
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 09/91/22. The contractual start date was in October 2011. The draft report began editorial review in July 2014 and was accepted for publication in April 2015. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Paul McNamara stated the following conflicts of interest: received personal fees and consultancy payments for assisting in setting up a study on new antiviral for respiratory syncytial virus disease in the UK 2013–15 from Alios BioPharma; and received personal fees and honorarium for advisory board attendance to discuss new respiratory syncytial virus antivirals and vaccines in February 2014 from Janssen Pharmaceuticals.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2015. This work was produced by Everard et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
Acute bronchiolitis is the most common reason for hospitalisation in infancy and childhood, with 1–3% of all infants being admitted to hospital during their first winter. 1–9 Common respiratory viruses infecting the lungs lead to severe difficulties in breathing by causing obstruction of the airways. 1–8,10–13 The age of peak incidence of babies admitted with this condition is between 1 and 6 months. 9 The acute illness is distressing for the infant and is associated with considerable stress for parents of an acutely unwell small infant.
The only intervention that has had a major impact on the survival of infants in the last 40 years of research is oxygen therapy, which has reduced mortality rates from around 20% to less than 1%. 14 Otherwise, current treatment consists of providing good supportive care until the infant recovers. 4,15,16 Admissions for bronchiolitis increased from 21,330 in 2004/5 to 33,472 in 2010/11,17 placing enormous strains on paediatric services and intensive care units (ICUs)4,8,18 which sometimes have to close because of the number of infants with acute bronchiolitis. 18–20 Despite many putative candidates, including antiviral agents, inhaled steroids and bronchodilators, no treatment other than oxygen therapy has been shown to have an impact on the course of the acute illness and an effective vaccine still appears some way off. The median duration of hospitalisation in the UK is around 3 days, which compares with a median of 1 day for all acute paediatric admissions. This relatively long duration of hospital stay combined with the large number of such infants admitted to hospital between November and March accounts for the substantial burden on hospital services resulting from the yearly winter epidemics.
At the time we were preparing the grant application for this project, a number of relatively small studies had suggested that nebulised hypertonic saline (HS) may reduce the duration of hospital stay for infants admitted with acute bronchiolitis. 21–24 A Cochrane review, published in 2008,25 undertook a systematic review of the literature, focusing on four trials which involved a total of 254 infants with acute viral bronchiolitis ,of whom 189 were inpatients and 65 patients were treated in the emergency department. The review concluded that ‘current evidence suggests nebulised 3% saline may significantly reduce the length of hospital stay and improve the clinical severity score in infants with acute viral bronchiolitis.’25 The conclusions do not go beyond suggesting that this intervention may have an impact because of the methodological limitations of the included studies and the potential for publication bias. The review noted that the conclusions are further limited by the small number of subjects included in each study and consequent low power; the different settings since inpatients and outpatients were included; and the failure to show a difference in some outcomes such as the failure to reduce hospitalisation in the outpatient groups. Despite these limitations, some hospitals in the UK had already adopted this approach to treatment by the time we started the SABRE (Saline in Acute Bronchiolitis RCT and Economic evaluation) trial. The subsequent development of the evidence base is discussed in Chapter 6, Strengths and weaknesses compared with earlier systematic reviews.
The suggested mode of action of HS is through an alteration in mucus rheology as a result of improved hydration and the breaking of ionic bonds within the mucus, leading to improvements in mucociliary clearance of secretions. 26,27 The observation that HS can increase ciliary beat frequency may further enhance clearance. 26 It is also suggested that this intervention reduces mucosal wall oedema through osmotic effects. There is good evidence that bronchoconstriction does not contribute significantly to the airways obstruction in infants with acute bronchiolitis, which explains why bronchodilators do not provide any benefit. 28–31 Instead, the obstruction appears to be in part because of oedema within the airway wall and, probably more importantly, accumulation of inflammatory exudates in the airways. Significant impairment of mucociliary clearance because of the shedding of ciliated cells compounds the problem and contributes to accumulation of secretions in the small airways. Hence an intervention leading to improvement in clearance of these secretions and reduced oedema of the airway wall may be of benefit in infants with acute bronchiolitis.
In contrast to these positive studies, a recent Canadian study based in an emergency department found no benefit when nebulised 3% HS was added to ‘usual care’,32 while a second study from Turkey,33 again involving non-hospitalised children, found that 3% HS offered no advantage over nebulised normal saline (NS) in addition to ‘usual’ care. In both cases the intervention was in addition to the use of bronchodilators. Understanding the differences between terminology for bronchiolitis used in North America and that used in the UK is key to understanding why these studies might be predicted to have a negative outcome and are not directly relevant to UK practice.
In North America, and a number of other countries, the term ‘acute bronchiolitis’ is generally used to describe an apparent viral infection in an infant or young child who is experiencing their first episode of wheeze. 2,5,34 In the UK, Australia and some northern European countries, the key feature of acute bronchiolitis is the presence of widespread crepitations on auscultations rather than wheeze, which may or may not be present. 2,4,34 In general, those patients with wheezing illnesses labelled as acute bronchiolitis are somewhat older (mean 9–15 months)35–37 than infants admitted in the UK with acute bronchiolitis, among whom the peak age for admissions is around 4 months of age. 35,38–41
The clinical phenotype of subjects included in a study has been shown to be important not just in the acute illness but in terms of subsequent morbidity, with those having the North American phenotype being much more likely to be subsequently shown to have asthma than those with acute bronchiolitis as defined in the UK, which is not associated with development of atopic asthma. 35,42
These subtle but very important differences in inclusion criteria are likely to explain some previous apparently contradictory results in this area. For example, some,36,37 but not all, studies assessing the possible efficacy of nebulised adrenaline in treating young children with acute bronchiolitis in North America have had a positive outcome, but when trialled in the UK and Australia nebulised adrenaline was found to be ineffective and was associated with potential side effects. 38,39 In contrast to experience with bronchodilators, it is possible that HS will be more effective in acute bronchiolitis as understood in the UK in that its putative modes of action would address the dominant mechanisms of airways obstruction in these patients, while it would be expected to have little or no effect in those with wheeze in whom there was significant bronchoconstriction. The studies from Canada and Turkey included children in an outpatient setting with an inclusion criterion of wheeze. Predictably, their mean age was significantly higher than that of the population admitted to UK units with ‘acute bronchiolitis’ and they represent a different phenotype, often referred to in the UK as wheezy bronchitis or virus-associated wheezing.
The observation that the greatest benefit from the use of nebulised HS appears to be in hospitalised infants with a similar age distribution (peak 2–6 months)28–30 to that seen in UK centres22–24 would support the potential of this approach in a well-defined UK population of infants admitted with acute bronchiolitis. Moreover, the mean duration of stay in the control group in all three studies was 3.5 days, again consistent with UK practice. The reduction in duration of inpatient stay in the three positive studies was 25–27%. 28–30
An inexpensive intervention that reduces length of hospital stay by 25% would be of considerable value to the NHS and is likely to have a significant impact on the levels of stress experienced by young parents with an acutely ill infant. Some paediatric units in the UK have already adopted this approach on the basis of the Cochrane review. However, it is possible that publication bias and poor study design has resulted in a false-positive outcome and, if this were the case, there is the potential for an ineffective therapy to creep into practice. Conversely, there is a significant risk that uncritical acceptance of the ambulatory studies may lead to discarding a potentially effective therapy through inclusion of a different patient population given the same clinical diagnosis.
In the light of the uncertainties surrounding this potential valuable therapy, it was essential to undertake an appropriately powered study in a clearly defined UK population to determine whether or not there is indeed reason for UK paediatric units to adopt this approach or if it should simply be added to the list of potential therapies known not to have a significant clinical impact. The importance of undertaking such a study had been recognised both by the Paediatric Respiratory Studies Group and by observers in countries with similar health-care systems and diagnostic criteria, such as Australia. 43
Research objectives
The purpose of the trial was to evaluate the clinical effectiveness and cost-effectiveness of nebulised HS in the treatment of acute bronchiolitis. The trial had one main objective and three secondary objectives.
Primary objective
The primary objective of the study was to assess whether or not the addition of nebulised 3% HS to usual supportive care resulted in a reduction in time to being declared fit for discharge.
Secondary objectives
-
Assessment of the impact of the intervention on other clinical outcomes and the quality of life of infants and carers at 28 days post randomisation.
-
Investigation of the impact on outcomes between those infants with human respiratory syncytial virus (RSV) infection and those with acute bronchiolitis due to other causes, including other viruses (non-RSV).
-
Assessment of the economic impact of the intervention on both the NHS and parents at 28 days post randomisation.
Chapter 2 Methods
There are three sections to this chapter: The SABRE study methods, Health economic methods and Systematic review methods.
This report is concordant with the 2010 Consolidated Standards of Reporting Trials (CONSORT) statement. 44
The SABRE study methods
Trial design
The SABRE study was a multicentre, parallel-group randomised controlled trial (RCT), with a one-to-one allocation ratio and economic evaluation. It aimed to determine whether or not the addition of nebulised 3% HS to usual care resulted in significant (25%) reduction in the duration of hospitalisation of infants with acute bronchiolitis. Subjects were recruited from the paediatric wards and assessment units of the participating centres in England and Wales between October 2011 and December 2013. A table of changes made to the protocol over the course of the project is presented in Appendix 1.
Important changes to methods after trial commencement
Substantial amendment 1
The primary outcome definition for fit for discharge was amended from 12 hours to 6 hours. After discussion with participating centres it was felt that 12 hours did not reflect current practice. This was agreed by the trial management group (TMG) and the chairperson of the trial steering committee (TSC). The time allowed to obtain full informed consent was also amended from 60 minutes to 90 minutes after agreement at a TMG that 90 minutes would be more practical in the clinical setting.
Minor amendment 3
It was clarified that ‘the saline will be discontinued once oxygen therapy has been discontinued’ as this had not been explicitly stated in the protocol previously. Clarification of one of the inclusion criteria was also provided, ‘Requiring supplemental oxygen therapy on admission’. Clarification of the wording for admissions to high-dependency unit (HDU)/ICU was provided, ‘Admission per se of a study participant to the HDU and/or ICU as a result of normal clinical diagnosis of acute bronchiolitis will be reported as an expected event not as [a serious adverse event]’.
Minor amendment 5
We further clarified when saline should be discontinued after discussion at a TMG meeting – the wording was amended from ‘the saline will be discontinued once oxygen therapy has been discontinued’ to ‘the saline will be discontinued once the “fit for discharge” criterion has been met – in air for 6 hours with oxygen saturations of at least 92% and feeding satisfactorily’.
Substantial amendment 6
The protocol was simplified to ensure that eligible babies who were admitted on oxygen were recruited within 90 minutes of admission. The web-based randomisation system was altered to request the time of admission in order to raise an alert if health professionals tried to randomise outside the 90-minute window.
Substantial amendment 7
Clarification of permitted medications for trial participants was provided, to reflect local practice and how these should be recorded. Inclusion criteria amended to clarify which staff can make ‘the decision to admit’ in order to begin the 90-minute consent/randomisation window.
Substantial amendment 8
The eligibility criteria for inclusion of patients in the trial were extended. Previously, in order for patients to be eligible for inclusion in the trial, they were required to be consented and randomised within 90 minutes of a decision to admit a patient to an inpatient ward. This amendment extended the time-based eligibility criteria to 4 hours. This change was proposed for two main reasons: first, a substantial number of included trial patients were retrospectively identified as protocol violations because of being randomised outside this 90-minute cut-off point (approximately 50 out of 172 patients randomised); and, second, there is no clinical rational basis for assuming that patients randomised in less than 90 minutes and patients randomised between 90 minutes and 4 hours are clinically different. Previous inpatient trials of saline in paediatric acute bronchiolitis have not included a strict time-based cut off for inclusion, as was operated in this study. This extension of eligibility was supported by the funder for the study [Health Technology Assessment (HTA) programme] and it also had the support of the study sponsor.
Participants and eligibility criteria
The target population was infants less than 12 months of age admitted to hospital with a clinical diagnosis of acute bronchiolitis and requiring supplemental oxygen as part of routine supportive care. Subjects were recruited from paediatric wards and assessment units from the 10 participating centres between October 2011 and December 2013.
Inclusion criteria
-
Previously healthy infants less than 1 year of age.
-
Admitted to hospital with a clinical diagnosis of acute bronchiolitis, following the UK definition of an infant with an apparent viral respiratory tract infection associated with airways obstruction manifest by hyperinflation, tachypnoea and subcostal recession with widespread crepitations on auscultation. 4 Admission was defined as the point the paediatrician, or paediatric advanced nursing practitioner, made the decision to admit. (The perspective at the time was that advanced nursing practitioners were equivalent of specialty trainee 1 – specialty trainee 3/senior house officer).
-
Requiring supplemental oxygen therapy on admission (either the infant was already in oxygen or oxygen was recommended at the point of admission).
-
Consented and randomised within 4 hours of admission.
Exclusion criteria
-
Had wheezy bronchitis or asthma – children with an apparent viral respiratory infection and wheeze with no or only occasional crepitations.
-
Had gastro-oesophageal reflux (if investigated and diagnosed in hospital).
-
Had previous lower respiratory tract infections (which required assessment in hospital).
-
Had risk factors for severe disease (gestation of < 32 weeks, immunodeficiency, neurological and cardiac conditions, chronic lung disease).
-
Subjects for whom the carer’s English was not fluent and translational services were not available.
-
Required admission to HDUs or ICUs at the time of recruitment.
The principal ethical consideration was that the subject group were unable to provide their own consent, but this is an issue for all paediatric studies involving infants and young children. For this study there were no known risks associated with the intervention and the participating units were very familiar with the ethical challenges in this population.
There was a small chance that participants were involved in other research studies and this question was asked during the informed consent process. Patients for whom the investigating team felt that it would have been inappropriate to include them in the study were excluded on this basis.
Every effort was made to avoid protocol non-compliances caused by the randomisation of participants outside the time window (see Important changes to methods after trial commencement). Participating centres were asked to note the following:
-
In the case that the person consenting was called away in an emergency before randomising, the centre was not to then attempt to randomise the patient if this would take place outside 4 hours from admission.
-
Patients were not to be randomised without written consent. The randomisation system requested that randomising clinicians confirm that the parent/carer has provided consent for participation in the study.
Settings and locations where the data were collected
The data used in this study came from the following sources:
-
for patients who were eligible for randomisation into the study (including patients who were randomised):
-
patient recruitment form, which contained the age, sex and details regarding entry criteria
-
-
for patients who were randomised into the study:
-
randomisation schedule, which contained randomisation codes and allocated intervention group
-
case report form, which contained patient demographics, characteristics at presentation, investigations and events during treatment, assessment of ‘fit for discharge’, resource use and adverse events (AEs) over subsequent 28 days
-
post-discharge data collection forms, comprised of the symptom and health service utilisation diary.
-
Interventions
Infants were randomly allocated to either standard supportive care (control) or standard supportive care plus nebulised 3% HS solution (intervention) using a remote access, computer-generated allocation algorithm.
Three per cent HS is licensed as a medical device in the UK under the brand name MucoClear® (PARI GmbH, Starnberg, Germany) and is indicated for the mobilisation of secretions in the lower respiratory tract in patients with persistent mucus accumulation such as those with acute bronchiolitis or cystic fibrosis. It was presented as a 4-ml plastic ampoule for nebulisation and came in packs of 20 or 60. The product contained no preservatives and had an expiry date of 3 years from manufacture. The product was administered via the PARI Sprint nebuliser (PARI Medical Ltd, Surrey, UK). The dose was 4 ml every 6 hours, in accordance with previously published studies. It was administered by a nurse, with infants inhaling the aerosolised saline. The saline was discontinued once the fit for discharge criteria had been met – in air for 6 hours with oxygen saturations of at least 92% and feeding satisfactorily.
For the purpose of this trial, MucoClear® 3% was sourced by and dispensed locally from each site’s pharmacy department. Although this was not a clinical trial of a medicinal product and therefore not subject to the Medicines for Human Use (Clinical Trials) Regulations 2004,45 the principal investigator in conjunction with his or her local pharmacy department ensured that the principles of good clinical practice (GCP) were applied to ensure robust accountability of the supply and administration of 3% HS.
Other medications
All concomitant medications were recorded. The use of antibiotics, saline nasal drops and HS given after an infant was declared fit for discharge was investigated in additional analyses to assess the impact of these possible confounding factors.
There is no proven drug therapy for the treatment of bronchiolitis. Antipyretics (paracetamol and/or ibuprofen) are recommended for pyrexia and pain management. Nappy rash cream, alginic acid, aluminium hydroxide, magnesium carbonate (Gaviscon®, Reckitt Benckiser), sodium chloride (Dioralyte, A Nattermann & Cie GmbH Cologne, Germany) and lactulose (and related generic medications) were permitted. Saline was continued if bronchiolitis remained the primary diagnosis. Antibiotics were permissible for suspected secondary bacterial infections (e.g. based on radiographic changes). It is routine clinical practice to discontinue antibiotics previously prescribed by general practitioners (GPs). Antibiotics are not recommended in any ‘bronchiolitis’ guideline, including the Scottish Intercollegiate Guidelines Network (SIGN) UK guideline,4 and published data have suggested their use has been associated with a significant level of AE with no discernible benefit.
Outcomes
Table 1 shows the timing of outcome assessments.
| Data collection time points | Before baseline | Baseline | Every 6 hours | After discharge | 28 days after randomisation |
|---|---|---|---|---|---|
| Eligibility criteria applied by doctors | ✓ | – | – | – | – |
| Participant information sheet offered by nurses | ✓ | – | – | – | – |
| Informed consent by doctors or delegated nursing staff | ✓ | – | – | – | – |
| Baseline demographic data | – | ✓ | – | – | – |
| Randomisation | – | ✓ | – | – | – |
| Apply fit for discharge criteria | – | ✓ | ✓ | – | – |
| Diary – symptoms and health service utilisation | – | – | – | ✓ | ✓ |
| ITQoL | – | – | – | – | ✓ |
| Nurse telephone call to confirm diary and health service utilisation | – | – | – | ✓ | ✓ |
| Safety assessments | – | – | ✓ | – | ✓ |
| Ongoing monitoring | – | ✓ | ✓ | ✓ | ✓ |
| Retrospective identification of additional AEs | – | – | – | ✓ | ✓ |
Primary end point
The primary outcome was time to ‘fit for discharge’, and was taken as the period of time from randomisation to when the infant was judged to be feeding adequately (taking > 75% of usual intake orally) and had been in air with oxygen saturations of at least 92% for 6 hours, to reflect clinical practice. 4 An oxygen saturation level of less than 92% as the trigger for starting supplemental oxygen is conservative by US standards. 5 The reasons for choosing time to fit for discharge as the primary end point were twofold: first, its objectivity [actual length of stay (LoS) is influenced by policy, the timing of ward rounds and other features not directly related to bronchiolitis]; and, second, because of its direct relevance both to patients and to service providers.
Secondary end points
-
Actual time to discharge, from randomisation.
-
Admission to ICU/HDU.
-
The number of readmissions and the reasons for readmission within 28 days of randomisation.
-
Duration of respiratory symptoms post discharge and within 28 days of randomisation (see Appendix 2).
-
Health-care utilisation, after discharge and within 28 days of randomisation (see Appendix 2).
-
Infant and parental quality of life, using the Infant Toddler Quality of Life Questionnaire (ITQoL) at 28 days following randomisation. 46
-
AEs.
Fitness for discharge was assessed at baseline, then six times hourly until ‘fit for discharge’ criteria had been met. Research nurses recorded baseline demographic data, co-interventions and outcome data on the case report form. Research nurses also collected basic anonymised details (e.g. age, reason for admission) on all eligible patients to allow completion of a CONSORT flow chart. To assess worsening of condition since discharge and maximise completeness of data, research nurses telephoned parents/guardians approximately 14 days after randomisation. Non-responders after the 28 days were contacted by the research nurse and sent a further ITQoL if appropriate. The involvement of parents/guardians and infants ended at this point.
Readmission was defined as any readmission to hospital within 28 days of randomisation. These have been tabulated by study group and are further classified according to whether or not the readmission was related to the original bronchiolitis or was a new presenting complaint.
The ITQoL is a standardised quality-of-life questionnaire that has been validated for use in infants aged 2 months to 5 years. 46 The questionnaire was completed by a parent or guardian. It covered nine domains – six for the child and three for the parent – each of which was scored from 0 (worst health) to 100 (best health). It had been used successfully in a respiratory illness sample similar to this study, with a response rate of 79.7%. 46
Site trial staff and delegated NHS staff were responsible for recording all AEs that occurred during routine clinical care and making them known to the principal investigator. AEs were recorded on the case report form and database, but did not need to be reported by fax to the sponsor and Clinical Trials Research Unit (CTRU). AEs and serious adverse events (SAEs) were reported regularly in data reports to the oversight committees. Admission per se of a study participant to the HDU and/or ICU as a result of normal clinical diagnosis of acute bronchiolitis was reported as an expected event not as a SAE. The sponsor and CTRU were responsible for assessing the seriousness and reporting to relevant regulatory bodies, where appropriate.
Changes to trial outcomes after the trial commenced, with reasons
Substantial amendment 1
Protocol version 1 (28 June 2011) was amended to update the primary outcome definition for fit for discharge from 12 hours to 6 hours. After discussion with participating centres, it was felt that 12 hours did not reflect current practice. This was agreed by the TMG and TSC chairperson, and implemented in protocol version 0.1 (28 June 2011) before the start of recruitment. In addition, protocol version 4 (17 November 2011) was amended to provide further clarification of when the saline should be discontinued after discussion at a TMG meeting – the wording was amended from ‘the saline will be discontinued once oxygen therapy has been discontinued’ to ‘the saline will be discontinued once the “fit for discharge” criteria have been met – in air for 6 hours with oxygen saturations of at least 92% and feeding satisfactorily’.
Sample size
Based on the current mean time to discharge of around 3 days, we felt that a 25% reduction would be the minimum clinically significant effect, and this was the magnitude of the effect observed in previous studies. 47 As LoS varies considerably across different settings, we used UK Hospital Episode Statistics data as the basis of our sample size justification. Assuming a log-normal distribution, the standard deviation (SD) was estimated at 32 hours. While a similar, or smaller, SD was expected for our primary outcome measure, a slightly inflated SD of 46 hours was used because of uncertainties over its derivation here. In order to have 90% power to detect a 25% difference in time to meeting discharge criteria, the study needed 139 patients per group at a two-sided α-level of 5%. The dropout rate was thought to be negligible for the analysis of the primary outcome measure and therefore a conservative estimate of sample size was 150 patients per group. Overall, 10 centres recruited patients to the study. Targets of 25 participants for district general hospitals and 40 participants for teaching hospitals were set.
Explanation of any interim analyses and stopping guidelines
The Data Monitoring and Ethics Committee (DMEC) charter allowed the study to be stopped prematurely on the grounds of safety or futility based on recommendations from the DMEC or the funder. No formal interim analyses were planned for efficacy and consequently no adjustment was initially made for multiplicity. At the request of the DMEC, one interim analysis was subsequently undertaken in July 2012 to assess efficacy, in order to inform a funding extension decision necessitated by recruitment being slower than expected. An O’Brien–Fleming stopping rule was retrospectively employed in which superiority would be declared if statistical significance was demonstrated at the 1% level in the interim analysis or at the 4.5% level at the final analysis. 48 The DMEC had the authority to recommend that recruitment to the study should be terminated if the primary outcome (time to fitness for discharge) was significantly shorter in the saline group at the α = 1% level of statistical significance.
Method used to generate the random allocation sequence
A central web-based randomisation service delivered by the CTRU was used after patient eligibility and written consent were confirmed. Patients were randomly allocated via the online system to receive either (1) nebulised HS with usual care (n = 150) or (2) usual care (n = 150). The randomisation schedule was computer generated prior to the study by the CTRU Randomisation Service.
Type of randomisation; details of any restriction (such as blocking and block size)
Participants were individually randomised using centralised, web-based randomisation system. Randomisation was conducted in randomly ordered blocks of size 2, 4 or 6, stratified by hospital.
Allocation concealment mechanism
The allocation schedule was concealed through the use of the centralised web-based randomisation service, which also allowed unblinding in the case of emergency. The randomisation sequence was not revealed to any person involved in patient recruitment. The data analysts were blind to treatment allocation until after the statistical analysis plan was finalised, the database locked and the data review completed. All unblinding (emergency and end of trial) would have been automatically logged by the CTRU randomisation system, which would have included the date, time and user responsible.
Sequence generation, enrolment and assignation
The randomisation sequence was computer generated and was not revealed to any person involved in patient recruitment. Recruitment was undertaken by GCP-trained trainee paediatricians, consultants or staff with equivalent training (e.g. GCP-trained paediatric nursing staff responsible for acute admissions). They approached parents/guardians to discuss the study at the point of first contact as soon as the infant had been identified as eligible for the study. Written information was then provided to parents/guardians willing to consider their infant taking part in the study. If agreed, written informed consent was obtained from the parents/guardians.
A flow chart detailing the consent and randomisation procedure can be found in Appendix 3. Owing to the acute nature of the condition, it was not possible to follow a usual time frame of at least 24 hours between receiving information and giving written informed consent. However, every opportunity was taken to ensure that parents/guardians fully understood the implications of taking part in our study. They were invited to ask questions and reflect prior to signing the informed consent document. No payment was offered to participants. The recruiter entered the participant’s details onto an online system and was asked the following:
-
to confirm written consent was received
-
to confirm the participant was in oxygen
-
to enter the time a paediatrician or paediatric advanced nursing practitioner made the decision to admit.
Participants were then randomly allocated via the online system to receive either (1) nebulised HS with usual care (n = 150) or (2) usual care (n = 150). The allocation was explained to participants by the hospital staff member obtaining consent.
Blinding
The study compared the intervention plus usual care with usual care alone, with no placebo. The use of placebo in this setting is ethically problematic as it would result in infants in the placebo arm receiving an intervention that may have a significant effect on outcome. The extra handling involved in nebulised therapy may have a deleterious effect, as has been shown to be associated with the increased handling associated with physiotherapy. 49 Similarly, the placebo agent may cause harm, as has been suggested in previous studies using nebulised distilled water, or may have an unexpected positive impact for an agent, such as NS. 32
The randomisation codes were stored electronically on the CTRU randomisation system. All other electronic data were held separately on the CTRU database system. Access to any data that would unblind the study was limited to members of the CTRU who were independent of the trial. All summaries presented to the DMEC were by treatment group. In order to maintain the blinding of the trial statistician, it was the responsibility of the CTRU to provide this by-treatment group information to the DMEC. No member of the study team had access to unblinded data sets or the unblinded reports until the final analyses.
Statistical methods
The detailed statistical analysis plan can be found in Appendix 4. As time to being declared ready for discharge could be regarded as a survival time, initial differences between groups were assessed using the log-rank test. In order to adjust for centre, a Cox proportional hazards regression model was used with centre fitted as a fixed effect (FE). One centre, Rotherham NHS Foundation Trust, randomised one patient: for the purposes of adjusting for centre, this patient was combined with those recruited from Doncaster and Bassetlaw Hospitals NHS Foundation Trust, given the similarities that exist between these two geographically proximate populations in south Yorkshire. The proportionality of the hazards was assessed by examining the scatter of scaled Schoenfeld residuals against time. 50 This model was further extended by inclusion of viral status (RSV vs. non-RSV) to allow an examination of whether or not viral status had an impact on the effectiveness of treatment. In order to answer this question, the coefficient for the interaction between infection group (RSV vs. non-RSV) and treatment group was calculated together with its 95% confidence interval (CI) and p-value. The collection of RSV test data was not a protocol requirement and such data were not collected at any particular centre except as part of routine practice.
The analysis of actual time to discharge was similar to the primary outcome measure, time to ‘fit to discharge’. Rates of admission to HDU/ICU and readmission rate within 28 days from randomisation were compared using Fisher’s exact test; although it is possible that children could be readmitted multiple times as a result of their single episode of bronchiolitis, it is much more likely that they will be readmitted just once. Thus, the percentage of children readmitted at least once was used for these analyses. In order to test the assumption that the results between groups differed according to RSV status, a logistic regression model was fitted and a coefficient for the interaction between infection group (RSV vs. non-RSV) and treatment group was calculated together with its 95% CIs and p-value. Each of the nine dimensions of the ITQoL used in this study was examined for differences between treatment groups, initially using a t-test. Where the assumptions underlying the t-test do not hold, a Mann–Whitney U-test was used.
It was originally envisaged that symptom duration would be analysed as a survival outcome. However, diary data were returned for only 108 individuals and, of these, data to 28 days were unavailable for almost one-quarter (25 out of 108: 1 at 15 days; 1 at 17 days; 1 at 18 days; 22 at 27 days) and almost one-quarter (24 out of 108) reported that they were still experiencing symptoms at 28 days. Although the majority of infants whose carers stopped sending in data before 28 days were no longer experiencing symptoms, because of the large amount of censoring at 28 days, we defined a new outcome variable that evaluated whether or not the patient was still experiencing symptoms at 28 days. This was analysed using the methods outlined above for binary outcomes.
The trial was originally designed to have a two-sided significance level of α = 5%. As a result of the aforementioned interim analysis, the significance threshold for the final analysis was changed to 4.5% in order to preserve the overall trial-wise significance level at 5%. All CIs remain two-sided 95% intervals. Analyses were conducted using IBM SPSS version 20 (IBM Corporation, Armonk, NY, USA) and SAS version 9.3 (SAS Institute Inc., Cary, NC, USA).
Analysis populations
Four populations were used in the analyses:
-
all screened patients: patients who were screened for eligibility to the study, including those randomised
-
full analysis set (FAS): all randomised patients, with the following exclusions:
-
patients who had previously been randomised, in which case only data relating to the first admission were analysed
-
patients for whom no recorded informed consent was obtained from carers (oral or written)
-
patients whose carers withdrew consent before any study medications were given
-
patients whose carers withdrew consent retrospectively (i.e. requested that all the patient’s data were removed)
-
-
per protocol (PP): the subset of patients in the FAS who did not deviate from the protocol
-
safety: all randomised patients, with the exception of those for whom there was no recorded informed consent.
Summaries based on the FAS and PP populations were on an intention-to-treat basis, with patients assigned to the treatment group as originally randomised. Summaries based on the safety population analysed patients by the actual treatment received. Aside from RSV status (see Statistical methods) no other subgroup analyses were planned.
All outputs presented to the DMEC were based on the FAS population unless otherwise requested.
Patient and public involvement
Study design
At the grant application stage, meetings involving parents confirmed the impression that this disease, affecting very young babies, has a tremendous impact on parents and families. The handling required to administer the nebuliser may occasionally cause the infant to cry, but feedback from the parental involvement meetings indicated that parents do not find this unduly stressful and that parents were keen for their children to participate in the study. Parental involvement meetings identified that the ITQoL was by far the most relevant of the available quality of life instruments. This questionnaire also includes questions relating to the well-being of parents and, as such, allowed us to develop the grant application by removing the parental stress index from our secondary outcome measures.
Study oversight
The independent TSC contained a patient and public involvement representative throughout.
Health economic methods
Background
Nebulised HS is a relatively cheap intervention: approximately £50 for the nebuliser plus 12 doses of saline. Consequently, a large reduction in LoS is not required to suggest that the introduction of nebulised HS may be cost neutral or even cost saving. However, a broader economic evaluation that captures a wider set of costs and also includes patient outcomes is preferred. For example, if the intervention is not cost neutral, it may still be cost-effective once the value of patient health effects are also considered. Alternatively, the intervention may be cost-saving from the perspective of the hospital, but worse symptoms and more associated care following discharge may show the intervention not to be cost-effective.
Overview
A cost–utility analysis (CUA) was undertaken from the NHS perspective, with a time frame of 36 days post randomisation. The economic evaluation was originally designed to have a 28-day follow-up, but one patient in the study had a LoS of 35.7 days, so the time frame was extended to avoid censoring. A longer time frame was relevant, as there are no long-term sequelae associated with acute bronchiolitis. Given the difficulties of measuring utilities in the very young, the CUA was supplemented with a cost–consequences analysis (CCA) which considers the secondary clinical outcome measures alongside costs to allow a more qualitative assessment of cost-effectiveness.
Resource use
The main items of resource use collected were LoS by type of ward and number of readmissions. Nursing time was not included because, although nurse time is required to set up the equipment in the intervention group, increased monitoring time may be needed in patients who are not receiving active treatment. Consequently, a detailed observational study would be required to measure these costs in both arms. It was considered that any cost difference would be small, and inconsequential compared with any difference in LoS, and that calculating any difference would incur huge costs . Furthermore, if there were no difference in LoS, then clinical practice would not change based on small differences in nursing time.
Although other costs were also considered to be negligible in comparison with the inpatient costs, ease of data collection led us to collect data on the use of nebulised saline, concomitant medications and use of primary care services in the 28 days following discharge (e.g. GP or emergency department visits). Inpatient resource use was collected by staff using the same case report forms as those used for the clinical evaluation. Primary care resource use was collected using a patient diary, which was to be completed by the parent or guardian on a daily basis; tick boxes were available for GP contacts, NHS Direct, NHS walk-in centres, minor injury units and the emergency department.
Unit costs
All unit costs used were at 2012/13 price levels. Costs for a day in hospital were based on 2012–13 NHS Reference Costs, nebuliser costs were supplied by one of the participating hospitals, saline and medication costs were taken from the British National Formulary (BNF)51 and primary care costs were taken from NHS reference costs52 and the Unit Costs of Health and Social Care publication produced by the Personal Social Services Research Unit. 53 These are summarised in Table 2.
| Resource | Unit cost (2012/13), £ | Source |
|---|---|---|
| Nebuliser kits | 44 | Study hospital |
| Saline (4 ml) | 0.45 | BNF 65.51 3%, 6% and 7% solutions all have identical prices (£27 for 60 doses) |
| Inpatient day on ward | 547 | Derived from NHS reference costs.52 Activity-weighted average across all non-elective admissions relating to PA15B |
| Inpatient day on ICU | 1946 | Derived from NHS reference costs.52 Activity-weighted average across all paediatric intensive care categories, excluding ECMO/ECLS |
| Inpatient day on HDU | 1061 | Derived from NHS reference costs.52 Activity-weighted average across all paediatric high-dependency care categories |
| GP attendance | 45 | Unit Costs of Health and Social Care 2013.53 Per-patient contact lasting 11.7 minutes, including direct care staff costs and costs of qualifications |
| NHS Direct | 25 | £24, 2010/11 prices.54 Inflated using Hospital and Community Health Care Pay and Prices Index53 |
| Walk-in centre | 41 | Derived from NHS reference costs.52 Activity-weighted average across service type 4, admitted and non-admitted. Category 1 investigation with category 3–4 treatment |
| Minor injuries unit | 88 | Derived from NHS reference costs.52 Activity-weighted average across service type 3, admitted and non-admitted. Category 1 investigation with category 3–4 treatment |
| Emergency department | 118 | Derived from NHS reference costs.52 Activity-weighted average across service types 1 and 2, admitted and non-admitted. Category 1 investigation with category 3–4 treatment |
The cost per day on a ward was derived using the NHS reference costs52 for an admission for paediatric acute bronchiolitis without complications (PA15B) and its associated number of bed-days. Separate costs per day for paediatric intensive care and high-dependency care are available directly from NHS reference costs. 52 The cost for paediatric acute bronchiolitis with complications or comorbidities (PA15A) was not considered relevant as our separate costing of paediatric intensive care and high-dependency care captures the costs of complications.
NHS Direct costs were taken from a report by the Medical Care Research Unit, which used a Department of Health contract value to derive a cost per call. 54 Emergency care unit costs (defined here as walk-in centres, minor injury units and emergency departments) relate to category 1 investigation (e.g. biochemistry) and category 3–4 treatment (e.g. oxygen).
Outcomes
For the CUA, it is usual for utilities to be calculated based on utility instruments, such as the EQ-5D, administered to patients. However, this is not possible for the participants in this trial, and even proxy completion by a parent would not be valid, as the descriptive systems of available utility instruments do not fit well with this patient group. Our original approach was to undertake a separate valuation study to estimate quality-adjusted life-year (QALY) losses related to hospitalisation and recovery and then apply these to the trial data. However, the National Institute for Health Research was not willing to fund this work, so we identified a published estimate relating to hospitalisation in paediatric populations that could be used. 55
Carroll et al. 55 estimated the utility decrement from full health during hospitalisation as 0.05 (with a standard error of 0.01) using the time trade-off technique. This then needs to be combined with the general population value for infants of the same age. The best method for combining comorbidities is considered to be through multiplication of the relevant values. 56 Following a search of the literature, the most relevant general population value using a paediatric utility instrument for the children recruited to the SABRE study was found to be a Health Utilities Index-II survey of 8-year-old Canadian children. The general population value in that study was 0.95 (compared with 0.82 for children of extremely low birthweight). 57 For the SABRE study, therefore, the non-hospitalised utility is 0.95 (taken from Saigal et al. 57), while the hospitalised utility is 0.9025 (calculated as the product of the Saigal et al. 57 and Carroll and Downs55 utility estimates).
Analysis
Differences in costs and QALYs were assessed using non-parametric permutation t-tests that allow robust comparisons of the means in the presence of non-normal data. A permutation test samples from the observed data to calculate the correct distribution of the test statistic under the null hypothesis. Paired cost and QALY data were then bootstrapped with 5000 replications and plotted on the cost-effectiveness plane. The focus of the economic analysis was the resultant cost-effectiveness acceptability curve and the probability that the intervention will be cost-effective at £20,000 per QALY.
The main cost driver was expected to be hospital costs. Deterministic sensitivity analyses were planned for the cost per day on a ward, in intensive care and in high-dependency care. Alternative plausible unit costs to our preferred ward estimate of £547 are £558 per day (with the latter also including complicated admissions) and £425 (derived using excess bed-days costs for uncomplicated admissions). Alternative plausible unit costs to our preferred intensive and high-dependency care estimates of £1946 and £1061 are £1743 and £886, respectively (derived using only the basic care categories within each type of unit).
Probabilistic sensitivity analysis is undertaken through the bootstrapping procedure described previously, thereby incorporating the sampling uncertainty related to all clinical effects and resource use items. Probabilistic sensitivity analysis using distributions around unit costs and utility values is not routinely undertaken in economic evaluations alongside trials.
Where appropriate, missing data were to be imputed using multiple imputation. This assessment would be based on the degree of missingness and the mechanism by which missing data were generated (e.g. random or non-random).
Systematic review methods
Rationale
To produce an overview of the clinical effectiveness of HS for the treatment of inpatients with acute bronchiolitis.
Objective
The objective was to systematically review the evidence relating to the use of nebulised HS in young children, hospitalised for the treatment of acute bronchiolitis. The primary outcome of consideration was whether or not the use of this intervention resulted in a reduced hospital LoS.
Protocol and registration
The PROSPERO registration (registered 3 March 2014 and revised 18 December 2014) outlining the study review protocol can be found in Appendix 5.
Eligibility criteria
Types of participants
Studies involving children up to the age of 2 years who had been hospitalised as the result of an episode of acute bronchiolitis were considered for the review. Confirmation of the presence of RSV was not required for study inclusion as not all cases of bronchiolitis are a result of RSV, and therefore all cases of bronchiolitis regardless of organism have been included.
Types of interventions
Studies evaluating nebulised HS with or without an adjunct bronchodilator treatment given compared with NS or no intervention (control) were considered for the review. These pre-specified groups of studies were as follows:
-
nebulised HS alone versus NS
-
nebulised HS plus a bronchodilator (e.g. salbutamol) versus NS
-
nebulised HS plus a bronchodilator (e.g. salbutamol) versus NS plus same bronchodilator
-
nebulised HS alone or plus a bronchodilator (e.g. salbutamol) versus no intervention.
Groups were based on those that were pre-specified in the Cochrane systematic review carried out by Zhang and colleagues. 58
No restrictions were applied in terms of the concentration, dose or the way the intervention (HS) or control (NS with or without adjunct treatment) was administered in the trials. Studies in which HS was not the principal intervention under review were excluded.
Types of studies
Published and unpublished RCTs and quasi-randomised trials which had completed participant accrual were considered for inclusion. Quasi-randomised trials are those in which the allocation of intervention may not be completely random. Observational studies were excluded.
Only studies available in English were considered, as defined in the protocol. Nine potentially relevant studies in other languages with no English abstract or an unclear English abstract were excluded at the abstract review stage. 59–67 In addition, two studies in other languages were excluded at the full paper review stage. 68,69 The main database searches were performed from the start date of the database to January 2015.
Outcome measures
There were no eligibility restrictions based on outcomes reported. The systematic review’s primary outcome of interest is length of hospital stay calculated via the mean (SD) number of days (LoS) for each arm of each trial, a relevant and meaningful outcome. 70 Secondary outcomes of interest were (1) rate of readmission to hospital; (2) any AEs however described, but particularly tachycardia, hypertension, pallor, tremor, nausea, vomiting and acute urinary retention; and (3) final clinical severity scale (CSS) scores.
Information sources
The electronic databases searched included MEDLINE (via Ovid) (1946 to January 2015), EMBASE (1974 to January 2015), the Cochrane Central Register of Controlled Trials, Google Scholar (Google Inc., Mountain View, CA, USA) (2010 to January 2015) and Web of Science (2010 to January 2015). The full search strategy used in each database is listed in Appendix 6. No restrictions or limits (e.g. age, language and publication date) were applied to any of the databases other than Google Scholar and Web of Science, which were searched for articles from 2010 onwards, when the Cochrane Systematic review by Zhang and colleagues71 was last updated.
The following trial registries were searched, using the terms ‘bronchiolitis’ and ‘hypertonic saline’, to identify any unpublished data: ClinicalTrials.gov; UK Clinical Trials Gateway; Centre for Reviews and Dissemination databases (Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database, HTA Database); controlled-trials.com; centrewatch.com. The National Research Register was searched using the terms ‘bronchiolitis and ‘hypertonic saline’ on 5 March 2014, as the website could not be accessed when the searches were updated. We also hand-searched the journals Chest, Paediatrics and Journal of Paediatrics in January 2015 using the terms HS and bronchiolitis. The reference lists of all eligible trial publications were checked to identify any further published trials.
Search
A range of search strategies were employed, including pearl growing and checking reference lists. 72,73 Pearl growing involved identifying medical subject headings from articles identified from the previous systematic review,25 which were then combined with initial search terms and the searches rerun again. The final search strategy is outlined in Appendix 6. All searches were performed between January 2013 and January 2015. Any duplicated articles were removed from the final list prior to applying the eligibility criteria to the trials to select the studies of interest.
Study selection
After removal of duplicates, two researchers (CM and HC) independently screened titles and abstracts for eligibility; differences were resolved by discussion with DH and ME. If the abstract suggested eligibility, if no abstract was available for a particular citation or if the title was vague and unclear, the full paper was retrieved. Full papers were screened for eligibility in the same way.
Unsuccessful attempts were made to contact trial investigators of two unpublished studies74,75 to request additional unreported data. Although studies would not have been excluded based on the primary outcome, all but three of the studies were appropriate to be included in the meta-analysis of the primary outcome of interest, LoS.
Data collection process
The Cochrane risk of bias assessment tool76 was modified after piloting on one study of known eligibility. Details of the modified data extraction tool can be found in Appendix 6 and Table 3. CM and HC then used the modified tool independently to extract data. Attempts were made to contact study teams for missing data, particularly for five studies74,75,84,88,89 for which data were available only on ClinicalTrials.gov or in abstract form. Owing to the constraints in time, it was not possible to contact all the study authors for each of the items of missing data, for example risk of bias assessment.
| Main author | Country | Patient demographic (infant age, sex, other) | Total number randomised | Baseline imbalances reviewed? | Illness severity | At least 80% allocated followed up in final analysis? | Inclusion/exclusion |
|---|---|---|---|---|---|---|---|
| Giudice et al., 201277 | One centre: Italy | Children aged under 24 months, clinical diagnosis of bronchiolitis | 109 | Yes: no significant differences | Severe; Wang et al.78 mean scores 8.8 (NS group) and 8.5 (HS group) | Yes: 106 of 109 patients followed up (97%) | Exclusion: pre-existing cardiac/pulmonary disease, premature birth (< 36 weeks), previous asthma diagnosis, initial oxygen saturation < 85% or respiratory distress requiring resuscitation |
| Inclusion: first episode of bronchiolitis, oxygen saturations of < 94% in room air, significant respiratory distress (Wang et al.78 CSS score) | |||||||
| Kuzik et al., 200724 | Three centres: one in the United Arab Emirates and two in Canada | Children aged under 18 months, diagnosed with bronchiolitis | 96 | Yes: no significant differences | Moderate severity; RDAI scale mean scores 8.1 (NS group) and 7.8 (HS group) | Yes: 100% included in ITT analysis | Exclusion: previous history of wheezing, cardiopulmonary disease or immunodeficiency, critical illness requiring admission to ICU, use of nebulised HS in last 12 hours or premature birth (< 34 weeks) |
| Inclusion: first episode of bronchiolitis | |||||||
| Tal et al., 200623 | Israel | Children aged up to 12 months, bronchiolitis | 44 | Yes: no significant differences | Moderate severity; baseline Wang et al.78 mean severity scores 7.6 (NS group) and 7.4 (HS group) | Yes: 41 of 44 patients (93%) | Exclusion: cardiac disease, chronic respiratory disease, previous wheezing episode, aged > 12 months, oxygen saturation < 85%, obtunded consciousness and/or progressive respiratory failure needing ventilation |
| Inclusion: clinical presentation of viral bronchiolitis that led to hospitalisation | |||||||
| Luo et al., 201079 | China | Children aged under 24 months (range 12–16.5 months), bronchiolitis | 93 | Yes: no significant differences | Mild to moderate; Wang et al.78 mean scores 5.7 (NS group) and 5.8 (HS group) | Yes: 100% included in final analysis | Exclusion: aged > 24 months, previous wheezing episode, chronic cardiac or pulmonary disease, immunodeficiency, accompanying respiratory failure, requiring mechanical ventilation, having intervention 12 hours before treatment, premature infants |
| Inclusion: first episode of viral bronchiolitis, mild to moderate bronchiolitis | |||||||
| Luo et al., 201180 | China | Children aged under 2 years, bronchiolitis | 126 | Yes: no significant differences | Moderate to severe; Wang et al.78 mean baseline scores 8.5 (NS group) and 8.8 (HS group) | Yes: 112 of 126 patients included in final analysis (89%) | Exclusion: age > 24 months, previous episode of wheezing, chronic cardiac and pulmonary disease, immunodeficiency, accompanying respiratory failure needing mechanical ventilation, inhaled 3% HS 12 hours before treatment, premature birth |
| Inclusion: infants aged < 24 months, first episode of wheezing, admitted to hospital for treatment of moderate to severe bronchiolitis | |||||||
| Mandelberg et al., 200322 | Israel | Children aged up to 12 months, bronchiolitis. Mean age 2.9 months (range 0.5–12 months) | 61 | Yes: no significant differences | Non-severe/moderate; Wang et al.78 mean scores 8.1 (NS group) and 8.29 (HS group) | Yes: 52 of 61 randomised included (85%) | Exclusion: cardiac or chronic respiratory disease, previous wheezing episode, saturation < 85% in room air, obtunded consciousness, progressive respiratory failure needing mechanical ventilation |
| Inclusion: viral bronchiolitis with temperature of > 38 °C leading to hospitalisation | |||||||
| Espelt, 201274 | Argentina | Children aged from 1 to 24 months, bronchiolitis | 100 | Insufficient evidence to say but similar numbers of each sex in each group | Moderate to severe (score of > 5) | Yes: 82 out of 100 randomised included (82%) | Exclusion: chronic respiratory or cardiovascular disease, respiratory failure |
| Inclusion: infants aged 1 to 24 months, first episode of bronchiolitis with CSS score of > 5 and O2 saturation of > 97% | |||||||
| Maheshkumar et al., 201381 | India | Children aged < 24 months: mean age 5.93 months (range 2–12 months). Enrolled within 24 hours of admission | 40 | Unclear: no baseline characteristics table provided | Mild to moderate severity); Wang et al.78 clinical scores between 4 and 8 | Yes: 100% included | Exclusion: pre-existing cardiac disease, previous wheezing episode, severe disease (score of > 8) needing mechanical ventilation, oxygen saturation < 85% on room air, cyanosis, obtunded consciousness and/or progressive respiratory failure |
| Inclusion: first episode of bronchiolitis, moderate distress | |||||||
| Sharma et al., 201382 | India | Children aged from 1 to 24 months | 250 | Yes: no significant differences | Moderate severity; Wang et al. clinical scores 3–6, median scores 6 (both groups) | Yes: 248 of 250 randomised included (99%) | Exclusions: obtunded consciousness, cardiac disease, chronic respiratory disease, previous wheezing episode, progressive respiratory distress needing respiratory support other than oxygen |
| Inclusion: first episode of acute bronchiolitis, hospitalised, CSS score of 3–6 | |||||||
| Al-Ansari et al., 201083 | Qatar | Children aged under 18 months diagnosed with acute bronchiolitis | 187 | Yes: ‘baseline characteristics before enrolment were similar in the three treatment arms’ | Moderate to severe bronchiolitis. BSS 5.77 (0.9% NS group) 6.16 (3% HS group) 5.65 (5% HS group) | Yes: 171 out of 187 included (91%) | Exclusion: < 34 weeks’ gestation, history of wheezing, steroid use within 48 hours, obtundation and progressive respiratory failure requiring ICU admission, apnoea within 24 hours, oxygen saturation < 85% in air, chronic lung disease, congenital heart disease, immunodeficiency |
| Inclusion: upper respiratory tract infection, wheezing and/or crackles on auscultation, BSS (> 4) | |||||||
| Nemsadze et al., 201384 | Georgia | Children aged 2–24 months with bronchiolitis | 42 | Yes: no significant differences | Mild to moderate (bronchiolitis clinical score) | Unclear – abstract only | Unclear – abstract only |
| Pandit et al., 201385 | One centre: India | Children aged 2–12 months diagnosed with acute bronchiolitis | 100 | Yes: no significant differences | Moderate/severe. Severity RDAI scale mean baseline scores 11.7 (NS group) and 12 (HS group) | Yes: 100% included | Exclusion: previous wheezing and respiratory distress, family history of asthma, atopy, congenital heart disease, ventilation as newborn infant, patients with shock, seizures, heart rate (> 180 beats per minute), respiratory rate (> 100 breaths per minute) and in respiratory failure, consolidation lung on radiograph |
| Inclusion: children age 2–12 months admitted with acute bronchiolitis | |||||||
| Teunissen et al., 201486 | 12 centres: the Netherlands | Children aged 0–24 months with viral bronchiolitis | 292 | Yes: no significant differences | Mild to moderate; Wang et al.78 mean scores at baseline 6.2 (0.9% NS group), 6.2 (3% HS group) and 6.2 (6% HS group) | Yes: 247 out of 292 analysed (85%) | Exclusion: excluded if Wang et al.78 score improved by at least two points after inhalation, congenital heart disease, chronic pre-existent lung disease, T-cell immunodeficiency, corticosteroid treatment and previous wheezing, eczema or food allergy |
| Inclusion: children aged 0–24 months admitted to hospital with viral bronchiolitis with a Wang et al.78 score of > 3 | |||||||
| Everard et al., 201487 | UK | Children aged up to 12 months with acute bronchiolitis | 317 | Yes: no significant differences | Severe bronchiolitis | Yes: 291 out of 317 included (92%) | Exclusion: wheezy bronchitis or asthma – children with an apparent viral respiratory infection and wheeze with no or occasional crepitations, reflux, previous lower respiratory tract infections (requiring assessment in hospital), risk factors for severe disease (< 32 weeks’ gestation, immunodeficiency, neurological and cardiac conditions, chronic lung disease), subjects where the carer’s English is not fluent and translational services are not available |
| Inclusion: previously healthy infants less than 1 year of age, admitted with acute bronchiolitis, consented and randomised within 4 hours of admission by a medical paediatrician | |||||||
| Silver, 201488 | USA | Children aged 0 to 12 months, with bronchiolitis | 227 | Yes: no significant differences | RDAI completed prior to first study treatment: mean 3.5 (NS group), 3.2 (HS group) | Yes: 190 out of 227 included (84%) | Exclusion: status asthmaticus, chronic cardiopulmonary disease, trisomy 21 and immunodeficiency or transplant recipient or neuromuscular disease. Admission directly to ICU, previous use of nebulised HS less than 12 hours prior to presentation and previous enrolment in the study in 72 hours prior to presentation |
| Inclusion: patients up to 12 months of age, admitted to hospital with bronchiolitis | |||||||
| Ozdogan et al., 201489 | Turkey | Infants aged 1 to 24 months, admitted with acute bronchiolitis | 69 | Yes: the demographic features were similar in each group | Respiratory assessment score, mild to moderate bronchiolitis | Unclear: abstract only | Exclusion: unclear – abstract only |
| Inclusion: infants 1–24 months of age admitted to hospital with acute bronchiolitis | |||||||
| Sosa-Bustamante, 201475 | Mexico | Children aged 2 to 24 months | Unclear: ClinicalTrials.gov only | Unclear: ClinicalTrials.gov only | Respiratory Distress Scale of the Sant Joan de Déu Hospital, moderate to severe bronchiolitis | Unclear: ClinicalTrials.gov only | Exclusion: subjects with history of previous wheezing, asthma, or who have received bronchodilator treatment before the present illness. Patients with chronic lung disease, heart disease, with congenital or acquired anatomic abnormalities of the airway |
| Inclusion: children aged 2–24 months, first episode of wheezing associated with respiratory distress, history of upper respiratory tract infections and evaluation of respiratory difficulty with the Respiratory Distress Scale of the Hospital Sant Joan de Déu from 6–16 at entry points | |||||||
| Ojha et al., 201490 | Nepal | Children aged 6 weeks to 24 months | 72 | Yes: baseline characteristic were comparable | Clinical scoring of respiratory distress of > 4. Mean scores at baseline 7.36 (0.9% NS group) and 8.08 (3% HS group) | Yes: 59 out of 72 included (82%) | Exclusion: previous episode of wheezing, chronic cardiac and pulmonary disease, immunodeficiency, respiratory failure, mechanical ventilation, inhaling nebulised 3% saline and salbutamol 12 hours before treatment, premature infants (less than 34 weeks), oxygen saturation < 85% on room air |
| Inclusion: children aged 6 weeks to 24 months presenting with bronchiolitis for the first time |
Data items
-
Study overview (country, year).
-
Participant characteristics (age, number randomised, baseline imbalances assessed by the authors in trials, withdrawals, % allocated completing follow-up, illness severity, eligibility).
-
Intervention and control group details (number randomised in each group, intervention details: duration, delivery, other drugs and compliance).
-
Outcomes data:
-
Continuous outcomes: LoS (mean LoS, SD and number of patients in each group, measured by who); CSS score (mean final CSS score, SD, number of patients for both groups).
-
AE data as available.
-
Risk of bias in individual studies
The Cochrane risk of bias assessment tool allows reported trial results to be differentiated from the methods used, with lower risk studies associated with more conservative effect estimates of risk. 91 CM and HC independently graded the quality of trials as ‘low’ where there is low risk, ‘high’ where there is high risk of bias or ‘unclear’ where the risk is unclear. Discrepancies were resolved by discussion. Risk of bias assessment was conducted at both study and outcome level. The Cochrane Handbook for Systematic Reviews of Interventions,76 chapter 8.5.2, was used as a guide to make judgements on the methodological quality of the trials for the following domains of bias: sequence generation; allocation concealment; blinding, completeness of outcome data; and selective reporting. A supporting statement for each bias domain was extracted where a grade is given. Where an unclear grade is given, effort was made to obtain further information to categorise the trial by contacting the trial authors or searching for the study protocol to identify sources of reporting bias. Risk of bias data were entered into the Cochrane Review Manager (RevMan version 5.2, Cochrane, The Nordic Cochrane Centre, Copenhagen, Denmark) tool for analysis. Both a risk of bias graph and risk of bias summary were derived from the data and are reported in the results section.
Summary measures
All outcomes were continuous; the summary measure was the median difference and its associated 95% CI.
Synthesis of results
Analyses were undertaken using both RevMan version 5.2 and Stata version 12.1 (StataCorp LP, College Station, TX, USA). A FE model was used to combine (unstandardised) median differences between HS and the control. The FE model assumes that the LoS outcome would estimate the same effect size in each of the studies and weights studies according to their precision (standard error), which in turn relates to the sample size: the greater the sample size, the greater the weight assigned to the trial. 92 In addition, a second analysis was undertaken using the random-effects (RE) approach of DerSimonian and Laird,79 which incorporates the variability among trial estimates into the weighting of the trials.
Three-armed trials, which compared more than one concentration of HS with a control, were included in the review. The Cochrane Handbook for Systematic Reviews of Interventions recommends combining the active arms, but notes that heterogeneity can be better assessed by keeping dose arms separate. 76 As high levels of heterogeneity were identified in our review (see Chapter 5, Primary outcome: results and synthesis), this was the approach we adopted. To avoid double-counting of control patients, in three-arm trials control group numbers were divided by 2.
Mean final (post-treatment) CSS scores for participants in each arm of the trial were also extracted. A scoring system devised by Wang et al. 78 was used to assign CSS scores at baseline. Some of the studies reported a final CSS score for both the intervention and the control groups.
Data on AEs were synthesised narratively.
We used the I2-statistic to measure statistical heterogeneity with the following guidance on interpretation from the Cochrane Handbook:76
-
I2-statistic of 0–40% = heterogeneity may not be important
-
I2-statistic of 30–60% = may mean moderate heterogeneity
-
I2-statistic of 50–90% = may mean substantial heterogeneity
-
I2-statistic of 75–100% = considerable heterogeneity
-
Overlapping CIs are an indication of lower heterogeneity within the trials.
Forest plots also display the chi-squared test to test for statistical heterogeneity, with corresponding p-values at or below the threshold of 0.1 conventionally indicating a statistically significant heterogeneity. 70
Risk of bias across studies
A funnel plot was generated to explore the possibility of publication bias in the primary outcome, LoS, for all trials. The main group was then split into subgroups to investigate reasons for heterogeneity.
Heterogeneity was explored by separating the group by comparator based on the following:
Intervention comparator:
-
nebulised HS alone versus NS alone (n = 4 trials)
-
nebulised HS plus a bronchodilator (e.g. salbutamol) versus NS alone (n = 0 trials)
-
nebulised HS plus a bronchodilator (e.g. salbutamol) versus NS plus same bronchodilator (n = 12 trials)
-
nebulised HS alone or plus a bronchodilator versus no treatment (n = 1 trial).
Further analyses were performed to separate the type of bronchodilator administered, for example beta-2 agonist compared with adrenaline. The subgroups for these are listed below:
-
nebulised HS alone versus NS alone (n = 4 trials)
-
nebulised HS plus a bronchodilator (e.g. salbutamol) versus NS alone (n = 0 trials)
-
NEBULISED HS plus a beta agonist versus NS plus same beta agonist (n = 7 trials)
-
nebulised HS plus adrenaline versus NS plus same adrenaline (n = 5 trials)
-
nebulised HS alone or plus a bronchodilator versus no treatment (n = 1 trial).
Outcome reporting bias arises from selectively picking outcome measures to report for the study based on the results achieved. 93 Smaller, lower-quality trials may in themselves introduce bias whereas larger, more expensive studies which may be more methodologically sound are more likely to be published even with negative results. 94 Using methods suggested by Dwan and colleagues,95 we also assessed the risk of outcome reporting bias.
Chapter 3 Trial results
Recruitment and participant flow
Participants who were randomly assigned, received intended treatment and were analysed for the primary outcome
Between 26 October 2011 and 23 December 2013, the trial recruited and randomised 317 participants, with 158 patients allocated to the nebulised 3% HS group and 159 allocated to usual care (Figures 1 and 2). There were five patients from the nebulised 3% HS group who did not receive the intended treatment. Of the 317 patients randomised, 26 of these patients were excluded because they were randomised when ineligible and for one patient primary outcome data were unavailable because their medical notes were lost; therefore, 290 patients were included in the primary outcome analysis.
FIGURE 1.
Participant recruitment curve.

FIGURE 2.
The CONSORT flow diagram. ITT, intention to treat.

Losses and exclusions after randomisation
The number of post-randomisation exclusions together with the reason for exclusion is displayed in Table 4. Overall, 26 patients were excluded from the study: 16 from the intervention group and 10 from the control group.
| Post-randomisation exclusion reasons | Standard care plus (intervention group) | Standard care (control group) |
|---|---|---|
| Randomised outside the 4-hour recruitment window | 2 | 4 |
| Received HS prior to randomisation | 3 | 0 |
| Verbal consent was obtained prior to randomisation with written consent being obtained after randomisation | 3 | 0 |
| No decision to admit prior to randomisation | 1 | 0 |
| No recommendation for oxygen at the point of admission | 1 | 0 |
| Previously investigated and diagnosed in hospital with reflux | 1 | 0 |
| Previous lower respiratory tract infection requiring assessment in hospital | 4 | 6 |
| Patient’s study documentation was lost | 1 | 0 |
Of the 317 randomised patients, 26 were excluded, as described previously. Of the remaining 291 patients, five were not included in the PP analysis for the following reasons: four withdrew before receiving any HS/intervention and one patient’s medical notes were lost (therefore no treatment data were available). Of the remaining patients in the PP analysis, 32 withdrew from treatment having received at least one dose of HS and three withdrew from the study, two from the standard care group and one from the standard care plus intervention group.
Dates defining the periods of recruitment and follow-up
The trial consisted of three recruitment seasons. The study recruited patients during winter 2011/12, winter 2012/13 and from October 2013 to December 2013. Patients were followed up for a period of 28 days after randomisation to collect data on readmissions, duration of respiratory symptoms post discharge, health-care utilisation post discharge and infant and parental quality of life.
Why the trial ended or was stopped
The trial closed to recruitment after reaching the accrual target on 23 December 2013.
Baseline data
Table 5 shows the characteristics of the recruited and non-recruited patients, the former being further subdivided according to whether or not the patient was subsequently excluded. There were no notable differences between these groups in terms of age or sex.
| Variable | Non-recruited | Recruited | ||
|---|---|---|---|---|
| Total (n = 455) | Total (n = 317) | Included (n = 291) | Excluded (n = 26) | |
| Site, n (%) | ||||
| Alder Hey Children’s NHS Foundation Trust | 66 (14.5) | 63 (18.8) | 58 (19.9) | 5 (19.2) |
| Bradford Teaching Hospitals NHS Foundation Trust | 67 (14.7) | 38 (12.0) | 34 (11.7) | 4 (15.4) |
| University Hospital of North Staffordshire NHS Trust | 21 (4.6) | 63 (19.9) | 59 (20.3) | 4 (15.3) |
| Doncaster and Bassetlaw Hospitals NHS Foundation Trust | 19 (4.1) | 12 (3.8) | 12 (4.0) | – |
| Calderdale and Huddersfield NHS Foundation Trust | 24 (5.2) | 18 (5.7) | 15 (5.2) | 3 (11.5) |
| Leeds Teaching Hospitals NHS Trust | 136 (29.2) | 38 (12.0) | 35 (12.0) | 3 (11.5) |
| Rotherham NHS Foundation Trust | 4 (0.8) | 1 (0.3) | 1 (0.3) | – |
| Sheffield Children’s NHS Foundation Trust | 41 (9.0) | 54 (17.0) | 48 (16.5) | 6 (23.1) |
| University Hospital of Wales | 8 (1.7) | 21 (6.6) | 20 (6.9) | 1 (3.9) |
| Oxford University Hospitals NHS Trust | 69 (15.2) | 9 (2.8) | 9 (3.1) | – |
| Age (months) | ||||
| Mean (SD) | 3.5 (2.8) | 3.5 (2.7) | 3.4 (2.7) | 4.7 (3.3) |
| Median (IQR); range | 2.5 (1.2–5.0); 0.2–12.0 | 2.5 (1.2–5.0); 0.2–11.5 | 2.5 (1.2–4.8); 0.2–11.5 | 4.1 (2.4–7.0); 0.2–11.5 |
| Sex, n (%) | ||||
| Male | 264a (58.4) | 172 (54.6) | 158 (54.3) | 14 (58.3) |
| Female | 188a (41.6) | 143 (45.4) | 133 (45.7) | 10 (41.7) |
Baseline characteristics are shown in Table 6. There were no notable differences between the study groups but the control group was slightly older, heavier and contained more males.
| Variable | Standard care plus intervention (n = 142) | Standard care (control) (n = 149) |
|---|---|---|
| Site, n (%) | ||
| Alder Hey Children’s NHS Foundation Trust | 30 (21.1) | 28 (18.8) |
| Bradford Teaching Hospitals NHS Foundation Trust University | 16 (11.3) | 18 (12.1) |
| Hospital of North Staffordshire NHS Trust | 29 (20.4) | 30 (20.1) |
| Doncaster and Bassetlaw Hospitals NHS Foundation Trust | 6 (4.2) | 6 (4.0) |
| Calderdale and Huddersfield NHS Foundation Trust | 6 (4.2) | 9 (6.0) |
| Leeds Teaching Hospitals NHS Trust | 18 (12.7) | 17 (11.4) |
| Rotherham NHS Foundation Trust | – | 1 (0.7) |
| Sheffield Children’s NHS Foundation Trust | 23 (16.2) | 25 (16.8) |
| University Hospital Wales | 10 (7.0) | 10 (6.7) |
| Oxford University Hospitals NHS Trust | 4 (2.8) | 5 (3.4) |
| Age (months) | ||
| Mean (SD) | 3.3 (2.6) | 3.4 (2.8) |
| Median (IQR); range | 2.3 (1.3–5.0); 0.3–11.5 | 2.5 (1.5–4.8); 0.3–11.5 |
| Birthweight (kg) | ||
| Mean (SD) | 3.31 (0.77) | 3.36 (0.78) |
| Median (IQR); range | 3.25 (2.90–3.70); 1.87–9.40 | 3.37 (3.0–3.70); 1.81–8.20 |
| Missing, n | 2 | 3 |
| Weight at presentation (kg) | ||
| Mean (SD) | 5.61 (1.84) | 5.98 (2.00) |
| Median (IQR); range | 5.10 (4.10–6.80); 2.20–11.20 | 5.70 (4.56–7.15); 2.60–11.9 |
| Gestation (weeks) | ||
| Mean (SD) | 39.3 (1.7) | 39.2 (1.8) |
| Median (IQR); range | 39.9 (38.0–40.3); (33.0–42.0) | 40.0 (38.0–40.6); (32.7–42.4) |
| Sex, n (%) | ||
| Male | 73 (51.4) | 85 (57.0) |
| Female | 69 (48.6) | 64 (43.0) |
| Ethnicity, n (%) | ||
| Caucasian | 117 (82.4) | 123 (83.1) |
| Asian or Asian British | 17 (12.0) | 13 (8.8) |
| Mixed | 4 (2.8) | 4 (2.7) |
| Black or Black British | – | 3 (2.0) |
| Other | 4 (2.8) | 5 (3.4) |
| Missing, n | – | 1 |
| Smoker in household | ||
| No, n (%) | 81 (58.3) | 86 (57.7) |
| Yes, n (%) | 58 (41.7) | 63 (42.3) |
| Missing, n | 3 | – |
| Infant in childcare | ||
| No, n (%) | 132 (94.3) | 133 (93.0) |
| Yes, n (%) | 8 (5.7) | 10 (7.0) |
| Missing, n | 2 | 6 |
| Feeding on admission | ||
| Breast, n (%) | 27 (19.0) | 24 (16.2) |
| Bottle, n (%) | 103 (72.5) | 120 (81.1) |
| Breast and bottle, n (%) | 12 (8.5) | 4 (2.7) |
| Missing, n | – | 1 |
| Number of siblings | ||
| 0, n (%) | 33 (23.2) | 27 (18.1) |
| 1, n (%) | 52 (36.6) | 62 (36.6) |
| 2, n (%) | 33 (23.2) | 29 (19.5) |
| 3, n (%) | 14 (9.9) | 15 (10.1) |
| 4, n (%) | 7 (4.9) | 10 (6.7) |
| 5 or more, n (%) | 3 (2.1) | 6 (4.0) |
| Mean (SD) | 1.45 (1.3) | 1.6 (1.4) |
| Median (IQR); range | 1 (1–2); 0–7 | 1 (1–2); 0–7 |
| First-degree relative with asthma | ||
| No, n (%) | 79 (57.7) | 77 (54.6) |
| Yes, n (%) | 58 (42.3) | 64 (45.4) |
| Missing, n | 5 | 8 |
| First-degree relative with eczema | ||
| No, n (%) | 91 (66.9) | 81 (57.4) |
| Yes, n (%) | 45 (33.1) | 60 (42.6) |
| Missing, n | 6 | 8 |
| First-degree relative with hay fever | ||
| No, n (%) | 89 (65.4) | 82 (58.6) |
| Yes, n (%) | 47 (34.6) | 58 (41.4) |
| Missing, n | 6 | 9 |
| Referral route, n (%) | ||
| GP (referral) | 60 (42.3) | 70 (47.0) |
| ED (walked in) | 41 (28.9) | 47 (31.5) |
| ED (ambulance) | 30 (21.1) | 25 (16.8) |
| Other | 11 (7.7) | 7 (4.7) |
| Previous conditions, n (%) | ||
| Bronchiolitis (non-hospital diagnosed) | 6 (4.2) | 6 (4.0) |
| Reflux (non-hospital diagnosed) | 4 (2.8) | 7 (4.7) |
| Respiratory problems | 2 (1.4) | 3 (2.0) |
| Heart murmur | 4 (2.8) | 2 (1.3) |
| Jaundice | 2 (1.4) | 5 (3.4) |
| Antibiotics, n (%) | ||
| Prior to admission | 14 (9.9) | 9 (6.0) |
| In hospital | 29 (20.4) | 24 (16.1) |
| Antireflux medications, n (%) | ||
| Prior to admission | 3 (2.1) | 7 (4.7) |
| In hospital | 3 (2.1) | 8 (5.4) |
| Ipratropium, n (%) | ||
| Prior to admission | 4 (2.8) | 4 (2.7) |
| In hospital | 6 (4.2) | 3 (2.0) |
| Oral steroids, n (%) | ||
| Prior to admission | 2 (1.4) | 0 (0) |
| In hospital | 3 (2.1) | 1 (0.7) |
| Beta-2 agonist, n (%) | ||
| Prior to admission | 4 (2.8) | 10 (6.7) |
| In hospital | 10 (7.0) | 4 (2.7) |
| Intravenous fluids in hospital | 9 (6.3) | 7 (4.7) |
| Induction therapy in hospital | 3 (2.1) | 2 (1.3) |
| Sedatives in hospital | 7 (4.9) | 4 (2.7) |
| Other medications in hospital | 10 (7.0) | 10 (6.7) |
Seven out of 10 centres collected RSV test data routinely, if not always completely. The viral status by season is shown in Table 7.
| Viral status | Season 1 (2011–12; N = 134) | Season 2 (2012–13; N = 111) | Season 3 (2013–14; N = 46) |
|---|---|---|---|
| RSV+, n (%) | 84 (63) | 64 (58) | 31 (67) |
| Non-RSV+, n (%) | 13 (10) | 12 (11) | 2 (4) |
| No virus identified, n (%) | 3 (2) | 3 (2) | 0 (0) |
| Not tested, n (%) | 34 (25) | 32 (28) | 13 (28) |
Numbers analysed
In the intention-to-treat (ITT) population, in which patients were analysed by their original assigned groups, there were 142 participants in the HS arm and 149 in the control arm. In one patient in the intervention group, date and time of fitness for discharge were not recorded and are not included in any of these analyses, but the patient was known to have been discharged on day 6 (i.e. between 120 and 144 hours); this LoS is similar enough to the LoS of the other trial participants that the missing data is unlikely to have had any material impact. In the PP analysis, five participants did not receive treatment as scheduled and so were removed, leaving 136 participants in the HS arm with complete data; 149 remained in the control arm.
Outcomes and estimation
There was no evidence of any difference between the two treatment arms in the primary outcome, the time to being declared fit for discharge [hazard ratio (HR) 0.95, 95% CI 0.75 to 1.20; p = 0.66], or in the time to actual discharge (HR 0.97, 95% CI 0.76 to 1.23) (Table 8 and Figures 3 and 4). In absolute terms, the median difference between HS and control (adjusted for site) was 2.5 hours (95% CI –13.8 to 18.7 hours) for time to discharge and 0.5 hours (95% CI –18.0 to 19.1 hours) for actual discharge. For both groups, the time to being declared fit for discharge was nearly 76 hours from admission and the time to actual discharge was nearly 89 hours.
| Variable | Intervention (n = 142) | Control (n = 149) | HR (95% CI) | p-value |
|---|---|---|---|---|
| Time to fit for discharge | ||||
| Mean (SD) | 90.4 (73.2) | 88.9 (67.9) | – | – |
| Median (IQR); range | 75.6 (46.1–113.3); 7.1–576.1 | 75.9 (45.5–121.0); 6.8–565.9 | – | – |
| Median (95% CI) | 75.6 (67.8 to 83.3) | 75.9 (65.7 to 86.1) | – | – |
| Univariate analysis (log-rank test) | – | – | 0.99 (0.79 to 1.24) | 0.97 |
| HR (Cox regression, adjusted for centre) | – | – | 0.95 (0.75 to 1.20) | 0.66 |
| Missing, n | 1 | 0 | – | – |
| Median (95% CI) time to fit for discharge by viral status | ||||
| RSV+ | 79.1 (66.3 to 98.2) | 80.3 (68.9 to 96.4) | 0.94 (0.70 to 1.26) | 0.68 |
| RSV– | 50.8 (27.1 to 75.8) | 48.8 (21.4 to 115.1) | 1.09 (0.50 to 2.39) | 0.83 |
| Time to discharge | ||||
| Mean (SD) | 100.6 (76.9) | 101.3 (84.4) | – | – |
| Median (IQR); range | 88.5 (51.6–120.9); 16.6–595.4 | 88.7 (50.9–123.6); 4.2–857.4 | – | – |
| Median (95% CI) | 88.5 (74.6 to 102.5) | 88.7 (76.9 to 100.5) | – | – |
| Univariate analysis (log-rank test) | – | – | 1.02 (0.81 to 1.28) | 0.86 |
| HR (Cox regression, adjusted for centre) | – | – | 0.97 (0.76 to 1.23) | 0.80 |
| Missing, n | 1 | 0 | – | – |
| Median (95% CI) time to fit for discharge by viral status | ||||
| RSV+ | 91.7 (72.7 to 100.2) | 90.8 (76.0 to 108.7) | 0.88 (0.64 to 1.21) | 0.43 |
| RSV– | 72.2 (42.3 to 101.0) | 70.2 (40.2 to 124.1) | 0.86 (0.34 to 2.17) | 0.75 |
FIGURE 3.
Cumulative survival plot for time to being declared fit for discharge.

FIGURE 4.
Cumulative survival plot for actual time to discharge.

Figures 5 and 6 show the time to discharge by treatment and RSV status [RSV positive (RSV+) vs. RSV negative (RSV–)]. RSV+ was associated with prolonged time to fitness to discharge and (less strongly) with time to actual discharge, but there was no indication that HS had a differential effect in relation to RSV status.
FIGURE 5.
Cumulative survival plot for time to being declared fit for discharge by study arm and viral status.

FIGURE 6.
Cumulative survival plot for time to being declared fit for discharge.
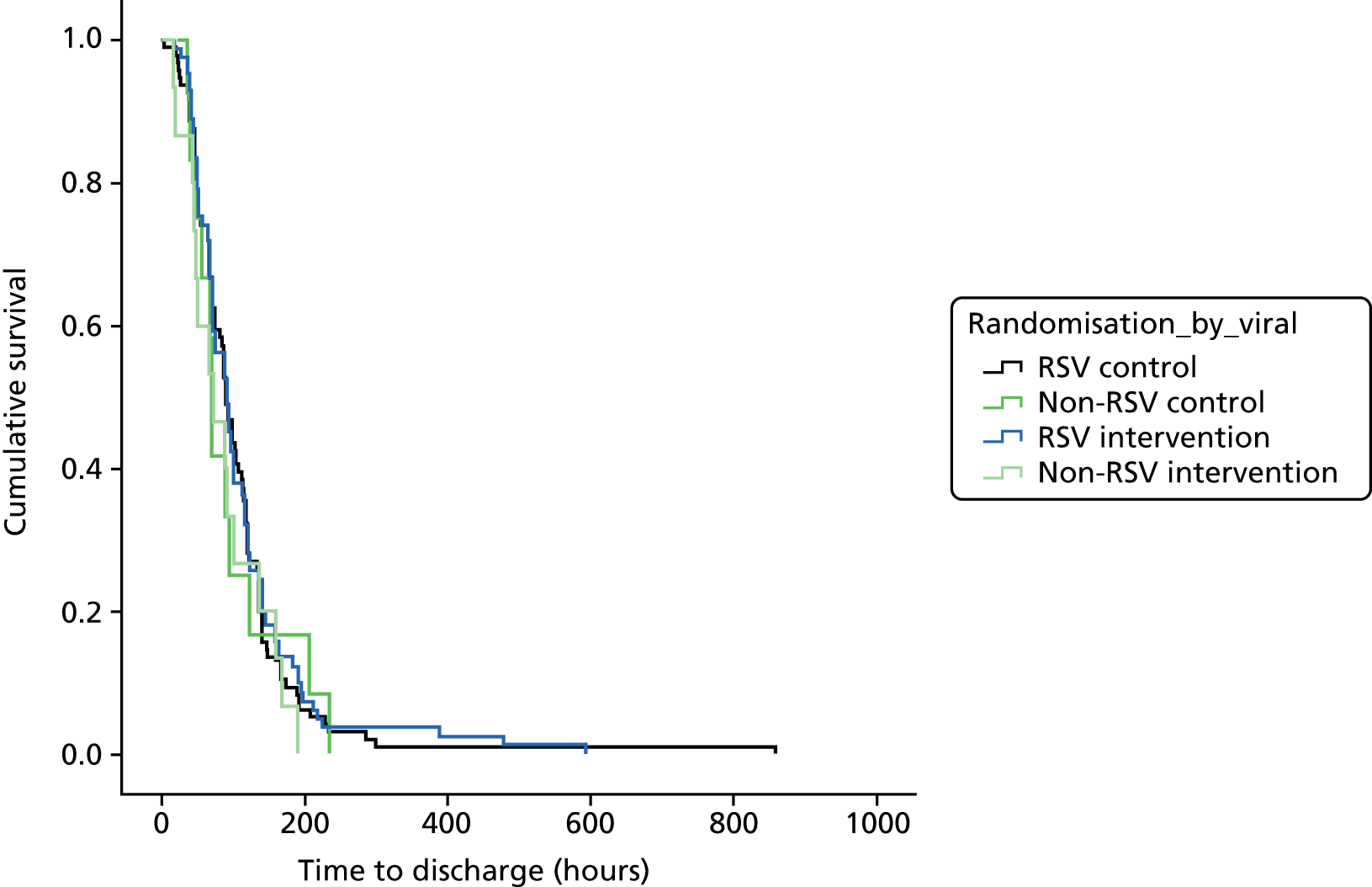
Admission to HDU/ICU and readmission rates are displayed in Table 9. There was a lack of evidence to suggest that there was a difference between treatment groups in terms of the numbers admitted to HDU/ICU or readmitted within 28 days of randomisation. In addition, as there was little evidence that the effect differed between treatment group by RSV status for readmission rates, there was some evidence of an interaction for admission to ICU/HDU, although the numbers were small and the study was not powered to detect this (RSV+ 3.7%, 95% CI –5.2% to 12.6% vs. non-RSV –26.7%, 95% CI –56.2% to 2.8%; p = 0.07).
| Variable | Intervention | Control | Difference (%), (95% CI) | Odds ratio (95% CI) | p-value | ||
|---|---|---|---|---|---|---|---|
| n/N | % admitted (95% CI) | n/N | % admitted (95% CI) | ||||
| Admitted to ICU/HDU | |||||||
| FAS | 12/142 | 8.5 (4.4 to 14.3) | 15/149 | 10.1 (5.7 to 16.1) | –1.6 (–8.3 to 5.0) | 0.96 (0.43 to 2.13) | 0.91 |
| PP | 12/137 | 8.8 (4.6 to 14.8) | 15/149 | 10.1 (5.7 to 16.1) | –1.3 (–8.1 to 5.5) | 0.85 (0.39 to 1.90) | 0.71 |
| RSV vs. non-RSV | |||||||
| RSV | 10/83 | 12.1 (5.9 to 21.0) | 8/96 | 8.3 (3.7 to 15.8) | 3.7 (–5.2 to 12.6) | 1.51 (0.57 to 4.02) | 0.41 |
| Non-RSV | 1/15 | 6.7 (0.2 to 32.0) | 4/12 | 33.3 (9.9 to 65.1) | –26.7 (–56.2 to 2.8) | 0.14 (0.01 to 1.51) | 0.11 |
| Readmitted with 28 days | |||||||
| FAS | 4/128 | 3.1 (0.9 to 7.8) | 7/140 | 5.0 (2.0 to 10.0) | –1.9 (–6.6 to 2.8) | 0.61 (0.18 to 2.14) | 0.44 |
| PP | 4/124 | 3.2 (0.9 to 8.1) | 7/140 | 5.0 (2.0 to 10.0) | –1.8 (–6.5 to 3.0) | 0.63 (0.18 to 2.22) | 0.48 |
| RSV vs. non-RSV | |||||||
| RSV | 2/72 | 3.8 (0.3 to 9.7) | 6/91 | 6.6 (2.5 to 13.8) | –3.8 (–10.2 to 2.5) | 0.40 (0.08 to 2.07) | 0.28 |
| Non-RSV | 2/15 | 13.3 (1.7 to 40.5) | 0/10 | 0.0 (0.0 to 30.9) | 13.3 (–3.9 to 30.5) | – | – |
| Symptoms to 28 days | |||||||
| FAS | 9/53 | 17.0 (6.9 to 27.1) | 15/55 | 27.3 (15.5 to 39.0) | –10.3 (–25.8 to 5.2) | 0.55 (0.22 to 1.38) | 0.20 |
| PP | 9/52 | 17.3 (7.0 to 27.6) | 15/55 | 27.3 (15.5 to 39.0) | –10.0 (–25.6 to 5.7) | 0.56 (0.22 to 1.42) | 0.22 |
| RSV vs. non-RSV | |||||||
| RSV | 4/29 | 13.8 (1.2 to 26.3) | 9/34 | 26.4 (11.6 to 41.3) | –12.7 (–32.1 to 6.8) | 0.44 (0.12 to 1.63) | 0.22 |
| Non-RSV | 1/3 | 33.3 (0.0 to 86.7) | 2/5 | 40.0 (0.0 to 82.9) | –6.7 (–75.2 to 61.8) | 0.75 (0.04 to 14.97) | 0.85 |
The ITQoL results are displayed in Table 10. There were no important differences between treatment group scores on any of the ITQoL dimensions and a lack of evidence that outcomes differed between groups according to RSV status (Figure 7).
| Variable | Intervention | Control | Difference (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| n (crude) | Mean (95% CI) | n (crude) | Mean (95% CI) | |||
| Overall health | ||||||
| FAS | 50 | 82.0 (77.5 to 86.5) | 52 | 77.7 (71.8 to 83.6) | 4.3 (–3.0 to 11.7) | 0.25 |
| PP | 49 | 81.9 (77.4 to 86.5) | 52 | 77.7 (71.8 to 83.6) | 4.3 (–3.1 to 11.6) | 0.26 |
| RSV vs. non-RSV | ||||||
| RSV | 28 | 83.9 (78.4 to 89.5) | 32 | 77.3 (69.7 to 85.0) | – | 0.34 |
| Non-RSV | 3 | 81.7 (31.5 to 131.9) | 6 | 88.3 (71.9 to 104.8) | – | – |
| Physical abilities | ||||||
| FAS | 24 | 90.0 (84.1 to 95.8) | 29 | 87.3 (79.1 to 95.5) | 2.7 (–7.6 to 12.9) | 0.60 |
| PP | 23 | 89.8 (83.7 to 96.0) | 29 | 87.3 (79.1 to 95.5) | – | 0.63 |
| RSV vs. non-RSV | ||||||
| RSV | 12 | 96.4 (91.8 to 101.0) | 18 | 86.5 (74.4 to 98.6) | 2.5 (–7.9 to 13.0) | 0.48 |
| Non-RSV | 3 | 85.1 (30.8 to 139.4) | 5 | 86.7 (62.5 to 110.8) | – | – |
| Growth and development | ||||||
| FAS | 56 | 90.7 (87.7 to 93.7) | 53 | 94.0 (91.0 to 97.1) | –3.4 (–7.6 to 0.9) | 0.12 |
| PP | 55 | 90.5 (87.5 to 93.5) | 53 | 94.0 (91.0 to 97.1) | – | 0.10 |
| RSV vs. non-RSV | ||||||
| RSV | 32 | 90.7 (86.4 to 94.9) | 34 | 92.4 (87.2 to 96.9) | –3.5 (–7.8 to 0.7) | 0.73 |
| Non-RSV | 4 | 95.0 (85.8 to 104.2) | 6 | 99.6 (98.5 to 100.7) | – | – |
| Bodily pain | ||||||
| FAS | 55 | 66.7 (60.2 to 73.1) | 53 | 68.6 (61.8 to 75.3) | –1.9 (–11.1 to 7.3) | 0.69 |
| PP | 54 | 66.7 (60.1 to 73.2) | 53 | 68.6 (61.8 to 75.3) | – | 0.69 |
| RSV vs. non-RSV | ||||||
| RSV | 32 | 65.1 (57.8 to 72.4) | 34 | 65.2 (56.5 to 73.9) | –1.9 (–11.2 to 7.4) | 1.00 |
| Non-RSV | 4 | 70.8 (19.5 to 122.1) | 6 | 70.8 (36.4 to 105.3) | – | – |
| Temperament | ||||||
| FAS | 56 | 70.6 (66.2 to 75.0) | 53 | 70.7 (66.2 to 75.1) | –0.06 (–6.3 to 6.2) | 0.98 |
| PP | 55 | 70.6 (66.1 to 75.1) | 53 | 70.7 (66.2 to 75.1) | – | 0.99 |
| RSV vs. non-RSV | ||||||
| RSV | 32 | 69.6 (64.5 to 74.7) | 34 | 71.0 (65.8 to 76.3) | –0.04 (–6.3 to 6.2) | 0.79 |
| Non-RSV | 4 | 63.1 (5.5 to 120.6) | 6 | 61.2 (31.5 to 91.0) | – | – |
| General health perceptions | ||||||
| FAS | 55 | 65.4 (61.1 to 15.8) | 51 | 63.2 (59.5 to 66.8) | 2.2 (–3.4 to 7.8) | 0.44 |
| PP | 54 | 65.2 (60.9 to 69.5) | 51 | 63.2 (59.5 to 66.8) | – | 0.48 |
| RSV vs. non-RSV | ||||||
| RSV | 31 | 63.1 (57.2 to 69.0) | 32 | 63.3 (59.3 to 67.3) | 2.0 (–3.6 to 7.7) | 0.51 |
| Non-RSV | 4 | 71.6 (38.1 to 105.1) | 5 | 65.0 (47.2 to 82.9) | – | – |
| Parental impact: emotional | ||||||
| FAS | 55 | 73.9 (67.3 to 80.5) | 50 | 76.5 (70.5 to 82.5) | –2.6 (–11.5 to 6.3) | 0.56 |
| PP | 54 | 73.6 (66.9 to 80.3) | 50 | 76.5 (70.5 to 82.5) | – | 0.52 |
| RSV vs. non-RSV | ||||||
| RSV | 32 | 74.0 (67.1 to 80.9) | 32 | 77.1 (69.8 to 84.5) | –2.9 (–11.8 to 6.1) | 0.26 |
| Non-RSV | 4 | 67.0 (–6.9 to 140.8) | 4 | 50.9 (–6.8 to 108.5) | – | – |
| Parental impact: time | ||||||
| FAS | 56 | 78.3 (72.2 to 84.3) | 50 | 73.2 (65.8 to 80.5) | 5.1 (–4.3 to 14.4) | 0.28 |
| PP | 55 | 79.1 (73.2 to 85.0) | 50 | 73.2 (65.8 to 80.5) | – | 0.21 |
| RSV vs. non-RSV | ||||||
| RSV | 32 | 76.0 (68.0 to 84.0) | 32 | 71.0 (61.4 to 80.7) | 5.9 (–3.3 to 15.2) | 0.59 |
| Non-RSV | 4 | 66.7 (5.7 to 127.6) | 4 | 51.2 (2.7 to 99.7) | – | – |
| Family cohesion | ||||||
| FAS | 56 | 90.5 (87.3 to 93.8) | 50 | 83.9 (77.7 to 90.1) | 6.6 (–0.1 to 13.3) | 0.05 |
| PP | 55 | 90.6 (87.4 to 93.9) | 50 | 83.9 (77.7 to 90.1) | – | 0.05 |
| RSV vs. non-RSV | ||||||
| RSV | 32 | 90.2 (85.8 to 94.6) | 32 | 90.2 (85.8 to 94.6) | 6.7 (–0.01 to 13.5) | 0.33 |
| Non-RSV | 4 | 88.8 (76.8 to 100.7) | 4 | 78.8 (25.8 to 131.7) | – | – |
FIGURE 7.
The ITQoL dimensions by study group: FAS.

All important harms or unintended effects in each group
Adverse events are reported in Tables 11–13. Six events were probably or possibly related to saline treatment, including one SAE. The SAE was bradycardia and desaturation during administration of the nebuliser, which resolved the following day. The remaining five non-SAEs, each of which occurred in different subjects, were bradycardia (self-correcting), desaturation, coughing fit, increased respiratory rate (all of which were resolved within 1 day) and a chest infection (which resolved after 6 days).
| Variable | Intervention (N = 142) | Control (N = 149) | ||
|---|---|---|---|---|
| Events | n (%) | Events | n (%) | |
| Overall | 51 | 35 (24.6) | 43 | 35 (23.5) |
| Secondary bronchiolitis | 2 | 2 (1.4) | 6 | 5 (3.4) |
| Cough | 7 | 7 (4.9) | 8 | 8 (5.4) |
| Chest infection | 3 | 3 (2.1) | 0 | 0 |
| LRTI | 7 | 7 (4.9) | 3 | 3 (2) |
| Other | 32 | 23 (16.2) | 26 | 20 (13.4) |
| Saline group | ||
|---|---|---|
| Category | Diagnosis | Frequency (related) |
| Secondary bronchiolitis | Bronchiolitis | 1 |
| Ongoing symptoms of bronchiolitis – readmission | 1 | |
| Chest infection | Chest infection | 1 (1) |
| Chest radiograph changes? Chest infection | 1 | |
| Secondary chest infection | 1 | |
| Cough | Attended GP with cough | 1 |
| Coughing fit | 2 (1) | |
| Difficulty feeding and sleeping, still coughing | 1 | |
| Increased work of breathing | 1 | |
| Ongoing cough | 1 | |
| Vomiting with cough | 1 | |
| LRTI | ? pneumonia | 1 |
| ? LRTI (chest radiograph showed right-sided collapse) | 1 | |
| LRTI | 1 | |
| Secondary bacterial LRTI | 2 | |
| Secondary LRTI | 1 | |
| Secondary LRTI with effusion | 1 | |
| Other | Accident/fall | 1 |
| Apnoea. Emergency visit to A&E. Discharged home | 1 | |
| Blood and protein in urine | 1 | |
| Bradycardia (self-correcting) during nebuliser | 1 (1) | |
| Chest radiograph showing upper lobe shadowing | 1 | |
| Chickenpox | 1 | |
| Choking episode | 1 | |
| Constipation | 2 | |
| Desaturation/bradycardias during the administration of 3% MucoClear and nebuliser | 1 (1a) | |
| Desaturation | 1 (1) | |
| Difficulty sleeping | 1 | |
| Fever | 1 | |
| Increase respiratory rate post nebuliser | 1 (1) | |
| Influenza | 1 | |
| Large vomit no cough mentioned post feed | 1 | |
| Lumbar puncture performed | 1 | |
| Pyrexia | 4 | |
| Pyrexia/discomfort | 2 | |
| Rash to left cheek | 1 | |
| Readmission as struggling to breathe | 1 | |
| Readmission | 1 | |
| Secondary bacterial infection | 2 | |
| Secondary infection | 1 | |
| Soft systolic murmur detected on USS | 1 | |
| Spherocytosis | 1 | |
| Thrush (eye) | 1 | |
| Standard treatment group | ||
|---|---|---|
| Category | Diagnosis | Frequency |
| Secondary bronchiolitis | Bronchiolitis | 1 |
| Bronchiolitis (readmission) | 1 | |
| Readmission to hospital with acute bronchiolitis | 1 | |
| Right lower respiratory tract infection | 1 | |
| RSV+ bronchiolitis | 1 | |
| RSV+ bronchiolitis right upper lobe collapse | 1 | |
| Cough | Cold | 2 |
| Cough | 2 | |
| Cough and breathing problems | 1 | |
| Cough and wheeze | 1 | |
| Uncomfortable following coughing bout | 1 | |
| Wheezy | 1 | |
| LRTI | Pneumonia | 1 |
| Secondary bacterial LRTI | 2 | |
| Other | Admission with measles | 1 |
| Apnoeic episode | 1 | |
| Bradycardias | 1 | |
| Conjunctivitis | 1 | |
| Constipation | 4 | |
| Large vomit | 1 | |
| Loose stools | 1 | |
| Not sleeping well | 1 | |
| Periorbital cellulitis | 1 | |
| Pyrexia | 3 | |
| Pyrexia/discomfort | 2 | |
| Rash on toes, and around mouth, torso and chin | 1 | |
| Rash to torso/arms/legs | 1 | |
| Reduced feeding | 1 | |
| Self-correcting bradycardias × 2 | 1 | |
| Snotty, coughing | 1 | |
| Urinary tract infection | 1 | |
| Viral illness | 1 | |
| Viral illness/upper respiratory tract infection | 1 | |
| Vitamin deficiency | 1 | |
Impact of post-randomisation exclusions
Of the 26 babies retrospectively excluded from analysis after randomisation, outcome data were available for 24. The time to fit for discharge and to actual discharge are summarised in Table 14. These times were similar to those of the 291 participants included in the trial, and the comparison of HS with control virtually unchanged if these were included.
| Variable | Intervention (n = 142) | Control (n = 149) | Post-randomisation exclusions (n = 26) |
|---|---|---|---|
| Time to fit for discharge | |||
| Mean (SD) | 90.4 (73.2) | 88.9 (67.9) | 113.6 (128.6) |
| Median (IQR); range | 75.6 (46.1–113.3); 7.1–576.1 | 75.9 (45.5–121.0); 6.8–565.9 | 74.9 (39.5–113.8); 7.0–573.9 |
| Missing, n | 1 | 0 | 2 |
| Time to fit for discharge | |||
| Mean (SD) | 100.6 (76.9) | 101.3 (84.4) | 132.1 (144.8) |
| Median (IQR); range | 88.5 (51.6–120.9); 16.6–595.4 | 88.7 (50.9–123.6); 4.2–857.4 | 84.2 (49.2–139.4); 3.0–619.2 |
| Missing, n | 1 | 0 | 2 |
Chapter 4 Health economic results
Complete hospital data were available for all patients, except for the duration of ward stay for one patient in the intervention group. Consequently, this patient was excluded from the analysis, but this is not thought to have any material effect on the results. Only small, non-statistically significant differences are seen in all measures of hospital stay (Table 15). A difference is seen in the number of doses of saline, with the intervention group receiving on average 10.4 doses of nebulised saline over their hospital stay, while no nebulised saline was used in the control group.
| Variable | Intervention (n = 142)a | Control (n = 149)a | Difference | p-value | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| Hours on ward | 91.8 | 62.9 | 94.5 | 80.4 | –2.7 | 0.752 |
| Hours in ICU | 4.8 | 30.5 | 1.67 | 11.8 | +3.1 | 0.257 |
| Hours in HDU | 4.0 | 17.7 | 5.2 | 19.8 | –1.2 | 0.589 |
| Day readmitted | 0.0 | 0.2 | 0.1 | 0.8 | –0.1 | 0.129 |
| Number of doses of saline | 10.4 | 7.6 | 0.0 | 0.0 | +10.4 | < 0.001 |
Concomitant medication data were analysed, but it was decided not to incorporate these into the economic evaluation. The two main reasons for this were that the medications were all low-cost treatments, for example salbutamol, and assumptions would be required to convert usage into costs, for example whether the patient was prescribed the full pack of tablets or it was taken from the ward supply, and what size of pack was used. It was decided, therefore, that given that the costs of the medications used were negligible in comparison to overall hospital costs, and additional assumptions were needed in their calculation, their inclusion would provide only spurious accuracy.
Resource use in primary care following discharge was self-completed by guardians. The overall response rate was low, with complete follow-up in 53 (37%) and 54 (36%) participants in the control and intervention groups, respectively. Of the remaining patients, the guardians of 57 (40%) and 72 (48%), respectively, were contacted by telephone and provided basic health resource usage; we include this information in Table 16 but have not included it in the economic analysis. One (control group) remained an inpatient at 28 days and their resource usage is included only in Table 15. The remaining 55 guardians were unable to be contacted after multiple attempts.
| (1) Participants completing detailed resource use questionnaire | ||||||
|---|---|---|---|---|---|---|
| Variable | Intervention (n = 53) | Control (n = 54) | Difference | p-value | ||
| Mean | SD | Mean | SD | |||
| GP contacts | 0.19 | 0.44 | 0.26 | 0.52 | –0.07 | 0.548 |
| NHS Direct contacts | 0.02 | 0.14 | 0.04 | 0.19 | –0.02 | 1.000 |
| A&E attendances | 0.02 | 0.14 | 0.06 | 0.23 | –0.04 | 0.610 |
| (2) Participants with brief resource use via telephone | ||||||
| Variable | Intervention (n = 57) | Control (n = 72) | Difference | p-value | ||
| n (%) with any GP contact | 6 (11%) | 6 (8%) | – | 0.764 | ||
| n (%) with any A&E attendance | 2 (4%) | 1 (1%) | – | 0.583 | ||
Use of primary care services was low and was similar in both patient groups (see Table 15). There were no reported attendances at NHS walk-in centres or minor injury units. In cases for which the brief resource information had been obtained by telephone, the resource use was lower than that obtained via questionnaire response, but again was similar between the randomised groups.
When individual cost components are combined with their unit costs, the mean hospital costs were £2595 in the control and £2727 in the intervention group (Table 17). The 95% CI around the difference of £132 is –£520 to £785. QALYs were calculated using the total LoS in hospital, then transformed into quality-adjusted life-days (QALDs) over the 36 days following randomisation in order to aid the presentation in the table of very small numbers. The difference in mean QALDs was 0.006 greater in the intervention group (or 0.0000175 QALYs) and was not statistically significant.
| Variable | Intervention (n = 141) | Control (n = 149) | Difference | p-value | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| Total hospital costs (£) | 2726 | 3204 | 2595 | 2345 | 132 | 0.675 |
| QALDs | 33.999 | 0.152 | 33.993 | 0.172 | 0.006 | 0.757 |
Mean primary care costs relating to the services described previously were £19 and £11 in the control and intervention groups, respectively (see Table 16). The difference in means is £8, with a 95% CI of –£21 to £6 (p = 0.25). Given the large number of missing data, it was decided not to impute missing values or combine them with the hospital costs.
With numerically higher costs (£132) and QALYs (0.0000175), an incremental cost-effectiveness ratio (ICER) can be produced, which is approximately £7.6M per QALY gained. The uncertainty around this is represented by plotting the incremental costs and QALYs on the cost-effectiveness plane from the non-parametric bootstrap of the trial data in Figure 8. This shows a cloud of observations centred, approximately, around the origin (representing zero cost difference and zero QALY difference).
FIGURE 8.
Cost-effectiveness plane of nebulised saline.
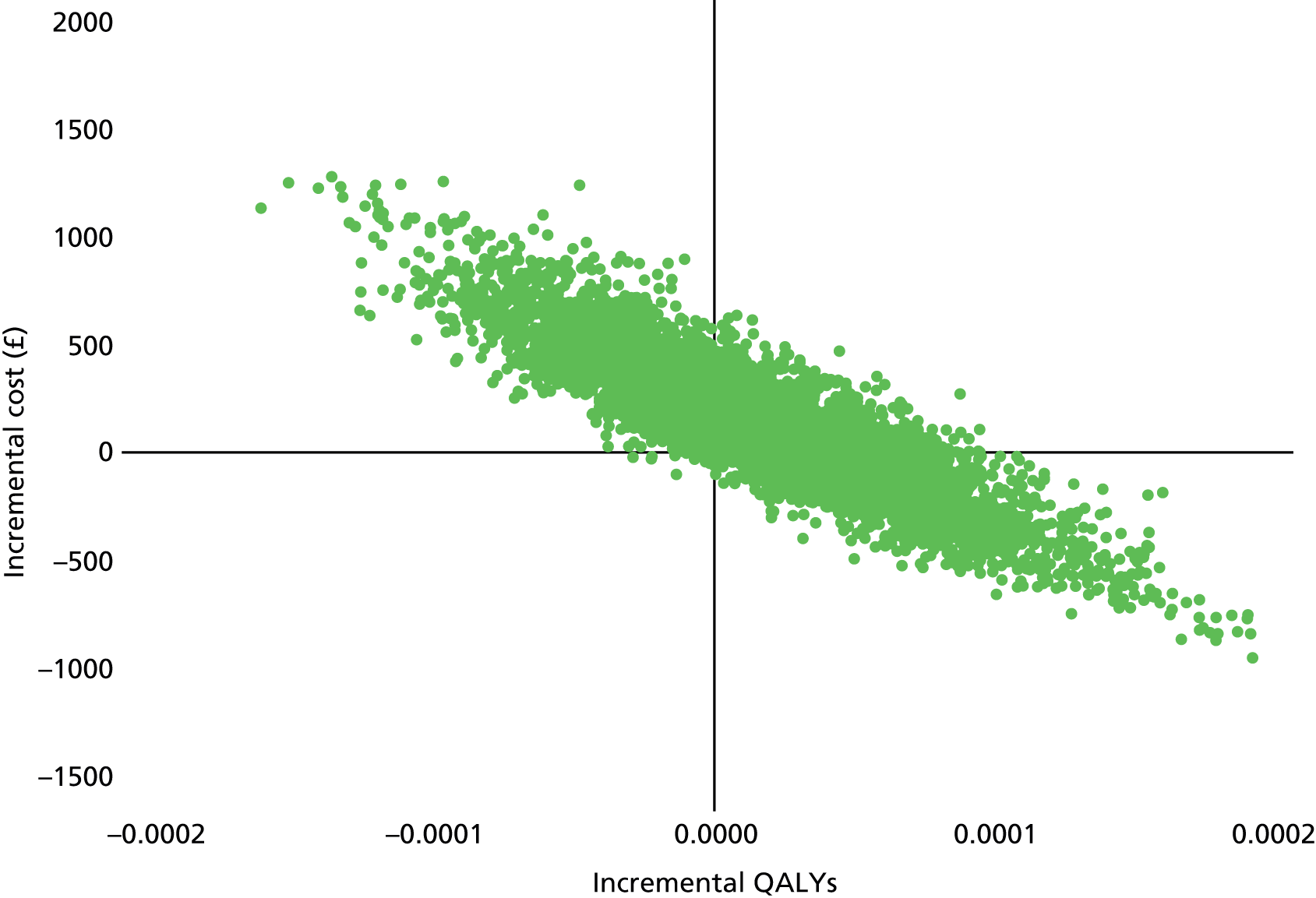
The estimates used to generate the cost-effectiveness plane are then transformed into a cost-effectiveness acceptability curve (Figure 9). This shows that there is a 34% probability of nebulised saline being cost-effective at a funding threshold of £20,000 per QALY. This probability is constant over the funding range that is typically considered by the NHS (£20,000–30,000). 96 One-way sensitivity analyses examining different unit costs for time spent on a ward, ICU or HDU did not alter the probability that nebulised saline would be cost-effective.
FIGURE 9.
Cost-effectiveness acceptability curve for the use of nebulised saline.

Chapter 5 Results of the systematic review
In this chapter we describe the results obtained from the review, incorporating the data from our study. The search strategy and identification is described in Study selection. The study characteristics of the individual studies are described in Study characteristics, followed by the results of the risk of bias assessment for individual studies in Risk of bias within studies. The results and meta-analyses of the effect of the intervention (HS) are summarised in Results and meta-analyses, Primary outcome: results and synthesis and Secondary outcomes: results and synthesis. Finally, the risk of bias across studies is considered in Risk of bias across studies.
Study selection
The search criteria outlined in Chapter 2, Systematic Review Methods (see also Appendix 6), and the number of titles retrieved from each search are outlined in Figure 10.
FIGURE 10.
Study flow diagram.
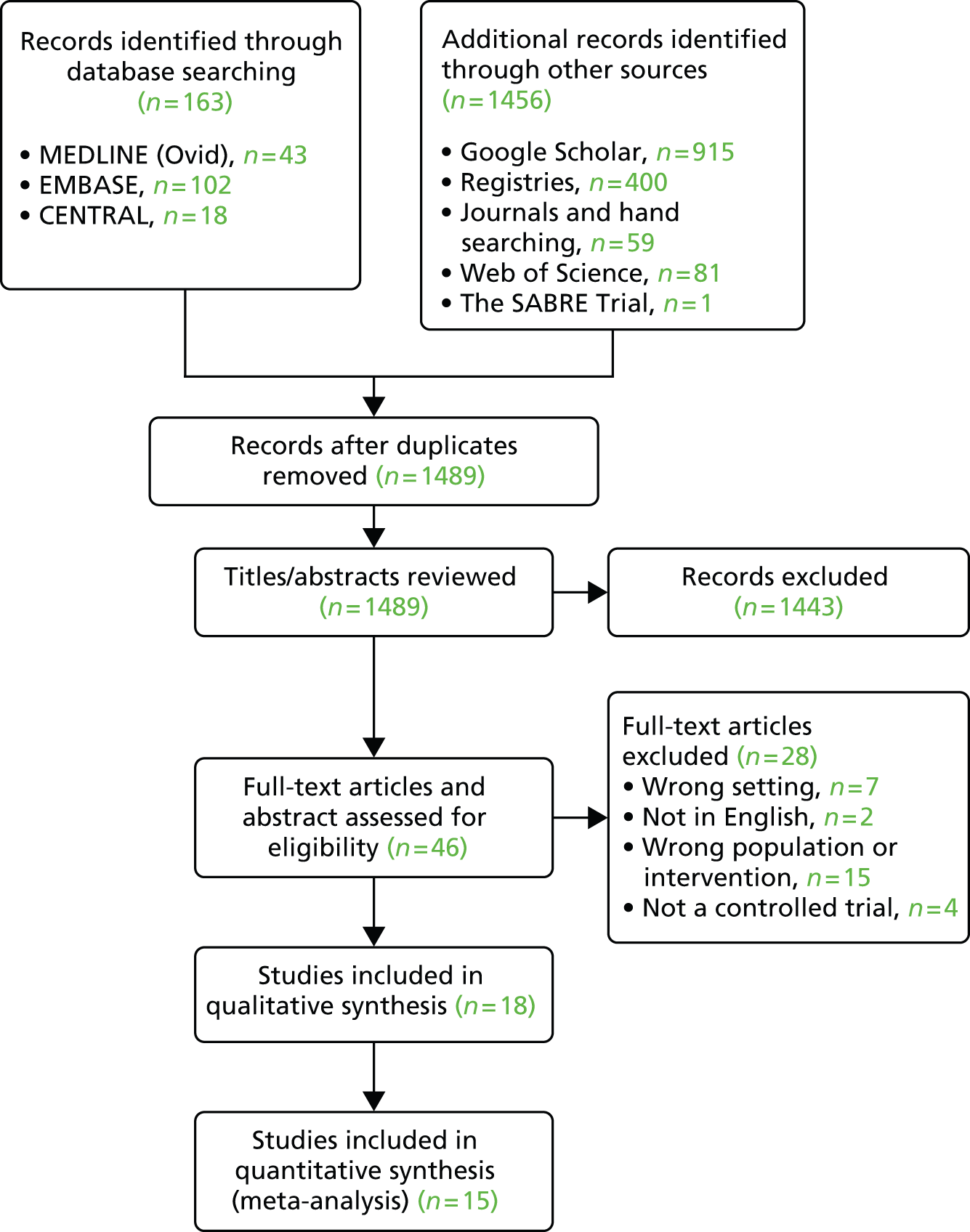
The searches identified a total of 1619 citations. After removal of duplicates, 1489 records were assessed at title and abstract stage. The main searches were all downloaded into reference manager software (RevMan version 5.2) and details of the search strategies can be found in Appendix 6. In total, 1443 records were excluded at this stage for reasons that include not being a RCT, not evaluating the intervention of interest and having the wrong population or intervention when this was apparent from the title and abstract. Nine potentially relevant studies were rejected at the abstract stage, because it was obvious that the full papers were not available in English. 59–67
In total, 46 full papers (including the SABRE study report) were retrieved for further review, and these included studies for which the title and abstract (if available) were ambiguous. Of these, 28 studies were excluded because they did not consider hospitalised infants (n = 7),32,33,97–101 they were not available in English (n = 2),68,69 they did not consider the intervention in the appropriate population of interest (n = 15)21,38,39,102–113 or they were not a controlled trial (n = 4)114–117 (Table 18).
| Studies excluded at full paper review | Reason for exclusion |
|---|---|
| Ipek et al., 2011101 | Wrong setting |
| Anil et al., 201033 | Wrong setting |
| Grewal et al., 200932 | Wrong setting |
| Kuzik et al., 201097 | Wrong setting |
| Sezer et al., 201098 | Wrong setting |
| Jacobs et al., 201499 | Wrong setting |
| Wu et al., 2014100 | Wrong setting |
| Park et al., 200568 | Not in English |
| Zheng et al., 201269 | Not in English |
| Gutpa et al., 2012102 | Wrong population or intervention |
| Khashabi et al., 2005103 | Wrong population or intervention |
| Lines et al., 1992104 | Wrong population or intervention |
| Milner et al., 1995105 | Wrong population or intervention |
| Patel et al., 2002106 | Wrong population or intervention |
| Tinsa et al., 2009107 | Wrong population or intervention |
| Wainwright et al., 200338 | Wrong population or intervention |
| Postiaux et al., 2011108 | Wrong population or intervention |
| Hariprakash et al., 200339 | Wrong population or intervention |
| Sarrell et al., 200221 | Wrong population or intervention |
| Chowdhury et al., 1995109 | Wrong population or intervention |
| Nenna et al., 2009110 | Wrong population or intervention |
| Bertrand et al., 2001111 | Wrong population or intervention |
| Nenna et al., 2013112 | Wrong population or intervention |
| Bueno Campaña et al., 2014113 | Wrong population or intervention |
| Mandelberg et al., 2010114 | Not a controlled trial |
| Principi et al., 2011115 | Not a controlled trial |
| Hom et al., 2011116 | Not a controlled trial |
| Sauvaget et al., 2012117 | Not a controlled trial |
Eighteen studies were accepted for final inclusion in the review (see Figure 10), all being methodologically appropriate. All but three of the studies were appropriate to be included in the meta-analysis of the primary outcome of interest: LoS.
Study characteristics
Eighteen trials comprising 2225 participants in total were identified via the searches described in Appendix 6; the number of recruited participants was unavailable for one trial. 75 All the trials included children under the age of 2 years, although five studies included only children under the age of 12 months. 22,23,85,87,88 Luo and colleagues79,80 studied bronchiolitis in separate children over two distinct winter seasons for each of the studies.
Data on the study overview are given in Table 3. Studies were performed across a variety of countries: India, Canada, Israel, Argentina, the United Arab Emirates, the People’s Republic of China, Italy, Qatar, Georgia, the Netherlands, Mexico, Turkey, the USA, Nepal and the UK. Overall, 11 of the trials described appropriate allocation sequence generation methods. 23,24,77,80–83,85,87,88,90 The number of participants ranged from 40 to 317. All studies included children hospitalised on their first episode of bronchiolitis only. Full details of the eligibility criteria are given in Appendix 5. Fifteen of the studies22–24,77,79,80,82–90 reviewed baseline characteristic variability between both arms; however, the unpublished data74,75 and one other study81 do not give sufficient evidence of this having been done.
The Wang et al. CSS system78 was used to classify disease severity in eight of the trials. 22,23,77,79–82,86 Three trials24,85,88 used the Respiratory Distress Assessment Instrument score,118 one trial used the Bronchiolitis Severity Score,83 one trial used the Bronchiolitis Clinical Score,84 one did not specify which scoring system was used74 and one did not assess disease severity. 87 In addition, one used the Respiratory Assessment Score,89 one used the Respiratory Distress Scale of the Sant Joan de Déu Hospital75 and another trial used a clinical scoring system. 90 The Wang scale uses different symptomatic characteristics (including wheezing, retractions and the general condition of the patient) to allocate a score: a score of 0–4.9 indicates mild distress, a score of 5–8.9 moderate distress and a score of 9–12 severe distress. 79 This scoring system was used by study authors to classify their cohort into one of the three groups, although trials included a combination of mild/moderate,89,84,79,81,86 moderate22–24,82 or moderate/severe74,75,77,80,83,85,87 participants. A severity classification was not provided by Ojha et al. 90 or Silver. 88 A simplified guide to interventions is shown in Table 19.
| Intervention Groups | Intervention | Control | Studies |
|---|---|---|---|
| 1 | Nebulised HS alone | NS | Luo et al., 201180 |
| Kuzik et al., 200724 | |||
| Silver, 201488 | |||
| Ojha et al., 201490 | |||
| 2 | Nebulised HS plus a bronchodilator (e.g. salbutamol) | NS | No trials |
| 3 (pre-specified subgroup) | Nebulised HS plus a bronchodilator (e.g. salbutamol) | NS plus same bronchodilator | Trials by pre-specified subgroup |
| 3.1 (post-hoc subgroup) | Nebulised HS plus a beta-2 agonist | NS plus same beta-2 agonist | Espelt, 201274 |
| Luo et al., 201079 | |||
| Maheshkumar et al., 201381 | |||
| Sharma et al., 201382 | |||
| Teunissen et al., 201486 | |||
| Ozdogan et al., 201489 | |||
| Sosa-Bustamante, 201475 | |||
| 3.2 (post-hoc subgroup) | Nebulised HS plus adrenaline | NS plus adrenaline | Al-Ansari et al., 201083 |
| Mandelberg et al., 200322 | |||
| Giudice et al., 201277 | |||
| Pandit et al., 201385 | |||
| Tal et al., 200623 | |||
| 4 | Nebulised HS alone or plus a bronchodilator (e.g. salbutamol) | No intervention | Everard et al., 201487 |
Nemsadze et al. ,84 Sosa-Bustamante75 and Ozdogan et al. 89 could not be included in the meta-analysis, as the data were available only in abstract form or on ClinicalTrials.gov and therefore the primary outcome LoS data were not available. We contacted the authors of Silver,88 who provided the mean and SD LoS, along with additional data requested.
Data on the intervention and comparators are given in Tables 20 and 21: study characteristics, intervention and comparator. All of the studies used 3% HS as the study intervention administered via a nebuliser, three of which were three-arm studies in which either 5% HS89,83 or 6% HS86 was also compared. The amount administered was stated for 15 out of 18 trials, and varied, being 3 ml74,81 in two studies, 4 ml22–24,75,79,80,82,85–88,90 in 12 studies and 5 ml for one study. 83 The flow rate of the nebulisers used to administer this was stated for 10 trials and ranged from 5 to 10 ml/minute. 22,23,77,81–83,85–88 There were clear differences in the additional drugs administered alongside the intervention in the studies: five trials gave HS alone,24,80,87,88,90 seven studies administered a beta-2 agonist (salbutamol, albuterol) alongside HS,74,75,79,81,82,86,89 five trials gave adrenaline alongside HS22,23,77,83,85 and one trial was reported in abstract form only and did not list additional drugs administered. 84 Only five of the trials drew any inference about compliance with treatment by stating that the nebulisers were administered until empty. 22,23,82,86,88 Studies are similar enough in population and interventions delivered to be comparable with each other.
| Main author | Intervention group | |||||
|---|---|---|---|---|---|---|
| Number randomised to intervention | Intervention name: volume and concentrationa | Duration of treatment/dosage | Delivery mechanism | Other drugs given | Compliance | |
| Giudice et al., 201277 | 53 | 3% HS | Every 6 hours | Nebuliser: flow rate 6 ml/minute | Aerosolised adrenaline (1.5 mg) | Not stated |
| Kuzik et al., 200724 | 47 | 3% HS (4 ml) | Every 2 hours (three doses), every 4 hours (five doses), every 6 hours until discharge | Nebuliser | None | Not stated |
| Tal et al., 200623 | 21 | 3% HS (4 ml) | Three treatments each day administered every 8 hours | Nebuliser: 6 ml/minute | 1.5 mg of adrenaline | Administered until nebuliser empty |
| Luo et al., 201079 | 50 | 3% HS (4 ml) | Three treatments every day every 8 hours until discharge | Nebuliser | 2.5 mg of salbutamol | Not stated |
| Luo et al., 201180 | 64 | 3% HS (4 ml) | Every 2 hours (three doses), every 4 hours (five doses), every 6 hours until discharge | Nebuliser | None | Not stated |
| Mandelberg et al., 200322 | 30 | 3% HS (4 ml) | Three times a day every 8 hours until ready for discharge | Aeromist nebuliser: flow rate 5 ml/minute | 1.5 mg of adrenaline | The nebuliser was administered until empty |
| Espelt, 201274 | 50 | 3% HS (3 ml) | Four times per day over 5 days | Nebuliser | 0.25 mg/kg of albuterol | Not stated |
| Maheshkumar et al., 201381 | 20 | 3% HS (3 ml) | Every 6 hours until fit for discharge | Nebuliser, 5–6 ml/minute | 0.15 mg/kg of salbutamol | Not stated |
| Sharma et al., 201382 | 125 | 3% HS (4 ml) | Every 4 hours, six times a day until ready for discharge | Nebuliser: flow rate 7 ml/minute | 2.5 mg of salbutamol | Administered until nebuliser empty |
| Al-Ansari et al., 201083 | 3% HS (n = 58), 5% HS (n = 57) | 3% HS (5 ml), 5% HS (5 ml) | Every 4 hours until they were ready for discharge | Nebuliser: flow rate 10 ml/minute | 1.5 ml of adrenaline | Not stated |
| Nemsadze et al., 201384 | Not stated – abstract only | 3% HS | Every 6 hours | Not stated – abstract only | Not stated – abstract only | Not stated – abstract only |
| Pandit et al., 201385 | 51 | 3% HS (4 ml) | The nebulisation was given three times with an interval of 1 hour, thereafter nebulisation was given every 6 hours until discharge | Nebuliser 6–8 ml/minute | 1 ml/1 mg of adrenaline | Not stated |
| Teunissen et al., 201486 | 3% HS (n = 97), 6% HS (n = 102) | 3% HS (4 ml), 6% HS (4 ml) | Every 8 hours until discharge | HOT Top Plus Nebuliser (Intersurgical Ltd, Wokingham, UK) 6–8 ml/minute | 2.5 mg of salbutamol | Administered until empty |
| Everard et al., 201487 | 158 | 3% HS (4 ml) | Every 6 hours until fit for discharge | LC® PARI Sprint nebuliser (PARI Medical Ltd, Surrey) 5 ml/minute | None | Not stated |
| Silver, 201488 | 113 | 3% HS (4 ml) | Every 4 hours until discharge | Standard pressurised hospital wall nebuliser flow rate 5 ml/minute | None | Administered until nebuliser treatment complete |
| Ozdogan et al., 201489 | Not stated – abstract only | 3% HS, 5% HS | Nebulisations given three times a day | Nebuliser | Albuterol | Not stated |
| Sosa-Bustamante, 201475 | Not stated – ClinicalTrials.gov only | 3% HS (4 ml) | Nebulised for three initial sessions lasting 20 minutes each and every 4 hours during hospital stay | Not stated – ClinicalTrials.gov only | 100 µg/kg of salbutamol | Not stated |
| Ojha et al., 201490 | 36 | 3% HS (4 ml) | Patients in each group received minimum of three nebulisations each day, delivered at 8-hour intervals until discharge | Nebuliser | None | Not stated |
| Main author | Control group | |||||
|---|---|---|---|---|---|---|
| Number randomised to control | Intervention name: volume and concentrationa | Duration of treatment | Delivery mechanism | Other drugs given | Compliance | |
| Giudice et al., 201277 | 56 | 0.9% NS | Every 6 hours | Nebuliser: flow rate 6 ml/minute | Aerosolised adrenaline (1.5 mg) | Not stated |
| Kuzik et al., 200724 | 49 | 0.9% NS (4 ml) | Every 2 hours (three doses), every 4 hours (5 doses), every 6 hours until discharge | Nebuliser | None | Not stated |
| Tal et al., 200623 | 20 | 0.9% NS (4 ml) | Three treatments each day administered every 8 hours | Nebuliser: 6 ml/minute | 1.5 mg adrenaline | Administered until nebuliser empty |
| Luo et al., 201079 | 43 | 0.9% NS (4 ml) | Three treatments every day every 8 hours until discharge | Nebuliser | 2.5 mg salbutamol | Not stated |
| Luo et al., 201180 | 62 | 0.9% NS (4 ml) | Every 2 hours (three doses), every 4 hours (five doses), every 6 hours until discharge | Nebuliser | None | Not stated |
| Mandelberg et al., 200322 | 30 | 0.9% NS (4 ml) | Three times a day every 8 hours until ready for discharge | Aeromist nebuliser: flow rate 5 ml/minute | 1.5 mg adrenaline | The nebuliser was administered until empty |
| Espelt, 201274 | 50 | 0.9% NS (3 ml) | Four times per day over 5 days | Nebuliser | 0.25 mg/kg albuterol | Not stated |
| Maheshkumar et al., 201381 | 20 | NS (3 ml) | Every 6 hours until fit for discharge | Nebuliser, 5–6 ml/minute | 0.15 mg/kg salbutamol | Not stated |
| Sharma et al., 201382 | 125 | 0.9% NS (4 ml) | Every 4 hours, 6 times a day until ready for discharge | Nebuliser: flow rate 7 ml/minute | 2.5 mg salbutamol | Administered until nebuliser empty |
| Al-Ansari et al., 201083 | 56 (0.9%) | 0.9% NS (5 ml) | Every 4 hours until patients were ready for discharge | Nebuliser: flow rate 10 ml/minute | 1.5 ml adrenaline | Not stated |
| Nemsadze et al., 201384 | Not stated – abstract only | NS | Every 6 hours | Not stated – abstract only | Not stated – abstract only | Not stated – abstract only |
| Pandit et al., 201385 | 49 | 0.9% NS (4 ml) | The nebulisation was given three times with 1 hour between treatment; thereafter nebulisation was given every 6 hours until discharge | Nebuliser | 1 ml/1 mg Adrenaline | Not stated |
| Teunissen et al., 201486 | 93 | 0.9% NS (4 ml) | Every 8 hours until discharge | HOT Top Plus Nebuliser 6–8 ml/minute | 2.5 mg salbutamol | Administered until empty |
| Everard et al., 201487 | 159 | Usual care | N/A | N/A | None | Not stated |
| Silver, 201488 | 114 | 0.9% NS (4 ml) | Every 4 hours until discharge | Nebuliser with 5 ml/l oxygen flow | None | Administered until nebuliser treatment complete |
| Ozdogan et al., 201489 | Not stated – abstract only | 0.9% NS | Nebulisations given three times per day | Not stated – abstract only | Albuterol | Not stated |
| Sosa-Bustamante, 201475 | Not stated – ClinicalTrials.gov only | 0.9% NS (4 ml) | Nebulised for three initial sessions lasting 20 minutes each and every 4 hours during hospital stay | Not stated – ClinicalTrials.gov only | 100 µg/kg salbutamol | Not stated |
| Ojha et al., 201490 | 36 | 0.9% NS (4 ml) | Patients in each group received minimum of three nebulisations each day delivered at 8-hour intervals until discharge | Nebuliser | None | Not stated |
Risk of bias within studies
The risk of bias assessments for each of the domains described above are summarised in Table 22. With the exception of four studies,74,75,84,89 the methodological quality of the included studies was generally high based on the Cochrane suggested domains for assessing internal study validity. All studies were RCTs; seven trials do not give a detailed description on random sequence generation,22,74,75,79,84,86,89 but none was adjudged clearly inappropriate.
| Main author | Risk of bias assessment – grade: low risk, high risk, unclear | ||||||
|---|---|---|---|---|---|---|---|
| Random sequence generation (selection bias) | Allocation concealment (selection bias) | Blinding of patients and personnel (performance bias) primary outcome | Blinding of patients and personnel (performance bias) control | Blinding of outcome assessment (detection bias) primary outcome | Incomplete outcome data addressed (attrition bias) primary outcome | Selective outcome reporting (reporting bias) | |
| Giudice et al., 201277 | Low: computer sequence | Low: study solutions prepared by local pharmacy | Low: double blind | Low: double blind | Unclear: no statement provided | Low: three withdrawals in total across both groups | Low: outcomes reported same as those listed on ClinicalTrials.gov |
| Kuzik et al., 200724 | Low: computer sequence | Low: identical appearance drug containers in sequence, research pharmacist prepared solutions | Low: double blind | Low: double blind | Unclear: no statement provided | Low: five withdrawn (two intervention and three control group). ITT used | Unclear: insufficient information to make this judgement |
| Tal et al., 200623 | Low: computer sequence, ‘code deposited with statistician’23 | Low: identical appearance drug containers in sequence | Low: double blind | Low: double blind | Low: attending physicians who made decision to discharge blinded to intervention | Low: three patients withdrawn | Unclear: insufficient information to make this judgement |
| Luo et al., 201079 | Unclear: no description given | Unclear: ‘patients were selected and randomly assigned’ | Low: double blind | Low: double blind | Low: attending physicians who made decision to discharge blinded to intervention | Low: all patients included in analysis | Unclear: insufficient information to make this judgement |
| Luo et al., 201180 | Low: computer sequence | Low: opaque, sealed envelope | Low: double blind | Low: double blind | Low: attending physicians who made decision to discharge blinded to intervention | Low: 14 patients excluded as discharged within 12 hours of enrolment | Unclear: insufficient information to make this judgement |
| Mandelberg et al., 200322 | Unclear: no description given although it mentions ‘the code was deposited with the statistician’22 | Unclear: ‘patients were selected and randomly assigned’ | Low: double blind | Low: double blind | Low: attending physicians who made decision to discharge blinded to intervention | Low: nine withdrawals (three from intervention and six from control; eight due to withdrawal of parental consent and one due to clinical deterioration) | Unclear: insufficient information to make this judgement |
| Espelt, 201274 | Unclear: no description given | Unclear: no description given | High: ‘open label’74 | High: ‘open label’74 | Unclear: no statement provided | High: 13 withdrawals from intervention group and five from control group, no reasons given for uneven distribution | Unclear: insufficient information to make this judgement |
| Maheshkumar et al., 201381 | Low: computer sequence | Unclear: ‘patients were recruited sequentially and randomised’ | Low: double blind | Low: double blind | Unclear: no statement provided | Low: no patients withdrawn, all included in analysis | Unclear: insufficient information to make this judgement |
| Sharma et al., 201382 | Low: computer sequence | Low: identical appearance containers | Low: double blind | Low: double blind | Unclear: no statement provided | Low: two withdrawals accounted for | High: final severity scores (secondary analysis) not fully reported, only stated ‘did not show statistically significant differences in [both] groups’82 |
| Al-Ansari et al., 201083 | Low: computer-generated list | Low: ‘computer generated list of random numbers was used by the enrolling physicians in consecutive order to identify a sealed envelope’83 | Low: double blind | Low: double blind | Unclear: no statement provided | High: 16 patients excluded from analysis, does not state which arm each of the patients were allocated to | Unclear: insufficient information to make this judgement |
| Nemsadze et al., 201384 | Unclear: no description given – abstract only | Unclear: ‘infants were randomly assigned’84 | Unclear: no statement provided – abstract only | Unclear: no statement provided – abstract only | Unclear: no statement provided – abstract only | Unclear: no description given – abstract only | Unclear: insufficient information to make this judgement – abstract only |
| Pandit et al., 201385 | Low: computer sequence | Low: ‘group allocation concealed in opaque envelope’ | High: ‘non-blinded study’ | High: ‘non-blinded study’ | Unclear: no statement provided | Low: no patients withdrawn, all included in analysis. ‘Out of 100 cases there were no drop outs’85 | Unclear: insufficient information to make this judgement |
| Teunissen et al., 201486 | Unclear: ‘randomisation was done per centre and clustered in blocks of six’86 | Low: identical appearance containers | Low: double blind | Low: double blind | Low: ‘all participants, care givers and medical staff were blinded’86 | Low: two withdrawn and 43 excluded from PP analysis, all accounted for | Low: outcomes reported same as those listed on The Dutch Trial Register |
| Everard et al., 201487 | Low: computer generated | Low: web-based randomisation system | High: non-blinded | High: non-blinded | Unclear: doctor making decision for discharge may not be aware of the treatment allocation | Low: 26 excluded from analysis, as randomised when ineligible, 16 of whom from the HS group and 10 from the control. All treatment withdrawals from HS group; however, the other arm was usual care. Three withdrawals from the study, two of whom were from the control group and one was from the HS group | Low: all outcomes reported as stated in the protocol |
| Silver, 201488 | Low: computer generated in blocks of eight | Low: web-based randomisation system | Low: double blind | Low: double blind | Low: outcome assessor blinded | Low: 20 withdrawn in HS group (10 for clinical worsening, three albuterol given, three parental requests and three protocol deviations. 17 withdrawn in NS group (nine for clinical worsening, five were given albuterol and three protocol deviations) | Unclear: insufficient information to make this judgement |
| Ozdogan et al., 201489 | Unclear: no description given – abstract only | Unclear: No description given – abstract only | Low: double blind | Low: double blind | Unclear: no description given – abstract only | Unclear: no description given – abstract only | Unclear: insufficient information to make this judgement |
| Sosa-Bustamante, 201475 | Unclear: no description given | Unclear: no description given | Low: double blind | Low: double blind | Unclear: no description given | Unclear: no results given | Unclear: insufficient information to make this judgement |
| Ojha et al., 201490 | Low: computer-generated random number table | Low: allocation kept in sealed envelopes relating to identical containers and labelled as solution A and B | Low: double blind | Low: double blind | Unclear: no description given | Low: five excluded from NS group and eight excluded from HS group, all accounted for | Unclear: insufficient information to make this judgement |
The local pharmacy prepared the solutions for two trials. 24,77 Use of sequential identical containers was employed by three studies,23,82,86 sealed envelopes by three studies80,83,85 and one study adopted both methods. 90 Four studies are not clear on their technique for allocation concealment but simply state that ‘patients were selected and randomly assigned’,79 ‘patients were recruited sequentially and randomised’81 or ‘infants were randomly assigned’. 22,84 A clear description was not given by three studies. 74,75,89 A web-based randomisation system was used for two of the studies. 87,88 All studies are stated as being double blind, except three which were open-label trials74,85,87 and one in which no statement was provided. 84
It was possible to assess detection bias for six of the trials in which the attending physicians who made a decision to discharge were stated as being blinded to the intervention,22,23,79,80,86,88 but no statement was provided in the other trials. Attrition bias was assessable in all studies as a description of withdrawals was provided apart from Sosa-Bustamante75, Ozdogan et al. 89 and Nemsadze et al. ,84 as information was available only in abstract form or on ClinicalTrials.gov. Potential bias due to selective outcome reporting was not possible to ascertain for 14 of the 18 trials. Of the remaining four, Giudice et al. 77 published their study protocol and made information available on ClinicalTrials.gov, Sharma et al. 82 did not fully report the final CSS score (secondary analysis), Teunissen et al. 86 made information available on The Dutch Trial Register and, finally, of course, the study protocol for our own trial87 is available.
The risk of bias summary outputs from RevMan can be seen in Figures 11 and 12. These relate to the review authors’ judgements about each risk of bias item presented as percentages across all included studies and then as a summary representing each risk of bias item for each included study.
FIGURE 11.
Risk of bias as a percentage across all studies.

FIGURE 12.
Risk of bias as a summary for all trials.

Fourteen of the 18 trials were double blind, allocation concealment was appropriately described in 11 trials and a random sequence was correctly generated in 11 studies. Conversely, an unclear judgement was made for the majority of trials in relation to selective reporting because of the unavailability of pre-specified outcomes making this difficult to judge. Four studies74,75,84,89 scored the lowest in terms of internal validity; however, all were available only in abstract form or on ClinicalTrials.gov. Other bias was mainly assessed as ‘unclear’ as there were no apparent other sources of bias observed in the study reports.
None of the trials was classified as being at 100% low risk of bias across each of the criteria. One of the trials74 was open label and also reported marked differences in withdrawal rates between groups, with no explanation given, and as such scored high risk of bias for both counts. Blinding of outcome measures was assessed as being at low risk of bias in 6 of the 18 trials. 22,23,79,80,86,88 Selective outcome reporting was difficult to assess without the availability of pre-specified outcomes, for example in the form of a published protocol, for 14 of the 18 trials, and it was high for one study82 as the secondary outcome was not fully reported. There were no other apparent sources of bias on examination of the literature from the studies.
Results and meta-analyses
Primary outcome: results and synthesis
The data extracted for the primary outcome are displayed in Table 23. Although all trials stated that LoS was being measured, a closer review of the methods revealed that most in fact had measured the time to being fit for discharge (variably defined). To avoid confusion among those familiar with the included trials, we will continue to use the term LoS for the remainder of this section, but will revisit this issue of nomenclature in Chapter 6, Discussion.
| Main author | Disease severity | Intervention | Control | ||||
|---|---|---|---|---|---|---|---|
| Mean (days) | SD (days) | Number of participants | Mean (days) | SD (days) | Number of participants | ||
| Giudice et al., 201277 | Severe | 4.9 | 1.3 | 52 | 5.6 | 1.6 | 54 |
| Kuzik et al., 200724 | Moderate | 2.6 | 1.9 | 45 | 3.5 | 2.9 | 46 |
| Tal et al., 200623 | Moderate | 2.6 | 1.4 | 21 | 3.5 | 1.7 | 20 |
| Luo et al., 201079 | Mild/moderate | 6 | 1.2 | 50 | 7.4 | 1.5 | 43 |
| Luo et al., 201180 | Moderate/severe | 4.8 | 1.2 | 57 | 6.4 | 1.4 | 55 |
| Mandelberg et al., 200322 | Moderate | 3 | 1.2 | 27 | 4 | 1.9 | 25 |
| Espelt, 201274 | Moderate | 5.8 | 2.7 | 37 | 5.47 | 2.1 | 45 |
| Maheshkumar et al., 201381 | Mild/moderate | 2.25 | 0.89 | 20 | 2.88 | 1.76 | 20 |
| Sharma et al., 201382 | Moderate | 2.64 | 0.88 | 125 | 2.66 | 0.93 | 123 |
| Al-Ansari et al., 201083 | Moderate to severe | 1.4 (3% HS) | 1.41 (3% HS) | 58 (3% HS) | 1.88 | 1.76 | 56 |
| 1.56 (5% HS) | 1.38 (5% HS) | 57 (5% HS) | |||||
| Nemsadze et al., 201384 | Mild to moderate | 4.4 | 1.1 | Unknown – abstract only | 4.9 | 1.2 | Unknown – abstract only |
| Pandit et al., 201385 | Moderate/severe | 3.92 | 1.72 | 51 | 4.08 | 1.90 | 49 |
| Teunissen et al., 201486 | Mild to moderate | 3.23 (3% HS) | 2.02 (3% HS) | 84 (3% HS) | 3.17 | 1.83 | 80 |
| 3.24 (6% HS) | 2.06 (6% HS) | 83 (6% HS) | |||||
| Everard et al., 201487 | Severe | 3.77 | 3.05 | 141 | 3.7 | 2.83 | 149 |
| Silver, 201488 | Unknown | 2.49 | 1.64 | 93 | 2.47 | 1.76 | 97 |
| Ozdogan et al., 201489 | Mild to moderate | Unknown – abstract only | Unknown – abstract only | Unknown – abstract only | Unknown – abstract only | Unknown – abstract only | Unknown – abstract only |
| Sosa-Bustamante, 201475 | Moderate to severe | Unknown – ClinicalTrials.gov only | Unknown – ClinicalTrials.gov only | Unknown – ClinicalTrials.gov only | Unknown – ClinicalTrials.gov only | Unknown – ClinicalTrials.gov only | Unknown – ClinicalTrials.gov only |
| Ojha et al., 201490 | Unknown | 1.87 | 0.96 | 28 | 1.82 | 1.18 | 31 |
The mean LoS, SD and sample size in each trial for each group. Data are provided for all of the trials; however, Nemsadze et al. did not provide the number of patients in each group and so cannot be included in the meta-analysis. 84 In addition, the mean, SD and sample size for LoS were not available for two trials, as information was available only in abstract form or on ClinicalTrials.gov. 75,89
To avoid double-counting of control patients, in the case of three-arm trials we divided control group numbers by two (see Chapter 2, Synthesis of results). Teunissen et al. 86 (3% HS group, n = 84; 6% HS group, n = 83; and control, n = 80) was included as two effective studies in the analysis [‘Teunissen (3% HS)’, n = 84 vs. 40; ‘Teunissen (6% HS)’, n = 83 vs. 40],86 and the same approach was taken with Al-Ansari83 [3% HS group, n = 58; 5% HS group, n = 57; and control, n = 56; ‘Al-Ansari (3% HS)’ n = 58 vs. 28; ‘Al-Ansari (5% HS)’ n = 57 vs. 28].
Figures 13 and 14 present the LoS for each trial, together with the pooled difference between HS and control, using both FE and RE methods. The weighted average of the effect sizes observed across 15 RCTs (n = 1922 participants) would indicate that HS, alone or in combination with other therapies, reduces the LoS in children with acute bronchiolitis by just over one-third of a day [mean difference (MD) –0.36, 95% CI –0.50 to –0.22] compared with no HS (see Figure 13). However, the level of statistical heterogeneity associated with this estimate is too high for it to be a safe basis for clinical decision-making, with over three-quarters of the between-study variability not attributable to the play of chance alone (I2 = 78%). Of particular note is that our own trial was one of the largest of the 15 trials included, yet it received a mere 4.2% weight in the FE analysis. The most probable reason for this apparent incongruity is the excessive heterogeneity among the studies, with differential case mix (e.g. patient severity or discharge policies) being typified by differences among the SDs and hence study weights. RE analyses have been advocated to address this; this method yields a larger MD (–0.48 days; 95% CI –0.81 to –0.15 days). Nevertheless, this does not overcome the problem of excessive heterogeneity; the between-trial variation dominates the within-trials variation and effectively allocates similar weights to each study irrespective of their size. 119,120 The same pattern was observed when combining standardised MDs (i.e. dividing the MD by the underlying SD78).
FIGURE 13.
Difference in length of hospital stay by intervention subgroup, 3% HS. D&L, DerSimonian and Laird; IV, inverse-variance; N/A, not applicable.

FIGURE 14.
Difference in LoS by intervention subgroup, all concentrations of HS. D&L, DerSimonian and Laird; IV, inverse-variance; N/A, not applicable.

Within the pre-planned, protocol-specified intervention subgroups (see Figure 13) there are lower levels of statistical heterogeneity, suggesting that clinical heterogeneity deriving from differences in care regimens could underlie some of the observed between-trial differences in treatment effect. However, the risk of bias from other sources (see Risk of biases across studies and Chapter 6, Strengths and weaknesses of the systematic review) means that the summary estimates even from the subgroup analyses should be treated with great caution.
The main analysis as presented in Figure 13 includes only comparisons of 3% concentrations of HS with control interventions, as in the SABRE study. Two trials also included third arms delivering higher doses of HS. When these arms were included, the pooled effect size and associated heterogeneity are reduced marginally without altering the results appreciably (MD –0.34, 95% CI –0.48 to –0.21, using a FE analysis; MD –0.44, 95% CI –0.74 to –0.14, using RE; see Figure 14).
What is clear is that a large amount of the heterogeneity is driven by two trials from the same team, led by Luo,79,80 with outlying results, relatively small sample sizes but narrow CIs (around 1 day, compared with a 1.5 days in the SABRE study87 and the other large northern European study, Teunissen et al. 86). Together, the Luo et al. trials contribute 14.5% of the weight in the main analysis (see Figure 13). Their central estimates of clinical effect, –1.4 days79 and –1.6 days,80 are included in the CIs of only four of the remaining trials. 22–24,81 The removal of these two studies from the main analysis considerably reduces the effect sizes and statistical significance in the analyses of HS versus NS alone (MD –0.07 days, 95% CI –0.41 to 0.27 days; p = 0.67) and HS + beta-2 agonist versus NS + beta-2 agonist (MD –0.03 days, 95% CI –0.24 to 0.17 days; p = 0.74). The elimination of Luo79 from the beta-2 agonist subgroup reduces the heterogeneity from I2 = 82% to I2 = 0%. The elimination of these two studies reduces the overall FE pooled effect size from 0.36 days (95% CI –0.50 to –0.22 days; p < 0.00001; test for heterogeneity p < 0.00001, I2 = 78%) down to –0.16 days (–0.31 to –0.01 days; p = 0.03; test for heterogeneity p = 0.02, I2 = 32%). On the other hand, the elimination of even the three most negative studies from the main analysis (see Figure 13) makes little difference. 74,86,87 In short, the Luo studies are major drivers of heterogeneity.
Secondary outcomes: results and synthesis
There was no mention of AEs in seven studies;74,75,77,81,84,89,90 however, Maheshkumar et al. 81 stated ‘HS is safe’, implying that no AEs were observed, and Nemsadze et al. 84 was published only as an abstract. Tal et al. 23 stated that no AEs were observed in either of the groups. 23 Kuzik et al. reported that there were no withdrawals because of AEs by medical staff; however, two patients in the intervention group were withdrawn on parental request (one because the infant cried vigorously during two inhalations and the other because of agitation on one inhalation). 24 Luo et al. 79 stated in the first season that all patients completed treatment and that wheezing and coughing did not worsen over course of treatment. In the second season, Luo et al. 80 highlight that no infants were withdrawn as a result of AEs and that coughing and wheezing never worsened during treatment. Five infants (two in the intervention group) experienced hoarse voices but this improved after 3 or 4 days. Mandelberg et al. 22 report that no AEs were observed. Pulse rate and room air saturation did not differ between the two groups on any day. One patient was excluded from analysis because of a need for mechanical ventilation. Sharma et al. 82 also concur that no AEs related to any of the patients were reported by parents, caregivers or treating medical attendants in either group. Al-Ansari et al. 83 also state that no safety concerns or adverse reactions were identified. Pandit et al. 85 reported that vomiting and diarrhoea occurred in four infants, all in the control group; in addition, they noted no tremors or paleness during treatment. Teunissen et al. 86 reported 95 AEs in the 3% HS group, 119 in the 6% HS group and 76 in the NS group. Silver88 stated that only nine participants in the 3% HS group and four in the NS group did not complete the study because of AEs. According to additional information provided by the author, 10 participants in the HS arm and nine in the control arm were transferred to ICU/clinical worsening. Finally, for the SABRE study,87 AEs are described in detail in Table 10; overall, there were 51 AEs recorded in the standard care plus intervention group and 43 in the standard care group.
Readmission rate was not reported in 15 trials. Al-Ansari et al. 83 state that short-stay readmission was required for 10 infants in the 5% HS group, eight infants in the 3% HS group and seven infants in the 0.9% saline group. 83 Silver88 reported that four participants in the 3% HS group and three in the 0.9% NS group were readmitted within 7 days. In addition, Everard et al. 87 showed a lack of evidence to suggest that there was a difference between treatment groups in terms of the numbers admitted to HDU/ICU and readmitted within 28 days of randomisation. The final mean CSS scores were also analysed for the five studies for which data were available23,77,79,80,86 (Table 24). The average change of –1.36 points (95% CI –1.52 to –1.20 points) on the CSS observed in participants given HS (Figure 15) should be treated with caution because of the number of studies contributing data to this analysis and the risk of bias discussed in Risk of biases across studies and Chapter 6, Strengths and weaknesses of the systematic review.
| Main author | Intervention | Control | ||||
|---|---|---|---|---|---|---|
| Mean | SD | Number of participants | Mean | SD | Number of participants | |
| Giudice et al., 201277 | 6.5 | 1.6 | 52 | 7.7 | 1.6 | 54 |
| Tal et al., 200623 | 5.35 | 1.3 | 21 | 6.45 | 1 | 20 |
| Luo et al., 201079 | 1.50 | 0.50 | 50 | 2.9 | 0.7 | 43 |
| Luo et al., 201180 | 1.7 | 0.6 | 57 | 3.1 | 0.7 | 55 |
| Teunissen et al., 201486 | 3.87 (3% HS) | 3.15 (3% HS) | 84 (3% HS) | 4.61 | 5.38 | 80 |
| 5.16 (6% HS) | 4.20 (6% HS) | 83 (6% HS) | ||||
FIGURE 15.
Mean final CSS scores. IV, inverse-variables.
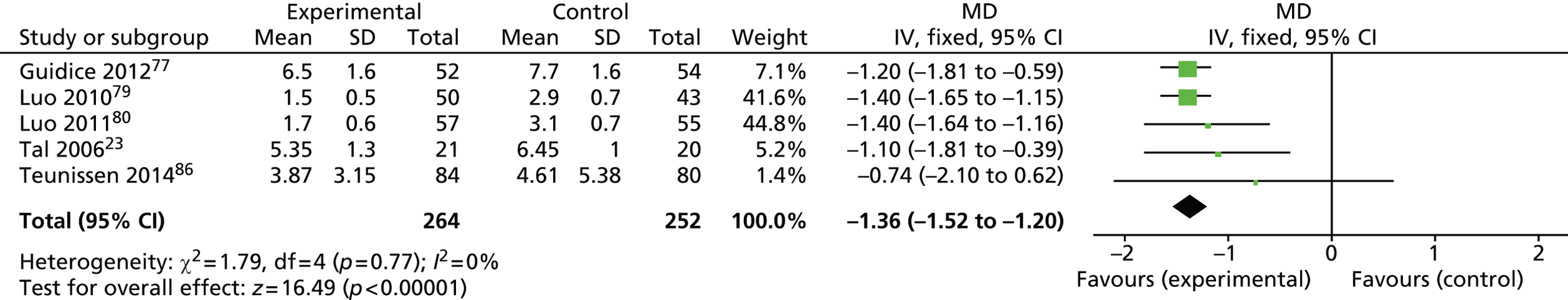
Risk of bias across studies
Publication bias
A funnel plot (plotting standard error against the MD) was generated to further assess the risk of bias across studies in relation to publication bias (Figure 16).
FIGURE 16.
Funnel plot, difference in length of hospital stay (whole group). SE, standard error.
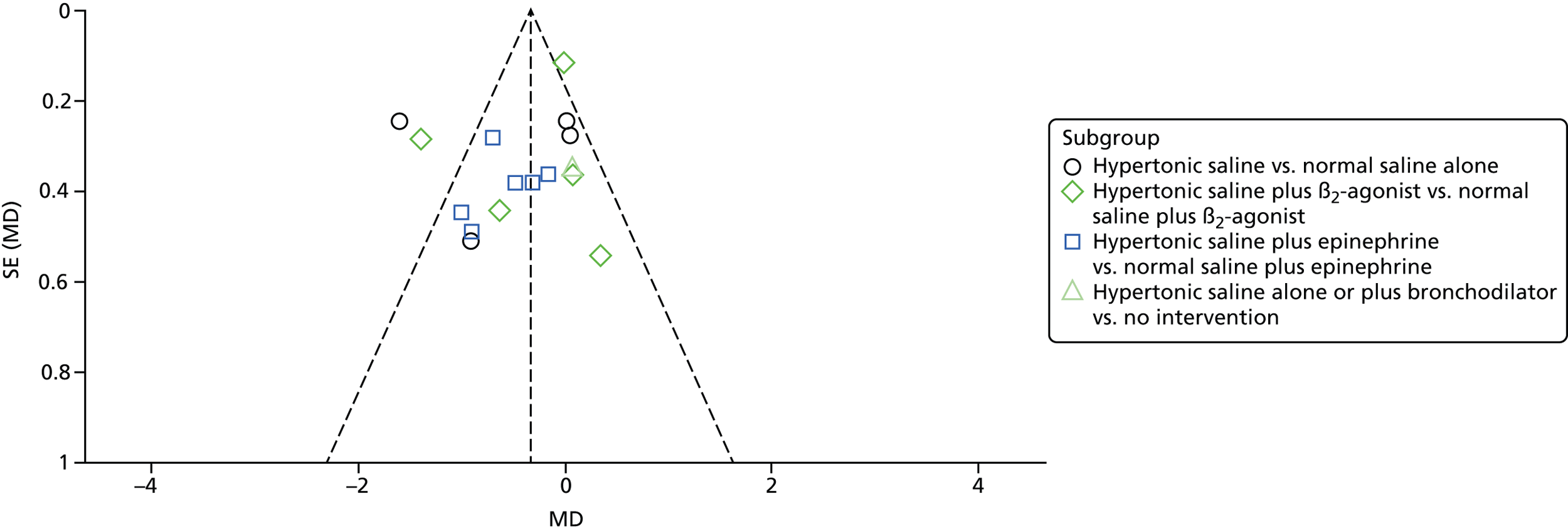
This is not a classically symmetrical funnel plot, in which the study with the smallest standard error (in this case Sharma et al. 82) sits at the top of the inverted funnel indicated by the dashed lines, which show the expected 95% CIs around the FE summary effect. But neither does it show the typical absence of small studies in the bottom right-hand corner, characteristic of meta-analyses affected by publication bias,121 although there are fewer studies to the right-hand side of the Sharma study82 than one would expect. Instead, we see four studies with comparatively small standard errors outside the dashed lines. These include the second and third largest studies (Sharma82 and Teunissen,86 respectively) as well as the two studies by Luo and colleagues. 79,80 A plausible explanation for this shape may be the high levels of heterogeneity discussed in Primary outcome: results and synthesis. 121
Outcome reporting bias
The results of the steps suggested by Dwan et al. 95 can be seen in Table 25.
| Main author | Primary outcome (LoS) | ORBIT classification | AEs | ORBIT classification | Hospital readmission rate | ORBIT classification | Final CSS score | ORBIT classification |
|---|---|---|---|---|---|---|---|---|
| Giudice et al., 201277 | X | N/A | O | F, low risk | O | I, no risk | X | N/A |
| Kuzik et al., 200724 | X | N/A | X | N/A | O | I, no risk | Partial (reported on different scale) | F, low risk |
| Tal et al., 200623 | X | N/A | X | N/A | O | I, no risk | X | N/A |
| Luo et al., 201079 | X | N/A | X | N/A | O | I, no risk | X | N/A |
| Luo et al., 201180 | X | N/A | X | N/A | O | I, no risk | X | N/A |
| Mandelberg et al., 200322 | X | N/A | X | N/A | O | I, no risk | Partial (only % decrease provided) | F, low risk |
| Espelt, 201274 | X | N/A | O | F, low risk | O | I, no risk | O | F, low risk |
| Maheshkumar et al., 201381 | X | N/A | Partial (concluded ‘it’s safe’) | F, low risk | O | I, no risk | Partial (final score not provided) | C, low risk |
| Sharma et al., 201382 | X | N/A | X | N/A | O | I, no risk | O | C, low risk |
| Al-Ansari et al., 201083 | X | N/A | X | N/A | X | N/A | O | I, no risk |
| Nemsadze et al., 201384 | Partial | C, low risk (abstract only) | O | F, low risk (abstract only) | O | H, low risk (abstract only) | O | H, low risk (abstract only) |
| Pandit et al., 201385 | X | N/A | X | N/A | O | I, no risk | O | I, no risk |
| Teunissen et al., 201486 | X | N/A | X | N/A | O | I, no risk | X | N/A |
| Everard et al., 201487 | X | N/A | X | N/A | X | N/A | O | I, no risk |
| Silver, 201488 | X | N/A | X | F, low risk (ClinicalTrials.gov only) | X | N/A | O | I, no risk |
| Ozdogan et al., 201489 | Partial | C, low risk (abstract only) | O | F, low risk (abstract only) | O | H, low risk (abstract only) | O | I, no risk |
| Sosa-Bustamante, 201475 | O | F, low risk (ClinicalTrials.gov only) | O | F, low risk (ClinicalTrials.gov only) | O | H, low risk (ClinicalTrials.gov only) | O | I, no risk |
| Ojha et al., 201490 | X | N/A | O | E, high risk | O | I, no risk | O | I, no risk |
The primary outcome was scored as ‘high risk’ for outcome reporting bias for one of the studies, owing to hospital LoS being stated as a secondary outcome, but no results were reported. 95 The majority returned a score of no risk (rate of readmission) or low risk/no risk where outcomes had been measured but partially reported, not necessarily analysed or not measured or analysed. Outcome reporting bias is concluded to be low for the review outcomes.
Chapter 6 Discussion
Main findings
The addition of nebulised 3% HS every 6 hours to usual care when treating infants admitted to hospital with acute bronchiolitis does not have discernible beneficial or harmful consequences when compared with usual care. In particular, the distributions of time taken for infants to be assessed as being fit for discharge were almost identical between the two arms, as was the actual time to discharge; the pre-specified minimum clinically important difference (18 hours or a 25% relative reduction) was at the very edge of the 95% CIs. Furthermore, the data showed no statistically significant differences in regards to readmission rates, admission to HDU/ICU or reported morbidity in the month following the acute illness.
There was considerable statistical heterogeneity in the analysis of the systematic review’s primary outcome, LoS (I2 = 75%), reflecting underlying clinical and methodological heterogeneity and indicating that formal pooling of studies is not a sound basis for decision-making in this instance.
The CUA estimated small and statistically insignificant differences in costs and QALYs between the control and intervention groups. The resultant ICER of £7.6M per QALY gained is the result of the very small difference in QALYs. When the uncertainty around the costs and QALYs is considered, nebulised saline has only a 34% chance of being cost-effective. This estimate may be considered high, given the very high ICER; however, it is completely in keeping with small differences associated with great uncertainty: there is approximately a 50% chance of it being cost-saving and approximately a 50% chance of it being effective. Based on this analysis, it is clear that the use of nebulised saline in this patient population is not cost-effective.
Strengths and weaknesses
Strengths and weaknesses of the trial
The sample size was calculated based on a difference in mean times but analysed using Cox regression. Although not ideal, doing so enabled us to calculate a minimum clinically important effect size in absolute units while analysing the data by methods which allow both for the likely skewed data and for any loss to follow-up. We presented both absolute and relative differences between time to fit for discharge and time to discharge. In the event, the use of Cox regression was unnecessary; only one patient had partial notes available.
Some readers may consider the absence of blinding a potential limitation of the study. A number of previous studies involving HS and other nebulised interventions such as the antiviral agent ribavirin have used a randomised blinded approach using either distilled water or NS as the nebulised placebo agent. These approaches have been criticised because hypo-osmolar distilled water can induce bronchoconstriction, thereby hindering the infant’s recovery, while proponents of hypertonic therapy have argued that failure to show a difference when compared with nebulised NS is because of the beneficial effects of the depositing NS in the lower airways. 26 The SABRE study team consider the use of an open pragmatic study design a strength, in that it avoids potential confounders and determines whether or not the use of nebulised HS may have a place in routine clinical practice. We believe that the primary outcome, assessed through routine records of hourly oxygen saturations, is robust against the risk of bias, regardless of the absence of blinding. To introduce systematic bias would have required multiple nurses across shifts and across 10 sites deliberately falsifying routine observations, which we do not believe is credible.
Among the strengths of this study was the multicentre design using teaching hospitals and district general hospitals, which ensures that the results are generalisable to all paediatric secondary care units admitting such infants while the numbers of subjects recruited are such that there can be considerable confidence in the conclusions drawn from the data. Moreover, the study was designed to assess the potential role of this intervention in the most severely affected infants, namely infants admitted to hospital requiring supplemental oxygen therapy. Such patients place a significant burden on the NHS and other health-care systems around the world. This is because of both the large numbers of infants admitted to hospital every winter and the duration of their hospitalisation which is significantly prolonged when compared with a median acute paediatric stay of around 24 hours. Hence they place a significant burden on paediatric units in terms of bed-days occupied. Nebulised 3% HS was used because this was the concentration used in the majority of previous studies and because there is a commercial product on the market being sold for this indication. For comparison, the concentration of salt in blood is 0.9% and that in sea water is 3.4% (range 3.2–3.8%).
Attrition rates were low for this study, although follow-up for some secondary outcomes was less satisfactory. Only 49 out of 149 participants returned the diary and ITQoL in the control group and only 54 out of 141 did in the intervention group. Of 142 participants from the control group, 40 were not RSV tested; in the intervention group 39 out of 149 were not tested.
Strengths and weaknesses of the economic analysis
There are some uncertainties in the health economic analysis. LoS data were missing for one patient, and after excluding those with only limited resource use we have 64% missing data relating to primary care use. We also elected not to include the costs of concomitant medications into estimates. However, it is not considered plausible that any of these will materially affect the results of the CUA. Uncertainties around unit costs for the major cost drivers were explored in sensitivity analyses and were found to have no impact on the results, owing to the lack of any differences in LoS.
There are also some uncertainties with the methods employed. The two most important relate to hospital cost calculations and the utility values. In terms of the hospital costs, the cost per day figures shown in Table 2 are derived from NHS Reference Costs 201352 by dividing the relevant total cost by the number of hospital days. It is well recognised that, in some circumstances, such average costs will not accurately represent the costs attributable to a marginal change in the LoS. In other words, the cost of a patient’s final day in hospital is different from the average because typically they are less unwell just prior to discharge. Although this argument is relevant to surgical and long-stay patients, it is not thought to be particularly pertinent to the patient population within this trial. It should also be recognised that these costs are economic, not financial; they represent the value of the resource rather than an identifiable expenditure.
The utility values used in the analysis are probably best described as notional. The generation of utility values for such a young population is extremely challenging and it would take a separate study to produce values that would be considered robust. Given the context of the study – a cheap intervention and large cost reductions associated with any reduction in LoS – the methods employed were considered appropriate. It could be argued that the use of a generic hospitalisation decrement might underestimate the impact of a severe, acute condition. However, the utility value for ICU hospitalisation was only slightly lower (0.87 vs. 0.94)57 and, so, even using this much more severe health state would have no significant effect on the results or conclusions. There is one further methodological uncertainty associated with the utility values from Saigal et al. ,57 which is that, while the authors describe their results as population values, the description of the elicitation task used does not make it clear whether the responses relate to those of a parent, those of a patient or a mixture of both. Consequently, in the light of the clinical results of the study, and the lack of robust evidence of a difference in LoS, it could be argued that the estimated QALY differences should be ignored.
The limitations of the utility estimates were highlighted at the design stage of the study and so, to guard against an over-reliance on the CUA and resultant ICER, we proposed that a CCA also be undertaken. In contrast to a CUA, a CCA does not focus on a single measure of outcome (i.e. the QALY) or produce a single definitive measure of cost-effectiveness (i.e. the ICER). A CCA is merely the comparison of costs and all relevant outcomes between the two arms of the study. As such, it requires readers to make their own assessment of whether or not the differences in outcomes are valued more highly than the costs. In the context of the SABRE study, the relevant outcomes are defined by the protocol as primary and secondary outcome measures: time to fit for discharge, actual time to discharge, admission to ICU/HDU, readmissions, duration of respiratory symptoms, health-care use, infant/parental quality of life and AEs. Across all these measures there is only one statistically significant difference between the study arms, relating to the family cohesion domain of the ITQoL. Given that no allowance has been made for multiple significance testing and the ITQoL result relates to a small self-selected subsample of the trial population, the robustness of this difference is open to question. Consequently, the CCA simplifies down to the cost difference reported in Chapter 4 (£132; p = 0.694) and no evidence of patient benefit.
Less importantly, another source of uncertainty is that the additional staff time required to manage a patient while on a nebuliser was not included in our analysis. Therefore, the costs in the intervention groups would be considered to be underestimated, albeit by a small amount.
Finally, the analysis employed a time frame of 36 days post randomisation, rather than the planned 28 days. This was because one patient had a time to discharge of 857 hours (35.7 days) and, thus, there were important costs beyond the time frame of the planned analysis. In order to conform to NICE economic evaluation guidelines, the time horizon of the analysis was adjusted to the first whole day following discharge of all patients. A sensitivity analysis was conducted that truncated time to discharge at 28 days and this did not have any noticeable impact on the results or conclusions.
Strengths and weaknesses of the systematic review
The results of the systematic review are problematic to interpret because of high levels of statistical heterogeneity. While it was not within the scope and resources of our review to investigate this between-study variation with a formal meta-regression, some notable sources of clinical and methodological heterogeneity may provide an explanation (Table 26). Across intervention subgroups, some of this heterogeneity may be explained by the presence of concomitant medications, with the effect of HS being largest in trials in which concomitant medications were not given. This notwithstanding, two trials conducted in China with small sample sizes (n = 9379 and n = 11280) but small standard errors appear to be driving the I2 value (see Chapter 5, Results and meta-analyses). As the quality of the studies seems adequate, it may be unfair to assume that there is some kind of fault with the individual trials. On the other hand, with the funnel plot (see Figure 16) suggestive of publication bias, it may be that there are a large number of trials missing as a result both of our restrictive eligibility criteria (the inclusion of only English-language publications) and of the documented publication bias associated with certain countries. 122
| Source | Example | Potential impact |
|---|---|---|
| Diagnostic |
|
|
| Management |
|
|
| Outcomes |
|
|
| Publication bias |
|
|
The systematic review was restricted to English-language articles because of the time and resource available for this review. The effect of this eligibility criterion has unknown consequences for the effect size, precision, heterogeneity and overall risk of bias associated with our meta-analysis. Although such restrictions can be seen as acceptable,123 we acknowledge the resulting exclusion of two trials of definite relevance at the full-paper stage68,69 and eight studies of possible relevance at the abstract stage59–64 is a limitation. 124 The use of language restrictions in systematic review-based meta-analyses is not always thought to cause systematic error, although it will potentially reduce the precision of pooled estimates. 125
The availability of adequate primary outcome data for all but one of the studies was a strength of the review, allowing for meta-analysis, subgroup analyses and investigation of publication bias. However, complete data were not available for all secondary outcomes or on features of trial design. The availability of more complete trial data may have enabled investigation of the underlying causes of statistical heterogeneity through metaregression,126 although this is not recommended when adequate data are only available for fewer than 10 studies. 76 It seems likely that disease severity and concomitant medications as well as local service configuration may be affecting between-study variation in effect size, but there are several other potential sources of heterogeneity which severely limit the use of aggregated level meta-regression analyses. In the absence of extensive individual patient data, the impact of these must remain speculative.
Unsuccessful attempts were made to contact trial investigators of one unpublished study74 to request additional unreported data. Although studies would not have been excluded based on the primary outcome, all but one of the studies were appropriate to be included in the meta-analysis of the primary outcome of interest, LoS. A number of the domains in the risk of bias assessment could not be assessed because of inadequate reporting. The biggest issue was with the ‘selective outcome reporting’ domain for which the absence of published protocols or responses from study teams made it difficult to assess pre-specified outcome measures. Attempts to contact the authors of one trial were unsuccessful,74 and the data retrieved from another author were of limited utility, being only in abstract form. 84 Similarly, incomplete and heterogeneous reporting the analysis of the final CSS score involved only five studies. Finally, there were two trials with three arms, in each case an arm with no HS, a 3% HS arm and an arm with a higher concentration of HS, either 5% (Al-Ansari) or 6% (Teunissen). We chose to compare both doses against control rather than combine them into one arm, since this might have helped in understanding and explaining some of the heterogeneity – although it seems unlikely that concentration of HS alone would explain all of the heterogeneity.
The synthesis of the systematic review’s primary outcome (LoS) is not in itself straightforward. The included studies were conducted in different countries and health-care settings with local guidelines on the assessment of infants as fit for discharge. No study reported on whether or not investigators adhered to these guidelines. We have no way of telling whether or not the difference between fitness for discharge and actual discharge (caused by the time and day of, consultant availability, etc.) was even across study arms, with only one study acknowledging this as a limitation. 23 Certainly, the definition of discharge fitness criteria differed across the trials – as indeed did the starting point, being sometimes from admission, sometimes from randomisation and sometimes from treatment start.
Of the 14 studies in the review, two of the studies reported hospital readmission rate. As previously discussed, this is an outcome of clinical relevance, certainly in the UK, as suggested by SIGN4 and NICE, but only two of the trials (both of which were non-UK based studies) reported on this outcome, which has potential health and economic implications. The Outcome Reporting Bias In Trials (ORBIT) classification (see Table 25) showed that the majority of readmission rates were classified as I (no risk of outcome bias), as this was potentially not considered important by trials included. However, lack of readmission data may mean that although the treatment is effective in the short term, patients are being readmitted and, therefore, the intervention may not be as economically beneficial as the summary result suggests. AEs may also be omitted by this scenario.
Overall, the defining feature of this systematic review is its heterogeneity, which limits any attempt to synthesise the evidence. The dissimilarity of the SDs of LoS indicates that the studies have taken place in very different settings. Two trials (n = 9679 and n = 11280) reported SDs of less than 1 day, indicating that their patients were typically discharged within a reasonably short time of each other. At the other extreme, the SABRE study and the other large northern European study86 reported SDs of over 3 days. This has consequences for the statistical analysis, as larger SDs lead to less study weight. The SABRE study87 contributed less than 4% of the total weight, despite being the largest of the 15 studies in the meta-analysis, and the Teunissen et al. study86 (n = 247) contributed 7.1% of the weight. The fact that large trials are contributing so little weight is counterintuitive and makes any overall summary of the 15 trials highly contentious. Of the possible sources of heterogeneity, the background care (in particular, concomitant medications) seems a likely explanation for at least some of these differences. In view of the number of ways by which the studies differed, however, we have not undertaken any formal meta-regression to attempt to explain this.
The question of whether to favour a FE or RE approach for the analysis – and for meta-analyses in general – is a controversial and unresolved issue. 92,127,128 The FE assumes a common underlying effect of treatment, which is highly questionable in the presence of heterogeneity. 92 The RE approach reweights studies to incorporate both the precision of the individual trial and the variability among trials into the weighting, but as a consequence all studies receive similar weights when between-trial variability is excessive and unexplained. 129 Neither approach seems satisfactory for situations such as this, and indeed we recommend against using an overall average however calculated. Rather, we suggest that the reader consider the specific setting within which each study was undertaken and apply findings only of those which appear consistent with their own setting.
Doing so, our recommendation for the UK NHS setting is unequivocal: hospital stay is not affected by adding HS to the care package. Although findings varied across the trials identified by the systematic review, the trials carried out in systems most comparable to the UK demonstrated almost zero difference between HS and the control treatments. The findings within our own trial were consistent across the 10 centres that recruited into the trial. Patients testing positive for RSV tended to remain in hospital longer than those testing negative, but HS did not appear to act differentially between the two subgroups.
The trial in context – other studies and differences in results
The SABRE study
This study was designed to understand the effects of regular nebulised 3% HS on reducing the time until an infant is ready for discharge and does not address another disputed and key question. We were precluded from assessing whether or not 3% HS provides short-term temporary symptomatic relief due to the lack of blinding and the subjective nature of current scoring systems; previous studies have shown a correlation between scores produced with, for example, the Wang score and objective physiological assessments such as those obtained with pulse oximetry. 78
The results presented in the SABRE study appear to conflict with the conclusions of a Cochrane review and the update to it presented herein. 25 The review reported a significant reduction in the LoS of patients admitted with acute bronchiolitis. 25 Of the five studies included, two were conducted by the same group (but with different patients), with Tal et al. 23 (n = 41) being an extension of the original study from Mandelberg et al. 22 (n = 52). The same centre has also published a positive study in a group of ambulatory subjects. Two studies followed from China,79,80 using 0.9% or 3% saline to deliver salbutamol in ‘mild to moderate subjects’ and 0.9% or 3% saline apparently without salbutamol in more severe patients. The severity of entry was assessed using the Wang score but there is no report of saturations, need for oxygen therapy or other assessments of severity. 78–80 Although the LoS was reduced in the group treated with 3% HS (twice hourly for three doses, four times hourly for five doses, then six times hourly) the mild to moderate group had a significantly longer stay than the moderate to severe group (7.4 days vs. 6.4 days in the 0.9% HS treated groups). 79,80 It is of interest to see how many studies use the Wang scoring system despite the conclusion in the abstract from the Wang et al. publication that ‘the limited agreement for clinical signs makes comparison of patient illness severity between studies difficult’. 78 This is one of the reasons that we did not use scoring systems, as they are not validated and almost certainly generate fewer robust data than, for example, the saturation recorded in room air and duration of oxygen therapy. One study from Italy reported a decrease in LoS when 0.9% or 3% HS was used to nebulise adrenaline but, interestingly, not a single patient was discharged prior to day 4. 77 The final study included 64 patients from the United Arab Emirates and 32 from Canada, with LoS being defined by a physician as being ready for discharge. 24
One study83 reporting a non-significant difference in LoS in those treated with 0.9%, 3% and 5% HS involving 165 patients was left out of the inpatient analysis in the Cochrane review58 for reasons that are unclear. These relatively mild patients were admitted to a short stay unit, with a short stay being defined as fewer than 8 days, so in effect they were hospitalised. 83 Two studies, which had been presented in 2011 as abstracts, that failed to identify a significant difference in LoS were also not included. One study from The Netherlands involving 160 subjects found no difference in LoS when 0.9%, 3% and 6% saline nebulised four times hourly were compared,130 while an Italian study involving 30 patients comparing 2.5 ml of 0.9% saline with 7% saline plus 0.1% hyaluronic acid at a dose of 2.5 ml twice a day for 3 days was also negative for the primary end point of LoS. 112
Since the Cochrane review25 was published, results from a number of studies have consistently found no reduction in hospitalisation rates from the use of HS. These include 250 participants from a comparison of 0.9% and 3% HS reported by Sharma from India, 82 participants from an Argentinian study comparing the same interventions and 40 participants from a second Indian study also comparing the two interventions. 74,81,82 Another study from Georgia was reported in abstract form in 2013, in which HS was associated with a reduction in LoS of half a day (albeit not statistically significant) in comparison with NS. 84 The abstract states that 42 patients were randomised, but it does not state the number in each group, or how many were included in the analysis. Since this study would account for at most 2% weight in a FE analysis, we have excluded it from this analysis. Thus, in total, 455 patients have participated in studies which have failed to show an impact on LoS, in addition to the 291 participants included in this study.
Sources of heterogeneity
In undertaking this systematic review it has become apparent that there are a number of semantic, methodological and cultural differences across the studies, all of which impact on the results obtained and the generalisability of an individual trial’s findings. We propose some of these factors and offer an explanation for how these may impact on the interpretation of the review’s findings.
1. The definition of ‘acute bronchiolitis’
The definition of ‘acute bronchiolitis’ differs between countries and indeed across clinicians in the same institution. Inevitably this diversity was reflected in the description of infants included, which variously specified wheeze and/or crackles (n = 4);24,83,86,87 a first episode of wheezing (n = 5);77,79,80,82,85 ‘bronchiolitis’ (n = 4);23,88,81,90 or bronchiolitis with a temperature > 38°C (n = 1);22 while information was absent in four others. 74,75,84,89 The term ‘wheeze’ is itself open to interpretation (and sometimes misinterpretation) within the medical profession;131–135 using this as a criterion may have led to the inclusion of asthmatic patients in whom bronchospasm is a feature. Pre-school children who are wheezing and who have a respiratory viral infection are more likely to have a ‘wheezy bronchitis’ than asthma, but among school-age children the opposite is true. Therefore, patients with clear wheeze and no (or few) crackles are likely to include a number of patients experiencing their first exacerbation of asthma and a much larger number of patients with what might be termed wheezy bronchitis – patients with a viral bronchitis who wheeze due to airways oedema and intraluminal secretions but without significant bronchoconstriction. The risk of including asthmatic subjects is therefore reduced by concentrating on the younger patients. Five studies limited inclusion to children under 12 months22,23,85,87,88 and a further two studies included up to 18 months;24,83 the remainder extended up to 24 months. 74,75,77,79–81,84,89,90,86 The mean age at study entry varied from 2.3 months to 8.61 months. This may have impacted the findings in the two Luo et al. studies79,80 (mean ages between 5.6 and 6.0 months), although positive results were also reported in the studies with the youngest mean age (mean ages between 2.3 and 3 months). 22,23
The patients included in this study met the definition of acute bronchiolitis as used in the UK, Australia and parts of Europe. In these health-care settings, the term ‘acute bronchiolitis’ refers to an infant with an apparent viral infection who develops signs of lower respiratory tract disease with airflow obstruction manifested as increased work of breathing, hyperinflation of the chest and widespread crackles; they may or may not intermittently exhibit wheeze. Clearly, however, there are considerable differences in setting and in the types of patients included in different studies, with many RCTs of HS involving patients seen in the emergency department and the use of wheeze as the key criteria for making a diagnosis of acute bronchiolitis.
By including both RSV+ and RSV– patients, we have been able to show that the viral status does not alter the response to therapy and hence the results can be generalised to all units looking after infants with acute bronchiolitis irrespective of whether or not the unit undertakes viral testing. Consistent with previous studies, more than 85% of tested infants were positive for RSV.
2. Variation among discharge criteria
The consistency of the outcomes – specifically ‘length of stay’ and ‘fit for discharge’ – is self-evidently defined and assessed in very different ways across the studies, as noted above. In our study, LoS was calculated as the time between admission and leaving the hospital but this was not the case elsewhere. Studies set (sometimes arbitrary) criteria regarding when the patient stay started, including ‘from study entry, which was within 12 hours of admission’ (n = 2);24,77 from hospital admission (n = 3);82,83,87 or from first dose of study medication (n = 2);88,86 information was absent for the remaining 11 studies. 22,23,74,75,79–81,84,85,89,90 The reported time to entry into study varied from 3 hours to 24 hours and generally did not specify whether or not ‘entry’ corresponded to consent or first treatment. Delays of up to 24 hours prior to entry into the study would represent a huge proportion of admissions in units with mean stays of 72 hours or less.
The restrictive eligibility criteria in our trial resulted in the exclusion of a large number of infants who would have been eligible in other trials. However, of much greater significance are the criteria used to determine the end of the LoS period. There is an understandable desire to separate the impact of the disease and/or treatment from extraneous social factors that may impact on a patient leaving the ward (lack of transport, carers are ill, etc.) Hence, the studies have determined that the inpatient stay is terminated at the point a clinician deems them to be fit for discharge.
This in turn carries two major components that will impact on LoS, namely how the decision is made that a patient is fit for discharge and how often it is possible to make that decision. In our study we used a definition which is calculated from the routine nursing observations recorded hourly for each patient, and which can thereby be corroborated. Elsewhere, discharge criteria are largely determined by the prevalent medical culture within their setting. In one study,87 a continuous discharge criteria was used, but in at least five studies the decision to discharge was made only once a day,22,23,79,82,85 meaning the time of discharge is effectively a discrete outcome which occurs at intervals of 24 hours. This inevitably overestimates the real time taken to be fit for discharge. It is far from obvious if this overestimation would affect the treatment groups differentially, but we note that most of the positive studies are based on a once-daily clinical assessment. In the remaining studies the frequency of assessment for discharge was unclear.
The criteria for discharge ranged from in air of oxygen saturation 92% or greater (equivalent to a severe rating – greater than 8.9 points on the Wang et al. scale) and oral feeding > 75% of usual intake87 to no respiratory signs or symptoms for the previous 12 hours. 79,80 In countries with relatively short LoS, discharge was based largely on in-air oxygen saturation 92% or greater87,136 or 93% or greater118 and taking sufficient oral feeds to maintain hydration. In contrast, other studies,87,137 presumably based on local medical cultural convention, specified variously that the saturation should be 95% or greater, although patients could go earlier on clinical grounds; that the air saturation should be 96% or greater; or that the patient should be completely well with no signs or symptoms. The criteria that patients should be free of any signs or symptoms is curious, as it has been well documented that the symptoms associated with acute bronchiolitis persists for many days or weeks. In the largest and most detailed follow-up of patients hospitalised for bronchiolitis,137 fewer than one-third were symptom free by 10 days and the figure for those most severely affected was less than 10%. Clearly, therefore, the criteria for discharge differ considerably across health-care settings, as previously noted by Becker et al. ,47 who observed a marked variation in LoS of patients admitted with RSV bronchiolitis with very short admissions (median approximately 72 hours) in North America, the UK and northern Europe compared with significantly longer admissions in Germany and southern Europe. These longer admissions were associated with increased comorbidities, such as diarrhoea, which may be as a result of nosocomial infection resulting from longer admission times. This cultural difference is again noted, with none of the Italian patients in the study of Giudice being discharged before 72 hours, a period beyond the mean ‘length of stay’ in the Dutch, UK and North American studies. 47,86,87 Finally, the subjects in the Luo studies with mild to moderate79 bronchiolitis remained in hospital longer than those with more severe disease,80 a finding which is somewhat difficult to explain.
3. Publication, generalisability and other biases
This difference in practice may also, in large part, explain the differences in observed treatment effects in the large UK, Dutch and North American studies which found no benefit compared with the apparently large effect observed in other studies. 86-88 While early indications of a potential benefit may have been attributable to publication bias,138,139 the positive effects of later large studies may be attributable to study design and cultural effects. The history of treatment of acute bronchiolitis is littered with therapies initially embraced enthusiastically only to be found to have no impact when subject to more rigorous scrutiny. Current guidelines, in essence, note that the management remains much as it did 50 years ago with good supportive care based on correcting hypoxia, minima handling and ensuring adequate fluid intake. The list of therapies suggested and subsequently abandoned include selective beta agonists, the non-selective beta agonist adrenaline, the antiviral agent ribavirin, theophylline, physiotherapy, French physiotherapy, inhaled and/or systemic corticosteroids and montelukast. 4 It is of note that all the recent studies of HS have failed to demonstrate any benefit, yet the meta-analysis still appears to favour the treatment. This effect is largely driven by the relatively large studies of Luo et al. 79,80 and it is likely that this is explicable when considering discharge criteria in more detail (see 2. Variation among discharge criteria).
Since completion of the SABRE study, a study conducted in Scotland140 found that the use of oxygen saturation of 90% for the initiation of oxygen therapy resulted in better outcomes and shorter LoS than 92% without any increase in readmissions or other complications, which makes studies in very different cultural settings such as China of doubtful relevance to the UK and similar countries.
Strengths and weaknesses compared with earlier systematic reviews
As the results of both the SABRE study and our systematic review and meta-analysis contradict the findings of the Cochrane review team, it is worth outlining the timeline over which these pieces of work have been conducted. The first, the 2008 version of Zhang et al. ’s Cochrane review25 included three inpatient trials that were comparable to the SABRE study: Mandelberg et al. in 2003,22 Tal et al. in 200623 and Kuzik et al. in 2007. 24 These trials suggested that HS reduced the MD in length of inpatient stay by –0.94 days (95% CI –1.48 to –0.40 days; p = 0.0006), with no heterogeneity. The SABRE study grant application was made on 8 January 2010. The Zhang et al. Cochrane review25 was assessed as up to date on 6 June 2010. The SABRE study commenced on 6 October 2011 and recruited its first participant on 26 October 2011. Although the Cochrane website does not register this, the Zhang review was updated in 201171 to contain one additional inpatient trial (a total of four trials) comparable to the SABRE study: Luo et al. 79 At this time, HS appeared to reduce the MD in length of inpatient stay by –1.16 days (95% CI –1.55 to –0.77 days; p < 0.00001), with no heterogeneity. Finally, the review was updated by Zhang et al. in 2013,141 including two additional inpatient trials, Luo et al. 201180 and Giudice et al. 201277 At this time they also included, in their review of studies recruiting in the emergency department, a study which we have classified as an inpatient trial (Al-Ansari et al. 201083). We found the following studies that could have been identified by Zhang et al. ’s searches in April, week 4, 2013:141 Espelt in 201274 and Maheshkumar et al. in January 2013. 81 In addition, the following studies were captured by our review but were published after the Zhang et al. 2013141 update: Pandit et al. in May 2013,85 Sharma et al. in 2013,82 Nemsadze et al. in 2013,84 Teunissen et al. in 2014,86 Silver in 2014,88 Ojha et al. in 2014,90 Ozdogan et al. in 2014,89 Sosa-Bustamante et al. in 201475 and Everard et al. in 2014. 87 Overall, 11 additional studies (n = 1367 participants, excluding Sosa-Bustamante et al. in 201475 as the number of patients randomised is unknown) were included in our review, eight of which were included in the meta-analysis in addition to those in the Cochrane review 2013 update. Our systematic review protocol was registered on PROSPERO on 3 March 2014 and the protocol was revised on 18 December 2014 following peer-review comments. The searches for this systematic review were run up to 14 January 2015.
The meta-analysis in the 2013 update of the Cochrane review by Zhang and colleagues contained six RCTs (n = 480) evaluating the effect of HS on length of inpatient stay, the largest of which randomised 112 participants. 58 Our meta-analysis contains 15 RCTs (n = 1922), including three much larger studies, all of which show null results. The eligibility criteria and search strategies of the two reviews differed, as a result of which our review picked up a number of publications which, we believe, are relevant to clinical decision-making and which might have been picked up the Cochrane review team’s searches in April–May 2013. With more than 10 studies, we were able to investigate the potential for publication bias. 76 Importantly, our meta-analysis also identified much higher levels of statistical heterogeneity than those reported by the Cochrane team, beyond what would be expected from the addition of this number of studies. 142 The Cochrane review provided what might have seemed a secure basis for clinical decision-making, suggesting that HS reduced inpatient stay by over 1 day (six studies, n = 480, MD –1.15 days, 95% CI –1.25 to –0.15 days; p< 0.0001, I2 = 30%). Our review suggests that the evidence base is far more equivocal, heterogeneous and, with publication and other biases at work, unsafe as a platform for decision-making (15 studies, n = 1922, MD –0.34 days, 95% CI –0.48 to –0.21 days; p< 0.00001, I2 = 75). In the long-run, the resources and standards of Cochrane may allow non-English-language and unpublished studies not identified by, or excluded from, our study to be incorporated into a research synthesis. Nonetheless, these salient differences, observed between closely contemporary reviews, confirm that duplicate systematic reviews can sometimes be valuable143,144 rather than wasteful. 145
Meaning of the study and implications for clinicians or policy-makers
The results from this study would suggest that there is no benefit to be gained by the introduction of nebulised 3% HS into the routine care of infants with acute bronchiolitis. As such, it joins a long list of other interventions that have been advanced as therapy only to be shown subsequently to be ineffective in reducing the morbidity of this condition. These include selective beta-2 agonists, adrenaline, anticholinergic agents, inhaled and systemic steroids, leukotriene antagonists, specific monoclonal antibodies and antiviral agents. Management remains essentially as was the case more than 50 years ago, with good supportive care, including oxygen when required, being the cornerstone of optimal care. A strength of this study is the comparison with usual care, as a criticism of negative studies by those who are advocates of this intervention is that nebulised NS in itself is a beneficial treatment. This cannot be argued in the case of this study and it is hard to argue bias because the data regarding fit for discharge were obtained from the routine nursing and clinical notes and if there were a bias it would be more likely be in favour of the intervention, as many are keen to find an effective intervention. Those advocating HS may argue that the current data suggest that HS is only beneficial when used with another agent such as adrenaline. Previous studies have been heterogeneous in the use of co-therapies either specifying the use of drugs such as adrenaline and salbutamol with the saline or permitting ‘physician’-ordered therapies. Studies of selective and non-selective beta-agonists in the same setting as this study in the UK and Australia have failed to show any benefit and, hence, one must argue that there is synergy producing a therapeutic effect when neither alone appears to work. In the setting of very young infants with acute bronchiolitis in whom bronchoconstriction does not appear to be a factor, it is hard to understand how this might work.
The 3% HS product used in this study, MucoClear® 3%, is currently on the market and being marketed as an effective treatment for bronchiolitis. The Pari website notes ‘this can afford patients with acute bronchiolitis or cystic fibrosis considerable relief’. 146 Interestingly, the product was licensed not as a drug but as a ‘device’, thereby avoiding the considerable regulatory hurdles posed by the drug regulators and highlighting the low threshold for devices to become registered.
The robust data generated in this study and other recent studies do not support the suggestion that this intervention be subject to any further studies either alone or in combination with other agents shown not to be effective. The quest for an effective therapy may be fraught with difficulties because of the intensity of the host neutrophil response at the time of presentation. For most infants the illness peaks at around 24 hours after admission, plateaus and then resolves spontaneously. Viral titres and the intensity of inflammation that viral replication induces appear to peak and then start to wane relatively soon after the onset of lower respiratory tract symptoms which in part probably accounts for the lack of efficacy of antiviral and anti-inflammatory agents. There is little to suggest that bronchospasm is a significant contributing factor in the airways obstruction experienced during acute bronchiolitis while the intense neutrophilia is unlikely to respond to steroid therapy. If an agent, be it antiviral or anti-inflammatory, were to be effective, it appears to be likely that it would need to be administered during the interval between developing upper respiratory tract symptoms and significant lower respiratory tract symptoms, the interval often being 2 to 3 days. However, this is challenging. If the agent had general effects on the neutrophilic-dominated inflammation then viral testing is unlikely to be required but if viral specific this would require rapid testing of all infants who develop upper respiratory symptoms. Moreover, the number needed to treat in order to prevent an admission is likely to be high, with less than 2% of all infants with a cold developing a lower respiratory tract illness of such severity as to require hospitalisation and the risk being greatest in the youngest patients. This is reflected in the median age of admission, in weeks, among patients recruited to this study. Much higher numbers develop a less severe LRTI and there may well be benefit in addressing this morbidity above that gained from preventing admissions. As yet no such strategy has been developed.
Recommendations for future research
-
There are a number of ongoing controlled trials evaluating the use of HS for acute bronchiolitis (ClinicalTrails.gov NCT02233985 and NCT02029040). Updated systematic reviews and meta-analysis will be required as results for these studies become available.
-
Meta-analysis of therapy trials in acute bronchiolitis is hampered by the use of different outcomes between trials. Further research is needed, using the methods promoted by the COMET (Core Outcome Measures in Effectiveness Trials) initiative,147 to produce a core outcome set for acute bronchiolitis. This should include a validated severity score. 148
-
The benefit of high-flow oxygen, used at one centre during this study, is still uncertain, although it is rapidly being adopted in many hospitals in the UK and worldwide. An emergency care-based feasibility study has confirmed that high-flow oxygen is safe and that a RCT is feasible. 149 High-quality RCTs are needed to definitively answer whether or not high-flow oxygen is clinically effective and cost-effective compared with standard oxygen therapy in acute settings.
-
Although neither HS nor adrenaline is as effective as monotherapy for the treatment of acute bronchiolitis, the meta-analysis indicates the possibility of a synergistic interaction. At present, statistical heterogeneity suggests that the evidence base is insufficient to inform commissioning. The next update of the Cochrane review will have access to non-English language publications and unpublished data excluded by our review; this may result in the inclusion of sufficient studies to reduce statistical heterogeneity to levels which are, by convention, considered satisfactory for decision-making (< I2 = 50%). If this is not the case, then the commissioning of a further large study evaluating oxygen and best supportive care with or without combination HS and adrenaline may be warranted.
Chapter 7 Conclusions
The results from this large pragmatic study indicate that the addition of nebulised HS to good supportive care when treating infants admitted to hospital with acute bronchiolitis does not appear to cause any harm but confers no benefit. The economic analysis used two forms of analysis to consider the cost-effectiveness of nebulised HS in this patient population. A CUA based on hospital costs and patient health-related quality of life suggests a 34% chance that the intervention is cost-effective and an ICER of £7.6M per QALY is gained. This figure is generated by an estimated additional cost associated with the intervention and an estimated incredibly small additional patient benefit, both of which are highly uncertain. Based on this analysis, the intervention cannot be considered either clinically effective or cost-effective in the UK health context. Taking a broader view of benefits that includes any of the primary and secondary outcomes measures does not appreciably alter this conclusion, as only one statistically significant difference between the study arms is apparent with that result being open to considerable biases. Our updated systematic review suggests that there is too much heterogeneity to make a pooled estimate of effect across 13 studies of inpatient care a useful basis for decision-making and shows that large studies, and those from northern Europe, have negative results which support the findings of the SABRE study.
Acknowledgements
We gratefully acknowledge the hard work, support and advice from the following: Kate Leech, Joanne Tomlinson, Marie Phipps, Ruth Jones, Eric Roe, Helen Parker (University Hospital of North Staffordshire NHS Trust); Louise Blackburn, Rebecca Mottram, Marjorie Allen, Nicola Balatoni (Leeds Teaching Hospitals NHS Trust); Kelly Young, Ruth Skelton, Louise Akeroyd, Heather Rostron (Bradford Teaching Hospitals NHS Foundation Trust); Janet Queen (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Rachel Swingler (Calderdale and Huddersfield NHS Foundation Trust); Caroline Amphlett, Pauline Jones (University Hospital of Wales); Jennifer McKenna, Louise Willis, Rebecca Beckley (Oxford University Hospitals NHS Trust); Ian Sinha, Gail Wallace, Hannah Leyland, Timothy Henderson, Karen Phelan (Alder Hey Children’s NHS Foundation Trust); Sarah Shortland (Sheffield Children’s NHS Foundation Trust, Doncaster and Bassetlaw Hospitals NHS Foundation Trust, Rotherham NHS Foundation Trust); John Bane, Susan George, Jayne Clements (Sheffield Children’s NHS Foundation Trust); Catrin Barker, Carol Craig, Gill Swift (Alder Hey Children’s NHS Foundation Trust); Christine Brighouse-Johnson (Calderdale and Huddersfield NHS Foundation Trust); Mark Fairweather (Doncaster and Bassetlaw Hospitals NHS Foundation Trust); Susan Brammer, Paul Hollinshead, Carole Beardmore (University Hospital of North Staffordshire NHS Trust); Kathryn Bethune, Annette Stone, Annie Rainey (University Hospital of Wales); Sophia Khan (Bradford Teaching Hospitals NHS Foundation Trust); Caroline Bedford, Paula Smalley, Helen Cunliffe, Sian Collins (Leeds Teaching Hospitals NHS Trust); Laura Watson (Oxford University Hospitals NHS Trust) for pharmacy involvement; Colin O’Keeffe and Kirsty Sprange for relief study management; Joseph Clark (University of Sheffield) for study monitoring; Amanda Loban for support on data management concerns; Kylie Cross for administrative and clerical support; and, Paul Dimitri (R&D Director, Sheffield Children’s NHS Foundation Trust) and Gillian Gatenby (Research Directorate Manager, Sheffield Children’s NHS Foundation Trust) for their support on ethical and governance issues.
We offer special thanks to the members of our two oversight committees, the TSC and the DMEC. TSC: Jonathan Grigg (independent chairperson, Barts and The London School of Medicine and Dentistry), Eirini Koutoumanou (independent statistician, University College London); Jeremy Hull (independent clinician, Oxford University Hospitals NHS Trust); Penny Broadley (independent parent representative, Sheffield Children’s NHS Foundation Trust); Mark L Everard (chief investigator, University of Western Australia); Kelechi Ugonna (chief investigator, Sheffield Children’s NHS Foundation Trust); Elizabeth Cross, Colin O’Keeffe, Kirsty Sprange (trial manager, University of Sheffield); Wendy Swann (sponsor representative, Sheffield Children’s NHS Foundation Trust); Alexa Cross (HTA representative); and Jennifer Freeman (trials statistician, University of Leeds). DMEC: Tim Cole (chairperson, University College London), Neil Gibson (Royal Hospital for Sick Children, Glasgow) and Jayesh Bhatt (Nottingham University Hospitals NHS Trust).
The views expressed in this report are those of the authors and not necessarily those of the National Institute for Health Research Health Technology Assessment programme. Any errors are the responsibility of the authors.
Contributions of authors
Mark L Everard (Professor Paediatric Respiratory Medicine, University of Western Australia), Daniel Hind (CTRU assistant director, University of Sheffield), Kelechi Ugonna (Consultant In Paediatric Respiratory Medicine, Sheffield Children’s NHS Foundation Trust), Jennifer Freeman (Associate Professor, statistics, University of Leeds), Mike Bradburn (senior statistician, University of Sheffield), Simon Dixon (Professor of Health Economics, University of Sheffield), Chin Maguire (Trial Manager, University of Sheffield) and Hannah Cantrill (Research Assistant, University of Sheffield) together produced the first draft of the report.
The following authors conceived of or designed the work: Mark L Everard, Daniel Hind, Jennifer Freeman, Simon Dixon, Chin Maguire, Warren Lenney (Consultant Respiratory Paediatrician; University Hospital of North Staffordshire NHS Trust), Heather Elphick (Consultant in Paediatric Respiratory Medicine, Sheffield Children’s NHS Foundation Trust), Philip AJ Chetcuti (Consultant Paediatrician; Leeds Teaching Hospitals NHS Trust), Eduardo F Moya (Consultant Paediatrician, Bradford Teaching Hospitals NHS Foundation Trust), Colin Powell (Consultant Respiratory Paediatrician, University Hospital of Wales), Jonathan P Garside (Consultant Paediatrician, Calderdale and Huddersfield NHS Foundation Trust), Matthew Kurian (Consultant Paediatrician, Doncaster and Bassetlaw Hospitals NHS Foundation Trust), Peter I MacFarlane (Consultant Paediatrician, Rotherham NHS Foundation Trust) and Cindy L Cooper (CTRU Director).
The following authors were involved in the acquisition of data for the work: Mark L Everard, Kelechi Ugonna, Jennifer Freeman, Simon Dixon, John Alexander (Consultant Paediatric Intensive Care, University Hospital of North Staffordshire NHS Trust), Warren Lenney, Paul McNamara (Consultant Respiratory Paediatrician, Alder Hey Children’s NHS Foundation Trust), Heather Elphick, Philip AJ Chetcuti, Eduardo F Moya, Colin Powell, Jonathan P Garside, Lavleen Kumar Chadha (Consultant Paediatrician, Doncaster and Bassetlaw Hospitals NHS Foundation Trust), Matthew Kurian, Ravinderjit S Lehal (Consultant Paediatrician, Oxford University Hospitals NHS Trust), Peter I MacFarlane and Cindy L Cooper.
The following authors were involved in the analysis of data: Jennifer Freeman, Mike Bradburn, Simon Dixon, Chin Maguire and Hannah Cantrill.
Mark L Everard, Daniel Hind, Kelechi Ugonna, Jennifer Freeman, Mike Bradburn, Simon Dixon, Chin Maguire and Hannah Cantrill were involved in the interpretation of data for the work.
Mark L Everard, Daniel Hind, Kelechi Ugonna, Jennifer Freeman, Mike Bradburn, Simon Dixon, Chin Maguire and Hannah Cantrill drafted the monograph.
Mark L Everard, Daniel Hind, Kelechi Ugonna, Jennifer Freeman, Mike Bradburn, Simon Dixon, Chin Maguire, Hannah Cantrill, John Alexander, Warren Lenney, Paul McNamara, Heather Elphick, Philip AJ Chetcuti, Eduardo F Moya, Colin Powell, Jonathan P Garside, Lavleen Kumar Chadha, Matthew Kurian, Ravinderjit S Lehal, Peter I MacFarlane, Cindy L Cooper and Elizabeth Cross (Trial Manager, University of Sheffield) revised the work critically for important intellectual content.
Mark L Everard, Daniel Hind, Kelechi Ugonna, Jennifer Freeman, Mike Bradburn, Simon Dixon, Chin Maguire, Hannah Cantrill, John Alexander, Warren Lenney, Paul McNamara, Heather Elphick, Philip AJ Chetcuti, Eduardo F Moya, Colin Powell, Jonathan P Garside, Lavleen Kumar Chadha, Matthew Kurian, Ravinderjit S Lehal, Peter I MacFarlane, Cindy L Cooper and Elizabeth Cross were involved in the final approval of the version to be published.
All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
The SABRE study research team
Writing group
Mark L Everard, Daniel Hind, Kelechi Ugonna, Jennifer Freeman, Mike Bradburn, Simon Dixon, Chin Maguire, Hannah Cantrill, John Alexander, Warren Lenney, Paul McNamara, Heather Elphick, Philip AJ Chetcuti, Eduardo F Moya, Colin Powell, Jonathan P Garside, Lavleen Kumar Chadha, Matthew Kurian, Ravinderjit S Lehal, Peter I MacFarlane, Cindy L Cooper and Elizabeth Cross.
Trial management group
Daniel Hind, Elizabeth Cross, Colin O’Keeffe (trial manager, University of Sheffield), Kirsty Sprange (trial manager, University of Sheffield), Chin Maguire, Jennifer Freeman, Mike Bradburn, Simon Dixon, Mark L Everard, Kelechi Ugonna, Heather Elphick, Sarah Shortland (Sheffield Children’s NHS Foundation Trust, Doncaster and Bassetlaw Hospitals NHS Foundation Trust, Rotherham NHS Foundation Trust), Wendy Swann (Sponsor Representative, Sheffield Children’s NHS Foundation Trust), John Alexander, Kate Leech (University Hospital of North Staffordshire NHS Trust), Joanne Tomlinson (University Hospital of North Staffordshire NHS Trust), Marie Phipps (University Hospital of North Staffordshire NHS Trust), Ruth Jones (University Hospital of North Staffordshire NHS Trust), Eric Roe (University Hospital of North Staffordshire NHS Trust), Helen Parker (University Hospital of North Staffordshire NHS Trust); Philip AJ Chetcuti, Louise Blackburn (Leeds Teaching Hospitals NHS Trust), Rebecca Mottram(Leeds Teaching Hospitals NHS Trust), Marjorie Allen (Leeds Teaching Hospitals NHS Trust), Nicola Balatoni (Leeds Teaching Hospitals NHS Trust), Eduardo F Moya, Kelly Young (Bradford Teaching Hospitals NHS Foundation Trust), Ruth Skelton (Bradford Teaching Hospitals NHS Foundation Trust), Louise Akeroyd (Bradford Teaching Hospitals NHS Foundation Trust), Heather Rostron (Bradford Teaching Hospitals NHS Foundation Trust), Lavleen Kumar Chadha, Mathew Kurian, Janet Queen (Doncaster and Bassetlaw Hospitals NHS Foundation Trust), Jonathan P Garside, Rachel Swingler (Calderdale and Huddersfield NHS Foundation Trust), Colin Powell, Caroline Amphlett (University Hospital of Wales), Pauline Jones (University Hospital of Wales); Ravinderjit S Lehal, Jennifer McKenna, Louise Willis (Oxford University Hospitals NHS Trust), Rebecca Beckley (Oxford University Hospitals NHS Trust); and Paul McNamara, Ian Sinha (Alder Hey Children’s NHS Foundation Trust), Gail Wallace (Alder Hey Children’s NHS Foundation Trust), Hannah Leyland (Alder Hey Children’s NHS Foundation Trust), Timothy Henderson (Alder Hey Children’s NHS Foundation Trust), Karen Phelan (Alder Hey Children’s NHS Foundation Trust) and Peter I MacFarlane.
Trial Steering Committee
Jonathan Grigg (independent chairperson, Barts and The London School of Medicine and Dentistry), Eirini Koutoumanou (independent statistician, University College London), Jeremy Hull (independent clinician, Oxford University Hospitals NHS Trust), Penny Broadley (independent parent representative, Sheffield Children’s NHS Foundation Trust), Mark L Everard (chief investigator), Kelechi Ugonna (chief investigator), Elizabeth Cross, Colin O’Keeffe, Kirsty Sprange, Wendy Swann, Alexa Cross (HTA representative) and Jennifer Freeman (trial statistician).
Data Monitoring and Ethics Committee
Tim Cole (chairperson, University College London), Neil Gibson (Royal Hospital for Sick Children, Glasgow) and Jayesh Bhatt (Nottingham University Hospitals NHS Trust).
Co-applicants
Mark L Everard, Cindy L Cooper, Simon Dixon, Jennifer Freeman, Warren Lenney, Philip AJ Chetcuti, Eduardo F Moya, Matthew Kurian, Jonathan P Garside, Peter I MacFarlane, Heather Elphick and Colin Powell.
Local investigators
Mark L Everard, Kelechi Ugonna, Heather Elphick, John Alexander, Philip AJ Chetcuti, Eduardo F Moya, Lavleen Kumar Chadha, Matthew Kurian, Jonathan P Garside, Colin Powell, Ravinderjit S Lehal, Paul McNamara and Peter I MacFarlane.
Systematic review team
Mark L Everard, Daniel Hind, Chin Maguire and Hannah Cantrill.
Publications
Everard ML, Hind D, Ugonna K, Freeman J, Bradburn M, Cooper CL, et al. SABRE: a multicentre randomised control trial of nebulised hypertonic saline in infants hospitalised with acute bronchiolitis. Thorax 2014;69:1105–12.
Data sharing statement
Please direct any requests to: ctru@sheffield.ac.uk.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Elliot S, Ray C, Taussig L, Landau L. Pediatric Respiratory Medicine. Philadelphia, PA: Mosby Elsevier; 2009.
- Everard M, Taussig L, Landau L. Paediatric Respiratory Medicine. St Louis, MO: Mosby; 2009.
- Smyth RL, Openshaw PJM. Bronchiolitis. Lancet 2006;368:312-22. http://dx.doi.org/10.1016/S0140-6736(06)69077-6.
- Bronchiolitis in Children – A National Clinical Guideline. Edinburgh: SIGN; 2006.
- Subcommittee on Diagnosis and Management of Bronchiolitis . Diagnosis and management of bronchiolitis. Pediatrics 2006;118:1774-93. http://dx.doi.org/10.1542/peds.2006-2223.
- Deshpande SA, Northern V. The clinical and health economic burden of respiratory syncytial virus disease among children under 2 years of age in a defined geographical area. Arch Dis Child 2003;88:1065-9. http://dx.doi.org/10.1136/adc.88.12.1065.
- Martin AJ, Gardner PS, Mcquillin J. Epidemiology of respiratory viral infection among paediatric inpatients over a six-year period in North-East England. Lancet 1978;312:1035-8. http://dx.doi.org/10.1016/S0140-6736(78)92351-6.
- Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med 2009;360:588-98. http://dx.doi.org/10.1056/NEJMoa0804877.
- Murray J, Bottle A, Sharland M, Modi N, Aylin P, Majeed A, et al. PLOS ONE. 2014.
- Centers for Disease Control and Prevention (CDC) . Respiratory Syncytial Virus Activity - United States, July 2008-December 2009. MMWR 2010;59:30-3. www.cdc.gov/mmwr/preview/mmwrhtml/mm5908a4.htm (accessed 5 March 2014).
- Fleming DM, Elliot AJ, Cross KW. Morbidity profiles of patients consulting during influenza and respiratory syncytial virus active periods. Epidemiol Infect 2007;135:1099-108. http://dx.doi.org/10.1017/S0950268807007881.
- Elliot AJ, Fleming DM. Influenza and respiratory syncytial virus in the elderly. Expert Rev Vaccines 2008;7:249-58. http://dx.doi.org/10.1586/14760584.7.2.249.
- Falsey A, Hennessey P, Formica M, Cox C, Walsh E. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med 2005;352:1749-59. http://dx.doi.org/10.1056/NEJMoa043951.
- Reynolds E, Cook C. The treatment of bronchiolitis. J Pediatr 1963;63:1205-7. http://dx.doi.org/10.1016/S0022-3476(63)80215-2.
- Panickar JR, Dodd SR, Smyth RL, Couriel JM. Trends in deaths from respiratory illness in children in England and Wales from 1968 to 2000. Thorax 2005;60:1035-8. http://dx.doi.org/10.1136/thx.2005.044750.
- Fleming DM, Pannell RS, Cross KW. Mortality in children from influenza and respiratory syncytial virus. J Epidemiol Community Health 2005;59:586-90. http://dx.doi.org/10.1136/jech.2004.026450.
- Hospital Episode Statistics 2009–10. Leeds: Health and Social Care Information Centre; 2010.
- Pelletier AJ, Mansbach JM, Camargo CA. Direct medical costs of bronchiolitis hospitalizations in the United States. Pediatrics 2006;118:2418-23. http://dx.doi.org/10.1542/peds.2006-1193.
- Buckingham SC, Quasney MW, Bush AJ, DeVincenzo JP. Respiratory syncytial virus infections in the pediatric intensive care unit: clinical characteristics and risk factors for adverse outcomes. Pediatr Crit Care Med 2001;2:318-23. http://dx.doi.org/10.1097/00130478-200110000-00006.
- Thorburn K, Kerr S, Taylor N, van Saene HKF. RSV outbreak in a paediatric intensive care unit. J Hosp Infect 2004;57:194-201. http://dx.doi.org/10.1016/j.jhin.2004.03.013.
- Sarrell EM, Tal G, Witzling M, Someck E, Houri S, Cohen HA, et al. Nebulized 3% hypertonic saline solution treatment in ambulatory children with viral bronchiolitis decreases symptoms. Chest 2002;122:2015-20. http://dx.doi.org/10.1378/chest.122.6.2015.
- Mandelberg A, Tal G, Witzling M, Someck E, Houri S, Balin A, et al. Nebulized 3% hypertonic saline solution treatment in hospitalized infants with viral bronchiolitis. Chest 2003;123:481-7. http://dx.doi.org/10.1378/chest.123.2.481.
- Tal G, Cesar K, Oron A, Houri S, Ballin A, Mandelberg A. Hypertonic saline/epinephrine treatment in hospitalized infants with viral bronchiolitis reduces hospitalization stay: 2 years experience. Isr Med Assoc J Imaj 2006;8:169-73.
- Kuzik BA, Al-Qadhi SA, Kent S, Flavin MP, Hopman W, Hotte S, et al. Nebulized hypertonic saline in the treatment of viral bronchiolitis in infants. J Pediatr 2007;151:266-70. http://dx.doi.org/10.1016/j.jpeds.2007.04.010.
- Zhang L, Mendoza-Sassi RA, Wainwright C, Klassen TP. Nebulized hypertonic saline solution for acute bronchiolitis in infants. Cochrane Database Syst Rev 2008;4. http://dx.doi.org/10.1002/14651858.cd006458.pub2.
- Mandelberg A, Amirav I. Hypertonic saline or high volume normal saline for viral bronchiolitis: mechanisms and rationale. Pediatr Pulmonol 2010;45:36-40. http://dx.doi.org/10.1002/ppul.21185.
- Boogaard R, de Jongste JC, Merkus PJFM. Pharmacotherapy of impaired mucociliary clearance in non-CF pediatric lung disease. A review of the literature. Pediatr Pulmonol 2007;42:989-1001. http://dx.doi.org/10.1002/ppul.20693.
- Gadomski AM, Bhasale A. Bronchodilators for bronchiolitis. Cochrane Database Syst Rev 2006;12. http://dx.doi.org/10.1002/14651858.cd001266.pub2.
- Hartling L, Wiebe N, Russell K, Patel H, Klassen TP. Epinephrine for bronchiolitis. Cochrane Database Syst Rev 2004;1. http://dx.doi.org/10.1002/14651858.cd003123.pub2.
- Brooks LJ, Cropp GJ. Theophylline therapy in bronchiolitis. A retrospective study. Am J Dis Child 1981;135:934-6. http://dx.doi.org/10.1001/archpedi.1981.02130340040014.
- Everard M, Bara A, Kurian M, N’Diaye T, Ducharme F, Mayowe V. Anticholinergic drugs for wheeze in children under the age of two years. Cochrane Database Syst Rev 2005;3. http://dx.doi.org/10.1002/14651858.cd001279.pub2.
- Grewal S, Ali S, McConnell DW, Vandermeer B, Klassen TP. A randomized trial of nebulized 3% hypertonic saline with epinephrine in the treatment of acute bronchiolitis in the emergency department. Arch Pediatr Adolesc Med 2009;163:1007-12. http://dx.doi.org/10.1001/archpediatrics.2009.196.
- Anil AB, Anil M, Saglam AB, Cetin N, Bal A, Aksu N. High volume normal saline alone is as effective as nebulized salbutamol-normal saline, epinephrine-normal saline, and 3% saline in mild bronchiolitis. Pediatr Pulmonol 2010;45:41-7. http://dx.doi.org/10.1002/ppul.21108.
- Everard ML. Acute bronchiolitis and croup. Pediatr Clin N Am 2009;56:119-33. http://dx.doi.org/10.1016/j.pcl.2008.10.007.
- Elphick H, Ritson S, Rigby A, Everard M. Phenotype of acute respiratory syncytial virus induced lower respiratory tract illness in infancy and subsequent morbidity. Acta Paediatr 2007;96:307-9. http://dx.doi.org/10.1111/j.1651-2227.2007.00082.x.
- Plint AC, Osmond MH, Klassen TP. The efficacy of nebulized racemic epinephrine in children with acute asthma: a randomized, double-blind trial. Acad Emerg Med 2000;7:1097-103. http://dx.doi.org/10.1111/j.1553-2712.2000.tb01258.x.
- Hartling L, Wiebe N, Russell K, Patel H, Klassen TP. A meta-analysis of randomized controlled trials evaluating the efficacy of epinephrine for the treatment of acute viral bronchiolitis. Arch Pediatr Adolesc Med 2003;157:957-64. http://dx.doi.org/10.1001/archpedi.157.10.957.
- Wainwright C, Altamirano L, Cheney M, Cheney J, Barber S, Price D, et al. A multicenter, randomized, double-blind, controlled trial of nebulized epinephrine in infants with acute bronchiolitis. N Engl J Med 2003;349:27-35. http://dx.doi.org/10.1056/NEJMoa022226.
- Hariprakash S, Alexander J, Carroll W, Ramesh P, Randell T, Turnbull F, et al. Randomized controlled trial of nebulized adrenaline in acute bronchiolitis. Pediatr Allergy Immunol 2003;14:134-9. http://dx.doi.org/10.1034/j.1399-3038.2003.00014.x.
- Cade A, Brownlee KG, Conway SP, Haigh D, Short A, Brown J, et al. Randomised placebo controlled trial of nebulised corticosteroids in acute respiratory syncytial viral bronchiolitis. Arch Dis Child 2000;82:126-30. http://dx.doi.org/10.1136/adc.82.2.126.
- Everard M, Swarbrick A, Rigby A, Milner A. The effect of ribavirin to treat previously healthy infants admitted with acute bronchiolitis on acute and chronic respiratory morbidity. Respir Med 2001;95:275-80. http://dx.doi.org/10.1053/rmed.2001.1033.
- Everard ML. The role of the respiratory syncytial virus in airway syndromes in childhood. Curr Allergy Asthma Rep 2006;6:97-102. http://dx.doi.org/10.1007/s11882-006-0046-z.
- Calogero C, Sly PD. Acute viral bronchiolitis: to treat or not to treat-that is the question. J Pediatr 2007;151:235-7. http://dx.doi.org/10.1016/j.jpeds.2007.05.041.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:1-28. http://dx.doi.org/10.1136/bmj.c869.
- Great Britain . The Medicines for Human Use (Clinical Trials) Regulations 2004 2004.
- Raat H, Landgraf J, Oostenbrink R, Moll H, Essink-bot M. Reliability and validity of the Infant and Toddler Quality of Life Questionnaire (ITQOL) in a general population and respiratory disease sample. Qual Life Res 2007;16:445-60. http://dx.doi.org/10.1007/s11136-006-9134-8.
- Behrendt CE, Decker MD, Burch DJ, Watson PH. International variation in the management of infants hospitalized with respiratory syncytial virus. International RSV Study Group. Eur J Pediatr 1998;157:215-20. http://dx.doi.org/10.1007/s004310050798.
- Elashoff J, Reedy T. Two-stage clinical trial stopping rules. Biometrics 1984;40:791-5. http://dx.doi.org/10.2307/2530923.
- Perrotta C, Ortiz Z, Roque M. Chest physiotherapy for acute bronchiolitis in paediatric patients between 0 and 24 months old. Cochrane Database Syst Rev 2005;2. http://dx.doi.org/10.1002/14651858.cd004873.pub2.
- Therneau TM, Grambsch P. Modeling Survival Data: Extending the Cox Model. New York, NY: Springer-Verlag; 2000.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2013.
- NHS Reference Costs 2013. London: DH; 2013.
- Curtis L. Unit Costs of Health and Social Care 2013. Canterbury: PSSRU, University of Kent; 2013.
- Turner J, O’Cathain A, Knowles E, Nicholl J, Tosh J, Sampson F, et al. Evaluation of NHS 111 pilot sites. Sheffield: University of Sheffield, School of Health and Related research (ScHARR); 2012.
- Carroll AE, Downs SM. Improving decision analyses: parent preferences (utility values) for pediatric health outcomes. J Pediatr 2009;155:21-5. http://dx.doi.org/10.1016/j.jpeds.2009.01.040.
- Ara R, Wailoo A. The Use of Health State Utility Values in Decision Models. NICE Decision Support Unit Technical Support Document 12. Sheffield: University of Sheffield, School of Health and Related Research (ScHARR); 2011.
- Saigal S, Feeny D, Furlong W, Rosenbaum P, Burrows E, Torrance G. Comparison of the health-related quality of life of extremely low birth weight children and a reference group of children at age eight years. J Pediatr 1994;125:418-25. http://dx.doi.org/10.1016/S0022-3476(05)83289-5.
- Zhang L, Mendoza-Sassi RA, Wainwright C, Klassen TP. Nebulised hypertonic saline solution for acute bronchiolitis in infants. Cochrane Database Syst Rev 2013;7. http://dx.doi.org/10.1002/14651858.cd006458.pub3.
- Liu J, Qiao B, Ma C, Zhang D, Zhang J. Treatment of bronchiolitis with inhaled nebulized hypertonic saline solution combined with budesonide. J Clin Pediatr 2013;31:440-2.
- Zhang GJ. Observation of the effects of 4.3% hypertonic saline and compound ipratropium bromide inhalation in treating bronchiolitis. Chinese J Med Guid 2013;10.
- Zhou Y, Lu YD, Tan XJ, Zhang CP. Impact of hypertonic saline combined with salbutamol inhalation on the infectious bronchiolitis. China Med 2013;8.
- Wang H. The mechanism of nebulized drugs in the treatment of patients with exacerbation of bronchiolitis. Int J Pediatr 2013:450-3.
- Zhang Y. Clinical effects and safety of hypertonic saline combined with budesonide via aerosol inhalation in treatment of bronchiolitis. Anhui Med J 2013;34:1333-5.
- Ramilo O, Mejias A. Novedades en el tratamiento de la bronquiolitis: perpectivas en el 2013. An Pediatr 2013;78:205-7. http://dx.doi.org/10.1016/j.anpedi.2013.02.017.
- Nüßlein T. Hypertone Salzlösung zur Inhalation bei akutem Giemen im Vorschulalter. Der Pneumol 2014;11:171-2. http://dx.doi.org/10.1007/s10405-014-0778-8.
- Xu J. Cost-effectiveness analysis of hypertonic saline combined with low-dose budesonide inhalation in mild to moderate bronchiolitis. J Pediatr Pharm 2013;19:38-40.
- Li G, Jing Z. Effectiveness of inhaled hypertonic saline in children with bronchiolitis. Chinese J Pediatr 2014;52:607-10.
- Park JY, Jeong YM, Jeong SJ, Seo SS. The efficacy of nebulized 3 percent hypertonic saline solution and fenoterol in infants with bronchiolitis. Korean J Pediatr 2005;48:518-22.
- Zheng W, Li L, Chengfung H, Yunmei H, Wei L. The effects of high frequency inhalation of the 3% hypertonic saline solution with ambroxol hydrochloride in the treatment of 43 bronchiolitis patients. J Pediatr Pharm 2012;18:19-21.
- Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Health Care. York: University of York; 2009.
- Zhang L, Mendoza-Sassi RA, Wainwright C, Klassen TP. Nebulized hypertonic saline solution for acute bronchiolitis in infants. Cochrane Database Syst Rev 2009;4. http://dx.doi.org/10.1002/14651858.CD006458.pub2.
- Papaioannou D, Sutton A, Carroll C, Booth A, Wong R. Literature searching for social science systematic reviews: consideration of a range of search techniques. Health Info Libr J 2010;27:114-22. http://dx.doi.org/10.1111/j.1471-1842.2009.00863.x.
- Schlosser RW, Wendt O, Bhavnani S, Nail-Chiwetalu B. Use of information-seeking strategies for developing systematic reviews and engaging in evidence-based practice: the application of traditional and comprehensive Pearl Growing. A review. Int J Lang Commun Disord 2006;41:567-82. http://dx.doi.org/10.1080/13682820600742190.
- Espelt M. Efficacy of Nebulized Hypertonic Saline in the Treatment of Acute Bronchiolitis (NCT01238848) 2012. www.clinicaltrials.gov/ct2/show/NCT01238848?term=espelt+bronchiolitis&rank=1 (accessed 31 January 2014).
- Sosa-Bustamante GP. Nebulized 3% Hypertonic Saline Solution Treatment of Bronchiolitis in Infants (NCT02233985) 2014. https://clinicaltrials.gov/ct2/show/study/NCT02233985?term=“Hypertonic+saline”+AND+“bronchiolitis”&lup_s=01/03/2014&rank=8 (accessed 19 January 2015).
- Higgins JP, Green S. Cochrane Handbook for Systematic Reviews of Interventions. London: Cochrane; 2011.
- Giudice M, Saitta F, Leonardi S, Capasso M, Niglio B, Chinellato I, et al. Effectiveness of nebulized hypertonic saline and epinephrine in hospitalized infants with bronchiolitis. Int J Immunopathol Pharmacol 2012;25:485-91.
- Wang EE, Milner RA, Navas L, Maj H. Observer agreement for respiratory signs and oximetry in infants hospitalized with lower respiratory infections. Am Rev Respir Dis 1992;145:106-9. http://dx.doi.org/10.1164/ajrccm/145.1.106.
- Luo Z, Liu E, Luo J, Li S, Zeng F, Yang X, et al. Nebulized hypertonic saline/salbutamol solution treatment in hospitalized children with mild to moderate bronchiolitis. Pediatr Int 2010;52:199-202. http://dx.doi.org/10.1111/j.1442-200X.2009.02941.x.
- Luo Z, Fu Z, Liu E, Xu X, Fu X, Peng D, et al. Nebulized hypertonic saline treatment in hospitalized children with moderate to severe viral bronchiolitis. Clin Microbiol Infect Off Eur Soc Clin Microbiol Infect 2011;17:1829-33. http://dx.doi.org/10.1111/j.1469-0691.2010.03304.x.
- Maheshkumar KB, Karunakara BP, Nagalli MM, Mallikarjuna HB. Aerosolised hypertonic saline in hospitalized young children with acute bronchiolitis: a randomized controlled clinical trial. J Pediatr Sci 2013;5.
- Sharma BS, Gupta MK, Rafik SP. Hypertonic (3%) saline vs 0.9% saline nebulization for acute viral bronchiolitis: a randomized controlled trial. Indian Pediatr 2013;50:743-7. http://dx.doi.org/10.1007/s13312-013-0216-8.
- Al-Ansari K, Sakran M, Davidson BL, El SR, Mahjoub H, Ibrahim K. Nebulized 5% or 3% hypertonic or 0.9% saline for treating acute bronchiolitis in infants. J Pediatr 2010;157:630-4. http://dx.doi.org/10.1016/j.jpeds.2010.04.074.
- Nemsadze K, Chkhaidze I, Kiknadze N. Nebulized 3% Hypertonic Saline in the Treatment of Acute Bronchiolitis in Infants. Barcelona: ERS Congress Abstract; 2013.
- Pandit S, Dhawan N, Thakur D. Utility of hypertonic saline in the management of acute bronchiolitis in infants: a randomised controlled study. Int J Clin Pediatr 2013;2:24-9. http://dx.doi.org/10.4021/ijcp96w.
- Teunissen J, Hochs AHJ, Vaessen-Verberne A, Boehmer ALM, Smeets CCJM, Brackel H, et al. The effect of 3% and 6% hypertonic saline in viral bronchiolitis: a randomised controlled trial. Eur Respir J 2014;44:913-21. http://dx.doi.org/10.1183/09031936.00159613.
- Everard ML, Hind D, Ugonna K, Freeman J, Bradburn M, Cooper CL, et al. SABRE: a multicentre randomised control trial of nebulised hypertonic saline in infants hospitalised with acute bronchiolitis. Thorax 2014;69:1105-12. http://dx.doi.org/10.1136/thoraxjnl-2014-205953.
- Silver A. A Study of Hypertonic Saline for Infants Hospitalized With Bronchiolitis (NCT01488448) 2014. https://clinicaltrials.gov/ct2/show/NCT01488448 (accessed 19 January 2015).
- Ozdogan S, Koker O, Kose G, Yildirmak Y. The efficacy of nebulized hypertonic saline in acute bronchiolitis in hospital setting: a randomized and double blind trial. Am J Resp Crit Care Med 2014;189.
- Ojha A, Mathema S, Sah S, Aryal U. A comparative study on use of 3% saline versus 0.9% saline nebulisation in children with bronchiolitis. J Nepal Heal Res Counc 2014;12:39-43.
- Hartling L, Ospina M, Liang Y, Dryden DM, Hooton N, Krebs Seida J, et al. Risk of bias versus quality assessment of randomised controlled trials: cross sectional study. BMJ 2009;339. http://dx.doi.org/10.1136/bmj.b4012.
- Egger M, Davey Smith G, Altman DG. Systematic Reviews in Health Care: Meta-Analysis in Context. London: BMJ Publishing Group; 2001.
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c365.
- Sterne JAC, Egger M, Smith GD. Systematic reviews in health care: Investigating and dealing with publication and other biases in meta-analysis. BMJ 2001;323:101-5. http://dx.doi.org/10.1136/bmj.323.7304.101.
- Dwan K, Gamble C, Kolamunnage-Dona R, Mohammed S, Powell C, Williamson PR. Assessing the potential for outcome reporting bias in a review: a tutorial. Trials 2010;11. http://dx.doi.org/10.1186/1745-6215-11-52.
- Guide to the Methods of Technology Appraisal 2013. London: NICE; 2013.
- Kuzik BA, Flavin MP, Kent S, Zielinski D, Kwan CW, Adeleye A, et al. Effect of inhaled hypertonic saline on hospital admission rate in children with viral bronchiolitis: a randomized trial. CJEM 2010;12:477-84.
- Sezer GR, Bozaykut A, Ipek IO, Uyur E, Seren PL, Paketci C. The efficacy of nebulized salbutamol, hypertonic saline and salbutamol / hypertonic saline combination in first bronchiolitis attack. Acta Paediatr 2010;99. http://dx.doi.org/10.1016/j.pupt.2011.09.004.
- Jacobs JD, Foster M, Wan J, Pershad J. 7% Hypertonic saline in acute bronchiolitis: a randomized controlled trial. Pediatrics 2014;133:e8-13. http://dx.doi.org/10.1542/peds.2013-1646.
- Wu S, Baker C, Lang ME, Schrager SM, Liley FF, Papa C, et al. Nebulized hypertonic saline for bronchiolitis: a randomized clinical trial. JAMA Pediatrics 2014;168:657-63. http://dx.doi.org/10.1001/jamapediatrics.2014.301.
- Ipek IO, Yalcin EU, Sezer RG, Bozaykut A. The efficacy of nebulized salbutamol, hypertonic saline and salbutamol/hypertonic saline combination in moderate bronchiolitis. Pulm Pharmacol Ther 2011;24:633-7. http://dx.doi.org/10.1016/j.pupt.2011.09.004.
- Gupta N, Puliyel A, Manchanda A, Puliyel J. Nebulized hypertonic-saline vs epinephrine for bronchiolitis: proof of concept study of cumulative sum (CUSUM) analysis. Indian Pediatr 2012;49:543-7. http://dx.doi.org/10.1007/s13312-012-0122-5.
- Khashabi J, Salari LS, Karamiyar M, Mussavi H. Comparison of the efficacy of nebulized L-epinephrine, salbutamol and normal saline in acute bronchiolitis: a randomized clinical trial. Med J Islam Repub Iran 2005;19:119-25.
- Lines DR, Bates ML, Rechtman AR, Sammartino LP. Efficacy of nebulised ipratropium bromide in acute bronchiolitis. Pediatr Rev Commun 1992;6:161-7.
- Milner A. The role of anticholinergic in acute bronchiolitis in infancy. Arch Pediatr 1995;2:159-62. http://dx.doi.org/10.1016/0929-693X(96)89885-X.
- Patel H, Platt RW, Pekeles GS, Ducharme FM. A randomized, controlled trial of the effectiveness of nebulized therapy with epinephrine compared with albuterol and saline in infants hospitalized for acute viral bronchiolitis. J Pediatr 2002;141:818-24. http://dx.doi.org/10.1067/mpd.2002.129844.
- Tinsa F, Rhouma AB, Ghaffari H, Boussetta K, Zouari B, Brini I, et al. A randomized, controlled trial of nebulized terbutaline for the first acute bronchiolitis in infants less than 12 months old. Tunis Med 2009;87:200-3.
- Postiaux G, Louis J, Labasse HC, Gerroldt J, Kotik AC, Lemuhot A, et al. Evaluation of an alternative chest physiotherapy method in infants with respiratory syncytial virus bronchiolitis. Respir Care 2011;56:989-94. http://dx.doi.org/10.4187/respcare.00721.
- Chowdhury D, al HM, Khalil M, al-Frayh AS, Chowdhury S, Ramia S. The role of bronchodilators in the management of bronchiolitis: a clinical trial. Ann Trop Paediatr 1995;15:77-84.
- Nenna R, Tromba V, Berardi R, De AD, Papoff P, Sabbatino G, et al. Recombinant human deoxyribonuclease treatment in hospital management of infants with moderate-severe bronchiolitis. Eur J Inflamm 2009;7:169-74.
- Bertrand P, Aranibar H, Castro E, Sanchez I. Efficacy of nebulized epinephrine versus salbutamol in hospitalized infants with bronchiolitis. Pediatr Pulmonol 2001;31:284-8. http://dx.doi.org/10.1002/ppul.1040.
- Nenna R, Papoff P, Moretti C, De Angelis D, Battaglia M, Papasso S, et al. Seven percent hypertonic saline-0.1% hyaluronic acid in infants with mild-to-moderate bronchiolitis. Pediatr Pulmonol 2013;46:919-25. http://dx.doi.org/10.1002/ppul.22935.
- Bueno Campaña M, Olivares Ortiz J, Notario Muñoz C, Rupérez Lucas M, Fernández Rincón A, Patiño Hernández O, et al. High flow therapy versus hypertonic saline in bronchiolitis: randomised controlled trial. Arch Dis Child 2014;12:1-5. http://dx.doi.org/10.1136/archdischild-2013-305443.
- Mandelberg A. Hypertonic saline in the treatment of acute bronchiolitis in the emergency department. Arch Pediatr Adolesc Med 2010;164:395-7. http://dx.doi.org/10.1001/archpediatrics.2010.37.
- Principi T, Komar L. J Popul Ther Clin Pharmacol 2011;18:e273-4.
- Hom J, Fernandes RM. When should nebulized hypertonic saline solution be used in the treatment of bronchiolitis?. Paediatr Child Health (Oxford) 2011;16:157-8.
- Sauvaget E, David M, Bresson V, Retornaz K, Bosdure E, Dubus JC. Nebulized hypertonic saline and acute viral bronchiolitis in infants: current aspects. Arch Pediatr 2012;19:635-41. http://dx.doi.org/10.1016/j.arcped.2012.03.018.
- Lowell DI, Lister G, Von Koss H, McCarthy P. Wheezing in infants: the response to epinephrine. Pediatrics 1987;79:939-45.
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta-analyses. Ann Intern Med 2001;135:982-9. http://dx.doi.org/10.7326/0003-4819-135-11-200112040-00010.
- Poole C, Greenland S. Random-effects meta-analyses are not always conservative. Am J Epidemiol 1999;150:469-75. http://dx.doi.org/10.1093/oxfordjournals.aje.a010035.
- Sterne JA, Egger M. Funnel plots for detecting bias in meta-analysis: guidelines on choice of axis. J Clin Epidemiol 2001;54:1046-55. http://dx.doi.org/10.1016/S0895-4356(01)00377-8.
- Vickers A, Goyal N, Harland R, Rees R. Do certain countries produce only positive results? A systematic review of controlled trials. Control Clin Trials 1998;19:159-66. http://dx.doi.org/10.1016/S0197-2456(97)00150-5.
- Egger M, Juni P, Bartlett C, Holenstein F, Sterne J. How important are comprehensive literature searches and the assessment of trial quality in systematic reviews? Empirical study. Health Technol Assess 2003;7.
- Smith V, Devane D, Begley CM, Clarke M. Methodology in conducting a systematic review of systematic reviews of healthcare interventions. BMC Med Res Methodol 2011;11. http://dx.doi.org/10.1186/1471-2288-11-15.
- Morrison A, Polisena J, Husereau D, Moulton K, Clark M, Fiander M, et al. The effect of English-language restriction on systematic review-based meta-analyses: a systematic review of empirical studies. Int J Technol Assess Health Care 2012;28:138-44. http://dx.doi.org/10.1017/S0266462312000086.
- Thompson SG, Higgins JPT. How should meta-regression analyses be undertaken and interpreted?. Stat Med 2002;21:1559-73. http://dx.doi.org/10.1002/sim.1187.
- Senn S. Some controversies in planning and analysing multi-centre trials. Stat Med n.d.:1753-65.
- Petitti D. Meta-Analysis, Decision Analysis, and Cost-Effectiveness Analysis: Methods for Quantitative Synthesis. Medicine: Methods for Quantitative Synthesis in Medicine. Oxford: Oxford University Press; 1999.
- Moreno SG, Sutton AJ, Thompson JR, Ades AE, Abrams KR, Cooper NJ. A generalized weighting regression-derived meta-analysis estimator robust to small-study effects and heterogeneity. Stat Med 2012;31:1407-17. http://dx.doi.org/10.1002/sim.4488.
- Teunissen J, Hochs A, Vaessen-Verberne AAP, Boehmer ALM, Smeets C, Brackel H, et al. Effect of Inhaled Hypertonic Saline Solution to Treat Infants Hospitalized With Viral Bronchiolitis 2011:293-4. www.ers-education.org/ersMade/abstract_print_11/files/Abstract_book_2011.pdf (accessed 15 April 2014).
- Elphick H, Everard M, David T. Recent Advances in Paediatrics. London: The Royal Society of Medicine; 2002.
- Elphick H, Ritson S, Rodgers H, Everard M. When a ‘wheeze’ is not a wheeze: acoustic analysis of breath sounds in infants. Eur Respir J 2000;16:593-7. http://dx.doi.org/10.1034/j.1399-3003.2000.16d04.x.
- Elphick HE, Lancaster GA, Solis A, Majumdar A, Gupta R, Smyth RL. Validity and reliability of acoustic analysis of respiratory sounds in infants. Arch Dis Child 2004;89:1059-63. http://dx.doi.org/10.1136/adc.2003.046458.
- Pasterkamp H. Nomenclature used by health care professionals to describe breath sounds in asthma. Chest J 1987;92. http://dx.doi.org/10.1378/chest.92.2.346.
- Spiteri MA, Cook DG, Clarke SW. Reliability of eliciting physical signs in examination of the chest. Lancet 1988;1:873-5. http://dx.doi.org/10.1016/S0140-6736(88)91613-3.
- American Academy of Pediatrics Subcommittee on Diagnosis and Management of Bronchiolitis . Diagnosis and management of bronchiolitis. Pediatrics 2006;118:1774-93. http://dx.doi.org/10.1542/peds.2006-2223 (accessed 21 February 2014).
- Bisgaard H, Flores-Nunez A, Goh A, Azimi P, Halkas A, Malice M-P, et al. Study of montelukast for the treatment of respiratory symptoms of post-respiratory syncytial virus bronchiolitis in children. Am J Respir Crit Care Med 2008;178:854-60. http://dx.doi.org/10.1164/rccm.200706-910OC.
- Rosenthal R. The file drawer problem and tolerance for null results. Psychol Bull 1979;86:638-41. http://dx.doi.org/10.1037/0033-2909.86.3.638.
- Cunningham S, McIntosh E, Lewis S. Effect of accepting lower oxygen saturation target (90%) on recovery from acute viral bronchiolitis (BIDS Trial): a multi-centre double-blind randomised equivalence trial. Am J Respir Crit Care Med 2014;189. http://dx.doi.org/10.1136/archdischild-2014-306237.90.
- Dickersin K. The existence of publication bias and risk factors for its occurrence. JAMA 1990;263:1385-9. http://dx.doi.org/10.1001/jama.1990.03440100097014.
- Zhang L, Mendoza-Sassi RA, Wainwright C, Klassen TP. Nebulised hypertonic saline solution for acute bronchiolitis in infants. Cochrane Database Syst Rev 2013;7. http://dx.doi.org/10.1002/14651858.cd006458.pub3.
- Turner RM, Davey J, Clarke MJ, Thompson SG, Higgins JPT. Predicting the Extent of Heterogeneity in Meta-analysis, Using Empirical Data from the Cochrane Database of Systematic Reviews. Oxford: Oxford University Press; 2012.
- Ziegler A. Clear redundancy in meta-analyses - overlapping meta-analyses can absolutely be useful. Dtsch Med Wochenschr 2013;138. http://dx.doi.org/10.1055/s-0032-1329061.
- Linden W. How many meta-analyses does it take to settle a question?. Psychosom Med 2013;75:332-4. http://dx.doi.org/10.1097/PSY.0b013e318295e046.
- Moher D. The problem of duplicate systematic reviews. BMJ 2013;347. http://dx.doi.org/10.1136/bmj.f5040.
- PARI Medical Ltd . MucoClear 3% Information n.d. www.parimedical.co.uk/products/pari_saline_solutions/product/detail/info/mucoclear_3_20_ampoules-1.html (accessed 15 April 2014).
- Gargon E, Williamson PR, Altman DG, Blazeby JM, Clarke M. The COMET Initiative database: progress and activities from 2011 to 2013. Trials 2014;15. http://dx.doi.org/10.1186/1745-6215-15-279.
- Van Miert C, Abbott J, Verheoff F, Lane S, Carter B, McNamara P. Development and validation of the Liverpool infant bronchiolitis severity score: a research protocol. J Adv Nurs 2014;70:2353-62. http://dx.doi.org/10.1111/jan.12387.
- Mayfield S, Bogossian F, O’Malley L, Schibler A. High-flow nasal cannula oxygen therapy for infants with bronchiolitis: pilot study. J Paediatr Child Health 2014;50:373-8. http://dx.doi.org/10.1111/jpc.12509.
- ICH Harmonised Tripartite Guideline: Statistical Principles for Clinical Trials E9. Geneva: ICH; 2010.
- DAMOCLES Study Group . A proposed charter for clinical trial data monitoring committees: helping them to do their job well. Lancet 2005;365:711-22. http://dx.doi.org/10.1016/S0140-6736(05)70939-9.
- SAS 9.1.3 Help and Documentation. Cary, NC: SAS Institute Inc.; 2007.
- Borenstein M, Hedges L, Rothstein H. Meta-Analysis Fixed Effect vs Random Effects. 2010.
Appendix 1 Changes to protocol
| Changes to protocol | Progress report | Date | Approved by |
|---|---|---|---|
| Protocol Version 0.1 (28 June 2011) | |||
The amendment related to the following:
|
1 (dated 1 February 2012) | 8 August 2011 | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 2 (28 September 2011) | |||
This amendment related to the following:
|
1 (dated 1 February 2012) | 7 October 2011 | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 3 (6 October 2011) | |||
This amendment related to the following:
|
1 (dated 1 February 2012) | 13 October 2011 (REC acknowledgement letter) | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 4 (17 November 2011) | |||
This amendment related to the following:
|
1 (dated 1 February 2012) | 24 January 2012 (REC acknowledgement letter) | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 5 (6 August 2012) | |||
This amendment related to the following:
|
2 (dated 1 October 2012) | 12 September 2012 | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 6 (1 November 2012) | |||
This amendment related to the following:
|
3 (dated 1 April 2013) | 21 November 2012 | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 7 (11 January 2013) | |||
This amendment related to the following:
|
3 (dated 1 April 2013) | 8 February 2012 | NRES Committee Yorkshire & Humber – South Yorkshire |
| Protocol Version 8 (4 June 2013) | |||
| This amendment related to the change in chief investigator from Professor Mark L Everard to Dr Kelechi Ugonna | 4 (dated 1 October 2013) | 23 July 2013 | NRES Committee Yorkshire & Humber – South Yorkshire |
Appendix 2 Symptom diary
Appendix 3 Flow chart for consent and randomisation
FIGURE 17.
Flow chart for consent and randomisation.

Appendix 4 Statistical analysis plan
Introduction, study design and objectives
Study background
The SABRE study is a parallel-group, RCT comparing whether or not the addition of 3% HS to usual supportive care results in an improvement in outcomes for infants admitted to hospital with acute bronchiolitis. It is funded by National Institute for Health Research HTA programme and sponsored by the Sheffield Children’s Hospital Trust.
The trial has one main objective and three secondary objectives.
Primary objective
Investigate whether or not the addition of 3% HS to usual supportive care results in an improvement in the time to being declared ‘fit for discharge’.
Secondary objectives
-
Assessment of the impact of the intervention on other clinical outcomes and the quality of life of infants and carers at 28 days post randomisation.
-
Investigate the impact on outcomes between those infants with human RSV infection and those with acute bronchiolitis due to other causes, including other viruses (non-RSV).
-
Assessment of the economic impact of the intervention on both the NHS and parents at 28 days post randomisation.
Following consent, infants admitted with acute bronchiolitis will be individually randomised to one of two groups:
-
standard supportive care
-
standard supportive care plus nebulised 3% HS solution.
This statistical analysis plan is written in conjunction with the International Conference for Harmonisation E9,150 applicable standard operating procedures from the Sheffield CTRU and trial documents (protocol and data capture forms).
Outcome measures
Primary end point
The primary end point is the time that the infants are declared ‘fit for discharge’, which will be judged to be when the infant is feeding adequately (taking > 75% of usual intake) and has been in air with a saturation of at least 92% for 6 hours.
Secondary end points
-
Actual time to discharge.
-
Admission to ICU/HDU.
-
Readmission within 28 days from randomisation.
-
Duration of respiratory symptoms post discharge and within 28 days from randomisation.
-
Infant and carer quality of life using the ITQoL at 28 days following randomisation.
-
Health-care utilisation, post-discharge and within 28 days from randomisation.
Sample size, randomisation, blinding
Sample size
Based on a current time to discharge criteria of around 3 days, a 25% reduction was considered to be the minimum clinically significant effect. This was thought to be a realistic effect, as it is the magnitude of the effect observed in previous studies. 47 Using Hospital Episode Statistics data relating to LoS, and assuming a log-normal distribution, the SD is estimated at 32 hours. While a similar, or smaller, SD is expected for our primary outcome measure, a slightly inflated SD of 46 hours is used because of uncertainties over its derivation here. In order to have 90% power to detect a 25% difference in time to meeting discharge criteria, the study will need 139 patients per group at a two-sided alpha level of 5%. The dropout rate is thought to be negligible for the analysis of the primary outcome measure and therefore a conservative estimate of sample size is 150 patients per group. It is expected that up to eight centres will recruit patients into the study. Assuming that all eight recruit a similar number of patients, each site would be expected to recruit approximately 38 patients.
Randomisation
A central web-based randomisation service delivered by the CTRU will be used after patient eligibility and written consent have been confirmed. Patients will be randomly allocated via the online system to receive either (1) HS with usual care (n = 150) or (2) usual care (n = 150). The randomisation schedule will be computer generated prior to the study by the CTRU randomisation service. The allocation schedule will be concealed through the use of the centralised web-based randomisation service, which also allows unblinding in the case of emergency. The randomisation sequence will not be revealed to any person involved in patient recruitment. The data analysts will be blind to treatment allocation until after the statistical analysis plan is finalised, the database locked and the data review completed. All unblinding (emergency and end of trial) will be automatically logged by the CTRU randomisation system, which will include the date, time and user responsible.
Blinding
The study will compare the addition of the intervention to usual care without placebo. The use of placebo in this setting is problematic in that this would involve an intervention that may have a significant effect on the outcome for subjects in the ‘placebo’ arm. The extra handling involved in nebulised therapy may have a deleterious effect, as has been shown to be associated with the increased handling associated with physiotherapy. 49 Similarly, the ‘placebo’ agent may cause harm as has been suggested in previous studies using nebulised distilled water or may have an unexpected positive impact for an agent such as a NS. 32
Interim analyses and study monitoring
Interim analyses
As the study was designed to be of short duration with the data collection period anticipated to last for only a single bronchiolitis season, no interim analyses were planned. However, poor recruitment during the first season resulted in a single interim analysis at the end of this first year, resulting in a decision to continue with the trial and a small reduction in the significance threshold as detailed in Level of significance.
Study monitoring
The trial will be monitored and audited in accordance with the monitoring standard operating procedures of the clinical research facility and the CTRU. Three committees will be established to govern the conduct of the trial:
-
TSC. The TSC will consist of an independent chair, the chief investigator, two independent members (statistician and clinician) and a parent representative.
-
DMEC. The DMEC will consist of an independent statistician and two independent experts. Its responsibilities will include reviewing the trial data at regular intervals and implementing stopping rules in accordance with MRC guidance. 151
-
TMG. The TMG will include the chief investigator, co-applicants, principal investigators, CTRU assistant director, trial manager, research nurses, trial statistician and health economist.
Data sources, data evaluability and analysis populations
Data sources
The data used in this study will come from the following sources:
For patients who are eligible for randomisation into the study (including patients who are randomised)
-
Patient recruitment form, which contains the age, sex and details regarding entry criteria.
For patients who are randomised into the study
-
Randomisation schedule, which contains randomisation codes and allocated intervention group
-
Case report form, which contains patient demographics, characteristics at presentation, investigations and events during treatment, assessment of ‘fit for discharge’, resource use and AEs over subsequent 28 days
-
Post-discharge data collection forms, comprising symptom and health-service utilisation diary, and ITQoL.
The randomisation codes are stored electronically on the CTRU randomisation system. All other electronic data are held separately on the CTRU database system. Access to any data that would unblind the study will be limited to members of the CTRU who are independent of the trial.
Protocol deviations
Patients who are deemed not to have adhered to the study procedures will be considered as deviations from the protocol. Deviations will be classified as either major or minor depending on their importance, and secondary analyses may be performed excluding patients who are considered to be major protocol deviations. These analyses are termed ‘per-protocol’ analyses, and the set of patients who are included in these analyses will be termed the PP analysis set. The definition of protocol deviations will be agreed and finalised prior to final database lock. In deciding upon protocol deviation, particular weight will be given to the following:
-
randomised despite not fitting the entry criteria
-
not receiving randomised treatment
-
receiving concomitant medications during treatment stage.
Analysis populations
Four analysis populations will be used in the analyses:
-
All screened patients: patients who are screened for eligibility to the study, including those randomised.
-
FAS: all randomised patients, with the following exclusions:
-
patients who have previously been randomised. Where this occurs, only data from the first admission will be analysed.
-
patients from whom there is no recorded informed consent obtained from carers (oral or written).
-
patients whose carers withdraw consent before any study medications are given.
-
patients whose carers withdraw consent retrospectively (i.e. request that their entire data are removed).
-
patients who were randomised despite not fulfilling the inclusion criteria.
-
-
Per-protocol (PP): the subset of the FAS who do not deviate from the protocol, as defined in Protocol deviations.
-
Safety: all randomised patients, with the exception of those who have no recorded informed consent.
Summaries based on the FAS and PP populations will be on an intention-to-treat basis with patients assigned to the treatment group as originally randomised. Summaries based on the safety population will analyse patients by the actual treatment received.
For the outputs presented to the DMEC, all outputs will be based on the FAS population unless otherwise requested.
Outline of the analyses
General considerations
Summary information
All summaries presented to the DMEC will be by treatment group. Additional summaries may be provided which report according to treatment centre, if requested. In order to maintain the blinding by the trial statistician, it will be the responsibility of the CTRU to provide this by-treatment group information to the DMEC. No member of the study team will have access to unblinded data sets or the unblinded reports until the final analyses. Summaries of continuous variables will include the number of observations used, mean, median, SD, interquartile range, minimum and maximum. Summaries of categorical variables will comprise the number of observations used, and the number and percentage in each category.
Number and timing of analyses – adjustments for multiplicity
The study may stop prematurely on grounds of safety or futility, based on recommendations from the DMEC or the funder. No formal interim analyses were planned for efficacy but due to poor recruitment in the first season a single interim analysis at the end of this first year was carried out, resulting in a decision to continue with the trial and a small reduction in the significance threshold as detailed in Level of significance. No other adjustments were made for multiplicity.
Level of significance
As a result of poor recruitment in the first season, there was an unplanned interim analysis at the end of the first year. The effect of this unplanned analysis on the overall significance level of the trial was investigated and as a result the significance threshold has been changed to 0.045 probability of declaring a false significance difference in the mean change in scores between the intervention arms will be tolerated. All CIs will remain two sided 95% intervals. No further adjustments will be made for multiple testing.
Statistical analysis packages used
All statistical analysis will be implemented in SAS version 9.1152 and Stata version 10.
Further details of data derivations are in Further details of statistical methods and calculations.
Data completeness
The following summary will be presented for all screened patients by centre and overall:
-
Enrolment: the number of days recruiting, the number of patients screened, the number of patients recruited, the number of patients recruited per day, the number of screened patients not recruited, and the reason for non-recruitment.
The following summary will be presented for the FAS:
-
Data completeness
-
by treatment group: the number of patients with complete data for key parameters
-
by centre: the number of patients with complete data for key parameters
-
-
Protocol deviations
-
By treatment group: number of patients who deviate from the protocol
-
By centre: the number of patients who deviate from the protocol
-
The inclusion of key parameters may be allowed to vary at the request of the DMEC during the trial. In order to allow time for data to be entered onto the system, the follow-up and questionnaire data will be considered complete if they have been entered within 30 days of the scheduled data collection time point.
Missing, spurious and unused data
Questionable data will be queried with the study manager and referred back to the site investigators where appropriate.
Demographics, baseline characteristics and concomitant medications
The following summaries will be presented:
-
demographics: centre, age, sex, ethnicity, age at admission, gestation, birth weight, current weight
-
background: smoker in household, child-care method, feeding method, number of siblings, family history of asthma, eczema and hay fever, previous illnesses, medical problems, length of current illness prior to admission
-
concomitant medications given by time period:
-
in the 2 weeks prior to admission
-
during admission.
-
Efficacy
The following summaries will be presented:
-
time to being declared fit for discharge
-
actual time to discharge
-
percentage transferred to HDU/ICU
-
percentage readmitted within 28 days from randomisation
-
time from randomisation to resolution of respiratory symptoms
-
health-care utilisation, post discharge and up to 28 days from randomisation.
Primary end point (time to being declared ‘ready for discharge’)
Time to being declared ‘ready for discharge’ can be regarded as a survival time and methods appropriate to survival data will be used to analyse these data, as these methods adjust for censoring in the observations. The primary analysis will compare differences between treatment groups, adjusted for centre using a cox-proportional hazards regression model. Further sensitivity analyses may be undertaken to include additional covariates if imbalances are noted. The proportionality of the hazards will be assessed by graphing scaled Schoenfeld residuals against time. 50 In addition to study group and centre, the model will include terms for any potential confounders in the baseline characteristics.
Secondary end points
The analysis of actual time to discharge will be similar to the primary outcome measure time to ‘fit to discharge’. For other secondary outcomes the statistical method appropriate to the data type will be used. For example, readmission rate will be treated as a binary outcome; although it is possible that children could be readmitted multiple times because of their single episode of bronchiolitis, it is much more likely that they will be readmitted just once. Thus, the percentage of children readmitted at least once will be calculated by study group and compared, initially with a Fisher’s exact test, and, if there is a need to adjust for covariates, using a logistic regression model. Each of the 13 dimensions of the ITQoL will be tabulated and examined for differences between treatment groups, initially using a t-test. Where the assumptions underlying the t-test do not hold, a Mann–Whitney U-test will be used. Where there is a need to adjust for covariates the relationship between the groups for each dimension of the ITQoL scale will also be compared using a linear regression model.
Safety outcomes
The following summaries will be based on the FAS population:
-
study medication usage
-
concomitant medications
-
AEs, overall and by type
-
SAEs, overall and by type
-
side effects, overall and by type
-
observations during trial treatment (supplemental oxygen, feeding method).
In summaries of AEs and medications, the number of patients experiencing any event will be presented by actual treatment received; patients will be counted only once in each row, even if more than one event is noted. Where no event is recorded the patient will be defined as having not experienced an event.
The following listings will also be provided. Note that each will contain the treatment, patient ID, event details, date/time and time to onset. For AEs the seriousness will also be reported. For SAEs the duration, expectedness, relationship to study drug, frequency, intensity, action taken and outcome will all be reported:
-
all AEs
-
all SAEs
-
all side effects.
Subgroup analysis
The study will investigate outcomes between those infants with human RSV infection and those with acute bronchiolitis due to one of the many other respiratory viruses that can cause the same clinical phenotype (non-RSV). There is currently no evidence about whether the response to treatment would differ between these two patient groups and if so what the probable magnitude might be. While the study has not been powered to investigate this, it is of interest to investigate and thus the coefficient for the interaction between infection group (RSV vs. non-RSV) and treatment group will be presented together with its 95% CIs and p-value. In addition, results will also be reported separately for the RSV and non-RSV groups in order to provide information for a power calculation for a future study, investigating the relative efficacy of nebulised HS for RSV compared with non-RSV patients.
Further details of statistical methods and calculations
Disposition and data completeness
Enrolment
The number of days recruiting will be defined for each centre as:
The overall number of recruiting days is defined as the total number of days recruiting, summed over all centres.
Summaries of non-recruitment will exclude patients who do not meet the inclusion criteria, unless they are recruited in error. Therefore, the recruitment rate will be:
This is assuming complete capture of all screened patients who meet the inclusion criteria. If it becomes apparent that some or all centres have not done so at the time of database lock, then alternative estimates will be investigated.
Data completion
Completeness (the appearance of any data) will be reported for selected variables, including primary outcome and all secondary outcomes. The percentage completeness of AE data will be defined as:
For all other variables reported completeness will be among all randomised patients.
Efficacy analyses
Primary end point
‘Fit to discharge’ will be measured every 6 hours, with the first evaluation at time of randomisation. Time to being declared ‘fit to discharge’ will be calculated as:
Secondary end point
Time to discharge will be calculated as:
Duration of respiratory symptoms post-randomisation (days) will be calculated as:
References
Trial documents
-
Trial protocol (V0.10, 28 June 2011).
-
Data collection forms:
-
Eligibility form (V1.0, 3 August 2011)
-
Baseline information form (V1.0, 3 August 2011)
-
Treatment data (V1.0 3 August 2011)
-
Discharge form (V1.0, 10 August 2011)
-
Follow-up forms including symptom diary (V1.0, 10 August 2011)
-
Study completion (V1.0, 3 August 2011)
-
AE recording form (V1.0, 3 August 2011)
-
1-month questionnaire (V1.0, 3 August 2011).
-
Standard operating procedures
-
Statistician input and the statistical analysis plan (Shef/CTRU/ST001, version 1.1, 29 October 2007).
-
Data evaluability (Shef/CTRU/ST003, version 1.0, 15 October 2007).
Appendix 5 PROSPERO registration
Nebulised hypertonic saline solution for acute bronchiolitis in infants: a systematic review and meta-analysis
Chin Maguire, Hannah Cantrill
How safe and effective is nebulised HS solution when used to treat acute bronchiolitis in hospitalised infants (under the age of 2 years).
SearchesThe following electronic databases will be searched: MEDLINE (via Ovid) (1946 to January 2015), EMBASE (1974 to January 2015), the Cochrane Central Register of Controlled Trials, Google Scholar (2010 to January 2015) and Web of Science (2010 to January 2015). The full search strategy used in each database is available in Appendix 6. No restrictions or limits (e.g. age, language or publication date) will be applied in any of the databases other than Google Scholar where a restriction of 2010 onwards will be applied.
Trial registries: other than electronic databases, individual trial registries were searched using the terms ‘bronchiolitis’ and ‘hypertonic saline’. These included: ClinicalTrials.gov; UK Clinical Trials Gateway; Centre for Reviews and Dissemination databases (Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database, HTA Database); controlled-trials.com; centrewatch.com and National Research Register, to identify any unpublished data.
Journals: the major journals identified for hand searching are Chest, Paediatrics, and Journal of Paediatrics because these are the journals where the current articles of choice were found. Each of the journals will be searched using the terms ‘hypertonic saline’ and ‘bronchiolitis’.
Other searches: the reference lists of all identified and suitable trials will be checked to identify any further trials with a view to obtaining any published data in order to minimise publication bias.
Types of study to be includedPublished and unpublished, RCTs and quasi-randomised trials; cohort and other observational studies will be excluded.
Only trials which have completed recruitment will be included in the review.
No language or publication restrictions will be applied at the search stage however only those trials published in English will be included in the review.
Condition or domain being studiedBronchiolitis (also known as RSV) is a very common respiratory tract infection in young children, most commonly aged between 2 and 5 months. Acute bronchiolitis results in symptoms of swelling of the airway wall, increased mucous production and an impairment of secretion clearance causing airway obstruction and a combination of gas trapping and ineffective gaseous exchange. Acute bronchiolitis is widely accepted to refer to the first episode of acute wheezing in infants younger than 24 months which may start as an upper respiratory tract viral infection. Overall, 1–2% of children diagnosed with bronchiolitis will require hospitalisation. Bronchiolitis is the main cause of hospital admission for respiratory tract infections with recurrent wheezing episodes seen in up to 50% of severely infected children years after their primary diagnosis.
Participants/populationResearch with children up to the age of 2 years who had been hospitalised as the result of an episode of acute bronchiolitis will be considered for the review. Criteria for inclusion include a first episode of acute wheezing associated with bronchiolitis. Not all cases of bronchiolitis are a result of RSV and as such all cases of bronchiolitis regardless of organism will be included.
Intervention(s), exposure(s)The intervention under consideration is nebulised HS with or without an adjunct treatment given versus NS or no intervention (control). These can be summarised in the following groups:
-
Nebulised HS alone vs. NS
-
Nebulised HS plus a bronchodilator (e.g. salbutamol) vs. NS
-
Nebulised HS plus a bronchodilator (e.g. salbutamol) vs. NS plus same bronchodilator
-
Nebulised HS alone or plus a bronchodilator (e.g. salbutamol) vs. no intervention
No restrictions will be applied in terms of the concentration, dose or the way the intervention (HS) or control (NS with or without adjunct treatment) is administered in the trials.
Comparator(s)/controlPlease see Intervention(s), exposure(s) above for outline of comparators used in the review.
ContextStudies will not be excluded based on the outcomes they measure; only the population and intervention will be used to screen trials for the review.
A systematic review by Zhang et al. conducted in 2010 suggests that HS results in improved clinical outcomes for infants with viral bronchiolitis. The review looked at children in a number of different settings (hospitalised, outpatients and those in the emergency department). This review will update the systematic review conducted in 2010 (with respect to hospitalised infants only) to incorporate both new published and unpublished data from recent clinical trials conducted since 2010.
Outcome(s) Primary outcomesThe primary objective of this review is to determine whether or not nebulised HS results in benefits to hospitalised children in terms of reducing the LoS typically defined as time to meeting discharge criteria.
A ‘summary of findings’ table will be included in the results section. For any dichotomous outcomes results will be expressed as risk ratios and 95% CI. For any continuous outcomes the MD and 95% CI will be used.
Secondary outcomesSecondary outcomes of interest include rate of readmission to hospital; any AEs however described but particularly tachycardia, hypertension, pallor, tremor, nausea, vomiting and acute urinary retention; and final CSS scores.
Data extraction (selection and coding)The titles and abstracts of all the studies identified by the search will be performed. The full articles of any studies that appear to meet the inclusion criteria or those where it is unclear, or where there is insufficient information to make a decision for their inclusion, will be retrieved. Papers that do not meet the inclusion criteria will be excluded.
Data will be extracted onto a standardised data extraction form to both assess the methodological quality of the studies and retrieve outcome data.
-
Key data to be extracted include:
-
study overview (country, year)
-
participant characteristics (age, number randomised, baseline imbalances assessed by the authors in trials, withdrawals, per cent allocated completing follow-up, illness severity, eligibility)
-
intervention and control group details (number randomised in each group, intervention details: duration, delivery, other drugs and compliance)
-
Outcomes data:
-
for continuous outcomes: LoS, typically defined as time to meeting discharge criteria (mean LoS typically defined as time to meeting discharge criteria, SD and number of patients in each group, measured by who); and CSS (mean final CSS score, SD, number of patients for both groups). Principal summary measure is weighted MD in LoS typically defined as time to meeting discharge criteria
-
-
qualitative AE data as available.
Risk of bias assessment will be performed on the extracted data based on the Cochrane Collaboration’s recommendations to assess the quality of the studies. Data will be summarised in the ‘risk of bias’ tables in the results of the review. Trials will be graded on their quality as ‘A’ low risk of bias, ‘B’ high risk of bias or ‘C’ risk of bias unclear. Where a ‘C’ grading is given, every effort will be made to obtain further information to categorise the trial by contacting the trial authors within the specified time frame of this piece of work. The funnel plot method will be used to investigate whether or not publication bias is present if sufficient studies are included in the meta-analysis. Any unpublished results that are obtained will be incorporated.
Risk of bias assessment data items will include: sequence generation; allocation concealment; blinding (outcome and personnel); incomplete outcome data and selective reporting
Data will be input into RevMan to generate summary statistics.
Strategy for data synthesisThe primary outcome data collected are the LoS typically defined as time to meeting discharge criteria SD and number of participants in both intervention and control groups. A FE model will be used to analyse these continuous data based on the assumption that LoS typically defined as time to meeting discharge criteria outcome would estimate the same effect size in each of the studies. 153 A weighted MD and associated 95% CI will be calculated (via RevMan) in order to generate a forest plot. The FE model allows the studies to be weighted depending on their sample size; the greater the sample size, the greater the weight assigned to the trial.
The secondary outcomes of interest include AEs (however reported), rates of hospital readmission and final CSS score. Data on AEs will be collected (however these are defined) and a descriptive narrative of the results will be undertaken. These form part of the qualitative synthesis of the results.
Analysis of subgroups or subsetsNone planned.
Dissemination plansThe review will form part of the HTA monograph for the SABRE clinical trial.
Contact details for further informationChin Maguire
ScHARR, University of Sheffield
c.maguire@sheffield.ac.uk
Organisational affiliation of the reviewScHARR, University of Sheffield, 30 Regent Street, Sheffield S1 4DA, UK.
Review teamMrs Chin Maguire, Clinical Trials Unit, University of Sheffield; Miss Hannah Cantrill, Clinical Trials Unit, University of Sheffield, Sheffield, UK.
CollaboratorsDr Daniel Hind, ScHARR, University of Sheffield, Sheffield, UK.
Details of any existing review of the same topic by the same authorsNone.
Anticipated or actual start date1 January 2013.
Anticipated completion date: 25 April 2014.
Funding sources/sponsorsThe results of the systematic review will be presented in the monograph for the ‘SABRE (hypertonic Saline in Acute Bronchiolitis RCT and Economic evaluation)’ trial funded by the HTA, reference 09/91/22.
Conflicts of interestNone known.
Other registration detailsNone.
LanguageEnglish.
CountryEngland.
Subject index terms statusSubject indexing to be assigned.
Subject index termsBronchiolitis; humans; infant; saline solution, hypertonic.
Reference and/or URL for protocolNone available.
Stage of reviewOngoing.
Date of registration in PROSPERO3 March 2014.
Date of publication of this revision18 December 2014.
| Stage of review at time of this submission | Started | Completed |
|---|---|---|
| Preliminary searches | No | Yes |
| Piloting of the study selection process | No | Yes |
| Formal screening of search results against eligibility criteria | No | Yes |
| Data extraction | Yes | No |
| Risk of bias (quality) assessment | Yes | No |
| Data analysis | Yes | No |
Appendix 6 Main literature search results
MEDLINE (via Ovid)
MEDLINE was searched from 1946 (inception) to present. No limits (e.g. dates, age) or restrictions were applied.
Search date: January 2015.
Search strategy
-
Bronchiolitis/ (2310 results)
-
Bronchiolitis.mp. [mp = title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] (9842 results)
-
1 OR 2 (9842 results)
-
Humans/ (13,622,085 results)
-
Humans.mp. [mp = title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] (13,701,574 results)
-
4 OR 5 (13,701,574 results)
-
3 AND 6 (8487 results)
-
Aerosols/ or “Nebulizers and Vaporizers”/ (30,740 results)
-
nebulize$.mp. [mp = title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] (11,059 results)
-
8 OR 9 (33,444 results)
-
7 AND 10 (238 results)
-
Sodium Chloride/ or Saline Solution, Hypertonic/ (54,351 results)
-
Hypertonic saline.mp. [mp = title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier] (4987 results)
-
12 OR 13 (56,498 results)
-
11 AND 14 (43 results)
EMBASE
EMBASE was searched from 1974 (inception) to present. No limits (e.g. dates, age) or restrictions were applied.
Search date: January 2015.
Search strategy
-
Bronchiolitis/ (9588 results)
-
Bronchiolitis.tw. (11,033 results)
-
1 OR 2 (14,695 results)
-
Human/ (15,229,989 results)
-
Human.mp. [mp = title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] (15981086 results)
-
4 OR 5 (15,981,086 results)
-
3 AND 6 (12,665 results)
-
Aerosol/ or nebulizer/ or nebulization/ (50,225 results)
-
Nebulize$.mp. [mp = title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] (11,285 results)
-
8 OR 9 (53,556 results)
-
7 AND 10 (413 results)
-
Sodium chloride/ (134,876 results)
-
Hypertonic saline.mp [mp = title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword] (6139 results)
-
12 OR 13 (136,836 results)
-
11 AND 14 (102 results)
The Cochrane Central Register of Controlled Trials
The Cochrane Central Register of Controlled Trials was searched from inception to present. No limits (e.g. dates, age) or restrictions were applied.
Search date: January 2015.
Search strategy
-
Medical subject heading (MeSH) descriptor: [Bronchiolitis] (279 results)
-
Bronchiolitis (749 results)
-
1 OR 2 (750 results)
-
MeSH descriptor: [Humans] (1100 results)
-
Human (504536 results)
-
4 OR 5 (504536 results)
-
3 AND 6 (444 results)
-
MeSH descriptor: [Nebulizers and Vaporizers] (1889 results)
-
Nebulize$ (13 results)
-
MeSH descriptor: [Aerosols] (2107 results)
-
8 OR 9 OR 10 (3713 results)
-
7 AND 11 (78 results)
-
MeSH descriptor: [Saline Solution, Hypertonic] (404 results)
-
Hypertonic saline (949 results)
-
13 OR 14 (949 results)
-
12 AND 15 (18 results)
Google Scholar
Bronchiolitis AND hypertonic saline performed from 2010 to present. 915 articles were found and reviewed.
Web of Science
Bronchiolitis AND hypertonic saline from 2010 to present. 81 articles were found and reviewed.
List of abbreviations
- AE
- adverse event
- BNF
- British National Formulary
- CCA
- cost–consequences analysis
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CSS
- clinical severity scale
- CTRU
- Clinical Trials Research Unit
- CUA
- cost–utility analysis
- DMEC
- Data Monitoring and Ethics Committee
- FAS
- full analysis set
- FE
- fixed effect
- GCP
- good clinical practice
- GP
- general practitioner
- HDU
- high-dependency unit
- HR
- hazard ratio
- HS
- hypertonic saline
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- ICU
- intensive care unit
- ITQoL
- Infant Toddler Quality of Life Questionnaire
- ITT
- intention to treat
- LoS
- length of stay
- MD
- mean difference
- MeSH
- medical subject heading
- NS
- normal saline
- PP
- per protocol
- QALD
- quality-adjusted life-day
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RE
- random effect
- RSV
- respiratory syncytial virus
- RSV–
- respiratory syncytial virus negative
- RSV+
- respiratory syncytial virus positive
- SABRE
- Saline in Acute Bronchiolitis RCT and Economic evaluation
- SAE
- serious adverse event
- SD
- standard deviation
- SIGN
- Scottish Intercollegiate Guidelines Network
- TMG
- trial management group
- TSC
- Trial Steering Committee