

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 16/15/03. The contractual start date was in September 2017. The draft report began editorial review in September 2020 and was accepted for publication in February 2021. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2021 Robson et al. This work was produced by Robson et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2021 Robson et al.
Chapter 1 Introduction
Scientific background
Nausea and vomiting in pregnancy (NVP) affects up to 85% of women in the first half of pregnancy. 1 Symptoms usually start at 6–8 weeks’ gestation, peak by 9 weeks’ gestation and, for many women, subside by 20 weeks’ gestation. 2 Symptoms are often mild, but 30% of sufferers experience more severe symptoms that require medical intervention. 2 The most severe form, hyperemesis gravidarum (HG), affects around 1% of women and is characterised by intractable vomiting, dehydration, ketonuria and weight loss. 1,3 HG can result in prolonged hospitalisation, multiple treatments and, where interventions fail, termination of pregnancy (TOP). NVP is associated with emotional and psychological distress and has a profound impact on quality of life (QoL), affecting all aspects of a woman’s life. 4 Women often feel unsupported; suffer higher rates of depression, anxiety and stress;5 and feel dissatisfied with care. 6 Women with HG are also more likely to deliver preterm and to have infants small for their gestational age, although there is no association with congenital anomalies or perinatal death. 7
The aetiology remains unclear, and there is no cure for NVP: treatment focuses on relieving symptoms and preventing morbidity. Many women with mild symptoms are able to self-manage the condition using lifestyle modifications. Some also access other patient-initiated interventions (e.g. ginger and vitamins). Many women with moderate or severe disease require clinician-initiated interventions in the form of intravenous (i.v.) fluids and antiemetic drugs, primarily antihistamines (e.g. cyclizine), dopamine antagonists (e.g. metoclopramide) and 5-hydroxytryptamine3 (5-HT3) antagonists (e.g. ondansetron).
Rationale
Nausea and vomiting in pregnancy incur substantial costs to sufferers (through the purchase of therapies, extra living or child-care costs or lost earnings8) and to the NHS. NVP resulted in > 30,000 admissions to hospital in England in 2013–14, with one-third for HG with metabolic disturbance. 9 Based on data collected between 2013 and 2016, the annual cost of NVP to the UK NHS was estimated at nearly £26M; however, owing to perceived underestimation of costs, the authors suggested that the cost could be as high as £62M. 8 Antiemetics are the most commonly dispensed drugs in early pregnancy, with over 10% of pregnant women in the US Medicaid Program prescribed promethazine or metoclopramide. 10
The assessment of the severity of NVP often lacks consistency, and the management varies substantially. This, combined with inappropriate treatment for presenting symptom severity, can leave women feeling unsupported, dissatisfied with care and experiencing negative interpersonal interactions with health-care providers. 6,11 This, in part, may be because of a previous lack of evidence-based guidelines to inform effective treatments in clinical practice. To address this, in June 2016 the Royal College of Obstetricians and Gynaecologists (RCOG) published guidelines on the management of NVP and HG. 12 Recommendations included the use of cyclizine, prochlorperazine, promethazine and chlorpromazine as first-line antiemetic therapies, and metoclopramide, domperidone and ondansetron as second-line antiemetic therapies.
In 2016, we reported a Health Technology Assessment (HTA)-funded evidence synthesis of the clinical effectiveness and cost-effectiveness of treatments for NVP or HG. 13 Seventy-three studies were identified and the results were narratively synthesised because the planned meta-analysis was not possible owing to the heterogeneity in the assessment of symptom severity, interventions, methods and outcomes, as well as incomplete reporting. There was variation in both the quality of the studies and the quality of reporting. For almost half of all randomised controlled trials (RCTs) identified, there was insufficient detail provided to permit clear judgement of the risk of bias in a range of key areas. There was also insufficient evidence to form a judgement on cost-effectiveness. Two Cochrane reviews were reported around the same time: Matthews et al. 14 reviewed interventions for nausea and vomiting in early pregnancy (41 trials) and Boelig et al. 15 reviewed interventions for treating HG (25 trials). Both reviews also highlighted that the evidence surrounding the effectiveness of treatments was both sparse and of poor quality. All three reviews13–15 concluded that, although there was some evidence that some treatments were better than the dummy for mild symptoms, there was little evidence on the effectiveness of treatments in more severe NVP/HG. The reviews also suggested that, in addition to the use of objective symptom scoring systems, clearly defined outcomes were required so that future trials could be compared using meta-analyses. Furthermore, although the available evidence indicated that these treatments were likely to be safe, more research was needed to clarify this.
Many women with severe NVP/HG who fail to respond to first-line antiemetic therapy (e.g. histamine receptor or dopamine receptor antagonists) will require a hospital review and second-line antiemetic medication. The RCOG recommends a 5-hydroxytryptamine (5-HT) receptor antagonist (ondansetron) or a dopamine receptor antagonist (metoclopramide or domperidone) as second-line therapy. 12 Our review identified no trials involving domperidone and one trial showing comparable improvements in nausea and vomiting with metoclopramide and promethazine (a histamine receptor antagonist). 16 The evidence available for ondansetron was predominantly at high or unclear risk of bias. Two trials were identified comparing ondansetron with metoclopramide. 17,18 Kashifard et al. 17 compared ondansetron [4 mg per os (p.o.) three times per day] with metoclopramide (10 mg p.o. three times per day) in 83 women. Over the 14 days of the trial, the ondansetron group had lower vomiting scores than the metoclopramide group, but there was no difference in nausea scores. Abas et al. 18 compared ondansetron (4 mg intravenously three times per day) with metoclopramide (10 mg intravenously three times per day) in 160 women. Symptom improvement was seen in both groups, with no evidence of difference between groups at 8, 16 or 24 hours.
The primary research recommendation from our HTA evidence synthesis was the need for a RCT, with an economic evaluation, to determine which second-line hospital-prescribed therapy, in addition to i.v. rehydration, should be adopted as mainstream provision in the UK NHS. In response to this recommendation, the HTA programme sought to commission research to determine what was the most effective drug regimen for women with severe NVP requiring secondary care. Specifically, the brief called for a trial comparing both 5-HT receptor antagonists and dopamine receptor antagonists (interventions) with dummy 5-HT receptor antagonists and dopamine receptor antagonists (comparators) in a double-dummy, double-blind controlled factorial trial. The EMPOWER (EMesis in Pregnancy – Ondansetron With mEtoClopRamide) trial was undertaken to answer this research question.
Aims and objectives
Main trial aims
This trial aimed to determine which hospital-prescribed therapy [metoclopramide (metoclopramide hydrochloride, Actavis UK Ltd, Barnstable, UK; IV Ratiopharm GmbH, Ulm, Germany) and ondansetron (ondansetron hydrochloride dehydrate, Wockhardt UK Ltd, Wrexham, UK; IV Hameln Pharma plus GmbH, Hameln)], in addition to i.v. rehydration, should be adopted as the mainstream second-line treatment of severe NVP by the UK NHS when standard first-line treatment has failed. To achieve these aims, we set the following objectives.
Primary objectives
To determine whether or not, in addition to i.v. rehydration, ondansetron compared with no ondansetron and metoclopramide compared with no metoclopramide reduced the rate of treatment failure up to 10 days after initiation.
Secondary objectives
To determine whether or not, in addition to i.v. rehydration, ondansetron compared with no ondansetron and metoclopramide compared with no metoclopramide:
-
improved symptom severity at 2, 5 and 10 days after intervention initiation
-
improved quality of life at 10 days after intervention initiation
-
had an acceptable side effect and safety profile.
In addition, we also aimed to estimate the incremental cost per treatment failure avoided and the net monetary benefits from the perspective of the NHS and women.
Internal pilot and qualitative trial objectives
Owing to uncertainty around the willingness of pregnant women to be recruited into a complex drug trial, an internal pilot phase was incorporated alongside a qualitative substudy. The main objectives of the pilot phase were to assess the feasibility of recruitment and rates of retention in those randomised. The qualitative component aimed to improve our understanding of why women did or did not consent to participation in the trial.
Chapter 2 Methods
Setting and conduct
Trial design
This was a multicentre, double-dummy, randomised, double-blinded, dummy-controlled 2 × 2 factorial trial with an internal pilot phase. Women attending hospital with severe NVP who experienced little or no improvement while taking first-line anti-sickness medication were invited to participate. Participants were randomised in a 1 : 1 : 1 : 1 ratio to receive active metoclopramide and dummy ondansetron; dummy metoclopramide and active ondansetron; active metoclopramide and active ondansetron; or dummy metoclopramide and dummy ondansetron. The trial aimed to compare whether or not, in addition to i.v. rehydration, ondansetron compared with no ondansetron and metoclopramide compared with no metoclopramide reduced the rate of treatment failure up to 10 days after initiation of treatment. The trial included an economic evaluation and a qualitative component.
Internal pilot
It was planned that there would be an internal pilot phase involving up to eight sites (subsequently extended to up to 15 sites) followed by a main phase with an additional seven to nine sites (22–24 sites in total).
To progress from the pilot trial to the main trial, 59 participants needed to be recruited within 6 months, with ≥ 40 participants retained to day 10.
Participants
The trial recruited women aged ≥ 18 years who were < 17 weeks’ gestation and had attended a secondary care service suffering from severe NVP or HG, and who had received previous treatment with a first-line antiemetic therapy in the current pregnancy with little or no sustained benefit. Potential participants were first approached by the clinical staff on duty when they attended hospital for treatment. Participants presented at gynaecology/early pregnancy clinics, maternity assessment units (MAUs) or accident and emergency (A&E) departments. The trial was introduced to the participant and permission was sought for someone in the research team to approach them and provide more information about the trial. Patients who expressed an interest in taking part were provided with a copy of the participant information sheet (PIS). Those still interested after reading the information provided were then assessed for eligibility.
Screening
Screening logs were completed at recruiting sites to record reasons for patient ineligibility and for declining participation. In August 2018, the screening logs were extended to collect information on prior use of trial drugs in the current pregnancy. Confirmation of eligibility was performed by a General Medical Council (GMC)-registered medically qualified doctor who was trained in good clinical practice (GCP).
Eligibility
The eligibility criteria that were initially used during the trial are listed below.
Inclusion criteria
-
Pregnant women suffering from severe NVP.
-
Gestation of < 17 weeks.
-
Had previously taken first-line antiemetic treatment (cyclizine, chlorpromazine, promethazine or prochlorperazine, as recommended by RCOG12) as prescribed, over at least 24 hours with no sustained improvement in symptoms.
-
Age ≥ 18 years.
-
Able to give informed consent.
-
Able to read/understand written English.
Exclusion criteria
-
Allergy/hypersensitivity to any of the study drugs.
-
Prior treatment with trial drugs in this pregnancy.
-
Pre-existing diagnosis of a medical condition: type 1 or 2 diabetes mellitus, chronic kidney disease stages 3–5, Graves’ disease, significant cardiac disease (including long QT syndrome), phaeochromocytoma, epilepsy (or other seizure disorder) or severe liver disease (alanine transaminase or aspartate transaminase of > 2.5 times the upper limit of normal pregnancy).
-
Moderate renal impairment diagnosed during pregnancy (definition: serum creatinine level of > 100 µmol/l).
-
Severe diarrhoea [definition: > 10 loose, watery stools in 1 day (24 hours)].
-
Hypokalaemia (definition: serum potassium level < 3 mmol/l).
-
Pre-existing diagnosis of hypomagnesaemia.
-
Vomiting caused by another underlying condition/infection.
-
Concomitant use of apomorphine or serotonergic drugs [e.g. selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors and lithium].
-
Confirmed diagnosis of severe lactose intolerance (e.g. patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency or glucose–galactose malabsorption).
Early in the period of screening and recruitment, it became apparent that the exclusion criterion ‘Prior treatment with the trial drugs in this pregnancy’ was causing high rates of ineligibility across all sites. After review by all trial oversight bodies, the wording of this criterion was changed to:
-
received either i.v. ondansetron or metoclopramide
-
received either p.o. ondansetron or metoclopramide for > 72 hours (with or without i.v. rehydration).
The updated eligibility criteria were implemented at sites on 20 February 2019.
Women who presented with severe diarrhoea and met the inclusion criteria but were subsequently found to have a serum potassium level of > 3 mmol/l could still be offered participation in the EMPOWER trial.
Women who presented with severe NVP routinely had urea and electrolytes assessed clinically. In the absence of severe diarrhoea, women could still be approached, consented and given trial treatments before urea and electrolyte results were available; if the serum potassium level was subsequently found to be low (< 3 mmol/l), the hypokalaemia had to be corrected quickly with i.v. supplementation, but there was no need to withdraw them from the trial.
Consent
A copy of the PIS was provided to eligible women by the local site team. Patients were first provided with a short PIS to provide them with an overview of the trial. Those interested after reading the short PIS were provided with a copy of the full PIS. Potential participants were given a minimum of 1 hour to review the PIS. During this time, an i.v. cannula could be sited, blood could be taken for necessary clinical tests and i.v. fluids could be commenced. Consent was received by a GMC-registered medically qualified doctor trained in GCP. Women who agreed to participate were asked if they would also be willing to take part in an optional qualitative interview.
Women who declined to participate in the trial itself were still asked if they would like to be approached to participate in the qualitative interviews.
Once consent had been received, randomisation was undertaken.
Schedule of events
The schedule of events for the trial is shown in Table 1 and a participant’s journey through the trial is shown in Figure 1.
| Procedures | Visit | |||||||
|---|---|---|---|---|---|---|---|---|
| Screening | Baseline | Treatment phase | Follow-up at 20 weeks’ gestation | Follow-up after delivery | ||||
| 12-hour review | Time point 1: 48 hours | Time point 2: 5 days | Time point 3: 10 days | |||||
| Eligibility assessment | ✗ | |||||||
| Blood tests (of urea, electrolytes and full blood count) | ✗ | |||||||
| Observations (temperature, pulse, blood pressure, weight and urinalysis) | ✗ | |||||||
| Informed consent | ✗ | |||||||
| Randomisation | ✗ | |||||||
| Demographics | ✗ | |||||||
| Medical history | ✗ | |||||||
| Dispensing of trial drug | ✗ | |||||||
| Review participant for responsiveness to trial treatment | ✗ | |||||||
| PUQE | ✗ | ✗ | ✗ | ✗ | ||||
| VAS for nausea | ✗ | ✗ | ✗ | |||||
| NVPQoL questionnaire | ✗ | ✗ | ||||||
| EPDS | ✗ | ✗ | ||||||
| Short STAI | ✗ | ✗ | ||||||
| Maternal Social Support Scale | ✗ | |||||||
| Health-care utilisation questionnaire | ✗ | |||||||
| Willingness-to-pay questionnaire | ✗ | |||||||
| Adverse events assessment | ✗ | ✗ | ✗ | |||||
| Assessment of IMP compliance | ✗ | ✗ | ✗ | |||||
| Concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Pregnancy outcomes | ✗ | ✗ | ||||||
FIGURE 1.
Trial flow chart. EOI, expression of interest; EPDS, Edinburgh Postnatal Depression Scale; IMP, investigational medicinal product; PUQE, Pregnancy Unique Quantification of Emesis; STAI, State–Trait Anxiety Inventory; VAS, visual analogue scale.
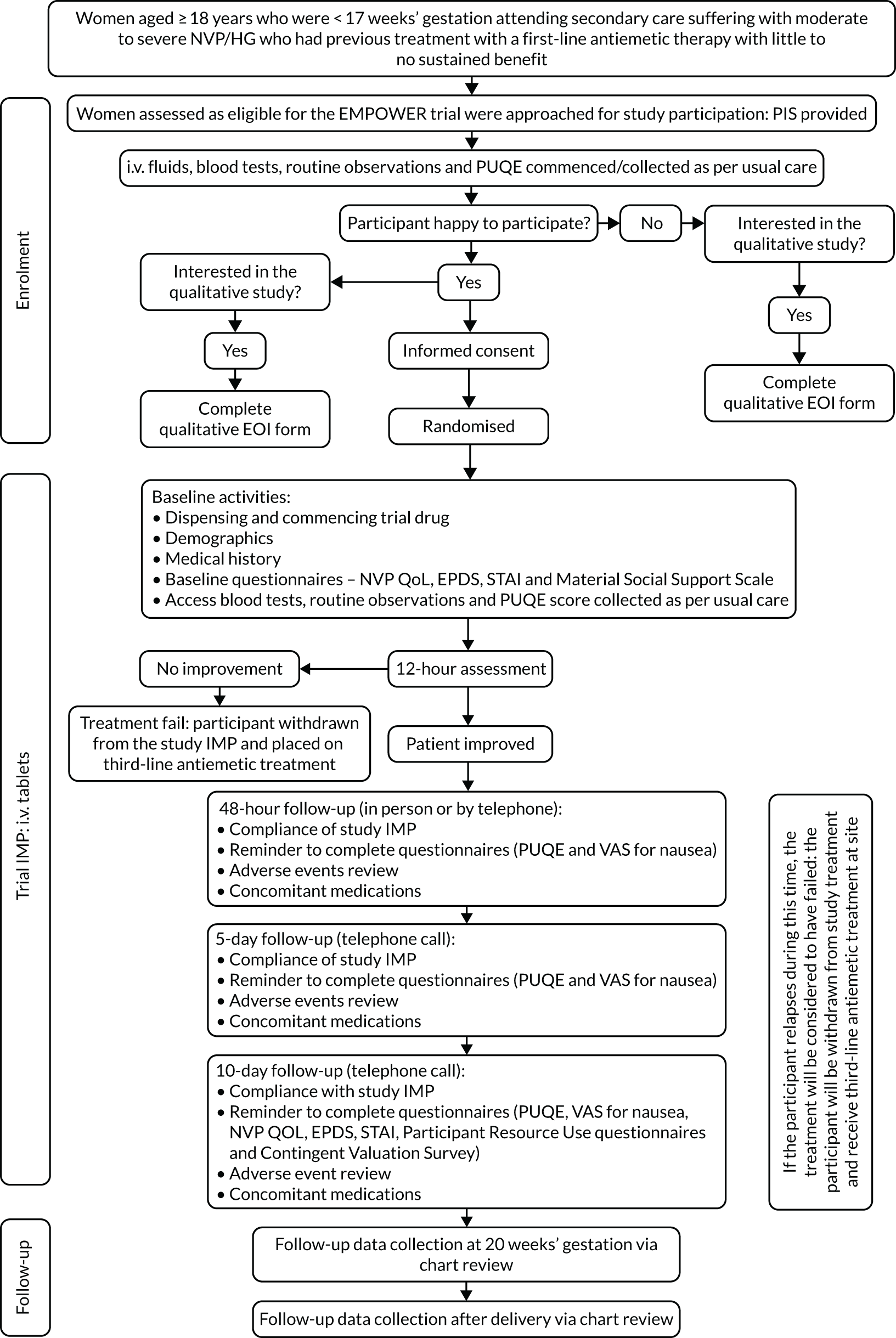
Once a participant was consented and randomised, the trial medication was dispensed by the research team and i.v. medication commenced as soon as possible. Baseline questionnaires were also completed by the participant.
While in hospital, participants were provided with a trial pack that included the questionnaires to be completed at 2, 5 and 10 days, a trial diary to record adverse events (AEs) and concomitant medications, and a trial identification (ID) card, including emergency contact details, for participants to carry with them while participating in the trial. All participants, unless they had been discharged from hospital, were reviewed at 12 hours to assess responsiveness to trial medication. If the participant had not responded to the trial medication within the first 12 hours of treatment, trial medication was stopped and the participant was placed on further treatment outside the trial.
Follow-up with the participants took place at 2 days post first dose of trial medication, at 5 days and at the final follow-up questionnaires at 10 days. These follow-ups were completed via telephone call by members of the research team. Participants were also followed up at 20 weeks’ gestation and post birth, but via medical chart review.
Participant expenses
No expenses were incurred by participants travelling for trial visits. Follow-up was completed via telephone or via review of the participant’s medical chart.
Details of the trial interventions
Participants were randomised to one of four groups. The groups that a participant could be allocated to are outlined below, along with the dose and route of administration:
-
metoclopramide [10 mg three times daily (i.v. and p.o.)] and dummy ondansetron (i.v. and p.o.)
-
ondansetron [4 mg three times daily (i.v. and p.o.)] and dummy metoclopramide (i.v. and p.o.)
-
metoclopramide [10 mg three times daily (i.v. and p.o.)] and ondansetron [4 mg three times daily (i.v. and p.o.)]
-
double dummy three times daily (i.v. and p.o.).
Delivery of the trial interventions
Participants initially received trial medication intravenously; trial medication could be administered intravenously for up to 4 days. Once the participant was able to tolerate fluids, they were converted to oral tablets. The oral medication was taken at the same frequency and dosage as the i.v. formulation. Both the i.v. and the p.o. medication were supplied in the same trial medication kit to prevent the necessity for a double prescription. The total duration of treatment for each participant was 10 days.
Treatment compliance
Participants took trial medication for a maximum of 10 days. Participants were contacted at 2, 5 and 10 days and compliance regarding taking oral medication was checked with participants at these time points.
Participants were asked to return all unused oral trial medication and all packaging (even if empty) to the trial team at each site. A stamped, addressed envelope was provided as part of the trial pack for participants to return medication after they had taken their final dose at day 10. Participants were reminded by the research team to post trial medication back to the site, but if they forgot there was the option to bring it with them to their next clinical appointment. The site team counted all returns (both tablet returns received from the participant and leftover ampoules returned to the pharmacy) and recorded these on the trial database.
Withdrawals
Participants had the right to withdraw from the trial at any time without having to give a reason; however, if provided, the reason for withdrawal was recorded. Participants remained in the trial unless they withdrew consent or the local principal investigator (PI) felt that it was no longer appropriate for the participant to continue. For participants who chose to withdraw, permission was sought to allow the research team to collect routinely collected data from their medical records. If the participant did not agree to this, only the data collected up to the point of withdrawal were retained. Participants who withdrew from the trial were not replaced.
Sites
The EMPOWER trial was conducted at 12 NHS sites in England. Site set-up commenced in October 2017.
Initially, seven sites were set up:
-
Newcastle upon Tyne NHS Foundation Trust
-
City Hospitals Sunderland Hospitals NHS Trust (now South Tyneside and Sunderland NHS Foundation Trust)
-
South Tees Hospitals NHS Foundation Trust
-
Bradford Teaching Hospitals NHS Foundation Trust
-
Leeds Teaching Hospitals NHS Trust
-
Birmingham Women’s and Children’s NHS Foundation Trust
-
Guy’s and St Thomas’ Foundation Trust.
Five further sites were set up during the extended internal pilot:
-
Pennine Acute Hospitals NHS Trust
-
University Hospitals of North Midlands NHS Trust
-
St George’s University Hospitals NHS Foundation Trust
-
Nottingham University Hospitals NHS Trust
-
Portsmouth Hospitals NHS Trust.
Outcome measurement
Primary outcome
The primary end point was the number of participants experiencing a treatment failure. Treatment failure was defined as the need for further treatment if a participant’s symptoms had worsened between 12 hours and 10 days post initiation of treatment. Where further treatment was required, a participant should have been placed on third-line antiemetic treatment (i.e. high-dose ondansetron or corticosteroids) unless the clinician considered further second-line treatment more appropriate.
Secondary outcomes
The following secondary patient-reported and clinical outcomes were collected:
-
Symptom severity, as measured by the Pregnancy Unique Quantification of Emesis (PUQE) score19–21 (see Appendix 1), collected at baseline and at 2, 5 and 10 days post initiation of treatment.
-
Severity of nausea, as measured by a visual analogue scale (VAS)17 for nausea, collected at 48 hours and at 5 and 10 days post initiation of treatment.
-
Quality of life, as measured by the health-related nausea and vomiting during pregnancy quality-of-life (NVPQoL)22 questionnaire (see Appendix 2), collected at baseline and 10 days post treatment commencing.
-
Depression, as measured by the Edinburgh Postnatal Depression Scale (EPDS)23 (see Appendix 3), collected at baseline and 10 days.
-
Anxiety, as measured by the six-item State–Trait Anxiety Inventory (STAI),24 collected at baseline and 10 days [see https://oml.eular.org/sysModules/obxOml/docs/ID_150/State-Trait-Anxiety-Inventory.pdf (accessed September 2021); the six items used were items 1, 3, 6, 15, 16 and 17].
-
Social support, as measured by the Maternity Social Support Scale (MSSS)25 (see Appendix 4), collected at baseline.
-
Clinical indicators of antiemetic effectiveness:
-
the number of participants experiencing a treatment failure at 2 days
-
relapse rate at 5 and 10 days (defined as a PUQE score of ≤ 6 at 2 days followed by an increase to > 12 at 5 or 10 days)
-
remission rate at 10 days [defined as a PUQE score of ≤ 6 at 2 days with return to persistent symptoms (PUQE score of ≥ 7) at 10 days]
-
readmission rates (the number of participants readmitted with NVP within 10 days of recruitment and between 10 days of recruitment and 20 weeks of pregnancy)
-
total inpatient days related to NVP between recruitment and 20 weeks of pregnancy and between 20 weeks of pregnancy and delivery
-
additional antiemetic use.
-
-
Side effects and AEs – participants were asked about side effects/AEs that they experienced at 2, 5 and 10 days post treatment commencing. Severity was graded using the Common Terminology Criteria for Adverse Events (CTCAE). 26
-
Pregnancy and neonatal outcomes – the numbers of women experiencing miscarriage, TOP and stillbirth were recorded. For each infant born, the mode of delivery, gestational age at delivery and birthweight were collected. Information on congenital anomalies detected prior to discharge was also collected. In the case of multiple births, this information was collected for each infant.
-
Key economic outcomes:
-
costs to the NHS and to participants (see Appendix 5)
-
willingness to pay (WTP) (see Appendix 6) for the benefits of antiemetic treatment
-
incremental cost per treatment failure avoided and net monetary benefits.
-
All data fields were locked on 5 December 2019, with the exception of fields relating to pregnancy outcome data, AEs/serious adverse events (SAEs), deviations/violations, investigational medicinal product (IMP) accountability and concomitant medication, which were locked on 9 April 2020.
Sample size calculation
As per the 2 × 2 factorial design, the intention was to have two main comparisons:
-
active metoclopramide (with either active or dummy ondansetron) and dummy metoclopramide (with either active or dummy ondansetron)
-
active ondansetron (with either active or dummy metoclopramide) and dummy ondansetron (with either active or dummy metoclopramide).
An antiemetic failure rate of 10% was assumed based on prior ondansetron studies (with rates of 13% at 48 hours27 and 7% at 5 days28) and our pilot RCT of midwife-led outpatient care,29 in which 10% of women had failure of a second-line antiemetic (primarily metoclopramide). With 266 participants in each comparison (i.e. 133 in each of four treatment combinations, 532 in total) and assuming no interaction between the two active treatments, it was estimated that we would have 90% power to detect an increase in failure rate from 10% in either antiemetic group to 20% in either dummy group (based on a two-sided test at the 5% level). This effect size was felt to be of clinical relevance to applicants and sufferers.
As the primary outcome was treatment failure between 12 hours and 10 days post treatment initiation, we anticipated obtaining outcome data from at least 90% of participants. Therefore, to account for a 10% dropout prior to assessment of the primary outcome, it was planned to recruit 600 patients (150 in each randomised group). Sample size calculations were performed using power twoprop in Stata® version 14 (StataCorp LP, College Station, TX, USA).
Randomisation
Randomisation was carried out centrally by the Newcastle Clinical Trials Unit (NCTU) using a secure web-based system. Patients were randomised on a 1 : 1 : 1 : 1 basis using computer-generated random permuted blocks of size four and stratified by site to receive (1) metoclopramide and dummy ondansetron; (2) ondansetron and dummy metoclopramide; (3) metoclopramide and ondansetron; or (4) double dummy. Randomisation was performed by local research staff, who were delegated this task via the delegation log. The randomisation system generated a unique participant trial identifier and allocated a pack number to be dispensed to the participant. Pre-populated prescriptions were generated by the randomisation system.
Unblinding
The EMPOWER study was a double-blind trial. It was not planned that participants would be routinely unblinded once they had completed trial treatment.
However, participants could be unblinded in a valid medical emergency or for safety reasons where it was necessary for a treating clinician to know which treatment the participant had been receiving. Emergency unblinding could be carried out by the site PI (or another member of the team delegated this responsibility via the delegation log) by accessing the 24-hour randomisation system. A set of back-up sealed code-break envelopes were also held at sites in case of system failure or lack of availability of a delegated individual.
Following discussions with the Trial Steering Committee (TSC), a decision was made to fully unblind all trial medications after the trial was stopped, allowing PIs to notify participants of the medication that they received during the trial.
Statistical analysis
A statistical analysis plan was drafted and finalised prior to data lock. Given that the trial closed to recruitment at the end of the pilot phase, only descriptive summaries of the data have been reported. No efficacy analyses have been undertaken owing to the small sample size. Had the internal pilot trial progressed to a full trial, the group comparisons would have been used as originally intended for this factorial trial. However, given that the pilot data are being reported, the focus of the results is on feasibility outcomes and, therefore, data have been summarised by randomised treatment group.
The progress of all participants through the trial is presented using a Consolidated Standard of Reporting Trials (CONSORT) flow diagram, including the number of screened and eligible patients. Rates of eligibility and recruitment are summarised by site. Reasons for ineligibility or non-participation have been tabulated.
Baseline data are summarised descriptively. Numbers (with percentages) for categorical variables and medians (with ranges) for continuous variables were summarised by randomised treatment group and overall.
A summary of trial medication received is reported using numbers (with percentages) by randomised treatment group and overall. Reasons for premature treatment discontinuation are described.
Primary and secondary outcome measures were summarised descriptively by randomised treatment group and overall. Data are presented following the intention-to-treat principle, with participants retained in the groups to which they were randomised. The primary outcome measure of treatment failure between 12 hours and 10 days was tabulated. The rate of treatment failure [with 95% confidence intervals (CIs)] was calculated out of those evaluable; participants choosing to withdraw from trial treatment before 12 hours and participants choosing to withdraw from the trial without allowing further use of routine data, between 12 hours and 10 days or without prior documented treatment failure were not considered evaluable. Readmissions for NVP, inpatient days related to NVP and additional antiemetic use were tabulated out of those evaluable. Relapse at day 5 or day 10 and remission at day 10, as defined by changes in the PUQE score, were tabulated out of those with completed PUQE questionnaires at day 2 and also at days 5 and 10. The completeness of participant-reported questionnaires was tabulated for each measure. Questionnaires were considered to be completed only if responses were available for all items. Questionnaire scores at each assessment point were summarised descriptively using numbers (with percentages) for categorical variables and medians (with ranges) for continuous variables. For each questionnaire, a score was calculated only if all items were completed. No imputation of missing data were used.
Pregnancy and neonatal outcomes were summarised descriptively, by randomised treatment group and overall, as numbers (with percentages) for categorical variables and medians (with ranges) for continuous variables. For multiple births, neonatal outcomes were summarised for each infant born.
Analyses were conducted in Stata® version 16.
Sources of bias
The primary outcome measure of treatment failure, defined as the need for further treatment because participants’ symptoms had worsened between 12 hours and 10 days, is likely to be partially subjective. This risk of ascertainment bias was reduced by the trial being double blinded, so neither the participant nor the clinical team were aware of the allocated treatment. Secondary outcome measures, including participant-reported questionnaires, were also completed or collected by participants, clinicians or research staff blinded to treatment allocation.
As far as possible, the reasons for eligible patients declining participation were recorded; however, patients were free to decline without giving a reason. It is possible that those who were too unwell to consider the trial information were more likely to decline than those with less severe symptoms; however, randomisation should have ensured that the treatment groups were balanced with respect to severity of NVP at baseline. The PIS illustrated the uncertainty and lack of high-quality evidence around the effectiveness of the trial drugs for women with NVP, and women had to be willing to be allocated to any treatment group, including double dummy.
Patients were not considered evaluable for the primary outcome measure if they withdrew from trial treatment prior to 12 hours or fully withdrew from the trial between 12 hours and 10 days without prior treatment failure. This could have introduced bias if participant withdrawal related to the effectiveness of trial treatment. Reasons for withdrawal were obtained where possible; however, participants were free to withdraw from the trial at any time without giving a reason.
Missing outcome data at follow-up, particularly for participant-reported outcome measures, could have introduced bias if data were not missing at random. Had the trial progressed to a main trial, patterns of missing data would have been explored and multiple imputation methods considered.
Health economic evaluation
The health economics analysis plan was finalised and approved before data lock. Given that the trial closed after the pilot phase, the proposed evaluation was no longer appropriate and the economic analysis was confined to two primary components:
-
Presentation of health service resource use data in the form of summary statistics.
-
Information on health-care resource use was collected via electronic case report forms (eCRFs) (for concomitant medication) and participant-completed health-care utilisation questionnaires (A&E attendance, receipt of primary care and other NHS/Personal Social Services care services), and was completed at 10 days post randomisation. Data on participant-related costs were also collected via the health-care utilisation questionnaire at the day 10 follow-up assessment. Participants were asked to provide information relating to the purchase of private health care or personal care. 30
An analysis of the completeness of the resource use data collected was conducted using descriptive statistics to calculate the number and percentage of participants, with data available for each resource item.
-
Presentation of WTP data in the form of summary statistics.
-
Participants were asked to directly express their WTP for a good service using a contingent valuation (CV) questionnaire. To ascertain their preferences, participants were presented with hypothetical scenarios and were asked how much they would be willing to pay to improve symptom severity for a 10-week period. The hypothetical scenarios were informed by input from women with experience of NVP, the literature and other experts. For a given level of income, a higher WTP value indicated that individuals derived greater benefit from a reduction in symptom severity.
Information gathered via the CV questionnaires is presented as mean [standard deviation (SD)] and median [interquartile range (IQR)] WTP amount to improve symptom severity over a 10-week period. The maximum and minimum WTP values for each of the four randomised treatment groups have also been reported.
All information was summarised by randomised treatment groups and overall. The analysis was conducted using Stata® version 15.
Qualitative evaluation
As part of the protocol for the EMPOWER clinical trial, an evaluation of women’s views of the acceptability of the trial design was carried out with the aim of improving our understanding of why women did, or did not, consent to randomisation and participation in the trial.
Recruitment to the qualitative study, as with the pilot trial, progressed slowly. The eligibility criteria were broadened in February 2019, as many women were found to be ineligible because of prior use of trial medication. As a result, the number of women available to approach for the qualitative study, particularly of decliners, was limited. In consultation with the TSC, it was therefore agreed to extend the qualitative component to include research staff to undertake a wider exploration of factors influencing trial recruitment.
Recruitment of study participants
Patient recruitment
Women who had been screened and were found to be eligible, and had accepted or declined to participate in the trial, were approached for recruitment to the qualitative substudy. Potential recruits were given a copy of the separate PIS and expression-of-interest (EOI) form. To be considerate to patients who were possibly still suffering from NVP, attempts by the qualitative researcher to contact and interview women were confined to the period between the 14th and the 28th day after the initial approach to participate in the EMPOWER trial. Brief 10- to 15-minute interviews were conducted with women who returned the form and were contactable (see Appendix 7). An option of an e-mail interview was also provided in case women were too ill to converse. Consent was obtained over the telephone, recorded and transcribed. A copy of their consent form, filled in by the researcher on their behalf, was sent to participants by e-mail or by post.
Research staff recruitment
Research staff contact details were accessed via NCTU. E-mails about the staff trial (with the PIS and consent form attached) were sent to staff who had screened or recruited women to the trial. Research midwives/nurses directly involved in trial recruitment, and site PIs who were interested in participating in the interviews, returned the consent form by e-mail or agreed to consent taken over the telephone. The large majority who were contacted were willing to participate and the telephone interviews lasted between 20 and 30 minutes (see Appendix 8).
Interview methods
To keep to the short period of time assigned for the patient interview, the topic guide was made up of a grid of questions and possible answers. This allowed the interviewer to quickly adapt the interview to the specific experiences of the woman being interviewed and allowed each participant’s priority topics to shape the conversation. The interview was semistructured, which allowed opportunities for women to expand on their answers wherever possible about positive and negative reasons for participating. The main questions in the grid were:
-
What did you think about when you were first asked to take part in the trial?
-
Why did you/did not you want to take part in the trial?
-
Did the way in which you were invited to take part affect your decision?
-
Was there anything about the trial itself that made you want/not want to take part?
Semistructured interviews were conducted with at least two research nurses/midwives and the PI from each trial site that had recruited at least one participant to the trial. Interviews covered the following topics: experiences of trial set-up and recruitment, difficulties and progress through the trial, and suggested improvements. They took place over the telephone at times convenient to the staff.
In the case of the research staff, a grounded theory approach with constant comparison during the data collection phase was adopted;31 although an interview topic guide was followed, this was adapted as findings from research staff interviews emerged and needed further exploration. A second topic guide was then designed based on these findings for the PIs because of their managerial responsibilities and oversight of the trial. The following were further topics that were explored in the course of the interviews with PIs:
-
proportion of women referred from A&E department
-
attempts to raise the profile of the trial with local general practitioners (GPs)
-
effect of the protocol amendment on recruitment
-
women with language difficulties
-
antidepressant use as a contraindication
-
involvement of research staff in the care of patients being recruited
-
awareness that trial drugs were being used in primary care.
An opportunity for triangulation with the patient data also arose as staff were able to describe particular cases of screening and recruitment that could be of interest to the trial. Recruitment to the patient group was more limited than originally anticipated. Nevertheless, data saturation was reached, that is the data obtained allowed us to reach the point at which no issues needed further confirmation or exploration, and no new themes emerged from either the patient or the trial staff data analysis.
Qualitative data management
Sound files of all interviews, Microsoft Word (Microsoft Corporation, Redmond, WA, USA) files of their transcriptions and Microsoft Word files of the four e-mails were stored on a password-secure server. The transcripts of patients and research staff were checked for accuracy, anonymised and entered into qualitative data management software NVivo 12 (QSR International, Warrington, UK) for indexing and retrieval. Using the software, cases were created of the eight trial sites, with patient and clinician interviews grouped together for cross-checking. The presentation of results based on site characteristics was discounted because of the risk of participants being identifiable.
Qualitative data analysis
The data were analysed using a generative thematic approach,32 in which codes were developed inductively from the reading and re-reading of transcripts, to form an initial coding framework that was revised, expanded or collapsed as the analysis progressed. The analytical process followed the first four stages described by Braun and Clarke:33
-
familiarisation with the data – reading and re-reading of a sample of transcripts to identify and agree descriptive codes (ML and RG: one meeting on patient data and another on trial staff data)
-
coding the data – coding the data according to an initial coding frame and revising the coding frame as further interviews were analysed (ML)
-
searching for themes – analysis of the text coded in descriptive codes to determine at an interpretive level the themes that would best capture the findings from the data (ML)
-
reviewing the themes – data analysis team meeting to review possible themes with a clinician chief investigator (SCR) and medical sociologist co-investigator (RG).
At a second data analysis team meeting, two broad themes were agreed for further analysis, that is the ‘hurdles’ to and ‘enablers’ of the trial process. Considering the complexity of the data with respect to the differences in the sample groups, it was decided that the best way to present the data was to chart the findings according to the framework approach. 34,35 The data were charted under the following groups:
-
patients who accepted participation
-
patients who declined participation
-
research midwives/nurses
-
PIs.
This enabled triangulation between the different types of experiential knowledge within the data sets overall.
In a third data analysis team meeting, the qualitative findings according to the two broad themes of ‘hurdles’ and ‘enablers’ were presented using illustrative quotations in a Microsoft Excel® spreadsheet (Microsoft Corporation, Redmond, WA, USA). A decision was made to present the key themes in the data with selected quotations from the Microsoft Excel® spreadsheet in the text itself as in a standard qualitative report. This allowed a clearer link between the discussion of the themes and the available evidence from the qualitative data.
This descriptive analysis was complemented by a more interpretative strand of analysis in which the qualitative data were presented according to sociological, concept-led themes relating to ‘pathways’ and ‘boundaries’, and ‘ethics of care’. The data findings under themes that related to process evaluation, such as recruitment complexity and staff morale, were also presented because they had emerged from the coding of research staff interviews.
Definition of end of trial
The end of trial was defined as the date that the last infant born to an EMPOWER trial participant was discharged from hospital.
Trial management
The Trial Management Group (TMG) was responsible for overseeing the day-to-day management of the trial. TMG meetings were well attended throughout and involved the chief investigator, statisticians, health economists, qualitative researchers, pharmacy representatives, sponsor representatives, trial management team members, patient contributors and local co-applicants. The TMG met regularly throughout the trial to ensure adherence to the trial protocol and monitor the conduct and progress of the trial.
Data management
The trial database was built by the trial data manager with input from the TMG using MACRO™ (Elsevier, Amsterdam, the Netherlands). The randomisation system and stock management were set up by the trial data manager using the NCTU randomisation system.
All visit data were entered into the MACRO™ database at each site by the local research team. Baseline questionnaire data were entered by local site staff. The 2-day, 5-day and 10-day questionnaires were returned to NCTU by participants and data entry was completed by the central trial team. The central team contacted participants by post after they had completed day 10 to remind them to return the questionnaires and unused IMP/packaging if they had not already done so. One further letter was sent approximately 2 weeks later if the questionnaires had still not been received by the central team.
Serious adverse event data were sent to the central trial team by secure e-mail and then entered centrally into the MACRO™ database.
Essential data will be retained for a period of at least 15 years following the close of the trial in line with sponsor policy and the latest directive on GCP (2005/28/EC). Data were handled, digitalised and stored in accordance with the Data Protection Act 199836 and the Data Protection Act 2018. 37 In line with GDPR (General Data Protection Regulation), the sponsor acted as the data controller and NCTU acted as the data processor for this trial.
It was planned to transfer outcome data at the end of the trial to the UK Teratology Information Service (UKTIS) national database for participants who had consented to this. This was to be retained (under section 25138) for the purpose of surveillance. However, given the limited data available, a decision was made by the TMG, in consultation with UKTIS, not to transfer the data.
Data monitoring, quality control and assurance
Monitoring of trial conduct was completed using a mix of central review and on-site monitoring visits to ensure that the trial was conducted in accordance with the protocol and GCP.
Data were monitored centrally throughout the trial by the data manager and checked regularly for completeness, and any discrepancies were queried with sites using the built-in data clarification request function in MACRO™. Validations built into the database flagged potential errors with dates and out-of-range values. A full list of built-in validations was detailed in the data validation plan. Source data verification was conducted by the trial manager during the on-site monitoring visits, as set out in the monitoring plan.
A data quality report was produced and reviewed at TMG meetings. Data from MACRO™ were compared with data from the NCTU randomisation system to ensure consistency.
An independent Data Monitoring Committee (iDMC) was convened to monitor safety. The iDMC met at the start of the trial and three times throughout the trial.
A TSC was established to provide overall supervision of the trial. The TSC consisted of an independent chairperson, two further independent clinicians, an independent statistician, an independent health economist, two independent lay representatives, a patient contributor and the chief investigator. Other members of the TMG attended as required. The committee met five times throughout the trial.
Serious adverse event reporting
All AEs and adverse reactions (ARs) occurring from the start of administration of the IMP through to 24 hours post last IMP dose were recorded via the eCRF AE page and documented in the participant’s medical records.
Each participant was assessed at baseline and any conditions or symptoms that they were currently experiencing were recorded in the participant’s medical records and graded using the CTCAE.
During follow-up, participants were asked about any new or ongoing symptoms, and sites followed the below process:
-
For symptoms previously reported at baseline –
-
If the CTCAE grading remained the same or better, the symptom did not need to be recorded as an AE or AR. Symptoms were still documented in the patient’s medical records.
-
If the CTCAE grading was worse, this was recorded as an AE or AR in the eCRF AE page and documented in the patient’s medical records.
-
-
For new symptoms not recorded at baseline, these were recorded as an AE or AR and graded using CTCAE. These were recorded via the eCRF AE page and were documented in the patient’s medical records.
All AEs and ARs that met the definition of serious were regarded as a SAE or serious adverse reaction (SAR) and needed to be reported as part of the trial. SAEs and SARs were also collected from the start of administration of the IMP through to 24 hours post last IMP dose. Thereafter, any SAR that came to the attention of the site team had to be reported up until the point of trial closure. All deaths were to be reported as SAEs irrespective of the cause.
All potential reactions were assessed for expectedness using the reference safety information. The reference safety information was contained in section 4.8 of the Summary of Product Characteristics for each of the four medications used in this trial (metoclopramide tablets, i.v. metoclopramide, ondansetron tablets and i.v. ondansetron).
Amendments to the trial protocol
Following receipt of a favourable opinion of the trial protocol from the National Research Ethics System, substantial amendments were submitted and received Health Research Authority (HRA) approval and favourable Research Ethics Committee (REC) opinion [and Medicines and Healthcare products Regulatory Agency (MHRA) approval where applicable].
In summary, these were as follows:
-
Amendment 1 – protocol V2.1, 26 February 2018. This was updated on advice from the TSC regarding treatment options for those who fail on EMPOWER trial treatment: some clarifications regarding eligibility criteria were also provided.
-
Amendment 2 – protocol V3.0, 11 September 2018. This was updated to include staff participants in qualitative interviews.
-
Amendment 3 – protocol V4.0, 15 November 2018. The protocol was updated to reflect the extension of the pilot phase and to update the eligibility criteria to include women who had received suboptimal treatment with ondansetron or metoclopramide based on findings during the pilot phase.
-
Amendment 4 – protocol V5.0, 19 March 2019. This included an update to the exclusion criteria (amended in the last update to provide clarity following feedback from sites); provision of the option to collect PUQE, VAS and Health Utilisation Questionnaire data by telephone at day 10; and clarification that participants needed to have been off SSRIs for at least 2 weeks to meet this eligibility criterion.
Patient and public involvement
Both public co-applicants had suffered from severe NVP during pregnancy. Both contributed to the design of the trial; specifically, they prioritised the primary outcome and contributed to the selection of secondary outcomes, the duration of intervention use before assessment for treatment failure and the overall maximum duration of intervention use. During the trial, both public co-applicants continued to provide extensive input, including reviewing the design and content of the PIS, designing a one-page short-form schematic of the PIS, reviewing the design of the WTP questionnaire, helping with the design and content of the EMPOWER trial website, and providing support to the team regarding the eligibility challenges faced during the pilot. One co-applicant facilitated a wider review of opinions on various design elements via an online group of women with previous experience of HG. The two lay members of the TSC were also engaged throughout and attended TSC meetings and teleconferences, as well as numerous additional telephone calls and e-mails with the chief investigator and lead research midwife.
Ethics and governance
The Newcastle upon Tyne Hospitals NHS Foundation Trust was the sponsor for the trial (reference 08367). The trial received clinical trial authorisation from the MHRA on 22 November 2017 and favourable ethics opinion from the North East – Newcastle & North Tyneside 1 Research Ethics Committee (reference number 17/NE/0325) on 30 November 2017. HRA approval was also received on 30 November 2017. Subsequent research and development approvals were granted by each participating site. Required approvals were sought and obtained prior to implementation of all substantive protocol amendments.
Trial registration and protocol availability
The trial was registered on the International Standard Randomised Controlled Trials Number (ISRCTN) registry as ISRCTN16924692 on 8 January 2018.
Chapter 3 Results
Participant flow
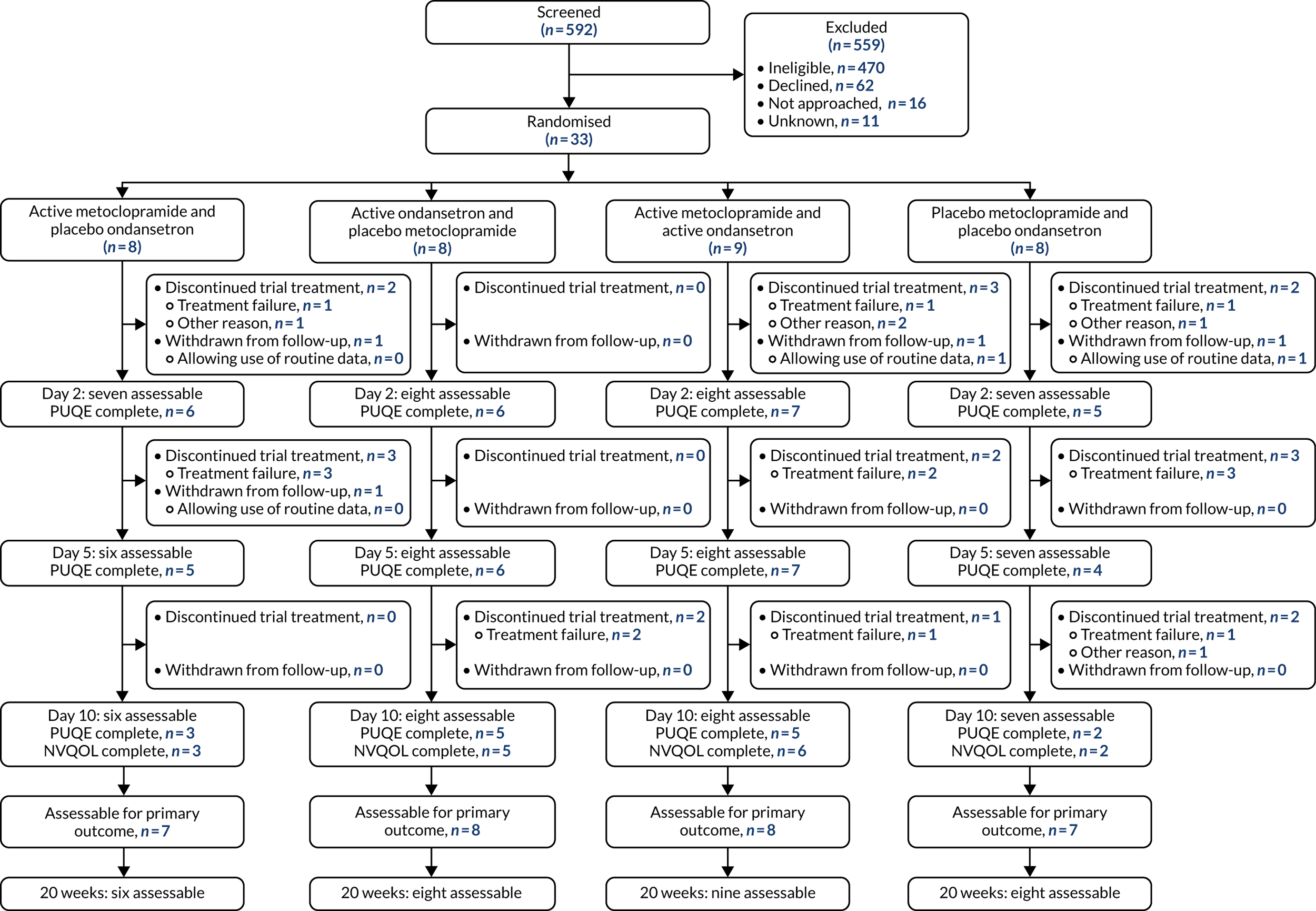
A CONSORT flow diagram of participant flow through the trial is shown in Figure 2.
FIGURE 2.
The CONSORT flow diagram.
Screening
Between 24 April 2018 and 12 August 2019, 592 patients were screened for eligibility. Table 2 summarises the number of patients screened by site. Overall, 122 (21%) patients were eligible. The proportion of eligible patients increased from 19% to 22% after the implementation of protocol version 4.0 to broaden the eligibility criteria around prior use of trial drugs.
| Site | Date recruitment opened | Overall, n (%) | Pre amendment (before 22 February 2019), n (%) | Post amendment (after 22 February 2019) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Screened | Eligible | Recruited | Screened | Eligible | Recruited | Screened | Eligible | Recruited | ||
| Newcastle | April 2018 | 93 | 26 (28) | 5 (19) | 60 | 15 (25) | 4 (27) | 33 | 11 (33) | 1 (9) |
| Sunderland | May 2018 | 43 | 16 (37) | 10 (63) | 32 | 9 (28) | 8 (89) | 11 | 7 (64) | 2 (29) |
| Leeds | May 2018 | 101 | 17 (17) | 5 (29) | 61 | 13 (21) | 5 (38) | 40 | 4 (10) | 0 (0) |
| Bradford | June 2018 | 88 | 9 (10) | 1 (11) | 59 | 6 (10) | 0 (0) | 29 | 3 (10) | 1 (33) |
| South Tees | June 2018 | 43 | 12 (28) | 6 (50) | 33 | 4 (12) | 1 (25) | 10 | 8 (80) | 5 (63) |
| Birmingham | July 2018 | 64 | 8 (13) | 2 (25) | 48 | 8 (17) | 2 (25) | 16 | 0 (0) | 0 (NA) |
| St Thomas’ | July 2018 | 81 | 26 (32) | 4 (15) | 20 | 6 (30) | 3 (50) | 61 | 20 (33) | 1 (5) |
| Nottingham | March 2019 | 34 | 1 (3) | 0 (0) | NA | NA | NA | 34 | 1 (3) | 0 (0) |
| Portsmouth | May 2019 | 25 | 2 (8) | 0 (0) | NA | NA | NA | 25 | 2 (8) | 0 (0) |
| St George’s | May 2019 | 16 | 3 (19) | 0 (0) | NA | NA | NA | 16 | 3 (19) | 0 (0) |
| Oldham | May 2019 | 3 | 2 (67) | 0 (0) | NA | NA | NA | 3 | 2 (67) | 0 (0) |
| Stoke | July 2019 | 1 | 0 (0) | 0 (NA) | NA | NA | NA | 1 | 0 (0) | 0 (NA) |
| Total | 592 | 122 (21) | 33 (27) | 313 | 61 (19) | 23 (38) | 279 | 61 (22) | 10 (16) | |
Reasons for ineligibility are shown by site in Table 3. The main reason for ineligibility was prior treatment with trial medication(s) in the current pregnancy (n = 320; 68%).
| Site | Ineligible (N) | Reason, n (%)a | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Prior treatment with trial drug(s) | Unable to read/understand written English | Aged < 18 years old | > 16+6 weeks’ gestation | Allergy/hyper-sensitivity to trial drug(s) | Pre-existing medical condition | Vomiting caused by another condition | Concomitant use of apomorphine or serotonergic drugs | Missing/unknown | ||
| Newcastle | 67 | 52 (78) | 4 (6) | 0 (0) | 6 (9) | 1 (1) | 2 (3) | 2 (3) | 0 (0) | 0 (0) |
| Sunderland | 27 | 17 (63) | 1 (4) | 0 (0) | 1 (4) | 0 (0) | 3 (11) | 3 (11) | 1 (4) | 1 (4) |
| Leeds | 84 | 37 (44) | 8 (10) | 1 (1) | 7 (8) | 1 (1) | 8 (10) | 1 (1) | 17 (20) | 4 (5) |
| Bradford | 79 | 62 (78) | 8 (10) | 2 (3) | 1 (1) | 0 (0) | 3 (4) | 3 (4) | 0 (0) | 0 (0) |
| South Tees | 31 | 27 (87) | 1 (3) | 0 (0) | 0 (0) | 0 (0) | 1 (3) | 1 (3) | 1 (3) | 0 (0) |
| Birmingham | 56 | 47 (84) | 6 (11) | 0 (0) | 0 (0) | 0 (0) | 3 (5) | 0 (0) | 0 (0) | 0 (0) |
| St Thomas’ | 55 | 26 (47) | 5 (9) | 2 (4) | 0 (0) | 0 (0) | 1 (2) | 0 (0) | 0 (0) | 21 (38) |
| Nottingham | 33 | 27 (82) | 1 (3) | 0 (0) | 1 (3) | 0 (0) | 2 (6) | 2 (6) | 0 (0) | 0 (0) |
| Portsmouth | 23 | 14 (61) | 0 (0) | 1 (4) | 1 (4) | 0 (0) | 2 (9) | 0 (0) | 4 (17) | 1 (4) |
| St George’s | 13 | 9 (69) | 1 (8) | 0 (0) | 0 (0) | 1 (8) | 1 (8) | 1 (8) | 0 (0) | 0 (0) |
| Oldham | 1 | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Stoke | 1 | 1 (100) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Total | 470 | 320 (68) | 35 (7) | 6 (1) | 17 (4) | 3 (1) | 26 (6) | 13 (3) | 23 (5) | 27 (6) |
Further detail on prior use of ondansetron or metoclopramide is shown in Table 4. This information was collected only from 29 August 2018, once prior treatment with trial drugs was identified as a barrier to recruitment. Of those ineligible owing to prior exposure to trial drug(s), 83% had received ondansetron and 17% had received metoclopramide. Prior to protocol version 4.0, in which data on route of administration were available, 41 (37%) participants had received oral treatment only; however, only 10 participants were known to have received oral treatment for ≤ 3 days. Where the source of prescription was known, 57 (24%) participants had received treatment from their GP and the remaining 179 (76%) participants from secondary care. In those receiving treatment from secondary care, the main reason for non-recruitment was the lack of availability of research staff (58%), mostly owing to patients attending out of hours (82%).
| Overall (N = 262), n (%) | Pre-protocol amendment (before 22 February 2019) (N = 128), n (%) | Post-protocol amendment (after 22 February 2019) (N = 134), n (%) | |
|---|---|---|---|
| Drug given | |||
| Ondansetron | 215 (83) | 107 (84) | 108 (82) |
| First line | 62 (36) | 18 (26) | 44 (43) |
| Second line | 108 (64) | 50 (74) | 58 (57) |
| Missing | 45 | 39 | 6 |
| Metoclopramide | 44 (17) | 20 (16) | 24 (18) |
| First line | 22 (58) | 8 (50) | 14 (64) |
| Second line | 16 (42) | 8 (50) | 8 (36) |
| Missing | 6 | 4 | 2 |
| Not recorded | 3 | 1 | 2 |
| Route of administration | |||
| Oral only | 82 (35) | 41 (37) | 41 (33) |
| Taken for ≤ 3 days | 12 (23) | 10 (48) | 2 (6) |
| Taken for > 3 days | 40 (77) | 11 (52) | 29 (94) |
| Missing duration | 30 | 20 | 10 |
| i.v. (± oral treatment) | 137 (59) | 55 (50) | 82 (67) |
| i.m. or PR | 14 (6) | 14 (13) | 0 (0) |
| Missing | 29 | 18 | 11 |
| Source of prescription | |||
| GP | 57 (24) | 28 (25) | 29 (24) |
| Secondary care | 179 (76) | 86 (75) | 93 (76) |
| A&E department | 39 (22) | 18 (22) | 21 (23) |
| Gynaecology/early pregnancy clinic | 61 (35) | 15 (18) | 46 (49) |
| Maternity | 75 (43) | 49 (60) | 26 (28) |
| Missing | 30 | 18 | 12 |
| Reason prescribed: if from secondary care | |||
| Woman’s request | 4 (3) | 3 (4) | 1 (1) |
| Doctor’s preference | 62 (40) | 27 (38) | 35 (40) |
| No research staff available | 93 (58) | 41 (58) | 52 (59) |
| Attended out of hours | 76 (82) | 33 (80) | 43 (83) |
| Other reason | 17 (18) | 8 (20) | 9 (17) |
| Missing | 20 | 15 | 5 |
Overall, 89 patients were eligible for the trial but were not recruited, with 62 (70%) of those declining participation. Reasons for eligible patients declining participation are shown in Table 5.
| Reason | Number of participants, n (%) |
|---|---|
| Reason that eligible participant was not recruited (N = 89) | |
| Declined | 62 (70) |
| Not approached | 16 (18) |
| Not recorded | 11 (12) |
| Reasons that participants declined (N = 62) | |
| Too unwell to consider information | 13 (21) |
| Worried care would be affected (e.g. chance of getting dummy)/does not want dummy | 14 (23) |
| Preference for specific treatment(s)/does not want (trial) drug(s) | 12 (19) |
| Too busy/other commitments/would find it difficult to participate | 7 (11) |
| Does not want to participate in research/sees no benefit in trial | 4 (6) |
| Worried subsequent care would be affected (e.g. would not know what drug received) | 1 (2) |
| No reason given | 7 (11) |
| Other reason | 4 (6) |
Recruitment
From the 122 eligible patients who were screened, 33 (27%) were randomised into the trial. As per the 2 × 2 factorial design, participants were randomised to receive active metoclopramide and dummy ondansetron (n = 8); active ondansetron and dummy metoclopramide (n = 8); active metoclopramide and active ondansetron (n = 9); or dummy metoclopramide and dummy ondansetron (n = 8) (see Figure 2).
Despite the proportion of eligible patients increasing slightly following the implementation of protocol version 4.0, the proportion of eligible patients randomised into the trial declined from 38% to 16% (see Table 2). Of the 12 trial centres open to recruitment, seven enrolled at least one participant into the trial.
Internal pilot
It was initially planned that there would be an internal pilot phase involving up to eight sites followed by a main phase with an additional 14–16 sites (22–24 sites in total).
The progression criterion to the main trial was set at 59 participants to be recruited within 6 months, with ≥ 40 participants retained for data collection at day 10. The trial opened to recruitment on 24 April 2018 and by the end of October 2018 seven sites were open to recruitment, with only 17 participants recruited.
Screening data showed that a larger than anticipated proportion of women were ineligible owing to prior treatment with ondansetron or metoclopramide in the current pregnancy. Following advice from the TSC, trial co-applicants, TMG and the HTA programme, the eligibility criteria were broadened in February 2019 to allow patients who had previously received suboptimal treatment with ondansetron or metoclopramide (orally for < 72 hours) in their current pregnancy to be included. Women who had received either of the trial drugs intravenously or orally for > 72 hours (with or without i.v. rehydration) were still ineligible.
The pilot phase was extended to the end of August 2019. The revised stop–go criterion required 96 participants to be recruited by the end of August 2019. This was based on a recruitment rate of one participant per site per month, with an additional six sites open to recruitment by May 2019.
Progress was reviewed with the iDMC, TSC and HTA programme in July 2019. At this time, 32 participants had been recruited and five additional sites had been opened. The main reason for ineligibility continued to be prior treatment with trial drugs, despite the protocol amendment to broaden the eligibility criteria. It was therefore decided that the trial would close and not progress to a main trial. Following the closure of the trial to recruitment in August 2019, follow-up of participants continued until the final pregnancy outcome was reported in April 2020.
Figure 3 shows the cumulative accrual rates originally anticipated for the pilot phase, the revised target for the pilot phase and the actual recruitment.
FIGURE 3.
Cumulative recruitment by month.

Withdrawals and ineligible participants
No participants were found to be ineligible after randomisation.
Two participants withdrew from the trial without allowing any use of routine data; both were allocated to receive active metoclopramide and dummy ondansetron. One participant withdrew on day 4 for personal reasons following treatment failure (this participant was evaluable for the primary outcome given that treatment failure was reported prior to withdrawal). The other participant wished to withdraw from the trial owing to perceived side effects of the medication on day 2. This participant was not evaluable for the primary outcome.
A further two participants withdrew from trial treatment and further follow-up assessments but allowed routine data to be collected (one metoclopramide and ondansetron, and one double dummy). Both of these participants withdrew prior to receiving 12 hours of trial drugs because they requested further medications or were not feeling better. Neither of these participants was evaluable for the primary outcome.
Numbers analysed
Thirty (91%) participants were evaluable for the primary outcome: seven (88%) active metoclopramide and dummy ondansetron; eight (100%) active ondansetron and dummy metoclopramide; eight (89%) metoclopramide and ondansetron; and seven (88%) double dummy. For information on those who were not evaluable for the primary outcome, see Withdrawals and ineligible participants.
Thirty-one (94%) participants were evaluable for data collected on readmissions for NVP and further antiemetic treatments prescribed up to 20 weeks’ gestation: six (75%) active metoclopramide and dummy ondansetron; eight (100%) active ondansetron and dummy metoclopramide; nine (100%) metoclopramide and ondansetron; and eight (100%) double dummy. Those who were not evaluable were those participants who withdrew from the trial without allowing any use of routine data. Between 20 weeks and delivery, 28 participants (85%) were evaluable, with three participants reporting a TOP prior to 20 weeks’ gestation.
Patient-reported outcome measures (PUQE, VAS for nausea, NVPQoL questionnaire, EPDS and six-item STAI) were expected from 29 (88%) participants at day 10, with four participants (two active metoclopramide and dummy ondansetron; one metoclopramide and ondansetron; and one double dummy) withdrawing from follow-up assessments prior to this time point.
Baseline data
Demographic and clinical baseline characteristics are presented by randomised treatment group in Table 6. Further baseline demographics, including baseline values of participant-reported outcome measures, can be found in Appendix 9, Table 18; Appendix 10, Table 19; and Appendix 11, Table 20.
| Characteristic | Treatment group | Overall (N = 33) | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Age (years), median (minimum, maximum) | 27 (18, 34) | 34 (21, 38) | 27 (21, 38) | 24 (18, 32) | 27 (18, 38) |
| Ethnicity | |||||
| White British, n (%) | 6 (75) | 6 (75) | 8 (89) | 6 (75) | 26 (79) |
| Other, n (%) | 2 (25) | 2 (25) | 1 (11) | 2 (25) | 7 (21) |
| Gestational age (weeks), median (minimum, maximum) | 76/7 (65/7, 133/7) | 86/7 (76/7, 162/7) | 76/7 (50/7, 125/7) | 94/7 (65/7, 155/7) | 85/7 (50/7, 162/7) |
| Multiple birth, n (%) | 1 (13) | 1 (13) | 1 (11) | 0 (0) | 3 (9) |
| First-line medication received,a n (%) | |||||
| Cyclizine | 7 (88) | 7 (88) | 7 (78) | 5 (63) | 26 (79) |
| Promethazine | 0 (0) | 1 (13) | 3 (33) | 1 (13) | 5 (15) |
| Prochlorperazine | 2 (25) | 3 (38) | 4 (44) | 4 (50) | 13 (39) |
| Gravida, n (%) | |||||
| 1 | 3 (38) | 1 (13) | 3 (33) | 5 (63) | 12 (36) |
| 2 | 1 (13) | 3 (38) | 4 (44) | 0 (0) | 8 (24) |
| 3 | 2 (25) | 0 (0) | 1 (11) | 0 (0) | 3 (9) |
| 4 | 2 (25) | 1 (13) | 0 (0) | 2 (25) | 5 (15) |
| 5–8 | 0 (0) | 3 (38) | 1 (11) | 1 (13) | 5 (15) |
| Parity, n (%) | |||||
| 0 | 4 (50) | 1 (13) | 4 (44) | 6 (75) | 15 (45) |
| 1 | 2 (25) | 4 (50) | 4 (44) | 1 (13) | 11 (33) |
| 2 | 2 (25) | 0 (0) | 0 (0) | 1 (13) | 3 (9) |
| 3 | 0 (0) | 3 (38) | 1 (11) | 0 (0) | 4 (12) |
| Past pregnancies | 5 (63) | 7 (88) | 6 (67) | 3 (38) | 21 (64) |
| If yes, NVP in any past pregnancy | 3 (60) | 6 (86) | 6 (100) | 1 (33) | 16 (76) |
The median gestation at the time of trial entry was 85/7 weeks and ranged from 5 weeks to 162/7 weeks. Three (9%) women were expecting more than one baby. Overall, 12 (36%) women had not had a previous pregnancy. Of the 21 women who had had a prior pregnancy, 16 (76%) had experienced NVP in a previous pregnancy. All of the women had received at least one first-line antiemetic medication, with 26 (79%) receiving cyclizine, 13 (39%) receiving prochlorperazine and five (15%) receiving promethazine.
Treatment received
All of the participants received at least one dose of their allocated trial drugs. One participant (allocated to double dummy) received only oral trial drugs at her request. All of the other participants received the trial drugs intravenously initially. The number of i.v. doses received is summarised in Table 7. One participant (allocated to active ondansetron) received three i.v. doses; however, two were received on day 4 following a readmission to hospital (for reasons other than NVP) and being nil by mouth (incidental paronychia; see Safety).
| Treatment | Treatment group, n (%) | Overall (N = 33), n (%) | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Number of i.v. doses | |||||
| 0 | 0 (0) | 0 (0) | 0 (0) | 1 (13) | 1 (3) |
| 1 | 8 (100) | 7 (88) | 8 (89) | 5 (63) | 28 (85) |
| 2 | 0 (0) | 0 (0) | 1 (11) | 2 (25) | 3 (9) |
| 3 | 0 (0) | 1 (13) | 0 (0) | 0 (0) | 1 (3) |
| Converted to oral medication | 8 (100) | 8 (100) | 8 (89) | 7 (88) | 31 (94) |
| Completed 10-day treatment period | |||||
| Yes | 3 (38) | 6 (75) | 3 (33) | 1 (13) | 13 (39) |
| No: treatment failure | 4 (50) | 2 (25) | 4 (44) | 5 (63) | 15 (45) |
| Time until treatment failure (hours) | |||||
| ≤ 48 | 1 (25) | 0 (0) | 1 (25) | 1 (20) | 3 (20) |
| 48.1–120 | 3 (75) | 0 (0) | 2 (50) | 3 (60) | 8 (53) |
| > 120 | 0 (0) | 2 (100) | 1 (25) | 1 (20) | 4 (27) |
| No: treatment discontinued for other reason | 1 (13) | 0 (0) | 2 (22) | 2 (25) | 5 (15) |
Thirteen (39%) participants completed the 10-day trial treatment period; however, one participant (who was allocated to receive active metoclopramide and active ondansetron) missed 16 doses of medication (equivalent to 5.3 days of treatment) because of side effects (drowsiness reported as an AE; see Safety). Fifteen (45%) participants discontinued treatment owing to treatment failure, as per the primary outcome. The remaining five (15%) participants chose to discontinue treatment prior to day 10. Two participants chose to discontinue treatment before 12 hours because they were not feeling better and requested further medications (one active metoclopramide and active ondansetron, and one double dummy); both received only one i.v. dose of the trial drugs and did not convert to oral medication. One participant (who was allocated to receive active metoclopramide alone) chose to discontinue treatment on day 2 owing to side effects (‘funny feeling in legs’, ‘increase in anxiety’ and ‘nightmares’ were reported as AEs). One participant (who was allocated to receive active metoclopramide and active ondansetron) chose to discontinue trial treatment on day 2 because she reported feeling better and decided to manage her symptoms without medication. Another participant (who was allocated to receive double dummy) chose to discontinue treatment on day 5 because she felt that the medication was not effective, but was feeling better so did not seek further review.
Primary outcome measure
Overall, 30 participants were evaluable for the primary outcome. Of those 30 participants, treatment failure between 12 hours and 10 days occurred in four (57%) participants allocated to active metoclopramide and dummy ondansetron, two (25%) participants allocated to active ondansetron and dummy metoclopramide, four (50%) participants allocated to active metoclopramide and active ondansetron, and five (71%) participants allocated to double dummy (Table 8). Three participants (one active metoclopramide and dummy ondansetron, one active metoclopramide and active ondansetron, and one double dummy) reported treatment failure within the first 48 hours. Further antiemetic treatments received are summarised in Table 8. One participant (who was allocated to receive active metoclopramide and dummy ondansetron) did not have information available on further treatments received because she withdrew from the trial, including the use of any routine data, following treatment failure. Thirteen (93%) of those participants reporting a treatment failure received oral antiemetics, four (29%) received i.v. antiemetics and five (36%) received intramuscular antiemetics.
| Treatment group | Overall (N = 33) | ||||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Number evaluable, n (%) | 7 (88) | 8 (100) | 8 (89) | 7 (88) | 30 (91) |
| Treatment failure between 12 hours and 10 days, n (%); 95% CI | 4 (57); 18% to 90% | 2 (25); 3% to 65% | 4 (50); 16% to 84% | 5 (71); 29% to 96% | 15 (50); 31% to 69% |
| Treatment failure by 48 hours, n (%) | 1 (14) | 0 (0) | 1 (13) | 1 (14) | 3 (10) |
| Further antiemetic treatment received, n (%) | |||||
| Ondansetron alone | 0 (0) | 1 (50) | 1 (25) | 1 (20) | 3 (21) |
| Cyclizine alone | 0 (0) | 1 (50) | 0 (0) | 1 (20) | 2 (14) |
| Metoclopramide alone | 0 (0) | 0 (0) | 0 (0) | 1 (20) | 1 (7) |
| Cyclizine and prochlorperazine | 1 (33) | 0 (0) | 1 (25) | 1 (20) | 3 (21) |
| Prochlorperazine and ondansetron | 1 (33) | 0 (0) | 1 (25) | 1 (20) | 3 (21) |
| Cyclizine and ondansetron | 1 (33) | 0 (0) | 0 (0) | 0 (0) | 1 (7) |
| Metoclopramide and ondansetron | 0 (0) | 0 (0) | 1 (25) | 0 (0) | 1 (7) |
| Unknown | 1 | 0 | 0 | 0 | 1 |
| Route(s) of further antiemetic treatment(s), n (%) | |||||
| Oral | 3 (100) | 2 (100) | 3 (75) | 5 (100) | 13 (93) |
| i.v. | 1 (33) | 1 (50) | 0 (0) | 2 (40) | 4 (29) |
| i.m. | 1 (33) | 0 (0) | 3 (75) | 1 (20) | 5 (36) |
| Unknown | 1 | 0 | 0 | 0 | 1 |
Secondary outcome measures
Clinical indicators of antiemetic effectiveness
Readmissions [including assessment unit (AU) visits] for NVP up to 20 weeks’ gestation are summarised in Table 9. Of those evaluable readmissions (n = 31), nine (29%) (including for a day visit) occurred within the first 10 days. Between day 10 and 20 weeks’ gestation, seven (23%) participants were readmitted: one allocated to active metoclopramide and dummy ondansetron, three allocated to active ondansetron and dummy metoclopramide, three allocated to active metoclopramide and active ondansetron, and no one allocated to double dummy. Between 20 weeks’ gestation and delivery, three participants were readmitted to hospital for NVP: one allocated to active ondansetron and dummy metoclopramide, and two allocated to active metoclopramide and active ondansetron.
| Treatment group, n (%) | Overall (N = 33), n (%) | ||||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Up to 20 weeks’ gestation | |||||
| Evaluable | 6 (75) | 8 (100) | 9 (100) | 8 (100) | 31 (94) |
| Number of readmissions within 10 days | |||||
| 0 | 4 (67) | 6 (75) | 5 (56) | 7 (88) | 22 (71) |
| 1 | 1 (17) | 1 (13) | 4 (44) | 1 (13) | 7 (23) |
| 2 | 0 (0) | 1 (13) | 0 (0) | 0 (0) | 1 (3) |
| 3 | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 1 (3) |
| Number of readmissions between 10 days and 20 weeks’ gestation | |||||
| 0 | 5 (83) | 5 (63) | 6 (67) | 8 (100) | 24 (77) |
| 1 | 0 (0) | 3 (38) | 2 (22) | 0 (0) | 5 (16) |
| 2 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| 3 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| 4 | 1 (17) | 0 (0) | 1 (11) | 0 (0) | 2 (6) |
| Number of inpatient days between trial entry and 20 weeks’ gestation | |||||
| 0 | 4 (67) | 5 (63) | 4 (44) | 7 (88) | 20 (65) |
| 1 | 1 (17) | 0 (0) | 2 (22) | 1 (13) | 4 (13) |
| 2 | 0 (0) | 1 (13) | 2 (22) | 0 (0) | 3 (10) |
| 3 | 0 (0) | 2 (25) | 0 (0) | 0 (0) | 2 (6) |
| 5 | 0 (0) | 0 (0) | 1 (11) | 0 (0) | 1 (3) |
| 9 | 1 (17) | 0 (0) | 0 (0) | 0 (0) | 1 (3) |
| 20 weeks’ gestation to delivery | |||||
| Number assessable | 6 (75) | 7 (88) | 8 (89) | 7 (88) | 28 (85) |
| Number of readmissions | |||||
| 0 | 6 (100) | 6 (86) | 6 (75) | 7 (100) | 25 (89) |
| 1 | 0 (0) | 1 (14) | 1 (13) | 0 (0) | 2 (7) |
| 2 | 0 (0) | 0 (0) | 1 (13) | 0 (0) | 1 (4) |
| Number of inpatient days | |||||
| 0 | 6 (100) | 6 (86) | 6 (75) | 7 (100) | 25 (89) |
| 1 | 0 (0) | 1 (14) | 1 (13) | 0 (0) | 2 (7) |
| 2 | 0 (0) | 0 (0) | 1 (13) | 0 (0) | 1 (4) |
Further antiemetic treatments prescribed up to 20 weeks’ gestation and between 20 weeks’ gestation and delivery are summarised in Table 10. Up to 20 weeks’ gestation, further antiemetic treatment was prescribed to 25 (81%) participants: four (67%) allocated to active metoclopramide and dummy ondansetron, seven (88%) allocated to active ondansetron and dummy metoclopramide, eight (89%) allocated to active metoclopramide and active ondansetron and six (75%) allocated to double dummy. Twenty (65%) participants were prescribed ondansetron, 10 (32%) cyclizine, eight (26%) prochlorperazine, seven (23%) metoclopramide and one (3%) promethazine. Between 20 weeks’ gestation and delivery, 12 (43%) participants received further antiemetic treatment. Further information on relapse and remission rates, as defined by changes in PUQE score, can be found in Appendix 12, Table 21.
| Treatment group, n (%) | Overall (N = 33), n (%) | ||||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Between trial entry and 20 weeks’ gestation | |||||
| Number assessable | 6 (75) | 8 (100) | 9 (100) | 8 (100) | 31 (94) |
| Number of different antiemetics prescribed | |||||
| 0 | 2 (33) | 1 (13) | 1 (11) | 2 (25) | 6 (19) |
| 1 | 0 (0) | 4 (50) | 2 (22) | 4 (50) | 10 (32) |
| 2 | 2 (33) | 3 (38) | 4 (44) | 2 (25) | 11 (35) |
| 3 | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 2 (6) |
| 4 | 0 (0) | 0 (0) | 2 (22) | 0 (0) | 2 (6) |
| Route of administrationa | |||||
| Oral | 4 (67) | 7 (88) | 8 (89) | 6 (75) | 25 (81) |
| i.v. | 2 (33) | 1 (13) | 1 (11) | 2 (25) | 6 (19) |
| i.m. | 1 (17) | 1 (13) | 3 (33) | 1 (13) | 6 (19) |
| Type of antiemetica | |||||
| Ondansetron | 3 (50) | 6 (75) | 8 (89) | 3 (38) | 20 (65) |
| Cyclizine | 3 (50) | 3 (38) | 2 (22) | 2 (25) | 10 (32) |
| Prochlorperazine | 3 (50) | 0 (0) | 3 (33) | 2 (25) | 8 (26) |
| Metoclopramide | 1 (17) | 1 (13) | 4 (44) | 1 (13) | 7 (23) |
| Promethazine | 0 (0) | 0 (0) | 1 (11) | 0 (0) | 1 (3) |
| Between 20 weeks’ gestation and delivery | |||||
| Number assessable | 6 (75) | 7 (88) | 8 (89) | 7 (88) | 28 (85) |
| Number of different antiemetics prescribed | |||||
| 0 | 4 (67) | 4 (57) | 3 (38) | 5 (71) | 16 (57) |
| 1 | 2 (33) | 2 (29) | 1 (13) | 1 (14) | 6 (21) |
| 2 | 0 (0) | 1 (14) | 3 (38) | 0 (0) | 4 (14) |
| 3 | 0 (0) | 0 (0) | 1 (13) | 1 (14) | 2 (7) |
| Route of administrationa | |||||
| Oral | 2 (33) | 3 (43) | 4 (50) | 2 (29) | 11 (39) |
| i.v. | 0 (0) | 0 (0) | 0 (0) | 1 (14) | 1 (4) |
| i.m. | 0 (0) | 1 (14) | 2 (25) | 1 (14) | 4 (14) |
| Type of antiemetica | |||||
| Ondansetron | 1 (17) | 3 (43) | 5 (63) | 2 (29) | 11 (39) |
| Cyclizine | 1 (17) | 0 (0) | 1 (13) | 1 (14) | 3 (11) |
| Prochlorperazine | 0 (0) | 1 (14) | 1 (13) | 0 (0) | 2 (7) |
| Metoclopramide | 0 (0) | 0 (0) | 3 (38) | 0 (0) | 3 (11) |
| Promethazine | 0 (0) | 0 (0) | 0 (0) | 1 (14) | 1 (4) |
Participant-reported outcomes
The number of fully completed participant-reported questionnaires at days 2, 5 and 10 is shown in Table 11. At day 2, of those evaluable (n = 30), 24 (80%) participants had completed a PUQE questionnaire; by day 5 and day 10 this figure had fallen to 22 (76%) and 15 (52%) of those evaluable (n = 29), respectively. Of the 15 PUQE questionnaires completed at day 10, three were from participants who had met the primary outcome of treatment failure, meaning that 12 participants in whom treatment had failed did not have a completed questionnaire. These figures were similar for other questionnaires (see Table 11).
| Time point | Treatment group, n (%) | Overall (N = 33), n (%) | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Day 2 | |||||
| PUQE | 6 (86) | 6 (75) | 7 (88) | 5 (71) | 24 (80) |
| VAS | 6 (86) | 7 (88) | 7 (88) | 5 (71) | 25 (83) |
| Withdrawn | 1 | 0 | 1 | 1 | 3 |
| Day 5 | |||||
| PUQE | 5 (83) | 6 (75) | 7 (88) | 4 (57) | 22 (76) |
| VAS | 5 (83) | 6 (75) | 7 (88) | 4 (57) | 22 (76) |
| Withdrawn | 2 | 0 | 1 | 1 | 4 |
| Day 10 | |||||
| PUQE | 3 (50) | 5 (63) | 5 (63) | 2 (29) | 15 (52) |
| Treatment failurea | 1 | 0 | 1 | 1 | 3 |
| VAS | 3 (50) | 5 (63) | 5 (63) | 2 (29) | 15 (52) |
| Treatment failurea | 1 | 0 | 1 | 1 | 3 |
| NVPQoL questionnaire | 3 (50) | 4 (50) | 5 (63) | 2 (29) | 14 (48) |
| Treatment failurea | 1 | 0 | 1 | 1 | 3 |
| EPDS | 3 (50) | 5 (63) | 5 (63) | 2 (29) | 15 (52) |
| Treatment failurea | 1 | 0 | 2 | 1 | 4 |
| STAI | 3 (50) | 5 (63) | 6 (75) | 2 (29) | 16 (55) |
| Treatment failurea | 1 | 0 | 2 | 1 | 4 |
| Withdrawn | 2 | 0 | 1 | 1 | 4 |
Summaries of the participant-reported outcome measures at each time point can be found in Appendix 10, Table 19 and Appendix 11, Table 20.
Pregnancy and neonatal outcomes
The pregnancy and neonatal outcomes are summarised descriptively in Table 12. Of those participants with follow-up data available (n = 31), 28 (90%) participants had a live birth and three (10%) participants had a TOP prior to 20 weeks’ gestation. One participant, who was allocated to active ondansetron and dummy metoclopramide, had a TOP because of fetal abnormalities (see Safety) and two participants, who were allocated to active ondansetron and active metoclopramide and double dummy, had a TOP for other reasons. The median gestation at delivery was 391/7 weeks and the median birthweight was 3.0 kg. All infants with a 5-minute Appearance, Pulse, Grimace, Activity, Respiration (Apgar) score available scored ≥ 7. Three infants (triplets born prematurely at 276/7 weeks gestation) did not have an Apgar score available. One infant, born to a mother who was allocated to receive double dummy, was reported to have a congenital abnormality at birth.
| Treatment group | Overall (N = 33) | ||||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Number assessable, n (%) | 6 (75) | 8 (100) | 9 (100) | 8 (100) | 31 (94) |
| Pregnancy outcome, n (%) | |||||
| Live born | 6 (100) | 7 (88) | 8 (89) | 7 (88) | 28 (90) |
| Termination for fetal abnormalitya | 0 (0) | 1 (13) | 0 (0) | 0 (0) | 1 (3) |
| Termination for other reasons | 0 (0) | 0 (0) | 1 (11) | 1 (13) | 2 (6) |
| If live born | |||||
| Number of infants | |||||
| 1, n (%) | 6 (100) | 6 (86) | 7 (88) | 7 (100) | 26 (93) |
| 2, n (%) | 0 (0) | 1 (14) | 0 (0) | 0 (0) | 1 (4) |
| 3, n (%) | 0 (0) | 0 (0) | 1 (13) | 0 (0) | 1 (4) |
| Total (n) | 6 | 8 | 10 | 7 | 31 |
| Gestational age (weeks) at delivery, median (minimum, maximum) | 386/7 (374/7, 411/7) | 392/7 (374/7, 405/7) | 386/7 (276/7, 414/7) | 374/7 (360/7, 411/7) | 391/7 (276/7, 414/7) |
| Per infant | |||||
| Birthweight (kg), median (minimum, maximum) | 3.1 (2.8, 3.9) | 3.6 (2.6, 4.3) | 2.8 (1.0, 4.6) | 2.6 (1.9, 3.7) | 3.0 (1.0, 4.6) |
| Apgar score at 5 minutes, n (%) | |||||
| ≥ 7 | 6 (100) | 8 (100) | 7 (70) | 7 (100) | 28 (90) |
| Not available | 0 (0) | 0 (0) | 3 (30) | 0 (0) | 3 (10) |
| Congenital abnormalityb | 0 (0) | 0 (0) | 0 (0) | 1 (14) | 1 (3) |
Health economic outcomes
The response rates relating to participant-completed health economic questionnaires are shown in Table 13. An equal number of the participants (n = 16, 48%) did not complete any part of the health-care utilisation and CV questionnaires. The pattern of non-response was similar across the four randomised treatment groups.
| Completion | Treatment group, n (%) | Overall (N = 33), n (%) | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Fully or partially completed health-care utilisation questionnaire | 3 (37) | 5 (63) | 6 (67) | 3 (37) | 17 (52) |
| Not completing any part of the health-care utilisation questionnaire | 5 (63) | 3 (37) | 3 (33) | 5 (67) | 16 (48) |
| Fully or partially completed CV questionnaire | 3 (37) | 5 (63) | 6 (67) | 3 (37) | 17 (52) |
| Not completing any part of the CV questionnaire | 5 (63) | 3 (37) | 3 (33) | 5 (63) | 16 (48) |
The main results of the WTP analysis are shown in Table 14. Overall, 13 participants (39%) provided an answer stating the maximum amount that they would be willing to pay to improve symptom severity for a 10-week period. The mean value across all groups was £674, with the double-dummy group reporting the lowest mean amount (£123). Participants across the other three randomised groups reported similar mean values. The minimum amount that a participant was willing to pay was £20 (reported by a participant in the double-dummy group) and the maximum stated amount was £2000 (reported by participants in the active metoclopramide and active ondansetron group, and the active ondansetron and dummy metoclopramide group).
| Treatment group (£) | Overall (n = 13) (£) | ||||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (n = 2) | Active ondansetron and dummy metoclopramide (n = 5) | Active metoclopramide and active ondansetron (n = 3) | Dummy metoclopramide and dummy ondansetron (n = 3) | ||
| Mean (SD) | 725 (742) | 740 (796) | 833 (1012) | 123 (117) | 617 (715) |
| Median (IQR) | 725 (200–1250) | 500 (100–1000) | 300 (200–2000) | 100 (20–250) | 250 (100–1000) |
| Maximum WTP value | 1250 | 2000 | 2000 | 250 | 2000 |
| Minimum WTP value | 200 | 100 | 200 | 20 | 20 |
Safety
The three SAEs that were reported during the trial are shown in Table 15. The TOP owing to fetal abnormalities was classed as a suspected unexpected serious adverse reaction (SUSAR), as the participant was identified as having received ondansetron following emergency unblinding. The causality of the event was indeterminable in relation to the IMP (ondansetron) and unexpected. The SUSAR underwent expedited reporting to the MHRA and REC and all site PIs were informed of the event.
| SAE ID | Treatment group | Event description | Severity | Type of event |
|---|---|---|---|---|
| 001 | Active metoclopramide and dummy ondansetron | Right calf deep-vein thrombosis | Moderate | SAE |
| 002 | Active ondansetron and dummy metoclopramide | Incidental paronychia | Mild | SAE |
| 003 | Active ondansetron and dummy metoclopramide | Termination of pregnancy owing to fetal abnormalitiesa | Severe | SUSAR |
Three further events, in two participants, were reported as being possibly related to trial treatment: one participant allocated to active metoclopramide and dummy ondansetron reported both constipation and diarrhoea; and one participant allocated to active metoclopramide and active ondansetron reported drowsiness. None of these events was serious.
Urgent safety measure
Immediately following the decision to close the extended internal pilot trial owing to recruitment challenges, the European Medicines Agency (EMA)’s Pharmacovigilance Risk Assessment Unit informed the trial of an urgent safety measure relating to ondansetron, recommending that it should not be used during the first trimester of pregnancy. Owing to the imminent closure of the trial, the urgent safety measure was implemented and recruitment to the pilot closed. All participants receiving trial medication were informed to discontinue their trial medication with immediate effect. Participants continued to be followed up until birth outcome. Following discussion with the TSC, the decision was subsequently taken to unblind all participants and inform them of the medication that they had received.
Chapter 4 Qualitative substudy: hurdles to and enablers of the EMPOWER trial
A total of 42 EOI forms (from 34% of all eligible women) were received. Between May 2018 and August 2019, 21 of the 42 women (50%) were interviewed, of whom seven (33%) had declined participation in the trial (Table 16). Most of the 21 women who were not interviewed did not respond to the researcher’s follow-up contact (between 14 and 28 days after the initial contact) despite their initial EOI. Two women agreed to be interviewed but did not respond subsequently, and two women declined to be interviewed when contacted. Seven women were interviewed after the protocol amendment. Ten out of the 21 patient participants were interviewed over the telephone at their first point of contact, and seven interviews took place at an arranged time following the initial contact. Four women completed the interview through an exchange of e-mails. The telephone interviews lasted a median of 8.2 (range 5.4–16.2) minutes.
| Site | EOIs (n) | Acceptors (n) | Decliners (n) | Interviewed (n) | Acceptors (n) | Decliners (n) |
|---|---|---|---|---|---|---|
| Newcastle | 9 | 4 | 5 | 4 | 2 | 2 |
| Sunderland | 10 | 9 | 1 | 4 | 4 | 0 |
| Leeds | 6 | 3 | 3 | 2 | 1 | 1 |
| Birmingham | 3 | 1 | 2 | 2 | 0 | 2 |
| London | 4 | 4 | 0 | 4 | 4 | 0 |
| Bradford | 5 | 1 | 4 | 2 | 0 | 2 |
| South Tees | 4 | 4 | 0 | 3 | 3 | 0 |
| Portsmouth | 1 | 0 | 1 | 0 | 0 | 0 |
| Total | 42 | 26 | 16 | 21 | 14 | 7 |
Most of the clinicians who were contacted were willing to participate; 22 research staff (research midwives or nurses, n = 16; PIs, n = 6) were interviewed between December 2018 and September 2019. The majority of clinicians (n = 16) were interviewed after the protocol was amended to broaden the eligibility criteria. Six of these interviews were conducted with site PIs towards the end of recruitment to the qualitative study. The telephone interviews with research staff lasted a median of 24.3 (range 12.1–44.5) minutes.
Altogether, 72 codes were generated from the NVivo12 coding, of which 36 nodes related to the patient interviews and 36 to research staff. The findings from these codes formed the framework describing the hurdles to and enablers of the process of conducting trial recruitment (see Appendix 13, Tables 22 and 23). These are presented under three headings: (1) requirements of the EMPOWER Clinical Trial of an Investigational Medicinal Product (CTIMP) in the context of the RCOG guidelines;12 (2) the presenting patient and pathways to care and to trial recruitment; and (3) the role of research staff. The key themes and subthemes are summarised in Figure 4.
FIGURE 4.
Summary of the themes in the qualitative evaluation.
Requirements of the EMPOWER trial (theme 1)
When staff were asked how the EMPOWER trial compared with other trials that they had worked on, the complexity of the trial was a recurrent theme in many responses. The TMG were aware of the trial’s complexities and attempted to take account of them in the trial design and site selection (i.e. the initial sites for the internal pilot were chosen because the research staff at the site had experience of recruiting pregnant women into CTIMPs). However, the demands of the CTIMP had a variable effect on recruitment across sites.
The trial question and adherence to the Royal College of Obstetricians and Gynaecologists guidelines
For the trial to succeed as intended, the rationale for the research needed to be understood and accepted by all those involved in delivering the trial. As the trial progressed, however, two emerging issues were identified by some site PIs: first, that the trial question had lost its relevance and second, that NVP had a lower priority than other pregnancy complications:
. . . if I’m honest with you I think the general feeling was that practice has moved on, and that the question that this trial was asking was now probably a historical question . . . You know we’d almost like we’d missed the boat.
TS19
. . . when we’re coming to nausea and vomiting the perceived, the sort of the perceived issues or problems coming out of it will not sort of rank alongside stillbirth or neonatal death.
TS18
However, other PIs, patients and midwives/nurses were of the opinion that research in this area was much needed and that the trial question remained important. This was in spite of the issue of randomisation to double dummy being controversial at the initial stages among patient and public involvement advisors:
I thought it was a well-designed study and it was worth definitely worth, a question worth asking . . . I don’t think the study design was a problem which was what we initially thought was going to be a problem.
TS18
There was evidence from both PIs and research midwives/nurses that there had been a change in the practice of prescribing antiemetic drugs since the decision was made to conduct the trial (February 2016), which impacted negatively on recruitment during the course of the trial:
I think because it took us a long while to get it going and I think the whole landscape changed almost just before we started recruiting [. . .] and my, my belief is that was largely to do with the guideline the RCOG guideline . . . the trial was an independent endeavour but meanwhile there were lots of us working in the background to improve care for women with hyperemesis, which meant enabling GPs and hospital doctors to prescribe appropriately for it, and ironically our success in doing that has led to the failure of the trial.
TS16
This shift in practice was evident in both primary and secondary care in spite of RCOG step-up guidelines12 recommending the use of the trial drugs as a second-line medication and confirmation from all trial sites that local guidelines on antiemetic use were consistent with RCOG guidelines. 12 However, participants indicated that not all clinicians at the study sites and in general practices in their localities adhered to these guidelines when they were treating women with severe NVP because they had become used to prescribing the trial drugs:
It was not easy a trial to start with because of all the bad practice which is all very well set where people would prescribe whatever antiemetic they would feel like at that time rather than follow RCOG standardised step-up protocol.
TS17
Some of the research staff at sites were unaware of the antiemetic-prescribing culture within which the trial was attempting to operate:
. . . we just hadn’t realised how many women were already on those study medications from GPs and things so it’s been a big eye opener to us about what kind of prescribing practices are out in primary care.
TS20
As a result of this culture of prescribing in primary and secondary care, women were frequently presenting with severe NVP having already been treated with one of the trial drugs, rendering them ineligible for the trial. In some regions, this practice was widespread among GPs, whereas in others the problem was in secondary care:
I did think initially when the trial started that a lot will be ineligible because the GPs prescribing. But I was surprised actually we could catch a lot who were eligible and it was, we were messing up their eligibility at secondary care level which was sorted later part. So I don’t think GPs were a major problem here.
TS17
Organising the delivery of the EMPOWER trial
The successful delivery of the trial required organisational co-operation from clinical staff across departments, that is liaison between maternity/obstetrics, gynaecology, emergency and pharmacy departments. Staff at one trial site were more familiar with trials that were based solely in maternity services, which meant additional effort working with gynaecology colleagues:
This trial was a little bit different in that it was more gynae[cology] than obstetrics which gave I think the trial team in every unit a little bit of challenge whereas we’re used to approaching women in pregnancy further on in pregnancy.
TS16
Nevertheless, there was evidence of good working relationships between clinical staff in maternity and clinical staff in gynaecology day units, and research staff, to facilitate recruitment:
We work with quite closely with the early pregnancy unit and also gynae[cology] emergency, and what we decided to do was get the clerical staff that work on reception to contact us, bleep us when the women were coming in. Cos we’ve got quite a good relationship with that area.
TS21
Where there were staff with both a clinical and a research role, this facilitated the recruitment process for the EMPOWER trial:
One of our research nurses works there in her substantive role as well. So she does some research and some early pregnancy and, so that’s brilliant because she knows all the systems she knows all the staff and consequently they’ve got to know us as well through her. We do, they are very good at referring people to us.
TS02
In some sites, the responsibility for the care of the patient was restricted to unit or ward staff, with research staff providing a minimal amount of help. This may be because the number of research staff was limited. In other sites, however, research staff contributed to the care of eligible women while they were considering the trial:
. . . what I’ve tended to do is not just give the information sheet and come away but I’ve usually kind of helped with starting i.v. fluids and you know making sure that if the patient has any questions that I’m there to answer, you know.
TS04
Good working relationships with the NCTU staff at Newcastle University were critical to the operational delivery of the trial. In the initial stages, there were several glitches, mainly to do with the unavailability of trial staff when they were urgently required:
. . . we’ve tried to call the EMPOWER office and there’s never been anybody who actually works on the trial available to talk to us.
TS05
. . . you’re usually be able to ring by phone if not but there was no back-up there was no way of getting a hold of anybody so we just had to scrap it which is not, not great.
TS20
Some staff commented that the trial database was not user-friendly and that it was unhelpful having separate log-ins to recruit and randomise women:
. . . the whole recruitment process and the databases, having separate databases for randomisation and things like that it can be very confusing.
TS08
Nevertheless, as the trial progressed, and as lines of communication were more firmly established, sites reported how helpful the NCTU staff were in supporting the management of the trial:
Yeah we’ve never had a problem we’re in contact regularly we get a lot of e-mails from them if there’s anything on the database that we haven’t done right, or if there’s any discrepancies they’re quite good at liaising with us and managing like that, so yeah all our contact with the team has been really positive, I think it’s a lovely trials team to work with.
TS11
Demands of the EMPOWER trial protocol
The trial protocol stipulated that women had to have taken first-line antiemetic treatment for a minimum of 24 hours before they could be considered eligible for the trial. Most site guidelines for the management of NVP did not recommend the admission of women, especially with moderate symptoms, who were started on first-line antiemetic therapy. Thus, trial staff reported that they were unable to easily determine if potentially eligible women responded to first-line treatment:
It’s, like we’ve got to get her in and out we can’t wait 3 hours and then see how she feels and then put her in EMPOWER because they don’t tend to like admitting them either.
TS03
So if they’ve never had anything before, usually in that time they’ll just have one dose of cyclizine and then go home. So it’s then waiting to see if they come back and then we can consider them.
TS05
Women could return at any time, including out of hours or at the weekend, when site research staff were not available. Nevertheless, sites were informed that women could be approached about future consideration of the trial when they were on first-line treatment. This advice was followed at some but not all sites:
We’ve also seen a few people who have come in and maybe just having their first-line treatment and just mentioned the study to them and just said we have a study running in the unit you wouldn’t be eligible for it now but bear it in mind if this treatment doesn’t work for you we may come and see you again. So just to raise a bit of awareness with these women who might be coming in.
TS13
One patient suggested that information should be provided about the trial at her first visit to the MAU:
I didn’t know whether you could like send something out after I’d been there in the first time, to say if you come back there is a trial. So I would have had prior knowledge.
EMP1003
The trial was perceived to be particularly challenging because it was a CTIMP with a complex design consisting of four groups, as well as requiring a trial population of pregnant women in a fragile physical and emotional state at the point of recruitment. For this reason, research staff were required to adopt a sensitive approach and give eligible women enough time (1 hour) to consider the trial information.
Women’s perspectives provided insight to the recruitment processes. Although one patient felt that she needed more time to think over her decision to participate, another patient declined because she thought that her wait for treatment would be lengthened because of the trial:
I think how it was explained was that obviously she would come back with, like in an hour, and then there was paperwork to be filled out so obviously that took, would have taken time and then that had to be uploaded in to the computer system to decide which treatment I would then go ahead with . . . I mean I think we’d had a chat about it so we’d pretty much made our mind up probably what 15 minutes after she’d left us. So had she maybe been around a little bit sooner than the hour that we were expecting, that might have helped a little bit.
EMP1003
Research staff reported that they felt that the recruitment process could be quite quick once women had agreed, whereas approaching the women and making sure that the information was understood took longer:
. . . obviously going to the patient making sure that they’re eligible, giving them the information sheet and obviously you want them to have the information sheet for the length of time so that they’re happy, that they understand and that I understand or know that they understand what we’re asking of them.
TS04
Some participants reported difficulties in accessing medical staff who were GCP trained. One group highlighted were GP trainees who were perceived to have less interest either in the specialty area or in contributing to the research:
I found I was quite successful with the doctors who were career trainees in obs[tetrics] and gynae[cology] but I didn’t have as much success with the GP trainees because they were here for 4 months or 6 months and I didn’t find that, they didn’t feel that for the time that it took that it was useful for their CVs.
TS19
On the other hand, other PIs reported that they had ensured that there were enough GCP-trained doctors available:
So most of our senior trainees have completed the GCP training and they are on the delegation log for a number of research trials.
TS22
If women agreed to participate in the trial, the protocol stated that an assessment of treatment efficacy had to be made at 12 hours to see if they were responding. However, some women felt that this time period was too long a time to endure:
I mean I do reassure them by saying after 12 hours if the medication wasn’t working you’d be withdrawn from the study and [ML – yeah] but I guess, you know, for the women who are feeling really bad, 12 hours probably seems like a very long time.
TS01
An additional issue was the chance that participants might receive the double dummy for 12 hours, an issue highlighted by both site staff and prospective participants:
And if they come in when they’re really, really ill when they come in, I could sort of see people not wanting to do that really just risking getting the placebo.
TS15
. . . I was really, really worried that I had two placebos.
EMP1702
The need for double blinding meant that participants were not aware of the trial drugs that they were taking. Concerns about unblinding in the event of treatment failure or trial withdrawal were dealt with through the initiatives of the research teams (e.g. by prescription of both trial drugs). However, this did not take away the desire of some participants to know what medication they had received after the trial was completed:
I just think sort of ethically I would although you sign up to do a trial you do have a right to know what you took because now I’m guessing I was on metoclopramide so in a way, or you know guessing that’s the best one for me to take erm yeah I would, I would like to know what I took.
EMP1702
Publicity and education about the trial
For the trial to be successful, relevant health-care professionals had to be made aware of the requirements of the trial so that appropriate referrals were made. Medical staff also had to be trained to approach patients, confirm eligibility, take written consent from the patients and prescribe the trial drugs. In addition, women with severe NVP needed to be made aware that there was an option for them to join a trial that might benefit them and other women with the same condition. If health-care professionals were not offering information about the trial and/or were not adhering to RCOG guidelines12 on antiemetic use, this could preclude potentially eligible women from participating in the EMPOWER trial.
Primary care and accident and emergency
Some attempts were made to publicise the trial in primary care by site investigators through local networks, but participants’ accounts suggested that the effect was felt to be minimal. However, at least one patient was referred from a GP who knew about the trial:
We also had some communication go out in a GP newsletter, we did get one GP phone us with a lady’s details who was interested in the trial so we’ve had one referral that way also.
TS02
Some sites engaged the help of community midwives to communicate information about the trial. However, for units who provided care for women outside their normal catchment area, this posed difficulties. For some sites, the proportion of patients in the ‘out-of-area’ category was significant:
. . . we have eight staff, well 7000 women have their babies here every year and half of the women are what we call ‘out of area’, they’re not our community midwives that look after them.
TS15
The task of publicising the trial and maintaining awareness of the trial was challenging, particularly in relation to the primary care setting, which involved many doctors working with many competing priorities:
You know if you’re going to go round and educate all the GPs in [site], they would have quite likely listened to it and then forgotten about it the next day, you know.
TS18
Accident and emergency department staff also needed to be made aware of the trial; however, this could be difficult if A&E departments were on a different site or part of a different health-care trust altogether. In some sites, research staff reported that the number of eligible women being seen in A&E departments was minimal. Research staff also reported that their A&E departments had a high turnover of staff and were extremely busy. Furthermore, many lacked dedicated research staff to remind colleagues about the EMPOWER trial:
A&E, it’s just very difficult because they have like quite a high turnover of staff and they’re a lot more busy with lots of other stuff.
TS13
But we don’t really have an active research team in our A&E department so it’s not something that A&E are used to really doing.
TS20
Some research staff made special efforts to liaise with staff in A&E departments, which had some positive effects. For example, at one site these efforts resulted in a new care pathway for women with NVP attending the A&E department:
A&E is also very good, we had an exchange of e-mails, we had written the pathways when they have to refer to us rather than give the antiemetic.
TS17
Clinical and non-clinical staff
Getting clinical staff on board was critical for the EMPOWER trial. However, there were participants who reported that there were some categories of staff who were less likely to be engaged with research, such as locums:
So you’re reliant on different doctors coming in, sometimes they’re locums in a very busy unit, haven’t been able to even think about, is there any research studies that these people might be eligible to.
TS02
Therefore, some staff were not as helpful in supporting recruitment to the trial as they might have been, especially if they were not part of the regular team. On the other hand, there were also accounts of good co-operation from clinical colleagues, which could have been a result of the constant reminders and training sessions provided by site research staff:
I think it just depends on the individual, like I say, some of them are really good and it’s there in their mind because they’ve got an interest in it.
TS01
So we did training sessions with the early pregnancy unit staff – they were regular sessions so I did, I started a couple but then the research nurse did regular training and kind of refresher update sessions as well.
TS19
Another way that clinical colleagues were supported to engage in the process was through existing networks and goodwill fostered between staff, including non-clinical staff:
I’ve got the help of somebody who’s looking for the patient and keeping a note of them for me. One of the, one of the clerks, the ward clerk is actually starting to look each day so hopefully I’ll track them a little bit better.
TS04
Research staff had to regularly remind colleagues about the EMPOWER trial, otherwise the profile of the trial among both clinical and non-clinical staff could be lost:
. . . where our offices are, is quite far away from the early pregnancy unit so I make sure I go over at least kind of once in the morning and once in the afternoon but then I do ring up as well quite regularly and just say have you got anybody and like I say they’ve got my telephone number written all over the department.
TS05
Research staff accounts indicated that the research culture at the trial sites was also important, but that there was always the odd clinician unaware of or not alert to the trial protocol:
So it’s, I mean the doctors are aware of the study but it’s just timing and things like that . . . And then you might get a doctor who for some reason doesn’t know about the study, so.
TS01
Even if all of the requirements of the clinical trial, including the need for publicity and education among staff, were in place, there was still the issue of patients, their pathway into care and their attitudes and preferences in relation to the trial.
Presentation of women to maternity services (theme 2)
There were multiple care pathways (Figure 5) that women with severe NVP could take depending on, for example, their gestation, their history of NVP and their knowledge of how to access care relevant to NVP. Two key points along the care pathways at which women could receive antiemetic drugs were at their GP surgery or at an A&E department (see Figure 5). Even with access to dedicated MAUs or gynaecological assessment units (GAUs), women could be given trial drugs because many of these units are closed out of hours. Furthermore, although some units had longer opening or weekend hours, staff working out of hours were unaware of the trial or research teams were not available.
FIGURE 5.
Care pathways of the EMPOWER trial patients. MAUs include maternity assessment centres, MAUs and antenatal day units. GAUs include early pregnancy units, early pregnancy assessment units, GAUs and gynaecology day units.
The impact of the unpredictability of presentation to secondary care
From staff accounts, there did not seem to be predictable patterns of when patients presented to sites at which recruitment would take place. Women could present with symptoms out of hours, at weekends or on bank holidays, perhaps because they could not find the time during the week when they were at work or they had other family commitments. Women might also have to wait until evening for transport to the hospital by their partners who are at work:
. . . if they’re at home and then someone else brings them cos they’ve been at work all day and then they’ll bring them in the evening.
TS13
Women who attended at the weekend as an emergency were often given a weekday follow-up appointment at a specialist MAU or GAU (TS16). Some staff interviewees made reference to the maternity assessment centre (MAC) or the antenatal day unit, whereas others referred to the early pregnancy unit or the early pregnancy assessment unit.
In addition, care pathways differed from hospital to hospital depending on the gestational age at presentation. This influenced when women were referred to specialist assessment units (AUs) at which they could be approached by trial staff. At some sites, to ease the workload at their MAU, women at lower gestations were seen within the gynaecology department (either in an early pregnancy unit or on the gynaecological ward). This meant that research staff had to try to adapt their recruitment strategies accordingly:
I think it depends on the way that your trust is set up so we, we have the obs[tetrics] med team and they go and see the ward every single day to see if anyone’s been admitted with hyperemesis.
TS08
In some units, it worked well that the local A&E department policy was to transfer patients with NVP directly to the specialist MAU or GAU. Some sites provided out-of-hours or 24-hour access to specialist assessment, but several staff made reference to the issue of women presenting out of hours leading to their inability to recruit:
But it just, it’s out of hours you know they tend to come in, I think they battle on through the day and come in on an evening or a weekend or maybe they’ve battled on at work and they they’ve come in out of hours then, you know, so. But it’s like all trials you’re going to miss you cannot be there 24 hours a day, 7 days a week, so it’s hard.
TS09
However, there were also pathways in which women were encouraged to attend in the daytime if they were able:
But we have a system where, doesn’t always happen but if somebody’s got hyperemesis and they’re not too unwell I think certainly from evenings onwards and overnight the appointment is made for them to come in the morning and they can have their treatment in daylight hours and they can speak to the research midwives.
TS15
On the other hand, some women presented with such severe symptoms that they were admitted as inpatients rather than being seen in an AU, which meant that they could potentially be missed by research staff if they focused efforts on screening women referred to the AU. When the protocol was changed to allow trial drugs to be taken orally, inpatients treated intravenously were likely to be missed:
I think once they’ve got to the point where they’ve being admitted to hospital and they’re in and they’re vomiting and they’re being rehydrated they’ve got i.v. access so they tend to get given i.v. drugs.
TS20
The impact of the infrequency of eligible women
Clinicians reported that the number of women who required specialist assessment and treatment for severe NVP was small. Familiarity with trial processes took some time to achieve, and research staff commented that the small number of eligible cases meant that they gained limited experience with the recruitment processes:
And I think the relatively infrequent presentation of women requiring treatment for nausea and vomiting in early pregnancy again makes it quite challenging . . . And I think that made it difficult for people to, for the clinical staff to always be on the ground, and always be on the ball if you like.
TS18
. . . it’s not like you can find your stride very quickly it takes, and then by the time you get another recruit you think what am I supposed to do again.
TS05
However, to some extent, features of help-seeking in the affected population offset this barrier. For example, some women attended the site several times, and staff reported that this gave opportunities for research staff to introduce the trial to them:
Yes to like attend you know the antenatal day unit because for 2 weeks I was constantly going in every other day because my, my, I was being dehydrated so nothing was working, sickness was at very peak. Then I kept going in to have some fluids because I was, my ketones in the urine was really high.
EMP1402
Women who were ineligible
Treatment with intravenous fluids and first-line treatment
Some patients who declined participation were clear that they wanted to try i.v. fluids first before considering the trial:
I did say that before erm she told me more about the study that I did say that if I tried the fluids and then if that didn’t work, I’d try this one.
EMP1501
Staff reported that several women improved considerably with hydration and had not required antiemetics, making them ineligible for the trial:
I think most women generally responded to the initial management of intravenous hydration itself.
TS22
However, there were women who were aware that hydration could fail and were appreciative of further options:
. . . it’s nice that people are looking into it more because this is like my second pregnancy and there wasn’t, my last pregnancy the drip and the normal process didn’t help but this time it did.
EMP1501
At times, staff were unable to recruit women not because they had not received first-line antiemetic treatment, but because they had not received it for long enough:
. . . that was one of the issues with the study that a lot of women either weren’t given the designated amount of time or we were seeing them before they’d had the actual amount of time they needed to be on the first-line treatment.
TS21
Previous prescription of trial drugs
There were women who reportedly would have been very willing to participate in the trial, except that they had already been treated with a trial drug. These may have been prescribed by the GP or in an A&E department:
I spoke to a lady the other day and she would have been interested in doing the trial but she’d had one of the study drugs . . . we’ve had quite a few of those.
TS02
A strong preference and confidence in a particular antiemetic drug was another reason for some women having received a trial drug.
When the first-line treatment was not effective, clinical staff on duty out of hours had little choice but to prescribe a trial drug, outside the trial, to women if research staff were not available to conduct the trial recruitment process there and then:
. . . obviously if you’ve weekends or bank holidays that’s not covered. So if it’s not working there’s no alternative but to give the metoclopramide or the ondansetron.
TS12
The protocol amendment allowed women to be considered if they had only received the study drugs orally. However, many women had been given study drugs intravenously because of the severity of their symptoms.
TS20
Use of concomitant medications
Some research staff accounts highlighted the concomitant use of other medications, in particular antidepressants, which rendered a number of women ineligible for recruitment to the trial:
I did a couple of ladies that weren’t eligible due to the fact that they were on certain antidepressant medication. I did sort of have a chat with them and say, would you, if you had been eligible would you have been willing to help and the two that I did ask did say yes they would.
TS21
However, one PI commented that this was a very unusual reason for ineligibility at his site, again indicating variability across sites:
So in the last 9 years I’ve only really come across, no two women who had mental health issues and were on some kind of antidepressants. So I don’t, it doesn’t feel like that was an issue.
TS19
Language difficulties
Staff from three sites reported that there were issues with women who had insufficient understanding of the English language to be able to be considered:
Yeah we’ve, I’ve had a few ladies that don’t speak English so we’ve kind of, which is a shame really we’ve not really been able to, speak to them as well.
TS05
However, for another site, language difficulties did not appear to pose a hurdle for the trial staff:
. . . generally it does not feel to me that ethnicity or language barriers have caused any problems for us. We’ve had a real mix of ethnicities that have been eligible for us.
TS06
Women who were eligible
The risk of the condition not improving
Women who experienced an improvement in their symptoms with an antiemetic in a previous pregnancy reported preferring to be on the same medication again, rather than taking the risk of not improving on the trial drugs:
. . . at the time you just sort of want the treatment, that had been recommended already rather than messing with and taking things out or, you know replacing it with a placebo and things like that. So I was more worried about it not working and getting any more ill than I already was because I was really, really ill.
EMP1502
According to some staff participants, patients were not reassured by the fact that they would receive i.v. fluids and were discouraged from participating in the trial because of the chance of receiving the double dummy. Some women resisted medication but reached a stage at which they were desperate for their symptoms to be alleviated:
People have been quite put off by the possibility of having a double placebo – that’s the only thing, even saying that they’ll still be getting fluids it’s just putting people off because I think they’re just, by the time they come to us.
TS05
However, this patient went ahead with participation despite being quite concerned:
Yeah, I mean yeah it’s worrying because you obviously, you’re obviously at sort of rock bottom, so you do need to take, you do feel that you need something.
EMP1702
Women who did not have a preference for a particular antiemetic drug were persuaded by a possible good outcome and the assurance that they could leave the trial at any time:
. . . obviously if it had been prescribed and they think it’s safe for me to take, then I’m going to take it if it’s going to make me better . . . obviously the midwife was like, the midwife was reassuring me that everything was going to be OK. So I was like, ‘OK then well let’s do it’.
EMP1202
I was aware I could withdraw if I still hadn’t improved which was very reassuring.
EMP1002
The impact of severe nausea and vomiting in pregnancy on trial participation
All of the sample groups were consistent in their views that the effects of severe NVP had a negative impact on trial participation. Women who declined felt that it was too much of a burden to try to deal with research paperwork. Staff corroborated this in their experience of recruiting patients:
Yeah I think at that time I was feeling like, I felt like I couldn’t do any extra than I am already doing. My condition hyperemesis is quite bad.
EMP1402
. . . the one lady that I had spoken too she just felt too poorly, I mean she was reading the information but half-way through she sort of like I just feel too unwell . . .
TS14
Research staff who were interviewed expressed concern that the process of trying to screen and recruit women could have an adverse effect on those they approached:
I think it’s hard sometimes approaching them cos they look how they feel, awful, you know. And sometimes you just feel like as if you’re hassling them, you know.
TS09
On the other hand, some women were motivated to join the trial to find a solution and for the good of other women as well:
I feel like there isn’t very much education or things to read about hyperemesis and I actually thought I want to do something that does inform other women because I just didn’t want anyone to go through er what I had, er you know I hope in the future.
EMP1702
Some staff noted that even women suffering from severe NVP were nevertheless still keen to participate:
I can’t imagine how women fill in these questionnaires and even focus on them but they’re very generous and they say if we can do anything to help people stop feeling as bad as we do then, that’s what they’ve all said, if this will help, to not feel as bad as I do then I’m happy to do it.
TS02
Reading and understanding the information
At least two women admitted that they could not be sure that they took in all of the information because of the way that they were feeling:
Some, some of the information was making sense but obviously some wasn’t because at this point I was still being sick.
EMP1104
I’d been struggling to concentrate at work and in life, so I think the one pager was quite good and the second page I looked at, at a later point. I did look at it in the hospital, but I don’t know how much information I actually processed.
EMP1702
This was corroborated by trial staff, who reported women rejecting the literature because they were too sick to read it:
Like a couple of women that I’ve had are like ‘d’you know, I can’t even read that leaflet that you’re asking me to read’. They’re curled up on the bed in a ball, being sick into a bowl next to them, they don’t want to look at anyone they don’t want to talk to anyone.
TS03
However, sickness was not always an unsurmountable barrier; some women commented that they received help going through the information, either from the research staff or from those who accompanied them to hospital:
. . . even though I was feeling really awful they were very good at going through the consent form and sheets with me and luckily I had a partner there who was able to read the sheets and sort of condense it down for me [. . .] make sure I knew what was going on.
EMP1703
So I had to get the, get my friend to read everything out so I understood everything. So that’s how we did it and that’s how we did all the interviews and how we managed to start the information for the trial.
EMP1704
. . . my mum was just sat there and did the paperwork ’cos I couldn’t do it.
EMP1103
Research staff reported that the flow chart provided by the TMG assisted understanding:
The pictorial, you know the flow chart diagram is particularly useful when it comes to trying to explain to women what the different treatment arms are cos I think they often understand it when they sort of see it as a diagram, you know they might get selected for one or both or none.
TS07
This information aid thus helped women to provide informed consent for the trial. When asked about their experience, one staff member described how they felt that the information could be made more accessible to sick patients:
There is a trial manager for a gynaecology trial and they did that they got somebody to read their patient information leaflet on a YouTube [YouTube, LLC, San Bruno, CA, USA] video and then they used to give out iPads [Apple Inc., Cupertino, CA, USA] and get them to watch it [ML – amazing] rather than them having to sit and read. Which yeah it doesn’t take long and once you’ve done it you don’t need to do it again do you so that was something they found really helpful.
TS20
Although the large amount of information to take in during the recruitment process could be a factor in discouraging participation, one woman said that she was persuaded by the information that she read to join the trial:
I just read the leaflet with all the information and then . . . it kind of persuaded me . . . When I was reading it I just felt like I could make, help other people and feel better myself with it.
EMP1701
Needing a quick, effective solution
Women with severe NVP were desperate for their condition to improve. Any delay in treatment that might improve symptoms discouraged women from joining the trial, a point also recognised by site staff. This was especially the case if they had children to take care of or work to get back to:
. . . the only reason I didn’t do the clinical trial was because I was going home after 8 hours, and I’ve got a little girl at home.
EMP1001
. . . because I have to be able to function for the kids at home and I was no further forward at that point I decided I just needed to know what I was taking to see, if I could get something to help at that point, so.
EMP1004
. . . some of the ladies have got other children to look after, some of them are not getting paid so they want to, want to get well and get back to work.
TS09
Women also reported concerns about having to seek further care and start another course of treatment if the trial drugs did not work:
I thought it was a good idea but, I mean when I read the, what the four categories may be and that it might not work and that I could be sick again or it could make me worse that’s what put me off?
EMP1401
However, there were reports about how smooth the process could be from both patients and staff:
All my sickness stopped and I think it was within an hour and a half later, I was home with my trial of sickness tablets.
EMP1201
She was feeling a bit better because she had the fluids going and then again the PI was around and she came and consented her and then we were able to pop her on to the computer and randomise her and then get her drugs like that. So that way worked quite well cos she felt that she was getting some treatment before she start, kind of consented.
TS05
Preferring not to take medication
Some women reported that they were concerned about how medication would affect their unborn baby:
I didn’t want to take the unnecessary medicines, I know they might have helped me but I just didn’t want to harm the unborn baby.
EMP1402
On the other hand, one woman agreed to participate despite being reluctant to take drugs during pregnancy because she reasoned that being so dehydrated could also adversely affect her pregnancy:
I’m generally one of those people who don’t tend to take paracetamol or ibuprofen but I think when I was worried that I was vomiting so much and so dehydrated that I was worried that I might do more damage to my body and for the baby if I didn’t try and control it, so.
EMP1703
Another patient was also reluctant to take any medication, but her desperation to find a solution persuaded her to participate:
I was very reluctant to take anything erm. But then I reached, obviously a point where I would have actually taken anything I think [laughs].
EMP1702
Perceived research burden
Some staff felt that women could be discouraged by what they had to do for the trial in terms of the information that they had to provide:
I just feel like when it’s easier for the patients it’s easier to recruit them and for them to take part but when there’s a lot for the patients to do it sort of puts patients off a bit.
TS10
However, among those who participated, their accounts suggested that they seemed able to manage the burden of completing trial questionnaires:
I could do the forms but I realise I was much slower and I was trying to read, you know, every word and you know I did manage to do the forms . . . it was struggle but it was not terrible.
EMP1702
Some staff observed that patients were appreciative that they would be monitored and able to access care more quickly than in the normal pathway:
. . . just someone to listen to your symptoms, and that was, that was really helpful for her.
TS03
. . . she could ring us and say I’m really not feeling well so we could organise her to go in to MAC so I think she found that quite nice that she didn’t have to ring them herself because sometimes they are reluctant to bring people in?
TS03
Trial staff roles (theme 3)
Because of the various care pathways that women could take, and their infrequency in presenting, research staff worked hard to catch women by ringing or visiting units within maternity, obstetrics and gynaecology (O&G) regularly to check and remind staff:
So I try and catch them as soon as I can because if you leave it later then there’s the risk that they might have already been given some medication . . . And if I went down and there was somebody on the ward to come I would just sort of keep popping up and down and like I say try and catch them as soon as they come in.
TS01
Apart from tracking, catching, screening and recruiting women, it was often necessary for research staff to work at liaising with their clinical colleagues, especially between maternity and gynaecology:
. . . because we had to work so hard to try and get any recruits, most of the research time was spent trying to find the women and liaising with the clinical staff about the eligibility log and reminding people.
TS16
Confidence and morale
The amount of paperwork, together with the slower than expected recruitment into the trial, had an impact on the confidence of research staff in carrying out trial processes to the standards required in a CTIMP. Accounts from staff on the delegation log indicated that they could be reluctant to take on the task of recruiting a patient because the process involved felt onerous:
. . . it sounds awful but you kind of dread actually recruiting someone because you know it’s so complicated, you know, it is really hard all the information you need to remember.
TS08
Failure to recruit was because of a number of administrative hurdles and in the early stages was a source of frustration:
And we had given our first recruit the medication orally because she asked for it that way and hadn’t realised that the first dose had to be i.v. so it was that, with that lady . . . and then there’s been some issues with sending things over different e-mail addresses . . . We have had to call to clarify at the point of recruitment on every single occasion.
TS02
Faced with the challenges of recruitment, enthusiasm for the EMPOWER trial waned. However, successfully recruiting new participants encouraged staff to press on:
. . . we’ve found, you know women, we were kind of getting a little bit, that we keep on looking and we can’t find women that are fitting the criteria but having the recent two recruits has been a real boost.
TS06
Morale was also boosted when comparisons were made with other trial sites through the regular updates provided by NCTU, and PIs reassured research staff that they were not alone:
. . . they feel better that it’s not just our site that isn’t doing as well. I mean they know that, you know, it’s a national thing it’s not just one particular site doing, not doing as well.
TS14
When staff encountered an AE, their morale was affected; however, when they saw participants benefiting from the care provided, they were motivated to carry on:
. . . the midwives have been on a shift where women have taken part it feels like they’ve got an enthusiasm for the study they can see, you know, these women kind of felt better in the short term. We know it might just be the fluids, we don’t know what really drug that we give them but it’s kind of a positive feeling which kind of helped, you know, remind the staff that it’s a good study.
TS06
Recruitment and trial processes
Recruiting vulnerable women suffering from severe NVP was frequently reported as challenging:
I would even say that like we’ve done studies on labour ward where you’re speaking to ladies in labour and sometimes that’s been easier than trying to speak to a lady who’s puking her guts up on the, on the assessment unit.
TS10
Trial staff were dependent on their clinical colleagues to assist, but they reported difficulties because of the amount of paperwork involved during recruitment that required them to be present. This made recruitment out of hours virtually impossible:
. . . it’s a little bit more time consuming that clinical staff aren’t able to fit that in. If it’s just a short period of time it’s not quite so bad and they will accommodate but this is a little bit more involved really I think.
TS04
. . . it’s always been really difficult to recruit patients out of hours when you’re relying on your medical teams unless it’s really straightforward and obviously there’s a lot of paperwork that goes along with EMPOWER recruitment isn’t there.
TS10
Staff also reported that data collection during the course of the trial could be demanding:
All the paper work you need to give, all the forms you need to fill out, you have to make sure the patients know, you know, when to fill it in and you know, you’ve got to call the patient, well if they’re discharged at 12 hours, they’re clearly fine at 48 hours, at you know 4 days, 10 days etc.
TS08
However, staff accounts indicated that good teamwork mitigated against the demands of the trial processes. Less confident staff reported that they felt supported by their colleagues:
But before I’d done those two I was a bit like if we get one can someone come with me so we can kind of go through it together.
TS03
Principal investigators in the trial valued their research midwives or nurses, especially if they did most of the liaising with fellow clinicians and co-ordinated the work of the trial:
I think the main staff on MAU they were aware of the study and I think I must give credit to [name 1] who was like the team leader and the co-ordinator there. So she used to liaise with [name 2] who was the research midwife and [name 3]. So [name 2] and [name 3] also co-ordinated closely with [name 1] and all the consultants too were aware of the study.
TS22
Patient counselling and staff empathy
There were reports describing how staff counselling about the risks involved in participating in the trial, particularly reassurance about patient care during and after the trial, was critical in the recruitment process:
. . . sometimes they worry about the placebo–placebo but once you explain to them that you are actually having the i.v. fluids they’re not leaving you with, you know, absolutely nothing and if it doesn’t work if you’re feeling unwell we can take you off straight away, they do feel more reassured.
TS08
The following observations from one of the PIs summarises the importance of staff counselling and care, not just from research staff but from clinical staff managing women at the point of admission:
. . . if, when a women came in with nausea and vomiting, the admitting midwife had a positive word to say for the study in ways, not just of course through the study, but other ways in which the woman would feel assured she was going to be taken care of, that makes it much easier for the recruiting team to come on board and make that, make that case effectively for taking part.
TS18
However, while providing information about the trial, convincing patients that rehydration with i.v. fluids was a form of treatment in itself could prove difficult.
At the same time, staff also expressed empathy for the women whom they were trying to recruit onto the trial, such as for the long wait that women had to endure for treatment:
I do sometimes feel that the women have a rough deal if you like, because they go to the GP and then they come to the hospital and it’s sometimes a while before they have, you know, the treatment.
TS04
If patients were given information about where they were in the queue to see a doctor, some participants felt that this could make women more open to considering the trial:
So we want to make sure that they know that their care isn’t being held up by us as soon as the doctor comes they’ll see them so, we just say we’re just seeing them while they’re waiting for the doctor anyway and I think that makes them feel a bit better cos they don’t feel like we’re like sort of delaying their treatment if you know what mean.
TS11
When approaching women, research staff had to be sensitive to how women were feeling at that point in time, and to approach them appropriately. One patient felt that the research was more important than the care that she was desperate to have:
But it was kind of, felt as though I’m waiting for some treatment which I’m not getting but the research information was more important, it felt to me, cos I had got a visit about three times.
EMP1401
On the other hand, many patients gave positive and appreciative accounts of how they were approached, whether they accepted or declined participation:
She explained everything in detail she gave me the time I needed to process. She, basically sat there and explained it all in all ways, in a way that I understood.
EMP1201
. . . when I spoke to them you know they said it was about my health and quality of life so they were very good at making sure I knew what my options were to withdraw from the trial if I needed to.
EMP1703
Principal investigators mostly had complete confidence in the abilities of their research midwives/nurses to counsel patients about their options:
They don’t have to do obviously you know sort of, in terms of consenting and everything but they’re happy to do all the talking, and you know very objective and unbiased and stuff so.
TS15
From other feedback from interviews that described empathy for women with NVP, it appeared that staff commitment to the trial could be compromised if they felt that participation had a negative effect on the well-being of their patients. To illustrate this, when a SUSAR, a fetal abnormality that could have had a link to one of the trial drugs, occurred research staff reported feeling distress and a sense of responsibility for the patient’s treatment with the trial drugs and the subsequent termination of the pregnancy.
Staffing levels and support
Appropriate levels of staffing were also critical to the trial; recruitment processes could have been affected by staff being on leave:
I only work 15 hours and because we’ve been short of staff in the, in the team, really everybody, although we’ve got about, we had about three staff until somebody just came back off long-term sick. Because we’re so short when we’re at work we’re just mainly focusing on our own studies.
TS10
Seen from the perspective of one PI, as recruitment numbers were small, the workload for midwives or nurses was relatively lighter than for busier trials with a large number of participants:
. . . there were few patients, even if we’d recruited to target it wasn’t many patients per centre and, therefore, the workload for the midwives [they were] working with the women, supporting women and the questionnaire . . .
TS16
From the perspective of other staff, however, much of their work involved keeping the EMPOWER trial on the agendas of their colleagues. Maintaining their familiarity with trial processes that were complex was crucial, and this was helped by the co-operation and support from fellow staff:
I’ve got lots of support though we’ve got a really good research team here and everybody kind of helps out each other. So always kind of felt like we’ve had plenty of support during the whole thing.
TS05
At some sites, because the AUs were so busy, research staff were involved in administering i.v. fluids and obtaining the medication. Good relations were fostered when research staff understood the pressures that clinical staff were under and the added burden placed on them by participation in trials:
. . . they’re working flat out and we’re constantly asking them to remember something new and something extra and doing extra jobs. So the more you can kind of give back and take off them in terms of their workload then yeah like you say the better the relations between the two are.
TS20
There were accounts that when trial staff did manage to recruit a woman to the trial, there was a real sense of achievement, despite the fact that it required a lot of effort and teamwork:
The other day we did get the woman in the study there were three members of the research team that stayed 2 hours late and one of the consultants who stayed an hour over to finally get somebody in. So we certainly feel like we’ve gone above and beyond . . .
TS20
Conclusions
The enablers of and hurdles to recruitment in the EMPOWER trial were both trial specific and site specific, and are summarised in Table 17.
| Themes | Hurdles | Enablers |
|---|---|---|
| Requirements of the EMPOWER trial | ||
| The trial question and adherence to RCOG guidelines12 |
|
|
| Organising the delivery of the EMPOWER trial |
|
|
| Demands of the EMPOWER trial protocol |
|
|
| Publicity and education about the trial |
|
|
| Presentation of women to maternity services |
|
|
| Trial staff roles |
|
|
Trial-specific factors
The issue of adherence to the RCOG guidelines12 was raised as a hurdle to recruitment, but this was not necessarily the case if women were approached by research staff before second-line treatment was prescribed. The problem arose when women attended AUs providing out-of-hours or 24-hour access at times when research staff were not available. One solution was to raise awareness with clinical teams along the care pathway so that women could be signposted to the trial. However, publicising the trial among primary care clinicians was generally not felt to be the way forward because of the scale of the task.
Operational difficulties at the start of the trial were to be expected, but the smooth working of the trial was generally facilitated by good staff and professional relationships between maternity and gynaecology departments, and with the Clinical Trials Unit. At most sites, there was a dogged determination by lead staff to work hard at liaising with colleagues across departments and educating them about the trial and the need to follow RCOG step-up guidelines. 12 Nevertheless, the key issues for all research staff were the unpredictability of women presenting to services and the infrequency of their presentation. These barriers affected the efforts of research staff to recruit women in the best way possible.
The demands of the complex trial design added to the recruitment challenges faced by staff. Staff spent most of their time finding ways and means of maintaining the profile of the trial; reminding their colleagues about trial process requirements; and tracking, identifying and screening women. Recruiting women was made more challenging because women with severe NVP were physically and emotionally vulnerable; therefore, time and patience were required. Recruitment to the EMPOWER trial involved more than just paperwork; it required high levels of empathy and patient care. Time was needed for the patient to consider the information carefully before making an informed decision about consenting to participate or not. Continuity of care for trial participants was provided and, where appropriate, trial drugs were prescribed as patients requested. In this way, the issue of unblinding did not seem to be as problematic an ethics issue as when first considered.
Women who were eligible to participate were discouraged at the thought of their condition not improving because of the risk of receiving a double dummy. Their experience of severe NVP had an impact on their decision; all that they could think of was a quick, effective solution. The effort and time required for participation, to read and understand the information, and to wait to be entered into the trial were significant for several women. However, for altruistic reasons, there were women who willingly participated to help others.
Site-specific factors
The care pathways that women took in seeking help for NVP were dependent on the trial site, the catchment area and the health service provision therein. Ethnic composition, health behaviour and antidepressant use were factors at each site. Trial sites that recruited more women were characterised by primary care providers that tended not to prescribe trial drugs as first- or second-line antiemetics. Sites that were unsuccessful were those that had to work hard to change the culture of prescribing antiemetics within secondary care. Relationships with A&E departments varied across trial sites; A&E departments in which research was not a priority was a factor. The availability and engagement of GCP-trained staff also varied across sites. Staff confidence and morale varied depending on recruitment rate, recruitment success, which ranged from 11% to 63% of eligible patients.
Chapter 5 Discussion
Severe NVP is a common condition that is associated with a range of adverse maternal and infant outcomes, and significant personal and health-care costs. Antiemetic prescribing and hospital admissions for NVP/HG increased continuously in the UK between 1998 and 2013; by 2013, 5.2% of pregnant women were prescribed antiemetic drugs in primary care and 2.1% were admitted. 39 The EMPOWER trial set out to determine the most clinically effective and cost-effective second-line hospital-prescribed antiemetic therapy that should be adopted by the NHS. The trial, commissioned by the National Institute for Health Research, followed the publication of management guidelines by the RCOG in 201612 that recommended the use of either a dopamine receptor antagonist (metoclopramide) or a 5-HT receptor antagonist (ondansetron or domperidone) as a second-line therapy. Three recent reviews13–15 on treatments for NVP highlight the poor quality of the available evidence and the heterogeneity of inclusion criteria and outcomes. Given these uncertainties, the EMPOWER trial was designed with an internal pilot phase to test key processes of the main trial, specifically recruitment. 40 However, despite the best efforts of those involved, including an extended pilot phase incorporating revised exclusion criteria to reflect changes in clinical practice, the required number of participants was not recruited. It was, therefore, decided, with the support of the TSC and HTA, not to progress to the main trial. To the best of our knowledge, no RCTs of second-line antiemetic therapy have been reported during the conduct of the EMPOWER trial and, therefore, the original question remains unanswered.
In designing the trial, we assumed a recruitment rate of 3 per 1000 pregnant women (based on 6 per 1000 women being eligible and 50% of eligible women agreeing to be randomised) based on our experience in a prior single-centre RCT (i.e. the Hyperemesis in Pregnancy Study29) and a review of women with severe NVP attending Newcastle upon Tyne Hospitals NHS Foundation Trust over a 6-month period. A subsequent large population-based cohort study from England, which utilised Hospital Episode Statistics from 1997 to 2012, reported an admission rate for HG of 14.8 per 1000 pregnancies. 41 Of more relevance to our trial population, 4.2 per 1000 pregnant women required two or more admissions. 41 Based on data from the seven initial pilot sites (that included 43,850 women in 2017–1842), 513 women who were assessed with severe NVP and who had been prescribed an antiemetic drug were screened for inclusion (≈ 11 per 1000 deliveries). The overall recruitment rate of eligible women was 29%, although this ranged from 11% to 63% at different sites. The key reason as to why recruitment was much lower than anticipated was the high rate of ineligibility (n = 399, 78%) that, in 67% of cases, was because of prior treatment with trial drugs, predominantly ondansetron. This was consistent across the seven sites (accounting for 44–87% of ineligible cases) and was not substantively altered by revising the inclusion criteria (to allow women with prior suboptimal use of the trial drug to be included). The evidence collected from the screening logs, which was amended after the problem was identified and confirmed in interviews, suggested that there were multiple reasons for this unexpectedly high rate of second-line antiemetic use.
We assumed that most women presenting with severe NVP would be managed by O&G services in accordance with RCOG guidelines12 after prescription of a first-line antiemetic drug. However, we found that nearly half of the women who were ineligible owing to prior treatment with ondansetron or metoclopramide had been prescribed a trial drug in primary care or the A&E department. Antiemetic prescribing by GPs is increasing; recent evidence suggests that 38–59% of women admitted to hospital with severe NVP/HG have been prescribed antiemetic drugs by their GP. 39,43 Fiaschi et al. ,39 in a large population-based cohort study from the UK between 1998 and 2014, reported that, of the 6390 (1.5%) women admitted before 20 weeks’ gestation, 7.8% had been prescribed metoclopramide, 1.3% had been prescribed domperidone and 0.4% had been prescribed ondansetron. In nearly 90% of cases, this was in addition to a first-line antiemetic. The evidence collected from general practices in the north-east of England between 2013 and 2016 (i.e. prior to the RCOG guidelines12) highlighted substantial variations in the use of antiemetics by GPs; of relevance, 15% of antiemetics prescribed were second-line drugs (despite that there was a year-on-year increase in the number of women admitted with NVP). 8 Of more concern, some current primary care prescribing guidelines still differ significantly from the RCOG recommendations. 44
An increasing proportion of women with NVP were being seen by A&E services, possibly reflecting changes in access to GPs and/or a lack of support/knowledge from GPs and community midwives on access to care. 45 A&E department guidelines for women with severe NVP vary; some refer directly to O&G services, whereas others treat women in short-stay units, reducing treatment delays and the need for admission. 45 Antiemetic guidelines often differ from RCOG management guidelines, and there is evidence that ondansetron and metoclopramide are preferred first-line therapies. 46,47 Interviews suggested that, having recognised the challenges, most research teams sought to increase the awareness of the trial (and the prescribing guidelines), especially in A&E departments, where liaison was easier. Indeed, at St George’s University Hospitals NHS Foundation Trust, where trial set-up was delayed, it was eventually agreed that the A&E department research team should lead the trial.
Even when referral pathways operated as predicted, experienced O&G research teams faced several recruitment challenges. Many women presented ‘out of hours’, when research staff were usually unavailable and there were fewer GCP-accredited medical staff. This often reflected their need to arrange child care and/or transport to hospital. However, there was also evidence that some medical staff in O&G services did not adhere to the RCOG antiemetic guidelines, tending to prescribe a trial drug (typically ondansetron) as the first-line treatment to resolve symptoms quickly. This was more of a problem than we had anticipated, given that all PIs confirmed adoption of RCOG guidelines12 prior to the start of the trial (and evidenced by submission of site guidelines). The challenges of implementing guidelines within O&G services are widely recognised,48,49 and the modification of practitioner prescribing practice often requires more complex interventions than the publication of guidelines alone. 50 Specific factors related to non-adherence to antiemetic prescribing may be the poor-quality evidence of effectiveness underpinning national recommendations (quoted as level 2++, i.e. mostly based on case–control or cohort studies12) or their recent publication with limited time to undertake audit and feed back to monitor/improve compliance. 49,51
The increased use of ondansetron to treat NVP, which is evident from the management of women screened for participation in the EMPOWER trial, is consistent with a recent report of antiemetic discharge prescriptions from one large UK trust between 2010 and 2015. 52 Following the first admission with HG, 45% of women were discharged with ondansetron [either alone (8.5%) or in combination with another antiemetic (37.0%)], compared with 27.5% with metoclopramide (3.7% alone and 23.8% in combination). The rates of ondansetron and metoclopramide prescription increased to 72.9% and 44.9%, respectively, for subsequent admissions. Prescribing practice appears to have changed even more dramatically in the USA, with one web-based questionnaire study suggesting that ondansetron is now the most commonly prescribed antiemetic. 53 Data from both the National Birth Defects Prevention Study and the Sloane Birth Defects Study suggest that the incidence of ondansetron use in women with first-trimester NVP increased from < 1% in 1997–98 to 12–14% in 2013–14. 54 The increased prescribing of ondansetron, especially in combination with other antiemetic drugs, would be a concern if the benefits do not outweigh the risks. Recent research has focused more on exploring the harms, with little consideration given to the benefits to women and their babies. Thus, there is little existing evidence of the benefits with which to formulate a robust assessment of the risk–benefit ratio.
Most eligible women felt very unwell: several commented on the need for a ‘quick and effective’ treatment. The protocol for the EMPOWER trial recommended rapid i.v. hydration based on limited evidence that i.v. fluids (without antiemetics) reduced nausea. 13,55 However, for some women the delay in initiating antiemetic treatment, even for the hour required for consideration of the trial information, and/or the chance of receiving no antiemetic treatment (i.e. the double dummy) was unacceptable. Both recruitment barriers, inherent in the trial design as specified in the HTA programme commissioning brief, were highlighted by the lay co-applicants and the lay members on the TSC. Considerable effort was made by the lay co-applicants to amend the information provided to women to ensure that it was as brief as possible, including the use of an initial short PIS. It is noteworthy that the trial information was commended by the REC. Most of the women who read the PIS felt that it provided a clear explanation of why the trial was needed and the risks and benefits of participation. However, interviews highlighted that many women rejected the PIS because they felt too unwell.
We incorporated the option of withdrawal from the IMP within the first 12 hours (if participants had not responded to trial medication) based on advice from the lay co-applicants and feedback from the women who had experienced NVP. Some women commented that this was reassuring. Trial staff were empathetic to women’s social circumstances and well-being; several highlighted the ethics ‘dilemmas’ inherent in delaying antiemetic treatment and offering an unknown antiemetic (owing to double blinding) as part of the trial, rather than one that had previously worked for the woman. The challenges of recruiting patients into dummy-controlled trials are well recognised. 56,57 A recent RCT of ursodeoxycholic acid compared with dummy in women with intrahepatic cholestasis of pregnancy reported that 57% of eligible women declined participation: 35% because they ‘wanted’ ursodeoxycholic acid and a further 13% because they ‘did not want to be randomly allocated’. 58 Interviews with women who were approached to participate in the EMPOWER trial confirmed two divergent themes: an explicit reluctance to take a dummy and an altruistic desire to help increase scientific knowledge. 57,59
Within O&G services there were variations in how women with severe NVP were managed. The EMPOWER trial was designed to acknowledge the shift in care from inpatient care to day care management. 12,13 A large trial60 published just before the start of the EMPOWER trial confirmed that outpatient ambulatory care is as effective as inpatient management. Recruiting women in busy assessment units created difficulties for the research teams, especially given the infrequent and unpredictable presentation of the condition and the complexity of the protocol. Interviews highlighted the need for staff at several sites to negotiate boundaries not just between maternity and gynaecological departments, but also between research and clinical care. 61 This ‘symbolic boundary’ that typically reflects the organisational management of professional responsibilities was negotiated differently at different sites;62 in some units these roles were combined with positive effects and in others role differentiation was maintained throughout. The qualitative study was unable to fully unpick some of the boundaries between research and clinical staff, but highlighted the contribution that empathetic working relationships across these boundaries could have on enhancing trial recruitment.
Previous systematic reviews of treatments for NVP/HG have highlighted the heterogeneity of inclusion criteria and outcome measures. 13–15 The EMPOWER trial was a pragmatic trial with simple inclusion criteria (i.e. the managing clinician felt that there was no sustained improvement in NVP despite taking a first-line antiemetic for at least 24 hours). Most women had been prescribed cyclizine (79%) and/or prochlorperazine (39%) in accordance with RCOG guidelines. 12 Although we opted not to use a predefined PUQE score as an inclusion criterion, 60% of the participants had severe symptoms (score of ≥ 13) and none had mild symptoms (score of < 7). High PUQE scores are known to be associated with greater hospitalisation and health-care costs, as well as lower well-being and QoL. 20,21 Consistent with this, recruited women had very poor QoL [with a mean baseline score on the NVPQoL questionnaire of 186 (out of a maximum of 210)]. This is more than two standard deviations above the mean for women in their first trimester of pregnancy. 22 Both objective measures confirm that, despite first-line antiemetic treatment, participants had severe NVP.
No core outcome set for trials on the treatment of NVP/HG was available in 2017. A subsequent review63 of outcome reporting in RCTs identified 34 distinct outcomes in 34 trials. The importance of taking account of patients’ perspectives when designing research outcomes is widely recognised. 64 We worked with lay co-applicants and previous sufferers who prioritised treatment failure (with the need for further treatment) as the primary outcome above several patient-reported outcomes (such as health-related QoL). 65 We also sought advice on the duration of trial drug administration before treatment failure declaration, acknowledging the balance between allowing enough time for trial drugs to have an antiemetic effect and the continued use of dummy in symptomatic participants. Although 12 hours was felt to be reasonable, two out of three withdrawals occurred before 12 hours because of persistent/worsening symptoms. A core outcome set involving 10 domains with 24 outcomes has subsequently been reported. 66 Treatment failure (as used in the EMPOWER trial) is relevant to two of the defined domains: ‘need for interventions to manage symptoms’ (with an outcome of ‘use of additional medication’) and ‘maternal service utilisation’ (with an outcome of ‘hospital treatment’). 66
No conclusions regarding the clinical effectiveness of ondansetron or metoclopramide nor the economic analysis can be drawn from the failed pilot trial. However, a couple of clinical findings are worth highlighting. Treatment failure was reported in a higher proportion of participants than expected (50%). However, those who completed the questionnaires reported improvements in symptom severity and QoL over time (including women in the double-dummy group). Consistent with high treatment failure, almost one-third of participants were readmitted with NVP within 10 days, and two women went on to request TOP. The findings are consistent with the 28% and 38% readmission rates reported by Fiaschi et al. 41 and Morris et al. ,67 respectively. Previous studies of ondansetron and metoclopramide have consistently documented improvements in mean symptom scores over 24–48 hours but none has reported the need for additional antiemetic treatment during the study period. 16–18,27,28 Abas et al. 18 reported that 14% of women randomised to ondansetron and 18% randomised to metoclopramide required open-label antiemetics after 24 hours of trial drug, although the duration of follow-up was not stated. The rates of TOP for NVP in the absence of fetal anomaly are poorly reported, and the factors associated with the decision are complex. Mazzotta et al. 4 reported that 16% of women considered TOP for NVP, and 3% reported terminating a pregnancy because of NVP. More recent studies have reported TOP rates of 8–9%. 68,69 Fejzo et al. 69 reported that women with HG who took ondansetron had a lower rate of TOP than those who did not take ondansetron (2.5% and 8.7%, respectively). The EMPOWER trial results suggest that no second-line antiemetic treatment is effective in all women with NVP/HG and that short-term symptom scores do not reflect the true personal and health-care costs of the condition.
The safety profile of antiemetic drugs used in pregnancy is critical. Assessing maternal drug-related AEs is made more complex because of the frequency of pregnancy-related symptoms, especially involving the gastrointestinal tract. For this reason, we opted to use the National Institutes of Health CTCAE26 and recorded a change in AE grade following initiation of trial drugs. Research teams reported that the criteria were easy to use but experience in pregnancy is still very limited; we could identify only one prior pregnancy-related study that has used these criteria, and that was conducted in women with gestational trophoblastic neoplasia. 45 Although one participant withdrew because of perceived side effects of the trial drugs, overall the limited evidence from the EMPOWER trial suggests that trial drugs were well tolerated.
Of particular concern to pregnant women is the risk of fetal AEs, notably congenital malformations. Several women who were interviewed expressed concerns about the effects of trial drugs on their unborn baby. Prior to commencing the EMPOWER trial, the available evidence suggested that metoclopramide was not associated with any adverse fetal outcomes. 70,71 Less information was available about the safety of ondansetron in pregnancy, but a systematic review published in 201672 concluded that the risk of malformations appeared to be low. During the conduct of the EMPOWER trial, three large studies from the USA were reported. 54,73,74 Parker et al. 54 analysed data from two case–control studies and reported a modest increase in the risk of cleft palate [odds ratio (OR) 1.6, 95% CI 1.1 to 2.3] in the National Birth Defects Prevention Study (1997–2011) but not in the Sloane Birth Defects Study (1997–2014). Huybrechts et al. 73 conducted a cohort study nested in the nationwide Medicaid Analytic eXtract (2000–13) and found no increase in the adjusted relative risk (aRR) of congenital malformations overall, but an increased risk of oral clefts (aRR 1.24, 95% CI 1.03 to 1.48). Zambelli-Weiner et al. 74 conducted a nested case–control study using the Truven Marketscan data (2000–14) and reported an increased risk of cardiac defects (OR 1.52, 95% CI 1.35 to 1.70) but not orofacial clefts. Based on the available evidence, and immediately after the decision was made not to progress the EMPOWER trial beyond the internal pilot phase, the EMA issued a signal of birth defects (specifically orofacial clefts) following first-trimester exposure to ondansetron. 75 The EMA recommended that ondansetron should not be used during the first trimester of pregnancy. 75 In response to the EMA’s signal, the UKTIS advised that patients must be adequately counselled regarding the benefits of ondansetron together with the small increase in risk of orofacial cleft that may exist (quantified as an excess absolute risk of 3 per 10,000 pregnancies), but that ondansetron should still be considered as an option for patients with severe NVP in whom first-line treatments have failed. 76 A very recent further study has reported an association between ondansetron use and neonatal ventricular septal defects. 77 Given these increasing safety concerns, the failure of the EMPOWER trial to provide useful evidence to guide practice is regrettable, especially because a key reason for failure was prescription of ondansetron.
It is widely recognised that one of the biggest threats to a RCT’s success is suboptimal recruitment, with only 56% of multicentre RCTs funded by the HTA programme achieving their recruitment target. 78 Although empirical research has helped to understand RCT recruitment issues,79 there are few effective interventions to facilitate RCT recruitment; one such intervention is the QuinteT (qualitative research integrated into trials) Recruitment Intervention (QRI),80 developed in 2016, and subsequently shown to increase recruitment rates in three of four RCTs experiencing difficulties. 81 Although we did not integrate a formal two-phase QRI-type intervention into our pilot trial, we did employ most of the methodologies designed to investigate recruitment processes (e.g. interviews with potential participants, mapping eligibility and recruitment pathways, and scrutinising trial documentation). Having identified that prior treatment with study drug was a key eligibility issue, there would have been benefits to extending the information collected in screening logs and undertaking interviews with research staff earlier to more quickly understand the contribution of care pathways. In retrospect, the ‘plan of action’,80 which focused on amending eligibility criteria, may have been too limited and greater consideration could have been given to providing earlier feedback about antiemetic prescribing to both clinical staff in the participating sites (in A&E departments and O&G services) and GPs referring into sites. However, it is unknown if earlier feedback would have had a positive and timely impact on recruitment and whether or not any impact would have been similar across care settings.
Strengths and limitations
The major strengths of the EMPOWER trial were the inclusion of an internal pilot, with a stop–go progression criterion based on a recruitment target, and the contribution of the public to the trial design and delivery. Internal pilot trials are perceived to be more cost-effective than an external pilot followed by a full trial; a recent analysis of HTA programme-funded trials reported that the proportion with an internal pilot phase had increased to 72%. 82 Given that this was a HTA-commissioned trial with the interventions and design stipulated in the brief, we acknowledged uncertainties about recruitment rate in the specified patient group and antiemetic failure rate. A further strength was the inclusion of a qualitative component to explore patient understanding of recruitment and participation, which is known to be particularly complex in trials involving pregnant women. 83
The obvious limitation of the trial was the failure to achieve the recruitment target. Recruitment problems were identified early and, with the support of the TSC and HTA, eligibility criteria were broadened. However, inclusion of research staff in the initial qualitative interviews would have identified the changes in care pathways and antiemetic prescribing earlier in the pilot. The value of capturing staff experiences has been highlighted previously,79,84 and inclusion of interviews with trial staff is recommended as part of phase 1 of the QRI. 80 Despite broadening the eligibility criteria (to allow inclusion of women who had received suboptimal treatment with study drug), we were unable to overcome the recruitment challenges related to unforeseen changes in care pathways and antiemetic prescribing, as well as the research culture in the trial sites. 85 This led us to conclude that the trial, as designed, was no longer feasible. Early closure minimised wastage of public funds.
Chapter 6 Conclusions
The EMPOWER trial was designed to provide guidance to health service providers on the most effective antiemetic drug for women with severe NVP who require secondary care. The trial incorporated an extended internal pilot trial, involving 12 sites experienced in delivering complex obstetric trials, to assess feasibility of recruitment. The extended pilot trial failed to achieve the revised recruitment target and did not progress to a main trial. A higher than anticipated proportion of women were ineligible for the trial; the main reason was prior prescription of trial medication(s) either outside O&G departments or when women presented to O&G departments out of hours (when there was no/limited access to research staff). This reflected a change in prescribing practice that was often inconsistent with RCOG antiemetic guidelines, with wider use of second-line drugs, particularly ondansetron. Owing to the small sample size it was not possible to draw meaningful conclusions from the main trial outcomes or the health economic outcomes. The qualitative evaluation provided valuable insight into both the barriers to and the facilitators of recruitment, which related to the requirements of the complex EMPOWER trial, the variable presentation of women to maternity services and the research staff roles. We did not anticipate the significant changes in ondansetron antiemetic prescribing practice, particularly outside O&G services. Had we embedded a more formal qualitative recruitment intervention, with earlier interviews with recruiters, it is possible that we could have identified key changes in care provision earlier, allowing a more impactful action plan, with greater potential to increase recruitment, aimed at clinicians’ adherence to antiemetic prescribing guidelines.
Implications for clinical practice
There are no implications for clinical practice given the failure to achieve the trial objectives. We are unable to provide improved evidence to support clinician discussions with patients about the best choice of second-line antiemetic for NVP, other than to highlight that treatment failure appears to be common and consideration should be given to ongoing support and follow-up to identify early those women who fail to respond to prescribed drug(s). The study also underscores the need for effective implementation strategies to embed guidelines into clinical practice, especially when care is multiprofessional and involves complex care pathways. 86
Recommendations for future research
-
There remains a lack of clear evidence of the clinical effectiveness and cost-effectiveness of treatment strategies in women with NVP who fail to achieve symptom improvement with first-line antiemetic drugs. The HTA programme should revisit this topic, especially given the recent safety concerns about the most widely used second-line antiemetic (ondansetron).
-
More research is required to understand the barriers to recruitment of complex RCTs involving pregnant women, especially when the condition and/or the care pathway result, in many women presenting out of hours. Understanding the impact of wider integration of clinical and research roles would be valuable. In addition, future trials should include resources to enable staff to be available out of hours with a mobile pager or mobile telephone, as well as a 24-hour trials helpline, to help overcome some of the difficulties reported by the EMPOWER trial staff.
-
Future trials of antiemetic drugs for NVP should acknowledge that short-term measurement of symptom scores provides limited information on clinical effectiveness; treatment failures (which result in the greatest impact on women and health-care services) frequently occur > 48 hours after treatment initiation. Incorporation of agreed core outcomes for HG research is vital. Furthermore, self-completion questionnaire burden is a problem in this population and alternative data collection methods should be considered.
-
Future RCTs in which there is uncertainty around recruitment rates should incorporate an integrated recruitment intervention, such as the QRI, into the pilot phase (or into a stand-alone pilot trial).
Acknowledgements
The authors would like to thank all of the staff from each of the contributing sites. Special thanks to the TSC (Amy Armstrong, Caitlin Dean, Pollyanna Hardy, Marjory MacLean, Gerry Richardson and Hora Soltari), particularly Susan Bewley (chairperson), and the iDMC (John Norrie and Deidre Murphy), particularly Mathew Coleman (chairperson). The authors would also like to thank Eva-Maria Holstein for her clerical support, Professor Luke Vale for his health economic advice and Jonathan Richardson and Laura Yates (UK Teratology Information Service) for their teratological advice. Finally, and most importantly, the authors would like to thank all of the women who participated in the study.
Contributions of authors
Professor Stephen Robson (https://orcid.org/0000-0001-7897-7987) was the lead applicant and made a substantial contribution to the conception, design and execution of the trial. He prepared the first and final draft of the report.
Dr Catherine McParlin (https://orcid.org/0000-0003-4144-8218) was a trial co-applicant and a member of the TMG. She made a substantial contribution to the design of the trial and the acquisition and interpretation of data and reviewed the report.
Ms Helen Mossop (https://orcid.org/0000-0002-6260-3734) was a member of the TMG and conducted the statistical analysis of the main clinical outcome measures, prepared the results for publication and contributed to the drafting and reviewing of the report.
Dr Mabel Lie (https://orcid.org/0000-0002-1703-283X) was a member of the TMG and conducted the qualitative evaluation. She contributed to the drafting and reviewing of the report.
Dr Cristina Fernandez-Garcia (https://orcid.org/0000-0002-7113-225X) wrote the health economics analysis plan, conducted the health economics analysis, interpreted the data and contributed to the drafting and reviewing of the report.
Ms Denise Howel (https://orcid.org/0000-0002-0033-548X) was a trial co-applicant and member of the TMG. She supervised the statistical analysis and contributed to the drafting and reviewing of the report.
Dr Ruth Graham (https://orcid.org/0000-0001-9432-760X) was a trial co-applicant and member of the TMG. She co-led on the qualitative component and contributed to the design, analysis and interpretation of qualitative data, and to the drafting and reviewing of the report.
Dr Laura Ternent (https://orcid.org/0000-0001-7056-298X) was a member of the TMG, contributed to the design of the health economic data collection tools, supervised the health economics research associate and contributed to data analysis and the drafting and reviewing of the report.
Dr Alison Steel (https://orcid.org/0000-0003-1279-1455) was a member of the TMG, contributed to protocol development, gave guidance on all aspects of governance and trial delivery and reviewed the final report.
Miss Nicola Goudie (https://orcid.org/0000-0003-1211-936X) was a member of the TMG, co-led the management of the trial and contributed to the drafting and reviewing of the report.
Ms Afnan Nadeem (https://orcid.org/0000-0002-2739-8130) was a member of the TMG, co-led on the management of the trial and reviewed the report.
Ms Julia Phillipson (https://orcid.org/0000-0002-1358-4223) was a member of the TMG and was responsible for the day-to-day management of the data and database and preparation and provision of data for analysis. She contributed to the drafting and reviewing of the report.
Dr Manjeet Shehmar (https://orcid.org/0000-0001-9390-1489) was a co-applicant, site PI and reviewed the report.
Mr Nigel Simpson (https://orcid.org/0000-0002-0758-7583) was a co-applicant, site PI and reviewed the report.
Professor Derek Tuffnell (https://orcid.org/0000-0001-7936-8876) was a trial co-applicant, site PI and reviewed the report.
Mr Ian Campbell (https://orcid.org/0000-0001-8002-6953) was a trial co-applicant, member of the TMG and provided advice on the design, packaging and labelling and storage of the IMP and dummy drugs. He also provided informal advice on research ethics.
Miss Rew Williams (https://orcid.org/0000-0002-9228-0972) was a trial co-applicant, contributed to the design of the study and reviewed the report.
Dr Margaret E O’Hara (https://orcid.org/0000-0001-9812-5183) was a trial co-applicant, contributed to the design of the study (in particular the patient information leaflets, main outcome measures and the health economic data collection tools) and reviewed the report.
Professor Elaine McColl (https://orcid.org/0000-0001-8300-3204) was a trial co-applicant, member of the TMG and reviewed the report.
Professor Catherine Nelson-Piercy (https://orcid.org/0000-0001-9311-1196) was a trial co-applicant, site PI and reviewed the final report.
Publication
An article titled ‘Emesis in Pregnancy – a qualitative study on trial recruitment failure’ has been submitted to BMC Pilot and Feasibility Studies for consideration.
MS: 8462574203716102.
Data-sharing statement
Anonymised data from this trial may be available to the scientific community subject to regulatory and ethics approval. Requests for data should be directed to the corresponding author.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Niebyl JR. Clinical practice. Nausea and vomiting in pregnancy. N Engl J Med 2010;363:1544-50. https://doi.org/10.1056/NEJMcp1003896.
- Jarvis S, Nelson-Piercy C. Management of nausea and vomiting in pregnancy. BMJ 2011;342. https://doi.org/10.1136/bmj.d3606.
- Einarson TR, Piwko C, Koren G. Quantifying the global rates of nausea and vomiting of pregnancy: a meta analysis. J Popul Ther Clin Pharmacol 2013;20:e171-83.
- Mazzotta P, Stewart DE, Koren G, Magee LA. Factors associated with elective termination of pregnancy among Canadian and American women with nausea and vomiting of pregnancy. J Psychosom Obstet Gynaecol 2001;22:7-12. https://doi.org/10.3109/01674820109049946.
- McCarthy FP, Khashan AS, North RA, Moss-Morris R, Baker PN, Dekker G, et al. A prospective cohort study investigating associations between hyperemesis gravidarum and cognitive, behavioural and emotional well-being in pregnancy. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0027678.
- Locock L, Alexander J, Rozmovits L. Women’s responses to nausea and vomiting in pregnancy. Midwifery 2008;24:143-52. https://doi.org/10.1016/j.midw.2006.12.001.
- Veenendaal MV, van Abeelen AF, Painter RC, van der Post JA, Roseboom TJ. Consequences of hyperemesis gravidarum for offspring: a systematic review and meta-analysis. BJOG 2011;118:1302-13. https://doi.org/10.1111/j.1471-0528.2011.03023.x.
- Gadsby R, Rawson V, Dziadulewicz E, Rousseau B, Collings H. Nausea and vomiting of pregnancy and resource implications: the NVP Impact Study. Br J Gen Pract 2019;69:e217-23. https://doi.org/10.3399/bjgp18X700745.
- NHS Digital . Hospital Episode Statistics, Admitted Patient Care, England – 2013–14 2015. https://digital.nhs.uk/data-and-information/publications/statistical/hospital-admitted-patient-care-activity/hospital-episode-statistics-admitted-patient-care-england-2013-14 (accessed 9 July 2020).
- Palmsten K, Hernández-Díaz S, Chambers CD, Mogun H, Lai S, Gilmer TP, et al. The most commonly dispensed prescription medications among pregnant women enrolled in the U.S. Medicaid program. Obstet Gynecol 2015;126:465-73. https://doi.org/10.1097/AOG.0000000000000982.
- Soltani H, Taylor GM. Changing attitudes and perceptions to hyperemesis gravidarum. RCM Midwives 2003;6:520-4.
- Royal College of Obstetricians and Gynaecologists . The Management of Nausea and Vomiting in Pregnancy and Hyperemesis Gravidarum 2016. www.rcog.org.uk/globalassets/documents/guidelines/green-top-guidelines/gtg69-hyperemesis.pdf (accessed 9 July 2020).
- O’Donnell A, McParlin C, Robson SC, Beyer F, Moloney E, Bryant A, et al. Treatments for hyperemesis gravidarum and nausea and vomiting in pregnancy: a systematic review and economic assessment. Health Technol Assess 2016;20. https://doi.org/10.3310/hta20740.
- Matthews A, Haas DM, O’Mathúna DP, Dowswell T. Interventions for nausea and vomiting in early pregnancy. Cochrane Database Syst Rev 2015;9. https://doi.org/10.1002/14651858.CD007575.pub4.
- Boelig RC, Barton SJ, Saccone G, Kelly AJ, Edwards SJ, Berghella V. Interventions for treating hyperemesis gravidarum. Cochrane Database Syst Rev 2016;5. https://doi.org/10.1002/14651858.CD010607.pub2.
- Tan PC, Khine PP, Vallikkannu N, Omar SZ. Promethazine compared with metoclopramide for hyperemesis gravidarum: a randomized controlled trial. Obstet Gynecol 2010;115:975-81. https://doi.org/10.1097/AOG.0b013e3181d99290.
- Kashifard M, Basirat Z, Kashifard M, Golsorkhtabar-Amiri M, Moghaddamnia A. Ondansetrone or metoclopromide? Which is more effective in severe nausea and vomiting of pregnancy? A randomized trial double-blind study. Clin Exp Obstet Gynecol 2013;40:127-30.
- Abas MN, Tan PC, Azmi N, Omar SZ. Ondansetron compared with metoclopramide for hyperemesis gravidarum: a randomized controlled trial. Obstet Gynecol 2014;123:1272-9. https://doi.org/10.1097/AOG.0000000000000242.
- Koren G, Piwko C, Ahn E, Boskovic R, Maltepe C, Einarson A, et al. Validation studies of the Pregnancy Unique-Quantification of Emesis (PUQE) scores. J Obstet Gynaecol 2005;25:241-4. https://doi.org/10.1080/01443610500060651.
- Lacasse A, Rey E, Ferreira E, Morin C, Berard A. Validity of a modified Pregnancy-Unique Quantification of Emesis and Nausea (PUQE) scoring index to assess severity of nausea and vomiting of pregnancy. Am J Obstet Gynecol 2008;198:e1-7. https://doi.org/10.1016/j.ajog.2007.05.051.
- Koren G, Boskovic R, Hard M, Maltepe C, Navioz Y, Einarson A. Motherisk-PUQE (pregnancy-unique quantification of emesis and nausea) scoring system for nausea and vomiting of pregnancy. Am J Obstet Gynecol 2002;186:228-31. https://doi.org/10.1067/mob.2002.123054.
- Lacasse A, Bérard A. Validation of the nausea and vomiting of pregnancy specific health related quality of life questionnaire. Health Qual Life Outcomes 2008;6. https://doi.org/10.1186/1477-7525-6-32.
- Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression: development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry 1987;150:782-6. https://doi.org/10.1192/bjp.150.6.782.
- Spielberger CD, Gorsuch RL, Lushene R, Vagg PR, Jacobs GA. Manual for the State Trait Anxiety Inventory. Palo Alto, CA: Consulting Psychologists Press; 1983.
- Webster J, Linnane JW, Dibley LM, Hinson JK, Starrenburg SE, Roberts JA. Measuring social support in pregnancy: can it be simple and meaningful?. Birth 2001;27:97-101. https://doi.org/10.1046/j.1523-536x.2000.00097.x.
- National Institutes of Health (NIH) . Common Terminology Criteria for Adverse Events (CTCAE) 2010. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf (accessed 9 July 2020).
- Sullivan CA, Johnson CA, Roach H, Martin RW, Stewart DK, Morrison JC. A pilot study of intravenous ondansetron for hyperemesis gravidarum. Am J Obstet Gynecol 1996;174:1565-8. https://doi.org/10.1016/S0002-9378(96)70607-5.
- Oliveira LG, Capp SM, You WB, Riffenburgh RH, Carstairs SD. Ondansetron compared with doxylamine and pyridoxine for treatment of nausea in pregnancy: a randomized controlled trial. Obstet Gynecol 2014;124:735-42. https://doi.org/10.1097/AOG.0000000000000479.
- McParlin C, Carrick-Sen D, Steen IN, Robson SC. Hyperemesis in pregnancy study: a pilot randomised controlled trial of midwife-led outpatient care. Eur J Obstet Gynecol Reprod Biol 2016;200:6-10. https://doi.org/10.1016/j.ejogrb.2016.02.016.
- Drummond MF, Sculpher MJ, Claxton K, Stoddart GL, Torrance GW. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2015.
- Charmaz K. Constructing Grounded Theory a Practical Guide Through Qualitative Analysis. London: SAGE Publications Ltd; 2006.
- Silverman D. Interpreting Qualitative Data: Methods for Analyzing Talk, Text and Interaction. London: SAGE Publications Ltd; 2006.
- Braun V, Clarke V. Using thematic analysis in psychology. Qual ResPsychol 2006;3:77-101. https://doi.org/10.1191/1478088706qp063oa.
- Smith J, Firth J. Qualitative data analysis: the framework approach. Nurse Res 2011;18:52-6. https://doi.org/10.7748/nr2011.01.18.2.52.c8284.
- Ritchie J, Spencer L, Bryman A, Burgess RG. Analyzing Qualitative Data. London: Routledge; 1994.
- Great Britain . Data Protection Act 1998 1998.
- Great Britain . Data Protection Act 2018 2018.
- Great Britain . National Health Service Act 2006 2006.
- Fiaschi L, Nelson-Piercy C, Deb S, King R, Tata LJ. Clinical management of nausea and vomiting in pregnancy and hyperemesis gravidarum across primary and secondary care: a population-based study. BJOG 2019;126:1201-11. https://doi.org/10.1111/1471-0528.15662.
- National Institute for Health Research (NIHR) . Definition of Feasibility Vs. Pilot Studies 2018. www.nihr.ac.uk/documents/nihr-research-for-patient-benefit-rfpb-programme-guidance-on-applying-for-feasibility-studies/20474 (accessed 9 July 2020).
- Fiaschi L, Nelson-Piercy C, Tata LJ. Hospital admission for hyperemesis gravidarum: a nationwide study of occurrence, reoccurrence and risk factors among 8.2 million pregnancies. Hum Reprod 2016;31:1675-84. https://doi.org/10.1093/humrep/dew128.
- NHS Digital . NHS Maternity Statistics, England 2017–18 2018. https://digital.nhs.uk/data-and-information/publications/statistical/nhs-maternity-statistics/2017-18 (accessed 9 July 2020).
- Trovik J, Vikanes AV. Antiemetics in hyperemesis gravidarum: unawareness or negligence?. BJOG 2019;126. https://doi.org/10.1111/1471-0528.15824.
- Nottinghamshire Area Prescribing Committee . Primary Care Management of Nausea and Vomiting In Early Pregnancy 2020. www.nottsapc.nhs.uk/media/1244/nausea-and-vomiting-in-early-pregnancy.pdf (accessed 9 July 2020).
- Havnen GC, Truong MB, Do MH, Heitmann K, Holst L, Nordeng H. Women’s perspectives on the management and consequences of hyperemesis gravidarum – a descriptive interview study. Scand J Prim Health Care 2019;37:30-4. https://doi.org/10.1080/02813432.2019.1569424.
- Skalley G, Denny J, Allen E, Rao S. Optimisation of hyperemesis gravidarum management through an emergency department setting. BMJ Open Qual 2018;7. https://doi.org/10.1136/bmjoq-2018-000330.
- Summers A. Emergency management of hyperemesis gravidarum. Emerg Nurse 2012;20:24-8. https://doi.org/10.7748/en2012.07.20.4.24.c9206.
- Bewley S. What inhibits obstetricians implementing reliable guidelines?. BJOG 2019;127.
- Chaillet N, Dubé E, Dugas M, Audibert F, Tourigny C, Fraser WD, et al. Evidence-based strategies for implementing guidelines in obstetrics: a systematic review. Obstet Gynecol 2006;108:1234-45. https://doi.org/10.1097/01.AOG.0000236434.74160.8b.
- Grol R, Grimshaw J. From best evidence to best practice: effective implementation of change in patients’ care. Lancet 2003;362:1225-30.
- Tuohy JF, Harding JE, Crowther CA, Bloomfield FH. Reported adherence to current antenatal corticosteroid guidelines in Australia and New Zealand. Aust N Z J Obstet Gynaecol 2019;59:416-21. https://doi.org/10.1111/ajo.12890.
- Fiaschi L, Housley G, Nelson-Piercy C, Gibson J, Raji A, Deb S, et al. Assessment of discharge treatment prescribed to women admitted to hospital for hyperemesis gravidarum. Int J Clin Pract 2019;73. https://doi.org/10.1111/ijcp.13261.
- Heitmann K, Holst L, Lupattelli A, Maltepe C, Nordeng H. Treatment of nausea in pregnancy: a cross-sectional multinational web-based study of pregnant women and new mothers. BMC Pregnancy Childbirth 2015;15. https://doi.org/10.1186/s12884-015-0746-2.
- Parker SE, Van Bennekom C, Anderka M, Mitchell AA. National Birth Defects Prevention Study . Ondansetron for treatment of nausea and vomiting of pregnancy and the risk of specific birth defects. Obstet Gynecol 2018;132:385-94. https://doi.org/10.1097/AOG.0000000000002679.
- Ditto A, Morgante G, la Marca A, De Leo V. Evaluation of treatment of hyperemesis gravidarum using parenteral fluid with or without diazepam. A randomized study. Gynecol Obstet Invest 1999;48:232-6. https://doi.org/10.1159/000010189.
- Wartolowska K, Collins GS, Hopewell S, Judge A, Dean BJ, Rombach I, et al. Feasibility of surgical randomised controlled trials with a placebo arm: a systematic review. BMJ Open 2016;6. https://doi.org/10.1136/bmjopen-2015-010194.
- Welton AJ, Vickers MR, Cooper JA, Meade TW, Marteau TM. Is recruitment more difficult with a placebo arm in randomised controlled trials? A quasirandomised, interview based study. BMJ 1999;318:1114-17. https://doi.org/10.1136/bmj.318.7191.1114.
- Chappell LC, Bell JL, Smith A, Linsell L, Juszczak E, Dixon PH, et al. Ursodeoxycholic acid versus placebo in women with intrahepatic cholestasis of pregnancy (PITCHES): a randomised controlled trial. Lancet 2019;394:849-60. https://doi.org/10.1016/S0140-6736(19)31270-X.
- Mattson ME, Curb JD, McArdle R. Participation in a clinical trial: the patients’ point of view. Control Clin Trials 1985;6:156-67. https://doi.org/10.1016/0197-2456(85)90121-7.
- Mitchell-Jones N, Farren JA, Tobias A, Bourne T, Bottomley C. Ambulatory versus inpatient management of severe nausea and vomiting of pregnancy: a randomised control trial with patient preference arm. BMJ Open 2017;7. https://doi.org/10.1136/bmjopen-2017-017566.
- Langley A, Lindberg K, Mork BE, Nicolini D, Raviola E, Walter L. Boundary work among groups, occupations and organizations: from cartography to process. Academy Management Annals 2019;13:704-36. https://doi.org/10.5465/annals.2017.0089.
- Lamont M, Molnar V. The study of boundaries in the social sciences. Annual Rev Sociol 2002;28:167-95.
- Koot MH, Boelig RC, Van’t Hooft J, Limpens J, Roseboom TJ, Painter RC, et al. Variation in hyperemesis gravidarum definition and outcome reporting in randomised clinical trials: a systematic review. BJOG 2018;125:1514-21. https://doi.org/10.1111/1471-0528.15272.
- Mercieca-Bebber R, King MT, Calvert MJ, Stockler MR, Friedlander M. The importance of patient-reported outcomes in clinical trials and strategies for future optimisation. Patient Relat Outcome Meas 2018;9:353-67. https://doi.org/10.2147/PROM.S156279.
- Heneghan C, Goldacre B, Mahtani KR. Why clinical trial outcomes fail to translate into benefits for patients. Trials 2017;18. https://doi.org/10.1186/s13063-017-1870-2.
- Jansen L, Koot MH, Van’t Hooft J, Dean CR, Duffy J, Ganzevoort W, et al. A core outcome set for hyperemesis gravidarum research: an international consensus study. BJOG 2020;127:983-92. https://doi.org/10.1111/1471-0528.16172.
- Morris ZH, Azab AN, Harlev S, Plakhy Y. Developing and validating a prognostic index predicting re-hospitalisation of patients with hyperemesis gravidarum. Eur J Obstet Gynecol Reprod Biol 2018;225:113-17. https://doi.org/10.1016/j.ejogrb.2018.04.028.
- Mangili G, Cioffi R, Danese S, Frigerio L, Ferrandina G, Cormio G, et al. Does methotrexate (MTX) dosing in a 8-day MTX/FA regimen for the treatment of low-risk gestational trophoblastic neoplasia affect outcomes? The MITO-9 study. Gynecol Oncol 2018;151:449-52. https://doi.org/10.1016/j.ygyno.2018.09.025.
- Fejzo MS, MacGibbon KW, Mullin PM. Ondansetron in pregnancy and risk of adverse fetal outcomes in the United States. Reprod Toxicol 2016;62:87-91. https://doi.org/10.1016/j.reprotox.2016.04.027.
- Matok I, Gorodischer R, Koren G, Sheiner E, Wiznitzer A, Levy A. The safety of metoclopramide use in the first trimester of pregnancy. N Engl J Med 2009;360:2528-35. https://doi.org/10.1056/NEJMoa0807154.
- Pasternak B, Svanström H, Mølgaard-Nielsen D, Melbye M, Hviid A. Metoclopramide in pregnancy and risk of major congenital malformations and fetal death. JAMA 2013;310:1601-11. https://doi.org/10.1001/jama.2013.278343.
- Carstairs SD. Ondansetron use in pregnancy and birth defects: a systematic review. Obstet Gynecol 2016;127:878-83. https://doi.org/10.1097/AOG.0000000000001388.
- Huybrechts KF, Hernández-Díaz S, Straub L, Gray KJ, Zhu Y, Patorno E, et al. Association of maternal first-trimester ondansetron use with cardiac malformations and oral clefts in offspring. JAMA 2018;320:2429-37. https://doi.org/10.1001/jama.2018.18307.
- Zambelli-Weiner A, Via C, Yuen M, Weiner DJ, Kirby RS. First trimester ondansetron exposure and risk of structural birth defects. Reprod Toxicol 2019;83:14-20. https://doi.org/10.1016/j.reprotox.2018.10.010.
- European Medicines Agency . PRAC Recommendations on Signals 2019. www.ema.europa.eu/en/documents/prac-recommendation/prac-recommendations-signals-adopted-8-11-july-2019-prac-meeting_en.pdf (accessed 9 July 2020).
- UK Teratology Information Service . EMA Pharmacovigilance Risk Assessment Committee (PRAC), Recommendations on Signals Adopted at the 8–11th July Meeting. Ondansetron; Signal of Birth Defects Following In-Utero Exposure During the First Trimester of Pregnancy Arising from Recent Publications 2019. www.uktis.org/docs/Ondansetron%20UKTIS%20Response%20Statement.pdf (accessed 9 July 2020).
- Lemon LS, Bodnar LM, Garrard W, Venkataramanan R, Platt RW, Marroquin OC, et al. Ondansetron use in the first trimester of pregnancy and the risk of neonatal ventricular septal defect. Int J Epidemiol 2020;49:648-56. https://doi.org/10.1093/ije/dyz255.
- Walters SJ, Bonacho Dos Anjos Henriques-Cadby I, Bortolami O, Flight L, Hind D, Jacques RM, et al. Recruitment and retention of participants in randomised controlled trials: a review of trials funded and published by the United Kingdom Health Technology Assessment Programme. BMJ Open 2017;7. https://doi.org/10.1136/bmjopen-2016-015276.
- Paramasivan S, Strong S, Wilson C, Campbell B, Blazeby JM, Donovan JL. A simple technique to identify key recruitment issues in randomised controlled trials: Q-QAT – Quanti-Qualitative Appointment Timing. Trials 2015;16. https://doi.org/10.1186/s13063-015-0617-1.
- Donovan JL, Rooshenas L, Jepson M, Elliott D, Wade J, Avery K, et al. Optimising recruitment and informed consent in randomised controlled trials: the development and implementation of the Quintet Recruitment Intervention (QRI). Trials 2016;17. https://doi.org/10.1186/s13063-016-1391-4.
- Rooshenas L, Scott LJ, Blazeby JM, Rogers CA, Tilling KM, Husbands S, et al. The QuinteT recruitment intervention supported five randomized trials to recruit to target: a mixed-methods evaluation. J Clin Epidemiol 2019;106:108-20. https://doi.org/10.1016/j.jclinepi.2018.10.004.
- Herbert E, Julios SA, Goodacre. Progression criteria in trials with an internal pilot: an audit of publically funded randomised controlled trials. Trials 2019;20. https://doi.org/10.1186/s13063-019-3578-y.
- Mohanna K, Tunna K. Withholding consent to participate in clinical trials: decisions of pregnant women. Br J Obstet Gynaecol 1999;106:892-7. https://doi.org/10.1111/j.1471-0528.1999.tb08426.x.
- Newington L, Metcalfe A. Factors influencing recruitment to research: qualitative study of the experiences and perceptions of research teams. BMC Med Res Methodol 2014;14. https://doi.org/10.1186/1471-2288-14-10.
- Huang GD, Bull J, Johnston McKee K, Mahon E, Harper B, Roberts JN. CTTI Recruitment Project Team . Clinical trials recruitment planning: a proposed framework from the Clinical Trials Transformation Initiative. Contemp Clin Trials 2018;66:74-9. https://doi.org/10.1016/j.cct.2018.01.003.
- Latibeaudiere M, Phillips J, Siassakos D, Winter C, Draycott T, Fox R. Implementation strategies – moving guidance into practice. Obstet Gynaecol 2013;15:51-7. https://doi.org/10.1111/j.1744-4667.2012.00141.x.
Appendix 1 Pregnancy Unique Quantification of Emesis questionnaire
Thinking about the last 24 hours, please rate how you have been by putting a tick in the relevant box.
| Pregnancy Unique Quantification of Emesis score | |||||
|---|---|---|---|---|---|
| 1. In the last 24 hours how long have you felt nauseated or sick to your stomach? | Not at all (1) | 1 hour or less (2) | 2–3 hours (3) | 4–6 hours (4) | More than 6 hours (5) |
| 2. In the last 24 hours have you vomited or thrown up? | 7 or more times (5) | 5–6 times (4) | 3–4 times (3) | 1–2 times (2) | I did not throw up (1) |
| 3. In the last 24 hours how many times have you had retching or dry heaves without bringing anything up? | No time (1) | 1–2 times (2) | 3–4 times (3) | 5–6 times (4) | 7 times or more (5) |
Total score______________
Appendix 2 Nausea and vomiting in pregnancy, quality-of-life questionnaire
This questionnaire has 30 questions and it has been designed to find out how you have been feeling during the last week. You will be asked about your symptoms related to your nausea and vomiting in pregnancy, how you have been affected in doing activities and how your mood has been. Please complete all of the questions and select only one response for each question.
| 1. How often did you have nausea in the last week? | ||||||
|---|---|---|---|---|---|---|
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 2. How often did you have vomiting in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 3. How often did you have dry heaves in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 4. How often did you experience sickness to your stomach in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 5. How often did it take you longer to get things done than usual as a result of your nausea and vomiting in pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 6. How often in the past week have you had difficulty or you have been limited or it has taken you extra effort to perform work and other activities as a result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 7. How often have you felt downhearted or blue as a result of your nausea and vomiting of pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 8. How often in the past week did you feel worn out and had lack of energy as a result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 9. How often in the past week did you have poor appetite as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 10. How often in the past week have you had difficulty maintaining your normal social activities with family, friends, neighbours or social groups as a result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 11. How often in the past week did you experience nausea and vomiting in pregnancy in the evening? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 12. How often have you felt frustrated as a result of your nausea and vomiting of pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 13. How often did you feel exhaustion as a result of your nausea and vomiting of pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 14. How often as result of your nausea and vomiting in pregnancy, in the past week have you had to rely on your partner to do things you would normally do for your family? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 15. How often in the past week have you felt fed up with being sick as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 16. How often in the past week have you had difficulty looking after the home as a result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 17. How often have you had difficulty shopping for food in the past week as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 18. How often did you feel tiredness as a result of your nausea and vomiting in pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 19. How often in the past week did you not eat for Ionger than you would like as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 20. How often in the past week did you feel reassured that your symptoms are part of normal pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 21. How often did you feel less interested in sex in the past week as a result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 22. How often did you feel fatigue as a result of your nausea and vomiting in pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 23. How often have you felt emotional as a result of your nausea and vomiting in pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 24, How often in the past week have you felt that you have accomplished less than you would like as a result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 25. How often have you cut down on the amount of time you spent at work or other activities in the past week as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 26. How often in the past week did you experience nausea and vomiting from being exposed to certain smells? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 27. How often in the past week have you felt that everything is an effort as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 28. How often have you felt that you cannot enjoy your pregnancy as a result of your nausea and vomiting in pregnancy in the past week? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 29. How often in the past week did you experience nausea and vomiting from being exposed to certain foods? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
| 30. How often in the past week have you had difficulty preparing or cooking meals as result of your nausea and vomiting in pregnancy? | ||||||
| 1. None of the time | 2. Hardly any of the time | 3. A little of the time | 4. Some of the time | 5. A good bit of the time | 6. Most of the time | 7. All of the time |
Appendix 3 Edinburgh Postnatal Depression Scale questionnaire
The Edinburgh Postnatal Depression Scale questionnaire has been reproduced with permission from Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression: development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry 1987;150:782–6.
Appendix 4 Maternity Social Support Scale
The Maternity Social Support Scale has been reproduced with permission from Webster J, Linnane JW, Dibley LM, Hinson JK, Starrenburg SE, Roberts JA. Measuring social support in pregnancy: can it be simple and meaningful? Birth 2001;27:97–101.
Appendix 5 Health-care utilisation questionnaire
Appendix 6 Willingness-to-pay questionnaire
Appendix 7 Interview guide
EMPOWER – telephone/e-mail interview topic guide – qualitative substudy.
Preamble:
-
Special thanks to participants for agreeing to take part in the research. Explain that one of the aims of the study is to understand better how participants experience the process of taking part in the trial. (For those not taking part, this includes understanding better why they decided not to take part at all.)
-
Make the point that they do not have to answer all questions.
-
Interviewer introduces self, outlines the substudy and explains that they will receive a summary of the results if they would wish to have one (at the end of the study).
-
(For telephone interviews.) Explain use of the audio-recorder – the interview is being audio-recorded so I have an accurate account of what participant has said and so the researcher doesn’t have to take handwritten notes. Interviews will be anonymised when they are typed up prior to analysis (i.e. their names and any other information that could identify them) are taken out. (For e-mail interviews, explain that the full e-mail correspondence will be used as the transcript. Explain that the e-mail interview would normally involve no more than five cycles of question response.)
-
Assure confidentiality. No information will be given to your GP without permission.
-
Ask whether they have any more questions about study?
-
Ask participant to sign their copy of the consent form. Make a note on researcher copy of consent form that consent form has been signed.
-
Explain that the interview can be ended or postponed at any time, and that this would not affect their care in anyway.
THEN MOVE TO MAIN QUESTION GRID – begin with the introductory questions in top left corner of grid – follow the direction of travel from participant’s response, i.e. (i) moving from left to right through the grid, to discuss key factors, or (ii) moving from top to bottom through the grid, to discuss reasons for taking part/not taking part.
| INTRODUCTORY QUESTIONS | Issues related to the patient | Issues related to the recruiting staff | Issues relating to the study design |
|---|---|---|---|
|
We are interested in your views about being invited to take part in the EMPOWER trial What did you think about when you were first asked to take part in the trial? |
Initial question: why did you/did not you want to take part in the study? | Initial question: did the way in which you were invited to part affect your decision? | Initial question: was there anything about the study itself that made you want/not want to take part? |
|
Positive reasons for recruitment Initial question: what factors made you want to take part in the study? |
|
|
|
|
Negative reasons for recruitment Initial question: what factors made you NOT want to take part in the study? |
|
|
|
| FOLLOW-UP QUESTIONS (if needed) | |||
| Could be either positive or negative for recruitment |
|
|
|
| MOP-UP QUESTIONS | |||
|
Could the researchers do anything else to make it easier for people to make their decision about whether to participant? What is the single biggest thing that made you want/not want to take part? Looking back on the experience now, would you make the decision? Why/why not? Is there anything else you want to tell us about your experiences? |
|||
|
Close interview: Thank you for agreeing to take part in the interview Confirm that it is still ok for the researchers to use the interview in the analysis Confirm arrangements for participant to send in the signed consent form Ask if the participant would like to receive a copy of the transcript Ask if the participant would like to receive a report on the outcomes at the end of the study |
|||
Appendix 8 Process evaluation topic guide for interviews with clinicians
Qualitative process evaluation
Topic guide for interviews with clinicians
Interviewer outlines the purpose of the interview, which is to identify problems of recruitment into the trial.
Interviewer explains that the participant will receive a copy of the interview transcript and a summary of the results if they would wish to have one:
-
Explain use of the audio-recorder – the interview is being audio-recorded to provide an accurate account of what the participant has said.
-
Explain that interviews will be anonymised when they are typed up prior to analysis (i.e. names and any other information that could identify them are taken out and replaced with pseudonyms or anonymous identifiers).
-
Assure confidentiality.
-
Ask whether they have any more questions about study?
-
Go through and sign consent form.
The semistructured interview approach adopted in this study focuses on encouraging the participant to explain the issues that they feel are relevant in an order that makes most sense to them. Apart from the introduction and conclusion, the questions listed below therefore act as an aide memoire rather than a definitive list of questions that are asked in a particular order.
Introduction
-
Can I just ask some background questions if you don’t mind? How long have you been in your present post?
-
Can you tell me about your experience of being involved in clinical trials?
-
What has your experience been of the set-up of the EMPOWER trial?
-
How does it compare with your previous experience of other trials?
-
Can you describe how you go about recruiting patients to this trial? (MAU, EPA unit, A&E department, pathway.)
-
What do you see are the main difficulties of recruiting patients in this trial?
-
Are there any improvements that you feel could be made to the trial processes?
-
What is your impression of patients’ understandings of a) the different treatment arms and b) being randomised in a trial?
-
How has the progress of the trial at your site been so far?
-
Have you had any feedback from patients about the trial?
-
Are there any particular cases/events you would like to highlight to the trial investigators?
-
Do you have anything else you would like to add?
Appendix 9 Additional baseline demographics
| Demographic | Treatment group | Overall (N = 33) | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Weight at booking (kg), median (minimum, maximum); n with data | 74 (45, 80); 7 | 79 (50, 113); 7 | 70 (60, 81); 7 | 67 (55, 108); 7 | 73 (45, 113); 28 |
| Highest education level, n (%) | |||||
| GCSEs or equivalent | 2 (25) | 3 (38) | 1 (11) | 3 (38) | 9 (27) |
| A level or equivalent | 1 (13) | 1 (13) | 1 (11) | 2 (25) | 5 (15) |
| Undergraduate | 2 (25) | 2 (25) | 2 (22) | 2 (25) | 8 (24) |
| Postgraduate | 1 (13) | 0 (0) | 1 (11) | 1 (13) | 3 (9) |
| NVQ or equivalent | 2 (25) | 1 (13) | 2 (22) | 0 (0) | 5 (15) |
| None of the above | 0 (0) | 0 (0) | 1 (11) | 0 (0) | 1 (3) |
| Not stated | 0 (0) | 1 (13) | 1 (11) | 0 (0) | 2 (6) |
| Employment, n (%) | |||||
| Full time | 3 (38) | 2 (25) | 3 (33) | 4 (50) | 12 (37) |
| Part time | 1 (13) | 2 (25) | 2 (22) | 0 (0) | 5 (15) |
| Unemployed | 4 (50) | 3 (38) | 3 (33) | 2 (25) | 12 (36) |
| Self-employed | 0 (0) | 1 (13) | 1 (11) | 0 (0) | 2 (6) |
| Student | 0 (0) | 0 (0) | 0 (0) | 2 (25) | 2 (6) |
| Marital status, n (%) | |||||
| Married | 3 (38) | 4 (50) | 2 (22) | 1 (13) | 10 (30) |
| Single | 1 (13) | 0 (0) | 0 (0) | 1 (13) | 2 (6) |
| Civil partnership | 0 (0) | 0 (0) | 0 (0) | 2 (25) | 2 (6) |
| Stable union | 4 (50) | 3 (38) | 6 (67) | 4 (50) | 17 (52) |
| Not stated | 0 (0) | 1 (13) | 1 (11) | 0 (0) | 2 (6) |
| Smoking, n (%) | |||||
| Never smoked | 6 (75) | 6 (75) | 4 (44) | 7 (88) | 23 (70) |
| Quit before pregnancy | 0 (0) | 1 (13) | 3 (33) | 0 (0) | 4 (12) |
| Quit during/owing to pregnancy | 1 (13) | 1 (13) | 0 (0) | 0 (0) | 2 (6) |
| Current smoker: < 5 times per day | 0 (0) | 0 (0) | 1 (11) | 1 (13) | 2 (6) |
| Current smoker: 5–10 times per day | 0 (0) | 0 (0) | 1 (11) | 0 (0) | 1 (3) |
| Not stated | 1 (13) | 0 (0) | 0 (0) | 0 (0) | 1 (3) |
| Alcohol use, n (%) | |||||
| Never drink | 2 (25) | 5 (63) | 3 (33) | 3 (38) | 13 (39) |
| Quit during/owing to pregnancy | 6 (75) | 3 (38) | 6 (67) | 5 (63) | 20 (61) |
| Illicit drug use, n (%) | |||||
| No | 8 (100) | 8 (100) | 9 (100) | 7 (88) | 32 (97) |
| Missing/not available | 0 (0) | 0 (0) | 0 (0) | 1 (13) | 1 (3) |
| Caring responsibilities for an adult or child, n (%) | |||||
| Yes | 4 (50) | 5 (63) | 4 (44) | 2 (25) | 15 (45) |
| No | 4 (50) | 2 (25) | 4 (44) | 6 (75) | 16 (48) |
| Not stated | 0 (0) | 1 (13) | 1 (11) | 0 (0) | 2 (6) |
| If yes, had help since symptoms started | |||||
| Yes | 4 (100) | 4 (80) | 3 (75) | 2 (100) | 13 (87) |
| Not stated | 0 (0) | 1 (20) | 1 (25) | 0 (0) | 2 (13) |
| Mother suffered from HG, n (%) | |||||
| Yes | 0 (0) | 2 (25) | 1 (11) | 2 (25) | 5 (15) |
| No | 4 (50) | 4 (50) | 5 (56) | 4 (50) | 17 (52) |
| Do not know | 4 (50) | 2 (25) | 3 (33) | 2 (25) | 11 (33) |
| Any sister who has been pregnant, n (%) | |||||
| Yes | 4 (50) | 1 (12.5) | 3 (33) | 2 (25) | 10 (30) |
| No | 2 (25) | 6 (75) | 1 (11) | 2 (25) | 11 (33) |
| No sister | 2 (25) | 1 (12.5) | 3 (33) | 4 (50) | 10 (30) |
| Do not know | 0 (0) | 0 (0) | 2 (22) | 0 (0) | 2 (6) |
| If yes, suffered from HG | |||||
| Yes | 2 (50) | 1 (100) | 1 (33) | 1 (50) | 5 (50) |
| No | 1 (25) | 0 (0) | 2 (67) | 1 (50) | 4 (40) |
| Do not know | 1 (25) | 0 (0) | 0 (0) | 0 (0) | 1 (10) |
| MSSS total score, median (minimum, maximum); n with data | 29 (25, 30); 8 | 29 (27, 30); 7 | 30 (27, 30); 8 | 29 (25, 30); 7 | 29 (25, 30); 30 |
Appendix 10 Participant-reported symptom scores over time
| Treatment group | Overall (N = 33) | ||||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| PUQE score, median (minimum, maximum); n with data | |||||
| Baseline | 11 (8, 15); 6 | 14 (10, 15); 8 | 13 (10, 15); 9 | 14 (7, 15); 7 | 13 (7, 15); 30 |
| Day 2 | 9 (3, 13); 6 | 5 (3, 13); 6 | 7 (3, 11); 7 | 8 (3, 12); 5 | 7 (3, 13); 24 |
| Day 5 | 7 (5, 8); 5 | 7 (3, 13); 6 | 7 (3, 11); 7 | 8 (4, 15); 4 | 7 (3, 15); 22 |
| Day 10 | 7 (6, 9); 3 | 8 (3, 14); 5 | 5 (3, 9); 5 | 6 (3, 9); 2 | 7 (3, 14); 15 |
| PUQE severity, n (%) | |||||
| Baseline | |||||
| Mild (3–6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Moderate (7–12) | 4 (67) | 3 (38) | 3 (33) | 2 (29) | 12 (40) |
| Severe (≥ 13) | 2 (33) | 5 (63) | 6 (67) | 5 (71) | 18 (60) |
| Day 2 | |||||
| Mild (3–6) | 2 (33) | 4 (67) | 3 (43) | 2 (40) | 11 (46) |
| Moderate (7–12) | 3 (50) | 1 (17) | 4 (57) | 3 (60) | 11 (46) |
| Severe (≥ 13) | 1 (17) | 1 (17) | 0 (0) | 0 (0) | 2 (8) |
| Day 5 | |||||
| Mild (3–6) | 2 (40) | 3 (50) | 2 (29) | 2 (50) | 9 (41) |
| Moderate (7–12) | 3 (60) | 2 (33) | 5 (71) | 1 (25) | 11 (50) |
| Severe (≥ 13) | 0 (0) | 1 (17) | 0 (0) | 1 (25) | 2 (9) |
| Day 10 | |||||
| Mild (3–6) | 1 (33) | 2 (40) | 3 (60) | 1 (50) | 7 (47) |
| Moderate (7–12) | 2 (67) | 2 (40) | 2 (40) | 1 (50) | 7 (47) |
| Severe (≥ 13) | 0 (0) | 1 (20) | 0 (0) | 0 (0) | 1 (7) |
| VAS for nausea, median (minimum, maximum); n with data | |||||
| Day 2 | 6 (0, 9); 6 | 0 (0, 8); 7 | 3 (0, 8); 7 | 5 (0, 6); 5 | 5 (0, 9); 25 |
| Day 5 | 5 (1, 5); 5 | 5 (0, 9); 6 | 6 (0, 10); 7 | 4 (2, 7); 4 | 5 (0, 10); 22 |
| Day 10 | 4 (3, 8); 3 | 5 (0, 7); 5 | 1 (0, 9); 5 | 2 (0, 3); 2 | 3 (0, 9); 15 |
| NVPQoL questionnaire, median (minimum, maximum); n with data | |||||
| Baseline | 178 (165, 189); 8 | 186 (168, 203); 7 | 186 (136, 205); 8 | 182 (132, 206); 7 | 186 (132, 206); 30 |
| Day 10 | 148 (88, 175); 3 | 160 (88, 196); 4 | 155 (40, 170); 5 | 105 (81, 128); 2 | 148 (40, 196); 14 |
Appendix 11 Participant-reported symptoms of depression and anxiety over time
| Scale | Treatment group, median (minimum, maximum); n with data | Overall (N = 33), median (minimum, maximum); n with data | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| EPDS | |||||
| Baseline | 14 (4, 21); 8 | 17 (4, 26); 8 | 14 (3, 20); 9 | 10 (5, 17); 7 | 15 (3, 26); 32 |
| Day 10 | 12 (11, 22); 3 | 14 (3, 18); 5 | 13 (10, 22); 5 | 8 (5, 11); 2 | 13 (3, 22); 15 |
| STAI | |||||
| Baseline | 17 (9, 20); 8 | 15 (12, 23); 7 | 14 (6, 24); 9 | 13 (8, 19); 7 | 15 (6, 24); 31 |
| Day 10 | 15 (13, 21); 3 | 16 (6, 24); 5 | 15 (6, 22); 6 | 9 (6, 12); 2 | 15 (6, 24); 16 |
Appendix 12 Clinical indicators of antiemetic effectiveness categorising Pregnancy Unique Quantification of Emesis questionnaire score as relapse or remission
| Clinical indicator | Treatment group, n (%) | Overall (N = 33), n (%) | |||
|---|---|---|---|---|---|
| Active metoclopramide and dummy ondansetron (N = 8) | Active ondansetron and dummy metoclopramide (N = 8) | Active metoclopramide and active ondansetron (N = 9) | Dummy metoclopramide and dummy ondansetron (N = 8) | ||
| Relapse at day 5a | |||||
| Number assessable | 5 (63) | 6 (75) | 7 (78) | 4 (50) | 22 (67) |
| No | 5 (100) | 6 (100) | 7 (100) | 4 (100) | 22 (100) |
| Relapse at day 10b | |||||
| Number assessable | 3 (38) | 5 (75) | 5 (56) | 2 (25) | 15 (45) |
| No | 3 (100) | 5 (100) | 5 (100) | 2 (100) | 15 (100) |
| Remission at day 10c | |||||
| Number assessable | 3 (38) | 5 (75) | 5 (56) | 2 (25) | 15 (45) |
| Yes | 0 (0) | 1 (20) | 1 (20) | 0 (0) | 2 (13) |
| No | 3 (100) | 4 (80) | 4 (80) | 2 (100) | 13 (87) |
Appendix 13 Hurdles to and enablers of recruitment
| Topic | Hurdles |
|---|---|
| Requirements of the clinical trial | Adherence to the RCOG guidelines12 |
| Second-line treatment adopted as normal practice | |
| Trial question out of step with current practice and not having a high enough profile with clinicians | |
| The liaison between maternity and gynaecology departments needed extra work | |
| CTU | Advice not available when required |
| Not available out of hours | |
| Data entry error | |
| Demands of a CTIMP protocol | Consent process takes time |
| GCP-trained doctor needs to consent and prescribe | |
| Waiting time for eligibility | |
| 12 hours to see if the IMP really does not work | |
| Publicity and education about the trial | GPs and community midwifery |
| A&E department staff | |
| Clinical staff on duty and out of hours | |
| Junior doctors have to buy in | |
| Not all aware of the trial | |
| Trial staff roles | Lacking confidence and motivation |
| Recruitment and trial paperwork demanding | |
| Databases not user-friendly | |
| Patient counselling and empathy | |
| Staffing levels and support | |
| Lack of confidence and familiarity | |
| Women presenting | Out of hours |
| Infrequently | |
| Not had first-line treatment or not had it for long enough | |
| Women ineligible | Had not failed first-line treatment |
| Had trial drugs | |
| After amendment, had trial drugs by i.v. | |
| On antidepressant medication or other | |
| Language ability | |
| Women eligible | Preferring previously known drug |
| Declining dummy | |
| So unwell | |
| Cannot read and understand | |
| A lot of information | |
| Needing a quick, definite solution | |
| Preferring not to take medication because of possible risk | |
| Perceived research burden |
| Topic | Enabler |
|---|---|
| Requirements of the clinical trial | Adherence to the RCOG guidelines12 |
| Trial question was seen as relevant | |
| Good staff relations and communication | |
| CTU | Staff available and responsive |
| Good liaison | |
| Demands of a CTIMP protocol | Counselling takes time, recruitment less so |
| GCP training was facilitated | |
| Waiting time for eligibility | |
| 12 hours to see if the IMP really does not work: was acceptable | |
| Publicity and education about the trial | GPs |
| A&E department staff | |
| Clinical staff on duty and out of hours | |
| Getting staff on board | |
| Engaging non-clinical staff | |
| Trial staff roles | Confidence and morale |
| Good teamwork and support for recruitment | |
| Patient counselling and empathy | |
| Staffing levels and workload | |
| Confidence and familiarity | |
| Women presenting | Out of hours |
| Frequently | |
| Had first-line treatment | |
| Women ineligible | Had not failed first-line treatment |
| On antidepressant medication | |
| Language ability | |
| Women eligible | Counselling about the risks of a double-blind trial |
| The experience of severe NVP | |
| Reading and understanding information | |
| Information | |
| Want a quick, definite solution | |
| Preferring not to take medication | |
| Extra care and attention as research participants |
List of abbreviations
- 5-HT
- 5-hydroxytryptamine
- A&E
- accident and emergency
- AE
- adverse event
- Apgar
- Appearance, Pulse, Grimace, Activity, Respiration
- AR
- adverse reaction
- aRR
- adjusted relative risk
- AU
- assessment unit
- CI
- confidence interval
- CONSORT
- Consolidated Standard of Reporting Trials
- CTCAE
- Common Terminology Criteria for Adverse Events
- CTIMP
- Clinical Trial of an Investigational Medicinal Product
- CV
- contingent valuation
- eCRF
- electronic case report form
- EMA
- European Medicines Agency
- EMPOWER
- EMesis in Pregnancy – Ondansetron With mEtoClopRamide
- EOI
- expression of interest
- EPDS
- Edinburgh Postnatal Depression Scale
- GAU
- gynaecological assessment unit
- GCP
- good clinical practice
- GMC
- General Medical Council
- GP
- general practitioner
- HG
- hyperemesis gravidarum
- HRA
- Health Research Authority
- HTA
- Health Technology Assessment
- ID
- identification
- iDMC
- independent Data Monitoring Committee
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ISRCTN
- International Standard Randomised Controlled Trials Number
- i.v.
- intravenous
- MAC
- maternity assessment centre
- MAU
- maternity assessment unit
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MSSS
- Maternity Social Support Scale
- NCTU
- Newcastle Clinical Trials Unit
- NVP
- nausea and vomiting in pregnancy
- NVPQoL
- nausea and vomiting during pregnancy quality of life
- O&G
- obstetrics and gynaecology
- OR
- odds ratio
- PI
- principal investigator
- PIS
- participant information sheet
- p.o.
- per os
- PUQE
- Pregnancy Unique Quantification of Emesis
- QoL
- quality of life
- QRI
- QuinteT Recruitment Intervention
- RCOG
- Royal College of Obstetricians and Gynaecologists
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SAR
- serious adverse reaction
- SD
- standard deviation
- SSRI
- selective serotonin reuptake inhibitor
- STAI
- State–Trait Anxiety Inventory
- SUSAR
- suspected unexpected serious adverse reaction
- TMG
- Trial Management Group
- TOP
- termination of pregnancy
- TSC
- Trial Steering Committee
- UKTIS
- UK Teratology Information Service
- VAS
- visual analogue scale
- WTP
- willingness to pay