VEGF concentrations as a percentage of the serum VEGF concentration for 426 patients with RCC. Results are shown for 426 patients rather than 430 as four of the most extreme values (210–474%) were omitted to be able to show the spread of the majority of samples optimally.
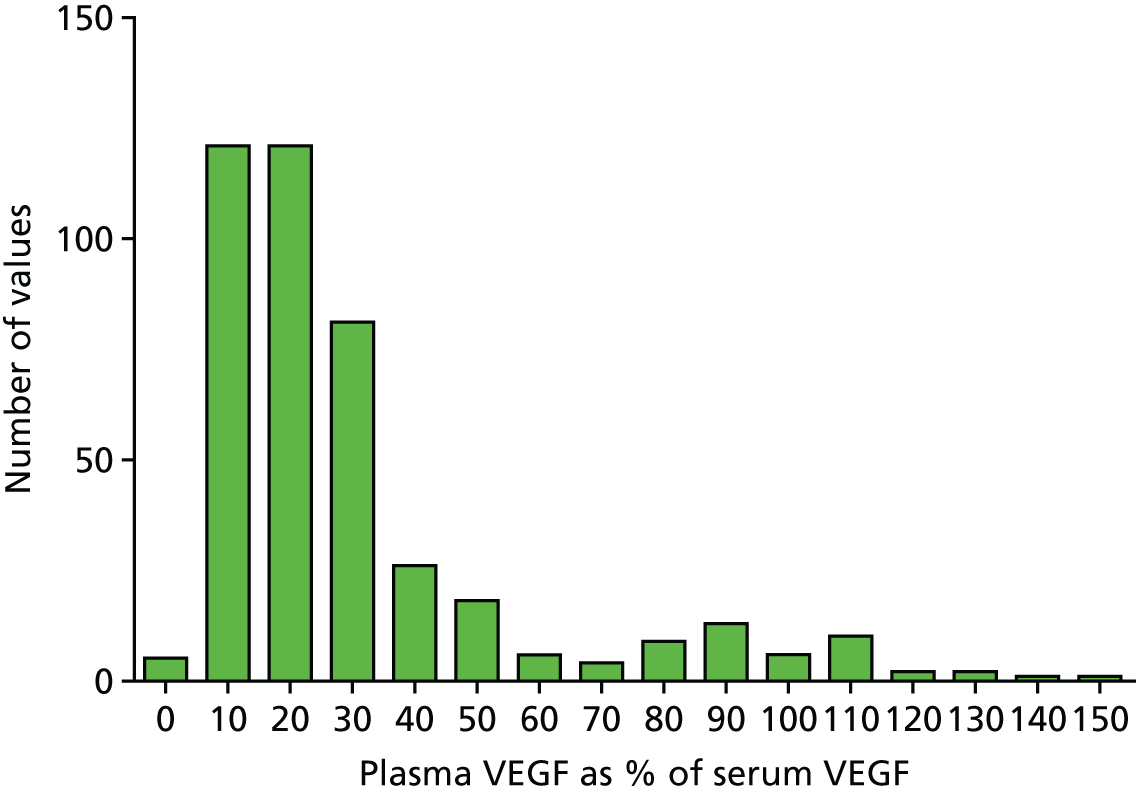
When the results were examined further, there were several apparent patterns. For four patients (not shown in Figure 47) it appeared that the plasma and serum aliquots may have been switched, either at the time of processing at the four specific sites involved or during analysis, as there were marked differences between them but in the opposite direction to that expected (e.g. 1480.9 pg/ml vs. 312.2 pg/ml for plasma and serum VEGF, respectively). For a further group of 48 patients (from around 80% upwards), plasma and serum samples were much more similar and in many cases differed by < 30 pg/ml (the range of the standard curve in this assay covers from 15.6 to 1000 pg/ml). This, together with (1) the relatively low values for serum VEGF concentration in this group (27.5–579 pg/ml, median 98.6 pg/ml) compared with the group as a whole, (2) the lack of a significant difference between serum VEGF concentrations in this group of 48 patients (27.5–579 pg/ml, median 98.6 pg/ml) and their matched plasma VEGF concentrations (25.0–621 pg/ml, median 92.4 pg/ml; p = 0.169), (3) the lack of recorded deviations from the protocol in terms of processing causing possible spurious results and (4) the fact that 44 of these 48 patients had been recruited at one specific site led to the hypothesis that most if not all of these patients had actually had no clotted sample collected for serum at recruitment, but two sets of anticoagulated blood for plasma collected. This is further supported by Figure 48. By taking the average of the two measurements (i.e. ‘serum’ and plasma) for each patient and subtracting it from the ‘serum’ result (in this case the ‘serum’ but the same result would be obtained for plasma), and expressing this difference as a percentage of the average, this could be examined in relation to the known variability of the assay. In Figure 48, essentially all of these 48 samples make up the columns from 10% down to –20%, as indicated by the arrows, the majority of which are, therefore, within the variability of the assay (9.1–9.5% inter-run CVs in our study) and are, therefore, likely to have been determined from two aliquots of the same sample type, that is, plasma in this case. The most extreme columns from –40% to –70% represent the four samples for which the most likely explanation is direct switching of serum and plasma at some point.
FIGURE 48.
Frequency distribution of the difference between the serum VEGF and the average of the matched serum and plasma VEGF results for each of 430 RCC patients expressed as a percentage of that average. When serum and plasma results are the same, the value on the x-axis would be zero. The green arrows indicate the bins containing the 48 samples where both samples may be plasma.
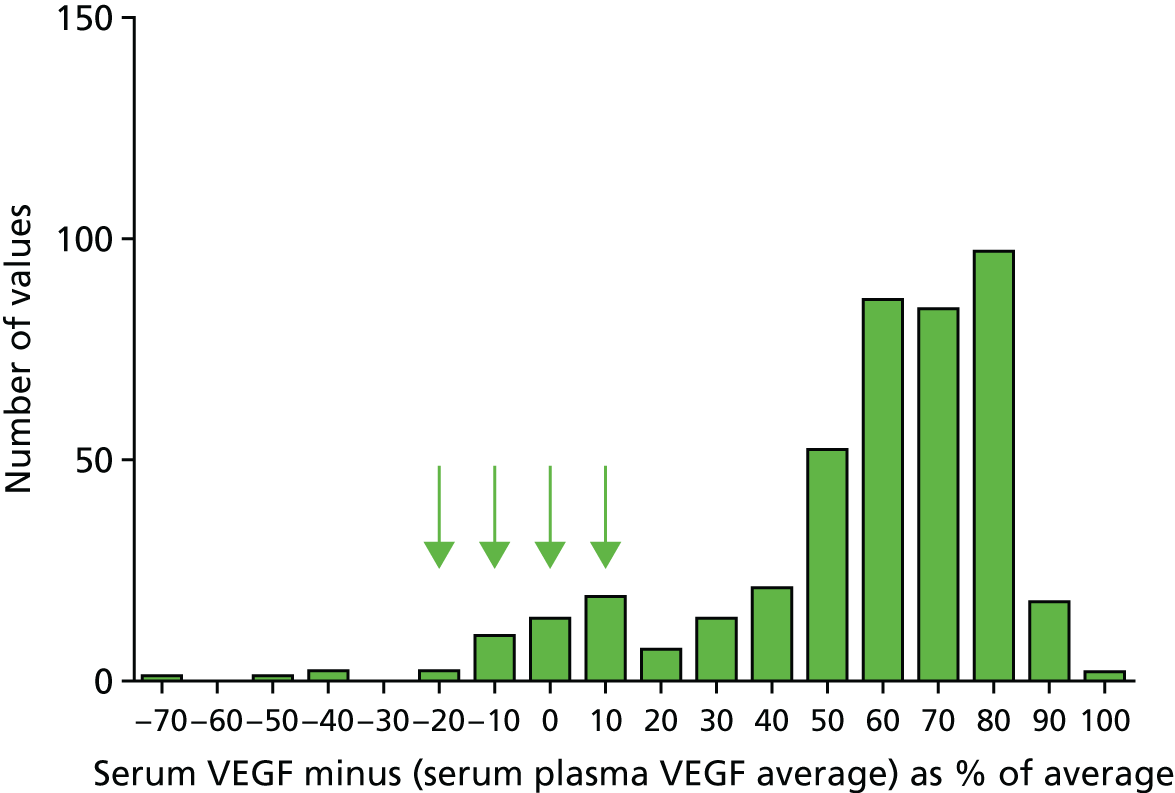
Biochemical analysis of some of the samples identified as having potential problems showed an absence of calcium in the ‘serum’ samples and high levels of potassium, consistent with the samples actually being EDTA plasma. Subsequent biochemical analysis of 235 serum samples from RCC patients for the prognostic study (run to provide some missing routinely measured analytes) found such results for calcium and potassium in 40 samples, all of which were from this recruitment site. Enquires at the site where the 44 patients had been recruited from established that staff had been unaware of any issues and had used Greiner tubes with purple and red tops for EDTA plasma and serum, respectively. However, on close inspection of the tubes used at that site, it was apparent that the tube with a purple top was a K2EDTA tube as expected but that the tube with a red top used was actually a K3EDTA tube rather than a red top Z serum clot activator tube; these two tubes differed purely by a purple band around the top of the patient label compared with a red band and the small print on the label showing K3EDTA, as shown in Figure 49.
FIGURE 49.
Greiner blood collection tubes showing the differences and overlap in colour closures and labelling (top) and the printed indication of additives (bottom). Tubes are (1) K3EDTA for crossmatch, (2) K2EDTA, (3) K3EDTA, (4) K3EDTA and (5) Z serum clot activator. Tubes 3 and 5 were the intended tubes to be used for EDTA plasma and serum collection, respectively, but one centre inadvertently used tube 4 instead of tube 5 for ‘serum’ collection.
Although we have definitive evidence for samples being collected in the wrong tubes for only 40 patients, a total of 46 patients recruited at this site had been included in the VEGF sample analysis for the prognostic study. In addition to the 44 samples known or suspected to be EDTA plasma rather than serum, one sample appeared to have actually been collected in the correct tube types, with values of 1118.0 and 42.5 pg/ml for serum and plasma VEGF, respectively (this was actually the first patient recruited at the site), and one sample was the sample with almost undetectable VEGF in both serum and plasma. At this stage, until all 46 samples are checked by analysing calcium and potassium levels, all ‘serum’ VEGF results from patients recruited at this site have been removed from the prognostic analysis described in the following chapter, as a precaution. The four samples from other sites that were part of the group of 48 samples in which plasma and serum concentrations were similar have been retained in the analysis at present as, in all cases, the serum VEGF concentration was higher than the plasma VEGF concentration, with plasma representing 76–93% of serum, and, therefore, there may not be an issue. Measurement of potassium and calcium concentrations for the sample with VEGF concentrations of 64.3 and 68.9 pg/ml for plasma and serum, respectively, confirmed the correctness of the plasma and serum attribution. The other three samples will also be checked.
For the 380 patients with no apparent issues with the plasma and serum VEGF measurements and matched results available, serum VEGF ranged from 61.6 to 4283.3 pg/ml (median 377.2 pg/ml) and was significantly different (p < 0.0001) from the plasma VEGF concentrations, with values of 9.2–419.5 pg/ml (median 67.5 pg/ml). As shown in Figure 50, the serum VEGF concentration was significantly correlated with the plasma VEGF concentration (Spearman’s r = 0.594; p < 0.0001) and markedly so with the serum minus plasma VEGF concentration (Spearman’s r = 0.977; p < 0.0001), which may not be completely unexpected given that, in 50% of the patients, the plasma VEGF component was ≤ 20% of the total, that is, the serum VEGF.
FIGURE 50.
Relationship between serum VEGF and plasma or serum minus plasma VEGF concentrations (n = 380). (a) Serum VEGF vs. plasma VEGF; (b) serum VEGF vs. plasma VEGF omitting eight samples with serum VEGF > 1500 pg/ml to allow expansion of the x-axis; (c) serum VEGF vs. serum – plasma VEGF; (d) serum VEGF vs. serum – plasma VEGF omitting eight samples with serum VEGF > 1500 pg/ml to allow expansion of the x-axis.




For 282 of these patients, platelet counts were available either on the same day as the day that the biomarker blood sample was collected or within 2 days of this. Platelet counts were highly significantly correlated with VEGF concentrations, particularly serum or serum minus plasma VEGF concentrations, with correlation coefficients of 0.57 and 0.56, respectively. Assuming that the serum minus plasma VEGF is derived from platelets, the calculated VEGF content per platelet is highly variable, ranging from 0.02 to 5.77 pg/106 platelets, with a median value of 1.14 pg/106 platelets. Of note, this was not corrected for the haematocrit, which we have previously advocated,630 as these data were not available at this time.
For the stability study, there was a significant difference between the frozen and freeze–thawed plasma samples, using the Wilcoxon matched-pairs signed-rank test (range 15–270 pg/ml compared with 11.0–231.0 pg/ml, respectively; median 77.0 pg/ml for both; p = 0.004), but not between the serum samples (p = 0.065) or the serum minus plasma samples (p = 0.447), as shown in Figure 51. However, although this was statistically significant, many plasma samples had concentrations of VEGF at the bottom end of the standard curve, where variability was higher, and only in six cases did the decrease in the freeze–thawed samples exceed 10%. In 6 out of 20 cases for plasma and 7 out of 20 cases for serum the results were either the same or higher in the freeze–thaw samples. This requires further investigation in a larger number of samples and with more samples with higher VEGF concentrations.
FIGURE 51.
Comparison of VEGF concentrations in matched plasma and serum samples stored frozen and thawed immediately prior to analysis with concentrations in paired aliquots that had been subjected to an additional freeze–thaw cycle. Samples from 20 patients with RCC were used, with storage at –80°C. Aliquots were thawed at room temperature. F/T, freeze–thaw.

Discussion
Pre-analytical considerations are of pivotal importance in clinical chemistry laboratories and represent an area of growing awareness and concern, as reviewed earlier in this chapter. With many biomarker studies being undertaken in research laboratories, such aspects are often overlooked and not considered either in the study design phase or during evaluation of the biomarkers, and this will increasingly contribute to the lack of consistency between published reports and will potentially be a major barrier to progress. A major finding here was the inadvertent use of the wrong blood tubes at one site, which resulted in no serum being banked but two samples of EDTA plasma being collected. This was detected both through VEGF measurements not fitting the usual pattern of serum compared with plasma concentrations and by the low concentration/absence of calcium and elevated potassium levels in those samples. A further three samples may also have been inadvertently switched and this will be investigated further. Although many analytes can be measured equally in plasma or serum, the findings here with VEGF illustrate the importance of using the correct tube type and specimen type if reliable biomarker results are to be achieved. If VEGF analysis of both serum and plasma samples had not been undertaken and if none of the clinical chemistry analysis had been necessary, this error may not have been apparent and the resultant data analysis could have led to additional inconsistent results appearing in the literature. This occurred despite site inductions being carried out to ensure that the correct tubes were used and, indeed, it appears as though the first sample taken at this site may have actually be taken using the correct tubes. Whether or not measurement of simple analytes such as potassium or calcium or fibrinogen should be undertaken routinely in samples associated with research biomarker studies in clinical trials, for example, should be considered. This could confirm sample types as being plasma or serum but unfortunately would still not control for deviations from sample processing protocols in terms of processing time delays or storage conditions, for example. CLSI standards and guidelines exist for many pre-analytical areas, including the identification of patients and samples, venepuncture and sample processing. 494,727,728 There is also a guideline covering all aspects of blood tubes, including construction material, additives and labels; although earlier versions also included aspects relating to the colour of the tube closures, this has been omitted from the current version. 726 Clearly, having similar colour tube caps for different tube types, and indicating the nature of the tube additives only in small print on the label and by using a small, different coloured band, can easily lead to errors. Given the heterogeneity in tube types, calls for harmonisation have been made by the EFLM and, although difficult given the multiple manufacturers, it is hoped that this will be achieved in the future. 497
The occurrence of thrombocytosis in many cancers has been recognised for over a hundred years and its association with shorter survival has been reviewed. 829 However, whether this is an epiphenomenon reflecting systemic elevation of cytokines, such as IL-6 or other tumour-derived thrombopoietic factors, or has a direct involvement in cancer progression, for example through physical interactions with tumour cells or the production of platelet-derived cytokines or growth factors, is still not clear. However, over the last few years it has become increasingly apparent that the sequestration of VEGF by platelets is a major determinant of the results seen in many studies examining VEGF as a potential biomarker. We found serum VEGF to be significantly correlated with platelet number, in line with many other studies, for example the studies by Verheul et al. 601 and Salgado et al. 604 In a meta-analysis it has been estimated that the total platelet concentration of VEGF far exceeds the circulating concentration in plasma in cancer patients by almost 30-fold, with leucocyte-associated VEGF accounting for a much smaller amount and tumour tissue being one of the main sources. 821 Interestingly, however, skeletal muscle was calculated as having the largest reservoir of VEGF. In an animal study investigating this further, similar findings were reported with regard to the importance of platelets in terms of their accumulation of VEGF, but VEGF-impregnated pellets implanted subcutaneously or microscopic xenografts also resulted in increased platelet VEGF concentrations but not plasma VEGF concentrations, supporting a role for platelets in actively sequestering such angiogenic factors, with implications for underlying biology and therapies. 830
The literature on VEGF as a biomarker is very mixed, with differences in whether serum or plasma has been used and which type of plasma has been used, the blood collection methods used and the sample processing/storage methods used; therefore, it is not surprising that there is a lack of consistency generally about the utility of VEGF as a biomarker. 821,831 Importantly, however, the majority of studies have used the same ELISA kit as in our studies, allowing studies to be more usefully compared. The consideration of which anticoagulant to use if circulating endogenous levels of VEGF are to be determined accurately is important. Our initial studies comparing serum and plasma were based on only four healthy volunteers; in terms of measuring endogenous circulating VEGF, we found citrate plasma to be optimal for measuring the lowest VEGF concentrations, presumably because of low levels of platelet activation,600 and this has been confirmed subsequently. 826 In most cases, little or no difference was seen comparing citrate plasma with EDTA plasma if samples were processed within 30 minutes or with a similar delay, with the samples kept on ice. 600,601,827 However, we reported that EDTA anticoagulated blood appeared to be less stable over time, with delayed processing up to 4 hours leading to markedly increased plasma VEGF concentrations in two out of four cases for EDTA plasma and to a lesser extent in one out of four cases for citrate plasma. This may represent the worst-case scenario, with blood being taken into a syringe before distribution into anticoagulant-containing tubes, because of the volumes needed, and, hence, possibly leading to more platelet activation than would have occurred if blood had been collected directly into anticoagulant-containing tubes. 600 In our biobanked RCC blood samples, overall, 97% were centrifuged within 2 hours of venepuncture, with a median time until centrifugation of 1 hour 11 minutes. Average increases in VEGF concentrations of 28–34% have been reported for EDTA blood left at room temperature for 1 hour before processing and of 64–80% for EDTA blood left at room temperature for 2 hours, although no changes were seen if samples were left at 4°C for prolonged periods. 826,827 Centrifugation speeds have also been highlighted as being important, presumably in terms of generating platelet-poor plasma, together with avoiding sampling the plasma immediately above the buffy coat, which we also adopt in our SOPs. 826,827
However, it is now apparent that, even with citrate, platelet activation occurs and VEGF is released. Studies adopting a very stringent protocol to avoid any platelet activation in vitro, monitored by concurrent measurement of platelet factor 4 (PF4), have found that plasma from blood collected into tubes containing either CTAD (sodium citrate, theophylline, adenosine and dipyridamole) or Edinburgh mixture (EDTA, prostaglandin E1 and theophylline), with rapid processing at 4°C, contained much lower VEGF concentrations. There was even more platelet activation if the citrated tubes were maintained at room temperature rather than at 4°C. 831 This study also reported that serum VEGF did not plateau until at least 2 hours after venepuncture, although plain glass tubes were used rather than clot activator tubes as in our study and we have shown that clotting in terms of peptide fragmentation is essentially complete within 60 minutes of venepuncture. 831,832 A study comparing samples collected without tourniquet in PECT tubes containing a mixture similar to that of the Edinburgh mixture at 4 C with citrated plasma collected with tourniquet and at room temperature, comparing healthy controls and patients with metastatic RCC or other cancers, found significantly higher VEGF levels in citrated plasma than in PECT plasma. 833 In addition, using citrated plasma, the VEGF concentration was higher in both the RCC group (and similar to the EDTA values in our study) and the non-RCC group than in the control group whereas using PECT plasma only the RCC group had a higher VEGF concentration. 833 PF4 concentrations were higher in all citrate samples than in the PECT samples, supporting the fact that, even in citrate samples, some platelet activation is occurring in vitro. 833 Interestingly, we have selected processing at ambient temperature within our biobanking protocols over many years to avoid the detrimental effects of cold temperatures on platelets, which are normally stored unrefrigerated, and yet clearly activation and degranulation appear to be inhibited at colder temperatures. 834
Comparing VEGF concentrations in serum from four patients with rheumatoid arthritis analysed prior to freezing with results following subsequent freeze–thaw cycles found a dramatic difference (average 67% reduction in the freeze–thaw sample, with almost total degradation seen in two samples) after just one freeze–thaw cycle and a continued decline with subsequent freeze–thaw cycles. 835 In absolute terms this meant a change from a mean of 352 pg/ml (SD 166 pg/ml) to 134 pg/ml (SD 184 pg/ml) after one cycle and 49 pg/ml (SD 18 pg/ml) after six cycles. Although we did not assay samples fresh (i.e. prior to freezing), our data examining samples after freezing or one further cycle of freeze–thaw still show very high serum values in all serum samples and, therefore, are unlikely to support their results, but this needs exploring further. One notable difference was the thawing of samples at 37°C in the study by Kisand et al. 835 Although EDTA plasma VEGF concentrations have been reported to decrease in samples (10 healthy controls and 10 patients with rheumatoid arthritis) that have been thawed more than once compared with once only (mean difference 20%), with no further decline until after 10 thaws, serum VEGF concentrations declined only after 10 thaws (mean difference 18%). 826 We did not find such changes and another study has reported no changes in EDTA plasma VEGF concentrations until after seven and nine freeze–thaw cycles. 827 Further study using a multiplex chip also found no effect on VEGF concentrations of freeze–thawing up to at least 10 times compared with immediate analysis using serum or heparinised plasma. 836 Similarly inconsistent results have been reported for storage, with stability for up to at least 2 years at –80°C reported for VEGF in EDTA plasma or serum826 compared with findings of significant degradation after periods of > 3 months at –75°C for VEGF in serum;835 however, the study by Kisand et al. 835 used accelerated stability tests at elevated temperatures and, given the possible effect of higher temperatures on VEGF during thawing, this needs to be revisited.
Systematic studies of biological variability of VEGF are few but consistent. In the first reported study comparing serum and plasma (citrated) we reported no effects of age, sex or menopausal status on plasma or serum VEGF630 and this has been confirmed since in a large (n = 306) reference range study using EDTA plasma and serum. 826 Some evidence of diurnal variation was reported and examination of intra-individual variability showed median CVs of 39–56% examined at several points in a month and repeated at 6 months and 1 year later and with exercise having a marked although short-term effect. 826 Similar, relatively large intra-individual variability has been reported in a further study using only EDTA plasma, with CVs of 69% and 57% for short- and long-term biological variation, respectively, compared with 51% for inter-individual variation. 827 Such biological variation may contribute to the lack of differences seen in studies using plasma between different groups of patients with breast diseases, which we reported;630 this has also been reported for patients with colorectal cancer and benign adenoma and disease-free groups. 827 Interestingly, platelet-associated VEGF has been reported to show only low levels of intra-individual variability over time, with CVs of only 17% and inter-subject CVs of 44%, although, interestingly, in this study the intra-subject variability of citrated platelet-poor plasma processed at ambient temperature was only 19%, although inter-subject variability was markedly higher at 148%. 837
Clearly, the clinical utility of VEGF as a biomarker is far from established and, given the critical effects of the anticoagulant used on ‘circulating endogenous VEGF’, it appears likely that studies employing EDTA or even citrated plasma are not truly representative of that VEGF component and may include variable contributions from platelet-derived VEGF fraction, which is largely covered by serum VEGF measurements. Similar to the findings here in RCC and with the caveat that background endogenous circulating VEGF came from measurement of EDTA plasma, there was a wide variation in the calculated platelet content of VEGF in breast cancer patients and healthy control subjects, with no significant difference between them, although, in a study examining patients with a range of advanced cancers, platelet VEGF was significantly increased compared with the VEGF level in healthy control subjects. 630,838 There was no relationship between either plasma or serum VEGF and clinicopathological parameters, although plasma VEGF discriminated more between control subjects and the various breast disease groups. 630 Although several groups did have elevated VEGF levels compared with normal control subjects, we did not find a clear trend in breast cancer for plasma or serum VEGF for local disease, remission or metastatic disease, but this was likely to be the result of a possible effect of tamoxifen on VEGF, both circulating and platelet derived. 630 The question of whether or not under standard consistent processing conditions EDTA or citrated plasma can act as a surrogate of cancer behaviour through essentially integrating platelet VEGF content with the activatability of platelets or whether or not serum provides a more stable indication of any cancer-associated properties remains to be determined and, under the conditions recorded in our biobanking activity, this should be possible to determine in RCC at least and is illustrated in the following chapter.
Interestingly, many of the published findings above regarding plasma VEGF and anticoagulant appear to have had little impact on the measurement of VEGF in clinical research studies, with variable procedures still being adopted, and it is important that this is highlighted in a large study. This may be in part because of the publication of many studies in clinical biochemistry-type journals; a greater impact may result if studies are published in cancer journals, for example, as many of the studies are led by people working in cancer research. This issue of continued publication of studies ignoring such facts has also been seen for other proteins, for example MMP-9; despite the importance of serum compared with plasma being highlighted and the effect on the interpretation of results, studies continue to measure MMP-9 inappropriately and neglect pre-analytical considerations. 839 We intend to publish the VEGF results from our studies as a separate paper also highlighting such aspects and additionally exploring further some of the discrepancies such as the stability of VEGF to freeze–thawing.
Overall conclusions
This chapter has shown several examples of the importance of verifying the performance of commercially available assays prior to use in biomarker studies and assessing the potential impact of pre-analytical factors. This illustrates how inconsistent results across studies can easily arise and, with studies employing certified assays on clinical chemistry platforms and research grade immunoassays being used at various stages in the biomarker pipeline, it is often difficult to interpret data across studies. Without assay characterisation and validation in an early phase of the biomarker translational pathway, progress in biomarker translation and adoption will continue to be slow and result in wasted resources. The value of the biobank can clearly be seen in this chapter and in the initial prognostic study in RCC described in the following chapter. Collection of samples in multicentre studies has to be pragmatic and take into account resource availability and considerations of cost and logistics when deciding on the possible sample types and frequencies of collection and of course consider future developments, maximising the value as long-term resource. 373,493 Of most importance is consistency of the processes adopted and recording of relevant information both at the biobank level and in publications, as proposed in the BRISQ recommendations. 375,724 Deviations can then be factored in, but the more complex the protocols the less likely that most centres will comply. Thought needs to be given to possible quality assurance checks on compliance with the sample types, processing timing steps and storage and this is being pursued by several groups, for example ISBER, although as yet there is no universal panel that can be used to provide the information needed. 723 However there will be no uniform protocol for all biomarkers and all fluid types as pre-analytical factors impact on different proteins in different ways. If a specific biomarker is the focus of studies then protocols can be evidence based and comply with any known necessary pre-analytical specifications to ensure that measurements are valid, in just the same way that the assays have to achieve the required technical performance criteria. By adopting high-standard evidence-based protocols with accurate record keeping and quality systems, it is likely that biobanks such as the one here will be of value for many years and many questions can be answered and the suitability of any specific biomarker can be determined based on the processes adopted and knowledge of pre-analytical impacts as studies evolve.
Chapter 14 Circulating prognostic biomarkers in renal cancer: clinical validation study of promising candidates
Introduction
As outlined in Chapter 10, there remains an urgent clinical need for the identification and validation of biomarkers that provide prognostic information for patients with localised RCC. It is recognised that following surgery to remove the primary tumour, around one-third of patients will relapse with distant metastatic disease. Accurately differentiating these patients from those who are likely to be cured by surgery alone allows for more rational use of finite NHS resources, in terms of intensity of follow-up, and stratifies patients for entry into ongoing trials of adjuvant therapy. Such treatments are likely to be both costly to the NHS and potentially toxic for patients, further highlighting the need to identify and target high-risk groups.
For a prognostic biomarker (or panel of markers) to be adopted into clinical practice, it must be shown to be superior, or add value, to currently employed prognostic scoring systems, which for RCC are based on standard clinicopathological criteria alone. 438 Such nomograms fail to adequately reflect individual tumour biology and the identification of molecular markers in RCC to improve risk stratification and the delivery of more personalised medicine is recognised as a research priority by both the European Association of Urology840 and the European Society for Medical Oncology. 841
The current study represents the culmination of the RCC-related work, to date, within workstream 2. It focuses on the clinical validation of candidate circulating biomarkers detectable in serum and/or plasma, collected pre nephrectomy/ablation, that have been reported in the literature by ourselves and/or others to carry prognostic value in patients with localised ccRCC, using the assembled multicentre prospective observational cohort and RTB described in Chapter 11. Markers were shortlisted based on the level of existing published evidence of their prognostic potential, in addition to the availability of suitable and robust assays. On this basis, the following proteins were selected for validation: (1) VEGF-A, referred to as VEGF, (2) OPN, (3) CAIX and (4) CRP. A number of routine laboratory variables were also included in the analysis, again based on existing supporting literature. 513,519,569,570,842,843
The aim of this study was to validate the prognostic utility of the selected markers individually or combined as a panel or index in a large multicentre cohort of UK patients with localised ccRCC. Furthermore, the ability of the markers to add value to the widely employed postoperative Leibovich score438 was examined, in particular among those patients deemed to be at high or intermediate risk of relapse by the score alone.
Methods
Patient population
Patients were identified retrospectively from the whole RCC cohort (described in full in Chapter 11). Inclusion criteria for the study were broad and included patients with (1) ccRCC, (2) radical/partial nephrectomy or tumour ablation, (3) localised disease (stages I–III), (4) preoperative serum/plasma sample availability and (5) preoperative clinical biochemistry/haematology measurement availability. All patients who fulfilled these criteria were included, except for patients with VHL disease (an exclusion criterion for the overall study) and coexistent other active cancers. For comparative descriptive purposes only, a subset of patients presenting with metastatic disease, who may or may not have undergone nephrectomy but who otherwise met the inclusion/exclusion criteria, were also included.
Vascular endothelial growth factor, osteopontin and carbonic anhydrase IX measurement
A full description of assay validation is provided in Chapter 13. Similarly, full details of sample collection, processing and storage are described in Chapter 11. Briefly, OPN and CAIX concentrations were quantified in EDTA plasma using commercially available ELISA Quantikine kits. Both serum and EDTA plasma VEGF were analysed, using the Human VEGF Quantikine kit, a sandwich ELISA specifically measuring VEGF-A. The difference between serum and plasma VEGF concentrations was also examined for its value as a prognostic variable, calculated as serum minus plasma VEGF concentration. All samples were measured blinded, in duplicate, and manufacturers’ QC samples were included on each plate. Assay runs not passing QC standards, that is, QC samples not meeting the specifications supplied by the manufacturer, were rerun (this applied to two plates only). Similarly, samples for which replicate CVs exceeded 10% were reanalysed. Other serum analytes, including CRP, were measured by the NHS clinical biochemistry laboratory at each participating centre, with assays having a CRP concentration of < 10 mg/l being reanalysed using the high-sensitivity CRP assay in the Leeds Teaching Hospitals Blood Sciences Laboratory.
Clinicopathological variables
Clinical factors examined included sex, age at diagnostic procedure, smoking history, alcohol consumption, BMI, symptoms (local/systemic/absent) and Eastern Cooperative Oncology Group performance status (ECOG PS). Pathological factors recorded were tumour size, TNM stage, Fuhrman grade, Leibovich score and presence or absence (if not commented, assumed to be absent) of histological necrosis, sarcomatoid change and microvascular invasion (MVI). Routine laboratory variables included haemoglobin, WBC count, neutrophil count, lymphocyte count, platelet count and serum measurements of sodium, potassium, urea, alanine transaminase (ALT), calcium, albumin and CRP. The derived parameter, NLR, was also examined on the basis of previous findings. 507,510,511
Statistical methods
Baseline concentrations of each marker were explored in terms of differences within demographic and clinical factors among all patients (stages I–IV) using the Spearman rank correlation coefficient and p-value if a single continuous variable or median (range) and p-value from the Wilcoxon–Mann–Whitney test or Kruskal–Wallis test if comparing two or more subgroups. Correlations between the markers were investigated using a correlogram, based on simple linear regression and the Spearman rank correlation coefficient.
Survival analyses were conducted exclusively in patients with stage I–III disease (i.e. non-metastatic). MFS formed the principal time-to-event end point, calculated as the period from the date of the procedure to the date of distant metastases, the definition also used in developing the Leibovich score. 438 Any patients without disease recurrence were censored at the date that they were last known to be recurrence free (for patients who died without recurrence this was the date of death). Secondary end points were OS and CSS, defined as the period from the date of the procedure to the date of (1) death from any cause (OS) or (2) death from RCC (CSS). Patients still alive at the time of analysis were censored at the date last known alive (or at the date of non-cancer-related death when considering CSS).
Cox proportional hazards models were used to determine the prognostic potential of the markers; survival functions were estimated using the Kaplan–Meier method and compared using the log-rank test. Markers were initially examined as continuous variables and then as dichotomised variables. The latter was accomplished by considering all possible cut-off points within the range of each marker and selecting the one that maximised Harrell’s concordance index (C-index). For each Cox proportional hazards model constructed, the proportional hazards assumption was tested by assessing Schoenfeld residuals. 844
The ability of the shortlisted markers to add prognostic utility to the Leibovich score was explored, particularly with respect to those patients in the intermediate- and high-risk scoring groups (score 3–5 and score ≥ 6, respectively). This was performed by sequentially including each marker into a Cox proportional hazards model with the Leibovich score as an existing predictor variable and MFS as the response variable.
Statistical analysis was carried out in the R Environment for Statistical Computing (R Core Team, Vienna, Austria) and reported according to REMARK criteria. 845 In making inferences, significance levels were adjusted for multiple testing, when appropriate. Tests for significance were two-sided and p-values of < 0.05 were considered significant.
Sample size
Sample size calculations were based on upper and lower extremes of relapse rates at 2 years for patients with localised ccRCC of 27.5% and 12.5%. Using the higher end of the relapse rate, we assumed a separation at 2 years of 15% between survival curves to be required for each marker (given a dichotomised split around a given point) to justify its inclusion in a multiplex marker model. This equates to a HR of approximately 0.5. Table 88 shows the sample sizes required to identify a HR of 0.5 with 80–95% power given a significance level (α) of 5% in a 5-year study at the higher and lower relapse rates and at an intermediate rate of 20%. Samples sizes are shown for unadjusted and adjusted (using Bonferroni correction) significance levels.
| Power | Relapse rate | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 27.5% | 20% | 12.5% | |||||||
| α1 | α2 | α3 | α1 | α2 | α3 | α1 | α2 | α3 | |
| 0.80 | 168 | 248 | 282 | 216 | 320 | 362 | 320 | 476 | 538 |
| 0.85 | 192 | 276 | 312 | 244 | 356 | 400 | 364 | 528 | 596 |
| 0.90 | 224 | 316 | 356 | 288 | 404 | 456 | 428 | 600 | 678 |
| 0.95 | 276 | 380 | 422 | 356 | 484 | 540 | 528 | 722 | 804 |
Results
In total, 706 patients were recruited into the full study between July 2011 and June 2014 across 11 UK centres. Among the 629 patients with a confirmed RCC, 481 (76.5%) had ccRCC, 59 (9.4%) papillary RCC, 46 (7.3%) chromophobe RCC and 27 (4.3%) oncocytoma; 12 (1.9%) were unclassified and the remaining four cases were made up of two translocation tumours, one cystic mixed chromophobe RCC and ccRCC and one mucinous tubular and spindle cell RCC.
Among patients with localised (stage I–III) ccRCC, the majority (n = 406; 94.2%) met the inclusion criteria for the current study. In addition, a subset of 30 out of the 50 patients with ccRCC presenting with stage IV disease was examined in parallel, selected to represent recruiting centres and distribution of metastatic site. These patients were included purely for descriptive purposes of the selected biomarkers, rather than for an assessment of their prognostic ability within this group. At the time of analysis, the median length of follow-up from diagnosis among patients still alive was 28.9 months (range 0.6–48.3 months). Among those presenting with localised RCC, 33 patients had relapsed with distant metastatic disease and 21 patients had died, of which six deaths were directly attributed to cancer. The small number of CSS events (n = 6) precluded the inclusion of this end point in the current analysis.
Patient and tumour characteristics
Patient characteristics for the 406 patients included in the current study are shown in Table 89. The male-to-female ratio was 1.9 : 1, confirming the known male preponderance of this tumour type. Based on BMI, approximately three-quarters of patients were classified as either obese (36%) or overweight (40%). The majority of patients had an ECOG PS of 0/1 (97%). One-third of patients were asymptomatic at the time of presentation. Among those patients reporting symptoms, 30% reported local symptoms (such as haematuria or flank pain), 14% reported systemic symptoms (such as weight loss or fatigue) and 22% reported both local and systemic symptoms.
| Characteristic | Patients with localised ccRCC (n = 406) |
|---|---|
| Sex, n (%) | |
| Male | 267 (66) |
| Female | 139 (34) |
| Age (years), median (range) | 63 (29–92) |
| BMI (kg/m2) | |
| < 18.5 (underweight) | 4 (1) |
| 18.5–24.9 (healthy) | 91 (22) |
| 25–29.9 (overweight) | 147 (36) |
| > 30 (obese) | 161 (40) |
| Missing | 3 (1) |
| ECOG PS, n (%) | |
| 0 | 319 (79) |
| 1 | 74 (18) |
| 2 | 11 (3) |
| 3 | 1 (< 1) |
| 4 | 1 (< 1) |
| Symptoms, n (%) | |
| Local | 120 (30) |
| Systemic | 57 (14) |
| Both | 88 (22) |
| None | 141 (34) |
| Pathological T stage, n (%) | |
| 1a | 122 (30) |
| 1b | 100 (25) |
| 2a | 38 (9) |
| 2b | 9 (2) |
| 3a | 106 (26) |
| 3b | 9 (2) |
| 3c | 1 (< 1) |
| Not applicablea | 21 (5) |
| Tumour size (mm), median (range) | 52 (11–180) |
| Fuhrman grade, n (%) | |
| 1 | 11 (3) |
| 2 | 142 (35) |
| 3 | 203 (50) |
| 4 | 49 (12) |
| Missing | 1 (< 1) |
| Necrosis, n (%) | |
| Present | 102 (25) |
| Absent | 283 (70) |
| Not applicablea | 21 (5) |
| Microvascular invasion, n (%) | |
| Present | 65 (16) |
| Absent | 320 (79) |
| Not applicablea | 21 (5) |
| Sarcomatoid change, n (%) | |
| Present | 18 (5) |
| Absent | 367 (90) |
| Not applicablea | 21 (5) |
| Leibovich risk group, n (%) | |
| Low | 147 (36) |
| Intermediate | 163 (40) |
| High | 71 (18) |
| Not applicableb | 25 (6) |
| TNM stage, n (%) | |
| I | 240 (59) |
| II | 44 (11) |
| III | 116 (29) |
| Missing | 6 (1) |
| Procedure, n (%) | |
| Radical nephrectomy | 286 (70) |
| Partial nephrectomy | 99 (24) |
| Radiofrequency ablation | 13 (3) |
| Cryoablation | 8 (2) |
| Relapsed, n (%) | |
| Yes | 33 (8) |
| No | 352 (87) |
| Missing | 21 (5) |
At the time of diagnosis, over half of tumours (55%) were pathological stage T1, of which 30% were pT1a, and 28% were locally advanced (pT3). Among the 381 patients with localised ccRCC who underwent a radical or partial nephrectomy with an evaluable Leibovich score, 39%, 43% and 18% were classified as low, intermediate and high risk, respectively.
Biomarker associations/correlations
Correlation of the markers with each other was examined. The correlation was strongest between plasma and serum VEGF values (r = 0.59, p < 0.001). In general, the markers were significantly correlated, with the exception that CAIX showed no correlation with either serum or plasma VEGF values (r = –0.02, p = 0.655, and r = 0.08, p = 0.112, respectively).
All four markers showed associations with several clinicopathological parameters. Increased preoperative plasma VEGF concentrations were consistently associated with poor prognostic tumour factors, such as increased pathological tumour size (r = 0.14, p = 0.005), stage IV disease (p < 0.001), grade 4 tumours (p = 0.025), high-risk Leibovich score (p = 0.031), the presence of sarcomatoid change (p < 0.001) and MVI (p = 0.046).
Among patients with localised disease, a trend towards increased plasma VEGF in relapsers compared with non-relapsers was observed (84.8 vs. 66.5 pg/ml; p = 0.054). Serum VEGF concentrations showed fewer significant associations, limited to Leibovich score (p = 0.008), presence of necrosis (p = 0.041) and overall TNM stage (p = 0.018). Serum minus plasma VEGF showed an association with Leibovich score and presence of necrosis only. Box and whisker plots are shown for the markers according to TNM stage (Figure 52) and Leibovich score (Figure 53).
FIGURE 52.
Box and whisker plots for preoperative circulating (a) CRP (mg/l), (b) OPN (ng/ml), (c) CAIX (pg/ml), (d) serum VEGF (pg/ml), (e) plasma VEGF (pg/ml) and (f) serum minus plasma VEGF (pg/ml) according to TNM stage (I–IV). Boxes correspond to the first and third quartiles (the 25th and 75th percentiles) with median concentration also shown. Whiskers extend to 1.5 × IQR, with data points beyond classed as outliers. 846

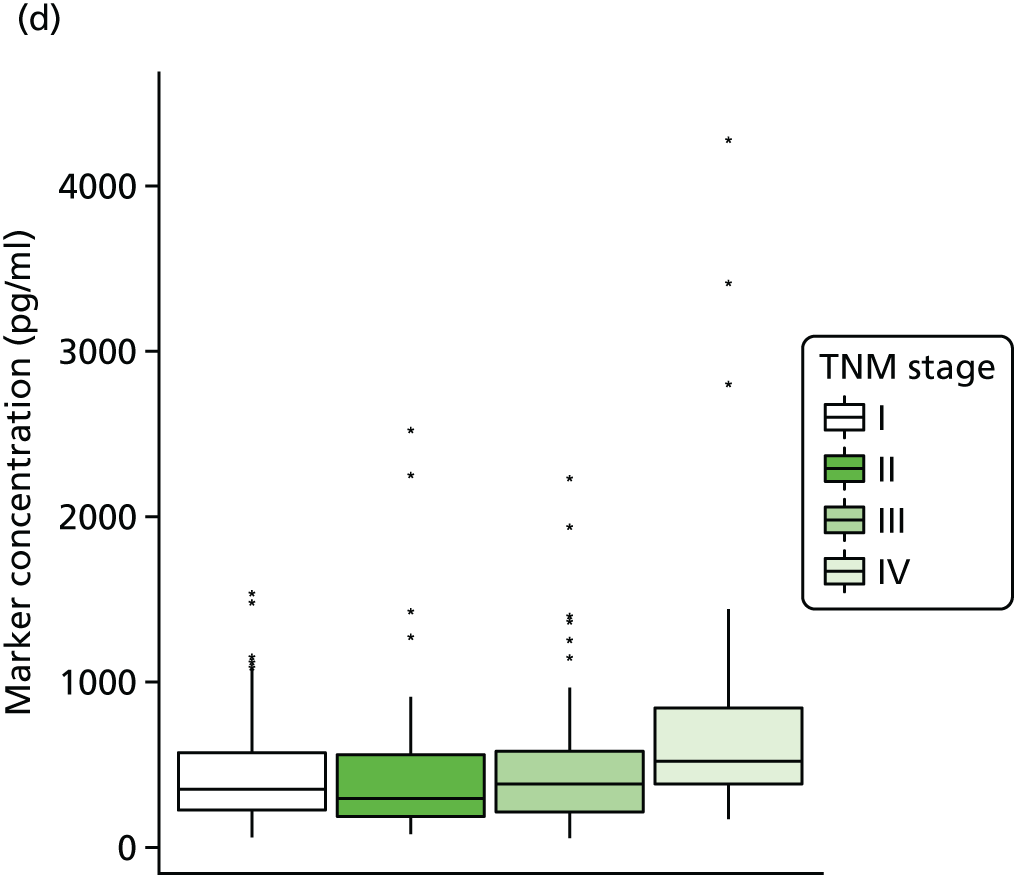
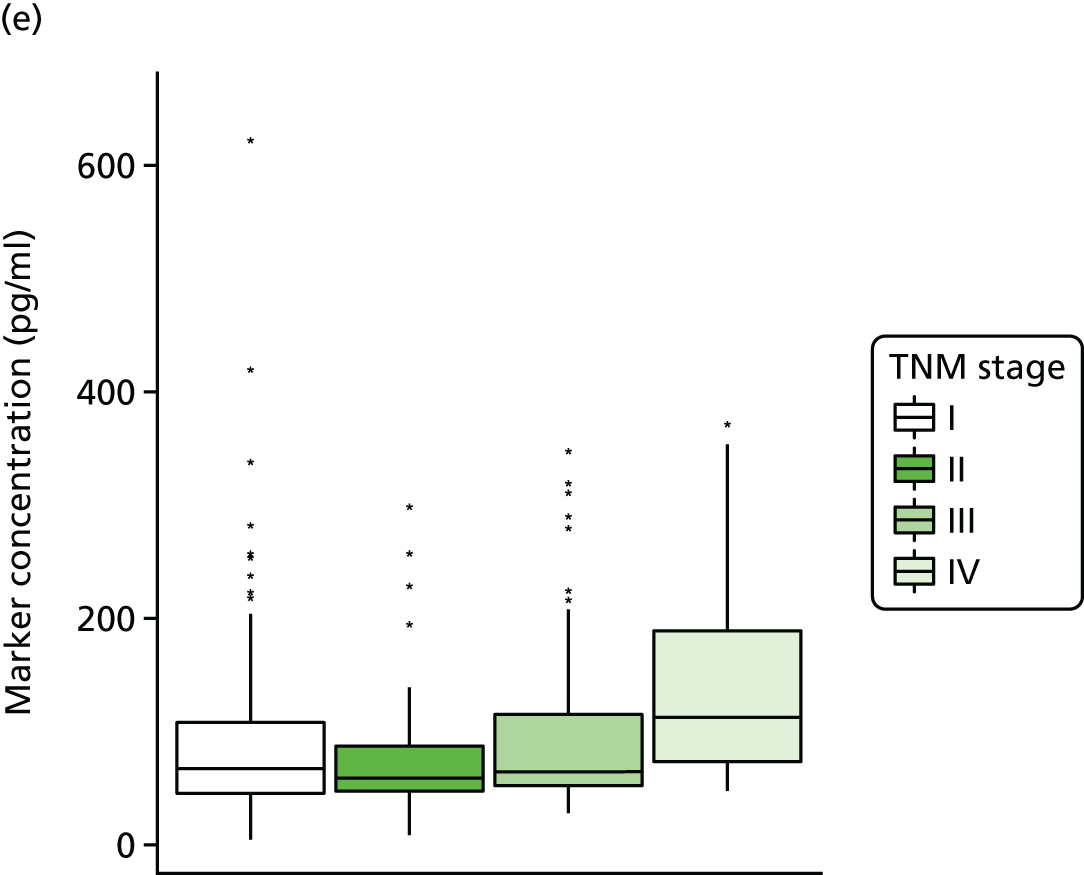
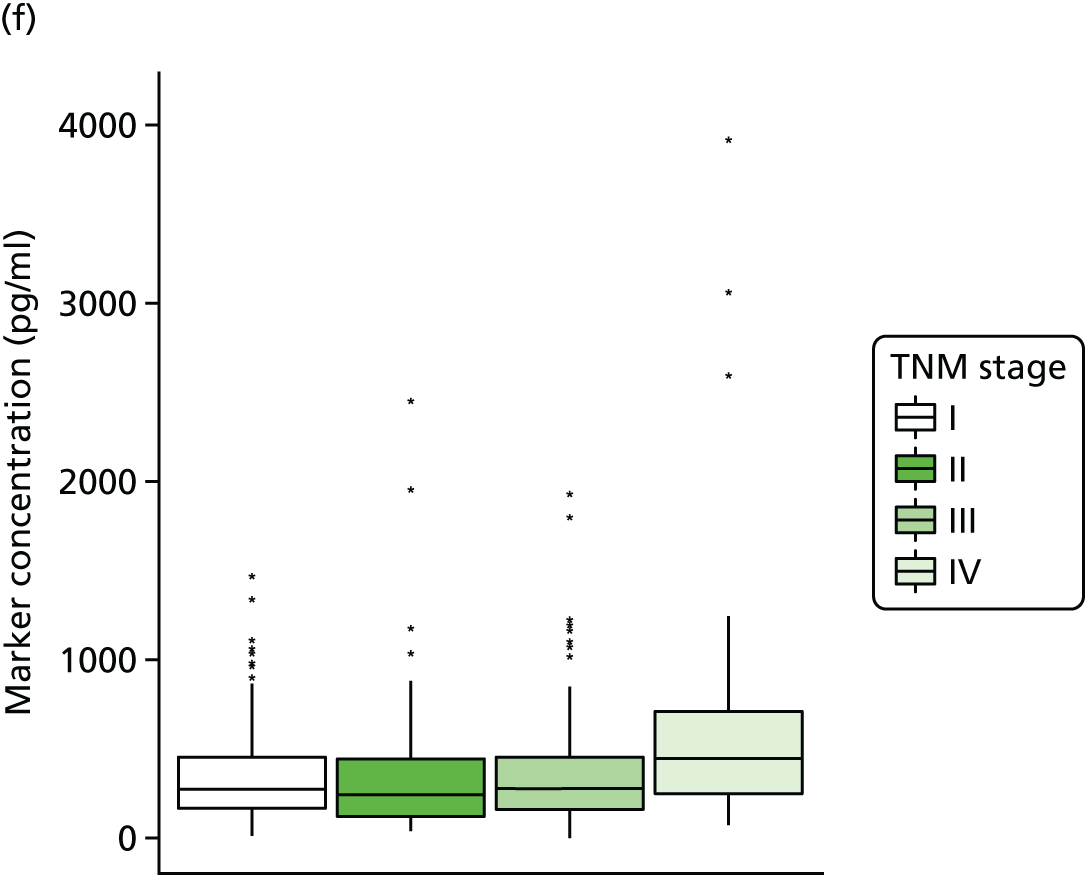
FIGURE 53.
Box and whisker plots for preoperative circulating (a) CRP (mg/l), (b) OPN (ng/ml), (c) CAIX (pg/ml), (d) serum VEGF (pg/ml), (e) plasma VEGF (pg/ml) and (f) serum minus plasma VEGF (pg/ml) according to Leibovich risk classification (low, intermediate or high). Boxes correspond to the first and third quartiles (the 25th and 75th percentiles) with median concentration also shown. Whiskers extend to 1.5 × IQR, with data points beyond classed as outliers. 846

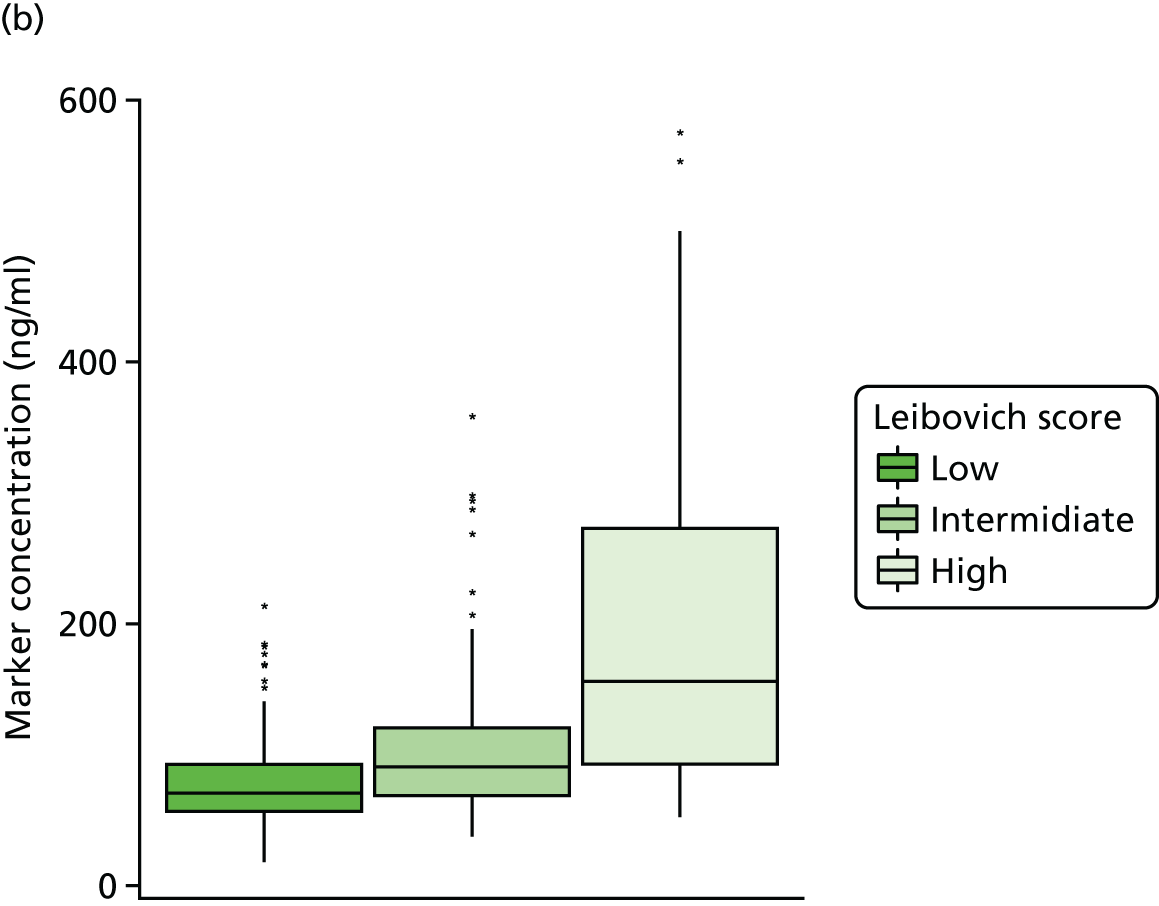

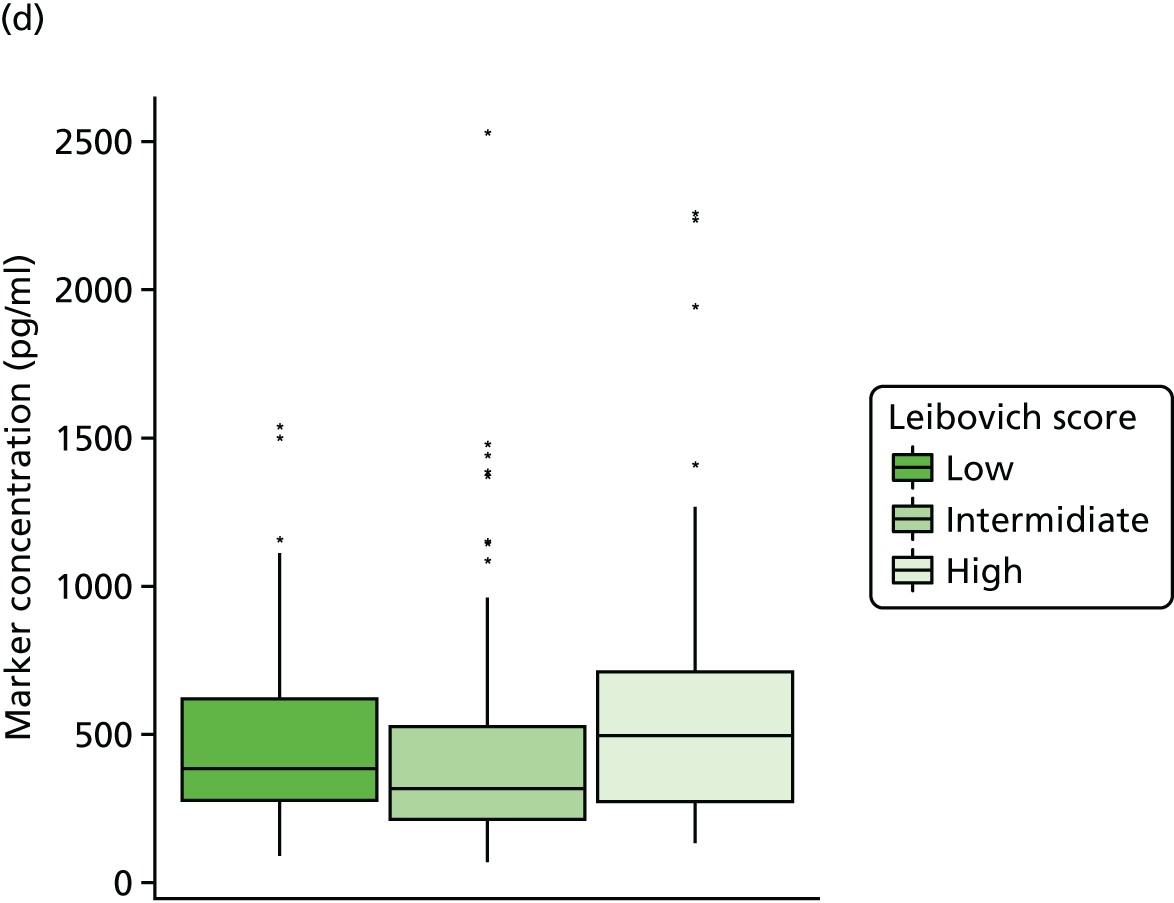
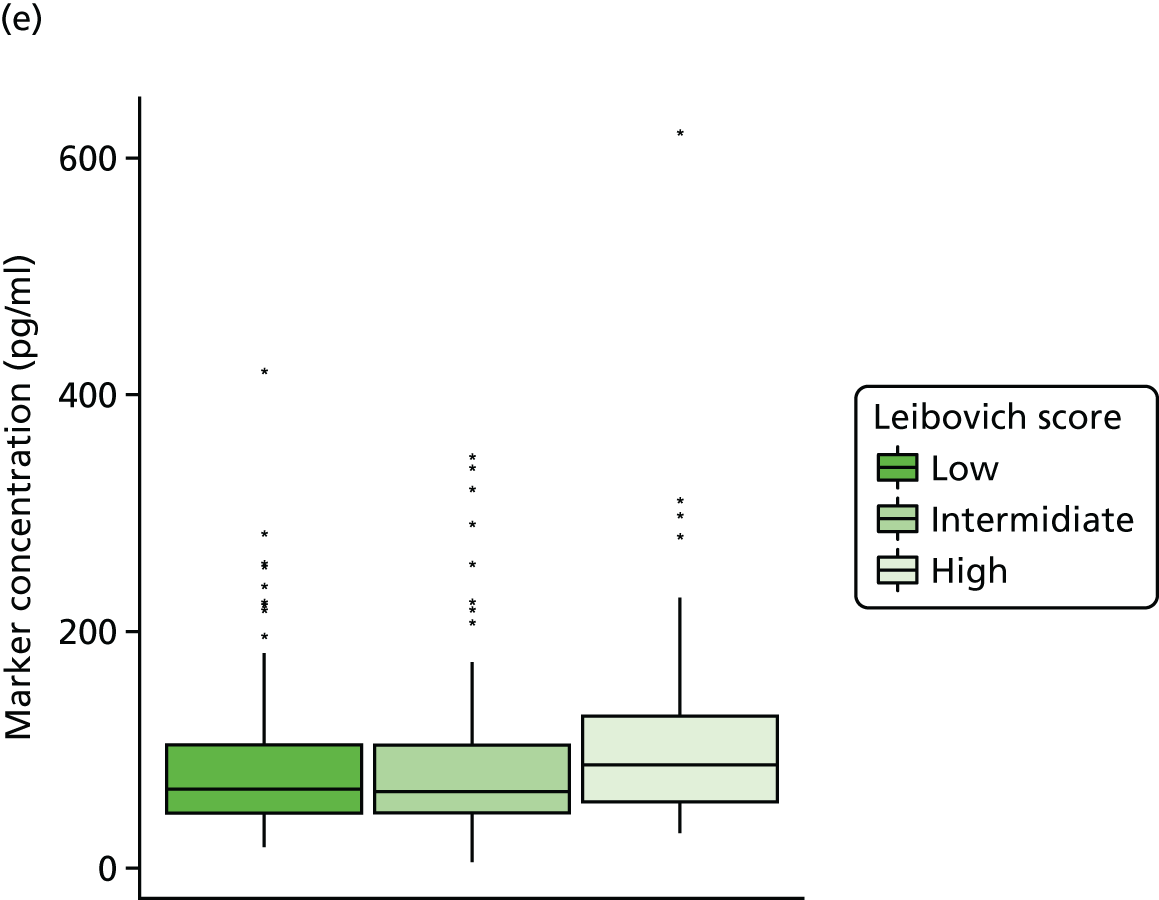
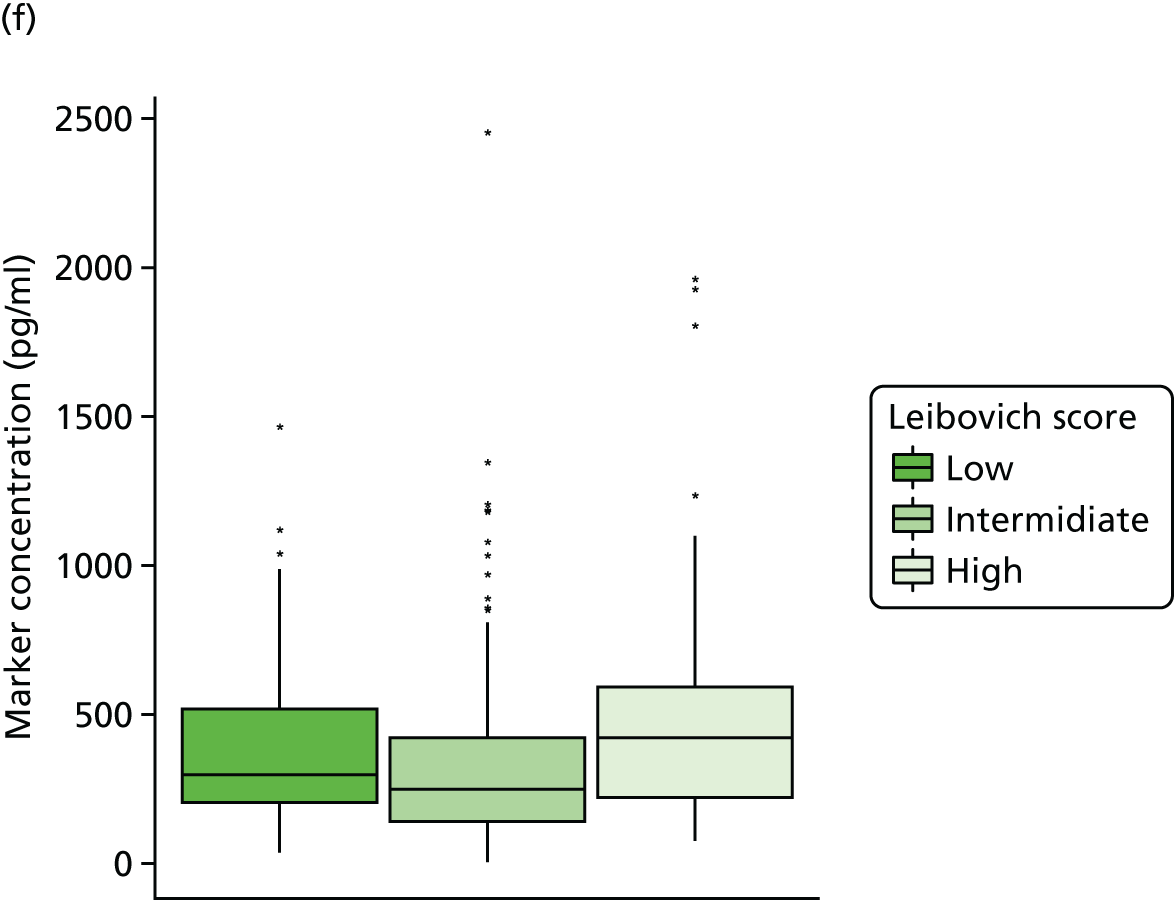
Similarly, higher baseline circulating concentrations of OPN, CRP and CAIX were consistently associated with poor prognostic features, including higher stage and grade, increased tumour size, presence of necrosis, MVI and sarcomatoid change. An association with Leibovich score was again observed (see Figure 53). Both CRP (p = 0.002) and OPN (p = 0.001) were elevated in relapsers compared with non-relapsers. In terms of clinical associations, higher circulating levels of all three biomarkers were associated with poorer ECOG PS. Circulating concentrations of both CRP and CAIX showed a significant association with BMI, with patients with a BMI of < 18.5 kg/m2 (underweight) having higher median serum/plasma levels of each biomarker. However, only six patients made up this group, two of whom had metastatic disease.
Univariate analysis of biomarkers and time-to-event end points
Metastasis-free survival
The following were statistically significantly associated with MFS (p < 0.05): sex, WBC count, neutrophil count, NLR, platelet count, potassium, albumin, CRP, OPN, serum VEGF, plasma VEGF, serum minus plasma VEGF, pathological tumour size, pathological T stage, Fuhrman grade, necrosis, MVI, sarcomatoid change, Leibovich score, CT tumour size, CT T stage and overall TNM stage (Table 90). Figure 54 shows Kaplan–Meier survival curves for Fuhrman grade and TNM stage as two of the most clinically relevant variables found to be significant in univariate analysis, in addition to the Leibovich score as a main focus of the study. Chi-squared and associated p-values from the log-rank test to compare survival curves are also shown and are significant in all three cases. In addition, there was weak evidence of an association with MFS for serum sodium concentration (HR 0.89, 95% CI 0.79 to 1.01; p = 0.064).
| Characteristic | Survival | |||
|---|---|---|---|---|
| MFS | OS | |||
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Sex | ||||
| Male | – | – | – | – |
| Female | 0.31 (0.11 to 0.90) | 0.031 | 0.46 (0.15 to 1.36) | 0.160 |
| WBC count | 1.18 (1.02 to 1.37) | 0.026 | 1.15 (0.99 to 1.35) | 0.069 |
| Neutrophil count | 1.23 (1.05 to 1.44) | 0.009 | 1.15 (0.96 to 1.39) | 0.136 |
| NLR | 1.13 (1.01 to 1.26) | 0.038 | 0.97 (0.78 to 1.21) | 0.770 |
| Platelet count | 1.00 (1.00 to 1.01) | 0.002 | 1.00 (1.00 to 1.01) | 0.073 |
| Sodium | 0.89 (0.79 to 1.01) | 0.064 | 0.8 (0.71 to 0.91) | 0.001 |
| Potassium | 2.41 (1.11 to 5.24) | 0.026 | 2.70 (1.13 to 6.48) | 0.026 |
| Albumin | 0.94 (0.90 to 0.98) | 0.006 | 0.93 (0.89 to 0.97) | 0.001 |
| CRP | 1.01 (1.00 to 1.01) | < 0.001 | 1.00 (1.00 to 1.01) | 0.342 |
| OPN | 1.01 (1.00 to 1.01) | < 0.001 | 1.00 (1.00 to 1.01) | 0.120 |
| CAIX | 1.00 (0.99 to 1.00) | 0.729 | 1.00 (1.00 to 1.01) | 0.001 |
| Serum VEGF | 1.00 (1.00 to 1.00) | 0.012 | 1.00 (1.00 to 1.00) | 0.573 |
| Plasma VEGF | 1.00 (1.00 to 1.01) | 0.032 | 1.00 (1.00 to 1.01) | 0.144 |
| Serum minus plasma VEGF | 1.00 (1.00 to 1.00) | 0.028 | 1.00 (1.00 to 1.00) | 0.672 |
| Tumour size | 1.01 (1.01 to 1.02) | 0.002 | 1.01 (0.99 to 1.02) | 0.271 |
| Pathological T stage | ||||
| T1 | – | – | – | – |
| T2 | 3.15 (0.89 to 11.16) | 0.076 | 1.01 (0.21 to 4.74) | 0.995 |
| T3/T4 | 6.22 (2.47 to 15.68) | < 0.001 | 2.23 (0.88 to 5.65) | 0.092 |
| Grade | ||||
| 1/2 | – | – | – | – |
| 3 | 2.62 (0.85 to 8.04) | 0.092 | 4.68 (1.05 to 20.91) | 0.043 |
| 4 | 11.39 (3.59 to 36.12) | < 0.001 | 12.25 (2.54 to 59.04) | 0.002 |
| Necrosis | ||||
| No | – | – | – | – |
| Yes | 5.29 (2.44 to 11.51) | < 0.001 | 2.19 (0.91 to 5.29) | 0.081 |
| Microvascular invasion | ||||
| No | – | – | – | – |
| Yes | 2.31 (1.02 to 5.24) | 0.046 | 1.93 (0.70 to 5.32) | 0.204 |
| Sarcomatoid change | ||||
| No | – | – | – | – |
| Yes | 7.08 (2.67 to 18.77) | < 0.001 | 6.64 (2.21 to 19.97) | 0.001 |
| Leibovich risk | ||||
| Low | – | – | – | – |
| Intermediate | 1.29 (0.36 to 4.59) | 0.691 | 0.69 (0.21 to 2.26) | 0.541 |
| High | 11.18 (3.78 to 33.04) | < 0.001 | 2.91 (1.03 to 8.19) | 0.043 |
| CT size | 1.18 (1.06 to 1.31) | 0.003 | 1.06 (0.92 to 1.21) | 0.431 |
| CT stage | ||||
| T1 | – | – | – | – |
| T2 | 2.62 (1.01 to 6.80) | 0.047 | 1.54 (0.57 to 4.17) | 0.393 |
| T3/T4 | 4.02 (1.55 to 10.41) | 0.004 | 1.44 (0.46 to 4.52) | 0.536 |
| TNM stage | ||||
| I | – | – | – | – |
| II | 3.61 (1.02 to 12.82) | 0.047 | 1.02 (0.22 to 4.74) | 0.977 |
| III | 6.83 (2.71 to 17.22) | < 0.001 | 2.22 (0.90 to 5.47) | 0.083 |
FIGURE 54.
Kaplan–Meier survival curves showing MFS by (a) TNM stage, (b) Fuhrman grade and (c) Leibovich risk score.



Optimally discriminative cut-off points in terms of maximised C-index were derived as follows: CRP, cut-off point 14.1 mg/l; OPN, cut-off point 120.8 ng/ml; CAIX, cut-off point 60.4 pg/ml; platelet count, cut-off point 333 × 109/l; and serum sodium, cut-off point 141 mmol/l). They were all significantly associated with MFS (p < 0.05) (Table 91). Corresponding Kaplan–Meier survival curves are shown in Figure 55.
| Biomarker | Survival | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MFS | OS | |||||||||
| Optimised cut-off point | N | Number of events | HR (95% CI) | p-value | Optimised cut-off point | N | Number of events | HR (95% CI) | p-value | |
| CRP (mg/l) | ≤ 14.1 | 294 | 11 | 1.00 | – | ≤ 3.9 | 174 | 5 | 1.00 | – |
| > 14.1 | 72 | 17 | 6.62 (3.10 to 14.16) | < 0.001 | > 3.9 | 199 | 15 | 2.65 (0.96 to 7.29) | 0.059 | |
| OPN (ng/ml) | ≤ 120.8 | 257 | 11 | 1.00 | – | ≤ 95.4 | 199 | 6 | 1.00 | – |
| > 120.8 | 86 | 15 | 4.28 (1.96 to 9.31) | < 0.001 | > 95.4 | 149 | 11 | 2.67 (0.99 to 7.22) | 0.053 | |
| CAIX (pg/ml) | ≤ 60.4 | 176 | 8 | 1.00 | – | ≤ 112.1 | 289 | 10 | 1.00 | – |
| > 60.4 | 199 | 20 | 2.37 (1.04 to 5.38) | 0.040 | > 112.1 | 93 | 11 | 3.51 (1.48 to 8.28) | 0.004 | |
| Serum VEGF (µg/ml) | ≤ 268.7 | 106 | 4 | 1.00 | – | ≤ 356.5 | 169 | 7 | 1.00 | – |
| > 268.7 | 223 | 20 | 2.56 (0.87 to 7.50) | 0.087 | > 356.5 | 167 | 11 | 1.72 (0.67 to 4.44) | 0.262 | |
| Plasma VEGF (µg/ml) | ≤ 60.0 | 147 | 7 | 1.00 | – | ≤ 132.1 | 322 | 14 | 1.00 | – |
| > 60.0 | 226 | 21 | 2.12 (0.90 to 5.01) | 0.086 | > 132.1 | 58 | 7 | 3.08 (1.24 to 7.66) | 0.015 | |
| Serum minus plasma VEGF (µg/ml) | ≤ 344.9 | 194 | 10 | 1.00 | – | ≤ 222.4 | 122 | 5 | 1.00 | – |
| > 344.9 | 135 | 14 | 2.12 (0.94 to 4.78) | 0.069 | > 222.4 | 214 | 13 | 1.68 (0.6 to 4.74) | 0.326 | |
| Platelet count (× 109/l) | ≤ 333 | 315 | 17 | 1.00 | – | ≤ 300 | 280 | 10 | 1.00 | – |
| > 333 | 61 | 10 | 3.16 (1.44 to 6.93) | 0.004 | > 300 | 103 | 11 | 2.84 (1.2 to 6.71) | 0.017 | |
| Serum sodium (mmol/l) | ≤ 141 | 276 | 27 | 1.00 | – | ≤ 137 | 71 | 9 | 1.00 | – |
| > 141 | 102 | 1 | 0.09 (0.01 to 0.66) | 0.018 | > 137 | 314 | 12 | 0.23 (0.1 to 0.56) | 0.001 | |
FIGURE 55.
Kaplan–Meier survival curves showing MFS for dichotomised markers, serum sodium and platelet count (only markers found to be significant in univariate analysis are shown). (a) CRP; (b) OPN; (c) CAIX; (d) serum sodium; and (e) platelet count.
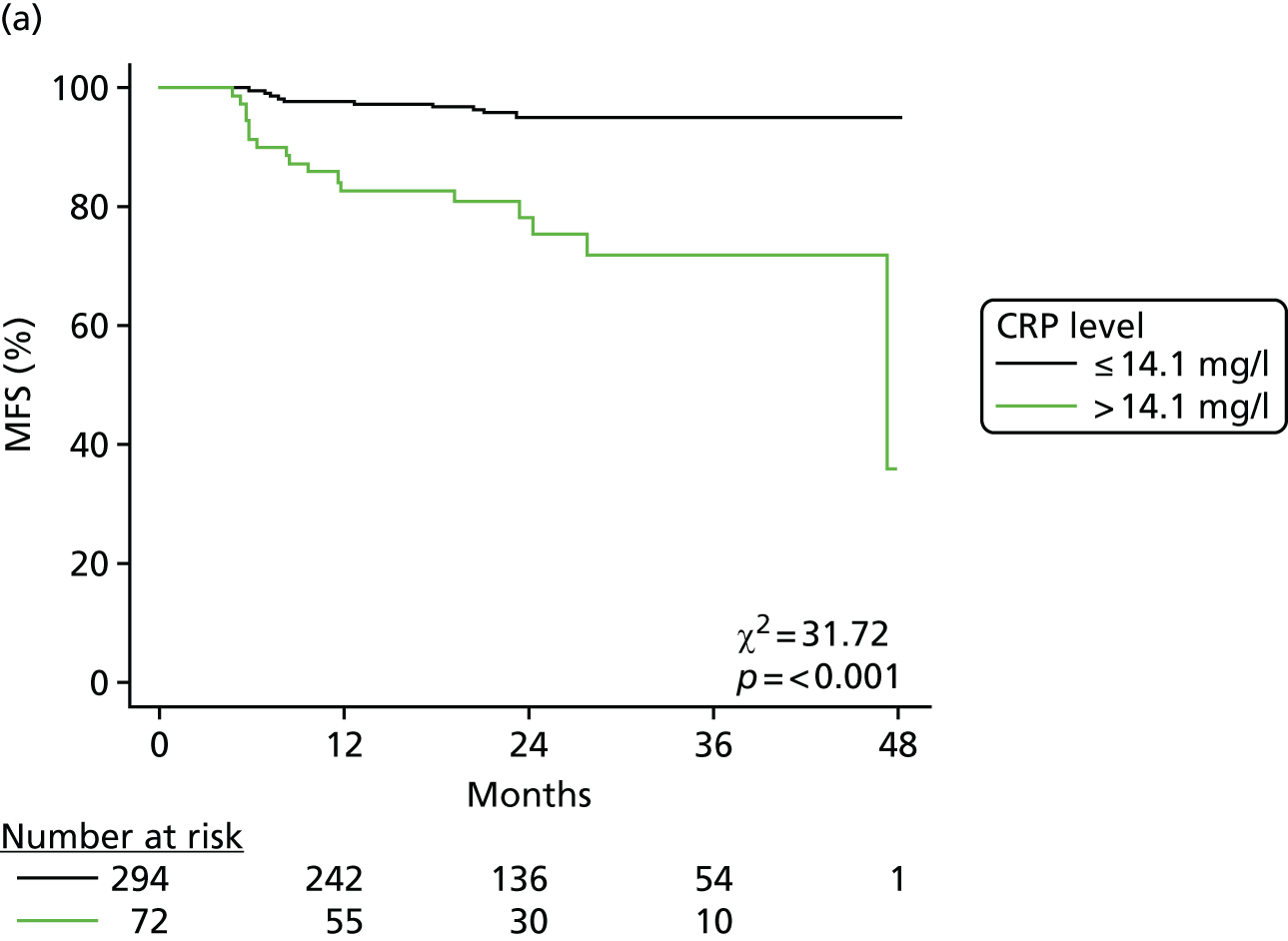



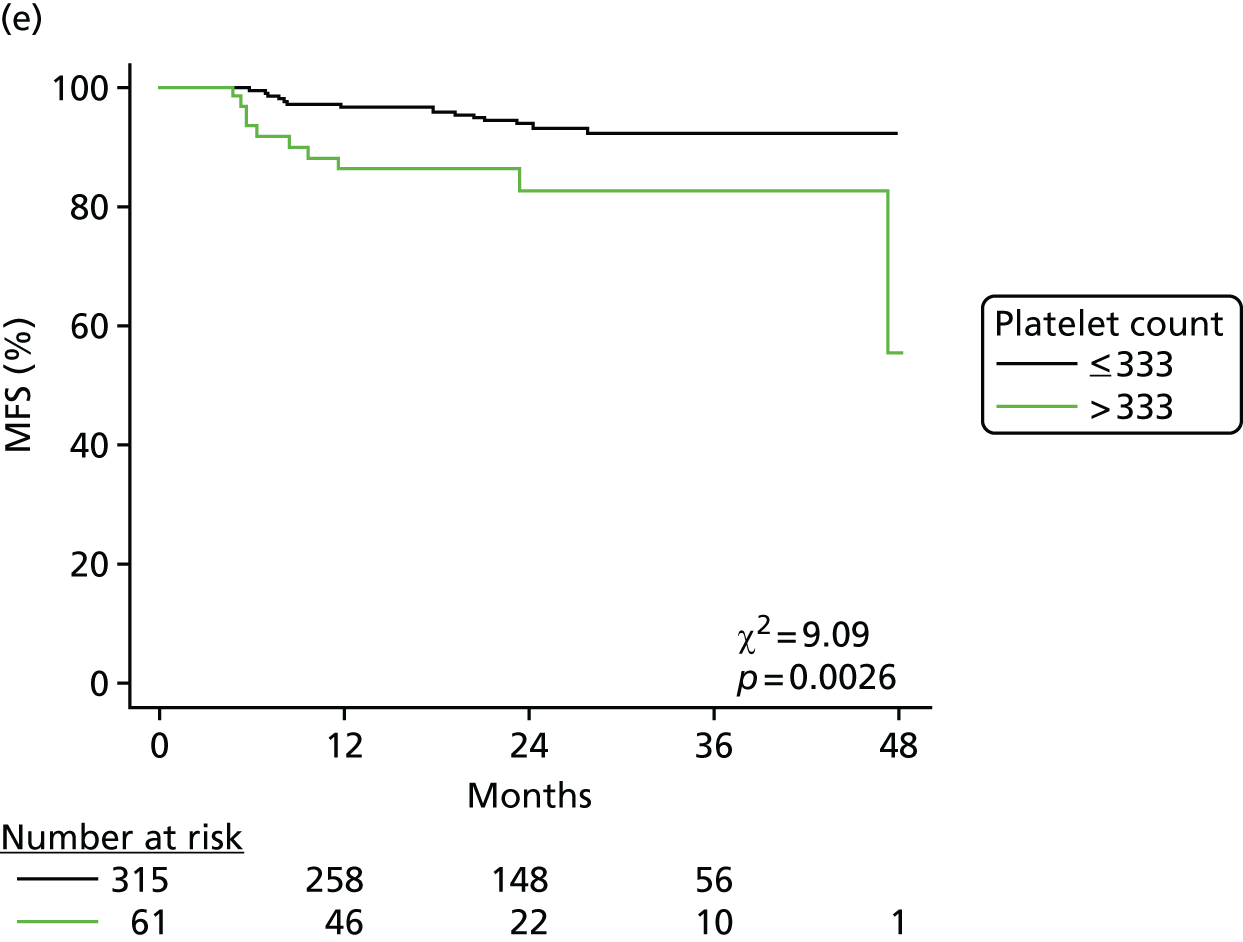
Overall survival
When considered as continuous variables, the following were statistically significantly associated with OS (p < 0.05): haemoglobin, serum sodium, serum potassium, serum albumin, CAIX, Fuhrman grade, sarcomatoid change and Leibovich score. When the markers serum sodium and platelet count were considered as dichotomised variables, the following were statistically significant: CAIX (cut-off point 112.1 pg/ml), plasma VEGF (cut-off point 132 pg/ml), platelet count (cut-off point 300 × 109/l) and serum sodium (cut-off point 137 mmol/l) (see Table 91).
Multivariable analysis of metastasis-free survival
The prognostic ability of the markers to substratify intermediate- and/or high-risk patients by the Leibovich score was examined by sequentially adding markers found to be significant at the univariate level, in addition to platelet count and serum sodium as predictor variables, into a Cox proportional hazards model, with the Leibovich score and MFS as the response variables. In the multivariable setting, for each of the markers considered, the intermediate and high Leibovich score patients had a greater risk of relapse than the low Leibovich score patients, although only the high-risk group was significantly different (Table 92). When markers were considered as continuous variables none was significant (results omitted). When dichotomised, the only biomarker significant in the multivariable models was CRP (HR 3.22, 95% CI 1.39 to 7.49; p = 0.007). Corresponding Kaplan–Meier survival curves are shown in Figure 56.
| Variable | HR (95% CI) | p-value |
|---|---|---|
| CRP (mg/l) | ||
| ≤ 14.1 | 1.00 | – |
| > 14.1 | 3.22 (1.39 to 7.49) | 0.007 |
| Leibovich risk | ||
| Low | 1.00 | – |
| Intermediate | 1.21 (0.34 to 4.29) | 0.770 |
| High | 6.44 (2.03 to 20.41) | 0.002 |
| OPN (ng/ml) | ||
| ≤ 120.8 | 1.00 | – |
| > 120.8 | 1.7 (0.73 to 3.97) | 0.223 |
| Leibovich risk | ||
| Low | 1.00 | – |
| Intermediate | 1.35 (0.32 to 5.69) | 0.680 |
| High | 11.29 (3.09 to 41.21) | < 0.001 |
| CAIX (pg/ml) | ||
| ≤ 60.4 | 1.00 | – |
| > 60.4 | 1.73 (0.74 to 4.05) | 0.205 |
| Leibovich risk | ||
| Low | 1.00 | – |
| Intermediate | 1.11 (0.31 to 4.02) | 0.873 |
| High | 9.31 (3.04 to 28.48) | < 0.001 |
| Serum sodium (mmol/l) | ||
| ≤ 141 | 1.00 | – |
| > 141 | 0.15 (0.02 to 1.14) | 0.067 |
| Leibovich risk | ||
| Low | 1.00 | – |
| Intermediate | 1.34 (0.38 to 4.75) | 0.651 |
| High | 9.17 (3.09 to 27.18) | < 0.001 |
| Platelet count (× 109/l) | ||
| ≤ 333 | 1.00 | – |
| > 333 | 1.60 (0.70 to 3.65) | 0.265 |
| Leibovich risk | ||
| Low | 1.00 | – |
| Intermediate | 1.71 (0.43 to 6.83) | 0.449 |
| High | 13.12 (3.78 to 45.52) | < 0.001 |
FIGURE 56.
Kaplan–Meier survival curves showing MFS for dichotomised markers, serum sodium and platelet count (only markers found to be significant in univariate analysis are shown) with the Leibovich score included as a further predictor variable. (a) CRP; (b) OPN; (c) CAIX; (d) serum sodium; and (e) platelet count. Optimised marker cut points were as follows: CRP = 14.1 mg/l; OPN = 120.8 ng/ml; CAIX = 60.4 pg/ml; serum sodium = 141 mmol/l; and platelet count = 333 × 109/l.


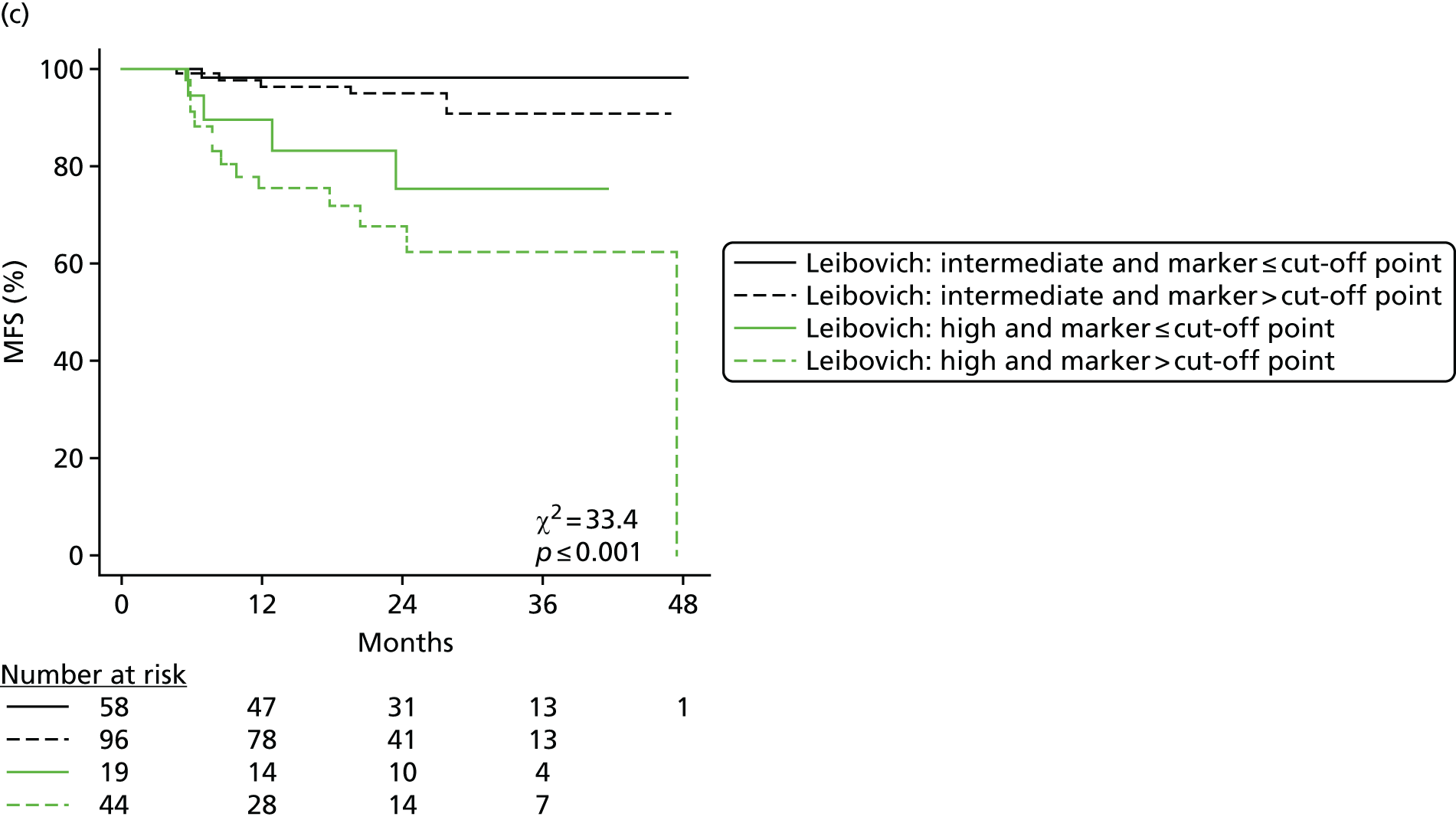

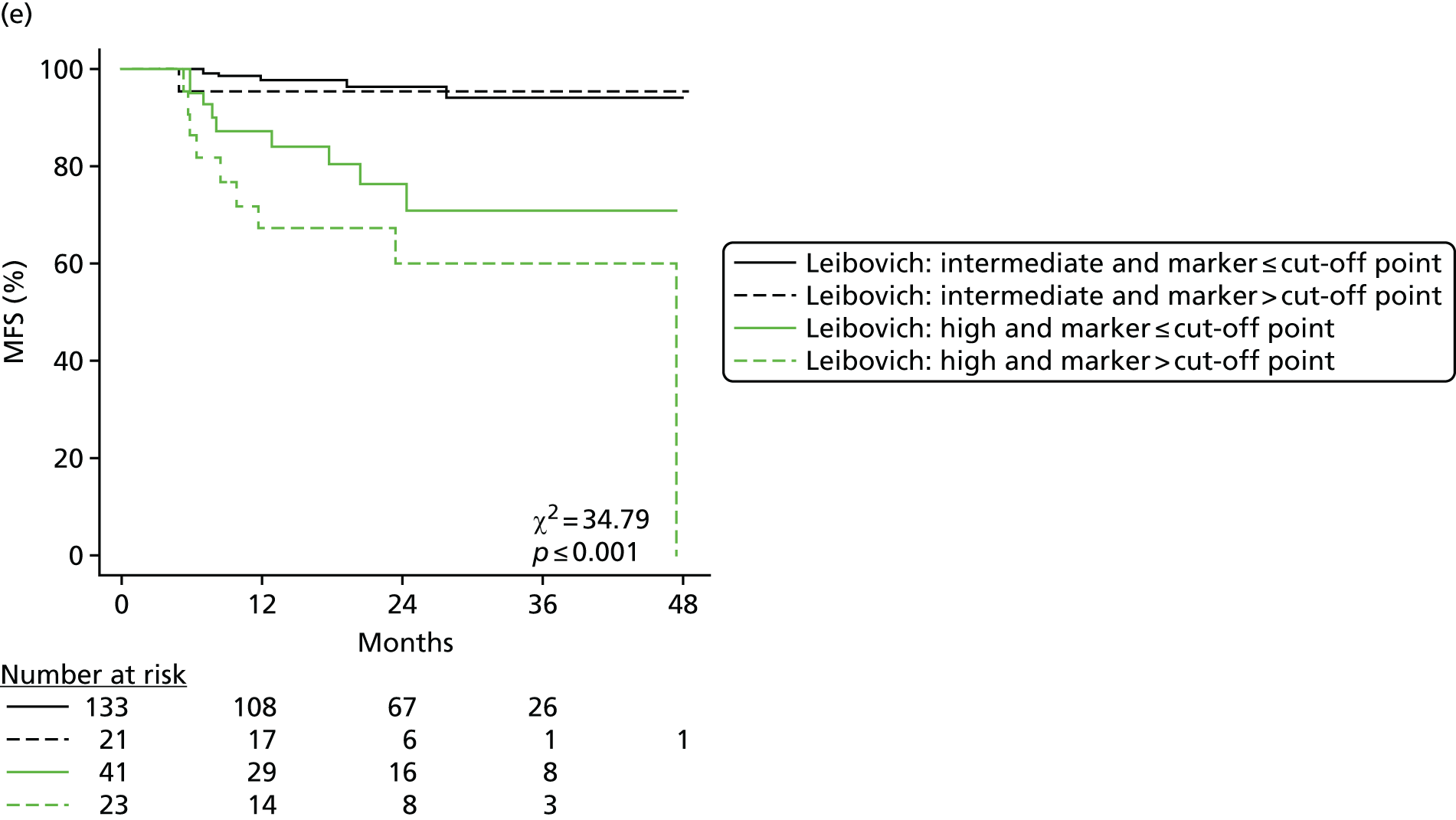
Discussion
The multicentre RCC biobank established within this programme represents a unique resource for biomarker validation in the UK. In this initial study, we have sought to validate a number of proposed circulating prognostic biomarkers within a large cohort of patients with localised ccRCC.
The characteristics of the current study population are in keeping with previous series. For example, in a Leeds cohort of 140 patients presenting with localised ccRCC between 1998 and 2005, 58%, 7% and 35% had a pT1, pT2 and pT3 tumour, respectively, compared with 58%, 12% and 40% in the current series. The prognostic nomogram proposed by Leibovich et al. 438 in 2003 was developed in a US study of 1671 patients with localised ccRCC. The tool classified patients into three risk groups, with 41%, 37% and 22% classified as having a low, intermediate and high risk for distant relapse, respectively. In the current UK cohort, equivalent figures were 39%, 43% and 18%, suggesting that this distribution has changed little over the past decade.
In the current study, almost one-third of patients were diagnosed with a small renal mass, defined as a mass of < 4 cm in maximal dimension. The incidental detection of patients with small renal masses has been rising in recent years because of the more widespread use of cross-sectional imaging. 847 Management of these small, typically low-risk tumours poses a significant challenge to clinicians, with the need to balance the risks of treatment against the chances of the tumour progressing within the lifetime of the patient. Biomarkers to allow stratification of these tumours by risk of progression remains a major unmet clinical need, which the current cohort of samples is well placed to help address in the future as the data mature. 848
Factors such as increasing tumour stage and grade, as well as the presence of necrosis and sarcomatoid change, are accepted poor prognosis factors in RCC and were associated with worse MFS in the current study. Reporting of MVI is variable and, in particular, is thought to be dependent on the meticulousness of the reviewing pathologist. 849 MVI is currently not recommended for inclusion in the TNM staging of RCC. 849 In a small study of 48 patients with T1/T2 RCCs (90% ccRCC), MVI was reported in 17% of patients and was an independent prognostic factor for DFS. 850 MVI was found in 29% of centrally reviewed cases in another study of 255 patients with pT1–pT3bN0M0 tumours (93% ccRCC) and was shown to have independent prognostic ability in terms of CSS and OS. 851 Furthermore, in a more recent study of 1754 patients with localised ccRCC, addition of MVI status was shown to improve the predictive accuracy of the Leibovich score by 1.4%. 852 Conversely, however, in a study of 2078 patients with ccRCC, although MVI (seen in 19.8%) was significantly associated with a worse CSS among localised disease patients on univariate analysis, this association was lost on multivariable testing. 853 In the current study, MVI was reported as present in 16% of cases, in keeping with previous series without central slide review, and showed a significant association with MFS (p = 0.046) on univariate analysis. The independent predictive ability of MVI was not examined in the current study because of the current small number of events limiting the power to detect differences in outcome, but will be investigated once the data have matured.
The current study confirms our previous finding from a smaller (n = 216), single-centre cohort of patients that higher circulating concentrations of OPN, CRP and CAIX are significantly associated with known poor prognostic factors such as higher stage and grade486 and, in addition, extends these findings to include associations with other features such as presence of tumour necrosis, sarcomatoid change and MVI at a univariate level. Both preoperative CRP and OPN, but not CAIX, were associated with MFS when considered as continuous variables. Conversely, only CAIX was associated with OS on univariate analysis. We previously reported an association of all three markers with DFS, CSS and OS. MFS was not examined in the previous study although each of these end points is an expected surrogate of survival. The differences here are likely to be because of the small number of events observed in the short period of follow-up.
Only CRP was found to be independently prognostic when considered in a multivariable model including the Leibovich risk classification (p = 0.007). Strikingly, the data suggest that patients in the intermediate-risk group can be substratified, with patients with a preoperative CRP of ≤ 14.1 mg/l associated with an excellent outcome, equivalent to a low-risk Leibovich group. Such patients could, therefore, be spared intensive follow-up and the necessity of considering adjuvant therapies. It must be acknowledged, however, that the relatively small number of events at the time of analysis limited the number of variables that could be included in multivariable modelling, meaning that the current analysis should be regarded as exploratory. Categorisation of continuous variables is more clinically applicable although does come at a cost, as information is lost, reducing statistical power. 854 How best to dichotomise the data is also debated, with studies variably using the median value or, as in the current study, determining an optimal cut-off point that gives the minimum p-value. Such data-driven approaches have been criticised as they can lead to overfitting and optimistic model performance. 855 Again, this is acknowledged by the authors and, in subsequent analyses, we will use additional methods for categorisation, such as our previously described simulation-based method. 856 Validation of our previously described optimal cut-off point for CRP (15 mg/l), as well as cut-off points defined by others (5 mg/l, 7.5 mg/l), will also be undertaken, although such cross-study comparisons must be performed with care. 486,511,570,572 Issues such as differences in the particular assay used and, when comparing older studies, the more recent availability of high-sensitivity CRP assays may confound results. Ultimately, whatever method and choice of cut-off point is used, these issues highlight the necessity to carefully validate initial results using adequately powered, independent data sets.
We were the first group to report preoperative serum sodium as being independently prognostic for DFS among patients with localised ccRCC both when considered as a continuous variable (n = 103) (HR 0.78, 95% CI 0.66 to 0.92; p = 0.003) and when dichotomised to above and below the median value (139 mmol/l) (n = 137) (HR 0.39, 95% CI 0.18 to 0.84; p = 0.012). 513 These findings have since been replicated in patients with metastatic RCC but, to our knowledge, have not been re-examined in patients with localised disease. 514–516 Here, we show that serum sodium considered as a continuous variable associates with MFS at the univariate level, although this does not reach significance at the p < 0.05 level (p = 0.064). As a dichotomised variable, using an optimal cut-off point, patients with a preoperative serum sodium of > 141 mmol/l had a HR for relapse of 0.09 (95% CI 0.01 to 0.66; p = 0.018), again suggesting that a higher serum sodium level is associated with a better outcome. The mechanism underlying this association remains unclear, but has even led some to suggest that correction of relative hyponatraemia may be worth exploring as a therapeutic strategy in RCC. 857
As outlined in Chapter 12 of this report, a number of studies have examined the prognostic utility of circulating VEGF concentrations in patients with RCC. 590,593,594,596,598,773 These studies have variably employed either serum or plasma, with no consistency in reported findings. The current study is unique in that we chose to examine both serum and plasma VEGF in parallel and, to our knowledge, is the largest to date to examine VEGF concentrations using either matrix among patients with localised ccRCC. Both serum and plasma VEGF concentrations were significantly elevated among patients with M1 compared with M0 disease, but were not different among stage I–III patients. However, among localised disease patients grouped by Leibovich score, preoperative VEGF concentrations were elevated among patients with high-risk compared with intermediate- or low-risk tumours, when measured in serum or plasma or when considering serum minus plasma concentrations. On univariate analysis, both serum and plasma VEGF associated with MFS when considered as a continuous variable, but not when dichotomised, although plasma VEGF was significant for OS (cut-off point 132 pg/ml). At present, therefore, it is difficult to conclude that either fluid is superior to the other in terms of clinical relevance to RCC outcomes and this requires further future analysis, which is planned.
It is recognised that both the choice and definition of time-to-event end points in clinical trials varies, making between-trial comparisons imprecise. 858 It is equally a potential issue when trying to compare biomarker studies. At the time of writing this report, in an effort to standardise reporting in RCC trials, a recent consensus view was published by the DATECAN (Definition for the Assessment of Time-to-event Endpoints in CANcer trials) renal cancer group. 858 Among patients with localised disease, MFS, DFS and locoregional-free recurrence were recommended as intermediate end points. MFS was defined as ‘death from kidney cancer or appearance of metastases, whichever comes first’. In the current study, we defined MFS as time to appearance of metastases only, not including deaths, as this was the definition of MFS on which the Leibovich score was developed. Although clearly OS and CSS represent constants, the otherwise lack of consistency among prognostic studies in choice and definition of intermediate end point is an important issue that biomarker reporting guidelines such as the REMARK guidelines367 should consider addressing.
A limitation of the current study is the relatively short median follow-up of approximately 2 years, meaning that relatively few events had occurred at the time of analysis. As most relapses occur within 18–24 months of nephrectomy, the currently reported biomarker associations may become more significant with greater length of follow-up and number of events. Future analyses are, therefore, planned and will be extended, for example by examining markers as continuous variables transformed using fractional polynomial methods and by looking at combinations of biomarkers and algorithms. The Leibovich score will be examined not just by risk group, but also by score (i.e. 0–11) and as individual elements, that is, T stage, N stage, tumour size, grade and necrosis. Furthermore, as certain elements, such as grade, are subjective and prone to interobserver variability, and the presence of necrosis is open to sampling error, the value of the selected markers to the score, excluding these elements, will be examined. 859,860
In conclusion, the multicentre RCC biobank established within this programme consists of a large cohort of patients with ccRCC with a typical distribution of clinicopathological characteristics and expected survival associations with known prognostic factors such as stage and grade. As such, it represents an excellent resource for validation studies of prioritised biomarkers. Despite the current relatively small number of survival events, this initial study has been able to demonstrate promising associations of the selected biomarkers with outcomes. Exploratory multivariable analysis suggests that, when dichotomised by optimal cut-off point, preoperative CRP may add value to the Leibovich score. The results justify further exploration in future analyses, which are planned and will be undertaken once median follow-up has been extended.
Chapter 15 Conclusions of the clinical translation workstream
The clinical translation workstream (workstream 2) was designed to evaluate approaches to streamline and speed up the central components of the biomarker pipeline. The pipeline runs from discovery to the implementation of the appropriate biomarker testing within the health-care system, generating benefits for patients and improvements in health-care quality and cost-effectiveness. The central components include consideration of analytical validity and clinical validity. Robust evaluation of these two aspects of the pipeline are essential before clinical utility and consequent benefits for patients and health-care services can be evaluated.
We identified modest amounts of literature in renal cancer and RT, with moderate numbers of candidate biomarkers, often identified only in single papers in mixed patient groups. Studies sometimes fail to distinguish the roles of candidate markers in, for example, prognosis or treatment selection. However, in both cases progress in evaluating the performance of the biomarkers and then taking them to clinical practice has been slow and few new biomarkers have been introduced in recent decades. Small study size and study heterogeneity are important factors in this. In liver disease, we were able to study the ELF test for which a substantial body of evidence for clinical utility existed. This presented us with the opportunity to take the ELF test into a formal randomised trial, the ELUCIDATE trial, described in subsequent chapters.
The investigators believed at the outset of this workstream that they would find that many of the candidate biomarkers lacked sufficient evidence for analytical and clinical validity to justify their evaluation in large prospective studies of clinical utility. They hypothesised that this would be the case because the acquisition of appropriate clinical samples, annotated with high-quality clinical data, is a slow process and studies are frequently carried out using samples of uncertain quality and inadequate numbers of samples and with insufficient attention to methodological considerations.
The acquisition of high-quality sample banks with appropriate clinical data was deemed to be one part of the solution to speed up the biomarker evaluation pipeline. The simple hypothesis was that a standing bank of samples, carefully curated and clinically annotated in adequate numbers, would provide a resource that would allow candidate biomarkers to be robustly evaluated to allow decisions to be made whether or not they should go through for full evaluation of clinical utility and their place in clinical practice.
The discipline of clinical biochemistry was strongly represented in the investigating team and these investigators contributed to the definition of the appropriate sample handling and curation requirements. The synergy between research scientists and clinical biochemists contributed to the rigour of test development and evaluation. The methodology teams from workstream 1 and workstream 3 advised on study design, cohort size and evaluation. The expertise of the CTRU, which was designing, delivering and analysing the ELUCIDATE trial in workstream 3, was used to establish a robust, prospective, high-quality clinical data annotation process.
The clinical translation workstream has delivered cohorts of patients with high-quality samples and clinical annotation. The performance of some candidate biomarkers has been evaluated in the immediate term and has provided a legacy for future studies. This resides in the sample banks and clinical data, which are a resource that will enable rapid validation of further biomarkers in these disease areas. However, we associate greater generic value with the outputs and learning points for the general aspects of the biomarker pipeline.
Several publications have already been generated from the programme, ranging from biomarker reviews through to exploration of technical issues of specific immunoassays and clearly much of the material described in the previous chapters will also result in further publications. These will include further technical pre-analytical papers and a commentary on the various aspects of the set-up process, which will be of considerable relevance when planning this type of activity going forwards, and, in addition, many biomarker studies are anticipated.
We would like to emphasise the generic learning points from the work described in Chapters 10–14. Chapters 10 and 11 describe the preparation and delivery of the multicentre sample banks in renal diseases. They demonstrate that with rigorous attention to detail it is possible in the NHS to generate high-quality sample banks and high-quality clinical annotations for biomarker evaluations. The research and innovation capacity of the NHS was harnessed across multiple centres to generate the samples, clinical data and infrastructure to rapidly evaluate candidate biomarkers in renal cancer and in patients after RT. The challenges faced were substantial and are often generalisable. Details of delays in study set-up and the challenges of quality assuring samples and clinical data are well illustrated by the data in Chapters 11 and 13. This work will serve as a useful exemplar for the strategic approach that we have advocated for biomarker evaluation. We have provided the NHS, the NIHR and the academic community and partners with access to materials that allow the prompt and robust evaluation of analytical and clinical validity. The challenges involved in multicentre studies, the characteristics of successful centres and the energy and commitment that are necessary to deliver this approach are clear.
In Chapter 12 the investigation team worked to review and prioritise the circulating biomarkers in renal cancer and RT that were identifiable from the literature. This chapter demonstrates that such candidate biomarkers exist in reasonable numbers but that the pace of innovation and new discovery, which is leading to biomarkers of robust analytical and clinical validity, is still slow. All of the investigation team were disappointed that, during the period of this study, exciting new biomarkers or biomarker panels, particularly protein biomarkers in body fluids, did not emerge. The review shows that many studies in the literature are small and inconclusive but the overview analysis clearly identified candidates that could be evaluated further. This led to the evaluation in Chapter 13 of appropriate assays with suitable analytical validity. Chapters 13 and 14 describe the delivery of tests of good analytical validity and clinical validity for the prioritised and selected biomarkers against the sample bank. Candidates for further tests of clinical utility were also demonstrated.
We cautiously conclude that this approach has merit and can provide an example of how this field can be streamlined. However, we have highlighted the considerable organisational and logistic challenges that must be overcome to effectively deliver development of the pipeline.
Critical to continued improvement in the biomarker pipeline will be the multidisciplinary nature of the approaches that must be taken. For the investigation within this programme we were fortunate to have enthusiastic inputs from research scientists, clinical biochemists, methodologists, clinicians and triallists. We believe that there is little prospect of success in individual studies or in continued improvement, streamlining and speeding up of the biomarker pipeline in the absence of consistent multidisciplinary inputs of this kind.
The organisational and logistical challenges have been highlighted. During the duration of this study we noted a steady improvement in set-up times following the hard work carried out by NIHR infrastructure organisations, including the NIHR CRN. To continue to deliver progress, partnerships between the NHS, universities and funders are essential. Each brings unique components of the expertise needed to deliver the improved biomarker pipeline that is sought by all.
Patients played a substantial part in the design, delivery and conduct of this work. Our PPI workstream and PPI commentaries were important at all stages. The engagement of patients in the methodology workstreams and of course their engagement as partners in the delivery of the cohorts and the provision of samples were essential components of the progress that has been identified in this workstream. We have continued to work very closely with UK industry and have developed strong working relationships, most notably with Randox Laboratories (Randox Laboratories Crumlin, County Antrim, Northern Ireland, UK) in a successful first-phase SBRI Healthcare bid.
The clinical translation workstream provided the basis for the application, including many of the investigators on the NIHR programme, to become a NIHR Diagnostic Evidence Co-operative (DEC), which was successful and began work in 2013. We will describe the DEC, the learning from the programme that underpinned its development and its operations and the extension of its scope beyond the programme to include our colleagues in musculoskeletal disease and in other aspects of oncology in Chapter 24.
Chapter 16 Introduction to the ELUCIDATE trial (including scientific background and explanation of rationale)
Workstream 3 aimed to conduct a RCT of an established panel of biomarkers (ELF) of potential value in CLD, to diagnose cirrhosis at an early stage when beneficial interventions to reduce dangerous complications are possible, which may lead to patient and NHS benefits. The rationale for selecting the ELF test is discussed in Chapter 1. Briefly, in the vast majority of cases, liver fibrosis is asymptomatic and cirrhosis develops insidiously with non-specific symptoms, so that opportunities for disease modification or cure are missed. Standard biochemical tests of liver function are not specific or sensitive. Liver biopsy is hazardous, inaccurate and subject to sampling error and variation in interpretation. 43–45 Imaging plays a major role in the detection and assessment of liver fibrosis. However, all imaging modalities including ultrasound, elastography, cross-sectional imaging with radiography or magnetic resonance imaging (MRI) require access to expensive technology and skilled operators. 46,47
Irrespective of the cause of CLD, progressive liver fibrosis culminates in architectural disruption of the liver by new collagen deposition termed cirrhosis. Once cirrhosis is established the most common and life-threatening complications of the cirrhotic state include portal hypertension and hepatocellular cancer. Treatment of the underlying cause of CLD may prevent or delay the onset of cirrhosis. However, once cirrhosis is established, whatever the cause, RCTs have demonstrated that a number of treatments (such as beta-blocker therapy for the treatment of varices and surgery for low-volume HCC) are effective at reducing the incidence of complications of cirrhosis. However, their effectiveness depends on cirrhosis being detected early enough to allow them to be delivered before disease is too advanced. Frequently, patients present for the first time when these life-threatening complications result in avoidable morbidity, mortality and cost. In workstream 3, we sought to identify a ‘pool’ of patients with progressive fibrosis, transitioning to cirrhosis, who could be treated early enough in the course of their disease to reduce the incidence of the serious complications of cirrhosis.
Evidence shows that early detection of varices and treatment with prophylactic use of beta-blockers to reduce portal hypertension, or band ligation, reduces morbidity and increases survival. Respected guidelines recommend surveillance for varices because of its benefits and health economic justification. 48–52 Similarly, early detection of ascites and treatment have been shown to reduce the morbidity associated with bacterial peritonitis from 17% to 2%. 53 The case for surveillance and early detection of HCC is more contentious, with some RCTs showing evidence of benefit and others showing none. International guidelines now advocate surveillance for HCC. 54–56 Retrospective analyses have identified criteria, essentially small tumours, that are associated with better outcomes for HCC resection and liver transplantation, but many patients are diagnosed after the growth of their tumours has ruled them out for curative resection or transplantation. 57,58
Blood tests for fibrosis and cirrhosis are highly attractive, having the potential to be automated, highly accurate and reproducible and repeatable at relatively shorter intervals than liver biopsy. Serum markers of liver fibrosis can be divided into those that are ‘indirect’, which measure liver biochemistry and haematological indices, and those that are ‘direct’, which measure constituents of liver matrix and enzymes involved in fibrogenesis and fibrolysis. 861,862 Indirect measures, although useful for some clinical purposes, are subject to the influence of inflammation, drug effects and other comorbidities. Direct markers of fibrosis have biological plausibility but theoretically may be affected by other fibrotic disorders; however, this has not been a major problem in clinical evaluation.
Studies and systematic reviews have demonstrated that single direct markers are less accurate than panels of markers in the detection of liver fibrosis. 59,60 One such panel of direct markers is the ELF test, the only CE-marked (EU regulatory approval) test for liver fibrosis, which measures constituents of liver matrix (HA and PIIINP) and a molecule critical to the regulation of matrix remodelling (TIMP-1), using sensitive automated ELISA assays designed and manufactured specifically for this purpose. 61 The three individual biomarkers were selected as optimal from among 11 ‘direct’ and 35 ‘indirect’ candidates. The results of the individual assays are combined in an algorithm derived and validated in > 1000 cases of liver fibrosis to generate a score that correlates with the severity of liver fibrosis on liver biopsy and subsequently fibroelastography. ELF values have been shown to be highly predictive of clinical outcomes, including variceal bleeding, ascites, HCC and mortality. Subsequent validation studies in hepatitis C, hepatitis B, fatty liver disease, HIV–HCV co-infection, primary sclerosing cholangitis and primary biliary cirrhosis have confirmed the performance of the test. 34–37 Although performance is best in the detection of advanced fibrosis and cirrhosis, the test can also detect mild and moderate degrees of fibrosis accurately, with AUC ROCs of 0.83 for Ishak fibrosis stage 0–3 compared with 4–6 and 0.86 for Ishak fibrosis stage 0–4 compared with 5–6.
The ELF test was developed by Siemens Medical Solutions (formerly Bayer Healthcare) in conjunction with the University of Southampton and iQur Ltd. We performed independent evaluations of the analytical validity of the ELF test.
Chapter 17 Verification of the analytical performance of the ADVIA Centaur Enhanced Liver Fibrosis test
The Siemens ELF test is an in vitro diagnostic assay that uses an algorithm combining quantitative measurements of serum HA, PIIINP and TIMP-1 to produce a single ELF value that reflects the degree of liver damage in patients with or at risk of cirrhosis. The ELF test is the subject of the clinical trial described in Chapters 18–22 and is available on the Siemens ADVIA Centaur automated analyser.
This chapter describes an independent verification of the ELF test’s analytical performance characteristics. It details two independent studies conducted within accredited NHS laboratories: an intra-laboratory study evaluating repeatability, intermediate imprecision and bias against control materials; and an inter-laboratory study evaluating reproducibility. In addition to verifying the manufacturer’s performance claims for precision and bias, recommendations are also made for further evaluation prior to routine clinical implementation. To the authors’ knowledge there have not been any published independent validation/verification studies of the analytical performance of the ELF test.
Introduction to analytical performance evaluation, precision and bias
In vitro diagnostic tests form the basis of ≈70% of clinical decision-making in the NHS. 863 The accuracy of, and associated uncertainty surrounding, diagnostic testing consequently has a major impact on the overall quality of clinical decisions and the subsequent effectiveness of this and other health-care systems.
Numerous pre-analytical, analytical and biological factors can contribute to uncertainty in diagnostic testing strategies (Figure 57). These uncertainties accumulate through the measurement system and may eventually affect patient outcomes. 291,864 The same factors can also introduce bias into clinical trials and contribute to lack of reproducibility in biomarker research studies. 865
FIGURE 57.
Feather diagram depicting biological, pre-analytical and analytical factors that may contribute to measurement uncertainty (UM).

Performance evaluation, in the form of method validation, is a legal requirement for both commercial and laboratory-developed diagnostic tests. Such evaluation is recognised as a critical step in mitigating risk as part of the development of new laboratory methods. 866 For commercial IVDs, such as the ELF test, it is the manufacturers’ responsibility to validate the analytical performance of the method and provide objective evidence that the tests meet the evidence requirements for their intended use, prior to seeking market approval This includes consideration of analytical sensitivity, specificity, accuracy, repeatability and reproducibility. 866
Before introducing any IVD into routine clinical practice, clinical laboratories should also, to fulfil national accreditation requirements, independently verify the analytical performance of the test. This is to confirm that the method is performing as claimed by the manufacturer in a routine setting and is necessary to safeguard patient safety as part of clinical governance.
The performance evaluations described here include verification of the ELF test’s analytical precision and bias (reflecting ‘trueness’), which together describe the accuracy of a measurement procedure, as illustrated in Figure 58. 867
FIGURE 58.
Schematic illustration of the relationships between precision, bias, trueness accuracy and uncertainty. Modified from Bailey and Barwick, LGC Ltd, 2007. 868

Precision and imprecision
Precision is defined as ‘the closeness of agreement between indications or measured quantity values obtained by replicate measurements on the same or similar objects under specified conditions’. 869 It is usually expressed in terms of SD and/or CV using the following equations:870
where σ is the SD, x¯ is the sample mean and V is the sample variance.
Precision reflects the random errors inherent in all measurement procedures and includes variability arising from M-Factors, including:
-
time – the time between measurements
-
calibration – how often the equipment is calibrated
-
operator – the number of staff carrying out the assay
-
equipment – whether or not the same equipment and batches of reagents are used.
Estimates of precision are strongly dependent on the conditions in which precision is assessed. Precision is generally evaluated with respect to repeatability, intermediate precision and reproducibility. 867,871,872 To assess repeatability, repeated measurements are made while keeping the factors above constant, so that they do not contribute to the imprecision. Reproducibility is assessed by comparing results for the same samples as measured in different laboratories, so that technical and additional factors (e.g. environmental, staff training) will also contribute to variation in results. Hence, repeatability and reproducibility represent the minimum and maximum extremes of investigation conditions for precision. It is often helpful to describe precision under conditions somewhere in between repeatability and reproducibility. Such conditions are referred to as intermediate precision conditions and are described in relation to the number of M-Factors that differ (M = 1, 2, 3 or 4). 872
Trueness and bias
Trueness is defined as ‘the closeness of agreement between the average of an infinite number of replicate measured quantity values and a reference quantity value’. 869 As trueness cannot be expressed numerically, it is usually expressed in terms of measurement bias. Measurement bias is defined as ‘an estimate of systematic measurement error’. 869 It is evaluated by comparing the difference between the mean of replicate measures made using a test method (x¯) and the assigned value of a reference material or method (x0) or the consensus mean result (x0) for a group of laboratories (e.g. those participating in an external quality assessment programme) using the following equations:
Analytical performance goals incorporating biological variation
To validate a method one must provide objective evidence that it fulfils the evidence requirements for a specific intended use and is ‘fit for purpose’. 869 However, defining these requirements remains a challenge, even after several decades of intensive efforts by members of the laboratory medicine community. 873 In 1999 a landmark conference in Stockholm agreed a hierarchical structure for setting analytical performance goals (APG). 874 A 2014 conference in Milan revised and refined these,875 suggesting three approaches based on:
-
the effect of analytical performance on clinical outcome (either directly or indirectly)
-
components of biological variation of the measurand or
-
‘state of the art’.
Analytical performance goals that incorporate biological variation can be calculated as follows:734
-
analytical imprecision (CVA) should be < 0.5 × CVI
-
analytical bias (BA) should be < 0.25 × (CVI + CVG),
where CVI is the within-individual variability, CVG is the between-individual variability and CVT is the total measurement variability [CVT = √(CVI2 + CVA2)].
Single-site evaluation of the Enhanced Liver Fibrosis test
As part of the regulatory submission for the ADVIA Centaur ELF test, Siemens undertook an evaluation of the imprecision of the ELF value. They performed a full validation of within-run (repeatability), between-run and total intra-laboratory (intermediate) imprecision according to the CLSI protocol EP05-A2 Evaluation of Precision Performance of Quantitative Measurement Methods. 876 Samples were assayed in triplicate, twice daily for 20 days (n = 120 replicates per sample). The intermediate imprecision validation was performed using one ADVIA Centaur XP and one ADVIA Centaur CP system, using one reagent lot and one calibrator lot; these data are presented in Table 93.
| ADVIA Centaur system | Mean ELF value | SD | CV | ||||
|---|---|---|---|---|---|---|---|
| Within-run | Between-run | Total intra-laboratory | Within run | Between-run | Total intra-laboratory | ||
| XP | 6.98 | 0.07 | 0.04 | 0.11 | 1.00 | 0.57 | 1.58 |
| XP | 7.12 | 0.04 | 0.03 | 0.08 | 0.56 | 0.42 | 1.12 |
| XP | 8.95 | 0.03 | 0.04 | 0.09 | 0.34 | 0.45 | 1.01 |
| XP | 11.05 | 0.03 | 0.04 | 0.08 | 0.27 | 0.36 | 0.72 |
| XP | 14.51 | 0.04 | 0.03 | 0.08 | 0.28 | 0.21 | 0.55 |
| CP | 7.09 | 0.04 | 0.07 | 0.11 | 0.56 | 0.99 | 1.55 |
| CP | 7.33 | 0.06 | 0.03 | 0.08 | 0.82 | 0.41 | 1.09 |
| CP | 9.08 | 0.05 | 0.04 | 0.08 | 0.55 | 0.44 | 0.88 |
| CP | 11.15 | 0.05 | 0.04 | 0.08 | 0.45 | 0.36 | 0.72 |
| CP | 14.59 | 0.06 | 0.04 | 0.09 | 0.41 | 0.27 | 0.62 |
Study aims
The primary aim of this study was to verify the claimed repeatability and intermediate imprecision of the ELF values (see Table 93) within a routine NHS clinical laboratory environment. Secondary objectives included verification of ELF value bias using control materials with assigned values and assessment of the bias, repeatability and intermediate imprecision of the individual assay components (HA, PIIINP and TIMP-1).
Study design
The imprecision and bias studies were designed according to the ACB guideline Measurement Verification in the Clinical Laboratory,877 which is based on the CLSI guideline EP15-A2 (User Verification of Performance and Trueness). 878 Five replicates were measured over 5 days using three clinically relevant concentrations (high, medium and low) of both patient pooled samples and control materials.
Study methods
Sample collection
Serum samples were collected (REC approval 10/H1306/88), processed and stored at –80°C according to our internal SOPs (SOP03S Serum Plasma Urine Processing v1.01004). The samples were then transferred on dry ice to Leeds Pathology Research and Development Department, Leeds General Infirmary, and stored at –80°C until required.
Sample preparation
Approximately 10 anonymised serum samples (Leeds General Infirmary, REC approval 10/H1306/88) were selected from patients with systemic sclerosis and combined to yield serum pools at three concentrations spanning the ELF test assay range (low, medium and high). Samples were selected to exclude any with physical or biochemical evidence of haemolysis, lipaemia and icterus, which could interfere with the assay. Each pool was mixed in a universal tube for approximately 5 minutes on a roller mixer at room temperature. Pools were subsequently subaliquoted in 250-µl volumes in cryotubes and immediately frozen at –80°C.
Measurement of pooled serum samples and reference controls
Study sample analysis was conducted during May and June 2015 in the Leeds Teaching Hospitals Blood Sciences Laboratory on one ADVIA Centaur XP system (serial number 8680). The analytical conditions (e.g. reagent and calibrator lot) remained unchanged between series. The same member of laboratory staff prepared and processed the samples. This reflects the analytical conditions of the intermediate imprecision studies reported by Siemens (see Table 93) (i.e. one reagent lot and one calibrator lot), in accordance with CLSI guideline EP05-A2. 876
Frozen serum samples were defrosted at room temperature, vortexed and centrifuged prior to analysis, as per local quality assurance procedures within the blood sciences laboratory. Three levels of each type of pooled serum sample and reference control material (Siemens) were analysed for each component assay and manually combined to produce the ELF value using the Centaur XP ELF algorithm:
Data analysis and verification
The ELF value and component analyte measurements were determined according to the ACB method verification protocol. 877 Imprecision data analysis was obtained using nested ANOVA to determine repeatability and intermediate imprecision using the ACB Spreadsheet A,879 which is based on the CLSI guideline EP15-A2. 878 Spreadsheet A was cross-validated with Analyse-it software (Method Validation Edition; Leeds, UK) and produced comparable results. A false-rejection rate of 5% was used.
The ELF value and component analyte imprecision was compared with published performance claims. Test performance was also reviewed in line with FDA bioanalytical guidelines880 and the tumour marker quality requirements guidelines of the National Academy of Clinical Biochemistry (NACB). 881 These specify intermediate imprecision performance goals of ≤ 15% CV for immunoassays and ≤ 5% CV for high-precision instruments, respectively, and goals for bias of ≤ 15% of the nominal value, except at the LLoQ, where it should not deviate by > 20%. 880,881 APGs for imprecision and bias were determined using estimates of total measurement variability (CVT) from the ELUCIDATE trial (see Table 16).
There is as yet no certified reference material or reference method for the ELF test. Analysis of bias was, therefore, verified by comparing the difference between the means (x¯) of five replicates of three manufacturers’ reference QC materials (Lot: Low-2418261, Mid-2418262, High-2418263), measured over 5 days, with their respective assigned values (x0). Data were analysed according to the ACB method verification protocol. 877 However, as Spreadsheet C882 enables inclusion of only two replicates per day, statistical analysis was performed as above using the Analyse-it software in accordance with the methods specified in CLSI guideline EP15-A2. 878 Tests for equality and equivalence were both performed. Equality tests assess whether or not the methods are producing identical results (average bias = 0), whereas equivalence tests assess whether or not the bias is within an allowable goal specified by the manufacturer. A false-rejection rate of 5% was used.
Results
Assessment of imprecision
The results shown in Tables 94 and 95 confirm that the manufacturer’s claims for ELF test imprecision are verifiable for both repeatability and intermediate imprecision when using control materials, but not for repeatability in two of three pooled serum samples.
| Reference materials | Mean (ng/ml) for HA, TIMP-1 and PIIINP | Repeatability (CV%) | Intermediate imprecision (CV%) | Claimed repeatability (CV%) | Claimed intermediate imprecision (CV%) | Verification claim (5% significance level) |
|---|---|---|---|---|---|---|
| ELF (Low) | 7.2 | 0.7 | 0.9 | 0.6 | 1.1 | Within claims |
| ELF (Mid) | 9.1 | 0.3 | 0.7 | 0.3 | 1.0 | Within claims |
| ELF (High) | 11.1 | 0.3 | 0.7 | 0.3 | 0.7 | Within claims |
| Reference materials | Mean (ng/ml) for HA, TIMP-1 and PIIINP | Repeatability (CV%) | Intermediate imprecision (CV%) | Claimed repeatability (CV%) | Claimed intermediate imprecision (CV%) | Verification claim (5% significance level) |
|---|---|---|---|---|---|---|
| ELF (Low) | 7.6 | 0.5 | 1.2 | 0.7 | 1.1 | Within claims |
| ELF (Mid) | 9.2 | 0.6 | 1.4 | 0.3 | 1.0 | Exceeds repeatability claim |
| ELF (High) | 11.1 | 0.5 | 0.9 | 0.3 | 0.7 | Exceeds repeatability claim |
The Levey–Jennings plots in Figure 59 demonstrate a low level of ELF test imprecision over time against an ELF value range spanning the clinical decision thresholds used within the ELUCIDATE trial (≥ 8.4 for randomisation and ≥ 9.5 for management of cirrhosis). The full range of ELF values observed in the ELUCIDATE trial was 6.41–17.84 (mean 9.304, median 9.11).
FIGURE 59.
Levey–Jennings charts of ELF values across 5 days. (a) Low QC; (b) mid QC; (c) high QC; (d) low serum pool; (e) mid serum pool; and (f) high serum pool. Dots represent within-run means; the solid line represents the between-run mean; and the dotted lines represent ±1 SD.




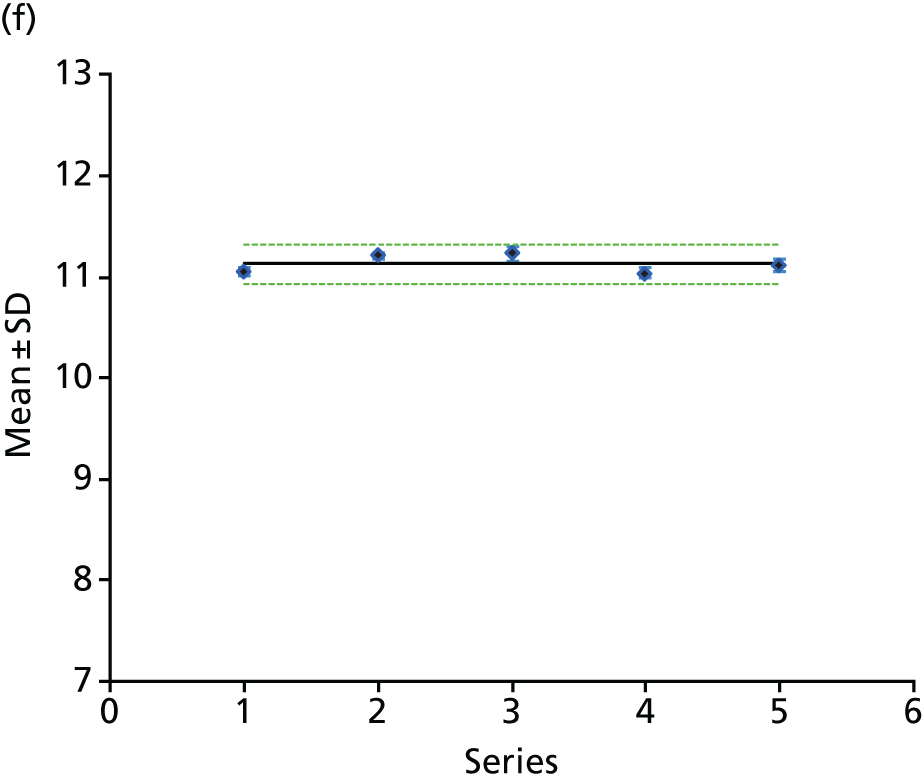
Tables 96 and 97 show the repeatability and intermediate imprecision of the ELF component analytes HA, PIIINP and TIMP-1 for control materials and pooled serum samples, respectively. The imprecision of the component analytes was worse than that of the ELF value, with all three components exceeding the NACB ≤ 5% CV criteria for at least one concentration of control materials and at least two concentrations of pooled serum samples. Furthermore, TIMP-1 exceeded the manufacturer’s intermediate imprecision claims in two of three control materials and three of three pooled serum samples, whereas PIINP exceeded both claims at the mid level and HA exceeded the repeatability claim at the high level in pooled serum samples.
| Variable | Lot | Mean concentration (ng/ml) for HA, TIMP-1 and PIIINP | Repeatability (CV%) | Intermediate imprecision (CV%) | Claimed repeatability (CV%) | Claimed intermediate imprecision (CV%) | Verification claim (5% significance level) |
|---|---|---|---|---|---|---|---|
| HA | |||||||
| Low | 2418261 | 19.7 | 3.3 | 4.5 | 4.5 | 7.5 | Within claims |
| Mid | 2418262 | 48.7 | 2.4 | 5.7 | 3.6 | 7.7 | Within claims |
| High | 2418263 | 201.7 | 2.6 | 4.8 | 3.9 | 6.6 | Within claims |
| TIMP-1 | |||||||
| Low | 2418261 | 89.3 | 2.8 | 9.4 | 2.5 | 5.1 | Exceeds intermediate claim |
| Mid | 2418262 | 272.3 | 1.8 | 7.3 | 1.9 | 6.0 | Within claims |
| High | 2418263 | 500.0 | 1.8 | 8.9 | 1.8 | 5.2 | Exceeds intermediate claim |
| PIIINP | |||||||
| Low | 2418261 | 2.3 | 3.8 | 4.7 | 5.0 | 6.6 | Within claims |
| Mid | 2418262 | 5.7 | 1.1 | 4.5 | 3.3 | 6.8 | Within claims |
| High | 2418263 | 12.5 | 2.3 | 6.0 | 2.2 | 4.4 | Within claims |
| Marker | Lot | Mean concentration (ng/ml) for HA, TIMP-1 and PIIINP | Repeatability (CV%) | Intermediate imprecision (CV%) | Claimed repeatability (CV%) | Claimed intermediate imprecision (CV%) | Verification claim (5% significance level) |
|---|---|---|---|---|---|---|---|
| HA | |||||||
| Low | 2418261 | 11.4 | 3.1 | 4.3 | 5.2 | 5.9 | Within claims |
| Mid | 2418262 | 41.4 | 3.3 | 5.5 | 3.6 | 7.7 | Within claims |
| High | 2418263 | 174.3 | 5.2 | 7.4 | 3.9 | 6.6 | Exceeds repeatability claim |
| TIMP-1 | |||||||
| Low | 2418261 | 141.3 | 4.6 | 7.4 | 1.8 | 3.3 | Exceeds both claims |
| Mid | 2418262 | 245.8 | 2.1 | 9.6 | 1.9 | 6.0 | Exceeds intermediate claim |
| High | 2418263 | 342.1 | 5.3 | 9.7 | 1.6 | 3.1 | Exceeds both claims |
| PIIINP | |||||||
| Low | 2418261 | 5.4 | 2.9 | 6.9 | 3.3 | 6.8 | Within claims |
| Mid | 2418262 | 7.8 | 4.7 | 7.8 | 1.9 | 2.9 | Exceeds both claims |
| High | 2418263 | 17.9 | 2.8 | 5.2 | 2.2 | 4.4 | Within claims |
Assessment of bias
The results of the assessment of bias are shown in Table 98 and Figure 60. These suggest that, although the ELF value bias was not equal to zero (p < 0.001), it was within the acceptable bias goals assigned by the manufacturer (p < 0.001). Similarly, the measured bias of two out of three HA, two out of three PIIINP and three out of three TIMP-1 reference QC materials was not equal to zero (p < 0.001), but all were within the acceptable bias goal (p < 0.001). However, it is interesting to note that, although it was within the manufacturer’s acceptable range, the percentage bias of TIMP-1 ranged from –21.19% to –20.26%, exceeding the FDA performance goal for a bias of < 20%.
| Reference material | Lot | Target (ng/ml) for HA, TIMP-1 and PIIINP | Low | High | Mean (ng/ml) for HA, TIMP-1 and PIIINP | Bias (ng/ml) for HA, TIMP-1 and PIIINP | Bias (%) | Equality test (5% significance level) | Equivalence test (5% significance level) |
|---|---|---|---|---|---|---|---|---|---|
| HA | |||||||||
| Low | 2418261 | 20.1 | 15.07 | 25.13 | 19.71 | –0.39 | –1.94 | Not equal to zero | Within goal |
| Mid | 2418262 | 50.8 | 38.1 | 63.5 | 48.72 | –2.08 | –4.10 | Not equal to zero | Within goal |
| High | 2418263 | 200 | 150 | 250 | 201.67 | 1.67 | 0.84 | Equal to zero | Within goal |
| PIIINP | |||||||||
| Low | 2418261 | 2.45 | 1.837 | 3.063 | 2.30 | –0.15 | –6.06 | Not equal to zero | Within goal |
| Mid | 2418262 | 5.96 | 4.47 | 7.45 | 5.68 | –0.28 | –4.68 | Not equal to zero | Within goal |
| High | 2418263 | 12.4 | 9.3 | 15.5 | 12.48 | 0.08 | 0.68 | Equal to zero | Within goal |
| TIMP-1 | |||||||||
| Low | 2418261 | 95.6 | 71.7 | 119.5 | 75.60 | –20.00 | –20.92 | Not equal to zero | Within goal |
| Mid | 2418262 | 296 | 222 | 370 | 236.02 | –59.98 | –20.26 | Not equal to zero | Within goal |
| High | 2418263 | 531 | 398 | 564 | 418.48 | –112.52 | –21.19 | Not equal to zero | Within goal |
| ELF | |||||||||
| Low | 2418261 | 7.3 | 6.73 | 7.75 | 7.21 | –0.09 | –1.26 | Not equal to zero | Within goal |
| Mid | 2418262 | 9.2 | 8.63 | 9.65 | 9.10 | –0.10 | –1.11 | Not equal to zero | Within goal |
| High | 2418263 | 11.2 | 10.6 | 11.6 | 11.13 | –0.07 | –0.61 | Not equal to zero | Within goal |
FIGURE 60.
Difference plot showing the bias of three manufacturers’ reference QC materials for (a) ELF; (b) HA; (c) PIIINP; and (d) TIMP-1.

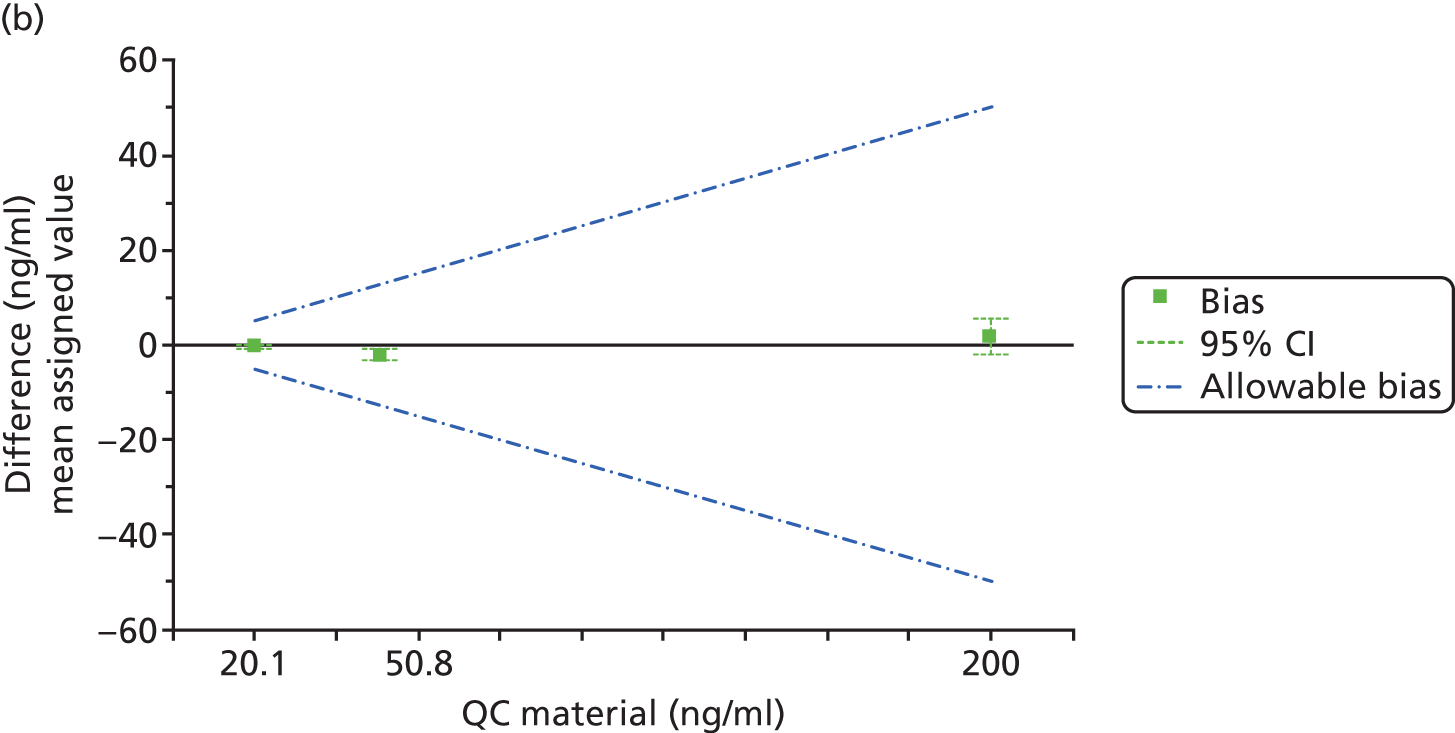

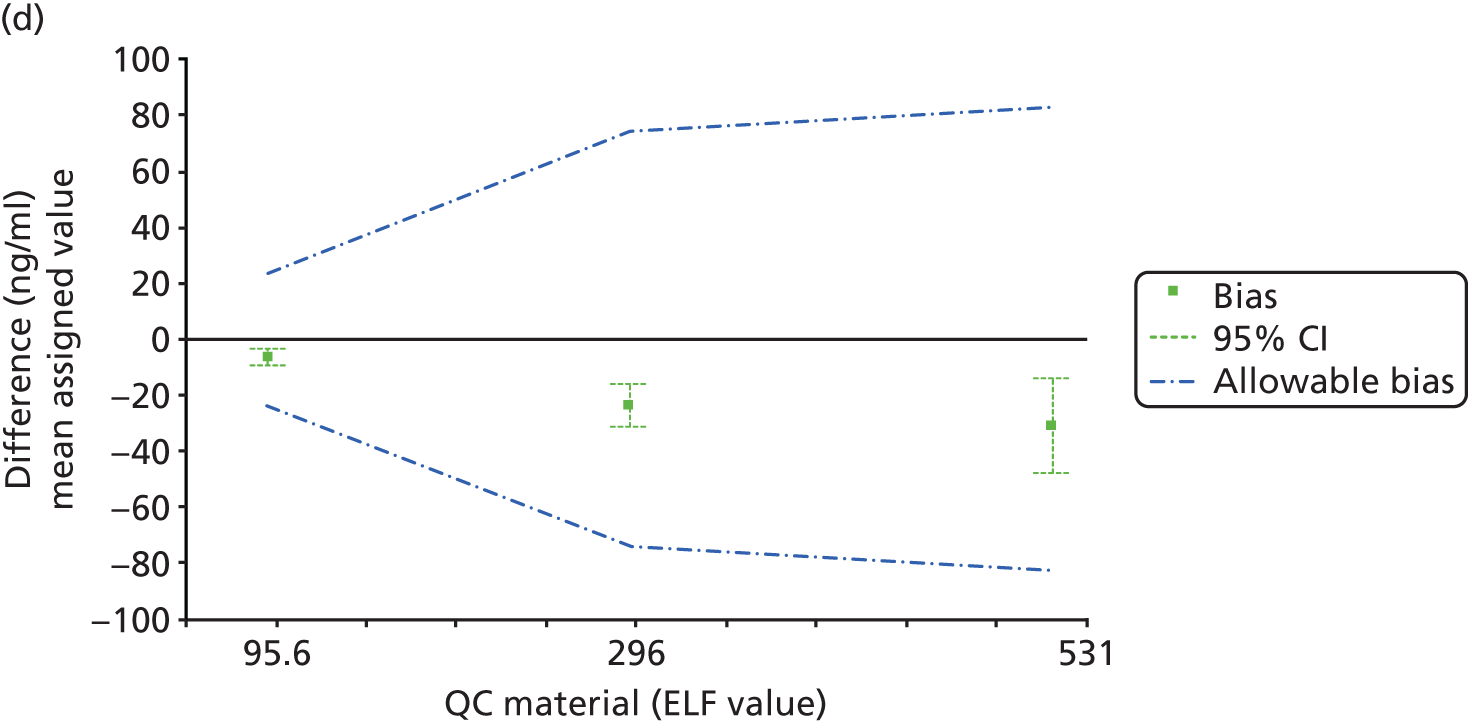
Determination of analytical performance goals
The APGs based on components of biological variation of the ELF value were determined to be as follows:
-
APG for CVA = 0.5 × 4.85 = 2.4%
-
APG for BA = 0.25 × (4.85 + 10) = 3.7%,
[CVI = √(0.47/9.3 × 100)2 – 1.42) = 4.85% (where σ total measurement variability is 0.47, CVA is < 1.4% and mean ELF value is 9.3); CVG = 0.93/9.3 × 100 = 10% (where σ between-individual variability is 0.93 and mean ELF value is 9.3].
Discussion
This study has verified the manufacturer’s intermediate imprecision and bias performance claims for the ELF test, in an accredited NHS laboratory. The results suggest that the ELF test is a precise and true assay with intermediate imprecision of < 1.4% and bias of < –1.26%. As the only assay factor that varied was time, the intermediate imprecision presented here is likely to underestimate the total intra-laboratory intermediate imprecision (M-Factor = 4).
In contrast, the manufacturer’s repeatability claims for the ELF test were not verified in two of three pooled serum samples. This may reflect specific characteristics of the pooled clinical samples as the manufacturer’s claimed repeatability at these concentrations is highly precise and the observed repeatability of < 1% is well below the specified requirements of the FDA and NACB of ≤ 15% and ≤ 5% CV, respectively. The ELF test imprecision was also within the APG for imprecision of 2.4% based on components of biological variation.
The imprecision of the individual components was less good and failed to meet all of the manufacturer’s claims and NACB criteria. However, all measurements had coefficients of variation of < 10% of the mean value and met the FDA criteria, so analytical performance is within ranges generally considered to be acceptable for clinical application.
This study has also verified the ELF test’s claims for bias using reference QC materials. The observed ELF test percentage bias was low across all three QC samples, with < –1.26% difference from the assigned value, well below the FDA goal for bias of < 20% and the APG for bias of 3.7% based on components of biological variation.
However, the comparable performance of the component analytes was less reassuring. TIMP-1 had the highest percentage bias of up to –21.19%, which, although within the manufacturer’s acceptable range, exceeded the FDA performance goal of < 20%.
It is interesting to note the comparative precision and bias of the ELF test for HA, PIIINP and TIMP-1. As might be expected, the logarithmic transformation of the biomarker concentrations within the ELF test also transforms the variance, greatly reducing the percentage CVs. Although this appears to be the main reason for the perceived improvements in precision, benefit may also be derived from the ‘averaging’ effect of the triple biomarker panel and the preferential weighting for HA, the most precise component. It is not immediately clear from the instructions for use whether clinical laboratories should undertake QC only for the ELF values or for the ELF values and each of the component analytes. Results of the verification reported here suggest that one or more individual component analytes might fail QC, even though the ELF values remained within acceptable limits. This might lead to rejection of an unnecessarily high number of tests. In view of the increasing number of complex decision algorithms that are being applied in laboratory medicine, this is an important issue that requires further research and guidance.
A limitation of this study was the use of pooled patient samples. Although this is not uncommon in precision studies, as the volume of available sample material is limited, the pooling of patient samples may dilute out any interfering substances and could also introduce interactions that would not naturally occur within an individual sample. The lack of available certified reference materials or reference methods was also a limitation of this study. Bias was, therefore, assessed using manufacturer’s reference QC materials in the intra-laboratory study, as recommended by Khatami et al. 877
A further potential limitation was that components of biological variation (e.g. CVI and CVG) were derived from the same study and not from a prospectively designed and powered biological variation study. 290,883 However, the data presented should provide a realistic estimation of the total measurement variability and are more likely to over- than underestimate variability.
Multisite evaluation of the Enhanced Liver Fibrosis test
When introducing new assays such as the ELF test to multiple sites within a health-care system, it is essential to demonstrate that good between-laboratory agreement can be achieved across multiple laboratories. Such inter-laboratory method performance studies can helpfully contribute to the validation of analytical methods as they incorporate assessment of the additional variance encountered when comparing results between laboratories. 884 Once a test has been accepted into clinical practice, continued performance surveillance is usually the responsibility of external quality assessment providers. 885
Inter-laboratory and external quality assessment studies usually involve distributing aliquoted samples of the same material (usually pooled human serum, plasma or urine) to laboratories participating in the study. Participants run the required test or tests in each of the specimens and return the results to the co-ordinating centre. Analysis of the submitted results enables calculation of between-laboratory agreement (and sometimes within-laboratory agreement). For heterogeneous analytes such as the ELF test components, the target values are usually consensus means.
Study aim
With the aim of assessing the feasibility of introducing the ELF test into NHS diagnostic laboratories, an inter-laboratory study was carried out to determine between-laboratory agreement (reproducibility) of all components of the ELF test.
Study design
Eight NHS diagnostic laboratories were invited to participate in the inter-laboratory study, for which availability of a Siemens ADVIA Centaur system was required. Specimens were sent to each participating laboratory together with 10 serum samples.
Test materials and distribution
Suitable anonymised left-over clinical samples from patients with liver disease were identified by staff in the Blood Sciences Laboratory at Leeds Teaching Hospitals. Ten 5.0-ml pools were prepared by combining two to three of the clinical samples and 0.5 ml of each pool was transferred to each of 10 prelabelled tubes (specimens E001–E010). Sets of 10 specimens were packaged according to UK NEQAS (Edinburgh) procedures and sent, together with a personalised results sheet (see Appendix 2), to each of the participating laboratories. Specimens were sent at ambient temperature to mimic routine clinical practice. Participants were requested to assay the specimens following the procedure recommended by Siemens for the ELF test and to return their results sheets and assay printouts by e-mail or fax to the UK NEQAS (Edinburgh) unit. They were asked to submit results for each individual component of the test as well as the final ELF value.
Laboratory procedures
The ELF test was set up on the individual analysers at the different sites with assistance from staff from Siemens, as is usual in clinical laboratories. Siemens also kindly supplied all reagents and controls required. The specimens were analysed in singlicate twice, in two separate runs. The manufacturer’s instructions were followed in all cases and three kit controls were included in each run.
Results
Results obtained from the individual sites are shown in full in Appendix 2 and summarised in Table 99. Between-run agreement for all analytes and all specimens was generally very good, with CVs for each pair of runs < 10% for all sites, except for site 6, where a hardware issue was identified with the analyser, which resulted in imprecise results for HA (see Appendix 2). Between-method agreement for the final ELF value was excellent, with between-method CVs for the 10 specimens as measured at eight sites ranging from 0.4% to 1.2% (see Table 99).
| Site | ELF value | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E001 | E002 | E003 | E004 | E005 | E006 | E007 | E008 | E009 | E010 | QC identifier | |||
| Low | Mid | High | |||||||||||
| 1 | 10.3 | 9.5 | 9.8 | 9.3 | 9.5 | 9.7 | 9.5 | 10.8 | 10.3 | 10.0 | 7.6 | 9.1 | 11.0 |
| 2 | 10.4 | 9.5 | 9.8 | 9.4 | 9.6 | 9.8 | 9.6 | 10.8 | 10.3 | 10.2 | 7.6 | 9.2 | 11.1 |
| 3 | 10.4 | 9.6 | 9.9 | 9.4 | 9.6 | 9.9 | 9.6 | 10.8 | 10.4 | 10.3 | 7.7 | 9.2 | 11.1 |
| 4 | 10.4 | 9.5 | 9.8 | 9.3 | 9.5 | 9.8 | 9.5 | 10.7 | 10.3 | 10.2 | 7.6 | 9.2 | 11.0 |
| 5 | 10.4 | 9.5 | 9.8 | 9.4 | 9.5 | 9.8 | 9.5 | 10.8 | 10.3 | 10.2 | 7.6 | 9.1 | 11.0 |
| 6 | 10.6 | 9.5 | 10.0 | 9.4 | 9.6 | 10.0 | 9.6 | 11.1 | 10.3 | 10.4 | 7.7 | 9.3 | 11.1 |
| 7 | 10.4 | 9.5 | 9.8 | 9.4 | 9.6 | 9.8 | 9.5 | 10.8 | 10.3 | 10.2 | 7.6 | 9.1 | 11.0 |
| 8 | 10.3 | 9.5 | 9.8 | 9.3 | 9.5 | 9.8 | 9.5 | 10.8 | 10.3 | 10.2 | 7.6 | 9.1 | 11.0 |
| Mean | 10.4 | 9.5 | 9.8 | 9.4 | 9.6 | 9.8 | 9.5 | 10.8 | 10.3 | 10.2 | 7.6 | 9.2 | 11.0 |
| SD | 0.10 | 0.04 | 0.08 | 0.05 | 0.05 | 0.09 | 0.05 | 0.13 | 0.04 | 0.12 | 0.05 | 0.08 | 0.05 |
| CV% | 0.9 | 0.4 | 0.8 | 0.6 | 0.6 | 0.9 | 0.6 | 1.2 | 0.4 | 1.2 | 0.7 | 0.8 | 0.5 |
Discussion
The multisite study described here considered primarily the verification of analytical imprecision. The results confirm that the ELF test is reproducible. The between-laboratory imprecision of < 1.2% observed across eight centres is well within the APG for imprecision of 2.4% based on components of biological variation and is also well within the specified requirements of the FDA and NACB of ≤ 15% and ≤ 5% CV, respectively. However, prior to its implementation into routine clinical practice, further work would be desirable to confirm the analytical specificity (including interference and cross-reactivity), analytical sensitivity, limits of detection/quantitation and measuring range of the test. Further characterisation of pre-analytical and biological factors, including within-individual variation, time from sample collection to analysis or stabilisation, type of collection tube, storage temperature, duration of storage, aliquot volume, number of freeze–thaw cycles and specific systematic factors (e.g. medications, fasting, alcohol and smoking), should also be conducted prior to implementation. These further studies are planned as part of an ongoing NIHR Career Development Fellowship (Dr Del Galdo), which will include assessment of the clinical utility of HA, PIIINP and TIMP-1 in patients with systemic sclerosis.
Study conclusions
The work presented here demonstrates that the ELF test performs as specified by the manufacturer and suggests that its transfer to routine use in NHS laboratories is feasible.
Provided the manufacturer’s instructions are followed and suitable QC procedures are implemented, the analytical performance of the ELF test should be appropriate for clinical use.
Further high-quality studies of pre-analytical and biological requirements should be conducted to determine the total ELF test measurement uncertainty.
Chapter 18 Design and set-up of the ELUCIDATE trial
Study design
The ELUCIDATE trial was a multicentre individually RCT that aimed to determine whether or not the use of the ELF test in addition to standard clinical monitoring significantly alters the diagnostic timing and subsequent management of cirrhosis of the liver, compared with standard clinical monitoring alone, in order to reduce the incidence and consequences of serious complications and improve outcomes for patients and service provision. In the trial the ELF test was used to test for liver cirrhosis in patients with CLD and pre-cirrhotic moderate to severe fibrosis (as classified by clinical, laboratory or histological evidence) as a result of viral liver disease, non-alcoholic fatty liver disease, alcoholic liver disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, haemochromatosis or combinations of these diseases.
Enhanced Liver Fibrosis test values have been evaluated in previous studies to relate biopsy evidence to ELF results (Table 100).
| Fibrosis stage (Ishak886) | ELF range |
|---|---|
| Normal/mild (F0–F2) | < 8.37 |
| Moderate (F3) | 8.37–8.73 |
| Moderate/severe (F4) | 8.74–9.12 |
| Severe (F5) | 9.13–9.49 |
| Cirrhosis (F6) | ≥ 9.5 |
It can be seen that ELF values of < 8.4 are not associated with fibrosis on biopsy and they carry only a very small risk of serious liver complications (see Figure 64). Scores of ≥ 9.5, however, are associated with cirrhosis on biopsy and a higher risk of serious complications (see Figure 64).
This trial aimed to answer the following questions:
-
Does the use of serum markers of liver fibrosis permit earlier detection of liver cirrhosis in patients with CLD to allow earlier interventions?
-
Does the use of serum markers of liver fibrosis affect the process of care, through (1) increased use of beta-blockers/band ligation of varices to prevent haemorrhage; (2) increased use of endoscopy and ultrasound/AFP assays to detect HCC at a surgically curable stage; and (3) effective early treatment to normalise liver function tests (LFTs) in patients with hepatitis B and hepatitis C?
-
Does the use of serum markers of liver fibrosis result in patient benefit through improved survival and reduced liver-related morbidity and mortality?
-
Does the use of serum markers of liver fibrosis improve the cost-effectiveness of the management of end-stage liver disease?
We also carried out a qualitative exit study to investigate patient understandings of clinical biomarkers; patient experiences and the acceptability and perceived utility of testing; and motivations for testing.
The design of the trial is summarised in Figure 61.
FIGURE 61.
The ELUCIDATE trial flow chart showing recruitment, randomisation and follow-up procedures.

The ELUCIDATE final trial protocol is provided as supplementary material (see Report Supplementary Material 1).
Ethics approval and research governance
Ethics approval for the trial was given by Leeds Central Research Ethics Committee, later known as Yorkshire and Humber – Bradford Leeds Committee (main REC) on 2 February 2010 (reference number 10/H1313/2). Participating sites were required to have obtained local management approvals and undertaken a site initiation meeting with the central co-ordinating clinical trials unit (CTRU, University of Leeds) prior to the start of recruitment into the trial. The trial was registered with the International Standard Randomised Controlled Trial (ISRCTN) register (ISRCTN74815110).
A summary of the changes made to the original protocol is given in Appendix 3.
Participants
The trial recruited patients with CLD from liver clinics in secondary care through the NIHR CRN Comprehensive Clinical Research Networks.
Inclusion criteria for registration
Patients were considered eligible for registration if they met all of the following criteria:
-
aged ≥ 18 years and < 75 years
-
had CLD due to any aetiology, including viral hepatitis C or B, non-alcoholic liver disease, alcoholic liver disease, primary biliary cirrhosis, primary sclerosing cholangitis, autoimmune hepatitis, haemochromatosis or combinations of these diseases, with no diagnosis of cirrhosis
-
had a life expectancy of > 6 months
-
were likely to comply with the follow-up schedule
-
were able to provide written informed consent.
Exclusion criteria for registration
Patients with any of the following criteria were not eligible for registration into the trial:
-
imaging, histological or laboratory (other than ELF) diagnosis of cirrhosis/portal hypertension as evidenced by any one of the following –
-
imaging evidence of portal hypertension (splenomegally, varices or ascites)
-
liver biopsy diagnostic of cirrhosis (Ishak fibrosis stage F6 or equivalent)
-
thrombocytopenia (platelet count of < 100 × 109/l)
-
hypoalbuminaemia (albumin level less than the lower limit of normal)
-
-
acute liver injury or acute liver failure (hepatic dysfunction of < 6 months in duration)
-
an ongoing or previous episode of hepatic decompensation (acute or chronic liver failure) including encephalopathy, variceal bleeding, ascites, jaundice or liver synthetic dysfunction
-
an established diagnosis of HCC or elevated AFP without investigation to exclude HCC
-
being treated with heparin (ELF test cannot be performed)
-
previously screened and found ineligible for the ELUCIDATE trial.
Screening and consent procedure
Nurses reviewed their caseload for potentially eligible participants. Subjects fulfilling the eligibility criteria were invited to participate in the study. Whenever possible, eligible patients were sent a patient information summary to consider prior to their next clinic appointment.
At their next clinic visit, patients were provided with the full patient information leaflet and further verbal details of the trial. Assenting patients were formally assessed for eligibility and invited to provide informed, written consent for registration, subsequent randomisation (if eligible at that time) and long-term follow-up via routine NHS data sources. Patients were permitted to have more time to consider trial participation and, if they subsequently assented, eligibility assessments and consent were undertaken at a later clinic visit. All participants were informed that they were free to withdraw at any time without reason and without it affecting the quality of their care.
Registration and baseline Enhanced Liver Fibrosis test
Consenting patients were registered into the ELUCIDATE trial via a 24-hour telephone registration system and provided a fasted serum sample for an ELF test (patients should have refrained from eating a large meal in the 2 hours prior to providing the sample). The ELF test was sent off to a central laboratory (iQur Ltd) for analysis.
Eligibility for randomisation
Only patients with an ELF value of above a predefined threshold (denoting at least moderate fibrosis), and who had no clinical, histological or laboratory diagnosis of cirrhosis were eligible for randomisation. As knowledge of ELF values may cause patients or clinicians to modify their behaviour, which might influence disease progression and result in confounding, to ensure equipoise, results were fed back to the investigator simply as (1) below the threshold and not eligible for randomisation or (2) equal to or above the threshold and eligible for randomisation.
In March 2011, after 43 patients had been randomised, the ELF test threshold for randomisation was amended from 11.0 to 8.4 to incorporate patients with an identified risk of progression to cirrhosis and severe complications (see Figure 64).
Randomisation
Patients with a registration ELF value equal to or above the threshold (originally 11.0, amended to 8.4 after March 2011) were invited back to clinic for a randomisation visit. The randomisation visit should have occurred as soon as possible following receipt of the registration test results and preferably within 6 weeks of the registration visit, but up to 12 weeks was permissible. If > 12 weeks had passed since registration, a repeat ELF test was taken to ensure that the patient remained eligible for the trial and had not progressed to cirrhosis. At the randomisation visit, patients were assessed to ensure that their liver disease had not progressed to clinically evident cirrhosis in the interval from their registration visit and were asked whether or not they were still happy to continue participating in the trial and were willing to be randomised. Assenting patients judged to still be pre-cirrhotic were individually randomised at the end of their baseline assessments.
Randomisation was undertaken using an automated 24-hour telephone randomisation system, which was administered remotely. The randomisation service was provided by the CTRU at the University of Leeds, a UK Clinical Research Collaboration-registered trials unit. A computer-generated minimisation program incorporating a random element was used to ensure that treatment groups were well balanced for the following characteristics:
-
centre
-
age (≥ 18 to < 40, ≥ 40 to < 65, ≥ 65 to < 75 years)
-
sex (male, female)
-
baseline ELF value [11–11.49, 11.5–11.99, 12–12.49 and 12.5+ or ≥ 8.4 to < 9.5, ≥ 9.5 to < 11.5, ≥ 11.5 to < 12.5 and ≥ 12.5 from protocol version 5.0 onwards (March 2011)]
-
history of high alcohol consumption (at any time), defined as > 6 units (60 g of alcohol) per day for ≥ 12 months for males and > 4 units per day for ≥ 12 months for females (yes, no)
-
current alcohol consumption per day [males: 0 units (teetotal), < 3 units (light), 3–6 units (moderate), > 6 units (high); females: 0 (teetotal), < 2 units (light), 2–4 units (moderate), > 4 units (high)]
-
type of CLD (alcoholic liver disease, viral, unknown/other, non-alcoholic fatty liver disease).
Patients were randomised to one of two treatment groups on a 1 : 1 ratio: standard clinical monitoring plus ELF test monitoring (intervention arm) or standard clinical monitoring alone (non-intervention arm). For patients randomised to the intervention arm, if their ELF value at registration was above the threshold for cirrhosis diagnosis, the randomisation system also notified the caller of that so that management of cirrhosis could begin.
Quality assurance of the Enhanced Liver Fibrosis test
All sites were issued with a sample processing SOP. Samples were used to determine eligibility for randomisation only if they had been kept at room temperature for no longer than 2 days between being taken and arriving at iQur Ltd. If shipping delays were anticipated (e.g. at a weekend), samples were stored in the fridge and shipped when delivery within 2 days was possible. If a sample had been kept at room temperature for > 2 days from the time that it was taken, then a repeat sample was requested.
Patients were requested to refrain from eating a large meal in the 2 hours prior to providing the blood sample for each ELF test. For the ELF sample collected at the randomisation visit, patients were requested to have fasted (gone without food for > 4 hours), to allow for glucose ± homeostatic model assessment – insulin resistance testing.
Treatment group allocation
Screening for cirrhosis with standard clinical monitoring
Patients allocated to the standard clinical monitoring arm were seen in clinic every 6 months and monitored as per standard practice. If a patient was deemed to be cirrhotic on clinical criteria [by examination, on the basis of laboratory tests (other than ELF) or through imaging], cirrhosis management commenced.
Screening for cirrhosis with standard clinical monitoring plus Enhanced Liver Fibrosis test
Patients allocated to the intervention arm were seen in clinic every 6 months and monitored as per standard practice. In addition, they also had their ELF value measured every 6 months by the central laboratory. If a patient was deemed to be cirrhotic on clinical criteria [by examination, on the basis of laboratory tests (other than ELF) or through imaging], cirrhosis management commenced. If a patient had an ELF value that was above a predefined threshold, the patient was deemed to be cirrhotic. The investigator was informed that the patient was above the threshold and the patient was recalled into clinic as soon as possible for cirrhosis management to commence.
The ELF threshold for cirrhosis was originally ≥ 12.5 but this was changed to ≥ 9.5 in protocol version 5.0 onwards (March 2011).
Data collection and management
Trial data were recorded by research staff on CRFs and submitted to the CTRU. Sites were provided with guidance on the schedule of CRFs, data to be collected and completion of CRFs. Data were entered into the trial database by CTRU staff using Infermed’s (London, UK) MACRO® Electronic Data Capture platform. A number of manual and in-built database cross-checks were routinely performed to check for missing and inconsistent data items, which were reported back to sites for resolving at the earliest opportunity.
Baseline registration assessment
Baseline assessments consisting of a physical examination, medical history and demographic details were conducted in the month prior to registration or at the registration visit. At the randomisation visit, a blood sample was taken for the ELF test and the patients completed the EuroQol-5 Dimensions (EQ-5D) and Short Form questionnaire-12 items (SF-12) version 2 health questionnaires.
Baseline randomisation assessment
At this visit, patients had another ELF sample taken and completed the EQ-5D/SF12v2 and Health Usage Questionnaire. Patients were not told their randomisation allocation until after they had completed the questionnaires. In addition, patients had the following assessments performed: LFTs, full blood count, international normalised ratio and glucose level.
Follow-up
From the date of randomisation, patients underwent follow-up assessments every 6 months until 30 months post randomisation, unless they were diagnosed as cirrhotic. At each follow-up visit all patients underwent a physical examination (weight, vital signs), medical history (including details of concomitant disease and medication) and blood tests (simple LFTs, platelets, albumin and clotting) and were required to complete the EQ-5D/SF-12v2 and Health Usage Questionnaire. In addition, patients randomised to the follow-up arm with ELF testing also underwent blood sample collection at each follow-up visit, with the samples sent to the central iQur Ltd laboratory for ELF testing. Patients were requested to refrain from eating a large meal in the 2 hours prior to providing the sample. In both treatment arms, when patients were diagnosed as cirrhotic (either by ELF testing or clinical means) within 30 months post randomisation, they were required to attend an initial post-cirrhotic follow-up assessment at 3 months post diagnosis and then every 6 months until 30 months post randomisation.
Follow-up and management of patients diagnosed with cirrhosis
The trial protocol included recommendations for the management of varices, ascites and HCC after a diagnosis of cirrhosis, based on appraisal of national and international guidelines, but sites were permitted to follow their own established protocols provided that these were documented and adhered to for all study participants and that they included as a minimum ultrasound scanning, oesophagogastroduodenoscopy (OGD) and measurement of AFP levels.
All patients with a diagnosis of cirrhosis were required to have an OGD as screening for varices within 3 months of their cirrhosis diagnosis, unless they had had an OGD in the previous 18 months, in which case the next OGD should have occurred within 18 months of the previous OGD, unless clinically indicated sooner. If the previous OGD did not identify oesophageal varices, subsequent OGDs were repeated every 18 months. If small oesophageal varices were identified on OGD, OGDs were repeated every 6 months to look for variceal progression. For large oesophageal varices that were being treated, the timing of subsequent OGDs was dictated by local guidance.
Moderate or large oesophageal varices should have been banded as primary prophylaxis, with banding repeated weekly until the varices were obliterated. Alternatively, patients could be treated with non-cardioselective beta-blockers as primary prophylaxis. As secondary prophylaxis, bleeding oesophageal varices should have been banded weekly until obliterated and, in addition, patients should have been considered for treatment with beta-blockers, unless contraindicated.
For prophylaxis of spontaneous bacterial peritonitis, all patients with ascites were treated with 400 mg of norfloxacin (Noroxin®, Merck Sharp & Dohme Corp., a subsidary of Merck & Co, Inc.) once daily or with an alternative antibiotic as per local protocol.
All patients diagnosed with cirrhosis were required to have their AFP measured and an ultrasound scan performed for HCC screening within 3 months of a diagnosis of cirrhosis, unless they had had an ultrasound scan or AFP test within the previous 6 months, in which case the next ultrasound scan or AFP test should have been performed within 6 months of the previous scan, unless clinically indicated sooner. The AFP test was repeated every 6 months. If the previous ultrasound scan did not identify any lesions and the patient’s AFP level remained stable, a subsequent ultrasound scan was repeated every 6 months. Any space-occupying lesions, equivocal ultrasound scans or rising AFP levels in the absence of a lesion on ultrasound were followed by triple-phase CT and/or MRI scans.
Suspected HCC was managed according to local, national and international guidelines and the management pathway was documented on the patient’s CRF.
Patients were considered for liver transplantation if they had a solitary lesion measuring < 5 cm in diameter or three lesions measuring < 3 cm in diameter, no evidence of extrahepatic manifestations and no evidence of vascular invasion. If a patient underwent liver transplantation, further 6-monthly follow-up ceased.
All other patients were considered for therapeutic interventions as per local protocols.
End points
Primary end point (according to protocol version 7.0, 30 January 2013)
The primary end point was time from randomisation to occurrence of the first severe complication. Severe complications were defined as:
-
variceal haemorrhage confirmed by one of the following –
-
visualisation through endoscopy
-
imaging
-
post mortem
-
-
spontaneous bacterial peritonitis –
-
ascites confirmed by –
-
imaging and/or
-
aspiration and
-
-
infection confirmed by –
-
microscopy and/or
-
culture
-
-
-
HCC beyond the Milan criteria57,887 (note that, for the purposes of the trial, cases of HCC falling within the Milan criteria were not regarded as end points as they are regarded as treatable)
-
encephalopathy – grade 3 or 4 defined using the West Haven criteria888 (Table 101)
-
liver-related mortality – any of the following:
-
any mention of liver disease in part 1 of the death certificate
-
death from HCC
-
death from liver failure
-
death from bleeding from portal hypertension
-
death from hepatorenal syndrome
-
death from sepsis occurring as a result of CLD
-
death from spontaneous bacterial peritonitis
-
death from encephalopathy.
-
| Grade | Criteria |
|---|---|
| 1 | Trivial lack of awareness |
| Euphoria or anxiety | |
| Shortened attention span | |
| Impaired performance of addition | |
| 2 | Lethargy or apathy |
| Minimal disorientation for time or place | |
| Subtle personality change | |
| Inappropriate behaviour | |
| Impaired performance of subtraction | |
| 3 | Somnolence to semistupor, but responsive to verbal stimuli |
| Confusion | |
| Gross disorientation | |
| 4 | Coma (unresponsive to verbal or noxious stimuli) |
Secondary end points (according to protocol version 7.0, 30 January 2013)
-
Time from diagnosis of cirrhosis (by ELF testing or clinical means) to incidence of first severe complication.
-
Time from randomisation to diagnosis of cirrhosis by ELF testing or clinical means (to allow instigation of prophylaxis and screening).
-
Process outcomes, namely:
-
treatment of varices with beta-blockers/band ligation
-
use of endoscopy and ultrasound/AFP tests
-
treatment to normalise LFTs in patients with hepatitis B and hepatitis C.
-
-
Detection and timing of complications following cirrhosis, including:
-
detection of small varices
-
detection of large varices
-
incidence of treatable HCC.
-
-
All causes of mortality.
-
Specific liver-related morbidity.
-
Economic evaluation of the ELF test in the early detection of cirrhosis and as such in the initiation of measures to reduce the incidence of severe complications following cirrhosis.
-
Quality of life.
-
Proportion of non-randomised patients (ELF value of < 8.4) who go on to develop cirrhosis (diagnosed by clinical means) within the follow-up period.
The process outcomes were added as secondary end points by way of a protocol amendment in January 2013, as part of the NIHR-approved trial extensions. The overall aim, and primary outcome, of the study was to reduce the incidence of severe complications and improve survival in this patient population through the use of ELF tests. For this to happen the use of such tests would have to affect clinical practice, that is, the process of care would have to change. This is a necessary step in order to expect an improvement in these primary outcomes and was by no means guaranteed.
Health-related quality of life
Health-related quality of life was assessed at registration, randomisation and 6-monthly intervals post randomisation for 30 months (five follow-up visits), using the SF-12v2.
Long-term follow-up
All patients who were registered into the trial (whether randomised or not) were flagged with the Health and Social Care Information Centre collection of for (now known as NHS Digital http://digital.nhs.uk/; accessed 22 May 2018) longer-term morbidity and mortality data.
Statistical analysis
The analysis and reporting of the trial was undertaken in accordance with CONSORT guidelines. 235 The original sample size calculation is given in detail in Appendix 4. This sample size calculation was not straightforward without the availability of the simulation approach, derived later on as part of the methodology workstream and described in Chapter 19. All statistical analysis was undertaken using SAS® software version 9.4 (SAS Institute Inc., Cary, NC, USA; SAS and all other SAS Institute Inc. product or service names are registered trademarks or trademarks of SAS Institute Inc. in the USA and other countries. ® indicates USA registration), following a predefined analysis plan agreed with the Trial Steering Committee. The primary comparative analyses between the randomised groups were conducted on an intention-to-treat basis without imputation of missing data.
Health economic methods
The main trial was supplemented with an economic evaluation consisting of two components:
-
a within-trial economic evaluation to compare the observed costs and outcomes of the cohort of patients randomised to ELF guided detection and management with those of a cohort of patients randomised to standard care, from the perspective of the NHS and Personal Social Services
-
a long-term economic evaluation using the clinical trial outcome and resource utilisation data to update the parameters of the pre-existing lifetime horizon cost-effectiveness model and subsequent calculation of the expected ICER for ELF guided detection and management, using a lifetime horizon.
At the outset of the trial, the primary outcome measure for the within-trial economic evaluation was QALYs. Quality of life weights were calculated using the EQ-5D algorithm. Life-years lived were obtained from the mortality data collected at the end of the clinical trial. Costs and outcomes will be discounted at 3.5% per annum. All costs will be indexed to the trial start year (2009) using the NHS Pay and Prices Index. 889 The primary result of the economic evaluation will be the ICER of ELF guided detection and management compared with standard care. This will be calculated as the difference in the mean cost of the interventions divided by the difference in the mean outcomes.
Parameter uncertainty will be examined using a non-parametric bootstrap simulation. 38
The results of the probabilistic sensitivity analysis will be presented as the expected ICER, a scatterplot on the cost-effectiveness plane and a cost-effectiveness acceptability curve. The expected net benefit of ELF-guided detection and management will be calculated for a range of values of lambda, including £5000, £15,000, £20,000 and £30,000.
Secondary within-trial economic evaluation
The secondary within-trial economic evaluation will substitute SF-12v2 quality of life weights for the EQ-5D quality of life weights used in the primary analysis. In all other regards, the secondary within-trial economic evaluation will be identical to the primary within-trial economic evaluation.
Long-term economic evaluation
The clinical trial outcome and resource utilisation data will be used. Parameter uncertainty will be examined using Monte Carlo simulation. The results of the probabilistic sensitivity analysis will be presented as the expected ICER, a scatterplot on the cost-effectiveness plane and a cost-effectiveness acceptability curve. The expected net benefit of ELF-guided detection and management will be calculated for a range of values of lambda, including £5000, £15,000, £20,000 and £30,000.
Delayed set-up time and initially slow recruitment resulted in two successful applications for 1-year, no-cost extensions. The analysis plan required evaluation of changes in the process of care of patients as a consequence of the use of ELF monitoring. Final evaluation of the impact on QALYs will follow the collection of long-term outcomes, including survival, from cancer registry and NHS information sources.
Chapter 19 Value of modelling development, modification and extension of the ELUCIDATE trial
Background
The ELUCIDATE trial was planned to run for 5 years, allowing sufficient time to randomise 1000 patients and follow them using the active monitoring intervention with ELF-based testing for 30 months. The trial began late because of delays in set-up and, by November 2012, 25 months after the first participant was recruited, only 530 patients had been randomised. At that time the Programme Grants for Applied Research Board conducted a site visit and raised doubts concerning the project team’s ability to complete adequate recruitment in the remaining period of grant funding and accepted the investigators’ request and recommendation for a 1-year extension and an additional new focus on process of care outcomes. The board recommended early closure of the trial.
The Trial Management Group (TMG) had identified the shortfall in recruitment and had worked with the 20 established sites to promote the trial, but also opened 17 additional sites bringing the total number of sites open to 37. Between the time of the site visit and the closure of recruitment, on the recommendation of the board, there was an upsurge in recruitment from new and established centres and a further 348 patients were randomised to the study in 4 months (39.6% of the total recruited in 14.8% of the recruitment period), bringing the total number of patients randomised and in follow-up to 878.
Rather than restricting the trial to investigating diagnostic performance and process outcomes only, this number of patients was sufficient to provide adequate power to enable completion of the trial in accordance with the original protocol and assessment of the clinical and health-care outcome end points identified in the original protocol, funded as part of the Programme Grants for Applied Research grant application. However, this required a further 12-month study extension to permit all enrolled patients to complete 30 months of exposure to the monitoring intervention in the trial (Figures 62 and 63). The case was made for the extension to complete the trial based on the rapid final surge in recruitment and a modelling of the likely impact of ELF testing allowing revised estimates of study power. This was approved by NIHR.
FIGURE 62.
A comparison of the model prediction of the identification of patients as cirrhotic over time (green line) with the observed patterns in the ELUCIDATE trial used to calibrate the model.
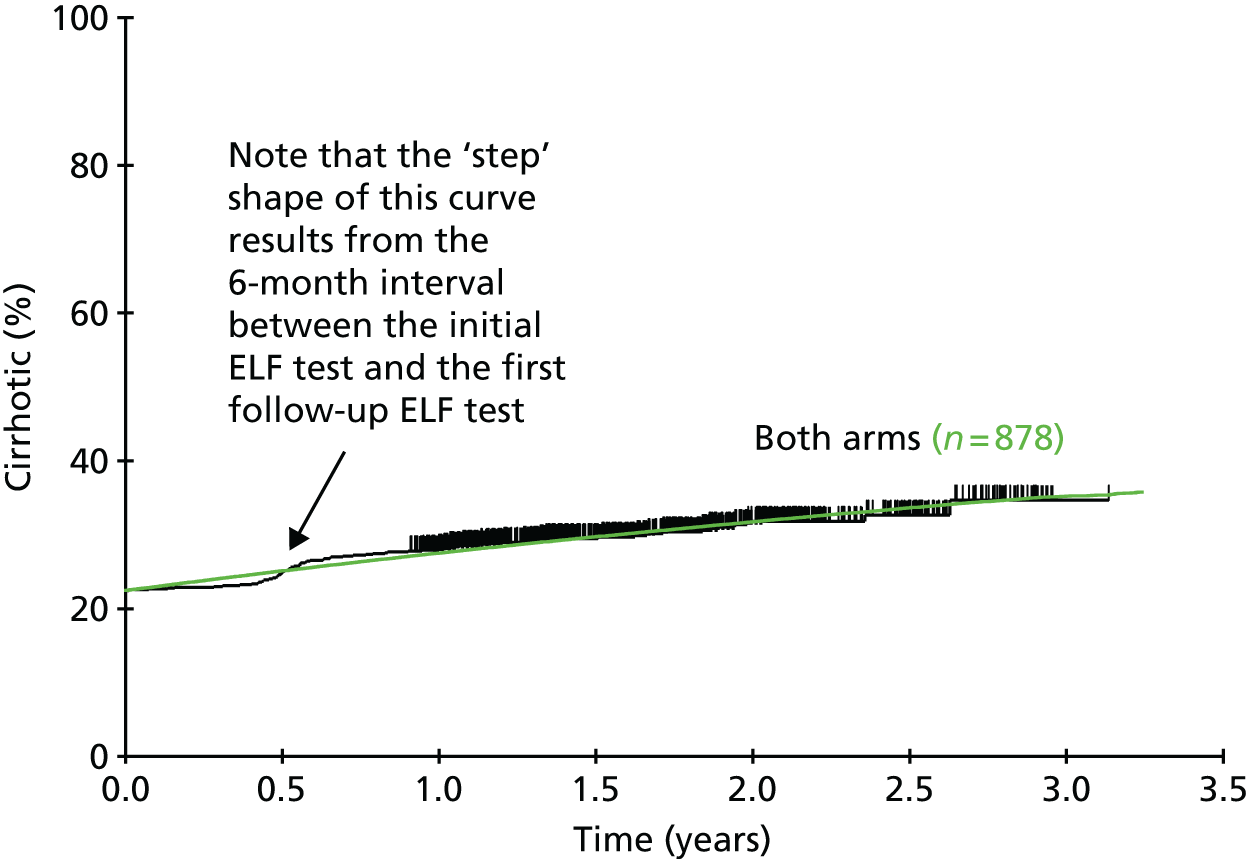
FIGURE 63.
Estimated pattern of development of cirrhosis without a trial extension, showing follow-up times assuming that the intervention finished at the end of October 2014.

It was recognised that delivery of the evaluation of the impact of ELF testing on the clinical and health-care outcomes will still (as originally approved) require the collection of ‘routine data’ from death certification, the Office for National Statistics (ONS), Hospital Episode Statistics (HES) and cancer and transplant registries at 5 years.
We performed modelling incorporating data from previous studies of disease progression in similar cohorts of patients with CLD investigated for CLD with ELF and this was the basis of all previous power calculations. However, importantly, at the point at which the extensions were approved, we were able to analyse preliminary data from the ELUCIDATE trial, allowing greater confidence in both the models themselves and the results and conclusions that can be drawn from them.
The model represents a significant innovation, allowing us to accurately predict the development of cirrhosis (see Figure 62).
The modelling also allowed us to demonstrate the impact on follow-up and monitoring using ELF tests that would occur with and without the 1-year extension (see Figure 63).
The ELUCIDATE trial, like all trials, depends for its statistical power on the number of events and the effect size. The 30-month period of active follow-up within the trial was both a period of case discovery and data collection and, vitally, the key monitoring intervention within the trial that could produce a clinical effect. We were testing the performance of the ELF test as a means of discovering cases of cirrhosis and thereby influencing the behaviour of clinicians in delivering, as a consequence of the identification of cirrhosis by ELF criteria, the appropriate investigations and treatments necessary for patients in the diagnostic cirrhotic category.
The shortening of the period of ‘active follow-up’ would, therefore, have had an effect on case discovery and would also have influenced the effect size by reducing the period of the intervention (i.e. ELF monitoring and treatment of cirrhosis once detected). We expected a reduction in effect size as well as reductions in case discovery if we were unable to complete the period of active follow-up/intervention.
We know from the existing literature that, when patients are diagnosed with cirrhosis, appropriate monitoring and treatment for major events such as bleeding or liver cancer result in a reduction of such events by 50% in RCTs. However, in the ELUCIDATE trial we always carefully considered whether or not clinical behaviours would change sufficiently and would result in the full effect in cirrhotic patients. Allowing for this in power calculations for the long-term clinical end points, we always mitigated the effect size from the 50% ceiling that is apparent in the literature to 40%, to allow for the possibility that clinicians would not implement the consequences of the diagnosis comprehensively and in a timely fashion in the real world.
In estimating the impact of the extension to 30 months in all patients to complete active follow-up/intervention (see Figures 62 and 63), we took account of a reduction in case discovery and a reduction in effect size. Using our established ability to model the trial, we calculated the loss of power as a consequence of loss of case discovery and very conservative estimates of reduction in effect size.
With the extension to enable the completion of the 30-month monitoring/intervention in all randomised patients, and retaining our previous (mitigated) effect size of 40%, the power of the study was 79.5%. Without the extension, if we made a conservative assumption of a reduced effect size to 35%, power dropped to 63%. A more likely reduction in effect size to 30% resulted in power of only 49%.
Such detailed modelling in the trial when preliminary data became available provided justification for the extension of the trial to complete the monitoring intervention for all enrolled patients based on the impact on the power calculations. The additional data generated will allow more robust economic analysis of the relationships between changes in the process of care (within the 30-month follow-up data) and the longer-term clinical and health-care outcomes (ONS and HES data).
At the end of the 30-month monitoring period we were able to conduct analyses and we report on the impact of ELF testing on the diagnosis of cirrhosis and on the process of care (AFP measurement, ultrasound scans, prescription of treatments, etc.). We will conduct (and have funded) the 5-year follow-up analysis using ONS and HES data as planned and approved.
Modelling methodology
The following gives details and assumptions for the simulation model on which the sample size estimate, and ultimately the extension request, was based.
The model relies on the data in Figure 64, from previously published data35 which show the likely occurrence of severe complications for different starting ELF ranges. It is then possible to fit a (parametric) model for each of the ELF ranges and to use this model to predict the subsequence occurrence of severe complication events. We decided to incorporate into our model a delay before events start to occur, followed by a negative exponential event-occurrence pattern, adopting a conservative approach. Therefore, if Si(t) (i = 1,6) are the times to incidence of severe complications for each of a range of six presentation ELF values (namely < 8.39, 8.4–9.49, 9.5–10.49, 10.5–11.49, 11.5–12.49, ≥ 12.5), then:
where αi (i = 1,6) are the exponential parameters and di(i = 1,6) are the delays before severe complications start to occur in each of the six groups.
FIGURE 64.
Expected incidence of severe complications as related to presentation ELF value.

For the purposes of the model patients were divided into those who present with an ELF value of > 9.5 and those who present with an ELF value of < 9.5. In the ELF arm patients with an ELF value of > 9.5 are classified as cirrhotic based on this ELF result. We can establish the proportions of patients who present with an ELF value of > 9.5, and with an ELF value in the ranges 9.5–10.49, 10.5–11.49, 11.5–12.49 and > 12.5, from the ELF arm and apply these proportions in the simulation to both arms, as, although we do not know these figures in the control arm, they would be expected to be the same.
In addition, it was necessary to consider the rate of development of cirrhosis in both arms for patients with a starting ELF value of < 9.5. These rates will be different as, in the ELF arm, this rate is based on the measured ELF value, whereas in the control arm it is based on clinical grounds only. As treatment is initiated only once cirrhosis has been diagnosed, any treatment effect was applied to patients diagnosed as cirrhotic.
We analysed the cumulative incidence curves for development of cirrhosis in the trial and assumed for simplicity that, for patients who were not already defined as cirrhotic by their starting ELF value, there was a uniform rate of development of cirrhosis, say λ, which fitted reasonably well with the observed data, as shown in Figure 62. This would imply that the development of cirrhosis for the population not already defined as cirrhotic by their starting ELF value follows an exponential distribution, specifically:
where Pc(t) is the probability of the patient becoming cirrhotic by time t and with different rates of development of cirrhosis in the ELF and control arms, as the ELF arm rate is dependent on the ELF level crossing the 9.5 threshold, whereas the rate is defined by clinical judgement in the control arm.
Refinements were considered, but the simulation results do not appear to be particularly sensitive to the assumption of a uniform rate of development of cirrhosis. Note that Figure 62 shows the time to development of cirrhosis for both arms combined (either defined by ELF value in the ELF arm or defined by clinical judgement in the control arm). These results, broken down by treatment arm, with associated model fits, were presented, in confidence, to the Data Monitoring and Ethics Committee (DMEC). Figure 63 provides a likely projection of Figure 62 without the trial extension, depicting graphically the cohort of patients who would not receive the full trial intervention (active monitoring for 30 months) in the event that the extension had not been granted.
Further refinements of the simulation model were possible looking at different models both for the development of cirrhosis and for subsequent occurrence of severe complications. For instance, in the latter case, we may consider models in which the incidence of severe complications plateaus after a particular period of time, for example (using previous terminology):
where pi is the proportion of patients in the ith group who never experience a severe complication.
To complete the model, we applied an assumed treatment effect, which applies a proportional hazards improvement in time to the development of severe complications for patients diagnosed as cirrhotic (in either arm). This simply changed the negative exponential parameter for the time to occurrence of severe complications in each of the different ELF ranges (9.5–10.49, 10.5–11.49, 11.5–12.49 and > 12.5). Again, using previous nomenclature, these times to occurrence of severe complications are, therefore, assumed to be:
where θ is the assumed HR. We assumed an effect size of 40%, equivalent to a HR of 0.6, for the trial design/sample size calculations for the primary outcome. We can readily estimate possible different effect sizes using this model.
Putting together all of these assumptions it is possible to simulate the trial. In the ELF arm patients are assumed to present with proportions in the different ELF ranges; in addition, patients with an ELF value of < 9.5 are assumed to have a rate of subsequent development of cirrhosis, with such patients developing cirrhosis at different ELF values, to match what we have seen in the trial to date. They then have a probability of developing a severe complication based on the negative exponential distributions described. The situation is similar in the control arm except that treatment is initiated somewhat later after the clinically based diagnosis of cirrhosis. For each simulated trial the times to severe events are generated given the actual recruitment times and a specified duration of follow-up (e.g. 5 years) and are then compared between the two arms using the log-rank test. Figure 65 provides a typical example and Figure 66 provides the theoretical, expected curves that result from these model assumptions (in this case with an effect size of 40%).
FIGURE 65.
Simulation of the development of severe liver complications in the trial until December 2019 by randomised arm.

FIGURE 66.
Simulated trial outcomes for each trial arm with follow-up continuing until December 2019.

The trial power was calculated for different type 1 error rates by simply counting the proportion of simulations in which the resulting p-value was less than the assumed type 1 error.
Note that it was practicable to apply such a model only when the trial was under way for a reasonable period of time, as we needed estimates of the rates of development of cirrhosis in the ELF and control arms, as well as the ELF ranges for the proportions of patients who do develop cirrhosis, at the point at which they develop cirrhosis. Using this type of simulation modelling approach, it would have been possible to apply this model prior to the start of the trial, but these rates would have had to be estimated based on few data. This should, for future trials, form part of the trial design sample size assumptions. It would perhaps have been possible to estimate these parameters from pilot data on sequential ELF values for a cohort of patients. For future designs, it would be helpful to collect such pilot data, which would be likely to be of fundamental importance to the trial design.
None of these estimated rates involved looking at unblinded primary event data.
Conclusions
These arguments and the modelling specifically allowed us to demonstrate our ability to complete the ELUCIDATE trial robustly and with good power for the principal clinical and health-care outcomes. Our approach has the potential to be generalisable to a variety of other monitoring trials and provides a basic framework for trial design in this area, which to date has been lacking. For instance, the model can be applied to situations in which patients are all initially below the threshold for intervention and who pass this threshold only as the study proceeds. We hope to be able to provide appropriate software so that these methods can be made widely available. This is in line with our remit in this programme grant to use the ELUCIDATE trial as an exemplar to aid in the design, analysis and interpretation of future monitoring trials. Note that the simulation approach described herein incorporates this variability in sequential ELF values as they change over time, through the models and parameters that relate to the rate of development of cirrhosis, as this is highly correlated with changing ELF values.
Chapter 20 Recruitment to and delivery of the ELUCIDATE trial
Trial organisation
Registration and randomisation
Eligible and consenting patients were registered and randomised into the study using an automated 24-hour telephone registration and randomisation system. Registered patients who met the ELF threshold criteria for randomisation were preferably randomised within 6 weeks of the registration visit.
Clinical queries
The Co-chief Investigators based at University College London Hospital (UCLH)/Royal Free Hospital, London (Professor William Rosenberg and Dr Sudeep Tanwar) were the contacts for clinical queries and the review of any related and unexpected serious adverse events (RUSAEs).
Project and trial management
The CTRU, Leeds Institute of Clinical Trials Research at the University of Leeds, was responsible for the overall project and data management of the study. The TMG, comprising the Chief Investigators, health economists, scientific advisors, trial co-ordinators and statistician, was responsible for the ongoing management and promotion of the study and for the interpretation of the results.
Trial Steering Committee
The Trial Steering Committee provided overall supervision of the trial, in particular relating to trial progress, adherence to the study protocol, patient safety and consideration of new information. The committee included an independent chairperson (Professor James Neuberger), an independent clinician (Dr Jonathan Fallowfield), an independent scientific adviser (Dr Christine Patch), a statistician (Dr Andrew Roddam), a health economist (Dr Simon Dixon), the Chief Investigator (Professor William Rosenberg) and members of the TMG. The PPI representatives were Tilly Hale and Joan Bedlington.
Leeds National Institute for Health Research Biomarker Research Tissue Bank
Patients who were recruited to the trial were asked on the patient information sheet if they would be happy to provide an additional blood sample for the Leeds NIHR Biomarker Bank at their randomisation visit. For all patients who consented, a serum sample was obtained, processed and stored on site within a –70 to –80°C freezer. On a site reaching 50 patients or at the end of the trial, these samples were collected for storage at the Leeds NIHR Biomarker Bank, for use in future research projects.
Enhanced Liver Fibrosis testing and the provision of Enhanced Liver Fibrosis and Enhanced Liver Fibrosis/National Institute for Health Research Biomarker Bank kits
Both the ELF/NIHR Biomarker Research Tissue Bank kits used at randomisation and the ELF kits to be used for the tests performed at patient registration, follow-up (patients in the ELF arm) and once patients had been diagnosed as cirrhotic (patients in the control arm) were prepared and dispatched by the Clinicaland Biomedical Proteomics Group at St James’s University Hospital, Leeds. The serum samples sent for ELF testing were processed at the iQur laboratory based at Royal Free Hospital, London. Results from these tests were disseminated to the CTRU. The CTRU then notified sites of a patient’s eligibility and current cirrhotic status (the latter pertaining only to those patients in the ELF screening arm).
Accrual
Thirty-seven centres across the UK were opened to recruitment between 23 September 2010 and 31 October 2012 (Table 102). In total, 1303 participants were registered into the trial, of whom 878 were subsequently randomised (Figure 67), 440 patients to the standard clinical monitoring arm and 438 to the standard clinical monitoring plus ELF test arm. Monthly and cumulative registrations and randomisations across the centres are shown in Tables 102 and 103. In each case it can be seen that the additional centres added late in the trial process added very substantial numbers of patients, frequently surpassing the achievements of centres open throughout the trial. Of the 425 registered patients who did not proceed to randomisation, the main reason was because their baseline ELF value was lower than the threshold required for randomisation (Table 104).
| Site | Date opened | 2011 | Year (n) | Accrual per centre | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2012 | 2013 | |||||||||||||||||
| January | February | March | April | May | June | July | August | September | October | November | December | January | February | March | ||||
| H1 | 23 September 2010 | 126 | 8 | 5 | 4 | 5 | 1 | 1 | 3 | 2 | 1 | 2 | 0 | 1 | 4 | 4 | 0 | 167 |
| H2 | 18 May 2011 | 104 | 14 | 13 | 10 | 6 | 13 | 5 | 8 | 6 | 7 | 5 | 10 | 2 | 8 | 12 | 0 | 223 |
| H3 | 15 June 2011 | 39 | 1 | 1 | 9 | 3 | 5 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 60 |
| H4 | 24 June 2011 | 5 | 0 | 0 | 6 | 5 | 4 | 0 | 1 | 4 | 4 | 2 | 6 | 0 | 1 | 1 | 0 | 39 |
| H5 | 13 July 2011 | 14 | 23 | 12 | 12 | 3 | 9 | 0 | 0 | 0 | 2 | 0 | 0 | 4 | 2 | 0 | 0 | 81 |
| H6 | 26 July 2011 | 4 | 6 | 18 | 5 | 5 | 8 | 1 | 10 | 0 | 0 | 2 | 4 | 0 | 0 | 3 | 0 | 66 |
| H7 | 26 July 2011 | 1 | 3 | 1 | 6 | 3 | 0 | 0 | 1 | 6 | 2 | 7 | 5 | 0 | 2 | 2 | 0 | 39 |
| H8 | 4 August 2011 | 2 | 2 | 3 | 5 | 3 | 3 | 0 | 0 | 3 | 4 | 1 | 4 | 1 | 0 | 0 | 2 | 33 |
| H9 | 14 September 2011 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 2 | 1 | 2 | 1 | 1 | 8 | 0 | 19 |
| H10 | 6 September 2011 | 3 | 2 | 5 | 2 | 1 | 2 | 2 | 0 | 2 | 1 | 5 | 2 | 0 | 3 | 3 | 0 | 33 |
| H11 | 17 October 2011 | 1 | 8 | 4 | 5 | 9 | 6 | 4 | 9 | 2 | 2 | 2 | 3 | 2 | 6 | 5 | 0 | 68 |
| H12 | 5 January 2012 | 0 | 0 | 0 | 0 | 4 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 10 | 0 | 17 | |
| H13 | 5 January 2012 | 0 | 0 | 0 | 3 | 1 | 0 | 2 | 1 | 2 | 2 | 0 | 0 | 1 | 0 | 0 | 12 | |
| H14 | 5 January 2012 | 0 | 0 | 2 | 2 | 2 | 1 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 11 | |
| H15 | 6 March 2012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 4 | 0 | 5 | |||
| H16 | 6 March 2012 | 0 | 0 | 0 | 0 | 2 | 6 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 10 | |||
| H17 | 27 March 2012 | 0 | 0 | 1 | 7 | 5 | 0 | 3 | 3 | 2 | 2 | 2 | 0 | 25 | ||||
| H18 | 3 April 2012 | 1 | 3 | 2 | 3 | 0 | 0 | 3 | 3 | 1 | 3 | 6 | 0 | 25 | ||||
| H19 | 19 April 2012 | 2 | 13 | 2 | 4 | 0 | 2 | 7 | 4 | 1 | 6 | 7 | 0 | 48 | ||||
| H20 | 20 April 2012 | 1 | 5 | 3 | 7 | 3 | 4 | 6 | 8 | 0 | 0 | 11 | 0 | 48 | ||||
| H21 | 26 April 2012 | 6 | 3 | 1 | 4 | 2 | 3 | 2 | 3 | 1 | 0 | 0 | 25 | |||||
| H22 | 24 May 2012 | 0 | 1 | 2 | 0 | 1 | 1 | 4 | 1 | 2 | 7 | 0 | 19 | |||||
| H23 | 14 May 2012 | 0 | 0 | 1 | 0 | 0 | 2 | 7 | 7 | 13 | 7 | 0 | 37 | |||||
| H24 | 28 May 2012 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |||||
| H25 | 7 June 2012 | 5 | 2 | 1 | 3 | 2 | 0 | 1 | 1 | 0 | 15 | |||||||
| H26 | 3 July 2012 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |||||||
| H27 | 30 July 2012 | 1 | 2 | 2 | 6 | 0 | 10 | 5 | 0 | 26 | ||||||||
| H28 | 21 June 2012 | 2 | 0 | 3 | 1 | 1 | 0 | 3 | 0 | 10 | ||||||||
| H29 | 25 July 2012 | 0 | 2 | 1 | 1 | 1 | 4 | 1 | 0 | 10 | ||||||||
| H30 | 25 July 2012 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | ||||||||
| H31 | 30 July 2012 | 0 | 0 | 0 | 0 | 0 | 3 | 4 | 0 | 7 | ||||||||
| H32 | 1 August 2012 | 0 | 0 | 0 | 0 | 2 | 0 | 7 | 0 | 9 | ||||||||
| H33 | 21 August 2012 | 0 | 0 | 7 | 5 | 7 | 12 | 19 | 0 | 50 | ||||||||
| H34 | 26 September 2012 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | |||||||||
| H35 | 10 October 2012 | 7 | 12 | 4 | 12 | 12 | 0 | 47 | ||||||||||
| H36 | 18 October 2012 | 0 | 2 | 2 | 1 | 3 | 2 | 10 | ||||||||||
| H37 | 31 October 2012 | 0 | 3 | 0 | 0 | 3 | ||||||||||||
| Monthly accrual | 67 | 62 | 66 | 52 | 86 | 28 | 72 | 50 | 45 | 78 | 99 | 44 | 101 | 147 | 6 | |||
| Cumulative accrual | 300 | 367 | 429 | 495 | 547 | 633 | 661 | 733 | 783 | 828 | 906 | 1005 | 1049 | 1150 | 1297 | 1303 | 1303 | |
FIGURE 67.
Monthly and cumulative accrual.
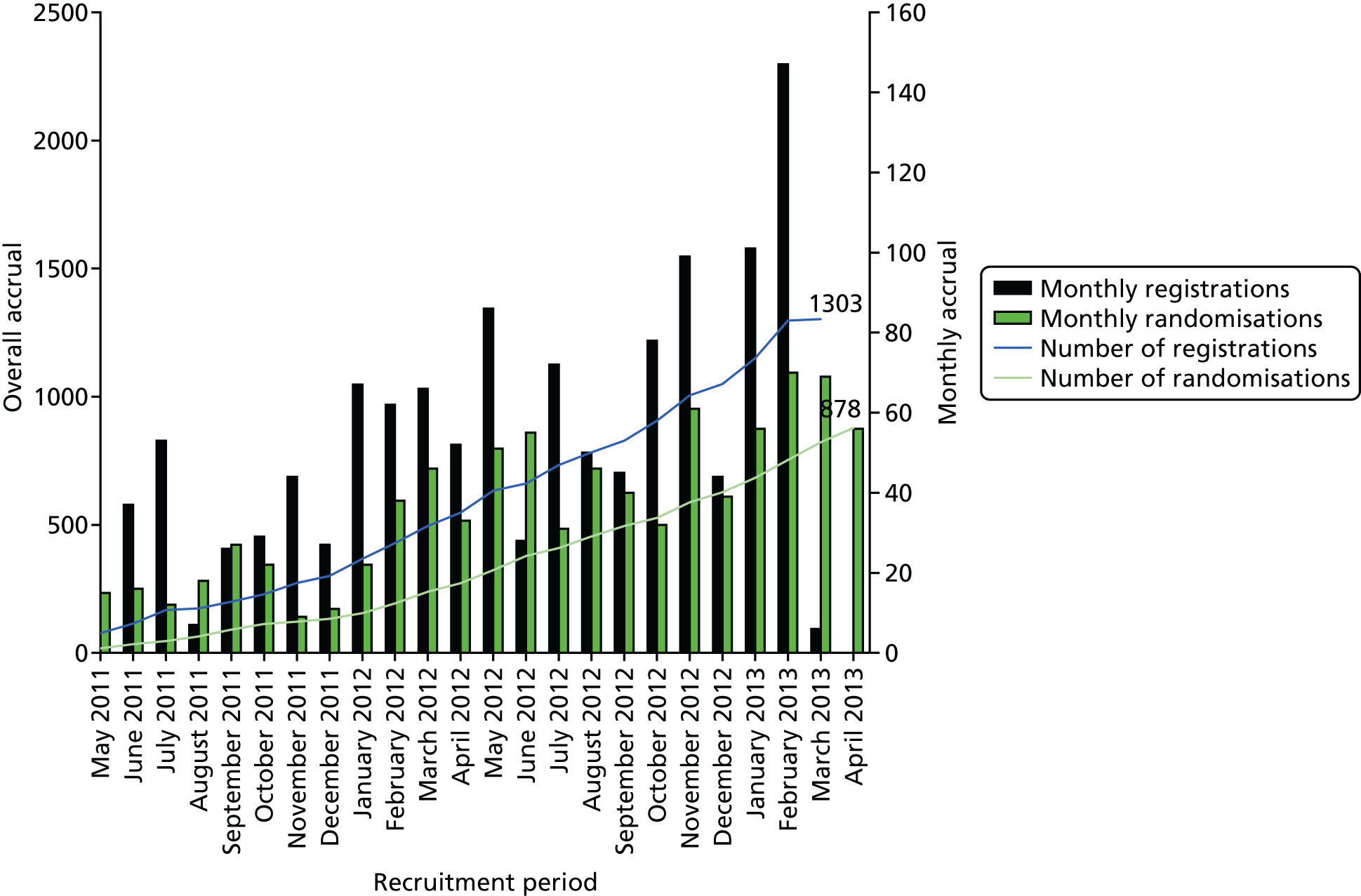
| Site | 2011 | Year (n) | Accrual per centre | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2012 | 2013 | |||||||||||||||||
| January | February | March | April | May | June | July | August | September | October | November | December | January | February | March | April | |||
| H1 | 62 | 3 | 3 | 2 | 5 | 2 | 6 | 0 | 1 | 5 | 0 | 0 | 0 | 1 | 1 | 2 | 1 | 94 |
| H2 | 44 | 5 | 7 | 15 | 4 | 11 | 11 | 1 | 3 | 8 | 2 | 6 | 2 | 5 | 6 | 3 | 3 | 136 |
| H3 | 19 | 8 | 2 | 1 | 7 | 4 | 5 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 47 |
| H4 | 4 | 0 | 0 | 0 | 2 | 6 | 5 | 0 | 2 | 2 | 5 | 3 | 2 | 1 | 1 | 1 | 0 | 34 |
| H5 | 0 | 0 | 4 | 7 | 4 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 20 |
| H6 | 1 | 2 | 10 | 12 | 3 | 5 | 3 | 2 | 6 | 0 | 2 | 1 | 0 | 2 | 0 | 1 | 0 | 50 |
| H7 | 0 | 0 | 1 | 3 | 1 | 1 | 2 | 1 | 3 | 2 | 1 | 7 | 1 | 1 | 1 | 1 | 0 | 26 |
| H8 | 1 | 1 | 4 | 0 | 3 | 4 | 0 | 0 | 2 | 3 | 3 | 2 | 2 | 2 | 0 | 1 | 1 | 29 |
| H9 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 2 | 0 | 1 | 2 | 2 | 4 | 1 | 15 |
| H10 | 1 | 1 | 4 | 2 | 1 | 2 | 1 | 1 | 0 | 2 | 0 | 4 | 1 | 0 | 3 | 0 | 2 | 25 |
| H11 | 0 | 2 | 3 | 3 | 2 | 6 | 1 | 3 | 6 | 0 | 1 | 3 | 2 | 1 | 6 | 1 | 1 | 41 |
| H12 | 0 | 0 | 0 | 0 | 1 | 3 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 4 | 16 | |
| H13 | 0 | 0 | 0 | 0 | 0 | 3 | 1 | 1 | 1 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 9 | |
| H14 | 0 | 0 | 1 | 1 | 1 | 1 | 2 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 9 | |
| H15 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 3 | 5 | |||
| H16 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 6 | |||
| H17 | 0 | 0 | 0 | 1 | 7 | 3 | 1 | 3 | 0 | 4 | 0 | 2 | 0 | 21 | ||||
| H18 | 0 | 1 | 2 | 1 | 2 | 0 | 0 | 3 | 1 | 1 | 1 | 2 | 3 | 17 | ||||
| H19 | 0 | 2 | 8 | 8 | 2 | 0 | 1 | 7 | 3 | 3 | 2 | 5 | 2 | 43 | ||||
| H20 | 0 | 0 | 1 | 1 | 1 | 6 | 0 | 1 | 0 | 5 | 0 | 1 | 5 | 21 | ||||
| H21 | 0 | 2 | 5 | 1 | 4 | 2 | 2 | 0 | 3 | 1 | 0 | 0 | 20 | |||||
| H22 | 0 | 0 | 0 | 2 | 0 | 1 | 0 | 1 | 0 | 0 | 3 | 7 | 14 | |||||
| H23 | 0 | 0 | 0 | 1 | 0 | 1 | 2 | 4 | 6 | 8 | 6 | 3 | 31 | |||||
| H24 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |||||
| H25 | 1 | 3 | 1 | 1 | 0 | 2 | 3 | 0 | 2 | 0 | 13 | |||||||
| H26 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | |||||||
| H27 | 0 | 0 | 1 | 2 | 2 | 0 | 7 | 3 | 0 | 15 | ||||||||
| H28 | 0 | 0 | 0 | 1 | 0 | 0 | 2 | 1 | 3 | 7 | ||||||||
| H29 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 6 | ||||||||
| H30 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 2 | ||||||||
| H31 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 2 | 5 | ||||||||
| H32 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 5 | 1 | 8 | ||||||||
| H33 | 0 | 0 | 0 | 4 | 2 | 10 | 14 | 7 | 4 | 41 | ||||||||
| H34 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | |||||||||
| H35 | 0 | 7 | 6 | 3 | 6 | 11 | 5 | 38 | ||||||||||
| H36 | 0 | 0 | 3 | 1 | 0 | 0 | 4 | 8 | ||||||||||
| H37 | 0 | 0 | 3 | 0 | 0 | 3 | ||||||||||||
| Monthly accrual | 22 | 38 | 46 | 33 | 51 | 55 | 31 | 46 | 40 | 32 | 61 | 39 | 56 | 70 | 69 | 56 | ||
| Cumulative accrual | 133 | 155 | 193 | 239 | 272 | 323 | 378 | 409 | 455 | 495 | 527 | 588 | 627 | 683 | 753 | 822 | 878 | 878 |
| Reason | Total (N = 425), n (%) |
|---|---|
| Patient’s baseline ELF value was lower than the threshold for eligibility | 317 (74.6) |
| Patient can no longer attend appointments | 14 (3.3) |
| Patient developed cirrhosis between registration and randomisation | 2 (0.5) |
| Other | 58 (13.6) |
| Missing | 34 (8.0) |
Chapter 21 Preliminary analysis of the ELUCIDATE trial
In this chapter, we present the analysis of the ELUCIDATE trial to date agreed by the DMEC. This includes patient characteristics, ELF values, compliance, time to cirrhosis-associated ELF values and changes in the process of care.
Baseline characteristics
The computerised minimisation in the ELUCIDATE trial worked as expected and the treatment arms were balanced with regard to all baseline characteristics (Tables 105 and 106).
| Characteristic | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438)a,b | Standard care (N = 440) | ||
| Age (years) (categorised) | |||
| 18–39 | 47 (10.7) | 51 (11.6) | 98 (11.2) |
| 40–64 | 306 (69.9) | 304 (69.1) | 610 (69.5) |
| 65–75 | 85 (19.4) | 85 (19.3) | 170 (19.4) |
| Age (years) | |||
| Mean (SD) | 54.0 (11.64) | 53.8 (11.19) | 53.9 (11.41) |
| Median (range) | 55.0 (23.0–74.0) | 54.0 (22.0–74.0) | 55.0 (22.0–74.0) |
| Missing | 0 | 0 | 0 |
| Sex | |||
| Male | 246 (56.2) | 246 (55.9) | 492 (56.0) |
| Female | 192 (43.8) | 194 (44.1) | 386 (44.0) |
| ELF value at registration (categorised) | |||
| 8.4–9.49 | 240 (54.8) | 242 (55.0) | 482 (54.9) |
| 9.5–11.49 | 174 (39.7) | 172 (39.1) | 346 (39.4) |
| 11.5–12.49 | 14 (3.2) | 15 (3.4) | 29 (3.3) |
| ≥ 12.5 | 10 (2.3) | 11 (2.5) | 21 (2.4) |
| Registration ELF value | |||
| Mean (SD) | 9.6 (1.07) | 9.7 (1.03) | 9.6 (1.05) |
| Median (range) | 9.4 (8.4–17.4) | 9.4 (8.4–14.2) | 9.4 (8.4–17.4) |
| Missing | 0 | 0 | 0 |
| History of alcohol consumption | |||
| Yes | 101 (23.1) | 107 (24.3) | 208 (23.7) |
| No | 337 (76.9) | 333 (75.7) | 670 (76.3) |
| Current alcohol consumption | |||
| Teetotal | 230 (52.5) | 231 (52.5) | 461 (52.5) |
| Light | 153 (34.9) | 151 (34.3) | 304 (34.6) |
| Moderate | 41 (9.4) | 41 (9.3) | 82 (9.3) |
| Heavy | 14 (3.2) | 17 (3.9) | 31 (3.5) |
| Primary diagnosis | |||
| Alcoholic liver disease | 35 (8.0) | 33 (7.5) | 68 (7.7) |
| Viral liver disease | 176 (40.2) | 177 (40.2) | 353 (40.2) |
| Other/unknown | 114 (26.0) | 117 (26.6) | 231 (26.3) |
| Non-alcoholic fatty liver disease | 113 (25.8) | 113 (25.7) | 226 (25.7) |
| Time from registration to randomisation (days) | |||
| Mean (SD) | 43.5 (32.42) | 42.2 (34.03) | 42.9 (33.23) |
| Median (range) | 35.0 (7.0–349.0) | 35.0 (6.0–328.0) | 35.0 (6.0–349.0) |
| Missing | 0 | 0 | 0 |
| Diagnosed with viral liver disease at randomisation | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||
| Yes | 176 (40.2) | 177 (40.2) | 353 (40.2) |
| No | 262 (59.8) | 263 (59.8) | 525 (59.8) |
As expected, the distribution of patients with and without viral disease varied across the sites (Table 107). Sites in areas where the patient population consists of a high proportion of patients with viral liver disease specialised in treating this group of patients, notably Royal Free Hospital, UCLH, Kings College Hospital, Royal Blackburn Hospital, Victoria Hospital Blackpool and Torbay General Hospital.
| Site | Liver disease, n (%) | Total, n (%) | |
|---|---|---|---|
| Viral | No viral | ||
| Royal Free Hospital | 81 (59.1) | 56 (40.9) | 137 (100.0) |
| UCLH | 85 (89.5) | 10 (10.5) | 95 (100.0) |
| Royal Liverpool University Hospital | 5 (10.2) | 44 (89.8) | 49 (100.0) |
| Royal Devon and Exeter Hospital | 14 (29.8) | 33 (70.2) | 47 (100.0) |
| Royal Blackburn Hospital | 43 (100.0) | 0 (0.0) | 43 (100.0) |
| St James’s University Hospital | 11 (26.8) | 30 (73.2) | 41 (100.0) |
| Bradford Royal Infirmary | 2 (4.9) | 39 (95.1) | 41 (100.0) |
| Singleton Hospital | 22 (57.9) | 16 (42.1) | 38 (100.0) |
| Royal Bournemouth Hospital | 6 (18.2) | 27 (81.8) | 33 (100.0) |
| Queen Alexandra Hospital | 4 (13.3) | 26 (86.7) | 30 (100.0) |
| Basingstoke and North Hampshire Hospital | 0 (0.0) | 29 (100.0) | 29 (100.0) |
| Southampton General Hospital | 2 (7.7) | 24 (92.3) | 26 (100.0) |
| Royal London Hospital | 11 (42.3) | 15 (57.7) | 26 (100.0) |
| James Cook University Hospital | 0 (0.0) | 21 (100.0) | 21 (100.0) |
| Nottingham Queen’s Medical Centre | 7 (33.3) | 14 (66.7) | 21 (100.0) |
| University Hospital Lewisham | 7 (33.3) | 14 (66.7) | 21 (100.0) |
| King’s College Hospital | 20 (100.0) | 0 (0.0) | 20 (100.0) |
| Torbay District General Hospital | 10 (58.8) | 7 (41.2) | 17 (100.0) |
| Derriford Hospital | 0 (0.0) | 16 (100.0) | 16 (100.0) |
| Royal Hallamshire Hospital | 4 (26.7) | 11 (73.3) | 15 (100.0) |
| Princess Alexandra Hospital | 0 (0.0) | 15 (100.0) | 15 (100.0) |
| Royal Albert Edward Infirmary | 0 (0.0) | 14 (100.0) | 14 (100.0) |
| Chelsea and Westminster Hospital | 2 (15.4) | 11 (84.6) | 13 (100.0) |
| Totals for other sitesa that randomised < 10 patients per site | 17 (24.3) | 53 (75.7) | 70 (100.0) |
Protocol violators
Protocol violators in the ELUCIDATE trial include patients who scored < 8.4 in any post-randomisation ELF test and patients who were randomised later than 12 weeks after registration.
Enhanced Liver Fibrosis test value of < 8.4 at follow-up
In total, 55 (25.5%) out of 216 patients with at least one reported ELF test after randomisation in the ELF arm and one (7.1%) out of 14 in the control arm had an ELF value of < 8.4 (Table 108). The proportion of patients who had an ELF value lower than this threshold value was largest (28.6%, 28/98) among patients with viral liver disease as the primary diagnosis (Table 109). Moreover, within this group of patients, the proportion of patients with an ELF value of < 8.4 was largest in those patients treated for hepatitis B or hepatitis C: 40.0% (8/20) and 42.3% (11/26), respectively, compared with 17.3% (9/52) in patients who had not received any treatment since randomisation (Table 110). Response to treatment might, therefore, play a role in the decrease in ELF values. This is also possibly the case for patients with non-alcoholic fatty liver disease. Here, weight loss might be the reason for a decrease in ELF values; however, we have not collected data on weight loss to explore this hypothesis.
| ELF value of < 8.4 at follow-up | Trial arm, n (%) | Total (N = 230), n (%) | |
|---|---|---|---|
| ELF (N = 216) | Standard care (N = 14) | ||
| Yes | 55 (25.5) | 1 (7.1) | 56 (24.3) |
| No | 160 (74.1) | 13 (92.9) | 173 (75.2) |
| Missing | 1 (0.5) | 0 (0.0) | 1 (0.4) |
| ELF value of < 8.4 at follow-up | Primary diagnosis, n (%) | Total (N = 230), n (%) | |||
|---|---|---|---|---|---|
| Alcoholic liver disease (N = 16) | Viral liver disease (N = 98) | Other/unknown (N = 62) | Non-alcoholic fatty liver disease (N = 54) | ||
| Yes | 1 (6.3) | 28 (28.6) | 13 (21.0) | 14 (25.9) | 56 (24.3) |
| No | 15 (93.8) | 70 (71.4) | 48 (77.4) | 40 (74.1) | 173 (75.2) |
| Missing | 0 (0.0) | 0 (0.0) | 1 (1.6) | 0 (0.0) | 1 (0.4) |
| ELF value of < 8.4 at follow-up | Treatment, n (%) | Total (N = 98), n (%) | ||
|---|---|---|---|---|
| Hepatitis B (N = 20) | Hepatitis C (N = 26) | No hepatitis (N = 52) | ||
| Yes | 8 (40.0) | 11 (42.3) | 9 (17.3) | 28 (28.6) |
| No | 12 (60.0) | 15 (57.7) | 43 (82.7) | 70 (71.4) |
More than 12 weeks between registration and randomisation
In total, 44 (10.0%) out of 438 patients in the ELF arm and 26 (5.9%) out of 440 patients in the control arm were not randomised within 12 weeks of registration (Table 111). For some of these patients, the ELF test was repeated before randomisation. Only 15 (3.4%) out of 438 patients in the ELF arm and 8 (1.8%) out of 440 patients in the standard arm were not randomised within 12 weeks of the (repeated) registration ELF test.
| Randomisation point | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||
| Randomisation within 12 weeks of registration | |||
| Yes | 394 (90.0) | 414 (94.1) | 808 (92.0) |
| No | 44 (10.0) | 26 (5.9) | 70 (8.0) |
| Randomisation within 12 weeks of registration ELF test | |||
| Yes | 421 (96.1) | 432 (98.2) | 853 (97.2) |
| No | 15 (3.4) | 8 (1.8) | 23 (2.6) |
| Missing | 2 (0.5) | 0 (0.0) | 2 (0.2) |
Visit compliance
Pre- and post-cirrhotic follow-up visit compliance in the ELUCIDATE trial was generally poor in both the ELF arm and the standard care arm.
Compliance with pre-cirrhotic follow-up visits
According to the protocol, patients in both the ELF arm and the standard care arm should have been seen every 6 months. If compliance is defined as being compliant with all visits, allowing visits to take place between 5 and 7 months after the previous visit, 67 (29.8%) out of 225 patients with expected pre-cirrhotic follow-up visits in the ELF arm and 81 (19.7%) out of 411 patients with expected pre-cirrhotic follow-up visits in the standard care arm were compliant (Table 112). The denominator in the ELF arm is substantially smaller than that in the standard care arm because many patients in the ELF arm were diagnosed with cirrhosis at randomisation and, thus, did not have any pre-cirrhotic follow-up visits.
| Compliance status | Trial arm, n (%) | Total (N = 636), n (%) | |
|---|---|---|---|
| ELF (N = 225) | Standard care (N = 411) | ||
| Compliant | 67 (29.8) | 81 (19.7) | 148 (23.3) |
| Non-compliant | 158 (70.2) | 330 (80.3) | 488 (76.7) |
Timing of pre-cirrhotic follow-up visits
A total of 597 pre-cirrhotic follow-up visits took place in the ELF arm, with on average 7.4 (SD 3.42) months between visits. In the standard care arm, there were 1424 reported pre-cirrhotic follow-up visits, with on average 7.0 (SD 2.89) months between visits (Table 113 and Figure 68).
| Time between follow-up visits (months) | Trial arm | Total (n = 2021) | |
|---|---|---|---|
| ELF (n = 597) | Standard care (n = 1424) | ||
| Mean (SD) | 7.4 (3.42) | 7.0 (2.89) | 7.1 (3.06) |
| Median (range) | 6.2 (1.8–33.2) | 6.2 (1.0–31.0) | 6.2 (1.0–33.2) |
| Missing | 0 | 0 | 0 |
FIGURE 68.
Time between pre-cirrhotic follow-up visits (for visits that were expected to take place 6 months after the previous visit).

Compliance with post-cirrhotic follow-up visits
According to the study protocol, all patients diagnosed as cirrhotic either by ELF testing or clinical means should attend an initial post-cirrhotic follow-up visit, at 3 months after the diagnosis of cirrhosis. All subsequent follow-up visits for the purposes of data collection should take place every 6 months. If compliance is defined as being compliant with all visits, 43 (16.2%) out of 266 patients with expected post-cirrhotic follow-up visits in the ELF arm and 4 (21.1%) out of 19 patients with expected post-cirrhotic follow-up visits in the standard care arm were compliant (Table 114). The denominator in the standard care arm is substantially smaller than that in the ELF arm because only a small number of patients in the standard care arm were diagnosed with cirrhosis.
| Compliance status | Trial arm, n (%) | Total (N = 285), n (%) | |
|---|---|---|---|
| ELF (N = 266) | Standard care (N = 19) | ||
| Compliant | 43 (16.2) | 4 (21.1) | 47 (16.5) |
| Non-compliant | 223 (83.8) | 15 (78.9) | 238 (83.5) |
Timing of post-cirrhotic follow-up visits
Looking only at post-cirrhotic visits that were expected to take place 6 months after the previous visit, the time between visits was on average 6.5 (SD 2.80) months in the ELF arm (n = 695). In the standard care arm the time between visits was on average 7.2 (SD 4.71) months (n = 35) (Table 115 and Figure 69).
| Time between follow-up visits (months) | Trial arm | Total (n = 729) | |
|---|---|---|---|
| ELF (n = 694) | Standard care (n = 35) | ||
| Mean (SD) | 6.5 (2.80) | 7.2 (4.71) | 6.5 (2.92) |
| Median (range) | 6.1 (0.7–23.4) | 5.9 (2.3–25.2) | 6.1 (0.7–25.2) |
| IQR | 5.2, 7.2 | 4.4, 7.4 | 5.2, 7.2 |
| Missing | 0 | 0 | 0 |
FIGURE 69.
Time between post-cirrhotic follow-up visits (for visits that were expected to take place 6 months after the previous visit).

Enhanced Liver Fibrosis test compliance
Patients in the ELF arm should have had an ELF test at every pre-cirrhosis follow-up visit. Patients in the standard care arm should have had one ELF test after diagnosis of cirrhosis.
Enhanced Liver Fibrosis test compliance in the Enhanced Liver Fibrosis arm
Compliance to ELF tests in the ELF arm was 72%, with compliance being defined as having an ELF test at every recorded pre-cirrhosis follow-up visit (Table 116). Note that the denominator includes only ELF arm patients who were expected to have at least one pre-cirrhotic follow-up visit and, for instance, does not include those patients who were diagnosed with cirrhosis at randomisation.
| Compliance status | Total (N = 225), n (%) |
|---|---|
| Compliant | 162 (72.0) |
| Non-compliant | 63 (28.0) |
Most ELF tests were not carried out because of administrative errors (n = 31) or lack of staff (n = 27) and most cases of non-compliance occurred at a small number of sites.
Enhanced Liver Fibrosis test compliance in the standard care arm
According to the protocol, patients in the standard care arm should have one ELF test after the confirmed clinical diagnosis of cirrhosis. In total, 12 out of 20 patients (60%) in the standard care arm who were expected to be tested were compliant with the ELF test; for one patient there was not enough information available to assess compliance (Table 117).
| Compliance status | Total (N = 20), n (%) |
|---|---|
| Compliant | 12 (60.0) |
| Non-compliant | 7 (35.0) |
| Insufficient information | 1 (5.0) |
Compliance with cirrhosis management
Compliance with the timing of AFP tests, scans (ultrasound, CT or MRI) and endoscopies (OGD) was assessed for patients who were diagnosed with cirrhosis.
Alpha-fetoprotein tests
Test compliance
According to the protocol, the timing of the initial AFP test following a diagnosis of cirrhosis depended on whether or not an AFP test was carried out in the 6 months prior to the diagnosis. If a patient had had an AFP test within 6 months of being diagnosed as cirrhotic, the next AFP measurement should be performed within 6 months of the previous test unless clinically indicated sooner. If the patient had not had an AFP test within 6 months of being diagnosed as cirrhotic, the next AFP measurement should be performed within 3 months of the diagnosis of cirrhosis. After the initial AFP test, the test should be repeated every 6 months. AFP test compliance was low in both the ELF arm and the standard care arm, at 12.6% and 11.8%, respectively (Table 118). Compliance was defined as being compliant with all visits so far and 1-month tolerance was applied.
| Compliance status | Trial arm, n (%) | Total (N = 248), n (%) | |
|---|---|---|---|
| ELF (N = 231) | Standard care (N = 17) | ||
| Compliant | 29 (12.6) | 2 (11.8) | 31 (12.5) |
| Non-compliant | 202 (87.4) | 15 (88.2) | 217 (87.5) |
Test timings
For all AFP measurements that were due to take place 6 months after the previous measurement, the median time between measurements was 6.0 (range 1.8–26.8) months in the ELF arm and 6.2 (range 3.0–25.3) months in the standard care arm (Table 119 and Figure 70).
| Time (months) | Trial arm | Total (n = 487) | |
|---|---|---|---|
| ELF (n = 465) | Standard care (n = 22) | ||
| Mean (SD) | 6.7 (3.56) | 7.9 (5.06) | 6.8 (3.64) |
| Median (range) | 6.0 (1.8–26.8) | 6.2 (3.0–25.3) | 6.0 (1.8–26.8) |
| IQR | 4.4, 7.4 | 5.1, 8.5 | 4.4, 7.6 |
| Missing | 0 | 0 | 0 |
FIGURE 70.
Overall AFP test timings (for all AFP measurements due to take place 6 months after the previous one).
Scans (ultrasound, computed tomography and magnetic resonance imaging)
If a cirrhotic patient had received a scan within 6 months of being diagnosed as cirrhotic, the next scan should have been performed within 6 months of the previous scan, unless clinically indicated sooner. If the patient had not had a scan within 6 months of being diagnosed as cirrhotic, he or she should have received a scan as screening for HCC within 3 months of the diagnosis of cirrhosis. Subsequent scans were to be repeated every 6 months from the timing of the previous scan.
Scan compliance
Compliance with scans was low in both the ELF arm and the standard care arm, at 29.5% and 14.3%, respectively (Table 120).
| Compliance status | ELF (N = 207), n (%) | Standard care (N = 14), n (%) | Total (N = 221), n (%) |
|---|---|---|---|
| Compliant | 61 (29.5) | 2 (14.3) | 63 (28.5) |
| Non-compliant | 146 (70.5) | 12 (85.7) | 158 (71.5) |
Scan timings
For all scans that were due to take place 6 months after the previous scan, the median time between measurements was 6.8 (range 0.0–30.0) months in the ELF arm and 7.5 (range 1.3–11.7) months in the standard care arm (Table 121 and Figure 71).
| Time (months) | Trial arm | Total (n = 267) | |
|---|---|---|---|
| ELF (n = 249) | Standard care (n = 18) | ||
| Mean (SD) | 7.9 (3.90) | 7.4 (2.65) | 7.8 (3.83) |
| Median (range) | 6.8 (0.0–30.0) | 7.5 (1.3–11.7) | 6.8 (0.0–30.0) |
| IQR | 5.7, 9.2 | 6.0, 9.1 | 5.7, 9.2 |
| Missing | 0 | 0 | 0 |
FIGURE 71.
Overall scan timings (for all scans due to take place 6 months after the previous one).

Endoscopies
Oesophagogastroduodenoscopy compliance
According to the protocol, all patients with a diagnosis of cirrhosis were to have an OGD as screening for varices within 3 months of diagnosis, unless they had had an OGD in the 18 months prior to the diagnosis of cirrhosis.
Compliance with the timing of endoscopies was fair in both the ELF arm and the standard care arm, at 54.9% and 50.0%, respectively (Table 122).
| Compliance status | Trial arm, n (%) | Total (N = 165), n (%) | |
|---|---|---|---|
| ELF (N = 153) | Standard care (N = 12) | ||
| Compliant | 84 (54.9) | 6 (50.0) | 90 (54.5) |
| Non-compliant | 69 (45.1) | 6 (50.0) | 75 (45.5) |
Withdrawals
In total, 32 patients in the ELF arm and 24 patients in the standard care arm withdrew from different aspects of the trial. Twenty-one patients in the ELF arm withdrew consent for further ELF testing. Of these 21 patients, 10 did not want to attend or were unable to attend for testing (with two specifically mentioning distance), five moved location, two did not want the ELF test [in one case because of difficulties with bleeding the patient and one case in which the patient mentioned that the ELF test indicated cirrhosis but a FibroScan (EchoSens, Paris, France) indicated that they were not cirrhotic and so they did not want more ELF tests] and four gave no reason for withdrawing consent. It can be seen that only two patients specifically mentioned issues with the ELF test itself. An additional 51 patients (28 in the ELF arm and 23 in the standard care arm) were no longer willing to be followed up as per the protocol schedule; 14 of these patients (ELF arm, n = 9; standard care arm, n = 5) were willing for further data to be collected at their standard visits, if available, and 27 patients (ELF arm, n = 16; standard care arm, n = 11) were willing to have their long-term data collected via a patient registry.
Disease progression to cirrhosis
In total, 281 (64.2%) out of 438 patients in the ELF arm and 20 (4.5%) out of 440 patients in the standard care arm were diagnosed with cirrhosis (Table 123). A total of 197 (70.1%) ELF arm patients were diagnosed at randomisation but none of the patients in the standard care arm was diagnosed at randomisation (Table 124). Overall, almost all ELF arm patients were diagnosed with an ELF test (99.6%), whereas the most frequent method of diagnosis in the standard care arm was a FibroScan (45.0%) (Table 125). Tables 126–128 provide the method of diagnosis of cirrhosis for patients who were not diagnosed at randomisation, the first method of diagnosis of cirrhosis for patients overall and the first method of diagnosis of cirrhosis for patients who were not diagnosed at randomisation, respectively.
| Diagnosis of cirrhosis during the trial | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||
| Yes | 281 (64.2) | 20 (4.5) | 301 (34.3) |
| No | 157 (35.8) | 420 (95.5) | 577 (65.7) |
| Diagnosis of cirrhosis after randomisation | Trial arm, n (%) | Total (N = 301), n (%) | |
|---|---|---|---|
| ELF (N = 281) | Standard care (N = 20) | ||
| Yes | 84 (29.9) | 20 (100.0) | 104 (34.6) |
| No | 197 (70.1) | 0 (0.0) | 197 (65.4) |
| Method of diagnosis | Trial arm, n (%) | Total (N = 301), n (%) | |
|---|---|---|---|
| ELF (N = 281) | Standard care (N = 20) | ||
| Liver biopsy | 1 (0.4) | 2 (10.0) | 3 (1.0) |
| Ultrasound scan | 2 (0.7) | 7 (35.0) | 9 (3.0) |
| Liver CT scan | 0 (0.0) | 3 (15.0) | 3 (1.0) |
| MRI scan | 1 (0.4) | 0 (0.0) | 1 (0.3) |
| Gastroscopy | 1 (0.4) | 7 (35.0) | 8 (2.7) |
| FibroScan | 2 (0.7) | 9 (45.0) | 11 (3.7) |
| Clinical judgement | 2 (0.7) | 6 (30.0) | 8 (2.7) |
| ELF test | 280 (99.6) | 0 (0.0) | 280 (93.0) |
| Method of diagnosis | Trial arm, n (%) | Total (N = 104), n (%) | |
|---|---|---|---|
| ELF (N = 84) | Standard care (N = 20) | ||
| Liver biopsy | 1 (1.2) | 2 (10.0) | 3 (2.9) |
| Ultrasound | 1 (1.2) | 7 (35.0) | 8 (7.7) |
| Liver CT scan | 0 (0.0) | 3 (15.0) | 3 (2.9) |
| Gastroscopy | 0 (0.0) | 7 (35.0) | 7 (6.7) |
| FibroScan | 1 (1.2) | 9 (45.0) | 10 (9.6) |
| Clinical judgement | 2 (2.4) | 6 (30.0) | 8 (7.7) |
| ELF test | 83 (98.8) | 0 (0.0) | 83 (79.8) |
| Method of diagnosis | Trial arm, n (%) | Total (N = 301), n (%) | |
|---|---|---|---|
| ELF (N = 281) | Standard care (N = 20) | ||
| Liver biopsy | 1 (0.4) | 2 (10.0) | 3 (1.0) |
| Ultrasound scan | 0 (0.0) | 6 (30.0) | 6 (2.0) |
| Liver CT scan | 0 (0.0) | 2 (10.0) | 2 (0.7) |
| Gastroscopy | 0 (0.0) | 3 (15.0) | 3 (1.0) |
| FibroScan | 1 (0.4) | 6 (30.0) | 7 (2.3) |
| Clinical judgement | 1 (0.4) | 1 (5.0) | 2 (0.7) |
| ELF test | 278 (98.9) | 0 (0.0) | 278 (92.4) |
| Method of diagnosis | Trial arm, n (%) | Total (N = 104), n (%) | |
|---|---|---|---|
| ELF (N = 84) | Standard care (N = 20) | ||
| Liver biopsy | 1 (1.2) | 2 (10.0) | 3 (2.9) |
| Ultrasound scan | 0 (0.0) | 6 (30.0) | 6 (5.8) |
| Liver CT scan | 0 (0.0) | 2 (10.0) | 2 (1.9) |
| Gastroscopy | 0 (0.0) | 3 (15.0) | 3 (2.9) |
| FibroScan | 1 (1.2) | 6 (30.0) | 7 (6.7) |
| Clinical judgement | 1 (1.2) | 1 (5.0) | 2 (1.9) |
| ELF test | 81 (96.4) | 0 (0.0) | 81 (77.9) |
Process of care outcomes
In this section the frequency of biopsy, AFP testing, imaging and endoscopy visits is summarised by treatment arm for both non-cirrhotic and cirrhotic patients. The difference in the proportion of patients undergoing/receiving each process end point between the treatment groups is compared using logistic regression, adjusting for the stratification factors. When the process outcome relates to the numbers of tests being performed (e.g. ultrasound, AFP), the two arms are compared using the Mann–Whitney non-parametric test.
Frequency of biopsies
In total, 12.1% of patients in the ELF arm had at least one biopsy post randomisation compared with 9.1% of patients in the standard care arm (Table 129). The odds ratio is 1.405, with higher odds for patients in the ELF arm to have a biopsy than patients in the standard care arm, but the 95% CI around this odds ratio (0.884 to 2.233) includes the critical value of 1 (equal odds) and the difference between the ELF arm and the standard care arm is, therefore, not statistically significant.
| Has the patient had at least one biopsy during the trial? | Trial arm, n (%) | Total (N = 878) | Odds ratioa | 95% CI | |
|---|---|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||||
| Yes | 53 (12.1) | 40 (9.1) | 93 (10.6) | 1.405 | 0.884 to 2.233 |
| No | 385 (87.9) | 400 (90.9) | 785 (89.4) | ||
Frequency of alpha-fetoprotein tests
Table 130 shows the frequency of AFP measurements post randomisation by arm. AFP measurements were more frequent in patients in the ELF arm, which can also be seen in the histogram in Figure 72, where the mass of the distribution of the number of AFP tests per randomised patient in the ELF arm is located to the right of the distribution in the standard care arm. The median number of AFP measurements was 2 (range 0–7) in the ELF arm compared with 1 (range 0–5) in the standard care arm; the difference between the two distributions was statistically significant, with a p-value of < 0.001 (Table 131). AFP measurements are a process of care in cirrhotic patients and this difference suggests that patients in the ELF arm not only were more likely to be diagnosed with cirrhosis (see Disease progression to cirrhosis) but also were more likely to receive the processes of care to monitor cirrhotic patients for major complications.
| Number of AFP tests | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||
| 0 | 89 (20.3) | 130 (29.5) | 219 (24.9) |
| 1 | 66 (15.1) | 92 (20.9) | 158 (18.0) |
| 2 | 68 (15.5) | 80 (18.2) | 148 (16.9) |
| 3 | 89 (20.3) | 67 (15.2) | 156 (17.8) |
| 4 | 67 (15.3) | 50 (11.4) | 117 (13.3) |
| 5 | 37 (8.4) | 21 (4.8) | 58 (6.6) |
| 6 | 16 (3.7) | 0 (0.0) | 16 (1.8) |
| 7 | 6 (1.4) | 0 (0.0) | 6 (0.7) |
FIGURE 72.
Number of AFP measurements per randomised patient by arm.
| Number of AFP tests | Trial arm | Total (N = 878) | p-value, Wilcoxon test (two-sided) | |
|---|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | |||
| Mean (SD) | 2.4 (1.82) | 1.7 (1.53) | 2.1 (1.72) | |
| Median (range) | 2.0 (0.0–7.0) | 1.0 (0.0–5.0) | 2.0 (0.0–7.0) | < 0.001 |
| Missing | 0 | 0 | 0 | |
The timing of post-diagnosis AFP testing followed the same pattern in the ELF arm as in the standard care arm (Figure 73). Moreover, before the diagnosis of cirrhosis, the average number of AFP tests per year was very similar in both arms, at 0.68 in the ELF arm and 0.56 in the standard care arm (Table 132). After the diagnosis of cirrhosis, this number was 0.90 in the ELF arm and 0.95 in the standard care arm (see Table 132). This is further evidence for the causal link between the diagnosis of cirrhosis and the higher frequency of AFP testing, independent of treatment allocation. Although we have seen in Disease progression to cirrhosis that participants in the ELF arm are more likely to be diagnosed with cirrhosis, we can see here that this diagnosis will also lead to a higher frequency of AFP testing in the ELF arm.
FIGURE 73.
Number of AFP measurements post diagnosis per 6-month period.
| Trial arm | Total number of AFP tests | Total years of follow-up | Average number of AFP tests per year of follow-up | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Any time | Before diagnosis | After diagnosis | Any time | Before diagnosis | After diagnosis | Any time | Before diagnosis | After diagnosis | |
| ELF | 1060 | 383 | 677 | 1322.19 | 567.28 | 754.91 | 0.80 | 0.68 | 0.90 |
| Standard care | 758 | 721 | 37 | 1326.63 | 1287.56 | 39.07 | 0.57 | 0.56 | 0.95 |
Frequency of ultrasound scans
Table 133 shows the frequency of ultrasound scans post randomisation by arm. Ultrasound scans were performed more frequently in patients in the ELF arm, which can also be seen in the histogram in Figure 74, where the mass of the distribution of the number of scans per randomised patient in the ELF arm is located to the right of the distribution in the standard care arm. The median number of ultrasound scans was 1 (range 0–6) in the ELF arm compared with 0 (range 0–5) in the standard care arm; the difference between the two distributions was statistically significant, with a p-value of < 0.001 (Table 134).
| Number of ultrasound scans | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||
| 0 | 151 (34.5) | 233 (53.0) | 384 (43.7) |
| 1 | 98 (22.4) | 111 (25.2) | 209 (23.8) |
| 2 | 77 (17.6) | 55 (12.5) | 132 (15.0) |
| 3 | 53 (12.1) | 30 (6.8) | 83 (9.5) |
| 4 | 40 (9.1) | 8 (1.8) | 48 (5.5) |
| 5 | 16 (3.7) | 3 (0.7) | 19 (2.2) |
| 6 | 3 (0.7) | 0 (0.0) | 3 (0.3) |
FIGURE 74.
Number of ultrasound scans per randomised patient by arm.

| Number of ultrasound scans | Trial arm | Total (n = 878) | p-value, Wilcoxon test (two-sided) | |
|---|---|---|---|---|
| ELF (n = 438) | Standard care (n = 440) | |||
| Mean (SD) | 1.5 (1.52) | 0.8 (1.08) | 1.2 (1.36) | |
| Median (range) | 1.0 (0.0–6.0) | 0.0 (0.0–5.0) | 1.0 (0.0–6.0) | < 0.001 |
| Missing | 0 | 0 | 0 | |
Ultrasound scans are a process of care in cirrhotic patients and this difference demonstrates that patients in the ELF arm not only were more likely to be diagnosed with cirrhosis (see Disease progression to cirrhosis) but also were more likely to receive the processes of care of cirrhotic patients. This is a really important finding as only regular and frequent ultrasound scans can detect HCC early.
The timing of post-diagnosis ultrasound scans followed the same pattern in the ELF arm as in the standard care arm (Figure 75). Moreover, before the diagnosis of cirrhosis, the average number of ultrasound scans per year was very similar in both arms, at 0.29 in the ELF arm and 0.26 in the standard care arm (Table 135). After the diagnosis of cirrhosis, this number was 0.66 in the ELF arm and 0.74 in the standard care arm. This is further evidence for the causal link between the diagnosis of cirrhosis and the higher frequency of ultrasound scans, independent of treatment allocation. Although we have seen in Disease progression to cirrhosis that participants in the ELF arm are more likely to be diagnosed with cirrhosis, we can see here that this diagnosis will also lead to a higher frequency of ultrasound scans in both trial arms. Table 135 shows an excess of 306 scans in the ELF arm compared with the standard care arm.
FIGURE 75.
Number of ultrasound scans post diagnosis per 6-month period.
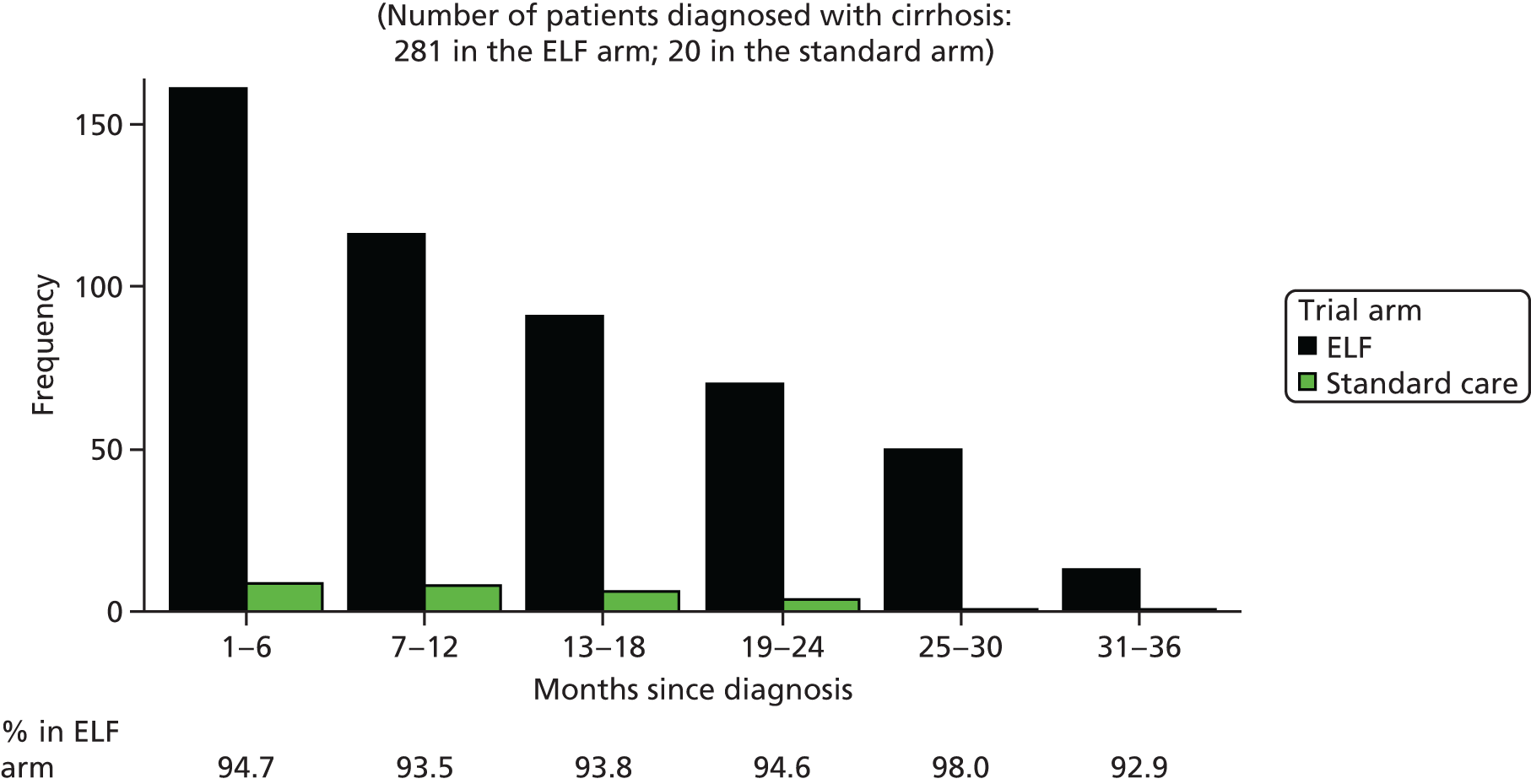
| Arm | Total number of ultrasound scans | Total years of follow-up | Average number of ultrasound scans per year of follow-up | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Any time | Before diagnosis | After diagnosis | Any time | Before diagnosis | After diagnosis | Any time | Before diagnosis | After diagnosis | |
| ELF | 664 | 163 | 501 | 1322.19 | 567.28 | 754.91 | 0.50 | 0.29 | 0.66 |
| Standard care | 358 | 329 | 29 | 1326.63 | 1287.56 | 39.07 | 0.27 | 0.26 | 0.74 |
Frequency of oesophagogastroduodenoscopy
In total, 34.9% of patients in the ELF arm had a least one endoscopy (OGD) post randomisation compared with 2.7% of patients in the standard care arm. The (adjusted) odds ratio is 83.9, with higher odds for patients in the ELF arm to have an endoscopy than patients in the standard care arm. The 95% CI around this odds ratio is 36.9 to 192.4; therefore, the difference between the ELF arm and the standard care arm is clearly significant (Table 136). This is likely to be because a larger proportion of patients are diagnosed with cirrhosis in the ELF arm than in the standard care arm and these patients have endoscopies as part of their cirrhosis management, as expected.
| Has the patient had at least one OGD? | ELF (N = 438), n (%) | Standard care (N = 440), n (%) | Total (N = 878), n (%) | Odds ratioa | 95% CI |
|---|---|---|---|---|---|
| Yes | 153 (34.9) | 12 (2.7) | 165 (18.8) | 83.894 | 36.573 to 192.443 |
| No | 285 (65.1) | 428 (97.3) | 713 (81.2) |
When looking only at the subset of patients who were diagnosed with cirrhosis, 54.4% of patients in the EFL arm had at least one OGD post diagnosis compared with 60% of patients in the standard care arm. The (adjusted) odds ratio (1.3) is still in favour of the ELF arm but the 95% CI (0.4 to 4.9) shows that this difference in odds is no longer significant (Table 137). This means that the odds of having an OGD post diagnosis of cirrhosis are equivalent, suggesting that patients received the same processes of care with regard to OGD in both arms. However, as a result, overall in the trial to date there were a total of 165 additional OGDs performed in the ELF arm, with only 14 OGDs performed in the standard care arm and 179 performed in the ELF arm (Table 138). This is probably the single largest difference in process of care tests between the arms.
| Has the patient had at least one OGD since diagnosis of cirrhosis? | ELF (N = 281), n (%) | Standard care (N = 20), n (%) | Total (N = 301), n (%) | Odds ratioa | 95% CI |
|---|---|---|---|---|---|
| Yes | 153 (54.4) | 12 (60.0) | 165 (54.8) | 1.304 | 0.350 to 4.861 |
| No | 128 (45.6) | 8 (40.0) | 136 (45.2) |
| Trial arm | Total (N = 193), n (%) |
|---|---|
| ELF | 179 (92.7) |
| Standard care | 14 (7.3) |
Frequency of beta-blocker/band ligation treatment
In total, nine (2.1%) patients in the ELF arm and seven (1.6%) in the standard care arm were diagnosed with varices (Table 139) and five (1.1%) patients in the ELF arm and three (0.7%) in the standard care arm were treated with beta-blockers or band ligation (Table 140). The adjusted odds ratio for treatment with beta-blockers or band ligation is 1.3 (95% CI 0.1 to13.8), showing that there is no significant difference in the likelihood of being treated between the ELF arm and the standard care arm.
| Has the patient developed varices? | Trial arm, n (%) | Total (N = 878), n (%) | |
|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||
| Yes | 9 (2.1) | 7 (1.6) | 16 (1.8) |
| No | 429 (97.9) | 433 (98.4) | 862 (98.2) |
| Has the patient been treated with beta-blockers or band ligation? | Trial arm, n (%) | Total (N = 878), n (%) | Odds ratioa | 95% CI | |
|---|---|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||||
| Yes | 5 (1.1) | 3 (0.7) | 8 (0.9) | 1.343 | 0.131 to 13.754 |
| No | 433 (98.9) | 437 (99.3) | 870 (99.1) | ||
Frequency of treatment to normalise liver function tests
In total, 57 (13%) patients in the ELF arm and 48 (10.9%) in the standard care arm received treatment to normalise LFTs (Table 141). The adjusted odds ratio is 1.5 (95% CI 0.9 to 2.7), suggesting that there is no statistically significant difference in the odds of receiving treatment to normalise LFTs between the two arms. However, again, the numbers are small and a few more ELF arm patients are receiving treatment to normalise LFTs, which is in the right direction, even though this difference is not significant.
| Has the patient received treatment to normalise LFTs? | Trial arm, n (%) | Total (N = 878), n (%) | Odds ratioa | 95% CI | |
|---|---|---|---|---|---|
| ELF (N = 438) | Standard care (N = 440) | ||||
| Yes | 57 (13.0) | 48 (10.9) | 105 (12.0) | 1.510 | 0.850 to 2.681 |
| No | 381 (87.0) | 392 (89.1) | 773 (88.0) | ||
Related unexpected serious adverse events
There were no RUSAEs reported in the ELUCIDATE trial.
Health economic consequences
The follow-up period for the ELUCIDATE trial has been extended to 5 years beyond the end of the NIHR programme grant, meaning that we are unable, at this stage, to access data on resources used and quality of life by arm. This prevented QALYs being estimated and the assessment of the cost-effectiveness of the ELF test in the early detection of cirrhosis.
Considering that one of the aims of the trial was to assess how the use of the ELF test affects the process of care, the aim of the health economic analysis at this stage was restricted to a descriptive analysis of the costs associated with the process of care outcomes. These are, namely, increased use of endoscopy, biopsy, ultrasound and AFP tests to detect HCC at a surgically curable stage and increased use of beta-blockers/band ligation of varices to prevent haemorrhage/HCC.
Descriptive analysis
Costs associated with process of care outcomes
For costing of the process of care outcomes we assigned a unit cost to the mean values for tests reported by several hospitals (as reported in the statistical analysis and Table 142) for the various procedures. Unit costs and their sources are presented in Table 142. Costs for endoscopy (OGD) and ultrasound scans were obtained from NHS Reference Costs 2013–2014. 890 When costs were not available from national databases they were obtained from the literature. This was the case for the AFP test and liver biopsy, whose costs were obtained from a study on antiviral therapy for mild chronic hepatitis C. 891 Finally, drug costs were obtained from the British National Formulary. 892 Costs were adjusted using 2015 prices and were discounted at 3.5%. Costs in each arm of the trial for liver biopsies, ultrasound scans, AFP testing, OGD and treatment with beta-blockers are given in Tables 143–147, respectively.
| Activity | Unit cost (£) | 2015 prices (£) | Comments | Source |
|---|---|---|---|---|
| Liver biopsy | 249.00 | 334.14 | Average across three hospitals | Wright et al.891 |
| AFP test | 6.03 | 8.09 | Average across three hospitals | Wright et al.891 |
| Ultrasound scan | 47.00 | 48.83 | – | NHS Reference Costs 2013–2014890 – diagnostic imaging |
| Endoscopy (OGD) | 406.00 | 421.80 | – | NHS Reference Costs 2013–2014890 – day case |
| Beta-blockers | ||||
| Propranolol (Inderal, AstraZeneca) | 1.45 | – | 40 mg, 28-tablet pack; dose: 40 mg twice daily | BNF892 |
| Carvedilol (Coreg, GlaxoSmithKline) | 1.26 | – | 12.5 mg, 28-tablet pack; dose: 12.5 mg once daily | BNF892 |
| Nadolol (Corgard, Pfizer) | 5.00 | – | 80 mg, 28-tablet pack; dose: 40 mg once daily | BNF892 |
| Variable | Trial arm | Total (n = 878) | |
|---|---|---|---|
| ELF (n = 438) | Standard care (n = 440) | ||
| Mean (SD) number of liver biopsies per patient | 0.1 (0.3) | 0.1 (0.3) | 0.1 (0.3) |
| Average cost (£) | 14,635 | 14,702 | 29,337 |
| Discounted cost (£) | 13,200 | 13,260 | 26,460 |
| Variable | Trial arm | Total (n = 878) | |
|---|---|---|---|
| ELF (n = 438) | Standard care (n = 440) | ||
| Mean (SD) number of ultrasound scans per patient | 1.5 (1.52) | 0.8 (1.08) | 1.2 (1.36) |
| Average cost (£) | 32,081 | 17,188 | 51,447 |
| Discounted cost (£) | 28,935 | 15,502 | 46,402 |
| Variable | Trial arm | Total (n = 878) | |
|---|---|---|---|
| ELF (n = 438) | Standard care (n = 440) | ||
| Mean (SD) number of AFP tests per patient | 2.4 (1.82) | 1.7 (1.53) | 2.1 (1.72) |
| Average cost (£) | 8504 | 6051 | 14,916 |
| Discounted cost (£) | 7670 | 5458 | 13,454 |
| Variable | Trial arm | Total (n = 878) | |
|---|---|---|---|
| ELF (n = 438) | Standard care (n = 440) | ||
| Mean (SD) number of OGDs per patient | 0.4 (0.6) | 0.03 (0.2) | 0.2 (0.5) |
| Average cost (£) | 73,899 | 5567 | 74,068 |
| Discounted cost (£) | 66,653 | 5022 | 66,805 |
| Treatment | Trial arm, 1-year cost (£) | |
|---|---|---|
| ELF (n = 5) | Standard care arm (n = 3) | |
| Propranolol | 170 | 102 |
| Carvedilol | 74 | 44 |
| Nadolol | 147 | 88 |
The statistical results showed that the average number of liver biopsies performed was very similar in the two arms of the trial, at 0.1 (SD 0.3). The discounted cost of liver biopsies was just over £13,000 in both arms.
The statistical results showed that diagnosis of cirrhosis (defined in the ELUCIDATE trial as an ELF value of ≥ 9.5) leads to a higher frequency of ultrasound scans, AFP tests and endoscopies (OGD) in both trial arms, as expected, given that these constitute processes of care in cirrhotic patients. However, ultrasound scans, AFP tests and endoscopies (OGD) are more frequent in patients in the ELF arm because they are more likely to be diagnosed with cirrhosis. This translates into a higher average cost of ultrasound scans in the ELF arm, specifically £28,935 compared with £15,503 in the standard care arm.
The same applies to AFP tests. AFP testing is a process of care in cirrhotic patients and, hence, these tests were administered more frequently in the ELF arm than on the standard care arm. On average, £7670 was spent on AFP tests in the ELF arm, whereas the average cost of AFP tests in the standard care arm was £5458.
The statistical results showed that there was a significant difference between the groups in the mean number of OGDs performed, at 0.4 (SD 0.6) in the ELF arm and 0.03 (SD 0.2) in the standard arm. This translates into a remarkable difference in cost, with £66,653 spent on endoscopies in the ELF arm compared with £5022 in the standard care arm.
It was not possible to distinguish between the use of beta-blockers or band ligation (it was recorded only if patients had been treated with either) and so we assumed that the first choice of treatment was drug therapy. This is supported by the UK guidelines on the management of variceal haemorrhage in cirrhotic patients,893 which advise towards pharmacological treatment with propranolol as first-line therapy (40 mg twice daily). Carvedilol (12.5 mg once daily) or nadolol (40 mg once daily) are suggested as alternatives to propranolol. Once initiated, the treatment continues indefinitely. In the ELF arm, five patients were treated for varices whereas in the standard care arm three patients were treated. We assumed that treatment started in the last year of follow-up and we calculated the cost for 1 year. The least expensive treatment is carvedilol, which cost £74 in the ELF arm and £44 in the standard care arm. The most expensive treatment is propranolol, with a cost of £170 in the ELF arm and £102 in the standard care arm.
Severe complications and deaths
The primary outcome of the trial is the incidence of severe complications; this will be analysed using registry (ONS/HES) data in the long-term follow-up analysis in 2021. In this report, the number of severe complications and the number of deaths are, therefore, not presented by arm.
In total, 16 (1.8%) of the randomised patients had at least one severe complication. When looking at the first identified severe complication in these 16 patients, HCC was the most common complication, with 10 cases (62.5%). Two patients had variceal haemorrhage and two had encephalopathy. Two patients with HCC and one patient with encephalopathy subsequently died of liver-related causes.
A total of 17 (1.9%) of 878 randomised patients died during follow-up. Five of these deaths (29.4%) were liver related (variceal haemorrhage, n = 1; HCC, n = 2; liver failure, n = 1; sepsis, n = 1); for one patient this information was missing.
Discussion
Although there was relatively low compliance to trial procedures, this was not unexpected in this group of patients. It should also be noted that fairly strict definitions of compliance were reported, with a tight window, usually of 1 month, in which to have the test performed; the ‘window’ graphs show that the majority of tests were still carried out, if often considerably later than would be ideal. Furthermore, in terms of looking at a series of tests, if one test at one particular time point was missed, that test would have been likely to have been carried out at the next visit, reducing the impact of the missed test.
The analysis of the process of care outcomes showed substantial differences in the care delivered between the ELF arm and the standard care arm, despite the apparent lack of compliance. Furthermore, when considering tests performed after the diagnosis of cirrhosis, which is defined by an ELF threshold value in the ELF arm and, therefore, includes vastly more patients in the ELF arm, differences in tests performed, such as AFP tests, ultrasound scans and OGDs, were very large (18 times as many AFP tests, 17 times as many ultrasound scans and 13 times as many OGDs were performed in the ELF arm) and so it seems clear that a very different treatment package was delivered in the two trial arms despite the low compliance under the definition used.
In terms of the design and outcome of the trial, the main effect of low compliance will be its potential impact on the effect size for the primary end point of occurrence of severe complications. For the intervention to be successful, it has to be successfully delivered, otherwise it cannot be expected to result in a difference in outcome. It is difficult to judge whether or not the magnitude of the low compliance would be sufficient to cause a substantial reduction in the effect size. However, the previous comments about the stringency of compliance reporting, combined with the large differences found in the use of tests such as AFP tests, ultrasound scans and OGDs, are crucial, and any reduction in effect size as a result of low compliance, as defined, would be expected to be relatively minor. We will be able to analyse effect size in relation to adherence to protocol guidelines in the final analysis, at which time it should be possible to quantify any such effect. This analysis will be included in the statistical analysis plan for analysis of the long-term follow-up for the primary end point.
The unsurprisingly higher cost associated with the ELF arm (£116,629 vs. £39,345) is the result of the additional care provided, mainly in the form of additional diagnostic investigations, as a consequence of more patients having an ELF value of ≥ 9.5 on ELF testing and, hence, being diagnosed with cirrhosis. This additional care is directed at preventing complications (in the case of beta-blocker therapy) or detecting complications early and at a curable stage. This should translate to improved survival rates and improved quality of life at the end of the extended follow-up period. The cost-effectiveness of the ELF test in the early detection of cirrhosis will ultimately depend on the impact that the additional process of care in the ELF arm has on survival rates, quality of care and health-care resource use.
The descriptive analysis has a number of limitations and assumptions. Information was not available on when the diagnostic investigations occurred during the 30-month follow-up and so we assumed that, for example, all ultrasound scans were carried out in the last year of follow-up and we discounted costs in the third year. With regard to the treatment of varices, patients can be treated surgically or with pharmacological treatment; we assumed that patients were all treated with beta-blockers, based on UK guidelines that advise the use of pharmacological treatment in first line. Finally, information on when treatment was initiated was not available; hence, we assumed that treatment starts in the last year of follow-up and we discounted costs in the third year. In addition, we did not know which particular drug was used; thus, we included the cost of both the most and the least costly treatment courses.
These data will allow us to complete the planned evaluation of the cost per QALY generated by the ELF monitoring strategy when the outcome data become available. In the meantime we will explore further modelling approaches to relate the cost of the strategy against potential improvements in outcomes that may be estimated from the changes in process of care.
Chapter 22 Workstream 3: next steps and preliminary conclusions
Summary and discussion of the results of the ELUCIDATE trial to date
At the time of compiling this report the data gathered during the conduct of the ELUCIDATE trial have been divided by randomisation arm only for the analysis of indicators of cirrhosis and changes in the process of care.
A number of observations can be made concerning two broad categories of data: those relating to the clinical aspects of the study and those relating to the conduct of the study.
Clinical aspects
Baseline demographics
Randomisation was effective in that there were no significant differences in the baseline characteristics between the two arms. The median age of participants was 55 years and there was a slight over-representation of men at 56%.
Enhanced Liver Fibrosis testing at registration
The median ELF value at registration was 9.4. Nearly 55% of those registered had an ELF value in the range 8.4–9.49, indicating that they were pre-cirrhotic and at low risk of liver-related complications of CLD (see Figure 64). Only 5.7% (n = 50) of participants had an ELF value that exceeded 11.49 and were, thus, at higher risk of liver-related complications within the next 5 years. This distribution of liver fibrosis among the participants means that only a relatively small proportion are likely to develop liver-related outcomes during the course of the trial. Longer-term follow-up will be required to capture these events and to determine if earlier detection of cirrhosis and management alters the course of disease and the incidence of morbidity and mortality.
The change to a randomisation threshold of 8.4 in March 2011 did not introduce bias but did reduce the risk of serious liver-related complications in the trial overall (see Figure 64).
Alcohol consumption
The levels of alcohol consumption in the cohort were relatively low compared with what was anticipated for patients with CLD. Only 9.3% reported moderately heavy levels of current consumption and only 3.5% reported current heavy alcohol consumption. Again, this lower than anticipated alcohol consumption is likely to reduce the incidence of liver-related events in the cohort during the trial and during subsequent follow-up. Patients may have under-reported alcohol use in both arms of the trial.
Aetiology of chronic liver disease
The distribution of aetiologies of CLD among the cohort differs from that seen in the majority of liver clinics, with alcoholic liver disease under-represented at 7.7% and viral hepatitis over-represented at 40.2%. Non-alcoholic fatty liver disease accounted for 25% of the cohort; this is similar to the rate of non-alcoholic fatty liver disease seen in most liver clinics. The course of the ELUCIDATE trial paralleled the introduction of highly effective therapies for HCV infection and more widespread treatment of HBV infection. As a consequence it is likely that patients recruited with these conditions will have been treated during the course of the trial and are likely to experience improvement in their liver fibrosis. Although this is likely to reduce the anticipated incidence of liver-related events, it will have created the opportunity to analyse the ability of ELF and other biochemical tests to monitor improvements in fibrosis consequent on control or eradication of hepatitis virus infection.
It is unfortunate that so few patients were recruited with alcoholic liver disease as their primary aetiology as these patients are at highest risk of liver-related events and are likely to be the group in which early detection of cirrhosis might be most beneficial. However, patients with alcoholic liver disease are often difficult to recruit and retain in clinical trials.
The significant representation of non-alcoholic fatty liver disease may be valuable. Fatty liver disease is increasingly recognised as an important cause of CLD and one in which the currently available therapeutic interventions (diet and exercise) have limited effect. However, new specific and general antifibrotic therapies are undergoing trials in non-alcoholic fatty liver disease and so it will be valuable to know what strategies are most effective in detecting cirrhosis in this condition.
The proportion of patients with viral hepatitis among those recruited varied considerably between sites, from 0% to 100%. This is likely to reflect the specialist interest of the local principal investigator, the representation of these patients in the local clinic population and the willingness of the investigators and patients to enrol these patients in the study.
Progression to cirrhosis
Inevitably, the proportion of patients progressing to ‘cirrhosis’, as defined by ELF testing, was greater in the ELF arm than in the standard care arm (64.2% vs. 4.5%). However, the definition of cirrhosis for each arm differed at this stage of the analysis, being defined by an ELF value of > 9.5 in the ELF arm and according to clinical criteria in the standard care arm. Patients with an ELF value of > 9.5 have a significant risk of serious complications (see Figure 64) and the investigators and steering group judged that was a sufficient justification for the initiation of measures to reduce the risk of serious complications.
In the ELF arm, 70% of the patients registered were diagnosed as cirrhotic at randomisation at the time of their first ELF test. None of the patients in the standard care arm was diagnosed as cirrhotic when they attended their randomisation visit. This difference supports the hypothesis that ELF will detect cirrhosis in patients who would not be recognised as cirrhotic using clinical criteria. However, this may merely reflect a difference in case definition or, at best, ‘lead time’ bias. Longer-term follow-up will determine what proportion of patients in each arm progressed to clinically relevant outcomes associated with cirrhosis.
Only nine patients in the ELF arm (0.4%) were diagnosed as cirrhotic using criteria other than ELF testing. The most common methods for diagnosing cirrhosis in the standard care arm were elastography (3.7%), ultrasound (3%), gastroscopy (2.7%) and clinical judgement (2.7%).
Following randomisation, 84 patients in the ELF arm and 20 patients in the standard care arm were subsequently diagnosed as cirrhotic during the course of the trial. Three patients in the ELF arm were diagnosed using clinical measures (one each of biopsy, elastography and clinical judgement). Ultrasound and elastography were the most common means of diagnosing cirrhosis in the standard care arm, with six patients diagnosed by each method.
Study conduct
Recruitment
Recruitment to the ELUCIDATE trial was slower than anticipated and the start-up time was longer than anticipated in many centres. However, 46 NHS trusts are participating in the ELUCIDATE trial, with a wide geographical distribution across the country. Many hospitals had little previous experience of recruitment into liver RCTs. The extension of the trial to additional centres ultimately allowed the recruitment of almost 90% of the target recruitment number but the delays have limited our ability to thus far analyse long-term outcomes.
Compliance
Pre-cirrhotic patients
Compliance with follow-up visit attendance was generally poor in both arms. This reflects clinical experience in liver clinics, where ‘did not attend’ rates vary between 5% and 25%. The ELUCIDATE protocol required 6-monthly attendance at clinics for pre-cirrhotic patients. It was apparent during the feasibility planning for the study that many clinics booked less frequent appointments for patients with CLD. It was decided that, rather than change the protocol, this variance in clinic practice would be captured in the conduct of the study. Thus, the low rates of compliance with the protocol were anticipated. Only 23.3% of patients complied with all planned visits and the rate of compliance was higher in the ELF arm at 29.8% than in the standard care arm at 19.7%.
Post-cirrhotic patients
Compliance with the protocol was even worse for patients following the diagnosis of cirrhosis. Again, this is likely to be because of local clinical practice deviating from national and international guidelines, as well as the ELUCIDATE protocol. Compliance with clinic visits was worse in the ELF arm at 16.2% than in the standard care arm at 21.1%. This may reflect greater concern for patients diagnosed as cirrhotic in the standard care arm, although the number of patients compliant with the protocol in the standard care arm was very small (n = 4).
Reasons for non-compliance
The most frequently reported reasons for non-compliance with clinic visits were administrative errors or lack of staff. Interestingly, the majority of these deviations occurred at UCLH and the Royal Free Hospital, where large numbers of patients were recruited, and at King’s College Hospital.
The ELUCIDATE trial provided no separate funding for research nurses to conduct the study. It was anticipated that NIHR-supported staff would recruit and consent patients to the study and this was certainly the case at the majority of sites. However, the relatively light research tasks involved in the conduct of the ELUCIDATE trial became ‘onerous’ at those centres recruiting large numbers of patients, when processing large numbers of blood samples and CRFs became time-consuming. These observations have implications for future NIHR studies that seek to rely on Portfolio adoption and access to NIHR faculty to conduct ‘non-onerous’ tasks critical to the completion of a study. Direct costing of the research component of observational studies should be considered but this will have significant impact on the cost of studies such as the ELUCIDATE trial.
Smaller centres with fewer competing studies performed particularly well and this must be recognised as a success of the NIHR goal to have more NHS trusts engaged in clinical research that is relevant to the NHS.
Compliance with cirrhosis management
Compliance with AFP testing was low in both arms, at 12.6% and 11.8% in the ELF and standard care arms, respectively.
Process of care outcomes
Frequency of biopsies
There was no significant difference in the number or proportion of patients undergoing liver biopsy between the different arms of the study. This suggests that clinicians did not regard an ELF-based diagnosis of cirrhosis as an indication for a liver biopsy.
Alpha-fetoprotein testing
A diagnosis of cirrhosis should be followed by monitoring for the development of HCC. Guidelines recommend the use of AFP measurement and ultrasound scanning every 6 months and this practice was incorporated into the ELUCIDATE trial protocol. Measurement of AFP was more frequent in the ELF arm than in the standard care arm, confirming that the investigators adhered to the protocol and conducted appropriate tests following the diagnosis of cirrhosis by ELF testing. Once cirrhosis was diagnosed the adherence to protocol was the same in both arms.
This suggests that if ELF testing correctly defines cirrhosis then clinicians will test for AFP as a screening test for HCC. This suggests that cancers may be diagnosed more frequently and earlier through the use of biochemical testing for cirrhosis and raises the possibility that more HCCs could be cured and that the outcomes in HCC could be improved. It remains to be determined whether or not this strategy will improve survival from HCC.
Ultrasound scans
More ultrasound scans were performed in the ELF arm than in the standard care arm confirming that the investigators adhered to the protocol and conducted appropriate tests following the diagnosis of cirrhosis by ELF testing. Once cirrhosis was diagnosed the adherence to protocol was the same in both arms.
This suggests that if ELF testing correctly defines cirrhosis then clinicians will screen for HCC appropriately. This suggests that cancers may be diagnosed more frequently and earlier through the use of biochemical testing for cirrhosis and raises the possibility that more HCCs could be cured and that the outcomes in HCC could be improved. It remains to be determined whether or not this strategy will improve survival from HCC.
Oesophagogastroduodenoscopy
The overall frequency of OGD was 34.9% in the ELF arm and 2.7% in the standard care arm. Following the diagnosis of cirrhosis the frequency of OGD was similar in both arms, with 54.4% of ELF arm cirrhotic patients and 60% of standard care arm cirrhotic patients undergoing endoscopy. This is a critical investigation for diagnosing treatable varices and so is an important step in reducing the incidence of life-threatening complications of cirrhosis. As more patients underwent OGD for a cirrhotic indication in the ELF arm (and RCT evidence has shown that treatment of varices reduces morbidity and mortality in CLD), the greater use of OGD in the ELF arm may translate into an overall benefit in terms of a reduction in morbidity and mortality in the ELF arm over time.
Diagnosis of varices
The number of patients deemed to have developed varices was greater in the ELF arm than in the standard care arm (n = 9 or 2.1% vs. n = 7 or 1.6%), but this difference was not statistically significant.
Of the patients diagnosed as cirrhotic who had an OGD to detect varices, 9 out of 153 (5.9%) in the ELF arm and 7 out of 12 (58.3%) in the standard care arm were found to have varices. At this stage of the analysis it is not possible to determine the diagnostic accuracy of either strategy but the number of cases detected was similar in each arm, suggesting that either strategy is equally effective at detecting varices.
Six of the 20 cases of cirrhosis in the standard care arm were detected using FibroScan. As an alternative method of non-invasive testing for fibrosis with similar effectiveness to ELF testing, the use of FibroScan in the standard care arm will have enriched for cirrhosis in this arm, reducing the measured effectiveness of the intervention (ELF testing). Access to FibroScan is limited in the UK and so once unblinding has occurred it will be possible to discern the impact of FibroScan on the detection of cirrhosis and treatment in the standard care arm.
Beta-blocker prescription or band ligation
The number of patients diagnosed and treated for portal hypertension and oesophageal varices was greater in the ELF arm (9 diagnosed, 5 treated) than in the control arm (7 diagnosed, 3 treated). Of the cases of diagnosed varices, five out of nine in the ELF arm and three out of seven in the standard care arm began treatment. The difference in use of beta-blockers and that of band ligation were not statistically significantly different. This suggests that the instigation of life-saving treatment was no greater in the ELF arm and suggests that the study is unlikely to provide evidence of benefit from the early diagnosis of cirrhosis. However, the numbers reaching this end point to date are small and, at this stage, the impact of the use of FibroScan in the standard care arm cannot be determined.
Treatment
A similar number of patients in each arm underwent treatment to normalise their liver function during the course of the ELUCIDATE trial, with 13% of ELF arm patients and 10.9% of standard care arm patients receiving treatment. This suggests that eradication or control of viral hepatitis is unlikely to account for the lower than anticipated incidence of complications of cirrhosis during the trial study period.
The trial outcomes
The ELUCIDATE trial was designed to test the hypothesis that earlier diagnosis of cirrhosis in patients with CLD through the use of serum testing would permit earlier and more widespread targeted screening for oesophageal varices and HCC linked to treatment and that this would result in reductions in morbidity and mortality.
More patients in the ELF arm than in the standard care arm were diagnosed with cirrhosis and then underwent screening for oesophageal varices and HCC. The excess of cases of cirrhosis in the ELF arm was anticipated as the criterion for diagnosing cirrhosis in the ELF arm was a universally applied biochemical threshold of an ELF value of > 9.5 whereas diagnosis of cirrhosis in the standard care arm relied on clinical recognition. Whether or not this early diagnosis proves to be of long-term clinical benefit remains to be determined. It is inevitable that earlier diagnosis will incur greater costs attributable to investigation and preventative treatment of complications. However, these costs may be exceeded by the costs of managing decompensated liver disease presenting de novo in patients who have been diagnosed with cirrhosis later.
The rate of screening among cirrhotic cases detected in each arm was similar, suggesting that the investigators adhered to the trial protocol and complied with guidelines for the management of cirrhosis equally for cirrhotic patients in each arm in accordance with the hypothesis underpinning the trial.
The number of cases of oesophageal varices diagnosed and treated was similar in each arm and, thus, the proportion of cirrhotic patients in each arm with varices and started on treatment was far higher in the standard care arm. This suggests that standard care may be more efficient and equally as effective as the use of ELF testing to detect and treat varices, although at present we do not know if cases of varices were missed in the standard care arm.
Six of the 20 cases of cirrhosis in the standard care arm were diagnosed following the use of FibroScan, a non-invasive test for cirrhosis that performs as effectively as ELF testing. Because the randomisation blinding has not been broken, we do not know at this stage what proportion of patients with varices in the standard care arm was diagnosed using FibroScan, but it is clear that one-third of the cirrhotic cases in the standard care arm were detected using this non-invasive test.
FibroScan is a technology that emerged into use in the NHS during the course of the ELUCIDATE trial. It was not CE marked when the trial was initiated and access to the technology remains limited within NHS trusts. The authors of the trial did not include FibroScan as part of the intervention arm of the trial because of the lack of regulatory approval at the start of the trial, limited access to the technology and the lack of consensus on thresholds for the diagnosis of cirrhosis. However, during the course of the study FibroScan has entered more widespread, but not universal, use. Although the use of an alternative novel non-invasive test for fibrosis may be regarded as ‘contaminating’ the control arm of the trial, the Chief Investigators took a pragmatic decision to not exclude the use of FibroScan but to record its use by investigators in both arms of the trial. It may be that the use of FibroScan in the standard care arm will have eliminated the intended difference between the arms and, hence, the power of the study to detect any benefit of the early detection of cirrhosis. Once the trial is unblinded it may be possible to determine the proportion of cases of cirrhosis diagnosed and managed in the standard care arm using the non-invasive test FibroScan as an alternative to ELF testing.
Another explanation for the high pick-up rate for varices among the cirrhotic patients in the standard care arm is a ‘Hawthorne effect’. It is likely that investigators were more vigilant about the onset of cirrhosis in patients in the standard care arm than for patients outside the trial. If one of the benefits of conducting the ELUCIDATE trial in a large number of NHS trusts is diffusion of best practice, then this should be seen as a benefit of conducting research in clinical services. This possibility could be explored through qualitative research conducted with the principal investigators.
If the use of ELF testing to detect cirrhosis is beneficial then those patients most likely to benefit will be those who are asymptomatic and have few clinical signs of cirrhosis. These patients are unlikely to be diagnosed in the standard care arm and are likely to be over-represented among the patients presenting for the first time with life-threatening variceal haemorrhage and inoperable HCC. It remains to be seen if complications will be detected and managed more effectively in this group of patients in the ELF arm compared with the standard care arm but this should be discernible with longer follow-up of the two arms once randomisation is unblinded.
The trial process
The trial process was ultimately successful in approaching the final target, but with important delays. The randomisation process was effective in that there were no significant differences between the patients recruited to the two arms of the trial. However, the recruitment process was slower than anticipated because of a number of problems with the regulatory processes, enforced changes to the protocol and problems at the participating sites. Much has been learned through the conduct of the study and some of the issues over regulatory processes have been addressed by the NIHR and will be addressed by the Health Research Authority. It is apparent that some of the smaller centres made proportionally greater contributions to recruitment than some of the larger centres. It was clear that site-specific visits by the Co-principal Investigator made a significant difference to recruitment.
The spectrum of aetiologies of CLDs among the participants was similar to that seen in the outpatient departments of specialist centres, but differs from the case mix presenting to accident and emergency departments and admitted to hospital wards. Thus, compared with hospital inpatients, the cohort under-represented alcoholic liver disease and over-represented viral hepatitis and non-alcoholic fatty liver disease. However, as the purpose of the study was to investigate the impact of detecting cirrhotic cases among hospital clinic attenders, this is a representative sample.
The spectrum of liver fibrosis among trial participants reflected the trial design and focused on pre-cirrhotic patients. Although this targeted the cohort of patients most likely to benefit from the early diagnosis of cirrhosis, a longer period of follow-up than allowed for in the period of the programme grant will be required to accumulate sufficient events for adequate statistical power. Thus, a later follow-up will be required. As this will not incorporate continued monitoring for progression of fibrosis, this will diminish any potential benefit from the intervention, limiting the main benefit to those patients reaching cirrhosis during the on-trial monitoring period of 30 months.
The patients most likely to benefit from monitoring for cirrhosis are those with progressive liver disease who develop cirrhosis during the study period. The modest levels of drinking (again possibly because of a Hawthorne effect) and the introduction of effective therapies for HCV and HBV infection during the conduct of the study are likely to reduce the proportion of patients with progressive fibrosis.
Compliance with trial visits was moderate and provides a good reflection of clinical practice. These data will permit more accurate estimates of feasibility and power when designing future monitoring and interventional studies in CLD.
Longer-term follow-up and analysis of the process of care, clinical events and outcomes by randomisation arm, as well as investigation of the impact of non-invasive screening for cirrhosis in the standard care arm will have to await unblinding of the trial and the passage of time. However, frameworks for the analysis of the data can now be developed to permit a full evaluation of the trial in due course.
Long-term follow-up studies
When the DMEC determines that the study can be fully unblinded we will conduct an analysis of outcomes in each arm as planned. Specifically, we will determine the numbers and proportions of patients in each arm who:
-
were diagnosed with cirrhosis
-
underwent OGD
-
had their AFP level measured
-
underwent ultrasonography
-
were diagnosed with HCC
-
the size and number of HCCs at the time of diagnosis
-
underwent treatment for HCC
-
were diagnosed with oesophageal varices
-
started therapy for oesophageal varices, including
-
band ligation
-
beta-blockers
-
-
presented with haematemesis as a result of portal hypertension
-
presented with ascites
-
presented with spontaneous bacterial peritonitis
-
presented with encephalopathy
-
presented with any other complication of cirrhosis
-
underwent liver transplantation
-
died from any cause
-
died from a liver-related cause.
In the ELF arm, cases diagnosed with cirrhosis will be analysed to determine the true- and false-positive rates for ELF testing by assigning a diagnosis of cirrhosis using clinical parameters, including:
-
non-invasive blood tests for cirrhosis, including platelet count, aspartate aminotransferase (AST)/ALT ratio, AST-to-platelet ratio index and fibrosis-4
-
fibroelastography, including FibroScan and acoustic radiation force impulse
-
imaging, including ultrasonography, CT and MRI
-
the clinical judgement of the clinician when recorded.
In cases diagnosed as having cirrhosis in the standard care arm, we will attempt to determine the impact of the use of FibroScan by comparing the number of patients diagnosed with cirrhosis and rate of diagnosis at those centres using FibroScan with the number and rate at those centres that had no access to FibroScan.
The accuracy of ELF testing and standard care in diagnosing clinically important cirrhosis will be determined by comparing the numbers and proportions of cases developing clinical signs of decompensated cirrhosis, HCC, transplantation or death in each arm at the time of censoring. These rates will be compared among those cases undergoing treatment for portal hypertension as well as among all cases per arm.
We will evaluate the impact of treatment for underlying CLDs by analysing the frequency of treatments of disease by aetiology in each arm. Specifically, we will investigate changes in ELF values in response to therapy in the ELF arm.
We will investigate the correlation between ELF value at randomisation and the incidence of complications of cirrhosis in all participants.
Feasibility of the long-term data collection: sustaining data collection from the end of the active period of follow-up to the long-term outcomes
There are few prospective inception cohort studies of patients with CLD investigated for long-term complications of CLD. In previous studies we have successfully monitored cohorts of patients with liver disease using ONS and HES data. In a former study we followed those patients initially recruited for the original ELF study of biomarkers of liver fibrosis, from which the ELF test was derived, using routine data sources, including death certification and detailed interrogation of clinical case notes. 35 For those patients lost to follow-up by the hospital services, we contacted their general practitioner. Using these methods we were able to obtain data on > 95% of the patients recruited into the study. Although examining the case notes of the participants was feasible, it proved to be labour intensive, necessitating one or two investigators spending 1–2 days extracting data at each participating site.
In a separate study of middle-aged women participating in screening for ovarian cancer [UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS)] who had provided prior consent for follow-up using routine data sources, we investigated the incidence of hospital admissions and deaths from liver-related conditions using routine data sources and patients’ NHS numbers. 894 In this study of 110,000 women we established a set of International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10)895 codes that could be used to define the incidence of liver-related outcomes and showed that NHS numbers could be used to access these data through routinely gathered, centrally stored HES data.
The cohort of patients with CLD recruited for the ELUCIDATE trial represent the range of CLDs commonly encountered in NHS clinical practice and were recruited at a similar stage of disease severity. From the outset we proposed to follow the cohort to assess their long-term morbidity and mortality to obtain a clearer picture of the course and consequences of CLD in the NHS.
It became apparent when the threshold for entry into the ELUCIDATE trial was reduced from an ELF value of 11.0 to an ELF value of 8.4 in March 2011 that a minority of patients recruited would develop liver-related morbidity or mortality during the course of the funded period of the study. Recognising this, the Trial Steering Group agreed to plan for a separately funded follow-up of the ELUCIDATE cohort at 5 and 10 years. We consulted with the NHS Information Centre (now known as the Health and Social Care Information Centre) to discuss data collection on the trial cohort at two time points, including exploration of process, costs and anticipated outputs. We were informed that this was a provided service and accessed by many health and research teams and that we should ensure that consent for such a follow-up was obtained from the patients at the time of recruitment to the trial and that 6 months prior to the censoring date a request should be submitted to the Health and Social Care Information Centre for mortality and morbidity data on the cohort of recruited patients.
The proposed methodology is to use routine data sources including death certification, cancer registries and HES data to gather clinical outcome data on the whole cohort. The NHS numbers of all participants who have provided informed consent for long-term follow-up will be provided to NHS Digital (formerly the Health and Social Care Information Centre) to obtain details of their clinical encounters with NHS secondary care providers. In addition to HES data, cancer registries and death certification registries will be searched for morbidity and mortality using participants’ NHS numbers.
Management
The long-term follow-up for clinical outcomes will be conducted at 5 years after the completion of the ELUCIDATE trial. A working group led by the Chief Investigator, Professor William Rosenberg, and comprising Professor Peter Selby, Professor Walter Gregory and Dr Julie Parkes will meet on an annual basis to ensure a sustained effort to deliver the additional data. The full protocol will be developed and renewed annually and will take into account the relevant processes for application to the Health and Social Care Information Centre at the time of censoring.
Planned analyses
Using NHS numbers, patients’ records will be surveyed for clinical outcomes associated with CLD, including the following ICD-10 codes: K70, K73, K74, K76, I850, I859, Z944 and C220. Codes K70, K73, K74 and K76 relate to CLD or cirrhosis. I850, I859, Z944 and C220 code for events associated with decompensation of CLD.
Cancer registries will be interrogated for incidence of HCC and cholangiocarcinoma. The UK Liver Transplant register will be interrogated for liver transplantations, indications for transplantation and the outcomes of transplantation.
Anticipated outcomes from the long-term follow-up
The long-term follow-up of the ELUCIDATE cohort is of considerable importance to the full evaluation of the trial’s impact and will permit us to:
-
Determine the relationship between change in ELF value and incidence of liver-related events. These analyses will provide the definitive assessment of the clinical effectiveness of ELF testing and will provide the data on which the full health economic analysis of the cost-effectiveness of the use of biochemical strategies for the early diagnosis and management of cirrhosis will be carried out.
-
Determine the prognostic performance of the ELF test in predicting liver disease morbidity and liver and all-cause mortality.
-
Determine the incidence of clinical events in patients diagnosed as having cirrhosis based on the ELF test.
-
Compare the incidence of liver-related events between patients diagnosed with cirrhosis based on ELF testing and patients diagnosed with cirrhosis based on standard clinical criteria.
Limitations
The ELUCIDATE trial is subject to several limitations. The slow start and slow initial recruitment (despite the ‘late surge’ in recruitment) mean that prolonged follow-up is now needed for us to be able to report the primary health-related end points. Our focus was always on an ‘end-to-end’ trial (from diagnostic test to patient outcomes and service outcomes), so this is disappointing. However, the lessons learned and the ‘process of care’ analysis will be very valuable as an exemplar trial. We run the risk that new technologies will supervene during the follow-up period. Compliance with the trial process was satisfactory but compliance with the interventions required by the test outcomes was lower than expected, which may also reduce the impact on the primary outcomes.
The ELUCIDATE trial is certainly of value as an example of an exemplar trial in an important area of health care. It should help those who are strategically planning the evaluation of new biomarkers to judge the feasibility of timely delivery of end-to-end trials and judge the pace of alternative strategies, which we discuss in Chapter 24.
Chapter 23 Patient and public perspectives
The multiple possible roles for biomarkers in patient management are likely to influence patient experience in many different ways. Monitoring of disease progression in CLD has the potential to cause anxiety, but also to stimulate possible lifestyle change by bringing home the reality of liver damage much earlier. Little research has been carried out into the psychosocial aspects of biomarkers, and the possible benefits and harms of their use need to be examined explicitly in future studies.
This chapter is in two parts. The first part presents the results of the research exit interviews with patient participants in the RCT reported in the ELUCIDATE trial workstream (workstream 3). The second part pulls together this information with the contributions made by PPI representatives consulted about the methodology work conducted in the methodology workstream (workstream 1) and reported in Chapter 9.
Part 1: patients’ experiences in the ELUCIDATE trial – a qualitative study about patient experiences of taking part in a trial to test biological fluid biomarkers for liver disease
This qualitative substudy of the ELUCIDATE trial aimed to explore the experiences and perspectives of patients who were enrolled in the ELUCIDATE trial, in order to provide additional insight about using the ELF test as part of patient care.
The starting point for the work is that patients may have very different experiences of having their disease monitored by the ELF test or similar tests. For some it may be reassuring to know that they are being tested regularly and may allow them to feel in control through knowing what is going on with their health. 322 Others may believe that their symptoms are not indicative of cancer or may be anxious about their condition potentially getting worse, even though this might never happen. If patients are anxious they might consequently utilise a number of coping strategies to cope with the associated distress. 322,896 Anxious patients might be hypervigilant with regard to potential symptoms, engage in information seeking and/or develop avoidant attitudes and behaviours. 896,897
Some patients might not adhere to monitoring because they fear its iatrogenic effects whereas older patients might believe that they are less likely to develop neoplasm because of their more advanced age or overall perceived lower susceptibility to cancer. 897–899 Systemic factors also importantly determine surveillance experiences. Health information given to patients, the role of health-care professionals and previous experiences with cancer importantly determine the acceptance and understanding of monitoring practices. 898,900
Patients’ understanding of clinical biomarkers and experiences of testing, their acceptability to the patients, their perceived utility and patient experiences and motivations for testing are, therefore, important factors of translation of biomarkers into clinical practice. By exploring patients’ experiences of being monitored by ELF test, this study will assess how acceptable the testing is to patients and so enable better support to be provided to liver patients undergoing monitoring in the future.
Method
We planned to approach 13 patients to take part in an in-depth semistructured interview that was guided by a semistructured topic guide.
Sites
It was planned to recruit participants from three sites, chosen to represent different sizes of institution and different types of catchment area: Leeds Teaching Hospitals NHS Trust, Bradford Teaching Hospitals NHS Foundation Trust and University College Hospitals NHS Trust. However, because of delays in obtaining permissions, it was possible to conduct the research at only two sites, Leeds and Bradford.
Sample
Patients were sampled purposively with the aim of recruiting eight participants from the standard care arm and eight participants from the intervention arm of the ELUCIDATE trial; other factors such as age and sex were considered. Participants had to be able to comply with the requirements of the study protocol and be able to provide written, informed consent. Patients were excluded if they were unable to comply with the requirements of the protocol or could not provide informed consent.
Recruitment
Eligible patients were first approached by a research nurse who was part of the hepatology team. The research nurse provided each patient with a patient information pack containing an information sheet, an opt-out form, a demographic form and a Freepost envelope. The research nurse asked for verbal consent to pass the patient’s contact details to the researchers from the University of Leeds who would conduct the interviews. All patients were given 7 days from the day that they were given the patient information pack to consider taking part. If after considering the information they did not want to participate, they could choose to opt out by completing the opt-out form and returning it in the Freepost envelope; after this no further contact with the patient was made. The use of an opt-out approach for recruitment was chosen as this is something that most patients find acceptable (particularly in this population of trial participants) and can minimise response bias. 901
Interviews
After 7 days the researchers from the University of Leeds contacted patients to confirm that they were still interested and to schedule an interview. Patients could either agree to the interview or request more time to consider the information and contact could be made at a later date. The interviews were 30–45 minutes in length and were carried out over the telephone or face-to-face in a location that was convenient for the patient. Several studies have provided evidence that there are minimal differences in the results of semistructured interviews that are conducted over the telephone or face-to-face. 902,903 Interviews were audio recorded with the consent of patients.
The interviews were guided by a semistructured topic guide that included questions about the patients’ understanding and experience of taking part in the ELUCIDATE trial and of the ELF test. Participants were also asked whether or not they would recommend this type of study to patients in the future.
Analysis
The interviews were professionally transcribed verbatim and managed using NVivo Version 10 (QSR International, Warrington, UK). The data were analysed inductively, with no prior hypotheses, using thematic analysis. Analysis was undertaken by two researchers who independently coded for emerging themes and then compared themes and codes. The analysis was further refined by using a constant comparison and contrastive approach and looking for negative cases in order to examine for similarities and differences within and between patients in different centres and within and between trial arms.
Results
Thirteen patients were contacted by the research nurses at the two sites. Of these, one patient opted out of the study, one was too ill to take part and two could not be contacted at their existing address. Nine interviews were completed, three with participants from the standard care arm and six with participants from the intervention arm.
Participant characteristics
All of the patients were of a similar ethnic origin. The mean age of participants was 63.4 years, with a range of 56–75 years. Six of the participants were female and three were male. Participants had the following conditions:
-
haemochromatosis (n = 1)
-
fatty liver (n = 1)
-
non-alcoholic fatty liver disease (n = 1)
-
methotrexate-related liver fibrosis (n = 1)
-
hepatitis C (n = 2)
-
autoimmune hepatitis (n = 1)
-
primary biliary cirrhosis (n = 1)
-
not stated (n = 1).
Participants demonstrated a range of educational abilities:
-
secondary school (n = 3)
-
college/diploma (n = 3)
-
university (n = 3).
Four participants were retired, three were in a professional role, one was unable to work because of ill health and one chose not to state their employment status (Table 148).
| Patient number | Site | Sex | Liver condition | Trial arm | Result | Education | Occupation | Ethnic origin | Religion |
|---|---|---|---|---|---|---|---|---|---|
| ELU01 | 1 | M | Fatty liver | Standard care | No cirrhosis | University degree | Professional | White British | Christian |
| ELU02 | 1 | M | Haemochromatosis | Standard care | No cirrhosis | University degree | Retired | White British | Christian |
| ELU03 | 1 | F | Methotrexate related | Standard care | No cirrhosis | College/diploma | Retired | White British | Christian |
| ELU04 | 1 | M | Not stated | ELF | No cirrhosis | Secondary school | Not stated | White British | Christian |
| ELU05 | 1 | F | Non-alcoholic fatty liver disease | ELF | No cirrhosis | University degree | Professional | White British | None |
| ELU06 | 2 | F | Hepatitis C | ELF | No cirrhosis | Secondary school | Professional | White British | Prefer not to disclose |
| ELU07 | 2 | F | Autoimmune hepatitis | ELF | ELF test indicated cirrhosis; followed cirrhosis pathway | College/diploma | Retired | White | Christian |
| ELU08 | 2 | F | Hepatitis C genotype 1 | ELF | ELF test indicated cirrhosis, which was later found not to be the case | College/diploma | Retired | White British | Buddhist |
| ELU09 | 2 | F | Primary billiary cirrhosis | ELF | No cirrhosis | Secondary school | Unable to work because of ill health | White British | None |
Key themes
The themes that were found tended to be related to the questions in the topic guide:
-
participants’ experience and understanding of the ELUCIDATE trial
-
participants’ experience and understanding of the ELF test
-
support and information offered to participants.
Generally, the participants interviewed had a good experience and found the ELF test acceptable compared with current available alternatives. An alternative view was presented by a patient who had been told that she was at risk of becoming cirrhotic. The use of language when communicating risk of cirrhosis to patients requires attention.
Participants’ experience and understanding of the ELUCIDATE trial
Participants’ understanding of the ELUCIDATE trial was generally clear. They appreciated the fact that the ELF test would indicate changes from fibrosis to cirrhosis:
my understanding is that, as biopsies are quite invasive, I found it quite invasive, and the trials are looking at different markers in the blood to see whether they could find the beginning of the cirrhosis so that you know, . . . people could start the treatment maybe sooner.
ELU03
However, some people struggled to remember what the trial entailed:
Yeah it was a bit long ago I can’t really remember.
ELU06
This probably reflected the length of time between taking part in the study and the qualitative exit interview. Most participants who took part in the trial appeared to do so for altruistic reasons, and partly to benefit themselves through being monitored and assessed more regularly:
as far as I’m concerned if you can help people by being a guinea pig it’s a common sense, and it might help me as well as other people you know.
ELU02
it gave me a little bit of confidence that I was involved, and people were looking out for what was going on with my liver so I was quite happy to do that.
ELU03
a closer eye would be kept on that when I had the specialist scans looking at the physical nature of the liver.
ELU05
One participant did refer to the fact that her treatment was expensive and so she wanted to give something back for that reason:
I just felt I had a very expensive treatment and it was the least I could do.
ELU06
The ELUCIDATE trial compared participants in two trial arms, one in which they were monitored with the ELF test and one in which they received routine expert care. Some participants were clear about the fact that there were two arms in the trial:
My understanding was some people had the ELF test and some people did not I think they were looking to compare the two, and the outcomes.
ELU01
However, others were less clear:
No, I was just told they were doing this trial, you know, obviously to see, to try and get some way of finding out if people were going to develop any other problems later on.
ELU07
Participants struggled to recall the difference between the arms because of length of time between the interviews and taking part in the trial. This was also because most perceived no negative impact and they were used to having blood tests as part of routine care. However, participants’ expectations of the study were quite high as the opportunity to have a less invasive test was appealing:
so obviously so yeah anything rather than going through a biopsy from my point of view, just having blood samples taken would be a lot better, so I said fine I’d go for that.
ELU01
I found the biopsy quite distressing, and apparently I’ve got really tough muscles! And it took a lot of getting into my muscle, it was awful, so I was quite happy with the bloods and weights and things.
ELU03
After considering the information most participants thought that taking part would be less of a problem than routine care:
I didn’t think there would be any detrimental effects, not any.
ELU06
Participants weighed up the potential risks and benefits and judged the ELUCIDATE trial to be of low risk and low effort, as demonstrated by the following quotation:
I think it was a positive thing really it certainly didn’t make me worry about it but it certainly gave me a bit more depth of understanding of what was going on I think really.
ELU01
The participants interviewed in this study were satisfied with the fact that they would be monitored more regularly:
No, in fact, I mean to some degree it helped me because it had to be done every 3 months, and X used to make sure I got an appointment. Some of my appointments, I would have waited for far longer.
ELU07
The main negative issues were reported in terms of having difficulty parking at the research unit and the cost of travelling in their own vehicles:
The only negative experience is travelling from X to [name of hospital site] and back you know and parking there.
ELU02
Participants’ experience and understanding of the Enhanced Liver Fibrosis test
Participants were not told the results of the ELF test unless there was a change in their condition:
No I did ask and he said normally, unless there was a major problem, where you needed treatment then they wouldn’t get back to you on your blood results.
ELU03
In most cases, this meant that no additional information was provided to participants. This was an acceptable situation for seven participants who were interviewed. With any diagnostic and monitoring test some patients will show a false-positive or a false-negative result. We interviewed a further participant, who said that:
I was told that I was cirrhotic . . . But I wasn’t. And that worried me considerably, being left, you know, just being told I was . . . cirrhotic, and just being told that, with no follow-up, if you see what I mean. So I had to do my own follow-up with my own doctor.
ELU08
The professional team was able to use other information to confirm that this participant was not cirrhotic. However, it had caused the participant to be concerned. They were reassured when other tests indicated that they were not cirrhotic:
The nurse told me, ‘Oh well, you know, it’s unlikely because, you know, your blood tests are fine’, but when you’re told something like that, it’s very shocking.
ELU08
This participant would have liked to have spoken to a doctor or been offered a further scan. Some discussion of why the professionals were certain that she did not have cirrhosis could have helped:
So, I’m not really sure, I think I would have liked to have talked to a doctor and then they would have said, ‘Look, this really is rubbish’, you know, ‘You’re absolutely fine’.
ELU08
In this case, the participant sought additional support from a patient forum. Being familiar with the internet and potential support options seemed to be of benefit for this participant, but such options might not readily be accessed by those with a lower level of health literacy:
Well yes, I mean, I was on a forum as well and we shared a lot of information on the forum about treatments that we were having at various hospitals through the country, so it was quite interesting, really.
ELU08
Seven of the nine participants interviewed did not have changes in their test results so were unable to comment on the question concerned with changes made in relation to test results. One of the two participants who had received above-threshold ELF test results reported that they were now following the cirrhosis pathway, but the other said:
Well, I’m not sure, [I’d make a change] especially as the test got me wrong.
ELU08
Again, attention should be paid to the way in which information about monitoring tests is communicated to patients; this includes the description of the test and what happens when a result is not correct. The issue of a test result indicating a diagnosis was also raised by this participant, who said:
Yeah I’d been seeing Dr X and he explained . . . [they could] possibly look at some alternatives to give me a good diagnosis, one of the things would have been a liver biopsy, which . . . I didn’t particularly fancy, and then he explained that they were then looking at this trial, and would be looking at alternative ways of helping make diagnoses and things and asked me if I’d be interested.
ELU05
Support and information received as part of the ELUCIDATE trial
Overall, participants had positive experiences whilst being enrolled on the ELUCIDATE trial and felt that the study was explained well:
No no not at all it was explained very well in the first place . . . it was very much, full detail was given and you know explanation all the way through as to what was happening and the whys and the wherefores so yeah it was good, yeah.
ELU01
Participants reported having a high level of trust in the professional team caring for them and carrying out the research. They appreciated that most of the study tasks and paperwork were completed alongside routine care:
That was usually what happened, yes, occasionally it didn’t work but usually it did, and we, we linked the two together so, I went for the appointment and did the study at the same time.
ELU01
Completing questionnaires and associated forms was found to be acceptable:
Well, it was mainly just filling out a form, so yes, it was absolutely fine.
ELU08
However, one participant had noted that the order of the questions had been changed and commented:
I personally found it quite tedious, filling them in every time but I know you needed the information.
ELU05
Participants did not remember being signposted to sources of information about research per se. Overall, those who took part in the study felt that it was important ‘that someone is the guinea pig’ and felt that they ‘wanted to give something back’ and did not expect any further reward, for example:
They did also offer to pay my expenses which I said no, I wasn’t bothered about that.
ELU03
Participants talked about the fact that the research nurses went above and beyond to help and support them. Most participants who took part in these interviews would recommend taking part in this type of research study. They wanted to be able to give something back as part of a reciprocal process. In general, they fitted the profile of those who usually agree to take part in research studies, including having a high level of trust in the professional team and wanting to give something back.
Participants’ perspectives of taking part in the ELUCIDATE trial: discussion points
Participants may have had very different experiences in the ELUCIDATE trial depending on a number of factors. The specific arm of the trial that participants were allocated to and whether or not they were told that they were at elevated risk of cirrhosis would have influenced their experience. Additional influencing factors may have been dependent on their personal circumstances, such as the condition they had, whether or not their employer would accommodate regular hospital appointments and the costs of taking part, including the costs of absence from work, travel to hospital sites and parking. Withdrawals from the trial may indicate that ongoing monitoring is not acceptable or convenient for some patients.
The trial information provided to participants as part of the ELUCIDATE trial was perceived to be sufficient, with few gaps noted, although some direction to professional support or peer support should be considered in future trials. The issue of information needing to be tailored to individuals makes this difficult to get right in every case. The research nurses at both of these sites (Leeds and Bradford) provided support to participants that was found to be personal and effective.
The process of having a blood test in addition to existing tests and in place of more invasive tests was found to be acceptable to participants. This was reflected in the experiences of patients who were not indicated to be at increased risk of cirrhosis according to the ELF test. One patient we spoke to had a different experience, having been told that she was cirrhotic based on a test result, which was resolved in the context of other information. Participants who were interviewed had a high level of trust in professionals and were comfortable in NHS environments. There were no reports here of participants who had problems with medical tests or who had had previous negative experiences, although the sample was limited.
There was only one example of a participant with what was perceived as an inaccurate result, so it would be good to know how well this reflected other ‘false-positive’ experiences in the trial. This participant experienced a level of shock and worry that should be considered further in terms of how this information is presented to patients and the type of support that could be offered (this is supported by PPI work).
When implementing this test in clinical practice, some attention needs to be paid to how the risk of cirrhosis is communicated to patients. A monitoring test is not necessarily the basis of a diagnosis; however, the language used for diagnostic and monitoring tests is often used interchangeably and can be confusing to both professionals and patients.
Limitations
The sample here was small and it is possible that other sites may have had different experiences. Most participants reported positive experiences and there may have been some bias in the selection and availability of participants for interview: none of those interviewed had undergone additional invasive tests in light of their ELF test result (e.g. endoscopy) and nor were those who dropped out of the study represented. This means that the generally benign nature of participants’ experiences may not fully reflect any potential for harm, as relevant accounts may have been missed. Additional information could have been collected if there had been time to develop the insight from this qualitative study into a questionnaire for a larger sample of study participants, to confirm these experiences.
Conclusion
As the alternative to a blood test is a more invasive biopsy, the ELF test proves to be an acceptable method of assessment. However, caution needs to be applied and some scrutiny given over how test results are communicated to patients, as part of a considered implementation strategy for any biomarker proven to be effective in a trial. Patients have the highest expectations that ethical methods are applied at all stages. Lessons from PSA testing demonstrate a need to fully understand all of the factors that affect test interpretation prior to implementation in clinical practice.
Part 2: drawing threads together
Part 1 in this chapter reports on a piece of qualitative research that specifically addressed participation in the ELUCIDATE trial. As will be recalled, more general issues about biomarker evaluation research were discussed with patient and public representatives in a consultation exercise, reported in Chapter 9.
Although fully recognising that these are two different types of activity, each generating very salient points of their own, a number of common themes can also be identified.
The patient interviews clearly indicate that adding ELF testing to an ongoing monitoring regime, as was performed in the intervention arm of the ELUCIDATE trial, was acceptable to participants and fitted well with routine care in this group of at-risk patients. Reassurance of continuing low scores (below the cut-off point) could be obtained by these means.
Individuals who were test/diagnosis positive were obviously in a different position. The term used in the trial protocol for participants testing positive in the intervention arm (and, hence, used by staff when communicating with participants) was ‘diagnosis’. A test score above the specified cut-off point triggered the initiation of a cirrhosis management protocol, which included further tests. The interviewee who had experienced this care pathway was told by staff that the subsequent tests had not supported this ‘diagnosis’, that is, that the ELF result had been a false-positive result, with upsetting as well as confusing consequences.
No quantitative argument is being made here. Clearly, interview numbers were small and many factors will have influenced who was available and willing to be interviewed. However, almost two-thirds of trial participants in the intervention arm were found to be test positive at some stage, and the interviewee’s experience does chime with concerns about false alarms expressed by members during the PPI consultation in workstream 1.
Clinicians’ desire not to miss preventable disease is very powerful, but the possibility of causing harm is real and must be considered. National screening programmes, for example, have been criticised for ignoring negative effects and a more balanced presentation of the pros and cons is now advocated.
The pathway from the introduction of a new monitoring strategy through to patient benefit is clearly complicated, but the health economic approach (see Chapter 8) lays out the fundamental trade-off very clearly: does the gain in ‘utility’ (quality of life multiplied by cost) achieved by successfully treating more cases outweigh the loss in utility incurred by unnecessarily treating more non-cases? Patients and family members can readily recognise the trade-off being made here and can relate it to their own circumstances and experience. Members of the PPI consultation group could also see that the numbers (e.g. test scores) on which decisions were based reflected professionals’ judgements and preferences as well as scientific ‘facts’.
Thinking first about judgements and preferences, members of the PPI consultation group could readily appreciate that a traditional clinical approach tends to focus on the benefits of successfully treating those patients who can benefit, and has relatively little to say about ‘the price’ – in every sense of the word – being paid for that. PPI members appreciated the application of a ‘precautionary principle’ to detecting the recurrence or progression of disease – assuming of course that early information is preventative – but they also emphasised the possibilities for harm. It could be additionally pointed out here (echoing a point previously made in Chapter 5) that the case for paying more attention to negative consequences when designing monitoring strategies is even stronger than for screening because monitoring offers multiple opportunities for harm.
The established health economic argument (see Chapter 8) here is that an inefficient use of resources will displace more efficient use of those same resources. This argument is more customarily thought of in terms of more and less effective treatments, but it also applies to more and less efficient use of follow-on testing resources. An additional point can also be made: if the eligibility criteria for enhanced monitoring are made too broad, then people at high risk of developing (or already having) the condition of interest will be on the same waiting lists for follow-on tests as people at much lower risk. PPI representatives familiar with an overburdened NHS, its resource constraints and waiting lists had no difficulty appreciating this point and wanted a more transparent and considered approach to be adopted. Simply widening the definition of who is eligible for enhanced care could be seen as seldom the best approach, even if the enhanced regime was acceptable and relatively benign from an individual patient’s perspective.
As for ‘scientific facts’, the existence of sizeable evidence gaps in the biomarker pipeline came as quite a surprise to members of the PPI consultation group. They were aware that very substantial sums continue to be spent worldwide on biomarker research and assumed that all components of the ACCE pipeline would already be of the highest methodological quality. When they realised that the quality of evidence sufficient to justify the use of a given test strategy in the NHS was not in fact uniformly high, they were rather shocked.
A number of other points with implications for patients are worth spelling out here, even though they were not discussed in detail with patients themselves. In the intervention arm of the trial, test-positive patients were managed as though they had been diagnosed with cirrhosis. The first management change was to implement more intensive monitoring, some of which entailed quite unpleasant procedures, for example endoscopy. One of the interviewed patients commented (see Part 1: patients’ experiences in the ELUCIDATE trial – a qualitative study about patient experiences of taking part in a trial to test biological fluid biomarkers for liver disease, Results) that being monitored with a blood test like ELF was an attractive alternative to having a biopsy, but the alternatives are actually more complicated than that, as the trial results (see Chapter 22) show. Many more endoscopies were conducted in the intervention arm, but there was no reduction in the biopsy rate. Inevitably, such management is also more costly.
Patient benefit cannot, of course, result from extra monitoring per se, but only from a timely response to information generated by the more frequent/intensive testing schedule. Process outcomes provide an early signal that changes to management are being triggered by test results, but it is important to maintain a distinction between process outcomes that reflect extra monitoring and process outcomes that reflect the initiation of treatment.
The clinical effectiveness of treatment at the particular stage of disease identified by the initial test (and any confirmatory tests) must next be evaluated, and here a robust randomised design is essential. Correctly identified early cases (as well, of course, as early identified false-positive results) may not be better off if early treatment does not lead to better outcomes than late treatment – and all will have spent more time under investigation with associated anxiety.
Here we come back to the crucial question asked earlier: does the gain in utility (quality of life times cost) achieved by successfully treating more cases outweigh the loss in utility incurred by unnecessarily treating more non-cases? A single RCT can answer this question in relation to a particular combination of measure, cut-off point, monitoring schedule, management protocols and patient population, but experience suggests that the choice of many of these study attributes is not evidence based. A research pipeline that operates in this way is inevitably inefficient, as it places no burden on investigators to justify many of their choices, still less to make some effort to optimise the combination selected. The implications for trial participants as well as for patients more generally are substantial.
Given the sums of money spent on biomarker research to date, and the lack of demonstrable patient benefit, some kind of change is clearly desirable. Patients and the public purse need all ACCE ingredients to be of the highest quality, not just those currently favoured by the research pipeline as it currently operates. This chapter concludes with an overview of the case for doing things better.
Guidelines to improve research quality
Guidelines for researchers about how best to conduct and report different kinds of biomarker studies have been available for some time. The REMARK guidelines,367 for example, offer reporting recommendations for tumour marker prognostic studies, and the STARD guideleines368 do a similar job for diagnostic accuracy studies. More recently, the MONITOR904 group have proposed a four-phase model for biomarker monitoring trials,904 and a position statement drawing together all of the main good practice guidelines has been issued by the European Group on Tumor Markers (EGTM). 342,904
In a welcome convergence, the four stages identified in the EGTM position statement, aimed at an oncology readership, are similar to the stages identified by the MONITOR group for triallists and by the test evaluation working group of the EFLM. 338 The first three stages all bear a close resemblance to the ACCE framework that was drawn on for the work reported in Chapter 9, but the last stage in the EGTM statement refers to regulatory approval, the last stage in the trial design proposal refers to audit and economic impact (including quality of life, assessed using established methods) and the last stage in the EFLM document refers to ‘the impact of testing on the patient, the organisation and society’. In 2015, IFCC took the emphasis on patients one step further in a report entitled ‘Current evidence and future perspectives on the effective practice of patient-centred laboratory medicine’. 905 The report argued that laboratory medicine specialists needed to work with multidisciplinary groups seeking to ‘optimise clinical outcomes and patient experiences in an efficient and cost-effective way’.
Gaps in the evidence
From a standard methodological perspective, the research pipeline on non-invasive liver markers can be seen to have gaps in it, but in more heavily researched areas, the same is also true. 345 Some gaps remain in territory that is well charted in other respects. In respect of a basic analytical validity question, for example, a study showing that a prompt repeat of PSA testing could reduce the number of unnecessary biopsies in men being screened for prostate cancer was published only in 2016. 906 Further down the pipeline, Bessen et al. 907 modelled the cost utility of different mammographic follow-up schedules and showed that they could be tailored according to risk of recurrence. Assumptions about the effects on patients of receiving reassuring test results may also not stand up to scrutiny. Rolfe and Burton908 found little evidence of psychological benefit for patients in relation to diagnostic tests that had essentially been ordered for reassurance purposes, although there may of course be benefits for the patients’ doctors. 909
There are also areas of relatively uncharted research territory. Further targeted methodological work could help to fill important evidence gaps, for example on the interdependency of patient mix, cut-off points and schedules in maximising overall patient benefit. The implications for both modelling and care of variation between patients in, for example, progression rates have been little studied in a monitoring context (compared with the screening context), and nor have the implications for research and service provision of omitting diagnostic testing from a monitoring care pathway. Traditionally, diagnosis is seen as informing treatment options, depending on the cause identified, and monitoring is seen as informing the timing of treatment, but these boundaries are blurring and no longer cover all possibilities. One important piece of work arising out of the present study will be the drawing together of the models described in Chapter 1, and another will be the further study of patient preferences for monitoring schedules with different properties and different implications for preventative behaviour and lifestyle change.
The work we presented to our PPI consultation group raises an important issue about the role of PPI in future ‘methodological’ studies. At the outset of this programme of work we were hesitant about trying to communicate ‘methodological’ knowledge to lay members and some colleagues took the view that it could not meaningfully be done. However, our meeting showed that the public have a more sophisticated palate, and a greater interest in this topic, than researchers have traditionally given them credit for. Participants demonstrated a genuine interest in these issues and asked pertinent questions, but we had scheduled to meet with the group only once. With hindsight, a series of four to five meetings with the group to facilitate a more wide-reaching discussion about the implications of the work and possible ways forward would have been more useful, both to them and to us.
Choosing wisely
In 2012 the American Board of Internal Medicine Foundation launched a campaign called Choosing Wisely. This campaign aims to reduce the number of unnecessary tests and procedures by promoting effective conversations between patients and their doctors. 910 In 2015, the Academy of Medical Royal Colleges publicised their initiative to bring the campaign to the UK. 911 The emphasis in both campaigns is on avoiding unnecessary treatments, but unnecessary tests and assessments are also addressed. In the UK campaign, doctors are encouraged to provide patients with resources to understand potential benefits and harms. They are encouraged to ask questions about the need for the test or procedure and about the alternative options. High-quality research evidence is clearly needed if clinicians are to be able to answer questions of this kind.
Personalised medicine, innovation and the funding of research
At the end of Chapter 9, the point was made that the acknowledged role for patient choice in treatment decisions also needs to be thought through in relation to testing decisions. For example, the choice of cut-off point for ELF testing – the case management threshold – was lowered in the ELUCIDATE trial from 12.5 to 9.5, primarily on the basis of judgements about which risk categories were likely to carry weight with clinicians. By increasing the numbers of test positives in this way, fewer people with incipient cirrhosis will have been missed, but inevitably this will have been accompanied by an increase in the number of people switched to an intensive monitoring regime unnecessarily. It seems likely that many patients would arrive at a different trade-off here, not least because of the need to take probabilities of benefits and harms into account as well as their potential magnitude.
Bossuyt and Parvin912 draw a similar conclusion in their paper about the evaluation of biomarkers used to guide treatment decisions – a not dissimilar function in principle from the monitoring markers under scrutiny in this report. After describing studies in which cancer patients were asked about the size of treatment benefit they would need to expect for adjuvant chemotherapy to be worthwhile, these authors concluded that, ‘for some the required gain may be fairly large, while for others extending survival is extremely important, and their threshold for accepting treatment is close to zero’. Bossuyt and Parvin912 went on to say, ‘This is definitely an area for personalized medicine: not in the abundant use of next-generation sequencing, but in the recognition that personal values and trade-offs differ.’ Although direct research evidence is not available, the patient perspectives described in this report make it likely that very similar conclusions could be drawn about the use of monitoring tests for disease progression.
As Bossuyt and Parvin912 note, in respect of test evaluation metrics, ‘The classical clinical performance measures, such as clinical sensitivity and specificity, can only be used in rare circumstances’. It can be argued, however, that research funding is much more likely to be available for studies of the sensitivity and specificity of innovative new markers than it is to fill gaps in our understanding of the real-world performance of existing ones, and funding for trials – even quite speculative ones – is much more widely available than funding for well-designed, descriptive longitudinal studies, or studies of patient perspectives on the quality and role of tests used in their care. Although the need to fill gaps in the evidence base for using biomarkers in patient care is scientifically and professionally convincing, as long as the current incentive structure for researchers remains, little is likely to change and the potential for patient benefit is unlikely to be realised.
Chapter 24 Programme conclusions and the framework for biomarker evaluation
General conclusions
In Chapters 9, 15 and 23 we have summarised and discussed the principal conclusions from each of the workstreams in methodology (workstream 1), clinical translation (workstream 2) and the ELUCIDATE trial (workstream 3).
Taken together they represent a substantial overview of the state of the art and the challenges in the introduction of known biomarkers into clinical practice and health-care systems. We have shared the results and the experience of this investigation with patients at the application, study design, study delivery, analysis and evaluation stages. In each workstream we have confirmed the importance of a rigorous and systematic approach to biomarker evaluation and made recommendations to help future work in this area. The incomplete and inadequate nature of some of the methodological approaches taken in this field was clearly identified in workstream 1; similar limitations of approaches in the laboratory and in sample preparation and study design in many cases were described in workstream 2. The challenges, both conceptual, logistic and organisational, of delivering ‘end-to-end’ clinical trials such as the ELUCIDATE trial to determine the place of new biomarkers in clinical practice were shown in workstream 3.
The investigators conclude that there are a number of important, generic and recurring themes in what we have learned in these studies:
-
Multidisciplinary research teams are essential to establish individual and portfolios of biomarker evaluation projects. All of the parts of the pipeline must be critically evaluated to a high standard using standardised approaches if the field is to move forward adequately.
-
The organisational and logistic challenges have to be addressed by well-trained teams with adequate resources to operate to the standards that are required.
-
Innovation in study design and research methodology is essential. ‘End-to-end’ RCTs will remain the gold standard for evaluating the place of a new biomarker or panel of biomarkers in clinical practice. Such study designs must be carefully and comprehensively grounded in appropriate prior knowledge of the performance of the test and the evaluation of the test in appropriate large current clinical populations. Statistical methods for the design of trials and the calculation of power have been developed in the course of this programme. To be useful at the beginning of a RCT, researchers require access to high-quality population-based cohorts of well-characterised patients with the disease under study, whose data reflect the current biological basis of disease (such as causative factors) and the current treatment environment.
-
New study designs and logistic solutions have to be deployed to streamline the biomarker pipeline. The methodology workstream (workstream 1) highlights these issues for monitoring studies.
-
For clinical translation (workstream 2), studies of analytical and clinical validity are crucial parts of the evaluation of tests but are beset by the challenge of having large numbers of good-quality samples with excellent annotated clinical data that are representative of the key clinical populations, are current and have adequate follow-up to answer long-term questions. Samples have to be carefully curated but managed, used, sustained and understood by specialised multidisciplinary research and innovation teams. Workstream 2 demonstrates this very clearly. Broadly, it took us 5 years to design and deliver the samples and clinical data sets for renal disease; it took us 5 months to review all of the literature on available biomarkers in RCC, find and validate the assays and report the findings in this report. Future strategies will require access to pre-existing sets of data and samples. We will curate these and sustain and use them as a resource for academic and commercial collaborators. This will be facilitated by the NIHR DEC. This should mean more ‘5-month’ study turnaround times. Even this would be at risk if our specialised multidisciplinary team, with nationwide clinical contacts, lost its energy or focus. Establishing and sustaining this approach in other topics will be challenging. Nevertheless, we have shown that with the appropriate infrastructure and planning, the analytical and clinical validity of tests may be evaluated promptly for the purposes for which they are intended – allowing robust initial evaluation of tests, which can then be evaluated for their impact on health-care outcomes using RCTs or other strategies.
-
The ELUCIDATE trial illustrates what can be achieved through the commitment and engagement of investigators in the NHS. It was slow in terms of set-up and initial recruitment. More centres were opened and existing centres often responded and recruitment flourished to bring in a large proportion of patients in the final months, even under threat of closure. To some extent the lessons are, therefore, conventional – work harder and faster, open more centres, drive the process by motivating the centres – and large RCTs get delivered.
-
We think, however, that the learning points from the ELUCIDATE trial are less conventional and the response needs to be more radical. The ‘end-to-end’ nature of this RCT, from the initial use of a biomarker to the consequent change in clinical behaviour to yield the desired changes in the process of care, through to long-term impacts on survival and other major clinical events, although desirable, is rarely going to be deliverable without radical changes in how we all work. The ELUCIDATE trial suffered from ‘conventional’ RCT challenges of scale, recruitment and set-up and the responses are listed above. It also suffered from changes that occurred in the disease population (more alcohol-related liver disease), changes in the diagnostic options (the availability of FibroScan in more centres) and changes in the therapeutic options (the availability of antiviral drugs for hepatitis). These changes happened between the studies of cohorts and during the conduct of the trial. They will continue during the planned long-term follow-up using NHS informatics in ONS and HES data. Therefore, the value of the trial to the NHS is restricted by its scale, duration and, of course, cost.
-
Our solution for the ELUCIDATE trial was rapid recruitment in the later phases; innovative modelling to support design and power calculations; analysis of process of care end points that reflect the impact of biomarker monitoring on clinical behaviour and practice; and longer-term follow-up using health informatics. This approach needs to be prospectively developed for future trials. Modern health informatics can provide large, current, relevant clinical data sets to complete the study design and early economic modelling quickly in the appropriate clinical populations. This means that trials may then be conducted in reasonably stable epidemiological, diagnostic and therapeutic environments. These will have to come from patient electronic records using carefully developed technical, confidentiality and governance routes, which are in the process of being put in place. The RCTs may then focus on changing clinical behaviour and the process of care. Long-term follow-up will come from health informatics (as in the ELUCIDATE trial) but will use Electronic Patient Record-based sources rather than the derived databases of ONS/HES origins.
-
Patient engagement has been constructive in the course of this programme and should remain a central feature of this field, as with all fields of biomedical and health research. We drew extensively on patient input not only in evaluating the methodological findings of workstream 1 but also in the design, delivery and evaluation of the preliminary results of workstream 3. Patients were involved in the clinical translation workstream in study design and advised at all stages of the delivery of the cohorts and have commented on the analysis and reporting of the three workstreams.
There is increasing concern about the volume of biomedical research undertaken that is irreproducible, with systematic reviews estimating that between 50% and 89% of preclinical research contains one or more errors, flaws, inadequacies or omissions that prevent the replication of results. 732 With growing financial pressures on researchers in the UK and elsewhere, research funders and publishers have a responsibility to ensure that the time and public money spent on research are spent wisely. This programme has highlighted the need for appropriate validation and verification of biomarker assays and diagnostic tests prior to conducting research studies (see also Chapter 13). The level of validation/verification should be appropriate to the stage of test development and its intended clinical use. If a test is used to inform clinical decision-making within a trial, or provides an end point or outcome measure within an interventional trial, it is essential that it is validated fully in a clinical laboratory as early as possible during its development. When a test or assay is to be used purely in an observational ‘research use only’ context, then less rigorous assay validation, as described in Chapter 13, may be sufficient. However, appropriate assay validation should be an essential requirement for funding and publication of biomarker studies.
This programme has identified new methodological approaches and new biomarkers that justify evaluation of clinical utility in kidney disease and shown that the deployment of the ELF test will alter clinical practice in ways that are likely to be associated with improved outcomes. It is disappointing that we do not have the final evidence of the impact of the ELF test on the important outcomes of liver disease, including cancer, haemorrhage and survival.
The experience of this programme has contributed to two broad aspects of a framework for the introduction of biomarkers into health-care systems:
-
a framework for introducing biomarkers into clinical practice through the NIHR DEC
-
a framework for the design and conduct of clinical trials for biomarker evaluation based on innovative study design and modern health informatics, and early engagement with health economics, discussed above.
The samples and clinical data accumulated in this programme will be an important asset for future studies, both for the programme investigators and collaborating centres who have delivered the samples and the data and guided the research and for new collaborations in academia and industry through the NIHR DEC and other routes.
The National Institute for Health Research Diagnostic Evidence Co-operative at Leeds
The IVD industry is the second largest UK medical technology sector by employment and the fifth largest by turnover, so is of fundamental importance to the UK economy and a huge area for growth, with a 17% increase between 2010 and 2011. The Leeds NIHR DEC was designed based substantially on the experience in this programme and takes four complementary strategic approaches to enhancing the evidence base for IVDs (Figure 76). We will:
-
Deploy and refine methods in IVD study design, health economics and health informatics to improve and speed up the way that IVDs can be evaluated for NHS use, drawing on our experience in this programme.
-
Sustain and strengthen our working networks of co-operating NHS sites to deliver studies effectively with their patients and samples. Drawing on the experience in workstream 2, we can sustain the capability and capacity to more rapidly evaluate new biomarkers in renal and liver diseases. This approach has been extended to musculoskeletal diseases and oncology/haematology.
-
Invite, select and prioritise specific IVD candidates in our clinical areas, from our own work partners and interested parties, and help them develop and deliver appropriate studies and evidence.
-
Create a strong stakeholder engagement group to work with our teams, patients and academic and commercial partners, to shape our strategies, research programmes and projects and identify new opportunities together.
FIGURE 76.
The Leeds NIHR DEC diagnostic evaluation pipeline and development areas.

Investigators in this applied programme are playing a major role in the national NIHR DEC developments. Jon Deeks leads on a joint methodology group for the four DECs (Imperial, Oxford, Newcastle and Leeds). Peter Selby, Mike Messenger and Steph Roberts organise and lead the Leeds DEC and other programme investigators (William Rosenberg, Cathie Sturgeon, Andrew Lewington, Naveen Vasudev, Claire Hulme, Carys Lippiatt, Chris McCabe, Roz Banks, Doug Altman and Walter Gregory) have leading roles.
In vitro diagnostics evaluation methodology research
Our NIHR DEC at Leeds is promoting a coherent philosophy based on continuous decision modelling through all phases of development. A central model for each project will draw on real-time NHS data mining to define the clinical pathway and key clinical decision points and their relationship with clinical outcomes and costs. Based on recent successful examples using these methods, time to adoption can be accelerated and research design efficiency can be promoted. Finding solutions to the challenges of evaluating diagnostics will be an important part of the DEC mission. Such challenges include the rapidly evolving nature of multiple competing technologies and real-world characterisation of diagnostic test properties and their impact on clinical and economic outcomes and optimisation of case definition thresholds. Members of and collaborators with the NIHR DEC at Leeds have been at the centre of recent methodology innovations aimed at addressing some of these challenges. The expertise is divided into three themes:
-
Study design and conduct. The gold standard for demonstrating clinical utility is the RCT. In response to the need for faster time to market, coupled with the complexities of evaluating diagnostic tests, new trial methods are required. Drawing on experience from our NIHR biomarkers programme and the ELUCIDATE trial, including recruiting almost 900 patients, we have developed concepts for RCTs to evaluate changes in care processes and modelling strategies to test long-term patient and NHS-centred outcomes. The DEC strategy will build on this to streamline randomised designs, making more use of early field trials, surrogates, modelling, routine linked data and integrated economic data collection.
-
Health economics. The gold standard for demonstrating cost-effectiveness is a model-based economic evaluation. Modelling will commence at the very start of IVD development under guidance from the DEC during selection and prioritisation. By introducing a model early, it is possible to characterise the potential impact of an IVD on the clinical pathway, clinical decision points and expected clinical end economic outcomes. The optimal case definition threshold for tests can be proposed for cost-effectiveness in addition to clinical validity alone. Models will be maintained as IVD development progresses, populated by meta-analysis of evidence generated both within and external to the DEC. Probabilistic modelling will be used to characterise areas of uncertainty in the evolving evidence for value between each phase of development, thus enabling iterative research design efficiency. Expected cost-effectiveness can be established as well as commercial headroom for the manufacturer. The trade-off between investment in a large RCT and alternative cheaper or quicker study designs can be described through the modelling process, including the use of Bayesian decision modelling and value of information analysis.
-
Health informatics. Central to improving the efficiency of IVD development will be early testing in real NHS settings. For this to be achievable, outcomes monitoring through pre-established clinical data collection is necessary. To demonstrate the potential impact of a new IVD, the standard care pathway with real-world outcomes needs first to be defined. A framework has been developed by Leeds researchers that allows the pathway (including event probabilities and patient characteristics) to be defined directly from observed clinical events held within clinical databases (the Leeds Patient Pathway Manager) using data-mining techniques. This allows a central decision model to be populated at the patient level with observed outcomes. Linkage with NHS finance and resource usage databases such as HES, Patient Level Information and Costing Systems and primary care databases allows a full economic model to be constructed. Applied examples and a set of modelling tools have been developed in Leeds in which standard care and IVD-specific clinical data feeds directly into a decision-analytic model to produce estimates of longer-term health impact and cost-effectiveness.
Qualify analytical validity
Independent verification of the technical performance of an IVD can be conducted by the DEC IVD Validation Group, including assessments of analytical sensitivity, specificity, precision, parallelism, recovery, selectivity, LoQ and vulnerability to interferences.
Qualify clinical validity
The IVD clinical performance (e.g. sensitivity, specificity, predictive values) will be reviewed and can be verified by the IVD Validation Group using samples from our established networks of co-operating NHS sites and biobanks, established as part of the NIHR Biomedical Research Unit, ELUCIDATE trial and NIHR biomarker applied programme. The DEC will identify funding (commercial consortia and/or public) to replenish biospecimen resources and support multicentre biobanking programmes alongside registries, providing low-cost high-value prospective samples and data for rapid IVD evidence generation in priority clinical areas.
Evaluation of clinical utility and cost-effectiveness
Decisions about the research design priorities will be decided by the DEC Methodology Group, following an update of the pathway model developed in Figure 76, stage 1, to include data gathered in Figure 76, stages 2 and 3; pathway outcomes from linked NHS data sets; and re-estimation of uncertainties and value of information. When further evidence is required the Methodology Group will make recommendations about research designs. When necessary, the DEC will then utilise the expertise of the Leeds CTRU on running trials of IVDs, incorporating the enhanced methodologies developed in DEC research programmes.
The interactions between workstreams
We have given clear examples in which the interaction between workstreams has been synergistic. The methodology work provided a literature-based and evidence-based framework for the development of workstream 2 and workstream 3. For instance, the simulation modelling made possible a radical redesign of the ELUCIDATE trial and provided a robust approach to power calculations that underpinned the completion of the trial and its extensions. The methodology workstream also emphasised the critical importance of the determination of analytical and clinical validity of tests in a meaningful way that can underpin evaluations of clinical utility and cost-effectiveness. This resulted in the re-evaluation of the ELF test as a laboratory test, reported in Chapter 17, and the rigorous reappraisals of the assays used throughout workstream 2, including those prioritised as a consequence of the literature reviews. The experience of developing the RCT informed the development of the cohorts for the workstream 2 renal cancer and RT work. Our ability to work with centres, promote trial set-up, open new centres and motivate our collaborators allowed delivery of the recruitment to target. However, it is important that we acknowledge again that the delay in delivering the ELUCIDATE trial meant that large bodies of information broken down by trial arm were not available for our methodology research.
Incorporating large clinical cohorts and RCTs into an applied programme is the attraction of the synergies of the kind described in the previous paragraph. However, delays in setting up and recruiting into those cohorts do have the potential to undermine the delivery of the programme as a whole and limit the synergy between the workstreams. There is no absolute ideal model, but we would urge caution in the integration of large RCTs into integrated programmes of work when other workstreams are dependent on the timely completion of those trials.
Future methodology research
To validate a method one must provide objective evidence that it fulfils the evidence requirements for a specific intended use and is ‘fit for purpose’. 869 However, defining the requirements remains a challenge, even after several decades of intensive efforts by members of the laboratory medicine community. 873 In 1999 a landmark conference in Stockholm agreed a hierarchical structure for setting APGs. 874 A more recent 2014 conference in Milan revised and refined this, suggesting three approaches based on:875
-
the effect of analytical performance on clinical outcome (either directly or indirectly)
-
components of biological variation of the measurand or
-
‘state of the art’.
However, although the Milan consensus provides a useful framework, it did not consider the interconnectivity of the approaches and the possibility that there may be a unified strategy for combining them. Furthermore, very few examples have been reported of APGs based on the effect on clinical outcomes, probably because of the complexity and cost of these approaches. 913 Whether or not it is pragmatic and appropriate to recommend that manufacturers, clinical scientists and researchers invest significant time and resources in striving to set APGs against the highest model, clinical outcomes, when a simpler solution may be ‘fit for purpose’, is, therefore, debatable.
The current statement also does not consider the role of cost-effectiveness, a major component of UK health-care decision-making. On an almost monthly basis, the NICE Diagnostic Assessment Programme produces health technology assessments and economic models that should, in principle, be adaptable to evaluating the impact of APGs on clinical outcomes and cost-effectiveness. Health economists and decision modelers have a wealth of methodological expertise that may be highly useful in developing APGs. Similarly, a better understanding of analytical and pre-analytical factors may prove useful in health technology assessments. Further methodological work in this area should be pursued.
Considering the demonstrable impact of pre-analytical and analytical factors on biomarker measurements, better utilisation of metrological concepts (e.g. measurement uncertainty) by medical statisticians may improve diagnostic test study and trial design, particularly in terms of power calculations, optimal cut-off thresholds and monitoring intervals. Furthermore, the specific impact of random (imprecision) and systematic (bias and specificity) factors on trial design should be considered. A recent simulation study of glycated haemoglobin measurement convincingly demonstrated that, when using a fixed guideline-based cut-off point, varying the bias and imprecision had very different effects on diagnostic accuracy, with bias having the most severe consequences. 864
Limitations
A programme with a responsibility for developing a framework built on excellent research innovative methodology, excellent clinical biochemistry and appraisal of the analytical and clinical validity of tests and also delivering a substantial RCT of a monitoring regimen was likely to be challenging. Our literature reviews demonstrate the scale of the challenge. There were likely to be disappointments and limitations. We feel that it is appropriate to highlight five of these:
-
The delays in set-up and recruitment into the study cohorts in workstream 2 and the ELUCIDATE trial had a negative impact on our ability to generate large and complete data sets with adequate long-term follow-up to answer questions conclusively. Further follow-up is required and will be carried out.
-
The low to moderate compliance rate in the ELUCIDATE trial may reduce the effect size of any benefit of ELF, and may possibly render the trial underpowered, although a different package of investigations was clearly delivered between the arms following the diagnosis of cirrhosis.
-
The absence of long-term follow-up data precludes conclusions being made about the true value of the ELF test to alter the serious consequences of liver disease.
-
The absence of long-term follow-up data at this stage means that there are limited opportunities for more methodological research.
-
The discovery pipeline in renal and liver disease has not yet provided exciting new molecular biomarkers to evaluate in our cohorts.
Final comments
In this programme, we have summarised what is known about monitoring tests using prominent examples and rigorous methodological appraisal and systematic overview; developed new approaches to evaluating the analytical and clinical validity of new biomarkers, particularly exploring the requirements for underpinning infrastructures; considered the products of modern proteomics; and delivered an exemplar RCT that has demonstrated changes in the process of care for a vitally important and growing area of morbidity and mortality in the UK. Incomplete follow-up as a consequence of delays in set-up and recruitment have limited our conclusions; these can be remedied by health informatic strategies, which we have outlined and planned and will robustly deliver.
The work of this programme was pivotal in our proposal for a NIHR DEC and the methodological, clinical biochemistry and clinical trials conclusions from the programme will continue to underpin the delivery of diagnostic evidence and the development of novel methods for delivering it in a more timely and cost-effective way for many years to come.
Acknowledgements
We are grateful to the patients who donated the samples and the clinical and nursing staff who contributed to the various cohorts of the programme. In addition, the authors would like to acknowledge the contributions of:
-
the members of LIVErNORTH for their contribution towards the PPI study
-
Ms Claire Davies, Senior Trial Manager, for help and support at the outset of the trial
-
Dr Sue Pavitt for assisting with aspects of translational medicine at the outset of the trial
-
Dr Barbara Potrata, Research Fellow, for help with design and set-up at the outset of the ELUCIDATE trial patient exit study
-
the ELUCIDATE trial DMEC – Dr Shahid Khan, Professor Elaine McColl, Professor Peter Mills and Professor Paula Williamson
-
the independent Trial Steering Committee members: Mrs Joan Bedlington (PPI), Professor Simon Dixon (Health Economist), Dr Jonathon Fallowfield (Hepatologist), Mrs Tilly Hale (PPI), Professor James Neuberger (Hepatologist), Dr Christine Patch (Clinical Geneticist) and Dr Andrew Roddam (Statistician)
-
Philip Akude and Mike Paulden for their contributions to the health economics study, with Professor Chris McCabe
-
Business Managers and Finance – Sandra Holliday, Donna Johnstone, Wendy Kennedy, Simon Revesai and Tom St David-Smith
-
sample banking and data management team: Joanne Brown, Andrew Bernard, Narinder Gahir, Damien Hindmarch, Leah Khazin, Norma Lister, Riitta Partanen, Lauren Tate, Emma Tidswell, Sharon Jackson and Gunnar Piho
-
the chairperson, Professor Phil Kalra, and committee members of the CRN Speciality Group for Renal Diseases and fellow committee members
-
the chairperson, Dr Stephen Ryder, and committee members of the CRN Speciality Group for Hepatology
-
all of the clinical laboratories that participated in the inter-laboratory study – Dr Allan Thompson and Mr James Dowd at Siemens Diagnostics and Helena Baker and colleagues at the Blood Sciences Laboratory at Leeds Teaching Hospitals – and Helena Baker for her involvement in the VEGF stability study, which will be extended and published
-
Dr Rebecca Kift and Miss Sophie Hepburn for their work on the analytical performance assessment of commercially available NGAL tests.
Contributions of authors
All authors made a substantial contribution to the concept and design of one or more of the studies in this programme or the acquisition of data, data analysis or interpretation of data and drafted the manuscript or revised it critically for important intellectual content. All senior programme leaders contributed to all chapters.
Peter J Selby (Programme Director and Clinical Lead on Renal Cancer) led on the management of the programme with the Programme Manager, was Co-chief Investigator of the ELUCIDATE trial and co-ordinated the completion of this report.
Rosamonde E Banks (Scientific Lead on Renal Disease Biomarkers) was a Senior Programme Leader, contributed to the programme application and led the clinical translation workstream (workstream 2) and the preparation of the workstream 2 chapters of this report.
Walter Gregory (Senior Lead, ELUCIDATE Trial Director and Principal Statistician) was a Senior Programme Leader, contributed to the programme application, led the ELUCIDATE trial workstream (workstream 3) and was Chief Statistician for the cohort and RCT design and analysis.
Jenny Hewison (Deputy Programme Director and Lead on Health Services Research) was a Senior Programme Leader, contributed to the programme application and led the methodology workstream (workstream 1) and preparation of the workstream 1 chapters of this report.
William Rosenberg (Clinical Lead on Hepatology) was a Senior Programme Leader and Chief Investigator of the ELUCIDATE trial and contributed to the programme application and drafting of this report.
Douglas G Altman (Strategic Lead on Methodology) was a programme co-applicant, contributed to the programme application and work on evidence-based monitoring and impact and contributed to Chapters 2–6 and 18.
Jonathan J Deeks (Strategic Lead on Methodology) was a programme co-applicant and senior lead on the monitoring and simulation aspects of the methodology workstream (workstream 1), advised on all aspects of the programme methodology and contributed to the programme application and drafting of this report (Chapters 2–7).
Christopher McCabe (Senior Lead on Health Economics) was a programme co-applicant, contributed to the programme application and drafting of the report, including Chapter 8, provided the health economic evaluation and contributed to the methodological considerations in the optimisation of monitoring biomarkers.
Julie Parkes (Senior Lead and Senior Lecturer in Public Health) was a programme co-applicant, contributed to the programme application and drafting of this report, including Chapters 4 and 22, contributed to the design of the RCT and was the designated lead for the follow-up of patients by the NHS Health and Social Care Information Centre.
Catharine Sturgeon (Strategic Lead on Clinical Biochemistry) was a programme co-applicant, contributed to the programme application and drafting of this report, including Chapters 13 and 17, advised on the clinical biochemistry work and contributed to the verification of the ELF test analytical performance.
Douglas Thompson (Operational Lead on Clinical Biochemistry) was a programme co-applicant, contributed to the programme application and clinical biochemistry work and contributed to the exploration of verification, validation and pre-analytical variables of biomarkers within the clinical translation workstream (Chapters 10, 13–15 and 17).
Maureen Twiddy (Senior Research Fellow) co-ordinated the patient and public evaluation of the methodology workstream (workstream 1) and ELUCIDATE trial (workstream 3) and contributed to the drafting of Chapters 9 and 23.
Janine Bestall (Senior Research Fellow) co-ordinated the patient and public evaluation of the methodology workstream (workstream 1) and ELUCIDATE trial (workstream 3) and contributed to the drafting of Chapters 9 and 23.
Joan Bedlington (Patient and Public Contributor) provided advice and guidance during the ELUCIDATE trial and assisted with the patient perspectives study and contributed to the drafting of Chapters 9 and 23.
Tilly Hale (Patient and Public Contributor) provided advice and guidance during the ELUCIDATE trial and assisted with the patient perspectives study and contributed to the drafting of Chapters 9 and 23. Sadly, Mrs Hale died in December 2015.
Jacqueline Dinnes (Senior Researcher, Methodology) was a theme operational lead, contributed to the methodology workstream (workstream 1) and led on the review of methods for monitoring and examination of case studies, led on Chapters 2–4 and 6 and contributed to Chapter 5 and the drafting of the report.
Marc Jones (Trial Co-Ordinator) was a theme operational lead, provided overall co-ordination and management of recruitment, compliance and follow-up for the ELUCIDATE trial and biomarker cohorts and contributed to the drafting of this report, including Chapters 11, 16, 18 and 20.
Andrew Lewington (Consultant Renal Physician and Honorary Associate Professor) was a clinical operational lead and Chief Investigator for the validation of biomarkers in the diagnosis of early kidney transplant complications and the prediction of chronic kidney transplant dysfunction, contributed to the review and prioritisation of circulating biomarkers in the renal transplantation cohort and contributed to the content of Chapters 10–12 and 15 and the overall drafting of the report.
Michael P Messenger (Principal Health-care Scientist) was a theme operational lead, led the set-up and co-ordination of centres for the prospective observational cohorts within the clinical translation workstream (workstream 2), contributed to the verification and validation of biomarker assays and the pre-analytical performance of the ELF test, led on Chapters 11 and 17 and contributed to the content of Chapters 12–15 and the overall drafting of the report.
Vicky Napp (Operations Director) was a theme operational lead, provided overall direction and management of the ELUCIDATE trial and contributed to Chapters 1, 16, 18, 20 and 22 and the overall drafting of this report.
Alice Sitch (Lecturer in Medical Statistics) was a theme operational lead, provided simulation modelling of patient benefit, led on Chapters 5 and 7 and contributed to Chapters 4 and 6 and the overall drafting of the report.
Sudeep Tanwar (ELUCIDATE Trial Co-Chief Investigator) was a clinical operational lead and Co-chief Investigator of the ELUCIDATE trial, directed the delivery of the later stages of the RCT and contributed to Chapters 20 and 21 and the overall drafting of the report.
Naveen S Vasudev (Clinical Associate Professor and Honorary Consultant in Medical Oncology) was a clinical operational lead and Co-chief Investigator of the validation/qualification of prognostic and monitoring biomarkers in RCC study and contributed to Chapters 10–12, 14 and 15 and the overall drafting of the report.
Paul Baxter (Associate Professor in Biostatistics) was a Co-investigator, provided health economic evaluation of the ELUCIDATE trial, contributed to the methodological considerations in the optimisation of monitoring biomarkers and contributed to Chapter 8 and the overall drafting of the report.
Sue Bell (Senior Trial Manager) co-ordinated and managed the ELUCIDATE trial, led on Chapter 16 and contributed to Chapters 18, 20 and 22 and the overall drafting of the report.
David A Cairns (Biostatistician) provided statistical analysis within the clinical translation workstream (workstream 2) and contributed to Chapter 14 and the drafting of the report.
Nicola Calder (Senior Research Officer) supported the co-ordination of the biomarker cohorts, contributed to the evaluation of promising renal cancer biomarker candidates and contributed to the content of Chapters 14 and 17 and the overall drafting of the report.
Neil Corrigan (Senior Medical Statistician) provided statistical support to the ELUDICATE trial and contributed to Chapter 18 and the drafting of the report.
Francesco Del Galdo (Senior Lecturer) contributed to the verification of the ELF test analytical performance in Chapter 17 and the drafting of the report.
Peter Heudtlass (Medical Statistician) provided statistical support to the ELUDICATE trial and contributed to Chapters 20 and 21 and the drafting of the report.
Nick Hornigold (Postdoctoral Researcher) designed and conducted the immunoassay work on biomarkers, which is reported in Chapter 14.
Claire Hulme (Professor of Health Economics) provided input to the health economic analysis of the ELUCIDATE trial in Chapter 21 and contributed to the drafting of the report.
Michelle Hutchinson (Biostatistician) – provided statistical analysis within the clinical translation workstream and contributed to Chapters 13 and 14 and the drafting of the report.
Carys Lippiatt (Clinical Lead, Pathology Research and Development) provided clinical biochemistry input and contributed to the drafting of Chapters 10 and 13–15 of the report.
Tobias Livingstone (Senior Research Officer) assisted with exploring the verification, validation and pre-analytical variables of biomarker assays and contributed to Chapter 13 and the drafting of the report.
Roberta Longo (Research Fellow, Health Economics) provided health economic evaluation of the ELUCIDATE trial (Chapter 21), led on the methodological considerations in the optimisation of monitoring biomarkers (Chapter 8) and contributed to the drafting of the report.
Matthew Potton (Data Management Assistant) provided data management for the ELUCIDATE trial and cohorts within the clinical translation workstream (workstream 2) and contributed to the drafting of the report.
Stephanie Roberts (Programme Manager) co-ordinated the programme, including the programme delivery and funding, drafted important parts of the manuscript and assisted in the co-ordination of the ELF analytical validation study (Chapter 17).
Sheryl Sim (Consultant Oncologist) had input into the clinical translation workstream (workstream 2) and the accrual of the renal cancer cohort and contributed to the drafting of Chapters 11 and 14 of the report.
Sebastian Trainor (Clinical Research Fellow) had input into the clinical translation workstream (workstream 2) and the accrual of the renal cancer cohort and contributed to the drafting of Chapter 14 of the report.
Matthew Welberry Smith (Renal Consultant) had input into the clinical translation workstream (workstream 2), the accrual of the renal transplant cohort and discovery of the ACY-1 biomarker and contributed to the drafting of Chapters 10–12 and 15.
James Neuberger (Professor of Hepatology) was the chairperson of the ELUCIDATE Trial Steering Committee and advised on the planning and delivery of the hepatology aspects of the programme, the design of the ELUCIDATE analysis and the drafting of the ELUCIDATE reports.
Douglas Thorburn (Consultant Hepatologist) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Royal Free London NHS Foundation Trust for the ELUCIDATE trial and contributed to the delivery of the trial and the drafting of the report.
Paul Richardson (Consultant Hepatologist) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Royal Liverpool and Broadgreen University Hospitals NHS Trust for the ELUCIDATE trial and contributed to the delivery of the trial and the drafting of the report.
John Christie (Consultant Gastroenterologist and Hepatologist) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Royal Devon and Exeter NHS Foundation Trust for the ELUCIDATE trial and contributed to the delivery of the trial and the drafting of the report.
Neil Sheerin (Professor of Nephrology) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Newcastle upon Tyne Hospitals NHS Foundation Trust for the renal transplant cohort and contributed to the delivery of the cohort and the drafting of the report.
William McKane (Consultant Nephrologist) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Sheffield Teaching Hospitals NHS Foundation Trust for the renal transplant cohort and contributed to the delivery of the cohort and the drafting of the report.
Paul Gibbs (Vascular and Transplant Surgeon) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Portsmouth Hospitals NHS Trust for the Renal Transplant Cohort and contributed to the delivery of the cohort and the drafting of the report.
Anusha Edwards (Kidney Transplant Surgeon) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the North Bristol NHS Trust for the renal transplant cohort and contributed to the delivery of the cohort and the drafting of the report.
Naeem Soomro (Consultant Urologist) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Newcastle upon Tyne Hospitals NHS Foundation Trust for the renal cancer cohort and contributed to the delivery of the cohort and the drafting of the report.
Adebanji Adeyoju (Consultant Urological Surgeon) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at the Stockport NHS Foundation Trust for the renal cancer cohort and contributed to the delivery of the cohort and the drafting of the report.
Grant D Stewart (Clinical Senior Lecturer and Honorary Consultant in Urological Surgery) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at NHS Lothian for the renal cancer cohort and contributed to the delivery of the cohort and the drafting of the report.
David Hrouda (Consultant Urologist) – top recruiting Hospital Principal Investigators and clinical team – was the Principal Investigator at Charing Cross Hospital, Imperial College Healthcare NHS Trust, for the renal cancer cohort and contributed to the delivery of the cohort and the drafting of the report.
Data sharing statement
Data can be obtained by contacting the corresponding author.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, CCF, NETSCC, PGfAR or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the PGfAR programme or the Department of Health and Social Care.
References
- Ransohoff DF. How to improve reliability and efficiency of research about molecular markers: roles of phases, guidelines, and study design. J Clin Epidemiol 2007;60:1205-19. https://doi.org/10.1016/j.jclinepi.2007.04.020.
- Zolg JW, Langen H. How industry is approaching the search for new diagnostic markers and biomarkers. Mol Cell Proteomics 2004;3:345-54. https://doi.org/10.1074/mcp.M400007-MCP200.
- Zolg W. The proteomic search for diagnostic biomarkers: lost in translation?. Mol Cell Proteomics 2006;5:1720-6. https://doi.org/10.1074/mcp.R600001-MCP200.
- Anderson NL. The roles of multiple proteomic platforms in a pipeline for new diagnostics. Mol Cell Proteomics 2005;4:1441-4. https://doi.org/10.1074/mcp.I500001-MCP200.
- Phillips KA, Van Bebber S, Issa AM. Diagnostics and biomarker development: priming the pipeline. Nat Rev Drug Discov 2006;5:463-9. https://doi.org/10.1038/nrd2033.
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol 2006;24:971-83. https://doi.org/10.1038/nbt1235.
- Wilson C, Schulz S, Waldman SA. Biomarker development, commercialization and regulation: individualisation of medicine lost in translation. Clin Pharmacol Ther 2007;81:153-5. https://doi.org/10.1038/sj.clpt.6100088.
- Lee JW, Figeys D, Vasilescu J. Biomarker assay translation from discovery to clinical studies in cancer drug development: quantification of emerging protein biomarkers. Adv Cancer Res 2007;96:269-98. https://doi.org/10.1016/S0065-230X(06)96010-2.
- Vitzthum F, Behrens F, Anderson NL, Shaw JH. Proteomics: from basic research to diagnostic application. A review of requirements & needs. J Proteome Res 2005;4:1086-97. https://doi.org/10.1021/pr050080b.
- Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics 2002;1:845-67. https://doi.org/10.1074/mcp.R200007-MCP200.
- National Institutes of Health . Office of Strategic Coordination – the Common Fund n.d. https://commonfund.nih.gov/grants/fundedresearch (accessed 19 December 2017).
- Evaluating and Introducing New Diagnostic Tests: The Need for a National Strategy. London: Royal College of Pathologists; 2006.
- On the State of the Public Health. Annual Report of the Chief Medical Officer. London: Department of Health and Social Care; 2001.
- Leon DA, McCambridge J. Liver cirrhosis mortality rates in Britain, 1950 to 2002. Lancet 2006;367. https://doi.org/10.1016/S0140-6736(06)68250-0.
- Griffiths C, Rooney C, Brock A. Leading causes of death in England and Wales – how should we group causes?. Health Stat Q 2005;28:6-17.
- Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology 2004;127:S87-96. https://doi.org/10.1053/j.gastro.2004.09.020.
- Bialecki ES, Di Bisceglie AM. Clinical presentation and natural course of hepatocellular carcinoma. Eur J Gastroenterol Hepatol 2005;17:485-9. https://doi.org/10.1097/00042737-200505000-00003.
- Meier-Kriesche HU, Ojo AO, Hanson JA, Cibrik DM, Punch JD, Leichtman AB, et al. Increased impact of acute rejection on chronic allograft failure in recent era. Transplantation 2000;70:1098-100. https://doi.org/10.1097/00007890-200010150-00018.
- Pallardó Mateu LM, Sancho Calabuig A, Capdevila Plaza L, Franco Esteve A. Acute rejection and late renal transplant failure: risk factors and prognosis. Nephrol Dial Transplant 2004;19:iii38-42. https://doi.org/10.1093/ndt/gfh1013.
- Irish WD, McCollum DA, Tesi RJ, Owen AB, Brennan DC, Bailly JE, et al. Nomogram for predicting the likelihood of delayed graft function in adult cadaveric renal transplant recipients. J Am Soc Nephrol 2003;14:2967-74. https://doi.org/10.1097/01.ASN.0000093254.31868.85.
- Brier ME, Ray PC, Klein JB. Prediction of delayed renal allograft function using an artificial neural network. Nephrol Dial Transplant 2003;18:2655-9. https://doi.org/10.1093/ndt/gfg439.
- Snyder JJ, Kasiske BL, Gilbertson DT, Collins AJ. A comparison of transplant outcomes in peritoneal and hemodialysis patients. Kidney Int 2002;62:1423-30. https://doi.org/10.1111/j.1523-1755.2002.kid563.x.
- Sola R, Alarcón A, Jiménez C, Osuna A. The influence of delayed graft function. Nephrol Dial Transplant 2004;19:iii32-7. https://doi.org/10.1093/ndt/gfh1012.
- Giral-Classe M, Hourmant M, Cantarovich D, Dantal J, Blancho G, Daguin P, et al. Delayed graft function of more than six days strongly decreases long-term survival of transplanted kidneys. Kidney Int 1998;54:972-8. https://doi.org/10.1046/j.1523–1755.1998.00071.x.
- American Society of Nephrology. American Society of Nephrology Renal Research Report . J Am Soc Nephrol 2005;16:1886-903. https://doi.org/10.1681/asn.2005030285.
- Drucker BJ. Renal cell carcinoma: current status and future prospects. Cancer Treat Rev 2005;31:536-45. https://doi.org/10.1016/j.ctrv.2005.07.009.
- Pantuck AJ, Zisman A, Belldegrun AS. The changing natural history of renal cell carcinoma. J Urol 2001;166:1611-23. https://doi.org/10.1016/S0022-5347(05)65640-6.
- Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med 2003;349:427-34. https://doi.org/10.1056/nejmoa021491.
- Ratain MJ, Eisen T, Stadler WM, Flaherty KT, Kaye SB, Rosner GL, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24:2505-12. https://doi.org/10.1200/jco.2005.03.6723.
- Motzer RJ, Michaelson MD, Rosenberg J, Bukowski RM, Curti BD, George DJ, et al. Sunitinib efficacy against advanced renal cell carcinoma. J Urol 2007;178:1883-7. https://doi.org/10.1016/j.juro.2007.07.030.
- Escudier B, Lassau N, Angevin E, Soria JC, Chami L, Lamuraglia M, et al. Phase I trial of sorafenib in combination with IFN alpha-2a in patients with unresectable and/or metastatic renal cell carcinoma or malignant melanoma. Clin Cancer Res 2007;13:1801-9. https://doi.org/10.1158/1078-0432.CCR-06-1432.
- McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics . Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst 2005;97:1180-4. https://doi.org/10.1093/jnci/dji237.
- Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. Towards complete and accurate reporting of studies of diagnostic accuracy: the STARD initiative. BMJ 2003;326:41-4. https://doi.org/10.1136/bmj.326.7379.41.
- Guha IN, Parkes J, Roderick P, Chattopadhyay D, Cross R, Harris S, et al. Noninvasive markers of fibrosis in nonalcoholic fatty liver disease: validating the European Liver Fibrosis Panel and exploring simple markers. Hepatology 2008;47:455-60. https://doi.org/10.1002/hep.21984.
- Parkes J, Roderick P, Wheatley M, Alexander G, Collier J, Day CP, et al. The European liver fibrosis panel of serum markers can predict clinical outcome in a cohort of patients from England with mixed aetiology chronic liver disease. Hepatology 2007;46.
- Parkes J, Bialek S, Bell B, Terrault N, Zaman A, Sofair A, et al. European liver fibrosis (ELF) markers accurately distinguish fibrosis severity in chronic hepatitis C (CHC); an external validation study in a population-based cohort. Hepatology 2006;44.
- Parkes J, Mayo M, Cross R, Harris S, Roderick P, Coombes B, et al. European liver fibrosis (ELF) markers accurately distinguish fibrosis severity in primary biliary cirrhosis; an external validation study. Hepatology 2006;44.
- Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Riley RD, Burchill SA, Abrams KR, Heney D, Lambert PC, Jones DR, et al. A systematic review and evaluation of the use of tumour markers in paediatric oncology: Ewing’s sarcoma and neuroblastoma. Health Technol Assess 2003;7. https://doi.org/10.3310/hta7050.
- Gagnon A, Ye B. Discovery and application of protein biomarkers for ovarian cancer. Curr Opin Obstet Gynecol 2008;20:9-13. https://doi.org/10.1097/GCO.0b013e3282f226a5.
- Lieberman R. Evidence-based medical perspectives: the evolving role of PSA for early detection, monitoring of treatment response, and as a surrogate end point of efficacy for interventions in men with different clinical risk states for the prevention and progression of prostate cancer. Am J Ther 2004;11:501-6. https://doi.org/10.1097/01.mjt.0000141604.20320.0c.
- Ulmert D, Serio AM, O’Brien MF, Becker C, Eastham JA, Scardino PT, et al. Long-term prediction of prostate cancer: prostate-specific antigen (PSA) velocity is predictive but does not improve the predictive accuracy of a single PSA measurement 15 years or more before cancer diagnosis in a large, representative, unscreened population. J Clin Oncol 2008;26:835-41. https://doi.org/10.1200/jco.2007.13.1490.
- Cadranel JF, Rufat P, Degos F. Practices of liver biopsy in France: results of a prospective nationwide survey. For the Group of Epidemiology of the French Association for the Study of the Liver (AFEF). Hepatology 2000;32:477-81. https://doi.org/10.1053/jhep.2000.16602.
- Bedossa P, Dargère D, Paradis V. Sampling variability of liver fibrosis in chronic hepatitis C. Hepatology 2003;38:1449-57. https://doi.org/10.1016/j.hep.2003.09.022.
- Bedossa P. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology 1994;20:15-20. https://doi.org/10.1002/hep.1840200104.
- Arena U, Vizzutti F, Corti G, Ambu S, Stasi C, Bresci S, et al. Acute viral hepatitis increases liver stiffness values measured by transient elastography. Hepatology 2007;47:380-4. https://doi.org/10.1002/hep.22007.
- Sagir A, Erhardt A, Schmitt M, Häussinger D. Transient elastography is unreliable for detection of cirrhosis in patients with acute liver damage. Hepatology 2008;47:592-5. https://doi.org/10.1002/hep.22056.
- D’Amico G, Pagliaro L, Bosch J. Pharmacological treatment of portal hypertension: an evidence-based approach. Semin Liver Dis 1999;19:475-50. https://doi.org/10.1055/s-2007-1007133.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey W. Practice Guidelines Committee of the American Association for the Study of Liver Diseases . Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007;46:922-38. https://doi.org/10.1002/hep.21907.
- Lo GH, Chen WC, Chen MH, Lin CP, Lo CC, Hsu PI, et al. Endoscopic ligation vs. nadolol in the prevention of first variceal bleeding in patients with cirrhosis. Gastrointest Endosc 2004;59:333-8. https://doi.org/10.1016/S0016-5107(03)02819-0.
- Schepke M, Kleber G, Nurnberg D, Willert J, Koch L, Veltzke-Schlieker W, et al. Ligation versus propranolol for the primary prophylaxis of variceal bleeding in cirrhosis. Hepatology 2004;40:65-72. https://doi.org/10.1002/hep.20284.
- Lay C-S, Tsai Y-T, Lee F-Y, Lai Y-L, Yu C-J, Chen C-B, et al. Endoscopic variceal ligation versus propranolol in prophylaxis of first variceal bleeding in patients with cirrhosis. J Gastroenterol Hepatol 2006;21:413-19. https://doi.org/10.1111/j.1440-1746.2005.04071.x.
- Grangé J-D, Roulot D, Pelletier G, Pariente É-A, Denis J, Ink O, et al. Norfloxacin primary prophylaxis of bacterial infections in cirrhotic patients with ascites: a double-blind randomized trial. J Hepatol 1998;29:430-6. https://doi.org/10.1016/s0168-8278(98)80061-5.
- Zhang BH, Yang BH, Tang ZY. Randomized controlled trial of screening for hepatocellular carcinoma. J Cancer Res Clin Oncol 2004;130:417-22. https://doi.org/10.1007/s00432-004-0552-0.
- Chen JG, Parkin DM, Chen QG, Lu JH, Shen QJ, Zhang BC, et al. Screening for liver cancer: results of a randomised controlled trial in Qidong, China. J Med Screen 2003;10:204-9. https://doi.org/10.1258/096914103771773320.
- Parikh S, Hyman D. Hepatocellular cancer: a guide for the internist. Am J Med 2007;120:194-202. https://doi.org/10.1016/j.amjmed.2006.11.020.
- Mazzaferro V, Regalia E, Doci R, Andreola S, Pulvirenti A, Bozzetti F, et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996;334:693-9. https://doi.org/10.1056/NEJM199603143341104.
- Regalia E, Coppa J, Pulvirenti A, Romito R, Schiavo M, Burgoa L, et al. Liver transplantation for small hepatocellular carcinoma in cirrhosis: analysis of our experience. Transplant Proc 2001;33:1442-4. https://doi.org/10.1016/s0041-1345(00)02546-x.
- Parkes J, Guha IN, Roderick P, Rosenberg W. Performance of serum marker panels for liver fibrosis in chronic hepatitis C. J Hepatol 2006;44:462-74. https://doi.org/10.1016/j.jhep.2005.10.019.
- Gebo KA, Herlong HF, Torbenson MS, Jenckes MW, Chander G, Ghanem KG, et al. Role of liver biopsy in management of chronic hepatitis C: a systematic review. Hepatology 2002;36:161-72. https://doi.org/10.1053/jhep.2002.36989.
- Rosenberg WM, Voelker M, Thiel R, Becka M, Burt A, Schuppan D, et al. Serum markers detect the presence of liver fibrosis: a cohort study. Gastroenterology 2004;127:1704-13. https://doi.org/10.1053/j.gastro.2004.08.052.
- Glasziou PP, Aronson JK, Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Fryback DG, Thornbury JR. The efficacy of diagnostic imaging. Med Decis Making 1991;11:88-94. https://doi.org/10.1177/0272989X9101100203.
- Bossuyt PM, Lijmer JG, Mol BW. Randomised comparisons of medical tests: sometimes invalid, not always efficient. Lancet 2000;356:1844-7. https://doi.org/10.1016/S0140-6736(00)03246-3.
- Bell KJ, Glasziou PP, Hayen A, Irwig L. Criteria for monitoring tests were described: validity, responsiveness, detectability of long-term change, and practicality. J Clin Epidemiol 2014;67:152-9. https://doi.org/10.1016/j.jclinepi.2013.07.015.
- Mant D, Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Irwig L, Glasziou PP, Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Ferrante di Ruffano L, Hyde CJ, McCaffery KJ, Bossuyt PM, Deeks JJ. Assessing the value of diagnostic tests: a framework for designing and evaluating trials. BMJ 2012;344. https://doi.org/10.1136/bmj.e686.
- Dinnes J, Hewison J, Altman DG, Deeks JJ. The basis for monitoring strategies in clinical guidelines: a case study of prostate-specific antigen for monitoring in prostate cancer. CMAJ 2012;184:169-77. https://doi.org/10.1503/cmaj.110600.
- Lilja H, Ulmert D, Vickers AJ. Prostate-specific antigen and prostate cancer: prediction, detection and monitoring. Nat Rev Cancer 2008;8:268-78. https://doi.org/10.1038/nrc2351.
- Cookson MS, Aus G, Burnett AL, Canby-Hagino ED, D’Amico AV, Dmochowski RR, et al. Variation in the definition of biochemical recurrence in patients treated for localized prostate cancer: the American Urological Association Prostate Guidelines for Localized Prostate Cancer Update Panel report and recommendations for a standard in the reporting of surgical outcomes. J Urol 2007;177:540-5. https://doi.org/10.1016/j.juro.2006.10.097.
- The AGREE Collaboration . Development and validation of an international appraisal instrument for assessing the quality of clinical practice guidelines: the AGREE project. Qual Saf Health Care 2003;12:18-23. https://doi.org/10.1136/qhc.12.1.18.
- The Appraisal of Guidelines for Research & Evaluation (AGREE) Instrument. London: The AGREE Research Trust; 2001.
- AGREE Next Steps Consortium . AGREE II Instrument. Update: December 2017 n.d. www.agreetrust.org/wp-content/uploads/2017/12/AGREE-II-Users-Manual-and-23-item-Instrument-2009-Update-2017.pdf (accessed 21 March 2018).
- British Association of Urological Surgeons . Guidelines on the management of prostate cancer. Clin Oncol (R Coll Radiol) 1999;11:S53-S88.
- Clinical Practice Guidelines: Evidence-based Information and Recommendations for the Management of Localised Prostate Cancer. Canberra, ACT: National Health and Medical Research Council; 2002.
- Guideline for the Management of Clinically Localized Prostate Cancer: 2007 Update. Linthicum, MD: American Urological Association Education and Research, Inc.; 2007.
- Heidenreich A, Bolla M, Joniau S, van der Kwast TH, Matveev V, Mason MD, et al. EAU Guidelines on Prostate Cancer. Arnhem: EAU Guidelines Office; 2009.
- PDQ® Prostate Cancer Treatment. Bethesda, MD: National Cancer Institute; 2008.
- Prostate Cancer: Diagnosis and Treatment. Full Guideline. Cardiff: National Collaborating Centre for Cancer; 2008.
- Heidenreich A BM, Joniau S, van der Kwast TH, Matveev VB, Mason MD, . EAU guidelines on prostate cancer. Arnhem: European Association of Urology; 2009.
- NCCN Clinical Practice Guidelines in Oncology: Prostate Cancer. Fort Washington, PA: National Comprehensive Cancer Network; 2009.
- Prostate-Specific Antigen Best Practice Statement: 2009 Update. Linthicum, MD: American Urological Association Education and Research, Inc.; 2009.
- Consensus statement: guidelines for PSA following radiation therapy . American Society for Therapeutic Radiology and Oncology Consensus Panel. Int J Radiat Oncol Biol Phys 1997;37:1035-41.
- Roach M, Hanks G, Thames H, Schellhammer P, Shipley WU, Sokol GH, et al. Defining biochemical failure following radiotherapy with or without hormonal therapy in men with clinically localized prostate cancer: recommendations of the RTOG-ASTRO Phoenix Consensus Conference. Int J Radiat Oncol Biol Phys 2006;65:965-74. https://doi.org/10.1016/j.ijrobp.2006.04.029.
- Kuban DA, Levy LB, Potters L, Beyer DC, Blasko JC, Moran BJ, et al. Comparison of biochemical failure definitions for permanent prostate brachytherapy. Int J Radiat Oncol Biol Phys 2006;65:1487-93. https://doi.org/10.1016/j.ijrobp.2006.03.027.
- Horwitz EM, Thames HD, Kuban DA, Levy LB, Kupelian PA, Martinez AA, et al. Definitions of biochemical failure that best predict clinical failure in patients with prostate cancer treated with external beam radiation alone: a multi-institutional pooled analysis. J Urol 2005;173:797-802. https://doi.org/10.1097/01.ju.0000152556.53602.64.
- Stephenson AJ, Kattan MW, Eastham JA, Dotan ZA, Bianco FJ, Lilja H, et al. Defining biochemical recurrence of prostate cancer after radical prostatectomy: a proposal for a standardized definition. J Clin Oncol 2006;24:3973-8. https://doi.org/10.1200/jco.2005.04.0756.
- Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. JAMA 1999;281:1591-7. https://doi.org/10.1001/jama.281.17.1591.
- Vicini FA, Vargas C, Abner A, Kestin L, Horwitz E, Martinez A. Limitations in the use of serum prostate specific antigen levels to monitor patients after treatment for prostate cancer. J Urol 2005;173:1456-62. https://doi.org/10.1097/01.ju.0000157323.55611.23.
- Cox JD, Gallagher MJ, Hammond EH, Kaplan RS, Schellhammer PF. Consensus statements on radiation therapy of prostate cancer: guidelines for prostate re-biopsy after radiation and for radiation therapy with rising prostate-specific antigen levels after radical prostatectomy. American Society for Therapeutic Radiology and Oncology Consensus Panel. J Clin Oncol 1999;17. https://doi.org/10.1200/JCO.1999.17.4.1155.
- Carroll P, Coley C, McLeod D, Schellhammer P, Sweat G, Wasson J, et al. Prostate-specific antigen best practice policy – part II: prostate cancer staging and post-treatment follow-up. Urology 2001;57:225-9. https://doi.org/10.1016/S0090-4295(00)00994-8.
- Aus G. Current status of HIFU and cryotherapy in prostate cancer – a review. Eur Urol 2006;50:927-34. https://doi.org/10.1016/j.eururo.2006.07.011.
- Bott SR. Management of recurrent disease after radical prostatectomy. Prostate Cancer Prostatic Dis 2004;7:211-16. https://doi.org/10.1038/sj.pcan.4500732.
- Catton C, Milosevic M, Warde P, Bayley A, Crook J, Bristow R, et al. Recurrent prostate cancer following external beam radiotherapy: follow-up strategies and management. Urol Clin North Am 2003;30:751-63. https://doi.org/10.1016/S0094-0143(03)00051-X.
- Edelman MJ, Meyers FJ, Siegel D. The utility of follow-up testing after curative cancer therapy. A critical review and economic analysis. J Gen Intern Med 1997;12:318-31. https://doi.org/10.1007/s11606-006-5070-0.
- Lee AK, D’Amico AV. Utility of prostate-specific antigen kinetics in addition to clinical factors in the selection of patients for salvage local therapy. J Clin Oncol 2005;23:8192-7. https://doi.org/10.1200/jco.2005.03.0007.
- Nelson JB, Lepor H. Prostate cancer: radical prostatectomy. Urol Clin North Am 2003;30:703-23. https://doi.org/10.1016/S0094-0143(03)00049-1.
- Polascik TJ, Oesterling JE, Partin AW. Prostate specific antigen: a decade of discovery – what we have learned and where we are going. J Urol 1999;162:293-306. https://doi.org/10.1016/S0022-5347(05)68543-6.
- Selley S, Donovan J, Faulkner A, Coast J, Gillatt D. Diagnosis, management and screening of early localised prostate cancer. Health Technol Assess 1997;1.
- Yao SL, Dipaola RS. An evidence-based approach to prostate cancer follow-up. Semin Oncol 2003;30:390-40. https://doi.org/10.1016/S0093-7754(03)00099-X.
- Albertsen PC, Hanley JA, Penson DF, Fine J. Validation of increasing prostate specific antigen as a predictor of prostate cancer death after treatment of localized prostate cancer with surgery or radiation. J Urol 2004;171:2221-5. https://doi.org/10.1097/01.ju.0000124381.93689.b4.
- Amling CL, Bergstralh EJ, Blute ML, Slezak JM, Zincke H. Defining prostate specific antigen progression after radical prostatectomy: what is the most appropriate cut point?. J Urol 2001;165:1146-51. https://doi.org/10.1016/S0022-5347(05)66452-X.
- Booker J, Eardley A, Cowan R, Logue J, Wylie J, Caress AL. Telephone first post-intervention follow-up for men who have had radical radiotherapy to the prostate: evaluation of a novel service delivery approach. Eur J Oncol Nurse 2004;8:325-33. https://doi.org/10.1016/j.ejon.2004.01.003.
- Buyyounouski MK, Hanlon AL, Eisenberg DF, Horwitz EM, Feigenberg SJ, Uzzo RG, et al. Defining biochemical failure after radiotherapy with and without androgen deprivation for prostate cancer. Int J Radiat Oncol Biol Phys 2005;63:1455-62. https://doi.org/10.1016/j.ijrobp.2005.05.053.
- Cathala N, Brillat F, Mombet A, Lobel E, Prapotnich D, Alexandre L, et al. Patient followup after radical prostatectomy by internet medical file. J Urol 2003;170:2284-7. https://doi.org/10.1097/01.ju.0000095876.39932.4a.
- Cheung R, Kamat AM, de Crevoisier R, Allen PK, Lee AK, Tucker SL, et al. Outcome of salvage radiotherapy for biochemical failure after radical prostatectomy with or without hormonal therapy. Int J Radiat Oncol Biol Phys 2005;63:134-40. https://doi.org/10.1016/j.ijrobp.2005.01.020.
- Crook JM, Bahadur YA, Bociek RG, Perry GA, Robertson SJ, Esche BA. Radiotherapy for localized prostate carcinoma. The correlation of pretreatment prostate specific antigen and nadir prostate specific antigen with outcome as assessed by systematic biopsy and serum prostate specific antigen. Cancer 1997;79:328-36. https://doi.org/10.1002/(SICI)1097-0142(19970115)79:2<328::AID-CNCR16>3.0.CO;2-2.
- D’Amico AV, Moul J, Carroll PR, Sun L, Lubeck D, Chen MH. Prostate specific antigen doubling time as a surrogate end point for prostate cancer specific mortality following radical prostatectomy or radiation therapy. J Urol 2004;172:S42-6.
- Eastham JA, Riedel E, Scardino PT, Shike M, Fleisher M, Schatzkin A, et al. Variation of serum prostate-specific antigen levels: an evaluation of year-to-year fluctuations. JAMA 2003;289:2695-700. https://doi.org/10.1001/jama.289.20.2695.
- Frazier HA, Robertson JE, Humphrey PA, Paulson DF. Is prostate specific antigen of clinical importance in evaluating outcome after radical prostatectomy. J Urol 1993;149:516-18. https://doi.org/10.1016/S0022-5347(17)36132-3.
- Klotz L. Active surveillance with selective delayed intervention using PSA doubling time for good risk prostate cancer. Eur Urol 2005;47:16-21. https://doi.org/10.1016/j.eururo.2004.09.010.
- Leibman BD, Dillioglugil O, Wheeler TM, Scardino PT. Distant metastasis after radical prostatectomy in patients without an elevated serum prostate specific antigen level. Cancer 1995;76:2530-4. https://doi.org/10.1002/1097-0142(19951215)76:12<2530::AID-CNCR2820761219>3.0.CO;2-F.
- Nielsen ME, Makarov DV, Humphreys E, Mangold L, Partin AW, Walsh PC. Is it possible to compare PSA recurrence-free survival after surgery and radiotherapy using revised ASTRO criterion – ’nadir + 2’?. Urology 2008;72:389-93. https://doi.org/10.1016/j.urology.2007.10.053.
- Niwakawa M, Tobisu K, Fujimoto H, Matsuoka N, Kakizoe T. Medically and economically appropriate follow-up schedule for prostate cancer patients after radical prostatectomy. Int J Urol 2002;9:134-40. https://doi.org/10.1046/j.1442-2042.2002.00435.x.
- Oefelein MG, Smith N, Carter M, Dalton D, Schaeffer A. The incidence of prostate cancer progression with undetectable serum prostate specific antigen in a series of 394 radical prostatectomies. J Urol 1995;154:2128-31. https://doi.org/10.1016/S0022-5347(01)66713-2.
- Patel R, Lepor H, Thiel RP, Taneja SS. Prostate-specific antigen velocity accurately predicts response to salvage radiotherapy in men with biochemical relapse after radical prostatectomy. Urology 2005;65:942-6. https://doi.org/10.1016/j.urology.2004.12.013.
- Pickles T. Prostate-specific antigen (PSA) bounce and other fluctuations: which biochemical relapse definition is least prone to PSA false calls? An analysis of 2030 men treated for prostate cancer with external beam or brachytherapy with or without adjuvant androgen deprivation therapy. Int J Radiat Oncol Biol Phys 2006;64:1355-9. https://doi.org/10.1016/j.ijrobp.2005.10.008.
- Ragde H, Blasko JC, Grimm PD, Kenny GM, Sylvester JE, Hoak DC, et al. Interstitial iodine-125 radiation without adjuvant therapy in the treatment of clinically localized prostate carcinoma. Cancer 1997;80:442-53. https://doi.org/10.1002/(SICI)1097-0142(19970801)80:3<442::AID-CNCR12>3.0.CO;2-X.
- Ray ME, Thames HD, Levy LB, Horwitz EM, Kupelian PA, Martinez AA, et al. PSA nadir predicts biochemical and distant failures after external beam radiotherapy for prostate cancer: a multi-institutional analysis. Int J Radiat Oncol Biol Phys 2006;64:1140-50. https://doi.org/10.1016/j.ijrobp.2005.07.006.
- Ritter MA, Messing EM, Shanahan TG, Potts S, Chappell RJ, Kinsella TJ. Prostate-specific antigen as a predictor of radiotherapy response and patterns of failure in localized prostate cancer. J Clin Oncol 1992;10:1208-17. https://doi.org/10.1200/JCO.1992.10.8.1208.
- Rose MA, Shrader-Bogen CL, Korlath G, Priem J, Larson LR. Identifying patient symptoms after radiotherapy using a nurse-managed telephone interview. Oncol Nurs Forum 1996;23:99-102.
- Sandler HM, Dunn RL, McLaughlin PW, Hayman JA, Sullivan MA, Taylor JM. Overall survival after prostate-specific-antigen-detected recurrence following conformal radiation therapy. Int J Radiat Oncol Biol Phys 2000;48:629-33. https://doi.org/10.1016/S0360-3016(00)00717-3.
- Sartor CI, Strawderman MH, Lin XH, Kish KE, McLaughlin PW, Sandler HM. Rate of PSA rise predicts metastatic versus local recurrence after definitive radiotherapy. Int J Radiat Oncol Biol Phys 1997;38:941-7. https://doi.org/10.1016/S0360-3016(97)00082-5.
- Stamey TA, Kabalin JN, McNeal JE, Johnstone IM, Freiha F, Redwine EA, et al. Prostate specific antigen in the diagnosis and treatment of adenocarcinoma of the prostate. II. Radical prostatectomy treated patients. J Urol 1989;141:1076-83. https://doi.org/10.1016/S0022-5347(17)41175-X.
- Stephan C, Klaas M, Müller C, Schnorr D, Loening SA, Jung K. Interchangeability of measurements of total and free prostate-specific antigen in serum with 5 frequently used assay combinations: an update. Clin Chem 2006;52:59-64. https://doi.org/10.1373/clinchem.2005.059170.
- Stephenson AJ, Shariat SF, Zelefsky MJ, Kattan MW, Butler EB, Teh BS, et al. Salvage radiotherapy for recurrent prostate cancer after radical prostatectomy. JAMA 2004;291:1325-32. https://doi.org/10.1001/jama.291.11.1325.
- Trapasso JG, deKernion JB, Smith RB, Dorey F. The incidence and significance of detectable levels of serum prostate specific antigen after radical prostatectomy. J Urol 1994;152:1821-5. https://doi.org/10.1016/S0022-5347(17)32394-7.
- Trock BJ, Han M, Freedland SJ, Humphreys EB, DeWeese TL, Partin AW, et al. Prostate cancer-specific survival following salvage radiotherapy vs. observation in men with biochemical recurrence after radical prostatectomy. JAMA 2008;299:2760-9. https://doi.org/10.1001/jama.299.23.2760.
- Ward JF, Zincke H, Bergstralh EJ, Slezak JM, Blute ML. Prostate specific antigen doubling time subsequent to radical prostatectomy as a prognosticator of outcome following salvage radiotherapy. J Urol 2004;172:2244-8. https://doi.org/10.1097/01.ju.0000145262.34748.2b.
- Zagars GK, Pollack A. Kinetics of serum prostate-specific antigen after external beam radiation for clinically localized prostate cancer. Radiother Oncol 1997;44:213-21. https://doi.org/10.1016/S0167-8140(97)00123-0.
- Soletormos G, Semjonow A, Sibley PE, Lamerz R, Petersen PH, Albrecht W, et al. Biological variation of total prostate-specific antigen: a survey of published estimates and consequences for clinical practice. Clin Chem 2005;51:1342-51. https://doi.org/10.1373/clinchem.2004.046086.
- Whiting P, Rutjes AW, Reitsma JB, Glas AS, Bossuyt PM, Kleijnen J. Sources of variation and bias in studies of diagnostic accuracy: a systematic review. Ann Intern Med 2004;140:189-202. https://doi.org/10.7326/0003-4819-140-3-200402030-00010.
- Parker E, Glasziou P. Use of evidence in hypertension guidelines: should we measure in both arms?. Br J Gen Pract 2009;59:e87-92. https://doi.org/10.3399/bjgp09X395012.
- Moschetti I, Brandt D, Perera R, Clarke M, Heneghan C. Adequacy of reporting monitoring regimens of risk factors for cardiovascular disease in clinical guidelines: systematic review. BMJ 2011;342. https://doi.org/10.1136/bmj.d1289.
- Cruse H, Winiarek M, Marshburn J, Clark O, Djulbegovic B. Quality and methods of developing practice guidelines. BMC Health Serv Res 2002;2. https://doi.org/10.1186/1472-6963-2-1.
- Savoie I, Kazanjian A, Bassett K. Do clinical practice guidelines reflect research evidence?. J Health Serv Res Policy 2000;5:76-82. https://doi.org/10.1177/135581960000500204.
- Campbell F, Dickinson HO, Cook JV, Beyer FR, Eccles M, Mason JM. Methods underpinning national clinical guidelines for hypertension: describing the evidence shortfall. BMC Health Serv Res 2006;6. https://doi.org/10.1186/1472-6963-6-47.
- McAlister FA, van Diepen S, Padwal RS, Johnson JA, Majumdar SR. How evidence-based are the recommendations in evidence-based guidelines?. PLOS Med 2007;4. https://doi.org/10.1371/journal.pmed.0040250.
- Burgers JS, Bailey JV, Klazinga NS, Van Der Bij AK, Grol R, Feder G. AGREE Collaboration . Inside guidelines: comparative analysis of recommendations and evidence in diabetes guidelines from 13 countries. Diabetes Care 2002;25:1933-9. https://doi.org/10.2337/diacare.25.11.1933.
- Bellera C, Hanley J, Joseph L, Albertsen P. A statistical evaluation of rules for biochemical failure after radiotherapy in men treated for prostate cancer. Int J Radiat Oncol Biol Phys 2009;75:1357-63. https://doi.org/10.1016/j.ijrobp.2009.01.013.
- Stevens RJ, Oke J, Perera R. Statistical models for the control phase of clinical monitoring. Stat Methods Med Res 2010;19:394-41. https://doi.org/10.1177/0962280209359886.
- Glasziou PP, Irwig L, Heritier S, Simes RJ, Tonkin A. LIPID Study Investigators . Monitoring cholesterol levels: measurement error or true change?. Ann Intern Med 2008;148:656-61. https://doi.org/10.7326/0003-4819-148-9-200805060-00005.
- Keenan K, Hayen A, Neal BC, Irwig L. Long term monitoring in patients receiving treatment to lower blood pressure: analysis of data from placebo controlled randomised controlled trial. BMJ 2009;338. https://doi.org/10.1136/bmj.b1492.
- Goldstein DE, Little RR, Lorenz RA, Malone JI, Nathan D, Peterson CM, et al. Tests of glycemia in diabetes. Diabetes Care 2004;27:1761-73. https://doi.org/10.2337/diacare.27.7.1761.
- Buclin T, Telenti A, Perera R, Csajka C, Furrer H, Aronson JK, et al. Development and validation of decision rules to guide frequency of monitoring CD4 cell count in HIV-1 infection before starting antiretroviral therapy. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0018578.
- The Guidelines Manual. London: NICE; 2009.
- Brouwers MC, Kho ME, Browman GP, Burgers JS, Cluzeau F, Feder G, et al. AGREE II: advancing guideline development, reporting and evaluation in health care. CMAJ 2010;182:E839-42. https://doi.org/10.1503/cmaj.090449.
- Hillier S, Grimmer-Somers K, Merlin T, Middleton P, Salisbury J, Tooher R, et al. FORM: an Australian method for formulating and grading recommendations in evidence-based clinical guidelines. BMC Med Res Methodol 2011;11. https://doi.org/10.1186/1471-2288-11-23.
- Clinical Practice Guidelines We Can Trust. Washington, DC: National Academies Press; 2011.
- Bossuyt PM, Reitsma JB, Linnet K, Moons KG. Beyond diagnostic accuracy: the clinical utility of diagnostic tests. Clin Chem 2012;58:1636-43. https://doi.org/10.1373/clinchem.2012.182576.
- Bossuyt PMM, Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Kronborg O, Jørgensen OD, Fenger C, Rasmussen M. Three randomized long-term surveillance trials in patients with sporadic colorectal adenomas. Scand J Gastroenterol 2006;41:737-43. https://doi.org/10.1080/00365520500442666.
- Smits JH, van der Linden J, Hagen EC, Modderkolk-Cammeraat EC, Feith GW, Koomans HA, et al. Graft surveillance: venous pressure, access flow, or the combination?. Kidney Int 2001;59:1551-8. https://doi.org/10.1046/j.1523-1755.2001.0590041551.x.
- Dember LM, Holmberg EF, Kaufman JS. Randomized controlled trial of prophylactic repair of hemodialysis arteriovenous graft stenosis. Kidney Int 2004;66:390-8. https://doi.org/10.1111/j.1523-1755.2004.00743.x.
- Rustin GJ, van der Burg ME, Griffin CL, Guthrie D, Lamont A, Jayson GC, et al. Early versus delayed treatment of relapsed ovarian cancer (MRC OV05/EORTC 55955): a randomised trial. Lancet 2010;376:1155-63. https://doi.org/10.1016/S0140-6736(10)61268-8.
- Severe P, Juste MA, Ambroise A, Eliacin L, Marchand C, Apollon S, et al. Early versus standard antiretroviral therapy for HIV-infected adults in Haiti. N Engl J Med 2010;363:257-65. https://doi.org/10.1056/NEJMoa0910370.
- Rosendahl K, Dezateux C, Fosse KR, Aase H, Aukland SM, Reigstad H, et al. Immediate treatment versus sonographic surveillance for mild hip dysplasia in newborns. Pediatrics 2010;125:e9-16. https://doi.org/10.1542/peds.2009-0357.
- Kanda Y, Yamashita T, Mori T, Ito T, Tajika K, Mori S, et al. A randomized controlled trial of plasma real-time PCR and antigenemia assay for monitoring CMV infection after unrelated BMT. Bone Marrow Transplant 2010;45:1325-32. https://doi.org/10.1038/bmt.2009.337.
- Davies AH, Hawdon AJ, Sydes MR, Thompson SG. Is duplex surveillance of value after leg vein bypass grafting? Principal results of the Vein Graft Surveillance Randomised Trial (VGST). Circulation 2005;112:1985-91. https://doi.org/10.1161/circulationaha.104.518738.
- Polkinghorne KR, Lau KK, Saunder A, Atkins RC, Kerr PG. Does monthly native arteriovenous fistula blood-flow surveillance detect significant stenosis – a randomized controlled trial. Nephrol Dial Transplant 2006;21:2498-506. https://doi.org/10.1093/ndt/gfl242.
- Pham MX, Teuteberg JJ, Kfoury AG, Starling RC, Deng MC, Cappola TP, et al. Gene-expression profiling for rejection surveillance after cardiac transplantation. N Engl J Med 2010;362:1890-900. https://doi.org/10.1056/NEJMoa0912965.
- Trinchet JC, Chaffaut C, Bourcier V, Degos F, Henrion J, Fontaine H, et al. Ultrasonographic surveillance of hepatocellular carcinoma in cirrhosis: a randomized trial comparing 3- and 6-month periodicities. Hepatology 2011;54:1987-97. https://doi.org/10.1002/hep.24545.
- Moist LM, Churchill DN, House AA, Millward SF, Elliott JE, Kribs SW, et al. Regular monitoring of access flow compared with monitoring of venous pressure fails to improve graft survival. J Am Soc Nephrol 2003;14:2645-53. https://doi.org/10.1097/01.ASN.0000089562.98338.60.
- van den Bent MJ, Afra D, de Witte O, Ben Hassel M, Schraub S, Hoang-Xuan K, et al. Long-term efficacy of early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults: the EORTC 22845 randomised trial. Lancet 2005;366:985-90. https://doi.org/10.1016/S0140-6736(05)67070-5.
- Koinberg IL, Fridlund B, Engholm GB, Holmberg L. Nurse-led follow-up on demand or by a physician after breast cancer surgery: a randomised study. Eur J Oncol Nurs 2004;8:109-17. https://doi.org/10.1016/j.ejon.2003.12.005.
- Lilleri D, Gerna G, Furione M, Bernardo ME, Giorgiani G, Telli S, et al. Use of a DNAemia cut-off for monitoring human cytomegalovirus infection reduces the number of preemptively treated children and young adults receiving hematopoietic stem-cell transplantation compared with qualitative pp65 antigenemia. Blood 2007;110:2757-60. https://doi.org/10.1182/blood-2007-03-080820.
- Tan BH, Low JG, Chlebicka NL, Kurup A, Cheah FK, Lin RT, et al. Galactomannan-guided preemptive vs. empirical antifungals in the persistently febrile neutropenic patient: a prospective randomized study. Int J Infect Dis 2011;15:e350-6. https://doi.org/10.1016/j.ijid.2011.01.011.
- Tessitore N, Lipari G, Poli A, Bedogna V, Baggio E, Loschiavo C, et al. Can blood flow surveillance and pre-emptive repair of subclinical stenosis prolong the useful life of arteriovenous fistulae? A randomized controlled study. Nephrol Dial Transplant 2004;19:2325-33. https://doi.org/10.1093/ndt/gfh316.
- Abraham WT, Adamson PB, Bourge RC, Aaron MF, Costanzo MR, Stevenson LW, et al. Wireless pulmonary artery haemodynamic monitoring in chronic heart failure: a randomised controlled trial. Lancet 2011;377:658-66. https://doi.org/10.1016/s0140-6736(11)60101-3.
- Bourge RC, Abraham WT, Adamson PB, Aaron MF, Aranda JM, Magalski A, et al. Randomized controlled trial of an implantable continuous hemodynamic monitor in patients with advanced heart failure: the COMPASS-HF study. J Am Coll Cardiol 2008;51:1073-9. https://doi.org/10.1016/j.jacc.2007.10.061.
- Armstrong DG, Holtz-Neiderer K, Wendel C, Mohler MJ, Kimbriel HR, Lavery LA. Skin temperature monitoring reduces the risk for diabetic foot ulceration in high-risk patients. Am J Med 2007;120:1042-6. https://doi.org/10.1016/j.amjmed.2007.06.028.
- Mant D, Gray A, Pugh S, Campbell H, George S, Fuller A. Southampton (UK): NIHR Journals Library; 2017.
- Lavery LA, Higgins KR, Lanctot DR, Constantinides GP, Zamorano RG, Armstrong DG, et al. Home monitoring of foot skin temperatures to prevent ulceration. Diabetes Care 2004;27:2642-7. https://doi.org/10.2337/diacare.27.11.2642.
- Sobhani I, Tiret E, Lebtahi R, Aparicio T, Itti E, Montravers F, et al. Early detection of recurrence by 18FDG-PET in the follow-up of patients with colorectal cancer. Br J Cancer 2008;98:875-80. https://doi.org/10.1038/sj.bjc.6604263.
- Barr H. Barrett’s Oesophagus Two Yearly Surveillance Versus Endoscopy At Need: A Randomised Controlled Trial to Estimate Effectiveness and Cost-effectiveness Study (BOSS). Protocol Version 13. Gloucester; 2011.
- Watanabe T, Ajioka Y, Matsumoto T, Tomotsugu N, Takebayashi T, Inoue E, et al. Target biopsy or step biopsy? Optimal surveillance for ulcerative colitis: a Japanese nationwide randomized controlled trial. J Gastroenterol 2011;46:11-6. https://doi.org/10.1007/s00535-010-0327-0.
- TOMBOLA Group . Cytological surveillance compared with immediate referral for colposcopy in management of women with low grade cervical abnormalities: multicentre randomised controlled trial. BMJ 2009;339. https://doi.org/10.1136/bmj.b2546.
- van der Aa MN, Steyerberg EW, Sen EF, Zwarthoff EC, Kirkels WJ, van der Kwast TH, et al. Patients’ perceived burden of cystoscopic and urinary surveillance of bladder cancer: a randomized comparison. BJU Int 2008;101:1106-10. https://doi.org/10.1111/j.1464-410X.2007.07224.x.
- Rustin GJ, Mead GM, Stenning SP, Vasey PA, Aass N, Huddart RA, et al. Randomized trial of two or five computed tomography scans in the surveillance of patients with stage I nonseminomatous germ cell tumors of the testis: Medical Research Council Trial TE08, ISRCTN56475197 – the National Cancer Research Institute Testis Cancer Clinical Studies Group. J Clin Oncol 2007;25:1310-15. https://doi.org/10.1200/jco.2006.08.4889.
- Gerna G, Baldanti F, Torsellini M, Minoli L, Vigano M, Oggionnis T, et al. Evaluation of cytomegalovirus DNAaemia versus pp65-antigenaemia cutoff for guiding preemptive therapy in transplant recipients: a randomized study. Antivir Ther 2007;12:63-72.
- Grossmann EM, Johnson FE, Virgo KS, Longo WE, Fossati R. Follow-up of colorectal cancer patients after resection with curative intent – the GILDA trial. Surg Oncol 2004;13:119-24. https://doi.org/10.1016/j.suronc.2004.08.005.
- Kim DY. Prospective, Randomized Study of PIVKA-II and AFP Measurement Every 3 Months Compared to AFP Every 6 Months in Surveillance Program for Early Detection of Hepatocellular Carcinoma 2007.
- Joffe J, Gabe R. Trial of Imaging and Schedule in Seminoma Testis (TRISST) 2010. www.isrctn.com/ISRCTN65987321.
- McCowan LM, Harding JE, Roberts AB, Barker SE, Ford C, Stewart AW. A pilot randomized controlled trial of two regimens of fetal surveillance for small-for-gestational-age fetuses with normal results of umbilical artery doppler velocimetry. Am J Obstet Gynacol 2000;182:81-6. https://doi.org/10.1016/S0002-9378(00)70494-7.
- Adamson PB, Abraham WT, Aaron M, Aranda J, Bourge RC, Smith A, et al. CHAMPION trial rationale and design: the long-term safety and clinical efficacy of a wireless pulmonary-artery pressure monitoring system. J Card Fail 2011;17:3-10. https://doi.org/10.1016/j.cardfail.2010.08.002.
- Adamson PB, Conti JB, Smith AL, Abraham WT, Aaron MF, Aranda JM, et al. Reducing events in patients with chronic heart failure (REDUCEhf ) study design: continuous hemodynamic monitoring with an implantable defibrillator. Clin Cardiol 2007;30:567-75. https://doi.org/10.1002/clc.20250.
- Braunschweig F, Ford I, Conraads V, Cowie MR, Jondeau G, Kautzner J, et al. Can monitoring of intrathoracic impedance reduce morbidity and mortality in patients with chronic heart failure? Rationale and design of the Diagnostic Outcome Trial in Heart Failure (DOT-HF). Eur J Heart Fail 2008;10:907-16. https://doi.org/10.1016/j.ejheart.2008.06.016.
- Burri H, Quesada A, Ricci RP, Boriani G, Davinelli M, Favale S, et al. The MOnitoring Resynchronization dEvices and CARdiac patiEnts (MORE-CARE) study: rationale and design. Am Heart J 2010;160:42-8. https://doi.org/10.1016/j.ahj.2010.04.005.
- Cao P. Comparison of surveillance vs aortic endografting for small aneurysm repair (CAESAR) trial: study design and progress. Eur J Vasc Endovasc Surg 2005;30:245-51. https://doi.org/10.1016/j.ejvs.2005.05.043.
- Cao P, De Rango P, Verzini F, Parlani G, Romano L, Cieri E, et al. Comparison of surveillance versus aortic endografting for small aneurysm repair (CAESAR): results from a randomised trial. Eur J Vasc Endovasc Surg 2011;41:13-25. https://doi.org/10.1016/j.ejvs.2010.08.026.
- De Rango P, Verzini F, Parlani G, Cieri E, Romano L, Loschi D, et al. Quality of life in patients with small abdominal aortic aneurysm: the effect of early endovascular repair versus surveillance in the CAESAR trial. Eur J Vasc Endovasc Surg 2011;41:324-31. https://doi.org/10.1016/j.ejvs.2010.11.005.
- Clarke MP, Hogan V, Buck D, Chen J, Powell C, Speed C, et al. An external pilot study to test the feasibility of a randomised controlled trial comparing eye muscle surgery against active monitoring for childhood intermittent distance exotropia [X(T)]. Health Technol Assess 2011;19:1-144. https://doi.org/10.3310/hta19390.
- Crossley GH, Chen J, Choucair W, Cohen TJ, Gohn DC, Johnson WB, et al. Clinical benefits of remote versus transtelephonic monitoring of implanted pacemakers. J Am Coll Cardiol 2009;54:2012-19. https://doi.org/10.1016/j.jacc.2009.10.001.
- Chen J, Wilkoff B, Choucair W, Cohen T, Crossley G, Johnson WB, et al. Design of the Pacemaker REmote Follow-up Evaluation and Review (PREFER) trial to assess the clinical value of the remote pacemaker interrogation in the management of pacemaker patients. Trials 2008;9. https://doi.org/10.1186/1745-6215-9-18.
- Crossley G, Boyle A, Vitense H, Sherfesee L, Mead RH. Trial design of the clinical evaluation of remote notification to reduce time to clinical decision: the Clinical evaluation Of remote NotificatioN to rEduCe Time to clinical decision (CONNECT) study. Am Heart J 2008;156:840-6. https://doi.org/10.1016/j.ahj.2008.06.028.
- Kirby PL, Brady AR, Thompson SG, Torgerson D, Davies AH. The Vein Graft Surveillance Trial: rationale, design and methods. VGST participants. Eur J Vasc Endovasc Surg 1999;18:469-74. https://doi.org/10.1053/ejvs.1999.0822.
- Ip J, Waldo AL, Lip GY, Rothwell PM, Martin DT, Bersohn MM, et al. Multicenter randomized study of anticoagulation guided by remote rhythm monitoring in patients with implantable cardioverter-defibrillator and CRT-D devices: rationale, design, and clinical characteristics of the initially enrolled cohort. The IMPACT study. Am Heart J 2009;158:364-70. https://doi.org/10.1016/j.ahj.2009.07.002.
- Kass MA, Heuer DK, Higginbotham EJ, Johnson CA, Keltner JL, Miller JP, et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol 2002;120:701-13. https://doi.org/10.1001/archopht.120.6.701.
- Gordon MO, Kass MA. Group ftOHTS . The Ocular Hypertension Treatment Study: design and baseline description of the participants. Arch Opthalmol 1999;117. https://doi.org/10.1001/archopht.117.5.573.
- Kass MA, Gordon MO, Gao F, Heuer DK, Higginbotham EJ, Johnson CA, et al. Delaying treatment of ocular hypertension: the ocular hypertension treatment study. Arch Ophthalmol 2010;128:276-87. https://doi.org/10.1001/archophthalmol.2010.20.
- Lederle FA, Wilson SE, Johnson GR, Reinke DB, Littooy FN, Acher CW, et al. Immediate repair compared with surveillance of small abdominal aortic aneurysms. N Engl J Med 2002;346:1437-44. https://doi.org/10.1056/NEJMoa012573.
- Lederle FA, Johnson GR, Wilson SE, Acher CW, Ballard DJ, Littooy FN, et al. Quality of life, impotence, and activity level in a randomized trial of immediate repair versus surveillance of small abdominal aortic aneurysm. J Vasc Surg 2003;38:745-52. https://doi.org/10.1016/S0741-5214(03)00423-3.
- Lund JN, Scholefield JH, Grainge MJ, Smith SJ, Mangham C, Armitage NC, et al. Risks, costs, and compliance limit colorectal adenoma surveillance: lessons from a randomised trial. Gut 2001;49:91-6. https://doi.org/10.1136/gut.49.1.91.
- Ouriel K, Clair DG, Kent KC, Zarins CK. Positive Impact of Endovascular Options for Treating Aneurysms Early (PIVOTAL) Investigators . Endovascular repair compared with surveillance for patients with small abdominal aortic aneurysms. J Vasc Surg 2010;51:1081-7. https://doi.org/10.1016/j.jvs.2009.10.113.
- Pham MX, Deng MC, Kfoury AG, Teuteberg JJ, Starling RC, Valantine H. Molecular testing for long-term rejection surveillance in heart transplant recipients: design of the Invasive Monitoring Attenuation Through Gene Expression (IMAGE) trial. J Heart Lung Transplant 2007;26:808-14. https://doi.org/10.1016/j.healun.2007.05.017.
- Ram SJ, Work J, Caldito GC, Eason JM, Pervez A, Paulson WD. A randomized controlled trial of blood flow and stenosis surveillance of hemodialysis grafts. Kidney Int 2003;64:272-80. https://doi.org/10.1046/j.1523-1755.2003.00070.x.
- Raymond J, Molyneux A, Fox A, Johnston SC, Collet JP, Rouleau I, et al. The TEAM trial: safety and efficacy of endovascular treatment of unruptured intracranial aneurysms in the prevention of aneurysmal hemorrhages: a randomized comparison with indefinite deferral of treatment in 2002 patients followed for 10 years. Trials 2008;9. https://doi.org/10.1186/1745-6215-9-43.
- Raymond J, Darsaut T, Molyneux A. TEAM Collaborative Group . A trial on unruptured intracranial aneurysms (the TEAM trial): results, lessons from a failure and the necessity for clinical care trials. Trials 2011;12. https://doi.org/10.1186/1745-6215-12-64.
- Robbin ML, Oser RF, Lee JY, Heudebert GR, Mennemeyer ST, Allon M. Randomized comparison of ultrasound surveillance and clinical monitoring on arteriovenous graft outcomes. Kidney Int 2006;69:730-5. https://doi.org/10.1038/sj.ki.5000129.
- Rodríguez MF, Saló J, Arcusa A, Boadas J, Piñol V, Bessa X, et al. Postoperative surveillance in patients with colorectal cancer who have undergone curative resection: a prospective, multicenter, randomized, controlled trial. J Clin Oncol 2006;24:386-93. https://doi.org/10.1200/JCO.2005.02.0826.
- Brurås KR, Aukland SM, Markestad T, Sera F, Dezateux C, Rosendahl K. Newborns with sonographically dysplastic and potentially unstable hips: 6-year follow-up of an RCT. Pediatrics 2011;127:e661-6. https://doi.org/10.1542/peds.2010-2572.
- Scaffaro LA, Bettio JA, Cavazzola SA, Campos BT, Burmeister JE, Pereira RM, et al. Maintenance of hemodialysis arteriovenous fistulas by an interventional strategy: clinical and duplex ultrasonographic surveillance followed by transluminal angioplasty. J Ultrasound Med 2009;28:1159-65. https://doi.org/10.7863/jum.2009.28.9.1159.
- Secco GB, Fardelli R, Gianquinto D, Bonfante P, Baldi E, Ravera G, et al. Efficacy and cost of risk-adapted follow-up in patients after colorectal cancer surgery: a prospective, randomized and controlled trial. Eur J Surg Oncol 2002;28:418-23. https://doi.org/10.1053/ejso.2001.1250.
- Cotton SC, Sharp L, Little J, Duncan I, Alexander L, Cruickshank ME, et al. Trial of management of borderline and other low-grade abnormal smears (TOMBOLA): trial design. Contemp Clin Trials 2006;27:449-71. https://doi.org/10.1016/j.cct.2006.04.001.
- Powell JT, Brown LC, Forbes JF, Fowkes FG, Greenhalgh RM, Ruckley CV, et al. Final 12-year follow-up of surgery versus surveillance in the UK Small Aneurysm Trial. Br J Surg 2007;94:702-8. https://doi.org/10.1002/bjs.5778.
- Mortality results for randomised controlled trial of early elective surgery or ultrasonographic surveillance for small abdominal aortic aneurysms . The UK Small Aneurysm Trial Participants. Lancet 1998;352:1649-55. https://doi.org/10.1016/S0140-6736(98)10137-X.
- The UK Small Aneurysm Trial: design, methods and progress . The UK Small Aneurysm Trial Participants. Eur J Vasc Endovasc Surg 1995;9:42-8. https://doi.org/10.1016/S1078-5884(05)80223-0.
- Karim ABMF, Afra D, Cornu P, Bleehan N, Schraub S, De Witte O, et al. Randomized trial on the efficacy of radiotherapy for cerebral low-grade glioma in the adult: European Organization for Research and Treatment of Cancer Study 22845 with the Medical Research Council study BRO4: an interim analysis. Int J Radiat Oncol Biol Phys 2002;52:316-24. https://doi.org/10.1016/S0360-3016(01)02692-X.
- van der Aa MN, Steyerberg EW, Bangma C, van Rhijn BW, Zwarthoff EC, van der Kwast TH. Cystoscopy revisited as the gold standard for detecting bladder cancer recurrence: diagnostic review bias in the randomized, prospective CEFUB trial. J Urol 2010;183:76-80. https://doi.org/10.1016/j.juro.2009.08.150.
- Varma N, Epstein AE, Irimpen A, Schweikert R, Love C. TRUST Investigators . Efficacy and safety of automatic remote monitoring for implantable cardioverter-defibrillator follow-up: the Lumos-T Safely Reduces Routine Office Device Follow-up (TRUST) trial. Circulation 2010;122:325-32. https://doi.org/10.1161/CIRCULATIONAHA.110.937409.
- Varma N. Rationale and design of a prospective study of the efficacy of a remote monitoring system used in implantable cardioverter defibrillator follow-up: the Lumos-T Reduces Routine Office Device Follow-Up Study (TRUST) study. Am Heart J 2007;154:1029-34. https://doi.org/10.1016/j.ahj.2007.07.051.
- Varma N, Michalski J, Epstein AE, Schweikert R. Automatic remote monitoring of implantable cardioverter-defibrillator lead and generator performance: the Lumos-T Safely RedUceS RouTine Office Device Follow-Up (TRUST) trial. Circ Arrhythm Electrophysiol 2010;3:428-36. https://doi.org/10.1161/CIRCEP.110.951962.
- Wilks J, Maw R, Peters TJ, Harvey I, Golding J. Randomised controlled trial of early surgery versus watchful waiting for glue ear: the effect on behavioural problems in pre-school children. Clin Otolaryngol Allied Sci 2000;25:209-14. https://doi.org/10.1046/j.1365-2273.2000.00319.x.
- UK Small Aneurysm Trial Participants . The long-term prognosis of patients with small abdominal aortic aneurysm following surgery or surveillance: 12-year final follow-up of patients enrolled in the UK Small Aneurysm Trial. The Vascular Society of Great Britain &Amp; Ireland Yearbook 2006;78.
- Tessitore N, Bedogna V, Poli A, Mantovani W, Lipari G, Baggio E, et al. Adding access blood flow surveillance to clinical monitoring reduces thrombosis rates and costs, and improves fistula patency in the short term: a controlled cohort study. Nephrol Dial Transplant 2008;23:3578-84. https://doi.org/10.1093/ndt/gfn275.
- Hopewell S, Loudon K, Clarke MJ, Oxman AD, Dickersin K. Publication bias in clinical trials due to statistical significance or direction of trial results. Cochrane Database Syst Rev 2009;1. https://doi.org/10.1002/14651858.MR000006.pub3.
- Delaney A, Angus DC, Bellomo R, Cameron P, Cooper DJ, Finfer S, et al. Bench-to-bedside review: the evaluation of complex interventions in critical care. Crit Care 2008;12. https://doi.org/10.1186/cc6849.
- Hawe P, Shiell A, Riley T. Complex interventions: how ‘out of control’ can a randomised controlled trial be?. BMJ 2004;328:1561-3. https://doi.org/10.1136/bmj.328.7455.1561.
- Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 2011;343. https://doi.org/10.1136/bmj.d5928.
- Akl EA, Sun X, Busse JW, Johnston BC, Briel M, Mulla S, et al. Specific instructions for estimating unclearly reported blinding status in randomized trials were reliable and valid. J Clin Epidemiol 2012;65:262-7. https://doi.org/10.1016/j.jclinepi.2011.04.015.
- Grimes DA, Schulz KF. Surrogate end points in clinical research: hazardous to your health. Obstet Gynecol 2005;105:1114-18. https://doi.org/10.1097/01.aog.0000157445.67309.19.
- Charles P, Giraudeau B, Dechartres A, Baron G, Ravaud P. Reporting of sample size calculation in randomised controlled trials: review. BMJ 2009;338. https://doi.org/10.1136/bmj.b1732.
- Kasenda B, von Elm EB, You J, Blumle A, Tomonaga Y, Saccilotto R, et al. Learning from failure – rationale and design for a study about discontinuation of randomized trials (DISCO study). BMC Med Res Methodol 2012;12. https://doi.org/10.1186/1471-2288-12-131.
- Schulz KF, Altman DG, Moher D. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340. https://doi.org/10.1136/bmj.c332.
- Ahdieh-Grant L. When to initiate highly active antiretroviral therapy: a cohort approach. Am J Epidemiol 2003;157:738-46. https://doi.org/10.1093/aje/kwg036.
- Bell KJ, Irwig L, Craig JC, Macaskill P. Use of randomised trials to decide when to monitor response to new treatment. BMJ 2008;336:361-5. https://doi.org/10.1136/bmj.39476.623611.25.
- Bell KJL, Hayen A, Macaskill P, Irwig L, Craig JC, Ensrud K, et al. Value of routine monitoring of bone mineral density after starting bisphosphonate treatment: secondary analysis of trial data. BMJ 2009;338. https://doi.org/10.1136/bmj.b2266.
- Bell KJL, Hayen A, Macaskill P, Craig JC, Neal BC, Irwig L. Mixed models showed no need for initial response monitoring after starting antihypertensive therapy. J Clin Epidemiol 2009;62:650-9. https://doi.org/10.1016/j.jclinepi.2008.07.018.
- Bell KJL, Kirby A, Hayen A, Irwig L, Glasziou P. Monitoring adherence to drug treatment by using change in cholesterol concentration: secondary analysis of trial data. BMJ 2011;342. https://doi.org/10.1136/bmj.d12.
- Bellera C, Hanley J, Joseph L, Albertsen P. Hierarchical changepoint models for biochemical markers illustrated by tracking postradiotherapy prostate-specific antigen series in men with prostate cancer. Ann Epidemiol 2008;18:270-82. https://doi.org/10.1016/j.annepidem.2007.10.006.
- Bellera CA, Hanley JA, Joseph L, Albertsen PC. Detecting trends in noisy data series: application to biomarker series. Am J Epidemiol 2008;167:1130-9. https://doi.org/10.1093/aje/kwn003.
- Cole SR, Li R, Anastos K, Detels R, Young M, Chmiel JS, et al. Accounting for leadtime in cohort studies: evaluating when to initiate HIV therapies. Stat Med 2004;23:3351-63. https://doi.org/10.1002/sim.1579.
- DeLong ER, Vernon WB, Bollinger RR. Sensitivity and specificity of a monitoring test. Biometrics 1985;41:947-58. https://doi.org/10.2307/2530966.
- Glasziou P, Chalmers I, Rawlins M, McCulloch P. When are randomised trials unnecessary? Picking signal from noise. BMJ 2007;334:349-51. https://doi.org/10.1136/bmj.39070.527986.68.
- Inoue LY, Etzioni R, Slate EH, Morrell C, Penson DF. Combining longitudinal studies of PSA. Biostatistics 2004;5:483-500. https://doi.org/10.1093/biostatistics/5.3.483.
- Li H, Gatsonis C. Dynamic optimal strategy for monitoring disease recurrence. Sci China Math 2012;55:1565-182. https://doi.org/10.1007/s11425-012-4475-y.
- Oke JL, Stevens RJ, Gaitskell K, Farmer AJ. Establishing an evidence base for frequency of monitoring glycated haemoglobin levels in patients with type 2 diabetes: projections of effectiveness from a regression model. Diabet Med 2012;29:266-71. https://doi.org/10.1111/j.1464-5491.2011.03412.x.
- Powers BJ, Olsen MK, Smith VA, Woolson RF, Bosworth HB, Oddone EZ. Measuring blood pressure for decision making and quality reporting: where and how many measures?. Ann Intern Med 2011;154:781-8. https://doi.org/10.7326/0003-4819-154-12-201106210-00005.
- Proust-Lima C, Taylor JMG. Development and validation of a dynamic prognostic tool for prostate cancer recurrence using repeated measures of posttreatment PSA: a joint modeling approach. Biostatistics 2009;10:535-49. https://doi.org/10.1093/biostatistics/kxp009.
- Proust-Lima C, Séne M, Taylor JM, Jacqmin-Gadda H. Joint latent class models for longitudinal and time-to-event data: a review. Stat Methods Med Res 2014;23:74-90. https://doi.org/10.1177/0962280212445839.
- Slate EH, Turnbull BW. Statistical models for longitudinal biomarkers of disease onset. Stat Med 2000;19:617-37. https://doi.org/10.1002/(SICI)1097-0258(20000229)19:4<617::AID-SIM360>3.0.CO;2-R.
- Sölétormos G, Hyltoft Petersen P, Dombernowsky P. Progression criteria for cancer antigen 15.3 and carcinoembryonic antigen in metastatic breast cancer compared by computer simulation of marker data. Clin Chem 2000;46:939-49.
- Subtil F, Rabilloud M. Robust non-linear mixed modelling of longitudinal PSA levels after prostate cancer treatment. Stat Med 2010;29:573-87. https://doi.org/10.1002/sim.3816.
- Takahashi O, Glasziou PP, Perera R, Shimbo T, Suwa J, Hiramatsu S, et al. Lipid re-screening: what is the best measure and interval?. Heart 2010;96:448-52. https://doi.org/10.1136/hrt.2009.172619.
- Takahashi O, Glasziou PP, Perera R, Shimbo T, Fukui T. Blood pressure re-screening for healthy adults: what is the best measure and interval?. J Hum Hypertens 2012;26:540-6. https://doi.org/10.1038/jhh.2011.72.
- Taylor JMG, Yu M, Sandler HM. Individualized predictions of disease progression following radiation therapy for prostate cancer. J Clin Oncol 2005;23:816-25. https://doi.org/10.1200/JCO.2005.12.156.
- Thiébaut R, Chêne G, Jacqmin-Gadda H, Morlat P, Mercié P, Dupon M, et al. Time-updated CD4+ T lymphocyte count and HIV RNA as major markers of disease progression in naive HIV-1-infected patients treated with a highly active antiretroviral therapy: the Aquitaine cohort, 1996–2001. J Acquir Immune Defic Syndr 2003;33:380-6. https://doi.org/10.1097/00126334-200307010-00013.
- Thompson SG, Pocock SJ. The variability of serum cholesterol measurements: implications for screening and monitoring. J Clin Epidemiol 1990;43:783-9. www.sciencedirect.com/science/article/pii/089543569090238K.
- Sterne JA, May M, Costagliola D, de Wolf F, Phillips AN, . When To Start Consortium . Timing of initiation of antiretroviral therapy in AIDS-free HIV-1-infected patients: a collaborative analysis of 18 HIV cohort studies. Lancet 2009;373:1352-63. https://doi.org/10.1016/S0140–6736(09)60612–7.
- Wolbers M, Babiker A, Sabin C, Young J, Dorrucci M, Chêne G, et al. Pretreatment CD4 cell slope and progression to AIDS or death in HIV-infected patients initiating antiretroviral therapy – the CASCADE collaboration: a collaboration of 23 cohort studies. PLOS Med 2010;7. https://doi.org/10.1371/journal.pmed.1000239.
- Baker SG. Identifying combinations of cancer markers for further study as triggers of early intervention. Biometrics 2000;56:1082-7. https://doi.org/10.1111/j.0006-341X.2000.01082.x.
- Baker SG, Kramer BS, McIntosh M, Patterson BH, Shyr Y, Skates S. Evaluating markers for the early detection of cancer: overview of study designs and methods. Clin Trials 2006;3:43-56. https://doi.org/10.1191/1740774506cn130oa.
- Baker SG. Improving the biomarker pipeline to develop and evaluate cancer screening tests. J Natl Cancer Inst 2009;101:1116-19. https://doi.org/10.1093/jnci/djp186.
- Lumbreras B, Porta M, Márquez S, Pollán M, Parker LA, Hernández-Aguado I. QUADOMICS: an adaptation of the Quality Assessment of Diagnostic Accuracy Assessment (QUADAS) for the evaluation of the methodological quality of studies on the diagnostic accuracy of ‘-omics’-based technologies. Clin Biochem 2008;41:1316-25. https://doi.org/10.1016/j.clinbiochem.2008.06.018.
- Parker LA, Gómez Saez N, Lumbreras B, Porta M, Hernández-Aguado I. Methodological deficits in diagnostic research using ‘-omics’ technologies: evaluation of the QUADOMICS tool and quality of recently published studies. PLOS ONE 2010;5. https://doi.org/10.1371/journal.pone.0011419.
- Pepe MS, Etzioni R, Feng Z, Potter JD, Thompson ML, Thornquist M, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst 2001;93:1054-61. https://doi.org/10.1093/jnci/93.14.1054.
- Pepe MS, Feng Z, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. J Natl Cancer Inst 2008;100:1432-8. https://doi.org/10.1093/jnci/djn326.
- Ransohoff DF, Gourlay ML. Sources of bias in specimens for research about molecular markers for cancer. J Clin Oncol 2010;28:698-704. https://doi.org/10.1200/JCO.2009.25.6065.
- Sturgeon CM, Lai LC, Duffy MJ. Serum tumour markers: how to order and interpret them. BMJ 2009;339. https://doi.org/10.1136/bmj.b3527.
- Sturgeon C, Hill R, Hortin GL, Thompson D. Taking a new biomarker into routine use – a perspective from the routine clinical biochemistry laboratory. Proteomics Clin Appl 2010;4:892-903. https://doi.org/10.1002/prca.201000073.
- Day NE, Walter SD. Simplified models of screening for chronic disease: estimation procedures from mass screening programmes. Biometrics 1984;40:1-14. http://europepmc.org/abstract/MED/6733223.
- Etzioni R, Shen Y. Estimating asymptomatic duration in cancer: the AIDS connection. Stat Med 1997;16:627-44. https://doi.org/10.1002/(SICI)1097-0258(19970330)16:6<627::AID-SIM438>3.0.CO;2-7.
- Frame PS, Frame JS. Determinants of cancer screening frequency: the example of screening for cervical cancer. J Am Board Fam Pract 1998;11:87-95. https://doi.org/10.3122/15572625-11-2-87.
- Lee SJ, Zelen M. Scheduling periodic examinations for the early detection of disease: applications to breast cancer. J Am Stat Assoc 1998;93:1271-81. https://doi.org/10.1080/01621459.1998.10473788.
- Lee S, Huang H, Zelen M. Early detection of disease and scheduling of screening examinations. Stat Methods Med Res 2004;13:443-56. https://doi.org/10.1191/0962280204sm377ra.
- McIntosh MW, Urban N, Karlan B. Generating longitudinal screening algorithms using novel biomarkers for disease. Cancer Epidemiol Biomarkers Prev 2002;11:159-66.
- McIntosh MW, Urban N. A parametric empirical Bayes method for cancer screening using longitudinal observations of a biomarker. Biostatistics 2003;4:27-40. https://doi.org/10.1093/biostatistics/4.1.27.
- Walter SD, Day NE. Estimation of the duration of a pre-clinical disease state using screening data. Am J Epidemiol 1983;118:865-86. https://doi.org/10.1093/oxfordjournals.aje.a113705.
- Zelen M. Optimal scheduling of examinations for the early detection of disease. Biometrika 1993;80. https://doi.org/10.2307/2337199.
- Cai T, Pepe MS, Zheng Y, Lumley T, Jenny NS. The sensitivity and specificity of markers for event times. Biostatistics 2006;7:182-97. https://doi.org/10.1093/biostatistics/kxi047.
- Etzioni R, Pepe M, Longton G, Goodman G. Incorporating the time dimension in receiver operating characteristic curves: a case study of prostate cancer. Med Decis Making 1999;19:242-51. https://doi.org/10.1177/0272989X9901900303.
- Parker CB, DeLong ER. ROC methodology within a monitoring framework. Stat Med 2003;22:3473-88. https://doi.org/10.1002/sim.1580.
- Pepe MS, Zheng Y, Jin Y, Huang Y, Parikh CR, Levy WC. Evaluating the ROC performance of markers for future events. Lifetime Data Anal 2008;14:86-113. https://doi.org/10.1007/s10985–007–9073-x.
- Subtil F, Pouteil-Noble C, Toussaint S, Villar E, Rabilloud M. A simple modeling-free method provides accurate estimates of sensitivity and specificity of longitudinal disease biomarkers. Methods Inf Med 2009;48:299-305. https://doi.org/10.3414/ME0583.
- Zheng Y, Heagerty PJ. Semiparametric estimation of time-dependent ROC curves for longitudinal marker data. Biostatistics 2004;5:615-32. https://doi.org/10.1093/biostatistics/kxh013.
- Biosca C, Ricós C, Lauzurica R, Petersen PH. Biological variation at long-term renal post-transplantation. Clin Chim Acta 2006;368:188-91. https://doi.org/10.1016/j.cca.2005.12.018.
- Clerico A, Emdin M. Diagnostic accuracy and prognostic relevance of the measurement of cardiac natriuretic peptides: a review. Clin Chem 2004;50:33-50. https://doi.org/10.1373/clinchem.2003.024760.
- Fraser CG, Peterson PH, Larsen ML. Setting analytical goals for random analytical error in specific clinical monitoring situations. Clin Chem 1990;36:1625-8. www.clinchem.org/content/36/9/1625.short.
- Fraser CG. Biological Variation: from Principles to Practice. Washington, DC: AACC Press; 2001.
- Klee GG. Establishment of outcome-related analytic performance goals. Clin Chem 2010;56:714-22. https://doi.org/10.1373/clinchem.2009.133660.
- Macaskill P, Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Omar F, van der Watt GF, Pillay TS. Reference change values: how useful are they?. J Clin Pathol 2008;61:426-7. https://doi.org/10.1136/jcp.2007.054833.
- Petersen PH. Making the most of a patient’s laboratory data: optimisation of signal-to-noise ratio. Clin Biochem Rev 2005;26:91-6. www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1320174&tool=pmcentrez&rendertype=abstract.
- Petersen PH, Jensen EA, Brandslund I. Analytical performance, reference values and decision limits. A need to differentiate between reference intervals and decision limits and to define analytical quality specifications. Clin Chem Lab Med 2012;50:819-31. https://doi.org/10.1515/cclm.2011.821.
- Smellie WSA. What is a significant difference between sequential laboratory results?. J Clin Pathol 2008;61:419-25. https://doi.org/10.1136/jcp.2007.047175.
- Sölétormos G, Schiøler V. Description of a computer program to assess cancer antigen 15.3, carcinoembryonic antigen, and tissue polypeptide antigen information during monitoring of metastatic breast cancer. Clin Chem 2000;46:1106-13.
- Gavit P, Baddour Y, Tholmer R. Use of change-point analysis for process monitoring and control. BioPharm Int 2009;22. www.biopharminternational.com/use-change-point-analysis-process-monitoring-and-control (accessed 1 May 2018).
- Tennant R, Mohammed MA, Coleman JJ, Martin U. Monitoring patients using control charts: a systematic review. Int J Qual Health Care 2007;19:187-94. https://doi.org/10.1093/intqhc/mzm015.
- Thor J, Lundberg J, Ask J, Olsson J, Carli C, Härenstam KP, et al. Application of statistical process control in healthcare improvement: systematic review. Qual Saf Health Care 2007;16:387-99. https://doi.org/10.1136/qshc.2006.022194.
- Baker RD. Use of a mathematical model to evaluate breast cancer screening policy. Health Care Manag Sci 1998;1:103-13. https://doi.org/10.1023/A:1019046619402.
- Karnon J, Goyder E, Tappenden P, McPhie S, Towers I, Brazier J, et al. A review and critique of modelling in prioritising and designing screening programmes. Health Technol Assess 2007;11. https://doi.org/10.3310/hta11520.
- Parmigiani G. Timing medical examinations via intensity functions. Biometrika 1997;84:803-16. https://doi.org/10.1093/biomet/84.4.803.
- Sutton AJ, Cooper NJ, Goodacre S, Stevenson M. Integration of meta-analysis and economic decision modeling for evaluating diagnostic tests. Med Decis Making 2008;28:650-67. https://doi.org/10.1177/0272989X08324036.
- Driffield T, Smith PC. A real options approach to watchful waiting: theory and an illustration. Med Decis Making 2007;27:178-88. https://doi.org/10.1177/0272989X06297390.
- Lasserre P, Moatti J-P, Soubeyran A. Early initiation of highly active antiretroviral therapies for AIDS: dynamic choice with endogenous and exogenous learning. J Health Econ 2006;25:579-98. https://doi.org/10.1016/j.jhealeco.2005.09.006.
- Meyer E, Rees R. Watchfully waiting: medical intervention as an optimal investment decision. J Health Econ 2012;31:349-58. https://doi.org/10.1016/j.jhealeco.2012.02.002.
- Palmer S, Smith PC. Incorporating option values into the economic evaluation of health care technologies. J Health Econ 2000;19:755-66. https://doi.org/10.1016/S0167-6296(00)00048-5.
- Shechter SM, Alagoz O, Roberts MS. Irreversible treatment decisions under consideration of the research and development pipeline for new therapies. IIE Transactions 2010;42:632-42. https://doi.org/10.1080/07408170903468589.
- Whynes DK. Optimal times of transfer between therapies: a mathematical framework. J Health Econ 1995;14:477-90. https://doi.org/10.1016/0167-6296(95)00014-9.
- Whiting P, Rutjes AWS, Reitsma JB, Bossuyt PMM, Kleijnen J. The development of QUADAS: a tool for the quality assessment of studies of diagnostic accuracy included in systematic reviews. BMC Med Res Methodol 2003;3:25-37. https://doi.org/10.1186/1471-2288-3-25.
- Batal I, Valizadegan H, Cooper GF, Hauskrecht M. A Temporal pattern mining approach for classifying electronic health record data. ACM Trans Intell Syst Technol 2013;4:1-36. https://doi.org/10.1145/2508037.2508044.
- Wright AP, Wright AT, McCoy AB, Sittig DF. The use of sequential pattern mining to predict next prescribed medications. J Biomed Inform 2015;53:73-80. https://doi.org/10.1016/j.jbi.2014.09.003.
- Witten I, Eibe F, Hall M, Pal C. Data Mining: Practical Machine Learning Tools and Techniques. London: Elsevier; 2017.
- Lijmer JG, Bossuyt PM. Various randomized designs can be used to evaluate medical tests. J Clin Epidemiol 2009;62:364-73. https://doi.org/10.1016/j.jclinepi.2008.06.017.
- Lin JS, Thompson M, Goddard KA, Piper MA, Heneghan C, Whitlock EP. Evaluating genomic tests from bench to bedside: a practical framework. BMC Med Inform Decis Mak 2012;12. https://doi.org/10.1186/1472-6947-12-117.
- Adriaensen WJ, Matheï C, Buntinx FJ, Arbyn M. A framework provided an outline toward the proper evaluation of potential screening strategies. J Clin Epidemiol 2013;66:639-47. https://doi.org/10.1016/j.jclinepi.2012.09.018.
- Lord SJ, Irwig L, Simes RJ. When is measuring sensitivity and specificity sufficient to evaluate a diagnostic test, and when do we need randomized trials?. Ann Intern Med 2006;144:850-5. https://doi.org/10.7326/0003-4819-144-11-200606060-00011.
- EP2 – Illness Labelling and Illness Experience. London: NICE; 2011.
- McCaffery K, Michie S, Glasziou PP, Irwig L, Aronson JK. Evidence-based Medical Monitoring: from Principles to Practice. Oxford: Blackwell Publishing; 2008.
- Lepage C, Adenis A, Bedenne L. Post-Operative Monitoring of Patients Operated for Stage II or III Colorectal Cancer With Intent to Cure Phase III Multicentre Prospective Trial. 2009.
- Jordens CF, Morrell B, Harnett P, Hobbs K, Mason C, Kerridge IH. Cancergazing? CA125 and post-treatment surveillance in advanced ovarian cancer. Soc Sci Med 2010;71:1548-56. https://doi.org/10.1016/j.socscimed.2010.07.033.
- Reitsma JB, Rutjes AW, Khan KS, Coomarasamy A, Bossuyt PM. A review of solutions for diagnostic accuracy studies with an imperfect or missing reference standard. J Clin Epidemiol 2009;62:797-806. https://doi.org/10.1016/j.jclinepi.2009.02.005.
- Glasziou P. How much monitoring?. Br J Gen Pract 2007;57:350-1. https://doi.org/10.1016/j.jacc.2006.10.081.6.
- A Framework for Development and Evaluation of RCTs for Complex Interventions to Improve Health. London: MRC; 2000.
- ISRCTN Registry . Enhanced Liver Fibrosis (ELF) Test to Uncover Cirrhosis As an Indication for Diagnosis and Action for Treatable Events n.d. www.isrctn.com/ISRCTN74815110?q=ELUCIDATE&filters=&sort=&offset=24&totalResults=35&page=3&pageSize=10&searchType=basic-search (accessed 12 March 2018).
- Poynard T, Bedossa P, Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997;349:825-32. https://doi.org/10.1016/S0140-6736(96)07642-8.
- Dancygier H. Clinical Hepatology. Berlin: Springer-Verlag; 2010.
- Parkes J, Roderick P, Harris S, Day C, Mutimer D, Collier J, et al. Enhanced liver fibrosis test can predict clinical outcomes in patients with chronic liver disease. Gut 2010;59:1245-51. https://doi.org/10.1136/gut.2009.203166.
- Longo R, Baxter P, Hall P, Hewison J, Afshar M, Hall G, et al. Methods for identifying the cost-effective case definition cut-off for sequential monitoring tests: an extension of Phelps and Mushlin. PharmacoEconomics 2014;32:327-34. https://doi.org/10.1007/s40273-014-0134-1.
- Levinson W, Kallewaard M, Bhatia RS, Wolfson D, Shortt S, Kerr EA. Choosing Wisely International Working Group . ‘Choosing Wisely’: a growing international campaign. BMJ Qual Saf 2015;24:167-74. https://doi.org/10.1136/bmjqs-2014-003821.
- Culyer AJ. Cost-effectiveness thresholds in health care: a bookshelf guide to their meaning and use. Health Economics, Policy and Law 2016;11:415-32. https://doi.org/10.17/S1744133116000049.
- Paulden M, O’Mahony J, McCabe C. Determinants of change in the cost-effectiveness threshold. Med Decis Making 2017;37:264-76. https://doi.org/10.1177/0272989X16662242.
- McCabe C, Paulden M, O’Mahony C, Edlin R, Culyer A. Life at a premium: considering an end-of-life premium in Value Based Reimbursement. Value Health 2015;18:6-7. https://doi.org/10.1007/978-3-319-28267-1_9.
- Phelps CE, Mushlin AI. Focusing technology assessment using medical decision theory. Med Decis Making 1988;8:279-89. https://doi.org/10.1177/0272989X8800800409.
- Novielli N, Cooper NJ, Sutton AJ. Evaluating the cost-effectiveness of diagnostic tests in combination: is it important to allow for performance dependency?. Value Health 2013;16:536-41. https://doi.org/10.1016/j.jval.2013.02.015.
- Felder S, Mayrhofer T. Medical Decision Making: a Health Economic Primer. Berlin: Springer-Verlag; 2011.
- Horvath AR, Lord SJ, St John A, Sandberg S, Cobbaert CM, Lorenz S, et al. From biomarkers to medical tests: the changing landscape of test evaluation. Clin Chim Acta 2014;427:49-57. https://doi.org/10.1016/j.cca.2013.09.018.
- Poste G. Bring on the biomarkers. Nature 2011;469:156-7. https://doi.org/10.1038/469156a.
- Parkinson DR, McCormack RT, Keating SM, Gutman SI, Hamilton SR, Mansfield EA, et al. Evidence of clinical utility: an unmet need in molecular diagnostics for patients with cancer. Clin Cancer Res 2014;20:1428-44. https://doi.org/10.1158/1078-0432.CCR-13-2961.
- Huber F, Montani M, Sulser T, Jaggi R, Wild P, Moch H, et al. Comprehensive validation of published immunohistochemical prognostic biomarkers of prostate cancer – what has gone wrong? A blueprint for the way forward in biomarker studies. Br J Cancer 2015;112:140-8. https://doi.org/10.1038/bjc.2014.588.
- Duffy MJ, Sturgeon CM, Sölétormos G, Barak V, Molina R, Hayes DF, et al. Validation of new cancer biomarkers: a position statement from the European group on tumor markers. Clin Chem 2015;61:809-20. https://doi.org/10.1373/clinchem.2015.239863.
- Haddow J, Palomaki GE, Khoury M, Little J, Burke W. Human Genome Epidemiology: A Scientific Foundation for Using Genetic Information to Improve Health and Prevent Disease. Oxford: Oxford University Press; 2003.
- Walley T. Evaluating laboratory diagnostic tests. BMJ 2008;336:569-70. https://doi.org/10.1136/bmj.39513.576701.80.
- Crossan C, Tsochatzis EA, Longworth L, Gurusamy K, Davidson B, Rodríguez-Perálvarez M, et al. Cost-effectiveness of non-invasive methods for assessment and monitoring of liver fibrosis and cirrhosis in patients with chronic liver disease: systematic review and economic evaluation. Health Technol Assess 2015;19. https://doi.org/10.3310/hta19090.
- Moher D, Liberati A, Tetzlaff J, Altman DG. The PRISMA Group . Preferred Reporting Items for Systematic Reviews and Meta-Analyses: the PRISMA Statement. BMJ 2009;339. https://doi.org/10.1136/bmj.b2535.
- Hawgood S, Hook-Barnard IG, O’Brien TC, Yamamoto KR. Precision medicine: beyond the inflection point. Sci Transl Med 2015;7. https://doi.org/10.1126/scitranslmed.aaa9970.
- Schleidgen S, Klingler C, Bertram T, Rogowski WH, Marckmann G. What is personalized medicine: sharpening a vague term based on a systematic literature review. BMC Med Ethics 2013;14. https://doi.org/10.1186/1472-6939-14-55.
- Matthews PM, Edison P, Geraghty OC, Johnson MR. The emerging agenda of stratified medicine in neurology. Nat Rev Neurol 2014;10:15-26. https://doi.org/10.1038/nrneurol.2013.245.
- Aronson N. Making personalized medicine more affordable. Ann N Y Acad Sci 2015;1346:81-9. https://doi.org/10.1111/nyas.12614.
- Montine TJ, Montine KS. Precision medicine: clarity for the clinical and biological complexity of Alzheimer’s and Parkinson’s diseases. J Exp Med 2015;212:601-5. https://doi.org/10.1084/jem.20150656.
- Rubin EH, Allen JD, Nowak JA, Bates SE. Developing precision medicine in a global world. Clin Cancer Res 2014;20:1419-27. https://doi.org/10.1158/1078–0432.CCR-14–0091.
- Audette J. Market Trends for Biomarker-Based IVD Tests (2003–2014): Realizing the Promise of Precision Medicine. Bend, OR: Amplion Inc.; 2015.
- Doble B, Tan M, Harris A, Lorgelly P. Modeling companion diagnostics in economic evaluations of targeted oncology therapies: systematic review and methodological checklist. Expert Rev Mol Diagn 2015;15:235-54. https://doi.org/10.1586/14737159.2014.929499.
- Wechsel HW, Feil G, Lahme S, Zumbragel A, Petri E, Bichler KH. Control of hepatic parameters in renal cell carcinoma (RCC) by interleukin-6 (IL-6)?. Anticancer Res 1999;19:2577-81.
- Poste G. Biospecimens, biomarkers, and burgeoning data: the imperative for more rigorous research standards. Trends Mol Med 2012;18:717-22. https://doi.org/10.1016/j.molmed.2012.09.003.
- Drucker E, Krapfenbauer K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA J 2013;4. https://doi.org/10.1186/1878-5085-4-7.
- Pavlou MP, Diamandis EP, Blasutig IM. The long journey of cancer biomarkers from the bench to the clinic. Clin Chem 2013;59:147-57. https://doi.org/10.1373/clinchem.2012.184614.
- Füzéry AK, Levin J, Chan MM, Chan DW. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics 2013;10. https://doi.org/10.1186/1559-0275-10-13.
- Diamandis EP. Cancer biomarkers: can we turn recent failures into success?. J Natl Cancer Inst 2010;102:1462-7. https://doi.org/10.1093/jnci/djq306.
- Heegaard NH, Østergaard O, Bahl JM, Overgaard M, Beck HC, Rasmussen LM, et al. Important options available – from start to finish – for translating proteomics results to clinical chemistry. Proteomics Clin Appl 2015;9:235-52. https://doi.org/10.1002/prca.201400137.
- Thongboonkerd V. Urinary proteomics: towards biomarker discovery, diagnostics and prognostics. Mol Biosyst 2008;4:810-15. https://doi.org/10.1039/b802534g.
- Jackson DH, Banks RE. Banking of clinical samples for proteomic biomarker studies: a consideration of logistical issues with a focus on pre-analytical variation. Proteomics Clin Appl 2010;4:250-70. https://doi.org/10.1002/prca.200900220.
- Findeisen P, Neumaier M. Mass spectrometry-based clinical proteomics profiling: current status and future directions. Expert Rev Proteomics 2009;6:457-9. https://doi.org/10.1586/epr.09.67.
- Koomen JM, Haura EB, Bepler G, Sutphen R, Remily-Wood ER, Benson K, et al. Proteomic contributions to personalized cancer care. Mol Cell Proteomics 2008;7:1780-94. https://doi.org/10.1074/mcp.R800002-MCP200.
- Parker CE, Pearson TW, Anderson NL, Borchers CH. Mass-spectrometry-based clinical proteomics – a review and prospective. Analyst 2010;135:1830-8. https://doi.org/10.1039/c0an00105h.
- McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics . REporting recommendations for tumour MARKer prognostic studies (REMARK). Eur J Cancer 2005;41:1690-6. https://doi.org/10.1016/j.ejca.2005.03.032.
- Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. Toward complete and accurate reporting of studies of diagnostic accuracy. The STARD initiative. Am J Clin Pathol 2003;119:18-22. https://doi.org/10.1309/8EXC-CM6Y-R1TH-UBAF.
- Riegman PHJ, de Jong BWD, Llombart-Bosch A. The organization of European Cancer Institute Pathobiology Working Group and its support of European biobanking infrastructures for translational cancer research. Cancer Epidemiol Biomarkers Prev 2010;19:923-6. https://doi.org/10.1158/1055-9965.EPI-10-0062.
- Navis GJ, Blankestijn PJ, Deegens J, De Fijter JW, Homan van der Heide JJ, Rabelink T, et al. The biobank of nephrological diseases in the Netherlands cohort: the string of pearls initiative collaboration on chronic kidney disease in the university medical centers in the Netherlands. Nephrol Dial Transplant 2014;29:1145-50. https://doi.org/10.1093/ndt/gft307.
- Kang B, Park J, Cho S, Lee M, Kim N, Min H, et al. Current status, challenges, policies, and bioethics of biobanks. Genomics Inform 2013;11:211-17. https://doi.org/10.5808/GI.2013.11.4.211.
- Ho TH, Nateras RN, Yan H, Park JG, Jensen S, Borges C, et al. A multidisciplinary biospecimen bank of renal cell carcinomas compatible with discovery platforms at Mayo Clinic, Scottsdale, Arizona. PLOS ONE 2015;10. https://doi.org/10.1371/journal.pone.0132831.
- Ellervik C, Vaught J. Preanalytical variables affecting the integrity of human biospecimens in biobanking. Clin Chem 2015;61:914-34. https://doi.org/10.1373/clinchem.2014.228783.
- Moore HM, Compton CC, Alper J, Vaught JB. International approaches to advancing biospecimen science. Cancer Epidemiol Biomarkers Prev 2011;20:729-32. https://doi.org/10.1158/1055-9965.EPI-11-0021.
- Moore HM, Kelly AB, Jewell SD, McShane LM, Clark DP, Greenspan R, et al. Biospecimen reporting for improved study quality (BRISQ). Cancer Cytopathol 2011;119:92-101. https://doi.org/10.1002/cncy.20147.
- Womack C, Mager SR. Human biological sample biobanking to support tissue biomarkers in pharmaceutical research and development. Methods 2014;70:3-11. https://doi.org/10.1016/j.ymeth.2014.01.014.
- Watson PH, Nussbeck SY, Carter C, O’Donoghue S, Cheah S, Matzke LA, et al. A framework for biobank sustainability. Biopreserv Biobank 2014;12:60-8. https://doi.org/10.1089/bio.2013.0064.
- Hofman V, Ilie M, Long E, Washetine K, Chabannon C, Figarella-Branger D, et al. Measuring the contribution of tumor biobanks to research in oncology: surrogate indicators and bibliographic output. Biopreserv Biobank 2013;11:235-44. https://doi.org/10.1089/bio.2013.0015.
- Olson JE, Bielinski SJ, Ryu E, Winkler EM, Takahashi PY, Pathak J, et al. Biobanks and personalized medicine. Clin Genet 2014;86:50-5. https://doi.org/10.1111/cge.12370.
- Simeon-Dubach D, Watson P. Biobanking 3.0: evidence based and customer focused biobanking. Clin Biochem 2014;47:300-8. https://doi.org/10.1016/j.clinbiochem.2013.12.018.
- Henderson GE, Cadigan RJ, Edwards TP, Conlon I, Nelson AG, Evans JP, et al. Characterizing biobank organizations in the US: results from a national survey. Genome Med 2013;5. https://doi.org/10.1186/gm407.
- Scudellari M. Biobank managers bemoan underuse of collected samples. Nat Med 2013;19. https://doi.org/10.1038/nm0313–253a.
- Rifai N, Watson ID, Miller WG. Commercial immunoassays in biomarkers studies: researchers beware!. Clin Chem 2012;58:1387-8. https://doi.org/10.1373/clinchem.2012.192351.
- Rodland KD. As if biomarker discovery isn’t hard enough: the consequences of poorly characterized reagents. Clin Chem 2014;60:290-1. https://doi.org/10.1373/clinchem.2013.216382.
- Williams PM, Lively TG, Jessup JM, Conley BA. Bridging the gap: moving predictive and prognostic assays from research to clinical use. Clin Cancer Res 2012;18:1531-9. https://doi.org/10.1158/1078-0432.CCR-11-2203.
- Prassas I, Diamandis EP. Translational researchers beware! Unreliable commercial immunoassays (ELISAs) can jeopardize your research. Clin Chem Lab Med 2014;52:765-6. https://doi.org/10.1515/cclm-2013-1078.
- Lee JW, Weiner RS, Sailstad JM, Bowsher RR, Knuth DW, O’Brien PJ, et al. Method validation and measurement of biomarkers in nonclinical and clinical samples in drug development: a conference report. Pharm Res 2005;22:499-511. https://doi.org/10.1007/s11095-005-2495-9.
- Tate JR. Troponin revisited 2008: assay performance. Clin Chem Lab Med 2008;46:1489-500. https://doi.org/10.1515/CCLM.2008.292.
- DeSilva B, Garofolo F, Rocci M, Martinez S, Dumont I, Landry F, et al. 2012 White Paper on recent issues in bioanalysis and alignment of multiple guidelines. Bioanalysis 2012;4:2213-26. https://doi.org/10.4155/bio.12.205.
- Bower J, Fast D, Garofolo F, Gouty D, Hayes R, Lowes S, et al. 8th GCC: consolidated feedback to US FDA on the 2013 draft FDA guidance on bioanalytical method validation. Bioanalysis 2014;6:2957-63. https://doi.org/10.4155/bio.14.287.
- Cummings J, Raynaud F, Jones L, Sugar R, Dive C. Fit-for-purpose biomarker method validation for application in clinical trials of anticancer drugs. Br J Cancer 2010;103:1313-17. https://doi.org/10.1038/sj.bjc.6605910.
- van de Merbel N, Savoie N, Yadav M, Ohtsu Y, White J, Riccio MF, et al. Stability: recommendation for best practices and harmonization from the Global Bioanalysis Consortium Harmonization Team. AAPS J 2014;16:392-9. https://doi.org/10.1208/s12248-014-9573-z.
- de Gramont A, Watson S, Ellis LM, Rodon J, Tabernero J, de Gramont A, et al. Pragmatic issues in biomarker evaluation for targeted therapies in cancer. Nat Rev Clin Oncol 2014;12:197-212. https://doi.org/10.1038/nrclinonc.2014.202.
- Hepburn S, Banks RE, Thompson D. Protein biomarker research in UK hospital clinical biochemistry laboratories: a survey of current practice and views. Clin Biochem Rev 2014;35:115-33.
- Mansfield EA. FDA perspective on companion diagnostics: an evolving paradigm. Clin Cancer Res 2014;20:1453-7. https://doi.org/10.1158/1078-0432.CCR-13-1954.
- Olsen D, Jørgensen JT. Companion diagnostics for targeted cancer drugs - clinical and regulatory aspects. Front Oncol 2014;4. https://doi.org/10.3389/fonc.2014.00105.
- National Institute for Health Research . NIHR Medtech and In Vitro Diagnostics Co-Operatives (MICs) n.d. www.nihr.ac.uk/about-us/how-we-are-managed/our-structure/infrastructure/Documents/medtech-and-in-vitro-diagnostic-co-operatives.htm (accessed 20 March 2018).
- Medicines Discovery Catapult n.d. https://md.catapult.org.uk/ (accessed 20 March 2018).
- Barker AD, Compton CC, Poste G. The National Biomarker Development Alliance accelerating the translation of biomarkers to the clinic. Biomark Med 2014;8:873-6. https://doi.org/10.2217/bmm.14.52.
- Poste G, Compton CC, Barker AD. The national biomarker development alliance: confronting the poor productivity of biomarker research and development. Expert Rev Mol Diagn 2015;15:211-18. https://doi.org/10.1586/14737159.2015.974561.
- International Agency for Research on Cancer . GLOBOCAN 2012: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012 n.d. http://globocan.iarc.fr/ (accessed February 2018).
- Cancer Research UK . Cancer Statistics for the UK n.d. www.cancerresearchuk.org/cancer-info/cancerstats/ (accessed 18 January 2018).
- Delahunt B, Srigley JR, Montironi R, Egevad L. Advances in renal neoplasia: recommendations from the 2012 International Society of Urological Pathology Consensus Conference. Urology 2014;83:969-74. https://doi.org/10.1016/j.urology.2014.02.004.
- Yap NY, Rajandram R, Ng KL, Pailoor J, Fadzli A, Gobe GC. Genetic and chromosomal aberrations and their clinical significance in renal neoplasms. Biomed Res Int 2015;2015. https://doi.org/10.1155/2015/476508.
- Srigley JR, Delahunt B, Eble JN, Egevad L, Epstein JI, Grignon D, et al. The International Society of Urological Pathology (ISUP) Vancouver Classification of Renal Neoplasia. Am J Surg Pathol 2013;37:1469-89. https://doi.org/10.1097/PAS.0b013e318299f2d1.
- Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A, et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol 2015;67:85-97. https://doi.org/10.1016/j.eururo.2014.04.029.
- Jonasch E, Gao J, Rathmell WK. Renal cell carcinoma. BMJ 2014;349. https://doi.org/10.1136/bmj.g4797.
- Laguna MP, Algaba F, Cadeddu J, Clayman R, Gill I, Gueglio G, et al. Current patterns of presentation and treatment of renal masses: a clinical research office of the endourological society prospective study. J Endourol 2014;28:861-70. https://doi.org/10.1089/end.2013.0724.
- Scelo G, Riazalhosseini Y, Greger L, Letourneau L, Gonzàlez-Porta M, Wozniak MB, et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat Commun 2014;5. https://doi.org/10.1038/ncomms6135.
- Young AC, Craven RA, Cohen D, Taylor C, Booth C, Harnden P, et al. Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res 2009;15:7582-92. https://doi.org/10.1158/1078-0432.CCR-09-2131.
- Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 2008;14:4726-34. https://doi.org/10.1158/1078-0432.CCR-07-4921.
- Frew IJ, Moch H. A clearer view of the molecular complexity of clear cell renal cell carcinoma. Annu Rev Pathol 2015;10:263-89. https://doi.org/10.1146/annurev-pathol-012414-040306.
- Srinivasan R, Ricketts CJ, Sourbier C, Linehan WM. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin Cancer Res 2015;21:10-7. https://doi.org/10.1158/1078-0432.CCR-13-2993.
- Maher ER. Genomics and epigenomics of renal cell carcinoma. Semin Cancer Biol 2013;23:10-7. https://doi.org/10.1016/j.semcancer.2012.06.003.
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015;373:1803-13. https://doi.org/10.1056/NEJMoa1510665.
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014;26:319-30. https://doi.org/10.1016/j.ccr.2014.07.014.
- Durinck S, Stawiski EW, Pavia-Jimenez A, Modrusan Z, Kapur P, Jaiswal BS, et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet 2015;47:13-21. https://doi.org/10.1038/ng.3146.
- Kovac M, Navas C, Horswell S, Salm M, Bardella C, Rowan A, et al. Recurrent chromosomal gains and heterogeneous driver mutations characterise papillary renal cancer evolution. Nat Commun 2015;6. https://doi.org/10.1038/ncomms7336.
- Albiges L, Guegan J, Le Formal A, Verkarre V, Rioux-Leclercq N, Sibony M, et al. MET is a potential target across all papillary renal cell carcinomas: result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin Cancer Res 2014;20:3411-21. https://doi.org/10.1158/1078-0432.CCR-13-2173.
- Marsaud A, Dadone B, Ambrosetti D, Baudoin C, Chamorey E, Rouleau E, et al. Dismantling papillary renal cell carcinoma classification: the heterogeneity of genetic profiles suggests several independent diseases. Genes Chromosomes Cancer 2015;54:369-82. https://doi.org/10.1002/gcc.22248.
- Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 1999;18:2343-50. https://doi.org/10.1038/sj.onc.1202547.
- Ma PC, Tretiakova MS, MacKinnon AC, Ramnath N, Johnson C, Dietrich S, et al. Expression and mutational analysis of MET in human solid cancers. Genes Chromosomes Cancer 2008;47:1025-37. https://doi.org/10.1002/gcc.20604.
- Choi JS, Kim MK, Seo JW, Choi YL, Kim DH, Chun YK, et al. MET expression in sporadic renal cell carcinomas. J Korean Med Sci 2006;21:672-7. https://doi.org/10.3346/jkms.2006.21.4.672.
- Atkins MB, Bukowski RM, Escudier BJ, Figlin RA, Hudes GH, Kaelin WG, et al. Innovations and challenges in renal cancer: summary statement from the Third Cambridge Conference. Cancer 2009;115:2247-51. https://doi.org/10.1002/cncr.24229.
- Oosterwijk E, Rathmell WK, Junker K, Brannon AR, Pouliot F, Finley DS, et al. Basic research in kidney cancer. Eur Urol 2011;60:622-33. https://doi.org/10.1016/j.eururo.2011.06.048.
- Richard PO, Jewett MA, Bhatt JR, Kachura JR, Evans AJ, Zlotta AR, et al. Renal tumor biopsy for small renal masses: a single-center 13-year experience. Eur Urol 2015;68:1007-13. https://doi.org/10.1016/j.eururo.2015.04.004.
- Schachter LR, Cookson MS, Chang SS, Smith JA, Dietrich MS, Jayaram G, et al. Second prize: frequency of benign renal cortical tumors and histologic subtypes based on size in a contemporary series: what to tell our patients. J Endourol 2007;21:819-23. https://doi.org/10.1089/end.2006.9937.
- Vasudev NS, Banks RE, Edelstein CL. Biomarkers of Kidney Diseases. Cambridge, MA: Academic Press; 2016.
- Morrissey JJ, London AN, Luo J, Kharasch ED. Urinary biomarkers for the early diagnosis of kidney cancer. Mayo Clin Proc 2010;85:413-21. https://doi.org/10.4065/mcp.2009.0709.
- Morrissey JJ, Kharasch ED. The specificity of urinary aquaporin 1 and perilipin 2 to screen for renal cell carcinoma. J Urol 2013;189:1913-20. https://doi.org/10.1016/j.juro.2012.11.034.
- Morrissey JJ, Mobley J, Song J, Vetter J, Luo J, Bhayani S, et al. Urinary concentrations of aquaporin-1 and perilipin-2 in patients with renal cell carcinoma correlate with tumor size and stage but not grade. Urology 2014;83:256.e9-14. https://doi.org/10.1016/j.urology.2013.09.026.
- Morrissey JJ, Mellnick VM, Luo J, Siegel MJ, Figenshau RS, Bhayani S, et al. Evaluation of urine aquaporin-1 and perilipin-2 concentrations as biomarkers to screen for renal cell carcinoma: a prospective cohort study. JAMA Oncol 2015;1:204-12. https://doi.org/10.1001/jamaoncol.2015.0213.
- Kim EH, Strope SA. Postoperative surveillance imaging for patients undergoing nephrectomy for renal cell carcinoma. Urol Oncol 2015;33:499-502. https://doi.org/10.1016/j.urolonc.2015.08.008.
- Sobin LH, Gospodarowicz MK, Wittekind C. TNM Classification of Malignant Tumours. Hoboken, NJ: Wiley-Blackwell; 2009.
- Edge SB, Byrd DR, Carducci MA. AJCC Cancer Staging Manual. New York, NY: Springer-Verlag New York; 2009.
- Crispen PL, Boorjian SA, Lohse CM, Leibovich BC, Kwon ED. Predicting disease progression after nephrectomy for localized renal cell carcinoma: the utility of prognostic models and molecular biomarkers. Cancer 2008;113:450-60. https://doi.org/DOI 10.1002/cncr.23566.
- Al-Aynati M, Chen V, Salama S, Shuhaibar H, Treleaven D, Vincic L. Interobserver and intraobserver variability using the Fuhrman grading system for renal cell carcinoma. Arch Pathol Lab Med 2003;127:593-6. https://doi.org/10.1043/0003-9985(2003)127<0593:IAIVUT>2.0.CO;2.
- Leibovich BC, Blute ML, Cheville JC, Lohse CM, Frank I, Kwon ED, et al. Prediction of progression after radical nephrectomy for patients with clear cell renal cell carcinoma: a stratification tool for prospective clinical trials. Cancer 2003;97:1663-71. https://doi.org/10.1002/cncr.11234.
- Heng DY, Xie W, Regan MM, Warren MA, Golshayan AR, Sahi C, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794-9. https://doi.org/10.1200/JCO.2008.21.4809.
- Heng DY, Xie W, Regan MM, Harshman LC, Bjarnason GA, Vaishampayan UN, et al. External validation and comparison with other models of the International Metastatic Renal-Cell Carcinoma Database Consortium prognostic model: a population-based study. Lancet Oncol 2013;14:141-8. https://doi.org/10.1016/S1470-2045(12)70559--.
- Karakiewicz PI, Suardi N, Capitanio U, Jeldres C, Ficarra V, Cindolo L, et al. A preoperative prognostic model for patients treated with nephrectomy for renal cell carcinoma. Eur Urol 2009;55:287-95. https://doi.org/10.1016/j.eururo.2008.07.037.
- Parker AS, Leibovich BC, Lohse CM, Sheinin Y, Kuntz SM, Eckel-Passow JE, et al. Development and evaluation of BioScore: a biomarker panel to enhance prognostic algorithms for clear cell renal cell carcinoma. Cancer 2009;115:2092-103. https://doi.org/10.1002/cncr.24263.
- Klatte T, Seligson DB, LaRochelle J, Shuch B, Said JW, Riggs SB, et al. Molecular signatures of localized clear cell renal cell carcinoma to predict disease-free survival after nephrectomy. Cancer Epidemiol Biomarkers Prev 2009;18:894-900. https://doi.org/Doi 10.1158/1055-9965.Epi-08-0786.
- Kim HL, Seligson D, Liu X, Janzen N, Bui MH, Yu H, et al. Using tumor markers to predict the survival of patients with metastatic renal cell carcinoma. J Urol 2005;173:1496-501. https://doi.org/10.1097/01.ju.0000154351.37249.f0.
- Brooks SA, Brannon AR, Parker JS, Fisher JC, Sen O, Kattan MW, et al. ClearCode34: a prognostic risk predictor for localized clear cell renal cell carcinoma. Eur Urol 2014;66:77-84. https://doi.org/10.1016/j.eururo.2014.02.035.
- Rini B, Goddard A, Knezevic D, Maddala T, Zhou M, Aydin H, et al. A 16-gene assay to predict recurrence after surgery in localised renal cell carcinoma: development and validation studies. Lancet Oncol 2015;16:676-85. https://doi.org/10.1016/S1470-2045(15)70167-1.
- Liyanage T, Ninomiya T, Jha V, Neal B, Patrice HM, Okpechi I, et al. Worldwide access to treatment for end-stage kidney disease: a systematic review. Lancet 2015;385:1975-82. https://doi.org/10.1016/S0140-6736(14)61601-9.
- Eckardt KU, Coresh J, Devuyst O, Johnson RJ, Köttgen A, Levey AS, et al. Evolving importance of kidney disease: from subspecialty to global health burden. Lancet 2013;382:158-69. https://doi.org/10.1016/S0140-6736(13)60439-0.
- Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, et al. Chronic kidney disease: global dimension and perspectives. Lancet 2013;382:260-72. https://doi.org/10.1016/S0140-6736(13)60687-X.
- Port FK, Wolfe RA, Mauger EA, Berling DP, Jiang K. Comparison of survival probabilities for dialysis patients vs cadaveric renal transplant recipients. JAMA 1993;270:1339-43. http://jama.jamanetwork.com/data/Journals/JAMA/9807/jama_270_11_036.pdf.
- Wolfe RA, Ashby VB, Milford EL, Ojo AO, Ettenger RE, Agodoa LY, et al. Comparison of mortality in all patients on dialysis, patients on dialysis awaiting transplantation, and recipients of a first cadaveric transplant. N Engl J Med 1999;341:1725-30. https://doi.org/10.1056/NEJM199912023412303.
- Meier-Kriesche HU, Schold JD, Srinivas TR, Reed A, Kaplan B. Kidney transplantation halts cardiovascular disease progression in patients with end-stage renal disease. Am J Transplant 2004;4:1662-8. https://doi.org/10.1111/j.1600-6143.2004.00573.x.
- National Kidney Foundation . Transplantation Cost-Effectiveness n.d. www.kidney.org.uk/archives/news-archive-2/campaigns-transplantation-trans-cost-effect/ (accessed 23 February 2018).
- Wong G, Howard K, Chapman JR, Chadban S, Cross N, Tong A, et al. Comparative survival and economic benefits of deceased donor kidney transplantation and dialysis in people with varying ages and co-morbidities. PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0029591.
- Reese PP, Boudville N, Garg AX. Living kidney donation: outcomes, ethics, and uncertainty. Lancet 2015;385:2003-13. https://doi.org/10.1016/S0140-6736(14)62484-3.
- Summers DM, Watson CJ, Pettigrew GJ, Johnson RJ, Collett D, Neuberger JM, et al. Kidney donation after circulatory death (DCD): state of the art. Kidney Int 2015;88:241-9. https://doi.org/10.1038/ki.2015.88.
- NHS Blood and Transplant . NHS Blood and Transplant Activity Report n.d. www.odt.nhs.uk/statistics-and-reports (accessed 29 March 2018).
- Crew RJ, Ratner LE. ABO-incompatible kidney transplantation: current practice and the decade ahead. Curr Opin Organ Transplant 2010;15:526-30. https://doi.org/10.1097/MOT.0b013e32833bfbba.
- Wongsaroj P, Kahwaji J, Vo A, Jordan SC. Modern approaches to incompatible kidney transplantation. World J Nephrol 2015;4:354-62. https://doi.org/10.5527/wjn.v4.i3.354.
- Gondos A, Dohler B, Brenner H, Opelz G. Kidney graft survival in Europe and the United States: strikingly different long-term outcomes. Transplantation 2013;95:267-74. https://doi.org/10.1097/TP.0b013e3182708ea8.
- Welberry Smith MP, Baker RJ. Assessment and management of a patient with a renal transplant. Br J Hosp Med 2007;68:656-62. https://doi.org/10.12968/hmed.2007.68.12.656.
- Siedlecki A, Irish W, Brennan DC. Delayed graft function in the kidney transplant. Am J Transplant 2011;11:2279-96. https://doi.org/10.1111/j.1600–6143.2011.03754.x.
- Yarlagadda SG, Coca SG, Formica RN, Poggio ED, Parikh CR. Association between delayed graft function and allograft and patient survival: a systematic review and meta-analysis. Nephrol Dial Transplant 2009;24:1039-47. https://doi.org/10.1093/ndt/gfn667.
- Chamberlain G, Baboolal K, Bennett H, Pockett RD, McEwan P, Sabater J, et al. The economic burden of posttransplant events in renal transplant recipients in Europe. Transplantation 2014;97:854-61. https://doi.org/10.1097/01.tp.0000438205.04348.69.
- Yarlagadda SG, Coca SG, Garg AX, Doshi M, Poggio E, Marcus RJ, et al. Marked variation in the definition and diagnosis of delayed graft function: a systematic review. Nephrol Dial Transplant 2008;23:2995-3003. https://doi.org/10.1093/ndt/gfn158.
- Gjertson DW. Impact of delayed graft function and acute rejection on kidney graft survival. Clin Transpl 2000:467-80.
- Sellers MT, Gallichio MH, Hudson SL, Young CJ, Bynon JS, Eckhoff DE, et al. Improved outcomes in cadaveric renal allografts with pulsatile preservation. Clin Transplant 2000;14:543-9. http://onlinelibrary.wiley.com/store/10.1034/j.1399-0012.2000.140605.x/asset/j.1399-0012.2000.140605.x.pdf?v=1&t=ihorcchh&s=ffbed3fc6150ad0bccd4bc30075fc79280c01995.
- Perico N, Cattaneo D, Sayegh MH, Remuzzi G. Delayed graft function in kidney transplantation. Lancet 2004;364:1814-27. https://doi.org/10.1016/S0140-6736(04)17406-0.
- Mallon DH, Summers DM, Bradley JA, Pettigrew GJ. Defining delayed graft function after renal transplantation: simplest is best. Transplantation 2013;96:885-9. https://doi.org/10.1097/TP.0b013e3182a19348.
- United Network for Organ Sharing . Data Resources n.d. https://unos.org/data/data-resources/ (accessed 23 February 2018).
- Irish WD, Ilsley JN, Schnitzler MA, Feng S, Brennan DC. A risk prediction model for delayed graft function in the current era of deceased donor renal transplantation. Am J Transplant 2010;10:2279-86. https://doi.org/10.1111/j.1600-6143.2010.03179.x.
- Rodrigo E, Miñambres E, Ruiz JC, Ballesteros A, Piñera C, Quintanar J, et al. Prediction of delayed graft function by means of a novel web-based calculator: a single-center experience. Am J Transplant 2012;12:240-4. https://doi.org/10.1111/j.1600-6143.2011.03810.x.
- Kayler LK, Srinivas TR, Schold JD. Influence of CIT-induced DGF on kidney transplant outcomes. Am J Transplant 2011;11:2657-64. https://doi.org/10.1111/j.1600-6143.2011.03817.x.
- Moers C, Kornmann NS, Leuvenink HG, Ploeg RJ. The influence of deceased donor age and old-for-old allocation on kidney transplant outcome. Transplantation 2009;88:542-52. https://doi.org/10.1097/TP.0b013e3181b0fa8b.
- Remuzzi G, Cravedi P, Perna A, Dimitrov BD, Turturro M, Locatelli G, et al. Long-term outcome of renal transplantation from older donors. N Engl J Med 2006;354:343-52. https://doi.org/10.1056/NEJMoa052891.
- Halloran PF, Aprile MA, Farewell V, Ludwin D, Smith EK, Tsai SY, et al. Early function as the principal correlate of graft survival. A multivariate analysis of 200 cadaveric renal transplants treated with a protocol incorporating antilymphocyte globulin and cyclosporine. Transplantation 1988;46:223-8. https://doi.org/10.1097/00007890-198808000-00007.
- Tapiawala SN, Tinckam KJ, Cardella CJ, Schiff J, Cattran DC, Cole EH, et al. Delayed graft function and the risk for death with a functioning graft. J Am Soc Nephrol 2010;21:153-61. https://doi.org/10.1681/ASN.2009040412.
- Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 2011;79:1361-9. https://doi.org/10.1038/ki.2011.42.
- Qureshi F, Rabb H, Kasiske BL. Silent acute rejection during prolonged delayed graft function reduces kidney allograft survival. Transplantation 2002;74:1400-4. https://doi.org/10.1097/01.TP.0000036053.99338.C4.
- Muhlberger I, Perco P, Fechete R, Mayer B, Oberbauer R. Biomarkers in renal transplantation ischemia reperfusion injury. Transplantation 2009;88:S14-19. https://doi.org/10.1097/TP.0b013e3181af65b5.
- Data Protection Act 1998. London: The Stationery Office; 1998.
- Kyzas PA, Denaxa-Kyza D, Ioannidis JP. Almost all articles on cancer prognostic markers report statistically significant results. Eur J Cancer 2007;43:2559-79. https://doi.org/10.1016/j.ejca.2007.08.030.
- Kyzas PA, Denaxa-Kyza D, Ioannidis JP. Quality of reporting of cancer prognostic marker studies: association with reported prognostic effect. J Natl Cancer Inst 2007;99:236-43. https://doi.org/10.1093/jnci/djk032.
- McShane LM, Altman DG, Sauerbrei W. Identification of clinically useful cancer prognostic factors: what are we missing?. J Natl Cancer Inst 2005;97:1023-5. https://doi.org/10.1093/jnci/dji193.
- Vasudev NS, Sim S, Cairns DA, Ferguson RE, Craven RA, Stanley A, et al. Pre-operative urinary cathepsin D is associated with survival in patients with renal cell carcinoma. Br J Cancer 2009;101:1175-82. https://doi.org/10.1038/sj.bjc.6605250.
- Sim SH, Messenger MP, Gregory WM, Wind TC, Vasudev NS, Cartledge J, et al. Prognostic utility of pre-operative circulating osteopontin, carbonic anhydrase IX and CRP in renal cell carcinoma. Br J Cancer 2012;107:1131-7. https://doi.org/10.1038/bjc.2012.360.
- Gregory WM, Reznek RH, Hallett M, Slevin ML. Using mathematical models to estimate drug resistance and treatment efficacy via CT scan measurements of tumour volume. Br J Cancer 1990;62:671-5. https://doi.org/10.1038/bjc.1990.354.
- Migdal C, Gregory W, Hitchings R. Long-term functional outcome after early surgery compared with laser and medicine in open-angle glaucoma. Ophthalmology 1994;101:1651-6. https://doi.org/10.1016/S0161-6420(94)31120-1.
- Human Tissue Act 2004. London: The Stationery Office; 2004.
- Dheensa S, Fenwick A, Shkedi-Rafid S, Crawford G, Lucassen A. Health-care professionals’ responsibility to patients’ relatives in genetic medicine: a systematic review and synthesis of empirical research. Genet Med 2016;18:290-301. https://doi.org/10.1038/gim.2015.72.
- Grady C, Eckstein L, Berkman B, Brock D, Cook-Deegan R, Fullerton SM, et al. Broad consent for research with biological samples: workshop conclusions. Am J Bioeth 2015;15:34-42. https://doi.org/10.1080/15265161.2015.1062162.
- Sylte MS, Wentzel-Larsen T, Bolann BJ. Estimation of the minimal preanalytical uncertainty for 15 clinical chemistry serum analytes. Clin Chem 2010;56:1329-35. https://doi.org/10.1373/clinchem.2010.146050.
- Kellogg MD, Ellervik C, Morrow D, Hsing A, Stein E, Sethi AA. Preanalytical considerations in the design of clinical trials and epidemiological studies. Clin Chem 2015;61:797-803. https://doi.org/10.1373/clinchem.2014.226118.
- Procedures for the Handling and Processing of Blood Specimens for Common Laboratory Tests; Approved Guideline – Fourth Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2010.
- Urinalysis; Approved Guideline – Third Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2009.
- Caboux E, Plymoth A, Hainault P. Common Minimum Technical Standards and Protocols for Biological Resource Centres Dedicated to Cancer Research. Lyon: International Agency for Cancer Research; 2007.
- Simundic AM, Cornes MP, Grankvist K, Lippi G, Nybo M, Ceriotti F, et al. Colour coding for blood collection tube closures – a call for harmonisation. Clin Chem Lab Med 2015;53:371-6. https://doi.org/10.1515/cclm-2014–0927.
- World Medical Association . WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects n.d. www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed 26 February 2018).
- University of Leeds . Leeds Multidisciplinary Research Tissue Bank n.d. www.multirtb.leeds.ac.uk (accessed 19 January 2018).
- Department of Health and Social Care . Eligibility Criteria for NIHR Clinical Research Network Support n.d. www.nihr.ac.uk/funding-and-support/documents/study-support-service/Eligibility/Eligibility-Criteria-for-NIHR-Clinical-Research-Network-Support.pdf (accessed 26 February 2018).
- Department of Health and Social Care . Attributing the Costs of Health and Social Care Research 2012. www.gov.uk/government/publications/guidance-on-attributing-the-costs-of-health-and-social-care-research (accessed 19 January 2018).
- National Institute for Health Research . The NIHR Performance in Initiating and Delivering Clinical Research (70 Day Benchmark) and the NIHR Clinical Research Network High Level Objectives: A Description of Purpose, Definition and Differences 2013. www.nihr.ac.uk/02-documents/policy-and-standards/Faster-easier-clinical-research/NIHR-Metrics-Comparison-CCF-December-2013.pdf (accessed 26 February 2018).
- National Institute for Health Research . Performance in Initiating and Delivering Clinical Research n.d. www.nihr.ac.uk/research-and-impact/nhs-research-performance/performance-in-initiating-and-delivering-research/ (accessed 26 February 2018).
- National Institute for Health Research . NIHR CRN High Level Objectives Quarterly Performance Report Quarter 1 2015–16 n.d. www.nihr.ac.uk/about-us/documents/CRN%20performance%20reports/Apr-Jun%2015%20NIHR_CRN_Q12015_16_PerformanceReport.pdf (accessed 26 February 2018).
- Wei Y, Jiang YZ, Qian WH. Prognostic role of NLR in urinary cancers: a meta-analysis. PLOS ONE 2014;9. https://doi.org/10.1371/journal.pone.0092079.
- Wen RM, Zhang YJ, Ma S, Xu YL, Chen YS, Li HL, et al. Preoperative neutrophil to lymphocyte ratio as a prognostic factor in patients with non-metastatic renal cell carcinoma. Asian Pac J Cancer Prev 2015;16:3703-8. https://doi.org/10.7314/APJCP.2015.16.9.3703.
- Pichler M, Hutterer GC, Stoeckigt C, Chromecki TF, Stojakovic T, Golbeck S, et al. Validation of the pre-treatment neutrophil–lymphocyte ratio as a prognostic factor in a large European cohort of renal cell carcinoma patients. Br J Cancer 2013;108:901-7. https://doi.org/10.1038/bjc.2013.28.
- de Martino M, Pantuck AJ, Hofbauer S, Waldert M, Shariat SF, Belldegrun AS, et al. Prognostic impact of preoperative neutrophil-to-lymphocyte ratio in localized nonclear cell renal cell carcinoma. J Urol 2013;190:1999-2004. https://doi.org/10.1016/j.juro.2013.06.082.
- Ohno Y, Nakashima J, Ohori M, Gondo T, Hatano T, Tachibana M. Followup of neutrophil-to-lymphocyte ratio and recurrence of clear cell renal cell carcinoma. J Urol 2012;187:411-17. https://doi.org/10.1016/j.juro.2011.10.026.
- Ohno Y, Nakashima J, Ohori M, Hatano T, Tachibana M. Pretreatment neutrophil-to-lymphocyte ratio as an independent predictor of recurrence in patients with nonmetastatic renal cell carcinoma. J Urol 2010;184:873-8. https://doi.org/10.1016/j.juro.2010.05.028.
- Jagdev SP, Gregory W, Vasudev NS, Harnden P, Sim S, Thompson D, et al. Improving the accuracy of pre-operative survival prediction in renal cell carcinoma with C-reactive protein. Br J Cancer 2010;103:1649-56. https://doi.org/10.1038/sj.bjc.6605973.
- Motzer RJ, Mazumdar M, Bacik J, Berg W, Amsterdam A, Ferrara J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J Clin Oncol 1999;17:2530-40. https://doi.org/10.1200/JCO.1999.17.8.2530.
- Vasudev NS, Brown JE, Brown SR, Rafiq R, Morgan R, Patel PM, et al. Prognostic factors in renal cell carcinoma: association of preoperative sodium concentration with survival. Clin Cancer Res 2008;14:1775-81. https://doi.org/10.1158/1078-0432.CCR-07-1721.
- Jeppesen AN, Jensen HK, Donskov F, Marcussen N, von der Maase H. Hyponatremia as a prognostic and predictive factor in metastatic renal cell carcinoma. Br J Cancer 2010;102:867-72. https://doi.org/10.1038/sj.bjc.6605563.
- Furukawa J, Miyake H, Kusuda Y, Fujisawa M. Hyponatremia as a powerful prognostic predictor for Japanese patients with clear cell renal cell carcinoma treated with a tyrosine kinase inhibitor. Int J Clin Oncol 2015;20:351-7. https://doi.org/10.1007/s10147-014-0713-3.
- Schutz FA, Xie W, Donskov F, Sircar M, McDermott DF, Rini BI, et al. The impact of low serum sodium on treatment outcome of targeted therapy in metastatic renal cell carcinoma: results from the International Metastatic Renal Cell Cancer Database Consortium. Eur Urol 2014;65:723-30. https://doi.org/10.1016/j.eururo.2013.10.013.
- Niedworok C, Dorrenhaus B, Vom Dorp F, Piotrowski JA, Tschirdewahn S, Szarvas T, et al. Renal cell carcinoma and tumour thrombus in the inferior vena cava: clinical outcome of 98 consecutive patients and the prognostic value of preoperative parameters. World J Urol 2014;33:1541-52. https://doi.org/10.1007/s00345-014-1449-4.
- Fahn HJ, Lee YH, Chen MT, Huang JK, Chen KK, Chang LS. The incidence and prognostic significance of humoral hypercalcemia in renal cell carcinoma. J Urol 1991;145:248-50. https://doi.org/10.1016/S0022-5347(17)38305-2.
- Magera JS, Leibovich BC, Lohse CM, Sengupta S, Cheville JC, Kwon ED, et al. Association of abnormal preoperative laboratory values with survival after radical nephrectomy for clinically confined clear cell renal cell carcinoma. Urology 2008;71:278-82. https://doi.org/10.1016/j.urology.2007.08.048.
- Yao M, Murakami T, Shioi K, Mizuno N, Ito H, Kondo K, et al. Tumor signatures of PTHLH overexpression, high serum calcium, and poor prognosis were observed exclusively in clear cell but not non clear cell renal carcinomas. Cancer Med 2014;3:845-54. https://doi.org/10.1002/cam4.270.
- Papworth K, Grankvist K, Ljungberg B, Rasmuson T. Parathyroid hormone-related protein and serum calcium in patients with renal cell carcinoma. Tumour Biol 2005;26:201-6. https://doi.org/10.1159/000086953.
- Kamai T, Arai K, Koga F, Abe H, Nakanishi K, Kambara T, et al. Higher expression of K-ras is associated with parathyroid hormone-related protein-induced hypercalcaemia in renal cell carcinoma. BJU Int 2001;88:960-6. https://doi.org/10.1046/j.1464-4096.2001.01294.x.
- Atlas I, Kwan D, Stone N. Value of serum alkaline phosphatase and radionuclide bone scans in patients with renal cell carcinoma. Urology 1991;38:220-2. https://doi.org/10.1016/S0090-4295(91)80348-B.
- Chuang YC, Lin AT, Chen KK, Chang YH, Chen MT, Chang LS. Paraneoplastic elevation of serum alkaline phosphatase in renal cell carcinoma: incidence and implication on prognosis. J Urol 1997;158:1684-7. https://doi.org/10.1016/S0022-5347(01)64095-3.
- Lee SE, Byun SS, Han JH, Han BK, Hong SK. Prognostic significance of common preoperative laboratory variables in clear cell renal cell carcinoma. BJU Int 2006;98:1228-32. https://doi.org/10.1111/j.1464-410X.2006.06437.x.
- Margulis V, McDonald M, Tamboli P, Swanson DA, Wood CG. Predictors of oncological outcome after resection of locally recurrent renal cell carcinoma. J Urol 2009;181:2044-51. https://doi.org/10.1016/j.juro.2009.01.043.
- Haddad AQ, Wood CG, Abel EJ, Krabbe LM, Darwish OM, Thompson RH, et al. Oncologic outcomes following surgical resection of renal cell carcinoma with inferior vena caval thrombus extending above the hepatic veins: a contemporary multicenter cohort. J Urol 2014;192:1050-6. https://doi.org/10.1016/j.juro.2014.03.111.
- Hofbauer SL, Stangl KI, de Martino M, Lucca I, Haitel A, Shariat SF, et al. Pretherapeutic gamma-glutamyltransferase is an independent prognostic factor for patients with renal cell carcinoma. Br J Cancer 2014;111:1526-31. https://doi.org/10.1038/bjc.2014.450.
- Dalpiaz O, Pichler M, Mrsic E, Reitz D, Krieger D, Venturino L, et al. Preoperative serum-gamma-glutamyltransferase (GGT) does not represent an independent prognostic factor in a European cohort of patients with non-metastatic renal cell carcinoma. J Clin Pathol 2015;68:547-51. https://doi.org/10.1136/jclinpath-2014-202683.
- Ohno Y, Nakashima J, Nakagami Y, Gondo T, Ohori M, Hatano T, et al. Clinical implications of preoperative serum total cholesterol in patients with clear cell renal cell carcinoma. Urology 2014;83:154-8. https://doi.org/10.1016/j.urology.2013.08.052.
- de Martino M, Leitner CV, Seemann C, Hofbauer SL, Lucca I, Haitel A, et al. Preoperative serum cholesterol is an independent prognostic factor for patients with renal cell carcinoma (RCC). BJU Int 2015;115:397-404. https://doi.org/10.1111/bju.12767.
- Ko K, Park YH, Lee JW, Ku JH, Kwak C, Kim HH. Influence of nutritional deficiency on prognosis of renal cell carcinoma (RCC). BJU Int 2013;112:775-80. https://doi.org/10.1111/bju.12275.
- Jeon HG, Choi DK, Sung HH, Jeong BC, Seo SI, Jeon SS, et al. Preoperative prognostic nutritional index is a significant predictor of survival in renal cell carcinoma patients undergoing nephrectomy. Ann Surg Oncol 2015;23:321-7. https://doi.org/10.1245/s10434-015-4614-0.
- Morgan TM, Tang D, Stratton KL, Barocas DA, Anderson CB, Gregg JR, et al. Preoperative nutritional status is an important predictor of survival in patients undergoing surgery for renal cell carcinoma. Eur Urol 2011;59:923-8. https://doi.org/10.1016/j.eururo.2011.01.034.
- Rasmuson T, Grankvist K, Ljungberg B. Serum beta 2-microglobulin and prognosis of patients with renal cell carcinoma. Acta Oncol 1996;35:479-82. https://doi.org/10.3109/02841869609109926.
- Grankvist K, Ljungberg B, Rasmuson T. Evaluation of five glycoprotein tumour markers (CEA, CA-50, CA-19–9, CA-125, CA-15–3) for the prognosis of renal-cell carcinoma. Int J Cancer 1997;74:233-6. https://doi.org/10.1002/(SICI)1097-0215(19970422)74:2<233::AID-IJC17>3.0.CO;2-E.
- Lucarelli G, Ditonno P, Bettocchi C, Vavallo A, Rutigliano M, Galleggiante V, et al. Diagnostic and prognostic role of preoperative circulating CA 15–3, CA 125, and beta-2 microglobulin in renal cell carcinoma. Dis Markers 2014;2014. https://doi.org/10.1155/2014/689795.
- Hotakainen K, Ljungberg B, Paju A, Rasmuson T, Alfthan H, Stenman UH. The free beta-subunit of human chorionic gonadotropin as a prognostic factor in renal cell carcinoma. Br J Cancer 2002;86:185-9. https://doi.org/10.1038/sj.bjc.6600050.
- Hotakainen K, Ljungberg B, Haglund C, Nordling S, Paju A, Stenman UH. Expression of the free beta-subunit of human chorionic gonadotropin in renal cell carcinoma: prognostic study on tissue and serum. Int J Cancer 2003;104:631-5. https://doi.org/10.1002/ijc.11000.
- Horstmann M, Merseburger AS, von der Heyde E, Serth J, Wegener G, Mengel M, et al. Correlation of bFGF expression in renal cell cancer with clinical and histopathological features by tissue microarray analysis and measurement of serum levels. J Cancer Res Clin Oncol 2005;131:715-22. https://doi.org/10.1007/s00432–005–0019-y.
- Takashi M, Sakata T, Kato K. Use of serum gamma-enolase and aldolase A in combination as markers for renal cell carcinoma. Jpn J Cancer Res 1993;84:304-9. https://doi.org/10.1111/j.1349-7006.1993.tb02871.x.
- Rasmuson T, Grankvist K, Ljungberg B. Serum gamma-enolase and prognosis of patients with renal cell carcinoma. Cancer 1993;72:1324-8. https://doi.org/10.1002/1097-0142(19930815)72:4<1324::AID-CNCR2820720429>3.0.CO;2-W.
- Rasmuson T, Grankvist K, Roos G, Ljungberg B. Neuroendocrine differentiation in renal cell carcinoma – evaluation of chromogranin A and neuron-specific enolase. Acta Oncol 1999;38:623-8. https://doi.org/10.1080/028418699431221.
- Xiao B, Ma LL, Zhang SD, Xiao CL, Lu J, Hong K, et al. Correlation between coagulation function, tumor stage and metastasis in patients with renal cell carcinoma: a retrospective study. Chin Med J (Engl) 2011;124:1205-8.
- Du J, Zheng JH, Chen XS, Yang Q, Zhang YH, Zhou L, et al. High preoperative plasma fibrinogen is an independent predictor of distant metastasis and poor prognosis in renal cell carcinoma. Int J Clin Oncol 2013;18:517-23. https://doi.org/10.1007/s10147-012-0412-x.
- Pichler M, Hutterer GC, Stojakovic T, Mannweiler S, Pummer K, Zigeuner R. High plasma fibrinogen level represents an independent negative prognostic factor regarding cancer-specific, metastasis-free, as well as overall survival in a European cohort of non-metastatic renal cell carcinoma patients. Br J Cancer 2013;109:1123-9. https://doi.org/10.1038/bjc.2013.443.
- Erdem S, Amasyali AS, Aytac O, Onem K, Issever H, Sanli O. Increased preoperative levels of plasma fibrinogen and D dimer in patients with renal cell carcinoma is associated with poor survival and adverse tumor characteristics. Urol Oncol 2014;32:1031-40. https://doi.org/10.1016/j.urolonc.2014.03.013.
- Miki S, Iwano M, Miki Y, Yamamoto M, Tang B, Yokokawa K, et al. Interleukin-6 (IL-6) functions as an in vitro autocrine growth factor in renal cell carcinomas. FEBS Lett 1989;250:607-10. https://doi.org/10.1016/0014-5793(89)80805-1.
- Dosquet C, Schaetz A, Faucher C, Lepage E, Wautier JL, Richard F, et al. Tumour necrosis factor-alpha, interleukin-1 beta and interleukin-6 in patients with renal cell carcinoma. Eur J Cancer 1994;30A:162-7. https://doi.org/10.1016/0959-8049(94)90079-5.
- Ljungberg B, Grankvist K, Rasmuson T. Serum interleukin-6 in relation to acute-phase reactants and survival in patients with renal cell carcinoma. Eur J Cancer 1997;33:1794-8. https://doi.org/10.1016/S0959-8049(97)00179-2.
- Yoshida N, Ikemoto S, Narita K, Sugimura K, Wada S, Yasumoto R, et al. Interleukin-6, tumour necrosis factor alpha and interleukin-1beta in patients with renal cell carcinoma. Br J Cancer 2002;86:1396-400. https://doi.org/10.1038/sj.bjc.6600257.
- Ramsey S, Lamb GW, Aitchison M, McMillan DC. The longitudinal relationship between circulating concentrations of C-reactive protein, interleukin-6 and interleukin-10 in patients undergoing resection for renal cancer. Br J Cancer 2006;95:1076-80. https://doi.org/10.1038/sj.bjc.6603387.
- Hrab M, Olek-Hrab K, Antczak A, Kwias Z, Milecki T. Interleukin-6 (IL-6) and C-reactive protein (CRP) concentration prior to total nephrectomy are prognostic factors in localized renal cell carcinoma (RCC). Rep Pract Oncol Radiother 2013;18:304-9. https://doi.org/10.1016/j.rpor.2013.06.002.
- Jabs WJ, Busse M, Krüger S, Jocham D, Steinhoff J, Doehn C. Expression of C-reactive protein by renal cell carcinomas and unaffected surrounding renal tissue. Kidney Int 2005;68:2103-10. https://doi.org/10.1111/j.1523-1755.2005.00666.x.
- Johnson TV, Ali S, Abbasi A, Kucuk O, Harris WB, Ogan K, et al. Intratumor C-reactive protein as a biomarker of prognosis in localized renal cell carcinoma. J Urol 2011;186:1213-17. https://doi.org/10.1016/j.juro.2011.06.014.
- Ljungberg B, Grankvist K, Rasmuson T. Serum acute phase reactants and prognosis in renal cell carcinoma. Cancer 1995;76:1435-9. https://doi.org/10.1002/1097-0142(19951015)76:8<1435::AID-CNCR2820760821>3.0.CO;2-Y.
- Dai J, Tang K, Xiao W, Yu G, Zeng J, Li W, et al. Prognostic significance of C-reactive protein in urological cancers: a systematic review and meta-analysis. Asian Pac J Cancer Prev 2014;15:3369-75. https://doi.org/10.7314/APJCP.2014.15.8.3369.
- Ito K, Asano T, Yoshii H, Satoh A, Sumitomo M, Hayakawa M. Impact of thrombocytosis and C-reactive protein elevation on the prognosis for patients with renal cell carcinoma. Int J Urol 2006;13:1365-70. https://doi.org/10.1111/j.1442-2042.2006.01563.x.
- Komai Y, Saito K, Sakai K, Morimoto S. Increased preoperative serum C-reactive protein level predicts a poor prognosis in patients with localized renal cell carcinoma. BJU Int 2007;99:77-80. https://doi.org/10.1111/j.1464-410X.2006.06497.x.
- Lamb GW, McArdle PA, Ramsey S, McNichol AM, Edwards J, Aitchison M, et al. The relationship between the local and systemic inflammatory responses and survival in patients undergoing resection for localized renal cancer. BJU Int 2008;102:756-61. https://doi.org/10.1111/j.1464-410X.2008.07666.x.
- García-Marchiñena P, Billordo-Perés N, Tobía-González I, Jurado A, Damia O, Gueglio G. High-sensitivity C-reactive protein as a predictor of locally advanced renal cell carcinoma. Arch Esp Urol 2012;65:601-7.
- Kawata N, Nagane Y, Yamaguchi K, Ichinose T, Hirakata H, Takahashi S. How do symptoms have an impact on the prognosis of renal cell carcinoma?. Int J Urol 2008;15:299-303. https://doi.org/10.1111/j.1442-2042.2008.01990.x.
- Tanaka M, Fujimoto K, Okajima E, Tanaka N, Yoshida K, Hirao Y. Prognostic factors of renal cell carcinoma with extension into inferior vena cava. Int J Urol 2008;15:394-8. https://doi.org/10.1111/j.1442–2042.2008.02017.x.
- Steffens S, Köhler A, Rudolph R, Eggers H, Seidel C, Janssen M, et al. Validation of CRP as prognostic marker for renal cell carcinoma in a large series of patients. BMC Cancer 2012;12. https://doi.org/10.1186/1471-2407-12-399.
- Ito K, Yoshii H, Sato A, Kuroda K, Asakuma J, Horiguchi A, et al. Impact of postoperative C-reactive protein level on recurrence and prognosis in patients with N0M0 clear cell renal cell carcinoma. J Urol 2011;186:430-5. https://doi.org/10.1016/j.juro.2011.03.113.
- Ramsey S, Lamb GW, Aitchison M, McMillan DC. Prospective study of the relationship between the systemic inflammatory response, prognostic scoring systems and relapse-free and cancer-specific survival in patients undergoing potentially curative resection for renal cancer. BJU Int 2008;101:959-63. https://doi.org/10.1111/j.1464-410X.2007.07363.x.
- Johnson TV, Abbasi A, Owen-Smith A, Young A, Ogan K, Pattaras J, et al. Absolute preoperative C-reactive protein predicts metastasis and mortality in the first year following potentially curative nephrectomy for clear cell renal cell carcinoma. J Urol 2010;183:480-5. https://doi.org/10.1016/j.juro.2009.10.014.
- Michigan A, Johnson TV, Master VA. Preoperative C-reactive protein level adjusted for comorbidities and lifestyle factors predicts overall mortality in localized renal cell carcinoma. Mol Diagn Ther 2011;15:229-34. https://doi.org/10.2165/11534900-000000000-00000.
- Karakiewicz PI, Hutterer GC, Trinh QD, Jeldres C, Perrotte P, Gallina A, et al. C-reactive protein is an informative predictor of renal cell carcinoma-specific mortality: a European study of 313 patients. Cancer 2007;110:1241-7. https://doi.org/10.1002/cncr.22896.
- Iimura Y, Saito K, Fujii Y, Kumagai J, Kawakami S, Komai Y, et al. Development and external validation of a new outcome prediction model for patients with clear cell renal cell carcinoma treated with nephrectomy based on preoperative serum C-reactive protein and TNM classification: the TNM-C score. J Urol 2009;181:1004-12. https://doi.org/10.1016/j.juro.2008.10.156.
- Nakayama T, Saito K, Ishioka J, Kawano K, Morimoto S, Matsuoka Y, et al. External validation of TNM-C score in three community hospital cohorts for clear cell renal cell carcinoma. Anticancer Res 2014;34:921-6. http://ar.iiarjournals.org/content/34/2/921.long.
- de Martino M, Klatte T, Seemann C, Waldert M, Haitel A, Schatzl G, et al. Validation of serum C-reactive protein (CRP) as an independent prognostic factor for disease-free survival in patients with localised renal cell carcinoma (RCC). BJU Int 2013;111:E348-53. https://doi.org/10.1111/bju.12067.
- Bedke J, Chun FK, Merseburger A, Scharpf M, Kasprzyk K, Schilling D, et al. Inflammatory prognostic markers in clear cell renal cell carcinoma – preoperative C-reactive protein does not improve predictive accuracy. BJU Int 2012;110:E771-7. https://doi.org/10.1111/j.1464-410X.2012.11642.x.
- Kimura M, Tomita Y, Imai T, Saito T, Katagiri A, Ohara-Mikami Y, et al. Significance of serum amyloid A on the prognosis in patients with renal cell carcinoma. Cancer 2001;92:2072-5. https://doi.org/10.1002/1097-0142(20011015)92:8<2072::AID-CNCR1547>3.0.CO;2-P.
- Ramankulov A, Lein M, Johannsen M, Schrader M, Miller K, Loening SA, et al. Serum amyloid A as indicator of distant metastases but not as early tumor marker in patients with renal cell carcinoma. Cancer Lett 2008;269:85-92. https://doi.org/10.1016/j.canlet.2008.04.022.
- Mittal A, Poudel B, Pandeya DR, Gupta SP, Sathian B, Yadav SK. Serum amyloid A as an independent prognostic factor for renal cell carcinoma – a hospital based study from the Western region of Nepal. Asian Pac J Cancer Prev 2012;13:2253-5. https://doi.org/10.7314/APJCP.2012.13.5.2253.
- Fischer K, Theil G, Hoda R, Fornara P. Serum amyloid A: a biomarker for renal cancer. Anticancer Res 2012;32:1801-4. http://ar.iiarjournals.org/content/32/5/1801.full.pdf.
- Wood SL, Rogers M, Cairns DA, Paul A, Thompson D, Vasudev NS, et al. Association of serum amyloid A protein and peptide fragments with prognosis in renal cancer. Br J Cancer 2010;103:101-11. https://doi.org/10.1038/sj.bjc.6605720.
- Essen A, Ozen H, Ayhan A, Ergen A, Tasar C, Remzi F. Serum ferritin: a tumor marker for renal cell carcinoma. J Urol 1991;145:1134-7. https://doi.org/10.1016/S0022-5347(17)38555-5.
- Partin AW, Criley SR, Steiner MS, Hsieh K, Simons JW, Lumadue J, et al. Serum ferritin as a clinical marker for renal cell carcinoma: influence of tumor volume. Urology 1995;45:211-17. https://doi.org/10.1016/0090-4295(95)80007-7.
- Kirkali Z, Güzelsoy M, Mungan MU, Kirkali G, Yörükoglu K. Serum ferritin as a clinical marker for renal cell carcinoma: influence of tumor size and volume. Urol Int 1999;62:21-5. https://doi.org/10.1159/000030349.
- Ozen H, Uygur C, Sahin A, Tekgül S, Ergen A, Remzi D. Clinical significance of serum ferritin in patients with renal cell carcinoma. Urology 1995;46:494-8. https://doi.org/10.1016/S0090-4295(99)80261-1.
- Sufrin G, Mirand EA, Moore RH, Chu TM, Murphy GP. Hormones in renal cancer. J Urol 1977;117:433-8. https://doi.org/10.1016/S0022-5347(17)58490-6.
- Ljungberg B, Rasmuson T, Grankvist K. Erythropoietin in renal cell carcinoma: evaluation of its usefulness as a tumor marker. Eur Urol 1992;21:160-3. https://doi.org/10.1159/000474825.
- Papworth K, Bergh A, Grankvist K, Ljungberg B, Rasmuson T. Expression of erythropoietin and its receptor in human renal cell carcinoma. Tumour Biol 2009;30:86-92. https://doi.org/10.1159/000216844.
- Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem 2013;153:13-9. https://doi.org/10.1093/jb/mvs136.
- Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med 2012;2. https://doi.org/10.1101/cshperspect.a006593.
- Dosquet C, Coudert MC, Lepage E, Cabane J, Richard F. Are angiogenic factors, cytokines, and soluble adhesion molecules prognostic factors in patients with renal cell carcinoma?. Clin Cancer Res 1997;3:2451-8.
- Ljungberg B, Jacobsen J, Haggstrom-Rudolfssson S, Rasmuson T, Lindh G, Grankvist K. Tumour vascular endothelial growth factor (VEGF) mRNA in relation to serum VEGF protein levels and tumour progression in human renal cell carcinoma. Urol Res 2003;31:335-40. https://doi.org/10.1007/s00240-003-0346-x.
- Schips L, Dalpiaz O, Lipsky K, Langner C, Rehak P, Puerstner P, et al. Serum levels of vascular endothelial growth factor (VEGF) and endostatin in renal cell carcinoma patients compared to a control group. Eur Urol 2007;51:168-73. https://doi.org/10.1016/j.eururo.2006.06.026.
- Tanimoto S, Fukumori T, El-Moula G, Shiirevnyamba A, Kinouchi S, Koizumi T, et al. Prognostic significance of serum hepatocyte growth factor in clear cell renal cell carcinoma: comparison with serum vascular endothelial growth factor. J Med Invest 2008;55:106-11. https://doi.org/10.2152/jmi.55.106.
- Prinsloo S, Wei Q, Scott SM, Tannir N, Jonasch E, Pisters L, et al. Psychological states, serum markers and survival: associations and predictors of survival in patients with renal cell carcinoma. J Behav Med 2015;38:48-56. https://doi.org/10.1007/s10865-014-9578-1.
- Guõbrandsdottir G, Hjelle KM, Frugård J, Bostad L, Aarstad HJ, Beisland C. Preoperative high levels of serum vascular endothelial growth factor are a prognostic marker for poor outcome after surgical treatment of renal cell carcinoma. Scand J Urol 2015;49:388-94. https://doi.org/10.3109/21681805.2015.1021833.
- Fujita N, Okegawa T, Terado Y, Tambo M, Higashihara E, Nutahara K. Serum level and immunohistochemical expression of vascular endothelial growth factor for the prediction of postoperative recurrence in renal cell carcinoma. BMC Res Notes 2014;7. https://doi.org/10.1186/1756-0500-7-369.
- Feldman AL, Alexander HR, Yang JC, Linehan WM, Eyler RA, Miller MS, et al. Prospective analysis of circulating endostatin levels in patients with renal cell carcinoma. Cancer 2002;95:1637-43. https://doi.org/10.1002/cncr.10845.
- Rioux-Leclercq N, Fergelot P, Zerrouki S, Leray E, Jouan F, Bellaud P, et al. Plasma level and tissue expression of vascular endothelial growth factor in renal cell carcinoma: a prospective study of 50 cases. Hum Pathol 2007;38:1489-95. https://doi.org/10.1016/j.humpath.2007.02.014.
- Klatte T, Böhm M, Nelius T, Filleur S, Reiher F, Allhoff EP. Evaluation of peri-operative peripheral and renal venous levels of pro- and anti-angiogenic factors and their relevance in patients with renal cell carcinoma. BJU Int 2007;100:209-14. https://doi.org/10.1111/j.1464-410X.2007.06871.x.
- Patard JJ, Rioux-Leclercq N, Masson D, Zerrouki S, Jouan F, Collet N, et al. Absence of VHL gene alteration and high VEGF expression are associated with tumour aggressiveness and poor survival of renal-cell carcinoma. Br J Cancer 2009;101:1417-24. https://doi.org/10.1038/sj.bjc.6605298.
- Yang H, Zhao K, Yu Q, Wang X, Song Y, Li R. Evaluation of plasma and tissue S100A4 protein and mRNA levels as potential markers of metastasis and prognosis in clear cell renal cell carcinoma. J Int Med Res 2012;40:475-85. https://doi.org/10.1177/147323001204000209.
- Banks RE, Forbes MA, Kinsey SE, Stanley A, Ingham E, Walters C, et al. Release of the angiogenic cytokine vascular endothelial growth factor (VEGF) from platelets: significance for VEGF measurements and cancer biology. Br J Cancer 1998;77:956-64. https://doi.org/10.1038/bjc.1998.158.
- Verheul HM, Hoekman K, Luykx-de Bakker S, Eekman CA, Folman CC, Broxterman HJ, et al. Platelet: transporter of vascular endothelial growth factor. Clin Cancer Res 1997;3:2187-90.
- Gunsilius E, Petzer A, Stockhammer G, Nussbaumer W, Schumacher P, Clausen J, et al. Thrombocytes are the major source for soluble vascular endothelial growth factor in peripheral blood. Oncology 2000;58:169-74. https://doi.org/10.1159/000012095.
- Niers TM, Richel DJ, Meijers JC, Schlingemann RO. Vascular endothelial growth factor in the circulation in cancer patients may not be a relevant biomarker. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0019873.
- Salgado R, Vermeulen PB, Benoy I, Weytjens R, Huget P, Van Marck E, et al. Platelet number and interleukin-6 correlate with VEGF but not with bFGF serum levels of advanced cancer patients. Br J Cancer 1999;80:892-7. https://doi.org/10.1038/sj.bjc.6690437.
- O’Byrne KJ, Dobbs N, Propper D, Smith K, Harris AL. Vascular endothelial growth factor platelet counts, and prognosis in renal cancer. Lancet 1999;353:1494-5. https://doi.org/10.1016/S0140-6736(99)00471-7.
- Oosterwijk E, Ruiter DJ, Hoedemaeker PJ, Pauwels EK, Jonas U, Zwartendijk J, et al. Monoclonal antibody G 250 recognizes a determinant present in renal-cell carcinoma and absent from normal kidney. Int J Cancer 1986;38:489-94. https://doi.org/10.1002/ijc.2910380406.
- Grabmaier K, Vissers JLM, De Weijert MCA, Oosterwijk-Wakka JC, Van Bokhoven A, Brakenhoff RH, et al. Molecular cloning and immunogenicity of renal cell carcinoma-associated antigen G250. Int J Cancer 2000;85:865-70. https://doi.org/Doi 10.1002/(Sici)1097-0215(20000315)85:6<865::Aid-Ijc21>3.0.Co;2-Q.
- Závada J, Závadová Z, Zat’ovicová M, Hyrsl L, Kawaciuk I. Soluble form of carbonic anhydrase IX (CA IX) in the serum and urine of renal carcinoma patients. Br J Cancer 2003;89:1067-71. https://doi.org/10.1038/sj.bjc.6601264.
- Li G, Feng G, Gentil-Perret A, Genin C, Tostain J. Serum carbonic anhydrase 9 level is associated with postoperative recurrence of conventional renal cell cancer. J Urol 2008;180:510-13. https://doi.org/10.1016/j.juro.2008.04.024.
- Papworth K, Sandlund J, Grankvist K, Ljungberg B, Rasmuson T. Soluble carbonic anhydrase IX is not an independent prognostic factor in human renal cell carcinoma. Anticancer Res 2010;30:2953-7.
- Miyata Y, Iwata T, Ohba K, Kanda S, Nishikido M, Kanetake H. Expression of matrix metalloproteinase-7 on cancer cells and tissue endothelial cells in renal cell carcinoma: prognostic implications and clinical significance for invasion and metastasis. Clin Cancer Res 2006;12:6998-7003. https://doi.org/10.1158/1078-0432.CCR-06-1626.
- Lu H, Yang Z, Zhang H, Gan M, Zhou T, Wang S. The expression and clinical significance of matrix metalloproteinase 7 and tissue inhibitor of matrix metalloproteinases 2 in clear cell renal cell carcinoma. Exp Ther Med 2013;5:890-6. https://doi.org/10.3892/etm.2012.859.
- Sarkissian G, Fergelot P, Lamy PJ, Patard JJ, Culine S, Jouin P, et al. Identification of pro-MMP-7 as a serum marker for renal cell carcinoma by use of proteomic analysis. Clin Chem 2008;54:574-81. https://doi.org/10.1373/clinchem.2007.090837.
- Ramankulov A, Lein M, Johannsen M, Schrader M, Miller K, Jung K. Plasma matrix metalloproteinase-7 as a metastatic marker and survival predictor in patients with renal cell carcinomas. Cancer Sci 2008;99:1188-94. https://doi.org/10.1111/j.1349-7006.2008.00802.x.
- Matusan K, Dordevic G, Stipic D, Mozetic V, Lucin K. Osteopontin expression correlates with prognostic variables and survival in clear cell renal cell carcinoma. J Surg Oncol 2006;94:325-31. https://doi.org/10.1002/jso.20447.
- Ramankulov A, Lein M, Kristiansen G, Meyer HA, Loening SA, Jung K. Elevated plasma osteopontin as marker for distant metastases and poor survival in patients with renal cell carcinoma. J Cancer Res Clin Oncol 2007;133:643-52. https://doi.org/10.1007/s00432-007-0215-z.
- Papworth K, Bergh A, Grankvist K, Ljungberg B, Sandlund J, Rasmuson T. Osteopontin but not parathyroid hormone-related protein predicts prognosis in human renal cell carcinoma. Acta Oncol 2013;52:159-65. https://doi.org/10.3109/0284186X.2012.693623.
- Zhang Q, Domenicucci C, Goldberg HA, Wrana JL, Sodek J. Characterization of fetal porcine bone sialoproteins, secreted phosphoprotein I (SPPI, osteopontin), bone sialoprotein, and a 23-kDa glycoprotein. Demonstration that the 23-kDa glycoprotein is derived from the carboxyl terminus of SPPI. J Biol Chem 1990;265:7583-9.
- King JD, Casavant BP, Lang JM. Rapid translation of circulating tumor cell biomarkers into clinical practice: technology development, clinical needs and regulatory requirements. Lab Chip 2014;14:24-31. https://doi.org/10.1039/C3lC50741f.
- Ueda T. Serum immunosuppressive acidic protein in renal cell carcinoma. Urol Res 1986;14:101-3. https://doi.org/10.1007/BF00257894.
- Gohji K, Ishii M, Nagata H, Matsumoto O, Kamidono S. Serum basic fetoprotein in patients with renal cell carcinoma. Cancer 1990;65:1405-11. https://doi.org/10.1002/1097-0142(19900315)65:6<1405::AID-CNCR2820650627>3.0.CO;2-5.
- Masuda H, Kurita Y, Suzuki K, Fujita K, Aso Y. Predictive value of serum immunosuppressive acidic protein for staging renal cell carcinoma: comparison with other tumour markers. Br J Urol 1997;80:25-9. https://doi.org/10.1046/j.1464-410X.1997.00244.x.
- Kawata N, Yamaguchi K, Hirakata H, Hachiya T, Yoshida T, Takimoto Y. Immunosuppressive acidic protein detects high nuclear grade localized renal cell carcinoma. Urology 2005;66:736-40. https://doi.org/10.1016/j.urology.2005.04.044.
- Igarashi T, Murakami S, Isaka S, Okano T, Shimazaki J, Matsuzaki O. Serum immunosuppressive acidic protein as a tumor marker for renal cell carcinoma. Eur Urol 1991;19:332-5. https://doi.org/10.1159/000473654.
- Miyata Y, Koga S, Nishikido M, Noguchi M, Kanda S, Hayashi T, et al. Predictive values of acute phase reactants, basic fetoprotein, and immunosuppressive acidic protein for staging and survival in renal cell carcinoma. Urology 2001;58:161-4. https://doi.org/10.1016/S0090-4295(01)01165-7.
- Araki K, Igarashi T, Tobe T, Mizoguchi K, Suzuki H, Furuya Y, et al. Serum immunosuppressive acidic protein doubling time as a prognostic factor for recurrent renal cell carcinoma after nephrectomy. Urology 2006;68:1178-82. https://doi.org/10.1016/j.urology.2006.08.1071.
- Oremek GM, Teigelkamp S, Kramer W, Eigenbrodt E, Usadel KH. The pyruvate kinase isoenzyme tumor M2 (Tu M2-PK) as a tumor marker for renal carcinoma. Anticancer Res 1999;19:2599-601.
- Wechsel HW, Petri E, Bichler KH, Feil G. Marker for renal cell carcinoma (RCC): the dimeric form of pyruvate kinase type M2 (Tu M2-PK). Anticancer Res 1999;19:2583-90.
- Nisman B, Yutkin V, Nechushtan H, Gofrit ON, Peretz T, Gronowitz S, et al. Circulating tumor M2 pyruvate kinase and thymidine kinase 1 are potential predictors for disease recurrence in renal cell carcinoma after nephrectomy. Urology 2010;76:513 e1-6. https://doi.org/10.1016/j.urology.2010.04.034.
- Adams J, Carder PJ, Downey S, Forbes MA, MacLennan K, Allgar V, et al. Vascular endothelial growth factor (VEGF) in breast cancer: comparison of plasma, serum, and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res 2000;60:2898-905.
- Luo P, He E, Eriksson S, Zhou J, Hu G, Zhang J, et al. Thymidine kinase activity in serum of renal cell carcinoma patients is a useful prognostic marker. Eur J Cancer Prev 2009;18:220-4. https://doi.org/10.1097/CEJ.0b013e328329d817.
- Matsumoto T, Furukawa A, Sumiyoshi Y, Akiyama KY, Kanayama HO, Kagawa S. Serum levels of soluble interleukin-2 receptor in renal cell carcinoma. Urology 1998;51:145-9. https://doi.org/10.1016/S0090-4295(97)00476-7.
- Masuda A, Arai K, Nishihara D, Mizuno T, Yuki H, Kambara T, et al. Clinical significance of serum soluble T cell regulatory molecules in clear cell renal cell carcinoma. Biomed Res Int 2014;2014. https://doi.org/10.1155/2014/396064.
- Fujimoto K, Ichimori Y, Kakizoe T, Okajima E, Sakamoto H, Sugimura T, et al. Increased serum levels of basic fibroblast growth factor in patients with renal cell carcinoma. Biochem Biophys Res Commun 1991;180:386-92. https://doi.org/10.1016/S0006-291X(05)81305-1.
- Fujimoto K, Ichimori Y, Yamaguchi H, Arai K, Futami T, Ozono S, et al. Basic fibroblast growth factor as a candidate tumor marker for renal cell carcinoma. Jpn J Cancer Res 1995;86:182-6. https://doi.org/10.1111/j.1349-7006.1995.tb03037.x.
- Rasmuson T, Grankvist K, Jacobsen J, Ljungberg B. Impact of serum basic fibroblast growth factor on prognosis in human renal cell carcinoma. Eur J Cancer 2001;37:2199-203. https://doi.org/10.1016/S0959-8049(01)00290-8.
- Rasmuson T, Grankvist K, Jacobsen J, Olsson T, Ljungberg B. Serum insulin-like growth factor-1 is an independent predictor of prognosis in patients with renal cell carcinoma. Acta Oncol 2004;43:744-8. https://doi.org/10.1080/02841860410017260.
- Paju A, Jacobsen J, Rasmuson T, Stenman UH, Ljungberg B. Tumor associated trypsin inhibitor as a prognostic factor in renal cell carcinoma. J Urol 2001;165:959-62. https://doi.org/10.1016/S0022-5347(05)66584-6.
- Muller DC, Scelo G, Zaridze D, Janout V, Holcatova I, Navratilova M, et al. Circulating 25-hydroxyvitamin D3 and survival after diagnosis with kidney cancer. Cancer Epidemiol Biomarkers Prev 2015;24:1277-81. https://doi.org/10.1158/1055–9965.EPI-14–1351.
- Stevens LA, Levey AS. Measurement of kidney function. Med Clin North Am 2005;89:457-73. https://doi.org/10.1016/j.mcna.2004.11.009.
- Waikar SS, Betensky RA, Bonventre JV. Creatinine as the gold standard for kidney injury biomarker studies?. Nephrol Dial Transplant 2009;24:3263-5. https://doi.org/10.1093/ndt/gfp428.
- Moran SM, Myers BD. Course of acute renal failure studied by a model of creatinine kinetics. Kidney Int 1985;27:928-37. https://doi.org/10.1038/ki.1985.101.
- Sirota JC, Klawitter J, Edelstein CL. Biomarkers of acute kidney injury. J Toxicol 2011;2011. https://doi.org/10.1155/2011/328120.
- Chen L-I, Guh J-Y, Wu K-D, Chen Y-M, Kuo M-C, Hwang S-J, et al. Modification of Diet in Renal Disease (MDRD) study and CKD Epidemiology Collaboration (CKD-EPI) equations for Taiwanese adults. PLOS ONE 2014;9. https://doi.org/10.1371/journal.pone.009964.
- Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976;16:31-4. https://doi.org/10.1159/000180580.
- Bicik Z, Bahcebasi T, Kulaksizoglu S, Yavuz O. The efficacy of cystatin C assay in the prediction of glomerular filtration rate. Is it a more reliable marker for renal failure?. Clin Chem Lab Med 2005;43:855-61. https://doi.org/10.1515/CCLM.2005.144.
- Lebkowska U, Malyszko J, Lebkowska A, Koc-Zorawska E, Lebkowski W, Malyszko JS, et al. Neutrophil gelatinase-associated lipocalin and cystatin C could predict renal outcome in patients undergoing kidney allograft transplantation: a prospective study. Transplant Proc 2009;41:154-7. https://doi.org/10.1016/j.transproceed.2008.10.092.
- Hall IE, Doshi MD, Poggio ED, Parikh CR. A comparison of alternative serum biomarkers with creatinine for predicting allograft function after kidney transplantation. Transplantation 2011;91:48-56. https://doi.org/10.1097/TP.0b013e3181fc4b3a.
- Fonseca I, Oliveira JC, Almeida M, Cruz M, Malho A, Martins LS, et al. Neutrophil gelatinase-associated lipocalin in kidney transplantation is an early marker of graft dysfunction and is associated with one-year renal function. J Transplant 2013;2013. https://doi.org/10.1155/2013/650123.
- Fonseca I, Reguengo H, Almeida M, Dias L, Martins L, Pedroso S, et al. Oxidative stress in kidney transplantation: malondialdehyde is an early predictive marker of graft dysfunction. Transplantation 2014;97:1058-65. https://doi.org/10.1097/01.TP.0000438626.91095.50.
- Fonseca I, Reguengo H, Oliveira JC, Martins S, Malheiro J, Almeida M, et al. A triple-biomarker approach for the detection of delayed graft function after kidney transplantation using serum creatinine, cystatin C, and malondialdehyde. Clin Biochem 2015;48:1033-8. https://doi.org/10.1016/j.clinbiochem.2015.07.007.
- Bataille A, Abbas S, Semoun O, Bourgeois E, Marie O, Bonnet F, et al. Plasma neutrophil gelatinase-associated lipocalin in kidney transplantation and early renal function prediction. Transplantation 2011;92:1024-30. https://doi.org/10.1097/TP.0b013e318230c079.
- Mahdavi-Mazdeh M, Amerian M, Abdollahi A, Hatmi ZN, Khatami MR. Comparison of serum neutrophil gelatinase-associated lipocalin (NGAL) with serum creatinine in prediction of kidney recovery after renal transplantation. Int J Organ Transplant Med 2012;3:176-82.
- Lee EY, Kim MS, Park Y, Kim HS. Serum neutrophil gelatinase-associated lipocalin and interleukin-18 as predictive biomarkers for delayed graft function after kidney transplantation. J Clin Lab Anal 2012;26:295-301. https://doi.org/10.1002/jcla.21520.
- Kusaka M, Iwamatsu F, Kuroyanagi Y, Nakaya M, Ichino M, Marubashi S, et al. Serum neutrophil gelatinase associated lipocalin during the early postoperative period predicts the recovery of graft function after kidney transplantation from donors after cardiac death. J Urol 2012;187:2261-7. https://doi.org/10.1016/j.juro.2012.01.033.
- Hollmen ME, Kyllonen LE, Merenmies J, Salmela KT. Serum neutrophil gelatinase-associated lipocalin and recovery of kidney graft function after transplantation. BMC Nephrol 2014;15. https://doi.org/10.1186/1471-2369-15-123.
- Buemi A, Musuamba F, Frederic S, Douhet A, De Meyer M, De Pauw L, et al. Is plasma and urine neutrophil gelatinase-associated lipocalin (NGAL) determination in donors and recipients predictive of renal function after kidney transplantation?. Clin Biochem 2014;47:68-72.
- Hollmen ME, Kyllonen LE, Inkinen KA, Lalla ML, Merenmies J, Salmela KT. Deceased donor neutrophil gelatinase-associated lipocalin and delayed graft function after kidney transplantation: a prospective study. Crit Care 2011;15. https://doi.org/10.1186/cc10220.
- Muller L, Nicolas-Robin A, Bastide S, Martinez O, Louart G, Colavolpe JC, et al. Assessment of neutrophil gelatinase-associated lipocalin in the brain-dead organ donor to predict immediate graft function in kidney recipients: a prospective, multicenter study. Anesthesiology 2015;122:96-105. https://doi.org/10.1097/ALN.0000000000000497.
- Welberry Smith MP, Zougman A, Cairns DA, Wilson M, Wind T, Wood SL, et al. Serum aminoacylase-1 is a novel biomarker with potential prognostic utility for long-term outcome in patients with delayed graft function following renal transplantation. Kidney Int 2013;84:1214-25. https://doi.org/10.1038/ki.2013.200.
- Blogowski W, Dolegowska B, Salata D, Budkowska M, Domanski L, Starzynska T. Clinical analysis of perioperative complement activity during ischemia/reperfusion injury following renal transplantation. Clin J Am Soc Nephrol 2012;7:1843-51. https://doi.org/10.2215/CJN.02200312.
- Steubl D, Hettwer S, Vrijbloed W, Dahinden P, Wolf P, Luppa P, et al. C-terminal agrin fragment – a new fast biomarker for kidney function in renal transplant recipients. Am J Nephrol 2013;38:501-8. https://doi.org/10.1159/000356969.
- Chapal M, Néel M, Le Borgne F, Meffray E, Carceles O, Hourmant M, et al. Increased soluble Flt-1 correlates with delayed graft function and early loss of peritubular capillaries in the kidney graft. Transplantation 2013;96:739-44. https://doi.org/10.1097/TP.0b013e31829f4772.
- Morales JM, Martinez-Flores JA, Serrano M, Castro MJ, Alfaro FJ, García F, et al. Association of early kidney allograft failure with preformed IgA antibodies to β2-glycoprotein I. J Am Soc Nephrol 2015;26:735-45. https://doi.org/10.1681/ASN.2014030228.
- Lauzurica R, Pastor MC, Bayés B, Hernandez JM, Bonet J, Doladé M, et al. Pretransplant inflammation: a risk factor for delayed graft function?. J Nephrol 2008;21:221-8.
- Alachkar N, Ugarte R, Huang E, Womer KL, Montgomery R, Kraus E, et al. Stem cell factor, interleukin-16, and interleukin-2 receptor alpha are predictive biomarkers for delayed and slow graft function. Transplant Proc 2010;42:3399-405. https://doi.org/10.1016/j.transproceed.2010.06.013.
- Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, Dinarello CA, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest 2001;107:1145-52. https://doi.org/10.1172/JCI12089.
- Fonseca I, Oliveira JC, Santos J, Malheiro J, Martins LS, Almeida M, et al. Leptin and adiponectin during the first week after kidney transplantation: biomarkers of graft dysfunction?. Metabolism 2015;64:202-7. https://doi.org/10.1016/j.metabol.2014.10.003.
- Oltean S, Pullerits R, Flodén A, Olausson M, Oltean M. Increased resistin in brain dead organ donors is associated with delayed graft function after kidney transplantation. J Transl Med 2013;11. https://doi.org/10.1186/1479-5876-11-233.
- Thorne-Tjomsland G, Hosfield T, Jamieson JC, Liu B, Nickerson P, Gough JC, et al. Increased levels of GALbeta1-4GLCNACalpha2-6 sialyltransferase pretransplant predict delayed graft function in kidney transplant recipients. Transplantation 2000;69:806-8. https://doi.org/10.1097/00007890-200003150-00022.
- Dołegowska B, Błogowski W, Domański L. Is it possible to predict the early post-transplant allograft function using 20-HETE measurements? A preliminary report. Transpl Int 2009;22:546-53. https://doi.org/10.1111/j.1432–2277.2008.00829.x.
- Dolegowska B, Blogowski W, Safranow K, Domanski L, Jakubowska K, Olszewska M. Lipoxygenase-derived hydroxyeicosatetraenoic acids – novel perioperative markers of early post-transplant allograft function?. Nephrol Dial Transplant 2010;25:4061-7. https://doi.org/10.1093/ndt/gfq320.
- Halazun KJ, Marangoni G, Hakeem A, Fraser SM, Farid SG, Ahmad N. Elevated preoperative recipient neutrophil–lymphocyte ratio is associated with delayed graft function following kidney transplantation. Transplant Proc 2013;45:3254-7. https://doi.org/10.1016/j.transproceed.2013.07.065.
- Nguyen MTJP, Fryml E, Sahakian SK, Liu SQ, Michel RP, Lipman ML, et al. Pretransplantation recipient regulatory T cell suppressive function predicts delayed and slow graft function after kidney transplantation. Transplantation 2014;98:745-53. https://doi.org/10.1097/Tp.0000000000000219.
- Nguyen MJ, Fryml E, Sahakian SK, Liu S, Cantarovich M, Lipman M, et al. Pretransplant recipient circulating CD4+CD127lo/– tumor necrosis factor receptor 2+ regulatory T cells: a surrogate of regulatory T cell-suppressive function and predictor of delayed and slow graft function after kidney transplantation. Transplantation 2016;100:314-24. https://doi.org/10.1097/TP.0000000000000942.
- Pianta TJ, Peake PW, Pickering JW, Kelleher M, Buckley NA, Endre ZH. Evaluation of biomarkers of cell cycle arrest and inflammation in prediction of dialysis or recovery after kidney transplantation. Transpl Int 2015;28:1392-404. https://doi.org/10.1111/tri.12636.
- Prassas I, Brinc D, Farkona S, Leung F, Dimitromanolakis A, Chrystoja CC, et al. False biomarker discovery due to reactivity of a commercial ELISA for CUZD1 with cancer antigen CA125. Clin Chem 2014;60:381-8. https://doi.org/10.1373/clinchem.2013.215236.
- Gutiérrez OM, Sun CC, Chen W, Babitt JL, Lin HY. Statement of concern about a commercial assay used to measure soluble hemojuvelin in humans. Am J Nephrol 2012;36:332-3. https://doi.org/10.1159/000342519.
- Berglund L, Björling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics 2008;7:2019-27. https://doi.org/10.1074/mcp.R800013-MCP200.
- Git A. Research tools: a recipe for disaster. Nature 2012;484:439-40. https://doi.org/10.1038/484439a.
- Rehfeld JF, Bardram L, Hilsted L, Poitras P, Goetze JP. Pitfalls in diagnostic gastrin measurements. Clin Chem 2012;58:831-6. https://doi.org/10.1373/clinchem.2011.179929.
- Clemmons DR. Consensus statement on the standardization and evaluation of growth hormone and insulin-like growth factor assays. Clin Chem 2011;57:555-9. https://doi.org/10.1373/clinchem.2010.150631.
- Myers GL, Miller WG, Coresh J, Fleming J, Greenberg N, Greene T, et al. Recommendations for improving serum creatinine measurement: a report from the laboratory working group of the National Kidney Disease Education Program. Clin Chem 2006;52:5-18. https://doi.org/10.1373/clinchem.2005.052514.
- Clinical Laboratory Standards Institute . Global Laboratory Standards for a Healthier World n.d. www.clsi.org (accessed 20 February 2018).
- Chesher D. Evaluating assay precision. Clin Biochem Rev 2008;29:23-6.
- Evaluation of Precision of Quantitative Measurement Procedures; Approved Guideline – Third Edition. Wayne, PA: CLSI; 2014.
- Evaluation of the Linearity of Quantitative Measurement Procedures: A Statistical Approach; Approved Guideline. Wayne, PA: CLSI; 2003.
- Interference Testing in Clinical Chemistry; Approved Guideline – Second Edition. Wayne, PA: CLSI; 2005.
- Measurement Procedure Comparison and Bias Estimation Using Patient Samples; Approved Guideline – Third Edition. Wayne, PA: CLSI; 2013.
- Preliminary Evaluation of Quantitative Clinical Laboratory Measurement Procedures; Approved Guideline – Third Edition. Wayne, PA: CLSI; 2014.
- User Protocol for Evaluation of Qualitative Test Performance; Approved Guideline – Second Edition. Wayne, PA: CLSI; 2008.
- Evaluation of Commutability of Processed Samples; Approved Guideline – Third Edition. Wayne, PA: CLSI; 2014.
- User Verification of Precision and Estimation of Bias; Approved Guideline – Third Edition. Wayne, PA: CLSI; 2014.
- Evaluation of Detection Capability for Clinical Laboratory Measurement Procedures; Approved Guideline – Second Edition. Wayne, PA: CLSI; 2012.
- Risk Management Techniques to Identify and Control Laboratory Error Sources; Approved Guideline – Second Edition. Wayne, PA: CLSI; 2009.
- Laboratory Quality Control Based on Risk Management; Workbook. Wayne, PA: CLSI; n.d.
- A Framework for Using CLSI Documents to Evaluate Clinical Laboratory Measurement Procedures, 2nd edition. Wayne, PA: CLSI; 2015.
- Estimation of Total Analytical Error for Clinical Laboratory Methods; Approved Guideline. Wayne, PA: CLSI; 2002.
- Laboratory Quality Control Based on Risk Management; Approved Guideline. Wayne, PA: CLSI; 2011.
- Laboratory Quality Control Based on Risk Management; Workbook. Wayne, PA: CLSI; n.d.
- Laboratory Quality Control Based on Risk Management; Worksheet Template. Wayne, PA: CLSI; n.d.
- Assessment of the Diagnostic Accuracy of Laboratory Tests Using Receiver Operating Characteristic Curves; Approved Guideline – Second Edition. Wayne, PA: CLSI; 2011.
- Evaluation of Stability of In Vitro Diagnostic Reagents; Approved Guideline. Wayne, PA: CLSI; 2009.
- User Evaluation of Between-Reagent Lot Variation; Approved Guideline. Wayne, PA: CLSI; 2013.
- How to Construct and Interpret an Error Grid for Quantitative Diagnostic Assays; Approved Guideline. Wayne, PA: CLSI; 2012.
- Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory; Approved Guideline – Third Edition. Wayne, PA: CLSI; 2010.
- Expression of Measurement Uncertainty in Laboratory Medicine; Approved Guideline. Wayne, PA: CLSI; 2012.
- Characterization and Qualification of Commutable Reference Materials for Laboratory Medicine; Approved Guideline. Wayne, PA: Clinical and Laboratory Standards Institute; 2010.
- Verification of Comparability of Patient Results Within One Health Care System; Approved Guideline (interim revision). Wayne, PA: Clinical and Laboratory Standards Institute; 2012.
- Metrological Traceability and Its Implementation; A Report. Wayne, PA: CLSI; 2006.
- Harmonization of Symbology and Equations. Wayne, PA: CLSI; 2015.
- Topic E, Nikolac N, Panteghini M, Theodorsson E, Salvagno GL, Miler M, et al. How to assess the quality of your analytical method?. Clin Chem Lab Med 2015;53:1707-18. https://doi.org/10.1515/cclm-2015-0869.
- Da Rin G. Pre-analytical workstations: a tool for reducing laboratory errors. Clin Chim Acta 2009;404:68-74. https://doi.org/10.1016/j.cca.2009.03.024.
- Plebani M, Sciacovelli L, Aita A, Padoan A, Chiozza ML. Quality indicators to detect pre-analytical errors in laboratory testing. Clin Chim Acta 2014;432:44-8. https://doi.org/10.1016/j.cca.2013.07.033.
- Green SF. The cost of poor blood specimen quality and errors in preanalytical processes. Clin Biochem 2013;46:1175-9. https://doi.org/10.1016/j.clinbiochem.2013.06.001.
- Cornes MP, Atherton J, Pourmahram G, Borthwick H, Kyle B, West J, et al. Monitoring and reporting of preanalytical errors in laboratory medicine: the UK situation. Ann Clin Biochem 2015;53:279-84. https://doi.org/10.1177/0004563215599561.
- Guder WG. History of the preanalytical phase: a personal view. Biochem Med 2014;24:25-30. https://doi.org/10.11613/BM.2014.005.
- Lippi G, Banfi G, Church S, Cornes M, De Carli G, Grankvist K, et al. Preanalytical quality improvement. In pursuit of harmony, on behalf of European Federation for Clinical Chemistry and Laboratory Medicine (EFLM) Working group for Preanalytical Phase (WG-PRE). Clin Chem Lab Med 2015;53:357-70. https://doi.org/10.1515/cclm-2014-1051.
- Plebani M. Exploring the iceberg of errors in laboratory medicine. Clin Chim Acta 2009;404:16-23. https://doi.org/10.1016/j.cca.2009.03.022.
- Plebani M, Sciacovelli L, Marinova M, Marcuccitti J, Chiozza ML. Quality indicators in laboratory medicine: a fundamental tool for quality and patient safety. Clin Biochem 2013;46:1170-4. https://doi.org/10.1016/j.clinbiochem.2012.11.028.
- Simundic AM, Church S, Cornes MP, Grankvist K, Lippi G, Nybo M, et al. Compliance of blood sampling procedures with the CLSI H3-A6 guidelines: an observational study by the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) working group for the preanalytical phase (WG-PRE). Clin Chem Lab Med 2015;53:1321-31. https://doi.org/10.1515/cclm-2014-1053.
- Lehmann S, Guadagni F, Moore H, Ashton G, Barnes M, Benson E, et al. Standard preanalytical coding for biospecimens: review and implementation of the Sample PREanalytical Code (SPREC). Biopreserv Biobank 2012;10:366-74. https://doi.org/10.1089/bio.2012.0012.
- Betsou F, Gunter E, Clements J, DeSouza Y, Goddard KA, Guadagni F, et al. Identification of evidence-based biospecimen quality-control tools: a report of the International Society for Biological and Environmental Repositories (ISBER) Biospecimen Science Working Group. J Mol Diagn 2013;15:3-16. https://doi.org/10.1016/j.jmoldx.2012.06.008.
- Simeon-Dubach D, Burt AD, Hall PA. Quality really matters: the need to improve specimen quality in biomedical research. J Pathol 2012;228:431-3. https://doi.org/10.1002/path.4117.
- Riondino S, Ferroni P, Spila A, Alessandroni J, D’Alessandro R, Formica V, et al. Ensuring sample quality for biomarker discovery studies – use of ICT tools to trace biosample life-cycle. Cancer Genomics Proteomics 2015;12:291-9.
- Tubes and Additives for Venous and Capillary Blood Specimen Collection; Approved Standard – Sixth Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2010.
- Accuracy in Patient and Sample Identification; Approved Guideline. Wayne, PA: Clinical and Laboratory Standards Institute; 2010.
- Procedures for the Handling and Processing of Blood Specimens by Venipuncture; Approved Standard – Sixth Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2007.
- Bowen RA, Remaley AT. Interferences from blood collection tube components on clinical chemistry assays. Biochem Med 2014;24:31-44. https://doi.org/10.11613/BM.2014.006.
- Bowen RA, Hortin GL, Csako G, Otanez OH, Remaley AT. Impact of blood collection devices on clinical chemistry assays. Clin Biochem 2010;43:4-25. https://doi.org/10.1016/j.clinbiochem.2009.10.001.
- Lima-Oliveira G, Lippi G, Salvagno GL, Montagnana M, Picheth G, Guidi GC. Preanalytical management: serum vacuum tubes validation for routine clinical chemistry. Biochem Med (Zagreb) 2012;22:180-6.
- Freedman LP, Cockburn IM, Simcoe TS. The economics of reproducibility in preclinical research. PLOS Biol 2015;13. https://doi.org/10.1371/journal.pbio.1002165.
- Bartlett WA, Braga F, Carobene A, Coşkun A, Prusa R, Fernandez-Calle P, et al. A checklist for critical appraisal of studies of biological variation. Clin Chem Lab Med 2015;53:879-85. https://doi.org/10.1515/cclm-2014–1127.
- Ricós C, Álvarez V, Perich C, Fernández-Calle P, Minchinela J, Cava F, et al. Rationale for using data on biological variation. Clin Chem Lab Med 2015;53:863-70. https://doi.org/10.1515/cclm-2014–1142.
- Westgard QC. Quality Requirements. Desirable Biological Variation Database Specifications n.d. www.westgard.com/biodatabase1.htm (accessed 21 February 2018).
- Perich C, Minchinela J, Ricós C, Fernández-Calle P, Alvarez V, Doménech MV, et al. Biological variation database: structure and criteria used for generation and update. Clin Chem Lab Med 2015;53:299-305. https://doi.org/10.1515/cclm-2014-0739.
- Westguard QC. Guest Essay. Biologic Variation and Desirable Specifications for QC n.d. www.westgard.com/guest17.htm (accessed 21 February 2018).
- Westguard QC. Guest Essay. Biological Variation Data for Setting Quality Specifications n.d. www.westgard.com/guest12.htm (accessed 21 February 2018).
- Carobene A. Reliability of biological variation data available in an online database: need for improvement. Clin Chem Lab Med 2015;53:871-7. https://doi.org/10.1515/cclm-2014-1133.
- Thue G, Sandberg S. Analytical performance specifications based on how clinicians use laboratory tests. Experiences from a post-analytical external quality assessment programme. Clin Chem Lab Med 2015;53:857-62. https://doi.org/10.1515/cclm-2014-1280.
- Siest G, Henny J, Gräsbeck R, Wilding P, Petitclerc C, Queraltó JM, et al. The theory of reference values: an unfinished symphony. Clin Chem Lab Med 2013;51:47-64. https://doi.org/10.1515/cclm-2012-0682.
- Kinders R, Ferry-Galow K, Wang L, Srivastava AK, Ji JJ, Parchment RE. Implementation of validated pharmacodynamic assays in multiple laboratories: challenges, successes, and limitations. Clin Cancer Res 2014;20:2578-86. https://doi.org/10.1158/1078-0432.CCR-14-0476.
- Armbruster DA, Pry T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev 2008;29:49-52.
- Bellahcene A, Castronovo V, Ogbureke KU, Fisher LW, Fedarko NS. Small integrin-binding ligand N-linked glycoproteins (SIBLINGs): multifunctional proteins in cancer. Nat Rev Cancer 2008;8:212-26. https://doi.org/10.1038/nrc2345.
- Anborgh PH, Mutrie JC, Tuck AB, Chambers AF. Role of the metastasis-promoting protein osteopontin in the tumour microenvironment. J Cell Mol Med 2010;14:2037-44. https://doi.org/10.1111/j.1582–4934.2010.01115.x.
- Kahles F, Findeisen HM, Bruemmer D. Osteopontin: a novel regulator at the cross roads of inflammation, obesity and diabetes. Mol Metab 2014;3:384-93. https://doi.org/10.1016/j.molmet.2014.03.004.
- Filia A, Elliott F, Wind T, Field S, Davies J, Kukalizch K, et al. Plasma osteopontin concentrations in patients with cutaneous melanoma. Oncol Rep 2013;30:1575-80. https://doi.org/10.3892/or.2013.2666.
- Lanteri P, Lombardi G, Colombini A, Grasso D, Banfi G. Stability of osteopontin in plasma and serum. Clin Chem Lab Med 2012;50:1979-84. https://doi.org/10.1515/cclm-2012-0177.
- Kon S, Maeda M, Segawa T, Hagiwara Y, Horikoshi Y, Chikuma S, et al. Antibodies to different peptides in osteopontin reveal complexities in the various secreted forms. J Cell Biochem 2000;77:487-98. https://doi.org/10.1002/(SICI)1097-4644(20000601)77:3<487::AID-JCB13>3.0.CO;2-8.
- Fedarko NS, Fohr B, Robey PG, Young MF, Fisher LW. Factor H binding to bone sialoprotein and osteopontin enables tumor cell evasion of complement-mediated attack. J Biol Chem 2000;275:16666-72. https://doi.org/10.1074/jbc.M001123200.
- Fedarko NS, Jain A, Karadag A, Van Eman MR, Fisher LW. Elevated serum bone sialoprotein and osteopontin in colon, breast, prostate, and lung cancer. Clin Cancer Res 2001;7:4060-6.
- Hermann N, Dressen K, Schildberg FA, Jakobs C, Holdenrieder S. Methodical and pre-analytical characteristics of a multiplex cancer biomarker immunoassay. World J Methodol 2014;4:219-31. https://doi.org/10.5662/wjm.v4.i4.219.
- Sennels HP, Jacobsen S, Jensen T, Hansen MS, Ostergaard M, Nielsen HJ, et al. Biological variation and reference intervals for circulating osteopontin, osteoprotegerin, total soluble receptor activator of nuclear factor kappa B ligand and high-sensitivity C-reactive protein. Scand J Clin Lab Invest 2007;67:821-35. https://doi.org/10.1080/00365510701432509.
- Cristaudo A, Foddis R, Bonotti A, Simonini S, Vivaldi A, Guglielmi G, et al. Comparison between plasma and serum osteopontin levels: usefulness in diagnosis of epithelial malignant pleural mesothelioma. Int J Biol Markers 2010;25:164-70.
- Vordermark D, Said HM, Katzer A, Kuhnt T, Hansgen G, Dunst J, et al. Plasma osteopontin levels in patients with head and neck cancer and cervix cancer are critically dependent on the choice of ELISA system. BMC Cancer 2006;6. https://doi.org/Artn 20710.1186/1471-2407-6-207.
- Isa S, Kawaguchi T, Teramukai S, Minato K, Ohsaki Y, Shibata K, et al. Serum osteopontin levels are highly prognostic for survival in advanced non-small cell lung cancer: results from JMTO LC 0004. J Thorac Oncol 2009;4:1104-10. https://doi.org/10.1097/JTO.0b013e3181ae2844.
- Mack PC, Redman MW, Chansky K, Williamson SK, Farneth NC, Lara PN, et al. Lower osteopontin plasma levels are associated with superior outcomes in advanced non-small-cell lung cancer patients receiving platinum-based chemotherapy: SWOG Study S0003. J Clin Oncol 2008;26:4771-6. https://doi.org/10.1200/JCO.2008.17.0662.
- Creaney J, Yeoman D, Musk AW, de Klerk N, Skates SJ, Robinson BW. Plasma versus serum levels of osteopontin and mesothelin in patients with malignant mesothelioma – which is best?. Lung Cancer 2011;74:55-60. https://doi.org/10.1016/j.lungcan.2011.02.007.
- Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A, et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799-809. https://doi.org/10.1074/jbc.M800938200.
- Barathova M, Takacova M, Holotnakova T, Gibadulinova A, Ohradanova A, Zatovicova M, et al. Alternative splicing variant of the hypoxia marker carbonic anhydrase IX expressed independently of hypoxia and tumour phenotype. Br J Cancer 2008;98:129-36. https://doi.org/10.1038/sj.bjc.6604111.
- Dorai T, Sawczuk IS, Pastorek J, Wiernik PH, Dutcher JP. The role of carbonic anhydrase IX overexpression in kidney cancer. Eur J Cancer 2005;41:2935-47. https://doi.org/10.1016/j.ejca.2005.09.011.
- Zatovicova M, Sedlakova O, Svastova E, Ohradanova A, Ciampor F, Arribas J, et al. Ectodomain shedding of the hypoxia-induced carbonic anhydrase IX is a metalloprotease-dependent process regulated by TACE/ADAM17. Br J Cancer 2005;93:1267-76. https://doi.org/10.1038/sj.bjc.6602861.
- Pastorekova S, Ratcliffe PJ, Pastorek J. Molecular mechanisms of carbonic anhydrase IX-mediated pH regulation under hypoxia. BJU Int 2008;101:8-15. https://doi.org/10.1111/j.1464-410X.2008.07642.x.
- Pastorek J, Pastorekova S, Callebaut I, Mornon JP, Zelnik V, Opavsky R, et al. Cloning and characterization of Mn, a human tumor-associated protein with a domain homologous to carbonic-anhydrase and a putative helix-loop-helix DNA-binding segment. Oncogene 1994;9:2877-88.
- Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 2000;60:7075-83.
- Divgi CR, Pandit-Taskar N, Jungbluth AA, Reuter VE, Gonen M, Ruan S, et al. Preoperative characterisation of clear-cell renal carcinoma using iodine-124-labelled antibody chimeric G250 (I-124-cG250) and PET in patients with renal masses: a phase I trial. Lancet Oncol 2007;8:304-10. https://doi.org/10.1016/S1470-2045(07)70044-X.
- Mahon BP, Pinard MA, McKenna R. Targeting carbonic anhydrase IX activity and expression. Molecules 2015;20:2323-48. https://doi.org/10.3390/molecules20022323.
- Zhou GX, Ireland J, Rayman P, Finke J, Zhou M. Quantification of carbonic anhydrase IX expression in serum and tissue of renal cell carcinoma patients using enzyme-linked immunosorbent assay: prognostic and diagnostic potentials. Urology 2010;75:257-61. https://doi.org/10.1016/j.urology.2009.09.052.
- Ilie M, Mazure NM, Hofman V, Ammadi RE, Ortholan C, Bonnetaud C, et al. High levels of carbonic anhydrase IX in tumour tissue and plasma are biomarkers of poor prognostic in patients with non-small cell lung cancer. Br J Cancer 2010;102:1627-35. https://doi.org/10.1038/sj.bjc.6605690.
- Hulick P, Zimmer M, Margulis V, Skates S, Hamel M, Dahl DM, et al. Blood levels of carbonic-anhydrase 9 correlate with clear cell renal cell carcinoma activity. Clin Proteomics 2009;5:37-45. https://doi.org/10.1007/s12014-008-9012-1.
- Woelber L, Mueller V, Eulenburg C, Schwarz J, Carney W, Jaenicke F, et al. Serum carbonic anhydrase IX during first-line therapy of ovarian cancer. Gynecol Oncol 2010;117:183-8. https://doi.org/10.1016/j.ygyno.2009.11.029.
- Hyrsl L, Zavada J, Zavadova Z, Kawaciuk I, Vesely S, Skapa P. Soluble form of carbonic anhydrase IX (CAIX) in transitional cell carcinoma of urinary tract. Neoplasma 2009;56:298-302. https://doi.org/10.4149/neo_2009_04_29.
- Pena C, Lathia C, Shan M, Escudier B, Bukowski RM. Biomarkers predicting outcome in patients with advanced renal cell carcinoma: results from sorafenib phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. Clin Cancer Res 2010;16:4853-63. https://doi.org/10.1158/1078-0432.CCR-09-3343.
- Wind TC, Messenger MP, Thompson D, Selby PJ, Banks RE. Measuring carbonic anhydrase IX as a hypoxia biomarker: differences in concentrations in serum and plasma using a commercial enzyme-linked immunosorbent assay due to influences of metal ions. Ann Clin Biochem 2011;48:112-20. https://doi.org/10.1258/acb.2010.010240.
- Pastoreková S, Závadová Z, Kostál M, Babusíková O, Závada J. A novel quasi-viral agent, MaTu, is a two-component system. Virology 1992;187:620-6. https://doi.org/10.1016/0042-6822(92)90464-Z.
- Rosser CJ, Ross S, Chang M, Dai Y, Mengual L, Zhang G, et al. Multiplex protein signature for the detection of bladder cancer in voided urine samples. J Urol 2013;190:2257-62. https://doi.org/10.1016/j.juro.2013.06.011.
- Chen LM, Chang M, Dai Y, Chai KX, Dyrskjot L, Sanchez-Carbayo M, et al. External validation of a multiplex urinary protein panel for the detection of bladder cancer in a multicenter cohort. Cancer Epidemiol Biomarkers Prev 2014;23:1804-12. https://doi.org/10.1158/1055-9965.EPI-14-0029.
- Rosser CJ, Chang M, Dai Y, Ross S, Mengual L, Alcaraz A, et al. Urinary protein biomarker panel for the detection of recurrent bladder cancer. Cancer Epidemiol Biomarkers Prev 2014;23:1340-5. https://doi.org/10.1158/1055-9965.EPI-14-0035.
- Opavský R, Pastoreková S, Zelník V, Gibadulinová A, Stanbridge EJ, Závada J, et al. Human MN/CA9 gene, a novel member of the carbonic anhydrase family: structure and exon to protein domain relationships. Genomics 1996;33:480-7. https://doi.org/10.1006/geno.1996.0223.
- Zat’ovicova M, Tarabkova K, Svastova E, Gibadulinova A, Mucha V, Jakubickova L, et al. Monoclonal antibodies generated in carbonic anhydrase IX-deficient mice recognize different domains of tumour-associated hypoxia-induced carbonic anhydrase IX. J Immunol Methods 2003;282:117-34. https://doi.org/10.1016/j.jim.2003.08.011.
- Závada J, Závadová Z, Pastorek J, Biesová Z, Jezek J, Velek J. Human tumour-associated cell adhesion protein MN/CA IX: identification of M75 epitope and of the region mediating cell adhesion. Br J Cancer 2000;82:1808-13. https://doi.org/10.1054/bjoc.2000.1111.
- Raiko I, Sander I, Weber DG, Raulf-Heimsoth M, Gillissen A, Kollmeier J, et al. Development of an enzyme-linked immunosorbent assay for the detection of human calretinin in plasma and serum of mesothelioma patients. BMC Cancer 2010;10. https://doi.org/10.1186/1471-2407-10-242.
- Larsen A, Bronstein IB, Dahl O, Wentzel-Larsen T, Kristoffersen EK, Fagerhol MK. Quantification of S100A12 (EN-RAGE) in blood varies with sampling method, calcium and heparin. Scand J Immunol 2007;65:192-201. https://doi.org/10.1111/j.1365-3083.2006.01875.x.
- Hill R. Verification and validation: whose responsibility, manufacturer or end user?. Ann Clin Biochem 2011;48:93-4. https://doi.org/10.1258/acb.2011.011008.
- Haase-Fielitz A, Haase M, Devarajan P. Neutrophil gelatinase-associated lipocalin as a biomarker of acute kidney injury: a critical evaluation of current status. Ann Clin Biochem 2014;51:335-51. https://doi.org/10.1177/0004563214521795.
- Ronco C, Legrand M, Goldstein SL, Hur M, Tran N, Howell EC, et al. Neutrophil gelatinase-associated lipocalin: ready for routine clinical use? An international perspective. Blood Purif 2014;37:271-85. https://doi.org/10.1159/000360689.
- Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem 1993;268:10425-32.
- Mishra J, Ma Q, Prada A, Mitsnefes M, Zahedi K, Yang J, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 2003;14:2534-43. https://doi.org/10.1097/01.ASN.0000088027.54400.C6.
- Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 2011;17:216-22. https://doi.org/10.1038/nm.2290.
- Mårtensson J, Bellomo R. The rise and fall of NGAL in acute kidney injury. Blood Purif 2014;37:304-10. https://doi.org/10.1159/000364937.
- Cai L, Rubin J, Han W, Venge P, Xu S. The origin of multiple molecular forms in urine of HNL/NGAL. Clin J Am Soc Nephrol 2010;5:2229-35. https://doi.org/10.2215/CJN.00980110.
- Nickolas TL, Forster CS, Sise ME, Barasch N, Solá-Del Valle D, Viltard M, et al. NGAL (Lcn2) monomer is associated with tubulointerstitial damage in chronic kidney disease. Kidney Int 2012;82:718-22. https://doi.org/10.1038/ki.2012.195.
- Mårtensson J, Xu S, Bell M, Martling CR, Venge P. Immunoassays distinguishing between HNL/NGAL released in urine from kidney epithelial cells and neutrophils. Clin Chim Acta 2012;413:1661-7. https://doi.org/10.1016/j.cca.2012.05.010.
- Siew ED, Ware LB, Ikizler TA. Biological markers of acute kidney injury. J Am Soc Nephrol 2011;22:810-20. https://doi.org/10.1681/ASN.2010080796.
- Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A. NGAL Meta-analysis Investigator Group . Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 2009;54:1012-24. https://doi.org/10.1053/j.ajkd.2009.07.020.
- Devarajan P. Review: neutrophil gelatinase-associated lipocalin: a troponin-like biomarker for human acute kidney injury. Nephrology (Carlton) 2010;15:419-28. https://doi.org/10.1111/j.1440-1797.2010.01317.x.
- Nickolas TL, Schmidt-Ott KM, Canetta P, Forster C, Singer E, Sise M, et al. Diagnostic and prognostic stratification in the emergency department using urinary biomarkers of nephron damage: a multicenter prospective cohort study. J Am Coll Cardiol 2012;59:246-55. https://doi.org/10.1016/j.jacc.2011.10.854.
- Hollmen ME, Kyllonen LE, Inkinen KA, Lalla MLT, Merenmies J, Salmela KT. Deceased donor neutrophil gelatinase-associated lipocalin and delayed graft function after kidney transplantation: a prospective study. Crit Care 2011;15. https://doi.org/ARTN R12110.1186/cc10220.
- Hollmen ME, Kyllonen LE, Inkinen KA, Lalla ML, Salmela KT. Urine neutrophil gelatinase-associated lipocalin is a marker of graft recovery after kidney transplantation. Kidney Int 2011;79:89-98. https://doi.org/10.1038/ki.2010.351.
- Grenier FC, Ali S, Syed H, Workman R, Martens F, Liao M, et al. Evaluation of the ARCHITECT urine NGAL assay: assay performance, specimen handling requirements and biological variability. Clin Biochem 2010;43:615-20. https://doi.org/10.1016/j.clinbiochem.2009.12.008.
- Lippi G, Aloe R, Storelli A, Cervellin G, Trenti T. Evaluation of NGAL Test (TM), a fully-automated neutrophil gelatinase-associated lipocalin (NGAL) immunoassay on Beckman Coulter AU 5822. Clin Chem Lab Med 2012;50:1581-4. https://doi.org/10.1515/Cclm.2011.839.
- Pedersen KR, Ravn HB, Hjortdal VE, Norregaard R, Povlsen JV. Neutrophil gelatinase-associated lipocalin (NGAL): validation of commercially available ELISA. Scand J Clin Lab Invest 2010;70:374-82. https://doi.org/10.3109/00365513.2010.486868.
- Cavalier E, Bekaert AC, Carlisi A, Legrand D, Krzesinski JM, Delanaye P. Neutrophil gelatinase-associated lipocalin (NGAL) determined in urine with the Abbott Architect or in plasma with the Biosite Triage? The laboratory’s point of view. Clin Chem Lab Med 2011;49:339-41. https://doi.org/10.1515/Cclm.2011.044.
- Kift RL, Messenger MP, Wind TC, Hepburn S, Wilson M, Thompson D, et al. A comparison of the analytical performance of five commercially available assays for neutrophil gelatinase-associated lipocalin using urine. Ann Clin Biochem 2013;50:236-44. https://doi.org/10.1258/acb.2012.012117.
- Legrand M, Collet C, Gayat E, Henao J, Giraudeaux V, Mateo J, et al. Accuracy of urine NGAL commercial assays in critically ill patients. Intensive Care Med 2013;39:541-2. https://doi.org/10.1007/s00134-012-2788-5.
- Cai LJ, Borowiec J, Xu SY, Han WY, Venge P. Assays of urine levels of HNL/NGAL in patients undergoing cardiac surgery and the impact of antibody configuration on their clinical performances. Clin Chim Acta 2009;403:121-5. https://doi.org/10.1016/j.cca.2009.01.030.
- Zhao C, Ozaeta P, Fishpaugh J, Rupprecht K, Workman R, Grenier F, et al. Structural characterization of glycoprotein NGAL, an early predictive biomarker for acute kidney injury. Carbohydr Res 2010;345:2252-61. https://doi.org/10.1016/j.carres.2010.07.024.
- Kjeldsen L, Koch C, Arnljots K, Borregaard N. Characterization of two ELISAs for NGAL, a newly described lipocalin in human neutrophils. J Immunol Methods 1996;198:155-64. https://doi.org/10.1016/S0022-1759(96)00153-6.
- Lindberg S, Jensen JS, Mogelvang R, Pedersen SH, Galatius S, Flyvbjerg A, et al. Plasma neutrophil gelatinase-associated lipocalinin in the general population: association with inflammation and prognosis. Artioscler Thromb Vasc Biol 2014;34:2135-42. https://doi.org/10.1161/ATVBAHA.114.303950.
- Rau S, Habicht A, Kauke T, Hillmer A, Wessely M, Stangl M, et al. Neutrophil gelatinase-associated lipocalin and end-stage renal disease: it is not all about the kidneys!. Eur J Clin Invest 2013;43:816-20. https://doi.org/10.1111/eci.12110.
- Hansen YB, Damgaard A, Poulsen JH. Evaluation of NGAL TestTM on Cobas 6000. Scand J Clin Lab Invest 2014;74:20-6. https://doi.org/10.3109/00365513.2013.855943.
- Itenov TS, Bangert K, Christensen PH, Jensen JU, Bestle MH. PASS Study Group . Serum and plasma neutrophil gelatinase associated lipocalin (NGAL) levels are not equivalent in patients admitted to intensive care. J Clin Lab Anal 2014;28:163-7. https://doi.org/10.1002/jcla.21662.
- Thrailkill KM, Moreau CS, Cockrell GE, Jo CH, Bunn RC, Morales-Pozzo AE, et al. Disease and gender-specific dysregulation of NGAL and MMP-9 in type 1 diabetes mellitus. Endocrine 2010;37:336-43. https://doi.org/10.1007/s12020-010-9308-6.
- Cullen MR, Murray PT, Fitzgibbon MC. Establishment of a reference interval for urinary neutrophil gelatinase-associated lipocalin. Ann Clin Biochem 2012;49:190-3. https://doi.org/10.1258/acb.2011.011105.
- Tomonaga Y, Szucs T, Ambühl P, Nock S, Risch M, Risch L. Insights on urinary NGAL obtained in a primary care setting. Clin Chim Acta 2012;413:733-9. https://doi.org/10.1016/j.cca.2012.01.001.
- Pennemans V, Rigo JM, Faes C, Reynders C, Penders J, Swennen Q. Establishment of reference values for novel urinary biomarkers for renal damage in the healthy population: are age and gender an issue?. Clin Chem Lab Med 2013;51:1795-802. https://doi.org/10.1515/cclm-2013-0157.
- van de Vrie M, Deegens JK, van der Vlag J, Hilbrands LB. Effect of long-term storage of urine samples on measurement of kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL). Am J Kidney Dis 2014;63:573-6. https://doi.org/10.1053/j.ajkd.2013.10.010.
- Parikh CR, Butrymowicz I, Yu A, Chinchilli VM, Park M, Hsu CY, et al. Urine stability studies for novel biomarkers of acute kidney injury. Am J Kidney Dis 2014;63:567-72. https://doi.org/10.1053/j.ajkd.2013.09.013.
- Lippi G, Plebani M. False myths and legends in laboratory diagnostics. Clin Chem Lab Med 2013;51:2087-97. https://doi.org/10.1515/cclm-2013–0105.
- Vempati P, Popel AS, Mac Gabhann F. Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning. Cytokine Growth Factor Rev 2014;25:1-19. https://doi.org/10.1016/j.cytogfr.2013.11.002.
- Kut C, Mac Gabhann F, Popel AS. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br J Cancer 2007;97:978-85. https://doi.org/10.1038/sj.bjc.6603923.
- Posadas EM, Limvorasak S, Sharma S, Figlin RA. Targeting angiogenesis in renal cell carcinoma. Expert Opin Pharmacother 2013;14:2221-36. https://doi.org/10.1517/14656566.2013.832202.
- Maroto P, Rini B. Molecular biomarkers in advanced renal cell carcinoma. Clin Cancer Res 2014;20:2060-71. https://doi.org/10.1158/1078-0432.CCR-13-1351.
- Mohle R, Green D, Moore MA, Nachman RL, Rafii S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci U S A 1997;94:663-8. https://doi.org/10.1073/pnas.94.2.663.
- Banks RE, Forbes MA, Searles J, Pappin D, Canas B, Rahman D, et al. Evidence for the existence of a novel pregnancy-associated soluble variant of the vascular endothelial growth factor receptor, Flt-1. Mol Hum Reprod 1998;4:377-86. https://doi.org/10.1093/molehr/4.4.377.
- Hetland ML, Christensen IJ, Lottenburger T, Johansen JS, Svendsen MN, Horslev-Petersen K, et al. Circulating VEGF as a biological marker in patients with rheumatoid arthritis? Preanalytical and biological variability in healthy persons and in patients. Dis Markers 2008;24:1-10. https://doi.org/10.1155/2008/707864.
- Svendsen MN, Brunner N, Christensen IJ, Ytting H, Bentsen C, Lomholt AF, et al. Biological variations in plasma VEGF and VEGFR-1 may compromise their biomarker value in colorectal cancer. Scand J Clin Lab Invest 2010;70:503-11. https://doi.org/10.3109/00365513.2010.521254.
- Feng CC, Ding GX, Song NH, Li X, Wu Z, Jiang HW, et al. Paraneoplastic hormones: parathyroid hormone-related protein (PTHrP) and erythropoietin (EPO) are related to vascular endothelial growth factor (VEGF) expression in clear cell renal cell carcinoma. Tumour Biol 2013;34:3471-6. https://doi.org/10.1007/s13277-013-0924-7.
- Buergy D, Wenz F, Groden C, Brockmann MA. Tumor-platelet interaction in solid tumors. Int J Cancer 2012;130:2747-60. https://doi.org/10.1002/ijc.27441.
- Klement GL, Yip TT, Cassiola F, Kikuchi L, Cervi D, Podust V, et al. Platelets actively sequester angiogenesis regulators. Blood 2009;113:2835-42. https://doi.org/10.1182/blood-2008-06-159541.
- Dittadi R, Meo S, Fabris F, Gasparini G, Contri D, Medici M, et al. Validation of blood collection procedures for the determination of circulating vascular endothelial growth factor (VEGF) in different blood compartments. Int J Biol Markers 2001;16:87-96. https://doi.org/10.1177/172460080101600202.
- Banks RE, Stanley AJ, Cairns DA, Barrett JH, Clarke P, Thompson D, et al. Influences of blood sample processing on low-molecular-weight proteome identified by surface-enhanced laser desorption/ionization mass spectrometry. Clin Chem 2005;51:1637-49. https://doi.org/10.1373/clinchem.2005.051417.
- Niers TM, Richel DJ, Meijers JC, Schlingemann RO. Vascular endothelial growth factor in the circulation in cancer patients may not be a relevant biomarker. PLOS ONE 2011;6. https://www.ncbi.nlm.nih.gov/m/pubmed/21637343.
- Egidi MG, D’Alessandro A, Mandarello G, Zolla L. Troubleshooting in platelet storage temperature and new perspectives through proteomics. Blood Transfus 2010;8:s73-81. https://doi.org/10.2450/2010.012S.
- Kisand K, Kerna I, Kumm J, Jonsson H, Tamm A. Impact of cryopreservation on serum concentration of matrix metalloproteinases (MMP)-7, TIMP-1, vascular growth factors (VEGF) and VEGF-R2 in Biobank samples. Clin Chem Lab Med 2011;49:229-35. https://doi.org/10.1515/CCLM.2011.049.
- Guo GH, Dong J, Yuan XH, Dong ZN, Tian YP. Clinical evaluation of the levels of 12 cytokines in serum/plasma under various storage conditions using evidence biochip arrays. Mol Med Rep 2013;7:775-80. https://doi.org/10.3892/mmr.2013.1263.
- Peterson JE, Zurakowski D, Italiano JE, Michel LV, Fox L, Klement GL, et al. Normal ranges of angiogenesis regulatory proteins in human platelets. Am J Hematol 2010;85:487-93. https://doi.org/10.1002/ajh.21732.
- Wiesner T, Bugl S, Mayer F, Hartmann JT, Kopp HG. Differential changes in platelet VEGF, Tsp, CXCL12, and CXCL4 in patients with metastatic cancer. Clin Exp Metastasis 2010;27:141-9. https://doi.org/10.1007/s10585-010-9311-6.
- Jung K, Mannello F, Lein M. Translating molecular medicine into clinical tools: doomed to fail by neglecting basic preanalytical principles. J Transl Med 2009;7. https://doi.org/10.1186/1479-5876-7-87.
- Ljungberg B, Bensalah K, Canfield S, Dabestani S, Hofmann F, Hora M, et al. EAU guidelines on renal cell carcinoma: 2014 update. Eur Urol 2015;67:913-24. https://doi.org/10.1016/j.eururo.2015.01.005.
- Escudier B, Porta C, Schmidinger M, Algaba F, Patard JJ, Khoo V, et al. Renal cell carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2014;25:iii49-56. https://doi.org/10.1093/annonc/mdu259.
- Brookman-May S, May M, Ficarra V, Kainz MC, Kampel-Kettner K, Kohlschreiber S, et al. Does preoperative platelet count and thrombocytosis play a prognostic role in patients undergoing nephrectomy for renal cell carcinoma? Results of a comprehensive retrospective series. World J Urol 2013;31:1309-16. https://doi.org/10.1007/s00345-012-0931-0.
- Manola J, Royston P, Elson P, McCormack JB, Mazumdar M, Negrier S, et al. Prognostic model for survival in patients with metastatic renal cell carcinoma: results from the international kidney cancer working group. Clin Cancer Res 2011;17:5443-50. https://doi.org/10.1158/1078-0432.CCR-11-0553.
- Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika 1994;81:515-26. https://doi.org/10.1093/biomet/81.3.515.
- Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting recommendations for tumor marker prognostic studies (REMARK): explanation and elaboration. BMC Med 2012;10. https://doi.org/10.1186/1741–7015–10–51.
- Tukey JW. Exploratory Data Analysis. Boston, MA: Addison–Wesley; 1977.
- Hollingsworth JM, Miller DC, Daignault S, Hollenbeck BK. Rising incidence of small renal masses: a need to reassess treatment effect. J Natl Cancer Inst 2006;98:1331-4. https://doi.org/10.1093/jnci/djj362.
- Uzzo RG. Renal masses – to treat or not to treat? If that is the question are contemporary biomarkers the answer?. J Urol 2008;180:433-4. https://doi.org/10.1016/j.juro.2008.04.124.
- Delahunt B, Cheville JC, Martignoni G, Humphrey PA, Magi-Galluzzi C, McKenney J, et al. The International Society of Urological Pathology (ISUP) grading system for renal cell carcinoma and other prognostic parameters. Am J Surg Pathol 2013;37:1490-504. https://doi.org/10.1097/PAS.0b013e318299f0fb.
- Madbouly K, Al-Qahtani SM, Ghazwani Y, Al-Shaibani S, Mansi MK. Microvascular tumor invasion: prognostic significance in low-stage renal cell carcinoma. Urology 2007;69:670-4. https://doi.org/10.1016/j.urology.2007.01.012.
- Lang H, Lindner V, Letourneux H, Martin M, Saussine C, Jacqmin D. Prognostic value of microscopic venous invasion in renal cell carcinoma: long-term follow-up. Eur Urol 2004;46:331-5. https://doi.org/10.1016/j.eururo.2004.03.020.
- Pichler M, Hutterer GC, Chromecki TF, Jesche J, Groselj-Strele A, Kampel-Kettner K, et al. Prognostic value of the Leibovich prognosis score supplemented by vascular invasion for clear cell renal cell carcinoma. J Urol 2012;187:834-9. https://doi.org/10.1016/j.juro.2011.10.155.
- Kroeger N, Rampersaud EN, Patard JJ, Klatte T, Birkhauser FD, Shariat SF, et al. Prognostic value of microvascular invasion in predicting the cancer specific survival and risk of metastatic disease in renal cell carcinoma: a multicenter investigation. J Urol 2012;187:418-23. https://doi.org/10.1016/j.juro.2011.10.024.
- Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ 2006;332. https://doi.org/10.1136/bmj.332.7549.1080.
- Collins GS, Reitsma JB, Altman DG, Moons KG. Transparent reporting of a multivariable prediction model for individual prognosis or diagnosis (TRIPOD): the TRIPOD statement. BJOG 2015;122:434-43. https://doi.org/10.1111/1471-0528.13244.
- Viprey VF, Gregory WM, Corrias MV, Tchirkov A, Swerts K, Vicha A, et al. Neuroblastoma mRNAs predict outcome in children with stage 4 neuroblastoma: a European HR-NBL1/SIOPEN study. J Clin Oncol 2014;32:1074-83. https://doi.org/10.1200/JCO.2013.53.3604.
- Bellmunt J, Leow JJ. Hyponatremia associated with worse outcomes in metastatic renal cell cancer: a potential target for intervention?. Eur Urol 2014;65:731-2. https://doi.org/10.1016/j.eururo.2013.10.057.
- Kramar A, Negrier S, Sylvester R, Joniau S, Mulders P, Powles T, et al. Guidelines for the definition of time-to-event end points in renal cell cancer clinical trials: results of the DATECAN project. Ann Oncol 2015;26:2392-8. https://doi.org/10.1093/annonc/mdv380.
- Ficarra V, Martignoni G, Maffei N, Brunelli M, Novara G, Zanolla L, et al. Original and reviewed nuclear grading according to the Fuhrman system: a multivariate analysis of 388 patients with conventional renal cell carcinoma. Cancer 2005;103:68-75. https://doi.org/10.1002/cncr.20749.
- Lang H, Lindner V, de Fromont M, Molinie V, Letourneux H, Meyer N, et al. Multicenter determination of optimal interobserver agreement using the Fuhrman grading system for renal cell carcinoma: Assessment of 241 patients with > 15-year follow-up. Cancer 2005;103:625-9. https://doi.org/10.1002/cncr.20812.
- Gressner OA, Weiskirchen R, Gressner AM. Biomarkers of liver fibrosis: clinical translation of molecular pathogenesis or based on liver-dependent malfunction tests. Clin Chim Acta 2007;381:107-13. https://doi.org/10.1016/j.cca.2007.02.038.
- Grigorescu M. Noninvasive biochemical markers of liver fibrosis. J Gastrointestin Liver Dis 2006;15:149-59.
- British In Vitro Diagnostics Association . Safe and Consistent? The Regulation of Pathology Testing 2013. www.bivda.co.uk/Portals/0/Documents/Bivda_in-house_testing_audit.pdf (accessed 4 January 2018).
- Hyltoft Petersen P, Klee GG. Influence of analytical bias and imprecision on the number of false positive results using guideline-driven medical decision limits. Clin Chim Acta 2014;430:1-8. https://doi.org/10.1016/j.cca.2013.12.014.
- Ioannidis JPA. Biomarker failures. Clin Chem 2013;59:202-4. https://doi.org/10.1373/clinchem.2012.185801.
- European Parliament and Council of the European Union . Directive 9879EC of the European Parliament and of the Council of 27 October 1998 on In Vitro Diagnostic Medical Devices 1998. http://eur-lex.europa.eu/legal-content/EN/TXT/HTML/?uri=CELEX:31998L0079&from=EN (accessed 4 January 2018).
- International Organization for Standardization . BS ISO 5725–1:1994: Accuracy (Trueness and Precision) of Measurement Methods and Results – Part 1: General Principles and Definitions 1994. www.iso.org/obp/ui/#iso:std:iso:5725:-1:ed-1:v1:en (accessed 5 March 2018).
- Bailey C, Barwick V. Laboratory Skills Training Handbook. Teddington: LGC Ltd; 2007.
- Joint Committee for Guides in Metrology . International Vocabulary of Metrology – Basic and General Concepts and Associated Terms (VIM) 2012. www.bipm.org/utils/common/documents/jcgm/JCGM_200_2012.pdf (accessed 5 March 2018).
- International Organization for Standardization . BS ISO 3534–1:2006: Statistics. Vocabulary and Symbols – Part 1: General Statistical Terms and Terms Used in Probability 2006. www.iso.org/obp/ui/#iso:std:iso:3534:-1:ed-2:v2:en (accessed 5 March 2018).
- International Organization for Standardization . BS ISO 5725–2:1994: Accuracy (Trueness and Precision) of Measurement Methods and Results – Part 2: Basic Methods for the Determination of Repeatability and Reproducibility of a Standard Measurement Method 1994. www.iso.org/obp/ui/#iso:std:iso:5725:-2:ed-1:v1:en (accessed 5 March 2018).
- International Organization for Standardization . BS ISO 5725–3:1994: Accuracy (Trueness and Precision) of Measurement Methods and Results – Part 3: Intermediate Measures of the Precision of a Standard Measurement Method 1994. www.iso.org/obp/ui/#iso:std:iso:5725:-3:ed-1:v1:en (accessed 5 March 2018).
- Panteghini M, Sandberg S. Defining analytical performance specifications 15 years after the Stockholm conference. Clin Chem Lab Med 2015;53:829-32. https://doi.org/10.1515/cclm-2015-0303.
- Kallner A, McQueen M, Heuck C. The Stockholm Consensus Conference on quality specifications in laboratory medicine, 25–26 April 1999. Scand J Clin Lab Invest 1999;59:475-6. https://doi.org/10.1080/00365519950185175.
- Sandberg S, Fraser CG, Horvath AR, Jansen R, Jones G, Oosterhuis W, et al. Defining analytical performance specifications: Consensus Statement from the 1st Strategic Conference of the European Federation of Clinical Chemistry and Laboratory Medicine. Clin Chem Lab Med 2015;53:833-5. https://doi.org/10.1515/cclm-2015-0067.
- Evaluation of Precision Performance of Quantitative Measurement Methods; Approved Guideline – Second Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2004.
- Khatami Z, Hill R, Sturgeon C, Kearney E, Breadon P, Kallner A. Measurement Verification in the Clinical Laboratory: a Guide to Assessing Analytical Performance During the Acceptance Testing of Methods (Quantitative Examination Procedures) and Or Analysers 2005. www.acb.org.uk/docs/default-source/committees/scientific/guidelines/measurement-verification/Measurement_verification_final_090608.pdf?sfvrsn=0 (accessed 4 January 2018).
- User Verification of Performance and Trueness; Approved Guideline – Second Edition. Wayne, PA: Clinical and Laboratory Standards Institute; 2005.
- Association for Clinical Biochemistry and Laboratory Medicine . Measurement Verification n.d. www.acb.org.uk/whatwedo/science/best_practice/MV_Terms1.aspx (accessed 9 March 2018).
- US Food and Drug Administration . Guidance for Industry – Bioanalytical Method Validation. Revision 2013. www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm368107.pdf (accessed 15 June 2015).
- Sturgeon CM, Hoffman BR, Chan DW, Ch’ng SL, Hammond E, Hayes DF, et al. National Academy of Clinical Biochemistry laboratory medicine practice guidelines for use of tumor markers in clinical practice: quality requirements. Clin Chem 2008;54:e1-e10. https://doi.org/10.1373/clinchem.2007.094144.
- Association for Clinical Biochemistry and Laboratory Medicine . Measurement Verification n.d. www.acb.org.uk/whatwedo/science/best_practice/MV_Terms3.aspx (accessed 9 March 2018).
- Roraas T, Petersen PH, Sandberg S. Confidence intervals and power calculations for within-person biological variation: effect of analytical imprecision, number of replicates, number of samples, and number of individuals. Clin Chem 2012;58:1306-13. https://doi.org/10.1373/clinchem.2012.187781.
- International Organization for Standardization, International Electrotechnical Commission . BS EN ISO IEC 17043:2010: Conformity Assessment – General Requirements for Proficiency Testing 2010. www.iso.org/obp/ui/#iso:std:iso-iec:17043:ed-1:v1:en (accessed 5 March 2018).
- Sturgeon C. External quality assessment schemes for immunoassays. Methods Mol Biol 2013;1065:291-305. https://doi.org/10.1007/978-1-62703-616-0_19.
- Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696-9. https://doi.org/10.1016/0168-8278(95)80226-6.
- Mazzaferro V, Bhoori S, Sposito C, Bongini M, Langer M, Miceli R, et al. Milan criteria in liver transplantation for hepatocellular carcinoma: An evidence-based analysis of 15 years of experience. Liver Transpl 2011;17:S44-S57. https://doi.org/10.1002/lt.22365.
- Ferenci P, Lockwood A, Mullen K, Tarter R, Weissenborn K, Blei AT. Hepatic encephalopathy–definition, nomenclature, diagnosis, and quantification: final report of the Working Party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 2002;35:716-21. https://doi.org/10.1053/jhep.2002.31250.
- Department of Health and Social Care . NHS Costing n.d. http://webarchive.nationalarchives.gov.uk/+/http://www.dh.gov.uk/en/Managingyourorganisation/NHScostingmanual/DH_129310 (accessed 31 May 2018).
- Department of Health and Social Care . NHS Reference Costs 2013 to 2014 n.d. www.gov.uk/government/publications/nhs-reference-costs-2013-to-2014 (accessed 31 May 2018).
- Wright M, Grieve R, Roberts J, Main J, Thomas HC. Health benefits of antiviral therapy for mild chronic hepatitis C: randomised controlled trial and economic evaluation. Health Technol Assess 2006;10. https://doi.org/10.3310/hta10210.
- Joint Formulary Committee . British National Formulary 2014.
- Tripathi D, Stanley AJ, Hayes PC, Patch D, Millson C, Mehrzad H, et al. UK guidelines on the management of variceal haemorrhage in cirrhotic patients. Gut 2015;64:1680-704. https://doi.org/10.1136/gutjnl-2015-309262.
- Trembling PM, Apostolidou S, Parkes J, Ryan A, Gentry-Maharaj A, Tanwar S, et al. Influence of BMI and alcohol on liver-related morbidity and mortality in a cohort of 108,000 women from the general population from UKCTOCS. J Hepatol 2013;58:S51-2. https://doi.org/10.1016/S0168-8278(13)60117-8.
- World Health Organisation . International Statistical Classification of Diseases and Related Health Problems, Tenth Revision n.d. www.apps.who.int/classification.icd10/browse/2016/en (accessed 24 May 2018).
- Wiener RS, Gould MK, Woloshin S, Schwartz LM, Clark JA. ‘The thing is not knowing’: patients’ perspectives on surveillance of an indeterminate pulmonary nodule. Health Expect 2015;18:355-65. https://doi.org/10.1111/hex.12036.
- McCaffery K, Borril J, Williamson S, Taylor T, Sutton S, Atkin W, et al. Declining the offer of flexible sigmoidoscopy screening for bowel cancer: a qualitative investigation of the decision-making process. Soc Sci Med 2001;53:679-91. https://doi.org/10.1016/S0277-9536(00)00375-0.
- Padgett DK, Yedidia MJ, Kerner J, Mandelblatt J. The emotional consequences of false positive mammography: African-American women’s reactions in their own words. Women Health 2001;33:1-14. https://doi.org/10.1300/J013v33n03_01.
- Remennick L. ‘I have no time for potential troubles’: Russian immigrant women and breast cancer screening in Israel. J Immigr Health 2003;5:153-63. https://doi.org/10.1023/A:1026163008336.
- Ryan PY, Graves KD, Pavlik EJ, Andrykowski MA. Abnormal ovarian cancer screening test result: women’s informational, psychological and practical needs. J Psychosoc Oncol 2007;25:1-18. https://doi.org/10.1300/J077v25n04_01.
- Hewison J, Haines A. Overcoming barriers to recruitment in health research. BMJ 2006;333:300-2. https://doi.org/10.1136/bmj.333.7562.300.
- Sturges JE, Hanrahan KJ. Comparing telephone and face-to-face qualitative interviewing: a research note. Qual Res 2004;4:107-18. https://doi.org/10.1177/1468794104041110.
- Novick G. Is there a bias against telephone interviews in qualitative research?. Res Nurs Health 2008;31:391-8. https://doi.org/10.1002/nur.20259.
- Sölétormos G, Duffy MJ, Hayes DF, Sturgeon CM, Barak V, Bossuyt PM, et al. Design of tumor biomarker–monitoring trials: a proposal by the European Group on Tumor Markers. Clin Chem 2013;59. https://doi.org/10.1373/clinchem.2011.180778.
- Hallworth MJ, Epner PL, Ebert C, Fantz CR, Faye SA, Higgins TN, et al. Current evidence and future perspectives on the effective practice of patient-centered laboratory medicine. Clin Chem 2015;61:589-99. https://doi.org/10.1373/clinchem.2014.232629.
- Lavallee LT, Binette A, Witiuk K, Cnossen S, Mallick R, Fergusson DA, et al. Reducing the harm of prostate cancer screening: repeated prostate-specific antigen testing. Mayo Clin Proc 2016;91:17-22. https://doi.org/10.1016/j.mayocp.2015.07.030.
- Bessen T, Keefe DM, Karnon J. Does one size fit all? Cost utility analyses of alternative mammographic follow-up schedules, by risk of recurrence. Int J Technol Assess Health Care 2015;31:281-8. https://doi.org/10.1017/s0266462315000598.
- Rolfe A, Burton C. Reassurance after diagnostic testing with a low pretest probability of serious disease: systematic review and meta-analysis. JAMA Intern Med 2013;173:407-16. https://doi.org/10.1001/jamainternmed.2013.2762.
- Redberg R, Katz M, Grady D. Diagnostic tests: another frontier for less is more: or why talking to your patient is a safe and effective method of reassurance. Arch Intern Med 2011;171. https://doi.org/10.1001/archinternmed.2010.465.
- Choosing Wisely: An Initiative of the ABIM Foundation . Choosing Wisely. Promoting Conversations Between Patients and Clinicians n.d. www.choosingwisely.org/ (accessed 1 May 2018).
- Malhotra A, Maughan D, Ansell J, Lehman R, Henderson A, Gray M, et al. Choosing Wisely in the UK: the Academy of Medical Royal Colleges’ initiative to reduce the harms of too much medicine. BMJ 2015;350. https://doi.org/10.1136/bmj.h2308.
- Bossuyt PM, Parvin T. Evaluating biomarkers for guiding treatment decisions. EJIFCC 2015;26:63-70.
- Horvath AR, Bossuyt PM, Sandberg S, John AS, Monaghan PJ, Verhagen-Kamerbeek WD, et al. Setting analytical performance specifications based on outcome studies – is it possible?. Clin Chem Lab Med 2015;53:841-8. https://doi.org/10.1515/cclm-2015-0214.
- Parkes J. Non Invasive Biomarkers in Chronic Liver Disease 2007.
- Collett D. Modelling Survival Data in Medical Research. London: Chapman and Hall; 1994.
- Freedman LS. Tables of numbers of patients required in clinical trials using the logrank test. Stat Med 1982;1:121-9. https://doi.org/10.1002/sim.4780010204.
Appendix 1 Appendices to Chapter 11
Appendix 2 Appendices to Chapter 17
| Sample ID | Site, HA (µg/l) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 87 | 83 | 85 | 2.9 | 3.3 | 83 | 91 | 87 | 5.4 | 6.2 | 88 | 102 | 95 | 9.9 | 10.4 | 88 | 86 | 87 | 1.5 | 1.7 |
| E002 | 54 | 51 | 52 | 1.8 | 3.5 | 51 | 52 | 52 | 0.6 | 1.2 | 51 | 59 | 55 | 6.2 | 11.3 | 53 | 51 | 52 | 1.5 | 2.8 |
| E003 | 76 | 76 | 76 | 0.5 | 0.6 | 76 | 80 | 78 | 3.2 | 4.1 | 78 | 89 | 83 | 7.6 | 9.1 | 70 | 83 | 76 | 8.9 | 11.7 |
| E004 | 35 | 36 | 36 | 0.3 | 1.0 | 39 | 39 | 39 | 0.3 | 0.8 | 38 | 44 | 41 | 4.3 | 10.6 | 37 | 36 | 36 | 0.7 | 1.8 |
| E005 | 51 | 51 | 51 | 0.3 | 0.6 | 51 | 55 | 53 | 2.4 | 4.5 | 55 | 57 | 56 | 1.8 | 3.2 | 50 | 51 | 50 | 0.6 | 1.1 |
| E006 | 68 | 65 | 66 | 2.1 | 3.2 | 69 | 73 | 71 | 2.4 | 3.3 | 80 | 86 | 83 | 4.4 | 5.4 | 70 | 72 | 71 | 1.2 | 1.6 |
| E007 | 41 | 43 | 42 | 1.3 | 3.1 | 42 | 44 | 43 | 1.9 | 4.5 | 44 | 47 | 46 | 2.6 | 5.7 | 41 | 43 | 42 | 1.4 | 3.5 |
| E008 | 97 | 100 | 98 | 2.7 | 2.8 | 97 | 102 | 100 | 4.0 | 4.0 | 109 | 100 | 104 | 5.7 | 5.5 | 98 | 99 | 99 | 0.7 | 0.7 |
| E009 | 65 | 62 | 64 | 2.3 | 3.6 | 65 | 63 | 64 | 1.5 | 2.4 | 71 | 65 | 68 | 3.7 | 5.5 | 66 | 65 | 65 | 1.1 | 1.7 |
| E010 | 85 | 95 | 90 | 6.8 | 7.5 | 94 | 102 | 98 | 5.4 | 5.5 | 120 | 103 | 111 | 11.6 | 10.4 | 101 | 101 | 101 | 0.0 | 0.0 |
| QC 1 | 20 | 20 | 20 | 0.1 | 0.5 | 22 | 20 | 21 | 1.4 | 6.6 | 25 | 23 | 24 | 1.6 | 6.9 | 21 | 20 | 21 | 0.8 | 3.9 |
| QC 2 | 46 | 43 | 45 | 2.2 | 5.0 | 48 | 47 | 48 | 1.2 | 2.6 | 51 | 52 | 51 | 0.5 | 0.9 | 49 | 46 | 48 | 2.5 | 5.3 |
| QC 3 | 175 | 161 | 168 | 10.0 | 6.0 | 180 | 173 | 176 | 5.1 | 2.9 | 191 | 195 | 193 | 3.2 | 1.6 | 169 | 160 | 164 | 6.4 | 3.9 |
| Sample ID | Site, HA (µg/l) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 84 | 82 | 83 | 1.7 | 2.1 | 121 | 99 | 110 | 15.4 | 14.1 | 84 | 83 | 83 | 0.4 | 0.5 | 83 | 85 | 84 | 1.4 | 1.6 |
| E002 | 52 | 49 | 51 | 2.0 | 3.9 | 39 | 63 | 51 | 16.8 | 33.0 | 51 | 51 | 51 | 0.1 | 0.2 | 51 | 53 | 52 | 1.3 | 2.5 |
| E003 | 73 | 69 | 71 | 2.9 | 4.1 | 105 | 83 | 94 | 15.4 | 16.3 | 74 | 71 | 72 | 1.7 | 2.4 | 73 | 75 | 74 | 1.4 | 1.8 |
| E004 | 35 | 37 | 36 | 1.1 | 3.2 | 50 | 27 | 39 | 15.9 | 41.4 | 35 | 36 | 35 | 0.6 | 1.6 | 36 | 36 | 36 | 0.2 | 0.6 |
| E005 | 47 | 52 | 49 | 3.7 | 7.4 | 64 | 42 | 53 | 15.9 | 30.0 | 49 | 50 | 50 | 0.1 | 0.1 | 50 | 52 | 51 | 1.6 | 3.2 |
| E006 | 71 | 66 | 68 | 4.0 | 5.8 | 93 | 71 | 82 | 15.5 | 18.8 | 68 | 69 | 68 | 1.1 | 1.6 | 73 | 73 | 73 | 0.4 | 0.5 |
| E007 | 41 | 42 | 42 | 0.7 | 1.6 | 60 | 32 | 46 | 20.0 | 43.4 | 42 | 44 | 43 | 1.6 | 3.7 | 43 | 43 | 43 | 0.2 | 0.4 |
| E008 | 100 | 101 | 100 | 0.8 | 0.8 | 153 | 146 | 150 | 5.0 | 3.3 | 101 | 104 | 102 | 2.0 | 2.0 | 105 | 103 | 104 | 1.6 | 1.6 |
| E009 | 60 | 69 | 64 | 5.9 | 9.2 | 45 | 95 | 70 | 35.3 | 50.7 | 68 | 61 | 65 | 5.1 | 7.9 | 65 | 66 | 66 | 0.8 | 1.2 |
| E010 | 102 | 105 | 104 | 2.6 | 2.5 | 145 | 114 | 129 | 22.2 | 17.2 | 101 | 101 | 101 | 0.1 | 0.1 | 95 | 109 | 102 | 9.6 | 9.4 |
| QC 1 | 21 | 21 | 21 | 0.3 | 1.3 | 24 | 25 | 25 | 0.8 | 3.5 | 20 | 20 | 20 | 0.2 | 0.8 | 23 | 22 | 22 | 0.6 | 2.9 |
| QC 2 | 45 | 43 | 44 | 1.8 | 4.1 | 62 | 52 | 57 | 6.8 | 12.0 | 45 | 44 | 45 | 0.4 | 0.9 | 44 | 44 | 44 | 0.0 | 0.1 |
| QC 3 | 156 | 167 | 162 | 8.0 | 5.0 | 141 | 212 | 177 | 50.7 | 28.7 | 173 | 167 | 170 | 4.2 | 2.4 | 167 | 167 | 167 | 0.6 | 0.4 |
| Sample ID | Site, PIIINP (µg/l) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 15.8 | 16.2 | 16.0 | 0.2 | 1.5 | 17.1 | 17.0 | 17.0 | 0.1 | 0.6 | 16.6 | 15.9 | 16.2 | 0.5 | 3.2 | 15.3 | 16.1 | 15.7 | 0.5 | 3.4 |
| E002 | 9.4 | 9.5 | 9.4 | 0.0 | 0.2 | 10.0 | 9.9 | 10.0 | 0.1 | 0.6 | 9.8 | 9.9 | 9.9 | 0.1 | 1.3 | 8.9 | 9.1 | 9.0 | 0.2 | 1.9 |
| E003 | 8.5 | 8.7 | 8.6 | 0.1 | 1.7 | 9.1 | 9.0 | 9.0 | 0.1 | 1.4 | 8.8 | 8.8 | 8.8 | 0.0 | 0.2 | 8.3 | 8.8 | 8.6 | 0.4 | 4.2 |
| E004 | 12.6 | 12.8 | 12.7 | 0.2 | 1.3 | 13.3 | 13.4 | 13.3 | 0.0 | 0.1 | 12.9 | 13.0 | 12.9 | 0.1 | 0.5 | 12.6 | 12.5 | 12.5 | 0.0 | 0.3 |
| E005 | 9.8 | 10.0 | 9.9 | 0.1 | 0.9 | 10.8 | 10.7 | 10.8 | 0.1 | 0.7 | 10.5 | 10.7 | 10.6 | 0.1 | 1.1 | 10.1 | 10.3 | 10.2 | 0.1 | 1.4 |
| E006 | 10.2 | 10.3 | 10.2 | 0.1 | 0.8 | 10.7 | 10.9 | 10.8 | 0.2 | 1.6 | 10.7 | 10.7 | 10.7 | 0.0 | 0.1 | 10.3 | 10.2 | 10.3 | 0.1 | 0.8 |
| E007 | 10.6 | 10.4 | 10.5 | 0.1 | 1.3 | 11.2 | 11.5 | 11.3 | 0.2 | 1.9 | 10.9 | 10.8 | 10.8 | 0.0 | 0.2 | 10.3 | 10.3 | 10.3 | 0.0 | 0.3 |
| E008 | 22.5 | 22.0 | 22.2 | 0.4 | 1.6 | 23.1 | 23.3 | 23.2 | 0.2 | 0.8 | 23.6 | 22.0 | 22.8 | 1.1 | 4.8 | 21.5 | 21.5 | 21.5 | 0.0 | 0.1 |
| E009 | 21.8 | 20.2 | 21.0 | 1.1 | 5.1 | 21.5 | 21.7 | 21.6 | 0.2 | 0.8 | 21.7 | 21.7 | 21.7 | 0.0 | 0.1 | 20.5 | 19.4 | 19.9 | 0.7 | 3.7 |
| E010 | 10.2 | 9.9 | 10.0 | 0.2 | 1.9 | 10.4 | 10.5 | 10.4 | 0.0 | 0.4 | 10.1 | 10.6 | 10.4 | 0.3 | 2.9 | 10.2 | 9.9 | 10.0 | 0.2 | 2.1 |
| QC 1 | 3.5 | 3.3 | 3.4 | 0.1 | 4.0 | 3.6 | 3.6 | 3.6 | 0.0 | 0.5 | 3.4 | 3.5 | 3.4 | 0.1 | 1.9 | 3.2 | 3.4 | 3.3 | 0.1 | 2.6 |
| QC 2 | 6.5 | 6.2 | 6.3 | 0.2 | 3.3 | 6.5 | 6.6 | 6.5 | 0.1 | 1.0 | 6.2 | 6.2 | 6.2 | 0.0 | 0.1 | 6.3 | 6.0 | 6.1 | 0.2 | 2.8 |
| QC 3 | 12.3 | 11.7 | 12.0 | 0.4 | 3.5 | 12.4 | 12.4 | 12.4 | 0.0 | 0.1 | 12.0 | 12.4 | 12.2 | 0.3 | 2.3 | 12.0 | 11.8 | 11.9 | 0.1 | 1.2 |
| Sample ID | Site, PIIINP (µg/l) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 16.3 | 16.7 | 16.5 | 0.2 | 1.5 | 16.7 | 16.7 | 16.7 | 0.0 | 0.0 | 16.7 | 16.0 | 16.3 | 0.4 | 2.7 | 16.6 | 16.3 | 16.5 | 0.2 | 1.2 |
| E002 | 9.7 | 10.2 | 9.9 | 0.3 | 3.4 | 9.7 | 9.8 | 9.7 | 0.0 | 0.1 | 9.8 | 10.0 | 9.9 | 0.1 | 1.4 | 9.9 | 9.6 | 9.8 | 0.2 | 2.4 |
| E003 | 9.3 | 8.7 | 9.0 | 0.4 | 4.5 | 9.1 | 8.9 | 9.0 | 0.1 | 1.1 | 9.3 | 9.0 | 9.1 | 0.2 | 1.7 | 8.9 | 9.0 | 8.9 | 0.0 | 0.4 |
| E004 | 12.8 | 13.8 | 13.3 | 0.7 | 5.1 | 13.2 | 13.4 | 13.3 | 0.2 | 1.3 | 13.1 | 13.3 | 13.2 | 0.1 | 0.9 | 13.8 | 13.5 | 13.6 | 0.2 | 1.6 |
| E005 | 9.8 | 11.2 | 10.5 | 0.9 | 9.0 | 10.9 | 10.9 | 10.9 | 0.0 | 0.5 | 10.7 | 10.9 | 10.8 | 0.2 | 1.5 | 10.7 | 10.6 | 10.6 | 0.1 | 0.8 |
| E006 | 10.4 | 11.2 | 10.8 | 0.6 | 5.3 | 10.8 | 10.8 | 10.8 | 0.0 | 0.3 | 11.1 | 10.8 | 10.9 | 0.2 | 2.3 | 10.9 | 10.9 | 10.9 | 0.0 | 0.1 |
| E007 | 11.1 | 11.3 | 11.2 | 0.1 | 1.1 | 11.1 | 11.3 | 11.2 | 0.2 | 1.5 | 11.2 | 11.3 | 11.3 | 0.1 | 1.1 | 11.1 | 11.2 | 11.2 | 0.1 | 0.8 |
| E008 | 23.6 | 22.6 | 23.1 | 0.7 | 2.9 | 22.9 | 23.4 | 23.2 | 0.4 | 1.6 | 22.7 | 22.3 | 22.5 | 0.3 | 1.5 | 22.8 | 23.2 | 23.0 | 0.3 | 1.4 |
| E009 | 21.1 | 21.7 | 21.4 | 0.4 | 1.9 | 21.2 | 21.6 | 21.4 | 0.3 | 1.3 | 21.2 | 22.3 | 21.7 | 0.8 | 3.7 | 21.4 | 21.5 | 21.5 | 0.1 | 0.4 |
| E010 | 10.9 | 10.5 | 10.7 | 0.2 | 2.3 | 10.6 | 10.6 | 10.6 | 0.0 | 0.1 | 10.3 | 10.5 | 10.4 | 0.1 | 1.3 | 10.8 | 11.2 | 11.0 | 0.2 | 2.2 |
| QC 1 | 3.3 | 3.3 | 3.3 | 0.0 | 0.2 | 3.2 | 3.3 | 3.3 | 0.1 | 1.7 | 3.4 | 3.5 | 3.4 | 0.0 | 1.2 | 3.2 | 3.3 | 3.3 | 0.1 | 3.2 |
| QC 2 | 6.2 | 6.4 | 6.3 | 0.1 | 2.3 | 6.5 | 6.4 | 6.4 | 0.1 | 1.8 | 6.2 | 6.3 | 6.3 | 0.1 | 1.1 | 6.2 | 6.0 | 6.1 | 0.1 | 2.2 |
| QC 3 | 11.9 | 12.9 | 12.4 | 0.7 | 5.9 | 12.3 | 12.1 | 12.2 | 0.1 | 1.0 | 12.3 | 12.5 | 12.4 | 0.1 | 1.0 | 12.2 | 12.2 | 12.2 | 0.0 | 0.1 |
| Sample ID | Site, TIMP-1 (µg/l) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 270 | 261 | 265 | 6.2 | 2.3 | 286 | 280 | 283 | 4.1 | 1.5 | 266 | 264 | 265 | 1.4 | 0.5 | 282 | 274 | 278 | 5.7 | 2.0 |
| E002 | 235 | 243 | 239 | 5.7 | 2.4 | 247 | 246 | 247 | 0.6 | 0.2 | 234 | 242 | 238 | 5.8 | 2.4 | 242 | 246 | 244 | 2.8 | 1.2 |
| E003 | 253 | 251 | 252 | 1.8 | 0.7 | 271 | 268 | 270 | 2.5 | 0.9 | 254 | 262 | 258 | 5.2 | 2.0 | 262 | 270 | 266 | 5.8 | 2.2 |
| E004 | 193 | 197 | 195 | 2.9 | 1.5 | 210 | 202 | 206 | 5.5 | 2.7 | 193 | 200 | 196 | 5.2 | 2.6 | 201 | 200 | 200 | 0.7 | 0.4 |
| E005 | 240 | 231 | 235 | 5.9 | 2.5 | 252 | 250 | 251 | 1.5 | 0.6 | 236 | 245 | 240 | 5.9 | 2.4 | 246 | 242 | 244 | 3.0 | 1.2 |
| E006 | 214 | 213 | 213 | 1.3 | 0.6 | 228 | 227 | 227 | 0.6 | 0.3 | 213 | 227 | 220 | 10.0 | 4.5 | 225 | 222 | 224 | 2.2 | 1.0 |
| E007 | 294 | 299 | 297 | 3.4 | 1.1 | 310 | 314 | 312 | 2.8 | 0.9 | 290 | 298 | 294 | 5.3 | 1.8 | 307 | 298 | 302 | 6.1 | 2.0 |
| E008 | 298 | 287 | 293 | 7.4 | 2.5 | 299 | 306 | 302 | 4.7 | 1.6 | 292 | 297 | 294 | 4.0 | 1.3 | 298 | 298 | 298 | 0.0 | 0.0 |
| E009 | 244 | 236 | 240 | 5.5 | 2.3 | 256 | 253 | 255 | 2.1 | 0.8 | 243 | 252 | 247 | 6.0 | 2.4 | 252 | 254 | 253 | 1.6 | 0.6 |
| E010 | 272 | 257 | 264 | 10.3 | 3.9 | 278 | 274 | 276 | 2.6 | 0.9 | 266 | 277 | 272 | 7.6 | 2.8 | 285 | 278 | 281 | 4.8 | 1.7 |
| QC 1 | 99 | 99 | 99 | 0.1 | 0.1 | 100 | 98 | 99 | 1.4 | 1.4 | 96 | 95 | 95 | 1.3 | 1.3 | 98 | 99 | 99 | 0.3 | 0.3 |
| QC 2 | 270 | 277 | 273 | 4.6 | 1.7 | 287 | 270 | 278 | 12.2 | 4.4 | 280 | 269 | 274 | 8.4 | 3.1 | 277 | 285 | 281 | 5.2 | 1.8 |
| QC 3 | 559 | 538 | 549 | 15.2 | 2.8 | 577 | 577 | 577 | 0.2 | 0.0 | 560 | 561 | 560 | 0.8 | 0.1 | 572 | 603 | 588 | 21.5 | 3.7 |
| Sample ID | Site, TIMP-1 (µg/l) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 272 | 278 | 275 | 4.5 | 1.6 | 279 | 280 | 280 | 1.0 | 0.4 | 267 | 262 | 265 | 3.3 | 1.2 | 274 | 278 | 276 | 2.8 | 1.0 |
| E002 | 239 | 238 | 239 | 0.9 | 0.4 | 246 | 247 | 246 | 0.1 | 0.1 | 235 | 230 | 232 | 3.2 | 1.4 | 234 | 240 | 237 | 4.0 | 1.7 |
| E003 | 260 | 267 | 263 | 5.0 | 1.9 | 266 | 270 | 268 | 3.1 | 1.2 | 255 | 250 | 253 | 3.5 | 1.4 | 257 | 261 | 259 | 2.6 | 1.0 |
| E004 | 194 | 202 | 198 | 5.2 | 2.6 | 206 | 199 | 202 | 4.7 | 2.3 | 190 | 187 | 189 | 2.1 | 1.1 | 196 | 200 | 198 | 2.5 | 1.3 |
| E005 | 241 | 245 | 243 | 2.9 | 1.2 | 252 | 256 | 254 | 2.5 | 1.0 | 230 | 239 | 234 | 6.5 | 2.8 | 245 | 239 | 242 | 4.4 | 1.8 |
| E006 | 221 | 223 | 222 | 1.8 | 0.8 | 224 | 233 | 228 | 6.0 | 2.6 | 217 | 216 | 216 | 1.3 | 0.6 | 219 | 223 | 221 | 3.3 | 1.5 |
| E007 | 294 | 291 | 293 | 2.3 | 0.8 | 301 | 310 | 305 | 6.3 | 2.1 | 289 | 285 | 287 | 3.3 | 1.1 | 301 | 309 | 305 | 5.6 | 1.8 |
| E008 | 293 | 295 | 294 | 1.0 | 0.3 | 300 | 302 | 301 | 1.2 | 0.4 | 287 | 291 | 289 | 2.8 | 1.0 | 281 | 293 | 287 | 8.4 | 2.9 |
| E009 | 250 | 248 | 249 | 1.6 | 0.6 | 255 | 253 | 254 | 1.6 | 0.6 | 246 | 240 | 243 | 4.0 | 1.7 | 256 | 249 | 253 | 4.9 | 2.0 |
| E010 | 272 | 271 | 272 | 1.2 | 0.4 | 279 | 281 | 280 | 1.0 | 0.4 | 263 | 263 | 263 | 0.1 | 0.1 | 275 | 263 | 269 | 8.4 | 3.1 |
| QC 1 | 94 | 101 | 98 | 5.1 | 5.2 | 97 | 100 | 99 | 2.2 | 2.2 | 96 | 96 | 96 | 0.1 | 0.1 | 100 | 101 | 100 | 0.3 | 0.3 |
| QC 2 | 272 | 274 | 273 | 1.3 | 0.5 | 278 | 275 | 276 | 2.4 | 0.9 | 269 | 267 | 268 | 1.4 | 0.5 | 272 | 286 | 279 | 9.6 | 3.4 |
| QC 3 | 565 | 579 | 572 | 10.3 | 1.8 | 582 | 584 | 583 | 1.2 | 0.2 | 573 | 560 | 567 | 9.0 | 1.6 | 583 | 579 | 581 | 2.4 | 0.4 |
| Sample ID | Site, ELF value | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 10.4 | 10.3 | 10.3 | 0.03 | 0.3 | 10.4 | 10.5 | 10.4 | 0.04 | 0.4 | 10.4 | 10.5 | 10.4 | 0.1 | 0.6 | 10.4 | 10.4 | 10.4 | 0 | 0 |
| E002 | 9.5 | 9.5 | 9.5 | 0.01 | 0.1 | 9.5 | 9.5 | 9.5 | 0.00 | 0.0 | 9.5 | 9.6 | 9.6 | 0.1 | 1.2 | 9.5 | 9.5 | 9.5 | 0 | 0 |
| E003 | 9.8 | 9.8 | 9.8 | 0.01 | 0.1 | 9.8 | 9.9 | 9.8 | 0.02 | 0.2 | 9.8 | 9.9 | 9.9 | 0.1 | 0.9 | 9.7 | 9.9 | 9.8 | 0.1 | 1.4 |
| E004 | 9.3 | 9.3 | 9.3 | 0.02 | 0.2 | 9.4 | 9.4 | 9.4 | 0.01 | 0.1 | 9.4 | 9.5 | 9.4 | 0.1 | 1.1 | 9.3 | 9.3 | 9.3 | 0.0 | 0.2 |
| E005 | 9.5 | 9.5 | 9.5 | 0.01 | 0.1 | 9.6 | 9.6 | 9.6 | 0.03 | 0.3 | 9.6 | 9.7 | 9.6 | 0.0 | 0.4 | 9.5 | 9.5 | 9.5 | 0.0 | 0.1 |
| E006 | 9.7 | 9.7 | 9.7 | 0.03 | 0.3 | 9.8 | 9.9 | 9.8 | 0.04 | 0.4 | 9.9 | 10.0 | 9.9 | 0.1 | 0.6 | 9.8 | 9.8 | 9.8 | 0.0 | 0.1 |
| E007 | 9.5 | 9.5 | 9.5 | 0.02 | 0.2 | 9.5 | 9.6 | 9.6 | 0.06 | 0.6 | 9.5 | 9.6 | 9.6 | 0.0 | 0.5 | 9.4 | 9.5 | 9.5 | 0.0 | 0.3 |
| E008 | 10.8 | 10.8 | 10.8 | 0.00 | 0.0 | 10.8 | 10.8 | 10.8 | 0.05 | 0.5 | 10.9 | 10.8 | 10.8 | 0.1 | 0.7 | 10.7 | 10.7 | 10.7 | 0.0 | 0.1 |
| E009 | 10.3 | 10.2 | 10.3 | 0.08 | 0.8 | 10.3 | 10.3 | 10.3 | 0.02 | 0.2 | 10.4 | 10.3 | 10.4 | 0.0 | 0.4 | 10.3 | 10.2 | 10.3 | 0.0 | 0.4 |
| E010 | 10.0 | 10.1 | 10.0 | 0.04 | 0.4 | 10.1 | 10.2 | 10.2 | 0.05 | 0.5 | 10.3 | 10.2 | 10.3 | 0.1 | 0.6 | 10.2 | 10.1 | 10.2 | 0.0 | 0.3 |
| QC 1 | 7.6 | 7.5 | 7.6 | 0.03 | 0.4 | 7.7 | 7.6 | 7.6 | 0.05 | 0.6 | 7.7 | 7.7 | 7.7 | 0.0 | 0.6 | 7.6 | 7.6 | 7.6 | 0.0 | 0.2 |
| QC 2 | 9.2 | 9.1 | 9.1 | 0.06 | 0.6 | 9.2 | 9.2 | 9.2 | 0.03 | 0.3 | 9.2 | 9.2 | 9.2 | 0.0 | 0.0 | 9.2 | 9.1 | 9.2 | 0.1 | 0.6 |
| QC 3 | 11.1 | 10.9 | 11.0 | 0.08 | 0.8 | 11.1 | 11.1 | 11.1 | 0.03 | 0.2 | 11.1 | 11.2 | 11.1 | 0.0 | 0.3 | 11.0 | 11.0 | 11.0 | 0.0 | 0.3 |
| Sample ID | Site, ELF value | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | |||||||||||||||||
| Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | Run 1 | Run 2 | Mean | SD | %CV | |
| E001 | 10.4 | 10.4 | 10.4 | 0.0 | 0.0 | 10.7 | 10.5 | 10.6 | 0.1 | 1.1 | 10.4 | 10.4 | 10.4 | 0.0 | 0.1 | 10.4 | 10.3 | 10.3 | 0.03 | 0.3 |
| E002 | 9.5 | 9.5 | 9.5 | 0.0 | 0.1 | 9.3 | 9.7 | 9.5 | 0.3 | 3.0 | 9.5 | 9.5 | 9.5 | 0.0 | 0.1 | 9.5 | 9.5 | 9.5 | 0.01 | 0.1 |
| E003 | 9.8 | 9.7 | 9.8 | 0.1 | 0.6 | 10.1 | 9.9 | 10.0 | 0.1 | 1.4 | 9.8 | 9.8 | 9.8 | 0.0 | 0.2 | 9.8 | 9.7 | 9.8 | 0.04 | 0.4 |
| E004 | 9.3 | 9.4 | 9.4 | 0.1 | 0.8 | 9.6 | 9.1 | 9.4 | 0.4 | 3.9 | 9.4 | 9.4 | 9.4 | 0.0 | 0.0 | 9.3 | 9.3 | 9.3 | 0.02 | 0.2 |
| E005 | 9.4 | 9.6 | 9.5 | 0.1 | 1.4 | 9.8 | 9.4 | 9.6 | 0.3 | 2.7 | 9.6 | 9.6 | 9.6 | 0.0 | 0.1 | 9.5 | 9.5 | 9.5 | 0.02 | 0.2 |
| E006 | 9.8 | 9.8 | 9.8 | 0.0 | 0.1 | 10.1 | 9.8 | 10.0 | 0.2 | 1.5 | 9.8 | 9.9 | 9.8 | 0.0 | 0.1 | 9.8 | 9.8 | 9.8 | 0.01 | 0.1 |
| E007 | 9.5 | 9.5 | 9.5 | 0.0 | 0.1 | 9.8 | 9.3 | 9.6 | 0.4 | 3.8 | 9.5 | 9.6 | 9.5 | 0.0 | 0.2 | 9.5 | 9.5 | 9.5 | 0.04 | 0.4 |
| E008 | 10.8 | 10.8 | 10.8 | 0.0 | 0.1 | 11.2 | 11.1 | 11.1 | 0.0 | 0.1 | 10.8 | 10.8 | 10.8 | 0.0 | 0.1 | 10.8 | 10.8 | 10.8 | 0.01 | 0.1 |
| E009 | 10.2 | 10.4 | 10.3 | 0.1 | 0.9 | 10.0 | 10.6 | 10.3 | 0.5 | 4.4 | 10.3 | 10.3 | 10.3 | 0.0 | 0.0 | 10.3 | 10.3 | 10.3 | 0.05 | 0.4 |
| E010 | 10.2 | 10.2 | 10.2 | 0.0 | 0.1 | 10.5 | 10.3 | 10.4 | 0.1 | 1.4 | 10.2 | 10.3 | 10.2 | 0.1 | 0.8 | 10.2 | 10.2 | 10.2 | 0.01 | 0.1 |
| QC 1 | 7.5 | 7.6 | 7.6 | 0.0 | 0.5 | 7.7 | 7.7 | 7.7 | 0.1 | 0.7 | 7.6 | 7.6 | 7.6 | 0.0 | 0.0 | 7.6 | 7.6 | 7.6 | 0.00 | 0.0 |
| QC 2 | 9.1 | 9.1 | 9.1 | 0.0 | 0.2 | 9.4 | 9.2 | 9.3 | 0.1 | 1.3 | 9.1 | 9.1 | 9.1 | 0.0 | 0.0 | 9.1 | 9.1 | 9.1 | 0.00 | 0.0 |
| QC 3 | 10.9 | 11.1 | 11.0 | 0.1 | 0.8 | 10.9 | 11.2 | 11.1 | 0.2 | 2.2 | 11.0 | 11.0 | 11.0 | 0.0 | 0.0 | 11.0 | 11.0 | 11.0 | 0.02 | 0.2 |
Appendix 3 Summary of changes to the original ELUCIDATE trial protocol
| Protocol version | Date approved | Summary of amendment |
|---|---|---|
| Version 1.0 | 2 February 2010 | Not applicable |
| Version 2.0 | 25 March 2010 |
|
| Version 3.0 | 30 April 2010 |
|
| Recruitment halted between 24 December 2010 and 29 March 2011 whilst protocol amendment approved. Forty-three patients registered up until 24 December 2010 | ||
| Version 4.0 | Not approved because of concerns regarding information provision for re-approached patients who had previously failed the eligibility criteria |
|
| Version 5.0 | 14 March 2011 |
|
| Version 6.0 | 30 May 2012 |
|
| Version 7.0 | 04 March 2013 |
|
Appendix 4 Original sample size calculation for the ELUCIDATE trial
Note that when these figures were derived, prior to the start of the trial, the ELF threshold for defining cirrhosis was set at 11 and, therefore, many fewer patients would have been defined as cirrhotic by ELF testing. The threshold was changed to 9.5 after the recruitment of three patients in March 2011, when it was realised that the threshold of 11 was too stringent.
The ELUCIDATE trial is testing the hypothesis that, if we monitor patients with CLD using the ELF value, we will detect liver cirrhosis earlier and, as a result, there will be fewer severe complications as well as improvements in other important patient outcomes. We are, therefore, aiming to show that the incidence of severe complications following cirrhosis is less in the ELF arm. This is the primary end point on which the trial is powered.
The trial will recruit over 18 months, with an additional 30 months of follow-up. Previous studies have led us to anticipate that, in the standard care arm, at 36 months, we will observe severe complications of the order of 2% for variceal bleeds, 1% for major bleeds and 0.7% for inoperable HCCs, giving a 3.7% incidence of potentially preventable undesirable clinical end points. 914
Previous data have illustrated that approximately 20% of patients will have varices suitable for therapy. 49 Such therapy has a large effect on the progression of these varices (for instance, a reduction from 37% progressing to 11% progressing); on the risk of bleeding from these varices (reduced from 30% to 14% in patients with medium to large varices); and on mortality at 36 months (reduced from 7% to 2% over 24 months). 49 Based on these data, we hypothesise that we could reduce the incidence of the undesirable clinical end points of cirrhosis by a half, or even two-thirds, in the ELF arm.
We anticipate that we will observe approximately twice as many patients developing cirrhosis in the ELF arm (approx. 20% of patients) than in the standard arm (approx. 10% of patients) over the 18 months of recruitment and 30 months of follow-up. Using this accrual and follow-up rate, along with the expected incidence rate, we can calculate the expected number of events in each arm using the method described by Collett915 for sample size estimates based on exponential survival distributions (more details of which can be seen in the statistical analysis plan). This gives expected numbers with severe complications of 19 and 6 in the control and experimental arms, respectively, assuming a two-thirds reduction in severe complications; or 12 and 6, respectively, assuming a reduction in severe complications of half.
With 1000 patients randomised, we would have > 99% power to detect this difference in numbers of patients encountering severe complications, subsequent to being detected with cirrhosis, with a 5% type 1 error, 18 months of recruitment and 30 months of follow-up. Sample size calculations are provided in the following paragraphs.
If it is assumed for simplicity that cirrhosis will be detected at uniform intervals throughout the 4 years of the trial, then we can estimate a pair of actuarial incidence curves (assuming exponentiality in generating these incidence curves) that give us the 19 and 6 events that we originally estimated, with 18 months of recruitment and 30 months of follow-up. These hypothetical curves are presented below.

The exponential parameters give medians of 2.85 years in the control arm and 27 years in the ELF arm (consistent with such a big, two-thirds reduction in severe complications). Carrying out sample size calculations with these figures gives a total sample size, say ns (within cirrhosis patients), of 150 (> 99% power). 916 For this calculation we are assuming that cirrhosis patients are identified over an 18-month period, with a further 30 months of follow-up, and that twice as many are identified in the ELF arm (20% of the total in the ELF arm vs. 10% of the total in the control arm). The power calculation for severe complications allows for this. If the total sample size of cirrhotic patients is nt, then ns = 20%(1/2nt) + 10%(1/2nt) = 15%(nt). So, nt = 100/15 and ns = 150. We, therefore, multiply 150 by 100/15 to obtain the total sample size of monitored patients, which is equal to 1000 (> 99% power).
Note that we have been plotting the likely incidence of severe complications following detection of cirrhosis, so that, even though the curve of the cumulative numbers detected with cirrhosis may have a shape that is concave then linear, with a slow start because of the initial 18-month accrual, the incidence of severe complications following cirrhosis might reasonably be expected to follow the exponential pattern assumed, where the likelihood of a severe complication that follows the detection of cirrhosis is unrelated to the time followed up after detection of cirrhosis.
There is a possibility that the trial itself will affect the control arm (contamination) positively. We have, therefore, gone on to look at this contamination issue. Suppose that the control arm receives more successful interventions, etc. and the difference in severe complications is reduced by only a half (instead of two-thirds). We therefore assume that there are in fact only 12 events in the control arm instead of 19, as in the following graph.
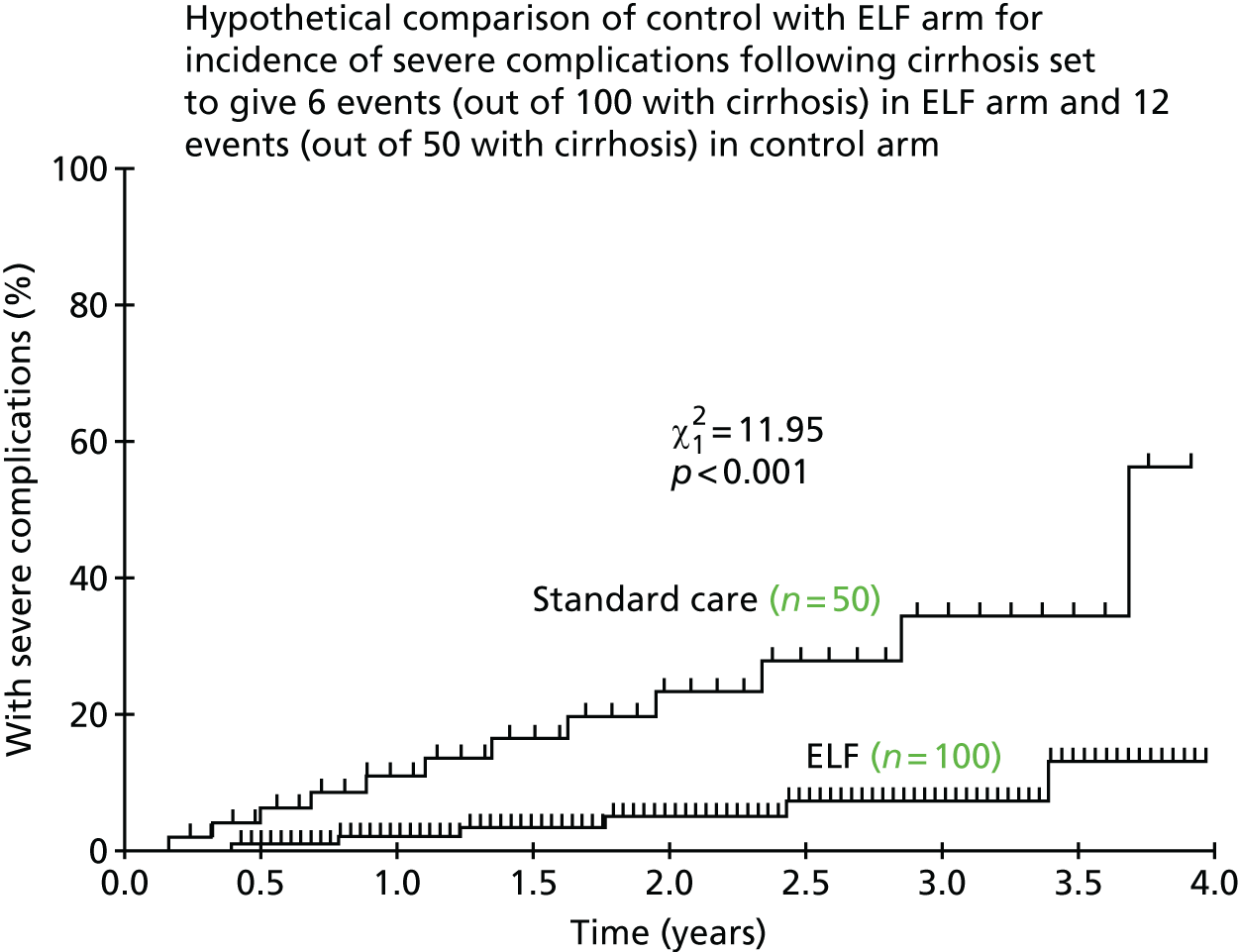
The exponential parameters in this case give medians of 5.5 years in the control arm and 27 years in the ELF arm. Carrying out sample size calculations with these figures gives a total sample size (within cirrhosis patients) of 143 (80% power) or 191 (90% power). We again multiply these values by 100/15 to obtain the total sample size of monitored patients of 953 (80% power) or 1273 (90% power).
This scenario might not be appropriate if we had a sizeable number of patients with severe complications before their cirrhosis is detected. However, if this happened, we would treat any patients having severe complications without prior detection of cirrhosis as having a zero time to incidence of severe complications following cirrhosis on the incidence curve. If this was similar in both arms the curves would simply start at the same probability (> 0) on the y-axis, and subsequent divergence of the curves would be more acute than previously to cause the overall twofold difference we are looking for.
The trial as its current size is, thus, well powered (at least 80% even under the assumption that we see only a 50% reduction in the incidence of severe complications) to show that this ELF monitoring policy will be of real clinical benefit.
List of abbreviations
- ACB
- Association for Clinical Biochemistry and Laboratory Medicine
- ACCE
- analytical validity; clinical validity; clinical utility; and ethical, legal and social considerations
- ACY-1
- serum aminoacylase-1
- AFP
- alpha-fetoprotein
- AGREE
- Appraisal of Guidelines for Research and Evaluation
- AIDS
- acquired immune deficiency syndrome
- AKI
- acute kidney injury
- ALP
- alkaline phosphatase
- ALT
- alanine transaminase
- ANOVA
- analysis of variance
- APG
- analytical performance goal
- AQP-1
- aquaporin-1
- AR
- acute rejection
- AST
- aspartate aminotransferase
- AUC
- area under the curve
- B7-H3
- B7 family ligand
- bFGF
- basic fibroblast growth factor
- BMI
- body mass index
- BRISQ
- Biospecimen Reporting for Improved Study Quality
- CA-15-3
- cancer antigen 15-3
- CA-125
- cancer antigen 125
- CAF
- C-terminal agrin fragment
- CAIX
- carbonic anhydrase IX
- ccRCC
- clear-cell renal cell carcinoma
- CCRN
- Comprehensive Clinical Research Network
- CD4
- cluster of differentiation 4
- CE
- Conformité Européene
- CEA
- carcinoembryonic antigen
- CENTRAL
- Cochrane Central Register of Controlled Trials
- CI
- confidence interval
- CIT
- cold ischaemic time
- CKD
- chronic kidney disease
- CLD
- chronic liver disease
- CLSI
- Clinical and Laboratory Standards Institute
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CRN
- Clinical Research Network
- CRP
- C-reactive protein
- CRR
- creatinine reduction ratio
- CSS
- cancer-specific survival
- CT
- computed tomography
- cTIMP
- clinical trial of investigational medicinal product
- CTRU
- Clinical Trials Research Unit
- CUZD1
- CUB and zona pellucida-like domains protein 1
- CV
- coefficient of variation
- CVG
- between-individual variability
- CV1
- within-individual variability
- DBD
- donation after brain death
- DCD
- donation after circulatory death
- DEC
- Diagnostic Evidence Co-operative
- DFS
- disease-free survival
- DGF
- delayed graft function
- DMEC
- Data Monitoring and Ethics Committee
- DNA
- deoxyribonucleic acid
- ECD
- expanded-criteria donor
- ECOG PS
- Eastern Cooperative Oncology Group performance status
- EDTA
- ethylenediaminetetraacetic acid
- EFLM
- European Federation of Clinical Chemistry and Laboratory Medicine
- eGFR
- estimated glomerular filtration rate
- EGTM
- European Group on Tumor Markers
- ELF
- Enhanced Liver Fibrosis
- ELISA
- enzyme-linked immunosorbent assay
- ELUCIDATE
- ELF to Uncover Cirrhosis as an Indication for Diagnosis and Action for Treatable Event
- EP
- Evaluation Protocol
- EPO
- erythropoietin
- EQ-5D
- EuroQol-5 Dimensions
- ESKD
- end-stage kidney disease
- ESRD
- end-stage renal disease
- EU
- European Union
- FDA
- Food and Drug Administration
- FFPE
- formalin-fixed paraffin-embedded
- GCP
- Good Clinical Practice
- GFR
- glomerular filtration rate
- HA
- hyaluronic acid
- HAS
- Haute Autorité de Santé
- HBV
- hepatitis B virus
- HCC
- hepatocellular carcinoma
- HCG
- human chorionic gonadotropin
- HCV
- hepatitis C virus
- HER2
- human epidermal growth factor receptor 2
- HES
- Hospital Episode Statistics
- HETE
- hydroxyeicosatetraenoic acid
- HIF
- hypoxia-inducible factor
- HIF-1α
- hypoxia-inducible factor-1α
- HIV
- human immunodeficiency virus
- HLA
- human leucocyte antigen
- HR
- hazard ratio
- HTA
- Human Tissue Authority
- IAP
- immunosuppressive acidic protein
- IARC
- International Agency for Research on Cancer
- ICD-10
- International Statistical Classification of Diseases and Related Health Problems, 10th Revision
- ICER
- incremental cost-effectiveness ratio
- IFCC
- International Federation of Clinical Chemistry and Laboratory Medicine
- IgA
- immunoglobulin A
- IgA-aβ2GP1-ab
- anti-beta-2-glycoprotein 1 antibody
- IGFBP7
- insulin-like growth factor-binding protein 7
- IL
- interleukin
- IL-6
- interleukin-6
- IL-16
- interleukin-16
- IL-18
- interleukin-18
- IQR
- interquartile range
- IRI
- ischaemia–reperfusion injury
- ISBER
- International Society for Biological and Environmental Repositories
- ISRCTN
- International Standard Randomised Controlled Trial
- IVD
- in vitro diagnostics
- LDT
- laboratory-developed test
- LFT
- liver function test
- LLoQ
- lower limit of quantification
- LoB
- limit of blank
- LoD
- limit of detection
- LoQ
- limit of quantification
- MDA
- malondialdehyde
- M-Factor
- multiple factor
- MET
- mesenchymal–epithelial transition
- MFS
- metastasis-free survival
- MMP-7
- matrix metalloproteinase-7
- MMP-9
- matrix metalloproteinase-9
- MRI
- magnetic resonance imaging
- MTA
- Material Transfer Agreement
- MVI
- microvascular invasion
- NACB
- National Academy of Clinical Biochemistry
- NGAL
- neutrophil gelatinase-associated lipocalin
- NICE
- National Institute for Health and Care Excellence
- NIH
- National Institutes of Health
- NIHR
- National Institute for Health Research
- NIHR CRN
- National Institute for Health Research Clinical Research Network
- NLR
- neutrophil–lymphocyte ratio
- NPV
- negative predictive value
- OCEI
- Organisation of European Cancer Institutes
- OGD
- oesophagogastroduodenoscopy
- ONS
- Office for National Statistics
- OPN
- osteopontin
- OS
- overall survival
- PAPP-A
- pregnancy-associated plasma protein A
- PF4
- platelet factor 4
- PFS
- progression-free survival
- PG
- proteoglycan
- PIIINP
- amino-terminal propeptide of type III procollagen
- PPI
- patient and public involvement
- PPV
- positive predictive value
- PSA
- prostate-specific antigen
- PTHLH
- parathyroid hormone-like hormone
- QALY
- quality-adjusted life-year
- QC
- quality control
- R&D
- research and development
- rCAIX
- recombinant carbonic anhydrase IX
- RCC
- renal cell carcinoma
- RCT
- randomised controlled trial
- RCV
- reference change value
- REC
- research ethics committee
- REMARK
- REporting recommendations for tumour MARKer prognostic studies
- RFS
- relapse-free survival
- rNGAL
- recombinant neutrophil gelatinase-associated lipocalin
- ROC
- receiver operating characteristic
- RRT
- renal replacement therapy
- RT
- renal transplantation
- RTB
- research tissue bank
- RTX
- radical radiotherapy
- RUSAE
- related and unexpected serious adverse event
- SAA
- serum amyloid A
- SCF
- stem cell factor
- SD
- standard deviation
- SF-12 v2
- Short Form questionnaire-12 items version 2
- sFlt-1
- soluble Fms-like tyrosine kinase-1
- SIBLING
- small integrin-binding ligand N-linked glycoprotein family
- sIL-2R
- soluble interleukin-2 receptor
- SOP
- standard operating procedure
- SPREC
- Sample PREanalytical Code
- SSI
- site-specific information
- SSIGN
- Stage, Size, Grade and Necrosis score
- SSOP
- study site operating procedure
- STARD
- STAndards for Reporting Diagnostic accuracy
- ST6Gall
- galbeta1, 4GIcNAcalpha2, 6-sialytransferase
- TIMP-1
- tissue inhibitor of metalloproteinase 1
- TMG
- Trial Management Group
- TNFR2
- tumour necrosis factor receptor 2
- TNM
- tumour, node, metastasis
- TuM2-PK
- tumour M2 pyruvate kinase
- UCLH
- University College London Hospital
- UISS
- UCLA Integrated Staging System
- UK NEQAS
- UK National External Quality Assessment Service
- UTI
- urinary tract infection
- VEGF
- vascular endothelial growth factor
- VEGF-A
- vascular endothelial growth factor-A
- VEGFR-1
- vascular endothelial growth factor receptor-1
- VHL/VHL
- von Hippel–Lindau
- WBC
- white blood cell
- WIT
- warm ischaemic time